SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| | | |
| (Mark One) | | |
þ | | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | | For the fiscal year ended December 31, 2007 |
OR |
o | | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | | For the transition period from to |
Commission File Number:000-50414
MIDDLEBROOK PHARMACEUTICALS, INC.
(Exact name of Registrant as specified in its Charter)
| | | |
| Delaware | | 52-2208264 |
(State or other jurisdiction of
incorporation or organization) | | (I.R.S. employer
identification number) |
| | | |
20425 Seneca Meadows Parkway
Germantown, Maryland
(Address of principal executive offices) | | 20876
(Zip Code) |
(301) 944-6600
(Registrant’s telephone number, including area code)
None
(Former name, former address and former
fiscal year — if changed since last report)
Securities registered pursuant to Section 12(b) of the Act:
None
Securities registered pursuant to Section 12(g) of the Act:
Common Stock, par value $0.01 per share
Indicate by check mark if the registrant is a well-known, seasoned issuer, as defined by Rule 405 of the Securities Act. Yes o No þ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Exchange Act. Yes o No þ
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes þ No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 ofRegulation S-K is not contained herein and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of thisForm 10-K or any amendment to thisForm 10-K. þ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| | | | | | | |
Large accelerated filer o | | Accelerated filer o | | Non-accelerated filer þ
(Do not check if a smaller reporting company) | | Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined inRule 12b-2 of the Exchange Act) Yes o No þ
As of June 30, 2007, the aggregate market value of the common stock held by non-affiliates of the registrant was approximately $54,765,164.
As of March 11, 2008, 55,676,625 shares of the registrant’s common stock were outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of MiddleBrook Pharmaceuticals, Inc.’s Notice of Annual Stockholders’ Meeting and Proxy Statement, to be filed within 120 days after the end of the registrant’s fiscal year, are incorporated by reference into Part III of this Annual Report.
MIDDLEBROOK PHARMACEUTICALS, INC.
INDEX
FORM 10-K
| | | | | | | | | |
| | | | | Page |
| |
PART I |
| | Item 1. | | | Business | | | 1 | |
| | Item 1A. | | | Risk Factors | | | 19 | |
| | Item 1B. | | | Unresolved Staff Comments | | | 32 | |
| | Item 2. | | | Properties | | | 32 | |
| | Item 3. | | | Legal Proceedings | | | 32 | |
| | Item 4. | | | Submission of Matters to a Vote of Security Holders | | | 33 | |
| |
PART II |
| | Item 5. | | | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities | | | 33 | |
| | Item 6. | | | Selected Financial Data | | | 36 | |
| | Item 7. | | | Management’s Discussion and Analysis of Financial Condition and Results of Operations | | | 37 | |
| | Item 7A. | | | Quantitative and Qualitative Disclosures About Market Risk | | | 58 | |
| | Item 8. | | | Financial Statements and Supplementary Data | | | 59 | |
| | Item 9. | | | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure | | | 59 | |
| | Item 9A. | | | Controls and Procedures | | | 59 | |
| | Item 9B. | | | Other Information | | | 60 | |
| |
PART III |
| | Item 10. | | | Directors, Executive Officers and Corporate Governance | | | 60 | |
| | Item 11. | | | Executive Compensation | | | 60 | |
| | Item 12. | | | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters | | | 60 | |
| | Item 13. | | | Certain Relationships and Related Transactions, and Director Independence | | | 61 | |
| | Item 14. | | | Principal Accountant Fees and Services | | | 61 | |
| |
PART IV |
| | Item 15. | | | Exhibits and Financial Statement Schedules | | | 61 | |
| | | | | Signatures | | | 65 | |
FORWARD-LOOKING STATEMENTS
This annual report onForm 10-K contains forward-looking statements, within the meaning of the Securities Exchange Act of 1934 and the Securities Act of 1933, that involve risks and uncertainties. In some cases, forward-looking statements are identified by words such as “believe,” “anticipate,” “expect,” “intend,” “plan,” “will,” “may” and similar expressions. You should not place undue reliance on these forward-looking statements, which speak only as of the date of this report. All of these forward-looking statements are based on information available to us at this time, and we assume no obligation to update any of these statements. Actual results could differ from those projected in these forward-looking statements as a result of many factors, including those identified in the sections titled “Risk Factors,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and elsewhere. We urge you to review and consider the various disclosures made by us in this report, and those detailed from time to time in our filings with the Securities and Exchange Commission, that attempt to advise you of the risks and factors that may affect our future results.
PART I
Corporate Overview
MiddleBrook Pharmaceuticals, Inc. (MiddleBrook or the Company) is a pharmaceutical company focused on developing and commercializing anti-infective drug products that fulfill substantial unmet medical needs in the treatment of infectious disease. We are developing a portfolio of drugs based on our novel biological finding that bacteria exposed to antibiotics in front-loaded, sequential bursts, or pulses, are killed more efficiently than those exposed to standard antibiotic treatment regimens. Based on this finding, we have developed a proprietary,once-a-day pulsatile delivery technology called PULSYS® (PULSYS). Our PULSYS technology is protected by a broad patent estate, with 26 patents currently issued in the U.S. and two foreign-issued patents, with the patent lives extending to 2020 and beyond. We own all rights to our PULSYS technology, and accordingly, there are no royalties due to any third party.
We have focused initially on developing PULSYS product candidates utilizing approved and marketed anti-infective drugs that no longer have patent protection or that have patents expiring in the next several years. Our lead PULSYS product candidate, based on the antibiotic amoxicillin, successfully completed a Phase III trial in 2006, and our New Drug Application (NDA) for the product under the trade name: MOXATAGtm (amoxicillin extended-release) Tablets was approved by the U.S. Food and Drug Administration (FDA) on January 23, 2008. MOXATAG is approved for the treatment of pharyngitis/tonsillitis (strep throat) for adults and pediatric patients age 12 and older. MOXATAG is the first and only once-daily amoxicillin treatment of pharyngitis/tonsillitis approved in the United States.
We have two PULSYS product candidates in clinical development. Our Keflex PULSYS product candidate, based on the antibiotic cephalexin, has gained FDA agreement for our Phase III clinical trial design and is ready for Phase III trials, subject to adequate financial resources. We believe the added convenience of improving Keflex from its typical two-to-four times per day dosing regimen to a once-daily product can create an attractive commercial opportunity. Our amoxicillin pediatric product candidate, which is a sprinkle formulation utilizing the antibiotic amoxicillin for use in pediatric patients under age 12, is ready to enter Phase II trials to determine the most efficient dosing regimen. The further development of both of these product candidates, as well as product candidates in preclinical development, is currently on hold, and will proceed only if we secure additional financial resources.
We currently market certain drug products which do not utilize our PULSYS technology and that are not protected by any other patents. We acquired the U.S. rights to the Keflex brand of cephalexin from Eli Lilly in 2004, which consisted primarily of the Keflex 250mg and 500mg capsules at that time. In May 2006, we received marketing approval from the FDA for two new strengths of Keflex, 750mg capsules and 333mg capsules. In July 2006, we began promoting Keflex 750mg capsules to targeted physicians through a dedicated contract sales force of 75 sales representatives and eight MiddleBrook district sales managers. In 2007, we reduced the number of
1
contract sales representatives to 30 and the number of district sales managers to three. Keflex product sales in 2007 were approximately $10.5 million, with $7.7 million attributable to Keflex 750mg. In November 2007, we entered into a transaction with Deerfield Management in which we sold our Keflex brand rights, including the trademark, and approved New Drug Applications for the Company’s existing, non-PULSYS Keflex products, to Deerfield. Under the transaction agreements, we have the right to repurchase the intangible assets sold at a future date, as well as to continue to utilize the Keflex trademark and other intangible assets in order to continue to operate the Keflex business, subject to certain consignment and royalty payments to Deerfield. We retained our rights to Keflex PULSYS intellectual property and technical data.
We are evaluating various strategic alternatives to further enhance shareholder value, and we retained an investment banking firm to assist us in this regard. Strategic alternatives we may pursue could include, but are not limited to, continued execution of our operating plan, licensing or development arrangements, the sale of some or all of our company’s assets, partnering or other collaboration agreements, or a merger or other strategic transaction. There can be no assurance that the exploration of strategic alternatives will result in any agreements or transactions, or that, if completed, any agreements or transactions will be successful or on attractive terms. The progress of our strategic alternatives process during 2008 will significantly affect the extent and timing of our MOXATAG commercial launch activities, as well as potential funding for research and development activities.
We were incorporated in Delaware in December 1999 and commenced operations in January 2000. Our principal executive offices are located at 20425 Seneca Meadows Parkway, Germantown, Maryland 20876. Our telephone number is(301) 944-6600. Our website is www.middlebrookpharma.com. Information contained on our website is not part of, and is not incorporated into, this annual report onForm 10-K. Our filings with the SEC are available without charge on our website as soon as reasonably practicable after filing. Our corporate name changed to MiddleBrook Pharmaceuticals, Inc. on June 27, 2007. Our previous corporate name was Advancis Pharmaceutical Corporation. MiddleBrook, MiddleBrook Pharmaceuticals, Inc., PULSYS, and MOXATAG are trademarks and trade names of MiddleBrook Pharmaceuticals, Inc. All other trademarks, trade names or service marks appearing in this annual report onForm 10-K are the property of their respective owners. All trademarks, trade names and logos that include the word “Advancis” were changed in June 2007. See Item 3,“Legal Proceedings”,below for more information.
Infectious Disease Market
Infectious diseases are caused by pathogens such as bacteria, viruses and fungi that enter the body through the skin or mucous membranes of the lungs, nasal passages and gastrointestinal tract, and overwhelm the body’s immune system. These pathogens establish themselves in various tissues and organs throughout the body and cause a number of serious and, in some cases, lethal infections.
We believe that the antibiotic market presents a highly attractive opportunity for the following reasons:
Substantial market. Antibiotics, along with antiviral medications and antifungal medications, constitute the primary categories of the anti-infectives market. According to sales data compiled by IMS Health, an independent pharmaceutical industry research firm, worldwide anti-infective sales were approximately $63 billion in 2006, including $20.8 billion in North America. Antibiotics accounted for approximately $33.3 billion of such 2006 worldwide sales, including more than $11 billion in North America (IMS World Review 2007).
Increased resistance to existing therapies. Certain medical, veterinary and agricultural practices and sociological factors have led to increased bacterial resistance to many currently available antibiotics. Bacterial resistance has been fostered through the erroneous prescription of anti-infective drugs for non-bacterial infections and unconfirmed infections and the administration of broad spectrum antibiotics before the specific disease-causing pathogen has been identified. In addition, the lack of patient compliance with prescribed course of therapies has contributed to bacterial resistance to currently marketed compounds. For example, it is estimated that one-third of all Streptococcus pneumoniae, a type of bacteria that can cause pneumonia, meningitis and ear infections are resistant to penicillin. The increased prevalence of resistant bacteria has resulted in prolonged hospitalizations, increased healthcare costs and higher mortality rates.
2
Growing need for improved new treatments. Social and demographic factors are contributing to the growth in the antibiotic market and the need for new, more effective therapies. The aging population of the United States is more likely to have suppressed immune systems and will require drugs that are effective against increasingly resistant strains of bacteria. Patients diagnosed with diseases that target the immune system, such as AIDS, increasingly require therapies that are more effective to combat infection. In addition, the pharmaceutical industry continues to develop therapeutics, such as cancer chemotherapy, that weaken the immune system as a side effect of the primary therapy. As a result, we believe there is a strong demand for new treatments that are more potent, more effective against resistant strains and that cause fewer side effects.
Difficulties in developing new classes of anti-infective compounds. We believe that the growing problem of resistance and other limitations of currently available antibiotics are not being adequately addressed. Moreover, many of the large pharmaceutical companies have reduced research and development efforts in this sector and others have stopped producing anti-infective products.
Limitations of standard treatment regimens. In addition to the increased incidence of antibiotic resistant bacteria, we believe that standard antibiotic treatment regimens have several other limitations, including multiple daily dosage requirements, lengthy treatment periods, limited effectiveness and severe side effects, all of which decrease patient compliance and ultimately, therapeutic efficacy.
Our Proprietary PULSYS® Technology
The significant unmet needs in the anti-infective market prompted our founders to search for a more efficient method to attack bacteria. In a series of seminal laboratory experiments, we observed that antibiotics such as amoxicillin can be more effective in killing bacteria when delivered in three to five discrete pulses of drug within the initial six to eight hours of a dosing interval. To take advantage of these experimental findings, we created a proprietary,once-a-day oral drug delivery technology called PULSYS®. PULSYS is designed to sequentially release specific portions of the drug dose, yielding a pulsatile pattern of antibiotic release. We believe that our novel finding, as implemented through our PULSYS technology, will potentially enable therapeutic advantages including:
| | |
| | • | Improved bactericidal activity, or bacteria killing efficiency. |
| |
| | • | Once-daily dosingand/or shorter length of treatment resulting in increased patient convenience and compliance. |
| |
| | • | Lower overall drug dose with a possibly reduced side effect profile. |
| |
| | • | Decreased emergence of antibiotic resistant bacteria. |
Our approach to improving antibiotic effectiveness represents a departure from traditional methods, which have focused on increasing drug dosages and searching for new classes of drugs. Our pulsatile dosing approach attempts to increase antibiotic effectiveness by better exploiting vulnerabilities in the growth cycle and natural defense mechanisms of bacteria.
3
The graph below conceptually illustrates drug concentration profiles in a patient’s bloodstream over a24-hour period comparing drugs administered through our PULSYS system with standard twice daily dosing. The standard dosing regimen reflects the administration of an immediate release tablet at the start of a day, followed by an additional immediate release tablet 12 hours later. The PULSYS profile reflects the administration of a single dose designed to release the drug in four front-loaded pulses, with no additional doses administered for the balance of the day.

PULSYS is a proprietary method of administering a pharmaceutical agent such that the active ingredient is given in a front loaded, sequential pulse fashion. PULSYS can be realized or practiced by administering a solid oral dosage form that may contain multiple units — for example, pellets or minitablets — with varying release profiles that are combined in a proportion to produce optimum medication levels during the first few hours after dosing. PULSYS can also be realized as other dosage forms such as topicals, transdermals, insertables, etc. We anticipate that our pulsatile drug products could each provide foronce-a-day dosing. We strive to utilize commonly-used inactive ingredients and common manufacturing processes when making PULSYS or any other type of anti-infective product. We are exploring the pulsatile administration of anti-infective agents in forms other than solid oral dosage forms.
PULSYS drug product candidates are evaluated using our proprietary design strategy. We are currently focusing on antibiotics, but also have proprietary positions in the antiviral, antifungal and oncology fields. This approach combines computer simulations with microbiology and other laboratory experiments to analyze the physical, chemical, biological and microbiological properties of each specific antibiotic in order to optimize selection and design of pulsatile drug candidates. This analysis includes an evaluation of the physicochemical properties and metabolism profiles of antibiotics as a function of position in the gastro-intestinal tract. We attempt to optimize overall antibiotic bioavailability by adjusting the timing and composition of pulses. By examining the bioavailability of antibiotics prior to the selection of PULSYS candidates, we believe that we will increase the likelihood of successful product development.
Our Strategy
We expect to use our novel finding and related proprietary technology to develop and commercialize more efficient, effective and convenient pharmaceutical products, with an initial focus on antibiotics. To achieve this objective, we have adopted the following product development and commercialization strategies:
Commercialize products with multiple advantages. We plan to develop PULSYS products that have multiple therapeutic advantages over currently available antibiotics, which may include once-daily dosing, lower doses, and in some cases, shorter treatment periods. We believe that these advantages will be further
4
reflected in at least some of our PULSYS products in fewer dose-related side effects, reduced incidence of resistance and improved efficacy.
Focus initially on existing antibiotics. We anticipate reducing development risk and expense and decreasing time to market for our drug candidates by focusing on improved versions of approved and marketed drugs, either delivered alone or in combination with other drugs. The additional benefits of developing improved formulations of existing and approved antibiotics include reasonable and predictable production costs and higher probability of market acceptance due to the use of well-known antibiotics. In addition, since these existing products have already been proven to be safe and effective, we anticipate being able to rely on existing approvals and existing safety and efficacy data, which would allow us to reduce the amount of new data that we will need to generate in order to support FDA approval of our products.
Focus on first-line, broad-spectrum antibiotics for community infections. We are pursuing a product development strategy focused primarily on first-line, broad spectrum antibiotics for community infections. Our pulsatile antibiotic products are expected to target upper respiratory tract infections and skin and skin structure infections in particular. The target indications for our current product candidates cover some of the top antibiotics-related diagnoses and are intended to compete against the top most-prescribed antibiotics. We believe products utilizing our front-loaded, pulsed dosing approach will support once-daily dosing where two-to-four times daily dosing is the norm, with a concomitant reduction in dose and treatment duration (in some cases) compared to current traditional therapies.
Multi-level patent strategy. We have implemented a multi-level patent strategy in order to protect our approved and potential future pulsatile drug products. The first level is composed of “umbrella” patents and patent applications to protect the PULSYS administration of general classes of anti-infective drugs, such as antibiotics, antivirals, and antifungals, as well as anti-cancer. The second level is composed of “sub-umbrella” patents and patent applications, protecting the PULSYS administration of subclasses of drugs, such as beta-lactam antibiotics with enzyme inhibitors. The third level includes patents and applications for specific anti-infective agents. We intend to continue to use and enhance this strategy in order to protect our intellectual property. We currently own 25 issued U.S. patents, 23 U.S. patent applications, and 2 issued foreign patents.
License or acquire antibiotic products. We continue to explore pulsatile formulations for a wide range of other antibiotics and antibiotic combinations and, assuming we have sufficient financial resources, we may in-license or acquire antibiotic products that we believe can be improved with our novel pulsatile dosing approach.
Our Approved and Marketed Products
| | | | | | | |
Products | | Key Indication(s) | | Status | | Marketing Rights |
| |
MOXATAGtm (amoxicillin extended-release) Tablets — 775 mg | | Pharyngitis/tonsillitis | | FDA-approved (January 23, 2008) | | Worldwide rights (100% ownership — no royalties due to any third party) |
Keflex® (cephalexin capsules, USP) — 250 mg, 333 mg, 500 mg, and 750 mg | | Skin and skin structure infections; upper respiratory tract infections | | Marketing | | U.S. and Puerto Rico rights (royalties to Eli Lilly and to Deerfield) |
Our Lead Product: MOXATAGtm (amoxicillin extended-release) Tablets
On January 23, 2008, we received FDA approval of our New Drug Application (NDA) for our once-daily amoxicillin PULSYS product, under the trade name, MOXATAGtm (amoxicillin extended-release) Tablets. MOXATAG is approved for the treatment of pharyngitisand/or tonsillitis secondary toStreptococcus pyogenes(strep throat) in adults and pediatric patients 12 years or older.
MOXATAG is aonce-a-day extended-release formulation of amoxicillin for oral administration consisting of three components: one immediate-release and two delayed-release. The three components are combined in a specific ratio to prolong the release of amoxicillin from MOXATAG compared to immediate-release amoxicillin.
5
MOXATAG is intended to provide a lower treatment dose, once-daily alternative to currently approved penicillin and amoxicillin regimens for the treatment of adults and pediatric patients 12 years and older with tonsillitisand/or pharyngitis. We utilized the Company’s proprietary PULSYS® once-daily pulsatile delivery technology to develop MOXATAG. We currently have a total of 26 issued U.S. patents and two issued foreign patents covering our PULSYS technology. Patents specifically relating to MOXATAG run to 2020.
MOXATAG is the first and only once-daily aminopenicillin therapy approved by the FDA to treat strep throat. According to prescription data from IMS Health, more than 30 million prescriptions were written for strep throat, pharyngitis and tonsillitis in the U.S. in 2007.
Clinical Development of MOXATAG
Our MOXATAG product successfully concluded a Phase III clinical trial for the treatment of pharyngitis/tonsillitis for adults and pediatric patients 12 years and older using a10-day treatment period in August 2006. We conducted our first Phase III clinical trial for adult and adolescent pharyngitis in 2005, using a shorter7-day duration of therapy, which failed to achieve its desired microbiological and clinical end points.
Our re-designed non-inferiority Phase III trial conducted in 2005/2006 enrolled 618 adult and adolescent patients in 50 centers in the U.S. and Canada. We compared our MOXATAG tablet for the treatment of pharyngitis/tonsillitis due to S. pyogenes (Group A streptococcus) delivered in a once-daily, 775 milligram tablet for a period of 10 days to 250 milligrams of penicillin dosed four times daily, for a total of one gram per day, for 10 days. MOXATAG demonstrated statistical non-inferiority to the comparator therapy in the trial’s primary endpoints, which were eradication of all bacteria as determined during the post-therapy “test-of-cure” visit for patients who fully complied with the trial protocol, as well as in a larger patient population. MOXATAG also demonstrated non-inferiority in the trial’s secondary endpoints, including clinical cure at the test-of-cure visit and bacterial eradication at the late post-therapy visit.
Based on the results of our successful Phase III trial, we submitted a New Drug Application (NDA) for MOXATAG on December 14, 2006. On February 12, 2007, we received a “refusal to file” letter from the FDA, indicating that they required additional data on our proposed commercial manufacturing process in order to accept our application for filing. In its letter, the FDA indicated that our application was not sufficiently complete in that it did not include a proposed commercial batch record or a detailed commercial process description with process parameters and in-process controls. We conducted a meeting with the FDA regarding our MOXATAG NDA on February 26, 2007. In that meeting, we reached agreement with the FDA on the additional information that is required for our NDA filing to be accepted by the FDA. Based on the outcome of the FDA meeting, we resubmitted the NDA to the FDA for our MOXATAG product on March 23, 2007. The NDA was accepted for filing and we received a FDA target action date in January 2008. On January 23, 2008, we received an approval letter from the FDA. With approval, MOXATAG is now the first and only once-daily amoxicillin treatment of pharyngitis/tonsillitis approved in the United States.
MOXATAG U.S. Market Opportunity
Amoxicillin is the most widely prescribed antibiotic drug in the United States. We believe the market opportunity for a once-daily amoxicillin product is substantial, with approximately 55 million prescriptions written for traditional multiple-times per day amoxicillin formulations in 2007 (IMS National Prescription Audit 2007).
Amoxicillin (marketed by GSK as Amoxil and marketed by other companies as a generic product) is a semi-synthetic antibiotic that is effective for the treatment of a variety of conditions, including ear, nose and throat infections, urinary tract infections, skin infections and lower respiratory infections. In 2007, amoxicillin had U.S. retail sales of approximately $1.1 billion, based on branded retail pricing of $20 per prescription. Approximately one-quarter of amoxicillin’s use is estimated to be for the treatment of pharyngitisand/or tonsillitis. Amoxicillin is generally recommended for dosing two or three times daily, for a period of ten to 14 days.
We believe MOXATAG will compete effectively in the strep throat segment of the antibiotic market due to its once-daily dosing and favorable side effect profile. We also expect MOXATAG to compete most directly against generic amoxicillin therapies and to a lesser degree against other common strep throat therapies such as penicillin,
6
cephalosporins, and amoxicillin/clavulanate. According to prescription data from IMS Health, more than 30 million prescriptions were written for strep throat, pharyngitis and tonsillitis in the U.S. in 2007.
Today in the United States, the most frequently prescribed pharyngitis prescription is for 500mg of amoxicillin three times daily for ten days, or 15 grams total over the course of therapy. In addition, amoxicillin is the most commonly mentioned antibiotic associated with the pharyngitis/tonsillitis diagnosis. Our MOXATAG product for adults and pediatric patients 12 years and older is dosed 775mg once-daily for ten days, or 7.75 grams total per course of therapy. Therefore, physicians prescribing MOXATAG would be able to dose approximately one-half the amount of amoxicillin, while also providing the convenience of once-daily dosing versus a typical amoxicillin therapy.
As part of our ongoing strategic evaluation process, we are currently evaluating commercialization options for our MOXATAG product. We believe the MOXATAG market opportunity would be best addressed through its direct promotion by a national sales force of at least 300 sales representatives. As such, a commercialization initiative would require significant resources and expertise, and we believe it would be in the Company’s best interests to seek potential acquirers or partners to capitalize on MOXATAG’s commercial potential. Potential sales and marketing strategies for MOXATAG include the acquisition of the Companyand/or MOXATAG by a larger pharmaceutical organization with an established commercialization infrastructure, working with contract sales organizations, developing our own internal sales organization, or co-promoting products with collaborative marketing partners. Through the anticipated commercialization efforts for MOXATAG, we would expect to target high-volume prescribers with a community-based contract sales force detailing physicians, including family practitioners and internists.
Even if we successfully conclude our strategic evaluation process and identify a third party to assist in the commercialization of MOXATAG, the earliest we could launch the product would be in the fourth quarter of 2008. In addition, in order for the Company to participate in the sales and marketing of MOXATAG, we would need to have sufficient financial resources, which will require us to raise additional capital. These forward-looking statements are based on information available to us in March 2008.
MOXATAG International Market Opportunity
We own the worldwide rights to MOXATAG. In addition to sales in the U.S., we believe there will be the opportunity for us to earn additional revenue from sales of MOXATAG in other countries. Our international commercialization strategy is currently being evaluated, and may include the outsourcing of the sales and marketing functions to others, in exchange for royalties or other financial consideration.
Marketed Products — Keflex (non-PULSYS)
Keflex is a first-generation cephalosporin approved for treatment of several types of bacterial infections. Keflex is most commonly used in the treatment of uncomplicated skin and skin structure infections and, to a lesser extent, upper respiratory tract infections. Keflex is among the most prescribed antibiotics in the U.S.; however, generic competition is intense, and a high percentage of all Keflex prescriptions are substituted by generic versions of cephalexin, the active ingredient in Keflex.
We have the exclusive U.S. rights to manufacture, sell and market Keflex pursuant to our purchase agreement with Eli Lilly and Company and pursuant to subsequent agreements with Deerfield Management. On June 30, 2004, we acquired the U.S. rights to the Keflex brand of cephalexin from Eli Lilly for a purchase price of $11.2 million, including transaction costs, which were paid in cash from our working capital. The asset purchase includes the exclusive rights to manufacture, sell and market Keflex in the United States (including Puerto Rico). We also acquired Keflex trademarks, technology and new drug applications (NDAs) supporting the approval of Keflex capsules and oral suspension. On December 9, 2004, we announced that we entered into a commercial supply agreement with Ceph International Corporation, a wholly owned subsidiary of Patheon’s MOVA Pharmaceutical Corporation, to secure a long-term supply for Keflex products beyond the transitional period.
On May 12, 2006, the FDA approved two new strengths of Keflex for marketing — 750mg and 333mg capsules. We decided to focus our commercialization efforts solely on Keflex 750mg capsules. We believe the
7
introduction of Keflex 750mg capsules allows physicians the flexibility to deliver higher doses of Keflex with fewer capsules per day. In July 2006, we began promoting Keflex 750mg capsules across the U.S. to targeted high-prescribing physicians through a dedicated national contract sales force of 75 sales representatives and eight MiddleBrook district sales managers. We market Keflex in the U.S. to healthcare practitioners, pharmacists, pharmaceutical wholesalers and retail pharmacy chains.
In addition to our ongoing sales and marketing responsibilities for non-PULSYS Keflex products, we have initiated a research program with the goal of developing aonce-a-day cephalexin product utilizing our proprietaryonce-a-day PULSYS dosing technology. In the event we are able to develop and commercialize a PULSYS-based Keflex product, other cephalexin products relying on the acquired NDAs, or other pharmaceutical products using the acquired trademarks, Eli Lilly will be entitled to royalties on these new products. Our Keflex 750 mg product (and our potential Keflex 333 mg product, should we decide to commercialize it) is subject to the royalty. Royalties are payable on a new product by new product basis for five years following the first commercial sale for each new product, up to a maximum aggregate royalty per calendar year. All royalty obligations with respect to any defined new product cease after the fifteenth anniversary of the first commercial sale of the first defined new product.
On November 7, 2007, we sold certain non-PULSYS Keflex assets and licensed certain non-PULSYS Keflex intellectual property to Deerfield Management. However, we continue to operate the Keflex business, subject to consignment and royalty payments to Deerfield. See“Keflex Agreements — Deerfield Transaction”below.
Our Product Pipeline
The following table summarizes the antibiotic compounds we have in clinical trials and preclinical development. We expect that these compounds will serve as the basis for drug products or, with additional clinical development, drug combination products. Each of our preclinical product candidates is still in the early stage of development, and their further clinical progress requires significant additional capital expenditures that would be completely dependent upon our ability to obtain additional financing. Due to our on-going research and development efforts, additional or alternative compounds may be selected to replace or supplement the compounds described below.
| | | | | | | | | |
PULSYS Product
| | | | | | Targeted PULSYS
| | |
Candidate/Program | | Key Indication(s) | | Current Therapy | | Added Value | | Program Status(1) |
| |
| Keflex (cephalexin) — Adult | | Skin and skin structure infections | | 7-14 days, two to four times daily | | 10-days, once-daily, lower dose | | Phase III-ready (on-hold) |
| Amoxicillin Pediatric Pharyngitis — sprinkle | | Pharyngitis/tonsillitis | | 10-14 days, two or three times daily | | Shorter course of therapy, once-daily | | Phase II-ready (on-hold) |
| | |
| (1) | | For an explanation of the terms Preclinical, Phase I, Phase I/II and Phase III, please refer to the information under the heading “Government Regulation” below. Each of the product candidates above is discussed in more detail in the next section below. |
A significant portion of our expenses may be related to research and development of investigational stage product candidates. Please see“Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations”for a detailed discussion of our research and development expenses. In the event that we are unable to raise additional capital, we may be forced to discontinue or alter our development programs.
Pulsatile Product Candidates
We intend to develop the pulsatile drugs listed above and those in preclinical development, with the intention of incorporating one or more of the following improvements:
| | |
| | • | Once-a-day formulation |
| |
| | • | Lower dose |
| |
| | • | Shorter duration of therapy |
8
| | |
| | • | Reduced side effect profile |
| |
| | • | Combination product with superior efficacy over either product alone |
| |
| | • | Improved pediatric dosage form |
Currently, our drug product candidates primarily represent improved versions of approved and marketed drugs, either delivered alone or in combination with other drugs. Since these existing drugs have already been approved for marketing by the FDA, we anticipate being able to rely, in part, on the FDA’s prior findings on the safetyand/or efficacy of these existing drugs in seeking FDA approval of our PULSYS products. For example, based on meetings with the FDA regarding the study program for our amoxicillin products, we filed our New Drug Application via the 505(b)(2) regulatory pathway for our MOXATAG product, which, in part, relied on the FDA’s prior findings regarding the safety and efficacy of amoxicillin.
Keflex (Cephalexin) PULSYS
We are developing a once-daily PULSYS version of Keflex, our first generation oral cephalosporin antibiotic. Our intent is to develop a once-daily Keflex PULSYS for uncomplicated skin and skin structure infections. Currently, Keflex (or, in its generic form, cephalexin) is the antibiotic most frequently prescribed by physicians in the treatment of uncomplicated skin and skin structure infections. Most commonly, Keflex is prescribed 500mg three times per day for a period of ten days. We believe a once-daily version of Keflex PULSYS may represent a substantial market opportunity. In 2007, cephalexin, the active ingredient in Keflex, was the third most prescribed antibiotic in the United States, with approximately 23 million prescriptions (IMS National Prescription Audit 2007). Assuming branded retail pricing of $30 per prescription, we estimate that the cephalexin market opportunity has a value of approximately $690 million.
We have completed a total of six Keflex PULSYS Phase I clinical studies, evaluating various pulsatile formulations of Keflex dosed in a total of more than 150 healthy volunteer subjects. Based on the results from our Phase I studies, we believe we have finalized the formulation development Phase I program for our Keflex PULSYS product candidate.
On June 25, 2007, we completed a meeting with the FDA’s Division of Anti-Infective and Ophthalmology Products to discuss our Phase III trial and regulatory strategy to support product approval for Keflex PULSYS for the treatment of uncomplicated skin and skin structure infections (uSSSIs) in adults and adolescents due to susceptibleStaphylococcus aureusand/orStreptococcus pyogenes. We believe our planned non-inferiority Phase III clinical trial design and regulatory strategy for Keflex PULSYS were acceptable to the FDA.
Our anticipated Phase III trial is designed as a two-arm, double-blind, non-inferiority trial with a minimum enrollment of 600 patients. We expect to compare our 1200 milligram Keflex PULSYS product administered once-daily for 10 days to 250 milligrams of Keflex dosed four-times daily, for a total daily dose of 1000 milligrams, for 10 days. These forward-looking statements are based on information available to us at this time. Actual results could differ because our trial results could be delayed or unsuccessful or due to delays in FDA approval, which may never occur.
Our once-daily Keflex PULSYS product candidate is designed to increase the convenience of cephalexin therapy, which is currently dosed two to four times daily for a period of seven to 14 days. Cephalexin is commonly prescribed as a first-line therapy for common uncomplicated skin infections such as impetigo (skin lesions), simple skin abscesses, and cellulitis (acute inflammation of connective tissue of the skin). There is currently no once-daily cephalexin product approved for marketing in the United States.
Amoxicillin Pediatric Pharyngitis Program
We have developed two amoxicillin PULSYS formulations, our MOXATAG tablet approved for adults and pediatric patients age 12 and older and a pediatric sprinkle. Our pediatric sprinkle product utilizes a similar formulation to the adult product; however, it is dosed in multiparticulate granules designed to be sprinkled over food. Survey results from patients and caregivers utilizing our pediatric sprinkle product suggest that its convenience and transportability may be beneficial features of our sprinkle formulation, and we expect to utilize our
9
sprinkle presentation as the method of dosing our amoxicillin pediatric product. We believe the market opportunity for a pediatric strep throat product is substantial, as more than half of the strep throat market is believed to be represented by pediatric patients.
In 2005, we concluded a Phase III clinical trial evaluating once-daily amoxicillin PULSYS in pediatric patients with pharyngitis/tonsillitis (strep throat) which failed to achieve its desired clinical endpoints. However, we believe there is potential for us to pursue a pulsatile version of amoxicillin for the treatment of pediatric patients with strep throat through a redesigned clinical trial program. In 2006, we completed a Phase I study evaluating the observed drug concentrations from various pulsatile sprinkle amoxicillin formulations in healthy volunteer subjects. Based on the results from the 2006 study along with our Phase I studies, we intend to evaluate the safety and efficacy of various daily doses and durations of treatment for our pediatric amoxicillin PULSYS product candidate in a Phase II study, should we have sufficient capital and other resources to do so.
As part of our FDA approval of MOXATAG on January 23, 2008, in adults and pediatric patients 12 years and older and in accordance with the requirements of the Pediatric Research Equity Act, we received from the FDA a deferral to further evaluate our product candidate for pediatric patients less than 12 years of age with pharyngitisand/or tonsillitis as part of a post-marketing commitment. Should the results of the Phase II study support proceeding into Phase III, we may design and conduct a Phase III trial in this population. We agreed to submit a completed study report and data set for MOXATAG in pediatric patients less than 12 years old within the next five years as part of this commitment. If the results of the Phase II study do not support proceeding into Phase III, we may file a request for a waiver for the assessment of the safety and effectiveness of the product in this population.
Other Possible Pulsatile Product Candidates
Our current focus is on the antibiotic product candidates that include amoxicillin and Keflex. We have also identified additional product candidates which we believe could be developed for delivery in a pulsatile manner. The timing of further development work on these candidates depends on our financial and other resources as well as our evaluation of the commercial potential of the products.
PULSYS Preclinical Development Programs
We have identified several additional opportunities to apply PULSYS technology to develop individual and combination antibiotic products which we believe could have application against the following indications:
| | |
| | • | Sinusitis |
| |
| | • | Chronic Bronchitis |
| |
| | • | Acute Otitis Media |
| |
| | • | Urinary Tract Infections |
| |
| | • | Community-acquired Methicillin Resistant Staphylococcus aureus (MRSA). |
We have conducted preclinical studies evaluating the bacterial killing efficiency of several antibiotics and antibiotic combinations dosed in a pulsatile manner. Based on these studies, along with the consultation of our scientific advisors, we believe we may be able to utilize our PULSYS technology in the creation of antibiotic product candidates that target some of the most common uses of antibiotics. These uses include: sinusitis, chronic bronchitis, acute otitis media, urinary tract infections, and community-acquired methicillin resistant Staphylococcus aureus (MRSA). We have currently placed all of these development programs on hold, and any plans for additional studies regarding these, or any other possible product candidates, will be dependent on our ability to raise sufficient capital to fund these types of studies.
We may also explore the use of our pulsatile dosing approach beyond antibiotics to other therapeutic categories, such as antivirals and antifungals, in the event that we have sufficient financial and other resources to do so. Although we have not tested the effectiveness of pulsatile dosing for these applications, we believe that our approach may yield benefits similar to those we have found for the treatment of bacterial infections.
Patent and Intellectual Property Protection
Our success depends in part on our ability to obtain patents, to protect trade secrets, to operate without infringing upon the proprietary rights of others and to prevent others from infringing on our proprietary rights. We
10
seek to protect our proprietary position by, among other methods, filing U.S. and foreign patent applications related to our proprietary technology, inventions and improvements that are important to the development of our business. Further, all of our employees have executed agreements assigning to us all rights to any inventions and processes they develop while they are employed by us. In addition, we may use license agreements to access external products and technologies, as well as to convey our own intellectual property to others. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary rights are covered by valid and enforceable patents or are effectively maintained as trade secrets. Protection of our intellectual property rights is subject to a number of risks.
Number of patents. We currently own 26 issued U.S. patents, 23 pending U.S. patent applications and 2 issued foreign patents. Our issued patents cover certain compositions and methods using pulsatile dosing. We also own 66 foreign-filed patent applications, which correspond to our U.S. patents and applications. We also own four International (PCT) patent applications, each of which International (PCT) patent applications we anticipate converting into several individually foreign-filed patent applications to further correspond to our U.S. patents and applications.
Multi-level patent strategy for PULSYS. We have implemented a multi-level patent strategy in order to protect our approved and potential future pulsatile drug products. The first level is composed of “umbrella” patents and patent applications to protect the PULSYS administration of general classes of anti-infective drugs, such as antibiotics, antivirals, antifungals and anti-cancer. The umbrella patents cover all drugs in class,once-a-day dosing, and all methods of dosing. The second level is composed of “sub-umbrella” patents and patent applications, protecting the PULSYS administration of subclasses of drugs, such as beta-lactam antibiotics with enzyme inhibitors. The third level includes patents and applications for specific anti-infective agents. We intend to continue to use and enhance this strategy in order to protect our intellectual property.
Expiration dates for key patents. Our general PULSYS antibiotic patents were issued in 2003 and 2004, and will continue to run for a number of years. The earliest patent expiration is in October 2020.
No royalties on PULSYS patents. We have developed all of our PULSYS technology in-house, and we have retained full ownership of the related patents. Thus, we owe no royalties to any third party for utilizing the PULSYS technology in our products.
Sales and Marketing
We received marketing approval for Keflex 750mg capsules from the FDA in May 2006. In June 2006, we entered into marketing agreements with Innovex, the commercialization division of Quintiles Transnational Corporation, to provide us contract sales and marketing services for the promotion of Keflex 750mg capsules. We launched Keflex 750mg capsules in July 2006.
We currently have a targeted and dedicated national contract sales force of approximately 30 sales representatives and three MiddleBrook district sales managers promoting Keflex 750mg capsules to high-prescribing physicians across the U.S. We are also selling Keflex 250mg capsules and 500mg capsules; however, we do not actively promote those Keflex strengths as their markets are dominated by generic equivalents. We are currently focusing our sales and marketing initiatives solely on the promotion of Keflex 750mg capsules in an effort to maximize the impact from what we believe to be our most significant Keflex market opportunity. We also have a small marketing staff supporting our sales of the Keflex brand of cephalexin. The selling and marketing of Keflex 750mg capsules is a substantial financial commitment for our company. As a result, we may consider alternative commercialization strategies for our Keflex 750mg capsules in 2008 in an effort to reduce the financial resources required to market the productand/or to maximize the profit derived from the product.
Keflex is primarily sold directly to pharmaceutical wholesalers. In the pharmaceutical industry there are a limited number of major wholesalers responsible for the majority of sales. Product sales of Keflex to Cardinal Health Inc., McKesson Corporation, and AmerisourceBergen Corporation represented approximately 94 percent of our net revenue from Keflex in 2007.
We believe that a significant percentage of prescriptions for first-line, broad-spectrum antibiotics is written by high-volume prescribers who can be reached by a community-based sales force. Over time, and if we are successful
11
in obtaining sufficient capital, we may expand our sales and marketing capabilities to provide greater support of our target audience. We may also enter into agreements with other parties to capitalize on their sales and marketing capabilities in order to optimally market our products.
We currently manage the distribution of our Keflex products, including warehousing and shipping, through Integrated Commercialization Solutions, a division of AmerisourceBergen Corporation.
If we successfully develop and receive regulatory approval to market additional product candidates, we believe we will have to substantially expand our sales and marketing capabilitiesand/or enter into partnerships with other pharmaceutical companies to successfully commercialize our product candidates. We will need to successfully recruit additional sales and marketing personnel and build our sales and marketing infrastructure to successfully commercialize MOXATAG and any additional products or product candidates that we develop, acquire or license. Additional financial resources would be required to expand our current sales and marketing capabilities. Our future profitability will depend in part on our ability to develop additional sales and marketing capabilities to commercialize our future products to our target audiences.
Competition
The pharmaceutical industry is highly competitive and characterized by a number of established, large pharmaceutical companies, as well as smaller emerging companies. Our main competitors are:
| | |
| | • | Large pharmaceutical companies, such as Pfizer, GlaxoSmithKline, Wyeth, Bristol-Myers Squibb, Merck, Johnson & Johnson, Roche, Schering-Plough, Novartis, sanofi-aventis, Abbott Laboratories, AstraZeneca, and Bayer, that may develop new drug compounds that render our drugs obsolete or noncompetitive. |
| |
| | • | Smaller pharmaceutical and biotechnology companies and specialty pharmaceutical companies engaged in focused research and development of anti-infective drugs, such as Trimeris, Vertex, Adams Respiratory Therapeutics (acquired by Reckitt Benckiser), Gilead Sciences, Cubist, Basilea, Replidyne, InterMune, Oscient, King, Advanced Life Sciences, and others. |
| |
| | • | Drug delivery companies, such as Johnson & Johnson’s Alza division, Biovail, DepoMed, Flamel Technologies, and SkyePharma, which may develop a dosing regimen that is more effective than pulsatile dosing. |
| | |
| | • | Generic drug companies, such as Teva, Ranbaxy, IVAX, Sandoz and Stada, which produce low-cost versions of antibiotics that may contain the same active pharmaceutical ingredients as our PULSYS product candidates. |
There are many approved antibiotics available to treat bacterial conditions in the United States. Our marketed Keflex products, our approved MOXATAG product, and products that are in development, will compete with other available products based primarily on:
| | |
| | • | efficacy |
| |
| | • | safety |
| |
| | • | tolerability |
| |
| | • | acceptance by doctors |
| |
| | • | patient compliance and acceptance |
| |
| | • | patent protection |
| |
| | • | convenience |
| |
| | • | price |
| |
| | • | insurance and other reimbursement coverage |
| |
| | • | distribution |
| |
| | • | marketing |
| |
| | • | adaptability to various modes of dosing. |
12
Our Keflex brand of cephalexin faces significant competition from generic distributors of cephalexin capsules and suspension. Currently, a significant portion of the prescriptions written for our Keflex 250mg and 500mg capsules are substituted at the pharmacy with generic versions of Keflex, supplied through leading generic drug manufacturers including Teva, Stada, IVAX, Ranbaxy, and others. In addition, our Keflex 750mg capsules are not covered by patent protection and thus would be subject to similar competition from generic versions of Keflex 750mg capsules if and when generic drug manufacturers decide to pursue the manufacture and marketing approvals required for a generic 750mg strength cephalexin product.
In some instances, our novel products that utilize our PULSYS technology may compete against non-pulsatile drug products that share the same active ingredient, but are less convenient or require more cumbersome administration schedules. A number of these non-pulsatile drug products are available in generic form, which are usually substantially less expensive than the branded version. Companies such as Teva, Ranbaxy, IVAX, Sandoz and Stada, and others are major manufacturers and distributors of generic versions of antibiotics with whom we may compete in the future.
New developments, including the development of methods of preventing the incidence of disease, such as vaccines, occur rapidly in the pharmaceutical industry. These developments may render our product candidates or technologies obsolete or noncompetitive.
Many of our competitors possess greater financial, managerial and technical resources and have established reputations for successfully developing and marketing drugs, all of which put us at a competitive disadvantage. Our competitors may be able to apply their resources and capabilities to develop and commercialize products that have distinct, enhanced, or perceived advantages versus our products. The competitors may be in a position to devote greater resources in the sales, marketing, and distribution of these products and therefore considerably impact our ability to successfully commercialize our own products.
Manufacturing
We currently rely on third-party contract manufacturers to produce sufficient quantities of our product candidates for use in our preclinical studies and clinical trials, and to produce sufficient quantities of commercial supplies of our marketed products. We believe that our initial focus on the production of improved formulations of approved and marketed drugs will reduce the risk and time involved in the development of manufacturing capabilities because production of these drugs involves well-established and well-accepted manufacturing techniques and processes. We intend to continue to rely upon third-party contract manufacturers for production of our clinical and commercial supplies for beta lactam products and product candidates. The use of third-parties for these activities allows us to minimize our initial capital investment and reduce the risk that would be associated with the establishment of our own commercial manufacturing and distribution operations. With the possible transition to non-beta lactam product candidates, we anticipate that our pilot facility could satisfy our drug production needs for clinical supplies through at least Phase II and, in some cases, through Phase III clinical trials.
In December 2004, we entered into a commercial supply agreement with Ceph International Corporation, a wholly owned subsidiary of Patheon’s MOVA Pharmaceutical Corporation, to secure a long-term supply for our Keflex brand of products. This agreement provides for commercial supply of our Keflex product beyond the transitional period agreed to by Eli Lilly as part of our June 2004 acquisition.
In April 2005, we entered into agreements under which Stada Production Ireland Limited (“SPI”), previously known as the manufacturing division of Clonmel Healthcare Limited, a subsidiary of Stada Arzneimittel AG, will provide us with commercial supply of our MOXATAG products. SPI has capacity in place to cover current projected needs for an initial commercial phase, with additional capacity for growth. In addition to commercial supply, MiddleBrook and SPI have also finalized an agreement for technology transfer, clinical/stability batches and commercialscale-up and validation, as well as an agreement covering MiddleBrook-funded facility build-out and equipment additions to support the commercial manufacturing program.
In connection with our manufacturing activities, we generate hazardous waste. We are subject to federal and state regulation regarding the disposal of hazardous and potentially hazardous waste. We may incur costs to comply with such regulations now or in the future.
13
Collaboration Agreements
We currently have no active collaboration agreements. We have previously been a party to collaborations with GSK and with Par Pharmaceuticals.
Termination of Our Collaboration with Par Pharmaceutical for Amoxicillin PULSYS
In May 2004, we entered into an agreement with Par Pharmaceutical to collaborate in the further development and commercialization of a PULSYS-based amoxicillin product. Under the terms of the agreement, we conducted the development program, including the manufacture of clinical supplies and the conduct of clinical trials, and were responsible for obtaining regulatory approval for the product. We were to own the product trademark and to manufacture or arrange for supply of the product for commercial sales. Par was to be the sole distributor of the product. Both parties were to share commercialization expenses, including pre-marketing costs and promotion costs, on an equal basis. Operating profits from sales of the product were also to be shared on an equal basis. Under the agreement, we received an upfront fee of $5.0 million and a commitment from Par to fund all further development expenses. Development expenses incurred by us were to be partially funded by quarterly payments aggregating $28 million over the period of July 2004 through October 2005, of which up to $14 million was contingently refundable.
On August 3, 2005, we were notified by Par of its decision to terminate the Amoxicillin PULSYS collaboration agreement. Under certain circumstances, the termination clauses of the agreement may entitle Par to receive a share of net profits up to one-half of their $23.25 million funding of the development of certain Amoxicillin PULSYS products, should a product covered by the agreement be successfully commercialized. Accordingly, we retained deferred revenue of $11.625 million related to the agreement.
Termination of the Collaboration with GlaxoSmithKline
In July 2003, we entered into a license agreement with GlaxoSmithKline (GSK) pursuant to which we licensed patents and PULSYS technology to GSK for use with its Augmentin (amoxicillin/clavulanate combination) products and with limited other amoxicillin products. Under the agreement, GSK was responsible, at its cost and expense, to use commercially reasonable efforts for the clinical development, manufacture and sale of the licensed products. We received an initial non-refundable payment of $5 million from GSK upon signing of the agreement, and a $3 million payment upon achievement of the first milestone. Our receipt of further milestone payments, royalty payments and sales milestone payments under the agreement depended on the ability of GSK to develop and commercialize the products covered by the agreement and was subject to certain conditions and limitations. The agreement could be terminated at any time by GSK, which it elected to do with an effective date of December 15, 2004. As a result of the termination, we accelerated the recognition of the remaining deferred revenue of approximately $3.2 million related to the collaboration during the fourth quarter of 2004. The termination had no other effects on our financial position.
Collaboration with Par Pharmaceutical for Generic Clarithromycin
In September 2003, we entered into an agreement pursuant to which we licensed to Par Pharmaceutical the distribution and marketing rights to our generic formulation of Abbott’s Biaxin XL (extended release clarithromycin). During the third quarter of 2004, we conducted bioequivalence studies on two revised formulations of the generic product, with both formulations failing to achieve bioequivalence. We concluded that due to the non-core nature of the product, the expense involved in the development of additional formulations, and the reduced market potential given the emergence of competing products, we would discontinue further development work on the product.
Keflex Agreements — Deerfield Transaction
On November 7, 2007, we entered into a series of agreements with Deerfield Management, a healthcare investment fund and one of our largest equity shareholders, which provided for a potential capital raise of up to $10 million in cash. The financing consisted of two potential closings, with the first closing occurring upon the signing of the agreements (for $7.5 million in gross proceeds, less $0.5 million in transaction expenses) and the
14
second closing (for an additional $2.5 million in gross proceeds) occurring at our option, contingent upon FDA approval of our New Drug Application for the Amoxicillin PULSYS adult product. The agreements were designed to provide us with financial flexibility.
First Closing
At the transaction’s first closing, we sold certain assets, including Keflex product inventories, and assigned certain intellectual property rights, relating only to our existing, non-PULSYS cephalexin business, to two Deerfield affiliates, Kef Pharmaceuticals, Inc. (Kef) and Lex Pharmaceuticals, Inc. (Lex). Under the terms of the agreement, $7.5 million was received by MiddleBrook on November 8, 2007 for the first closing, and MiddleBrook reimbursed Deerfield $0.5 million for transaction-related expenses. Approximately $4.6 million of those proceeds was used to fully repay the outstanding Merrill Lynch Capital loan balance, with the remainder available for general corporate purposes. Pursuant to a consignment of those assets and license of those intellectual property rights back to the Company, the Company will continue to operate its existing cephalexin business, subject to consignment and royalty payments to Deerfield of 20% of net sales, which decline to 15% should the Company elect to make an extension payment of $1.35 million ($1.8 million if the second closing has occurred as described below) by June 30, 2008, subject to a minimum quarterly payment of $400,000. In addition, we granted to Deerfield a six-year warrant to purchase 3.0 million shares of the Company’s common stock at $1.38,the closing market price on November 7, 2007.
Second Closing
The agreements provided for a second closing, at the Company’s option. In the event that the we received approval (or an acceptable approvable letter) of our Amoxicillin PULSYS New Drug Application from the FDA, then we could require Deerfield to acquire and license certain intellectual property rights relating only to our cephalexin PULSYS business for a payment of $2.5 million. Pursuant to a required sublicense of those intellectual property rights back to us, we would continue to operate our cephalexin PULSYS activities. Cephalexin PULSYS is not approved for marketing by the FDA. To date, we have not exercised this option. This option expires June 30, 2008.
Repurchase Right
Deerfield also granted us the right to repurchase all of the assets and rights sold and licensed by us to Deerfield by purchasing all of the outstanding capital stock of both Kef and Lex. If we exercise this right prior to November 7, 2008, then the purchase price for all of the outstanding capital stock of Kef and Lex is a total of $11.0 million, if we have not elected the second closing (which would have required Deerfield to acquire and license certain intellectual property rights relating to the Company’s cephalexin PULSYS business), or $14.0 million if we did elect the second closing, in which we would have received $2.5 million in cash and Deerfield would have acquired and licensed certain rights to our cephalexin PULSYS business (in each case subject to certain adjustments). Those purchase prices will increase by $2.0 million on each subsequent anniversary of that date until the right is exercised or expires.
Our purchase rights expire on June 30, 2008, unless an extension payment of $1.35 million ($1.8 million, if a second closing has occurred) is made to extend the expiration date to December 31, 2008. If an extension payment of $4.5 million ($6.0 million, if a second closing has occurred) is made by December 31, 2008, the expiration date is extended to September 30, 2009. If an extension payment of $2.2 million ($2.9 million if a second closing has occurred) is made by September 30, 2009, then the expiration date for the right to purchase the capital stock of Lex is extended to November 1, 2012. We may not exercise our right to purchase the capital stock of Lex without first exercising our right to purchase the capital stock of Kef. Our exercise of this purchase right is mandatory upon the change of control of the Company.
Government Regulation
We are subject to extensive pre- and post-market regulation by the FDA, including regulations that govern the testing, manufacturing, safety, efficacy, labeling, storage, record keeping, advertising, and promotion of drugs
15
promulgated under the Federal Food, Drug, and Cosmetic Act and the Public Health Service Act and by comparable agencies in foreign countries. FDA approval is required before any new drug can be marketed in the United States.
New Drug Application Process
The process required by the FDA before a new drug may be marketed in the United States generally involves:
| | |
| | • | Completion of preclinical laboratory and animal testing. |
| |
| | • | Submission of an investigational new drug application (IND) which must become effective before the commencement of clinical trials. |
| |
| | • | Performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug product’s intended use. |
| |
| | • | Submission to and approval by the FDA of a New Drug Application (NDA) which includes inspection of manufacturing facilities. |
PRECLINICAL: Preclinical studies generally include laboratory evaluation of product chemistry, formulation and stability, as well as animal studies, to assess the safety and efficacy of the product. Preclinical trials also provide a basis for design of human clinical studies.
Human clinical trials are typically conducted in three sequential phases which may overlap:
PHASE I: During typical Phase I studies, the drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion.
PHASE II: During Phase II studies, the drug is introduced to patients that have the medical condition that the drug is intended to treat. Phase II studies are intended to identify possible adverse effects and safety risks, to determine the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. Phase II studies are sometimes combined with Phase I studies (referred to as Phase I/II studies) in certain instances when safety issues and questions of absorption, metabolism, distribution and excretion are well-established.
PHASE III: When Phase II evaluations suggest that a dosage range of the product is effective and has an acceptable safety profile, Phase III trials are undertaken to further evaluate dosage, clinical efficacy and to further test for safety in an expanded patient population, often at geographically dispersed clinical study sites.
The drug sponsor, the FDA or the institutional review board at each institution at which a clinical trial is being performed may suspend a clinical trial at any time for various reasons, including a concern that the subjects are being exposed to an unacceptable health risk.
The results of product development, preclinical studies and human studies are submitted to the FDA as part of the NDA. The NDA also must contain extensive manufacturing information. The FDA may approve or disapprove the NDA if applicable FDA regulatory criteria are not satisfied or it may require additional data, including clinical data, to continue to evaluate the NDA.
As an alternate path to FDA approval for new or improved formulations of previously approved products, a company may file a Section 505(b)(2) NDA. Section 505(b)(2) of the Federal Food, Drug, & Cosmetic Act permits the filing of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. In other words, the applicant can rely upon FDA’s previous findings of safety and efficacy for an approved product and/or published literature for related products, whether or not approved, and only perform those additional studies or measurements that are needed to support the change from the referenced product. In our NDA submissions, we intend to rely, in part, on prior FDA approvals of the antibiotic ingredients used in our products and on data generated by other parties which help to demonstrate the safety and effectiveness of those ingredients. In the case of products that we may develop in conjunction with sponsors of previously approved products, we expect that we will have a specific right of reference to the data contained in the prior applications and submit a traditional NDA. In any case in which we do not have a specific right of reference from the sponsor of the previously approved product, we anticipate that we will submit
16
Section 505(b)(2) NDAs All data necessary to satisfy the FDA of the safety and effectiveness of our own versions of these products will have to be generated by or for us and submitted to the FDA in support of our applications. These data are expected to include data establishing the safety and efficacy of the pulsatile dosage form and any other differences between the dosage form and the conditions for use of our products and the dosage form and conditions for use of the previously approved products.
To the extent that the Section 505(b)(2) applicant is relying on previous FDA findings for an already approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book. Specifically, the applicant must certify that: (i) the required patent information has not been filed; (ii) the listed patent has expired; (iii) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (iv) the listed patent is invalid or will not be infringed by the new product. A certification that the new product will not infringe the already approved product’s listed patents or that such patents are invalid is called a paragraph iv certification.
Absent a paragraph iv certification, the Section 505(b)(2) NDA will not be approved until all the listed patents claiming the referenced product have expired. The Section 505(b)(2) application also will not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product has expired. These procedures and limitations will not apply to the products in our development pipeline which contain active ingredients that are classified by FDA as antibiotics and that were the subject of marketing applications submitted to FDA prior to November 21, 1997. The only active ingredients which may be considered in future development projects which do not fall within this exempt antibiotic category are metronidazole, and fluoroquinolones such as ciprofloxacin. Of these, at the present time, we do not believe that the applicable limitations will have any effect on any potential future metronidazole development projects. With respect to products containing fluoroquinolones such as ciprofloxacin, in the absence of a licensing agreement, any application for approval that we submit to FDA may need to include statutorily-required certifications regarding our non-infringement of certain patents covering previously approved products, we may be required to notify the original NDA holder and patent holder of those filings, and we may be subject to approval delays of up to30-months, or longer, in the event that the patent holder brings suit against us for patent infringement within 45 days of such notifications.
In the case of antibiotic ingredients not previously approved in the combinations that we propose, it will also be necessary for us to satisfy the FDA’s combination drug policy with data establishing that each active component contributes to the effectiveness of the combination and that the dosage of each component is such that the combination is safe and effective for a significant patient population requiring such concurrent therapy. This policy typically requires very large clinical trials that test each antibiotic alone and in combination. In its review of our NDA submissions, the FDA will have broad discretion to require us to generate data on these matters. No assurance can be given that NDAs submitted for our products will receive FDA approval on a timely basis, or at all.
For all of the products that we have in development that contain antibiotic ingredients that were submitted to the FDA for approval prior to November 21, 1997, we will not, under current law, be able to submit to the FDA patent information covering those products. Therefore, once approved, the FDA’s Orange Book, which lists patent information on drug products, will not include patent information on those products. As a consequence, potential competitors who submit 505(b)(2) or ANDA applications for generic versions of those products will not have to provide certifications regarding any of our patents that they may infringe or to provide us notice if they intend to market their products prior to expiration of those patents. Additionally, if we bring a patent infringement action against any such applicants, there will be no automatic30-month stay of approval of those potentially infringing products. However, we would be entitled to pursue traditional patent-law procedures and remedies, such as preliminary and permanent injunctions. In the case of potential generic versions of any of our products that are not classified as exempt antibiotics, such as those containing only metronidazole or fluoroquinolone ingredients such as ciprofloxacin, we would be entitled to list our applicable patents in the Orange Book, potential competitors who submit 505(b)(2) or ANDA applications for generic version of those products would be subject to the certification and notice requirements, and there could be automatic30-month stays of approval of the generic products while we pursue patent infringement actions against the applicants.
17
Under the Prescription Drug User Fee Act (PDUFA) generally, the submission of an NDA is subject to substantial application user fees, currently $1,178,000, and the manufacturerand/or sponsor under an approved NDA are also subject to annual product and establishment user fees, currently $65,030 per product and $392,700 per establishment. These fees are typically increased annually. In addition, the PDUFA statute has been subject to significant amendments in connection with its regular reauthorization. We are not in a position to predict whether and how the user fee requirements will be interpreted and applied to us and our products in the future.
Other Regulatory Constraints
In addition to the results of product development, preclinical animal studies and clinical human studies, an NDA also must contain extensive information on the chemistry, manufacturing and controls that relate to the planned routine production and testing of the drug. An NDA must also contain proposed prescribing information for the product, supported by available clinical and other testing data, describing how the product may properly be used. The FDA may approve, deny approval or grant conditional approval depending on whether it finds that information provided sufficiently addresses all issues regarding the manufacture and proposed use of the product candidate. Both prior to and subsequent to approval, the Federal Food, Drug, and Cosmetic Act and FDA regulations require that the manufacture and testing of any drug for investigational use or for commercial use in humans be manufactured in accordance with current Good Manufacturing Practice (cGMP). Failure to follow cGMP requirements, as well as other regulatory requirements, can subject a sponsor and its products to various sanctions, including civil and criminal penalties, injunctions against the distribution of products and seizure of violative products. cGMP requirements are complex, are not always clearly defined, and can evolve over time. We have used, and intend to continue to use, third-party firms that we believe are knowledgeable and qualified in compliance with cGMP requirements to manufacture and test our product candidates and, to the extent that we engage in these activities on our own behalf, intend to utilize cGMP-compliant procedures and controls. There can be no assurance, however, that we or our contractors will be and remain at all times in full compliance with all cGMP requirements. In addition, the FDA retains authority to object to promotional activities engaged in by the applicant, including promotional activities that exceed the scope of approved prescribing information. Under certain circumstances, the FDA may also impose post-marketing testing requirements and may propose to withdraw or suspend approval of products based on new information about their safety and effectiveness for their approved uses.
Foreign Regulatory Approval
Outside the United States, our ability to market our products will also be contingent upon receiving marketing authorizations from the appropriate regulatory authorities. The foreign regulatory approval process includes all the risks associated with FDA approval described above. The requirements governing conduct of clinical trials and marketing authorization vary widely from country to country.
Under European Union regulatory systems, marketing authorizations may be submitted either under a centralized or decentralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the application and assessment report, each member state must decide whether to recognize approval. We plan to choose the European regulatory filing procedure that we believe will allow us to obtain regulatory approvals quickly. However, the chosen regulatory strategy may not secure regulatory approvals or approvals of the chosen product indications. In addition, these approvals, if obtained, may take longer than anticipated.
Obtaining foreign regulatory approvals would require additional financial resources and in the event we choose to seek those approvals, we would need to raise additional capital. We cannot assure you that any of our product candidates will prove to be safe or effective, will receive regulatory approvals, or will be successfully commercialized.
18
Employees
As of February 29, 2008, we had 33 employees, 9 of whom are senior management, 17 are in supervisory positions and 7 are non-management. Of the 33 employees, 11 perform scientific and research activities and 25 hold advanced degrees.
There are a number of important factors that could cause our actual results to differ materially from those that are indicated by forward-looking statements. Those factors include, without limitation, those listed below and elsewhere herein.
Risks Related to our Business
We have a history of losses, we expect to incur losses for the foreseeable future and we may never become profitable.
From the date we began operations in January 2000 through December 31, 2007, we have incurred losses of approximately $195.3 million, including a loss of approximately $42.2 million for the fiscal year ended December 31, 2007. Our losses to date have resulted principally from research and development costs related to the development of our product candidates, the purchase of equipment and establishment of our facilities and selling, general and administrative costs related to our operations.
We expect to incur substantial losses in 2008 and for the foreseeable future thereafter. Among other things, in 2008 we expect to incur significant expenses in anticipation of commercialization of our MOXATAG product that was approved for marketing by the FDA in January 2008. We have postponed further development of our Keflex PULSYS product candidate and our amoxicillin pediatric PULSYS product candidate in order to reduce our expenses. We may also incur losses in connection with the continued sales and marketing of our 750 mg Keflex product that was approved for marketing by the FDA in May 2006. In addition, we expect to incur additional expenses as a result of other research and development costs and regulatory compliance activities.
Our chances for achieving profitability depend on numerous factors, including success in:
| | |
| | • | obtaining additional financing; |
| |
| | • | commercializing our MOXATAG (extended-release amoxicillin) Tablets product, which was approved for marketing by the FDA on January 23, 2008; |
| |
| | • | maintaining sales of our Keflex 750 mg product; and |
| |
| | • | successfully developing, gaining FDA approval for, and commercializing other product candidates. |
We may never become profitable.
If we are unable to complete a strategic transaction, we may, if possible, enter into arrangements to raise additional capital which may dilute the ownership of our equity investors.
We are evaluating various strategic alternatives to further enhance shareholder value, and in February 2008 we retained Morgan Stanley, an investment banking firm, to assist us in this regard. Strategic alternatives we may pursue could include, but are not limited to, continued execution of our operating plan, licensing or development arrangements, the sale of some or all of our company’s assets, partnering or other collaboration agreements, or a merger or other strategic transaction. There can be no assurance that the exploration of strategic alternatives will result in any agreements or transactions, or that, if completed, any agreements or transactions will be successful or on attractive terms.
Revenues we currently generate from our Keflex 750 mg product and other Keflex product sales are insufficient to cover our expenses. We expect to incur a loss from operations in 2008. However, we believe that our existing cash resources, including the net proceeds from the private placement transaction in January 2008, will be sufficient to fund our operations into the first quarter of 2009 at our planned levels of research, development,
19
sales and marketing activities, barring unforeseen developments, and excluding the costs of a commercial launch of MOXATAG in 2008.
We are currently evaluating commercialization options for our MOXATAG product, which was approved for marketing by the FDA on January 23, 2008. We believe the MOXATAG market opportunity would be best addressed through its direct promotion by a national sales force of at least 300 sales representatives. As such, a commercialization initiative would require significant resources and expertise, and we believe it would be in the Company’s best interests to seek potential acquirers or partners to capitalize on MOXATAG’s commercial potential. Potential sales and marketing strategies for MOXATAG include the acquisition of the Company by a larger pharmaceutical organization with an established commercialization infrastructure, working with contract sales organizations, developing our own internal sales organization, or co-promoting products with collaborative marketing partners. Even if we successfully conclude our strategic evaluation process and identify a third party to assist in the commercialization of MOXATAG, the earliest we could launch the product would be in the fourth quarter of 2008. In addition, in order for the Company to participate in the sales and marketing of MOXATAG, we would need to have sufficient financial resources, which may require us to raise additional capital.
Our requirements for additional capital are substantial. We currently have no committed source of capital, although we are evaluating a number of alternatives. We cannot assure you that financing will be available to us or, if available, that it will be on favorable terms. To the extent we raise additional capital through the sale of equity securities, the issuance of those securities could result in substantial dilution to our existing stockholders. In addition, if we obtain debt financing, we may be required to pledge all or a significant portion of our assets to secure such debt financing, a substantial portion of our operating cash flow may be dedicated to the payment of principal and interest on such indebtedness and we may become subject to financial covenants and other terms that restrict the manner in which we can operate our business. If we are unable to raise additional capital, we may need to sell assets, pursue other strategic alternativesand/or curtail significantly our development and commercialization activities.
Even if we are able to raise capital in the near term, the amount of funds we are able to raise may not be sufficient to allow us to commercially launch MOXATAG product. We may need to raise capital on more than one occasion to meet our anticipated expenses for the next twelve months as well as commercially launch MOXATAG, should we choose to launch the product in the fourth quarter of 2008. We further anticipate that we will also need to raise additional capital in the future to allow us to continue to develop Keflex PULSYS and our other products under development.
If we are unable to maintain sales of our 750 mg Keflex product, our liquidity will be further adversely affected.
We launched our Keflex 750 mg product in July 2006. While we have invested considerable resources in the launch of this product, to date, sales have not met our expectations. We had previously anticipated that this product would generate revenues in excess of related expenses and would contribute additional cash flow to help fund our other operations by the end of 2007. During 2007, we reduced the number of sales representatives and marketing costs associated with Keflex 750 in order to more closely match the monthly sales level. While we believe that the product now generates monthly revenues slightly in excess of related expenses, we cannot assure you that this will continue, as this product may not gain sufficient market acceptance among physicians, the medical community and patients. The extent to which this product is accepted by physicians is dependent upon a number of factors, including the recognition of the potential advantages, despite higher cost, over alternative generic dosage strengths of cephalexin and the effectiveness of our marketing and distribution capabilities. In addition, this product faces significant competition from other dosage strengths of cephalexin manufactured by generic pharmaceutical companies, as well as from other dosage strengths of Keflex marketed by us. Although there is no current 750 mg dosage strength of generic cephalexin, the product is not protected by patents and we expect to have a limited window of opportunity to market this product, should generic pharmaceutical manufacturers choose to compete with us. If our 750 mg Keflex product does not achieve greater market acceptance, our liquidity will further suffer. Even if sales of this product increase in the short-term, we expect that the amount of revenues from this product could decline significantly within a few years due to competition from generic formulations of the product.
20
Our PULSYS technology is based on a finding that could ultimately prove to be incorrect, or could have limited applicability.
Our PULSYS product candidates are based on our finding that bacteria exposed to antibiotics in front-loaded, rapid sequential bursts are eliminated more efficiently and effectively than those exposed to presently available treatment regimens. Ultimately, our finding may be incorrect, in which case our pulsatile drugs would not differ substantially from competing drugs and may be inferior to them. If these products are substantially identical or inferior to products already available, the market for our pulsatile drugs will be reduced or eliminated.
Even if pulsatile dosing is more effective than traditional dosing, we may be unable to apply this finding successfully to a substantial number of products in the anti-infective market. Our preliminary studies indicate that pulsatile dosing may not provide superior performance for all types of antibiotics. Additionally, we have not conducted any studies with anti-viral or anti-fungal medications. If we cannot apply our technology to a wide variety of antibiotics or other anti-infectives, our potential market will be substantially reduced.
Our PULSYS delivery technology may not be effective for product candidates other than MOXATAG, which would prevent us from commercializing products that are more effective than those of our competitors.
Even if we are correct that pulsatile dosing is more effective than traditional dosing of antibiotics, our PULSYS delivery technology must be effective in humans such that the pulsatile administration of drugs are at levels that prove effective in curing infections. If our PULSYS delivery technology is not effective in delivering rapid bursts of antibiotics, or is unable to do so at an appropriate concentration and we are not able to create an alternative delivery method for pulsatile dosing that proves to be effective, we will be unable to capitalize on any advantage of our discovery. Should this occur, our pulsatile product candidates may not be more effective than the products of our competitors, which may decrease or eliminate market acceptance of our products.
If a competitor produces and commercializes an antibiotic that is superior to our PULSYS antibiotics, the market for our potential products would be reduced or eliminated.
We have devoted a substantial amount of our research efforts and capital to the development of pulsatile antibiotics. Competitors are developing or have developed new drugs that may compete with our pulsatile A number of pharmaceutical companies are also developing new classes of compounds, such as oxazolidinones, that may also compete against our pulsatile antibiotics. In addition, other companies are developing technologies to enhance the efficacy of antibiotics by adding new chemical entities that inhibit bacterial metabolic function. If a competitor produces and commercializes an antibiotic or method of delivery of antibiotics that provides superior safety, effectiveness or other significant advantages over our pulsatile antibiotics, the value of our pulsatile drugs would be substantially reduced. As a result, we would need to conduct substantial new research and development activities to establish new product targets, which would be costly and time consuming. In the event we are unable to establish new product targets, we will be unable to generate sources of revenue.
We have not conducted an extensive third party patent infringement, invalidity and enforceability investigation on pulsatile dosing and our PULSYS technology, and we are aware of at least one issued patent covering pulsatile delivery.
Our patents, prior art and infringement investigations were primarily conducted by our senior management and other employees. Although our patent counsel has consulted with management in connection with management’s intellectual property investigations, our patent counsel has not undertaken an extensive independent analysis to determine whether our pulsatile technology infringes upon any issued patents or whether our issued patents or patent applications covering pulsatile dosing could be invalidated or rendered unenforceable for any reason. We are aware of one issued patent owned by a third party that covers certain aspects of delivering drugs by use of two delayed release pulses. The patent covers a drug delivery system employing two delayed release pulses using two polymers. The claims made by this patent could be argued to cover certain aspects of our technology. However, we believe that we will be able to manufacture and market formulations of our pulsatile products without infringing any valid claims under this patent. Any reformulation of our products, if required, could be costly and time-consuming
21
and may not be possible. We cannot assure you that a claim will not be asserted by such patent holder or any other holder of an issued patent that any of our products infringe their patent or that our patents are invalid or unenforceable. We may be exposed to future litigation by third parties based on claims that our products or activities infringe the intellectual property rights of others. We cannot assure you that, in the event of litigation, any claims would be resolved in our favor. Any litigation or claims against us, whether or not valid, may result in substantial costs, could place a significant strain on our financial resources, divert the attention of management and harm our reputation. In addition, intellectual property litigation or claims could result in substantial damages and force us to do one or more of the following:
| | |
| | • | cease selling, incorporating or using any of our products that incorporate the challenged intellectual property; |
| |
| | • | obtain a license from the holder of the infringed intellectual property right, which license may be costly or may not be available on reasonable terms, if at all; or |
| |
| | • | redesign our products, which would be costly and time-consuming and may not be possible. |
We have not sought patent protection for certain aspects of the technology used in our PULSYS product candidates.
We have not filed for patent protection with respect to all of our specific formulations, materials (including inactive ingredients) or manufacturing process approaches that are incorporated in our PULSYS product candidates, and we may not seek such patent coverage in the future. In producing our PULSYS products, we expect to use general formulation techniques used in the industry that would be modified by us and which would, therefore, include know-how and trade secrets that we have developed. We cannot be certain that a patent would issue to cover such intellectual property, and currently, we would prefer to keep such techniques and know-how as our trade secrets. In the event a competitor is able to develop technology substantially similar to ours and patent that approach, we may be blocked from using certain of our formulations or manufacturing process approaches, which could limit our ability to develop and commercialize products.
If we are unable to develop and successfully commercialize our PULSYS product candidates, we may never achieve profitability.
We have not commercialized any PULSYS products or recognized any revenue from PULSYS product sales. With the exception of MOXATAG, which was approved for marketing by the FDA on January 23, 2008, all of our pulsatile drugs are in early stages of development. We must obtain regulatory approval for our products before we are able to commercialize these products and generate revenue from their sales. We expect that we must conduct significant additional research and development activities on our other PULSYS product candidates and successfully complete preclinical, Phase I, Phase I/II or Phase II, and Phase III clinical trials before we will be able to receive final regulatory approval to commercialize these pulsatile products. Even if we succeed in developing and commercializing one or more of our PULSYS products, we may never generate sufficient or sustainable revenue to enable us to be profitable.
If clinical trials for our products are unsuccessful or delayed, we will be unable to meet our anticipated development and commercialization timelines.
We must demonstrate through preclinical testing and clinical trials that our product candidates are safe and effective for use in humans before we can obtain regulatory approvals for their commercial sale. In addition, we will also need to demonstrate through clinical trials any claims we may wish to make that our product candidates are comparable or superior to existing products. For drug products which are expected to contain active ingredients in fixed combinations that have not been previously approved by the FDA, we may also need to conduct clinical studies in order to establish the contribution of each active component to the effectiveness of the combination in an appropriately identified patient population.
Conducting clinical trials is a lengthy, time-consuming and expensive process. With the exception of our MOXATAG product that has completed Phase III clinical trials, we have not completed preclinical studies and
22
initial clinical trials (Phase I, Phase I/II or Phase II) to extrapolate proper dosage for our product candidates for Phase III clinical efficacy trials in humans. In the event we incorrectly identify a dosage as appropriate for human clinical trials, any results we receive from such trials may not properly reflect the optimal efficacy or safety of our products and may not support approval in the absence of additional clinical trials using a different dosage.
The commencement and rate of completion of clinical trials for our products may be delayed by many factors, including:
| | |
| | • | lack of efficacy during the clinical trials; |
| |
| | • | unforeseen safety issues; |
| |
| | • | slower than expected rate of patient recruitment; or |
| |
| | • | government or regulatory delays. |
The results from preclinical testing and early clinical trials are often not predictive of results obtained in later clinical trials. Although a new product may show promising results in preclinical and initial clinical trials, it may subsequently prove unfeasible or impossible to generate sufficient safety and efficacy data to obtain necessary regulatory approval. Data obtained from preclinical and clinical studies are susceptible to varying interpretations, which may delay, limit or prevent regulatory approval. In addition, we may encounter regulatory delays or rejections as a result of many factors, including results that do not support our claims, perceived defects in the design of clinical trials and changes in regulatory policy during the period of product development. Our business, financial condition and results of operations may be materially adversely affected by any delays in, or termination of, our clinical trials or a determination by the FDA that the results of our trials are inadequate to justify regulatory approval.
Although our MOXATAG product has been approved for commercial sale, we will not be successful if the product is not accepted by the market.
Even though we have obtained regulatory approval to market MOXATAG, it, or any of our other potential PULSYS products, may not gain market acceptance among physicians, patients, healthcare payors and the medical community. The degree of market acceptance of any pharmaceutical product that we develop will depend on a number of factors, including:
| | |
| | • | demonstration of clinical efficacy and safety; |
| |
| | • | cost-effectiveness; |
| |
| | • | potential advantages over alternative therapies; |
| |
| | • | reimbursement policies of government and third-party payors; and |
| |
| | • | effectiveness of our marketing and distribution capabilities and the effectiveness of such capabilities of our collaborative partners. |
Our products will compete with a number of products manufactured and marketed by major pharmaceutical companies, biotechnology companies and manufacturers of generic drugs. Our products may also compete with new products currently under development by others. Physicians, patients, third-party payors and the medical community may not accept and use any product candidates that we or our collaborative partners develop. To the extent current antibiotics already successfully treat certain infections, physicians may not be inclined to prescribe our pulsatile drugs for the same indications. If our products do not achieve significant market acceptance, we will not be able to generate significant revenues or become profitable.
23
We have limited sales, marketing, and distribution capabilities, and currently depend in large part on a third party contract sales force for the marketing of our Keflex 750 mg product. If we fail to develop our own sales, marketing and distribution capabilities or fail to enter into arrangements with third parties, we will not be able to successfully commercialize our products.
We have limited sales, marketing, and distribution capabilities. We currently rely in large part on a third party contract sales force provided to us by Innovex, the commercialization division of Quintiles Transnational Corporation, for sales and marketing services relating to our Keflex 750 mg product. In order to commercialize our MOXATAG product which was approved for marketing by the FDA on January 23, 2008, or any other product candidates, we will need to considerably expand our commercial capabilities or make arrangements with third parties to perform these services for us. In order to market any of our product candidates directly, we must considerably expand our commercial infrastructure, including distribution, marketing and sales personnel. The expansion or contracting of a sales and distribution infrastructure would require substantial resources, which may divert the attention of our management and key personnel and defer our product development efforts. To the extent that we enter into sales and marketing arrangements with other companies, our revenues will depend on the efforts of others. These efforts may not be successful. If we fail to expand sales, marketing and distribution capabilities, or fail to enter into arrangements with third parties, we will experience delays in product sales and incur increased costs.
We rely upon a limited number of pharmaceutical wholesalers and distributors, which could impact our ability to sell our Keflex products or MOXATAG.
We rely largely upon specialty pharmaceutical distributors and wholesalers to deliver Keflex to end users, including physicians, hospitals, and pharmacies. Product sales to the three major pharmaceutical wholesalers, Cardinal Health Inc., McKesson Corporation and AmerisourceBergen Corporation, represented approximately 94% of our net revenue from Keflex in 2007. There can be no assurance that our distributors and wholesalers will adequately fulfill the market demand for Keflex or, after commercial launch, MOXATAG. Given the high concentration of sales to certain pharmaceutical distributors and wholesalers, we could experience a significant loss if one of our top customers were to declare bankruptcy or otherwise become unable to pay its obligations to us.
The potential success of our products and product candidates, including our Keflex 750 mg product and, our recently-approved MOXATAG product, will be dependent upon successfully pricing the products in the marketplace.
While we believe that physicians make antibiotic prescribing decisions based primarily on efficacy, safety, and compliance, we also believe that, when deciding whether to prescribe a modified-release drug or its immediate release generic analog, physicians also weigh patient co-pay and patient preferences. As a result, we believe that price will be an important driver of the adoption of our products and product candidates. In addition, we believe it will be important to carefully manage against resistance from payor organizations by pricing our products in such a way as to minimize the incremental payor cost burden relative to generic analogs.
Generic pricing plans, such as that implemented by Wal-Mart and other retailers, may affect the market for our products.
In September 2006, Wal-Mart began offering certain generic drugs at $4 per prescription. Amoxicillin and cephalexin are on the list of drugs that Wal-Mart intends to provide at $4 per prescription. Wal-Mart has significant market presence. As a result, there can be no assurance that Wal-Mart’s generic pricing plan,and/or similar plans adopted by others, will not have a material adverse effect on the market for our products.
Our products are subject to therapeutic equivalent substitution, Medicaid reimbursement and price reporting.
The cost of pharmaceutical products continues to be a subject of investigation and action by governmental agencies, legislative bodies and private organizations in the U.S. and other countries. In the U.S., most states have enacted legislation requiring or permitting a dispensing pharmacist to substitute a generic equivalent to the
24
prescribed drug. Federal legislation requires pharmaceutical manufacturers to pay to state Medicaid agencies prescribed rebates on drugs to enable them to be eligible for reimbursement under Medicaid programs. Federal and state governments continue to pursue efforts to reduce spending in Medicaid programs, including imposing restrictions on amounts agencies will reimburse for certain products. We also must give discounts or rebates on purchases or reimbursements of our products by certain other federal and state agencies and programs. Our ability to earn sufficient returns on our products will depend, in part, on the availability of reimbursements from third party payers, such as health insurers, governmental health administration authorities and other organizations and the amount of rebates payable under Medicaid programs.
We are dependent on our contract manufacturers and suppliers to provide us with active pharmaceutical ingredients and finished products.
We do not maintain commercial scale manufacturing facilities. Our Keflex products are manufactured for us by Ceph International Corporation (Ceph), a wholly-owned subsidiary of Patheon’s MOVA Pharmaceutical Corporation. MOXATAG is expected to be manufactured for us by Stada Production Ireland Limited (SPI), previously known as the manufacturing division of Clonmel Healthcare Limited, a subsidiary of STADA Arzneimittel AG, pursuant to a contract manufacturing arrangement we have entered into with them.
Although we believe that the active pharmaceutical ingredients and finished Keflex and MOXATAG products could be potentially obtained from several suppliers, our applications for regulatory approval currently authorize only Ceph as our source for Keflex, and SPI is identified as our only source for MOXATAG. In the event that Cephand/or SPI is unable to supply the products to us in sufficient quantities on a timely basis or at a commercially reasonable price, or in the event either of them breaches their agreement with us, or if Cephand/or SPI loses its regulatory status as an acceptable source, we would need to locate another source. A change to a supplier not previously approved or an alteration in the procedures or product provided to us by an approved supplier may require formal approval by the FDA before the product could be sold and could result in significant disruption to our business. These factors could limit our ability to sell Keflexand/or MOXATAG and could materially adversely affect our business, financial condition and results of operations.
In addition, we obtain active pharmaceutical ingredients (APIs) and finished products from certain specialized manufacturers for use in clinical studies. Although the antibiotics and finished products we use in our clinical studies may generally be obtained from several suppliers, the loss of a supplier could result in delays in conducting or completing our clinical trials and could delay our ability to commercialize products.
Our ability to conduct clinical trials will be impaired if we fail to qualify our clinical supply manufacturing facility and we are unable to maintain relationships with current clinical supply manufacturers or enter into relationships with new manufacturers.
We currently rely on several contractors to manufacture product samples for our clinical studies. In the fourth quarter of 2003, we completed construction of a manufacturing facility in Germantown, Maryland for production of clinical supplies sufficient for use through our Phase II and, in some cases, Phase III clinical trials. This facility has the potential to be qualified and operational in the future, although we currently have no plans to qualify the facility.
We intend to rely on third parties to manufacture products that we intend to sell through our own commercialization and sales efforts. We believe that there are a variety of manufacturers that we may retain to produce these products. However, once we retain a manufacturing source, if we are unable to maintain our relationship with such manufacturer, qualifying a new manufacturing source will be time consuming and expensive, and may cause delays in the development of our products.
Clinical trials for our product candidates may be delayed due to our dependence on third parties for the conduct of such trials.
We have limited experience in conducting and managing clinical trials. We rely, and will continue to rely, on third parties, including contract research organizations and outside consultants, to assist us in managing and monitoring clinical trials. Our reliance on these third parties may result in delays in completion of, or the failure to complete, these trials if they fail to perform their obligations under our agreements with them.
25
Our success may depend on our ability to successfully attract and retain collaborative partners.
For certain product candidates, we may enter into collaborative arrangements with third parties. Collaborations may be necessary in order for us to:
| | |
| | • | fund our research and development activities; |
| |
| | • | fund manufacturing by third parties; |
| |
| | • | seek and obtain regulatory approvals; and |
| |
| | • | successfully commercialize our product candidates. |
We cannot assure you that we will be able to enter into collaborative agreements with partners on terms favorable to us, or at all, and any future agreement may expose us to risks that our partner might fail to fulfill its obligations and delay commercialization of our products. We also could become involved in disputes with partners, which could lead to delays in or terminations of our development and commercialization programs and time consuming and expensive litigation or arbitration. Our inability to enter into additional collaborative arrangements with other partners, or our failure to maintain such arrangements, may limit the number of product candidates we can develop and ultimately, decrease our sources of any future revenues. For a discussion of certain collaboration arrangements to which we were previously a party and which have been terminated, see the information under the caption“Collaboration Agreements”in Item 1 above.
If we cannot enter into new licensing arrangements or otherwise gain access to products, our ability to develop a diverse product portfolio could be limited.
A component of our business strategy may involve in-licensing or acquiring drug compounds developed by other pharmaceutical and biotechnology companies or academic research laboratories that may be marketed and developed or improved upon using our novel technologies. Competition for promising compounds can be intense and currently we have not entered into any arrangement to license or acquire any drugs from other companies. If we are not able to identify licensing or acquisition opportunities or enter into arrangements on acceptable terms, we may be unable to develop a diverse portfolio of products. Any product candidate that we acquire may require significant additional research and development efforts prior to seeking regulatory approval and commercial sale, including extensive preclinicaland/or clinical testing. All product candidates are prone to the risks of failure inherent in pharmaceutical product development, including the possibility that the product candidate will not be safe, non-toxic and effective or approved by regulatory authorities. In addition, we cannot assure you that any approved products that we develop or acquire will be: manufactured or produced economically; successfully commercialized; widely accepted in the marketplace or that we will be able to recover our significant expenditures in connection with the development or acquisition of such products. In addition, proposing, negotiating and implementing an economically viable acquisition is a lengthy and complex process. Other companies, including those with substantially greater financial, sales and marketing resources, may compete with us for the acquisition of product candidates and approved products. We may not be able to acquire the rights to additional product candidates and approved products on terms that we find acceptable, or at all. In addition, if we acquire product candidates from third parties, we may be dependent on third parties to supply such products to us for sale. We could be materially adversely affected by the failure or inability of such suppliers to meet performance, reliability and quality standards.
We could be forced to pay substantial damage awards if product liability claims that may be brought against us are successful.
The use of any of our product candidates in clinical trials, and the sale of any approved products, may expose us to liability claims and financial losses resulting from the use or sale of our products. We have obtained limited product liability insurance coverage for our clinical trials, which we believe is adequate to cover our present activities. However, such insurance may not be adequate to cover any claims made against us. In addition, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts or scope to protect us against losses.
26
Our executive officers and other key personnel are critical to our business and our future success depends on our ability to retain them.
We are highly dependent on the principal members of our scientific and management teams, especially Edward M. Rudnic, our president and chief executive officer. In order to pursue our product development, marketing and commercialization plans, we may need to hire additional personnel with experience in clinical testing, government regulation, manufacturing, marketing and business development. We may not be able to attract and retain personnel on acceptable terms given the intense competition for such personnel among biotechnology, pharmaceutical and healthcare companies, universities and non-profit research institutions. We are not aware of any present intention of any of our key personnel to leave our company or to retire. However, although we have employment agreements with our executive officers, these employees may terminate their services upon 90 days advance notice. The loss of any of our key personnel, or the inability to attract and retain qualified personnel, may significantly delay or prevent the achievement of our research, development or business objectives and could materially adversely affect our business, financial condition and results of operations. Although we maintain key man life insurance on Dr. Rudnic, such insurance may not be sufficient to cover the costs of the loss of his services and the expense of recruiting and hiring a new president and chief executive officer.
Risks Related to our Industry
Any inability to protect our intellectual property could harm our competitive position.
Our success will depend in part on our ability to obtain patents and maintain adequate protection of other intellectual property for our technologies and products in the U.S. and other countries. If we do not adequately protect our intellectual property, competitors may be able to use our technologies and erode or negate our competitive advantage. Further, the laws of some foreign countries do not protect our proprietary rights to the same extent as the laws of the U.S., and we may encounter significant problems in protecting our proprietary rights in these foreign countries.
The patent positions of pharmaceutical and biotechnology companies, including our patent positions, involve complex legal and factual questions and, therefore, validity and enforceability cannot be predicted with certainty. Patents may be challenged, deemed unenforceable, invalidated or circumvented. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that we cover our proprietary technologies with valid and enforceable patents or we effectively maintain such proprietary technologies as trade secrets. We will apply for patents covering both our technologies and product candidates as we deem appropriate. We may fail to apply for patents on important technologies or products in a timely fashion, or at all, and in any event, the applications we do file may be challenged and may not result in issued patents. Any future patents we obtain may not be sufficiently broad to prevent others from practicing our technologies or from developing competing products. Furthermore, others may independently develop similar or alternative technologies or design around our patented technologies. In addition, if challenged, our patents may be declared invalid. Even if valid, our patents may fail to provide us with any competitive advantages.
We rely upon trade secrets protection for our confidential and proprietary information. We have taken measures to protect our proprietary information; however, these measures may not provide adequate protection. We seek to protect our proprietary information by entering into confidentiality agreements with employees, collaborators and consultants. Nevertheless, employees, collaborators or consultants may still disclose our proprietary information, and we may not be able to meaningfully protect our trade secrets. In addition, others may independently develop substantially equivalent proprietary information or techniques or otherwise gain access to our trade secrets.
If we do not compete successfully in the development and commercialization of products and keep pace with rapid technological change, we will be unable to capture and sustain a meaningful market position.
The biotechnology and pharmaceutical industries are highly competitive and subject to significant and rapid technological change. While we are not aware of any company using rapid bursts of antibiotics as a treatment method, there are numerous companies actively engaged in the research and development of anti-infectives.
27
Our main competitors are:
| | |
| | • | Large pharmaceutical companies, such as Pfizer, GlaxoSmithKline, Wyeth, Bristol-Myers Squibb, Merck, Johnson & Johnson, Roche, Schering-Plough, Novartis, sanofi-aventis, Abbott Laboratories, AstraZeneca, and Bayer, that may develop new drug compounds that render our drugs obsolete or noncompetitive. |
| |
| | • | Smaller pharmaceutical and biotechnology companies and specialty pharmaceutical companies engaged in focused research and development of anti-infective drugs, such as Trimeris, Vertex, Adams Respiratory Therapeutics, Gilead Sciences, Cubist, Basilea, Replidyne, InterMune, Oscient, King, Advanced Life Sciences, and others. |
| |
| | • | Drug delivery companies, such as Johnson & Johnson’s Alza division, Biovail, DepoMed, Flamel Technologies, and SkyePharma, which may develop a dosing regimen that is more effective than pulsatile dosing. |
| |
| | • | Generic drug companies, such as Teva, Ranbaxy, IVAX, Sandoz and Stada, which produce low-cost versions of antibiotics that may contain the same active pharmaceutical ingredients as our PULSYS product candidates. |
Many of these competitors, either alone or together with their collaborative partners, have substantially greater financial resources and larger research and development staffs than we do. In addition, many of these competitors, either alone or together with their collaborative partners, have significantly greater experience than we do in:
| | |
| | • | developing products; |
| |
| | • | undertaking preclinical testing and human clinical trials; |
| |
| | • | obtaining approvals of products from the FDA and other regulatory agencies; and |
| |
| | • | manufacturing and marketing products. |
Developments by others may render our product candidates or technologies obsolete or noncompetitive. We face and will continue to face intense competition from other companies for collaborative arrangements with pharmaceutical and biotechnology companies, for establishing relationships with academic and research institutions, and for licenses of products or technology. These competitors, either alone or with their collaborative partners, may succeed in developing technologies or products that are more effective than ours.
If we experience delays in obtaining regulatory approvals, or are unable to obtain or maintain regulatory approvals, we may be unable to commercialize any products.
Our product candidates are subject to extensive and rigorous domestic government regulation. The FDA regulates, among other things, the development, testing, manufacture, safety, efficacy, record-keeping, labeling, storage, approval, advertising, promotion, sale and distribution of pharmaceutical products. If our products are marketed abroad, they will also be subject to extensive regulation by foreign governments. None of our PULSYS product candidates has been approved for sale in any foreign market. The regulatory review and approval process takes many years, requires the expenditure of substantial resources, involves post-marketing surveillance and may involve ongoing requirements for post-marketing studies. The actual time required for satisfaction of FDA pre-market approval requirements may vary substantially based upon the type, complexity and novelty of the product or the medical condition it is intended to treat. Government regulation may delay or prevent marketing of potential products for a considerable period of time and impose costly procedures upon a manufacturer’s activities. Delays in obtaining regulatory approvals may:
| | |
| | • | adversely affect the commercialization of any drugs that we or our collaborative partners develop; |
| |
| | • | impose costly procedures on us or our collaborative partners; |
| |
| | • | diminish any competitive advantages that we or our collaborative partners may attain; and |
| |
| | • | adversely affect our receipt of revenues or royalties. |
Success in early stage clinical trials does not assure success in later stage clinical trials. Data obtained from clinical activities is not always conclusive and may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval.
28
Any required approvals, once obtained, may be withdrawn. Further, if we fail to comply with applicable FDA and other regulatory requirements at any stage during the regulatory process, we may encounter difficulties including:
| | |
| | • | delays in clinical trials or commercialization; |
| |
| | • | product recalls or seizures; |
| |
| | • | suspension of productionand/or distribution; |
| |
| | • | withdrawals of previously approved marketing applications; and |
| |
| | • | fines, civil penalties and criminal prosecutions. |
We may rely on future collaborative partners to file investigational new drug applications and generally direct the regulatory approval process for some of our products. These collaborative partners may not be able to conduct clinical testing or obtain necessary approvals from the FDA or other regulatory authorities for any product candidates. If we fail to obtain required governmental approvals, we or our collaborative partners will experience delays in, or be precluded from, marketing products developed through our research.
We and our contract manufacturers also are required to comply with applicable FDA good manufacturing practice regulations. Good manufacturing practice regulations include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation. Manufacturing facilities are subject to inspection by the FDA. These facilities must be approved before we can use them in commercial manufacturing of our products. We or our contract manufacturers may not be able to comply with the applicable good manufacturing practice requirements and other FDA regulatory requirements. If we or our contract manufacturers fail to comply, we could be subject to fines or other sanctions, or be precluded from marketing our products.
The manufacture and storage of pharmaceutical and chemical products is subject to environmental regulation and risk.
Because of the chemical ingredients of pharmaceutical products and the nature of their manufacturing process, the pharmaceutical industry is subject to extensive environmental regulation and the risk of incurring liability for damages or the costs of remedying environmental problems. We use a number of chemicals and drug substances that can be toxic to humans. These chemicals include acids, solvents and other reagents used in the normal course of our chemical and pharmaceutical analysis, and other materials, such as polymers, inactive ingredients and drug substances, used in the research, development and manufacture of drug products. If we fail to comply with environmental regulations to use, discharge or dispose of hazardous materials appropriately or otherwise to comply with the conditions attached to our operating licenses, the licenses could be revoked and we could be subject to criminal sanctionsand/or substantial liability or could be required to suspend or modify our operations.
Environmental laws and regulations can require us to undertake or pay for investigation,clean-up and monitoring of environmental contamination identified at properties that we currently own or operate or that we formerly owned or operated. Further, they can require us to undertake or pay for such actions at offsite locations where we may have sent hazardous substances for disposal. These obligations are often imposed without regard to fault. In the event we are found to have violated environmental laws or regulations, our reputation will be harmed and we may incur substantial monetary liabilities. We currently have insurance coverage that we believe is adequate to cover our present activities. However, this insurance may not be available or adequate to cover any losses arising from contamination or injury resulting from our use of hazardous substances.
Market acceptance of our products will be limited if users of our products are unable to obtain adequate reimbursement from third-party payors.
The commercial success of our products and product candidates will depend in part on the availability of reimbursement from third-party payors, including government health administrators, managed care providers and private health insurers. We cannot assure you that third-party payors will consider our products cost-effective or provide reimbursement in whole or in part for their use.
29
Significant uncertainty exists as to the reimbursement status of newly approved health care products. Third-party payors may conclude that our products are less safe, effective or cost-effective than existing products. Therefore, third-party payors may not approve our products for reimbursement.
If third-party payors do not approve our products for reimbursement or fail to reimburse them adequately, sales will suffer as some physicians or their patients will opt for a competing product that is approved for reimbursement or is adequately reimbursed. Even if third-party payors make reimbursement available, reimbursement levels may not be sufficient for us to realize an appropriate return on our investment in product development.
Moreover, the trend toward managed healthcare in the United States, the growth of organizations such as health maintenance organizations, and legislative proposals to reform healthcare and government insurance programs could significantly influence the purchase of healthcare services and products, resulting in lower prices and reduced demand for our products. In addition, legislation and regulations affecting the pricing of pharmaceuticals may change in ways adverse to us. While we cannot predict the likelihood of any of these legislative or regulatory proposals, if any government or regulatory agencies adopt these proposals, they could materially adversely affect our business, financial condition and results of operations.
Potential regulatory changes and the billing and reimbursement process applicable to underlying conditions may cause price erosion and reduce sales revenue for our products and our products may not be accepted by health care providers.
Government and private healthcare programs currently are under financial stress due to overall medical cost increases. Federal and state governments are taking steps to ease the burden on healthcare programs in ways that could affect the pricing of pharmaceuticals. Any such federal and state laws and regulations can have a negative impact on the pricing of prescription drugs, including Medicare, Medicaid, pharmaceutical importation laws and other laws and regulations that directly or indirectly impose controls on pricing.
Market acceptance of our products may depend on the availability of reimbursement by government and private third-party payors. In recent years, there have been numerous proposals to change the healthcare system in the United States. The growth of managed care organizations (“MCOs”) (e.g., medical insurance companies, medical plan administrators, hospital alliances and pharmaceutical benefit managers) has placed increase pressure on drug prices and on overall healthcare expenditures. MCOs and government and other private third-party payors increasingly are attempting to contain health care costs by limiting both the coverage and the level of reimbursement of drug products. Consequently, the reimbursement status of our products is highly uncertain and we cannot assure that third-party coverage will be available or that available third-party coverage or payment will be adequate.
Other Risks
HealthCare Ventures V, L.P., HealthCare Ventures VI, L.P., and HealthCare Ventures VII, L.P. have significant influence over our business, and the interests of the HealthCare Ventures partnerships may not be consistent with the interests of our other stockholders.
HealthCare Ventures V, L.P., HealthCare Ventures VI, L.P. and HealthCare Ventures VII, L.P. currently beneficially own an aggregate of 21.2% of our outstanding common stock. James H. Cavanaugh and Harold R. Werner, members of our board of directors, are general partners of HealthCare Partners V, L.P., HealthCare Partners VI, L.P., and HealthCare Partners VII, L.P., which are the general partners of HealthCare Ventures V, L.P., HealthCare Ventures VI, L.P. and HealthCare Ventures VII, L.P., respectively. Accordingly, the HealthCare Ventures partnerships are able to exert significant influence over all matters requiring stockholder approval, including the election and removal of directors and any merger, consolidation or sale of all or substantially all of our assets, as well as over the day-to-day management of our business. The HealthCare Ventures partnerships may direct our affairs in a manner that is not consistent with the interests of our other stockholders. In addition, this concentration of ownership could have the effect of delaying, deferring or preventing a change in control, or impeding a merger or consolidation, takeover or other business combination or a sale of all or substantially all of our assets.
30
Deerfield Management may have significant influence over our ability to raise capital or to enter into certain major transactions, and Deerfield’s interests may not be consistent with the interests of our other shareholders.
In November 2007, we issued warrants for the purchase of 3,000,000 shares of our common stock to affiliates of Deerfield Management in connection with the sale of certain non-PULSYS tangible and intangible assets. The warrant agreement contains provisions granting rights to Deerfield upon the occurrence of certain major transactions, as defined in the agreement. Major transactions include a change in control of the Company, a sale of assets of the Company exceeding a purchase price of $25.0 million or representing more than 60% of the Company’s total assets, the issuance of shares by the Company, without the approval of Deerfield, in excess of 25% of the Company’s outstanding common stock, and liquidation or bankruptcy of the Company. In the event of a major transaction, Deerfield has the right to require the Company to redeem its warrants in cash, at an amount calculated in accordance with the Black-Scholes value as defined in the agreement. Deerfield can also require that this amount be placed into escrow in advance of the closing of a major transaction. In the event the Company attempts to consummate a major transaction without placing the redemption amount in escrow, Deerfield has the right to apply for an injunction.
To the extent our capital resources are insufficient to meet our future capital requirements, we will need to raise additional capital, incur indebtedness, or consider the sale of company assets in order to fund our operations. If Deerfield does not pre-approve the transaction or waive its rights to the redemption of its warrants, and if the Company is unable to fund the cash value of the Deerfield warrants into escrow in advance of a major transaction, the Company may be prevented from closing a financing transaction that it believes is in the interests of a majority of its shareholders.
Future sales of our common stock, or the perception that these sales may occur, could depress our stock price.
Sales of substantial amounts of our common stock in the public market, or the perception in the public markets that these sales may occur, could cause the market price of our common stock to decline. This could also impair our ability to raise additional capital through the sale of our equity securities. Selling of a large number of shares by any of our existing shareholders or management shareholders could cause the price of our common stock to decline. Furthermore, if we file a registration statement to offer additional shares of our common stock and have to include shares held by those holders, it could impair our ability to raise needed capital by depressing the price at which we could sell our common stock. As a result of private placements by us in 2005, 2006, 2007 and 2008, we currently have four outstanding shelf registration statements permitting investors in these private placements to publicly resell shares of our common stock.
Our certificate of incorporation and provisions of Delaware law could discourage a takeover you may consider favorable or could cause current management to become entrenched and difficult to replace.
Provisions in our certificate of incorporation and Delaware law may have the effect of delaying or preventing a merger or acquisition of us, or making a merger or acquisition less desirable to a potential acquirer, even when the stockholders may consider the acquisition or merger favorable. Under the terms of our certificate of incorporation, we are authorized to issue 25 million shares of “blank check” preferred stock, and to determine the price, privileges, and other terms of these shares. The issuance of any preferred stock with superior rights to our common stock could reduce the value of our common stock. In particular, specific rights we may grant to future holders of preferred stock could be used to restrict an ability to merge with or sell our assets to a third party, preserving control by present owners and management and preventing you from realizing a premium on your shares.
In addition, we are subject to provisions of the Delaware corporation law that, in general, prohibit any business combination with a beneficial owner of 15% or more of our common stock for five years unless the holder’s acquisition of our stock was approved in advance by our board of directors. These provisions could affect our stock price adversely.
31
The price of our common stock has been and will likely continue to be volatile.
Prior to our October 2003 initial public offering, there was no public market for our common stock. The initial public offering price of our common stock was $10.00 per share. Since our initial public offering, the price of our common stock has been as high as $10.30 and as low as $0.86 per share. Some companies that have had volatile market prices for their securities have been subject to securities class action suits filed against them. If a suit were to be filed against us, regardless of the outcome, it could result in substantial costs and a diversion of our management’s attention and resources. This could have a material adverse effect on our business, results of operations and financial condition.
We could be forced to pay liquidated damages if we do not maintain the effectiveness of ourS-3 registration statements.
In November 2007, we issued warrants for the purchase of 3,000,000 shares of our common stock at a price of $1.38 to affiliates of Deerfield Management. In April 2007, we completed a private placement of 10,155,000 shares of common stock and warrants to purchase 7,616,250 shares of common stock, at a price of $2.36375 per unit. In December 2006, we completed a private placement of 6,000,000 shares of common stock at a price of $3.00 per share, resulting in gross proceeds of $18.0 million. In April 2005, we completed a private placement of 6,846,735 shares of our common stock at a price of $3.98 per share and warrants to purchase a total of 2,396,357 shares of common stock at an exercise price of $4.78 per share, resulting in gross proceeds of $27.25 million. Pursuant to the terms of the registration rights agreements for each of these transactions, we filed with the SEC shelf registration statements onForm S-3 covering resales of common stock. The registration rights agreement in each transaction provides that if a registration statement is not effective within a specified number of days of closing, or if we do not subsequently maintain the effectiveness of the registration statement, then in addition to any other rights the investor may have, we will be required to pay the investor liquidated damages in cash.
The SEC declared each of ourForm S-3s effective within the specified number of days of closing. However, if we fail to maintain the effectiveness of the registration statements in the future, liquidated damages could be substantial.
| |
| Item 1B. | Unresolved Staff Comments |
Not applicable.
Our principal executive offices are located in an approximately 62,000 square foot facility in Germantown, Maryland. We moved into this facility in May 2003 and completed the transfer of our laboratory function to this facility in December 2003. The lease for this facility expires in June 2013.
In August 2004, we entered into a lease for approximately 53,000 square feet of additional research and development space, in a building adjacent to the Company’s existing headquarters in Germantown, Maryland. The lease for this facility expires in May 2013. We ceased use of this facility in 2007 and are currently marketing the facility for sublease to other companies.
We previously had an approximately 8,432 square foot lab and office facility in Gaithersburg, Maryland, the lease for which expired in November 2005. Also, in September 2004 we rented an office of approximately 6,681 square feet for engineering space in Bridgewater, New Jersey under a short-term lease that expired in September 2006.
We believe that our facilities are suitable and adequate to meet our current needs.
| |
| Item 3. | Legal Proceedings |
We are not a party to any material pending legal proceedings, other than ordinary routine litigation incidental to our business, except as discussed below.
32
In December 2003, Aventis and Aventis Pharmaceuticals Inc., now part of sanofi-aventis, brought an action against MiddleBrook Pharmaceuticals, Inc., then named Advancis Pharmaceutical Corp, alleging, in essence, that the Advancis corporate name was infringing the plaintiff’s trademark and sought injunctive relief. A trial was held in May 2005, and the Court’s decision, dated September 26, 2006, ruled in favor of sanofi-aventis. On June 28, 2007 the name change was completed pursuant to the Company’s jointly submitted Permanent Injunction and Order with sanofi-aventis of October 27, 2006, whereby the Company agreed to cease using the Advancis name by June 30, 2007. No monetary damages were associated with the decision, and the Company agreed to cease using the Advancis name by June 30, 2007. The Company implemented the name change on June 28, 2007, and there was no significant financial impact resulting from the change.
In August 2007, Eli Lilly and Company provided notice of a legal matter relating to Keflex to MiddleBrook. A product liability claim was filed by Jamie Kaye Moore against Eli Lilly, Teva Pharmaceuticals, Inc. and Teva Pharmaceuticals Industries Ltd. on March 28, 2007. The claim alleges injury from ingestion of some form of “Keflex.” Lilly has filed preliminary objections to the complaint, and has also requested prescription and other records, in order to determine whether the plaintiff ingested brand or generic cephalexin and which manufacturer might be involved. Since the identity of the manufacturer is not known, Lilly is not currently requesting indemnification from MiddleBrook.
| |
| Item 4. | Submission of Matters to a Vote of Security Holders |
No matters were submitted to a vote of our security holders during the fourth quarter of the fiscal year ended December 31, 2007.
PART II
| |
| Item 5. | Market for Registrant’s Common Equity and Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
Our common stock has been traded on The Nasdaq National Market under the symbol MBRK (previously, AVNC until June 29, 2007) since October 17, 2003. The following table sets forth the quarterly high and low sales prices per share of our common stock as reported by Nasdaq for each quarter during the last two fiscal years, commencing on January 1, 2006:
| | | | | | | | | |
| | | High | | | Low | |
| |
| December 31, 2007: | | | | | | | | |
| Fourth quarter | | $ | 2.47 | | | $ | 1.00 | |
| Third quarter | | | 2.62 | | | | 1.71 | |
| Second quarter | | | 4.50 | | | | 2.20 | |
| First quarter | | | 3.99 | | | | 2.00 | |
| December 31, 2006: | | | | | | | | |
| Fourth quarter | | $ | 5.90 | | | $ | 3.23 | |
| Third quarter | | | 6.70 | | | | 2.79 | |
| Second quarter | | | 3.98 | | | | 2.50 | |
| First quarter | | | 3.72 | | | | 1.23 | |
Holders
As of March 11, 2008, there were 115 holders of record of our common stock. This figure does not represent the actual number of beneficial owners of our common stock because shares are generally held in “street name” by securities dealers and others for the benefit of individual owners who may vote the shares.
33
Dividends
We have never declared or paid any cash dividends on our common stock. We currently intend to retain our future earnings, if any, to finance the further development and expansion of our business and do not intend to pay cash dividends for the foreseeable future. Any future determination to pay dividends will be at the discretion of our board of directors and will depend on our financial condition, results of operations, capital requirements, restrictions contained in current or future financing instruments and such other factors as our board of directors deems relevant.
Recent Sales of Unregistered Securities
In November 2007, we issued warrants to purchase 3,000,000 common shares to affiliates of Deerfield Management. The Company filed a registration statement onForm S-3 covering the resale of the common stock to be acquired upon exercise of the warrants. The registration statement was declared effective by the SEC on December 12, 2007.
34
Corporate Performance Graph
The following Performance Graph and related information shall not be deemed to be “soliciting material” or to be “filed” with the SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933 or Securities Exchange Act of 1934, each as amended, except to the extent that the Company specifically incorporates it be reference into such filing.
The following graph shows the cumulative total return resulting from a hypothetical $100 investment in our common stock on October 16, 2003, the date of our initial public offering, through February 29, 2008. MiddleBrook stock price performance over this period is compared to the same amount invested in the Nasdaq Stock Market (U.S.) Index and the Nasdaq Pharmaceutical Index over the same period (in each case, assuming reinvestment of dividends). This graph is presented as required by SEC rules. Past performance might not be indicative of future results. While total stockholder return can be an important indicator of corporate performance, we believe it is not necessarily indicative of our corporation’s degree of success in executing our business plan, particularly over short periods.
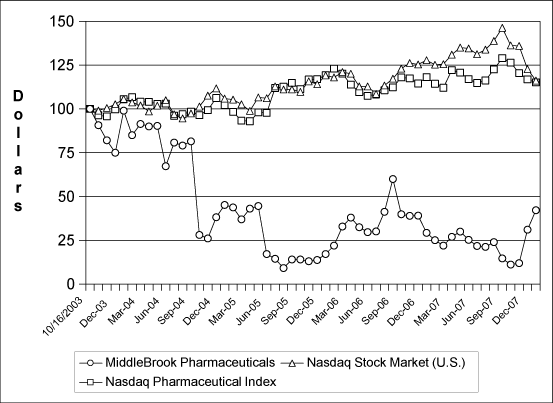
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | 10/16/03 | | | 12/31/03 | | | 12/31/04 | | | 12/31/05 | | | 12/31/06 | | | 12/31/07 |
| MiddleBrook Pharmaceuticals | | | $ | 100.00 | | | | $ | 75.00 | | | | $ | 38.20 | | | | $ | 13.80 | | | | $ | 39.10 | | | | $ | 12.00 | |
| Nasdaq Stock Market (U.S.) | | | $ | 100.00 | | | | $ | 102.66 | | | | $ | 111.72 | | | | $ | 114.10 | | | | $ | 125.36 | | | | $ | 135.93 | |
| Nasdaq Pharmaceutical Index | | | $ | 100.00 | | | | $ | 99.75 | | | | $ | 106.24 | | | | $ | 116.99 | | | | $ | 114.52 | | | | $ | 120.43 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
35
| |
| Item 6. | Selected Financial Data |
The following selected financial information has been derived from the audited financial statements. The information below is not necessarily indicative of results of future operations and should be read in conjunction with Item 7, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” of thisForm 10-K and the financial statements and related notes thereto included in Item 8 of thisForm 10-K in order to fully understand factors that may affect the comparability of the information presented below.
| | | | | | | | | | | | | | | | | | | | | |
| | | For the Years Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | | | 2004 | | | 2003 | |
| |
Statements of Operations Data | | | | | | | | | | | | | | | | | | | | |
| Total revenue | | $ | 10,456,700 | | | $ | 4,810,410 | | | $ | 16,847,690 | | | $ | 11,358,032 | | | $ | 3,625,000 | |
| | | | | | | | | | | | | | | | | | | | | |
| Cost and expenses: | | | | | | | | | | | | | | | | | | | | |
| Cost of product sales | | | 2,576,954 | | | | 899,601 | | | | 562,009 | | | | 169,854 | | | | — | |
| Research and development | | | 21,957,708 | | | | 25,973,844 | | | | 39,729,441 | | | | 33,642,930 | | | | 16,594,629 | |
| Selling, general and administrative | | | 26,043,711 | | | | 21,288,968 | | | | 10,515,302 | | | | 12,219,409 | | | | 6,427,453 | |
| | | | | | | | | | | | | | | | | | | | | |
| Total expenses | | | 50,578,373 | | | | 48,162,413 | | | | 50,806,752 | | | | 46,032,193 | | | | 23,022,082 | |
| | | | | | | | | | | | | | | | | | | | | |
| Loss from operations | | | (40,121,673 | ) | | | (43,352,003 | ) | | | (33,959,062 | ) | | | (34,674,161 | ) | | | (19,397,082 | ) |
| Interest income (expense), net | | | (40,834 | ) | | | 385,034 | | | | 954,193 | | | | 669,448 | | | | 88,565 | |
| Beneficial conversion feature — deemed interest | | | — | | | | — | | | | — | | | | — | | | | (1,666,667 | ) |
| Other income or (expense) | | | (2,249,048 | ) | | | 976,815 | | | | 16,292 | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | | |
| Net loss including noncontrolling interest | | | (42,411,555 | ) | | | (41,990,154 | ) | | | (32,988,577 | ) | | | (34,004,713 | ) | | | (20,975,184 | ) |
| Loss attributable to noncontrolling interest | | | 162,189 | | | | — | | | | — | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | | |
| Net loss applicable to common shareholders | | | (42,249,366 | ) | | | (41,990,154 | ) | | | (32,988,577 | ) | | | (34,004,713 | ) | | | (20,975,184 | ) |
| Accretion of issuance costs of mandatorily redeemable convertible preferred stock | | | — | | | | — | | | | — | | | | — | | | | (209,173 | ) |
| Beneficial conversion feature — deemed dividend to preferred shareholders | | | — | | | | — | | | | — | | | | — | | | | (20,907,620 | ) |
| | | | | | | | | | | | | | | | | | | | | |
| Net loss applicable to common stockholders | | $ | (42,249,366 | ) | | $ | (41,990,154 | ) | | $ | (32,988,577 | ) | | $ | (34,004,713 | ) | | $ | (42,091,977 | ) |
| | | | | | | | | | | | | | | | | | | | | |
| Basic and diluted net loss per share | | $ | (0.96 | ) | | $ | (1.38 | ) | | $ | (1.20 | ) | | $ | (1.50 | ) | | $ | (7.58 | ) |
| | | | | | | | | | | | | | | | | | | | | |
| Shares used in computing net loss per share, basic and diluted | | | 43,816,145 | | | | 30,535,965 | | | | 27,421,516 | | | | 22,684,410 | | | | 5,554,773 | |
| | | | | | | | | | | | | | | | | | | | | |
Balance Sheet Data at Year-End: | | | | | | | | | | | | | | | | | | | | |
| Unrestricted cash, cash equivalents and marketable securities | | $ | 1,951,715 | | | $ | 15,379,461 | | | $ | 29,431,058 | | | $ | 30,051,937 | | | $ | 65,087,122 | |
| Total assets | | | 23,665,795 | | | | 42,005,769 | | | | 57,796,892 | | | | 61,142,140 | | | | 84,174,843 | |
| Long-term debt, including current portion | | | — | | | | 6,963,889 | | | | 1,567,412 | | | | 2,577,387 | | | | 2,440,588 | |
| Noncontrolling interest | | | 7,337,811 | | | | — | | | | — | | | | — | | | | — | |
| Accumulated deficit | | | (195,334,828 | ) | | | (153,085,462 | ) | | | (111,095,308 | ) | | | (78,106,731 | ) | | | (44,102,018 | ) |
| Total stockholders’ equity (deficit) | | | (5,848,152 | ) | | | 11,872,020 | | | | 33,342,011 | | | | 39,738,379 | | | | 70,149,920 | |
36
| |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
The following discussion of our financial condition and results of operations should be read in conjunction with the condensed financial statements and the related notes included elsewhere in this annual report onForm 10-K. This discussion may contain forward-looking statements, the accuracy of which involve risks and uncertainties. As a result of many factors, such as those set forth under the“Forward-Looking Statements”and“Factors that May Affect our Business” sections in Part 1, Item 1 and elsewhere in this annual report onForm 10-K, our actual results may differ materially from those anticipated in these forward-looking statements.
Our Business
MiddleBrook Pharmaceuticals, Inc. was incorporated in Delaware in December 1999 and commenced operations on January 1, 2000. On June 28, 2007, we changed our corporate name from Advancis Pharmaceutical Corporation to MiddleBrook Pharmaceuticals, Inc. We are a pharmaceutical company focused on developing and commercializing anti-infective drug products that fulfill unmet medical needs in the treatment of infectious disease. We are developing a portfolio of drugs based on the novel biological finding that bacteria exposed to antibiotics in front-loaded, sequential bursts, or pulses, are killed more efficiently than those exposed to standard antibiotic treatment regimens. We currently have 26 issued U.S. patents and two issued foreign patent covering our proprietaryonce-a-day pulsatile delivery technology called PULSYS. We have initially focused on developing PULSYS product candidates utilizing approved and marketed drugs that no longer have patent protection or that have patents expiring in the next several years. Our lead pulsatile product candidate, based on the antibiotic amoxicillin, received U.S. Food and Drug Administration (FDA) approval for marketing on January 23, 2008, under the trade name MOXATAGtm, and our Keflex PULSYS product candidate, based on the antibiotic cephalexin, is currently under evaluation in Phase I clinical trials. We also have a number of additional pulsatile product candidates in preclinical development, although further development of these candidates will only occur if we secure additional capital resources. We acquired the U.S. rights to Keflex (cephalexin) from Eli Lilly in 2004. We currently sell our line of Keflex products to wholesalers in both capsule and powder formulations, and received FDA approval in 2006 for two additional Keflex strengths — 333 mg capsules and 750 mg capsules. We have focused our commercialization initiatives solely on the Keflex 750 mg capsules. In support of the launch of the Keflex 750 mg capsules, we entered into an agreement with a contract sales organization and currently deploy approximately 30 contract sales representatives across the United States. We have also entered into agreements with third-party contract manufacturers for the commercial supply of our products. In March 2007, we announced that we are evaluating various strategic alternatives to further enhance shareholder value and in February 2008, announced that we retained Morgan Stanley as our strategic advisor to assist us in this regard. Strategic alternatives we may pursue could include, but are not limited to, continued execution of our operating plan, licensing or development arrangements, the sale of some or all of our company’s assets, partnering or other collaboration agreements, or a merger or other strategic transaction.
General
Our future operating results will depend largely on our ability to successfully commercialize our lead PULSYS product, MOXATAG, and our ability to successfully commercialize our launched Keflex 750 mg product, and the progress of other product candidates currently in our research and development pipeline. The results of our operations will vary significantly from year to year and quarter to quarter and depend on a number of factors, including risks related to our business, risks related to our industry, and other risks which are detailed in this Annual Report onForm 10-K.
Management Overview of the Key Developments in 2007
The following is a summary of key events that occurred in 2007.
Amoxicillin PULSYS product development — MOXATAGtmapproval
| | |
| | • | In August 2006, we announced that our Amoxicillin PULSYS Phase III clinical trial for the treatment of adolescents and adults with acute pharyngitisand/or tonsillitis achieved its desired clinical and |
37
| | |
| | | microbiological endpoints. The trial demonstrated statistical non-inferiority of Amoxicillin PULSYS therapy versus the penicillin comparator therapy for the trial’s primary endpoints of bacterial eradication rates for two distinct patient populations. The trial also demonstrated Amoxicillin PULSYS reached 85 percent bacterial eradication for the “per protocol” group of patients, in accordance with FDA guidance for product approval as first-line pharyngitis therapy. |
| | |
| | • | Based on the successful Phase III trial data, we submitted a New Drug Application (NDA) for Amoxicillin PULSYS on December 14, 2006. On February 12, 2007, we received a “refusal to file” letter from the FDA for our Amoxicillin PULSYS NDA, requesting additional information on our planned commercial manufacturing processes. The FDA did not raise any clinical or other issues in its communication. |
| |
| | • | We conducted a meeting with the FDA regarding our Amoxicillin PULSYS NDA on February 26, 2007, and obtained clarification on the additional information that would be required for the FDA to accept our NDA for filing. We resubmitted our Amoxicillin PULSYS NDA on March 23, 2007, and were notified that the NDA was accepted for filing on May 22, 2007. In the notification letter, we received a Prescription Drug User Fee Act (PDUFA) target action date of January 23, 2008. |
| |
| | • | We received FDA approval of our NDA on January 23, 2008, for our once-daily Amoxicillin PULSYS product under the trade name MOXATAGtm (amoxicillin extended-release) Tablets for the treatment of adults and pediatric patients 12 years and older with pharyngitisand/or tonsillitis secondary toStreptococcus pyogenes (commonly referred to as strep throat). With the FDA approval of MOXATAG, physicians now have available the first once-daily product in the aminopenicillin class for the treatment of pharyngitis. |
Marketed Products — Keflex Capsules (Cephalexin USP)
| | |
| | • | In 2007, net sales of our branded Keflex product line were approximately $10.5 million, an increase from $4.8 million in 2006. |
| |
| | • | During 2007, we continued our commercialization efforts for our 750 mg strength of Keflex capsules through a targeted and dedicated national contract sales force, which currently consists of approximately 30 sales representatives and three MiddleBrook district sales managers. Our contract sales representatives began directly promoting Keflex 750 mg capsules to targeted physicians as well as providing patient starter samples in late July 2006. |
Investment Bank Retained to Explore Strategic Alternatives
| | |
| | • | In March 2007, we announced that we had initiated a process to explore various strategic alternatives to further enhance shareholder value. Subsequent to receiving FDA approval for our MOXATAG product in January 2008, we announced that the strategic review process was ongoing and that we had retained Morgan Stanley as our strategic advisor to assist us in this regard. Strategic alternatives we may pursue could include, but are not limited to, continued execution of our operating plan, licensing or development arrangements, the sale of some or all of our company’s assets, partnering or other collaboration agreements, or a merger or other strategic transaction. There can be no assurance that the exploration of strategic alternatives will result in any agreements or transactions, or that, if completed, any agreements or transactions will be successful or on attractive terms. We do not intend to disclose developments with respect to this process unless and until the evaluation of strategic alternatives has been completed. |
Cost Reduction Initiatives for 2007
| | |
| | • | During 2007, we implemented a cost reduction program including personnel reductions, postponement of PULSYS clinical development programs other than Amoxicillin PULSYS for adults and adolescents, and elimination of other discretionary spending during the year. Additionally, future development efforts for our PULSYS product candidates other than Amoxicillin PULSYS will be dependent upon our ability to secure additional capital or to find a partner to help fund their continued development. |
38
Private Placement Transactions
| | |
| | • | In April 2007, we closed a private placement transaction of common stock and warrants, which provided $22.4 million in net cash proceeds. |
| |
| | • | Subsequent to our December 31, 2007 fiscal yearend, we closed a private placement transaction of common stock and warrants on January 28, 2008, which provided $19.9 million in net cash proceeds. |
Agreement For the Sale and Repurchase Rights to Our Cephalexin Assets
| | |
| | • | On November 8, 2007, we announced an agreement with Deerfield Management, a healthcare investment fund and one of the Company’s largest equity shareholders, for the sale and license of certain of our cephalexin assets and intellectual property. Under the terms of the agreement, we raised $7.5 million upon closing of the transaction, and an additional $2.5 million was available, at our option, upon the approval of our MOXATAG NDA. Proceeds from the transaction were used to retire our outstanding debt of approximately $4.6 million and were also used for general corporate purposes. We also retained the right to repurchase all such assets and intellectual property at a future date. See “Keflex Agreements — Deerfield Transaction,” in Part I above. |
Focus for 2008
Our primary focus for the 2008 will be on the manufacturing process for our MOXATAG product for adults and pediatric patients 12 years and older, along with the continued commercialization of our Keflex 750 mg capsules. Our NDA supporting MOXATAG was approved by the FDA on January 23, 2008, and we believe MOXATAG could be marketed to healthcare professionals by as soon as the fourth quarter of 2008, should we successfully conclude our strategic evaluation process. We intend to continue promoting Keflex 750 mg capsules through our approximately 30 contract sales representatives and three MiddleBrook district sales managers to targeted U.S. physicians throughout 2008, assuming there are no generic competitors that enter the market during the year. In order to minimize our financing requirements in 2008, we expect to maintain our reduced cost structure implemented during 2007, including personnel reductions, postponement of PULSYS clinical development programs, and the elimination of other discretionary spending.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses. On an ongoing basis, we evaluate our estimates and judgments, including those related to accrued expenses, fair valuation of stock related to stock-based compensation and income taxes. We based our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our financial statements.
Revenue Recognition
We recognize revenue for the sale of pharmaceutical products and for payments received, if any, under collaboration agreements for licensing, milestones, and reimbursement of development costs as follows:
Product Sales. Revenue from product sales, net of estimated provisions, is recognized when there is persuasive evidence that an arrangement exists, delivery has occurred, the selling price is fixed or determinable, and collectibility is reasonably probable. Our customers consist primarily of large pharmaceutical wholesalers who sell directly into the retail channel. Provisions for sales discounts, and estimates for chargebacks, rebates, and product returns are established as a reduction of product sales revenue at the time revenues are recognized, based on historical experience adjusted to reflect known changes in the factors that impact these reserves. Factors include current contract prices and terms, estimated wholesaler inventory levels,
39
remaining shelf life of product, and historical information for similar products in the same distribution channel. These revenue reductions are generally reflected either as a direct reduction to accounts receivable through an allowance, or as an addition to accrued expenses if the payment is due to a party other than the wholesaler.
Chargebacks and Rebates. We record chargebacks and rebates based on the difference between the prices at which we sell our products to wholesalers and the sales price ultimately paid under fixed price contracts by third party payers, including governmental agencies. We record an estimate at the time of sale to the wholesaler of the amount to be charged back to us or rebated to the end user. We have recorded reserves for chargebacks and rebates based upon various factors, including current contract prices, historical trends, and our future expectations. The amount of actual chargebacks and rebates claimed could be either higher or lower than the amounts we accrued. Changes in our estimates would be recorded in the income statement in the period of the change.
Product Returns. In the pharmaceutical industry, customers are normally granted the right to return product for a refund if the product has not been used prior to its expiration date, which for our Keflex product is typically three years from the date of manufacture (two years, in the case of oral suspension products). Our return policy typically allows product returns for products within an eighteen-month window from six months prior to the expiration date and up to twelve months after the expiration date. We estimate the level of sales which will ultimately be returned pursuant to our return policy, and record a related reserve at the time of sale. These amounts are deducted from our gross sales to determine our net revenues. Our estimates take into consideration historical returns of our products and our future expectations. We periodically review the reserves established for returns and adjust them based on actual experience. The amount of actual product returns could be either higher or lower than the amounts we accrued. Changes in our estimates would be recorded in the income statement in the period of the change. If we over or under estimate the quantity of product which will ultimately be returned, there may be a material impact to our financial statements.
Contract Revenue. We use the milestone payment method of revenue recognition when all milestones in respect of payments to be received under contractual arrangements are determined to be substantive, at-risk and the culmination of an earnings process. Substantive milestones are payments that are conditioned upon events requiring substantive effort, when the amounts of the milestones are reasonable relative to the time, effort and risk involved in achieving them and when the milestones are reasonable relative to each other and the amount of any up-front payment. If these criteria are not met, the timing of the recognition of revenue from the milestone payment may vary. Up-front payments are recorded as deferred revenue. We estimate the length of the remaining development period and amortize an up-front payment over that development period.
Reimbursement of Development Costs. We record revenue for reimbursement of development costs as the actual costs to perform the work are incurred. We are required to use judgment in recognizing reimbursement revenue in cases where the agreement provides for funding to us that is not dependent on actual costs we incur within a specific fiscal period. Our policy is to limit revenue recognized to the minimum amounts expected under a specific collaboration agreement and to exclude amounts contingent on future events, such as successful commercialization and future profit-sharing, and amounts that are contingently refundable. Revenue recognized is limited to cumulative amounts under each contract such that, at any time, if a termination of the agreement were to occur, revenue previously recognized would not need to be reversed. Cash received in excess of revenue recognized is recorded as deferred revenue, with the deferred revenue recognized as revenue at the time future events occur that remove the contingencies.
Inventories
Inventory is stated at the lower of cost or market with cost determined under thefirst-in, first-out method. Inventory consists of Keflex finished capsules and finished oral suspension powder. We purchase our Keflex products from third-party manufacturers only at the completion of the manufacturing process, and accordingly have no raw material orwork-in-process inventories. At least on a quarterly basis, we review our inventory levels and write down inventory that has become obsolete or has a cost basis in excess of its expected net realizable value or is in excess of expected requirements. Inventory levels are evaluated by management relative to product demand,
40
remaining shelf life, future marketing plans and other factors, and reserves for obsolete and slow-moving inventories are recorded for amounts which may not be realizable.
Intangible Assets
Acquired Intangible Assets. We acquired the U.S. rights to the Keflex brand of cephalexin in 2004. We may acquire additional pharmaceutical products in the future that include license agreements, product rights and other identifiable intangible assets. When intangible assets are acquired, we review and identify the individual intangible assets acquired and record them based on relative fair values. Each identifiable intangible asset is then reviewed to determine if it has a definite life or indefinite life, and definite-lived intangible assets are amortized over their estimated useful lives.
Impairment. We assess the impairment of identifiable intangible assets on an annual basis or when events or changes in circumstances indicate that the carrying value may not be recoverable. Some factors we consider important which could trigger an impairment review include significant underperformance compared to historical or projected future operating results, significant changes in our use of the acquired assets or the strategy for our overall business, or significant negative industry or economic trends. If we determine that the carrying value of intangible assets may not be recoverable based upon the existence of one or more of these factors, we first perform an assessment of the asset’s recoverability based on expected undiscounted future net cash flow, and if the amount is less than the asset’s value, we measure any impairment based on a projected discounted cash flow method using a discount rate determined by our management to be commensurate with the risk inherent in our current business model.
Accrued Expenses
As part of the process of preparing financial statements, we are required to estimate accrued expenses for services performed and liabilities incurred. This process involves identifying services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for such service as of each balance sheet date in our financial statements. Examples of estimated accrued expenses for services include professional service fees, such as lawyers and accountants, contract service fees, such as amounts paid to clinical monitors, data management organizations and investigators in conjunction with clinical trials, fees paid to our contract sales organization, and fees paid to contract manufacturers in conjunction with the production of clinical materials. In connection with such service fees, our estimates are most affected by our understanding of the status and timing of services provided relative to the actual levels of services incurred by such service providers. The majority of our service providers invoice us monthly in arrears for services performed. In the event that we do not identify certain costs that have begun to be incurred or we under- or over-estimate the level of services performed or the costs of such services, our reported expenses for such period would be too low or too high. The date on which certain services commence, the level of services performed on or before a given date and the cost of such services are often judgmental. We make these judgments based upon the facts and circumstances known to us in accordance with generally accepted accounting principles. We also make estimates for other liabilities incurred, including health insurance costs for our employees. We are self-insured for claims made under our health insurance program and record an estimate at the end of a period for claims not yet reported. Our risk exposure is limited, as claims over a maximum amount are covered by an aggregate stop loss insurance policy.
Stock-Based Compensation
Effective January 1, 2006, we adopted Statement of Financial Accounting Standards No. 123R,“Share-Based Payment”(SFAS 123R). We adopted SFAS 123R using the modified prospective transition method, which requires the recognition of compensation expense under the Statement on a prospective basis only. Accordingly, prior period financial statements were not restated. Under this transition method, stock-based compensation cost for the years ended December 31, 2007 and 2006 includes (a) compensation cost for all share-based awards granted prior to, but not yet vested as of, January 1, 2006, based on the grant date fair value estimated in accordance with the original provisions of SFAS 123, and (b) compensation cost for all share-based awards granted subsequent to January 1, 2006 based on the grant-date fair value estimated in accordance with the fair value provisions of SFAS 123R.
41
SFAS 123R also requires us to estimate forfeitures in calculating the expense related to share-based compensation rather than recognizing forfeitures as a reduction in expense as they occur. To the extent actual forfeitures differ from our estimates, such amounts will be recorded as a cumulative adjustment in the period that the estimates are revised. We plan to refine our estimated forfeiture rate as we obtain more historical data.
We determine the value of stock option grants using the Black-Scholes option-pricing model. Our determination of fair value of share-based payment awards on the date of grant is affected by our stock price as well as assumptions regarding a number of highly complex and subjective variables. These variables include, but are not limited to, our expected stock price volatility over the term of the awards and projected employee stock option exercise behaviors. This model requires that we estimate our future expected stock price volatility as well as the period of time that we expect the share-based awards to be outstanding.
| | |
| | • | We have elected to determine the expected term of share-based awards granted subsequent to January 1, 2006 using the transition approach provided by Staff Accounting Bulletin No. 107, under which an expected term of 6.25 years may be used for four-year grants with ten-year contractual terms. We plan to refine our estimate of expected term in the future as we obtain more historical data. A shorter expected term would result in lower compensation expense. |
| |
| | • | To estimate expected future volatility, we considered several factors and data sets, including available information from our limited trading history, as well as the reported volatility rates of other comparable public companies. We have no implied volatility data since we have no publicly traded options or other financial instruments from which implied volatility can be derived. For the expected future volatility factor computed for input to the Black-Scholes model, we based our estimate of expected future volatility upon a combination of our historical volatility together with the average of volatility rates of comparable public companies. Using a higher volatility input to the Black-Scholes model would result in a higher compensation expense. |
| |
| | • | The risk-free rate is based on U.S. Treasury yields in effect at the time of grant corresponding with the expected term of the options. Estimates of fair value are not intended to predict actual future events or the value ultimately realized by the employees who receive equity awards. |
Income Taxes
As part of the process of preparing our financial statements we are required to estimate our income taxes in each of the jurisdictions in which we operate. We account for income taxes by the liability method. Under this method, deferred income taxes are recognized for tax consequences in future years of differences between the tax bases of assets and liabilities and their financial reporting amounts at each year-end, based on enacted laws and statutory tax rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are provided if, based upon the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized. We have not recorded any tax provision or benefit for the years ended December 31, 2007, 2006 and 2005. We have provided a valuation allowance for the full amount of our net deferred tax assets since realization of any future benefit from deductible temporary differences and net operating loss carry forwards cannot presently be sufficiently assured. At December 31, 2007 and 2006, we had federal and state net operating loss carryforwards of approximately $161.5 million and $120.6 million, respectively, available to reduce future taxable income, which will begin to expire in 2020. Under the provisions of Section 382 of the Internal Revenue Code, certain substantial changes in our ownership may result in a limitation on the amount of net operating loss and research and experimentation tax credit carry forwards which can be used in future years. During 2001 and 2005, we may have experienced such ownership changes. Ownership changes in 2001 and 2005 may have created annual limitations of approximately $0.9 million and $3.8 million, respectively. There were no ownership changes under Section 382 in other years.
In June 2006, the FASB issued FASB Interpretation No. 48,“Accounting for Uncertainty in Income Taxes — an Interpretation of FASB Statement No. 109” (FIN 48). FIN 48 clarifies the accounting for uncertainty in income taxes recognized in an enterprise’s financial statements in accordance with SFAS No. 109. FIN 48 prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken on a tax return. FIN 48 also provides guidance on derecognition,
42
classification, interest and penalties, accounting in interim periods, disclosure and transition. We applied the provisions of FIN 48 effective January 1, 2007. The implementation of FIN 48 had no impact on the Company’s financial condition, results of operations, or cash flows, as the Company has no unrecognized tax benefits.
Warrants
Freestanding financial instruments, such as detachable warrants, must be evaluated under the authoritative accounting literature to determine whether they should be classified as assets or liabilities (derivative accounting), temporary equity, or permanent equity. Management initially evaluates whether the instruments are covered by SFAS 150,“Accounting for Certain Financial Instruments with Characteristics of both Liabilities and Equity.”If the instrument is not governed by SFAS 150, then management determines whether it meets the definition of a derivative under SFAS 133,“Accounting for Derivative Instruments and Hedging Activities.”To determine whether a specific warrant agreement would follow derivative accounting under SFAS 133, management must first evaluate whether the warrant would meet the definition of equity under the provisions ofEITF 00-19,“Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company’s Own Stock.”Financial instruments such as warrants that are classified as permanent or temporary equity are excluded from the definition of a derivative for purposes of SFAS 133. Financial instruments, including warrants, that are classified as assets or liabilities are considered derivatives under SFAS 133, and are marked to market at each reporting date, with the change in fair value recorded in the income statement.
UnderEITF 00-19, contracts that require physical settlement or net-share settlement and contracts that give the Company the choice of settlement (in cash or shares) are classified as equity. Contracts that require net-cash settlement or that give the counterparty a choice which includes net-cash settlement are classified as assets or liabilities, not equity. If a transaction is outside the control of the company and there is the possibility that the Company could net-cash settle, then for purposesEITF 00-19 it is assumed that the Company will have to net-cash settle, which may preclude accounting for a contract as equity of the company except in certain circumstances where the existing common stockholders would also receive cash.
Management judgment is required in evaluating the terms of freestanding instruments, such as warrants, and the application of authoritative accounting literature. In November 2007, the Company issued 3,000,000 warrants to affiliates of Deerfield Management in connection with the sale and license of certain non-PULSYS Keflex tangible and intangible assets. The warrant agreement contains provisions for cash settlement under certain conditions, including a major asset sale or acquisition in certain circumstances, which is available to the warrant holders at their option. As a result, management concluded that the warrants should be classified as a liability at their contractual fair value in the consolidated balance sheet.
Registration Payment Arrangements
The Company views a registration rights agreement containing a liquidated damages provision as a separate freestanding contract which has nominal value, and the Company has followed that accounting approach, consistent with FASB Staff PositionNo. EITF 00-19-2,“Accounting for Registration Payment Arrangements.”Under this approach, the registration rights agreement is accounted for separately from the financial instrument. Under FSPNo. EITF 00-19-2, registration payment arrangements are measured in accordance with SFAS No. 5,“Accounting for Contingencies.”Should the Company conclude that it is more likely than not that a liability for liquidated damages will occur, the Company would record the estimated cash value of the liquidated damages liability at that time.
Consolidation of Variable Interest Entities
FASB Interpretation No. 46 (revised 2003),“Consolidation of Variable Interest Entities”(FIN 46R) clarifies the application of Accounting Research Bulletin No. 51,“Consolidated Financial Statements”,to certain entities in which equity investors do not have the characteristics of a controlling financial interest or do not have sufficient equity at risk for the entity to finance its activities without additional subordinated financial support from other parties. The usual condition for a controlling financial interest is ownership of a majority of voting interest;
43
however, there may situations where the controlling financial interest may be achieved through arrangements that do not involve voting interests.
Variable interest entities are entities that are subject to consolidation because they meet the provisions of FIN 46R. Management judgment is required in identifying potential variable interest entities and in evaluating the application of the provisions of FIN 46R to those potential variable interest entities. Paragraph 5 of FIN 46R specifies that an entity shall be subject to consolidation under FIN 46R if (a) the total equity investment at risk is not sufficient to permit the entity to finance its activities without additional subordinated financial support from other parties or (b) the equity investment holders lack (1) the direct or indirect ability to make decisions about an entity’s activities, (2) the obligation to absorb the expected losses of the entity, or (3) the right to receive the expected residual returns of the entity. If management concludes that an entity is a variable interest entity under FIN 46R, an analysis must then be made to determine if there is a primary beneficiary, using either a qualitative or quantitative approach.
In accordance with FIN 46R, MiddleBrook management evaluated whether the Deerfield affiliates Kef and Lex are variable interest entities and, if so, whether there is a primary beneficiary with a controlling financial interest. Since MiddleBrook is making the important decisions with respect to the ongoing activities involving the assets owned by Kef and Lex, the Kef and Lex entities were determined to be variable interest entities for this characteristic. Since MiddleBrook has a fixed price repurchase option, the equity holders in Kef and Lex do not have rights to all of the residual returns of the entities, so Kef and Lex were determined to be variable interest entities for this characteristic. Management used a qualitative analysis to determine whether Deerfield or MiddleBrook was the primary beneficiary of the entities. MiddleBrook was determined to be the primary beneficiary, since it is the party exposed to the majority of the risks. Thus, MiddleBrook consolidates the financial condition and results of operations of Kef and Lex in accordance with FIN 46R.
Recent Accounting Pronouncements
In February 2007, the Financial Accounting Standards Board (FASB) issued SFAS No. 159,“The Fair Value Option for Financial Assets and Financial Liabilities”(SFAS 159), which is effective for financial statements issued for fiscal years beginning after November 15, 2007. SFAS 159 provides companies with an option to report selected financial assets and liabilities at fair value. The Statement also establishes presentation and disclosure requirements designed to facilitate comparisons between companies that choose different measurement attributes for similar types of assets and liabilities. SFAS 159 requires companies to provide additional information that will help investors and other users of financial statements to more easily understand the effect of the company’s choice to use fair value on its earnings. It also requires entities to display the fair value of those assets and liabilities for which the company has chosen to use fair value on the face of the balance sheet. Adoption of SFAS 159 is not expected to have a material effect on our results of operations and financial condition.
In June 2007, the Emerging Issues Task Force (EITF) reached a consensus on EITF IssueNo. 07-03,“Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities,”(EITF 07-03).EITF 07-03 concludes that nonrefundable advance payments for future research and development activities should be deferred and capitalized until the goods have been delivered or the related services have been performed. If an entity does not expect the goods to be delivered or services to be rendered, the capitalized advance payment should be charged to expense. This consensus is effective for fiscal years beginning after December 15, 2007. Adoption ofEITF 07-03 is not expected to have a material effect on our results of operations and financial condition.
In December 2007, the EITF reached a consensus on EITF IssueNo. 07-01,“Accounting for Collaborative Arrangements,”(EITF 07-01).EITF 07-01 requires collaborators to present the result of activities for which they act as the principal on a gross basis and report any payments received from (made to) other collaborators based on other applicable GAAP or, in the absence of other applicable GAAP, based on analogy to authoritative accounting literature or a reasonable, rational, and consistently applied accounting policy election. In addition, a participant in a collaborative arrangement should provide the following disclosures separately for each collaborative arrangement: (a) the nature and purpose of the arrangement, (b) its rights and obligations under the collaborative arrangement, (c) the accounting policy for the arrangement in accordance with APB Opinion 22, “Disclosure of Accounting
44
Policies,” and (d) the income statement classification and amounts arising from the collaborative arrangement between participants for each period an income statement is presented.EITF 07-01 will be effective for annual periods beginning after December 15, 2008. Adoption ofEITF 07-01 is not expected to have a material effect on our consolidated financial statements.
In December 2007, the FASB issued SFAS No. 141 (revised 2007),“Business Combinations”(SFAS 141 R), which is effective for financial statements issued for fiscal years beginning on or after December 15, 2008. SFAS 141R establishes principles and requirements for how an acquirer recognizes and measures in its financial statements the identifiable assets acquired, the liabilities assumed, any noncontrolling interest in the acquiree, and the goodwill acquired in the business combination. SFAS 141R also establishes disclosure requirements to enable the evaluation of the nature and financial effects of the business combination. SFAS 141R will be applied prospectively. We are currently evaluating the effect that the adoption of SFAS 141R will have on our results of operations and financial condition.
In December 2007, the FASB issued SFAS No. 160,“Noncontrolling Interests in Consolidated Financial Statements, an amendment of ARB No. 51,”(SFAS 160). SFAS 160 changes the accounting and reporting for minority interests, which will be recharacterized as noncontrolling interests (NCI) and classified as a component of equity. The Statement also requires that entities provide sufficient disclosures that clearly identify and distinguish between the interests of the parent and the interests of the noncontrolling owners. SFAS 160 is effective for fiscal years beginning on or after December 15, 2008. Earlier adoption is prohibited. SFAS 160 will be applied prospectively as of the beginning of the fiscal year in which the Statement is initially applied, except for the presentation and disclosure requirements, which shall be applied retrospectively for all periods presented. We are currently evaluating the effect that the adoption of SFAS 160 will have on our results of operations and financial condition.
In February 2008, the FASB issued a FASB Staff Position, or FSP, to defer the effective date of SFAS No. 157,“Fair Value Measurements,”(SFAS 157), for nonfinancial assets and nonfinancial liabilities, except for items that are recognized or disclosed at fair value in the financial statements on a recurring basis. The FSP defers the effective date of SFAS 157 to fiscal years beginning after November 15, 2008. The delay is intended to provide the Board additional time to consider the effect of certain implementation issues that have arisen from the application of SFAS 157 to these assets and liabilities. SFAS 157 defines fair value, establishes a framework for measuring fair value in accordance with generally accepted accounting principles, and expands disclosures about fair value measurements. We are currently evaluating the effect that the adoption of SFAS 157 will have on our results of operations and financial condition.
Research and Development Expenses
We expect our research and development expenses to be significant as we continue to develop our product candidates. These expenses consist primarily of salaries and related expenses for personnel, fees paid to professional service providers in conjunction with independently monitoring our clinical trials and acquiring and evaluating data in conjunction with our clinical trials, costs of contract manufacturing services, costs of materials used in clinical trials and research and development, depreciation of capital resources used to develop our products, and costs of facilities. We expense research and development costs as incurred. We believe that significant investment in product development is a competitive necessity and plan to continue these investments, assuming sufficient financial resources are available, in order to be in a position to realize the potential of our product candidates and proprietary technologies.
45
Summary of Product Development Initiatives. The following table summarizes our product development initiatives for the fiscal years ended December 31, 2007, 2006 and 2005. Included in this table is the research and development expense recognized in connection with each product candidate currently in clinical development and all preclinical product candidates as a group.
See“Our Product Pipeline”above for our current priority product candidates.
| | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | Total Expense
| | | |
| | | | | | | | | | | | Incurred From
| | | |
| | | | | | | | | | | | Inception
| | | |
| | | | | | | | | | | | (January 1, 2000)
| | | Clinical
|
| | | Year Ended December 31, | | | to December 31,
| | | Development
|
| | | 2007 | | | 2006 | | | 2005 | | | 2007 | | | Phase |
| |
Direct Project Costs(1) | | | | | | | | | | | | | | | | | | |
| Amoxicillin PULSYS(2) | | $ | 10,615,000 | | | $ | 12,354,000 | | | $ | 24,294,000 | | | $ | 71,099,000 | | | NDA approved |
| Keflex Product Development(3) | | | 3,324,000 | | | | 5,424,000 | | | | 5,360,000 | | | | 14,330,000 | | | Phase III-ready
(on hold) |
| Generic Clarithromycin(4) | | | — | | | | — | | | | 79,000 | | | | 15,579,000 | | | Discontinued |
| Other Product Candidates | | | 147,000 | | | | 863,000 | | | | 1,289,000 | | | | 16,255,000 | | | Preclinical |
| | | | | | | | | | | | | | | | | | | |
| Total Direct Project Costs | | | 14,086,000 | | | | 18,641,000 | | | | 31,022,000 | | | | 117,263,000 | | | |
| | | | | | | | | | | | | | | | | | | |
Indirect Project Costs(1) | | | | | | | | | | | | | | | | | | |
| Facility | | | 3,468,000 | | | | 3,136,000 | | | | 3,603,000 | | | | 15,569,000 | | | |
| Depreciation | | | 2,854,000 | | | | 2,441,000 | | | | 2,610,000 | | | | 11,173,000 | | | |
| Other Indirect Overhead | | | 1,550,000 | | | | 1,756,000 | | | | 2,494,000 | | | | 11,177,000 | | | |
| | | | | | | | | | | | | | | | | | | |
| Total Indirect Expense | | | 7,872,000 | | | | 7,333,000 | | | | 8,707,000 | | | | 37,919,000 | | | |
| | | | | | | | | | | | | | | | | | | |
Total Research & Development Expense | | $ | 21,958,000 | | | $ | 25,974,000 | | | $ | 39,729,000 | | | $ | 155,182,000 | | | |
| | | | | | | | | | | | | | | | | | | |
| | |
| (1) | | Many of our research and development costs are not attributable to any individual project because we share resources across several development projects. We record direct costs, including personnel costs and related benefits and stock-based compensation, on aproject-by-project basis. We record indirect costs that support a number of our research and development activities in the aggregate. |
| |
| (2) | | On January 23, 2008, we received approval for marketing from the FDA of our Amoxicillin PULSYS adult product, with the trade name MOXATAG. See“Our Lead Product: MOXATAG (amoxicillin extended-release) Tablets”above for clinical development background. We previously had an agreement under which Par Pharmaceutical was to be responsible for funding the anticipated future development costs of this product. See“Termination of Our Collaboration with Par Pharmaceutical for Amoxicillin PULSYS”above. Our amoxicillin pediatric sprinkle product is ready for Phase II clinical trials. See“Amoxicillin Pediatric Pharyngitis Program”above. |
| |
| (3) | | Direct Project Costs for Keflex product development include development costs for the non-pulsatile Keflex 750 mg and Keflex 333 mg line extension products, which commercially launched in July 2006, as well as research and development costs for aonce-a-day Keflex PULSYS product, currently in Phase I clinical trials. Additional development of Keflex PULSYS is on hold, until we have sufficient financial resources. |
| |
| (4) | | We have discontinued development efforts for this product. See“Our Collaboration with Par Pharmaceutical for Generic Clarithromycin” above. |
Net Losses
We have a limited history of operations. We anticipate that our results of operations will fluctuate for the foreseeable future due to several factors, including progress of our research and development efforts, approval and commercial launch of new products, and the timing and outcome of regulatory approvals. Our limited operating history makes predictions of future operations difficult or impossible. Since our inception, we have incurred
46
significant losses. As of December 31, 2007, we had an accumulated deficit of approximately $195.3 million. We anticipate incurring additional annual losses, perhaps at higher levels, for the foreseeable future.
Results of Operations
Fiscal Year Ended December 31, 2007 Compared to Fiscal Year Ended December 31, 2006
Revenues. We recorded revenues of $10.5 million during the fiscal year ended December 31, 2007 compared to $4.8 million during the fiscal year ended December 31, 2006, composed of Keflex product sales as follows:
| | | | | | | | | |
Product Sale Revenues: | | 2007 | | | 2006 | |
| |
| Keflex 750mg capsules | | $ | 7,717,000 | | | $ | 2,680,000 | |
| Keflex 250mg and 500mg capsules | | | 2,740,000 | | | | 2,130,000 | |
| | | | | | | | | |
| Total | | $ | 10,457,000 | | | $ | 4,810,000 | |
| | | | | | | | | |
Prior to the third quarter of 2006, net product sales consist primarily of shipments of the Keflex 250 and 500 milligram strengths to wholesalers. In July 2006, we launched a 750 milligram strength capsule, supported by a targeted and dedicated national contract sales force of 75 sales representatives and eight Advancis district sales managers. Sales of 750mg in 2007 reflected a full 12 months of shipments as compared to six months in 2006. Sales of Keflex 250mg and 500mg capsules increased in 2007 primarily due to price increases implemented during the year.
Cost of Product Sales. Cost of product sales represents the purchase cost of the Keflex products sold during the year, provisions for obsolescence, as well as royalties, if applicable. The following table discloses the major components of cost of product sales:
| | | | | | | | | |
Cost of Product Sales: | | 2007 | | | 2006 | |
| |
| Product manufacturing costs | | $ | 903,000 | | | $ | 459,000 | |
| Obsolescence provisions | | | 864,000 | | | | 140,000 | |
| Royalty to Lilly | | | 810,000 | | | | 301,000 | |
| | | | | | | | | |
| | | $ | 2,577,000 | | | $ | 900,000 | |
| | | | | | | | | |
The increase in product manufacturing costs and Lilly royalty expense reflect a full year of sales activity for the Keflex 750mg product in 2007 versus six months in 2006. Only the Keflex 750mg product is currently subject to Lilly royalty cost. Consignment and royalty payments due to affiliates of Deerfield Management from MiddleBrook based on sales of all Keflex non-PULSYS products, beginning in November 2007, which approximated $333,000 for the period from November 8, 2007 through December 31, 2007, are eliminated in the consolidated statement of operations in accordance with FIN 46R. Obsolescence provisions result from projections of future sales compared to inventory levels, and a determination that a portion of inventory may not be sold prior to expiration date.
Research and Development Expenses. Research and development expenses decreased $4.0 million, or 15%, to $22.0 million for the fiscal year ended December 31, 2007 from $26.0 million for the fiscal year ended December 31, 2006. Research and development expenses consist of direct costs which include salaries and related costs of research and development personnel, and the costs of consultants, materials and supplies associated with research and development projects, as well as clinical studies. Indirect research and development costs include facilities, depreciation, and other indirect overhead costs.
47
The following table discloses the components of research and development expenses reflecting our project expenses.
| | | | | | | | | |
| | | Year Ended December 31, | |
Research and Development Expenses | | 2007 | | | 2006 | |
| |
| Direct project costs: | | | | | | | | |
| Personnel, benefits and related costs | | $ | 6,766,000 | | | $ | 6,252,000 | |
| Stock-based compensation | | | 718,000 | | | | 1,435,000 | |
| Consultants, supplies, materials and other direct costs | | | 6,530,000 | | | | 4,916,000 | |
| Clinical studies | | | 72,000 | | | | 6,038,000 | |
| | | | | | | | | |
| Total direct costs | | | 14,086,000 | | | | 18,641,000 | |
| Indirect project costs | | | 7,872,000 | | | | 7,333,000 | |
| | | | | | | | | |
| Total | | $ | 21,958,000 | | | $ | 25,974,000 | |
| | | | | | | | | |
Personnel, benefits and related costs increased due to an increased bonus payout of $0.2 million resulting from our successfully achieving FDA approval of the MOXATAG NDA, increased healthcare and other benefits costs of $0.2 million, and normal annual pay increases. Stock-based compensation decreased $0.7 million as certain grants awarded in prior years became fully amortized, and as the total number of new options awarded declined due to the decline in employee headcount.
Consultants, supplies, materials and other direct costs increased $1.6 million as we continued developing our manufacturing capability in anticipation of an eventual launch of MOXATAG. Clinical trials expense decreased $5.9 million overall, as we conducted a single Phase III amoxicillin trial in 2006, and none in 2007.
Indirect project costs increased by $0.5 million, primarily due to costs related to our ceasing to use one of our Germantown, Maryland research facilities.
Selling, General and Administrative Expenses. Selling, general and administrative expenses increased $4.8 million, or 22%, to $26.0 million for the year ended December 31, 2007 from $21.3 million for the year ended December 31, 2006.
| | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | |
| |
| Salaries, benefits and related costs | | $ | 3,284,000 | | | $ | 2,710,000 | |
| Severance costs | | | 534,000 | | | | (359,000 | ) |
| Stock-based compensation | | | 1,213,000 | | | | 1,970,000 | |
| Legal and consulting expenses | | | 2,212,000 | | | | 2,142,000 | |
| Other expenses | | | 6,526,000 | | | | 6,175,000 | |
| Marketing costs | | | 4,746,000 | | | | 3,786,000 | |
| Contract sales expenses | | | 7,529,000 | | | | 4,865,000 | |
| | | | | | | | | |
| Total | | $ | 26,044,000 | | | $ | 21,289,000 | |
| | | | | | | | | |
Selling, general and administrative expenses consist of salaries and related costs for executive and other administrative personnel, selling and product distribution costs, professional fees and facility costs. Major increases in 2007 include costs of promoting sales of our Keflex 750mg line extension for a full year in 2007 vs. six months in 2006.
Salaries, benefits and related costs increased $0.6 million versus 2006, due in part to having a full year of sales managers in place compared to six months in 2006, and to increased bonus and benefits costs compared to the prior year.
Severance costs related to a reduction in the number of administrative employees in the fourth quarter, as compared to 2006 when a credit to expense was recognized upon the rehire of an executive who had been terminated in 2005.
48
Stock-based compensation decreased $0.8 million as certain grants awarded in prior years became fully amortized, and as the total number of new options awarded declined due to the decline in employee headcount.
Other expenses increased $0.4 million as we incurred a regulatory filing fee of $0.9 million in connection with our Amoxicillin PULSYS NDA, versus a prior year fee of $0.3 million, and other regulatory fees of $0.2 million.
Marketing and contract sales expenses totaled $12.3 million versus $8.7 million in 2006 reflecting costs to promote sales of our Keflex 750 milligram product for a full year, versus part year in 2006.
Net Interest Income (Expense). Net interest income was $41,000 for the year ended December 31, 2007 compared to net interest income of $385,000 for the year ended December 31, 2006. Interest income overall declined $353,000 due to reduced cash balances available for investing during the year.
Interest expense increased from 2005 as we incurred interest costs related to our Merrill Lynch term debt facility, which was in place for approximately six months in 2006 versus over 10 months in 2007 before being paid off in November.
| | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | |
| |
| Interest income | | $ | 543,000 | | | $ | 896,000 | |
| Interest expense | | | (584,000 | ) | | | (511,000 | ) |
| | | | | | | | | |
| Total, net | | $ | (41,000 | ) | | $ | 385,000 | |
| | | | | | | | | |
Warrant expense. Warrant expense in 2007 of $2.1 million resulted from an initial charge of $2.6 million, reduced by a credit at year end of $0.5 million as the options were revalued based upon the year end stock price, which had declined since the warrants were issued on November 7, 2007.
Other Income. Other income of $0.1 million in 2007 represents forgiveness of our debt to Montgomery County. During 2007 we received notice from Montgomery County that we had met the conditions required for our development loan to be forgiven, and accordingly the full amount was recognized as Other Income in the quarter.
Other income of $1.0 million in 2006 represents the recognition in income of an advance payment received in 2005 from a potential buyer of our Keflex brand. We did not enter into a definitive agreement for the asset sale, and in January 2006 we decided to retain the Keflex assets. The agreement in principle expired on February 28, 2006. Under the circumstances, the advance payment of $1,000,000 was not refundable and was therefore recognized as income in 2006.
Noncontrolling Interest. Pursuant to the agreements that we entered into with Deerfield Management in November 2007, we consolidate the financial condition and results of operations of Kef Pharmaceuticals, Inc. and Lex Pharmaceuticals, Inc. in accordance with FIN 46R. Accordingly, we have deducted the losses of $162,000 attributable to the noncontrolling interest (the losses of Kef and Lex) from our net loss in the consolidated statement of operations, and we have also reduced the noncontrolling interest holders’ ownership interest in Kef and Lex in the consolidated balance sheet by the losses of Kef and Lex.
Fiscal Year Ended December 31, 2006 Compared to Fiscal Year Ended December 31, 2005
Revenues. We recorded revenues of $4.8 million during the fiscal year ended December 31, 2006 compared to $16.8 million during the fiscal year ended December 31, 2005, as follows:
| | | | | | | | | |
| | | 2006 | | | 2005 | |
| |
| Keflex product sales — net | | $ | 4,810,000 | | | $ | 4,809,000 | |
| Amortization of upfront license fee — Par amoxicillin | | | — | | | | 4,028,000 | |
| Reimbursement of development costs — Par amoxicillin | | | — | | | | 8,011,000 | |
| | | | | | | | | |
| Total | | $ | 4,810,000 | | | $ | 16,848,000 | |
| | | | | | | | | |
Prior to the third quarter of 2006, net product sales consist primarily of shipments of the Keflex 250 and 500 milligram strengths to wholesalers. In July 2006, we launched a 750 milligram strength capsule, supported by a
49
targeted and dedicated national contract sales force of 75 sales representatives and eight Advancis district sales managers. During the year, we recognized $2.7 million of revenue related to sales of the 750mg product, which includes initial stocking to wholesalers and pharmacies. Net revenue for other Keflex products was $2.1 million, consisting entirely of net product sales of Keflex 250 and 500 capsules, as compared to $4.8 million in 2005. The decrease in Keflex sales compared to prior periods was mainly due to a net reduction in orders received from our wholesaler customers for our Keflex 250 and 500 products. In addition, there is increasing substitution of generic cephalexin for prescriptions of existing 250mg and 500mg Keflex brand capsules. Net product sales of our oral suspension Keflex product were insignificant for the periods presented.
Revenues recognized in 2005 for amortization of upfront licensing fees represent the amortization of a $5.0 million upfront payment received from Par Pharmaceutical in May 2004 in connection with our collaboration for Amoxicillin PULSYS, which was being amortized into revenue on a straight-line basis over the estimated development period. On August 3, 2005, Par terminated the Amoxicillin PULSYS collaboration, at which time we recognized the remaining deferred revenue balance of $3.2 million.
Reimbursement of development costs by Par was recognized based on the related costs incurred. As a result of the contract termination by Par on August 3, 2005, we accelerated the revenue recognition of $2.4 million in the third quarter, which was the remaining deferred revenue balance in excess of the amount retained for future contingent liability to Par.
Cost of Product Sales. Cost of product sales represents the purchase cost of the Keflex products sold during the year as well as royalties, if applicable. Cost of product sales increased from $0.6 million in 2005 to $0.9 million in 2006, as a result of purchase and royalty costs associated with the 750 milligram product launched in 2006.
Research and Development Expenses. Research and development expenses decreased $13.7 million, or 35%, to $26.0 million for the fiscal year ended December 31, 2006 from $39.7 million for the fiscal year ended December 31, 2005. Research and development expenses consist of direct costs which include salaries and related costs of research and development personnel, and the costs of consultants, materials and supplies associated with research and development projects, as well as clinical studies. Indirect research and development costs include facilities, depreciation, and other indirect overhead costs.
The following table discloses the components of research and development expenses reflecting our project expenses.
| | | | | | | | | |
| | | Year Ended December 31, | |
Research and Development Expenses | | 2006 | | | 2005 | |
| |
| Direct project costs: | | | | | | | | |
| Personnel, benefits and related costs | | $ | 6,252,000 | | | $ | 10,716,000 | |
| Stock-based compensation | | | 1,435,000 | | | | 160,000 | |
| Consultants, supplies, materials and other direct costs | | | 4,916,000 | | | | 7,912,000 | |
| Clinical studies | | | 6,038,000 | | | | 12,234,000 | |
| | | | | | | | | |
| Total direct costs | | | 18,641,000 | | | | 31,022,000 | |
| Indirect project costs | | | 7,333,000 | | | | 8,707,000 | |
| | | | | | | | | |
| Total | | $ | 25,974,000 | | | $ | 39,729,000 | |
| | | | | | | | | |
Personnel, benefits and related costs decreased $4.5 million in 2006 primarily due to severance charges of $2.8 million incurred in 2005, versus zero in 2006, and a benefit of $1.5 million due to lower staffing levels throughout 2006 attributable to reductions in staff implemented in 2005. Stock-based compensation costs increased $1.3 million, of which $0.9 million is related to employees and results primarily from the impact of adoption of SFAS 123R in 2006, and the remaining increase of $0.4 million relates to expense for non-employees, which increased from the impact of a higher stock price in 2006 versus 2005 as well as grants for the first time to contracted sales representatives.
Consultants, supplies, materials and other direct costs decreased $3.0 million, a result of decreased spending of $1.7 million on Amoxicillin PULSYS materials, a reduction in pediatric amoxicillin costs of $0.9 million as we
50
refocused on passing the adult trial, and reductions in other projects of $0.4 million. Clinical trials expense decreased $6.2 million overall, as we conducted a single phase III amoxicillin trial in 2006, versus two trials in 2005.
Indirect project costs decreased by $1.4 million, primarily due to decreases in facility-related costs of $0.5 million from vacating the Gaithersburg facility in 2005, consulting costs of $0.6 million, and equipment depreciation of $0.2 million as some equipment became fully depreciated while new acquisitions were minimal.
Selling, General and Administrative Expenses. Selling, general and administrative expenses increased $10.8 million, or 102%, to $21.3 million for the year ended December 31, 2006 from $10.5 million for the year ended December 31, 2005.
| | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2006 | | | 2005 | |
| |
| Salaries, benefits and related costs | | $ | 2,351,000 | | | $ | 3,387,000 | |
| Stock-based compensation | | | 1,970,000 | | | | 376,000 | |
| Legal and consulting expenses | | | 2,142,000 | | | | 1,342,000 | |
| Other expenses | | | 6,175,000 | | | | 5,410,000 | |
| Marketing costs | | | 3,786,000 | | | | — | |
| Contract sales expenses | | | 4,865,000 | | | | — | |
| | | | | | | | | |
| Total | | $ | 21,289,000 | | | $ | 10,515,000 | |
| | | | | | | | | |
Selling, general and administrative expenses consist of salaries and related costs for executive and other administrative personnel, selling and product distribution costs, professional fees and facility costs. Major increases in 2006 include costs of promoting sales of our Keflex 750mg line extension, including the addition of eight district sales managers, engaging a third party sales organization, and other marketing activities in support of the product line extension.
Salaries, benefits and related costs decreased $1.0 million versus 2005, due to severance costs for a workforce reduction in 2005 of $1.1 million and lower compensation costs in 2006 related to the reduced number of employees of $0.3 million, partly offset by the additional costs of the sales management team of $0.5 million in 2006 as compared to 2005.
Stock-based compensation increased $1.6 million primarily from the effect of SFAS 123R, which was effective on January 1, 2006. Also, prior year expense was relatively low due to the reversal of prior period expense of approximately $0.5 million for the forfeiture of stock options resulting from the workforce reduction in 2005.
Legal and consulting costs increased $0.8 million as we engaged outside organizations to conduct extensive market research in support of its Keflex line extension and potential future products.
Other expenses increased $0.8 million as we incurred regulatory license and filing fees of $0.5 million in connection with our Keflex product line and Amoxicillin PULSYS NDA, and increased patent costs of $0.4 million.
Marketing costs of $3.8 million in 2006 reflect activities in support of the initial launch and continuing marketing of our Keflex line extension. Contract sales expenses consist of the direct costs of 75 sales representatives we have engaged through a third-party contract sales organization, to promote our Keflex 750mg product to physicians.
Net Interest Income (Expense). Net interest income was $0.4 million for the year ended December 31, 2006 compared to net interest income of $1.0 million for the year ended December 31, 2005. While the yield on investments improved in 2006 due to modestly higher interest rates, interest income overall declined $0.2 million due to reduced cash balances available for investing during the year. Interest expense increased from 2005 as we incurred interest costs related to our Merrill Lynch term debt facility, which originated at the end of the second quarter 2006.
51
| | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2006 | | | 2005 | |
| |
| Interest income | | $ | 896,000 | | | $ | 1,075,000 | |
| Interest expense | | | (511,000 | ) | | | (121,000 | ) |
| | | | | | | | | |
| Total, net | | $ | 385,000 | | | $ | 954,000 | |
| | | | | | | | | |
Other Income. Other income in 2006 primarily consists of the recognition in income of a non-refundable advance payment of $1.0 million, received in 2005 for the potential sale of the Keflex brand rights, as the sale was not completed.
Liquidity and Capital Resources
We have funded our operations principally with the proceeds of $54.5 million from a series of five preferred stock offerings and one issue of convertible notes over the period 2000 through 2003, the net proceeds of $54.3 million from our initial public offering in October 2003, and private placements of common stock for net proceeds of $25.8 million, $16.7 million and $22.4 million in April 2005, December 2006, and April 2007, respectively. In addition, we have received funding of $8.0 million and $28.25 million from GlaxoSmithKline and Par Pharmaceutical, respectively, as a result of collaboration agreements for the development of new products. Since July 2004, we have also received cash of approximately $25.8 million from sales of our Keflex products. We received a $1.0 million advance payment in 2005 from a potential buyer of our Keflex brand, which we recognized in income in 2006 as the sale was not completed. In the second quarter of 2006, we received proceeds of $6.9 million from a term loan, net of costs and the payoff of existing debt. In November 2007, we sold certain of our Keflex assets in exchange for $7.5 million (not reflecting a $500,000 payment to the purchaser), while retaining the right to continue operating the Keflex business subject to certain consignment and royalty payments to the purchaser as well as the right to repurchase the assets at a future date at predetermined prices.
We are evaluating various strategic alternatives to further enhance shareholder value, and we retained an investment banking firm to assist us in this regard. Strategic alternatives we may pursue could include, but are not limited to, continued execution of our operating plan, licensing or development arrangements, the sale of some or all of our company’s assets, partnering or other collaboration agreements, or a merger or other strategic transaction. There can be no assurance that the exploration of strategic alternatives will result in any agreements or transactions, or that, if completed, any agreements or transactions will be successful or on attractive terms.
Cash and Marketable Securities
At December 31, 2007, unrestricted cash, cash equivalents and marketable securities were $2.0 million compared to $15.4 million at December 31, 2006.
| | | | | | | | | |
| | | As of December 31, | |
| | | 2007 | | | 2006 | |
| |
| Cash and cash equivalents | | $ | 1,952,000 | | | $ | 14,857,000 | |
| Marketable securities | | | — | | | | 522,000 | |
| | | | | | | | | |
| Total | | $ | 1,952,000 | | | $ | 15,379,000 | |
| | | | | | | | | |
Our cash and cash equivalents are highly-liquid investments with a maturity of 90 days or less at date of purchase and consist of time deposits, investments in money market funds with commercial banks and financial institutions, and commercial paper of high-quality corporate issuers. At December 31, 2007 we held no auction rate securities in our portfolio. Our marketable securities are highly-liquid investments and are classified as available-for-sale, as they can be utilized for current operations. Our investment policy requires the selection of high-quality issuers, with bond ratings of AAA to A1+/P1. Our objective is to limit the investment portfolio to a maximum average duration of approximately one year, with no individual security exceeding a two-year duration. At December 31, 2007 and 2006, no security was held with a maturity greater than 6 months from those dates.
52
Also, we maintain cash balances with financial institutions in excess of insured limits. We do not anticipate any losses with respect to such cash balances.
Cash Flow
The following table summarizes our sources and uses of cash and cash equivalents for fiscal years ending December 31, 2007, 2006, and 2005.
| | | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| |
| Net cash used in operating activities | | $ | (35,261,000 | ) | | $ | (36,331,000 | ) | | $ | (24,890,000 | ) |
| Net cash provided by (used in) investing activities | | | (834,000 | ) | | | 10,793,000 | | | | 7,676,000 | |
| Net cash provided by financing activities | | | 23,190,000 | | | | 22,278,000 | | | | 24,935,000 | |
| | | | | | | | | | | | | |
| Net increase (decrease) in cash and cash equivalents | | $ | (12,905,000 | ) | | $ | (3,260,000 | ) | | $ | 7,721,000 | |
| | | | | | | | | | | | | |
Operating Activities
Net cash used in operating activities for the three years ended December 31, 2007 is presented in the following table, which displays cash received and cash disbursed by major element.
| | | | | | | | | | | | | |
| | | Year Ended December 31, | |
Operating Activities | | 2007 | | | 2006 | | | 2005 | |
| |
| Cash receipts: | | | | | | | | | | | | |
| Cash received from product sales | | $ | 10,707,000 | | | $ | 6,120,000 | | | $ | 5,159,000 | |
| Cash received from collaboration partners | | | — | | | | — | | | | 14,250,000 | |
| Interest income received and other | | | 785,000 | | | | 1,568,000 | | | | 1,622,000 | |
| | | | | | | | | | | | | |
| Total cash receipts | | | 11,492,000 | | | | 7,688,000 | | | | 21,031,000 | |
| | | | | | | | | | | | | |
| Cash disbursements: | | | | | | | | | | | | |
| Cash paid for employee compensation and benefits | | | 11,260,000 | | | | 9,969,000 | | | | 11,432,000 | |
| Cash paid to vendors, suppliers, and other | | | 35,493,000 | | | | 34,050,000 | | | | 34,489,000 | |
| | | | | | | | | | | | | |
| Total cash disbursements | | | 46,753,000 | | | | 44,019,000 | | | | 45,921,000 | |
| | | | | | | | | | | | | |
| Net cash used in operating activities | | $ | (35,261,000 | ) | | $ | (36,331,000 | ) | | $ | (24,890,000 | ) |
| | | | | | | | | | | | | |
Cash received from product sales in 2007 of $10.7 million exceeded product sales cash receipts in 2006 of $6.1 million, reflecting the positive impact of a full year of sales of our Keflex 750mg product as compared to a half-year in 2006. 2005 sales included only 250mg and 500mg product. Cash received from collaboration partners relates to our previous collaboration agreements with Par Pharmaceutical for amoxicillin PULSYS, for which we received $14.25 million in 2005 prior to the contract being terminated by Par. Cash paid for employee compensation and benefits increased in 2007 from 2006, as we employed sales managers throughout 2007 as compared to a part year in 2006, and we added certain staff positions to prepare the amoxicillin PUSLYS manufacturing facility for FDA approval and eventual launch. Cash paid to vendors reflects a full year of marketing and external sales force costs to market Keflex 750mg capsules, as well as payments to complete the buildout of our manufacturing facility in Clonmel, Ireland. Clinical trials spending decreased in 2007 as compared to 2006 and 2005, as we did not conduct any Phase III trials in the year.
53
Investing Activities
Net cash provided by / used in investing activities for the three years ended December 31, 2007 is presented in the following table, which displays cash received and cash disbursed by major element.
| | | | | | | | | | | | | |
| | | Year Ended December 31, | |
Investing Activities | | 2007 | | | 2006 | | | 2005 | |
| |
| Cash receipts: | | | | | | | | | | | | |
| Sale of marketable securities, net of purchases | | $ | 563,000 | | | $ | 10,590,000 | | | $ | 8,176,000 | |
| Advance payment received for potential sale of Keflex | | | — | | | | — | | | | 1,000,000 | |
| Sale of fixed assets, restricted cash and other | | | — | | | | 754,000 | | | | 423,000 | |
| | | | | | | | | | | | | |
| Total cash receipts | | | 563,000 | | | | 11,344,000 | | | | 9,599,000 | |
| | | | | | | | | | | | | |
| Cash disbursements: | | | | | | | | | | | | |
| Property and equipment purchases and deposits | | | 1,397,000 | | | | 551,000 | | | | 1,923,000 | |
| | | | | | | | | | | | | |
| Net cash provided by (used in) investing activities | | $ | (834,000 | ) | | $ | 10,793,000 | | | $ | 7,676,000 | |
| | | | | | | | | | | | | |
Property and equipment purchases in 2007 were primarily to complete the buildout and equipping of the amoxicillin PULSYS manufacturing facility in Ireland.
The most significant investing activities in 2006 included net purchases and sales of marketable securities of $10.6 million, the release of restricted cash of $0.7 million, and purchases and deposits on property and equipment of $0.6 million
The most significant investing activities in 2005 included net purchases and sales of marketable securities of $8.2 million, receipt of a $1.0 million advance payment pursuant to the potential sale of Keflex assets (which we retained, as theagreement-in-principle expired without the sale of the business), and purchases of and deposits on property and equipment of $1.9 million.
Financing Activities
Net cash provided by financing activities for the three years ended December 31, 2007 is presented in the following table, which displays cash received and cash disbursed by major element.
| | | | | | | | | | | | | |
| | | Year Ended December 31, | |
Financing Activities | | 2007 | | | 2006 | | | 2005 | |
| |
| Cash receipts: | | | | | | | | | | | | |
| Cash received from private placement | | $ | 22,412,000 | | | $ | 16,736,000 | | | $ | 25,844,000 | |
| Cash received from term debt | | | — | | | | 7,793,000 | | | | — | |
| Cash received from Deerfield | | | 7,500,000 | | | | — | | | | — | |
| Cash received from exercise of stock options | | | 167,000 | | | | 353,000 | | | | 101,000 | |
| | | | | | | | | | | | | |
| Total cash receipts | | | 30,079,000 | | | | 24,882,000 | | | | 25,945,000 | |
| | | | | | | | | | | | | |
| Cash disbursements: | | | | | | | | | | | | |
| Cash paid for debt | | | 6,889,000 | | | | 2,604,000 | | | | 1,010,000 | |
| | | | | | | | | | | | | |
| Total cash disbursements | | | 6,889,000 | | | | 2,604,000 | | | | 1,010,000 | |
| | | | | | | | | | | | | |
| Net cash provided by financing activities | | $ | 23,190,000 | | | $ | 22,278,000 | | | $ | 24,935,000 | |
| | | | | | | | | | | | | |
The major financing activities in 2007 were a private placement of common stock that occurred in April which generated $22.4 million of net proceeds, and the November sale of our inventory and non-PULSYS Keflex intangibles to Deerfield, which generated $7.5 million, of gross proceeds, not reflecting a $500,000 payment to the purchaser. The proceeds from the Deerfield transaction were used to pay off the remaining outstanding debt.
54
The major financing activities in 2006 were a private placement of common stock, which provided $16.7 million net of issuance costs, and a debt facility with Merrill Lynch which provided financing of $7.8 million. Additionally, repayments on lines of credit totaled $2.6 million during the period.
The major financing activity in 2005 was the private placement of common stock, which provided $25.8 million net of issuance costs. Additionally, repayments on lines of credit totaled $1.0 million during the period.
Borrowings
In November, the remaining outstanding Merrill Lynch term debt was paid in full from the proceeds of the Deerfield financing. As of December 31, 2007 we have no other debt outstanding, nor any debt facilities available.
Stock Issuances
In April 2007, we completed a private placement of 10,155,000 shares of our common stock at a price of $2.36375 per share, and warrants to purchase a total of 7,616,250 shares of common stock at an exercise price of $2.27 per share, resulting in net proceeds after commissions and expenses of $22.4 million.
In December 2006, we completed a private placement of 6,000,000 shares of our common stock at a price of $3.00 per share, resulting in net proceeds after commissions and expenses of $16.7 million. There were no warrants associated with the transaction.
In April 2005, we completed a private placement of 6,846,735 shares of our common stock at a price of $3.98 per share and warrants to purchase a total of 2,396,357 shares of common stock at an exercise price of $4.78 per share, resulting in net proceeds, after commissions and expenses, of $25.8 million. The warrants are exercisable for five years.
Subsequent to December 31, 2007, we completed in January 2008 a private placement of 8,750,001 shares of our common stock and warrants to purchase 3,500,001 shares of common stock at a price of $2.40 per unit, resulting in net proceeds after commissions and expenses of $19.9 million.
Contractual Obligations
The following table summarizes our contractual obligations at December 31, 2007 and the effects such obligations are expected to have on our liquidity and cash flows in future periods.
Payments Due by Period
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | After
| |
Contractual Obligations(1),(2) | | Total | | | 2008 | | | 2009 | | | 2010 | | | 2011 | | | 2012 | | | 2012 | |
| | | (In thousands) | |
| |
| Minimum purchase commitments(3) | | $ | 1,244 | | | $ | 1,244 | | | $ | — | | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| Minimum royalty commitment(4) | | | 800 | | | | 800 | | | | — | | | | — | | | | — | | | | — | | | | — | |
| Operating lease obligations | | | 11,828 | | | | 2,139 | | | | 2,156 | | | | 2,214 | | | | 2,155 | | | | 2,220 | | | | 944 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Total contractual cash obligations | | $ | 13,872 | | | $ | 4,183 | | | $ | 2,156 | | | $ | 2,214 | | | $ | 2,155 | | | $ | 2,220 | | | $ | 944 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Other commercial commitments(5) | | $ | 10,486 | | | $ | 8,239 | | | $ | 2,247 | | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | |
| (1) | | This table does not include potential royalty payments, at a rate of 10% of sales value, to Eli Lilly and Company, which may be due on product line extensions of Keflex, including Keflex 750 mg. Any such royalties cannot be estimated at this time. |
| |
| (2) | | This table does not include a contingent liability to Par Pharmaceutical under our amoxicillin development and commercialization agreement that was terminated by Par in August 2005. Under certain circumstances, the termination clauses of the agreement may entitle Par to receive a share of future net profits, if any, up to one-half of Par’s total $23.25 million investment in the development of certain amoxicillin PULSYS products, should a |
55
| | |
| | product covered by the agreement be successfully commercialized. Accordingly, we retained $11.625 million of deferred revenue in recognition of this contingent liability to Par. |
| |
| (3) | | In the event we were to terminate our Innovex sales contract, we would incur minimum costs to exit of approximately $1.2 million. The remaining purchase commitments include costs of our product manufacturing capability under development in Ireland and Puerto Rico. |
| |
| (4) | | Effective November 7, 2007, we pay consignment payments and royalties to Deerfield at 20% of net Keflex revenues, with a minimum combined quarterly payment of $400,000. The combined 20% rate declines to 15% if we elect to make an extension payment, as defined in the agreements with Deerfield, of $1.35 million ($1.8 million if a second closing has occurred) to Deerfield by June 30, 2008. The related agreements expire June 30, 2008, unless extension payments are made, at the Company’s option, to extend the expiration dates of the repurchase rights. |
| |
| (5) | | In addition to the minimum purchase commitments required under our contracts, we expect to incur additional amounts under the contractual arrangement. These amounts represent the remaining contractual amount due over the remaining Innovex contract term, for on on-going commercialization efforts related to MOXATAG, and for manufacturing and research related to our Keflex products, |
In addition to the minimum purchase commitments and contractual obligations in the above table, we may incur funding liabilities for additional obligations which we enter into on a discretionary basis. These discretionary obligations could include additional facilities or equipment, investments in new technologies or products, acquisitions, funding of clinical trials, or similar events. Due to the approval of MOXATAG in January 2008, we authorized the re-start of pre-production development work at the Clonmel facility, to prepare for commercial production of MOXATAG.
Off-Balance Sheet Arrangements
We have not entered into any transactions, agreements or other contractual arrangements that meet the definition of off-balance sheet arrangements, with the exception of our private placements of common stock and warrants in April 2005 and April 2007. Warrants are instruments that meet the definition of a derivative under SFAS 133, although they may qualify for the scope exception under paragraph 11 of SFAS 133. In the April 2005 private placement, warrants were issued to purchase a total of 2,396,357 shares of common stock at an exercise price of $4.78 per share. In the April 2007 private placement, warrants were issued to purchase a total of 7,616,250 shares of common stock at an exercise price of $2.27 per share. In November 2007, warrants were issued to affiliates of Deerfield Management to purchase a total of 3,000,000 shares of common stock at an exercise price of $1.38; these warrants have been determined to be a derivative and have therefore been recorded as a liability in our December 31, 2007 balance sheet.
In August 2004, the Company leased additional space adjacent to its Germantown, Maryland, facility. We ceased the use of this facility during the third quarter of 2007. Effective April 2008, another company leased approximately 40 percent of the facility directly from the landlord, with the landlord amending our lease to reflect a rent reduction for the amount of rent the landlord will receive each month from the other company. We remain contingently liable for the other company’s rental payments under a financial guarantee to the landlord. Due to the financial guarantee, we have included 100 percent of the full building rent in our contractual obligations table above for operating lease obligations.
Prospective Information — Risks and Uncertainties related to Our Future Capital Requirements
At December 31, 2007, unrestricted cash, cash equivalents and marketable securities were $1.9 million, and in January 2008, we closed a private placement of common stock and warrants for net cash proceeds of $19.9 million.
We are evaluating various strategic alternatives to further enhance shareholder value, and in February 2008 we retained Morgan Stanley, an investment banking firm, to assist us in this regard. Strategic alternatives we may pursue could include, but are not limited to, continued execution of our operating plan, licensing or development arrangements, the sale of some or all of our company’s assets, partnering or other collaboration agreements, or a merger or other strategic transaction. There can be no assurance that the exploration of strategic alternatives will result in any agreements or transactions, or that, if completed, any agreements or transactions will be successful or on attractive terms.
56
We expect to incur a loss from operations in 2008. However, we believe that our existing cash resources, including the net proceeds from the private placement transaction in January 2008, will be sufficient to fund our operations at least into the first quarter of 2009 at our planned levels of research, development, sales and marketing activities, barring unforeseen developments. Furthermore, due to the approval of our Amoxicillin PULSYS NDA on January 23, 2008, we have the right, if we choose to give notice by June 30, 2008, to require a second closing of the Deerfield transaction, in which we would receive an additional payment of $2.5 million, net, in exchange for the acquisition and license by Deerfield of certain intellectual property rights relating only to the Company’s cephalexin PULSYS business; we would retain the right to operate that business (subject to certain future consignment and royalty payments to Deerfield, should a cephalexin PULSYS product be successfully developed and commercialized) as well as the right to reacquire those intellectual property rights at some point in the future.
To minimize our cash requirements, we have continued our program of cost reductions including personnel reductions, postponement of PULSYS clinical development programs, and elimination of other discretionary spending. Our net cash requirements for 2008 will depend, among other things, on the cash received from sales of our existing non-PULSYS products (primarily sales of Keflex capsules in 250 mg, 500 mg and 750 mg strengths) and the cash expended for (1) cost of products sold, including royalties due to Eli Lilly on Keflex 750 net revenues and consignment and royalty payments due to Deerfield on all Keflex net revenues, (2) research and development spending, (3) sales and marketing expenses for Keflex 750 and MOXATAG, and (4) general and administrative expenses. Our cash receipts and cash expenditures assumptions for 2008 include the following: (1) continuation of Keflex 750 monthly prescriptions at the current 20,000 to 25,000 prescriptions per month rate (end-user demand), assuming no generic competitive product enters the market in 2008, (2) validation and manufacturingscale-up activities at our contract manufacturing site in Clonmel, Ireland, in preparation for the MOXATAG product launch, (3) research and development programs for PULSYS product candidates basically on hold unless and until additional finance is obtained, (4) a sales force of approximately 30 representatives, excluding the sales and marketing cost of a commercial launch of MOXATAG, and (5) continued focus on reductions in discretionary spending. We expect to incur a significant loss in 2008, as we expect that revenues from product sales will not be sufficient to fully fund our operating costs. These 2008 estimates are forward-looking statements that involve risks and uncertainties, and actual results could vary.
Our cash projections for 2008 do not include the cost of selling and marketing activities for the commercial launch of MOXATAG. We are currently evaluating commercialization options for our MOXATAG product. We believe the MOXATAG market opportunity would be best addressed through its direct promotion by a national sales force of at least 300 sales representatives. As such, a commercialization initiative would require significant resources and expertise, and we believe it would be in the Company’s best interests to seek potential acquirers or partners to capitalize on MOXATAG’s commercial potential. Potential sales and marketing strategies for MOXATAG include the acquisition of the Company by a larger pharmaceutical organization with an established commercialization infrastructure, working with contract sales organizations, developing our own internal sales organization, or co-promoting products with collaborative marketing partners. Even if we successfully conclude our strategic evaluation process and identify a third party to assist in the commercialization of MOXATAG, the earliest we could launch the product would be in the fourth quarter of 2008. In addition, in order for the Company to participate in the sales and marketing of MOXATAG, we would need to have sufficient financial resources, which may require us to raise additional capital.
We have experienced significant losses since our inception in 2000, and as of December 31, 2007, we had an accumulated deficit of $195.3 million. The process of developing and commercializing our products requires significant research and development work, preclinical testing and clinical trials, as well as regulatory approvals, significant marketing and sales efforts, and manufacturing capabilities. These activities, together with our general and administrative expenses, are expected to continue to result in significant operating losses for the foreseeable future. To date, the revenues we have recognized from our non-PULSYS products have been limited and have not been sufficient for us to achieve or sustain profitability. Our product revenues are unpredictable in the near term and may fluctuate due to many factors, many of which we cannot control, including the market acceptance of our products. If our products fail to achieve market acceptance, we would have lower product revenues which may increase our capital requirements.
57
Our estimates of future capital requirements are uncertain and will depend on a number of factors, including the progress of our research and development of product candidates, the timing and outcome of regulatory approvals, cash received from sales of our existing non-PULSYS products, payments received or made under any future collaborative agreements, the costs involved in preparing, filing, prosecuting, maintaining, defending and enforcing patent claims and other intellectual property rights, the acquisition of licenses for new products or compounds, the status of competitive products, the availability of financing and our or our partners’ success in developing markets for our product candidates. Changes in our commercialization plans, partnering activities, regulatory activities and other developments may increase our rate of spending and decrease the period of time our available resources will fund our operations. Insufficient funds may require us to delay, scale back or eliminate some or all of our research, development or commercialization programs, or may adversely affect our ability to operate as a going concern.
We have no unused credit facility, or other committed sources of capital, except for the $2.5 million we would be entitled to receive in the event that we request a second closing from Deerfield and require Deerfield to license certain intellectual property rights relating to cephalexin PULSYS, as described above. To the extent our capital resources are insufficient to meet our future capital requirements, we will need to raise additional capital, incur indebtedness, or consider the sale of company assets in order to fund our operations. There is no assurance additional debt or equity financing will be available on acceptable terms, if at all. Our ability to raise capital by issuing additional equity may require the prior approval of Deerfield Management, under the terms of its warrant agreement, to the extent the number of common shares to be issued exceeds 25% of the outstanding common shares; should Deerfield not approve the transaction, it may require the redemption of its warrants in cash, at the contractual fair value calculated in accordance with the Black-Scholes formula. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate our research and development programs, reduce our commercialization efforts, effect changes to our facilities or personnel, or obtain funds through arrangements with collaborative partners or others that may require us to relinquish rights to certain product candidates that we might otherwise seek to develop or commercialize independently. Any future funding may dilute the ownership of our equity investors.
Safe Harbor Statement under the Private Securities Litigation Reform Act of 1995
Certain statements contained in “Business” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. The forward-looking statements are based on our current intent, belief and expectations. These statements are not guarantees of future performance and are subject to certain risks and uncertainties that are difficult to predict. Actual results may differ materially from these forward-looking statements because of our unproven business model, our dependence on new technologies, the uncertainty and timing of clinical trials, our ability to develop and commercialize products, our dependence on collaborators for services and revenue, our lease obligations, our changing requirements and costs associated with planned facilities, intense competition, the uncertainty of patent and intellectual property protection, our dependence on key management and key suppliers, the uncertainty of regulation of products, the impact of future alliances or transactions and other risks described in this filing and our other filings with the Securities and Exchange Commission. Existing and prospective investors are cautioned not to place undue reliance on these forward-looking statements, which speak only as of today’s date. We undertake no obligation to update or revise the information contained in this announcement whether as a result of new information, future events or circumstances or otherwise.
| |
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
Market Risk
Our exposure to market risk is currently confined to our cash and cash equivalents, marketable securities, and restricted cash that generally have maturities of less than one year. We currently do not hedge interest rate exposure. We have not used derivative financial instruments for speculation or trading purposes. Because of the short-term maturities of our cash, cash equivalents and marketable securities, we do not believe that an increase in market rates would have any significant impact on the realized value of our investments.
58
Fair Value of Warrants (Derivative Liabilities)
As of December 31, 2007, the estimated fair value of warrant liabilities recorded on our balance sheet, which are related to the Deerfield transaction, was $2.1 million. We estimate the fair value of these instruments using the Black-Scholes option pricing model which takes into account a variety of factors, including stock price volatility, risk-free interest rates, remaining term, and the closing price of our common stock. The determination of the fair value of the Deerfield warrants is most significantly affected by the change in the closing price of our common stock. The stock price volatility factor for the Deerfield warrants is not a risk exposure, as the volatility factor is fixed in the agreement. The following table illustrates the potential effect of changes in the assumptions used to calculate fair value:
| | | | | |
| | | Increase (Decrease) in
| |
| | | Fair Value of Derivative | |
| |
| 10% increase in stock price | | $ | 270,000 | |
| 20% increase in stock price | | $ | 570,000 | |
| 20% increase in risk-free interest rate | | $ | 30,000 | |
| 10% decrease in stock price | | $ | (300,000 | ) |
| 20% decrease in stock price | | $ | (570,000 | ) |
| 20% decrease in risk-free interest rate | | $ | (30,000 | ) |
The Company’s stock price at the market close on December 31, 2007 was $1.20 per share. On January 24, 2008, the Company publicly announced the approval for marketing by the FDA of its MOXATAG product, and the Company’s stock price closed at $2.99 per share. As of March 20, 2008, the Company’s stock price was $4.16 per share. The significant increase in the Company’s stock price would be the primary reason for a significant increase in the fair value of the warrant liabilities, from $2.1 million as of December 31, 2007 to approximately $9.9 million as of March 20, 2008. We cannot predict our future stock prices nor can we predict the future fair value of the warrant liabilities.
Inflation
Our most liquid assets are cash, cash equivalents and marketable securities. Because of their liquidity, these assets are not directly affected by inflation. We also believe that we have intangible assets in the value of our intellectual property. In accordance with generally accepted accounting principles, we have not capitalized the value of this intellectual property on our balance sheet. Due to the nature of this intellectual property, we believe that these intangible assets are not affected by inflation. Because we intend to retain and continue to use our equipment, furniture and fixtures and leasehold improvements, we believe that the incremental inflation related to replacement costs of such items will not materially affect our operations. However, the rate of inflation affects our expenses, such as those for employee compensation and contract services, which could increase our level of expenses and the rate at which we use our resources.
| |
| Item 8. | Financial Statements and Supplementary Data |
The information required by this item is set forth on pages F-1 to F-35.
| |
| Item 9. | Changes and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| |
| Item 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
Our management, including our principal executive and principal financial officers, has evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2007. Our disclosure controls and procedures are designed to provide reasonable assurance that the information required to be disclosed in this annual report onForm 10-K has been appropriately recorded, processed, summarized and reported. Based on that
59
evaluation, our principal executive and principal financial officers have concluded that our disclosure controls and procedures are effective at the reasonable assurance level.
Changes in Internal Control over Financial Reporting during the Quarter
Our management, including our principal executive and principal financial officers, has evaluated any changes in our internal control over financial reporting that occurred during the quarter ended December 31, 2007, and has concluded that there was no change that occurred during the quarter ended December 31, 2007 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Controls over Financial Reporting
The Company’s management is responsible for establishing and maintaining adequate internal controls over financial reporting, as defined inRule 13a-15(f) under the Securities Exchange Act of 1934. The Company’s system of internal controls over financial reporting was designed to provide reasonable assurance to the Company’s management and Board of Directors regarding the preparation and fair presentation of published financial statements.
All internal control systems, no matter how well designed, have inherent limitations. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation and may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Management, including the chief executive officer and chief financial officer, assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2007. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) inInternal Control — Integrated Framework. Based on our assessment, management concluded that the Company maintained effective internal control over financial reporting as of December 31, 2007.
| |
| Item 9B. | Other Information |
None.
PART III
| |
| Item 10. | Directors, Executive Officers and Corporate Governance |
We incorporate herein by reference the information concerning directors and executive officers in our Notice of Annual Stockholders’ Meeting and Proxy Statement to be filed within 120 days after the end of our fiscal year (the “2008 Proxy Statement”).
| |
| Item 11. | Executive Compensation |
We incorporate herein by reference the information concerning executive compensation to be contained in the 2008 Proxy Statement.
| |
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
We incorporate herein by reference the information concerning security ownership of certain beneficial owners and management to be contained in the 2008 Proxy Statement.
60
| |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
We incorporate herein by reference the information concerning certain relationships and related transactions, and Director independence, to be contained in the 2008 Proxy Statement.
| |
| Item 14. | Principal Accountant Fees and Services |
We incorporate herein by reference the information concerning certain relationships and related transactions to be contained in the 2008 Proxy Statement.
PART IV
| |
| Item 15. | Exhibits, Financial Statements Schedule, and Reports onForm 8-K |
(a) The following documents are filed as part of this Annual Report:
(1) Index to Financial Statements
| | | | | |
| | | Page
|
| | | Number |
| |
| Report of Independent Registered Public Accounting Firm | | | F-2 | |
| Consolidated Balance Sheets at December 31, 2007 and 2006 | | | F-3 | |
| Consolidated Statements of Operations for the Years Ended December 31, 2007, 2006 and 2005 | | | F-4 | |
| Consolidated Statement of Changes in Stockholders’ Equity (Deficit) for the Years Ended December 31, 2007, 2006 and 2005 | | | F-5 | |
| Consolidated Statements of Cash Flows for the Years Ended December 31, 2007, 2006 and 2005 | | | F-6 | |
| Notes to Consolidated Financial Statements | | | F-7 | |
(2) Financial Statement Schedule
The following schedule is filed as part of thisForm 10-K:
Exhibit II — Valuation and Qualifying Accounts for the Years Ended December 31, 2007, 2006, and 2005
(3) Exhibits
| | | | | |
Exhibit No. | | |
| |
| | 2 | .1(3)†+ | | Asset Purchase Agreement dated as of June 30, 2004, by and between the Registrant and Eli Lilly and Company |
| | 2 | .2(15) | | Asset Purchase Agreement, dated November 7, 2007, by and between the Registrant and Kef Pharmaceuticals, Inc. |
| | 2 | .3(15) | | Asset Purchase Agreement, dated November 7, 2007, by and between the Registrant and Lex Pharmaceuticals, Inc. |
| | 3 | .1(1) | | Certificate of Incorporation |
| | 3 | .2(1) | | Bylaws |
| | 3 | .3(13) | | Certificate of Ownership and Merger Merging MiddleBrook Pharmaceuticals, Inc. Into Advancis Pharmaceutical Corporation |
| | 4 | .1(1) | | Specimen Stock Certificate |
| | 4 | .2(10) | | Form of Registration Rights Agreement dated April 9, 2007 |
| | 4 | .3(15) | | Form of Warrant of the Registrant |
| | 4 | .4(15) | | Registration Rights Agreement, dated November 7, 2007, by and among the Registrant, Deerfield Private Design International Fund, L.P., Deerfield Special Situations Fund, L.P., Deerfield Special Situation International Limited and Deerfield Private Design Fund, L.P. |
| | 4 | .5(17) | | Form of Registration Rights Agreement dated January 24, 2008 |
| | 4 | .6(17) | | Form of Warrant Agreement dated January 28, 2008 |
61
| | | | | |
Exhibit No. | | |
| |
| | 10 | .1(1) | | Executive Employment Agreement between the Registrant and Edward M. Rudnic dated January 7, 2000 |
| | 10 | .2(1) | | Executive Employment Agreement between the Registrant and Sandra E. Wassink dated August 13, 2003 |
| | 10 | .3(1) | | Executive Employment Agreement between the Registrant and Beth A. Burnside dated August 13, 2003 |
| | 10 | .4(1) | | Executive Employment Agreement between the Registrant and Darren Buchwald dated September 1, 2003 |
| | 10 | .5(4) | | Executive Employment Agreement between the Registrant and Donald Treacy dated March 19, 2004 |
| | 10 | .6(8) | | Executive Employment Agreement between the Registrant and Robert C. Low dated December 12, 2005 |
| | 10 | .7(1) | | Form of Indemnification Agreement |
| | 10 | .8(1) | | Stock Incentive Plan |
| | 10 | .9(1) | | Form of Incentive Stock Option Agreement |
| | 10 | .10(1) | | Form of Non-qualified Stock Option Agreement |
| | 10 | .11(1) | | Form of Stock Restriction Agreement |
| | 10 | .12(2) | | Employee Stock Purchase Plan |
| | 10 | .13(1) | | Form of Employment Agreement on Ideas, Inventions and Confidential Information |
| | 10 | .14(1) | | Construction Services Agreement between the Registrant and Barclay White Skanska, Inc. dated July 12, 2002 |
| | 10 | .15(1) | | Amendment No. 1 dated April 7, 2003 to Agreement between Owner and Construction Manager dated July 12, 2002 between the Registrant and Skanska USA Building, Inc. successor by merger to Barclay White Skanska, Inc. |
| | 10 | .16(1) | | Standard Form of Agreement between Owner and Architect with Standard Form of Architect’s Services between the Registrant and Gaudreau, Inc. dated July 8, 2002 |
| | 10 | .17(1) | | Lease Agreement between the Registrant and Seneca Meadows Corporate Center II LLC dated August 1, 2002 |
| | 10 | .18(1) | | Fourth Amended and Restated Stockholders’ Agreement |
| | 10 | .19(1) | | Omnibus Addendum and Amendment to Series E Convertible Preferred Stock Purchase Agreement and Fourth Amended and Restated Stockholders’ Agreement |
| | 10 | .20(1)+ | | Supply, Distribution and Marketing Agreement between the Registrant and Par Pharmaceutical, Inc. dated September 4, 2003 |
| | 10 | .21(3)+ | | Manufacturing Agreement dated as of June 30, 2004, by and between the Registrant and Eli Lilly and Company |
| | 10 | .22(3)+ | | Transition Services Agreement dated as of June 30, 2004, by and between the Registrant and Eli Lilly and Company |
| | 10 | .23(4)+ | | Development and Commercialization Agreement between the Registrant and Par Pharmaceutical, Inc. dated May 28, 2004 |
| | 10 | .24(5)+ | | Commercial Supply Agreement between the Registrant and Ceph International Corporation dated December 3, 2004 |
| | 10 | .25(5)+ | | First Amendment to Development and Commercialization Agreement between the Registrant and Par Pharmaceutical Corporation dated December 14, 2004 |
| | 10 | .26(7)+ | | Manufacturing and Supply Agreement between the Registrant and Clonmel Healthcare Limited, dated as of April 19, 2005 |
| | 10 | .27(7)+ | | Development and Clinical Manufacturing Agreement between the Registrant and Clonmel Healthcare Limited, dated as of April 19, 2005 |
62
| | | | | |
Exhibit No. | | |
| |
| | 10 | .28(7)+ | | Facility Build-Out Agreement between the Registrant and Clonmel Healthcare Limited, dated as of April 19, 2005 |
| | 10 | .29(6)+ | | Form of Purchase Agreement dated April 26, 2005, including the form of Warrant attached thereto |
| | 10 | .30(9) | | Credit and Security Agreement, dated June 30, 2006 between the Registrant and Merrill Lynch Capital |
| | 10 | .31(10) | | Securities Purchase Agreement dated April 9, 2007, including the form of Warrant attached thereto |
| | 10 | .32(11) | | Letter Agreement, dated May 9, 2007, between the Registrant and Merrill Lynch Capital, a division of Merrill Lynch Business Financial Services, Inc. |
| | 10 | .33(12) | | Form of Amendment to the Form of Incentive Stock Option Agreement |
| | 10 | .34(12) | | Form of Amendment to the Form of Non-Qualified Stock Option Agreement |
| | 10 | .35(14) | | Letter Agreement, dated August 14, 2007, between the Registrant and Merrill Lynch Capital, a division of Merrill Lynch Business Financial Services, Inc. |
| | 10 | .36(15) | | Stock Purchase Agreement, dated November 7, 2007, by and among Deerfield Private Design International Fund, L.P., Deerfield Special Situations Fund, L.P., Deerfield Special Situation International Limited, Deerfield Private Design Fund, L.P., Deerfield Management, L.P., Kef Pharmaceuticals, Inc. and the Registrant |
| | 10 | .37(15) | | Stock Purchase Agreement, dated November 7, 2007, by and among Deerfield Private Design International Fund, L.P., Deerfield Special Situations Fund, L.P., Deerfield Special Situation International Limited, Deerfield Private Design Fund, L.P., Deerfield Management, L.P., Lex Pharmaceuticals, Inc. and the Registrant |
| | 10 | .38(15) | | Inventory Consignment Agreement, dated November 7, 2007, by and between the Registrant and Kef Pharmaceuticals, Inc. |
| | 10 | .39(15) | | Registration and Trademark License Agreement, dated November 7, 2007, by and between the Registrant and Lex Pharmaceuticals, Inc. |
| | 10 | .40(15) | | Form of Patent License Agreement, by and between the Registrant and Kef Pharmaceuticals, Inc. |
| | 10 | .41(15) | | Form of Patent Sublicense Agreement, by and between the Registrant and Kef Pharmaceuticals, Inc. |
| | 10 | .42(15) | | Regulatory Responsibility Agreement, dated November 7, 2007, by and between the Registrant and Lex Pharmaceuticals, Inc. |
| | 10 | .43(15) | | Keflex Products Transition Agreement, dated November 7, 2007, by and between the Registrant and Kef Pharmaceuticals, Inc. |
| | 10 | .44(15) | | Contingent Manufacturing Assignment, dated November 7, 2007, by and between the Registrant and Lex Pharmaceuticals, Inc. |
| | 10 | .45(16) | | First Amendment to Executive Employment Agreement Amendment between the Registrant and Edward M. Rudnic, dated November 19, 2007 |
| | 10 | .46(16) | | Second Amendment to Executive Employment Agreement Amendment between the Registrant and Robert C. Low, dated November 19, 2007 |
| | 10 | .47(16) | | Second Amendment to Executive Employment Agreement Amendment between the Registrant and Darren Buchwald, dated November 19, 2007 |
| | 10 | .48(16) | | Second Amendment to Executive Employment Agreement Amendment between the Registrant and Beth A. Burnside, dated November 19, 2007 |
| | 10 | .49(16) | | Second Amendment to Executive Employment Agreement Amendment between the Registrant and Donald Treacy, dated November 19, 2007 |
| | 10 | .50(16) | | Second Amendment to Executive Employment Agreement Amendment between the Registrant and Sandra E. Wassink, dated November 19, 2007 |
| | 10 | .51(17) | | Form of Purchase Agreement dated January 24, 2008 |
| | 23 | .1 | | Consent of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm |
| | 31 | .1 | | Rule 13a-14(a) Certification of Principal Executive Officer |
| | 31 | .2 | | Rule 13a-14(a) Certification of Principal Financial Officer |
63
| | | | | |
Exhibit No. | | |
| |
| | 32 | .1 | | Section 1350 Certification of Chief Executive Officer |
| | 32 | .2 | | Section 1350 Certification of Chief Financial Officer |
| | |
| (1) | | Incorporated by reference to our Registration Statement onForm S-1 (FileNo. 333-107599). |
| |
| (2) | | Incorporated by reference to our Registration Statement onForm S-8 (FileNo. 333-109728). |
| |
| (3) | | Incorporated by reference to our Current Report onForm 8-K filed July 15, 2004. |
| |
| (4) | | Incorporated by reference to our Quarterly Report onForm 10-Q filed August 6, 2004. |
| |
| (5) | | Incorporated by reference to our Annual Report onForm 10-K filed March 10, 2005. |
| |
| (6) | | Incorporated by reference to our Current Report onForm 8-K dated April 27, 2005. |
| |
| (7) | | Incorporated by reference to our Quarterly Report onForm 10-Q filed August 15, 2005. |
| |
| (8) | | Incorporated by reference to our Annual Report onForm 10-K filed March 29, 2006. |
| |
| (9) | | Incorporated by reference to our Quarterly Report onForm 10-Q filed August 9, 2006. |
| |
| (10) | | Incorporated by reference to our Current Report onForm 8-K filed April 13, 2007. |
| |
| (11) | | Incorporated by reference to our Quarterly Report onForm 10-Q filed May 11, 2007. |
| |
| (12) | | Incorporated by reference to our Current Report onForm 8-K filed May 22, 2007. |
| |
| (13) | | Incorporated by reference to our Current Report onForm 8-K filed June 28, 2007. |
| |
| (14) | | Incorporated by reference to our Current Report onForm 8-K filed August 20, 2007. |
| |
| (15) | | Incorporated by reference to our Current Report onForm 8-K filed November 13, 2007. |
| |
| (16) | | Incorporated by reference to our Current Report onForm 8-K filed November 26, 2007. |
| |
| (17) | | Incorporated by reference to our Current Report onForm 8-K filed January 30, 2008. |
| |
| † | | The Schedules and certain of the Exhibits to this Asset Purchase Agreement have been omitted in reliance upon the rules of the Securities and Exchange Commission. A copy will be delivered to the Securities and Exchange Commission upon request. |
| |
| + | | Confidential treatment requested for certain portions of this Exhibit pursuant to Rule 406 under the Securities Act, which portions are omitted and filed separately with the Securities and Exchange Commission. |
64
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
MIDDLEBROOK PHARMACEUTICALS, INC.
Edward M. Rudnic, Ph.D.
President and Chief Executive Officer
Dated: March 25, 2008
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and the dates indicated:
| | | | | | | |
Signature | | Title | | Date |
| |
| | | | | |
/s/ R. Gordon Douglas
R. Gordon Douglas, M.D. | | Chairman of the Board of Directors | | March 25, 2008 |
| | | | | |
/s/ Edward M. Rudnic
Edward M. Rudnic, Ph.D. | | President, Chief Executive Officer and Director (Principal Executive Officer) | | March 25, 2008 |
| | | | | |
/s/ Robert C. Low
Robert C. Low | | Vice President — Finance, Chief Financial Officer, and Treasurer (Principal Financial and Accounting Officer) | | March 25, 2008 |
| | | | | |
/s/ James H. Cavanaugh
James H. Cavanaugh, Ph.D. | | Director | | March 25, 2008 |
| | | | | |
/s/ Richard W. Dugan
Richard W. Dugan | | Director | | March 25, 2008 |
| | | | | |
/s/ Wayne T. Hockmeyer
Wayne T. Hockmeyer, Ph.D. | | Director | | March 25, 2008 |
| | | | | |
/s/ Martin A. Vogelbaum
Martin A. Vogelbaum | | Director | | March 25, 2008 |
| | | | | |
/s/ Harold R. Werner
Harold R. Werner | | Director | | March 25, 2008 |
65
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| | | | | |
| | | Page
|
| | | Number |
| |
| Report of PricewaterhouseCoopers LLP, Independent Registered Public Accounting Firm | | | F-2 | |
| Consolidated Balance Sheets at December 31, 2007 and 2006 | | | F-3 | |
| Consolidated Statements of Operations for the Years ended December 31, 2007, 2006 and 2005 | | | F-4 | |
| Consolidated Statement of Changes in Stockholders’ Equity (Deficit) for the Years ended December 31, 2007, 2006 and 2005 | | | F-5 | |
| Consolidated Statements of Cash Flows for the Years ended December 31, 2007, 2006 and 2005 | | | F-6 | |
| Notes to Consolidated Financial Statements | | | F-7 | |
F-1
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Shareholders
of MiddleBrook Pharmaceuticals, Inc.:
In our opinion, the consolidated financial statements listed in the index appearing under Item 15(a)(1) present fairly, in all material respects, the financial position of MiddleBrook Pharmaceuticals, Inc. at December 31, 2007 and December 31, 2006, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2007 in conformity with accounting principles generally accepted in the United States of America. In addition, in our opinion, the financial statement schedule listed in the index appearing under Item 15(a)(2) presents fairly, in all material respects, the information set forth therein when read in conjunction with the related consolidated financial statements. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2007, based on criteria established inInternal Control — Integrated Frameworkissued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company’s management is responsible for these financial statements and financial statement schedule, for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting, appearing under Item 9A. Our responsibility is to express opinions on these financial statements, on the financial statement schedule, and on the Company’s internal control over financial reporting based on our integrated audits. We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
As discussed in Note 17 to the financial statements, the Company changed the manner in which it accounts for share-based compensation in 2006.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
/s/ PricewaterhouseCoopers LLP
Baltimore, Maryland
March 25, 2008
F-2
MIDDLEBROOK PHARMACEUTICALS, INC.
| | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
| |
ASSETS |
| Current assets: | | | | | | | | |
| Cash and cash equivalents | | $ | 1,951,715 | | | $ | 14,856,738 | |
| Marketable securities | | | — | | | | 522,723 | |
| Accounts receivable, net | | | 687,787 | | | | 303,514 | |
| Inventories, net | | | 687,933 | | | | 2,077,390 | |
| Prepaid expenses and other current assets | | | 1,142,905 | | | | 1,682,685 | |
| | | | | | | | | |
| Total current assets | | | 4,470,340 | | | | 19,443,050 | |
| | | | | | | | | |
| Property and equipment, net | | | 10,928,659 | | | | 11,764,627 | |
| Restricted cash | | | 872,180 | | | | 872,180 | |
| Deposits and other assets | | | 174,965 | | | | 1,548,585 | |
| Intangible assets, net | | | 7,219,651 | | | | 8,377,327 | |
| | | | | | | | | |
| Total assets | | $ | 23,665,795 | | | $ | 42,005,769 | |
| | | | | | | | | |
| |
LIABILITIES, NONCONTROLLING INTEREST AND STOCKHOLDERS’ EQUITY (DEFICIT) |
| Current liabilities: | | | | | | | | |
| Accounts payable | | $ | 1,659,752 | | | $ | 2,285,736 | |
| Accrued expenses and advances | | | 5,613,544 | | | | 7,817,224 | |
| Lines of credit and short term debt | | | — | | | | 6,888,889 | |
| Note payable | | | — | | | | 75,000 | |
| Deferred product revenue | | | — | | | | 189,000 | |
| | | | | | | | | |
| Total current liabilities | | | 7,273,296 | | | | 17,255,849 | |
| Warrant liability | | | 2,100,000 | | | | — | |
| Deferred contract revenue | | | 11,625,000 | | | | 11,625,000 | |
| Deferred rent and credit on lease concession | | | 1,177,840 | | | | 1,252,900 | |
| | | | | | | | | |
| Total liabilities | | | 22,176,136 | | | | 30,133,749 | |
| | | | | | | | | |
| Noncontrolling interest | | | 7,337,811 | | | | — | |
| | | | | | | | | |
| Commitments and contingencies | | | | | | | | |
| Stockholders’ equity (deficit): | | | | | | | | |
| Preferred stock, $0.01 par value; 25,000,000 shares authorized, no shares issued or outstanding at December 31, 2007 and 2006 | | | — | | | | — | |
| Common stock, $0.01 par value; 225,000,000 shares authorized, and 46,748,748 and 36,362,447 shares issued and outstanding at December 31, 2007 and 2006, respectively | | | 467,488 | | | | 363,625 | |
| Capital in excess of par value | | | 189,019,188 | | | | 164,593,930 | |
| Accumulated deficit | | | (195,334,828 | ) | | | (153,085,462 | ) |
| Accumulated other comprehensive loss | | | — | | | | (73 | ) |
| | | | | | | | | |
| Total stockholders’ equity (deficit) | | | (5,848,152 | ) | | | 11,872,020 | |
| | | | | | | | | |
| Total liabilities, noncontrolling interest and stockholders’ equity (deficit) | | $ | 23,665,795 | | | $ | 42,005,769 | |
| | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-3
MIDDLEBROOK PHARMACEUTICALS, INC.
| | | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| |
| Revenues: | | | | | | | | | | | | |
| Product sales | | $ | 10,456,700 | | | $ | 4,810,410 | | | $ | 4,809,222 | |
| Contract revenue | | | — | | | | — | | | | 4,027,778 | |
| Reimbursement of development costs | | | — | | | | — | | | | 8,010,690 | |
| | | | | | | | | | | | | |
| Total revenue | | | 10,456,700 | | | | 4,810,410 | | | | 16,847,690 | |
| | | | | | | | | | | | | |
| Cost and expenses: | | | | | | | | | | | | |
| Cost of product sales | | | 2,576,954 | | | | 899,601 | | | | 562,009 | |
| Research and development | | | 21,957,708 | | | | 25,973,844 | | | | 39,729,441 | |
| Selling, general and administrative | | | 26,043,711 | | | | 21,288,968 | | | | 10,515,302 | |
| | | | | | | | | | | | | |
| Total expenses | | | 50,578,373 | | | | 48,162,413 | | | | 50,806,752 | |
| | | | | | | | | | | | | |
| Loss from operations | | | (40,121,673 | ) | | | (43,352,003 | ) | | | (33,959,062 | ) |
| Interest income | | | 543,442 | | | | 895,685 | | | | 1,075,084 | |
| Interest expense | | | (584,276 | ) | | | (510,651 | ) | | | (120,891 | ) |
| Early extinguishment of debt | | | (224,048 | ) | | | — | | | | — | |
| Warrant expense | | | (2,100,000 | ) | | | — | | | | — | |
| Other income | | | 75,000 | | | | 976,815 | | | | 16,292 | |
| | | | | | | | | | | | | |
| Loss including noncontrolling interest | | $ | (42,411,555 | ) | | $ | (41,990,154 | ) | | $ | (32,988,577 | ) |
| Loss attributable to noncontrolling interest | | | 162,189 | | | | — | | | | — | |
| | | | | | | | | | | | | |
| Net loss | | $ | (42,249,366 | ) | | $ | (41,990,154 | ) | | $ | (32,988,577 | ) |
| | | | | | | | | | | | | |
| Basic and diluted net loss per share applicable to common stockholders | | $ | (0.96 | ) | | $ | (1.38 | ) | | $ | (1.20 | ) |
| | | | | | | | | | | | | |
| Shares used in calculation of basic and diluted net loss per share | | | 43,816,145 | | | | 30,535,965 | | | | 27,421,516 | |
| | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-4
MIDDLEBROOK PHARMACEUTICALS, INC.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | Accumulated
| | | Total
| |
| | | | | | | | | Capital in
| | | Deferred
| | | | | | Other
| | | Stockholders’
| |
| | | Common
| | | Par
| | | Excess of
| | | Stock-Based
| | | Accumulated
| | | Comprehensive
| | | Equity
| |
| | | Shares | | | Value | | | Par Value | | | Compensation | | | Deficit | | | Income (Loss) | | | (Deficit) | |
| |
| Balance at December 31, 2004 | | | 22,706,679 | | | $ | 227,067 | | | $ | 120,315,949 | | | $ | (2,607,247 | ) | | $ | (78,106,731 | ) | | $ | (90,659 | ) | | $ | 39,738,379 | |
| Exercise of stock options | | | 171,155 | | | | 1,712 | | | | 98,783 | | | | — | | | | — | | | | — | | | | 100,495 | |
| Issuance of restricted stock | | | 40,570 | | | | 406 | | | | 24,305 | | | | — | | | | — | | | | — | | | | 24,711 | |
| Issuance and remeasurement of stock options for services | | | — | | | | — | | | | (123,149 | ) | | | — | | | | — | | | | — | | | | (123,149 | ) |
| Amortization of deferred stock-based compensation | | | — | | | | — | | | | — | | | | 1,522,554 | | | | — | | | | — | | | | 1,522,554 | |
| Reversal of deferred stock-based compensation and related amortization due to forfeited options | | | — | | | | — | | | | (1,325,261 | ) | | | 461,642 | | | | — | | | | — | | | | (863,619 | ) |
| Proceeds from private placement of common stock and warrants, net of issuance expenses | | | 6,846,735 | | | | 68,467 | | | | 25,775,586 | | | | — | | | | — | | | | — | | | | 25,844,053 | |
| Comprehensive income (loss): | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss | | | — | | | | — | | | | — | | | | — | | | | (32,988,577 | ) | | | — | | | | (32,988,577 | ) |
| Unrealized gain on marketable securities, net | | | — | | | | — | | | | — | | | | — | | | | — | | | | 87,164 | | | | 87,164 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | | | | | (32,901,413 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance at December 31, 2005 | | | 29,765,139 | | | | 297,652 | | | | 144,766,213 | | | | (623,051 | ) | | | (111,095,308 | ) | | | (3,495 | ) | | | 33,342,011 | |
| Exercise of stock options | | | 558,377 | | | | 5,584 | | | | 347,153 | | | | — | | | | — | | | | — | | | | 352,737 | |
| Issuance of restricted stock | | | 38,931 | | | | 389 | | | | 23,748 | | | | — | | | | — | | | | — | | | | 24,137 | |
| Issuance and remeasurement of stock options for services | | | — | | | | — | | | | 323,273 | | | | — | | | | — | | | | — | | | | 323,273 | |
| Stock-based employee compensation expense | | | — | | | | — | | | | 3,080,790 | | | | — | | | | — | | | | — | | | | 3,080,790 | |
| Elimination of deferred stock-based compensation due to adoption of SFAS 123R | | | — | | | | — | | | | (623,051 | ) | | | 623,051 | | | | — | | | | — | | | | — | |
| Proceeds from private placement of common stock, net of issuance expenses | | | 6,000,000 | | | | 60,000 | | | | 16,675,804 | | | | — | | | | — | | | | — | | | | 16,735,804 | |
| Comprehensive income (loss): | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss | | | — | | | | — | | | | — | | | | — | | | | (41,990,154 | ) | | | — | | | | (41,990,154 | ) |
| Unrealized gain on marketable securities, net | | | — | | | | — | | | | — | | | | — | | | | — | | | | 3,422 | | | | 3,422 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | | | | | (41,986,732 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance at December 31, 2006 | | | 36,362,447 | | | | 363,625 | | | | 164,593,930 | | | | — | | | | (153,085,462 | ) | | | (73 | ) | | | 11,872,020 | |
| Exercise of stock options | | | 201,249 | | | | 2,013 | | | | 164,777 | | | | — | | | | — | | | | — | | | | 166,790 | |
| Vesting of unvested restricted stock | | | 30,052 | | | | 300 | | | | 18,332 | | | | — | | | | — | | | | — | | | | 18,632 | |
| Issuance and remeasurement of stock options for services | | | — | | | | — | | | | 16,594 | | | | — | | | | — | | | | — | | | | 16,594 | |
| Stock-based employee compensation expense | | | — | | | | — | | | | 1,914,845 | | | | — | | | | — | | | | — | | | | 1,914,845 | |
| Proceeds from private placement of common stock and warrants, net of issuance expenses | | | 10,155,000 | | | | 101,550 | | | | 22,310,710 | | | | — | | | | — | | | | — | | | | 22,412,260 | |
| Comprehensive income (loss): | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss | | | — | | | | — | | | | — | | | | — | | | | (42,249,366 | ) | | | — | | | | (42,249,366 | ) |
| Unrealized gain on marketable securities, net | | | — | | | | — | | | | — | | | | — | | | | — | | | | 73 | | | | 73 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | | | | | (42,249,293 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance at December 31, 2007 | | | 46,748,748 | | | $ | 467,488 | | | $ | 189,019,188 | | | $ | 0 | | | $ | (195,334,828 | ) | | $ | 0 | | | $ | (5,848,152 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-5
MIDDLEBROOK PHARMACEUTICALS, INC.
| | | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| |
| Cash flows from operating activities: | | | | | | | | | | | | |
| Net loss | | $ | (42,249,366 | ) | | $ | (41,990,154 | ) | | $ | (32,988,577 | ) |
| Adjustments to reconcile net income to net cash used in operating activities: | | | | | | | | | | | | |
| Loss attributed to non-controlling interest | | | (162,189 | ) | | | — | | | | — | |
| Depreciation and amortization | | | 4,460,069 | | | | 3,919,267 | | | | 4,044,419 | |
| Warrant liability | | | 2,100,000 | | | | — | | | | — | |
| Stock-based compensation | | | 1,931,439 | | | | 3,404,063 | | | | 535,786 | |
| Deferred rent and credit on lease concession | | | (75,060 | ) | | | (15,957 | ) | | | 47,629 | |
| Amortization of premium on marketable securities | | | (39,687 | ) | | | 204,525 | | | | 253,483 | |
| (Gain) or loss on disposal of fixed assets | | | — | | | | 23,185 | | | | (16,292 | ) |
| Advance payment for sale of Keflex | | | — | | | | (1,000,000 | ) | | | — | |
| Changes in: | | | | | | | | | | | | |
| Accounts receivable | | | (384,273 | ) | | | 574,751 | | | | (406,648 | ) |
| Inventories | | | 1,389,457 | | | | (1,857,939 | ) | | | (39,713 | ) |
| Prepaid expenses and other current assets | | | 539,780 | | | | (885,432 | ) | | | 247,136 | |
| Deposits other than on property and equipment, and other assets | | | 304,151 | | | | (30,096 | ) | | | (62,394 | ) |
| Accounts payable | | | (625,984 | ) | | | 599,249 | | | | (2,200,076 | ) |
| Accrued expenses and advances | | | (2,260,048 | ) | | | 534,236 | | | | 3,483,910 | |
| Deferred product and contract revenue | | | (189,000 | ) | | | 189,000 | | | | 2,211,532 | |
| | | | | | | | | | | | | |
| Net cash used in operating activities | | | (35,260,711 | ) | | | (36,331,302 | ) | | | (24,889,805 | ) |
| | | | | | | | | | | | | |
| Cash flows from investing activities: | | | | | | | | | | | | |
| Advance payment for potential sale of Keflex intangible assets | | | — | | | | — | | | | 1,000,000 | |
| Purchase of marketable securities | | | (5,867,519 | ) | | | (13,764,736 | ) | | | (15,029,229 | ) |
| Sale and maturities of marketable securities | | | 6,430,000 | | | | 24,355,000 | | | | 23,205,000 | |
| Purchases of property and equipment | | | (1,396,954 | ) | | | (550,929 | ) | | | (1,922,881 | ) |
| Proceeds from sale of fixed assets | | | — | | | | 25,000 | | | | 111,163 | |
| Change in restricted cash | | | — | | | | 728,744 | | | | 312,390 | |
| | | | | | | | | | | | | |
| Net cash provided by (used in) investing activities | | | (834,473 | ) | | | 10,793,079 | | | | 7,676,443 | |
| | | | | | | | | | | | | |
| Cash flows from financing activities: | | | | | | | | | | | | |
| Proceeds from private placement of common stock and warrants, net of issuance costs | | | 22,412,260 | | | | 16,735,804 | | | | 25,844,053 | |
| Proceeds from purchase of noncontrolling interest | | | 7,500,000 | | | | — | | | | — | |
| Proceeds from issuance of debt, net of issue costs | | | — | | | | 7,792,976 | | | | — | |
| Payments on lines of credit | | | (6,888,889 | ) | | | (2,603,524 | ) | | | (1,009,975 | ) |
| Proceeds from exercise of common stock options | | | 166,790 | | | | 352,737 | | | | 100,495 | |
| | | | | | | | | | | | | |
| Net cash provided by financing activities | | | 23,190,161 | | | | 22,277,993 | | | | 24,934,573 | |
| | | | | | | | | | | | | |
| Net increase (decrease) in cash and cash equivalents | | | (12,905,023 | ) | | | (3,260,230 | ) | | | 7,721,211 | |
| Cash and cash equivalents, beginning of period | | | 14,856,738 | | | | 18,116,968 | | | | 10,395,757 | |
| | | | | | | | | | | | | |
| Cash and cash equivalents, end of period | | $ | 1,951,715 | | | $ | 14,856,738 | | | $ | 18,116,968 | |
| | | | | | | | | | | | | |
| Supplemental disclosure of cash flow information: | | | | | | | | | | | | |
| Cash paid for interest | | $ | 566,726 | | | $ | 386,454 | | | $ | 120,891 | |
| | | | | | | | | | | | | |
| Supplemental disclosure of non-cash transactions: | | | | | | | | | | | | |
| Reclassification of liability related to early exercises of restricted stock to equity upon vesting of the restricted stock | | $ | 18,632 | | | $ | 24,137 | | | $ | 24,711 | |
| | | | | | | | | | | | | |
| Warrants issued | | $ | 2,580,000 | | | $ | — | | | $ | — | |
| | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-6
MIDDLEBROOK PHARMACEUTICALS, INC.
| |
| 1. | Nature of the Business |
MiddleBrook Pharmaceuticals, Inc. (MiddleBrook or the Company) was incorporated in Delaware in December 1999 and commenced operations on January 1, 2000. The Company is focused on developing and commercializing anti-infective drug products that fulfill unmet medical needs in the treatment of infectious disease. The Company is developing a portfolio of drugs based on the novel biological finding that bacteria exposed to antibiotics in front-loaded, sequential bursts, or pulses, are killed more efficiently than those exposed to standard antibiotic treatment regimens. Based on this finding, the Company has developed a proprietary,once-a-day pulsatile delivery technology called PULSYS® (PULSYS). The Company has initially focused on developing pulsatile formulations of approved and marketed drugs that no longer have patent protection or that have patents expiring in the next several years. The Company’s lead PULSYS product candidate, based on the antibiotic amoxicillin, successfully completed a Phase III trial in 2006, and its New Drug Application (NDA) for the product, under the trade name: MOXATAGtm (amoxicillin extended-release) Tablets, was approved by the U.S. Food and Drug Administration on January 23, 2008. The Company currently markets certain drug products which do not utilize its PULSYS technology and that are not protected by any other patents. In 2004, the Company acquired the U.S. rights to Keflex (cephalexin capsules, USP) from Eli Lilly and commenced product sales. In November 2007, the Company entered into a transaction with Deerfield Management in which the Company sold its Keflex brand rights, including the trademark and approved New Drug Applications for the Company’s existing, non-PULSYS Keflex products, to Deerfield. Under the transaction agreements, the Company has the right to repurchase the Keflex assets at a future date, as well as to continue to utilize the Keflex trademark and other intangible assets in order to continue to operate its Keflex business, subject to certain consignment and royalty payments to Deerfield. The Company retained its rights to Keflex PULSYS intellectual property and technical data.
The Company has experienced significant operating losses since its inception in 2000, including a net loss of $42.2 million in 2007. As of December 31, 2007, the Company had an accumulated deficit of $195.3 million. The process of developing and commercializing the Company’s products requires significant research and development work, preclinical testing and clinical trials, as well as regulatory approvals, significant marketing and sales efforts, and manufacturing capabilities. These activities, together with the Company’s general and administrative expenses, require significant investments and are expected to continue to result in significant operating losses for the foreseeable future. In January 2008, the Company received approval for marketing from the FDA for its lead product, MOXATAG (amoxicillin extended-release) Tablets, and it expects to incur significant expenses in preparing for the commercial launch of the product. To date, revenues recognized from non-PULSYS products have been limited and have not been sufficient for the Company to achieve or sustain profitability. The Company expects to incur a loss from operations in 2008. The Company believes its existing cash resources, including the net proceeds of $19.9 million from a private placement transaction completed in January 2008, will be sufficient to fund its operations at least into the first quarter of 2009 at its planned levels of research, development, sales and marketing activities, barring unforeseen developments. However, the Company does not currently have the cash resources to fully fund the commercial launch of MOXATAG in late 2008 or in 2009 or to fund clinical trials of additional PULSYS product candidates. The Company is currently exploring various strategic alternatives, including licensing or development arrangements, the sale of some or all of the Company’s assets, partnering or other collaboration agreements, or a merger or other strategic transaction. Execution of the Company’s future strategies is dependent on the outcome of the strategic alternatives process. Should a strategic transaction not be completed, the Company may, if possible, enter into arrangements to raise additional capital which may dilute the ownership of its equity investors. The Company believes that additional financing may be available to it, but there can be no guarantee financing will be available on acceptable terms or at all. If adequate funds are not available, the Company may be required to delay, reduce the scope of or eliminate its research and development programs, reduce its commercialization efforts, or effect changes to its facilities or personnel. The Company’s future operations are dependent on the success of the Company in commercializing new products, and there is no assurance that profitable operations can be achieved or sustained on a continuing basis.
F-7
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| |
| 2. | Summary of Significant Accounting Policies |
Basis of Presentation
The consolidated financial statements include the accounts of MiddleBrook Pharmaceuticals, Inc., together with the accounts of Kef Pharmaceuticals, Inc. (Kef) and Lex Pharmaceuticals, Inc. (Lex), two variable interest entities for which MiddleBrook is the primary beneficiary as defined by FASB Interpretation No. 46 (revised 2003),“Consolidation of Variable Interest Entities”(FIN 46R). Kef and Lex are legal entities that were formed in November 2007 by Deerfield Management, a stockholder and affiliate of MiddleBrook, which purchased certain non-PULSYS Keflex assets from the Company. See Note 13,“Noncontrolling Interest — Deerfield Transaction”for a discussion of the transaction. As Deerfield and MiddleBrook are related parties, no gain or loss was recognized by the Company on the transaction and the initial measurement of assets and liabilities transferred to the variable interest entities remained at the amounts at which they were carried in the accounts of MiddleBrook, in accordance with FIN 46R, paragraph 20. Expenses recognized in the consolidated statement of operations for cost of product sales and amortization of intangible assets have been calculated on a basis consistent with the calculations that would have been made had the related inventory and intangible assets remained with MiddleBrook. All significant intercompany accounts and transactions between MiddleBrook and the two variable interest entities, Kef and Lex, have been eliminated.
Use of Estimates
The preparation of financial statements in conformity with generally accepted accounting principles in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Revenue Recognition
Product sales revenue, net of estimated provisions, is recognized when persuasive evidence that an arrangement exists, delivery has occurred, the selling price is fixed or determinable, and collectibility is reasonably assured. Provisions for sales discounts, and estimates for chargebacks, rebates, and product returns are established as a reduction of product sales revenue at the time revenues are recognized, based on historical experience adjusted to reflect known changes in the factors that impact these reserves. These factors include current contract prices and terms, estimated wholesaler inventory levels, remaining shelf life of product, and historical information for similar products in the same distribution channel.
Deferred product revenuerepresents goods shipped under guaranteed sales arrangements in connection with initial stocking for a new product launch or other product sale arrangements containing terms that may differ significantly from the Company’s customary terms and conditions. For such arrangements, the risk of loss has not passed to the customer and, accordingly, products delivered under guaranteed sales arrangements or certain incentive terms are accounted for as consignment sales. The Company recognizes revenue when the product is sold by its customer or at the expiration of the consignment period if the product has not been returned.
Contract revenuesinclude license fees and milestone payments associated with collaborations with third parties. Revenue from non-refundable, upfront license fees where the Company has continuing involvement is recognized ratably over the development or agreement period.
Deferred contract revenuerepresents cash received in excess of revenue recognized. See“Note 3 — Revenue”for discussion of deferred contract revenue related to the terminated collaboration with Par Pharmaceutical.
F-8
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Research and Development
The Company expenses research and development costs as incurred. Research and development costs primarily consist of salaries and related expenses for personnel, fees paid to consultants and outside service providers, including clinical research organizations for the conduct of clinical trials, costs of materials used in clinical trials and research and development, development costs for contract manufacturing prior to FDA approval of products, depreciation of capital resources used to develop products, and costs of facilities, including costs to modify third-party facilities.
Cash and Cash Equivalents
Cash equivalents are highly liquid investments with a maturity of three months or less at date of purchase and consist of time deposits, investments in money market funds with commercial banks and financial institutions, commercial paper and high-quality corporate bonds. At December 31, 2007 and 2006, the Company maintained all of its cash and cash equivalents in three financial institutions. Deposits held with banks may exceed the amount of insurance provided on such deposits. Generally, these deposits may be redeemed upon demand, and the Company believes there is minimal risk of losses on such cash balances. At December 31, 2007, the Company did not have any investments in auction-rate securities.
Restricted Cash
In conjunction with the lease of its corporate, research and development facilities, the Company provided the landlord with letters of credit that were collateralized with restricted cash deposits in the amounts of $872,180 at December 31, 2007, and 2006 (see Note 20). These deposits are recorded as non-current restricted cash at December 31, 2007 and 2006.
Marketable Securities
The Company classifies all of its marketable securities as available-for-sale. Available-for-sale securities are carried at fair value, with the unrealized gains and losses reported as a component of stockholders’ equity in accumulated other comprehensive loss. Marketable securities available for current operations are classified in the balance sheet as current assets; marketable securities held for long-term purposes are classified as noncurrent assets. Interest income, net of amortization of premium on marketable securities, and realized gains and losses on securities are included in “Interest income” in the statements of operations.
Fair Value of Financial Instruments
The carrying amounts of the Company’s financial instruments, which include cash and cash equivalents, marketable securities, notes payable and line of credit borrowings, approximate their fair values due to their short maturities. Warrants classified as liabilities are recorded at their fair value, based on the Black-Scholes option-pricing model.
Accounts Receivable
Accounts receivable represent amounts due from wholesalers for sales of pharmaceutical products. Allowances for estimated product discounts and chargebacks are recorded as reductions to gross accounts receivable. Amounts due for returns and estimated rebates payable to third parties are included in accrued liabilities.
Inventories
Inventories consist of finished products purchased from third-party contract manufacturers and are stated at the lower of cost or market. Cost is determined on thefirst-in, first-out (FIFO) method. Reserves for obsolete or slow-moving inventory are recorded as reductions to inventory cost. The Company periodically reviews its product
F-9
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
inventories on hand. Inventory levels are evaluated by management relative to product demand, remaining shelf life, future marketing plans and other factors, and reserves for obsolete and slow-moving inventories are recorded for amounts which may not be realizable. As discussed above under“Basis of Presentation,”inventories were sold on November 7, 2007 to affiliates of Deerfield Management; however, the Deerfield affiliates are consolidated with MiddleBrook in accordance with FIN 46R, and there was no change in the accounting policies or basis for inventories.
Property and Equipment
Property and equipment are stated at cost and depreciated over their estimated useful lives using the straight-line method. Leasehold improvements are capitalized and amortized over the shorter of their economic life or the lease term. Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are removed from the accounts and any resulting gain or loss is credited or charged to operations. Repairs and maintenance costs are expensed as incurred.
Intangible Assets
Identifiable intangible assets with definite lives are amortized on a straight-line basis over their estimated useful lives. The Keflex brand rights are amortized over 10 years, the Keflex non-compete agreement with Eli Lilly and Company is amortized over five years, and certain acquired patents are amortized over 10 years. The Company does not have identifiable intangible assets with indefinite lives. The Keflex brand name and other intangible assets were acquired for marketing purposes, and the related amortization is charged to selling expense. In November 2007, the Company sold its Keflex brand rights to affiliates of Deerfield Management, as discussed further in Note 13. The Company retained the right to repurchase at a predetermined price the intangible assets sold at a future date, as well as to continue to utilize the Keflex trademark and other intangible assets in order to continue to operate its Keflex business. As discussed above under“Basis of Presentation,”the Deerfield affiliates are consolidated with MiddleBrook in accordance with FIN 46R, and there was no change in the accounting policies or basis for intangible assets.
Patents are carried at cost less accumulated amortization which is calculated on a straight-line basis over the estimated useful lives of the patents. The Company periodically reviews the carrying value of patents to determine whether the carrying amount of the patent is recoverable. For the years ended December 31, 2007, 2006 and 2005, there were no adjustments to the carrying values of patents. The Company is amortizing the cost of the patent applications over a period of 10 years.
Impairment of Long-Lived Assets
SFAS No. 144,“Accounting for the Impairment or Disposal of Long-Lived Assets,”establishes accounting standards for the impairment of long-lived assets. The Company reviews its long-lived assets, including property and equipment and intangible assets owned by variable interest entities included in the consolidated balance sheet, for impairment whenever events or circumstances indicate that the carrying amount of an asset may not be recoverable. If this review indicates that the asset will not be recoverable based on the expected undiscounted net cash flows of the related asset, an impairment loss is recognized. There were no indications of impairment through December 31, 2007, and consequently there were no impairment losses recognized in 2007, or prior years. If the Company is not able to carry out its business plans, there is the potential that this will be an indicator of an event or change in circumstances under SFAS 144 that would require the Company to perform an impairment analysis, and ultimately may result in impairment of the long-lived assets.
Leases
The Company leases its office and laboratory facilities under operating leases. Lease agreements may contain provisions for rent holidays, rent escalation clauses or scheduled rent increases, and landlord lease concessions such
F-10
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
as tenant improvement allowances. The effects of rent holidays and scheduled rent increases in an operating lease are recognized over the term of the lease, including the rent holiday period, so that rent expense is recognized on a straight-line basis. For lease concessions such as tenant improvement allowances, the Company records a deferred rent liability included in “Deferred rent and credit on lease concession” on the balance sheet and amortizes the deferred liability on a straight-line basis as a reduction to rent expense over the term of the lease. The tenant improvements are capitalized as leasehold improvements and are amortized over the shorter of the economic life of the improvement or the lease term (excluding optional renewal periods). Amortization of leasehold improvements is included in depreciation expense. The Company’s leases do not include contingent rent provisions. For leased facilities where the company has ceased use for a portion or all of the space, the Company accrues a loss if the cost of the leased space is in excess of market rates for potential sublease income. In the year ended December 31, 2007 the Company accrued a loss of $590,000 for leased facility space for which it had ceased use.
Income Taxes
The Company accounts for income taxes under the liability method. Under this method, deferred income taxes are recognized for tax consequences in future years of differences between the tax bases of assets and liabilities and their financial reporting amounts at each year-end, based on enacted laws and statutory tax rates applicable to the periods in which the differences are expected to affect taxable income. Valuation allowances are provided if, based upon the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
Warrant Liabilities
Warrants may be classified as assets or liabilities (derivative accounting), temporary equity, or permanent equity, depending on the terms of the specific warrant agreement. Warrants are evaluated under SFAS 150,“Accounting for Certain Financial Instruments with Characteristics of both Liabilities and Equity.”If the instrument is not governed by SFAS 150, then it is reviewed to determine whether it meets the definition of a derivative under SFAS 133,“Accounting for Derivative Instruments and Hedging Activities” or whether the warrant would meet the definition of equity under the provisions ofEITF 00-19,“Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company’s Own Stock.”Financial instruments such as warrants that are classified as permanent or temporary equity are excluded from the definition of a derivative for purposes of SFAS 133. Financial instruments, including warrants, that are classified as assets or liabilities are considered derivatives under SFAS 133, and are marked to market at each reporting date, with the change in fair value recorded in the income statement. The Company has determined that the warrants it issued in November 2007 should be accounted for as liabilities and marked to market at each reporting date.
Registration Payment Arrangements
The Company views a registration rights agreement containing a liquidated damages provision as a separate freestanding contract which has nominal value, and the Company has followed that accounting approach, consistent with FASB Staff PositionNo. EITF 00-19-2,“Accounting for Registration Payment Arrangements.”Under this approach, the registration rights agreement is accounted for separately from the financial instrument. Under FSPNo. EITF 00-19-2, registration payment arrangements are measured in accordance with SFAS No. 5,“Accounting for Contingencies.”Should the Company conclude that it is more likely than not that a liability for liquidated damages will occur, the Company would record the estimated cash value of the liquidated damages liability at that time.
Comprehensive Income
SFAS No. 130,“Reporting Comprehensive Income,”requires a full set of general-purpose financial statements to include the reporting of “comprehensive income.” Comprehensive income is composed of two components, net
F-11
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
income and other comprehensive income. For the years ended December 31, 2007, 2006 and 2005, other comprehensive income of $73, $3,422 and $87,164 respectively, consists of unrealized gains and losses on available-for-sale marketable securities.
Earnings Per Share
Basic earnings per share is computed based on the weighted average number of common shares outstanding during the period. Diluted earnings per share is computed based on the weighted average shares outstanding adjusted for all dilutive potential common shares. The dilutive impact, if any, of potential common shares outstanding during the period, including outstanding stock options, is measured by the treasury stock method. Potential common shares are not included in the computation of diluted earnings per share if they are antidilutive. The Company incurred net losses for 2007, 2006 and 2005 and, accordingly, did not assume exercise of any of the Company’s outstanding stock options, or warrants, because to do so would be antidilutive.
The following are the securities that could potentially dilute basic earnings per share in the future that were not included in the computation of diluted earnings per share because to do so would have been antidilutive for the periods presented:
| | | | | | | | | | | | | |
| | | December 31, | |
(Number of Underlying Common Shares) | | 2007 | | | 2006 | | | 2005 | |
| |
| Stock options | | | 4,774,206 | | | | 4,378,578 | | | | 4,095,417 | |
| Nonvested restricted stock | | | — | | | | 30,052 | | | | 71,032 | |
| Warrants | | | 13,012,607 | | | | 2,396,357 | | | | 2,396,357 | |
| | | | | | | | | | | | | |
| Total | | | 17,786,813 | | | | 6,804,987 | | | | 6,562,806 | |
| | | | | | | | | | | | | |
Segment and Geographic Information
In accordance with SFAS No. 131,“Disclosure about Segments of an Enterprise and Related Information,”the Company has determined that it operates in one business segment. The Company is organized along functional lines of responsibility and does not utilize a product, divisional or regional organizational structure. The Company is managed and operated as one business. The entire business is managed by a single management team that reports to the chief executive officer.
The Company sells its products to a limited number of pharmaceutical wholesalers, and all product sales occur in the United States. Long-lived assets, consisting of property and equipment, are located both in the United States and Ireland.
| | | | | | | | | |
| | | | | | Long-Lived
| |
Geographic Information | | Product Sales | | | Assets | |
| |
| United States | | $ | 10,456,700 | | | $ | 8,415,184 | |
| Ireland | | | — | | | | 2,513,475 | |
| | | | | | | | | |
| Total | | $ | 10,456,700 | | | $ | 10,928,659 | |
| | | | | | | | | |
Recent Accounting Pronouncements
In February 2007, the Financial Accounting Standards Board (FASB) issued SFAS No. 159,“The Fair Value Option for Financial Assets and Financial Liabilities”(SFAS 159), which is effective for financial statements issued for fiscal years beginning after November 15, 2007. SFAS 159 provides companies with an option to report selected financial assets and liabilities at fair value. The Statement also establishes presentation and disclosure requirements designed to facilitate comparisons between companies that choose different measurement attributes for similar types of assets and liabilities. SFAS 159 requires companies to provide additional information that will
F-12
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
help investors and other users of financial statements to more easily understand the effect of the company’s choice to use fair value on its earnings. It also requires entities to display the fair value of those assets and liabilities for which the company has chosen to use fair value on the face of the balance sheet. Adoption of SFAS 159 is not expected to have a material effect on the Company’s results of operations and financial condition.
In June 2007, the Emerging Issues Task Force (EITF) reached a consensus on EITF IssueNo. 07-03,“Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities,”(EITF 07-03).EITF 07-03 concludes that nonrefundable advance payments for future research and development activities should be deferred and capitalized until the goods have been delivered or the related services have been performed. If an entity does not expect the goods to be delivered or services to be rendered, the capitalized advance payment should be charged to expense. This consensus is effective for fiscal years beginning after December 15, 2007. Adoption ofEITF 07-03 is not expected to have a material effect on the Company’s results of operations and financial condition.
In December 2007, the EITF reached a consensus on EITF IssueNo. 07-01,“Accounting for Collaborative Arrangements,”(EITF 07-01).EITF 07-01 requires collaborators to present the result of activities for which they act as the principal on a gross basis and report any payments received from (made to) other collaborators based on other applicable GAAP or, in the absence of other applicable GAAP, based on analogy to authoritative accounting literature or a reasonable, rational, and consistently applied accounting policy election. In addition, a participant in a collaborative arrangement should provide the following disclosures separately for each collaborative arrangement: (a) the nature and purpose of the arrangement, (b) its rights and obligations under the collaborative arrangement, (c) the accounting policy for the arrangement in accordance with APB Opinion 22, “Disclosure of Accounting Policies,” and (d) the income statement classification and amounts arising from the collaborative arrangement between participants for each period an income statement is presented.EITF 07-01 will be effective for annual periods beginning after December 15, 2008. Adoption ofEITF 07-01 is not expected to have a material effect on the Company’s consolidated financial statements.
In December 2007, the FASB issued SFAS No. 141 (revised 2007),“Business Combinations”(SFAS 141 R), which is effective for financial statements issued for fiscal years beginning on or after December 15, 2008. SFAS 141R establishes principles and requirements for how an acquirer recognizes and measures in its financial statements the identifiable assets acquired, the liabilities assumed, any noncontrolling interest in the acquiree, and the goodwill acquired in the business combination. SFAS 141R also establishes disclosure requirements to enable the evaluation of the nature and financial effects of the business combination. SFAS 141R will be applied prospectively. The Company is currently evaluating the effect that the adoption of SFAS 141R will have on its results of operations and financial condition.
In December 2007, the FASB issued SFAS No. 160,“Noncontrolling Interests in Consolidated Financial Statements, an amendment of ARB No. 51,”(SFAS 160). SFAS 160 changes the accounting and reporting for minority interests, which will be recharacterized as noncontrolling interests (NCI) and classified as a component of equity. The Statement also requires that entities provide sufficient disclosures that clearly identify and distinguish between the interests of the parent and the interests of the noncontrolling owners. SFAS 160 is effective for fiscal years beginning on or after December 15, 2008. Earlier adoption is prohibited. SFAS 160 will be applied prospectively as of the beginning of the fiscal year in which the Statement is initially applied, except for the presentation and disclosure requirements, which shall be applied retrospectively for all periods presented. The Company is currently evaluating the effect that the adoption of SFAS 160 will have on its results of operations and financial condition.
In February 2008, the FASB issued a FASB Staff Position, or FSP, to defer the effective date of SFAS No. 157, “Fair Value Measurements,”(SFAS 157), for nonfinancial assets and nonfinancial liabilities, except for items that are recognized or disclosed at fair value in the financial statements on a recurring basis. The FSP defers the effective date of SFAS 157 to fiscal years beginning after November 15, 2008. The delay is intended to provide the Board additional time to consider the effect of certain implementation issues that have arisen from the application of
F-13
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
SFAS 157 to these assets and liabilities. SFAS 157 defines fair value, establishes a framework for measuring fair value in accordance with generally accepted accounting principles, and expands disclosures about fair value measurements. The Company is currently evaluating the effect that the adoption of SFAS 157 will have on its results of operations and financial condition.
The Company records revenue from sales of pharmaceutical products (Keflex brand) and, in 2005, from the recognition of revenue earned under collaboration agreements.
Product Sales. The Company’s largest customers are large wholesalers of pharmaceutical products. Cardinal Health, McKesson, and AmerisourceBergen accounted for approximately 49.3%, 33.3%, and 10.9% of the Company’s net revenues from product sales in the year ended December 31, 2007; 43.6%, 33.5%, and 13.8% of the Company’s net revenues from product sales in the year ended December 31, 2006, and 59.7%, 20.9% and 13.7% of the Company’s net revenues from product sales in the year ended December 31, 2005.
Contract Revenue. Revenue recognized for upfront payments and milestones under collaboration agreements is as follows:
| | | | | | | | | | | | | |
| | | December 31, | |
Contract Revenue | | 2007 | | | 2006 | | | 2005 | |
| |
| Upfront payment — Par — amortization | | $ | — | | | $ | — | | | $ | 796,783 | |
| Upfront payment — Par — acceleration upon termination | | | — | | | | — | | | | 3,230,995 | |
| | | | | | | | | | | | | |
| Total | | $ | — | | | $ | — | | | $ | 4,027,778 | |
| | | | | | | | | | | | | |
The GSK and Par collaborations have been terminated, and the Company currently has no other collaborations in place that would provide future funding or revenue.
Reimbursement of Development Costs. Revenue recognized for reimbursement by third parties of research and development costs is as follows:
| | | | | | | | | | | | | |
| | | December 31, | |
Reimbursement of Development Costs | | 2006 | | | 2006 | | | 2005 | |
| |
| Reimbursement by Par of period costs incurred | | $ | — | | | $ | — | | | $ | 5,635,690 | |
| Acceleration upon termination of Par collaboration | | | — | | | | — | | | | 2,375,000 | |
| | | | | | | | | | | | | |
| Total | | $ | — | | | $ | — | | | $ | 8,010,690 | |
| | | | | | | | | | | | | |
Collaboration with Par Pharmaceutical for Amoxicillin PULSYS. In May 2004, the Company entered into an agreement with Par Pharmaceutical to collaborate in the further development and commercialization of a PULSYS-based amoxicillin product. Under the terms of the agreement, the Company conducted the development program, including the manufacture of clinical supplies and the conduct of clinical trials, and was responsible for obtaining regulatory approval for the product. The Company was to own the product trademark and was to manufacture or arrange for supplies of the product for commercial sales. Par was to be the sole distributor of the product. Both parties were to share commercialization expenses, including pre-marketing costs and promotion costs, on an equal basis. Operating profits from sales of the product were also to be shared on an equal basis. Under the agreement, the Company received an upfront fee of $5,000,000 and a commitment from Par to fund all further development expenses. Development expenses incurred by the Company were to be partially funded by quarterly payments aggregating $28 million over the period of July 2004 through October 2005, of which up to $14 million would have been contingently refundable.
Revenue related to the receipt of the quarterly payments from Par was recognized based on actual costs incurred as the work was performed, limited to the minimum amounts expected to be received under the agreement
F-14
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
and excluding amounts contingent on future events or that were contingently refundable, with the balance of cash received in excess of revenue recognized recorded as deferred revenue. The excess of the development costs incurred by the Company over the quarterly payments made by Par was to be funded subsequent to commercialization, by the distribution to the Company of Par’s share of operating profits until the excess amount had been reimbursed. The Company did not record any amounts as revenue on a current basis that were dependent on achievement of future operating profits.
On August 3, 2005, the Company was notified by Par that Par decided to terminate the companies’ Amoxicillin PULSYS collaboration agreement. Advancis received from Par the $4,750,000 development funding quarterly payment due in July 2005 and expects no further payments under the collaboration. Under certain circumstances, the termination clauses of the agreement may entitle Par to receive a share of net profits up to one-half of their cumulative $23,250,000 funding of the development costs of certain Amoxicillin PULSYS products, should a product covered by the agreement be successfully commercialized. Accordingly, in 2005 the Company retained deferred revenue of $11,625,000 related to the agreement, and accelerated the recognition into current revenue of the remaining balance of $2,375,000 of deferred reimbursement revenue. The Company received approval for marketing of MOXATAG from the FDA on January 23, 2008. If MOXATAG is successfully commercialized and if it generates net profits, as defined, the balance of deferred revenue would be reduced as payments for a share of net profits are made to Par.
Revenue related to the $5,000,000 upfront fee was being amortized into contract revenue on a straight-line basis over the estimated development period. As a result of the termination, the Company recognized the remaining deferred revenue balance of $3,230,995 related to the upfront fee as revenue in 2005.
The Company held no marketable securities at December 31, 2007.
Marketable securities, including accrued interest, at December 31, 2006 were as follows:
| | | | | | | | | | | | | | | | | |
| | | December 31, 2006 | |
| | | | | | Gross
| | | Gross
| | | | |
| | | Amortized
| | | Unrealized
| | | Unrealized
| | | Fair
| |
Available-for-Sale | | Cost | | | Gains | | | Losses | | | Value | |
| |
| Marketable securities: | | | | | | | | | | | | | | | | |
| Corporate debt securities: | | | | | | | | | | | | | | | | |
| -In unrealized loss position under 12 months | | $ | 522,796 | | | $ | — | | | $ | (73 | ) | | $ | 522,723 | |
| | | | | | | | | | | | | | | | | |
At December 31, 2006, there was one security in an unrealized loss position for less than 12 months, and no securities in an unrealized loss position for greater than 12 months. The security matured in 2007 without a realized loss.
Accounts receivable, net, consists of the following:
| | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
| |
| Accounts receivable for product sales, gross | | $ | 1,290,630 | | | $ | 520,444 | |
| Allowances for discounts, chargebacks and rebates | | | (602,843 | ) | | | (216,930 | ) |
| | | | | | | | | |
| Accounts receivable for product sales, net | | $ | 687,787 | | | $ | 303,514 | |
| | | | | | | | | |
F-15
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The Company’s largest customers are large wholesalers of pharmaceutical products. Three of these large wholesalers accounted for approximately 53.3%, 30.0% and 10.6% of the Company’s accounts receivable for product sales as of December 31, 2007, and 48.9%, 24.0% and 18.3% as of December 31, 2006.
Inventories, net, consist of the following:
| | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
| |
| Finished goods | | $ | 1,692,334 | | | $ | 2,371,346 | |
| Reserve for obsolete and slow-moving inventory | | | (1,004,401 | ) | | | (293,956 | ) |
| | | | | | | | | |
| Inventories, net | | $ | 687,933 | | | $ | 2,077,390 | |
| | | | | | | | | |
The Company periodically reviews product inventories on hand. Inventory levels are evaluated by management relative to product demand, remaining shelf life, future marketing plans and other factors, and reserves for obsolete and slow-moving inventories are recorded for amounts which may not be realizable. The Company recorded provisions for excess inventory of $864,401, $140,000 and $154,367 in the years ended December 31, 2007, 2006 and 2005, respectively.
As discussed above in Note 2 under“Basis of Presentation,” inventories were sold on November 7, 2007 to affiliates of Deerfield Management; however, the Deerfield affiliates are consolidated with MiddleBrook in accordance with FIN 46R, and there was no change in the accounting policies or basis for inventories.
| |
| 7. | Prepaids and Other Current Assets |
Prepaid expenses and other current assets consist of the following:
| | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
| |
| Prepaid insurance | | $ | 836,334 | | | $ | 804,790 | |
| New Drug Application filing fee — refundable | | | — | | | | 672,150 | |
| Other prepaid costs | | | 306,571 | | | | 205,745 | |
| | | | | | | | | |
| Prepaid expenses and other current assets | | $ | 1,142,905 | | | $ | 1,682,685 | |
| | | | | | | | | |
At December 31, 2006 prepaid expenses and other current assets included the refundable portion of a filing fee paid to the FDA for a New Drug Application (NDA) the Company submitted for Amoxicillin PULSYS in 2006. The amount was refunded in 2007.
F-16
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| |
| 8. | Property and Equipment |
Property and equipment consist of the following:
| | | | | | | | | | | |
| | | Estimated Useful Life
| | December 31, | |
| | | (Years) | | 2007 | | | 2006 | |
| |
| Construction in progress | | n/a | | $ | 46,752 | | | $ | 554,673 | |
| Computer equipment | | 3 | | | 1,038,543 | | | | 1,024,149 | |
| Furniture and fixtures | | 3-10 | | | 1,405,918 | | | | 1,405,918 | |
| Equipment | | 3-10 | | | 11,401,691 | | | | 9,140,957 | |
| | | Shorter of economic life | | | | | | | | |
| Leasehold improvements | | or lease term | | | 9,292,903 | | | | 8,738,230 | |
| | | | | | | | | | | |
| Subtotal | | | | | 23,185,807 | | | | 20,863,927 | |
| Less — accumulated depreciation | | | | | (12,257,148 | ) | | | (9,099,300 | ) |
| | | | | | | | | | | |
| Property and equipment, net | | | | $ | 10,928,659 | | | $ | 11,764,627 | |
| | | | | | | | | | | |
The Company ceased use of one of its facilities during the third quarter of 2007. As a result, the Company abandoned leasehold improvements it had made to the property. Accordingly, the Company revised its depreciation estimate, which resulted in accelerating the depreciation of the remaining balance of $512,000 during the third quarter, with $461,000 charged to research and development expense and $51,000 charged to selling, general and administrative expense. For the same facility, the Company also accrued a loss for the cost of the leased space in excess of potential sublease income in the amount of $592,000, with $533,000 charged to research and development expense and $59,000 charged to selling, general and administrative expense.
Depreciation expense for the years ended December 31, 2007, 2006 and 2005 was $3,157,848, $2,699,111 and $2,886,743, respectively.
Intangible assets at December 31, 2007 and December 31, 2006 consist of the following:
| | | | | | | | | | | | | |
| | | December 31, 2007 | |
| | | Gross Carrying
| | | Accumulated
| | | Net Carrying
| |
| | | Amount | | | Amortization | | | Amount | |
| |
| Keflex brand rights | | $ | 10,954,272 | | | $ | (3,834,012 | ) | | $ | 7,120,260 | |
| Keflex non-compete agreement | | | 251,245 | | | | (175,854 | ) | | $ | 75,391 | |
| Patents acquired | | | 120,000 | | | | (96,000 | ) | | $ | 24,000 | |
| | | | | | | | | | | | | |
| Intangible assets | | $ | 11,325,517 | | | $ | (4,105,866 | ) | | $ | 7,219,651 | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | December 31, 2006 | |
| | | Gross Carrying
| | | Accumulated
| | | Net Carrying
| |
| | | Amount | | | Amortization | | | Amount | |
| |
| Keflex brand rights | | $ | 10,954,272 | | | $ | (2,738,580 | ) | | $ | 8,215,692 | |
| Keflex non-compete agreement | | | 251,245 | | | | (125,610 | ) | | $ | 125,635 | |
| Patents acquired | | | 120,000 | | | | (84,000 | ) | | $ | 36,000 | |
| | | | | | | | | | | | | |
| Intangible assets | | $ | 11,325,517 | | | $ | (2,948,190 | ) | | $ | 8,377,327 | |
| | | | | | | | | | | | | |
On June 30, 2004, the Company acquired the U.S. rights to the Keflex brand of cephalexin from Eli Lilly and Company. The purchase price was $11.2 million, including transaction costs, which was paid in cash from the
F-17
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Company’s working capital. The identified intangible assets acquired consisted of the Keflex brand and a non-compete agreement with Lilly. The Company did not acquire customer lists or sales personnel from Lilly.
In the event the Company is able to develop and commercialize a PULSYS-based Keflex product, another cephalexin product relying on the acquired NDAs, or other pharmaceutical products using the acquired trademarks, Eli Lilly will be entitled to royalties on these new products. Royalties, at 10 percent of sales value, are payable on a new product by new product basis for five years following the first commercial sale for each new product, up to a maximum aggregate royalty per calendar year. In 2006 the Company launched its Keflex 750mg product, which is covered by the agreement and is subject to the royalty. All royalty obligations with respect to any defined new product cease after the fifteenth anniversary of the first commercial sale of the first defined new product.
Identifiable intangible assets with definite lives are amortized on a straight-line basis over their estimated useful lives. The Keflex brand rights are amortized over 10 years, the non-compete agreement with Lilly is amortized over five years, and certain acquired patents are amortized over 10 years.
Amortization expense for acquired intangible assets with definite lives was $1,157,676 for the years ending December 31, 2007, 2006 and 2005. For the next five years, annual amortization expense for acquired intangible assets is expected to be approximately $1,200,000 for 2008, and approximately $1,100,000 for 2009, 2010, 2011, and 2012.
In November 2007, the Company sold its Keflex brand rights to affiliates of Deerfield Management, as discussed further in Note 13. The Company retained the right to repurchase at a predetermined price the intangible assets sold at a future date, as well as to continue to utilize the Keflex trademark and other intangible assets in order to continue to operate its Keflex business. As discussed in Note 2 under“Basis of Presentation,”the Deerfield affiliates are consolidated with MiddleBrook in accordance with FIN 46R, and there was no change in the accounting policies or basis for intangible assets.
The Company’s obligations on borrowings are as follows:
| | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
| |
| Merrill Lynch Capital term loan | | $ | — | | | $ | 6,888,889 | |
| Montgomery County note payable | | | — | | | | 75,000 | |
| | | | | | | | | |
| Total | | $ | — | | | $ | 6,963,889 | |
| | | | | | | | | |
Merrill Lynch Capital Term Loan
On June 30, 2006, the Company entered into a $12 million senior secured credit facility with Merrill Lynch Capital, consisting of an $8 million term loan (“Term Loan”) and a $4 million revolving loan facility (“Revolving Loan”). The entire $8 million Term Loan was borrowed at closing. The Company never utilized the Revolving Loan. On November 8, 2007, the Company repaid the outstanding Merrill Lynch Capital loan balance in full, using a portion of the proceeds from the transaction with Deerfield Management, as discussed further in Note 13,“Noncontrolling Interest — Deerfield Transaction.”
The Term Loan was to mature on June 30, 2009 and was payable in 36 equal monthly payments of principal. Interest on the outstanding balance of the Term Loan was payable monthly at an annual rate equal to one-month LIBOR plus 5.0 percent, which at December 31, 2006 equaled 10.35 percent. The Company borrowed the entire Term Loan commitment of $8 million at closing on June 30, 2006, and received net proceeds of $7,792,976. From the net loan proceeds, $987,008 was used to fully repay existing bank loans. The Company incurred $207,024 in debt issuance costs, which were included as a component of Deposits and Other Assets and were amortized using
F-18
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
the effective interest method as additional interest expense over the expected 36 month loan term. If the Term Loan was prepaid, the Company would be required to pay a prepayment fee of 2.0 percent, 1.25 percent, or 0.75 percent, if the prepayment is made within the first, second, or third years after closing (June 30, 2006), respectively. On November 8, 2007, the Company prepaid the Term Loan and incurred prepayment fees of $158,770, as well as expensed the remaining balance of the debt issuance costs of $65,278, which were charged to Other Expenses as early extinguishment of debt.
Montgomery County Note Payable
In December 2001, the Company entered into an Economic Development Fund Agreement with Montgomery County, Maryland. The primary purpose of the Economic Development Fund is to assist private employers who are located, planning to locate or substantially expand operations in Montgomery County. In September 2002, the Company received a $75,000 loan from the County. According to the agreement, the County would permanently forgive part or all of the $75,000 loan principal balance together with the accrued interest if certain conditions relating to employment levels and capital investment are met. During the first quarter of 2007, the Company received notice from Montgomery County that it had met the conditions required for the debt to be forgiven, and accordingly the full amount of $75,000 was recognized as Other Income.
| |
| 11. | Accrued Expenses and Advances |
Accrued expenses and advances consist of the following:
| | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
| |
| Product returns | | $ | 1,414,507 | | | $ | 937,044 | |
| Bonus | | | 1,255,357 | | | | 1,081,503 | |
| Research and development expenses | | | 731,273 | | | | 1,695,628 | |
| Accrued loss on leased facility | | | 589,587 | | | | — | |
| Professional fees | | | 475,392 | | | | 734,250 | |
| Insurance and benefits | | | 240,577 | | | | 228,009 | |
| Product royalties | | | 231,211 | | | | 102,414 | |
| Severance — current portion | | | 190,317 | | | | 1,094,375 | |
| Sales and marketing expense | | | 127,890 | | | | 1,598,437 | |
| Liability for exercised unvested stock options | | | — | | | | 18,632 | |
| Other expenses | | | 357,433 | | | | 326,932 | |
| | | | | | | | | |
| Total accrued expenses | | $ | 5,613,544 | | | $ | 7,817,224 | |
| | | | | | | | | |
Accrued Severance
In 2007, the Company reduced its workforce in order to reduce its operating expenses, and recorded a charge of $533,696 for salaries and benefits. In July and September 2005, the Company reduced its workforce a total of approximately 38% as part of a cost-saving initiative. It recorded a charge of $3,973,265 for severance costs related to salaries and benefits, a non-cash benefit of $512,488 for the reversal of cumulative amortization of deferred stock-based compensation related to forfeited stock options, and a charge of $140,366 for the remaining cost of the New Jersey office lease.
F-19
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Severance and related expenses incurred in connection with the workforce reductions in 2007 and 2005 were recorded as follows (no severance expense was recorded in 2006):
| | | | | | | | | | | | | |
| | | Expense for
| |
| | | Year Ended December 31, 2007 | |
| | | Research &
| | | Selling, General &
| | | | |
| | | Development
| | | Administrative
| | | | |
| | | Expense | | | Expense | | | Total | |
| |
| Severance — salaries and benefits | | $ | — | | | $ | 533,696 | | | $ | 533,696 | |
| Stock-based compensation — forfeitures | | | — | | | | (19,229 | ) | | | (19,229 | ) |
| | | | | | | | | | | | | |
| Total | | $ | — | | | $ | 514,467 | | | $ | 514,467 | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | Expense for
| |
| | | Year Ended December 31, 2005 | |
| | | Research &
| | | Selling, General &
| | | | |
| | | Development
| | | Administrative
| | | | |
| | | Expense | | | Expense | | | Total | |
| |
| Severance — salaries and benefits | | $ | 2,847,220 | | | $ | 1,126,045 | | | $ | 3,973,265 | |
| Stock-based compensation — forfeitures | | | (182,581 | ) | | | (329,907 | ) | | | (512,488 | ) |
| Accrued rent for closed office location | | | 105,275 | | | | 35,091 | | | | 140,366 | |
| | | | | | | | | | | | | |
| Total | | $ | 2,769,914 | | | $ | 831,229 | | | $ | 3,601,143 | |
| | | | | | | | | | | | | |
The following table summarizes the activity in 2007 and 2006 for the liability for the cash portion of severance costs related to thereductions-in-force:
| | | | | | | | | | | | | | | | | |
| | | December 31, 2007 | |
| | | Beginning
| | | | | | | | | Ending
| |
Accrued Severance — 2007 Activity | | Balance | | | Charge | | | Cash Paid | | | Balance | |
| |
| 2007 Workforce reduction | | $ | — | | | $ | 533,696 | | | $ | (343,379 | ) | | $ | 190,317 | |
| 2005 Workforce reduction | | $ | 1,094,375 | | | | | | | | (1,094,375 | ) | | $ | — | |
| | | | | | | | | | | | | | | | | |
| | | $ | 1,094,375 | | | $ | 533,696 | | | $ | (1,437,754 | ) | | $ | 190,317 | |
| | | | | | | | | | | | | | | | | |
| Current portion | | $ | 1,094,375 | | | | | | | | | | | $ | 190,317 | |
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| | | December 31, 2006 | |
| | | Beginning
| | | | | | | | | Ending
| |
Accrued Severance — 2006 Activity | | Balance | | | Reversal | | | Cash Paid | | | Balance | |
| |
| 2005 Workforce reduction | | $ | 3,105,873 | | | $ | (358,530 | ) | | $ | (1,652,968 | ) | | $ | 1,094,375 | |
| | | | | | | | | | | | | | | | | |
| Current portion | | $ | 1,870,479 | | | | | | | | | | | $ | 1,094,375 | |
| Noncurrent portion | | | 1,235,394 | | | | | | | | | | | | — | |
| | | | | | | | | | | | | | | | | |
| | | $ | 3,105,873 | | | | | | | | | | | $ | 1,094,375 | |
| | | | | | | | | | | | | | | | | |
In 2006, an executive was rehired for whom a severance charge had previously been recorded upon his termination in 2005. Upon his rehiring, the remaining accrued severance applicable to that individual of $358,530 that was no longer payable was reversed, and the benefit was recorded as a reduction in Selling, General and Administrative Expense.
F-20
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Advance Payment for Potential Sale of Keflex Assets
In August 2005, Advancis entered into an agreement in principle with a private company for the potential sale of its Keflex assets, including the rights to the U.S. brand and inventories. As part of the agreement, the potential buyer made a $1,000,000 payment to Advancis, which provided it with exclusive negotiating rights through December 31, 2005. The payment was recorded as an advance, since, under certain conditions, the payment could become refundable or, if the sale were to have been completed, the $1,000,000 payment would have been applied to the purchase price. The two parties did not enter into a definitive agreement for the asset sale, and in January 2006, Advancis decided to retain the Keflex assets. The agreement in principle expired on February 28, 2006. Accordingly, the advance payment of $1,000,000 was recognized as other income in 2006.
In November 2007, the Company issued warrants for the purchase of 3,000,000 shares of its common stock to Deerfield Management in connection with the sale of certain non-PULSYS Keflex tangible and intangible assets (see Note 13). The warrants are exercisable immediately upon issuance for a period of six years. The warrant agreement contains provisions for cash settlement under certain conditions, including a major asset sale or acquisition in certain circumstances, which is available to the warrant holders at their option. As a result, the warrants cannot be classified as permanent equity under the requirements ofEITF 00-19,“Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company’s Own Stock,”and are instead classified as a liability at their contractual fair value in the accompanying consolidated balance sheet. Upon issuance of the warrants in November 2007, the Company recorded the warrant liability at is initial fair value of $2,580,000 based on the Black-Scholes option-pricing model, using the following assumptions: exercise price of $1.38, expected life of 6.0 years, expected volatility of 65.0% (contractual volatility rate fixed for the life of the warrant agreement), risk-free interest rate of 3.90%, and dividend yield of 0%.
Equity derivatives not qualifying for permanent equity accounting are recorded at fair value and are remeasured at each reporting date until the warrants are exercised or expire. Changes in the fair value of the warrants issued in November 2007 are reported in the consolidated statement of operations as non-operating income or expense. At December 31, 2007, the aggregate fair value of these warrants decreased to $2,100,000, using the Black-Scholes option pricing model, from their initial fair value of $2,580,000 at the issuance date of November 7, 2007, resulting in a noncash gain of $480,000 and a corresponding reduction in the recorded value of the warrant liability as of December 31, 2007. The gain was attributable to a decrease in the Company’s stock price and a reduction in the remaining term of the warrants.
| |
| 13. | Noncontrolling Interest — Deerfield Transaction |
On November 7, 2007, the Company entered into a series of agreements with Deerfield Management, a healthcare investment fund and one of the Company’s largest equity shareholders, which provided for a potential capital raise of up to $10 million in cash. The financing consisted of two potential closings, with the first closing occurring upon the signing of the agreements (for $7.5 million in gross proceeds, less $0.5 million in transaction expenses) and the second closing (for an additional $2.5 million in gross proceeds) occurring at our option, contingent upon FDA approval of the Company’s New Drug Application for the Amoxicillin PULSYS adult product. The agreements were designed to provide the Company with financial flexibility.
First Closing
At the transaction’s first closing, the Company sold certain assets, including Keflex product inventories, and assigned certain intellectual property rights, relating only to its existing, non-PULSYS cephalexin business, to two Deerfield affiliates, Kef Pharmaceuticals, Inc. (Kef) and Lex Pharmaceuticals, Inc. (Lex). Under the terms of the agreement, $7.5 million was received by the Company on November 8, 2007 for the first closing, and the Company reimbursed Deerfield $0.5 million for transaction-related expenses. Approximately $4.6 million of those proceeds
F-21
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
was used to fully repay the outstanding Merrill Lynch Capital loan balance, with the remainder available for general corporate purposes. Pursuant to a consignment of those assets and license of those intellectual property rights back to the Company, the Company will continue to operate its existing cephalexin business, subject to consignment and royalty payments to Deerfield of 20% of net sales, which decline to 15% should the Company elect to make an extension payment, of $1.35 million ($1.8 million, if the second closing has occurred as described below) by June 30, 2008 subject to a minimum quarterly payment of $400,000. In addition, the Company granted to Deerfield a six-year warrant to purchase 3.0 million shares of the Company’s common stock at $1.38, the closing market price on November 7, 2007.
Second Closing
The agreements provided for a second closing, at the Company’s option. In the event that the Company received approval (or an acceptable approvable letter) of its Amoxicillin PULSYS New Drug Application from the FDA, then it could require Deerfield to acquire and license certain intellectual property rights relating only to the Company’s cephalexin PULSYS business for a payment of $2.5 million. Pursuant to a required sublicense of those intellectual property rights back to the Company, the Company would continue to operate its cephalexin PULSYS activities. Cephalexin PULSYS is not approved for marketing by the FDA. To date, the Company has not exercised the option for a second closing. This option expires June 30, 2008.
Repurchase Right
Deerfield also granted the Company the right to repurchase all of the assets and rights sold and licensed by the Company to Deerfield by purchasing all of the outstanding capital stock of both Kef and Lex. If the Company exercises this right prior to November 7, 2008, then the purchase price for all of the outstanding capital stock of Kef and Lex is a total of $11.0 million, if the Company has not elected the second closing (which would have required Deerfield to acquire and license certain intellectual property rights relating to the Company’s cephalexin PULSYS business), or $14.0 million if the Company did elect the second closing, in which it would have received $2.5 million in cash and Deerfield would have acquired and licensed certain rights to the Company’s cephalexin PULSYS business (in each case subject to certain adjustments). Those purchase prices will increase by $2.0 million on each subsequent anniversary of that date until the right is exercised or expires.
The Company’s purchase rights expire on June 30, 2008, unless an extension payment of $1.35 million ($1.8 million, if a second closing has occurred) is made to extend the expiration date to December 31, 2008. If an extension payment of $4.5 million ($6.0 million, if a second closing has occurred) is made by December 31, 2008, the expiration date is extended to September 30, 2009. If an extension payment of $2.2 million ($2.9 million if a second closing has occurred) is made by September 30, 2009, then the expiration date for the right to purchase the capital stock of Lex is extended to November 1, 2012. The Company may not exercise its right to purchase the capital stock of Lex without first exercising its right to purchase the capital stock of Kef. The Company’s exercise of this purchase right is mandatory upon the change of control of the Company.
Variable Interest Entities and FIN 46R Consolidation
In connection with the transaction, Deerfield Management established two new legal entities, Kef Pharmaceuticals, Inc. (Kef) and Lex Pharmaceuticals, Inc. (Lex) to hold the Keflex tangible and intangible assets. Affiliates of Deerfield own 100 percent of the voting interests in the two entities. In accordance with FIN 46R, MiddleBrook management evaluated whether Kef and Lex are variable interest entities and, if so, whether there is a primary beneficiary with a controlling financial interest. A key characteristic of a controlling financial interest is the equity holder’s ability to make important decisions with respect to the ongoing activities. Since MiddleBrook is making the important decisions with respect to the ongoing activities involving the assets owned by Kef and Lex, the Kef and Lex entities were determined to be variable interest entities for this characteristic. Another characteristic of a controlling financial interest is whether the equity holders of the entities have the obligation to absorb the expected
F-22
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
losses of the entity or to receive the expected residual returns of the entity. Since MiddleBrook has a fixed price repurchase option, the equity holders in Kef and Lex do not have rights to all of the residual returns of the entities and Kef and Lex were determined to be variable interest entities for this characteristic. Management used a qualitative analysis to determine whether Deerfield or MiddleBrook was the primary beneficiary of the entities. MiddleBrook was determined to be the primary beneficiary, since it is the party exposed to the majority of the risks. Thus, MiddleBrook consolidates the financial condition and results of operations of Kef and Lex in accordance with FIN 46R. Accordingly, the losses of $162,000 attributable to the noncontrolling interest (the losses of Kef and Lex) have been deducted from the net loss in the consolidated statement of operations, and the noncontrolling interest holders’ ownership interest in Kef and Lex in the consolidated balance sheet has been reduced by the losses of Kef and Lex.
| |
| 14. | Preferred Stock — Undesignated |
On October 22, 2003, the Company’s certificate of incorporation was amended to authorize the issue of up to 25,000,000 shares of undesignated preferred stock. The Company’s Board of Directors, without any further action by the Company’s stockholders, is authorized to issue shares of undesignated preferred stock in one of more classes or series. The Board may fix the rights, preferences and privileges of the preferred stock. The preferred stock could have voting or conversion rights that could adversely affect the voting power or other rights of common stockholders. As of December 31, 2007 and 2006, no shares of preferred stock have been issued.
| |
| 15. | Common Stock and Warrants |
Effective with the Company’s initial public offering on October 22, 2003, the Company’s certificate of incorporation was amended to increase the number of authorized shares of common stock to 225,000,000.
Each share of common stock is entitled to one vote. The holders of common stock are also entitled to receive dividends whenever funds are legally available and when declared by the Board of Directors, subject to the prior rights of holders of all classes of stock outstanding.
Private Placements of Common Stock
In April 2007, the Company closed a private placement of 10,155,000 shares of its common stock and warrants to purchase 7,616,250 shares of common stock, at a price of $2.36375 per unit. Each unit consists of one share of the Company’s common stock and a warrant to purchase 0.75 shares of common stock. The warrants have a five-year term and an exercise price of $2.27 per share. The transaction raised approximately $24.0 million in gross proceeds. Net proceeds to the Company after deducting commissions and expenses were approximately $22.4 million. Pursuant to the terms of the registration rights agreement, the Company filed with the SEC a registration statement covering the resale of common stock. The registration rights agreement provides that if the initial registration statement is not effective within 120 days of closing, or if the Company does not subsequently maintain the effectiveness of the initial registration statement or any additional registration statements, then in addition to any other rights the investor may have, the Company will be required to pay the investor liquidated damages, in cash, equal to one percent per month of the aggregate purchase price paid by such investor. Maximum aggregate liquidated damages payable to an investor are 20% of the aggregate amount paid by the investor. The SEC declared the Company’sForm S-3 effective on May 23, 2007, which was within 120 days of closing.
In December 2006, the Company completed a private placement of 6,000,000 shares of its common stock at a price of $3.00 per share, resulting in gross proceeds to the Company of $18.0 million. Net proceeds to the Company after deducting commissions and expenses were approximately $16.7 million. No warrants were issued in the transaction. Pursuant to the terms of the registration rights agreement, the Company filed with the SEC a registration statement onForm S-3 covering the resale of common stock. The registration rights agreement provides that if a registration statement is not effective within 90 days of closing, or if the Company does not subsequently maintain the effectiveness of the registration statement, then in addition to any other rights the investor may have, the Company will
F-23
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
be required to pay the investor liquidated damages, in cash, equal to one percent per month of the aggregate purchase price paid by such investor. The SEC declared the Company’sForm S-3 effective on December 29, 2006, which was within 90 days of closing.
In April 2005, the Company completed a private placement of 6,846,735 shares of its common stock at a price of $3.98 per share and warrants to purchase a total of 2,396,357 shares of common stock at an exercise price of $4.78 per share, resulting in gross proceeds to the Company of $27.25 million. Net proceeds to the Company after deducting commissions and expenses were approximately $25.8 million. The warrants are exercisable for five years. Pursuant to the terms of the registration rights agreement, the Company filed with the SEC a registration statement onForm S-3 covering the resale of common stock. The registration rights agreement provides that if a registration statement is not effective within 60 days of closing, or if the Company does not subsequently maintain the effectiveness of the registration statement, then in addition to any other rights the investor may have, the Company will be required to pay the investor liquidated damages, in cash, equal to one percent per month of the aggregate purchase price paid by such investor. The SEC declared the Company’sForm S-3 effective on June 1, 2005, which was within 60 days of closing.
Subsequent to December 31, 2007, the Company closed a private placement in January 2008 of 8,750,001 shares of its common stock and warrants to purchase 3,500,001 shares of common stock for net proceeds of $19.9 million. See Note 23,“Subsequent Event”.
The Company views the registration rights agreement containing the liquidated damages provision as a separate freestanding contract which has nominal value, and the Company has followed that accounting approach, consistent with FASB Staff PositionNo. EITF 00-19-2,“Accounting for Registration Payment Arrangements.”Under this approach, the registration rights agreement is accounted for separately from the financial instrument. Accordingly, the classification of the warrants has been determined underEITF 00-19,“Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company’s Own Stock,” and the warrants have been accounted for as permanent equity. Under FSPNo. EITF 00-19-2, registration payment arrangements are measured in accordance with SFAS No. 5,“Accounting for Contingencies.”Should the Company conclude that it is more likely than not that a liability for liquidated damages will occur, the Company will record the estimated cash value of the liquidated damages liability at that time.
FSPNo. EITF 00-19-2 requires disclosure of the maximum potential amount of consideration, undiscounted, that an issuer could be required to transfer under the registration payment arrangement. The Company’s registration rights agreements generally require that the Company undertake to file a registration statement within a specified number of days, to have the SEC declare the registration statement effective within a specified number of days, and that it maintain the effectiveness of the registration statement for a period of time, for example, until the date as of which all investors as selling stockholders may sell the registered securities without restriction pursuant to Rule 144 (or any successor rule thereto) or the actual date that the securities have been sold. These agreements also specify liquidated damages for each day the Company fails to comply with these obligations. For each of the four private placements completed by the Company and for the Deerfield transaction, the Company has met the required deadlines for filing and for achieving effectiveness, and the Company has never had a period in which any of its registration statements was not continuously effective. Nevertheless, solely for purposes of the calculation required by FSPEITF 00-19-2 of the maximum possible liquidated damages that the Company might incur, the following assumptions have been made: (a) in addition to the original investment, all warrants are exercised for cash at the latest possible date before expiration; (b) no shares are sold during the entire period; and (c) the registration statements are not effective for the entire term of the agreement, including one year after the last potential warrant exercise (a total of 6 years for the private placements and 7 years for the Deerfield transaction). The April 2007 and January 2008 private placements have caps on maximum liquidated damages, and the caps have been used as the maximum potential liquidated damages for those two private placements. For the private placements that closed in January 2008, April 2007, December 2006, and April 2005, the Company’s maximum potential liquidated damages are approximately $6.3 million, $8.3 million, $10.4 million, and $14.6 million, respectively, as of March 20, 2008. For the Deerfield warrants that were issued in November 2007, the Company’s maximum potential liquidated damages are approximately $12.0 million as of March 20, 2008.
F-24
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Warrants
The Company has granted warrants to purchase shares of common stock in connection with certain of our financing transactions, as well as the transaction with Deerfield. At December 31, 2007, the following warrants to purchase common stock were outstanding and exercisable:
| | | | | | | | | | | |
| | | Exercise
| | | Expiration
| | Number of
| |
Date Issued | | Price | | | Date | | Shares | |
| |
| April 29, 2005 | | $ | 4.78 | | | April 29, 2010 | | | 2,396,357 | |
| April 12, 2007 | | $ | 2.27 | | | April 12, 2012 | | | 7,616,250 | |
| November 7, 2007 | | $ | 1.38 | | | November 7, 2013 | | | 3,000,000 | |
| | | | | | | | | | | |
| | | | | | | | | | 13,012,607 | |
| | | | | | | | | | | |
In January 2008, in connection with a private placement transaction, the Company granted 3,500,001 warrants to purchase shares of common stock at an exercise price of $3.00. These warrants expire July 28, 2013.
Shares Reserved for Future Issuance
At December 31, 2007, common stock reserved for future issuance is as follows:
| | | | | |
| | | Number of Shares | |
| |
| Outstanding common stock options | | | 4,774,206 | |
| Common stock available for future grant under our stock option plans | | | 2,659,604 | |
| Common stock available for future grant under our Employee Stock Purchase Plan | | | 100,000 | |
| Warrants | | | 13,012,607 | |
| | | | | |
| | | | 20,546,417 | |
| | | | | |
The Company currently grants stock options under the Stock Incentive Plan (the “Plan”). The number of shares available for issuance under the Plan is 9,348,182.
Options granted under the Plan may be incentive stock options or non-statutory stock options. Stock purchase rights may also be granted under the Plan. Incentive stock options may only be granted to employees. The compensation committee of the Board of Directors determines the period over which options become exercisable. Options granted to employees and consultants normally vest over a4-year period. Options granted to directors, upon their initial appointment or election, vest monthly over periods of 36 or 48 months. Annual director and advisor grants vest monthly over 12 months. Director and advisor grants are exercisable on the date of grant but are restricted, subject to repurchase until vested. The exercise price of incentive stock options and non-statutory stock options shall be no less than 100% of the fair market value per share of the Company’s common stock on the grant date. The term of all options outstanding is 10 years. As of December 31, 2007 and 2006, there were 2,659,604 and 1,756,481 shares of common stock available for future option grants, respectively.
F-25
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The following table summarizes the activity of the Company’s stock option plan for the years ended December 31, 2007, 2006 and 2005:
| | | | | | | | | | | | | | | | | |
| | | | | | | | | Weighted-Average
| | | | |
| | | | | | | | | Remaining
| | | Aggregate
| |
| | | Number of
| | | Weighted-Average
| | | Contractual
| | | Intrinsic
| |
| | | Options | | | Exercise Price | | | Term (Yrs) | | | Value | |
| |
| Outstanding, December 31, 2004 | | | 3,736,726 | | | $ | 5.43 | | | | | | | | | |
| Granted | | | 1,926,350 | | | | 3.24 | | | | | | | | | |
| Exercised | | | (171,155 | ) | | | 0.59 | | | | | | | | | |
| Cancelled | | | (1,396,504 | ) | | | 4.89 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Outstanding, December 31, 2005 | | | 4,095,417 | | | | 4.79 | | | | | | | | | |
| Granted | | | 1,322,870 | | | | 2.24 | | | | | | | | | |
| Exercised | | | (558,377 | ) | | | 0.63 | | | | | | | | | |
| Cancelled | | | (481,332 | ) | | | 6.86 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Outstanding, December 31, 2006 | | | 4,378,578 | | | $ | 4.30 | | | | | | | | | |
| Granted | | | 1,075,150 | | | | 2.56 | | | | | | | | | |
| Exercised | | | (201,249 | ) | | | 0.83 | | | | | | | | | |
| Cancelled | | | (478,273 | ) | | | 3.12 | | | | | | | | | |
| | | | | | | | | | | | | | | | | |
| Outstanding, December 31, 2007 | | | 4,774,206 | | | $ | 4.18 | | | | 7.3 | | | $ | 205,434 | |
| | | | | | | | | | | | | | | | | |
| Exercisable, December 31, 2007 | | | 3,472,204 | | | $ | 4.74 | | | | 6.9 | | | $ | 204,127 | |
| | | | | | | | | | | | | | | | | |
The total intrinsic value of options exercised during the years ended December 31, 2007, 2006, and 2005 were $306,191, $787,345, and $223,876, respectively. Cash received by the Company in the years ended December 31, 2007, 2006, and 2005 upon the issuance of shares from option exercises were $166,790, $352,737, and $100,495, respectively. The Company’s policy is to issue new shares of common stock to satisfy stock option exercises.
A summary of the Company’s nonvested options as of and for the year ended December 31, 2007 is presented below:
| | | | | | | | | |
| | | Number of
| | | Weighted
| |
| | | Nonvested Stock
| | | Average Grant Date
| |
| | | Options | | | Fair Value | |
| |
| Outstanding, December 31, 2006 | | | 1,919,440 | | | $ | 3.48 | |
| Granted | | | 1,075,150 | | | | 1.79 | |
| Vested | | | (1,169,931 | ) | | | 3.70 | |
| Forefeited | | | (451,983 | ) | | | 2.13 | |
| | | | | | | | | |
| Outstanding, December 31, 2007 | | | 1,372,676 | | | $ | 1.85 | |
| | | | | | | | | |
The fair value of options vested during the years ended December 31, 2007 and 2006 was $4,328,745 and $3,639,235 respectively.
F-26
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The following table summarizes information about stock options outstanding, and exercisable at December 31, 2007:
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | Weighted-
| | | Weighted-
| | | | | | Weighted-
| |
Range of
| | Number
| | | Average Remaining
| | | Average
| | | Number
| | | Average
| |
Exercise Prices | | Outstanding | | | Contractual Life | | | Exercise Price | | | Exercisable | | | Exercise Price | |
| |
| December 31, 2007 | | | | | | | | | | | | | | | | | | | | |
| $0.35 to $0.93 | | | 499,462 | | | | 6.5 | | | $ | 0.79 | | | | 494,623 | | | $ | 0.79 | |
| $1.28 to $1.79 | | | 1,125,055 | | | | 7.5 | | | | 1.52 | | | | 684,692 | | | | 1.49 | |
| $2.03 to $3.59 | | | 1,144,381 | | | | 8.9 | | | | 2.66 | | | | 530,348 | | | | 2.71 | |
| $4.05 to $5.57 | | | 767,365 | | | | 7.3 | | | | 4.36 | | | | 554,822 | | | | 4.36 | |
| $8.40 to $10.00 | | | 1,237,943 | | | | 6.0 | | | | 9.24 | | | | 1,207,719 | | | | 9.26 | |
| | | | | | �� | | | | | | | | | | | | | | | |
| | | | 4,774,206 | | | | 7.3 | | | $ | 4.18 | | | | 3,472,204 | | | $ | 4.74 | |
| | | | | | | | | | | | | | | | | | | | | |
| |
| 17. | Stock-Based Compensation |
The Company has recorded stock-based compensation expense for the grant of stock options to employees and to nonemployees as follows:
| | | | | | | | | | | | | |
| | | December 31, | |
Stock-Based Compensation Expense: | | 2007 | | | 2006 | | | 2005 | |
| |
| Employees: | | | | | | | | | | | | |
| SFAS 123R fair-value method | | $ | 1,914,845 | | | $ | 3,080,790 | | | $ | — | |
| APB 25 intrinsic value method | | | — | | | | — | | | | 658,935 | |
| | | | | | | | | | | | | |
| Subtotal — Employees | | | 1,914,845 | | | | 3,080,790 | | | | 658,935 | |
| Nonemployees: | | | | | | | | | | | | |
| Fair value method | | | 16,594 | | | | 323,273 | | | | (123,149 | ) |
| | | | | | | | | | | | | |
| Total | | $ | 1,931,439 | | | $ | 3,404,063 | | | $ | 535,786 | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | December 31, | |
Included in Income Statement Captions as Follows: | | 2007 | | | 2006 | | | 2005 | |
| |
| Research and development expense | | $ | 718,257 | | | $ | 1,434,664 | | | $ | 159,699 | |
| Selling, general and administrative expense | | | 1,213,182 | | | | 1,969,399 | | | | 376,087 | |
| | | | | | | | | | | | | |
| Total | | $ | 1,931,439 | | | $ | 3,404,063 | | | $ | 535,786 | |
| | | | | | | | | | | | | |
Employees. Prior to January 1, 2006, the Company’s share-based awards were accounted for by the intrinsic value method in accordance with Accounting Principles Board Opinion No. 25,“Accounting for Stock Issued to Employees,”(APB 25) and related interpretations. Effective January 1, 2006, the Company adopted Statement of Financial Accounting Standards No. 123R,“Share-Based Payment”(SFAS 123R). This Statement replaces SFAS No. 123 and supersedes APB 25. SFAS 123R requires that employee share-based compensation be measured using a fair value method and that the resulting compensation cost be recognized in the financial statements. The Company adopted SFAS 123R using the modified prospective transition method, which requires the recognition of compensation expense under the Statement on a prospective basis only. Accordingly, prior period financial statements have not been restated. Under this transition method, stock-based compensation cost for 2006 includes (a) compensation cost for all share-based awards granted prior to, but not yet vested as of, January 1, 2006, based on the grant date fair value estimated in accordance with the original provisions of SFAS 123, and (b) compensation
F-27
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
cost for all share-based awards granted subsequent to January 1, 2006 based on the grant-date fair value estimated in accordance with the fair value provisions of SFAS 123R.
Under APB 25, the Company had recorded deferred stock-based compensation for certain stock options granted in prior years with an exercise price below the estimated fair market value at the date of grant. The deferred stock-based compensation was amortized as a charge to expense over the related vesting periods of the options. In accordance with SFAS 123R, effective January 1, 2006, the Company reversed the remaining balance of deferred stock-based compensation of $623,051 against Capital in Excess of Par Value.
SFAS 123R also requires the Company to estimate forfeitures in calculating the expense related to share-based compensation rather than recognizing forfeitures as a reduction in expense as they occur. Prior to the adoption of SFAS 123R, the Company accounted for forfeitures as they occurred as permitted under previous accounting standards. The cumulative effect adjustment of adopting the change in estimating forfeitures was not considered material to the financial statements upon implementation as of January 1, 2006 or for the year ended December 31, 2006.
The Company records compensation expense over the requisite service period, which is equal to the vesting period. For awards granted prior to January 1, 2006, compensation expense is recognized on a graded-vesting basis over the vesting term. For awards granted on or after January 1, 2006, compensation is recognized on a straight-line basis over the vesting term. All share-based compensation costs are charged to expense and not capitalized.
As of December 31, 2007, the total unrecognized compensation cost related to nonvested stock awards was $1,913,292, and the related weighted-average period over which it is expected to be recognized is approximately 2.4 years.
As a result of the adoption of SFAS 123R, the Company’s net loss for 2006 was approximately $2,552,000 higher than it would have been under the Company’s previous intrinsic value method of accounting for share-based compensation. Basic and diluted net loss per common share for the year ended December 31, 2006 were negatively impacted by the change in accounting method by approximately $0.08 per share. The adoption of SFAS 123R had no effect on the Company’s operating cash flows or financing cash flows, as the Company has not realized the benefits of tax deductions in excess of recognized compensation costs due to its net operating loss position.
F-28
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
The following table illustrates the effect on net loss and net loss per share if the Company had applied the fair value recognition provision of SFAS 123 to options granted under the Company’s stock-based compensation plan for the year ended December 31, 2005. For purposes of this pro forma disclosure, the value of the options is estimated using the Black-Scholes option-pricing model and amortized to expense using a graded vesting schedule with forfeitures recognized as a reduction in expense as they occur.
| | | | | |
| | | Year Ended
| |
| | | December 31, | |
| | | 2005 | |
| |
| Net loss, as reported | | $ | (32,988,577 | ) |
| Add — Stock-based employee compensation | | | | |
| expense determined under the intrinsic | | | | |
| value method | | | 658,935 | |
| Less — Stock-based employee compensation | | | | |
| expense determined under the fair value | | | | |
| based method | | | (3,298,472 | ) |
| | | | | |
| Pro forma net loss applicable to common | | | | |
| stockholders | | $ | (35,628,114 | ) |
| | | | | |
| Net loss per share: | | | | |
| Basic and diluted, as reported | | $ | (1.20 | ) |
| | | | | |
| Basic and diluted, pro forma | | $ | (1.30 | ) |
| | | | | |
The weighted average fair value of options granted to employees during 2007, 2006 and 2005 was $1.79, $1.51 and $2.13 per share, respectively. The fair value of each option grant was estimated on the date of grant using the Black-Scholes options pricing model with the following assumptions for grants in 2007, 2006 and 2005:
| | | | | | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| |
| Expected term (in years) | | | 6.25 | | | | 6.25 | | | | 5 | |
| Risk-free interest rate | | | 4.79 | % | | | 4.57 | % | | | 3.81 | % |
| Volatility | | | 75.0 | % | | | 75.0 | % | | | 79.6 | % |
| Dividend yield | | | 0 | % | | | 0 | % | | | 0 | % |
The Company has elected to determine the expected term of share-based awards granted subsequent to January 1, 2006 using the transition approach provided by Staff Accounting Bulletin No. 107, under which an expected term of 6.25 years may be used for four-year grants with a ten-year contractual term. Due to the limited Company-specific historical and implied volatility data, the Company has based its estimate of expected future volatility upon a combination of its historical volatility together with the average of volatility rates of comparable public companies. The risk-free rate is based on U.S. Treasury yields in effect at the time of grant corresponding with the expected term of the options. Estimates of fair value are not intended to predict actual future events or the value ultimately realized by the employees who receive equity awards.
In May 2007, the Company amended its Form of Incentive Stock Option Agreement and its Form of Non-Qualified Stock Option Agreement to provide that all outstanding unvested stock options will automatically become fully vested upon a Change in Control (as that term is defined in the Amended and Restated MiddleBrook Pharmaceuticals, Inc. Stock Incentive Plan). This modification resulted in no additional compensation charge as the modification to vesting did not change the assumptions underlying the fair value of the options granted.
F-29
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Nonemployees. The Company has recorded stock-based compensation expense for options granted to nonemployees, including consultants, Scientific Advisory Board (SAB) members and contract sales representatives based on the fair value of the equity instruments issued. Stock-based compensation for options granted to non employees is periodically remeasured as the underlying options vest in accordance with Emerging Issues Task Force IssueNo. 96-18, “Accounting for Equity Instruments that are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services.”The Company recognizes an expense for such options throughout the performance period as the services are provided by the nonemployees, based on the fair value of the options at each reporting period. The options are valued using the Black-Scholes option pricing model. For graded-vesting options, a final measurement date occurs as each tranche vests. As of December 31, 2007, the balance of unamortized stock-based compensation for options granted to non-employees was approximately $64,000. This amount will be adjusted based on changes in the fair value of the options at the end of each reporting period. As of December 31, 2007, 151,328 options are outstanding and exercisable for non employees.
Restricted Stock
Certain of the Company’s directors, consultants and employees (and/or immediate family members or related entities to which certain of those individuals have transferred their options or shares of common stock) have entered into the Company’s standard form of stock restriction agreement as a condition to their exercise of options to acquire common stock pursuant to the Plan. These agreements provide, among other things, for a right of first refusal to the Company in connection with the option holder’s sale of the common stock, as well as the right for the Company to purchase the stockholder’s common stock in the event that the stockholder’s relationship with the Company is terminated under certain circumstances. Shares issued under non-statutory stock options exercised prior to vesting are subject to forfeiture in accordance with the vesting schedule of the granted stock options. Prior to 2004, certain of the Company’s employees, board members and consultants exercised unvested stock options, awarded under the Company’s Stock Incentive Plan, to acquire a total of 162,281 shares of restricted common stock. There were no such exercises in 2005, 2006 or 2007. At December 31, 2006, there were 30,052 shares of restricted common stock remain unvested pursuant to awards, and none at December 31, 2007.
For all exercises of stock options into unvested restricted stock after March 2002, the Company recorded a liability for the amount of the proceeds received, which is reclassified to equity upon the vesting of the restricted stock. As of December 31, 2006, $18,632 related to 30,052 shares of restricted stock were recorded as a liability. There were none at December 31, 2007.
The Company has not recorded any tax provision or benefit for the years ended December 31, 2007, 2006 and 2005. The Company has provided a valuation allowance for the full amount of its net deferred tax assets since realization of any future benefit from deductible temporary differences and net operating loss carryforwards cannot be sufficiently assured at December 31, 2007 and 2006.
F-30
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
Deferred tax assets consist of the following:
| | | | | | | | | |
| | | December 31, | |
| | | 2007 | | | 2006 | |
| |
| Net operating loss carryforwards | | $ | 62,627,851 | | | $ | 47,842,353 | |
Start-up costs | | | 324,279 | | | | 999,992 | |
| Deferred revenue | | | 4,503,358 | | | | 4,704,335 | |
| Depreciation and amortization | | | 1,348,447 | | | | 80,837 | |
| Stock-based compensation | | | 642,494 | | | | 653,822 | |
| Loss on sale of intangible assets | | | (667,921 | ) | | | — | |
| Accrued research and development, accrued returns and other items | | | 1,325,741 | | | | 1,632,071 | |
| Patent costs | | | 613,806 | | | | 531,175 | |
| Research and experimentation tax credit | | | 4,262,057 | | | | 6,126,272 | |
| | | | | | | | | |
| Deferred tax assets | | | 74,980,112 | | | | 62,570,857 | |
| Valuation allowance | | | (74,980,112 | ) | | | (62,570,857 | ) |
| | | | | | | | | |
| Net deferred tax assets | | $ | — | | | $ | — | |
| | | | | | | | | |
During 2007, the Company reviewed its deferred tax asset for research and experimentation tax credits, based on an analysis of qualifying costs for the tax credit. As a result, the deferred tax asset was reduced by approximately $2.4 million. A corresponding adjustment for the same amount was made to the valuation allowance for deferred tax assets, so there was no net effect on the Company’s balance sheet, income statement or cash flows.
The effective tax rate differs from the U.S. federal statutory tax rate of 34% due to the following:
| | | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| |
| U.S. federal statutory income tax rate | | | (34.0 | )% | | | (34.0 | )% | | | (34.0 | )% |
| State income taxes, net of federal tax benefit | | | (0.9 | )% | | | (5.8 | )% | | | (5.8 | )% |
| Permanent items, primarily stock-based compensation | | | 1.4 | % | | | 2.9 | % | | | 0.2 | % |
| Research and experimentation tax credit | | | (1.4 | )% | | | (2.8 | )% | | | (6.3 | )% |
| Change in valuation allowance | | | 34.9 | % | | | 39.7 | % | | | 45.9 | % |
| | | | | | | | | | | | | |
| Effective tax rate | | | (0.0 | )% | | | (0.0 | )% | | | (0.0 | )% |
| | | | | | | | | | | | | |
At December 31, 2007 and 2006, the Company had federal and state net operating loss carryforwards of approximately $161.7 million and $120.6 million, respectively, available to reduce future taxable income, which will begin to expire in 2020. At December 31, 2007, the Company had federal research and experimentation tax credit carryforwards of approximately $3.7 million which begin to expire in 2020 and state tax credit carryforwards of $0.6 million which begin to expire in 2018.
Under the provisions of Section 382 of the Internal Revenue Code, certain substantial changes in the Company’s ownership may result in a limitation on the amount of net operating loss and research and experimentation tax credit carryforwards which can be utilized in future years. During 2005 and prior years, the Company may have experienced such ownership changes. Ownership changes in 2001 and 2005 may have created annual limitations of approximately $900,000 and $3,800,000, respectively. There were no ownership changes under Section 382 in other years.
In June 2006, the FASB issued FASB Interpretation No. 48,“Accounting for Uncertainty in Income Taxes — an Interpretation of FASB Statement No. 109”(FIN 48). FIN 48 clarifies the accounting for uncertainty in income
F-31
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
taxes recognized in an enterprise’s financial statements in accordance with SFAS No. 109. FIN 48 prescribes a recognition prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken on a tax return. FIN 48 also provides guidance on derecognition, classification, interest and penalties, accounting in interim periods, disclosure and transition. The Company applied the provisions of FIN 48 effective January 1, 2007. The implementation of FIN 48 had no impact on the Company’s financial condition, results of operations, or cash flows, as the Company has no unrecognized tax benefits.
The Company is primarily subject to U.S. and Maryland state corporate income tax. All tax years from the Company’s inception in 2000 remain open to examination by U.S. federal and state authorities.
The Company’s policy is to recognize interest related to income tax matters, if any, in interest expense and penalties related to income tax matters, if any, in operating expenses. As of January 1, 2007 and December 31, 2007, the Company had no accruals for interest or penalties related to uncertain tax positions or other income tax matters.
The Company is unaware of any positions for which it is reasonably possible that the total amounts of unrecognized tax benefits will significantly increase or decrease within the next 12 months.
| |
| 19. | 401(k) Savings Plan and Employee Stock Purchase Plan |
During 2000, the Company established a defined contribution savings plan under Section 401(k) of the Internal Revenue Code. This plan covers substantially all employees who meet minimum age and service requirements and allows participants to defer a portion of their annual compensation on a pre-tax basis. The Company’s Board of Directors has discretion to match contributions made by the Company’s employees. To date, no matching contributions have been made by the Company.
During 2003, the Company adopted an employee stock purchase plan which provides for the issuance of up to 100,000 shares of common stock. This plan, which is intended to qualify under Section 423 of the Internal Revenue Code, provides the Company’s employees with an opportunity to purchase shares of its common stock through payroll deductions. Options to purchase the common stock may be granted to each eligible employee periodically. The purchase price of each share of common stock will not be less than the lesser of 85% of the fair market value of the common stock at the beginning or end of the option period. Participation is limited so that the right to purchase stock under the purchase plan does not accrue at a rate which exceeds $25,000 of the fair market value of the Company’s common stock in any calendar year. To date, the plan has not been activated, and no shares have been issued under this plan.
| |
| 20. | Commitments and Contingencies |
Leases
In August 2002, the Company entered into a10-year lease for its corporate, research and development facility in Germantown, Maryland, which is renewable for two periods of five consecutive years each at the end of the original term. The Company took possession of the lease space during 2003. In conjunction with the execution of the lease agreement, the Company provided the landlord with a letter of credit, which the Company collateralized with a restricted cash deposit in the amount of $566,180 at December 31, 2007 and 2006 (see Note 2). The lease includes scheduled base rent increases over the term of the lease. The total amount of the base rent payments will be charged to expense on the straight-line method over the term of the lease (excluding renewal periods). In 2004 and 2003, the Company received $87,078 and $830,010, respectively, in cash from the landlord in connection with the build-out of the facility. These amounts were recorded as deferred rent and are being amortized on a straight-line basis as a reduction to rent expense over the term of the lease.
In August 2004, the Company leased additional space adjacent to its Germantown, Maryland, facility. This lease, which includes a rent holiday and scheduled rent increases annually over its term, is being charged to expense
F-32
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
on a straight-line basis over the entire term of the lease, which expires May 31, 2013. In conjunction with the execution of the lease agreement, the Company provided the landlord with a letter of credit, which the Company collateralized with a restricted cash deposit in the amount of $306,000 at December 31, 2007 and 2006. (see Note 2,“Summary of Significant Accounting Policies”).
The Company ceased the use of this facility during the third quarter of 2007. For leased facilities where the company has ceased use for a portion or all of the space, the Company accrues a loss if the cost of the leased space is in excess of market rates for potential sublease income. In 2007, the Company accrued a loss for the cost of the leased space in excess of potential sublease income in the amount of $590,000, with $531,000 charged to research and development expense and $59,000 charged to selling, general and administrative expense. Effective April 2008, another company leased approximately 40 percent of the facility directly from the landlord, with the landlord amending the Company’s lease to reflect a rent reduction for the amount of rent the landlord will receive each month from the other company. The Company remains contingently liable for the other company’s rental payments under a financial guarantee to the landlord. The contingent rentals due under the financial guarantee have been included in the table of future minimum lease payments below.
Rent expense under all leases, including deferred rent adjustments as well as the accrual of the remaining rent of $140,366 for the New Jersey office closed in 2005, was $2,586,101, $2,019,610, and $2,472,819 for the years ended December 31, 2007, 2006 and 2005, respectively.
Future minimum lease payments under noncancelable operating leases at December 31, 2007 are as follows:
| | | | | |
| | | Operating
| |
Year Ending December 31, | | Leases | |
| |
| 2008 | | $ | 2,139,250 | |
| 2009 | | | 2,156,210 | |
| 2010 | | | 2,214,399 | |
| 2011 | | | 2,155,021 | |
| 2012 | | | 2,219,670 | |
| Thereafter | | | 943,669 | |
| | | | | |
| Total | | $ | 11,828,219 | |
| | | | | |
Royalties
In the event the Company is able to develop and commercialize a PULSYS-based Keflex product, another cephalexin product relying on the acquired NDAs, or other pharmaceutical products using the acquired trademarks, Eli Lilly will be entitled to royalties on these new products. In 2006 the Company launched its Keflex 750mg product, which is covered by the agreement and is subject to a royalty on net sales, as defined, of 10 percent. Royalties are payable on a new product by new product basis for five years following the first commercial sale for each new product, up to a maximum aggregate royalty per calendar year. All royalty obligations with respect to any defined new product cease after the fifteenth anniversary of the first commercial sale of the first defined new product.
On November 7, 2007, the Company closed an agreement with Deerfield Management, a healthcare investment fund and one of the Company’s largest equity shareholders. The Company sold certain assets, and assigned certain intellectual property rights, relating only to its existing cephalexin business, excluding cephalexin PULSYS, to Deerfield for $7.5 million (less a $500,000 payment to Deerfield). Pursuant to an inventory consignment agreement and license of those intellectual property rights back to the Company, the Company will continue to operate its existing cephalexin business, subject to consignment and royalty payments to Deerfield of 20% of net sales, which decline to 15% should the Company elect to make an extension payment of $1.35 million ($1.8 million if a second closing has occurred) by June 30, 2008. Regardless of the level of net sales, the minimum
F-33
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
combined consignment and royalty payment is $400,000 for each calendar quarter. Consignment and royalty payments due to affiliates of Deerfield Management from MiddleBrook are eliminated in the consolidated balance sheet and consolidated statement of operations in accordance with FIN 46R.
Legal Proceedings
The Company is a party to legal proceedings and claims that arise during the ordinary course of business.
In December 2003, Aventis and Aventis Pharmaceuticals Inc., now part of sanofi-aventis, brought an action against MiddleBrook Pharmaceuticals, Inc., then named Advancis Pharmaceutical Corp, alleging, in essence, that the Advancis corporate name was infringing the plaintiff’s trademark and sought injunctive relief. A trial was held in May 2005, and the Court’s decision, dated September 26, 2006, ruled in favor of sanofi-aventis. On June 28, 2007 the name change was completed pursuant to the Company’s jointly submitted Permanent Injunction and Order with sanofi-aventis of October 27, 2006, whereby the Company agreed to cease using the Advancis name by June 30, 2007. No monetary damages were associated with the decision, and the Company agreed to cease using the Advancis name by June 30, 2007. The Company implemented the name change on June 28, 2007, and there was no significant financial impact resulting from the change.
In August 2007, Eli Lilly and Company provided notice of a legal matter relating to Keflex to MiddleBrook. A product liability claim was filed by Jamie Kaye Moore against Eli Lilly, Teva Pharmaceuticals, Inc. and Teva Pharmaceuticals Industries Ltd. on March 28, 2007. The claim alleges injury from ingestion of some form of “Keflex.” Lilly has filed preliminary objections to the complaint, and has also requested prescription and other records, in order to determine whether the plaintiff ingested brand or generic cephalexin and which manufacturer might be involved. Since the identity of the manufacturer is not known, Lilly is not currently requesting indemnification from MiddleBrook.
| |
| 21. | Related Party Transactions |
Deerfield Transaction
On November 7, 2007, the Company entered into a series of agreements with Deerfield Management, a healthcare investment fund and one of the Company’s largest equity shareholders. The transaction and related agreements are described in Note 13,“Noncontrolling Interest — Deerfield Transaction.”
Loans to Executive Officer
In October 2001, the Company provided loans to Dr. Edward Rudnic, the Company’s president, chief executive officer and a director, and two trusts affiliated with Dr. Rudnic, that were evidenced by full recourse notes in the aggregate principal amount of $121,500. The notes accrued interest at a fixed annual interest rate of 5.5%, with the interest payable annually, and were fully repaid together with accrued interest upon maturity in October 2006.
Consulting Arrangements
Effective May 1, 2004, Mr. James D. Isbister retired as the chairman of the board of directors. At that time, the Company entered into an agreement with Mr. Isbister which provides for a payment to him of up to $100,000 per year in exchange for consulting services. These consulting services include tactical advice and planning with regard to corporate operations, financing approaches, and product development and commercialization strategies. The initial term of the agreement is for 40 months, and it could be renewed by mutual agreement. No payments were made to Mr. Isbister nor did the Company recognize any expense for his services during 2007. The agreement terminated on August 31, 2007 and was not renewed.
F-34
MIDDLEBROOK PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
| |
| 22. | Quarterly Financial Data (Unaudited) |
The following table presents unaudited quarterly financial data of the Company. The Company’s quarterly results of operations for these periods are not necessarily indicative of future results of operations.
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | Net Loss
| |
| | | | | | | | | | | | Net Loss
| | | Per Share
| |
| | | | | | | | | | | | Applicable to
| | | Applicable to
| |
| | | | | | Operating
| | | | | | Common
| | | Common
| |
| | | Revenue | | | Loss | | | Net Loss | | | Stockholders | | | Stockholders | |
| |
Year ended | | | | | | | | | | | | | | | | | | | | |
| December 31, 2007: | | | | | | | | | | | | | | | | | | | | |
| First quarter | | $ | 1,773,037 | | | $ | (13,678,122 | ) | | $ | (13,662,990 | ) | | $ | (13,662,990 | ) | | $ | (0.38 | ) |
| Second quarter | | | 2,680,558 | | | | (9,523,430 | ) | | | (9,477,927 | ) | | $ | (9,477,927 | ) | | | (0.21 | ) |
| Third quarter | | | 3,144,532 | | | | (10,024,075 | ) | | | (10,056,779 | ) | | $ | (10,056,779 | ) | | | (0.22 | ) |
| Fourth quarter | | | 2,858,573 | | | | (6,896,046 | ) | | | (9,051,670 | ) | | | (9,051,670 | ) | | | (0.19 | ) |
Year ended | | | | | | | | | | | | | | | | | | | | |
| December 31, 2006: | | | | | | | | | | | | | | | | | | | | |
| First quarter | | $ | 860,231 | | | $ | (8,866,141 | ) | | $ | (7,597,350 | ) | | $ | (7,597,350 | ) | | $ | (0.25 | ) |
| Second quarter | | | 336,357 | | | | (10,909,876 | ) | | | (10,725,547 | ) | | | (10,725,547 | ) | | | (0.35 | ) |
| Third quarter | | | 2,369,975 | | | | (9,876,760 | ) | | | (9,897,162 | ) | | | (9,897,162 | ) | | | (0.33 | ) |
| Fourth quarter | | | 1,243,847 | | | | (13,699,226 | ) | | | (13,770,095 | ) | | | (13,770,095 | ) | | | (0.44 | ) |
In January 2008, the Company closed a private placement of 8,750,001 shares of its common stock and warrants to purchase 3,500,001 shares of common stock, at a price of $2.40 per unit. Each unit consists of one share of the Company’s common stock and a warrant to purchase 0.40 shares of common stock. The transaction raised approximately $21.0 million in gross proceeds and $19.9 million in net proceeds, net of expenses. The warrants have afive-year term and an exercise price of $3.00 per share. The warrant agreement contains provisions for cash settlement, at the warrant holders’ option, should certain transactions occur as defined in the agreement, including certain reorganizations or mergers. Pursuant to the terms of the registration rights agreement, the Company filed with the SEC a registration statement covering the resale of common stock. The registration rights agreement provides that if the initial registration statement is not effective within 120 days of closing, or if the Company does not subsequently maintain the effectiveness of the initial registration statement or any additional registration statements, then in addition to any other rights the investor may have, the Company will be required to pay the investor liquidated damages, in cash, equal to 1.0 percent per month of the aggregate purchase price paid by such investor. Maximum aggregate liquidated damages payable to an investor are 20 percent of the aggregate amount paid by the investor. The SEC declared the Company’sForm S-3 effective on February 11, 2008, which was within 120 days of closing.
F-35
SCHEDULE II
MIDDLEBROOK PHARMACEUTICALS, INC.
VALUATION AND QUALIFYING ACCOUNTS
Years Ended December 31, 2007, 2006, and 2005
| | | | | | | | | | | | | | | | | |
| | �� | Balance at
| | | | | | | | | | |
| | | Beginning of
| | | | | | | | | Balance at End of
| |
| | | Period | | | Additions | | | Deductions(1) | | | Period | |
| |
Accounts receivable allowances: | | | | | | | | | | | | | | | | |
| Year Ended December 31, 2007 | | $ | 216,930 | | | $ | 1,104,515 | | | $ | (718,602 | ) | | $ | 602,843 | |
| Year Ended December 31, 2006 | | $ | 352,920 | | | $ | 821,165 | | | $ | (957,155 | ) | | $ | 216,930 | |
| Year Ended December 31, 2005 | | $ | 128,569 | | | $ | 690,463 | | | $ | (466,112 | ) | | $ | 352,920 | |
Inventory reserves: | | | | | | | | | | | | | | | | |
| Year Ended December 31, 2007 | | $ | 293,956 | | | $ | 864,404 | | | $ | (153,959 | ) | | $ | 1,004,401 | |
| Year Ended December 31, 2006 | | $ | 154,367 | | | $ | 140,000 | | | $ | (411 | ) | | $ | 293,956 | |
| Year Ended December 31, 2005 | | $ | — | | | $ | 154,367 | | | $ | — | | | $ | 154,367 | |
Deferred tax asset valuation reserves: | | | | | | | | | | | | | | | | |
| Year Ended December 31, 2007 | | $ | 62,570,857 | | | $ | 12,409,255 | | | $ | — | | | $ | 74,980,112 | |
| Year Ended December 31, 2006 | | $ | 48,030,603 | | | $ | 14,540,254 | | | $ | — | | | $ | 62,570,857 | |
| Year Ended December 31, 2005 | | $ | 31,870,513 | | | $ | 16,160,090 | | | $ | — | | | $ | 48,030,603 | |
| | |
| (1) | | Deductions represent utilization of the allowances. For accounts receivable, these include the deduction by customers of prompt pay discounts from their payments, chargebacks made by wholesalers to the Company, wholesaler rebates and writeoffs of bad debts, if any. |
S-1