Resmetirom has been well-tolerated with mostly mild AEs and some moderate AEs, the numbers of which are balanced between placebo and drug-treatment groups. Fewer than seven percent of patients did not complete the study, and patients who discontinued for AEs, all mild to moderate, were balanced between drug-treated and placebo patients. There were two serious AEs in the study, both considered unrelated to treatment, one in a placebo and one in a drug-treated patient.
Lead Product
Candidate—MGL-3196
(resmetirom)
We believe that resmetirom may be the first
THR-agonist
product candidate in development for NASH that selectively targets the
THR-ß
pathway. Active thyroid hormone, known as T3, interacts with two nuclear receptors,
THR-α,
which is the predominant receptor expressed in most human tissues, including heart and bone, and
THR-ß,
which has more restricted tissue expression, and is the predominant receptor responsible for metabolic actions in the liver, including both cholesterol- and
TG-lowering.
Selective activation of the
THR-ß
receptor in liver tissue is believed to favorably affect cholesterol and lipoprotein levels via multiple mechanisms, which may be complementary to those of other lipid-lowering therapies such as statin drugs. We believe that these characteristics of
THR-ß
activation by resmetirom will in turn lead to clinically meaningful reductions in
LDL-C,
and plasma and liver TGs.
We believe that resmetirom is the first selective small molecule
THR-ß
agonist compound. Resmetirom, along with
MGL-3745,
a potential backup compound to resmetirom, was discovered at
Hoffmann-La
Roche, or Roche, in Nutley, New Jersey, by utilizing a novel functional assay that, unlike a simple receptor binding assay, assessed the functional activity of compounds which interacted with thyroid hormone receptors. In a published study by Madrigal and Roche in the Journal of Medicinal Chemistry using this functional assay, resmetirom was shown to be highly selective for the
THR-ß
receptor, with almost no effect on
THR-α,
unlike other compounds purported in published studies to be ß -selective based on binding affinity, but which were shown to equally activate
THR-α
and
THR-ß
in the novel functional assay.
We believe that the ß-selectivity and liver-targeting properties of resmetirom are critically important for resmetirom’s beneficial metabolic actions in the liver, and enable avoidance of safety issues associated with
THR-α
activation by thyroid hormone and/or less selective THR agonists in tissues such as heart and bone. In a variety of preclinical animal model studies, resmetirom showed enhanced safety relative to T3 or other thyroid agonists. In animal models, resmetirom demonstrated cholesterol lowering, liver TG lowering, and reduction of markers of NASH-related liver inflammation and fibrosis at drug levels similar to those that lowered
LDL-C
in human clinical trials, providing data to support the advancement of resmetirom into NASH and FH clinical trials. In chronic animal toxicology studies in dogs and rats, no effects on bone or cartilage histology were seen at any resmetirom dose in either species.
Resmetirom did not increase liver enzymes in Phase 1 studies and there were no bone and cartilage histologic findings in chronic animal toxicology studies.
MGL-3196
(resmetirom) Clinical and
Non-Clinical
Development Program
To date, we have completed a series of Phase 1 and 2 clinical studies,
Phase 2-enabling
preclinical good laboratory practice, or GLP, toxicology studies, and drug manufacturing studies to support further clinical development, including active pharmaceutical ingredient, or API, manufacturing and drug product development studies, drug metabolism studies, acute, subchronic and chronic animal toxicology studies, and other safety pharmacology and toxicology studies.
We have completed Phase 1 studies with resmetirom in a total of 252 subjects to evaluate the safety, tolerability, pharmacokinetics and pharmacodynamic effects of resmetirom. Our Phase 1 studies included randomized, placebo-controlled, double-blind, single and
14-day
multiple-dose escalation studies, drug-interaction studies with statins, with the antidiabetic agent pioglitazone, and with the antiplatelet agent clopidogrel. In addition, a study using radiolabeled resmetirom, a study of a tablet versus capsule formulation of resmetirom, and a multiple dose drug interaction and food effect study have also been completed. In Phase 1 studies, resmetirom appeared safe and was well-tolerated up to 200 mg once daily. The results of these studies suggest that resmetirom is suitable for once-daily oral dosing. Currently there is an ongoing Phase 1 hepatic impairment study to characterize the safety and pharmacokinetics of resmetirom in patients with hepatic impairment, a Phase 1 drug-interaction study with warfarin, and a drug interaction study of resmetirom and pravastatin and simvastatin.
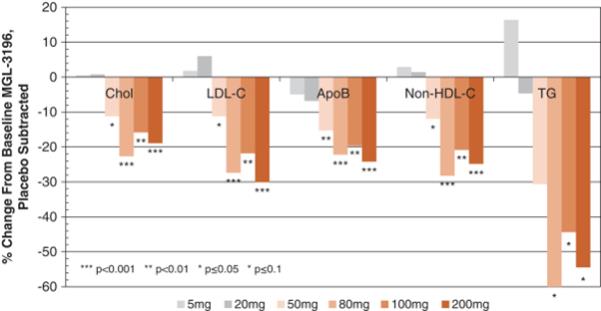
In a Phase 1 multiple ascending dose study in healthy subjects, the effects of resmetirom on lipid parameters were assessed as initial markers of pharmacodynamic activity (Atherosclerosis
230:373-380,
2013). As illustrated in the figure below, daily doses of resmetirom ranging from 50 to 200 mg showed highly statistically significant reductions relative to placebo of up to 30% for
LDL-C
(range,
p=.05-<0.0001),
28% for
non-high-density
lipoprotein cholesterol, or
(range,
p =0.027-p<0.0001)
and 24% for ApoB (range,
p =0.008-0.0004),
and statistical trends of up to 60% reduction in TG (range,
p =0.13-0.016).
The near maximal lipid effects were observed at a resmetirom dose of 80 mg once-daily. Resmetirom was well-tolerated at all doses, with no dose-related