Table of Contents
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
| þ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2006
OR
| o | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File No. 001-32157
ADVENTRX Pharmaceuticals, Inc.
(Exact name of registrant as specified in its charter)
| Delaware | 84-1318182 | |
| (State or other jurisdiction of incorporation or organization) | (I.R.S. Employer Identification No.) | |
| 6725 Mesa Ridge Road, Ste 100 San Diego CA | 92121 | |
| (Address of principal executive offices) | (Zip Code) |
(858) 552-0866
(Registrant’s telephone number, including area code)
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class: | Name of each exchange on which registered: | |
| Common Stock, par value $0.001 per share | The American Stock Exchange |
Securities registered pursuant to Section 12(g) of the Act:
None
None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yeso Noþ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yeso Noþ
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter periods that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. YESþ NOo
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statement incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K.o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one):
Large accelerated filero Accelerated filerþ Non-accelerated filero
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). YESo NOþ
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant as of June 30, 2006 was approximately $189,214,000, based upon the closing price on the American Stock Exchange reported for such date. Shares of common stock held by each officer and director and by each person who is known to own 5% or more of the outstanding common stock have been excluded in that such persons may be deemed to be affiliates of the Company. This determination of affiliate status is not necessarily a conclusive determination for other purposes.
89,676,739 shares of the registrant’s common stock were issued and outstanding as of March 12, 2007.
DOCUMENTS INCORPORATED BY REFERENCE
Certain information required to be disclosed in Part III of this report is incorporated by reference from the registrant’s definitive Proxy Statement, which will be filed with the Securities and Exchange Commission in connection with the registrant’s Annual Meeting of Stockholders to be held on May 23, 2007.
Table of Contents
Table of Contents
PART I
This Annual Report onForm 10-K, particularly in Item 1 “Business,” and Item 7 “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and the documents incorporated by reference, include forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. All statements, other than statements of historical fact, are statements that could be deemed forward-looking statements, including, but not limited to, statements regarding our future financial position, business strategy and plans and objectives of management for future operations. When used in this report, the words “believe,” “may,” “could,” “will,” “estimate,” “continue,” “anticipate,” “intend,” “expect” and similar expressions are intended to identify forward-looking statements.
We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends that we believe may affect our financial condition, results of operations, business strategy, short-term and long-term business operations and objectives, and financial needs. These forward-looking statements are subject to certain risks and uncertainties that could cause our actual results to differ materially from those reflected in these forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those discussed in this report, and in particular, the risks discussed in Item 1A “Risk Factors,” and those discussed in other documents we file with the Securities and Exchange Commission. Except as required by law, we do not intend to update these forward-looking statements publicly or to update the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future.
In light of these risks, uncertainties and assumptions, the forward-looking events and circumstances discussed in this report and in the documents incorporated in this report may not occur and actual results could differ materially and adversely from those anticipated or implied in such forward-looking statements. Accordingly, readers are cautioned not to place undue reliance on such forward-looking statements.
Item 1. Business
Overview
We are a biopharmaceutical research and development company focused on commercializing proprietary product candidates for the treatment of cancer and infectious diseases. We seek to improve the performance and safety of existing treatments by addressing significant problems such as drug metabolism, bioavailability, excessive toxicity and treatment resistance.
Our product candidates are modeled on currently marketed drugs, either as chemical modifications, or formulation modifications, with the intent of successfully exploiting the already well-defined therapeutic mechanism of action of the parent molecule, while improving performance to a clinically meaningful degree. Specifically, we seek to improve the performance of currently marketed products for cancer and infectious diseases, and to improve the commercial potential of such products, by:
| • | Enhancing their effectiveness; | ||
| • | Improving their safety and reducing the incidence of adverse effects; | ||
| • | Increasing their convenience and cost-effectiveness to patients and medical personnel, including through less invasive methods of administration and/or reduced administration time; and | ||
| • | Discovering new uses and synergies with other products. |
We have patent and marketing rights to a number of clinical and preclinical product candidates, including two product candidates that are in marketing-enabling clinical trials; that is, clinical trials sufficient to support a New Drug Application, or NDA, for marketing approval by the United States Food and Drug Administration, or FDA. Our lead product candidate, ANX-510 (fotrexorin calcium in the U.S.), or CoFactor, is in Phase III and Phase IIb clinical trials for the treatment of metastatic colorectal cancer, as well as in a Phase II clinical trial for the treatment of advanced breast cancer. ANX-530 (vinorelbine emulsion), a new formulation of vinorelbine tartrate, is in a marketing-enabling clinical trial for the treatment of non-small cell lung cancer.
1
Table of Contents
By the end of 2007, we anticipate initiating a Phase I/II clinical trial of ANX-201, or Thiovir™, for the treatment of human immunodeficiency virus, or HIV. Additionally, pending positive results from on-going preclinical pharmacokinetic testing and agreement on clinical protocol design with the FDA, we expect to initiate a marketing-enabling clinical trial of either ANX-016 (vancomycin emulsion) or ANX-015 (clarithromycin emulsion) for the treatment of certain bacterial infections, as well as a marketing-enabling clinical trial of ANX-514 (docetaxel emulsion) for the treatment of certain cancers. We have additional compounds in our portfolio that we currently are evaluating for future preclinical and clinical development. We intend to continue to build a portfolio of product candidates for the treatment of cancer and infectious diseases, with a focus on making improvements to currently marketed products with well-known mechanisms of action, that appear to have additional commercial potential if we are successful in our development efforts.
In October 2006, we licensed U.S. rights to ANX-211 (chitosan gel) to Theragenex, LLC in consideration of a licensing fee, milestone payments and royalty payments of 15% to 20% on licensed product sales, depending on sales levels. We anticipate Theragenex will launch a licensed product in late 2007 for the 2007/2008 cold and influenza season.
We are also pursuing the development of emulsion formulations for several currently marketed products that may allow us, under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act, or FDCA, to obtain marketing approval of such product candidates on timelines shorter than those associated with developing new chemical entities. For example, the FDA has indicated that a single 28-patient clinical study of ANX-530 indicating bioequivalence between our emulsion formulation of vinorelbine and the currently marketed product should be sufficient to support an NDA filing with the FDA.
Our overall strategy is to continue developing and commercializing our current and future product candidates, either independently or through license arrangements and co-development and/or co-marketing partnerships, and to enhance the commercial value of existing product candidates by improving their performance and safety profiles.
Because we are a developmental stage company that has not yet marketed any products or generated any significant revenue, we will seek to maintain adequate capital to fund operations through fees, milestones and royalties from license arrangements, or through other forms of financing such as debt financing, royalty-based financing, or sales of shares of common or preferred stock.
Our business was incorporated in Delaware in December 1995. In October 2000, we merged our wholly-owned subsidiary, Biokeys Acquisition Corp., with and into Biokeys, Inc. and changed our name to Biokeys Pharmaceuticals, Inc. In May 2003, we merged Biokeys, Inc., our wholly-owned subsidiary, with and into us and changed our name to ADVENTRX Pharmaceuticals, Inc. In July 2004, we formed a wholly-owned subsidiary ADVENTRX (Europe) Ltd. in the United Kingdom, primarily to facilitate conducting clinical trials in the European Union. In April 2006, we acquired SD Pharmaceuticals, Inc. as a wholly-owned subsidiary. Our principal executive offices are located at 6725 Mesa Ridge Road, Suite 100, San Diego, California 92121, and our telephone number is (858) 552-0866. Our corporate website is located at www.adventrx.com.
Our trademark CoFactor® is registered in the United States Patent and Trademark Office (in the Supplemental Register) under Registration No. 2,946,934, for use in connection with chemotherapy modulators derived from folic acid. We are developing commercial names for our other product candidates. All other trademarks, service marks or trade names appearing in this report, including but not limited to Avastin®, Camptosar®, Cremophor®, Eloxatin®, Erbitux®, Isovorin®, Navelbine®, Retrovir®, Taxol®, Taxotere®, Viread®, Abraxane® and Xeloda® are the property of their respective owners. Use or display by us of other parties’ trademarks, trade dress or products is not intended to and does not imply a relationship with, or endorsements or sponsorship of, us by the trademark or trade dress owners.
2
Table of Contents
Strategy
Our goal is to be a leading biopharmaceutical company focused on commercializing proprietary product candidates for the treatment of cancer and infectious diseases. Our near-term strategy is to focus on obtaining regulatory approval of our existing product candidates. Longer term, we intend to develop internally and/or acquire additional product candidates that fit our areas of expertise in cancer and infectious diseases. Specifically, we intend to:
| • | Increase the value of our lead product candidate, ANX-510, or CoFactor. We are applying the expertise of our clinical development team to conduct and complete our Phase III and Phase IIb clinical trials of CoFactor in the treatment of metastatic colorectal cancer. We are pursuing a clinical development strategy designed to facilitate regulatory approval and optimize marketing claims for this product candidate. To this end, in May 2006, we reached a written agreement with the FDA through the Special Protocol Assessment, or SPA, process regarding the design and planned analysis of our Phase III clinical trial. | ||
| • | Partner with leading pharmaceutical organizations. We plan to draw on the development, regulatory and commercial expertise of pharmaceutical companies in those instances where we believe our product candidates would benefit from such expertise. We believe CoFactor will benefit from a strategic alliance to gain competitive access to the oncology provider market and we are in active discussions with potential strategic partners. We also are evaluating partners for our other product candidates and would enter into partnering or license arrangements if the terms are attractive to us. | ||
| • | Pursue additional indications and commercial opportunities for our product candidates. We seek to increase the value of our current and future product candidates by pursuing other indications and commercial opportunities. For instance, in December 2006 we initiated a Phase II clinical trial of CoFactor for the treatment of advanced breast cancer. | ||
| • | Pursue the clinical development of our other product candidates. By the end of 2007, we anticipate initiating a Phase I/II clinical trial of ANX-201, or Thiovir™, for the treatment of HIV. We have a broad portfolio of product candidates and intend to evaluate, and if appropriate, seek regulatory approval for some of our product candidates under Section 505(b)(2) of the FDCA, which may accelerate our time-to-market. For example, in January 2007, we initiated a marketing-enabling clinical trial of ANX-530 (vinorelbine emulsion). In addition, pending positive results from on-going preclinical pharmacokinetic testing and agreement on clinical protocol design with the FDA, we expect to initiate a marketing-enabling clinical trial of either ANX-016 (vancomycin emulsion) or ANX-015 (clarithromycin emulsion) for the treatment of certain bacterial infections and a marketing-enabling clinical trial of ANX-514 (docetaxel emulsion) for the treatment of certain cancers. | ||
| • | Expand our portfolio of products and product candidates. We will seek additional opportunities to acquire product candidates to more fully exploit our clinical and regulatory capabilities and to increase the value of our business. |
3
Table of Contents
Product and Product Candidate Portfolio (a)
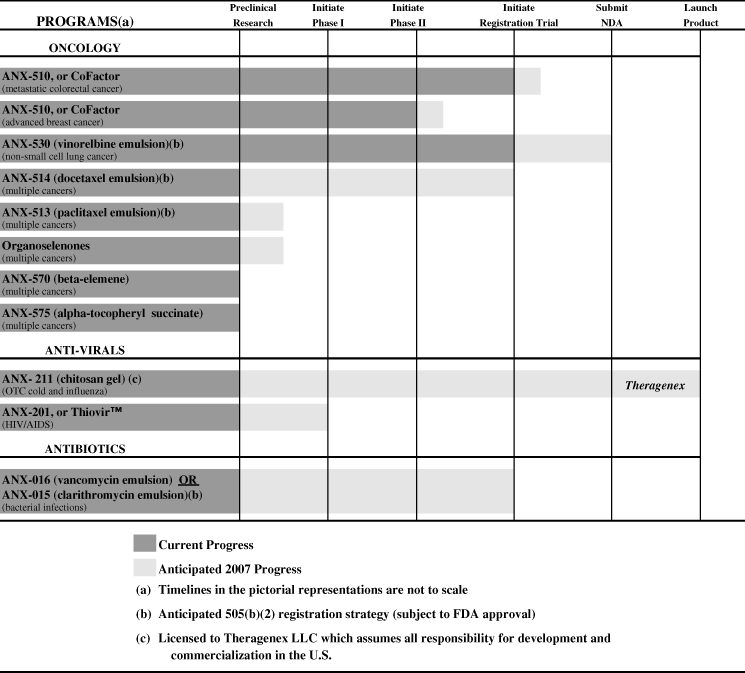
OUR ONCOLOGY PROGRAM CANDIDATES
Cancer Market and Current Therapies
Each year, more than 11 million people worldwide are diagnosed with and over 7 million people die from cancer. According to the American Cancer Society, cancer is the second most common cause of death in the U.S., accounting for 1 of every 4 deaths. Over 1.4 million new cancer cases will be diagnosed and over 500,000 people will die of cancer in the U.S. in 2007.
Treatment choices for cancer patients depend on the stage of the cancerous tumor, and whether and/or how far the cancer has spread. Treatment options include surgery, radiation, chemotherapy, hormone therapy and immunotherapy. Treatment of cancer with chemicals is referred to as chemotherapy. Cancer chemotherapies generated over $7.9 billion in revenues in the seven major pharmaceutical markets (the U.S., Japan, France, Germany, Italy, Spain and the United Kingdom) in 2005.
4
Table of Contents
Chemotherapy is highly individualized, depending on the type of disease and its progression, the action of the chemotherapeutic agent(s) used and the side effects in the patient, and may be used alone or in combination with other cancer therapies, such as surgery or radiation. Most chemotherapy drugs are chemical agents that are extremely toxic and are generally not curative.
Cancer therapies, or regimens, historically are defined in terms of lines (e.g., first-line, second-line, etc.), and the first treatment regimen a patient undertakes is typically termed “first line” therapy. What delineates one line from another varies based on the cancer. Typically, however, when a current regimen ceases to work and the cancer progresses, a new treatment regimen is undertaken and the patient is said to have moved to a subsequent line of therapy.
Antimetabolites are one class of chemotherapy drugs. The antimetabolite 5-fluorouracil, or 5-FU, is a widely used chemotherapeutic agent. Primary use of 5-FU includes treatment of colorectal, breast, gastric and hepatic cancers. 5-FU is sometimes used to treat other cancers, such as ovarian, pancreatic, prostate, bladder, cervical and head and neck.
ANX-510, or CoFactor
Metastatic Colorectal Cancer Market and Treatment Summary
The incidence of colorectal cancer in the seven major pharmaceutical markets is estimated to be over 450,000 in 2006. Colorectal cancer claims more than 170,000 lives annually in the U.S. and EU.
Multiple drugs and drug regimens are available to treat metastatic colorectal cancer; however, 5-FU and leucovorin represent the backbone of chemotherapy. 5-FU is associated with multiple toxicities, including hematological and gastrointestinal toxicities. Leucovorin, a folate-based compound, is often administered prior to the administration of 5-FU in order for 5-FU to work more effectively. Results from multiple clinical trials have shown that leucovorin in combination with 5-FU is effective in improving clinical outcomes in cancer patients relative to 5-FU alone. As a result, survey data of treating oncologists have shown that leucovorin is administered along with 5-FU in 73% of metastatic colorectal cancer patients in the first line setting, in 60% of metastatic colorectal cancer patients in the second line setting and in 68% of colorectal cancer patients when chemotherapy is administered in conjunction with a surgical resection of localized cancer. Based on data available from the most recent meta analysis of 19 previously published studies using 5-FU plus leucovorin in the first line treatment of metastatic colorectal cancer, or the historical comparator data, median overall survival was determined to be 11.7 months, with tumor response rate determined to be 21% (The Meta-Analysis Group in Cancer, J Clin Oncol 22:3766-3775, 2004, errata J Clin Oncol 23:1337-1338, 2005).
Chemotherapy regimens for metastatic colorectal cancer often include the addition of other cytotoxic agents to 5-FU/leucovorin-based therapies. For example, irinotecan (Camptosar) and oxaliplatin (Eloxatin) are drugs that may be added to 5-FU/leucovorin-based therapies. Newer, antibody-based drugs, such as cetuximab (Erbitux) and bevacizumab (Avastin) may also be added to 5-FU and leucovorin or be used as a monotherapy (as in the case of Erbitux). These drugs are used in a diverse set of therapeutic regimens, most of which include 5-FU and leucovorin, and are associated with multiple toxicities, including hematological and gastrointestinal toxicities, among others.
Recent data suggest the order of treatment regimens may reduce overall toxicity, improve the likelihood of the patient continuing treatment and thereby increase patient quality of life. The following tables, excerpted from a recent publication, provide some detail of these regimens. (Goldberg, et. al., The Oncologist 12:38-50, 2007) The decision to elect intensive versus less intensive therapy is influenced by the health, age and stamina of the patient. Notably, 5-FU/leucovorin is a component in almost all of these treatment regimens.
5
Table of Contents
Table A — Intensive Therapy for Metastatic Colorectal Cancer

FOLFOX consists of 5-FU, leucovorin and oxaliplatin
FOLFIRI consists of 5-FU, leucovorin and irinotecan
FOLFIRI consists of 5-FU, leucovorin and irinotecan
Table B — Less Intensive Therapy for Metastatic Colorectal Cancer
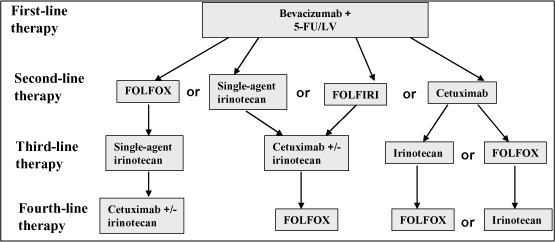
FOLFOX consists of 5-FU, leucovorin and oxaliplatin
FOLFIRI consists of 5-FU, leucovorin and irinotecan
FOLFIRI consists of 5-FU, leucovorin and irinotecan
The Use of Leucovorin in 5-FU Based Chemotherapy
The enzyme thymidylate synthase, or TS, acts in cells to convert deoxyuridine to deoxythymidine for incorporation into newly-replicating deoxyribonucleic acid, or DNA. Inhibition of TS is a well-established and effective method of killing rapidly dividing cells, such as tumor cells. Inhibition of TS is most frequently accomplished via use of 5-FU, the metabolite which binds to TS and disrupts DNA replication. This binding event between TS and the metabolite of 5-FU requires the action of a specific folate: 5,10-methylenetetrahydrofolate, or MTHF.
Currently, the source of this folate in the clinical setting is intravenous, or i.v., leucovorin calcium, which is administered prior to the administration of 5-FU. However, leucovorin efficiency is limited by the requirement that it undergo as many as four metabolic conversions to become the active folate MTHF. Leucovorin is an indirect source of MTHF and is often insufficient to achieve desired levels of TS inhibition in tumor cells. Even in high doses, leucovorin may not reach the desired concentration in the tumor tissue to be effective in helping 5-FU achieve its anti-tumor potential.
6
Table of Contents
Global sales of leucovorin, including the single-isomer formulation of leucovorin, exceeded $500 million in 2005.
The Opportunity for CoFactor
Our goal is to develop CoFactor as a replacement for leucovorin in 5-FU/leucovorin-based therapies in multiple cancers, including metastatic colorectal cancer. We believe that 5-FU/leucovorin will continue to be the backbone of chemotherapy in metastatic colorectal cancer and that CoFactor is well positioned to replace leucovorin as the biomodulator of 5-FU. Importantly, although there are a number of drugs being investigated to treat colorectal cancer, most of these compounds are being tested as additions to 5-FU/leucovorin-containing regimens. With an ongoing need for significant improvement in the clinical response of tens of thousands of cancer patients, especially those with metastatic colorectal cancer for which treatment regimens can be highly toxic, we believe CoFactor will successfully compete against or be used in conjunction with other therapies.
CoFactor, a biomodulator of 5-FU and a stable preparation of MTHF, is a folate-based compound that is the active form of leucovorin. We are developing CoFactor as a direct source of the active form of folate needed to achieve inhibition of TS. Unlike leucovorin, which must undergo chemical conversion prior to binding, CoFactor directly delivers the active form of folate and improves 5-FU’s ability to kill cancer cells while reducing 5-FU-associated toxicity.
CoFactor has been studied in the clinical setting as part of a treatment regimen for metastatic colorectal, breast, gastric and pancreatic cancers and has demonstrated enhancement of the antitumor effects of 5-FU. Two clinical studies have shown that 5-FU/CoFactor was a safe and well-tolerated regimen, with objective response, time to tumor progression and survival results that surpassed historical comparator data from multiple 5-FU/leucovorin clinical trials. In addition, we have performed preclinical studies that show benefits of CoFactor use with capecitabine (Xeloda), an orally-delivered compound that converts to 5-FU, over that of capecitabine alone.
We know of no other company that is developing a metabolite of leucovorin to enhance 5-FU activity, though leucovorin is marketed by more than a dozen companies as a generic drug for i.v. dosing in conjunction with 5-FU. Oral formulations of leucovorin are also available.
Clinical Development History
Phase I/II Clinical Trial in Metastatic Colorectal Cancer
In 1997, results were published from a Phase I/II dose-ranging clinical trial in 62 patients with colorectal, breast, gastric, pancreatic or gallbladder cancer. Varying doses of 5-FU and CoFactor were administered on a weekly bolus schedule with an endpoint of overall safety. Response rate, time–to-tumor progression, or TTP, and survival data were also captured. The trial indicated that 5-FU/CoFactor was a safe and well-tolerated regimen. Objective responses were observed in colorectal (21%), breast (56%), gastric (33%) and pancreatic (40%) cancer. Of the 24 patients who received 5-FU plus CoFactor for first-line treatment of metastatic colorectal cancer, 29% had an objective response.
Phase II Clinical Trial in Metastatic Colorectal Cancer
In 2005, we released data from a Phase II clinical trial in 50 patients which evaluated tumor response, safety, TTP and overall survival in first-line treatment of metastatic colorectal cancer patients using 5-FU and CoFactor. The study followed an open-label, single-arm, Simon two-stage design.
The primary endpoint for the study, objective response, exceeded the 25% target originally established as the lower boundary of interest. Blinded third-party radiology reviewers determined that 35% of patients achieved an objective response with 5-FU plus CoFactor, based on 46 patients that could be evaluated for response. Objective response consists of either a complete (100% regression of tumors) or partial (at least 50% regression of tumors) response lasting at least 4 weeks. According to the historical comparator data, the tumor response rate was determined to be 21% for first line treatment of metastatic colorectal cancer using 5-FU plus leucovorin.
7
Table of Contents
Median overall survival was reported as 459 days or approximately 15.1 months as estimated by Kaplan-Meier projections. Overall survival is defined as the time from the start of patient dosing until death. Median survival is the point at which 50% of patients in the study are still alive. According to the historical comparator data, median overall survival was determined to be 11.7 months for first line treatment of metastatic colorectal cancer using 5-FU plus leucovorin.
Median TTP was 162 days, or approximately 5.3 months. TTP is defined as the time from the start of patient dosing until objective tumor progression. This duration compares favorably to historical data from other clinical trials which used 5-FU and leucovorin, including the control arms for the pivotal registration trials of irinotecan and capecitabine.
CoFactor was well tolerated, with zero incidence of drug-related grade 3 or grade 4 gastrointestinal or hematological toxicity observed. A single case of grade 4 hematological toxicity occurred after the completion of the trial and subsequent to treatment with new chemotherapy. Because this event occurred within 30 days of the CoFactor study, it was reported as an adverse event, though it was attributed by the study investigator to a subsequent course of FOLFOX therapy.
In June 2006, we announced results from a longer term follow-up evaluation of the 50 patients who completed the clinical trial. The new information captures clinical responses to therapies for metastatic colorectal cancer that were selected by the clinical trial investigators in 33 patients after disease progression using 5-FU/CoFactor.
Of the 33 patients who went on to second-line treatment, four underwent partial liver resection for potential cure and 29 patients received chemotherapy. Chemotherapy consisted of either irinotecan or oxaliplatin, alone or in combination with 5-FU/leucovorin, 5-FU/leucovorin alone, as well as other agents. Seventeen patients did not receive any second-line treatment. Of the 29 patients who received post-study chemotherapy, four patients (13.8%) had an objective response, including one complete response. Median overall survival, measured from the initiation of first-line treatment, was 15.1 months for the whole population and was 23.0 months for the 33 patients that received second-line treatment, which includes the four patients who underwent surgical resection.
These results suggest that patients who initiated therapy with 5-FU/CoFactor had similar long term survival to patients who initiated therapy with oxaliplatin or irinotecan containing regimens, and that 5-FU/CoFactor could be a viable first-line treatment in a sequential treatment strategy.
Clinical Development Plan
Phase IIb Clinical Trial in Metastatic Colorectal Cancer
In September 2006, we completed enrollment in our 300-patient, Phase IIb, multi-national, randomized clinical trial. The study is designed to evaluate 5-FU/leucovorin versus 5-FU/CoFactor with the 5-FU administered over 46 hours by intravenous infusion (the de Gramont regimen). The primary endpoint of this study is a reduction of grade 3 and grade 4 gastrointestinal and hematological toxicity. Additionally, response rate, time to tumor progression and overall survival will be monitored.
Phase III Clinical Trial in Metastatic Colorectal Cancer
In June 2006, we initiated a pivotal Phase III trial, under a Special Protocol Assessment with the FDA. This clinical trial is sufficient to support a New Drug Application for marketing approval with the FDA in the U.S. This 1,200 patient, randomized clinical trial is being conducted in up to as many as 100 sites across the U.S. in patients with metastatic colorectal cancer who are receiving therapy in a first-line setting. Patients are randomized to either a leucovorin control arm or CoFactor experimental arm with a bolus regimen of 5-FU (the Roswell Park regimen) and bevacizumab. A primary endpoint of progression-free survival will be evaluated with secondary endpoints of severity of adverse events, response rate and overall survival. We expect that patient enrollment in this study will be completed in 2008.
8
Table of Contents
Phase II Clinical Trial in Advanced Breast Cancer
In December 2006, we initiated a Phase II clinical study of CoFactor for the treatment of advanced breast cancer. This trial is a single arm, multicenter study to evaluate the safety and efficacy of treatment with CoFactor plus 5-FU in advanced breast cancer patients who have failed anthracycline and taxane chemotherapies. Patients will be treated with CoFactor followed by 5-FU administered by i.v. bolus weekly for 6 weeks, with tumor and safety assessments every 8 weeks. The primary endpoint for the study is objective response rate, and secondary endpoints are duration of response, progression free survival, overall survival and incidence and severity of adverse events. We expect to enroll a total of 31 patients at several clinical sites in 2007. If we determine to proceed with the development of CoFactor in this disease indication, the outcome of this trial will help us determine a protocol for a Phase III pivotal study in advanced breast cancer.
ANX-530 (vinorelbine emulsion)
Background and Market Opportunity
Vinorelbine is a generic drug approved for use alone or in combination with cisplatin, a chemotherapy drug used to treat various types of cancers, for treatment of advanced non-small cell lung cancer. Vinorelbine works by disrupting microtubule formation and is a member of the vinca alkaloid class of antineoplastic agents.
In 2006, worldwide vinorelbine sales were approximately $200 million. Recently published clinical studies (ANITA trial results, presented by J. Douillard at 2005 ASCO Annual Meeting and Winton, et al, published in NEJM, June 23, 2005), showed a statistically significant improvement (p=0.0131) in overall survival in patients treated with vinorelbine plus cisplatin following tumor resection (this form of chemotherapy is commonly known as adjuvant therapy). Consequently, we believe that the use of vinorelbine in the adjuvant setting may increase. Vinorelbine has also shown activity in breast, ovarian and other cancers.
Vinorelbine tartrate is an approved product marketed under the brand name Navelbine and has been in use in Europe since 1989 and in the U.S. since 1994. Navelbine is approved to treat non-small cell lung cancer (in the U.S. and EU) and metastatic breast cancer (in the EU). The current formulation of vinorelbine can cause injection site reactions. Adverse events were reported in the Navelbine product insert from a Phase III clinical trial in patients treated with Navelbine as a single agent as an i.v. injection on a weekly basis. Adverse events reported for the non-small cell lung cancer subgroup from this trial include injection site reactions, consisting of erythema, pain at injection site and vein discoloration, and occurred in approximately one-third of patients. Five percent of these reactions were severe. Furthermore, chemical phlebitis along the vein proximal to the injection site was reported in ten percent of patients.
Advantages of ANX-530
ANX-530 is a novel emulsion formulation of vinorelbine tartrate. ANX-530 is designed to reduce the incidence and severity of vein irritation from i.v. delivery of the drug. This formulation emulsifies vinorelbine into a homogeneous suspension of nanoparticles and is designed to protect the venous endothelium during administration into a peripheral vein, thereby reducing associated vein irritation caused by the drug.
Preclinical Efficacy
In preclinical testing, ANX-530 was compared with vinorelbine reference products, such as Navelbine. In this setting, ANX-530 demonstrated less vein irritation than Navelbine. Vein irritation was examined in rabbits following repeated i.v. injections in the marginal ear vein. In two cancer model studies in rodents, ANX-530 demonstrated comparable efficacy to vinorelbine tartrate. In addition, preclinical results in a rat pharmacokinetic model demonstrated that the pharmacokinetics of ANX-530 were unchanged as compared to Navelbine.
Clinical Development Plan
We are conducting a single marketing-enabling clinical trial of ANX-530. In December 2006, the FDA accepted our Investigational New Drug Application, or IND, for ANX-530 and indicated that a single 28-patient clinical trial of ANX-530 indicating bioequivalence between our emulsion formulation of vinorelbine and the currently marketed product is sufficient to support an NDA filing with the FDA. Accordingly, we initiated a clinical trial that will compare the pharmacokinetics of ANX-530 with that of the approved form of vinorelbine in 28 patients
9
Table of Contents
with advanced solid tumors. Patient recruitment began in January 2007 and is on-going. While we intend to further develop ANX-530 as a form of vinorelbine that reduces vein irritation, if approved for commercialization, we intend to initially market the drug as equivalent to generic vinorelbine. Though we intend to conduct subsequent future clinical studies with the objective of demonstrating that our formulation of vinorelbine reduces vein irritation relative to the generic form of vinorelbine, we will not be able to promote ANX-530 as having any clinical advantages over currently marketed formulations of vinorelbine unless and until human clinical data supports such claims.
ANX-514 (docetaxel emulsion)
ANX-514 is a novel nano-emulsion formulation of the chemotherapy drug docetaxel, an approved product marketed under the brand name Taxotere, that is designed to eliminate the need for multi-day immunosuppressant premedication. Immunosuppressant premedication is recommended for docetaxel therapy to reduce the incidence and severity of allergic reactions. Docetaxel is an anti-cancer agent that acts by disrupting the cellular microtubular network that is essential for cell division. Docetaxel is approved to treat breast, non-small cell lung, prostate and gastric cancers. Global sales of Taxotere were 1.75 billion euros in 2006 or approximately U.S. $2.2 billion.
All taxanes, including docetaxel and paclitaxel, are highly water-insoluble. Polysorbate 80, a detergent that is used to solubilize docetaxel, can cause severe hypersensitivity reactions. Premedication with corticosteroids is recommended for patients treated with docetaxel. Severe hypersensitivity reactions may still occur despite pretreatment with corticosteroids.
ANX-514 is formulated without polysorbate 80 or other detergents and is designed to reduce the severity and/or incidence of hypersensitivity reactions. As a result, the need for multi-day immunosuppressant premedication could be eliminated. Currently, we are conducting additional preclinical pharmacokinetic testing of ANX-514 to compare this product candidate with the approved version of the product and plan to seek guidance from the FDA in 2007 with respect to the appropriateness of a Section 505(b)(2) NDA regulatory path for ANX-514.
ANX-513 (paclitaxel emulsion)
ANX-513 is a novel formulation of the chemotherapy drug paclitaxel, an approved product marketed under the brand name Taxol, that is intended to be non-allergenic and to reduce the need for immunosuppressant premedication. Immunosuppressant premedication is recommended for paclitaxel therapy to reduce the incidence and severity of hypersensitivity reactions. Paclitaxel is an antimicrotubule agent that inhibits the normal dynamic reorganization of the cellular microtubule network that is essential for cellular division. Paclitaxel is approved to treat breast, ovarian, Kaposi’s sarcoma and non-small cell lung cancers. Sales of paclitaxel, including branded (Taxol) and generic forms, exceeded $900 million in the 7 major markets in 2005. U.S. sales of Abraxane, an albumin-bound form of paclitaxel, totaled $174.9 million in 2006.
Cremophor (polyoxyl castor oil) is used to solubilize paclitaxel and is an active compound that can cause severe hypersensitivity reactions. It is recommended that all patients be premedicated with corticosteroids or anti-histamines prior to paclitaxel administration in order to prevent severe hypersensitivity reactions. Despite premedication, fatal reactions may still occur.
Our emulsion formulation of paclitaxel is formulated without Cremophor or detergents and is designed to reduce the severity and/or incidence of hypersensitivity reactions. As a result, the need for multi-day immunosuppressant premedication could be eliminated. Currently, we are conducting additional preclinical pharmacokinetic testing of ANX-513 to compare this product candidate with the approved version of the product.
Organoselenones
Organoselenones are selenium-containing drugs that belong to the alkylating class of chemotherapy. Alkylating agents are the most broadly used anticancer agents in the world. However, the effectiveness of current alkylating agents is limited since a majority of cancers develop drug resistance. Common drug-resistance mechanisms to alkylating agents include thiol resistance, alkyl transferase elevation and multidrug resistance. Accordingly, we believe there is potential for new products to address drug resistance in cancer therapy.
We are developing various compounds in our organoselenones program based on their broad anticancer activity and potentially lower susceptibility to drug resistance mechanisms. Preclinical studies have shown effectiveness of organoselenones against multiple tumor types, including breast, lung, ovarian, head/neck cancer, leukemias and lymphomas. Organoselenones have also shown anti-tumor activity against tumor cell lines with prior drug resistant profiles, including tumor cell lines resistant to other alkylating agents.
10
Table of Contents
Under a license from the University of Southern California, we have exclusive rights to a patent claiming a method of treating drug resistant cancers by administering one or more organoselenone compounds. We intend to undertake further preclinical testing of the organoselenones during 2007 in order to determine a potential lead compound from this family of drugs that may be moved into human testing in the future.
Other Oncology Program Product Candidates
We have a number of other compounds in our oncology program development portfolio that we are currently evaluating for future preclinical and clinical development. These compounds include ANX-570, a novel formulation of beta-elemene, a small molecule anticancer agent belonging to the triterpene family, and ANX-575, an emulsion formulation of alpha-tocopheryl succinate, which has been shown in preclinical studies to selectively facilitate cell death in cancer cells.
OUR ANTI-VIRAL PROGRAM CANDIDATES
ANX-211 (chitosan gel)
Chitosan gel is an intranasal/topical antiviral that, in preclinical studies, has demonstrated efficacy against viruses responsible for the common cold, influenza and other respiratory tract viral infections. We acquired chitosan gel in April 2006 as a part of our acquisition of SD Pharmaceuticals, Inc.
Each year there are an estimated 20-50 million cases of influenza and 500 million cases of the common cold in the U.S. alone. Nearly $3 billion is spent each year in the U.S. for over-the-counter, or OTC, drug products to fight the common cold, influenza and allergies.
In October 2006, we entered into a license agreement with Theragenex, LLC, a life science and technology company focusing on commercializing therapies across a number of different therapeutic areas. Under the agreement, we granted Theragenex exclusive rights to develop and commercialize chitosan gel in the U.S., in exchange for a licensing fee of $1.0 million ($500,000 of which we received in January 2007, with the remainder due in June 2007), a $1.0 million milestone payment that will be due upon the launch of each licensed product, and royalties of 15% to 20% on licensed wholesale product sales, depending on sales levels.
We anticipate Theragenex will launch a licensed product during the 2007/2008 cold and influenza season. Currently, the initial chitosan gel offering is intended to be sold as an OTC drug product. OTC drug products may be marketed for use by consumers without the intervention of a health care professional to obtain the product. Marketing pre-clearance of OTC drug products is not required if the standards of the OTC monograph are met. OTC monographs represent regulatory standards for the marketing of non-prescription drug products not covered by an NDA. These standards provide the marketing conditions for some OTC drug products, including the active ingredients, labeling and other general requirements.
ANX-201, or Thiovir™
Background and Market Opportunity
The World Health Organization and the Centers for Disease Control report there are 1.5 million HIV-positive individuals in the U.S. and the EU, where the vast majority of HIV drugs are used. According to the United Nations Program on HIV/AIDS, an estimated 39.5 million adults and children in the world are currently living with HIV and there were 4.3 million people newly infected with HIV in 2006.
Significant advancements have been made in the treatment of asymptomatic HIV positive patients with highly active antiretroviral therapy, or HAART, consisting of a three or four drug “cocktail” that can reduce HIV viral load to below detectable levels. However, studies have shown that poor patient treatment compliance, due to toxic side effects, number of pills and cost, will likely continue to cause problems of viral resistance, rendering many drugs ineffective. HIV has the ability to mutate into forms that are resistant to existing drug treatments. No single combination of drugs is effective for all patients and therapies are continually modified based upon patient progress.
The current HIV market consists of four different classes of drugs: nucleoside reverse transcriptase inhibitors, or NRTIs, non-nucleoside reverse transcriptase inhibitors, or NNRTIs, and protease inhibitors, which are dosed orally in various forms, as well as one viral entry inhibitor, which
11
Table of Contents
was approved in March 2003 and is dosed by injection. HIV entry inhibitors and maturation inhibitors are also currently under development by various pharmaceutical companies. Reverse transcriptase inhibitor sales were approximately $4.9 billion in the six major markets (U.S., Germany, UK, France, Italy and Spain) in 2005. The global commercial market for HIV treatments is expected to grow to almost $12 billion by 2012.
Limitations on Current Therapies
HIV replicates rapidly and can readily mutate to eventually evade inhibitor drugs and drug cocktails. Therefore, new drugs that target novel areas of the virus or overcome drug resistance are needed. We believe there is opportunity for ANX-201 since its mechanism of action is different from other classes on the market or that we are aware are in development. ANX-201 does not to bind reverse transcriptase in the same manner as currently-marketed NNRTIs.
Advantages of ANX-201
ANX-201 is a broad spectrum antiviral compound that has been shownin vitroto inhibit HIV, herpes and influenza A viruses. ANX-201 is a pyrophosphate analog that binds in the active site of viral polymerases, including reverse transcriptase, inhibiting viral DNA or RNA chain elongation and therefore viral replication. ANX-201 has a different mode of action and unique resistance profile, as shown in preclinical tests, from other HIV reverse transcriptase inhibitors. ANX-201 is being developed as a broad spectrum antiviral and novel reverse transcriptase inhibitor designed for oral delivery and as a component of HAART for HIV/AIDS.
ANX-201 is an active antiviral drug and is a precursor for the i.v.-delivered antiviral drug foscarnet (phosphonoformic acid). ANX-201 was designed to have increased bioavailability, enabling oral delivery. ANX-201, like foscarnet, is a pyrophosphate analog and inhibits a novel target on the reverse transcriptase molecule. The HIV resistance profile of ANX-201 and foscarnet is unique among reverse transcriptase inhibitors reflecting the distinct mechanism of action of this novel drug class.
Foscarnet is an effective, broad-spectrum antiviral we believe has limitations from a commercial perspective because it must be delivered by protracted infusion. We believe that ANX-201 can serve as an effective oral antiviral drug as part of HAART for HIV/AIDS. Preclinical studies have demonstrated that ANX-201 is equivalent to foscarnet as a reverse transcriptase inhibitor and has a dosage profile similar to foscarnet to inhibit HIV polymerase, with less effect on human DNA polymerases.
Preclinical Efficacy
In July 2005, we announced results from anin vitrostudy indicating that ANX-201 demonstrated effectiveness against HIV-1 which is resistant to other NNRTIs and NRTIs. ANX-201 also exhibited a slightly higher level of antiviral activity against HIV-1 than foscarnet. In combination testing with zidovudine, an NRTI marketed under the brand name Retrovir, ANX-201 was highly synergistic while foscarnet was only slightly synergistic to antagonistic. We have demonstrated synergy of ANX-201 with tenofovir, an NRTI marketed under the brand name Viread, in preclinical studies, and we are planning additional preclinical studies to test the synergy of ANX-201 with other NRTIs and NNRTIs in 2007.
In April 2006, we presented preclinical data for ANX-201 that showed strong antiviral synergy, without added cytotoxicity, in combination testing with zidovudine in a series ofin vitrotests serving as models for HIV infection. These preclinical results suggest that ANX-201 and zidovudine would be compatible as part of an antiviral therapy.
In June 2006 we presented preclinical results which demonstrated ANX-201 activity against HIV-1 and HIV-2 and against complex NRTI- and NNRTI-resistant viruses. Additional studies using ANX-201 with zidovudine showed synergistic activity against HIV strains, but without synergistic toxicity in human cells. Furthermore, ANX-201 re-sensitized zidovudine-resistant HIV strains to zidovudine, a finding that has potential important clinical implications.
Other preclinical tests
In preclinical tests with influenza virus, ANX-201 demonstrated antiviral activity against multiple subtypes of influenza B and influenza A, including a hybrid H5N1 avian influenza virus. These tests were conducted using assays measuring specific influenza virus antigen. ANX-201 was also found to be active in micro-molar concentrations against herpes simplex virus-1, or HSV-1, and HSV-2 in preclinical testing as measured by virus infection assays in human cell lines.
12
Table of Contents
In January 2006, we announced that a series of independent preclinical tests confirmed inhibition of influenza A virus by ANX-201. We conducted preliminary preclinical research on strains of influenza A, which includes the H5N1 avian flu strain. Additional preclinical studies are planned during 2007 to study ANX-201 against strains of influenza A, including the H5N1 avian flu.
These preclinical results demonstrate ANX-201 antiviral activity against a variety of important human viruses in the preclinical setting, and encourage further study of the antiviral efficacy of ANX-201 with multiple viruses.
Clinical Development Plan
We plan to file an IND with the FDA in 2007 and to initiate a Phase I/II clinical trial of ANX-201 in HIV-infected patients who have resistance to NRTIs. We anticipate the clinical trial will enroll patients who are resistant to one or more NRTIs.
OUR ANTIBIOTICS PROGRAM CANDIDATES
ANX-016 (vancomycin emulsion)
Vancomycin emulsion is a novel formulation of vancomycin, a glycopeptide antibiotic approved to treat gram-positive bacterial infections. Our emulsion formulation of vancomycin is designed to reduce the vein irritation and phlebitis associated with the i.v.-delivered drug. Vancomycin is approved to treat severe bacterial infections, including most Gram-positive bacteria such asStreptococci,Corynebacteria,ClostridiaandListeria. Commercially available i.v. vancomycin is highly irritating to tissue and can cause infusion-related events including erythema and pruritus. Phlebitis occurs in up to 13% of patients.
In 2005, the market for vancomycin for injection was approximately $130 million in the U.S. and over $470 million worldwide. Due to the rise of significant hospital infections, vancomycin use has increased. According to IMS Health, U.S. vancomycin unit sales increased by 14.3% compounded annual growth from 2000 to 2005.
Currently, we are conducting additional preclinical pharmacokinetic testing of ANX-016 to compare this product candidate with the approved version of the product and plan to seek guidance from the FDA in 2007 with respect to the appropriateness of a Section 505(b)(2) NDA regulatory path for our emulsion formulation of vancomycin.
ANX-015 (clarithromycin emulsion)
ANX-015 is a proprietary intravenous emulsion formulation of clarithromycin, a macrolide antibiotic. ANX-015 is designed to reduce vein irritation associated with i.v.-delivery of the drug. The most frequently reported infusion-related adverse events in clinical studies with i.v. clarithromycin were injection-site inflammation, tenderness, phlebitis and pain. Clarithromycin is highly potent against a variety of aerobic and anaerobic Gram-positive and Gram-negative organisms. Clarithromycin-administered i.v. is indicated whenever parenteral therapy is required for treatment of sensitive microorganisms in upper respiratory tract infections, lower respiratory tract infections and skin and soft tissue infections.
Clarithromycin is approved for mild to moderate bacterial infections such as in community-acquired pneumonia. Worldwide sales of i.v. clarithromycin, excluding the U.S. where an i.v. formulation is not available, exceeded 1.9 million units in 2005.
Currently, we are evaluating potential clinical development of ANX-015 and could pursue clinical development pending the outcome of our studies of ANX-016.
13
Table of Contents
Competition
If we receive regulatory approval to manufacture, market, distribute and sell any of our product candidates, we will face significant and long-term competition from pharmaceutical companies, pharmaceutical divisions of chemical companies and biotechnology, biopharmaceutical and specialty pharmaceuticals companies. This competition will likely become more intense as commercial applications for biotechnology products increase. Most of our competitors, particularly large pharmaceutical companies, have greater clinical, regulatory, manufacturing, marketing, distribution and financial resources and experience than we have. Many of these companies have commercial arrangements with other companies to supplement their internal research capabilities.
The introduction of new products or processes by our competitors or new information about our existing products or product candidates may impact potential pricing of our products or cause us to discontinue the development of one or more of our products or product candidates, even for products and or product candidates protected by patents. For example, Xeloda, an orally-delivered compound that converts to 5-FU and that may be used without leucovorin, could compete against sales of CoFactor, if CoFactor is approved. In addition, orally-administered leucovorin, even if inferior to CoFactor in terms of safety and effectiveness, has a differentiating quality relative to CoFactor, which currently is being developed as an i.v. drug. Furthermore, with respect to ANX-530 (vinorelbine emulsion), companies have developed novel formulation technologies, such as those utilizing liposomal carriers which may be applied to vinorelbine. There is also an oral formulation of vinorelbine approved for use in the EU against which we would compete if our emulsion formulation of vinorelbine were approved for use in the EU.
Many of our product candidates, if approved, initially will compete against generic products. As a result, our products may compete primarily on the basis of price, quality of product and consumer awareness and perception. Most of our competitors will likely have substantially greater financial, marketing and other resources, longer operating histories, larger product portfolios and greater brand recognition than us. With our limited resources, our success may be tied to emphasizing the unique claims regarding our products and providing consumers with innovative delivery systems; however, we will be limited to marketing those of our product candidates that are approved under a Section 505(b)(2) NDA as equivalent to their generic counterparts. Our ability to differentiate our products will be limited to claims we can support with data from additional clinical trials that the FDA approves.
Over the longer term, our ability to work with our collaborators to successfully manufacture, market, distribute and sell any of our or their approved products, expand their usage and bring new products to the marketplace will depend on many factors, including, but not limited to, the effectiveness and safety of these products, FDA and foreign regulatory agencies’ approvals of new products and indications, the degree of patent protection afforded to particular products and the effect of managed care as a purchaser of these products.
Commercialization Strategy
We have not received the necessary regulatory approval from the FDA or any other similar government agency to commercially market, distribute or sell any of our products. We currently do not have significant internal sales, distribution or marketing capabilities. To commercialize any of our product candidates, we must either acquire or internally develop sales, distribution and marketing capabilities, or enter into collaborations with partners to perform these services for us, or both. If we approach the point at which we anticipate receiving regulatory approval to commercially market, distribute or sell any of our products, we will likely arrange with third parties, such as pharmaceutical companies, to market, distribute and sell our products.
In October 2006, we entered into a license agreement with Theragenex, LLC, a life science and technology company focusing on commercializing therapies across a number of different therapeutic areas. Under the agreement, we granted Theragenex exclusive rights to develop and commercialize chitosan gel in the U.S. in exchange for a licensing fee of $1.0 million ($500,000 of which we received in January 2007, with the remainder due in June 2007), a $1.0 million milestone payment that will be due within 45 days after the launch of each licensed product, and royalties of 15% to 20% on licensed product sales, depending on sales levels. The agreement remains in effect through the later of the latest date on which the last licensed product is covered by a valid claim or 20 years from the date of the first commercial sale of the last licensed product by Theragenex. Either party may terminate the agreement if the other materially breaches or materially defaults in the performance or observance of any of the
14
Table of Contents
provisions of the agreement. In addition, either party may, upon notice, terminate the agreement, if the other party admits in writing that it is generally unable to meet its debts when due, or upon the filing of bankruptcy, reorganization, liquidation or receivership proceedings involving such party. Theragenex may terminate the agreement at any time, upon 90 days’ written notice, if Theragenex concludes in good faith, based on technical information learned by it following execution of the agreement, that there is no reasonable likelihood of a commercially viable licensed product.
Manufacturing
We do not have our own manufacturing facilities. We currently contract with numerous outside manufacturers in order to produce our product candidates, including our clinical trial material. We rely on these suppliers for all aspects of the production process, including manufacture and milling of active pharmaceutical ingredient, or API, formulation and assembly of the final drug product, labeling, testing and release, packaging and storage of API and finished drug product. Each manufacturer is responsible for sourcing all raw materials used in its production of our product candidates from third party suppliers, which are widely available. We rely on individual proposals and purchase orders to meet our needs from these suppliers and typically rely on terms and conditions proposed by the manufacturer or us to govern our rights and obligations under each order (including provisions with respect to intellectual property, if any). We do not have agreements with any of our outside manufacturers for the long-term or commercial supply of any of our product candidates, including our clinical trial material. We currently anticipate negotiating more comprehensive contracts with certain manufacturers. We have some flexibility in securing other manufacturers to produce our product candidates, though in some circumstances we may be limited in our alternatives due to proprietary technologies or methods used in the manufacture of some of our product candidates.
Intellectual Property
Patents
We own or have exclusive rights under 12 issued patents (9 U.S. and 3 foreign) and 87 applications pending (11 U.S. and 76 foreign (including PCT)). Patents and patent applications owned by or exclusively licensed to us include compositions comprising 5,10-methylenetetrahydrofolate and methods of formulation and their use for treating cancer (which relate to CoFactor), methods for the production of thiophosphonoformic acid and its analogs and their use for treating HIV (which relate to ANX-201), antiviral compositions comprising chitosan and zinc (which relate to ANX-211), vinca alkaloid emulsions for treating cancer (which relate to ANX-530), paclitaxel and docetaxel emulsions for treating cancer (which relate to ANX-513 and ANX-514), clarithromycin and vancomycin emulsions for treating bacterial infections (which relate to ANX-015 and ANX-016), bete-elemene emulsions for treating cancer and other indications (which relate to ANX-570), pharmaceutical compositions comprising alpha tocopheryl succinate (which relate to ANX-575), and methods for treating drug resistant cancers using organoselenone compounds.
| Issued Patents and Pending Applications | ||||||||||||||||||||||||||||||||
| United States | Foreign | |||||||||||||||||||||||||||||||
| Expiration | Expiration | Expiration | Expiration | |||||||||||||||||||||||||||||
| Subject: | Issued | Date | Pending | Date | Issued | Date | Pending | Date | ||||||||||||||||||||||||
| 5,10-methylenetetrahydrofolate | 2 | 2011-13 | 1 | 2026 | 1 | 2011 | 10 | 2025-26 | ||||||||||||||||||||||||
| Thiophosphonoformic acid | 6 | 2009-20 | 2 | 2026 | 2 | 2019-20 | 4 | 2020-26 | ||||||||||||||||||||||||
| Chitosan-zinc antiviral | — | 1 | 2024 | — | 25 | 2024 | ||||||||||||||||||||||||||
| Vinca alkaloid emulsion | — | 1 | 2024 | — | 24 | 2024 | ||||||||||||||||||||||||||
| Alpha tocopheryl succinate | — | 1 | 2025 | — | 9 | 2024 | ||||||||||||||||||||||||||
| Paclitaxel/docetaxel emulsions | — | 2 | 2024-25 | — | 1 | 2024-25 | ||||||||||||||||||||||||||
| Clarithromycin emulsion | — | 1 | 2024 | — | 3 | 2024 | ||||||||||||||||||||||||||
| Vancomycin emulsion | — | 1 | 2026 | — | — | |||||||||||||||||||||||||||
| Beta-elemene emulsion | — | 1 | 2026 | — | — | |||||||||||||||||||||||||||
| Organoselenone | 1 | 2014 | — | — | — | |||||||||||||||||||||||||||
| Total | 9 | 11 | 3 | 76 | ||||||||||||||||||||||||||||
We cannot provide assurance that our pending patent applications will issue as patents, that any issued patents will provide us with significant competitive advantages, or that the validity or enforceability of any of our patents will not be challenged or, if instituted, that these challenges will not be successful. The cost of litigation to uphold the validity and prevent infringement of our patents could be substantial. Furthermore, we cannot provide assurance
15
Table of Contents
that others will not independently develop similar technologies or duplicate our technologies or design around the patented aspects of our technologies. We can provide no assurance that our proposed technologies will not infringe patents or rights owned by others, the licenses to which might not be available to us.
In addition, the approval process for patent applications in foreign countries may differ significantly from the process in the U.S. The patent authorities in each country administer that country’s laws and regulations relating to patents independently of the laws and regulations of any other country and the patents must be sought and obtained separately. Therefore, approval in one country does not necessarily indicate that approval can be obtained in other countries.
Trademarks
Our trademark CoFactor is registered in the U.S. Patent and Trademark Office (in the Supplemental Register) under Registration No. 2,946,934, for use in connection with chemotherapy modulators derived from folic acid.
Research and Development
Our research and development expenses were $12.0 million in 2006, $8.7 million in 2005 and $2.7 million for 2004. Our research and development expenses consist primarily of salaries and related employee benefits, costs associated with clinical trials managed by our clinical research organizations, or CROs, and costs associated with non-clinical activities, such as regulatory expenses. Our most significant costs are for clinical trials. These expenses include payments to vendors such as CROs, investigators, clinical suppliers and related consulting.
Licensing Agreements
University of Southern California Agreements
ANX-510, or CoFactor
Under an option and license agreement with the University of Southern California, or USC, entered into in January 1998 and amended in August 2000, we hold exclusive rights to a number of patents that have issued in the U.S. and Canada covering our CoFactor product candidate and its use in connection with cancer chemotherapy. An additional patent included in the agreement relates to compounds in our organoselenones program that we are currently evaluating for future preclinical and clinical development.
This agreement terminates on the last to expire of the licensed patents, which is expected to occur in March 2014. Upon breach or default under the agreement, the non-breaching party may terminate the agreement by 45 days’ written notice. USC may terminate the agreement upon 20 days’ notice if we fail to obtain and maintain the insurance required by the agreement and may terminate the agreement immediately upon notice if we attempt to use, sublicense, transfer or assign our rights or obligations under the agreement in any manner contrary to its terms or in derogation of USC’s proprietary rights and upon bankruptcy, reorganization, liquidation or receivership proceedings involving us. We may terminate the agreement at any time by providing USC 30 days’ written notice.
This agreement provides for the payment to USC of a 3% royalty on net sales by us or a sublicensee of licensed products, as well as a prepaid royalty of $100,000 within 30 days of approval of an NDA by the FDA for any product covered by the claims of the licensed patents (which prepaid royalty is deductible from future royalty payments). In addition, we are required to reimburse all reasonable legal expenses incurred by USC in filing, prosecuting and maintaining the licensed patents. No royalties have been paid to date under this agreement.
ANX-201, or Thiovir™
Under an option and license agreement with USC entered into in August 2000 and amended in April 2003 and January 2007, we hold exclusive rights to a number of patents that have issued in the U.S. and the EU covering methods for the manufacture of our ANX-201 product candidate and of various analogs and derivatives thereof, and the use of ANX-201 in connection with the treatment of HIV.
16
Table of Contents
This agreement terminates on the last to expire of the licensed patents, which is expected to occur in November 2020. Upon breach or default under the agreement, the non-breaching party may terminate the agreement by 45 days’ written notice. USC may terminate the agreement immediately upon notice if we attempt to use, sublicense, transfer or assign our rights or obligations under the agreement in any manner contrary to its terms or in derogation of USC’s propriety rights, we fail to obtain and maintain the insurance required by the agreement and upon bankruptcy, reorganization, liquidation or receivership proceedings involving us. In addition, if we fail to achieve the milestones set forth in the agreement, as amended, USC has the option to terminate the agreement but only by providing written notice of termination to us within one (1) month of the applicable milestone deadline. We may terminate the agreement at any time by providing USC 30 days’ written notice and reimbursing the reasonable legal expenses incurred by USC for up to one (1) month from the date written notification of termination is sent by us.
This agreement provides for the payment to USC of a 1% royalty on net sales by us of licensed products and milestone payments on each licensed product upon entering Phase I clinical trials ($75,000), reaching Phase II clinical trials ($100,000), reaching Phase III clinical trials ($125,000) and upon receiving market approval from the FDA or other government regulatory agency ($250,000). In addition, if any licensed product is manufactured and sold under sublicense from us, we will pay USC a royalty based on a percentage of all of the revenue that we receive from the sublicense (including all earned royalties, prepaid royalties and license fees). Furthermore, we are required to reimburse all reasonable legal expenses incurred by USC in filing, prosecuting and maintaining the licensed patents. No royalties have been paid to date under this agreement.
Though the licenses granted to us under both USC agreements are ostensibly exclusive, because the technologies developed by USC were developed in part through funding provided by the U.S. government, our license is subject to a non-exclusive, non-transferable, royalty-free right of the U.S. government and USC to practice the licensed technologies for research purposes and, in the case of the U.S. government, other governmental purposes on behalf of the U.S. and on behalf of any foreign government or international organization pursuant to any existing or future treaty or agreement with the U.S., but only to the extent the government funded the research. The government also reserves the right to require us to grant sublicenses to third parties when necessary to fulfill public health and safety needs or if we do not reasonably satisfy government requirements for public use of the technology. In addition, USC has the right to use all improvements to the licensed technology for research and educational purposes. In addition, licenses of technology developed through funding provided by the U.S. government, including the USC licenses, require that licensees — in this case, us — and our affiliates and sub-licensees agree that products covered by the licenses will be manufactured substantially in the U.S. If we are unable to contract for manufacturing facilities in the U.S. or obtain an appropriate waiver, we risk losing our rights under the USC agreements.
SD Pharmaceuticals Agreement
In April 2006 we acquired SD Pharmaceuticals, Inc. Under a prior license agreement between SD Pharmaceuticals, Latitude Pharmaceuticals, Inc. and Andrew X. Chen, the sole owner of Latitude Pharmaceuticals, Dr. Chen had assigned to SD Pharmaceuticals all right and interest of Dr. Chen and Latitude Pharmaceuticals to certain patents throughout the world other than in China, Taiwan, Hong Kong and Macau. Under this agreement, SD Pharmaceuticals granted back to Latitude Pharmaceuticals a worldwide, exclusive, royalty-free and irrevocable license to use the assigned patents in all fields of use other than certain excluded fields as specified in the agreement. Our rights in ANX-015 (clarithromycin emulsion), ANX-016 (vancomycin emulsion), ANX-211 (chitosan gel), ANX-513 (paclitaxel emulsion), ANX-514 (docetaxel emulsion), ANX-530 (vinorelbine emulsion), ANX-570 (beta elemene), ANX-575 (alpha-tocopheryl succinate) arise through our interest in SD Pharmaceuticals. Accordingly, we have no rights in these product candidates in China, Taiwan, Hong Kong and Macau, and our rights under the assigned patents in the rest of the world are limited to the following fields:
| • | For ANX-015, clarithromycin intravenous emulsion formulation for infectious disease and any other disease indication. | ||
| • | For ANX-016, vancomycin intravenous emulsion formulation for infectious disease and any other disease indication. | ||
| • | For ANX-211, chitosan in combination with zinc for topical anti-viral use. | ||
| • | For ANX-513, paclitaxel intravenous emulsion formulation for cancer treatment and any other disease indication. |
17
Table of Contents
| • | For ANX-514, docetaxel intravenous emulsion formulation for cancer treatment and any other disease indication. | ||
| • | For ANX-530, vinca alkyloid intravenous emulsion formulation for cancer treatment and any other disease indication. | ||
| • | For ANX-570,b-elemene intravenous emulsion formulation for cancer treatment and any other disease indication. | ||
| • | For ANX-575, alpha-tocopheryl succinate intravenous emulsion formulations for cancer and any other disease indication. |
Government Regulations
FDA Approval Process
The manufacture, distribution, marketing and sale of therapeutic drugs are subject to government regulation in the U.S. and in various foreign countries. In the U.S., we must follow rules and regulations established by the FDA requiring the presentation of data indicating that our product candidates are safe and effective and are manufactured in accordance with current good manufacturing practice, or cGMP, regulations.
The steps required to be taken before a new prescription drug may be marketed in the U.S. include (i) preclinical laboratory and animal tests, (ii) the submission to the FDA of an IND, which must be evaluated and not subject to a “hold” by the FDA before human clinical trials may commence, (iii) adequate and well-controlled human clinical trials to establish the safety and effectiveness of the drug, (iv) the submission of an NDA to the FDA and (v) FDA approval of the NDA. Prior to obtaining FDA approval of an NDA, the facilities that will be used to manufacture the drug must undergo a pre-approval inspection to ensure compliance with the cGMP regulations.
Preclinical Testing
Preclinical tests include laboratory evaluation of product chemistry or biology and animal studies to assess the safety and effectiveness of the drug and its formulation. The results of the preclinical tests and any human data are submitted to the FDA as part of an IND, and unless the FDA objects, the IND will become effective 30 days following its receipt by the FDA, after which human clinical trials may begin. If the FDA has concerns about the proposed clinical trial, it may delay the trial and require modifications to the trial protocol prior to permitting the trial to begin.
Clinical Trials
Clinical trials involve the administration of the drug to healthy volunteers or to patients identified as having the condition for which the drug is being tested. The drug is administered under the supervision of a qualified principal investigator. Clinical trials are conducted in accordance with protocols previously submitted to the FDA as part of the IND that detail the objectives of the trial, the parameters used to monitor safety and the efficacy criteria that are being evaluated. Each clinical trial is conducted under the auspices of an institutional review board. This review board considers, among other things, ethical factors, the safety of the human subjects and the possible liability risk for the institution.
Clinical trials are typically conducted in three sequential phases that may or may not overlap. In Phase I, the emphasis is on testing for safety (adverse effects), dosage tolerance, metabolism, distribution, excretion and clinical pharmacology. Study populations in Phase I clinical trials are typically healthy human volunteers. Phase II involves trials in a limited patient population to determine the effectiveness of the drug for specific targeted indications, to further refine dosage tolerance and optimal dosage and to identify possible adverse side effects and safety risks.
In clinical trials of cancer indications, Phase II typically denotes an uncontrolled study which addresses primary response rate. Phase IIb studies are generally larger, and controlled, and serve to finalize dosing regimens for Phase III trials.
In serious diseases, such as HIV and cancer, patients suffering from the disease rather than healthy volunteers are included in Phase I trials. In addition, Phase I trials may be divided between Phase Ia, in which single doses of the drug are given, and Phase Ib, in which multiple doses are given. In the latter instance, some efficacy data may be obtained if the subjects are patients suffering from the disease rather than healthy volunteers, and these trials are referred to as “Phase Ib/IIa.”
18
Table of Contents
After a drug has been shown in Phase II trials to have an acceptable safety profile and probable effectiveness, Phase III trials are undertaken to evaluate clinical effectiveness further and to further test for safety within an expanded patient population at multiple clinical study sites. The FDA reviews both the clinical trial plans and the results of the trials at each phase, and may discontinue the trials at any time if there are significant safety issues.
Special Protocol Assessment
The sponsor of a clinical trial may request a special protocol assessment from the FDA. If an SPA is requested, the FDA will evaluate within 45 days certain protocols, including Phase III clinical trial protocols, and issues relating to the protocols to assess whether they are adequate to meet scientific and regulatory requirements identified by the sponsor. In a Phase III clinical protocol SPA request, the sponsor may request that data used in a Phase III clinical trial form the primary basis for an efficacy claim. The clinical protocols for Phase III trials can relate to efficacy claims that will be part of an original NDA or that will be part of an efficacy supplement to an approved NDA.
The results of the preclinical tests and clinical trials are submitted to the FDA in the form of an NDA for marketing approval. The testing and approval process requires substantial time and effort, and FDA approval may not be granted on a timely basis or at all. The approval process is affected by a number of factors, including the severity of the disease, the availability of alternative treatments and the risks and benefits demonstrated in clinical trials. Additional animal studies or clinical trials may be requested during the FDA review process that may delay marketing approval.
Section 505(b)(1) New Drug Applications
The approval process described above is premised on the applicant being the owner of, or having obtained a right of reference to, all of the data required to prove the safety and effectiveness of a drug. This type of marketing application, sometimes referred to as a “full” or “stand-alone” NDA, is governed by Section 505(b)(1) of the FDCA. A Section 505(b)(1) NDA contains full reports of investigations of safety and effectiveness, which includes the results of preclinical studies and clinical trials, together with detailed information on the manufacture and composition of the product, in addition to other information. We intend to submit a Section 505(b)(1) application for ANX-201 and CoFactor.
Section 505(b)(2) New Drug Applications
Section 505(b)(2) of the FDCA allows the FDA to approve a follow-on drug on the basis of data in the scientific literature or conclusions regarding safety or effectiveness made by the FDA in the approval of other drugs. Section 505(b)(2) of the FDCA was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Act. This procedure potentially makes it easier for drug manufacturers to obtain rapid approval of new forms of drugs based on the FDA’s approval of the original drug. Some examples of products that may be allowed to follow a 505(b)(2) path to approval are drugs that have a new dosage form, strength, route of administration, formulation or indication.
To the extent that the Section 505(b)(2) applicant is relying on the FDA’s findings for an already-approved drug, the applicant is required to certify to the FDA concerning any patents listed for the approved drug in the FDA’s Orange Book publication. Specifically, the applicant must certify that: (1) the required patent information has not been filed; (2) the listed patent has expired; (3) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (4) the listed patent is invalid or will not be infringed by the manufacture, use or sale of the new drug. A certification that the new drug will not infringe the already approved drug’s Orange Book-listed patents or that such patents are invalid is called a paragraph IV certification. If the applicant does not challenge the listed patents, the Section 505(b)(2) application will not be approved until all the listed patents claiming the referenced drug have expired. The Section 505(b)(2) application may also not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced drug has expired.
19
Table of Contents
If the applicant has provided a paragraph IV certification to the FDA, the applicant must also send notice of the paragraph IV certification to the NDA and patent holders once the NDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a legal challenge to the paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of their receipt of a paragraph IV certification automatically prevents the FDA from approving the Section 505(b)(2) NDA until the earliest of 30 months, expiration of the patent, settlement of the lawsuit or a decision in the infringement case that is favorable to the Section 505(b)(2) applicant. For drugs with five-year exclusivity, if an action for patent infringement is initiated after year four of that exclusivity period, then the 30-month stay period is extended by such amount of time so that 7.5 years has elapsed since the approval of the NDA with five-year exclusivity. This period could be extended by six months if the NDA sponsor obtains pediatric exclusivity. Thus, the Section 505(b)(2) applicant may invest a significant amount of time and expense in the development of its drugs only to be subject to significant delay and patent litigation before its drugs may be commercialized. Alternatively, if the listed patent holder does not file a patent infringement lawsuit within the required 45-day period, the applicant’s NDA will not be subject to the 30-month stay.
Notwithstanding the approval of many products by the FDA pursuant to Section 505(b)(2), over the last few years, certain brand-name pharmaceutical companies and others have objected to the FDA’s interpretation of Section 505(b)(2) and one pharmaceutical company has sued the FDA on the matter. Although the issues in that litigation are specific to the products involved, if the FDA does not prevail, it may be required to change its interpretation of Section 505(b)(2), which could delay or even prevent the FDA from approving any Section 505(b)(2) NDA that we submit. We intend to submit an NDA under Section 505(b)(2) for vinorelbine emulsion.
Upon approval, a drug may be marketed only for the FDA-approved indications in the approved dosage forms. Further clinical trials are necessary to gain approval for the use of the product for any additional indications or dosage forms. The FDA may also require post-market reporting and may require surveillance programs to monitor the side effects of the drug, which may result in withdrawal of approval after marketing begins.
The Hatch-Waxman Act
Under the Hatch-Waxman Act, newly-approved drugs and indications benefit from a statutory period of non-patent marketing exclusivity. The Hatch-Waxman Act provides five-year marketing exclusivity to the first applicant to gain approval of an NDA for a new chemical entity, meaning that the FDA has not previously approved any other new drug containing the same active moiety. Hatch-Waxman prohibits the submission of an abbreviated new drug application, or ANDA, or a Section 505(b)(2) NDA for another version of such drug during the five-year exclusive period; however, as explained above, submission of an ANDA or Section 505(b)(2) NDA containing a paragraph IV certification is permitted after four years, which may trigger a 30-month stay of approval of the ANDA or Section 505(b)(2) NDA. Protection under Hatch-Waxman will not prevent the submission or approval of another full NDA; however, the applicant would be required to conduct its own preclinical and adequate and well-controlled clinical trials to demonstrate safety and effectiveness. The Hatch-Waxman Act also provides three years of marketing exclusivity for the approval of new and supplemental NDAs, including Section 505(b)(2) NDAs, for, among other things, new indications, dosages or strengths of an existing drug, if new clinical investigations that were conducted or sponsored by the applicant are essential to the approval of the application.
The FDA and other jurisdictions have developed several regulatory procedures to accelerate the clinical testing and approval of, or to confer certain commercial benefits to, drugs intended to treat serious or life-threatening and under-served illnesses.
Orphan Drug Designation and Exclusivity
The U.S., as well as other jurisdictions (including the EU), may designate drugs for relatively small patient populations as orphan drugs. The FDA grants orphan drug designation to drugs intended to treat a rare disease or condition that affects fewer than 200,000 individuals in the U.S. or more than 200,000 individuals in the U.S. and for which there is no reasonable expectation that the cost of developing and making available in the U.S. a drug for this type of disease or condition will be recovered from sales in the U.S. for that drug. In the U.S., orphan drug
20
Table of Contents
designation must be requested before submitting an application for marketing approval. Orphan drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process. If, however, a drug which has an orphan drug designation subsequently receives the first FDA approval for the indication for which it has such designation, the drug is entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan exclusivity. Also, competitors may receive approval of different drugs or biologics for the indications for which the orphan product has exclusivity. We have submitted to and received from the FDA an orphan drug designation for CoFactor for the treatment of pancreatic cancer. If the FDA approves our marketing application and CoFactor subsequently receives the first FDA approval for pancreatic cancer, we will be granted seven years of orphan drug exclusivity. This period of exclusivity will run concurrently with any three-year period of exclusivity applicable to this product candidate awarded upon FDA approval.
Under EU medicines laws, criteria for designation as an “orphan medicine” are similar but somewhat different from those in the U.S. A drug is designated as an orphan drug if the sponsor can establish that the drug is intended for a life-threatening or chronically debilitating condition affecting no more than five in 10,000 persons in the EU or that is unlikely to be profitable, and if there is no approved satisfactory treatment or if the drug would be a significant benefit to those persons with the condition. Orphan medicines are entitled to ten years of market exclusivity, except under certain limited circumstances comparable to U.S. law. During this period of market exclusivity, no “similar” product, whether or not supported by full safety and efficacy data, will be approved unless a second applicant can establish that its product is safer, more effective or otherwise clinically superior. This period may be reduced to six years if the conditions that originally justified orphan designation change or the sponsor makes excessive profits. We have submitted and received in the EU an orphan drug designation for CoFactor for the treatment of pancreatic cancer.
Other Regulatory Requirements
In addition to FDA restrictions on marketing of drugs, several other types of state and federal laws have been applied to restrict certain marketing practices in the pharmaceutical industry in recent years. These laws include anti-kickback statutes and false claims statutes. The federal health care program anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any health care item or service reimbursable under Medicare, Medicaid or other federally financed health care programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Violations of the anti-kickback statute are punishable by imprisonment, criminal fines, civil monetary penalties and exclusion from participation in federal health care programs. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. Recently, several pharmaceutical and other health care companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws. The majority of states also have statutes or regulations similar to the federal anti-kickback law and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
Also, as part of the sales and marketing process, pharmaceutical companies frequently provide samples of approved drugs to physicians. This practice is regulated by the FDA and other governmental authorities, including, in particular, requirements concerning record-keeping and control procedures.
21
Table of Contents
Outside of the U.S., our ability to market our products will depend on receiving marketing authorizations from the appropriate regulatory authorities. The foreign regulatory approval process includes all of the risks associated with the FDA approval process described above. The requirements governing the conduct of clinical trials and marketing authorization vary widely from country to country. In addition to the import requirements of foreign countries, a company must also comply with U.S. laws governing the export of FDA-regulated products.
We and our manufacturers and clinical research organizations may also be subject to regulations under other federal, state and local laws, including the Occupational Safety and Health Act, the Environmental Protection Act, the Clean Air Act and import, export and customs regulations as well as the laws and regulations of other countries.
Third Party Reimbursement and Pricing Controls
In the U.S. and elsewhere, sales of prescription pharmaceuticals are dependent in large part on the availability of reimbursement to the consumer from third party payors, such as government and private insurance plans. We and other pharmaceutical companies are affected by the efforts of governments and third party payors to contain or reduce the cost of health care through various means, and third-party payors are increasingly challenging the prices charged for medical goods and services. A number of legislative and regulatory proposals aimed at changing the health care system have been proposed in recent years including the Medicare Prescription Drug Improvement and Modernization Act of 2003. In addition, increasing emphasis on managed care in the U.S. has and will likely continue to increase pressures on drug prices. While we cannot predict whether legislative or regulatory proposals will be adopted or the effect such proposals or managed care efforts may have on our business, the announcement or adoption of such proposals or efforts could have a material and adverse effect on us. In many foreign markets, including the countries of the EU, pricing of pharmaceutical products is subject to government control.
Employees
As of March 1, 2007 we employed 24 persons, including 14 engaged in research and development activities, including preclinical research, clinical development, and regulatory affairs, and 10 in selling, general and administrative functions such as marketing, accounting, legal, purchasing and investor relations. Our staff includes 5 employees with Ph.D. or M.D. degrees. None of our employees are unionized and we believe that our relationship with our employees is good.
Available Information
Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and all amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 are available free of charge on our corporate website (www.adventrx.com) as soon as reasonably practicable after they are filed with, or furnished to, the Securities and Exchange Commission.
22
Table of Contents
Item 1A. Risk Factors
An investment in our securities involves a high degree of risk. You should carefully consider the following risk factors together with all other information contained or incorporated by reference in this report before you decide to invest in our common stock. The risks and uncertainties described below are not the only ones facing us. Additional risks and uncertainties that management is not aware of or focused on or that management currently deems immaterial may also impair our business operations. This report is qualified in its entirety by these risk factors. If any of these risks or uncertainties actually occur, our business, financial condition and results of operations could be materially and adversely affected. If this were to happen, the value of our securities could decline significantly, and you could lose all or part of your investment.
Risks Related to Our Company
We have never generated revenues or profits from operations and we may not be able to generate revenues sufficient to achieve profitability.
We are a development stage company and have not generated revenues from operations or been profitable since inception, and it is possible we will never achieve profitability. We have devoted our resources to developing a new generation of therapeutic products, but such products cannot be marketed until clinical testing is completed and governmental approvals have been obtained. Accordingly, there is no current source of revenues from operations, much less profits, to sustain our present activities, and no revenues from operations will likely be available until, and unless, our product candidates are clinically tested, approved by the FDA or other regulatory agencies and successfully marketed, either by us or a partner, an outcome which we are not able to guarantee.
We have limited capital resources and will need to raise additional funds to support our operations.
We have experienced significant operating losses in funding our research, development and clinical testing of product candidates, accumulating operating losses totaling over $89 million as of December 31, 2006, and we expect to continue to incur substantial operating losses for the foreseeable future. As of December 31, 2006, we had approximately $51.7 million in cash and cash equivalents and short-term investments in securities and we do not expect to generate positive net cash flows for the foreseeable future.
We will need to raise significant amounts of additional capital to finance our ongoing operations. We cannot be certain we will be able to obtain such financing on satisfactory terms, if at all, or that it will be sufficient to meet our cash requirements. If adequate funds are not available, we may be required to delay or reduce the scope of our research and development programs or attempt to continue development by entering into arrangements with partners or others that, if available at all, may not be on favorable terms and may require us to relinquish some or all of our rights to our product candidates or the financial benefits thereof.
Based on our current loss rate and existing capital resources as of the date of the filing of this report, we estimate that we have sufficient funds to sustain our operations at their current levels for at least the next twelve months; however, we cannot provide any assurance that we will not require additional funds earlier. Because we do not know whether our clinical research and development programs will progress at the rates expected, it is difficult to estimate our projected capital needs beyond our current spending levels.
We will seek to raise additional capital and may do so at any time and may do so through various financing alternatives, including selling shares of our common or preferred stock and rights to acquire our common or preferred stock, licensing or selling our technologies and product candidates, or through the issuance of one or more forms of senior or subordinated debt. Each of these financing alternatives carries certain risks. Raising capital through the issuance of common stock may depress the market price of our stock and may substantially dilute our existing stockholders. If we instead seek to raise capital through licensing transactions or sales of one or more of our technologies or product candidates, then we will likely need to share a significant portion of future revenues from these product candidates with our licensees. Additionally, the development of any product candidates licensed or sold to third parties will no longer be in our control and thus we may not realize the full value of any such relationships. Debt financing would likely involve covenants that restrict our operations. These restrictive covenants may include limitations on additional borrowing and specific restrictions on the use of our assets, as well as
23
Table of Contents
prohibitions on our ability to create liens or make investments and may, among other things, preclude us from making distributions to stockholders (either by paying dividends or redeeming stock) and taking other actions beneficial to our stockholders.
Our ability to timely raise capital may be impaired if we became ineligible to file shelf registration statements on Form S-3. We will become ineligible if we fail to comply with all applicable requirements of Form S-3, including filing in a timely manner all reports required to be filed by us. Though we are a small company with limited resources, we are subject to the wide-ranging laws and regulations applicable to public companies, including the provisions of the Sarbanes-Oxley Act of 2002, which may impair our ability to timely and completely comply with the requirements of Form S-3.
If we are unable to raise additional capital to fund future operations, then we may be required to reduce operations or defer or abandon one or more of our clinical or pre-clinical research programs.
Further testing of our product candidates will be required and there is no assurance of FDA approval.
Human pharmaceutical products are subject to rigorous preclinical testing and clinical trials and other approval procedures mandated by the FDA and foreign regulatory authorities. Various federal and foreign statutes and regulations also govern or influence the manufacturing, safety, labeling, storage, record keeping and marketing of pharmaceutical products. The process of obtaining these approvals and the subsequent compliance with appropriate U.S. and foreign statutes and regulations are time-consuming and require the expenditure of substantial resources. In addition, these requirements and processes vary widely from country to country.
The effect of government regulation and the need for FDA approval will delay commercialization of our product candidates for a considerable period of time, impose costly procedures upon our activities, and provide an advantage to larger companies that compete with us. There can be no assurance that the FDA or other regulatory approval for any products developed by us will be granted on a timely basis, or at all. Even though we have an agreement under the SPA process for our Phase III clinical trial of CoFactor in the treatment of metastatic colorectal cancer, the FDA may still require additional studies or data before granting marketing approval for CoFactor, if such approval is ever granted. Any delay in obtaining, or failure to obtain, approvals would materially and adversely affect the marketing of any contemplated products and the ability to earn product revenue. Further, regulation of manufacturing facilities by state, local, and other authorities is subject to change. Any additional regulation could result in limitations or restrictions on our ability to utilize any of our technologies, thereby adversely affecting our operations.
Our product candidates may cause undesirable side effects or have other properties that could delay or prevent their regulatory approval or commercialization.
Undesirable side effects caused by our product candidates could interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or other regulatory authorities for any or all targeted indications, and in turn prevent us from commercializing our product candidates and generating revenues from their sale.
In addition, if any of our product candidates receive marketing approval and we or others later identify undesirable side effects caused by the product:
| • | regulatory authorities may require the addition of labeling statements, such as a “black box” warning or a contraindication; | ||
| • | regulatory authorities may withdraw their approval of the product; | ||
| • | we may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product; and | ||
| • | our reputation may suffer. |
24
Table of Contents
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the product candidate, which in turn could delay or prevent us from generating significant revenues from its sale.
Even if our product candidates receive regulatory approval, they may still face future development and regulatory difficulties.
Even if U.S. regulatory approval is obtained, the FDA may still impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies. Our product candidates will also be subject to ongoing FDA requirements related to the labeling, packaging, storage, advertising, promotion, record-keeping and submission of safety and other post-market information on the product. In addition, approved products, manufacturers and manufacturers’ facilities are subject to continual review and periodic inspections. If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, a regulatory agency may impose restrictions on that product or us, including requiring withdrawal of the product from the market. If our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
| • | issue warning letters or untitled letters; | ||
| • | impose civil or criminal penalties; | ||
| • | suspend regulatory approval; | ||
| • | suspend any ongoing clinical trials; | ||
| • | refuse to approve pending applications or supplements to approved applications filed by us; | ||
| • | impose restrictions on operations, including costly new manufacturing requirements; or | ||
| • | seize or detain products or require a product recall. |
Even if our product candidates receive regulatory approval in the United States, we may never receive approval or commercialize our products outside of the United States.
In order to market any products outside of the United States of America, or the U.S., we must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks detailed above regarding FDA approval in the U.S. as well as other risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval could have the same adverse effects detailed above regarding FDA approval in the U.S. As described above, such effects include the risks that our product candidates may not be approved for all indications requested, which could limit the uses of our product candidates and have an adverse effect on potential royalties and product sales, and that such approval may be subject to limitations on the indicated uses for which the product may be marketed or require costly, post-marketing follow-up studies.
Positive results in our preclinical testing and clinical trials do not ensure that future clinical trials will be successful or that our product candidates will receive the regulatory approvals necessary for their commercialization.
Before obtaining regulatory approvals for the commercial sale of any of our product candidates, we must demonstrate through preclinical testing and clinical trials that each product is safe and effective for use in each target indication. Success in preclinical testing and clinical trials does not ensure that large-scale clinical trials will be
25
Table of Contents
successful. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals. The length of time necessary to complete clinical trials and to submit an application for marketing approval for a final decision by a regulatory authority varies significantly and may be difficult to predict. In addition, delays or rejections may be encountered based upon changes in FDA policy for drug approval during the period of product development and FDA regulatory review of each submitted new drug application, or NDA. There is a significant risk that any of our product candidates could fail to show satisfactory results in continued trials, and would not justify further development. A failure to obtain requisite regulatory approvals or to obtain approvals of the scope requested will delay or preclude us from marketing our products or limit the commercial use of the products, and would have a material adverse effect on our business, financial condition and results of operations.
We expect intense competition in the marketplace for CoFactor and in the target markets for our other product candidates.
The industry in which we operate is highly competitive and rapidly changing. If successfully developed and approved, all of our product candidates will likely compete with existing and new products and therapies and our competitors may succeed in commercializing products more rapidly or effectively than us, which would have a material and adverse effect on our results of operations and financial condition. ANX-510, or CoFactor, our leading product candidate, would likely compete against a well-established generic product, leucovorin, as well as isovorin, which is marketed primarily in Japan. In addition, there are numerous companies with a focus in oncology and/or anti-viral therapeutics that are pursuing the development of pharmaceuticals that target the same diseases as are targeted by the products being developed by us. We anticipate that we will face intense and increasing competition in the future as new products enter the market and advanced technologies become available. There is no assurance that existing products or new products developed by competitors will not be more effective, or more effectively marketed and sold, than those we may market and sell. Competitive products may render our products and product candidates obsolete or noncompetitive.
Companies likely to have products that will compete with CoFactor, such as Wyeth and Roche, and our other product candidates have significantly greater financial, technical and human resources and are better equipped to develop, manufacture, market and distribute products. Many of these companies have extensive experience in preclinical testing and clinical trials, obtaining FDA and other regulatory approvals and manufacturing and marketing products and have products that have been approved or are in late-stage development and operate large, well-funded research and development programs. Other companies, such as Merck Eprova, which manufactures folates, may be developing products which compete with CoFactor.
Smaller companies may also prove to be significant competitors, particularly through collaborative arrangements with large pharmaceutical and biotechnology companies. Furthermore, academic institutions, government agencies and other public and private research organizations are becoming increasingly aware of the commercial value of their inventions and are actively seeking to commercialize the technology they have developed.
If any of our product candidates for which we receive regulatory approval do not achieve broad market acceptance, the revenues we generate from their sales will be limited.
Our success will depend in substantial part on the extent to which our products for which we obtain marketing approval from the FDA and comparable foreign regulatory authorities are accepted by the medical community and reimbursement of them by third-party payors, including government payors. The degree of market acceptance will depend upon a number of factors, including, among other things:
| • | limitations or warnings in a product’s approved labeling; | ||
| • | the establishment and demonstration in the medical community of the safety and efficacy of our products and our ability to provide acceptable evidence of safety and efficacy; | ||
| • | availability of alternative treatments; | ||
| • | the product’s perceived advantages over existing treatment methods (including relative convenience and ease of administration and prevalence and severity of any adverse side effects); |
26
Table of Contents
| • | pricing and cost-effectiveness; | ||
| • | reimbursement and coverage policies of government and third-party payors; and | ||
| • | the prevalence of off-label substitution of chemically equivalent products. |
We cannot predict or guarantee that physicians, patients, healthcare insurers or maintenance organizations, or the medical community in general, will accept or utilize any of our products. If our products are approved but do not achieve an adequate level of acceptance by these parties, we may not generate sufficient revenue from these products to become or remain profitable. In addition, our efforts to educate the medical community and third-party payors regarding the benefits of our products may require significant resources and may never be successful.
We are subject to uncertainty relating to healthcare reform measures and reimbursement policies that, if not favorable to our products, could hinder or prevent our products’ commercial success.
Our ability to commercialize our products successfully will depend in part on the extent to which reimbursement for the costs of such products and related treatments will be available from government health administration authorities, private health insurers and other third-party payors. Significant uncertainty exists as to the reimbursement status of newly approved medical products. The continuing efforts of the government, insurance companies, managed care organizations and other payors of healthcare services to contain or reduce costs of healthcare may adversely effect:
| • | our ability to set a price we believe is fair for our products; | ||
| • | our ability to generate revenues or achieve or maintain profitability; | ||
| • | the future revenues and profitability of our potential customers, suppliers and collaborators; and | ||
| • | the availability to us of capital. |
If we are successful in getting FDA approval for CoFactor, we will compete with leucovorin, a generic drug, which has a lower cost and a long, established history of reimbursement. Our ability to commercialize CoFactor will depend in part on the extent to which governmental authorities, private health insurers and other organizations establish appropriate coverage and reimbursement levels for the cost of our products and related treatments. These payors are increasingly attempting to contain healthcare costs by limiting both coverage and the level of reimbursement, particularly for new therapeutic products. Accordingly, even if coverage and reimbursement are provided, market acceptance of our products would be adversely affected if the amount of coverage and/or reimbursement available for the use of our products proved to be unprofitable for healthcare providers.
There have been federal and state proposals to subject the pricing of healthcare goods and services, including prescription drugs, to government control and to make other changes to the U.S. healthcare system. For example, the Medicare Prescription Drug Improvement Act of 2003 provides a new Medicare prescription drug benefit, which became effective January 1, 2006, and mandates other reforms. While we cannot predict the full outcome of the implementation of this legislation, it is possible that the new Medicare prescription drug benefit, which will be managed by private health insurers and other managed care organizations, will result in additional government reimbursement for prescription drugs, which may make some prescription drugs more affordable but may further exacerbate industry-wide pressure to reduce prescription drug prices. In addition, in certain foreign markets, the pricing of prescription drugs is subject to government control and reimbursement may in some cases be unavailable or insufficient. It is uncertain if future legislative proposals, whether domestic or abroad, will be adopted that might affect the product candidates in our programs or what actions federal, state, or private payors for healthcare treatment and services may take in response to any such healthcare reform proposals or legislation. Any such healthcare reforms could have a material adverse effect on the marketability of any products for which we ultimately require or receive FDA approval.
27
Table of Contents
We may not achieve our projected development goals in the time frames we announce. Delays in the commencement or completion of preclinical testing or clinical trials could result in increased costs to us and delay or limit our ability to generate revenues.
We set goals for and make public statements regarding our estimates of the timing of the accomplishment of objectives material to our success. The actual timing of these events can vary dramatically due to any number of factors, including delays or failures in our preclinical testing and clinical trials and the uncertainties inherent in the regulatory approval process.
We have an active preclinical program that we use to assess the merits of potential product candidates and future research and development activities. Delays in our preclinical program could occur for a number of reasons, including:
| • | delays in reaching agreement on acceptable terms with prospective clinical research organizations, or CROs, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs; | ||
| • | failures on the part of our CROs in developing study procedures or otherwise conducting the studies on timeframes requested by us; | ||
| • | changes in regulatory requirements or other standards or guidance relating to preclinical testing, including testing of pharmaceutical products in animals; | ||
| • | a lack of availability of animals that are suitable for the types of studies we plan to conduct; and | ||
| • | unforeseen results of preclinical testing that require us to amend study designs or delay future preclinical testing, clinical trials and related regulatory filings. |
In addition, we do not know whether planned clinical trials or timelines for enrollment in our Phase III clinical trial of CoFactor for the treatment of metastatic colorectal cancer will commence on time or be completed on schedule, if at all. The commencement and completion of clinical trials can be delayed for a variety of reasons, including delays related to:
| • | obtaining regulatory approval to commence a clinical trial; | ||
| • | identifying appropriate trial sites and reaching agreement on acceptable terms with prospective contract research organizations, or CROs, trial sites and clinical investigators, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs, trial sites and clinical investigators; | ||
| • | manufacturing sufficient quantities of a product candidate; | ||
| • | obtaining institutional review board approval to conduct a clinical trial at a prospective site; | ||
| • | recruiting and enrolling patients to participate in clinical trials for a variety of reasons, including competition from other clinical trial programs for the same indication as our product candidates; and | ||
| • | retaining patients who have initiated a clinical trial but may be prone to withdraw due to side effects from the therapy, lack of efficacy or personal issues, or who are lost to further follow-up. |
For example, we are seeking to enroll 1,200 patients in our Phase III clinical trial of CoFactor for the treatment of metastatic colorectal cancer. Currently, because bevacizumab, a component of the study, is typically not reimbursed outside the U.S., we are seeking to enroll patients only in the U.S. and, accordingly, the potential pool of patients for our study is correspondingly limited. If we chose to enroll patients outside the U.S., we will most likely be required to, among other things, reimburse the cost of bevacizumab and engage CROs to assist with these overseas trials, all of which will add substantial cost to the study.
28
Table of Contents
In addition, a clinical trial may be suspended or terminated by us, the FDA or other regulatory authorities due to a number of factors, including:
| • | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; | ||
| • | inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; | ||
| • | unforeseen safety issues; or | ||
| • | lack of adequate funding to continue the clinical trial. |
Additionally, changes in regulatory requirements and guidance relating to clinical trials may occur and we may need to amend clinical trial protocols to reflect these changes. Amendments may require us to resubmit our clinical trial protocols to institutional review boards for reexamination or renegotiate terms with CROs, trial sites and clinical investigators, all of which may impact the costs, timing or successful completion of a clinical trial.
There can be no assurance that our preclinical testing and clinical trials will commence or be completed, that we will make regulatory submissions or receive regulatory approvals as planned or that we will be able to adhere to our current schedule for the launch of any of our products. If we experience delays in completion of, or if we terminate, our clinical trials or preclinical testing, the commercial prospects for our product candidates will be harmed, and our ability to generate product revenues will be delayed. In addition, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials or preclinical testing may also ultimately lead to the denial of regulatory approval of a product candidate. Even if we are able to ultimately commercialize our product candidates, other therapies for the same indications may have been introduced to the market and established a competitive advantage.
We rely in part on third parties to conduct our clinical trials and other aspects of our research and development programs.
We do not possess research and development facilities necessary to conduct all of the activities associated with our research and development programs. We engage consultants, advisors and CROs to design and conduct clinical trials in connection with the development of our product candidates. As a result, these important aspects of our product candidates’ development are outside our direct control. In addition, there can be no assurance that such third parties will perform all of their obligations under arrangements with us or will perform those obligations satisfactorily.
The CROs with which we contract for execution of our clinical trials play a significant role in the conduct of the trials and subsequent collection and analysis of data, and we will likely depend on other CROs and clinical investigators to conduct our future clinical trials or assist with our on-going clinical trials. For instance, for our CoFactor phase III clinical trial, we rely on Synteract, Inc., for data management, biostatistics and pharmacovigilance, and Pharmatech, Inc., for site management and enrollment support, both of which are CROs. Individuals working at these companies, as well as clinical investigators at the sites at which our clinical trials are conducted, are not our employees, and we cannot control the amount or timing of resources that they devote to our programs. If these CROs fail to devote sufficient time and resources to our clinical trials, or if their performance is substandard, it will delay the approval of our applications to regulatory agencies and our introduction of our products. Failure of these CROs to meet their obligations could adversely affect clinical development of our product candidates. Moreover, these CROs may have relationships with other commercial entities, some of which may compete with us. If they assist our competitors at our expense, it could harm our competitive position.
We do not have manufacturing capabilities and may not be able to effectively develop manufacturing capabilities or contract for such services from third parties on commercially acceptable terms, or at all.
We do not have any manufacturing capability. We meet our manufacturing requirements by establishing relationships with third-party manufacturers for the manufacture of clinical trial material and we anticipate establishing relationships with third-party manufacturers for the commercial production of our products, though we
29
Table of Contents
do not have any long-term agreements or commitments for the supply of these materials or products. We cannot ensure that we will be able to establish relationships with third-party manufacturers on commercially acceptable terms, or at all. Any failure to establish relationships with third parties for our manufacturing requirements on commercially acceptable terms would have a material and adverse effect on us. Even if we successfully establish relationships with third-party manufacturers on commercially acceptable terms, our manufacturers may not perform as agreed or may terminate their agreements with us.
In addition, all manufacturers of our products and product candidates must comply with current good manufacturing practice, or cGMP, requirements enforced by the FDA through its facilities inspection program, as well as applicable requirements of foreign regulatory authorities. These requirements include quality control, quality assurance and the maintenance of records and documentation. Manufacturers of our products and product candidates may be unable to comply with these cGMP requirements and with other FDA, state and foreign regulatory requirements. We have little control over our manufacturers’ compliance with these regulations and standards. A failure to comply with these requirements may result in fines and civil penalties, suspension of production, suspension or delay in product approval, product seizure or recall, or withdrawal of product approval.
Furthermore, the manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls. Manufacturers of pharmaceutical products often encounter difficulties in production, particularly in scaling up initial production. These problems include difficulties with production costs and yields, quality control, including stability of the product candidate and quality assurance testing and shortages of qualified personnel. If our manufacturers were to encounter any of these difficulties or otherwise fail to comply with their contractual obligations, our ability to provide product candidates to patients in our clinical trials may be jeopardized.
Any delay or interruption in the supply of clinical supplies could delay the completion of our clinical trials, increase the costs associated with maintaining our research and development programs and, depending upon the period of delay, require us to commence new trials at significant additional expense or terminate the trials completely. We cannot ensure that manufacturing or quality control problems will not arise in connection with the manufacture of our products or product candidates, or that third-party manufacturers will be able to maintain the necessary governmental licenses and approvals to continue manufacturing such products or product candidates. Any of the above factors could cause us to delay or suspend clinical trials, regulatory submissions, required approvals or commercialization of our product candidates, entail higher costs or result in our being unable to effectively commercialize our products. Our dependence upon third parties for the manufacture of our products and product candidates may adversely affect our future costs and our ability to develop and commercialize our products and product candidates on a timely and competitive basis.
Our success will depend on patents and other protection we and our licensors obtain on our product candidates and proprietary technology.
Our success will depend in part on our ability and, in certain cases, our licensors’ ability to:
| • | obtain and maintain patent protection with respect to our products; | ||
| • | maintain our licenses; | ||
| • | prevent third parties from infringing upon our proprietary rights; | ||
| • | maintain trade secrets; | ||
| • | operate without infringing upon the patents and proprietary rights of others; and | ||
| • | obtain appropriate licenses to patents or proprietary rights held by third parties if infringement would otherwise occur, both in the U.S. and in foreign countries. |
The patent and intellectual property positions of biopharmaceutical companies, including ours, are uncertain and involve complex legal and factual questions. There is no guarantee that we or our licensors have or will develop or
30
Table of Contents
obtain the rights to products or processes that are patentable, that patents will issue from any pending applications or that claims allowed will be sufficient to protect the technology we develop or have developed or that is licensed to us. In addition, we cannot be certain that patents issued or licensed to us will not be challenged, invalidated, infringed or circumvented, including by our competitors, or that the rights granted thereunder will provide competitive advantages to us. Furthermore, patent applications in the U.S. are confidential for a period of time until they are published, and publication of discoveries in scientific or patent literature typically lags actual discoveries by several months. As a result, we cannot be certain that the inventors of any patent or patent application owned or licensed to us were the first to conceive of the inventions covered by such patents and patent applications or that such inventors were the first to file patent applications for such inventions.
We may also rely on unpatented trade secrets and know-how and continuing technological innovation to develop and maintain our competitive position, which we seek to protect, in part, by confidentiality agreements with employees, consultants, collaborators and others. We also have invention or patent assignment agreements with our employees and certain consultants. There can be no assurance, however, that binding agreements will not be breached, that we will have adequate remedies for any breach, or that trade secrets will not otherwise become known or be independently discovered by competitors. In addition, there can be no assurance that inventions relevant to us will not be developed by a person not bound by an invention assignment agreement with us.
Our market opportunity for CoFactor may be limited by the lack of composition-of-matter patents in territories outside the United States and Canada.
We do not hold composition-of-matter patents covering the active pharmaceutical ingredients of CoFactor outside the United States and Canada. Composition-of-matter patents are widely viewed as the strongest form of intellectual property protection for pharmaceutical products as they apply without regard to any method of use or other type of limitation. As a result, competitors who obtain the requisite regulatory approval can offer products with the same active ingredients as our products so long as the competitors do not infringe any method of use or formulation patents that we may hold.
The principal patent protection that covers, or that we expect will cover, CoFactor outside the United States and Canada is method-of-use patents. This type of patent protects the product only when used or sold for the specified method. This type of patent does not limit a competitor from making and marketing a product that is identical to our product for an indication that is outside of the patented method. Moreover, physicians may prescribe such a competitive identical product for off-label indications that are covered by the applicable patents. Although such off-label prescriptions may infringe or contribute to the infringement of method of use patents, the practice is common and such infringement is difficult to prevent or prosecute.
We have licensed several of our product candidates from third parties and, if we default on any of our obligations, we could lose rights to our product candidates.
We have licensed rights to our product candidates that are important to our business, and we expect to enter into similar licenses in the future. For instance, the license agreement pursuant to which we license our lead product candidate, CoFactor, which is also the agreement pursuant to which we license ANX-540, or Selone, and the license agreement pursuant to which we license ANX-201, or Thiovir™, permit the licensor, the University of Southern California, or USC, to terminate the agreement under certain circumstances, such as our failure to use our reasonable (for CoFactor and the compounds in our organoselenones program) or diligent (for ANX-201) efforts to commercialize the licensed technology or the occurrence of any other uncured breach by us. In addition, in January 2006, we further amended the license agreement pursuant to which we license ANX-201 such that, among other things, we became subject to certain development milestone obligations that, if not achieved, provide USC a 30-day right to terminate the underlying license. These license agreements also provide that the licensor is primarily responsible for obtaining patent protection for the technology licensed, and we are required to reimburse the licensor for the costs it incurs in performing these activities. These license agreements also require the payment of specified royalties. Any inability or failure to observe these terms or pay these costs or royalties could result in the termination of the applicable license agreement in certain cases. The termination of any license agreement could have a material and adverse effect on us.
31
Table of Contents
The United States government and the University of Southern California retain certain rights in the technologies we have licensed from the University of Southern California.
The technologies developed by the University of Southern California were developed in part through funding provided by the U.S. government. Therefore, in addition to the University of Southern California’s termination rights described above, our licenses are subject to a non-exclusive, non-transferable, royalty-free right of the U.S. government and the University of Southern California to practice the licensed technologies for research purposes and, in the case of the U.S. government, other governmental purposes on behalf of the U.S. and on behalf of any foreign government or international organization pursuant to any existing or future treaty or agreement with the U.S., but only to the extent that the government funded the research. The government also reserves the right to require us to grant sublicenses to third parties when necessary to fulfill public health and safety needs or if we do not reasonably satisfy government requirements for public use of the technology. In addition, the University of Southern California has the right to use all improvements to the licensed technology for research and educational purposes. Although we are currently the only parties licensed to actively develop the technology, we cannot assure you that the government will not in the future require us to sublicense the technology. Any action by the government to force us to issue such sublicenses or development activities pursuant to its reserved rights in the technology would erode our ability to exclusively develop our products and product candidates based on the technology and could materially harm our financial condition and operating results.
Licenses of technology developed through funding provided by the U.S. government, including the University of Southern California licenses, require that licensees — in this case, us — and our affiliates and sub-licensees agree that products covered by the licenses will be manufactured substantially in the U.S. We cannot assure you that we will be able to contract for manufacturing facilities in the U.S. on favorable terms or obtain waivers of such requirement, or that such requirement will not impede our ability to license our products or product candidates to others. If we are unable to contract for manufacturing facilities in the U.S. or obtain an appropriate waiver, we risk losing our rights under the University of Southern California licenses, which could materially harm our financial condition and operating results.
If we are sued for infringing the proprietary rights of third parties, it will be costly and time consuming, and an unfavorable outcome would have an adverse effect on our business.
Our commercial success depends on our ability and the ability of our future collaborators to develop, manufacture, market and sell our products and product candidates and use our proprietary technologies without infringing the proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties, exist in the fields in which we and our future collaborators are or may be developing products. As the biotechnology and pharmaceutical industry expands and more patents are issued, the risk increases that our products and product candidates may give rise to claims that our products or product candidates infringe the rights of others. Because patent applications can take many years to publish and issue, there may be currently pending applications, unknown to us, that may later result in issued patents that our products, product candidates or technologies infringe.
We may be exposed to, or threatened with, future litigation by third parties having patent or other intellectual property rights alleging that our products, product candidates and/or technologies infringe their intellectual property rights. If one of these patents was found to cover our products, product candidates, technologies or their uses, we or our future collaborators could be required to pay damages and could be unable to commercialize our products or use our technologies or methods unless we or they are able to obtain a license to the patent or intellectual property right. A license may not be available to us or our future collaborators on acceptable terms, if at all. In addition, during litigation, a patent holder could obtain a preliminary injunction or other equitable remedy that could prohibit us from making, using or selling our products, technologies or methods.
There is a substantial amount of litigation involving patent and other intellectual property rights in the biotechnology and pharmaceutical industries generally. If a third party claims that we or our collaborators infringe its intellectual property rights, we may face a number of issues, including, but not limited to:
| • | infringement and other intellectual property claims which, with or without merit, may be expensive and time consuming to litigate and may divert our management’s attention from our core business; |
32
Table of Contents
| • | substantial damages for infringement, including treble damages and attorneys’ fees, which we may have to pay if a court decides that the product at issue infringes on or violates the third party’s rights; | ||
| • | a court prohibiting us from selling or licensing the product unless the third party licenses its product rights to us, which it is not required to do; | ||
| • | if a license is available from the third party, we may have to pay substantial royalties, fees and/or grant cross-licenses to our products; and | ||
| • | redesigning our products or processes so they do not infringe, which may not be possible or may require substantial funds and time. |
No assurance can be given that patents do not exist, have not been filed, or could not be filed or issued, which contain claims covering our products, technology or methods. Because of the number of patents issued and patent applications filed in our field, we believe there is a risk that third parties may allege they have patent rights encompassing our products, technology or methods.
In addition, it may be necessary for us to enforce patents under which we have rights, or to determine the scope, validity and unenforceability of other parties’ proprietary rights, which may affect our rights. There can be no assurance that our owned or licensed patents would be held valid by a court or administrative body or that an alleged infringer would be found to be infringing. The uncertainty resulting from the mere institution and continuation of any technology-related litigation or interference proceeding could have a material and adverse effect on us.
We currently have no sales capability, and limited marketing capability.
We currently do not have sales personnel. We have limited marketing and business development personnel. To commercialize our products, we will have to acquire or develop sales, marketing and distribution capabilities, or rely on marketing partners or other arrangements with third parties for the marketing, distribution and sale of products. There is no guarantee that we will be able to establish marketing, distribution or sales capabilities or make arrangements with third parties to perform those activities on terms satisfactory to us, or that any internal capabilities or third party arrangements will be cost-effective. The acquisition or development of a sales and distribution infrastructure will require substantial resources, which may divert the attention of our management and key personnel and negatively impact our product development efforts.
In addition, any third parties with which we establish marketing, distribution or sales arrangements may have significant control over important aspects of the commercialization of our products, including market identification, marketing methods, pricing, composition of sales force and promotional activities. There can be no assurance that we will be able to control the amount and timing of resources that any third party may devote to our products or prevent any third party from pursuing alternative technologies or products that could result in the development of products that compete with, or the withdrawal of support for, our products.
We will need to increase the size of our organization, and we may experience difficulties in managing growth.
As of March 1, 2007, we had 24 full-time employees. We will need to continue to expand our managerial, operational, financial and other resources in order to manage our operations and clinical trials, continue our research and development programs and commercialize our product candidates. Our management and personnel, systems and facilities currently in place may not be adequate to support this growth. Our need to effectively manage our operations, growth and various projects requires that we:
| • | manage our clinical trials effectively, including our Phase III clinical trial for CoFactor, which is being conducted at numerous distinct clinical trial sites; | ||
| • | manage our internal development efforts effectively while carrying out our contractual obligations to collaborators and other third parties; |
33
Table of Contents
| • | continue to improve our operational, financial and management controls, reporting systems and procedures; and; | ||
| • | attract and retain sufficient numbers of talented employees. |
We may be unable to successfully implement these tasks on a larger scale and, accordingly, may not achieve our development and commercialization goals.
We have engaged in and may continue to engage in opportunistic acquisitions of companies and intellectual property, which could negatively affect our business and operations.
In April 2006, we acquired SD Pharmaceuticals, Inc., including its portfolio of product candidates. We intend to continue to be opportunistic in acquiring products, businesses or technologies that we believe are a strategic fit with our business or complement our existing product candidates. There are risks associated with such activities. These risks include, among others, incorrectly assessing the asset quality of a prospective merger partner, encountering greater than anticipated costs in integrating acquired businesses, facing resistance from customers or employees, and being unable to profitably deploy assets acquired in the transaction. Additional country- and region-specific risks are associated with transactions outside the U.S. To the extent we issue securities in connection with additional transactions, these transactions and related issuances may have a dilutive effect on earnings per share and our ownership.
Our operations, financial condition, and prospects after a merger or acquisition depend in part on our ability to successfully integrate the operations of the acquired products, business or technologies. We may be unable to integrate operations successfully or to achieve expected cost savings. Any cost savings which are realized may be offset by losses in revenues or other charges to operations.
We may be unable to retain skilled personnel and maintain key relationships.
The success of our business depends, in part, on our ability to attract and retain highly qualified management, scientific and other personnel, and on our ability to develop and maintain important relationships with leading research institutions and consultants and advisors. Competition for these types of personnel and relationships is intense from numerous pharmaceutical and biotechnology companies, universities and other research institutions, particularly in the San Diego, California area. We are currently dependent upon our scientific staff, which has a deep background in our product candidates and our research and development programs. Recruiting and retaining senior employees with relevant product development experience in cancer and infections diseases is costly and time-consuming. There can be no assurance that we will be able to attract and retain such individuals on an uninterrupted basis and on commercially acceptable terms, and the failure to do so could have a material and adverse effect on us by significantly delaying one or more of our research and development programs. The loss of any of our executive officers, including our chief executive officer, president/chief medical officer, chief scientific officer or our vice president, medical affairs, in particular, could have a material and adverse effect on us and the market for our common stock, particularly if such loss was abrupt or unexpected. None of our employees is obligated to provide services to us for any particular period of time. We do not have non-competition agreements with any of our employees. Furthermore, even if we successfully attract and retain qualified personnel, we may not select individuals with the appropriate skills for the jobs for which they are hired or that integrate well with our existing personnel. Underperforming employees and internal friction may divert the attention of our management and key personnel and negatively impact our product development efforts. In addition, we may incur costs and liabilities terminating our employment relationship with unsatisfactory employees.
We face potential product liability exposure and, if successful claims are brought against us, we may incur substantial liability for a product or product candidate and may have to limit its commercialization. In the future, we anticipate that we will need to obtain additional or increased product liability insurance coverage and it is uncertain that such increased or additional insurance coverage can be obtained on commercially reasonable terms.
Our business (in particular, the use of our product candidates in clinical trials and the sale of our products for which we obtain marketing approval) will expose us to product liability risks. Product liability claims might be
34
Table of Contents
brought against us by consumers, health care providers, pharmaceutical companies or others selling our products. If we cannot successfully defend ourselves against these claims, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| • | decreased demand for our products or product candidates; | ||
| • | impairment of our business reputation; | ||
| • | withdrawal of clinical trial participants; | ||
| • | costs of related litigation; | ||
| • | substantial monetary awards to patients or other claimants; | ||
| • | loss of revenues; and | ||
| • | the inability to commercialize our products and product candidates. |
We maintain $10 million in limited product liability insurance for our clinical trials, but our insurance coverage may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses.
We intend to expand our insurance coverage to include the sale of commercial products if we obtain marketing approval of any of our product candidates, but we may be unable to obtain product liability insurance on commercially acceptable terms or that we will be able to maintain such insurance at a reasonable cost or in sufficient amounts to protect us against potential losses. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us could cause our stock price to fall and, if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
If a trademark infringement action is commenced against us regarding the use of our corporate name, we could be required to pay monetary damages and/or change our name.
In March of 2005, we received correspondence from Aventis Pharmaceuticals, Inc. and its parent, Sanofi-Aventis, or collectively Sanofi, in which Sanofi asserted that our use of the word “ADVENTRX” infringes upon their trademark “AVENTIS” and demanded that we discontinue use of the word ADVENTRX. In May of 2005, we responded with a letter in which we outlined reasons why we believe that our name, ADVENTRX, does not infringe on Sanofi’s trademark, AVENTIS. Since our response, counsels for both parties have exchanged further communications and Sanofi has made further inquiries regarding our use of the “ADVENTRX” mark. In June 2006, we received a letter from counsel to Sanofi that, based on the fact that we do not own any registrations or applications for the ADVENTRX name and that Sanofi is not aware of any instances of actual confusion in the marketplace, Sanofi has decided not to take any further action. Sanofi indicated that, if we attempt to secure trademark/service mark registration protection for the ADVENTRX name or should instances of actual confusion come to Sanofi’s attention, it will reevaluate its position. Accordingly, Sanofi may take legal action in the future, including proceeding with an action for trademark infringement. Depending upon the circumstances, an adverse result in a trademark infringement action could require the payment of monetary damages by us and/or changing our corporate name.
Changes in laws and regulations that affect the governance of public companies have increased our operating expenses and may continue to do so.
Recently enacted changes in the laws and regulations affecting public companies, including the provisions of the Sarbanes-Oxley Act of 2002 and AMEX listing requirements, as well as disclosure requirements related to executive and director compensation, have imposed new duties on us and on our executives, directors, attorneys and independent accountants. In order to comply with these new rules, we have hired additional personnel (and may hire additional personnel) and engaged outside legal, accounting and advisory services, which have increased and are likely to continue increasing our operating expenses. In particular, we expect to incur additional administrative expenses as we continue to comply with the requirements of Section 404 of the Sarbanes-Oxley Act, which requires management to extensively evaluate and report on, and our independent registered public accounting firm to attest to, our internal controls. For example, we have incurred significant expenses, and expect to incur additional expenses, in connection with the evaluation, implementation, documentation and testing of our existing and newly
35
Table of Contents
implemented control systems. Management time associated with these compliance efforts necessarily reduces time available for other operating activities, which could adversely affect operating results. If we are unable to achieve full and timely compliance with these regulatory requirements, we could be required to incur additional costs and expend additional money and management time on additional remedial efforts, all of which could adversely affect our results of operations.
Risks Related to Our Common Stock
The market price of our common stock has been and is likely to continue to be highly volatile.
Market prices for our securities and the securities of other biotechnology and biopharmaceutical companies have historically been highly volatile, and the market has from time to time experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. The market price of our common stock may fluctuate significantly in response to a number of factors, many of which are beyond our control, including:
| • | changes in the regulatory status of CoFactor and our other product candidates, including results of our clinical trials and other research and development programs; | ||
| • | FDA or international regulatory actions and regulatory developments in the United States and foreign countries; | ||
| • | announcements of new products or technologies, commercial relationships or other events (including clinical trial results) by us or our competitors; | ||
| • | market conditions in the pharmaceutical, biopharmaceutical and biotechnology sectors; | ||
| • | developments concerning intellectual property rights generally or those of our or our competitors; | ||
| • | litigation or public concern about the safety of our products or product candidates; | ||
| • | changes in securities analysts’ estimates of our financial performance or deviations in our business and the trading price of our common stock from the estimates of securities analysts; | ||
| • | events affecting our existing in-license agreements and any future collaborations, commercial agreements and grants; | ||
| • | fluctuations in stock market prices and trading volumes of similar companies; | ||
| • | sales of large blocks of our common stock, including sales by our executive officers, directors and significant stockholders or pursuant to effective shelf registration statements that register shares of our common stock that may be sold by certain of our current stockholders; | ||
| • | discussion of us or our stock price by the financial and scientific press and in online investor communities; | ||
| • | additions or departures of key personnel; and | ||
| • | third party reimbursement policies. |
The realization of any of the foregoing could have a dramatic and adverse impact on the market price of our common stock. In addition, class action litigation has often been instituted against companies whose securities have experienced periods of volatility in market price. Any such litigation brought against us could result in substantial costs and a diversion of management’s attention and resources, which could hurt our business, operating results and financial condition.
36
Table of Contents
Sales of substantial amounts of our common stock or the perception that such sales may occur could cause the market price of our common stock to drop significantly, even if our business is performing well.
The market price of our common stock could decline as a result of sales by, or the perceived possibility of sales by, our existing stockholders of shares of our common stock. These sales might also make it more difficult for us to sell equity securities at a time and price that we deem appropriate. In addition, we have filed shelf registration statements to register shares of our common stock that may be sold by certain of our stockholders, which may increase the likelihood of sales by, or the perception of an increased likelihood of sales by, our existing stockholders of shares of our common stock.
Anti-takeover provisions in our charter documents and under Delaware law may make an acquisition of us, which may be beneficial to our stockholders, more difficult, which could depress our stock price. Alternatively, prohibitions on anti-takeover provisions in our charter documents may restrict us from acting in the best interests of our stockholders.
We are incorporated in Delaware. Certain anti-takeover provisions of Delaware law and our charter documents as currently in effect may make a change in control of our company more difficult, even if a change in control would be beneficial to our stockholders. Our bylaws limit who may call a special meeting of stockholders and establish advance notice requirements for nomination for election to our Board of Directors or for proposing matters that can be acted upon at stockholders’ meetings. Delaware law also prohibits corporations from engaging in a business combination with any holders of 15% or more of their capital stock until the holder has held the stock for three years unless, among other possibilities, the Board of Directors approves the transaction. Our Board of Directors may use these provisions to prevent changes in the management and control of our company. Also, under applicable Delaware law, our Board of Directors may adopt additional anti-takeover measures in the future. In addition, provisions of certain contracts, such as stock option agreements under our 2005 Equity Incentive Plan and employment agreements with our executive officers, may have an anti-takeover effect. In particular, we agreed with each of our president/chief medical officer and chief financial officer that, among other things, in the event of our acquisition, 50% of any unvested portion of an option we granted to them would vest upon such acquisition, with the remaining unvested portion vesting monthly over the 12 months following such acquisition. As a result, if an acquirer desired to retain the services of our president/chief medical officer or our chief financial officer following an acquisition, it may be required to further incentive each with additional options or other securities, which may deter or affect the terms of an acquisition or potential acquisition.
In connection with a July 2005 private placement, we agreed with the investors in that transaction that we would not implement certain additional measures that would have an anti-takeover effect. As a result, under our amended and restated certificate of incorporation, we are prohibited from dividing our Board of Directors into classes and adopting or approving any “rights plan,” “poison pill” or other similar plan or device. A classified board of directors could serve to protect our stockholders against unfair treatment in takeover situations, by making it more difficult and time-consuming for a potential acquirer to take control of our Board of Directors. A company may also adopt a classified board of directors to ensure stability in the board of directors and thereby improve long-term planning, which may benefit stockholders. A “poison pill” or similar plan or device may encourage potential acquirers to discuss their intentions with the board of directors of a company and avoid the time, expense and distraction of a hostile take-over. Any benefit to us and our stockholders from instituting a classified board or adopting or approving a “poison pill” or similar plan or device in these and other circumstances would be unavailable unless and until we amend our amended and restated certificate of incorporation.
Concentration of ownership of our common stock among our existing executive officers, directors and principal stockholders may prevent new investors from influencing significant corporate decisions.
Our executive officers and directors and the beneficial owners of 5% or more of our common stock and their affiliates, in aggregate, beneficially own approximately 22.2% of our outstanding common stock as of December 31, 2006. These persons, if acting together, will be able to exercise significant influence over all matters requiring stockholders’ approval, including the election and removal of directors and any merger, consolidation or sale of all or substantially all of our assets. In addition, these persons, acting together, may have the ability to control our management and affairs. Further, the interests of significant stockholders may be different than yours and they may support transactions that you feel are not in your best interest. This concentration of ownership may harm the market price of our common stock by delaying or preventing a change in control of our company at a premium price even if beneficial to our other stockholders.
37
Table of Contents
Because we do not expect to pay dividends in the foreseeable future, you must rely on stock appreciation for any return on your investment.
We have paid no cash dividends on any of our capital stock to date, and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. As a result, we do not expect to pay any cash dividends in the foreseeable future, and payment of cash dividends, if any, will also depend on our financial condition, results of operations, capital requirements and other factors and will be at the discretion of our Board of Directors. Furthermore, we are subject to various laws and regulations that may restrict our ability to pay dividends and we may in the future become subject to contractual restrictions on, or prohibitions against, the payment of dividends. Accordingly, the success of your investment in our capital stock will likely depend entirely upon any future appreciation and there is no guarantee that our capital stock will appreciate in value.
Item 1B. Unresolved Staff Comments
Not applicable.
Item 2. Properties
Our principal offices are located at 6725 Mesa Ridge Road, San Diego, California 92121. Our principal offices consist of 12,038 square feet of office and lab space, which we use pursuant to a lease which will expire on August 31, 2009. The base rent for this space is currently $249,475 annually, excluding incremental operating cost adjustments. We may lease additional space to accommodate our anticipated 2007 growth.
Item 3. Legal Proceedings
We are not currently a party to any material legal proceedings.
Item 4. Submission of Matters to a Vote of Security Holders
Not applicable.
38
Table of Contents
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information
Our common stock trades under the symbol “ANX” on the American Stock Exchange, or AMEX. The following table sets forth the high and low closing prices for our common stock in each of the quarters over the past two years, as reported by AMEX.
| Common Stock Price | ||||||||||||||||
| 2006 | 2005 | |||||||||||||||
| High | Low | High | Low | |||||||||||||
| First Quarter | $ | 5.00 | $ | 3.30 | $ | 1.69 | $ | 0.90 | ||||||||
| Second Quarter | $ | 5.28 | $ | 3.05 | $ | 3.12 | $ | 1.61 | ||||||||
| Third Quarter | $ | 3.65 | $ | 2.40 | $ | 4.13 | $ | 2.18 | ||||||||
| Fourth Quarter | $ | 3.37 | $ | 2.33 | $ | 3.65 | $ | 2.68 | ||||||||
As of March 1, 2007, we had approximately 240 holders of record of our common stock. We believe that the number of beneficial owners is substantially greater than the number of record holders because a large portion of our common stock is held of record through brokerage firms in “street name.”
Dividend Policy
We have never declared or paid any cash dividends on our capital stock and do not anticipate paying any cash dividends on our capital stock in the foreseeable future. We expect to retain all available funds and future earnings, if any, to support operations and fund the development and growth of our business. Our board of directors will determine future dividends, if any.
39
Table of Contents
Cumulative Total Return to Stockholders
The following graph compares the cumulative 5-year total return to stockholders on our common stock relative to the cumulative total returns of the Russell 2000 index, the AMEX Composite index, the AMEX Biotechnology index and the NASDAQ Biotechnology Index. The graph assumes that the value of the investment in our common stock, and in each index (including reinvestment of dividends) was $100 on December 31, 2001 and tracks it through December 31, 2006.
COMPARISON OF 5 YEAR CUMULATIVE TOTAL RETURN*
Among ADVENTRX Pharmaceuticals, Inc., The Russell 2000 Index,
The AMEX Composite Index, The Amex Biotechnology Index
And The NASDAQ Biotechnology Index
Among ADVENTRX Pharmaceuticals, Inc., The Russell 2000 Index,
The AMEX Composite Index, The Amex Biotechnology Index
And The NASDAQ Biotechnology Index
| * | $100 invested on December 31, 2001 in stock or index-including reinvestment of dividends. |
| Years ending December 31, | 2001 | 2002 | 2003 | 2004 | 2005 | 2006 | ||||||||||||||||||
| ADVENTRX Pharmaceuticals, Inc. | 100.00 | 22.17 | 48.89 | 63.67 | 186.47 | 167.71 | ||||||||||||||||||
| Russell 2000 | 100.00 | 79.52 | 117.09 | 138.55 | 144.86 | 171.47 | ||||||||||||||||||
| AMEX Composite | 100.00 | 100.08 | 144.57 | 178.46 | 220.35 | 262.17 | ||||||||||||||||||
| AMEX Biotechnology | 100.00 | 65.69 | 102.69 | 115.46 | 151.89 | 145.42 | ||||||||||||||||||
| NASDAQ Biotechnology | 100.00 | 62.08 | 90.27 | 99.08 | 111.81 | 110.06 | ||||||||||||||||||
| The stock price performance included in this graph is not necessarily indicative of future stock price performance. |
40
Table of Contents
For the comparison of 5-year cumulative total return, we added the Russell 2000 index, which we joined in 2006, and we will be dropping the NASDAQ Biotechnology index that we included in prior reports. We believe that the Russell 2000 is a more appropriate benchmark to assess our performance relative to our peers. We have retained two indices from last year – the AMEX Composite, which reflects a major market, and the AMEX Biotechnology, which reflects our line of business or industry.
Recent Sales of Unregistered Securities
During the fiscal year ended December 31, 2006, we did not issue any securities that were not registered under the Securities Act of 1933, as amended, except:
| • | as disclosed in previous filings with the Securities and Exchange Commission (including our annual report on Form 10-K filed on March 16, 2006); and | ||
| • | during the fourth quarter, we issued an aggregate of 1,060,596 shares of common stock to certain of our warrant holders for gross proceeds of approximately $769,000 in connection with their exercise of outstanding warrants. |
We issued the shares subject to outstanding warrants in reliance on the exemption from registration under Section 4(2) of the Securities Act of 1933, as amended, as transactions by an issuer not involving any public offering. The recipients of such common stock represented their respective intentions to acquire the common stockfor investment only and not with a view to or for sale in connection with any distribution thereof and appropriate legends were affixed to the common stock.
Item 6. Selected Financial Data
The selected consolidated financial data set forth below at December 31, 2006 and 2005, and for the years ended December 31, 2006, 2005 and 2004, are derived from our audited consolidated financial statements included elsewhere in this report. This information should be read in conjunction with those consolidated financial statements, the notes thereto, and the report of independent registered public accounting firm thereon, and with “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” The selected consolidated financial data set forth below at December 31, 2004, 2003 and 2002, and for the years ended December 31, 2003 and 2002, are derived from our audited consolidated financial statements that are contained in reports previously filed with the United States Securities and Exchange Commission, or SEC.
Summary Financial Information
| Years Ended December 31, | ||||||||||||||||||||
| Statement of operations data: | 2006 | 2005 | 2004 | 2003 | 2002 | |||||||||||||||
| Loss from operations | $ | (29,836,467 | ) | $ | (13,699,045 | ) | $ | (6,804,090 | ) | $ | (2,339,960 | ) | $ | (2,053,303 | ) | |||||
| Net loss | $ | (29,331,773 | ) | $ | (24,782,646 | ) | $ | (6,701,048 | ) | $ | (2,332,077 | ) | $ | (2,105,727 | ) | |||||
| Net loss applicable to common stock | $ | (29,331,773 | ) | $ | (24,782,646 | ) | $ | (6,701,048 | ) | $ | (2,369,917 | ) | $ | (2,347,927 | ) | |||||
| Basic and diluted net loss per common share (1) | $ | (0.40 | ) | $ | (0.41 | ) | $ | (0.13 | ) | $ | (0.07 | ) | $ | (0.15 | ) | |||||
| Basic and diluted weighted average number of shares of common stock outstanding (1) | 73,988,206 | 59,828,357 | 50,720,180 | 31,797,986 | 15,681,743 | |||||||||||||||
| Cash dividends declared per share | $ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||
41
Table of Contents
| December 31, | ||||||||||||||||||||
| Balance sheet data: | 2006 | 2005 | 2004 | 2003 | 2002 | |||||||||||||||
| Cash and cash equivalents | $ | 25,974,041 | $ | 14,634,618 | $ | 13,032,263 | $ | 4,226,397 | $ | 103,928 | ||||||||||
| Short-term investments | 25,771,406 | 7,958,458 | — | — | — | |||||||||||||||
| Total cash, cash equivalents and short-term investments | 51,745,447 | 22,593,076 | 13,032,263 | 4,226,397 | 103,928 | |||||||||||||||
| Working capital (deficit) | 19,532,149 | (8,534,219 | ) | 12,047,819 | 4,091,730 | (822,274 | ) | |||||||||||||
| Total assets | 52,798,385 | 23,621,773 | 13,608,787 | 4,283,356 | 130,345 | |||||||||||||||
| Long-term obligations | 35,674 | 57,078 | — | — | 56,873 | |||||||||||||||
| Total liabilities | 32,840,637 | 31,450,389 | 1,218,396 | 163,043 | 983,075 | |||||||||||||||
| Stockholders’ equity (deficit) | 19,957,748 | (7,828,616 | ) | 12,390,391 | 4,120,313 | (852,730 | ) | |||||||||||||
| (1) | See Note 2 of the Notes to Consolidated Financial Statements,Significant Accounting Policies — Computation of Net Loss per Common Share, for an explanation of the method used to calculate the net loss per common share and the number of shares used to compute the per share amount. |
42
Table of Contents
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with the information set forth under the caption “Selected Consolidated Financial Data” and our financial statements and related notes appearing elsewhere in this report. In addition to historical information, this discussion and analysis contains forward-looking statements that involve risks, uncertainties, and assumptions. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of certain factors, including but not limited to those set forth under the caption “Risk Factors.”
Overview
We are a biopharmaceutical research and development company focused on commercializing proprietary product candidates for the treatment of cancer and infectious diseases. We seek to improve the performance and safety of existing treatments by addressing significant problems such as drug metabolism, bioavailability, excessive toxicity and treatment resistance. Our lead product candidate, ANX-510, or CoFactor, is in Phase III and Phase IIb clinical trials for the treatment of metastatic colorectal cancer, as well as in a Phase II clinical trial for the treatment of advanced breast cancer.
The following are some of our recent highlights:
| • | April 2006 – we completed the acquisition of SD Pharmaceuticals, Inc., or SDP, a privately-held drug development company. In connection with the SDP acquisition, we acquired SDP’s rights to certain oncology and infectious disease product candidates, including rights to a product candidate that we licensed from SDP in October 2005. See Note 3,Acquisition of SDP, in Notes to Consolidated Financial Statements. | ||
| • | June 2006 – we initiated a Phase III pivotal clinical trial of CoFactor in the treatment of metastatic colorectal cancer. Related to this, in May 2006, we reached an agreement with the FDA on a special protocol assessment, or SPA, regarding the design and planned analysis of this clinical trial. The SPA provides an understanding between the FDA and us that the protocol for and data obtained from our Phase III clinical trial may represent the basis for an efficacy claim. | ||
| • | September 2006 – we completed patient enrollment in our Phase IIb clinical trial of CoFactor in the treatment of metastatic colorectal cancer. This trial is designed to compare the safety of 5-fluorouracil, or 5-FU, plus CoFactor to 5-FU plus leucovorin in the treatment of metastatic colorectal cancer. | ||
| • | October 2006 – we licensed the U.S. rights to ANX-211 (chitosan gel), one of our proprietary antiviral products, to Theragenex LLC, or Theragenex, a life science and technology company focused on commercializing therapies across a number of different therapeutic areas. We anticipate Theragenex will launch a licensed product in the 2007/2008 cold and influenza season. | ||
| • | November 2006 – we raised approximately $37.1 million from the sale of common stock to institutional investors in a registered direct offering at a price of $2.75 per share. | ||
| • | December 2006 – we initiated a marketing-enabling clinical study of ANX-530 (vinorelbine emulsion), which is designed to establish the bioequivalence of ANX-530 and vinorelbine tartrate. Vinorelbine tartrate is an anticancer agent approved for use in non-small cell lung cancer. | ||
| • | December 2006 – we initiated a Phase II clinical trial of CoFactor in the treatment of advanced metastatic breast cancer. | ||
| • | August through December 2006 – we made a number of significant additions and changes to our management team, including the appointment of a president and chief medical officer, a chief financial officer, a vice president of medical affairs and a general counsel. |
43
Table of Contents
We have historically sought to maintain flexibility in our cost structure by actively managing several outsourced functions, such as clinical trials, documentation and testing of internal controls, pre-clinical development work and manufacturing, rather than maintaining these functions in house. We believe the benefits of outsourcing, including being flexible and able to rapidly respond to program delays or successes, outweigh the higher costs often associated with outsourcing at this stage of our development.
Two of our product candidates were in the clinical trial phase of development as of December 31, 2006. We intend to continue to seek partnerships with pharmaceutical and other companies to help fund our research and development programs and commercialize those of our product candidates that may be approved in exchange for rights in our products and product candidates. We may acquire other product candidates to leverage our current infrastructure for research and development. We are unable to determine if and when we might reach profitability until we know the outcome of future development of our products and product candidates and decisions by the FDA related to our research and development programs. Trends in various types of expenses are discussed further in the “Results of Operations.”
We will need to raise additional capital if we continue our Phase III pivotal clinical trial of CoFactor in the treatment of metastatic colorectal cancer. We may seek to raise this additional capital at any time through various financing alternatives, including selling shares of our common or preferred stock and rights to acquire our common or preferred stock, licensing or selling our technologies and product candidates, or through the issuance of one or more forms of senior or subordinated debt. Our future capital needs will also depend on the economic terms and the timing of any new partnerships or collaborative arrangements with pharmaceutical companies we may enter into in the future. If we are unable to raise capital as needed to fund our operations, or if we are unable to enter into any such partnerships or collaborative arrangements, we may need to slow the rate of development of some of our research and development programs, in particular our Phase III clinical trial of CoFactor.
CRITICAL ACCOUNTING POLICIES AND ESTIMATES
Our discussion and analysis of our financial condition and results of operations is based upon audited financial statements that we have prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of these financial statements requires management to make a number of assumptions and estimates that affect the reported amounts of assets, liabilities, revenues and expenses in our consolidated financial statements and accompanying notes. On an on-going basis, we evaluate these estimates and assumptions, including those related to recognition of expenses in research contracts, expenses in research and development, expenses in share-based compensation and loss on fair value of warrants. Management bases its estimates on historical information and assumptions believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
Our critical accounting policies are those that affect our financial statements materially and involve a significant level of judgment by management. Our critical accounting policies regarding revenue recognition involve license agreements and grant revenues; however, since inception we have not recognized a material amount of revenue. Our critical accounting policies also address recognition of expenses in research contracts, expenses in research and development, and expenses in share-based compensation.
Revenue Recognition
We recognize revenue in accordance with the SEC’s Staff Accounting Bulletin Topic 13, or Topic 13,Revenue Recognitionand Emerging Issues Task Force Issue, or EITF, No. 00-21,Revenue Arrangements with Multiple Deliverables. Revenue is recognized when all of the following criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services have been rendered; (3) the seller’s price to the buyer is fixed and determinable; and (4) collectibility is reasonably assured.
Revenue from licensing agreements is recognized based on the performance requirements of the agreement. Revenue is deferred for fees received before earned. Nonrefundable upfront fees that are not contingent on any future performance by us are recognized as revenue when revenue recognition criteria are met and the license term commences. Nonrefundable upfront fees, where we have an ongoing involvement or performance obligations, are recorded as deferred revenue and recognized as revenue over the life of the contract, the period of the performance obligation or the development period, whichever is appropriate in light of the circumstances.
44
Table of Contents
Payments related to substantive, performance-based milestones in an agreement are recognized as revenue upon the achievement of the milestones as specified in the underlying agreements when they represent the culmination of the earnings process. Royalty revenue from licensed products will be recognized when earned in accordance with the terms of the license agreements.
Recognition of Expenses in Research Contracts
Pursuant to management’s assessment of the services that have been performed on clinical trials and other contracts, we recognize expenses as the services are provided. Such management assessments generally consist of, but are not limited to, an evaluation by the project manager of the work that has been completed during the period, measurement of progress prepared internally and/or provided by the third-party service provider, analysis of data that justifies the progress, and finally, management’s judgment. Several of our contracts extend across multiple reporting periods, including our largest contract, with a CRO, representing a $9.0 million commitment over the life of the contract. A 3% variance in our estimate of the work completed in our largest contract could increase or decrease our operating expenses by approximately $270,000.
Research and Development Expenses
Research and development, or R&D, expenses consist of expenses incurred in performing research and development activities, including salaries and benefits, facilities and other overhead expenses, clinical trials, contract services and other outside expenses. Research and development expenses are charged to operations as they are incurred.
License fees. Payments made in connection with in-licensed technology or product candidates are expensed as incurred when there is uncertainty in receiving future economic benefits from the licensed technology or product candidates. We consider the future economic benefits from the licensed technology or product candidates to be uncertain until such licensed technology or product candidates are approved by the FDA or when other significant risk factors are abated. For expense accounting purposes, management has viewed future economic benefits for all of our licensed technology or product candidates to be uncertain.
Purchased In-Process Research and Development
In accordance with Statement of Financial Accounting Standards, or FAS, No. 141,Business Combinations, we immediately charge the costs associated with purchased in-process research and development, or IPR&D, to statement of operations upon acquisition. These amounts represent an estimate of the fair value of purchased IPR&D for projects that, as of the acquisition date, had not yet reached technological feasibility, had no alternative future use, and had uncertainty in receiving future economic benefits from the purchased IPR&D. We determine the future economic benefits from the purchased IPR&D to be uncertain until such technology is approved by the FDA or when other significant risk factors are abated. We incurred significant IPR&D expense related to the SDP acquisition.
Share-based Compensation Expenses
Effective January 1, 2006, we adopted the provisions of revised FAS No. 123,Share-Based Payment,or FAS 123R, including the provisions of Staff Accounting Bulleting No. 107. Under FAS 123R, share-based compensation cost is measured at the grant date, based on the estimated fair value of the award, and is recognized as expense over the employee’s requisite service period. We have no awards with market or performance conditions. We adopted the provisions of FAS 123R using the modified prospective transition method. Accordingly, prior periods have not been revised for comparative purposes.
On November 10, 2005, the Financial Accounting Standards Board, or FASB, issued FASB Staff Position No. FAS 123(R)-3,Transition Election Related to Accounting for Tax Effects of Share-Based Payment Awards. We have elected to adopt the alternative transition method provided in FAS 123R. The alternative transition method includes a simplified method to establish the beginning balance of the additional paid-in capital pool related to the tax effects of employee share-based compensation, which is available to absorb tax deficiencies recognized subsequent to the adoption of FAS 123R.
45
Table of Contents
We grant share-based awards to our employees, consultants and directors under our 2005 Equity Incentive Plan, or the 2005 Plan. We recognize the share-based compensation costs on a straight-line basis over the requisite service period of the award, which is generally four years; however, the 2005 Plan allows for other vesting periods and we have granted employees options where the requisite service period is three years, and we grant our directors options where the requisite service period is one year. The valuation provisions of FAS 123R apply to new awards and to awards that are outstanding on the effective date, January 1, 2006, which are subsequently modified or cancelled. Prior to 2006, we accounted for share-based compensation under the recognition and measurement principles of FAS No. 123,Accounting for Stock-Based Compensation, or FAS 123. Estimated compensation expense for awards outstanding at January 1, 2006 is recognized over the remaining service period using the compensation cost calculated for recognition purposes under FAS 123.
We estimate the fair value of stock option awards on the date of grant using the Black-Scholes option-pricing model, or Black-Scholes model. The determination of the fair value of share-based payment awards on the date of grant using an option-pricing model is affected by our stock price as well as assumptions regarding a number of complex and subjective variables. These variables include, but are not limited to, our expected stock price volatility over the term of the awards, actual and projected employee stock option exercise behaviors, a risk-free interest rate and expected dividends.
If factors change or we employ different assumptions in the application of FAS 123R in future periods, the compensation expense that we record under FAS 123R may differ significantly from what is reflected in this report. Option-pricing models were developed for use in estimating the value of traded options that have no vesting or hedging restrictions, are fully transferable and do not cause dilution. Because our share-based payments have characteristics significantly different from those of freely-traded options, and because changes in the subjective input assumptions can materially affect our estimates of fair values, in our opinion, existing valuation models, including the Black-Scholes model, may not provide reliable measures of the fair values of our share-based compensation. There currently is not a market-based mechanism or other practical application to verify the reliability and accuracy of the estimates stemming from these valuation models, nor is there a means to compare and adjust the estimates to actual values. Although the fair value of employee share-based awards is determined in accordance with FAS 123R and the SEC’s Staff Accounting Bulletin No. 107, using an option-pricing model, that value may not be indicative of the fair value observed in a willing buyer/willing seller market transaction. In addition, there are significant differences among valuation models, and there is a possibility that we will adopt different valuation models in the future. This may result in a lack of consistency in future periods and materially affect the fair value estimate of share-based payments. It may also result in a lack of comparability with other companies that use different models, methods and/or assumptions.
Estimates of share-based compensation expenses are significant to our financial statements, but these expenses are based on option-pricing models, and by the terms of our outstanding options and will not result in the payment of cash by us. For this reason, and because we do not view share-based compensation as related to our operational performance, we exclude estimated share-based compensation expense when evaluating our business performance.
We account for share-based compensation awards granted to non-employees in accordance with EITF No. 96-18,Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services, or EITF 96-18. Under EITF 96-18, we determine the fair value of the share-based compensation awards granted as either the fair value of the consideration received or the fair value of the equity instruments issued, whichever is more reliably measurable. If the fair value of the equity instruments issued is used, it is measured using the stock price and other measurement assumptions as of the earlier of either of (1) the date at which a commitment for performance by the counterparty to earn the equity instruments is reached or (2) the date at which the counterparty’s performance is complete.
46
Table of Contents
Registration Payment Arrangement
We account for contingent obligations in a registration payment arrangement in accordance with EITF Issue No. 00-19,Accounting for Derivative Financial Instruments Indexed To, and Potentially Settled in a Company’s Own Stock, or EITF 00-19, and the SEC’s December 2005 interpretation. In connection with a sale of shares of our common stock in July 2005, we entered into a registration payment arrangement which requires us to use our best efforts to (a) file a registration statement with the SEC, (b) have it declared effective by the end of a certain period and (c) maintain effectiveness of the registration statement for a certain period of time. See Note 7 in Notes to Consolidated Financial Statements,Warrant Liability, for a detailed discussion. In the event we fail to meet the registration requirements, the arrangement requires us to make payments to the purchasers until the registration payment obligations no longer exist.
Because the arrangement requires payments to be settled in cash, we recorded the fair value of the arrangement as a liability, with an offsetting reduction to additional paid-in capital as of the closing date of the sale. At the end of each reporting period, the value of the arrangement will be re-measured based on the fair market value of the underlying shares, and changes to the liability and related gain or loss will be made appropriately. In addition, the shares issued that are subject to the registration payment arrangement are reported as temporary equity. The liability and temporary equity will be reclassified to equity when the registration payment obligations no longer exist.
In December 2006, the Financial Accounting Standards Board, or FASB, issued FASB Staff Position on No. EITF 00-19-2,Accounting for Registration Payment Arrangements, or FSP EITF 00-19-2. FSP EITF 00-19-2 provides that the contingent obligation to make future payments or otherwise transfer consideration under a registration payment arrangement should be separately recognized and measured in accordance with FAS No. 5,Accounting for Contingencies, which defines that loss contingencies should be recognized as liabilities if they are probable and reasonably estimable. The guidance in FSP EITF 00-19-2 is effective immediately for registration payment arrangements and the financial instruments subject to those arrangements that are entered into or modified subsequent to the date of issuance of FSP EITF 00-19-2. For registration payment arrangements and financial instruments subject to those arrangements that were entered into prior to the issuance of FSP EITF 00-19-2, this guidance shall be effective for financial statements issued for years beginning after December 15, 2006, and interim periods within those years.
Effective January 1, 2007, we will apply new guidance under FSP EITF 00-19-2 to account for this registration payment arrangement. We are in the process of evaluating the impact of FSP EITF 00-19-2 on our financial position and results of operations.
The above listing is not intended to be a comprehensive list of all of our accounting policies. In most cases, the accounting treatment of a particular transaction is specifically dictated by GAAP.
NATURE OF OPERATING EXPENSES
Our total operating expenses are influenced substantially by the amount of spending devoted to our research and development programs. During the last three years, we have expanded our product candidate pipeline, which requires that we allocate significant amounts of our resources to such programs, including increased spending on clinical trials as those programs advance. We expect research and development expenses will represent at least 60% of our operating expenses for 2007. We expect that selling, general and administrative expenses for 2007 will represent less than 40% of our operating expenses.
Our business is exposed to significant risks, as discussed in the section entitled “Risk Factors,” which may result in additional expenses, delays and lost opportunities that could have a material adverse effect on our results of operations and financial condition.
47
Table of Contents
RESULTS OF OPERATIONS
We operate our business on the basis of a single reportable segment – commercializing proprietary product candidates for the treatment of cancer and infectious diseases.
| Operating Expenses | ||||||||||||
| Years Ended December 31, | ||||||||||||
| 2006 | 2005 | 2004 | ||||||||||
| Research and development | 40 | % | 63 | % | 40 | % | ||||||
| In-process research and development | 35 | % | 0 | % | 0 | % | ||||||
| Selling, general and administrative | 24 | % | 36 | % | 59 | % | ||||||
| Depreciation and amortization | 1 | % | 1 | % | 1 | % | ||||||
| Total operating expenses | 100 | % | 100 | % | 100 | % | ||||||
Comparison of 2006 and 2005
Operating Expenses
Total operating expenses amounted to $29.8 million in 2006, compared to $13.7 million in 2005. The $16.1 million or 118% increase in operating expenses was due to a $3.3 million or 38% increase in research and development expenses, a one-time charge of $10.4 million in connection with purchased IPR&D related to the acquisition of SDP and a $2.3 million or 48% increase in selling, general and administrative expenses. Explanations of operating expenses in both 2006 and 2005 are described more fully in the paragraphs that follow.
R&D Expenses.R&D expenses increased to $12.0 million in 2006 from $8.7 million in 2005. The $3.3 million or 38% increase is primarily due to a $2.0 million increase in spending for launching a Phase III clinical trial of CoFactor and continuing a Phase IIb CoFactor clinical trial for the treatment of metastatic colorectal cancer, a $902,000 increase in compensation expenses related to an increase in clinical personnel, a $330,000 increase in outside services expense related to clinical support efforts and a $144,000 increase in preclinical expenditures related to CoFactor, ANX-530 and ANX-201.
IPR&D.In 2006, we recorded a charge of $10.4 million in connection with purchased IPR&D related to the acquisition of SDP in April 2006.
Selling, General and Administrative, or SG&A, Expenses.SG&A expenses increased to $7.2 million in 2006 from $4.9 million in 2005. The $2.3 million or 47% increase is primarily due to a $1.3 million increase in personnel and employee-related expense, a $579,000 increase in professional and consulting fees and a $226,000 increase in insurance costs related to an increase in liability coverage.
Loss on Fair Value of Warrants.For 2006, we recorded a loss of $660,000 on the fair value of warrants issued in July 2005 compared to a loss of $11.6 million for 2005, a decrease of $10.9 million or 94%. The loss recorded in 2006 was less than the loss recorded in 2005 because the fair value of the warrants did not increase as much in 2006 as compared to 2005, the initial year the fair value of the warrant was recorded.
Interest Income.Interest income increased by $669,000, or 135%, to $1.2 million in 2006 from $496,000 in 2005. The increase is primarily due to higher invested balances resulting from an equity financing in November 2006 with net proceeds of $37.1 million and from higher interest rate yields on these balances in 2006 compared to 2005.
Net Loss.Net loss was $29.3 million or $0.40 per share in 2006 compared to a net loss of $24.8 million or $0.41 per share in 2005.
We expect to continue to pursue our product development strategy focused on the development of CoFactor, ANX-530 and ANX-201 followed by other programs in earlier stages of development. To help fund and develop our product development efforts, we may elect to license certain of our technologies and product candidates to third parties. These potential license arrangements could materially change our outlook for future revenues and costs. However, the timing of such potential arrangements is unpredictable.
48
Table of Contents
Comparison of 2005 and 2004
Operating Expenses
Total operating expenses amounted to $13.7 million in 2005, compared to $6.8 million in 2004. Explanations of operating expenses in both 2005 and 2004 are described more fully in the paragraphs that follow.
R&D Expenses.R&D expenses increased to $8.7 million in 2005 from $2.7 million in 2004. The increase is primarily due to a $3.5 million increase in expenses for our Phase II and Phase IIb CoFactor clinical trials, a $1.2 million increase in preclinical expenditures related to CoFactor, ANX-530 and ANX-201, a $1.1 million increase in compensation expenses related to an increase in clinical personnel and employee-related expenses and a $225,000 increase in outside services expense related to clinical support efforts.
SG&A Expenses.SG&A expenses increased to $4.9 million in 2005 from $4.0 million in 2004. The $882,000 increase is primarily due to a $300,000 increase in compensation expenses related to an increase in SG&A personnel, a $280,000 increase in outside consulting fees related to efforts to comply with the Sarbanes-Oxley Act of 2002 and related system implementation efforts and a $115,000 increase in facilities cost due to an increase in the amount of space leased.
Loss on Fair Value of Warrants.In July 2005, we issued warrants to purchase 10,810,809 shares of our common stock in conjunction with a private placement. Pursuant to EITF 00-19, we recorded a liability for registration payment obligations associated with these warrants based on the fair value of the warrants on the closing date of the transaction. The fair value of these warrants is then re-measured at each reporting date with a resulting gain or (loss) charged to our consolidated statement of operations. For 2005, we recorded a total loss of $11.6 million associated with these warrants.
Interest Income.Interest income increased by $393,000, or 381%, to $496,000 in 2005 from $103,000 in 2004. The increase is primarily due to the investment of the proceeds of an equity financing which occurred in July 2005 with net proceeds of $19.0 million which resulted in an increase in the average balance of investments in 2005, compared to 2004. In addition, we experienced a rise in interest rates in the second half of 2005.
Net Loss.Net loss was $24.8 million or $0.41 per share in 2005 compared to a net loss of $6.7 million or $0.13 per share in 2004.
LIQUIDITY AND CAPITAL RESOURCES
Historically, we have funded our operations primarily through sales of our equity securities. As of December 31, 2006, we had cash, cash equivalents and short-term investments in securities totaling $51.7 million, including cash and cash equivalents of $26.0 million and short-term investments of $25.7 million. Our net working capital balance as of December 31, 2006 was $19.5 million. As of December 31, 2005, we had cash, cash equivalents and short-term investments totaling $22.6 million. Our net working capital deficit balance as of December 31, 2005 was $8.5 million. Explanations of net cash provided by or used in operating, investing and financing activities are provided below.
| Increase | ||||||||||||
| December 31, | (Decrease) | December 31, | ||||||||||
| 2006 | During 2006 | 2005 | ||||||||||
| Cash, cash equivalents and investments in securities | $ | 51,745,447 | $ | 29,152,371 | $ | 22,593,076 | ||||||
| Net working capital (deficit) | $ | 19,532,149 | $ | 28,066,368 | $ | (8,534,219 | ) | |||||
| Year Ended | Change | Year Ended | ||||||||||
| December 31, | Between | December 31, | ||||||||||
| 2006 | Periods | 2005 | ||||||||||
| Net cash used in operating activities | $ | (15,773,544 | ) | $ | (4,126,715 | ) | $ | (11,646,829 | ) | |||
| Net cash used in investing activities | (17,774,585 | ) | (9,688,580 | ) | (8,086,005 | ) | ||||||
| Net cash provided by financing activities | 44,887,552 | 23,552,363 | 21,335,189 | |||||||||
| Net increase in cash and cash equivalents | $ | 11,339,423 | $ | 9,737,068 | $ | 1,602,355 | ||||||
49
Table of Contents
Operating activities.Net cash used in operating activities was $15.8 million in 2006, compared to $11.6 million in 2005 and $5.2 million in 2004. The increase in cash used in operating activities in 2006 and 2005 was mainly due to the increase in our research and development and selling, general and administrative expenses.
Investing activities.Net cash used in investing activities was $17.8 million in 2006, compared to $8.1 million in 2005 and $306,000 in 2004. The increase in 2006 was mainly due to net purchases of short-term investments of $17.6 million. The increase in 2005 as compared to 2004 was mainly due to net purchases of short-term investments of $7.8 million.
Financing activities.Net cash provided by financing activities was $44.9 million in 2006, consisting primarily of $37.1 million in net proceeds from sales of our common stock through private placements and $7.7 million in net proceeds from exercises of warrants to purchase our common stock. Net cash provided by financing activities was $21.3 million in 2005, consisting primarily of $18.1 million in net proceeds from sales of our common stock and warrants to purchase our common stock through private placements and $3.1 million from exercises of warrants to purchase our common stock. Net cash provided by financing activities amounted to $14.3 million in the 2004, consisting of $14.3 million received from the sale of our common stock and warrants to purchase our common stock.
Contractual Obligations. As of December 31, 2006, we have contractual obligations for operating leases and purchase obligations, as summarized in the table that follows. We anticipate being able to satisfy the obligations described below out of cash, cash equivalents and investments in securities held by us as of December 31, 2006. We do not have any off balance sheet arrangements and no commitments for any significant additional capital expenditures.
| Payments Due by Period | ||||||||||||||||||||
| Less than | More than | |||||||||||||||||||
| Total | 1 Year | 1-3 Years | 3-5 Years | 5 years | ||||||||||||||||
| Operating lease obligations | $ | 689,339 | $ | 255,630 | $ | 433,709 | $ | — | $ | — | ||||||||||
| Purchase obligations | 28,862,313 | 10,940,450 | 17,921,863 | — | — | |||||||||||||||
| Total | $ | 29,551,652 | $ | 11,196,080 | $ | 18,355,572 | $ | — | $ | — | ||||||||||
Purchase obligations presented above represent contractual commitments entered into for goods and services in the normal course of our business. The amount includes all known contracts and open purchase orders related to our clinical and preclinical activities. In many cases, amounts related to service contracts may be cancelled based on contract provisions prior to completion. The allocation of the obligations by year is based on our best estimate of the timing of the expenditures.
The following contingent payments are excluded from the contractual obligations presented above:
| • | Royalties (including prepaid royalties), milestone payments, payments resulting from sublicensing activities and reimbursement of legal expenses due USC. | ||
| • | Milestone payments (payable in cash and stock) to a consultant based on the clinical success of CoFactor. |
Management Outlook
We believe that cash, cash equivalents, and short-term investments of approximately $51.7 million at December 31, 2006, should be sufficient to sustain our planned level of operations for at least the next twelve months. We expect that capital necessary to fund operations in 2007 will be higher than the $15.8 million used in 2006, as we continue developing our existing product candidates and pipeline. In order to maintain sufficient cash and investments to fund future operations longer term, and to continue developing our existing product candidates, we will need to raise additional capital from time to time, and may do so through various financing alternatives, including selling shares of our common or preferred stock and rights to acquire our common or preferred stock, licensing or selling our technologies and product candidates, or through the issuance of one or more forms of senior or subordinated debt. The balance of securities available for sale under our existing shelf registration was approximately $60.0 million as of December 31, 2006. If we are unable to raise capital as needed to fund future operations, then we may defer or abandon one or more of our clinical or preclinical research programs and may need to take additional cost-cutting measurements.
50
Table of Contents
During 2007, we expect to recognize $2.0 million from the licensing of ANX-211 to Theragenex, and may recognize royalties based on Theragenex’s success in selling licensed products. Additionally, we are in active discussions with potential partners regarding our product candidates, though some of our product candidates could take several more years of development before they reach the stage of being partnerable with other companies. If we successfully consummate a partnering deal, we may be entitled to license fees and milestone payments that we may recognize in 2007. Of course, any such fees and payments will depend on successfully consummating a deal and achieving milestones under such arrangements.
For information regarding the risks associated with our need to raise capital to fund our ongoing and planned operations, please see “Risk Factors.”
Recent Accounting Pronouncements
See Note 2, “Summary of Significant Accounting Policies – Recent Accounting Pronouncements,” in the Notes to Consolidated Financial Statements for a discussion of recent accounting pronouncements and their effect, if any, on the Company.
Item 7A. Quantitative and Qualitative Disclosures about Market Risk
As described below, we are exposed to market risk related to changes in interest rates. Because substantially all our expenses and capital purchasing activities are transacted in U.S. dollars, our exposure to foreign currency exchange rates is immaterial. Until such time as we are faced with material amounts of foreign currency exchange rate risks, we do not plan to use derivative financial instruments, which can be used to hedge such risks. We will evaluate the use of derivative financial instruments to hedge our exposures as the needs and risks should arise.
Interest Rate Sensitivity
Our investment portfolio consists primarily of government or investment grade fixed income instruments with an average duration of under 60 days. The primary objective of our investments in debt securities is to preserve principal while achieving attractive yields, without significantly increasing risk. We classify our investments in securities as of December 31, 2006 as available-for-sale. These available-for-sale securities are subject to interest rate risk. Due to the short average duration as of December 31, 2006 the interest rate risks were not significant.
Item 8. Financial Statements and Supplementary Data
The consolidated financial statements and supplementary financial information required by this item are filed with this report as described under Item 15.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure
None.
Item 9A. Controls and Procedures
Disclosure Controls and Procedures
We maintain disclosure controls and procedures that are designed to ensure that information required to be disclosed in our reports under the Securities Exchange Act of 1934, as amended, or the Exchange Act, is recorded, processed, summarized and reported within the timelines specified in the SEC’s rules and forms, and that such information is accumulated and communicated to our management, including our chief executive officer and chief financial officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognized that any controls and procedures, no matter how well designed and operated, can only provide reasonable assurance of achieving the desired control objectives, and
51
Table of Contents
in reaching a reasonable level of assurance, management necessarily was required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Under the supervision and with the participation of our management, including our chief executive officer and chief financial officer, we have evaluated the effectiveness of our disclosure controls and procedures (as defined under Exchange Act Rule 13a-15(e)), as of December 31, 2006. Based on that evaluation, our chief executive officer and chief financial officer have concluded that these disclosure controls and procedures were effective as of December 31, 2006.
Changes in Internal Control over Financial Reporting
There was no change in our internal control over financial reporting that occurred during the fiscal quarter ended December 31, 2006 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in Exchange Act Rules 13a-15(f). Under the supervision and with the participation of our management, including our chief executive officer and chief financial officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting based on the framework inInternal Control — Integrated Frameworkissued by the Committee of Sponsoring Organizations of the Treadway Commission. Based on our evaluation under the framework inInternal Control — Integrated Framework, our management concluded that our internal control over financial reporting was effective as of December 31, 2006.
Our assessment of the effectiveness of our internal control over financial reporting as of December 31, 2006 has been attested to by J.H. Cohn LLP, our independent registered public accounting firm, as stated in their report, which is set forth below in this Item 9A.
Report of Independent Registered Public Accounting Firm
To Board of Directors and Stockholders
ADVENTRX Pharmaceuticals, Inc.
ADVENTRX Pharmaceuticals, Inc.
We have audited management’s assessment, included in Item 9A, Management’s Report on Internal Control over Financial Reporting, that ADVENTRX Pharmaceuticals, Inc. and Subsidiaries maintained effective internal control over financial reporting as of December 31, 2006 based on criteria established in “Internal Control — Integrated Framework” issued by the Committee of Sponsoring Organizations of the Treadway Commission. ADVENTRX Pharmaceuticals, Inc. and Subsidiaries’ management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting. Our responsibility is to express an opinion on management’s assessment and an opinion on the effectiveness of the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, evaluating management’s assessment, testing and evaluating the design and operating effectiveness of internal control, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial
52
Table of Contents
statements in accordance with accounting principles generally accepted in the United States of America, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, management’s assessment that ADVENTRX Pharmaceuticals, Inc. and Subsidiaries maintained effective internal control over financial reporting as of December 31, 2006, is fairly stated, in all material respects, based on the criteria established in “Internal Control - Integrated Framework” issued by the Committee of the Sponsoring Organizations of the Treadway Commission. Also, in our opinion, ADVENTRX Pharmaceuticals, Inc. and Subsidiaries maintained, in all material respects, effective internal control over financial reporting as of December 31, 2006, based on the criteria established in “Internal Control — Integrated Framework” issued by the Committee of the Sponsoring Organizations of the Treadway Commission.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheet of ADVENTRX Pharmaceuticals, Inc. and Subsidiaries as of December 31, 2006, and the related consolidated statements of operations, stockholders’ equity (deficit) and cash flows for the year then ended, and our report dated February 23, 2007 expressed an unqualified opinion.
/s/ J. H. Cohn LLP
San Diego, California
February 23, 2007
February 23, 2007
Item 9B. Other Information
On October 10, 2006, we terminated a consulting agreement with our former chief financial officer, who also served as our treasurer, vice president, finance and secretary, that we entered into in connection with her separation from us in September 2006.
PART III
Certain information required by Part III is omitted from this report because we will file a definitive proxy statement within 120 days after the end of our fiscal year pursuant to Regulation 14A, or the Proxy Statement, for our annual meeting of stockholders to be held on May 23, 2007, and such information included in the Proxy Statement is incorporated herein by reference.
Item 10. Directors, Executive Officers and Corporate Governance
Code of Ethics
We have adopted a code of ethics that applies to our principal executive officer, principal financial officer, principal accounting officer or persons performing similar functions, as well as all of our other officers, directors and employees. This code of ethics is a part of our conduct of business conduct and ethics and available on our corporate website at www.adventrx.com. We intend to disclose future amendments to, or waivers of, certain provisions of our code of ethics that apply to our principal executive officer, principal financial officer, principal accounting officer or persons performing similar functions on the above website within four business days following such amendment or waiver.
The other information required by this item will be set forth in the Proxy Statement and is incorporated into this report by reference.
53
Table of Contents
Item 11. Executive Compensation
The information required by this item will be set forth in the Proxy Statement and is incorporated into this report by reference.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The information required by this item will be set forth in the Proxy Statement and is incorporated into this report by reference.
Item 13. Certain Relationships and Related Transactions and Director Independence
The information required by this item will be set forth in the Proxy Statement and is incorporated into this report by reference.
Item 14. Principal Accountant Fees and Services
The information required by this item will be set forth in the Proxy Statement and is incorporated into this report by reference.
PART IV
Item 15. Exhibits, Financial Statement Schedules
| (a) | Documents Filed. The following documents are filed as part of this report: |
| (1) | Financial Statements. The following report of J.H. Cohn LLP and financial statements: |
| • | Report of J.H. Cohn LLP, Independent Registered Public Accounting Firm | ||
| • | Consolidated Balance Sheets as of December 31, 2006 and 2005 | ||
| • | Consolidated Statements of Operations for the years ended December 31, 2006, 2005 and 2004 and from inception through December 31, 2006 | ||
| • | Consolidated Statements of Stockholders’ Equity (Deficit) from inception through December 31, 2006 | ||
| • | Consolidated Statements of Cash Flows for the years ended December 31, 2006, 2005 and 2004 and from inception through December 31, 2006 | ||
| • | Notes to Consolidated Financial Statements |
| (2) | Financial Statement Schedules. See subsection (c) below. | ||
| (3) | Exhibits. See subsection (b) below. |
| (b) | Exhibits. |
| Exhibit | Description | |
| 2.1 (1) | Agreement and Plan of Merger, dated April 7, 2006, among the registrant, Speed Acquisition, Inc., SD Pharmaceuticals, Inc. and certain individuals named therein (including exhibits thereto) | |
| 3.1 (2) | Amended and Restated Certificate of Incorporation of the registrant | |
| 3.2 (3) | Amended and Restated Bylaws of the registrant (formerly known as Biokeys Pharmaceuticals, Inc.) | |
| 4.1(4) | Form of Registration Rights Agreement entered into in October and November 2001 (including the original schedule of holders) |
54
Table of Contents
| Exhibit | Description | |
| 4.2 (5) | $2.50 Warrant to Purchase Common Stock issued on April 12, 2002 to Emisphere Technologies, Inc. | |
| 4.3 (4) | Form of $0.60 Warrant to Purchase Common Stock issued May 28, 2003 (including the original schedule of holders) | |
| 4.4 (4) | Form of $1.25 Warrant to Purchase Common Stock issued between October 15, 2003 and December 29, 2003 (including the original schedule of holders) | |
| 4.5 (4) | Common Stock and Warrant Purchase Agreement, dated as of April 5, 2004, among the registrant and the Investors (as defined therein) | |
| 4.6 (4) | Registration Rights Agreement, dated April 5, 2004, among the registrant and the Investors (as defined therein) | |
| 4.7 (4) | Form of $2.00 A-1 Warrant to Purchase Common Stock issued April 8, 2004 (including the original schedule of holders) | |
| 4.8 (4) | Form of $2.50 A-2 Warrant to Purchase Common Stock issued April 8, 2004 (including the original schedule of holders) | |
| 4.9 (6) | Common Stock and Warrant Purchase Agreement, dated April 8, 2004, between the registrant and CD Investment Partners, Ltd. | |
| 4.10 (6) | Registration Rights Agreement, dated April 8, 2004, between the registrant and CD Investment Partners, Ltd. | |
| 4.11 (6) | $2.00 A-1 Warrant to Purchase Common Stock issued on April 8, 2004 to CD Investment Partners, Ltd. | |
| 4.12 (6) | $2.00 A-1 Warrant to Purchase Common Stock issued on April 8, 2004 to Burnham Hill Partners | |
| 4.13 (6) | $2.00 A-1 Warrant to Purchase Common Stock issued on April 8, 2004 to Ernest Pernet | |
| 4.14 (6) | $2.00 A-1 Warrant to Purchase Common Stock issued on April 8, 2004 to W.R. Hambrecht + Co., LLC | |
| 4.15 (7) | Common Stock and Warrant Purchase Agreement, dated April 19, 2004, between the registrant and Franklin M. Berger | |
| 4.16 (8) | Registration Rights Agreement, dated April 19, 2004, between the registrant and Franklin M. Berger | |
| 4.17 (9) | $2.00 A-1 Warrant to Purchase Common Stock issued on April 19, 2004 to Franklin M. Berger | |
| 4.18 (8) | Securities Purchase Agreement, dated July 21, 2005, among the registrant and the Purchasers (as defined therein) | |
| 4.19 (8) | Rights Agreement, dated July 27, 2005, among the registrant, the Icahn Purchasers and Viking (each as defined therein) | |
| 4.20 (9) | First Amendment to Rights Agreement, dated September 22, 2006, among the registrant and the Icahn Purchasers (as defined therein) | |
| 4.21 (8) | Form of $2.26 Common Stock Warrant issued on July 27, 2005 (including the original schedule of holders) | |
| 4.22 (8) | Form of $2.26 Common Stock Warrant issued on July 27, 2005 (including the original schedule of holders) | |
| 4.23 | $0.50 Warrant (WC-291) to Purchase Common Stock transferred on June 15, 2005 to S. Neborsky and R Neborsky TTEE Robert J. Neborsky MD Inc Comb Retirement Trust | |
| 4.24 (10) | $0.50 Warrant (WC-292) to Purchase Common Stock transferred on June 15, 2005 to S. Neborsky and R Neborsky TTEE Robert J. Neborsky MD Inc Comb Retirement Trust | |
| 4.25 (10) | $2.50 Warrant to Purchase Common Stock issued on October 22, 2004 to Thomas J. DePetrillo | |
| 10.1# (11) | 2005 Equity Incentive Plan | |
| 10.2# (12) | Form of Stock Option Agreement under the 2005 Equity Incentive Plan | |
| 10.3# (2) | Form of Restricted Share Award Agreement under the 2005 Equity Incentive Plan |
55
Table of Contents
| Exhibit | Description | |
| 10.4# (12) | 2005 Employee Stock Purchase Plan | |
| 10.5# (12) | Form of Subscription Agreement under the 2005 Employee Stock Purchase Plan | |
| 10.6* (13) | Option and License Agreement, dated January 23, 1998, between the registrant and the University of Southern California | |
| 10.7 (3) | First Amendment to License Agreement, dated August 16, 2000, between the registrant and the University of Southern California | |
| 10.8* (13) | Option and License Agreement, dated August 17, 2000, between the registrant and the University of Southern California | |
| 10.9* (14) | Amendment to Option and License Agreement, dated April 21, 2003, between the registrant and the University of Southern California | |
| 10.10* (2) | Agreement, effective as of May 1, 2005, between the registrant and Pharm-Olam International Ltd. | |
| 10.11 (2) | Amendment dated July 19, 2005 to the Agreement between the registrant and Pharm-Olam International Ltd. | |
| 10.12 (15) | License Agreement, dated October 20, 2006, between the registrant, through its wholly-owned subsidiary SD Pharmaceuticals, Inc., and Theragenex, LLC | |
| 10.13 | License Agreement, dated December 10, 2005, between SD Pharmaceuticals, Latitude Pharmaceuticals and Andrew Chen | |
| 10.14 (16) | Standard Multi-Tenant Office Lease — Gross, dated June 3, 2004, between the registrant and George V. Casey & Ellen M. Casey, Trustees of the Casey Family Trust dated June 22, 1998 | |
| 10.15 (2) | First Amendment to the Standard Multi-Tenant Office Lease — Gross, dated June 3, 2004 between the registrant and George V. & Ellen M. Casey, Trustees of the Casey Family Trust dated June 22, 1998 | |
| 10.16# (17) | Offer letter, dated March 5, 2003, to Joan M. Robbins | |
| 10.17# (18) | Offer letter, dated November 15, 2004, to Brian M. Culley | |
| 10.18# (18) | Offer letter, dated November 17, 2004, to Carrie Carlander | |
| 10.19# (19) | Severance Agreement and Release of All Claims, dated September 7, 2006, with Carrie Carlander | |
| 10.20# (19) | Consulting Agreement, dated September 7, 2006, with Carrie Carlander | |
| 10.21# (19) | Offer letter, dated September 7, 2006, to James A. Merritt | |
| 10.22# (19) | Form of Stock Option Agreement between the registrant and James A. Merritt (included in Exhibit 10.21) | |
| 10.23# (20) | Offer letter, dated December 13, 2006, to Gregory P. Hanson | |
| 10.24# (20) | Stock Option Agreement, effective December 20, 2006, between the registrant and Gregory P. Hanson | |
| 10.25 (21) | Form of Director and Officer Indemnification Agreement | |
| 10.26# (22) | Director compensation policy | |
| 10.27 (23) | Placement Agency Agreement, dated November 2, 2006, among the registrant, ThinkEquity Partners LLC and Fortis Securities LLC | |
| 14.1 (24) | Code of Business Conduct and Ethics | |
| 21.1 | List of Subsidiaries | |
| 23.1 | Consent of J.H. Cohn LLP, Independent Registered Public Accounting Firm | |
| 31.1 | Certification of chief executive officer pursuant to Rule 13a-14(a)/15d-14(a) | |
| 31.2 | Certification of chief financial officer pursuant to Rule 13a-14(a)/15d-14(a) | |
| 32.1 ± | Certification of chief executive officer and chief financial officer pursuant to 18 U.S.C. 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| * | Indicates that confidential treatment has been requested or granted to certain portions, which portions have been omitted and filed separately with the Securities and Exchange Commission |
56
Table of Contents
| # | Indicates management contract or compensatory plan | |
| ± | These certifications are being furnished solely to accompany this report pursuant to 18 U.S.C. 1350, and are not being filed for purposes of Section 18 of the Securities Exchange Act of 1934 and are not to be incorporated by reference into any filing of the registrant, whether made before or after the date hereof, regardless of any general incorporation language in such filing. | |
| (1) | Filed with the registrant’s Amendment No. 1 to Current Report on Form 8-K/A on May 1, 2006 | |
| (2) | Filed with the registrant’s Annual Report on Form 10-K on March 16, 2006 | |
| (3) | Filed with the registrant’s Registration Statement on Form 10SB on October 2, 2001 | |
| (4) | Filed with the registrant’s Registration Statement on Form S-3 on June 30, 2004 | |
| (5) | Filed with the registrant’s Amendment No. 1 to Quarterly Report on Form 10-Q/A on October 30, 2006 | |
| (6) | Filed with the registrant’s Current Report on Form 8-K/A on April 13, 2004 | |
| (7) | Filed with the registrant’s Quarterly Report on Form 10-QSB on May 12, 2005 | |
| (8) | Filed with the registrant’s Quarterly Report on Form 10-Q on August 12, 2005 | |
| (9) | Filed with the registrant’s Current Report on Form 8-K on September 22, 2006 | |
| (10) | Filed with the registrant’s Registration Statement on Form S-3 on August 26, 2005 | |
| (11) | A copy of the registrant’s 2005 Equity Incentive Plan was filed with the registrant’s Registration Statement on Form S-8 on July 13, 2005 but contained a typographical error. A correct copy of the registrant’s 2005 Equity Incentive Plan is filed with this report. | |
| (12) | Filed with the registrant’s Registration Statement on Form S-8 on July 13, 2005 | |
| (13) | Filed with the registrant’s Registration Statement on Form 10SB/A on January 14, 2002 | |
| (14) | Filed with the registrant’s Quarterly Report on Form 10-QSB on August 14, 2003 | |
| (15) | Filed with the registrant’s Current Report on Form 8-K on October 23, 2006 | |
| (16) | Filed with the registrant’s Quarterly Report on Form 10-QSB on August 10, 2004 | |
| (17) | Filed with the registrant’s Annual Report on Form 10-KSB on April 16, 2003 | |
| (18) | Filed with the registrant’s Annual Report on Form 10-KSB on March 31, 2005 | |
| (19) | Filed with the registrant’s Current Report on Form 8-K on September 8, 2006 | |
| (20) | Filed with the registrant’s Current Report on Form 8-K on December 20, 2006 | |
| (21) | Filed with the registrant’s Current Report on Form 8-K on October 23, 2006 | |
| (22) | Filed with the registrant’s Current Report on Form 8-K on June 23, 2006 | |
| (23) | Filed with the registrant’s Current Report on Form 8-K on November 3, 2006 | |
| (24) | Filed with the registrant’s Current Report on Form 8-K on January 23, 2007 | |
| (c) | Financial Statement Schedules. All schedules are omitted because they are not applicable, the amounts involved are not significant or the required information is shown in the financial statements or notes thereto. |
57
Table of Contents
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| ADVENTRX Pharmaceuticals, Inc. | ||||
| By: | /s/ Evan M. Levine | |||
| Evan M. Levine | ||||
| Chief Executive Officer | ||||
Date: March 15, 2007
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Evan M. Levine and Gregory P. Hanson, jointly and severally, as his true and lawful attorneys-in-fact and agents, each with full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this Annual Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents, or their substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Signature | Title | Date | ||
/s/ Evan M. Levine | Chief Executive Officer (Principal Executive Officer) | March 15, 2007 | ||
| /s/ Gregory P. Hanson | Chief Financial Officer, Senior Vice President, Finance, and Treasurer (Principal Financial and Accounting Officer) | March 15, 2007 | ||
| /s/ M. Ross Johnson | Chairman of the Board | March 15, 2007 | ||
| /s/ Mark N.K. Bagnall | Director | March 15, 2007 | ||
| | Director | March 15, 2007 | ||
| /s/ Michael M. Goldberg | Director | March 15, 2007 | ||
| /s/ Jack Lief | Director | March 15, 2007 | ||
| /s/ Mark J. Pykett | Director | March 15, 2007 |
58
Table of Contents
Index to Consolidated Financial Statements
| Page | ||
| F-2 | ||
| Financial Statements: | ||
| F-3 | ||
| F-4 | ||
| F-5 - F-9 | ||
| F-10 - F-11 | ||
| F-12 - F-32 | ||
| Financial Statement Schedules: | ||
| Financial statement schedules have been omitted for the reason that the required information is presented in financial statements or notes thereto, the amounts involved are not significant or the schedules are not applicable |
F-1
Table of Contents
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders
ADVENTRX Pharmaceuticals, Inc.
ADVENTRX Pharmaceuticals, Inc.
We have audited the accompanying consolidated balance sheets of ADVENTRX Pharmaceuticals, Inc. and Subsidiaries (a development stage enterprise) as of December 31, 2006 and 2005, and the related consolidated statements of operations, stockholders’ equity (deficit) and cash flows for each of the three years in the period ended December 31, 2006 and for the period from June 12, 1996 (date of inception) to December 31, 2006. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits. The consolidated financial statements for the period from June 12, 1996 (date of inception) to December 31, 2001 were audited by other auditors whose report, dated April 10, 2002, expressed an unqualified opinion and included an explanatory paragraph concerning the Company’s ability to continue as a going concern. Our opinion on the consolidated statements of operations, stockholders’ equity and cash flows for the period from June 12, 1996 (date of inception) to December 31, 2006, insofar as it relates to amounts for the period for June 12, 1996 (date of inception) to December 31, 2001, is based solely on the report of the other auditors.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, based on our audits and the report of the other auditors, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of ADVENTRX Pharmaceuticals, Inc. and Subsidiaries (a development stage enterprise) as of December 31, 2006 and 2005, and their results of operations and its cash flows for each of the years in the three-year period ended December 31, 2006 and for the period from June 12, 1996 (date of inception) to December 31, 2006, in conformity with accounting principles generally accepted in the United States of America.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the effectiveness of ADVENTRX Pharmaceuticals, Inc. and Subsidiaries’ internal control over financial reporting as of December 31, 2006, based on criteria established in “Internal Control Integrated Framework” issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated February 23, 2007 expressed an unqualified opinion on management’s assessment of internal control over financial reporting and an unqualified opinion of the effectiveness of internal control over financial reporting.
/s/ J.H. Cohn LLP
San Diego, California
February 23, 2007
February 23, 2007
F-2
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
(A Development Stage Enterprise)
Consolidated Balance Sheets
| December 31, | ||||||||
| 2006 | 2005 | |||||||
Assets | ||||||||
| Current assets: | ||||||||
| Cash and cash equivalents | $ | 25,974,041 | $ | 14,634,618 | ||||
| Short-term investments | 25,771,406 | 7,958,458 | ||||||
| Interest receivable | 80,338 | 10,214 | ||||||
| Prepaid expenses | 511,327 | 255,802 | ||||||
| Total current assets | 52,337,112 | 22,859,092 | ||||||
| Property and equipment, net | 402,968 | 407,544 | ||||||
| Other assets | 58,305 | 355,137 | ||||||
| Total assets | $ | 52,798,385 | $ | 23,621,773 | ||||
Liabilities and Stockholders’ Equity (Deficit) | ||||||||
| Current liabilities: | ||||||||
| Accounts payable | $ | 480,402 | $ | 593,228 | ||||
| Accrued liabilities | 1,675,226 | 930,274 | ||||||
| Accrued compensation and payroll taxes | 292,896 | 173,398 | ||||||
| Warrant liability | 30,356,439 | 29,696,411 | ||||||
| Total current liabilities | 32,804,963 | 31,393,311 | ||||||
| Long-term liabilities | 35,674 | 57,078 | ||||||
| Total liabilities | 32,840,637 | 31,450,389 | ||||||
| Commitments and contingencies | ||||||||
| Temporary equity: | ||||||||
| Common stock subject to continuing registration, $0.001 par value; 10,810,809 shares issued and outstanding | — | — | ||||||
| Stockholders’ equity (deficit): | ||||||||
| Common stock, $0.001 par value; 200,000,000 shares authorized; 78,865,930 and 56,529,388 shares issued and outstanding at December 31, 2006 and 2005, respectively | 89,678 | 67,364 | ||||||
| Additional paid-in capital | 109,166,773 | 52,105,329 | ||||||
| Deficit accumulated during the development stage | (89,296,613 | ) | (59,964,840 | ) | ||||
| Accumulated other comprehensive loss | (2,090 | ) | (1,722 | ) | ||||
| Treasury stock, 23,165 shares at December 31, 2005, at cost | — | (34,747 | ) | |||||
| Total stockholders’ equity (deficit) | 19,957,748 | (7,828,616 | ) | |||||
| Total liabilities and stockholders’ equity (deficit) | $ | 52,798,385 | $ | 23,621,773 | ||||
See accompanying notes to consolidated financial statements.
F-3
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
(A Development Stage Enterprise)
Consolidated Statements of Operations
| Inception | ||||||||||||||||
| (June 12, 1996) | ||||||||||||||||
| through | ||||||||||||||||
| Years Ended December 31, | December 31, | |||||||||||||||
| 2006 | 2005 | 2004 | 2006 | |||||||||||||
| Net sales | $ | — | $ | — | $ | — | $ | 174,830 | ||||||||
| Cost of goods sold | — | — | — | 51,094 | ||||||||||||
| Gross margin | — | — | — | 123,736 | ||||||||||||
| Grant revenue | — | — | — | 129,733 | ||||||||||||
| Total revenue | — | — | — | 253,469 | ||||||||||||
| Operating expenses: | ||||||||||||||||
| Research and development | 12,001,212 | 8,682,498 | 2,744,328 | 28,157,964 | ||||||||||||
| In-process research and development | 10,422,130 | — | — | 10,422,130 | ||||||||||||
| Selling, general and administrative | 7,236,437 | 4,901,002 | 4,018,453 | 24,570,736 | ||||||||||||
| Depreciation and amortization | 176,688 | 115,545 | 41,309 | 10,432,249 | ||||||||||||
| Impairment loss — write-off of goodwill | — | — | — | 5,702,130 | ||||||||||||
| Equity in loss of investee | — | — | — | 178,936 | ||||||||||||
| Total operating expenses | 29,836,467 | 13,699,045 | 6,804,090 | 79,464,145 | ||||||||||||
| Loss from operations | (29,836,467 | ) | (13,699,045 | ) | (6,804,090 | ) | (79,210,676 | ) | ||||||||
| Interest income | 1,164,722 | 496,059 | 103,042 | 1,863,059 | ||||||||||||
| Loss on fair value of warrants | (660,028 | ) | (11,579,660 | ) | — | (12,239,688 | ) | |||||||||
| Interest expense | — | — | — | (179,090 | ) | |||||||||||
| Loss before income taxes | (29,331,773 | ) | (24,782,646 | ) | (6,701,048 | ) | (89,766,395 | ) | ||||||||
| Provision for income taxes | — | — | — | — | ||||||||||||
| Loss before cumulative effect of change in accounting principle | (29,331,773 | ) | (24,782,646 | ) | (6,701,048 | ) | (89,766,395 | ) | ||||||||
| Cumulative effect of change in accounting principle | — | — | — | (25,821 | ) | |||||||||||
| Net loss | (29,331,773 | ) | (24,782,646 | ) | (6,701,048 | ) | (89,792,216 | ) | ||||||||
| Preferred stock dividends | — | — | — | (621,240 | ) | |||||||||||
| Net loss applicable to common stock | $ | (29,331,773 | ) | $ | (24,782,646 | ) | $ | (6,701,048 | ) | $ | (90,413,456 | ) | ||||
| Loss per common share — basic and diluted | $ | (0.40 | ) | $ | (0.41 | ) | $ | (0.13 | ) | |||||||
| Weighted average shares outstanding — basic and diluted | 73,988,206 | 59,828,357 | 50,720,180 | |||||||||||||
See accompanying notes to consolidated financial statements.
F-4
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
(A Development Stage Enterprise)
Consolidated Statements of Stockholders’ Equity (Deficit)
Inception (June 12, 1996) through December 31, 2006
| Deficit | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Accumulated | accumulated | Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cumulative convertible | Cumulative convertible | Cumulative convertible | Additional | other | during the | Treasury | stockholders’ | |||||||||||||||||||||||||||||||||||||||||||||||||
| preferred stock, series A | preferred stock, series B | preferred stock, series C | Common stock | paid-in | comprehensive | development | stock, | equity | Comprehensive | |||||||||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | capital | loss | stage | at cost | (deficit) | loss | |||||||||||||||||||||||||||||||||||||||||||
| Balances at June 12, 1996 (date of incorporation) | — | $ | — | — | $ | — | — | $ | — | — | $ | — | $ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||||||||||||||||||||||||||
| Sale of common stock without par value | — | — | — | — | — | — | 503 | 5 | 5 | — | — | — | 10 | |||||||||||||||||||||||||||||||||||||||||||
| Change in par value of common stock | — | — | — | — | — | — | — | (4 | ) | 4 | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock and net liabilities assumed in acquisition | — | — | — | — | — | — | 1,716,132 | 1,716 | 3,224 | — | (18,094 | ) | — | (13,154 | ) | |||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock | — | — | — | — | — | — | 2,010,111 | 2,010 | 456 | — | (2,466 | ) | — | — | ||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (259,476 | ) | — | (259,476 | ) | $ | (259,476 | ) | ||||||||||||||||||||||||||||||||||||||
| Balances at December 31, 1996 | — | — | — | — | — | — | 3,726,746 | 3,727 | 3,689 | — | (280,036 | ) | — | (272,620 | ) | $ | (259,476 | ) | ||||||||||||||||||||||||||||||||||||||
| Sale of common stock, net of offering costs of $9,976 | — | — | — | — | — | — | 1,004,554 | 1,004 | 1,789,975 | — | — | — | 1,790,979 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock in acquisition | — | — | — | — | — | — | 375,891 | 376 | 887,874 | — | — | — | 888,250 | |||||||||||||||||||||||||||||||||||||||||||
| Minority interest deficiency at acquisition charged to the Company | — | — | — | — | — | — | — | — | — | — | (45,003 | ) | — | (45,003 | ) | |||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (1,979,400 | ) | — | (1,979,400 | ) | $ | (1,979,400 | ) | ||||||||||||||||||||||||||||||||||||||
| Balances at December 31, 1997 | — | — | — | — | — | — | 5,107,191 | 5,107 | 2,681,538 | — | (2,304,439 | ) | — | 382,206 | $ | (1,979,400 | ) | |||||||||||||||||||||||||||||||||||||||
| Rescission of acquisition | — | — | — | — | — | — | (375,891 | ) | (376 | ) | (887,874 | ) | — | 561,166 | — | (327,084 | ) | |||||||||||||||||||||||||||||||||||||||
| Issuance of common stock at conversion of notes payable | — | — | — | — | — | — | 450,264 | 451 | 363,549 | — | — | — | 364,000 | |||||||||||||||||||||||||||||||||||||||||||
| Expense related to stock warrants issued | — | — | — | — | — | — | — | — | 260,000 | — | — | — | 260,000 | |||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (1,204,380 | ) | — | (1,204,380 | ) | $ | (1,204,380 | ) | ||||||||||||||||||||||||||||||||||||||
| Balances at December 31, 1998 | — | — | — | — | — | — | 5,181,564 | 5,182 | 2,417,213 | — | (2,947,653 | ) | — | (525,258 | ) | $ | (1,204,380 | ) | ||||||||||||||||||||||||||||||||||||||
| Sale of common stock | — | — | — | — | — | — | 678,412 | 678 | 134,322 | — | — | — | 135,000 | |||||||||||||||||||||||||||||||||||||||||||
| Expense related to stock warrants issued | — | — | — | — | — | — | — | — | 212,000 | — | — | — | 212,000 | |||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (1,055,485 | ) | — | (1,055,485 | ) | $ | (1,055,485 | ) | ||||||||||||||||||||||||||||||||||||||
| Balances at December 31, 1999 | — | — | — | — | — | — | 5,859,976 | 5,860 | 2,763,535 | — | (4,003,138 | ) | — | (1,233,743 | ) | $ | (1,055,485 | ) | ||||||||||||||||||||||||||||||||||||||
| Sale of preferred stock, net of offering costs of $76,500 | 3,200 | 32 | — | — | — | — | — | — | 3,123,468 | — | — | — | 3,123,500 | |||||||||||||||||||||||||||||||||||||||||||
See accompanying notes to consolidated financial statements.
F-5
Table of Contents
| Deficit | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Accumulated | accumulated | Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cumulative convertible | Cumulative convertible | Cumulative convertible | Additional | other | during the | Treasury | stockholders’ | |||||||||||||||||||||||||||||||||||||||||||||||||
| preferred stock, series A | preferred stock, series B | preferred stock, series C | Common stock | paid-in | comprehensive | development | stock, | equity | Comprehensive | |||||||||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | capital | loss | stage | at cost | (deficit) | loss | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock at conversion of notes and interest payable | — | — | — | — | — | — | 412,487 | 412 | 492,085 | — | — | — | 492,497 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock at conversion of notes payable | — | — | — | — | — | — | 70,354 | 70 | 83,930 | — | — | — | 84,000 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock to settle obligations | — | — | — | — | — | — | 495,111 | 496 | 1,201,664 | — | — | — | 1,202,160 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock for acquisition | — | — | — | — | — | — | 6,999,990 | 7,000 | 9,325,769 | — | — | — | 9,332,769 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of warrants for acquisition | — | — | — | — | — | — | — | — | 4,767,664 | — | — | — | 4,767,664 | |||||||||||||||||||||||||||||||||||||||||||
| Stock issued for acquisition costs | — | — | — | — | — | — | 150,000 | 150 | 487,350 | — | — | — | 487,500 | |||||||||||||||||||||||||||||||||||||||||||
| Expense related to stock warrants issued | — | — | — | — | — | — | — | — | 140,000 | — | — | — | 140,000 | |||||||||||||||||||||||||||||||||||||||||||
| Dividends payable on preferred stock | — | — | — | — | — | — | — | — | (85,000 | ) | — | — | — | (85,000 | ) | |||||||||||||||||||||||||||||||||||||||||
| Cashless exercise of warrants | — | — | — | — | — | — | 599,066 | 599 | (599 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (3,701,084 | ) | — | (3,701,084 | ) | $ | (3,701,084 | ) | ||||||||||||||||||||||||||||||||||||||
| Balances at December 31, 2000 | 3,200 | 32 | — | — | — | — | 14,586,984 | 14,587 | 22,299,866 | — | (7,704,222 | ) | — | 14,610,263 | $ | (3,701,084 | ) | |||||||||||||||||||||||||||||||||||||||
| Dividends payable on preferred stock | — | — | — | — | — | — | — | — | (256,000 | ) | — | — | — | (256,000 | ) | |||||||||||||||||||||||||||||||||||||||||
| Repurchase of warrants | — | — | — | — | — | — | — | — | (55,279 | ) | — | — | — | (55,279 | ) | |||||||||||||||||||||||||||||||||||||||||
| Sale of warrants | — | — | — | — | — | — | — | — | 47,741 | — | — | — | 47,741 | |||||||||||||||||||||||||||||||||||||||||||
| Cashless exercise of warrants | — | — | — | — | — | — | 218,493 | 219 | (219 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock to pay preferred dividends | — | — | — | — | — | — | 93,421 | 93 | 212,907 | — | — | — | 213,000 | |||||||||||||||||||||||||||||||||||||||||||
| Detachable warrants issued with notes payable | — | — | — | — | — | — | — | — | 450,000 | — | — | — | 450,000 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of warrants to pay operating expenses | — | — | — | — | — | — | — | — | 167,138 | — | — | — | 167,138 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock to pay operating expenses | — | — | — | — | — | — | 106,293 | 106 | 387,165 | — | — | — | 387,271 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of preferred stock to pay operating expenses | 137 | 1 | — | — | — | — | — | — | 136,499 | — | — | — | 136,500 | |||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (16,339,120 | ) | — | (16,339,120 | ) | $ | (16,399,120 | ) | ||||||||||||||||||||||||||||||||||||||
| Balances at December 31, 2001 | 3,337 | 33 | — | — | — | — | 15,005,191 | 15,005 | 23,389,818 | — | (24,043,342 | ) | — | (638,486 | ) | $ | (16,399,120 | ) | ||||||||||||||||||||||||||||||||||||||
| Dividends payable on preferred stock | — | — | — | — | — | — | — | — | (242,400 | ) | — | — | — | (242,400 | ) | |||||||||||||||||||||||||||||||||||||||||
| Repurchase of warrants | — | — | — | — | — | — | — | — | — | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||||
| Sale of warrants | — | — | — | — | — | — | 240,000 | 240 | 117,613 | — | — | — | 117,853 | |||||||||||||||||||||||||||||||||||||||||||
| Cashless exercise of warrants | — | — | — | — | — | — | 100,201 | 100 | (100 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||
See accompanying notes to consolidated financial statements.
F-6
Table of Contents
| Deficit | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Accumulated | accumulated | Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cumulative convertible | Cumulative convertible | Cumulative convertible | Additional | other | during the | Treasury | stockholders’ | |||||||||||||||||||||||||||||||||||||||||||||||||
| preferred stock, series A | preferred stock, series B | preferred stock, series C | Common stock | paid-in | comprehensive | development | stock, | equity | Comprehensive | |||||||||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | capital | loss | stage | at cost | (deficit) | loss | |||||||||||||||||||||||||||||||||||||||||||
| Exercise of warrants | — | — | — | — | — | — | 344,573 | 345 | 168,477 | — | — | — | 168,822 | |||||||||||||||||||||||||||||||||||||||||||
| Sale of preferred stock at $1.50 per share | — | — | 200,000 | 2,000 | — | — | — | — | 298,000 | — | — | — | 300,000 | |||||||||||||||||||||||||||||||||||||||||||
| Sale of preferred stock at $10.00 per share | — | — | — | — | 70,109 | 701 | — | — | 700,392 | — | — | — | 701,093 | |||||||||||||||||||||||||||||||||||||||||||
| Conversion of preferred stock into common stock | (3,000 | ) | (30 | ) | — | — | — | — | 1,800,000 | 1,800 | (1,770 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||
| Preferred stock dividends forgiven | — | — | — | — | — | — | — | — | 335,440 | — | — | — | 335,440 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of warrants to pay operating expenses | — | — | — | — | — | — | — | — | 163,109 | — | — | — | 163,109 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock to pay operating expenses | — | — | — | — | — | — | 6,292 | 6 | 12,263 | — | — | — | 12,269 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of preferred stock to pay operating expenses | 136 | 1 | — | — | — | — | — | — | 6,000 | — | — | — | 6,001 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of stock options to employees | — | — | — | — | — | — | — | — | 329,296 | — | — | — | 329,296 | |||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (2,105,727 | ) | — | (2,105,727 | ) | $ | (2,105,727 | ) | ||||||||||||||||||||||||||||||||||||||
| Balances at December 31, 2002 | 473 | 4 | 200,000 | 2,000 | 70,109 | 701 | 17,496,257 | 17,496 | 25,276,138 | — | (26,149,069 | ) | — | (852,730 | ) | $ | (2,105,727 | ) | ||||||||||||||||||||||||||||||||||||||
| Dividends payable on preferred stock | — | — | — | — | — | — | — | — | (37,840 | ) | — | — | — | (37,840 | ) | |||||||||||||||||||||||||||||||||||||||||
| Conversion of Series C preferred stock into common stock | — | — | — | — | (70,109 | ) | (701 | ) | 14,021,860 | 14,022 | (13,321 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock to pay interest on Bridge Notes | — | — | — | — | — | — | 165,830 | 165 | 53,326 | — | — | — | 53,491 | |||||||||||||||||||||||||||||||||||||||||||
| Sale of common stock at $0.40 per share, net of issuance costs | — | — | — | — | — | — | 6,640,737 | 6,676 | 2,590,656 | — | — | — | 2,597,332 | |||||||||||||||||||||||||||||||||||||||||||
| Sale of common stock at $1.00 per share, net of issuance costs | — | — | — | — | — | — | 3,701,733 | 3,668 | 3,989,181 | — | — | — | 3,992,849 | |||||||||||||||||||||||||||||||||||||||||||
| Exchange of warrants | — | — | — | — | — | — | 235,291 | 235 | 49,486 | — | — | — | 49,721 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock to pay operating expenses | — | — | — | — | — | — | 230,000 | 230 | 206,569 | — | — | — | 206,799 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of warrants to pay operating expenses | — | — | — | — | — | — | — | — | 156,735 | — | — | — | 156,735 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of stock options to employees | — | — | — | — | — | — | — | — | 286,033 | — | — | — | 286,033 | |||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (2,332,077 | ) | — | (2,332,077 | ) | $ | (2,332,077 | ) | ||||||||||||||||||||||||||||||||||||||
| Balances at December 31, 2003 | 473 | 4 | 200,000 | 2,000 | — | — | 42,491,708 | 42,492 | 32,556,963 | — | (28,481,146 | ) | — | 4,120,313 | $ | (2,332,077 | ) | |||||||||||||||||||||||||||||||||||||||
See accompanying notes to consolidated financial statements.
F-7
Table of Contents
| Deficit | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Accumulated | accumulated | Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cumulative convertible | Cumulative convertible | Cumulative convertible | Additional | other | during the | Treasury | stockholders’ | |||||||||||||||||||||||||||||||||||||||||||||||||
| preferred stock, series A | preferred stock, series B | preferred stock, series C | Common stock | paid-in | comprehensive | development | stock, | equity | Comprehensive | |||||||||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | capital | loss | stage | at cost | (deficit) | loss | |||||||||||||||||||||||||||||||||||||||||||
| Extinguishment of dividends payable on preferred stock | — | — | — | — | — | — | — | — | 72,800 | — | — | — | 72,800 | |||||||||||||||||||||||||||||||||||||||||||
| Conversion of Series A cumulative preferred stock | (473 | ) | (4 | ) | — | — | — | — | 236,500 | 236 | (232 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||
| Conversion of Series B preferred stock | — | — | (200,000 | ) | (2,000 | ) | — | — | 200,000 | 200 | 1,800 | — | — | — | — | |||||||||||||||||||||||||||||||||||||||||
| Cashless exercise of warrants | — | — | — | — | — | — | 464,573 | 465 | (465 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||
| Exercise of warrants | — | — | — | — | — | — | 23,832 | 23 | 27,330 | — | — | — | 27,353 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of warrants in settlement of a claim | — | — | — | — | — | — | — | — | 86,375 | — | — | — | 86,375 | |||||||||||||||||||||||||||||||||||||||||||
| Sale of common stock at $1.50 per share | — | — | — | — | — | — | 10,417,624 | 10,419 | 15,616,031 | — | — | — | 15,626,450 | |||||||||||||||||||||||||||||||||||||||||||
| Payment of financing and offering costs | — | — | — | — | — | — | — | — | (1,366,774 | ) | — | — | — | (1,366,774 | ) | |||||||||||||||||||||||||||||||||||||||||
| Issuance of stock options to employees | — | — | — | — | — | — | — | — | 524,922 | — | — | — | 524,922 | |||||||||||||||||||||||||||||||||||||||||||
| Acquisition of treasury stock | — | — | — | — | — | — | — | — | 34,747 | — | — | (34,747 | ) | — | ||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (6,701,048 | ) | — | (6,701,048 | ) | $ | (6,701,048 | ) | ||||||||||||||||||||||||||||||||||||||
| Balances at December 31, 2004 | — | — | — | — | — | — | 53,834,237 | 53,835 | 47,553,497 | — | (35,182,194 | ) | (34,747 | ) | 12,390,391 | $ | (6,701,048 | ) | ||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (24,782,646 | ) | — | (24,782,646 | ) | $ | (24,782,646 | ) | ||||||||||||||||||||||||||||||||||||||
| Effect of change in fair value of available for sale securities | — | — | — | — | — | — | — | — | — | (1,722 | ) | — | — | (1,722 | ) | (1,722 | ) | |||||||||||||||||||||||||||||||||||||||
| Par value of shares issued in conjunction with mezzanine financing | — | — | — | — | — | — | 10,810,809 | 10,811 | (10,811 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||
| Cashless exercise of warrants | — | — | — | — | — | — | 149,613 | 149 | (149 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||
| Exercise of warrants | — | — | — | — | — | — | 2,258,703 | 2,259 | 3,071,179 | — | — | — | 3,073,438 | |||||||||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | — | — | 185,000 | 185 | 144,815 | — | — | — | 145,000 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of stock options to employees | — | — | — | — | — | — | — | — | 994,874 | — | — | — | 994,874 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of stock options to non-employee | — | — | — | — | — | — | — | 93,549 | — | — | — | 93,549 | ||||||||||||||||||||||||||||||||||||||||||||
| Issuance of common stock to vendor | — | — | — | — | — | — | 125,000 | 125 | 258,375 | — | — | — | 258,500 | |||||||||||||||||||||||||||||||||||||||||||
| Balances at December 31, 2005 | — | — | — | — | — | — | 67,363,362 | 67,364 | 52,105,329 | (1,722 | ) | (59,964,840 | ) | (34,747 | ) | (7,828,616 | ) | $ | (24,784,368 | ) | ||||||||||||||||||||||||||||||||||||
See accompanying notes to consolidated financial statements.
F-8
Table of Contents
| Deficit | ||||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Accumulated | accumulated | Total | ||||||||||||||||||||||||||||||||||||||||||||||||||||||
| Cumulative convertible | Cumulative convertible | Cumulative convertible | Additional | other | during the | Treasury | stockholders’ | |||||||||||||||||||||||||||||||||||||||||||||||||
| preferred stock, series A | preferred stock, series B | preferred stock, series C | Common stock | paid-in | comprehensive | development | stock, | equity | Comprehensive | |||||||||||||||||||||||||||||||||||||||||||||||
| Shares | Amount | Shares | Amount | Shares | Amount | Shares | Amount | capital | loss | stage | at cost | (deficit) | loss | |||||||||||||||||||||||||||||||||||||||||||
| Net loss | — | — | — | — | — | — | — | — | — | — | (29,331,773 | ) | — | (29,331,773 | ) | $ | (29,331,773 | ) | ||||||||||||||||||||||||||||||||||||||
| Effect of change in fair value of available for sale securities | — | — | — | — | — | — | — | — | — | (368 | ) | — | — | (368 | ) | (368 | ) | |||||||||||||||||||||||||||||||||||||||
| Cashless exercise of warrants | — | — | — | — | — | — | 420,161 | 420 | (420 | ) | — | — | — | — | ||||||||||||||||||||||||||||||||||||||||||
| Exercise of warrants, net of financing costs | — | — | — | — | — | — | 5,103,746 | 5,104 | 7,686,486 | — | — | — | 7,691,590 | |||||||||||||||||||||||||||||||||||||||||||
| Acquisition of SD Pharmaceuticals. Inc. | — | — | — | — | — | — | 2,099,990 | 2,100 | 10,161,852 | — | — | — | 10,163,952 | |||||||||||||||||||||||||||||||||||||||||||
| Sale of common stock at $2.75 per share, net of offering costs | — | — | — | — | — | — | 14,545,000 | 14,545 | 37,055,666 | — | — | — | 37,070,211 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of stock for severance agreement | — | — | — | — | — | — | 60,145 | 60 | 196,614 | — | — | — | 196,674 | |||||||||||||||||||||||||||||||||||||||||||
| Exercise of stock options | — | — | — | — | — | — | 92,500 | 93 | 125,658 | — | — | — | 125,751 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of restricted stock to non-employees | — | — | — | — | — | — | 15,000 | 15 | 68,635 | — | — | — | 68,650 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of stock options to employees | — | — | — | — | — | — | — | — | 1,697,452 | — | — | — | 1,697,452 | |||||||||||||||||||||||||||||||||||||||||||
| Issuance of stock options to non-employee | — | — | — | — | — | — | — | — | 104,225 | — | — | — | 104,225 | |||||||||||||||||||||||||||||||||||||||||||
| Cancellation of treasury stock shares | — | — | — | — | — | — | (23,165 | ) | (23 | ) | (34,724 | ) | — | — | 34,747 | — | ||||||||||||||||||||||||||||||||||||||||
| Balances at December 31, 2006 | — | $ | — | — | $ | — | — | $ | — | 89,676,739 | $ | 89,678 | $ | 109,166,773 | $ | (2,090 | ) | $ | (89,296,613 | ) | $ | — | $ | 19,957,748 | $ | (29,332,141 | ) | |||||||||||||||||||||||||||||
See accompanying notes to consolidated financial statements.
F-9
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
(A Development Stage Enterprise)
Consolidated Statements of Cash Flows
| Inception | ||||||||||||||||
| (June 12, 1996) | ||||||||||||||||
| through | ||||||||||||||||
| Years ended December 31, | December 31, | |||||||||||||||
| 2006 | 2005 | 2004 | 2006 | |||||||||||||
Cash flows from operating activities: | ||||||||||||||||
| Net loss | $ | (29,331,773 | ) | $ | (24,782,646 | ) | $ | (6,701,048 | ) | $ | (89,792,216 | ) | ||||
| Adjustments to reconcile net loss to net cash used in operating activities: | ||||||||||||||||
| Depreciation and amortization | 176,688 | 115,545 | 41,309 | 9,982,249 | ||||||||||||
| Loss on fair value of warrant liability | 660,028 | 11,579,660 | — | 12,239,688 | ||||||||||||
| Amortization of debt discount | — | — | — | 450,000 | ||||||||||||
| Forgiveness of employee receivable | — | — | — | 30,036 | ||||||||||||
| Impairment loss — write-off of goodwill | — | — | — | 5,702,130 | ||||||||||||
| Expenses paid by issuance of warrants | — | — | 86,375 | 573,357 | ||||||||||||
| Expenses paid by issuance of preferred stock | — | — | — | 142,501 | ||||||||||||
| Expenses paid by issuance of common stock | 343,658 | 101,833 | — | 1,263,039 | ||||||||||||
| Expenses related to stock warrants issued | — | — | — | 612,000 | ||||||||||||
| Expenses related to employee stock options issued | 1,697,452 | 994,874 | 524,922 | 3,832,577 | ||||||||||||
| Expenses related to options issued to non-employees | 104,225 | 93,549 | — | 197,774 | ||||||||||||
| Equity in loss of investee | — | — | — | 178,936 | ||||||||||||
| In-process research and development | 10,422,130 | — | — | 10,422,130 | ||||||||||||
| Write-off of license agreement | — | — | — | 152,866 | ||||||||||||
| Write-off assets available for sale | — | 108,000 | — | 108,000 | ||||||||||||
| Cumulative effect of change in accounting principle | — | — | — | 25,821 | ||||||||||||
| Accretion of discount on investments in securities | (242,681 | ) | (111,960 | ) | — | (354,641 | ) | |||||||||
| Changes in assets and liabilities, net of effect of acquisitions: | ||||||||||||||||
| Increase in prepaid and other assets | (107,151 | ) | (281,266 | ) | (255,101 | ) | (819,006 | ) | ||||||||
| Increase in accounts payable and accrued liabilities | 525,284 | 478,504 | 1,128,153 | 2,625,231 | ||||||||||||
| Increase (decrease) in long-term liabilities | (21,404 | ) | 57,078 | — | 35,674 | |||||||||||
| Net cash used in operating activities | (15,773,544 | ) | (11,646,829 | ) | (5,175,390 | ) | (42,391,854 | ) | ||||||||
Cash flows from investing activities: | ||||||||||||||||
| Purchase of certificate of deposit | — | — | — | (1,016,330 | ) | |||||||||||
| Maturity of certificate of deposit | — | — | — | 1,016,330 | ||||||||||||
| Purchases of property and equipment | (172,112 | ) | (237,785 | ) | (305,773 | ) | (838,139 | ) | ||||||||
| Purchases of short-term investments | (32,600,411 | ) | (13,123,220 | ) | — | (45,723,631 | ) | |||||||||
| Proceeds from sales and maturities of short-term investments | 15,029,776 | 5,275,000 | — | 20,304,776 | ||||||||||||
| Cash paid for acquisitions, net of cash acquired | (31,838 | ) | — | — | 32,395 | |||||||||||
| Payment on obligation under license agreement | — | — | — | (106,250 | ) | |||||||||||
| Issuance of note receivable — related party | — | — | — | (35,000 | ) | |||||||||||
| Payments on note receivable | — | — | — | 405,993 | ||||||||||||
| Advance to investee | — | — | — | (90,475 | ) | |||||||||||
| Cash transferred in rescission of acquisition | — | — | — | (19,475 | ) | |||||||||||
| Cash received in rescission of acquisition | — | — | — | 230,000 | ||||||||||||
| Net cash used in investing activities | (17,774,585 | ) | (8,086,005 | ) | (305,773 | ) | (25,839,806 | ) | ||||||||
Cash flows from financing activities: | ||||||||||||||||
| Proceeds from sale of common stock | 39,998,749 | 19,999,997 | 15,626,450 | 84,151,342 | ||||||||||||
| Proceeds from exercise of stock options | 125,751 | 145,000 | — | 270,751 | ||||||||||||
| Proceeds from sale or exercise of warrants | 7,897,866 | 3,073,438 | 27,353 | 11,382,894 | ||||||||||||
| Proceeds from sale of preferred stock | — | — | — | 4,200,993 | ||||||||||||
| Repurchase of warrants | — | — | — | (55,279 | ) | |||||||||||
| Payments for financing and offering costs | (3,134,814 | ) | (1,883,246 | ) | (1,366,774 | ) | (6,483,809 | ) | ||||||||
F-10
Table of Contents
| Inception | ||||||||||||||||
| (June 12, 1996) | ||||||||||||||||
| through | ||||||||||||||||
| Years ended December 31, | December 31, | |||||||||||||||
| 2006 | 2005 | 2004 | 2006 | |||||||||||||
| Payments on notes payable and long-term debt | — | — | — | (605,909 | ) | |||||||||||
| Proceeds from issuance of notes payable and detachable warrants | — | — | — | 1,344,718 | ||||||||||||
| Net cash provided by financing activities | 44,887,552 | 21,335,189 | 14,287,029 | 94,205,701 | ||||||||||||
| Net increase in cash and cash equivalents | 11,339,423 | 1,602,355 | 8,805,866 | 25,974,041 | ||||||||||||
| Cash and cash equivalents at beginning of period | 14,634,618 | 13,032,263 | 4,226,397 | — | ||||||||||||
| Cash and cash equivalents at end of period | $ | 25,974,041 | $ | 14,634,618 | $ | 13,032,263 | $ | 25,974,041 | ||||||||
See accompanying notes to consolidated financial statements.
F-11
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements
December 31, 2006
| (1) | Description of Business | |
| ADVENTRX Pharmaceuticals, Inc., a Delaware corporation (“ADVENTRX,” “we” or the “Company”), is a biopharmaceutical research and development company focused on commercializing proprietary product candidates for the treatment of cancer and infectious diseases. Our business is in the development stage; we have not yet marketed any products or generated any significant revenue. Through our license agreements with the University of Southern California (“USC”) and our acquisition of SD Pharmaceuticals, Inc. (“SDP”), we have rights to product candidates in varying stages of development. | ||
| In October 2000, we merged our wholly-owned subsidiary, Biokeys Acquisition Corp., with and into Biokeys, Inc. and changed our name to Biokeys Pharmaceuticals, Inc. In May 2003, we merged Biokeys Inc., our wholly-owned subsidiary, with and into us and changed our name to ADVENTRX Pharmaceuticals, Inc. The merger had no effect on our financial statements. In July 2004, we formed a wholly-owned subsidiary, ADVENTRX (Europe) Ltd., in the United Kingdom primarily to facilitate conducting clinical trials in the European Union. In April 2006, we acquired all of the outstanding capital stock of SDP through a merger with our newly created wholly-owned subsidiary, Speed Acquisition, Inc. (the “Merger Sub”) and changed the name of the Merger Sub to SD Pharmaceuticals, Inc. | ||
| (2) | Summary of Significant Accounting Policies | |
| Basis of Presentation | ||
| The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiaries, SDP and ADVENTRX (Europe) Ltd. All intercompany accounts and transactions have been eliminated in consolidation. Certain amounts in the prior year financial statements have been reclassified to conform to the current year presentation. | ||
| Use of Estimates | ||
| The preparation of financial statements in conformity with accounting principles generally accepted in the U. S. requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and accompanying notes. Actual results could differ from those estimates. | ||
| Purchase Price Allocation | ||
| The allocation of purchase price for an acquisition requires extensive use of accounting estimates and judgments in allocating the purchase price to the identifiable tangible and intangible assets acquired, including in-process research and development, and liabilities assumed based on their respective fair values. In 2006, we completed the acquisition of SDP. See Note 3,Acquisition of SDP, for a detailed discussion. | ||
| Cash Equivalents | ||
| Cash equivalents consist of highly liquid investments with original maturities of three months or less at the date of purchase. | ||
| Short-term Investments | ||
| We account for and report our investments in accordance with Statement of Financial Accounting Standards (“FAS”) No. 115,Accounting for Certain Investments in Debt and Equity Securities. Investments are comprised of marketable securities consisting primarily of certificates of deposit, federal, state and municipal government obligations and corporate bonds. Short-term investments are marketable securities with maturities of less than one year from the balance sheet date. All marketable securities are held in our name and primarily under the custodianship of two major financial institutions. Our policy is to protect the principal value of our investment portfolio and minimize principal risk. |
F-12
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| Our marketable securities are classified as “available-for-sale” and stated at fair value, with net unrealized gains or losses recorded as a component of accumulated other comprehensive income (loss). The amortized cost of debt securities is adjusted for amortization of premiums and accretion of discounts to maturity with all amortization and accretion included in interest income. Realized gains and losses on available-for-sale securities are included in other income (loss). The cost of securities sold is based on the specific identification method. Interest on securities classified as available-for-sale is included in interest income. Marketable securities are evaluated periodically for impairment. If it is determined that a decline of any investment is other than temporary, then the investment basis would be written down to fair value and the write-down would be included in earnings as a loss. | ||
| Concentrations | ||
| Financial instruments that potentially subject us to concentrations of credit risk consist principally of cash, cash equivalents and investment securities. Our cash and cash equivalents are in excess of the Federal Deposit Insurance Corporation limit at year end. We invest our excess cash primarily in marketable debt securities of corporations, financial institutions and government agencies with strong credit ratings. We have adopted an investment policy that includes guidelines related to diversification and maturities to maintain safety and liquidity. | ||
| During 2006 and 2005, approximately 16% and 20%, respectively, of our total vendor payments were made to a contract research organization that is assisting us in our clinical trial administration and data management. If we were to lose this vendor, we could experience delays in continuing our clinical trial efforts which would result in increased costs as well as delays in obtaining FDA approvals. | ||
| Fair Value of Financial Instruments | ||
| At December 31, 2006 and 2005, our financial instruments included cash and cash equivalents, short-term investments, accounts payable, accrued expenses, accrued compensation and payroll taxes and warrant liability. The carrying amounts of cash and cash equivalents, accounts payable, accrued expenses and accrued compensation and payroll taxes approximate fair value due to the short-term maturities of these instruments. Our short-term investments in securities are carried at fair value based on quoted market prices. Warrant liability is carried at fair value of the underlying shares. | ||
| Property and Equipment | ||
| Property and equipment are stated at cost. Depreciation and amortization are calculated using the straight-line method over the estimated useful lives of the assets. The costs of improvements that extend the lives of the assets are capitalized. Repairs and maintenance are expensed as incurred. | ||
| Impairment of Long-lived Assets | ||
| Long-lived assets with finite lives are evaluated for impairment whenever events or changes in circumstances indicate that their carrying value may not be recoverable. If the review indicates that intangibles or long-lived assets are not recoverable (i.e. carrying amount is less than the future projected undiscounted cash flows), their carrying amount would be reduced to fair value. Since inception through December 31, 2006, we recognized an impairment loss of the value of goodwill in the amount of $5.7 million, which was recorded in the year ended December 31, 2001. |
F-13
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| Revenue Recognition | ||
| We recognize revenue in accordance with the Securities and Exchange Commission’s (“SEC”) Staff Accounting Bulletin Topic 13,Revenue Recognition(“Topic 13”) and Emerging Issues Task Force Issue (“EITF”) No. 00-21,Revenue Arrangements with Multiple Deliverables. Revenue is recognized when all of the following criteria are met: (1) persuasive evidence of an arrangement exists; (2) delivery has occurred or services have been rendered; (3) the seller’s price to the buyer is fixed and determinable; and (4) collectibility is reasonably assured. | ||
| Revenue from licensing agreements is recognized based on the performance requirements of the agreement. Revenue is deferred for fees received before earned. Nonrefundable upfront fees that are not contingent on any future performance by us are recognized as revenue when revenue recognition criteria are met and the license term commences. Nonrefundable upfront fees, where we have an ongoing involvement or performance obligations, are recorded as deferred revenue and recognized as revenue over the life of the contract, the period of the performance obligation or the development period, whichever is appropriate in light of the circumstances. | ||
| Payments related to substantive, performance-based milestones in a collaborative agreement are recognized as revenue upon the achievement of the milestones as specified in the underlying agreements when they represent the culmination of the earnings process. Royalty revenue from licensed products will be recognized when earned in accordance with the terms of the license agreements. | ||
| Recognition of Expenses in Research Contracts | ||
| Pursuant to management’s assessment of the services that have been performed on clinical trials and other contracts, we recognize expenses as the services are provided. Such management assessments generally consist of, but are not limited to, an evaluation by the project manager of the work that has been completed during the period, measurement of progress prepared internally and/or provided by the third-party service provider, analysis of data that justifies the progress, and finally, management’s judgment. Several of our contracts extend across multiple reporting periods, including our largest contract, representing a $9.0 million clinical trial contract as of December 31, 2006. A 3% variance in our estimate of the work completed in our largest contract could increase or decrease our operating expenses by approximately $270,000. | ||
| Research and Development Costs | ||
| All research and development costs are expensed as incurred, including Company-sponsored research and development and costs of technology rights under license agreements that have no alternative future use when incurred. | ||
| License fees. Payments made in connection with in-licensed technology or product candidates are expensed as incurred when there is uncertainty in receiving future economic benefits from the licensed technology or product candidates. We consider the future economic benefits from the licensed technology or product candidates to be uncertain until such licensed technology or product candidates are approved by the FDA or when other significant risk factors are abated. For expense accounting purposes, management has determined future economic benefits for all of our licensed technology or product candidates to be uncertain. | ||
| Purchased In-Process Research and Development | ||
| In accordance with FAS No. 141,Business Combinations, we immediately charge the costs associated with purchased in-process research and development (“IPR&D”) to statement of operations upon acquisition. These amounts represent an estimate of the fair value of purchased IPR&D for projects that, as of the acquisition date, had not yet reached technological feasibility, had no alternative future use and had uncertainty in receiving future economic benefits from the purchased IPR&D. We determine the future economic benefits from the purchased IPR&D to be uncertain until such technology is approved by the FDA or when other significant risk factors are abated. We incurred significant IPR&D expense related to the SDP acquisition. | ||
| Accounting for Share-Based Compensation | ||
| Effective January 1, 2006, we adopted the provisions of revised FAS No. 123,Share-Based Payment(“FAS 123R”), including the provisions of Staff Accounting Bulleting No. 107 (“SAB 107”). Under FAS 123R, share-based compensation cost is measured at the grant date, based on the estimated fair value of the award, and is |
F-14
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| recognized as expense over the employee’s requisite service period. We have no awards with market or performance conditions. We adopted the provisions of FAS 123R using the modified prospective transition method. Accordingly, prior periods have not been revised for comparative purposes. | ||
| On November 10, 2005, the Financial Accounting Standards Board (“FASB”) issued FASB Staff Position No. FAS 123(R)-3,Transition Election Related to Accounting for Tax Effects of Share-Based Payment Awards. We have elected to adopt the alternative transition method provided in FAS 123R. The alternative transition method includes a simplified method to establish the beginning balance of the additional paid-in capital pool (“APIC pool”) related to the tax effects of employee share-based compensation, which is available to absorb tax deficiencies recognized subsequent to the adoption of FAS 123R. | ||
| The valuation provisions of FAS 123R apply to new awards and to awards that are outstanding on the effective date, January 1, 2006, which are subsequently modified or cancelled. Prior to 2006, we accounted for share-based compensation under the recognition and measurement principles of FAS No. 123,Accounting for Stock-Based Compensation(“FAS 123”).Estimated compensation expense for awards outstanding at January 1, 2006 is recognized over the remaining service period using the compensation cost calculated for recognition purposes under FAS 123. | ||
| Share-based compensation expense recognized in our consolidated statement of operations for the year ended December 31, 2006 included compensation expense for share-based payment awards granted prior to, but not yet vested as of, December 31, 2005 based on the grant date fair value estimated in accordance with the recognition provisions of FAS 123 and share-based payment awards granted subsequent to December 31, 2005 based on the grant date fair value estimated in accordance with FAS 123R. For share awards granted during the year ended December 31, 2006, expenses are amortized under the straight-line method. For share awards granted prior to 2006, expenses are amortized under the straight-line method prescribed by FAS 123. As share-based compensation expense recognized in the consolidated statement of operations for the year ended December 31, 2006 is based on awards ultimately expected to vest, it has been reduced for estimated forfeitures. FAS 123R requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. In the years ended December 31, 2005 and 2004, we accounted for forfeitures as they occurred in accordance with the recognition provisions of FAS 123. | ||
| We account for share-based compensation awards granted to non-employees in accordance with EITF No. 96-18,Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services(“EITF 96-18”). Under EITF 96-18, we determine the fair value of the share-based compensation awards granted as either the fair value of the consideration received or the fair value of the equity instruments issued, whichever is more reliably measurable. If the fair value of the equity instruments issued is used, it is measured using the stock price and other measurement assumptions as of the earlier of either of (1) the date at which a commitment for performance by the counterparty to earn the equity instruments is reached or (2) the date at which the counterparty’s performance is complete. | ||
| Registration Payment Arrangement | ||
| We account for contingent obligations in a registration payment arrangement in accordance with EITF Issue No. 00-19,Accounting for Derivative Financial Instruments Indexed To, and Potentially Settled in a Company’s Own Stock(“EITF 00-19”), and the SEC’s December 2005 interpretation. In connection with a sale of shares of our common stock in July 2005, we entered into a registration payment arrangement which requires us to use our best efforts to (a) file a registration statement with the SEC, (b) have it declared effective by the end of a certain period and (c) maintain effectiveness of the registration statement for a certain period of time. See Note 7,Warrant Liability, for a detailed discussion. In the event we fail to meet the registration requirements, the arrangement requires us to make payments to the purchasers until the registration payment obligations no longer exist. |
F-15
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| Because the arrangement requires payments to be settled in cash, we recorded the fair value of the arrangement as a liability, with an offsetting reduction to additional paid-in capital as of the closing date of the sale. At the end of each reporting period, the value of the arrangement will be re-measured based on the fair market value of the underlying shares, and changes to the liability and related gain or loss will be made appropriately. In addition, the shares issued that are subject to the registration payment arrangement are reported as temporary equity. The liability and temporary equity will be reclassified to equity when the registration payment obligations no longer exist. | ||
| In December 2006, the FASB issued FASB Staff Position on No. EITF 00-19-2,Accounting for Registration Payment Arrangements(“FSP EITF 00-19-2”). FSP EITF 00-19-2 provides that the contingent obligation to make future payments or otherwise transfer consideration under a registration payment arrangement should be separately recognized and measured in accordance with FAS No. 5,Accounting for Contingencies,which defines that loss contingencies should be recognized as liabilities if they are probable and reasonably estimable. The guidance in FSP EITF 00-19-2 is effective immediately for registration payment arrangements and the financial instruments subject to those arrangements that are entered into or modified subsequent to the date of issuance of FSP EITF 00-19-2. For registration payment arrangements and financial instruments subject to those arrangements that were entered into prior to the issuance of FSP EITF 00-19-2, this guidance shall be effective for financial statements issued for fiscal years beginning after December 15, 2006, and interim periods within those fiscal years. | ||
| Effective January 1, 2007, we will apply new guidance under FSP EITF 00-19-2 to account for this registration payment arrangement. We are in the process of evaluating the impact of FSP EITF 00-19-2 on our consolidated financial position and results of operations. | ||
| Patent Costs | ||
| Legal costs in connection with approved patents and patent applications are expensed as incurred and classified as selling, general and administrative expense in our consolidated statement of operations. | ||
| Income Taxes | ||
| We account for income taxes and the related accounts under the liability method. Deferred tax assets and liabilities are determined based on the differences between the financial statement carrying amounts and the income tax bases of assets and liabilities. A valuation allowance is applied against any net deferred tax asset if, based on available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. | ||
| Comprehensive Loss | ||
| Comprehensive loss is defined as the change in equity of a business enterprise during a period from transactions and other events and circumstances from non-owner sources, including foreign currency translation adjustments and unrealized gains and losses on marketable securities. We present an accumulated other comprehensive loss in our consolidated statements of stockholders’ equity (deficit) and comprehensive loss. | ||
| Computation of Net Loss per Common Share | ||
| We calculate basic and diluted net loss per share in accordance with the FAS No. 128,Earnings Per Share. Basic net loss per share was calculated by dividing the net loss by the weighted-average number of common shares outstanding during the period. Diluted net loss per share was calculated by dividing the net loss by the weighted-average number of common stock equivalents outstanding during the period. For purposes of this calculation, options and warrants are considered to be common stock equivalents and are only included in the calculation of diluted earnings per share when their effect is dilutive. We have excluded the following options |
F-16
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| and warrants from the calculation of diluted net loss per common share for 2006, 2005 and 2004 because their effect is anti-dilutive: |
| 2006 | 2005 | 2004 | ||||||||||
| Warrants | 13,458,549 | 19,629,933 | 11,154,964 | |||||||||
| Options | 3,767,103 | 2,457,000 | 1,625,000 | |||||||||
| 17,225,652 | 22,086,933 | 12,779,964 | ||||||||||
| Supplemental Cash Flow Information |
| Inception | ||||||||||||||||
| (June 12, 1996) | ||||||||||||||||
| Years ended December 31, | through | |||||||||||||||
| 2006 | 2005 | 2004 | December 31, 2006 | |||||||||||||
| Supplemental disclosures of cash flow information: | ||||||||||||||||
| Interest paid | $ | — | $ | — | $ | — | $ | 179,090 | ||||||||
| Income taxes paid | — | — | — | — | ||||||||||||
| Supplemental disclosures of non-cash investing and financing activities: | ||||||||||||||||
| Issuance of warrants, common stock and preferred stock for: | ||||||||||||||||
| Conversion of notes payable and accrued interest | $ | — | $ | — | $ | — | $ | 1,213,988 | ||||||||
| Prepaid services to consultants | 258,500 | — | 1,482,781 | |||||||||||||
| Conversion of preferred stock | — | — | 2,004 | 2,705 | ||||||||||||
| Acquisitions | 10,163,952 | — | — | 24,781,555 | ||||||||||||
| Payment of dividends | — | — | — | 213,000 | ||||||||||||
| Financial advisor services in conjunction with private placement | — | — | 1,137,456 | 1,137,456 | ||||||||||||
| Acquisition of treasury stock in settlement of a claim | — | — | 34,747 | 34,747 | ||||||||||||
| Cancellation of treasury stock | (34,747 | ) | — | — | (34,747 | ) | ||||||||||
| Assumptions of liabilities in acquisitions | 226,340 | — | — | 1,235,907 | ||||||||||||
| Acquisition of license agreement for long-term debt | — | — | — | 161,180 | ||||||||||||
| Cashless exercise of warrants | 420 | 150 | 465 | 4,312 | ||||||||||||
| Dividends accrued | — | — | — | 621,040 | ||||||||||||
| Trade asset converted to available for sale asset | — | — | 108,000 | 108,000 | ||||||||||||
| Dividends extinguished | — | — | 72,800 | 408,240 | ||||||||||||
| Trade payable converted to note payable | — | — | — | 83,948 | ||||||||||||
| Issuance of warrants for return of common stock | — | — | — | 50,852 | ||||||||||||
| Detachable warrants issued with notes payable | — | — | — | 450,000 | ||||||||||||
| Unrealized loss on short-term investments | 368 | 1,722 | — | 2,090 | ||||||||||||
| New Accounting Pronouncements | ||
| In June 2006, the FASB issued Interpretation No. 48,Accounting for Uncertainty in Income Taxes — an interpretation of FAS No. 109(“FIN 48”), which clarifies the accounting for uncertainty in income taxes. Currently, the accounting for uncertainty in income taxes is subject to significant and varied interpretations that have resulted in diverse and inconsistent accounting practices and measurements. Addressing such diversity, FIN 48 prescribes a consistent recognition threshold and measurement attribute, as well as clear criteria for subsequently recognizing, derecognizing and measuring changes in such tax positions for financial statement |
F-17
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| purposes. FIN 48 also requires expanded disclosure with respect to the uncertainty in income taxes. FIN 48 is effective for fiscal years beginning after December 15, 2006. We have not yet determined the impact of FIN 48 on our consolidated financial position, results of operations, cash flows or financial statement disclosures. | ||
| In September 2006, FASB issued FAS No. 157,Fair Value Measurements(“FAS 157”), which defines fair value, establishes a framework for measuring fair value under GAAP and expands disclosures about fair value measurements. FAS 157 is effective for fiscal years beginning after November 15, 2007. We do not expect the adoption of FAS 157 will have a material impact on our consolidated results of operations or financial position. | ||
| In September 2006, the SEC issued SAB No. 108 (“SAB 108”). Due to diversity in practice among registrants, SAB 108 expresses SEC staff views regarding the process by which misstatements in financial statements are evaluated for purposes of determining whether financial statement restatement is necessary. SAB 108 is effective for fiscal years ending after November 15, 2006. We do not believe SAB 108 will have a material impact on our consolidated results from operations or financial position. | ||
| In December 2006, the FASB issued FSP EITF 00-19-2,Accounting for Registration Payment Arrangements. See Note 2,Summary of Significant Accounting Policies – Registration Payment Arrangement, for a detailed discussion. | ||
| (3) | Acquisition of SDP | |
| On April 26, 2006, we completed the acquisition of all of the outstanding capital stock of SDP, a Delaware corporation, a privately-held drug development company, for a total purchase price of $10,195,790. We accounted for the acquisition as a purchase of net assets and not as a business combination since SDP had no revenue-producing operations, no employee base or self-sustaining operations, among other things, at the acquisition date. We acquired SDP’s rights to certain oncology and infectious disease product candidates (the “SDP Product Candidates”), including rights to a product candidate that we licensed from SDP in October 2005. The results of operations of SDP have been included in the consolidated financial statements since the date of acquisition. | ||
| The aggregate purchase price of $10,195,790 consisted of 2,099,990 shares of common stock valued at $10,163,952 and transaction costs of $31,838. The value of the common shares issued was determined based on the average market price of our common shares over the two-day period before and after the terms of the acquisition were agreed to and announced. | ||
| We determined that the assets acquired consisted principally of incomplete in-process research and development assets and that these assets had no alternative future uses in their current state. The estimated fair values of assets acquired and liabilities assumed are as follows: |
| Intangible assets – In-process research and development | $ | 10,422,130 | ||
| Accounts payable, net of cash acquired | (226,340 | ) | ||
| $ | 10,195,790 | |||
| The estimated fair value of the in-process research and development was determined based on the use of a discounted cash flow model using an income approach for the acquired SDP Product Candidates. Estimated revenues were adjusted to take into account the stage of completion and the risks surrounding the successful development and commercialization. The estimated after-tax cash flows were then discounted to a present value using a discount rate of 14%. Solely for the purpose of estimating the fair value of SDP Product Candidates, we assumed that we would incur future research and development costs of approximately $7.75 million from the date of acquisition through and including the year when commercialization is expected to occur. |
F-18
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| The major risks and uncertainties associated with the timely and successful completion of the acquired in-process projects consist of the ability to confirm the safety and efficacy of the product candidates based on the data from clinical trials and obtaining necessary regulatory approvals. No assurance can be given that the underlying assumptions used to forecast the cash flows or the timely and successful completion of the product candidates will materialize, as estimated. For these reasons, among others, actual results may vary significantly from the estimated results. | ||
| The following unaudited financial information presents the pro forma results of operations and gives effect to the SDP acquisition as if the acquisition was consummated at the beginning of 2005. This information is presented for informational purposes only, and is not intended to be indicative of any expected results of operations for future periods, or the results of operations that actually would have been realized if the acquisition had in fact occurred as of the beginning of 2005. |
| 2006 (1) | 2005 | |||||||
| Pro forma net revenues | $ | — | $ | — | ||||
| Pro forma net loss before cumulative effect of change in accounting principle (2) | $ | (29,390,573 | ) | $ | (35,381,176 | ) | ||
| Pro forma net loss (2) | $ | (29,390,573 | ) | $ | (35,381,176 | ) | ||
| Pro forma loss per basic and diluted share: | ||||||||
| Loss before cumulative effect of change in accounting principle | $ | (0.39 | ) | $ | (0.57 | ) | ||
| Net loss | $ | (0.39 | ) | $ | (0.57 | ) | ||
| Shares used for basic and diluted computation (3) | 76,088,196 | 61,928,347 | ||||||
| (1) | SDP’s results of operations for the period from January 1, 2006 through acquisition date are not available; therefore, the amounts are estimated using 2005 actual results on a pro rata basis. | ||
| (2) | Includes a non-recurring charge of $10,422,130 in each year for purchased in-process research and development costs as if the transaction occurred on the first day of each year presented. | ||
| (3) | Includes 2,099,990 shares of our common stock issued as part of consideration for the acquisition. | ||
| (4) | Short-term investments | |
| The following table summarizes our investments in securities, all of which are classified as available for sale: |
| 2006 | ||||||||||||
| Gross Unrealized | ||||||||||||
| Cost | Gains (Losses) | Fair Value | ||||||||||
| Government debt securities | $ | 4,079,400 | $ | 1,104 | $ | 4,080,504 | ||||||
| Commercial paper | 16,800,966 | (1,906 | ) | 16,799,060 | ||||||||
| Corporate bonds | 4,893,130 | (1,288 | ) | 4,891,842 | ||||||||
| $ | 25,773,496 | $ | (2,090 | ) | $ | 25,771,406 | ||||||
| 2005 | ||||||||||||
| Gross Unrealized | ||||||||||||
| Cost | Gains (Losses) | Fair Value | ||||||||||
| Government debt securities | $ | 1,443,845 | $ | 165 | $ | 1,444,010 | ||||||
| Commercial paper | 6,215,397 | (1,690 | ) | 6,213,707 | ||||||||
| Corporate bonds | 300,938 | (197 | ) | 300,741 | ||||||||
| $ | 7,960,180 | $ | (1,722 | ) | $ | 7,958,458 | ||||||
F-19
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| (5) | Property and Equipment | |
| Property and equipment at December 31, 2006 and 2005 were as follows: |
| Useful lives | 2006 | 2005 | ||||||||||
| Office furniture, computer and lab equipment | 3 - 5 years | $ | 651,012 | $ | 513,222 | |||||||
| Computer software | 3 years | 98,881 | 64,559 | |||||||||
| 749,893 | 577,781 | |||||||||||
| Less accumulated depreciation and amortization | (346,925 | ) | (170,237 | ) | ||||||||
| $ | 402,968 | $ | 407,544 | |||||||||
| Depreciation and amortization expense was $176,688, $115,545 and $41,309 for the years ended December 31, 2006, 2005 and 2004, respectively. | ||
| (6) | Income Taxes | |
| Due to our net loss position for the years ended December 31, 2006, 2005 and 2004, and as we have recorded a full valuation allowance against deferred tax assets, there was no provision or benefit for income taxes recorded. There were no components of current or deferred federal, state or foreign tax provisions for the years ended December 31, 2006, 2005 and 2004. | ||
| The income tax provision is different from that which would be obtained by applying the statutory Federal income tax rate (34%) to income before income tax expense. The items causing this difference for the period are as follows: |
| 2006 | 2005 | 2004 | ||||||||||
| Income tax benefit at federal statutory rate | $ | 9,973,000 | $ | 4,489,000 | $ | 2,278,000 | ||||||
| State tax on continuing operations | (2,000 | ) | (1,000 | ) | (1,000 | ) | ||||||
| In-process research and development (SDP acquisition) | (3,544,000 | ) | — | — | ||||||||
| Other | (593,000 | ) | 373,000 | (19,000 | ) | |||||||
| Change in federal valuation allowance | (5,834,000 | ) | (4,861,000 | ) | (2,146,000 | ) | ||||||
| Stock options previously expensed | — | — | (112,000 | ) | ||||||||
| $ | — | $ | — | $ | — | |||||||
| Deferred income taxes reflect the net tax effect of temporary differences between the carrying amount of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of deferred tax assets and liabilities at December 31, 2006 and 2005 are as follows: |
| December 31, | ||||||||
| 2006 | 2005 | |||||||
| Deferred tax assets: | ||||||||
| Accrued expenses | $ | 115,453 | $ | 47,028 | ||||
| Stock options expense under FAS 123 | 1,636,685 | 772,919 | ||||||
| Net operating loss carryforwards | 16,693,749 | 11,068,149 | ||||||
| Income tax credit carryforwards | 720,067 | 844,509 | ||||||
| Property, plant and equipment | 20,295 | 5,628 | ||||||
| Intangibles | 811,597 | 600,162 | ||||||
| Other | 577 | 577 | ||||||
| Total deferred tax assets | 19,998,423 | 13,338,972 | ||||||
| Less: valuation allowance | (19,998,423 | ) | (13,338,972 | ) | ||||
| Total deferred tax assets, net of valuation allowance | $ | — | $ | — | ||||
F-20
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| We have established a valuation allowance against our deferred tax asset due to the uncertainty surrounding the realization of such assets. Management periodically evaluates the recoverability of the deferred tax asset. At such time as it is determined that it is more likely than not that the deferred tax assets are realizable, the valuation allowance will be reduced. We have recorded a valuation allowance of $19,998,000 as of December 31, 2006 to reflect the estimated amount of deferred taxes that may not be realized. We increased the valuation allowance by $6,659,000 for the year ended December 31, 2006. The valuation allowance includes approximately $116,000 related to stock option deductions, the benefit of which may eventually be credited to equity. | ||
| At December 31, 2006, we had federal and California tax loss carryforwards of approximately $45,360,000 and $21,567,000, respectively. The federal and California net operating loss carryforwards begin to expire in 2011 and 2013 respectively, if unused. At December 31, 2006, we had federal and state tax credit carryforwards of approximately $482,000 and $361,000, respectively. The federal credits will begin to expire in 2024. | ||
| The utilization of net operating loss carryforwards and tax credit carryforwards is dependent on our future profitability. Furthermore, the Internal Revenue Code imposes a substantial restriction on the utilization of net operating loss and tax credit carryforwards in the event of an “ownership change” of more than 50% during any three-year period. As a result of the “change in ownership” provisions, utilization of net operating loss and tax credit carryforwards may be subject to an annual limitation in future periods. As a result of an annual limitation, a portion of these carryforwards may expire before ultimately becoming available to reduce future taxable income or income tax. The extent of such limitations, if any, is not known. | ||
| (7) | Warrant Liability | |
| On July 21, 2005 (the “Closing Date”), we entered into a Securities Purchase Agreement with certain accredited institutional investors (the “Purchasers”) for the sale of 10,810,809 shares of our common stock (the “Shares”) at a purchase price of $1.85 per share for aggregate gross proceeds of $19,999,997. In connection with this financing, we issued the Purchasers seven-year warrants to purchase 10,810,809 shares of our common stock (the “Warrant Shares”) at an exercise price of $2.26 per share. We received net proceeds of $18,313,751, after deducting commissions and offering fees and expenses, which included cash payments of $1,403,000 to placement agents and $283,246 in legal and accounting fees. | ||
| Pursuant to the terms of the Securities Purchase Agreement, if (i) a registration statement covering (A) all of the Shares and the Warrant Shares and (B) any other shares of common stock issued or issuable in respect to the Shares and the Warrant Shares because of stock splits, stock dividends, reclassifications, recapitalizations or similar events (together, the “Registrable Shares”) required to be covered thereby and required to be filed by us is (A) not filed with the SEC on or before 45 days after the closing date (a “Filing Failure”) or (B) if such registration statement is not declared effective by the SEC on or before (1) 90 days after the closing date (an “Effectiveness Failure”) or (ii) on any day after the effective date of the registration statement sales of all the Registrable Shares required to be included on such registration statement cannot be made (other than as permitted during a suspension pursuant to this agreement) pursuant to such registration statement (including, without limitation, because of a failure to keep such Registration Statement effective, to disclose such information as is necessary for sales to be made pursuant to such Registration Statement or to register sufficient shares of Shares) (a “Maintenance Failure”), then, we will be obligated, without limiting any other remedies of any Purchaser, to pay as liquidated damages (the “Liquidated Damages”) for such failure and not as a penalty to any Purchaser an amount in cash determined in accordance with the formula set forth below: |
| • | For each 30-day period that a Filing Failure, Effectiveness Failure or Maintenance Failure remains uncured, we will pay an amount equal to the purchase price paid to us for all Shares then held by such Purchaser multiplied by 1% for the first 30-day period or any portion thereof and increasing by an additional 1% with regard to each additional 30-day period until such Filing Failure, Effectiveness Failure or Maintenance Failure is cured. | ||
| • | For any partial 30-day period in which a Filing Failure, Effectiveness Failure or Maintenance Failure exists but is cured prior to the end of the 30-day period, we will pay the Purchasers a pro rata portion of |
F-21
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| the amount which would be due if the failure continued for the entire 30-day period. For example, if the purchase price paid for all Shares then held by a Purchaser is $5,000,000, then, (a) at the end of the 30th day, the Liquidated Damages would be 1% or $50,000, (b) at the end of the 60th day, the Liquidated Damages for the first 30-day period would have been 1% or $50,000 and for the second 30-day period would be 2% or $100,000, and (c) at the end of the 105th day, the Liquidated Damages for the first 30-day period would have been 1% or $50,000, for the second 30-day period 2% or $100,000, for the third 30-day period 3% or $150,000, and for the final 15-day period, 4% applied pro rata to such 15 days, or $100,000. |
| There is no cap to the amount of Liquidated Damages that we may be obligated to pay. Payments to be made pursuant to this Securities Purchase Agreement will be due and payable to the Purchasers at the end of each calendar month during which Liquidated Damages will have accrued. No Liquidated Damages will be due or payable to a Purchaser in any event if as of the date of the Filing Failure, Effectiveness Failure or Maintenance Failure such Purchaser could sell all of the Registrable Shares such Purchaser then holds without registration by reason of Rule 144(k) of the Securities Act. | ||
| The registration statement was filed and declared effective by the SEC on September 2, 2005, which was within the allowed time. As of December 31, 2006, we have not yet been required to pay any Liquidated Damages in connection with the filing or effectiveness of the registration. | ||
| At the Closing Date, the Shares and amount of proceeds that are subject to Liquidated Damages payments were reclassified to temporary equity on our consolidated balance sheet, because Liquidated Damages, if any, are required to be settled in cash. In addition, we recorded a liability for the registration payment arrangement based on the fair value of the warrants at the Closing Date, with an offsetting reduction to temporary equity. The liability is included in warrant liability on our consolidated balance sheet. The fair value of the warrants was estimated to be $19,439,185 at the Closing Date. The difference of $1,125,434 between the fair value of the warrants of $19,439,185 and the net proceeds from the offering was classified as loss on fair value of warrants in our consolidated statement of operations. | ||
| The fair value of the warrants is re-measured at each reporting date and any changes in fair value are reported as gain (loss) on fair value of warrants in the period of the change in our consolidated statement of operations. At December 31, 2006 and 2005, the fair value was estimated to be $30,356,439 and $29,696,411, respectively, with increases in fair values due to the increases in the market value of our common stock. In the years ended December 31, 2006 and 2005, we recorded $660,028 and $11,579,660, respectively, in loss on fair value of warrants in our consolidated statements of operations. | ||
| The fair value of the warrants was estimated using the Black-Scholes option-pricing model with the following assumptions at December 31, 2006 and 2005: no dividends; risk-free (10-year U.S. Treasury yield) interest rate of 4.7% and 4.4%, respectively; the contractual life of seven years and volatility of 139% and 90%, respectively. | ||
| (8) | Capital Stock | |
| Preferred Stock | ||
| In November 2005, at a special meeting of our stockholders, the stockholders approved a proposal to increase the number of shares of common stock we are authorized to issue to 200,000,000 shares. The number of authorized shares of preferred stock remains unchanged at 1,000,000 shares. The Series A, Series B and Series C preferred stock were eliminated, and we are no longer authorized to issue any such series of preferred stock as previously designated. We have no present plans to issue any new shares or designate any series of preferred stock. |
F-22
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| Common Stock | ||
| 2006. In April 2006, we issued 2,099,990 shares of common stock at $4.84 per share for a fair value of $10,163,952 to acquire SDP. See Note 3,Acquisition of SDP,for a detailed discussion. | ||
| In September 2006, we ended an employment relationship with our former chief financial officer who also served as treasurer, vice president, finance and secretary. In connection with the separation from us, a severance agreement was entered into wherein the former chief financial officer’s outstanding vested and unvested options were cancelled upon the separation and we issued 60,145 shares of common stock with a fair value of $196,674 and paid employment taxes totaling $109,434. The entire severance amount of $306,108 was charged to selling, general and administrative expense for the year ended December 31, 2006. | ||
| In November 2006, we issued and sold to certain accredited institutional investors 14,545,000 shares of common stock in a registered direct offering at a price of $2.75 per share, for aggregate offering proceeds of approximately $40.0 million and net offering proceeds of approximately $37.1 million, after deducting commissions and offering fees and expenses. The offering was made pursuant to our shelf registration statement on Form S-3, filed with the SEC on May 1, 2006. | ||
| During 2006, we issued an aggregate of 420,161 shares of our common stock upon the cashless exercises of warrants to purchase an aggregate of 527,528 shares of common stock at the weighted average exercise price of $0.57 per share. | ||
| Also during 2006, we issued an aggregate of 5,196,246 shares of our common stock in connection with the exercises of stock purchase warrants (5,103,746 shares at a weighted average price of $1.55 per share for cash in the aggregate amount of $7,691,590, net of $206,274 in commissions) and employee stock options (92,500 shares at a weighted average price of $1.36 per share for cash in the aggregate amount of approximately $125,751). We also issued 15,000 shares of restricted stock to our consultants for services performed with a fair value of $68,650. | ||
| 2005. In April 2005, we issued 25,000 shares of common stock as partial payment for services rendered by a consulting firm. Those shares were recognized at fair market value as of the date of obligation and resulted in compensation expense of $23,500 in the year ended December 31, 2005, when the services were performed. | ||
| In July 2005, we issued 100,000 shares of common stock, with a fair market value at the date of issuance of $235,000, pursuant to a consulting agreement entered into in January 2005. The compensation cost related to those shares is recognized over the three-year service period at an annual amortization of $78,333. | ||
| In July 2005, we issued and sold to certain accredited institutional investors 10,810,809 shares of common stock at $1.85 per share, for aggregate gross proceeds of $19,999,997 and net proceeds of $18,313,751, after deducting commissions and offering costs. In connection with this transaction, we issued warrants to purchase 10,810,809 shares of common stock at an exercise price of $2.26 per share. See Note 7,Warrant Liability, for a detailed discussion. | ||
| During 2005, we issued an aggregate of 149,613 shares of our common stock upon the cashless exercises of warrants to purchase an aggregate of 252,049 shares of common stock at the weighted average exercise price of $1.18 per share. | ||
| Also during 2005, we issued an aggregate of 2,443,703 shares of our common stock in connection with the exercises of stock purchase warrants (2,258,703 shares at a weighted average price of $1.37 per share for cash in the aggregate amount of $3,073,439) and employee stock options (185,000 shares at a weighted average price of $0.78 per share for cash in the aggregate amount of $145,000). | ||
| 2004. In March 2004, 473 shares of Series A cumulative convertible preferred stock, representing all of the Series A cumulative convertible preferred stock then outstanding, were converted into 236,500 shares of |
F-23
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| common stock. In conjunction with the conversion, dividends payable of $72,800 at December 31, 2003, were extinguished. | ||
| In March 2004, 200,000 shares of Series B convertible preferred stock, representing all of the Series B convertible preferred stock then outstanding, were converted into 200,000 shares of common stock. | ||
| In April 2004, we sold 10,417,624 shares of common stock at $1.50 per share and issued warrants to purchase 3,125,272 shares of common stock at $2.00 and warrants to purchase 2,083,518 shares of common stock at $2.50 per share in a private placement for aggregate gross proceeds of $15,626,450 in cash. In connection with the private placement, we paid cash commissions of $900,452 and other related expenses of $466,322 and issued warrants to purchase 632,547 shares of common stock at $2.00 per share to two placement agents, having a fair market value of $890,963 on the date of issuance. | ||
| During 2004, we issued an aggregate of 464,573 shares of our common stock upon the cashless exercises of warrants to purchase 502,528 and 110,000 shares of common stock at the exercise prices of $0.49 and $0.50 per share, respectively. | ||
| Also during 2004, we issued an aggregate of 23,832 shares of our common stock in connection with the exercises of stock purchase warrants at a weighted average price of $1.15 per share for cash in the aggregate amount of approximately $27,353. | ||
| (9) | Warrants | |
| In July 2005, we issued warrants to purchase 10,810,809 shares of common stock at an exercise price of $2.26 per share in connection with the sale of 10,810,809 shares of common stock in July 2005. | ||
| In October 2004, we issued a warrant to purchase 300,000 shares of common stock at an exercise price of $2.50 in settlement of a claim. The warrant had a value of $86,375 on the date of issuance. | ||
| In April 2004, we issued to the investors warrants to purchase 3,125,272 shares of common stock at $2.00 and warrants to purchase 2,083,518 shares of common stock at $2.50 per share in connection with the April 2004 private placement We engaged W.R. Hambrecht + Co., LLC for financial advisory and investment banking services related to the April 2004 private placement, and in connection with that engagement, issued a warrant to purchase 175,000 shares of common stock at $2.00 per share, having a fair market value of $246,493 on the date of issuance. | ||
| At December 31, 2006, outstanding warrants to purchase shares of common stock are as follows: |
| Warrants | Exercise Price | Expiration Date | ||||||||
| 50,000 | $ | 2.50 | Apr-07 | |||||||
| 35,000 | $ | 2.50 | Oct-07 | |||||||
| 1,872,693 | $ | 1.98 | Apr-09 | |||||||
| 117,000 | $ | 2.38 | Apr-09 | |||||||
| 573,047 | $ | 1.98 | Jun-09 | |||||||
| 10,810,809 | $ | 2.26 | Jul-12 | |||||||
| 13,458,549 | ||||||||||
(10) Equity Incentive Plans
| At December 31, 2006, we had the 2005 Equity Incentive Plan (the “2005 Plan”) and the 2005 Employee Stock Purchase Plan (the “Purchase Plan”), which are described below. The share-based compensation expense from |
F-24
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| all share-based awards that has been charged to our consolidated statements of operations in the years ended December 31, 2006, 2005 and 2004 was comprised of the following: |
| Years Ended December 31, | ||||||||||||
| 2006 | 2005 | 2004 | ||||||||||
| Selling, general and administrative expense | $ | 1,635,369 | $ | 572,263 | $ | 445,755 | ||||||
| Research and development expense | 509,966 | 594,493 | 79,167 | |||||||||
| Share-based compensation expense before taxes | 2,145,335 | 1,166,756 | 524,922 | |||||||||
| Related income tax benefits | — | — | — | |||||||||
| Share-based compensation expense | $ | 2,145,335 | $ | 1,166,756 | $ | 524,922 | ||||||
| Net share-based compensation expense per common share – basic and diluted | $ | 0.03 | $ | 0.02 | $ | 0.01 | ||||||
| Share-based compensation expense from: | ||||||||||||
| Stock options | $ | 1,801,677 | $ | 1,088,423 | $ | 524,922 | ||||||
| Share grant | 275,008 | 78,333 | — | |||||||||
| Restricted stock awards | 68,650 | — | — | |||||||||
| $ | 2,145,335 | $ | 1,166,756 | $ | 524,922 | |||||||
| Since we accounted for employee share-based awards using the recognition method under the provisions of FAS 123 prior to 2006, the adoption of FAS 123R did not have a material impact on our consolidated results of operations. Since we have a net operating loss carry-forward as of December 31, 2006, no excess tax benefits for the tax deductions related to share-based awards were recognized in the consolidated statement of operations. Additionally, no incremental tax benefits were recognized from stock options exercised in the year ended December 31, 2006 that would have resulted in a reclassification to reduce net cash provided by operating activities with an offsetting increase in net cash provided by financing activities. | ||
| 2005 Equity Incentive Plan | ||
| The 2005 Plan, which is stockholder-approved, is intended to encourage ownership of shares of common stock by our directors, officers, employees, consultants and advisors and to provide additional incentive for them to promote the success of our business through the grant of share-based awards. The 2005 Plan provides for the grant of incentive and non-statutory stock options as well as share appreciation rights, restricted shares, restricted share units, performance units, shares and other share-based awards. Share-based awards are subject to terms and conditions established by the Board of Directors or the Compensation Committee of our Board of Directors. Our policy is to issue new common shares upon the exercise of stock options, conversion of share units or issuance of shares or restricted stock. | ||
| The maximum aggregate number of shares of common stock which may be issued pursuant to or subject to the foregoing types of awards granted under the 2005 Plan is 6,673,634 as of December 31, 2006. This maximum number is subject to an annual automatic increase beginning on January 1, 2006 equal to the lesser of (i) 1% of the number of outstanding shares of common stock on such day, (ii) 750,000 or (iii) such other amount as our board of directors may specify. The 2005 Plan is intended to comply with applicable securities law requirements, permit performance-based awards that qualify for deductibility under Section 162(m) of the Internal Revenue Code and allow for the issuance of incentive stock options. As of December 31, 2006 and 2005, 2,453,886 and 3,258,000 shares of common stock, respectively, remained available for issuance under the 2005 Plan. On January 1, 2007, the number of shares of common stock available for issuance under the 2005 Plan increased by 750,000 shares in accordance with the provisions for annual increases under the 2005 Plan. | ||
| Stock options are typically granted with an exercise price equal to the current market price of our common stock at the grant date and have ten-year contractual terms. Option awards generally vest over four years based on |
F-25
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| continuous service; however, our equity compensation plan allows for other vesting periods and we have granted employees options where the requisite service period is three years and we grant our directors options where the requisite service period is one year. During the years ended December 31, 2006 and 2005, we granted stock options and issued stock under the 2005 Plan. | ||
| During January through April 2004, which was prior to the adoption of the 2005 Plan and prior to listing our common stock on AMEX, we granted employees options to purchase an aggregate of 310,000 shares of common stock at a purchase price of $1.50 per share. The total value of all the options on the dates of grant was $395,403. | ||
| Subsequent to listing our common stock on AMEX, in the period of May 2004 through August 2004 we granted employees options to purchase an aggregate of 66,000 shares of common stock at purchase prices of $1.20 to $1.80 per share. AMEX listing requirements prohibit granting equity without a stockholder vote or an approved stock option plan; therefore, the options were rescinded in February 2005. Accordingly, the financial statement effect of the options granted was reversed in 2004. | ||
| In July 2005, we granted 1,625,000 options to employees under the 2005 Plan to replace pre-existing options that were not issued under the 2005 Plan or any other incentive plan approved by our stockholders. In addition in July 2005, we granted 1,103,000 new options to employees and board members under the 2005 Plan. | ||
| In December 2005, the exercise prices on 743,000 of the 1,103,000 options were increased to equal the fair market value of common stock on the date of grant in July 2005. In addition, the exercise prices on 730,000 of the pre-existing options were increased to equal the fair market value of common stock on the original grant dates. There was no material impact to the compensation expense as a result of this change. There were 14 employees affected by this change. | ||
| We cancelled 413,397, 200,000 and 1,665,000 options in the years ended December 31, 2006, 2005 and 2004, respectively, related to terminated employees, and the shares underlying such options were returned to and are available for re-issuance under the 2005 Plan. |
F-26
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| A summary of all of our option activity as of December 31, 2006 and of changes in options outstanding under the plans during the year ended December 31, 2006 are as follows: |
| Weighted- | ||||||||||||||||
| average | ||||||||||||||||
| Weighted- | Remaining | Aggregate | ||||||||||||||
| average | Contractual | Intrinsic | ||||||||||||||
| Shares | Exercise Price | Term | Value | |||||||||||||
| Outstanding at January 1, 2004 | 2,980,000 | $ | 0.38 | |||||||||||||
| Granted | 310,000 | $ | 1.50 | |||||||||||||
| Exercised | — | $ | — | |||||||||||||
| Canceled/forfeited/expired | (1,665,000 | ) | $ | 0.23 | ||||||||||||
| Options outstanding at December 31, 2004 | 1,625,000 | $ | 0.75 | |||||||||||||
| Granted | 1,217,000 | $ | 2.34 | |||||||||||||
| Exercised | (185,000 | ) | $ | 0.78 | ||||||||||||
| Canceled/forfeited/expired | (200,000 | ) | $ | 1.41 | ||||||||||||
| Options outstanding at December 31, 2005 | 2,457,000 | $ | 1.45 | |||||||||||||
| Granted | 1,816,000 | $ | 3.84 | |||||||||||||
| Exercised | (92,500 | ) | $ | 1.36 | ||||||||||||
| Canceled/forfeited/expired | (413,397 | ) | $ | 3.37 | ||||||||||||
| Options outstanding at December 31, 2006 | 3,767,103 | $ | 2.39 | 6.56 | $ | 3,525,025 | ||||||||||
| Options vested and expected to vest in the future, December 31, 2006 | 3,343,915 | $ | 2.28 | 6.20 | $ | 3,451,036 | ||||||||||
| Options exercisable at December 31, 2006 | 1,996,460 | $ | 1.46 | 4.26 | $ | 3,211,778 | ||||||||||
| Options exercisable at December 31, 2005 | 1,557,503 | $ | 1.03 | |||||||||||||
| Options exercisable at December 31, 2004 | 1,072,502 | $ | 1.28 | |||||||||||||
| The weighted-average grant-date fair value of options granted during the years ended December 31, 2006, 2005 and 2004 was $2.97, $2.14 and $1.89, respectively. As of December 31, 2006, there was $4.2 million of unamortized compensation cost related to unvested stock option awards, which is expected to be recognized over a weighted-average remaining period of approximately two years. | ||
| The total intrinsic value of options exercised during the years ended December 31, 2006, 2005 and 2004 was $153,850, $395,000 and $0, respectively, based on the differences in the market prices on the dates of exercise and the option exercise prices. During the years ended December 31, 2006, 2005 and 2004, we received a total of $125,751, $145,000 and $0, respectively, in cash from exercised options under all share-based payment arrangements. No tax benefit was realized for the tax deductions from option exercises of the share-based payment arrangements in the years ended December 31, 2006, 2005 and 2004. | ||
| Our determination of fair value is affected by our stock price as well as a number of assumptions that require judgment. The fair value of each option award is estimated on the date of grant using the Black-Scholes option-valuation model. The assumptions used in the Black-Scholes model for option grants during the years ended December 31, 2006, 2005 and 2004 are as follows: |
| Years ended December 31, | ||||||||||||
| 2006 | 2005 | 2004 | ||||||||||
| Risk-free interest rate | 4.1 - 5.2 | % | 3.7 - 4.3 | % | 2.8 - 4.3 | % | ||||||
| Dividend yield | 0.0 | % | 0.0 | % | 0.0 | % | ||||||
| Expected volatility | 85-142 | % | 90 | % | 81 - 90 | % | ||||||
| Weighted-average volatility | 111 | % | 90 | % | 87 | % | ||||||
| Expected term (in years) | 5-6.1 years | 5 years | 3 - 5 years | |||||||||
| The risk-free interest rate assumption is based on the U.S. Treasury yield for a period consistent with the expected term of the option in effect at the time of the grant. We have not paid any dividends on common stock |
F-27
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| since our inception and do not anticipate paying dividends on our common stock in the foreseeable future. The expected option term is computed using the “simplified” method as permitted under the provisions of SAB 107. The expected volatility is based on the historical volatility of our common stock and other factors. In 2006, we used an alternative historical volatility based on the daily close price of our common stock, which we determined was a better indicator of volatility than the method used in the prior years. The effect of this change on share-based compensation was immaterial. | ||
| In 2005, we granted 114,000 options, respectively, to consultants. No options were granted to consultants in 2006 and 2004. These option grants were valued as of the date at which the consultants’ performance is complete using the Black-Scholes pricing model. The assumptions used in the Black-Scholes model for non-employee option grants for the years ended December 31, 2006 and 2005 are as follows: |
| 2006 | 2005 | |||||||
| Risk-free interest rate | 4.7 | % | 4.4 | % | ||||
| Dividend yield | 0.0 | % | 0.0 | % | ||||
| Expected volatility | 139 | % | 90 | % | ||||
| Contractual term (in years) | 3.5 – 6.1 years | 3.5 – 6.1 years | ||||||
| We recognized $104,225 and $93,549 in share-based compensation expense associated with non-employee options in the years ended December 31, 2006 and 2005, respectively. | ||
| The following table summarizes information concerning our outstanding and exercisable stock options as of December 31, 2006: |
| Options Outstanding | Options Exercisable | |||||||||||||||||||
| Weighted- | ||||||||||||||||||||
| Number | Average | Weighted- | Number | Weighted- | ||||||||||||||||
| Outstanding at | Remaining | Average | Exercisable at | Average | ||||||||||||||||
| December 31, 2006 | Contractual | Exercise | December 31, 2006 | Exercise | ||||||||||||||||
| Range of Exercise Price | in 000’s | Life | Price | in 000’s | Price | |||||||||||||||
| $0.50 to $1.64 | 1,280 | 2.00 | $ | 0.70 | 1,280 | $ | 0.70 | |||||||||||||
| $2.04 to $2.86 | 1,461 | 8.62 | $ | 2.51 | 580 | $ | 2.37 | |||||||||||||
| $2.99 to $4.89 | 1,026 | 9.30 | $ | 4.34 | 136 | $ | 4.63 | |||||||||||||
| 3,767 | 6.56 | $ | 2.39 | 1,996 | $ | 1.46 | ||||||||||||||
| Restricted Stock Awards.Restricted stock awards are grants that entitle the holder to acquire shares of restricted common stock at no cost. The shares of the restricted stock awards cannot be sold, pledged or otherwise disposed of until the award vests and any unvested shares may be transferred back to us following the awardee’s termination of service. During the year ended December 31, 2006, we granted 15,000 shares of restricted stock awards to consultants for services performed. These restricted stock awards vest monthly over twelve months of service. No restricted stock awards were granted in the years ended December 31, 2005 and 2004. | ||
| A summary of our unvested restricted share awards as of December 31, 2006 and changes during the year then ended are presented below: |
| Weighted Average | ||||||||
| Number of shares | Grant Date Fair Value | |||||||
| Unvested, January 1, 2006 | — | $ | — | |||||
| Granted | 15,000 | $ | 4.58 | |||||
| Vested | (15,000 | ) | $ | 4.58 | ||||
| Forfeited | — | $ | — | |||||
| Unvested, December 31, 2006 | — | $ | — | |||||
F-28
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| The fair value of restricted stock awards granted in the year ended December 31, 2006 was $68,650. As of December 31, 2006, there are no unrecognized compensation costs related to restricted stock awards. | ||
| Stock Grants.Stock grants are grants of shares of our common stock not subject to restrictions or other forfeiture conditions. During the year ended December 31, 2006, we granted an employee 60,145 shares of common stock with a grant-date fair value of $196,674. During the year ended December 31, 2005, we granted a consultant 100,000 shares of common stock with a fair value of $235,000. No stock grants were granted under the 2005 Plan in the year ended December 31, 2004. As of December 31, 2006, there was $78,334 unrecognized compensation cost related to stock grants, which is expected to be recognized in 2007. | ||
| Employee Stock Purchase Plan | ||
| The Purchase Plan was approved by our stockholders in 2005; however, we have not implemented the Purchase Plan. The Purchase Plan allows all eligible employees to purchase shares of common stock at 85% of the lower of the fair market value on the first or the last day of each offering period. Employees may authorize us to withhold up to 15% of their compensation during any offering period, subject to certain limitations. The maximum aggregate number of shares of common stock which may be issued under the Purchase Plan is 1,673,634 as of December 31, 2006. This maximum number is subject to an annual automatic increase beginning on January 1, 2006 equal to the lesser of (i) 1% of the number of outstanding shares of common stock on such day, (ii) 750,000 or (iii) such other amount as our board of directors may specify. At December 31, 2006, no shares of common stock have been issued under the Purchase Plan. On January 1, 2007, the number of shares of common stock available for issuance under the Purchase Plan increased by 750,000 in accordance with the provisions for annual increases under the Purchase Plan. | ||
| (11) | License Agreements | |
| USC Agreements | ||
| Under an option and license agreement with the University of Southern California (“USC”) entered into in January 1998 and amended in August 2000, we hold exclusive rights to a number of patents that have issued in the U.S. and Canada covering our CoFactor product candidate and its use in connection with cancer chemotherapy. An additional patent included in the agreement relates to compounds in our organoselenones program that we are currently evaluating for future preclinical and clinical development. | ||
| This agreement terminates on the last to expire of the licensed patents, which is expected to occur in March 2014. Upon breach or default under the agreement, the non-breaching party may terminate the agreement by 45 days’ written notice. USC may terminate the agreement upon 20 days’ notice if we fail to obtain and maintain the insurance required by the agreement and may terminate the agreement immediately upon notice if we attempt to use, sublicense, transfer or assign our rights or obligations under the agreement in any manner contrary to its terms or in derogation of USC’s propriety rights and upon bankruptcy, reorganization, liquidation or receivership proceedings involving us. We may terminate the agreement at any time by providing USC 30 days’ written notice. | ||
| This agreement provides for the payment to USC of a 3% royalty on net sales by us or a sublicensee of licensed products, as well as a prepaid royalty of $100,000 within 30 days of approval of an NDA by the FDA for any product covered by the claims of the licensed patents (which prepaid royalty is deductible from future royalty payments). In addition, we are required to reimburse all reasonable legal expenses incurred by USC in filing, prosecuting and maintaining the licensed patents. No royalties have been paid to date under this agreement. | ||
| Under another option and license agreement with USC entered into in August 2000 and amended in April 2003 and January 2007, we hold exclusive rights to a number of patents that have issued in the U.S. and the EU covering methods for the manufacture of our ANX-201 product candidate and of various analogs and derivatives thereof, and the use of ANX-201 in connection with the HIV. |
F-29
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| This agreement terminates on the last to expire of the licensed patents, which is expected to occur in November 2020. Upon breach or default under the agreement, the non-breaching party may terminate the agreement by 45 days’ written notice. USC may terminate the agreement immediately upon notice if we attempt to use, sublicense, transfer, or assign our rights or obligations under the agreement in any manner contrary to its terms or in derogation of USC’s propriety rights, we fail to obtain and maintain the insurance required by the agreement and upon bankruptcy, reorganization, liquidation or receivership proceedings involving us. In addition, if we fail to achieve the milestones set forth in the agreement, as amended, USC has the option to terminate the agreement but only by providing written notice of termination to us within one (1) month of the applicable milestone deadline. We may terminate the agreement at any time by providing USC 30 days’ written notice and reimbursing the reasonable legal expenses incurred by USC for up to one (1) month from the date written notification of termination is sent by us. | ||
| This agreement provides for the payment to USC of a 1% royalty on net sales by us of licensed products and milestone payments on each licensed product upon entering Phase I clinical trials ($75,000), reaching Phase II clinical trials ($100,000), reaching Phase III clinical trials ($125,000) and upon receiving market approval from the FDA or other government regulatory agency ($250,000). In addition, if any licensed product is manufactured and sold under sublicense from us, we will pay USC a royalty based on a percentage of all of the revenue we received from the sublicense (including all earned royalties, prepaid royalties and license fees). Furthermore, we are required to reimburse all reasonable legal expenses incurred by USC in filing, prosecuting and maintaining the licensed patents. No royalties have been paid to date under this agreement. | ||
| Theragenex Agreement | ||
| In October 2006, we entered into a license agreement with Theragenex, LLC, a life science and technology company. Under the agreement, we granted Theragenex exclusive rights to develop and commercialize chitosan gel in the U.S. in exchange for a licensing fee of $1.0 million ($500,000 of which we received in January 2007, with the remainder due in June 2007), a $1.0 million milestone payment that will be due with 45 days after the launch of each licensed product, and royalties of 15% to 20% on licensed product sales, depending on sales levels. The agreement remains in effect through the later of the latest date on which the last licensed product is covered by a valid claim or 20 years from the date of the first commercial sale of the last licensed product by Theragenex. Either party may terminate the agreement if the other materially breaches or materially defaults in the performance or observance of any of the provisions of the agreement. In addition, either party may, upon notice, terminate the agreement, if the other party admits in writing that it is generally unable to meet its debts when due, or upon the filing of bankruptcy, reorganization, liquidation or receivership proceedings involving such party. Theragenex may terminate the agreement at any time, upon 90 days’ written notice, if Theragenex concludes in good faith, based on technical information learned by it following execution of the agreement, that there is no reasonable likelihood of a commercially viable licensed product. For the year ended December 31, 2006, none of the license fee had been paid and no revenue had been recognized under the agreement. | ||
| M.D. Anderson | ||
| Pursuant to a patent and technology license agreement dated June 14, 1996 between M.D. Anderson and us (the “M.D. Anderson License Agreement”), we acquired a license to seven patents and patent applications related to technology for HIV/AIDS therapy and prevention. Under the M.D. Anderson License Agreement, we were obligated to pay M.D. Anderson for all out-of-pocket expenses incurred in filing, prosecuting, enforcing, and maintaining the licensed patent rights and all future patent-related expenses paid by M.D. Anderson as long as the M.D. Anderson License Agreement remained in effect. In 2005, we terminated the M.D. Anderson License Agreement. |
F-30
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| NIH Agreement | ||
| In December 2002, we entered into a worldwide exclusive patent license agreement with the Public Health Service National Institutes of Health (“NIH”) concerning composition of matter for BlockAide/CR, a product we were previously developing. Under the terms of the agreement, we agreed to pay minimum royalty payments during the first year of the license and minimum annual royalties thereafter or the higher amount based upon a percentage of net sales. In addition, there were benchmark royalties based upon: initiation of Phase I trials, initiation of Phase II trials, initiation of Phase III trials, and upon first approval of a Product License Application for an HIV therapeutic or vaccine in the U.S. and for first approval in Europe. No material amount was paid under this agreement. In 2005, we terminated the NIH agreement. | ||
| (12) | Commitments | |
| Operating Leases | ||
| We are obligated under operating leases for office space and equipment. In July 2004, we entered into a lease for our current office space in San Diego, California. In June 2005, we leased additional space in the same facility. Based on a straight-line basis, the lease requires a monthly payment of $20,729. The lease expires in August 2009. Rent expense was $246,000, $220,517 and $118,966 during the years ended December 31, 2006, 2005 and 2004, respectively. | ||
| Future rental commitments under all operating leases are as follows: |
| Year Ending December 31, | ||||
| 2007 | $ | 255,630 | ||
| 2008 | 259,011 | |||
| 2009 | 174,698 | |||
| Total | $ | 689,339 | ||
| (13) | Litigation | |
| In the normal course of business, we may become subject to lawsuits and other claims and proceedings. Such matters are subject to uncertainty. Management is not aware of any pending or threatened lawsuit or proceedings that would have a material adverse effect on our consolidated financial position, results of operations or cash flows. | ||
| (14) | 401(k) Plan | |
| In January 2005, we adopted a plan intended to qualify as a qualified cash or deferred arrangement under Section 401(k) of the Internal Revenue Code of 1986, as amended. Under the provisions of our 401(k) Plan, we are required to make matching contributions in the amount of 100% of salary deferrals up to 3% and 50% of salary deferrals between 3% and 5% of the annual salary of the contributing employee. We incurred total expenses of $80,393 and $61,354 in employer matching contributions in 2006 and 2005, respectively. |
F-31
Table of Contents
ADVENTRX Pharmaceuticals, Inc. and Subsidiaries
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
(A Development Stage Enterprise)
Notes to Consolidated Financial Statements (continued)
December 31, 2006
| (15) | Summary of Quarterly Financial Data (unaudited) | |
| The following is a summary of the unaudited quarterly results of operations for the years ended December 31, 2006 and 2005: |
| Quarters Ended | ||||||||||||||||
| Quarterly statement of operations data | September 30, | |||||||||||||||
| for 2006 (unaudited): | March 31, 2006 | June 30, 2006 (1) | 2006 | December 31, 2006 | ||||||||||||
| Loss from operations | $ | (4,256,143 | ) | $ | (15,451,711 | ) | $ | (5,328,321 | ) | $ | (4,800,292 | ) | ||||
| Net income (loss) (2) | $ | (21,046,681 | ) | $ | 2,763,714 | $ | (4,609,181 | ) | $ | (6,439,625 | ) | |||||
| Basic net income (loss) per share | $ | (0.31 | ) | $ | 0.04 | $ | (0.06 | ) | $ | (0.08 | ) | |||||
| Diluted net income (loss) per share | $ | (0.31 | ) | $ | 0.03 | $ | (0.06 | ) | $ | (0.08 | ) | |||||
| Basic weighted average number of shares of common stock outstanding | 67,976,352 | 71,214,523 | 73,435,715 | 83,092,233 | ||||||||||||
| Diluted weighted average number of shares of common stock outstanding | 67,976,352 | 81,797,928 | 73,435,715 | 83,092,233 | ||||||||||||
| Quarters Ended | ||||||||||||||||
| Quarterly statement of operations data | September 30, | |||||||||||||||
| for 2005 (unaudited): | March 31, 2005 | June 30, 2005 | 2005 | December 31, 2005 | ||||||||||||
| Loss from operations | $ | (2,882,256 | ) | $ | (3,395,151 | ) | $ | (3,641,848 | ) | $ | (3,779,790 | ) | ||||
| Net loss (2) | $ | (2,844,934 | ) | $ | (3,330,554 | ) | $ | (16,454,867 | ) | $ | (2,152,291 | ) | ||||
| Basic and diluted net loss per share | $ | (0.05 | ) | $ | (0.06 | ) | $ | (0.26 | ) | $ | (0.03 | ) | ||||
| Basic and diluted weighted average number of shares of common stock outstanding | 53,967,933 | 54,821,480 | 63,255,407 | 67,194,366 | ||||||||||||
| (1) | Includes a charge of $10,442,130 for purchased in-process research and development in connection with our acquisition of SDP in the quarter ended June 30, 2006. | |
| (2) | Includes gain (loss) on fair value of warrant liability commencing in the quarter ended September 30, 2005. |
F-32
Table of Contents
Exhibit Index
| Exhibit | Description | |
| 2.1 (1) | Agreement and Plan of Merger, dated April 7, 2006, among the registrant, Speed Acquisition, Inc., SD Pharmaceuticals, Inc. and certain individuals named therein (including exhibits thereto) | |
| 3.1 (2) | Amended and Restated Certificate of Incorporation of the registrant | |
| 3.2 (3) | Amended and Restated Bylaws of the registrant (formerly known as Biokeys Pharmaceuticals, Inc.) | |
| 4.1(4) | Form of Registration Rights Agreement entered into in October and November 2001 (including the original schedule of holders) | |
| 4.2 (5) | $2.50 Warrant to Purchase Common Stock issued on April 12, 2002 to Emisphere Technologies, Inc. | |
| 4.3 (4) | Form of $0.60 Warrant to Purchase Common Stock issued May 28, 2003 (including the original schedule of holders) | |
| 4.4 (4) | Form of $1.25 Warrant to Purchase Common Stock issued between October 15, 2003 and December 29, 2003 (including the original schedule of holders) | |
| 4.5 (4) | Common Stock and Warrant Purchase Agreement, dated as of April 5, 2004, among the registrant and the Investors (as defined therein) | |
| 4.6 (4) | Registration Rights Agreement, dated April 5, 2004, among the registrant and the Investors (as defined therein) | |
| 4.7 (4) | Form of $2.00 A-1 Warrant to Purchase Common Stock issued April 8, 2004 (including the original schedule of holders) | |
| 4.8 (4) | Form of $2.50 A-2 Warrant to Purchase Common Stock issued April 8, 2004 (including the original schedule of holders) | |
| 4.9 (6) | Common Stock and Warrant Purchase Agreement, dated April 8, 2004, between the registrant and CD Investment Partners, Ltd. | |
| 4.10 (6) | Registration Rights Agreement, dated April 8, 2004, between the registrant and CD Investment Partners, Ltd. | |
| 4.11 (6) | $2.00 A-1 Warrant to Purchase Common Stock issued on April 8, 2004 to CD Investment Partners, Ltd. | |
| 4.12 (6) | $2.00 A-1 Warrant to Purchase Common Stock issued on April 8, 2004 to Burnham Hill Partners | |
| 4.13 (6) | $2.00 A-1 Warrant to Purchase Common Stock issued on April 8, 2004 to Ernest Pernet | |
| 4.14 (6) | $2.00 A-1 Warrant to Purchase Common Stock issued on April 8, 2004 to W.R. Hambrecht + Co., LLC | |
| 4.15 (7) | Common Stock and Warrant Purchase Agreement, dated April 19, 2004, between the registrant and Franklin M. Berger | |
| 4.16 (8) | Registration Rights Agreement, dated April 19, 2004, between the registrant and Franklin M. Berger | |
| 4.17 (9) | $2.00 A-1 Warrant to Purchase Common Stock issued on April 19, 2004 to Franklin M. Berger | |
| 4.18 (8) | Securities Purchase Agreement, dated July 21, 2005, among the registrant and the Purchasers (as defined therein) | |
| 4.19 (8) | Rights Agreement, dated July 27, 2005, among the registrant, the Icahn Purchasers and Viking (each as defined therein) | |
| 4.20 (9) | First Amendment to Rights Agreement, dated September 22, 2006, among the registrant and the Icahn Purchasers (as defined therein) | |
| 4.21 (8) | Form of $2.26 Common Stock Warrant issued on July 27, 2005 (including the original schedule of holders) | |
| 4.22 (8) | Form of $2.26 Common Stock Warrant issued on July 27, 2005 (including the original schedule of holders) |
Table of Contents
| Exhibit | Description | |
| 4.23 | $0.50 Warrant (WC-291) to Purchase Common Stock transferred on June 15, 2005 to S. Neborsky and R Neborsky TTEE Robert J. Neborsky MD Inc Comb Retirement Trust | |
| 4.24 (10) | $0.50 Warrant (WC-292) to Purchase Common Stock transferred on June 15, 2005 to S. Neborsky and R Neborsky TTEE Robert J. Neborsky MD Inc Comb Retirement Trust | |
| 4.25 (10) | $2.50 Warrant to Purchase Common Stock issued on October 22, 2004 to Thomas J. DePetrillo | |
| 10.1# (11) | 2005 Equity Incentive Plan | |
| 10.2# (12) | Form of Stock Option Agreement under the 2005 Equity Incentive Plan | |
| 10.3# (2) | Form of Restricted Share Award Agreement under the 2005 Equity Incentive Plan | |
| 10.4# (12) | 2005 Employee Stock Purchase Plan | |
| 10.5# (12) | Form of Subscription Agreement under the 2005 Employee Stock Purchase Plan | |
| 10.6* (13) | Option and License Agreement, dated January 23, 1998, between the registrant and the University of Southern California | |
| 10.7 (3) | First Amendment to License Agreement, dated August 16, 2000, between the registrant and the University of Southern California | |
| 10.8* (13) | Option and License Agreement, dated August 17, 2000, between the registrant and the University of Southern California | |
| 10.9* (14) | Amendment to Option and License Agreement, dated April 21, 2003, between the registrant and the University of Southern California | |
| 10.10* (2) | Agreement, effective as of May 1, 2005, between the registrant and Pharm-Olam International Ltd. | |
| 10.11 (2) | Amendment dated July 19, 2005 to the Agreement between the registrant and Pharm-Olam International Ltd. | |
| 10.12 (15) | License Agreement, dated October 20, 2006, between the registrant, through its wholly-owned subsidiary SD Pharmaceuticals, Inc., and Theragenex, LLC | |
| 10.13 | License Agreement, dated December 10, 2005, between SD Pharmaceuticals, Latitude Pharmaceuticals and Andrew Chen | |
| 10.14 (16) | Standard Multi-Tenant Office Lease — Gross, dated June 3, 2004, between the registrant and George V. Casey & Ellen M. Casey, Trustees of the Casey Family Trust dated June 22, 1998 | |
| 10.15 (2) | First Amendment to the Standard Multi-Tenant Office Lease — Gross, dated June 3, 2004 between the registrant and George V. & Ellen M. Casey, Trustees of the Casey Family Trust dated June 22, 1998 | |
| 10.16# (17) | Offer letter, dated March 5, 2003, to Joan M. Robbins | |
| 10.17# (18) | Offer letter, dated November 15, 2004, to Brian M. Culley | |
| 10.18# (18) | Offer letter, dated November 17, 2004, to Carrie Carlander | |
| 10.19# (19) | Severance Agreement and Release of All Claims, dated September 7, 2006, with Carrie Carlander | |
| 10.20# (19) | Consulting Agreement, dated September 7, 2006, with Carrie Carlander | |
| 10.21# (19) | Offer letter, dated September 7, 2006, to James A. Merritt | |
| 10.22# (19) | Form of Stock Option Agreement between the registrant and James A. Merritt (included in Exhibit 10.21) | |
| 10.23# (20) | Offer letter, dated December 13, 2006, to Gregory P. Hanson | |
| 10.24# (20) | Stock Option Agreement, effective December 20, 2006, between the registrant and Gregory P. Hanson | |
| 10.25 (21) | Form of Director and Officer Indemnification Agreement | |
| 10.26# (22) | Director compensation policy | |
| 10.27 (23) | Placement Agency Agreement, dated November 2, 2006, among the registrant, ThinkEquity Partners LLC and Fortis Securities LLC | |
| 14.1 (24) | Code of Business Conduct and Ethics |
Table of Contents
| Exhibit | Description | |
| 21.1 | List of Subsidiaries | |
| 23.1 | Consent of J.H. Cohn LLP, Independent Registered Public Accounting Firm | |
| 31.1 | Certification of chief executive officer pursuant to Rule 13a-14(a)/15d-14(a) | |
| 31.2 | Certification of chief financial officer pursuant to Rule 13a-14(a)/15d-14(a) | |
| 32.1 ± | Certification of chief executive officer and chief financial officer pursuant to 18 U.S.C. 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| * | Indicates that confidential treatment has been requested or granted to certain portions, which portions have been omitted and filed separately with the Securities and Exchange Commission | |
| # | Indicates management contract or compensatory plan | |
| ± | These certifications are being furnished solely to accompany this report pursuant to 18 U.S.C. 1350, and are not being filed for purposes of Section 18 of the Securities Exchange Act of 1934 and are not to be incorporated by reference into any filing of the registrant, whether made before or after the date hereof, regardless of any general incorporation language in such filing. | |
| (1) | Filed with the registrant’s Amendment No. 1 to Current Report on Form 8-K/A on May 1, 2006 | |
| (2) | Filed with the registrant’s Annual Report on Form 10-K on March 16, 2006 | |
| (3) | Filed with the registrant’s Registration Statement on Form 10SB on October 2, 2001 | |
| (4) | Filed with the registrant’s Registration Statement on Form S-3 on June 30, 2004 | |
| (5) | Filed with the registrant’s Amendment No. 1 to Quarterly Report on Form 10-Q/A on October 30, 2006 | |
| (6) | Filed with the registrant’s Current Report on Form 8-K/A on April 13, 2004 | |
| (7) | Filed with the registrant’s Quarterly Report on Form 10-QSB on May 12, 2005 | |
| (8) | Filed with the registrant’s Quarterly Report on Form 10-Q on August 12, 2005 | |
| (9) | Filed with the registrant’s Current Report on Form 8-K on September 22, 2006 | |
| (10) | Filed with the registrant’s Registration Statement on Form S-3 on August 26, 2005 | |
| (11) | A copy of the registrant’s 2005 Equity Incentive Plan was filed with the registrant’s Registration Statement on Form S-8 on July 13, 2005 but contained a typographical error. A correct copy of the registrant’s 2005 Equity Incentive Plan is filed with this report. | |
| (12) | Filed with the registrant’s Registration Statement on Form S-8 on July 13, 2005 | |
| (13) | Filed with the registrant’s Registration Statement on Form 10SB/A on January 14, 2002 | |
| (14) | Filed with the registrant’s Quarterly Report on Form 10-QSB on August 14, 2003 | |
| (15) | Filed with the registrant’s Current Report on Form 8-K on October 23, 2006 | |
| (16) | Filed with the registrant’s Quarterly Report on Form 10-QSB on August 10, 2004 | |
| (17) | Filed with the registrant’s Annual Report on Form 10-KSB on April 16, 2003 | |
| (18) | Filed with the registrant’s Annual Report on Form 10-KSB on March 31, 2005 | |
| (19) | Filed with the registrant’s Current Report on Form 8-K on September 8, 2006 | |
| (20) | Filed with the registrant’s Current Report on Form 8-K on December 20, 2006 | |
| (21) | Filed with the registrant’s Current Report on Form 8-K on October 23, 2006 | |
| (22) | Filed with the registrant’s Current Report on Form 8-K on June 23, 2006 | |
| (23) | Filed with the registrant’s Current Report on Form 8-K on November 3, 2006 | |
| (24) | Filed with the registrant’s Current Report on Form 8-K on January 23, 2007 |