Exhibit 99.1
YM BIOSCIENCES INC.
ANNUAL INFORMATION FORM
YEAR ENDED JUNE 30, 2007
September 24, 2007
YM BIOSCIENCES INC.
ANNUAL INFORMATION FORM
TABLE OF CONTENTS
| DOCUMENTS INCORPORATED BY REFERENCE | 2 | |||
| GLOSSARY OF TERMS AND PROPER NAMES | 3 | |||
| FORWARD LOOKING STATEMENTS | 6 | |||
| CORPORATE STRUCTURE | 6 | |||
| GENERAL DEVELOPMENT OF THE BUSINESS | 7 | |||
| NARRATIVE DESCRIPTION OF THE BUSINESS | 8 | |||
| DIVIDENDS | 37 | |||
| CAPITAL STRUCTURE | 38 | |||
| COMMON SHARES | 38 | |||
| MARKET FOR SECURITIES | 38 | |||
| DIRECTORS AND OFFICERS | 39 | |||
| AUDIT FEES | 46 | |||
| LEGAL PROCEEDINGS | 46 | |||
| TRANSFER AGENT AND REGISTRAR | 46 | |||
| MATERIAL CONTRACTS | 46 | |||
| INTERESTS OF EXPERTS | 47 | |||
| ADDITIONAL INFORMATION | 47 |
DOCUMENTS INCORPORATED BY REFERENCE
YM BioSciences’ “Management’s Discussion and Analysis of Financial Condition and Results of Operations” (the “MD&A”) and the audited consolidated balance sheets as at June 30, 2007 and June 30, 2006 and the audited consolidated statements of earnings and retained earnings and changes in financial position for each of the years in the three year period ended June 30, 2007 (the “Financial Statements”) previously filed.
The MD&A and the Financial Statements, in their entirety, are incorporated by reference in, and form part of, this Annual Information Form.
All of the documents referred to above have been filed via SEDAR (System for Electronic Document Analysis and Retrieval) and are available to the public at SEDAR’s website, www.sedar.com. Further information may also be found on the Corporation’s website www.ymbiosciences.com.
- 2 -
GLOSSARY OF TERMS AND PROPER NAMES
This glossary contains general terms used in the discussion of the biopharmaceutical industry, as well as specific technical terms used in the descriptions of our technology and business.
Adjuvant - Substance added to a vaccine to enhance its immunogenicity (i.e. its ability to stimulate an immune response)
Amgen - Amgen Incorporated
ASCO - The American Society of Clinical Oncology
AstraZeneca - AstraZeneca PLC
BMS - Bristol Myers Squibb Company
Cancer Vaccine - Vaccines or candidate vaccines designed to treat cancer
cGMP - current good manufacturing practices, as mandated from time to time by health regulatory authorities
CIM - Centro de Inmunología Molecular (Center for Molecular Immunology), Havana, Cuba
CIMAB - CIMAB S.A., a Cuban company responsible for commercializing products developed at CIM
CIMYM BioSciences - CIMYM BioSciences Inc., an 80% owned subsidiary of YM
CR - Complete Response, the disappearance of a tumour
CTA - Clinical Trial Application - previously known as an Investigational New Drug application which must be filed and accepted by the regulatory agency of Health Canada before each phase of human clinical trials may begin
Cyclophosphamide - Approved chemotherapeutic agent
Cytoprotective - Having the capacity to protect cells
Cytotoxic - Having capacity to kill cells
DEA - Drug Enforcement Administration (United States)
DELEX - DELEX Therapeutics Inc., a wholly-owned subsidiary of YM which was wound up on April 30, 2006 into the Corporation
Doxorubicin - Approved chemotherapeutic agent
DSMB - Data Safety Monitoring Board
EGFr - A protein known as Epidermal Growth Factor Receptor
Eli Lilly - Eli Lilly and Company
EMEA - The Europe Agency for the Evaluation of Medicinal Products - the European health regulatory authority
- 3 -
Epithelial - Derived from epithelium which is the layer of cells forming the epidermis of the skin and the surface layer of the serous and mucous membranes
Eximias - Eximias Pharmaceutical Corporation
5-FU - Approved chemotherapeutic agent, Fluorouracil
FDA - United States Food and Drug Administration
Fusion protein - Two or more proteins genetically engineered to be produced as a single protein
Genentech - Genentech Incorporated
Genmab - Genmab A/S
Glioma - A form of brain cancer involving the malignant transformation of a glial cell
GMP - good manufacturing practices, i.e. guidelines established by the governments of various countries, including Canada and the United States, to be used as a standard in accordance with the World Health Organization's Certification Scheme on the quality of pharmaceutical products
GnRH - Gonadotrophin Releasing Hormone; controlling the circulating levels of the sex hormones
HER-1 positive tumors - Tumors expressing/producing the EGF receptor
Humanized - The process whereby an antibody derived from murine cells is altered to resemble a human antibody.
ImClone - ImClone Systems Incorporated
IND - Investigational New Drug application which must be filed and accepted by the FDA before each phase of human clinical trials may begin
Irinotecan - An approved chemotherapeutic agent
In vivo - In the living body or organism. A test performed on a living organism
ISIS - ISIS Pharmaceuticals
Ligand - Used herein to describe a protein or peptide that binds to a particular receptor
Lorus - Lorus Therapeutics Inc.
Merck - Merck KGaA
Metastatic - A term used to describe a cancer where tumor cells have migrated from the primary tumor to a secondary site (e.g. from prostate to bone)
Monoclonal antibody (“MAb”) - Antibodies of exceptional purity and specificity derived from hybridoma cells
Murine - adjective for mouse
NCE - A new chemical entity
Neoplastic - New and abnormal growth of tissue (neoplasm), which may be benign or cancerous
- 4 -
NSCLC - Non Small Cell Lung Cancer
OFAC - Office of Foreign Assets Control of the United States Department of the Treasury
Oncoscience - Oncoscience AG of Germany
Orange Book - A reference to the Hatch/Waxman Act
Orphan Drug - A drug aimed at treating a condition with an incidence of less than 200,000 per year in the United States (often given a seven year market exclusivity by the FDA
OSI - OSI Pharmaceuticals, Inc.
Overall Survival - For patients who have died, overall survival was calculated in months from the day of randomization to date of death. Otherwise, survival was censored at the last day the patient is known alive
PR - Partial Response, the shrinkage of a tumour measured by decrease by at least 30% as measured by a decrease in the sums of the longest diameter according to RECIST criteria
Passive Immunotherapy - Immunologically active material transferred into the patient as a passive recipient. Monoclonal antibodies are considered Passive Immunotherapy since antibodies are generated outside the body and given to the patient
Phosphorylation - Addition/donation of a phosphate group to a particular amino acid which can lead to tumor growth
RECIST - Response Evaluation Criteria in Solid Tumours, a US standard
Roche - F.Hoffmann-LaRoche Ltd.
SD - Stable Disease
TAP - TAP Pharmaceuticals
TGFα - Transforming growth factor alpha
Therapeutic vaccine - An approach to the treatment of cancer utilizing “active immunotherapy”
Tyrosine kinase - An enzyme that catalyzes the phosphorylation of tyrosine residues in proteins with nucleotides as phosphate donors
YM (USA) - YM BioSciences USA Inc., a wholly-owned subsidiary of YM
YM US Operations - YM BioSciences US Operations Inc., an indirect wholly-owned subsidiary of YM
- 5 -
FORWARD LOOKING STATEMENTS
Statements contained in this annual information form that are not based on historical fact, including without limitation statements containing the words "believes," "may," “likely,” "plans," "will," "estimate," "continue," "anticipates," "intends," "expects" and similar expressions, constitute "forward-looking statements" within the meaning of the United States Private Securities Litigation Reform Act of 1995. Such forward-looking statements involve known and unknown risks, uncertainties and other factors that may cause the actual results, events or developments to be materially different from any future results, events or developments expressed or implied by such forward-looking statements. Such factors include, without limitation, changing market conditions, our ability to obtain patent protection and protect our intellectual property rights, commercialization limitations imposed by intellectual property rights owned or controlled by third parties, intellectual property liability rights and liability claims asserted against us, the successful and timely completion of clinical studies, the impact of competitive products and pricing, new product development, uncertainties related to the regulatory approval process, product development delays, our ability to attract and retain business partners and key personnel, future levels of government funding, our ability to obtain the capital required for research, operations and marketing and other risks detailed elsewhere in this annual information form and in the documents incorporated by reference herein. These forward-looking statements are based on our beliefs and expectations on the date the statements are made, and subject to the requirements of applicable securities laws, we undertake no obligation to publicly update or revise any forward-looking statements, whether as a result of new information, future events or otherwise. In light of these risks, uncertainties and assumptions, the forward-looking events discussed in this annual information form might not occur and you should not place undue reliance on forward-looking statements. Forward-looking statements are subject to known and unknown risks, uncertainties and other factors that may cause our actual results, level of activity, performance or achievements to be materially different from those expressed or implied by such forward-looking statements, including:
| • | the risk of our inability to profitably commercialize our products |
| • | the extent of any future losses |
| • | the risk of our inability to establish or manage manufacturing, development or marketing collaborations |
| • | the risk of delay of regulatory approvals and, ultimately, product launches |
| • | dependence on third parties for successful commercialization of our products |
| • | insufficient quota of active ingredient supply to complete clinical trials or to meet commercial demand |
| • | the risk of the termination or conversion to non-exclusive licenses or our inability to enforce our rights under our licenses |
| • | the uncertainty of recovery of advances to joint venture subsidiaries |
| • | other factors discussed under “Risk Factors”. |
Unless otherwise indicated, or the context requires otherwise, the information appearing in this annual information form is stated as at June 30, 2007 and references in this annual information form to “$” or “dollars” are to Canadian dollars. Information contained on our website is not part of this annual information form.
In this annual information form, “YM BioSciences”, “YM”, “we”, “us”, “our” and the “Corporation” refer to YM BioSciences Inc. and its subsidiaries.
CORPORATE STRUCTURE
YM BioSciences Inc. was incorporated under the laws of the Province of Ontario on August 17, 1994 under the name “York Medical Inc.”. On February 7, 2001 we changed our name to “YM BioSciences Inc.” and on December 11, 2001 were continued into the Province of Nova Scotia under the Nova Scotia Companies Act.
Our head office and principal place of business is 5045 Orbitor Drive, Building 11, Suite 400, Mississauga, Ontario, L4W 4Y4. Our registered head office is 1959 Upper Water Street, Suite 800, Halifax, Nova Scotia, B3J 2X2
Organizational Structure
We currently have three material subsidiaries, shown in the following diagram:
- 6 -
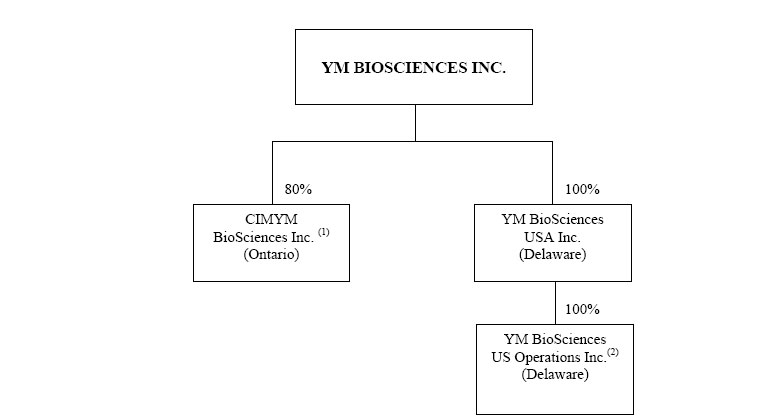
| (1) | 20% owned by CIMAB S.A. |
| (2) | US operating subsidiary (formerly Eximias Pharmaceutical Corporation) |
On June 30, 2006 CIMYM Inc., an Ontario company was amalgamated under the laws of Ontario with CIMYM Inc., a Barbados company to form CIMYM BioSciences Inc (CIMYM). CIMYM is 80% owned by the Corporation and 20% owned by CIMAB.
On May 2, 2005, the Corporation acquired all of the issued and outstanding shares of capital stock of DELEX Therapeutics Inc. YM issued to the DELEX shareholders common shares of YM in consideration for their DELEX shares and additional shares of YM in consideration for working capital in DELEX. On April 30, 2006 DELEX was dissolved and wound up.
YM BioSciences USA Inc. (YM USA) was incorporated on November 23, 2005 under the laws of Delaware. YM US Operations was incorporated on April 10, 2006 under the laws of Delaware. On May 9, 2006 YM US Operations was merged with Eximias Pharmaceutical Corporation.
Unless otherwise noted, “YM BioSciences”, “YM”, and the “Corporation” includes YM BioSciences Inc. and its subsidiaries.
GENERAL DEVELOPMENT OF THE BUSINESS
We were founded in 1994 to acquire rights to develop drug products. We are principally focused on products for the treatment of patients with cancer.
In 1995, we secured our first drug licenses and our initial financing. We initially licensed a range of drug products at various stages of assessment and development, including certain of our current anti-cancer products. In 1998, we concentrated our activities on anti-cancer products. We have used funds raised in our initial financing and subsequent financings to advance certain of our licensed drug products through clinical trials in Canada, the United States, Europe and elsewhere, and to expand our portfolio of anti-cancer products by licensing additional cancer drug and cancer-related products in later stages of development. In addition, we have licensed certain drug products that were pre-clinical. See “Narrative Description of the Business - Products in Clinical Development” and “-- Products in Pre-Clinical Development”.
We have three product candidates currently in the clinical stage of development:
- 7 -
o NIMOTUZUMAB (previously known as TheraCIM hR3), a humanized monoclonal antibody, targeting the protein known as Epidermal Growth Factor Receptor ("EGFr"), is designed to treat epithelial cancers and to be administered prior to, simultaneously with, or subsequent to, chemotherapy and radiotherapy. In various Phase II trials, the drug has significantly improved the reported complete response rate to radiation in head-and-neck tumors and demonstrated clinical benefit in adult and pediatric glioma. The drug has reportedly been approved for sale in the People’s Republic of China (PRC) for nasopharangeal cancer and for head and neck cancer in Argentina, Columbia, and India. Certain of our rights to nimotuzumab have been sub-licensed to Daiichi-Sankyo Co. Ltd in Japan, Oncoscience AG in Europ, to Kuhnil Pharmaceutical Company for Korea and to Innogene Kalbiotech Ltd. of Singapore for certain Pacific-rim countries and certain African countries.
o AeroLEF™, a proprietary formulation of both free and liposome-encapsulated fentanyl administered by pulmonary inhalation, is being developed for the treatment of severe and moderate acute pain including cancer pain. AeroLEF™ has been developed to provide both rapid onset and extended relief of pain while recognizing the need for interpersonal variability as well as inter-episode variability in the amount of drug needed. A 120-patient (99 randomized) Phase IIb trial reported positive results in 2007 and has been cleared by the US FDA in 2007 to commence a US trial.
o TESMILIFENE is a small molecule drug with multiple modes of action that demonstrated enhancement of the activity of traditional chemotherapy. On January 30, 2007, we stopped our pivotal Phase III trial of tesmilifene in patients with metastatic or recurrent breast cancer on the basis that an interim analysis of 351 events indicated it was very unlikely significant differences in the overall survival rate would be shown between treatment arms. In a previous Phase III trial, tesmilifene was administered with doxorubicin and demonstrated a greater than 50% increase in survival in women with metastatic and recurrent breast cancer compared with the patients treated with doxorubicin alone. As at June 30, 2007, tesmilifene continues to be studied in a cooperative trial together with Sanofi-Aventis in which tesmilifene is added to Taxotere regimen for the treatment of women with metastatic breast cancer and we have announced the intention of conducting no additional clinical development.
There is no indication of any public takeover offers by third parties in respect of YM's shares or by YM in respect of other companies' shares which have occurred during the last and current financial year.
NARRATIVE DESCRIPTION OF THE BUSINESS
We are a biopharmaceutical company engaged in the development of products primarily for the treatment of patients with cancer (see “Business Overview”). We own or in-license substances designed for anti-cancer use in order to advance them along the regulatory and clinical pathways toward commercial approval. Our licenses generally cover the major market countries of the developed world (including Canada, the United States, Japan and Europe) or are world-wide. We use our expertise to manage and perform what we believe are the most critical aspects of the drug development process which include the design and conduct of clinical trials, the development and execution of strategies for the protection and maintenance of intellectual property rights and the interaction with drug regulatory authorities internationally. We concentrate on drug development and do not engage in drug discovery, avoiding the investment of time and capital that is generally required before a compound is identified as appropriate for clinical trials. We have previously in-licensed certain preclinical products which have been relevant to our clinical programs. We both conduct and out-source clinical trials, and we out-source the manufacture of clinical materials to third parties.
Our current portfolio of products in clinical development includes two anti-cancer agents (a monoclonal antibody and a small molecule) in a number of formulations currently targeting more than ten different tumors and/or stages of cancer as well as a proprietary inhalation delivery approach for fentanyl to treat acute pain including cancer pain. We also have a financial interest in two additional anti-cancer immunotherapies in pre-clinical development. We principally intend to license the rights to manufacture and market our drug products to other pharmaceutical companies in exchange for license fees and royalty payments and to continue to seek other in-licensing opportunities in pursuing our business strategy. We do not currently intend to manufacture or market products although we may, if the opportunity is available on terms that are considered attractive, participate in ownership of manufacturing facilities or retain marketing or co-development rights to specific products.
- 8 -
Risk Factors
An investment in our securities is speculative and involves a high degree of risk. Prospective investors should carefully consider, together with other matters referred to in this Annual Information Form, the following risk factors. If any event arising from these risks occurs, our business, prospects, financial condition, results of operations and cash flows could be materially adversely affected.
Risks Related To Our Business
We are in the early stages of development and, as a result, are unable to predict whether we will be able to profitably commercialize our products.
Since our incorporation in 1994, none of our products, licensed or owned, have received regulatory approval for sale in any jurisdiction in which we have an economic interest in a product. Accordingly, we have not generated any revenues from the product sales. A significant commitment of resources to conduct clinical trials and for additional product development will be required to commercialize most of the products. There can be no assurance that our remaining products will meet applicable regulatory standards, be capable of being produced in commercial quantities at reasonable cost or be successfully marketed, or that the investment made by us in the commercialization of the products will be recovered through sales, license fees or related royalties.
We have a lack of revenues and a history of losses and, therefore, are unable to predict the extent of any future losses or when or if we will become profitable.
Up to June 30, 2007, we recognized as revenue approximately $6.3 million from a number of licensing agreements including the July 2004 agreement signed with Tarcanta Inc. (a subsidiary of Cancervax Corporation, now Micromet Inc.) with respect to products relating to HER-1 and TGFa; a January 2005 agreement with Shin Poong Pharmaceutical Co., Ltd. to which we licensed the commercial rights for tesmilifene for the South Korean market; an August 2005 agreement with Kuhnil Pharmaceutical Co., Ltd. to which the Company licensed the commercial rights for nimotuzumab for the South Korean market; a January 2006 agreement with Innogene Kalbiotech Private Limited to which the Company licensed the commercial rights for nimotuzumab for several countries in Asia and Africa; and a July 2006 agreement among our subsidiary CIMYM BioSciences, CIMAB and Daiichi Pharmaceutical Co., Ltd. under which CIMYM licensed certain development and marketing rights for nimotuzumab in Japan. As at June 30, 2007, we have an accumulated deficit of $118.3 million. We expect expenditures and the accumulated deficit to increase as we proceed with our commercialization programs until such time as sales, license fees and royalty payments, if any, may generate sufficient revenues to fund our continuing operations. There can be no assurance that the revenues from the commercialization of our products will be sufficient to offset increases in expenditures and the accumulated deficit and therefore there can be no assurance of when or if we will become profitable.
We depend upon being able to identify promising molecules for licensing or acquisition and successfully completing the acquisitions or licensing on economic terms. There is no assurance that we can continue to identify and license molecules for development.
We do not conduct basic research of our own. Basic research on a particular drug product is conducted by biopharmaceutical companies, scientific and academic institutions and hospitals, or scientists affiliated with those institutions. Once the basic research is complete, we enter into license agreements to in-license the right to develop and market the products or acquire them. We have negotiated certain in-licensing agreements with the University of Manitoba, CancerCare Manitoba, Vincent Research and Consulting, and CIMAB. In addition, AeroLEF™ technology was developed at Dalhousie University.
- 9 -
We depend upon others for the manufacture, development and sale of our products. If we are unable to establish or manage collaborations in the future, there could be a delay in the manufacture, development and sale of our products.
We enter into arrangements with and are dependent on others with respect to the manufacture, development and sale of our in-licensed products. Product development includes, but is not limited to, pre-clinical testing, regulatory approval processes, clinical testing, and the development of additional regulatory and marketing information. Our ability to successfully develop and commercialize our in-licensed products is dependent on our ability to make arrangements with others on commercially acceptable terms. The product development process may be delayed or terminated if we cannot secure or maintain such arrangements on terms acceptable to us or at all. We do not have long-term material third party manufacture, formulation or supply agreements, except with respect to one of our licensed products, nimotuzumab, which is manufactured in quantities sufficient for clinical testing by CIMAB or a related party, subject to certain terms and conditions of the licensing agreements between us and CIMAB and CIMAB shall supply commercial quantities or will source such supply if, as and when approval for sale has been granted. The formulation of AeroLEF™ is manufactured for us by Dalton Pharma Services in Toronto, Canada in quantities sufficient for clinical trials. Tesmilifene is manufactured for us by Abraxis Bioscience Inc. in Chicago, Illinois in quantities sufficient for clinical trials.
We expect to enter into out-licensing agreements with others with respect to the manufacturing and marketing of our drug products. We may retain co-development and marketing rights if management determines it appropriate to do so.
On November 12, 2003, we entered into the first out-licensing agreement through our subsidiary, CIMYM Inc., an Ontario corporation, now CIMYM BioSciences Inc. On such date, CIMYM BioSciences out-licensed the rights for nimotuzumab in most of Europe to Oncoscience. Under the terms of the agreement, CIMYM BioSciences is entitled to receive up to US$30 million as a share of any amounts received by Oncoscience in relation to the development or sublicensing of the product and/or as a royalty on. Once CIMYM BioSciences has received US$30 million, CIMYM BioSciences will continue to receive royalties on net sales of nimotuzumab but at a lesser percentage.
YM and CIMYM (Barbados) (now CIMYM BioSciences Inc.) entered into the second out-licensing agreement with Tarcanta and CIMAB relating to the licensing of TGFa and HER-1 to Tarcanta from CIMAB. CancerVax (now, Micromet Inc.) received a license from the United States Department of Treasury authorizing Tarcanta to enter into the transactions with CIMAB and us. On July 13, 2004, the Corporation, CIMYM (Barbados), CIMAB and Tarcanta entered into a License, Development, Manufacturing and Supply Agreement. Under the terms of this agreement, the 2001 CIMYM License has been suspended until such time, if at all, that there is a default under the agreement with Tarcanta. Under the terms of the new agreement and in consideration for the suspension of the 2001 CIMYM License, we were entitled to receive an aggregate payment of $1,000,000 payable in four equal instalments, with the final payment being made on December 31, 2005. In addition, under the new agreement we may receive 35% of an aggregate of $16,350,000 in milestone payments to be paid by Tarcanta upon the successful completion of certain research and development activities. We have no continuing involvement in these research and development activities and have no future obligations under the development plan established by the out-licensing arrangement between CIMAB and Tarcanta. Finally, we retain an interest in the revenues from the manufacture, marketing and sub-licensing of the drugs.
On July 25, 2006 CIMYM BioSciences and CIMAB licensed development and marketing rights in Japan for nimotuzumab to Daiichi Pharmaceutical Co., Ltd. (a wholly owned subsidiary of Daiichi Sankyo Company, Ltd., one of Japan’s largest pharmaceutical companies). Under the agreement, CIMYM received an up-front payment of US$14.5 million and will receive certain milestone payments at certain stages of development for each of a number of indications as well as payments based on supply of nimotuzumab and sales performance in several cancer indications.
There can be no assurance that we will be successful in maintaining our relationships with research institutions or others or in negotiating additional in-licensing or out-licensing agreements on terms acceptable to us or at all, or that any such arrangements will be successful. In addition, there can be no assurance that other parties will not enter into arrangements with such entities for the development or commercialization of similar products or that the parties with whom we have made such arrangements will not pursue alternative technologies or develop products on their own or in collaboration with others, including our competitors. If we do not establish sufficient in-licensing and out-licensing arrangements, we may encounter delays in product introductions or may find that the development, manufacture or sale of our licensed products could be materially adversely affected.
- 10 -
We have no experience in commercial manufacturing of our products and may encounter problems or delays in making arrangements for products to be commercially manufactured, which could result in delayed development, regulatory approval and marketing.
We have not commercially launched any of our licensed or owned products and have no commercial manufacturing experience with respect to our products. To be successful, the products must be manufactured in commercial quantities in compliance with regulatory requirements and at acceptable costs. We do not have and do not intend to acquire facilities for the production of our products although we may invest in the ownership of production facilities if appropriate opportunities are available.
Nimotuzumab is expected to be manufactured in quantities sufficient for clinical testing by CIMAB or a related party, subject to certain terms and conditions of the licensing agreements between us and CIMAB. Currently these expectations are being met. There can be no assurance, however, that such entities will be able to develop adequate manufacturing capabilities for commercial scale quantities in a commercially reasonable manner. Tesmilifene (which is licensed to us) and AeroLEF™ (which is owned by us) have been manufactured, finished and filled in small quantities for testing by third parties. The manufacturing processes for these drugs are such that we expect that commercial quantities of these drugs can be manufactured. If current suppliers cannot manufacture commercial quantities or we otherwise experience a problem with current suppliers, it will be necessary for us to obtain these drugs from new suppliers. We do not have supply agreements with the third party suppliers of tesmilifene or AeroLEF™, but suppliers have produced quantities for us to specification on purchase order. We expect that we could find new suppliers for AeroLEF™, if necessary. There can be no assurance, however, that we will be able to reach satisfactory arrangements with our current suppliers or, if necessary, new suppliers or that such arrangements will be successful. All manufacturing facilities must comply with applicable regulations in their jurisdiction or where products are to be sold. In addition, production of the licensed and owned products may require raw materials for which the sources and amount of supply are limited. An inability to obtain adequate supplies of such raw materials could significantly delay the development, regulatory approval and marketing of our licensed and owned products.
We are dependent on devices from third parties in order to successfully commercialize AeroLEF™.
Third-party devices will be an important element for successful commercialization of AeroLEF™ in both the inpatient and outpatient settings.
We have selected the AeroEclipse(R) inhalation device for our Phase II clinical studies for the inpatient indications for AeroLEF™ and anticipate using the AeroEclipse(R) for our planned Phase III clinical studies for the inpatient market opportunity. Material changes to the AeroEclipse(R) device by the manufacturer or a decision to switch to an alternative inhalation device would likely delay the initiation of Phase III clinical trials. Switching after the initiation of Phase III studies, however, would require additional clinical trials and could significantly delay the commercialization of AeroLEF™. Currently DELEX purchases the AeroEclipse(R) and it does not have a defined supply agreement.
While inpatient use of AeroLEF™, in the hospital or hospice setting, is accomplished with existing equipment such as the AeroEclipse(R), outpatient use will require a portable nebulization device. Several devices currently exist and are available for use with approved respiratory agents (bronchodilators, antibiotics, steroids). We have an active development program to evaluate and identify the best devices for use with AeroLEF™ and other pipeline products. Although we would prefer to access these devices on arms-length basis from the manufacturer, we will explore the benefits of a strategic partnership that could provide for custom adaptations to optimize product delivery, lower prices or other benefits.
- 11 -
The Drug Enforcement Administration limits the availability of the active ingredients in certain of our current drug candidates and, as a result, our quota may not be sufficient to complete clinical trials, or to meet commercial demand or may result in clinical delays.
The DEA regulates chemical compounds as Schedule I, II, III, IV and V substances, with Schedule I substances considered to present the highest risk of substance abuse and Schedule V substances the lowest risk. Certain active ingredients in AeroLEF™, such as fentanyl, are listed by the DEA as Schedule II under the Controlled Substances Act of 1970. Consequently, their manufacture, research, shipment, storage, sale and use are subject to a high degree of oversight and regulation. For example, all Schedule II drug prescriptions must be signed by a physician, physically presented to a pharmacist and may not be refilled without a new prescription. Further, the amount of Schedule II substances we can obtain for clinical trials and commercial distribution is limited by the DEA and our quota may not be sufficient to complete clinical trials or meet commercial demand. There is a risk that DEA regulations may interfere with the supply of the drugs used in our clinical trials, and, in the future, our ability to produce and distribute our products in the volume needed to meet commercial demand.
We are dependent on licenses from third parties and the maintenance of licenses is necessary for our success.
We have obtained our rights to the licensed products under license agreements from various third party licensors as follows:
(a) License Agreement between CIMAB and us dated May 3, 1995 as amended with respect to nimotuzumab; and
(b) License Agreement between our Corporation, the University of Manitoba and The Manitoba Cancer Treatment and Research Foundation, carrying on its undertaking as CancerCare Manitoba, dated November 2, 2000 with respect to tesmilifene.
As we own AeroLEF™, we do not have to license it.
We depend upon the license rights to the licensed products and commercialization of the licensed products. While we believe we are in compliance with our obligations under the licenses, certain licenses may be terminated or converted to non-exclusive licenses by the licensors if there is a breach of the terms of the licenses. There can be no assurance that the licenses are enforceable or will not be terminated or converted. The termination or conversion of the licenses or our inability to enforce our rights under the licenses would have a material adverse effect on our business as we would not have the rights to the products that we are developing. To the extent that management considers a particular license to be material to our undertaking, we have entered into a signed license agreement for that license. Terms of certain remaining licenses are to be determined at a later date. The in-license agreements to which we are currently a party require us to maintain and defend the patent rights that we in-license against third parties.
Although our current licenses are governed by the laws of Ontario, the enforcement of certain of them may necessitate pursuing legal proceedings and obtaining orders in other jurisdictions, including the United States and the Republic of Cuba. There can be no assurance that a court judgment or order obtained in one jurisdiction will be enforceable in another. In international venture undertakings it is standard practice to attorn to a neutral jurisdiction to seek remedy for unresolved commercial disputes. These arrangements are usually negotiated as part of the original business agreement. In the case of the license agreements with us, the parties have agreed that the law governing the agreements is Ontario law and the parties will attorn to the courts of Ontario or the Federal court of Canada to resolve any dispute regarding the agreements.
One of our products is licensed from Cuba, a developing country. As is the case in many developing countries, the commercial and legal environment in Cuba is in a formative stage and may be subject to greater political risk. It is possible that we may not be able to enforce our legal rights in Cuba or against Cuban entities to the same extent that we would be able to do in a country with a more developed commercial and legal system. Termination of our license arrangements or difficulties in enforcement of such arrangements could have a material adverse effect on our ability to continue development of our licensed products from that country.
- 12 -
We have a number of license agreements with CIMAB. CIMAB is a corporation owned by an institution of the Government of Cuba that purportedly operates at arms-length from the state bureaucracy with regard to its business, scientific and administrative decision-making. CIMAB is akin to a "crown corporation" in Canada. CIMAB's management is purportedly both autonomous and responsible for the success of their business decisions. Despite the fact that CIMAB's management is purportedly both autonomous and responsible for business decisions and that the license agreements with us declare Ontario law as the governing law, because of the fact that CIMAB is ultimately a state-owned entity, we will not be able to force CIMAB to comply with any judgment if CIMAB or the Government of Cuba refuses to comply.
We have advanced funds to our joint venture subsidiaries which we are only entitled to recover when the joint venture's net income exceeds the amount of cumulative advances.
YM and CIMAB entered into a funding agreement with CIMYM (now CIMYM BioSciences) in November 1995 (the "Funding Agreement") in connection with the 1995 CIMYM License as amended. The Funding Agreement provides that we will arrange for the appropriate studies and clinical trials for the licensed products held by CIMYM BioSciences and will fund the cost of such studies and trials provided that doing so would not be commercially or scientifically unreasonable. Accordingly, we make the final determination as to whether or not a clinical trial expense is justified with respect to any given product.
We are entitled to reimbursement of all advances made by us pursuant to the Funding Agreement, subject to the successful development of the licensed products and generation of income. CIMYM BioSciences repays such advances out of a portion of its revenues in priority to any revenue or profit sharing arrangements under the 1995 CIMYM License.
As at June 30, 2007, we have advanced $26.5 million to CIMYM BioSciences and we have expensed the total amount. Any reimbursement of such advances would be considered to be income by us.
We are reliant on licensors for research on new products.
We do not conduct our own basic research with respect to the identification of new products. Instead, we rely upon research and development work conducted by others as a primary source for new products. While we expect that we will be able to continue to identify licensable products or research suitable for licensing and commercialization by us, there can be no assurance that this will occur.
We conduct our business internationally and are subject to laws and regulations of several countries which may affect our ability to access regulatory agencies and may affect the enforceability and value of our licenses.
Clinical trials on our products in development have been conducted by us in more than 20 countries including Canada, the United Kingdom, Japan, Germany, India, Russia and the United States and we intend to, and may, conduct future clinical trials in these and other jurisdictions. There can be no assurance that any sovereign government, including Canada's, will not establish laws or regulations that will be deleterious to our interests. There is no assurance that we, as a Canadian corporation, will continue to have access to the regulatory agencies in any jurisdiction where we might want to conduct clinical trials or obtain final regulatory approval, and there can be no assurance that we will be able to enforce our licenses in foreign jurisdictions. Governments have, from time to time, established foreign exchange controls which could have a material adverse effect on our business and financial condition, since such controls may limit our ability to flow funds into a particular country to meet our obligations under in-licensing agreements, and to flow funds which we may be entitled to, in the form of royalty and milestone payments, under out-licensing agreements out of a particular country In addition, the value of our licenses will depend upon the absence of punitive or prohibitive legislation in respect of biological materials.
- 13 -
We also conduct our business internationally and we currently license products and technologies from sources in Canada and Cuba. We have previously licensed, and intend to and may license, products from sources in other jurisdictions.
We have licensed nimotuzumab from CIMAB, a corporation representing an academic institute in Cuba. The United States has maintained an embargo against Cuba, administered by the United States Department of Treasury. The laws and regulations establishing the embargo have been amended from time to time, most recently by the passage of the Cuban Liberty and Democratic Solidarity Act (the "Helms-Burton Act"). The embargo applies to almost all transactions involving Cuba or Cuban enterprises, and it bars from such transactions any US persons unless such persons obtain specific licenses from the United States Department of Treasury authorizing their participation in the transactions. There is Canadian legislation (the Foreign Extraterritorial Measures Act) which provides generally that judgments against Canadian companies under the Helms-Burton Act will not be enforceable in Canada. The US embargo could have the effect of limiting our access to US capital, US financing, US customers and US suppliers. In particular, our products licensed from Cuban sources, noted above, are likely to be prohibited from sale in the United States unless the United States Department of Treasury issues a license or the embargo is lifted.
The Helms-Burton Act authorizes private lawsuits for damages against anyone who "traffics" in property confiscated, without compensation, by the Government of Cuba from persons who at the time were, or have since become, nationals of the United States. We do not own any real property in Cuba and, to the best of our knowledge, and based upon the advice of the Cuban government, none of the properties of the scientific centers of the licensors in which the licensed products were developed and are or may be manufactured was confiscated by the Government of Cuba from persons who at the time were, or have since become, nationals of the United States. However, there can be no assurance that this is correct.
Risks Related To Our Financial Results And Need For Financing
We may be a "passive foreign investment company" which could result in adverse US tax consequences for US investors.
We may be deemed to be a "passive foreign investment company" ("PFIC"). A PFIC is a non-US corporation that meets an income test and/or an asset test. The income test is met if 75% or more of our gross income is "passive income" (generally, dividends, interest, rents, royalties, and gains from the disposition of assets producing passive income) in any taxable year. The asset test is met if at least 50% of the average value of our assets produce, or are held for the production of, passive income. Based on our current income, assets and activities, we may be a PFIC. As a result, a US holder of our common shares could be subject to increased tax liability, possibly including an interest charge, upon the sale or other disposition of the US holder's common shares or upon the receipt of "excess distributions".
We may not be able to obtain necessary funding from sales or license fees or royalties and, as a result, may need to try to obtain future capital through the public market or private financing which may not be available on acceptable terms or at all.
We may require additional funding for the commercialization of our products, licensed and owned, and if new products are licensed or acquired and put into development. The amount of additional funding required depends on the status of each project or new opportunity at any given time. Our business strategy is to in-license rights to promising drug products, further develop those products by progressing the products toward regulatory approval by conducting and managing clinical trials, and finally to out-license rights to manufacture and/or market resulting drug products to other pharmaceutical firms in exchange for royalties and license fees. Due to the in- and out-licensing arrangements and our dependence on others for the manufacture, development and sale of our in-licensed products, we do not have consistent monthly or quarterly expenditures and cannot determine the amount and timing of required additional funding with any certainty. As at June 30, 2007 we had cash and short-term deposits totalling $75.6 million and payables of $3.3 million.
- 14 -
We assess our additional funding needs on a project-by-project basis from time-to-time. To the extent that we are unable to fund our expenditures from sales, license fees and royalties, it may be necessary to reconsider whether to continue existing projects or enter into new projects, or it may be necessary to access either the public markets or private financings whenever conditions permit. In addition, we have no established bank financing arrangements and there can be no assurance that we will be able to establish such arrangements on satisfactory terms or at all. Such financing, if required and completed, may have a dilutive effect on the holders of our common shares. There is no assurance that such financing will be available if required, or that it will be available on favorable terms.
Our operating results and stock price may fluctuate significantly.
The trading price of our common shares, as with many emerging biopharmaceutical companies, is likely to be highly volatile. Factors such as the efficacy of our products or the products of our competitors, announcements of technological innovations by us or our competitors, governmental regulations, developments in our patents or other proprietary rights, our licensors or our competitors, litigation, fluctuations in our operating results, thin capitalization, market conditions for biopharmaceutical stocks and general market and economic conditions could have a significant impact on the future trading price of our common shares. In addition, our common shares are highly volatile since it may take years before any of our licensed products will receive final regulatory approval to be marketed in Canada, the United States or other territories.
There is no assurance that an active trading market in our common shares will be sustained.
Our common shares are listed for trading on the TSX, AMEX and AIM. However, there can be no assurance that an active trading market in our common shares on these stock exchanges will be sustained.
Risks Related To Our Industry
If our pre-clinical and clinical testing of drug products do not produce successful results, we will not be able to commercialize our products.
Each of our products, licensed or owned, must be subjected to additional pre-clinical and/or clinical testing in order to demonstrate the safety and efficacy of our products in humans. Our ability to commercialize our products will depend on the success of currently ongoing pre-clinical and clinical trials and subsequent pre-clinical and clinical trials that have not yet begun.
We are not able to predict the results of pre-clinical and clinical testing of our drug products. It is not possible to predict, based on studies or testing in laboratory conditions or in animals, whether a drug product will prove to be safe or effective in humans. Further, preclinical data may not be sufficient for regulators to accept positive clinical data for approval to commercialize a product. In addition, success in one stage of testing is not necessarily an indication that the particular drug product will succeed in later stages of testing and development. There can be no assurance that the pre-clinical or clinical testing of our products will yield satisfactory results that will enable us to progress toward commercialization of such products. Unsatisfactory results may have a material adverse effect on our business, financial condition or results of operations as they could result in us having to reduce or abandon future testing or commercialization of particular drug products.
If our competitors develop and market products that are more effective than our existing product candidates or any products that we may develop, or obtain marketing approval before we do, our products may be rendered obsolete or uncompetitive.
Technological competition from pharmaceutical companies, biotechnology companies and universities is intense and is expected to increase. Many of our competitors and potential competitors have substantially greater product development capabilities and financial, scientific, marketing and human resources than we have. Our future success depends in part on our ability to maintain a competitive position, including our ability to further progress our products, licensed or owned, through the necessary pre-clinical and clinical trials towards regulatory approval for sale and commercialization. Other companies may succeed in commercializing products earlier than we are able to commercialize our products or they may succeed in developing products that are more effective than our products. We consider our main competitors to be: Amgen Inc., AstraZeneca PLC, Bristol-Myers Squibb Company, F. Hoffmann-LaRoche Ltd., Genentech, Genmab A/S, ImClone Systems Inc. Merck KGaA, and OSI Pharmaceuticals, Inc. with respect to nimotuzumab. The main competitors, to our knowledge, for the AeroLEF™ product are Cephalon, Inc., Endo Pharmaceuticals Holdings Inc., Akela Inc., Alexza Molecular Delivery Corporation, Javelin Pharmaceuticals, Inc. (formerly IDDS, Inc.), Barr Pharmaceuticals, Inc., CeNeS Pharmaceuticals plc and Alza Corporation.
- 15 -
Our success depends in part on developing and maintaining a competitive position in the development and commercialization of our products, licensed or owned, and technological capabilities in our areas of expertise. The biotechnology and pharmaceutical industries are subject to rapid and substantial technological change. While we will seek to expand our technological capabilities in order to remain competitive, there can be no assurance that developments by others will not render our products non-competitive or that we or our licensors will be able to keep pace with technological developments. Competitors have developed technologies that could be the basis for competitive products. Some of those products may have an entirely different approach or means of accomplishing the desired therapeutic effect than our products and may be more effective or less costly than our products. In addition, other forms of medical treatment may offer competition to the products. The success of our competitors and their products and technologies relative to our technological capabilities and competitiveness could have a material adverse effect on the future pre-clinical and clinical trials of our products, including our ability to obtain the necessary regulatory approvals for the conduct of such trials.
We are subject to extensive government regulation that increases the cost and uncertainty associated with gaining final regulatory approval of our product candidates.
Securing final regulatory approval for the manufacture and sale of human therapeutic products in Canada and our other markets, including the United States, is a long and costly process that is controlled by that particular country’s national regulatory agency. The national regulatory agency in Canada is Health Canada, and in the United States it is the FDA. Other national regulatory agencies have similar regulatory approval processes, but each is slightly different. Approval in either Canada or the United States does not assure approval by other national regulatory agencies, although often test results from one country may be used in applications for regulatory approval in another country.
Prior to obtaining final regulatory approval to market a drug product, every national regulatory agency has a variety of statutes and regulations which govern the principal development activities. These laws require controlled research and testing of products, government review and approval of a submission containing pre-clinical and clinical data establishing the safety and efficacy of the product for each use sought, approval of manufacturing facilities including adherence to good manufacturing practices during production and storage, and control of marketing activities, including advertising and labelling.
None of our products have been completely developed or tested and, therefore, we are not yet in a position to seek final regulatory approval to market any of our products. To date we have obtained various regulatory approvals to develop and test our products. Nimotuzumab has been approved for testing in the US, Canada, Europe, Japan, Korea, and Indonesia/Malaysia/Singapore and has been designated as an orphan drug in Europe and the United States. It is in Phase II and III trials in certain of those territories. Trials of AeroLEF™ have concluded a Phase I, IIa and IIb in Canada, a Phase II has been cleared for initiation in the US and a Phase III is currently being designed.
Nimotuzumab, which is being developed in Canada, Europe, Japan and other Southeast Asian countries sub-licensed by YM is also being separately developed or tested in India, China, Argentina, Columbia, and Cuba. The US established an embargo against Cuba in 1961, reinforced by the Helms-Burton Act in 1996 and is among several nations which have been identified by the US Department of State as being a state sponsoring terrorism. As such the US Government has put in place certain limitations on conduct of business with Cuba and anti-terrorism legislation against Cuba. Although as of the date of this filing such anti-terrorism controls have not had any adverse effect on our operations, because of the anti-terrorism controls and the Helms-Burton Act there is no assurance that the Corporation will be able to complete clinical testing in the United States or obtain final regulatory approval in order to successfully commercialize our nimotuzumab in the United States. We were successful in September 2006 in our application for a Special License to import nimotuzumab for a clinical trial in the US and received clearance for this trial from the FDA in certain of these territories following the fiscal 2007 year end.
- 16 -
There can be no assurance that the licensed products will be successfully commercialized. The process of completing clinical testing and obtaining final regulatory approval to market the licensed products is likely to take a number of years for most of the licensed products and require the expenditure of substantial resources. Any failure to obtain, or a delay in obtaining, such approvals could adversely affect our ability to develop the product and delay commercialization of the product. Further, there can be no assurance that our licensed products will prove to be safe and effective in clinical trials under the standards of the regulations in our territories or receive applicable regulatory approvals from applicable regulatory bodies.
Changes in government regulations although beyond our control could have an adverse effect on our business.
We have, or have had, licenses with, or clinical trials at, various academic organizations, hospitals and companies in Canada, Cuba, Italy, Japan, Germany, the United States and the United Kingdom and numerous other countries and we depend upon the validity of our licenses and access to the data for the timely completion of clinical research in those jurisdictions. Any changes in the drug development regulatory environment or shifts in political attitudes of a government are beyond our control and may adversely affect our business.
Our business may also be affected in varying degrees by such factors as government regulations with respect to intellectual property, regulation or export controls. Such changes remain beyond our control and the effect of any such changes cannot be predicted.
These factors could have a material adverse effect on our ability to further develop our licensed products.
Risks Related To Intellectual Property And Litigation
Our success depends upon our ability to protect our intellectual property and our proprietary technology.
Our success will depend, in part, on our ability and our licensors’ ability to obtain patents, maintain trade secrets protection, and operate without infringing on the proprietary rights of third parties or having third parties circumvent our rights. Certain licensors and the institutions that they represent, and in certain cases, us on behalf of the licensors and the institutions that they represent, have filed and are actively pursuing certain applications for Canadian and foreign patents. The patent position of pharmaceutical and biotechnology firms is uncertain and involves complex legal and financial questions for which, in some cases, certain important legal principles remain unresolved. There can be no assurance that the patent applications made in respect of the owned or licensed products will result in the issuance of patents, that the term of a patent will be extendable after it expires in due course, that the licensors or the institutions that they represent will develop additional proprietary products that are patentable, that any patent issued to the licensors or us will provide us with any competitive advantages, that the patents of others will not impede our ability to do business or that third parties will not be able to circumvent or successfully challenge the patents obtained in respect of the licensed products. The cost of obtaining and maintaining patents is high. Furthermore, there can be no assurance that others will not independently develop similar products which duplicate any of the licensed products, or, if patents are issued, design around the patent for the product. There can be no assurance that our processes or products or those of our licensors do not or will not infringe upon the patents of third parties, or that the scope of our patents or those of our licensors will successfully prevent third parties from developing similar and competitive products.
Much of our know-how and technology may not be patentable, though they may constitute trade secrets. There can be no assurance, however, that we will be able to meaningfully protect our trade secrets. To help protect our intellectual property rights and proprietary technology we require employees, consultants, advisors and collaborators to enter into confidentiality agreements. There can be no assurance that these agreements will provide meaningful protection for our trade secrets, know-how or other proprietary information in the event of any unauthorized use or disclosure.
- 17 -
We maintain patents in connection with nimotuzumab, AeroLEF™ and tesmilifene. The following is a description of our key current and pending patents in connection with these drug products.
Nimotuzumab
CIMYM is the exclusive licensee for particular territories including the United States under a patent estate that includes composition of matter coverage for nimotuzumab, and further includes coverage for nimotuzumab-based formulations and end-uses in the treatment of EGFr-dependent cancers. The composition-of-matter patents are granted in the United States, in Europe, are allowable in Japan, and are pending in Canada.
CIMYM's key US patent, US 5,891,996 expires in November 2015, and term extensions of up to five years may be available under the Patent Term Restoration Act. The same term and extension apply also to the key European patent, EP 712863.
We are aware of US 5,770,195, a patent granted to Genentech, for the anti-cancer use of EGFr MAbs in combination with a cytotoxic agent. We are also aware of US patents granted to others in this field. In April 2001 Rorer International (Overseas) ("Rorer") was issued the US 6,217,866 which includes claims to any antibody targeting the EGFr administered with any anti-neoplastic agent. We believe that the Rorer patents are exclusively licensed to ImClone A counterpart patent has been granted in Europe. We have filed an opposition to the grant of the European patent. The opposition proceedings in Europe have been suspended pending the outcome of cases in the U.K. and Germany related to inventorship claims filed by Yeda Research and Development Corporation, Ltd. (“Yeda”). A September 19, 2006 decision in the United States District Court of Southern New York granted sole inventorship of the ‘866 patent to scientists from Weizmann Institute of Science (Rehovet, Israel) represented by Yeda. Yeda now has the right to grant non-exclusive licenses in the United States. In addition, we are aware of a separate series of national patent applications filed by ImClone, and represented by EP1080113, claiming the anti-cancer use of radiation in combination with any inhibitor of any receptor tyrosine kinase that is involved in the genesis of tumours. ImClone has also filed a US and PCT applications covering the use of EGFr MAbs to treat patients having tumors that do not respond to treatment with conventional therapies. We continue to monitor Imclone’s pending radiation-related patent applications. A European patent application claiming the use of radiation in combination with tyrosine kinase receptors was withdrawn due to prior art brought to the attention of the examiner. We plan on virgorously challenging ImClone’s claims in respect of the radiation-related patent applications by having filed additional prior art in the EU and Japan. The outcome of these challenges cannot be predicted, and there can be no assurance that we will succeed in challenging the validity or scope of patent claims by ImClone or any other patent applicant. If our challenges are not successful, this may have a material adverse effect on our business.
The manufacturing of nimotuzumab may fall within the scope of process patents owned by Protein Design Labs Inc., Genentech, and the Medical Research Council of the United Kingdom. We are aware that some of these process patents are currently being challenged by companies other than us. In the event any of the applicable process patents are upheld, we believe we will be able to obtain licenses under such patents on commercially reasonable terms, though there can be no assurance of this.
There may also be risks related to nimotuzumab as our license originates from Cuba. Cuba is a socialist country and, under the current patent law, ownership of the inventions of the Cuban inventors for which patent applications have been filed rests with the State. The material license agreement for our Cuban sourced products is a license agreement between us and CIMAB, dated May 3, 1995, as awarded, with respect to nimotuzumab. There is no guarantee that, in the event of a change in the political regime, the Cuban government will continue to honour such license agreement.
- 18 -
AeroLEF™
The AeroLEF™ product is described in four patent families. We own key patents, expiring in 2014, claiming a method of administering systemic analgesia by inhaling free and liposome-encapsulated opioid analgesic. North American coverage includes a reissued US patent and a Canadian patent. We own two US applications with counterpart PCT applications, expiring in 2024, claiming the formulation for use in a method comprised of continuously inhaling the formulation to deposit at least one rapid-onset opioid and one sustained-effect opioid in the lungs to avoid the onset of side effects. A pending PCT application entitled "Stable Compositions" claims the manufacturing method and other physical characteristics of the formulation.
We are aware of US patents owned by Phares Pharmaceutical Research NV related to a method of manufacturing liposome compositions. These patents expire in 2008 and are not expected to adversely affect our commercial activities.
Tesmilifene
We are the exclusive licensee to patents and patent applications from the University of Manitoba for tesmilifene. Patents that claim the use of tesmilifene in combination with chemotherapeutic agents have been issued in the United States, Europe, Japan, Canada and Australia. US patent 5,859,065 broadly claims the use of tesmilifene and structurally related analogs in combination with any chemotherapeutic for the treatment of any cancer. Although the twenty-year term of this patent expires in December 2010, we plan to take full advantage of patent terms extensions of up to five additional years granted under the Patent Term Restoration Act in the United States. Other issued patents US 6,284,799 and US 5,747,543 expire in 2014 and 2015 respectively. In addition to these granted patents, patent applications relevant to the use of tesmilifene in neoadjuvant treatment are pending.
In addition to patent protection, we intend to rely upon the available term of data exclusivity in the US and other countries for NCEs. Furthermore, full advantage will be taken of the Orange Book provisions in the United States and equivalent provision in Canada and other countries, as a means for delaying generic competition.
Our potential involvement in intellectual property litigation could negatively affect our business.
Our future success and competitive position depend in part upon our ability to maintain our intellectual property portfolio. There can be no assurance that any patents will be issued on any existing or future patent applications. Even if such patents are issued, there can be no assurance that any patents issued or licensed to us will not be challenged. Our ability to establish and maintain a competitive position may be achieved in part by prosecuting claims against others who we believe are infringing our rights and by defending claims brought by others who believe that we are infringing their rights. In addition, enforcement of our patents in foreign jurisdictions will depend on the legal procedures in those jurisdictions. Even if such claims are found to be invalid, our involvement in intellectual property litigation could have a material adverse effect on our ability to out-license any products that are the subject of such litigation. In addition, our involvement in intellectual property litigation could result in significant expense, which could materially adversely affect the use or licensing of related intellectual property and divert the efforts of our valuable technical and management personnel from their principal responsibilities, whether or not such litigation is resolved in our favor.
Product liability claims are an inherent risk of our business, and if our clinical trial and product liability insurance prove inadequate, product liability claims may harm our business.
Human therapeutic products involve an inherent risk of product liability claims and associated adverse publicity. We currently maintain clinical trial liability insurance with an ultimate net loss value of up to $10 million per claim and a policy aggregate of $10 million (except for AeroLEF which has a limit of $5 million per claim and a policy aggregate of $5 million). We currently have no other product liability insurance and there can be no assurance that we will be able to obtain or maintain product liability insurance on acceptable terms or with adequate coverage against potential liabilities. Such insurance is expensive, difficult to obtain and may not be available in the future on acceptable terms, or at all. An inability to obtain sufficient insurance coverage on reasonable terms or to otherwise protect against potential product liability claims could have a material adverse effect on our business by preventing or inhibiting the commercialization of our products, licensed and owned, if a product is withdrawn or a product liability claim is brought against us.
- 19 -
Risks Related To Being A Canadian Entity
We are governed by the corporate laws in Nova Scotia, Canada which in some cases have a different effect on shareholders than the corporate laws in Delaware, United States.
The material differences between the Nova Scotia Companies Act (the "NSCA") as compared to the Delaware General Corporation Law ("DGCL") which may be of most interest to shareholders include the following: (i) for material corporate transactions (such as amalgamations, other extraordinary corporate transactions, amendments to the memorandum of association and amendments to the articles of association) the NSCA generally requires three-quarter majority vote by shareholders which in most instances requires a confirmatory resolution by a majority of the shareholders (and, in addition, especially where the holders of a class of shares is being affected differently from others, approval will be required by holders of two-thirds of the shares of such class voting in a meeting called for the purpose), whereas DGCL generally only requires a majority vote of shareholders for similar material corporate transactions; (ii) quorum for shareholders meetings is not prescribed under the NSCA and is only 5% under our articles of association, whereas under DGCL, quorum requires the holders of a majority of the shares entitled to vote to be present; and (iii) our articles of association require a special resolution and the Corporations Miscellaneous Provisions Act (Nova Scotia) requires three-quarters of all shareholders entitled to vote to pass a resolution for one or more directors to be removed, whereas DGCL only requires the affirmative vote of a majority of the shareholders.
Business Overview
Overview
We are a biopharmaceutical company engaged in the development of products primarily for the treatment of patients with cancer. We generally in-license, or acquire, substances designed to be marketed for anti-cancer use in order to advance them along the regulatory and clinical pathways toward commercial approval. Our licenses generally cover the major market countries of the developed world (including Canada, the United States, Japan and Europe) or are world-wide. We use our expertise to manage and perform what we believe are the most critical aspects of the drug development process which include the design and conduct of clinical trials, the development and execution of strategies for the protection and maintenance of intellectual property rights and the interaction with drug regulatory authorities internationally. We concentrate on drug development and do not engage in drug discovery, avoiding the significant investment of time and capital that is generally required before a compound is identified in clinical trials. We have in-licensed certain preclinical products which have been related to our clinical programs. We both conduct and out-source clinical trials, and out-source the manufacture of clinical materials to third parties.
Our current portfolio of products in clinical development includes two anti-cancer agents (a monoclonal antibody and a small molecule) in a number of formulations currently targeting more than 10 different tumors and/or stages of cancer as well as a proprietary inhalation delivery approach for fentanyl to treat acute pain including cancer pain. We also have a financial interest in two additional anti-cancer immunotherapies in pre-clinical development. We principally intend to license the rights to manufacture and/or market our drug products to other pharmaceutical companies in exchange for license fees and royalty payments and to continue to seek other in-licensing opportunities in pursuing our business strategy. We do not currently intend to manufacture or market products although we may, if the opportunity is available on terms that are considered attractive, participate in ownership of manufacturing facilities or retain marketing or co-development rights to specific products. We intend to generally license the rights to manufacture and/or market our drug products to other pharmaceutical companies in exchange for license fees and royalty payments and to continue to seek other in-licensing opportunities in pursuing our business strategy.
Business Strategy
We are principally focused on development of products for the treatment of cancer or cancer-related conditions. Our strategy is to license rights to promising products or acquire such products, further develop those products by conducting and managing clinical research and trials and progressing the products toward regulatory approval, and, generally, sub-license or out-license rights to manufacture and/or market resulting drug products to other pharmaceutical firms in exchange for royalties and license fees. We seek to use our product development capabilities to bridge discoveries and research from scientific/academic institutions or other biopharmaceutical companies, on the one hand, with commercial manufacturing and marketing of biopharmaceutical products, on the other hand.
- 20 -
The main elements of our business strategy are described below:
Identification of Product Candidates: We directly perform scientific evaluation and market assessment of biopharmaceutical products and research developed by scientific/academic institutions and other biopharmaceutical companies. As part of this process, we evaluate the related scientific research and pre-clinical and clinical research, if any, and the intellectual property rights in such products and research, with a view to determining the therapeutic and commercial potential of the applicable product candidates.
In-Licensing: Upon identifying a promising biopharmaceutical product, we seek to negotiate a license to the rights for the product from the holder of those rights, the developer or researcher. The terms of such licenses vary, but generally our goal is to secure licenses that permit us to engage in further development, clinical trials, intellectual property protection (on behalf of the licensor or otherwise) and further licensing of manufacturing and marketing rights to any resulting products. This process of securing license rights to products is commonly known as "in-licensing".
Further Development: Upon in-licensing a cancer-related product, our strategy is to apply our skills and expertise to progress the products toward regulatory approval and commercial production and sale in major markets. These activities include implementing intellectual property protection and registration strategies, performing or having performed for us, pre-clinical research and testing, the formulating or reformulating of drug products, making regulatory submissions, performing or managing clinical trials in target jurisdictions, and undertaking or managing the collection, collation and interpretation of clinical and field data and the submission of such data to the relevant regulatory authorities in compliance with applicable protocols and standards.
Out-Licensing: We generally plan to further license manufacturing and marketing rights to our licensed products to other pharmaceutical firms. This is commonly known as "out-licensing". Under our business model, licensees would be expected, to the extent necessary, to participate in the remaining clinical development required to obtain final regulatory approval for the product. We expect that out-licensing would result in a pharmaceutical company or other licensee marketing or manufacturing the product in return for licensing fees in addition to royalties on any sales of the product. Management believes this model is consistent with current biotechnology and pharmaceutical industry licensing practices. In addition, although out-licensing is a primary strategy of ours, we may retain co-development or marketing rights to particular products or territories. To date, we have out-licensed one of our products in certain European countries, two anti-cancer pre-clinical products to two wholly-owned subsidiaries of a United States corporation, one product in Japan, one product in several jurisdictions in South East Asia and Africa and two products in South Korea. See "Business - Licensing Arrangements - Out-Licensing".
We actively search for new product opportunities using the relationships of our management and advisory team and continuous monitoring of the academic and biotechnology environment in cancer treatment developments. Our staff analyses and evaluates opportunities and continuously reviews them. In addition, we have existing rights of first refusal in certain of our existing license agreements for certain additional products and extensions to existing products. We intend to seek other in-licensing opportunities in pursuing our business strategy.
Cancer And Cancer Therapeutic Market
According to World Health Organization (April 2003) more than 10 million people are diagnosed with cancer every year. The organization projects a 50% increase in cancer rates from year 2000 to 2020 to reach 15 million new cases being diagnosed annually. According to the American Cancer Society, there were approximately 1.4 million new cancer cases diagnosed in the United States in 2006. Cancer is the second leading cause of disease-related death in North America, behind cardiovascular disease which it is predicted to surpass in the next few years. The principal reasons for this projection appear to be the aging population in more developed countries, environmental issues related to industrial development, and improvements in the treatment of cardiovascular disease. North America, Europe and Japan are the principal markets for cancer therapies because of the established healthcare and payor systems.
- 21 -
The principal types of cancer in the United States, accounting for approximately 57% of the incidence of all cancers, based on management's analysis, are prostate (17%), breast (15%), lung (12%) and colorectal (11%). These four types of cancer are also responsible for the highest combined mortality, accounting for over 50% of all cancer deaths in the United States. Bladder, ovarian, brain and oral cancer, as well as lymphoma, leukemia and melanoma account for the majority of the balance of cancer deaths. The incidence of a particular cancer varies greatly between continents, principally because of diet and habit.
Surgery, radiation and chemotherapy remain the principal effective treatments for cancer. Although there is an ongoing debate about the value of chemotherapeutics with regards to prolongation of life, their palliative value has resulted in significant improvements in quality of life for cancer sufferers. In addition, although the reason is not clearly understood, current cancer drugs are effective in only a subpopulation of individuals with the same disease. Notwithstanding this, revenues across seven major oncology markets were reported to be approximately US$34.6 billion in 2005 (Datamonitor, 2006) and are expected to increase to over US$45 billion by 2011. The use of cancer therapies is forecast to increase as diagnostic methods improve (as already demonstrated in prostate cancer) and, particularly, as more effective treatments are developed.
Numerous new approaches to cancer are currently in clinical trials. As targets become validated and technologies improve, research is beginning to yield therapeutic approaches that appear to be more effective than existing ones. Monoclonal antibodies were first described in 1978, and are now beginning to yield commercially viable therapeutic products, such as Rituxan(R), the first monoclonal treatment for cancer, approved by the FDA in 1998. The Corporation is aware of only six naked monoclonal antibodies approved in the United States for the treatment of cancer, Rituxan® , Campath®, Herceptin®, Avastin®, Erbitux® and Vectebix® although many more are in development A second approach to cancer treatment, therapeutic cancer vaccines, has been under development for many years, and the first such vaccine, Melacine(R), was approved in 1999 in Canada.
We are also developing a novel formulation of the opioid, fentanyl, for the treatment of severe and moderate pain, including cancer pain. In the United States cancer pain is suffered by more than 1.5 million patients. We are aware of several other companies that are pursuing approaches to the delivery of fentanyl including Cephalon, Inc. (“Cephalon”), Endo Pharmaceuticals Holdings Inc. (“Endo”), Akela Inc. (“Akela”), Alexza Molecular Delivery Corporation (“Alexza”), Aradigm Corporation (“Aradigm”), Barr Pharmaceuticals, Inc. (“Barr”), CeNeS Pharmaceuticals plc (“CeNeS”) and Alza Corporation (“Alza”).
Products In Clinical Development
Our current portfolio of products in clinical development includes two anti-cancer agents (a monoclonal antibody and a small molecule) in a number of formulations currently targeting more than 10 different tumors and/or stages of cancer as well as a proprietary inhalation delivery approach for fentanyl to treat acute pain including cancer pain. We also have a financial interest in two additional anti-cancer immunotherapies in pre-clinical development. A number of the Corporation’s products involve newer approaches to the treatment of cancer and include a novel monoclonal antibody, nimotuzumab. Another product, tesmilifene, is a chemical that has been clinically reported to enhance the activity of certain known chemotherapeutics although its clinical failure in 2007 makes its effect uncertain. Our principal targets are of the most ubiquitous cancer indications, including breast, non-small cell lung cancer, and colorectal cancer as well as head-and-neck cancer, brain cancers and certain indications with orphan drug designations. We expect, based on clinical trials done to date, to develop all of our clinical stage candidates beyond their respective initial indications except for tesmilifene for which no additional clinical development is planned.
Nimotuzumab
Background:
Nimotuzumab is a humanized MAb targeting EGF Receptor (EGF-R). The EGF-R is present in high concentrations on the surface of many cancer cells and it is postulated that the binding of ligands to this receptor is important in the continuing growth of cancer cells. Nimotuzumab appears to block this binding, resulting in the potential for inhibition of cell growth or, possibly, cell destruction by the immune system. Improved tumor responses have been reported when EGF-R targeting agents are combined with other anti-cancer treatments. Our EGF-R MAb is being developed to be administered alone, or in combination with other anti-cancer treatments.
- 22 -
Clinical Experience And Development Plans:
Nimotuzumab is reported to have been administered to more than 900 cancer patients worldwide and shown to be well tolerated. The product has been approved for use in numerous clinical trials by various regulatory agencies including the EMEA, Health Canada and the FDA. Trials that were or are being conducted with the drug include:
| a) | Phase I safety and PK/PD trial in 12 patients with epithelial-derived cancers conducted by CIMAB; |
| b) | Phase II clinical trial in patients with locally advanced head-and-neck cancer designed to evaluate safety and efficacy in this indication. Complete tumour response was observed in 17 out of 24 evaluable patients and 14 patients are still alive more than 5.5 years after the start of the study, conducted by CIMYM; |
| c) | Phase II randomized trial of nimotuzumab + radiation vs. radiation alone designed to evaluate efficacy and safety in 78 patients with hormone refractory prostate cancers, conducted by CIMAB; |
| d) | Phase II randomized trial of nimotuzumab + radiation + chemotherapy vs. radiation + chemotherapy designed to evaluate efficacy and safety in 68 patients with esophageal cancers, conducted by CIMAB |
| e) | Phase I safety, PK/PD and MTD trial in 15 patients with breast cancer receiving nimotuzumab + chemotherapy, conducted by CIMAB; |
| f) | Phase I PK/PD trial in 10 patients with head-and neck cancers receiving nimotuzumab + radiation, conducted by CIMAB; |
| g) | Phase II/III randomized trial in 148 patients with uterine cervix cancers conducted by CIMAB. The study is designed to assess efficacy and safety in patients receiving either nimotuzumab + radiation + chemotherapy or radiation + chemotherapy; |
| h) | Phase II/III randomized trial in patients with head-and-neck cancers conducted by CIMAB. The study is designed to assess local control, safety and survival in 112 patients receiving either nimotuzumab + radiation or radiation alone; |
| i) | Phase II randomized trial of nimotuzumab + radiation vs. radiation alone in 30 patients with brain metastases from NSCLC, conducted by CIMAB; |
| j) | Phase III randomized trial of nimotuzumab + radiation vs. radiation alone in 80 patients with high grade malignancy astrocytic tumors conducted by CIMAB. The study is designed to assess survival, local control and safety; |
| k) | Phase I/II trial in patients with head-and-neck cancers conducted by CIMAB enrolled 24 fully evaluable patients receiving nimotuzumab with radiation. This trial demonstrated a greater than 60% complete response rate compared to approximately 30% complete response rate expected with radiation alone; |
| l) | Phase I safety, PK/PD and MTD trial of nimotuzumab + chemotherapy in up to 24 patients with transarterial immunochemoembolization in hepatocellular carcinoma conducted by CIMAB; |
| m) | Phase I/II safety, PK/PD and MTD trial of nimotuzumab plus chemotherapy in up to 24 patients with liver metastasis from colorectal cancer conducted by CIMAB; |
| n) | Phase II study in pediatric patients with high grade gliomas conducted by Oncoscience. The trial demonstrated a 38% clinical benefit rate in 45 evaluable patients. This included two partial responses in a patient population where no such responses have ever been documented. |
- 23 -
| o) | Phase III study in which nimotuzumab and the current standard of care is compared to the current standard of care in patients with glioblastoma multiforme (GBM) conducted by Oncoscience. The primary end-point for this first-line trial is progression-free survival with response rate and symptom control among the secondary endpoints. |
| p) | Phase II/III study in patients with locally advanced or metastatic pancreatic cancer who will be treated with either gemcitabine plus nimotuzumab or gemcitabine plus placebo conducted by Oncoscience and CIMYM. The primary end-points for this trial are time to tumour progression and overall survival with quality of life and response rate among the secondary endpoints. We anticipate we will extend the European trial in pancreatic cancer into Canada by submitting the protocol to Canadian health regulatory authorities and adding Canadian sites to accelerate recruitment. |
| q) | Phase II pivotal trial conducted by CIMAB and Biotech Pharmaceuticals Limited (China) assessing efficacy and safety of nimotuzumab combined with radiation compared to radiation alone in locally advanced Stage III-IV nasopharyngeal carcinoma, a subset of head-and-neck cancer. Of the 130 patients in the intent-to-treat analysis, those in the combination arm had a 90.6% complete response rate compared to 51.5% in the radiation-alone group; |
| r) | Phase I pharmaco-dynamic study by CIMYM in patients with solid tumors. The study is investigating EGFR-related signaling in tumor and skin biopsies before and after treatment with 100, 200, 400 and 800 mg doses of nimotuzumab. Of the 16 evaluable patients enrolled in the trial, confirmed stable disease was reported in six patients, a confirmed partial response in one patient and prolonged progression-free status observed in three patients; |
| s) | Phase I/II trial in Canada and Korea in patients with stage IIB, III and IV NSCLC, who are not sufficiently fit to be able to tolerate the standard chemotherapy regimen, conducted by CIMYM. Study designed to assess safety response rate and survival in these patients. |
| t) | Phase III single arm trial of nimotuzumab in combination with radiation in children newly diagnosed with diffuse brain stem glioma. The trial is conducted by Oncoscience in several European countries and intended to serve as a registration trial in EMEA countries; |
| u) | A Phase II randomized 4-arm safety and efficacy trial in 92 patients with head-and-neck cancers comparing nimotuzumab + chemotherapy + radiation to chemotherapy + radiation and nimotuzumab + radiation to + radiation, conducted by Biocon Biopharm Pvt. Ltd.; |
| v) | Phase I clinical trial of nimotuzumab monotherapy for the treatment of solid tumours conducted by Daiichi Sankyo Inc in Japan. The study will enroll a maximum of 20 patients with various solid tumours and will evaluate safety of nimotuzumab in Japanese population. |
| w) | Phase II trial investigating nimotuzumab in pediatric patients with recurrent diffuse intrinsic pontine glioma (DIPG), a form of inoperable, treatment-resistant brain cancer. This study is being conducted by YM BioSciences USA Inc and will include eight leading US pediatric clinical centers. The single-arm trial will enrol 44 patients with DIPG who will be treated with nimotuzumab as monotherapy. |
| x) | Phase II trial investigating nimotuzumab in colorectal cancer patients who have failed previous irinotecan-containing regimens. The single-arm trial conducted by CIMYM will enroll approximately 100 patients in Canada who will be treated with irinotecan plus nimotuzumab. Equal cohorts investigating two dosing schedules will be enrolled. |
Several more trials are in planning stage but have not yet received regulatory approval.
In July 2004, nimotuzumab was designated an orphan drug by EMEA.
- 24 -
In November 2004, nimotuzumab was designated an orphan drug by the FDA in the United States.
In April 2005, we were advised that nimotuzumab was approved for sale in China.
In May 2005, study results on 24 evaluable adult patients treated with antibody and radiation for high-grade malignant gliomas were presented at the 2005 Annual Meeting of ASCO. Reported complete response was achieved by 16.7% and partial response by 20.8% of patients. 66.8% of patients were reported to have achieved disease stabilization for a total 87.5% clinical benefit rate.
In August 2005, Health Canada approved a Clinical Trial Application (CTA) for a multi-center Phase I/II trial with nimotuzumab. The randomized NSCLC trial will compare the effects of the combination of nimotuzumab with radiation against radiation alone in patients with stage IIB, III and IV disease, who are not sufficiently fit to be able to tolerate the standard chemotherapy regimen. The lead-in dose escalation part of this study was initiated in Canada and has been extended to Korea, where YM’s licensee Kuhnil Pharmaceutical Co. will fund the development of nimotuzumab for this territory. The randomized phase of the trial is expected to enroll approximately 100 patients and complete recruitment in approximately 18 months after initiation. The endpoint of the Phase II trial is an increase in local tumor control. First patient of the lead-in part of this study was enrolled in March 2006.
In March 2006, the FDA approved the use of nimotuzumab as a monotherapy in the treatment of a child with advanced glioma under an investigator-initiated IND. A total of eight investigator-initiated or compassionate use patients have been treated up to the date of this form.
In June 2006, we were advised that India’s health regulatory body, The Drug Control General, had granted initial marketing approval to nimotuzumab for the treatment of head-and-neck cancers. Biocon Biopharmaceuticals Ltd, a joint venture between Biocon (NSE:BIOCON) and CIMAB, has rights to the drug from CIMAB for the Indian sub-continent.
In September 2006, YM USA received a Special License from OFAC for the importation of nimotuzumab for a US trial in pediatric pontine glioma.
In June 2007, final data for the Phase II trial of nimotuzumab as a monotherapy in children with high-grade glioma, were presented at the 2007 Annual Meeting of ASCO. Of 45 evaluable patients with various forms of glioma at the end of the six week induction phase, 21 were reported to have diffuse intrinsic pontine glioma (DIPG). Eight of the DIPG patients were reported to have clinical benefit (1 PR, 7 SD). Those eight patients continued on to maintenance treatment and were reassessed at week 21 at which point three patients were reported to have PR and one SD.
In June 2007, Oncoscience reported that final data at the 2007 Annual Meeting of ASCO Phase II trial of nimotuzumab in children with high-grade glioma, as a monotherapy achieved its clinical endpoint and that six of 17 evaluable patients (35.3%) demonstrated a clinical benefit. Data from that trial were presented at the European High-Grade Glioma Meeting in February 2005. The primary endpoint is progression-free survival.
In July 2007, we initiated a trial in colorectal cancer in Canada. The single-arm trial will enroll approximately 100 patients in Canada. The trial is designed to enroll two 50-patient cohorts consecutively, with the first cohort receiving irinotecan on one of the conventional dosing schedules with weekly dosing of nimotuzumab and the second cohort receiving irinotecan on one of the conventional dosing schedules with nimotuzumab every two weeks.
In August 2007, Oncoscience announced completion of enrollment in a Phase III single arm trial of nimotuzumab in combination with radiation in children with diffuse pontine glioma initiated in March 2006. The clinical trial was also reviewed by the EMEA and is designed as a prospectively registrable study on the basis of a single arm trial, because of the absence of treatment options for children suffering from pontine glioma. It is anticipated that marketing authorization subsequent to a successful trial would be sought under the EMEA centralized procedure. The trial enrolled 40 children with diffuse pontine glioma who will be treated concomitantly with radiation and nimotuzumab. The primary clinical endpoints in the trial will be Progression-Free Survival at six months with Median Survival as secondary endpoint. Clinical sites were located in Germany, Italy, Belarus and Russia.
- 25 -
In August 2007, Oncoscience advises completion of recruitment. Based on the historical median survival for this form of cancer of approximately 8.5 months, the trial could be completed in the third quarter of 2008.
In August 2007, Oncoscience advised that it had initiated a Phase IIa/IIIb trial in patients with metastatic pancreatic cancer and a Phase III trial in adult glioma patients.
In August 2007, the FDA cleared an IND for a Phase II clinical trial for the treatment with nimotuzumab of children with progressive pediatric pontine glioma.
In September 2007, preliminary data on the use of nimotuzumab in NSCLC was contained in a poster at the 12th World Conference on Lung Cancer. Preliminary results were as follows: of the six patients enrolled in the 1st cohort (100mg), four partial responses and two stable diseases were reported. Median overall survival of the group was 41.5 weeks. Of the seven patients enrolled in the second cohort (200mg) of the study, two partial responses and five stable diseases were reported. Median overall survival of the group has not been reached but as at September 4th, 2007 exceeded 25 weeks.
Manufacturing:
Currently, CIMAB supplies nimotuzumab in quantities sufficient to facilitate the clinical development of these products. The license agreement with CIMAB requires that CIMAB will manufacture and supply, or will contract for the manufacture and supply of, commercial quantities of nimotuzumab in accordance with the then-current licensing agreements at such time and stage of product development as commercial quantities of these products are required. There is a risk that CIMAB may experience difficulties obtaining or producing commercially viable quantities of these products. Product from CIMAB's manufacturing plant has been approved for use in a clinical trial in Canada and Europe. The plant operates according to GMP principles and its cGMP compliance status has been reviewed on behalf of the Corporation by industry experts and was inspected and approved by the Paul Erlich Institute that is responsible for approvals of biological manufacturing facilities in Germany. The approval covers 300, 500 and 1000 litre ferementers. The manufacturing facility has to continue to meet GMP standards in order to supply product for commercial use. In 1999, CIMAB’s process of 500 litres was accepted by Health Canada for commercial scale manufacturing.
Our license agreement for nimotuzumab contemplates manufacturing of the product by CIMAB or a supplier contracted by CIMAB. Should CIMAB agree to alternative manufacturing arrangements, such as a sub-licensee of CIMYM manufacturing the product, the loss of manufacturing benefits to CIMAB may be reflected in a lower license fee and royalty payable to CIMYM than if manufacturing remains with CIMAB. See "Business - Licensing Arrangements".
Marketing:
Nimotuzumab is licensed by us from a Cuban source, CIMAB, and as such is likely to be prohibited from sale in the United States unless OFAC issues a license or the US embargo against Cuba is lifted. YM USA has received a Special License from OFAC to the importation of nimotuzumab for a clinical trial that has been cleared by the FDA to proceed.
Intellectual Property:
CIMYM is the exclusive licensee for the major market territories, including the United States. The patent estate includes coverage for the composition of matter, claiming the amino acid sequence of the nimotuzumab and variants thereof, and end-uses in the treatment of EGFr-dependent cancers. These patents are granted in the United States, Europe, Canada, and Japan. The patents US 5,891,966 and US 6,506,883 expire November 2015.
- 26 -
We are aware of the patent US 5,770,195, a patent granted to Genentech, for the anti-cancer use antibodies directed toward the her1 receptor in combination with cytotoxic factors. We are also aware of other US patents granted to others in this field. In April 2001, Rhone-Poulenc Rorer International ("Rorer") was issued US patent 6,217,866 claiming any antibody targeting the EGFr administered in combination with any anti-neoplastic agent. A counterpart application was granted in Europe. We have filed an opposition to the grant of the European patent citing prior art and other factors related to the lack of written description for the claimed subject matter. The opposition proceedings have been suspended pending the outcome of court cases filed by Yeda Research and Development Corp. in multiple jurisdictions claiming ownership of the Rorer patent. A September 19, 2006 decision in the United States District Court of Southern New York granted sole inventorship of the patent to scientists from Weizmann Institute of Science (Rehovet, Israel) represented by Yeda. Yeda now has the right to grant non-exclusive licenses to patent 6,217,866 in the United States. We are aware of national patent applications filed by Imclone claiming the use of tyrosine kinase receptor inhibitors in combination with radiation. The European application EP1080113 was recently withdrawn due to prior art. We plan on taking appropriate actions in additional jurisdictions as needed. Imclone has also filed patent applications claiming the use of EGFr antibodies to treat refractory tumors. We are monitoring these applications and will take the appropriate actions as needed. The outcome of these challenges cannot be predicted, and there can be no assurance that we will succeed in challenging the validity or scope of patent claims by ImClone or any other patent application.
We are aware of certain patents and patent applications related to the manufacture of humanized monoclonal antibodies owned by Genentech, Protein Design Lab, and Medical Research Council. Several of these patents expire prior to our anticipated market entry. In addition, certain of these process patents are currently being challenged by companies other than us. In the event that any of the applicable product by process claims are upheld, we believe that non-exclusive licenses under such patents can be obtained on commercially reasonable terms, though there can be no assurance of this.
Competitive Position:
To our knowledge, other companies that are involved in the development of monoclonal antibody cancer therapeutics directly related to our efforts include Amgen, Genmab, ImClone/BMS, and Merck KGaA.
We understand that OSI, in concert with Genentech and Roche, and AstraZeneca, have small molecules designed to target the tyrosine kinase domains of EGF receptors. We understand that Iressa(R), from AstraZeneca, has been approved in thirty-five countries, including Japan and the United States for third line monotherapy of NSCLC. OSI reported that it has positive survival data in a Phase III monotherapy study in refractory lung cancer. TARCEVA® monotherapy is now approved for the treatment of patients with locally advanced or metastatic NSCLC. In addition, TARCEVA® in combination with gemcitabine is approved for the first-line treatment of patients with locally advanced, unresectable or metastatic pancreatic cancer.
OSI's product, Tarceva®, is in co-development with Roche and Genentech and is reported to be in numerous trials in various indications, including Phase III trials in ovarian cancer, colorectal cancer, and NSCLC (1st line and adjuvant). TarcevaTM has been approved in the United States for NSCLC. See "-- Competition".
Erbitux(R), developed by ImClone/BMS and Merck, is approved in the United States, Canada, Germany, Austria and Switzerland for metastatic colorectal cancer in combination with irinotecan in irinotecan-refractory patients. Management understands that Erbitux(R) is under review by other regulatory agencies including EMEA for this and additional indications.
- 27 -
AeroLEFTM
Background:
AeroLEFTM is a proprietary formulation of fentanyl, an opioid analgesic, that is administered by inhalation and permits self-titration by patients. The development of AeroLEFTM as a combination of pulmonarily-delivered free and liposomal dosage form takes advantage of (1) the lung’s large absorptive surface and thin barrier to absorption to permit rapid transport of the free fentanyl fraction (loading dose) into the systemic circulation and (2) the capacity of liposomes to function as reservoirs for the regulated release over time of the encapsulated fentanyl. AeroLEFTM is being developed to provide both rapid and extended opioid analgesic levels for patients with severe and moderate acute pain and breakthrough cancer pain.
Clinical Experience:
The clinical experience with AeroLEFTM includes two Phase I studies with healthy volunteers, one completed Phase II acute pain study in post-surgical patients carried out in Canada, one Phase IIb randomized trial in 120 patients (99 randomized, 21 open label) under IND submissions approved by Health Canada and clearance by the FDA in June 2007 to initiate a Phase II trial in the US.
The preliminary results of a Phase II acute pain study using AeroLEFTM as the primary postoperative analgesic were reported at the annual meeting of the American Society of Anesthesiologists in October 2004. The study involved a unique “dose-to-effective-analgesia” following patient-controlled administration of AeroLEFTM as the primary analgesic treatment in adult patients experiencing severe and moderate pain following elective orthopedic surgery. This Phase II study demonstrated that 95% of patients successfully achieved analgesia via self-titration with AeroLEFTM as the primary medication. Eighteen (18) subjects rapidly achieved perceptible analgesia soon after commencing nebulization (median 2.7 min) and continued self-titration to a median time to effective/adequate analgesia of 17 minutes.
On August 30, 2005 YM received permission from Health Canada to initiate a randomized Phase IIb study with AeroLEF, the first patient was recruited in January 2006, and the final patient in March 2007. The trial succeeded in meeting its primary endpoint - the Summed Pain Relief plus Pain Intensity Difference (SPRID).
Manufacturing:
The AeroLEFTM formulation is manufactured through a controlled process that has been developed to produce a targeted ratio of liposome encapsulated fentanyl along with free fentanyl. AeroLEFTM clinical product supply for the two Phase I studies and the Phase IIa study was produced by a manufacturer based in the Netherlands. During 2004, the manufacturing process was transferred to a company located in Ontario, Canada. AeroLEFTM drug product produced at the Canadian manufacturer was used to support the Phase IIb trial, and is expected to be used for future clinical trials and other development activities. As AeroLEFTM advances to Phase III trials, we are seeking a second manufacturing source in the United States to provide pivotal trial material and future commercial supply. Fentanyl, the active pharmaceutical ingredient in the AeroLEFTM formulation, is commercially available from multiple vendors holding Drug Master Files (DMF) with the FDA, and licensed to synthesize controlled substances.
Intellectual Property:
The AeroLEFTM product is described in four patent families. We own key patents, expiring in 2014, claiming a method of administering systemic analgesia by inhaling free and liposome-encapsulated opioid analgesic. North American coverage includes a reissued US patent and a granted patent in Canada. We own two US applications with counterpart PCT applications, expiring in 2024, claiming the formulation for use in a method comprised of continuously inhaling the formulation to deposit at least one rapid-onset opioid and one sustained-effect opioid in the lungs to avoid side effects. A pending PCT application entitled “Stable Compositions” claims the manufacturing methods related to liposomal composition and other physical characteristics.
- 28 -
We are aware of US patents owned by Phares Pharmaceutical Research NV related to liposome compositions. These patents expire in 2008 and are not expected to hamper our commercial activities.
Competitive Position:
The opioid analgesic market which includes products based on morphine, fentanyl, oxycodone, or hydromorphone is currently dominated by several pharmaceuticals companies such as Johnson & Johnson Inc., Abbott Laboratories, Baxter International Inc., AstraZeneca PLC, Purdue Pharma L.P., Cephalon, Inc., and Endo Pharmaceutical Holdings Inc. The fentanyl segment of the opioid analgesic market includes three approved routes of administration:
| (a) | intravenous administration of fentanyl citrate available as generic products from various suppliers |
| (b) | transdermal administration via the Duragesic® patch and Ionsys® patch |
| (c) | transmucosal administration via the Actiq® lollipop and Fentora® |
Several competitors are developing non-invasive alternatives for enhanced delivery of fentanyl, including Endo (Rapinyl for sublingual delivery), BDSI (BEMA for buccal delivery), Archimedes (intranasal delivery), Sosei (sublingual spray), Alexza (pulmonary delivery) and Akela (pulmonary delivery). All the competitors, to our knowledge, will deliver fixed dosage forms of the drug.
The biopharmaceutical industry is intensely competitive. Many companies, including other biopharmaceutical companies and biotechnology companies, are actively engaged in activities similar to ours, including research and development of drugs for the treatment of cancer. More specifically, competitors for the development of new therapeutic products to treat cancer also focus on MAb-based cancer therapeutics, cancer vaccines and other approaches that are based on both active and passive immunotherapies and small molecule discovery and development. A 2001 survey by the Pharmaceutical Research and Manufacturers of America ("PhRMA") listed 399 new treatments for cancer that are currently being tested by researchers.
To our knowledge, other companies that are involved in the development of monoclonal antibody cancer therapeutics directly related to our efforts include Amgen, Genmab, ImClone, and Merck. We understand that OSI in concert with Genentech and Roche and AstraZeneca have small molecules designed to target the tyrosine kinase domains of EGF receptors. Iressa(R) has been approved in twenty countries, including Japan and the United States for third line monotherapy of NSCLC. OSI reported that is has positive survival data in a phase III monotherapy study in treatment refractory NSCLC. TarcevaTM has been approved in the United States for NSCLC. Erbitux(R) is approved in United States, Austria, Canada, Germany and Switzerland for metastatic colorectal in combination with irinotecan in irinotecan-refractory patients. Erbitux(R) is under review by other regulatory agencies including EMEA the European regulatory agency.
Several competitors are developing non-invasive alternatives for enhanced delivery of fentanyl, including Endo (Rapinyl for sublingual delivery), BDSI (BEMA for buccal delivery), Archimedes (intranasal delivery), Sosei (sublingual spray), Alexza (pulmonary delivery) and Akela (pulmonary delivery). All the competitors, to our knowledge, will deliver fixed dosage forms of the drug.
We expect to encounter significant competition for the pharmaceutical products we are developing and plan to develop in future. Many of our competitors have substantially greater financial and other resources, larger research and development capabilities and more extensive marketing and manufacturing organizations than we have. In addition, some such companies have considerable experience in pre-clinical testing, clinical trials and other regulatory approval procedures. There are also academic institutions, governmental agencies and other research organizations which are conducting research in areas in which we are working and they may also market commercial products, either on their own or through collaborative efforts. If any of these competitors were to complete clinical trials, obtain required regulatory approvals and commence commercial sales of their products before us they may achieve a significant competitive advantage.
- 29 -
Norelin TM and Products in Preclinical Development from the University of Saskatchewan
During fiscal 2007 licenses for Norelin and the preclinical products licensed from the University of Saskatchewan were returned to the University.
Tesmilifene
Background:
Tesmilifene is a small molecule drug with multiple modes of action that appears to enhance the activity of traditional chemotherapy agents. Its chemical designation is N,N-diethyl-2-[4-(phenylmethyl)phenoxy]ethanamine hydrochloride. It has demonstrated synergistic effects with anthracyclines in late-stage clinical trials and with taxanes, 5-FU vinca alkaloids and platins in earlier-stage clinical and pre-clinical studies.
Clinical Experience:
On January 30, 2007 we announced that the independent DSMB for the pivotal Phase III trial of tesmilifene in patients with metastatic or recurrent breast cancer completed its third planned safety and efficacy analysis. The DSMB advised the Company to stop the trial based on an interim analysis of 351 events, indicating it was very unlikely significant differences in overall survival would be shown between treatment arms as the data matured. The trial was not stopped due to safety concerns relating to the product. We plan to submit data from this trial to an appropriate medical meeting after we complete our review.
The pivotal Phase III trial compared the survival of patients treated with tesmilifene combined with epirubicin/cyclophosphamide to epirubicin/cyclophosphamide alone in women with rapidly progressing metastatic and/or recurrent breast cancer. The study, which completed enrollment of 723 patients in September, 2005, was the subject of a Special Protocol Assessment and a Fast Track designation for advanced breast cancer by the FDA.
The trial was conducted according to a sequential design that permitted a number of planned interim analyses while the trial continued until one of two specific statistical conditions was satisfied. At each analysis, survival for the tesmilifene-containing treatment arm and the control arm was calculated and then reviewed by the DSMB. The trial was to be concluded if either the tesmilifene-containing treatment arm was superior to the control by a specified margin or it was determined that such evidence was not going to be found. After the third planned analysis, the DSMB concluded that the trial was highly unlikely to achieve a pre-specified survival benefit.
In March 2004, we entered into a Clinical Research Services Agreement with Pharm-Olam International (“POI”), a clinical research organization ("CRO"), to conduct this Phase III trial internationally. POI has received the majority of its entitlement under the contract and immaterial amounts remain payable on completion of certain remaining reports.
In January 2005, we formed a joint development team with Shin Poong Pharmaceutical Company of Seoul, Korea (“Shin Poong”) to oversee the expansion of the development program for tesmilifene into gastric cancer with the clinical failure of tesmilifene it is expected that this expansion program will terminate.
In January 2006 we entered into a collaborative agreement with Sanofi-Aventis to test tesmilifene together with Taxotere® in women with aggressive metastatic breast cancer. Thirty-three patients have been recruited completing enrollment; pharmcokinetics data are anticipated by calendar year-end 2007 and overall survival data by calendar year-end 2008.
Intellectual Property:
We obtained an exclusive license to patent rights covering tesmilifene from the University of Manitoba. Aspects of tesmilifene, including its anti-cancer and cytoprotective uses, are the subject of patents that have issued in the United States, Europe, Japan, Canada and Australia.
- 30 -
The patent estate comprises numerous layers of patent protection. A key patent among these is US Patent No. 5,859,065 claiming the use of tesmilifene, including certain structural analogs, in combination with any chemotherapeutic agents for the treatment of any cancer. The twenty year term of this patent expires in 2010. Still other issued patents US 6,284,799 and 5,747,543, expire in 2014 and 2015 respectively. It is anticipated that tesmilifene will qualify for patent term extension under the Patent Term Restoration Act which could provide additional protection of up to five years. We intend to take full advantage of the available term extension.
In addition, international patent applications are pending based upon our clinical development program. This series of patent applications focused on the survival advantage demonstrated following the analysis of the earlier phase III trail and relates to the selection of patient populations that will most benefit from the chemopotentiating and cytoprotective properties of tesmilifene. Patents that result from these filings should expires in 2022 in the US and other major markets.
In addition to patents, we intend to rely on the available term of data exclusivity in the US and other countries given that tesmilifene qualifies as a NCE. Furthermore, full advantage will be taken of the Orange Book provisions in the United States and equivalent provision in Canada and other countries, as a means for delaying generic competition.
Clinical, Pre-Clinical And Basic Research
We design, fund and manage clinical and some pre-clinical research, and may support, but do not conduct, basic research. We manage the development of products that we in-license through our own team of clinical, regulatory, licensing and business development executives and through a number of research and medical collaborations. We are responsible for filing applications with the relevant authorities for regulatory approval for clinical trials and conducts, or has conducted on our behalf, clinical trials to progress products in development toward regulatory approval and possible out-licensing for commercial sale. Our current licenses generally provide that we will conduct, or cause to be conducted, the tests and clinical studies necessary to progress products in development toward regulatory approval with a view to obtaining the approval for sale of the licensed drug from appropriate regulatory authorities. We have received regulatory approvals for clinical trials in a number of countries, including in Canada, the United States, the United Kingdom, Europe and South Africa from Phase I through Phase III. Some basic research is conducted at the facilities of our licensors, and we pay for certain amounts of this research.
Licensing Arrangements
In-Licensing
Licenses For Nimotuzumab
In May 1995, YM acquired an exclusive, sub-licensable license (as amended, the "1995 CIMYM License") from CIMAB, acting on behalf of CIM, to products for passive immunotherapy of cancer directed toward EGF and EGFr as targets, including hR3, a humanized MAb targeting the EGFr. CIMAB is the company responsible for the commercialization of products developed at CIM. The 1995 CIMYM License is in respect of Europe, Canada, the United States, Japan, Australia, Taiwan, Singapore, Thailand, Hong Kong, South Korea, Malaysia, Indonesia and the Philippines. As a term of the 1995 CIMYM License, YM has a right of first refusal with respect to licensing any other products derived from the EGF and EGFr programs of CIMAB except its anti-EGFr monoclonal antibody for psoriasis in Europe.
Pursuant to the 1995 CIMYM License, in 1995 we incorporated CIMYM and assigned the 1995 CIMYM License to CIMYM. Pursuant to the terms of the 1995 CIMYM License, CIMAB acquired a 20% equity interest in CIMYM as partial consideration for the 1995 CIMYM License. In addition to that 20% equity interest in CIMYM, CIMAB is entitled to receive 10% of net revenues received by CIMYM. In addition, YM and CIMYM, pursuant to the terms of the 1995 CIMYM License, paid US$2,750,000 for certain product development costs for nimotuzumab and US$330,000 for certain product development costs for RadioTheraCIM.
- 31 -
The terms of the 1995 CIMYM License provide for CIMYM to conduct or cause to be conducted pre-clinical and clinical trials to evaluate the licensed products and to work with CIMAB to select sites, develop protocols and instruct investigators for pre-clinical and clinical trials. CIMYM is to decide after the end of each stage of trials whether to proceed with further development or to terminate the 1995 CIMYM License with respect to that product. In addition, the 1995 CIMYM License provides that, where commercially reasonable, CIMYM shall file applications for regulatory approval to market the licensed products in the applicable territory. Pursuant to the 1995 CIMYM License, CIMAB has the right, subject to certain terms and conditions, to supply the related drug substances (i.e., nimotuzumab) for commercial sale. CIMAB shall sell the product manufactured by it in Cuba to CIMYM at 85% of the sales price that CIMYM sets for the sale of the product to sub-licensees, thereby entitling CIMYM to the 15% difference. CIMYM shall use its best efforts to obtain a sub-license agreement in which CIMAB retains the right to manufacture the product. YM will be responsible for any failure of CIMYM to fulfill its obligations under the 1995 CIMYM License. This license agreement shall be in force as long as any patents thereunder are valid, or until such time as the license agreement is terminated by either party because of a default by the other party, or by CIMYM with written notice within 90 days after the end of a stage of pre-clinical trials or after each stage of clinical trials.
As at September 30, 2004 we had advanced US$24 million to CIMYM and CIMYM (Barbados), collectively, for the licensing and development of the products licensed by CIMYM. We were given the right to recover all funds advanced to CIMYM and CIMYM (Barbados), collectively, from either CIMYM and CIMYM (Barbados). To the extent that the net revenues of CIMYM are less than or equal to the advanced amounts, we are only permitted to recover such advances from 30% of the net revenues. At this time none of the advances have been repaid. There have been no revenues to date.
On June 30, 2006 CIMYM amalgamated with CIMYM (Barbados) to form CIMYM BioSciences Inc., an Ontario company. CIMAB owns a 20% equity interest in CIMYM BioSciences and is entitled to receive 10% of net revenues received by CIMYM.
Licenses For AeroLEFTM
The technology related to AeroLEFTM formulation and delivery is owned not licensed.
License For Tesmilifene
In November 2000, YM was granted an exclusive worldwide license by the University of Manitoba and The Manitoba Cancer Treatment and Research Foundation (now CancerCare Manitoba) (the "Original Licensor") for all products and formulations of tesmilifene pursuant to which the Corporation undertook the responsibility for the clinical development of the product and its commercialization.
We must pay to the Original Licensor a specified minority percentage of revenues received from sub-licensing the product, after our recovery of certain specified development and attributed overhead costs. If we manufacture and sell tesmilifene itself rather than through sub-licensing, we must pay a specified lesser minority percentage of net sales, after our recovery of certain specified development and attributed overhead costs, to the Original Licensor. We believe these royalties are consistent with general industry practice for similar arrangements. No royalties have been paid to date, and future royalties cannot be quantified because they are dependent on net sales, net royalties and net revenues which have not yet materialized. There can be no assurance as to if or when we may sell the licensed product nor enter into sub-licensing arrangements for the product. Under the terms of this license agreement, we have paid US$300,000 over the years 2000, 2001 and 2002 for sponsored research. We must make reasonable efforts to ensure that the licensed product is efficiently marketed and distributed by November 2005. We may sub-license the product. This license agreement shall be in force as long as any patents thereunder are valid, or until such time as the license agreement is terminated by either party because of a default by the other party, by either party if the other party enters into liquidation or reorganization proceedings or receivership or bankruptcy, or by YM on 90 days written notice if there are no sub-licensees. In 2003, we acquired certain additional patent rights related to a method of selecting patients demonstrating an enhanced survival benefit. Vincent Research and Consulting transferred assignment of the patent applications in exchange for a small share of YM's future royalty revenues. We do not consider the agreement with Vincent Research and Consulting to be material to us as of the date hereof.
- 32 -
Out-Licensing
We generally plan to out-license our licensed drugs to pharmaceutical companies for manufacturing and marketing under license, although we may retain co-development or marketing rights if management considers it appropriate to do so. Under our business model, licensees would be expected, to the extent necessary, to participate in the remaining clinical development required to obtain final regulatory approval for the product. We expect that out-licensing would result in a pharmaceutical company or other licensee marketing or manufacturing the product in return for licensing fees and royalties on the sale of the product. We believe this model is consistent with current biotechnology and pharmaceutical industry licensing practices.
Our objectives in seeking to out-license products include:
| o | obtaining long term revenue streams from royalty payments on the sale of the products; |
| o | providing access to the resources and experience of large pharmaceutical companies; |
| o | obtaining up-front payments for product sub-licensing rights; and |
| o | minimizing development expenditures through cost sharing programmes (especially late-stage clinical trials and regulatory approval applications). |
We believe that out-licensing arrangements could be attractive to pharmaceutical corporations because they would provide the prospective partner with access to new products without the initial research risk or earlier clinical development costs. Since partners are expected to be sought only at the later stages of a product's development, we anticipate that prospective licensees would view our products as having a reduced risk of failure to achieve regulatory approval.
YM does not intend to develop our own manufacturing, marketing or distribution programs although we may wish to participate in ownership of manufacturing facilities if appropriate opportunities become available. We intend to remain principally focused on the identification, further development and commercialization of in-licensed products.
Nimotuzumab
On November 12, 2003, our subsidiary, CIMYM (now CIMYM BioSciences), licensed the rights for nimotuzumab (known as “Theraloc” in Europe) in most of Europe to Oncoscience of Germany. Under the terms of the agreement, CIMYM BioSciences is entitled to receive up to US$30 million as a share of any amounts received by Oncoscience in relation to development or sublicensing of the product and as a royalty on initial net sales. After CIMYM BioSciences has received US$30 million, CIMYM BioSciences continues to receives royalties on net sales but at a lesser percentage. Oncoscience has agreed to use diligent and reasonable efforts to develop and commercially exploit nimotuzumab in the licensed territory. No amounts or royalties have been received as of the date hereof by CIMYM BioSciences from Oncoscience, since no sublicensing fees or net sales amounts have been received by Oncoscience. This license agreement may be terminated by either party in the event of specified breaches and insolvency events, if a Phase II trial of nimotuzumab has not commenced in Europe within two years of licensing, or if certain regulatory approvals for marketing nimotuzumab in Europe have not been obtained within five years. In addition, Oncoscience may terminate the agreement at any time on 90 days notice.
In May 2005, we licensed the rights to nimotuzumab in South Korea to Kuhnil. They formed a joint development team to oversee the expansion of the development program for nimotuzumab, its monoclonal antibody against the EGF receptor, for a specific population of patients with non-small cell lung cancer. Kuhnil will fully fund development costs and will provide up-front, milestone and royalty payments.
On October 31, 2005, IGK, a subsidiary of P.T. Kalbe Farma TBK of Jakarta Indonesia paid CIMYM BioSciences Inc. a license fee of US $1,000,000. The agreement also entitles the CIMYM BioSciences Inc. to receive milestone payments on the occurrence of regulatory approval and royalties on the commercial sale of the developed product.
- 33 -
In July 2006, CIMYM BioSciences licensed development and marketing rights for nimotuzumab in Japan to Daiichi Pharmaceutical Co., Ltd., a wholly owned subsidiary of Daiichi Sankyo Co., Ltd., one of Japan’s largest pharmaceutical companies. Under the agreement, CIMYM BioSciences will receive an up-front payment of US$14.5 million and significant milestone payments at certain states of development for each of a number of indications as well as payments based on supply of nimotuzumab and sales performance in the territory. Daiichi will develop nimotuzumab for the Japanese market in several cancer indications.
Regulatory Approval
Securing final regulatory approval for the manufacture and sale of human therapeutic products in Canada and our other territories, including the United States, is a long and costly process that is controlled by that particular territory's national regulatory agency. The national regulatory agency in Canada is Health Canada, and in the United States it is the FDA. Other national regulatory agencies have similar regulatory approval processes, but each national regulatory agency has its own approval processes. Approval in either Canada or the United States does not assure approval by other national regulatory agencies, although often test results from one country may be used in applications for regulatory approval in another country.
Prior to obtaining final regulatory approval to market a drug product, every national regulatory agency has a variety of statutes and regulations which govern the principal development activities. These laws require controlled research and testing of products, government review and approval of a submission containing pre-clinical and clinical data establishing the safety and efficacy of the product for each use sought, approval of manufacturing facilities including adherence to GMP during production and storage, and control of marketing activities, including advertising and labeling.
None of our products has been completely developed or tested and, therefore, we are not yet in a position to seek final regulatory approval to market any of our in-licensed products. To date we have obtained various regulatory approvals to develop and test our in-licensed products. Currently we are conducting an international Phase III trial of tesmilifene in metastatic and recurrent breast cancer in 700 patients. We have received regulatory approvals for the tesmilifene study in several countries, including Canada and the United States, and approval is pending in a few other countries. In addition, nimotuzumab has been designated an orphan drug in Europe and by the FDA in the United States. See "Products in Clinical Development".
Canadian Approval Process
The manufacture, distribution and consumption of medical products, drugs and equipment is regulated by a variety of industry-specific statutes and regulations in Canada and the countries to which YM has rights for the licensed products. Drugs sold in Canada are regulated by the Food and Drugs Act (Canada) and the regulations made under that Act.
Even though a drug, medical product or device may be approved for use in another jurisdiction, it may not be sold in Canada until approved by Health Canada. Outside Canada, the regulatory approval process for the manufacture and sale of pharmaceuticals varies from country to country and the time required may be longer or shorter than that required by Health Canada.
The Canadian Food and Drug Regulations require licensing of manufacturing facilities, carefully controlled research and testing of products, governmental review and approval of test results prior to marketing of therapeutic products, and adherence to GMP, as defined by each licensing jurisdiction, during production.
The principal activities which must be completed prior to obtaining approval for marketing of a therapeutic drug product are essentially the same in Canada as in most major markets of the world and are as follows:
Pre-clinical Animal Studies. Pre-clinical studies are conducted in animals to test pharmacology and toxicology and to do formulation work based on in vivo results.
- 34 -
Phase I Clinical Trials. Phase I clinical trials consist of testing a product in a small number of humans for its safety (toxicity), dose tolerance and pharmacokinetic properties.
Phase II Clinical Trials. Phase II clinical trials usually involve a larger patient population than is required for Phase I trials and are conducted to evaluate the efficacy of a product in patients having the disease or medical condition for which the product is indicated. These trials also serve to further identify side effects and risks in a larger group of patients.
Phase III Clinical Trials. Phase III clinical trials involve “conducting tests in an expanded patient population at geographically dispersed test sites (multi-center trials) in a controlled and/or uncontrolled environment to gather information about clinical safety and efficacy.” These trials also generate information from which the overall benefit-risk relationship of the drug can be determined and provide a basis for drug labeling.
Two key factors influencing the progression of clinical trials are the rate at which patients can be recruited into clinical trials and whether effective treatments are currently available for the disease the drug is intended to treat. Patient recruitment is largely dependent upon the incidence and severity of the disease and the alternative treatments available, as well as alternate research studies.
A Clinical Trial Application must be filed and accepted by either the Therapeutic Products Directorate ("TPD") or the Biologics and Genetic Therapies Directorate ("BGTD") of Health Canada before each phase of human clinical trials may begin. The CTA application must contain specified information including the results of the pre-clinical or clinical tests completed at the time of the CTA application. In addition, since the method of manufacture may affect the efficacy and safety of a drug, information on chemistry and manufacturing methods must be presented. Health Canada conducts inspections to determine compliance with GMP. Good manufacturing practices and quality control procedures must be in place.
Upon completion of all clinical studies, the results are submitted to the TPD or BGTD as part of a New Drug Submission ("NDS"). A notice of compliance ("NOC") which permits marketing of the product typically takes between 12 and 24 months from the date a NDS is submitted.
Even after marketing approval has been obtained, further studies may be required to provide additional data on safety and efficacy in order to gain approval for the use of a drug as a treatment for clinical indications other than those for which the product was initially tested. Also, Health Canada conducts post-market surveillance programmes to monitor a product's side effects. Results of post-marketing programmes may limit or expand the further marketing of products. A serious safety or efficacy problem involving an approved drug or medical device may result in Health Canada action requiring withdrawal of the product from the market.
United States Approval Process
In the United States, the FDA, a federal government agency, is responsible for the drug approval process. The FDA's mission is to ensure that all medications on the market are safe and are effective. The FDA's approval process examines potential drugs; only those that meet strict requirements are approved.
The U.S food and drug regulations require licensing of manufacturing facilities, carefully controlled research and testing of products, governmental review and approval of test results prior to marketing of therapeutic products, and adherence to GMP, as defined by each licensing jurisdiction, during production.
The drug approval process begins with the discovery of a potential drug. Pharmaceutical companies then test the drug extensively. A description of the different stages in the drug approval process in the US follows.
Stage 1: Preclinical Research After an experimental drug is discovered, research is conducted to help determine its potential for treating or curing an illness. This is called preclinical research. Animal studies are conducted to determine if there are any harmful effects of the drug and to help understand how the drug works. Information from these experiments is submitted to the FDA in an Investigational New Drug Application. The FDA reviews information in an IND Application and decides if the drug is safe to study in humans.
- 35 -
Stage 2: Clinical Research In Stage 2, the experimental drug is studied in humans. The studies are known as clinical trials. Clinical trials are carefully designed and controlled experiments in which the experimental drug is administered to patients to test its safety and to determine the effectiveness of an experimental drug. The four general phases of clinical research are described below.
Phase I Clinical Studies. Phase I clinical studies are generally conducted with healthy volunteers who are not taking other medicines; patients with the illness that the drug will treat are not tested at this stage. Ultimately, Phase I studies demonstrate how an experimental drug affects the body of a healthy individual. Phase I consists of a series of small studies consisting of "tens" of volunteers. Tests are done on each volunteer throughout the study to see how the person's body processes, responds to, and is affected by the drug. Low doses and high doses of the drug are usually studied, resulting in the determination of the safe dosage range in volunteers by the end of Phase I. This information will determine whether the drug proceeds to Phase II.
Phase II Clinical Studies. Phase II clinical studies are conducted in order to determine how an experimental drug affects people who have the disease to be treated. Phase II usually consists of a limited number of studies that help determine the drug's short-term safety, side effects, and general effectiveness. The studies in Phase II are often controlled investigations, involving comparison between the experimental drug and a placebo, or between the experimental drug and an existing drug. Information gathered in Phase II studies will determine whether the drug proceeds to Phase III.
Phase III Clinical Studies. Phase III clinical studies are “expanded controlled and uncontrolled trials that are used to more fully investigate the safety and effectiveness of the drug”(CFR). These trials differ from Phase II trials because a larger number of patients are studied (sometimes in the thousands) and because the studies are usually of longer duration. As well, Phase III studies can include patients who have more than one illness and are taking medications in addition to the experimental drug used in the study. Therefore, the patients in Phase III studies more closely reflect the general population. The information from Phase III forms the basis for most of the drug's initial labeling, which will guide physicians on how to use the drug.
Phase IV Clinical Studies. Phase IV clinical studies are conducted after a drug is approved. Companies often conduct Phase IV studies to more fully understand how their drug compares to other drugs. Also, the FDA may require additional studies after the drug is approved. FDA-required Phase IV studies often investigate the drug in specific types of patients that may not have been included in the Phase III studies. FDA-required Phase IV studies can also involve very large numbers of patients to further assess the drug's safety.
Stage 3: FDA Review and Approval Following Phase III, the pharmaceutical company prepares reports of all studies conducted on the drug and submits the reports to the FDA in a New Drug Application ("NDA"). The FDA reviews the information in the NDA to determine if the drug is safe and effective for its intended use. Occasionally, the FDA will ask experts for their opinion of the drug; this occurs at advisory committee meetings. If the FDA determines that the drug is safe and effective, the drug will be approved.
Stage 4: Marketing After the FDA has approved the experimental drug, the pharmaceutical company can make it available to physicians and their patients. A company may also continue to conduct research to discover new uses for the drug. Each time a new use for a drug is discovered, the drug is once again subject to the entire FDA approval process before it an be marketed for that purpose.
Arrangements With Subsidiaries
YM and CIMAB entered into a funding agreement with CIMYM in November 1995 in connection with the 1995 CIMYM License. The Funding Agreement provides that YM will arrange for the appropriate studies and clinical trials for the licensed products held by CIMYM and will fund the cost of such studies and trials, provided that doing so would not be commercially or scientifically unreasonable. Accordingly, YM makes the final determination as to whether or not a clinical trial expense is justified with respect to any given product. YM is entitled to be reimbursed for all funds we provide pursuant to the Funding Agreement out of revenue generated from the exploitation of the relevant license, subject to the successful development of the licensed products and adequate generation of revenue.
- 36 -
YM and CIMAB, contemporaneously with the assignment of the 1995 CIMYM License, entered into a joint-venture shareholders agreement (the "Shareholders Agreement") with CIMYM relating to its operation. Pursuant to the Shareholders Agreement, CIMYM is required to include nominees of CIMAB both as board members and as members of operating management. The Shareholder Agreement provides that: (i) issued and outstanding shares of CIMYM may not be sold or transferred without the consent of both YM and CIMAB; (ii) the issue of additional shares of CIMYM shall first be offered to each of YM and CIMAB in proportion to their holdings, and thereafter, with the consent of both YM and CIMAB, to any other person; and (iii) the boards of directors of CIMYM will consist of five directors, three of whom are nominees of YM and two of whom are nominees of CIMAB. All material and out-of-the-ordinary-course-of-business contracts of CIMYM, including those relating to the borrowing of money, issuing guarantees, entering into non arm's-length agreements, paying dividends and pledging of property, must be approved by four-fifths of the Board of Directors.
CIMYM (Barbados) was incorporated in Barbados in May 1996 to market the licensed products under the 1995 CIMYM License outside of Canada. On June 30, 2006, CIMYM (Barbados) was amalgamated with CIMYM to form CIMYM BioSciences Inc.
Property, Plants And Equipment
Facilities
We currently occupy 5,800 square feet of space in Mississauga, Ontario pursuant to a sub-lease agreement dated July 31, 1997 (the "Sub-Lease") and a lease amending and extension agreement dated February 1, 2003 (the "Lease Amending Agreement"), such Lease Amending Agreement extended the initial terms of the Sub-Lease for a term of five years commencing on February 1, 2003 and expiring on January 31, 2008. The average annual costs, including operating expenses, are approximately $120,000.
We also occupy 7,200 square feet of space at 6535 Millcreek Drive in Mississauga, Ontario pursuant to a lease amending and extension agreement dated February 28, 2007. The term of the lease is based on a month to month tenancy and expires on February 28, 2008. Either the lessor or lessee may terminate this lease at least 90 days written notice. The annual cost, including operating expenses is approximately $71,000 under this lease.
YM USA currently occupies 9,000 square feet of space in Wayne, Pennsylvania pursuant to a lease agreement dated July 27, 2006 and expiring on February 15, 2012. Average annual fixed rent, excluding operating costs is approximately US$228,000.
There are no environmental issues associated with any of our facilities and we currently have no plans to construct, expand or improve our facilities.
Equipment And Other Property
As at June 30, 2007, we owned tangible fixed assets with a book value of approximately $325,000 consisting primarily of office and lab equipment.
Employees
As of June 30, 2007, we employed 37 permanent employees. 26 employees are currently located in Mississauga, Ontario and 11 employees are in Wayne, Pennsylvania. Other than administrative staff, our employees conduct our licensing and product development activities.
DIVIDENDS
We have not paid any dividends since its incorporation. We will consider paying dividends in future as our operational circumstances may permit having regard to, among other things, our earnings, cash flow and financial requirements. It is the current policy of our Board of Directors to retain all earnings to finance our business plan.
- 37 -
CAPITAL STRUCTURE
Our authorized share capital consists of 500,000,000 common shares without nominal or par value, 500,000,000 Class A non-voting common shares without nominal or par value, 500,000,000 Class A preferred shares without nominal or par value and 500,000,000 Class B preferred shares, issuable in series, without nominal or par value. As at June 30, 2007, there were 58,216,309 common shares (including 2,380,953 common shares held in escrow for contingent additional payment related to the acquisition of Delex), no Class A non-voting common shares and no preferred shares outstanding.
Common Shares
All of the common shares rank equally as to voting rights, participation in a distribution of our assets on a liquidation, dissolution or winding-up and the entitlement to dividends. The holders of the common shares are entitled to receive notice of all meetings of shareholders and to attend and vote the common shares at the meetings. Each common share carries with it the right to one vote.
In the event of the liquidation, dissolution or winding-up of our company, the holders of our common shares will be entitled, subject to the rights, privileges, restrictions and conditions attaching to any other class of shares of the Corporation, to receive, on a pro rata basis, share for share, with the Class A non-voting common shares, all of our remaining property. There are no pre-emptive or conversion rights and no provisions for redemption, retraction, purchase for cancellation or surrender or sinking or purchase funds.
Stock Option Plan
We have a stock option plan (the “Plan”) pursuant to which stock options to purchase our common shares (“Options”) may be granted to eligible persons, including officers, directors, employees and service providers of the Corporation. The aggregate number of common shares reserved for issuance upon the exercise of all Options granted under the Plan is a “rolling” amount which, under the current Plan, may not exceed 12.5% of the issued and outstanding common shares of the Corporation as at the date of each grant. We consider the granting of stock options important to attract and maintain the most qualified directors, senior management and other employees in the Canadian and US healthcare technology industry. In our view, the ability to grant stock options is an important means of compensating such persons for their contributions to the Corporation’s overall financial performance. The number of Common Shares that may be reserved for issuance to our insiders (as defined in the Securities Act (Ontario)) and any affiliate or subsidiary thereof (collectively, “Insiders”) pursuant to the Plan, may not exceed 10% of the then outstanding issue. In any one-year period, options that may be granted to any Insider, and such Insider’s associates, shall not exceed 5% of the outstanding issue. Terms and vesting provisions in respect of the Options may be decided at the date of grant but, unless otherwise specified, Options are exercisable for a period of 10 years from the date of grant and vest over a period of five years, at 20% per year. Recently, we made certain house-keeping amendments to the Plan in order to permit “cashless exercise” by Optionholders.
MARKET FOR SECURITIES
Our common shares have been listed on the Toronto Stock Exchange (“TSX”) and the Alternative Investment Market (“AIM”) operated by the London Stock Exchange since June 11, 2002. Initially, we listed our Class B Preferred Shares, Series 1 on those stock exchanges. On June 12, 2003, the Class B Preferred Shares, Series 1, were automatically converted on a one-for-one basis into our common shares, which became listed on the TSX and AIM on that date.
Our common shares have traded on the TSX since June 12, 2003 under the symbol “YM”, were admitted to trading on the AIM on June 12, 2003 under the symbol “YMBA” and have been traded on the American Stock and Options Exchange (“AMEX”) since October 1, 2004 under the symbol “YMI”.
The following table shows the high and low sale prices and the trading volume of our common shares on the TSX on a monthly basis during fiscal 2007:
- 38 -
Price Range | |||
| High | Low | Volume | |
| June 2006 | 5.86 | 3.90 | 1,350,186 |
| July 2006 | 4.40 | 3.40 | 768,379 |
| August 2006 | 4.09 | 2.91 | 1,995,022 |
| September 2006 | 4.01 | 3.47 | 1,302,722 |
| October 2006 | 3.81 | 2.98 | 1,148,992 |
| November 2006 | 3.90 | 3.26 | 1,026,694 |
| December 2006 | 3.50 | 3.11 | 1,431,491 |
| January 2007 | 4.88 | 1.84 | 10,566,222 |
| February 2007 | 2.13 | 1.63 | 3,578,901 |
| March 2007 | 2.04 | 1.55 | 1,546,035 |
| April 2007 | 2.39 | 1.80 | 1,970,243 |
| May 2007 | 2.23 | 1.77 | 1,347,370 |
| June 2007 | 2.25 | 1.84 | 1,795,473 |
The closing price of our common shares on the TSX on June 30, 2007 was $1.91 per share and, as at September 21, 2007, was $1.62 per share.
DIRECTORS AND OFFICERS
Name | Position | Period Served |
David G.P. Allan Toronto, Canada | Chairman, Chief Executive Officer and Director | Since 1994 |
Thomas I.A. Allen (1)(2)(3) Toronto, Canada | Director | Since 1996 |
James Barrett(3) Germantown, USA | Director | Since May 2006 |
Lisa DeLuca Berwyn, USA | Officer | Since May 2006 |
Mark Entwistle (3) Ottawa, Canada | Director | Since 1997 |
Gary Floyd Berwyn, USA | Officer | Since May 2006 |
John Friedman(3) New York, USA | Director | Since 2004 |
Henry Friesen (1) Winnipeg, Manitoba | Director | Since 2001 |
Paul M. Keane Mississauga, Ontario | Officer | Since 1996 |
Diana Pliura Mississauga, Ontario | Officer | Since 2005 |
Vincent Salvatori Victoria, British Columbia | Officer | Since 2002 |
Leonard Vernon Nobleton, Ontario | Officer | Since 1997 |
Julius Vida (2) Greenwich, USA | Director | Since 2001 |
Gilbert Wenzel(1) Zurich, Switzerland | Director | Since 2001 |
Tryon M. Williams (1)(2) London, England | Director | Since 1995 |
(1) Member of Audit Committee.
(2) Member of Corporate Governance and Nominating Committee.
(3) Member of Compensation Committee.
- 39 -
Share Ownership Of Directors And Executive Officers
The following table sets out details of our shares and options that are directly or indirectly owned or controlled by directors and executive officers as at June 30, 2007, based on 58,216,309 common shares issued and outstanding on such date.
Name | Number of Common Shares | Percentage of Common Shares Outstanding | Common Shares Held Under Option | Exercise Price | Expiration Date |
| David G.P. Allan | 789,6591 | 1.2% | 900,000 | $1.75 - $4.50 | 2007 - 2016 |
| Thomas I.A. Allen | - | - | 73,160 | $1.75 - $4.50 | 2007 - 2016 |
| James Barrett | 1,349,130 2 | 2.4%2 | 0 | ||
| Lisa DeLuca | - | - | 70,000 | US$ 5.74 | 2016 |
| Mark Entwistle | - | - | 98,160 | $1.75 - $4.50 | 2007 - 2016 |
| Gary Floyd | - | - | 70,000 | US$ 5.74 | 2016 |
| John Friedman | 294,999 | * | 67,500 | $2.10 - $3.61 | 2014 - 2016 |
| Henry Friesen | - | - | 98,160 | $1.75 - $4.50 | 2011 - 2016 |
| Paul M. Keane | 72,500 | * | 128,100 | $1.75 - $4.50 | 2007 - 2016 |
| Diana Pliura | - | - | 300,000 | $3.15 - $4.36 | 2015 - 2016 |
| Vincent Salvatori | - | - | 220,000 | $1.75 - $4.36 | 2008 - 2016 |
| Gail Schulze | - | - | 266,667 | US$5.74 | 2008 |
| Igor Sherman | - | - | 70,000 | $2.75 - $4.36 | 2014 - 2016 |
| Sean Thompson | 6,500 | * | 165,375 | $1.75 - $4.50 | 2010 - 2016 |
| Leonard Vernon | - | - | 243,000 | $1.75 - $4.50 | 2008 - 2016 |
| Julius Vida | - | - | 93,160 | $1.75 - $4.50 | 2011 - 2016 |
| Gilbert Wenzel | - | - | 93,160 | $1.75 - $4.50 | 2011 - 2016 |
| Tryon M. Williams | 20,100 | * | 138,098 | $1.75 - $4.50 | 2010 - 2016 |
* Less than one percent
Note 1: Of such shares, 80,000 common shares are held through private holding companies over which Mr. Allan exercises direction and control.
Note 2: Shares are owned by New Enterprise Associates 11, Limited Partnership. Dr. Barrett is a manager of NEA 11 GP, LLC which is the general partner of NEA Partners 11, Limited Partnership which is the general partner of New Enterprise Associates 11, Limited Partnership.
- 40 -
As of the date of hereof, the directors and senior officers of YM BioSciences as a group beneficially owned or controlled, directly or indirectly, 2,604,888 common shares of YM, representing approximately 4.477% of the issued and outstanding voting shares of the Corporation.
David G.P. Allan - Chairman, Chief Executive Officer And Director
Mr. Allan has been Chief Executive Officer of the Corporation since April 1998 and Chairman of the board of directors of the Corporation since 1994. In addition, Mr. Allan is a Director of Synthemed Inc. (medical devices) USA and DiaMedica Inc. (diabetes therapeutics) Canada. In 1992 he founded the Knowledge-Based Industries Group of a Canadian investment banking firm, organized as the first in Canada involved in financing, analyzing and creating strategic alliances for life sciences and information technology companies, where he was Executive Director until 1998. Mr. Allan was formerly a governor of The Toronto Stock Exchange, a member, and working group Chair, of the Ontario Biotechnology Advisory Board, the Awards Selection Committee for the Networks of Centres of Excellence in Canada and of the Board of Trustees for the Ontario College of Art and Design. Mr. Allan is currently a member of BioteCanada’s Emerging Companies Advisory Board.
Thomas I.A. Allen, Q.C., F.C.I.Arb - Director
Mr. Allen is counsel to Ogilvy Renault, a Canadian law firm, and Chairman of Westwind Capital Corporation, the parent of an institutional investment dealer with offices in Canada and England. Mr. Allen was the initial Chairman of the Accounting Standards Oversight Council of Canada and was a member of the Advisory Board of the Office of the Superintendent of Financial Institutions of Canada. He is currently a director of a number of public companies including Finavera Renewables Inc., Terra Nova Gold Inc., Mundoro Mining Inc., Middlefield Bancorp Limited and Longview Capital Partners Incorporated. Mr. Allen recently actied as Chairman of the Task Force to Modernize Securities Legislation in Canada.
James Barrett - Director
Dr. Barrett is a General Partner at New Enterprise Associates, a leading venture capital firm, where he specializes in biotechnology and other healthcare investments. Dr. Barrett is a director of GlycoMimetics, Inc., Inhibitex, Inc., Iomai Corporation, MedImmune, Inc., Peptimmune, Pharmion, Inc. and Targacept, Inc.
Lisa Deluca - Vice-President Of Global Regulatory Affairs
Dr. DeLuca has more than 15 years of experience in the pharmaceutical industry, including her roles as Vice President of Regulatory Affairs at Eximias (joining in 2004) and Director of Regulatory Affairs at AstraZeneca, from 1999 to 2004, where she was responsible for the regulatory launch of IRESSA(R). She has also held regulatory positions at Centocor and Wyeth (1993-1999) and drug discovery positions at SmithKline Beecham (now GlaxoSmithKline) and The Upjohn Company (now Pfizer, Inc.) (1990-1993). Dr. DeLuca received a PhD in biochemistry from the University of Toledo in 1988.
- 41 -
Mark Entwistle, M.A. - Director
Prior to founding his own consulting practice in 1997 in international trade, political business intelligence and strategic communications, Mr. Entwistle was an Ambassador for Canada in the Caribbean from 1993 to 1997. Mr. Entwistle was previously a career diplomat with the Canadian Department of Foreign Affairs and International Trade in a variety of embassy positions from 1982 to 1997, and served as Press Secretary and Director of Communications to the Prime Minister of Canada from 1991-1993. He is a Fellow of the Canadian Defence and Foreign Affairs Institute. Mr. Entwistle has been a director of the Corporation since October 1997.
Gary Floyd - Vice-President Of Operations
Mr. Floyd is a seasoned executive with an extensive background and 36 years of experience in the global pharmaceutical industry. Prior to joining Eximias in February 2005, Mr. Floyd served as President of Pharmavene LLC, a pharmaceutical consulting firm. Prior to this, he was Senior Vice President, Worldwide Logistics and Procurement at Aventis Behring where he was responsible for Supply Chain Management of the company’s global business. He has also held senior positions in manufacturing, information systems, and sales and marketing at Aventis Behring and its predecessor companies. Mr. Floyd holds a Bachelors degree in Zoology and Chemistry from Olivet Nazarene University
John Friedman - Director
Mr. Friedman launched the Easton Capital Group (“Easton”) in 1993, with Easton Capital Corporation. In 1999, Easton Hunt Capital Partners was added to the Group. Prior to Easton, Mr. Friedman was a founder of Atrium Capital Corporation, which he helped manage from 1991-1993, and also the founder and Managing General Partner of Security Pacific Capital Investors from 1989 through 1991. Security Pacific Capital Investors was a $200-million private equity fund geared towards expansion financings and recapitalizations. Prior to joining Security Pacific, Mr. Friedman was a Managing Director and Partner at E.M. Warburg, Pincus & Co., Inc., where he spent eight and a half years from 1981-1989. Prior thereto, he worked at Shearson Loeb Rhoades and was an attorney with Sullivan and Cromwell from 1978 through 1980. He holds a JD degree from Yale Law School and a BA degree from Yale College. Mr. Friedman currently serves on the Boards of Renovis, Trellis Bioscience, and Assistive Technology, and is on the President's Council at the Cold Spring Harbor Laboratory. Mr. Friedman has been a director of the Corporation since April 2004.
Henry Friesen, C.C., M.D., F.R.S.C. - Director
Dr. Friesen currently serves as Chair of the Gairdner Foundation whose international awards are Canada's most prestigious prizes in the biomedical sciences. He was most recently Chair, Genome Canada, 2000-2005; a $600 million budget non-profit organization that supports genomics/proteomics programs to position Canada as a world leader in selected areas in this important sector. From 1991 to 2000 Dr. Friesen was President of the Medical Research Council of Canada and was instrumental in transforming it into the Canadian Institute of Health Research, an organization with an annual budget of over $650 million per year dedicated to supporting Canadian researchers as well as industry participants. Dr. Friesen is noted for his discoveries about the human hormone prolactin and as Head of the Department of Physiology and Distinguished Professor Emeritus of Medicine at the University of Manitoba. Dr. Friesen is a Fellow of the Royal Society of Canada, a Companion of the Order of Canada and also sits on the board of directors of Sanofi Pasteur Canada and Spectral Diagnostics Inc. Dr. Friesen has been a director of the Corporation since November 2001.
Paul M. Keane, M.D., F.R.C.P.C., F.A.C.P., F.R.C. Path - Director, Medical Affairs
Dr. Keane has been an officer of the Corporation since January 1996. Dr. Keane was Director of Clinical Research at Miles Canada Inc. (now Bayer Canada) from 1989 to 1995, prior to which he was Professor of Medicine at University of Calgary and Professor of Pathology at McMaster University. Dr. Keane has authored numerous scientific publications in peer review journals, has acted as a reviewer of research proposals for the Medical Research Council of Canada and has acted in an editorial capacity for a number of scientific journals.
- 42 -
Diana Pliura, Ph.D. - Executive Vice President, AeroLEF™
Dr. Pliura has over 25 years of experience in the pharmaceutical, biopharmaceutical and venture capital sectors. Since June 2001, she has served as the founding President and CEO of DELEX Therapeutics. From 1998 through 2001, she served as Company Creator for the Eastern Technology Seed Investment Fund, responsible for assessment of biotechnology investment opportunities and serving in executive management roles for selected investments. In 1997, she founded a consulting firm focused on providing scientific and management expertise to biotechnology companies and their investors. Previously Dr. Pliura was a Scientific Advisor to MDS Health Ventures from 1996 to 1997 and Vice-President Research at Hemosol Inc. from 1992 through 1995. Prior to that, she held various positions at Syntex (Canada) Inc. from 1981 to 1991 including serving as Head of the Division of General Biochemistry in the drug discovery unit from 1989-1991. Dr. Pliura is a co-inventor or co-author on numerous patents and peer-reviewed articles. She received her doctoral degree in Chemistry from the University of Toronto and completed post-doctoral training at Harvard University.
Vincent Salvatori, Ph.D. - Executive Vice President, President And CEO, CIMYM Biosciences
Dr. Salvatori has been an officer of the Corporation since December 2002. Dr. Salvatori is an experienced drug development executive with an accomplished background in the pharmaceutical and biotechnology industry. He has more than 24 years of experience in all aspects of drug development, corporate operations and external collaborations. Dr. Salvatori most recently held the position of Senior Vice President of Clinical Operations for Bioniche Life Sciences Inc. from May 1998 to July 2002. He was previously at StressGen Biotechnologies Corporation from January 1995 to April 1998 where he held the positions of Chief Operating Officer and Vice President of Research and Development, subsequently appointed to Senior Vice President. In this capacity, Dr. Salvatori was responsible for corporate operations, strategic management and clinical/regulatory development. Prior to joining StressGen, Dr. Salvatori was the Senior Director of Program Management at QLT PhotoTherapeutics Inc. from June 1990 to December 1994 and held various positions at Boehringer Ingelheim (Canada) Ltd. from April 1982 to June 1990.
Gail Schulze - Director
Ms. Schulze has more than 25 years of pharmaceutical industry experience. She was Chief Executive Officer of Eximas Pharmaceutical Corporation from 2005 until its acquisition by YM in May 2006. Thereafter she served as President of YM and Chief Executive Officer of YM (USA) until February 2007. Prior to joining Eximias, she was with Aventis Behring, one of the world’s leading pharmaceutical companies, from 1997 to 2004. She joined Aventis Behring as Senior Executive Vice President and Chief Marketing Officer in 1997 and became COO of one of its joint ventures in 2001. Ms. Schulze received a BS in Psychobiology from the University of California, was an NIH Fellow in Neurophysiology at the University of Wisconsin and received an MBA from Stanford’s Graduate School of Business.
Sean Thompson, B.Sc. - Vice President, Corporate Development
Mr. Thompson is responsible for all aspects of corporate development for YM, including licensing, identifying targets for M&A and development partnerships. He has a solid background in clinical development and big Pharma marketing experience, and was previously Director of Clinical Research for YM. Prior to YM he worked for Roche, sitting on the Global Business Team in virology, and was Clinical Research Manager. Prior to Roche, he was a Clinical Scientist for SmithKline Beecham developing products for oncology, respiratory and CNS disorders. He is currently a member of the Ontario Cancer Research Networks’ Industry Advisory Board and a Board member for Variation Biotechnologies Inc. Mr. Thompson holds a Bachelor of Science degree from the University of Waterloo.
- 43 -
Leonard Vernon, B.Sc., C.A. - Vice-President, Finance And Administration
Mr. Vernon earned a B.Sc. in 1968 and was awarded his C.A. in 1972 with Clarkson Gordon & Co. (now Ernst & Young llp). He has held senior financial positions with a number of organizations both public and private. Prior to joining YM as an officer in July 1997, Mr. Vernon was an independent consultant working with senior management in a variety of industries. Prior to 1992 he was Vice-President, Finance and Administration of Unitel Inc., now Allstream Inc., a major Canadian telecommunications company.
Julius Vida, Ph.D., M.B.A. - Director
Dr. Vida has been the President of Vida International Pharmaceutical Consultants, a consulting firm advising pharmaceutical and biotechnology companies, since 1993. Previously Dr. Vida was Director of Licensing and subsequently Vice President, Business Development, Licensing and Strategic Planning at Bristol-Myers Squibb, from 1975 to 1993. Dr. Vida is a director of a number of biotechnology firms including Medarex, Inc., FibroGen, Inc., Spectrum Pharmaceuticals Inc. and Chemical Synthesis Services Albachem (UK). Dr. Vida has been a director of the Corporation since September 2001.
Gilbert Wenzel, Ph.D. - Director
Dr. Wenzel is currently Chairman of QUISISANA HOLDING AG and Chief Executive Officer, a pharma company focused on importing, registering and commercializing generics for private label use by pharmacy chains and insurance companies in Europe.. Prior to founding Quisisana in January 2003, Dr. Wenzel joined Novartis Group, a global pharmaceutical manufacturer, in November 2000 where he served as Head of Strategic Planning and a Member of its Executive Committee until January 2003. Prior to joining Novartis in November 2000, Dr. Wenzel spent 15 years with McKinsey & Co., an international management consulting firm, and was a member of the European Leadership Group of its Pharma/Healthcare Sector and of the European New Venture Initiative. From 1981 to 1985, Dr. Wenzel was at Hoechst AG in Germany and developed global strategies for generics and over-the-counter medicines. Dr. Wenzel has been a director of the Corporation since March 2001.
Tryon M. Williams, B.Sc. (Math) - Director
Mr. Williams is the Chairman, CEO and director of CellStop Systems, Inc., an automobile security device manufacturer, and CEO and director of Bingo.com, Ltd., an internet technology company. Since 1993, Mr. Williams has been Adjunct Professor, Sauder School of Business, The University of British Columbia. Mr. Williams is also a director of several other private corporations. Mr. Williams has been a director of the Corporation since November 1995.
Clinical And Scientific Advisory Board
We maintain a Clinical and Scientific Advisory Board (“CSAB”) composed of internationally recognized clinicians and scientists. Management meets with members of the CSAB periodically to review operational aspects of our clinical and scientific programme and make recommendations with regard to the perceived trends and direction of medical and biopharmaceutical technologies and the industry generally. Each member of the CSAB has signed a confidentiality agreement with us. CSAB members receive honoraria paid by us of varying amounts per year. The current composition of the CSAB is as follows:
Lorne J. Brandes, B.Sc., M.D., C.R.C.P.C.
Professor, Departments of Medicine and Pharmacology/Therapeutics, University of Manitoba, Winnipeg, Manitoba, Canada; Section of Hematology/Oncology, CancerCare Manitoba, Winnipeg, Manitoba, Canada. Dr. Brandes has been an advisor since November 2000.
- 44 -
Robert S. Kerbel, Ph.D.
Professor of Medical Biophysics, University of Toronto, Toronto, Ontario, Canada; Canada Research Chair in Molecular Medicine; Director, Molecular and Cell Biology Research, Sunnybrook and Women's College Health Science Centre, Toronto, Ontario, Canada. Dr. Kerbel has been an advisor since April 1999.
Leonard Saltz, M.D
Professor of Medicine, Weill College of Medicine, Cornell University, New York, New York, New York; Member of Memorial Sloan-Kettering Cancer Center, New York, New York, United States; Attending Physician, Memorial Hospital for Cancer and Allied Diseases, New York, New York, United States. Dr. Saltz has been an advisor since March, 2006.
Niclas Stiernholm, Ph.D.
Chief Executive Officer, Trillium Therapeutics Inc., Toronto, Ontario, Canada. Dr. Stiernholm was an executive vice-president of the Corporation until he resigned in December 2002 at which time he became an advisor.
Mark Vincent, M.D., M.R.C.P., F.R.C.P.C. - Chair
Associate Professor, Department of Oncology, University of Western Ontario, London, Ontario, Canada; Staff Medical Oncologist, London Regional Cancer Centre, London, Ontario, Canada. Dr. Vincent has been an advisor since October 1998.
Daniel D. Von Hoff, M.D., F.A.C.P.
Professor of Medicine, University of Arizona and Executive Vice President, Translational Genomics Research Institute and Director, Translational Drug Development Program, Tucson, Arizona, United States. Dr. Von Hoff has been an advisor since July 2001.
Audit Committee
The members of the Corporation's Audit Committee are Thomas I.A. Allen, Henry Friesen, Tryon M. Williams, and Gilbert Wenzel.
The principal functions of the Audit Committee are to appoint, compensate and oversee the external auditors; to review and approve annual and quarterly financial statements and all legally required continuous and public disclosure documents containing financial information about the Corporation before they are submitted to the Board of Directors for approval; to review and approve the adequacy of internal accounting controls and the quality of financial reporting procedures and systems; to examine the presentation and impact of key financial and other significant risks that may be material to the Corporation's financial reporting; and to review and approve the nature and scope of the annual audit and review the results of the external auditor's examination. The Audit Committee reports its findings with respect to such matters to the Board of Directors.
Corporate Governance And Nominating Committee
The members of the Corporation's Corporate Governance and Nominating Committee are Thomas I.A. Allen, Julius Vida and Tryon M. Williams.
The mandate of the Corporation's Corporate Governance and Nominating Committee is to develop and monitor the Corporation's system of corporate governance in the context of the Toronto Stock Exchange Report on Corporate Governance, and the rules and regulations promulgated by the Ontario Securities Commission and the Securities and Exchange Commission, including reviewing the mandate of the Board of Directors and its committees; periodically reviewing and evaluating the performance of all directors, committees and the Board as a whole; selecting new candidates for Board memberships, making recommendations to the Board and ensuring that appropriate orientation and education programmes are available for new Board members; establishing procedures to ensure that the Board may meet independent of Management and reviewing annually the membership and chairs of all committees. All members of this Committee are unrelated and non-executive directors of the Corporation.
- 45 -
Compensation Committee
The members of the Corporation's Compensation Committee are Thomas I.A. Allen, James Barrett, John Friedman and Mark Entwistle.
The mandate of the Compensation Committee is to establish and monitor our policies for attracting, retaining, developing and motivating senior employees. The compensation policies are designed to support our strategic objectives, ensure that incentive programmes are designed to motivate senior managers to achieve or exceed corporate objectives and to enhance shareholder value and to ensure that there is reasonable consistency in the application of the compensation policies. The Committee's responsibilities include reviewing annually the performance of the Chief Executive Officer (or more frequently if deemed necessary by the Compensation Committee), setting the Chief Executive Officer's compensation and, in consultation with the Chief Executive Officer, establishing his personal objectives, reviewing the performance and approving the compensation of executive officers of the Corporation on the recommendation of the Chief Executive Officer, establishing incentive compensation programmes and monitoring their effectiveness and developing and documenting the compensation policy and philosophy of the Corporation for approval by the Board of Directors. All members of this Committee are unrelated and non-executive directors of the Corporation.
AUDIT FEES
During the years ended June 30, 2006 and 2005, we were billed the following fees by our external auditors, KPMG LLP:
Fees Incurred | ||
Service | 2007 | 2006 |
| Audit and Audit-Related Fees | $260,000 | $420,000 |
| Tax Fees | $82,200 | $50,600 |
| All Other Fees | Nil | Nil |
Total Fees Paid | 342,200 | $470,600 |
The Board of Directors have established a written mandate for the audit committee, a copy of which is attached hereto as Schedule “A”. The Audit Committee follows the policies and procedures for the pre-approval of services to be provided by our external auditors set out in the mandate.
LEGAL PROCEEDINGS
We are not a party to any material pending legal or arbitration proceedings and is not aware of any material contemplated legal proceedings to which we may be a party.
TRANSFER AGENT AND REGISTRAR
The registrar and transfer agent for our common shares in Canada is CIBC Mellon Trust Company at its principal offices in Toronto, Canada and in the United States is Mellon Investor Services LLC at its principal offices in Ridgefield Park, New Jersey.
MATERIAL CONTRACTS
Except for contracts entered into in the ordinary course of business, the only material contract which we entered into prior to the date hereof is anEscrow Agreement dated as of May 2, 2005 among YM, the Business Development Bank of Canada, New Generation Biotech (Equity) Fund Inc., Eastern Technology Seed Investment Fund Limited Partnership and Equity Transfer Services Inc., pursuant to which certain common shares of YM are held in escrow for the benefit of the former DELEX shareholders to be released in tranches over time and upon completion of certain milestones. See “General Development of the Business”.
- 46 -
In the ordinary course of our business, we enter into licenses for products which we develop and out-license. The licenses for these products are more fully described in this annual information form under the heading “Business Overview - Licensing Arrangements”.
INTERESTS OF EXPERTS
KPMG LLP, the Corporation’s external auditor, has reported on the consolidated financial statements of the Corporation for each of the years in the three-year period ended June 30, 2007. KPMG LLP is independent of the Corporation in accordance with the applicable Rules of Professional Conduct/Code of Ethics of the Institute of Chartered Accountants of Ontario, and within the meaning of the Securities Acts administered by the United States Securities and Exchange Commission and the requirements of the Independence Standards Board.
ADDITIONAL INFORMATION
Additional information, including directors’ remuneration and indebtedness, principal holders of the Corporation’s securities, options to purchase securities and interests of insiders in material transactions, if any, is contained in the Corporation’s information circular for its most recent annual meeting of shareholders that involved the election of directors and that additional financial information is provided in the Corporation’s comparative financial statements for its most recently completed year.
When securities of the Corporation are in the course of distribution pursuant to a short form prospectus, or when a preliminary short form prospectus has been filed in respect of the Corporation’s securities, the Corporation will provide the following documents to any person or company upon request to the Corporate Secretary of the Corporation:
| 1. | a copy of this annual information form, together with a copy of any document or the pertinent pages of any document incorporated by reference in this annual information form; |
| 2. | a copy of our Financial Statements, together with the accompanying auditors’ report as well as copies of any subsequent interim financial statements that we have filed; |
| 3. | a copy of our information circular in respect of our most recent annual meeting of shareholders that involved the election of directors; and |
| 4. | a copy of any other document that is incorporated by reference into the preliminary short form prospectus or the short form prospectus. |
At any other time, a copy of the documents referred to in subsections 1, 2, 3 and 4 above may be obtained from our Corporate Secretary, however, a reasonable fee may be charged if the request is made by a person or company who is not a shareholder of YM. These documents and additional information may also be found at www.sedar.com.
All requests for the above-mentioned documents must be addressed to:
YM BioSciences Inc.
5045 Orbitor Drive
Building 11, Suite 400
Mississauga, Ontario
L4W 4Y4
- 47 -
Attention: Secretary
Telephone: (905) 629-9761
Fax: (905) 629-4959
e-mail: ir@ymbiosciences.com
Web Page: www. ymbiosciences.com
- 48 -
Schedule “A”
YM BIOSCIENCES INC.
AUDIT COMMITTEE MANDATE
1. General
The board of directors (the “Board”) of YM BioSciences Inc. (the “Corporation”) has delegated the responsibilities, authorities and duties described below to the audit committee (the “Audit Committee”). For the purpose of these terms of reference, the term “Corporation” shall include the Corporation and its subsidiaries.
The Audit Committee shall be directly responsible for overseeing the accounting and financial reporting processes of the Corporation and audits of the financial statements of the Corporation, and the Audit Committee shall be directly responsible for the appointment, compensation, and oversight of the work of any registered external auditor employed by the Corporation (including resolution of disagreements between management of the Corporation and the external auditor regarding financial reporting) for the purpose of preparing or issuing an audit report or related work. In so doing, the Audit Committee will comply with all applicable Canadian and United States securities laws, rules and guidelines, any applicable stock exchange requirements or guidelines and any other applicable regulatory rules.
2. Members
The Audit Committee shall be composed of a minimum of three members. Members of the Audit Committee shall be appointed by the Board. Each member shall serve until such member’s successor is appointed, unless that member resigns or is removed by the Board or otherwise ceases to be a director of the Corporation. The Board shall fill any vacancy if the membership of the Committee is less than three directors. The Chair of the Committee may be designated by the Board or, if it does not do so, the members of the Committee may elect a Chair by vote of a majority of the full Committee membership.
All members of the Audit Committee must satisfy the independence, financial literacy and experience requirements of applicable Canadian and United States securities laws, rules and guidelines, any applicable stock exchange requirements or guidelines and any other applicable regulatory rules. In particular:
| (a) | each member shall be “independent” and “financially literate” within the meaning of Multilateral Instrument 52-110 “Audit Committees”; |
| (b) | at least one member must be “financially sophisticated” under the rules of the American Stock Exchange; and |
| (c) | at least one member must be an “audit committee financial expert” within the meaning of that term under the United States Securities Exchange Act of 1934, as amended, and the rules adopted by the United States Securities and Exchange Commission thereunder. |
3. Meetings
The Audit Committee shall meet at least quarterly at such times and at such locations as the Chair of the Audit Committee shall determine, provided that meetings shall be scheduled so as to permit the timely review of the Corporation’s quarterly and annual financial statements and related management discussion and analysis. The external auditor or any two members of the Audit Committee may also request a meeting of the Audit Committee. The Chair of the Audit Committee shall hold in camera sessions of the Audit Committee, without management present, at every meeting.
- 49 -
The Audit Committee shall submit the minutes of all meetings to the Board, and when requested to, shall discuss the matters discussed at each Audit Committee meeting with the Board.
4. Committee Charter
The Audit Committee shall have a written charter that sets out its mandate and responsibilities and the Audit Committee shall review and reassess the adequacy of such charter at least annually or otherwise, as it deems appropriate, and propose recommended changes to the Board.
5. Duties of the Audit Committee:
The Audit Committee shall have the following duties:
Financial Information and Reporting
| 1. | The Audit Committee shall review with management and the external auditor, and recommend to the Board for approval, the annual and interim financial statements of the Corporation and related financial reporting, including management’s discussion and analysis and earnings press releases. |
| 2. | The Audit Committee shall review with management and the external auditor, and recommend to the Board for approval, any financial statements of the Corporation which have not previously been approved by the Board and which are to be included in a prospectus or other public disclosure document of the Corporation. |
| 3. | The Audit Committee shall consider and be satisfied that adequate policies and procedures are in place for the review of the Corporation’s disclosure of financial information extracted or derived from the Corporation’s financial statements (other than disclosure referred to in clause (a)(i) above), and periodically assess the adequacy of such procedures. |
Internal Controls
| 4. | The Audit Committee shall review, as appropriate, the Corporation’s internal system of audit controls and the results of internal audits. |
| 5. | The Audit Committee shall establish procedures for the receipt, retention and treatment of any complaint regarding accounting, internal accounting controls or auditing matters; and the confidential, anonymous submissions by employees of concerns regarding questionable accounting or auditing matters. |
External Auditors
| 6. | The Audit Committee shall be directly responsible for overseeing the work of the external auditor engaged for the purpose of preparing or issuing an auditor’s report or performing other audit, review or attest services for the Corporation, including the resolution of disagreements between management and the external auditor regarding financial reporting. |
| 7. | The external auditor shall report directly to the Audit Committee and the Audit Committee should have a clear understanding with the external auditor that such external auditor must maintain an open and transparent relationship with the Audit Committee, and that the ultimate accountability of the external auditor is to the shareholders of the Corporation. |
| 8. | The Audit Committee shall recommend to the Board the external auditor to be nominated for the purpose of preparing or issuing an auditor’s report or performing other audit, review or attest services for the Corporation; and the compensation of the external auditor. |
- 50 -
| 9. | The Audit Committee will ensure the rotation of partners on the audit engagement team of the external auditor in accordance with applicable law. |
| 10. | The Audit Committee shall meet with the external auditor, as the Audit Committee may deem appropriate, to consider any matter which the Audit Committee or external auditor believes should be brought to the attention of the Board or the shareholders of the Corporation. |
| 11. | The Audit Committee shall meet with the external auditor, as the Audit Committee may deem appropriate, to review and discuss a report from the external auditor at least quarterly regarding: |
| (a) | all critical accounting policies and practices to be used |
| (b) | all alternative treatments within generally accepted accounting principles for policies and practices related to material items that have been discussed with management, including the ramifications of the use of such alternative disclosures and treatments, and the treatment preferred by the external auditor, and |
| (c) | other material written communications between the external auditor and management, such as any management letter or schedule of unadjusted differences. |
Pre Approval of Non-Audit Services
| 12. | The Audit Committee shall pre-approve all non-audit services to be provided to the Corporation or its subsidiary entities by the Corporation’s external auditor. |
Complaints procedure
| 13. | The Audit Committee shall establish procedures for the receipt, retention and treatment of complaints received by the Corporation regarding accounting, internal accounting controls, or auditing matters; and the confidential, anonymous submission by employees of the Corporation of concerns regarding questionable accounting or auditing matters. |
| 14. | The Audit Committee shall review and approve the Corporation’s hiring policies regarding partners, employees and former partners and employees of the present and former external auditor of the Corporation. |
Reporting
| 15. | The Audit Committee shall report regularly to the Board about any issues that arise with respect to the quality or integrity of the Corporation’s financial statements, the Corporation’s compliance with legal or regulatory requirements, the performance and independence of the external auditor, or the internal audit function. |
6. Authority to engage independent counsel and advisors
The Audit Committee has the authority to engage independent counsel and other advisors as it determines necessary to carry out its duties, to set and pay the compensation for any advisors employed by the audit committee, and to communicate directly with the internal and external auditors.
The Corporation shall provide appropriate funding, as determined by the Audit Committee, in its capacity as a committee of the board of directors, for payment of compensation (a) to the external auditors employed by the issuer for the purpose of rendering or issuing an audit report, and (b) to any advisers employed by the Audit Committee.
- 51 -