Table of Contents
As filed with the Securities and Exchange Commission on January 20, 2005
Registration No. 333-120757
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT NO. 2
To
FORM S-1
REGISTRATION STATEMENT UNDER THE SECURITIES ACT OF 1933
AlgoRx Pharmaceuticals, Inc.
(Exact name of Registrant as specified in its charter)
| Delaware | 2834 | 943392501 | ||
| (State or other jurisdiction of incorporation or organization) | (Primary Standard Industrial Classification Code Number) | (I.R.S. Employer Identification No.) |
500 Plaza Drive, 2nd Floor
Secaucus, New Jersey 07094-3619
(201) 325-6900
(Address, including zip code and telephone number,
including area code, of Registrant’s principal executive offices)
Ronald M. Burch, M.D., Ph.D.
Chief Executive Officer
AlgoRx Pharmaceuticals, Inc.
500 Plaza Drive, 2nd Floor
Secaucus, New Jersey 07094-3619
(201) 325-6900
(Name, address, including zip code and telephone number,
including area code, of agent for service)
Copies to:
| Stephen M. Davis, Esq. Peter DiIorio, Esq. Heller Ehrman White & McAuliffe LLP 120 West 45th Street New York, New York 10036 Phone: (212) 832-8300 Fax: (212) 763-7600 | Paul M. Kinsella, Esq. Paul Laurino, Esq. Ropes & Gray LLP One International Place Boston, MA 02110-2624 Phone: (617) 951-7000 Fax: (617) 951-7050 |
Approximate date of commencement of proposed sale to public: As soon as practicable after this Registration Statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box: o
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering: o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering: o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering: o
If delivery of the prospectus is expected to be made pursuant to Rule 434, please check the following box: o
CALCULATION OF REGISTRATION FEE
| Proposed Maximum | ||||
| Title of Each Class of | Aggregate | Amount of | ||
| Securities to be Registered | Offering Price(1) | Registration Fee(2) | ||
| Common Stock, $0.001 par value per share | $93,840,000 | $11,721 | ||
| (1) | Estimated solely for the purpose of calculating the registration fee pursuant to Rule 457(o) of the Securities Act of 1933, as amended. |
| (2) | A registration fee of $9,503 has previously been paid in connection with this Registration Statement based on an estimate of the aggregate offering price. Accordingly, the Registrant has paid the difference of $2,218 with this filing. |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
Table of Contents
| The information in this prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted. |
SUBJECT TO COMPLETION, DATED JANUARY 20, 2005
6,800,000 Shares

Common Stock
Prior to this offering, there has been no public market for our common stock. The initial public offering price of our common stock is expected to be between $10 and $12 per share. We have applied for quotation of our common stock on the Nasdaq National Market under the symbol “AGRX”.
The underwriters have an option to purchase a maximum of 1,020,000 shares of common stock to cover over-allotments of shares.
Investing in our common stock involves risks. See “Risk Factors” beginning on page 8.
| Underwriting | ||||||||||||
| Price to | Discounts and | Proceeds to | ||||||||||
| Public | Commissions | AlgoRx | ||||||||||
| Per Share | $ | $ | $ | |||||||||
| Total | $ | $ | $ | |||||||||
Delivery of the shares of common stock will be made on or about , 2005.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
| Credit Suisse First Boston | Citigroup |
| Piper Jaffray | Lazard |
The date of this prospectus is , 2005.
TABLE OF CONTENTS
| Page | ||||||||
| 1 | ||||||||
| 6 | ||||||||
| 8 | ||||||||
| 25 | ||||||||
| 26 | ||||||||
| 26 | ||||||||
| 27 | ||||||||
| 28 | ||||||||
| 30 | ||||||||
| 32 | ||||||||
| 42 | ||||||||
| 68 | ||||||||
| 77 | ||||||||
| 79 | ||||||||
| 82 | ||||||||
| 86 | ||||||||
| 88 | ||||||||
| 90 | ||||||||
| 93 | ||||||||
| 94 | ||||||||
| 94 | ||||||||
| 94 | ||||||||
| F-1 | ||||||||
| COLLABORATION, DEVELOPMENT AND LICENSE AGREEMENT | ||||||||
| CONSENT OF ERNST & YOUNG LLP | ||||||||
You should rely only on the information contained in this document or to which we have referred you. We have not authorized anyone to provide you with information that is different. This document may only be used where it is legal to sell these securities. The information in this document may only be accurate on the date of this document.
Dealer Prospectus Delivery Obligation
Until (25 days after commencement of the offering), all dealers that effect transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to the dealer’s obligation to deliver a prospectus when acting as an underwriter and with respect to unsold allotments or subscriptions.
Table of Contents
PROSPECTUS SUMMARY
You should read the following summary together with the entire prospectus, including the more detailed information in our financial statements and related notes appearing in the back of this prospectus. You should carefully consider, among other things, the matters discussed in “Risk Factors.” References in this prospectus to “we,” “us” and “our” refer to AlgoRx Pharmaceuticals, Inc. and its subsidiaries, unless the context requires otherwise.
Our Business
Overview
We are an emerging pharmaceutical company focused on developing and commercializing a diversified portfolio of pharmaceutical product candidates to address pain, a large and under-served market. We have three product candidates that we are developing in four clinical programs and one preclinical program. Two of our product candidates are in late-stage clinical development, including ALGRX 3268 which is being evaluated in two phase III clinical trials.
ALGRX 4975 (capsaicin for injection and capsaicin gel for intraoperative use) is being developed in three programs for pain: severe postsurgical pain, post-trauma neuropathic pain, a form of pain resulting from direct injury to nerves that remains long after traumatic injuries, and musculoskeletal pain, including osteoarthritis and tendonitis. In these programs, we have completed one phase I clinical trial and three phase II clinical trials. Our phase II clinical trials have yielded statistically significant improvements in pain scores and have provided us with favorable safety data. We are continuing to evaluate ALGRX 4975 in five phase II clinical trials across the three programs. ALGRX 3268, our most advanced product candidate, is a powder formulation of the local anesthetic lidocaine delivered into the skin using our proprietary needle-free dispenser. We are developing ALGRX 3268 to reduce the pain associated with needle-sticks from blood draws or intravenous line insertions in children. We have recently commenced two phase III clinical trials for ALGRX 3268. We recently in-licensed a third product candidate, ALGRX 1207, a new molecular entity in a new class of anesthetics that is undergoing preclinical development as a topical local anesthetic. We expect to file an investigational new drug application for ALGRX 1207 with the U.S. Food and Drug Administration in the second half of 2005.
The Market Opportunity
Pain management is a large, under-served market for which currently used therapies often provide insufficient efficacy or cause significant side effects. Even with these shortcomings, drugs that treat pain, known as analgesics, generated an estimated $28 billion of worldwide prescription sales in 2003 according to Global Industry Analysts, Inc. Several analgesics that were commercialized during the past decade have achieved high sales volume even though they generally represented only minor improvements over existing therapies and are often associated with patient and physician dissatisfaction. We believe there is a large market opportunity for analgesics that address the shortcomings of current therapies, which include:
| • | insufficient efficacy; | |
| • | lack of site specificity; | |
| • | occurrence of side effects; | |
| • | need for frequent dosing; | |
| • | slow onset of action; and | |
| • | potential to cause physical dependence. |
1
Table of Contents
Our Product Candidates
| ALGRX 4975 (capsaicin for injection and capsaicin gel for intraoperative use) |
ALGRX 4975 is our product candidate for the treatment of site-specific severe or intractable pain. These types of pain are poorly treated by existing drugs such as opioids. We are developing ALGRX 4975 to treat patients in three broad areas:
| • | severe postsurgical pain following a variety of surgical procedures, including bunion removal surgery, total knee replacement and abdominal surgery, such as hernia repair, hysterectomy and gall bladder removal; | |
| • | post-trauma neuropathic pain; and | |
| • | pain resulting from musculoskeletal diseases, such as osteoarthritis and tendonitis. |
During a surgical procedure, ALGRX 4975 is applied directly onto the cut surfaces of skin, muscle and bone. For post-trauma neuropathic pain and musculoskeletal pain, ALGRX 4975 is delivered to the site of pain using a needle and syringe. Although the active ingredient in ALGRX 4975, capsaicin, has been available for many years in over-the-counter products, we believe we are the first to evaluate formulations of capsaicin for use inside the body. We believe that by harnessing capsaicin’s ability to cause changes in nerve endings for a period of 12 weeks or more, ALGRX 4975, if approved, may represent a novel method of providing long lasting, site-specific analgesia without many of the significant side effects associated with opioids and other methods of treating pain.
ALGRX 4975 has been studied in four AlgoRx-sponsored clinical trials that have enrolled a total of 250 patients. An additional five ongoing phase II clinical trials involving a total of 254 patients are designed to study the safety and efficacy of a variety of doses in a broad range of pain conditions. In addition, we believe that ALGRX 4975 may provide long lasting pain relief with infrequent dosing, potentially only four times per year, for patients with certain chronic pain conditions.
| ALGRX 3268 (PowderJect® Dermal Lidocaine) |
ALGRX 3268 is our product candidate that delivers lidocaine powder into the skin using our proprietary needle-free dispenser. It is intended to provide rapid, easy-to-administer local analgesia to reduce the pain associated with needle-sticks from blood draws or the insertion of intravenous lines in patients. Existing products for this use are characterized by a lengthy onset of action from at least 10 minutes to over an hour. This delay makes the use of these products inconvenient for both health care providers and patients. Current topical anesthetic products also tend to be messy and bulky since they are often formulated as creams and may require patches to be applied over the treatment area. By comparison, ALGRX 3268 has shown efficacy in one minute following application and is administered using a convenient, disposable needle-free dispenser.
The primary target market for ALGRX 3268 would be the estimated 42 million needle-stick procedures that are performed in children in the United States each year. We believe that this market is highly under-served by existing products. Based on our company estimates of unit sales, EMLA® and L.M.X.4®, two leading products, are used in fewer than 15% of pediatric needle-stick procedures. We believe that, if approved, ALGRX 3268’s rapid onset of action, convenience of use and efficacy in reducing pain would be welcomed by health care professionals and would be important factors for increasing physician use of analgesia prior to needle-stick procedures.
We have completed our clinical program through phase II involving 1,139 adults and children to evaluate the ability of ALGRX 3268 to reduce pain when used prior to needle-stick procedures. In clinical studies designed to assess efficacy, ALGRX 3268 achieved statistically significant reductions in pain associated with needle-sticks when compared to placebo, with no serious side effects. We have initiated a phase III clinical program to confirm the safety and efficacy of ALGRX 3268. If this phase III clinical program is successful, we believe that we will submit a new drug application to the FDA for ALGRX 3268 by the end of 2005.
2
Table of Contents
Preclinical Product Candidate
ALGRX 1207, our preclinical product candidate, is a new molecular entity that we are developing as a topical local anesthetic to potentially treat patients with certain types of neuropathic pain and for pre-procedural administration to reduce the pain associated with procedures on the skin. Certain types of neuropathic pain, including pain associated with postherpetic neuralgia, painful HIV polyneuropathy and painful diabetic polyneuropathy, affect almost two million patients in the United States and are not well-treated by current therapies. We believe that a simple-to-use, topical, local anesthetic with a long duration of action and deep penetration into the skin would represent an improvement over existing therapies. Based on studies in animals, ALGRX 1207 has been shown to provide analgesia more rapidly and with a longer-lasting effect than currently available topical anesthetics following direct administration to the skin. In addition, we believe that ALGRX 1207, if approved, could address a wide variety of procedures involving the skin, including dermatological surgery, cosmetic skin treatments and catheter placement, as well as pain arising from surgical incisions that does not subside after surgery has taken place.
Our Strategy
Our goal is to become a leading pharmaceutical company focused on the commercialization of drugs to treat various types of pain. The key elements of our strategy for achieving this goal are to:
| • | Continue to build, through acquisition or in-licensing, a balanced portfolio of product candidates that leverages our understanding of the mechanisms of pain and the pain management market; | |
| • | Complete clinical development and obtain regulatory approval for our product candidates utilizing our management team’s extensive experience in developing and commercializing drugs for pain management; | |
| • | Develop a specialized sales and marketing team to commercialize our product candidates, if approved, in the United States; and | |
| • | Establish corporate partnerships to assist in the clinical development, regulatory approval process and commercialization of our products in the United States and major foreign markets. |
Risks
Our business is subject to a number of risks, which you should be aware of before making an investment decision. These risks are discussed more fully in “Risk Factors.” All of our product candidates are in clinical or preclinical stages of development. Accordingly, we have not received regulatory approval for, or commercial revenues from, any of our product candidates. It is possible that we may never successfully commercialize any of our product candidates. As of September 30, 2004, we had incurred $50.3 million in net losses since inception. We expect to continue to incur increasing losses over the next several years, and we may never become profitable.
Recent Results
As of December 31, 2004, we had cash and cash equivalents of approximately $39.9 million. In the fourth quarter of 2004, we did not realize any revenues and we continued to incur significant losses. In the fourth quarter of 2004, we recorded stock-based compensation expense related to stock options to purchase 177,840 shares of common stock granted in the fourth quarter, and we also incurred research and development expenses relating to a $1 million up-front cash license fee and the issuance of 160,000 shares of common stock, paid in October 2004 in connection with the in-license of the technology for ALGRX 1207.
3
Table of Contents
Company Information
We were incorporated in Delaware in March 2001. Our principal executive offices are located at 500 Plaza Drive, 2nd Floor, Secaucus, New Jersey 07094-3619, and our telephone number is (201) 325-6900. Our website is located atwww.algorx.com. The information contained in or that can be accessed through our website is not part of this prospectus.
“AlgoRx” is our registered trademark. Other service marks, trademarks and trade names referred to in this prospectus are the property of their respective owners.
4
Table of Contents
THE OFFERING
| Common stock offered by AlgoRx: | 6,800,000 shares, or 7,820,000 shares if the underwriters’ over-allotment option is fully exercised | |
| Common stock to be outstanding after this offering: | 22,168,995 shares, or 23,188,995 shares if the underwriters’ over- allotment option is fully exercised | |
| Proposed Nasdaq National Market symbol: | AGRX | |
| Use of proceeds: | For general corporate purposes, including conducting clinical trials, research and development expenses, general and administrative expenses, manufacturing expenses and potential acquisitions of companies, products and technologies that complement our business. See “Use of Proceeds.” |
The number of shares of common stock to be outstanding after this offering is based on 15,368,995 shares of common stock outstanding as of December 31, 2004, and excludes:
| • | 2,352,345 shares of common stock issuable upon exercise of outstanding options at a weighted average exercise price of $1.30 per share; | |
| • | 3,408 shares of common stock reserved for future grants under our 2001 Equity Incentive Plan; and | |
| • | 69,265 shares of common stock issuable upon the exercise of an outstanding warrant at an exercise price of $5.925 per share that, by its terms, terminates upon completion of this offering. | |
Except as otherwise indicated, all information in this prospectus:
| • | gives effect to our amended and restated certificate of incorporation, which we will file immediately prior to the closing of this offering; | |
| • | gives effect to a 1-for-10 reverse stock split of our common stock to be effected prior to the closing of this offering; | |
| • | gives effect to the automatic conversion of all outstanding shares of our preferred stock into 14,275,747 shares of common stock upon the closing of this offering; and | |
| • | assumes no exercise by the underwriters of their option to purchase up to 1,020,000 shares of our common stock in this offering to cover over-allotments. | |
5
Table of Contents
SUMMARY FINANCIAL DATA
The following table summarizes our financial data for the period from March 6, 2001, the date of our inception, to December 31, 2001 and for the years ended December 31, 2002 and 2003, and is derived from our audited financial statements included in this prospectus. We have also included data from our unaudited financial statements for the nine months ended September 30, 2003 and 2004 and for the period from March 6, 2001 to September 30, 2004. You should read this data together with our financial statements and related notes and the information under “Selected Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations.”
| Period from | Period from | ||||||||||||||||||||||||
| March 6, 2001 | March 6, 2001 | ||||||||||||||||||||||||
| (Inception) to | Nine Months Ended | (Inception) to | |||||||||||||||||||||||
| December 31, | Years Ended December 31, | September 30, | September 30, | ||||||||||||||||||||||
| 2001 | 2002 | 2003 | 2003 | 2004 | 2004 | ||||||||||||||||||||
| (Unaudited) | (Unaudited) | (Unaudited) | |||||||||||||||||||||||
| (In thousands, except share and per share data) | |||||||||||||||||||||||||
Statement Of Operations Data: | |||||||||||||||||||||||||
| Contract revenues | $ | — | $ | 149 | $ | 100 | $ | 100 | $ | — | $ | 249 | |||||||||||||
| Operating expenses: | |||||||||||||||||||||||||
| Research and development | 365 | 11,745 | 12,191 | 10,939 | 10,682 | 34,983 | |||||||||||||||||||
| General and administrative | 1,096 | 3,076 | 3,477 | 2,320 | 2,914 | 10,564 | |||||||||||||||||||
| Acquired in-process research and development | — | 5,716 | — | — | — | 5,716 | |||||||||||||||||||
| Total operating expenses | 1,461 | 20,537 | 15,668 | 13,259 | 13,596 | 51,263 | |||||||||||||||||||
| Income (loss) from operations | (1,461 | ) | (20,388 | ) | (15,568 | ) | (13,159 | ) | (13,596 | ) | (51,014 | ) | |||||||||||||
| Gain (loss) on sale of assets | — | (36 | ) | 103 | 102 | — | 67 | ||||||||||||||||||
| Interest expense | (2 | ) | (4 | ) | (107 | ) | (72 | ) | (24 | ) | (137 | ) | |||||||||||||
| Interest income | 47 | 237 | 86 | 70 | 429 | 799 | |||||||||||||||||||
| Net income (loss) | $ | (1,416 | ) | $ | (20,191 | ) | $ | (15,486 | ) | $ | (13,059 | ) | $ | (13,191 | ) | $ | (50,285 | ) | |||||||
| Basic and diluted net loss per share | $ | (79.27 | ) | $ | (110.36 | ) | $ | (59.75 | ) | $ | (51.03 | ) | $ | (17.14 | ) | ||||||||||
| Shares used to compute basic and diluted net loss per share | 17,866 | 182,949 | 259,182 | 255,909 | 769,582 | ||||||||||||||||||||
| Pro forma basic and diluted net loss per share (unaudited) | $ | (5.23 | ) | $ | (1.01 | ) | |||||||||||||||||||
| Shares used to compute pro forma basic and diluted net loss per share (unaudited) | 2,960,028 | 12,946,630 | |||||||||||||||||||||||
6
Table of Contents
| As of September 30, 2004 | ||||||||||||
| Pro Forma | ||||||||||||
| Actual | Pro Forma | As Adjusted | ||||||||||
| (Unaudited, in thousands) | ||||||||||||
Balance Sheet Data: | ||||||||||||
| Cash, cash equivalents and short-term investments | $ | 47,573 | $ | 47,573 | $ | 115,075 | ||||||
| Total assets | 49,929 | 49,929 | 117,431 | |||||||||
| Deficit accumulated during the development stage | (50,285 | ) | (50,285 | ) | (50,285 | ) | ||||||
| Total stockholders’ equity | 46,412 | 46,412 | 113,914 | |||||||||
Pro forma basic and diluted net loss per common share is calculated assuming, and the pro forma balance sheet data as of September 30, 2004 gives effect to the conversion of all outstanding shares of our preferred stock into 14,275,747 shares of common stock upon completion of this offering as if such conversion had occurred at the date of original issuance.
The pro forma as adjusted balance sheet data as of September 30, 2004 also gives effect to the sale of 6,800,000 shares of common stock offered by this prospectus at an assumed initial public offering price of $11 per share, after deducting underwriting discounts and commissions and estimated offering expenses.
7
Table of Contents
RISK FACTORS
Before you invest in our common stock, you should understand the high degree of risk involved. You should consider carefully the following risks and other information in this prospectus, including our historical financial statements and related notes, before you decide to purchase shares of our common stock. If any of the following risks actually occurs, our business, financial condition and operating results could be adversely affected. As a result, the trading price of our common stock could decline and you could lose part or all of your investment.
Risks Related to Our Company
| Clinical trials may fail to adequately demonstrate the safety or efficacy of ALGRX 3268, ALGRX 4975 or any future product candidates, preventing or significantly delaying their regulatory approval. |
Before obtaining regulatory approval to commercialize any of our product candidates, we must subject them to extensive preclinical and clinical testing. We have recently commenced two phase III clinical trials for ALGRX 3268, and we are currently conducting phase II clinical trials for ALGRX 4975. These clinical trials are designed to provide both efficacy and safety data, which will help determine the steps we need to take to continue development of these product candidates.
Clinical trial programs are expensive, time-consuming and typically take years to complete. In particular, we do not expect to receive approval from the FDA to market ALGRX 3268 until 2007 at the earliest, if at all. We believe that ALGRX 4975 will not receive approval for marketing for any indication until 2009 at the earliest, if at all. We are developing an injectable formulation of ALGRX 4975 to potentially treat patients with pain resulting from musculoskeletal diseases, such as osteoarthritis and tendonitis. No drug has ever been approved by the FDA for the treatment of tendonitis or many other musculoskeletal diseases. Thus, it is difficult to estimate the types of clinical trials that the FDA might require and the time it will take to complete such trials.
We are developing ALGRX 4975 in both a gel and an injectable formulation. In our clinical trials to date, we have used only a liquid form of ALGRX 4975, though we plan to use the gel formulation in some of our later trials. To date, no clinical trials have been completed using the gel formulation. We may encounter unexpected difficulties in developing the gel formulation, which may delay the development of ALGRX 4975 to treat severe postsurgical pain. This may occur for various reasons, including efficacy or safety issues arising from the use of the gel formulation in our later clinical trials and concerns that may be raised by the FDA about the gel formulation. Such difficulties may delay or prevent our efforts to develop a gel formulation of ALGRX 4975.
In connection with clinical trials, we face the risks that:
| • | a product candidate may not prove to be efficacious; | |
| • | we may discover that a product candidate causes harmful side effects; | |
| • | patients may die or suffer other adverse medical effects for reasons that may or may not be related to the product candidate being tested; | |
| • | the results may not meet the level of statistical significance required by the FDA or other regulatory agencies; | |
| • | the results may not confirm positive results of earlier trials; and | |
| • | the FDA may place a clinical hold on studies of our product candidates. |
Both ALGRX 4975 and ALGRX 3268 were placed on clinical hold by the FDA at separate times during 2003. In the case of ALGRX 3268, the FDA requested the results from several nonclinical studies regarding the ways in which particles were ejected from the device. In the case of ALGRX 4975, the FDA requested that we provide them with the results of an ongoing osteoarthritis clinical trial then underway in the United Kingdom under the clinical trials exemption. In both cases, the FDA subsequently
8
Table of Contents
lifted each hold to allow us to initiate or continue the trials after we submitted the information they had requested. Imposition of additional clinical holds could substantially delay our current product development efforts. The FDA may place our clinical trials on clinical hold due to a variety of factors, including safety concerns, or results obtained from other trials of the same product candidate. If a clinical trial of ours is placed on hold, we would have to suspend enrollment or cancel the trial, and would be unable to enroll any patients unless and until the FDA allowed us to continue.
The results from the phase II clinical trials for ALGRX 3268 may not be predictive of results that may be obtained in our phase III clinical trials. The results from the phase I and II clinical trials for ALGRX 4975 for injection may not be predictive of results obtained in additional phase II and phase III clinical trials. Future clinical trials may indicate previously undetected side effects or fail to demonstrate efficacy in a statistically significant manner. If our ALGRX 4975 or ALGRX 3268 clinical trials are not completed, significantly delayed or fail to demonstrate that the product candidates are safe and efficacious, our business and reputation would be harmed and our stock price would be negatively affected.
| To obtain regulatory approval to market our product candidate ALGRX 1207, we will need to conduct nonclinical studies in animals, and the results of these nonclinical studies may not demonstrate adequate efficacy and, even if they do, the results may not necessarily be predictive of results in human trials. |
As part of the regulatory approval process, we must conduct, at our own expense, nonclinical studies in laboratory animals and clinical trials in humans. Nonclinical studies for ALGRX 1207 were commenced by Bridge Pharma, Inc. prior to our in-license of this product candidate in October 2004, and we will be required to continue nonclinical studies for this product candidate. The number of nonclinical trials that the regulatory authorities will require varies depending on the product candidate, the disease or condition the product candidate is being developed to address and regulations applicable to the particular product candidate. We may need to perform multiple nonclinical studies using various doses and formulations of ALGRX 1207 before we can begin clinical trials, which could result in delays in our ability to develop ALGRX 1207. Furthermore, nonclinical results in animal studies are not necessarily predictive of outcomes in human clinical trials. After we have conducted nonclinical studies in animals for ALGRX 1207, we must demonstrate in clinical trials that ALGRX 1207 is safe and efficacious for use on humans in order to receive regulatory approval for commercial sale. Even if initial results of nonclinical studies for ALGRX 1207 are positive, we may obtain different results in later stages of drug development, including failure to show desired safety and efficacy.
| Failure to enroll patients for clinical trials may cause delays in developing our product candidates, and delays in the commencement of clinical testing of our current product candidates could result in increased cost to us and delay our ability to generate revenues. |
We are currently enrolling or seeking to enroll patients in five clinical trials for our phase II studies of ALGRX 4975 and in two phase III clinical trials for ALGRX 3268. We will encounter delays or possibly regulatory rejections if we are unable to enroll enough patients to complete clinical trials. Patient enrollment depends on many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites and the eligibility criteria for the trial. While we have not experienced significant delays in patient enrollment in our completed or ongoing clinical trials, any delays in planned patient enrollment in the future may result in increased costs and delays, which could harm our ability to develop product candidates.
Delays in the commencement of clinical testing could significantly increase our product development costs and delay product commercialization. In addition, many of the factors that may cause, or lead to, a delay in the commencement of clinical trials may also ultimately lead to denial of regulatory approval of a product candidate. The commencement of clinical trials can be delayed for a variety of reasons, including delays in:
| • | demonstrating sufficient safety and efficacy to obtain regulatory approval to commence a clinical trial; |
9
Table of Contents
| • | reaching agreement on acceptable terms with prospective contract research organizations and trial sites; | |
| • | manufacturing sufficient quantities of a product candidate; | |
| • | obtaining approval of an IND application from the FDA; and | |
| • | obtaining institutional review board approval to conduct a clinical trial at a prospective site. |
| There may be delays in developing ALGRX 4975 because we need to conduct studies to evaluate the potential of the product candidate to cause cancer and drug interaction studies. |
In addition to our clinical trials, we are required to conduct studies to evaluate the potential of the product candidate to cause cancer in laboratory rodents in support of our planned NDA to market ALGRX 4975 for the treatment of post-trauma neuropathic pain and for pain resulting from musculoskeletal diseases. Such studies require about three years to complete and report. The time frame of our development program for these indications is also affected by the International Conference on Harmonisation guidelines that dictate that at least 1,500 patients must be exposed to the drug prior to submission of a registration application and at least 500 patients be exposed to a new drug for one year. Thus, the length of the development program for ALGRX 4975 for post-trauma neuropathic pain and for pain resulting from musculoskeletal diseases is expected to be longer than our development program seeking approval of ALGRX 4975 for severe postsurgical pain. In addition to the time required to conduct these studies, the results of such studies may demonstrate harmful side effects of ALGRX 4975 which would impair or prevent our ability to develop this product candidate. Assuming there are no delays with our development program for ALGRX 4975 for post-trauma neuropathic pain, we could submit an NDA to the FDA by the end of 2008 at the earliest.
Further, if we are unsuccessful for any reason in developing a gel formulation of ALGRX 4975 for use during surgical procedures to treat severe postsurgical pain, and instead rely on the liquid formulation to address this indication, the FDA may raise concerns of significant off-label use of ALGRX 4975 and may require that we complete studies to evaluate the potential of the product candidate to cause cancer prior to submission of the NDA for this indication. For instance, the FDA may be concerned that the liquid formulation of ALGRX 4975 may be prescribed for patients with osteoarthritis, a disease which would otherwise require drug interaction studies and a lengthier approval process as discussed below. If such a program is required, then submission of the NDA for ALGRX 4975 for use during surgical procedures to treat severe postsurgical pain will be delayed considerably.
Osteoarthritis is a chronic disease that mostly afflicts elderly patients. Since these patients often take many drugs for a variety of ailments, clinical development of new products for osteoarthritis tends to require many drug interaction studies and studies of elderly patients with other disease conditions, such as liver failure or kidney failure. Consequently, the development of a new drug for osteoarthritis requires exposure of far more patients to the drug than for many other indications. We expect that a submission of an NDA for treatment of osteoarthritis would require several years and expect that an NDA submission may not occur before 2010, if at all. In addition to the time required to conduct such drug interaction studies and the studies to evaluate the potential of the product candidate to cause cancer described above, the results of such studies may demonstrate adverse side effects of ALGRX 4975, which would harm our ability to develop this product candidate to treat osteoarthritis.
| The independent clinical investigators and contract research organizations that we rely on to conduct clinical trials may not be diligent, careful or timely, and may not allocate adequate resources to our trials which could result in incomplete collection of data that would require us to perform additional trials thereby increasing cost and time to regulatory approval. |
We depend on several independent clinical investigators and contract research organizations to conduct our clinical trials under their agreements with us. We are not employing these investigators, and we cannot control the amount of time or resources that they devote to our programs. Our contracts with
10
Table of Contents
these investigators may involve fixed fees. If the costs of performing the clinical trials exceed estimates, these investigators may fail to devote sufficient time and resources to our drug development programs, fail to enroll patients as rapidly as expected, or otherwise fail to perform in a satisfactory manner, and therefore potential regulatory approval and introduction of new products would be delayed. We contract with contract research organizations for execution of our clinical trials for our product candidates. Failure of the contract research organizations to meet their obligations could adversely affect clinical development of our product candidates. Moreover, these independent investigators and contract research organizations may also have relationships with other commercial entities, some of which may compete with us. Failure to perform by these third parties may increase our development costs, delay our ability to obtain regulatory approval and prevent the commercialization of our drug candidates. We currently use approximately 20 contract research organizations to perform services for our preclinical and clinical trials. While we believe that there are numerous alternative sources to provide these services, in the event we seek alternative sources, we may not be able to enter into replacement arrangements without delays or additional expenditures. We cannot estimate these costs or delays with certainty, but do not expect them to be material. To date, we have not experienced any significant difficulties with these independent investigators or contract research organizations and do not currently expect that their future performance will be less than satisfactory.
| If we fail to obtain U.S. regulatory approvals for product candidates under development, we will not be able to generate revenue in the U.S. market. |
We must receive FDA approval for ALGRX 3268, ALGRX 4975 and ALGRX 1207 before we can commercialize or sell these product candidates in the United States. Even if one of our product candidates is approved by the FDA, the approval may be significantly limited to specific disease indications, patient populations and dosages. For instance, we will likely need separate FDA approvals before ALGRX 4975 can be commercialized to treat each of the three indications for which this product candidate is being developed: severe postsurgical pain, post-trauma neuropathic pain and musculoskeletal pain. The FDA can limit or deny its approval for many reasons, including:
| • | a product candidate may be found to be unsafe or ineffective; | |
| • | regulators may interpret data from preclinical testing and clinical trials differently and less favorably than we do; | |
| • | regulators may not approve the manufacturing processes or facilities that we use; and | |
| • | regulators may change their approval policies or adopt new regulations. |
Failure to obtain FDA approval or any delay or setback in obtaining such approval would:
| • | adversely affect our ability to market any drugs that we develop and generate product revenues; and | |
| • | impose additional costs and diminish any competitive advantages that we may attain. |
Even if we obtain FDA approval, our product candidates may not be approved for all indications that we request, which could limit the uses of our products and adversely impact our potential product sales and royalties. If FDA approval of a product is granted, such approval will be subject to limitations on the indicated uses for which the product may be marketed and could require costly, post-marketing follow-up studies. As to any product for which marketing approval is obtained, the labeling, packaging, adverse event reporting, storage, advertising, promotion and record keeping related to the product will be subject to extensive regulatory requirements. The subsequent discovery of previously unknown problems with the product, such as an adverse side effect, may result in restrictions on the product, including withdrawal of the product from the market. We may be slow to adapt, or we may never adapt, to changes in existing requirements or adoption of new requirements or policies.
If we fail to comply with applicable U.S. regulatory requirements, we may be subject to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
11
Table of Contents
| If we fail to obtain regulatory approvals in other countries for our product candidates under development, we will not be able to generate revenue in such countries. |
In order to market our products outside of the United States, we must establish and comply with numerous and varying regulatory requirements of other countries. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval in the United States. The regulatory approval process in other countries may include all of the risks detailed above regarding FDA approval. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval could have the same adverse effects detailed above regarding FDA approval in the United States, including the risk that our product candidate may not be approved for all indications that we request, which could limit the uses of our product and adversely impact our potential royalties and product sales, and such approval may be subject to limitations on the indicated uses for which the product may be marketed or require costly, post-marketing follow-up studies.
If we fail to comply with applicable foreign regulatory requirements, we may be subject to fines, suspension or withdrawal of regulatory approvals, product recalls, seizure of products, operating restrictions and criminal prosecution.
| Competition in the biotechnology and pharmaceutical industries is intense, and even if our product candidates are approved and commercialized, competitive products may impede market acceptance of our products. |
Our business is characterized by extensive research and development efforts, rapid developments and intense competition. Our competitors may have or may develop superior technologies or approaches, which may provide them with competitive advantages. Our potential products may not compete successfully. If our competitors get to the marketplace before we do with better or less expensive drugs, our product candidates, if approved for commercialization, may not be profitable to sell or worthwhile to continue to develop. Technology in the pharmaceutical industry has undergone rapid and significant change, and we expect that it will continue to do so. Any compounds, products or processes that we develop may become obsolete or uneconomical before we recover any expenses incurred in connection with their development. The success of our product candidates will depend upon factors such as product efficacy, safety, reliability, availability, timing, scope of regulatory approval, acceptance and price, among other things. Other important factors to our success include speed in developing product candidates, completing clinical development and laboratory testing, obtaining regulatory approvals and manufacturing and selling commercial quantities of potential products to the market.
Our product candidates are intended to compete directly or indirectly with existing drugs. Even if approved and commercialized, our products may fail to achieve market acceptance with hospitals, physicians or patients. Hospitals, physicians or patients may conclude that our potential products are less safe or effective or otherwise less attractive than these existing drugs. If our product candidates do not receive market acceptance for any reason, our revenue potential would be diminished, which would materially adversely affect our ability to become profitable.
ALGRX 4975, if approved and commercialized, will face significant competition. For severe postsurgical pain, morphine administered by infusion pump is a common treatment method. Several other oral, injectable and patch opioids are also used, including Vicodin® (Abbott Labs), OxyContin® (Purdue Pharma), and Duragesic® (Johnson & Johnson). For localized neuropathic pain, Neurontin® (Pfizer) and tricyclic antidepressants are used to treat neuropathic pain. For later-stage osteoarthritis, hyaluronic acid products, including Synvisc® (Genzyme), a market leader in 2003, are injected locally and several oral opioids, most prominently OxyContin® (Purdue Pharma) and Duragesic® (Johnson & Johnson) are used. For the treatment of tendonitis, glucocorticosteroids are used. VR1, which is involved in the transmission of pain signals to the brain and which is affected by ALGRX 4975, has become a popular target for the
12
Table of Contents
pharmaceutical industry. VR1 inhibitors that may also compete with ALGRX 4975 are being developed by several companies, including Merck-Neurogen, Amgen, Schwarz Pharma-Amore Pacific, Purdue Pharma, and PainCeptor. These VR1 inhibitors are expected to advance to clinical evaluation shortly. We believe there are other products that are in development that may compete with our current product candidates.
ALGRX 3268, if approved and commercialized, will face significant competition. During 2003, two leading products for local anesthesia prior to needle-stick procedures were L.M.X.4®, a cream-based product (formerly ELA-MAX, Ferndale Labs), and EMLA®, a cream-based product sold by AstraZeneca. EMLA® has historically been the market leader, and in 2003, several generic versions of EMLA® that are manufactured by Fougera, Atrix, Geneva, and Hi-Tech Pharmaceuticals were approved by the FDA. These products already have established distribution channels and are well known to physicians and hospitals. There are additional products including Numby Stuff® (Iomed) and LidoSite® (Braun-Vyteris) with more rapid onset than the cream-based products above, and two other products, including S-Caine® Patch (ZARS), for which an NDA has been submitted that may also compete with ALGRX 3268. If ALGRX 3268 is unable to deliver lidocaine to patients without pain, the product will not be readily adopted by hospitals, physicians or patients, if at all.
ALGRX 1207, if approved and commercialized, will face competition from existing products, including LidoDerm® (Endo), which is a lidocaine patch, and a variety of local anesthetic creams and products with alternative means of delivering lidocaine, including EMLA® cream (AstraZeneca) and its generic equivalents, L.M.X.4® (Ferndale Labs), S-Caine® Patch (ZARS) and LidoSite® (Braun-Vyteris). There are also capsaicin products in development by NeurogesX and Winston Labs, which would be applied to the skin and which may be approved prior to ALGRX 1207. If approved, ALGRX 1207, which will also likely be formulated as a cream or patch, will compete with existing products based on factors such as efficacy, convenience and onset time of pain relief.
Most of our competitors, including many of those listed above, have substantially greater capital resources, research and development staffs, facilities and experience in conducting clinical trials and obtaining regulatory approvals, as well as in manufacturing and marketing pharmaceutical products. As a result, they may achieve product commercialization or patent protection earlier than we can.
| We have no experience selling, marketing or distributing products and have minimal capabilities to do so. |
If we receive regulatory approval to commence commercial sales of any of our product candidates, we will have to establish a sales and marketing organization with appropriate technical expertise and distribution capability. At present, we have no sales and only one marketing employee. Factors that may inhibit our efforts to commercialize our products without strategic partners or licensees include:
| • | difficulty recruiting and retaining adequate numbers of effective sales and marketing personnel; | |
| • | the inability of sales personnel to obtain access to, or persuade adequate numbers of, physicians to prescribe our products; | |
| • | the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage against companies with broader product lines; and | |
| • | unforeseen costs associated with creating an independent sales and marketing organization. |
As an alternative to establishing our own sales and marketing organization, we may engage other pharmaceutical or health care companies with an existing distribution system and direct sales organization to assist us. We may not be able to negotiate favorable distribution partnering arrangements, if at all. To the extent we enter co-promotion or other licensing arrangements, any revenues we receive will depend on the efforts of third parties and will not be under our control.
13
Table of Contents
| If third party manufacturers of our products fail to devote sufficient time and resources to our concerns, or if their performance is substandard, our clinical trials and product introductions may be delayed and our costs may rise. |
We have no manufacturing facilities, and we have no experience in the commercial manufacturing of drugs or in designing drug manufacturing processes. We have contracted with third party manufacturers to produce, in collaboration with us, our product candidates for clinical trials. We intend to rely on third party contract manufacturers to manufacture, supply, store and distribute any resulting products. The BOC Group plc acts as the sole supplier for the cylinder of compressed helium gas, a key component in the dispenser for ALGRX 3268. While we have not experienced any significant problems with our third party manufacturers to date, our reliance on these third party manufacturers, particularly The BOC Group plc, the sole supplier of cylinders used in our ALGRX 3268 product candidate, exposes us to the following risks, any of which could delay or prevent the completion of our clinical trials, the approval of our product candidates by the FDA or other regulatory agencies, or the commercialization of our products, result in higher costs, or deprive us of potential product revenues:
| • | Contract manufacturers are obliged to operate in accordance with FDA-mandated current good manufacturing practices, or cGMPs. A failure of any of our contract manufacturers to establish and follow cGMPs or to document their adherence to such practices may lead to significant delays in the availability of material for clinical trials, may delay or prevent filing or approval of marketing applications for our products and may impair our ability to commercialize our products. | |
| • | Changing contract manufacturers may be difficult, and the number of potential manufacturers is limited. Changing manufacturers may require re-validation of the manufacturing processes and procedures in accordance with FDA-mandated cGMPs. Such re-validation may be costly and time-consuming. It may be difficult or impossible for us to find replacement manufacturers on acceptable terms quickly, if at all. |
Drug manufacturers are subject to ongoing, periodic unannounced inspections by the FDA and corresponding state and foreign agencies to ensure strict compliance with cGMPs, other government regulations and corresponding foreign standards. We are not aware of any violations by our third party manufacturers of any of these regulations or standards. While we are obligated to audit the performance of third party contractors, we do not have control over our third party manufacturers’ compliance with these regulations and standards. Failure by our third party manufacturers or us to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of the government to grant approval of product candidates, delays, suspension or withdrawal of approvals, seizures or recalls of products, operating restrictions and criminal prosecutions.
| Our third party manufacturers may not be able to manufacture our product candidates in commercial quantities, which would prevent us from commercializing our product candidates. |
To date, our product candidates have been manufactured in small quantities by third party manufacturers for preclinical and clinical trials. If any of these product candidates are approved by the FDA or other regulatory agencies for commercial sale, we will need to manufacture them in larger quantities. Our third party manufacturers may not be able to successfully increase their manufacturing capacity for any of our product candidates in a timely or economic manner, or at all. Significant scale-up of manufacturing may require additional validation studies, which the FDA must review and approve. If we are unable to successfully increase the manufacturing capacity for a product candidate, the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage in the supply of the product candidate. Our product candidates require precise, high quality manufacturing. The failure of our third party manufacturers to achieve and maintain these high manufacturing standards, including the incidence of manufacturing errors, could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could seriously harm our business.
14
Table of Contents
| Materials necessary to make ALGRX 3268 may not be available on commercially reasonable terms, or at all, which may delay our development and commercialization of the drug. |
There are a small number of suppliers of the materials which are necessary to manufacture ALGRX 3268 and, in the case of the cylinder used in ALGRX 3268, we rely on a sole supplier. The cylinder of compressed helium gas is a key component in the dispenser for ALGRX 3268. We acquire the cylinders for our ALGRX 3268 product candidate from PowderJect Technologies Limited under a long-term supply agreement. PowderJect Technologies Limited is currently our sole supplier and source of cylinders, which are manufactured for PowderJect Technologies Limited by The BOC Group plc, and to date we have not identified an alternative source. If we are required to seek an alternative source for the cylinders, we might not be successful in establishing an alternative commercial arrangement with a supplier, or, if we were successful in finding an alternate supplier, it could be on terms which are less favorable than our current supply agreement with PowderJect Technologies Limited.
Our contract manufacturers for ALGRX 3268 need to purchase the materials required for ALGRX 3268 for our clinical trials and they or we will need to purchase these materials for commercial distribution if we obtain regulatory approval for ALGRX 3268. Suppliers may not sell us these materials at the time we need them or on commercially reasonable terms. If our manufacturers are unable to obtain these materials for our clinical trials, product testing and potential regulatory approval of ALGRX 3268 would be delayed, significantly impacting our ability to develop the product candidate and potentially increasing our costs. If we obtain regulatory approval for ALGRX 3268 and our manufacturers or we are unable to purchase these materials, the commercial launch of ALGRX 3268 would be delayed or there would be a shortage in supply of ALGRX 3268, which would harm our ability to generate revenues from the sale of ALGRX 3268. If suppliers increase the price of these materials, the price for ALGRX 3268 may increase which may make ALGRX 3268 a less competitive product for the relief of needle-stick pain. If we change suppliers for any of these materials or any of our suppliers experiences a shutdown or disruption in the facilities used to produce these materials, due to technical, regulatory or other problems, it could harm our ability to manufacture our products. While we have not suffered any shortages to date of any of the materials required for ALGRX 3268, our inability to obtain these materials for any reason could substantially impair our development activities or the production, marketing and distribution of ALGRX 3268.
| If we are unable to retain and recruit qualified personnel or if any of our key executives, Ronald M. Burch, M.D., Ph.D, Paul R. Hamelin, R.Ph., or Jeffrey D. Lazar, M.D., Ph.D, discontinues his employment with us, it may delay our development efforts. |
One of our key strategies for successfully building our company is utilizing the extensive expertise and experience of our senior management and scientific staff, especially of our Chief Executive Officer, Ronald M. Burch, M.D., Ph.D., our President and Chief Operating Officer, Paul R. Hamelin, R.Ph. and our Executive Vice President of Clinical and Regulatory Affairs, Jeffrey D. Lazar, M.D., Ph.D. Therefore, we consider our business to be highly dependent on these individuals. The loss of any of our key employees could impede the achievement of our development objectives. We do not expect to lose the services of any of our key employees in the near future. We have no key-man insurance on any members of our management team. Furthermore, recruiting and retaining qualified personnel to perform research and development work in the future is critical to our success. We may be unable to attract and retain personnel on acceptable terms given the competition among biotechnology and pharmaceutical companies, universities and non-profit research institutions for experienced scientists. To date, we have been able to attract and hire qualified candidates to meet our personnel needs.
| We have a history of losses, and we may never achieve sustained profitability. |
Since our inception, we have incurred significant net losses, including net losses of $13.2 million and $15.5 million for the nine months ended September 30, 2004 and the year ended December 31, 2003, respectively. As of September 30, 2004, our cumulative net loss was $50.3 million. We have not yet completed the development, including obtaining regulatory approvals, of any of our product candidates and,
15
Table of Contents
consequently, have not generated revenues from the sale of products. Even if we succeed in developing and commercializing one or more of our product candidates, we expect to incur substantial losses for the foreseeable future. We also expect to continue to incur significant operating and capital expenditures and anticipate that our expenses will increase substantially in the foreseeable future as we:
| • | continue the development and the preparation for possible commercialization of ALGRX 4975 and ALGRX 3268, including establishing sales and marketing capabilities; | |
| • | continue the preclinical development and commence the clinical development of ALGRX 1207 which we recently in-licensed in October 2004; | |
| • | expand our development programs; and | |
| • | increase our administrative functions. |
We also expect to experience negative cash flow for the foreseeable future as we fund our operating losses and capital expenditures. We will need to generate significant revenues to achieve profitability. We may not be able to generate these revenues, and we may never achieve profitability. Our failure to achieve profitability could negatively impact the market price of our common stock. Even if we do become profitable, we may not be able to sustain or increase profitability on a quarterly or annual basis.
| If we cannot raise additional capital on acceptable terms, we may be unable to complete planned clinical trials, obtain regulatory approvals or commercialize our product candidates. |
We believe that existing cash reserves, together with proceeds from this offering, will fund our planned activities through at least 2007. However, we will require substantial future capital in order to continue to conduct the research and development, preclinical and clinical programs and regulatory activities necessary to bring our product candidates to market and to establish commercial manufacturing, sales and marketing capabilities. During the nine months ended September 30, 2004, our net cash used in operating activities was $10.7 million and we had capital expenditures of $48,000 for the same period. Our future capital requirements depend on many factors, including:
| • | the progress of preclinical development and clinical trials; | |
| • | the time and costs involved in obtaining regulatory approvals; | |
| • | delays that may be caused by evolving requirements of regulatory agencies; | |
| • | the number of product candidates we pursue; | |
| • | the costs involved in filing and prosecuting patent applications and enforcing or defending patent claims; | |
| • | our plans to establish sales and/or marketing capabilities; | |
| • | our ability to establish and maintain selected strategic alliances required for product commercialization; | |
| • | the acquisition of technologies or products and other business opportunities that require financial commitments; and | |
| • | our revenues, if any, from successful development and commercialization of our products. |
We may seek additional funding through strategic collaborations and may seek funding through private or public sales of our securities or by licensing all or a portion of our technology. This funding may significantly dilute existing stockholders or may limit our rights to our technology. We may not be able to obtain additional funding on favorable terms, or at all. If we cannot obtain adequate funds, we may:
| • | terminate or delay preclinical development or clinical trials for one or more of our product candidates; | |
| • | delay establishment, or fail to establish, manufacturing, sales, and/or marketing capabilities; |
16
Table of Contents
| • | curtail significant product development programs that are designed to identify new product candidates; | |
| • | not be in a position to acquire technologies or pursue other business opportunities that require financial commitments; and/or | |
| • | relinquish rights to our product candidates. |
| If we lose our licenses from PowderMed Limited for ALGRX 3268, certain individuals for ALGRX 4975 or Bridge Pharma, Inc. for ALGRX 1207, we will not be able to continue development of our current products. |
We are party to three significant license agreements relating to patents, patent applications and know-how covering the technology relating to ALGRX 4975, ALGRX 3268 and ALGRX 1207, our three product candidates. These license agreements impose various diligence, commercialization, royalty and other obligations on us. If we fail to comply with the obligations in the license agreements, the licensor may have the right to terminate the license and we may not be able to market products that were covered by the license. Our license agreement with James N. Campbell, M.D., Richard A. Meyer, M.S. and Marco Pappagallo, M.D. relates to the steps of administering capsaicin for pain reduction that we use in our product candidate, ALGRX 4975, and our rights under this agreement can be terminated on 10 days’ written notice if we fail to make a payment or fulfill any material obligation under the agreement and the failure is not cured by us within 180 days of receiving notice of such failure. Our license agreement with PowderMed Limited relates to technology underlying our product candidate, ALGRX 3268. Our agreement with PowderMed Limited can be terminated immediately by either party if the other party ceases to do business in the ordinary course, or assigns all or substantially all of its assets for the benefit of creditors. Either party can also terminate for material breach if not cured within 60 days of notice or if not cured within 30 days of notice if the breach relates to payment provisions. Our agreement with Bridge Pharma, Inc. relates to proprietary analgesic and local anesthetic pharmaceutical agents and technology underlying our product candidate ALGRX 1207, and our rights under this agreement can be terminated for cause by Bridge Pharma, Inc. if we breach any material provision of the agreement and fail to cure the breach within 60 days of the receipt of the notice of breach. Either party may terminate the agreement immediately in the event the other party suffers certain insolvency events. To date, we believe we have met our obligations under all of these agreements.
| If we fail to enter into additional in-licensing agreements or if these arrangements are unsuccessful, our business and operations might be adversely affected, and if we engage in any acquisition or in-licensing, we will incur a variety of costs, and we may never realize the anticipated benefits of the transaction. |
Since our inception, we have acquired three product candidates, ALGRX 4975, ALGRX 3268 and ALGRX 1207, through in-licensing. One of our strategies for expanding our business is the acquisition of products and additional product candidates. The success of this strategy depends upon our ability to identify, select and acquire the right pharmaceutical product candidates and products. We may be unable to enter into any additional in-licensing agreements because suitable product candidates that are within our expertise may not be available to us on terms that are acceptable to us or because competitors with greater resources seek to in-license the same product candidates. Product candidates that we would like to develop may not be available to us because they are controlled by competitors who are unwilling to license the rights to the drug compound or candidate to us. We may also need to in-license drug delivery or other technology in order to develop future drug candidates. If we are unable to enter into additional agreements to license drug candidates, drug delivery technology or other technology or if these arrangements are unsuccessful, our development efforts could be adversely affected.
We may attempt to acquire product candidates, or other potentially beneficial technologies, through in-licensing or the acquisition of businesses, services, products or programs that we believe are a strategic fit with our business. Although we currently have no commitments or agreements with respect to any acquisitions, if we undertake an acquisition, the process of integrating the acquired business, technology,
17
Table of Contents
services, product or programs may result in unforeseen operating difficulties and expenditures and may divert significant management attention from our ongoing business operations. Moreover, we may fail to realize the anticipated benefits of any acquisition for a variety of reasons, such as an acquired product candidate proving to not be safe or effective in later clinical trials. We may fund any future acquisition by issuing equity or equity-linked securities, which could dilute the ownership percentage of our equity holders. We may also incur debt to pay for an acquisition. The holders of the debt may have priority in the event of a subsequent liquidation or bankruptcy, which would impair value for our equity holders. Acquisition efforts can require substantial expenditures, which could detract from our other programs. In addition, we may devote resources to potential acquisitions that are never completed.
| We may not be able to succeed in our business model of seeking to enter into collaborations with partners at the early stages of developing our product candidates, and even if we do our collaborations would be subject to many risks that could prevent us from developing and commercializing our product candidates. |
Our business strategy includes entering into collaborations or marketing and distribution arrangements with corporate partners, primarily pharmaceutical companies, for the development, commercialization, marketing and distribution of certain of our product candidates. No such arrangements presently exist. We may enter into a significant corporate collaboration, or we may not be able to enter into any. If we are able to enter into a collaboration, the terms may be onerous and we may be compelled to give up a large portion of our interest in our products. We do not know at this time what the extent of such interest may be. We may be dependent on a corporate collaboration to fund clinical testing, to make certain regulatory filings and to manufacture and market products resulting from the collaboration. Risks that we face in connection with entering into collaborations include the following:
| • | collaborators may underfund or not commit sufficient resources to the testing, marketing, distribution or development of our products; | |
| • | collaborators may not properly maintain or defend our intellectual property rights, or they may utilize our proprietary information in such a way as to invite litigation that could jeopardize or potentially invalidate our proprietary information or expose us to potential liability; | |
| • | collaborators may pursue alternative technologies or develop alternative products either on their own or in collaboration with others; | |
| • | collaborators may encounter conflicts of interest, changes in business strategy or other business issues, which could adversely affect their willingness or ability to fulfill their obligations to us; for example, pharmaceutical and biotechnology companies historically have re-evaluated their priorities following mergers and consolidations, which have been common in recent years in these industries; and | |
| • | disputes may arise between us and our collaborators delaying or terminating the research, development or commercialization of our product candidates, resulting in significant litigation or arbitration that could be time-consuming and expensive, or causing collaborators to act in their own self-interest and not in the interest of our stockholders. |
A reduction in sales efforts or a discontinuance of sales of any developed products by any collaborative partner could result in reduced revenues and have a material adverse effect on our business, financial position and results of operations.
Our current strategy for developing and commercializing our product candidates includes securing collaborations with pharmaceutical and biotechnology companies relatively early in the drug development process and for these collaborators to undertake the advanced clinical development and commercialization of our product candidates. It may be difficult for us to find third parties that are willing to enter into collaborations for our other product candidates at an early stage of development or on favorable economic terms. If we are not able to enter into collaborations for our product candidates at relatively early stages, we could be required to undertake and fund further development, clinical trials, manufacturing and
18
Table of Contents
marketing activities solely at our own expense. Alternatively, we would have to substantially reduce our development efforts which would delay the commercialization of our product candidates.
| Because we have operated as a private company, we have no experience attempting to comply with public company obligations, including recently enacted changes in securities laws and regulations; attempting to comply with these requirements will increase our costs and require additional management resources and we still may fail to comply. |
As directed by Section 404 of the Sarbanes-Oxley Act of 2002, the SEC adopted rules requiring public companies to include a report of management on the company’s internal controls over financial reporting in their annual reports on Form 10-K. In addition, the public accounting firm auditing the company’s financial statements must attest to and report on management’s assessment of the effectiveness of the company’s internal controls over financial reporting. This requirement will first apply to our annual report on Form 10-K for our fiscal year ending December 31, 2005. If we are unable to conclude that we have effective internal controls over financial reporting or, if our independent auditors are unable to provide us with an unqualified report as to the effectiveness of our internal controls over financial reporting as of December 31, 2005 and future year ends as required by Section 404 of the Sarbanes-Oxley Act of 2002, investors could lose confidence in the reliability of our financial statements, which could result in a decrease in the value of our securities.
We are a small company with limited resources. We have operated as a private company, not subject to many of the requirements applicable to public companies. The number and qualifications of our finance and accounting staff are consistent with those of a private company. While we plan to expand our staff if we become public, we may encounter substantial difficulty attracting qualified staff with requisite experience due to the high level of competition for experienced financial professionals. Furthermore, we have not begun a formal process to evaluate our internal controls over financial reporting. Given the status of our efforts, coupled with the fact that guidance from regulatory authorities in the area of internal controls continues to evolve, substantial uncertainty exists regarding our ability to comply by applicable deadlines.
Risks Related to Our Industry
| Claims that we infringe a third party’s intellectual property may give rise to burdensome litigation, result in potential liability for damages or stop our development and commercialization efforts. |
Third parties may assert patent or other intellectual property infringement claims against us with respect to technologies used in our potential products. While we have conducted patent searches to determine whether our products infringe patents held by third parties, we expect that numerous patent applications are currently pending and may be filed in the future for technologies generally related to our technologies, including many patent applications that remain confidential after filing. Due to these factors and the inherent uncertainty in conducting patent searches, we may violate third party patent rights.
There may also be patent applications filed by these or other parties in the United States and various foreign jurisdictions that are in the same fields as some of our product candidates. These patent applications, if issued, could subject us to infringement actions. The owners or licensees of these and other patents may file one or more infringement actions against us. Patent litigation can involve complex factual and legal questions, and its outcome is uncertain. Any claim relating to infringement of patents that is successfully asserted against us may require us to pay substantial damages. Even if we were to prevail, any litigation could be costly and time-consuming and would divert the attention of our management and key personnel from our business operations. Furthermore, as a result of a patent infringement suit brought against us, we may be forced to stop or delay developing, manufacturing or selling potential products that are claimed to infringe a third party’s intellectual property unless that party grants us rights to use its intellectual property. In such cases, we may be required to obtain licenses to patents or proprietary rights of others in order to continue to commercialize our products. However, we may not be able to obtain any required licenses on acceptable terms, or at all. Even if we were able to obtain rights to the third party’s
19
Table of Contents
intellectual property, these rights may be non-exclusive, thereby giving our competitors access to the same intellectual property. Ultimately, we may be unable to commercialize some of our potential products or may have to discontinue development of a product candidate or cease some of our business operations as a result of patent infringement claims, which could severely diminish our commercial prospects.
Third parties may also challenge the validity of our issued patents on various grounds. In fact, one of our issued European patents covering capsaicin for injection has been challenged by Grunenthal, a German pharmaceutical company, in the European Patent Court. In response, we submitted proposed modifications to the patent, which the patent court approved and published in November 2004. Any party, including Grunenthal, can object to the amended patent within the two month period after the amended patent is published by the court. If any future challenge by Grunenthal or any other party is ultimately successful in invalidating the patent, then the ability of third parties to market competing technologies to ALGRX 4975 in Europe could be enhanced. Also, because the Grunenthal opposition resulted in our amending the claims of one of our patents in Europe, a competitor could seek to exploit that result by using arguments similar to Grunenthal’s to challenge claims in issued patents, including U.S. patents, licensed by us and in the same patent family as the amended European patent.
| We may be subject to damages resulting from claims that we or our employees have wrongfully used or disclosed alleged trade secrets of their former employers. |
Many of our employees were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail in defending such claims, in addition to paying money claims, we may lose valuable intellectual property rights or personnel. A loss of key personnel or their work product could hamper or prevent our ability to commercialize certain product candidates, which would adversely affect our commercial development efforts.
| If we are unable to protect our intellectual property rights, our competitors may develop and market products with similar features that may reduce demand for our potential products. |
The following factors are important to our success:
| • | receiving patent protection for our product candidates; | |
| • | preventing others from infringing our intellectual property rights; and | |
| • | maintaining our patent rights and trade secrets. |
We will be able to protect our intellectual property rights in patents and trade secrets from unauthorized use by third parties only to the extent that such intellectual property rights are covered by valid and enforceable patents or are effectively maintained as trade secrets.
We try to protect our proprietary position by filing U.S. and foreign patent applications related to our important proprietary technology, inventions and improvements. Because the patent position of pharmaceutical companies involves complex legal and factual questions, the issuance, scope and enforceability of patents cannot be predicted with certainty. For instance, the issued patent underlying ALGRX 4975 covers “injection” of capsaicin. If that patent were determined not to cover application of ALGRX 4975 for use during surgical procedures in circumstances where the capsaicin is applied by means other than through a syringe, we may not have an exclusive right to that application of capsaicin unless and until, for instance, we were issued a valid claim covering non-injection internal use of capsaicin or covering the gel formulation that we are developing for use during surgical procedures. Patents, if issued, may be challenged, invalidated or circumvented. U.S. patents and patent applications may also be subject to interference proceedings, and U.S. patents may be subject to reexamination proceedings in the U.S. Patent and Trademark Office and foreign patents may be subject to opposition or comparable
20
Table of Contents
proceedings in corresponding foreign patent offices, which proceedings could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, reexamination and opposition proceedings may be costly. Thus, any patents that we own or license from others may not provide any protection against competitors. Furthermore, an adverse decision in an interference proceeding can result in a third party receiving the patent rights sought by us, which in turn could affect our ability to market a potential product to which that patent filing was directed. Our pending patent applications, those we may file in the future, or those we may license from third parties may not result in patents being issued. If issued, they may not provide us with proprietary protection or competitive advantages against competitors with similar technology. Furthermore, others may independently develop similar technologies or duplicate any technology that we have developed. We rely on third party payment services for the payment of foreign patent annuities and other fees. Non-payment or delay in payment of such fees, whether intentional or unintentional, may result in loss of patents or patent rights important to our business. Many countries, including certain countries in Europe, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties. For example, compulsory licenses may be required in cases where the patent owner has failed to “work” the invention in that country, or the third party has patented improvements. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent. Moreover, the legal systems of certain countries, particularly certain developing countries, do not favor the aggressive enforcement of patent and other intellectual property protection which makes it difficult to stop infringement.
In addition, our ability to enforce our patent rights depends on our ability to detect infringement. It is difficult to detect infringers who do not advertise the compounds that are used in their products. Also, in the case of ALGRX 4975, it is not the composition of the capsaicin that is covered by the issued patent, but rather its method of use. This means that a manufacturer of an injectable form of capsaicin may not infringe the patent except indirectly, which is more difficult to prove, and any claim for direct infringement might be against only a clinician injecting the product. Any litigation to enforce or defend our patent rights, even if we were to prevail, could be costly and time-consuming and would divert the attention of our management and key personnel from our business operations.
We also rely on trade secrets, know-how and technology, which are not protected by patents, to maintain our competitive position. We try to protect this information by entering into confidentiality agreements with parties that have access to it, such as our strategic partners, collaborators, employees and consultants. Any of these parties may breach these agreements and disclose our confidential information or our competitors might learn of the information in some other way. If any trade secret, know-how or other technology not protected by a patent was to be disclosed to or independently developed by a competitor, our business, financial condition and results of operations could be materially adversely affected.
| Governmental and third party payors may impose sales and pharmaceutical pricing restrictions or controls on our potential products that could limit our future product revenues and adversely affect profitability. |
The commercial success of our potential products is substantially dependent on whether third party reimbursement is available for the ordering of our products by the medical profession for use by their patients. Medicare, Medicaid, health maintenance organizations and other third party payors may not cover or provide adequate payment for our potential products. They may not view our potential products as cost-effective and reimbursement may not be available to consumers or may not be sufficient to allow our potential products to be marketed on a competitive basis. Reimbursement will most likely not be available for ALGRX 3268. Hospitals are currently reimbursed a fixed amount of money for the needle-stick procedures in which ALGRX 3268 would be used. The cost of ALGRX 3268 would be an expense associated with that procedure and therefore would reduce the hospital’s profit. Hospitals may determine that the cost of ALGRX 3268 is not worth the benefit and decline to use it. Likewise, legislative or regulatory efforts to control or reduce health care costs or reform government health care programs could
21
Table of Contents
result in lower prices or rejection of our potential products that we may commercialize. Changes in reimbursement policies or health care cost containment initiatives that limit or restrict reimbursement for our products may cause our revenue to decline.
Another development that may affect the pricing of drugs is Congressional action regarding drug reimportation into the United States. The Medicare Prescription Drug Plan legislation, which became law in December 2003, requires the Secretary of Health and Human Services to promulgate regulations for drug reimportation from Canada into the United States under some circumstances, including when the drugs are sold at a lower price than in the United States. The Secretary retains the discretion not to implement a drug reimportation plan if he finds that the benefits do not outweigh the cost. Proponents of drug reimportation may attempt to pass legislation that would directly allow reimportation under certain circumstances. If legislation or regulations were passed allowing the reimportation of drugs, they could decrease the price we receive for any products that we may commercialize, negatively affecting our anticipated revenues and prospects for profitability.
| We face potential product liability exposure far in excess of our limited insurance coverage. |
The use of any of our product candidates in clinical trials, and the sale of any approved products, may expose us to product liability claims. These claims might be made directly by consumers, health care providers, pharmaceutical companies or others selling our products. We have obtained limited product liability insurance coverage for our clinical trials in the amount of $5 million per occurrence and $5 million in the aggregate. However, our insurance may not reimburse us or may not be sufficient to reimburse us for expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. If we obtain marketing approval for any of our product candidates in development, we intend to expand our insurance coverage to include the sale of commercial products, but we may be unable to obtain commercially reasonable product liability insurance for any products approved for marketing. On occasion, juries have awarded large judgments in class action lawsuits based on drugs that had unanticipated side effects. In addition, the pharmaceutical and biotechnology industries, in general, have been subject to significant product liability litigation. A successful product liability claim or series of claims brought against us would decrease our cash reserves and could cause our stock price to fall.
Risks Related to Our Common Stock
| We expect that our stock price will fluctuate significantly. |
Prior to this offering, you could not buy or sell our common stock publicly. An active public market for our common stock may not develop or be sustained after this offering. We will negotiate and determine the initial public offering price with the underwriters based on several factors. This price may vary from the market price of our common stock after this offering. You may be unable to sell your shares of common stock at or above the initial offering price. The stock market, particularly in recent years, has experienced significant volatility particularly with respect to pharmaceutical and biotechnology stocks. The volatility of pharmaceutical and biotechnology stocks often does not relate to the operating performance of the companies represented by the stock. Factors that could cause this volatility in the market price of our common stock include:
| • | the timing and the results from our clinical trial programs, including the initiation and completion of phase II clinical trials for ALGRX 4975 and phase III clinical trials for ALGRX 3268; | |
| • | the timing and results from our preclinical and future clinical studies relating to ALGRX 1207; | |
| • | the in-licensing or acquisition of additional product candidates; | |
| • | FDA or international regulatory actions; | |
| • | failure of any of our product candidates, if approved, to achieve commercial success; | |
| • | announcements of new products by our competitors; |
22
Table of Contents
| • | market conditions in the pharmaceutical and biotechnology sectors; | |
| • | developments concerning intellectual property rights; | |
| • | litigation or public concern about the safety of our potential products; | |
| • | comments by securities analysts; | |
| • | actual and anticipated fluctuations in our quarterly operating results; | |
| • | deviations in our operating results from the estimates of securities analysts; | |
| • | rumors relating to us or our competitors; | |
| • | additions or departures of key personnel; | |
| • | third party reimbursement policies; and | |
| • | developments concerning current or future strategic alliances. |
These and other factors may cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent investors from readily selling their shares of common stock and may otherwise negatively affect the liquidity of our common stock. In addition, in the past, when the market price of a stock has been volatile, holders of that stock have instituted securities class action litigation against the company that issued the stock. If any of our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit. Such a lawsuit could also divert the time and attention of our management.
| Future sales of common stock by our existing stockholders may cause our stock price to fall. |
The market price of our common stock could decline as a result of sales by our existing stockholders of shares of common stock in the market after this offering, or the perception that these sales could occur. These sales might also make it more difficult for us to sell equity securities at a time and price that we deem appropriate. The lock-up agreements delivered by our executive officers and directors and substantially all of our stockholders and optionholders provide that Credit Suisse First Boston LLC and Citigroup Global Markets Inc., in their sole discretion, may release those parties, at any time or from time to time and without notice, from their obligation not to dispose of shares of common stock for a period of 180 days (which period could be extended by those underwriters for up to an additional 34 days under certain circumstances) after the date of this prospectus. Credit Suisse First Boston LLC and Citigroup Global Markets Inc. have no pre-established conditions to waiving the terms of the lock-up agreements, and any decision by them to waive those conditions would depend on a number of factors, which may include market conditions, the performance of the common stock in the market and our financial condition at that time. After this offering, approximately 14,505,012 shares of our common stock will be subject to registration rights. Please see “Shares Eligible for Future Sale.”
| Our directors and management will exercise significant control over our company which may delay, prevent or deter corporate actions that may be in the best interest of our stockholders. |
After this offering, our directors and executive officers and their affiliates will collectively control approximately 38.9% of our outstanding common stock, assuming that the underwriters’ over-allotment option is not exercised. As a result, these stockholders will collectively be able to significantly influence all matters requiring approval of our stockholders including the election of directors and approval of significant corporate transactions. The concentration of ownership may delay, prevent or deter a change in control of our company even when such a change may be in the best interest of all the stockholders, could deprive stockholders of an opportunity to receive a premium for their common stock as part of a sale of our company or assets and might affect the prevailing market price of our common stock.
23
Table of Contents
| We will have broad discretion in how we use the proceeds of this offering, and we may not use these proceeds effectively, which could affect our results of operations and cause our stock price to decline. |
We will have considerable discretion in the application of the net proceeds of this offering. We currently intend to use the net proceeds of this offering for general corporate purposes, including clinical trials, research and development expenses, general and administrative expenses, and potential acquisitions of companies, products and technologies that complement our business. We may use the net proceeds for corporate purposes that do not yield a significant return or any return at all for our stockholders.
| Provisions of Delaware law or our charter documents could delay or prevent an acquisition of our company, even if the acquisition would be beneficial to our stockholders, and could make it more difficult for you to change management. |
Provisions of our certificate of incorporation and bylaws which we plan to adopt immediately prior to this offering may discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. This is because these provisions may prevent or frustrate attempts by stockholders to replace or remove our current management or members of our board of directors.
These provisions include:
| • | a classified board of directors; | |
| • | a prohibition on stockholder action through written consent; | |
| • | a requirement that special meetings of stockholders be called only by the Chairman of the board of directors, the Chief Executive Officer, or the board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors; | |
| • | advance notice requirements for stockholder proposals and nominations; | |
| • | a requirement that 66 2/3% of stockholders approve certain amendments to our certificate of incorporation and amendments to our by-laws; and | |
| • | the authority of the board of directors to issue preferred stock with such terms as the board of directors may determine. |
As a result, these provisions and others available under Delaware law could limit the price that investors are willing to pay in the future for shares of our common stock.
| We have never paid dividends on our capital stock, and we do not anticipate paying any cash dividends in the foreseeable future, and, consequently, any gains from an investment in our common stock will depend on whether the price of our common stock increases. |
We have paid no cash dividends on any of our classes of capital stock to date and we currently intend to retain our future earnings, if any, to fund the development and growth of our businesses. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future. Consequently, in the foreseeable future, you will only experience a gain from your investment in our common stock if the price of our common stock increases.
| Investors in this offering will pay a much higher price than the book value of our common stock and will incur immediate dilution. |
If you purchase common stock in this offering, you will pay more for your shares than the amounts paid by existing stockholders for their shares. You will incur immediate and substantial dilution of $5.82 per share, representing the difference between our pro forma net tangible book value per share after giving effect to this offering and an assumed initial public offering price of $11 per share. In the past, we issued options and warrants to acquire common stock at prices significantly below the assumed initial public offering price. To the extent these outstanding options or warrants are ultimately exercised, you will sustain further dilution.
24
Table of Contents
FORWARD-LOOKING STATEMENTS
This prospectus, including the sections entitled “Prospectus Summary,” “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business,” contains forward-looking statements.
Forward-looking statements include, but are not limited to, statements about:
| • | our ability to in-license or acquire new technology or product candidates; | |
| • | the progress, timing and completion of our research, development and clinical trials for product candidates; | |
| • | our ability to receive regulatory approvals to develop and commercialize our product candidates; | |
| • | our ability to build an internal sales and marketing infrastructure; | |
| • | our ability to contract for and maintain third party manufacturing services; | |
| • | our ability to achieve market acceptance for our product candidates and successfully compete in the market; | |
| • | our ability to enter into collaborative or strategic relationships to assist in the clinical development and commercialization of our product candidates; | |
| • | our ability to protect our intellectual property and operate our business without infringing upon the intellectual property rights of others; | |
| • | our estimates for future performance; and | |
| • | our estimates regarding anticipated operating losses, use of proceeds from this offering, future revenues, capital requirements and our needs for additional financing. |
These statements relate to future events or our future financial performance, and involve known and unknown risks, uncertainties and other factors that may cause our actual results, levels of activity, performance or achievements to be materially different from any future results, levels of activity, performance or achievements expressed or implied by these forward-looking statements. These risks and other factors include those listed under “Risk Factors” and elsewhere in this prospectus. In some cases, you can identify forward-looking statements by terminology such as “may,” “will,” “should,” “expects,” “intends,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “potential,” “continue” or the negative of these terms or other comparable terminology. Although we believe that the expectations reflected in the forward-looking statements are reasonable, we cannot guarantee future results, levels of activity, performance or achievements. We do not intend to update any of the forward-looking statements after the date of this prospectus or to conform these statements to actual results. Neither the Private Securities Litigation Reform Act of 1995 nor Section 27A of the Securities Act of 1933 provides any protection for statements made in this prospectus.
25
Table of Contents
USE OF PROCEEDS
We estimate that we will receive net proceeds from the sale of shares of our common stock in this offering of approximately $67.5 million, or $77.9 million if the underwriters fully exercise their over-allotment option, based upon an assumed initial public offering price of $11 per share and after deducting underwriting discounts and commissions and estimated offering expenses payable by us.
We estimate that we will use approximately $19 million of the net proceeds of this offering to continue the development of our product candidate ALGRX 3268, including the continuation of our clinical trial program. We expect, with these proceeds, to file our NDA with the FDA and fund our pre-commercialization activities.
We estimate that we will use approximately $22 million of the net proceeds of this offering to continue the development of our product candidate ALGRX 4975, including the continuation of our clinical trial program and toxicology program. We expect, with these proceeds, to complete multiple phase II clinical trials and begin our phase III program.
We estimate that we will use approximately $14 million of the net proceeds of this offering to develop our most recently in-licensed candidate, ALGRX 1207. We expect, with these proceeds, to complete multiple phase I and phase II clinical trials.
We intend to use the balance of the net proceeds of this offering to fund operations, to provide working capital and for other general corporate purposes, which may include in-licensing or acquiring additional product candidates. We have no current agreements or commitments with respect to any future acquisitions or in-licensing, and we are not currently engaged in any negotiations with respect to any transactions of that nature.
As of the date of this prospectus, we cannot specify with certainty all of the particular uses for the net proceeds to be received upon the completion of this offering. The amounts and timing of our actual expenditures may vary significantly from our expectations depending upon numerous factors, including the progress of our development and commercialization efforts, the progress of our clinical trials, and our operating costs and capital expenditures. Accordingly, our management will have broad discretion in the application of the net proceeds, and investors will be relying on the judgment of our management regarding the application of the proceeds of this offering. Pending these uses, we plan to invest the net proceeds in short-term, interest-bearing obligations, investment-grade instruments, certificates of deposit or direct or guaranteed obligations of the United States. The goal with respect to the investment of these net proceeds is capital preservation and liquidity so that funds are readily available to fund our research and development operations. In March 2004, our board of directors adopted our written investment policy. The investment policy imposes restrictions on our investments designed to ensure that we preserve our principal and maintain our liquidity. Any investment of the net proceeds from this offering will be made in accordance with the terms of our investment policy.
DIVIDEND POLICY
We have never declared or paid cash dividends on our capital stock. We do not anticipate paying any cash dividends on our capital stock in the foreseeable future. We currently intend to retain all available funds and any future earnings to fund the development and growth of our business.
26
Table of Contents
CAPITALIZATION
The following table sets forth our capitalization as of September 30, 2004:
| • | on an actual basis; | |
| • | on a pro forma basis, reflecting the conversion of all of our preferred stock into an aggregate of 14,275,747 shares of common stock immediately prior to the closing of this offering; and | |
| • | on a pro forma as adjusted basis giving effect to the sale of 6,800,000 shares of common stock in this offering at an assumed initial public offering price of $11 per share, after deducting underwriting discounts and commissions and estimated offering expenses payable by us. | |
You should read this table in conjunction with the sections of this prospectus entitled “Selected Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and with our financial statements and related notes.
| As of September 30, 2004 | ||||||||||||
| Pro Forma | ||||||||||||
| Actual | Pro Forma | As Adjusted | ||||||||||
| (Unaudited) | ||||||||||||
| (In thousands, except shares) | ||||||||||||
| Convertible preferred stock, $0.001 par value; actual, 137,405,754 shares authorized and 130,546,942 shares issued and outstanding; pro forma, no shares authorized, issued or outstanding | $ | 87,484 | $ | — | $ | — | ||||||
| Preferred stock, $0.001 par value; actual, no shares authorized, issued or outstanding; pro forma, 25,000,000 shares authorized and no shares issued or outstanding | — | — | — | |||||||||
| Common stock, $0.001 par value; actual, 17,384,027 shares authorized, 960,331 shares issued and 933,248 shares outstanding; pro forma, 17,384,027 shares authorized, 15,236,078 shares issued and 15,208,995 shares outstanding; pro forma as adjusted, 150,000,000 shares authorized, 22,036,078 shares issued and 22,008,995 shares outstanding | 1 | 15 | 22 | |||||||||
| Additional paid-in capital | 17,656 | 105,126 | 172,621 | |||||||||
| Accumulated other comprehensive income | (26 | ) | (26 | ) | (26 | ) | ||||||
| Deferred compensation | (8,418 | ) | (8,418 | ) | (8,418 | ) | ||||||
| Treasury stock, at cost, 27,083 shares | — | — | — | |||||||||
| Deficit accumulated during the development stage | (50,285 | ) | (50,285 | ) | (50,285 | ) | ||||||
| Total stockholders’ equity | 46,412 | 46,412 | 113,914 | |||||||||
| Total capitalization | $ | 46,412 | $ | 46,412 | $ | 113,914 | ||||||
The actual number of shares of common stock shown as issued and outstanding in the table above excludes:
| • | 2,242,505 shares of common stock issuable upon the exercise of options outstanding as of September 30, 2004, with exercise prices ranging from $1.00 to $10.00 per share and a weighted average exercise price of $1.20 per share; | |
| • | 113,248 additional shares of common stock reserved for future grants under our 2001 Equity Incentive Plan as of September 30, 2004; and | |
| • | 69,265 shares of common stock issuable upon the exercise of an outstanding warrant at $5.925 per share, or $410,000 in the aggregate, that, by its terms, terminates upon completion of this offering. | |
27
Table of Contents
DILUTION
Our historical net tangible book value as of September 30, 2004 was approximately $46.4 million, or $49.73 per share, based on 933,248 shares of common stock outstanding as of September 30, 2004. Historical net tangible book value per share is determined by dividing our total tangible assets less total liabilities by the actual number of outstanding shares of our common stock. Our pro forma net tangible book value as of September 30, 2004 was approximately $46.4 million, or approximately $3.05 per share, based on 15,208,995 shares of common stock outstanding after giving effect to the conversion of all outstanding shares of our convertible preferred stock into common stock upon the closing of this offering. Pro forma net tangible book value per share represents the amount of our total tangible assets less total liabilities, divided by the pro forma number of shares of common stock outstanding before giving effect to this offering.
After giving effect to our sale of 6,800,000 shares of common stock in this offering based on an assumed initial public offering price of $11 per share, less underwriting discounts and commissions and offering expenses payable by us, our pro forma net tangible book value as of September 30, 2004 would have been $5.18 per share. This represents an immediate increase in pro forma net tangible book value per share of $2.12 to existing stockholders and immediate dilution in pro forma net tangible book value of $5.82 per share to new investors purchasing our common stock in the offering at the initial public offering price. Dilution per share to new investors is determined by subtracting pro forma net tangible book value per share after this offering from the assumed initial public offering price per share paid by a new investor. The following table illustrates the per share dilution without giving effect to the over-allotment option granted to the underwriters:
| Assumed initial public offering price per share | $ | 11.00 | |||||||
| Historical net tangible book value per share as of September 30, 2004 | 49.73 | ||||||||
| Decrease per share due to the conversion of all shares of preferred stock | 46.68 | ||||||||
| Pro forma net tangible book value per share as of September 30, 2004 | 3.05 | ||||||||
| Increase per share attributable to this offering | 2.13 | ||||||||
| Pro forma net tangible book value per share after the offering | 5.18 | ||||||||
| Dilution per share to new investors | $ | 5.82 | |||||||
The following table summarizes as of September 30, 2004 the number of shares of our common stock purchased from us, the total cash consideration paid to us and the average price per share paid to us by existing stockholders, to be paid by new investors purchasing shares of our common stock in this offering, before deducting underwriting discounts and commissions and estimated offering expenses payable by us:
| Shares Purchased | Total Consideration | Average | |||||||||||||||||||
| Price Per | |||||||||||||||||||||
| Number | Percent | Amount | Percent | Share | |||||||||||||||||
| Existing stockholders | 15,208,995 | 69.1 | $ | 89,350,000 | 54.4 | $ | 5.87 | ||||||||||||||
| New investors | 6,800,000 | 30.9 | 74,800,000 | 45.6 | $ | 11.00 | |||||||||||||||
| Total | 22,008,995 | 100.0 | $ | 164,150,000 | 100.0 | ||||||||||||||||
The above discussion and tables are based on 933,248 shares of common stock outstanding as of September 30, 2004 and also reflects the automatic conversion of all of our preferred stock into an aggregate of 14,275,747 shares of common stock and excludes:
| • | 2,242,505 shares of common stock issuable upon the exercise of options outstanding as of September 30, 2004, with exercise prices ranging from $1.00 to $10.00 per share and a weighted average exercise price of $1.20 per share; | |
| • | 113,248 additional shares of common stock reserved for future grants under our 2001 Equity Incentive Plan, as of September 30, 2004; and | |
28
Table of Contents
| • | 69,265 shares of common stock issuable upon the exercise of an outstanding warrant that, by its terms, terminates upon completion of this offering. |
To the extent the outstanding options are exercised, you will experience further dilution. In addition, we may choose to raise additional capital due to market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans. To the extent that additional capital is raised through the sale of equity or convertible debt securities, the issuance of these securities could result in further dilution to our stockholders.
29
Table of Contents
SELECTED FINANCIAL DATA
The following selected financial data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” following this section and our financial statements and related notes included in the back of this prospectus. The selected financial data for the period from March 6, 2001, the date of our inception, to December 31, 2001 and for the years ended December 31, 2002 and 2003 and as of December 31, 2002 and 2003 are derived from our audited financial statements included in the back of this prospectus. The selected financial data for the nine months ended September 30, 2003 and 2004 and for the period from March 6, 2001 to September 30, 2004 are derived from our unaudited financial statements included in the back of this prospectus. The unaudited financial statements include, in the opinion of management, all adjustments, consisting only of normal, recurring adjustments, that management considers necessary for a fair statement of the results for those aforementioned periods. The historical results are not necessarily indicative of results to be expected in any future period, and the results for the nine months ended September 30, 2004 should not be considered indicative of results expected for the full fiscal year.
The pro forma net loss per share information is computed using the weighted average number of common shares outstanding, after giving pro forma effect to the conversion of all outstanding shares of our convertible preferred stock into, and the exercise in full of all outstanding warrants for, shares of our common stock effective upon the completion of this offering, as if such conversion had occurred at the date of original issuance.
| Period from | Period from | ||||||||||||||||||||||||
| March 6, 2001 | Nine Months Ended | March 6, 2001 | |||||||||||||||||||||||
| (Inception) to | Years Ended December 31, | September 30, | (Inception) to | ||||||||||||||||||||||
| December 31, | September 30, | ||||||||||||||||||||||||
| 2001 | 2002 | 2003 | 2003 | 2004 | 2004 | ||||||||||||||||||||
| (Unaudited) | (Unaudited) | (Unaudited) | |||||||||||||||||||||||
| (In thousands, except shares and per share data) | |||||||||||||||||||||||||
| Contract revenues | $ | — | $ | 149 | $ | 100 | $ | 100 | $ | — | $ | 249 | |||||||||||||
| Operating expenses: | |||||||||||||||||||||||||
| Research and development | 365 | 11,745 | 12,191 | 10,939 | 10,682 | 34,983 | |||||||||||||||||||
| General and administrative | 1,096 | 3,076 | 3,477 | 2,320 | 2,914 | 10,564 | |||||||||||||||||||
| Acquired in-process research and development | — | 5,716 | — | — | — | 5,716 | |||||||||||||||||||
| Total operating expenses | 1,461 | 20,537 | 15,668 | 13,259 | 13,596 | 51,263 | |||||||||||||||||||
| Income (loss) from operations | (1,461 | ) | (20,388 | ) | (15,568 | ) | (13,159 | ) | (13,596 | ) | (51,014 | ) | |||||||||||||
| Gain (loss) on sale of assets | — | (36 | ) | 103 | 102 | — | 67 | ||||||||||||||||||
| Interest expense | (2 | ) | (4 | ) | (107 | ) | (72 | ) | (24 | ) | (137 | ) | |||||||||||||
| Interest income | 47 | 237 | 86 | 70 | 429 | 799 | |||||||||||||||||||
| Net income (loss) | $ | (1,416 | ) | $ | (20,191 | ) | $ | (15,486 | ) | $ | (13,059 | ) | $ | (13,191 | ) | $ | (50,285 | ) | |||||||
| Basic and diluted net loss per share | $ | (79.27 | ) | $ | (110.36 | ) | $ | (59.75 | ) | $ | (51.03 | ) | $ | (17.14 | ) | ||||||||||
| Shares used to compute basic and diluted net loss per share | 17,866 | 182,949 | 259,182 | 255,909 | 769,582 | ||||||||||||||||||||
| Pro forma basic and diluted net loss per share (unaudited) | $ | (5.23 | ) | $ | (1.01 | ) | |||||||||||||||||||
| Shares used to compute pro forma basic and diluted net loss per share (unaudited) | 2,960,028 | 12,946,630 | |||||||||||||||||||||||
30
Table of Contents
| As of | ||||||||||||||||
| As of December 31, | September 30, | |||||||||||||||
| 2001 | 2002 | 2003 | 2004 | |||||||||||||
| (Unaudited) | ||||||||||||||||
| (In thousands) | ||||||||||||||||
Balance Sheet Data: | ||||||||||||||||
| Cash, cash equivalents and short-term investments | $ | 7,914 | $ | 8,873 | $ | 4,546 | $ | 47,573 | ||||||||
| Total assets | 8,053 | 12,681 | 7,401 | 49,929 | ||||||||||||
| Deficit accumulated during the development stage | (1,416 | ) | (21,607 | ) | (37,093 | ) | (50,285 | ) | ||||||||
| Total stockholders’ equity (net capital deficiency) | 7,708 | 10,917 | (4,368 | ) | 46,412 | |||||||||||
31
Table of Contents
MANAGEMENT’S DISCUSSION AND ANALYSIS
OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion of our financial condition and results of operations in conjunction with the financial statements and the notes to those statements included elsewhere in this prospectus. This discussion contains forward-looking statements that involve risks and uncertainties. As a result of many factors, such as those set forth under the section entitled “Risk Factors” and elsewhere in this prospectus, our actual results may differ materially from those anticipated in these forward-looking statements.
Overview
We are an emerging pharmaceutical company focused on developing and commercializing drugs to treat pain. We have three product candidates that we are developing in four clinical programs and one preclinical program. Two product candidates, ALGRX 4975 and ALGRX 3268, are in clinical development, and one product candidate, ALGRX 1207, is in preclinical development. We commenced operations in March 2001. Since our inception, we have generated significant losses. As of September 30, 2004, we had an accumulated deficit of $50.3 million. We expect to incur substantial and increasing losses for the next several years as we:
| • | continue the development and the preparation for commercialization of ALGRX 4975 and ALGRX 3268, including establishing sales and marketing capabilities; | |
| • | continue the preclinical, and commence the clinical development of ALGRX 1207, which we in-licensed in October 2004; | |
| • | expand our research and development programs; and | |
| • | advance new product candidates into clinical development from our existing research programs. |
We have a limited history of operations. To date, we have funded our operations primarily through the private placement of equity securities. As of September 30, 2004, our only revenue since inception has been $0.2 million derived from a collaborative research program and the one-time upfront payment from an out-license of technology that we do not plan to develop ourselves. We have had no other income since inception other than interest income from short-term investments.
We hold exclusive worldwide rights to commercialize ALGRX 4975, ALGRX 3268 and ALGRX 1207, and, if approved, we intend to market these product candidates ourselves or with a partner in the United States and through strategic collaborations outside of the United States. We currently contract with outside firms to manufacture our product candidates and have no plans to establish internal manufacturing capabilities.
In August 2001, we licensed the intellectual property underlying ALGRX 4975 from James N. Campbell, M.D., Richard A. Meyer, M.S. and Marco Pappagallo, M.D. In consideration for the license, we paid Drs. Campbell and Pappagallo and Mr. Meyer an up-front fee and are obligated to pay additional fees upon achieving certain milestones with respect to ALGRX 4975. We also granted them options which vest based on AlgoRx achieving predetermined milestones. If ALGRX 4975 is commercialized, we will owe Drs. Campbell and Pappagallo and Mr. Meyer royalties on any future sales of ALGRX 4975.
In March 2002, we acquired the powder injection delivery business and a license to certain intellectual property for our product candidate ALGRX 3268 from subsidiaries of PowderJect Pharmaceuticals plc, in a transaction accounted for as an acquisition of assets. The purchase price for this acquisition was approximately $9.9 million. In October 2004, we in-licensed the intellectual property underlying ALGRX 1207 from Bridge Pharma, Inc. In consideration for the license, we paid Bridge Pharma, Inc. an up-front license fee consisting of a cash payment of $1.0 million and 160,000 shares of our common stock. We are also obligated to make additional payments and royalties to Bridge Pharma, Inc. if we achieve certain clinical, regulatory and commercial milestones.
32
Table of Contents
Revenue
We do not expect to generate revenue from product sales or royalties until at least 2007, if at all. We intend to seek to generate revenue from a combination of product sales, up-front fees and milestone payments in connection with collaborative or strategic relationships, and royalties resulting from the license of our intellectual property. We expect that any revenue we generate will fluctuate from quarter to quarter as a result of the nature, timing and amount of milestone payments we may receive from our collaborative or strategic relationships, as well as revenue we may receive upon the sale of our products, to the extent any are successfully commercialized.
Research and Development Expenses
Our research and development expenses consist primarily of:
| • | salaries and related expenses for personnel; | |
| • | costs of facilities and equipment; | |
| • | fees paid to consultants and clinical research organizations in conjunction with independently monitoring our clinical trials and acquiring and evaluating data in conjunction with our clinical trials; | |
| • | fees paid to research organizations in conjunction with nonclinical animal studies; | |
| • | costs of materials used in research and development; | |
| • | upfront and milestone payments under in-licensing agreements; | |
| • | consulting fees paid to third parties; and | |
| • | depreciation of capital resources used to develop our products. |
We expense both internal and external research and development costs as incurred. We expect our research and development expense to increase as we continue to develop our product candidates.
We use our employee and infrastructure resources across several projects, and some of our costs are not attributable to an individually-named project but rather are directed across these research projects. Accordingly, we make certain allocations for these internal research and development costs on a project-by-project basis. As a result, we cannot state precisely the total costs incurred for each of our clinical and preclinical projects on a project-by-project basis. The following table shows, from inception through September 30, 2004, the total costs associated with ALGRX 4975 and ALGRX 3268:
| Period from | Period from | ||||||||||||||||||||||||
| March 6, 2001 | Year Ended | Nine Months Ended | March 6, 2001 | ||||||||||||||||||||||
| (Inception) to | December 31, | September 30, | (Inception) | ||||||||||||||||||||||
| December 31, | September 30, | ||||||||||||||||||||||||
| 2001 | 2002 | 2003 | 2003 | 2004 | 2004 | ||||||||||||||||||||
| (In thousands) | |||||||||||||||||||||||||
| ALGRX 4975 | $ | 365 | $ | 4,047 | $ | 4,697 | $ | 4,268 | $ | 5,446 | $ | 14,555 | |||||||||||||
| ALGRX 3268 | — | 6,877 | 7,129 | 6,308 | 5,008 | 19,014 | |||||||||||||||||||
| Other R&D | — | 821 | 365 | 363 | 228 | 1,414 | |||||||||||||||||||
| Total | $ | 365 | $ | 11,745 | $ | 12,191 | $ | 10,939 | $ | 10,682 | $ | 34,983 | |||||||||||||
We expect that a large percentage of our research and development expenses in the future will be incurred in support of our current and future preclinical and clinical development programs. These expenditures are subject to numerous uncertainties in timing and cost to completion. We test our product candidates in numerous preclinical studies for toxicology, safety and efficacy. We then conduct early stage clinical trials for each drug candidate. If we are not able to engage a collaboration partner prior to the commencement of later stage clinical trials, we may fund these trials ourselves. As we obtain results from clinical trials, we may elect to discontinue or delay clinical trials for certain product candidates or
33
Table of Contents
programs in order to focus our resources on more promising product candidates or programs. Completion of clinical trials by us or our future collaborators may take several years or more, but the length of time generally varies according to the type, complexity, novelty and intended use of a drug candidate. The cost of clinical trials may vary significantly over the life of a project as a result of differences arising during clinical development, including, among others:
| • | the number of sites included in the trials; | |
| • | the length of time required to enroll suitable patient subjects; | |
| • | the number of patients that participate in the trials; | |
| • | the number of doses that patients receive; | |
| • | the duration of patient follow-up; | |
| • | the phase of development the product is in; and | |
| • | the efficacy and safety profile of the product. |
Our expenses related to clinical trials are based on estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and clinical research organizations that conduct and manage clinical trials on our behalf. The financial terms of these agreements are subject to negotiation and vary from contract to contract and may result in uneven payment flows. Generally, these agreements set forth the scope of work to be performed at a fixed fee or unit price. Payments under the contracts depend on factors such as the successful enrollment of patients or the completion of clinical trial milestones. Expenses related to clinical trials generally are accrued based on contracted amounts applied to the level of patient enrollment and activity according to the protocol. If timelines or contracts are modified based upon changes in the clinical trial protocol or scope of work to be performed, we modify our estimates of accrued expenses accordingly on a prospective basis.
None of our drug candidates has received FDA or foreign regulatory marketing approval. In order to grant marketing approval, the FDA or foreign regulatory agencies must conclude that our and our collaborators’ clinical data establishes the safety and efficacy of our drug candidates. Furthermore, our strategy includes entering into collaborations with third parties to participate in the development and commercialization of our products. In the event that third parties have control over the preclinical development or clinical trial process for a product candidate, the estimated completion date would largely be under control of that third party rather than under our control. We cannot forecast with any degree of certainty which of our drug candidates will be subject to future collaborations or how such arrangements would affect our development plan or capital requirements.
As a result of the uncertainties discussed above, we are unable to determine the duration and completion costs of our research and development projects or when and to what extent we will receive cash inflows from the commercialization and sale of a product.
General and Administrative Expenses
General and administrative expenses consist primarily of compensation for employees in executive and operational functions, including finance, business development and corporate development. Other significant costs include facilities costs and professional fees for accounting and legal services, including legal services associated with obtaining and maintaining patents. After completion of the offering made by this prospectus, we anticipate incurring increases in general and administrative expenses, such as increased costs for insurance and investor relations associated with operating as a public company. These increases will also likely include the hiring of additional personnel and payment to outside consultants, lawyers and accountants. We also expect to incur significant costs to comply with the requirements set forth under the new regulations for corporate governance and internal controls.
34
Table of Contents
Critical Accounting Policies
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses and related disclosure of contingent assets and liabilities. We review our estimates on an ongoing basis. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions. While our significant accounting policies are described in more detail in the notes to our financial statements included in this prospectus, we believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our financial statements.
| Stock-Based Compensation |
Stock-based compensation represents the difference between the exercise price of options granted to employees and directors and the fair value of our common stock on the date of grant for financial statement purposes in accordance with Accounting Principles Board Opinion No. 25 and its related interpretations. We recognize this compensation charge over the vesting periods of the shares purchasable upon exercise of options. Should our assumptions of fair value change, the amount recorded as intrinsic value will increase or decrease in the future.
We recorded deferred stock-based compensation related to stock options granted to employees and directors of $0.2 million and related amortization of $13,000 during 2003, and deferred stock-based compensation of $8.7 million and related amortization of $0.4 million during the nine-month period September 30, 2004. We expect to amortize approximately $8.5 million of deferred stock-based compensation with respect to stock options granted through September 30, 2004 in future periods, including $0.5 million during the fourth quarter of 2004, $2.2 million during 2005, $2.2 million during 2006, $2.2 million during 2007 and $1.4 million during 2008.
| Acquired In-Process Research and Development |
Acquired in-process research and development relates primarily to in-licensed technology, intellectual property and know-how. We evaluate the stage of development of acquired projects, taking into account the level of effort, time and estimated cost associated with further developing the in-process technology and producing a commercial product. The nature of the remaining efforts for completion of acquired in-process research and development projects generally include completion of clinical trials, completion of manufacturing validation, interpretation of clinical and preclinical data and obtaining marketing approval from the FDA and other regulatory bodies, the cost, length and success of which are extremely difficult to determine. Numerous risks and uncertainties exist with timely completion of development projects, including clinical trial results, manufacturing process development results, and ongoing feedback from regulatory authorities, including obtaining marketing approval. In addition, acquired products under development may never be successfully commercialized due to the uncertainties associated with the pricing of new pharmaceuticals, the cost to produce these products in a commercial setting, changes in the reimbursement environment, or the introduction of new competitive products. As a result of the uncertainties noted above, we expense such acquired in-process research and development projects when incurred.
| Revenue Recognition |
Our revenue recognition policies are in accordance with Securities and Exchange Commission Staff Accounting Bulletins No. 101 and No. 104, or SAB 101 and 104, and EITF 00-21,Revenue Recognition in Financial Statements, which provides guidance on revenue recognition in financial statements and is based on the interpretations and practices developed by the SEC. SAB 101 requires that four basic criteria be met before revenue can be recognized: (1) persuasive evidence exists of an arrangement; (2) delivery
35
Table of Contents
has occurred or services have been rendered; (3) the fee is fixed and determinable; and (4) collectibility is reasonably assured. Determination of criteria (3) and (4) are based on our management’s judgments regarding the fixed nature of the fees charged for services rendered and products delivered and the collectibility of those fees. Accordingly, revenues from licensing agreements are recognized based on the performance requirements of the agreement. If we have an ongoing involvement or performance obligation, non-refundable up-front fees are generally recorded as deferred revenue in the balance sheet and amortized into license fees in the statement of operations over the term of the performance obligation. If we have no ongoing involvement or performance obligation, non-refundable up-front fees are generally recorded as revenue in the period in which the rights are transferred. Should changes in conditions cause our management to determine that these criteria are not met for certain future transactions, revenue recognition for those transactions will be delayed and our revenues could be adversely affected.
Results of Operations
| Comparison of the Nine Months Ended September 30, 2004 and 2003 |
Revenue. For the nine months ended September 30, 2004, we did not recognize any revenues. We recognized $100,000 of revenue during the nine months ended September 30, 2003. This revenue was derived from the licensing of technology acquired as part of the PowderJect acquisition that we did not intend to develop ourselves. Under the terms of the agreement, the licensee paid us a one-time fee of $100,000 upon the execution of the agreement, and the licensee is obligated to pay royalties to us equal to a percentage of sales. To date, no royalty payments have been paid nor do we expect to receive any in the near future. We do not expect any future royalty payments under the agreement to be material.
Research and Development Expenses. Research and development expenses were $10.7 million for the nine months ended September 30, 2004, compared with $10.9 million for the nine months ended September 30, 2003. The $0.2 million decrease in our research and development expenses reflects an increase of $1.2 million for ALGRX 4975 offset by a decrease of $1.3 million for the ALGRX 3268 program, a decrease of $0.3 million in other program expenses, and an increase of $0.2 million for amortization of deferred compensation. The $1.2 million increase in costs for the ALGRX 4975 program for the first nine months of 2004 was primarily attributable to an increase of $1.4 million in clinical costs, salary and other costs necessary to support the increased clinical activity as we completed two phase II clinical trials begun in 2003, offset by a decrease of $0.2 million in drug safety costs that were incurred through 2003 and early 2004 to support the IND for ALGRX 4975 and the initiation and planning of five other clinical trials for ALGRX 4975. The $1.3 million decrease in costs for the ALGRX 3268 program for the first nine months of 2004 was primarily attributable to decreased personnel costs of $1.7 million and decreased clinical costs of $0.6 million, offset by an increase of $1.0 million in manufacturing, quality assurance and other costs. The decreased personnel costs were due to the company’s reduction of force of 28 employees in March 2003, all of whom were involved with the ALGRX 3268 program. The increase in manufacturing, quality assurance and other costs of $1.0 million was due to a variety of projects necessary to prepare for phase III clinical trials and potential commercialization of the product candidate.
In March 2003, we announced a reorganization plan intended to reduce operating costs at our Fremont, California facility in response to a clinical hold placed on ALGRX 3268 by the FDA. The primary focus of the workforce at the Fremont facility was ALGRX 3268. We reduced our staff by 28 employees though no research projects were discontinued. The 28 employees represented approximately 82% of total employees at the Fremont facility and 61% of total AlgoRx employees overall as of the date of reorganization. During the clinical hold period, which lasted three months, we began an outsourcing program that has since replaced all of the necessary functions with outside vendors and consultants. The total restructuring cost of $1.1 million was expensed during 2003. Severance and related costs were $0.8 million. The vesting of certain options held by the terminated employees was accelerated in connection with this reorganization, and we recorded a charge of $66,240 related to this acceleration of vesting. The reorganization resulted in an impairment of the assembled workforce intangible asset that had been recorded as a result of the March 2002 acquisition from PowderJect Pharmaceuticals plc. This
36
Table of Contents
impairment of $0.2 million was recorded as accelerated amortization expense during 2003 and was classified under research and development expenses for the nine months ended September 30, 2003.
General and Administrative Expenses. General and administrative expenses were $2.9 million for the nine months ended September 30, 2004, compared with $2.3 million for the nine months ended September 30, 2003. The $0.6 million increase was primarily attributable to the increase in administration, salary and facility costs.
Interest Expense. Interest expense was $24,000 for the nine months ended September 30, 2004, compared with $72,000 for the nine months ended September 30, 2003. The $48,000 decrease was due to the shorter period of time in 2004 during which the $9.8 million of convertible notes were outstanding.
Interest Income. Interest income was $0.4 million for the nine months ended September 30, 2004, compared with $70,000 for the nine months ended September 30, 2003. The increase was due to higher average cash and investment balances.
| Comparison of the Years Ended December 31, 2003 and 2002 and the Period From March 6, 2001 (Inception) Through December 31, 2001 |
Research and Development Expenses. Research and development expenses were $12.2 million in 2003, $11.7 million in 2002 and $0.4 million in 2001. The 2001 period reflects the period from our inception on March 6, 2001 through December 31, 2001. The expenses are associated with research and development programs that we conducted during these periods in the areas of pain management.
The $0.5 million increase in our research and development expenses from 2002 to 2003 reflects an increase of $0.7 million for ALGRX 4975 and an increase of $0.3 million for ALGRX 3268, offset by a decrease of $0.5 million in other program expenses. The increase in costs associated with ALGRX 4975 was primarily attributable to an increase in clinical activity in which we completed a phase I safety trial and initiated two phase II clinical trials. In 2002, we initiated our first clinical trial in the second half of the year and only incurred a small amount of costs. The increase of costs associated with ALGRX 3268 in 2003 was primarily attributable to higher salary costs, severance costs for 28 employees at our California facility in March 2003, increased clinical activity, and an increase in facility operating costs, together totaling $0.9 million, offset by a decrease of $0.6 million in drug safety studies.
The $11.3 million increase in our research and development expenses from 2001 to 2002 was primarily attributable to a significant increase in personnel and external spending following the in-licensing of ALGRX 4975 in the third quarter of 2001 and the purchase and licensing of technology relating to ALGRX 3268 in the first quarter of 2002. This $11.3 million increase in 2002 resulted from an increase of $3.6 million in the costs of our ALGRX 4975 program related to the preparation of the IND application and the start of our first clinical trial, an increase of $6.9 million due to the initiation of our efforts in the ALGRX 3268 program which we acquired at the end of the first quarter of 2002, and additional increases of $0.8 million related to other uses of our in-licensed PowderJect device. The ALGRX 3268 program costs of $6.9 million in 2002 were primarily attributable to $2.4 million in salary costs and related expenses, $1.5 million related to drug safety studies, $1.0 million in clinical studies and $1.9 million in facility and other operating costs. The other costs of $0.8 million were $0.4 million for salary and related expenses and $0.4 million for drug safety costs.
General and Administrative Expenses. General and administrative expenses were $3.5 million in 2003, $3.1 million in 2002 and $1.1 million in 2001. The $0.4 million increase in expenses from 2002 to 2003 was primarily attributable to termination costs and business development costs partially offset by lower personnel costs. The $2.0 million increase in expenses from 2001 to 2002 was primarily attributable to increased staffing and other personnel costs associated with supporting our programs, particularly due to the purchase and license of ALGRX 3268 in the first quarter of 2002.
Acquired In-Process Research and Development. Generally, we determine the amount of any in-process research and development expense based on an analysis using risk-adjusted cash flows expected to be generated by products that may result from the in-process technologies acquired. We review the
37
Table of Contents
acquired project rights to determine the stage of their development, the probability of demonstrating in clinical trials sufficient safety and efficacy for FDA approval and product risk factors inherent in the drug development process. The product-specific risk factors include the type of drug under development, likelihood of regulatory approval, manufacturing process capability, scientific rationale, preclinical safety and efficacy data, target product profile and development plans. The valuation used to estimate in-process research and development expense requires us to use significant estimates and assumptions that, if changed, may result in a different valuation for in-process technology.
In connection with our license agreement and acquisition of assets from subsidiaries of PowderJect Pharmaceuticals plc, we reviewed the rights and assets that we acquired to determine the stage of development, the probability of demonstrating in our clinical trials sufficient safety and efficacy for FDA approval and the technical milestones needed to be reached before commercialization would be possible. We determined, as of the acquisition date, there was significant risk that our clinical trials may not demonstrate the levels of safety and efficacy needed for FDA approval and that the project had significant milestones to reach before commercialization. We also determined that all of the projects had no alternative future uses if they were not successful. Accordingly, we classified a portion of the ALGRX 3268 and associated projects as in-process research and development and expensed $5.7 million immediately. The remaining purchase price was allocated to tangible net assets of $3.7 million and acquired workforce of $0.5 million. We valued the assets acquired based on the value of the consideration that PowderJect Pharmaceuticals plc and three individuals received in this transaction, which included 6,166,154 shares of Series B convertible preferred stock and 152,615 shares of common stock. The Series B preferred stock was valued at $8.0 million based on the arm’s length Series B preferred stock financing completed concurrently with the acquisition. The common stock issued in the acquisition was valued at $0.2 million using the estimated fair value used for option grants to our employees. The purchase price for the acquisition also included cash consideration of $0.8 million for a minority interest’s share in the acquired company and assumed liabilities of $0.2 million. The total purchase price for this acquisition was $9.9 million, including transaction costs of $0.7 million.
Interest Expense. Interest expense was $0.1 million in 2003, $4,000 in 2002 and $2,000 in 2001. The increase in interest expense from 2002 to 2003 was due to non-cash accrued interest expense for the $9.8 million of convertible notes.
Interest Income. Interest income was $86,000 in 2003, $0.2 million in 2002 and $47,000 in 2001. The decrease in interest income from 2002 to 2003 was primarily attributable to lower average cash balances and the lower interest rate environment. The increase in interest income from 2001 to 2002 was primarily attributable to the higher level of cash available during 2002 as proceeds from the sale of our Series B preferred stock were received in March 2002.
Liquidity and Capital Resources
To date, we have funded our operations primarily through the sale of equity securities. As of September 30, 2004, we had received approximately $87.8 million of cash proceeds from the sale of equity securities, including the notes that were converted into preferred stock, net of offering expenses.
As of September 30, 2004, we had $47.6 million in cash, cash equivalents and marketable securities as compared to $4.5 million as of December 31, 2003, an increase of $43.1 million. This increase resulted primarily from the issuance of $65 million of Series C preferred stock for $53.8 million of cash proceeds, net of $1.4 million of related transaction costs and conversion into Series C preferred stock of $9.8 million of convertible promissory notes that were issued in April 2003.
Net cash used in operating activities amounted to $10.7 million for the nine months ended September 30, 2004 and $11.4 million for the nine months ended September 30, 2003. The primary use of cash was to fund our net losses for these periods, adjusted for non-cash expenses, including depreciation and amortization of $0.8 million for the nine months ended September 30, 2004 and $1.1 million for the nine months ended September 30, 2003, non-cash stock based compensation of $0.5 million for the nine months ended September 30, 2004 and $82,000 for the nine months ended September 30, 2003 and an
38
Table of Contents
increase in net operating assets and liabilities of $1.2 million for the nine months ended September 30, 2004 and $0.6 million for the nine months ended September 30, 2003. Our losses were primarily derived from our clinical development activities. During the nine months ended September 30, 2004, these activities primarily consisted of clinical trials for ALGRX 4975 and ALGRX 3268, the preparation for our end of Phase II meeting with the FDA in the fourth quarter of 2004 for ALGRX 3268 and the initiation of our Phase III program for ALGRX 3268. During the nine months ended September 30, 2003, these activities primarily consisted of clinical trials for ALGRX 4975, outsourcing activities for ALGRX 3268 resulting from the reduction in force at our facility in Fremont, California in the first quarter of 2003 and clinical trials for ALGRX 3268.
Net cash used in investing activities, excluding net proceeds from purchases, sales and maturities of short-term investments, for the nine months ended September 30, 2004 amounted to $47,000 and $0.3 million for the nine months ended September 30, 2003. We paid approximately $47,000 during the nine months ended September 30, 2004 for leasehold improvements and capital equipment necessary to support our anticipated future growth, net of proceeds from the disposal of equipment.
Net cash provided by financing activities amounted to $53.8 million for the nine months ended September 30, 2004, and $9.8 million for the nine months ended September 30, 2003. These activities consisted primarily of private sales of our Series C preferred stock and convertible promissory notes convertible into our Series C preferred stock, which were offset in part by stock issuance costs.
Net cash used in operating activities was $14.0 million in 2003. The primary use of cash was to fund our net loss, adjusted for non-cash expenses including depreciation and amortization of $1.3 million in 2003, non-cash stock-based compensation of $88,000 in 2003 and non-cash interest expense of $0.1 million in 2003. In 2003, the net cash used in operating activities resulted primarily from the conduct of clinical trials for ALGRX 4975, outsourcing activities for ALGRX 3268 resulting from the reduction in force at our facility in Fremont, California in the first quarter of 2003 and the conduct of clinical trials for ALGRX 3268.
Net cash used in investing activities was $0.2 million in 2003. These activities consisted primarily of purchases, offset by sales, of property and equipment.
Net cash provided by financing activities was $9.8 million in 2003. This amount was from the sale of convertible promissory notes to institutional investors, the proceeds from which were offset by issuance costs.
We had a deferred tax asset of $14.4 million as of December 31, 2003 offset by a valuation allowance of $14.4 million. The valuation allowance is based on the high level of uncertainty regarding our ability to generate net income in the future and utilize the deferred tax asset.
The following table summarizes our long-term contractual obligations as of September 30, 2004:
| Remainder of 2004 | 2005 | 2006 | 2007 | 2008 | Thereafter | |||||||||||||||||||
| (In thousands) | ||||||||||||||||||||||||
| Operating leases | $ | 222 | $ | 299 | $ | 246 | $ | 246 | $ | 246 | $ | 123 | ||||||||||||
| R & D obligations | 0 | 1,500 | 1,000 | 1,000 | 500 | 0 | ||||||||||||||||||
| Total | $ | 222 | $ | 1,799 | $ | 1,246 | $ | 1,246 | $ | 746 | $ | 123 | ||||||||||||
We have four license agreements, two of which relate to ALGRX 4975 and one of which relates to ALGRX 3268, our two product candidates in clinical trials. The other license agreement relates to our preclinical product candidate, ALGRX 1207 that we in-licensed in October 2004, subsequent to the date of the information reflected in the table above. Under these four license agreements, we could be required to pay up to a total of $6.7 million in milestone payments through product approval, in addition to sales milestones and royalties on commercial sales if any occur. Under our license with PowderMed Limited, if we use the licensed technology to develop cytokines, which are proteins that regulate blood cells, we are obligated to spend $1.0 million on research and development associated with the licensed technology during the period July 7, 2003 to July 7, 2005 and to spend an additional $1.0 million on
39
Table of Contents
research and development for every 12 month period from July 7, 2005 to July 7, 2008. To date, we have chosen not to use the licensed technology to develop cytokines. We have no material commitments for capital expenditures. Under our license agreement with Bridge Pharma, Inc., we are obligated to spend a minimum of $1 million for product development in each calendar year during the term of the agreement commencing in 2005 and ending on the first commercial sale of a product using the licensed technology. We are also responsible under the Bridge Pharma agreement for paying expenses associated with any patent prosecution and maintenance relating to the underlying technology and for certain costs associated with the research, development, regulatory filings and approvals and commercialization of products using the underlying technology.
Our future capital uses and requirements depend on numerous forward-looking factors. These factors include, but are not limited to, the following:
| • | the progress of preclinical development and laboratory testing and clinical trials; | |
| • | the time and costs involved in obtaining regulatory approvals; | |
| • | delays that may be caused by evolving requirements of regulatory agencies; | |
| • | the number of product candidates we pursue; | |
| • | the costs involved in filing and prosecuting patent applications and enforcing or defending patent claims; | |
| • | our plans to establish sales, marketing and/or manufacturing capabilities; | |
| • | our ability to establish, enforce and maintain selected strategic alliances and activities required for product commercialization; | |
| • | the acquisition of technologies, products and other business opportunities that require financial commitments; and | |
| • | our revenues, if any, from successful development and commercialization of our products. |
We believe that our existing cash and cash equivalents, together with proceeds from this offering, will be sufficient to meet our projected operating requirements for at least the next 24 months.
Until we can generate significant cash from our operations, we expect to continue to fund our operations with existing cash resources. If we need to raise funds in the future, we may be required to raise those funds through public or private financings, strategic relationships or other arrangements.
As of September 30, 2003 and 2004 and December 31, 2001, 2002 and 2003, we did not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we do not engage in trading activities involving non-exchange traded contracts. Therefore, we are not materially exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in these relationships.
Recent Accounting Pronouncements
In January 2003, the FASB issued FIN No. 46,Consolidation of Variable Interest Entities. This interpretation requires existing unconsolidated variable interest entities to be consolidated by their primary beneficiaries if the entities do not effectively disperse risks among parties involved. It explains how to identify variable interest entities and how an enterprise assesses its interest in a variable interest entity to decide whether to consolidate that entity. As amended, this interpretation applies in the first fiscal year or interim period beginning after December 15, 2003, to variable interest entities in which an enterprise holds a variable interest that it acquired before February 1, 2003. The adoption of FIN No. 46 did not have a material impact on our financial position, results of operations or cash flows.
40
Table of Contents
In May 2003, the FASB issued Statement of Financial Accounting Standards No. 150,Accounting for Certain Financial Instruments with Characteristics of both Liabilities and Equity, or SFAS 150. This statement establishes standards for classifying and measuring as liabilities certain financial instruments that embody obligations of the issuer and have characteristics of both liabilities and equity. The scope of this pronouncement includes mandatorily redeemable equity instruments. Application to pre-existing instruments should be recognized as the cumulative effect of a change in accounting principle. Application by retroactive restatement is precluded. Exceptions to the transition requirements were provided for mandatorily redeemable instruments of non-public companies. Subsequent to the issuance of SFAS 150, the FASB issued FSP 150-3, which further deferred the transition requirements for nonpublic companies that also are not SEC registrants for instruments that are mandatorily redeemable on fixed dates that are for fixed amounts. The classification, measurement, and disclosure provisions of SFAS 150 are effective for fiscal periods beginning after December 15, 2004. The adoption of this statement is not expected to impact the classification of the Company’s convertible preferred stock, which will convert into common stock on the completion of this offering.
In December 2004, the FASB issued FASB Statement No. 123,Share-Based Payment. This statement, referred to as Statement 123R, was an amendment of FASB Statements No. 123 and 95. The change in accounting would replace existing requirements under FAS 123,Accounting for Stock-Based Compensation, and APB Opinion No. 25,Accounting for Stock Issued to Employees.
The statement covers a wide range of equity-based compensation arrangements. Under this FASB statement, all forms of share-based payments to employees, including employee stock options, would be treated the same as other forms of compensation by recognizing the related cost in the income statement. The expense of the award would generally be measured at fair value at the grant date.
In October 2004, the FASB concluded that Statement 123R would be effective for public companies for interim or annual periods beginning after June 15, 2005 except small business issuers as defined in SEC Regulation S-B. Retroactive application of the requirements of Statement 123, not Statement 123R, to the beginning of the fiscal year that includes the effective date would be permitted but not required. Early adoption of Statement 123R is encouraged.
A calendar-year company therefore would be required to apply Statement 123R beginning July 1, 2005, and could choose to apply Statement 123R retroactively from January 1, 2005 to June 30, 2005. The cumulative effect of adoption, if any, would be measured and recognized on July 1, 2005. Further, we could choose to early adopt the proposed statement at the beginning of our first quarter ended March 31, 2005, or even in the fourth quarter of 2004 if the FASB issues the final statement prior to the issuance of those financial statements.
Quantitative and Qualitative Disclosure About Market Risk
Our primary exposure to market risk is interest income sensitivity, which is affected by changes in the general level of U.S. interest rates, particularly because the majority of our investments are in short-term marketable securities. Due to the nature of our short-term investments, we believe that we are not subject to any material market risk exposure. We do not have any foreign currency or other derivative financial instruments.
41
Table of Contents
BUSINESS
Overview
We are an emerging pharmaceutical company focused on developing and commercializing a diversified portfolio of pharmaceutical product candidates to address pain, a large and under-served market. Our pipeline consists of two clinical product candidates, ALGRX 4975 and ALGRX 3268, and one preclinical product candidate, ALGRX 1207. ALGRX 4975 (capsaicin for injection and capsaicin gel for intraoperative use) is being developed as a long lasting, site-specific treatment for severe or intractable pain in three programs and is currently being evaluated in five phase II clinical trials. ALGRX 3268 (PowderJect® Dermal Lidocaine) is a lidocaine powder delivered into the skin by means of our proprietary needle-free dispenser. We recently initiated two phase III clinical trials for ALGRX 3268. We are also developing ALGRX 1207, a new molecular entity, in preclinical trials as a topical local anesthetic for use in a variety of pain conditions.
We are developing ALGRX 4975 in three programs, which address site-specific, severe or intractable pain in acute or chronic settings:
| • | for use during surgical procedures to treat patients who are at risk for developing severe postsurgical pain; | |
| • | to treat patients with post-trauma neuropathic pain; and | |
| • | to treat patients with chronic pain associated with musculoskeletal diseases. |
To date, we have tested ALGRX 4975, of which the active ingredient is capsaicin, using an injectable liquid. We will continue using this liquid formulation in the latter two programs, but for the postsurgical pain program, we plan to transition to a gel formulation, which would provide greater ease of use during surgical procedures.
We have evaluated ALGRX 4975 in one phase I clinical trial and three phase II clinical trials that have yielded favorable safety and efficacy data. We have six phase II clinical trials underway to further evaluate ALGRX 4975. We recently completed a 182-patient phase II clinical trial for severe postsurgical pain. We plan to begin additional clinical trials in several indications in early 2005.
Our most advanced product candidate, ALGRX 3268, is a powder formulation of the local anesthetic lidocaine delivered into the skin using our proprietary needle-free dispenser. We are evaluating the use of ALGRX 3268 to reduce the pain experienced by patients from needle-sticks in procedures where blood is drawn or intravenous lines are inserted. We recently commenced two phase III clinical trials for ALGRX 3268 with a goal of submitting a new drug application, or NDA, with the FDA for ALGRX 3268 by the end of 2005.
ALGRX 1207 is a new molecular entity that we in-licensed in October 2004 and are developing in preclinical trials as a topical local anesthetic. We intend to develop ALGRX 1207 as a therapy for chronic pain stemming from skin diseases and to reduce the pain associated with surgical procedures on the skin. We plan to file an investigational new drug, or IND, application covering ALGRX 1207 with the FDA in the second half of 2005.
Our goal is to commercialize drugs for the treatment of acute and chronic pain, which we believe are poorly served by existing therapies. We believe that our understanding of the mechanisms of pain combined with our management’s clinical development expertise in pain therapeutics will continue to enable us to identify, in-license or acquire, and develop promising potential products for treating painful conditions that are not adequately treated with existing drugs or other therapies.
Pain Description and Classification
Pain is an unpleasant sensation that occurs as a result of injury to the body or as a manifestation of a diseased state. Pain can be classified in many ways. For example, pain can be classified based on its
42
Table of Contents
duration (acute or chronic), its underlying cause (nociceptive or neuropathic) or its severity (mild, moderate or severe).
Duration. Pain can be classified as either acute or chronic depending on how long the pain lasts. Acute pain is brief in nature. Key features of acute pain are that it is generally experienced in proportion to the degree of injury or damage to the body and that it resolves or subsides as the injury heals. Examples of acute pain include pain resulting from a broken bone, a surgical procedure, or accidental trauma. Acute pain may serve a protective function by alerting an individual to tissue damage and by causing the individual to alter activity to prevent additional injury. By contrast, chronic pain is pain extending 3 to 6 months beyond onset or beyond the expected period of healing and may have undetermined causes or may persist inexplicably after acute pain should have subsided. Importantly, chronic pain can be accompanied by irritability, social withdrawal, a depressed mood, disruption of work and social relationships and symptoms such as changes in sleep and appetite. Examples of chronic pain include pain associated with cancer or arthritis.
Underlying cause. Pain can be classified as either pain that results directly from local tissue injury, which is referred to as nociceptive pain, or pain that follows nerve injury, which is referred to as neuropathic pain. Nociceptive pain results from direct tissue damage. Key features of nociceptive pain are that it can be experienced as a sharp, dull or aching pain. Examples of nociceptive pain include pain from surgical incisions, bone pain from fractures or metastatic cancer and pain from joint diseases such as osteoarthritis and rheumatoid arthritis.
Neuropathic pain occurs as a result of damage to, or dysfunction of, the nervous system. Neuropathic pain is frequently described as burning, tingling or having an electrical shock-like feeling. Another key feature of this type of pain is its occurrence upon stimulation that otherwise would not be expected to cause pain. For example, a condition called trigeminal neuralgia may cause patients to feel extreme pain upon a light touch on the cheek. Examples of neuropathic pain include the pain resulting from diabetes and HIV infection, and postherpetic neuralgia, which is a painful skin condition caused by the chicken pox virus long after the initial infection has healed, in many cases years later. Neuropathic pain frequently coexists with or follows nociceptive pain, as for example when a patient that has had a surgical procedure continues to experience pain long after the wound has healed.
Severity. Pain can be classified into three categories of severity: mild, moderate and severe. It is important to determine a patient’s particular level of pain since doing so will influence the type of drugs or other treatments prescribed by physicians and other health care providers. Because there are no clear differentiating factors among the three categories of pain severity, health care providers frequently determine severity by gauging the extent to which pain interferes with a patient’s daily function or by relying on a patient’s own opinion of the severity of his or her pain. In clinical trials, for example, the severity of pain in patients is sometimes measured by patients on a numerical scale with ratings of 1 to 4 corresponding to mild pain, 4 to 6 corresponding to moderate pain, and 6 to 10 corresponding to severe pain. Examples of mild pain include headaches or joint pain, and examples of moderate pain include the types of pain often associated with minor surgery or some forms of arthritis. Patients experiencing severe pain often suffer from a serious underlying illness, such as AIDS or cancer. Severe pain can also result from major surgery, nerve damage, advanced arthritis or cancer.
Pain Management Market
Pain is a worldwide problem with serious health and economic consequences. The medical effort to treat pain, known as pain management, addresses a large and under-served market. Global Industry Analysts, Inc. estimates that the worldwide prescription market for pain drugs totaled over $28 billion in 2003. IMS Health estimates that nearly $18 billion was spent in 2003 on prescription pain drugs in the United States. In the United States:
| • | medical economists estimate that the economic impact of pain is approximately $100 billion annually. Pain in the hospital is associated with increased length of stay, longer recovery times and poorer patient outcomes, all of which have health care quality and cost implications; |
43
Table of Contents
| • | approximately 25 million Americans experience acute pain each year due to injury or surgery, according to the American Pain Society; and | |
| • | approximately 48 million Americans suffer chronic pain, according to the National Pain Survey published in 1999 by Ortho-McNeil Pharmaceutical, Inc. |
According to a Global Strategic Business Report published in 2004 by Global Industry Analysts, Inc., the prescription pain management market is anticipated to grow at a compounded annual growth rate of 9% through 2010 due to a number of factors, including:
| • | a rapidly aging population with an increasing need and desire to address pain-related ailments; | |
| • | longer survival times for patients with painful chronic conditions, such as cancer and AIDS; | |
| • | patients’ increased demand for effective pain relief; and | |
| • | increasing recognition of the therapeutic and economic benefits of effective pain management by physicians, other health care providers and payors. |
Analgesic Drugs
Drugs that treat pain are referred to as analgesics, and the type of analgesic selected for treatment depends principally upon the severity of the pain. For mild pain, weak analgesics such as acetaminophen or nonsteroidal antiinflammatory drugs, or NSAIDs, such as ibuprofen are used. For moderate pain, NSAIDs, weak opioids such as codeine or short-acting formulations of strong opioids may be used. Severe pain requires strong opioids such as morphine, oxycodone, hydrocodone or fentanyl.
The table below sets forth selected major classes of analgesics, examples of product brands within each class and the corresponding prescription revenues in 2003 for each class as prescribed to treat pain, as reported by IMS Health:
| Class | Example | 2003 US Revenues | ||||
| (millions) | ||||||
| NSAIDs (including COX-2) | Celebrex®, Bextra®, diclofenac | $ | 6,271 | |||
| Opioids (all delivery methods) | OxyContin®, Percocet®, Duragesic®, injectable morphine, Ultram® | $ | 5,037 | |||
| Antiepileptics (prescribed for pain) | Neurontin® | $ | 928 | |||
| Local anesthetics (injectable and topical) | lidocaine | $ | 503 | |||
Source: IMS Health, IMS National Sales Perspectives™; Retail and Provider, December 2003.
Shortcomings of Current Pain Management
Despite widespread clinical use of drugs for pain, pain management remains less than optimal due to a variety of factors, including:
| • | Insufficient efficacy. Opioids, the current standard of care for severe nociceptive pain, reduce pain less than 50% in a majority of situations. Neuropathic pain is difficult to treat with existing analgesics because of the differing types of nerves and organs involved in, and types of injuries causing, this kind of pain. Neuropathic pain does not respond to treatment with NSAIDs and responds poorly to treatment with opioids at doses that do not impair the ability of patients to live reasonably active lifestyles. | |
| • | Lack of site specificity. Most analgesics, including opioids and NSAIDs, are given orally or by intravenous infusion and thereby subject the patient to high circulating concentrations of drug, even though most types of pain are experienced in discrete parts of the body. Opioids must be given by mouth or infusion because they provide pain relief by acting on nerves all over the body: in the spinal cord, in the brain and at the site of injury. As a consequence, opioids do not provide site- |
44
Table of Contents
| specific pain relief because their action is not targeted specifically to the area of the body that is experiencing pain. Moreover, circulating drugs cause side effects at parts of the body unrelated to the perception of pain. Although there are currently means of delivering site-specific analgesia, such as by injection of short-acting anesthetics into joints such as the ankle or knee, these techniques are reserved to provide relatively short-term anesthesia prior to surgery and are not appropriate for long-term pain relief. | ||
| • | Occurrence of side effects. NSAIDs may cause gastrointestinal ulcers, and between 10,000 and 20,000 patients die each year from gastrointestinal bleeding believed to be related to the use of NSAIDs. Use of opioids is associated with nausea and vomiting in many patients. High-dose opioids cause sedation and may also cause respiratory depression, or a decrease in the ability to breathe spontaneously. Opioids used chronically can cause severe constipation that leads many patients to stop using them, and opioids may sometimes cause severe itching. All of the drugs used to treat neuropathic pain frequently cause problems with coordination and sedation. | |
| • | Need for frequent dosing. Drugs used to treat neuropathic pain require frequent dosing that makes their use inconvenient, often leading to reduced patient compliance. | |
| • | Slow onset of action. Local anesthetics that are used prior to procedures involving manipulation of the skin, such as needle-sticks or skin surgery, are typically formulated as patches or creams and have a slow onset of pain relief. This slow onset, as well as poor efficacy, is due to the poor penetration of skin by the anesthetics used in these products. | |
| • | Potential to cause physical dependence. Opioids, when used chronically, can cause physical dependence. Fear of physical dependence often influences clinicians to prescribe less than adequate doses of opioid analgesics. Similar fears lead many patients to refuse opioid analgesics. |
Given doctors’ and patients’ desire to achieve adequate control of pain, and the significant shortcomings associated with existing treatments, doctors and patients often struggle to find an appropriate balance between pain relief and adverse side effects. With both over- and under-treatment of pain, patients may be suffering unnecessarily, have poor quality of life and have difficulty meeting their social, familial and work-related commitments.
Our Solution
Based on our preclinical and clinical trials to date, we believe our three product candidates, if approved, will be able to offer patients tangible benefits over existing treatments for the indications targeted by each of our product candidates.
ALGRX 4975: ALGRX 4975 is our product candidate for the treatment of site-specific, severe or intractable pain. We believe ALGRX 4975 represents a novel approach to the treatment of pain because it provides long-lasting analgesia with a single administration to a discrete part of the body and because it acts via a different mechanism than any current prescription analgesic. As a result, ALGRX 4975 avoids many of the side effects of existing pain therapies. In addition, whereas many existing opioids are not particularly effective for certain types of severe pain or require frequent dosing, ALGRX 4975, by harnessing capsaicin’s ability to cause changes in nerve endings lasting up to 16 weeks, has been shown in clinical trials to provide meaningful, long-lasting pain relief following a single administration. We expect that if ALGRX 4975 does provide more meaningful pain relief with fewer side effects than opioid analgesics, treatment with ALGRX 4975 can result in improved quality of life for many patients.
ALGRX 3268:ALGRX 3268 is intended to provide rapid, easy-to-administer local analgesia to reduce the pain associated with needle-stick procedures in children and in adults who have fear of needle insertion. Existing therapeutic products have an onset of analgesia of at least 10 minutes, although the most frequently used products must be applied 30 to 60 minutes prior to a needle-stick procedure. This delay creates logistical difficulties in administering needle-stick procedures and creates a significant barrier to use in busy emergency rooms, oncology suites and pediatric offices. Furthermore, existing products are formulated as creams and patches, which may be perceived by health care staff and patients as messy and
45
Table of Contents
cumbersome to utilize. By contrast, ALGRX 3268 uses a convenient application procedure, and our clinical trials to date have shown that it is effective in one minute of application. We believe these are important factors for increasing physician use of analgesia prior to needle-stick procedures.
ALGRX 1207: ALGRX 1207, our preclinical product candidate, is the first new molecular entity from a new class of local anesthetics that will potentially treat patients with certain types of neuropathic pain and be used for pre-procedural administration to reduce the pain associated with surgical procedures on the skin. These types of pain are not well-treated by current analgesic therapy. The most common existing treatments for these conditions consist of patches or creams containing a local anesthetic. These patches and creams have a slow onset of pain relief, and the desired efficacy may not be achieved due to the poor penetration into the skin by the anesthetics used in these products. In addition, these treatments are often inconvenient to use. Based on nonclinical studies in animals, ALGRX 1207 has been shown to provide analgesia more rapidly following direct administration to skin and with a longer-lasting effect than currently available topical anesthetics. In addition, we believe that ALGRX 1207, if approved, could address the pain associated with a wide variety of procedures involving the skin, including dermatological surgery, cosmetic skin treatments and catheter placement, as well as the pain arising from surgical incisions that does not subside after the surgery has taken place.
Strategy
Given the shortcomings of current therapies and the economic impact of pain on society, our goal is to become a leading pharmaceutical company focused on the commercialization of drugs to treat acute and chronic pain. The key elements of our strategy for achieving this goal are as follows:
| Continue to build, through acquisition or in-licensing, a balanced portfolio of product candidates that leverages our understanding of the mechanisms of pain and the pain management market. |
We currently have five distinct programs in various stages of research and development in the areas of acute and chronic pain management. Each of our product candidates is in a different stage of development, has a different mechanism of action and seeks to treat different types of pain conditions. In building our product pipeline, we intend to leverage our management’s experience and understanding of the unmet medical needs in treating pain, the mechanisms of action of existing therapies and the mechanisms of pain. We intend to acquire or in-license additional products or product candidates from a wide range of sources, including academic centers and biotechnology and pharmaceutical companies, in both domestic and international markets.
| Complete clinical development and obtain regulatory approval for our product candidates utilizing our management team’s extensive experience in developing and commercializing drugs for pain management. |
We believe that our scientific expertise is broadly applicable across a wide range of drugs. Our management team has over 100 years of cumulative experience in the pharmaceutical industry, including having previously led the research and development, approval and commercialization of numerous products for pain. Our Chief Executive Officer, Ronald M. Burch, M.D., Ph.D., has spent over 16 years developing drugs for pain management, and our President and Chief Operating Officer, Paul R. Hamelin, R.Ph., has spent over seven years marketing, developing and commercializing pain drugs, including participating in the development and launch of Celebrex® in several countries. We believe that our management team’s experience in the planning and management of clinical trials will help us maximize the relevance and utility of our clinical trial programs. We plan to rely on this experience in seeking to obtain regulatory approval for our current product candidates in the United States and, where appropriate, the rest of the world.
| Develop a specialized sales and marketing team to commercialize our product candidates, if approved, in the United States. |
We currently retain all global commercialization rights to our product candidates. We intend to build a specialized sales and marketing organization to market our products to physicians and health care
46
Table of Contents
professionals who are prescribers of pain medications within the United States. We will focus our marketing efforts on concentrated market segments for our products. For example, in the management of severe postsurgical pain with ALGRX 4975, we believe that our target market will be hospital pain clinics, general surgeons and orthopedic surgeons that are primarily hospital-based and concentrated in major metropolitan areas. We believe that with such a specialized sales and marketing organization we will be able to market ALGRX 4975 effectively, if approved.
| Establish corporate partnerships to assist in the clinical development, regulatory approval process and commercialization of our products in the United States and major foreign markets. |
We intend to form collaborations and partnerships with leading pharmaceutical, biotechnology or specialty pharmaceutical companies, both domestically and internationally, to augment our internal clinical and regulatory resources and to accelerate the implementation of our strategies. We believe these selective partnerships will extend and ensure our ability to market products in a resource-efficient manner in certain markets in the United States and in large foreign markets such as Europe and Japan.
Our Product Pipeline
Our product pipeline consists of two clinical product candidates, ALGRX 4975 and ALGRX 3268, and one preclinical product candidate, ALGRX 1207. ALGRX 4975 is currently in several phase II clinical trials and is being developed as two formulations in three clinical development programs. We have recently commenced two phase III clinical trials for ALGRX 3268, and we expect to file an IND application for ALGRX 1207 with the FDA in the second half of 2005. The following table summarizes the current status of our product candidates in clinical and preclinical development and of our research program:
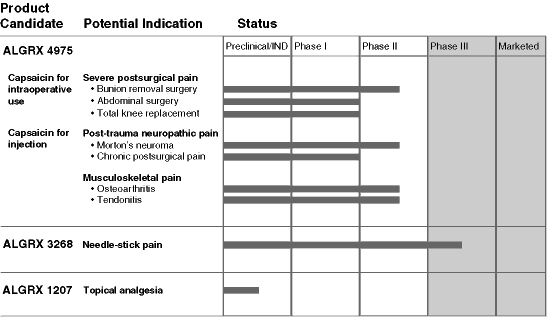
| ALGRX 4975 (capsaicin for injection and capsaicin gel for intraoperative use) |
ALGRX 4975 is our product candidate for the treatment of site-specific severe or intractable pain. These types of pain are poorly treated with existing drugs, many of which have well-documented and severe side effects. We are developing two formulations of ALGRX 4975 to potentially treat patients with severe pain: a gel formulation for use during a variety of surgical procedures, including bunion removal surgery, total knee replacement and abdominal surgeries, such as hernia repair or hysterectomy; and an
47
Table of Contents
injectable formulation for post-trauma neuropathic pain and pain resulting from musculoskeletal diseases, such as osteoarthritis and tendonitis. During a surgical procedure, ALGRX 4975 is delivered directly onto the cut surfaces of skin, muscle and bone. For post-trauma neuropathic pain and pain resulting from musculosketal diseases, it is delivered to the site of pain using a needle and syringe. Prior to injection with ALGRX 4975, these patients receive an injection of a local anesthetic to alleviate the transient pain experienced upon injection of ALGRX 4975. We are currently evaluating ALGRX 4975 in a variety of phase II clinical trials.
| Capsaicin |
The active ingredient in ALGRX 4975 is capsaicin. Capsaicin is currently marketed over-the-counter for topical administration in the form of low dose, non-sterile creams and patches, which tend to be poorly absorbed into the skin. There are many brands of capsaicin creams and patches, including Capzasin-P® (Chattem) and Zostrix® (Rodlen Laboratories). These formulations are generally crude preparations of capsaicin that may contain other chemical entities. Over-the-counter capsaicin creams and patches are used topically by consumers to relieve pain in conditions such as osteoarthritis, postherpetic neuralgias, psoriasis and diabetic neuropathy.
ALGRX 4975, if approved, would be used in different clinical settings than current over-the-counter products. Currently, there are no FDA-approved uses of capsaicin for analgesia or other indications inside the body. Results of our clinical trials suggest that capsaicin, when used in the manner and doses we are investigating, may have analgesic properties previously unknown to clinical investigators. We currently own or license three patents and 12 patent applications related to our capsaicin technology, compounds and their applications in pharmaceutical development or their use as pharmaceuticals. We believe these issued patents and pending patents, if and when issued, will provide us with intellectual property protection in the methods of purification, manufacture, medical usage and formulation of capsaicin.
| Mechanism of Action |
Three major classes of nerves, or neurons, Ad, Ab and C, transmit sensory information through the body’s nervous system. Pain signals are transmitted by two of those classes: Ad and C neurons. Ad neurons transmit signals rapidly giving rise to sharp pain sensations, whereas C neurons transmit signals slowly giving rise to severe dull, aching or throbbing pain sensations. For example, if a person hits his thumb with a hammer, there is an immediate, sharp pain that lasts only for an instant, which is transmitted by Ad neurons, followed by a dull, aching, throbbing pain that may last for a considerable time, which is transmitted by C neurons.
Capsaicin works to relieve pain by causing localized degradation of the C neuron endings and is the only analgesic known to relieve pain by this mechanism. Capsaicin’s activity results from its ability to bind to and activate a type of ion channel on the surface of neurons called vanilloid receptor 1, or VR1, which is involved in the transmission of pain signals to the brain. Under normal circumstances, when the VR1 ion channel is activated, usually by changes in pH, it opens for a short time, causing the C neurons to transmit a pain signal toward the brain. When capsaicin binds to and activates VR1, it causes a series of events within the cell that degrade the pain-sensing endings of the C neuron, thereby preventing the neuron from transmitting pain signals. Our animal and human studies have demonstrated that following capsaicin treatment, the C neuron terminals usually regenerate over a period of 12 to 16 weeks. This unique action is the basis for what we believe will be ALGRX 4975’s ability, if approved, to provide meaningful, long-lasting pain relief following a single administration. Because VR1 is found only on C neurons, capsaicin reduces only the long-term noxious pain associated with transmission by these neurons. Capsaicin does not affect the short-lasting, sharp adaptive pain carried by Ad neurons, or the fine touch, temperature and place perception signals carried by Ab neurons. As a consequence, ALGRX 4975 may be a highly specific pain therapeutic that provides long-lasting analgesia.
We believe the effects of capsaicin are confined exclusively to the region of application because of low distribution to other areas of the body after ALGRX 4975 is administered. After injection into a joint
48
Table of Contents
space or after application in a surgical procedure to the cut surfaces of skin, muscle and bone, capsaicin enters the blood slowly by diffusion from its site of initial application. Thereafter, capsaicin is highly metabolized, or broken down, by the liver into various inactive compounds, none of which retains any of capsaicin’s analgesic properties. As a consequence, capsaicin does not usually act at sites in the body distant from its initial application, nor is the body exposed to any derivatives of capsaicin that could act in a similar manner. By contrast, opioids and many other analgesics must be given orally or by intravenous injection, thereby subjecting the patient to circulation of high concentrations of drug. These high circulating concentrations may cause undesirable side effects by acting on parts of the body unrelated to pain perception. For example, opioids may cause constipation when used chronically. Opioids also may cause alteration of mood or alertness, and may cause patients to feel drowsy, euphoric, or sleepy. These effects, when experienced by patients in the hospital, tend to increase rehabilitation time because patients are often sedated and therefore unable to begin the recovery process.
Based on our clinical trial results to date, we believe that ALGRX 4975 may provide significant pain relief to patients in postsurgical situations by acting only at the site of application and without causing sedation or other side effects, thereby potentially reducing rehabilitation time following some procedures. We also believe ALGRX 4975 may provide significant pain relief to patients with post-trauma neuropathic pain or who suffer from severe musculoskeletal pain. We expect that if ALGRX 4975 provides more meaningful pain relief with fewer side effects than opioid analgesics, it may result in improved quality of life for many patients.
ALGRX 4975 — Capsaicin for the Treatment of Severe Postsurgical Pain
| Overview |
We are developing ALGRX 4975 for use during a variety of surgical procedures to treat severe postsurgical pain. Opioid analgesics are commonly used for the treatment of severe postsurgical pain. The common delivery method for in-hospital pain relief is a patient controlled morphine pump that provides opioid analgesic infusions directly into the spinal fluid. The process is expensive, may cause infections, and since the pain relief from opioid analgesics is relatively brief, the patient goes through cycles of pain relief followed by increased pain. We believe that ALGRX 4975, by providing long-lasting analgesia without sedation, may mitigate the cyclical nature of patient-controlled morphine and also alleviate the increase in pain suffered by many patients following discharge from the hospital, as they transition from opioid analgesic infusions to oral opioids.
Use of opioids following certain types of surgery such as total knee replacement may also extend rehabilitation time. For such patients, walking as soon as possible after surgery is an important factor for both preventing blood clots from forming in the veins of the legs and for maintaining range of motion of the knee. Patients taking opioid analgesics immediately after surgery may be too sedated or drowsy to walk, thereby diminishing the ability to get out of bed. We believe that ALGRX 4975, if approved, will provide strong analgesia that could reduce the amount of opioids taken by patients and therefore decrease rehabilitation time.
| Clinical Trials |
The following table describes the status of our clinical trials for ALGRX 4975 to treat severe postsurgical pain:
| Number of | ||||||||
| Phase of Clinical Trial | Status | Protocol | Subjects | |||||
| Phase II | Completed | Bunion removal surgery | 40 | |||||
| Phase II | Completed | Bunion removal surgery | 182 | |||||
| Phase II | Ongoing | Hernia repair | 48 | |||||
In our trials to date, we have only used a liquid form of ALGRX 4975, but we plan to use the gel formulation in later trials because we believe that a gel formulation will offer improvements over the liquid
49
Table of Contents
form during surgical procedures. For instance, we expect that a gel formula will be easier to apply, and will bind more effectively to cut surfaces of skin, muscle and bone.
| Phase II Clinical Trial — Bunion Removal Surgery |
In November 2003, we completed a randomized, double-blind, placebo-controlled phase II clinical trial of ALGRX 4975 in 40 patients evaluating postsurgical pain following bunion removal surgery at a single center in the United Kingdom. Just prior to closure of the surgical wound, either 1,000 ug of ALGRX 4975 or placebo was instilled into the wound. The primary endpoint of this trial was to record the use of opioid rescue medication. The opioid rescue medication consisted of opioids given to a patient in the absence of adequate pain relief from the placebo or ALGRX 4975 during the first 24 hours following surgery. Secondary endpoints included the use of rescue medication during the first 72 hours following surgery, the quantity of rescue medication used, and the patient pain scores measured on a visual analog scale, or VAS, a commonly used chart that patients are given to rate the severity of their pain, from zero representing no pain to 10 representing extreme pain. Several safety endpoints were also evaluated, including pain on administration and wound healing.
This trial demonstrated a statistically significant reduction in the use of rescue medication during the first 72 hours following surgery in patients receiving ALGRX 4975 as compared to patients receiving placebo. During the first 72 hours, 45% of patients who received ALGRX 4975 requested rescue medication compared to 90% in patients who received placebo. Further, this trial indicated that VAS pain scores were reduced in patients who received ALGRX 4975 as compared to patients who received placebo. No safety concerns were identified in this study.
| Phase II Clinical Trial — Bunion Removal Surgery |
In December 2004, we completed a second randomized, double-blind, placebo-controlled phase II clinical trial of ALGRX 4975 in 182 patients evaluating postsurgical pain following bunion removal surgery at two centers in the United States. In this trial, three dose levels of ALGRX 4975, 100 ug, 500 ug and 1,000 ug, were compared to placebo. The primary endpoint of this clinical trial was the magnitude and duration of pain relief in those patients receiving ALGRX 4975 compared to those receiving placebo. Secondary endpoints included the use of rescue medication from the time of surgery to awakening the next morning, a period usually lasting 18 to 20 hours, and from the time of surgery to discharge, a period usually lasting approximately 32 hours, and the quantity of rescue medication used. Several safety endpoints were also evaluated, including pain on administration and wound healing.
This trial demonstrated a statistically significant reduction in the magnitude of pain suffered during the first 32 hours following surgery by those subjects who received ALGRX 4975 in the 500 ug or 1,000 ug doses, compared to subjects who received placebo. Furthermore, during the first 18 to 20 hours, fewer patients who received ALGRX 4975 requested rescue medication compared to patients who received placebo; these results were statistically significant. No safety concerns were identified in this study.
| Phase II Clinical Trial — Hernia Repair |
We are currently conducting a randomized, double-blind, placebo-controlled phase II clinical trial in 48 patients to evaluate the safety and efficacy of ALGRX 4975 in the treatment of postsurgical pain following hernia repair at multiple centers in Denmark. In this trial, 1,000 ug of ALGRX 4975 or placebo will be applied to the cut surfaces of connective tissue, muscle and skin prior to closing the surgical wound. The primary endpoint will be the use of rescue medications following surgery. Secondary endpoints will include the amount of rescue medication used and VAS pain scores. A variety of safety endpoints will be evaluated in this trial, including pain on administration and wound healing.
| Regulatory Pathway |
We filed a clinical trials exemption, or CTX, for ALGRX 4975 with the Medicine and Healthcare Products Regulatory Agency in the United Kingdom in June 2003 and commenced our first clinical trial in
50
Table of Contents
severe postsurgical pain in August 2003. This study was completed in January 2004. We submitted an IND application for ALGRX 4975 with the FDA in May 2004 and initiated our first U.S.-based phase II clinical trial to study pain following bunion removal surgery in July 2004. Our clinical program will consist of phase II clinical trials in a variety of surgical procedures, followed by several intended pivotal clinical trials in several surgical procedures. We are developing this program with the intention that it will satisfy the requirements for an NDA.
ALGRX 4975 — Capsaicin for Injection for Post-Trauma Neuropathic Pain
| Overview |
We are developing an injectable formulation of ALGRX 4975 to potentially treat patients with post-trauma neuropathic pain. Post-trauma neuropathic pain is a form of neuropathic pain that remains long after traumatic injuries such as limb amputations, accidents or injuries caused by repeated physical stresses applied to the same area of the body. Post-trauma neuropathic pain is not well-treated with currently available drugs, which include tricyclic antidepressants and antiepileptic agents that are prescribed “off-label” for this type of pain. These drugs demonstrate meaningful efficacy in only a minority of patients. When drugs are unsuccessful in providing meaningful pain relief, patients suffering from post-trauma neuropathic pain sometimes consider other procedures that may alleviate pain. One option is surgery to remove the offending nerve, although this procedure results in side effects such as the lack of sensation and compromised motor function. A second option involves killing nerves by injecting them with alcohol or formaldehyde, which also has side effects similar to surgical removal. We believe that ALGRX 4975 may provide long-lasting, effective pain relief in these patients without the side effects caused by damaging or killing nearby nerves.
| Clinical Trials |
The following table describes the status of our clinical trial for the injectable formulation of ALGRX 4975 for post-trauma neuropathic pain:
| Number of | ||||||||
| Phase of Clinical Trial | Status | Protocol | Subjects | |||||
| Phase II | Ongoing | Morton’s neuroma | 60 | |||||
| Phase II Clinical Trial — Morton’s Neuroma |
We are currently enrolling patients in a randomized, double-blind, placebo-controlled phase II clinical trial of ALGRX 4975 in patients with Morton’s neuroma or related neuromas at two centers in the United States. Morton’s neuroma is an abnormal enlargement of two nerves that are located between the bones of the feet. This enlargement is typically seen in women who wear high-heeled shoes and in long-distance runners, all of whom subject their feet to repetitive stresses. The primary endpoint of this trial will be to evaluate the magnitude of pain relief for 1,000 ug of ALGRX 4975 compared to placebo, measured for each of the four weeks following administration. Safety endpoints to be evaluated in this trial will include pain on administration. Pharmacokinetics, which measures the time required for the administered drug to reach the bloodstream, and the time over which the drug is then removed from the blood due to metabolism, will also be assessed.
| Regulatory Pathway |
Our clinical trial program for post-trauma neuropathic pain is being conducted under the IND application that we submitted to the FDA in May 2004. Our first clinical trial under the IND application is a phase II clinical trial studying Morton’s neuroma. In addition to our phase II clinical trial studying Morton’s neuroma, we expect to initiate phase II clinical trials to study other types of post-trauma neuropathic pain. Based on the results of those studies, we will develop a phase III program.
51
Table of Contents
ALGRX 4975 — Capsaicin for Injection for Musculoskeletal Diseases
| Overview |
We are developing an injectable formulation of ALGRX 4975 to potentially treat patients with pain resulting from musculoskeletal diseases such as osteoarthritis and tendonitis. Early-stage osteoarthritis is usually treated using acetaminophen and, as pain increases, COX-2 inhibitors. As the disease progresses and joint damage increases, most patients reach a point at which they do not experience sufficient pain relief from COX-2 inhibitors. At this point, patients have several options. Some patients opt for hyaluronic acid injections directly into the joint space. The injected hyaluronic acid acts as a lubricant to enhance joint mobility, thereby reducing pain. Other patients may begin using opioid analgesics, which provide better pain relief than the COX-2 inhibitors, but still may not provide adequate pain relief. We believe that by administering ALGRX 4975 directly into the joint space, approximately once every three to four months, patients may experience meaningful pain relief with few side effects. Because joint replacement is usually the only remaining option for patients whose pain from osteoarthritis becomes intolerable, the potential ability to delay these procedures through more adequate pain relief could represent a substantial advance over the current range of therapeutic options.
| Clinical Trials |
The following table describes the status of our clinical trials for the injectable formulation of ALGRX 4975 for musculoskeletal diseases:
| Number of | ||||||||
| Phase of Clinical Trial | Status | Protocol | Subjects | |||||
| Phase I | Completed | End-stage osteoarthritis | 16 | |||||
| Phase II | Completed | End-stage osteoarthritis | 12 | |||||
| Phase II | Ongoing | Osteoarthritis of the knee | 52 | |||||
| Phase II | Ongoing | Osteoarthritis of the knee | 54 | |||||
| Phase II | Ongoing | Tendonitis | 40 | |||||
| Phase I Clinical Trial — End-stage Osteoarthritis |
In April 2003, we completed a randomized, double-blind, placebo-controlled phase I clinical trial of ALGRX 4975 in 16 patients in one center in the United Kingdom, the first study of ALGRX 4975 in humans. In this study, ALGRX 4975 was injected directly into the knee joint space of patients with end-stage osteoarthritis who were scheduled for knee replacement surgery. Patients were allocated to one of four treatment groups based on the time between the administration of ALGRX 4975 or placebo and the date scheduled for the patient’s surgery, which were two days, four days, 10 days or 14 days after injection of ALGRX 4975. Patients received either a 10 ug, 100 ug, or 300 ug dose of ALGRX 4975 or placebo. Each patient received an injection of the local anesthetic lidocaine directly into the joint space 10 minutes prior to injection of ALGRX 4975 in order to alleviate the transient pain experienced by patients upon injection of capsaicin. Since this was a phase I clinical trial, the primary endpoints evaluated safety of ALGRX 4975. Secondary endpoints included pharmacokinetics and pain relief assessed using a VAS pain scale. Furthermore, because all patients in this trial subsequently underwent surgery, this study allowed for the direct examination of the knee to evaluate side effects caused by the drug.
This phase I clinical trial demonstrated that ALGRX 4975 was well-tolerated. Although dose-related pain on administration was found despite pretreatment with lidocaine, pain was adequately managed in all patients with placement of an icepack on the knee. Pain reduction was also observed in this trial following the administration of ALGRX 4975. When assessed on the scheduled day of surgery, pain was found to be reduced on average by 64% when compared to patients’ baseline scores. The pain reduction data from this trial was not statistically significant because this trial was designed to demonstrate safety rather than efficacy.
52
Table of Contents
| Phase II Clinical Trial — End-stage Osteoarthritis |
In December 2003, we completed a randomized, double-blind, placebo-controlled phase II clinical trial of ALGRX 4975 in 12 patients with end-stage osteoarthritis of the knee at one center in the United Kingdom. In this study, patients received treatment with either a single dose of 1,000 ug of ALGRX 4975 or placebo. Knee replacement surgery was scheduled between three and six weeks following the administration of ALGRX 4975. As in the previous study, injection of ALGRX 4975 was preceded by injection of lidocaine into the joint space. The primary endpoint of this trial was to evaluate the magnitude of pain and duration of reduction in pain at week three following administration of ALGRX 4975 since no patient had undergone surgery at that time.
In this study, ALGRX 4975 was shown to reduce pain at each of the assessed time points (weeks one, two, three, four, five and six) compared to the pain experienced by patients prior to the administration of ALGRX 4975. This trial demonstrated a statistically significant reduction in pain at week three. At all time points, pain was found to have been reduced by approximately 50% to 60% in the patients treated with ALGRX 4975. Pain was not meaningfully reduced in patients who received placebo. Most patients who received injections of ALGRX 4975 experienced transient burning pain (the median lasting 20 minutes) upon administration of ALGRX 4975. In most of these patients, ice packs were used to manage the transient pain.
| Phase II Clinical Trials — Osteoarthritis of the Knee |
Two clinical trials are ongoing in osteoarthritis of the knee. In the first, a randomized, double-blind, placebo-controlled trial in 52 patients at one center in the United States, either 300 ug of ALGRX 4975 or placebo is injected into the knee joint space, following lidocaine, in patients who no longer receive adequate pain relief from NSAIDs. The primary efficacy endpoint is the magnitude and length of time that arthritis pain is reduced in the treated knee following a single administration, which will be evaluated at one and two weeks following administration, then monthly until treatment failure, or the point in time at which patients cease to experience analgesia. A variety of safety endpoints will be evaluated, including pain on administration.
We are conducting a second, open-label, phase II clinical trial in 54 patients at two centers in Poland. Lidocaine will be injected into the joint and, at various times thereafter, either a single dose of ALGRX 4975 or placebo will be injected. In other subjects, a set dose of lidocaine will be injected, followed by a daily dose of ALGRX 4975 at three weekly intervals. The primary endpoint of this trial will be the efficacy of lidocaine administration to reduce pain after administration of ALGRX 4975.
| Phase II Clinical Trial — Tendonitis |
We are currently conducting a randomized, double-blind, placebo-controlled phase II trial of ALGRX 4975 in 40 patients with tennis elbow at two centers in Slovakia. In this trial, 100 ug of ALGRX 4975 is being compared to placebo. The primary endpoint of this trial will be magnitude and duration of pain relief compared to placebo. There will be several safety endpoints of this trial including pain on administration.
| Regulatory Pathway |
We initiated our first clinical trial, a phase I clinical trial in end stage osteoarthritis, in November 2002 under approval from the Institutional Review Board of Charing Cross Hospital in London, England. The trial was designed to provide safety data on ALGRX 4975. We then filed a CTX in the United Kingdom and initiated a phase II clinical trial in end-stage osteoarthritis. In September 2003, we submitted an IND application to the FDA. In August 2004, we initiated a phase II clinical trial in osteoarthritis in the United States. We also initiated a phase II clinical trial in tendonitis in November 2004 in Slovakia, also under the IND. Because osteoarthritis is a long term chronic condition, often suffered by the elderly, the FDA typically requires longer trials with a larger amount of patients for osteoarthritis drugs. We expect that the next phase II clinical trial for osteoarthritis will include multiple
53
Table of Contents
administrations over a period of at least a year per patient. We will develop our phase III plan based on the results of that trial.
| Commercialization Plan |
Currently, we retain worldwide marketing rights to ALGRX 4975, and if we receive regulatory approval to market this product, we intend to create a sales and marketing team to launch the product in the United States. Because we expect that ALGRX 4975, if approved, would have two formulations, address multiple indications and be utilized across different physician specialties, we plan to pursue multiple sales and marketing strategies tailoring our efforts to specific market segments. We expect that our own sales and marketing infrastructure would target certain markets for ALGRX 4975 in the United States, and we may also seek a development and marketing partner to maximize the opportunity for this product candidate in other market segments in the United States. In major markets abroad, we intend to collaborate with or create co-development or co-marketing partnerships with pharmaceutical companies who have greater sales and marketing capabilities than we do. We expect that these partners would help fund the development costs of ALGRX 4975 and the costs associated with the launch of ALGRX 4975 outside the United States.
ALGRX 3268 (PowderJect®Dermal Lidocaine)
ALGRX 3268 (PowderJect® Dermal Lidocaine) is our product candidate that delivers a powder formulation of the local anesthetic lidocaine into the skin using our proprietary needle-free dispenser. ALGRX 3268 is intended to provide rapid, easy-to-administer local analgesia to reduce the pain associated with needle-stick procedures in children and also in adults who have fear of needle insertion. The primary market for ALGRX 3268 is the estimated 42 million needle-stick procedures that are performed on children in the United States each year. We believe that this market is highly under-served by existing products, and we believe that the medical community is interested in reducing the pain associated with needle-stick procedures. In fact, a joint recommendation from the American Academy of Pediatrics and American Pain Society has urged consideration of local anesthetics and strategies to soothe and minimize distress even for simple procedures such as blood draws. We also believe that there is an opportunity to serve the adult market, which consists of an estimated 315 million procedures annually where blood is drawn or intravenous lines are inserted.
Existing therapeutic products have an onset of analgesia of at least 10 minutes, although the most frequently used products direct the health care provider to apply the products at least 30 to 60 minutes prior to a needle-stick procedure. This delay requires that a patient first be seen by a nurse or other caregiver in order to have the analgesic product applied; the patient must then wait until appropriate analgesia ensues, after which the needle-stick procedure can be done. In practice, this creates logistical difficulties and other inconveniences as patients and health care providers must wait for analgesia to set in. These inconveniences are a significant barrier to use in busy emergency rooms, oncology suites and pediatric offices. Furthermore, existing products are formulated as creams and patches, which may be perceived by health care providers and patients as messy and cumbersome to use. By contrast, ALGRX 3268 uses a convenient application procedure, and our clinical trials to date have shown that it is effective in one minute following administration. We believe these are important factors for increasing physician use of analgesia prior to needle-stick procedures.
| Method of Application |
ALGRX 3268 contains lidocaine, a currently used, commercially approved drug, in powder form. ALGRX 3268 is designed to deliver lidocaine powder exclusively into the skin with a sterile, single-use, needle-free disposable dispenser. Pressing a button on the dispenser releases compressed helium gas from an enclosed cylinder, which ruptures the cassette that contains the lidocaine powder. The lidocaine powder is accelerated in the gas stream and penetrates the skin, where it provides analgesia to the skin at the site of application. Based on the results of our clinical trials to date, ALGRX 3268 is effective in one minute following administration. We believe this rapid onset will allow the health care provider to prepare and
54
Table of Contents
sterilize the injection site, administer ALGRX 3268, and prepare the needle set and insert it, all without having to wait between steps. We believe that ALGRX 3268, if approved, would eliminate much of the inconvenience that is currently associated with the administration of cream- and patch-based formulations and provide a more efficient and satisfactory experience to health care providers and patients.
| Clinical Trials |
Development of ALGRX 3268 was initiated by Chiroscience Group plc and PowderJect Pharmaceuticals plc. Our license to this technology was acquired by us in connection with our acquisition of certain of PowderJect’s assets in 2002. In a series of clinical trials conducted by Chiroscience and PowderJect, nearly 2,000 administrations of this product were given to adults and children using various doses of lidocaine powder and various helium pressures which are measured in units called bars. These trials were intended to help refine the powder injection device. Since in-licensing this product candidate, we have completed our clinical program for ALGRX 3268 through phase II in approximately 1,100 adults and children using various doses of lidocaine powder and various helium pressures. We had an end of phase II meeting with the FDA in November 2004, and we recently commenced two phase III clinical trials for ALGRX 3268. We anticipate an NDA filing at the end of 2005.
The following table describes the status of our clinical trials for ALGRX 3268:
| Number of | ||||||||||
| Phase of Clinical Trial | Status | Age Group | Subjects | |||||||
| Phase I | Completed | Adult | 272 | |||||||
| Phase I | Completed | Adult | 38 | |||||||
| Phase I | Completed | Adult | 183 | |||||||
| Phase II | Completed | Children | 195 | |||||||
| Phase II | Completed | Children | 145 | |||||||
| Phase II | Completed | Children | 306 | |||||||
| Phase III | Ongoing | Children | 500 | |||||||
| Phase III | Ongoing | Children | 500 | |||||||
The clinical development program for ALGRX 3268 consisted of evaluation of safety and efficacy in adults and children following application at either the antecubital fossa located at the inside of the elbow or back of the hand, after which pain was elicited by means of inserting a needle into a vein and drawing blood. The studies in adults were phase I clinical trials performed in normal volunteers, whereas the studies in children were phase II clinical trials performed in children visiting clinics for scheduled blood draws.
| Phase I Clinical Trial — Adult Antecubital Fossa |
In October 2002, we completed a randomized, double-blind, placebo-controlled phase I clinical trial that studied the safety and efficacy of ALGRX 3268 in adults at one center in the United States. Three configurations of the device were used in this study: 0.25 mg of lidocaine and 20 bar helium pressure; 0.50 mg and 20 bar; and 0.50 mg and 40 bar. For each configuration of the device, we evaluated skin irritation caused by the lidocaine particle injection. We examined the patients’ skin for the appearance of redness, swelling, and pinpoint blood spots. Each of these effects was measured using a five-point scale, on which scores of three or higher are considered clinically significant. Neither of the configurations with 20 bar helium pressure caused clinically significant redness, swelling or pinpoint blood spots, whereas the configuration with 40 bar helium pressure caused scores of three or greater in several subjects. Thus, we concluded that a device using 40 bar pressure was not acceptable.
�� Following administration of ALGRX 3268 at the antecubital fossa, it was found that each of the three configurations of ALGRX 3268 used in this trial reduced pain on needle insertion in a statistically significant manner compared with placebo when administered one, three or five minutes prior to needle insertion. With all configurations, efficacy was not apparent when ALGRX 3268 was administered
55
Table of Contents
10 minutes or more prior to needle insertion. From the study, we concluded that a dose of 0.25 mg lidocaine was suboptimal and that the configuration with 0.50 mg lidocaine delivered at 20 bar pressure was the most suitable for further development to provide procedural analgesia at the antecubital fossa.
| Phase I Clinical Trial — Adult Back of Hand |
In July 2003, we completed a randomized, double-blind, placebo-controlled phase I clinical trial that studied the safety and efficacy of ALGRX 3268 in adults at one center in the United States. Two configurations of the device were used in this study: 0.50 mg at 20 bar; and 0.50 mg at 40 bar. Skin irritation caused by lidocaine particle injection was evaluated for each configuration. We examined patients’ skin for the appearance of redness, swelling, and pinpoint blood spots. Each of these was measured using a five-point scale, on which scores of three or higher are considered clinically significant. The configuration with 20 bar helium pressure caused no clinically significant redness, swelling or pinpoint blood spots, whereas the configuration with 40 bar helium pressure caused scores of three or greater in several subjects. Thus, we concluded that a device using 40 bar pressure was not acceptable.
Following administration at the back of the hand, it was found that both of the configurations of ALGRX 3268 used in this trial reduced pain on needle insertion in a statistically significant manner compared with placebo when administered one, three or five minutes prior to needle insertion. With each configuration, efficacy was not apparent when ALGRX 3268 was administered 10 minutes or more prior to needle insertion. We were also able to conclude from this study that the configuration with 0.50 mg of lidocaine delivered at 20 bar pressure was the most suitable for further development to provide procedural analgesia at the back of the hand.
| Phase I Clinical Trial — Pharmacokinetics Following Administration to the Antecubital Fossa in Adults |
In December 2002, we completed a randomized, double-blind, placebo-controlled phase I clinical trial at one center in the United States that studied the circulation of ALGRX 3268 in the blood of adult subjects after administration. Lidocaine circulating in the blood at concentrations of approximately 100 ng/ml or more may be associated with heart rhythm abnormalities and seizures. Thus, we investigated the amount of lidocaine circulating in the blood following administration of ALGRX 3268 in the antecubital fossa in adults. We found that following administration of a dose of 0.50 mg of ALGRX 3268 at 20 bar helium pressure, the concentration of lidocaine in each subject’s blood plasma was below five ng/ml at every time point of the study. Thus, this trial demonstrated that, following administration of ALGRX 3268, the concentration of lidocaine in the blood was at a level unlikely to cause adverse side effects. The low blood concentrations of lidocaine in this trial resulted from the very small dose necessary to achieve local anesthesia with this highly-targeted method.
| Phase II Clinical Trial — Child Antecubital Fossa |
In July 2003, we completed a randomized, double-blind, placebo-controlled phase II clinical trial that studied the safety and efficacy of ALGRX 3268 in children at one center in Poland. Two configurations of the device were used in the trial: 0.25 mg at 20 bar; and 0.50 mg at 20 bar. Skin irritation caused by particle injection was evaluated in this trial. We examined patients’ skin for the appearance of redness, swelling, and pinpoint blood spots. Each of these was measured using a five-point scale, on which scores of three or higher are considered clinically significant. Neither of the device configurations used in this trial caused clinically significant redness, swelling or the appearance of pinpoint blood spots.
Following administration at the antecubital fossa, it was found that a dose of 0.25 mg of ALGRX 3268 at 20 bar helium pressure did not reduce pain on needle insertion in a statistically significant manner when administered one to three minutes prior to needle insertion. In contrast, a dose of 0.50 mg of ALGRX 3268 at 20 bar helium pressure was found to reduce pain on needle insertion in a statistically significant manner in children aged three to 12 years and in children 13 to 18 years of age when compared to placebo. Therefore, we determined that the configuration of ALGRX 3268 most suitable for further
56
Table of Contents
development, when assessed at the antecubital fossa in children, was 0.50 mg at 20 bar helium pressure, the same configuration as in adults.
| Phase II Clinical Trial — Child Back of Hand |
In January 2004, we completed a randomized, double-blind, placebo-controlled phase II clinical trial that studied the safety and efficacy of ALGRX 3268 in children at one center in Poland. Skin irritation caused by lidocaine particle injection was evaluated in this study. We examined the patients’ skin for the appearance of redness, swelling and pinpoint blood spots. Each of these was measured using a five-point scale, on which scores of three or higher are considered clinically significant. In this study, ALGRX 3268 did not cause clinically significant redness, swelling or pinpoint blood spots.
Following administration at the antecubital fossa, it was found that a dose of 0.25 mg of ALGRX 3268 at 20 bar helium pressure did not reduce pain on needle insertion in a statistically significant manner when administered one to three minutes prior to needle insertion. In contrast, a dose of 0.5 mg of ALGRX 3268 at 20 bar helium pressure was shown to reduce pain on needle insertion in a statistically significant manner in children aged three to seven when compared to placebo. Children aged eight to 18 years of age reported only mild pain on needle insertion, and ALGRX 3268 did not further reduce pain in a statistically significant manner. Given the lack of reports of pain in the older age groups, we decided to conduct an additional clinical study.
| Phase II Clinical Trial — Child Back of Hand |
In August 2004, we completed a randomized, double-blind, placebo-controlled phase II clinical trial that studied the safety and efficacy of ALGRX 3268 in children at one center in the United States. Skin irritation caused by lidocaine particle injection was evaluated in this study. We examined the patients’ skin for the appearance of redness, swelling and pinpoint blood spots. Each of these was measured using a five-point scale, on which scores of three or higher are considered clinically significant. In this study, ALGRX 3268 did not cause clinically significant redness, swelling or pinpoint blood spots.
In this trial, a dose of 0.5 mg of ALGRX 3268 at 20 bar helium pressure administered one to three minutes prior to needle insertion, was shown to reduce pain in a statistically significant manner in children aged three to 12 and in children aged 13 to 18 when compared to placebo.
| Phase III Clinical Trials — Child Antecubital Fossa and Back of Hand |
In November 2004, we initiated a randomized, double-blind, placebo-controlled phase III clinical trial to study the safety and efficacy of ALGRX 3268 in up to five centers in the United States. In January 2005, we initiated a separate identical phase III clinical trial in up to five centers in the United States. In each trial, we will study the reduction in needle-stick pain caused by 0.50 mg of lidocaine at 20 bar helium pressure in 500 children aged three to 18, when applied three minutes prior to needle-stick procedures at the antecubital fossa or back of hand. The safety indices to be measured in these studies include redness, swelling and the appearance of pinpoint blood spots.
| Regulatory Pathway |
In March 2002, the IND application for ALGRX 3268 was transferred to us by PowderJect Technologies Inc. All of our clinical trials to date have been conducted under this IND application. In November 2004, we had an end of phase II meeting with the FDA to discuss our results to date and to discuss our proposed phase III clinical program and manufacturing plan. Based on the results of this meeting, we initiated two phase III clinical trials. If our phase III clinical trial program is successful, we expect to submit an NDA with the FDA at the end of 2005 seeking marketing approval for ALGRX 3268.
57
Table of Contents
| Commercialization Plan |
In preparation for the potential approval and launch of ALGRX 3268, our most advanced product candidate, we plan to build a sales and marketing team to target the most highly concentrated U.S. market segments for this product candidate. We believe such a sales team will be able to market ALGRX 3268 to hospital- and medical center-based pediatric centers, emergency rooms, urgent care centers and pediatric and adult oncology centers within the top 50 to 100 major metropolitan areas in the United States. In addition to building our own sales and marketing team within the United States, we will also evaluate partnering with one or more pharmaceutical companies to help penetrate primary care markets and other specialty health care audiences not highly concentrated in hospitals or major medical centers within the United States. We expect that such partners would also have marketing responsibility for ALGRX 3268 in key regions outside the United States, including Europe and Japan.
ALGRX 1207 — Topical local anesthetic for painful conditions of the skin
| Overview |
ALGRX 1207, our preclinical product candidate, is a new molecular entity we are developing as a topical local anesthetic to potentially treat patients with certain types of neuropathic pain and for pre-procedural administration to reduce the pain associated with surgical procedures on the skin. Nearly two million patients in the United States suffer from the types of neuropathic pain that are readily treatable by topical application of drugs. Among the various types of neuropathic pain, the most severe is postherpetic neuralgia, which has a prevalence of approximately 200,000 patients in the United States. The most common neuropathic pain condition results from diabetic polyneuropathy, which is caused by nerve damage in patients with diabetes as a result of high circulating levels of blood sugar. Symptoms of diabetic polyneuropathy include numbness, pain, tingling or burning in the hands, lower legs and feet, and of the more than 3.5 million patients in the United States who suffer from diabetic polyneuropathy, approximately 14% of these patients experience pain as one of the symptoms. Neuropathic pain is also experienced in patients with HIV. Nearly one million people in the United States are infected with HIV, and, among those people, approximately 20% experience pain as a result of nerve damage caused by HIV infection.
Postherpetic neuralgia and diabetic polyneuropathy are not well-treated by current analgesic therapy. In the United States, the most common treatment involves application of patches containing a local anesthetic to the painful area. Lidoderm® (Endo) is the most commonly used treatment for postherpetic neuralgia. As with other existing topical treatments, Lidoderm® is inconvenient to use and requires that large patches be worn for 12 hours and then removed for 12 hours in order to avoid harmful side effects. For some neuropathic pain diseases, such as painful HIV polyneuropathy and painful diabetic polyneuropathy, application of patches is difficult. To treat the pain associated with such diseases, creams containing local anesthetics must first be applied, followed by application of a dressing. Both patches and creams containing local anesthetics have a slow onset of pain relief, and efficacy may be poor due to the poor penetration of skin by the local anesthetics that are currently available. We believe that a simple-to-use local anesthetic with a long duration of action and deep penetration into the skin would provide significant analgesia and would therefore represent an improvement over existing patches and creams. Based on nonclinical studies in animals, ALGRX 1207 has been shown to provide analgesia following direct administration to skin more rapidly and with a longer-lasting effect than currently available topical anesthetics. In addition, we believe that ALGRX 1207, if approved, could address the pain associated with a wide variety of procedures involving the skin, including dermatological surgery, cosmetic skin treatments, and catheter placement, as well as the pain arising from surgical incisions that does not subside after the surgery has taken place.
| Nonclinical Results |
To date, ALGRX 1207 has been studied in nonclinical trials. In these animal studies, analgesia following the administration of ALGRX 1207 was demonstrated within ten minutes of application,
58
Table of Contents
compared to more than 30 minutes following lidocaine. These studies also demonstrated that ALGRX 1207 provides analgesia lasting about five hours compared to less than one-half hour for lidocaine.
| Regulatory Pathway |
ALGRX 1207 is currently undergoing drug safety evaluation and formulation development in nonclinical trials. We plan to file an IND covering ALGRX 1207 with the FDA in the second half of 2005 and, thereafter, evaluate the safety and effectiveness of ALGRX 1207 in humans in phase I clinical trials.
Intellectual Property
The following factors are important to our success:
| • | receiving patent protection for our product candidates; | |
| • | not infringing on the intellectual property rights of others; | |
| • | preventing others from infringing our intellectual property rights; and | |
| • | maintaining our patent rights and trade secrets. |
We actively seek, when appropriate, protection for our products, technologies and proprietary information through U.S. and foreign patents. In addition, we rely upon trade secrets and contractual arrangements to protect our proprietary information.
As of September 30, 2004, we own or license approximately 10 U.S. patents, 30 U.S. patent applications, 90 foreign patents and 140 foreign patent applications related to our technologies, compounds and their applications in pharmaceutical development or their use as pharmaceuticals. We face the risk that one or more of the above patent applications may be denied. We also face the risk that our issued patents may be challenged or circumvented or may otherwise not provide protection for any commercially viable products we develop. We also note that U.S. patents and patent applications may be subject to interference proceedings, and U.S. patents may be subject to reexamination proceedings in the U.S. Patent and Trademark Office and foreign patents may be subject to opposition or comparable proceedings in the corresponding foreign patent office, which could result in either loss of the patent or denial of the patent application or loss or reduction in the scope of one or more of the claims of the patent or patent application. In addition, such interference, reexamination and opposition proceedings may be costly. Furthermore, an adverse decision in an interference proceeding can result in a third party receiving the very patent rights sought by us, which in turn could affect our ability to market a potential product to which that patent filing was directed. In the event that we seek to enforce any of our owned or exclusively licensed patents against an infringing party, it is likely that the party defending the claim will seek to invalidate the patents we assert, which, if successful, would result in the entire loss of our patent or the relevant portion of our patent and not just with respect to that particular infringer. Any litigation to enforce or defend our patent rights, even if we were to prevail, could be costly and time-consuming and would divert the attention of our management and key personnel from our business operations.
In addition, our ability to assert our patents against a potential infringer depends on our ability to detect the infringement in the first instance. Many countries, including certain European countries, have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties in some circumstances. For example, compulsory licenses may be required in cases where the patent owner has failed to “work” the invention in that country, or the third party has patented improvements. In addition, many countries limit the enforceability of patents against government agencies or government contractors. In these countries, the patent owner may have limited remedies, which could materially diminish the value of the patent. Compulsory licensing of life saving drugs is also becoming increasingly popular in developing countries either through direct legislation or international initiatives. Such compulsory licenses could be extended to include some of our product candidates, which could limit our potential revenue opportunities. Moreover, the legal systems of certain countries, particularly certain
59
Table of Contents
developing countries, do not favor the aggressive enforcement of patent and other intellectual property protection.
Our success will also depend in part upon our not infringing patents issued to others. If our product candidates are found to infringe the patents of others, our development, manufacture and sale of such potential products could be severely restricted or prohibited. In fact, one of our issued European patents covering capsaicin for injection has been challenged by Grunenthal, a German pharmaceutical company, in the European Patent Court. In response to this challenge, we submitted proposed modifications to the patent which the patent court approved and published in November 2004. The amended patent can be objected to by Grunenthal or any other third party within two months following publication of the amended patent by the court. If any future challenge by Grunenthal or any other party is ultimately successful in invalidating the patent, the ability of third parties to market competing technologies to ALGRX 4975 in Europe could be enhanced.
Patent litigation can involve complex factual and legal questions and its outcome is uncertain. Any claim relating to infringement of patents that is successfully asserted against us may require us to pay substantial damages. Even if we were to prevail, any litigation could be costly and time-consuming and would divert the attention of our management and key personnel from our business operations. Furthermore, if a patent infringement suit were brought against us or our future strategic partners or licensees, if any, we or they may be forced to stop or delay developing, manufacturing or selling potential products that are claimed to infringe a third party’s intellectual property unless that party grants us or our strategic partners or licensees rights to use its intellectual property. In such cases, we may be required to obtain licenses to patents or proprietary rights of others in order to continue to commercialize our products. However, we may not be able to obtain any licenses required under any patents or proprietary rights of third parties on acceptable terms, or at all. Even if our strategic partners, licensees or we were able to obtain rights to the third party’s intellectual property, these rights may be non-exclusive, thereby giving our competitors access to the same intellectual property. Ultimately, we may be unable to commercialize some of our potential products or may have to cease some of our business operations as a result of patent infringement claims, which could severely harm our business.
Much of our technology and many of our processes depend upon the knowledge, experience and skills of our scientific and technical personnel. To protect rights to our proprietary know-how and technology, we generally require all employees, contractors, consultants, advisors, visiting scientists and collaborators as well as potential collaborators to enter into confidentiality agreements that prohibit the disclosure of confidential information. The agreements with employees and consultants also require disclosure and assignment to us of ideas, developments, discoveries and inventions. These agreements may not effectively prevent disclosure of our confidential information or provide meaningful protection for our confidential information.
Many of our employees were previously employed by other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management. If we fail in defending such claims, in addition to paying money claims, we may lose valuable intellectual property rights or personnel.
License Agreements
| License Agreement with James N. Campbell, M.D., Richard A. Meyer, M.S. and Marco Pappagallo, M.D. |
In August 2001, we entered into an agreement with James N. Campbell, M.D., Richard A. Meyer, M.S. and Marco Pappagallo, M.D. to acquire the exclusive, worldwide license to U.S. Patent Application No. 09/041294 (U.S. Patent No. 5,962,532) and all applications and products relating thereto directed to methods and kits for relieving pain using capsaicin and an anesthetic. The technology licensed under the
60
Table of Contents
agreement relates to the steps of administering capsaicin for pain reduction that we use in our product ALGRX 4975. This license excludes topical applications of capsaicin and analogues to the skin. Upon execution of the agreement, we paid to the licensees an aggregate up-front license fee of approximately $42,000, granted options to the licensees for an aggregate of 21,667 shares of our common stock and reimbursed the licensees for expenses associated with filing, prosecution and maintenance of the patent. We are obligated to pay Drs. Campbell and Pappagallo and Mr. Meyer royalties on any future sales of ALGRX 4975 by us and any of our sublicensees. We are also obligated to pay up to $775,000 in milestone payments under the agreement, of which, as of September 30, 2004, we have paid an aggregate of $200,000. Of the remaining milestone payments, we are obligated to pay $25,000 upon the grant of a Japanese patent using the licensed technology, $200,000 upon the first administration of licensed technology in a phase III clinical trial and $350,000 upon approval of the licensed technology for commercial use by the FDA. The license terminates on March 12, 2018, the date of expiration of the patent (U.S. Patent No. 5,962,532), or earlier upon the date of the invalidation of the patent. Our rights under this agreement can be terminated on 10 days’ written notice if we fail to fulfill any material obligation under the agreement and the failure is not cured by us within 180 days of receiving notice of such failure. We can terminate the agreement upon 30 days’ prior notice for any reason or upon 10 days prior notice for the failure of any counterparty to fulfill a material obligation not cured within 90 days of our giving notice of the failure. The license is subject to a license granted by Drs. Campbell and Pappagallo and Mr. Meyer to Johns Hopkins University for non-profit purposes. The license is subject to a sublicense to the inventors for research and development, with no right to commercialization.
| License Agreement with Marco Pappagallo, M.D. |
In August 2001, we entered into a non-exclusive, worldwide license agreement with Marco Pappagallo, M.D. for U.S. Provisional Patent Application No. 60/006,385 and U.S. Utility Patent Application No. 08/746,207 (U.S. Patent No. 6,248,788) directed to methods of treating neuropathic pain using capsaicin anesthetic, and all applications and patents relating thereto. The licensed technology relates to the use of capsaicin for pain relief. The primary patent underlying the license expires on November 6, 2016. This license agreement makes reference to the August 2001 license agreement between us and Drs. Campbell and Pappagallo and Mr. Meyer and provides that if Dr. Pappagallo develops or has any right to any technology under U.S. Patent No. 6,248,788 relating to an injectable product or service using capsaicin and its analogues for pain relief, the technology will be licensed to us pursuant to the terms of the August 2001 license agreement with Drs. Campbell and Pappagallo and Mr. Meyer. We are also obligated to pay up to $222,000 in milestone payments, and we have made no milestone payments to date. Of the $222,000 in milestone payments, $40,000 is payable upon the first administration to a subject using licensed technology in a phase I clinical trial, $66,000 is payable upon the first administration to a subject using licensed technology in a phase III clinical trial and $116,000 is payable upon FDA approval of the first product using licensed technology. With respect to the licensed technology, we are obligated to pay Dr. Pappagallo royalties on any future sales by us or our sublicensees of transdermal or topical products or services developed from the licensed technology. If at any time Dr. Pappagallo becomes the exclusive owner of the licensed technology, the royalty payments that we are obligated to pay will increase and we will be obligated to make milestone payments of up to $666,000. Our rights under the agreement can be terminated on 10 days’ written notice if we fail to fulfill any material obligation under the agreement and the failure is not cured by us within 180 days of receiving notice of such failure. We can terminate the agreement upon 30 days’ prior notice for any reason or upon 10 days’ prior notice for the failure of any counterparty to fulfill a material obligation not cured within 90 days of our giving notice of the failure. The license is subject to a sublicense to the inventors for research and development, with no right to commercialization.
| License with PowderMed Limited (formerly with PowderJect Research Limited) |
In March 2002, we acquired from PowderJect Research Limited a license to intellectual property consisting of over 150 patents and applications relating to the methods and apparatus for the delivery of powder forms of medications. The technology licensed under this agreement with PowderJect includes the
61
Table of Contents
technology underlying our product ALGRX 3268. The license is exclusive worldwide with respect to products delivered by powder injection into the space between cells under the skin, except for certain immune products and cytokine drugs and except for products to which PowderJect retained the exclusive right for delivery in dental procedures to the extracellular space within the oral cavity. PowderJect Research Limited is part of the Chiron group of companies operating under the Chiron Corporation. In May 2004, PowderJect Research Limited assigned its rights and obligations under the license agreement to PowderMed Limited, except that any royalties under the license for any future sales by us or sublicencees of ALGRX 3268 or other products derived from, or produced with the licensed technology will be payable by us to Chiron Vaccines Holdings Limited. With respect to ALGRX 3268, we are required to pay royalties to Chiron Vaccines Holdings Limited on any future direct sales and any future sales effected by any sublicensee. For products other than ALGRX 3268 resulting from the licensed technology, we are also obligated to pay Chiron royalties on any future direct sales. We must also pay royalties on licensing fees, milestone payments and other consideration that we receive from any sublicensees, if any. To date, we have received no milestone payments from any sublicensees. Under the agreement, if we use the licensed technology to develop cytokines, which are proteins that regulate blood cells, we are obligated to spend $1,000,000 researching and developing cytokines during the period July 7, 2003 to July 7, 2005, plus an additional $1,000,000 for every 12 month period from July 7, 2005 to July 7, 2008. To date, we have chosen not to use the licensed technology to develop cytokines.
The term of the license commenced on March 22, 2002 and continues until the expiration of the last patent to expire licensed under the agreement unless the agreement is otherwise terminated. The primary patents licensed under the agreement and used by us in connection with ALGRX 3268 expire in 2014. The agreement can be terminated by either party if the other party ceases to do business in the ordinary course, or assigns all or substantially all of its assets for the benefit of creditors. Either party can also terminate for material breach if not cured within 60 days of notice or if not cured within 30 days of notice if the breach relates to payment provisions. The license agreement also implemented an intellectual property sharing arrangement pursuant to which we and PowderMed Limited are obligated to share with one another any improvements and modifications to the licensed technology. The sharing arrangement expires on March 22, 2007.
| Collaboration, Development and License Agreement with Bridge Pharma, Inc. |
In October 2004, we entered into an agreement with Bridge Pharma, Inc. under which we acquired the exclusive worldwide license to proprietary technology relating to certain analgesic and local anesthetic pharmaceutical agents and compounds. The licensed technology relates to our product candidate, ALGRX 1207. The agreement also grants us the right to research, develop, sell, import or otherwise commercialize products based on such compounds, provided such products are an analgesic and/or local anesthetic for human or animals in any route of administration, including without limitation, dermal, mucosal, dental, ophthalmic or injection. Upon execution of the agreement, we paid Bridge Pharma, Inc. an up-front license fee consisting of a cash payment of $1 million and the issuance of 160,000 shares of our common stock. We are obligated to pay Bridge Pharma, Inc. royalties on any future sales by us or our sublicensees and additional payments if we achieve certain clinical, regulatory and commercial milestones. We are required to pay milestone payments upon the commencement of phase I, II and III clinical trials and upon the occurrence of certain events including the filing of a new drug application, the regulatory approval for each of the first and second products using the licensed technology and the reaching certain revenue thresholds from sales of products using the licensed technology. We may be obligated to pay up to an aggregate of $2.5 million in milestone payments prior to product approval, plus additional amounts up to an aggregate of $3.0 million payable upon the regulatory approval of a licensed product for each of the first, second and third indications. To date, we have paid no milestone payments. We are obligated to spend a minimum of $1 million for product development in each calendar year during the term of the agreement commencing in 2005 and ending on the first commercial sale of a product using the licensed technology. We are also responsible under the Bridge Pharma agreement for paying expenses associated with any patent prosecution and maintenance relating to the underlying technology and for certain costs associated with the research, development, regulatory filings and approvals and commercialization of
62
Table of Contents
products using the underlying technology. The term of the agreement commenced on October 28, 2004 and continues until our obligation to pay royalties to Bridge Pharma, Inc. expires, or earlier if terminated by either party. Either party may terminate the agreement for material breach if not cured within 60 days of notice, or with immediate effect if the other party makes an assignment to benefit creditors, files an insolvency petition in bankruptcy or commences any similar action such as a liquidation or reorganization.
Government Regulation
The FDA and comparable regulatory agencies in foreign countries, as well as local and state or other regional governmental agencies, impose substantial requirements upon the clinical development, manufacture and marketing of pharmaceutical products. These agencies and other federal, state and local entities regulate research and development activities and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising and promotion of our products.
The process required by the FDA before product candidates may be marketed in the United States generally involves the following:
| • | preclinical laboratory and animal tests; | |
| • | submission of an investigational new drug application, or IND application, which must become effective before clinical trials may begin; | |
| • | adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use; | |
| • | pre-approval inspection of manufacturing facilities, company regulatory files, and selected clinical investigators; and | |
| • | FDA approval of a new drug application, or NDA, or FDA approval of an NDA supplement in the case of a new indication if the product is already approved for another indication. | |
The testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any new approvals for our product candidates will be granted on a timely basis, if at all.
Prior to commencing the first human clinical trial, we must submit an IND application to the FDA. The IND application automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the preclinical drug testing or nonclinical safety evaluation in animals, or the design or conduct of the first proposed clinical trial and places the study on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial may begin. Our submission of an IND application may result in the FDA’s denial of authorization to commence a clinical trial. In fact, with respect to our first IND application for ALGRX 4975 and our IND application for ALGRX 3268, the FDA placed our clinical trials on hold. In the case of ALGRX 3268, the FDA requested the results from several nonclinical studies regarding the ways in which particles were ejected from the device. In the case of ALGRX 4975, the FDA requested that we provide them with the results of an ongoing osteoarthritis clinical trial then underway in the United Kingdom under the clinical trials exemption. In both cases, the FDA subsequently lifted each hold to allow us to initiate or continue the trials after we submitted the information they had requested. A separate submission to the existing IND application must be made for each successive clinical trial conducted during product development, and the FDA must not object to the submission before each clinical trial may start and continue. Further, an independent institutional review board for investigation in human subjects within each medical center in which an investigator wishes to participate in the clinical trial must review and approve the preclinical drug testing and nonclinical safety evaluation and efficacy in animals or prior human trials as well as the design and goals of the proposed clinical trial before the clinical trial commences at that center. Regulatory authorities or an institutional review board or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
63
Table of Contents
For purposes of NDA approval, human clinical trials are typically conducted in three sequential phases that may overlap.
| • | Phase I: Phase I clinical studies are initially conducted in a limited patient population to evaluate the product candidate for safety, dosage tolerance, absorption, metabolism, distribution and excretion in healthy humans or patients. | |
| • | Phase II: Phase II clinical studies are conducted in a limited patient population to further identify and measure possible adverse effects or other safety risks, to determine the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. Multiple phase II clinical trials may be conducted by the sponsor to obtain information prior to beginning larger and more expensive phase III clinical trials. The first, limited phase II clinical trials may be termed “phase IIa” trials to denote that only a few patients are enrolled in order to obtain preliminary safety and efficacy data. Later, larger phase II clinical trials may be termed “phase IIb” trials to denote that preliminary evidence of safety and efficacy has already been obtained, and the current trial is intended to confirm the finding of the earlier trial, as well as to enroll additional subjects in order to further refine the estimate of the optimal dose and to detect less common side effects. In some instances, a phase IIb trial may be declared acceptable by a regulatory agency, such as the FDA, as a “pivotal” trial necessary for purposes of obtaining marketing authorization for a drug. | |
| • | Phase III: When phase II clinical evaluations demonstrate that a dosage range of the product appears to be effective and has an acceptable safety profile, phase III clinical trials are undertaken in large patient populations to further evaluate dosage, to confirm clinical efficacy and to evaluate safety in yet larger and more diverse patient populations at multiple clinical trial sites. |
The FDA may require, or companies may pursue, additional clinical trials after a product is approved. These so-called phase IV clinical studies may be made a condition to be satisfied after a drug receives approval. The results of phase IV clinical studies may confirm the effectiveness of a product and may provide important safety information to augment the FDA’s voluntary adverse drug reaction reporting system.
The results of product development, preclinical studies and clinical trials are submitted to the FDA as part of an NDA, or as part of an NDA supplement. The FDA may deny approval of an NDA or NDA supplement if the applicable regulatory criteria are not satisfied, or it may require additional clinical data or an additional pivotal phase III clinical trial. Even if such data are submitted, the FDA may ultimately decide that the NDA or NDA supplement does not satisfy the criteria for approval. The FDA may withdraw product approval, once issued, if ongoing regulatory standards are not met or if safety problems occur after the product reaches the market. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved products which have been commercialized, and the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs.
Satisfaction of FDA requirements or similar requirements of state, local and foreign regulatory agencies typically takes several years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease. Typically, if a drug product is intended to treat a chronic disease, as is the case with some of the product candidates we are developing, safety and efficacy data must be gathered over an extended period of time, which can range from one to three years or more. Government regulation may delay or prevent marketing of product candidates for new indications for a considerable period of time and impose costly procedures upon our activities. The FDA or any other regulatory agency may not grant approvals for new indications for our product candidates on a timely basis, if at all. Success in early stage clinical trials does not ensure success in later stage clinical trials. Data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. Even if a product candidate receives regulatory approval, the approval may be significantly limited to specific disease states, patient populations and dosages. Further, even after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. Delays in obtaining, or failures to obtain, additional regulatory approvals for our
64
Table of Contents
products would harm our business. In addition, we cannot predict what adverse governmental regulations may arise from future U.S. or foreign governmental action.
Any products manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including record-keeping requirements and reporting of adverse experiences with the drug. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with good manufacturing practices, which impose certain procedural and documentation requirements upon us and our third party manufacturers. We cannot be certain that we or our present or future suppliers will be able to comply with the good manufacturing practices regulations and other FDA regulatory requirements. If our present or future suppliers are not able to comply with these requirements, the FDA may halt our clinical trials, require us to recall a drug from distribution, or withdraw approval of the NDA for that drug.
The FDA closely regulates the marketing and promotion of drugs. A company is permitted to make only those claims relating to safety and efficacy that are approved by the FDA. Failure to comply with these requirements can result in adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available drugs for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturers’ communications on the subject of off-label use.
The FDA’s policies may change, and additional government regulations may be enacted which could prevent or delay regulatory approval of our product candidates or approval of new diseases for our product candidates. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the United States or abroad.
Supply Agreement
In March 2002, we entered into a supply agreement with PowderJect Technologies Limited to supply us with the cylinders that are a key component in the dispenser for ALGRX 3268. PowderJect Technologies Limited is part of the Chiron group of companies operating under the Chiron Corporation. PowderJect Technologies Limited is currently our sole supplier of cylinders, and we currently have no alternate supplier or source of cylinders. The cylinders are exclusively manufactured for PowderJect Technologies Limited by The BOC Group plc pursuant to supply and manufacturing agreements by and between PowderJect Technologies Limited and the third party manufacturer. We are not a party to any agreements with that third party manufacturer. If The BOC Group plc breaches its obligations to PowderJect Technologies Limited and the breach results in PowderJect Technologies Limited’s inability to supply us with cylinders, PowderJect Technologies Limited, under the terms of its supply agreement with us, must take all reasonable steps to enforce its rights under its agreements with The BOC Group plc and seek remedies that are in the best interest of AlgoRx and PowderJect Technologies Limited, including without limitation, seeking specific performance or injunctive relief. Although we have not experienced any shortages of cylinders to date, our inability to obtain the cylinders for any reason could substantially impair our development activities or the production, marketing and distribution of ALGRX 3268.
The term of our supply agreement with PowderJect Technologies Limited commenced on March 22, 2002 and continues until the later of March 22, 2012 and the termination of the agreement with the third party manufacturer, unless terminated earlier. We have an option to extend the term of the agreement until March 2014, if the agreement with the third party manufacturer terminates on mutually agreeable terms before March 2010. We can terminate our supply agreement with PowderJect Technologies Limited on account of a material uncured breach. If we materially breach our payment obligations under the supply agreement with PowderJect Technologies Limited, PowderJect Technologies Limited can suspend its supply obligations to us if we fail to cure the breach within 60 days of the date of notice of such
65
Table of Contents
breach. Our cost per cylinder is based on a number of factors including, PowderJect Technologies Limited’s direct cost of materials and labor and fixed costs, such as utilities and rent, allocated to us by PowderJect Technologies Limited.
Manufacturing
We have no manufacturing facilities. We have entered into arrangements with various third parties for the formulation and manufacture of our clinical supplies. These supplies and the manufacturing facilities must comply with regulations and current good laboratory practices or cGLPs, and current good manufacturing practices or cGMPs, enforced by the FDA. We plan to continue to outsource formulation and manufacturing for our clinical trials and potential commercialization. Other than for the cylinder used in ALGRX 3268 described above, we believe that there are alternate manufacturers available to produce our clinical supplies and, if our product candidates are approved by the FDA, commercial supplies of our product components.
Competition
We compete in the segment of the pharmaceutical market that treats pain, which is highly competitive. We face significant competition from most pharmaceutical companies as well as biotechnology companies that are also researching and selling products designed to treat pain. Many of our competitors have significantly greater financial, manufacturing, marketing and product development resources than we do. Large pharmaceutical companies in particular have extensive experience in clinical testing and in obtaining regulatory approvals for drugs. These companies also have significantly greater research capabilities than we do. In addition, many universities and private and public research institutes are active in neurological research, some in direct competition with us. We also must compete with these organizations to recruit scientists and clinical development personnel.
We believe that our ability to successfully compete will depend on, among other things:
| • | the efficacy, safety and reliability of our product candidates; | |
| • | the timing and scope of regulatory approval; | |
| • | the speed at which we develop product candidates; | |
| • | our ability to manufacture and sell commercial quantities of a product to the market; | |
| • | product acceptance by physicians and other health care providers; | |
| • | the quality and breadth of our technology; | |
| • | the skills of our employees and our ability to recruit and retain skilled employees; | |
| • | the protection of our intellectual property; and | |
| • | the availability of substantial capital resources to fund development and commercialization activities. |
ALGRX 4975. If ALGRX 4975 is approved and commercialized, it will face significant competition. For treatment of severe postsurgical pain, morphine administered by infusion pump is a leading product. Several other oral, injectable and patch opioid formulations are also used, including Vicodin® (Abbott Labs), OxyContin® (Purdue Pharma) and Duragesic® (Johnson & Johnson). For treatment of localized neuropathic pain, Neurontin® (Pfizer) and tricyclic antidepressants (several manufacturers) are used. For treatment of later-stage osteoarthritis, Synvisc® (Genzyme) and other hyaluronic acid products are injected locally into the knee, and several opioids, most prominently OxyContin® (Purdue Pharma) and Duragesic® (Johnson & Johnson) are also used. VR1 inhibitors are being developed by several companies. Among these, products being developed by Merck-Neurogen, Amgen, Schwarz Pharma-Amore Pacific, Purdue Pharma and PainCeptor are expected to advance to clinical evaluation in late 2004 through 2005.
ALGRX 3268. If ALGRX 3268 is approved and commercialized, it will face significant competition. During 2003, the leading products for local anesthesia prior to needle-stick procedures were L.M.X.4®, a cream-based product (formerly ELA-MAX®, Ferndale Labs), and EMLA®, a cream-based product sold
66
Table of Contents
by AstraZeneca. EMLA® has historically been the market leader. Additionally, beginning in 2003, several generic versions of EMLA® manufactured by companies including Fougera, QLT, Geneva, and Hi-Tech Pharmacal were approved by the FDA. Products with more rapid onset than the cream-based products listed above include Numby Stuff® (Iomed) and two products for which NDAs have been submitted, LidoSite® (Braun-Vyteris) and S-Caine® Patch (ZARS).
ALGRX 1207. If ALGRX 1207 is approved and commercialized, it will face significant competition from existing products, including LidoDerm® (Endo), which is a lidocaine patch, and a variety of local anesthetic creams and products with alternative means of delivering lidocaine, including EMLA® cream (AstraZeneca) and its generic equivalents, L.M.X.4® (Ferndale Labs), S-Caine® Patch (ZARS) and LidoSite® (Braun-Vyteris). There are capsaicin products in development by NeurogesX and Winston Labs, which could be applied to the skin and which may be approved prior to ALGRX 1207. Another VR1 agonist, resiniferatoxin, which is currently in phase II clinical trials, has similar attributes as capsaicin, is being developed for topical delivery by ICOS and may compete with ALGRX 1207. If approved, ALGRX 1207, which will also likely be formulated as a cream or patch, will compete with existing products based on factors such as efficacy, convenience and onset time of pain relief.
Legal Proceedings
We are not involved in any material legal proceedings.
Facilities
We currently lease and occupy approximately 14,900 square feet of office space in Secaucus, New Jersey. The lease expires on July 6, 2009, although we have an option to extend the lease for an additional five years at the then market rate. We also lease 24,800 square feet of office and laboratory space in Fremont, California, but have since scaled back operations in California. The Fremont, California lease expires on January 31, 2005, and we are currently seeking to lease approximately 2,300 square feet of new office space near our current Fremont, California facility to occupy upon the expiration of our current Fremont lease. We believe our facilities are adequate for our present operations.
Employees
As of September 30, 2004, we had 23 full-time employees. We believe our relations with our employees are good.
67
Table of Contents
MANAGEMENT
Executive Officers, Directors and Key Employees
Our executive officers, directors and key employees, as of January 17, 2005, are as follows:
| Name | Age | Positions | ||||
| Ronald M. Burch, M.D., Ph.D. | 50 | Chief Executive Officer and Director | ||||
| Paul R. Hamelin, R.Ph. | 50 | President and Chief Operating Officer | ||||
| Jeffrey D. Lazar, M.D., Ph.D. | 58 | Senior Vice President, Clinical Research and Regulatory Affairs | ||||
| Jeffrey A. Rona | 36 | Vice President, Finance and Chief Financial Officer | ||||
| Badri N. Dasu | 41 | Vice President, Manufacturing and Process Development | ||||
| Diana Davidson | 49 | Vice President, Marketing | ||||
| Charles M. Cohen, Ph.D.(1) | 54 | Chairman of the Board of Directors | ||||
| Rodney A. Ferguson, J.D., Ph.D.(1) | 48 | Director | ||||
| Arnold L. Oronsky, Ph.D.(2) | 63 | Director | ||||
| Michael F. Powell, Ph.D.(1)(2) | 50 | Director | ||||
| Carter H. Eckert(2) | 62 | Director | ||||
| (1) | Member of the audit and compensation committees. |
| (2) | Member of the nominating and corporate governance committee. |
Ronald M. Burch, M.D., Ph.D. co-founded AlgoRx Pharmaceuticals, Inc. in March 2001, and served as our President until September 2004 and as Chief Executive Officer since inception. Prior to joining AlgoRx, Dr. Burch was employed at Purdue Pharma, a privately held pharmaceutical company, from 1995 until 2001, serving in a number of managerial positions, including Vice President, Scientific Evaluations and Immunotherapeutics and Project Manager and Medical Safety Officer for several pain development programs. From 1993 to 1995, Dr. Burch served as Director, Pharmacology at Zeneca Pharmaceuticals, a pharmaceutical company. In 1992, Dr. Burch served as Director, Immunology and Bone Metabolism at Rhone Poulenc-Rorer, a global pharmaceutical company, and as Director, Pain and Inflammation at Nova Pharmaceutical Corp., a pharmaceutical company. From 1987 to 1991, Dr. Burch served in various capacities at Nova Pharmaceutical, including as Director, Pain and Inflammation, Research Technology. Dr. Burch obtained a Ph.D. in Pharmacology and M.D. from the Medical University of South Carolina, and served as a Medical Staff Fellow at the National Institute of Health.
Paul R. Hamelin, R.Ph.has served as our President and Chief Operating Officer since September 2004. Prior to joining us, Mr. Hamelin served as President and Chief Executive Officer of Elitra Pharmaceuticals, a biotechnology company with a focus on antimicrobial drugs, from 2002 to 2004. From 2000 to 2002, he served as Senior Vice President of Global Commercial Operations at Millennium Pharmaceuticals, a biopharmaceutical company. Prior to joining Millennium Pharmaceuticals, Mr. Hamelin served as Senior Vice President at Pharmacia/ Searle, a pharmaceutical company, from 1995 to 2000. Prior to Pharmacia/ Searle, Mr. Hamelin spent 6 years at Abbott Laboratories, a pharmaceutical company, and 10 years at Eli Lilly and Company, a pharmaceutical company. At these pharmaceutical companies, Mr. Hamelin had many different sales and marketing roles, including the responsibility of launching several products. Mr. Hamelin obtained a Bachelor’s Degree in Pharmacy — Clinical Pharmacist from Ferris State University in 1980 and a B.S. in Zoology from Michigan State University in 1976.
Jeffrey D. Lazar, M.D., Ph.D.has served as our Senior Vice President, Clinical Research and Regulatory Affairs since 2002. From 1999 to 2002, he served as President, Lazar Associates, LLC, a consulting firm to pharmaceutical companies. Prior to his work with Lazar Associates, Dr. Lazar served as Senior Medical Director at Purdue Pharma from 1997 to 1999. From 1996 to 1997, he served as Director
68
Table of Contents
of the Clinical Trials Management Office for the Greenville Hospital System. From 1992 to 1995, he served as Executive Vice President of Pharmaco Dynamics Research, Inc. From 1985 to 1992, he served as Director of Clinical Pharmacology at Pfizer. Dr. Lazar received his M.D. from the University of Michigan in 1974 and his Ph.D. from the University of Heidelberg in 1973. He was also a fellow at Vanderbilt University.
Jeffrey A. Ronahas served as our Chief Financial Officer since February 2004. Prior to becoming our Chief Financial Officer, he served as our Vice President of Finance from September of 2002 to February of 2004. Prior to joining us, Mr. Rona was in the Investment Banking Department at UBS Warburg, a global securities and investment banking firm, from 2000 to 2002. From 1998 to 2000, Mr. Rona served as the Director of Finance and Corporate Development at Antigenics Inc., a biotechnology firm developing products to treat cancer, infectious diseases and autoimmune disorders. In 1998, Mr. Rona was employed by Carr & Company, a private equity firm. From 1990 to 1997, Mr. Rona was with Coopers and Lybrand and its wholly-owned subsidiary Coopers & Lybrand Securities, serving in a variety of capacities. Mr. Rona received a B.S. in Accounting from Case Western Reserve University in 1990.
Badri N. Dasuhas served as our Vice President, Manufacturing and Process Development since 2002. Prior to joining us, he was employed at PowderJect Technologies Inc., a pharmaceutical company, from 2000 until 2002, serving in a number of positions including Vice President, Manufacturing, Process Development and Device Development. From 1996 to 2002, Mr. Dasu served in a variety of engineering positions including Director, Process Development and Product Transfer at Metrika Inc., a maker of micro optical, disposable medical diagnostics. From 1992 to 1996, Mr. Dasu was an Engineer at Cygnus Inc., a maker of drug delivery systems. Mr. Dasu has a M.S. in Chemical Engineering from the University of Tulsa.
Diana Davidsonhas served as our Vice President, Marketing since July 2003. Prior to joining us, she served in a variety of positions at Elan Corporation, a pharmaceutical company developing products for neurological and autoimmune diseases, from 1994 to 2003, including Vice President of Marketing for Elan Drug Delivery from 1994 to 2003. From 1986 to 1994, she worked at Pharmacia Deltec and held a number of positions including Director of Marketing and Director of Strategic Planning. Ms. Davidson received a B.S. in Laboratory Technology from the University of Oklahoma in 1977.
Charles M. Cohen, Ph.D.has served as a member of our board of directors since February 2004 and has served as the Chairman of our board of directors since December 2004. Since May 2003, Dr. Cohen has been a partner at Advent International, a global venture capital firm. Currently Dr. Cohen is the Chairman, Supervisory Board of Cellzome AG, a post-genomics biopharmaceutical company. From 2000 to 2002, Dr. Cohen was the Chief Executive Officer of Cellzome AG. Before this, Dr. Cohen co-founded Creative BioMolecules, Inc., a biotechnology company, in 1982 and was a director and its President and Chief Executive Officer from 1985 to 1995. Dr. Cohen serves on the board of directors of Exelixis, Inc. and Anadys Pharmaceuticals, Inc. He has been the Chief Executive Officer of several companies. He received his Ph.D. from New York University School of Medicine.
Rodney A. Ferguson, J.D., Ph.D. has served as a member of our board of directors since March 2001. Dr. Ferguson is a Partner on the Life Sciences venture capital team at JPMorgan Partners, a global private equity firm. Prior to joining JPMorgan Partners in January 2001, Dr. Ferguson was a Partner at InterWest Partners, a venture capital firm, focusing on investments in medical technology, from July 1999 to December 2000. Prior to joining InterWest, Dr. Ferguson was Senior Director of Business and Corporate Development at Genentech, Inc., a biotechnology company. Prior to joining the business and corporate development department at Genentech in 1993, Dr. Ferguson was Senior Corporate Counsel in Genentech’s legal department. Prior to joining Genentech in 1988, Dr. Ferguson was an associate at McCutchen, Doyle, Brown & Enersen in San Francisco. Dr. Ferguson is also a director and chairman of Corgentech Inc. and a director of Santarus, Inc. Dr. Ferguson holds a Ph.D. in Biochemistry from the State University of New York at Buffalo and a J.D. from Northwestern University.
Arnold L. Oronsky, Ph.D. has served as a member of our board of directors since March 2001. Dr. Oronsky is General Partner with InterWest Partners, a venture capital firm focusing on investments in
69
Table of Contents
medical technology. Dr. Oronsky joined InterWest in a full-time capacity in 1994 after serving as a special limited partner since 1989. In addition to the position of General Partner at InterWest, he also serves as a senior lecturer in the Department of Medicine at Johns Hopkins Medical School. From 1980 to 1993, Dr. Oronsky was the Vice President for Discovery Research at the Lederle Laboratories division of American Cyanamid Company, a pharmaceutical company. From 1970 to 1972, Dr. Oronsky was assistant professor at Harvard Medical School, where he also served as a research fellow from 1968 to 1970. From 1973 to 1976, Dr. Oronsky was the head of the Inflammation, Allergy, and Immunology Research program for Ciba-Geigy Pharmaceutical Company. Dr. Oronsky is also a director of Myogen Inc., Corixa Corporation, Metabasis Therapeutics, Inc. and Dynavax Technologies Corp. Dr. Oronsky holds a Ph.D. from Columbia University’s College of Physicians and Surgeons.
Michael F. Powell, Ph.D. has served as a member of our board of directors since March 2001. He has served as Managing Director of Sofinnova Ventures, a venture capital firm, since 1998. Previously, he was a Group Leader at Genentech from December 1990 to June 1997 and Director of Product Development for Cytel Corporation, a biotechnology company, from September 1987 to December 1990. In 1993, Dr. Powell was honored as a Fellow by the American Association of Pharmaceutical Scientists. Dr. Powell is the author of nearly 100 publications and books, including a treatise on vaccine design. Dr. Powell is also a director of Seattle Genetics, Inc. Dr. Powell received a Ph.D. in Chemistry from the University of Toronto in 1981 and was a postdoctoral fellow in Bio-Organic Chemistry at the University of California, Berkeley.
Carter H. Eckert has served as a member of our board of directors since December 2004. From February 2003 to March 2004, Mr. Eckert was the Chairman of the Board of Directors and Chief Executive Officer of IMPATH Inc., a cancer information company. In September 2003, IMPATH filed voluntary petitions for reorganization under Chapter 11 of the U.S. Bankruptcy Code. IMPATH Inc. subsequently sold certain of its assets to IMPAC Medical Systems, Inc. in November 2003 and sold its remaining assets to Genzyme Corporation in May 2004. From 1995 to 2001, Mr. Eckert served as President of Knoll Pharmaceutical Company and as President of the Americas for Knoll’s parent company, BASF Pharma. During that period, Mr. Eckert also was a member of BASF Pharma’s Pharmaceutical Board, where he was responsible for global therapeutic franchises and corporate transactions. Prior to joining Knoll and BASF Pharma in 1995, Mr. Eckert was President and Chief Executive Officer of Boots Pharmaceuticals, Inc. where he was responsible for North American operations. Mr. Eckert joined Boots Pharmaceuticals in 1985 as Executive Vice President and Chief Operating Officer after more than a decade at Baxter Travenol Laboratories, where he served as President of the Pharmaceutical Products Division. Mr. Eckert also serves as a director of Orasure Technologies, Inc. and Andrx Corporation and a trustee of Caldwell College. Mr. Eckert received his B.S. in Chemical Engineering from the Illinois Institute of Technology and his M.B.A. from Northwestern University.
Scientific and Clinical Advisory Board
The following individuals are members of our Scientific and Clinical Advisory Board:
| Name | Current Positions | |
| James N. Campbell, M.D. | Professor of Neurosurgery and Vice Chairman, Department of Neurosurgery, The Johns Hopkins University School of Medicine and Director of the Blaustein Pain Treatment Program, The Johns Hopkins Hospital; President and Chairman, American Pain Foundation | |
| Richard B. Carter, Ph.D. | Executive Director, Neuroscience, Global Pharmaceutical Development, Novartis | |
| Ronald Dubner, D.D.S., Ph.D. | Professor and Chair, Department of Bio-Medical Sciences, University of Maryland Dental School; Director, University of Maryland Pain Center |
70
Table of Contents
| Name | Current Positions | |
| Michael G. Ehrlich, M.D. | Vincent Zecchino Professor and Chairman, Surgeon-in- Chief, Department of Orthopedics, Brown Medical School | |
| Henrik Kehlit, M.D., Ph.D. | Professor of Surgery, University of Copenhagen School of Medicine; Department of Surgical Gastroenterology, Hvidovre Hospital, Hvidovre, Denmark | |
| Kenneth A. Selzer, M.D. | General Partner, Neuropractice; Founder and Director, INC Research, Inc. | |
| Davis L. Temple, Jr., Ph.D. | President, Temple Consulting; Director, Cortex Pharmaceutical; Director, Arcaris Genetics; Director, InternetSound.com |
Composition of our Board of Directors
Immediately prior to this offering, our board of directors will be divided into three staggered classes of directors of the same or nearly the same number and each of our directors will be assigned to one of the three classes. At each annual meeting of stockholders, a class of directors will be elected for a three-year term to succeed the directors of the same class whose terms are then expiring. The terms of the directors will expire upon election and qualification of successor directors at the annual meeting of stockholders to be held during the years 2005 for the Class I directors, 2006 for the Class II directors and 2007 for the Class III directors.
Our certificate of incorporation and bylaws provide that the number of our directors shall be fixed from time to time by a resolution of the majority of our board of directors. Any additional directorships resulting from an increase in the number of directors will be distributed among the three classes so that, as nearly as possible, each class shall consist of one third of the directors. There are no family relationships among any of our directors or executive officers.
The division of our board of directors into three classes with staggered three-year terms may delay or prevent stockholder efforts to effect a change of our management or a change in control.
Committees of our Board of Directors
Our board of directors has established an audit committee and a compensation committee and intends, in conjunction with this offering, to establish a nominating and corporate governance committee.
Audit Committee. Our audit committee is a standing committee of our board of directors and operates under a written charter adopted by our board. Our audit committee consists of Dr. Charles M. Cohen (Chairman), Dr. Rodney A. Ferguson and Dr. Michael F. Powell. The audit committee reviews and monitors our accounting practices and financial statements; appoints, determines funding for, and oversees our independent auditors; reviews the results and scope of audits; approves the retention of the independent auditors to perform any proposed permissible non-audit services; and reviews and evaluates our audit and control functions. Dr. Cohen is an audit committee financial expert under the rules and regulations of the Securities and Exchange Commission.
Compensation Committee. Our compensation committee is a standing committee of, and operates under a written charter adopted by, our board of directors. Our compensation committee consists of Dr. Charles M. Cohen, Dr. Rodney A. Ferguson (Chairman) and Dr. Michael F. Powell. The compensation committee makes decisions and recommendations regarding salaries, benefits and incentive compensation for our directors and executive officers, and administers our incentive compensation and benefit plans, including our 2001 Equity Incentive Plan.
Nominating and Corporate Governance Committee. Our nominating and corporate governance committee is a standing committee of, and operates under a written charter adopted by, our board of directors. Our nominating and corporate governance committee consists of Carter H. Eckert, Dr. Michael
71
Table of Contents
F. Powell and Dr. Arnold L. Oronsky (Chairman). The nominating and corporate governance committee identifies and approves individuals qualified to serve as members of our board of directors, selects director nominees for our annual meetings of stockholders, evaluates our board’s performance and develops and recommends to our board corporate governance guidelines and provides oversight with respect to corporate governance and ethical conduct.
Other Committees. Our board of directors may establish other committees as it deems necessary or appropriate from time to time.
Compensation Committee Interlocks and Insider Participation
None of the members of our compensation committee has at any time been one of our officers or employees. None of our executive officers currently serves, or in the past fiscal year has served, as a member of the board of directors or compensation committee of any entity that has one or more executive officers serving on our board of directors or compensation committee.
Director Compensation
We reimburse our directors who are not employees for their reasonable expenses incurred in attending meetings of the board of directors. Our directors are eligible to participate in our 2001 Equity Incentive Plan.
Limitation of Liability and Indemnification of Officers and Directors
Our certificate of incorporation limits the liability of directors to the maximum extent permitted by Delaware law. Delaware law provides that directors of a corporation will not be personally liable for monetary damages for breach of their fiduciary duties as directors, except liability for:
| • | any breach of their duty of loyalty to the corporation or its stockholders; | |
| • | acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law; | |
| • | unlawful payments of dividends or unlawful stock repurchases or redemptions; or | |
| • | any transaction from which the director derived an improper personal benefit. |
Our bylaws provide that we will indemnify our directors and officers to the fullest extent permitted by law, including if he or she is serving as a director, officer, employee or agent of another company at our request. We believe that indemnification under our bylaws covers at least negligence and gross negligence on the part of indemnified parties. Our bylaws also permit us to secure insurance on behalf of any officer, director, employee or other agent for any liability arising out of his or her actions in connection with their services to us.
In conjunction with this offering, we intend to enter into separate indemnification agreements with our directors and executive officers, in addition to the indemnification provided for in our bylaws. These agreements, among other things, will provide that we will indemnify our directors and executive officers for certain expenses including attorneys’ fees, judgments, fines and settlement amounts incurred by a director or executive officer in any action or proceeding arising out of such person’s services as one of our directors or executive officers, or any of our subsidiaries or any other company or enterprise to which the person provides services at our request. We believe that these provisions and agreements are necessary to attract and retain qualified persons as directors and executive officers.
There is no pending litigation or proceeding involving any of our directors or executive officers as to which indemnification is required or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
72
Table of Contents
Executive Compensation
The following table sets forth information regarding the compensation that we paid to our Chief Executive Officer and each of our other most highly compensated executive officers during the years ended December 31, 2003 and December 31, 2004. We refer to these officers in this prospectus as the “named executive officers.”
Summary Compensation Table
| Long-Term | |||||||||||||||||||||||||
| Compensation | |||||||||||||||||||||||||
| Annual Compensation | |||||||||||||||||||||||||
| Securities | |||||||||||||||||||||||||
| Other Annual | Underlying | All Other | |||||||||||||||||||||||
| Name and Principal Position | Year | Salary($) | Bonus($) | Compensation(1) | Options | Compensation($) | |||||||||||||||||||
| Ronald M. Burch, M.D., Ph.D. | 2004 | 307,300 | 41,700 | — | 567,598 | 3,500 | (2) | ||||||||||||||||||
| Chief Executive Officer | 2003 | 278,000 | — | — | — | 2,940 | (2) | ||||||||||||||||||
| Paul R. Hamelin, R.Ph. | 2004 | 91,250 | 50,000 | 32,172 | (4) | 525,000 | 3,500 | (2) | |||||||||||||||||
| President and Chief Operating | |||||||||||||||||||||||||
| Officer(3) | |||||||||||||||||||||||||
| Jeffrey D. Lazar, M.D., Ph.D. | 2004 | 267,000 | 28,700 | — | 201,045 | 3,500 | (2) | ||||||||||||||||||
| Senior Vice President, Clinical | 2003 | 255,000 | — | — | — | 3,236 | (2) | ||||||||||||||||||
| Research and Regulatory Affairs | |||||||||||||||||||||||||
| Jeffrey A. Rona | 2004 | 197,000 | 25,000 | — | 182,078 | 3,500 | (2) | ||||||||||||||||||
| Vice President, Finance and | 2003 | 177,000 | 13,000 | — | — | 2,246 | (2) | ||||||||||||||||||
| Chief Financial Officer | |||||||||||||||||||||||||
| (1) | In accordance with the rules of the SEC, the compensation described in this table does not include medical, group life insurance or other benefits which are available generally to all of our salaried employees and certain perquisites and other personal benefits received which do not exceed the lesser of $50,000 or 10% of any named executive officer’s salary and bonus disclosed in this table. |
| (2) | Figures in this column represent 401(k) matching contributions. |
| (3) | Mr. Hamelin joined the Company as President and Chief Operating Officer in September 2004. |
| (4) | This amount represents an estimate of the Company’s reimbursement obligation for Mr. Hamelin’s relocation expenses. |
73
Table of Contents
Stock Option Grants in 2004
The following table sets forth information regarding grants of stock options to each of our named executive officers during the fiscal year ended December 31, 2004. The percentage of options granted was based on aggregate grants of options to purchase common stock totaling 1,902,440 shares during 2004. Potential realizable values are net of exercise price before taxes, and are based on the assumption that our common stock appreciates at the annual rate shown, compounded annually, from the date of grant until expiration of the 10 year term.
| Individual Grants | ||||||||||||||||||||||||
| Potential Realizable Value at | ||||||||||||||||||||||||
| Number of | Percentage of | Assumed Annual Rates of Stock | ||||||||||||||||||||||
| Shares of | Total Options | Price Appreciation for Option | ||||||||||||||||||||||
| Common Stock | Granted to | Terms | ||||||||||||||||||||||
| Underlying | Employees | Exercise Price | Expiration | |||||||||||||||||||||
| Name | Option Granted | in FY 2004 | Per Share(1) | Date | 5% | 10% | ||||||||||||||||||
| Ronald M. Burch, M.D., Ph.D. | 4,170 | (2) | 0.2 | % | $ | 1.50 | 01/15/14 | $ | 68,463 | $ | 112,718 | |||||||||||||
| 535,428 | (3) | 28.1 | % | $ | 1.00 | 03/23/14 | $ | 9,058,317 | $ | 14,740,707 | ||||||||||||||
| 28,000 | (4) | 1.5 | % | $ | 2.50 | 12/08/14 | $ | 431,701 | $ | 728,860 | ||||||||||||||
| Paul R. Hamelin, R.Ph. | 525,000 | (2) | 27.6 | % | $ | 1.00 | 09/29/14 | $ | 8,881,898 | $ | 14,453,617 | |||||||||||||
| Jeffrey D. Lazar, M.D., Ph.D. | 2,868 | (2) | 0.2 | % | $ | 1.50 | 01/15/14 | $ | 47,087 | $ | 77,524 | |||||||||||||
| 198,177 | (3) | 10.4 | % | $ | 1.00 | 03/23/14 | $ | 3,352,739 | $ | 5,455,952 | ||||||||||||||
| Jeffrey A. Rona | 2,500 | (2) | 0.1 | % | $ | 1.50 | 01/15/14 | $ | 41,045 | $ | 67,577 | |||||||||||||
| 143,974 | (2) | 7.6 | % | $ | 1.00 | 03/23/14 | $ | 2,435,738 | $ | 3,963,705 | ||||||||||||||
| 31,604 | (3) | 1.7 | % | $ | 1.00 | 03/23/14 | $ | 534,673 | $ | 870,080 | ||||||||||||||
| 4,000 | (4) | 0.2 | % | $ | 2.50 | 12/08/14 | $ | 61,672 | $ | 104,123 | ||||||||||||||
| (1) | There was no public trading market for our common stock during fiscal 2004. Accordingly, these exercise prices are based on our board of directors’ determination of the fair market value of the underlying shares as of the dates of grant. |
| (2) | A total of 25% of these options vest after one year from the date of grant and the remaining options vest monthly in equal installments over the following 36 months. |
| (3) | These options vest monthly in equal installments over the 48 months following the date of grant. |
| (4) | These options vested immediately at the date of grant. |
The potential realizable value is calculated based on the 10 year term of the stock option at the time of grant. The assumed 5% and 10% rates of stock price appreciation are provided in accordance with the rules of the SEC based on an assumed initial public offering price of $11 per share and do not represent our estimate or projection of our future stock price. The potential realizable values at 5% and 10% appreciation are calculated by:
| • | multiplying the number of shares of common stock subject to a given stock option by the assumed initial public offering price of $11 per share; | |
| • | assuming that the aggregate stock value derived from that calculation compounds at the annual 5% or 10% rate shown in the table until the expiration of the option; and | |
| • | subtracting from that result the aggregate option exercise price. |
74
Table of Contents
Aggregated Option Exercises in Last Fiscal Year and Fiscal Year-End Option Values
The following table sets forth the number and value of securities underlying options held as of December 31, 2004 by each of our named executive officers. Options shown as exercisable in the table below are immediately exercisable.
| Number of Shares | Value of Unexercised In-the- | |||||||||||||||||||||||
| Underlying Unexercised | Money Options at Fiscal | |||||||||||||||||||||||
| Number of | Options at Fiscal Year-End | Year-End(1) | ||||||||||||||||||||||
| Shares | Value | |||||||||||||||||||||||
| Name | Acquired | Realized | Exercisable | Unexercisable | Exercisable | Unexercisable | ||||||||||||||||||
| Ronald M. Burch, M.D., Ph.D. | — | — | 304,448 | 439,206 | $ | 2,914,452 | $ | 4,389,975 | ||||||||||||||||
| Paul R. Hamelin, R.Ph. | — | — | — | 525,000 | $ | 0 | $ | 5,250,000 | ||||||||||||||||
| Jeffrey D. Lazar, M.D., Ph.D. | — | — | 132,160 | 163,887 | $ | 1,275,099 | $ | 1,637,436 | ||||||||||||||||
| Jeffrey A. Rona | — | — | 23,325 | 172,153 | $ | 220,550 | $ | 1,720,280 | ||||||||||||||||
| (1) | These values have been calculated based on an assumed initial public offering price of $11 per share, less the applicable exercise price per share, multiplied by the underlying shares, without taking into account any taxes that may be payable in connection with the transaction. |
Benefit Plans
| 2001 Equity Incentive Plan |
We maintain a stock incentive plan which allows for the grant of incentive stock options, nonqualified stock options, stock bonuses and rights to acquire restricted stock to our employees, directors and consultants. The plan has a 10 year term that commenced on March 30, 2001 and terminates on March 30, 2011 and may be administered by our board of directors or any committee of one or more directors appointed by the board. Currently, the plan is administered by our board of directors. We have reserved 2,355,753 shares of our common stock for issuance pursuant to awards under this plan. As of December 31, 2004, options to purchase a total of 2,352,345 shares of common stock, with a weighted average price of $1.30 per share, were outstanding under this plan.
The administrative committee selects the individuals to receive awards under the stock incentive plan and sets the terms and conditions of each award. The administrative committee has plenary authority to interpret the stock incentive plan and to make all determinations relating to the plan. After the date that awards under this plan are no longer exempt from the provisions of Section 162(m) of the Code, the maximum number of shares of our common stock that may be subject to awards of options or stock appreciation rights for any single individual in any year cannot exceed 125,000 shares.
Options granted under our stock incentive plan may be incentive stock options or nonqualified stock options. The exercise price and vesting schedules for options will be set by the administrative committee at the time of grant, provided that the per share exercise price for incentive stock options and nonqualified stock options intended to be exempt from the provisions of Section 162(m) of the Code cannot be less than the fair market value of a share of our common stock on the date of grant. Fair market value is defined in the stock incentive plan as the closing price of our common stock on the Nasdaq National Market, or such national securities exchange upon which our common stock is listed, on the trading date immediately prior to the date of grant. The exercise price for nonqualified stock options cannot be less than the par value of our common stock. The term of each option will be set by the administrative committee, provided no term can exceed 10 years from the date of grant. Options may expire earlier upon an optionee’s termination of employment. Upon exercise, the exercise price for an option may be paid in cash or by bank check or, in the discretion of the administrative committee, through delivery of shares of our common stock or other property having an aggregate value equal to the aggregate exercise price, or through a brokered exercise procedure not in violation of any law. The administrative committee may allow for the voluntary surrender of options in exchange for the grant of new options with similar or different terms.
75
Table of Contents
Shares of restricted stock granted under the stock incentive plan will be non-transferable and subject to forfeiture upon the termination of employment. The holder of a restricted stock award will generally have the rights and privileges of a stockholder, including the right to vote such shares. Cash and stock dividends on such shares, if any, may be distributed to the holder of a restricted stock award or held for the account of the holder, as determined by the administrative committee.
The administrative committee may grant other cash, stock or stock-related awards under the stock incentive plan, including stock appreciation rights, limited stock appreciation rights, phantom stock awards, the bargain purchase of our common stock and stock bonuses. The terms and conditions of any such other stock-based awards will be determined by the administrative committee, in its sole discretion.
All outstanding awards under the stock incentive plan, the maximum number of shares available under the stock incentive plan and the maximum number of shares of our common stock available pursuant to the grant of options and stock appreciation rights to any single person in any year, if applicable, are subject to adjustment or substitution, as determined by the administrative committee in the event of certain corporate transactions.
The stock incentive plan may be terminated or amended at any time by our board of directors, provided that without stockholder approval no such amendment may:
| • | materially increase the number of shares of our common stock available or the formula for automatic increase of shares under the stock incentive plan; | |
| • | extend the maximum term of any option beyond ten years; | |
| • | extend the expiration date of the plan; or | |
| • | change the class of persons eligible to receive awards under the plan without stockholder approval. |
Except for adjustments subject to certain corporate transactions, alterations to outstanding awards under the stock incentive plan may be made only with the consent of the award recipient.
| 401(k) Plan |
We have established a tax-qualified employee savings and retirement plan for all employees who satisfy certain eligibility requirements, including requirements relating to age and length of service. Under our 401(k) plan, employees may elect to reduce their current compensation by up to 15% or the statutory limit, $13,000 in 2004, whichever is less, and have us contribute the amount of this reduction to the 401(k) plan. In addition, we match a percentage of an employee’s contribution that we establish from time to time. As of September 30, 2004, we had 23 employees eligible for participation in our 401(k) plan. We made matching contributions of $40,052 in 2003 and $34,825 in the nine month period ended September 30, 2004.
We intend for the 401(k) plan to qualify under Section 401 of the Internal Revenue Code of 1986, as amended, so that contributions by employees or by us to the 401(k) plan, and income earned on plan contributions, are not taxable to employees until withdrawn from the 401(k) plan. Our contributions, if any, will be deducted by us when made.
76
Table of Contents
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
Since March 6, 2001, there has not been, nor is there currently planned, any transaction or series of similar transactions to which we were or are a party in which the amount involved exceeds $60,000 and in which any director, executive officer or holder of more than 5% of our capital stock or any member of such person’s immediate family had or will have a direct or indirect material interest other than agreements which are described under the caption “Management” and the transactions described below.
Purchases of Securities
Since March 6, 2001, the following executive officers, directors, holders of more than 5% of our capital stock and members of such person’s immediate family purchased securities in the amounts and as of the dates shown below.
| Number of | Number of Shares | Number of Shares | Number of Shares | ||||||||||||||
| Shares of | of Series A | of Series B | of Series C | ||||||||||||||
| Name of Purchaser | Common Stock | Preferred Stock | Preferred Stock | Preferred Stock | |||||||||||||
Entities Affiliated with Directors: | |||||||||||||||||
| Entities affiliated with InterWest Partners(1) | — | 3,000,000 | 4,676,923 | 16,877,637 | |||||||||||||
| Entities affiliated with J.P. Morgan Partners(2) | — | 3,000,000 | 4,676,923 | 16,877,637 | |||||||||||||
| Entities affiliated with Sofinnova Ventures(3) | — | 3,000,000 | 2,338,462 | 6,683,544 | |||||||||||||
Other 5% Stockholders: | |||||||||||||||||
| Entities affiliated with Advent International Corporation(4) | — | — | — | 11,814,345 | |||||||||||||
| Entities affiliated with Index Ventures(5) | — | — | — | 9,282,700 | |||||||||||||
| Entities affiliated with Pacific Rim(6) | — | — | — | 9,282,700 | |||||||||||||
| S.R. One, Limited | — | — | — | 9,282,700 | |||||||||||||
Directors and Executive Officers: | |||||||||||||||||
| Ronald M. Burch, M.D., Ph.D. | 100,000 | 100,000 | — | — | |||||||||||||
| Paul R. Hamelin, R.Ph. | — | — | — | — | |||||||||||||
| Jeffrey D. Lazar, M.D., Ph.D. | — | — | — | — | |||||||||||||
| Jeffrey A. Rona | — | — | — | — | |||||||||||||
| Charles M. Cohen, Ph.D. | — | — | — | — | |||||||||||||
| Rodney A. Ferguson, J.D., Ph.D. | — | — | — | — | |||||||||||||
| Arnold L. Oronsky, Ph.D. | — | — | — | — | |||||||||||||
| Michael F. Powell, Ph.D. | — | — | — | — | |||||||||||||
| Carter H. Eckert | — | — | — | — | |||||||||||||
| Price Per Share: | $ | 0.01 | $ | 1.00 | $ | 1.30 | $ | 0.5925 | |||||||||
| Date(s) of Purchases: | 03/2001 | 04-09/2001 | 03/2002 | 02/2004 | |||||||||||||
| (1) | Represents shares held by InterWest Partners VIII, L.P., InterWest Investors Q VIII, L.P., and InterWest Investors VIII, L.P. Includes the conversion of convertible promissory notes, issued to the InterWest Funds in April 2003, in the aggregate principal amount of $3,920,000. The convertible promissory notes were converted into Series C preferred stock in February 2004 at a rate of $0.5925 per share. Dr. Arnold L. Oronsky, one of our directors, is a Managing Director of InterWest Management Partners VIII, LLC, the general partner of InterWest Partners VIII, L.P., InterWest Investors Q VIII, L.P., and InterWest Investors VIII, L.P. |
77
Table of Contents
| (2) | Represents shares held by J.P. Morgan Partners (SBIC), LLC, J.P. Morgan Partners Global Investors, L.P., J.P. Morgan Partners Global Investors A, L.P., J.P. Morgan Partners Global Investors (Cayman), L.P., and J.P. Morgan Partners Global Investors (Cayman) II, L.P. Includes the conversion of convertible promissory note, issued to J.P. Morgan Partners (SBIC), LLC in April 2003, in the aggregate principal amount of $3,920,000. The convertible promissory note was converted into Series C preferred stock in February 2004 at a rate of $0.5925 per share. Dr. Rodney A. Ferguson, one of our directors, is a Managing Director of J.P. Morgan Partners (SBIC), LLC, and JPMP Capital Corp. |
| (3) | Represents shares held by Sofinnova Partners V, LP, Sofinnova Affiliates V, LP. and Sofinnova Venture Principals V, LP. Includes the conversion of convertible promissory notes, issued to Sofinnova Partners V, LP, Sofinnova Affiliates V, LP. and Sofinnova Venture Principals V, LP. in April 2003, in the aggregate principal amount of $3,920,000. The convertible promissory notes were converted into Series C preferred stock in February 2004 at a rate of $0.5925 per share. Dr. Michael F. Powell, one of our directors, is a Managing Director of each of these partnerships. |
| (4) | Represents shares held by Advent Healthcare and Life Science III LP, Advent Healthcare and Life Sciences III-A, LP, Advent Partners HLS III LP and Advent Partners II LP. |
| (5) | Represents shares held by Index Ventures II (Jersey) II LP, Index Ventures II (Delaware) LP, Index Ventures II GmbH Co. KG, Index Ventures II Parallel Entrepreneur Fund (Jersey-A) LP, Index Ventures II Parallel Entrepreneur Fund (Jersey-A) LP and Index Ventures Management SA on behalf of Index Employee Investment Plan. |
| (6) | Represents shares held by Pacific Rim AQUA Life Science No. 1 Investment Partnership, Pacific Rim AQUA Life Science No. 2 Investment Partnership, Pacific Rim AQUA Life Science No. 3 Investment Partnership, Pacific Rim AQUA Life Science No. 4 Investment Partnership and Pacific Rim AQUA Life Science No. 5 Investment Partnership. |
Indemnification Agreements
As permitted by the Delaware General Corporation Law, we have adopted provisions in our certificate of incorporation and bylaws that authorize and require us to indemnify our officers and directors to the full extent permitted under Delaware law, subject to limited exceptions. See “Management — Limitation of Liability and Indemnification of Officers and Directors.”
Stock Option Grants
We have granted options to purchase shares of our common stock to our executive officers and directors. See “Principal Stockholders.”
Amended and Restated Investor Rights Agreement
We and our preferred stockholders have entered into an agreement under which our preferred stockholders have registration rights with respect to their shares of common stock following this offering. Upon closing of this offering, all our currently outstanding shares of preferred stock will be converted into shares of our common stock. See “Description of Capital Stock — Registration Rights” for a further description of the terms of this agreement.
78
Table of Contents
PRINCIPAL STOCKHOLDERS
The following table presents information concerning the beneficial ownership of the shares of our common stock as of December 31, 2004 by:
| • | each person we know to be the beneficial owner of 5% or more of our outstanding shares of our capital stock; | |
| • | each of our named executive officers listed on the Summary Compensation Table; | |
| • | each of our directors; and | |
| • | all of our executive officers and directors as a group. |
The information set forth in the table gives effect to the conversion of all of our preferred stock and the exercise of an outstanding warrant that, by its terms, terminates upon completion of this offering.
Beneficial ownership is determined under the rules of the SEC and generally includes voting or investment power over securities. Except in cases where community property laws apply or as indicated in the footnotes to this table, we believe that each stockholder identified in the table possesses sole voting and investment power over all shares of common stock shown as beneficially owned by the stockholder. Percentage of beneficial ownership is based on 15,368,995 shares of common stock outstanding as of December 31, 2004, and 22,168,995 shares of common stock outstanding after the completion of this offering. Shares of common stock subject to options that are currently exercisable or exercisable within 60 days of December 31, 2004, are considered outstanding and beneficially owned by the person holding the options for the purpose of computing the percentage ownership of that person but are not treated as outstanding for the purpose of computing the percentage ownership of any other person. Unless indicated below, the address of each individual listed below is c/o AlgoRx Pharmaceuticals, Inc., 500 Plaza Drive, 2nd floor, Secaucus, New Jersey 07094-3619.
| Percentage of Shares | ||||||||||||
| Number of | Outstanding | |||||||||||
| Shares | ||||||||||||
| Beneficially | Prior to the | After the | ||||||||||
| Name and Address of Beneficial Owner | Owned(1) | Offering | Offering | |||||||||
Stockholders owning approximately 5% or more | ||||||||||||
| Entities affiliated with InterWest Partners(2) | 2,914,840 | 19.0 | % | 13.1 | % | |||||||
| Entities affiliated with JPMorgan Partners(3) | 2,914,840 | 19.0 | 13.1 | |||||||||
| Entities affiliated with Sofinnova Ventures(4) | 1,497,892 | 9.7 | 6.8 | |||||||||
| Entities affiliated with Advent International(5) | 1,181,434 | 7.7 | 5.3 | |||||||||
| Entities affiliated with Index Ventures(6) | 928,270 | 6.0 | 4.2 | |||||||||
| Entities affiliated with Pacific Rim(7) | 928,270 | 6.0 | 4.2 | |||||||||
| S.R. One, Limited(8) | 928,270 | 6.0 | 4.2 | |||||||||
Directors and Executive Officers | ||||||||||||
| Ronald M. Burch, M.D., Ph.D.(9) | 442,287 | 2.8 | 2.0 | |||||||||
| Paul R. Hamelin, R.Ph. | 0 | 0 | 0 | |||||||||
| Jeffrey D. Lazar, M.D., Ph.D.(10) | 141,194 | * | * | |||||||||
| Jeffrey A. Rona(11) | 25,319 | * | * | |||||||||
| Charles M. Cohen, Ph.D.(5) | 1,181,434 | 7.7 | 5.3 | |||||||||
| Rodney A. Ferguson, J.D., Ph.D.(3) | 2,914,840 | 19.0 | 13.1 | |||||||||
| Arnold L. Oronsky, Ph.D.(2) | 2,914,840 | 19.0 | 13.1 | |||||||||
| Michael F. Powell, Ph.D.(4) | 1,497,892 | 9.7 | 6.8 | |||||||||
| Carter H. Eckert | 0 | 0 | 0 | |||||||||
| All directors and executive officers as a group (9 persons)(12) | 9,117,806 | 57.5 | % | 40.2 | % | |||||||
79
Table of Contents
| * | Less than 1%. |
| (1) | Includes shares of common stock issuable pursuant to options exercisable within 60 days of December 31, 2004. |
| (2) | Includes 2,811,947 shares held by InterWest Partners VIII, L.P., 80,449 shares held by InterWest Investors Q VIII, L.P., and 22,444 shares held by InterWest Investors VIII, L.P., collectively, the InterWest Funds. Dr. Arnold L. Oronsky, one of our directors, is a Managing Director of InterWest Management Partners VIII, LLC, the general partner of the InterWest Funds. Dr. Oronsky has shared voting and dispositive powers over the shares held by the InterWest Funds. He disclaims beneficial ownership of the shares held by the InterWest Funds, except to the extent of his pecuniary interest therein. The address for InterWest Partners is 2710 Sand Hill Road, Second Floor, Menlo Park, CA 94025. |
| (3) | Includes 2,630,415 shares held by J.P. Morgan Partners (SBIC), LLC, or JPMP SBIC, 138,715 shares held by J.P. Morgan Partners Global Investors, L.P., 21,314 shares held by J.P. Morgan Partners Global Investors A, L.P., 69,641 shares held by J.P. Morgan Partners Global Investors (Cayman), L.P., 46,967 shares held by J.P. Morgan Partners Global Investors (Selldown) L.P. and 7,788 shares held by J.P. Morgan Partners Global Investors (Cayman) II, L.P. JPMP SBIC is a wholly owned subsidiary of J.P. Morgan Partners (BHCA), L.P., the general partner of which is JPMP Master Fund Manager, L.P. or “MF Manager”. JPMP Global Investors, L.P., or Global Investors is the general partner of each of J.P. Morgan Partners Global Investors, L.P., J.P. Morgan Partners Global Investors A, L.P., J.P. Morgan Partners Global Investors (Cayman), L.P., J.P. Morgan Partners Global Investors (Selldown) L.P. and J.P. Morgan Partners Global Investors (Cayman) II, L.P. JPMP Capital Corp., or “JPMP Capital”, a wholly owned subsidiary of J.P. Morgan Chase & Co., or “JPM Chase”, is the general partner of each of MF Manager and Global Investors. As a result thereof, each of MF Manager, JPMP Capital and JPM Chase may be deemed to beneficially own the shares held by JPMP SBIC, and each of Global Investors, JPMP Capital and JPM Chase may be deemed to beneficially own the share held by each of J.P. Morgan Partners Global Investors, L.P., J.P. Morgan Partners Global Investors A, L.P., J.P. Morgan Partners Global Investors (Cayman), L.P., J.P. Morgan Partners Global Investors (Selldown) L.P. and J.P. Morgan Partners Global Investors (Cayman) II, L.P. Rodney A Ferguson, one of our directors, is a managing director of JPMP SBIC and JPMP Capital and as a result, Dr. Ferguson may be deemed to be the beneficial owner of the shares held by JPMP SBIC and each of J.P. Morgan Partners Global Investors, L.P., J.P. Morgan Global Investors A, L.P. Morgan Partners Global Investors (Cayman), L.P., J.P. Morgan Partners Global Investors (Selldown) L.P. and J.P. Morgan Partners Global Investors (Cayman) II, L.P. The foregoing, however, shall not be an admission that Dr. Ferguson, MF Manager, Global Investors, JPMP Capital or JPM Chase are the beneficial owners of such shares and each disclaims beneficial ownership except to the extent of their pecuniary interest in each of the applicable entities. The address for J.P. Morgan is 1221 Avenue of the Americas, New York, NY 10020. |
| (4) | Includes 1,428,968 shares held by Sofinnova Partners V, LP, 47,013 shares held by Sofinnova Affiliates V, LP. and 21,911 shares held by Sofinnova Venture Principals V, LP. Dr. Michael F. Powell is a Managing Director of each of these partnerships, shares voting and dispositive power with respect to the shares held by each of Sofinnova Partners V, LP, Sofinnova Affiliates V, LP. and Sofinnova Venture Principals V, LP and disclaims beneficial ownership of such shares in which he has no pecuniary interest. The address for Sofinnova Venture Partners is 140 Geary Street, 10th Floor, San Francisco, CA 94106. |
| (5) | Includes the ownership by the following venture capital funds managed by Advent International Corporation: 404,725 shares held by Advent Healthcare and Life Science III LP, 758,987 shares held by Advent Healthcare and Life Sciences III-A, LP, 14,346 shares held by Advent Partners HLS III LP and 3,376 shares held by Advent Partners II LP. In its capacity as manager or general partner of these funds, Advent International Corporation exercises sole voting and investment power with respect to all shares held by these funds. Dr. Charles M. Cohen, a partner at Advent |
80
Table of Contents
| International, may be deemed to be a beneficial owner of the shares held by these Advent funds. Dr. Cohen disclaims beneficial ownership of these shares held by Advent except to the extent of his pecuniary interest therein. The address for Advent International Corporation is 75 State Street, 29th Floor, Boston, Massachusetts, 02109. | |
| (6) | Includes 284,069 shares held by Index Ventures (Jersey) II LP, 522,291 shares held by Index Ventures II (Delaware) LP, 83,507 shares held by Index Ventures II GmbH & Co. KG, 9,534 shares held by Index Ventures II Parallel Entrepreneur Fund (Jersey-A) LP, 14,945 shares held by Index Ventures II Parallel Entrepreneur Fund (Jersey-B) LP and 13,924 shares held by Index Ventures Management SA on behalf of Index Employee Investment Plan. Index Venture Associates II Limited is the general partner for each of Index Ventures II (Jersey) II LP, Index Ventures II (Delaware) LP, Index Ventures II Parallel Entrepreneur Fund (Jersey-A) LP, Index Ventures II Parallel Entrepreneur Fund (Jersey-B) LP. Index Ventures II (SLP) Limited is the special limited partner of Index Ventures II Gmbh KG. The address for the Index funds is No. 1 Seaton Place, St. Helier, Jersey, Channel Islands JE4 8YJ. |
| (7) | Includes 161,480 shares held by Pacific Rim AQUA Life Science No. 1 Investment Partnership, 190,660 shares held by Pacific Rim AQUA Life Science No. 2 Investment Partnership, 109,280 shares held by Pacific Rim AQUA Life Science No. 3 Investment Partnership, 213,690 shares held by Pacific Rim AQUA Life Science No. 4 Investment Partnership and 253,160 shares held by Pacific Rim AQUA Life Science No. 5 Investment Partnership. Pacific Rim Ventures Co., Ltd is the general partner of each of the above referenced Pacific Rim entities and exercises sole voting and investment power with respect to all shares held by these funds. The address for Pacific Rim Ventures Co. Ltd is Green Plaza, 2nd flr., 3-7- Komazawa, Setagaya-ku, Tokyo 154-0012 Japan. |
| (8) | The address for S.R. One, Limited is Four Tower Bridge, 200 Barr Harbor Drive, Suite 250, W. Conshohocken, PA 19428. |
| (9) | Includes 327,887 shares of common stock issuable upon the exercise of stock options exercisable within 60 days of December 31, 2004. |
| (10) | Includes 141,194 shares of common stock issuable upon the exercise of stock options exercisable within 60 days of December 31, 2004. |
| (11) | Includes 25,319 shares of common stock issuable upon the exercise of stock options exercisable within 60 days of December 31, 2004. |
| (12) | This total includes 494,400 shares of common stock issuable upon exercise of stock options exercisable within 60 days of December 31, 2004. This total also includes the shares described in notes (2) through (5) above. |
81
Table of Contents
DESCRIPTION OF CAPITAL STOCK
Upon the closing of this offering, our authorized capital stock will consist of 150,000,000 shares of common stock, par value $0.001 per share, and 25,000,000 shares of preferred stock, par value $0.001 per share. As of December 31, 2004, there were 15,368,995 shares of common stock outstanding. As of December 31, 2004, we had approximately 62 record holders of our capital stock. All of our outstanding shares of preferred stock will automatically convert into shares of common stock immediately prior to the closing of this offering. After the closing of this offering, we will have 22,168,995 shares of common stock and no shares of preferred stock outstanding. In addition, as of December 31, 2004, 3,408 shares of our common stock were reserved for future grants under our 2001 Equity Incentive Plan and options to purchase 2,352,345 shares of our common stock were outstanding. Also, as of December 31, 2004, 69,265 shares of our common stock were issuable under an outstanding warrant that, by its terms, terminates upon completion of this offering.
The following description of our capital stock and provisions of our certificate of incorporation and bylaws are summaries of material terms and provisions and are qualified by reference to our certificate of incorporation and bylaws, copies of which have been filed with the SEC as exhibits to the registration statement of which this prospectus is a part. The descriptions of the common stock and preferred stock reflect amendments to our certificate of incorporation and bylaws that will become effective upon closing of this offering.
Common Stock
Upon the closing of this offering, we will be authorized to issue one class of common stock. Stockholders will be entitled to one vote for each share of our common stock held of record on all matters on which stockholders are entitled or permitted to vote. Our common stock will not have cumulative voting rights in the election of directors. As a result, holders of a majority of the shares of our common stock voting for the election of directors can elect all the directors standing for election. Holders of our common stock will be entitled to receive dividends out of legally available funds when and if declared from time to time by our board of directors. See “Dividend Policy.” In the event of our liquidation, dissolution or winding up, the holders of our common stock will be entitled to share ratably in all assets remaining after payment of liabilities, subject to the rights of any then outstanding preferred stock. The rights, preferences and privileges of holders of our common stock will be subject to, and may be adversely affected by, the rights of holders of shares of any series of preferred stock that we may designate and issue in the future. All outstanding shares of our common stock are fully paid and nonassessable, and the shares of common stock offered hereby will be fully paid and nonassessable.
Preferred Stock
Upon the closing of this offering, all outstanding shares of our preferred stock will be converted into an aggregate of 14,275,747 shares of common stock. Under our certificate of incorporation, upon the closing of this offering, we will be authorized, subject to the limits imposed by the Delaware General Corporation Law, to issue 25,000,000 shares of preferred stock in one or more series, to establish from time to time the number of shares to be included in each series, to fix the rights, preferences and privileges of the shares of each wholly unissued series and any of its qualifications, limitations, restrictions. Our board of directors can also increase or decrease the number of shares of any series of our preferred stock, but not below the number of shares of that series then outstanding, without any further vote or action by our stockholders.
Our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of our common stockholders. The issuance of preferred stock, while providing flexibility in connection with possible acquisitions and other corporate purposes, could have the effect of delaying or preventing our change in control and may cause the market price of our common stock to decline or impair the voting and other rights of the holders of our common stock. We have no current plans to issue any shares of preferred stock.
82
Table of Contents
Warrant
In February 2004, in connection with our Series C preferred stock financing transaction, we issued a warrant to purchase an aggregate of 692,658 shares of our Series C preferred stock at an exercise price of $0.5925 per share to an investment adviser. The termination date of this warrant is the earlier of February 19, 2009 and the closing of this offering. The exercise price and the number of shares of common stock issuable upon exercise of the warrant are subject to adjustment upon the occurrence of any stock dividend or stock split. Because all of our preferred stock, including our Series C preferred stock, will be converted into common stock upon the completion of this offering, our outstanding warrant will become exercisable for shares of common stock immediately prior to the closing of this offering and the warrant will terminate at the closing if not exercised by the closing. In addition, as a result of our 1-for-10 pre-offering reverse stock split, the number of shares of common stock underlying the warrant will be proportionately decreased to 69,265 shares and the exercise price of the warrant will be proportionately increased to $5.925 per share.
Registration Rights
Upon completion of this offering, assuming the expected exercise of an outstanding warrant that, by its terms, terminates upon completion of this offering, the holders of approximately 14,505,012 shares of common stock will be entitled to rights with respect to the registration of these shares under the Securities Act as described below. We have delivered notice to the holders of registration rights informing them of the exercise of the underwriters’ right to exclude all sales by such holders from this offering. In addition, all holders with registration rights have agreed not to exercise their registration rights without the prior written consent of Credit Suisse First Boston LLC and Citigroup Global Markets Inc. for a period of 180 days after the date of this prospectus. This 180-day period could be extended by those underwriters for up to an additional 34 days under certain circumstances.
| Demand Registration Rights |
At any time 180 days after the closing of this offering, the holders of a majority of the shares having demand registration rights can demand that we file a registration statement for those shares so long as the demand covers at least a majority of the shares subject to these registration rights or the aggregate offering price of the shares is greater than $10.0 million. We will effect the registration as requested, unless the underwriters decide to limit the number of shares that may be included in the registration due to marketing factors. We are only obligated to satisfy two demand registrations, and we may defer a registration for up to 120 days under specified circumstances not more than twice in any 12-month period. We are not obligated to satisfy a demand registration if the registrable shares may be immediately registered pursuant to a registration statement on Form S-3, as described below.
| Piggyback Registration Rights |
If we register any securities for public sale, the holders of the shares having piggyback registration rights may include their shares in the registration statement. If the registration relates to an underwritten offering, the underwriters have the right to limit the number of shares having registration rights that may be included in the registration statement, subject to certain limitations.
| Form S-3 Registration Rights |
If we are eligible to file a registration statement on Form S-3, any holders of the shares having Form S-3 registration rights can demand that we file a registration statement on Form S-3 or any similar short-form registration statement, so long as (i) the aggregate amount of securities to be sold under the registration statement on Form S-3 or any similar short-form registration statement is at least $5.0 million and (ii) we have not already effected two registrations on Form S-3. We may defer a Form S-3 registration by up to 90 days under specified circumstances once per 12-month period.
83
Table of Contents
| Expenses of Registration |
We will pay for all registration expenses, including in specified situations the reasonable legal expenses of a single counsel for the holders not to exceed $25,000, relating to any demand, piggyback or Form S-3 registration, other than the underwriting discount and selling commissions applicable to the sale. However, we are generally not required to pay for the expenses of any demand or Form S-3 registration if the request is subsequently withdrawn by the holders of a majority of the shares having registration rights.
| Expiration of Registration Rights |
The registration rights described above will expire five years after the date of the closing of this offering. The registration rights will terminate earlier for a particular stockholder if (i) we have completed our initial public offering and are subject to the Securities Exchange Act of 1934, as amended, (ii) such holder, together with its affiliates, partners and former partners, holds less than 1% of our outstanding common stock assuming the conversion of any outstanding preferred stock into common stock and (iii) such holder can resell all of its securities in a 90-day period under Rule 144 of the Securities Act.
Antitakeover Effects of Delaware Law and Provisions of our Certificate of Incorporation and Bylaws
| Delaware Takeover Statute |
We are subject to Section 203 of the Delaware General Corporation Law. This statute regulating corporate takeovers prohibits a Delaware corporation from engaging in any business combination with any interested stockholder for three years following the date that the stockholder became an interested stockholder, unless:
| • | prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder; | |
| • | upon completion of the transaction that resulted in the interested stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the number of shares outstanding (a) shares owned by persons who are directors and also officers, and (b) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or | |
| • | on or subsequent to the date of the transaction, the business combination is approved by the board and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 66 2/3% of the outstanding voting stock which is not owned by the interested stockholder. |
Generally, a business combination includes a merger, asset or stock sale, or other transaction resulting in a financial benefit to the interested stockholder. An interested stockholder is a person who, together with affiliates and associates, owns or, within three years prior to the determination of interested stockholder status, did own 15% or more of a corporation’s outstanding voting securities. We expect the existence of this provision to have an anti-takeover effect with respect to transactions our board of directors does not approve in advance. We also anticipate that Section 203 may discourage takeover attempts that might result in a premium over the market price for the shares of common stock held by stockholders.
| Certificate of Incorporation and Bylaw Provisions |
Provisions of our certificate of incorporation and bylaws, which will become effective upon the closing of this offering, may have the effect of making it more difficult for a third party to acquire, or discourage a third party from attempting to acquire, control of our company by means of a tender offer, a proxy contest or otherwise. These provisions may also make the removal of incumbent officers and directors more
84
Table of Contents
difficult. These provisions are intended to discourage certain types of coercive takeover practices and inadequate takeover bids and to encourage persons seeking to acquire control of AlgoRx to first negotiate with us. These provisions could also limit the price that investors might be willing to pay in the future for shares of our common stock. These provisions may make it more difficult for stockholders to take specific corporate actions and could have the effect of delaying or preventing a change in control.
In particular, our certificate of incorporation and bylaws, which will become effective upon the closing of this offering, will provide for the following:
Staggered Board of Directors.Our board of directors is divided into three classes of the same or nearly the same number of directors, each serving staggered three-year terms, which means that only one class of directors may be elected at each annual meeting or special meeting in lieu of such annual meeting. These provisions may make the removal of incumbent directors difficult and may discourage third parties from attempting to circumvent the anti-takeover effects of our certificate of incorporation and bylaws by removing our incumbent directors.
No Written Consent of Stockholders. Any action to be taken by our stockholders must be effected at a duly called annual or special meeting and may not be effected by written consent.
Special Meetings of Stockholders. Special meetings of our stockholders may be called only by (i) the Chairman of the board of directors, (ii) the Chief Executive Officer, or (iii) the board of directors pursuant to a resolution adopted by a majority of the total number of authorized directors.
Advance Notice Requirement. Stockholder proposals to be brought before an annual meeting of our stockholders must comply with advance notice procedures. These advance notice procedures require timely notice and apply in several situations, including stockholder proposals relating to the nominations of persons for election to the board of directors. Generally, to be timely, notice must be received at our principal executive offices not less than 90 days nor more than 120 days prior to the first anniversary date of the annual meeting for the preceding year.
Issuance of Undesignated Preferred Stock. Our board of directors is authorized to issue, without further action by the stockholders, up to 25,000,000 shares of undesignated preferred stock with rights and preferences, including voting rights, designated from time to time by the board of directors. The existence of authorized but unissued shares of preferred stock enables our board of directors to render more difficult or to discourage an attempt to obtain control of us by means of a merger, tender offer, proxy contest or otherwise.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is American Stock Transfer & Trust Company.
Listing
We have applied for our common stock to be quoted on the Nasdaq National Market under the trading symbol “AGRX.”
85
Table of Contents
UNITED STATES FEDERAL INCOME TAX CONSEQUENCES
TO NON-UNITED STATES HOLDERS
The following is a general discussion of the material United States federal income tax consequences of the ownership and disposition of our common stock to a non-United States holder. For the purpose of this discussion, a non-United States holder is any beneficial holder of our common stock that for United States federal income tax purposes is not a United States person. For purposes of this discussion, the term United States person means:
| • | an individual citizen or resident of the United States; | |
| • | a corporation or other entity taxable as a corporation or a partnership or entity taxable as a partnership created or organized in the United States or under the laws of the United States or any political subdivision thereof; | |
| • | an estate whose income is subject to United States federal income tax regardless of its source; or | |
| • | a trust (i) whose administration is subject to the primary supervision of a United States court and which has one or more United States persons who have the authority to control all substantial decisions of the trust or (ii) which has made an election to be treated as a United States person. |
If a partnership holds common stock, the tax treatment of a partner will generally depend on the status of the partner and upon the activities of the partnership. Accordingly, we urge partnerships which hold our common stock and partners in such partnerships to consult their tax advisors.
This discussion assumes that non-United States holders will hold our common stock issued pursuant to the offering as a capital asset (generally, property held for investment). This discussion does not address all aspects of United States federal income taxation that may be relevant in light of a non-United States holder’s special tax status or special tax situations. United States expatriates, life insurance companies, tax-exempt organizations, dealers in securities or currency, banks or other financial institutions, holders whose functional currency is other than the United States dollar, and investors that hold common stock as part of a hedge, straddle or conversion transaction are among those categories of potential investors that are subject to special rules not covered in this discussion. This discussion does not address any tax consequences arising under the laws of any state, local or non-United States taxing jurisdiction. Furthermore, the following discussion is based on current provisions of the Internal Revenue Code of 1986, as amended, legislative history and Treasury Regulations and administrative and judicial interpretations thereof, all as in effect on the date hereof, and all of which are subject to change, possibly with retroactive effect. Accordingly, we urge each non-United States Holder to consult a tax advisor regarding the United States federal, state, local and non-United States income and other tax consequences of acquiring, holding and disposing of shares of our common stock.
Dividends
We have not paid any dividends on our common stock, and we do not plan to pay any dividends for the foreseeable future. However, if we do pay dividends on our common stock, those payments will constitute dividends for United States tax purposes to the extent paid from our current and accumulated earnings and profits, as determined under United States federal income tax principles. To the extent those dividends exceed our current and accumulated earnings and profits, the dividends will constitute a return of capital and will first reduce a holder’s basis, but not below zero, and then will be treated as gain from the sale of stock.
Any dividend (out of earnings and profits) paid to a non-United States holder of common stock generally will be subject to United States withholding tax either at a rate of 30% of the gross amount of the dividend or such lower rate as may be specified by an applicable tax treaty. In order to receive a reduced treaty rate, a non-United States holder must provide us with an IRS Form W-8BEN certifying, under penalty of perjury, the holder’s status as a non-United States person and qualification for the reduced rate.
86
Table of Contents
Dividends received by a non-United States holder that are effectively connected with a United States trade or business conducted by the non-United States holder are exempt from such withholding tax. In order to obtain this exemption, a non-United States holder must provide us with an IRS Form W-8ECI properly certifying such exemption. Such effectively connected dividends, although not subject to withholding tax, are taxed at the same graduated rates applicable to United States persons, net of certain deductions and credits. In addition to the graduated tax described above, dividends received by a corporate non-United States holder that are effectively connected with a United States trade or business of the corporate non-United States holder may also be subject to a branch profits tax at a rate of 30% or such lower rate as may be specified by an applicable tax treaty.
A non-United States holder of common stock that is eligible for a reduced rate of withholding tax pursuant to a tax treaty may obtain a refund of any excess amounts currently withheld if an appropriate claim for refund is filed with the Internal Revenue Service, or IRS.
Gain on Disposition of Common Stock
A non-United States holder generally will not be subject to United States federal income tax on any gain realized upon the sale or other disposition of our common stock unless:
| • | the gain is effectively connected with a United States trade or business of the non-United States holder (which gain, in the case of a corporate non-United States holder, must also be taken into account for branch profits tax purposes); | |
| • | the non-United States holder is an individual who holds his or her common stock as a capital asset (generally, an asset held for investment purposes) and who is present in the United States for a period or periods aggregating 183 days or more during the calendar year in which the sale or disposition occurs and certain other conditions are met; or | |
| • | our common stock constitutes a United States real property interest by reason of our status as a “United States real property holding corporation” for United States federal income tax purposes at any time within the shorter of the five-year period preceding the disposition or the holder’s holding period for our common stock. We believe that we are not currently, and that we will not become, a “United States real property holding corporation” for United States federal income tax purposes. |
Backup Withholding and Information Reporting
Generally, we must report annually to the IRS the amount of dividends paid, the name and address of the recipient, and the amount, if any, of tax withheld. Subject to certain exceptions, a similar report is sent to the holder. Pursuant to tax treaties or other agreements, the IRS may make its reports available to tax authorities in the recipient’s country of residence.
Payments of dividends or of proceeds on the disposition of stock made to a non-United States holder may be subject to backup withholding unless the non-United States holder establishes an exemption, for example, by properly certifying its non-United States status on a Form W-8BEN or another appropriate version of Form W-8. Notwithstanding the foregoing, backup withholding may apply if either we or our paying agent has actual knowledge, or reason to know, that the holder is a United States person.
Backup withholding is not an additional tax. Rather, the United States income tax liability of persons subject to backup withholding will be reduced by the amount of tax withheld. If withholding results in an overpayment of taxes, a refund may be obtained, provided that the required information is furnished to the IRS.
87
Table of Contents
SHARES ELIGIBLE FOR FUTURE SALE
Prior to this offering, there has been no public market for our common stock, and there can be no assurance that a significant public market for the common stock will develop or be sustained after this offering. Future sales of substantial amounts of our common stock, including shares issued upon exercise of outstanding options, in the public market following this offering could adversely affect market prices prevailing from time to time and could impair our ability to raise capital through the sale of our equity securities.
Upon completion of this offering and based on shares outstanding at December 31, 2004, we will have outstanding 22,168,995 shares of common stock. The 6,800,000 shares sold in this offering, plus any shares issued upon exercise of the underwriters’ option to purchase additional shares from us, will be freely tradable without restriction under the Securities Act, unless purchased by our “affiliates” as that term is defined in Rule 144 under the Securities Act.
The remaining shares of common stock outstanding are “restricted securities” within the meaning of Rule 144 under the Securities Act. Restricted securities may be sold in the public market only if registered or if they qualify for an exemption from registration under Rule 701 or meet the safe harbor qualifications under Rule 144 under the Securities Act as summarized below.
Our directors, officers and substantially all of our stockholders, who combined hold approximately 99% of our stock, have entered into lock-up agreements with the underwriters of this offering generally providing that they will not offer, sell, contract to sell or grant any option to purchase or otherwise dispose of our shares of common stock or any securities exercisable for or convertible into our common stock owned by them prior to this offering for a period of 180 days after the date of this prospectus (which period could be extended by those underwriters for up to an additional 34 days under certain circumstances) without the prior written consent of Credit Suisse First Boston LLC and Citigroup Global Markets Inc. on behalf of our underwriters. As a result of these contractual restrictions, notwithstanding possible earlier eligibility for sale under the provisions of Rules 144, 144(k) and 701, shares subject to lock-up agreements may not be sold until such agreements expire or are waived by Credit Suisse First Boston LLC and Citigroup Global Markets Inc. on behalf of our underwriters. Credit Suisse First Boston LLC and Citigroup Global Markets Inc. have advised us that it has no current intention to shorten or release any of the shares subject to the lock-up agreements prior to the expiration of the lock-up period. Based on shares outstanding as of December 31, 2004, taking into account the lock-up agreements, and assuming Credit Suisse First Boston LLC and Citigroup Global Markets Inc. do not release stockholders from these agreements prior to the expiration of the 180-day lock-up period, the following shares will be eligible for sale in the public market at the following times:
| • | beginning on the date of this prospectus, the 6,800,000 shares sold in this offering will be immediately available for sale in the public market; | |
| • | beginning 180 days after the date of this prospectus (which period could be extended by those underwriters for up to an additional 34 days under certain circumstances), approximately 15,208,995 additional shares will become eligible for sale under Rule 144 or 701, subject to volume restrictions as described below; and | |
| • | the remainder of the restricted securities will be eligible for sale from time to time thereafter, subject in some cases to compliance with Rule 144. |
In general, under Rule 144 as currently in effect, a person who has beneficially owned restricted shares for at least one year, including the holding period of any prior owner except an affiliate, would be entitled to sell within any three-month period a number of shares that does not exceed the greater of:
| • | 1% of the number of shares of common stock then outstanding, which will equal approximately 221,169 shares immediately after this offering; or | |
| • | the average weekly trading volume of our common stock during the four calendar weeks preceding the date on which notice of the sale is filed. |
88
Table of Contents
Sales under Rule 144 are also subject to certain manner of sale provisions and notice requirements and to the availability of current public information about us. Under Rule 144(k), a person who is not deemed to have been an affiliate of us at any time during the three months preceding a sale, and who has beneficially owned the shares proposed to be sold for at least two years, including the holding period of any prior owner except an affiliate, is entitled to sell such shares without complying with the manner of sale, public information, volume limitation or notice provisions of Rule 144.
Any of our employees, officers, directors or consultants who purchased shares under a written compensatory plan or contract may be entitled to rely on the resale provisions of Rule 701. Rule 701 permits affiliates to sell their Rule 701 shares under Rule 144 without complying with the holding period requirements of Rule 144. Rule 701 further provides that non-affiliates may sell such shares in reliance join on Rule 144 without having to comply with the holding period, public information, volume limitation or notice provisions of Rule 144. All holders of Rule 701 shares are required to wait until 90 days after the effective date of this offering before selling such shares. However, substantially all of the Rule 701 shares are subject to lock-up agreements and will only become eligible for sale upon the expiration of the 180-day lock-up (or of any extension of up to 34 additional days of that lock-up period) agreements unless Credit Suisse First Boston LLC and Citigroup Global Markets Inc. waive the lock-up on behalf of the underwriters. Credit Suisse First Boston LLC and Citigroup Global Markets Inc. have advised us that they have no current intention to shorten or release any of the shares subject to the lock-up agreements prior to the expiration of the lock-up period.
89
Table of Contents
UNDERWRITING
Under the terms and subject to the conditions contained in an underwriting agreement dated , 2005, we have agreed to sell to the underwriters named below, for whom Credit Suisse First Boston LLC, Citigroup Global Markets Inc., Piper Jaffray & Co. and Lazard Frères & Co. LLC are acting as representatives, the following respective numbers of shares of common stock:
| Number | |||||
| of Shares | |||||
| Underwriter | |||||
| Credit Suisse First Boston LLC | |||||
| Citigroup Global Markets Inc. | |||||
| Piper Jaffray & Co. | |||||
| Lazard Frères & Co. LLC | |||||
| Total | 6,800,000 | ||||
Credit Suisse First Boston LLC and Citigroup Global Markets Inc. are acting as joint book-running managers for this offering.
The underwriting agreement provides that the underwriters are obligated to purchase all the shares of common stock in the offering if any are purchased, other than those shares covered by the over-allotment option described below. The underwriting agreement also provides that if an underwriter defaults the purchase commitments of non-defaulting underwriters may be increased or the offering may be terminated.
We have granted to the underwriters a 30-day option to purchase on a pro rata basis up to additional shares at the initial public offering price less the underwriting discounts and commissions. The option may be exercised only to cover any over-allotments of common stock.
The underwriters propose to offer the shares of common stock initially at the public offering price on the cover page of this prospectus and to selling group members at that price less a selling concession of $ per share. The underwriters and selling group members may allow a discount of $ per share on sales to other broker/ dealers. After the initial public offering, the representatives may change the public offering price and concession and discount to broker/ dealers.
The following table summarizes the compensation and estimated expenses we will pay:
| Per Share | Total | |||||||||||||||
| Without Over- | With Over- | Without Over- | With Over- | |||||||||||||
| allotment | allotment | allotment | allotment | |||||||||||||
| Underwriting Discounts and Commissions paid by us | $ | $ | $ | $ | ||||||||||||
| Expenses payable by us | $ | $ | $ | $ | ||||||||||||
The underwriters have informed us that they do not expect sales to accounts over which the underwriters have discretionary authority to exceed 5% of the shares of common stock being offered.
We have agreed that we will not offer, sell, contract to sell, pledge or otherwise dispose of, directly or indirectly, or file with the Securities and Exchange Commission a registration statement under the Securities Act of 1933 relating to, any shares of our common stock or securities convertible into or exchangeable or exercisable for any shares of our common stock, or publicly disclose the intention to make any offer, sale, pledge, disposition or filing, without the prior written consent of Credit Suisse First Boston LLC and Citigroup Global Markets Inc. for a period of 180 days after the date of this prospectus. However, in the event that either (1) during the last 17 days of the “lock-up” period, we release earnings results or material news or a material event relating to us occurs or (2) prior to the expiration of the “lock-up” period, we announce that we will release earnings results during the 16-day period beginning on the last day of the “lock-up” period, then in either case the expiration of the “lock-up” will be extended until the expiration of the 18-day period beginning on the date of the release of the earnings results or the
90
Table of Contents
occurrence of the material news or event, as applicable, unless Credit Suisse First Boston LLC and Citigroup Global Markets Inc. waive, in writing, such an extension.
Our officers, directors and security holders, who combined hold approximately 99% of our stock, have agreed that they will not offer, sell, contract to sell, pledge or otherwise dispose of, directly or indirectly, any shares of our common stock or securities convertible into or exchangeable or exercisable for any shares of our common stock, enter into a transaction that would have the same effect, or enter into any swap, hedge or other arrangement that transfers, in whole or in part, any of the economic consequences of ownership of our common stock, whether any of these transactions are to be settled by delivery of our common stock or other securities, in cash or otherwise, or publicly disclose the intention to make any offer, sale, pledge or disposition, or to enter into any transaction, swap, hedge or other arrangement, without, in each case, the prior written consent of Credit Suisse First Boston LLC and Citigroup Global Markets Inc. for a period of 180 days after the date of this prospectus. However, in the event that either (1) during the last 17 days of the “lock-up” period, we release earnings results or material news or a material event relating to us occurs or (2) prior to the expiration of the “lock-up” period, we announce that we will release earnings results during the 16-day period beginning on the last day of the “lock-up” period, then in either case the expiration of the “lock-up” will be extended until the expiration of the 18-day period beginning on the date of the release of the earnings results or the occurrence of the material news or event, as applicable, unless Credit Suisse First Boston LLC and Citigroup Global Markets Inc. waive, in writing, such an extension.
We have agreed to indemnify the underwriters against liabilities under the Securities Act, or contribute to payments that the underwriters may be required to make in that respect.
We have applied for quotation of our common stock on the Nasdaq National Market under the symbol “AGRX.”
In February 2004, Piper Jaffray Healthcare Fund IV, L.P. purchased from us for $2.4 million 4,050,632 shares of our Series C preferred stock, the underlying common shares of which have registration rights described under the section of this prospectus entitled “Description of Capital Stock — Registration Rights.” At the time of its investment, Piper Jaffray Healthcare Fund IV, L.P. was an affiliate of Piper Jaffray & Co., an underwriter of this offering. On December 31, 2004, SightLine Partners LLC, an independent entity not affiliated with Piper Jaffray & Co., replaced a subsidiary of Piper Jaffray & Co. as the manager of the general partner of Piper Jaffray Healthcare Fund IV, L.P., thereby assuming responsibility for managing Piper Jaffray Healthcare Fund IV, L.P. Also on December 31, 2004, Piper Jaffray Healthcare Fund IV, L.P. changed its name to SightLine Healthcare Fund IV, L.P. Following this transaction, Piper Jaffray & Co. retains a minority investment in the general partner of SightLine Healthcare Fund IV, L.P.
Prior to the offering, there has been no public market for the shares. The initial public offering price has been negotiated among the company and the underwriters. Among the factors to be considered in determining the initial public offering price of the shares, in addition to prevailing market conditions, will be the company’s historical performance, estimates of the business potential and earnings prospects of the company, an assessment of the company’s management and the consideration of the above factors in relation to market valuation of companies in related businesses. We cannot be sure that the initial public offering price will correspond to the price at which the common stock will trade in the public market following this offering or that an active trading market for the common stock will develop and continue after this offering.
In connection with the offering, the underwriters may engage in stabilizing transactions, over-allotment transactions, syndicate covering transactions and penalty bids in accordance with Regulation M under the Securities Exchange Act of 1934.
| • | Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specified maximum. |
91
Table of Contents
| • | Over-allotment involves sales by the underwriters of shares in excess of the number of shares the underwriters are obligated to purchase, which creates a syndicate short position. The short position may be either a covered short position or a naked short position. In a covered short position, the number of shares over-allotted by the underwriters is not greater than the number of shares that they may purchase in the over-allotment option. In a naked short position, the number of shares involved is greater than the number of shares in the over-allotment option. The underwriters may close out any covered short position by either exercising their over-allotment option and/or purchasing shares in the open market. | |
| • | Syndicate covering transactions involve purchases of the common stock in the open market after the distribution has been completed in order to cover syndicate short positions. In determining the source of shares to close out the short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through the over-allotment option. If the underwriters sell more shares than could be covered by the over- allotment option, a naked short position, the position can only be closed out by buying shares in the open market. A naked short position is more likely to be created if the underwriters are concerned that there could be downward pressure on the price of the shares in the open market after pricing that could adversely affect investors who purchase in the offering. | |
| • | Penalty bids permit the representatives to reclaim a selling concession from a syndicate member when the common stock originally sold by the syndicate member is purchased in a stabilizing or syndicate covering transaction to cover syndicate short positions. |
These stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market price of our common stock or preventing or retarding a decline in the market price of the common stock. As a result, the price of our common stock may be higher than the price that might otherwise exist in the open market. These transactions may be effected on the Nasdaq National Market or otherwise and, if commenced, may be discontinued at any time.
A prospectus in electronic format will be made available on the web sites maintained by one or more of the underwriters, or selling group members, if any, participating in this offering and one or more of the underwriters participating in this offering may distribute prospectuses electronically. The representatives may agree to allocate a number of shares to underwriters and selling group members for sale to their online brokerage account holders. Internet distributions will be allocated by the underwriters and selling group members that will make internet distributions on the same basis as other allocations.
Each underwriter has represented, warranted and agreed that: (i) it has not offered or sold and, prior to the expiry of a period of six months from the closing of the offering, will not offer or sell any shares to persons in the United Kingdom except to persons whose ordinary activities involve them in acquiring, holding, managing or disposing of investments (as principal or agent) for the purposes of their businesses or otherwise in circumstances which have not resulted and will not result in an offer to the public in the United Kingdom within the meaning of the Public Offers of Securities Regulations 1995; (ii) it has only communicated or caused to be communicated and will not communicate or cause to be communicated any invitation or inducement to engage in investment and will only communicate or cause to be communicated any invitation or inducement to engage in investment activity (within the meaning of section 21 of the Financial Services and Markets Act 2000, or the FSMA), received by it in connection with the issue or sale of any shares in circumstances in which section 21(1) of the FSMA does not apply to Foundation Coal Holdings, Inc.; and (iii) it has complied and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to the shares in, from or otherwise involving the United Kingdom.
92
Table of Contents
NOTICE TO CANADIAN RESIDENTS
Resale Restrictions
The distribution of our common stock in Canada is being made only on a private placement basis exempt from the requirement that we prepare and file a prospectus with the securities regulatory authorities in each province where trades of common stock are made. Any resale of our common stock in Canada must be made under applicable securities laws which will vary depending on the relevant jurisdiction, and which may require resales to be made under available statutory exemptions or under a discretionary exemption granted by the applicable Canadian securities regulatory authority. Purchasers are advised to seek legal advice prior to any resale of shares of common stock.
Representations of Purchasers
By purchasing our common stock in Canada and accepting a purchase confirmation, a purchaser is representing to us and the dealer from whom the purchase confirmation is received that:
| • | the purchaser is entitled under applicable provincial securities laws to purchase the shares of common stock without the benefit of a prospectus qualified under those securities laws, | |
| • | where required by law, that the purchaser is purchasing as principal and not as agent, and | |
| • | the purchaser has reviewed the text above under Resale Restrictions. |
Rights of Action — Ontario Purchasers Only
Under Ontario securities legislation, a purchaser who purchases a security offered by this prospectus during the period of distribution will have a statutory right of action for damages, or while still the owner of the shares of common stock, for rescission against us in the event that this prospectus contains a misrepresentation. A purchaser will be deemed to have relied on the misrepresentation. The right of action for damages is exercisable not later than the earlier of 180 days from the date the purchaser first had knowledge of the facts giving rise to the cause of action and three years from the date on which payment is made for the shares. The right of action for rescission is exercisable not later than 180 days from the date on which payment is made for the shares. If a purchaser elects to exercise the right of action for rescission, the purchaser will have no right of action for damages against us. In no case will the amount recoverable in any action exceed the price at which the shares of common stock were offered to the purchaser and if the purchaser is shown to have purchased the securities with knowledge of the misrepresentation, we will have no liability. In the case of an action for damages, we will not be liable for all or any portion of the damages that are proven to not represent the depreciation in value of the shares of common stock as a result of the misrepresentation relied upon. These rights are in addition to, and without derogation from, any other rights or remedies available at law to an Ontario purchaser. The foregoing is a summary of the rights available to an Ontario purchaser. Ontario purchasers should refer to the complete text of the relevant statutory provisions.
Enforcement of Legal Rights
All of our directors and officers as well as the experts named herein may be located outside of Canada and, as a result, it may not be possible for Canadian purchasers to effect service of process within Canada upon us or those persons. All or a substantial portion of our assets and the assets of those persons may be located outside of Canada and, as a result, it may not be possible to satisfy a judgment against us or those persons in Canada or to enforce a judgment obtained in Canadian courts against us or those persons outside of Canada.
Taxation and Eligibility for Investment
Canadian purchasers of our common stock should consult their own legal and tax advisors with respect to the tax consequences of an investment in our common stock in their particular circumstances
93
Table of Contents
and about the eligibility of the shares of common stock for investment by the purchaser under relevant Canadian legislation.
VALIDITY OF SECURITIES
The validity of the common stock offered hereby will be passed upon for us by Heller Ehrman White & McAuliffe LLP and for the underwriters by Ropes & Gray LLP. As of December 31, 2004, persons and entities affiliated with Heller Ehrman White & McAuliffe LLP beneficially owned 8,438 shares of our common stock immediately prior to the closing of this offering.
EXPERTS
Ernst & Young LLP, Independent Registered Public Accounting Firm, have audited our financial statements at December 31, 2001, 2002 and 2003, for the period from March 6, 2001 (inception) to December 31, 2001, for the years ended December 31, 2002 and 2003 and for the period March 6, 2001 (inception) to December 31, 2003, as set forth in their report. We have included our financial statements in the prospectus and elsewhere in the registration statement in reliance of Ernst & Young LLP’s report, given on their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1 under the Securities Act that registers the shares of our common stock to be sold in this offering. This prospectus does not contain all of the information set forth in the registration statement and the exhibits and schedules filed as part of the registration statement. For further information with respect to us and our common stock, we refer you to the registration statement and the exhibits and schedules filed as a part of the registration statement. Statements contained in this prospectus concerning the contents of any contract or any other document are not necessarily complete. If a contract or document has been filed as an exhibit to the registration statement, we refer you to the copy of the contract or document that has been filed. Each statement in this prospectus relating to a contract or document filed as an exhibit is qualified in all respects by the filed exhibit. The reports and other information we file with the SEC can be read and copied at the SEC’s Public Reference Room at 450 Fifth Street, N.W., Washington D.C. 20549. Copies of these materials can be obtained at prescribed rates from the Public Reference Section of the SEC at the principal offices of the SEC, 450 Fifth Street, N.W., Washington D.C. 20549. You may obtain information regarding the operation of the public reference room by calling 1(800) SEC-0330. The SEC also maintains a web site (http://www.sec.gov) that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC.
Upon completion of this offering, we will become subject to the reporting and information requirements of the Exchange Act and, as a result, will file periodic reports, proxy statements and other information with the SEC. These periodic reports, proxy statements and other information will be available for inspection and copying at the SEC’s public reference room and the web site of the SEC referred to above.
94
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
CONTENTS
| Report of Independent Registered Public Accounting Firm | F-2 | |||
| Consolidated Balance Sheets as of December 31, 2002 and 2003 | F-3 | |||
| Consolidated Statements of Operations for the period from March 6, 2001 (inception) to December 31, 2001, the years ended December 31, 2002 and 2003 and the period from March 6, 2001 (inception) to December 31, 2003 | F-4 | |||
| Consolidated Statements of Stockholders’ Deficiency for the period from March 6, 2001 (inception) to December 31, 2001, the years ended December 31, 2002 and 2003 and the period from March 6, 2001 (inception) to December 31, 2003 | F-5 | |||
| Consolidated Statements of Cash Flows for the period from March 6, 2001 (inception) to December 31, 2001, the years ended December 31, 2002 and 2003 and the period from March 6, 2001 (inception) to December 31, 2003 | F-6 | |||
| Notes to Audited Consolidated Financial Statements | F-7 | |||
| Consolidated Balance Sheets as of December 31, 2003 and September 30, 2004 (unaudited) | F-22 | |||
| Consolidated Statements of Operations for the nine months ended September 30, 2003 and 2004 and the period from March 6, 2001 (inception) to September 30, 2004 (unaudited) | F-23 | |||
| Consolidated Statements of Cash Flows for the nine months ended September 30, 2003 and 2004 and the period from March 6, 2001 (inception) to September 30, 2004 (unaudited) | F-24 | |||
| Notes to Unaudited Consolidated Financial Statements | F-25 |
All other schedules for which provision is made in the applicable accounting regulation of the Securities and Exchange Commission are not required under the related instructions or are inapplicable and therefore have been omitted.
F-1
Table of Contents
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and Stockholders
AlgoRx Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of AlgoRx Pharmaceuticals, Inc. (a development stage company) as of December 31, 2002 and 2003, and the related consolidated statements of operations, stockholders’ equity (deficiency) and cash flows for the period from March 6, 2001 (inception) to December 31, 2001, and for the years ended December 31, 2002 and 2003, and for the period from March 6, 2001 (inception) to December 31, 2003. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position of AlgoRx Pharmaceuticals, Inc. at December 31, 2002 and 2003, and the consolidated results of their operations and their cash flows for the period from March 6, 2001 (inception) to December 31, 2001 and for the years ended December 31, 2002 and 2003, and for the period from March 6, 2001 (inception) to December 31, 2003, in conformity with U.S. generally accepted accounting principles.
| Ernst & Young LLP |
MetroPark, New Jersey
November 23, 2004, except as to Note 13, as to which the date is January 19, 2005.
The foregoing report is in the form that will be signed upon completion of the restatement of capital accounts described in Note 13 to the consolidated financial statements.
| /s/ Ernst & Young LLP |
F-2
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
CONSOLIDATED BALANCE SHEETS
| December 31, | ||||||||||
| 2002 | 2003 | |||||||||
| ASSETS | ||||||||||
| Current assets: | ||||||||||
| Cash and cash equivalents | $ | 8,873,270 | $ | 4,545,976 | ||||||
| Prepaid expenses and other current assets | 162,709 | 183,961 | ||||||||
| Total current assets | 9,035,979 | 4,729,937 | ||||||||
| Property and equipment, net | 3,155,598 | 2,373,840 | ||||||||
| Acquired workforce, net | 274,161 | — | ||||||||
| Deferred financing costs | — | 82,377 | ||||||||
| Other assets | 214,993 | 214,993 | ||||||||
| Total assets | $ | 12,680,731 | $ | 7,401,147 | ||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIENCY) | ||||||||||
| Current liabilities: | ||||||||||
| Accounts payable | $ | 274,476 | $ | 493,530 | ||||||
| Accrued compensation | 128,533 | 273,399 | ||||||||
| Other accrued liabilities | 1,360,848 | 1,202,453 | ||||||||
| Convertible notes | — | 9,800,000 | ||||||||
| Total current liabilities | 1,763,857 | 11,769,382 | ||||||||
| Stockholders’ equity (deficiency): | ||||||||||
| Series A convertible preferred stock, $0.001 par value; 9,150,000 shares authorized, issued and outstanding at December 31, 2002 and 2003, respectively (aggregate liquidation preference of $9,150,000 at December 31, 2002 and 2003) | 9,099,428 | 9,099,428 | ||||||||
| Series B convertible preferred stock, $0.001 par value; 26,350,000 shares authorized at December 31, 2002 and 2003; 17,858,462 issued and outstanding at December 31, 2002 and 2003, respectively (aggregate liquidation preference of $23,216,000 at December 31, 2002 and 2003) | 23,094,410 | 23,094,410 | ||||||||
| Common stock, $0.001 par value, 5,000,000 shares authorized at December 31, 2002 and 2003; 336,565 and 341,290 shares issued at December 31, 2002 and 2003, respectively; 336,565 and 314,207 shares outstanding at December 31, 2002 and 2003, respectively | 337 | 341 | ||||||||
| Additional paid-in capital | 330,007 | 673,857 | ||||||||
| Deferred compensation | — | (142,564 | ) | |||||||
| Treasury stock, at cost; 0 and 27,083 shares at December 31, 2002 and 2003, respectively | — | (271 | ) | |||||||
| Deficit accumulated during the development stage | (21,607,308 | ) | (37,093,436 | ) | ||||||
| Total stockholders’ equity (deficiency) | 10,916,874 | (4,368,235 | ) | |||||||
| Total liabilities and stockholders’ equity (deficiency) | $ | 12,680,731 | $ | 7,401,147 | ||||||
See accompanying notes.
F-3
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
CONSOLIDATED STATEMENTS OF OPERATIONS
| Period from | Period from | ||||||||||||||||
| March 6, 2001 | Year Ended December 31, | March 6, 2001 | |||||||||||||||
| (inception) to | (inception) to | ||||||||||||||||
| December 31, 2001 | 2002 | 2003 | December 31, 2003 | ||||||||||||||
| Contract revenues | $ | — | $ | 149,216 | $ | 100,000 | $ | 249,216 | |||||||||
| Costs and expenses: | |||||||||||||||||
| Research and development | 365,303 | 11,745,089 | 12,190,813 | 24,301,205 | |||||||||||||
| General and administrative | 1,096,671 | 3,075,615 | 3,477,329 | 7,649,615 | |||||||||||||
| Acquired in-process research and development and other | — | 5,716,247 | — | 5,716,247 | |||||||||||||
| Total costs and expenses | 1,461,974 | 20,536,951 | 15,668,142 | 37,667,067 | |||||||||||||
| Loss from operations | (1,461,974 | ) | (20,387,735 | ) | (15,568,142 | ) | (37,417,851 | ) | |||||||||
| Gain (loss) on sale of assets | — | (36,101 | ) | 103,324 | 67,223 | ||||||||||||
| Interest expense | (1,577 | ) | (4,353 | ) | (107,310 | ) | (113,240 | ) | |||||||||
| Interest and other income, net | 47,333 | 237,099 | 86,000 | 370,432 | |||||||||||||
| Net loss | $ | (1,416,218 | ) | $ | (20,191,090 | ) | $ | (15,486,128 | ) | $ | (37,093,436 | ) | |||||
| Basic and diluted net loss per share | $ | (79.27 | ) | $ | (110.36 | ) | $ | (59.75 | ) | ||||||||
| Weighted average shares outstanding — basic and diluted | 17,866 | 182,949 | 259,182 | ||||||||||||||
| Unaudited pro forma basic and diluted net loss per share | $ | (5.23 | ) | ||||||||||||||
| Unaudited pro forma weighted average shares outstanding — basic and diluted | 2,960,028 | ||||||||||||||||
See accompanying notes.
F-4
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIENCY)
Period from March 6, 2001 (inception) to December 31, 2003
| Deficit | |||||||||||||||||||||||||||||||||||||||||||||||||
| Series A Convertible | Series B Convertible | Accumulated | Total | ||||||||||||||||||||||||||||||||||||||||||||||
| Preferred Stock | Preferred Stock | Common Stock | Additional | Treasury Stock | During the | Stockholders’ | |||||||||||||||||||||||||||||||||||||||||||
| Paid-In | Deferred | Development | Equity | ||||||||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Capital | Compensation | Shares | Amount | Stage | (Deficiency) | ||||||||||||||||||||||||||||||||||||||
| Balance at March 6, 2001 (inception) | — | $ | — | — | $ | — | — | $ | — | $ | — | $ | — | — | $ | — | $ | — | $ | — | |||||||||||||||||||||||||||||
| Issuance of common stock to founders for $0.001 per share in March 2001 | — | — | — | — | 150,000 | 150 | 1,350 | — | — | — | — | 1,500 | |||||||||||||||||||||||||||||||||||||
| Issuance of common stock upon exercise of stock options in April 2001 | — | — | — | — | 23,800 | 24 | 23,776 | — | — | — | — | 23,800 | |||||||||||||||||||||||||||||||||||||
| Issuance of Series A convertible preferred stock at $1.00 per share, net of issuance costs of $50,572, in April 2001 | 9,150,000 | 9,099,428 | — | — | — | — | — | — | — | — | — | 9,099,428 | |||||||||||||||||||||||||||||||||||||
| Net loss and comprehensive loss | — | — | — | — | — | — | — | — | — | — | (1,416,218 | ) | (1,416,218 | ) | |||||||||||||||||||||||||||||||||||
| Balance at December 31, 2001 | 9,150,000 | 9,099,428 | — | — | 173,800 | 174 | 25,126 | — | — | — | (1,416,218 | ) | 7,708,510 | ||||||||||||||||||||||||||||||||||||
| Issuance of common stock upon exercise of stock options in March 2002 | — | — | — | — | 10,150 | 10 | 10,140 | — | — | — | — | 10,150 | |||||||||||||||||||||||||||||||||||||
| Issuance of Series B convertible preferred stock at $1.30 per share, net of issuance costs of $121,590, in March 2002 | — | — | 11,692,308 | 15,078,410 | — | — | — | — | — | — | — | 15,078,410 | |||||||||||||||||||||||||||||||||||||
| Issuance of Series B convertible preferred stock at $1.30 per share for the acquisition of PowderJect Technologies, Inc. in March 2002 | — | — | 6,166,154 | 8,016,000 | — | — | — | — | — | — | — | 8,016,000 | |||||||||||||||||||||||||||||||||||||
| Issuance of common stock for acquisition of PowderJect Technologies, Inc. in March 2002 | — | — | — | — | 152,615 | 153 | 228,770 | — | — | — | — | 228,923 | |||||||||||||||||||||||||||||||||||||
| Stock-based compensation resulting from stock options granted to nonemployees | — | — | — | — | — | — | 65,971 | — | — | — | — | 65,971 | |||||||||||||||||||||||||||||||||||||
| Net loss and comprehensive loss | — | — | — | — | — | — | — | — | — | — | (20,191,090 | ) | (20,191,090 | ) | |||||||||||||||||||||||||||||||||||
| Balance at December 31, 2002 | 9,150,000 | 9,099,428 | 17,858,462 | 23,094,410 | 336,565 | 337 | 330,007 | — | — | — | (21,607,308 | ) | 10,916,874 | ||||||||||||||||||||||||||||||||||||
| Issuance of common stock upon exercise of stock options in 2003 | — | — | — | — | 4,725 | 4 | 5,583 | — | — | — | — | 5,587 | |||||||||||||||||||||||||||||||||||||
| Accrued interest costs | — | — | — | — | — | — | 107,310 | — | — | — | — | 107,310 | |||||||||||||||||||||||||||||||||||||
| Deferred compensation related to stock options | — | — | — | — | — | — | 155,856 | (155,856 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||
| Noncash compensation | — | — | — | — | — | — | 66,240 | — | — | — | — | 66,240 | |||||||||||||||||||||||||||||||||||||
| Repurchase of common stock | — | — | — | — | — | — | — | 27,083 | (271 | ) | (271 | ) | |||||||||||||||||||||||||||||||||||||
| Amortization of deferred compensation | — | — | — | — | — | — | — | 13,292 | — | — | — | 13,292 | |||||||||||||||||||||||||||||||||||||
| Stock-based compensation resulting from stock options granted to nonemployees | — | — | — | — | — | — | 8,861 | — | — | — | — | 8,861 | |||||||||||||||||||||||||||||||||||||
| Net loss and comprehensive loss | — | — | — | — | — | — | — | — | — | — | (15,486,128 | ) | (15,486,128 | ) | |||||||||||||||||||||||||||||||||||
| Balance at December 31, 2003 | 9,150,000 | $ | 9,099,428 | 17,858,462 | $ | 23,094,410 | 341,290 | $ | 341 | $ | 673,857 | $ | (142,564 | ) | 27,083 | $ | (271 | ) | $ | (37,093,436 | ) | $ | (4,368,235 | ) | |||||||||||||||||||||||||
See accompanying notes.
F-5
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
CONSOLIDATED STATEMENTS OF CASH FLOWS
| Period from | Period from | |||||||||||||||||
| March 6, | March 6, | |||||||||||||||||
| 2001 | 2001 | |||||||||||||||||
| (inception) to | Year Ended December 31, | (inception) to | ||||||||||||||||
| December 31, | December 31, | |||||||||||||||||
| 2001 | 2002 | 2003 | 2003 | |||||||||||||||
Operating activities | ||||||||||||||||||
| Net loss | $ | (1,416,218 | ) | $ | (20,191,090 | ) | $ | (15,486,128 | ) | $ | (37,093,436 | ) | ||||||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||||||||
| Depreciation and amortization | 9,920 | 956,200 | 1,059,889 | 2,026,009 | ||||||||||||||
| Noncash stock-based compensation | — | 65,971 | 88,393 | 154,364 | ||||||||||||||
| Non-cash interest expense | — | — | 107,310 | 107,310 | ||||||||||||||
| Acquired in-process research and development | — | 5,716,247 | — | 5,716,247 | ||||||||||||||
| Amortization of intangible assets | — | 174,138 | 274,161 | 448,299 | ||||||||||||||
| Loss on disposal of equipment | — | 36,101 | — | 36,101 | ||||||||||||||
| Gain on disposal of equipment | — | — | (103,324 | ) | (103,324 | ) | ||||||||||||
| Changes in operating assets and liabilities: | ||||||||||||||||||
| Prepaid expenses and other current assets | (41,169 | ) | (75,012 | ) | (21,252 | ) | (137,433 | ) | ||||||||||
| Other assets | (16,594 | ) | — | (82,377 | ) | (98,971 | ) | |||||||||||
| Accounts payable | 203,742 | 53,358 | 219,053 | 476,153 | ||||||||||||||
| Accrued compensation | 103,814 | (62,806 | ) | 144,866 | 185,874 | |||||||||||||
| Other accrued liabilities | — | 1,271,766 | (161,775 | ) | 1,109,991 | |||||||||||||
| Net cash used in operating activities | (1,156,505 | ) | (12,055,127 | ) | (13,961,184 | ) | (27,172,816 | ) | ||||||||||
Investing activities | ||||||||||||||||||
| Purchases of property and equipment | (48,071 | ) | (529,889 | ) | (394,373 | ) | (972,333 | ) | ||||||||||
| Proceeds from disposal of equipment | — | 30,000 | 222,947 | 252,947 | ||||||||||||||
| Acquisition of PowderJect Technologies, Inc. | — | (1,442,029 | ) | — | (1,442,029 | ) | ||||||||||||
| Other acquisition related expenditures | — | (96,613 | ) | — | (96,613 | ) | ||||||||||||
| Net cash used in investing activities | (48,071 | ) | (2,038,531 | ) | (171,426 | ) | (2,258,028 | ) | ||||||||||
Financing activities | ||||||||||||||||||
| Repayment of capital lease obligations | (5,998 | ) | (37,313 | ) | — | (43,311 | ) | |||||||||||
| Proceeds from issuance of convertible preferred stock, net of issuance costs | 9,099,428 | 15,078,410 | — | 24,177,838 | ||||||||||||||
| Proceeds from issuances of common stock | 25,300 | 11,677 | 5,587 | 42,564 | ||||||||||||||
| Funds used to repurchase common stock | — | — | (271 | ) | (271 | ) | ||||||||||||
| Proceeds from debt | — | — | 9,800,000 | 9,800,000 | ||||||||||||||
| Net cash provided by financing activities | 9,118,730 | 15,052,774 | 9,805,316 | 33,976,820 | ||||||||||||||
| Net (decrease) increase in cash and cash equivalents | 7,914,154 | 959,116 | (4,327,294 | ) | 4,545,976 | |||||||||||||
| Cash and cash equivalents, beginning of period | — | 7,914,154 | 8,873,270 | — | ||||||||||||||
| Cash and cash equivalents, end of period | $ | 7,914,154 | $ | 8,873,270 | $ | 4,545,976 | $ | 4,545,976 | ||||||||||
Supplemental disclosure of cash flow information | ||||||||||||||||||
| Cash paid during the year for interest | $ | 1,577 | $ | 4,353 | $ | — | $ | 5,930 | ||||||||||
Supplemental cash flow information | ||||||||||||||||||
| Issuance of $8,016,000 of convertible preferred stock and $228,923 of common stock in connection with acquisition of PowderJect Technologies, Inc. | $ | — | $ | 8,244,923 | $ | — | $ | 8,244,923 | ||||||||||
| Equipment acquired under capital leases | $ | 43,311 | $ | — | $ | — | $ | 43,311 | ||||||||||
See accompanying notes.
F-6
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2003
| 1. | Summary of Significant Accounting Policies |
| Organization, Description of Business and Basis of Presentation |
AlgoRx Pharmaceuticals, Inc. (the “Company” or “AlgoRx”) was incorporated on March 6, 2001 in Delaware. During 2003, the Company was headquartered in Cranbury, New Jersey, with facilities also in Fremont, California. In July of 2004, the Company moved its headquarters to Secaucus, New Jersey. AlgoRx is focused on building a diversified portfolio of pharmaceutical products and technologies to address the pain therapeutic market. The Company’s activities since inception have consisted principally of acquiring product and technology rights, raising capital, establishing facilities and performing research and development. Accordingly, the Company is considered to be in the development stage as defined by Statement of Financial Accounting Standards No. 7,Accounting and Reporting by Development Stage Enterprises. The Company operates in one business segment.
The Company expects to continue to incur substantial losses over the next several years during its development phase. To fully execute its business plan, the Company will need to complete certain research and development activities and clinical trials. Further, the Company’s product candidates will require regulatory approval prior to commercialization. These activities may span many years and require substantial expenditures to complete and may ultimately be unsuccessful. Any delays in completing these activities could adversely impact the Company. The Company plans to meet its capital requirements primarily through issuances of equity securities, research and development contract revenue, and in the longer term, revenue from product sales (see Note 13).
| Use of Estimates |
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
| Principles of Consolidation |
The consolidated financial statements include the accounts of AlgoRx and, beginning on March 22, 2002, its wholly owned subsidiary, AlgoRx Technologies, Inc. (formerly PowderJect Technologies, Inc.), located in Fremont, California. Intercompany accounts and transactions have been eliminated.
| Cash and Cash Equivalents |
Cash and cash equivalents consist of highly liquid instruments purchased with an original maturity of three months or less.
| Property and Equipment |
Property and equipment are stated at cost, less accumulated depreciation. Depreciation is provided over the estimated useful lives of the respective assets, which range from three to five years, using the straight-line method.
Leasehold improvements are amortized over the lives of the related leases or their estimated useful lives, whichever is shorter, using the straight-line method.
F-7
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| Intangible Asset |
Intangible asset consists of the assembled workforce acquired in the acquisition of PowderJect Technologies, Inc. (see Note 2). The acquired workforce is being amortized on a straight-line basis over two years, which represents the estimated retention period for the related employees. Acquired workforce amortization is recorded in research and development expense.
| Long-Lived Assets |
Long-lived assets and certain identifiable intangible assets to be held and used are reviewed for impairment when events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. Determination of recoverability is based on an estimate of undiscounted future cash flows resulting from the use of the asset and its eventual disposition. In the event that such cash flows are not expected to be sufficient to recover the carrying amount of the assets, the assets are written down to their estimated fair values. Long-lived assets and certain identifiable assets to be disposed of are reported at the lower of carrying amount or fair value less cost to sell.
| Fair Value of Financial Instruments |
The carrying values of the Company’s financial instruments, which include cash and cash equivalents, accounts payable and accrued expenses, approximates their fair values.
| Revenue Recognition |
The Company’s revenue recognition policies are in accordance with Securities and Exchange Commission (SEC) Staff Accounting Bulletins No. 101 and No. 104, or SAB 101 and 104, and EITF 00-21,Revenue Recognition in Financial Statements, which provides guidance on revenue recognition in financial statements and is based on the interpretations and practices developed by the SEC. SAB 101 requires that four basic criteria be met before revenue can be recognized: (1) persuasive evidence exists of an arrangement; (2) delivery has occurred or services have been rendered; (3) the fee is fixed and determinable; and (4) collectibility is reasonably assured. Determination of criteria (3) and (4) are based on management’s judgments regarding the fixed nature of the fees charged for services rendered and products delivered and the collectibility of those fees. Accordingly, revenues from licensing agreements are recognized based on the performance requirements of the agreement. If the Company has an ongoing involvement or performance obligation, non-refundable up-front fees are generally recorded as deferred revenue in the balance sheet and amortized into license fees in the statement of operations over the term of the performance obligation. If the Company has no ongoing involvement or performance obligation, non-refundable up-front fees are generally recorded as revenue in the period in which the rights are transferred.
| Research and Development Costs |
Research and development costs are expensed as incurred. Research and development costs consist of salaries, employee benefits, laboratory supplies, consulting services, clinical services and facility costs.
Acquired in-process research and development relates primarily to in-licensed technology, intellectual property and know-how. The Company evaluates the stage of development of acquired projects, taking into account the level of effort, time and estimated cost associated with further developing the in-process technology and producing a commercial product. The nature of the remaining efforts for completion of acquired in-process research and development projects generally include completion of clinical trials, completion of manufacturing validation, interpretation of clinical and preclinical data and obtaining
F-8
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
marketing approval from the FDA and other regulatory bodies, the cost, length and success of which are extremely difficult to determine. Numerous risks and uncertainties exist with timely completion of development projects, including clinical trial results, manufacturing process development results, and ongoing feedback from regulatory authorities, including obtaining marketing approval. In addition, acquired products under development may never be successfully commercialized due to the uncertainties associated with the pricing of new pharmaceuticals, the cost to produce these products in a commercial setting, changes in the reimbursement environment, or the introduction of new competitive products. As a result of the uncertainties noted above, the Company expenses such acquired in-process research and development projects when incurred.
| Concentration of Credit Risk |
The Company’s financial instruments that are exposed to credit risks consist primarily of cash and cash equivalents. The Company maintains its cash and cash equivalents in bank accounts, which, at times, exceed federally insured limits. The Company has not experienced any losses in such accounts. The Company believes it is not exposed to significant credit risk related to cash and cash equivalents.
| Income Taxes |
The Company accounts for income taxes under Statement of Financial Accounting Standards No. 109, Accounting for Income Taxes. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates that will be in effect when the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized.
| Comprehensive Loss |
The Company’s total comprehensive loss is the same as its net loss for all periods presented.
| Net Loss Per Share |
The Company computes net loss per share in accordance with Statement of Financial Accounting Standards No. 128, “Earnings per Share” (“SFAS 128”). Under the provisions of SFAS 128, basic net loss per common share (“Basic EPS”) is computed by dividing net loss by the weighted-average number of common shares outstanding (excluding unvested founders’ shares subject to repurchase). Diluted net loss per common share (“Diluted EPS”) is computed by dividing net loss by the weighted-average number of common shares and dilutive common shares equivalents then outstanding. Common equivalent shares consist of the incremental common shares issuable upon the conversion of preferred stock, convertible debt, shares issuable upon the exercise of stock options, and unvested founders’ shares subject to repurchase. Diluted EPS is identical to Basic EPS since common equivalent shares are excluded from the calculation, as their effect is anti-dilutive.
| Unaudited Pro Forma Information |
The unaudited pro forma net loss per share is computed using the weighted average number of common shares outstanding, including the pro forma effects of the automatic conversion of all outstanding convertible preferred stock into shares of the Company’s common stock effective upon the assumed closing of the Company’s proposed initial public offering, as if such conversion had occurred at the date of original issuance.
F-9
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| Stock-Based Compensation |
As permitted by the Financial Accounting Standards Board (“FASB”) Statement No. 123, Accounting for Stock-Based Compensation (“SFAS 123”), as amended, the Company accounts for employee stock-based compensation in accordance with Accounting Principles Board Opinion No. 25, Accounting for Stock Issued to Employees (“APB 25”), and related interpretations, in accounting for its stock-based compensation plans. Under APB 25, when the exercise price of the Company’s employee stock options equals or exceeds the estimated fair value of the underlying stock on the date of grant, no compensation expense is recorded.
Pro forma information regarding net loss and net loss per share has been determined as if the Company had accounted for its employee stock options under the fair value method proscribed by SFAS 123. The resulting effect on pro forma net loss disclosed is not likely to be representative of the effects of loss on a pro forma basis in future years due to additional grants and years of vesting in subsequent years. Pro forma compensation related to stock option grants is expensed over their respective vesting periods.
Had compensation cost for options granted under the Company’s stock option plan been determined based on the fair value at the grant dates for the awards under a method prescribed by SFAS 123, the Company’s net loss would have been adjusted to the following pro forma amounts:
| Period from | ||||||||||||
| March 6, 2001 | ||||||||||||
| (inception) to | Year Ended December 31, | |||||||||||
| December 31, | ||||||||||||
| 2001 | 2002 | 2003 | ||||||||||
| Net loss, as reported | $ | (1,416,218 | ) | $ | (20,191,090 | ) | $ | (15,486,128 | ) | |||
| Add: Noncash employee compensation as reported | — | — | 13,292 | |||||||||
| Deduct: Stock-based employee compensation expense under the fair value based method for all awards, net of tax | (5,464 | ) | (140,391 | ) | (199,622 | ) | ||||||
| Net loss, pro forma | $ | (1,421,682 | ) | $ | (20,331,481 | ) | $ | (15,672,458 | ) | |||
| Basic and diluted loss attributable to common stockholders per share, as reported | $ | (79.27 | ) | $ | (110.36 | ) | $ | (59.75 | ) | |||
| Basic and diluted loss attributable to common stockholders per share, SFAS 123, pro forma | $ | (79.57 | ) | $ | (111.13 | ) | $ | (60.47 | ) | |||
| Unaudited pro forma basic and diluted net loss per share, SFAS 123, pro forma (Note 1) | $ | (5.29 | ) | |||||||||
The fair values of stock options granted to employees for the period from March 6, 2001 (inception) to December 31, 2001 and for the years ended December 31, 2002 and 2003 were estimated
F-10
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
on the respective dates of grant using the minimum value option-pricing model with the following weighted-average assumptions:
| Period from | ||||||||||||
| March 6, 2001 | Year Ended | |||||||||||
| (inception) to | December 31, | |||||||||||
| December 31, | ||||||||||||
| 2001 | 2002 | 2003 | ||||||||||
| Risk-free interest rate | 4.50 | % | 4.61 | % | 4.02 | % | ||||||
| Expected life (in years) | 9.0 | 9.0 | 9.0 | |||||||||
| Dividend yield | — | — | — | |||||||||
| Fair value | $ | 0.90 | $ | 1.40 | $ | 9.90 | ||||||
The Company has also granted stock options to purchase stock to consultants, advisors, and other vendors. The Company accounts for stock awards issued to such nonemployees in accordance with the provisions of SFAS 123 and Emerging Issues Task Force (“EITF”) Issue No. 96-18, Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services (“Issue No. 96-18”). Under SFAS 123 and Issue No. 96-18, stock awards to nonemployees are accounted for at their respective fair values using the Black-Scholes option-pricing model unless a more readily determinable fair value is available. The fair value of options granted to nonemployees is remeasured during the performance period as the underlying options vest or as milestones are reached.
| Recently Issued Accounting Standards |
In January 2003, the FASB issued Interpretation No. 46 (“FIN 46”),Consolidation of Variable Interest Entities, which addresses consolidation of variable interest entities (“VIEs”). FIN 46 requires a VIE to be consolidated by a parent company if that company is subject to a majority of the risk of loss from the variable interest entity’s activities or entitled to receive a majority of the entity’s residual returns or both. A VIE is a corporation, partnership, trust or any other legal structure used for business purposes that either does not have equity investors with voting rights or has equity investors that do not provide sufficient financial resources for the entity to support its activities. The consolidation requirements of FIN 46 apply immediately to VIEs created after January 31, 2003. For entities created prior to February 1, 2003, the effective date of these requirements has been deferred so as not to apply until the first period ending after December 15, 2004. The adoption of the provisions applicable to special purpose entities (“SPEs”) and all other variable interests obtained after January 31, 2003 had no impact on the Company’s financial statements. The Company is currently evaluating the impact of adopting FIN 46-R applicable to non-SPEs created prior to February 1, 2003, but does not expect a material impact on its financial position, results of operations or cash flows.
In May 2003, the FASB issued Statement of Financial Accounting Standards No. 150,Accounting for Certain Financial Instruments with Characteristics of both Liabilities and Equity (“SFAS 150”). This statement establishes standards for classifying and measuring as liabilities certain financial instruments that embody obligations of the issuer and have characteristics of both liabilities and equity. The scope of this pronouncement includes mandatorily redeemable equity instruments. Application to pre-existing instruments should be recognized as the cumulative effect of a change in accounting principle (application by retroactive restatement is precluded). Exceptions to the transition requirements were provided for mandatorily redeemable instruments of non-public companies. Subsequent to the issuance of SFAS 150, the FASB issued FSP 150-3, which further deferred the transition requirements for nonpublic companies that also are not SEC registrants for instruments that are mandatorily redeemable on fixed dates that are for fixed amounts. The classification, measurement, and disclosure provisions of SFAS 150 are effective for
F-11
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
fiscal periods beginning after December 15, 2004. The adoption of this statement is not expected to impact the classification of the Company’s convertible preferred stock.
| 2. | Acquisition of PowderJect Technologies, Inc. |
On March 22, 2002, the Company acquired all of the outstanding shares of PowderJect Technologies, Inc. (“PowderJect”), a development stage company, for a total purchase price of approximately $9.9 million. This purchase price included 6,166,154 shares of the Company’s Series B convertible preferred stock, 152,615 shares of common stock, cash consideration of $718,150 for a minority interest’s share in the acquired company, acquisition related costs of $723,879 and assumed liabilities of $192,458. The Series B convertible preferred stock was valued based on the arm’s length Series B financing completed during that year and the common stock consideration was valued using the Company’s estimated fair value used for option grants made to employees. PowderJect was in the business of developing drug applications for a patented, needle-free, delivery system developed by its parent company, PowderJect Pharmaceuticals plc. The Company also entered into a supply agreement with PowderJect Technologies Limited (subsequently purchased by Chiron Corporation) for cylinders. The cylinders are a key component of the product candidate, ALGRX 3268, and PowderJect Technologies Limited is the sole supplier. No other supplier has been identified by the Company.
The acquisition was completed to provide the Company with the ability to exploit the opportunities associated with its in-process lidocaine technology, as well as gain access to the patented drug delivery technology. The purchase price was determined in accordance with Statement of Financial Accounting Standards No. 141,Business Combinations(“SFAS 141”), and Statement of Financial Accounting Standards No. 142,Goodwill and Other Intangible Assets (“SFAS 142”). A summary of the determination of the purchase price during 2002 was as follows:
| 6,166,154 shares — Series B convertible preferred stock | $ | 8,016,000 | ||
| 152,615 shares — common stock | 228,923 | |||
| Cash for minority interest buyout | 718,150 | |||
| Acquisition related costs | 723,879 | |||
| Assumed liabilities | 192,458 | |||
| Total purchase price | $ | 9,879,410 | ||
The acquisition was accounted for as an acquisition of assets rather than as a business combination as PowderJect was a development stage company that had not commenced its planned principal operations. PowderJect lacked the necessary elements of a business because it did not have completed products and, therefore, no ability to access customers. The PowderJect operating results have been included in the Company’s consolidated results of operations since March 23, 2002.
The Company allocated the purchase price in accordance with the provisions of SFAS 142 related to the purchase of a group of assets. SFAS 142 provides that the cost of a group of assets acquired in a transaction other than a business combination shall be allocated to the individual assets acquired based on their relative fair values and shall not give rise to goodwill.
In accordance with the provisions of SFAS 142, all identifiable intangible assets and acquired in-process research and development were assigned a portion of the purchase price based on their relative fair values. To this end, a third party valuation was used to assist management in determining the fair value of
F-12
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
the identifiable assets and acquired in-process research and development. Based on this valuation, the Company allocated the total consideration as follows:
| Acquired in-process research and development and other | $ | 5,716,247 | ||
| Tangible net assets | 3,714,864 | |||
| Acquired workforce | 448,299 | |||
| Total purchase price | $ | 9,879,410 | ||
The income approach was used to estimate the fair value of the patent license and the acquired in-process research and development based on projected cash flows, assuming a 30% discount rate. This discount rate is a significant assumption and is based on the Company’s estimated weighted average cost of capital taking into account the risks associated with the projects acquired. The projected cash flows from such projects were based on estimates of revenues and operating profits related to such projects considering the stage of development of each potential product acquired, the time and resources needed to complete each product, the estimated life of each potential commercialized product and associated risks which included the inherent difficulties and uncertainties in developing a drug compound, obtaining FDA and other regulatory approvals, and risks related to the viability of, and potential alternative treatments in, any future target markets. Approximately $4.5 million of the purchase price was allocated to acquired in-process research and development due to PowderJect’s incomplete research and development programs that had not yet reached technological feasibility as of March 2002 and had no alternative future use as of that date. In light of the number of years required to achieve product commercialization, the associated technology risk, and therefore the risk that the asset value will not be realized, the Company has recorded, as an expense, the value of the favorable patent license, which is $1.2 million, similar to the treatment afforded the acquired in-process research and development.
The cost approach was used to determine the value of the acquired workforce. The value allocated to the acquired workforce is attributable to 31 employees hired by the Company from PowderJect following the asset acquisition, which eliminated the need to hire replacement employees. The value of the acquired workforce was determined by estimating the cost of assembling a new workforce, including costs of salaries, benefits, training, and recruiting.
As of December 31, 2002 and 2003, the Company’s intangible assets are as follows:
| December 31, | ||||||||
| 2002 | 2003 | |||||||
| Acquired workforce | $ | 448,299 | $ | 448,299 | ||||
| Accumulated amortization | (174,138 | ) | (448,299 | ) | ||||
| Net balance | $ | 274,161 | $ | — | ||||
Amortization expense was $174,138, $274,161 and $448,299 for the years ended December 31, 2002 and 2003 and from March 6, 2001 (inception) to December 31, 2003, respectively. Included in the 2003 amortization was $227,461 related to the acquired workforce that was written off in connection with the restructuring described in Note 3.
| 3. | Restructuring |
In March 2003, the Company announced a reorganization plan intended to reduce operating costs and reduced its staff by 28 employees (approximately 61% of total employees as of the date of reorganization); however, no research projects were discontinued. The reorganization resulted in an impairment of the
F-13
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
assembled workforce intangible asset of $227,461 recorded as accelerated amortization expense during 2003. This expense is classified under research and development expenses for the year ended December 31, 2003. In addition, the vesting of certain options held by such employees at the time of termination was accelerated. The Company recorded a charge of $66,240 related to this acceleration of vesting. The total restructuring cost of $1,122,444 was charged to research and development expense.
| 4. | Property and Equipment |
Property and equipment consist of the following:
| December 31, | ||||||||
| 2002 | 2003 | |||||||
| Leasehold improvements | $ | 2,025,500 | $ | 1,987,027 | ||||
| Computer and office equipment | 234,426 | 127,048 | ||||||
| Lab equipment | 894,030 | 850,081 | ||||||
| Construction-in-process | 801,324 | 562,616 | ||||||
| Furniture and fixtures | 87,165 | 87,165 | ||||||
| 4,042,445 | 3,613,937 | |||||||
| Less accumulated depreciation and amortization | (886,847 | ) | (1,240,097 | ) | ||||
| Property and equipment, net | $ | 3,155,598 | $ | 2,373,840 | ||||
Depreciation and amortization expense was $9,920, $956,200, $1,059,889 and $2,026,009 for the period from March 6, 2001 (inception) to December 31, 2001, for the years ended December 31, 2002 and 2003 and for the period from March 6, 2001 (inception) to December 31, 2003, respectively. During 2003, the Company sold certain computer and lab equipment that was determined to be unnecessary due to the reduction in force. The sale resulted in a recognized gain on disposal of approximately $103,000.
| 5. | Other Accrued Liabilities |
Other accrued liabilities consist of the following:
| December 31, | ||||||||
| 2002 | 2003 | |||||||
| Accrued patent costs | $ | 150,000 | $ | 294,000 | ||||
| Accrued research and development costs | 684,558 | 341,158 | ||||||
| Accrued legal | 41,411 | 141,356 | ||||||
| Accrued vacation | 122,557 | 130,611 | ||||||
| Other accrued liabilities | 362,322 | 295,328 | ||||||
| Total | $ | 1,360,848 | $ | 1,202,453 | ||||
| 6. | Commitments |
| Leases |
The Company leased certain office facilities and office equipment under operating and capital leases during the year ended December 31, 2003. As of December 31, 2003, the Company had satisfied its
F-14
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
obligation under its capital leases. The future minimum payments for all noncancelable operating leases as of December 31, 2003 are as follows:
| Years ending December 31, | |||||
| 2004 | $ | 665,397 | |||
| 2005 | 51,530 | ||||
| Thereafter | — | ||||
| Total | $ | 716,927 | |||
See Note 13 for information concerning leases entered into after December 31, 2003.
Rent expense under operating leases was approximately $184,000, $500,000, $660,000 and $1,344,000, for the period from March 6, 2001 (inception) to December 31, 2001, for the years ended December 31, 2002 and 2003 and for the period from March 6, 2001 (inception) to December 31, 2003, respectively.
| Other Commitments |
Under the Company’s license agreements, the Company could be required to pay up to a total of $1,366,000 in payments for milestones such as the initiation of clinical trials and the granting of patents. As of December 31, 2003, we had paid $75,000 in milestone payments for the execution of agreements, patent approvals and the initiation of U.S. clinical trials. Milestone payments will also be due upon the first administration to a subject using licensed technology in a phase I clinical trial, the first administration to a subject using licensed technology in a phase III clinical trial and FDA approval of ALGRX 4975. Phase III clinical trials and product approval of ALGRX 4975 in addition to sales milestones and royalties payable on commercial sales if any occur. The Company has no material commitments for capital expenditures.
Under our license with PowderMed Limited we are obligated and have spent $1,000,000 on research and development associated with the licensed technology during the period July 7, 2003 to July 7, 2005. We are also obligated to spend an additional $1,000,000 on research and development for every 12 month period from July 7, 2005 to July 7, 2008.
| 7. | Capital Structure |
| Common Stock |
The Company is authorized to issue 5,000,000 shares of common stock.
Dividends on common stock will be paid when, and if, declared by the Board of Directors. Each holder of common stock is entitled to vote on all matters and is entitled to one vote for each share held. The Company will, at all times, reserve and keep available, out of its authorized but unissued shares of common stock, sufficient shares to affect the conversion of the shares of the convertible preferred stock and stock options. As of December 31, 2003, the Company has reserved 3,161,254 shares of common stock for future issuance. Common shares issued to the founders of the Company vest over a four-year period. Unvested shares are subject to repurchase by the Company at the original issuance price. As of December 31, 2003, there were 42,187 founders shares subject to repurchase at an aggregate price of $422.
| Convertible Preferred Stock |
The Company is authorized to issue 35,500,000 shares of preferred stock. In April 2001, the Company issued 9,150,000 shares of Series A convertible preferred stock (“Series A”). In March 2002, the
F-15
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Company issued 17,858,462 shares of Series B convertible preferred stock (“Series B”). Such stock is subject to the following rights and privileges:
| Voting |
Series A and B stockholders are entitled to the number of votes equal to the number of shares of common stock into which each share of preferred stock is convertible.
| Dividends |
The holders of Series A and B are entitled to receive annual dividends at a rate of 8% of the original purchase price in advance of any distributions to common shareholders. Dividends are payable when, and as, declared by the Board of Directors and are noncumulative. No dividends have been declared through December 31, 2003.
| Conversion |
Series A and B stockholders are entitled, at any time, to cause their shares to be converted into fully paid and nonassessable shares of common stock. Shares of Series A and B will be converted into common stock based on a one-for-ten basis, subject to adjustment for antidilution. The antidilution rights go into effect if stock is sold at a price less than what was paid by the Series A or B stockholders. Additionally, the preferred stock will convert automatically (i) upon the affirmative election of the holders of at least a majority of the outstanding shares of preferred stock, or (ii) immediately upon the closing of a public offering pursuant to an effective registration statement under the Securities Act of 1933 covering the offer and sale of common stock, which results in aggregate net proceeds to the Company of at least $25,000,000 and a per share price of at least $5.00 (appropriately adjusted for any stock dividend, stock split or recapitalization).
| Liquidation |
In the event of any liquidation, dissolution or winding up of the Company, including a change of control, either voluntary or involuntary, the holders of Series A and B are entitled to receive, in preference to common stock, an amount equal to the purchase price per share, plus all declared but unpaid dividends (appropriately adjusted for any stock dividend, stock split or recapitalization). After payment of these preferential amounts, the remaining assets of the Company shall be distributed among the holders of common and preferred stock (assuming conversion of preferred stock).
| Convertible Notes |
In April 2003, the Company entered into several loan agreements with various financial institutions, whereby the financial institutions agreed to loan the Company an aggregate principal amount of $9,800,000 that upon closing would be converted into Series C preferred stock at the price at which the Series C preferred stock was sold. The interest on these loans was 1.46% per annum and was payable on December 31, 2004 if the notes were held and not converted on such date. As required by the terms of the loans, they were converted into Series C preferred stock at the Series C preferred stock price of $0.5925 per share, for a total of 16,540,084 shares of Series C preferred stock in February 2004 (see Note 13) and no interest was paid to the financial institutions in accordance with the loan agreements. In accordance with EITF 85-17:Accrued Interest upon Conversion of Convertible Debt, the interest cost of $107,310 was accrued through December 31, 2003 and the corresponding credit was recorded as a component of additional paid-in capital.
F-16
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| 8. | Stock Option Plan |
On March 30, 2001, the Company’s Board of Directors and shareholders approved the Company’s 2001 Equity Incentive Plan (the “2001 Plan”). The 2001 Plan provides for the granting of options to purchase common stock in the Company to employees and consultants at a price not less than the estimated fair value at the date of grant, or 110% of the estimated fair value at the date of grant if the optionee is a 10% owner of the Company. Under the provisions of the 2001 Plan, no option will have a term in excess of ten years.
In connection with the granting of employee stock options in 2003, the Company recorded deferred compensation of approximately $156,000. Deferred compensation is being amortized over the vesting period of the options resulting in non-cash stock-based compensation expense of approximately $13,000 for the year ended December 31, 2003.
The 2001 Plan is intended to encourage ownership of stock by employees and consultants of the Company and to provide additional incentives for them to promote the success of the Company’s business. The Board of Directors is responsible for determining the individuals to receive option grants, the number of options each individual will receive, the option price per share and the exercise period of each option. Options granted pursuant to the 2001 Plan generally vest over four years and have been granted at the estimated fair value of the Company’s common stock, as determined by the Board of Directors, as of each grant date. The 2001 Plan allows the option holders to exercise their options early, which are then subject to repurchase by the Company at the original exercise price of such options. No early exercise of options occurred through December 31, 2003.
The following table summarizes stock option activity for the Company:
| Options Outstanding | |||||||||||||||||
| Shares | Weighted- | ||||||||||||||||
| Available for | Number of | Exercise Price | Average | ||||||||||||||
| Grant | Shares | Per Share | Exercise Price | ||||||||||||||
| Balance at March 6, 2001 (inception) | — | — | $ | — | $ | — | |||||||||||
| Shares authorized | 271,666 | — | — | — | |||||||||||||
| Granted | (72,616 | ) | 72,616 | 1.00 - 10.00 | 3.70 | ||||||||||||
| Exercised | — | (23,800 | ) | 1.00 | 1.00 | ||||||||||||
| Balance at December 31, 2001 | 199,050 | 48,816 | 1.00 - 10.00 | 5.00 | |||||||||||||
| Shares authorized | 520,030 | — | — | ||||||||||||||
| Granted | (674,847 | ) | 674,847 | 1.00 - 1.50 | 1.50 | ||||||||||||
| Exercised | — | (10,150 | ) | 1.00 | 1.00 | ||||||||||||
| Forfeited | 92,004 | (92,004 | ) | 1.50 | 1.50 | ||||||||||||
| Balance at December 31, 2002 | 136,237 | 621,509 | 1.00 - 10.00 | 1.80 | |||||||||||||
| Granted | (19,200 | ) | 19,200 | 1.50 | 1.50 | ||||||||||||
| Exercised | — | (4,725 | ) | 1.00 - 1.50 | 1.10 | ||||||||||||
| Forfeited | 109,022 | (109,022 | ) | 1.50 | 1.50 | ||||||||||||
| Balance at December 31, 2003 | 226,059 | 526,962 | 1.00 - 10.00 | 1.80 | |||||||||||||
F-17
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The following table summarizes information about vested stock options outstanding:
| December 31, | ||||||||||||
| 2001 | 2002 | 2003 | ||||||||||
| Vested Stock Options | 9,500 | 18,166 | 203,872 | |||||||||
| Weighted average exercise price | $ | 3.80 | $ | 2.50 | $ | 1.50 | ||||||
The following table summarizes information about stock options outstanding at December 31, 2003:
| Weighted Average | ||||||||||||
| Options | Options | Remaining | ||||||||||
| Exercise Price | Outstanding | Vested | Contractual Life | |||||||||
| $ 1.00 | 30,500 | 21,218 | 7.9 | |||||||||
| $ 1.50 | 474,796 | 179,654 | 8.6 | |||||||||
| $10.00 | 21,666 | 3,000 | 7.7 | |||||||||
| 526,962 | 203,872 | 8.5 | ||||||||||
The Company also grants options to purchase common stock to consultants, advisors, and others (collectively, the “nonemployees”). The Company has granted options to purchase 47,491 shares of common stock to nonemployees during the period from March 6, 2001 (inception) to December 31, 2003. Of these options, 28,825 vest over time periods ranging from immediate vesting to 24 months and 18,666 have performance-based vesting criteria. The Black-Scholes option-pricing model is used to calculate the fair values of the stock options to nonemployees as the vesting periods of respective awards lapse. The Company recognized stock-based compensation expense of approximately $66,000 and $9,000 during the years ended December 31, 2002 and 2003, respectively, related to the fair value of the vested portions of nonemployee stock option awards. Stock-based compensation expense was insignificant for the period from March 6, 2001 (inception) to December 31, 2001. Additional stock-based compensation expense will be recorded in future periods for the remaining unvested portion of the stock option awards.
| 9. | Employee Benefit Plan |
The Company maintains a defined contribution 401(k) plan available to employees. Employee contributions are voluntary and are determined on an individual basis, limited by the maximum amounts allowable under federal tax regulations. Company contributions totaled $0, $27,738, $40,052 and $67,790, for the period from March 6, 2001 (inception) to December 31, 2001, for the years ended December 31, 2002 and 2003 and for the period from March 6, 2001 (inception) to December 31, 2003, respectively.
| 10. | Income Taxes |
As of December 31, 2002 and 2003, the Company had deferred tax assets of $6,566,000 and $14,425,000, respectively. Realization of the deferred tax assets is dependent upon the Company generating future taxable income, if any, the amount and timing of which are uncertain. Accordingly, the deferred tax assets have been fully offset by a valuation allowance at December 31, 2002 and 2003. The net valuation allowance increased by approximately $600,000, $5,966,000 and $7,859,000 for the period from March 6, 2001 (inception) to December 31, 2001, and for the years ended December 31, 2002 and 2003, respectively.
F-18
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Deferred income taxes reflect the net effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The Company’s deferred tax assets relate primarily to net operating loss carryforwards. Significant components of the Company’s deferred tax assets are as follows:
| December 31, | |||||||||
| 2002 | 2003 | ||||||||
| Deferred tax assets: | |||||||||
| Net operating loss carryforwards | $ | 6,098,000 | $ | 12,472,000 | |||||
| Research credits | 100,000 | 657,000 | |||||||
| Depreciation and amortization | 221,000 | 1,232,000 | |||||||
| Other temporary differences | 147,000 | 64,000 | |||||||
| Total gross deferred tax assets | 6,566,000 | 14,425,000 | |||||||
| Valuation allowance | (6,566,000 | ) | (14,425,000 | ) | |||||
| Net deferred tax assets | $ | — | $ | — | |||||
A reconciliation of the statutory tax rates and the effective tax rates for the period from March 6, 2001 (inception) to December 31, 2001, and for the years ended December 31, 2002 and 2003, is as follows:
| 2001 | 2002 | 2003 | ||||||||||
| Statutory rate | (34 | )% | (34 | )% | (34 | )% | ||||||
| State and local income taxes (net of federal tax benefit) | (6 | ) | (6 | ) | (6 | ) | ||||||
| Change in valuation allowance | 40 | 40 | 40 | |||||||||
| Effective tax rates | 0 | % | 0 | % | 0 | % | ||||||
As of December 31, 2003, the Company had federal net operating loss carryforwards of approximately $31,000,000. The Company also had federal research and development tax credit carryforwards of approximately $657,000. The federal net operating loss and tax credit carryforwards will expire at various dates beginning in 2021, if not utilized.
Utilization of the net operating loss carryforwards and credits may be subject to a substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code of 1986, as amended, and similar state provisions. The Company has not performed a detailed analysis to determine whether an ownership change under Section 382 of the Internal Revenue Code has occurred. The effect of an ownership change would be the imposition of an annual limitation on the use of net operating loss carryforwards attributable to periods before the change.
F-19
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| 11. | Loss Per Share |
The following table sets forth the computation of basic and diluted net loss attributable to common stockholders per share.
| Period from | ||||||||||||
| March 6, 2001 | ||||||||||||
| (Inception) to | Year Ended December 31, | |||||||||||
| December 31, | ||||||||||||
| 2001 | 2002 | 2003 | ||||||||||
| Numerator for basic and diluted net loss per share — net loss | $ | (1,416,218 | ) | $ | (20,191,090 | ) | $ | (15,486,128 | ) | |||
| Denominator for basic and dilutive net loss per share — weighted average shares | 17,866 | 182,949 | 259,182 | |||||||||
| Basic and diluted net loss per share | $ | (79.27 | ) | $ | (110.36 | ) | $ | (59.75 | ) | |||
The following table shows dilutive common share equivalents outstanding, which are not included in the above historical calculations, as the effect of their inclusion is anti-dilutive during each period. Restricted stock that is not yet vested is included as dilutive common share equivalents because the Company considers such securities as the equivalent of stock options.
| Period from | ||||||||||||
| March 6, 2001 | ||||||||||||
| (Inception) to | Year Ended December 31, | |||||||||||
| December 31, | ||||||||||||
| 2001 | 2002 | 2003 | ||||||||||
| Preferred stock | 915,000 | 2,700,846 | 2,700,846 | |||||||||
| Restricted stock | 150,000 | 91,406 | 42,187 | |||||||||
| Convertible notes | — | — | 1,654,008 | |||||||||
| Options | 48,816 | 621,510 | 526,962 | |||||||||
| 1,113,816 | 3,413,762 | 4,924,003 | ||||||||||
| 12. | Selected Quarterly Financial Data (Unaudited) |
| Quarter Ended | ||||||||||||||||
| March 31 | June 30 | September 30 | December 31 | |||||||||||||
2002 | ||||||||||||||||
| Contract revenues | $ | — | $ | 112,586 | $ | 36,630 | $ | — | ||||||||
| Net loss | (7,115,358 | ) | (3,202,117 | ) | (4,851,854 | ) | (5,021,761 | ) | ||||||||
| Basic and diluted net loss per common share* | (248.62 | ) | (14.14 | ) | (20.78 | ) | (20.88 | ) | ||||||||
2003 | ||||||||||||||||
| Contract revenues | $ | — | $ | — | $ | 100,000 | $ | — | ||||||||
| Net loss | (6,720,543 | ) | (3,418,805 | ) | (2,919,406 | ) | (2,427,374 | ) | ||||||||
| Basic and diluted net loss per common share* | (27.15 | ) | (13.36 | ) | (11.05 | ) | (9.03 | ) | ||||||||
| * | Per common share amounts for the quarters and full years have been calculated separately. Accordingly, quarterly amounts do not add to the annual amount because of differences in the weighted average common shares outstanding during each period principally due to the effect of the Company’s issuing shares of its common stock during the year. |
F-20
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Diluted EPS is identical to Basic EPS since common equivalent shares are excluded from the calculation, as their effect is anti-dilutive.
| 13. | Subsequent Events |
| Financing |
In February 2004, the Company completed a Series C convertible preferred stock financing. The Company issued 109,704,634 shares of Series C preferred stock at a price of $0.5925 per share for gross consideration of approximately $65 million. The Company also issued a warrant to purchase 692,658 shares of Series C preferred stock to the placement agent. The Company valued the warrant at $285,000 utilizing the Black-Scholes model and reflected the value as an addition to additional paid-in capital and a deduction to Series C convertible preferred stock in the balance sheet. This consideration included cash proceeds of approximately $55 million which was offset by approximately $1.4 million of issuance costs and the conversion of $9.8 million of promissory notes, issued in April 2003, into 16,540,084 shares of Series C preferred stock. In addition, this financing required an adjustment to the conversion prices for the Series A and B convertible preferred stock as a result of antidilution provisions.
Certain holders of the Company’s Series B preferred shares did not participate in the Series C financing. As a result, their holdings, totaling 6,166,154 shares of Series B preferred stock converted to 616,615 shares of common stock.
| Office Lease |
In May 2004, the Company entered into a lease for office space in Secaucus, New Jersey which will be the Company’s new headquarters. The lease commitment for 2004 is $81,980 and is $245,939 each year for 2005, 2006, 2007 and 2008 and $122,970 for 2009. The entire commitment is $1,188,706 for the five year lease.
| License Agreement |
In October 2004, the Company licensed the intellectual property underlying ALGRX 1207 from Bridge Pharma, Inc. In consideration for the license, the Company paid Bridge Pharma, Inc. an up-front license fee consisting of a cash payment and shares of its common stock. Such amounts were expensed during the fourth quarter of 2004. The Company is also obligated to pay additional fees to Bridge Pharma, Inc. if it achieves certain clinical, regulatory and commercial milestones.
| Reverse Stock Split |
At its Board meeting on January 19, 2005, the Board approved a one-for-ten reverse stock split that will be effective prior to the time the Company’s registration statement on Form S-1 relating to the initial public offering of the Company’s common stock becomes effective. In connection with the reverse stock split, every ten shares of the Company’s common stock will be replaced with one share of the Company’s common stock. All references to common stock, common shares outstanding, average number of common shares outstanding and per share amounts in these consolidated financial statements and notes to consolidated financial statements prior to the effective date of the reverse stock split have been restated to reflect the one-for-ten reverse stock split on a retroactive basis.
F-21
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
CONSOLIDATED BALANCE SHEETS
| December 31, | September 30, | September 30, 2004 | |||||||||||
| 2003 | 2004 | Pro forma (Note 1) | |||||||||||
| (Unaudited) | |||||||||||||
| ASSETS | |||||||||||||
| Current assets: | |||||||||||||
| Cash and cash equivalents | $ | 4,545,976 | $ | 14,875,314 | $ | 14,875,314 | |||||||
| Marketable securities | — | 32,697,490 | 32,697,490 | ||||||||||
| Prepaid expenses and other current assets | 183,961 | 573,286 | 573,286 | ||||||||||
| Total current assets | 4,729,937 | 48,146,090 | 48,146,090 | ||||||||||
| Property and equipment, net | 2,373,840 | 1,582,329 | 1,582,329 | ||||||||||
| Deferred financing costs | 82,377 | — | — | ||||||||||
| Other assets | 214,993 | 201,046 | 201,046 | ||||||||||
| Total assets | $ | 7,401,147 | $ | 49,929,465 | $ | 49,929,465 | |||||||
| LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIENCY) | |||||||||||||
| Current liabilities: | |||||||||||||
| Accounts payable | $ | 493,530 | $ | 126,659 | $ | 126,659 | |||||||
| Accrued compensation | 273,399 | 78,078 | 78,078 | ||||||||||
| Other accrued liabilities | 1,202,453 | 3,312,258 | 3,312,258 | ||||||||||
| Convertible notes | 9,800,000 | — | — | ||||||||||
| Total current liabilities | 11,769,382 | 3,516,995 | 3,516,995 | ||||||||||
| Stockholders’ equity (deficiency): | |||||||||||||
| Series A convertible preferred stock, $0.001 par value; 9,150,000 shares authorized, issued and outstanding at December 31, 2003 and September 30, 2004, respectively; and no shares issued and outstanding pro forma (aggregate liquidation preference of $9,150,000 at December 31, 2003 and September 30, 2004) | 9,099,428 | 9,099,428 | — | ||||||||||
| Series B convertible preferred stock, $0.001 par value; 26,350,000 and 17,858,462 shares authorized at December 31, 2003 and September 30, 2004, respectively; 17,858,462 and 11,692,308 shares issued and outstanding at December 31, 2003 and September 30, 2004, respectively; and no shares issued and outstanding pro forma (aggregate liquidation preference of $23,216,000 at December 31, 2003 and $15,200,000 at September 30, 2004) | 23,094,410 | 15,078,409 | — | ||||||||||
| Series C convertible preferred stock, $0.001 par value; 0 and 110,397,292 shares authorized at December 31, 2003 and September 30, 2004, respectively; 0 and 109,704,634 issued and outstanding at December 31, 2003 and September 30, 2004, respectively; and no shares issued and outstanding pro forma (aggregate liquidation preference of $0 and $97,500,000 at December 31, 2003 and September 30, 2004, respectively) | — | 63,306,585 | — | ||||||||||
| Common stock, $0.001 par value, 5,000,000, 17,384,027 and 17,384,027 shares authorized at December 31, 2003, September 30, 2004 and pro forma, respectively; 341,292, 960,331 and 15,236,078 shares issued at December 31, 2003, September 30, 2004 and pro forma, respectively; 314,209, 933,248 and 15,208,995 shares outstanding at December 31, 2003, September 30, 2004 and pro forma, respectively | 341 | 960 | 15,236 | ||||||||||
| Additional paid-in capital | 673,857 | 17,655,643 | 105,125,789 | ||||||||||
| Accumulated other comprehensive loss | — | (26,183 | ) | (26,183 | ) | ||||||||
| Deferred compensation | (142,564 | ) | (8,417,565 | ) | (8,417,565 | ) | |||||||
| Treasury stock, at cost, 27,083 shares at December 31, 2003 and September 30, 2004 | (271 | ) | (271 | ) | (271 | ) | |||||||
| Deficit accumulated during the development stage | (37,093,436 | ) | (50,284,536 | ) | (50,284,536 | ) | |||||||
| Total stockholders’ equity (deficiency) | (4,368,235 | ) | 46,412,470 | 46,412,470 | |||||||||
| Total liabilities and stockholders’ equity (deficiency) | $ | 7,401,147 | $ | 49,929,465 | $ | 49,929,465 | |||||||
See accompanying notes.
F-22
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
CONSOLIDATED STATEMENTS OF OPERATIONS
| Period from | |||||||||||||
| Nine Months Ended | March 6, 2001 | ||||||||||||
| September 30, | (Inception) to | ||||||||||||
| September 30, | |||||||||||||
| 2003 | 2004 | 2004 | |||||||||||
| (Unaudited) | (Unaudited) | (Unaudited) | |||||||||||
| Contract revenues | $ | 100,000 | $ | — | $ | 249,216 | |||||||
| Costs and expenses: | |||||||||||||
| Research and development | 10,939,257 | 10,681,730 | 34,982,935 | ||||||||||
| General and administrative | 2,319,821 | 2,914,695 | 10,564,310 | ||||||||||
| Acquired in-process research and development and other | — | — | 5,716,247 | ||||||||||
| Total costs and expenses | 13,259,078 | 13,596,425 | 51,263,492 | ||||||||||
| Loss from operations | (13,159,078 | ) | (13,596,425 | ) | (51,014,276 | ) | |||||||
| Gain on sale of assets | 102,150 | 261 | 67,484 | ||||||||||
| Interest expense | (71,540 | ) | (23,847 | ) | (137,087 | ) | |||||||
| Interest and other income, net | 69,714 | 428,911 | 799,343 | ||||||||||
| Net loss | $ | (13,058,754 | ) | $ | (13,191,100 | ) | $ | (50,284,536 | ) | ||||
| Basic and diluted net loss per share | $ | (51.03 | ) | $ | (17.14 | ) | |||||||
| Weighted average shares outstanding — basic and diluted | 255,909 | 769,582 | |||||||||||
| Pro forma basic and diluted net loss per share | $ | (1.01 | ) | ||||||||||
| Pro forma weighted average shares outstanding — basic and diluted | 12,946,630 | ||||||||||||
See accompanying notes.
F-23
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
CONSOLIDATED STATEMENTS OF CASH FLOWS
| Period from | ||||||||||||||
| Nine Months Ended | March 6, 2001 | |||||||||||||
| September 30, | (Inception) to | |||||||||||||
| September 30, | ||||||||||||||
| 2003 | 2004 | 2004 | ||||||||||||
| (Unaudited) | (Unaudited) | (Unaudited) | ||||||||||||
Operating activities | ||||||||||||||
| Net loss | $ | (13,058,754 | ) | $ | (13,191,100 | ) | $ | (50,284,536 | ) | |||||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||||
| Depreciation and amortization | 792,860 | 837,746 | 2,863,755 | |||||||||||
| Noncash stock-based compensation | 81,746 | 461,402 | 615,766 | |||||||||||
| Non-cash interest expense | 71,540 | 23,847 | 131,157 | |||||||||||
| Acquired in-process research and development | — | — | 5,716,247 | |||||||||||
| Amortization of intangible assets | 274,161 | — | 448,299 | |||||||||||
| Loss on disposal of equipment | — | — | 36,101 | |||||||||||
| Gain on disposal of equipment | (102,150 | ) | (261 | ) | (103,585 | ) | ||||||||
| Other non-cash expenses | — | 3,631 | 3,631 | |||||||||||
| Changes in operating assets and liabilities: | ||||||||||||||
| Prepaid expenses and other current assets | (24,333 | ) | (389,325 | ) | (526,758 | ) | ||||||||
| Other assets | — | 13,947 | (85,024 | ) | ||||||||||
| Accounts payable | (110,592 | ) | (366,871 | ) | 109,282 | |||||||||
| Accrued compensation | (79,962 | ) | (195,321 | ) | (9,447 | ) | ||||||||
| Other accrued liabilities | 768,826 | 2,109,805 | 3,219,796 | |||||||||||
| Net cash used in operating activities | (11,386,658 | ) | (10,692,500 | ) | (37,865,316 | ) | ||||||||
Investing activities | ||||||||||||||
| Purchases of property and equipment | (345,044 | ) | (47,696 | ) | (1,020,029 | ) | ||||||||
| Proceeds from disposal of equipment | 66,223 | 500 | 253,447 | |||||||||||
| Purchases of marketable securities | — | (37,801,272 | ) | (37,801,272 | ) | |||||||||
| Sales of marketable securities | — | 5,077,600 | 5,077,600 | |||||||||||
| Acquisition of PowderJect Technologies, Inc. | — | — | (1,442,029 | ) | ||||||||||
| Other acquisition related expenditures | — | — | (96,613 | ) | ||||||||||
| Net cash used in investing activities | (278,821 | ) | (32,770,868 | ) | (35,028,896 | ) | ||||||||
Financing activities | ||||||||||||||
| Repayment of capital lease obligations | — | — | (43,311 | ) | ||||||||||
| Proceeds from issuance of convertible preferred stock, net of issuance costs | — | 53,791,581 | 77,969,419 | |||||||||||
| Proceeds from issuances of common stock | 5,587 | 1,125 | 43,689 | |||||||||||
| Funds used to repurchase common stock | (271 | ) | — | (271 | ) | |||||||||
| Proceeds from debt | 9,800,000 | — | 9,800,000 | |||||||||||
| Net cash provided by financing activities | 9,805,316 | 53,792,706 | 87,769,526 | |||||||||||
| Net increase (decrease) in cash and cash equivalents | (1,860,163 | ) | 10,329,338 | 14,875,314 | ||||||||||
| Cash and cash equivalents, beginning of period | 8,873,270 | 4,545,976 | — | |||||||||||
| Cash and cash equivalents, end of period | $ | 7,013,107 | $ | 14,875,314 | $ | 14,875,314 | ||||||||
Supplemental disclosure of cash flow information | ||||||||||||||
| Cash paid during the year for interest | $ | — | $ | — | $ | 5,529 | ||||||||
Supplemental cash flow information | ||||||||||||||
| Issuance of $8,016,000 of convertible preferred stock and $228,923 of common stock in connection with acquisition of PowderJect Technologies, Inc. | $ | — | $ | — | $ | 8,244,923 | ||||||||
| Conversion of convertible preferred stock to common stock | $ | — | $ | 8,016,000 | $ | 8,016,000 | ||||||||
| Conversion of convertible notes to preferred stock | $ | — | $ | 9,800,000 | $ | 9,800,000 | ||||||||
| Equipment acquired under capital leases | $ | — | $ | — | $ | 43,311 | ||||||||
See accompanying notes.
F-24
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS
September 30, 2004
| 1. | Summary of Significant Accounting Policies |
| Organization, Description of Business and Basis of Presentation |
AlgoRx Pharmaceuticals, Inc. (the “Company” or “AlgoRx”) was incorporated on March 6, 2001 in Delaware. During 2003, the Company’s headquarters were located in Cranbury, New Jersey, with facilities also in Fremont, California. In July of 2004, the Company moved its headquarters to Secaucus, New Jersey. AlgoRx is focused on building a diversified portfolio of pharmaceutical products and technologies to address the pain therapeutic market. The Company’s activities since inception have consisted principally of acquiring product and technology rights, raising capital, establishing facilities and performing research and development. Accordingly, the Company is considered to be in the development stage as defined by Statement of Financial Accounting Standards No. 7,Accounting and Reporting Development Stage Enterprise. The Company operates in one business segment.
The Company expects to continue to incur substantial losses over the next several years during its development phase. To fully execute its business plan, the Company will need to complete certain research and development activities and clinical trials. Further, the Company’s future products may require regulatory approval prior to commercialization. These activities may span many years and require substantial expenditures to complete and may ultimately be unsuccessful. Any delays in completing these activities could adversely impact the Company. The Company plans to meet its capital requirements primarily through issuances of equity securities, research and development contract revenue, and in the longer term, revenue from product sales.
In February 2004, the Company completed a Series C convertible preferred stock financing. The Company issued 109,704,634 shares of its Series C preferred stock at a price of $0.5925 per share for gross consideration of approximately $65 million that are convertible on a one to one basis. The Series C preferred stock contained provisions which provided for antidilution protection, a liquidation preference of 1.5 times the issuance price, protective voting provisions, the right to vote on an as-if-converted basis and the right to appoint one member to the Board of Directors. The Company also issued a warrant to purchase 692,658 shares of Series C preferred stock to the placement agent. The Company valued the warrant at $285,000 utilizing the Black-Scholes model and reflected the value as an addition to additional paid-in capital and a deduction to Series C convertible preferred stock in the balance sheet. This consideration included cash proceeds of approximately $55 million which was offset by approximately $1.4 million of issuance costs and the conversion of $9.8 million of promissory notes held by certain Series C investors, issued to them in April 2003, into 16,540,084 shares of Series C convertible preferred stock. In addition, this financing required an adjustment to the conversion prices of the Series A convertible preferred stock and the Series B convertible preferred stock as a result of antidilution provisions.
Certain holders of the Company’s Series B preferred shares did not participate in the Series C financing. As a result, their holdings, totaling 6,166,154 shares of Series B preferred stock, converted to 616,615 shares of common stock.
| Interim Financial Statements |
The financial statements as of September 30, 2004 and for the nine months ended September 30, 2003 and 2004 have been prepared by the Company without audit. In the opinion of management, all adjustments (which include only normal recurring adjustments) necessary to present fairly the financial position and the results of operations and cash flows for the nine months ended September 30, 2003 and 2004 and the period from March 6, 2001 (inception) to September 30, 2004 have been made. Certain
F-25
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
information and footnote disclosures normally included in financial statements prepared in accordance with U.S. generally accepted accounting principles have been condensed or eliminated.
| Use of Estimates |
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
| Principles of Consolidation |
The consolidated financial statements include the accounts of AlgoRx and, beginning on March 22, 2002, its wholly owned subsidiary, AlgoRx Technologies, Inc. (formerly PowderJect Technologies, Inc.), located in Fremont, California. Intercompany accounts and transactions have been eliminated.
| Cash and Cash Equivalents |
Cash and cash equivalents consist of highly liquid instruments purchased with an original maturity of three months or less.
| Marketable Securities |
Investments classified as available for sale are carried at estimated fair value with unrealized gains and losses recorded as a component of accumulated other comprehensive income. Management determines the appropriate classification of its investment in debt and equity securities at the time of purchase and reevaluates such determination at each balance sheet date.
| Property and Equipment |
Property and equipment are stated at cost, less accumulated depreciation. Depreciation is provided over the estimated useful lives of the respective assets, which range from three to five years, using the straight-line method.
Leasehold improvements and assets under capital leases are amortized over the lives of the related leases or their estimated useful lives, whichever is shorter, using the straight-line method.
| Intangible Asset |
Intangible asset consists of the acquired workforce acquired in the acquisition of PowderJect Technologies, Inc. (see Note 2). The acquired workforce is being amortized on a straight-line basis over two years, which represents the estimated retention period for the related employees. Acquired workforce amortization is recorded in research and development expense.
| Long-Lived Assets |
Long-lived assets and certain identifiable intangible assets to be held and used are reviewed for impairment when events or changes in circumstances indicate that the carrying amount of such assets may not be recoverable. Determination of recoverability is based on an estimate of undiscounted future cash flows resulting from the use of the asset and its eventual disposition. In the event that such cash flows are not expected to be sufficient to recover the carrying amount of the assets, the assets are written down to their estimated fair values. Long-lived assets and certain identifiable assets to be disposed of are reported at the lower of carrying amount or fair value less cost to sell.
F-26
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| Fair Value of Financial Instruments |
The carrying values of the Company’s financial instruments, which include cash and cash equivalents, investments, accounts payable, accrued expenses and convertible notes approximates their fair values.
| Revenue Recognition |
The Company’s revenue recognition policies are in accordance with Securities and Exchange Commission (SEC) Staff Accounting Bulletins No. 101 and No. 104, or SAB 101 and 104, and EITF 00-21,Revenue Recognition in Financial Statements, which provides guidance on revenue recognition in financial statements and is based on the interpretations and practices developed by the SEC. SAB 101 requires that four basic criteria be met before revenue can be recognized: (1) persuasive evidence exists of an arrangement; (2) delivery has occurred or services have been rendered; (3) the fee is fixed and determinable; and (4) collectibility is reasonably assured. Determination of criteria (3) and (4) are based on management’s judgments regarding the fixed nature of the fees charged for services rendered and products delivered and the collectibility of those fees. Accordingly, revenues from licensing agreements are recognized based on the performance requirements of the agreement. If the Company has an ongoing involvement or performance obligation, non-refundable up-front fees are generally recorded as deferred revenue in the balance sheet and amortized into license fees in the statement of operations over the term of the performance obligation. If the Company has no ongoing involvement or performance obligation, non-refundable up-front fees are generally recorded as revenue in the period in which the rights are transferred.
| Research and Development Costs |
Research and development costs are expensed as incurred. Research and development costs consist of salaries, employee benefits, laboratory supplies, consulting services, clinical services and facility costs.
Acquired in-process research and development relates primarily to in-licensed technology, intellectual property and know-how. The Company evaluates the stage of development of acquired projects, taking into account the level of effort, time and estimated cost associated with further developing the in-process technology and producing a commercial product. The nature of the remaining efforts for completion of acquired in-process research and development projects generally include completion of clinical trials, completion of manufacturing validation, interpretation of clinical and preclinical data and obtaining marketing approval from the FDA and other regulatory bodies, the cost, length and success of which are extremely difficult to determine. Numerous risks and uncertainties exist with timely completion of development projects, including clinical trial results, manufacturing process development results, and ongoing feedback from regulatory authorities, including obtaining marketing approval. In addition, acquired products under development may never be successfully commercialized due to the uncertainties associated with the pricing of new pharmaceuticals, the cost to produce these products in a commercial setting, changes in the reimbursement environment, or the introduction of new competitive products. As a result of the uncertainties noted above, the Company expenses such acquired in-process research and development projects when incurred.
| Concentration of Credit Risk |
The Company’s financial instruments that are exposed to credit risks consist primarily of cash and cash equivalents. The Company maintains its cash and cash equivalents in bank accounts, which, at times, exceed federally insured limits. The Company has not experienced any losses in such accounts. The Company believes it is not exposed to significant credit risk related to cash and cash equivalents.
F-27
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| Income Taxes |
The Company utilizes the asset and liability method of accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial reporting and tax bases of assets and liabilities and are measured using the enacted tax rates that will be in effect when the differences are expected to reverse. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized.
| Comprehensive Loss |
The Company reports comprehensive loss in accordance with Statement of Financial Accounting Standards No. 130,Reporting Comprehensive Income(“SFAS 130”). SFAS 130 establishes rules for the reporting and display of comprehensive loss and its components. SFAS 130 requires unrealized loss on available for sale securities to be included in other comprehensive loss.
| Net Loss Per Share |
The Company computes net loss per share in accordance with Statement of Financial Accounting Standards No. 128, “Earnings per Share” (“SFAS 128”). Under the provisions of SFAS 128, basic net loss per common share (“Basic EPS”) is computed by dividing net loss by the weighted-average number of common shares outstanding (excluding unvested founders’ shares subject to repurchase). Diluted net loss per common share (“Diluted EPS”) is computed by dividing net loss by the weighted-average number of common shares and dilutive common shares equivalents then outstanding. Common equivalent shares consist of the incremental common shares issuable upon the conversion of preferred stock, shares issuable upon the exercise of stock options, the conversion of preferred stock upon the exercise of warrants and unvested founders’ shares subject to repurchase. Diluted EPS is identical to Basic EPS since common shares equivalent are excluded from the calculation, as their effect is anti-dilutive.
| Unaudited Pro Forma Information |
The unaudited pro forma balance sheet as of September 30, 2004 reflects the automatic conversion of all outstanding shares of the Company’s convertible preferred stock into an aggregate of 14,275,747 shares of common stock upon completion of the Company’s initial public offering.
Unaudited pro forma net loss per share is computed using the weighted average number of common shares outstanding, including the pro forma effects of the automatic conversion of all outstanding convertible preferred stock into shares of the Company’s common stock effective upon the assumed closing of the Company’s proposed initial public offering, as if such conversion had occurred at the date of the original issuance.
| Stock-Based Compensation |
As permitted by the Financial Accounting Standards Board (“FASB”) Statement No. 123, Accounting for Stock-Based Compensation (“SFAS 123”), as amended, the Company accounts for employee stock-based compensation in accordance with Accounting Principles Board Opinion No. 25, Accounting for Stock Issued to Employees (“APB 25”), and related interpretations, in accounting for its stock-based compensation plans. Under APB 25, when the exercise price of the Company’s employee stock options equals or exceeds the estimated fair value of the underlying stock on the date of grant, no compensation expense is recorded.
In connection with the granting of employee stock options in 2003 and for the nine months ended September 30, 2004, the Company recorded deferred compensation of approximately $156,000 and
F-28
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
$8,712,000, respectively. Deferred compensation is being amortized over the vesting period of the options resulting in non-cash stock-based compensation expense of approximately $4,000 and $437,000 for the nine months ended September 30, 2003 and 2004, respectively.
Pro forma information regarding net loss and net loss per share has been determined as if the Company had accounted for its employee stock options under the fair value method proscribed by SFAS 123. The resulting effect on pro forma net loss disclosed is not likely to be representative of the effects of loss on a pro forma basis in future years due to additional grants and years of vesting in subsequent years. Pro forma compensation related to stock option grants is expensed over their respective vesting periods.
Had compensation cost for options granted under the Company’s stock option plan been determined based on the fair value at the grant dates for the awards under a method prescribed by SFAS 123, the Company’s net loss would have been adjusted to the following pro forma amounts:
| Nine Months Ended September 30, | ||||||||
| 2003 | 2004 | |||||||
| (Unaudited) | ||||||||
| Net loss, as reported | $ | (13,058,754 | ) | $ | (13,191,100 | ) | ||
| Add: Noncash employee compensation as reported | 3,551 | 437,390 | ||||||
| Deduct: Stock-based employee compensation expense under the fair value based method for all awards, net of tax | (144,765 | ) | (655,177 | ) | ||||
| Net loss, pro forma | (13,199,968 | ) | (13,408,887 | ) | ||||
| Basic and diluted loss per share, as reported | $ | (51.03 | ) | $ | (17.14 | ) | ||
| Basic and diluted loss per share, SFAS 123 pro forma | $ | (51.58 | ) | $ | (17.42 | ) | ||
| Unaudited pro forma basic and diluted net loss per share (Note 1) SFAS 123, pro forma | $ | (1.04 | ) | |||||
The fair values of stock options granted to employees for the nine months ended September 30, 2003 and 2004 were estimated on the respective dates of grant using the minimum value option-pricing model with the following weighted-average assumptions:
| Nine Months | ||||||||
| Ended | ||||||||
| September 30, | ||||||||
| 2003 | 2004 | |||||||
| (Unaudited) | ||||||||
| Risk-free interest rate | 4.0 | % | 4.0 | % | ||||
| Expected life (in years) | 9.0 | 9.0 | ||||||
| Dividend yield | — | — | ||||||
| Fair value | $ | 9.90 | $ | 5.90 | ||||
The Company has also granted stock options to purchase stock to consultants, advisors, and other vendors. The Company accounts for stock awards issued to such nonemployees in accordance with the provisions of SFAS 123 and Emerging Issues Task Force (“EITF”) Issue No. 96-18, Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services (“Issue No. 96-18”). Under SFAS 123 and Issue No. 96-18, stock awards to nonemployees are accounted for at their respective fair values using the Black-Scholes option-pricing model unless a more readily determinable fair value is available. The fair value of options granted to
F-29
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
nonemployees is remeasured during the performance period as the underlying options vest or as milestones are reached.
| Recently Issued Accounting Standards |
In January 2003, the FASB issued Interpretation No. 46 (“FIN 46”),Consolidation of Variable Interest Entities, which addresses consolidation of variable interest entities (“VIEs”). FIN 46 requires a VIE to be consolidated by a parent company if that company is subject to a majority of the risk of loss from the variable interest entity’s activities or entitled to receive a majority of the entity’s residual returns or both. A VIE is a corporation, partnership, trust or any other legal structure used for business purposes that either does not have equity investors with voting rights or has equity investors that do not provide sufficient financial resources for the entity to support its activities. The consolidation requirements of FIN 46 apply immediately to VIEs created after January 31, 2003. For entities created prior to February 1, 2003, the effective date of these requirements has been deferred so as not to apply until the first period ending after December 15, 2004. The adoption of the provisions applicable to special purpose entities (“SPEs”) and all other VIEs obtained after January 31, 2003 had no impact on the Company’s financial statements. The Company is currently evaluating the impact of adopting FIN 46-R applicable to non-SPEs created prior to February 1, 2003, but does not expect a material impact on its financial position, results of operations or cash flows.
In May 2003, the FASB issued Statement of Financial Accounting Standards No. 150,Accounting for Certain Financial Instruments with Characteristics of both Liabilities and Equity. (“SFAS 150”) This statement establishes standards for classifying and measuring as liabilities certain financial instruments that embody obligations of the issuer and have characteristics of both liabilities and equity. The scope of this pronouncement includes mandatorily redeemable equity instruments. Application to pre-existing instruments should be recognized as the cumulative effect of a change in accounting principle (application by retroactive restatement is precluded). Exceptions to the transition requirements were provided for mandatorily redeemable instruments of non-public companies. Subsequent to the issuance of SFAS 150, the FASB issued FSP 150-3, which further deferred the transition requirements for nonpublic companies that also are not SEC registrants for instruments that are mandatorily redeemable on fixed dates that are for fixed amounts. The classification, measurement, and disclosure provisions of SFAS 150 are effective for fiscal periods beginning after December 15, 2004. The adoption of this statement is not expected to impact the classification of the Company’s convertible preferred stock.
In December 2004, the FASB issued FASB Statement No. 123,Share-Based Payment. This statement, referred to as Statement 123R, was an amendment of FASB Statements No. 123 and 95. The change in accounting would replace existing requirements under FAS 123,Accounting for Stock-Based Compensation, and APB Opinion No. 25,Accounting for Stock Issued to Employees.
The statement covers a wide range of equity-based compensation arrangements. Under this FASB statement, all forms of share-based payments to employees, including employee stock options, would be treated the same as other forms of compensation by recognizing the related cost in the income statement. The expense of the award would generally be measured at fair value at the grant date.
On October 13, 2004, the FASB concluded that Statement 123R would be effective for public companies for interim or annual periods beginning after June 15, 2005 except small business issuers as defined in SEC Regulation S-B. Retroactive application of the requirements of Statement 123, not Statement 123R, to the beginning of the fiscal year that includes the effective date would be permitted but not required. Early adoption of Statement 123R is encouraged.
F-30
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
A calendar-year company therefore would be required to apply Statement 123R beginning July 1, 2005, and could choose to apply Statement 123R retroactively from January 1, 2005 to June 30, 2005. The cumulative effect of adoption, if any, would be measured and recognized on July 1, 2005. The Company could choose to early adopt the proposed statement at the beginning of our first quarter ended March 31, 2005, or even in the fourth quarter of 2004 if the FASB issues the final statement prior to the issuance of those financial statements.
2. Acquisition of PowderJect Technologies, Inc.
On March 22, 2002, the Company acquired all of the outstanding shares of PowderJect Technologies, Inc. (“PowderJect”), a development stage company, for a total purchase price of approximately $9.9 million. This purchase price included 6,166,154 shares of the Company’s Series B convertible preferred stock, 152,615 shares of common stock, cash consideration of $718,150 for a minority interest’s share in the acquired company, acquisition related costs of $723,879 and assumed liabilities of $192,458. The Series B convertible preferred stock was valued based on the arm’s length Series B financing completed during that year and the common stock consideration was valued using the Company’s estimated fair value used for option grants made to employees. PowderJect was in the business of developing drug applications for a patented, needle-free delivery system developed by its parent company, PowderJect Pharmaceuticals plc. The Company also entered into a supply agreement with PowderJect Technologies Limited (subsequently purchased by Chiron Corporation) for cylinders. The cylinders are a key component of the product candidate, ALGRX 3268, and PowderJect Technologies Limited is the sole supplier. No other supplier has been identified by the Company.
The acquisition was completed to provide the Company with the ability to exploit the opportunities associated with its in-process lidocaine technology, as well as gain access to the patented drug delivery technology. The purchase price was determined in accordance with Statement of Financial Accounting Standards No. 141,Business Combinations(“SFAS 141”), and Statement of Financial Accounting Standards No. 142,Goodwill and Other Intangible Assets (“SFAS 142”). A summary of the determination of the purchase price during 2002 was as follows:
| 6,166,154 shares — Series B convertible preferred stock | $ | 8,016,000 | ||
| 152,615 shares — common stock | 228,923 | |||
| Cash for minority interest buyout | 718,150 | |||
| Acquisition related costs | 723,879 | |||
| Assumed liabilities | 192,458 | |||
| Total purchase price | $ | 9,879,410 | ||
The acquisition was accounted for as an acquisition of assets rather than as a business combination as PowderJect was a development stage company that had not commenced its planned principal operations. PowderJect lacked the necessary elements of a business because it did not have completed products and, therefore, no ability to access customers. The PowderJect operating results have been included in the Company’s consolidated results of operations since March 23, 2002.
The Company allocated the purchase price in accordance with the provisions of SFAS 142 related to the purchase of a group of assets. SFAS 142 provides that the cost of a group of assets acquired in a transaction other than a business combination shall be allocated to the individual assets acquired based on their relative fair values and shall not give rise to goodwill.
F-31
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
In accordance with the provisions of SFAS 142, all identifiable intangible assets and acquired in-process research and development were assigned a portion of the purchase price based on their relative fair values. To this end, a third party valuation was used to assist management in determining the fair value of the identifiable assets and acquired in-process research and development. Based on this valuation, the Company allocated the total consideration as follows:
| Acquired in-process research and development and other | $ | 5,716,247 | ||
| Tangible net assets | 3,714,864 | |||
| Acquired workforce | 448,299 | |||
| Total purchase price | $ | 9,879,410 | ||
The income approach was used to estimate the fair value of the patent license and the acquired in-process research and development based on projected cash flows, assuming a 30% discount rate. This discount rate is a significant assumption and is based on the Company’s estimated weighted average cost of capital taking into account the risks associated with the projects acquired. The projected cash flows from such projects were based on estimates of revenues and operating profits related to such projects considering the stage of development of each potential product acquired, the time and resources needed to complete each product, the estimated life of each potential commercialized product and associated risks which included the inherent difficulties and uncertainties in developing a drug compound, obtaining FDA and other regulatory approvals, and risks related to the viability of, and potential alternative treatments in, any future target markets. Approximately $4.5 million of the purchase price was allocated to acquired in-process research and development due to PowderJect’s incomplete research and development programs that had not yet reached technological feasibility as of March 2002 and had no alternative future use as of that date. In light of the number of years required to achieve product commercialization, the associated technology risk, and therefore the risk that the asset value will not be realized, the Company has recorded, as an expense, the value of the favorable patent license, which is $1.2 million, similar to the treatment afforded the acquired in-process research and development.
The cost approach was used to determine the value of the acquired workforce. The value allocated to the acquired workforce is attributable to 31 employees hired by the Company from PowderJect following the asset acquisition, which eliminated the need to hire replacement employees. The value of the acquired workforce was determined by estimating the cost of assembling a new workforce, including costs of salaries, benefits, training, and recruiting.
Amortization expense was $46,700 and $0 for the nine months ended September 30, 2003 and 2004, respectively, and $448,299 from March 6, 2001 (inception) to September 30, 2004. Included in the 2003 amortization was $227,461 related to the acquired workforce that was written off in connection with the restructuring described in Note 3 during the first quarter of 2003 and, accordingly, the net intangible assets as of December 31, 2003 and September 30, 2004 was $0.
| 3. | Restructuring |
In March 2003, the Company announced a reorganization plan intended to reduce operating costs and reduced its staff by 28 employees (approximately 61% of total employees as of the date of reorganization); however, no research projects were discontinued. The reorganization resulted in an impairment of the assembled workforce intangible asset of $227,461 recorded as accelerated amortization expense during 2003. This expense is classified under research and development expenses for the nine months ended September 30, 2003. In addition, the vesting of certain options held by such employees was accelerated.
F-32
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The Company recorded a charge of $66,240 related to this acceleration of vesting. The total restructuring cost of $1,122,444 was charged to research and development expense.
| 4. | Available for Sale Investments |
The following is a summary of available for sale investments as of September 30, 2004:
| Gross | Gross | ||||||||||||||||
| Unrealized | Unrealized | ||||||||||||||||
| Cost | Gains | Losses | Fair Value | ||||||||||||||
| Maturities within one year: | |||||||||||||||||
| Commercial paper | $ | 6,090,858 | $ | — | $ | (2,062 | ) | $ | 6,088,796 | ||||||||
| U.S. Treasury Notes | 13,977,481 | — | (12,587 | ) | 13,964,894 | ||||||||||||
| 20,068,339 | — | (14,649 | ) | 20,053,690 | |||||||||||||
| Maturities between one to five years: | |||||||||||||||||
| U.S. Treasury Notes | 12,655,334 | — | (11,534 | ) | 12,643,800 | ||||||||||||
| Total | $ | 32,723,673 | $ | — | $ | (26,183 | ) | $ | 32,697,490 | ||||||||
| 5. | Other Accrued Liabilities |
Other accrued liabilities consist of the following:
| December 31, | September 30, | |||||||
| 2003 | 2004 | |||||||
| (Unaudited) | ||||||||
| Accrued patent costs | $ | 294,000 | $ | 366,000 | ||||
| Accrued research and development costs | 341,158 | 2,373,095 | ||||||
| Accrued legal | 141,356 | — | ||||||
| Accrued vacation | 130,611 | 165,159 | ||||||
| Other accrued liabilities | 295,328 | 408,004 | ||||||
| Total | $ | 1,202,453 | $ | 3,312,258 | ||||
| 6. | Commitments |
| Leases |
In May 2004, the Company entered into a lease for office space in Secaucus, New Jersey which is the Company’s new headquarters. The entire commitment is $1,188,706 for the five year lease. The future minimum payments for all noncancelable operating leases as of September 30, 2004 are as follows:
| Remainder of 2004 | $ | 222,300 | ||
| 2005 | 299,039 | |||
| 2006 | 245,939 | |||
| 2007 | 245,939 | |||
| 2008 | 245,939 | |||
| Thereafter | 122,970 | |||
| Total | $ | 1,382,126 | ||
F-33
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Rent expense under operating leases was $498,094, $527,762 and $1,871,762, for the nine months ended September 30, 2003 and 2004 and for the period from March 6, 2001 (inception) to September 30, 2004, respectively.
| Other Commitments |
Under the Company’s license agreements, the Company could be required to pay up to a total of $6.2 million in milestone payments through product approval, in addition to sales milestones and royalties on commercial sales if any occur. The Company has no material commitments for capital expenditures.
Under our license with PowderMed Limited we are obligated and have spent $1,000,000 on research and development associated with the licensed technology during the period July 7, 2003 to July 7, 2005. We are also obligated to spend an additional $1,000,000 on research and development for every 12 month period from July 7, 2005 to July 7, 2008.
| 7. | Stock Option Plan |
On March 30, 2001, the Company’s Board of Directors and shareholders approved the Company’s 2001 Equity Incentive Plan (the “2001 Plan”). The 2001 Plan provides for the granting of options to purchase common stock in the Company to employees and consultants at a price not less than the estimated fair value at the date of grant, or 110% of the estimated fair value at the date of grant if the optionee is a 10% owner of the Company. Under the provisions of the 2001 Plan, no option will have a term in excess of ten years.
In connection with the granting of employee stock options in 2004, the Company recorded deferred compensation of approximately $8,712,000. Deferred compensation is being amortized over the vesting period of the options resulting in non-cash compensation expense of approximately $4,000 and $437,000 for the nine months ended September 30, 2003 and 2004, respectively.
The 2001 Plan is intended to encourage ownership of stock by employees and consultants of the Company and to provide additional incentives for them to promote the success of the Company’s business. The Board of Directors is responsible for determining the individuals to receive option grants, the number of options each individual will receive, the option price per share and the exercise period of each option. Options granted pursuant to the 2001 Plan generally vest over four years and have been granted at the estimated fair value of the Company’s common stock, as determined by the Board of Directors, as of each grant date. The 2001 Plan allows the option holders to early exercise their options which are then subject to repurchase by the Company at the original exercise price. No early exercise of options occurred through September 30, 2004.
F-34
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The following table summarizes stock option activity for the Company.
| Options Outstanding | |||||||||||||||||
| Shares | Weighted- | ||||||||||||||||
| Available for | Number of | Exercise Price | Average | ||||||||||||||
| Grant | Shares | Per Share | Exercise Price | ||||||||||||||
| Balance at March 6, 2001 (inception) | — | — | $ | — | $ | — | |||||||||||
| Shares authorized | 271,666 | — | — | — | |||||||||||||
| Granted | (72,616 | ) | 72,616 | 1.00-10.00 | 3.70 | ||||||||||||
| Exercised | — | (23,800 | ) | 1.00 | 1.00 | ||||||||||||
| Balance at December 31, 2001 | 199,050 | 48,816 | 1.00-10.00 | 5.00 | |||||||||||||
| Shares authorized | 520,030 | — | — | ||||||||||||||
| Granted | (674,847 | ) | 674,847 | 1.00-1.50 | 1.50 | ||||||||||||
| Exercised | — | (10,150 | ) | 1.00 | 1.00 | ||||||||||||
| Forfeited | 92,004 | (92,004 | ) | 1.50 | 1.50 | ||||||||||||
| Balance at December 31, 2002 | 136,237 | 621,509 | 1.00-10.00 | 1.80 | |||||||||||||
| Granted | (19,200 | ) | 19,200 | 1.50 | 1.50 | ||||||||||||
| Exercised | — | (4,725 | ) | 1.00-1.50 | 1.10 | ||||||||||||
| Forfeited | 109,022 | (109,022 | ) | 1.50 | 1.50 | ||||||||||||
| Balance at December 31, 2003 | 226,059 | 526,962 | 1.00-10.00 | 1.80 | |||||||||||||
| Shares authorized | 1,605,160 | — | — | ||||||||||||||
| Granted | (1,724,608 | ) | 1,724,608 | 1.00-1.50 | 1.00 | ||||||||||||
| Exercised | — | (2,428 | ) | 1.00-1.50 | 1.50 | ||||||||||||
| Forfeited | 6,637 | (6,637 | ) | 1.00-1.50 | 1.30 | ||||||||||||
| Balance at September 30, 2004 (unaudited) | 113,248 | 2,242,505 | 1.00-10.00 | 1.20 | |||||||||||||
The following table summarizes information about vested stock options outstanding:
| December 31, 2003 | September 30, 2004 | |||||||
| Vested Stock Options | 203,873 | 435,456 | ||||||
| Weighted average exercise price | $1.50 | $1.50 | ||||||
The following table summarizes information about stock options outstanding at September 30, 2004:
| Weighted Average | ||||||||||||
| Remaining | ||||||||||||
| Exercise Price | Options Outstanding | Options Vested | Contractual Life | |||||||||
| $1.00 | 1,732,893 | 137,896 | 9.6 | |||||||||
| $1.50 | 487,945 | 288,338 | 7.9 | |||||||||
| $10.00 | 21,667 | 9,222 | 6.9 | |||||||||
| 2,242,505 | 435,456 | 9.2 | ||||||||||
The Company also grants options to purchase common stock to consultants, advisors, and others (collectively, the “nonemployees”). The Company has granted options to purchase 47,491 shares of common stock to nonemployees during the period from March 6, 2001 (inception) to September 30, 2004. Of these options, 28,825 vest over time periods ranging from immediate vesting to 24 months and
F-35
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
18,666 have performance-based vesting criteria. The Black-Scholes option-pricing model is used to calculate the fair values of the stock options to nonemployees as the vesting periods of respective awards lapse. The Company recognized stock-based compensation expense of approximately $7,000, $24,000 and $99,000 during the nine months ended September 30, 2003 and 2004 and the period from March 6, 2001 (inception) to September 30, 2004, respectively, related to the fair value of the vested portions of non-employee stock option awards. Additional stock-based compensation expense will be recorded in future periods for the remaining unvested portion of the non-employee stock option awards.
| 8. | Income Taxes |
As of December 31, 2003 and September 30, 2004, the Company had deferred tax assets of approximately $14,425,000 and $19,389,000, respectively. Realization of the deferred tax assets is dependent upon the Company generating future taxable income, if any, the amount and timing of which are uncertain. Accordingly, the deferred tax assets have been fully offset by valuation allowances at December 31, 2003 and September 30, 2004. The net valuation allowance increased by approximately $4,964,000 for the nine months ended September 30, 2004. Deferred tax assets primarily relate to net operating loss and tax credit carryforwards.
Deferred income taxes reflect the net effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. The Company’s deferred tax assets relate primarily to net operating loss carryforwards. Significant components of the Company’s deferred tax assets are as follows:
| December 31, | September 30, | ||||||||
| 2003 | 2004 | ||||||||
| Deferred tax assets: | |||||||||
| Net operating loss carryforwards | $ | 12,472,000 | $ | 16,900,000 | |||||
| Research credits | 657,000 | 470,000 | |||||||
| Depreciation and amortization | 1,232,000 | 777,000 | |||||||
| Other temporary differences | 64,000 | 1,242,000 | |||||||
| Total gross deferred tax assets | 14,425,000 | 19,389,000 | |||||||
| Valuation allowance | (14,425,000 | ) | (19,389,000 | ) | |||||
| Net deferred tax assets | $ | — | $ | — | |||||
A reconciliation of the statutory tax rates and the effective tax rates for the nine months ended September 30, 2002 and 2003 is as follows:
| 2003 | 2004 | |||||||
| (Unaudited) | ||||||||
| Statutory rate | (34 | )% | (34 | )% | ||||
| State and local income taxes (net of federal tax benefit) | (6 | ) | (6 | ) | ||||
| Change in valuation allowance | 40 | 40 | ||||||
| Effective tax rates | 0 | % | 0 | % | ||||
As of September 30, 2004, the Company had federal net operating loss carryforwards of approximately $42,000,000. The Company also had federal research and development tax credit carryforwards of approximately $470,000. The federal net operating loss and tax credit carryforwards will expire at various dates beginning in 2022, if not utilized.
F-36
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Utilization of the net operating loss carryforwards and credits may be subject to a substantial annual limitation due to the ownership change limitations provided by the Internal Revenue Code of 1986, as amended, and similar state provisions. The Company has not performed a detailed analysis to determine whether an ownership change under Section 382 of the Internal Revenue Code has occurred. The effect of an ownership change would be the imposition of an annual limitation on the use of net operating loss carryforwards attributable to periods before the change.
| 9. | Loss Per Share |
The following table sets forth the computation of basic and diluted net loss attributable to common stockholders per share.
| Nine Months Ended September 30, | |||||||||
| 2003 | 2004 | ||||||||
| (Unaudited) | |||||||||
| Numerator for basic and diluted net loss per share — net loss | $ | (13,058,754 | ) | $ | (13,191,100 | ) | |||
| Denominator: | |||||||||
| Denominator for basic and dilutive net loss per share–weighted — average shares | 255,909 | 769,582 | |||||||
| Basic and diluted net loss per share | $ | (51.03 | ) | $ | (17.14 | ) | |||
The following table shows dilutive common share equivalents outstanding, which are not included in the above historical calculations, as the effect of their inclusion is anti-dilutive during each period. Restricted stock that is not yet vested is included as a dilutive common share equivalent because the Company considers such securities as the equivalent of options.
| Nine Months Ended | ||||||||
| September 30, | ||||||||
| 2003 | 2004 | |||||||
| (Unaudited) | ||||||||
| Preferred stock | 2,700,846 | 13,054,694 | ||||||
| Restricted stock | 46,875 | 28,125 | ||||||
| Convertible notes | 1,654,008 | — | ||||||
| Warrants | — | 69,265 | ||||||
| Options | 526,963 | 2,242,505 | ||||||
| Total | 4,928,692 | 15,394,589 | ||||||
F-37
Table of Contents
AlgoRx Pharmaceuticals, Inc.
(a development stage company)
NOTES TO UNAUDITED CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| 10. | Subsequent Events |
| License Agreement |
In October 2004, the Company licensed the intellectual property underlying ALGRX 1207 from Bridge Pharma, Inc. In consideration for the license, the Company paid Bridge Pharma, Inc. an up-front license fee consisting of a cash payment of $1,000,000 and 160,000 shares of its common stock. Such amounts were expensed during the fourth quarter of 2004. The Company is also obligated to pay additional fees to Bridge Pharma, Inc. if it achieves certain clinical, regulatory and commercial milestones. The Company is required to pay such milestone payments upon the commencement of phase I, II and III clinical trials and upon the occurrence of certain events including the filing of a new drug application with the FDA, the regulatory approval for each of the first and second products using the licensed technology and reaching certain revenue thresholds. We may be obligated to pay up to an aggregate of $2.5 million in milestone payments prior to product approval, plus additional amounts up to an aggregate of $3.0 million payable upon the regulatory approval of a licensed product for each of the first, second and third indications. To date, the Company has paid no milestone payments. We are obligated to spend a minimum of $1,000,000 for product development in each calendar year during the term of the agreement commencing in 2005 and ending on the first commercial sale of a product using the licensed technology.
| Reverse Stock Split |
At its Board meeting on January 19, 2005, the Board approved a one-for-ten reverse stock split that will be effective prior to the time the Company’s registration statement on Form S-1 relating to the initial public offering of the Company’s common stock becomes effective. In connection with the reverse stock split, every ten shares of the Company’s common stock will be replaced with one share of the Company’s common stock. All references to common stock, common shares outstanding, average number of common shares outstanding and per share amounts in these consolidated financial statements and notes to consolidated financial statements prior to the effective date of the reverse stock split have been restated to reflect the one-for-ten reverse stock split on a retroactive basis.
F-38
Table of Contents
Table of Contents
PART II.
INFORMATION NOT REQUIRED IN PROSPECTUS
| Item 13. | Other Expenses of Issuance and Distribution |
The following table sets forth all expenses, other than the underwriting discounts and commissions, payable by the registrant in connection with the sale of the common stock being registered. All the amounts shown are estimates except the registration fee and the NASD filing fee.
| Total | |||||
| SEC registration fee | $ | 11,721 | |||
| NASD filing fee | 9,884 | ||||
| Nasdaq National Market initial listing fee | 100,000 | ||||
| Blue sky qualification fees and expenses | 10,000 | ||||
| Printing and engraving expenses | 500,000 | ||||
| Legal fees and expenses | 800,000 | ||||
| Accounting fees and expenses | 500,000 | ||||
| Transfer agent and registrar fees | 10,000 | ||||
| Miscellaneous | 120,000 | ||||
| Total | $ | 2,061,605 | |||
| * | To be supplied by amendment. |
| Item 14. | Indemnification of Officers and Directors |
Section 145 of the Delaware General Corporation Law authorizes a court to award, or a corporation’s board of directors to grant, indemnity to directors and officers in terms sufficiently broad to permit such indemnification under certain circumstances for liabilities (including reimbursement for expenses incurred) arising under the Securities Act of 1933, as amended (the “Securities Act”).
As permitted by the Delaware General Corporation Law, our amended and restated certificate of incorporation includes a provision that eliminates the personal liability of our directors for monetary damages for breach of fiduciary duty as a director, except for liability (1) for any breach of the director’s duty of loyalty to us or our stockholders, (2) for acts or omissions not in good faith or that involve intentional misconduct or a knowing violation of law, (3) under section 174 of the Delaware General Corporation Law (regarding unlawful dividends and stock purchases) or (4) for any transaction from which the director derived an improper personal benefit.
As permitted by the Delaware General Corporation Law, our amended and restated bylaws provide that (1) we are required to indemnify our directors and officers to the fullest extent permitted by the Delaware General Corporation Law, subject to certain very limited exceptions, (2) we may indemnify our other employees and agents as set forth in the Delaware General Corporation Law, (3) we are required to advance expenses, as incurred, to our directors and executive officers in connection with a legal proceeding to the fullest extent permitted by the Delaware General Corporation Law, subject to certain very limited exceptions and (4) the rights conferred in the amended and restated bylaws are not exclusive.
We also intend to enter into indemnification agreements with each of our directors and executive officers to give such directors and officers additional contractual assurances regarding the scope of the indemnification set forth in our amended and restated certificate of incorporation and to provide additional procedural protections. We also intend to enter into indemnification agreements with any new directors and executive officers in the future.
II-1
Table of Contents
The Underwriting Agreement to be filed as Exhibit 1.1 to this Registration Statement will provide for indemnification by the underwriters of our officers, directors and controlling persons against certain liabilities, including liabilities arising under the Securities Act, in connection with matters specifically provided in writing by the underwriters for inclusion in the Registration Statement.
The indemnification provisions in our restated certificate of incorporation, restated bylaws and the indemnification agreements to be entered into between us and each of our directors and executive officers may be sufficiently broad to permit indemnification of our directors and executive officers for liabilities arising under the Securities Act.
We also intend to maintain director and officer liability insurance, if available on reasonable terms, to insure our directors and officers against the cost of defense, settlement or payment of a judgment under certain circumstances.
| Item 15. | Recent Sales of Unregistered Securities |
Since March 6, 2001, the registrant has sold and issued the following unregistered securities:
| (1) Since March 6, 2001, the registrant granted stock options and stock awards to employees, directors and consultants under its 2001 Equity Incentive Plan covering an aggregate of 2,755,246 shares of common stock, at exercise prices ranging from $1.00 to $10.00 per share. Of these options, an aggregate of 207,055 were canceled without being exercised. | |
| (2) Since March 6, 2001, the registrant sold an aggregate of 41,103 shares of its common stock to employees, directors and consultants for cash consideration in the aggregate amount of $43,180 upon the exercise of stock options under its 2001 Equity Incentive Plan. | |
| (3) In April 2001 and September 2001, the registrant sold a total of 9,150,000 of Series A preferred stock to nine purchasers at $1.00 per share, for an aggregate purchase price of $9,150,000. Shares of Series A preferred stock were originally convertible into shares of common stock at the rate of one share of common stock for each share of Series A preferred stock owned. As a result of subsequent antidilution adjustments, the shares of Series A preferred stock are convertible at the rate of 0.144 shares of common stock for each share of Series A preferred stock. | |
| (4) In March 2002, the registrant sold 11,692,308 shares of Series B preferred stock to seven purchasers, at $1.30 per share, for an aggregate purchase price of $15,200,000. Shares of Series B preferred stock were originally convertible into shares of common stock at the rate of one share of common stock for each share of Series B preferred stock owned. As a result of subsequent anti-dilution adjustments, the shares of Series B preferred stock are convertible at the rate of 0.170 shares of common stock for each share of Series B preferred stock. | |
| (5) In April 2003, the registrant issued convertible promissory notes in the aggregate principal amount of $9,800,000 convertible into Series C preferred stock to seven investors in connection with a bridge loan. The convertible promissory notes converted at the rate of $0.5925 per share. | |
| (6) In February 2004, the registrant sold 109,704,634 shares of Series C preferred stock to 45 purchasers, at $0.5925 per share, for an aggregate purchase price of $64,999,996. Shares of Series C preferred stock are convertible into shares of common stock at the rate of one share of common stock for each share of Series C preferred stock owned. | |
| (7) In February 2004, the registrant issued a warrant to purchase 692,658 shares of Series C preferred stock to an investor for financial services rendered total registrant in connection with its Series C Preferred Stock financing. The warrant has an exercise price of $0.5925 per share. The warrant is currently exercisable in whole or in part and shall terminate on the earlier of February 19, 2009 or the consummation of our initial public offering. The warrant shall terminate unless exercised prior to the closing of this offering. | |
II-2
Table of Contents
| (8) In October 2004, the registrant issued 160,000 shares of common stock to one party in connection with a licensing transaction. |
The registrant claimed exemption from registration under the Securities Act for the sales and issuances of securities in the transactions described in paragraphs (1) and (2) above under Section 4(2) under the Securities Act in that such sales and issuances did not involve a public offering or under Rule 701 promulgated under the Securities Act, in that they were offered and sold either pursuant to written compensatory plans or pursuant to a written contract relating to compensation, as provided by Rule 701.
The registrant claimed exemption from registration under the Securities Act for the sale and issuance of securities in the transactions described in paragraphs (3) through (7) by virtue of Section 4(2) and/or Regulation D promulgated thereunder as transactions not involving any public offering. All of the purchasers of unregistered securities for which the registrant relied on Regulation D and/or Section 4(2) represented that they were accredited investors as defined under the Securities Act. The registrant claimed such exemption on the basis that (a) the purchasers in each case represented that they intended to acquire the securities for investment only and not with a view to the distribution thereof and that they either received adequate information about the registrant or had access, through employment or other relationships, to such information and (b) appropriate legends were affixed to the stock certificates issued in such transactions.
| Item 16. | Exhibits and Financial Statement Schedules |
| Exhibit No. | Description | |||
| 1.1 | * | Form of Underwriting Agreement | ||
| 3.1 | + | Third Amended and Restated Certificate of Incorporation, as currently in effect | ||
| 3.2 | * | Form of Amended Restated Certificate of Incorporation (to be filed in connection with the closing of this offering) | ||
| 3.3 | + | Bylaws, as currently in effect | ||
| 3.4 | * | Form of Amended and Restated Bylaws (to be effective upon the closing of this offering) | ||
| 4.1 | + | Second Amended and Restated Investor Rights Agreement, entered into as of February 17, 2004, by and among AlgoRx Pharmaceuticals, Inc. and certain investors named therein | ||
| 4.2 | + | Investor Rights Agreement entered into as of October 28, 2004, by and between AlgoRx Pharmaceuticals, Inc. and Bridge Pharma, Inc. | ||
| 4.3 | * | Specimen Common Stock Certificate | ||
| 5.1 | * | Opinion of Heller Ehrman White & McAuliffe LLP | ||
| 10.1 | + | 2001 Equity Incentive Plan, as amended and restated | ||
| 10.2 | + | Form of Stock Option Grant Notice under AlgoRx Pharmaceuticals, Inc. 2001 Equity Incentive Plan | ||
| 10.4 | + | Form of Stock Option Agreement for Employee under AlgoRx Pharmaceuticals, Inc. 2001 Equity Incentive Plan | ||
| 10.5 | **+ | Form of Stock Option Agreement for Senior Management under AlgoRx Pharmaceuticals, Inc. 2001 Equity Incentive Plan | ||
| 10.6 | **+ | Form of Stock Option Agreement for Senior Management under AlgoRx Pharmaceuticals, Inc. 2001 Equity Incentive Plan with acceleration provisions | ||
| 10.7 | + | Form of Early Exercise Stock Purchase Agreement under AlgoRx Pharmaceuticals, Inc. 2001 Equity Incentive Plan | ||
| 10.8 | + | Lease Agreement dated August 27, 1997, by and between AlgoRx Technologies, Inc. (formerly PowderJect Technologies, Inc.) and John Arrillaga, or his Successor Trustee, UTA dated 07/20/77 (the John Arrillaga Survivor’s Trust) as amended and Richard T. Peery, Trustee, or his Successor Trustee, UTA 7/20/77 (Richard T. Peery Separate Property Trust) as amended | ||
II-3
Table of Contents
| Exhibit No. | Description | |||
| 10.9 | + | Lease dated May 10, 2004, between AlgoRx Pharmaceuticals, Inc. and 500 Plaza Drive Corp. | ||
| 10.1 | 0+ | License Agreement entered into as of August 28, 2001, among AlgoRx Pharmaceuticals, Inc. and James N. Campbell, M.D., Richard Meyer, M.S. and Marco Pappagallo, M.D. | ||
| 10.1 | 1+ | License Agreement entered into as of August 28, 2001, between AlgoRx Pharmaceuticals, Inc. and Marco Pappagallo, M.D. | ||
| 10.1 | 2†+ | License Agreement entered into as of March 22, 2002, by and between AlgoRx Pharmaceuticals, Inc. and PowderJect Research Limited | ||
| 10.1 | 3+ | First Amendment to License Agreement entered into as of July 7, 2003, between AlgoRx Pharmaceuticals, Inc. and PowderJect Research Limited | ||
| 10.1 | 4+ | Assignment, Assumption and Consent Agreement made as of May 14, 2004 by and among PowderMed Limited, PowderJect Research Limited, PowderJect Technologies Limited, AlgoRx Pharmaceuticals, Inc. and AlgoRx Technologies, Inc. | ||
| 10.1 | 5+ | Letter Agreement entered into as of September 30, 2004, between AlgoRx Pharmaceuticals, Inc. and PowderJect Research Limited | ||
| 10.1 | 6†+ | Supply Agreement entered into as of March 22, 2002, between AlgoRx Pharmaceuticals, Inc. and PowderJect Research Ltd. | ||
| 10.1 | 7†+ | First Amendment to Supply Agreement entered into as of July 7, 2003, between AlgoRx Pharmaceuticals, Inc. and PowderJect Technologies Limited. | ||
| 10.1 | 8† | Collaboration, Development and License Agreement made as of October 28, 2004, between AlgoRx Pharmaceuticals, Inc. and Bridge Pharma, Inc. | ||
| 21.1 | + | List of subsidiaries | ||
| 23.1 | Consent of Ernst & Young LLP, independent auditors | |||
| 23.2 | * | Consent of Heller Ehrman White & McAuliffe LLP (included in Exhibit 5.1) | ||
| 24.1 | + | Power of Attorney (included on signature page) | ||
| * | To be filed by Amendment. |
| ** | Indicates a management contract or compensatory plan. |
| † | Application has been made to the Securities and Exchange Commission to seek confidential treatment of certain provisions of this exhibit under Rule 406 of the Securities Act of 1933. Omitted material for which confidential treatment has been requested has been filed separately with the Securities and Exchange Commission. |
| + | Previously filed. |
| Item 17. | Undertakings |
The undersigned registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers, and controlling persons of the registrant pursuant to the foregoing provisions described in Item 14 or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act of 1933 and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer, or controlling person of the registrant in the successful defense of any action, suit, or proceeding) is asserted by such director, officer, or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act of 1933 and will be governed by the final adjudication of such issue.
II-4
Table of Contents
The undersigned registrant undertakes that:
| (1) for purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in the form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective, and | |
| (2) for the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initialbona fideoffering thereof. |
II-5
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant has caused this Amendment No. 2 to the Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized in the City of Secaucus, State of New Jersey, on the 19th day of January, 2005.
| ALGORX PHARMACEUTICALS, INC. |
| By: | /s/ RONALD M. BURCH |
| Ronald M. Burch, M.D., Ph.D. | |
| Chief Executive Officer |
Pursuant to the requirements of the Securities Act of 1933, this Amendment No. 2 to the Registration Statement has been signed below by the following persons in the capacities and on the dates indicated.
| Signature | Title | Date | ||||
| /s/ RONALD M. BURCH Ronald M. Burch, M.D., Ph.D. | Chief Executive Officer & Director (Principal Executive Officer) | January 19, 2005 | ||||
| /s/ JEFFREY A. RONA Jeffrey A. Rona | Vice President, Finance & Chief Financial Officer (Principal Financial & Accounting Officer) | January 19, 2005 | ||||
| /s/ CHARLES M. COHEN Charles M. Cohen, Ph.D | Director | January 19, 2005 | ||||
| * Arnold L. Oronsky, Ph.D | Director | January 19, 2005 | ||||
| /s/ RODNEY A. FERGUSON Rodney A. Ferguson, J.D., Ph.D. | Director | January 19, 2005 | ||||
| * Michael F. Powell, Ph.D. | Director | January 19, 2005 | ||||
| /s/ CARTER H. ECKERT Carter H. Eckert | Director | January 19, 2005 | ||||
| *By: | /s/ JEFFREY A. RONA Attorney in Fact | January 19, 2005 | ||||
II-6
Table of Contents
EXHIBIT INDEX
| Exhibit No. | Description | |||||
| 1 | .1* | Form of Underwriting Agreement | ||||
| 3 | .1+ | Third Amended and Restated Certificate of Incorporation, as currently in effect | ||||
| 3 | .2* | Form of Amended Restated Certificate of Incorporation (to be filed in connection with the closing of this offering) | ||||
| 3 | .3+ | Bylaws, as currently in effect | ||||
| 3 | .4* | Form of Amended and Restated Bylaws (to be effective upon the closing of this offering) | ||||
| 4 | .1+ | Second Amended and Restated Investor Rights Agreement, entered into as of February 17, 2004, by and among AlgoRx Pharmaceuticals, Inc. and certain investors named therein | ||||
| 4 | .2+ | Investor Rights Agreement entered into as of October 28, 2004, by and between AlgoRx Pharmaceuticals, Inc. and Bridge Pharma, Inc. | ||||
| 4 | .3* | Specimen Common Stock Certificate | ||||
| 5 | .1* | Opinion of Heller Ehrman White & McAuliffe LLP | ||||
| 10 | .1+ | 2001 Equity Incentive Plan, as amended and restated | ||||
| 10 | .2+ | Form of Stock Option Grant Notice under AlgoRx Pharmaceuticals, Inc. 2001 Equity Incentive Plan | ||||
| 10 | .4+ | Form of Stock Option Agreement for Employee under AlgoRx Pharmaceuticals, Inc. 2001 Equity Incentive Plan | ||||
| 10 | .5**+ | Form of Stock Option Agreement for Senior Management under AlgoRx Pharmaceuticals, Inc. 2001 Equity Incentive Plan | ||||
| 10 | .6**+ | Form of Stock Option Agreement for Senior Management under AlgoRx Pharmaceuticals, Inc. 2001 Equity Incentive Plan with acceleration provisions | ||||
| 10 | .7+ | Form of Early Exercise Stock Purchase Agreement under AlgoRx Pharmaceuticals, Inc. 2001 Equity Incentive Plan | ||||
| 10 | .8+ | Lease Agreement dated August 27, 1997, by and between AlgoRx Technologies, Inc. (formerly PowderJect Technologies, Inc.) and John Arrillaga, or his Successor Trustee, UTA dated 07/20/77 (the John Arrillaga Survivor’s Trust) as amended and Richard T. Peery, Trustee, or his Successor Trustee, UTA 7/20/77 (Richard T. Peery Separate Property Trust) as amended | ||||
| 10 | .9+ | Lease dated May 10, 2004, between AlgoRx Pharmaceuticals, Inc. and 500 Plaza Drive Corp. | ||||
| 10 | .10+ | License Agreement entered into as of August 28, 2001, among AlgoRx Pharmaceuticals, Inc. and James N. Campbell, M.D., Richard Meyer, M.S. and Marco Pappagallo, M.D. | ||||
| 10 | .11+ | License Agreement entered into as of August 28, 2001, between AlgoRx Pharmaceuticals, Inc. and Marco Pappagallo, M.D. | ||||
| 10 | .12†+ | License Agreement entered into as of March 22, 2002, by and between AlgoRx Pharmaceuticals, Inc. and PowderJect Research Limited | ||||
| 10 | .13+ | First Amendment to License Agreement entered into as of July 7, 2003, between AlgoRx Pharmaceuticals, Inc. and PowderJect Research Limited | ||||
| 10 | .14+ | Assignment, Assumption and Consent Agreement made as of May 14, 2004, by and among PowderMed Limited, PowderJect Research Limited, PowderJect Technologies Limited, AlgoRx Pharmaceuticals, Inc. and AlgoRx Technologies, Inc. | ||||
| 10 | .15+ | Letter Agreement entered into as of September 30, 2004, between AlgoRx Pharmaceuticals, Inc. and PowderJect Research Limited | ||||
| 10 | .16†+ | Supply Agreement entered into as of March 22, 2002, between AlgoRx Pharmaceuticals, Inc. and PowderJect Research Ltd. | ||||
| 10 | .17†+ | First Amendment to Supply Agreement entered into as of July 7, 2003, between AlgoRx Pharmaceuticals, Inc. and PowderJect Technologies Limited. | ||||
| 10 | .18† | Collaboration, Development and License Agreement made as of October 28, 2004, between AlgoRx Pharmaceuticals, Inc. and Bridge Pharma, Inc. | ||||
| 21 | .1+ | List of subsidiaries | ||||
Table of Contents
| Exhibit No. | Description | |||||
| 23 | .1 | Consent of Ernst & Young LLP, independent auditors | ||||
| 23 | .2* | Consent of Heller Ehrman White & McAuliffe LLP (included in Exhibit 5.1) | ||||
| 24 | .1+ | Power of Attorney (included on signature page) | ||||
| * | To be filed by Amendment. |
| ** | Indicates a management contract or compensatory plan. |
| † | Application has been made to the Securities and Exchange Commission to seek confidential treatment of certain provisions of this exhibit under Rule 406 of the Securities Act of 1933. Omitted material for which confidential treatment has been requested has been filed separately with the Securities and Exchange Commission. |
| + | Previously filed. |