Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
(Mark One)
x QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the quarterly period ended September 30, 2012
OR
o TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from to
Commission file number: 001-33277
SYNTA PHARMACEUTICALS CORP.
(Exact name of registrant as specified in its charter)
Delaware (State or other jurisdiction of incorporation or organization) | | 04-3508648 (I.R.S. Employer Identification No.) |
| | |
45 Hartwell Avenue Lexington, Massachusetts (Address of principal executive offices) | | 02421 (Zip Code) |
Registrant’s telephone number, including area code: (781) 274-8200
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer o | | Accelerated filer x |
| | |
Non-accelerated filer o | | Smaller reporting company o |
(Do not check if a smaller reporting company) | | |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x
As of November 1, 2012, the registrant had 61,900,896 shares of common stock outstanding.
Table of Contents
PART I - FINANCIAL INFORMATION
Item 1. Financial Statements.
SYNTA PHARMACEUTICALS CORP.
Condensed Consolidated Balance Sheets
(in thousands, except share and per share amounts)
(unaudited)
| | September 30,
2012 | | December 31,
2011 | |
Assets | | | | | |
Current assets: | | | | | |
Cash and cash equivalents | | $ | 29,442 | | $ | 30,075 | |
Marketable securities | | 25,697 | | 9,650 | |
Prepaid expenses and other current assets | | 803 | | 561 | |
Total current assets | | 55,942 | | 40,286 | |
Property and equipment, net | | 1,205 | | 1,407 | |
Other assets | | 466 | | 631 | |
Total assets | | $ | 57,613 | | $ | 42,324 | |
Liabilities and Stockholders’ Equity | | | | | |
Current liabilities: | | | | | |
Accounts payable | | $ | 3,690 | | $ | 3,467 | |
Accrued contract research costs | | 3,719 | | 2,841 | |
Other accrued liabilities | | 3,643 | | 4,594 | |
Current portion of capital lease obligations | | 13 | | 12 | |
Current portion of term loans | | 7,900 | | 4,234 | |
Total current liabilities | | 18,965 | | 15,148 | |
Long-term liabilities: | | | | | |
Capital lease obligations, net of current portion | | 4 | | 14 | |
Term loans, net of current portion | | 6,454 | | 12,388 | |
Total long-term liabilities | | 6,458 | | 12,402 | |
Total liabilities | | 25,423 | | 27,550 | |
| | | | | |
Stockholders’ equity: | | | | | |
Preferred stock, par value $0.0001 per share Authorized: 5,000,000 shares at September 30, 2012 and December 31, 2011; no shares issued and outstanding at September 30, 2012 and December 31, 2011 | | — | | — | |
Common stock, par value $0.0001 per share Authorized: 100,000,000 shares at September 30, 2012 and December 31, 2011; 61,881,097 and 49,539,808 shares issued and outstanding at September 30, 2012 and December 31, 2011, respectively | | 6 | | 5 | |
Additional paid-in-capital | | 475,273 | | 413,196 | |
Accumulated other comprehensive income | | 8 | | 3 | |
Accumulated deficit | | (443,097 | ) | (398,430 | ) |
Total stockholders’ equity | | 32,190 | | 14,774 | |
Total liabilities and stockholders’ equity | | $ | 57,613 | | $ | 42,324 | |
See accompanying notes to consolidated financial statements.
1
Table of Contents
SYNTA PHARMACEUTICALS CORP.
Condensed Consolidated Statements of Operations
(in thousands, except share and per share amounts)
(unaudited)
| | Three Months Ended
September 30, | | Nine Months Ended
September 30, | |
| | 2012 | | 2011 | | 2012 | | 2011 | |
Revenues: | | | | | | | | | |
Collaboration revenues: | | | | | | | | | |
License and milestone revenue | | $ | — | | $ | 1,143 | | $ | — | | $ | 3,429 | |
Total collaboration revenues | | — | | 1,143 | | — | | 3,429 | |
Grant revenues | | — | | 521 | | 147 | | 732 | |
Total revenues | | — | | 1,664 | | 147 | | 4,161 | |
Operating expenses: | | | | | | | | | |
Research and development | | 11,743 | | 10,751 | | 35,061 | | 30,605 | |
General and administrative | | 2,796 | | 3,131 | | 8,324 | | 8,749 | |
Total operating expenses | | 14,539 | | 13,882 | | 43,385 | | 39,354 | |
Loss from operations | | (14,539 | ) | (12,218 | ) | (43,238 | ) | (35,193 | ) |
Interest expense, net | | (457 | ) | (516 | ) | (1,429 | ) | (1,444 | ) |
Net loss | | $ | (14,996 | ) | $ | (12,734 | ) | $ | (44,667 | ) | $ | (36,637 | ) |
Net loss per common share: | | | | | | | | | |
Basic and diluted net loss per common share | | $ | (0.25 | ) | $ | (0.26 | ) | $ | (0.77 | ) | $ | (0.79 | ) |
Basic and diluted weighted average number of common shares outstanding | | 60,661,720 | | 49,403,589 | | 58,235,263 | | 46,446,328 | |
See accompanying notes to condensed consolidated financial statements.
2
Table of Contents
SYNTA PHARMACEUTICALS CORP.
Condensed Consolidated Statements of Comprehensive Loss
(in thousands)
(unaudited)
| | Three Months
Ended September 30, | | Nine Months
Ended September 30, | |
| | 2012 | | 2011 | | 2012 | | 2011 | |
Net loss | | $ | (14,996 | ) | $ | (12,734 | ) | $ | (44,667 | ) | $ | (36,637 | ) |
Other comprehensive (loss) income: | | | | | | | | | |
Unrealized gain (loss) on available-for-sale securities | | 4 | | (9 | ) | 5 | | 4 | |
Comprehensive loss | | $ | (14,992 | ) | $ | (12,743 | ) | $ | (44,662 | ) | $ | (36,633 | ) |
See accompanying notes to condensed consolidated financial statements.
3
Table of Contents
SYNTA PHARMACEUTICALS CORP.
Condensed Consolidated Statements of Cash Flows
(in thousands)
(unaudited)
| | Nine Months Ended
September 30, | |
| | 2012 | | 2011 | |
Cash flows from operating activities: | | | | | |
Net loss | | $ | (44,667 | ) | $ | (36,637 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | |
Stock-based compensation expense | | 2,359 | | 2,506 | �� |
Depreciation and amortization | | 623 | | 1,173 | |
Changes in operating assets and liabilities: | | | | | |
Collaboration receivable | | — | | 116 | |
Prepaid expenses and other current assets | | (242 | ) | (931 | ) |
Other assets | | 165 | | (164 | ) |
Accounts payable | | 223 | | 997 | |
Accrued contract research costs | | 878 | | 499 | |
Other accrued liabilities | | (951 | ) | (945 | ) |
Deferred collaboration revenue | | — | | (3,428 | ) |
Net cash used in operating activities | | (41,612 | ) | (36,814 | ) |
Cash flows from investing activities: | | | | | |
Purchases of marketable securities | | (45,887 | ) | (46,994 | ) |
Maturities of marketable securities | | 29,845 | | 36,644 | |
Purchases of property and equipment | | (421 | ) | (308 | ) |
Net cash used in investing activities | | (16,463 | ) | (10,658 | ) |
Cash flows from financing activities: | | | | | |
Proceeds from the issuance of common stock, excluding to related parties, net of transaction costs | | 28,960 | | 27,494 | |
Proceeds from the issuance of common stock to related parties | | 30,759 | | 7,734 | |
Proceeds from term loans | | — | | 2,000 | |
Payment of term loans | | (2,268 | ) | (233 | ) |
Payment of capital lease obligations | | (9 | ) | (190 | ) |
Net cash provided by financing activities | | 57,442 | | 36,805 | |
Net decrease in cash and cash equivalents | | (633 | ) | (10,667 | ) |
Cash and cash equivalents at beginning of period | | 30,075 | | 31,310 | |
Cash and cash equivalents at end of period | | $ | 29,442 | | $ | 20,643 | |
Supplemental disclosure of cash flow information: | | | | | |
Cash paid for interest | | $ | 1,335 | | $ | 1,484 | |
See accompanying notes to condensed consolidated financial statements.
4
Table of Contents
SYNTA PHARMACEUTICALS CORP.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(1) Nature of Business
Synta Pharmaceuticals Corp. (the Company) was incorporated in March 2000 and commenced operations in July 2001. The Company is a biopharmaceutical company focusing on discovering, developing and commercializing small molecule drugs to extend and enhance the lives of patients with severe medical conditions, including cancer and chronic inflammatory diseases.
The Company is subject to risks common to emerging companies in the drug development and pharmaceutical industry including, but not limited to, uncertainty of product development and commercialization, lack of marketing and sales history, dependence on key personnel, uncertainty of market acceptance of products, product liability, uncertain protection of proprietary technology, potential inability to raise additional financing and compliance with the U.S. Food and Drug Administration and other government regulations.
(2) Summary of Significant Accounting Policies
The accompanying condensed consolidated financial statements are unaudited, have been prepared on the same basis as the annual financial statements and, in the opinion of management, reflect all adjustments, which include only normal recurring adjustments necessary to present fairly the Company’s financial position as of September 30, 2012 and the consolidated results of operations, comprehensive loss and cash flows for the three months and nine months ended September 30, 2012 and 2011. The preparation of financial statements in conformity with accounting principles generally accepted in the United States (GAAP) requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from these estimates. The results of operations for the three months and nine months ended September 30, 2012 are not necessarily indicative of the results to be expected for the year ending December 31, 2012 or for any other interim period or any other future year. For more complete financial information, these condensed financial statements, and the notes hereto, should be read in conjunction with the audited financial statements for the year ended December 31, 2011 included in the Company’s Annual Report on Form 10-K.
Principles of Consolidation
The consolidated financial statements include the financial statements of the Company and its wholly owned subsidiaries. All significant intercompany balances and transactions have been eliminated in consolidation.
Reclassification in the Preparation of Financial Statements
Certain amounts in prior years’ financial statements have been reclassified to conform to the current period presentation. The Company reclassified $7.7 million from proceeds from the issuance of common stock to proceeds from the issuance of common stock to related parties on the statement of cash flows for the nine months ended September 30, 2011. The reclassification had no effect on the Company’s previously reported consolidated balance sheet and results of operations as of and for the nine months ended September 30, 2011.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect certain reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting periods. Significant items subject to such estimates and assumptions include contract research accruals, recoverability of long-lived assets, measurement of stock-based compensation, and the periods of performance under its collaborative research and development agreements. The Company bases its estimates on historical experience and various other assumptions that management believes to be reasonable under the circumstances. Changes in estimates are recorded in the period in which they become known. Actual results could differ from those estimates.
5
Table of Contents
Cash and Cash Equivalents
The Company considers all highly liquid investments with original maturities of three months or less at the date of purchase and an investment in a money market fund to be cash equivalents. Changes in cash and cash equivalents may be affected by shifts in investment portfolio maturities, as well as actual cash disbursements to fund operations.
The primary objective of the Company’s investment activities is to preserve its capital for the purpose of funding operations and the Company does not enter into investments for trading or speculative purposes. The Company invests in money market funds and high-grade, short-term commercial paper, which are subject to minimal credit and market risk. The Company’s cash is deposited in a highly rated financial institution in the United States. Declines in interest rates, however, would reduce future investment income.
Marketable Securities
Marketable securities consist of investments in high-grade corporate obligations, and government and government agency obligations that are classified as available-for-sale. Since these securities are available to fund current operations they are classified as current assets on the consolidated balance sheets.
The Company adjusts the cost of available-for-sale debt securities for amortization of premiums and accretion of discounts to maturity. The Company includes such amortization and accretion in interest and investment income. Realized gains and losses and declines in value, if any, that the Company judges to be other-than-temporary on available-for-sale securities are reported in interest and investment income. To determine whether an other-than-temporary impairment exists, the Company considers whether it intends to sell the debt security and, if the Company does not intend to sell the debt security, it considers available evidence to assess whether it is more likely than not that it will be required to sell the security before the recovery of its amortized cost basis. During the three months and nine months ended September 30, 2012 and 2011 the Company determined that no securities were other-than-temporarily impaired.
Marketable securities are stated at fair value, including accrued interest, with their unrealized gains and losses included as a component of accumulated other comprehensive loss, which is a separate component of stockholders’ equity. The fair value of these securities is based on quoted prices and observable inputs on a recurring basis. Realized gains and losses are determined on the specific identification method. During the three months and nine months ended September 30, 2012 and 2011 the Company recorded no realized gains or losses on marketable securities.
Fair Value of Financial Instruments
The carrying amounts of the Company’s financial instruments, which include cash equivalents, marketable securities, accounts payable and capital lease and term loan obligations, approximate their fair values. The fair value of the Company’s financial instruments reflects the amounts that would be received upon sale of an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The fair value hierarchy has the following three levels:
Level 1—quoted prices in active markets for identical assets and liabilities.
Level 2—observable inputs other than Level 1 inputs. Examples of Level 2 inputs include quoted prices in active markets for similar assets or liabilities and quoted prices for identical assets or liabilities in markets that are not active.
Level 3—unobservable inputs that reflect the Company’s own assumptions about the assumptions market participants would use in pricing the asset or liability.
Financial assets and liabilities are classified in their entirety based on the lowest level of input that is significant to the fair value measurement. The Company measures the fair value of its marketable securities by taking into consideration valuations obtained from third-party pricing sources. The pricing services utilize industry standard valuation models, including both income and market based approaches, for which all significant inputs are
6
Table of Contents
observable, either directly or indirectly, to estimate fair value. These inputs include reported trades of and broker-dealer quotes on the same or similar securities, issuer credit spreads, benchmark securities and other observable inputs. As of September 30, 2012, the Company’s financial assets valued based on Level 1 inputs consisted of cash and cash equivalents in a money market fund and its financial assets valued based on Level 2 inputs consisted of high-grade corporate and government-sponsored agency bonds and commercial paper. In the three months and nine months ended September 30, 2012, there were no transfers of financials assets between Levels 1 and 2. As of September 30, 2012, the Company had no financial liabilities that were recorded at fair value on the balance sheet. The fair value of the Company’s term loan obligations is determined using current applicable rates for similar instruments as of the balance sheet date. The carrying value of the Company’s term loan obligations approximates fair value as the Company’s interest rate yield is near current market rate yields. The Company’s term loan obligations are Level 3 liabilities within the fair value hierarchy.
Revenue Recognition
Collaboration and License Agreements
The Company’s principal source of revenue to date has been generated primarily through its former collaborative research and development agreements with Hoffman-La Roche (Roche) and GlaxoSmithKline, which included upfront license payments, development milestones, reimbursement of research and development costs, and potential profit sharing payments, commercial and sales-based milestones and royalties. The application of accounting rules requires subjective analysis and requires management to make estimates and assumptions about whether deliverables within multiple-element arrangements are separable from the other aspects of the contractual arrangement into separate units of accounting and to determine the fair value to be allocated to each unit of accounting.
For multiple-element arrangements entered into or materially modified after January 1, 2011, the Company follows the provisions of ASU No. 2009-13 Multiple-deliverable Revenue Arrangements (ASU No. 2009-13). This standard addresses the determination of the unit(s) of accounting for multiple-element arrangements and how the arrangement’s consideration should be allocated to each unit of accounting.
Pursuant to this standard, each required deliverable is evaluated to determine if it qualifies as a separate unit of accounting. For the Company this determination includes an assessment as to whether the deliverable has “stand-alone value” to the customer separate from the undelivered elements. The arrangement’s consideration is then allocated to each separate unit of accounting based on the relative selling price of each deliverable. The estimated selling price of each deliverable is determined using the following hierarchy of values: (i) vendor-specific objective evidence of fair value, (ii) third-party evidence of selling price, and (iii) the Company’s best estimate of the selling price (BESP). The BESP reflects the Company’s best estimate of what the selling price would be if the deliverable was regularly sold by it on a stand-alone basis. The Company expects, in general, to use BESP for allocating consideration to each deliverable in future collaboration agreements. In general, the consideration allocated to each unit of accounting is then recognized as the related goods or services are delivered limited to the consideration not contingent upon future deliverables.
For multiple-element arrangements entered into prior to January 1, 2011 and not materially modified thereafter, the Company continued to apply its prior accounting policy with respect to such arrangements. Under this policy, in general, revenue from non-refundable, upfront fees related to intellectual property rights/licenses where the Company had continuing involvement was recognized ratably over the estimated period of ongoing involvement because there was no objective and reliable evidence of fair value for certain of the undelivered item to allow the delivered item to be considered a separate unit of accounting. This requirement with respect to the fair value of undelivered items was eliminated in the newly issued accounting standard. In general, the consideration with respect to the other deliverables was recognized when the goods or services were delivered.
The Company’s deliverables under its former collaboration agreement with Roche, including the related rights and obligations, contractual cash flows and performance periods, are more fully described in Note 8. Certain of the deliverables were combined as a single unit of accounting.
The cash flows associated with the single unit of accounting from the research and development portions of the Company’s collaborations were recognized as revenue using a time-based model. Under this model, cash flow streams were recognized as revenue over the estimated performance period. Upon achievement of milestones, as
7
Table of Contents
defined in the collaboration agreements, revenue was recognized to the extent the accumulated service time, if any, had occurred. The remainder was deferred and recognized as revenue ratably over the remaining estimated performance period. A change in the period of time expected to complete the deliverable was accounted for as a change in estimate on a prospective basis. Revenue was limited to amounts that were non-refundable and that the Company’s collaborators were contractually obligated to pay to the Company.
Effective January 1, 2011, the Company adopted ASU No. 2009-13 which codified a method of revenue recognition that has been common practice. Under this method, contingent consideration from research and development activities that is earned upon the achievement of a substantive milestone is recognized in its entirety in the period in which the milestone is achieved. At the inception of each arrangement that includes milestone payments, the Company evaluates whether each milestone is substantive. This evaluation includes an assessment of whether (a) the consideration is commensurate with either (1) the entity’s performance to achieve the milestone, or (2) the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity’s performance to achieve the milestone, (b) the consideration relates solely to past performance and (c) the consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. The Company evaluates factors such as the scientific, clinical, regulatory, commercial and other risks that must be overcome to achieve the respective milestone, the level of effort and investment required and whether the milestone consideration is reasonable relative to all deliverables and payment terms in the arrangement in making this assessment. From the effective date of the adoption of this standard, the Company did not achieve any developmental, commercial or sales-based milestones pursuant to its research and collaboration agreement with Roche. Upon the effectiveness of the termination of the collaboration agreement with Roche on February 16, 2012, as more fully described in Note 8, the Company has no ongoing research and collaboration agreements under which milestones may be achieved.
Royalty revenues are based upon a percentage of net sales. Royalties from the sales of products will be recorded on the accrual basis when results are reliably measurable, collectibility is reasonably assured and all other revenue recognition criteria are met. Commercial and sales-based milestones, which are based upon the achievement of certain agreed-upon sales thresholds, will be recognized in the period in which the respective sales threshold is achieved and collectibility is reasonably assured.
Grant Revenue
In March 2011, the Company received a grant from the Department of Defense, in the approximate amount of $1 million, for the development of STA-9584 in advanced prostate cancer. The Company conducted work on this study during the one year grant period from April 2011 through March 2012. Reimbursements were based on actual costs agreed upon in the proposal (salary, fringe benefits, overhead, and direct costs such as materials and subcontractors). The Company recognized $0 and $521,000 of grant revenue under this grant in the three months ended September 30, 2012 and 2011, respectively, and $147,000 and $732,000 of grant revenue under this grant in the nine months ended September 30, 2012 and 2011, respectively, and $1 million of grant revenue during the one year grant period.
Deferred Collaboration Revenue
Consistent with the Company’s policy on revenue recognition, deferred collaboration revenue represents cash received and amounts earned and invoiced for licensing and option fees and milestones, as well as cash received and amounts invoiced for research and development services to be performed by the Company. Such amounts are reflected as deferred collaboration revenue until revenue can be recognized under the Company’s revenue recognition policy. Deferred collaboration revenue is classified as current if management believes the Company will complete the earnings process and be able to recognize the deferred amount as revenue within 12 months of the balance sheet date.
Stock-Based Compensation
The Company recognizes stock-based compensation expense based on the fair value of stock options granted to employees, officers and directors. The Company uses the Black-Scholes option pricing model as it is the most appropriate valuation method for its option grants. The Black-Scholes model requires inputs for risk-free interest rate, dividend yield, volatility and expected lives of the options. Since the Company has a limited history of stock activity, expected volatility was based upon the weighted average historical volatility data of the Company’s
8
Table of Contents
common stock and the historical volatility data from several guideline public biotechnology companies similar in size and value to the Company that also have stock compensation plans with similar terms. The Company estimates the forfeiture rate based on historical data. Based on an analysis of historical forfeitures, the Company has applied a forfeiture rate of 10% to all options that vest upon completion of the first year of service following the date of grant. The analysis is re-evaluated at least annually and the forfeiture rate is adjusted as necessary. The risk-free rate for periods within the expected life of the option is based on the U.S. Treasury yield curve in effect at the time of the grant. The expected lives for options granted represent the period of time that options granted are expected to be outstanding. The Company uses the simplified method for determining the expected lives of options.
For awards with graded vesting, the Company allocates compensation costs on a straight-line basis over the requisite service period. The Company amortizes the fair value of each option over each option’s service period, which is generally the vesting period.
Certain of the employee stock options granted by the Company are structured to qualify as incentive stock options (ISOs). Under current tax regulations, the Company does not receive a tax deduction for the issuance, exercise or disposition of ISOs if the employee meets certain holding requirements. If the employee does not meet the holding requirements, a disqualifying disposition occurs, at which time the Company may receive a tax deduction. The Company does not record tax benefits related to ISOs unless and until a disqualifying disposition is reported. In the event of a disqualifying disposition, the entire tax benefit is recorded as a reduction of income tax expense. The Company has not recognized any income tax benefit for the share-based compensation arrangement due to the fact that the Company does not believe it is more likely than not it will recognize any deferred tax assets from such compensation cost recognized in the current period.
Comprehensive Loss
Comprehensive loss is defined as the change in equity of a business enterprise during a period from transactions, and other events and circumstances from non-owner sources. Changes in unrealized gains and losses on marketable securities represent the only difference between the Company’s net loss and comprehensive loss.
In the first quarter of 2012, the Company adopted ASU No. 2011-05, Comprehensive Income (Topic 220): Presentation of Comprehensive Income (ASU No. 2011-05). ASU No. 2011-05 requires companies to present the components of net income and other comprehensive income either as one continuous statement or as two consecutive statements, eliminating the option to present components of other comprehensive income as part of the statement of changes in stockholders’ equity. This update does not change the items which must be reported in other comprehensive income, how such items are measured or when they must be reclassified to net income. Upon adoption, the Company elected to present comprehensive income in two separate but consecutive statements as part of the condensed consolidated financial statements included in this Quarterly Report on Form 10-Q.
Segment Reporting
Operating segments are determined based on the way management organizes its business for making operating decisions and assessing performance. The Company has only one operating segment, the discovery, development and commercialization of drug products.
Basic and Diluted Loss Per Common Share
Basic net loss per share is computed using the weighted average number of common shares outstanding during the period, excluding restricted stock that has been issued but is not yet vested. Diluted net loss per common share is computed using the weighted average number of common shares outstanding and the weighted average dilutive potential common shares outstanding using the treasury stock method. However, for the three months and nine months ended September 30, 2012 and 2011, diluted net loss per share is the same as basic net loss per share as the inclusion of weighted average shares of unvested restricted common stock and common stock issuable upon the exercise of stock options would be anti-dilutive.
9
Table of Contents
The following table summarizes outstanding securities not included in the computation of diluted net loss per common share as their inclusion would be anti-dilutive:
| | September 30, | |
| | 2012 | | 2011 | |
Common stock options | | 6,007,123 | | 5,918,559 | |
Unvested restricted common stock | | 58,936 | | 86,871 | |
(3) Cash, Cash Equivalents and Marketable Securities
A summary of cash, cash equivalents and available-for-sale marketable securities held by the Company as of September 30, 2012 and December 31, 2011 was as follows:
| | September 30, 2012 | |
| | Cost | | Unrealized
gains | | Unrealized
losses | | Fair
value | |
| | (in thousands) | |
Cash and cash equivalents: | | | | | | | | | |
Cash and money market funds (Level 1) | | $ | 23,194 | | $ | — | | $ | — | | $ | 23,194 | |
Corporate debt securities due within 3 months of date of purchase (Level 2) | | 6,248 | | — | | — | | 6,248 | |
Total cash and cash equivalents | | $ | 29,442 | | $ | — | | $ | — | | $ | 29,442 | |
Marketable securities (due within 1 year of date of purchase): | | | | | | | | | |
Corporate debt securities (Level 2) | | 23,191 | | 7 | | — | | 23,198 | |
Government-sponsored entities (Level 2) | | 2,498 | | 1 | | — | | 2,499 | |
Total marketable securities | | 25,689 | | 8 | | — | | 25,697 | |
Total cash, cash equivalents and marketable securities | | $ | 55,131 | | $ | 8 | | $ | — | | $ | 55,139 | |
| | December 31, 2011 | |
| | Cost | | Unrealized
gains | | Unrealized
losses | | Fair
value | |
| | (in thousands) | |
Cash and cash equivalents: | | | | | | | | | |
Cash and money market funds (Level 1) | | $ | 25,326 | | $ | — | | $ | — | | $ | 25,326 | |
Government-sponsored entities and corporate debt securities due within 3 months of date of purchase (Level 2) | | 4,749 | | — | | — | | 4,749 | |
Total cash and cash equivalents | | $ | 30,075 | | $ | — | | $ | — | | $ | 30,075 | |
Marketable securities: | | | | | | | | | |
Corporate debt securities due within 1 year of date of purchase (Level 2) | | 9,647 | | 3 | | — | | 9,650 | |
Total cash, cash equivalents and marketable securities | | $ | 39,722 | | $ | 3 | | $ | — | | $ | 39,725 | |
(4) Property and Equipment
Property and equipment consist of the following:
| | September 30,
2012 | | December 31,
2011 | |
| | (in thousands) | |
Laboratory equipment | | $ | 12,531 | | $ | 12,468 | |
Leasehold improvements | | 4,939 | | 4,847 | |
Computers and software | | 2,546 | | 2,315 | |
Furniture and fixtures | | 1,170 | | 1,135 | |
| | 21,186 | | 20,765 | |
Less accumulated depreciation and amortization | | (19,981 | ) | (19,358 | ) |
| | $ | 1,205 | | $ | 1,407 | |
10
Table of Contents
Depreciation and amortization expenses of property and equipment, including equipment purchased under capital leases, were approximately $0.2 million and $0.4 million for the three months ended September 30, 2012 and 2011, respectively, and $0.6 million and $1.2 million for the nine months ended September 30, 2012 and 2011, respectively.
(5) Stockholders’ Equity
Registered Direct Offering
In July 2012, the Company entered into subscription agreements with certain directors, including its largest existing stockholder, pursuant to which the Company sold 3,976,702 shares of its common stock in a registered direct offering at a purchase price of $6.49 per share. These shares were sold directly to these directors without a placement agent, underwriter, broker or dealer. The net proceeds to the Company were approximately $25.8 million after deducting estimated offering expenses payable by the Company.
Public Offering
In January 2012 and February 2012, the Company raised approximately $35.4 million in gross proceeds from the sale of an aggregate 8,050,000 shares of its common stock in a public offering at $4.40 per share, including 7,000,000 shares in the initial closing in January 2012 and 1,050,000 shares in a second closing in February 2012 upon the full exercise of the over-allotment option granted to the underwriters. One of the Company’s directors, who is its largest stockholder, purchased 1,136,363 shares in this offering. The net offering proceeds to the Company were approximately $33.0 million after deducting underwriters’ discounts, fees and commissions, and other offering expenses payable by the Company.
At-The-Market Issuance Sales Agreement
On May 2, 2012, the Company entered into an at-the-market issuance sales agreement (Sales Agreement) with MLV & Co. LLC (MLV), pursuant to which the Company may issue and sell shares of its common stock having an aggregate offering price of up to $35 million from time to time, at the Company’s option, through MLV as its sales agent. Sales of common stock through MLV, if any, will be made by any method that is deemed an “at-the-market” offering as defined in Rule 415 promulgated under the Securities Act of 1933, as amended, including by means of ordinary brokers’ transactions at market prices, in block transactions or as otherwise agreed by the Company and MLV. Subject to the terms and conditions of the Sales Agreement, MLV will use commercially reasonable efforts to sell the common stock based upon the Company’s instructions (including any price, time or size limits or other customary parameters or conditions the Company may impose). The Company is not obligated to make any sales of its common stock under the Sales Agreement. Any shares sold will be sold pursuant to the Company’s effective shelf registration statement on Form S-3. The Company will pay MLV a commission of up to 3% of the gross proceeds. The Sales Agreement will terminate upon the earlier of the sale of all common stock subject to the Sales Agreement or termination of the Sales Agreement by the Company or MLV. To date, no shares have been sold under the Sales Agreement.
(6) Stock-Based Compensation
The Company’s 2006 Stock Plan provides for the grant of incentive stock options, nonstatutory stock options and non-vested stock to employees, officers, directors and consultants to the Company. In January 2012, the number of shares of common stock reserved for issuance under the 2006 Stock Plan was increased from 6,400,000 to 7,700,000 pursuant to an “evergreen” provision, which provides for an annual increase based on the lesser of 1,300,000 shares, 5% of the Company���s then outstanding shares of common stock, or such other amount as the board of directors may determine. This increase was approved by the board of directors in November 2011. The administration of the 2006 Stock Plan is under the general supervision of the compensation committee of the board of directors. The exercise price of the stock options is determined by the compensation committee of the board of directors, provided that incentive stock options are granted at not less than fair market value of the common stock on the date of grant and expire no later than ten years from the date the option is granted. Options generally vest over four years. As of September 30, 2012, the Company had options outstanding to purchase 6,007,123 shares of its
11
Table of Contents
common stock, which includes options outstanding under its 2001 Stock Plan that was terminated in March 2006, had 58,936 outstanding restricted shares of common stock and had 1,954,674 shares available for future issuance.
The following table summarizes stock option activity during the nine months ended September 30, 2012:
| | Shares | | Weighted average
exercise price | |
Outstanding at January 1 | | 5,821,073 | | $ | 7.54 | |
Options granted | | 1,736,256 | | 5.05 | |
Options exercised | | (269,344 | ) | 3.35 | |
Options cancelled | | (1,280,862 | ) | 8.83 | |
Outstanding at September 30 | | 6,007,123 | | $ | 6.73 | |
Exercisable at September 30 | | 3,502,865 | | $ | 7.96 | |
The weighted-average grant date fair values of options granted during the three months ended September 30, 2012 and 2011 were $6.00 and $3.93, respectively, and during the nine months ended September 30, 2012 and 2011 were $4.04 and $4.27, respectively.
Non-Vested (“Restricted”) Stock Awards With Service Conditions
The Company’s share-based compensation plan provides for awards of restricted shares of common stock to employees, officers, directors and consultants to the Company. Restricted stock awards are subject to forfeiture if employment or service terminates during the prescribed retention period. Restricted shares vest over the service period.
The following table summarizes unvested restricted shares during the nine months ended September 30, 2012:
| | Shares | | Weighted
average
grant date
fair value | |
Outstanding at January 1 | | 82,450 | | $ | 4.94 | |
Vested | | (68,757 | ) | 5.27 | |
Granted | | 45,243 | | 5.47 | |
Outstanding at September 30 | | 58,936 | | $ | 4.95 | |
Stock-Based Compensation Expense
For the three months and nine months ended September 30, 2012 and 2011, the fair value of each employee stock option award was estimated on the date of grant based on the fair value method using the Black-Scholes option pricing valuation model with the following weighted average assumptions:
| | Three Months
Ended September 30, | | Nine Months
Ended September 30, | |
| | 2012 | | 2011 | | 2012 | | 2011 | |
Risk-free interest rate | | .92 | % | 2.05 | % | 1.11 | % | 2.49 | % |
Expected life in years | | 6.23 | | 6.25 | | 6.24 | | 6.25 | |
Volatility | | 102 | % | 100 | % | 101 | % | 101 | % |
Expected dividend yield | | — | | — | | — | | — | |
Stock-based compensation during the three and nine months ended September 30, 2012 and 2011 was as follows (in thousands):
12
Table of Contents
| | Three Months
Ended September 30, | | Nine Months
Ended September 30, | |
| | 2012 | | 2011 | | 2012 | | 2011 | |
Stock-based compensation expense by type of award: | | | | | | | | | |
Employee stock options | | $ | 765 | | $ | 774 | | $ | 2,222 | | $ | 2,196 | |
Restricted stock | | 36 | | 93 | | 137 | | 310 | |
Total stock-based compensation expense | | $ | 801 | | $ | 867 | | $ | 2,359 | | $ | 2,506 | |
| | | | | | | | | |
Effect of stock-based compensation expense by line item: | | | | | | | | | |
Research and development | | $ | 612 | | $ | 648 | | $ | 1,797 | | $ | 1,861 | |
General and administrative | | 189 | | 219 | | 562 | | 645 | |
Total stock-based compensation expense included in net loss | | $ | 801 | | $ | 867 | | $ | 2,359 | | $ | 2,506 | |
Unrecognized stock-based compensation expense as of September 30, 2012 was as follows (in thousands):
| | Unrecognized
stock
compensation
expense as of
September 30,
2012 | | Weighted
average
remaining
period
(in years) | |
Employee stock options | | $ | 8,305 | | 2.76 | |
Restricted stock | | 278 | | 0.87 | |
Total | | $ | 8,583 | | 2.71 | |
7) Other Accrued Liabilities
Other accrued liabilities consist of the following:
| | September 30,
2012 | | December 31,
2011 | |
| | (in thousands) | |
Compensation and benefits | | $ | 1,979 | | $ | 2,914 | |
Professional fees | | 851 | | 1,069 | |
Other | | 813 | | 611 | |
| | $ | 3,643 | | $ | 4,594 | |
(8) License and Development Agreements
Roche
In December 2008, as amended in February 2010, February 2011 and July 2011, the Company and Roche entered into a collaborative license agreement (the Roche Agreement) to discover, develop, and commercialize small-molecule drugs targeting calcium release-activated calcium modulator (CRACM) channels. The goal of this alliance was to develop a novel category of oral, disease-modifying agents for the treatment of rheumatoid arthritis and other autoimmune diseases and inflammatory conditions. The Roche Agreement consisted of the following funding streams: an upfront license payment, reimbursements of certain research and development costs, product development milestones, sales milestones and product royalty payments.
Pursuant to the Roche Agreement, the Company received a non-refundable upfront license payment of $16 million in January 2009. Roche reimbursed all of the Company’s research and certain early development costs over the two year research term that concluded on December 31, 2010. The Company received approximately $21.2 million in research and development support under the Roche Agreement.
Roche terminated the Roche Agreement effective February 16, 2012. All rights to certain products, referred to as Licensed Compounds, which were identified and studied prior to the end of the two year research term, reverted to the Company upon the effectiveness of the termination. The Company may pay Roche a low single-digit royalty on any potential future sales of licensed products. The Company did not incur any termination costs or penalties as a result of the termination of the Roche Agreement. No development milestones were achieved under the Roche Agreement.
13
Table of Contents
The $16 million non-refundable upfront license payment was being recognized ratably using the time-based model over the estimated performance period through June 2012. In the fourth quarter of 2011, upon notification of Roche’s election to terminate the Roche Agreement, the Company accelerated the recognition of approximately $2.1 million of remaining deferred revenue from the upfront payment because the Company had no remaining substantial performance obligations. Under the Roche Agreement, the Company recognized $0 and $1.1 million of license revenue in the three months ended September 30, 2012 and 2011, respectively, and $0 and $3.4 million in the nine months ended September 30, 2012 and 2011, respectively.
Co-Development Agreement
In July 2011, the Company entered into a co-development agreement with a clinical research organization (CRO) for the conduct of certain company-sponsored clinical trials. Under the co-development agreement, this CRO will perform clinical research services under a reduced fee structure in exchange for a share of licensing payments and commercial revenues, if any, resulting from the product under development up to a specified maximum payment, which is defined as a multiple of the fee reduction realized.
(9) Term Loans
General Electric Capital Corporation
In September 2010, the Company entered into a $15 million loan and security agreement, as amended in November 2010, March 2011, July 2011, January 2012 and July 2012, with General Electric Capital Corporation (GECC) and one other lender, all of which was funded at the closing in September 2010 (the GECC Term Loan). Interest on the borrowings under the GECC Term Loan accrues at an annual rate of 9.75%.
The Company made interest-only payments through June 2012. Beginning in July 2012, the Company began making 25 equal monthly payments of principal plus accrued interest on the outstanding balance. In addition to the interest payable under the GECC Term Loan, the Company paid origination and amendment fees in the amount of $373,000 and is obligated to pay an exit fee of $525,000 at the time of the final payment of the outstanding principal.
Origination and exit fees are being amortized and accreted, respectively, to interest expense over the term of the GECC Term Loan. The Company paid approximately $264,000 of legal fees and expenses in connection with the GECC Term Loan. These expenses have been deferred and, together with the origination fees, are included in other assets, and are being expensed over the term of the GECC Term Loan. The Company recognized approximately $82,000 and $72,000 in the three months ended September 30, 2012 and 2011, respectively, and $239,000 and $202,000 in the nine months ended September 30, 2012 and 2011, respectively, in interest expense in connection with these origination, exit and transaction fees and expenses. The Company recognized approximately $341,000 and $382,000 in the three months ended September 30, 2012 and 2011, respectively, and $1,076,000 and $1,109,000 in the nine months ended September 30, 2012 and 2011, respectively, in interest expense related to the outstanding principal under the GECC Term Loan. No warrants were issued in connection with the GECC Term Loan. The Company may prepay the full amount of the GECC Term Loan, subject to prepayment premiums under certain circumstances.
The GECC Term Loan is secured by substantially all of the Company’s assets, except its intellectual property. The Company has granted GECC a springing security interest in its intellectual property in the event the Company is not in compliance with certain cash usage covenants, as defined therein. The GECC Term Loan contains restrictive covenants, including the requirement for the Company to receive the prior written consent of GECC to enter into loans, other than up to $4.0 million of equipment financing, restrictions on the declaration or payment of dividends, restrictions on acquisitions, and customary default provisions that include material adverse events, as defined therein. The Company has determined that the risk of subjective acceleration under the material adverse events clause is remote and therefore has classified the outstanding principal in current and long-term liabilities based on the timing of scheduled principal payments. In addition, at the time of the closing of the GECC Term Loan, the Company repaid approximately $787,000 of remaining principal outstanding under its existing equipment leases with GECC.
Oxford Finance Corporation
In March 2011, the Company entered into a $2 million loan and security agreement with Oxford Finance Corporation (Oxford), all of which was funded in March 2011 (the Oxford Term Loan). Interest on the borrowings
14
Table of Contents
under the Oxford Term Loan accrues at an annual rate of 13.35%. Beginning in May 2011, the Company began making 36 equal monthly payments of principal plus accrued interest on the outstanding balance. The Company recognized approximately $40,000 and $61,000 in the three months ended September 30, 2012 and 2011, respectively, and $137,000 and $136,000 in the nine months ended September 30, 2012 and 2011, respectively, in interest expense related to the outstanding principal under the Oxford Term Loan. In addition to the interest payable under the Oxford Term Loan, the Company paid approximately $66,000 of administrative and legal fees and expenses in connection with the Oxford Term Loan. These expenses have been deferred, are included in other assets and are being expensed over the term of the Oxford Term Loan. No warrants were issued in connection with the Oxford Term Loan. The Company may prepay the full amount of the Oxford Term Loan, subject to prepayment premiums under certain circumstances. Oxford has the right to require the Company to prepay the full amount of the Oxford Term Loan if the Company prepays the full amount of the GECC Term Loan under certain circumstances.
The Oxford Term Loan is secured by certain laboratory and office equipment, furniture and fixtures acquired through September 30, 2010. In connection with the Oxford Term Loan, Oxford and GECC entered into a Lien Subordination Agreement, whereby GECC granted Oxford a first priority perfected security interest in the loan collateral. The Oxford Term Loan contains restrictive covenants, including the requirement for the Company to receive the prior written consent of Oxford to enter into acquisitions in which the Company incurs more than $2.0 million of related indebtedness, and customary default provisions that include material adverse events, as defined therein. The Company has determined that the risk of subjective acceleration under the material adverse events clause is remote and therefore has classified the outstanding principal in current and long-term liabilities based on the timing of scheduled principal payments.
Future principal payments under the GECC and Oxford Term Loans as of September 30, 2012 are approximately as follows (in thousands):
Year Ending December 31, | | | |
2012 | | $ | 1,966 | |
2013 | | 7,924 | |
2014 | | 4,464 | |
| | $ | 14,354 | |
15
Table of Contents
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
You should read this discussion together with the consolidated financial statements, related notes and other financial information included elsewhere in this Quarterly Report on Form 10-Q. The following discussion may contain predictions, estimates and other forward-looking statements that involve a number of risks and uncertainties, including those discussed under “Risk Factors” in our Annual Report on Form 10-K for the year ended December 31, 2011 filed with the Securities and Exchange Commission. These risks could cause our actual results to differ materially from any future performance suggested below.
Overview
Synta Pharmaceuticals Corp. is a biopharmaceutical company focused on discovering, developing, and commercializing small molecule drugs to extend and enhance the lives of patients with severe medical conditions, including cancer and chronic inflammatory diseases. We have two drug candidates in clinical trials for treating multiple types of cancer and several drug candidates in the preclinical stage of development. Each of our drug candidates was discovered and developed internally using our proprietary, unique chemical compound library and integrated discovery engine. We retain full ownership of all of our drug candidates.
We were incorporated in March 2000 and commenced operations in July 2001. Since that time, we have been principally engaged in the discovery and development of novel drug candidates. As of September 30, 2012, we have funded our operations principally with $405.4 million in net proceeds from private and public offerings of our equity, as well as $17 million in gross proceeds from two term loans, including $15 million from a term loan that was executed in September 2010 with General Electric Capital Corporation, or GECC, and one other lender, and $2 million from a term loan that was executed in March 2011 with Oxford Finance Corporation, or Oxford.
In January and February 2012, we raised approximately $33.0 million in net proceeds from the sale of an aggregate of 8,050,000 shares of our common stock in a public offering at a public offering price of $4.40 per share, including 7,000,000 shares in the initial closing in January 2012 and 1,050,000 shares in a second closing in February 2012 following the full exercise of the over-allotment option granted to the underwriters. In July 2012, we raised approximately $25.8 million in net proceeds from a registered direct offering of 3,976,702 shares of our common stock at a price of $6.49 per share to certain directors, including our largest existing stockholder.
On May 2, 2012, we entered into an at-the-market issuance sales agreement, or Sales Agreement, with MLV & Co. LLC, or MLV, pursuant to which we may issue and sell shares of our common stock having an aggregate offering price of up to $35 million from time to time, at our option, through MLV as our sales agent, subject to certain terms and conditions. To date, no shares have been sold under the Sales Agreement.
In addition to raising capital from financing activities, we have also received substantial capital from partnering activities. In October 2007, we entered into a global collaborative development, commercialization and license agreement with GlaxoSmithKline, or GSK, for the joint development and commercialization of elesclomol. This collaboration was terminated in September 2009. In December 2008, we entered into a collaborative license agreement with Hoffman-La Roche, or Roche, for our CRACM inhibitor program. This collaboration was terminated effective on February 16, 2012. As of September 30, 2012, we have received $167.2 million in nonrefundable partnership payments under these agreements with GSK and with Roche, including $96 million in upfront payments, $50 million in operational milestones and $21.2 million in research and development funding. As of September 30, 2012, these nonrefundable partnership payments together with the net cash proceeds from equity financings, the term loans from GECC and Oxford, and the exercise of common stock warrants and options, provided aggregate net cash proceeds of approximately $592.4 million. We have also generated funds from government grants, equipment lease financings and investment income. We are engaged in preliminary partnership discussions for a number of our programs, which may provide us with additional financial resources if consummated.
We have devoted substantially all of our capital resources to the research and development of our drug candidates. Since our inception, we have had no revenues from product sales. As of September 30, 2012, we had an accumulated deficit of $443.1 million. We expect to incur significant operating losses for the foreseeable future as we advance our drug candidates from discovery through preclinical development and clinical trials, and seek regulatory approval and eventual commercialization. We will need to generate significant revenues from product sales to achieve future profitability and may never do so.
Oncology Programs
We have two clinical-stage programs and one preclinical-stage program in oncology:
16
Table of Contents
Ganetespib (Hsp90 Inhibitor)
Summary
Ganetespib is a novel, potent, small molecule inhibitor of heat shock protein 90 (Hsp90), a molecular chaperone which is required for the proper folding and activation of many cancer-promoting proteins. Inhibition of Hsp90 by ganetespib leads to the simultaneous degradation of many of its client proteins and the subsequent death or cell cycle arrest of cancer cells dependent on those proteins. A number of Hsp90 client proteins are also involved in the resistance of cancer cells to other anti-cancer treatments, such as chemotherapy. The ability to reduce cancer-cell drug resistance suggests the combination of ganetespib with chemotherapies or other agents may provide greater benefit than those agents administered alone. In preclinical studies, ganetespib has shown potent anti-cancer activity in a broad range of solid and hematologic cancers, both as a monotherapy and in combination with certain widely used anti-cancer agents.
Ganetespib is currently being evaluated in over 20 clinical trials, including trials evaluating monotherapy administration in certain genetically-defined targeted patient populations, such as our trials in ALK+ lung cancer, HER2+ breast cancer, and triple-negative breast cancer, as well as trials evaluating combination treatment in a broader patient population, such as our GALAXY lung cancer trial. The safety profile across these trials, involving over 600 patients treated with ganetespib to date, has been consistent and favorable. Ganetespib has shown no evidence of the serious liver or common ocular toxicities reported with other Hsp90 inhibitors, or the neurotoxicity, bone marrow toxicities, and alopecia characteristic of many chemotherapies. The most common adverse event reported with ganetespib has been transient, mild or moderate diarrhea, which can be prevented or effectively managed with standard supportive care.
In clinical trials, ganetespib has shown promising activity in a broad range of cancers, both as a monotherapy and in combination:
· Monotherapy:
· Objective responses or anti-tumor activity have been seen in patients with ALK+ lung cancer, mutant BRAF lung cancer, mutant KRAS lung cancer, mutant KRAS gastric cancer, HER2+ breast cancer, triple-negative breast cancer, renal cancer, colorectal cancer, and melanoma. One patient with ALK+ lung cancer and one patient with mutant KRAS gastric cancer have remained on ganetespib therapy for two years.
· Combination: At the European Society for Medical Oncology 2012 Congress, investigators presented encouraging results from the second interim efficacy analysis of our ongoing, randomized, Phase 2b/3 GALAXY trial evaluating ganetespib plus docetaxel vs. docetaxel alone in second-line advanced non-small cell lung cancer (NSCLC).
· In the 172 adenocarcinoma patients enrolled as of the September 10 data cutoff, an increase in overall survival was observed in patients treated with ganetespib plus docetaxel. A median overall survival of 7.4 months was observed in the docetaxel control arm, while median overall survival had not been reached in the ganetespib arm. Results for docetaxel were consistent with results from prior second line NSCLC therapy trials.
· Objective response rate and progression-free survival in adenocarcinoma patients were also improved: from 8% to 16%, and from 2.8 months to 4.2 months, in the control arm vs. ganetespib arm, respectively. Overall response and progression-free survival rates in the control arm were consistent with results from prior trials with docetaxel in this setting.
· Results in several GALAXY patient subpopulations, defined by pre-specified clinical and biomarker characteristics, showed a substantially improved survival difference between the control arm and ganetespib arm, as compared with the difference in the all-comer (intent-to-treat) adenocarcinoma patient population. These findings have been incorporated into the design of the Phase 3 portion of the GALAXY program, with the objective of enriching for patients likely to derive the greatest benefit from ganetespib treatment.
· Clinical and preclinical results were presented that suggest potent ganetespib anti-angiogenic activity. Analyses of tumor samples from patients treated with ganetespib showed a reduction of levels of hypoxia induced factor (HIF) and vascular endothelial growth factor (VEGF). In addition, preclinical experiments demonstrated strong inhibition of tumor vasculature by ganetespib. These results suggest ganetespib offers a novel way to inhibit angiogenesis: reducing production of angiogenesis factors, rather than targeting those signaling factors directly with an antibody (such as bevacizumab) or a kinase inhibitor.
· A favorable safety profile was observed with the ganetespib plus docetaxel combination in adenocarcinoma patients. Transient, mild-to-moderate diarrhea was the most common adverse event, consistent with observations from other clinical trials evaluating ganetespib.
17
Table of Contents
The results observed with ganetespib monotherapy administration suggest promising potential for treating specific, targeted populations such as patients with cancers driven by increased expression or mutations in genes encoding “strong” Hsp90 clients. This represents a sizable unmet need and commercial opportunity. Our CHIARA trial, for example, evaluates ganetespib in patients with ALK+ lung cancer. There are an estimated 40,000-70,000 new patients diagnosed worldwide each year with this cancer type. Our ENCHANT trial evaluates ganetespib in patients with HER2+ or triple-negative metastatic breast cancer. Each of these subpopulations is estimated at 15-20% of the 1.4 million patients diagnosed with breast cancer worldwide each year.
The results observed to date in our GALAXY trial suggest an even broader unmet need and commercial opportunity for the combination therapy approach. Across the US, UK, Germany, France, Spain, Italy, and Japan an estimated 160,000 new patients each year progress following first-line treatment for advanced NSCLC adenocarcinoma, and receive subsequent treatment. This is the patient population being addressed in our GALAXY trial. In addition, over 500,000 patients receive taxanes each year, across all cancer indications. The ability to combine with taxanes with minimal additional toxicity and possible enhanced activity represents a promising opportunity not only in lung cancer but in breast, prostate, ovarian, gastric, bladder, and head and neck cancers, where taxanes are commonly used. In preclinical models, ganetespib has shown ability to enhance the activity of a number of other widely used anti-cancer agents, in addition to the taxanes, including pemetrexed, gemcitabine, bevacizumab, cytarabine, irinotecan, etoposide, doxorubicin, carboplatin, cisplatin, vincristine, tamoxifen, fulvestrant, temsirolimus, lapatinib, crizotinib, vemurafenib, selumetinib, and bortezomib. Combination trials with a number of these agents have recently been initiated.
Ganetespib Mechanism of Action and Preclinical Results
Ganetespib is a novel, small-molecule inhibitor of Hsp90 structurally unrelated to first-generation, ansamycin-family compounds, such as 17-AAG or 17-DMAG. In preclinical studies, ganetespib has shown 10-100 times greater potency than 17-AAG across a broad range of cancer cell types as well as activity in animal models that are resistant to treatment with 17-AAG.
Hsp90 is a molecular chaperone required for the proper folding and activation of many cancer-promoting proteins. Many of the client proteins of Hsp90, such as ALK, AKT, BCR-ABL, BRAF, KIT, MET, EGFR, FLT3, HER2, HIF-1alpha, PDGFRA, and VEGFR, are the targets of clinically validated cancer drugs such as Avastin, Erbitux, Gleevec, Herceptin, Nexavar, Sutent, Tarceva, Votrient, Xalkori, and Zelboraf. In preclinical studies, inhibition of Hsp90 by ganetespib results in simultaneous degradation of these client proteins, resulting in cancer cell death or cell cycle arrest.
Ganetespib also inhibits known mechanisms by which cancer cells evade or recover from other anti-cancer treatments. For example, cancer cells can minimize DNA damage caused by chemotherapy or radiation therapy through modification of cell cycle dynamics and activation of DNA repair processes. Many of the cell cycle and DNA repair components — such as ATM, ATR, CHK1, BRCA1, and WEE1 — are Hsp90 client proteins.
Ganetespib has shown activity both as a monotherapy and in combination in a broad range of in vitro and in vivo models of cancer. Combination activity has been observed in models of ALK+ NSCLC with Xalkori, KRAS mutant NSCLC with Taxotere, EGFR mutant NSCLC with Avastin, HER2+ breast cancer with Tykerb, colorectal cancer with radiation or platinum therapy, BRAF mutant melanoma with Zelboraf, hormone refractory prostate cancer with mTOR inhibitors, and AML with cytarabine.
Results published in Molecular Cancer Therapeutics in December 2011 highlighted certain physicochemical properties of ganetespib believed to contribute to its improved safety and activity relative to other Hsp90 inhibitors. These include smaller size, greater potency, improved ability to passively enter cells, improved
18
Table of Contents
interaction with the drug target, absence of a molecular component known to cause liver toxicity, and ability to penetrate deep into tumor tissues.
Results presented at the AACR-EORTC-NCI meeting in November 2011 and at the American Society of Clinical Oncology (ASCO) meeting in June 2012 demonstrated that common ocular toxicities seen with some Hsp90 inhibitors, but not observed in clinical trials with ganetespib or with 17-AAG, are associated with physicochemical properties that affect drug distribution to the eye.
Ganetespib Clinical Trials
Ganetespib is being evaluated in over 20 clinical trials ongoing, currently initiating, or recently completed, including trials in lung, breast, colorectal, gastric, prostate, melanoma, and pancreatic cancers, as well as in certain types of blood cancers. Many of these trials are sponsored by individual investigators or groups of investigators. We are sponsoring three principal ongoing trials evaluating ganetespib activity:
· GALAXY: a randomized Phase 2b/3 trial evaluating ganetespib in combination with docetaxel versus docetaxel alone as second-line therapy in patients with advanced NSCLC (Ganetespib Assessment in Lung cAncer with docetaXel);
· CHIARA: a Phase 2 trial evaluating ganetespib monotherapy in patients whose tumors have a genetic profile characterized by rearrangement of the ALK gene (ALK+) (Chaperone Inhibition in ALK Rearranged lung cAncer), and
· ENCHANT: a Phase 2 trial evaluating ganetespib monotherapy in patients with newly diagnosed HER2+ and triple-negative metastatic breast cancer (EvaluatiNg Chaperone inhibition by gANetespib in breasT cancer)
Ganetespib in combination with chemotherapy: the GALAXY Trial
Cancer treatments are often given in combination in order to maximize benefit to patients. A challenge with combination therapy is that the added toxicities from combining two or more anti-cancer agents may not be tolerable, particularly if the toxicity profiles overlap. The favorable safety profile seen to date with ganetespib and the non-overlapping toxicities with many standard-of-care agents support such a combination therapy approach.
Results to date suggest potential for combining ganetespib and taxanes. These include a strong scientific rationale based on multiple mechanisms of synergistic anti-cancer activity, strong synergistic results in in vitro and in vivo experiments, and the encouraging safety profile seen in our Phase 1 and Phase 2b/3 combination studies of ganetespib and docetaxel.
GALAXY Trial Design
In 2011 we initiated the GALAXY trial, a Phase 2b/3 program in patients with advanced NSCLC who have received one prior treatment for advanced disease, i.e., a second-line treatment setting. The GALAXY trial compares treatment with docetaxel alone, which is approved for second-line treatment, versus treatment with ganetespib plus docetaxel. The first stage, Phase 2b portion is designed to establish the clinical benefit and safety profile of ganetespib in combination with docetaxel relative to docetaxel alone, and to identify the patient populations, by biomarker or other disease characteristics, which may be most responsive to combination treatment. The first stage of this program is intended to build the clinical and operational experience needed to optimize the design and execution of the second stage, Phase 3 portion.
Patients in both arms receive a standard regimen of docetaxel 75 mg/m2 on day 1 of a 21-day cycle. Patients in the combination arm also receive ganetespib 150 mg/m2 on days 1 and 15. Treatment continues until disease progression per Response Evaluation Criteria in Solid Tumors (RECIST) criteria. Enrollment is stratified by ECOG performance status, LDH, smoking status, and time since diagnosis of metastatic disease to ensure balance of these prognostic factors between the two arms.
The Phase 2b portion of the trial was designed to enroll 240 second-line advanced NSCLC patients in order to evaluate several pre-specified hypotheses on which patients might be most responsive to combination treatment. On initial design the co-primary endpoints were progression-free survival in all patients (the ITT or intent-to-treat
19
Table of Contents
population) and overall survival in patients with elevated baseline level of serum LDH. Several months after trial initiation, but before any substantial patient enrollment, the trial was amended to elevate improvement in progression-free survival in patients with mutant KRAS (the mKRAS population) from a secondary endpoint to a co-primary endpoint, based on clinical results observed in a separate ganetespib trial around that time. Both LDH and mutant KRAS were pre-specified for evaluation from blood and tumor tissue, respectively, by an independent, central laboratory.
The GALAXY trial was designed to enroll patients with all histologies — including both adenocarcinoma and squamous cell. Earlier this year enrollment of patients with non-adenocarcinoma histologies (which consists primarily of squamous cell carcinomas) was terminated based on the lack of benefit observed in this population; possible safety concerns, including risk of bleeding; a trend towards survival disadvantage; and the consistency of the emerging ganetespib profile with known anti-angiogenic agents, for which patients with squamous cell carcinoma histology are commonly excluded from clinical trials or labeled indications. The trial was amended at that time to enroll 240 patients with adenocarcinoma histology only.
The current co-primary endpoints of the first-stage, Phase 2b portion are: PFS in patients with elevated LDH and PFS in patients with mutant KRAS. Both of these represent patient populations with particularly high unmet medical need and for which there are encouraging preclinical and early clinical results supporting the use of ganetespib. Key secondary endpoints, to be evaluated with the statistical gatekeeping methodology, include OS and PFS in the all adenocarcinoma population. The Phase 2b stage is 90% powered to detect a PFS improvement from 6 to 12 weeks in patients with elevated LDH and from 5 weeks to 10 weeks in patients with mutant KRAS. For all adenocarcinoma patients, GALAXY is 88% powered to detect an improvement in PFS from 3 to 4.5 months, and 73% powered to detect an improvement in overall survival from 6 to 8.5 months. All powering assumptions are based on a 1-sided alpha of 0.05.
GALAXY Interim Results
In a June 2012 press release we reported top line results from a planned interim analysis of the GALAXY trial. The analysis was planned for when approximately 50% of patients had been enrolled. At the time of this analysis, completed in June, a total of 114 adenocarcinoma and 69 non-adenocarcinoma patients had been enrolled.
On September 29, 2012, we reported results from the second interim efficacy analysis at the European Society for Medical Oncology 2012 Congress (ESMO). This analysis was planned for when approximately 80% of patients had been enrolled. At the time of the September 10 data cutoff, 172 adenocarcinoma patients had been entered into the clinical database. There were 88 patients in the docetaxel control arm and 84 patients in the ganetespib arm. Baseline characteristics were generally well balanced between both arms. An additional analysis was planned for patients who would have a minimum of 6 months of follow-up, defined by those patients enrolled before March 20. There were 38 patients in the docetaxel control arm and 39 patients in the ganetespib arm in this group. Baseline characteristics were also generally well balanced between both arms in this group. Overall survival results are described in the table below. Median survival for the docetaxel control arm in both groups was consistent with comparable historical results. Median survival had not yet been reached for the combination arm.
Overall survival, all adenocarcinoma patients
| | All patients in database
(N=172) | | All patients enrolled more than 6
months prior to data cutoff
(N=77) | |
| | | | | |
HR | | 0.688 | | 0.568 | |
C.I. (90%) | | (0.417, 1.135) | | (0.312, 1.032) | |
p-Value | | 0.183 | | 0.056 | |
Median (D vs G+D) | | 7.4 mo vs. NR | | 7.4 mo vs NR | |
HR: Hazard ratio, C.I.: confidence interval, NR: not reached
20
Table of Contents
Hazard ratio (HR) represents the odds that a patient in the experimental treatment arm will experience the event of interest (such as death or disease progression) before a patient in the control arm. A hazard ratio of 1.0 corresponds to no treatment effect, while a hazard ratio of less than 1.0 signifies that the treatment is working better than the control.
Progression free survival was 2.8 months vs. 4.2 months (p=0.076) and overall response rate was 8% vs. 16% (p=0.078) for docetaxel vs. ganetespib plus docetaxel, respectively. All p-values are calculated using the 1-sided stratified log-rank test for survival endpoints and using Fisher’s Exact test for response rate.
Shown below are the Kaplan-Meier analyses of overall survival. The Y-axes represent the fraction of patients alive in each arm of the study.
Overall survival, all adenocarcinoma patients (n=172)

21
Table of Contents
Overall survival, patients enrolled > 6 months prior to data cutoff (n=77)
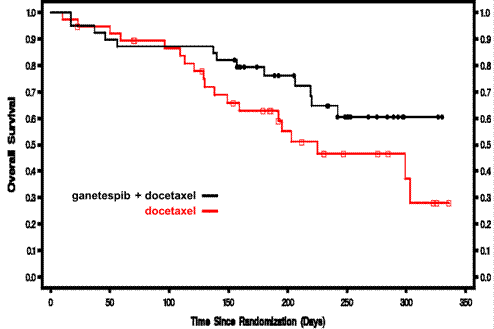
In the GALAXY Phase 2b trial there are four pre-specified stratification factors and one additional biomarker defined primary endpoint subpopulation (mutant KRAS patients). Results for these patient populations are shown below.
Overall survival, pre-specified subpopulations and stratification groups
LDH | | Elevated | | Normal | |
N | | 49 | | 123 | |
HR | | 0.67 | | 0.69 | |
C.I. (90%) | | (0.33,1.37) | | (0.33,1.40) | |
p-Value | | 0.18 | | 0.19 | |
KRAS | | Mutant | | Wild-type/ND | |
N | | 38 | | 94 | |
HR | | 0.41 | | 0.72 | |
C.I. (90%) | | (0.15,1.16) | | (0.36,1.45) | |
p-Value | | 0.07 | | 0.22 | |
Time since diagnosis of advanced
disease | | >6 mo | | <=6 mo | |
N | | 108 | | 51 | |
HR | | 0.37 | | 1.83 | |
C.I. (90%) | | (0.18,0.77) | | (0.80,4.19) | |
p-Value | | 0.01 | | 0.89 | |
Smoking status | | Never/past | | Current | |
N | | 130 | | 42 | |
HR | | 0.48 | | 1.61 | |
C.I. (90%) | | (0.26,0.90) | | (0.67,3.89) | |
p-Value | | 0.02 | | 0.81 | |
ECOG Performance Status | | 0 | | 1 | |
N | | 80 | | 92 | |
HR | | 0.75 | | 0.72 | |
C.I. (90%) | | (0.33,1.73) | | (0.38,1.35) | |
p-Value | | 0.29 | | 0.19 | |
ND: not determined
22
Table of Contents
Results in several of these patient subpopulations showed a substantially improved survival difference between the control arm and ganetespib arm, reflected in a substantially lower hazard ratio as compared with the all-comers, or intent-to-treat, patient population. These findings have been used to inform the design of the Phase 3 portion of the GALAXY program, in order to enrich for patients likely to derive the greatest benefit from ganetespib treatment.
The adverse event profile was comparable between both arms. The proportion of adenocarcinoma patients with at least one adverse event (AE) was 69% vs. 90%; with grade 3 or 4 AEs was 37% vs. 56%; with AEs leading to treatment discontinuation was 8% vs. 15%; and with AEs with outcome of death were 8% vs. 7%, for D (N=86) vs. G+D (N=81), respectively. The most common AEs, all grades were neutropenia (50% vs. 49%), diarrhea (12% vs. 42%) and fatigue (20% vs. 31%), for D vs. G+D, respectively. Diarrhea and fatigue were predominantly grade 1 and grade 2; the incidence of grade 3 or 4 diarrhea was 0% vs. 4% and grade 3 or 4 fatigue was 2% vs. 5% in D vs. G+D, respectively. The most common grade 3 or 4 AEs were neutropenia (34% vs. 35%), febrile neutropenia (2% vs. 10%), and fatigue (2% vs. 5%). Compared to other Hsp90 inhibitors, there were relatively few reported incidences of ocular toxicity, 4 (5%) in the G+D arm and 1 (1%) in the D arm, all of which were transient and grade 1 or 2. None of the ocular toxicity cases were described as visual impairment.
Changing the biology of tumors: ganetespib inhibition of hypoxia-induced factor (HIF)
HIF-1alpha is a cellular protein whose expression in cells rises rapidly as oxygen supply to the cell is reduced. Preclinical experiments have shown that this protein transcribes (activates) genes involved in properties that drive the growth and spread of tumors including metastasis, angiogenesis, suppression of the immune response, survival of stem or tumor-initiating cells, and resistance to treatment with other therapies including chemotherapy, radiotherapy, and immunotherapy. HIF-1alpha is a known client protein of Hsp90. Experiments by us and our collaborators have shown the treatment with ganetespib strongly reduces the level of HIF-1alpha in cancer cells. Inhibition of HIF-1alpha has been shown in preclinical experiments to reduce the ability of tumor cells to spread (metastasize), grow new blood vessels (angiogenesis), suppress the anti-tumor immune response, and resist chemotherapy.
Our collaborators at Emory University have demonstrated elements of these findings in patients. Tumor samples from rectal cancer patients treated with ganetespib were shown to have significantly lower levels of HIF-1alpha and VEGF, a driver of angiogenesis, after treatment with ganetespib as compared to before treatment. Preclinical experiments confirmed that treatment with ganetespib disrupts blood vessels surrounding tumors comparable to traditional anti-angiogenic agents, and that the reduction in VEGF levels induced by ganetespib is likely to be due to its inhibition of HIF-1alpha.
Reduced angiogenesis is an example of how inhibiting Hsp90 offers a unique approach to changing the biology of tumors. Monoclonal antibodies and kinase inhibitors bind directly to signaling factors that drive cancer cell survival and proliferation to inhibit their activity. Herceptin, for example, binds HER2, Erbitux binds EGFR, and Avastin binds VEGF. The kinase inhibitor Gleevec binds to BCR-ABL, Tarceva binds to EGFR, and Sutent binds to VEGFR, PDGFR and others. Ganetespib, on the other hand, inhibits the production and decreases the expression of many of these growth factors by targeting their reliance on the Hsp90 chaperone. This has the advantages of simultaneously suppressing multiple growth factors, reducing common resistance mechanisms, and providing a distinct and complementary approach to direct growth factor inhibition by antibodies or kinase inhibtors.
Summary of GALAXY Trial Interim Findings
· In the 172 adenocarcinoma patients enrolled as of the September 10 data cutoff, an increase in overall survival was observed in patients treated with ganetespib plus docetaxel. A median overall survival of 7.4 months was observed in the docetaxel control arm, while median overall survival had not been reached in the ganetespib arm. Results for docetaxel were consistent with results from prior second line NSCLC therapy trials.
23
Table of Contents
· Objective response rate and progression-free survival in adenocarcinoma patients were also improved: from 8% to 16%, and from 2.8 months to 4.2 months, in the control arm vs. ganetespib arm, respectively. Overall response and progression-free survival rates in the control arm were consistent with results from prior trials with docetaxel in this setting.
· Results in several GALAXY patient subpopulations, defined by pre-specified clinical and biomarker characteristics, showed a substantially improved survival difference between the control arm and ganetespib arm, as compared with the difference in the all-comer (intent-to-treat) adenocarcinoma patient population. These findings have been incorporated into the design of the Phase 3 portion of the GALAXY program, with the objective of enriching for patients likely to derive the greatest benefit from ganetespib treatment.
· Clinical and preclinical results were presented that suggest potent ganetespib anti-angiogenic activity. Analyses of tumor samples from patients treated with ganetespib showed a reduction of levels of hypoxia induced factor (HIF) and vascular endothelial growth factor (VEGF). In addition, preclinical experiments demonstrated strong inhibition of tumor vasculature by ganetespib. These results suggest ganetespib offers a novel way to inhibit angiogenesis: reducing production of angiogenesis factors, rather than targeting those signaling factors directly with an antibody (such as bevacizumab) or a kinase inhibitor.
· A favorable safety profile was observed with the ganetespib plus docetaxel combination in adenocarcinoma patients. Transient, mild-to-moderate diarrhea was the most common adverse event, consistent with observations from other clinical trials evaluating ganetespib.
GALAXY Trial Plans
Enrollment
We recently completed enrollment of the planned 240 adenocarcinoma patients for the Phase 2b portion of the GALAXY program. The clinical trial protocol specifies that additional patients with elevated LDH or mutant KRAS may be enrolled up to a prespecified maximum number of patients with each of these baseline characteristics. We have elected to continue with the trial and enroll these additional patients. We expect to enroll up to 60 additional patients in these subpopulations, which will bring the total GALAXY Phase 2b enrollment to approximately 300 adenocarcinoma patients.
Transition to Phase 3
We recently completed an End-of-Phase 2 (EOP2) meeting with the Food and Drug Administration (FDA) to review plans for the Phase 3 portion of the GALAXY program. We have incorporated comments from the EOP2 meeting into the Phase 3 protocol and are currently initiating this trial. We expect enrollment to begin early next year.
The Phase 3 trial has the same design as the Phase 2b trial. Adenocarcinoma patients with advanced NSCLC who have received one prior chemotherapy regimen will be randomized 1:1 to treatment with either docetaxel plus ganetespib or docetaxel alone. The same dose and schedule used in the Phase 2b trial will be used in the Phase 3 trial. Patients on both arms will receive docetaxel generally for four to six 21-day cycles, as per standard practice at their treatment center. After completion of docetaxel treatment, patients on the ganetespib arm are eligible to continue to receive ganetespib monotherapy as maintenance treatment. The trial will be conducted in many of the 60 centers across Europe and North America that participated in the Phase 2b trial, together with up to 60 additional centers.
The Phase 3 trial will enroll approximately 500 adenocarcinoma patients and overall survival will be the primary endpoint. Based on results from the Phase 2b trial, the Phase 3 trial will exclude patients who experienced rapidly progressing disease, an estimated 30 to 40% of the total eligible population. The resulting population, which excludes rapidly progressing patients, showed a substantially enhanced survival difference between the ganetespib arm and the control arm in the Phase 2b trial as compared to the survival difference observed in the total population.
Data timing
Based on our current plans and projections, we anticipate final PFS and updated overall survival data from the Phase 2b portion of GALAXY in the first half of 2013, and final overall survival data in the second half of 2013. Two event-driven interim analyses of the overall survival primary endpoint of the Phase 3 trial have been specified. Based on current projections and statistical assumptions, we expect these analyses, together with the final overall survival analysis, to occur in 2014. Additional elements of the Phase 3 trial design will be announced following the start of enrollment
Ganetespib as Monotherapy
ALK+ NSCLC: In June and July 2011 we presented results from a Phase 2 trial of ganetespib administered as a monotherapy in patients with advanced NSCLC at the ASCO Annual Meeting and the International Association for the Study of Lung Cancer (IASLC) 14th World Conference on Lung Cancer, respectively. Patients in this trial had failed to respond to, or experienced disease progression following, numerous prior therapies. In this trial, as in other trials, ganetespib treatment was associated with favorable safety.
24
Table of Contents
Encouraging evidence of clinical activity was also observed in this trial, as evident by the durable objective tumor responses achieved in certain patients, as evaluated by RECIST. The disease control rate, using the standard definition of complete response plus partial response plus stable disease, was 54%. This rate compares favorably with disease control rates observed in trials for approved and experimental agents in a similar broad, pre-treated, advanced NSCLC patient population.
Results presented at these meetings also showed a connection between single-agent ganetespib clinical activity and certain tumor genetic profiles. Four of eight patients who were ALK+, i.e., for whom tumor genetic testing revealed rearrangements in the ALK gene, experienced confirmed partial responses following treatment with ganetespib (a 50% objective response rate, using the standard definition of complete response plus partial response). These responses were durable, with the responding patients remaining on therapy an average of about one year (range 7 to >24 months). Six of these eight patients experienced tumor shrinkage in target lesions, and seven of these eight patients (88%) achieved disease control for eight weeks or more. These results are encouraging when compared to results typically seen with chemotherapy and other agents in these advanced NSCLC treatment settings, for which objective response rates have been in the range of 5-10%.
Although only eight crizotinib-untreated, ALK+ patients were reported in this trial, these results are comparable to those seen with the direct ALK inhibitor Xalkori® (crizotinib), which was granted accelerated approval in August 2011 by the FDA for the treatment of ALK+ NSCLC. In a Phase 1 trial enrolling 136 ALK+ patients and in a single-arm Phase 2 trial in 119 ALK+ patients, crizotinib demonstrated a 50% and a 61% objective response rate, respectively, by investigator review, and a 42% and 51% objective response rate, respectively, by independent review.
Ganetespib has also been shown to be active as monotherapy and in combination with crizotinib in preclinical models of ALK+ NSCLC. Importantly, ALK inhibition via direct inhibition of Hsp90 supports a complementary, rather than competitive, mechanism with crizotinib and other direct ALK inhibitors. Combined with clinical observations so far, these results present strong evidence that Hsp90 inhibition with ganetespib is a promising approach for treating ALK+ NSCLC patients.
To further characterize ganetespib activity in this treatment setting, we recently initiated the CHIARA trial to evaluate ganetespib monotherapy in ALK+ NSCLC patients who have not been previously treated with a direct ALK inhibitor. Based on our current estimates, a pre-specified interim analysis in the first 20 patients enrolled in CHIARA is expected to occur in the first half of 2013.
In addition to CHIARA, a number of cancer centers and cooperative groups have approached us with proposals to support trials evaluating ganetespib in combination with other agents in ALK+ disease. An investigator-sponsored Phase 1/2 trial evaluating ganetespib and crizotinib combinations in patients with ALK+ NSCLC that have not been previously treated with an ALK inhibitor began enrolling patients at Memorial Sloan-Kettering Cancer Center (MSKCC) in New York City earlier this year.
HER2+ and triple negative metastatic breast cancer: At the San Antonio Breast Cancer Symposium in December 2011, researchers from MSKCC presented results from a Phase 2 trial evaluating ganetespib monotherapy in patients with metastatic breast cancer who had been previously treated with multiple lines of chemotherapy or other anti-cancer agents. Results showed that 15% (2/13) of the HER2+ patients experienced a confirmed partial response and an additional 46% (6/13) achieved stable disease. These results with ganetespib in HER2+ disease are consistent with results from an earlier Phase 2 study of 17-AAG, a first-generation Hsp90 inhibitor, in patients who had progressed following treatment with one line of trastuzumab (Herceptin). In that trial 22% (6/27) of patients achieved a partial response and an additional 37% (10/27) achieved stable disease. While in the latter study 17-AAG was given in combination with trastuzumab, in the former study ganetespib was given as a monotherapy. Together, these studies present strong evidence that Hsp90 inhibition is a promising approach for treating HER2+ breast cancer.
Results with ganetespib in patients with triple-negative breast cancer (TNBC) were also reported in December 2011. One of three evaluable patients in the Phase 2 clinical trial experienced significant tumor shrinkage following three doses of ganetespib. An objective response was also reported in a patient with TNBC participating in a ganetespib Phase 1 trial. TNBC represents a difficult-to-treat disease, for which no targeted therapies are currently approved. These results are encouraging, and suggest that ganetespib is active in TNBC.
25
Table of Contents
We recently initiated the ENCHANT trial designed to evaluate ganetespib monotherapy as first-line treatment for both metastatic HER2+ breast cancer and TNBC. Patients in both cohorts will be assessed at baseline and at week 3, 6, and 12 with a combination of PET and CT scans. The primary endpoint of this study is overall response rate at week 12. Up to 35 patients will be enrolled in each of the HER2+ and TNBC cohorts. We expect preliminary results from the ENCHANT trial in the first half of 2013.
In addition to evaluating monotherapy administration of ganetespib in breast cancer, we and our collaborators believe that combination therapy with ganetespib has promise. MSKCC has announced that it will initiate a Phase 1/2 trial evaluating ganetespib in combination with paclitaxel and Herceptin in HER2+ breast cancer, and ganetespib in combination with paclitaxel in TNBC.
Additional oncology indications
In addition to the clinical trials we plan to initiate and continue in 2012, a number of ganetespib trials sponsored by third parties, including cooperative groups, foundations, and individual investigators, have recently initiated or are expected to initiate in 2012. These include
· the trials evaluating ganetespib in breast cancer and in ALK+ lung cancer sponsored by MSKCC described above
· a randomized trial evaluating the combination of fulvestrant and ganetespib in patients with hormone receptor-positive, metastatic breast cancer, being conducted at the Dana-Farber Cancer Institute, which began enrolling patients earlier this year
· a trial evaluating the combination of ganetespib with capecitabine and radiation in patients with locally advanced rectal cancer being conducted at Emory University, which began enrolling patients earlier this year
· a trial evaluating both ganetespib monotherapy and the combination of ganetespib and bortezomib in multiple myeloma, supported by a grant of up to $1 million by the Multiple Myeloma Research Foundation, which began enrolling patients earlier this year
· a randomized trial evaluating the combination of ganetespib and low dose ara-C chemotherapy in elderly patients with acute myeloid leukemia (AML), which began enrolling patients earlier this year.
· a trial evaluating ganetespib in combination with pemetrexed and cisplatin in patients with malignant pleural mesothelioma, being sponsored by Cancer Research UK, which is expected to initiate later this year
Additional ongoing investigator-sponsored trials include trials in prostate cancer, pancreatic cancer, liver cancer, melanoma, and ocular melanoma. In addition to the trials described above, a European cooperative group plans to initiate a randomized trial comparing paclitaxel with and without ganetespib in patients with advanced ovarian cancer in 2013.
Elesclomol (Mitochondria-Targeting Agent)
Elesclomol is a first-in-class, investigational drug candidate that triggers programmed cell death (apoptosis) in cancer cells through a novel mechanism: disrupting cancer cell mitochondrial metabolism. In preclinical experiments, anti-cancer activity of elesclomol has been shown to correlate with certain biomarkers, including LDH, which can distinguish between active mitochondria (sufficient oxygen) and inactive mitochondria (insufficient oxygen). Consistent with these findings in three randomized clinical trials, LDH was an important predictor of elesclomol treatment outcome.
Our current clinical program for elesclomol includes a clinical trial of elesclomol as a monotherapy in AML. In December 2009, we presented results at the American Society for Hematology (ASH) meeting showing that elesclomol was highly active against AML cell lines and primary blast cells from AML patients. In February 2011, we announced that the first patient had been treated in a Phase 1 dose escalation study of elesclomol as a single agent in patients with AML. This trial will enroll up to 36 patients with relapsed or refractory AML and total baseline serum LDH level less than 0.8 times ULN. Patients will be treated with elesclomol sodium on a once-weekly schedule at a starting dose of 200 mg/m2, with dose escalation planned based on safety, tolerability and
26
Table of Contents
pharmacokinetic considerations. The trial is being conducted at Princess Margaret Hospital in Toronto, Canada and at MSKCC in New York.
We are also evaluating the use of elesclomol in combination with paclitaxel in ovarian cancer. In March 2011, the Gynecological Oncology Group (GOG), initiated a Phase 2 clinical trial of elesclomol in combination with paclitaxel for the treatment of persistent or recurrent ovarian, fallopian tube or primary peritoneal cancer for patients with total baseline serum LDH level less than 0.8 times ULN. The GOG is a non-profit organization with the purpose of promoting excellence in the quality and integrity of clinical and basic scientific research in the field of gynecologic malignancies. The National Cancer Institute is providing financial support of up to approximately $300,000 for the trial through its Cancer Therapy Evaluation Program.
STA-9584 (Vascular Disrupting Agent)
STA-9584 is a novel, injectable, small molecule compound that appears to disrupt the blood vessels that supply tumors with oxygen and essential nutrients, and is in preclinical development. In March 2011, we received a $1 million grant from the United States Department of Defense (DoD) for the development of STA-9584 in advanced prostate cancer.
Inflammatory Disease Programs
We have two preclinical-stage programs focusing on treatments for inflammatory diseases. Both of our inflammatory disease programs focus on oral, disease- modifying drug candidates that act through novel mechanisms and could potentially target multiple indications.
CRACM Ion Channel Inhibitors
We have developed novel, small molecule inhibitors of CRACM ion channels expressed on immune cells. Our CRACM ion channel inhibitors have shown strong anti-inflammatory activity in preclinical studies both in vitro and in vivo, inhibiting T cell and mast cell activity, including cytokine release, degranulation, and immune cell proliferation. Potential applications include a wide range of inflammatory diseases and disorders for which modulating T cell and mast cell function has been shown to be critical, including rheumatoid arthritis (RA), asthma, chronic obstructive pulmonary disease (COPD), allergy, transplant rejection, and other autoimmune diseases and inflammatory conditions. We have several promising CRACM inhibitors in preclinical development. Because there are a number of CRACM ion channel targets on immune cells, we believe that CRACM inhibitor compounds can be developed that target different diseases.
Roche CRACM Inhibitor Alliance
In December 2008, as amended in February 2010, February 2011 and July 2011, we formed a strategic alliance with Roche to discover, develop, and commercialize small-molecule drugs targeting CRACM channels, which we refer to as the Roche Agreement. The goal of this alliance was to develop a novel category of oral, disease-modifying agents for the treatment of RA and other autoimmune diseases and inflammatory conditions. The Roche Agreement was terminated by Roche effective on February 16, 2012.
As a result of termination of the Roche Agreement, the research, development and commercialization licenses granted to Roche by us have terminated. Ownership of all rights to all Licensed Compounds (as defined in the agreement) (including the scientific data relating to those compounds) has reverted to us. We have also received an exclusive license to use Roche’s patent rights and know-how to research, develop, manufacture, commercialize and import any collaboration compound, including the Licensed Compounds. We are obligated to pay a low single digit royalty on a country-by-country and Licensed Product-by-Licensed Product (as defined in the agreement) basis upon commercialization of any Licensed Product.
IL-12/23 Inhibitors
We have identified several small molecule IL-12/23 inhibitors that represent a promising opportunity to develop drug candidates that could be administered orally and potentially address a wide range of serious inflammatory diseases with high unmet medical needs.
27
Table of Contents
Financial Operations Overview
Revenue
We have not yet generated any product revenue and do not expect to generate any product revenue in the foreseeable future, if at all. Our revenues to date have been generated primarily through our former collaboration agreements with GSK and Roche. The terms of these agreements included payment to us of upfront license fees, milestone payments, research and development cost sharing and royalties. We will seek to generate revenue from product sales and from future collaborative or strategic relationships. In the future, we expect any revenue we generate will fluctuate from quarter-to-quarter as a result of the timing and amount of payments received and expenses incurred under future collaborations or strategic relationships, and the amount and timing of payments we receive upon the sale of our drug candidates, to the extent any are successfully commercialized.
Research and Development
Research and development expense consists of costs incurred in connection with developing and advancing our drug discovery technology and identifying and developing our drug candidates. We charge all research and development expenses to operations as incurred.
Our research and development expense consists of:
· internal costs associated with research, preclinical and clinical activities;
· payments to third party contract research organizations, investigative sites and consultants in connection with our preclinical and clinical development programs;
· costs associated with drug formulation and supply of drugs for clinical trials;
· personnel related expenses, including salaries, stock-based compensation, benefits and travel; and
· overhead expenses, including rent and maintenance of our facilities, and laboratory and other supplies.
We do not know if we will be successful in developing our drug candidates. We believe that accurately projecting total program-specific expenses through commercialization is not possible at this time. The timing and amount of these expenses will depend upon the costs associated with potential future clinical trials of our drug candidates, and any expansion of our research and development organization, regulatory requirements, advancement of our preclinical programs and product manufacturing costs, many of which cannot be determined with accuracy at this time based on the stage of development of our drug candidates. This is due to the numerous risks and uncertainties associated with the duration and cost of clinical trials, which vary significantly over the life of a project as a result of unanticipated events arising during clinical development, including with respect to:
· the number of clinical sites included in the trial;
· the length of time required to enroll suitable subjects;
· the number of subjects that ultimately participate in the trials; and
· the efficacy and safety results of our clinical trials and the number of additional required clinical trials.
Our expenditures are subject to additional uncertainties, including the terms and timing of regulatory approvals. In addition, we may obtain unexpected or unfavorable results from our clinical trials. We may elect to discontinue, delay or modify clinical trials of some drug candidates or focus on others. A change in the outcome of any of the foregoing variables in the development of a drug candidate could mean a significant change in the costs and timing associated with the development of that drug candidate. For example, if the FDA or other regulatory authority were to require us to conduct clinical trials beyond those that we currently anticipate, or if we experience significant delays in any of our clinical trials, we would be required to expend significant additional financial resources and time on the completion of clinical development. Additionally, future commercial and regulatory factors beyond our control will evolve and therefore impact our clinical development programs and plans over time.
28
Table of Contents
Beyond our current lead drug candidates, we anticipate that we will select drug candidates and research projects for further development on an ongoing basis in response to their preclinical and clinical success, as well as commercial potential.
General and Administrative
General and administrative expense consists primarily of salaries and related expenses for personnel in executive, finance, business and commercial development, investor and medical community relations, human resources and administrative functions. Other costs include stock-based compensation costs, directors’ and officers’ liability insurance premiums, legal costs of pursuing patent protection of our intellectual property, fees for general legal, accounting, public-company requirements and compliance, and other professional services, as well as overhead-related costs not otherwise included in research and development.
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our financial statements which have been prepared in accordance with U.S. generally accepted accounting principles, or GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reported periods. We are required to make estimates and judgments with respect to research contract accruals, the recoverability of long-lived assets, measurement of stock-based compensation and the periods of performance under collaborative research and development agreements. We base our estimates on historical experience, known trends and events, and various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources and the reported amounts of revenues and expenses. Actual results may differ from these estimates under different assumptions or conditions.
You should read the following discussion of our reported financial results in conjunction with the critical accounting policies disclosed in our Annual Report on Form 10-K for the year ended December 31, 2011, as filed with the Securities and Exchange Commission on February 22, 2012. There have been no significant changes to our critical accounting policies in 2012.
Consolidated Results of Operations
Three Months Ended September 30, 2012 Compared with Three Months Ended September 30, 2011
Revenue
| | Three Months Ended
September 30, | | 2012 to 2011 Change | |
| | 2012 | | 2011 | | $ | | % | |
| | (dollars in millions) | | | | | |
Collaboration revenue | | | | | | | | | |
License and milestone revenue—Roche | | $ | — | | $ | 1.1 | | $ | (1.1 | ) | (100 | )% |
Total collaboration revenue | | — | | 1.1 | | (1.1 | ) | (100 | )% |
Grant revenue | | — | | 0.5 | | (0.5 | ) | (100 | )% |
Total revenue | | $ | — | | $ | 1.6 | | $ | (1.6 | ) | (100 | )% |
Roche
In 2012 as compared to 2011, license and milestone revenue under the Roche Agreement decreased by $1.1 million. Roche terminated the Roche Agreement effective February 16, 2012. In the fourth quarter of 2011, upon notification of Roche’s election to terminate the Roche Agreement, we accelerated the recognition of approximately $2.1 million of remaining deferred revenue from the upfront payment because we had no remaining significant performance obligations.
29
Table of Contents
Grant revenue
In 2012 as compared to 2011, grant revenue decreased by $0.5 million. In March 2011, we received a grant from the DoD in the approximate amount of $1 million, for the development of STA-9584 in advanced prostate cancer. We conducted work on this study during the one year grant period from April 2011 through March 2012. We recognized $0 and $0.5 million of grant revenue under this grant in the three months ended September 30, 2012 and 2011, respectively, and $1 million of grant revenue during the one year grant period.
Research and Development Expense
| | Three Months Ended
September 30, | | 2012 to 2011 Change | |
| | 2012 | | 2011 | | $ | | % | |
| | (dollars in millions) | | | | | |
Clinical-stage drug candidates | | | | | | | | | |
Ganetespib | | $ | 11.1 | | $ | 7.8 | | $ | 3.3 | | 42 | % |
Elesclomol | | 0.1 | | 0.9 | | (0.8 | ) | (89 | )% |
Total clinical-stage drug candidates | | 11.2 | | 8.7 | | 2.5 | | 29 | % |
CRACM | | 0.5 | | 1.6 | | (1.1 | ) | (69 | )% |
STA-9584 | | — | | 0.4 | | (0.4 | ) | (100 | )% |
Total research and development | | $ | 11.7 | | $ | 10.7 | | $ | 1.0 | | 9 | % |
Ganetespib
In 2012 as compared to 2011, costs incurred under our ganetespib program increased by $3.3 million, including increases of $1.5 million for personnel-related costs, related research supplies, operational overhead and stock compensation, and $1.8 million for external costs. These increases were principally due to the near completion of patient enrollment in the Phase 2b portion of the GALAXY trial that was initiated in the second quarter of 2011, start-up activities conducted in support of the CHIARA and ENCHANT trials that initiated in 2012, and increases related to the conduct of supporting drug supply and other non-clinical activities. We anticipate that the overall costs under our ganetespib program in 2012 as compared to 2011 will continue to increase as we further advance clinical development, including the transition of the ongoing Phase 2b portion of the GALAXY trial to the planned Phase 3 portion and the conduct of the CHIARA and ENCHANT trials, as well as the conduct of non-clinical supporting activities.
Elesclomol
In 2012 as compared to 2011, costs incurred under our elesclomol program decreased by $0.8 million, including decreases of $0.5 million for personnel-related costs, related research supplies, operational overhead and stock compensation, and $0.3 million for external costs. These decreases were principally related to timing differences in the conduct of the Phase 2 clinical trial of elesclomol in combination with paclitaxel in ovarian cancer that is being conducted by the GOG and the Phase 1 clinical trial of elesclomol as a single agent in AML that were initiated in the first quarter of 2011, as well as supporting clinical drug supply. We anticipate that the overall costs under our elesclomol program in 2012 as compared to 2011 will continue to decrease.
CRACM
In 2012 as compared to 2011, costs incurred under our CRACM program decreased by $1.1 million, principally due to a decrease of $1.1 million for personnel-related costs, related research supplies, operational overhead and stock compensation. This decrease was the result of a lower investment in CRACM research following the conclusion of the Roche Agreement on February 16, 2012. We anticipate that the overall costs under our CRACM program in 2012 as compared to 2011 will continue to decrease.
30
Table of Contents
STA-9584
In 2012 as compared to 2011, costs incurred under our STA-9584 program decreased by $0.4 million, including decreases of $0.1 million for personnel-related costs, related research supplies, operational overhead and stock compensation, and $0.3 million for external costs. In March 2011, we received a $1 million grant from the DoD for the development of STA-9584 in advanced prostate cancer. We conducted work on this study during the one year grant period from April 2011 through March 2012. As our main focus continues to be advancing our ganetespib program, additional investments in our STA-9584 program are dependent upon obtaining funding from additional grants or partnerships.
General and Administrative Expense
| | Three Months Ended
September 30, | | 2012 to 2011 Change | |
| | 2012 | | 2011 | | $ | | % | |
| | (dollars in millions) | | | | | |
General and administrative | | $ | 2.8 | | $ | 3.1 | | $ | (0.3 | ) | (10 | )% |
| | | | | | | | | | | | |
In 2012 as compared to 2011, general and administrative expenses decreased by $0.3 million principally due to an increase of $0.1 million for personnel-related costs, operational overhead and stock compensation, offset by a $0.4 million decrease for external costs. In 2012 as compared to 2011, we anticipate that general and administrative expenses will continue to remain at levels similar to 2011.
Interest Expense, net
| | Three Months Ended
September 30, | | 2012 to 2011 Change | |
| | 2012 | | 2011 | | $ | | % | |
| | (dollars in millions) | | | | | |
Interest expense, net | | $ | 0.4 | | $ | 0.5 | | $ | (0.1 | ) | (20 | )% |
| | | | | | | | | | | | |
In 2012 as compared to 2011, interest expense decreased due to the commencement of principal payments under the GECC Term Loan that began in July 2012. In 2012 as compared to 2011, we anticipate that interest expense will continue to decrease as principal continues to be paid under the GECC Term Loan.
Nine Months Ended September 30, 2012 Compared with Nine Months Ended September 30, 2011
Revenue
| | Nine Months Ended
September 30, | | 2012 to 2011 Change | |
| | 2012 | | 2011 | | $ | | % | |
| | (dollars in millions) | | | | | |
Collaboration revenue | | | | | | | | | |
License and milestone revenue—Roche | | $ | — | | $ | 3.4 | | $ | (3.4 | ) | (100 | )% |
Total collaboration revenue | | — | | 3.4 | | (3.4 | ) | (100 | )% |
Grant revenue | | 0.1 | | 0.7 | | (0.6 | ) | (86 | )% |
Total revenue | | $ | 0.1 | | $ | 4.1 | | $ | (4.0 | ) | (98 | )% |
Roche
In 2012 as compared to 2011, license and milestone revenue under the Roche Agreement decreased by $3.4 million. Roche terminated the Roche Agreement effective February 16, 2012. In the fourth quarter of 2011, upon notification of Roche’s election to terminate the Roche Agreement, we accelerated the recognition of approximately $2.1 million of remaining deferred revenue from the upfront payment because we had no remaining significant performance obligations.
31
Table of Contents
Grant revenue
In 2012 as compared to 2011, grant revenue decreased by $0.6 million. In March 2011, we received a grant from the DoD in the approximate amount of $1 million, for the development of STA-9584 in advanced prostate cancer. We conducted work on this study during the one year grant period from April 2011 through March 2012. We recognized $0.1 million and $0.7 million of grant revenue under this grant in the nine months ended September 30, 2012 and 2011, respectively, and $1 million of grant revenue during the one year grant period.
Research and Development Expense
| | Nine Months Ended
September 30, | | 2012 to 2011 Change | |
| | 2012 | | 2011 | | $ | | % | |
| | (dollars in millions) | | | | | |
Clinical-stage drug candidates | | | | | | | | | |
Ganetespib | | $ | 31.7 | | $ | 21.7 | | $ | 10.0 | | 46 | % |
Elesclomol | | 0.8 | | 3.1 | | (2.3 | ) | (74 | )% |
Total clinical-stage drug candidates | | 32.5 | | 24.8 | | 7.7 | | 31 | % |
CRACM | | 2.2 | | 4.9 | | (2.7 | ) | (55 | )% |
STA-9584 | | 0.2 | | 0.7 | | (0.5 | ) | (71 | )% |
Early stage programs and other | | 0.2 | | 0.2 | | — | | — | % |
Total research and development | | $ | 35.1 | | $ | 30.6 | | $ | 4.5 | | 15 | % |
Ganetespib
In 2012 as compared to 2011, costs incurred under our ganetespib program increased by $10.0 million, including increases of $4.0 million for personnel-related costs, related research supplies, operational overhead and stock compensation, and $6.0 million for external costs. These increases were principally due to the near completion of patient enrollment in the Phase 2b portion of the GALAXY trial that was initiated in the second quarter of 2011, start-up activities conducted in support of the CHIARA and ENCHANT trials that initiated in 2012, and increases related to the conduct of supporting drug supply and other non-clinical activities.
Elesclomol
In 2012 as compared to 2011, costs incurred under our elesclomol program decreased by $2.3 million, including decreases of $1.5 million for personnel-related costs, related research supplies, operational overhead and stock compensation, and $0.8 million for external costs. These decreases were principally related to timing differences in the conduct of the Phase 2 clinical trial of elesclomol in combination with paclitaxel in ovarian cancer that is being conducted by the GOG and the Phase 1 clinical trial of elesclomol as a single agent in AML that were initiated in the first quarter of 2011, as well as supporting clinical drug supply.
CRACM
In 2012 as compared to 2011, costs incurred under our CRACM program decreased by $2.7 million, principally due to a decrease of $2.7 million for personnel-related costs, related research supplies, operational overhead and stock compensation. This decrease was the result of a lower investment in CRACM research following the conclusion of the Roche Agreement on February 16, 2012.
STA-9584
In 2012 as compared to 2011, costs incurred under our STA-9584 program decreased by $0.5 million, principally due to decreases of $0.1 million for personnel-related costs, related research supplies, operational overhead and stock compensation, and $0.4 million for external costs. In March 2011, we received a $1 million grant from the DoD for the development of STA-9584 in advanced prostate cancer. We conducted work on this study during the one year grant period from April 2011 through March 2012.
32
Table of Contents
General and Administrative Expense
| | Nine Months Ended
September 30, | | 2012 to 2011 Change | |
| | 2012 | | 2011 | | $ | | % | |
| | (dollars in millions) | | | | | |
General and administrative | | $ | 8.3 | | $ | 8.7 | | $ | (0.4 | ) | (5 | )% |
| | | | | | | | | | | | |
In 2012 as compared to 2011, general and administrative expenses decreased by $0.4 million principally due to an increase of $0.3 million for personnel-related costs, operational overhead and stock compensation, offset by a $0.7 million decrease for external costs.
Interest Expense, net
| | Nine Months Ended
September 30, | | 2012 to 2011 Change | |
| | 2012 | | 2011 | | $ | | % | |
| | (dollars in millions) | | | | | |
Interest expense, net | | $ | 1.4 | | $ | 1.4 | | $ | — | | — | % |
| | | | | | | | | | | | |
Liquidity and Capital Resources
Cash Flows
The following table provides information regarding our cash position, cash flows and capital expenditures for the nine months ended September 30, 2012 and 2011.
| | Nine Months Ended
September 30, | |
| | 2012 | | 2011 | |
| | (dollars in millions) | |
Cash, cash equivalents and marketable securities | | $ | 55.1 | | $ | 50.7 | |
Working capital | | 37.0 | | 34.9 | |
Cash flows (used in) provided by: | | | | | |
Operating activities | | (41.6 | ) | (36.8 | ) |
Investing activities | | (16.5 | ) | (10.7 | ) |
Financing activities | | 57.4 | | 36.8 | |
| | | | | | | |
Our operating activities used cash of $41.6 million and $36.8 million in 2012 and 2011, respectively. The use of cash in these periods principally resulted from our losses from operations, as adjusted for non-cash charges for depreciation and stock-based compensation, and changes in our working capital accounts.
In 2012, our investing activities used cash of $16.5 million, including the purchases of marketable securities in the amount of $45.9 million and purchases of property and equipment in the amount of $0.4 million, offset by maturities of marketable securities in our investment portfolio in the amount of $29.8 million. In 2011, our investing activities used cash of $10.7 million, including the purchases of marketable securities in the amount of $47.0 million and purchases of property and equipment in the amount of $0.3 million, offset by maturities of marketable securities in our investment portfolio in the amount of $36.6 million.
Our financing activities provided cash of $57.4 million and $36.8 million in 2012 and 2011, respectively. In 2012, we raised approximately $59.7 million in net cash proceeds, including $33.0 million in net proceeds from the sale of 8,050,000 shares of our common stock in a public offering in January 2012 and February 2012, $25.8 million in net proceeds from the sale of 3,976,702 shares of our common stock in a registered direct offering in July 2012 and $0.9 million from the exercise of common stock options. In 2011, we raised $37.2 million in net cash proceeds, including $34.8 million in net proceeds from the sale of 7,191,731 shares of our common stock in an issuer-directed registered direct offering in April 2011, $2.0 million in gross proceeds from the Oxford Term Loan that was executed in March 2011 and $0.4 million from the exercise of common stock options. We repaid $2.3 million and $0.2 million in principal payments in 2012 and 2011, respectively, in connection with the GECC Term Loan and the Oxford Term Loan. In July 2012, we began making 25 equal monthly payments of principal under the GECC Term Loan. We repaid $0.2 million in capital equipment leases in 2011.
33
Table of Contents
Contractual Obligations and Commitments
As of September 30, 2012, there have been no material changes to the contractual obligations and commitments included in our Annual Report on Form 10-K for the fiscal year ended December 31, 2011.
Registered Direct Offering
In July 2012, we entered into subscription agreements with certain directors, including our largest existing stockholder, pursuant to which we sold 3,976,702 shares of our common stock in a registered direct offering at a purchase price of $6.49 per share. These shares were sold directly to these directors without a placement agent, underwriter, broker or dealer. The net proceeds to us were approximately $25.8 million after deducting estimated offering expenses payable by us.
Public Offering
In January and February 2012, we raised approximately $35.4 million in gross proceeds from the sale of an aggregate of 8,050,000 shares of our common stock in a public offering at a public offering price of $4.40 per share, including 7,000,000 shares in the initial closing in January 2012 and 1,050,000 shares in a second closing in February 2012 upon the full exercise of the over-allotment option granted to the underwriters. One of our directors, who is our largest stockholder, purchased 1,136,363 shares in this offering. The net offering proceeds to us were approximately $33.0 million after deducting underwriters’ discounts, fees and commissions, and other offering expenses payable by us.
At-The-Market Issuance Sales Agreement with MLV
On May 2, 2012, we entered into an at-the-market issuance sales agreement, or Sales Agreement, with MLV pursuant to which we may issue and sell shares of our common stock having an aggregate offering price of up to $35 million from time to time, at our option, through MLV as our sales agent, subject to certain terms and conditions. Any shares sold will be sold pursuant to our effective shelf registration statement on Form S-3. We will pay MLV a commission of up to 3% of the gross proceeds of the sale of any shares sold through MLV. To date, no shares have been sold under the Sales Agreement.
Term Loans
General Electric Capital Corporation (GECC)
In September 2010, as amended in November 2010, March 2011, July 2011, January 2012 and July 2012, we entered into a $15 million loan and security agreement with GECC and one other lender, all of which was funded at the closing in September 2010, which we refer to herein as the GECC Term Loan. Interest on the borrowings under the GECC Term Loan accrues at an annual rate of 9.75%. We made interest-only payments through June 2012. In July 2012, we began making 25 equal monthly payments of principal plus accrued interest on the outstanding balance and will pay an exit fee of $525,000 upon the conclusion of the GECC Term Loan. (See Note 9 of the accompanying consolidated financial statements.)
Oxford Finance Corporation (Oxford)
In March 2011, we entered into a $2 million loan and security agreement with Oxford, all of which was funded at the closing, which we refer to herein as the Oxford Term Loan. Interest on the borrowings under the Oxford Term Loan accrues at an annual rate of 13.35%. Beginning in May 2011, we began making 36 equal monthly payments of principal plus accrued interest on the outstanding balance. (See Note 9.)
34
Table of Contents
Liquidity
Funding Requirements
We expect to continue to incur significant operating expenses and capital expenditures and anticipate that our expenses and losses may increase substantially in the foreseeable future as we:
· complete the ongoing clinical trials of ganetespib in solid tumors, including the ongoing Phase 2b portion and the planned Phase 3 portion of the GALAXY trial, and the CHIARA and ENCHANT trials that initiated in 2012, and initiate additional clinical trials of ganetespib if supported by trial results;
· complete preclinical development of an additional Hsp90 inhibitor and initiate clinical trials of this compound, if supported by the preclinical data;
· complete the ongoing clinical trials of elesclomol in AML and ovarian cancers, and initiate additional clinical trials of elesclomol, if supported by trial results;
· complete preclinical development of STA-9584 and initiate clinical trials, if supported by preclinical data;
· advance our CRACM inhibitor into preclinical development and initiate clinical trials, if supported by preclinical data;
· discover, develop, and seek regulatory approval for backups of our current drug candidates and other new drug candidates;
· identify additional compounds or drug candidates and acquire rights from third parties to those compounds or drug candidates through licenses, acquisitions or other means; and
· commercialize any approved drug candidates.
Our funding requirements will depend on a number of factors, including:
· the progress and results of our ongoing clinical trials of ganetespib and elesclomol, and any additional clinical trials we may initiate in the future based on the results of these clinical trials;
· the results of our preclinical studies of any additional Hsp90 inhibitors we may develop, our CRACM inhibitor and STA-9584, and our decision to initiate clinical trials, if supported by the preclinical and other test results;
· uncertainty associated with costs, timing, and outcome of regulatory review of our drug candidates;
· the scope, progress, results, and cost of preclinical development, clinical trials, and regulatory review of any new drug candidates we may discover or acquire;
· the costs of preparing, filing, and prosecuting patent applications and maintaining, enforcing, and defending intellectual property-related claims;
· our ability to establish additional strategic collaborations and licensing or other arrangements on terms favorable to us;
· the costs to satisfy our obligations under potential future collaborations; and
· the timing, receipt, and amount of sales or royalties, if any, from ganetespib, elesclomol, STA-9584, our CRACM inhibitors, our IL-12/23 inhibitors and our other potential products.
35
Table of Contents
As of September 30, 2012, we had $55.1 million in cash, cash equivalents and marketable securities, an increase of $15.4 million from $39.7 million as of December 31, 2011. This increase principally reflects the $58.8 million in net proceeds raised from the public and registered direct offerings of our common stock in 2012, offset by our cash used in operations as discussed under “Cash Flows” above.
We do not anticipate that we will generate product revenue in the foreseeable future, if at all. We expect our continuing operations to use cash over the next several years and such cash use may increase significantly from year to year. While we are engaged in multiple preliminary partnership discussions for each of our currently unpartnered programs, including ganetespib, elesclomol, STA-9584, CRACM, and our IL-12/23 inhibitors, which could result in one or more new partnership agreements that may include upfront payments and cost-sharing provisions, there is no guarantee we will be successful in entering into any such partnership agreements on commercially reasonable terms, if at all, or that we will receive any other revenue through these partnership efforts in the future. Based on our current operating levels, we expect our cash resources will be sufficient to fund operations into the second half of 2013. This estimate assumes that the timing and nature of activities contemplated for 2013 will be conducted subject to the availability of sufficient financial resources. We continue to evaluate additional potential sources of funding, including partnership agreements, cost or risk-sharing arrangements, equity financings, use of our $35 million at-the-market issuance sales agreement with MLV or other sources.
We may require significant additional funds earlier than we currently expect in order to conduct additional clinical trials and conduct additional preclinical and discovery activities. Because of the numerous risks and uncertainties associated with the development and commercialization of our drug candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures associated with our current and anticipated clinical trials.
To the extent our capital resources are insufficient to meet our future capital requirements, we will need to finance our future cash needs through public or private equity offerings, collaboration agreements, debt financings or licensing arrangements. However, additional funding may not be available to us on acceptable terms or at all. In addition, the terms of any financing may adversely affect the holdings or the rights of our stockholders. For example, if we raise additional funds by issuing equity securities or by selling convertible debt securities, further dilution to our existing stockholders may result. If we raise funds through collaboration agreements or licensing arrangements, we may be required to relinquish rights to our technologies or drug candidates, or grant licenses on terms that are not favorable to us.
If adequate funds are not available, we may be required to terminate, significantly modify or delay our research and development programs, reduce our planned commercialization efforts, or obtain funds through collaborators that may require us to relinquish rights to our technologies or drug candidates that we might otherwise seek to develop or commercialize independently. Conversely, we may elect to raise additional funds even before we need them if the conditions for raising capital are favorable, including through offerings of securities pursuant to our shelf registration statement on Form S-3, under which we currently have up to $88.8 million in securities available for issuance, including up to $35 million in shares of common stock that we may offer and sell under the at-the-market issuance sales agreement with MLV.
36
Table of Contents
Certain Factors That May Affect Future Results of Operations
The Securities and Exchange Commission, or SEC, encourages companies to disclose forward-looking information so that investors can better understand a company’s future prospects and make informed investment decisions. This Quarterly Report on Form 10-Q contains such “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995.
Words such as “may,” “anticipate,” “estimate,” “expects,” “projects,” “intends,” “plans,” “believes” and words and terms of similar substance used in connection with any discussion of future operating or financial performance, identify forward-looking statements. All forward-looking statements are management’s present expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those described in the forward-looking statements. These risks include, but are not limited to those set forth under the heading “Risk Factors” contained in Item 1A of our Annual Report on Form 10-K for the year ended December 31, 2011 that we have filed with the SEC.
In light of these assumptions, risks and uncertainties, the results and events discussed in the forward-looking statements contained in this Quarterly Report on Form 10-Q might not occur. Stockholders are cautioned not to place undue reliance on the forward-looking statements, which speak only as of the date of this Quarterly Report on Form 10-Q. We are not under any obligation, and we expressly disclaim any obligation, to update or alter any forward-looking statements, whether as a result of new information, future events or otherwise. All subsequent forward-looking statements attributable to Synta or to any person acting on its behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section.
37
Table of Contents
Item 3. Quantitative and Qualitative Disclosures About Market Risk.
Interest Rate Sensitivity. As of September 30, 2012, we had cash, cash equivalents and marketable securities of $55.1 million consisting of cash deposited in a highly rated financial institution in the United States and in a short-term money market fund, as well as high-grade corporate and government-sponsored agency bonds and commercial paper. The primary objective of our investment activities is to preserve our capital for the purpose of funding operations and we do not enter into investments for trading or speculative purposes. We believe that we did not have material exposure to high-risk investments such as mortgage-backed securities, auction rate securities or other special investment vehicles within our money-market fund investments. We believe that we do not have any material exposure to changes in fair value as a result of changes in interest rates. Declines in interest rates, however, would reduce future investment income.
Capital Market Risk. We currently have no product revenues and depend on funds raised through other sources. One possible source of funding is through further equity offerings. Our ability to raise funds in this manner depends upon capital market forces affecting our stock price.
Item 4. Controls and Procedures.
(a) Evaluation of Disclosure Controls and Procedures. Our principal executive officer and principal financial officer evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act) as of the end of the period covered by this Quarterly Report on Form 10-Q. Based on this evaluation, our principal executive officer and principal financial officer have concluded that our disclosure controls and procedures were effective to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms, and is accumulated and communicated to our management, including our principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure.
(b) Changes in Internal Controls. There were no changes in our internal control over financial reporting, identified in connection with the evaluation of such internal control that occurred during our last fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
38
Table of Contents
PART II - OTHER INFORMATION
Item 1. Legal Proceedings.
We are currently not a party to any material legal proceedings.
Item 1A. Risk Factors.
There have been no material changes to the risk factors included in our Annual Report on Form 10-K for the fiscal year ended December 31, 2011.
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds.
None.
Item 3. Defaults Upon Senior Securities.
None.
Item 4. Mine Safety Disclosures.
Not applicable.
Item 5. Other Information.
None.
Item 6. Exhibits.
(a) Exhibits
31.1 Certification of principal executive officer under Section 302(a) of the Sarbanes-Oxley Act of 2002.
31.2 Certification of principal financial officer under Section 302(a) of the Sarbanes-Oxley Act of 2002.
32.1 Certifications of the principal executive officer and the principal financial officer under Section 906 of the Sarbanes-Oxley Act of 2002.
101* The following materials from Synta Pharmaceuticals Corp.’s Quarterly Report on Form 10-Q for the quarter ended September 30, 2012, formatted in XBRL (eXtensible Business Reporting Language): (i) the Unaudited Condensed Consolidated Balance Sheets, (ii) the Unaudited Condensed Consolidated Statements of Operations, (iii) the Unaudited Condensed Consolidated Statements of Comprehensive Loss, (iv) the Unaudited Condensed Consolidated Statements of Cash Flows, and (v) Notes to Unaudited Condensed Consolidated Financial Statements.
* Users of the XBRL data are advised pursuant to Rule 406T of Regulation S-T that this interactive data file is deemed not filed or part of a registration statement or prospectus for purposes of sections 11 or 12 of the Securities Act of 1933, is deemed not filed for purposes of section 18 of the Securities Exchange Act of 1934, and otherwise is not subject to liability under these sections.
39
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| SYNTA PHARMACEUTICALS CORP. |
| | |
| | |
Date: November 6, 2012 | By: | /s/ Safi R. Bahcall |
| | Safi R. Bahcall, Ph.D. |
| | President and Chief Executive Officer |
| | (principal executive officer) |
| | |
| | |
Date: November 6, 2012 | By: | /s/ Keith S. Ehrlich |
| | Keith S. Ehrlich, C.P.A. |
| | Vice President Finance and Administration, |
| | Chief Financial Officer |
| | (principal accounting and financial officer) |
40