UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
(Mark One)
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2007.
Or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to .
Commission File Number 000-51171
COMBINATORX, INCORPORATED
(Exact name of registrant as specified in its charter)
| | |
| Delaware | | 04-3514457 |
(State or other jurisdiction of incorporation or organization) | | (IRS Employer Identification Number) |
| 245 First Street | | |
| Sixteenth Floor | | |
| Cambridge, Massachusetts | | 02142 |
| (Address of Principal Executive Offices) | | (Zip Code) |
Registrant’s telephone number, including area code: (617) 301-7000
Securities registered pursuant to Section 12(b) of the Exchange Act:
| | |
Title of Each Class | | Name of Exchange on Which Registered |
| Common Stock, par value $0.001 | | The Nasdaq Global Market |
Securities registered pursuant to Section 12(g) of the Exchange Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Note—Checking the box above will not relieve any registrant required to file reports pursuant to Section 13 or Section 15(d) of the Act from their obligations under those Sections.
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer”, “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. Check One:
| | |
| Large Accelerated Filer: ¨ | | Accelerated Filer: x |
| Non-Accelerated Filer: ¨ | | Smaller Reporting Company: ¨ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). ¨
The aggregate market value of voting common equity of the registrant held by non-affiliates of the registrant was approximately $142,890,209, on June 29, 2007. For purposes of the foregoing sentence, the term “affiliate” includes each director and executive officer of the registrant and each holder of more than 5% of the registrant’s common stock. The computation of the aggregate market value is based upon the closing price of the common stock as reported on the NASDAQ Global Market on June 29, 2007.
As of March 12, 2008, the registrant had 34,844,351 shares of common stock, par value $0.001 per share, outstanding.
Specified portions of the registrant’s definitive Proxy Statement relating to the registrant’s Annual Meeting of Stockholders to be held on May 29, 2008, which is to be filed pursuant to Regulation 14A within 120 days after the end of the registrant’s fiscal year ended December 31, 2007 are incorporated by reference in Part III of this Annual Report on Form 10-K.
COMBINATORX, INCORPORATED
ANNUAL REPORT
ON FORM 10-K
INDEX
2
PART I
FORWARD-LOOKING STATEMENTS
This annual report on Form 10-K contains forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995 that involve risks and uncertainties, as well as assumptions that, if they never materialize or prove incorrect, could cause the results of CombinatoRx to differ materially from those expressed or implied by such forward-looking statements. All statements other than statements of historical fact are statements that could be deemed forward-looking statements, including any projections of financing needs, revenue, expenses, earnings or losses from operations, or other financial items; any statements of the plans, strategies and objectives of management for future operations; any statements concerning product candidate research, development and commercialization plans and timelines; any statements regarding safety and efficacy of product candidates, any statements of expectation or belief; and any statements of assumptions underlying any of the foregoing. In addition, forward-looking statements may contain the words “believe,” “anticipate,” “expect,” “estimate,” “intend,” “plan,” “project,” “will be,” will continue,” “will result,” “seek,” “could,” “may,” “might,” or any variations of such words or other words with similar meanings.
The risks, uncertainties and assumptions referred to above include risks that are described in “Business—Risk Factors That May Affect Future Results” and elsewhere in this annual report and that are otherwise described from time to time in our Securities and Exchange Commission reports filed after this report.
The forward-looking statements included in this annual report represent our estimates as of the date of this annual report. We specifically disclaim any obligation to update these forward-looking statements in the future. These forward-looking statements should not be relied upon as representing our estimates or views as of any date subsequent to the date of this annual report.
Overview
We are a biopharmaceutical company pioneering the new field of synergistic combination pharmaceuticals, with a broad portfolio of product candidates in phase 2 clinical development. Going beyond traditional combinations, we create product candidates with novel mechanisms of action striking at the biological complexities of human disease. The lead programs in our portfolio are advancing into later stage clinical trials based on the strength of multiple positive phase 2a results. This portfolio is internally generated from our proprietary combination high throughput screening, or cHTSTM technology, which provides a renewable and previously untapped source of novel drug candidates. We are currently developing a clinical portfolio of product candidates targeting multiple diseases including immuno-inflammatory diseases, topical dermatoses and metabolic disease. We also have a broad pipeline of preclinical product candidates in development for immuno-inflammatory diseases, topical dermatology, infectious diseases, oncology, neurodegenerative diseases, medical devices, ophthalmic conditions and inherited diseases.
We are developing our combination drugs in response to the understanding that many diseases affect the body through multiple biological pathways. Traditional drug discovery has focused on agents that target a single biological pathway. However, the activity of a therapeutic compound against a single pathway can be insufficiently effective because biological systems often compensate by using a secondary pathway. We believe that by targeting multiple pathways, our combination drug candidates may create synergistic therapeutic effects, which could result in improved treatments for many diseases.
We use our cHTS technology to systematically screen pair-wise combinations from our library, which includes over 2,000 drugs approved in the United States, Europe and Japan, selected development-stage small molecules, mechanistic probes and biologics in cell-based assays corresponding to major diseases such as cancer, rheumatoid arthritis, psoriasis, hepatitis C, cystic fibrosis, neurodegenerative diseases and diabetes. Using these
3
cell-based assays, our cHTS technology screens the effects of millions of possible dose-specific combinations of existing and development-stage drugs and small molecules in each of our selected disease models.
We have discovered pairs of approved drugs which, in preclinical studies, exhibit a combination therapeutic effect against a model for a target disease, even though neither drug is indicated for such disease on its own. We have also discovered pairs of drugs where our preclinical studies suggest the effectiveness, safety or tolerability of one drug in its primary disease indication may be improved by combining it with another drug that is not indicated for that disease.
For example, our lead product candidate, CRx-102, a novel dissociated glucocorticoid product candidate in phase 2b clinical development for osteoarthritis and rheumatoid arthritis, was discovered using our cHTS technology. CRx-102 combines the cardiovascular agent dipyridamole with a very low dose of the glucocorticoid prednisolone. CRx-102 works through a novel multi-target mechanism of action in which dipyridamole selectively amplifies prednisolone’s anti-inflammatory and immunomodulatory activities without an associated increase in its side effects. In phase 2 proof-of-concept clinical trials, CRx-102 demonstrated a powerful anti-inflammatory effect and rapid onset of action in patients with osteoarthritis and rheumatoid arthritis, and was generally well tolerated. CRx-102 is being developed in a uniquely engineered commercial formulation for the treatment of rheumatoid arthritis and osteoarthritis.
In addition to CRx-102, we are currently advancing three other product candidates, CRx-191, CRx-197 and CRx-401, into or through clinical research and development. We also have two other clinical product candidates in our portfolio, CRx-139 for immuno-inflammatory diseases and CRx-170 for chronic pain, which have completed phase 2a clinical trials but are not currently being advanced into later-stage clinical trials, and we have a number of product candidates in preclinical development. Our product candidate CRx-191 is a topical synergistic combination drug candidate with a novel multi-target mechanism that inhibits TNF-a and interferon-gamma, key cell mediators of inflammation. CRx-191 contains a mid-potency glucocorticoid, mometasone, and a low dose of the tricyclic anti-depressant, nortriptyline, co-formulated as a topical cream for the treatment of psoriasis and other steroid-responsive dermatoses. We have conducted a phase 2a clinical trial of CRx-191 in subjects with psoriasis. Our product candidate CRx-197 is a selective cytokine modulator containing low concentrations of the antihistamine loratadine and the tricyclic anti-depressant nortriptyline, neither of which is approved for the treatment of topical dermatoses. This combination has been co-formulated as a topical cream for the treatment of atopic dermatitis, psoriasis and other inflammatory dermatoses. We are currently planning to conduct a phase 2a clinical trial of CRx-197 in subjects with select inflammatory dermatoses during 2008. Our product candidate CRx-401 is a novel oral anti-diabetic in development to treat Type 2 diabetes. CRx-401 is a synergistic combination containing sustained-release bezafibrate, an anti-cholesterol agent approved outside the United States and a low dose of diflunisal, an analgesic. CRx-401 is currently being studied in a phase 2a clinical trial in subjects with Type 2 diabetes.
We believe that our historical focus on combinations of approved drugs has enhanced and will continue to enhance the speed, efficiency and yield of our drug discovery and development process. Because the active pharmaceutical ingredients in our current portfolio of product candidates are themselves approved drugs with existing safety profiles, we have been able to move some of our product candidates expeditiously into human proof-of-concept clinical studies without the need to first complete extensive preclinical toxicology and pharmacology studies as is generally required for new chemical entities. We believe that this approach will continue to allow us to make early development decisions based on human proof-of-concept clinical data, rather than only studies in animals. Based on this proof-of-concept clinical data, our product candidates may take different development paths based on the combination biological effect we observe in patients and the commercial potential of the combination. Further development may involve developing a novel formulation for the delivery of the combination, using medicinal chemistry to create the structural analog of a component or obtaining unapproved compounds that may be able to exploit the same biologic effect identified by the original combination of approved drugs.
4
Because we have developed a portfolio of product candidates and believe that our drug discovery technology will enable us to identify additional product candidates, we will continue to use objective commercial and scientific criteria to select which product candidates to advance to later stage clinical trials. As we obtain results from our other clinical trials, we may elect to discontinue or delay trials for certain product candidates in order to refocus our resources on more promising product candidates.
We are developing proprietary formulations for our CRx-102 and CRx-401 product candidates in connection with further phase 2 clinical development. We have also developed topical formulations of our product candidates CRx-191 and CRx-197 for use in proof-of-concept clinical trials. For proof-of-concept clinical trials of our most advanced product candidates, we have typically used a controlled regimen of commercially available dosages of the active pharmaceutical ingredients of our product candidate designed to simulate our expected commercial formulation. We plan to develop and commercialize our product candidates using formulations whose pharmacology, dosage strength and route of delivery are determined on the basis of the observed activity of their active pharmaceutical ingredients when administered in combination.
Our Strategy
We are focused on discovering, developing, and commercializing novel therapeutics with multi-target mechanisms of action that are built from combinations of drugs that utilize novel biology to treat a number of conditions including immuno-inflammatory diseases, cancer, metabolic diseases, neurodegenerative diseases, infectious diseases and other diseases. The key elements of our strategy are to:
| | • | | Advance selected product candidates into later stage clinical development. We plan to selectively advance product candidates into later stage clinical trials, based on the results of proof-of-concept clinical trials and our assessment of their market potential. |
| | • | | Develop and commercialize our product candidates through collaborations with pharmaceutical and biotechnology companies and to potentially develop a specialty sales force within appropriate niche indications. We plan to determine after phase 2 clinical trials which product candidates we will retain for internal development and which product candidates we will seek to develop and commercialize with others. We may independently commercialize product candidates that have a development plan and target market size that is manageable for our company, which may include developing a specialty sales force to serve niche indications. We expect to seek development and commercialization partners for our other product candidates to obtain access to additional development, commercial or financial resources. |
| | • | | Continue to deploy our technology and approach to drug discovery in the field of combination drugs. We plan to continue to use our proprietary drug discovery technology and approach to identify additional new combination drug candidates with multi-target mechanisms by: |
| | • | | applying our drug discovery technology to new disease indications, such as infectious diseases, neurodegenerative diseases, other central nervous system disorders, inherited diseases, and other diseases, and the potential enhancement of medical devices and other interventional medicine products. |
| | • | | using medicinal chemistry to find mechanistic and structural analogs of the components of our combination drug candidates that can be developed as new chemical entities, either as combination drugs or on their own. These new chemical entities may be second or third generation product candidates that extend the life cycle of the original combination drug or may be first generation product candidates. |
| | • | | screening combinations of approved drugs and development-stage compounds that we believe target specific biological pathways that may play an important role in one or more major diseases. |
| | • | | capitalizing on our combination drug discovery and development capabilities in an effort to discover or obtain additional product candidates. We seek to accomplish this by entering into |
5
| | selected discovery research collaborations, such as our collaboration agreements with Angiotech, Fovea, Cystic Fibrosis Foundation Therapeutics, CHDI, Inc., Charley’s Fund and the Spinal Muscular Atrophy Foundation, our formation and funding of CombinatoRx Singapore with BioMedical Sciences Investment Fund Pte Ltd, or through in-licensing or other arrangements. |
Clinical Status of Our Product Candidates
The following table summarizes the clinical status of our principal clinical programs. Our clinical trials are conducted and planned to be conducted both in the United States and outside of the United States. Clinical trials conducted in the United States are subject to an investigational new drug application, or IND, with the FDA and outside the United States are subject to a clinical trial application, or CTA, with an appropriate regulator:
| | | | |
Product Candidate | | Product Description (Components) | | Clinical Status |
Immuno-Inflammatory | | | | |
CRx-102 | | Dissociated glucocorticoid (prednisolone, dipyridamole) | | Two phase 2b studies ongoing in rheumatoid arthritis and knee osteoarthritis Phase 2a complete in rheumatoid arthritis, hand osteoarthritis and biomarker study |
| | |
CRx-139 | | Selective steroid amplifier (prednisolone, paroxetine) | | Gated program—phase 2a complete in rheumatoid arthritis |
| | |
Unnamed | | Multiple product candidates | | Preclinical |
| | |
Topical Dermatology | | | | |
CRx-191 | | Topical dissociated glucocorticoid (mometasone, nortriptyline) | | Phase 2a complete in psoriasis |
| | |
CRx-197 | | Topical selective cytokine modulator (loratadine, nortriptyline) | | Phase 2a planned to be initiated in 2008 in psoriasis and atopic dermatitis |
| | |
Unnamed | | Multiple topical product candidates | | Preclinical |
| | |
Metabolic | | | | |
CRx-401 | | Anti-diabetic agent (bezafibrate, diflunisal) | | Phase 2a ongoing in Type 2 diabetes |
| | |
Chronic Pain | | | | |
CRx-170 | | Novel dual-action analgesic (prednisolone, nortriptyline) | | Gated program—phase 2a complete in asthma* |
| * | Clinical trial conducted using the corticosteroid budesonide instead of prednisolone. |
Our Product Candidates
All of our product candidates are focused on diseases with continuing medical need and potentially large commercial markets. Our principal drug development programs are in the areas of immuno-inflammatory disease, topical dermatology and metabolic disease.
6
Our Immuno-Inflammatory Product Candidates
We currently have two clinical stage product candidates, CRx-102 and CRx-139, targeting immuno-inflammatory diseases and multiple additional preclinical product candidates. Immuno-inflammatory diseases include rheumatoid arthritis, osteoarthritis, inflammatory bowel disease, asthma, lupus, polymyalgia rheumatica, fibromyalgia and multiple sclerosis.
Rheumatoid Arthritis Background. Rheumatoid arthritis is a chronic disease, mainly characterized by inflammation of the lining, or synovium, of the joints. According to the Arthritis Foundation, rheumatoid arthritis affects one percent of the United States population, or 2.1 million Americans. Rheumatoid arthritis can lead to long-term joint damage, resulting in chronic pain, loss of function and disability. Because it is a chronic disease, rheumatoid arthritis continues indefinitely and frequent flares in disease activity can occur. Rheumatoid arthritis is also a systemic disease, which means it can affect other organs in the body. Studies have shown that early aggressive treatment of rheumatoid arthritis can limit joint damage, which in turn limits loss of movement, decreased ability to work, higher medical costs and potential surgery.
Osteoarthritis Background.Osteoarthritis is one of the most common degenerative joint diseases and a frequent cause of physical disability among older adults. In the United States more than 21 million people suffer from osteoarthritis. Osteoarthritis affects the hands, lower back, neck, and weight-bearing joints such as the knees, hips, and feet. Symptoms of osteoarthritis range from stiffness and intermittent mild pain to severe joint pain and impaired biomechanical function. Although there is no cure for most forms of osteoarthritis, various therapies can help patients manage symptoms such as non-steroidal anti-inflammatory drugs, COX-2 inhibitors, local analgesics, opioids, intra-articular corticosteroid injection and surgery.
CRx-102—A Dissociated Glucocorticoid
Background. With CRx-102, we are seeking to create a dissociated glucocorticoid that selectively amplifies the potent immunomodulatory activity of prednisolone, without the associated increase in side effects. Steroids of the glucocorticoid class are prescribed for the treatment of many chronic immuno-inflammatory diseases such as rheumatoid arthritis. The utility of these drugs, however, is limited by their substantial, dose-dependent adverse side effects.
CRx-102 CRx-102 is our novel dissociated glucocorticoid product candidate designed to enhance the anti-inflammatory benefits of glucocorticoids, without associated side effects. CRx-102 contains the cardiovascular agent dipyridamole and a very low dose of the glucocorticoid prednisolone and is being developed in a uniquely engineered formulation. In proof-of-concept clinical trials, CRx-102 demonstrated a powerful anti-inflammatory effect and rapid onset of action in patients with osteoarthritis and rheumatoid arthritis and was generally well-tolerated. CRx-102 is in phase 2b clinical development for the treatment of rheumatoid arthritis and osteoarthritis.
We believe CRx-102 works through a novel multi-target mechanism of action in which dipyridamole selectively amplifies prednisolone’s anti-inflammatory and immunomodulatory activities without increases in side effects typically associated with higher doses of glucocorticoids. The anti-inflammatory activity of the glucocorticoid prednisolone is achieved by binding and activating the glucocorticoid receptor; the glucocorticoid/receptor complex then moves into the nucleus of the cell where it blocks the activity of transcription factors such as AP-1 and NFkB that would normally promote the synthesis of pro-inflammatory proteins. In CRx-102 the anti-inflammatory effect of prednisolone is enhanced by dipyridamole, which modulates the activity of multiple molecular targets. These targets act to increase intracellular levels of cyclic adenosine monophosphate (cAMP) that activates a number of pathways, which ultimately lead to suppression of transcription factors such as NFkB and NFAT that promote the transcription of pro-inflammatory genes. We believe that the anti-inflammatory effects of CRx-102 are achieved through cellular pathways co-regulated by the glucocorticoid receptor and cAMP. We believe CRx-102 activates these pathways selectivity by minimally affecting other cellular pathways that lead to high-dose steroid side effects. We also believe that additional cellular mechanisms contribute to the selective immunomodulatory activity of CRx-102, and these mechanisms are under active investigation.
7
Rheumatoid Arthritis Clinical Results.Prior to initiating our ongoing phase 2b clinical trial of CRx-102 in rheumatoid arthritis, we studied CRx-102 in a multi-center randomized, blinded, placebo-controlled phase 2a clinical trial of 59 patients with rheumatoid arthritis. The clinical trial compared CRx-102 plus a disease-modifying anti-rheumatic drug (DMARD) to placebo plus DMARD in subjects with rheumatoid arthritis. In this trial, CRx-102 demonstrated a statistically significant improvement on the primary endpoint of reduction of C-reactive protein, or CRP, with a 50% median reduction from baseline to day 42 compared to 19% with control (p=0.024). Importantly, CRx-102 demonstrated statistically significant improvements in two clinically meaningful measures of efficacy, ACR20 and DAS28. In this study CRx-102 demonstrated a statistically significant 63% ACR 20 response at day 42 compared to 30% with placebo (p=0.025) and a statistically significant DAS28 score, with a -1.6 mean change from baseline to day 42 compared to -0.7 with control (p=0.016). ACR 20 is a standard measure developed by the American College of Rheumatology to rate rheumatoid arthritis disease improvement and DAS 28, a composite disease activity score using 28 joint counts that is used to monitor disease activity in rheumatoid arthritis patients. Patients are classified as ACR20 responders if they demonstrate at least a 20% improvement from baseline in tender and swollen joint count and at least 3 of 5 other symptom-related criteria. Data provided for the rheumatoid arthritis trial of CRx-102 are for the per protocol population; statistical significance remained consistent in the intent-to-treat population. In this rheumatoid arthritis trial, CRx-102 was generally well tolerated, and there were no serious adverse events reported for subjects treated with CRx-102. The most common adverse events observed with CRx-102 that occurred with a frequency of greater than 5% were headache, gastro-intestinal symptoms and dizziness, known side effects of dipyridamole.
Patients were enrolled in this study with established rheumatoid arthritis and moderate disease activity as determined by DAS28 scores of greater than 4.5 and CRP levels of greater than 2.2mg/L. Patients were required to be on a DMARD therapy (such as methotrexate or sulfasalazine) for at least 3 months and be on a stable dose of DMARD therapy for a minimum of 28 days prior to enrollment. CRx-102 was dosed in this trial using 3mg of prednisolone plus 200 mg dipyridamole for the first week of treatment and 3 mg prenisolone plus 400 mg of dipyridamole for the following five weeks of treatment.
Hand Osteoarthritis Clinical Results. Prior to initiating our ongoing phase 2b clinical trial of CRx-102 in knee osteoarthritis, we studied CRx-102 in a randomized, blinded, placebo-controlled phase 2a clinical trial in 83 patients with moderate to severe osteoarthritis of the hand in Norway. The study met its primary endpoint of improvement in joint pain using the Australian Canadian Osteoarthritis, or AUSCAN, index. The AUSCAN Osteoarthritis Index is a composite patient-reported outcome tool developed specifically for hand osteoarthritis, similar to the Western Ontario and McMaster University Osteoarthritis, or WOMAC, index which is used for knee and hip osteoarthritis. Analysis of the primary endpoint shows mean change from baseline in the CRx-102 group was a 31% improvement in pain, compared to mean change from baseline in the placebo group of a 7% improvement in pain (p=0.007). CRx-102 demonstrated improvements in clinical secondary endpoints including stiffness, the AUSCAN physical function subscale, joint pain and patient global assessment scores.
| | | | | | | | | | | | |
Clinical Measure | | CRx-102
Mean
Improvement | | Placebo
Mean
Improvement | | CRx-102
P Value | | | CRx-102
Mean
Baseline | | CRx-102
Improvement
from
Baseline*** | |
Pain* | | 102.4 mm | | 31 mm | | 0.006 | ** | | 309.3 mm | | 33 | % |
Stiffness | | 20.3 mm | | 8.3 mm | | 0.023 | ** | | 62.9 mm | | 32 | % |
Physical Function | | 115.8 mm | | 53.1 mm | | 0.081 | | | 584.2 mm | | 20 | % |
Joint Pain | | 23.5 mm | | 6.3 mm | | 0.002 | ** | | 59.8 mm | | 39 | % |
Patient Global | | 23.4 mm | | 4.6 mm | | <0.001 | ** | | 61.5 mm | | 38 | % |
| ** | Statistically significant |
| *** | Calculation (mean improvement/mean baseline x 100) |
8
Patients were enrolled in the clinical trial that had moderate to severe hand osteoarthritis as determined by American College of Rheumatology criteria and a score on the pain dimension of the AUSCAN scale above a prespecified minimum. CRx-102 was dosed in this trial using the same dosing regimen as the rheumatoid arthritis clinical trial. Patients received the lower ratio for the first week of treatment and the higher ratio for the following five weeks. CRx-102 was generally well tolerated, and there were no serious adverse events reported from patients taking CRx-102. As with the rheumatoid arthritis trial, the most common adverse events observed with CRx-102 were headache and nausea, known side effects of dipyridamole.
Our Topical Dermatology Product Candidates
We currently have two product candidates, CRx-191 and CRx-197, that we are advancing or planning to advance into clinical trials for the topical treatment of inflammatory dermatoses.
Psoriasis Background. Psoriasis is a chronic inflammatory skin disease affecting between 6 and 7.5 million people in the United States characterized by skin thickening, redness and scaling. Topical drugs, such as glucocorticoids, Vitamin D3, Vitamin A derivatives and anthralin are usually the first line treatment for psoriasis. Dermatologists favor the use of local topical delivery in all but the most severe cases of psoriasis to limit systemic exposure and avoid potential systemic side effects. Glucocorticoids are the most commonly prescribed class of topical treatment used in psoriasis. Existing topical glucocorticoids are available in a wide range of potencies. Currently available high-potency topical glucocorticoids are associated with local skin toxicities, including thinning following only short periods of exposure and irreversible skin atrophy after longer treatment periods. A topical selective glucocorticoid amplifier that enhances potency without enhancing side effects may provide a significantly improved treatment option for psoriasis patients.
CRx-191 CRx-191 is a topical synergistic combination drug candidate with a novel multi-target mechanism that inhibits TNF-a and interferon-gamma, key cell mediators of dermal inflammation. CRx-191 contains a mid-potency glucocorticoid, mometasone, and a very low dose of the tricyclic anti-depressant, nortriptyline. CRx-191 is thought to work through a novel mechanism of action in which nortriptyline amplifies mometasone’s anti-inflammatory activities without enhancing side effects, to provide the efficacy of a high-potency topical glucocorticoid with a mid-potency glucocorticoid safety profile. We have developed a novel topical cream formulation of CRx-191 which provides the first topical formulation of nortriptyline. We have demonstrated that the CRx-191 class of combinations of tricyclic anti-depressants and glucocorticoids are effective in multiple experimental and preclinical models of inflammation. CRx-191 is under development for psoriasis and other glucocorticoid responsive dermatoses.
Clinical Status and Results. We investigated CRx-191 in a 12-day, randomized, placebo-controlled, phase 2a clinical trial in plaque psoriasis, conducted in Germany. The clinical trial was a study of 21 patients, with endpoints of psoriatic infiltrate dermal band thickness, as measured by high-frequency ultrasound, erythema (redness) as measured by chromometry, a clinical skin condition score and the measurement of inflammatory biomarkers. In this trial, each of the patients had one psoriatic plaque treated with two different concentrations of CRx-191, two different concentrations of nortriptyline, one concentration of mometasone and a placebo cream, resulting in the potential to evaluate the effect of all six preparations on the same patient. As a result, the clinical trial was designed to characterize the activity of CRx-191 in psoriasis as compared to the placebo as well as the activity of each of its components.
In this clinical trial, the high dose of CRx-191 demonstrated an 81% reduction in psoriatic infiltrate from baseline to day 12, compared to 11% for placebo. This reduction demonstrated with CRx-191 was statistically significant (P<0.0001). Similarly, CRx-191 demonstrated a 58% reduction in erythema (redness) from baseline to day 12, as compared to 6% with placebo. This effect was also statistically significant (p<0.0001). In clinical assessments of skin condition, CRx-191 demonstrated clinically significant improvements from baseline to day 12 in 100% of all test fields, as compared to 0% for placebo. The high and low doses of CRx-191 performed
9
similarly, although CRx-191 high dose was numerically superior to low dose on most measures. CRx-191 was also compared to its individual components on multiple measures such as psoriatic infiltrate thickness, erythema and clinical skin condition and produced greater improvements than mometasone and nortriptyline alone. Although this trial was not powered to achieve statistical significance in comparison to its components, CRx-191 was superior in all aspects and achieved statistical significance on the erythema endpoint. The mean reduction in erythema for CRx-191 compared to mometasone was statistically significant (p=0.017) and numerically greater on infiltrate thickness reduction and clinical skin assessment scores. CRx-191 induced statistically significant reductions compared with nortriptyline on infiltrate thickness (P<0.0001) and erythema (p<0.0001) and improved clinical skin assessment scores compared with nortriptyline. In the study, CRx-191 was well tolerated and, there were no adverse events reported for any subjects treated in this trial.
We also studied CRx-191 in a 28-day randomized, vehicle-controlled tolerability study in 20 healthy volunteers to evaluate the potential of CRx-191 to induce skin thinning, a key side effect of many potent glucocorticoids. This study was single-center study, conducted in Germany with safety endpoints of skin thickness, as determined by ultrasound, and visual assessments of adverse reactions, such as irritation, dilated blood vessels and clinical atrophy. In this study, each of the 20 subjects were topically administered two different concentrations of CRx-191, one concentration of commercially available clobetasol as a positive control, which is a high potency topical steroid, one concentration of nortriptyline, one concentration of mometasone and a placebo cream. The conclusion of this study was that CRx-191 was generally safe and well tolerated and that the CRx-191 combination of mometasone and nortriptyline did not induce skin thinning above mometasone alone.
Atopic Dermatitis Background. Atopic dermatitis is a chronic inflammatory skin disease affecting approximately 15 million people in the United States. Atopic dermatitis is treated primarily with topical glucocorticoids but their use is limited by glucocorticoid-associated side effects. As an alternative to glucocorticoids, the approved topical immunomodulatory calcineurin inhibitors represent an effective treatment option. However, the use of topical calcineurin inhibitors has been diminished as a result of a 2005 FDA health advisory and black box warning on the approved products Elidel and Protopic. As a result, we believe significant medical need and commercial opportunity exists for topical immunomodulatory agents with novel mechanisms of action.
CRx-197 CRx-197 is a novel topical anti-inflammatory product candidate that we plan to enter into clinical development in 2008 as a topical cream for the treatment of psoriasis, atopic dermatitis and other inflammatory dermatoses. CRx-197 is a selective cytokine modulator containing low concentrations of the antihistamine loratadine, and the anti-depressant nortriptyline, neither of which is indicated for the treatment of atopic dermatitis on its own but which have been shown to act synergistically in preclinical models of inflammation. We have developed a topical formulation of CRx-197 for use in our planned proof-of-concept clinical trials, initially in atopic dermatitis and psoriasis.
Our Metabolic Disease Product Candidate
Diabetes Background. Since 2001, the incidence of Type 2 diabetes in the United States has increased by 54%. Type 2 diabetes is characterized by a deregulation in the secretion of insulin or a decreased response of peripheral tissues to insulin, known as insulin resistance. While the cause of Type 2 diabetes remains unclear, epidemiologic studies suggest that this form of diabetes results from a collection of multiple genetic defects or polymorphisms, each contributing its own predisposing risks which are modified by environmental factors, including excess weight, poor diet, inactivity and excess alcohol consumption. Type 2 diabetic patients often have a characteristic lipid profile involving elevated triglycerides, low HDL levels and small particle size LDL with relatively normal overall LDL levels. Returning patients’ blood glucose levels to near normal levels is the recognized goal for the treatment of diabetes. Multiple third party clinical trials have demonstrated clinical outcome benefits from maintaining control of a patient’s glycemic parameters. However, achieving and maintaining control of plasma glucose levels can be difficult in Type 2 diabetes. An estimated 63% of diagnosed
10
Type 2 diabetics have not achieved an HbA1c level below the 7% level that the American Diabetes Association (ADA) recognizes as diabetic. This is the case even though an estimated 75% of Type 2 diabetics take multiple therapeutics in order to try to achieve their glycemic goals. Numerous drug therapies exist for the treatment of diabetes, such as insulin, metformin, sulfonylureas, thiazolidinediones, or TZDs, and GLP-1 agonists. Unfortunately, many of these approved therapies are sub-optimal from an efficacy, cost, or risk-benefit perspective.
CRx-401 CRx-401 is a novel oral anti-diabetic in development to treat Type 2 diabetes. CRx-401 is a synergistic combination drug candidate containing sustained-release bezafibrate, an anti-cholesterol agent approved outside the United States, and a low dose of diflunisal, a widely available analgesic. CRx-401 is thought to have a novel mechanism of action that reduces hyperglycemia and improves HDL and triglyceride levels without promoting weight gain. A phase 2a clinical trial evaluating the efficacy of CRx-401 in Type 2 diabetes is currently underway.
Clinical Status. We are investigating CRx-401 in a 90-day, randomized, placebo-controlled, phase 2a clinical trial in Type 2 diabetes, being conducted in Canada. The clinical trial is a study of approximately 80 patients comparing CRx-401 to bezafibrate, with a primary endpoint of change in levels of fasting plasma glucose, with other secondary endpoints relevant to Type 2 diabetes, including the measurement of changes in HbA1c levels (a measure of glycosylated hemoglobin in the blood, where elevated levels are present in Type 2 diabetics), triglycerides, high density lipoprotein cholesterol (HDL) and insulin resistance using the HOMA-IR score. This clinical trial is designed to characterize the activity of CRx-401 in Type 2 diabetes as compared to its potentially active component, bezafibrate. Enrollment in the study is ongoing. Results from this study will enhance our understanding of CRx-401 and will serve as the basis for determining the development path for CRx-401 in Type 2 diabetes or other metabolic diseases.
Gated Clinical Programs
Our gated clinical programs have completed early stage proof-of-concept phase 2a clinical trials but are not currently being advanced into later-stage clinical trials.
CRx-139 CRx-139 is another dissociated glucocorticoid product candidate we have been developing to treat immuno-inflammatory diseases. CRx-139 is a synergistic combination of low doses of the steroid prednisolone and the anti-depressant paroxetine.
Clinical Results. We studied CRx-139 in a phase 2a clinical trial in rheumatoid arthritis. The clinical trial was a study of 210 patients, with a primary endpoint of signs and symptoms of disease activity as measured by ACR 20 and secondary endpoints of biomarker response. The clinical trial was designed to characterize the activity of CRx-139 in rheumatoid arthritis compared to 3mg of prednisolone. While the activity of CRx-139 was not statistically significant as compared to 3 mg prednisolone as measured by ACR 20 at day 70, CRx-139 did achieve statistical significance compared to 3 mg prednisolone alone as measured by ACR 20 and ACR 50 at earlier time points. In this rheumatoid arthritis trial, CRx-139 was generally well tolerated and there were no drug-related serious adverse events reported for subjects treated with CRx-139. The most common adverse events observed with CRx-139 that occurred with a frequency of greater than 5% were headache and nausea, known side effects of paroxetine.
CRx-170 CRx-170 is an oral synergistic combination drug candidate containing low doses of the steroid prednisolone and the tricyclic anti-depressant nortriptyline. We have been evaluating CRx-170 for the potential treatment of chronic pain conditions. The potential development of CRx-170 in the area of chronic pain is supported by preclinical data that suggests, when administered together in CRx-170, the analgesic activity of nortriptyline and the immunomodulatory activity of the low dose prednisolone synergize to allow lower dosing of the components to reduce dose-dependent side effects and to create a broader therapeutic window. Preclinical data indicates that CRx-170 may have a superior risk-to-benefit ratio compared to other analgesic drugs.
11
Clinical Results. We investigated a version of CRx-170 with the glucocorticoid budesonide in a phase 2a clinical trial in the United Kingdom in 17 asthma patients. In the asthma clinical trial, CRx-170 provided benefit on important clinical measures such as FEV1 (forced expiratory volume in 1 second), FVC (forced vital capacity) and PEF (peak expiratory flow) and demonstrated activity on immunomodulatory markers. As measured by FEV1, the standard clinical measure of breathing capacity in asthma and chronic obstructive pulmonary disease (COPD), the CRx-170 combination containing budesonide, after one week of treatment, demonstrated a statistically significant mean percentage improvement (increase) in FEV1 from the treatment baseline of the study of approximately 6% (p=0.045). Neither low dose nortriptyline as a single agent nor low dose budesonide as a single agent showed significant improvement in FEV1 from the separate study baseline used to evaluate the single agents (approximately 2% decrease for low-dose nortriptyline, p=0.466; approximately 5% decrease for low dose budesonide, p=0.251). CRx-170 did not show a modulation of the inflammatory marker, CD163, from the treatment baseline, although nortriptyline clearly did decrease CD163 on its own. CRx-170 was generally well tolerated and there were no serious adverse events reported. The most common adverse events related to CRx-170 were dry mouth, drowsiness, constipation and headache, known effects of nortriptyline.
Preclinical Programs
Our preclinical pipeline includes multiple product candidates targeted for potential development in multiple immuno-inflammatory diseases, topical dermatoses, infectious diseases, including hepatitis C, oncology, including B-cell malignancies, neurodegenerative diseases, inherited diseases and other therapeutic areas. We have entered into a research and license agreement with Angiotech Pharmaceuticals, Inc., under which we have agreed to provide access to our clinical and preclinical product candidates and our proprietary Chalice database and analysis platform for Angiotech to develop compounds they select for use in medical devices and other interventional medicine products. Under the agreement we are also screening combinations of compounds for Angiotech to potentially develop for use with medical devices and other interventional medicine products, and we may develop any combinations discovered through the joint research project outside of the medical device field. We are also screening in the area of infectious disease, beginning with hepatitis C, through our subsidiary, CombinatoRx Singapore. We are screening in the area of neurodegenerative disease through our collaboration with CHDI, Inc. We also plan to develop product candidates for ophthalmic diseases through our collaboration with Fovea and for the treatment of muscular dystrophy through our collaboration with Charley’s Fund and the Nash Avery Foundation and in the area of cystic fibrosis through our collaboration with Cystic Fibrosis Foundation Therapeutics, Inc. In addition, we are working to discover and develop potential therapeutics for anthrax toxin with the National Institute of Allergy and Infectious Disease.
Our Drug Discovery Technology
Our combination high throughput screening technology, or cHTS™, is a robotic high throughput screening system, including both customized hardware and software elements, that screens millions of concentration-specific combinations including, but not limited to, the pharmacopeia of approved drugs in cell-based assays for the diseases we are targeting. Chalice, our integrated database and analysis platform, enables the selection and characterization of combination drug hits generated by cHTS for further research and development. We deploy this drug discovery platform to identify synergistic combinations of drugs and other small molecules whose active pharmaceutical ingredients have what we believe to be desirable chemical, pharmacological and therapeutic properties, which may then be advanced to preclinical and clinical testing.
Our library of over 2,000 drugs approved in the United States, Europe and Japan and other selected development-stage small molecules and biologics generate over 2,000,000 possible unique binary combinations. Combinations of the approved drugs in this library that are identified by our drug discovery technology can take many forms. In order to identify and analyze potentially valuable combination effects, cHTS generates a dose matrix for each chemical combination. The dose matrix captures the combined activity of two compounds over a broad range of single agent concentrations. cHTS is capable of generating hundreds of thousands of data points per day in order to efficiently screen in a dose matrix format.
12
The dose matrix data generated by our cHTS technology requires specialized analyses. We have developed Chalice, our proprietary visualization and analysis tool to collect and merge similar dose matrices before quantitatively benchmarking them to expected combination response patterns. We believe that the comparison models are useful in determining the drug candidate synergy, which helps us determine the novelty of a combination therapy or to gain insight into the biological mechanism of action of a drug combination. Using these tools, combinations are analyzed, quantitatively scored and visualized in a comprehensive combination effect report, which provides links to available internal and external data on the combination and its constituent compounds.
Product candidate selection includes external information about the compounds drawn from the Chalice database. Published chemical, therapeutic, and pharmacological data on the drug library or proprietary collections of compounds is incorporated into our database to assist us in assessing each compound’s suitability as a component in a new combination drug candidate. Before proceeding into animal studies, we require that new combinations first pass in silico tests, where candidate compounds are compared against a database that aggregates published safety and pharmacology information and data about the compounds in our library. This in silico step is intended to ensure, to the extent possible based on published information, that the active pharmaceutical ingredients in our potential combinations have safety profiles we believe to be appropriate for the disease indication we are interested in, are compatible from a drug-drug interaction perspective, can potentially be formulated in the appropriate route of administration and that the combination has not previously been discovered and meets additional key development and commercial criteria.
Collaborations
We intend to seek collaborations with pharmaceutical and biotechnology companies to support the full development and commercialization of selected product candidates to obtain access to additional development, commercial or financial resources or a large sales force. We also plan to engage in selected discovery research collaborations to explore new therapeutic areas. We intend to seek these collaborations to expand our product pipeline and diversify our therapeutic reach and compound diversity.
Angiotech Pharmaceuticals, Inc.
In October 2005, we entered into a research and license agreement with Angiotech Pharmaceuticals, Inc., or Angiotech, under which we have granted Angiotech an exclusive, royalty-bearing license to up to ten compounds to be selected by Angiotech from our portfolio of clinical and preclinical product candidates or Chalice database, as well as an option to purchase the same rights to an additional five compounds. This license is for Angiotech’s research, development and potential commercialization of the licensed compounds as drug components to be used in Angiotech’s field with medical devices or interventional medicine products to treat conditions in specific areas of the human body. In addition, we have agreed to use our cHTS technology in a joint research project with Angiotech to screen in different disease-specific assays combinations of compounds that may be developed and commercialized by Angiotech for use in combination with medical devices or with interventional medicine products in Angiotech’s field. We and Angiotech will jointly own the intellectual property that results from the joint research project. We have granted Angiotech an exclusive, royalty-bearing license to the intellectual property from the joint research in Angiotech’s field of use in combination with medical devices and interventional medicine products to treat conditions in specific areas of the human body, and we have an exclusive, non-royalty bearing license to such intellectual property for use outside of Angiotech’s field.
Under the research and license agreement, Angiotech paid us a $27.0 million up-front license execution fee. As contemplated by the original agreement, on June 8, 2007, Angiotech agreed to extend the research project beyond the original 30-month term to a total term of five years for an additional license execution fee of $7.0 million, which we have received. We may also receive payments from Angiotech of up to $10.0 million upon Angiotech’s election to receive a license to up to five additional compounds, beyond the initial ten
13
compounds, from our portfolio of clinical and preclinical product candidates or Chalice database for development. In addition, for each compound licensed to Angiotech that is discovered through the research project or through Angiotech’s selection of compounds from our portfolio of clinical and preclinical product candidates or Chalice database, we may also receive up to $30.0 million in milestone payments if certain development and regulatory approval milestones are met, as well as royalties on any future product sales incorporating the compounds.
During the period of the research project and for one year thereafter, or, if the Angiotech research and license agreement is terminated, for one year after such termination, we have agreed not to enter into any agreement to license or grant rights to any compound to a third party in defined areas of Angiotech’s field. We may enter into an agreement that grants rights in Angiotech’s field other than these defined areas if such grant of rights is materially broader than or different from Angiotech’s field and such agreement is not entered into for the purpose of granting rights to use our intellectual property in Angiotech’s field. In addition, during such period of time, we have also agreed not to perform cHTS screening using the same screens performed pursuant to the Angiotech research project, or grant a license or other rights to use our intellectual property to third parties operating primarily in Angiotech’s field.
The research and license agreement will remain in effect during the five-year period of the research project as long as Angiotech is either conducting preclinical research, doing clinical development or commercializing a licensed compound that requires the payment of a royalty to us. Angiotech may terminate the research and license agreement without cause upon not less than 45 days prior written notice, with the research period not ending until six months following the notice of termination or the end of the period of the research project, whichever is earlier. Either of us may terminate the collaboration relationship upon the bankruptcy of the other party or the commitment by the other party of an uncured material default, as defined in the research and license agreement. We may terminate the agreement upon 180 days prior written notice if Angiotech materially defaults, and fails to cure such default, with respect to its obligations to develop and commercialize the compounds selected from our portfolio of clinical and preclinical product candidates or our Chalice database. Upon termination of the agreement, we remain entitled to milestone and royalty payments accrued prior to the termination.
Upon a change of control of us, as defined in the research and license agreement, the agreement would remain in effect, although Angiotech would have the right to terminate the agreement or the research project in the six months after a change of control if we were acquired by an entity operating primarily in Angiotech’s field.
In connection with the research and license agreement with Angiotech, we also entered into a stock purchase agreement with Angiotech under which in exchange for $15.0 million we issued to Angiotech 1,363,636 shares of Series E preferred stock that automatically converted into 1,948,051 shares of our common stock upon the closing of our initial public offering.
CombinatoRx Singapore
On August 16, 2005, we formed a subsidiary in Singapore, CombinatoRx (Singapore) Pte Ltd, for the purpose of conducting our discovery and development of product candidates to treat infectious diseases. We own 51% of the subsidiary’s capital stock. Pursuant to a Subscription and Shareholders Agreement, BioMedical Sciences Investment Fund Pte Ltd, an affiliate of Bio*One Capital Pte Ltd, a biomedical sciences investment management company associated with Singapore’s Economic Development Board (EDB), has invested $2.5 million in shares of convertible, redeemable preferred stock of the subsidiary, which represents on an as-converted basis 49% of the subsidiary’s capitalization. BioMedical Sciences further committed to invest up to an additional $17.5 million in the subsidiary through the purchase of a series of convertible promissory notes through December 31, 2009, provided our subsidiary achieves certain milestones related to the development of infectious disease product candidates. A Series 1 convertible note with a principal amount of $5.5 million was
14
purchased concurrently with the initial investment in the subsidiary’s preferred stock on August 30, 2005, a $3.5 million Series 2 convertible note was purchased on June 8, 2006 and a $3.5 million Series 3 convertible note was purchased on May 30, 2007.
In addition to the funding from BioMedical Sciences, on April 19, 2006, the subsidiary received approval for a grant from the Singapore EDB Biomedical Sciences Group for up to approximately $5.8 million to support infectious disease drug research and development. The grant covers a percentage of qualifying costs of the research and development project on a cost reimbursement basis. Qualifying costs include salaries, equipment, scientific consumables and intellectual property costs. Reimbursement for these costs under the grant is subject to the satisfaction of certain conditions by the subsidiary, including completion of the development project for infectious disease within a specified timeline, spending specified amounts on the project, the completion of other development milestones and the maintenance of specified levels of employment in Singapore. Subject to agreed upon audit rights by the EDB, cumulative qualifying costs are reimbursed upon application until 70% of the initial grant amount has been submitted by the subsidiary. The remaining 30% of the award may be paid by the EDB once the subsidiary completes the research and development project. The grant extends through September 30, 2010. If the subsidiary breaches a condition of the grant, after good faith negotiations, the EDB may recover previously released grant funds from the subsidiary. In addition, the EDB retains the right to change the terms and conditions of the grant as deemed necessary by the EDB. Through December 31, 2007, we have received approximately $1.1 million in funding under the EDB grant.
Under a Services Agreement between us and our subsidiary, we have agreed to provide assay development and screening services for the subsidiary aimed at the discovery and development of combination therapies for the treatment of infectious disease, over a four year period. The subsidiary employs clinical research and development personnel in Singapore to conduct research based on the results of our initial screening efforts. The Services Agreement provides for the assignment to the subsidiary of all intellectual property rights covering novel therapeutic combination therapies for infectious disease discovered through our work under the Agreement, in exchange for cash payments to us from the subsidiary of up to approximately $7.3 million over four years and a 2.5% royalty on sales of all products covered by any patent right assigned to the subsidiary. The Services Agreement provides for the grant back to us by the subsidiary of a fully paid, exclusive license under all intellectual property rights assigned by us to the subsidiary for use outside of the infectious disease field. As defined in the Services Agreement, the infectious disease field does not include biodefense applications or any topical application for the treatment of acne or impetigo, with the result that we retain all rights for these applications.
Under the Subscription and Shareholders Agreement, we have agreed to provide assay development and screening services in the infectious disease field, as defined, exclusively to the subsidiary during the term of the Services Agreement and not to compete with the subsidiary in the infectious disease field, as defined, in substantially all markets until the earlier of August 19, 2009 or one year after we cease to hold any stock in the subsidiary.
Under the Subscription and Shareholders Agreement, the subsidiary is to be governed by a five person board of directors, two of whom are appointed by us, two by BioMedical Sciences and one by agreement between us and BioMedical Sciences. Operations of the subsidiary are conducted pursuant to a business plan and budget agreed to by us and BioMedical Sciences. Significant corporate actions by the subsidiary require both our consent and the consent of BioMedical Sciences, as do modifications of any agreement between the subsidiary and us or between the subsidiary and BioMedical Sciences. Neither we nor BioMedical Sciences may sell or transfer our shares in the subsidiary prior to August 19, 2009, except to affiliates. Thereafter, such shares may be transferred only after they are first offered to the subsidiary and to the other party. We also have granted to BioMedical Sciences, and BioMedical Sciences has granted to us, tag along rights to participate in any proposed sale to a third party of shares in the subsidiary.
15
The preferred stock of our subsidiary held by BioMedical Sciences is entitled to an annual 5% dividend payable upon redemption or liquidation of the subsidiary, and is subject to redemption by the subsidiary for a cash payment equal to 125% of the purchase price of the shares plus accrued, but unpaid, dividends. The notes bear interest at an annual rate of 5% and become due and payable on December 31, 2009, unless we elect through the subsidiary to prepay the notes before that date. If we elect to prepay, the prepayment amount would equal 125% of the outstanding principal balance of the notes plus accrued, but unpaid, interest. The notes are secured by a security interest in all of the non-intellectual property assets of the subsidiary, and by a negative pledge by the subsidiary with respect to its intellectual property rights. We have pledged our shares in the subsidiary as additional collateral for our subsidiary’s obligations to BioMedical Sciences under the notes. We are not obligated to make any cash payment on the notes if the subsidiary fails to do so, but on default by the subsidiary, BioMedical Sciences has the right to convert the notes into our common stock.
BioMedical Sciences has the option to convert its shares of preferred stock of our subsidiary into our common stock. In addition, upon the proposed redemption of the shares of preferred stock or the prepayment or repayment of any note, BioMedical Sciences may elect to receive in lieu of some or all of the cash otherwise payable to it, shares of our common stock. The notes are convertible into our common stock at the option of BioMedical Sciences only upon maturity, acceleration or default or any proposed prepayment. For any conversion of currently outstanding preferred stock or the originally issued Series 1 note into or payment satisfied by the issuance of our common stock (other than in the case of default by us or our subsidiary), the price of our common stock for conversion purposes will be $10.80 per share, in the case of the Series 2 note issued on June 8, 2006, $11.57 per share and in the case of the Series 3 note issued on May 30, 2007, $9.11 per share. Additional notes issued will convert at a price obtained by dividing the aggregate principal balance of such notes by a 40% premium to the volume-weighted average of our common stock price based on the trading price of its common stock over the 20 trading days immediately prior to the time such notes issued. If at any time the volume-weighted average price of our common stock exceeds $13.50 over the prior 20 consecutive trading days, we may require BioMedical Sciences to convert its shares of preferred stock or the initially issued Series 1 note into shares of our common stock. The volume-weighted average price of our common stock must exceed $14.47 for 20 consecutive trading days for us to be able to require BioMedical Sciences to convert the Series 2 note issued to it on June 8, 2006 into our common stock. The volume-weighted average price of our common stock must exceed $11.39 for 20 consecutive trading days for us to be able to require BioMedical Sciences to convert the Series 3 note issued to it on May 30, 2007 into our common stock. If we or our subsidiary are in default at the time of any conversion, the premium will not apply. BioMedical Sciences may not convert its convertible preferred stock or convertible promissory notes to the extent that after such conversion it would own more than 19.9% of our common stock then outstanding.
We have agreed to file, on BioMedical Sciences’s request, at any time after November 9, 2006, a registration statement covering the resale by them of any of our common stock they acquire through conversion or redemption. BioMedical Sciences has agreed not to sell in any calendar quarter more than 25% of the shares of our common stock acquired by them at any time under the conversion provisions, provided that this restriction ceases to apply to any shares one year after the shares are first issued.
Fovea Pharmaceuticals SA
On January 30, 2006, we entered into a research and license agreement with Fovea Pharmaceuticals SA, or Fovea, of Paris, France. Under the terms of the agreement, Fovea agreed to conduct, at its own expense, preclinical and clinical development of combination drug candidates it selected from our portfolio of product candidates for certain ophthalmic indications, including creating ophthalmic formulations for these selected drug candidates.
On June 12, 2007, we and Fovea amended and restated the agreement. Under the amended and restated research and license agreement, Fovea will continue to conduct, at its own expense, preclinical and clinical
16
development for certain ophthalmic indications of combination drug candidates it has selected from our portfolio of product candidates. Fovea is obligated to develop selected combination candidates pursuant to specified development criteria through the end of phase 2b clinical trials.
We and Fovea will continue to jointly own new intellectual property and data generated by Fovea regarding the selected combination candidates through phase 2a clinical trials. We retain the rights to develop and commercialize the combination candidates licensed to Fovea in North America and certain other countries and we granted Fovea exclusive rights to commercialize selected combination candidates that are developed through phase 2b clinical trials for specified ophthalmic indications in Europe and all other countries that are not retained by us. The parties have co-exclusive rights in Japan and Taiwan.
Under the agreement, in exchange for Fovea’s development investment, we also granted Fovea an exclusive worldwide license to certain preclinical drug combinations to treat allergic and inflammatory diseases of the front of the eye. For these licensed combinations, we have received payments totaling approximately $0.8 million, and are eligible to receive up to approximately $20.0 million in development and regulatory milestone payments for the first combination successfully developed, and an additional $10.0 million milestone payment for the approval of a combination in a specified additional indication. We are also eligible to receive royalties for each product commercialized by Fovea in connection with the agreement.
The license agreement has no definite term; however, Fovea’s royalty payment obligations terminate on the later of 15 years from the date of the first commercial sale of a licensed combination and the expiration of all patents covering a royalty bearing product under the license agreement, each on a country-by-country basis. The agreement may be terminated on a product by product basis by either party upon an unremedied material breach; provided that, upon the second occurrence of an unremedied material breach by Fovea, we may terminate the agreement in its entirety and all of our intellectual property rights would return to us. In addition, if Fovea fails to develop a product candidate it selects pursuant to specified diligence milestones, after discussions between the parties, the agreement may be terminated by us for such class of combination product candidates. We may terminate the agreement if Fovea fails to make required undisputed payments and either party may terminate the agreement upon the insolvency of the other party.
Cystic Fibrosis Foundation Therapeutics
On May 31, 2006, we entered into a research, development and commercialization agreement with Cystic Fibrosis Foundation Therapeutics Incorporated, or CFFT, the nonprofit drug discovery and development affiliate of the Cystic Fibrosis Foundation, to discover and develop novel therapeutics built from synergistic drug combinations to treat cystic fibrosis. Under the terms of the agreement, CFFT has agreed to award us up to approximately $13.8 million in research funding and expenses over time during the term of the research and development project and until the filing of an investigational new drug application for the first product candidate developed under the agreement. In addition, CFFT has agreed to fund up to 75% of the clinical development expenses we incur through a phase 2a clinical trial of the first potential product candidate, provided both parties have agreed to commence clinical development of the product candidate. We have received approximately $3.0 million of research funding from CFFT through December 31, 2007. We retain worldwide commercialization rights for any product candidates discovered or developed under the agreement and we will own all new intellectual property and data generated by the research and development project. We are eligible to receive payments from CFFT upon successful completion of specified clinical and regulatory milestones for each product candidate developed under the agreement. CFFT will be eligible to receive variable royalties from us on the net sales of any approved products that are discovered under the agreement.
The agreement has no definite term, but the research and development project will terminate upon the earlier of the completion of one phase 2a clinical trial of the first product candidate developed under the agreement and seven years after the initiation of research under the agreement. Our royalty payment obligations
17
to CFFT do not terminate, but our royalties in the field of cystic fibrosis and certain other pulmonary diseases may be reduced on a country-by-country basis upon the expiration of all valid patents covering a royalty bearing product under the agreement. The agreement may be terminated by either party upon an unremedied material breach. CFFT may terminate its funding of the research and development project upon 90-days’ notice only on each anniversary of the initiation of the research and development project and prior to the filing of an investigational new drug application with the FDA for the first product candidate discovered pursuant to the agreement. In addition, CFFT may terminate its funding of the research and development project if we fail to use commercially reasonable efforts directed toward the development and commercialization of product candidates to treat cystic fibrosis and certain other pulmonary diseases for a period of time, provided CombinatoRx is given a period of time to respond or accept such assertion and further disagreement may be resolved by arbitration. In the case of such termination, CFFT will be granted an exclusive license to the intellectual property for each product candidate or product selected for development to date pursuant to the agreement.
If we and CFFT agree to commence clinical development of a product candidate, until the earlier of the fifth anniversary of the termination of the agreement by CFFT for an interruption as described above, or the third anniversary of the FDA approval of a product developed under the agreement, all of our research, development and marketing efforts directed at the identification, development and commercialization of products that have as their principal mode of action the modulation of a specific protein related to cystic fibrosis will be under the agreement, provided, however, we may engage in such activity with a third party as long as CFFT is paid a royalty by us or the third party that is equal to the royalty payable under the agreement.
CHDI, Inc.
In August 2005, we entered into a research agreement with CHDI, Inc. a foundation aimed at preventing and treating Huntington’s disease, to perform joint research and development to discover and perform preclinical development of product candidates for the treatment of Huntington’s disease. Under the terms of the research agreement, as amended and restated in February 2007, subject to satisfaction of conditions, we will receive up to approximately $6.7 million in research and development funding from CHDI over four years, of which approximately $3.7 million has been received through December 31, 2007. We and CHDI will jointly own the resulting intellectual property on product candidates discovered in the collaboration, and the products may be developed and commercialized as mutually agreed by the parties. In addition, we may receive milestone or revenue sharing payments under certain circumstances if a product candidate discovered through the collaboration is commercialized. CHDI may terminate the agreement if the underlying research is interrupted, if we do not achieve specified scientific milestones, for material uncured breaches of the agreement or after we provide notice that the research is unlikely to yield scientifically valid or useful results. We may terminate the agreement for material uncured breaches of the agreement by CHDI.
Charley’s Fund and the Nash Avery Foundation
In November 2007, we entered into a sponsored research collaboration agreement with an entity formed by Charley’s Fund and the Nash Avery Foundation, two nonprofit organizations founded to support Duchenne muscular dystrophy, or DMD, research. Under the agreement we will seek to identify novel disease-modifying multi-targeted treatments for DMD, the most common childhood form of muscular dystrophy. Under the terms of the agreement, we are eligible to receive up to $3.0 million in research funding and reimbursement of additional expenses during the term of the DMD research and development project, of which approximately $0.1 million has been received through December 31, 2007. We retain worldwide commercialization rights for any product candidates discovered or developed under the agreement, and we will own all new intellectual property and data generated by the research and development project. In the event that we either enter into a license agreement with a third party granting the rights to make, use or sell a product developed under the agreement to treat DMD or we or any of our affiliates or licensees first sells a product developed under the agreement to treat DMD, we will pay the DMD foundations a payment equal to 100% of the research funding provided to us under the agreement. In addition, on the first anniversary of the first commercial sale of a product developed under the agreement, we
18
will pay the DMD foundations an additional payment equal to 100% of the research funding provided to us under the agreement. Finally, if a product developed under the agreement to treat DMD achieves cumulative net sales of at least $100 million, within 90 days of such occurrence, we will pay the DMD foundations an additional payment equal to 200% of the research funding provided to us under the agreement.
The agreement with respect to research and development collaboration terminates upon the expiration of the research and development project, which is currently planned to last for two years from the commencement of the agreement. The agreement may be terminated by either party after 60 days’ notice upon an unremedied material breach. In addition, if we intend to discontinue pre-clinical or clinical development activities with respect to a DMD product candidate and do not intend to license such candidate to a third party for pre-clinical or clinical development, within one year after such determination, we shall notify the foundations, who may then exercise their rights to an exclusive, fully-paid and sublicensable license to the intellectual property developed under the collaboration in the field of DMD.
NIAID
In April 2005, we were awarded an approximately $4.4 million research grant from the National Institutes of Allergy and Infectious Diseases, or NIAID, which will be payable over five years to perform research and preclinical development in the area of bioterror defense, subject to annual United States government appropriations and the submission of annual progress reports and development plans to NIAID demonstrating the achievement of milestones to be agreed upon for the funding year. This grant was renewed in March 2007 and is subject to annual renewals for two subsequent annual periods. Through December 31, 2007, we have received approximately $2.3 million in funding under this grant.
HenKan Pharmaceutical Company
In May 2005, we entered into a license agreement with HenKan Pharmaceutical Company of Taiwan, or HenKan, under which HenKan received the exclusive right to develop and commercialize our cancer product candidate CRx-026 in Taiwan, China, and South Korea. We received a $0.5 million up-front payment and under the license agreement we are eligible to receive up to approximately $23.0 million in development and commercialization milestone payments, plus royalties on sales within the territory. Under the license agreement, HenKan agreed under certain circumstances to fund additional phase 2 clinical trials with our support targeting oncology indications characterized by high unmet medical need in the Asian market. We also have the option to buy back the licensed intellectual property from HenKan on a country-by-country basis, provided that such buy-back occurs prior to the first sale of a product in a country in the territory of the license. The license agreement has no definite term; however, HenKan’s royalty payment obligations terminate on the later of 15 years from the date of the license agreement or the expiration of all patents covering a royalty bearing product under the license agreement. The license agreement may be terminated on a country-by-country basis by either party upon an unremedied material breach. We may terminate the license agreement if HenKan fails to make required payments, if HenKan does not use commercially reasonable efforts to develop and commercialize CRx-026 in the territory, if HenKan fails to perform, or upon a change in control of HenKan. HenKan may terminate the license agreement upon 120 days notice to allow us to find a substitute collaboration partner for CRx-026 in the territory.
Spinal Muscular Atrophy Foundation
In August 2004, we entered into a sponsored research agreement with the Spinal Muscular Atrophy Foundation, or SMA Foundation. Pursuant to the agreement, we were obligated to perform research aimed at identifying and advancing to the initial new drug application stage at least one combination drug candidate for the treatment of spinal muscular atrophy, a fatal neuromuscular disease. Under the agreement, we screened a library of compounds and cooperated with SMA Foundation to select and test promising candidates in preclinical models. The agreement terminated in 2007 at the conclusion of the research after the parties mutually determined
19
not to advance any product candidates into clinical testing. Through December 31, 2007, we received approximately $1.6 million in research funding from the SMA Foundation.
SAIC—National Institute of Neurological Disorders and Stroke
In addition to the collaboration with the SMA Foundation, in August of 2005 we entered into a subcontract with Science Applications International Corporation, or SAIC, where we acted as thein vitro bioassay screening facility for the Spinal Muscular Atrophy Project established by the National Institute of Neurological Disorders and Stroke, or NINDS. Under the terms of the subcontract, we were eligible to receive two years of research and development funding of up to approximately $1.9 million for the performance of agreed upon services, such as program management support, compound screening, information support and development of new assays. We performed the required assay development, screening, and support services under the subcontract until its expiration in September 2007. As of December 31, 2007, we received approximately $1.1 million in funding under the SAIC subcontract.
Patents and Other Proprietary Rights
Our success depends on our ability to protect our intellectual property and other proprietary rights. We rely upon a combination of patent, trademark, trade secret, copyright and unfair competition laws, assignment of inventions and non-disclosure agreements and other contractual provisions to protect our intellectual property and other proprietary rights.
As of February 29, 2008, our patent estate, on a worldwide basis, includes 70 issued patents and approximately 470 pending patent applications, with claims covering all of our current clinical stage product candidates and select preclinical and research programs, including research programs with our collaborators. Of the 70 issued patents, eight are issued in the United States. Of the 470 pending patent applications, 43 are United States non-provisional applications and 16 are United States provisional applications.
One issued United States patent, which expires in October 2022, covers the method of use of CRx-102 to treat certain immuno-inflammatory diseases, such as rheumatoid arthritis. We also have pending United States applications relating to CRx-102 which, if issued as patents, would be expected to expire between 2022 and 2029. These applications include claims covering the pharmaceutical composition, other methods of use, mechanism of action and formulation of CRx-102.
We also have a pending United States patent application with claims covering CRx-401, which, if issued as a patent, would be expected to expire in 2025. This application includes claims covering the pharmaceutical composition, method of use and formulation of CRx-401.
One issued United States patent, which expires in 2022, covers the pharmaceutical composition and methods of use of CRx-191.
We also have a pending United States patent application with claims covering the pharmaceutical composition, methods of use, mechanism of action and formulation of CRx-197, which, if issued as a patent, would expire in 2024.
We have two issued United States patents, which cover the composition of matter and method of use of CRx-170 to treat immuno-inflammatory diseases, both expiring in 2022. We also have a pending United States patent application relating to CRx-170 which, if issued as a patent, would be expected to expire in 2028. This application includes claims covering methods of use of CRx-170.
20
We also have pending United States patent applications relating to CRx-139, which, if issued as patents, would expire between 2023 and 2028. These applications include claims covering the pharmaceutical composition, methods of use, mechanism of action and formulation of CRx-139.
We have also been issued a total of 24 patents worldwide related to our drug discovery technology and three of our pending United States patent applications and 17 of our pending foreign patent applications relate to our drug discovery technology. The issued patents relating to our drug discovery technology have expiration dates between 2021 and 2024.
It is our current practice to seek the issuance of extensive claims in our patent applications that cover the combination drug candidates we develop, including claims directed to the following:
| | • | | pharmaceutical compositions comprising the active pharmaceutical ingredients in the combination; |
| | • | | pharmaceutical compositions comprising structural, functional, or mechanistic analogs of the active pharmaceutical ingredients in the combination; |
| | • | | methods of treating diseases by administering the active pharmaceutical ingredients in the combination or their analogs; |
| | • | | pharmaceutical compositions or kits or packages, including the active pharmaceutical ingredients in the combination or their analogs and instructions for the treatment of diseases; and |
| | • | | compositions and methods of use for formulations, preferred routes of administration, dosages and other properties for our more advanced product candidates. |
In addition to seeking patent protection in the United States, we generally file patent applications in European countries, Canada, Japan and additional foreign countries on a selective basis in order to further protect the inventions that we or our collaboration partners consider important to the development of our potential foreign business. As we develop novel formulations of our product candidates and learn more about the most promising dose ratios, pharmacokinetic and pharmacodynamic parameters and mechanism of action information for our drug candidates, we intend to file additional patent applications to augment the core composition of matter and method of use patents we have been issued and are currently seeking.
We also intend to pursue patents covering our product discovery platform. To date, our drug discovery technology directed patent applications have included claims directed to the protection of our screening methodologies, the accompanying informatics and computational biology techniques, and various research applications of the discovery platform.
In all of our activities, we rely on proprietary materials and information, trade secrets, and know-how to conduct research and development activities and to attract and retain collaborative partners, licensees, and customers. We attempt to protect our trade secrets by entering into confidentiality agreements with third parties, employees, and consultants. Our employees and consultants are also asked to sign agreements requiring that they assign to us their interests in patents and other intellectual property arising from their work for us. We also require all employees to sign an agreement not to engage in any conflicting employment or activity during their employment with us, and not to disclose or misuse our confidential information.
In certain instances, a patent term can be extended to recapture a portion of the term effectively lost as a result of the FDA regulatory review period. The duration of foreign patents varies in accordance with provisions of applicable local law, but typically is 20 years from the earliest effective filing date. Our patent estate, based on patents existing now and expected by us to issue based on pending applications, will expire on dates ranging from 2020 to 2029.
The actual protection afforded our product candidates by a patent varies from country to country and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patent.
21
Government Regulation
FDA Regulation of Drugs and Biologics
In the United States, federal and state statutes and regulations govern, among other things, the research, development, testing, manufacture, storage, record keeping, reporting, labeling, distribution, promotion, and marketing of pharmaceutical products. At the federal government level, the FDA is principally responsible for regulating drugs and biologics, including the product candidates we have under development. Failure to comply with applicable regulatory requirements may subject a company to administrative or judicially imposed sanctions, such as warning letters, product recalls, product seizure, injunctions, civil penalties, disgorgement of past or future profits, criminal prosecution, suspension of production, license suspension or revocation, withdrawal of an approval, or FDA refusal to approve pending marketing applications.
The steps ordinarily required before a new pharmaceutical product may be marketed in the United States begin primarily with preclinical testing. Preclinical tests include laboratory evaluation of product chemistry, toxicology and other characteristics. Animal studies are used to assess the potential safety of the product. Many preclinical studies are regulated by the FDA and must comply with good laboratory practice, or GLP, regulations. Violations of these regulations can, in some cases, lead to invalidation of the studies, requiring such studies to be replicated if the data are to be submitted to the FDA in support of a marketing application for a new drug.
In March 2006, the FDA releasedGuidance for Industry: Nonclinical Safety Evaluation of Drug Combinations.The guidance discusses what preclinical studies are appropriate to support the clinical study and approval of new combination products and therapies. In the case of new products composed of previously marketed drugs, the guidance states that generally the FDA believes sufficient clinical and preclinical data will exist for each drug component separately. Therefore, in such a case, the issues to be resolved before the new product is tested in humans generally relate to possible interactions between the components of the proposed product. The guidance identifies specific potential interaction issues to be considered and suggests the type of testing that may be appropriate to resolve any issues that require such testing.
The results of the preclinical development work, together with other information as required by the FDA, are summarized in an investigational new drug application, or IND, which must be submitted to the FDA before the drug may be provided to clinical investigators for use in humans in clinical trials. An IND also sets forth the plan for investigating the drug, including the protocols for each planned study. FDA regulations provide that human clinical trials may begin 30 days following submission of an IND, unless the FDA advises otherwise or requests additional information, clarification, or additional time to review the application. Clinical trials cannot begin until any concerns raised by the FDA have been resolved.
Each clinical trial must also be approved by an independent institutional review board, or IRB, which is typically associated with the institution or research facility at which the investigator will conduct the trial, before the trial may begin. The IRB must approve the protocol and the procedures for obtaining the informed consent of the study participants. An IRB will consider, among other things, ethical factors, the safety of human subjects, and the possible liability of the institution in which the study will be conducted. The IRB is required to conduct continuous review of the trials at intervals appropriate to the degree of risk involved and may suspend or terminate its approval if the trials are not being conducted in accordance with the IRB’s approval or there has been unexpected serious harm to subjects.
During the conduct of a clinical trial, a company is required to monitor the investigators’ compliance with the clinical study protocol and other FDA requirements, including the requirements to submit reports to the sponsor, the IRB, and the FDA, and to keep detailed records regarding study findings and use and disposition of the study drug. Although monitoring can help reduce the risk of inadequate compliance by study investigators, it cannot eliminate this risk entirely. Inadvertent regulatory noncompliance by the investigator, or intentional investigator misconduct, can jeopardize the usefulness of study results and, in rare circumstances, require a
22
company to repeat a study. A company must report to the FDA any adverse event that is both unexpected and serious and for which there is a reasonable possibility that the event may have been caused by the investigational drug. In addition, a company must within seven days report to the FDA any unexpected fatal or life-threatening event that may have been caused by the drug. The FDA may stop the trials by placing a “clinical hold” on such trials because of concerns about, for example, the safety of the product being tested. Such holds can cause substantial delay and in some cases may require abandonment of a product candidate.
Clinical testing in humans involves the administration of the investigational drug to healthy volunteers or to patients under the supervision of a qualified principal investigator, usually a physician, pursuant to an FDA-reviewed protocol. Human clinical trials typically are conducted in three sequential phases, but the phases may overlap. Phase 1 clinical trials consist of testing the product in a small number of patients or normal volunteers, primarily to evaluate the drug’s safety, at one or more dosage levels, as well as to study the drug’s pharmacokinetic and/or pharmacodynamic profile. In phase 2 clinical trials, in addition to safety, the efficacy of multiple dose levels of the product is evaluated in a patient population. Phase 3 clinical trials typically involve additional testing for safety and clinical efficacy in an expanded population at multiple geographically dispersed sites.
When two or more drugs are combined in a single dosage form, as many of our product candidates will be, the data submitted to FDA must ordinarily show that each component makes a contribution to the claimed effects and that the dosage of each component (amount, frequency, duration) is such that the combination is safe and effective for a significant patient population requiring such concurrent therapy as defined in the labeling for the drug. This FDA policy may necessitate more elaborate and expensive clinical trials than would be required for a single-agent pharmaceutical because the trials may need to be designed to study the combined agent, each drug as a single agent and a placebo.
When FDA approval is sought for a new use of a previously approved drug, the sponsor must demonstrate that the drug is safe and effective for the proposed use. However, because pre-existing information on the drug’s safety is available, the safety data required for FDA approval of a previously approved drug is ordinarily less than the safety data required to support approval of a new drug. Since our products are combinations of previously approved products, the FDA may not require us to submit some types of safety data, such as data from certain types of animal and human pharmacokinetic studies. The FDA’s specific requirements will be determined on a case-by-case basis for each product candidate. It is possible that our product candidates could present new safety issues because the previously approved drugs are being used in combinations or because the proposed combination products are being used under different circumstances than the components are used as single agents. For example, the combination might be proposed for long-term use for a chronic condition while the single agents are used short-term for acute conditions. In such a case, the FDA may require additional animal or human studies to address any safety issues.
Upon completion of clinical trials, a company seeking FDA approval to market a new drug must file a new drug application, or NDA, with the FDA, or in the case of a biological product, a biological license application, a BLA. To approve an NDA, the FDA must determine, based on the information submitted in the application, that the drug is safe and effective for its intended uses. To approve a BLA, the FDA must determine that the product is safe, pure, and potent and that the facilities in which the product is manufactured or otherwise handled meet the applicable standards. In addition to reports of the preclinical and clinical trials conducted under IND, the NDA or BLA includes information pertaining to the product’s safety and efficacy, preparation of the drug substance, analytical methods, drug product formulation, manufacturing details, and proposed product packaging and labeling. In addition, the manufacturing facility must also pass an FDA current Good Manufacturing Practices, or cGMP, inspection before the marketing application can be approved.
Submission of a NDA or BLA does not assure FDA approval for marketing. After the application is submitted, the FDA initially determines whether all pertinent data and information have been submitted before
23
accepting the application for filing. After the application is accepted for filing, the FDA begins its substantive review. The FDA typically will request a review of the data in the NDA or BLA and recommendation regarding approval by an advisory committee consisting of outside experts. The FDA may accept or reject the advisory committee’s recommendations, or accept them with modifications. The application review process generally takes a year or longer to complete, although reviews of drugs that meet a medical need for serious or life-threatening diseases may be accelerated or prioritized for a six-month review. The FDA may deny approval of an application. Any such denial may require extensive additional testing, which could take years to complete, in order to make the application approvable, or the denial may be based on considerations that cannot be favorably resolved through additional testing. In some circumstances, the FDA may approve an application even though some unanswered questions remain about the product, if the applicant agrees to conduct post-marketing studies. The FDA may impose other conditions of approval as well. Expedited or accelerated approvals may require additional larger confirmatory clinical studies to be conducted following approval.
Product approval may be withdrawn if compliance with regulatory requirements is not maintained or if post-marketing adverse events associated with the product are reported that cannot be addressed satisfactorily through changes to the product’s labeling or warnings to healthcare professionals. The FDA requires reporting of certain safety and other information that becomes known to a manufacturer of an approved product. A company may become aware of such information from reports of adverse events suspected to be related to the product, voluntarily provided to the company and/or to the FDA by physicians and other healthcare professionals, or from published scientific data. In some circumstances, the FDA may require the company to make changes to its approved product labeling or to issue safety warnings to healthcare professionals or the public, which may have a negative impact on product sales. In addition, the recently enacted Amendments Act of 2007 provides the FDA with expanded authority over drug products after approval, including the authority to require post-approval studies and clinical trials, labeling changes based on new safety information, and compliance with risk evaluation and mitigation strategies approved by the FDA. The FDA’s exercise of this authority could result in delays or increased costs during the period of product candidate development, clinical trials and regulatory review and approval, increased costs to assure compliance with new post-approval regulatory requirements, and potential restrictions on the sale of approved products, which could lead to lower product revenues to us or our collaborators. Manufacturing and sales may also be disrupted or delayed in the event of failure to comply with all required cGMP, as determined by FDA investigators in periodic inspections of manufacturing facilities. Upon approval, a drug or biological product may only be marketed for the approved indications, in the approved dosage forms, and at the approved dosage. The nature of marketing claims that we will be permitted to make in the labeling and advertising of our products will be limited to those specified in an FDA approval.
Foreign Regulations
Outside the United States, our ability to market a product is contingent upon receiving marketing authorization from the appropriate regulatory authorities. The requirements governing the conduct of clinical trials, marketing authorization, pricing, and reimbursement vary widely from country to country. At present, foreign marketing authorizations are applied for at a national level, although within the European Union, centralized procedures are available to companies wishing to market a product in more than one European Union member state. If the regulatory authorities are satisfied that adequate evidence of safety, quality, and efficacy has been presented, a marketing authorization will be granted. This foreign regulatory approval process includes all of the risks and potential delays associated with FDA approval set forth above.
Other Regulations
In addition to regulations enforced by the FDA, we also are subject to regulation under the Occupational Safety and Health Act, the Toxic Substances Control Act, the Resource Conservation and Recovery Act, and other present and potential future federal, state, and local statutes and regulations. Our research and development involves the controlled use of hazardous materials, chemicals, and various radioactive compounds. Although we
24
believe that our safety procedures for storing, handling, using, and disposing of such materials comply with the standards prescribed by applicable regulations, the risk of accidental contaminations or injury from these materials cannot be completely eliminated. In the event of such an accident, we could be held liable for any damages that result, and any such liability could materially affect our ongoing business.
Competition
The development and commercialization of pharmaceutical products and medical devices is highly competitive. We will be competing against a wide range of pharmaceutical, biotechnology, life science, medical device and interventional medicine companies that have greater resources than us, including existing research and development programs in the markets we plan to target. We must compete with these companies both in regard to the discovery technology we use to identify potential product candidates and in regard to the development and commercialization of our product candidates themselves.
In regard to our discovery technology, we file patent applications for our technology and protect our trade secrets in order to give us the exclusive right to use our technologies. These efforts may be limited if we do not succeed in achieving patent or other intellectual property protection, such protection is limited in scope, or to the extent that third parties can utilize different high throughput screening methods not covered by any issued patents we may receive. Many companies have already developed and employ high throughput screening technologies. Should these companies seek to apply these technologies to the discovery of combination drugs, our drug discovery technology may be rendered obsolete or noncompetitive.
In regard to our product candidates, we file patent applications on the composition and use of the drug combinations we discover. If we obtain the patent protection we are seeking for our product candidates, we believe that this will give us the exclusive rights to market products covered by our patents. We also believe that, if obtained, we should be able to use our patents to prevent the makers of either of the drugs included in our combination products from marketing their drug for use together with the other drug that comprises the product. We are also developing or plan to develop, for each of our product candidates that we advance beyond proof-of-concept clinical studies, a customized formulation that will be selected for its pharmacology, dosage strength, and route of delivery based on the activity and pharmacology of the drugs when delivered together in combination. Where appropriate, we will seek to protect these formulations by patent applications or as trade secrets. We intend to seek regulatory approval for our product candidates as new drugs, and the expense and time involved in seeking regulatory approval for a new drug may deter potential competitors.
Our ability to deter competitors will be limited to the extent that we are unable to obtain patent protection for our product candidates or patent or trade secret protection for our formulations. Competitors may also be able to use similar component drugs or different combinations of our component drugs to develop combination products that are not covered by our patents. In addition, the approved drugs that are combined to produce our product candidates are likely to be commercially available at lower prices, so physicians may be able to prescribe the individual drugs already approved and marketed by other companies instead of our combination products, and it would be difficult or impossible for us to enforce our patents, if obtained, to prevent this practice.
In addition to potential competition from other combination drugs, all of our product candidates will face competition from single agent pharmaceuticals. The target markets for our product candidates and those of our collaborators, including immuno-inflammatory diseases, oncology, metabolic diseases, neurodegenerative diseases, and medical devices or interventional medicine products, are all very competitive, with existing approved products holding substantial market share and other product candidates being developed by other pharmaceutical, biotechnology or medical device companies.
Principal competitive factors impacting drug development and commercialization include:
| | • | | improved patient outcomes; |
| | • | | demonstratable safety of product candidates; |
25
| | • | | acceptance of products by physicians and other healthcare providers; |
| | • | | research and drug development capabilities; |
| | • | | intellectual property positions; |
| | • | | sales and marketing capabilities; and |
| | • | | availability of capital resources to fund research, development and commercialization activities. |
Many of the companies competing against us have financial and other resources substantially greater than our own. In addition, many of our competitors have significantly greater experience in clinical testing, obtaining FDA and other regulatory approvals and in the manufacture and commercialization of products.
Manufacturing
We have no manufacturing capabilities. We rely and plan to continue to rely on third parties to manufacture bulk compounds for research, development, preclinical and clinical trials. We believe that there are several manufacturing sources available to us on commercially reasonable terms to meet our clinical requirements.
We plan to rely on third parties to manufacture commercial quantities of products we successfully develop, if any. Among the conditions for FDA approval of a pharmaceutical product is the requirement that the manufacturer’s quality control and manufacturing procedures conform to current Good Manufacturing Practice, or cGMP, which must be followed at all times. The FDA typically inspects manufacturing facilities every two years. In complying with cGMP regulations, pharmaceutical manufacturers must expend resources and time to ensure compliance with product specifications as well as production, record keeping, quality control, reporting, and other requirements. We plan to seek suitable third-party manufacturing arrangements for the commercial production of a product candidate.
Employees
As of February 29, 2008, we employed 164 persons, of whom 21 hold Ph.D. or M.D. degrees. Approximately 139 employees are engaged in research and development, and 25 employees are engaged in business development, intellectual property, finance, legal, and other administrative functions. Our workforce is non-unionized, and we believe that our relations with employees are good.
Corporate and Available Information
We were incorporated in Delaware in 2000. Our principal executive offices are located at 245 First Street, Sixteenth Floor, Cambridge, Massachusetts 02142. We have two subsidiaries, CombinatoRx (Singapore) Pte Ltd and CombinatoRx Securities Corp. Our Internet website is www.combinatorx.com. We make available free of charge through our website our annual report on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and amendments to those reports filed or furnished pursuant to Sections 13(a) and 15(d) of the Securities Exchange Act of 1934, as amended. We have made these reports available through our website during the period covered by this report and at the same time that they become available on the Securities and Exchange Commission’s website. The public may read and copy any materials that we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains an Internet site (http://www.sec.gov) that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC.
Our code of conduct and ethics, corporate governance guidelines, and the charters of the Audit Committee, Compensation Committee and Nominating and Corporate Governance Committee are all available on the corporate governance section of our website at www.combinatorx.com/investors. Stockholders may request a free copy of any of these documents by writing to Investor Relations, CombinatoRx, Incorporated, 245 First Street, Sixteenth Floor, Cambridge, Massachusetts 02142.
26
RISK FACTORS THAT MAY AFFECT FUTURE RESULTS
Investment in our common stock involves a high degree of risk and uncertainty. You should carefully consider each of the risks and uncertainties described below before you decide to invest in our common stock. If any of the following risks and uncertainties actually occurs, our business, financial condition, and results of operations could be severely harmed. This could cause the trading price of our common stock to decline, and you could lose all or part of your investment.
Risks Related to Our Discovery, Development and Commercialization of Combination Drugs
Our approach to the discovery and development of combination drugs is unproven and may never lead to commercially viable products.
Our approach to drug discovery and development is complex and unproven and has not been successfully used or, to our knowledge, attempted by any company. Previously unrecognized or unexpected defects or limitations to our drug discovery technology or drug development strategy may emerge, which we may also be unable to overcome or mitigate. None of the product candidates identified or developed to date, through the application of our business model and drug discovery technology, has been approved by any regulatory agency for commercial sale or been commercialized.
While we have often advanced and continue to advance our product candidates into phase 2a proof-of-concept and phase 2 clinical trials on the basis of their approved drug components, these product candidates could fail in these or future clinical trials. In addition, in some cases we have been required to conduct additional preclinical and phase 1 clinical studies for our product candidates prior to phase 2a proof-of-concept clinical trials, and we may be required to conduct these studies in connection with the formulations of our product candidates we have developed or plan to develop. If the additional preclinical or phase 1 clinical studies required for a product candidate or its new formulation are extensive, it could delay or prevent further development of the product candidate. Regulatory approval for a combination drug generally requires clinical trials to compare the activity of each component drug with the combination. As a result, it may be more difficult and costly to obtain regulatory approval of a combination drug than of a new drug containing only a single active pharmaceutical ingredient. In some instances, we may choose to advance product candidates where one or more of the compounds of the combination are not approved drugs, which may lead to longer clinical development timelines for these types of product candidates.
Our business model is dependent on the ability of our proprietary high throughput discovery technology to identify additional promising product candidates. High throughput screening involves testing large numbers of compounds in cell-based assays using automated systems that measure the biological activity of the compounds and provide detailed data regarding the results. Because our high throughput discovery technology is unproven in identifying drugs that can be approved, we cannot be certain that we will be able to discover additional drug combinations that show promising effects in our cell-based disease models and preclinical studies, which we can advance into clinical trials. As a result, we may not be able to identify additional product candidates. Many other companies with substantially greater resources than ours use high throughput screening for drug discovery. These or other companies may have developed or could in the future develop combination screening technology equal or superior to our technology.
For these and other reasons, our approach to drug discovery and development may not be successful and our business model may not generate viable products or revenue. Even if our approach is theoretically viable, we may not complete the significant research and development or obtain the financial resources and personnel required to further develop and apply our discovery technology, advance promising product candidates to and through clinical trials, and obtain required regulatory approvals.
27
Our results to date provide no basis for predicting whether any of our product candidates will be safe or effective, or receive regulatory approval.
All of our product candidates are in an early stage of development and their risk of failure is high. To date, the data supporting our drug discovery and development programs is derived from either laboratory and preclinical studies and limited early stage clinical trials that were not all designed to be statistically significant or proof-of-concept clinical trials, some of which are exploratory in nature. We cannot predict when or if any one of our product candidates will prove effective or safe in humans or will receive regulatory approval. If we are unable to discover or successfully develop drugs that are effective and safe in humans, we will not have a viable business.
The approved drugs included in our product candidates may have adverse or unacceptable side effects and we may not be able to achieve an acceptable level of side effect risks, compared to the potential therapeutic benefits, for our product candidates.
The approved drugs included in our combination product candidates have known adverse side effects. These side effects may be acceptable when an ingredient is used in its approved dosage to achieve a therapeutic benefit for its currently approved indications, but the side effect risk compared to the therapeutic benefit may not be acceptable when used for the intended indications for the product candidate. These side effects may also make it difficult for us to obtain regulatory or other approval to initiate clinical trials of our product candidates. In addition, the therapeutic effect of an approved drug in its currently approved indications may be inappropriate or undesirable in the intended indication for our product candidate. Also, the adverse side effects of an approved drug may be enhanced when it is combined with the other approved drug in the product candidate or other drugs patients are taking, or the combined drugs in a product candidate may produce additional side effects. Adverse side effects of the approved drugs could, in any of these situations, prevent successful development and commercialization of some or all of our product candidates, because the risks associated with the approved drug component may outweigh the therapeutic benefit of the combination.
The development of a product candidate could be adversely affected by safety or efficacy issues that subsequently arise or become the focus of increased attention or publicity regarding use of either of the approved drugs which comprise the product candidate or similar drugs. We could be forced to abandon a product candidate or an approved product due to adverse side effects from long-term or other use of one of the active pharmaceutical ingredients in the product candidate or product, even if such long-term or other use is not contemplated for such product candidate or product.
Several of our product candidates seek to increase the therapeutic effect of a reduced-dose oral glucocorticoid or mid-potency topical glucocorticoid by the combination of such glucocorticoid with a second pharmaceutical ingredient that serves as an enhancer agent. The adverse side effects of the steroids and the antidepressant or other enhancer agent included in these drug candidates are significant and generally increase as their dosage and/or duration of therapy increases. As a result, the success of these product candidates depends upon the ability of an acceptable dose of the candidate’s enhancer agent to selectively amplify the therapeutic effect of a reduced-dose glucocorticoid or mid-potency glucocorticoid, without causing unacceptable expected or unexpected adverse side effects. To achieve sufficient efficacy in humans, we may need to include higher doses of the glucocorticoid, or of the enhancer agent, than we expected to include based on our screening procedures, preclinical trials and limited clinical trials. As a result, our product candidates could have greater adverse side effects than anticipated and could fail to achieve risk-to-benefit profiles that would justify their continued development.
Certain antidepressants, including the antidepressants in our product candidates, CRx-139, CRx-170, CRx-191 and CRx-197, carry a black box warning and expanded warning statements that alert health care providers to an increased risk of suicidality in children and adolescents being treated with these drugs. Several
28
scientific publications also suggest the possibility of an increased risk for suicidal behavior in adults, in addition to children and adolescents, who are being treated with antidepressant medications. In particular, studies and publications suggest that selective serotonin reuptake inhibitors, including the antidepressant included in CRx-139, may increase the risk of suicidal behavior in adults. In 2006, the FDA reviewed all available data and determined there is an increased risk of suicidality in adults being treated with antidepressant medications, particularly young adults. Additional product labeling or even suspension of the use of some or all of the anti-depressants included in our product candidates would delay or prevent the continuation of our clinical trials, development and eventual commercialization of these product candidates. The occurrence of the adverse side effects described in the black box warning during the development of the CRx-197 product candidate could lead to product liability claims.
Diflunisal, a component of our CRx-401 product candidate, is a member of the NSAID (non steroidal anti-inflammatory drug) class, which carries an FDA-required black box warning alerting physicians to the potential for increased thrombotic cardiovascular events (including heart attacks and stroke) which may increase with duration of use, an increased risk of serious gastrointestinal adverse events including bleeding, ulceration, and perforation of the stomach or intestines, which could be fatal and the fact that diflunisal is contraindicated for the treatment of peri-operative pain in the setting of coronary artery bypass graft (CABG) surgery. The occurrence of the adverse side effects described in the diflunisal black-box warning during the development of our CRx-401 product candidate could lead to difficulty in obtaining regulatory approval or other approval for clinical trials, the termination of our clinical trials or could result in product liability claims.
Significant adverse side effects of the component drugs included in our clinical stage product candidates include: in the case of our product candidate CRx-102, headache, nausea, dizziness, diarrhea, muscle and bone loss, diabetes, dyslipidemia, osteoporosis, fractures, weakness, adrenal suppression, infections, abdominal distress, peptic ulceration, arrhythmias, cataracts, glaucoma and myopathy; in the case of our product candidates CRx-191 and CRx-197 burning, skin atrophy, stinging, adrenal suppression, suicidality, headache, drowsiness, fatigue, nervousness, constipation, nausea, blurred vision, abdominal distress, tachycardia, and arrhythmias; in the case of our product candidate CRx-401 heart attack, stroke, hypertension, bleeding, ulceration, and perforation of the stomach or intestines, diarrhea, constipation, flatulence, liver effects, renal impairment, drowsiness, insomnia, dizziness, headache, fatigue, nausea, muscular pain, weakness or cramps and alopecia; and in the case of our product candidates CRx-139 and CRx-170, drowsiness, muscle and bone loss, fractures, adrenal suppression, cataracts, glaucoma, nausea, birth defects, suicidality, infections, constipation and abdominal distress. These side effects are not the only side effects of the components of our clinical stage product candidates, but are provided based on their severity and expected frequency of occurrence. The occurrence of these or other significant adverse side effects such as electrocardiogram changes, hepatic or renal dysfunction, infections, weight gain, immunosuppression, tachycardia and agranulocytosis could lead to difficulty in obtaining regulatory or other approval for clinical trials of our product candidates, the termination of our clinical trials or could result in product liability claims.
The active pharmaceutical ingredients in our product candidates may produce adverse side effects when delivered in combination.
While an active pharmaceutical ingredient in one of our product candidates may be safe, or have an acceptable risk-to-benefit profile when administered as a single agent for its approved indications, the same active pharmaceutical ingredient when delivered in combination with the other active pharmaceutical ingredient in the product candidate or other drugs being taken by patients we are seeking to treat may cause serious unexpected or unacceptable adverse side effects. Our discovery technology is not designed to and does not detect adverse side effects that may result from the combination of the two drugs, and these side effects may not be detected in any preclinical toxicology studies we conduct. Side effects resulting from the delivery in combination of our product candidates or the interaction with other drugs could be discovered in the course of performing clinical trials of our product candidates. In addition, the active pharmaceutical ingredients in a product candidate
29
may not be approved for treatment of the product candidate’s targeted disease and may result in additional adverse side effects not typically associated with products for treatment of such a disease. The FDA or other regulators could require preclinical and phase 1 studies testing for combination side effects before we advance product candidates to further clinical trials.
Our product candidates are generally combinations of approved drugs that are commercially available and marketed by other companies. As a result, our products may be subject to substitution and competition.
We anticipate that the approved drugs that are combined to produce our product candidates are likely to be commercially available at prices lower than the prices at which we would seek to market our product candidates. Even with new formulations, we cannot be sure that physicians will view our products as sufficiently superior to a treatment regimen of the individual active pharmaceutical ingredients as to justify the significantly higher cost we expect to seek for our combination products, and they may prescribe the individual drugs already approved and marketed by other companies instead of our combination products. Even if we are issued patents covering the composition of matter, method of use or formulations of our combination products, those patents may be ineffective as a practical matter to protect against physicians prescribing the individual drugs marketed by other companies instead of our combination products. To the extent that the price of our products is significantly higher than the prices of the individual components as marketed by other companies, physicians may have a greater incentive to write prescriptions for the individual components instead of for our combination products, and this may limit how we price our products. Similar concerns could also limit the reimbursement amounts private health insurers or government agencies in the United States are prepared to pay for our products, which could also limit market and patient acceptance of our products, and could negatively impact our revenues and net income, if any. Physicians might also prescribe the individual components of a product candidate prior to its approval, which could adversely affect our development of the product candidate due to our lack of control over the administration to patients of the combination of active pharmaceutical ingredients in our product candidate, the occurrence of adverse effects, and other reasons. Such pre-approval use could also adversely affect our ability to market and commercialize any such product candidate which we successfully develop.
In many countries where we plan to market our products, including Europe, Japan and Canada, the pricing of prescription drugs is controlled by the government or regulatory agencies. Regulatory agencies in these countries could determine that the pricing for our products should be based on prices for their active pharmaceutical ingredients when sold separately, rather than allowing us to market our products at a premium as new drugs.
We must create commercially viable pharmaceutical formulations for our product candidates.
The success of our product candidates will depend on our ability to develop a formulation of the product candidate that is superior to a treatment regimen of the two approved drugs included in the product candidate taken separately. In some instances, to be commercially successful, this formulation must have a different method of administration than the approved drugs. We have developed or are developing novel formulations for our most advanced product candidates. Developing such novel formulations is costly and difficult, and we have limited experience in developing formulations ourselves. We are relying on and expect to rely on third-party suppliers to develop the pharmaceutical formulations, delivery methods or packaging for our product candidates and they may not be successful in doing so or may experience delays in doing so that could delay our clinical trials, and ultimately our ability to obtain approval of our product candidates. Defects in the formulation, delivery method or packaging of any of our product candidates could delay our ability to conduct clinical trials or require us to repeat clinical trials using a revised formulation, delivery method or packaging. If we are unsuccessful in creating commercially viable formulations, delivery methods or packaging, we may never generate product revenue or be profitable.
We are also undertaking some efforts to utilize medicinal chemistry to potentially develop new product candidates or to potentially create next-generation versions of our existing product candidates. We have only
30
limited experience with medicinal chemistry and research and development regarding new chemical entities. We may be completely unsuccessful in finding new product candidates or in discovering potential next-generation product candidates. In addition, development and regulatory approval timelines for these product candidates will be longer in duration that the timelines for our combination product candidates.
We may not be able to initiate and complete clinical trials for our product candidates.
Conducting clinical studies for any of our drug candidates requires finding appropriate clinical sites and clinical investigators, securing approvals for such studies from the independent review board at each such site and local regulatory authorities and enrolling sufficient numbers of patients. We may not be able to arrange for appropriate clinical trials for our product candidates, secure the necessary approvals or enroll the necessary number of participants. In particular, we have experienced delays in the enrollment of some of our historical phase 2 rheumatoid arthritis clinical trials, primarily due to competing therapies otherwise available to the relevant patient population or due to our enrollment criteria that often requires the discontinuance of glucocorticoid use by subjects. In addition, we cannot guarantee that outside clinical investigators conducting our clinical trials will conduct them in compliance with applicable United States or foreign regulations. Clinical sites may fail the FDA’s or other regulatory agencies’ inspections or reviews, and our trials could be halted for these or other reasons. We are conducting and planning to conduct additional proof-of-concept clinical trials as well as phase 2b clinical trials for some of our product candidates. These clinical trials may be in indications where we do not have prior experience or may be of a size and scope greater than what we have undertaken before, and this lack of experience or size may lead to delays in enrollment and other aspects of the trials. We have contracted with third-party clinical research organizations to conduct some of our phase 2b and other phase 2 proof-of-concept clinical trials for our product candidates and plan to continue to do so. These organizations may not completely perform their contractual obligations regarding the trial, or may not diligently or completely perform their tasks with respect to clinical trials under their supervision. As a result of these risks, our clinical trials may be extended, delayed or terminated, which could delay the receipt of clinical results for our product candidates, which could delay, impede or stop the development, regulatory approval or successful commercialization of our product candidates.
We may be unable to find safe and effective doses and dose ratios for our product candidates without extensive clinical trials and substantial additional costs, if at all.
We must select the doses, including the amount, frequency and duration, of each of the two active pharmaceutical ingredients included in our product candidates, and the relative amounts, or dose ratios, of these doses. Our clinical trials in humans may show that the doses or dose ratios we select based on our high throughput screening, animal testing or early clinical trials do not achieve the desired therapeutic effect in humans, or achieve this effect only in a small part of the population. Even if the doses or dose ratios we select show efficacy in humans, the resulting doses or dose ratios of our active pharmaceutical ingredients may not have acceptable safety profiles for our targeted indications. Furthermore, even if we believe that our preclinical and clinical studies adequately demonstrate that the doses or dose ratios we select for our product candidates are safe and effective in humans, the FDA or other regulatory agencies in foreign jurisdictions may conclude that our clinical trials do not support our conclusion. We may be required to conduct additional long-term clinical studies and provide more evidence substantiating the safety and effectiveness of the doses or dose ratios we select in a significant patient population. If we need to adjust the doses or dose ratios, we may need to conduct new clinical trials. We may also be required to make different doses or dose ratios available for different types of patients. All of this may result in significant delays and additional costs or prevent commercialization of our product candidates.
31
We may fail to select or capitalize on the most scientifically, clinically or commercially promising or profitable product candidates.
We may make incorrect determinations as to which of our product candidates should proceed to initial clinical trials, later stage clinical development and potential commercialization. Our decisions to allocate our research, management and financial resources toward particular product candidates or therapeutic areas may not lead to the development of viable commercial products and may divert resources from better opportunities. Similarly, our decisions to delay or terminate drug development programs may also be incorrect and could cause us to miss valuable opportunities.
We have no sales or distribution capabilities and may not obtain the collaboration, development, commercialization, manufacturing or other third-party relationships that we will require to develop, commercialize and manufacture some or all of our product candidates.
We have no sales or distribution capabilities and lack the resources, capabilities, and experience necessary to clinically develop, formulate, test, market and sell pharmaceuticals. As a result, to succeed in our business plan, we will be dependent on the efforts of third parties. We depend on collaborators, licensees, clinical research organizations and other third parties to formulate product candidates and to conduct clinical trials for some or all of our product candidates. We also rely on obtaining sufficient quantities of the approved drugs in our product candidates from sources acceptable to the FDA and other regulators for early stage clinical trials.
We expect to be able to develop and commercialize many of our product candidates only with the participation of pharmaceutical or biotechnology company collaborators or by out-licensing rights to the product candidates. Pharmaceutical and biotechnology companies and others may be reluctant to collaborate with us or to license rights to our product candidates due to the unproven nature of our drug discovery and development approach, the fact that the active pharmaceutical ingredients in our product candidates are approved drugs marketed by other companies, the risk that healthcare providers may substitute the component active pharmaceutical ingredients for our proposed combination products, concerns regarding the pricing of and reimbursement for our product candidates if they are successfully developed, or other factors.
We cannot guarantee that we will be able to successfully negotiate agreements for relationships with collaborators, partners, licensees, clinical investigators, manufacturers and other third parties on favorable terms, if at all. If we are unable to obtain these agreements, we may not be able to clinically develop, formulate, manufacture, test, obtain regulatory approvals for or commercialize our product candidates.
We expect to expend substantial funds and management time and effort to enter into relationships with these third parties and, if we successfully enter into such relationships, to manage these relationships. However, we cannot control the amount or timing of resources our contract partners will devote to our research and development programs, product candidates or potential product candidates, and we cannot guarantee that these parties will succeed in a timely fashion, if at all.
Because we have limited manufacturing experience, we depend on third-party manufacturers to manufacture product candidates for us. Failure of these manufacturers to perform could lead to delays in the clinical trials of our product candidates or costs and delays associated with contracting with new manufacturers.
We do not have any manufacturing experience, nor do we have any manufacturing facilities. We plan to rely upon third-party manufacturers to manufacture all clinical quantities of our product candidates. We depend on these third-party manufacturers to perform their obligations in a timely manner and in accordance with applicable governmental regulations. In many cases, we only have a single contract manufacturer making one of our product candidates, in which case there is a concentration of risk of non-performance with that single manufacturer that
32
would be costly to mitigate or could lead to the delay or suspension of one of our clinical trials if the manufacturer is unable to perform.
Our third-party manufacturers may encounter difficulties with meeting our requirements, including problems involving:
| | • | | inconsistent production yields; |
| | • | | poor quality control and assurance or inadequate process controls; and |
| | • | | lack of compliance with regulations set forth by the FDA or other foreign regulatory agencies. |
These contract manufacturers may not be able to manufacture our product candidates at a cost or in quantities necessary to make them commercially viable. We also have no control over whether third-party manufacturers breach their agreements with us or whether they may terminate or decline to renew agreements with us. To date, our third-party manufacturers have met our manufacturing requirements, but we cannot assure you that they will continue to do so. Furthermore, changes in the manufacturing process or procedure, including a change in the location where the drug is manufactured or a change of a third-party manufacturer, require prior FDA review and approval in accordance with the FDA’s current Good Manufacturing Practices, or cGMPs or comparable foreign requirements. This review may be costly and time-consuming and could delay or prevent the use of the product candidate in clinical trials or the launch of a product. The FDA or similar foreign regulatory agencies at any time may also implement new standards, or change their interpretation and enforcement of existing standards for manufacture, packaging or testing of products. If we or our contract manufacturers are unable to comply, we or they may be subject to regulatory action, civil actions or penalties.
If we are unable to enter into agreements with additional manufacturers on commercially reasonable terms, or if there is poor manufacturing performance on the part of our third-party manufacturers, we may not be able to complete development of, or market, our product candidates.
Financing arrangements for our subsidiary CombinatoRx (Singapore) Pte Ltd, which we refer to as CombinatoRx Singapore, may cause substantial dilution to our stockholders, limit our control over important development decisions, divert resources away from other projects, prevent us from entering into relationships with third parties for infectious disease indications and result in the loss of significant intellectual property rights.
Exercise by BioMedical Sciences of the rights granted to it to convert its shares of preferred stock in our Singapore subsidiary and convertible promissory notes issued by our Singapore subsidiary that they hold into shares of our common stock could result in substantial dilution to our stockholders. BioMedical Sciences may convert its shares of convertible preferred stock and convertible promissory notes in our Singapore subsidiary issued on August 30, 2005 into our common stock at a conversion price of $10.80, in the case of the convertible promissory note issued by our Singapore subsidiary with a $3.5 million principal amount on June 8, 2006, the conversion price is $11.57, and in the case of the convertible promissory note issued by our Singapore subsidiary with a $3.5 million principal amount on May 30, 2007, the conversion price is $9.11. These prices may be substantially less than the market price of our shares at the time of conversion or the price per share paid by investors in our common stock.
As a condition of the financing provided to our subsidiary by BioMedical Sciences, we agreed to enter into a Services Agreement requiring us to provide substantial assay development and screening services to our subsidiary over the next three years and to assign to our subsidiary ownership of all intellectual property rights covering combination therapies for the treatment of infectious disease discovered by us through our work under the Services Agreement. We have also agreed to provide screening services in the field of the therapeutic treatment of infectious disease through combination therapies exclusively to the subsidiary and not to compete
33
with the subsidiary, which will prevent us from doing work in infectious disease indications for third parties. We will commit substantial personnel resources to support operations of the subsidiary, both directly and under the Services Agreement, which could adversely affect our other operations. The performance of this work for the subsidiary may limit our ability to perform work on other disease indications. Assignment of the intellectual property rights to the subsidiary exposes us to the risk that the subsidiary may be unable to successfully develop promising therapies.
Our Singapore subsidiary will need substantially greater resources to commercialize any promising combination therapy than the resources provided by the BioMedical Sciences financing and may not be able to obtain such financing on terms acceptable to the subsidiary, us or BioMedical Sciences. BioMedical Sciences has extensive control over the operations of the subsidiary and we will not control development decisions regarding the combination therapies assigned to the subsidiary or other major decisions to be made in regard to the subsidiary.
If we or our subsidiary defaults on our obligations to BioMedical Sciences, we could lose our entire interest in the subsidiary and have no further ability to commercialize the intellectual property assigned to the subsidiary for the therapeutic treatment of infectious diseases.
Our equity interest in the subsidiary is illiquid and cannot be transferred or sold for at least four years from inception. In addition, our equity interest in the Singapore subsidiary is junior to the preferred stock and secured convertible promissory notes of the subsidiary held by Biomedical Sciences, and would likely be worthless in the event of any liquidation or forced sale of the subsidiary or its assets.
A material component of our business strategy is to establish and maintain collaborative relationships to fund research and possible development and commercialization of combination product candidates, by us or by collaborators. If we or any collaborator terminates or fails to perform any obligations under our collaboration agreements, the development and commercialization of product candidates under these agreements could be delayed or terminated.
A material component of our business strategy is to establish and maintain collaborative arrangements with pharmaceutical, biotechnology and medical device companies, research institutions and foundations, and to seek grants from agencies of the United States government, to fund research and possible development and commercialization of combination drug products for the treatment of diseases. We also intend to seek to establish collaborative relationships to obtain domestic or international sales, marketing and distribution capabilities if any of our combination product candidates receive regulatory approval.
The process of establishing collaborative relationships is difficult, time-consuming and involves significant uncertainty. Moreover, even if we do establish collaborative relationships, it may be difficult for us to maintain or perform under such collaboration arrangements, as our funding resources may be limited or our collaborators may seek to renegotiate or terminate their relationships with us due to unsatisfactory research or clinical results, a change in business strategy, a change of control or other reasons. If we or any collaborator fails to fulfill any responsibilities in a timely manner, or at all, our research, clinical development or commercialization efforts related to that collaboration could be delayed or terminated. Additionally it may become necessary for us to assume responsibility for activities that would otherwise have been the responsibility of our collaborator. Further, if we are unable to establish and maintain collaborative relationships on acceptable terms, we may have to delay or discontinue further development of one or more of our product candidates, undertake development and commercialization activities at our own expense or find alternative sources of funding.
Our collaborations are generally new, and we have only a short history of working together with Angiotech, Fovea, CFFT, CHDI, HenKan, the DMD foundations or our other collaborators and cannot predict the success of any of our collaborations. Our collaborations typically involve a complex allocation of responsibilities, costs and
34
benefits and provide for milestone payments to us upon the achievement of specified clinical and regulatory milestones. Our collaborations also may provide us with royalty-based revenue if product candidates are successfully commercialized. Under the Angiotech and other collaborations, we will rely on our collaborators to provide resources to develop new product candidates and to potentially achieve these milestones and commercialize any new products. We may not be able to achieve any of the milestones provided in our Angiotech or other collaboration agreements or derive any license or royalty revenue with respect to these collaborations.
Disputes under key agreements could delay or prevent development or commercialization of our product candidates.
Any agreements we have or may enter into with third parties, such as collaboration, license, formulation supplier, manufacturing, testing, clinical research organization or clinical trial agreements, may give rise to disputes regarding the rights and obligations of the parties. Disagreements could develop over rights to ownership or use of intellectual property, the scope and direction of research and development, the approach for regulatory approvals or commercialization strategy. We intend to conduct research programs in a range of therapeutic areas, but our pursuit of these opportunities could result in conflicts with the other parties to these agreements who may be developing or selling pharmaceuticals or conducting other activities in these same therapeutic areas. Any disputes or commercial conflicts could lead to the termination of our agreements, delay progress of our product development programs, compromise our ability to renew agreements or obtain future agreements, lead to the loss of intellectual property rights or result in costly litigation.
We may not be able to gain market acceptance of our product candidates, which would prevent us from becoming profitable.
We cannot be certain that any of our product candidates, if approved, will gain market acceptance among physicians, patients, healthcare payors, pharmaceutical companies or others. Demonstrating the safety and efficacy of our product candidates and obtaining regulatory approvals will not guarantee future revenue. Sales of medical products largely depend on the reimbursement of patients’ medical expenses by government healthcare programs and private health insurers. Governments and private insurers closely examine medical products to determine whether they should be covered by reimbursement and if so, the level of reimbursement that will apply. We cannot be certain that third-party payors will sufficiently reimburse sales of our products, or enable us to sell our products at profitable prices. Sales of medical products also depend on physicians’ willingness to prescribe the treatment, which is likely to be based on a determination by these physicians that the products are safe, therapeutically effective and cost-effective. We cannot predict whether physicians, other healthcare providers, government agencies or private insurers will determine that our products are safe, therapeutically effective and cost effective relative to competing treatments, including a treatment regimen of the individual approved drugs included in our combination products.
If we fail to manage our growth, our operations and financial results could be adversely affected.
Since our inception in 2000, we have grown to approximately 164 employees and advanced multiple product candidates into clinical trials. We have expanded our operations and strategic relationships at a rapid pace through numerous collaborations, including with Angiotech, Fovea, CFFT, the DMD foundations, the Spinal Muscular Atrophy Foundation, HenKan, CHDI, Inc., the Liverpool School of Tropical Medicine, or LSTM, National Institutes of Health and through our subsidiary CombinatoRx Singapore. We have experienced a period of rapid growth that has placed a strain on our administrative and operational infrastructure as well as additional demands on our financial resources. We expect this strain to continue as we continue to grow and seek to obtain and manage collaborations and relationships with third parties. We must continue to refine and expand our research and development capabilities, our management of the clinical trial process, access to financing resources and technology. As we grow, we must continue to hire, train, supervise and manage new employees.
35
We may not be successful in hiring sufficient research, development, clinical, regulatory and administrative personnel and management to manage our expansion effectively. If we are unable to manage our growth, our operations and financial results could be adversely affected.
If we are not able to retain our current senior management team or attract and retain qualified scientific, technical and business personnel, our business will suffer.
We are dependent on the members of our management team for our business success. In addition, an important element of our strategy is to take advantage of the development, regulatory and commercialization expertise of our current management. Our employment agreements with our executive officers are terminable on short notice or no notice. The loss of any one of our executive officers could result in a significant loss in the knowledge and experience that we, as an organization, possess and could cause significant delays, or outright failure, in the development and further commercialization of our product candidates.
To grow, we will need to hire a significant number of qualified commercial, scientific and administrative personnel. However, there is intense competition for human resources, including management in the technical fields in which we operate, and we may not be able to attract and retain qualified personnel necessary for the successful development and commercialization of our product candidates. As we grow, the inability to attract new employees when needed or to retain existing employees could limit our growth and harm our business.
Risks Related to Our Financial Results and Need for Additional Financing
We have a history of operating losses, expect to incur significant and increasing operating losses and may never be profitable. Our stock is a highly speculative investment.
We commenced operations in March 2000 and have a limited operating history for you to evaluate our business. We have no approved products and have generated no product revenue. We have incurred operating losses since our inception in 2000. As of December 31, 2007, we had an accumulated deficit of $186.2 million. We have spent, and expect to continue to spend, significant resources to fund research and development of our product candidates and to enhance our drug discovery technology. We expect to incur substantial and increasing operating losses over the next several years as our research, development, preclinical testing, and clinical trial activities increase. As a result, our accumulated deficit will also increase significantly.
Our product candidates are in the early stages of development and may never result in any revenue. We will not be able to generate product revenue unless and until one of our product candidates successfully completes clinical trials and receives regulatory approval. As our most advanced product candidates are in or recently emerged from the proof-of-concept stage, we do not expect to receive revenue from any product candidate for the next four years. We may seek to obtain revenue from collaboration or licensing agreements with third parties. Our current collaboration and license agreements may not provide us with material, sustainable ongoing future revenue, and we may not be able to enter into additional collaboration agreements. Even if we eventually generate product revenues, we may never be profitable, and if we do achieve profitability, we may not be able to sustain or increase profitability on a quarterly or annual basis.
We may be unable to raise the substantial additional capital that we will need to sustain our operations.
We will need substantial additional funds to support our planned operations. Based on our current operating plans, we expect our existing resources to be sufficient to fund our planned operations through approximately the fourth quarter of 2009. We may, however, need to raise additional funds before that time if our research and development expenses exceed our current expectations or our collaboration funding is less than our current expectations. This could occur for many reasons, including:
| | • | | some or all of our product candidates fail in clinical or preclinical studies or prove to be less commercially promising than we expect and we are forced to seek additional product candidates; |
36
| | • | | our product candidates require more extensive clinical or preclinical testing or our clinical trials take longer to complete than we currently expect; |
| | • | | we advance more of our product candidates than expected into costly later stage clinical trials; |
| | • | | we advance more preclinical product candidates than expected into early stage clinical trials; |
| | • | | we determine or are required to conduct more high throughput screening than expected against current or additional disease targets to develop additional product candidates; |
| | • | | we are required, or consider it advisable, to acquire or license rights from one or more third parties; or |
| | • | | we determine to acquire or license rights to additional product candidates or new technologies. |
While we expect to seek additional funding through public or private financings, we may not be able to obtain financing on acceptable terms, or at all. In addition, the terms of our financings may be dilutive to, or otherwise adversely affect, holders of our common stock. We may also seek additional funds through arrangements with collaborators or others. These arrangements would generally require us to relinquish rights to some of our technologies, product candidates or products, and we may not be able to enter into such agreements, on acceptable terms, if at all. The arrangements also may include issuances of equity, which may also be dilutive to, or otherwise adversely affect, holders of our common stock. If we are unable to obtain additional funding on a timely basis, we may be required to curtail or terminate some or all of our research or development programs, including some or all of our product candidates.
Risks Related to Regulatory Approvals
The regulatory approval process is costly and lengthy and we may not be able to successfully obtain all required regulatory approvals.
The preclinical development, clinical trials, manufacturing, marketing, testing and labeling of pharmaceuticals and medical devices are all subject to extensive regulation by numerous governmental authorities and agencies in the United States and other countries. We or our collaborators must obtain regulatory approval for product candidates before marketing or selling any of them. The approval process is typically lengthy and expensive, and approval is never certain. It is not possible to predict how long the approval processes of the FDA or any other applicable federal or foreign regulatory authority or agency for any of our products will take or whether any such approvals ultimately will be granted. The FDA and foreign regulatory agencies have substantial discretion in the drug and medical device approval process, and positive results in preclinical testing or early phases of clinical studies offer no assurance of success in later phases of the approval process. Generally, preclinical and clinical testing of products and medical devices can take many years and require the expenditure of substantial resources, and the data obtained from these tests and trials can be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. Any delay in obtaining, or failure to obtain, approvals could prevent or adversely affect the marketing of our products or our collaborator’s products and our ability to generate product revenue. The risks associated with the approval process include delays or rejections in the regulatory approval process based on the failure of clinical or other data to meet expectations, or the failure of the product or medical device to meet a regulatory agency’s requirements for safety, efficacy and quality. In addition, regulatory approval, if obtained, may significantly limit the indicated uses for which a product may be marketed.
We or our collaborators may delay, suspend or terminate clinical trials to obtain marketing authorization of any of our product candidates or their associated medical devices or products at any time for reasons including:
| | • | | ongoing discussions with the FDA or comparable foreign authorities regarding the scope or design of our clinical trials; |
37
| | • | | delays or the inability to obtain required approvals from institutional review boards or other governing entities at clinical sites selected for participation in our clinical trials; |
| | • | | delays in enrolling patients and volunteers into clinical trials; |
| | • | | lower than anticipated retention rates of patients and volunteers in clinical trials; |
| | • | | the need to repeat clinical trials as a result of inconclusive or negative results or poorly executed testing; |
| | • | | lack of effectiveness of a product candidate in other clinical trials; |
| | • | | lack of sufficient funds for further clinical development; |
| | • | | insufficient supply or deficient quality of product candidate materials or other materials necessary to conduct our clinical trials; |
| | • | | unfavorable regulatory inspection of a manufacturing, testing, labeling or packaging facility for drug substance or drug product; |
| | • | | unfavorable regulatory inspection and review of a clinical or preclinical trial site or records of any clinical or preclinical investigation; |
| | • | | serious and unexpected drug-related side effects or serious adverse safety events experienced by participants in our clinical trials or by patients following commercialization; or |
| | • | | the placement of a clinical hold on a product candidate in an ongoing clinical trial. |
Positive or timely results from preclinical studies and early clinical trials do not ensure positive or timely results in late stage clinical trials or product approval by the FDA or any other regulatory authority. Product candidates that show positive preclinical or early clinical results often fail in later stage clinical trials. Data obtained from preclinical and clinical activities is susceptible to varying interpretations, which could delay, limit, or prevent regulatory approvals.
We have limited experience in conducting the clinical trials required to obtain regulatory approval. We may not be able to conduct clinical trials at preferred sites, enlist clinical investigators, enroll sufficient numbers of patients, or begin or successfully complete clinical trials in a timely fashion, if at all. Any failure to perform may delay or terminate the trials. Our current clinical trials may be insufficient to demonstrate that our potential products are active, safe, or effective and as a result we may decide to abandon further development of such product candidates. Additional clinical trials may be required if clinical trial results are negative or inconclusive, which will require us to incur additional costs and significant delays. If we do not receive the necessary regulatory approvals, we will not be able to generate product revenues and will not become profitable. We may encounter significant delays in the regulatory process that could result in excessive costs that may prevent us from continuing to develop our product candidates. In addition, the failure to comply with applicable regulatory requirements may result in criminal prosecution, civil penalties, product recalls, withdrawal of product approval, mandatory restrictions and other actions that could impair our ability to conduct our business.
The FDA and other regulatory agencies may require additional preclinical and phase 1 clinical data for our combination products.
Our proposed products are composed of drugs previously approved as single agents, and as a result, there is significant pre-existing information on the safety of those drugs as single agents for their approved indications in the form of animal studies, phase 1 pharmacokinetic and other clinical studies and actual clinical experience. Nonetheless, new safety issues may arise when the previously approved drugs are used in combination in our products or when the proposed combination products are used in different treatment regimens for disease indications different than the disease indications for which the components are used as single agents. For
38
example, the combination might be proposed for long-term use for a chronic condition, while the single agents are currently approved for short-term use in acute conditions. In addition, if a component has not been approved in all jurisdictions in which approval of the combination is sought, the regulatory agencies having authority over the combination in the jurisdictions that had not approved each component as a single agent may require us to submit data that would not otherwise be required. If any of these issues arises, we may be required to conduct additional nonclinical and phase 1 clinical trials for our product candidates. If the additional nonclinical or phase 1 clinical trials required for a product candidate or its formulation are extensive, it could delay or prevent further development of the product candidate.
The FDA and other regulatory agencies may require more extensive or expensive trials for our combination product candidates than may be required for single agent pharmaceuticals.
To obtain regulatory approval for our combination product candidates, we are typically required to show that each active pharmaceutical ingredient in the product candidate makes a contribution to the combined product candidate’s claimed effects and that the dosage of each component, including amount, frequency and duration, is such that the combination is safe and effective for a significant patient population requiring such concurrent therapy. As a result, we are typically required to conduct clinical trials comparing each component drug with the combination. This could require us to conduct more extensive and more expensive clinical trials than would be the case for many single agent pharmaceuticals. The need to conduct such trials could make it more difficult and costly to obtain regulatory approval of a combination drug than of a new drug containing only a single active pharmaceutical ingredient.
Even if we receive regulatory approvals for marketing our product candidates, if we fail to comply with continuing regulatory requirements, we could lose our regulatory approvals, and our business would be adversely affected.
The FDA and other regulatory authorities continue to review therapeutic products and medical devices even after they receive initial approval. If we receive approval to commercialize any product candidates, the manufacturing, testing, marketing, sale and distribution of these drugs and medical devices will be subject to continuing regulation, including compliance with quality systems regulations, good manufacturing practices, adverse event reporting requirements and prohibitions on promoting a product for unapproved uses. Furthermore, heightened Congressional scrutiny on the adequacy of the FDA’s drug approval process and the agency’s efforts to assure the safety of marketed drugs has resulted in the enactment of new legislation, the FDA Amendments Act of 2007, addressing, among other things, drug safety issues. This new law provides the FDA with expanded authority over drug products after approval, including the authority to require post-approval studies and clinical trials, labeling changes based on new safety information, and compliance with risk evaluation and mitigation strategies approved by the FDA. The FDA’s exercise of this authority could result in delays or increased costs during the period of product candidate development, clinical trials and regulatory review and approval, increased costs to assure compliance with new post-approval regulatory requirements, and potential restrictions on the sale of approved products, which could lead to lower product revenues to us or our collaborators. Enforcement actions resulting from failure to comply with government requirements could result in fines, suspension of approvals, withdrawal of approvals, recalls of products, product seizures, operating restrictions, and civil or criminal penalties. These enforcement actions could affect the manufacturing, testing, marketing, sale and distribution of our products.
Healthcare reform measures could adversely affect our business.
The efforts of governmental and third-party payors to contain or reduce the costs of healthcare may adversely affect the business and financial condition of pharmaceutical, biotechnology and medical device companies. In the United States and in foreign jurisdictions there have been, and we expect that there will continue to be, a number of legislative and regulatory proposals aimed at changing the healthcare system. For
39
example, in some countries other than the United States, pricing of prescription drugs and medical devices is subject to government control, and we expect proposals to implement similar controls in the United States to continue. The pendency or approval of such proposals could result in a decrease in our common stock price or limit our ability to raise capital or to enter into collaborations or license rights to our products.
Federal law may increase the pressure to reduce prices of pharmaceutical products paid for by Medicare, which could adversely affect our revenues, if any.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or MMA, expanded Medicare coverage for drug purchases by the elderly and disabled beginning in 2006. Under the MMA, private insurance plans subsidized by the government offer prescription drug coverage to Medicare beneficiaries who elect to enroll in their plans. Although almost all prescription drugs are potentially available to plan enrollees, the plans are allowed to use formularies, preferred drug lists and similar mechanisms to favor selected drugs and limit access to other drugs except in certain circumstances. The price of a drug as negotiated between the manufacturer and a plan is a factor that the plan can consider in determining its availability to enrollees.
As a result, we expect that there will be increased pressure to reduce prices for drugs to obtain favorable status for them under the plans offering prescription drug coverage to Medicare beneficiaries. This pressure could decrease the coverage and price that we receive for our products in the future and could seriously harm our business. It is possible that our products, if successfully developed, could be particularly subject to price reduction initiatives because they are based on combinations of lower priced existing drugs.
In addition, some members of Congress advocate that the federal government should negotiate directly with manufacturers for lower prices for drugs in the Medicare program, rather than rely on private plans. If the law were changed to allow or require such direct negotiation, there could be additional reductions in the coverage of and prices that we receive for our products.
Federal laws or regulations on drug importation could make lower cost versions of our future products available, which could adversely affect our revenues, if any.
The prices of some drugs are lower in other countries than in the United States because of government regulation and market conditions. Under current law, importation of drugs into the United States is generally not permitted unless the drugs are approved in the United States and the entity that holds that approval consents to the importation. Various proposals have been advanced to permit the importation of drugs from other countries to provide lower cost alternatives to the products available in the United States. In addition, the MMA requires the Secretary of Health and Human Services to promulgate regulations for drug reimportation from Canada into the United States under some circumstances, including when the drugs are sold at a lower price than in the United States.
If the laws or regulations are changed to permit the importation of drugs into the United States in circumstances not now permitted, such a change could have an adverse effect on our business by making available lower priced alternatives to our future products.
Failure to obtain regulatory and pricing approvals in foreign jurisdictions could delay or prevent commercialization of our products abroad.
If we succeed in developing any products, we intend to market them in the European Union and other foreign jurisdictions. In order to do so, we must obtain separate regulatory approvals and comply with numerous and varying regulatory requirements. The approval procedure varies among countries and can involve additional testing. The time required to obtain approval abroad may differ from that required to obtain FDA approval. The foreign regulatory approval process may include all of the risks associated with obtaining FDA approval and additional risks because there may be additional variations between how our combinations of approved drugs and
40
single agent drugs are treated in foreign jurisdictions. We may not obtain foreign regulatory approvals on a timely basis, if at all. Approval by the FDA does not ensure approval by regulatory authorities in other countries, and approval by one foreign regulatory authority does not ensure approval by regulatory authorities in other foreign countries or by the FDA. We and our collaborators may not be able to file for regulatory approvals and may not receive necessary approvals to commercialize our products in any market outside the United States. The failure to obtain these approvals could materially adversely affect our business, financial condition and results of operations. Even if we are successful at obtaining these approvals, regulatory agencies in foreign countries where pricing of prescription drugs or medical devices is controlled by the government, could determine that pricing for our products should be based on prices for the existing drugs that comprise the active pharmaceutical ingredients in our combination products instead of allowing us to price our products at a premium as novel medicines.
Risks Related to Our Intellectual Property
Our success depends upon our ability to obtain and maintain intellectual property protection for our products and technologies.
Our success will depend on our ability to obtain and maintain adequate protection of our intellectual property, including our proprietary drug discovery technology and any products or product candidates we plan to develop. In addition to taking other steps to protect our intellectual property, we intend to apply for patents with claims covering our technologies, processes, products and product candidates when and where we deem it appropriate to do so. We have applied for patent protection covering our clinical and preclinical product candidates and our drug discovery technologies in the United States, and some, but not all, foreign countries. In countries where we have not and do not seek patent protection, third parties may be able to manufacture and sell our products without our permission, and we may not be able to stop them from doing so.
Similarly to other biotechnology companies, our patent position is generally uncertain and involves complex legal and factual questions. In addition, the laws of some foreign countries do not protect proprietary rights to the same extent as the laws of the United States, and other biotechnology companies have encountered significant problems in protecting and defending their proprietary rights in foreign jurisdictions. Whether filed in the United States or abroad, our patent applications may be challenged or may fail to result in issued patents. In addition, our existing patents and any future patents we obtain may not be sufficiently broad to prevent others from practicing our technologies or from developing or commercializing competing products. Furthermore, others may independently develop or commercialize similar or alternative technologies or drugs, or design around our patents. Our patents may be challenged, invalidated or fail to provide us with any competitive advantages.
The United States Patent and Trademark Office and similar agencies in foreign jurisdictions may not agree with our view that our combination product candidates and our drug discovery technologies are patentable or novel and non-obvious, and on this basis may deny us patent protection. Even if we receive patent protection, others, including those who own patent or trade secret rights associated with the approved drugs that are active pharmaceutical ingredients of our product candidates, may attempt to invalidate our patent or trade secret rights. Even if our patent or trade secret rights are not directly challenged, disputes among third parties could lead to the weakening or invalidation of our intellectual property rights.
If we do not obtain or we are unable to maintain adequate patent or trade secret protection for our products in the United States, competitors could duplicate them without repeating the extensive testing that we will be required to undertake to obtain approval of the products by the FDA and other regulatory authorities. Regardless of any patent protection, under the current statutory framework the FDA is prohibited by law from approving any generic version of any of our combination products for three years after it has approved our product. Upon the expiration of that period, or if that time period is altered, the FDA could approve a generic version of our product unless we have patent protection sufficient for us to block that generic version. Without sufficient patent protection, the applicant for a generic version of our product would be required only to conduct a relatively
41
inexpensive study to show that its product is bioequivalent to our product and would not have to repeat the studies that we conducted to demonstrate that the product is safe and effective. In the absence of adequate patent protection in other countries, competitors may similarly be able to obtain regulatory approval of products that duplicate our products.
We may not be able to develop or commercialize our product candidates due to intellectual property rights held by third parties.
If a third party holds a patent to a composition or method of use of an approved drug that is a component of one or more of our product candidates or a formulation technology related to our planned formulation of a product candidate, we may not be able to develop or commercialize such product candidates without first obtaining a license to such patent, or waiting for the patent to expire. In particular, for two of our product candidates, CRx-139 and CRx-170, some of the various formulations and methods of use of one drug in the combination are covered by third-party patents which could pose an impediment to our ability to develop and commercialize these products. We believe that other formulations and methods of use of these drugs, which are not covered by any third-party patents, are available. However, we cannot be sure that the unpatented formulations or methods of use of these drugs will be the optimal formulation, or that a feasible formulation for these product candidates will be available. Additionally, there are three United States patents that may cover therapeutic uses of CRx-401, our Type 2 diabetes product candidate. We may not be able to develop or commercialize our CRx-401 product candidate without first obtaining a license to these patents, or waiting for them to expire. Our business will be harmed if we are unable to use the optimal formulation or methods of use of the component drugs that comprise our product candidates. This may occur because the formulations or methods of use are covered by one or more third-party patents, and a license to such patents is unavailable or is available on terms that are unacceptable to us.
We may be unable to in-license intellectual property rights or technology necessary to develop and commercialize our products.
Depending on its ultimate formulation and method of use, before we can develop, make, use, or sell a particular product candidate, we may need to obtain a license from one or more third parties who have patent or other intellectual property rights covering components of our product candidate or its method of use. There can be no assurance that such licenses will be available on commercially reasonable terms, or at all. Because our product candidates are based on combinations of existing drugs, there may be a significant number of patents covering both the active pharmaceutical ingredients in our product candidates and their method of use. If a third party does not offer us a necessary license or offers a license only on terms that are unattractive or unacceptable to us, we might be unable to develop and commercialize one or more of our product candidates.
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information.
In our activities, we rely substantially upon proprietary materials, information, trade secrets and know-how to conduct our research and development activities, and to attract and retain collaborators, licensees and customers. We take steps to protect our proprietary rights and information, including the use of confidentiality and other agreements with our employees and consultants and in our academic and commercial relationships. However, these steps may be inadequate, agreements may be violated, or there may be no adequate remedy available for a violation of an agreement. Our proprietary information may be inadvertently disclosed or we may lose the protection of our trade secrets. Our competitors may independently develop substantially equivalent proprietary information or may otherwise gain access to our trade secrets, which could adversely affect our ability to compete in the market.
42
Litigation or third-party claims of intellectual property infringement could require substantial time and money to resolve. Unfavorable outcomes in these proceedings could limit our intellectual property rights and our activities.
We may need to resort to litigation to enforce or defend our intellectual property rights, including any patents issued to us. If a competitor or collaborator files a patent application claiming technology also invented by us, in order to protect our rights, we may have to participate in an expensive and time consuming interference proceeding before the United States Patent and Trademark Office. We cannot guarantee that our product candidates will be free of claims by third parties alleging that we have infringed their intellectual property rights. Third parties may assert that we are employing their proprietary technologies without authorization and they may resort to litigation to attempt to enforce their rights. Third parties may have or obtain patents in the future and claim that the use of our technology or any of our product candidates infringes their patents. We may not be able to develop or commercialize combination product candidates because of patent protection others have. Our business will be harmed if we cannot obtain a necessary or desirable license, can obtain such a license only on terms we consider to be unattractive or unacceptable, or if we are unable to redesign our product candidates or processes to avoid actual or potential patent or other intellectual property infringement.
Our efforts to obtain, protect and defend our patent and other intellectual property rights, whether we are successful or not, can be expensive and may require us to incur substantial costs, including the diversion of management and technical personnel. An unfavorable ruling in patent or intellectual property litigation could subject us to significant liabilities to third parties, require us to cease developing, manufacturing or selling the affected products or using the affected processes, require us to license the disputed rights from third parties, or result in awards of substantial damages against us. In addition, defending patent or other intellectual property litigation, whether we are successful or not, can be very expensive and may require us to incur substantial costs, including the diversion of management and technical personnel. During the course of any patent litigation, there may be public announcements of the results of hearings, motions, and other interim proceedings or developments in the litigation. If securities analysts or investors regard these announcements as negative, the market price of our common stock may decline. General proclamations or statements by key public figures may also have a negative impact on the perceived value of our intellectual property.
There can be no assurance that we would prevail in any intellectual property infringement action, will be able to obtain a license to any third-party intellectual property on commercially reasonable terms, successfully develop non-infringing alternatives on a timely basis, or license non-infringing alternatives, if any exist, on commercially reasonable terms. Any significant intellectual property impediment to our ability to develop and commercialize our products could seriously harm our business and prospects.
Risks Related to Our Industry
Our competitors and potential competitors may develop products and technologies that make ours less attractive or obsolete.
Many companies, universities, and research organizations developing competing product candidates have greater resources and significantly greater experience in research and development, manufacturing, marketing, sales, distribution, financial and technical regulatory matters than we have. In addition, many competitors have greater name recognition and more extensive collaborative relationships. Our competitors could commence and complete clinical testing of their product candidates, obtain regulatory approvals, and begin commercial-scale manufacturing of their products faster than we are able to for our products. They could develop drug discovery technology or products that would render our drug discovery technology and product candidates, and those of our collaborators, obsolete and noncompetitive. They may also employ high throughput screening technologies to the discovery of combination drugs, which may render our technologies or our approach to drug discovery and development obsolete or noncompetitive. Our drug discovery technology will compete against well-established techniques to discover new drugs. If we are unable to compete effectively against these existing techniques and
43
the companies that support them, then we may not be able to commercialize our product candidates or achieve a competitive position in the market. This would adversely affect our ability to generate revenues.
We may have significant product liability exposure which may harm our business and our reputation.
We face exposure to product liability and other claims if our product candidates, products or processes are alleged to have caused harm. These risks are inherent in the testing, manufacturing, and marketing of human therapeutic products and medical devices. We maintain product liability insurance covering our clinical trials of our product candidates. We may not have sufficient insurance coverage, and we may not be able to obtain sufficient coverage at a reasonable cost, if at all. Our inability to obtain product liability insurance at an acceptable cost or to otherwise protect against potential product liability claims could prevent or inhibit the commercialization of any products or product candidates that we develop. If we are sued for any injury caused by our products, product candidates or processes, our liability could exceed our product liability insurance coverage and our total assets. Claims against us, regardless of their merit or potential outcome, may also generate negative publicity or hurt our ability to obtain physician endorsement of our products or expand our business.
We use and generate materials that may expose us to expensive and time-consuming legal claims.
Our development programs involve the use of hazardous materials, chemicals, and biological materials. We are subject to foreign, federal, state and local laws and regulations governing the use, manufacture, storage, and disposal of materials and waste products. We believe that our safety procedures for handling these materials comply with the standards prescribed by laws and regulations. However, we may incur significant costs to comply with current or future environmental laws and regulations. In addition, we cannot completely eliminate the risk of contamination or injury from hazardous materials. In the event of an accident, an injured party may seek to hold us liable for any damages that result. Any liability could exceed the limits or fall outside the coverage of our insurance, and we may not be able to maintain insurance on acceptable terms, if at all.
Risks Related to an Investment in Our Common Stock
Our common stock may have a volatile public trading price.
The market prices for securities of companies comparable to us have been highly volatile. Often, these stocks have experienced significant price and volume fluctuations for reasons unrelated to the operating performance of the individual companies. Factors giving rise to this volatility may include:
| | • | | disclosure of actual or potential clinical results with respect to product candidates we are developing or their compenents; |
| | • | | regulatory developments in both the United States and abroad; |
| | • | | developments concerning proprietary rights, including patents and litigation matters; |
| | • | | public concern about the safety or efficacy of our product candidates or technology, their components, or related technology, or new technologies generally; |
| | • | | public announcements by our competitors or others; and |
| | • | | general market conditions and comments by securities analysts and investors. |
Fluctuations in our operating losses could adversely affect the price of our common stock.
Our operating losses may fluctuate significantly on a quarterly basis. Some of the factors that may cause our operating losses to fluctuate on a period-to-period basis include the status of our preclinical and clinical development programs, level of expenses incurred in connection with our preclinical and clinical development
44
programs, implementation or termination of collaboration, licensing, manufacturing or other material agreements with third parties, non-recurring revenue or expenses under any such agreement, and compliance with regulatory requirements. Period-to-period comparisons of our historical and future financial results may not be meaningful, and investors should not rely on them as an indication of future performance. Our fluctuating losses may fail to meet the expectations of securities analysts or investors. Our failure to meet these expectations may cause the price of our common stock to decline.
Our product development strategy may cause volatility in our stock price, and we may incur significant costs from class action litigation.
Our strategy of initiating proof-of-concept clinical trials for multiple product candidates and making development decisions based on the results of these trials may result in a greater number of public announcements regarding the progress of our development efforts than would be true for a company developing fewer products or advancing products less quickly into proof-of-concept clinical trials. These announcements, including announcements regarding decisions to terminate further development of one or more product candidates, may cause significant volatility in our stock price.
Recently, when the market price of a stock has been volatile, as our stock price may be, holders of that stock have occasionally brought securities class action litigation against the company that issued the stock. If any of our stockholders were to bring a lawsuit of this type against us, even if the lawsuit is without merit, we could incur substantial costs defending the lawsuit. A stockholder lawsuit could also divert the time and attention of our management.
Anti-takeover provisions in our charter documents and provisions of Delaware law may make an acquisition more difficult and could result in the entrenchment of management.
We are incorporated in Delaware. Anti-takeover provisions of Delaware law and our charter documents may make a change in control or efforts to remove management more difficult. Also, under Delaware law, our board of directors may adopt additional anti-takeover measures.
Our charter authorizes our board of directors to issue up to 5,000,000 shares of preferred stock and to determine the terms of those shares of stock without any further action by our stockholders. If the board of directors exercises this power to issue preferred stock, it could be more difficult for a third party to acquire a majority of our outstanding voting stock and vote the stock they acquire to remove management or directors.
Our charter also provides staggered terms for the members of our board of directors. Under Section 141 of the Delaware General Corporation Law, our directors may be removed by stockholders only for cause and only by vote of the holders of a majority of voting shares then outstanding. These provisions may prevent stockholders from replacing the entire board in a single proxy contest, making it more difficult for a third party to acquire control of us without the consent of our board of directors. These provisions could also delay the removal of management by the board of directors with or without cause. In addition, our directors may only be removed for cause and our bylaws limit the ability our stockholders to call special meetings of stockholders.
Our equity incentive plans generally permit our board of directors to provide for acceleration of vesting of options granted under these plans in the event of certain transactions that result in a change of control. If our board of directors uses its authority to accelerate vesting of options, this action could make an acquisition more costly, and it could prevent an acquisition from going forward.
Under Section 203 of the Delaware General Corporation Law, a corporation may not engage in a business combination with any holder of 15% or more of its capital stock until the holder has held the stock for three years unless, among other possibilities, the board of directors approves the transaction. Our board of directors could
45
use this provision to prevent changes in management. The existence of the foregoing provisions could limit the price that investors might be willing to pay in the future for shares of our common stock.
Under our research and license agreement with Angiotech, we have agreed that upon a change of control of us, as defined in the research and license agreement, the agreement would remain in effect, although Angiotech would have the right to terminate the agreement in the six months after a change of control if we were acquired by an entity operating primarily in Angiotech’s field.
| Item 1B. | Unresolved Staff Comments |
None.
We currently lease and occupy approximately 63,000 square feet of laboratory and office space in Cambridge, Massachusetts and approximately 4,800 square feet of office and laboratory space in Singapore for the operations of our subsidiary, CombinatoRx Singapore. We believe that our Cambridge office and laboratory space and our space in Singapore are adequate for our needs through 2008. We also believe we will be able to obtain additional space, as needed, on commercially reasonable terms.
We are currently not a party to any material legal proceedings.
| Item 4. | Submission of Matters to a Vote of Security Holders |
None.
46
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
(a) Market Price of and Dividends on CombinatoRx Common Stock and Related Stockholder Matters.
Our common stock is listed for quotation on the NASDAQ Global Market under the symbol “CRXX.” Trading of our common stock commenced following our initial public offering on November 9, 2005. The following table sets forth the high and low sale prices per share of our common stock as reported on the NASDAQ Global Market for the periods indicated.
| | | | | | |
| | | Common Stock Price |
| | | High | | Low |
Fiscal year ended December 31, 2006 | | | | | | |
First quarter | | $ | 14.50 | | $ | 8.16 |
Second quarter | | $ | 12.20 | | $ | 7.71 |
Third quarter | | $ | 9.38 | | $ | 5.50 |
Fourth quarter | | $ | 9.91 | | $ | 5.70 |
Fiscal year ended December 31, 2007 | | | | | | |
First quarter | | $ | 9.55 | | $ | 6.69 |
Second quarter | | $ | 7.30 | | $ | 5.54 |
Third quarter | | $ | 7.37 | | $ | 5.77 |
Fourth quarter | | $ | 6.91 | | $ | 4.11 |
On March 12, 2008, the reported last sale price of our common stock on the NASDAQ Global Market was $4.13 per share. As of March 12, 2008, there were approximately 71 holders of record of our common stock.
We have never paid cash dividends on our common stock. We currently do not anticipate paying cash dividends on our common stock in the foreseeable future. We currently intend to retain earnings, if any, to finance the growth and development of our business. Payment of future dividends, if any, will be at the discretion of our board of directors and will depend on our financial condition, results of operations, capital requirements, restrictions contained in current or future financing instruments and other factors that our board of directors deems relevant.
47
Securities Authorized For Issuance Under Equity Compensation Plans
| | | | | | | |
Plan Category | | Number of Securities
to be Issued upon
Exercise of
Outstanding
Options, Warrants
or Rights(1) | | Weighted Average
Exercise Price of
Outstanding
Options,
Warrants or Rights | | Number of
Securities
Remaining
Available for
Future Issuance
Under Equity
Compensation
Plan (Excluding
Securities
Reflected in
Column(a) (1)(2) |
| | | (a) | | (b) | | (c) |
Equity compensation plans approved by security holders | | 5,163,492 | | $ | 6.30 | | 1,409,026 |
Equity compensation plans not approved by security holders | | — | | | — | | — |
| | | | | | | |
Total | | 5,163,492 | | $ | 6.30 | | 1,409,026 |
| | | | | | | |
| (1) | As of December 31, 2007. |
| (2) | Our Amended and Restated 2004 Incentive Plan (the “2004 Plan”) contains an “evergreen provision” that allows for an annual increase in the number of shares of common stock available for issuance under the 2004 Plan, which annual increase is and will be added on the first day of each fiscal year from 2007 through 2011, inclusive, and will be equal to the least of: (i) 2,000,000 shares of common stock, (ii) 4% of the outstanding shares on that date or (iii) such lesser amount determined by the board of directors. On January 15, 2008, the Compensation Committee of the board of directors ratified the amount of the annual increase for 2008 as 1,392,887 shares of common stock, or 4% of the outstanding common stock on January 1, 2008. |
48
Comparative Stock Performance Graph
The comparative stock performance graph below compares the cumulative total stockholder return (assuming reinvestment of dividends, if any) from investing $100 on November 9, 2005, the date on which CombinatoRx common stock was first publicly traded, and plotted at the close of the last trading day of the fiscal year ended December 31, 2007, in each of (i) CombinatoRx common stock, (ii) the Nasdaq Global Stock Market Index, which is referred to as the Nasdaq Stock Market Index, and (iii) the Nasdaq Global Stock Market Biotechnology Index, which is referred to as the Nasdaq Biotechnology Index; except, in the case of the Nasdaq Stock Market Index and the Nasdaq Biotechnology Index, the stock performance graph below reflects an investment date of October 31, 2005.
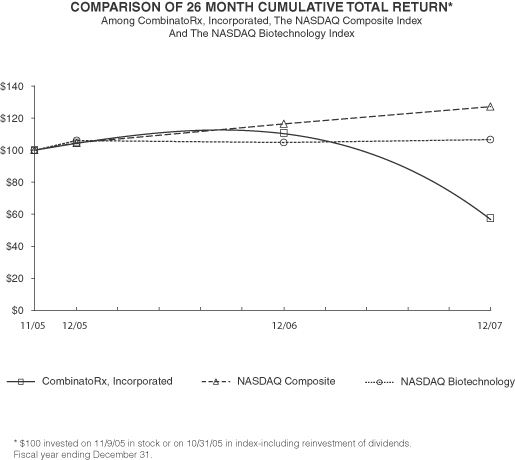
| | | | | | | | |
| | | Cumulative Total Return |
| | | 11/9/05 | | 12/31/05 | | 12/31/06 | | 12/31/07 |
COMBINATORX, INCORPORATED | | 100.00 | | 104.20 | | 110.32 | | 56.56 |
NASDAQ COMPOSITE | | 100.00 | | 104.50 | | 116.42 | | 127.20 |
NASDAQ BIOTECHNOLOGY | | 100.00 | | 106.02 | | 104.76 | | 106.55 |
49
| Item 6. | Selected Financial Data |
The historical financial data set forth below should be read in conjunction with our “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and the related notes thereto appearing elsewhere in this Annual Report on Form 10-K. The selected financial data in this section are not intended to replace the financial statements. We have derived the statement of operations data for the years ended December 31, 2007, 2006 and 2005 and the balance sheet data as of December 31, 2007 and 2006 from our consolidated financial statements included elsewhere in this annual report, which have been audited by Ernst & Young LLP, our independent registered public accounting firm. We derived the statement of operations data for the years ended December 31, 2004 and 2003 and the balance sheet data as of December 31, 2005, 2004 and 2003 from our audited consolidated financial statements which are not included herein. See the notes to the financial statements for an explanation of the method used to determine the number of shares used in determining basic and diluted net loss per common share.
| | | | | | | | | | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | | | 2004 | | | 2003 | |
| | | (in thousands, except share and per share amounts) | |
Statement of Operations Data: | | | | | | | | | | | | | | | | | | | | |
Revenue: | | | | | | | | | | | | | | | | | | | | |
Collaborations | | $ | 12,226 | | | $ | 11,725 | | | $ | 4,143 | | | $ | 178 | | | $ | — | |
Government contracts and grants | | | 2,712 | | | | 1,548 | | | | 515 | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | |
Total revenue | | | 14,938 | | | | 13,273 | | | | 4,658 | | | | 178 | | | | — | |
| | | | | | | | | | | | | | | | | | | | |
Operating expenses: | | | | | | | | | | | | | | | | | | | | |
Research and development | | | 55,434 | | | | 34,094 | | | | 24,059 | | | | 15,896 | | | | 12,145 | |
General and administrative | | | 16,879 | | | | 18,641 | | | | 10,576 | | | | 6,757 | | | | 4,501 | |
| | | | | | | | | | | | | | | | | | | | |
Total operating expenses | | | 72,313 | | | | 52,735 | | | | 34,635 | | | | 22,653 | | | | 16,646 | |
| | | | | | | | | | | | | | | | | | | | |
Loss from operations | | | (57,375 | ) | | | (39,462 | ) | | | (29,977 | ) | | | (22,475 | ) | | | (16,646 | ) |
Interest income | | | 5,391 | | | | 5,913 | | | | 1,296 | | | | 620 | | | | 499 | |
Interest expense | | | (1,304 | ) | | | (722 | ) | | | (834 | ) | | | (403 | ) | | | (176 | ) |
Other (expense) income | | | (9 | ) | | | 34 | | | | — | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | |
Net loss before provision for income taxes | | | (53,297 | ) | | | (34,237 | ) | | | (29,515 | ) | | | (22,258 | ) | | | (16,323 | ) |
Provision for income taxes | | | (46 | ) | | | (51 | ) | | | — | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | |
Net loss | | $ | (53,343 | ) | | $ | (34,288 | ) | | $ | (29,515 | ) | | $ | (22,258 | ) | | $ | (16,323 | ) |
| | | | | | | | | | | | | | | | | | | | |
Net loss per share applicable to common stockholders—basic and diluted | | $ | (1.78 | ) | | $ | (1.26 | ) | | $ | (8.53 | ) | | $ | (33.23 | ) | | $ | (24.13 | ) |
Weighted-average number of common shares used in net loss per share calculation—basic and diluted net loss per common share | | | 30,025,830 | | | | 27,223,319 | | | | 4,169,355 | | | | 869,581 | | | | 860,166 | |
| |
| | | As of December 31, | |
| | | 2007 | | | 2006 | | | 2005 | | | 2004 | | | 2003 | |
| | | (in thousands) | |
Balance Sheet Data: | | | | | | | | | | | | | | | | | | | | |
Cash, cash equivalents and short-term investments | | $ | 108,584 | | | $ | 117,089 | | | $ | 99,747 | | | $ | 35,115 | | | $ | 20,911 | |
Working capital | | | 96,409 | | | | 100,956 | | | | 89,088 | | | | 31,675 | | | | 18,216 | |
Total assets | | | 132,243 | | | | 138,335 | | | | 108,301 | | | | 39,395 | | | | 24,586 | |
Convertible notes payable of subsidiary | | | 13,404 | | | | 9,301 | | | | 5,362 | | | | — | | | | — | |
Long-term debt, less current maturities | | | 5,415 | | | | 2,527 | | | | 816 | | | | 1,729 | | | | 560 | |
Minority interest in subsidiary | | | 2,792 | | | | 2,669 | | | | 2,542 | | | | — | | | | — | |
Convertible preferred stock and redeemable convertible preferred stock | | | — | | | | — | | | | — | | | | 103,843 | | | | 65,230 | |
Accumulated deficit | | | (186,213 | ) | | | (132,748 | ) | | | (98,333 | ) | | | (68,658 | ) | | | (43,445 | ) |
Total stockholders’ equity (deficit) | | | 75,235 | | | | 87,050 | | | | 68,350 | | | | (70,268 | ) | | | (44,178 | ) |
50
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations in conjunction with our financial statements and their notes appearing elsewhere in this annual report. The following discussion contains forward-looking statements that involve risks and uncertainties. Our actual results and the timing of certain events could differ materially from those anticipated in these forward-looking statements as a result of certain factors, including those discussed below and elsewhere in this annual report, particularly under the heading “Risk Factors That May Affect Future Results.”
Overview
We are a biopharmaceutical company focused on developing synergistic combinations of approved drugs, with a portfolio of product candidates in phase 2 clinical development, including our lead product candidate CRx-102. To date, we have devoted substantially all of our resources to the development of our drug discovery technology and the research and development of our drug candidates, including conducting preclinical and clinical trials and seeking protection for our technology and product candidates. Since our inception in March 2000, we have had no revenue from product sales, and have funded our operations principally through the sale of equity securities, debt financings and revenue from license, research and collaboration agreements as well as government contracts and grants.
We have never been profitable and, as of December 31, 2007, we have an accumulated deficit of $186.2 million. We had net losses of $53.3 million for the year ended December 31, 2007, $34.3 million for the year ended December 31, 2006 and $29.5 million for the year ended December 31, 2005.
We are currently advancing four product candidates, CRx-102, CRx-401, CRx-191 and CRx-197 into or through clinical research and development. We have two other product candidates in our portfolio, CRx-139 for immuno-inflammatory diseases and CRx-170 for chronic pain, which have completed phase 2a clinical trials, but are not currently being advanced into later-stage clinical trials, and we have a number of product candidates in preclinical development. Our most advanced product candidate, CRx-102, is a novel dissociated glucocorticoid product candidate being developed to treat immuno-inflammatory disorders. We are currently conducting two phase 2b clinical trials of CRx-102, one in knee osteoarthritis, which started in September 2007, and one in rheumatoid arthritis, which started in 2008. Our product candidate CRx-191 is a topical synergistic combination drug candidate with a novel multi-target mechanism that inhibits TNF-a and interferon-gamma, key cell mediators of inflammation. CRx-191 contains the mid-potency glucocorticoid, mometasone, and a low dose of the tricyclic anti-depressant, nortriptyline, co-formulated as a topical cream for the treatment of psoriasis and other steroid-responsive dermatoses. We conducted a skin atrophy clinical study of CRx-191 during 2007 and conducted a phase 2a clinical trial of CRx-191 in subjects with psoriasis. Our product candidate CRx-197 is a selective cytokine modulator containing low concentrations of the antihistamine loratadine, and the tricyclic anti-depressant nortriptyline, neither of which is approved for the treatment of topical dermatoses. This combination has been co-formulated as a topical cream for the treatment of atopic dermatitis and other inflammatory dermatoses. We are currently planning to conduct a phase 2a clinical trial of CRx-197 in subjects with selected inflammatory dermatoses during 2008. Our product candidate CRx-401 is a novel oral anti-diabetic that is currently being studied in a phase 2a clinical trial in subjects with Type 2 diabetes.
We expect to incur significant and increasing operating losses for the foreseeable future as we advance our product candidates from discovery through preclinical and clinical trials and seek regulatory approval and eventual commercialization. We will need to raise additional capital and generate significant revenues to achieve profitability and may never do so.
51
Our management currently uses consolidated financial information in determining how to allocate resources and assess performance. We have determined that we conduct operations in one business segment. The majority of our revenues since inception have been generated in the United States, and the majority of our long-lived assets are located in the United States.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with United States generally accepted accounting principles, or GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. On an ongoing basis, we evaluate our estimates and judgments, including those related to revenue, stock-based compensation and accrued expenses. We base our estimates on historical experience, known trends and events and various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe that several accounting policies are important to understanding our historical and future performance. We refer to these policies as “critical” because these specific areas generally require us to make judgments and estimates about matters that are uncertain at the time we make the estimate, and different estimates—which also would have been reasonable—could have been used, which would have resulted in different financial results. We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our financial statements. It is important that the discussion of our operating results that follows be read in conjunction with the critical accounting policies discussed below.
Revenue Recognition
We have entered into collaborative research and development agreements with other pharmaceutical and biotechnology companies, government agencies and charitable foundations. These agreements are generally in the form of research and development and license agreements. The agreements are for early-stage compounds and are generally focused on specific disease areas. The agreements provide for nonrefundable up-front payments, milestone payments upon achieving significant milestone events and in some cases ongoing research funding. The agreements also contemplate royalty payments on sales if and when the product receives marketing approval by the FDA or other regulatory agency.
We recognize revenue in accordance with Emerging Issues Task Force, Issue No. 00-21, “Revenue Arrangements with Multiple Deliverables” (“EITF 00-21”) and Securities and Exchange Commission Staff Accounting Bulletin No. 104, “Revenue Recognition in Financial Statements” (“SAB 104”). Revenue arrangements with multiple deliverables are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered elements. The consideration received is allocated among separate elements based on their respective fair values. Revenue is recognized when there is persuasive evidence of an arrangement, delivery has occurred, the price is fixed or determinable and collection is reasonably assured. License fees or other amounts received in advance of performance obligations, or in cases where we have a continuing obligation to perform services, are deferred and recognized over the performance period. Revenues from milestone payments that are deemed to be substantive and represent the culmination of a separate earnings process are recorded when the milestone is achieved. Contract revenues are recorded as the services are performed. The periods over which revenue should be recognized are subject to estimates by management and may change over the course of the collaborative agreement. Such a change could have a material impact on the amount of revenue we record in future periods.
52
Revenue under government grants or cost reimbursement contracts is recognized as we perform the underlying research and development activities.
Stock-Based Compensation
We adopted Statement of Financial Accounting Standards No. 123 (revised 2004), “Share-Based Payment” (“SFAS 123R”) on January 1, 2006, using the prospective method for stock option grants prior to January 1, 2005 and the modified-prospective transition method for stock option grants and restricted stock issued after January 1, 2005. As a result, the fair value of unamortized compensation expense from stock options granted prior to January 1, 2005 is not included in the statement of operations. Stock options granted after January 1, 2005 are included due to the fact that once we were in registration for our initial public offering, we included a volatility factor in our Black-Scholes calculations for purposes of the proforma footnote disclosures under SFAS 123.
Prior to January 1, 2006, we accounted for our stock-based compensation plans under the intrinsic-value method prescribed in Accounting Principles Board Opinion No. 25 “Accounting for Stock Issued to Employees,” (“APB 25”) and related interpretations as permitted by Statement of Financial Accounting Standards No. 123, “Accounting for Stock-Based Compensation.” (“SFAS 123”). Under APB 25, when the exercise price of stock options granted to employees equals the market price of the common stock on the date of grant, no compensation expense is recorded. When the exercise price of options granted to employees is less than the market price of the common stock on the date of grant, compensation expense is recognized over the vesting period.
SFAS 123R requires share-based payments to employees to be measured at fair value. However, the valuation of employee stock options is an inherently subjective process, since market values are generally not available for long-term, non-transferable employee stock options. Accordingly, an option pricing model is utilized to derive an estimated fair value. We use the Black-Scholes pricing model in order to calculate the estimated fair value for our stock options. The Black-Scholes calculation requires input values for the following variables:
| | • | | the stock option exercise price, |
| | • | | the expected term of the option, |
| | • | | the grant date price of our common stock, |
| | • | | the expected volatility of our common stock, |
| | • | | expected dividends on our common stock, and |
| | • | | the risk-free interest rate for the expected option term. |
Making estimates for the values required by Black-Scholes is more easily accomplished if a company has a substantial public market trading history. Since we have a limited history as a publicly traded entity, we relied on data from several peer companies similar to ours in generating our assumptions. Of the variables mentioned above, the estimates for expected term and stock price volatility are the most subjective. In accordance with the guidance provided in Staff Accounting Bulletin No. 107, “Share-Based Payment” (“SAB 107”), we generated our estimates by performing an analysis of these peer companies’ expected term and volatility data as reported in their filings with the SEC. Our peers had a similar amount of in-the-money stock options (options with exercise prices below the current market price of the underlying stock), a trading history of sufficient duration (at least as long as its expected term) and stock option life and vesting terms similar to ours (10 year options with approximately 4 year vesting).
The expected term represents the period of time that options granted are expected to be outstanding prior to being exercised. We believe the expected term of our stock options will follow the average term of our peer group. As a result, our weighted-average term was 5.8 years for the year ended December 31, 2007. The range of
53
our weighted-average expected terms was between 5.6 and 6.1 years for the year ended December 31, 2007. Volatility represents the expected change in the price of our common stock during the expected term of our stock options. To estimate our volatility, we calculated the average historical stock volatility of our peer group (over a period equal to our expected term) including a percentage applied to our own historical volatility and calculated our weighted- average volatility to be 61.2% for the year ended December 31, 2007. As we gain further trading history, we will continue to increase the weight applied to the actual volatility of our common stock in the overall volatility calculation.
The risk-free interest rates used in the Black-Scholes model are based on the United States Treasury yield curve in effect for periods corresponding with the expected term of the stock option. We have not issued dividends in the past and have no plans to issue dividends for the foreseeable future.
The specific valuation assumptions discussed above have been applied to stock options that we granted subsequent to our adoption of SFAS 123R on January 1, 2006. However, approximately 30% of the stock-based compensation expense recorded in the year ended December 31, 2007 relates to the continued vesting of stock options that were granted in 2005 and the amortization of the stock-based compensation expense for stock options granted with exercise prices below fair value in 2003 and 2004. In accordance with the transition provisions of SFAS 123R, the grant date fair value estimates for these stock options have not been changed. The specific valuation assumptions that were utilized for purposes of deriving an estimate for the Black-Scholes value of stock options granted in 2005 are disclosed in our consolidated financial statements.
Upon the adoption of SFAS 123R, we also began to estimate the level of stock-based award forfeitures expected to occur and record compensation cost only for those awards that are ultimately expected to vest. This requirement applies to all awards that are not yet vested, including awards granted prior to January 1, 2006. We believe that using an average of our own historical data for percentage of option cancellations and actual employee turnover rates derived from periods of business activity most likely to be representative of future periods to be the most appropriate measure of forfeitures. Changes in estimated forfeitures are recognized through a cumulative catch-up adjustment in the period of change and will also impact the amount of compensation expense to be recognized in future periods. Our average forfeiture rate was 5.8% for the year ended December 31, 2007.
Beginning in 2006, we began to make grants of restricted stock in addition to grants of stock options. As of the year ended December 31, 2007 there were 165,938 shares of unvested restricted stock outstanding that had been granted to certain employees and consultants at $0.001 per share.
As of December 31, 2007, there was approximately $13.9 million of total stock-based compensation expense not yet recognized relating to non-vested awards granted under our option plans and restricted stock agreements as calculated under APB No. 25 and SFAS 123R. This expense is net of estimated forfeitures and is expected to be recognized over a weighted-average period of approximately 2.4 years.
The amount of stock-based compensation expense to be recorded in any future period cannot be accurately predicted due to the uncertainty of future grant levels and actual forfeitures to be recorded. Additionally, changes to the assumptions used in the Black-Scholes model could cause a material change in the amount of stock-based compensation expense to be recorded in future reporting periods.
Accrued Expenses
As part of the process of preparing our consolidated financial statements, we are required to estimate certain accrued expenses. This process involves identifying services that third parties have performed on our behalf and estimating the amount of service performed and the associated cost incurred for these services as of the balance sheet date in our consolidated financial statements. Examples of estimated accrued expenses for our business are contract service fees, such as amounts due to clinical research organizations who are supporting clinical trials for
54
our product candidates CRx-102, CRx-401 and CRx-191, preclinical and toxicology research services providers and formulation development providers, professional service fees, such as attorneys and accountants, and investigators in conjunction with our clinical trials. In connection with these service fees, our estimates are most affected by our understanding of the status and timing of services provided relative to the actual level of services incurred by the service providers. In the event that we do not identify certain costs that have been incurred or we under- or over-estimate the level of services or the costs of such services, our reported expenses for a reporting period could be understated or overstated.
Financial Operations Overview
Revenue. We have not generated any product revenue since our inception and do not expect to generate any revenue from the sale of products for the foreseeable future. All of our revenue to date has been derived from license and research and development payments we have received from our collaborators and from grants we received from the National Institutes of Allergy and Infectious Disease (NIAID) and the Singapore Economic Development Board. We will seek to generate revenue from a combination of research funding, up-front license fees, milestone payments, royalties and product sales in future periods.
Research and Development. Our research and development expenses consist primarily of costs associated with the clinical, preclinical and pharmaceutical development of our lead product candidates, CRx-102, CRx-191, CRx-197 and CRx-401, and our other product candidates such as CRx-139 and CRx-170, as well as expenses incurred in connection with developing and advancing our drug discovery technology and identifying new product candidates. These expenses consist primarily of compensation and related expenses for research and development personnel, supplies and materials, facility costs and depreciation as well as direct external costs such as costs for clinical trials including related contract research, formulation and manufacturing and fees paid to other contract research organizations or external consultants. We charge all research and development expenses to operations as incurred.
At this time, due to the risks inherent in the clinical trial process and given the earlier stage of development of our product candidates, we are unable to estimate with any certainty the costs we will incur in the continued development of our product candidates. Clinical development timelines, likelihood of success and total costs of clinical trials vary widely. We are currently focused primarily on advancing the clinical development of the following product candidates:
| | • | | CRx-102 to treat immuno-inflammatory disorders, by conducting two phase 2 clinical trials of CRx-102, one in knee osteoarthritis and one in rheumatoid arthritis; |
| | • | | CRx-191 to treat psoriasis and other steroid-responsive dermatoses, by conducting a skin atrophy clinical study and a phase 2 clinical trial in subjects with psoriasis; |
| | • | | CRx-197 to treat atopic dermatitis and other inflammatory dermatoses, by preparing to conduct a phase 2 clinical trial in subjects with selected inflammatory dermatoses; and |
| | • | | CRx-401 to treat metabolic diseases, by conducting a phase 2a clinical trial in subjects with Type 2 diabetes. |
In addition, during the year ended December 31, 2007, we completed phase 2 clinical trials in rheumatoid arthritis for our product candidates CRx-150 and CRx-139. We also made significant investments in the preclinical development of our product candidate CRx-170, as well as in the discovery and development of other new product candidates.
During the first quarter of 2007, we implemented a project costing methodology and system which enabled us to allocate external direct costs and internal direct costs such as personnel costs, supplies and materials for certain research and development departments directly to projects for periods after January 1, 2007. The table below summarizes our allocation of research and development expenses using this costing methodology to our
55
clinical programs CRx-102, CRx-191, CRx-401, CRx-150 and CRx-139 for the year ended December 31, 2007. As initially implemented in 2007, our project costing methodology did not allocate the personnel and other indirect costs from all of our research and development departments to specific clinical and preclinical programs, and such unallocated costs are further summarized in the table below. Unallocated clinical program costs consist primarily of the personnel and other expenses for our clinical operations, medical affairs, biostatistics, data systems, medical writing and clinical program leadership departments, all of which supported the development of our clinical product candidates CRx-102, CRx-191, CRx-401, CRx-150 and CRx-139 during 2007. Preclinical program costs consist of the personnel and other expenses allocated to our internally funded preclinical programs, including CRx-170 and CRx-197 and the research activities of CombinatoRx Singapore, as well as the direct costs allocated to all of our research collaborations, including the personnel costs of our alliance management department. Unallocated clinical and preclinical program costs consist primarily of the personnel and other expenses for our formulations, pharmacology, regulatory, quality, new products and discovery departments, all of which supported the development of both our clinical product candidates CRx-102, CRx-401, CRx-191, CRx-150 and CRx-139 as well as our preclinical product candidates. Infrastructure and support costs consist of facility costs, depreciation and amortization and costs for research and development support personnel such as our informatics and facilities departments.
| | | |
| | | Year Ended
December 31, 2007
(in thousands) |
CRx-102 | | $ | 14,175 |
CRx-191 | | | 1,761 |
CRx-150 | | | 937 |
CRx-139 | | | 719 |
CRx-401 | | | 648 |
Unallocated clinical program costs | | | 3,924 |
Preclinical program costs | | | 10,857 |
Unallocated clinical and preclinical program costs | | | 10,289 |
Infrastructure and support costs | | | 8,492 |
Non-cash employee and non-employee stock-based compensation expense | | | 3,632 |
| | | |
Total research and development expense | | $ | 55,434 |
| | | |
Prior to January 1, 2007, we did not track our internal direct and indirect research and development costs on an individual product candidate basis. We used our research and development resources, including certain employees and our drug discovery technology, across multiple drug development programs. However, direct expenses for clinical trials associated with CRx-102, CRx-150, CRx-139, CRx-170, CRx-026 and other product candidates were $3.2 million for the year ended December 31, 2006 and $4.6 million for the year ended December 31, 2005. Our total research and development expenses for the years ended December 31, 2006 and 2005 were $34.1 million and $24.1 million, respectively.
We expect our research and development costs to be substantial and to increase as we advance our product candidate CRx-102 through later stage clinical trials and as we plan to advance our product candidates CRx-191, CRx-197 and CRx-401 into and through phase 2a clinical trials. In addition, we anticipate that we will select product candidates and research projects for further development on an ongoing basis in response to their preclinical and clinical success and commercial potential. Due to the fact that our most advanced product candidates are in the earlier stages of development, we cannot estimate anticipated completion dates and when we might receive material net cash inflows from future collaboration agreements with sponsored research funding, up-front payments, milestones or royalties. Further, we expect increased research and development expenses as a result of the activities of CombinatoRx Singapore and the activities under our collaboration agreements with Angiotech, CHDI, CFFT and the DMD foundations.
56
General and Administrative. General and administrative expense consists primarily of salaries and related expenses for personnel in administrative, finance, investor relations, business development, commercialization, marketing, intellectual property and human resource functions. Other costs include legal costs of pursuing patent protection of our intellectual property, unallocated facility costs and professional fees for legal and other consulting services. After our initial public offering in November 2005, we have experienced increases in general and administrative expense relating to continuing to operate as a public company with a greater number of employees. These increases include legal fees, accounting fees and directors’ and officers’ insurance premiums as well as fees for investor and public relations services.
Results of Operations
Years Ended December 31, 2007 and 2006
Revenue. For the year ended December 31, 2007 we recorded $14.9 million of revenue from our research and collaboration agreements with Angiotech, CFFT, CHDI, Fovea, the DMD foundations and LSTM and from government contracts and grants from NIAID, SAIC (NINDS) and the Singapore Economic Development Board. For the year ended December 31, 2006 we recorded $13.3 million of revenue from our research and collaboration agreements with Angiotech, CHDI, CFFT and the Spinal Muscular Atrophy Foundation and Sirtris Pharmaceuticals and from grants from NIAID, SAIC (NINDS) and the Singapore Economic Development Board. In April 2007, we received an interim indirect cost negotiation agreement, or interim rate agreement, from the United States Department of Health and Human Services related to the rates we are allowed to charge under our NINDS and NIAID grants. The rates provided in this interim agreement are retroactive to January 2005 and extend through June 2008. We are currently in the process of finalizing our provisional rates for the 2005 and 2006 periods. As a result, revenue recorded in the year ended December 31, 2007 is based upon the interim rate agreement and may be subject to additional adjustments in future periods should our final rate agreement vary from our interim rate agreement.
The $1.6 million increase in revenue is primarily due to our receipt of the approved interim rates from our SAIC (NINDS) and NIAID grants which resulted in $0.8 million of cumulative incremental revenue that we recorded upon obtaining evidence of approval of the rates, the amendment of our collaboration with Fovea which led to the recognition of $0.7 million of deferred revenue, as well as $2.7 million in new research and development collaboration revenue with CFFT, LSTM and the DMD foundations. These increases are offset by a decrease of $2.4 million in revenue recognized under our research and license agreement with Angiotech. In June 2007, Angiotech agreed to extend the research project under our research and license agreement beyond the original 30-month term to a total term of five years for an additional license execution fee of $7.0 million, which we received in October 2007. As a result of the extension of the research project, we revised our revenue recognition based on this change in estimate from approximately $2.3 million in revenue recognized per quarter to approximately $1.2 million in revenue recognized per quarter. This resulted in $6.6 million of revenue being recognized from the Angiotech agreement for the year ended December 31, 2007, compared to revenue recognized of $9.0 million under the Angiotech agreement for the year ended December 31, 2006.
Research and Development. Research and development expense for the year ended December 31, 2007 was $55.4 million compared to $34.1 million for the year ended December 31, 2006. The $21.3 million increase from the 2006 period to the 2007 period was primarily due to an increase of $6.5 million for personnel-related expenses in order to support expanded activities in our research, clinical development, and regulatory departments, an $8.8 million increase in formulation development, pharmacology and toxicology costs, a $2.7 million increase in external clinical trial costs, a $1.8 million increase in depreciation expense and a $1.4 million increase in clinical and regulatory consulting to support a higher number of research and development collaborations.
General and Administrative. General and administrative expense for the year ended December 31, 2007 was $16.9 million compared to $18.6 million for the year ended December 31, 2006. The $1.7 million decrease from the 2006 period to the 2007 period was primarily due to a decrease of $0.6 million relating to external general
57
and patent related legal expenses, a $0.4 million decrease in consulting, a $0.2 million decrease in bonus expense, a $0.2 million decrease in recruiting expenses and a $0.1 million decrease in travel expenses.
Interest Income. Interest income decreased to $5.4 million for the year ended December 31, 2007 from $5.9 million for the year ended December 31, 2006. The decrease in interest income was primarily caused by decreases in our average cash and short-term investments balances and lower average interest rates for the securities held in our investment portfolio.
Interest Expense. Interest expense increased to $1.3 million for the year ended December 31, 2007 from $0.7 million for the year ended December 31, 2006. The $0.6 million increase was primarily due to higher average debt balances on our equipment lines of credit with GE Capital and the issuance of additional convertible notes by CombinatoRx Singapore.
Years Ended December 31, 2006 and 2005
Revenue. For the year ended December 31, 2006 we recorded $13.3 million of revenue from our research and collaboration agreements with Angiotech, CHDI, CFFT, the Spinal Muscular Atrophy Foundation and Sirtris Pharmaceuticals and from government contracts and grants from NIAID, SAIC (NINDS) and the Singapore Economic Development Board. For the year ended December 31, 2005 we recorded $4.7 million of revenue from our research and development collaborations with Angiotech, Sirtris, CHDI, Novartis, the Spinal Muscular Atrophy Foundation, and ABC2 and from government contracts and grants from NIAID and SAIC (NINDS). This increase is primarily due to a full year of revenue recognition under the Angiotech research and license agreement compared to one quarter of revenue recognized in the year ended December 31, 2005.
Research and Development. Research and development expense for the year ended 2006 was $34.1 million compared to $24.1 million for the year ended December 31, 2005. The $10.0 million increase from the 2005 period to the 2006 period was primarily due to an increase of $3.5 million for personnel, outside services and travel related expenses in order to support expanded activities in our research, clinical development, and regulatory departments, a $2.3 million increase in laboratory and clinical related expenses incurred to support a higher number of research and development collaborations and $4.1 million of facilities and informatics related expenses resulting from our move and expansion into new facilities in Cambridge, Massachusetts, and an increase of $0.1 million in stock-based compensation expense.
General and Administrative. General and administrative expense for the year ended December 31, 2006 was $18.6 million compared to $10.6 million for the year ended 2005. The $8.0 million increase from the 2005 period to the 2006 period was primarily due to a $5.1 million increase in personnel and professional services related expenses to support our operation as a publicly traded company as well as the expansion of our commercial development capabilities, a $1.8 million increase relating to non-cash stock-based compensation expense and a $1.1 million increase in facilities, informatics, insurance and permitting related expenses resulting from our move and expansion into new facilities in Cambridge, Massachusetts.
Interest Income. Interest income increased to $5.9 million for the year ended December 31, 2006 from $1.3 million for the year ended December 31, 2005. The increase in interest income was primarily caused by increases in our average cash and short-term investments balances. The increase in our average cash balance for the year ended December 31, 2006 as compared to the year ended December 31, 2005 resulted from the completion of our initial public offering in November 2005, payments and investments from our collaboration with Angiotech, the closing of a private placement of common stock in March 2006 as well as reimbursement of leasehold improvement expenses from the landlord of our Cambridge facility.
Interest Expense. Interest expense decreased to $0.7 million for the year ended December 31, 2006 from $0.8 million for the year ended December 31, 2005. In 2005, we recorded $0.4 million in non-cash interest expense associated with the write-off of the value of warrants issued in connection with a terminated debt financing, partially offset by higher average debt balances throughout 2006.
58
Liquidity and Capital Resources
Since our inception in March of 2000 until our initial public offering on November 9, 2005, we have funded our operations principally through the private placement of equity securities, which provided aggregate net cash proceeds of approximately $104.9 million. Our initial public offering provided aggregate net cash proceeds of $43.8 million, a private placement of our common stock in March 2006 provided net proceeds to us of $45.4 million and a public offering of our common stock in October 2007 provided net proceeds of $33.0 million. We have also generated funds from debt financing and payments from our collaboration partners. As of December 31, 2007, we had cash, cash equivalents and short-term investments of approximately $112.6 million, which includes $4.1 million of restricted cash. Our funds are primarily currently invested in short-term investment grade securities and money market funds. As a matter of investment policy, we do not invest in auction rate securities, and we do not have any exposure to mortgage-backed securities.
Based on our current operating plans, we expect our existing resources to be sufficient to fund our planned operations through approximately the fourth quarter of 2009. However, we may require significant additional funds earlier than we currently expect if our research and development expenses exceed our current expectations or our collaboration funding is less than our current expectations. We may seek additional funding through collaboration agreements and public or private financings of debt or equity capital. Additional funding may not be available to us on acceptable terms or at all. If we are unable to obtain funding on a timely basis, we may be required to significantly curtail one or more of our research or development programs. We also could be required to seek funds through arrangements with collaborators or others that may require us to relinquish rights to some of our technologies or product candidates which we would otherwise pursue on our own.
Our operating activities used cash of $43.3 million in the year ended December 31, 2007, used cash of $21.7 million in the year ended December 31, 2006 and provided cash of $2.4 million in the year ended December 31, 2005. Our increase in the net use of cash in operating activities in the 2007 period is primarily attributed to our $19.1 million increase in net loss for the 2007 period, as well as adjustments for changes in working capital experienced during the periods. The change in the use of cash in operations for the 2006 period as compared to the 2005 period was due primarily to year over year increases of $1.9 million in non-cash stock-based compensation expense, deferred rent of $2.0 million, prepaid and other assets of $2.0 million and accounts payable and accrued expenses of $4.9 million. These favorable working capital items were offset by our $33.5 million year over year decrease in the deferred revenue balance. We received a $27.0 million up-front payment from Angiotech in the fourth quarter of 2005, which is being amortized over the three-year term of the agreement. In 2006, we recorded a full year of amortization of this payment compared to three months in 2005. The net cash provided in the 2005 period was primarily the result of the receipt of the up-front license fee from Angiotech.
Our investing activities provided cash of $4.5 million for the year ended December 31, 2007 and used cash of $30.1 million and $61.7 million for the years ended December 31, 2006 and 2005, respectively. The cash provided by investing activities in 2007 was due to sales and maturities of short-term investments. The cash used in investing activities in all periods was primarily due to purchases of short term investments. Our investing activities in all periods consisted of purchases of property and equipment, purchases and maturities of marketable securities and additions of restricted cash to secure standby letters of credit for our Cambridge, Massachusetts facility lease. We incurred $6.6 million in capital expenditures for the year ended December 31, 2007 principally related to leasehold improvements, lab and computer equipment and furniture and fixtures for our new office and lab facility in Cambridge, Massachusetts.
Our financing activities since inception consisted primarily of the sale of preferred stock to private investors in the net amount of $89.9 million, financing associated with the formation of CombinatoRx Singapore of $11.5 million, net proceeds from our equipment lines of credit of $15.9 million, net proceeds from our initial public offering of $43.8 million, net proceeds from the private placement of our common stock in March 2006 of $45.4 million and net proceeds from a public offering of our common stock in October 2007 of $33.0 million. Our
59
financing activities provided $41.2 million of cash proceeds as of December 31, 2007 compared to $52.0 million and $66.2 million in the years ended December 31, 2006 and 2005, respectively. The financing activities in the year ended December 31, 2007 consisted of net proceeds of $33.0 million from a public offering of our common stock in October 2007, a $3.5 million convertible promissory note issued by CombinatoRx Singapore to Biomedical Sciences and promissory notes of $6.8 million issued by us or CombinatoRx Singapore to General Electric Capital Corporation or its affiliates for capital equipment purchases, offset by $2.7 million in notes payable repayments. The financing activities in the year ended December 31, 2006 consisted of net proceeds of $45.4 million from a private placement of our common stock in March 2006, a $3.5 million convertible promissory note issued by CombinatoRx Singapore to Biomedical Sciences and promissory notes of $3.9 million issued by us to General Electric Capital Corporation for capital equipment purchases, offset by $1.4 million in notes payable repayments. The cash provided by financing activities in the year ended December 31, 2005 was due to the receipt of proceeds of $43.8 million related to the issuance of common stock in our initial public offering, the issuance of Series E preferred stock to Angiotech for $15.0 million and the issuance of a convertible promissory note and preferred stock to BioMedical Sciences by CombinatoRx Singapore for proceeds of $8.0 million.
We have received approximately $106.4 million in payments through December 31, 2007, from our collaborations and research and development agreements with Angiotech, Fovea Pharmaceuticals, HenKan Pharmaceutical Company, CHDI, CFFT, the DMD foundations, LSTM, Novartis, Sirtris, the SMA Foundation, Accelerate Brain Cancer Cure, Inc. and from government contracts and grants from NIAID, SAIC (NINDS) and the Singapore Economic Development Board, including the $15.0 million equity investment by Angiotech. We expect that our sources of funding for the next several years will also include, subject to our satisfying conditions, additional research funding, license fees, potential milestone payments and royalties relating to our collaboration and research and development agreements with Angiotech, CFFT, CHDI, Fovea, HenKan Pharmaceutical Company, LSTM, the DMD foundations and government grants from NIAID and the Singapore Economic Development Board, and any other collaborative agreements into which we may enter.
In July 2004, we refinanced our equipment line of credit with General Electric Capital Corporation, or GE Capital, and borrowed $3.0 million under the new arrangement. In June 2005, the 2004 agreement with GE Capital was amended to establish a new line of credit which enabled us to borrow an additional $1.0 million through June 2006, and we borrowed approximately $1.0 million under this amended secured equipment line of credit with GE Capital prior to its expiration. Amounts borrowed in 2004 through 2006 under the line of credit were repayable over 36 months.
In June 2006, we amended the secured equipment line of credit to increase the line of credit by $3.3 million, and in June 2006, we borrowed approximately $1.6 million under the amended agreement. In December 2006, we borrowed an additional $1.5 million under the secured equipment line of credit. In March 2007, we amended the secured line of credit with GE Capital to increase the line of credit by $0.9 million, and in March 2007, we borrowed the remaining $1.1 million available under the amended line of credit.
In June 2007, we further amended the secured equipment line of credit with GE Capital to increase the line of credit by an additional $4.0 million, of which we borrowed approximately $2.9 million in June 2007 and $0.8 million in September 2007. In December 2007, we amended the secured equipment line of credit with GE Capital to increase the line of credit by $0.1 million, and we also borrowed approximately $0.4 million under the amended line of credit. Additional amounts borrowed in 2006 through 2007 under the amended line of credit are repayable over 48 months in the case of laboratory and scientific equipment, 36 months in the case of other equipment and 24 months in the case of software. Borrowings under the equipment line of credit with GE Capital are collateralized by the financed assets. Once drawdowns under the equipment line of credit with GE Capital are repaid, they may not be reborrowed. The equipment line of credit with GE Capital contains a subjective acceleration clause which provides GE Capital with the ability to demand repayment of the loan early upon a material adverse event, as defined in the agreement with GE Capital. As of December 31, 2007, there was approximately $8.3 million outstanding under this line of credit, and no funds remained available for borrowing.
60
The following table depicts our history of borrowings with GE Capital and includes the remaining principal and interest balances at December 31, 2007:
Company Borrowings
| | | | | | | | | | | | |
Borrowing Date | | Principal Amount
(in thousands) | | Interest
Rate | | | Remaining Principal
Balance at 12/31/07
(in thousands) | | Remaining Interest
at 12/31/07
(in thousands) |
July 2004 | | $ | 3,000 | | 8.42 | % | | $ | — | | $ | — |
June 2005 | | | 159 | | 9.76 | % | | | 34 | | | 1 |
March 2006 | | | 806 | | 10.52 | % | | | 389 | | | 30 |
June 2006 | | | 1,607 | | 10.68 | % | | | 1,004 | | | 121 |
December 2006 | | | 1,479 | | 9.98 | % | | | 1,098 | | | 137 |
March 2007 | | | 1,101 | | 10.12 | % | | | 908 | | | 135 |
June 2007 | | | 2,912 | | 10.46 | % | | | 2,603 | | | 445 |
September 2007 | | | 781 | | 10.46 | % | | | 749 | | | 142 |
December 2007 | | | 442 | | 10.46 | % | | | 442 | | | 87 |
| | | | | | | | | | | | |
| | $ | 12,287 | | | | | $ | 7,227 | | $ | 1,098 |
| | | | | | | | | | | | |
In February 2007, CombinatoRx Singapore entered into a $2.1 million secured equipment line of credit with GE Capital Services Pte. Ltd. and borrowed $1.2 million under the line of credit in February 2007 and $0.4 million under the line of credit in November 2007. The line of credit was available through November 30, 2007 and is secured by a fixed charge security interest in Singapore over the equipment financed. We also provided a corporate guaranty of payment in connection with the line of credit. Amounts borrowed under the line of credit are repayable over 48 months in the case of laboratory and scientific equipment and over 36 months in the case of other equipment. As of December 31, 2007, there was $1.5 million outstanding under this line of credit.
The following table depicts the history of CombinatoRx Singapore’s borrowings under its line of credit and includes the remaining principal and interest balance as of December 31, 2007:
CombinatoRx Singapore Borrowings
| | | | | | | | | | | | |
Borrowing Date | | Principal Amount
(in thousands) | | Interest
Rate | | | Remaining Principal
Balance at 12/31/07
(in thousands) | | Remaining Interest
at 12/31/07
(in thousands) |
February 2007 | | $ | 779 | | 10.42 | % | | $ | 622 | | $ | 108 |
February 2007 | | | 457 | | 10.45 | % | | | 330 | | | 39 |
November 2007 | | | 10 | | 10.10 | % | | | 10 | | | 1 |
November 2007 | | | 339 | | 10.13 | % | | | 325 | | | 68 |
| | | | | | | | | | | | |
| | $ | 1,585 | | | | | $ | 1,287 | | $ | 216 |
| | | | | | | | | | | | |
On March 9, 2006, we entered into an amendment to the lease for our Cambridge, Massachusetts office and laboratory facility in order to secure additional space. Because the amendment increases the total space we rent at the Cambridge location, we were obligated to increase the amount of the standby letter of credit required under the lease from $2.5 to $4.0 million. In addition, under the lease agreement as amended, we were entitled to receive $6.9 million in tenant improvement funds from our landlord. We initially submitted approximately $4.0 million in tenant improvement reimbursements to the landlord for which we received payment in July 2006. We submitted approximately $2.9 million in tenant improvements during the first quarter of 2007. We received approximately $1.5 million in February 2007, approximately $1.3 million in April 2007 and the remaining $0.1 million in August 2007.
61
Contractual Obligations and Commitments
The following table summarizes our contractual obligations at December 31, 2007 and the effects such obligations are expected to have on our liquidity and cash flows in future periods.
| | | | | | | | | | | | | | | |
Contractual Obligations | | Total | | 2008 | | 2009
through
2010 | | 2011
through
2012 | | After
2012 |
Short and long-term debt: | | | | | | | | | | | | | | | |
General Electric Capital(1) | | $ | 9,828 | | $ | 3,836 | | $ | 5,414 | | $ | 578 | | $ | — |
Notes payable issued to BioMedical Sciences(2) | | | 14,770 | | | — | | | 14,770 | | | — | | | — |
Operating lease obligations: | | | | | | | | | | | | | | | |
Cambridge facility(3) | | | 26,302 | | | 2,715 | | | 5,430 | | | 5,777 | | | 12,380 |
Singapore facility(4) | | | 353 | | | 147 | | | 206 | | | — | | | — |
| | | | | | | | | | | | | | | |
Total contractual obligations | | $ | 51,253 | | $ | 6,698 | | $ | 25,820 | | $ | 6,355 | | $ | 12,380 |
| | | | | | | | | | | | | | | |
| (1) | This amount represents indebtedness incurred by us and CombinatoRx Singapore and includes approximately $1.3 million in interest payments. Our equipment lines of credit with General Electric Capital Corporation and its affiliates contain a subjective acceleration clause which provides the lender the ability to demand repayment of the loan upon a material adverse event. |
| (2) | Represents $12.5 million in principal amount of convertible promissory notes issued by CombinatoRx Singapore to BioMedical Sciences on August 30, 2005, June 8, 2006 and May 30, 2007 and interest of $2.3 million. The notes bear interest at an annual rate of 5% and are due and payable on December 31, 2009, unless we elect to prepay the notes before that date through CombinatoRx Singapore. The notes are secured by a security interest in the non-intellectual property assets of the subsidiary and by a negative pledge by the subsidiary with respect to its intellectual property rights. We have pledged our shares in CombinatoRx Singapore as additional collateral for the subsidiary’s obligations under the notes. The notes are convertible into our common stock at the option of BioMedical Sciences only upon maturity, acceleration or default of any proposed prepayment. |
| (3) | On October 18, 2005, we entered into a lease agreement for approximately 40,000 square feet of office and laboratory space in Cambridge, Massachusetts. On March 9, 2006, we entered into an amendment to the lease agreement for an additional approximately 23,000 square feet of laboratory space. The lease term, as amended, extends through January 2017. Our obligations under the original lease and under the March 2006 lease amendment are reflected as operating lease obligations in the table above. Our payment obligations under the amended lease are supported by standby letters of credit totaling $4.0 million. |
| (4) | On February 16, 2006, we entered into a lease agreement for approximately 4,800 square feet of office and laboratory space in Singapore. The lease term extends until April 2010. Our obligations under the lease are reflected as operating lease obligations in the table above. Our payment obligations are supported by a security deposit equal to one month of rent. |
In connection with our research and license agreement with CFFT, in the event we commercialize a product candidate developed under the agreement, we will owe CFFT specified royalties on product sales. Because of the uncertainty over when, if ever, a product candidate would be commercialized, the amount and timing of such royalty payment obligations are highly uncertain and are not included in the above table.
In connection with our agreement with the DMD foundations, in the event that we either enter into a license agreement with a third party granting the rights to make, use or sell a product developed under the agreement to treat DMD, or we or any of our affiliates or licensees first sells a product developed under the agreement to treat DMD, we will pay the DMD foundations a payment equal to 100% of the research funding provided to us under the agreement. In addition, on the first anniversary of the first commercial sale of a product developed under the agreement, we will pay the DMD foundations an additional payment equal to 100% of the research funding provided to us under the agreement. Finally, if a product developed under the agreement to treat DMD achieves
62
cumulative net sales of at least $100 million, within 90 days of such occurrence, we will pay the DMD foundations an additional payment equal to 200% of the research funding provided to us under the agreement. As of December 31, 2007, based on the $125,000 of research funding that we have received as of that date, the maximum contingent obligation that we may have to pay under this agreement is $0.5 million, but will be expected to increase as we receive approximately $2.9 million of additional research funding as contemplated under the agreement. Because of the uncertainty over when, if ever, such payments must be made upon outlicensing or product sales, the amount and timing of such payment obligations are highly uncertain and are not included in the above table.
On April 19, 2006, CombinatoRx Singapore received approval for a grant from the Economic Development Board of Singapore (EDB) Biomedical Sciences Group for up to approximately $5.8 million to support infectious disease drug research and development. The grant covers a percentage of qualifying costs of the research and development project on a reimbursement basis. Qualifying costs include salaries, equipment, scientific consumables and intellectual property costs. Reimbursement for these costs under the grant is subject to the satisfaction of certain conditions by CombinatoRx Singapore, including completion of the development project for infectious disease within a specified timeline, spending specified amounts on the project, the completion of other development milestones and the maintenance of specified levels of employment in Singapore. Subject to agreed upon audit rights by the EDB, cumulative qualifying costs are reimbursed upon application until 70% of the initial grant amount has been submitted by the subsidiary. The remaining 30% of the award may be paid by the EDB once we complete the research and development project. The grant extends through September 30, 2010. If the subsidiary breaches a condition of the grant, after good faith negotiations, the EDB may recover previously released grant funds from the subsidiary. As of December 31, 2007, we have received approximately $1.1 million under the grant. In addition, the EDB retains the right to change the terms and conditions of the grant as deemed necessary by the EDB.
In order for us and CombinatoRx Singapore to screen for combinations of drugs to treat Hepatitis C, we have entered into a non-exclusive license agreement with Novartis Vaccines and Diagnostics, Inc. to provide us with access to certain proprietary technologies relevant to Hepatitis C drug research and development. We may, under certain circumstances, be liable to Novartis Vaccines for payments associated with the achievement of milestones and for royalties on product sales. Because of the uncertainty over whether a Hepatitis C product candidate would trigger these payment obligations, and uncertainty as to when, if ever, a product candidate would be commercialized, the amount and timing of such milestone and royalty payment obligations are highly uncertain and are not included in the above table.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements or relationships with unconsolidated entities of financial partnerships, such as entities often referred to as structured finance or special purpose entities.
Tax Loss Carryforwards and Other Deferred Tax Assets
As of December 31, 2007, we had net operating loss carryforwards available to offset future federal and state taxable income of approximately $78.0 million and $81.7 million, respectively; federal and state research and development tax credit carryforwards of approximately $4.2 million and $3.0 million, respectively; and a foreign net operating loss carryforward of approximately $8.6 million available to offset future taxes. The net operating loss related to stock option deductions that will be booked through additional paid-in capital is approximately $2.7 million. The net operating loss and credit carryforwards expire at various dates through 2027. Under the provisions of the Internal Revenue Code of 1986, as amended, certain substantial changes in the company’s ownership may result in a limitation on the amount of net operating loss carryforwards and research and development carryforwards which could be utilized annually to offset future taxable income and taxes payable.
We have provided a valuation allowance for the full amount of these net operating loss carryforwards and tax credit carryforwards, as well as for the full amount of our other deferred tax assets, since realization of any future benefit from these deferred tax assets cannot be sufficiently assured.
63
We adopted the provisions of Financial Accounting Standards Board, or FASB, Interpretation No. 48, “Accounting for Uncertainty in Income Taxes–an interpretation of FASB Statement No. 109,” or FIN 48, on January 1, 2007. FIN 48 clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements in accordance with FASB Statement 109, “Accounting for Income Taxes”, and prescribes a recognition threshold and measurement process for financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. FIN 48 also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition. The implementation of FIN No. 48 did not have a material impact on our consolidated financial statements, results of operations or cash flows. At the adoption date of January 1, 2007, and also at December 31, 2007, we had no unrecognized tax benefits. We have not, as yet, conducted a study of our research and development credit carryforwards. This study may result in an increase or decrease to our research and development credit carryforwards; however, until a study is completed and any adjustment is known, no amounts are being presented as an uncertain tax position under FIN 48. A full valuation allowance has been provided against our research and development credits, and if an adjustment is required, this adjustment would be offset by an adjustment to the valuation allowance. As a result, there would be no impact to the consolidated balance sheet, statement of operations or cash flows if an adjustment were required.
Recently Issued Accounting Pronouncements
In February 2007, Statement of Financial Standard No. 159, “The Fair Value Option for Financial Assets and Financial Liabilities, Including an Amendment of FASB Statement No. 115”, or SFAS 159, was issued. SFAS 159 permits us to choose to measure many financial instruments and certain other items at fair value. It also establishes presentation and disclosure requirements. SFAS 159 was effective January 1, 2008 for us. We are evaluating the impact, if any, SFAS 159 will have on our consolidated financial statements.
In June 2007, FASB issued EITF Issue No. 07-3, “Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities,” or EITF 07-3. EITF 07-3 requires that nonrefundable advance payments for goods or services to be received in the future for use in research and development activities should be deferred and capitalized. The capitalized amounts should be expensed as the related goods are delivered or the services are performed. EITF 07-3 is effective for new contracts entered into during fiscal years beginning after December 15, 2007. We are currently analyzing the effect, if any, EITF 07-3 will have on our consolidated financial position and results of operations.
| Item 7A. | Quantitative and Qualitative Disclosure about Market Risk |
We are exposed to market risk related to changes in interest rates and changes in the exchange rate of the United States dollar to the Singapore dollar. As of December 31, 2007, we had unrestricted cash, cash equivalents and marketable securities of $108.6 million consisting of cash and highly liquid, short-term and long-term investments. Our cash is deposited in and invested through highly rated financial institutions in North America and Singapore. Our marketable securities are subject to interest rate risk and will decrease in value if market interest rates increase. If market interest rates were to increase immediately and uniformly by 10% from levels at December 31, 2007, we estimate that the fair value of our investments will decline by an immaterial amount, and therefore, our exposure to interest rate changes is immaterial. Our outstanding notes payable are at fixed interest rates and therefore have minimal exposure to changes in interest rates.
Transactions by our subsidiary, CombinatoRx Singapore, may be denominated in a currency other than the entity’s functional currency, which is the United States dollar. Exchange gains or losses resulting from the translation between the currency in which a transaction is denominated and CombinatoRx Singapore’s functional currency are included in net loss for our consolidated financial statements. Fluctuations in exchange rates, primarily between the United States dollar and the Singapore dollar, may adversely affect our results of operations, financial position and cash flows.
64
| Item 8. | Financial Statements and Supplementary Data |
The information called for by this item is indexed on page F-1 of this Annual Report on Form 10-K and is contained on pages F-2 through F-31.
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
| (a) | Evaluation of Disclosure Controls and Procedures |
As required by Rule 13a-15(b) of the Securities Exchange Act of 1934, as amended (the “1934 Act”), the company’s management, including the Chief Executive Officer and the Chief Financial Officer, conducted an evaluation as of the end of the period covered by this Annual Report on Form 10-K of the effectiveness of the design and operation of the company’s disclosure controls and procedures. Based on that evaluation, the company’s Chief Executive Officer and Chief Financial Officer concluded that the company’s disclosure controls and procedures are effective at the reasonable assurance level in ensuring that information required to be disclosed by the company in the reports that it files or submits under the 1934 Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms.
| (b) | Management’s Report on Internal Control Over Financial Reporting |
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the 1934 Act as a process designed by, or under the supervision of, the Company’s principal executive and principal financial officers and effected by the company’s board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
| | • | | Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of the assets of the company; |
| | • | | Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and |
| | • | | Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements. |
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2007. In making this assessment, the company’s management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework. Based on this assessment, our management concluded that, as of December 31, 2007, our internal control over financial reporting is effective based on those criteria.
Our independent registered public accounting firm has issued an audit report on our assessment of our internal control over financial reporting. This report appears below.
65
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
CombinatoRx, Incorporated
We have audited CombinatoRx, Incorporated’s internal control over financial reporting as of December 31, 2007, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). CombinatoRx, Incorporated’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on the effectiveness of the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion CombinatoRx, Incorporated maintained, in all material respects, effective internal control over financial reporting as of December 31, 2007, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets as of December 31, 2007 and 2006, and the related statements of operations, convertible preferred stock, redeemable convertible preferred stock and stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2007 of CombinatoRx, Incorporated and our report dated March 7, 2008 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Boston, Massachusetts
March 7, 2008
66
| (c) | Changes in Internal Control |
As required by Rule 13a-15(d) of the 1934 Act, the company’s management, including the Chief Executive Officer and the Chief Financial Officer, conducted an evaluation of the internal control over financial reporting to determine whether any changes occurred during the period covered by this Annual Report on Form 10-K that have materially affected, or are reasonably likely to materially affect, the company’s internal control over financial reporting. Based on that evaluation, the Chief Executive and Chief Financial Officer concluded that there was no such change during the last quarter of the fiscal year covered by this Annual Report on Form 10-K that has materially affected, or is reasonably likely to materially affect, the company’s internal control over financial reporting.
| Item 9B. | Other Information |
None.
PART III
| Item 10. | Directors and Executive Officers |
Information concerning our directors and executive officers will appear in our Proxy Statement for the 2008 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before April 29, 2008, under the captions “Election of Directors” and “Executive Officers.” Such information is incorporated herein by reference.
Information concerning compliance with Section 16(a) of the Act of 1934 will appear in the Company’s Proxy Statement for the 2008 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before April 29, 2008, under the caption “Section 16(a) Beneficial Ownership Reporting Compliance.” Such information is incorporated herein by reference.
Information about our Audit Committee, including the members of the Committee, and our Audit Committee financial experts, will appear in our Proxy Statement for the 2008 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before April 29, 2008, under the captions “The Audit Committee” and “Audit Committee Financial Experts.” Such information is incorporated herein by reference.
Information concerning our Code of Ethics and Conduct will appear in our Proxy Statement for the 2008 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before April 29, 2008, under the caption “Code of Ethics and Conduct.” Such information is incorporated herein by reference.
| Item 11. | Executive Compensation |
Information in response to this item will appear in our Proxy Statement for the 2008 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before April 29, 2008, under the captions “Executive Compensation,” “Director Compensation,” and “Report of the Compensation Committee on Executive Compensation.” Such information is incorporated herein by reference.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
Information concerning security ownership of certain beneficial owners and management will appear in our Proxy Statement for the 2008 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before April 29, 2008, under the caption “Security Ownership of Certain Beneficial Owners and Management.” Such information is incorporated herein by reference.
67
| Item 13. | Certain Relationships and Related Transactions |
Information concerning certain relationships and related transactions will appear in the Company’s Proxy Statement for the 2008 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before April 29, 2008, under the caption “Certain Relationships and Related Transactions.” Such information is incorporated herein by reference.
| Item 14. | Principal Accountant Fees and Services |
Information concerning principal accounting fees and services will appear in our Proxy Statement for the 2008 Annual Meeting of Stockholders, to be filed pursuant to Regulation 14A on or before April 29, 2008, under the caption “Independent Public Accountants.” Such information is incorporated herein by reference.
PART IV
| Item 15. | Exhibits, Financial Statement Schedules |
| | (a)(1) | Financial Statements. |
The consolidated financial statements filed as part of this Annual Report on Form 10-K are listed and indexed at page F-1.
| | (a)(2) | Financial Statement Schedules. |
Certain schedules are omitted because they are not applicable, or not required, or because the required information is included in the consolidated financial statements or notes thereto.
The Exhibits listed in the Exhibit Index immediately preceding the Exhibits are filed as a part of this Annual Report on Form 10-K.
68
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | |
| COMBINATORX, INCORPORATED |
| |
| By: | | /s/ ALEXIS BORISY |
| | Alexis Borisy |
| | President and Chief Executive Officer |
Date: March 13, 2008
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the date indicated.
| | | | |
Signature | | Title | | Date |
| | |
/s/ ALEXIS BORISY Alexis Borisy | | President and Chief Executive Officer (Principal Executive Officer) | | March 13, 2008 |
| | |
/s/ ROBERT FORRESTER Robert Forrester | | Executive Vice President and Chief Financial Officer (Principal Financial and Accounting Officer) | | March 13, 2008 |
| | |
/s/ SALLY CRAWFORD Sally Crawford | | Director | | March 13, 2008 |
| | |
/s/ BARBARA DEPTULA Barbara Deptula | | Director | | March 13, 2008 |
| | |
/s/ PATRICK FORTUNE Patrick Fortune | | Director | | March 13, 2008 |
| | |
/s/ FRANK HAYDU Frank Haydu | | Director | | March 13, 2008 |
| | |
/s/ MICHAEL KAUFFMAN Michael Kauffman | | Director | | March 13, 2008 |
| | |
/s/ W. JAMES O’SHEA W. James O’Shea | | Director | | March 13, 2008 |
| | |
/s/ RICHARD POPS Richard Pops | | Director | | March 13, 2008 |
69
CombinatoRx, Incorporated
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
F-1
Report of Independent Registered Public Accounting Firm
The Board of Directors and Stockholders
CombinatoRx, Incorporated
We have audited the accompanying consolidated balance sheets of CombinatoRx, Incorporated (the “Company”) as of December 31, 2007 and 2006, and the related consolidated statements of operations, convertible preferred stock, redeemable convertible preferred stock and stockholders’ equity (deficit), and cash flows for each of the three years in the period ended December 31, 2007. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of CombinatoRx, Incorporated at December 31, 2007 and 2006, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2007, in conformity with U.S. generally accepted accounting principles.
As discussed in Note 2 to the consolidated financial statements, effective January 1, 2006, the Company adopted Statement of Financial Accounting Standards No. 123R, “Share-Based Payment” using the prospective transition method for stock options granted prior to January 1, 2005 and the modified prospective transition method for stock options and restricted stock granted after January 1, 2005.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), CombinatoRx, Incorporated’s internal control over financial reporting as of December 31, 2007, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission, and our report dated March 7, 2008 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
Boston, Massachusetts
March 7, 2008
F-2
CombinatoRx, Incorporated
Consolidated Balance Sheets
(in thousands except per share data)
| | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
Assets | | | | | | | | |
Current assets: | | | | | | | | |
Cash and cash equivalents | | $ | 11,585 | | | $ | 9,194 | |
Restricted cash | | | 50 | | | | — | |
Short-term investments | | | 96,999 | | | | 107,895 | |
Accounts receivable | | | 397 | | | | 83 | |
Unbilled accounts receivable | | | 746 | | | | 1,206 | |
Prepaid expenses and other current assets | | | 2,526 | | | | 3,451 | |
| | | | | | | | |
Total current assets | | | 112,303 | | | | 121,829 | |
| | |
Property and equipment, net | | | 15,933 | | | | 12,506 | |
Restricted cash and other assets | | | 4,007 | | | | 4,000 | |
| | | | | | | | |
Total assets | | $ | 132,243 | | | $ | 138,335 | |
| | | | | | | | |
Liabilities and stockholders’ equity | | | | | | | | |
Current liabilities: | | | | | | | | |
Accounts payable | | $ | 1,964 | | | $ | 4,489 | |
Accrued expenses | | | 4,751 | | | | 4,336 | |
Deferred revenue | | | 5,431 | | | | 9,548 | |
Current portion of notes payable, net of discount | | | 3,099 | | | | 1,851 | |
Current portion of lease incentive obligation | | | 649 | | | | 649 | |
| | | | | | | | |
Total current liabilities | | | 15,894 | | | | 20,873 | |
| | |
Convertible notes payable of subsidiary | | | 13,404 | | | | 9,301 | |
Notes payable, net of current portion and discount | | | 5,415 | | | | 2,527 | |
Deferred revenue, net of current portion | | | 12,068 | | | | 8,011 | |
Deferred rent | | | 2,190 | | | | 2,244 | |
Lease incentive obligation, net of current portion | | | 5,245 | | | | 5,660 | |
| | |
Commitments (Note 12) | | | | | | | | |
| | |
Minority interest in subsidiary | | | 2,792 | | | | 2,669 | |
| | |
Stockholders’ equity: | | | | | | | | |
Preferred stock, $0.001 par value; 5,000 shares authorized; no shares issued and outstanding | | | — | | | | — | |
Common stock, $0.001 par value; 60,000 shares authorized; 34,822 and 28,828 shares issued and outstanding at December 31, 2007 and 2006, respectively | | | 35 | | | | 29 | |
Additional paid-in capital | | | 261,187 | | | | 219,730 | |
Accumulated other comprehensive income | | | 226 | | | | 39 | |
Accumulated deficit | | | (186,213 | ) | | | (132,748 | ) |
| | | | | | | | |
Stockholders’ equity | | | 75,235 | | | | 87,050 | |
| | | | | | | | |
Total liabilities and stockholders’ equity | | $ | 132,243 | | | $ | 138,335 | |
| | | | | | | | |
See accompanying notes.
F-3
CombinatoRx, Incorporated
Consolidated Statements of Operations
(in thousands, except share and per share amounts)
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
Revenue: | | | | | | | | | | | | |
Collaborations | | $ | 12,226 | | | $ | 11,725 | | | $ | 4,143 | |
Government contracts and grants | | | 2,712 | | | | 1,548 | | | | 515 | |
| | | | | | | | | | | | |
Total revenue | | | 14,938 | | | | 13,273 | | | | 4,658 | |
| | | | | | | | | | | | |
Operating expenses: | | | | | | | | | | | | |
Research and development | | | 55,434 | | | | 34,094 | | | | 24,059 | |
General and administrative | | | 16,879 | | | | 18,641 | | | | 10,576 | |
| | | | | | | | | | | | |
Total operating expenses | | | 72,313 | | | | 52,735 | | | | 34,635 | |
| | | | | | | | | | | | |
Loss from operations | | | (57,375 | ) | | | (39,462 | ) | | | (29,977 | ) |
Interest income | | | 5,391 | | | | 5,913 | | | | 1,296 | |
Interest expense | | | (1,304 | ) | | | (722 | ) | | | (834 | ) |
Other (expense) income | | | (9 | ) | | | 34 | | | | — | |
| | | | | | | | | | | | |
Net loss before provision for income taxes | | | (53,297 | ) | | | (34,237 | ) | | | (29,515 | ) |
Provision for income taxes | | | (46 | ) | | | (51 | ) | | | — | |
| | | | | | | | | | | | |
Net loss | | $ | (53,343 | ) | | $ | (34,288 | ) | | $ | (29,5155 | ) |
| | | | | | | | | | | | |
Net loss per share applicable to common stockholders—basic and diluted | | $ | (1.78 | ) | | $ | (1.26 | ) | | $ | (8.53 | ) |
| | | | | | | | | | | | |
Weighted average number of common shares used in net loss per share calculation—basic and diluted | | | 30,025,830 | | | | 27,223,319 | | | | 4,169,355 | |
| | | | | | | | | | | | |
See accompanying notes.
F-4
CombinatoRx, Incorporated
Consolidated Statements of Convertible Preferred Stock, Redeemable Convertible Preferred Stock,
Stockholders’ Equity (Deficit)
(in thousands, except share amounts)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Convertible
Preferred
Stock and
Redeemable
Convertible
Preferred Stock | | | Common Stock | | Additional
Paid-in
Capital | | | Deferred
Compensation | | | Accumulated
Other
Comprehensive
Loss | | | Accumulated
Deficit | | | Total | |
| | | Shares | | | Par
Value | | | Shares | | Par
Value | | | | | |
Balance at December 31, 2004 | | 22,907,015 | | | $ | 103,843 | | | 906,872 | | $ | 1 | | $ | 12,352 | | | $ | (13,918 | ) | | $ | (45 | ) | | $ | (68,658 | ) | | $ | (70,268 | ) |
Net loss | | — | | | | — | | | — | | | — | | | — | | | | — | | | | — | | | | (29,515 | ) | | | (29,515 | ) |
Unrealized loss on investments | | — | | | | — | | | — | | | — | | | — | | | | — | | | | (20 | ) | | | — | | | | (20 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Comprehensive loss | | — | | | | — | | | — | | | — | | | — | | | | — | | | | — | | | | — | | | | (29,535 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Exercise of stock options | | — | | | | — | | | 163,504 | | | — | | | 145 | | | | | | | | — | | | | — | | | | 145 | |
Issuance of warrants | | — | | | | — | | | — | | | — | | | 372 | | | | — | | | | — | | | | (114 | ) | | | 258 | |
Write-off of deferred compensation and recapture of compensation expense for terminated employees | | — | | | | — | | | — | | | — | | | (1,659 | ) | | | 1,475 | | | | — | | | | — | | | | (184 | ) |
Deferred stock-based compensation associated with issuance of stock options | | — | | | | — | | | — | | | — | | | 2,647 | | | | (2,647 | ) | | | — | | | | — | | | | — | |
Amortization of deferred compensation | | — | | | | — | | | — | | | — | | | — | | | | 3,976 | | | | — | | | | — | | | | 3,976 | |
Amortization of deferred compensation expense in connection with prior stock option modification | | — | | | | — | | | — | | | — | | | — | | | | 36 | | | | — | | | | — | | | | 36 | |
Stock-based compensation expense related to non-employees | | — | | | | — | | | — | | | — | | | 295 | | | | 26 | | | | — | | | | — | | | | 321 | |
Stock-based compensation expense in connection with stock option modification | | — | | | | — | | | — | | | — | | | 960 | | | | — | | | | — | | | | — | | | | 960 | |
Issuance of Series E preferred stock | | 1,363,636 | | | | 15,000 | | | — | | | — | | | — | | | | — | | | | — | | | | — | | | | — | |
Accretion of dividends on redeemable convertible preferred stock | | — | | | | 5,985 | | | — | | | — | | | (5,985 | ) | | | — | | | | — | | | | (46 | ) | | | (6,031 | ) |
Issuance of common stock in connection with initial public offering, net of offering costs of $1,077 | | — | | | | — | | | 6,900,000 | | | 7 | | | 43,836 | | | | — | | | | — | | | | — | | | | 43,843 | |
Automatic conversion of preferred stock into common stock upon initial public offering | | (24,270,651 | ) | | | (124,828 | ) | | 15,314,695 | | | 15 | | | 124,814 | | | | — | | | | — | | | | — | | | | 124,829 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2005 | | — | | | | — | | | 23,285,071 | | | 23 | | | 177,777 | | | | (11,052 | ) | | | (65 | ) | | | (98,333 | ) | | | 68,350 | |
See accompanying notes.
F-5
CombinatoRx, Incorporated
Consolidated Statements of Convertible Preferred Stock, Redeemable Convertible Preferred Stock,
Stockholders’ Equity (Deficit) (Continued)
(in thousands, except share amounts)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Convertible
Preferred
Stock and
Redeemable
Convertible
Preferred Stock | | Common Stock | | Additional
Paid-in
Capital | | | Deferred
Compensation | | | Accumulated
Other
Comprehensive
Income (Loss) | | | Accumulated
Deficit | | | Total | |
| | | Shares | | Par
Value | | Shares | | | Par
Value | | | | | |
Balance at December 31, 2005 | | — | | | — | | 23,285,071 | | | | 23 | | | 177,777 | | | | (11,052 | ) | | | (65 | ) | | | (98,333 | ) | | | 68,350 | |
Net loss | | — | | | — | | — | | | | — | | | — | | | | | | | | — | | | | (34,288 | ) | | | (34,288 | ) |
Unrealized gain on investments | | — | | | — | | — | | | | — | | | — | | | | | | | | 104 | | | | — | | | | 104 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Comprehensive loss | | — | | | — | | — | | | | — | | | — | | | | — | | | | — | | | | — | | | | (34,184 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Exercise of stock options | | — | | | — | | 561,768 | | | | 1 | | | 599 | | | | — | | | | — | | | | — | | | | 600 | |
Exercise of warrants | | — | | | — | | 33,249 | | | | — | | | — | | | | — | | | | — | | | | — | | | | — | |
Issuance of restricted stock to employees | | — | | | — | | 255,000 | | | | — | | | — | | | | | | | | — | | | | — | | | | — | |
Issuance of restricted stock to non-employees | | — | | | — | | 10,000 | | | | — | | | 111 | | | | | | | | — | | | | — | | | | 111 | |
Issuance of common stock in connection with private placement offering, net of issuance costs of $2,627 | | — | | | — | | 4,682,942 | | | | 5 | | | 45,368 | | | | | | | | — | | | | — | | | | 45,373 | |
Reclassification of deferred stock-based compensation | | — | | | — | | — | | | | — | | | (11,052 | ) | | | 11,052 | | | | — | | | | — | | | | — | |
Accretion of dividends on redeemable convertible preferred stock | | — | | | — | | — | | | | — | | | — | | | | — | | | | — | | | | (127 | ) | | | (127 | ) |
Stock-based compensation expense | | — | | | — | | — | | | | — | | | 6,365 | | | | — | | | | — | | | | — | | | | 6,365 | |
Stock-based compensation expense related to non-employees | | — | | | — | | — | | | | — | | | 562 | | | | — | | | | — | | | | — | | | | 562 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2006 | | — | | $ | — | | 28,828,030 | | | $ | 29 | | $ | 219,730 | | | $ | — | | | $ | 39 | | | $ | (132,748 | ) | | $ | 87,050 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | — | | | — | | — | | | | — | | | — | | | | — | | | | | | | | (53,343 | ) | | | (53,343 | ) |
Unrealized gain on investments | | — | | | — | | — | | | | — | | | — | | | | — | | | | 187 | | | | — | | | | 187 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Comprehensive loss | | — | | | — | | — | | | | — | | | — | | | | — | | | | — | | | | — | | | | (53,156 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Exercise of stock options | | — | | | — | | 420,390 | | | | — | | | 519 | | | | — | | | | — | | | | — | | | | 519 | |
Cancellation of restricted stock | | — | | | — | | (26,250 | ) | | | — | | | — | | | | — | | | | — | | | | — | | | | — | |
Issuance of common stock in connection with public offering, net of issuance costs of $1,961 | | — | | | — | | 5,600,000 | | | | 6 | | | 33,033 | | | | — | | | | — | | | | — | | | | 33,039 | |
Accretion of dividends on redeemable convertible preferred stock | | — | | | — | | — | | | | — | | | — | | | | — | | | | — | | | | (122 | ) | | | (122 | ) |
Stock-based compensation expense | | — | | | — | | — | | | | — | | | 7,730 | | | | — | | | | — | | | | — | | | | 7,730 | |
Stock-based compensation expense related to non-employees | | — | | | — | | — | | | | — | | | 175 | | | | — | | | | — | | | | — | | | | 175 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2007 | | — | | $ | — | | 34,822,170 | | | $ | 35 | | $ | 261,187 | | | $ | — | | | $ | 226 | | | $ | (186,213 | ) | | $ | 75,235 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
See accompanying notes.
F-6
CombinatoRx, Incorporated
Consolidated Statements of Cash Flows
(in thousands)
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
Operating activities | | | | | | | | | | | | |
Net loss | | $ | (53,343 | ) | | $ | (34,288 | ) | | $ | (29,515 | ) |
Adjustments to reconcile net loss to net cash (used in) provided by operating activities: | | | | | | | | | | | | |
Depreciation and amortization | | | 3,139 | | | | 1,865 | | | | 1,126 | |
Noncash interest expense | | | 662 | | | | 462 | | | | 627 | |
Noncash rent expense | | | (649 | ) | | | (365 | ) | | | — | |
Stock-based compensation expense | | | 7,905 | | | | 7,038 | | | | 5,109 | |
(Decrease) increase in deferred rent | | | (54 | ) | | | 2,135 | | | | 109 | |
Changes in assets and liabilities: | | | | | | | | | | | | |
Increase in accounts receivable | | | (314 | ) | | | (83 | ) | | | — | |
Decrease (increase) in unbilled accounts receivable | | | 460 | | | | (976 | ) | | | (230 | ) |
(Increase) decrease in prepaid expenses and other assets | | | (1,751 | ) | | | 1,234 | | | | (724 | ) |
(Decrease) increase in accounts payable | | | (2,525 | ) | | | 3,259 | | | | (471 | ) |
Increase in accrued expenses | | | 380 | | | | 2,024 | | | | 900 | |
(Decrease) increase in deferred revenue | | | (60 | ) | | | (8,025 | ) | | | 25,518 | |
Proceeds from landlord under tenant improvements | | | 2,900 | | | | 4,015 | | | | — | |
| | | | | | | | | | | | |
Net cash (used in) provided by operating activities | | | (43,250 | ) | | | (21,705 | ) | | | 2,449 | |
| | | |
Investing activities | | | | | | | | | | | | |
Purchases of property and equipment | | | (6,566 | ) | | | (11,669 | ) | | | (1,534 | ) |
Purchases of short-term investments | | | (543,199 | ) | | | (641,505 | ) | | | (295,930 | ) |
Sales and maturities of short-term investments | | | 554,282 | | | | 624,382 | | | | 238,202 | |
Increase in restricted cash | | | (50 | ) | | | (1,352 | ) | | | (2,448 | ) |
| | | | | | | | | | | | |
Net cash provided by (used in) investing activities | | | 4,467 | | | | (30,144 | ) | | | (61,710 | ) |
| | | |
Financing activities | | | | | | | | | | | | |
Proceeds from issuance of convertible preferred stock and redeemable convertible preferred stock, net of issuance costs | | | — | | | | — | | | | 15,000 | |
Proceeds from issuance of common stock | | | 33,039 | | | | 45,373 | | | | 43,843 | |
Proceeds from exercise of stock options | | | 519 | | | | 600 | | | | 145 | |
Proceeds from subsidiary’s convertible note payable | | | 3,500 | | | | 3,500 | | | | 5,500 | |
Proceeds from subsidiary’s preferred stock | | | — | | | | — | | | | 2,500 | |
Proceeds from notes payable | | | 6,778 | | | | 3,892 | | | | 159 | |
Repayment of notes payable | | | (2,662 | ) | | | (1,401 | ) | | | (963 | ) |
| | | | | | | | | | | | |
Net cash provided by financing activities | | | 41,174 | | | | 51,964 | | | | 66,184 | |
| | | | | | | | | | | | |
Net increase in cash and cash equivalents | | | 2,391 | | | | 115 | | | | 6,923 | |
Cash and cash equivalents at beginning of the period | | | 9,194 | | | | 9,079 | | | | 2,156 | |
| | | | | | | | | | | | |
Cash and cash equivalents at end of the period | | $ | 11,585 | | | $ | 9,194 | | | $ | 9,079 | |
| | | | | | | | | | | | |
Supplemental disclosure of cash flow information | | | | | | | | | | | | |
Cash paid for interest | | $ | 642 | | | $ | 260 | | | $ | 205 | |
| | | | | | | | | | | | |
Cash paid for income taxes | | $ | 98 | | | $ | — | | | $ | — | |
| | | | | | | | | | | | |
Supplemental disclosure of noncash investing and financing activities | | | | | | | | | | | | |
Lease incentive obligation | | $ | 415 | | | $ | 5,361 | | | $ | 948 | |
| | | | | | | | | | | | |
Write-off of deferred compensation | | $ | — | | | $ | 11,052 | | | $ | 1,475 | |
| | | | | | | | | | | | |
Issuance of warrants in connection with debt issuances | | $ | — | | | $ | — | | | $ | 258 | |
| | | | | | | | | | | | |
Issuance of warrants in connection with equity financing | | $ | — | | | $ | — | | | $ | 114 | |
| | | | | | | | | | | | |
See accompanying notes.
F-7
CombinatoRx, Incorporated
Notes to Consolidated Financial Statements
(in thousands, except share and per share amounts)
CombinatoRx, Incorporated (the “Company”) was formed as a Delaware corporation on March 28, 2000. The Company is a biopharmaceutical company focused on developing synergistic combinations of approved drugs, with a portfolio of product candidates in phase 2 clinical development. To date, the Company has devoted substantially all of its resources to the development of its drug discovery technology and the research and development of its drug candidates, including conducting preclinical and clinical trials and seeking intellectual property protection for its technology and product candidates.
CombinatoRx is subject to risks common to companies in the life science industry. The Company has not received regulatory approval for, or generated revenues from, any of its product candidates. All of its current product candidates are in preclinical or clinical development. If it does not successfully commercialize any of its product candidates, it will be unable to generate product revenue or achieve profitability.
The Company has a limited operating history and has incurred losses from operations since inception, resulting in an accumulated deficit of $186,213 at December 31, 2007. The Company expects its existing cash resources to be sufficient to fund its planned operations, including continued research and drug development through approximately the fourth quarter of 2009. However, the Company may require significant additional funds earlier than currently expected if research and development expenses exceed the Company’s current expectations or if anticipated funding from collaboration agreements is less than the Company’s current expectations. The Company may seek additional funding through public or private equity or debt financings and collaboration agreements. Additional funding, if needed, may not be available to the Company on acceptable terms or at all. Any additional equity financing would be dilutive to existing stockholders and any debt financing, if available, may involve restrictive covenants that could adversely impact how the Company conducts its business. If the Company is unable to obtain funding on a timely basis, it may be required to significantly curtail one or more of its research or development programs. The Company also could be required to seek funds through arrangements with collaborators or others that may require the Company to relinquish rights to some of its technologies or product candidates which the Company would otherwise develop and pursue on its own.
| 2. | Summary of Significant Accounting Policies |
Principles of Consolidation
In connection with the establishment of CombinatoRx Singapore Pte Ltd (“CombinatoRx Singapore”) in August 2005 as discussed in Note 6, the accompanying financial statements as of December 31, 2007 include the accounts of the Company and its majority owned subsidiary, CombinatoRx Singapore. The minority interest in CombinatoRx Singapore is held by BioMedical Sciences Investment Fund Pte Ltd (“Bio One”) and is represented by their $2.5 million investment in shares of the subsidiary’s convertible, redeemable preferred stock. The preferred stock of the subsidiary is entitled to an annual 5% dividend payable upon redemption or liquidation of the subsidiary, and is subject to redemption by the subsidiary for a cash payment equal to 125% of the purchase price of the shares plus accrued, but unpaid, dividends. As a result of the redemption and dividend rights of the preferred stock, the minority interest will not be reduced for Bio One’s share of the net loss of the subsidiary. Accordingly, the Company’s 2007 and 2006 consolidated statement of operations includes 100% of the net loss of CombinatoRx Singapore. In addition, the accounts of the Company’s wholly-owned subsidiary, CombinatoRx Securities Corp., are included in the consolidated financial results. All significant intercompany accounts and transactions have been eliminated.
The functional currency of CombinatoRx Singapore is the United States dollar. Foreign currency transaction gains and losses are recorded in the consolidated statement of operations. Net losses of $1 and net gains of $26 were recorded in other (expense) income in 2007 and 2006, respectively.
F-8
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ from those estimates.
Research and Development
Research and development costs include all direct costs, including cash compensation, stock-based compensation and benefits for research and development personnel, supplies and materials, direct external costs including costs of clinical trials, formulation manufacturing, pre-clinical programs, collaboration expenses, external consultants, other outside costs and infrastructure and overhead related to the development of drug candidates. These costs have been charged to research and development expense as incurred.
Comprehensive Loss
SFAS No. 130,Reporting Comprehensive Income, establishes standards for reporting and displaying comprehensive income (loss) and its components in the financial statements. Comprehensive income (loss) is the change in equity of a company during a period from transactions and other events and circumstances, excluding transactions resulting from investments by owners and distributions to owners. Comprehensive loss includes net loss and unrealized gain (loss) on investments for all periods presented.
Revenue Recognition
Collaborations
The Company has entered into collaborative research and development agreements with other pharmaceutical and biotechnology companies, government agencies and charitable foundations. These agreements are generally in the form of research and development and license agreements. The agreements are for early-stage compounds and are generally focused on specific disease areas. The agreements provide for nonrefundable up-front payments, milestone payments upon achieving significant milestone events and in some cases ongoing research funding. The agreements also contemplate royalty payments on sales if and when the product receives marketing approval by the FDA or other regulatory agency.
The Company recognizes revenue in accordance with Emerging Issues Task Force, Issue No. 00-21, “Revenue Arrangements with Multiple Deliverables” (“EITF 00-21”) and Securities and Exchange Commission Staff Accounting Bulletin No. 104, “Revenue Recognition in Financial Statements” (“SAB 104”). Revenue arrangements with multiple deliverables are divided into separate units of accounting if certain criteria are met, including whether the delivered element has stand-alone value to the customer and whether there is objective and reliable evidence of the fair value of the undelivered elements. The consideration received is allocated among separate elements based on their respective fair values. Revenue is recognized when there is persuasive evidence that an arrangement exists, delivery has occurred, the price is fixed or determinable, and collection is reasonably assured. Under arrangements where the license fees and research and development activities can be accounted for as a separate unit of accounting, nonrefundable up-front license fees are deferred and recognized as revenue on a straight-line basis over the expected term of the Company’s continued involvement in the research and development process. Revenues from the achievement of research and development milestones, if deemed substantive, are recognized as revenue when the milestones are achieved, and the milestone payments are due and collectible. Milestones are considered substantive if all of the following conditions are met: (1) the milestone is nonrefundable; (2) achievement of the milestone was not reasonably assured at the inception of the arrangement; (3) substantive effort is involved to achieve the milestone; and (4) the amount of the milestone appears reasonable in relation to the effort expended, the other milestones in the arrangement and the related risk associated with achievement of the milestone. If any of these conditions are not met, the Company recognizes a proportionate amount of the milestone payment upon receipt as revenue that correlates to work already performed and the remaining portion of the milestone payment will be deferred and recognized as revenue as the Company completes its performance obligations.
F-9
Government Contracts and Grants
Revenue under government grants or cost reimbursement contracts is recognized as the Company performs the underlying research and development activities.
Concentrations of Credit Risk
Financial instruments that potentially expose the Company to concentrations of credit risk consist of cash, cash equivalents, short-term investments, accounts receivable and unbilled receivables. Short-term investments consist of money market funds, corporate debt securities and asset-backed securities. The Company maintains its cash, cash equivalents and marketable securities at high-quality financial institutions. The Company limits the amount of investment in any one type of investment, thereby reducing credit risk concentrations. The Company does not believe there is significant concentration of credit risk related to accounts receivable and unbilled receivables since its customers are large well-capitalized pharmaceutical companies, foundations or government agencies.
Cash and Cash Equivalents
The Company considers all highly liquid investments with an original or remaining maturity of three months or less at the date of purchase to be cash equivalents, except for those funds managed by the Company’s investment manager, which are classified as short-term investments. Cash equivalents consist primarily of money market instruments.
Short-term Investments
Short-term investments consist primarily of investments with original maturities greater than ninety days and less than one year when purchased. The Company classifies these investments as available-for-sale as defined by SFAS No. 115,Accounting for Certain Investments in Debt and Equity Securities. Unrealized gains and losses are included in other comprehensive income (loss).
Available-for-sale securities at December 31, 2007 and 2006 consist of the following:
| | | | | | | | | | | | | |
| | | Amortized
Cost | | Unrealized
Gains | | Unrealized
Losses | | | Fair
Value |
December 31, 2007— | | | | | | | | | | | | | |
Corporate debt securities | | $ | 63,451 | | $ | 204 | | $ | (5 | ) | | $ | 63,650 |
Asset-backed securities | | | 25,868 | | | 27 | | | — | | | | 25,895 |
Money market funds | | | 7,454 | | | — | | | — | | | | 7,454 |
| | | | | | | | | | | | | |
| | $ | 96,773 | | $ | 231 | | $ | (5 | ) | | $ | 96,999 |
| | | | | | | | | | | | | |
December 31, 2006— | | | | | | | | | | | | | |
Corporate debt securities | | $ | 83,710 | | $ | 37 | | $ | — | | | $ | 83,747 |
Asset-backed securities | | | 15,718 | | | 2 | | | — | | | | 15,720 |
Money market funds | | | 8,428 | | | — | | | — | | | | 8,428 |
| | | | | | | | | | | | | |
| | $ | 107,856 | | $ | 39 | | $ | — | | | $ | 107,895 |
| | | | | | | | | | | | | |
The amortized cost and estimated fair value of investments in debt securities, which excludes money market funds, at December 31, 2007 and 2006, by contractual maturity, were as follows:
| | | | | | | | | | | | |
| | | December 31, 2007 | | December 31, 2006 |
| | | Cost | | Estimated
Fair Value | | Cost | | Estimated
Fair Value |
Maturing in one year or less | | $ | 89,319 | | $ | 89,545 | | $ | 99,428 | | $ | 99,467 |
| | | | | | | | | | | | |
F-10
The cost of securities sold is determined based on the specific identification method for purposes of recording realized gains and losses. Gross realized gains and losses on the sales of investments have not been material to the Company’s results of operations for all periods presented. As a matter of investment policy, the Company does not invest in auction rate securities.
Fair Value of Financial Instruments
The carrying amounts of the Company’s financial instruments, which include cash equivalents, short-term investments, accounts payable, accrued expenses, convertible notes payable of subsidiary and notes payable, approximate their fair values due to their short maturities.
Property and Equipment
Property and equipment are recorded at cost and depreciated over their estimated useful lives using the straight-line method. Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are removed from the accounts, and any resulting gain or loss is credited or charged to the statement of operations. Repairs and maintenance costs are expensed as incurred.
Impairment of Long-Lived Assets
In accordance with SFAS No. 144,Accounting for the Impairment or Disposal of Long-Lived Assets, the Company continually monitors whether events or circumstances have occurred that indicate that the estimated remaining useful life of its long-lived assets may warrant revision or that the carrying value of these assets may be impaired. The carrying value for long-lived assets with finite lives is reviewed for impairment when events or changes in circumstances indicate the book value of the assets may not be recoverable. An impairment loss would be recognized when estimated undiscounted future cash flows from the use of the asset and its eventual disposition is less than its carrying amount. Any write-downs are treated as permanent reductions in the carrying amount of the assets. Based on this evaluation, the Company believes that, as of each of the balance sheet dates presented, none of the Company’s long-lived assets were impaired.
Capitalized Software
The Company capitalizes certain internal and external costs incurred to develop internal use software in accordance with the American Institute of Certified Public Accountants (“AICPA”) Statement of Position 98-1,Accounting for the Costs of Computer Software Developed or Obtained for Internal Use. Capitalized software development costs are included in property and equipment and are depreciated over estimated useful lives (five years) when development is complete. The net book value of the Company’s capitalized software was $149 and $267 at December 31, 2007 and 2006, respectively.
Costs of Postponed Initial Public Offering
The Company expensed $1,551 of costs associated with its prior registration statement on Form S-1 in the year ended December 31, 2005. The Company’s initial public offering was postponed for a period in excess of 90 days, and as a result, it was deemed an aborted offering in accordance with Staff Accounting Bulletin Topic 5A. These costs are included in general and administrative expenses in the Statements of Operations for the year ended December 31, 2005.
Net Loss Per Share
The Company calculates net loss per share in accordance with SFAS No. 128,Earnings Per Share. Basic and diluted net loss per common share was determined by dividing net loss applicable to common stockholders by the weighted average common shares outstanding during the period. The Company’s potentially dilutive shares, which include outstanding common stock options, convertible preferred stock and notes payable of subsidiary, warrants and unvested restricted stock, have not been included in the computation of diluted net loss per share for all periods, as the result would be anti-dilutive.
F-11
Net loss applicable to common stockholders and net loss per share applicable to common stockholders are as follows:
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
Net loss | | $ | (53,343 | ) | | $ | (34,288 | ) | | $ | (29,515 | ) |
Accretion of convertible preferred and redeemable convertible preferred stock | | | (122 | ) | | | (127 | ) | | | (6,031 | ) |
| | | | | | | | | | | | |
Net loss applicable to common stockholders | | $ | (53,465 | ) | | $ | (34,415 | ) | | $ | (35,546 | ) |
| | | | | | | | | | | | |
Weighted-average common shares outstanding | | | 30,025,830 | | | | 27,223,319 | | | | 4,169,355 | |
Net loss per share applicable to common stockholders—basic and diluted | | $ | (1.78 | ) | | $ | (1.26 | ) | | $ | (8.53 | ) |
| | | | | | | | | | | | |
Amounts for 2007 and 2006 relate to the accretion of dividends on shares of redeemable, convertible preferred stock of CombinatoRx (Singapore) Pte Ltd as discussed in Note 6.
The following potentially dilutive securities outstanding prior to the use of the treasury stock method have been excluded from the computation of diluted weighted-average shares outstanding for the years ended December 31, 2007, 2006 and 2005, as they would be anti-dilutive.
| | | | | | |
| | | As of December 31, |
| | | 2007 | | 2006 | | 2005 |
Options outstanding | | 5,067,240 | | 4,219,277 | | 3,585,876 |
Unvested restricted stock | | 165,938 | | 248,750 | | — |
Warrants outstanding | | 96,252 | | 96,252 | | 197,395 |
Convertible notes payable | | 1,195,733 | | 811,589 | | 509,136 |
Stock-Based Compensation
The Company adopted Statement of Financial Accounting Standards No. 123, “Share-Based Payment” (“SFAS 123R”) effective January 1, 2006. Under SFAS 123R, the Company is required to recognize, as expense, the estimated fair value of all share-based payments to employees. The Company adopted SFAS 123R under the prospective method for stock options granted prior to January 1, 2005, since stock options granted prior to that date were valued under the minimum value method with no volatility factor. The Company applied the modified prospective method for stock options and restricted stock awards granted after January 1, 2005, since once the Company was in registration for its initial public offering in 2005, it began to value the stock options granted in 2005 under the Black-Scholes Method using a volatility factor for purposes of proforma footnote disclosure under SFAS 123. Under the modified prospective method, the Company recognized stock-based compensation expense for all share-based payments granted to employees in 2007 and 2006 based on the grant date estimate of fair value for those awards beginning on January 1, 2006 and the unvested portion of awards granted in 2005. Prior period financial information has not been restated.
For periods prior to the adoption of SFAS 123R, the Company had elected to follow Accounting Principles Board Opinion No. 25 “Accounting for Stock Issued to Employees,” (APB 25) and related interpretations in accounting for its share-based payment awards. Under APB 25, since the exercise price of certain of the Company’s employee stock options was less than the deemed fair value of the underlying stock on the date of the grant in 2003, 2004 and 2005, the Company had recorded stock-based compensation expense in its consolidated financial statements.
F-12
The following table illustrates the effect on net loss and net loss per share as if the Company had applied the fair value recognition provisions of SFAS 123 to its stock-based employee compensation:
| | | | |
(in thousands, except per share amounts) | | Year Ended
December 31, 2005 | |
Net loss applicable to common shareholders, as reported | | $ | (29,515 | ) |
Add: Employee stock-based compensation expense included in reported net loss | | | 3,796 | |
Less: Stock-based compensation expense determined under fair value-based method for all employee awards | | | (4,227 | ) |
| | | | |
Pro forma net loss | | $ | (29,946 | ) |
| | | | |
Net loss per share applicable to common stockholders, as reported-basic and diluted: | | $ | (8.53 | ) |
| | | | |
Pro forma net loss per share applicable to common stockholders-basic and diluted: | | $ | (7.18 | ) |
| | | | |
Upon adoption of SFAS 123R, the Company recognized the stock-based compensation expense associated with awards granted after January 1, 2006 and the unamortized compensation expense related to the unvested portion of 2005 awards outstanding as of January 1, 2006. During the years ended December 31, 2007 and 2006, the Company recognized stock-based compensation expense of $7,286 and $6,328, respectively, for stock options and $619 and $710, respectively, for awards of restricted stock using the straight-line method. Upon the adoption of SFAS 123R, the Company reclassified deferred stock-based compensation expense of $11,052 to additional paid-in-capital. No cumulative catch-up adjustment related to forfeitures was recorded. As a result of the adoption of SFAS 123R, loss from operations, net loss and net loss per basic and diluted share for the year ended December 31, 2006 increased by $3,389, $3,389 and $0.12, respectively. The adoption of SFAS 123R had no effect on the statement of cash flows due to the Company’s net loss position.
Stock Options Granted to Non-Employees
The Company accounts for transactions in which services are received from non-employees in exchange for equity instruments based on the fair value of such services received or of the equity instruments issued, whichever is more reliably measured, in accordance with SFAS 123 and the Emerging Issues Task Force EITF Issue No. 96-18, Accounting for Equity Instruments that Are Issued to Other than Employees for Acquiring, or in Conjunction with Selling, Goods or Services.
Income Taxes
Deferred taxes are determined based on the difference between the financial statement and tax basis of assets and liabilities using enacted tax rates in effect in the years in which the differences are expected to reverse. Valuation allowances are provided, if, based upon the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
The Company adopted the provisions of Financial Accounting Standards Board (“FASB”) Interpretation No. 48, “Accounting for Uncertainty in Income Taxes—an interpretation of FASB Statement No. 109” (“FIN 48”), on January 1, 2007. FIN 48 clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements in accordance with FASB Statement 109, “Accounting for Income Taxes”, and prescribes a recognition threshold and measurement process for financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. FIN 48 also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition.
F-13
The Company concluded that there are no significant uncertain tax positions requiring recognition in the consolidated financial statements. The Company’s evaluation was performed for the tax years ended December 31, 2004, 2005, 2006 and 2007, the tax years which remain subject to examination by major tax jurisdictions as of December 31, 2007.
The Company may from time to time be assessed interest or penalties by major tax jurisdictions, although any such assessments historically have not impacted the financial results of the Company. In the event the Company would receive an assessment for interest and/or penalties, it would be classified in the consolidated financial statements as general and administrative expense.
Recently Issued Accounting Pronouncements
In February 2007, Statement of Financial Standard No. 159, “The Fair Value Option for Financial Assets and Financial Liabilities, Including an Amendment of FASB Statement No. 115”, or SFAS 159, was issued. SFAS 159 permits the Company to choose to measure many financial instruments and certain other items at fair value. It also establishes presentation and disclosure requirements. SFAS 159 was effective January 1, 2008 for the Company. The Company is evaluating the impact, if any, SFAS 159 will have on its financial statements.
In June 2007, FASB issued EITF Issue No. 07-3, “Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities” (“EITF 07-3”). EITF 07-3 requires that nonrefundable advance payments for goods or services to be received in the future for use in research and development activities should be deferred and capitalized. The capitalized amounts should be expensed as the related goods are delivered or the services are performed. EITF 07-3 is effective for new contracts entered into during fiscal years beginning after December 15, 2007. The Company is currently analyzing the effect, if any, EITF 07-3 will have on its consolidated financial position and results of operations.
Reclassifications
The Company reclassified $2,900 and $4,015 of proceeds from landlord under tenant improvements from financing activities to operating activities at December 31, 2007 and 2006, respectively.
Property and equipment consist of the following:
| | | | | | | | | | |
| | | Estimated Useful Life (Years) | | December 31, | |
| | | | 2007 | | | 2006 | |
Leasehold improvements | | Lesser of useful life or life of lease | | $ | 10,393 | | | $ | 5,402 | |
Laboratory equipment | | 5 | | | 8,527 | | | | 5,231 | |
Computer equipment | | 3 | | | 2,895 | | | | 2,179 | |
Construction in progress | | — | | | 546 | | | | 3,392 | |
Capitalized software | | 5 | | | 591 | | | | 591 | |
Furniture and fixtures | | 3 | | | 1,361 | | | | 952 | |
| | | | | | | | | | |
| | | | | 24,313 | | | | 17,747 | |
Less: accumulated depreciation | | | | | (8,380 | ) | | | (5,241 | ) |
| | | | | | | | | | |
| | | | $ | 15,933 | | | $ | 12,506 | |
| | | | | | | | | | |
Depreciation expense for the years ended December 31, 2007, 2006 and 2005 was approximately $3,139, $1,865, and $1,126, respectively.
F-14
In July 2004, the Company entered into a loan agreement (the “2004 GE Agreement”) with General Electric Capital Corporation (GECC). The Company borrowed all $3,000 available under the 2004 GE Agreement during 2004. In June 2005, the 2004 GE Agreement was amended (the “Amended 2004 GE Agreement”) to establish a new line of credit which enabled the Company to borrow an additional $1,000 through June 2006. The Company borrowed $965 under this amended line of credit prior to its expiration. Amounts borrowed under the 2004 GE Agreement and the Amended 2004 GE Agreement are repayable over 36 months.
In June 2006, the Company amended the Amended 2004 GE Agreement (the “2006 Amendment”) to increase the line of credit by $3,310. On June 2006, the Company borrowed $1,607 under the 2006 Amendment. In December 2006, the Company borrowed an additional $1,479 under the 2006 Amendment. In March 2007, the Company further amended the 2004 GE Agreement and 2006 Amendment to increase the line of credit available by $877 and then concurrently borrowed the remaining $1,101 available under this amended line of credit.
In June 2007, the Company further amended the 2004 Amended GE Agreement by increasing the available secured line of credit by an additional $4,000 (the “2007 Amendment”). The Company borrowed $2,912 in June 2007 and an additional $781 in September 2007. In December 2007, the Company further amended the 2007 Amendment to increase the line of credit by $135, and the Company borrowed the remaining $442 available. Amounts borrowed under the 2006 and 2007 amendments are repayable over 48 months in the case of laboratory and scientific equipment, 36 months in the case of other equipment and 24 months in the case of software. Borrowings are collateralized by substantially all of the Company’s tangible assets. At December 31, 2007 and 2006, $8,325 and $4,399, respectively, were payable under the Amended 2004 GE Agreement, 2006 Amendment and 2007 Amendment, and no additional funds were available to be borrowed. The amended line of credit with GECC contains a subjective acceleration clause which provides GECC the ability to demand repayment of the loan early upon a material adverse event, as defined.
In connection with the 2004 GE Agreement, GECC received a warrant to purchase 15,561 shares of Series D redeemable convertible preferred stock which converted into the right to purchase 8,892 shares of common stock upon the Company’s initial public offering, with an exercise price of $6.7476 per share of common stock. The warrant has a term of ten years. The Company recorded the fair value of this warrant of $75 as a discount to the note payable to GECC. The discount is being amortized to interest expense over the three-year period that the note to GECC is outstanding. The fair value of the warrant was calculated using the Black-Scholes option pricing model with the following assumptions: deemed fair market value of the Series D redeemable convertible preferred stock of $5.24, 100% volatility, risk free interest rate of 4.05%, a ten year term and no dividend yield.
In connection with the Amended 2004 GE Agreement, GECC received a warrant to purchase 471 shares of common stock with an exercise price of $6.75 per share of common stock. The warrant has a term of ten years. The Company recorded the fair value of this warrant of $5 as a discount to the note payable to GECC. The discount is being amortized to interest expense over the three-year period that the note to GECC is outstanding. The fair value of the warrant was calculated using the Black-Scholes option pricing model with the following assumptions: deemed fair market value of common stock of $11.00, 80% volatility, risk free interest rate of 3.92%, a ten year term and no dividend yield.
F-15
The following table depicts the Company’s history of borrowings with GECC and includes the remaining principal and interest balances at December 31, 2007:
Company Borrowings
| | | | | | | | | | | | |
Borrowing Date | | Principal Amount | | Interest
Rate | | | Remaining Principal
Balance at 12/31/07 | | Remaining Interest
at 12/31/07 |
July 2004 | | $ | 3,000 | | 8.42 | % | | $ | — | | $ | — |
June 2005 | | | 159 | | 9.76 | % | | | 34 | | | 1 |
March 2006 | | | 806 | | 10.52 | % | | | 389 | | | 30 |
June 2006 | | | 1,607 | | 10.68 | % | | | 1,004 | | | 121 |
December 2006 | | | 1,479 | | 9.98 | % | | | 1,098 | | | 137 |
March 2007 | | | 1,101 | | 10.12 | % | | | 908 | | | 135 |
June 2007 | | | 2,912 | | 10.46 | % | | | 2,603 | | | 445 |
September 2007 | | | 781 | | 10.46 | % | | | 749 | | | 142 |
December 2007 | | | 442 | | 10.46 | % | | | 442 | | | 87 |
| | | | | | | | | | | | |
| | $ | 12,287 | | | | | $ | 7,227 | | $ | 1,098 |
| | | | | | | | | | | | |
In February 2007, CombinatoRx Singapore entered into a $2,100 secured equipment line of credit with GE Capital Services Pte. Ltd., (the “Singapore line of credit”). CombinatoRx Singapore borrowed $1,236 under the Singapore line of credit in February 2007 and $349 in November 2007. The Singapore line of credit was available through November 30, 2007 and is secured by a fixed charge security interest over the equipment financed in Singapore. The Company also provided a corporate guaranty of payment in connection with the Singapore line of credit. Amounts borrowed under the Singapore line of credit for laboratory and scientific equipment are repayable over 48 months, and amounts borrowed for other equipment are repayable over 36 months. As of December 31, 2007, there was $1,503 payable under the Singapore line of credit, and no additional funds were available to be borrowed.
The following table depicts the history of CombinatoRx Singapore’s borrowings under the Singapore line of credit and includes the remaining principal and interest balance as of December 31, 2007:
CombinatoRx Singapore Borrowings
| | | | | | | | | | | | |
Borrowing Date | | Principal Amount | | Interest
Rate | | | Remaining Principal
Balance at 12/31/07 | | Remaining Interest
at 12/31/07 |
February 2007 | | $ | 779 | | 10.42 | % | | $ | 622 | | $ | 108 |
February 2007 | | | 457 | | 10.45 | % | | | 330 | | | 39 |
November 2007 | | | 10 | | 10.10 | % | | | 10 | | | 1 |
November 2007 | | | 339 | | 10.13 | % | | | 325 | | | 68 |
| | | | | | | | | | | | |
| | $ | 1,585 | | | | | $ | 1,287 | | $ | 216 |
| | | | | | | | | | | | |
Future principal and interest payments under the loan agreements with GECC and GE Capital Services Pte. Ltd. at December 31, 2007, are as follows:
| | | |
Year Ending December 31, | | | |
2008 | | $ | 3,836 |
2009 | | | 3,462 |
2010 | | | 1,952 |
2011 | | | 570 |
2012 | | | 8 |
| | | |
| | $ | 9,828 |
| | | |
F-16
In connection with a 2004 financing, the Company issued a warrant to purchase shares of Series D redeemable convertible preferred stock which converted into the right to purchase 51,870 shares of common stock upon the Company’s initial public offering at $6.74765 per share of common stock. The warrant has a term of seven years. The Company recorded the fair value of this warrant of $515 as deferred financing costs in other assets and began to amortize the balance to interest expense over the five-year period that amounts borrowed under the line of credit were to be outstanding. Since no amounts were borrowed under the line of credit and the line of credit is no longer available, the value of the warrants was charged to interest expense in October 2005. The warrant remains outstanding at December 31, 2007.
The Company has reserved 61,233 shares of common stock for the exercise of warrants issued in connection with the 2004 GE Agreement, the Amended 2004 GE Agreement, and the 2004 financing discussed above. These shares are included in the total shares of common stock reserved for the exercise of stock options and warrants at December 31, 2007, as discussed in Note 8.
| 5. | Research and Development Agreements |
2007 Agreements
On November 7, 2007, the Company entered into a sponsored research collaboration agreement with an entity formed by Charley’s Fund and the Nash Avery Foundation (the “DMD Foundations”), two nonprofit organizations founded to support Duchenne muscular dystrophy, or DMD research. Under the agreement, the Company will seek to identify novel disease-modifying multi-targeted treatments for DMD, the most common childhood form of muscular dystrophy. Under the terms of the agreement, the Company is eligible to receive up to $3,000 in research funding and reimbursement of additional expenses during the term of the DMD research and development project, of which $125 has been received through December 31, 2007. The Company retains worldwide commercialization rights for any product candidates discovered or developed under the agreement, and the Company will own all new intellectual property and data generated by the research and development project. In the event that the Company either enters into a license agreement with a third party granting the rights to make, use or sell a product developed under the agreement to treat DMD, or the Company or any of its affiliates or licensees first sells a product developed under the agreement to treat DMD, the Company will pay the DMD Foundations a payment equal to 100% of the research funding provided to the Company under the agreement. In addition, on the first anniversary of the first commercial sale of a product developed under the agreement, the Company will pay the DMD Foundations an additional payment equal to 100% of the research funding provided to the Company under the agreement. Finally, if a product developed under the agreement to treat DMD achieves cumulative net sales of at least $100 million, within 90 days of such occurrence, the Company will pay the DMD Foundations an additional payment equal to 200% of the research funding provided to the Company under the agreement. The Company concluded that the research funding fees of $3,000 are fixed or determinable despite the potential payments it might make to the DMD Foundations that are based upon percentages of research funding received since the research is in the early stages and thus these payments are not probable upon execution of the agreement.
The agreement with respect to research and development collaboration terminates upon the expiration of the research and development project, which is currently planned to last for two years. The agreement may be terminated by either party after sixty days’ notice upon an unremedied material breach. In addition, if the Company intends to discontinue pre-clinical or clinical development activities with respect to a DMD product candidate and does not intend to license such candidate to a third party for pre-clinical or clinical development, within one year after such determination, the Company shall notify the DMD Foundations, who may then exercise their rights to an exclusive, fully-paid and sublicensable license to the intellectual property developed under the collaboration in the field of DMD. During the year ended December 31, 2007, the Company received payments of $125 and recognized $7 of revenue under this agreement.
F-17
On April 25, 2007, the Company and CombinatoRx Singapore entered into an Interim Consortium Agreement with the Liverpool School of Tropical Medicine (“LSTM”) and other parties, under which the Company and CombinatoRx Singapore will act as subcontractors to perform assay development and screening services relating to a grant awarded to LSTM by the Bill and Melinda Gates Foundation focusing on the development of treatment regimens against filaraisis, or African river blindness. Under the terms of the interim agreement, CombinatoRx Singapore could receive up to $3,968 in research and development funding over a five-year period. The interim agreement expires on March 31, 2008 unless a definitive consortium agreement is entered into between the Company, CombinatoRx Singapore, LSTM and the other parties. The Company received funding of $561 in 2007 and recorded $498 of revenue in 2007, based upon research services performed and costs incurred. The Company recorded $63 of deferred revenue for this agreement at December 31, 2007.
2006 Agreements
On January 30, 2006, the Company entered into a research and license agreement (the “Original Fovea Agreement”) with Fovea Pharmaceuticals SA (“Fovea”). Under the terms of the Original Fovea Agreement, Fovea agreed to fund and conduct pre-clinical and clinical development in ophthalmology of combination drug candidates it selects from the Company’s portfolio, including creating ophthalmic formulations. Additionally, Fovea agreed to develop selected combination candidates up to the start of phase 3 clinical trials. In exchange for Fovea’s development investment, the Company granted to Fovea an exclusive license to commercialize selected products in Europe and certain additional countries. The Company will retain exclusive rights to commercialize selected products in North America. The parties will have co-exclusive rights in Japan and Taiwan. Under the Original Fovea Agreement, the Company also granted to Fovea an exclusive worldwide license to certain preclinical drug combinations to treat specified diseases of the front of the eye. In consideration for the license of these combinations to treat specified diseases of the front of the eye, the Company is entitled to receive license execution fees, development milestones and royalties from Fovea if certain conditions within the Original Fovea Agreement are satisfied.
On June 12, 2007, the Company and Fovea amended and restated the Original Fovea Agreement (the “Amended Fovea Agreement”). Under the Amended Fovea Agreement, Fovea will continue to conduct, at its own expense, preclinical and clinical development for certain ophthalmic indications of combination drug candidates it has selected from the Company’s portfolio of product candidates. Fovea is obligated to develop selected combination candidates pursuant to specified development criteria through the end of phase 2b clinical trials.
The Company and Fovea will continue to jointly own new intellectual property and data generated by Fovea regarding the selected combination candidates through phase 2a clinical trials. The Company retains the rights to develop and commercialize the combination candidates licensed to Fovea in North America and certain other countries and the Company granted Fovea exclusive rights to commercialize selected combination candidates that are developed to through phase 2b clinical trials for specified ophthalmic indications in Europe and all other countries that are not retained by the Company. The parties have co-exclusive rights in Japan and Taiwan. The grant by the Company to Fovea of an exclusive milestone and royalty-bearing worldwide license to certain preclinical drug combinations to treat allergic and inflammatory diseases of the front of the eye was retained in the Amended Fovea Agreement.
Under the Amended Fovea Agreement, the Company may also receive up to approximately $20,000 in development milestone payments for each licensed combination successfully developed and approved by the regulatory authorities in the European Union, United States and Japan. The Company could receive an additional milestone payment of $10,000 for the approval by any regulatory authority of a licensed combination developed to treat a specifically identified indication within the agreement. The Company is also eligible to receive royalties for product(s) commercialized by Fovea. On December 30, 2006, Fovea selected the licensed combination compounds as provided in the Original Fovea Agreement. Beginning January 1, 2007, the Company began to recognize the $750 in previously received non-refundable license execution fees ratably over seven years, which
F-18
was the Company’s estimate of its period of significant continuing involvement. In connection with the Amended Fovea Agreement, the Company determined that it no longer had any significant continuing involvement in the collaboration from an accounting perspective and as a result recorded the remaining deferred revenue balance of $705 as revenue in June 2007.
On May 31, 2006, the Company entered into a research, development and commercialization agreement (the “CF Agreement”) with Cystic Fibrosis Foundation Therapeutics Incorporated (“CFFT”). Under the terms of the CF Agreement, the Company was awarded up to $13,825 in research funding and expenses. In addition, CFFT has agreed to fund up to 75% of the clinical development expenses incurred by the Company through a phase 2a clinical trial of the first potential product candidate, provided both parties have agreed to commence clinical development of the product candidate. The Company is eligible to receive milestone payments from CFFT upon successful completion of specified clinical and regulatory events for each product candidate developed under the CF Agreement. CFFT will be eligible to receive variable royalties from the Company on the net sales of any approved products that are discovered under the CF Agreement. The Company retains worldwide commercialization rights for any product candidates discovered or developed under the agreement, and the Company will own all new intellectual property and data generated by the research and development project.
The CF Agreement has no definite term, but the research and development project will terminate upon the earlier of the completion of one phase 2a clinical trial of the first product candidate developed under the CF Agreement and seven years after the initiation of research under the CF Agreement. The Company’s royalty payment obligations to CFFT do not terminate, but its royalties in the field of cystic fibrosis and certain other pulmonary diseases may be reduced on a country-by-country basis upon the expiration of all valid patents covering a royalty bearing product under the agreement. During the year ended December 31, 2007, the Company received payments of $2,365 and recognized $2,214 of revenue under the CF Agreement, which represented 14.8% of total revenue in 2007. During the year ended December 31, 2006, the Company received payments of $600 and recognized $222 of revenue under the CF Agreement. The Company had $530 of deferred revenue at December 31, 2007 and $19 of unbilled receivables for this agreement at December 31, 2006.
2005 Agreements
In February 2005, the Company entered into an agreement and a statement of work with Novartis Pharmaceuticals, Inc. (“Novartis”), under which the Company agreed to test certain compounds provided by Novartis in combination with other compounds in cancer cell lines determined by Novartis. The statement of work provided for $500 of research funding, all of which was received by the Company in 2005. The Company recognized $500 of revenue ratably over the performance period in 2005, which represented 10.7% of total revenue in 2005.
In April 2005, the Company received a grant from the National Institutes of Allergy and Infectious Diseases to perform research and preclinical development in the area of bioterror defense. The Company received funding of $1,312, $655 and $285 in 2007, 2006 and 2005, respectively, and recorded $1,137, $730 and $414 of revenue in 2007, 2006 and 2005, respectively, based upon allowable costs incurred during the year. The Company had $29, $204 and $129 of unbilled receivables for this agreement at December 31, 2007, 2006 and 2005, respectively. In April 2007, the Company received an interim indirect cost negotiation agreement (the “interim rate agreement”) from the United States Department of Health and Human Services related to the rates the Company is allowed to charge under its National Institutes of Allergy and Infectious Diseases (“NIAID”) grant. As a result, during the second quarter of 2007, the Company recorded an incremental $414 in revenue under this grant. The rates provided by this interim agreement are retroactive to January 2005 and extend through June 2008. The Company is currently in the process of obtaining final approval of its provisional rates for the 2005 and 2006 periods. The Company will provide the documentation required to support its 2007 rates in 2008. As a result, revenue recorded in the year ended December 31, 2007 is based upon the interim rate agreement and may be subject to additional adjustments in future periods should the Company’s final rate agreement vary from its interim rate agreement. Such adjustments will be recorded in revenue in the period in which they become fixed or determinable.
F-19
In May 2005, the Company entered into a license agreement with HenKan Pharmaceutical Company (“HenKan”), under which HenKan received the exclusive right to develop and commercialize CRx-026 in Taiwan, China and South Korea. The Company received a $500 up-front license payment and could receive up to approximately $23,000 in development and commercial milestone payments, and royalties on sales within the territory. The up-front payment is creditable against future licenses in the event that development of the compound is unsuccessful. As a result, the license fee will not be recognized as revenue until this contingency is resolved. At that time, the license fee is expected to be recognized as revenue over any remaining performance period.
In August 2005, the Company entered into a subcontract with Science Applications International Corporation (“SAIC”) under which the Company will be the in vitro bioassay screening facility for the Spinal Muscular Atrophy Project established by the National Institute of Neurological Disorders and Stroke (“NINDS”). Under the terms of the agreement, the Company could receive up to $1,917 in research and development funding over a two-year period. The Company received funding of $761 and $316 in 2007 and 2006 respectively and recorded $885, $376 and $102 of revenue in 2007, 2006 and 2005, respectively, based upon research services performed and costs incurred. The Company had $286 and $162 of receivables for this agreement at December 31, 2007 and 2006, respectively, and $102 of unbilled receivables for this agreement at December 31, 2005. In April 2007, the Company received an interim indirect cost negotiation agreement (the “interim rate agreement”) from the United States Department of Health and Human Services related to the rates the Company is allowed to charge under its SAIC-NINDS subcontract. The Company will provide the documentation required to support its 2007 rates in 2008. As a result, during the second quarter of 2007, the Company recorded an incremental $366 in revenue under this subcontract. The rates provided by this interim agreement are retroactive to January 2005 and extend through June 2008. The Company is currently in the process of obtaining final approval of its provisional rates for the 2005 and 2006 periods. As a result, revenue recorded in the year ended December 31, 2007 is based upon the interim rate agreement and may be subject to additional adjustments in future periods should the Company’s final rate agreement vary from its interim rate agreement. Such adjustments will be recorded in revenue in the period in which they become fixed or determinable.
In August 2005, the Company entered into a research agreement with CHDI, Inc. to perform joint research and development to discover and perform preclinical development of product candidates for the treatment of Huntington’s disease. Under the terms of this agreement as amended and restated in February 2007, subject to satisfaction of conditions, the Company could receive up to $6,695 in research funding over a four-year period and may receive milestone and revenue sharing payments under certain circumstances if a product candidate is commercialized. The Company received payments of $2,148 in 2007 and recorded $2,067 of revenue based upon research services performed and costs incurred, which represented 13.8% of total revenue in 2007. The Company received payments of $899 in 2006 and recorded $1,696 of revenue based upon research services performed and costs incurred, which represented 12.8% of total revenue in 2006. The Company received payments of $669 in 2005 and recorded $482 of revenue based upon research services performed and costs incurred, which represented 10.4% of total revenue in 2005. The Company had $529 and $610 of unbilled receivables for the agreement at December 31, 2007 and 2006 respectively.
In September 2005, the Company entered into a screening agreement with Sirtris Pharmaceuticals, Inc. (“Sirtris”), wherein the Company will provide access to its Chalice database over a 13.5-month period for screening against selected targets to evaluate activity and then potentially develop and commercialize human therapeutic products. The Company received a $175 access fee as consideration for providing access to the Chalice database. The Company recognized the access fee as revenue ratably over the 13.5-month term of the agreement. Accordingly, the Company recorded $147 and $28 of revenue in 2006 and 2005, respectively. The Company could receive up to $1,075 in development milestone payments and royalty payments based on sales of products developed by Sirtris.
In October 2005, the Company entered into a research and license agreement (the “R&L Agreement”) with Angiotech Pharmaceuticals, Inc. (“Angiotech”), under which the Company granted Angiotech a royalty-bearing license for up to ten compounds to be selected by Angiotech from the Company’s portfolio of clinical and
F-20
preclinical product candidates or Chalice database, as well as an option to purchase the same rights to an additional five compounds. This license is for Angiotech’s research, development and potential commercialization of the licensed compounds as drug components to be used in Angiotech’s field with medical devices or interventional medicine products to treat conditions in specific areas of the human body. In addition, the Company agreed to use its combination High Throughput Screening technology in a joint research project to screen combinations of compounds that may be developed and commercialized by Angiotech for use in combination with medical devices or with interventional medicine products in Angiotech’s field. The Company received a $27,000 up-front license fee upon execution of the R&L Agreement in 2005. As contemplated by the original agreement, on June 8, 2007, Angiotech agreed to extend the research project beyond the original 30-month term to a total term of five years for an additional license execution fee of $7,000. The original three-year research project performance period included a six-month period beyond the 30 months where the Company must provide Angiotech with all reasonable assistance required in order to transfer the licensed information to Angiotech. As a result of the extension of the research project, the Company revised its revenue recognition based on this change in estimate from $2,250 recognized per quarter to $1,239 recognized per quarter. The Company may also receive up to an additional $10,000 upon Angiotech’s election to receive a license to up to five additional compounds, beyond the initial ten compounds, from the Company’s portfolio of clinical and preclinical product candidates or Chalice database for development. In addition, for each compound licensed to Angiotech that is discovered through the research project or through Angiotech’s selection of compounds from the Company’s portfolio of clinical and preclinical product candidates or Chalice database for development, the Company may also receive up to $30,000 in milestone payments if certain development and regulatory approval milestones are met, as well as royalties on any future product sales incorporating the compounds. The Company recognized $6,641, $9,000 and $2,250 of revenue under this agreement in 2007, 2006 and 2005, respectively, which represented 44.5%, 67.8% and 48.3% of total revenue in 2007, 2006 and 2005, respectively. The Company had deferred revenue of $16,109 and $15,750 at December 31, 2007 and 2006, respectively.
| 6. | CombinatoRx (Singapore) Pte Ltd |
In August 2005, the Company formed a subsidiary in Singapore, CombinatoRx (Singapore) Pte Ltd (the “Subsidiary”), for the purpose of conducting discovery and development of product candidates to treat infectious diseases. The Company owns 51% of the Subsidiary’s capital stock. The Company has agreed to provide assay development and screening services for the Subsidiary over a four year period. BioMedical Sciences Investment Fund Pte Ltd (“BioMedical Sciences”) invested $2,500 in 2,500,000 shares of redeemable, convertible preferred stock (the “Subsidiary Preferred Stock”) of the Subsidiary and committed to invest up to an additional $17,500 in the Subsidiary through the purchase of a series of convertible promissory notes (“Notes”), $5,500 of which were purchased concurrently with its investment in the Subsidiary Preferred Stock (the “Series 1 Note”). The remaining $12,000 in funding was to be provided through the purchase of additional series of Notes over the next four years, provided that the Subsidiary achieved certain milestones related to the development of infectious disease product candidates. On June 8, 2006, upon the achievement of a milestone, BioMedical Sciences invested an additional $3,500 in the Subsidiary for which it was issued a new $3,500 convertible promissory note (the “Series 2 Note”). On May 30, 2007, upon the achievement of a milestone, BioMedical Sciences invested an additional $3,500 in the Subsidiary for which it was issued a new $3,500 convertible promissory note (the “Series 3 Note”).
The holder of the Subsidiary Preferred Stock is entitled to an annual 5% dividend payable upon redemption or liquidation of the Subsidiary. The Subsidiary Preferred Stock is subject to redemption by the Subsidiary for a cash payment equal to 125% of the purchase price of the shares plus accrued, but unpaid, dividends. The Company had accumulated accretion of $292 and $169 of dividends on the Subsidiary Preferred Stock for the years ended December 31, 2007 and 2006, respectively. The Subsidiary Preferred Stock is convertible into common stock of the Company at a conversion price obtained by dividing the aggregate amount paid for such Subsidiary Preferred Stock by a variable premium to the weighted average of the Company’s common stock price based on the trading price of its common stock over the 20 trading days after the Company’s initial public offering in November 2005, or $10.80. The initial premium to the volume-weighted average stock price is 40%.
F-21
The Notes bear interest at an annual rate of 5% and are due and payable on December 31, 2009, unless the Company elects to prepay the Notes before that date through the Subsidiary. The Notes are secured by a security interest in the non-intellectual property assets of the Subsidiary and by a negative pledge by the Subsidiary with respect to its intellectual property rights. The Company has pledged its shares in the Subsidiary as additional collateral for the Subsidiary’s obligations under the Notes. The Notes are convertible into the Company’s common stock at the option of BioMedical Sciences only upon maturity, acceleration or default or any proposed prepayment. The Company recorded $545, $381 and $94 of cumulative accrued interest on the Series 1, Series 2 and Series 3 Notes in the year ended December 31, 2007, on the Series 1 and Series 2 Notes in the year ended December 31, 2006 and on the Series 1 Note in the year ended December 31, 2005, respectively. Upon maturity or any proposed prepayment, the Series 1 Note is convertible at a price obtained by dividing the aggregate principal balance of such Note by $10.80, the Series 2 Note is convertible at a price obtained by dividing the aggregate principal balance of such Note by $11.57 and the Series 3 Note is convertible at a price obtained by dividing the aggregate principal balance of such Note by $9.11.
Upon a default by the Company or Subsidiary, the Notes and the Subsidiary Preferred Stock are convertible at the option of BioMedical Sciences as described with respect to a conversion upon maturity or prepayment except that (i) the conversion price of the Notes would include a 10% default interest rate accrued from the date of issuance and the Subsidiary Preferred Stock would also include a 10% dividend accrual accrued from the date of issuance and (ii) no conversion premium would apply with respect to conversions occurring after the Company’s initial public offering.
In connection with the formation of the Subsidiary, BioMedical Sciences received a warrant to purchase 25,000 shares of the Company’s common stock at an exercise price of $11.00 per share, exercisable after August 19, 2006 through August 19, 2010. The Company allocated the fair value of this warrant to the originally issued Note and the Subsidiary Preferred Stock based upon their relative fair values. This resulted in a $126 allocation of discount on the originally issued Note, which will be amortized to interest expense over the period that the originally issued Note is outstanding and a $57 allocation to the Subsidiary Preferred Stock which was charged to accumulated deficit. The allocation of the value of the warrants to the originally issued Note and the Subsidiary Preferred Stock resulted in a beneficial conversion feature under EITF 98-5,Accounting for Convertible Securities with Beneficial Conversion Features or Contingently Adjustable Conversion Ratios. As a result, the Company recorded an additional discount on the Series 1 Note of $126 and a charge to accumulated deficit of $57. The Company recorded $58, $58 and $19 of interest expense in 2007, 2006 and 2005, respectively. The fair value of the warrant was calculated using the Black-Scholes option pricing model with the following assumptions: deemed fair market value of common stock of $11.00 per share, 80% volatility, risk-free interest rate of 4.12%, no dividend yield and a five-year term.
On April 19, 2006, the Subsidiary received approval for a grant from the Economic Development Board of Singapore (EDB) Biomedical Sciences Group for up to approximately $5,830 to support infectious disease drug research and development. The grant covers a percentage of qualifying costs of the research and development project on a reimbursement basis. Qualifying costs include salaries, equipment, scientific consumables and intellectual property costs. Reimbursement for these costs under the grant is subject to the satisfaction of certain conditions by the Subsidiary, including completion of the development project for infectious disease within a specified timeline, spending specified amounts on the project, the completion of other development milestones and the maintenance of specified levels of employment in Singapore. Subject to agreed upon audit rights by the EDB, cumulative qualifying costs are reimbursed upon application until 70% of the initial grant amount has been submitted by the Subsidiary. The remaining 30% of the award may be paid by the EDB once the Company completes the research and development project. The grant extends through September 30, 2010. If the Subsidiary breaches a condition of the grant, after good faith negotiations, the EDB may recover previously released grant funds from the Subsidiary. In addition, the EDB retains the right to change the terms and conditions of the grant as deemed necessary by the EDB. The Company recognizes revenue under the grant as qualifying costs are incurred up to a maximum of 70% of the initial grant amount (approximately $4,081). Reimbursements for qualifying costs in excess of 70% of the initial grant amount will be recognized once the
F-22
reimbursements are deemed to be fixed or determinable. The Company recognizes revenue for equipment costs that are reimbursed ratably over the remaining term of the grant, which approximates the estimated useful life of the equipment. The Company recorded $643 and $426 of revenue based upon qualifying costs incurred and deferred $180 and $162 of revenue pertaining to equipment reimbursements in 2007 and 2006 respectively. The Company had $186 and $228 of unbilled receivables for this agreement at December 31, 2007 and 2006, respectively. The Company received $705 and $360 in 2007 and 2006, respectively.
The registration statement for the Company’s initial public offering was declared effective on November 9, 2005. In connection with the initial public offering, the Company issued 6,900,000 shares of common stock for net proceeds of $43,843.
On March 24, 2006, the Company completed a private placement (the “Private Placement”) of 4,682,942 shares of common stock (the “Shares”) at a price of $10.25 per share for net proceeds of approximately $45,373. The Company entered into a Securities Purchase Agreement (the “Purchase Agreement”) with the purchasers of Shares in the Private Placement in which the Company agreed to file a Registration Statement on Form S-1 with the Securities and Exchange Commission (the “Commission”) by April 24, 2006 to register the resale of the Shares sold through the Private Placement. The Company filed a resale Registration Statement on Form S-1 with the Commission on April 24, 2006. The Company also agreed to use its commercially reasonable efforts to have the Registration Statement declared effective within 90 days after the closing of the Private Placement if the Registration Statement receives no Commission review or 150 days after the closing of the Private Placement if the Registration Statement receives Commission review. The Registration Statement was declared effective by the Commission on May 23, 2006. In addition, the Company agreed to use its commercially reasonable efforts to keep the Registration Statement effective until the earlier of two years after the effective date of the Registration Statement, the date on which the Shares may be resold pursuant to Rule 144 under the Securities Act of 1933, as amended (the “Securities Act”). If, after the Registration Statement is declared effective, the Company suspends the use of the Registration Statement by the purchasers for the resale of the Shares, the Company has agreed to pay each purchaser as liquidated damages an amount equal to 1.0% of the purchase price paid by each such purchaser in the Private Placement for each month that the use of the Registration Statement is suspended in excess of 30 consecutive days or more than 60 days in any 12-month period, subject to the aggregate limit on liquidated damages. Under the Purchase Agreement, the maximum aggregate amount of liquidated damages payable to each purchaser is limited to 10% of the purchase price paid by each such purchaser in the Private Placement. As of December 31, 2007, the Company has not incurred any liquidated damages relating to these provisions.
On October 10, 2007, the Company entered into a placement agent agreement, relating to the offering, issuance and sale of an aggregate of 5,600,000 shares of the Company’s common stock, par value $0.001 per share, at a price of $6.25 per share to selected institutional investors (the “Direct Offering”). The Direct Offering closed on October 16, 2007, with net proceeds to the Company of $33,039 after deducting all offering expenses and placement agency fees payable by the Company. The Direct Offering was made under the Company’s Registration Statement on Form S-3 (File No. 333-139260) filed with the Commission on December 11, 2006 and declared effective on December 20, 2006. After the Direct Offering, the Registration Statement permits the Company to issue, in one or more offerings, shares of common stock, preferred stock, warrants or debt securities at an aggregate initial offering price not to exceed $40,000.
Each share of common stock is entitled to one vote. The holders of common stock are also entitled to receive dividends whenever funds are legally available and when declared by the Board of Directors, subject to the prior rights of holders of all classes of stock outstanding.
The Company has reserved a total of 5,163,492 shares of common stock for the exercise of stock options and warrants at December 31, 2007. In addition to the warrants disclosed in Note 4 and Note 6, the Company has also issued a warrant to purchase 10,019 shares of common stock to Silicon Valley Bank with an exercise price of $7.87 per share and a term that expires on April 25, 2011.
F-23
| 8. | Stock Compensation Plans |
In 2000, the Company adopted the 2000 Stock Plan (“2000 Plan”), as amended, under which 3,028,571 shares of the Company’s common stock were reserved for issuance to employees, officers, directors, advisors and consultants. Options granted under the 2000 Plan may be incentive stock options or non-statutory stock options. In December 2004, the Board of Directors and stockholders adopted the 2004 Incentive Plan, which was effective upon the Company’s initial public offering on November 9, 2005. On June 1, 2006, the Company’s stockholders approved the Amended and Restated 2004 Incentive Plan (“2004 Plan”). The Company has reserved 3,714,286 shares of the Company’s common stock for issuance under the 2004 Plan. The 2004 Plan includes an “evergreen provision” that allows for an annual increase in the number of shares of common stock available for issuance under the 2004 Plan, which annual increase will be added on the first day of each fiscal year from 2007 through 2011, inclusive, and will be equal to the least of (i) 2,000,000 shares of common stock, (ii) 4% of the outstanding shares on that date or (iii) such lesser amount determined by the Board of Directors. On January 17, 2007, the Compensation Committee of the Board of Directors ratified the amount of the annual increase for 2007 as 1,153,121 shares of common stock, or 4% of the outstanding common stock on January 1, 2007. On January 15, 2008, the Compensation Committee of the Board of Directors ratified the amount of the annual increase for 2008 as 1,392,887 shares of common stock, or 4% of the outstanding common stock on January 1, 2008. The 2004 Plan provides for the grant of incentive stock options, nonstatutory stock options, restricted stock and unrestricted stock awards, stock appreciation rights, cash awards, performance awards and stock units. Awards under the 2004 Plan may be granted to employees, directors, consultants and advisors. Generally, stock options and restricted stock granted to employees pursuant to the 2000 and 2004 Plans fully vest four years from the grant date, with 25% of the award vesting after one year and 6.25% of the award vesting quarterly thereafter. Stock options have a contractual term of 10 years.
The Board of Directors, or the Compensation Committee of the Board of Directors, administers the 2000 Plan and the 2004 Plan and has sole discretion to grant options to purchase shares of the Company’s common stock and other stock-based awards or to delegate to certain officers of the Company the ability to make specified grants. The Compensation Committee or the respective officers of the Company determine the exercise price and the period over which options become exercisable. However, incentive stock options may not be granted at less than 100% of the fair market value of the Company’s common stock as determined by the Compensation Committee at the time of grant, or for a term in excess of ten years. For holders of more than 10% of the Company’s total combined voting power of all classes of stock, incentive stock options may not be granted at less than 110% of the fair market value of the Company’s common stock at the date of grant, and for a term not to exceed five years.
As of December 31, 2007, there were 1,409,026 shares available for future issuance under the plans.
A summary of the status of the Company’s stock option plans at December 31, 2007 and changes during the year then ended are presented in the table and narrative below:
| | | | | | | | | | | |
| | | Options | | | Weighted-
Average
Exercise Price | | Weighted
Average
Remaining
Contractual
Term | | Aggregate
Intrinsic Value |
Outstanding at December 31, 2006 | | 4,219,277 | | | $ | 5.26 | | | | | |
Granted | | 1,797,071 | | | | 7.83 | | | | | |
Exercised | | (420,390 | ) | | | 1.23 | | | | | |
Cancelled | | (528,718 | ) | | | 7.46 | | | | | |
| | | | | | | | | | | |
Outstanding at December 31, 2007 | | 5,067,240 | | | $ | 6.27 | | 7.78 | | $ | 4,733 |
| | | | | | | | | | | |
Vested or expected to vest at December 31, 2007 | | 4,679,193 | | | $ | 6.06 | | 7.71 | | $ | 4,688 |
| | | | | | | | | | | |
Exercisable at December 31, 2007 | | 2,138,393 | | | $ | 4.34 | | 6.84 | | $ | 4,038 |
| | | | | | | | | | | |
F-24
The aggregate intrinsic value in the table above represents the value (the difference between the Company’s closing common stock price on the last trading day of the year ended December 31, 2007 and the exercise price of the options, multiplied by the number of in-the-money options) that would have been received by the option holders had all option holders exercised their options on December 31, 2007. As of December 31, 2007, there was $11,575 of total unrecognized stock-based compensation expense related to stock options granted under the plans. The expense is expected to be recognized over a weighted-average period of 2.6 years. Additionally, the Company has approximately $1,417 of stock-based compensation expense related to stock options granted below fair market value prior to January 1, 2005 not yet recognized. This expense is net of estimated forfeitures and is expected to be recognized over a weighted-average period of approximately .8 years. The weighted-average grant date fair value of options for the years ended December 31, 2007, 2006 and 2005 was $4.69, $5.16 and $6.21, respectively. The intrinsic value of stock options exercised for the years ended December 31, 2007, 2006 and 2005 was $2,389, $4,197 and $450, respectively, and represents the difference between the exercise price of the option and the market price of the Company’s common stock on the dates exercised.
In January, May and June of 2007, the Company modified certain stock option and restricted stock grants for two employees and one Board member who received an acceleration of service vesting in connection with their termination agreements or resignation. In accordance with SFAS 123R, the Company recorded stock-based compensation expense of $412 at the modification date as no additional service was required.
In September and October of 2006, the Company modified certain stock option grants for two employees who received an acceleration of service vesting in connection with their termination agreements. In accordance with SFAS 123R, the Company recorded stock-based compensation expense of $54 at the modification date as no additional service was required.
In August 2005, the Company modified certain stock option grants to an employee upon a change in status to a non-employee. In accordance with EITF No. 00-23,Issues Related to the Accounting for Stock Compensation under APB Opinion No. 25 and FASB Interpretation No. 44 and FIN 44, Accounting for Certain Transactions Involving Stock Compensation, the stock options were accounted for as a new grant and were valued using the Black-Scholes method. The Company recorded stock-based compensation expense of $960 in the year ended December 31, 2005. The non-employee ceased performing services for the Company effective December 31, 2005.
The Company valued stock options using a Black-Scholes method of valuation and has applied the weighted-average assumptions set forth in the following table. The resulting fair value is recorded as compensation cost on a straight line basis over the requisite service period, which generally equals the option vesting period. Since the Company completed its initial public offering in November 2005, it did not have sufficient history as a publicly traded company to evaluate its volatility factor and expected term. As such, the Company analyzed the volatilities and expected terms of several peer companies to support the assumptions used in its calculations. The Company averaged the volatilities and expected terms of these peer companies with a similar amount of in-the-money options, sufficient trading history and similar vesting terms to generate the assumptions detailed above. The Company also began to include its own historical volatility in the expected volatility calculation once it was available and has continued to increase the weight applied to its own historical volatility over time. The risk-free interest rates used in the analysis are based on the United States Treasury yield curve in effect for periods corresponding with the expected life of the stock option. The Company has estimated forfeitures based upon an average of its historical data of option cancellations and employee turnover rates. Changes in estimated forfeitures are recognized through a cumulative true-up adjustment in the period of change. The Company’s estimated forfeiture rates were 4.65%, 4.85%, 6.31% and 7.33% for the first, second, third and fourth quarters of 2007, respectively.
F-25
During the years ended December 31, 2007, 2006 and 2005, respectively, the weighted-average assumptions used in the Black-Scholes model were as follows:
| | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
Volatility factor | | 61.17 | % | | 63.15 | % | | 80.00 | % |
Risk-free interest rate | | 4.47 | % | | 4.71 | % | | 4.39 | % |
Dividend yield | | — | % | | — | % | | — | % |
Expected term (in years) | | 5.8 | | | 5.6 | | | 5.0 | |
Restricted Stock
Prior to January 1, 2006, the Company had not granted awards of restricted stock. A summary of the status of non-vested restricted stock as of December 31, 2007 is as follows:
| | | | | | |
| | | Restricted
Stock | | | Weighted-
Average Grant
Date Fair Value |
Non-vested at December 31, 2006 | | 248,750 | | | $ | 9.86 |
Granted | | — | | | | — |
Vested | | (56,562 | ) | | | 9.72 |
Canceled | | (26,250 | ) | | | 8.14 |
| | | | | | |
Non-vested at December 31, 2007 | | 165,938 | | | $ | 10.18 |
| | | | | | |
As of December 31, 2007, there was $873 of total unrecognized stock-based compensation expense related to non-vested restricted stock arrangements granted under the 2004 Plan. The expense is expected to be recognized over a weighted-average period of 1.5 years. The total fair value of shares vested was $550 for the year ended December 31, 2007.
Stock Option Grants to Non-Employees
During the year ended December 31, 2007, the Company granted 42,500 stock options to non-employees. Certain stock options vest immediately and others vest over periods of up to two years. The unvested portion of the stock options will be remeasured at each reporting period. Total stock-based compensation expense for non-employee stock option grants for the years ended December 31, 2007, 2006 and 2005 was $175, $562 and $321, respectively.
| 9. | Segment and Geographic Information |
SFAS No. 131,Disclosures about Segments of an Enterprise and Related Information, established standards for reporting information about operating segments in annual financial statements and requires selected information about operating segments to be presented in interim financial reports issued to stockholders. It also established standards for disclosures about products and services and geographic areas. Operating segments are defined as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision maker, or decision-making group, in deciding how to allocate resources and in assessing performance. The Company views its operations and manages its business in one operating segment. The Company operates in two geographic segments: the United States and Singapore. In 2007, the Company recorded revenue of $13,797 and $1,141 in the United States and Singapore, respectively. As of December 31, 2007, $14,643 and $1,290 of the Company’s long-lived assets were located in the United States and Singapore, respectively.
F-26
The components of loss before provision for income taxes are as follows:
| | | | | | | | | | | | |
| | | December 31,
2007 | | | December 31,
2006 | | | December 31,
2005 | |
Domestic | | $ | (49,038 | ) | | $ | (30,689 | ) | | $ | (28,604 | ) |
Foreign | | | (4,259 | ) | | | (3,548 | ) | | | (911 | ) |
| | | | | | | | | | | | |
Loss before provision for income taxes | | $ | (53,297 | ) | | $ | (34,237 | ) | | $ | (29,515 | ) |
| | | | | | | | | | | | |
The Company has provided current state income tax expense of $46 and $51 for the years ended December 31, 2007 and 2006, respectively.
A reconciliation of the expected income tax (benefit) computed using the federal statutory income tax rate to the Company’s effective income tax rate is as follows for the years ended December 31, 2007, 2006 and 2005:
| | | | | | |
| | | December 31,
2007 | | December 31,
2006 | | December 31,
2005 |
Income tax computed at federal statutory tax rate | | (34.0)% | | (34.0)% | | (34.0)% |
State taxes, net of federal benefit | | 0.1 % | | 0.1 % | | — % |
Change in valuation allowance | | 35.7 % | | 32.4 % | | 36.6 % |
Stock-based compensation | | 2.1 % | | 4.4% | | — % |
Research and development credits | | (3.9) % | | (2.8)% | | — % |
Permanent differences | | 0.1 % | | — % | | (2.5)% |
Other | | — % | | — % | | (0.1)% |
| | | | | | |
Total | | 0.1% | | 0.1% | | — % |
| | | | | | |
The Company has incurred net operating losses from inception. At December 31, 2007, the Company had domestic federal and state net operating loss carryforwards of approximately $78,037 and $81,705, respectively, available to reduce future taxable income, which expire at various dates beginning in 2006 through 2027. The Company also had federal and state research and development tax credit carryforwards of approximately $4,232 and $2,990, respectively, available to reduce future tax liabilities and which expire at various dates beginning in 2015 through 2027. The Company also had foreign net operating loss carryforwards of approximately $8,596 as of December 31, 2007. The net operating loss carryfowards included $2,671 of federal and state net operating losses that are attributable to stock option exercises which will be recorded as an increase in additional paid-in-capital once they are “realized” in accordance with SFAS 123R.
Under the provisions of the Internal Revenue Code, certain substantial changes in the Company’s ownership may result in a limitation on the amount of net operating loss carryforwards and research and development credit carryforwards which could be utilized annually to offset future taxable income and taxes payable.
F-27
Deferred taxes consist of the following:
| | | | | | | | |
| | | As of December 31, | |
| | | 2007 | | | 2006 | |
Net operating loss carryforwards . | | $ | 32,471 | | | $ | 20,983 | |
Research and development credits | | | 6,206 | | | | 3,948 | |
Capitalized start-up costs | | | 2,057 | | | | 3,231 | |
Capitalized research and development costs | | | 7,973 | | | | 12,421 | |
Depreciation and amortization | | | 592 | | | | 301 | |
Deferred revenue | | | 6,902 | | | | 6,343 | |
Stock-based compensation | | | 2,517 | | | | 1,561 | |
Other | | | 1,189 | | | | 217 | |
| | | | | | | | |
Deferred tax asset | | | 59,907 | | | | 49,005 | |
Deferred tax asset valuation allowance | | | (59,907 | ) | | | (49,005 | ) |
| | | | | | | | |
Net deferred tax asset | | $ | — | | | $ | — | |
| | | | | | | | |
As required by SFAS No. 109, management of the Company has evaluated the positive and negative evidence bearing upon the realizability of its deferred tax assets. Management has determined at this time that it is more likely than not that the Company will not recognize the benefits of its federal and state deferred tax assets and, as a result, a valuation allowance of $59.9 million and $49.0 million has been established at December 31, 2007 and 2006, respectively.
In June 2006, the FASB issued Interpretation No. 48, “Accounting for Uncertainty in Income Taxes, an interpretation of FAS 109” (“FIN 48”). This statement clarifies the criteria that an individual tax position must satisfy for some or all of the benefits of that position to be recognized in a company’s financial statements. The Company adopted FIN No. 48 on January 1, 2007. The implementation of FIN No. 48 did not have a material impact on the Company’s consolidated financial statements, results of operations or cash flows. At the adoption date of January 1, 2007, and also at December 31, 2007, the Company had no unrecognized tax benefits. The Company has not, as yet, conducted a study of its research and development credit carryforwards. This study may result in an increase or decrease to the Company’s research and development credit carryforwards; however, until a study is completed and any adjustment is known, no amounts are being presented as an uncertain tax position under FIN 48. A full valuation allowance has been provided against the Company’s research and development credits, and if an adjustment is required, this adjustment would be offset by an adjustment to the valuation allowance. As a result, there would be no impact to the consolidated balance sheet, statement of operations or cash flows if an adjustment were required.
The tax years 2000 through 2006 remain open to examination by major taxing jurisdictions to which the Company is subject, which are primarily in the United States, as carryforward attributes generated in years past may still be adjusted upon examination by the Internal Revenue Service or state tax authorities if they have or will be used in a future period. The Company is currently not under examination by the Internal Revenue Service or any other jurisdictions for any tax years. The Company has not recorded any interest and penalties on any unrecognized tax benefits since its inception.
F-28
Accrued expenses consist of the following:
| | | | | | |
| | | As of December 31, |
| | | 2007 | | 2006 |
Accrued payroll and related benefits | | $ | 918 | | $ | 1,065 |
Accrued construction costs | | | — | | | 1,508 |
Accrued clinical trial costs | | | 2,186 | | | 934 |
Accrued professional fees | | | 491 | | | 527 |
Accrued other expenses | | | 1,156 | | | 302 |
| | | | | | |
| | $ | 4,751 | | $ | 4,336 |
| | | | | | |
On October 18, 2005, the Company entered into a lease agreement for approximately 40,000 square feet of office and laboratory space located in Cambridge, Massachusetts. The initial term of the lease commenced on September 1, 2006 for the office space and December 1, 2006 for the laboratory space and was to extend until November 30, 2016 with two five-year renewal options. The Company had the right to use and controlled physical access to the leased premises beginning on December 6, 2005. Thus, the effective lease term began on that date. In March 2006, the Company amended (the “First Amendment”) the October 18, 2005 operating lease agreement. The First Amendment provided for 23,199 square feet of additional laboratory space. The Company has committed to lease this additional laboratory space through January 2017. In addition, the First Amendment extended the original lease term of the existing space an additional two months though January 2017. Additionally, the lease, as amended, contains rent escalation, rent holiday, and leasehold improvement incentives. Rent escalation and rent holiday are being accounted for as rent expense under the straight-line method. In connection with the lease, the Company received approximately $6.9 million in leasehold improvement incentives from the landlord. The Company received $2.9 million in leasehold improvement incentives during 2007 and $4.0 million in 2006. These leasehold improvement incentives are being accounted for as a reduction in rent expense ratably over the lease term. The balance from these leasehold improvement incentives is included in current portion of lease incentive obligation and lease incentive obligation, net of current portion in the balance sheets at December 31, 2007 and 2006. Leasehold improvements are amortized using the straight-line method over the estimated useful lives of the assets or the term of the lease, whichever is shorter. The Company has provided a $4.0 million standby letter of credit as security for the amended lease as of December 31, 2007 and 2006. The certificate of deposit that secures the letter of credit is included in non-current restricted cash on the balance sheet in the amount of $4.0 million at December 31, 2007 and 2006, respectively.
The Company formerly subleased its prior office and laboratory space in Boston, Massachusetts, under a noncancelable operating lease which expired in August 2006.
The Company also leases certain office equipment under various operating leases. Total rent expense was $2,846, $2,959 and $1,304 for the years ended December 31, 2007, 2006 and 2005, respectively.
Future minimum lease payments under noncancelable operating leases at December 31, 2007, are as follows:
| | | |
Year ending December 31, | | | |
2008 | | $ | 2,862 |
2009 | | | 2,875 |
2010 | | | 2,762 |
2011 | | | 2,754 |
2012 | | | 3,022 |
Thereafter | | | 12,380 |
| | | |
| | $ | 26,655 |
| | | |
F-29
| 13. | Employee Benefit Plans |
In May 2001, the Company adopted the CombinatoRx, Incorporated 401(k) Plan (“401(k) Plan”). The 401(k) Plan allows employees to make pre-tax contributions up to the maximum allowable amount set by the IRS. Under the 401(k) Plan, the Company may make discretionary contributions as approved by the Board of Directors. During 2007, 2006 and 2005, the Company made contributions of $571, $432 and $250, respectively.
Effective December 1, 2007, the Company approved the CombinatoRx Nonqualified Deferred Compensation Plan (the “NQ Plan”), a non-qualified tax-deferred compensation plan in which certain senior managers and officers of the Company may participate. The NQ Plan provides a tax-favorable vehicle for deferring cash compensation, including base salary and bonus awards. Under the NQ Plan, each year a participant may defer up to 25% of his or her base salary and up to 100% of his or her annual cash bonus pay. The participant will at all times be vested in the portion of his or her account attributable to the compensation the participant has elected to defer under the NQ Plan. The Company has established a special account for each participant, however, the Company’s obligation to pay the balance credited to such account will at all times be an unfunded and unsecured obligation of the Company and rank on parity with other unsecured and unsubordinated indebtedness of the Company from time to time outstanding.
The Company may also credit to the account of each eligible participant who makes deferrals a matching contribution in an amount equal to 100% of the deferrals contributed by the participant for such plan year, up to a maximum amount equal to: (i) four percent (4%) of each such participant’s cash compensation for such year, less (ii) the amount of matching contributions made to the Company’s qualified 401(k) Plan for such year on behalf of such participant. In order to be eligible for a matching contribution for a given year, a participant must be employed by the Company on the date the matching contribution is credited to the NQ Plan, which is currently planned to be the January following a participant’s election, and have deferred the maximum amount permitted under the Company’s tax-qualified Section 401(k) Plan for such year. A participant will become 100% vested in any employer contributions credited to his or her account upon the participant’s death, disability or a change in control (as defined in the NQ Plan). Deferred balances are credited to each participant’s account under the NQ Plan and will be credited, at periodic intervals, with earnings that track the actual rate of return for such period realized by the investment fund or funds or index or indices selected by such participant from the range of investment vehicles offered under the NQ Plan. Deferred amounts are paid, at the participant’s option, either in a lump sum or in annual installments over a period of up to ten years upon separation from service or up to five years for scheduled in-service withdrawals. The Company did not contribute to the NQ Plan as of December 31, 2007.
F-30
| 14. | Quarterly Financial Information (unaudited) |
| | | | | | | | | | | | | | | | |
| | | First Quarter
Ended
March 31,
2007 | | | Second Quarter
Ended
June 30,
2007 | | | Third Quarter
Ended
September 30,
2007 | | | Fourth Quarter
Ended
December 31,
2007 | |
Revenue | | $ | 3,634 | | | $ | 5,287 | | | $ | 3,002 | | | $ | 3,015 | |
Operating expenses: | | | | | | | | | | | | | | | | |
Research and development | | | 12,762 | | | | 12,714 | | | | 15,874 | | | | 14,084 | |
General and administrative | | | 4,507 | | | | 4,083 | | | | 4,116 | | | | 4,173 | |
| | | | | | | | | | | | | | | | |
Total operating expenses | | | 17,269 | | | | 16,797 | | | | 19,990 | | | | 18,257 | |
Loss from operations | | | (13,635 | ) | | | (11,510 | ) | | | (16,988 | ) | | | (15,242 | ) |
Interest income | | | 1,427 | | | | 1,321 | | | | 1,207 | | | | 1,436 | |
Interest expense | | | (245 | ) | | | (309 | ) | | | (375 | ) | | | (375 | ) |
Other income | | | 0 | | | | 0 | | | | 0 | | | | (9 | ) |
| | | | | | | | | | | | | | | | |
Net loss before provision for income taxes | | $ | (12,453 | ) | | $ | (10,498 | ) | | $ | (16,156 | ) | | $ | (14,190 | ) |
Provision for income taxes | | | (21 | ) | | | 0 | | | | 0 | | | | (25 | ) |
| | | | | | | | | | | | | | | | |
Net loss | | $ | (12,474 | ) | | $ | (10,498 | ) | | $ | (16,156 | ) | | $ | (14,215 | ) |
| | | | | | | | | | | | | | | | |
Net loss per share applicable to common stockholders—basic and diluted | | $ | (0.44 | ) | | $ | (0.36 | ) | | $ | (0.56 | ) | | $ | (0.42 | ) |
| | | | | | | | | | | | | | | | |
Weighted-average number of common shares used in net loss per share calculation—basic and diluted | | | 28,635,258 | | | | 28,837,552 | | | | 28,971,559 | | | | 33,615,652 | |
| | | | |
| | | First Quarter
Ended
March 31,
2006 | | | Second Quarter
Ended
June 30,
2006 | | | Third Quarter
Ended
September 30,
2006 | | | Fourth Quarter
Ended
December 31,
2006 | |
Revenue | | $ | 3,247 | | | $ | 3,396 | | | $ | 3,182 | | | $ | 3,448 | |
Operating expenses: | | | | | | | | | | | | | | | | |
Research and development | | | 7,151 | | | | 8,703 | | | | 8,526 | | | | 9,714 | |
General and administrative | | | 3,985 | | | | 5,090 | | | | 4,488 | | | | 5,078 | |
| | | | | | | | | | | | | | | | |
Total operating expenses | | | 11,136 | | | | 13,793 | | | | 13,014 | | | | 14,792 | |
Loss from operations | | | (7,889 | ) | | | (10,397 | ) | | | (9,832 | ) | | | (11,344 | ) |
Interest income | | | 1,083 | | | | 1,526 | | | | 1,689 | | | | 1,615 | |
Interest expense | | | (130 | ) | | | (154 | ) | | | (211 | ) | | | (227 | ) |
Other income | | | — | | | | — | | | | — | | | | 34 | |
| | | | | | | | | | | | | | | | |
Net loss before provision for income taxes | | $ | (6,936 | ) | | $ | (9,025 | ) | | $ | (8,354 | ) | | $ | (9,922 | ) |
Provision for income taxes | | | — | | | | — | | | | — | | | | 51 | |
| | | | | | | | | | | | | | | | |
Net loss | | $ | (6,936 | ) | | $ | (9,025 | ) | | $ | (8,354 | ) | | $ | (9,973 | ) |
| | | | | | | | | | | | | | | | |
Net loss per share applicable to common stockholders—basic and diluted | | $ | (0.29 | ) | | $ | (0.32 | ) | | $ | (0.29 | ) | | $ | (0.35 | ) |
| | | | | | | | | | | | | | | | |
Weighted-average number of common shares used in net loss per share calculation—basic and diluted | | | 23,739,960 | | | | 28,120,203 | | | | 28,442,946 | | | | 28,526,650 | |
F-31
EXHIBIT INDEX
| | | | | | | | | | |
| | | | | Incorporated by Reference to: |
| | | | | | | | | Filing
Date with
SEC | | |
Exhibit No. | | | | Form or
Schedule | | Exhibit
No. | | | SEC File
Number |
| | Description | | | | |
| | | | | |
�� 3.1 | | Sixth Amended and Restated Certificate of Incorporation. | | S-1/A | | 3.2 | | 11/4/2005 | | 333-121173 |
| | | | | |
3.2 | | Amended and Restated By-Laws. | | 8-K | | 3.1 | | 6/4/2007 | | 000-51171 |
| | | | | |
4.1 | | Specimen Common Stock Certificate. | | S-1/A | | 4.1 | | 1/19/2005 | | 333-121173 |
| | | | | |
4.2 | | Warrant issued to General Electric Capital Corporation on September 15, 2004 to purchase up to 8,892 shares of the Registrant’s Common Stock. | | S-1 | | 10.7 | | 12/10/2004 | | 333-121173 |
| | | | | |
4.3 | | Warrant issued to General Electric Capital Corporation on June 28, 2005 to purchase 471 shares of the Registrant’s Common Stock. | | 10-K | | 4.5 | | 3/20/2006 | | 000-51171 |
| | | | | |
4.4 | | Warrant issued to Silicon Valley Bank on April 25, 2001 to purchase up to 10,019 of the Registrant’s Common Stock. | | S-1 | | 10.9 | | 12/10/2004 | | 333-121173 |
| | | | | |
4.5 | | Registration Rights Agreement dated as of April 25, 2001 by and between Silicon Valley Bank and the Registrant. | | S-1 | | 10.10 | | 12/10/2004 | | 333-121173 |
| | | | | |
4.6 | | Form of Warrant to purchase shares of the Registrant’s Stock, together with a schedule of warrant holders. | | S-1 | | 10.11 | | 12/10/2004 | | 333-121173 |
| | | | | |
4.7 | | Form of Warrant issued to BioMedical Sciences Investment Fund Pte Ltd on August 19, 2005 to purchase up to 25,000 shares of the Registrant’s Common Stock. | | S-1/A | | 10.45 | | 8/19/2005 | | 333-121173 |
| | | | | |
4.8 | | Second Amended and Restated Investors’ Rights Agreement, dated as of February 18, 2004, by and between the Registrant and the investors named therein, as amended. | | S-1 | | 10.17 | | 12/10/2004 | | 333-121173 |
| | | | | |
4.9 | | Amendment to the Second Amended and Restated Investors’ Rights Agreement by and between the Registrant and the investors named therein, dated as of December 8, 2004. | | S-1 | | 10.18 | | 12/10/2004 | | 333-121173 |
| | | | | |
4.10 | | Omnibus Amendment Agreement to Second Amended and Restated Investors’ Right Agreement and by and between the Registrant and the investors named therein and the Registration Rights Agreement by and between Silicon Valley Bank and the Registrant | | 8-K | | 10.1 | | 12/6/2006 | | 000-51171 |
| | | | | |
#10.1 | | 2000 Stock Option Plan, as amended. | | S-1 | | 10.1 | | 12/10/2004 | | 333-121173 |
| | | | | |
#10.2 | | Amended and Restated 2004 Incentive Plan. | | 8-K | | 10.1 | | 6/5/2006 | | 000-51171 |
| | | | | |
#10.3 | | Form Incentive Stock Option Agreement under the 2004 Incentive Plan. | | 10-K | | 10.3 | | 3/20/2006 | | 000-51171 |
| | | | | |
#10.4 | | Form Non-Qualified Option Agreement under the 2004 Incentive Plan. | | 10-K | | 10.4 | | 3/20/2006 | | 000-51171 |
| | | | | | | | | | |
| | | | | Incorporated by Reference to: |
| | | | | | | | | Filing
Date with
SEC | | |
Exhibit No. | | | | Form or
Schedule | | Exhibit
No. | | | SEC File
Number |
| | Description | | | | |
| #10.5 | | Nonqualified Deferred Compensation Plan, effective December 1, 2007. | | S-8 | | 4.1 | | 11/30/2007 | | 333-147745 |
| | | | | |
| 10.6 | | Master Security Agreement, dated as of July 20, 2004, by and between the Registrant and General Electric Capital Corporation. | | S-1 | | 10.12 | | 12/10/2004 | | 333-121173 |
| | | | | |
| 10.7 | | Form of Promissory Note issued by the Registrant to General Electric Capital Corporation. | | S-1 | | 10.13 | | 12/10/2004 | | 333-121173 |
| | | | | |
| 10.8 | | Revised Loan Proposal, dated as of May 3, 2005, between General Electric Capital Corporation and the Registrant. | | 10-K | | 10.8 | | 3/20/2006 | | 000-51171 |
| | | | | |
| 10.9 | | Revised Loan Proposal, dated as of April 19, 2006, between General Electric Capital Corporation and the Registrant. | | 8-K | | 10.2 | | 6/30/2006 | | 000-51171 |
| | | | | |
| 10.10 | | Promissory Note, dated as of March 29, 2007, between General Electric Capital Corporation and the Registrant. | | 8-K | | 10.1 | | 4/4/2007 | | 000-51171 |
| | | | | |
| 10.11 | | Loan Proposal, dated as of June 27, 2007, between General Electric Capital Corporation and the Registrant. | | 8-K | | 10.1 | | 6/28/2007 | | 000-51171 |
| | | | | |
| 10.12 | | Promissory Note, dated as of December 21, 2007, between General Electric Capital Corporation and the Registrant. | | 8-K | | 10.1 | | 12/21/2007 | | 000-51171 |
| | | | | |
| 10.13 | | Loan Proposal, dated as of November 20, 2006, between CombinatoRx (Singapore) Pte. Ltd. and GE Capital Services Pte. Ltd. | | 8-K | | 10.1 | | 2/21/2007 | | 000-51171 |
| | | | | |
| 10.14 | | Facility Letter, dated as of February 14, 2007, between CombinatoRx (Singapore) Pte. Ltd. and GE Capital Services Pte. Ltd. | | 8-K | | 10.1 | | 2/21/2007 | | 000-51171 |
| | | | | |
| 10.15 | | Deed of Debenture, dated as of February 14, 2007, between CombinatoRx (Singapore) Pte. Ltd. and GE Capital Services Pte. Ltd. | | 8-K | | 10.1 | | 2/21/2007 | | 000-51171 |
| | | | | |
| 10.16 | | Guaranty, dated as of February 14, 2007, between the Registrant and GE Capital Services Pte. Ltd. | | 8-K | | 10.1 | | 2/21/2007 | | 000-51171 |
| | | | | |
| #10.17 | | First Amended Employment, Confidentiality and Non-Competition Agreement with Alexis Borisy, dated as of July 1, 2004. | | S-1 | | 10.22 | | 12/10/2004 | | 333-121173 |
| | | | | |
| #10.18 | | Employment, Confidentiality and Non-Competition Agreement with Robert Forrester, dated as of February 23, 2004. | | S-1 | | 10.23 | | 12/10/2004 | | 333-121173 |
| | | | | |
| #10.19 | | Employment Letter Agreement with Daniel Grau, dated as of March 1, 2005. | | 10-K | | 10.15 | | 3/20/2006 | | 000-51171 |
| | | | | |
| #10.20 | | Third Amended Employment, Confidentiality and Non-Competition Agreement with Curtis Keith, dated as of December 1, 2006 | | S-3 | | 10.12 | | 12/11/2006 | | 333-139260 |
| | | | | | | | | | |
| | | | | Incorporated by Reference to: |
| | | | | | | | | Filing
Date with
SEC | | |
Exhibit No. | | | | Form or
Schedule | | Exhibit
No. | | | SEC File
Number |
| | Description | | | | |
| #10.21 | | Employment Letter Agreement with Jason F. Cole, dated as of January 23, 2006. | | 10-K | | 10.15 | | 3/20/2006 | | 000-51171 |
| | | | | |
| #10.22 | | Employment Letter Agreement with Lynn Baird, dated as of March 31, 2006 | | 8-K | | 10.1 | | 4/24/2006 | | 000-51171 |
| | | | | |
| #10.23 | | Amended and Restated Restricted Stock Award Agreement, dated January 17, 2007, by and between Alexis Borisy and the Registrant. | | 8-K | | 10.1 | | 1/22/2007 | | 000-51171 |
| | | | | |
| #10.24 | | Amended and Restated Restricted Stock Award Agreement, dated January 17, 2007, by and between Robert Forrester and the Registrant. | | 8-K | | 10.2 | | 1/23/2007 | | 000-51171 |
| | | | | |
| #10.25 | | Amended and Restated Restricted Stock Award Agreement, dated January 17, 2007, by and between Curtis Keith and the Registrant. | | 8-K | | 10.4 | | 1/22/2007 | | 000-51171 |
| | | | | |
| #10.26 | | Amended and Restated Restricted Stock Award Agreement, dated January 17, 2007, by and between Daniel Grau and the Registrant. | | 10-K | | 10.22 | | 3/15/2007 | | 000-51171 |
| | | | | |
| #10.27 | | Amended and Restated Restricted Stock Award Agreement, dated January 17, 2007, by and between Lynn Baird and the Registrant. | | 10-K | | 10.23 | | 3/15/2007 | | 000-51171 |
| | | | | |
| 10.28 | | Form of Indemnification Agreement for directors. | | S-1/A | | 10.32 | | 1/19/2005 | | 333-121173 |
| | | | | |
| 10.29 | | Sponsored Research Agreement, dated as of August 19, 2004, by and between Spinal Muscular Atrophy Foundation and the Registrant. | | S-1 | | 10.19 | | 12/10/2004 | | 333-121173 |
| | | | | |
| 10.30 | | License Agreement, dated as of May 4, 2005, by and between HenKan Pharmaceutical Company and the Registrant. | | S-1/A | | 10.34 | | 8/19/2005 | | 333-121173 |
| | | | | |
| 10.31 | | Amended and Restated Research Agreement, dated as of February 22, 2007, by and between the Registrant and CHDI, Inc. | | 10-K | | 10.29 | | 3/15/2007 | | 000-51171 |
| | | | | |
| 10.32 | | Subcontract Agreement, effective August 10, 2005, between Science Application International Corporation and the Registrant. | | S-1/A | | 10.36 | | 8/19/2005 | | 333-121173 |
| | | | | |
| 10.33 | | Research Project Cooperative Agreement, dated April 10, 2005, between the National Institutes of Health and the Registrant. | | S-1/A | | 10.37 | | 8/19/2005 | | 333-121173 |
| | | | | |
| 10.34 | | Form of Subscription and Shareholders Agreement, among CombinatoRx (Singapore) Pte Ltd, BioMedical Sciences Investment Fund Pte Ltd and the Registrant. | | S-1/A | | 10.38 | | 8/19/2005 | | 333-121173 |
| | | | | |
| 10.35 | | Form of Terms and Conditions of the Notes, of CombinatoRx (Singapore) Pte Ltd. | | S-1/A | | 10.39 | | 8/19/2005 | | 333-121173 |
| | | | | |
| 10.36 | | Form of Debenture between CombinatoRx (Singapore) Pte Ltd and BioMedical Sciences Investment Fund Pte Ltd. | | S-1/A | | 10.40 | | 8/19/2005 | | 333-121173 |
| | | | | | | | | | |
| | | | | Incorporated by Reference to: |
| | | | | | | | | Filing
Date with
SEC | | |
Exhibit No. | | | | Form or
Schedule | | Exhibit
No. | | | SEC File
Number |
| | Description | | | | |
| 10.37 | | Form of Services Agreement, by and between the Registrant and CombinatoRx (Singapore) Pte Ltd. | | S-1/A | | 10.41 | | 8/19/2005 | | 333-121173 |
| | | | | |
| 10.38 | | Form of Registration Rights Agreement, by and among the Registrant and BioMedical Sciences Investment Fund Pte Ltd. | | S-1/A | | 10.42 | | 8/19/2005 | | 333-121173 |
| | | | | |
| 10.39 | | Form of Swap-up Agreement, among CombinatoRx (Singapore) Pte Ltd, BioMedical Sciences Investment Fund Pte Ltd and the Registrant. | | S-1/A | | 10.43 | | 8/19/2005 | | 333-121173 |
| | | | | |
| 10.40 | | Form of Share Charge, between the Registrant and BioMedical Sciences Investment Fund Pte Ltd. | | S-1/A | | 10.44 | | 8/19/2005 | | 333-121173 |
| | | | | |
| †10.41 | | Research and License Agreement, dated as of October 3, 2005, between Angiotech Pharmaceuticals, Inc. and the Registrant. | | S-1/A | | 10.46 | | 10/5/2005 | | 333-121173 |
| | | | | |
| 10.42 | | Stock Purchase Agreement, dated as of October 3, 2005, by and among the Registrant and Angiotech Pharmaceuticals, Inc. | | S-1/A | | 10.47 | | 10/3/2005 | | 333-121173 |
| | | | | |
| 10.43 | | Amended and Restated Research and License Agreement, dated June 12, 2007, between Fovea Pharmaceuticals SA and the Registrant. | | 8-K | | 10.1 | | 6/12/2007 | | 000-51171 |
| | | | | |
| 10.44 | | Office and Laboratory Lease Agreement, as of October 18, 2005, by and between MA-Riverview, 1245 First Street, L.L.C. and the Registrant. | | S-1/A | | 10.48 | | 10/24/2005 | | 333-121173 |
| | | | | |
| 10.45 | | First Amendment to Office and Laboratory Lease Agreement, as of March 9, 2006, by and between MA-Riverview, 245 First Street, L.L.C. and the Registrant. | | 10-K | | 10.44 | | 3/20/2006 | | 000-51171 |
| | | | | |
| †10.46 | | Research, Development and Commercialization Agreement, dated May 31, 2006, between the Cystic Fibrosis Foundation Therapeutics Incorporated and the Registrant. | | 8-K | | 10.1 | | 6/5/2006 | | 000-51171 |
| | | | | |
| 10.47 | | Sponsored Research Collaboration Agreement, dated November 7, 2007, between Dart Therapeutics, LLC. | | 8-K | | 10.1 | | 11/8/2007 | | 000-51171 |
| | | | | |
| *21.1 | | List of subsidiaries. | | | | | | | | |
| | | | | |
| *23.1 | | Consent of Ernst & Young LLP. | | | | | | | | |
| | | | | |
| *31.1 | | Certification of Chief Executive Officer pursuant to Exchange Act rules 13a-14 or 15d-14. | | | | | | | | |
| | | | | |
| *31.2 | | Certification of Chief Financial Officer pursuant to Exchange Act rules 13a-14 or 15d-14. | | | | | | | | |
| | | | | |
| *32.1 | | Certifications of Chief Executive Officer and Chief Financial Officer pursuant to Exchange Act rules 13a-14(b) or 15d-14(b) and 18 U.S.C. Section 1350. | | | | | | | | |
| † | Confidential treatment requested as to certain portions, which portions are omitted and filed separately with the Securities and Exchange Commission. |
| # | Management contract or compensatory plan or arrangement filed as an Exhibit to this report pursuant to Items 15(a) and 15(c) of Form 10-K. |