Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-Q
(Mark One)
x | QUARTERLY REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the quarterly period ended March 31, 2014
OR
o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission file number: 001-33277
SYNTA PHARMACEUTICALS CORP.
(Exact name of registrant as specified in its charter)
Delaware (State or other jurisdiction of incorporation or organization) 45 Hartwell Avenue Lexington, Massachusetts (Address of principal executive offices) | | 04-3508648 (I.R.S. Employer Identification No.) 02421 (Zip Code) |
Registrant’s telephone number, including area code: (781) 274-8200
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filer o | | Accelerated filer x |
| | |
Non-accelerated filer o (Do not check if a smaller reporting company) | | Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No x
As of May 2, 2014, the registrant had 93,274,506 shares of common stock outstanding.
Table of Contents
PART I - FINANCIAL INFORMATION
Item 1. Financial Statements.
SYNTA PHARMACEUTICALS CORP.
Condensed Consolidated Balance Sheets
(in thousands, except share and per share amounts)
(unaudited)
| | March 31,
2014 | | December 31,
2013 | |
Assets | | | | | |
Current assets: | | | | | |
Cash and cash equivalents | | $ | 52,034 | | $ | 48,490 | |
Marketable securities | | 26,753 | | 42,986 | |
Prepaid expenses and other current assets | | 1,605 | | 765 | |
Total current assets | | 80,392 | | 92,241 | |
Property and equipment, net | | 1,408 | | 1,553 | |
Other assets | | 384 | | 1,409 | |
Total assets | | $ | 82,184 | | $ | 95,203 | |
Liabilities and Stockholders’ Equity | | | | | |
Current liabilities: | | | | | |
Accounts payable | | $ | 5,810 | | $ | 6,589 | |
Accrued contract research costs | | 12,488 | | 10,407 | |
Other accrued liabilities | | 4,765 | | 5,718 | |
Current portion of capital lease obligations | | 41 | | 42 | |
Current portion of term loans | | 9,261 | | 9,451 | |
Total current liabilities | | 32,365 | | 32,207 | |
Long-term liabilities: | | | | | |
Capital lease obligations, net of current portion | | 75 | | 85 | |
Term loans, net of current portion | | 11,520 | | 13,820 | |
Total long-term liabilities | | 11,595 | | 13,905 | |
Total liabilities | | 43,960 | | 46,112 | |
Stockholders’ equity: | | | | | |
Preferred stock, par value $0.0001 per share Authorized: 5,000,000 shares at March 31, 2014 and December 31, 2013; no shares issued and outstanding at March 31, 2014 and December 31, 2013 | | — | | — | |
Common stock, par value $0.0001 per share Authorized: 200,000,000 shares at March 31, 2014 and December 31, 2013; 87,593,991 and 85,232,506 shares issued and outstanding at March 31, 2014 and December 31, 2013, respectively | | 9 | | 9 | |
Additional paid-in-capital | | 613,180 | | 600,477 | |
Accumulated other comprehensive income | | 4 | | 17 | |
Accumulated deficit | | (574,969 | ) | (551,412 | ) |
Total stockholders’ equity | | 38,224 | | 49,091 | |
Total liabilities and stockholders’ equity | | $ | 82,184 | | $ | 95,203 | |
See accompanying notes to condensed consolidated financial statements.
3
Table of Contents
SYNTA PHARMACEUTICALS CORP.
Condensed Consolidated Statements of Operations
(in thousands, except share and per share amounts)
(unaudited)
| | Three Months Ended
March 31, | |
| | 2014 | | 2013 | |
Revenues: | | | | | |
Total revenues | | $ | — | | $ | — | |
Operating expenses: | | | | | |
Research and development | | 17,583 | | 16,380 | |
General and administrative | | 5,324 | | 3,878 | |
Total operating expenses | | 22,907 | | 20,258 | |
Loss from operations | | (22,907 | ) | (20,258 | ) |
Interest expense, net | | (650 | ) | (470 | ) |
Net loss | | $ | (23,557 | ) | $ | (20,728 | ) |
Net loss per common share: | | | | | |
Basic and diluted net loss per common share | | $ | (0.28 | ) | $ | (0.30 | ) |
Basic and diluted weighted average number of common shares outstanding | | 85,438,127 | | 68,991,371 | |
See accompanying notes to condensed consolidated financial statements.
4
Table of Contents
SYNTA PHARMACEUTICALS CORP.
Condensed Consolidated Statements of Comprehensive Loss
(in thousands)
(unaudited)
| | Three Months Ended
March 31, | |
| | 2014 | | 2013 | |
Net loss | | $ | (23,557 | ) | $ | (20,728 | ) |
Other comprehensive income: | | | | | |
Unrealized (loss) gain on available-for-sale securities | | (13 | ) | 5 | |
Comprehensive loss | | $ | (23,570 | ) | $ | (20,723 | ) |
See accompanying notes to condensed consolidated financial statements.
5
Table of Contents
SYNTA PHARMACEUTICALS CORP.
Condensed Consolidated Statements of Cash Flows
(in thousands)
(unaudited)
| | Three Months Ended
March 31, | |
| | 2014 | | 2013 | |
Cash flows from operating activities: | | | | | |
Net loss | | $ | (23,557 | ) | $ | (20,728 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | |
Stock-based compensation expense | | 2,586 | | 1,216 | |
Depreciation and amortization | | 168 | | 100 | |
Changes in operating assets and liabilities: | | | | | |
Prepaid expenses and other current assets | | 153 | | (689 | ) |
Other assets | | 32 | | (23 | ) |
Accounts payable | | (780 | ) | 58 | |
Accrued contract research costs | | 2,081 | | 140 | |
Other accrued liabilities | | (953 | ) | (2,288 | ) |
Net cash used in operating activities | | (20,270 | ) | (22,214 | ) |
Cash flows from investing activities: | | | | | |
Purchases of marketable securities | | (3,548 | ) | (55,290 | ) |
Maturities of marketable securities | | 19,768 | | 18,950 | |
Purchases of property and equipment | | (23 | ) | (276 | ) |
Net cash provided by (used in) investing activities | | 16,197 | | (36,616 | ) |
Cash flows from financing activities: | | | | | |
Proceeds from issuance of common stock, net of transaction costs, and exercise of common stock options | | 10,118 | | 1,045 | |
Proceeds from term loans | | — | | 13,210 | |
Payment of term loans | | (2,490 | ) | (1,972 | ) |
Payment of capital lease obligations | | (11 | ) | (3 | ) |
Net cash provided by financing activities | | 7,617 | | 12,280 | |
Net increase (decrease) in cash and cash equivalents | | 3,544 | | (46,550 | ) |
Cash and cash equivalents at beginning of period | | 48,490 | | 81,512 | |
Cash and cash equivalents at end of period | | $ | 52,034 | | $ | 34,962 | |
Supplemental disclosure of cash flow information: | | | | | |
Cash paid for interest | | $ | 560 | | $ | 789 | |
See accompanying notes to condensed consolidated financial statements.
6
Table of Contents
SYNTA PHARMACEUTICALS CORP.
Notes to Condensed Consolidated Financial Statements
(unaudited)
(1) Nature of Business
Synta Pharmaceuticals Corp. (the Company) was incorporated in March 2000 and commenced operations in July 2001. The Company is a biopharmaceutical company focusing on discovering, developing and commercializing small molecule drugs to extend and enhance the lives of patients with severe medical conditions, including cancer and chronic inflammatory diseases.
The Company is subject to risks common to emerging companies in the drug development and pharmaceutical industry including, but not limited to, uncertainty of product development and commercialization, lack of marketing and sales history, dependence on key personnel, uncertainty of market acceptance of products, product liability, uncertain protection of proprietary technology, potential inability to raise additional financing and compliance with the U.S. Food and Drug Administration and other government regulations.
The Company may require significant additional funds earlier than it currently expects in order to conduct additional clinical trials and continue to fund its operations. There can be no assurances, however, that additional funding will be available on favorable terms, or at all. If adequate funds are not available, the Company may be required to delay, significantly modify or terminate its research and development programs or reduce its planned commercialization efforts.
(2) Summary of Significant Accounting Policies
The accompanying condensed consolidated financial statements are unaudited, have been prepared on the same basis as the annual financial statements and, in the opinion of management, reflect all adjustments, which include only normal recurring adjustments necessary to present fairly the Company’s financial position as of March 31, 2014 and the consolidated results of operations, comprehensive loss and cash flows for the three months ended March 31, 2014 and 2013. The preparation of financial statements in conformity with accounting principles generally accepted in the United States (GAAP) requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from these estimates. The results of operations for the three months ended March 31, 2014 are not necessarily indicative of the results to be expected for the year ending December 31, 2014 or for any other interim period or any other future year. For more complete financial information, these condensed consolidated financial statements, and the notes hereto, should be read in conjunction with the audited financial statements for the year ended December 31, 2013 included in the Company’s Annual Report on Form 10-K as filed with the Securities and Exchange Commission on March 11, 2014.
Principles of Consolidation
The condensed consolidated financial statements include the financial statements of the Company and its wholly owned subsidiaries. All significant intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect certain reported amounts of assets and liabilities, disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting periods. Significant items subject to such estimates and assumptions include contract research accruals, recoverability of long-lived assets, measurement of stock-based compensation, and the periods of performance under collaborative research and development agreements. The Company bases its estimates on historical experience and various other assumptions that management believes to be reasonable under the circumstances. Changes in estimates are recorded in the period in which they become known. Actual results could differ from those estimates.
Cash and Cash Equivalents
The Company considers all highly liquid investments with original maturities of three months or less at the date of purchase and an investment in a money market fund to be cash equivalents. Changes in the level of cash and cash equivalents may be affected by changes in investment portfolio maturities, as well as actual cash disbursements to fund operations.
The primary objective of the Company’s investment activities is to preserve its capital for the purpose of funding operations and the
7
Table of Contents
Company does not enter into investments for trading or speculative purposes. The Company invests in money market funds and high-grade, short-term commercial paper and corporate bonds, which are subject to minimal credit and market risk. The Company’s cash is deposited in a highly rated financial institution in the United States. Declines in interest rates, however, would reduce future investment income.
Marketable Securities
Marketable securities consist of investments in high-grade corporate obligations, and government and government agency obligations that are classified as available-for-sale. Since these securities are available to fund current operations they are classified as current assets on the consolidated balance sheets.
The Company adjusts the cost of available-for-sale debt securities for amortization of premiums and accretion of discounts to maturity. The Company includes such amortization and accretion as a component of interest expense, net. Realized gains and losses and declines in value, if any, that the Company judges to be other-than-temporary on available-for-sale securities are reported as a component of interest expense, net. To determine whether an other-than-temporary impairment exists, the Company considers whether it intends to sell the debt security and, if the Company does not intend to sell the debt security, it considers available evidence to assess whether it is more likely than not that it will be required to sell the security before the recovery of its amortized cost basis. During the three months ended March 31, 2014 and 2013, the Company determined it did not have any securities that were other-than-temporarily impaired.
Marketable securities are stated at fair value, including accrued interest, with their unrealized gains and losses included as a component of accumulated other comprehensive income or loss, which is a separate component of stockholders’ equity. The fair value of these securities is based on quoted prices and observable inputs on a recurring basis. Realized gains and losses are determined on the specific identification method. During the three months ended March 31, 2014 and 2013, the Company did not have any realized gains or losses on marketable securities.
Fair Value of Financial Instruments
The carrying amounts of the Company’s financial instruments, which include cash equivalents, marketable securities and term loan obligations, approximate their fair values. The fair value of the Company’s financial instruments reflects the amounts that would be received upon sale of an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. The fair value hierarchy has the following three levels:
Level 1—quoted prices in active markets for identical assets and liabilities.
Level 2—observable inputs other than Level 1 inputs. Examples of Level 2 inputs include quoted prices in active markets for similar assets or liabilities and quoted prices for identical assets or liabilities in markets that are not active.
Level 3—unobservable inputs that reflect the Company’s own assumptions about the assumptions market participants would use in pricing the asset or liability.
Financial assets and liabilities are classified in their entirety based on the lowest level of input that is significant to the fair value measurement. The Company measures the fair value of its marketable securities by taking into consideration valuations obtained from third-party pricing sources. The pricing services utilize industry standard valuation models, including both income and market based approaches, for which all significant inputs are observable, either directly or indirectly, to estimate fair value. These inputs include reported trades of and broker-dealer quotes on the same or similar securities, issuer credit spreads, benchmark securities and other observable inputs. As of March 31, 2014, the Company’s financial assets valued based on Level 1 inputs consisted of cash and cash equivalents in a money market fund and its financial assets valued based on Level 2 inputs consisted of high-grade corporate bonds and commercial paper. During the three months ended March 31, 2014 and 2013, the Company did not have any transfers of financials assets between Levels 1 and 2. As of March 31, 2014, the Company did not have any financial liabilities that were recorded at fair value on the balance sheet. The disclosed fair value of the Company’s term loan obligations is determined using current applicable rates for similar instruments as of the balance sheet date. The carrying value of the Company’s term loan obligations approximates fair value as the Company’s interest rate yield is near current market rate yields. The disclosed fair value of the Company’s term loan obligations is based on Level 3 inputs.
8
Table of Contents
Revenue Recognition
Collaboration and License Agreements
The Company’s principal source of revenue to date has been its former collaboration and license agreements, which included upfront license payments, development milestones, reimbursement of research and development costs, potential profit sharing payments, commercial and sales-based milestones and royalties. The accounting for collaboration and license agreements requires subjective analysis and requires management to make estimates and assumptions about whether deliverables within multiple-element arrangements are separable from the other aspects of the contractual arrangement into separate units of accounting and to determine the arrangement consideration to be allocated to each unit of accounting.
For multiple-element arrangements entered into or materially modified after January 1, 2011, the Company follows the provisions of Financial Accounting Standards Board (FASB) Accounting Standards Update (ASU) No. 2009-13— Multiple-deliverable Revenue Arrangements ( ASU No. 2009-13). This standard addresses the determination of the unit(s) of accounting for multiple-element arrangements and how an arrangement’s consideration should be allocated to each unit of accounting.
Pursuant to this standard, each required deliverable is evaluated to determine if it qualifies as a separate unit of accounting. For the Company this determination includes an assessment as to whether the deliverable has “stand-alone value” to the customer separate from the undelivered elements. The arrangement’s consideration is then allocated to each separate unit of accounting based on the relative selling price of each deliverable. The estimated selling price of each deliverable is determined using the following hierarchy of values: (i) vendor-specific objective evidence of fair value, (ii) third-party evidence of selling price, or (iii) the Company’s best estimate of the selling price (BESP). The BESP reflects the Company’s best estimate of what the selling price would be if the deliverable was regularly sold by it on a stand-alone basis. The Company expects, in general, to use BESP for allocating consideration to each deliverable in future collaboration agreements. In general, the consideration allocated to each unit of accounting is then recognized as the related goods or services are delivered limited to the consideration not contingent upon future deliverables.
The Company accounts for development milestones under collaboration and license agreements pursuant to ASU No. 2010-17Milestone Method of Revenue Recognition (ASU No. 2010-17). ASU No. 2010-17 codified a method of revenue recognition that has been common practice. Under this method, contingent consideration from research and development activities that is earned upon the achievement of a substantive milestone is recognized in its entirety in the period in which the milestone is achieved. At the inception of each arrangement that includes milestone payments, the Company evaluates whether each milestone is substantive. This evaluation includes an assessment of whether (a) the consideration is commensurate with either (1) the entity’s performance to achieve the milestone, or (2) the enhancement of the value of the delivered item(s) as a result of a specific outcome resulting from the entity’s performance to achieve the milestone, (b) the consideration relates solely to past performance and (c) the consideration is reasonable relative to all of the deliverables and payment terms within the arrangement. The Company evaluates factors such as the scientific, clinical, regulatory, commercial and other risks that must be overcome to achieve the respective milestone, the level of effort and investment required and whether the milestone consideration is reasonable relative to all deliverables and payment terms in the arrangement in making this assessment. The Company does not have any ongoing collaboration and license agreements under which milestones may be achieved.
Royalty revenues are based upon a percentage of net sales. Royalties from the sales of products will be recorded on the accrual basis when results are reliably measurable, collectability is reasonably assured and all other revenue recognition criteria are met. Commercial and sales-based milestones, which are based upon the achievement of certain agreed-upon sales thresholds, will be recognized in the period in which the respective sales threshold is achieved and collectability is reasonably assured. The Company does not have any ongoing collaboration and license agreements under which royalties or commercial and sales-based milestones may be achieved.
Stock-Based Compensation
The Company recognizes stock-based compensation expense based on the grant date fair value of stock options granted to employees, officers and directors. The Company uses the Black-Scholes option pricing model to determine the grant date fair value as it is the most appropriate valuation method for its option grants. The Black-Scholes model requires inputs for risk-free interest rate, dividend yield, volatility and expected lives of the options. Expected volatility is based upon the weighted average historical volatility data of the Company’s common stock. The risk-free rate for periods within the expected life of the option is based on the U.S. Treasury
9
Table of Contents
yield curve in effect at the time of the grant. The expected lives for options granted represent the period of time that options granted are expected to be outstanding. The Company uses the simplified method for determining the expected lives of options. The Company estimates the forfeiture rate based on historical data. This analysis is re-evaluated at least annually and the forfeiture rate is adjusted as necessary.
For awards with graded vesting, the Company allocates compensation costs on a straight-line basis over the requisite service period. The Company amortizes the fair value of each option over each option’s service period, which is generally the vesting period.
Certain of the employee stock options granted by the Company are structured to qualify as incentive stock options (ISOs). Under current tax regulations, the Company does not receive a tax deduction for the issuance, exercise or disposition of ISOs if the employee meets certain holding requirements. If the employee does not meet the holding requirements, a disqualifying disposition occurs, at which time the Company may receive a tax deduction. The Company does not record tax benefits related to ISOs unless and until a disqualifying disposition is reported. In the event of a disqualifying disposition, the entire tax benefit is recorded as a reduction of income tax expense. The Company has not recognized any income tax benefit for its share-based compensation arrangements due to the fact that the Company does not believe it is more likely than not it will realize the related deferred tax assets.
Comprehensive Loss
Comprehensive loss is defined as the change in equity of a business enterprise during a period from transactions and other events and circumstances from non-owner sources. Changes in unrealized gains and losses on marketable securities represent the only difference between the Company’s net loss and comprehensive loss.
Segment Reporting
Operating segments are determined based on the way management organizes its business for making operating decisions and assessing performance. The Company has a single operating segment, the discovery, development and commercialization of drug products.
Basic and Diluted Loss Per Common Share
Basic net loss per share is computed using the weighted average number of common shares outstanding during the period, excluding restricted stock that has been issued but is not yet vested. Diluted net loss per common share is computed using the weighted average number of common shares outstanding and the weighted average dilutive potential common shares outstanding using the treasury stock method. However, for the three months ended March 31, 2014 and 2013, diluted net loss per share is the same as basic net loss per share as the inclusion of weighted average shares of unvested restricted common stock and common stock issuable upon the exercise of stock options would be anti-dilutive.
The following table summarizes outstanding securities not included in the computation of diluted net loss per common share as their inclusion would be anti-dilutive:
| | March 31, | |
| | 2014 | | 2013 | |
Common stock options | | 8,179,958 | | 7,084,855 | |
Unvested restricted common stock | | 35,000 | | 98,814 | |
(3) Cash, Cash Equivalents and Marketable Securities
A summary of cash, cash equivalents and available-for-sale marketable securities held by the Company as of March 31, 2014 and December 31, 2013 was as follows (see Note 2):
| | March 31, 2014 | |
| | Cost | | Unrealized
gains | | Unrealized
losses | | Fair
value | |
| | (in thousands) | |
Cash and cash equivalents: | | | | | | | | | |
Cash and money market funds (Level 1) | | $ | 48,664 | | $ | — | | $ | — | | $ | 48,664 | |
Corporate debt securities due within 3 months of date of purchase (Level 2) | | 3,370 | | — | | — | | 3,370 | |
Total cash and cash equivalents | | $ | 52,034 | | $ | — | | $ | — | | $ | 52,034 | |
Marketable securities: | | | | | | | | | |
Corporate debt securities due within 1 year of date of purchase (Level 2) | | 26,749 | | 5 | | (1 | ) | 26,753 | |
Total cash, cash equivalents and marketable securities | | $ | 78,783 | | $ | 5 | | $ | (1 | ) | $ | 78,787 | |
10
Table of Contents
| | December 31, 2013 | |
| | Cost | | Unrealized
gains | | Unrealized
losses | | Fair
value | |
| | (in thousands) | |
Cash and cash equivalents: | | | | | | | | | |
Cash and money market funds (Level 1) | | $ | 40,586 | | $ | — | | $ | — | | $ | 40,586 | |
Corporate debt securities due within 3 months of date of purchase (Level 2) | | 7,904 | | — | | — | | 7,904 | |
Total cash and cash equivalents | | $ | 48,490 | | $ | — | | $ | — | | $ | 48,490 | |
Marketable securities: | | | | | | | | | |
Corporate debt securities due within 1 year of date of purchase (Level 2) | | 42,969 | | 18 | | (1 | ) | 42,986 | |
Total cash, cash equivalents and marketable securities | | $ | 91,459 | | $ | 18 | | $ | (1 | ) | $ | 91,476 | |
(4) Property and Equipment
Property and equipment consist of the following:
| | March 31,
2014 | | December 31,
2013 | |
| | (in thousands) | |
Laboratory equipment | | $ | 12,656 | | $ | 12,681 | |
Leasehold improvements | | 4,958 | | 4,958 | |
Computers and software | | 3,069 | | 3,220 | |
Furniture and fixtures | | 1,170 | | 1,170 | |
| | 21,853 | | 22,029 | |
Less accumulated depreciation and amortization | | (20,445 | ) | (20,476 | ) |
| | $ | 1,408 | | $ | 1,553 | |
Depreciation and amortization expenses of property and equipment, including equipment purchased under capital leases, were approximately $0.2 million and $0.1 million for the three months ended March 31, 2014 and 2013, respectively.
(5) Stockholders’ Equity
Common Stock
Each common stockholder is entitled to one vote for each common share of stock held. The common stock will vote together with all other classes and series of stock of the Company as a single class on all actions to be taken by the Company’s stockholders. Each share of common stock is entitled to receive dividends, as and when declared by the Company’s board of directors.
The Company has never declared cash dividends on its common stock and does not expect to do so in the foreseeable future.
11
Table of Contents
At-The-Market Issuance Sales Agreement
On May 2, 2012, the Company entered into an at-the-market issuance sales agreement, as amended, (Sales Agreement) with MLV & Co. LLC (MLV), pursuant to which the Company may issue and sell shares of its common stock having an aggregate offering price of up to $28 million from time to time, at the Company’s option, through MLV as its sales agent. Sales of common stock through MLV may be made by any method that is deemed an “at-the-market” offering as defined in Rule 415 promulgated under the Securities Act of 1933, as amended, including by means of ordinary brokers’ transactions at market prices, in block transactions or as otherwise agreed by the Company and MLV. Subject to the terms and conditions of the Sales Agreement, MLV will use commercially reasonable efforts to sell the common stock based upon the Company’s instructions (including any price, time or size limits or other customary parameters or conditions the Company may impose). The Company is not obligated to make any sales of its common stock under the Sales Agreement. Any shares sold will be sold pursuant to the Company’s effective shelf registration statement on Form S-3 (File No. 333-176022). The Company will pay MLV a commission of up to 3% of the gross proceeds. The Sales Agreement will terminate upon the earlier of the sale of all common stock subject to the Sales Agreement or termination of the Sales Agreement by the Company or MLV. During March and April 2014, the Company sold an aggregate of 6,588,875 shares of common stock pursuant to the Sales Agreement for an aggregate of approximately $28.0 million in gross proceeds at an average selling price of $4.25 per share. Net proceeds to the Company were approximately $27.3 million after deducting commissions and other transactions costs, including approximately $9.3 million from the sale of 2,158,360 shares in March 2014 and approximately $18.0 million from the sale of 4,430,515 shares in April 2014, following which the Sales Agreement was terminated.
(6) Stock-Based Compensation
The Company’s 2006 Stock Plan provides for the grant of incentive stock options, non-statutory stock options and non-vested restricted stock to employees, officers, directors and consultants of the Company. In January 2014, the number of shares of common stock reserved for issuance under the 2006 Stock Plan was increased from 9,000,000 to 10,300,000 pursuant to an “evergreen” provision, which provides for an annual increase based on the lesser of 1,300,000 shares, 5% of the Company’s then outstanding shares of common stock, or such other amount as the board of directors may determine. This increase was approved by the board of directors in December 2013. The administration of the 2006 Stock Plan is under the general supervision of the compensation committee of the board of directors. The exercise price of the stock options is determined by the compensation committee of the board of directors, provided that incentive stock options are granted at not less than fair market value of the common stock on the date of grant and expire no later than ten years from the date the option is granted. Options generally vest over four years. As of March 31, 2014, the Company had options outstanding to purchase 8,179,958 shares of its common stock, which includes options outstanding under its 2001 Stock Plan that was terminated in March 2006, and had 35,000 restricted shares of common stock outstanding. As of March 31, 2014, 1,250,918 shares were available for future issuance.
The following table summarizes stock option activity during the three months ended March 31, 2014:
| | Shares | | Weighted average
exercise price | |
Outstanding at January 1, 2014 | | 6,814,417 | | $ | 6.90 | |
Options granted | | 1,743,946 | | 6.15 | |
Options exercised | | (203,125 | ) | 4.16 | |
Options cancelled | | (175,280 | ) | 7.05 | |
Outstanding at March 31, 2014 | | 8,179,958 | | $ | 6.80 | |
Exercisable at March 31, 2014 | | 4,590,267 | | $ | 6.86 | |
The total cash received by the Company as a result of stock option exercises during the three months ended March 31, 2014 and 2013 was $0.8 million and $1.0 million, respectively. The weighted-average grant date fair values of options granted during the three months ended March 31, 2014 and 2013 were $5.02 and $7.77, respectively.
Non-Vested (“Restricted”) Stock Awards With Service Conditions
The Company’s share-based compensation plan provides for awards of restricted shares of common stock to employees, officers, directors and consultants to the Company. Restricted stock awards are subject to forfeiture if employment or service terminates during the prescribed retention period. Restricted shares vest over the service period. The total fair value of restricted stock that vested during the three months ended March 31, 2014 and 2013 was $0.1 million and $0.1 million, respectively.
12
Table of Contents
The following table summarizes unvested restricted share activity during the three months ended March 31, 2014:
| | Shares | | Weighted
average
grant date
fair value | |
Outstanding at January 1, 2014 | | 45,000 | | $ | 4.63 | |
Vested | | (10,000 | ) | 5.00 | |
Outstanding at March 31, 2014 | | 35,000 | | $ | 4.53 | |
Stock-Based Compensation Expense
For the three months ended March 31, 2014 and 2013, the fair value of each employee stock option award was estimated on the date of grant based on the fair value method using the Black-Scholes option pricing valuation model with the following weighted average assumptions:
| | Three Months
Ended March 31, | |
| | 2014 | | 2013 | |
Risk-free interest rate | | 1.84 | % | 1.10 | % |
Expected life in years | | 6.25 | | 6.25 | |
Volatility | | 104 | % | 101 | % |
Expected dividend yield | | — | | — | |
Stock-based compensation expense during the three months ended March 31, 2014 and 2013 was as follows (in thousands):
| | Three Months
Ended March 31, | |
| | 2014 | | 2013 | |
Stock-based compensation expense by type of award: | | | | | |
Employee stock options | | $ | 2,529 | | $ | 1,131 | |
Restricted stock | | 57 | | 85 | |
Total stock-based compensation expense | | $ | 2,586 | | $ | 1,216 | |
| | | | | |
Effect of stock-based compensation expense by line item: | | | | | |
Research and development | | $ | 1,131 | | $ | 629 | |
General and administrative | | 1,455 | | 587 | |
Total stock-based compensation expense included in net loss | | $ | 2,586 | | $ | 1,216 | |
Unrecognized stock-based compensation expense as of March 31, 2014 was as follows (dollars in thousands):
| | Unrecognized
stock
compensation
expense as of
March 31,
2014 | | Weighted
average
remaining
period
(in years) | |
Employee stock options | | $ | 16,538 | | 3.08 | |
Restricted stock | | 151 | | 1.90 | |
Total | | $ | 16,689 | | 3.07 | |
13
Table of Contents
7) Other Accrued Liabilities
Other accrued liabilities consist of the following:
| | March 31,
2014 | | December 31,
2013 | |
| | (in thousands) | |
Compensation and benefits | | $ | 2,162 | | $ | 3,137 | |
Professional fees | | 1,621 | | 1,585 | |
Other | | 982 | | 996 | |
| | $ | 4,765 | | $ | 5,718 | |
(8) Co-Development Agreement
In July 2011, the Company entered into a co-development agreement with a clinical research organization (CRO) for the conduct of certain company-sponsored clinical trials. Under the co-development agreement, this CRO was performing clinical research services under a reduced fee structure in exchange for a share of licensing payments and commercial revenues, if any, resulting from the product under development up to a specified maximum payment, which is defined as a multiple of the fee reduction realized. Research and development expenses were being recognized based on the reduced fee structure and expected payments will be recorded in the future if and when payment is probable. The maximum amount of the service fee discount was realized in the year ended December 31, 2013.
(9) Term Loans
General Electric Capital Corporation
In March 2013, the Company amended its loan and security agreement entered into in September 2010 with General Electric Capital Corporation (GECC) and another lender (the GECC Term Loan) and obtained $12.9 million in additional loan funding and, as a result, increased the principal balance to $22.5 million at March 31, 2013. This amendment was accounted for as a loan modification. Interest on the borrowings under the GECC Term Loan remains at the annual rate of 9.75%. The Company made interest-only payments for the period from April 2013 through December 2013. In January 2014, the Company began making 30 equal monthly payments of principal plus accrued interest on the outstanding balance. During the period from July 2012 through March 2013, the Company made equal monthly payments of principal plus accrued interest on the outstanding balance. Prior to July 2012, the Company made interest-only payments.
The Company has paid various transaction fees and expenses in connection with the GECC Term Loan, which are deferred and are being amortized as interest expense over the remaining term of the GECC Term Loan. In addition, the Company is obligated to pay an exit fee of $788,000 at the time of the final principal payment which is being accreted and expensed as interest over the remaining term of the GECC Term Loan. In the three months ended March 31, 2014 and 2013, the Company recognized GECC Term Loan interest expense of $0.6 million and $0.4 million, respectively, of which $0.1 million and $0.2 million, respectively, was in connection with these transaction and exit fees and expenses. The Company may prepay the full amount of the GECC Term Loan, subject to prepayment premiums under certain circumstances. The Company did not issue any warrants in connection with the GECC Term Loan.
The GECC Term Loan is secured by substantially all of the Company’s assets, except its intellectual property. The Company has granted GECC a springing security interest in its intellectual property in the event the Company is not in compliance with certain cash usage covenants, as defined therein. The GECC Term Loan contains restrictive covenants, including the requirement for the Company to receive the prior written consent of GECC to enter into loans, other than up to $4.0 million of equipment financing, restrictions on the declaration or payment of dividends, restrictions on acquisitions, and customary default provisions that include material adverse events, as defined therein. The Company has determined that the risk of subjective acceleration under the material adverse events clause is remote and therefore has classified the outstanding principal in current and long-term liabilities based on the timing of scheduled principal payments.
14
Table of Contents
Oxford Finance Corporation
In March 2011, the Company entered into a loan and security agreement with Oxford Finance Corporation (Oxford) and received $2.0 million in loan funding (the Oxford Term Loan). Interest on the borrowings under the Oxford Term Loan accrues at an annual rate of 13.35%. Beginning in May 2011, the Company began making 36 equal monthly payments of principal plus accrued interest on the outstanding balance. In December 2012, the Company entered into a loan modification agreement, as amended, under which the Company could elect to draw down up to an additional $0.6 million in equipment financing until June 30, 2013 that would be payable in 36 equal monthly payments of principal plus accrued interest on the outstanding balance. As of June 30, 2013, the Company had fully utilized the $0.6 million in additional equipment financing. The Company recognized approximately $20,000 and $29,000 in the three months ended March 31, 2014 and 2013, respectively, in interest expense related to the outstanding principal under the Oxford Term Loan. In addition to the interest payable under the Oxford Term Loan, the Company paid approximately $108,000 of administrative and legal fees and expenses in connection with the Oxford Term Loan. These expenses have been deferred and are being expensed over the term of the Oxford Term Loan. The Company did not issue any warrants in connection with the Oxford Term Loan. The Company may prepay the full amount of the Oxford Term Loan, subject to prepayment premiums under certain circumstances. Oxford has the right to require the Company to prepay the full amount of the Oxford Term Loan if the Company prepays the full amount of the GECC Term Loan under certain circumstances.
The Oxford Term Loan is secured by certain laboratory and office equipment, furniture and fixtures. In connection with the Oxford Term Loan, Oxford and GECC entered into a Lien Subordination Agreement, whereby GECC granted Oxford a first priority perfected security interest in the loan collateral. The Oxford Term Loan contains restrictive covenants, including the requirement for the Company to receive the prior written consent of Oxford to enter into acquisitions in which the Company incurs more than $2.0 million of related indebtedness, and customary default provisions that include material adverse events, as defined therein. The Company has determined that the risk of subjective acceleration under the material adverse events clause is remote and therefore has classified the outstanding principal in current and long-term liabilities based on the timing of scheduled principal payments.
Future principal payments under the GECC and Oxford Term Loans as of March 31, 2014 are approximately as follows (in thousands):
Years ending December 31, | | | |
2014 | | $ | 6,961 | |
2015 | | 9,214 | |
2016 | | 4,606 | |
Total principal payments | | 20,781 | |
Less current portion | | (9,261 | ) |
Long term portion | | $ | 11,520 | |
(10) Subsequent Events
Registered Direct Offering
In April 2014, the Company sold 1,250,000 shares of its common stock at a purchase price of $4.01 per share in a registered direct offering to an affiliate of a director who is its largest stockholder. These shares were sold directly without a placement agent, underwriter, broker or dealer. The net proceeds to the Company were approximately $5.0 million after deducting offering expenses payable by the Company.
New At-The-Market Issuance Sales Agreement
In May 2014, the Company entered into a new at-the-market issuance sales agreement, or New Sales Agreement, with MLV pursuant to which it may issue and sell shares of its common stock having an aggregate offering price of up to $40 million from time to time, at its option, through MLV as its sales agent, subject to similar terms and conditions. The shares would be sold pursuant to its effective shelf registration statement on Form S-3 (File No. 333-187242). The Company would pay MLV a commission of up to 3% of the gross proceeds of the sale of any shares sold through MLV. To date, no shares have been sold under the New Sales Agreement.
15
Table of Contents
License Arrangement
In May 2014, the Company entered into a license arrangement for its CRACM program, including two lead candidates and the associated intellectual property portfolio, with PRCL Research Inc. (PRCL), a company funded by TVM Life Science Venture VII and the Fonds de Solidarité des Travailleurs du Québec, based in Montreal, Canada. PRCL plans to develop to proof-of-concept, one of the two lead candidates licensed from us. Synta was granted a minority interest in PRCL in exchange for its contribution of know-how and intellectual property and will also hold a seat on PRCL’s Board of Directors. Synta will not be required to provide any research funding or capital contributions to PRCL. Synta will be reimbursed by PRCL for any ongoing intellectual property management costs in connection with the contributed intellectual property and may conduct preclinical research activities which would be reimbursed by PRCL. If and when proof-of-concept is reached with either drug candidate, Eli Lilly and Company, which is an investor in TVM, will manage the development program through one of its divisions and will have an option to acquire PRCL or its assets at the then fair value.
16
Table of Contents
Item 2. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
You should read this discussion together with the condensed consolidated financial statements, related notes and other financial information included elsewhere in this Quarterly Report on Form 10-Q. The following discussion may contain predictions, estimates and other forward-looking statements that involve a number of risks and uncertainties, including those discussed under “Risk Factors” in our Annual Report on Form 10-K for the year ended December 31, 2013 filed with the Securities and Exchange Commission on March 11, 2014. These risks could cause our actual results to differ materially from any future performance suggested below.
Overview
Synta Pharmaceuticals Corp. is a biopharmaceutical company focused on discovering, developing, and commercializing small molecule drugs to extend and enhance the lives of patients with severe medical conditions, including cancer and chronic inflammatory diseases. We have two drug candidates in clinical trials for treating multiple types of cancer and a novel proprietary drug discovery platform. All of our drug candidates have been discovered and developed internally using our proprietary, unique chemical compound library and integrated discovery engine. We retain full ownership of all of our drug candidates.
We were incorporated in March 2000 and commenced operations in July 2001. Since that time, we have been principally engaged in the discovery and development of novel drug candidates. As of March 31, 2014, we have raised an aggregate of approximately $734.5 million in cash proceeds to fund operations, including $531.5 million in net proceeds from private and public offerings of our equity, $30.5 million in gross proceeds from term loans and $167.2 million in non-refundable payments from partnering activities under prior collaborations, as well as $5.3 million from the exercise of common stock warrants and options. We have also generated funds from government grants, equipment lease financings and investment income. We are engaged in preliminary partnership discussions for a number of our programs, which may provide us with additional financial resources if consummated.
In April 2014, we sold an aggregate of 5,680,515 shares of our common stock for an aggregate of approximately $23.0 million in net proceeds, including $18.0 million in net proceeds from 4,430,515 shares of our common stock pursuant to the at-the-market issuance sales agreement with MLV & Co. LLC (MLV) and $5.0 million in net proceeds from 1,250,000 shares of our common stock in a registered direct offering to an affiliate of a director who is our largest stockholder.
We have devoted substantially all of our capital resources to the research and development of our drug candidates. Since our inception, we have had no revenues from product sales. As of March 31, 2014, we had an accumulated deficit of $575.0 million. We expect to incur significant operating losses for the foreseeable future as we advance our drug candidates from discovery through preclinical development and clinical trials, and seek regulatory approval and eventual commercialization. We will need to generate significant revenues from product sales to achieve future profitability and may never do so.
17
Table of Contents
Oncology Programs
We have two clinical-stage programs in oncology (ganetespib and elesclomol) and a novel, proprietary small molecule cancer drug development program (the HDC platform).
Ganetespib (Hsp90 Inhibitor)
Summary
Ganetespib is a novel, potent, small molecule inhibitor of Hsp90, a molecular chaperone which is required for the proper folding and activation of many cancer-promoting proteins. Inhibition of Hsp90 by ganetespib leads to the simultaneous degradation of many of these client proteins and the subsequent death or cell cycle arrest of cancer cells dependent on those proteins. A number of Hsp90 client proteins are also involved in the resistance of cancer cells to other anti-cancer treatments, such as chemotherapy. The ability to reduce cancer-cell drug resistance suggests that the combination of ganetespib with chemotherapies or other anti-cancer agents may provide greater benefit than those agents administered alone. In preclinical studies, ganetespib has shown potent anti-cancer activity against a broad range of solid and hematologic cancers, both as a monotherapy and in combination with certain widely used anti-cancer agents.
Ganetespib is currently being evaluated in a broad range of cancer clinical trials including our GALAXY NSCLC program (GALAXY-1 and GALAXY-2) in combination with docetaxel chemotherapy, and as monotherapy in certain genetically-defined targeted patient populations. A favorable safety profile has been consistently observed across clinical trials, involving over 1,000 patients treated with ganetespib to date. Ganetespib has not shown the serious liver or common ocular toxicities reported with other Hsp90 inhibitors, or the neurotoxicity, bone marrow toxicities, and alopecia characteristic of many chemotherapies. The most common adverse event reported with ganetespib has been transient, mild or moderate diarrhea, which can be prevented or effectively managed with standard supportive care. In the clinical trials conducted to date, ganetespib has shown promising activity both in combination with chemotherapy and as a monotherapy.
The results observed to date in our GALAXY program suggest a significant potential commercial opportunity for use of ganetespib in combination with docetaxel as second-line treatment of patients with NSCLC. Across the United States, United Kingdom, Germany, France, Spain, Italy, and Japan, there are an estimated 160,000 patients each year who have progressed on first line therapy and are eligible for subsequent treatment of non-small cell lung adenocarcinoma. Approximately 90,000 of these eligible patients are estimated to be chemosensitive and negative for both EGFR mutation and ALK translocation.
Ganetespib in lung cancer: The GALAXY program
GALAXY-1 Phase 2b Trial
In 2011, we initiated the GALAXY-1 trial in patients with advanced NSCLC who received one prior treatment for advanced disease, i.e., a second-line treatment setting. GALAXY-1 compares treatment with docetaxel alone, which is approved for second-line treatment, vs. treatment with ganetespib plus docetaxel. The aims of this study were to:
· Evaluate clinical benefit and establish the safety profile of ganetespib in combination with docetaxel relative to docetaxel alone;
· Identify the patient populations, by biomarker or other disease characteristics, which may be most
18
Table of Contents
responsive to combination treatment; and
· Build the clinical and operational experience needed to optimize the design and execution of the pivotal GALAXY-2 Phase 3 trial.
Patients in both arms of GALAXY-1 receive a standard regimen of docetaxel 75 mg/m2 on day 1 of a 21-day treatment cycle. Patients in the combination arm also receive ganetespib 150 mg/m2 on days 1 and 15. Treatment continues until disease progression or until treatment intolerance. To ensure balance of prognostic factors between the two arms, patients were stratified by ECOG performance status, baseline LDH level, smoking status, and time since diagnosis of advanced disease.
Rate of disease progression during or following first line chemotherapy is a common stratification factor in salvage-setting (after first-line treatment) lung cancer clinical trials to ensure balance and evaluate any difference in treatment benefit between refractory and chemosensitive patients. Commonly used measures include time since completion of first line chemotherapy, best response to first line therapy, time since initiation of first line therapy, as well as time since diagnosis of advanced disease. The latter was chosen for GALAXY-1 in order to reduce ambiguity introduced by the recent approvals of maintenance therapy following first line treatment, as well as to avoid possible subjectivity in assessment of tumor response in the first-line setting.
GALAXY-1 was originally designed to enroll 240 second-line advanced NSCLC patients of all histologies in order to evaluate several hypotheses on which patients might be most responsive to combination treatment. Co-primary endpoints were PFS in all patients (the ITT population) and OS in patients with elevated baseline level of serum LDH (eLDH). During the course of the trial, the co-primary endpoints were changed to PFS in patients with eLDH and PFS in patients with mutant KRAS (mKRAS). Key secondary endpoints are OS and PFS in the adenocarcinoma patient population.
In early 2012, enrollment of patients with non-adenocarcinoma histologies (which consists primarily of squamous cell carcinoma) was terminated based on possible safety concerns, including risk of bleeding and a trend towards inferior survival. The trial was amended at that time to enroll 240 patients with adenocarcinoma histology only. To ensure the specified number of eLDH and mKRAS patients were included, a total of 385 patients were enrolled in GALAXY-1. Enrollment in GALAXY-1 was completed in May 2013.
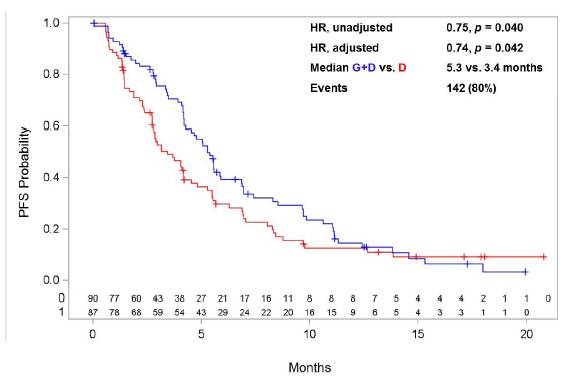
The final analysis of GALAXY-1 data was conducted in early May 2014. Publication of the final data from GALAXY-1 is expected in the second half of 2014. A summary of key efficacy data is presented in the tables and figures below:
| | Hazard Ratio
G+D vs. D | | eLDH
N=87 | | mKRAS
N=89 | | Chemosensitive*
N=177 | | Adenocarcinoma
N=253 |
| | | | | | | | | | |
| | | | | | | | | | |
OS | | Unadjusted | | 0.88
p=0.300 | | 1.18
p=0.755 | | 0.71
p=0.023 | | 0.87
p=0.150 |
| | | | | | | | | |
| Adjusted | | 0.75
p=0.118 | | 1.23
p=0.204 | | 0.69
p=0.019 | | 0.84
p=0.114 |
| | | | | | | | | | |
PFS | | Unadjusted | | 1.06
p=0.595 | | 0.93
p=0.387 | | 0.75
p=0.040 | | 0.85
p=0.112 |
| | | | | | | | | |
| Adjusted | | 0.88
p=0.295 | | 1.11
p=0.338 | | 0.74
p=0.042 | | 0.82
p=0.078 |
* Population selected for phase 3 GALAXY-2 trail
P-values are 1-sided
Hazard ratios were calculated using Cox proportional hazards model
19
Table of Contents
Unadjusted: univariate analysis
Adjusted: pre-specified analysis adjusting for multiple prognostic variables such as gender, smoking status, LDH, ECOG performance status, interval since diagnosis of advanced disease, age, total baseline target lesion size, and geographic region
G+D vs. D | | | | eLDH
N=87 | | mKRAS
N=89 | | Chemosensitive*
N=177 | | Adenocarcinoma
N=253 |
| | | | | | | | | | |
| | | | | | | | | | |
OS | | Median (months) | | 6.0 vs. 5.1 | | 7.6 vs. 6.4 | | 11.0 vs. 7.4 | | 10.2 vs. 8.4 |
| | | | | | | | | | |
| | Events | | 72 (83%) | | 68 (76%) | | 132 (75%) | | 190 (75%) |
| | | | | | | | | | |
PFS | | Median (months) | | 2.8 vs. 2.7 | | 3.9 vs. 3.0 | | 5.3 vs. 3.4 | | 4.5 vs. 3.2 |
| | | | | | | | | | |
| | Events | | 70 (80%) | | 73 (82%) | | 142 (80%) | | 205 (81%) |
* Population selected for Phase 3 GALAXY-2 trial
Figure 1: OS Kaplan Meier plot for the chemosensitive patient population of GALAXY-1 selected for evaluation in the GALAXY-2 Phase 3 trial

Figure 2: PFS Kaplan Meier plot for the chemosensitive patient population of GALAXY-1 selected for evaluation in the GALAXY-2 Phase 3 trial
20
Table of Contents
Safety
The safety profile of adenocarcinoma patients treated with the combination of ganetespib (G) and docetaxel (D) was favorable, consistent with previously reported results. The most common adverse events (AEs), all grades, were neutropenia (46% vs. 45%), diarrhea (50% vs. 17%) and fatigue (35% vs. 24%), for G+D vs. D, respectively. Diarrhea was effectively prevented or managed with standard supportive care; the incidence of grade 3 or 4 diarrhea was 4% (G+D) vs. 0% (D). Fatigue was predominantly grade 1 and grade 2; grade 3 or 4 fatigue was 6% (G+D) vs. 4% (D). The most common grade 3 or 4 AEs were neutropenia (41% vs. 42%), febrile neutropenia (9% vs. 5%), and leukopenia (10% vs 6%). Only one case of visual impairment was reported in this study, which was mild (Grade 1) and transient. The safety profile of patients in the chemosensitive population being evaluated in Phase 3 was comparable to the profile in the adenocarcinoma population.
GALAXY-2 Phase 3 Trial
In early 2013, we initiated the GALAXY-2 trial, a global, randomized, multi-center study comparing the same treatments as in GALAXY-1 in the 2 nd -line non-small cell adenocarcinoma patient population, with overall survival as the primary endpoint. Patients are required to be chemosensitive and have tumors that are negative for both EGFR mutation and ALK translocation.
Patients on both arms receive docetaxel generally for four to six 21-day cycles, according to standard practice at their treatment center. After completion of docetaxel treatment, patients on the ganetespib arm are eligible to continue to receive ganetespib monotherapy as maintenance treatment.
21
Table of Contents
The GALAXY-2 trial is expected to enroll approximately 850 patients, of which it is estimated that a minimum of 700 will be negative for both ALK translocations and EGFR mutations. Assuming a median overall survival of 7 months in the control arm and 9.3 months in the combination arm (a hazard ratio of 0.75), 5 months of follow up, and a two-sided overall Type I error rate of 0.05, GALAXY-2 has an 87% or higher power to detect a statistically significant treatment difference at the final analysis. Two event-driven interim analyses of the overall survival primary endpoint of GALAXY-2 have been pre-specified.
Based on current projections and statistical assumptions, we expect that the two GALAXY-2 interim overall survival analyses will be conducted in the second half of 2015, and we expect that the final overall survival analysis will be conducted in the first half of 2016.
Clinical trial of ganetespib and crizotinib combination in ALK positive, crizotinib-naïve NSCLC patients
This clinical trial is sponsored by Memorial Sloan Kettering Cancer Center in NYC. In the first stage, initiated in 2012, the safety profile of escalating doses of the combination was successfully evaluated and the trial is now proceeding to Phase 2 evaluation of activity.
Ganetespib in breast cancer
ENCHANT-1 Trial
Interim results from the ENCHANT-1 trial were presented in an oral presentation at the European Breast Cancer Conference (EBCC) in March 2014. These results have confirmed early signals of activity of ganetespib in breast cancer patients. The strength of the scientific rationale and evidence of clinical activity have led to the selection of ganetespib into the I-SPY 2 program (described further below). In this randomized Phase 2 trial, safety and efficacy of ganetespib will be evaluated in combination with standard chemotherapy initially in patients with HER2-negative breast cancer, and if positive, results from this trial will provide a robust proof of concept for ganetespib in this indication. In light of the inclusion of ganetespib in the I-SPY 2 program, we have decided to close the ENCHANT-1 trial and direct our resources in breast cancer towards the I-SPY 2 trial.
I-SPY 2 Trial
Ganetespib has been selected for study in the I-SPY 2 TRIAL (Investigation of Serial Studies to Predict Your Therapeutic Response with Imaging And moLecular Analysis 2). I-SPY 2 is a standing phase 2 randomized, controlled, multicenter trial for women with newly diagnosed, locally advanced breast cancer (Stage 2 or higher) that is designed to test whether adding investigational drugs to standard chemotherapy is better than standard chemotherapy alone in the neo-adjuvant setting (prior to surgery).
I-SPY 2 employs a unique adaptive trial design to match experimental therapies with patients. Genetic or biological markers (“biomarkers”) from individual patients’ tumors are used to screen promising new treatments, identifying which treatments are most effective in specific patient subgroups.
22
Table of Contents
Regimens that have a high Bayesian predictive probability of showing superiority in a 300 patient Phase 3 confirmatory trial in at least one of 10 predefined signatures may “graduate” from I-SPY 2. A regimen can graduate early and at any time after having 60 patients assigned to it, and exits the trial after a maximum of 120 patients. This high efficacy bar and rapid turnaround time allows the trial to match the most promising drug with the right patient in the most expeditious fashion.
I-SPY 2 was initiated as a pre-competitive consortium that brings together the Food and Drug Administration (FDA), National Cancer Institute (NCI), pharmaceutical companies, leading academic medical centers, and patient advocacy groups under its umbrella. I-SPY 2 is sponsored by QuantumLeap Healthcare Collaborative (QLHC), a non-profit 501(3)C foundation dedicated to accelerating healthcare solutions. QLHC shares a unique partnership with the Foundation for the National Institutes of Health Biomarkers Consortium, who manages intellectual property that emerges from the trial. The trial was developed by principal investigators, Laura J. Esserman, M.D., M.B.A., Professor of Surgery and Radiology and Director of the Carol Frank Buck Breast Care Center at UCSF Helen Diller Family Comprehensive Cancer Center in San Francisco, and Donald A. Berry, Ph.D., Professor in the Department of Biostatistics at The University of Texas MD Anderson Cancer Center, and founder of Berry Consultants.
Enrollment in the ganetespib arm of I-SPY 2 is expected to begin in 2014. Ganetespib will initially be available to patients with HER2-negative disease, with the intent to expand its eligibility to all biomarker subtypes after safety testing with trastuzumab is completed.
Clinical trial of ganetespib and fulvestrant in patients with hormone receptor positive metastatic breast cancer
This randomized Phase 2 trial is evaluating safety and activity of the fulvestrant and ganetespib combination in patients with hormone receptor positive metastatic breast cancer who are experiencing progression after initial treatment with hormonal therapy. At present, patient recruitment is ongoing. The trial is sponsored by Dana Farber Cancer Institute in Boston.
Ganetespib in Acute Myeloid Leukemia (AML) and Myelodysplastic Syndrome (MDS)
AML is a rapidly progressing hematologic cancer characterized by uncontrolled proliferation of immature blast cells in the bone marrow. The American Cancer Society estimates approximately 14,590 new cases of AML and approximately 10,370 deaths in the U.S. in 2013. MDS is a hematopoietic stem cell neoplasm characterized by disordered and ineffective hematopoiesis which results in irreversible decline in the number and quality of blood-forming cells. In most cases, progressive bone marrow failure results in neutropenia and thrombocytopenia, and in about one third of patients the disease progresses into AML, usually within a few years.
AML is a biologically heterogeneous disease, and therefore represents a major challenge in the advancement of treatment. Treatment choice and outcome are substantially decided by age, yet current long term remission rates remain poor, with only 40% of younger patients (age <60 years) and less than 10% of older patients (age > 60 years) achieving complete remissions. AML patients with relapsed or refractory disease and newly diagnosed AML patients over 60 years of age with poor prognostic risk factors typically die within one year, resulting in an acute need for new treatment options for these patients.
Starting in 2011, the Leukemia & Lymphoma Research Fund and Cancer Research UK sought to fund and initiate three large, multicenter, randomized trials to evaluate different investigational treatments, alone or in combination with chemotherapy, in patients with first-line AML and high risk MDS. These trials are being conducted under the sponsorship of Cardiff University, UK, and under the auspices of the UK NCRI Haematological Oncology Study Group, with investigators in Denmark, France, New Zealand, and the United Kingdom. Ganetespib, in combination with chemotherapy, has been selected for investigation in all three of these studies, which have initiated or are expected to initiate in 2014:
· The AML-LI (Less Intensive)-1 Phase 2/3 trial is ongoing, and is evaluating the combination of ganetespib with low dose cytarabine (Ara-C) vs. low dose Ara-C alone in patients who are not
23
Table of Contents
eligible for intensive chemotherapy and are traditionally not included in most trials. Up to 50 patients will be enrolled in the ganetespib arm, after which an interim analysis will be conducted to evaluate whether to proceed with full Phase 3 enrollment. This interim analysis is expected to be conducted in mid-2014.
· The AML-18 trial, expected to begin enrolling patients in 1H 2014, will evaluate ganetespib with standard DA (daunorubicin and Ara-C) in patients over 60 years old who can tolerate intensive chemotherapy vs. treatment with standard DA alone. Up to 200 patients are expected to be enrolled in the ganetespib arm. Results from a pilot study conducted in the UK in 2012 under the auspices of the Cardiff Experimental Cancer Medicine Centre confirmed the feasibility and safety of combining ganetespib with intensive chemotherapy in older patients with AML.
· The AML-19 trial, expected to begin enrolling patients in 2H 2014, will evaluate ganetespib in combination with conventional chemotherapy vs chemotherapy alone in younger patients with AML. The trial is expected to enroll up to 200 patients in the ganetespib arm and will be conducted by the UK NCRI Group, a network of over 100 institutions. Patients will receive ganetespib sequentially to standard intensive therapy, followed by ganetespib maintenance treatment. The objective is to identify if ganetespib reduces the risk of relapse in the overall population or in key subgroups, and as a result, improves overall survival, the primary endpoint.
The selection of ganetespib for these studies was supported by preclinical results generated by Synta and its academic collaborators, including Alan K. Burnett of Cardiff University, principal investigator of the LI-1 study, and Sanjay Bansal of the UT Health Science Center at San Antonio. Results from these studies show that ganetespib inhibits a number of cancer-promoting factors believed to contribute to the proliferation of leukemic cells and renders them more vulnerable to treatment with chemotherapy.
Ganetespib in ovarian cancer
GANNET53 Trial
Each year, approximately 230,000 new cases of ovarian cancer are diagnosed worldwide. Ovarian cancer is the most deadly of the gynecologic cancers, causing approximately 140,000 deaths annually, including 41,900 deaths in Europe and 14,000 deaths in the US. The serous ovarian cancer subtype, a particularly aggressive form driven by mutations of p53, an Hsp90 client protein found in greater than 50% of all human cancers, makes up 75 to 80% of diagnoses, with approximately 70% of all cases diagnosed in stage III or IV. Platinum-based chemotherapy remains the mainstay of therapy in ovarian cancer and results in a 5-year survival rate of only 30%, which is diminished to 10% for stages III and IV.
GANNET53, a Seventh Framework Programme (FP7) research project funded by the European Commission, is a pan-European randomized trial designed to evaluate the combination of ganetespib and paclitaxel vs. paclitaxel alone in over 200 patients with metastatic, predominantly p53 mutant, platinum-resistant ovarian cancer. Preclinical models have shown that mutant p53 is critical to the growth and proliferation of these cancers. Many mutations render p53 unable to fold appropriately, leaving the protein highly dependent on Hsp90 for stability. Inhibition of Hsp90 destroys the complex between Hsp90 and mutant p53, leading to the degradation of the protein and cancer cell death. This anti-cancer activity is substantially stronger in cells with mutant p53 than in cells with non-mutated p53, suggesting potential as a predictive biomarker for Hsp90 inhibitors such as ganetespib.
Hsp90 inhibition has also been shown to sensitize mutant p53 cancer cells to treatment with chemotherapies, as has been seen in preclinical studies evaluating ganetespib in other tumor types, supporting the planned trial design evaluating the combination of ganetespib and paclitaxel vs. paclitaxel
24
Table of Contents
alone.
The safety lead-in Phase 1 portion of GANNET53 is expected to begin enrollment in mid-2014, with centers in Austria, Belgium, France, and Germany will participating. The study’s consortium consists of national clinical trial groups in gynecological oncology and high-volume university centers as well as noted p53 scientists and three innovative small and medium sized companies (SMEs).
Elesclomol (Mitochondria-Targeting Agent)
Elesclomol is a first-in-class, investigational drug candidate that triggers programmed cell death (apoptosis), in cancer cells through a novel mechanism: disrupting cancer cell mitochondrial metabolism. In preclinical experiments, anti-cancer activity of elesclomol has been shown to correlate with certain biomarkers, including LDH, which can distinguish between active mitochondria (sufficient oxygen present) and inactive mitochondria (insufficient oxygen present). Consistent with these findings in three randomized clinical trials, LDH was an important predictor of elesclomol treatment outcome.
We are evaluating the use of elesclomol in combination with paclitaxel in ovarian cancer. In March 2011, the Gynecological Oncology Group (GOG) initiated a Phase 2 clinical trial of elesclomol in combination with paclitaxel for the treatment of persistent or recurrent ovarian, fallopian tube or primary peritoneal cancer for patients with total baseline serum LDH level less than 0.8 times the upper limit of normal (ULN). The GOG is a non-profit organization with the purpose of promoting excellence in the quality and integrity of clinical and basic scientific research in the field of gynecologic malignancies. The National Cancer Institute is providing financial support of up to approximately $300,000 for the trial through its Cancer Therapy Evaluation Program. The ovarian cancer trial met the pre-specified efficacy requirement to advance to stage 2, indicating potential activity in this difficult-to-treat patient population with limited treatment options. Enrollment of stage 2 of this study is ongoing.
Hsp90-inhibitor Drug Conjugate (HDC) Platform: improving the delivery of small molecule anti-cancer therapies to tumors
In September 2013, we announced the launch of a novel, proprietary small molecule cancer drug development program: the HDC Platform. This innovative approach to tumor targeted delivery capitalizes on the prolonged retention of Hsp90 inhibitors in tumors to trap an active agent of interest inside cancer cells. The HDC program builds on our extensive expertise in the science of Hsp90.
The HDC platform stemmed from the observation that small molecule inhibitors of Hsp90 are retained in tumors for as much as 20 times longer than in blood or normal tissue. Our researchers have shown that ganetespib can persist in tumor cells for over a week, while it is cleared from blood and normal tissues in a matter of hours. Several other research groups have published results demonstrating this characteristic is shared by first-generation inhibitors such as 17-AAG and its derivatives, as well as other classes of Hsp90 inhibitors. One group in particular has provided clinical validation of the observation by imaging tumors in patients using an 124 I radiolabeled form of their Hsp90 inhibitor (PU-H71).
This property of the Hsp90 inhibitor class is believed to be due to overexpression of an active form of Hsp90 in cancer cells that preferentially binds Hsp90 inhibitors, as compared to normal tissues. Even weak Hsp90 inhibitors that do not engage degradation of Hsp90 client proteins can be retained for days by cancer cells, enabling use of this property purely as a targeting mechanism to deliver an anticancer drug into cancer cells.
HDCs are drug candidates consisting of an Hsp90 inhibitor (targeting moiety) joined to an anti-
25
Table of Contents
cancer agent (payload) via a cleavable chemical linker optimized for controlled release of payload drug inside cancer cells. Unlike antibody-drug conjugates (ADCs), HDCs are small molecules that do not require cell surface antigens for targeting or endocytosis for cellular uptake. Instead, HDCs home in on an intracellular target (Hsp90) that is present in a wide range of cancers.
HDCs can deliver micromolar concentrations of an active payload to tumor cells for extended periods of time, eliminating the need for using ultra-high potency toxins in the conjugates and opening the door to a wide range of possibilities for enhancement of approved anticancer agents and promising development candidates. By directing sustained, high concentrations of active payload drug to cancer cells, HDCs enable greater cancer cell killing than can be achieved with administration of unconjugated chemotherapy or other payloads.
The HDC platform enables the rapid creation of an extensive proprietary pipeline of novel anticancer drugs that we may elect to develop independently or co-develop with selected partners.
Figure 3: The HDC Platform: using the preferential retention of Hsp90 inhibitors by tumor cells to selectively deliver anti-cancer payloads.
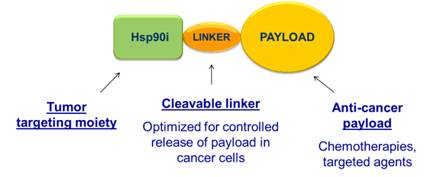
We have developed HD-Conjugated chemotherapeutics, kinase inhibitors, hormone therapies, immunomodulators, and epigenetic modifiers, creating the potential for next-generation compounds in each of these categories. Examples include HD-Conjugated bendamustine, temozolomide, doxorubicin, 5-FU, pemetrexed, SN-38, topotecan, vorinostat, panobinostat, fulvestrant, abiraterone, lenalidomide, pomalidomide, docetaxel, carboplatin, bortezomib, sunitinib, and sorafenib.
In October 2013, we announced the publication of the first key patent application covering our proprietary HDC technology, PCT/US2013/036783, published as International Patent Application No. WO/2013/158644, including composition of matter claims covering HDC compounds, methods for identifying therapeutically effective compounds, and methods of use against a wide range of diseases and conditions. Any resulting patent will expire no earlier than 2034.
Our Inflammatory Disease Program
CRACM Ion Channel Inhibitors
We have developed novel, small molecule inhibitors of CRACM ion channels expressed on immune cells. Our CRACM ion channel inhibitors have shown strong anti-inflammatory activity in preclinical studies both in vitro and in vivo, inhibiting T cell and mast cell activity, including cytokine release, degranulation, and immune cell proliferation. Potential applications include a wide range of inflammatory diseases and disorders for which modulating T cell and mast cell function has been shown
26
Table of Contents
to be critical, including rheumatoid arthritis (RA), asthma, chronic obstructive pulmonary disease (COPD), allergy, transplant rejection, and other autoimmune diseases and inflammatory conditions. We have several promising CRACM inhibitors in preclinical development. Because there are a number of CRACM ion channel targets on immune cells, we believe that CRACM inhibitor compounds can be developed that target different diseases.
In May 2014, we entered into a license arrangement for our CRACM program, including two lead candidates and the associated intellectual property portfolio, with PRCL Research Inc. (PRCL), a company funded by TVM Life Science Venture VII and the Fonds de Solidarité des Travailleurs du Québec, based in Montreal, Canada. PRCL plans to develop to proof-of-concept, one of the two lead candidates licensed from us. We were granted a minority interest in PRCL in exchange for our contribution of know-how and intellectual property and we will also hold a seat on PRCL’s Board of Directors. We will not be required to provide any research funding or capital contributions to PRCL. We will be reimbursed by PRCL for intellectual property management costs in connection with the contributed intellectual property and may conduct preclinical research activities which would be reimbursed by PRCL. If and when proof-of-concept is reached with either drug candidate, Eli Lilly and Company, which is an investor in TVM, will help manage the development program through one of its divisions and will have an option to acquire PRCL or its assets at the then fair value.
27
Table of Contents
Financial Operations Overview
Revenue
We have not yet generated any product revenue and do not expect to generate any product revenue in the foreseeable future, if at all. Our revenues to date have been generated primarily through our former collaboration and license agreements. The terms of these agreements included payment to us of upfront license fees, milestone payments, research and development cost sharing and royalties. We will seek to generate revenue from product sales and from future collaborative or strategic relationships. In the future, we expect any revenue we generate will fluctuate from quarter-to-quarter as a result of the timing and amount of payments received and expenses incurred under future collaborations or strategic relationships, if consummated, and the amount and timing of payments we receive upon the sale of our drug candidates, to the extent any are successfully commercialized.
Research and Development
Research and development expense consists of costs incurred in connection with developing and advancing our drug discovery technology and identifying and developing our drug candidates. We charge all research and development expenses to operations as incurred.
28
Table of Contents
Our research and development expense consists of:
· internal costs associated with research, preclinical and clinical activities;
· payments to third party contract research organizations, investigative sites and consultants in connection with our preclinical and clinical development programs;
· costs associated with drug formulation and supply of drugs for clinical trials;
· personnel related expenses, including salaries, stock-based compensation, benefits and travel; and
· overhead expenses, including rent and maintenance of our facilities, and laboratory and other supplies.
We do not know if we will be successful in developing our drug candidates. We believe that accurately projecting total program-specific expenses through commercialization is not possible at this time. The timing and amount of these expenses will depend upon the costs associated with potential future clinical trials of our drug candidates, and any expansion of our research and development organization, regulatory requirements, advancement of our preclinical programs and product manufacturing costs, many of which cannot be determined with accuracy at this time based on the stage of development of our drug candidates. This is due to the numerous risks and uncertainties associated with the duration and cost of clinical trials, which vary significantly over the life of a project as a result of unanticipated events arising during clinical development, including with respect to:
· the number of clinical sites included in the trial;
· the length of time required to enroll suitable subjects;
· the number of subjects that ultimately participate in the trials; and
· the efficacy and safety results of our clinical trials and the number of additional required clinical trials.
Our expenditures are subject to additional uncertainties, including the terms and timing of regulatory approvals. In addition, we may obtain unexpected or unfavorable results from our clinical trials. We may elect to discontinue, delay or modify clinical trials of some drug candidates or focus on others. A change in the outcome of any of the foregoing variables in the development of a drug candidate could mean a significant change in the costs and timing associated with the development of that drug candidate. For example, if the FDA or other regulatory authority were to require us to conduct clinical trials beyond those that we currently anticipate, or if we experience significant delays in any of our clinical trials, we would be required to expend significant additional financial resources and time on the completion of clinical development. Additionally, future commercial and regulatory factors beyond our control will evolve and therefore impact our clinical development programs and plans over time.
In 2014, we anticipate that the overall research and development costs, principally under the ganetespib program, may increase as we further advance the GALAXY-2 trial, our Phase 3 trial in second-line advanced NSCLC, and conduct non-clinical supporting activities.
Beyond our current lead drug candidates, we anticipate that we will select drug candidates and research projects for further development on an ongoing basis in response to their preclinical and clinical success, as well as commercial potential.
General and Administrative
General and administrative expense consists primarily of salaries and related expenses for personnel in executive, finance, business and commercial development, investor and medical community relations, human resources and administrative functions. Other costs include stock-based compensation costs, directors’ and officers’ liability insurance premiums, legal costs of pursuing patent protection of our intellectual property, fees for general legal, accounting, public-company requirements and compliance, and other professional services, as well as overhead-related costs not otherwise included in research and development. In 2014, we anticipate that general and administrative expenses may increase depending upon the rate that we expand our pre-commercialization activities.
Critical Accounting Policies and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our financial statements which have been prepared in accordance with U.S. generally accepted accounting principles, or GAAP. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reported periods. We are required to make estimates and judgments with respect to contract research accruals, the
29
Table of Contents
recoverability of long-lived assets, measurement of stock-based compensation and the periods of performance under collaboration and license agreements. We base our estimates on historical experience, known trends and events, and various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources and the reported amounts of revenues and expenses. Actual results may differ from these estimates under different assumptions or conditions.
You should read the following discussion of our reported financial results in conjunction with the critical accounting policies disclosed in our Annual Report on Form 10-K for the year ended December 31, 2013, as filed with the Securities and Exchange Commission on March 11, 2014. There have been no significant changes to our critical accounting policies in 2014.
Consolidated Results of Operations
Three Months Ended March 31, 2014 Compared with Three Months Ended March 31, 2013
Revenues
There were no revenues in each of 2014 and 2013.
Research and Development Expense
| | Three Months Ended
March 31, | | 2014 to 2013 Change | |
| | 2014 | | 2013 | | $ | | % | |
| | (dollars in millions) | | | | | |
Clinical-stage drug candidates | | | | | | | | | |
Ganetespib | | $ | 14.1 | | $ | 14.8 | | $ | (0.7 | ) | (5 | )% |
Elesclomol | | 0.2 | | 0.1 | | 0.1 | | 100 | % |
Total clinical-stage drug candidates | | 14.3 | | 14.9 | | (0.6 | ) | (4 | )% |
CRACM | | 0.1 | | 0.4 | | (0.3 | ) | (75 | )% |
Early stage programs and other | | 3.2 | | 1.1 | | 2.1 | | 191 | % |
Total research and development | | $ | 17.6 | | $ | 16.4 | | $ | 1.2 | | 7 | % |
Ganetespib
In 2014 as compared to 2013, costs incurred under our ganetespib program decreased by $0.7 million, including decreases of $0.3 million for personnel-related costs, related research supplies, operational overhead and stock compensation, and $0.4 million for external costs. The decreases in external costs were principally due to a full quarter of patient-related costs that were incurred in the first quarter of 2014 following the commencement of enrollment in April 2013 in connection with the GALAXY-2 trial, our Phase 3 trial in second-line advanced NSCLC, offset by lower costs incurred in 2014 related to the wind-down of the GALAXY-1 trial, our Phase 2 trial in second-line advanced NSCLC, and other company-sponsored trials. In addition, costs were incurred in the first quarter of 2013 for the conduct of NDA-supporting clinical pharmacology studies that were not incurred in the first quarter of 2014. In 2014, we anticipate that the overall costs under our ganetespib program may increase as we further advance the GALAXY-2 trial and conduct additional non-clinical supporting activities.
Elesclomol
In 2014 as compared to 2013, costs incurred under our elesclomol program increased by $0.1 million, principally due to an increase of $0.1 million for external costs. We anticipate that future costs under our elesclomol program will remain at low levels due to the pace of the ongoing clinical trial in ovarian cancer being conducted by the GOG.
30
Table of Contents
CRACM
In 2014 as compared to 2013, costs incurred under our CRACM program decreased by $0.3 million, including decreases of $0.2 million for personnel-related costs, related research supplies, operational overhead and stock compensation, and $0.1 million for external costs. These decreases were the result of a continued lower investment in the CRACM program as we sought to seek a corporate partner. In May 2014, we entered into a license arrangement with PRCL under which we may conduct preclinical research activities in the future that would be reimbursed by PRCL.
Early-stage programs
In 2014 as compared to 2013, costs incurred under our early stage programs increased by $2.1 million, including increases of $1.8 million for personnel-related costs, related research supplies, operational overhead and stock compensation, and $0.3 million for external costs. These increases were principally the result of our investment in the HDC program that was announced in September 2013. In 2014, we anticipate that costs under the HDC program may increase as we seek to develop a drug candidate for pre-clinical development.
General and Administrative Expense
| | Three Months Ended
March 31, | | 2014 to 2013 Change | |
| | 2014 | | 2013 | | $ | | % | |
| | (dollars in millions) | | | | | |
| | | | | | | | | |
General and administrative | | $ | 5.3 | | $ | 3.9 | | $ | 1.4 | | 36 | % |
| | | | | | | | | | | | |
In 2014 as compared to 2013, general and administrative expenses increased by $1.4 million, including an increase of $1.8 million for personnel-related costs, related overhead and stock compensation, offset by $0.4 million for net decreases in external professional fees. In March 2014, our former President and Chief Executive Officer, who was a member of the Board of Directors, resigned and we entered into a separation agreement with him. In the first quarter of 2014, we recognized approximately $2.0 million in costs in connection with this separation agreement, including approximately $1.0 million in cash compensation to be paid over two years and approximately $1.0 million in non-cash stock compensation expense related to the accelerated vesting and extended vesting period of certain of his stock options. In 2014, we anticipate that other general and administrative expenses may increase depending upon the rate we expand our pre-commercialization activities.
Interest Expense, net
| | Three Months Ended
March 31, | | 2014 to 2013 Change | |
| | 2014 | | 2013 | | $ | | % | |
| | (dollars in millions) | | | | | |
Interest expense, net | | 0.7 | | $ | 0.5 | | $ | 0.2 | | 40 | % |
| | | | | | | | | | | |
In 2014 as compared to 2013, interest expense increased as a result of the approximate $13.5 million in aggregate additional funding that was obtained in March 2013 and June 2013 in connection with the GECC and Oxford Term Loans. In 2014, we anticipate that interest expense will decrease due to the start of principal payments in January 2014 under the GECC Term Loan.
Liquidity and Capital Resources
Cash Flows
The following table provides information regarding our cash position, cash flows and capital expenditures for the three months ended March 31, 2014 and 2013.
31
Table of Contents
| | Three Months Ended
March 31, | |
| | 2014 | | 2013 | |
| | (dollars in millions) | |
Cash, cash equivalents and marketable securities | | $ | 78.8 | | $ | 90.4 | |
Working capital | | 48.0 | | 75.3 | |
Cash flows (used in) provided by: | | | | | |
Operating activities | | (20.3 | ) | (22.2 | ) |
Investing activities | | 16.2 | | (36.6 | ) |
Financing activities | | 7.6 | | 12.3 | |
| | | | | | | |
Our operating activities used cash of $20.3 million and $22.2 million in 2014 and 2013, respectively. The use of cash in these periods principally resulted from our losses from operations, as adjusted for non-cash charges for depreciation and stock-based compensation, and changes in our working capital accounts.
In 2014, our investing activities provided cash of $16.2 million, including the purchases of marketable securities in the amount of $3.5 million and purchases of property and equipment in the amount of $0.1 million, offset by maturities of marketable securities in our investment portfolio in the amount of $19.8 million. In 2013, our investing activities used cash of $36.6 million, including the purchases of marketable securities in the amount of $55.3 million and purchases of property and equipment in the amount of $0.3 million, offset by maturities of marketable securities in our investment portfolio in the amount of $19.0 million.
Our financing activities provided cash of $7.6 million and $12.3 million in 2014 and 2013, respectively. In 2014, we raised approximately $10.1 million in net cash proceeds, including $9.3 million in net proceeds from sales of our common stock under the at-the-market issuance sales agreement with MLV and $0.8 million from the exercise of common stock options. In 2013, we raised approximately $14.3 million in net cash proceeds, including $13.2 million in gross proceeds from additional funding under the GECC Term Loan and Oxford Term Loan and $1.1 million from the exercise of common stock options. We repaid $2.5 million and $2.0 million in principal payments in 2014 and 2013, respectively, in connection with the GECC Term Loan and the Oxford Term Loan.
Contractual Obligations and Commitments
There were no material changes to the contractual obligations and commitments included in our Annual Report on Form 10-K for the fiscal year ended December 31, 2013.
Registered Direct Offering
In April 2014, we sold 1,250,000 shares of our common stock at a purchase price of $4.01 per share in a registered direct offering to an affiliate of a director who is our largest stockholder. These shares were sold directly without a placement agent, underwriter, broker or dealer. The net proceeds to us were approximately $5.0 million after deducting offering expenses payable by us.
At-The-Market Issuance Sales Agreements with MLV
In May 2012, as amended, we entered into an at-the-market issuance sales agreement, or Sales Agreement, with MLV pursuant to which we may issue and sell shares of our common stock having an aggregate offering price of up to $28 million from time to time, at our option, through MLV as our sales agent, subject to certain terms and conditions. Any shares sold will be sold pursuant to our effective shelf registration statement on Form S-3 (File No. 333-176022). We will pay MLV a commission of up to 3% of the gross proceeds of the sale of any shares sold through MLV. During March and April 2014, we sold 6,588,875 shares of common stock pursuant to the Sales Agreement for an aggregate of approximately $28.0 million in gross proceeds at an average selling price of $4.25 per share. Net proceeds to us were approximately $27.3 million after deducting commissions and other transactions costs, including approximately $9.3 million from the sale of 2,158,360 shares in March 2014 and approximately $18.0 million from the sale of 4,430,515 shares in April 2014, following which the Sales Agreement was terminated.
In May 2014, we entered into a new at-the-market issuance sales agreement, or New Sales Agreement, with MLV pursuant to which we may issue and sell shares of our common stock having an aggregate offering price of up to $40 million from time to time, at our option, through MLV as our sales agent, subject to similar terms and conditions. The shares would be sold pursuant to our effective shelf registration statement on Form S-3 (File No. 333-187242). We would pay MLV a commission of up to 3% of the gross proceeds of the sale of any shares sold through MLV. To date, no shares have been sold under the New Sales Agreement.
32
Table of Contents
Term Loans
General Electric Capital Corporation (GECC)
In March 2013, we amended our loan and security agreement entered into in September 2010 with GECC and one other lender, or the GECC Term Loan, and obtained $12.9 million in additional loan funding and, as a result, increased the principal balance to $22.5 million at March 31, 2013. Interest on the borrowings under the GECC Term Loan remains at the annual rate of 9.75%. We made interest-only payments for the period from April 2013 through December 2013. In January 2014, we began making 30 equal monthly payments of principal plus accrued interest on the outstanding balance. We are obligated to pay an exit fee of $788,000 at the time of the final principal payment. (See Note 9 of the accompanying condensed consolidated financial statements.)
Oxford Finance Corporation (Oxford)
In March 2011, we entered into a loan and security agreement with Oxford and received $2.0 million in loan funding, which we refer to herein as the Oxford Term Loan. Interest on the borrowings under the Oxford Term Loan accrues at an annual rate of 13.35%. Beginning in May 2011, we began making 36 equal monthly payments of principal plus accrued interest on the outstanding balance. In December 2012, we entered into a loan modification agreement with Oxford, as amended, under which we could elect to draw down up to an additional $0.6 million in equipment financing until June 30, 2013. As of June 30, 2013, the Company had fully utilized the $0.6 million in additional equipment financing. (See Note 9 of the accompanying condensed consolidated financial statements.)
Liquidity
Funding Requirements
We expect to continue to incur significant operating expenses and capital expenditures and anticipate that our expenses and losses may increase substantially in the foreseeable future as we:
· complete the ongoing clinical trials of ganetespib in solid tumors, including the GALAXY-1, GALAXY-2, ENCHANT-1 and I-SPY 2 trials, and initiate additional clinical trials of ganetespib if supported by trial results;
· complete preclinical development of an additional Hsp90 inhibitor and initiate clinical trials of this compound, if supported by the preclinical data;
· complete the ongoing clinical trial of elesclomol in ovarian cancer, and initiate additional clinical trials of elesclomol, if supported by trial results;
· advance our HDC program into preclinical development and initiate clinical trials, if supported by preclinical data;
· discover, develop, and seek regulatory approval for backups of our current drug candidates and other new drug candidates;
· identify additional compounds or drug candidates and acquire rights from third parties to those compounds or drug candidates through licenses, acquisitions or other means; and
· commercialize any approved drug candidates.
Our funding requirements will depend on a number of factors, including:
· the progress and results of our ongoing clinical trials of ganetespib and elesclomol, and any additional clinical trials we may initiate in the future based on the results of these clinical trials;
· the results of our preclinical studies of any additional Hsp90 inhibitors we may develop and our HDC program, and our decision to initiate clinical trials, if supported by the preclinical and other test results;
· uncertainty associated with costs, timing, and outcome of regulatory review of our drug candidates;
· the scope, progress, results, and cost of preclinical development, clinical trials, and regulatory review of any new drug
33
Table of Contents
candidates we may discover or acquire;
· the costs of preparing, filing, and prosecuting patent applications and maintaining, enforcing, and defending intellectual property-related claims;
· our ability to establish additional strategic collaborations and licensing or other arrangements on terms favorable to us
· the costs to satisfy our obligations under potential future collaborations; and
· the timing, receipt, and amount of sales or royalties, if any, from ganetespib, our HDC program, elesclomol, our CRACM inhibitors and our other potential products.
As of March 31, 2014, we had $78.8 million in cash, cash equivalents and marketable securities, a decrease of $12.7 million from $91.5 million as of December 31, 2013. This decrease principally reflects cash used in operations and term loan principal payments as discussed under “Cash Flows” above, offset by a total of $10.1 million in net cash proceeds, including $9.3 million in net proceeds from sales of our common stock under the at-the-market issuance sales agreement with MLV and $0.8 million from the exercise of common stock options.
We do not anticipate that we will generate product revenue in the foreseeable future, if at all. We expect our continuing operations to use cash over the next several years and such cash use may increase significantly from year to year. While we are engaged in multiple preliminary partnership discussions for each of our currently unpartnered programs, including ganetespib, the HDC platform and elesclomol, which could result in one or more new partnership agreements that may include upfront payments and cost-sharing provisions, there is no guarantee we will be successful in entering into any such partnership agreements on commercially reasonable terms, if at all, or that we will receive any other revenue through these partnership efforts in the future. Based on our current operating levels, we expect our $78.8 million in cash resources as of March 31, 2014, together with an aggregate of $23.0 million raised in net cash proceeds from additional sales of our common stock in April 2014 under the at-the-market issuance sales agreement with MLV and in a registered direct offering to an affiliate of a director who is our largest stockholder, will be sufficient to fund operations at least into the first half of 2015. This estimate assumes that the timing and nature of activities contemplated for the remainder of 2014 and 2015 will be conducted subject to the availability of sufficient financial resources. We continue to evaluate additional potential sources of funding, including partnership agreements, cost or risk-sharing arrangements, equity financings, use of our $40 million new at-the-market issuance sales agreement with MLV or other sources. We have an effective shelf registration statement on Form S-3 (File No. 333-187242), under which we currently have up to $234.6 million in securities available for future issuance, including up to $40 million in shares of common stock that we have reserved and that may be offered and sold under the New Sales Agreement with MLV.
We may require significant additional funds earlier than we currently expect in order to conduct additional clinical trials and conduct additional preclinical and discovery activities. Because of the numerous risks and uncertainties associated with the development and commercialization of our drug candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures associated with our current and anticipated clinical trials.
To the extent our capital resources are insufficient to meet our future operating and capital requirements, we will need to finance our future cash needs through public or private equity offerings, collaboration agreements, debt financings or licensing arrangements. However, additional funding may not be available to us on acceptable terms or at all. In addition, the terms of any financing may adversely affect the holdings or the rights of our stockholders. For example, if we raise additional funds by issuing equity securities or by selling convertible debt securities, further dilution to our existing stockholders may result. If we raise funds through collaboration agreements or licensing arrangements, we may be required to relinquish rights to our technologies or drug candidates, or grant licenses on terms that are not favorable to us.
If adequate funds are not available, we may be required to terminate, significantly modify or delay our research and development programs, reduce our planned commercialization efforts, or obtain funds through collaborators that may require us to relinquish rights to our technologies or drug candidates that we might otherwise seek to develop or commercialize independently. Conversely, we may elect to raise additional funds even before we need them if the conditions for raising capital are favorable.
Certain Factors That May Affect Future Results of Operations
The Securities and Exchange Commission, or SEC, encourages companies to disclose forward-looking information so that investors can better understand a company’s future prospects and make informed investment decisions. This Quarterly Report on Form 10-Q contains such “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995.
34
Table of Contents
Words such as “may,” “anticipate,” “estimate,” “expects,” “projects,” “intends,” “plans,” “believes” and words and terms of similar substance used in connection with any discussion of future operating or financial performance, identify forward-looking statements. All forward-looking statements are management’s present expectations of future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially and adversely from those described in the forward-looking statements. These risks include, but are not limited to those set forth under the heading “Risk Factors” contained in Item 1A of our Annual Report on Form 10-K for the year ended December 31, 2013 that we filed with the SEC on March 11, 2014.
In light of these assumptions, risks and uncertainties, the results and events discussed in the forward-looking statements contained in this Quarterly Report on Form 10-Q might not occur. Stockholders are cautioned not to place undue reliance on the forward-looking statements, which speak only as of the date of this Quarterly Report on Form 10-Q. We are not under any obligation, and we expressly disclaim any obligation, to update or alter any forward-looking statements, whether as a result of new information, future events or otherwise. All subsequent forward-looking statements attributable to Synta or to any person acting on its behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section.
Item 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Sensitivity. As of March 31, 2014, we had cash, cash equivalents and marketable securities of $78.8 million consisting of cash deposited in a highly rated financial institution in the United States and in a short-term U.S. Treasury money market fund, as well as high-grade corporate bonds and commercial paper. The primary objective of our investment activities is to preserve our capital for the purpose of funding operations and we do not enter into investments for trading or speculative purposes. We believe that we do not have material exposure to high-risk investments such as mortgage-backed securities, auction rate securities or other special investment vehicles within our money-market fund investments. We believe that we do not have any material exposure to changes in fair value as a result of changes in interest rates. Declines in interest rates, however, would reduce future investment income.
Capital Market Risk. We currently have no product revenues and depend on funds raised through other sources. One possible source of funding is through further equity offerings. Our ability to raise funds in this manner depends upon capital market forces affecting our stock price.
Item 4. Controls and Procedures.
(a) Evaluation of Disclosure Controls and Procedures. Our principal executive officer and principal financial officer evaluated the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e) under the Securities Exchange Act of 1934, as amended, or the Exchange Act) as of the end of the period covered by this Quarterly Report on Form 10-Q. Based on this evaluation, our principal executive officer and principal financial officer have concluded that our disclosure controls and procedures were effective to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms, and is accumulated and communicated to our management, including our principal executive and principal financial officers, or persons performing similar functions, as appropriate to allow timely decisions regarding required disclosure.
(b) Changes in Internal Controls. There were no changes in our internal control over financial reporting, identified in connection with the evaluation of such internal control that occurred during our last fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
PART II - OTHER INFORMATION
Item 1. Legal Proceedings.
We are currently not a party to any material legal proceedings.
Item 1A. Risk Factors.
There have been no material changes to the risk factors included in our Annual Report on Form 10-K for the fiscal year ended December 31, 2013.
35
Table of Contents
Item 2. Unregistered Sales of Equity Securities and Use of Proceeds.
None.
Item 3. Defaults Upon Senior Securities.
None.
Item 4. Mine Safety Disclosures.
Not applicable.
Item 5. Other Information.
On May 7, 2014, we entered into a letter agreement with MLV & Co. LLC, or MLV, pursuant to which we terminated the at-the-market issuance sales agreement dated May 2, 2012.
On May 7, 2014, we entered into a new at-the-market issuance sales agreement, or the New Sales Agreement, with MLV pursuant to which we may issue and sell shares of our common stock having an aggregate offering price of up to $40.0 million from time to time through MLV as our sales agent.
Any sales of shares of our common stock pursuant to the New Sales Agreement will be made under our previously filed and currently effective shelf registration statement on Form S-3 (File No. 333-187242), or the Registration Statement, and the related prospectus supplement dated May 1, 2013 and filed on May 3, 2013. MLV may sell the shares of common stock by any method that is deemed to be an “at-the-market offering” as defined in Rule 415 promulgated under the Securities Act of 1933, as amended, including by means of ordinary brokers’ transactions at market prices, in block transactions or as otherwise agreed by MLV and us. MLV may also sell the shares of common stock in negotiated transactions, subject to our prior approval. Subject to the terms and conditions of the New Sales Agreement, MLV will use its commercially reasonable efforts consistent with its normal trading and sales practices and applicable laws, rules and regulations to sell the shares of our common stock from time to time, based upon our instructions (including any price, time or size limits or other parameters or conditions we may impose). We will pay MLV a commission of up to 3.0% of the gross proceeds of the sale of any shares of common stock sold through MLV as agent under the New Sales Agreement. We have also provided MLV with customary indemnification rights.
We are not obligated to make any sales of common stock under the New Sales Agreement and no assurance can be given that we will sell any shares under the New Sales Agreement, or, if we do, as to the price or amount of shares that we will sell, or the dates on which any such sales will take place. The New Sales Agreement will terminate upon the earlier of the sale of all common stock subject to the New Sales Agreement or termination of the New Sales Agreement by us or MLV.
The foregoing description of the New Sales Agreement is not complete and is qualified in its entirety by reference to the full text of the New Sales Agreement, a copy of which is filed as Exhibit 10.3 to this Quarterly Report on Form 10-Q and is incorporated herein by reference. The New Sales Agreement is also incorporated by reference into the Registration Statement. A copy of the opinion of Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C., relating to the legality of the shares of common stock issuable under the New Sales Agreement, is filed as Exhibit 5.1 to this Quarterly Report on Form 10-Q and is also incorporated by reference into the Registration Statement.
The above disclosure shall not constitute an offer to sell or the solicitation of an offer to buy the securities discussed herein, nor shall there be any offer, solicitation, or sale of the securities in any state in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such state.
Item 6. Exhibits.
(a) Exhibits
5.1 Opinion of Opinion of Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C.
10.1* Separation Agreement between the Company and Dr. Bahcall, dated March 19, 2014 (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K filed March 20, 2014 (File No. 001-33277)).
10.2* Amended and Restated Director Compensation Policy, effective March 18, 2014.
36
Table of Contents
10.3 New At-the-Market Issuance Sales Agreement, dated May 7, 2014, by and between the Registrant and MLV & Co. LLC.
10.4 Letter Agreement, dated May 7, 2014, by and between the Registrant and MLV & Co. LLC, terminating the At the Market Issuance Sales Agreement dated as of May 2, 2012.
23.1 Consent of Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C. (included in the opinion filed as Exhibit 5.1).
31.1 Certification of principal executive officer under Section 302(a) of the Sarbanes-Oxley Act of 2002.
31.2 Certification of principal financial officer under Section 302(a) of the Sarbanes-Oxley Act of 2002.
32.1 Certifications of the principal executive officer and the principal financial officer under Section 906 of the Sarbanes-Oxley Act of 2002.
101 The following materials from Synta Pharmaceuticals Corp.’s Quarterly Report on Form 10-Q for the quarter ended March 31, 2014, formatted in XBRL (eXtensible Business Reporting Language): (i) the Unaudited Condensed Consolidated Balance Sheets, (ii) the Unaudited Condensed Consolidated Statements of Operations, (iii) the Unaudited Condensed Consolidated Statements of Comprehensive Loss, (iv) the Unaudited Condensed Consolidated Statements of Cash Flows, and (v) Notes to Unaudited Condensed Consolidated Financial Statements.
* Management contract, compensatory plan or arrangement.
37
Table of Contents
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| SYNTA PHARMACEUTICALS CORP. |
| |
| |
Date: May 8, 2014 | By: | /s/ Keith R. Gollust |
| | Keith R. Gollust |
| | Executive Chairman |
| | (principal executive officer) |
| | |
| | |
Date: May 8, 2014 | By: | /s/ Keith S. Ehrlich |
| | Keith S. Ehrlich, C.P.A. |
| | Vice President Finance and Administration, |
| | Chief Financial Officer |
| | (principal accounting and financial officer) |
38