Item 1. Business
Overview
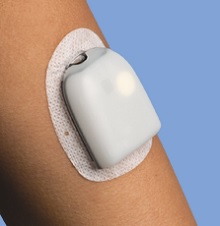
We are primarily engaged in the development, manufacturing and sale of our proprietary OmniPod Insulin Management System (the “OmniPod System”), an innovative, discreet and easy-to-use continuous insulin delivery system for people with insulin-dependent diabetes. The OmniPod System features a small, lightweight, self-adhesive disposable tubeless OmniPod device which is worn on the body for approximately three days at a time and its wireless companion, the handheld Personal Diabetes Manager (“PDM”). Conventional insulin pumps require people with insulin-dependent diabetes to learn to use, manage and wear a number of cumbersome components, including up to 42 inches of tubing. In contrast, the OmniPod System features only two discreet, easy-to-use devices that eliminate the need for a bulky pump, tubing and separate blood glucose meter, provides for virtually pain-free automated cannula insertion, communicates wirelessly and integrates a blood glucose meter. We believe that the OmniPod System’s unique proprietary design and features allow people with insulin-dependent diabetes to manage their diabetes with unprecedented freedom, comfort, convenience, and ease.
We began commercial sale of the OmniPod System in the United States in 2005. We sell the OmniPod System and other diabetes management supplies in the United States through direct sales to customers or through our distribution partners. The OmniPod System is currently available in multiple countries in Europe, Canada and Israel. In July 2015, we executed an asset purchase agreement with GlaxoSmithKline ("GSK") whereby we acquired assets associated with the Canadian distribution of our products and we assumed the distribution, sales, marketing, training and support activities for the OmniPod system in Canada. Additional information regarding this acquisition is provided in note 3 to the consolidated financial statements.
In addition to using the OmniPod® for insulin delivery, we also partner with global pharmaceutical and biotechnology companies to tailor the OmniPod technology platform for the delivery of subcutaneous drugs across multiple therapeutic areas.
In June 2011, we acquired Neighborhood Holdings, Inc. and its wholly-owned subsidiaries (collectively, “Neighborhood Diabetes”). Through Neighborhood Diabetes, we provided customers with blood glucose testing supplies, traditional insulin pumps, pump supplies and pharmaceuticals, processing claims as either durable medical equipment or through pharmacy benefits. In February 2016, we sold Neighborhood Diabetes to Liberty Medical LLC ("Liberty Medical"). Additional information regarding the sale of Neighborhood Diabetes is provided in note 18 to the consolidated financial statements.
Insulet Corporation is a Delaware corporation formed in 2000. Our principal offices are located at 600 Technology Park Drive, Suite 200, Billerica, Massachusetts 01821, and our telephone number is (978) 600-7000. Our website address is http://www.insulet.com. We make available, free of charge, on or through our website, our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, proxy statements and any amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, as soon as reasonably practicable after such material is electronically filed with or furnished to the Securities and Exchange Commission. The information on our website is not part of this Current Report on Form 8-K.
Our Market
Diabetes is a chronic, life-threatening disease for which there is no known cure. Diabetes is caused by the body’s inability to produce or effectively utilize the hormone insulin. This inability prevents the body from adequately regulating blood glucose levels. Glucose, the primary source of energy for cells, must be maintained at certain concentrations in the blood in order to permit optimal cell function and health. In people with diabetes, blood glucose levels fluctuate between very high levels, a condition known as hyperglycemia, and very low levels, a condition called hypoglycemia. Hyperglycemia can lead to serious short-term complications, such as confusion, vomiting, dehydration and loss of consciousness and long-term complications, such as blindness, kidney disease, nervous system disease, occlusive vascular diseases, stroke and cardiovascular disease, or death. Hypoglycemia can lead to confusion, loss of consciousness or death.
Diabetes is typically classified as either Type 1 or Type 2:
| |
| • | Type 1 diabetes is characterized by the body’s nearly complete inability to produce insulin. It is frequently diagnosed during childhood or adolescence. Individuals with Type 1 diabetes require daily insulin therapy, typically administered via injections or continuous infusion through pump therapy, to survive. |
| |
| • | Type 2 diabetes, the more common form of diabetes, is characterized by the body’s inability to either properly utilize insulin or produce enough insulin. Historically, Type 2 diabetes has occurred in later adulthood, but its incidence is increasing among the younger population, due primarily to increasing childhood obesity. Initially, many people with Type 2 diabetes attempt to manage their diabetes with improvements in diet, exercise and/or oral medications. As their diabetes advances, some patients progress to multiple drug therapy, which often includes insulin therapy. Guidelines, including those published by the American Diabetes Association in 2014, suggest more aggressive treatment for people with Type 2 diabetes, including the early adoption of insulin therapy and more frequent testing. It is now becoming more accepted for insulin therapy to be started earlier in people with Type 2 diabetes, and, in some cases, as part of the initial treatment. |
Throughout this Current Report on Form 8-K, we refer to both Type 1 diabetes and insulin-requiring Type 2 diabetes as insulin-dependent diabetes.
The OmniPod Delivery System is an automated drug delivery platform. In addition to using the Pod for insulin delivery we have also partnered with multiple pharmaceutical and biotechnology companies that utilize a customized form of the OmniPod system to deliver a drug over a specified interval of time, at a certain administered volume.
Managing Diabetes
Diabetes Management Challenges
Diabetes is often frustrating and difficult for patients to manage. Blood glucose levels can be affected by the carbohydrate and fat content of meals, exercise, stress, illness or impending illness, hormonal releases, variability in insulin absorption and changes in the effects of insulin on the body. For people with insulin-dependent diabetes, many corrections, consisting of the administration of additional insulin or ingestion of additional carbohydrates, are needed throughout the day in order to maintain blood glucose levels within normal ranges. Achieving this result can be very difficult without multiple daily injections of insulin or the use of continuous subcutaneous insulin infusion (“CSII”) therapy. Patients attempting to control their blood glucose levels tightly to prevent the long-term complications associated with fluctuations in blood glucose levels are at greater risk for overcorrection and the resultant hypoglycemia. As a result, many patients have difficulty managing their diabetes optimally. Additionally, the time spent in managing diabetes, the swings in blood glucose levels and the fear of hypoglycemia can all render diabetes management overwhelming to patients and their families.
Current Insulin Therapy
People with insulin-dependent diabetes need a continuous supply of insulin, known as basal insulin, to provide for background metabolic needs. In addition to basal insulin, people with insulin-dependent diabetes require supplemental insulin, known as bolus insulin, to compensate for carbohydrates ingested during meals or snacks or for a high blood glucose level.
There are three primary types of insulin therapy practiced today: conventional therapy; multiple daily injection (“MDI”) therapy using syringes or insulin pens; and CSII therapy using insulin pumps. Both MDI and CSII therapies are considered intensive insulin management therapies.
Many healthcare professionals believe that intensive insulin management therapies are superior to conventional therapies in delaying the onset and reducing the severity of diabetes-related complications. As a result, we believe that the use of intensive insulin management therapies has significantly expanded over the past decade, and that many Type 1 patients manage their diabetes using an intensive insulin management therapy. A significantly smaller percentage of people with insulin-requiring Type 2 diabetes manage their diabetes using an intensive insulin management therapy.
The OmniPod System
The OmniPod Insulin Management System is an innovative continuous insulin delivery system that provides all the proven benefits of CSII therapy in a way no conventional insulin pump can. The System's innovative design and features allows people with insulin-dependent diabetes to live their life, and manage their diabetes, with unprecedented freedom, comfort, convenience, and ease.
The long-term health benefits of better blood glucose control are well known. Maintaining near-normal blood glucose levels can help people with insulin-dependent diabetes live a longer, healthier life with fewer diabetes-related complications. The OmniPod System also has many practical, everyday benefits, including convenience, freedom, flexibility and ease of use.
Continuous insulin delivery at preset rates eliminates the need for injections and the interruptions that come with them. In addition, with the OmniPod System, insulin delivery can be changed with the press of a button to adapt to snacks or unexpected changes in daily routine.
The OmniPod System works much like the pancreas of a person without diabetes by delivering insulin in two ways:
| |
| • | A small, constant background supply of insulin (called a basal rate) is delivered automatically at a programmed rate, all day and night. |
| |
| • | An extra dose of insulin (called a bolus) can be delivered when a patient needs it to match the carbohydrates in a meal or snacks or to correct high blood glucose. |
The OmniPod System is a discreet two part design, the OmniPod (Pod) and the PDM, that eliminates the need for the external tubing required with conventional pumps.
| |
| • | The Pod is a small, lightweight, self-adhesive device that the patient fills with insulin and wear directly on the body. The Pod delivers precise, personalized doses of insulin into the body through a small flexible tube (called a cannula), based on instructions that the patient programs into the Pod's wireless companion, the PDM. |
| |
| • | The PDM is a wireless, handheld device that programs the Pod with the patient's personalized insulin-delivery instructions, wirelessly monitors the Pod's operation and includes a FreeStyle® blood glucose meter. |
We have designed the OmniPod System to fit within the normal daily routines of patients. The OmniPod System requires the fewest steps to start insulin delivery of all CSII therapies on the market by automating much of the process. In addition, the OmniPod System consists of just two devices, as opposed to up to seven for conventional insulin pumps. As a result, the OmniPod System is easy for patients to use, which reduces the training burden on healthcare professionals. We believe that the OmniPod System’s overall ease of use makes it very attractive to people with insulin-dependent diabetes. We also believe that the OmniPod System’s ease of use and substantially lower training burden helps to redefine which diabetes patients are appropriate for CSII therapy, enabling healthcare professionals to prescribe CSII therapy to a broader pool of patients.
The OmniPod System’s unique patented design and proprietary manufacturing process have enabled us to provide CSII therapy at a relatively low up-front investment compared to conventional insulin pumps. We believe that our pricing model reduces the risk of investing in CSII therapy for third-party payors and makes CSII therapy much more accessible for people with insulin-dependent diabetes.
Research and Development
Our current research and development efforts are primarily focused on the development of mobile applications for the OmniPod, including a Bluetooth-enabled PDM, integration with continuous glucose monitoring technology, an artificial pancreas platform, and development to support the use of concentrated insulin for Type I and Type II patients with higher insulin-requirements. In addition to insulin delivery, we continue to work with multiple pharmaceutical and biotechnology companies on alternative uses for our OmniPod System technology to use our technology as a delivery platform for a range of different pharmaceuticals.
Manufacturing and Quality Assurance
We believe a key contributing factor to the overall attractiveness of the OmniPod System is the disposable OmniPod continuous insulin delivery device. In order to manufacture sufficient volumes and achieve a cost-effective per unit production price for the OmniPod, we have designed the OmniPod to be manufactured through a semi-automated process.
We are currently producing the OmniPod on varying degrees of semi-automated manufacturing lines at a facility in China, operated by a subsidiary of Flextronics International Ltd. (“Flextronics”). We purchase OmniPods pursuant to our agreement with Flextronics. Under the agreement, Flextronics agrees to supply us with OmniPods at a price reflective of the forecast that we provide pursuant to the agreement. The current term of the agreement expires in December 2017 and is subject to automatic renewal for one-year successive terms subsequently. It may be terminated by either party upon compliance with certain advance written notice provisions that are intended to provide the parties with sufficient time to make alternative arrangements.
We seek to increase manufacturing capacity and reduce the per-unit production cost for the OmniPod. We continue to invest in our manufacturing capacity in order to meet our expected 2016 demand and beyond for the OmniPod.
We rely on outside vendors for the supply of components, sub-assemblies, and various services used in the manufacture of the OmniPod System. Although a number of these suppliers are sole-source suppliers, we continue to focus on identifying alternate supply sources and duplicate custom tooling.
All outside vendors produce the components to our specifications and they are audited periodically by our Quality Assurance Department to ensure conformity with the specifications, policies and procedures for the OmniPods. Our Quality Assurance Department also inspects and tests the OmniPods at various steps in the manufacturing cycle to facilitate compliance with our stringent specifications. We have received approval of our Quality Management System from the BSI Group London, U.K., an accredited Notified Body for CE Marking and the International Standards Organization (“ISO”). Processes utilized in the manufacture, test and release of the OmniPod have been verified and validated as required by the U.S. Federal Food and Drug Administration ("FDA") and other regulatory bodies. As a medical device manufacturer and distributor, our manufacturing facilities and the facilities of our suppliers and sterilizer are subject to periodic inspection by the FDA, our notified body and certain corresponding state agencies.
Intellectual Property
We believe that to maintain a competitive advantage, we must develop and preserve the proprietary aspect of our technologies. We rely on a combination of copyright, patent, trademark, trade secret and other intellectual property laws, non-disclosure agreements and other measures to protect our proprietary rights. Currently, we require our employees, consultants and advisors to execute non-disclosure agreements in connection with their employment, consulting or advisory relationships with us, where appropriate. We also require our employees, consultants and advisors who we expect to work on our current or future products to agree to disclose and assign to us all inventions conceived during their work with us that are developed using our property or which relate to our business. Despite any measures taken to protect our intellectual property, unauthorized parties may attempt to copy aspects of the OmniPod System or to obtain and use information that we regard as proprietary.
Patents. As of December 31, 2015, we had obtained 15 issued United States patents, and had 10 additional pending United States patent applications. We believe it will take up to four years, and possibly longer, for the most recent of these U.S. patent applications to result in issued patents. We are also seeking patent protection for our proprietary technology in other countries and regions throughout the world. The issued patents and pending patent applications cover, among other things:
| |
| • | the basic architecture of the OmniPod System, including the pump and the PDM; |
| |
| • | the OmniPod shape memory alloy drive system; |
| |
| • | the OmniPod System cannula insertion system; |
| |
| • | communication features between system components; |
| |
| • | software for controlling the OmniPod System; and |
| |
| • | various novel aspects of the OmniPod System and potential future generations of OmniPod Systems. |
Trademarks. We have registered various trademarks associated with our business, including INSULET, OMNIPOD and the OMNIPOD design with the United States Patent and Trademark Office on the Principal Register and in other appropriate jurisdictions.
Markets and Distribution Methods
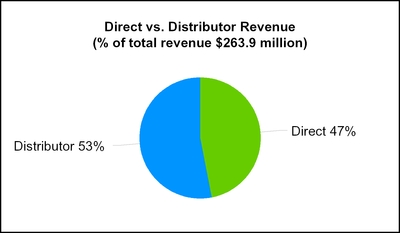
We sell our OmniPod System through a combination of direct sales representatives and independent distributors in both the United States and outside of the United States. Independent distributors can represent as much as 40% of our total sales in the United States. We have been distributing the OmniPod System in certain countries in Europe, through Ypsomed Distribution AG ("Ypsomed"), since 2010. In Canada, we had historically sold our product through an independent distributor, however we acquired that business in July 2015.
For the year ending December 31, 2015 the percentage of our total consolidated revenue from direct sales and independent distributors was as follows:
Comprehensive approach across three interrelated constituencies. Our sales and marketing effort for the OmniPod System is focused on patient retention and growing patient, clinician and payor demand for the OmniPod System. We have a uniform sales and marketing approach, aligned across patients, physicians and providers, to capitalize on the unique benefits of our OmniPod technology. We have three areas of focus:
| |
| • | First, build patient awareness about the features and benefits that the OmniPod System provides. |
| |
| • | Second, build physician support by increasing the clinical evidence that clearly demonstrates the benefits that the OmniPod System provides. |
| |
| • | Third, provide payors with the clinical and economic justification of why the OmniPod System is a greater benefit for the patients whom they insure. |
Training. We believe that patient training is critical to ensure successful outcomes and retain patients on the OmniPod System. We have streamlined our new patient training by developing improved online resources, a standardized approach as well as increasing our field clinician team to directly train our new patients.
Customer Support. We seek to provide our customers with high quality customer support, from product ordering to insurance investigation, order fulfillment and ongoing support. We have integrated our customer support systems with our sales, reimbursement and billing processes and also offer support by telephone and through our website to provide customers with seamless and reliable customer support.
Competition
The medical device industry is intensely competitive, subject to rapid change and significantly affected by new product introductions and other market activities of industry participants. The majority of our patients have previously undertaken MDI therapy, which is substantially less expensive than CSII therapy. The OmniPod System competes with a number of existing insulin delivery devices as well as other methods for the treatment of diabetes. Medtronic MiniMed, a division of Medtronic has historically held the majority share of the conventional insulin pump market in the United States. Other significant competitors in the United States are Animas Corporation, a division of Johnson & Johnson, and Tandem Diabetes Care, Inc. We also compete with drug delivery device companies such as West Pharmaceuticals.
Several of our competitors are large, well-capitalized companies with significantly more market share and resources than we have. They are able to spend aggressively on product development, marketing, sales and other product initiatives. Some of these competitors have:
| |
| • | significantly greater name recognition; |
| |
| • | established relations with healthcare professionals, customers and third-party payors; |
| |
| • | larger and more established sales forces and distribution networks; |
| |
| • | greater experience in conducting research and development, manufacturing, clinical trials, marketing and obtaining regulatory approval for products; and |
| |
| • | greater financial and human resources for product development, sales and marketing and patent litigation. |
In addition to the established insulin pump competitors, several companies are working to develop and market new insulin “patch” pumps and other methods for the treatment of diabetes, such as inhaled insulin. These companies are at various stages of development. The companies working in this area of which we are aware include Medtronic, Johnson & Johnson, Valeritas Inc., Cellnovo Limited, VinCentra, Debiotech S.A., Becton Dickinson and Co., Enable Injections, Sensile Medical and Unilife.
Government Regulation
Domestic Regulation. The OmniPod System is a medical device subject to extensive and ongoing regulation by the FDA and other federal, state, and local regulatory bodies. FDA regulations govern, among other things, product design and development, pre-clinical and clinical testing, manufacturing, labeling, post-market adverse event reporting, post-market surveillance, complaint handling, repair or recall of products, product storage, record keeping, pre-market clearance or approval, advertising and promotion, and sales and distribution.
FDA’s Pre-Market Notification (510(k)) and Pre-Market Approval Requirements. Unless an exemption applies, each medical device we seek to commercially distribute in the United States will require either prior 510(k) clearance or pre-market approval (“PMA”) from the FDA. The FDA classifies medical devices into one of three classes. Devices deemed to pose low to moderate risk are placed in either class I or II, which, absent an exemption, requires the manufacturer to submit to the FDA a premarket notification requesting permission for commercial distribution. This process is known as 510(k) clearance. Some low risk devices are exempt from this requirement. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, or devices deemed not substantially equivalent to a previously cleared 510(k) device, are placed in class III, requiring approval of a PMA application. We have obtained 510(k) clearance for the OmniPod System. Both the 510(k) clearance and PMA processes can be expensive and lengthy and entail significant user fees, unless an exemption is available.
In order to obtain pre-market approval and, in some cases, a 510(k) clearance, a product sponsor must conduct well-controlled clinical trials designed to test the safety and effectiveness of the product. Conducting clinical trials generally entails a long, costly and uncertain process that is subject to delays and failure at any stage. The data obtained from clinical trials may be inadequate to support approval or clearance of a submission. In addition, the occurrence of unexpected findings in connection with clinical trials may prevent or delay obtaining approval or clearance. If we conduct clinical trials, they may be delayed or halted, or be inadequate to support approval or clearance.
| |
| • | 510(k) Clearance. To obtain 510(k) clearance for any of our potential future devices (or for certain modifications to devices that have previously received 510(k) clearance), we must submit a pre-market notification demonstrating that the proposed device is substantially equivalent to a previously cleared 510(k) device or a pre-amendment device that was in commercial distribution before May 28, 1976 for which the FDA has not yet called for the submission of a PMA application. The FDA’s 510(k) clearance pathway generally takes from three to twelve months from the date the application is completed, but can take significantly longer. After a medical device receives 510(k) clearance, any modification that could significantly affect its safety or effectiveness, or that would constitute a significant change in its intended use, requires a new 510(k) clearance or, depending on the modification, could require a PMA application. The FDA requires each manufacturer to make this determination initially, but the FDA can review any such decision and can disagree with a manufacturer’s determination. As further described below, as part of an inspection conducted by the FDA in December of 2015, we agreed to submit a 510(k) for modifications previously made to the OmniPod System. In addition, we also agreed to submit a 510(k) associated with the field action described below that we initiated in October 2015. |
If the FDA disagrees with a manufacturer’s determination regarding whether a new premarket submission is required for the modification of an existing device, the FDA can, at its discretion, require the manufacturer to cease marketing and/or recall the modified device until 510(k) clearance or approval of a PMA application is obtained. In addition, in these circumstances, we may be subject to significant regulatory fines or penalties for failure to submit the requisite PMA application(s).
| |
| • | PMA. Devices deemed by the FDA to pose the greatest risk, such as life-sustaining, life-supporting or implantable devices, devices deemed not substantially equivalent to a previously cleared 510(k) device or devices in commercial distribution before May 28, 1976 for which PMAs have not been required, generally require a PMA before they can be commercially distributed. A PMA application must be supported by extensive data, including technical information, pre-clinical and clinical trials, manufacturing and labeling to demonstrate the safety and effectiveness of the device to the FDA’s satisfaction. After a PMA application is complete, the FDA begins an in-depth review of the submitted information, which generally takes between one and three years, but may take significantly longer. During this review period, the FDA may request additional information or clarification of information already provided. Also during the review period, an advisory panel of experts from outside the FDA may be convened to review and evaluate the application and provide recommendations to the FDA as to the approvability of the device. In addition, the FDA will conduct a pre-approval inspection of the manufacturing facility to ensure compliance with Quality System Regulations, or QSRs, which impose elaborate design development, testing, control, documentation and other quality assurance procedures in the design and manufacturing process. The FDA may approve a PMA application with post-approval conditions intended to ensure the safety and effectiveness of the device including, among other things, restrictions on labeling, promotion, sale and distribution and collection of long-term follow-up data from patients in the clinical study that supported approval. Failure to comply with the conditions of approval can result in materially adverse enforcement action, including the loss or withdrawal of the approval. After any pre-market approval, a new pre-market approval application or application supplement may be required in the event of modifications to the device, its labeling, intended use or indication or its manufacturing process. PMA supplements often require submission of the same type of information as a PMA application, except that the supplement is limited to information needed to support any changes from the device covered by the original PMA application, and may not require as extensive clinical data or the convening of an advisory panel. |
Ongoing Regulation by FDA. Even after a device is placed on the market, regardless of its classification or premarket pathway, numerous regulatory requirements apply. These include, but are not limited to:
| |
| • | establishment registration and device listing; |
| |
| • | quality system regulation, or QSR, which requires manufacturers, including third party manufacturers, to follow stringent design, testing, control, documentation and other quality assurance procedures during all aspects of the manufacturing process; |
| |
| • | labeling regulations and FDA prohibitions against the promotion of products for uncleared, unapproved or “off-label” uses, and other requirements related to promotional activities; |
| |
| • | medical device reporting regulations, which require that manufacturers report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if the malfunction were to recur; |
| |
| • | corrections and removals reporting regulations, which require that manufacturers report to the FDA field corrections and product recalls or removals if undertaken to reduce a risk to health posed by the device or to remedy a violation of the Federal Food, Drug and Cosmetic Act that may present a risk to health. In addition, FDA may order a mandatory recall if there is a reasonable probability that the device would cause serious adverse health consequences or death; and |
| |
| • | post-market surveillance regulations, which apply when necessary to protect the public health or to provide additional safety and effectiveness data for the device. |
With respect to corrections and removals, in July 2015 we implemented a field removal of certain lots due to the possibility that some OmniPod Systems had a higher rate of failure than its current manufacturing standards. In September 2015, as part of our product quality monitoring process, we identified that certain lots of the OmniPod® had a slight increase (1% - 2%) in the reported cases in which the Pod’s cannula failed to deploy. On October 29, 2015, we implemented a field correction to advise patients of the possibility of a needle deployment failure and provided recommendations on how to manage such an event. Both field actions were initiated with the knowledge of the FDA and were reported to the agency in accordance with the requirements of 21 C.F.R. Part 806.
Failure to comply with applicable regulatory requirements can result in enforcement actions by the FDA and other regulatory agencies, which may include any of the following sanctions: untitled letters or warning letters, fines, injunctions, consent decrees, civil or criminal penalties, recall or seizure of our current or future products, operating restrictions, partial suspension or total shutdown of production, refusal of or delay in granting 510(k) clearance or PMA approval of new products or modified products, rescinding previously granted 510(k) clearances or withdrawing previously granted PMA approvals, or refusal to grant export approval of our products.
We are subject to announced and unannounced inspections by the FDA, and these inspections may include the manufacturing facilities of our subcontractors. If, as a result of these inspections, the FDA determines that our equipment, facilities, laboratories or processes do not comply with applicable FDA regulations and conditions of product approval, the FDA may seek civil, criminal or administrative sanctions and/or remedies against us, including the suspension of our manufacturing operations. Since approval of the OmniPod System, we have been subject to FDA inspections of our facility on multiple occasions. Our facility located at 600 Technology Park Drive, Suite 200, Billerica, MA 01821 was inspected by the FDA between March 11, 2015 and March 27, 2015, which resulted in four inspectional observations (FDA Form 483) and a subsequent Warning Letter dated June 5, 2015. We have completed all of the commitments from the Form 483 and Warning Letter responses, but have not yet received notification that the FDA has closed the Warning Letter. More recently, our facility located in Billerica, MA was re-inspected by the FDA between November 30, 2015 and December 11, 2015. This inspection also resulted in four inspectional observations (FDA Form 483). We responded to the most recent inspectional observations on December 31, 2015.
International Regulation. International sales of medical devices are subject to foreign government regulations, which may vary substantially from country to country. The time required to obtain approval by a foreign country may be longer or shorter than that required for FDA clearance or approval, and the requirements may differ. There is a trend towards harmonization of quality system standards among the European Union, United States, Canada and various other industrialized countries. In April 2009, we received CE Mark approval for the original OmniPod System, and in August 2011, we received CE Mark approval for our OmniPod product. The CE Mark gives us authorization to distribute the OmniPod System throughout the European Union and in other countries that recognize the CE Mark. In September 2009, we received Health Canada approval to distribute the original OmniPod System throughout Canada, and in March 2013, we received Health Canada approval for our new OmniPod product. We have been distributing the OmniPod System in certain countries in Europe, through Ypsomed, since 2010. In Canada, we had historically sold our product through a distributor, however as a result of our acquisition in July 2015, we now sell the OmniPod System direct.
Licensure. Several states require that durable medical equipment (“DME”) providers be licensed in order to sell products to patients in that state. Certain of these states require, among other things, that DME providers maintain an in-state location. Although we believe we are in compliance with all applicable state regulations regarding licensure requirements, if we were found to be noncompliant, we could lose our licensure in that state, which could prohibit us from selling our current or future products directly to patients in that state.
In addition, we are subject to certain state laws regarding professional licensure. We believe that our certified diabetes educators are in compliance with all such state laws. However, if our educators or we were to be found non-compliant in a given state, we may need to modify our approach to providing education, clinical support and customer service.
Federal Anti-Kickback and Self-Referral Laws. The Federal Anti-Kickback Statute prohibits the knowing and willful offer, payment, solicitation or receipt of any form of remuneration in return for, or to induce the:
| |
| • | furnishing or arranging for the furnishing of items or services reimbursable under Medicare, Medicaid or other governmental programs; or |
| |
| • | purchase, lease, or order of, or the arrangement or recommendation of the purchasing, leasing, or ordering of any item or service reimbursable under Medicare, Medicaid or other governmental programs. |
We provide the initial training to patients necessary for appropriate use of the OmniPod System either through our own diabetes educators or by contracting with outside diabetes educators that have completed a Certified Pod Trainer training course. Outside diabetes educators are reimbursed for their services at contracted rates deemed to be consistent with the market. Although we believe that these arrangements do not violate the law, regulatory authorities may determine otherwise, especially as enforcement of this law historically has been a high priority for the federal government. In addition, because we may provide some coding and billing information to purchasers of the OmniPod System, and because we cannot assure that the government will regard any billing errors that may be made as inadvertent, the federal anti-kickback legislation may apply to us. Noncompliance with the federal anti-kickback legislation can result in exclusion from Medicare, Medicaid or other governmental programs, restrictions on operating in certain jurisdictions, as well as civil and criminal penalties, any of which could have an adverse effect on our business and results of operations.
Federal law also includes a provision commonly known as the “Stark Law,” which prohibits a physician from referring Medicare or Medicaid patients to an entity providing “designated health services,” including a company that furnishes durable medical equipment, in which the physician has an ownership or investment interest or with which the physician has entered into a compensation arrangement. Violation of the Stark Law could result in denial of payment, disgorgement of reimbursements received under a noncompliant arrangement, civil penalties, and exclusion from Medicare, Medicaid or other governmental programs. Although we believe that we have structured our provider arrangements to comply with current Stark Law requirements, these arrangements may not expressly meet the requirements for applicable exceptions from the law.
Additionally, as some of these laws are still evolving, we lack definitive guidance as to the application of certain key aspects of these laws as they relate to our arrangements with providers with respect to patient training. We cannot predict the final form that these regulations will take or the effect that the final regulations will have on us. As a result, our provider and training arrangements may ultimately be found not to be in compliance with applicable federal law.
Federal False Claims Act. The Federal False Claims Act provides, in part, that the federal government may bring a lawsuit against any person whom it believes has knowingly presented, or caused to be presented, a false or fraudulent request for payment from the federal government, or who has made a false statement or used a false record to get a claim approved. In addition, amendments in 1986 to the Federal False Claims Act have made it easier for private parties to bring “qui tam” whistleblower lawsuits against companies under the Federal False Claims Act. Penalties include fines ranging from $5,500 to $11,000 for each false claim, plus three times the amount of damages that the federal government sustained because of the act of that person. In any event, we believe that we are in compliance with the federal government’s laws and regulations concerning the filing of reimbursement claims.
Civil Monetary Penalties Law. The Federal Civil Monetary Penalties Law prohibits the offering or transferring of remuneration to a Medicare or Medicaid beneficiary that the person knows or should know is likely to influence the beneficiary’s selection of a particular supplier of Medicare or Medicaid payable items or services. Noncompliance can result in civil money penalties of up to $10,000 for each wrongful act, assessment of three times the amount claimed for each item or service and exclusion from the federal healthcare programs. We believe that our arrangements comply with the requirements of the Federal Civil Monetary Penalties Law.
State Fraud and Abuse Provisions. Many states have also adopted some form of anti-kickback and anti-referral laws and a false claims act. We believe that we are in conformance with such laws. Nevertheless, a determination of liability under such laws could result in fines and penalties and restrictions on our ability to operate in these jurisdictions.
Administrative Simplification of the Health Insurance Portability and Accountability Act of 1996. The Health Insurance Portability and Accountability Act of 1996 (“HIPAA”) mandated the adoption of standards for the exchange of electronic health information in an effort to encourage overall administrative simplification and enhance the effectiveness and efficiency of the healthcare industry. Ensuring privacy and security of patient information is one of the key factors driving the legislation. We believe we are in substantial compliance with the applicable HIPAA regulations.
Patient Protection and Affordable Care Act. The Patient Protection and Affordable Care Act (“ACA”) enacted significant changes to the provision of and payment for healthcare in the United States. Under the ACA and related laws and regulations, federal and state government initiatives are focused on limiting the growth of healthcare costs and implementing changes to healthcare delivery structures. These reforms are intended in part to put increased emphasis on the delivery to patients of more cost-effective therapies. While uncertainty exists regarding some aspects of the ACA, we expect that the ACA will continue to have a significant impact on the delivery of healthcare in the United States and on our business.
Physician Payments Sunshine Act. The Physician Payments Sunshine Act (“Sunshine Act”) seeks to increase the transparency of relationships between medical device, pharmaceutical and other companies and healthcare professionals (“HCPs”). Under the Sunshine Act, we are required to track and publicly report many types of payments made and items of value provided to HCPs. Moreover, several states have imposed similar or more restrictive requirements. In addition, we have adopted policies and codes of conduct regarding our interactions with HCPs. Our failure to adhere to these requirements could materially adversely impact our business and financial results.
Third-Party Reimbursement
In the United States, our products are generally reimbursed by third-party payors, and we bill those payors for products provided to patients. Our fulfillment and reimbursement systems are fully integrated such that product is generally shipped only after confirmation of a physician’s valid statement of medical necessity and current health insurance information. We maintain an insurance benefits investigation department that works to simplify and expedite claims processing and to assist patients in obtaining third-party reimbursement.
We continue to work with third-party payors in the United States to establish coverage and payment for the OmniPod System and other diabetes management supplies. Our coverage contracts with third-party payors typically have a term of between one and three years and set coverage amounts during that term. Typically, coverage contracts automatically renew for specified incremental periods upon expiration, unless one of the parties terminates the contract.
Third-party payors may decline to reimburse for procedures, supplies or services determined not to be “medically necessary” or “reasonable.” In a limited number of cases, some third-party payors have declined to reimburse us for a particular patient because such patient failed to meet its criteria, most often because the patient already received reimbursement for an insulin pump from that payor within the warranty period, which is generally four years, or because the patient did not meet their medical criteria for an insulin infusion device. Common medical criteria for third-party payors approving reimbursement for CSII therapy include a patient having elevated A1c levels, a history of recurring hypoglycemia, fluctuations in blood glucose levels prior to meals or upon waking or, severe glycemic variability.
As part of our international distribution agreements, our distribution partners establish appropriate reimbursement contracts with third-party payors in countries and provinces in which they distribute the OmniPod System prior to distributing the OmniPod System in each territory.
Currently, there is not an established mechanism for Medicare coverage for the majority of the OmniPod System. However, we are continuing a dialogue with Centers for Medicare & Medicaid Services ("CMS") about Medicare coverage for the OmniPod System.
Employees
As of December 31, 2015, we had 647 full-time employees. None of our employees are represented by a collective bargaining agreement, and we have never experienced any work stoppage. We believe that our employee relations are good.