SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 8-K
CURRENT REPORT
Pursuant to Section 13 or 15(d) of theSecurities Exchange Act of 1934
Date of Report (Date of earliest event reported): July 21, 2004
Email Real Estate.com, Inc.
(Exact Name of Registrant as Specified in Charter)
| Colorado | 0-50782 | 84-158841 |
| (State or Other Jurisdiction of Incorporation) | (Commission File Number) | (IRS Employer Identification Number) |
400 Oyster Point Boulevard, Suite 215
South San Francisco, California 94080
(Address of Principal Executive Offices, Zip Code)
South San Francisco, California 94080
(Address of Principal Executive Offices, Zip Code)
Registrant’s telephone number, including area code: (650) 588-6404
21 Wilcox Street #C, Castle Rock, CO 80104
(Former Name or Former Address, if Changed Since Last Report)
(Former Name or Former Address, if Changed Since Last Report)
Forward-Looking Statements
This Current Report contains forward-looking statements regarding, among other items, expected sales penetration, our growth strategy and anticipated trends in the industry and our business. These forward-looking statements are based largely on our expectations and are subject to a number of risks and uncertainties, including the risks described herein in Part (F) of Item 5. Many of these risks are beyond our control. Actual results could likely differ materially from these forward-looking statements. In light of these risks and uncertainties, there can be no assurance that any of the forward-looking information contained in this Current Report will, in fact, transpire or prove to be accurate.
ITEM 1. CHANGES IN CONTROL OF REGISTRANT
(A) Merger Transaction with Hana Health Sciences, Inc.
Pursuant to an Agreement and Plan of Merger dated as of June 17, 2004 (the “Merger Agreement”), by and among Email Real Estate.com, Inc. (the “Registrant” or the “Company”), EMLR Acquisition Corp., a Delaware corporation and wholly owned subsidiary of the Registrant (“Acquisition Co.”), and Hana Biosciences, Inc. (formerly, Hudson Health Sciences, Inc.), a Delaware corporation (“Hana”), Acquisition Co. merged with and into Hana, with Hana remaining as the surviving company and a wholly owned operating subsidiary of the Registrant (the “Merger”). The Merger was effective as of July 21, 2004, upon the filing of a Certificate of Merger with the Delaware Secretary of State (the “Certificate of Merger,” and together with the Merger Agreement, the “Plan of Merger”). A copy of the press release dated July 22, 2004 announcing the completion of the Merger is attached to this Form 8-K as Exhibit 99.1 and incorporated herein by reference.
Immediately prior to the effective time of the Merger, Hana had outstanding 6,179,829 shares of its common stock (“Hana Common Stock”) and 2,395,210 shares of its Series A Convertible Preferred Stock (“Hana Series A”). In accordance with the Plan of Merger, each share of Hana Common Stock automatically converted into and is exchangeable for one (1) share of the Registrant’s newly-designated Series B Convertible Preferred Stock (the “EMLR Series B”), and each share of Hana Series A converted into and is exchangeable for one (1) share of the Registrant’s newly-designated Series A Convertible Preferred Stock (the “EMLR Series A”). Each share of EMLR Series A and EMLR Series B is convertible into 16.920814 shares of the Registrant’s common stock. A more complete description of the terms, rights and preferences of the EMLR Series A and EMLR Series B is set forth in this Form 8-K under the heading “Description of Capital Stock” in Item 5. Hana also had outstanding options and warrants to purchase an aggregate of 1,311,065 shares of Hana Common Stock at the time of the Merger which, as a result of the Plan of Merger, now represent the right to purchase an aggregate of approximately 22,184,287 shares of the Registrant’s common stock. Accordingly, on a fully-diluted basis, after giving effect to the Merger, the Registrant has approximately 192,276,931 shares of common stock outstanding. On all matters submitted to the holders of the Registrant’s common stock, the holders of the EMLR Series A and EMLR Series B stock are entitled to such number of votes as is equal to the number of the Registrant’s common shares issuable upon conversion of such preferred stock. Accordingly, the former Hana stockholders, who now collectively own all of the EMLR Series A and EMLR Series B shares, together hold approximately 75 percent of the Registrant’s outstanding voting power.
In accordance with the Plan of Merger, the Registrant’s Board of Directors was reconstituted immediately following the effective time of the Merger. Specifically, prior to July 21, 2004, the Registrant’s Board of Directors consisted of Dan O’Meara and Sharon Leach. Prior to the Merger on July 21, 2004, Sharon Leach resigned, leaving Mr. O’Meara as the sole director. Immediately following the Merger, in accordance with the Registrant’s bylaws and the Colorado Business Corporation Act, Mr. O’Meara appointed Mark J. Ahn, Ph.D., Arie Belldegrun, M.D., Isaac Kier, Leon Rosenberg, M.D., and Michael Weiser, M.D., Ph.D., each of whom was an existing director of Hana. Following their appointment, the Registrant’s newly-constituted board of directors appointed new officers of the Registrant, as follows: Dr. Ahn, President & CEO; Jonathan P. Serbin, Chief Financial Officer (until August 15, 2004); John P. Iparraguirre, Chief Financial Officer (on an interim basis, beginning August 15, 2004) and Secretary; and Fred Vitale, Vice President – Business Development. More complete biographical information concerning each of the Registrant’s new officers and directors is set forth in Item 5 of this Form 8-K under the heading “Management.”
| 1 | ||
(B) Sale of Common Stock by Principal Shareholder
As previously disclosed in the Registrant’s Form 8-K dated June 17, 2004, pursuant to a Stock Purchase Agreement dated June 17, 2004, as amended (the “Stock Purchase Agreement”), among the Registrant, R&R Biotech, LLC, Turquoise Partners, LLC, Chase Financing, Inc. (collectively, the “Purchasers”) and The Washington Trust (the “Seller” or “Trust”), the Trust sold to the Purchasers an aggregate of 20,684,618 shares of the Registrant’s common stock, which represented approximately 83 percent of the Registrant’s outstanding common shares. The Trust received an aggregate purchase price of approximately $388,000 in cash. The Stock Purchase Agreement is included as an exhibit to this Form 8-K.
ITEM 2. ACQUISITION OR DISPOSITION OF ASSETS
See Item 1(A) of this Form 8-K.
ITEM 5. OTHER EVENTS AND REQUIRED FD DISCLOSURE
(A) HANA BIOSCIENCES PRIVATE PLACEMENT
In conjunction with the Merger, Hana completed a private placement of 2,395,210 shares of its Series A Convertible Preferred Stock at a per share price of $3.34 for aggregate gross proceeds of $8 million. Upon the effective time of the Merger, the Hana Series A shares automatically converted into and became exchangeable for EMLR Series A shares, as described in Item 1(A) of this Form 8-K.
As a result of the Merger, the Registrant is now obligated to perform Hana’s obligations under its agreements with the investors in the private placement. Accordingly, within 30 days following the closing of the Merger, the Registrant is required to file with the SEC a registration statement on Form SB-2 (or another available form) under the Securities Act of 1933, as amended (the “Registration Statement”), covering the resale of the common shares issuable upon exercise of the EMLR Series A shares (the “Series A Conversion Shares”). The Registrant has further agreed to use its best efforts to cause the Registration Statement to be declared effective by the SEC on the earlier of (1) 180 days following the closing of the Merger, or (2) five (5) trading days following the date on which the Company is notified by the SEC that the Registration Statement will not be reviewed or is no longer subject to further review. In the event the Registration Statement is not filed within such 30-day period, or is not effective within such 5 or 180-day period, as applicable, the Company is required to pay to each investor a cash amount equal to 1.5 percent of the purchase price paid for the Hana Series A shares by such investor for each monthly period, or portion thereof, in which the Company is in default of our obligation to file or make effective the Registration Statement.
| 2 | ||
(B) CHANGES IN MANAGEMENT
As described in Item 1(A) of this Current Report, immediately following the effective time of the Merger, the Registrant’s board of directors was reconstituted and new officers were appointed. Biographical information for the Registrant’s new officers and directors is set forth below.
Name | Age | Position |
| Mark Ahn, Ph.D. | 41 | President, Chief Executive Officer and Director |
| Jonathan Serbin, J.D. | 34 | Chief Financial Officer* |
| Fred Vitale | 48 | Vice President, Business Development |
| John P. Iparraguirre | 28 | Controller and Secretary* |
| Arie Belldegrun, M.D. | 54 | Director |
| Isaac Kier, J.D. | 51 | Director |
| Leon Rosenberg, M.D. | 71 | Director |
| Michael Weiser, M.D., Ph.D. | 41 | Director |
_____________
* Mr. Serbin has informed the Company of his resignation, effective as of August 15, 2004. Upon his departure, Mr. Iparraguirre will serve as the Company’s Chief Financial Officer on an interim basis.
Mark J. Ahn, Ph.D.Dr. Ahn joined Hana in November 2003 as its President and Chief Executive Officer. Prior to joining Hana, from December 2001 to November 2003, he served as Vice President, Hematology and corporate officer at Genentech, Inc. where he was responsible for commercial and clinical development of the Hematology franchise. From February 1991 to February 1997 and from February 1997to December 2001, Dr. Ahn was employed by Amgen and Bristol-Myers Squibb Company, respectively, holding a series of positions of increasing responsibility in strategy, general management, sales & marketing, business development, and finance. He has also served as an officer in the U.S. Army. Dr. Ahn is a Henry Crown Fellow at the Aspen Institute, founder of the Center for Non-Profit Leadership, and a member of the Board of Trustees for the MEDUNSA (Medical University of South Africa) Trust. Dr. Ahn received a BA in History/Economics and an MBA in Finance from Chaminade University. He was a graduate fellow in Economics at Essex University, and has a Ph.D. in Business Administration from the University of South Australia.
Jonathan P. Serbin.Mr. Serbin has served as Chief Financial Officer of Hana since January 2004. From May 2000 to January 2004,he served as President of Sinosure, Inc., an international financial advisory firm, based in New York and Shanghai. Prior to Sinosure, from August 1999 to May 2000, Mr. Serbin was an investment banker at Lehman Brothers in New York, focusing on mergers & acquisitions. From March 1995 to May 1997, he was an attorney at Coudert Brothers, an international law firm, focusing on corporate finance, M&A and corporate governance. Mr. Serbin received an MBA from Columbia Business School in New York, a JD with honors from Boston University and a BA in Economics from Washington University in St. Louis.
Fred Vitale.Mr. Vitale has served as Vice President, Business Development of Hana since January 2004. From April 2001 to January 2004, Mr. Vitale was employed by Genentech, where he served as head of commercial Rituxan (rituximab) and pre-launch medical education for Avastin (bevacizumab). From December 1998 to April 2001, Mr. Vitale was Director, Global Oncology Marketing at Bristol-Myers Squibb where he was responsible for pipeline development, licensing, and life cycle management for cancer products including Taxol (paclitaxel); as well as Director of Operations and Planning for Japan and China. Prior to that, Mr. Vitale held several roles of increasing responsibility in sales, marketing and general management at Amgen from January 1990 to December 1998. Mr. Vitale received a Bachelor of Science in Biology from The Citadel in Charleston, South Carolina and a Physician Assistant degree from the Medical University of South Carolina.
| 3 | ||
John P. Iparraguirre.Mr. Iparraguirre, who has served as controller of Hana since May 2004, will serve as the Company’s interim chief financial officer commencing August 15, 2004. Prior to joining Hana, Mr. Iparraguirre was the Accounting Manager at Discovery Toys, Inc. from April 2002 until May 2004where he held several roles of responsibility in Finance Management. Mr. Iparraguirre was primarily responsible for maintaining the integrity of the company’s financial reporting as well as coordinating all aspects of its SEC regulatory filings. Prior to Discovery Toys, Mr. Iparraguirre was a Senior Audit Associate at BDO Seidman, LLP, an international accounting firm, from September 1998 until April 2002, focusing on publicly traded companies and their related SEC compliance. In addition, he was appointed the West Coast administrator for BDO’s Computer Assisted Auditing Tools. Mr. Iparraguirre received a Bachelor of Science degree in Business Economics with an Emphasis in Accounting from the University of California, Santa Barbara.
Arie Belldegrun, M.D., FACS.Dr. Belldegrun served on Hana’s Board of Directors since April 2004. He has served as Professor of Urology since 1994, Chief of the Division of Urologic Oncology since 1996 at the David Geffen School of Medicine at the University of California, Los Angeles (UCLA). He has also held the Roy and Carol Doumani Chair in Urologic Oncology at UCLA since 2000. Dr. Belldegrun completed his medical degree at the Hebrew University Hadassah Medical School in Jerusalem, his post at the Weizmann Institute of Science and his residency in Urology at Harvard Medical School. Prior to UCLA, Dr. Belldegrun served as a research fellow in surgical oncology at the National Cancer Institute/NIH under Steven A. Rosenberg, MD, Ph.D. from 1985 to 1988. He is certified by the American Board of Urology and is a Fellow of the American College of Surgeons. Dr. Belldegrun is on the scientific boards of several biotechnology and pharmaceutical companies and serves as a reviewer for many medical journals and granting organizations. In addition to holding several patents, Dr. Belldegrun is the author of several books on prostate and kidney cancers, and has written over 350 scientific publications with an emphasis on urologic oncology, particularly kidney, prostate and bladder cancers. He is the founder of Agensys, Inc., a company focused on the development of fully human monoclonal antibodies to treat solid tumor cancers based on Agensys’ propriety targets. Dr. Belldegrun served as founding Chairman of Agensys from 1997-2002 and currently serves on the Board of Directors and as a consultant. Dr. Belldegrun is also Vice Chairman of the Board of Directors and Chairman of the Scientific Advisory Board of Cougar Biotechnology, Inc., established to in-license and develop early clinical stage drugs, with a specific focus on the field of oncology.
Isaac Kier.Mr. Kier has served on Board of Directors of Hana since February 2004. Since February 2000, Mr. Kier has been a general partner of Coqui Capital Partners, a venture capital firm licensed by the Small Business Administration as a small business investment company having investments in the telecommunications, media and biotechnology industries. Since February 2004, he has served as treasurer and director of Tremisis Energy Acquisition Corporation (TEGYU.OB), a special purpose acquisition company. He served as President, Chief Executive Officer and Chairman of the Board of Lida, Inc (Nasdaq: LIDA) from 1981 until 1995. He was a lead investor in eDiets.com (Nasdaq:DIET) in 1999 and has continued as director. Mr. Kier has served on the board of directors of private comp anies such as Montebello Brand Liquors, Inc. since 2001, and Caribbean Storage, Inc. since 2000. Since April 1997, he has been a principal of First Americas Partners, LLC, an investment partnership focusing on real estate investments in North and South America. From 1987 to 1997, he also served as the Managing Partner of Dana Communications Limited, a non-wire-line cellular licensee.
Leon E. Rosenberg, M.D. Dr. Rosenberg, a director of Hana since February 2004, has been a Professor in the Princeton University Department of Molecular Biology and the Woodrow Wilson School of Public and International Public Affairs since September 1997. Since July 1999, he has also been Professor Adjunct of Genetics at Yale University School of Medicine. From January 1997 to March 1998, Dr. Rosenberg served as Senior Vice President, Scientific Affairs of Bristol-Myers Squibb Company, and from September 1991 to January 1997, Dr. Rosenberg served as President of the Bristol-Myers Squibb Pharmaceutical Research Institute, where he was responsible for the company’s worldwide pharmaceutical research and development. Prior to Bristol-Myers Squibb, Dr. Rosenberg was Dean of the Yale University School of Medicine fr om July 1984 to September 1991. Dr. Rosenberg also serves on the Boards of Directors of Lovelace Respiratory Research Institute (since 1997), Karo Bio AB (since 2000), and Medicines for Malaria Venture (since 2000).
| 4 | ||
Michael Weiser, M.D., Ph.D.Dr. Weiser, a director of Hana since its inception in December 2002, is the Director of Research of Paramount BioCapital Asset Management, Inc., New York. Dr. Weiser also currently serves on the boards of directors of Manhattan Pharmaceuticals, Inc. (OTCBB: MHTT), a company engaged in developing pharmaceutical technologies, since February 2003, Chiral Quest, Inc. (OTCBB: CQST) since February 2003, Innovative Drug Delivery Systems, Inc. and several other privately-held biotechnology companies. Dr. Weiser completed his Ph.D. in Molecular Neurobiology at Cornell University Medical College and received his M.D. from New York University School of Medicine, where his also completed a Postdoctoral Fellowship in the Department of Physiology and Neuroscience.
(C) EMPLOYMENT AGREEMENTS WITH EXECUTIVES
Mark J. Ahn, Ph.D.
Dr. Ahn’s employment with Hana was governed by an employment agreement dated November 1, 2003, which will be assigned to and assumed by the Registrant. The agreement provides for a term of 3 years with an annual base salary of $250,000. Dr. Ahn is also eligible to receive milestone bonus payments, as follows: (i) $50,000 upon the dosing of the first patient in a Phase II clinical trial of PT-523; (ii) $75,000 upon the dosing of the first patient in a Phase III clinical trial of PT-523; (iii) $75,000 upon the Company’s licensing of a Phase II clinical compound introduced to the Company by Dr. Ahn; (iv) $100,000 upon the Company’s licensing of a Phase III clinical compound introduced to the Company by Dr. Ahn; (v) $50,000 following the successful completion of an initial public offering; and (vi) $100,000 in the event that the Company’s market capitalization is at least $100 million for a period of 90 consecutive days. Dr. Ahn is further eligible to receive an annual bonus in an amount up to 75 percent of his base salary, as determined by the Board of Directors.
In the event the Company terminates Dr. Ahn’s employment for “cause” (as defined in the employment agreement), it is only obligated to pay his compensation through the date of termination and all unvested options then held by Dr. Ahn immediately terminate. In the event Dr. Ahn’s employment is terminated upon the occurrence of a “change of control” or for a reason other than for “cause,” the Company is obligated to continue paying to Dr. Ahn his base salary for a period of one year, as well as any accrued and unpaid bonus through the date of termination; provided, that the Company’s obligation to pay Dr. Ahn such amounts shall be reduced by amounts he otherwise earns during the 1-year period following termination. In the event his employment is terminated upon a change of control, all of Dr. Ahn’s stock options that have not yet vested will accelerate and be deemed to have vested upon such termination; otherwise, the unvested portion of such options will terminate and he will have 90 days to exercise the vested portions of any options.
Fred Vitale
Mr. Vitale’s employment with Hana was governed by an employment agreement dated January 25, 2004, which will be assigned to and assumed by the Company. The agreement provides for a term of 2 years, subject to 1-year renewals as mutually agreed upon by the Company and Mr. Vitale, with an annual base salary of $175,000. Mr. Vitale also received a signing bonus of $40,000 upon execution of the employment agreement and is eligible to receive periodic incentive bonuses upon the achievement of milestones to be determined by the chief executive officer in an amount not to exceed $50,000. In connection with his employment agreement, Mr. Vitale was also granted stock options relating to 100,000 shares of Hana common stock, which vest in two equal installments on January 15, 2005 and January 15, 2006. The options are exercisable at a price equal to $0.475 per share. As a resultof the Merger, the Hana options now represent the right to purchase approximately 1,692,081 shares of the Company’s common stock at a price of $0.028 per share.
| 5 | ||
In the event Mr. Vitale’s employment is terminated for “cause” (as defined in the employment agreement), the Company is only obligated to pay his compensation through the date of termination and all stock options held by Mr. Vitale that have not yet vested immediately terminate. In the event the Company terminates Mr. Vitale’s employment upon a “change of control,” then Mr. Vitale is entitled to continue receiving his base salary for a period of six months. All stock options that have not yet vested as of such date will be accelerated and deemed to have vested. If the Company terminates Mr. Vitale’s employment agreement for a reason other than for cause or upon a change of control, he is entitled to receive his base salary for a period of one year, which amount may be reduced by any amounts earned by Mr. Vitale from other employment during such one- year period.
(D) DESCRIPTION OF BUSINESS OF HANA BIOSCIENCES, INC.
Upon completion of the Merger, the Registrant ceased all operations relating to its historical business and adopted the business plan of Hana, which is now a wholly-owned subsidiary of the Registrant. Set forth below in this section entitled “Description of Business of Hana Biosciences, Inc.” is a summary of Hana’s business plan, including a description of its product candidates. References to the terms “we”, “our” and “us” refer to Hana Biosciences, Inc.
Overview
We are a development stage, South San Francisco-based biopharmaceutical company that aims to acquire, develop, and commercialize innovative products for the treatment of important unmet medical needs in cancer and immunological diseases. We currently have the rights to and are developing two product candidates, PT-523 and IPdR. We are committed to creating value by building a world-class team, accelerating the development of lead product candidates, expanding our pipeline by being the alliance partner of choice, and nurturing a favorable company culture.
Cancer Overview
Cancer is a group of diseases characterized by either the runaway growth of cells or the failure of cells to die normally. Often, cancer cells spread to distant parts of the body, where they can form new tumors. Cancer can arise in any organ of the body and strikes one of every two American men and one of every three American women at some point in their lives.
Each year, nearly 1.4 million new cases of cancer are diagnosed in the United States, a figure that does not include the 900,000 cases of skin cancer diagnosed annually. Cancer is the second leading cause of death (after heart disease) in the United States, accounting for 560,000 deaths every year.
There are more than 100 different varieties of cancer, which can be divided into six major categories. Carcinomas, the most common type of cancer, originate in tissues that cover a surface or line a cavity of the body. Sarcomas begin in tissue that connects, supports or surrounds other tissues and organs. Lymphomas are cancers of the lymph system, the circulatory system that bathes and cleanses the body's cells. Leukemias involve blood-forming tissues and blood cells. As their name indicates, brain tumors are cancers that begin in the brain, and skin cancers, including dangerous melanomas, originate in the skin. Cancers are considered metastatic if they spread via the blood or lymphatic system to other parts of the body to form secondary tumors.
| 6 | ||
Cancer is caused by a series of mutations, or alterations, in genes that control cells' ability to grow and divide. Some mutations are inherited; others arise from environmental factors such as smoking or exposure to chemicals, radiation, or viruses that damage cells’ DNA. The mutations cause cells to divide relentlessly or lose their normal ability to die.
This year, one in four deaths in the US is expected to be due to cancer. For all forms of cancer combined, the 5-year relative survival rate is 62%.Despite the fact that the cancer mortality rate in the U.S. has risen steadily for the past 50 years, scientific advances appear to have begun to turn the tide. 1997 was the first year in the past half century in which fewer Americans died of cancer than the year before-the start of what researchers hope will be a long-term decline in cancer deaths.
The cost of cancer to the healthcare system is significant. The National Institute of Health (NIH) estimates that the overall cost of cancer in 2002 was $171.6 billion. This cost includes $60.9 billion in direct medical expenses, $15.5 billion in indirect morbidity costs, and $95.2 billion in indirect mortality costs.
Cancer Treatment is a Significant Unmet Medical Need and Promising Market Opportunity
Normal cells grow and die in a controlled way. When cancer occurs, cells in the body that are not normal keep dividing and forming more cells without control. Major treatments for cancer include surgery, radiotherapy, and chemotherapy. There are many different drugs that are used to treat cancer, including cytotoxics or antineoplastics, hormones, and biologics. There are also many experimental treatments under investigation including radiation sensitizers, vaccines, gene therapy and immunotoxins.
- Radiotherapy, also called radiation therapy, is the treatment of cancer and other diseases with ionizing radiation. Ionizing radiation deposits energy that injures or destroys cells in the area being treated (the "target tissue") by damaging their genetic material, making it impossible for these cells to continue to grow. Although radiation damages both cancer cells and normal cells, the latter are able to repair themselves and function properly. Radiotherapy may be used to treat localized solid tumors, such as cancers of the skin, tongue, larynx, brain, breast, or uterine cervix. It can also be used to treat leukemia and lymphoma (cancers of the blood-forming cells and lymphatic system, respectively).
Scientists also are looking for ways to increase the effectiveness of radiation therapy. Two types of investigational drugs are being studied for their effect on cells undergoing radiation. Radiosensitizers, such as IPdR by Hana Health Sciences, potentially make the tumor cells more likely to be damaged; and radioprotectors protect normal tissues from the effects of radiation. - Cytotoxics, or anticancer drugs, destroy cancer cells by stopping them from growing or multiplying. Healthy cells can also be harmed, especially those that divide quickly. Harm to healthy cells is what causes side effects. These cells usually repair themselves after chemotherapy. Chemotherapy can be used for different goals including to cure the cancer (shen the patient remains free of evidence of cancer cells), control the cancer (by keeping the cancer from spreading), and to relieve symptoms of cancer (such as pain to help patients live more comfortably).
Cytotoxic agents act primarily on macromolecular synthesis, repair or activity which affects the production or function of DNA, RNA or protein. PT-523 by Hana Health Sciences, for example, is a novel non-classical antifolate or antimetabolite being developed for use as a cytotoxic agent for the treatment of solid tumors. Thus, although there are many cytotoxic agents, there is a considerable amount of overlap in their mechanisms of action. As such, the choice of a particular agent or group of agents is generally not a consequence of a prior prediction of antitumor activity by the drug, but instead the result of empirical clinical trials.
| 7 | ||
In addition, according to Datamonitor the global cancer market is estimated at $33.5 billion in 2003. Cytotoxics or antineoplastics account for 28% or $9.5 billion of the total global cancer drug market. Predominant classes of cytotoxic agents are antimetabolites, alkylating agents, cytotoxic antibiotics, vinca alkaloids, platinum compounds, and taxanes.
Global Cancer Market Overview
The global cancer market was estimated at $33.5 billion in 2003. Cytotoxics or antineoplastics account for 28% or $9.5 billion of the total global cancer drug market. Predominant classes of cytotoxic agents are antimetabolites, alkylating agents, cytotoxic antibiotics, vinca alkaloids, platinum compounds, and taxanes. A segmentation of the global cancer drug market can be found below in Figure 1.
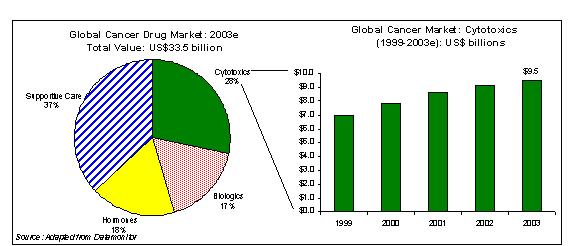
Fig. 1
| 8 | ||
PT-523: Product Description
PT-523 is a novel non-classical antifolate that is a water-soluble, nonpolyglutamatable analogue of aminopterin developed at the Dana-Farber Cancer Institute (DFCI) and the National Cancer Institute (NCI) as part of a program to develop products with improved efficacy, tolerability and resistance. PT-523 is ten times more tightly bound to DHFR (dihydrofolate reductase) than MTX (methotrexate); has ten higher affinity to the membrane-bound transporter utilized by MTX; and is 10 to 100 times more potent than MTX against a wide variety of cell lines and animal models. An integrated battery of preclinical mechanistic studies, including efficacy studies in both in vitro cell culture and in vivo human tumor xenograft models, as well as tissue distribution, pharmacokinetic and systemic toxicity studies have been conducted with PT-523. The combined results of these studies suggest that PT-523 has the potential to significantly enhance the treatment of patients with cancer and other autoimmune diseases. As a result, we are currently conducting a Phase I clinical study of PT-523 at the Dana-Farber Cancer Institute (DFCI), Massachusetts General Hospital, and Beth-Israel Deaconess Hospital.
Antifolates are an important class of cytotoxic or antineoplastic agents, which are antimetabolites. Antifolates, also known as folic acid analogs, have been used clinically for more than 30 years to treat both solid and hematological cancers (such as breast cancer and acute lymphocytic leukemia), as well as inflammatory diseases (such as rheumatoid arthritis). Antimetabolites, such as methotrexate, are structurally related to folic acid and act as antagonists to this vitamin by inhibiting the enzyme that converts folic acid to its active form. Rapidly dividing cells, such as cancer cells, need folic acid to multiply. The decreased level of this active vitamin leads to depressed DNA, RNA, and protein synthesis which in turn leads to apoptosis, or cell death.
Antifolates may be classified as polytglutamatable, or classical antifolates, as well as Type A and Type B non-polyglutamatable antifolates as described below:
Classification Scheme for Dihydrofolate Reductase Inhibitors
Polyglutamatable Inhibitors | Historically referred to as “classical” antifolates, this group includes AMT and MTX analogs modified in the B-ring, the bridge, and/or the p-aminobenzoic acid (PABA) moiety, but with an intact glutamate side chain as required for folylpolyglutamate synthetase (FPGS) activity. |
Nonpolyglutamatable Inhibitors (Type A) | Traditionally referred to as “nonclassical” or “small molecule” antifolates, this group includes very lipophilic drugs like DDMP, TMX, and PTX, which are not substrates for the reduced folate carrier transport system used by classical antifolates, and are presumably taken up by passive and/or facilitated diffusion. |
Nonpolyglutamatable Inhibitors (Type B) | This group consists of compounds that are more polar than those of Type A and retain most of the basic structural motifs of classical antifolates, including the alpha-COOH group. However, they cannot form polyglutamates because the gamma-COOH is blocked or the distal end of the side-chain is otherwise altered. Mechanistically, these “hybrid” inhibitors may be viewed as nonclassical, although they differ from Type A inhibitors in that they are water-soluble, are cleared rapidly from the circulation, and enter cells via the reduced folate carrier transport system (RFC). Moreover, unlike some Type A inhibitors (e.g. TMX), their activity is not influenced by P-glycoprotein over expression. |
| 9 | ||
Classical antifolates contain a glutamic acid side chain which is converted to polyglutamates once the antifolate enters the cell. These polyglutamate metabolites have a prolonged intracellular half life and allow for prolonged drug action in malignant cells. These metabolites are also potent, direct inhibitors of several folate-dependent enzymes involved in de novo purine synthesis. However, cancer cells lose their ability to convert classical antifolates into the more active polyglatamated form over time and consequently reduce their therapeutic effect.
Nonclassical antifolates are primarily lipophilic and do not contain a glutamic acid side chain. As such, they cannot be converted to polyglutamates once they enter the cell. Nonclassical antifolates presumably enter the cell by passive and/or facilitated diffusion.
PT-523’s side chain, illustrated in Figure 2 below, contains a hemiphthaloylorithine moiety in place of glutamate. These features impart an added lipophilic character which improves uptake as well as tighter binding to DHFR. At the same time, the retention of the two COOH groups provides good water solubility at physiological pH, and allows efficient use of the reduced folate carrier transport system (RFC) for active transport across the cell membrane.
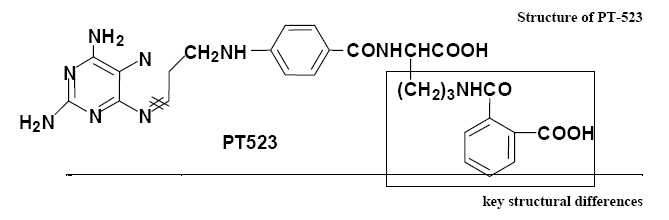
Fig. 2. PT 523 has three key structural differences from AMT and MTX, specifically, the presence of additional methylene, amide, and phenyl moieties. These features enhance the lipophilic character of the molecule which was expected to improve its uptake into cells as well as binding affinity to DHFR. Retention of the two carboxylic acid functional groups provides good water solubility at physiological pH and allows efficient use the RFC pathway for active transport across the cell membrane.
In addition, pharmacology studies have demonstrated that PT-523 inhibits de novo pyrimidine and purine synthesis more potently than either methotrexate or trimetrexate. PT-523 produces cytotoxicity via inhibition of dihydrofolate reductase (DHFR), binding to DHFR much more tightly than classical antifolates such as methotrexate. We believe that PT-523 enters cells by the reduced folate carrier transport system pathway and may be transported up to 10-times more efficiently than methotrexate. PT-523 has exhibited promising anti-tumor activity in multiple animalmodels using a wide variety of cell lines and tumor types. In addition, PT-523 is active in anti-tumor models known to be resistant to classical polyglutamatable antifolates such as methotrexate. We believe that the high potency of PT-523 observed in anti-tumor models is consistent with the combined effects of its enhanced affinity for DHFR and more efficient cellular uptake.
| 10 | ||
Interest in PT-523 was first sparked by the discovery that this compound administered in the low nanomolar range had excellent potency against tumor cells that had shown a clinically relevant level of resistance to MTX. Table 1 exhibits human head-and-neck squamous carcinoma cells with acquired resistance to MTX involving either a defect in transport and polyglutamation or an increase in DHFR expression.
Table 1
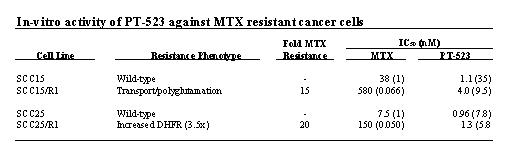
The activity of PT-523 does not require polyglutamation and therefore it may be more efficacious against tumors. As such, the efficacy of PT-523 and MTX was assessed in a tumor growth delay (TGD) mouse model by Wright (1998) as depicted in Fig. 3, which shows that PT-523 was more effective than MTX on either treatment schedule.
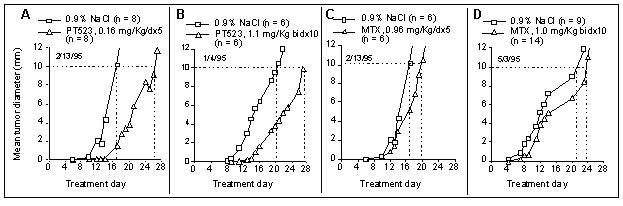
Fig.3. Effect of PT523 on growth of SCC VII murine squamous cell carcinoma. Cells (2x106) were implanted IM in the hind leg of C3D mice on d0, and treatment was started on d1. The endpoint for the experiment was an RMS diameter of 10 mm.Panel A (PT523, 5d infusion): 1/6 mice died on d13 with no tumor; the rest were euthanized between d24 and d28 (10 mm reached).Panel B: (PT523, bidx10): all mice were euthanized between d26 and d28 (10 mm reached).Panel C (MTX, 5d infusion): 1/6 mice died on d17 and 1/6 on d18; 3/6 mice were euthanized on d21 and 1/6 on d24.Panel D(MTX, bidx10): 1/14 mice died on d11,1/6 on d/13, 1/6 on d/16; 7/14 mice were euthanized on d26 and 4/14 on d31 (Wright, 1998).
| 11 | ||
PT-523: Potential Advantages Over Existing Therapies
Potential advantages of PT-523 over existing therapies include increased targeting to tumor cells, better tolerability, and a superior resistance profile:
- Increased Targeting to Tumor Cells:PT-523 produces cytotoxicity via inhibition of dihydrofolate reductase (DHFR). It binds to DHFR much more tightly than other antifolates, allows PT-523 to inhibit DNA synthesis more potently than other antifolates including MTX (Table 2). Additionally, we believe that PT-523 may enter cells up to 10 times more efficiently than MTX by way of the reduced folate carrier transport system pathway. Nonclassical antifolates are not able to enter through this pathway. We believe that these characteristics allow for potent, targeted delivery of PT-523.

Table 2
- Better Tolerability:We believe that PT-523 may be more tolerable than other existing cancer drugs. Classical compounds such as MTX are polyglutamated once they enter the cells. This polyglutamation allows the drug to stay in the cell and act on its target more effectively. However, while healthy cells maintain their ability to make polyglutamates, cancer cells lose their ability to form polyglutamates. Consequently, the drug stays in the healthy cells, causing toxicity, but it is not able to exert its effect on the cancer cell to kill it. PT-523 solves this problem by blocking and altering the side chain of the compound. We believe this alteration allows the drug to stay in the cancer cell and act on its target.
- Superior Resistance Profile:In preclinical studies, PT-523 has demonstrated efficacy against tumor cells that were resistant to other antifolates. Human head-and-neck squamous carcinoma cells that were resistant to MTX were shown to be highly sensitive to the effects of PT-523 (see Table 2).
PT-523: Clinical Development Plan
In March 2003, Hana submitted its Investigational New Drug application (“IND”) with the United States Food and Drug Administration (“FDA”) to seek approval to commence Phase I clinical trials of PT-523 for the treatment of cancer in humans. On May 30, 2003 Hana received notice from the FDA granting the IND, thereby enabling the Company to initiate clinical trials in the U.S. The Company has received institutional review board (IRB) approval to begin Phase I clinical studies at the Dana Farber Cancer Institute (DFCI), Massachusetts General Hospital, and Beth-Israel Deaconess Hospital. Joseph Paul Eder, M.D., Assistant Professor of Medicine at Harvard Medical School and the Clinical Director of the Experimental Therapeutics Program, will be the primary investigator for the Phase I clinical study.
| 12 | ||
The primary objectives of this study are to evaluate the safety of PT-523 when administered intravenously to patients with solid tumors who have failed curative or survival prolonging therapy or for whom no such therapies exist; and to establish the maximum tolerated dose (MTD) and identify the dose limiting toxicities (DLT) of PT-523. The secondary objectives of this study are to determine the pharmacokinetics and to evaluate preliminary efficacy of PT-523 in patients receiving a bolus intravenous injection of PT-523. It is estimated that 20-40 patients will be required to determine the MTD. Upon completion of this study, Hana will evaluate each of several clinical oncology indications in attempting to achieve the most rapid approval by the FDA for use in the treatment of cancer.
PT-523 Development Plan
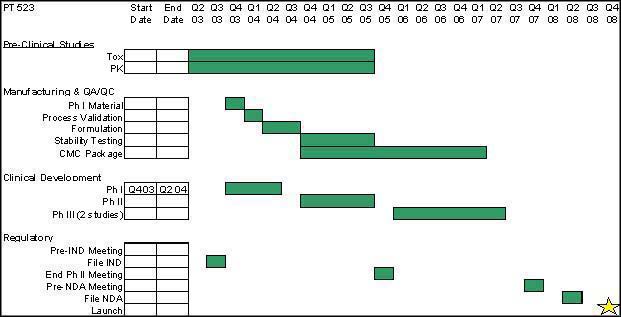
Additional Potential Indications for PT-523
While PT-523 continues in clinical development for oncology, Hana will also begin to evaluate its potential in non-oncology indications, such as rheumatoid arthritis, psoriasis, inflammatory bowel disease, and multiple sclerosis. After initial preclinical studies are evaluated in each of the indications, the Company’s scientific advisory board, consultants, and management team will evaluate its potential in these indications. Hana will then continue the clinical testing of PT-523 in the non-oncology indications in which it shows the most potential.
There are several theories about how antifolates work to treat inflammatory diseases. One hypothesis is based on the idea that antifolates promote the formation of a natural substance in the body called adenosine, which has anti-inflammatory properties. Antifolates, such as methotrexate (MTX), have been shown to inhibit neutrophil function by stimulating adenosine release from connective tissue cells. Another hypothesis states that antifolates inhibit the production of tumor necrosis factor and nitric oxide in the body. Both tumor necrosis factor (TNF) and nitric oxcide (NO) have been shown to cause inflammation. TNF has been shown to play a central role in the pathogenesis of rheumatoid arthritis. NO production has been shown to be decreased in mice treated with MTX.
| 13 | ||
PT-523: Competition
The efficacy and safety profile of PT-523 will potentially make it an attractive alternative to existing antifolate therapies for oncology and inflammatory diseases. The Company’s intends to achieve market share at the expense of existing and established products, as well as future products in the relevant target markets. Some of Hana’s competitors include, but are not limited to Pfizer (trimetrexate),Eli Lilly & Co. (pemetrexed),Novartis (edatrexate), and Allos (PDX) respectively.
Currently Available Antifolates
Cytotoxic agents such as antifolates have been in use for many years. Originally used as a chemotherapy drug to treat certain kinds of cancer, methotrexate, a classical antifolate, was also found to be beneficial in those with inflammatory arthritis and/or psoriasis. In cancer, MTX has been used in breast, head and neck, lung, acute lymphocytic leukemia, gestational trophoblastic disease, lymphoma, and bone tumors. It is also used to treat rheumatoid arthritis and psoriasis. Trimetrexate is a methotrexate analog originally developed by Pfizer, which was approved in 1993 for the treatment of moderate-to-severe pneumocystis carinii pneumonia for immunocompromised patients. Pfizer is also developing a topical and oral formulation of trimetrexate, which it believes may be useful in treating psoriasis and rheumatoid arthritis.
Antifolates in Development
- Eli Lilly is developing pemetrexed for the treatment of mesothelioma and other solid tumors. Recently, pemetrexed has shown efficacy in clinical studies for mesothelioma and Eli Lilly recently received FDA approval of the drug (Alimta®) for this indication.
- Novartis is developing edatrexate for the treatment of cancer. Clinical trials have been conducted in patients with non-small-cell lung cancer, cervical cancer, colon cancer, and breast cancer. The current development status for edatrexate is not known.
- Allos, under license from Memorial Sloan-Kettering Cancer Center, is development PDX (10-propargyl-10-deazaaminopterin). A Phase II trial in non-small-cell lung cancer has been completed and studies are ongoing in mesothelioma and lymphoma.
PT-523 will compete with other cytotoxic, or anticancer, therapies such as antimetabolites, alkyltaing agents, cytotoxic antibiotics, vinca alkaloids, platinum compounds, and taxanes. Since there is a considerable amount of overlap in their mechanisms of action, the choice of a particular agent or group of agents is generally not a consequence of a prior prediction of antitumor activity by the drug, but instead the result of empirical clinical trials.
IPdR: Product Description
Radiotherapy, also called radiation therapy, is the treatment of cancer and other diseases with ionizing radiation. Ionizing radiation deposits energy that injures or destroys cells in the area being treated (the "target tissue") by damaging their genetic material, making it impossible for these cells to continue to grow. Although radiation damages both cancer cells and normal cells, the latter are able to repair themselves and function properly. Radiotherapy may be used to treat localized solid tumors, such as cancers of the skin, tongue, larynx, brain, breast, or uterine cervix. It can also be used to treat leukemia and lymphoma (cancers of the blood-forming cells and lymphatic system, respectively).
| 14 | ||
Two types of investigational drugs are being studied for their effect on cells undergoing radiation.Radiosensitizers, such as IPdR, potentially make the tumor cells more likely to be damaged, andradioprotectors, whichprotect normal tissues from the effects of radiation. Hyperthermia, the use of heat, is also being studied for its effectiveness in sensitizing tissue to radiation.
Halogenated dThd analogues such as IUdR and BUdR are known radiosensitizers with demonstrated clinical efficacy, but poor tolerability
HalogenateddThd analogues such as intravenously (iv) administered IUdR and BUdR are a promising class of radiosensitizers. These halogenateddThd analogues are incorporated into DNA in place of dThd and are thought to radiosensitize cells by forming reactive uracil radicals following irradiation that are capable of inducing strand breaks. Unrepaired or misrepaired DNA double strand breaks result in cell death. The results of recent Phase I/II clinicaltrials suggest an improved outcome compared to radiotherapy alone in patientswith anaplastic astrocytomas and possibly in patients with glioblastomamultiforme. dThd analogue use may also augment treatment for othertypes of clinically radioresistant cancers, including locallyadvanced cervical cancer, head and neck cancers, unresectablehepatic metastases from colorectal cancers, and locally advancedsarcomas, based on the results of other recent Phase I/II clinicaltrials. However, systemic toxicitywith i.v. IUdR to rapidly proliferating normal tissues (principally bone marrowand intestine) can limit the duration and dose rate of the druginfusion and consequently may limit the extent of human tumorradiosensitization.
IPdR is a novel oral prodrug of IUdR being developed as a radiation therapy sensitizer for the treatment of certain types of brain cancers
IPdR (5-iodo-2-pyrimidinone-2'-deoxyribose)is a novel orally administered prodrug for 5-iodo-2'-deoxyuridine(IUdR) for tumor radiosensitization. IPdR is converted to IUdR by aldehyde oxidase in the liver. The IPdR-aldehyde oxidase activity levels are at least one log higher in normal liver compared with other normal tissues. Because only a low amount of aldehyde oxidase is present in the small intestine and bone marrow, IPdR treatment may avoid previously observed toxicities with i.v. IUdR. Preclinical studies with IPdR in several animal models demonstrated that IPdR and radiation is superior in terms of safety and efficacy versus IUdR and radiation. The following sections summarize results from these studies.

Fig.4. IPdR: Structures of 5-substituted 2-pyrmidine analogs. Abbreviations: MPdR, 5-methyl-2-pyrimidinone; IPdR, 5-iodo-2-pyrimidinone; EPdR, 5-ethynyl-2-pyrimidinone; and PPdR, 5-propynyl-2-pyrimidinone
| 15 | ||
IPdR conversion to IUdR results in effective DNA incorporation and enhanced tumor radiation sensitivity. DNA incorporation is a prerequisite for radiosensitization ofhuman tumors by the halogenated dThd analogues, and the extentof radiosensitization correlates directly with the percentagedThd replacement in DNA. Using two different human colon cancer cell lines (HT-29 and HCT 116; Kinsella et al, 1994, Cancer Res 54: 2695; Kinsella et al, 1998, Clinical Cancer Research 4: 99) and one human glioblastoma cell line (U251) as s.c. xenografts in athymic mice, Kinsella et al found a2-fold increasein percentage IUdR-DNA in tumor cells and a2-fold decreasein percentage IUdR-DNA incorporation in proliferating normaltissues (Table 3) after oral IPdR compared toeither oral or continuous infusion IUdR. In addition, compared to radiation alone, a significant sensitizer enhancement ratio (SER) was found with the combination of oral IPdR + radiation but not for the combination of continuous infusion IUdR + radiation in the U251 xenografts (Kinsella et al, 2000, Clinical Cancer Research 6: 1468) (Fig.5).
Table 3. Percentage IUdR-DNA replacement in cellular DNA from normal mouse tissues and U251 tumor xenografts after 14-day treatment with oral IPdR or continuous infusion IUdR. (Kinsella et al, 2000, Clinical Cancer Research 6: 1468)

| 16 | ||
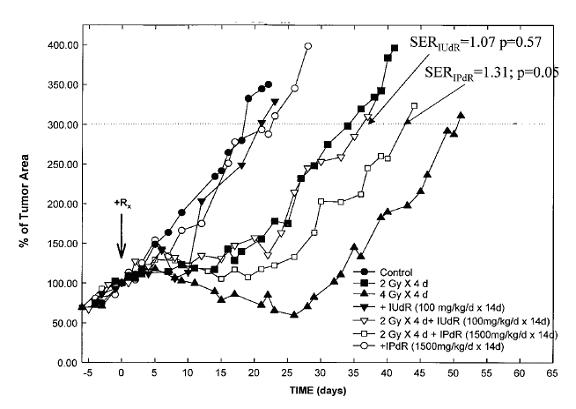
Fig.5. Tumor growth of U251 s.c. xenografts after treatment with XRT alone, oral IPdR alone, continuous infusion with IUdR alone, the combination of IPdR and XRT, and the combination of IUdR and XRT. Control xenografts received no treatment. The time to reach 300% tumor volume was determined to be the end point for these tumor regrowth assays. Group of six mice were included in each experiment group.SeeKinsella et al, 2000, Clinical Cancer Research 6: 1468.
Oral IPdR provides the radiation sensitizing effect of IUdR—with potentially reduced gastrointestinal and hematological side effects associated with intravenously administered IUdR. It is well establishedfrom the Phase I and II clinical trials of continuous or prolongedintermittent i.v. infusions of IUdR and its related analogueBUdR that the systemic toxicities to the bone marrow (myelosuppression)and intestine (diarrhea) limit the duration and dose rate ofa continuous infusion, which may also limit the extent of radiosensitization. In summary, the combination of preclinical studies in a variety of models (including three different human tumor xenografts in nude mice, as well as systemic toxicity and pharmacokinetic studies in non-rodent ferrets and rhesus monkeys) comparing IPdR and IUdR as radiation sensitizer demonstrated that IPdR administrated orally has superior efficacy and toxicity profile than IUdR administrated orally or by continuous infusion.
It is hypothesized that the improved therapeutic indexof oral IPdR compared to continuous infusion IUdR results inpart from the 10–100-fold lower aldehyde oxidase activityfound in most normal mouse tissues including intestine, bonemarrow, lung, brain, and kidney. In thestudies of oral IPdR in athymic mice with or without human tumorxenografts, marked reduction in percentageIUdR-DNA incorporation in normal bone marrow and to a lesserextent in normal small intestine compared to continuous infusionIUdR was found (Table ) (Kinsella et al, 2000, Clinical Cancer Research 6: 1468).
| 17 | ||
Similarly,using the bone marrow as a surrogate of a proliferating normaltissue and liver as a surrogate of a nonproliferating normaltissue in ferrets, the % IUdR-DNA incorporation in the liver was found very low andbelow detection (<0.05%) in IPdR-treated male and femaleferrets. The % IUdR-DNA incorporation in ferret bonemarrow increased in a dose-dependent fashion for the three IPdRdosage groups (Fig.6) which was similar to the % IUdR-DNA incorporationfound in normal bone marrow in athymic mice using the same dosageschedule. In both animal species, no changes in peripheralblood counts were found after the 14-day treatment. In contrast,myelosuppression (both low WBC and platelets) is one of thedose-limiting systemic toxicities in the clinical trials ofthe active drug, IUdR, when given as a prolonged continuousi.v. infusion.
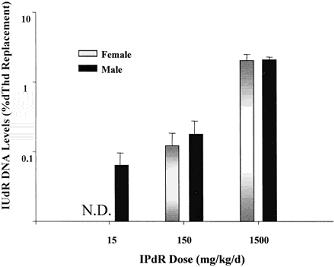
Fig.6.Percentage of IUdR-DNA levels in male and female ferret bone marrow after the 14-day IPdR treatment based on the three dosage groups. The levels represent the bone marrow samples from three, same-gender ferrets in each IPdR dose group; bars, SE. N.D., none detected
The low toxicity profile of oralIPdR in athymic mice may result in part from its pharmacokineticproperties. After a single oral administration ofIPdR at 250-1500 mg/kg, IPdR was efficiently converted to IUdR, the active metabolite, within15–20 min, resulting in peak IUdR plasma levels of 40–75 µM and IUdR plasma levels persisting at >20 µM for up to 90 min. Additionalpharmacokinetic studies in rhesus monkeys showed IPdR was cleared rapidly from plasma (Fig.7).
| 18 | ||
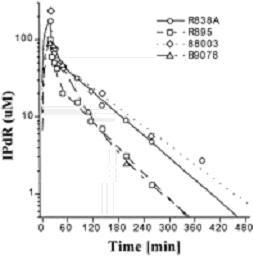
Fig. 7.Plasma IPdR concentration-time profiles in the four Rhesus monkeys after a 50-mg/kg dose of IPdR given as a 20-min i.v. infusion. The lines are the model fit to the data for each monkey (listed by number on figure) using a two-compartment model with first-order elimination from the central compartment.
The clinical development strategy for IPdR is to improve the therapeuticindex of halogenated dThd analogue radiosensitization in poorlyresponsive (clinically radioresistant) human tumors. To achieve this, we propose to usea less systemically toxic halogenanted analogue that can be metabolizedin vivo to the active drug by tumor tissue or a normal tissue.Based on preclinical studies, the use of orally administered IPdR as a prodrug for IUdR-mediatedtumor radiosensitization should achieve this goal.
The first potential indication for the use of IPdR will be for malignant brain tumors. Glioblastoma multiforme and anaplastic astrocytoma are the two most common forms of malignant brain tumor. They are highly aggressive, locally invasive, and poorly responsive to most treatments. They account for more than 50% of the estimated 20,765 primary brain tumors diagnosed each year in the United States. Overall, the incidence of anaplastic astrocytoma and glioblastoma multiforme in the United States is 12,000 new cases each year. Malignant gliomas are typically diagnosed later in life, with a median age at diagnosis of 62 years. The incidence of malignant glioma has been increasing in the elderly population in recent years.
There are several reasons behind initially targeting malignant gliomas. Radiation therapy is standard treatment for this cancer. Additionally, these tumors are highly aggressive. Consequently, standard endpoints such as increase in survival can be replaced by increased tumor response or decreased toxicity in these patients.
Current treatments for glioblastoma and anaplastic astrocytoma involve surgical resection, radiation therapy, and chemotherapy. The prevailing standard of care for primary gliomas is surgical resection, followed by radiation therapy and either carmustine chemotherapy alone, or combination regimen of PCV (procarbazine, lomustine, vincristine). Guilford’s Gliadel wafer, an implantable, biodegradable from of carmustine, has also shown promise. In addition, there are several drugs in development for the treatment of glioblastoma including thalidomide, EGFR (epithelial growth factor receptor), and the anti-tenascin monoclonal antibody.
| 19 | ||
The anticipated timeline for the clinical development milestones for IPdR are shown below. Initially, we plan on having a pre-IND meeting with the FDA to confirm that we have sufficient data to file the IND in order to begin clinical studies. After filing and approval of the IND, we will begin our Phase I studies, which are estimated to take 9 months to complete. After the completion of this study, we will begin a Phase II study for malignant gliomas. After the completion of that study and meeting with the FDA, we plan on initiating several additional Phase II/III studies. We envision these studies being done for primary brain tumor and for the treatment of brain metastases. Upon completion of these studies, and having a Pre-NDA Meeting with the FDA, we hope to use data from at least one of these studies to file a NDA. We estimate NDA approval and market launch in 2 009. Also, development timelines may potentially be shorter through FDA Fast Track review due to the likelihood of orphan drug designation, high unmet medical need, and high rate of disease progression.

Additional Potential Indications for IPdR
Indications beyond brain tumor and metastases to the brain will need further evaluation and preclinical testing. Additional potential uses of IPdR include, but are not limited to, radiosensitive tumors such lymphoma, as well as cancers of the breast, prostate, head and neck, esophagus, bladder, cervix, uterus, soft tissue sarcomas, and retina.
IPdR: Competition
Various pharmacological approaches have been tried experimentallyand clinically to improve the therapeutic gain of halogenateddThd analogue radiosensitization in poorly radioresponsive humantumors. Selective intra-arterial infusions to increasetumor bed drug concentrations has been used clinically for primarybrain tumors and hepatic metastases with a suggested modestimprovement in the therapeutic gain. Experimentally,biochemical modulation of the key enzymes involved in dThd analoguemetabolism (TK) or in the maintenance of cellular deoxyribonucleotidetriphosphate pools (both thymidylate synthase and ribonucleotidereductase) have been studied usingin vitro andin vivo human tumorsystems. Biochemical modulation of thymidy latesynthase has also been tried in clinical Phase I trials usingconcomitant continuous infusions of IUdR with either 5-fluoro-2'-deoxyuridineor folinic acid (leucovorin), but no improvements in the therapeuticgain were found.
| 20 | ||
There are several radiation sensitizers in clinical development including RSR13 and gadolinium texaphyrin. Currently, there are no approved radiation sensitizers on the market.
- RSR13, being developed by Allos, is a synthetic small molecule that enhances the diffusion of oxygen to hypoxic tumor tissues from hemoglobin. Results have been announced from a subgroup analysis of patients with metastatic breast cancer in a phase III, randomized study of radiation therapy in patients with brain metastases.Based on these findings from the Phase III trial, the company is currently submitting a rolling New Drug Application (NDA) for RSR13 as an adjunct to radiation therapy for the treatment of brain metastases originating from breast cancer to the FDA.
- Xcytrin (gadolinium texaphyrin) is being developed by Pharmacyclics as a radiosensitizer for the potential treatment of cancers including brain metastases, primary brain tumors, and non-small cell lung cancer. Pharmacyclics has initiated a confirmatory phase III trial in patients with non-small cell lung cancer whose cancer has spread to the brain. Xcytrin has just recently been granted fast-track status by the U.S. Food and Drug Administration.
License Agreements and Intellectual Property
General
Hana’s goal is to obtain, maintain and enforce patent protection for our products, formulations, processes, methods and other proprietary technologies, preserve its trade secrets, and operate without infringing on the proprietary rights of other parties, both in the United States and in other countries. Hana’s policy is to actively seek to obtain, where appropriate, the broadest intellectual property protection possible for its product candidates, proprietary information and proprietary technology through a combination of contractual arrangements and patents, both in the U.S. and abroad. However, even patent protection may not always afford complete protection against competitors who seek to circumvent patents.See “Risk Factors – Risks Relating to Hana’s Business – If we fail to adequately protect or enforce our intellectual property rights or secure rights to patents of others, the value of our intellectual property rights would diminish,” above.
We also depend upon the skills, knowledge and experience of our scientific and technical personnel, as well as that of our advisors, consultants and other contractors, none of which is patentable. To help protect our proprietary know-how which is not patentable, and for inventions for which patents may be difficult to enforce, we rely and intend to rely in the future on trade secret protection and confidentiality agreements to protect our interests.
| 21 | ||
License Agreements
PT-523. In December 2002, we entered into an exclusive worldwide royalty bearing license agreement with Dana-Farber Cancer Institute and Ash Stevens, Inc., including the right to grant sublicenses, for rights to the intellectual property and know-how relating to PT-523 and all of its uses. Patents related to this technology include: (i) a composition of matter patent, which has been granted, (ii) a utility application, which has been filed; and (iii) provisional use patents for multiple indications. The technology licensed to us include one United States patent issued in 1988 and one pending patent application filed in April 2002.
The license agreement requires us to make future payments totaling up to $6 million upon the achievement of certain milestones, including a $5 million payment upon approval by the FDA of a New Drug Application for PT-523. To date, we have made one of these milestone payments in the amount of $100,000 following commencement of the Phase I clinical trial. Additionally, we are obligated to pay royalties in the amount of 3.5 percent of “net sales” (as defined in the license agreement) of PT-523. We are also required to pay to the licensors 20 percent of fees or non-royalty consideration (e.g., milestone payments, license fees) received by us in connection with any sublicense of PT-523 granted prior to the start of a Phase II trial, and 15 percent of such fees after initiation of a Phase II clinical trial.
The license agreement includes certain other covenants, which require us to, among other things, maintain and prosecute patents related to PT-523; use our commercially reasonable efforts to bring the licensed product to market as soon as reasonably practicable; and prepare and provide to the licensors certain reports concerning our development and commercialization efforts. In the event we fail to carry out our responsibilities under the license agreement, the licensors may terminate the license. The license agreement may also be terminated in the event we fail to make a scheduled milestone or royalty payment, we otherwise materially breach the license agreement, or if we become involved in a bankruptcy, insolvency or similar proceeding, provided that we are entitled to notice of such intention to terminate and an opportunity to cure.
IPdR.In February 2004, we entered into an exclusive worldwide, royalty-bearing license agreement with Yale University and The Research Foundation of State University of New York, including the right to grant sublicenses, for the rights to the intellectual property relating to IPdR. The licensed intellectual property includes patent rights relating to IPdR that do not expire until 2017, at the earliest. In addition to license fees paid to date, Hana is required to make additional license payments in the aggregate amount of $500,000 upon the completion of a Phase IIb clinical trial and upon NDA approval by the FDA. As further consideration for the license, Hana is required to pay royalties to Yale and SUNY equal to 3 percent of net sales (as defined in the license agreement) from IPdR.
The license agreement also includes certain other covenants which obligate us to, among other things, initiate a Phase I trial for IPdR by February 2006 and a Phase II trial within 18 months of successful completion of the Phase I trial; file an NDA for FDA approval by February 2011; maintain and prosecute the patents relating to IPdR; and use our reasonable commercial efforts to implement a mutually-agreed upon plan for developing and commercializing IPdR. In the event we commit a material breach of our obligations under the license agreement, Yale and SUNY have the right to terminate the license agreement following notice to us and an opportunity to cure such breach (if capable of being cured).
| 22 | ||
Hana Financing Transactions and Available Capital
Hana was founded and initially financed by Paramount BioCapital Investments LLC. In February 2004, Hana completed a private placement of 1,987,846 shares of its common stock for aggregate gross proceeds of approximately $4.7 million.
In conjunction with and immediately prior to the completion of the Merger, discussed in Item 1 and 2 above, on July 21, 2004 Hana completed a private placement of 2,395,210 shares of its Series A Convertible Preferred Stock at a per share price of $3.34, resulting in gross proceeds of $8 million. The shares sold in the private placement were then immediately converted into and exchangeable for the same number of EMLR Series A shares. Pursuant to a lending arrangement with Paramount BioCapital and as a result of the private placement, Hana is required to repay approximately $800,000 of outstanding principal and accrued interest owed to Paramount BioCapital. The remaining proceeds will be used for working capital and general corporate purposes.
Additionally, Hana has secured grants for both of its initial product candidates, valued in the aggregate at approximately $12 million. Hana has received grants for the development of PT-523 totaling approximately $8 million from the National Cancer Institute, Harvard University and Dana-Farber Cancer Institute. These grants include the exclusive RAID (Rapid Access to Intervention Development) from the Developmental Therapeutics Program at the National Cancer Institute/National Institute of Health. This grant covered the cost of all pre-clinical efficacy and safety testing of PT-523. The RAID grant also supported manufacturing of sufficient material for the Phase I/II studies. Further, our Phase I/II study at the Dana-Farber Cancer Consortium, is being sponsored by the CTEP (Cancer Therapy Evaluation Program) at the NCI. This grant covers all of the expenses of the Phase I/II study. Grants for IPdR, totaling over an estimated $4 million in value, have been administered by the NCI, including RAID funding. Additionally, Case Western University has funded extensive pre-clinical studies relating to the technology.
Legal Proceedings
We are not subject to any pending legal proceeding, nor are we aware of any threatened claims against us.
Properties
We lease 5,942 square feet of office space in South San Francisco, California. This lease currently requires us to make monthly payments of approximately $10,100, which increase to approximately $10,400 in fiscal 2005. The lease expires December 31, 2005. We do not own any real property. We believe that our existing facilities are adequate to meet our needs for the foreseeable future.
Employees
We currently employee 9 persons, all of whom are based at our South San Francisco office. We believe the relationships with our employees is satisfactory.
| 23 | ||
(E) RISK FACTORS
In General. The purchase of shares of the Registrant’s common stock is very speculative and involves a very high degree of risk. An investment in the Registrant is suitable only for the persons who can afford the loss of their entire investment. Accordingly, investors should carefully consider the following risk factors, as well as other information set forth herein, in making an investment decision with respect to securities of the Registrant.
The market price of our common shares may fluctuate significantly in response to factors, some of which are beyond our control, such as:
- the announcement of new products or product enhancements by us or our competitors;
- developments concerning intellectual property rights and regulatory approvals;
- quarterly variations in our and our competitors’ results of operations;
- changes in earnings estimates or recommendations by securities analysts;
- developments in our industry; and
- general market conditions and other factors, including factors unrelated to our own operating performance.
Further, the stock market in general has recently experienced extreme price and volume fluctuations. Continued market fluctuations could result in extreme volatility in the price of our common shares, which could cause a decline in the value of our common shares. You should also be aware that price volatility might be worse if the trading volume of our common shares is low.
Because Hana Biosciences became public by means of a reverse merger, we may not be able to attract the attention of major brokerage firms.
Additional risks may exist since we became public through a “reverse merger.” Security analysts of major brokerage firms may not cover us since there is no incentive to brokerage firms to recommend the purchase of our common stock. No assurance can be given that brokerage firms will want to conduct any secondary offerings on our behalf in the future.
Trading of our common stock is conducted on the National Association of Securities Dealers’ Over-the-Counter Bulletin Board, or “OTC Bulletin Board.” This has adversely effected the liquidity of our securities, not only in terms of the number of securities that can be bought and sold at a given price, but also through delays in the timing of transactions and reduction in security analysts' and the media's coverage of us. This may result in lower prices for our common stock than might otherwise be obtained and could also result in a larger spread between the bid and asked prices for our common stock.
| 24 | ||
Because it is a “penny stock,” it will be more difficult for you to sell shares of our common stock.
In addition, our common stock is a “penny stock.” Broker-dealers who sell penny stocks must provide purchasers of these stocks with a standardized risk-disclosure document prepared by the SEC. This document provides information about penny stocks and the nature and level of risks involved in investing in the penny-stock market. A broker must also give a purchaser, orally or in writing, bid and offer quotations and information regarding broker and salesperson compensation, make a written determination that the penny stock is a suitable investment for the purchaser, and obtain the purchaser’s written agreement to the purchase. The penny stock rules may make it difficult for you to sell your shares of our stock. Because of the rules, there is less trading in penny stocks. Also, many brokers choose not to participate in penny-stock transactions. Accordingly, you may not always be able to resell shares of our common stock publicly at times and prices that you feel are appropriate.
Upon the effective time of the Registration Statement, there will be a significant number of shares of our common stock eligible for sale, which could depress the market price of our stock.
Following the effective date of the Registration Statement that we are required to file on behalf of the investors in the July 2004 Hana private placement (see Item 5A, above), a large number of our shares of common stock will become available for sale in the public market, which could harm the market price of our stock. Further, some or all of our shares may be offered from time to time in the open market pursuant to Rule 144, and these sales may have a depressive effect on the market for our common stock. In general, a person who has held restricted shares for a period of one year may, upon filing with the SEC a notification on Form 144, sell into the market common stock in an amount equal to the greater of 1 percent of the outstanding shares or the average weekly number of shares sold in the last four weeks prior to such sale. Such sales may be repeated once each three months, and any of the restricted shares may be sold by a non-affiliate after they have been held two years.
Risks Related to Our New Business
We currently have no product revenues and will need to raise additional capital to operate our business.
To date, we have generated no product revenues. Until we receive approval from the U.S. Federal Drug Administration, or “FDA,” and other regulatory authorities for our product candidates, we cannot sell our drugs and will not have product revenues. Therefore, for the foreseeable future, we will have to fund all of our operations and capital expenditures from the net proceeds of this Offering, cash on hand, licensing fees and grants. We will need additional financing in addition to the proceeds of the Offering, which may not be available on favorable terms, if at all. Assuming we sell the Minimum number of Shares offered hereby, we expect that we will have sufficient cash to fund our operations for at least the next 12 months. However, changes may occur that would consume our existing capital prior to that time, including the progress of our research and development efforts, changes in governmental regulation and acquisitions of additional product candidates. If we are unable to raise additional funds in the future on acceptable terms, or at all, we may be unable to complete planned pre-clinical and clinical trials or obtain approval of our product candidates from the FDA and other regulatory authorities. In addition, we could be forced to discontinue product development, reduce or forego sales and marketing efforts and forego attractive business opportunities. Any additional sources of financing will likely involve the sale of our equity securities, which will have a dilutive effect on our stockholders.
We are not currently profitable and may never become profitable.
We have a history of losses and expect to incur substantial losses and negative operating cash flow for the foreseeable future, and we may never achieve or maintain profitability. Even if we succeed in developing and commercializing one or more of our product candidates, we expect to incur substantial losses for the foreseeable future and may never become profitable. We also expect to continue to incur significant operating and capital expenditures and anticipate that our expenses will increase substantially in the foreseeable future as we:
| 25 | ||
- continue to undertake pre-clinical development and clinical trials for our current and new product candidates;
- seek regulatory approvals for our product candidates;
- implement additional internal systems and infrastructure; and
- hire additional personnel.
We also expect to experience negative cash flow for the foreseeable future as we fund our operating losses and capital expenditures. As a result, we will need to generate significant revenues in order to achieve and maintain profitability. We may not be able to generate these revenues or achieve profitability in the future. Our failure to achieve or maintain profitability could negatively impact the value of our stock.
We have a limited operating history upon which to base an investment decision.
Hana is a development-stage company that was founded in 2002. To date, we have not demonstrated an ability to perform the functions necessary for the successful commercialization of any of our product candidates. The successful commercialization of our product candidates will require us to perform a variety of functions, including:
- continuing to undertake pre-clinical development and clinical trials;
- participating in regulatory approval processes;
- formulating and manufacturing products; and
- conducting sales and marketing activities.
Our operations have been limited to organizing and staffing our company, acquiring, developing and securing our proprietary technology and undertaking, through third parties, pre-clinical trials and clinical trials of our principal product candidates. These operations provide a limited basis for you to assess our ability to commercialize our product candidates and the advisability of investing in our securities.
We may not obtain the necessary U.S. or worldwide regulatory approvals to commercialize our product candidates.
We will need FDA approval to commercialize our product candidates in the U.S. and approvals from the FDA equivalent regulatory authorities in foreign jurisdictions to commercialize our product candidates in those jurisdictions. In order to obtain FDA approval of any of our product candidates, we must submit to the FDA a New Drug Application, or NDA, demonstrating that the product candidate is safe for humans and effective for its intended use. This demonstration requires significant research and animal tests, which are referred to as pre-clinical studies, as well as human tests, which are referred to as clinical trials. Satisfaction of the FDA’s regulatory requirements typically takes many years, depends upon the type, complexity and novelty of the product candidate and requires substantial resources for research, development and testing. We cannot predict whether our research and clinical approaches will result in drugs that the FDA considers safe for humans and effective for indicated uses. The FDA has substantial discretion in the drug approval process and may require us to conduct additional pre-clinical and clinical testing or to perform post-marketing studies. The approval process may also be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals may:
| 26 | ||
- delay commercialization of, and our ability to derive product revenues from, our product candidates;
- impose costly procedures on us; and
- diminish any competitive advantages that we may otherwise enjoy.
Even if we comply with all FDA requests, the FDA may ultimately reject one or more of our New Drug Applications (NDAs). We cannot be sure that we will ever obtain regulatory clearance for our product candidates. Failure to obtain FDA approval of any of our product candidates will severely undermine our business by reducing our number of salable products and, therefore, corresponding product revenues.
In foreign jurisdictions, we must receive approval from the appropriate regulatory authorities before we can commercialize our drugs. Foreign regulatory approval processes generally include all of the risks associated with the FDA approval procedures described above. We cannot assure you that we will receive the approvals necessary to commercialize our product candidates for sale outside the United States.
Our product candidates are in early stages of clinical trials.
Our product candidate is in an early stage of development and requires extensive clinical testing. In 2003, the FDA approved our Investigational New Drug application, or “IND,” for PT-523 and we have only recently initiated a Phase I clinical trial at Dana-Farber Cancer Institute, Massachusetts General Hospital and Beth-Israel Deaconess Hospital. The IND for IPdR is still under development and we do not expect to commence a Phase I trial until at least late 2004. We cannot predict with any certainty if or when we might submit an NDA for regulatory approval of our product candidates or whether any such NDA will be accepted.
Clinical trials are very expensive, time-consuming and difficult to design and implement.
Human clinical trials are very expensive and difficult to design and implement, in part because they are subject to rigorous regulatory requirements. The clinical trial process is also time consuming. We estimate that clinical trials of our product candidates will take at least several years to complete. Furthermore, failure can occur at any stage of the trials, and we could encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed by several factors, including:
- unforeseen safety issues;
- determination of dosing issues;
- lack of effectiveness during clinical trials;
- slower than expected rates of patient recruitment;
- inability to monitor patients adequately during or after treatment; and
- inability or unwillingness of medical investigators and institutional review boards to follow our clinical protocols.
| 27 | ||
In addition, we or the FDA may suspend our clinical trials at any time if it appears that we are exposing participants to unacceptable health risks or if the FDA finds deficiencies in our IND submissions or the conduct of these trials.
The results of our clinical trials may not support our product candidate claims.
Even if our clinical trials are completed as planned, we cannot be certain that their results will support our product candidate claims. Success in pre-clinical testing and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the results of later clinical trials will replicate the results of prior clinical trials and pre-clinical testing. The clinical trial process may fail to demonstrate that our product candidates are safe for humans and effective for indicated uses. This failure would cause us to abandon a product candidate and may delay development of other product candidates. Any delay in, or termination of, our clinical trials will delay the filing of our NDAs with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues. In addition, our clinical trials involve a small patient population. Because of the small sample size, the results of these early clinical trials may not be indicative of future results.
Physicians and patients may not accept and use our drugs.
Even if the FDA approves our product candidates, physicians and patients may not accept and use them. Acceptance and use of our product will depend upon a number of factors including:
- perceptions by members of the health care community, including physicians, about the safety and effectiveness of our drugs;
- pharmacological benefit and cost-effectiveness of our product relative to competing products;
- availability of reimbursement for our products from government or other healthcare payers; and
- effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any.
Because we expect sales of our current product candidates, if approved, to generate substantially all of our product revenues for the foreseeable future, the failure of any of these drugs to find market acceptance would harm our business and could require us to seek additional financing.
Our drug-development program depends upon third-party researchers who are outside our control.
We depend upon independent investigators and collaborators, such as universities and medical institutions, to conduct our pre-clinical and clinical trials under agreements with us. These collaborators are not our employees and we cannot control the amount or timing of resources that they devote to our programs. These investigators may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking such programs ourselves. If outside collaborators fail to devote sufficient time and resources to our drug-development programs, or if their performance is substandard, the approval of our FDA applications, if any, and our introduction of new drugs, if any, will be delayed. These collaborators may also have relationships with other commercial entities, some of whom may compete with us. If our collaborators assist our competitors at our expense, our competitive position would be harmed.
| 28 | ||
We rely exclusively on third parties to formulate and manufacture our product candidate.
We have no experience in drug formulation or manufacturing and do not intend to establish our own manufacturing facilities. We lack the resources and expertise to formulate or manufacture our own product candidates. We currently have no contract for the manufacture of our product candidates. We intend to contract with one or more manufacturers to manufacture, supply, store and distribute drug supplies for our clinical trials.If any of our product candidates receive FDA approval, we will rely on one or more third-party contractors to manufacture our drugs. Our anticipated future reliance on a limited number of third-party manufacturers, exposes us to the following risks:
- We may be unable to identify manufacturers on acceptable terms or at all because the number of potential manufacturers is limited and the FDA must approve any replacement contractor. This approval would require new testing and compliance inspections. In addition, a new manufacturer would have to be educated in, or develop substantially equivalent processes for, production of our products after receipt of FDA approval, if any.
- Our third-party manufacturers might be unable to formulate and manufacture our drugs in the volume and of the quality required to meet our clinical needs and commercial needs, if any.
- Our future contract manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store and distribute our products.
- Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA, the DEA, and corresponding state agencies to ensure strict compliance with good manufacturing practice and other government regulations and corresponding foreign standards. We do not have control over third-party manufacturers’ compliance with these regulations and standards, but we will be ultimately responsible for any of their failures.
- If any third-party manufacturer makes improvements in the manufacturing process for our products, we may not own, or may have to share, the intellectual property rights to the innovation.
Each of these risks could delay our clinical trials, the approval, if any, of our product candidates by the FDA, or the commercialization of our product candidates or result in higher costs or deprive us of potential product revenues.
We have no experience selling, marketing or distributing products and no internal capability to do so.
We currently have no sales, marketing or distribution capabilities. While we intend to have a role in the commercialization of our products, we do not anticipate having the resources in the foreseeable future to globally develop sales and marketing capabilities for all of our proposed products. Our future success depends, in part, on our ability to enter into and maintain collaborative relationships with other companies having sales, marketing and distribution capabilities, the collaborator’s strategic interest in the products under development and such collaborator’s ability to successfully market and sell any such products. We intend to pursue collaborative arrangements regarding the sales and marketing of our products, however, there can be no assurance that we will be able to establish or maintain such collaborative arrangements, or if able to do so, that they will have effective sales forces. To the extent that we decide not to, or are unable to, enter into collaborative arrangements with respect to the sales and marketing of our proposed products, significant capital expenditures, management resources and time will be required to establish and develop an in-house marketing and sales force with technical expertise. There can also be no assurance that we will be able to establish or maintain relationships with third party collaborators or develop in-house sales and distribution capabilities. To the extent that we depend on third parties for marketing and distribution, any revenues we receive will depend upon the efforts of such third parties, as well as the terms of our agreements with such third parties, which cannot be predicted at this early stage of our development. There can be no assurance that such efforts will be successful. In addition, there can also be no assurance that we will be able to market and sell our products in the United States or overseas.
| 29 | ||
If we cannot compete successfully for market share against other drug companies, we may not achieve sufficient product revenues and our business will suffer.
The market for our product candidates is characterized by intense competition and rapid technological advances. If our product candidates receive FDA approval, they will compete with a number of existing and future drugs and therapies developed, manufactured and marketed by others. Existing or future competing products may provide greater therapeutic convenience or clinical or other benefits for a specific indication than our products, or may offer comparable performance at a lower cost. If our products fail to capture and maintain market share, we may not achieve sufficient product revenues and our business will suffer.
We will compete against fully integrated pharmaceutical companies and smaller companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. In addition, many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs and have substantially greater financial resources than we do, as well as significantly greater experience in:
- developing drugs;
- undertaking pre-clinical testing and human clinical trials;
- obtaining FDA and other regulatory approvals of drugs;
- formulating and manufacturing drugs; and
- launching, marketing and selling drugs.
Developments by competitors may render our products or technologies obsolete or non-competitive.
Companies that currently sell both generic and proprietary compounds for the treatment of cancer include, among others, Pfizer (trimetrexate),Eli Lilly & Co. (pemetrexed),Novartis (edatrexate), and Allos (PDX). Alternative technologies are being developed to treat cancer and immunological disease, several of which are in advanced clinical trials. (See: “ITEM 5(D) - Description of Business of Hana Biosciences, Inc. - PT-523: Competition;” “- IPDR Competition”). In addition, companies pursuing different but related fields represent substantial competition. Many of these organizations have substantially greater capital resources, larger research and development staffs and facilities, longer drug development history in obtaining regulatory approvals and greater manufacturing and marketing capabilities than we do. These organizations also compete with us to attract qualified personnel, parties for acquisitions, joint ventures or other collaborations.
| 30 | ||
If we fail to adequately protect or enforce our intellectual property rights or secure rights to patents of others, the value of our intellectual property rights would diminish.
Our success, competitive position and future revenues will depend in part on our ability and the abilities of our licensors to obtain and maintain patent protection for our products, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties.
To date, through our license agreements for PT-523 and IPdR, we hold certain exclusive patent rights, including rights under U.S. patents and U.S. patent applications. We also have patent applications pending in several foreign jurisdictions. We anticipate filing additional patent applications both in the U.S. and in other countries, as appropriate. However, we cannot predict:
- the degree and range of protection any patents will afford us against competitors, including whether third parties will find ways to invalidate or otherwise circumvent our licensed patents;
- if and when patents will issue;
- whether or not others will obtain patents claiming aspects similar to those covered by our licensed patents and patent applications; or
- whether we will need to initiate litigation or administrative proceedings which may be costly whether we win or lose.
Our success also depends upon the skills, knowledge and experience of our scientific and technical personnel, our consultants and advisors as well as our licensors and contractors. To help protect our proprietary know-how and our inventions for which patents may be unobtainable or difficult to obtain, we rely on trade secret protection and confidentiality agreements. To this end, we require all of our employees to enter into agreements which prohibit the disclosure of confidential information and, where applicable, require disclosure and assignment to us of the ideas, developments, discoveries and inventions important to our business. These agreements may not provide adequate protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure or the lawful development by others of such information. If any of our trade secrets, know-how or other proprietary information is disclosed, the value of our trade secrets, know-how and other proprietary rights would be significantly impaired and our business and competitive position would suffer.
If we infringe the rights of third parties we could be prevented from selling products, forced to pay damages, and defend against litigation.
If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we may have to:
- obtain licenses, which may not be available on commercially reasonable terms, if at all;
- redesign our products or processes to avoid infringement;
- stop using the subject matter claimed in the patents held by others, which could cause us to lose the use of one or more of our product candidates;
- pay damages; or
- defend litigation or administrative proceedings which may be costly whether we win or lose, and which could result in a substantial diversion of our valuable management resources.
| 31 | ||
Our license agreements relating to our product candidates are subject to termination in the event we commit a material breach.
Our license agreements relating to PT-523 and IPdR are subject to termination by our licensors in the event we materially breach those agreements. With respect to the PT-523 license, our licensor may terminate the agreement, after giving us notice and an opportunity to cure, if we commit a material breach, including failing to make a scheduled milestone or other payment when due. The agreement also provides that it may be terminated if we become involved in a bankruptcy, insolvency or similar proceeding. Our license agreement for IPdR contains similar provisions. In the event these license agreements are terminated, we will lose all of our rights to develop and commercialize the applicable product candidate covered by such license, which would significantly harm our business and future prospects. See “ITEM 5(D) – License Agreements and Intellectual Property – License Agreements.”
Our ability to generate product revenues will be diminished if our drugs sell for inadequate prices or patients are unable to obtain adequate levels of reimbursement.
Our ability to commercialize our drugs, alone or with collaborators, will depend in part on the extent to which reimbursement will be available from:
- government and health administration authorities;
- private health maintenance organizations and health insurers; and
- other healthcare payers.
Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Healthcare payers, including Medicare, are challenging the prices charged for medical products and services. Government and other healthcare payers increasingly attempt to contain healthcare costs by limiting both coverage and the level of reimbursement for drugs. Even if our product candidates are approved by the FDA, insurance coverage may not be available, and reimbursement levels may be inadequate, to cover our drugs. If government and other healthcare payers do not provide adequate coverage and reimbursement levels for any of our products, once approved, market acceptance of our products could be reduced.
We may not successfully manage our growth.
Our success will depend upon the expansion of our operations and the effective management of our growth, which will place a significant strain on our management and on our administrative, operational and financial resources. To manage this growth, we must expand our facilities, augment our operational, financial and management systems and hire and train additional qualified personnel. If we are unable to manage our growth effectively, our business would be harmed.
We may be exposed to liability claims associated with the use of hazardous materials and chemicals.
Our research and development activities may involve the controlled use of hazardous materials and chemicals. Although we believe that our safety procedures for using, storing, handling and disposing of these materials comply with federal, state and local laws and regulations, we cannot completely eliminate the risk of accidental injury or contamination from these materials. In the event of such an accident, we could be held liable for any resulting damages and any liability could materially adversely effect our business, financial condition and results of operations. In addition, the federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous or radioactive materials and waste products may require us to incur substantial compliance costs that could materially adversely affect our business, financial condition and results of operations.
| 32 | ||
We rely on key executive officers and scientific and medical advisors, and their knowledge of our business and technical expertise would be difficult to replace.
We are highly dependent on our principal scientific, regulatory and medical advisors. We do not have “key person” life insurance policies for any of our officers. The loss of the technical knowledge and management and industry expertise of any of our key personnel could result in delays in product development, loss of customers and sales and diversion of management resources, which could adversely affect our operating results.
If we are unable to hire additional qualified personnel, our ability to grow our business may be harmed.
We will need to hire additional qualified personnel with expertise in pre-clinical testing, clinical research and testing, government regulation, formulation and manufacturing and sales and marketing. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for such individuals, particularly in the San Francisco Bay Area, is intense, and we cannot be certain that our search for such personnel will be successful. Attracting and retaining qualified personnel will be critical to our success.
We may incur substantial liabilities and may be required to limit commercialization of our products in response to product liability lawsuits.
The testing and marketing of medical products entail an inherent risk of product liability. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our products. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop, alone or with collaborators. We currently do not carry clinical trial insurance or product liability insurance. Although we intend to obtain clinical trial insurance prior to the commencement of any clinical trials, we, or any collaborators, may not be able to obtain insurance at a reasonable cost, if at all. Even if our agreements with any future collaborators entitle us to indemnification against losses, such indemnification may not be available or adequate should any claim arise.
(F) DESCRIPTION OF CAPITAL STOCK
As discussed in Item 1(A), above, in order to facilitate the Merger, the Registrant created two new classes of preferred stock, designated as Series A Convertible Preferred Stock and Series B Convertible Preferred Stock. A description of the terms of each class is set forth below.
EMLR Series A Convertible Preferred Stock
Each EMLR Series A share shall have one vote on all matters submitted to the holders of common stock for each share of common stock which such preferred stock would be voted on an as-converted basis as of the date of such vote. No amendment to the preferences or rights of the EMLR Series A shares may be made without the affirmative vote of the holders of at least a majority of outstanding shares of such class. Each EMLR Series A share shall be convertible into one share of our common stock at any time at the option of the holder, subject to adjustments for stock splits, recapitalizations and the like.
| 33 | ||
The EMLR Series A shares shall rank senior to our common stock and shall have a liquidation preference equal to $3.34 per share. In the event of any liquidation, dissolution or winding up of the Company, holders of the EMLR Series A shares will be entitled to a one-time liquidation preference before any distribution is made to common stockholders in an amount equal to $3.34 per share. The EMLR Series A shares shall automatically convert upon the effective date of the Registration Statement covering the resale under the Securities Act of the common shares issuable upon conversion of the EMLR Series A shares.
Holders of EMLR Series A shares are not entitled to any dividends. As long as there is any Preferred Stock outstanding, no dividends may be declared on our common stock without the consent of the holders of a majority of the Preferred Stock.
The Certificate of Designation attached to this Form 8-K as Exhibit 4.1, and incorporated by reference herein, contains a complete description of the rights, terms and preferences of the EMLR Series A shares.
EMLR Series B Convertible Preferred Stock
In connection with the Merger, the holders of Hana’s common stock received, in exchange for such shares, one share of EMLR’s Series B Preferred Stock for each share of Hana common stock held by such holder. Each share of EMLR Series B Preferred Stock shall have one vote on all matters submitted to the holders of EMLR common shares for each share of EMLR common stock issuable upon conversion of the EMLR Series B shares as of the date of such vote and assuming all conditions to conversion are satisfied. No amendment to the preferences or rights of the EMLR Series B Preferred Stock may be made without the affirmative vote of the holders of at least a majority of outstanding shares of EMLR Series B Preferred Stock.
Holders of EMLR Series B Preferred Stock will not be entitled to any dividends. The EMLR Series B Preferred Stock shall rank junior to the EMLR Series A Preferred Stock and senior to EMLR’s common shares. In the event of any liquidation, dissolution or winding up of EMLR, holders of the EMLR Series B Preferred Stock will be entitled to a one-time liquidation preference before any distribution is made to the common stockholders in an amount equal to $3.34 per share, but only after satisfaction of the liquidation preference payable to holders of the EMLR Series A Preferred Stock. The EMLR Series B Preferred Stock shall automatically convert into EMLR common stock upon the effective date of a recapitalization event that provides a sufficient number of authorized shares of EMLR common stock as to allow for the issuance of common shares upon the conversion of both the EMLR Series A and Series B Preferred Stock.
The Certificate of Designation attached to this Form 8-K as Exhibit 4.2, and incorporated by reference herein, contains a complete description of the rights, terms and preferences of the EMLR Series B Preferred Stock.
(G) REINCORPORATION
The Registrant intends to call a special meeting of shareholders as soon as practicable for the purpose of considering reincorporating under the laws of the State of Delaware. If approved, it is expected that such reincorporation would be effected by merging the Registrant with and into its wholly-owned subsidiary, Hana Biosciences, Inc., a Delaware corporation. In connection with such reincorporation merger, the surviving company would be named Hana Biosciences, Inc.
| 34 | ||
(H) SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth information known to the Registrant with respect to the beneficial ownership of each class of the Registrant’s capital stock as of July 21, 2004 for (1) each person known by the Registrant to beneficially own more than 5% of each class of the Registrant’s voting securities, (2) each executive officer, (3) each of the Registrant’s directors and (4) all of the Registrant’s executive officers and directors as a group. The number of shares beneficially owned is determined under rules promulgated by the SEC, and the information is not necessarily indicative of beneficial ownership for any other purpose. Under those rules, beneficial ownership includes any shares as to which the individual has sole or shared voting power or investment power and also any shares which the individual has the right to acquire within 60 days of the date hereof, through the exercise or conversion of any stock option, convertible security, warrant or other right. Including those shares in the tables does not, however, constitute an admission that the named stockholder is a direct or indirect beneficial owner of those shares. The table also presents the number of votes each person is entitled to cast at the Annual Meeting. Unless otherwise indicated, each person or entity named in the table has sole voting power and investment power (or shares that power with that person’s spouse) with respect to all shares of capital stock listed as owned by that person or entity. Unless otherwise indicated, the address of each of the following persons is 400 Oyster Point Blvd, Suite 215, South San Francisco, CA 94080.
Common Stock | Series A Preferred | Series B Preferred | Voting Power | ||||||
Name | Number | Pct. | Number | Pct. | Number | Pct. | Number | Pct. | |
Mark J. Ahn | 0 | — | 0 | — | 0 | — | 0 | — | |
Jonathan P. Serbin | 0 | — | 0 | — | 0 | — | 0 | — | |
Fred Vitale | 0 | — | 0 | — | 0 | — | 0 | — | |
John P. Iparraguirre | 0 | — | 0 | — | 0 | — | 0 | — | |
Arie J. Belldegrun | 0 | — | 0 | — | 0 | — | 0 | — | |
Isaac Kier(1) | 0 | — | 179,641 | 7.5 | 284,210 | 4.6 | 7,848,428 | 4.6 | |
Leon Rosenberg | 0 | — | 0 | — | 0 | — | 0 | — | |
Michael Weiser | 0 | — | 0 | — | 390,525 | 6.3 | 6,608,001 | 3.9 | |
All directors and officers as a group | 0 | — | 179,641 | 7.5 | 674,735 | 10.9 | 14,456,429 | 8.5 | |
R&R Biotech, LLC 330 Madison Avenue New York, NY 10017 | 16,547,694 | 66.2 | — | — | — | — | 16,547,694 | 9.7 | |
J. Jay Lobell(2) 365 West End Avenue New York, NY 10024 | 0 | — | 0 | — | 1,921,987 | 31.1 | 32,521,584 | 19.1 | |
Atlas Equity I, Ltd.(3) 181 W. Madison, Ste 3600 Chicago, IL 60602 | 0 | — | 935,000 | 3.9 | 42,105 | * | 16,533,412 | 9.7 | |
__________
| * | less than 1 percent |
| (1) | Includes (a) 119,761 shares of Series A Preferred Stock and 200,000 shares of Series B Preferred Stock held by Coqui Capital Partners, LP, of which Mr. Kier is the general partners, (b) 29,940 shares of Series A Preferred Stock held by JIJ Investments, of which Mr. Kier is a partner, and (c) 84,210 shares of Series B Preferred Stock held by Kier Family Partners, L.P., of which Mr. Kier is general partner. |
| (2) | Includes 1,912,224 shares of Series B Convertible Stock held by various trusts established for the benefit of Lindsay A. Rosenwald, M.D. and members of Dr. Rosenwald’s family, of which Mr. Lobell is trustee or investment manager. |
| (3) | Includes 42,105 shares of Series A Preferred Stock held by Jacob Gotlieb, a principal of an entity affiliated with Atlas Equity I, Ltd. |
| 35 | ||
ITEM 7. FINANCIAL STATEMENTS, PRO FORMA FINANCIAL INFORMATION AND EXHIBITS
(a) Financial Statements of Business Acquired. The financial statements of Hana Biosciences, Inc. and will be filed by amendment to this Current Report on Form 8-K on or before October 4, 2004.
(b) Pro forma financial statements will be filed by amendment to this Current Report on or before October 4, 2004.
(c) Exhibits.
2.1 Agreement and Plan of Merger by and among the Registrant, EMLR Acquisition Corp. and Hana Biosciences, Inc. (f/k/a Hudson Health Sciences, Inc.) dated June 17, 2004 (incorporated by reference to Exhibit 2.0 to the Registrant’s Form 8-K filed on June 24, 2004).
2.2 Stock Purchase Agreement dated June 17, 2004 by and among the Registrant, R&R Biotech, LLC, Turquoise Partners, LLC, Chase Financing, Inc. and The Washington Trust (incorporated by reference to Exhibit 2.1 to the Registrant’s Form 8-K filed on June 24, 2004).
2.3 Amendment No. 1 dated July 16, 2004 to Stock Purchase Agreement dated June 17, 2004 by and among the Registrant, R&R Biotech, LLC, Turquoise Partners, LLC, Chase Financing, Inc. and The Washington Trust.
4.1 Certificate of Designation of Series A Convertible Preferred Stock.
4.2 Certificate of Designation of Series B Convertible Preferred Stock.
99.1 Press Release dated July 21, 2004.
______________________________________________
SIGNATURES
Pursuant to the requirements of the Securities and Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned hereunto duly authorized.
EMAIL REAL ESTATE.COM, INC. | ||
| | | |
| Date: August 4, 2004 | By: | /s/ Mark J. Ahn |
| Mark J. Ahn, Ph.D. President and Chief Executive Officer | ||
| 36 | ||
EXHIBIT INDEX
| 2.3 | Amendment No. 1 dated July 16, 2004 to Stock Purchase Agreement dated June 17, 2004 by and among the Registrant, R&R Biotech, LLC, Turquoise Partners, LLC, Chase Financing, Inc. and The Washington Trust. |
| 4.1 | Certificate of Designation of Series A Convertible Preferred Stock. |
| 4.2 | Certificate of Designation of Series B Convertible Preferred Stock. |
| 99.1 | Press Release dated July 21, 2004. |
| 37 | ||