UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
_____________________________________________________________________________________________
Form 10-K
_____________________________________________________________________________________________ | | | | | |
| ☑ | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2024
OR | | | | | |
| ☐ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the transition period from to
Commission File Number 001-36407
_____________________________________________________________________________________________
ALNYLAM PHARMACEUTICALS, INC.
(Exact Name of Registrant as Specified in Its Charter)
_____________________________________________________________________________________________ | | | | | | | | |
Delaware (State or Other Jurisdiction of Incorporation or Organization) | | 77-0602661 (I.R.S. Employer Identification No.) |
675 West Kendall Street, Henri A. Termeer Square Cambridge, MA 02142
(Address of Principal Executive Offices) (Zip Code)
Registrant’s telephone number, including area code: (617) 551-8200
Securities registered pursuant to Section 12(b) of the Act: | | | | | | | | | | | | | | |
| Title of Each Class | | Trading Symbol(s) | | Name of Each Exchange on Which Registered |
| Common Stock, $0.01 par value per share | | ALNY | The Nasdaq Stock Market LLC |
Securities registered pursuant to Section 12(g) of the Act: None
_____________________________________________________________________________________________
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ☑ No ☐
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ☐ No ☑
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ☑ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit such files). Yes ☑ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company,” and “emerging growth company” in Rule 12b-2 of the Exchange Act.
| | | | | | | | | | | | | | |
| Large accelerated filer | ☑ | | Accelerated filer | ☐ |
| Non-accelerated filer | ☐ | | Smaller reporting company | ☐ |
| | | Emerging growth company | ☐ |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any new or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. Yes ☑ No ☐
If securities are registered pursuant to Section 12(b) of the Act, indicate by check mark whether the financial statements of the registrant included in the filing reflect the correction of an error to previously issued financial statements. ☐
Indicate by check mark whether any of those error corrections are restatements that required a recovery analysis of incentive-
based compensation received by any of the registrant’s executive officers during the relevant recovery period pursuant to §240.10D-1(b). ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ☐ No ☑
The aggregate market value of the registrant’s common stock, $0.01 par value per share (“Common Stock”), held by non-affiliates of the registrant, based on the last sale price of the Common Stock at the close of business on June 30, 2024, was approximately $30.99 billion. For the purpose of the foregoing calculation only, all directors and executive officers of the registrant are assumed to be affiliates of the registrant.
At February 6, 2025, the registrant had 129,457,156 shares of Common Stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the registrant’s definitive proxy statement for its 2025 annual meeting of stockholders, which the registrant intends to file pursuant to Regulation 14A with the Securities and Exchange Commission not later than 120 days after the registrant’s fiscal year end of December 31, 2024, are incorporated by reference into Part II, Item 5 and Part III of this Form 10-K.
ALNYLAM PHARMACEUTICALS, INC.
ANNUAL REPORT ON FORM 10-K
For the Year Ended December 31, 2024
TABLE OF CONTENTS
“Alnylam,” ONPATTRO®, AMVUTTRA®, GIVLAARI®, OXLUMO®, Alnylam Act® and IKARIA™ are trademarks and registered trademarks of Alnylam Pharmaceuticals, Inc. Our logo, trademarks and service marks are property of Alnylam. All other trademarks or service marks appearing in this Annual Report on Form 10-K are the property of their respective holders.
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. We intend these forward-looking statements to be covered by the safe harbor provisions for forward-looking statements contained in the Private Securities Litigation Reform Act of 1995 and are including this statement for purposes of complying with those safe harbor provisions. All statements other than statements of historical fact contained in this Annual Report on Form 10-K are forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as “may,” “will,” “should,” “could,” “expects,” “plans,” “intends,” “anticipates,” “believes,” “estimates,” “predicts,” “potential,” “continue,” or the negative of these terms or other comparable terminology. These forward-looking statements include, but are not limited to, statements about:
•our views with respect to the potential for our approved and investigational RNAi therapeutics and those of our collaborators, including ONPATTRO, AMVUTTRA, GIVLAARI, OXLUMO, Leqvio® (inclisiran), fitusiran, zilebesiran, mivelsiran and nucresiran;
•our plans for additional global regulatory filings and the continuing product launches of ONPATTRO, AMVUTTRA, GIVLAARI, OXLUMO and our collaborators’ plans with respect to Leqvio and fitusiran;
•our ability to obtain regulatory approval of AMVUTTRA (vutrisiran) for the treatment of ATTR amyloidosis with cardiomyopathy in the United States, or U.S., and other jurisdictions;
•our expectations regarding the potential market size for, and the successful commercialization of, ONPATTRO, AMVUTTRA, GIVLAARI, OXLUMO, Leqvio, fitusiran or any future products;
•our ability to obtain and maintain regulatory approvals and pricing and reimbursement for ONPATTRO, AMVUTTRA, GIVLAARI, OXLUMO or any future products, and our collaborators’ ability with respect to Leqvio and fitusiran;
•the progress of our research and development programs, including programs across a broad range of disease areas and indications;
•the potential for improved product profiles to emerge from our technologies and our ability to expand our clinical development pipeline to include additional tissue types and disease indications;
•our current and anticipated clinical trials and expectations regarding the reporting of data from these trials;
•the number and timing of regulatory filings and interactions with, or actions or advice of, regulatory authorities, which may affect the design, initiation, timing, continuation and/or progress of clinical trials, or result in the need for additional preclinical and/or clinical testing or the timing or likelihood of regulatory approvals;
•the status of our manufacturing operations and any delays, interruptions or failures in the manufacture and supply of ONPATTRO, AMVUTTRA, GIVLAARI, OXLUMO or any of our product candidates (or other products or product candidates being developed and commercialized by our collaborators), by our or their contract manufacturers or by us or our collaborators;
•the impact of any future pandemics or public health emergencies on, among other things, our financial performance, business and operations, including manufacturing, supply chain, research and development activities and pipeline programs, and other potential impacts to our business;
•our progress continuing to build and leverage our global commercial infrastructure;
•the possible impact of any competing products on the commercial success of ONPATTRO, AMVUTTRA, GIVLAARI, OXLUMO, Leqvio and fitusiran, as well as our product candidates, and, our, or with respect to Leqvio or fitusiran, our collaborators’, ability to compete against such products;
•our ability to manage our growth and operating expenses;
•our views and plans with respect to our 5-year Alnylam P5x25 strategy and our intentions to achieve the metrics associated with this strategy, including to become a top-tier biotech company by the end of 2025, and our ability to successfully execute on our Alnylam P5x25 strategy;
•our belief that our current cash balance should enable us to achieve a self-sustainable profile without the need for future equity financing;
•our expectations regarding the length of time our current cash, cash equivalents and marketable equity and debt securities will support our operations based on our current operating plan;
•our dependence on third parties for development, manufacture and distribution of our products;
•our ability to maintain our existing collaborations and our expectations regarding potential future research and development funding, licensing fees and milestone and royalty payments that we may receive under existing or future collaboration agreements;
•our ability to obtain, maintain and protect our intellectual property;
•our ability to attract and retain qualified key management and scientists, development, medical and commercial staff, consultants and advisors;
•the outcome of litigation, including our patent infringement suits against Pfizer, Inc., BioNTech SE and Moderna, Inc., or of other legal proceedings or government investigations;
•regulatory developments in the U.S. and other jurisdictions;
•the impact of laws and regulations;
•developments relating to our competitors and our industry;
•our ability to satisfy our payment obligations, and to service the interest on or to refinance our indebtedness, including our convertible notes, or to make cash payments in connection with any conversion of our convertible notes, to the extent required;
•our expectations regarding the effect of the capped call transactions and the anticipated market activities of the option counterparties and/or their respective affiliates; and
•other risks and uncertainties, including those listed under the caption Part I, Item 1A, “Risk Factors” of this Annual Report on Form 10-K.
Any forward-looking statements in this Annual Report on Form 10-K reflect our current views with respect to future events and with respect to our business and future financial performance, and involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by these forward-looking statements. Factors that may cause actual results to differ materially from current expectations include, among other things, those described under Part I, Item 1A, “Risk Factors” and elsewhere in this Annual Report on Form 10-K. Moreover, we operate in a very competitive and rapidly changing environment. New risk factors emerge from time to time and it is not possible for management to predict all risk factors, nor can we assess the impact of all risk factors on our business or the extent to which any factor, or combination of factors, may cause actual results to differ materially from those contained in any forward-looking statements. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future. You are advised, however, to consult any further disclosure we make in our reports filed with the Securities and Exchange Commission, or SEC.
This Annual Report on Form 10-K may include data that we obtained from industry publications and third-party research, surveys and studies. Industry publications and third-party research, surveys and studies generally indicate that their information has been obtained from sources believed to be reliable, although they do not guarantee the accuracy or completeness of such information. This Annual Report on Form 10-K also may include data based on our own internal estimates and research, which have not been verified by any independent source and, while we believe any data obtained from industry publications and third-party research, surveys and studies are reliable, we have not independently verified such data. Any such third-party data, as well as our internal estimates and research, are subject to a high degree of uncertainty and risk due to a variety of factors, including those described in Part I, Item 1A, “Risk Factors” and elsewhere in this Annual Report on Form 10-K. These and other factors could cause our results to differ materially from those expressed in this Annual Report on Form 10-K.
PART I
ITEM 1. BUSINESS
Overview
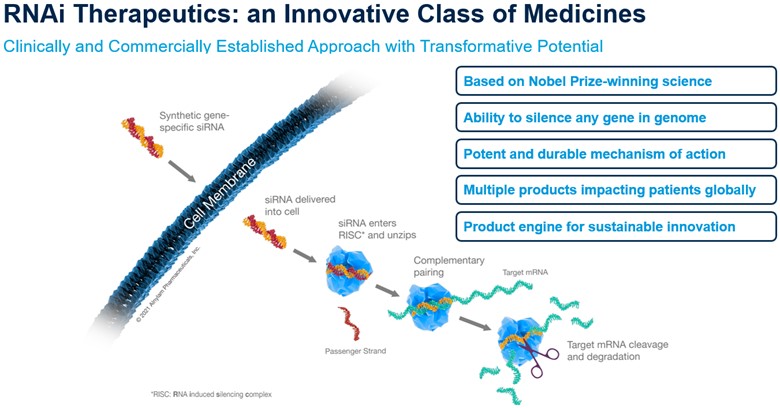
Alnylam Pharmaceuticals, Inc. (also referred to as Alnylam, the Company, we, our or us) is a global commercial-stage biopharmaceutical company developing novel therapeutics based on ribonucleic acid interference, or RNAi. RNAi is a naturally occurring biological pathway within cells for sequence-specific silencing and regulation of gene expression. By harnessing the RNAi pathway, we have developed a new class of innovative medicines, known as RNAi therapeutics. RNAi therapeutics are comprised of small interfering RNA, or siRNA, that function upstream of conventional medicines by potently silencing messenger RNA, or mRNA, that encode for proteins implicated in the cause or pathway of disease, thus preventing them from being made. We believe this is a revolutionary approach with the potential to transform the care of patients across a broad range of disease areas and indications. To date, our efforts to advance this revolutionary approach have yielded the approval of five first-in-class RNAi-based medicines, ONPATTRO® (patisiran), AMVUTTRA® (vutrisiran), GIVLAARI® (givosiran), OXLUMO® (lumasiran) and Leqvio® (inclisiran).
Our research and development strategy is to target genetically validated genes that have been implicated in the cause or pathway of human disease. We utilize a N-acetylgalactosamine (GalNAc) conjugate approach or lipid nanoparticle (LNP) to enable hepatic delivery of siRNAs. For delivery to the central nervous system, or CNS, and the eye (ocular delivery), we are utilizing an alternative conjugate approach based on a hexadecyl (C16) moiety as a lipophilic ligand. We are also advancing approaches for heart, skeletal muscle and adipose tissue delivery of siRNAs. Our focus is on clinical indications where there is a high unmet need, a genetically validated target, early biomarkers for the assessment of clinical activity in Phase 1 clinical trials, and a definable path for drug development, regulatory approval, patient access and commercialization.
In early 2021, we launched our Alnylam P5x25 strategy, which focuses on our planned transition to a top-tier biotech company by the end of 2025. With Alnylam P5x25, we aim to deliver transformative medicines in both rare and common diseases benefiting patients around the world through sustainable innovation and exceptional financial performance, resulting in a leading biotech profile.
We ended 2024 making considerable progress on these goals, and currently have five marketed products and more than 20 clinical programs, including several in late-stage development, across a broad range of disease areas and indications.
ONPATTRO is approved in the U.S. for the treatment of the polyneuropathy of hATTR amyloidosis in adults and has also been approved in the European Union, or EU, for the treatment of hATTR amyloidosis in adult patients with stage 1 or stage 2 polyneuropathy, in Japan for the treatment of TTR-type familial amyloidosis with polyneuropathy, and in multiple additional countries. In February 2025, ONPATTRO received regulatory approval from the Brazilian Health Regulatory Agency, or ANVISA, for the treatment of ATTR amyloidosis with cardiomyopathy.
AMVUTTRA is approved in the U.S. for the treatment of hereditary transthyretin-mediated amyloidosis, or hATTR amyloidosis, with polyneuropathy in adults, in the EU, and the United Kingdom, or UK, for the treatment of hATTR amyloidosis in adult patients with stage 1 or stage 2 polyneuropathy, in Japan for the treatment of transthyretin, or TTR, type familial amyloidosis with polyneuropathy, and in multiple additional countries. Regulatory reviews continue in other territories. In June 2024, we reported positive topline results from the HELIOS-B clinical trial of vutrisiran (the generic name of AMVUTTRA) in patients with ATTR amyloidosis with cardiomyopathy and announced that we planned to proceed with global regulatory filings seeking approval of AMVUTTRA as a potential treatment for ATTR amyloidosis with cardiomyopathy. In August and September 2024, we reported detailed results from the HELIOS-B clinical trial. On October 9, 2024, we announced that we had filed a supplemental New Drug Application, or sNDA, with the United States Food and Drug Administration, or the FDA, using a Priority Review Voucher. In November 2024, the FDA accepted our sNDA submission for review for vutrisiran, with an action date set for March 23, 2025 under the Prescription Drug User Fee Act, or PDUFA. Vutrisiran is also under regulatory review by the European Medicines Agency, or the EMA, ANVISA, the Japanese Health Authority, or PMDA, and the Colombian Health Authority, or INVIMA.
GIVLAARI is approved in the U.S. for the treatment of adults with acute hepatic porphyria, or AHP, in the EU for the treatment of AHP in adults and adolescents aged 12 years and older, and in several other countries. Regulatory filings for givosiran (the generic name of GIVLAARI) in additional territories are pending or planned during 2025 and beyond.
OXLUMO is approved in the U.S. for the treatment of primary hyperoxaluria type 1, or PH1, to lower urinary and plasma oxalate levels in pediatric and adult patients, and in the EU and the UK for the treatment of PH1 in all age groups. OXLUMO has also been approved in several other countries and regulatory filings for lumasiran (the generic name of OXLUMO) in additional territories are pending or planned during 2025 and beyond.
Leqvio (inclisiran), our fifth product, is being developed and commercialized by our collaborator Novartis AG, or Novartis, and has received marketing authorization from the European Commission, or EC, for the treatment of adults with hypercholesterolemia or mixed dyslipidemia and from the FDA as an adjunct to diet and maximally tolerated statin therapy for the treatment of adults with heterozygous familial hypercholesterolemia, or HeFH, or clinical atherosclerotic cardiovascular
disease, or ASCVD, who require additional lowering of low-density lipoprotein cholesterol, or LDL-C. In July 2023, the FDA approved an expanded indication for Leqvio to include treatment of adults with high LDL-C and who are at increased risk of heart disease. Leqvio has also been approved in China and Japan, and as of the end of January 2025, Leqvio had been registered in more than 100 countries.
In addition to our marketed products, as part of our Alnylam P5x25 strategy, we have multiple drivers of future growth, including the development of transformative medicines to treat prevalent disease. In addition to Leqvio, we are advancing zilebesiran, an investigational, subcutaneously administered RNAi therapeutic targeting angiotensinogen in development for the treatment of hypertension. In 2023, we entered into a Collaboration and License Agreement, or the Roche Collaboration and License Agreement, with F. Hoffmann-La Roche Ltd. and Genentech, Inc. or, collectively, Roche, pursuant to which we established a worldwide, strategic collaboration for the joint development and commercialization of zilebesiran. In March 2024, we reported positive topline results from our KARDIA-2 clinical trial, which is designed to evaluate the safety and efficacy of zilebesiran administered biannually as a concomitant therapy in patients whose blood pressure is not adequately controlled by a standard of care antihypertensive medication. In April 2024, we dosed the first patient in our KARDIA-3 Phase 2 clinical trial, which is designed to evaluate the efficacy and safety of zilebesiran as an add-on therapy in adult patients with high cardiovascular risk and uncontrolled hypertension despite treatment with two to four standard of care antihypertensive medications, and we expect to report results from the KARDIA-3 Phase 2 clinical trial in the second half of 2025. In January 2025, we announced that we expect to initiate a Phase 3 cardiovascular outcomes trial in the second half of 2025.
We are advancing nucresiran (formerly ALN-TTRsc04), an investigational RNAi therapeutic in development for the treatment of ATTR amyloidosis. In November 2024, we announced positive new results from the ongoing Phase 1 study of nucresiran in healthy volunteers. These results demonstrated that a single dose of nucresiran at 300 mg or higher resulted in mean reductions of serum TTR of greater than 90% from baseline achieved at Day 15 and sustained through at least Day 180. We expect to initiate a Phase 3 clinical trial of nucresiran in patients with ATTR amyloidosis with cardiomyopathy in the first half of 2025.
We are also advancing mivelsiran (formerly ALN-APP), an investigational RNAi therapeutic targeting amyloid precursor protein in development for the treatment of Alzheimer’s disease, or AD, and cerebral amyloid angiopathy, or CAA. In October 2024, we announced positive initial results from the multiple dose portion of the Phase 1 clinical trial of mivelsiran in patients with early-onset AD. We expect to initiate a Phase 2 clinical trial of mivelsiran in patients with AD in the second half of 2025. In July 2024, we initiated dosing in the cAPPRicorn-1 Phase 2 clinical trial of mivelsiran in patients with CAA.
We have additional late-stage investigational programs advancing toward potential commercialization, including fitusiran for the treatment of hemophilia, which is being advanced by our collaborator Genzyme Corporation, a Sanofi Company, or Sanofi, and for which a New Drug Application, or NDA, has been filed with the FDA with a target action date of March 28, 2025, and cemdisiran for the treatment of complement-mediated diseases, which our collaborator, Regeneron Pharmaceuticals, Inc., or Regeneron, is advancing in combination with pozelimab in Phase 3 clinical trials in myasthenia gravis and geographic atrophy and as a monotherapy in a Phase 3 clinical trial in paroxysmal nocturnal hemoglobinuria.
In further support of our Alnylam P5x25 strategy and in view of our evolving risk profile, we remain focused on the continued evolution of our global infrastructure, including key objectives such as optimizing our global structure for execution in key markets, enhancing performance consistent with our values, and continuing to strengthen our culture. We continue to build our global compliance program to drive its evolution and enhancement through the launch of new systems and leveraging data analytics to strengthen the efficiency and effectiveness of our program. Building from our global Code of Business Conduct and Ethics, our compliance program is designed to empower our employees and those with whom we work to execute on our strategy consistent with our values and in compliance with applicable laws and regulations, and to mitigate risk. Comprised of components such as risk assessment and monitoring; policies, procedures, and guidance; training and communications; dedicated resources; and systems and processes supporting activities such as third party engagements and investigations and remediation, and the Company’s enterprise risk management program; our program and related controls are built to enhance our business processes, structures, and controls across our global operations, and to empower ethical decision making.
Based on our expertise in RNAi therapeutics and broad intellectual property estate, we have formed collaborations with leading pharmaceutical and life sciences companies to support our development and commercialization efforts, including Roche, Regeneron, Sanofi, and Novartis (which acquired our collaborator The Medicines Company, or MDCO, in 2020).
Key 2024 and Recent Highlights
TTR Franchise
•ONPATTRO (patisiran) and AMVUTTRA (vutrisiran)– hATTR Amyloidosis with Polyneuropathy
◦Achieved global net product revenues for ONPATTRO and AMVUTTRA for the full year 2024 of $252.9 million and $970.5 million, respectively
•Vutrisiran – ATTR Amyloidosis with Cardiomyopathy
◦Submitted an sNDA to the FDA using a Priority Review Voucher for vutrisiran for the treatment of ATTR amyloidosis with cardiomyopathy. Parallel filings for regulatory approval have also been submitted in all major regions, including Europe and Japan
▪The FDA has accepted the sNDA and set an action goal date of March 23, 2025, under PDUFA
•Nucresiran (formerly ALN-TTRsc04) – ATTR Amyloidosis
◦Received Orphan Drug Designation from the FDA and announced positive interim Phase 1 data of nucresiran in patients with ATTR amyloidosis
Commercial/Late-Stage Pipeline
•GIVLAARI (givosiran) – Acute Hepatic Porphyria
◦Recognized GIVLAARI global net revenue of $255.9 million for the year ended December 31, 2024
•OXLUMO (lumasiran) – Primary Hyperoxaluria Type 1
◦Recognized OXLUMO global net revenue of $167.1 million for the year ended December 31, 2024
•Leqvio (inclisiran) – Hypercholesterolemia (in collaboration with Novartis)
◦Our collaborator, Novartis, continued the launch of Leqvio, with a focus on increasing account and patient adoption, and continuing medical education
•Fitusiran – Hemophilia (in collaboration with Sanofi)
◦Our collaborator, Sanofi, continued to advance fitusiran, an investigational RNAi therapeutic in development for the treatment of hemophilia A and B, with or without inhibitors, and the FDA has set a PDUFA target action date of March 28, 2025
Early-Stage and Preclinical Pipeline
•Zilebesiran – Hypertension
◦Initiated Phase 1 studies of zilebesiran + REVERSIR for hypertension
•Mivelsiran – Cerebral Amyloid Angiopathy and Alzheimer’s Disease
◦Announced positive initial results from the multiple dose portion of the Phase 1 study of mivelsiran in patients with Alzheimer’s disease
•ALN-HTT02 – Huntington’s Disease
◦Initiated a Phase 1 study of ALN-HTT02 in adult patients with Huntington’s disease
Corporate Highlights
•Finance
◦Ended 2024 with $2.69 billion in cash, cash equivalents and marketable securities
RNAi Therapeutics – A Class of Innovative Medicines
Overview of RNAi Therapeutics
Alnylam was founded in 2002 to translate the discovery of RNAi into a new class of medicines, which we refer to as RNAi therapeutics. One of the biggest scientific challenges with RNAi therapeutics has been delivering the therapeutic to its destination in the body. In recent years, we have made tremendous progress on delivery of RNAi therapeutics, beginning with delivery to the liver and more recently with delivery to the CNS and other tissues. We believe this success will enable us to sustainably and organically create and commercialize transformative medicines that benefit patients around the world across a broad range of disease areas and indications while also delivering strong financial performance, which will result in a leading biotech profile by the end of 2025.
Early RNAi delivery efforts focused on delivery of RNAi therapeutics utilizing LNPs, which encapsulate siRNA molecules in specific lipid-based formulations. This technology enables systemic delivery with intravenous drug administration and is associated with potent, rapid and durable target gene silencing and an encouraging tolerability profile. Our first commercial product, ONPATTRO, is formulated utilizing LNPs.
In parallel, we advanced a proprietary technology that conjugates a sugar molecule, referred to as GalNAc, to the siRNA molecule. This delivery approach enables more convenient, subcutaneous administration of our drug candidates directed to liver cells expressing target genes. Results from our Enhanced Stabilization Chemistry, or ESC, GalNAc-conjugate delivery platform have demonstrated a durability of effect that provides increased potency and durability, as well as a wide therapeutic index, and has the potential to support once-monthly, once-quarterly, and bi-annual subcutaneous dose regimens. Our more recently approved medicines, GIVLAARI, OXLUMO, Leqvio, and AMVUTTRA, utilize this conjugate platform.
Our next generation Enhanced Stabilization Chemistry-Plus, or ESC+ GalNAc-conjugates utilize advanced design features to further improve specificity, while maintaining potency and durability, further improving our therapeutic index by up to six-fold. Our first RNAi therapeutics based on this ESC+ design, zilebesiran and elebsiran, are currently in clinical development.
Additional advancements include our IKARIA platform harboring chemistry innovations that enable robust target knockdown with the potential for bi-annual or annual dosing regimens. We believe that this platform could have potential applications in a wide range of diseases. In November 2024, we announced positive interim results from the Phase 1 clinical trial of nucresiran, our investigational RNAi therapeutic in development for the treatment of ATTR amyloidosis which utilizes our IKARIA technology, demonstrating rapid knockdown of TTR that was sustained for six months following a single dose.
Our platform enhancements have also provided a strong foundation for pursuing a conjugate-based approach to extrahepatic delivery, including delivery to the brain and spinal cord, as well as ocular delivery. In July 2022, our publication in Nature Biotechnology showcased data utilizing an alternative conjugate approach based on a hexadecyl (C16) moiety as a lipophilic ligand, with proof-of-concept demonstrated in rodent and non-human primates. C16 conjugates provide robust CNS knockdown with wide biodistribution and long duration of action. In 2023, we announced positive interim results from the Phase 1 clinical trial of mivelsiran, the first investigational RNAi therapeutic that leverages our C16 conjugate technology, in
early onset AD. Mivelsiran targets amyloid precursor protein and is currently in development for the treatment of AD and CAA. We are also advancing our C16 technology as part of our collaboration with Regeneron, including work on ALN-HTT02, our investigational RNAi therapeutic in development for the treatment of Huntington’s disease, or HD, which is a devastating, fatal neurodegenerative disease for which there are currently no approved disease-modifying therapies.
We are continuing to advance other extrahepatic delivery approaches, including delivery to muscle and adipose cells. In addition, we are exploring peptide and antibody-based approaches for targeted siRNA delivery to new tissues as part of our collaboration with PeptiDream, Inc., which aims to discover and develop novel conjugates for targeted delivery of RNAi therapeutics to a broader range of tissues. As we seek to address more complex conditions with multiple comorbidities, we are also advancing our GEMINI technology, which combines conjugate siRNAs for the simultaneous silencing of two transcripts using a single chemical entity.
Finally, we continue to leverage human genetics to advance our efforts to bring innovative medicines to patients. We have established five major collaborations—including with the UK BioBank and Our Future Health—to support the sourcing of novel, genetically validated targets and to secure access to databases of genetic information.
Our Product Pipeline
Our broad pipeline includes five approved products and multiple late and early-stage investigational RNAi therapeutics across a broad range of disease areas and indications. We describe our commercial and clinical-stage pipeline in more detail below. The clinical-stage therapeutics described below are in various stages of clinical development and the scientific information included about these therapeutics is preliminary and investigative. These clinical-stage therapeutics have not been approved by the FDA, EMA, or any other health authority and no conclusions can or should be drawn regarding the safety or efficacy of these investigational therapeutics.
The table below represents our commercial products and late- and early-stage development programs as of February 1, 2025.
As indicated in the table above, to date we have received marketing approval for ONPATTRO, AMVUTTRA, GIVLAARI and OXLUMO, and Novartis has received approval for Leqvio, in each case in certain territories for the specific indications approved in each such territory, with additional regulatory submissions pending.
Our TTR Franchise
About Transthyretin Amyloidosis (ATTR)
ATTR amyloidosis is an underdiagnosed, rapidly progressive and fatal disease that affects multiple parts of the body including the heart, nerves and digestive system. There are two forms of ATTR – hATTR and wild-type ATTR, which can manifest in a variety of ways. ATTR with cardiomyopathy – including hereditary and wild-type forms – affects greater than 300,000 people worldwide, while hATTR with polyneuropathy affects fewer than 30,000 people worldwide.
ONPATTRO (patisiran)
ONPATTRO (patisiran) is an intravenously administered RNAi therapeutic targeting TTR. It is designed to target and silence TTR mRNA, thereby reducing the production of TTR protein before it is made. ONPATTRO blocks the production of TTR in the liver, reducing its accumulation in the body’s tissues to halt or improve the progression of disease.
Patisiran has received orphan drug designations in the U.S., EU and Japan; specific orphan drug designations vary by country/region.
ONPATTRO (patisiran) – hATTR Amyloidosis with Polyneuropathy
ONPATTRO is the first ever FDA-approved RNAi therapeutic and was our first product to receive marketing approval. In the U.S. and Canada, ONPATTRO is indicated for the treatment of hATTR amyloidosis with polyneuropathy in adults. In the EU, Switzerland, Brazil and Israel, ONPATTRO is indicated for the treatment of hATTR amyloidosis in adults with stage 1 or stage 2 polyneuropathy, and in Japan, ONPATTRO is indicated for the treatment of TTR type familial amyloidosis with polyneuropathy.
Patisiran was also evaluated in a Phase 4 clinical trial in hATTR amyloidosis patients with polyneuropathy due to a T60A or V122I variant.
ONPATTRO (patisiran) - ATTR Amyloidosis with Cardiomyopathy
In February 2025, ONPATTRO received regulatory approval from the Brazilian Health Regulatory Agency, or ANVISA, for the treatment of ATTR amyloidosis with cardiomyopathy. We do not plan to pursue label expansions for ONPATTRO in other regions.
AMVUTTRA (vutrisiran)
AMVUTTRA (vutrisiran) is a subcutaneously administered RNAi therapeutic targeting TTR. With its rapid knockdown, it targets and silences TTR mRNA, thereby reducing the production of TTR protein before it is made. AMVUTTRA utilizes our ESC+ delivery platform, and designed for increased potency and high metabolic stability to allow for quarterly subcutaneous administration.
Vutrisiran has received orphan drug designation in the U.S., EU and Japan; specific orphan drug designations vary by country/region.
AMVUTTRA (vutrisiran) – hATTR Amyloidosis with Polyneuropathy
In June 2022, AMVUTTRA was approved by the FDA for the treatment of hATTR amyloidosis with polyneuropathy in adults. AMVUTTRA was subsequently approved in the EU, UK, Argentina, Switzerland and Canada for the treatment of hATTR amyloidosis in adult patients with stage 1 or stage 2 polyneuropathy, in Japan for the treatment of TTR type familial amyloidosis with polyneuropathy, and in Brazil for the treatment of hATTR amyloidosis in adults. AMVUTTRA has launched in several additional countries and regulatory filings in additional territories are currently under review and additional filings are planned for 2025.
Vutrisiran – ATTR Amyloidosis with Cardiomyopathy
Vutrisiran is also in development as a potential treatment for patients with ATTR amyloidosis with cardiomyopathy in the ongoing HELIOS-B Phase 3 clinical trial. In June 2024, we reported positive topline results from the HELIOS-B clinical trial of vutrisiran in patients with ATTR amyloidosis with cardiomyopathy. In August and September 2024, we reported detailed results from the HELIOS-B clinical trial, and on October 9, 2024, we announced that we had filed an sNDA with the FDA, using a Priority Review Voucher. We later announced on October 16, 2024, that we had submitted a Type II Variation to the EMA. In November 2024, the FDA accepted our sNDA submission for review for vutrisiran, with an action date set for March 23, 2025 under PDUFA. Vutrisiran is also under regulatory review by the EMA, ANVISA, PMDA, and INVIMA. Additional filings are planned for 2025 and beyond.
HELIOS-B Phase 3 Study
The HELIOS-B Phase 3 clinical trial, initiated in late 2019, was a randomized, double-blind, placebo-controlled, multicenter global study designed and powered to evaluate the efficacy and safety of vutrisiran on the reduction of all-cause mortality and recurrent cardiovascular events as a primary composite endpoint in patients with ATTR amyloidosis
with cardiomyopathy. The secondary endpoints evaluated included change from baseline in 6-minute walk test, or 6-MWT, change from baseline in Kansas City Cardiomyopathy Questionnaire, or KCCQ-OS, all-cause mortality, and change from baseline in New York Heart Association, or NYHA, class, with additional exploratory endpoints evaluated. The study randomized 655 adult patients with ATTR amyloidosis (hereditary or wild-type) with cardiomyopathy. Patients were randomized 1:1 to receive vutrisiran 25mg or placebo subcutaneously once every three months during a double-blind treatment period of up to 36 months. After the double-blind period, all eligible patients remaining on the study were able to receive vutrisiran in an open-label extension period of HELIOS-B.
Efficacy and Safety Results
In June 2024, we announced positive topline results, noting that study met the primary endpoint, demonstrating a statistically significant reduction in the composite of all-cause mortality and recurrent cardiovascular events during the double-blind period in both the overall population and in the monotherapy population. In the overall population, vutrisiran reduced the risk of all-cause mortality and recurrent cardiovascular events by 28%, with similar reductions in both the mortality and cardiovascular events components of the endpoint. Mortality in the overall population was reduced by 31% during the double-blind period and by 36% up to 42 months. In the monotherapy population, vutrisiran reduced the risk of all-cause mortality and recurrent cardiovascular events by 33% and reduced the risk of mortality by 35% up to 42 months. As a component of the primary endpoint, a non-significant reduction of 30% in mortality was observed in the monotherapy population during the double-blind period. The study also demonstrated statistically significant improvements across all secondary endpoints in both the overall and monotherapy populations. This includes key measures of disease progression: 6-MWT, KCCQ and NYHA Class at Month 30.
In the HELIOS-B clinical trial, the safety and tolerability profiles of vutrisiran were consistent with what had been established in the currently approved patient population, as well as earlier clinical studies.
Nucresiran (formerly ALN-TTRsc04) – ATTR Amyloidosis
Nucresiran is an investigational RNAi therapeutic in development for the treatment of ATTR amyloidosis that utilizes our IKARIA technology and provides the potential to achieve deeper and more durable rapid knockdown of TTR, allowing for less frequent dosing. In December 2023 and November 2024, we announced positive initial results in the Phase 1 study of nucresiran in healthy volunteers. Single doses of 300 mg or higher of nucresiran achieved rapid knockdown with a mean TTR reduction of greater than 90% at Day 15. A peak mean TTR reduction of 96% was achieved at Day 29 and a mean TTR reduction of greater than 90% was sustained at Day 180. At all dose levels evaluated to date, single doses of nucresiran have been well tolerated. The majority of adverse events across doses have been mild and none have been considered to be related to treatment. There have been no safety signals identified, including no injection site reactions and no liver-related signals.
These Phase 1 results suggest nucresiran has the potential to offer over 90% max TTR reduction and to be administered once annually. The Phase 1 study is ongoing, and in January 2025, we announced that we expect to initiate a Phase 3 study of nucresiran in ATTR amyloidosis with cardiomyopathy in the first half of 2025.
Nucresiran has received orphan drug designation in the U.S.
Our Other Marketed Products
GIVLAARI (givosiran) — Acute Hepatic Porphyria (AHP)
AHP refers to a family of ultra-rare, genetic diseases characterized by potentially life-threatening attacks and, for some patients, chronic manifestations that negatively impact daily functioning and quality of life. AHP is comprised of four types: acute intermittent porphyria, hereditary coproporphyria, variegate porphyria, and aminolevulinic acid dehydratase-deficiency porphyria. We estimate there are approximately 3,000 AHP patients diagnosed in the U.S. and EU with active disease. Each type of AHP results from a genetic defect leading to deficiency in one of the enzymes of the heme biosynthesis pathway in the liver. AHP disproportionately impacts women of working and childbearing age, and symptoms of the disease vary widely. Severe, unexplained abdominal pain is the most common symptom, which can be accompanied by limb, back or chest pain, nausea, vomiting, confusion, anxiety, seizures, weak limbs, constipation, diarrhea, or dark or reddish urine. The nonspecific nature of AHP signs and symptoms can often lead to misdiagnoses of other more common conditions such as viral gastroenteritis, irritable bowel syndrome and appendicitis. Consequently, patients with AHP can wait up to 15 years for a confirmed diagnosis. In addition, long-term complications and comorbidities of AHP can include hypertension, chronic kidney disease, or liver disease including hepatocellular carcinoma.
Our RNAi therapeutic, GIVLAARI is the first GalNAc-conjugate RNA therapeutic to be approved. GIVLAARI works by specifically reducing induced liver aminolevulinic acid synthase 1 mRNA, leading to reduction of toxins associated with attacks and other disease manifestations of AHP and is administered by subcutaneous injection.
GIVLAARI is approved in the U.S. for the treatment of adults with AHP and in the EU for the treatment of AHP in adults and adolescents aged 12 years and older. GIVLAARI has also received marketing authorizations for the treatment of AHP in adults in Brazil and Canada, and for the treatment of AHP in adults and adolescents in Japan, the UK, Argentina, Australia, Switzerland, Israel and Taiwan. We have also filed for regulatory approval for givosiran (the non-branded drug name for
GIVLAARI) in Colombia, Mexico, South Korea and Kuwait, and additional regulatory filings are pending or planned in 2025 and beyond.
OXLUMO (lumasiran)— Primary Hyperoxaluria Type 1 (PH1)
PH1 is an ultra-rare genetic disease that affects an estimated one to three individuals per million in the U.S. and Europe. PH1 is characterized by oxalate overproduction in the liver. The excess oxalate results in the deposition of calcium oxalate crystals in the kidneys and urinary tract and can lead to the formation of painful and recurrent kidney stones and nephrocalcinosis. Renal damage is caused by a combination of tubular toxicity from oxalate, calcium oxalate deposition in the kidneys, and urinary obstruction by calcium oxalate stones. PH1 is associated with a progressive decline in kidney function, which exacerbates the disease as the excess oxalate can no longer be effectively excreted, resulting in subsequent accumulation and deposition of oxalate in bones, eyes, skin, and heart, leading to severe illness and death. Management options prior to availability of OXLUMO were limited to hyperhydration, crystallization inhibitors and, in a minority of patients with a specific genotype, pyridoxine (vitamin B6). These measures do not adequately address oxalate overproduction but instead help to delay inevitable progression to kidney failure and the need for intensive dialysis as a bridge to a dual or sequential liver/kidney transplant. Liver transplantation is the only intervention that addresses the underlying metabolic defect, but is associated with high morbidity and mortality, and life-long immunosuppression.
OXLUMO is an RNAi therapeutic targeting hydroxyacid oxidase 1, or HAO1, developed for the treatment of PH1. HAO1 encodes glycolate oxidase, or GO, an enzyme upstream of the disease-causing defect in PH1. OXLUMO works by degrading HAO1 mRNA and reducing the synthesis of GO, which inhibits hepatic production of oxalate, the toxic metabolite responsible for the clinical manifestations of PH1. OXLUMO utilizes our ESC-GalNAc-conjugate technology, which enables subcutaneous dosing with increased potency and durability and a wide therapeutic index.
OXLUMO is the first approved pharmaceutical therapy for PH1. OXLUMO is approved in the U.S. for the treatment of PH1 to lower urinary oxalate levels in pediatric and adult patients, and has received marketing authorization in the EU for the treatment of PH1 in all age groups. OXLUMO has also received additional marketing authorizations in Argentina, Australia, Brazil, the UK, Switzerland, Canada, Israel, Taiwan, Oman and Qatar, and regulatory filings in other territories are pending and additional filings are planned for 2025 and beyond.
Leqvio (inclisiran) — Hypercholesterolemia
Hypercholesterolemia, also known as high cholesterol, is a lipid disorder in which there are abnormally high levels of fat in the blood, which increases the risk of heart disease and stroke. Approximately 100 million people worldwide suffer from hypercholesterolemia and are treated with lipid lowering therapies, predominantly statins, to reduce LDL-C and the associated risk of death, nonfatal myocardial infarction and nonfatal stroke or associated events. Nonetheless, statins are associated with well-known limitations, residual risk for cardiovascular events remains, and current therapies for the management of elevated LDL-C remain insufficient in some subjects. This is particularly the case in patients with pre-existing coronary heart disease and/or diabetes or a history of familial hypercholesterolemia who are at the highest risk and require the most intensive management.
Our RNAi therapeutic Leqvio, developed and commercialized by our collaborator, Novartis, is the first and only siRNA therapy (or RNAi therapeutic) to lower LDL-C, and is the first RNAi therapeutic approved for a prevalent disease. Leqvio is a subcutaneously administered RNAi therapeutic targeting proprotein convertase subtilisin/kexin type 9, or PCSK9, to reduce LDL-C levels via an RNAi mechanism of action and could help improve outcomes for patients with ASCVD, a deadly form of cardiovascular disease.
Leqvio is approved in the U.S. as an adjunct to diet and statin therapy for the treatment of adults with primary hyperlipidemia, including HeFH, to reduce LDL-C. Leqvio is approved in the EU for the treatment of adults with primary hypercholesterolemia (heterozygous familial and non-familial) or mixed dyslipidemia, as an adjunct to diet: in combination with a statin or statin with other lipid-lowering therapies in patients unable to reach LDL-C goals with the maximally tolerated dose of a statin, or alone or in combination with other lipid-lowering therapies in patients who are statin-intolerant, or for whom a statin is contraindicated. As of the end of January 2025, Leqvio had been approved in over 100 countries. Multiple additional Phase 3 clinical trials of Leqvio are currently ongoing, including two cardiovascular outcomes trials in secondary prevention, ORION-4 co-sponsored by Novartis and Oxford University and the Novartis-sponsored VICTORION-2-PREVENT.
In February 2013, we and MDCO (acquired by Novartis in January 2020) entered into a license and collaboration agreement pursuant to which we granted to MDCO an exclusive, worldwide license to develop, manufacture and commercialize RNAi therapeutics targeting PCSK9 for the treatment of hypercholesterolemia and other human diseases. Following its acquisition of MDCO, Novartis has all of the rights and obligations under the MDCO agreement. A description of the MDCO agreement is included below under the heading “Our Collaboration and Licensing Strategy.”
Additional Late-Stage Clinical Development Programs
Fitusiran — Hemophilia
Hemophilia is a hereditary bleeding disorder characterized by an underlying defect in the ability to generate adequate levels of thrombin needed for effective fibrin clot formation, thereby resulting in recurrent bleeds into joints, muscles, and major internal organs. There are approximately 200,000 people living with hemophilia A and hemophilia B worldwide. Standard treatment for people with hemophilia currently involves replacement of the deficient clotting factor either as prophylaxis or on-demand therapy, which can lead to a temporary restoration of thrombin generation capacity. However, with current factor replacement treatments people with hemophilia are at risk of developing neutralizing antibodies, or inhibitors, to their replacement factor, a very serious complication affecting as many as one third of people with severe hemophilia A and a smaller fraction of people with hemophilia B. People who develop inhibitors become refractory to replacement factor therapy and are twice as likely to be hospitalized for a bleeding episode.
Lowering antithrombin, or AT, may promote the generation of sufficient levels of thrombin needed to form an effective fibrin clot and prevent bleeding. This rationale is supported by human genetic data suggesting that co-inheritance of thrombophilic mutations, including AT deficiency, may ameliorate bleeding in hemophilia. We believe this approach is a unique and innovative strategy for preventing bleeding in people with hemophilia.
Fitusiran is an investigational, subcutaneously administered RNAi therapeutic targeting AT for the treatment of hemophilia A and B, with and without inhibitors, that is being advanced by our collaborator, Sanofi. Fitusiran is designed to lower levels of AT with the goal of promoting sufficient thrombin generation to prevent bleeding. AT acts by inactivating thrombin and other coagulation factors, and plays a key role in normal hemostasis by helping to limit the process of fibrin clot formation.
Fitusiran is currently being evaluated in the ATLAS Phase 3 program. Sanofi presented positive results from the ATLAS-A/B and ATLAS-INH Phase 3 clinical trials of fitusiran in December 2021, and in July 2022, Sanofi presented positive results from the Phase 3 ATLAS-PPX study evaluating the efficacy and safety of once-monthly fitusiran (80 mg) in adults and adolescents with severe hemophilia A or B who were previously treated with prior factor or bypassing agent prophylaxis. In 2023, Sanofi presented positive results from the ATLAS-OLE Phase 3 extension clinical trial of fitusiran, demonstrating a substantially improved safety profile and consistent bleed protection in people with hemophilia A or B, with or without inhibitors. Specifically, the risk of thrombosis was reduced, with rates comparable to those reported in the general hemophilia population. In June 2024, Sanofi presented new ATLAS Phase 3 clinical trial data that reinforce the potential of fitusiran to provide prophylaxis for people with hemophilia A or B, with or without inhibitors, and announced that their NDA for fitusiran had been accepted for review by the FDA with a PDUFA action date of March 28, 2025. Regulatory submissions for fitusiran for the treatment of hemophilia A or B in adults and adolescents with or without inhibitors have been completed in China and Brazil. The FDA granted fitusiran Breakthrough Therapy Designation for hemophilia B with inhibitors in December 2023.
We have granted Sanofi global rights to develop and commercialize fitusiran and any back-up products. A description of our Sanofi collaboration, as well as the amended and restated AT3 License Terms, is included under the heading “Our Collaboration and Licensing Strategy.”
Cemdisiran — Complement-Mediated Diseases
The complement system plays a central role in immunity as a protective mechanism for host defense, but its dysregulation results in a broad range of human diseases including myasthenia gravis, paroxysmal nocturnal hemoglobinuria, and geographic atrophy, among others.
Cemdisiran is a subcutaneously administered, investigational RNAi therapeutic targeting the C5 component of the complement pathway in development for the treatment of complement-mediated diseases. In June 2024, we granted Regeneron global rights to develop, manufacture and commercialize cemdisiran as a monotherapy in addition to the license we previously granted Regeneron to investigate cemdisiran in combination with anti-C5 antibodies. Regeneron is now solely responsible for development, manufacturing, and commercialization of cemdisiran as a monotherapy and in combination with anti-C5 antibodies. Regeneron is evaluating cemdisiran in combination with its anti-C5 monoclonal antibody, pozelimab, in a Phase 3 clinical trial in paroxysmal nocturnal hemoglobinuria and as a monotherapy and in combination with pozelimab in Phase 3 clinical trials in myasthenia gravis and geographic atrophy.
Early-Stage Clinical Development Programs
Zilebesiran — Hypertension
Hypertension is a complex multifactorial disease clinically defined by most major guidelines as a systolic blood pressure of above 140 mm Hg and/or a diastolic blood pressure greater than 90 mm Hg, although the American College of Cardiology/American Heart Association guidelines define hypertension as a systolic blood pressure above 130 mm Hg and/or a diastolic blood pressure greater than 80 mm Hg. More than one billion people worldwide live with hypertension. In the U.S. alone, approximately 47 percent of adults live with hypertension, with more than half of patients on medication remaining above the blood pressure target level. Despite the availability of anti-hypertensive medications, there remains a significant unmet medical need, especially given the poor rates of adherence to existing daily oral medications and daily peak and trough effects, resulting
in inconsistent blood pressure control and an increased risk for stroke, heart attack and premature death. In particular, there are a number of high unmet need settings where novel approaches to hypertension warrant additional development focus, including patients with poor medication adherence and in patients with high cardiovascular risk.
Zilebesiran is an investigational, subcutaneously administered RNAi therapeutic targeting angiotensinogen, or AGT, a protein that plays an important role in regulating blood pressure, that is in development for the treatment of hypertension in high unmet need populations. AGT is the most upstream precursor in the renin-angiotensin-aldosterone system, a cascade which has a demonstrated role in blood pressure regulation and its inhibition has well-established anti-hypertensive effects. Zilebesiran inhibits the synthesis of AGT in the liver, potentially leading to durable reductions in AGT protein and ultimately, in the vasoconstrictor angiotensin II.
The KARDIA-1 Phase 2 clinical trial is designed to evaluate zilebesiran as a monotherapy across different doses administered quarterly and biannually. In September 2023, we reported positive topline results from the KARDIA-1 clinical trial, with zilebesiran meeting the primary endpoint and demonstrating greater than 15mmHg reduction of systolic blood pressure at three months of treatment compared to placebo at the two highest single doses evaluated. The KARDIA-2 Phase 2 clinical trial is evaluating the safety and efficacy of zilebesiran administered biannually as a concomitant therapy in patients whose blood pressure is not adequately controlled by a standard of care antihypertensive medication. In April 2024, we reported positive topline results from the KARDIA-2 clinical trial, with zilebesiran meeting the primary endpoint and demonstrating additive, placebo-adjusted reductions of up to 12.1 mmHg in 24-hour mean systolic blood pressure at Month 3 when zilebesiran was added to a thiazide-like diuretic, calcium channel blocker or angiotensin receptor blocker. At Month 6, zilebesiran demonstrated clinically significant and placebo-adjusted, time-adjusted reductions in office systolic blood pressure when added to indapamide, amlodipine and olmesartan, despite the addition of rescue antihypertensives at Month 3. In addition, zilebesiran resulted in clinically significant placebo-adjusted, time-adjusted reductions in 24-hour mean systolic blood when added to indapamide and amlodipine, sustained to Month 6. A non-statistically significant result was observed when zilebesiran was added to the maximum dose of olmesartan when evaluated by time-adjusted change from baseline in 24-hour mean systolic blood pressure at Month 6.
In April 2024, we initiated the KARDIA-3 Phase 2 clinical trial, which is evaluating zilebesiran as an add-on therapy in high cardiovascular risk patients with uncontrolled hypertension despite receiving two or more antihypertensives. We plan to announce topline results from KARDIA-3, and to initiate a Phase 3 cardiovascular outcomes trial of zilebesiran, in the second half of 2025.
In July 2023, we announced a collaboration with Roche to co-develop and co-commercialize zilebesiran. As part of our collaboration with Roche we are also developing ALN-AGT01 RVR-001, a reversal agent for zilebesiran using our REVERSIR platform. We initiated a Phase 1 clinical trial of ALN-AGT01 RVR-001 in healthy volunteers in November 2024. A description of the Roche Collaboration and License Agreement is included below under the heading “Our Collaboration and Licensing Strategy.”
Elebsiran (formerly ALN-HBV02 (VIR-2218)) – Chronic Hepatitis B and D Virus Infection
Elebsiran (formerly ALN-HBV02 (VIR-2218)) is a subcutaneously administered, investigational RNAi therapeutic targeting the HBV genome for the treatment of chronic HBV infection, which is being advanced by our collaborator, Vir. Elebsiran is designed to inhibit expression of all HBV proteins, including hepatitis B surface antigen. Almost one-third of the world’s population have previous or current HBV infection. Worldwide, more than 250 million people are chronically infected with HBV, and an estimated 1 million people die each year from complications of chronic HBV such as cirrhosis and hepatocellular carcinoma. Current treatment options include life-long suppressive antiviral therapies. There is a significant need for safe and convenient novel therapeutics that restore the host immune response, leading to control of the virus after a finite duration of therapy, which is the definition of a functional cure.
The safety and efficacy of elebsiran as part of a treatment regimen for chronic HBV infection is being assessed in the ongoing Phase 2 MARCH trial. Vir has also evaluated elebsiran in combination with tobevibart for the treatment of chronic hepatitis D virus, or CHD, and Vir expects to initiate a Phase 3 registrational study to evaluate the combination of elebsiran and tobevibart in CHD in the first half of 2025. We have the right to opt into a profit-sharing arrangement for elebsiran prior to the start of a Phase 3 study.
Mivelsiran (formerly ALN-APP) – Alzheimer’s Disease and Cerebral Amyloid Angiopathy
Mivelsiran is an investigational, intrathecally administered RNAi therapeutic targeting amyloid precursor protein, or APP, in development in collaboration with Regeneron for the treatment of AD, and CAA. Genetic mutations that increase production of APP or alter its cleavage cause early-onset AD, early-onset CAA, or both. Mivelsiran is designed to decrease APP mRNA in the CNS to decrease synthesis of APP protein and all downstream intracellular and extracellular APP-derived cleavage products, including amyloid beta (Aβ). Reducing APP protein production is expected to reduce the secretion of Aβ peptides that aggregate into extracellular amyloid deposits in AD and CAA and reduce the intraneuronal APP cleavage products that may trigger the formation of neurofibrillary tangles and cause neuronal dysfunction in AD. Mivelsiran is the first program utilizing our C16 conjugate technology, which enables enhanced delivery to cells in the CNS, to enter clinical development.
In early 2022, we initiated a Phase 1 clinical trial of mivelsiran in patients with early-onset AD, and in April 2023 and July 2023, we reported positive interim results from the ongoing single ascending dose part of the Phase 1 clinical trial. Further exploration of single doses of mivelsiran is ongoing in Part A of the Phase 1 clinical trial. In addition, the multiple-dose part of the study, Part B is now enrolling. In February 2024, we announced that the FDA had provided clearance to initiate Part B of the Phase 1 clinical trial at sites in the U.S. The FDA confirmed that multiple-dosing in the Phase 1 clinical trial may proceed at doses up to 180 mg given every six months, which covers all dose regimens planned to be explored in Part B. A partial clinical hold remains in place in the U.S. for higher or more frequent dosing regimens.
In October 2024, we announced positive initial results from the multiple-dose Part B portion of the Phase 1 clinical trial, and we expect to report additional interim results from Part B of the Phase 1 clinical trial in AD in the second half of 2025. We also expect to initiate a Phase 2 clinical trial of mivelsiran in patients with AD in the second half of 2025.
In July 2024, we initiated dosing in the cAPPricorn-1 Phase 2 clinical trial of mivelsiran in patients with CAA.
In May 2024, Regeneron notified us of its decision to opt-out of the further co-development of mivelsiran. As a result of Regeneron’s opt-out, we now have full global development and commercialization rights to mivelsiran in all indications, and we are responsible for all development and commercialization costs of mivelsiran other than Regeneron’s share of the Phase 1 budget.
ALN-HTT02 – Huntington’s Disease
HD is an autosomal dominantly inherited, fully penetrant, severely debilitating, and fatal neurodegenerative disease, caused by an abnormal expansion of cytosine-adenine-guanine repeats in exon 1 of the HTT gene. Typically, HD is an adult-onset neurodegenerative disease with a protracted but relentlessly progressive course. No disease modifying treatments have been developed or approved for HD, and treatment options are limited to management of symptoms and optimization of quality of life.
ALN-HTT02 is an investigational, intrathecally administered RNAi therapeutic targeting huntingtin, or HTT, that is in development in collaboration with Regeneron for the treatment of HD. ALN-HTT02 is designed to target a conserved sequence in exon 1 of the HTT messenger RNA, thereby reducing the expression of all isoforms of HTT protein, including shorter exon1-containing HTT fragments which have been implicated in disease pathology but are missed by many other HTT-lowering therapeutic strategies. Beyond the more inclusive exon 1 targeting strategy, our underlying C16-siRNA delivery platform may offer an opportunity to fully explore the potential efficacy of HTT-lowering via widespread distribution, deep and sustained lowering, and infrequent dosing regimens.
In September and November 2024, we presented nonclinical data supporting the tolerability of deep and sustained HTT-lowering in wild-type nonhuman primates after single and repeated intrathecal administration of ALN-HTT02. We also presented the design of a Phase 1b clinical trial. The Phase 1b clinical trial of ALN-HTT02 in adult patients with HD was initiated in late 2024, with the first patient dosed in November.
Additional Early-Stage and Preclinical Programs
During 2025, we plan to file four or more new investigational new drug applications, or INDs, or CTAs from our organic product engine. We also intend to continue to build on our progress with extrahepatic delivery during 2025, advancing our CNS programs under our collaboration with Regeneron, as well as continuing to advance our other extrahepatic delivery initiatives, including delivery to muscle and adipose tissues.
Our Collaboration and Licensing Strategy
Our business strategy is to develop and commercialize a broad pipeline of RNAi therapeutic products directed across a broad range of disease areas and indications. As part of this strategy, we have entered into, and expect to enter into additional, collaboration and licensing agreements as a means of obtaining resources, capabilities and funding to advance our investigational RNAi therapeutic programs. Our collaboration strategy is to form collaborations that create significant value for ourselves and our collaborators in the advancement of RNAi therapeutics. We expect these collaborations to provide us with financial support in the form of upfront cash payments, license fees, equity investments, research, development, and sales and marketing support and/or funding, milestone payments and/or royalties or profit sharing based on sales of RNAi therapeutics.
Below is a brief description of our key collaborations.
Product Collaborations
Roche. In July 2023, we entered into the Roche Collaboration and License Agreement, pursuant to which we and Roche established a worldwide, strategic collaboration for the joint development of pharmaceutical products containing zilebesiran. Under the Roche Collaboration and License Agreement, we granted to Roche (i) co-exclusive rights to develop zilebesiran worldwide and commercialize zilebesiran in the U.S., (ii) exclusive rights to commercialize zilebesiran outside of the U.S., and (iii) non-exclusive rights to manufacture zilebesiran for the development and commercialization of zilebesiran outside of the U.S. Roche made an upfront payment of $310.0 million. We achieved the development milestone associated with the dosing of the first patient in the KARDIA-3 Phase 2 clinical trial in April 2024 and received a $65.0 million development milestone
payment from Roche. In addition, we are eligible to receive up to an additional $2.45 billion in contingent payments based on the achievement of specified development, regulatory and sales-based milestones. We are responsible for forty percent (40%), and Roche is responsible for sixty percent (60%), of development costs incurred in the conduct of development activities that support regulatory approval of zilebesiran globally. We and Roche share equally (50/50) all costs incurred in connection with development activities that are conducted to support regulatory approval of zilebesiran in the U.S. Roche will be solely responsible for costs incurred in connection with commercialization of zilebesiran outside of the U.S. and will pay us tiered, low double digit royalties based on net sales of zilebesiran on a country-by-country basis outside of the U.S. during the royalty term. We and Roche will share equally (50/50) profits and losses (including commercialization costs) of zilebesiran in the U.S.
For more information regarding the Roche Collaboration and License Agreement, including the ongoing or expected financial and accounting impact on our business, please see Note 4, Net Revenues from Collaborations, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K.
Regeneron. In April 2019, we entered into a global, strategic collaboration with Regeneron to discover, develop and commercialize RNAi therapeutics for a broad range of diseases by addressing therapeutic targets expressed in the eye and CNS, in addition to a select number of targets expressed in the liver, which we refer to as the Regeneron Collaboration. The Regeneron Collaboration is governed by a Master Agreement, referred to as the Regeneron Master Agreement, which became effective in May 2019.
Under the terms of the Regeneron Collaboration, we are working exclusively with Regeneron to discover RNAi therapeutics for eye and CNS diseases for an initial research period of up to seven years, which we refer to as the Initial Research Term. Regeneron has an option to extend the Initial Research Term for up to an additional five years by paying a research term extension fee of $300.0 million. The Regeneron Collaboration also covers a select number of RNAi therapeutic programs designed to target genes expressed in the liver. We retain broad global rights to all of our liver-directed clinical and preclinical pipeline programs that are not part of the Regeneron Collaboration.
Regeneron leads development and commercialization for all programs targeting eye diseases (subject to limited exceptions), entitling us to certain potential milestone and royalty payments pursuant to the terms of a license agreement, the form of which has been agreed upon by the parties. We and Regeneron are alternating leadership on CNS and liver programs, with the lead party retaining global development and commercial responsibility.
In August 2019, in connection with the Regeneron Master Agreement, we and Regeneron entered into (i) a co-co collaboration agreement covering the development of cemdisiran, our C5 siRNA, as a monotherapy for C5 complement-mediated diseases, or the C5 Co-Co Collaboration Agreement, and (ii) a license agreement to evaluate anti-C5 antibody-siRNA combinations for C5 complement-mediated diseases including evaluating the combination of Regeneron’s pozelimab and cemdisiran, or the C5 License Agreement. In June 2024, we entered into an amended and restated C5 License Agreement, or the Amended C5 License Agreement, which terminated the C5 Co-Co Collaboration Agreement and granted Regeneron a worldwide license to cemdisiran as a monotherapy in addition to the license to cemdisiran in combination with anti-C5 antibodies. Through the Amended C5 License Agreement, Regeneron is now solely responsible for development, manufacturing, and commercialization of cemdisiran as a monotherapy and in combination with anti-C5 antibodies. Regeneron provided us with an upfront payment of $10.0 million and we will receive certain milestone payments upon receipt of regulatory approval for cemdisiran as a monotherapy, and tiered, double-digit royalties on net sales. The Amended C5 License Agreement did not change our rights to receive low double-digit royalties and commercial milestones of up to $325.0 million on any potential product sales if cemdisiran is used as part of a combination product.
In May 2024, Regeneron notified us of its decision to opt out of the further co-development of mivelsiran, an investigational RNAi therapeutic in development for the treatment of hereditary CAA and autosomal dominant Alzheimer’s Disease under our co-co collaboration agreement with respect to mivelsiran. As a result of Regeneron’s opt-out, we now have full global development and commercialization rights to mivelsiran in all indications, and we are responsible for all development and commercialization costs of mivelsiran other than Regeneron’s share of the then-current Phase 1 budget. Regeneron will no longer share potential future profits from sales of mivelsiran with us, although we remain subject to certain financial obligations to Regeneron under the mivelsiran co-co collaboration agreement. We continue to advance multiple other programs with Regeneron.
For more information regarding the Regeneron Collaboration, including the ongoing or expected financial and accounting impact on our business, please see Note 4, Net Revenues from Collaborations, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K.
Sanofi. We formed a broad strategic alliance with Sanofi in 2014. In January 2018, we and Sanofi amended our 2014 collaboration and entered into the Exclusive License Agreement, referred to as the Exclusive TTR License, under which we were granted exclusive rights to pursue the further global development and commercialization of all TTR products, including ONPATTRO, AMVUTTRA and any back-up products, and the ALN-AT3 Global License Terms, referred to as the AT3 License Terms, under which Sanofi has the exclusive right to pursue the further global development and commercialization of fitusiran and any back-up products. Under the Exclusive TTR License, Sanofi is eligible to receive (i) royalties up to 25%
increasing over time, based on annual net sales of ONPATTRO in territories excluding the U.S., Canada and Western Europe, provided royalties on annual net sales of ONPATTRO in Japan were set at 25% beginning at the effective date of the Exclusive TTR License and (ii) tiered royalties on global annual net sales of AMVUTTRA across all indications in the following tiers: 15% of global annual net sales of $0 to $150.0 million; 17.5% of global annual net sales greater than $150.0 million to $300.0 million; 20% of global annual net sales greater than $300.0 million to $500.0 million; 25% of global annual net sales greater than $500.0 million to $1.50 billion; and 30% of global annual net sales in excess of $1.50 billion. In April 2019, we and Sanofi amended and restated the AT3 License Terms to modify certain of the business terms. The material collaboration terms for fitusiran were unchanged. Under the amended and restated AT3 License Terms, we are eligible to receive tiered royalties of 15% to 30% based on global annual net sales of fitusiran by Sanofi, its affiliates and its sublicensees.
Novartis. In February 2013, we entered into an exclusive, worldwide license with MDCO (acquired by Novartis AG in January 2020) pursuant to which MDCO was granted the right to develop, manufacture and commercialize RNAi therapeutics targeting proprotein convertase subtilisin/kexin type 9 for the treatment of hypercholesterolemia and other human diseases, including Leqvio.
For more information regarding the MDCO agreement, including its ongoing financial and accounting impact on our business, please see Note 4, Net Revenues from Collaborations, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K.
Strategic Financing Collaboration
The Blackstone Group Inc. In April 2020, we entered into a strategic financing collaboration with certain affiliates of Blackstone to accelerate our advancement of RNAi therapeutics. In connection with the collaboration, Blackstone agreed to provide us up to $2.00 billion in financing, including $1.00 billion in committed payments to acquire 50% of royalties and 75% of commercial milestones payable to us in connection with sales of Leqvio, up to $750.0 million in a first lien senior secured term loan, and up to $150.0 million towards the development of vutrisiran and zilebesiran pursuant to a co-development agreement that was executed in August 2020. In November 2021, Blackstone opted in to Phase 2 clinical trial funding of zilebesiran and has funded $26.0 million in Phase 2 development costs of zilebesiran. Blackstone also has the right, but is not obligated, to fund up to $54.0 million for development costs related to a Phase 3 clinical trial of zilebesiran. As part of the strategic financing collaboration, Blackstone also purchased an aggregate of $100.0 million of our common stock. Please see Note 9, Liability Related to the Sale of Future Royalties, and Note 10, Development Derivative Liability, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K for additional details on our transaction with Blackstone, including its ongoing financial and accounting impact on our business.
Other Collaboration and License Agreements
We intend to continue to evaluate and explore opportunities through collaboration and licensing arrangements, and may enter into new collaborations to advance certain products or disease areas. We also have entered into license agreements to obtain rights to intellectual property in the field of RNAi. In addition, because delivery of RNAi therapeutics has historically been an important objective of our research activities, we have entered into various collaboration and licensing arrangements with other companies and academic institutions to gain access to delivery technologies, including various LNP delivery technologies, and we may enter into such agreements in the future to gain access to products or technologies.
Below is a brief description of certain other collaboration and license agreements we have entered into.
PeptiDream, Inc. In July 2021, we entered into a license and collaboration agreement with PeptiDream to discover and develop peptide-siRNA conjugates to create multiple opportunities to deliver RNAi therapeutics to tissues outside the liver. Through this collaboration, we are collaborating with PeptiDream to select and optimize peptides for targeted delivery of small siRNA molecules to a wide range of cell types and tissues via specific interactions with receptors expressed on the target cells.
Dicerna Pharmaceuticals, Inc. In April 2020, we and Dicerna entered into a Patent Cross-License Agreement, pursuant to which each party agreed to cross-license its respective intellectual property related to our lumasiran program and Dicerna’s nedosiran program, each for the treatment of PH.
Ionis Pharmaceuticals, Inc. In January 2015, we and Ionis Pharmaceuticals, Inc., or Ionis, entered into a second amended and restated strategic collaboration and license agreement, which we further amended in July 2015, or the 2015 Ionis agreement. The 2015 Ionis agreement provides for certain exclusive cross-licenses of intellectual property on eight disease targets, providing each company with exclusive RNA therapeutic license rights for four programs, and extended the parties’ existing non-exclusive technology cross-license through April 2019. Pursuant to the 2015 Ionis agreement, Ionis granted to us an exclusive, low single-digit royalty-bearing license to its chemistry, motif, mechanism and target-specific intellectual property for oligonucleotide therapeutics against four targets. In exchange, we granted to Ionis an exclusive, low single-digit royalty-bearing license to our chemistry, motif, mechanism and target-specific intellectual property for oligonucleotide therapeutics against four targets. Under the original agreement, Ionis licensed to us its patent estate related to antisense motifs and mechanisms and oligonucleotide chemistry for double-stranded RNAi products in exchange for a technology access fee, participation in fees for our collaboration programs and future milestone and royalty payments from us for programs that
incorporate Ionis’ intellectual property. We have the right to use Ionis’ intellectual property in our development programs or in collaborations, and Ionis agreed not to grant licenses under these patents to any other organization for the discovery, development and commercialization of double-stranded RNA products designed to work through an RNAi mechanism, except in the context of a collaboration in which Ionis plays an active role. In turn, in exchange for option fees, and future milestone and royalty payments from Ionis for RNAi programs that incorporate certain of our intellectual property, we non-exclusively licensed to Ionis our patent estate relating to antisense motifs and mechanisms and oligonucleotide chemistry to research, develop and commercialize single-stranded antisense therapeutics, single stranded RNAi therapeutics and to research double-stranded RNAi compounds. Ionis also received a license to develop and commercialize double-stranded RNAi drugs targeting a limited number of therapeutic targets on a non-exclusive basis.
Intellectual Property, Proprietary Rights and Exclusivities
We have devoted considerable effort and resources through both in-licensing and filing patent applications on our own inventions, as well as protecting our trade secrets and know-how to establish what we believe to be a strong intellectual property position relevant to RNAi therapeutic products and delivery technologies. In this regard, we have amassed a portfolio of patents, patent applications and other intellectual property covering:
•fundamental aspects of the structure and uses of siRNAs, including their use as therapeutics, and RNAi-related mechanisms;
•chemical modifications to siRNAs that improve their suitability for therapeutic and other uses;
•compositions of siRNAs directed to specific targets as well as their methods of use, including as therapeutics and diagnostics;
•delivery technologies, such as in the fields of siRNA conjugates, including carbohydrate, lipophilic and other conjugates as well as cationic liposomes and other delivery vehicles; and
•all aspects of our development candidates and marketed products, with an additional level of protection for trademarks related to our marketed products.
In addition to patents and trademarks for our marketed products, we seek to obtain all available regulatory exclusivities for our marketed products, including data and orphan exclusivities in the relevant jurisdictions.
Key Patents and Regulatory Exclusivities
We typically obtain protection of our product candidates with patents and patent applications directed to compositions of matter and their uses. Below is a summary of selected granted patents that we own or control covering products marketed by us in the U.S. and Europe.
ONPATTRO
| | | | | | | | | | | | | | |
| Patent Number | Country/Region* | Patent Type | Expiration Date** | Owner/Licensor |
| 8168775 | United States | Compositions of Matter & Methods of Use | 8/10/2032 | Alnylam |
| 8334373 | United States | Compositions of Matter & Methods of Use | 5/27/2025 | Alnylam |
| 8741866 | United States | Compositions of Matter & Methods of Use | 10/20/2029 | Alnylam |
| 9234196 | United States | Compositions of Matter & Methods of Use | 10/20/2029 | Alnylam |
| 8802644 | United States | Compositions of Matter & Methods of Use | 10/21/2030 | Arbutus Biopharma |
| 8158601 | United States | Compositions of Matter & Methods of Use | 11/10/2030 | Arbutus Biopharma |
| 2937418 | Europe | Compositions of Matter & Methods of Use | 8/28/2033 | Alnylam |
| 2344639 | Europe | Compositions of Matter & Methods of Use | 10/20/2029 | Alnylam |
| 2440183 | Europe | Compositions of Matter | 10/21/2030 | Arbutus Biopharma |
_________________________________________
* Shown here are selected granted patents in the U.S. and Europe. Additional granted and pending patents in the U.S., Europe and other countries may be available.
** Expiration dates listed here include any granted or anticipated patent term extensions and supplemental protection certificates but exclude any pediatric extensions that may be available.
In addition, in connection with our FDA approval on August 10, 2018, the FDA granted ONPATTRO orphan drug exclusivity, or ODE, until August 10, 2025. In connection with our EMA approval on August 26, 2018, the EMA granted ONPATTRO marketing exclusivity and ODE until August 26, 2028.
AMVUTTRA
| | | | | | | | | | | | | | |
| Patent Number | Country/Region* | Patent Type | Expiration Date** | Owner/Licensor |
| 8106022 | United States | Compositions of Matter & Methods of Use | 12/12/2029 | Alnylam |
| 8828956 | United States | Compositions of Matter & Methods of Use | 12/4/2028 | Alnylam |
| 9370581 | United States | Compositions of Matter & Methods of Use | 12/4/2028 | Alnylam |
| 9399775 | United States | Compositions of Matter & Methods of Use | 11/16/2032 | Alnylam |
| 10131907 | United States | Compositions of Matter & Methods of Use | 8/24/2028 | Alnylam |
| 10208307 | United States | Compositions of Matter & Methods of Use | 7/28/2036 | Alnylam |
| 10570391 | United States | Compositions of Matter | 11/16/2032 | Alnylam |
| 10612024 | United States | Compositions of Matter & Methods of Use | 8/14/2035 | Alnylam |
| 10683501 | United States | Methods of Use | 7/28/2036 | Alnylam |
| 10806791 | United States | Compositions of Matter | 12/4/2028 | Alnylam |
| 11286486 | United States | Methods of Use | 7/28/2036 | Alnylam |
| 11401517 | United States | Compositions of Matter & Methods of Use | 8/14/2035 | Alnylam |
| 3301177 | Europe | Compositions of Matter & Methods of Use | 11/16/2032 | Alnylam |
| 3329002 | Europe | Compositions of Matter & Methods of Use | 7/28/2036 | Alnylam |
_________________________________________
* Shown here are selected granted patents in the U.S. and Europe. Additional granted and pending patents in the U.S., Europe and other countries may be available.
** Expiration dates listed here do not account for any patent term extensions, supplemental protection certificates or pediatric extensions that may be available.
In addition, in connection with our FDA approval on June 13, 2022, the FDA granted AMVUTTRA new chemical entity, or NCE, exclusivity until June 13, 2027. In connection with our EMA approval on September 15, 2022, the EMA granted AMVUTTRA marketing exclusivity and ODE until September 15, 2032.
GIVLAARI
| | | | | | | | | | | | | | |
| Patent Number | Country/Region* | Patent Type | Expiration Date** | Owner/Licensor |
| 8106022 | United States | Compositions of Matter & Methods of Use | 12/12/2029 | Alnylam |
| 8828956 | United States | Compositions of Matter & Methods of Use | 12/4/2028 | Alnylam |
| 9133461 | United States | Compositions of Matter & Methods of Use | 5/14/2033 | Alnylam/Icahn School of Medicine at Mount Sinai |
| 9150605 | United States | Compositions of Matter | 8/28/2025 | Ionis Pharmaceuticals |
| 9631193 | United States | Methods of Use | 3/15/2033 | Alnylam/Icahn School of Medicine at Mount Sinai |
| 10119143 | United States | Compositions of Matter & Methods of Use | 10/3/2034 | Alnylam/Icahn School of Medicine at Mount Sinai |
| 10125364 | United States | Compositions of Matter & Methods of Use | 3/15/2033 | Alnylam/Icahn School of Medicine at Mount Sinai |
| 10131907 | United States | Compositions of Matter & Methods of Use | 8/24/2028 | Alnylam |
| 2836595 | Europe | Compositions of Matter & Methods of Use | 4/10/2033 | Alnylam/Icahn School of Medicine at Mount Sinai |
_________________________________________
* Shown here are selected granted patents in the U.S. and Europe. Additional granted and pending patents in the U.S., Europe and other countries may be available.
** Expiration dates listed here do not account for any patent term extensions, supplemental protection certificates or pediatric extensions that may be available.
In addition, in connection with our FDA approval on November 20, 2019, the FDA granted GIVLAARI ODE until November 20, 2026. In connection with our EMA approval on March 2, 2020, the EMA granted GIVLAARI Marketing Exclusivity and ODE until March 2, 2030.
OXLUMO
| | | | | | | | | | | | | | |
| Patent Number | Country/Region* | Patent Type | Expiration Date** | Owner/Licensor |
| 8106022 | United States | Compositions of Matter & Methods of Use | 12/12/2029 | Alnylam |
| 8828956 | United States | Compositions of Matter & Methods of Use | 12/4/2028 | Alnylam |
| 9828606 | United States | Compositions of Matter | 12/26/2034 | Dicerna Pharmaceuticals |
| 10131907 | United States | Compositions of Matter & Methods of Use | 8/24/2028 | Alnylam |
| 10435692 | United States | Methods of Use | 12/26/2034 | Dicerna Pharmaceuticals |
| 10465195 | United States | Compositions of Matter & Methods of Use | 12/26/2034 | Dicerna Pharmaceuticals |
| 10478500 | United States | Compositions of Matter & Methods of Use | 10/9/2035 | Alnylam |
| 10487330 | United States | Compositions of Matter & Methods of Use | 12/26/2034 | Dicerna Pharmaceuticals |
| 10612024 | United States | Compositions of Matter | 8/14/2035 | Alnylam |
| 10612027 | United States | Compositions of Matter & Methods of Use | 8/14/2035 | Alnylam |
| 3087184 | Europe | Compositions of Matter | 12/26/2034 | Dicerna Pharmaceuticals |
_________________________________________
* Shown here are selected granted patents in the U.S. and Europe. Additional granted and pending patents in the U.S., Europe and other countries may be available.
** Expiration dates listed here do not account for any patent term extensions, supplemental protection certificates or pediatric extensions that may be available.
In addition, in connection with our FDA approval on November 23, 2020, the FDA granted OXLUMO NCE exclusivity until November 23, 2025, and ODE until November 23, 2027. In connection with our EMA approval on November 19, 2020, the EMA granted OXLUMO Marketing Exclusivity and ODE until November 19, 2030.
Trademarks
We file trademarks to protect our corporate brand and our products. Typically, we file trademark applications in the U.S., Europe and elsewhere in the world as appropriate. In addition to multiple pending trademark applications in the U.S. and other major countries, we have registered trademarks in the U.S., including but not limited to Alnylam® and the Alnylam logo, as well as ONPATTRO® and the ONPATTRO logo, AMVUTTRA® and the AMVUTTRA logo, GIVLAARI® and the GIVLAARI logo and OXLUMO® and the OXLUMO logo.
Competition
Competition in the pharmaceutical marketplace remains intense, with hundreds of companies competing to discover, develop, and market new drugs. For our marketed and discovery assets, we face a broad spectrum of current and potential competitors, ranging from very large, global pharmaceutical companies with significant resources, to biotechnology companies with resources and expertise comparable to our own, to smaller biotechnology companies with fewer resources and less expertise than those we currently possess. We believe that for most or all of our drug development programs, there will be one or more competing programs being marketed and/or under development at other companies.
The competition we face can be grouped into three broad categories:
•other companies working to develop RNAi and microRNA therapeutic products;
•companies developing technology known as antisense, which, like RNAi, attempts to silence the activity of specific genes by targeting the mRNAs copied from them; and
•marketed products and development programs for therapeutics that treat the same diseases for which we are marketing products or developing treatments.
As RNAi therapies expand beyond rare diseases to other more common conditions, an increasing number of other companies are investing in and developing RNAi therapeutic products. Some of these companies are seeking, as we are, to develop chemically synthesized siRNAs as drugs. Others are following a gene therapy approach, with the goal of treating patients with synthetic, exogenously introduced genes designed to produce siRNA-like molecules within cells.
Companies working on chemically synthesized siRNAs include Arrowhead Pharmaceuticals, Inc., or Arrowhead, and its collaborators, Takeda Pharmaceutical Company Ltd., Janssen Pharmaceuticals, Inc., GlaxoSmithKline plc, and Amgen Inc.; Quark Pharmaceuticals, Inc.; F. Hoffmann-La Roche Ltd.; Silence Therapeutics plc and its collaborators, AstraZeneca plc, Jiangsu Hansoh Pharmaceuticals Group Co., Ltd., and Mallinckrodt plc; Arbutus Biopharma Corp., or Arbutus; Sylentis, S.A.U., or Sylentis; Novo Nordisk and its collaborators, Aro Biotherapeutics, Boehringer Ingelheim, and Eli Lilly and Company; and our collaborators Regeneron, Sanofi and Vir.
The competitive landscape continues to expand, and we expect that additional companies will initiate programs focused on the development of RNAi therapeutic products using the approaches described above, as well as potentially new approaches that may result in more rapid development of RNAi therapeutics or more effective technologies for RNAi drug development or delivery.
Competing Drugs for Our Marketed Products and Late-Stage Investigational RNAi Therapeutics
ATTR Amyloidosis Franchise. ATTR amyloidosis is an underdiagnosed, rapidly progressive, debilitating and fatal disease caused by misfolded TTR proteins, which accumulate as amyloid deposits in various parts of the body, including the nerves, heart and gastrointestinal tract. Patients may present with polyneuropathy, cardiomyopathy, or both manifestations of disease.
hATTR Amyloidosis with Polyneuropathy (hATTR-PN). In addition to ONPATTRO and AMVUTTRA, currently approved therapies to treat hATTR-PN include TEGSEDI (inotersen, not available in the U.S.) marketed by Ionis and WAINUA (eplontersen) marketed by Ionis and AstraZeneca plc. There are a number of product candidates in clinical development for the treatment of hATTR-PN, including nexiguran ziclumeran (nex-z, formerly NTLA-2001) which is being developed by Intellia Therapeutics, Inc., or Intellia, and Regeneron and is in Phase 1 clinical development; and AT0-2 which is being developed by Attralus, Inc. and is in Phase 2 clinical development.
ATTR Amyloidosis with Cardiomyopathy (ATTR-CM). Currently approved therapies to treat ATTR-CM include VYNDAQEL/VYNDAMAX (tafamidis) marketed by Pfizer Inc. and recently approved ATTRUBY (acoramidis) marketed by BridgeBio Pharma, Inc., or BridgeBio. We believe that these approved drugs will compete with AMVUTTRA for the treatment of ATTR-CM in the event that vutrisiran receives regulatory approval in the U.S. and other jurisdictions. Additionally, there are a number of other product candidates in clinical development, including WAINUA (eplontersen), which is being developed by AstraZeneca and Ionis and is in Phase 3 clinical development, nexiguran ziclumeran (nex-z, formerly NTLA-2001) which is being developed by Intellia and Regeneron and is in Phase 3 clinical development; ALXN2220 (formerly NI006) which is being developed by Neurimmune AG and Alexion, AstraZeneca Rare Disease and is in Phase 3 clinical development; coramitug (NNC-6019-0001) which is being developed by Novo Nordisk and is in Phase 2 clinical development; and AT0-2 which is being developed by Attralus, Inc. and is in Phase 2 clinical development.
Acute Hepatic Porphyria. GIVLAARI is currently the only approved therapy for prophylactic treatment of AHP in the U.S. and EU. Nevertheless, Recordati S.p.A has two products, PANHEMATIN and NORMOSANG, that are approved in the U.S. and EU, respectively, for the treatment of acute porphyria attacks, and some physicians may prescribe these therapies off-label for the prophylactic treatment of AHP.
Primary Hyperoxaluria. In addition to OXLUMO, Novo Nordisk’s RIFVLOZA (nedosiran) is approved in the U.S. for treatment of PH1 to lower urinary oxalate levels in children nine years of age and older and adults with relatively preserved kidney function. Other currently used treatments include hyper-hydration, vitamin B6, oral citrate, and dual liver/kidney transplantation. Transplantation is a costly and invasive procedure and carries significant morbidity and mortality risk In addition, several companies have investigational drugs in clinical development for the treatment of primary hyperoxaluria, including Biocodex, Inc. in collaboration with M8 Pharmaceuticals, Inc., and YolTech Therapeutics (China only). We believe that these approved products and product candidates, if approved, could compete with OXLUMO.
Hypercholesterolemia. In addition to Leqvio, several companies have approved products for the treatment of hypercholesterolemia, including Amgen Inc. (REPATHA), Sanofi S.A. (PRALUENT), Amarin Corporation (VASCEPA), Esperion Therapeutics, Inc. (NEXLETOL), and Regeneron (EVKEEZA). In addition, LIB Therapeutics, LLC submitted a BLA to the FDA for lerodalcibep for the treatment of primary hyperlipidemia, including heterozygous and homozygous familial hypercholesterolemia (HeFH / HoFH), There are also several companies with investigational drugs in varying stages of clinical development for the treatment of hypercholesterolemia, including Merck & Co., Inc., Jiangsu Hengrui Pharmaceuticals Co., Ltd., Arrowhead, and Verve Therapeutics, Inc.
Hemophilia. There are several approved products for the treatment of hemophilia, including: Factor VIII replacement products, Factor IX replacement products, factor replacement products with extended half-lives, and a bispecific antibody that mimics Factor VIII. For the treatment of individuals with inhibitors, there is an approved Factor VIIa replacement product and an activated prothrombin complex concentrate, as well as a bispecific antibody that mimics Factor VIII. In addition, new, innovative molecules have been recently approved or are currently in development for treatment of hemophilia A and B, both with and without inhibitors. BioMarin’s ROCTAVIAN is approved for the treatment of Hemophilia A. ROCTAVIAN is a gene therapy that uses virus-like particles to deliver a functional section of a particular gene into the liver cells of a person with hemophilia. A number of companies are also actively developing similar gene therapy products for the treatment of hemophilia. We believe that these approved products and product candidates could compete with fitusiran if it receives regulatory approval for the treatment of hemophilia.
Regulatory Matters
U.S. Regulatory Considerations
The research, testing, approval, manufacture and marketing of drug products and their delivery systems, as well as their pricing (including discounts and rebates) and price reporting are extensively regulated in the U.S. and the rest of the world. In the U.S., drugs are subject to rigorous regulation by the FDA. The Federal Food, Drug, and Cosmetic Act, or FDCA, and other federal and state statutes and regulations govern, among other things, the research, development, testing, approval, manufacture, storage, record keeping, reporting, labeling, advertising, promotion, marketing, import, export, and distribution of drug products. Regulations and administrative guidance often are revised or reinterpreted by the agencies in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted or if FDA or comparable ex-U.S. regulations, guidance or interpretations will change. Failure to comply with the applicable regulatory requirements may subject a company to a variety of administrative or judicially-imposed sanctions and the inability to obtain or maintain required approvals to test or market drug products. These sanctions could include, among other things, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, exclusion from participation in government programs or contracts, clinical holds, injunctions, fines, civil penalties or criminal prosecution.
New Drug Development Process
The steps ordinarily required before a new drug product may be marketed in the U.S. include:
•completion of nonclinical laboratory tests, animal tests and formulation studies conducted according to good laboratory practices, or GLP, and applicable regulations;
•the submission to the FDA of an IND, which must become effective prior to commencement of clinical testing in the U.S.;
•approval by an institutional review board, or IRB, representing each clinical site before each trial may be initiated;
•completion of adequate and well-controlled clinical trials in accordance with good clinical practices, or GCP, to establish that the drug product is safe and effective for the indication and other conditions of use for which FDA approval is sought;
•submission to the FDA of an NDA (or supplemental NDA for approved products);
•satisfactory completion of a pre-approval FDA inspection of the manufacturing facility or facilities at which the product will be produced to assess compliance with current good manufacturing practices, or cGMP; and
•FDA review and approval of the NDA (or supplemental NDA for approved products.
Satisfaction of the FDA’s pre-market approval requirements typically takes several years, but may vary substantially depending upon the complexity of the product and the nature of the disease. Government regulation may delay, limit or prevent marketing of product candidates for a considerable period of time and impose costly procedures on a company’s activities. Success in early-stage clinical trials does not necessarily assure success in later-stage clinical trials. Data obtained from clinical activities, including but not limited to the data derived from our clinical trials for product candidates, are not always conclusive and may be subject to alternative interpretations that could delay, limit or even prevent regulatory approval. Even if a product receives regulatory approval, later discovery of previously unknown problems with a product, including new safety risks from commercial use, clinical or nonclinical data or manufacturing issues, may result in restrictions on the product or even complete withdrawal of the product from the market.
Once a drug candidate is selected for development, it enters the nonclinical testing stage. Nonclinical tests include laboratory evaluation of product chemistry and formulation, as well as animal pharmacology and toxicology studies. The conduct of the nonclinical tests and formulation of compounds for testing must comply with applicable regulations and requirements, including in some cases the FDA’s GLP requirements and the Animal Welfare Act. The results of nonclinical testing are submitted to the FDA as part of an IND, together with chemistry, manufacturing and controls information, analytical and stability data, a proposed clinical trial protocol and other information. Clinical testing in humans may not commence until an IND is in effect. Nonclinical testing typically continues even after the IND is submitted.
An IND becomes effective 30 days after receipt by the FDA unless the FDA notifies the sponsor that the proposed investigation(s) are subject to a clinical hold. If the FDA imposes a clinical hold, or partial clinical hold, the FDA’s concerns must be resolved prior to the commencement of clinical trials, or the FDA can enforce other changes to the clinical development program or clinical trial(s).The IND review process can result in substantial delay and expense. If the FDA accepts the IND, the drug can then be studied in human clinical trials to determine if the product candidate is safe and effective.
Clinical trials involve the administration of an investigational new drug to healthy volunteers or patients under the supervision of a qualified investigator. Clinical studies are conducted under protocols detailing, among other things, the objectives of the trial and the safety and effectiveness criteria to be evaluated. Each protocol involving testing on human subjects in the U.S. must be submitted to the FDA as part of the IND. In addition, clinical trials must be conducted in compliance with federal regulations and requirements, including GCP, to assure data integrity and protect the rights, safety and well-being of trial participants. These requirements include, among other things, that all research subjects must provide their informed consent prior to participating in any clinical study and that each clinical trial must be reviewed and approved by an IRB either centrally or individually at each institution at which the clinical trial will be conducted. The IRB must review and approve, among other things, the study protocol and informed consent information to be provided to study subjects. An IRB must operate in compliance with FDA regulations.
Clinical trials to support NDAs are typically conducted in three sequential phases, which may overlap or be combined.
•In Phase 1, the initial introduction of the drug into a limited number of healthy human subjects or patients, the drug is tested primarily to assess safety, tolerability, pharmacokinetics, pharmacological actions and metabolism associated with increasing doses.
•Phase 2 usually involves trials in a limited patient population with the specified disease or condition the drug is intended to treat to assess the optimum dosage and dose regimen, identify possible adverse effects and safety risks, and to preliminarily evaluate the efficacy of the drug in the indication being studied.
•Phase 3 clinical trials further evaluate the drug’s clinical efficacy, side effects and safety in an expanded patient population, typically at geographically dispersed clinical trial sites, to establish the overall benefit-risk relationship of the drug and to provide an adequate basis for regulatory approval and labeling of the drug.
Phase 1, Phase 2 or Phase 3 testing of any drug candidates may not be completed successfully within any specified time period, if at all. Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and more frequently in other situations, and the occurrence of serious adverse events must also be reported. The FDA, IRB, or the clinical trial sponsor may suspend or discontinue a clinical trial at any time for various reasons, including a finding that the subjects or patients are being exposed to an unacceptable health risk. Finally, sponsors are required to publicly disseminate information about certain ongoing and completed clinical trials on ClinicalTrials.gov, a government website administered by the National Institutes of Health, or NIH.
Post-approval trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication and are commonly intended to generate additional safety data regarding use of the product in a clinical setting. In certain instances, the FDA may mandate the performance of Phase 4 clinical trials as a condition of approval of an NDA or BLA.
New Drug Applications
We believe that any RNAi product candidate we develop, whether for the treatment of ATTR amyloidosis, AHP, PH1, hypercholesterolemia or the various indications targeted in our clinical development or nonclinical discovery programs, will be regulated by the FDA as a new drug that is not considered to be a biologic, and thus will require an NDA rather than a biologics license agreement, or BLA. FDA approval of an NDA is required before commercial distribution of a new drug may begin in the U.S. An NDA must include the results of extensive nonclinical, clinical and other testing, as described above, a compilation of data relating to the product’s pharmacology, information related to the product’s chemistry and manufacturing, proposed labeling and other information.
In addition, under the Pediatric Research Equity Act of 2023, or PREA, an NDA for a new active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration typically must contain data assessing the safety and effectiveness for the claimed indication in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective, although deferrals or full or partial waivers may be available in some circumstances.
The cost of preparing and submitting an NDA is substantial. Under the PDUFA, as amended, each NDA must be accompanied by an application fee. For fiscal year 2024, the application fee for each NDA requiring clinical data is approximately $4.0 million. The PDUFA also imposes an annual program fee for each approved prescription drug product, which has been set at approximately $417,000 for fiscal year 2024. The FDA adjusts the PDUFA user fees on an annual basis. Fee waivers, reductions and exceptions are available in certain circumstances. Additionally, no application fees are assessed on NDAs for products designated as orphan drugs, unless the NDA also includes a non-orphan indication.
The FDA conducts a preliminary review of all NDAs within the first 60 days after submission before accepting them for filing to determine whether they are sufficiently complete to permit substantive review. During that time, the FDA may request additional information before deciding whether to accept an NDA for filing. If the FDA determines that an NDA is not sufficiently complete to permit substantive review, it will issue a refuse to file determination and the NDA will not be substantively reviewed by the FDA. If the submission is accepted for filing, the FDA begins an in-depth review of the NDA. The FDA has agreed to specified performance goals regarding the timing of the completion of its review of NDAs, although the goals are not binding and the FDA does not always meet these goals. Under the goals and policies agreed to by the FDA under PDUFA, the FDA has ten months from filing in which to complete its initial review of a standard application and respond to the sponsor, and six months for a priority review application. A major amendment to an NDA submitted at any time during the review cycle, including in response to a request from the FDA, may extend the goal date by three months.
For novel drug products or drug products that present difficult questions of safety or efficacy, the FDA may refer the application to an advisory committee, which is typically in the form of a panel that includes independent clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it carefully considers and often follows such recommendations. In addition, before approving an NDA, the FDA may inspect the facility or facilities where the drug is manufactured and tested to gain assurance that the manufacturing facility or facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity, and are in compliance with regulations governing cGMP requirements. The FDA will also often conduct a bioresearch monitoring inspection of select clinical trial sites to assure data integrity and compliance with applicable GCP requirements, and could also conduct GCP inspections of the sponsor.
If the FDA’s evaluation of an NDA is favorable, the FDA may issue an approval letter, which authorizes commercial marketing of the drug with specific prescribing information for a specific indication. The approved indication may be narrower than what was proposed by the applicant or for a narrower patient population than the population studied in clinical trials. As a condition of NDA approval, the FDA may require post-approval evaluations, including Phase 4 trials, or other surveillance to monitor the drug’s safety or effectiveness and may impose other conditions, including labeling restrictions, such as a Boxed Warning, and/or distribution and use restrictions through a Risk Evaluation and Mitigation Strategy, or REMS, all of which can materially affect the potential market and profitability of the product. Once granted, product approvals may be further limited or withdrawn if compliance with regulatory requirements is not maintained or safety or other problems are identified following initial marketing.
The FDA will issue a complete response letter, or CRL, if the agency decides not to approve the NDA in its present form. The complete response letter describes the deficiencies that the FDA has identified in an application and may recommend actions that the applicant can take to address the deficiencies. Such actions may include, among other things, conducting additional safety or efficacy studies. Even with the completion of this additional testing or the submission of additional requested information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. With limited exceptions, the FDA may withhold approval of an NDA regardless of prior advice it may have provided or commitments it may have made to the sponsor.
Post-Approval Regulation
Once an NDA is approved, a product will be subject to extensive post-approval regulatory requirements, including requirements for manufacturing establishment registration and product listing, adverse event reporting, submission of other periodic reports, field alerts, recordkeeping, product sampling and distribution. Additionally, the FDA strictly regulates the labeling and advertising of prescription drug products. Among other things, the FDA generally prohibits pharmaceutical companies from promoting their drugs for uses that are not approved by the FDA as reflected in the product’s approved labeling, although a physician may prescribe a drug for such off-label uses in accordance with the practice of medicine. The FDA and other regulatory agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses. We must also notify the FDA of any change in an approved product beyond variations in the approved application. Certain changes to the product, its labeling or its manufacturing require prior FDA approval and may require the conduct of further clinical investigations to support the change. Such approvals may be expensive and time-consuming and, if not approved, the FDA will not allow the product to be commercially distributed as modified.
Additionally, the quality control and manufacturing procedures for our products must continually conform with cGMP. Manufacturers must devote substantial time, money and effort in the areas of production, quality control, and quality assurance to maintain cGMP compliance. The FDA, and other regulatory agencies, conduct periodic inspections following the initial approval of a product. If a manufacturing facility is not in substantial compliance with the applicable regulations and requirements imposed when the product was approved, regulatory or judicial enforcement action may be initiated, which may include a warning letter, suspension of manufacturing, product seizure, or an injunction against shipment of products from the facility and/or recall of products previously shipped.
Some of our product candidates may need to be administered using specialized drug delivery systems that are considered to be medical devices and may necessitate compliance with regulatory laws applicable to medical devices, including those governing the testing, manufacture, approval, distribution, and marketing of medical devices.
Abbreviated Applications and 505(b)(2) Applications
Once an NDA is approved, the product covered thereby becomes a reference listed drug that can, in turn, be relied upon by potential competitors in support of approval of an abbreviated NDA, or ANDA, or a 505(b)(2) application. An ANDA generally provides an abbreviated approval pathway for a drug product that has the same active ingredients in the same strength, dosage form and route of administration as the listed product, has been shown to be bioequivalent to the listed product, and has the same labeling as the listed product (subject to certain exceptions). The application is “abbreviated” because it need not include nonclinical or clinical data to demonstrate safety and effectiveness and may instead rely on the U.S. FDA’s previous finding that the brand drug, or reference drug, is safe and effective. Drugs approved via an ANDA are commonly referred to as generics and can often be substituted by pharmacists under prescriptions written for the original listed drug. A 505(b)(2) application is a type of NDA that contains full reports of investigations of safety and effectiveness, where at least some of the information required for approval comes from studies not conducted by or for the applicant, and for which the applicant has not obtained a right of reference. Such applications often are submitted for changes to previously approved drug products.
The acceptance and approval of ANDAs and 505(b)(2) applications can be delayed by patents and non-patent exclusivity covering the listed drug. Federal law provides a five-year period of NCE exclusivity following approval of a drug that contains a new chemical entity, or NCE, that has not been previously approved by the FDA. If a listed drug has NCE exclusivity, ANDAs and 505(b)(2) applications referencing the listed drug cannot be submitted to the FDA for five years following the approval of the listed drug unless the application contains a certification challenging a listed patent, i.e., a paragraph IV certification (discussed further below), in which case the ANDA or 505(b)(2) application may be submitted four years following approval of the listed drug. Federal law also provides for a period of three years of exclusivity following approval of a listed product that is not an NCE, if the application contains the results of new clinical investigations, other than bioavailability studies, conducted or sponsored by the applicant and essential to the approval of the application. This three-year exclusivity covers only the conditions of approval for which the new clinical investigations were essential, such as a new dosage form or indication. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the nonclinical studies and clinical trials necessary to demonstrate safety and effectiveness.
NDA applicants and NDA holders must provide information about certain patents claiming their drugs, or methods of use of the drug that is the subject of the NDA, for listing in FDA’s publication Approved Drug Products with Therapeutic Equivalence Evaluations, commonly referred to as the Orange Book. When an ANDA or 505(b)(2) application is submitted, it must contain one of several possible certifications regarding each of the patents listed in the Orange Book for the reference drug. For a method-of-use patent, however, the applicant can submit a statement that it is not seeking approval of a use claimed by the patent instead of making a certification. These certifications (and statements) affect when the FDA can approve the ANDA or 505(b)(2) application. If the ANDA or 505(b)(2) applicant certifies that it does not intend to market its product before a listed patent expires (i.e., a Paragraph III certification), then the FDA will not grant effective approval of the ANDA or 505(b)(2) application until the relevant patent expires. An ANDA or 505(b)(2) applicant may also certify that a listed patent is invalid, unenforceable, or will not be infringed by its proposed product, and thus that it is seeking approval prior to patent
expiration (i.e., a Paragraph IV certification). Within 20 days of FDA’s acceptance of an ANDA or 505(b)(2) application containing a Paragraph IV certification, the applicant must notify the NDA holder and patent owner that the application has been submitted, and provide the factual and legal basis for the applicant’s opinion that the patent is invalid, unenforceable, or not infringed. The NDA holder or patent holder may then initiate a patent infringement suit in response to the Paragraph IV notice. If this is done within 45 days of receiving notice of the Paragraph IV certification, a 30-month stay of the U.S. FDA’s ability to approve the ANDA or 505(b)(2) application is triggered. The FDA may approve the proposed product before the expiration of the 30-month stay only if a court finds the patent invalid or not infringed, and the court may shorten or lengthen the 30-month stay under certain limited circumstances.
Orphan Drug Designation
Under the Orphan Drug Act, as amended, the FDA may grant orphan drug designation to a drug intended to treat a rare disease or condition, which is a disease or condition that affects fewer than 200,000 individuals in the U.S. or affects more than 200,000 individuals and for which there is no reasonable expectation of recovering drug development costs in the U.S. from sales in the U.S. Orphan drug designation must be requested before submitting an NDA or an sNDA for the orphan indication. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential use for an orphan indication are disclosed publicly by the FDA. We have received orphan drug designation for certain of our products and intend to request orphan drug designation for certain of our product candidates in the future, if applicable. For example, the FDA granted orphan drug designation for patisiran, vutrisiran and nucresiran as therapeutic approaches for the treatment of ATTR amyloidosis, givosiran as a therapeutic approach for AHP, lumasiran as a therapeutic approach for PH1, and inclisiran as a therapeutic approach for homozygous familial hypercholesterolemia.
If a product that has orphan drug designation subsequently receives the first FDA approval for that product for the disease for which it has such designation, the product is entitled to orphan drug exclusivity, which means that the FDA may not approve for seven years any other applications, including a full NDA, from another sponsor to market the “same drug” for the same indication, except in limited circumstances. For purposes of small molecule drugs, the FDA defines “same drug” as a drug that contains the same active moiety and is intended for the same use as the previously approved orphan drug. For purposes of large molecule drugs, the FDA defines “same drug” as a drug that contains the same principal molecular structural features, but not necessarily all of the same structural features, and is intended for the same use as the previously approved drug. However, a drug that is “clinically superior” to an orphan drug will not be considered the “same drug” and thus will not be blocked by orphan drug exclusivity. To demonstrate a drug is “clinically superior” to the previously approved orphan drug, a sponsor must show that the drug provides a significant therapeutic advantage over and above the previously approved product in terms of greater efficacy, greater safety, or by providing a major contribution to patient care.
Under the FDA’s regulations, a designated orphan drug may not receive orphan drug exclusivity for a use that is broader than the indication for which it received orphan drug designation and regulatory approval. However, a 2021 decision by the U.S. Court of Appeals for the Eleventh Circuit in Catalyst Pharmaceuticals, Inc. v. Becerra adopted a broader interpretation of the scope of orphan drug exclusivity, holding that orphan drug exclusivity prevents the FDA from approving another marketing application for the same drug for the same orphan-designated disease or condition for a period of seven years. Although the FDA announced in January 2023 that it will not apply the Catalyst decision beyond the facts at issue in that case, Catalyst could serve as a precedent for future challenges to FDA’s orphan drug-related decisions. Legislation has been introduced, but has not been passed, that would codify the scope of orphan drug exclusivity set forth in the FDA’s regulations, rather than the interpretation adopted by the Eleventh Circuit in Catalyst.
In addition, orphan drug exclusivity may be lost if the FDA later determines that the orphan drug designation request was materially defective or if the manufacturer is unable to ensure the availability of sufficient quantities of the drug to meet the needs of patients with the rare disease or condition, or if the manufacturer chooses to provide consent to approval of other applications.
Pediatric Study Plans
The FDCA requires that a sponsor planning to submit an NDA for a drug that includes a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration must submit a pediatric study plan to FDA outlining the proposed pediatric study or studies they plan to conduct, including study objectives and design, any deferral or waiver requests, and other information required by regulation. The FDA must then review the information submitted, consult with the sponsor, and agree upon a final plan. The FDA or the sponsor may request an amendment to the plan at any time. Drugs with orphan drug designation are exempt from these requirements to the extent that the indication being sought under the marketing application is within the scope of the designated orphan use.
For NDAs, BLAs, or supplemental applications subject to PREA, sponsors must include an agreed initial PSP in the application when a deferral of pediatric studies is requested. A final decision about granting or denying such requests is made by the review division at the time of approval of the marketing application. Failure to fulfill the requirement to submit a pediatric study plan with the application may be grounds for refusal to file an application. If, under certain situations, the agreed initial PSP included nonclinical and/or pediatric clinical studies that were expected to have been completed before submission
of the NDA or supplement, failure of the sponsor to complete these agreed studies in a timely manner may result in a refusal to file.
Fast Track, Breakthrough Therapy and Priority Review Designation
The FDA has several programs designed to expedite the development and approval of drugs intended to treat serious or life-threatening diseases or conditions. These programs include fast track designation, breakthrough therapy designation, and priority review designation. These designations are not mutually exclusive, and a product may qualify for one or more of these programs. While these programs are intended to expedite product development and approval, they do not alter the standards for FDA approval.
The FDA may grant a product fast track designation if it is intended for the treatment of a serious or life-threatening disease or condition, and nonclinical or clinical data demonstrate the potential to address an unmet medical need for such disease or condition. For fast track products, sponsors may have greater interactions with the FDA, and the FDA may initiate review of sections of a fast track product’s application before the application is complete in some circumstances. Fast track designation may be rescinded if FDA believes that the product no longer meets the qualifying criteria.
A product may be designated as a breakthrough therapy if it is intended, either alone or in combination with one or more products, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the product may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. The FDA may take certain actions with respect to products designated as breakthrough therapies, including holding meetings with the sponsor throughout the development process; providing timely advice to the product sponsor regarding development and approval; involving more senior staff in the review process; assigning a cross-disciplinary project lead for the review team; and taking other steps to aid sponsors in designing the clinical trials in an efficient manner. Breakthrough designation may be rescinded if a product no longer meets the qualifying criteria.
FDA may designate a product for priority review if it is a product that treats a serious condition and, if approved, would provide a significant improvement in safety or effectiveness of the treatment, prevention, or diagnosis of such condition. A priority designation is intended to direct overall attention and resources to the evaluation of such applications, and it shortens the FDA’s goal for taking action on a marketing application from ten months to six months from filing.
Accelerated Approval
The FDA may grant accelerated approval to a drug product for a serious or life-threatening condition that provides meaningful therapeutic advantage to patients over existing treatments based upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit. The FDA may also grant accelerated approval for such a condition when the product has an effect on an intermediate clinical endpoint that can be measured earlier than an effect on irreversible morbidity or mortality, or IMM, and that is reasonably likely to predict an effect on IMM or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments.
The accelerated approval pathway is most often used in settings in which the course of a disease is long and an extended period of time is required to measure the intended clinical benefit of a product, even if the effect on the surrogate or intermediate clinical endpoint occurs rapidly.
For products granted accelerated approval, FDA generally requires sponsors to conduct, in a diligent manner, additional post-approval confirmatory studies to verify and describe the product’s clinical benefit. The Food and Drug Omnibus Reform Act of 2022 gave FDA the authority to require, as appropriate, a post-approval study to be underway prior to granting accelerated approval. Failure to conduct required post-approval studies with due diligence, failure to confirm a clinical benefit during the post-approval studies, or dissemination of false or misleading promotional materials would allow the FDA to withdraw the product approval on an expedited basis. All promotional materials for products approved under accelerated approval are subject to prior review by the FDA unless FDA informs the sponsor otherwise.
Rare Pediatric Disease Designation and Priority Review Voucher
The FDCA provides a rare pediatric disease priority review voucher, or PRV, to sponsors under a program that is intended to incentivize the development of new drug and biological products for the prevention and treatment of “rare pediatric diseases.” A rare pediatric disease is any disease that is a rare disease and is serious or life-threatening with the serious or life-threatening manifestations primarily affecting individuals from birth to 18. Under this program, the sponsor of an application for a rare pediatric disease drug may be eligible to obtain a voucher that can be used to obtain a priority review for a subsequent human drug application.
In order to receive a rare pediatric disease priority review voucher, the product must receive designation from the FDA as a drug for a rare pediatric disease prior to submission of the marketing application. In addition to receiving rare pediatric disease designation, in order to receive a rare pediatric disease priority review voucher upon NDA approval, the application must be granted priority review, rely on clinical data derived from studies examining a pediatric population and dosages of the drug intended for that population, not seek approval for a different adult indication in the original rare pediatric disease product
application and be for a drug that does not include a previously approved active ingredient. Rare pediatric disease designation does not guarantee that the sponsor will receive a PRV. If awarded, the PRV may be transferred unlimited times before the PRV is used. The rare pediatric disease PRV program was initially created in 2012, and Congress has extended the PRV program through September 30, 2024, with the potential for PRVs to be granted through September 30, 2026 for a drug that receives rare pediatric disease designation by September 30, 2024. Although legislation to extend the rare pediatric disease PRV program has been proposed, Congress has not yet, and may never, pass a bill to reauthorize the program and extend the sunset dates. The FDA awarded a rare pediatric disease PRV to us upon approval of the NDA for lumasiran in November 2020, and we utilized this PRV in connection with the sNDA filing for vutrisiran for the treatment of ATTR amyloidosis with cardiomyopathy.
Pharmaceutical Coverage, Pricing and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any products for which we obtain regulatory approval. In the U.S. and other countries, sales of any products for which we may receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third-party payors. Third-party payors include government healthcare programs, managed care providers, private health insurers and other reimbursement organizations. The coverage and reimbursement status of any drug products for which we obtain regulatory approval may vary significantly across these third-party payors. Third-party payors may limit coverage to specific drug products on an approved list, or formulary, which might not include all of the FDA-approved drugs for a particular disease or condition. Third-party payors may provide coverage, but place stringent limitations on such coverage, such as requiring alternative treatments to be tried first. The process for determining whether a payor will provide coverage for a drug product may be separate from the process for setting the price or reimbursement rate that the payor will pay for the drug product. Eligibility for coverage does not imply that any drug will be reimbursed in all cases or at a rate that covers a manufacturer’s costs, including research, development, manufacture, sale and distribution, or that covers a particular provider’s cost of acquiring the drug. Third-party payors are increasingly scrutinizing the price and examining the medical necessity and cost-effectiveness of medical products and services, in addition to their safety and efficacy. Factors payors may consider in determining reimbursement include, among others, the extent to which the product and/or the use of the product is:
•a covered benefit under a health plan;
•safe, effective and medically necessary;
•appropriate for the specific patient;
•clinically superior or therapeutically advantageous compared to other products;
•cost-effective; and
•neither experimental nor investigational.
A manufacturer may need to conduct additional research, including healthcare economic studies, in order to demonstrate the clinical value and cost-effectiveness of products, separate and apart from the studies required to obtain FDA approvals. Product candidates may not be considered medically reasonable or necessary or cost-effective. Increasingly, the third-party payors are requiring that drug companies provide them with predetermined discounts from list prices, and are seeking to reduce the prices charged or the amounts reimbursed for drug products. Even if a product is covered, a payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be provided. Lack of adequate third-party reimbursement may prevent price levels sufficient to sell current or future product(s) on a competitive basis or realize an appropriate return on investment in product development.
Some of the drugs we market need to be administered under the supervision of a physician or other healthcare professional on an outpatient basis, including ONPATTRO, AMVUTTRA, GIVLAARI and OXLUMO. Under currently applicable U.S. law, certain drugs that are not usually self-administered (including injectable drugs) may be eligible for coverage under the Medicare Part B program if:
•they are incident to a physician’s services;
•they are reasonable and necessary for the diagnosis or treatment of the illness or injury for which they are administered according to accepted standards of medical practice; and
•they have been approved by the FDA and meet other statutory requirements.
Federal, state and local governments in the U.S. and foreign governments have established and continue to consider policies to limit the growth of healthcare costs, including the cost of prescription drugs. Specifically, there have been several recent U.S. Congressional inquiries into prescription drug pricing, and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to drug pricing, reduce the cost of prescription drugs under Medicare, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drugs.
At the federal level, for example, in August 2022, the Inflation Reduction Act of 2022, or IRA, was signed into law. Key provisions of the IRA include the following, among others:
•The IRA requires manufacturers to pay rebates for Medicare Part B and Part D drugs whose net price increases exceed inflation;
•The IRA eliminates the “donut hole” under Medicare Part D beginning in 2025 by significantly lowering the beneficiary maximum out-of-pocket cost and requiring manufacturers to subsidize, through a manufacturer discount program, 10% of Part D enrollees’ prescription costs for brand drugs below the out-of-pocket maximum and 20% once the out-of-pocket maximum has been reached.
•The IRA delays the rebate rule that would require pass through of pharmacy benefit manager rebates to beneficiaries.
•The IRA directs allows the Centers for Medicare and Medicaid Services, or CMS, to engage in price-capped negotiation for certain Medicare Part B and Part D products. Specifically, the IRA’s Price Negotiation Program applies to high-expenditure single-source drugs and biologics that have been approved for at least seven or 11 years, respectively, among other negotiation selection criteria, beginning with ten high-cost drugs paid for by Medicare Part D starting in 2026, followed by 15 Part D drugs in 2027, 15 Part B or Part D drugs in 2028, and 20 Part B or Part D drugs in 2029 and beyond. The negotiated prices will be capped at a statutorily determined ceiling price. There are certain statutory exemptions from the IRA’s Price Negotiation Program, such as for a drug that has only a single orphan drug designation and is approved only for an indication or indications within the scope of such designation. The IRA’s Price Negotiation Program is currently the subject of legal challenges.
Manufacturers that fail to comply with the IRA may be subject to various penalties, including civil monetary penalties or a potential excise tax. The IRA permits the Secretary of Health and Human Services, or HHS Secretary, to implement many of the IRA’s provisions through guidance, as opposed to regulation, for the initial years. The IRA is anticipated to have significant effects on the pharmaceutical industry and may reduce the prices pharmaceutical manufacturers can charge and reimbursement pharmaceutical manufacturers can receive for approved products, among other effects.
Further, in the U.S., manufacturers are required to provide discounts or rebates on purchases of pharmaceuticals under various federal and state healthcare programs, including Medicare, Medicaid, the Veterans Administration and the 340B program. The Medicaid Drug Rebate Program requires pharmaceutical manufacturers to enter into and have in effect a national rebate agreement with the HHS Secretary, among other requirements, as a condition for payment for any of the manufacturer’s drugs under Medicaid or Medicare Part B. The minimum basic Medicaid rebate on most branded prescription drugs and biologic products is 23.1% of average manufacturer price, or AMP. The Medicaid Drug Rebate Program requires participating manufacturers to report certain drug-related product and pricing information to CMS monthly and quarterly.
In order for a drug product to receive federal reimbursement under the Medicare Part B and Medicaid programs, the manufacturer also must offer its innovator products on the Federal Supply Schedule for purchase at legally prescribed prices and extend discounts on its products to entities eligible to participate in the 340B drug pricing program. The required 340B discount on a given product is calculated based on the AMP and Medicaid rebate amounts reported by the manufacturer. Manufacturers must report and certify prices and other data to various government entities under these programs. The related calculations are complex, and any inaccuracies or delays in reporting can result in enforcement measures or other liability. Changes to these government programs could affect our business. For instance, the 340B program continues to be subject to legal and regulatory activity, including litigation, at the federal and state levels, and any related developments could impact the scope of the program and our obligation to offer discounts. Continued expansion of the 340B program and growth of entities claiming entitlement to 340B pricing, including in ways that may be inconsistent with the statutory scheme, could impact our revenue. Changes to the calculation of rebates under the Medicaid program could increase our Medicaid rebate obligations and decrease the prices charged to 340B covered entities. On September 20, 2024, CMS issued a final rule specifying penalties for misclassification of drugs, and otherwise altering manufacturer obligations, under the Medicaid Drug Rebate Program.
Other executive or administrative action could alter the landscape for coverage, reimbursement, and pricing. For instance, the Center for Medicare and Medicaid Innovation is developing, new models intended to lower drug costs under Medicare and Medicaid, including designing new payment methods for drugs approved under accelerated approval, in consultation with the FDA, to encourage timely confirmatory trial completion and improve access to post-market safety and efficacy data with the goal of reducing Medicare spending on drugs that have no confirmed clinical benefit; creating a list of generic drugs for which the out-of-pocket Part D costs will be capped at $2 a month per drug; and establishing new approach for administering outcomes-based agreements for cell and gene therapies.
A 2021 Executive Order affirmed the Biden administration’s policy to, among other things, support legislative reforms that enhance competition and allow for the development and market entry of lower-cost generic drugs and biosimilars. Among other things, the executive order directed the FDA to work with states and Indian Tribes that propose to develop section 804 Importation Programs in accordance with the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, and the FDA’s implementing regulations. The FDA released such implementing regulations on September 24, 2020, which went into effect on November 30, 2020, providing guidance for states to build and submit importation plans for drugs from Canada.
In response, authorities in Canada have passed rules designed to safeguard the Canadian drug supply from shortages. On January 5, 2024, the FDA authorized Florida’s Agency for Health Care Administration’s drug importation proposal, the first step toward Florida facilitating importation of certain prescription drugs from Canada. We cannot predict how further developments of or changes to these policies and rules will affect our business.
Insurers are increasingly adopting programs and policies that limit access to medications and increase out-of-pocket costs for patients. In the U.S., to help patients access and afford our approved product(s), pharmaceutical manufacturers may utilize programs to assist them, including patient assistance programs and co-pay coupon programs for eligible patients. On April 25, 2019, CMS published a regulation clarifying that where no medically appropriate generic equivalent is available, amounts paid toward cost sharing using any form of direct support offered by drug manufacturers must be counted by applicable insurers toward the Affordable Care Act’s annual limitation on cost sharing. On May 4, 2020, CMS published a regulation allowing applicable insurers flexibility to determine whether to include or exclude support provided by drug manufacturers from the Affordable Care Act’s annual limitation on cost sharing. On September 29, 2023, the U.S. District Court for the District of Columbia set aside the 2020 regulation. Further, on April 2, 2024, CMS released a final rule codifying that all prescription drugs covered by a health plan are to be considered Essential Health Benefits and therefore are subject to the limitation on cost sharing and annual and lifetime dollar limits established by the Affordable Care Act. It is possible that further changes in insurer policies regarding co-pay coupons and patient assistance programs and/or new legislation or regulatory action, or decisions regarding enforcement of such action, could restrict or otherwise negatively affect these co-pay coupon programs and patient support programs, which could result in fewer patients using affected products, and therefore could have a material adverse effect on pharmaceutical manufacturers’ sales, business, and financial condition.
At the state level, governments have enacted and continue to consider legislation and regulations focused on pharmaceutical product pricing. Some of these measures include considering upper payment limits on state-regulated payers; regulating product access, copayment assistance. and marketing; imposing drug price, cost, and marketing disclosure and transparency requirements; permitting importation from other countries; and encouraging bulk purchasing.
Further, there has been significant consolidation in the health insurance industry, increasing the leverage of large insurers and PBMs in pricing and other negotiations and potentially impacting product sales, business and results of operations. The federal government continues to consider legislative reforms that could alter the role and operations of PBMs in the marketplace, and such reforms could have downstream implications on pharmaceutical manufacturers. We expect continued scrutiny on drug pricing and health care from Congress, agencies, and other bodies at the federal and state levels, and potential further alterations to the coverage, reimbursement, and pricing landscape for pharmaceutical products.
Healthcare Fraud and Abuse
Federal and state laws generally prohibit the payment or receipt of kickbacks, bribes or other remuneration in exchange for the referral of patients or other healthcare-related business. For example, the Federal Anti-Kickback Statute prohibits anyone from, among other things, knowingly and willfully offering, paying, soliciting or receiving any bribe, kickback or other remuneration intended to induce the referral of patients for, or the purchase, order or recommendation of, healthcare products and services reimbursed by a federal healthcare program, including Medicare and Medicaid. Violations of this federal law can result in significant penalties, including imprisonment, monetary fines and assessments, and exclusion from Medicare, Medicaid and other federal healthcare programs. Exclusion of a manufacturer would preclude any federal healthcare program from paying for its products. In addition to the Federal Anti-Kickback Statute, many states have their own laws that are analogous to the Federal Anti-Kickback Statute, but may apply regardless of whether any state healthcare program business is involved.
In addition, federal and state false claims laws prohibit anyone from presenting, or causing to be presented, claims for payment to third-party payers that are false or fraudulent. For example, the federal False Claims Act, or FCA, imposes liability on any person or entity who, among other things, knowingly and willfully presents, or causes to be presented, a false or fraudulent claim for payment by a federal healthcare program, including Medicaid and Medicare. Some suits filed under the FCA, known as “qui tam” actions, can be brought by a “whistleblower” or “relator” on behalf of the government, and such individuals may share in any amounts paid by the entity to the government in fines or settlement. Manufacturers can be held liable under false claims laws, even if they do not submit claims to the government, where they are found to have caused submission of false claims by, among other things, providing incorrect coding or billing advice about their products to customers that file claims, providing inaccurate reporting regarding pricing or other product data in connection with their government price reporting, or by engaging in kickback arrangements or off-label promotion with customers that file claims. In addition, the government may assert that a claim including items and services resulting from a violation of the U.S. federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the FCA. A number of states also have false claims laws, and some of these laws may apply to claims for items or services reimbursed under Medicaid and/or commercial insurance. Sanctions under these federal and state fraud and abuse laws may include civil monetary penalties and criminal fines, exclusion from government healthcare programs and imprisonment.
The U.S. Foreign Corrupt Practices Act of 1977, as amended, or FCPA, and similar anti-bribery laws in non-U.S. jurisdictions generally prohibit companies and their officers, directors, employees and intermediaries from offering or making
improper payments to non-U.S. officials for the purpose of obtaining or retaining business. Violation of the FCPA could result in substantial civil and criminal penalties and remedies, including fines, disgorgement, and imprisonment.
The federal Sunshine Act requires manufacturers to report certain payments to healthcare providers to CMS. Many state laws require drug manufacturers to report similar information related to payments and other transfers of value provided to other healthcare providers. Some states prohibit these expenditures altogether. Laws in a number of states also require companies to adopt marketing codes of conduct, to disclose pricing information about their products, or to license pharmaceutical sales representatives.
As described above, we maintain a global compliance program designed to support the execution of our business strategy and operations in compliance with these laws.
Possible Change in Laws or Policies
From time to time, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of drug products. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency or reviewing courts in ways that may significantly affect our business and development of our product candidates and any products that we may commercialize. It is impossible to predict whether additional legislative changes will be enacted, or FDA regulations, guidance or interpretations will be changed, or what the impact of any such changes may be. Federal budget uncertainties or spending reductions may reduce the capabilities of the FDA, extend the duration of required regulatory reviews, and reduce the availability of clinical research grants.
Our present and future business has been and will continue to be subject to various other laws and regulations. Various laws, regulations and recommendations relating to safe working conditions, laboratory practices, the experimental use of animals, and the purchase, storage, movement, import, export and use and disposal of hazardous or potentially hazardous substances are or may be applicable to our activities. As noted above, the extent of government regulation, which might result from future legislation or administrative action, cannot accurately be predicted.
EU Regulatory Considerations
In the EU, medicinal products are subject to extensive pre- and post-market regulation by regulatory authorities at both the EU and national levels.
Clinical Trials
Clinical trials of medicinal products conducted in the EU must comply with the harmonized regulatory framework provided by Regulation (EU) No 536/2014, or the Clinical Trials Regulation, and the International Conference on Harmonization, or ICH, guidelines on GCP. Where the sponsor of the clinical trial is not established within the EU, it must appoint an entity within the EU to act as its legal representative. The legal representative is responsible for ensuring compliance with the sponsor’s obligations under the EU Regulation on Clinical Trials and assumes the role as the addressee for all communications with the sponsor provided for under the Regulation. The sponsor must provide proof of insurance cover or indemnification arrangement for personal injury claims arising through participation in a clinical trial.
A clinical trial to be conducted in an EU Member State must be authorized by the competent authority of the Member State concerned, and positively reviewed by an independent ethics committee. The application for a clinical trial authorization, or CTA, must include, among other things, a copy of the trial protocol and an investigational medicinal product dossier containing information about the manufacture and quality of the medicinal product under investigation, and data derived from non-clinical studies and from its clinical use. Any substantial changes to the trial protocol or other information submitted with the CTAs with a significant impact on the safety of the approved clinical trial must be notified to or approved by the relevant competent authorities and ethics committees in respect of the nature of and reason for such a change.
The Clinical Trials Regulation, which became applicable on January 31, 2022, establishes the procedure to enable sponsors to submit one CTA application for it to be approved in a more streamlined and coordinated manner by two or more countries in the EU/EEA where the clinical trial is to be performed. From January 31, 2023, sponsors are required submit an on-line application through a single on-line platform known as Clinical Trials Information System, or CTIS, for approval. To make it more efficient to carry out such multinational trials, one national authority assumes the role as the Reporting Member State to lead the scientific review of the application for consideration by the Member States concerned by the application for agreement within the timeframe stipulated by the Clinical Trials Regulation. Each concerned member state is responsible for assessing country-specific issues concerning the clinical trial including those related to adequacy of the trial sites, facilities, recruitment details.
One of the key objectives of the Clinical Trial Regulation is to strengthen the transparency of clinical trial related information. All data and information in connection with clinical trials entered into the CTIS must be publicly accessible, unless the information benefits from being excluded from disclosure under the Regulation.
Manufacturers are afforded the opportunity to seek scientific advice from regulatory authorities to guide their research and development programs. A request for scientific advice can be made nationally by engaging the national competent authorities or
centrally which is coordinated by the EMA at different stages of product development in relation to questions concerning an assessment of the product quality, non-clinical testing and clinical development. Advice is generally not viewed as binding with regard to any future marketing authorization application, or MAA, of the product concerned and departure from the adopted advice should be justified in the MAA.
Marketing Authorizations
After completion of the required clinical testing, we must obtain a marketing authorization before we can place a medicinal product on the market in the EU. There are various application procedures available, depending on the type of product involved. All application procedures require an application to be presented in the internationally harmonized Common Technical Document format, which includes the submission of detailed information about the manufacturing and quality of the product, and nonclinical testing and clinical trial to inform the benefit-risk assessment.
The centralized procedure gives rise to marketing authorizations that are valid throughout the EU and, by extension (after national implementing decisions), in Norway, Iceland and Liechtenstein, which, together with the EU member states, comprise the European Economic Area, or EEA. Applicants file MAAs with the EMA, where they are reviewed by relevant scientific committees, including the Committee for Medicinal Products for Human Use, or CHMP. The EMA forwards CHMP opinions to the EC, for ratification to adopt an implementing decision for granting a marketing authorization. The centralized procedure is compulsory for medicinal products that (1) are developed by biotechnology processes including advanced therapy medicinal products such as gene and cell therapies, (2) contain a new active substance indicated for the treatment of certain diseases, such as HIV/AIDS, cancer, diabetes, neurodegenerative disorders, viral diseases or autoimmune diseases and other immune dysfunctions or (3) are orphan medicinal products. For medicines that do not fall within these categories, an applicant may voluntarily submit an application for a centralized marketing authorization to the EMA, as long as the CHMP agrees that (i) the medicine concerned contains a new active substance, (ii) the medicine is a significant therapeutic, scientific, or technical innovation, or (iii) the authorization of the medicine under the centralized procedure would be in the interest of public health.
For those medicinal products for which the centralized procedure is not available, the applicant must submit MAAs to the national medicines regulators through one of three procedures: (1) a national procedure, which results in a marketing authorization in a single EU member state; (2) the decentralized procedure, in which applications are submitted simultaneously in two or more EU member states and a reference member state is appointed to lead the scientific assessment; and (3) the mutual recognition procedure, which must be used if the product has already been nationally authorized in at least one EU member state which assumes the role as the reference member state, and the marketing authorization holder wishes to obtain a marketing authorization for the same product in at least one other member state. The concerned member states may refuse to accept the assessment of the reference member state in the decentralized procedure and the mutual recognition procedure where there are grounds related to a serious risk to public health. A national procedure applies where the medicinal product is to be marketed in only one member state and the product does not fall within the mandatory centralized assessment procedure.
Under the centralized procedure in the EU, the maximum timeframe for the evaluation of an MAA is 210 days. However, this timeline excludes clock stops, when additional written or oral information is to be provided by the applicant in response to questions asked by the CHMP, so the overall process typically may take a year or more. Accelerated evaluation might be granted by the CHMP in exceptional cases, when a medicinal product is expected to be of a major interest for public health and therapeutic innovation. Typically, the justification could present the arguments to support the claim that the medicinal product addresses a significant extent the unmet medical needs where there exists the absence or insufficiency of an appropriate alternative therapeutic approach for the disease to be treated and anticipation of high therapeutic benefit of the new product. In this circumstance, the EMA ensures that the opinion of the CHMP is given within 150 days. The EMA granted an accelerated assessment for patisiran, which was approved in the EU in August 2018 under the centralized procedure.
There is an increasing trend in the EU towards greater transparency in the decision-making to allow for greater access to documents held by regulatory authorities in relation to evaluation of medicinal products. While the manufacturing or quality information is currently generally protected against disclosure as it is treated as confidential information, there exists a presumptive public interest right to access the nonclinical and clinical information contained in marketing authorization dossiers, including the full clinical study reports, in response to freedom of information requests after the marketing authorization has been granted. As of October 2016, the EMA began publishing clinical data (including clinical study reports) on the agency’s website following the grant, denial or withdrawal of an MAA for a centralized marketing authorization, subject to procedures for limited redactions and protection against unfair commercial use.
Data Exclusivity
MAAs for generic medicinal products do not need to include the results of preclinical studies and clinical trials, but instead can refer to such data included in the marketing authorization of a reference product for which regulatory data exclusivity has expired. Where a marketing authorization is granted for a medicinal product containing a new active substance, that product benefits from eight years of data exclusivity, during which generic MAAs referring to the data of that product will not be accepted by the regulatory authorities, and a further two years of marketing protection, during which such generic products, even if approved, may not be placed on the market until the full 10-year protection period has expired. The 10-year protection
period may be extended to 11 years if during the first eight years of the product’s authorization, one or more new therapeutic indications with significant clinical benefit over existing therapies are approved.
There is a special regime for biosimilars, or biological medicinal products that are similar to a reference medicinal product but that do not meet the definition of a generic medicinal product, for example, because of differences in raw materials or manufacturing processes. For such products, the results of appropriate preclinical studies or clinical trials must be provided, and guidelines from the EMA detail the type of supplementary data to be provided for different types of biological product. There are no such guidelines for complex biological products, such as gene or cell therapy medicinal products, and so it is unlikely that biosimilars of those products will currently be approved in the EU. However, guidance from the EMA states that they will be considered in the future in light of the scientific knowledge and regulatory experience gained at the time.
Orphan Medicinal Products
The EMA’s Committee for Orphan Medicinal Products, or COMP, may recommend orphan medicinal product designation to promote the development of products that are intended for the diagnosis, prevention or treatment of life-threatening or chronically debilitating conditions affecting not more than five in 10,000 persons in the EU. Additionally, designation is granted for products intended for the diagnosis, prevention or treatment of a life-threatening, seriously debilitating or serious and chronic condition when, without incentives, it is unlikely that sales of the product in the EU would be sufficient to justify the necessary investment in developing the medicinal product. The COMP may only recommend orphan medicinal product designation when the product in question offers a significant clinical benefit over existing approved products for the relevant indication or where no satisfactory method of diagnosis, prevention or treatment of such condition exists. Following a positive opinion by the COMP, the EC adopts a decision granting orphan status. The COMP will reassess orphan status in parallel with EMA review of an MAA and orphan status may be withdrawn at that stage if it no longer fulfills the orphan criteria (for instance because in the meantime a new product was approved for the indication and no convincing data are available to demonstrate a significant benefit over that product). Orphan medicinal product designation entitles a party to financial incentives such as reduction of fees or fee waivers and ten years of market exclusivity is granted following marketing authorization. During this period, the competent authorities may not accept or approve a similar medicinal product for the same approved therapeutic indication, unless (i) the second medicinal product is safer, more effective or otherwise clinically superior to the authorized orphan product; (ii) the marketing authorization holder for the authorized product consents to a second orphan medicinal product application; or (iii) the marketing authorization holder for the authorized product cannot supply enough orphan medicinal product. This period may be reduced to six years if the orphan medicinal product designation criteria are no longer met, including where it is shown that the product is sufficiently profitable not to justify maintenance of orphan designation. Patisiran, approved in the EU in August 2018, givosiran, approved in the EU in March 2020, lumasiran, approved in the EU in November 2020, as well as vutrisiran, approved in the EU in September 2022 and fitusiran have been granted orphan medicinal product designation.
Post-Approval Controls
The holder of a marketing authorization must establish and maintain a pharmacovigilance system and appoint an individual qualified person for pharmacovigilance who is responsible for oversight of that system. Key obligations include expedited reporting of suspected serious adverse reactions and submission of periodic safety update reports, or PSURs.
All new MAAs must include a risk management plan, or RMP, describing the risk management system that the company will put in place and documenting measures to prevent or minimize the risks associated with the product. The regulatory authorities may also impose specific obligations as a condition of the marketing authorization. Such risk-minimization measures or post-authorization obligations may include additional safety monitoring, more frequent submission of PSURs, or the conduct of additional clinical trials or post-authorization safety studies. RMPs and PSURs are routinely available to third parties requesting access, subject to limited redactions.
All advertising and promotional activities for the product must be consistent with the approved summary of product characteristics, and therefore all off-label promotion is prohibited. Direct-to-consumer advertising of prescription medicines is also prohibited in the EU. Although general requirements for advertising and promotion of medicinal products are established under EU directives, the details are governed by regulations in each member state and can differ from one country to another.
Manufacturing
Medicinal products may only be manufactured in the EU, or imported into the EU from another country, by the holder of a manufacturing authorization from the competent national authority. The manufacturer or importer must have a qualified person who is responsible for certifying that each batch of product has been manufactured in accordance with EU standards of cGMP before releasing the product for commercial distribution in the EU or for use in a clinical trial. Manufacturing facilities are subject to periodic inspections by the competent authorities for compliance with cGMP. Testing and certification on importation is exempted where there exists a Mutual Recognition Agreement between the exporting country and the EU.
Pricing and Reimbursement
Governments influence the price of medicinal products in the EU through their pricing and reimbursement rules and control of national healthcare systems that fund a large part of the cost of those products to consumers. Some jurisdictions operate positive and negative list systems under which products may only be marketed once a reimbursement price has been agreed. To obtain reimbursement or pricing approval, some of these countries may require the completion of clinical trials that compare the cost-effectiveness of a particular product candidate to currently available therapies. Other member states allow companies to fix their own prices for medicines, but monitor and control company profits. The downward pressure on healthcare costs in general, particularly prescription medicines, has become very intense. In addition to an assessment of cost-effectiveness, the national health systems may consider whether market access is affordable. As a result, increasingly high barriers are being erected to the entry of new products.
Hazardous Materials
Our research, development and manufacturing processes involve the controlled use of hazardous materials, chemicals and radioactive materials and produce waste products. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous materials and waste products. We do not expect the cost of complying with these laws and regulations to be material.
Manufacturing
To date, we have manufactured limited supplies of drug substance for use in IND-enabling toxicology studies in animals and clinical trials at our own facilities, as well as patisiran formulated bulk drug product. We have contracted with several third-party contract manufacturing organizations, or CMOs, for the supply of drug substance, drug product and finished goods to meet our needs for preclinical toxicology studies, clinical and commercial supply. We expect to continue to rely on third-party CMOs for the supply of drug substance and drug product, including for ONPATTRO, AMVUTTRA, GIVLAARI and OXLUMO, as well as other product candidates, for the next several years, including the launch of our additional product candidates and to supply the needs of our collaborators. In 2015, we amended our manufacturing services agreement with Agilent Technologies, Inc., or Agilent, to provide for Agilent to supply, subject to any conflicting obligations under our third-party agreements, a specified percentage of the active pharmaceutical ingredients required for certain of our products in clinical development, as well as other products the parties may agree upon in the future. Under this agreement, we are required to provide rolling forecasts for products on a quarterly basis, a portion of which will be considered a binding, firm order. Agilent is required to reserve sufficient capacity to ensure that it can supply products in the amounts specified under such firm orders, as well as up to a certain percentage of the remaining, non-binding portions of each forecast. Subject to any conflicting obligations under our third-party agreements, we have also agreed to negotiate in good faith to enter into separate commercial manufacturing supply agreements with Agilent for certain products, consistent with certain specified terms, including a specified minimum purchase commitment. Currently, Agilent is the sole manufacturer of the active pharmaceutical ingredient for ONPATTRO, AMVUTTRA and GIVLAARI, and we have entered into manufacturing services agreements with Agilent for such supply of ONPATTRO, AMVUTTRA and GIVLAARI. Pursuant to the Agilent supply agreements, we are required to provide rolling forecasts on a quarterly basis, a portion of which will be considered a binding, firm order. Agilent is required to reserve sufficient capacity to ensure that it can supply our commercial and clinical products in the amounts specified under such firm orders, including a certain percentage of the remaining, non-binding portions of each forecast, as well as a specified number of batches each year.
In 2012, we established a manufacturing facility in Cambridge, Massachusetts and have developed GMP capabilities and processes for the manufacture of patisiran formulated bulk drug product for late-stage clinical trial use and commercial supply. We expect to continue to manufacture commercial supply for formulated bulk drug product for ONPATTRO in this facility for the near future. Commercial quantities of ONPATTRO and any other drugs that we may seek to develop will have to be manufactured in facilities, and by processes, that comply with FDA regulations and other federal, state and local regulations, as well as comparable foreign regulations.
During 2020, we completed construction and qualification of our GMP manufacturing facility in Norton, Massachusetts, where we currently manufacture drug substance for clinical programs and, eventually, intend to manufacture drug substance for commercial use. In December 2020, we began GMP operations, and we believe this facility will enable us to initiate manufacturing for multiple new early-stage programs over the next few years, as well as provide us the manufacturing capabilities to support select late-stage clinical and commercial programs in the future.
We believe we have sufficient manufacturing capacity through our third-party CMOs and our current internal manufacturing facilities to meet our current, clinical development and commercial needs and the needs of our collaborators. We believe that the current supply capacity we have established externally, together with the internal capabilities we developed to support preclinical development, our existing facility for patisiran formulated bulk drug product and our Norton manufacturing facility, will be sufficient to meet our and our collaborators’ anticipated needs for the next several years. We monitor the capacity availability for the manufacture of drug substance and drug product and believe that our supply agreements with our CMOs and the lead times for new supply agreements would allow us to access additional capacity to meet our and our
collaborators’ currently anticipated needs. We also believe that our products can be manufactured at a scale and with production and procurement efficiencies that will result in commercially competitive costs.
Commercial Operations
We have built and continue to scale global commercial operations designed to sequentially manage product launches across multiple geographies. Over the last several years, we have been building commercial capabilities and leveraging the internal knowledge we have accumulated as well as hiring talented people with broad industry experience to enable us to commercialize our products ourselves and with collaborators in key countries globally. The conduct of these commercial activities will continue to be dependent upon regulatory approvals and on agreements that we have made or may make in the future with strategic collaborators, currently as follows with respect to our first five approved products and our late-stage clinical programs:
•With respect to our ATTR amyloidosis franchise, we have global rights to develop and commercialize both ONPATTRO and AMVUTTRA;
•With respect to our Global Rare franchise, we have global rights to develop and commercialize GIVLAARI and OXLUMO;
•We granted MDCO, which was acquired by Novartis in January 2020, global rights to develop and commercialize Leqvio; and
•We granted Sanofi global rights to develop and commercialize fitusiran and any back-ups.
Throughout the development of our product candidates, we have remained focused on keeping patients at the center of everything we do. This patient focus has continued as we have transitioned into commercialization. ONPATTRO, AMVUTTRA, GIVLAARI and OXLUMO, as well as the other programs we are advancing to commercialization are focused on a broad range of disease areas and indications, and we have been executing on our strategy to make ONPATTRO, AMVUTTRA, GIVLAARI, OXLUMO and any future products successful, including through efforts to increase awareness and diagnosis. In addition, consistent with our Alnylam P5x25 strategy, we are now advancing RNAi therapeutics beyond rare diseases across a broad range of disease areas and indications. Beginning with the approval of Leqvio, which was the first RNAi therapeutic approved for a prevalent disease (hypercholesterolemia), we believe the RNAi therapeutic profile supports the potential for expansion to prevalent diseases, including hypertension, NASH and diabetes, where there remains significant unmet need. We are scaling our global commercial organization and infrastructure to support our eventual expansion to commercialize RNAi therapeutics in prevalent diseases.
We have a proactive market access strategy that includes entering into value-based agreements, or VBAs, with commercial payers in the U.S. and certain state Medicaid programs. We have entered into VBAs with multiple commercial payers, including for ONPATTRO, AMVUTTRA, GIVLAARI and OXLUMO. In our VBAs for GIVLAARI and OXLUMO we offer a prevalence-based adjustment that provides that we will pay a rebate if the number of patients identified within a plan population exceeds the expected disease prevalence. For OXLUMO, we offer a VBA component called a patient need adjustment that provides payers with greater budget certainty for medicines administered across a broad range of patient age groups by paying a rebate if the average number of vials utilized by a plan member exceeds an established threshold. These adjustment seek to address an unknown that exists in the context of an ultra-rare disease. Discussions with additional payers continue for our marketed products. Outside of the U.S. we believe we have made strong progress with patient access and have established availability of ONPATTRO, AMVUTTRA, OXLUMO and GIVLAARI in more than 60 markets through direct reimbursement or in partnership with our distributors. In addition, we have been encouraged by the strength of the adoption of AMVUTTRA in hATTR amyloidosis with polyneuropathy in key markets and expect continued expansion across global markets.
We are continuing to augment the key components of our global commercial organization with a focus on successfully launching our commercially approved products around the world and preparing for the anticipated commercial launches of additional RNAi therapeutics, assuming successful development and regulatory approval. With respect to our approved products, throughout 2024, we continued to build our commercial capabilities and to expand these capabilities to additional countries globally. We are building a focused commercial team with broad experience in marketing, sales, patient access, patient services, distribution and product reimbursement, and incorporating and enhancing appropriate quality systems, compliance policies, systems and procedures, as well as implementing internal systems and infrastructure to support global commercial sales, and the establishment of patient-focused programs.
We intend to leverage our existing commercial infrastructure to also support the launch of vutrisiran in ATTR-CM, assuming regulatory approval. In preparation for potential regulatory approvals, we have implemented a compliance program to ensure that our promotional activities remain focused solely on AMVUTTRA in hATTR amyloidosis with polyneuropathy until we obtain approval to market vutrisiran for ATTR amyloidosis with cardiomyopathy in each market. Our Ethics & Compliance team has partnered with our Commercial and Medical Affairs leadership teams in the implementation of this program, which is focused on proactive messaging and training, field monitoring, and data analysis. For many territories/countries, we may also utilize strategic partners, distributors or contract sales forces to assist in the commercialization of our products. Our objective is to execute successful product launches leveraging our positive experience with the launches of our approved products.
Medical Affairs
Our Medical Affairs organization facilitates communications with the medical and scientific community through the publication of clinical trial data and participation in medical congresses, provides healthcare professionals with accurate information about our products, responds to medical inquiries, and generates data to demonstrate product value, cost-effectiveness and supporting data for market access and reimbursement. Finally, our Medical Affairs organization generates real world evidence to address knowledge gaps. These capabilities ultimately enable patient diagnosis and improved patient care, including through support for independent third party genetic testing programs, including Alnylam Act. With a scalable framework for these capabilities, we believe our Medical Affairs organization is well positioned to support potential launches of our products in new indications and to expand our addressable indications to include prevalent diseases.
Human Capital Management
As of December 31, 2024, we employed approximately 2,230 full-time employees, of whom approximately 1,760 were employed in the U.S. and approximately 470 were employed outside of the U.S. None of our employees in the U.S. are represented by a labor union or covered by collective bargaining agreements, and we believe our relationship with our employees is good. During 2024, we enhanced our capabilities by adding approximately 200 new full-time employees. The new employees were hired to support a variety of functions and key initiatives, including extending our research, clinical and preclinical pipeline development, as well as our medical affairs and commercialization capabilities, with hires in commercial, compliance, legal, clinical development and operations, research, medical affairs and other general and administrative functions. We expect to continue to add additional employees in 2025, with a focus on further enhancing our capabilities and increasing our capacities in these areas, as well as expanding our geographic reach as we continue with global launches of our products and prepare for the launch of vutrisiran for the treatment of ATTR amyloidosis with cardiomyopathy, assuming regulatory approval.
We consider the intellectual capital, skills and experience of our employees to be an essential driver of our business and key to our future prospects. We face intense competition for qualified individuals from numerous pharmaceutical and biotechnology companies, universities, governmental entities and other research institutions, and we believe that our future success will depend in large part on our continued ability to attract and retain highly skilled employees. To attract qualified applicants to the Company and retain our employees, we offer a total rewards package consisting of base salary and cash target bonus, a comprehensive benefit package and equity compensation for every employee. Our total rewards package for each employee is based on geography, the position, peer group information and overall market competitiveness. Annual cash bonus targets are based on grade level and are communicated as a percentage of base salary. Equity compensation awards are based on grade level, geography, and performance. Any actual bonus payout is based solely on our performance against our corporate goals in the case of our executive leadership team and is based on a combination of individual performance and corporate performance (or regional or national commercial performance metrics, as applicable) in the case of all other employees.
As a global commercial-stage biopharmaceutical company, we value our talented and diverse workforce around the world and believe it contributes to our long-term success and ability to deliver innovative, safe and effective medicines to patients. We pride ourselves on an open culture in which we respect co-workers, value employees’ health and well-being, and foster professional development. We support employee growth and development in a variety of ways, including with group training, individual mentoring and coaching, conference attendance and tuition reimbursement. Our management conducts annual employee engagement surveys and reports to our board of directors on human capital management topics including corporate culture, employee development and retention, succession planning, and compensation and benefits. Similarly, our board of directors regularly provides input on important decisions relating to these matters, including with respect to employee compensation and benefits, succession planning, talent retention and development.
Corporate Information
Alnylam Pharmaceuticals, Inc. is a Delaware corporation that was formed in May 2003. Alnylam U.S., Inc., one of our wholly owned subsidiaries, is also a Delaware corporation that was formed in June 2002 as our initial corporate entity. Our principal executive office is located at 675 West Kendall Street, Henri A. Termeer Square, Cambridge, Massachusetts 02142, and our telephone number is (617) 551-8200.
Investor Information
We maintain an internet website at http://www.alnylam.com. The information on our website is not incorporated by reference into this Annual Report on Form 10-K and should not be considered to be a part of this Annual Report on Form 10-K or any other report or document we file with the SEC. Our website address is included in this Annual Report on Form 10-K as an inactive technical reference only. Our reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended, including our annual reports on Form 10-K, our quarterly reports on Form 10-Q and our current reports on Form 8-K, and amendments to those reports, are accessible through our website, free of charge, as soon as reasonably practicable after these reports are filed electronically with, or otherwise furnished to, the SEC. We also make available on our website the charters of our audit committee, people, culture and compensation committee, nominating and corporate governance committee, and science and technology committee, as well as our corporate governance guidelines and
our code of business conduct and ethics. We intend to disclose on our website any amendments to, or waivers from, our code of business conduct and ethics that are required to be disclosed pursuant to SEC rules.
The SEC maintains an Internet website that contains reports, proxy and information statements, and other information regarding Alnylam and other issuers that file electronically with the SEC. The SEC’s Internet website address is http://www.sec.gov.
ITEM 1A. RISK FACTORS
Investing in our securities involves a high degree of risk. You should carefully consider the following risk factors in addition to the other information set forth or incorporated by reference in this Annual Report on Form 10-K, including our consolidated financial statements and the related notes and “Management’s Discussion of Financial Condition and Results of Operations,” in evaluating our company and our business. If any of the following risks, or any additional risk not currently known to us or that we currently deem immaterial, actually occurs, our business, prospects, operating results or financial condition could be materially and adversely affected. In these circumstances, the trading price of our common stock could decline, and you may lose all or part of your investment.
SUMMARY OF MATERIAL RISKS ASSOCIATED WITH OUR BUSINESS
Our business is subject to numerous risks and uncertainties, discussed in more detail in the following section. These risks include, among others, the following key risks:
Business Related Risks – Risks Related to Our Financial Results
•The marketing and sale of our approved products or any future products may be unsuccessful or less successful than anticipated and we may be unable to expand the approved indications for AMVUTTRA.
•We have a history of losses and may not become and remain profitable.
•We will require substantial funds to continue our research, development and commercialization activities.
Risks Related to Our Dependence on Third Parties
•We may be unable to maintain existing or enter into new collaborations with other companies that can provide business and scientific capabilities and funds for the development and commercialization of certain of our product candidates.
•If any collaborator materially amends, terminates or fails to perform its obligations under agreements with us, the development and commercialization of certain of our product candidates could be delayed or terminated.
•We expect to incur significant costs as we continue to grow our manufacturing capabilities and resources and develop manufacturing expertise; in the meantime, we rely, and expect to continue to rely, on third parties to manufacture our products.
•We rely on third parties to conduct our clinical trials, and if such third parties fail to fulfill their obligations, our development plans may be adversely affected.
Risks Related to Managing Our Operations
•If we are unable to attract and retain qualified key management and scientists, development, medical and commercial staff, consultants and advisors, our ability to implement our business plan may be adversely affected.
•We may have difficulty expanding our operations successfully as we continue our evolution from a U.S.- and Europe-based company primarily involved in discovery, preclinical testing and clinical development into a global company that develops and commercializes multiple drugs in multiple geographies.
Industry Related Risks – Risks Related to Development, Clinical Testing and Regulatory Approval of Our Product Candidates and the Commercialization of Our Approved Products
•Any product candidate we or our collaborators develop may fail in development or experience significant delays.
•If any of our current or future products or product candidates causes undesirable side effects or has other unexpected adverse properties, such side effects or properties could delay or prevent regulatory approval, limit the commercial potential or result in significant negative consequences following any potential marketing approval.
•We or our collaborators may be unable to obtain regulatory approval for our or our collaborated product candidates, and, as a result, we or our collaborators may be unable to commercialize such product candidates.
•Even if we or our collaborators obtain regulatory approvals, our products will be subject to ongoing regulatory oversight.
•We may incur significant liability if enforcement authorities allege or determine that we are engaging in commercial activities with respect to our unapproved product candidates or promoting our commercially approved products in a way that violates applicable regulations.
•Even if we or our collaborators receive regulatory approval to market our product candidates, the market may not be receptive to such product candidates upon their commercial introduction.
•We are a multi-product commercial company and expect to continue to invest significant financial and management resources to continue to build our marketing, sales, market access and distribution capabilities and further establish our global infrastructure, and our efforts may not be successful.
•Any products we currently market or may develop in the future may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives.
Risks Related to Patents, Licenses and Trade Secrets
•If we are not able to obtain and enforce patent protection for our discoveries, our ability to develop and commercialize our product candidates will be harmed.
•We license patent rights from third-party owners. If such owners do not properly or successfully obtain, maintain or enforce the patents underlying such licenses, our competitive position and business, prospects, operating results and financial condition may be harmed.
•Other companies or organizations may challenge our patent rights or may assert patent rights that prevent us from developing and commercializing our products.
•If we become involved in intellectual property litigation or other proceedings related to a determination of rights, including our ongoing patent infringement litigation against Pfizer, Inc., or Pfizer, and Moderna, Inc., or Moderna, we could incur substantial costs and expenses, and in the case of such litigation or proceedings against us, substantial liability for damages or be required to stop our product development and commercialization efforts.
•If we fail to comply with our obligations under any licenses or related agreements, we may be required to pay damages and could lose license or other rights that are necessary for developing, commercializing and protecting our current and future products and product candidates.
Risks Related to Competition
•The pharmaceutical market is intensely competitive. If we or our collaborators are unable to compete effectively with existing drugs, new treatment methods and new technologies, we or our collaborators may be unable to commercialize successfully any drugs that we or our collaborators develop.
•We and our collaborators face competition from other companies that are working to develop novel drugs and technology platforms using technology similar to ours, as well as from companies utilizing emerging technologies.
Risks Related to Our Common Stock
•Our stock price has been and may in the future be volatile, and an investment in our common stock could suffer a decline in value.
•We expect that results from our and our collaborators’ clinical development activities and the clinical development activities of our competitors will continue to be released periodically and may result in significant volatility in the price of our common stock.
Risks Related to Our Convertible Notes
•We may not have sufficient cash flow from our business to pay our indebtedness.
•We may not have the ability to raise the funds necessary to settle for cash conversions of our 1% Convertible Senior Notes due 2027, or the Notes, or to repurchase the Notes for cash upon a fundamental change.
•The conditional conversion feature of the Notes, if triggered, may adversely affect our liquidity.
Risks Related to Our Business
Risks Related to Our Financial Results
The marketing and sale of our approved products or any future products may be unsuccessful or less successful than anticipated, and we may be unable to expand the approved indications for certain of our commercial products, including AMVUTTRA.
Although we have commercially launched four products and have an additional product being commercialized by a collaborator, we cannot predict whether we will successfully market and sell our approved products, or successfully expand the
approved indications of certain of our commercial products, including AMVUTTRA. For example, in August and September 2022, we reported positive safety and efficacy results from the APOLLO-B Phase 3 clinical trial of patisiran, which was designed and powered to evaluate the effects of patisiran on functional capacity and quality of life in patients with ATTR amyloidosis with cardiomyopathy. Despite positive safety and efficacy results from our APOLLO-B clinical trial, in October 2023, the FDA issued a CRL for our sNDA for patisiran for the treatment of ATTR amyloidosis with cardiomyopathy.
To execute our business plan of building a profitable, top-tier biotech company by the end of 2025 and achieving our Alnylam P5x25 strategy and the metrics associated with such strategy, in addition to successfully marketing, selling and expanding the approved indications of our approved products, we will need to successfully:
•execute product development activities and continue to leverage new technologies related to both RNAi and to the delivery of siRNAs to the relevant tissues and cells, including the liver, CNS, eye, lung, adipose and muscle;
•build and maintain a strong intellectual property portfolio;
•gain regulatory acceptance for the development and commercialization of our product candidates and successfully market our approved products, as well as any other products we commercialize;
•attract and retain customers for our products;
•enter into and maintain successful collaborations; and
•manage our spending as our costs and expenses increase due to, among other things, an increase in the number and size of our clinical trials, and the expansion of our commercialization activities.
If we are unsuccessful in accomplishing the objectives set forth above, we may not be able to develop product candidates, successfully commercialize our approved products or any future products, raise capital, if needed, repay our indebtedness, achieve financial self-sustainability or continue our operations.
We have a history of losses and may not become and remain profitable.
We have experienced significant operating losses since our inception. As of December 31, 2024, we had an accumulated deficit of $7.29 billion. Although to date we have launched four products in the U.S., EU and various other countries globally, and expect to launch our commercially approved products in additional countries during 2025 and beyond, and have one marketed product that is commercialized by a collaborator, we may not achieve or sustain profitability or positive cash flow from operations. For the year ended December 31, 2024, we recognized $1.65 billion in net product revenues from sales of ONPATTRO, AMVUTTRA, GIVLAARI and OXLUMO. We may continue to incur annual operating losses, and will require substantial resources over the next several years as we expand our efforts to discover, develop and commercialize RNAi therapeutics, and aim to achieve sustainable profitability by the end of 2025. While we believe our current cash, cash equivalents and marketable equity and debt securities, as well as the revenue we expect to generate from product sales and under our existing collaborations, including milestones and royalties on Leqvio sales, should enable us to achieve a self-sustainable profile by the end of 2025 without the need for future equity financing, we will depend on our ability to generate product, collaboration and royalty revenues to achieve this goal. In addition to revenues derived from sales of our current and future, if any, commercially approved products, we anticipate that a portion of any revenues we generate over the next several years will continue to be from collaborations with pharmaceutical and biotechnology companies, including Roche, Regeneron, Sanofi and Novartis, and we cannot be certain that we will be able to maintain our existing collaborations, secure and maintain new collaborations, meet our obligations under collaboration agreements, or achieve any milestones that we may be required to meet or achieve to receive payments under our existing or new collaborations. Moreover, we cannot be certain that our collaborators, including Roche and Novartis, will continue to successfully execute their obligations under our collaboration agreements and generate collaboration and royalty revenues for us.
To remain profitable, we must succeed in discovering, developing and commercializing novel product candidates with significant market potential. This will require us to build upon the success we have had in a range of challenging activities, including continued platform innovation, preclinical testing and clinical trial stages of development, obtaining regulatory approval and reimbursement for our novel product candidates and manufacturing, marketing and selling our approved products. We may not be able to sustain or increase profitability on a quarterly or annual basis. If we cannot remain consistently profitable, the market price of our common stock could decline. In addition, we may be unable to raise capital, expand our business, develop and commercialize additional product candidates or continue our operations.
We will require substantial funds to continue our research, development and commercialization activities, and if we require greater funds than we have estimated, we may need to critically limit, significantly scale back or cease certain activities.
We have used substantial funds to develop our RNAi technologies and will require substantial funds to conduct further research and development activities, including preclinical testing and clinical trials of our product candidates, and to manufacture, market and sell our approved products and any other products that are approved for commercial sale. Because the length of time or scope of activities associated with successful development of our product candidates may be greater than we anticipate, we may be unable to estimate the actual funds needed to develop and commercialize our product candidates.
Our future capital requirements and the period for which our existing resources will support our operations may vary from what we currently expect. We have based our expectations on a number of factors, many of which are difficult to predict or are outside of our control, including:
•progress in our research and development programs, including programs across a broad range of disease areas and indications, as well as what may be required by regulatory authorities to advance these programs;
•the timing, receipt and amount of milestone, royalty, research and development funding and other payments, if any, from present and future collaborators, if any, including milestone, royalty and research and development funding payments from Roche with respect to the development and commercialization of zilebesiran, as well as milestone and royalty payments from Novartis related to the commercialization of Leqvio;
•our ability to establish and maintain existing and additional collaborations and/or new business initiatives;
•the potential for improved product profiles to emerge from our new technologies and our ability to successfully advance our delivery efforts in extrahepatic tissues;
•the resources, time and costs required to successfully initiate and complete our preclinical studies and clinical trials, obtain regulatory approvals, prepare for global commercialization of our product candidates and obtain and maintain licenses to third-party intellectual property;
•our ability to establish, maintain and operate our own manufacturing facilities in a timely and cost-effective manner;
•our ability to manufacture, or contract with third parties for the manufacture of, our product candidates for clinical testing and our approved products for commercial sale;
•the impact of any future pandemics or public health emergencies or the ongoing conflicts in the Middle East and Ukraine on the initiation or completion of preclinical studies or clinical trials and the supply of our products or product candidates;
•the resources, time and cost required for the preparation, filing, prosecution, maintenance and enforcement of patent claims;
•the costs associated with legal activities, including litigation and government investigations, arising in the course of our business activities and our ability to prevail or reach a satisfactory result in any such legal disputes and investigations;
•the timing, receipt and amount of sales milestones and royalties, if any, from our approved products and our product candidates, if and when approved; and
•the outcome of the regulatory review process and commercial success of our products, including AMVUTTRA for the treatment of ATTR amyloidosis with cardiomyopathy, and products for which we are entitled to receive royalties, including Leqvio and fitusiran, assuming regulatory approval.
If our estimates, predictions and financial guidance relating to these factors are incorrect, we may need to modify our operating plan and may be required to seek additional funding in the future. We may do so through either collaborative arrangements, public or private equity offerings or debt financings, royalty or other monetization transactions or a combination of one or more of these funding sources. Additional funds may not be available to us on acceptable terms or at all.
The terms of any financing we may be required to pursue in the future may adversely affect the holdings or the rights of our stockholders. If we raise additional funds by issuing equity securities, dilution to our existing stockholders will result. In addition, as a condition to providing additional funding to us, future investors may demand, and may be granted, rights superior to those of existing stockholders.
If we require additional funding and are unable to obtain such funding on a timely basis, we may be required to significantly delay or curtail one or more of our research or development programs, or delay or curtail the further development of our global commercial infrastructure, and our ability to achieve our long-term strategic goals may be delayed or diminished. We also could be required to seek funds through arrangements with collaborators or others that may require us to relinquish rights to some of our technologies, product candidates or products that we would otherwise pursue on our own.
Although we sold a portion of the royalty stream and commercial milestones from the global sales of Leqvio by Novartis, we are entitled to retain the remaining portions of the future royalties and commercial milestone payments on Leqvio, and any negative developments related to Leqvio could have a material adverse effect on our receipt of those payments.
In April 2020, we sold to BX Bodyguard Royalties L.P. (an affiliate of The Blackstone Group Inc.), or Blackstone Royalties, 50% of the royalties payable to us with respect to net sales by Novartis, its affiliates or sublicensees of Leqvio and 75% of the commercial milestone payments payable to us under the MDCO License Agreement. If Blackstone does not receive royalty payments in respect of global sales of Leqvio equaling at least $1.00 billion by December 31, 2029, Blackstone Royalties’ interest in Leqvio royalties will increase to 55% (and our interest will decrease to 45%) effective January 1, 2030. As
a result, any factor that has an adverse impact on sales of Leqvio could affect our ability to meet the $1.00 billion repayment threshold in this timeframe, which in turn would have a negative impact on the percentage of the Leqvio royalty stream that we are entitled to retain.
Factors that could have an adverse impact on Leqvio sales include:
•competitors may develop new therapies or alternative formulations of products for HeFH and ASCVD;
•lack of acceptance of Leqvio by patients, the medical community or third party payors;
•any negative developments relating to Leqvio, such as safety, efficacy, or reimbursement issues;
•any disputes concerning patents or proprietary rights, or under license and collaboration agreements;
•foreign currency exchange rate fluctuations; and
•adverse regulatory or legislative developments that limit or prohibit the sale of Leqvio, such as restrictions on the use of Leqvio or safety-related label changes, including enhanced risk management programs.
If the revenues generated by sales of Leqvio are lower than expected, we may not receive commercial milestone payments and/or royalties in the amount we are currently anticipating, and our business, prospects, operating results and financial condition could be materially and adversely affected.
If the estimates we make, or the assumptions on which we rely, in preparing our financial statements and/or our projected guidance prove inaccurate, our actual results may vary from those reflected in our projections and accruals.
Our consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America, or GAAP. The preparation of these consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of our assets, liabilities, revenues and expenses, the amounts of charges accrued by us and related disclosure of contingent assets and liabilities. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. We cannot assure you, however, that our estimates, or the assumptions underlying them, will be correct.
Further, from time to time we issue financial guidance relating to our expectations regarding our combined product sales, collaboration and royalty revenues, and GAAP and non-GAAP combined research and development and selling, general and administrative expenses, which guidance is based on estimates and the judgment of our management. If, for any reason, our product sales, revenues and/or expenses differ materially from our guidance, we may have to adjust our publicly announced financial guidance. If we fail to meet, or if we are required to change or update any element of, our publicly disclosed financial guidance or other expectations about our business, our stock price could decline.
The investment of our cash, cash equivalents and marketable securities is subject to risks which may cause losses and affect the liquidity of these investments.
As of December 31, 2024, we had $2.69 billion in cash, cash equivalents and marketable securities. We historically have invested these amounts in money market funds, certificates of deposit, commercial paper, corporate notes, U.S. government-sponsored enterprise securities and U.S. treasury securities through highly rated financial institutions. Corporate notes may also include foreign bonds denominated in U.S. dollars. These investments are subject to general credit, liquidity, market and interest rate risks. We may realize losses in the fair value of these investments or a complete loss of these investments, which would have a negative effect on our financial condition. In addition, should our investments cease paying or reduce the amount of interest paid to us, our interest income would decline. The market risks associated with our investment portfolio may have an adverse effect on our operating results, liquidity and financial condition.
Volatility in foreign currency exchange rates could have a material adverse effect on our operating results.
Our revenue from outside of the U.S. is expected to increase as our products, whether commercialized by us or our collaborators, gain marketing approval in such jurisdictions. Our primary foreign currency exposure relates to movements in the Japanese yen, Euro and British pound. If the U.S. dollar weakens against a specific foreign currency, our revenues will increase, having a positive impact on net income, but our overall expenses will increase, having a negative impact. Conversely, if the U.S. dollar strengthens against a specific foreign currency, our revenues will decrease, having a negative impact on net income, but our overall expenses will decrease, having a positive impact. Any future volatility in foreign exchange rates is likely to impact our operating results and financial condition.
Changes in tax laws could adversely affect our business, prospects, operating results and financial condition.
Our business is subject to numerous international, federal, state, and other governmental laws, rules, and regulations that may adversely affect our operating results, including, taxation and tax policy changes, tax rate changes, new tax laws, or revised tax law interpretations, which individually or in combination may cause our effective tax rate to increase. In the U.S., the rules dealing with federal, state, and local income taxation are constantly under review by persons involved in the legislative process and by the Internal Revenue Service and the U.S. Treasury Department. Changes to tax laws (which changes may have
retroactive application) could adversely affect us or holders of our common stock. In recent years, many such changes have been made and changes are likely to continue to occur in the future. Future changes in tax laws could have a material adverse effect on our business, prospects, operating results and financial condition.
Additionally, the Organization for Economic Co-operation and Development, or the OECD, the EC, and individual taxing jurisdictions where we and our affiliates do business have recently focused on issues related to the taxation of multinational corporations. The OECD has released its comprehensive plan to create an agreed set of international rules for fighting base erosion and profit shifting. In addition, the OECD, the EC and individual countries are examining changes to how taxing rights should be allocated among countries considering the digital economy. As a result, tax laws in the U.S. and other countries in which we and our affiliates do business could change on a prospective or retroactive basis and any such changes could materially adversely affect our business, prospects, operating results and financial condition.
We may incur additional tax liabilities related to our operations.
We are subject to income tax in the U.S. and the foreign jurisdictions in which we operate. Significant judgment is required in determining our worldwide tax liabilities, and our effective tax rate is derived from the applicable statutory tax rates and relative earnings in each taxing jurisdiction. We record liabilities for uncertain tax positions that involve significant management judgment as to the application of law. Domestic or foreign taxing authorities may disagree with our interpretation of tax law as applied to our and our subsidiaries’ operations or with the positions we may take with respect to particular tax issues on our tax returns. Consequently, tax assessments or judgments in excess of accrued amounts that we have estimated in preparing our financial statements may materially and adversely affect our reported effective tax rate or our cash flows. Further, other factors may adversely affect our effective tax rate, including changes in the mix of our profitability from country to country, tax effects of stock-based compensation (which depend in part on the price of our stock and, therefore, are beyond our control), and changes in tax laws or regulations. For example, the OECD Global Anti-Base Erosion Model have influenced tax laws in countries in which we operate, including the implementation of minimum taxes. Changes to these or other laws and regulations or their interpretations could materially and adversely impact our effective tax rate or cash flows.
Any future outbreaks of pandemics or public health emergencies, may directly or indirectly adversely affect our business, results of operations and financial condition.
In the future, we may experience disruptions from a pandemic or public health emergency that could impact our business and operations, including our ability to obtain regulatory approval for and successfully commercialize our approved products, and we may not be able to meet expectations with respect to commercial sales as a result. In addition, we may experience decreased patient demand for our approved products if current or potential patients decide to delay treatment as a result of a pandemic or public health emergency. Business interruptions from pandemics or public health emergencies, including staffing shortages, raw material or other supply chain shortages, production slowdowns and disruptions in delivery systems, may also adversely impact the third parties we or our collaborators rely on in the U.S. and abroad to sufficiently manufacture our approved products and to produce product candidates in quantities we require, which may impair our commercialization efforts, our research and development activities and the potential commercialization of our product candidates.
Additionally, timely completion of preclinical activities and initiation of planned clinical trials are dependent upon the availability of, for example, preclinical and clinical trial sites, researchers and investigators, patients or healthy volunteer subjects available for recruitment and enrollment, and regulatory agency personnel, which may be adversely affected by global health matters, such as any pandemic or public health emergency. Health regulatory agencies globally may also experience disruptions in their operations as a result of a pandemic or future public health emergency, which could impact review, inspection and approval timelines.
While the ultimate impact of any pandemic or public health emergency on our business is uncertain, any negative impacts of such pandemic or public health emergency, alone or in combination with others, could exacerbate other risk factors discussed herein. The full extent to which any pandemic or public health emergency, will negatively affect our operations, financial performance, and stock price will depend on future developments that are highly uncertain and cannot be predicted.
Risks Related to Our Dependence on Third Parties
If we are unable to maintain our existing collaborations, or enter into new collaborations with other companies that can provide business and scientific capabilities and funds for the development and commercialization of our product candidates, it may have a negative impact on our business, prospects, operating results and financial condition.
We do not currently have adequate capacity or capabilities to advance all opportunities arising from our growing pipeline of RNAi therapeutics. Accordingly, we have entered into collaborations with third party collaborators we believe can provide such capacity and capabilities in certain territories and/or for certain product candidates, and we intend to enter into additional such collaborations in the future. Specifically, we currently have active collaborations with, among others, Roche, Regeneron, Sanofi and Novartis covering various products and product candidates in our pipeline.
In such collaborations, we expect our current, and may expect any future, collaborators to provide substantial capabilities in clinical development, regulatory affairs, and/or marketing, sales and distribution. Under certain of our collaborations, we also
expect our collaborators to develop, market and/or sell certain of our product candidates in certain territories or globally, and we have limited or no control over the development, sales, marketing and distribution activities of these collaborators. Our future revenues may depend heavily on the success of the efforts of these third parties. For example, we will rely entirely on (i) Regeneron for the development and commercialization of all programs targeting eye diseases (subject to limited exceptions), and potentially other CNS and liver programs; (ii) Novartis for the development and commercialization of Leqvio worldwide; (iii) Sanofi for the development and commercialization of fitusiran worldwide; and (iv) Roche for the commercialization of zilebesiran outside of the U.S. In the case of each collaboration referenced in clauses (i)-(iv) above, we are entitled to royalties, and in some instances commercial milestone payments, on the sales of the applicable product. If our collaborators are not successful in their development and/or commercialization efforts, our future revenues from the relevant product or product candidate may be adversely affected. For example, in December 2020 Novartis received a CRL from the FDA stating that the FDA could not approve the NDA by the PDUFA action date due to unresolved inspection-related conditions at a third party manufacturing facility. While Leqvio was ultimately approved by the FDA in December 2021, the resolution of the CRL resulted in a delay in the payment of an approval milestone and potential U.S. royalties. As discussed above, under our agreement with Blackstone Royalties, if the revenues generated by the royalties received by Blackstone Royalties from us with respect to Leqvio sales do not reach a certain level by the end of 2029, Blackstone Royalties will be entitled to a higher royalty percentage beginning in 2030, which would have an adverse impact on our royalty revenues beginning in 2030.
We may not be successful in entering into future collaborations on terms favorable to us due to various factors, including our ability to demonstrate improved product profiles from our new technologies, our ability to successfully demonstrate proof-of-concept for our technology in humans in certain tissues or disease areas, our ability to demonstrate the safety and efficacy of our specific product candidates, our ability to manufacture or have third parties manufacture RNAi therapeutics, the strength of our intellectual property portfolio and/or concerns around challenges or potential challenges to our intellectual property portfolio. Even when we succeed in securing such new collaborations, we may not be able to maintain them, or they may not be successful, if, for example, development or approval of a product candidate is delayed, challenges are raised as to the validity or scope of our intellectual property, we are unable to secure adequate reimbursement from payors, sales of an approved drug are lower than we expected, or our collaborator changes its strategic focus or otherwise determines not to move forward with a product or product candidate or to continue its collaboration with us.
Furthermore, any delay in entering into new collaboration agreements would have the potential to prevent or delay the development and commercialization of certain product candidates, or reduce the competitiveness such product candidates if they ultimately reach the market, which in turn could adversely affect our business, prospects, operating results and financial condition.
For certain product candidates, we have formed collaborations to fund all or part of the costs of drug development and commercialization, such as our collaborations with Roche, Regeneron, Sanofi and Novartis. We may not, however, be able to enter into additional collaborations for certain other programs, and the terms of any collaboration agreements we do secure may not be favorable to us. If we are not successful in our efforts to enter into future collaboration arrangements with respect to one or more of our product candidates, we may not have sufficient funds or other resources to develop these product candidates or other product candidates on our own, or to bring such product candidates to market. In these circumstances, we will not be able to generate revenues from these product candidates, and this will substantially harm our business, prospects, operating results and financial condition.
If any collaborator materially amends, terminates or fails to perform its obligations under agreements with us, the development and commercialization of our product candidates could be delayed or terminated.
Our dependence on collaborators for capabilities and funding means that our business could be adversely affected if any collaborator materially amends or terminates its collaboration agreement with us, in whole or in part, or fails to perform its obligations under that agreement. Our current or future collaborations, if any, may not be scientifically or commercially successful. Disputes may arise in the future with respect to the ownership of rights to technology or products developed with our collaborators, which could have an adverse effect on our ability to develop and commercialize any affected product candidate. Our current collaborations allow, and we expect that any future collaborations will allow, either party to terminate the collaboration for a material breach by the other party.
In addition, our collaborators may have additional termination rights for convenience with respect to the collaboration as a whole or a particular program under the collaboration, under certain circumstances. For example, in August 2024, we announced that Regeneron had opted out of further co-development and co-commercialization of mivelsiran for portfolio prioritization reasons. As a result of Regeneron’s opt-out, we have full development and commercialization rights to mivelsiran in all indications but we will be responsible for funding further development and commercialization of mivelsiran, including the ongoing Phase 2 development program, without funding from Regeneron. Regeneron will be eligible to receive low double-digit royalties on sales of mivelsiran, if approved.
Our collaboration agreement with Roche requires that Roche pay us a milestone payment at the initiation of a Phase 3 clinical trial of zilebesiran. In the event that Roche determines to terminate our collaboration prior to the initiation of a Phase 3 clinical trial of zilebesiran, whether because of data from the earlier clinical development of zilebesiran or for any other reason,
we would not receive the Phase 3 milestone from Roche, which could have a material adverse effect on our operating results and financial condition and make it more difficult for us to achieve profitability in 2025. In these circumstances, we would also need to determine whether to continue the clinical development of zilebesiran in the absence of funding and other support from Roche.
Our agreement with Novartis relating to the development and commercialization of inclisiran worldwide may be terminated by Novartis at any time upon four months’ prior written notice, provided if the agreement is terminated by Novartis for convenience, Novartis must grant a license to us under certain technology developed in the course of its (or MDCO’s) activities under the agreement, subject to a royalty to be negotiated between the parties. Moreover, any adverse actions by Novartis with respect to the MDCO License Agreement or disputes with Novartis regarding the MDCO License Agreement could adversely impact our ability to comply with our obligations under our agreements with Blackstone Royalties. If we were to lose a commercialization collaborator, we would have to attract a new collaborator (potentially on less favorable terms for us than we have with our existing collaborator) or develop expanded sales, distribution and marketing capabilities internally, which would require us to invest significant amounts of financial and management resources.
In addition, if we have a dispute with a collaborator over the ownership of technology or other matters, or if a collaborator terminates its collaboration with us, for breach or otherwise, or determines not to pursue the research, development and/or commercialization of the affected product or product candidate, it could delay our development of product candidates, result in the need for additional company resources to develop our product candidates, require us to expend time and resources to develop expanded sales and marketing capabilities on a more expedited timeline, make it more difficult for us to attract new collaborators and adversely affect how we are perceived in the business and financial communities.
Moreover, a collaborator, or in the event of a change in control of a collaborator or the assignment of a collaboration agreement to a third party, the successor entity or assignee, as in the case of MDCO and Novartis, could determine that it is in its interests to:
•pursue alternative technologies or develop alternative products, either on its own or jointly with others, that may be competitive with the products on which it is collaborating with us or which could affect its commitment to its collaboration with us;
•pursue higher-priority programs or change the focus of its development programs, which could affect the collaborator’s commitment to us; or
•if it has marketing rights, choose to devote fewer resources to the marketing of our product candidates, if any are approved for marketing, than it does for product candidates developed without us.
If any of these occur, the development and commercialization of one or more products or product candidates could be delayed, curtailed or terminated because we may not have sufficient financial resources or capabilities to continue such development and commercialization on our own.
We expect to incur significant costs as we continue to grow our manufacturing capabilities and resources and develop manufacturing expertise; in the meantime, we rely, and expect to continue to rely, on third parties to manufacture our products and product candidates. The loss of these or future third-party suppliers, or their inability to provide us with sufficient supply, could harm our business.
We have been expanding our manufacturing capabilities, and in order to continue to commercialize our approved products, continue to develop our current product candidates, apply for regulatory approvals and, if approved, commercialize future products, we will need to continue to develop our internal manufacturing capabilities and/or contract or otherwise arrange for any necessary external manufacturing capabilities. During 2020, we completed construction and qualification of our manufacturing facility in Norton, Massachusetts where we manufacture drug substances for early-stage clinical development and have the possibility to manufacture drug substances for late-stage clinical development and commercial use, in the future.
At the present time, we only have the capacity to manufacture limited quantities of clinical trial drug substance ourselves, and otherwise we continue to rely on third party CMOs to manufacture additional drug substance, and we rely on third party CMOs for all of our drug product requirements for clinical and commercial use. There are a limited number of CMOs worldwide with the expertise to manufacture our siRNA therapeutic products, and we currently rely on a limited number of CMOs in North America, Europe and Asia to manufacture our products and product candidates. There are risks inherent in pharmaceutical manufacturing that could affect the ability of our CMOs to meet our delivery time requirements or provide adequate amounts of material to meet our needs, and if our CMOs fail to do these things it could delay our clinical trials and potentially put our commercial supply at risk, as well as result in additional expense to us. To fulfill our future requirements, we will likely need to contract with additional CMOs, and such alternative suppliers may be limited, not be readily available, or we may be unable to enter into agreements with them on reasonable terms and in a timely manner, or at all.
In addition to the manufacture of synthetic siRNAs, we may have additional manufacturing requirements related to the technology required to deliver the siRNA to the relevant cell or tissue type, such as LNPs or conjugates or other drug delivery technologies. In some cases, the delivery technology we utilize is highly specialized or proprietary, and for technical and/or
legal reasons, we may have access to only one or a limited number of potential manufacturers for such delivery technology. In addition, the scale-up of our delivery technologies could be very difficult and/or take significant time. We also have limited experience in such scale-up and manufacturing, requiring us to depend on a limited number of third parties, who might not be able to deliver in a timely manner, or at all. Failure by manufacturers to properly manufacture our delivery technology and/or formulate our siRNAs for delivery could result in unusable product, supply delays and drug shortages. Furthermore, competition for supply from our manufacturers from other companies, a breach by such manufacturers of their contractual obligations or a dispute with such manufacturers would disrupt our clinical and/or commercial supply, cause delays in our discovery and development efforts, and result in additional expense to us.
In developing manufacturing capabilities by building our own manufacturing facilities, we have incurred substantial expenditures, and expect to incur significant additional expenditures in the future. Also, we have had to, and will likely need to continue to, recruit, hire, and train qualified employees to staff our facilities. If we are unable to manufacture sufficient quantities of material or if we encounter problems with our facilities in the future, we may also need to secure alternative suppliers, and such alternative suppliers may not be available, or we may be unable to enter into agreements with them on reasonable terms and in a timely manner, or at all. Given our dependence on a limited number of CMOs to supply our commercial products and clinical candidates, and the ongoing utilization of our own facilities, any delay or setback in the manufacture of our products could impede ongoing clinical and commercial supply, which could materially and adversely impact our business, prospects, operating results and financial condition. In addition, to the extent we or our collaborators rely on CMOs to supply our product candidates, any delays or disruptions in supply could have a material adverse impact on the research and development activities and potential commercialization of our or our collaborators’ product candidates.
The manufacturing processes for our products and any other product candidates that we may develop is subject to the FDA and foreign regulatory authority approval processes and we will need to meet, and will need to contract with CMOs who can meet, all applicable FDA and foreign regulatory authority requirements on an ongoing basis. The failure of any CMO to meet required regulatory authority requirements could result in the delayed submission of regulatory applications, or delays in receiving regulatory approval for any of our or our current or future collaborators’ product candidates. For example, in April 2022, due to an amendment to our vutrisiran NDA submission to address a pending inspection classification at a third-party secondary packaging and labeling facility, the FDA extended the review timeline of the NDA. In addition, if we receive the necessary regulatory approval for any product candidate, we also expect to rely on third parties, including potentially our commercial collaborators, to produce materials required for commercial supply.
Additionally, in January 2024, the U.S. House of Representatives introduced the BIOSECURE ACT (H.R. 7085), which was subsequently amended on May 15, 2024 and passed by the U.S. House of Representatives on September 9, 2024, and the Senate advanced a substantially similar bill (S.3558), both of which would prohibit U.S. federal executive agencies from contracting with any entity where the biotechnology equipment or services of a “biotechnology company of concern” would be used in the performance of that contract. Generally, a “biotechnology company of concern” is a biotechnology company that is subject to the jurisdiction, direction, control, or operates on behalf of a foreign adversary’s government and poses a risk to the national security of the U.S. The final language, pathway and timing for either of these bills or their provisions to become law remain uncertain and, although the bill was passed in the House on September 9, 2024, the Senate did not pass the bill before the end of the 118th Congress’s term. Nonetheless, if these bills are re-introduced in the House and Senate and become law, or similar laws are passed, they would have the potential to severely restrict our ability to purchase services or products from, or otherwise collaborate with, certain Chinese “biotechnology companies of concern” without losing the ability to contract with, or otherwise receive reimbursement from, the U.S. government. We do business with companies in China and it is possible some of our contractual counterparties could be impacted by the legislation described above and alternative arrangements may need to be made.
The new administration has substantially altered prior U.S. government international trade policy and has commenced activities to renegotiate, or potentially terminate, certain existing bilateral or multi-lateral trade agreements and treaties with foreign countries. In addition, the new administration has initiated or is considering imposing tariffs on certain foreign goods. Related to this action, certain foreign governments, including China, have instituted or are considering imposing tariffs on certain U.S. goods. It remains unclear what the new administration or foreign governments will or will not do with respect to tariffs or other international trade agreements and policies. A trade war or other governmental action related to tariffs or international trade agreements or policies has the potential to disrupt our research activities, affect our suppliers, increase the cost of materials purchased to manufacture our products, impact our ability to sell our products outside the U.S. or to sell our products outside the U.S. at competitive prices and/or to affect the U.S. or global economy or certain sectors thereof and, thus, could adversely impact our business.
If the third parties we engage to supply materials or manufacture product candidates or products for preclinical testing or clinical or commercial supply should cease to do so for any reason, we would likely experience delays in advancing these preclinical tests and clinical trials and/or interruptions in commercial supply while we identify and qualify replacement suppliers or manufacturers, and we may be unable to obtain replacement supplies on terms that are favorable to us, or at all. If we are not able to obtain adequate supplies of our product candidates or products or the substances used to manufacture them, it could materially and adversely impact our business, prospects, operating results or financial condition.
To the extent that we have existing, or enter into future, manufacturing arrangements with third parties, we depend, and will depend in the future, on these third parties to perform their obligations in a timely manner and consistent with contractual and regulatory requirements, including those related to quality control and quality assurance. The failure of any CMO to perform its obligations as expected, or, to the extent we manufacture all or a portion of our product candidates ourselves, our failure to execute on our manufacturing requirements, could adversely affect our business in a number of ways, including:
•we or our current or future collaborators may not be able to initiate or continue clinical trials of product candidates that are under development;
•we or our current or future collaborators may be delayed in submitting regulatory applications, or receiving regulatory approvals, for our product candidates;
•we may lose the cooperation of our collaborators;
•our facilities and those of our CMOs, and our products could be the subject of inspections by regulatory authorities that could have a negative outcome and result in supply delays;
•we may be required to cease distribution or recall some or all batches, of our products or take action to recover clinical trial material from clinical trial sites; and
•ultimately, we may not be able to meet the clinical and commercial demands for our product candidates and products.
We rely on third parties to conduct our clinical trials, and if such third parties fail to fulfill their obligations, our development plans may be adversely affected.
We rely on independent clinical investigators, CROs, and other third-party service providers to assist us in managing, monitoring and otherwise carrying out our clinical trials. We have contracted with, and we plan to continue to contract with, certain third parties to provide certain services for our clinical trials, including site selection, enrollment, monitoring, auditing and data management services. These investigators and CROs are not our employees, and we have limited control over the amount of time and resources they dedicate to our programs. These third parties may have contractual relationships with other entities, some of which may be our competitors, which may draw their time and resources away from our programs. Although we depend heavily on these parties, we control only limited aspects of their activity and therefore, we cannot be assured that these third parties will adequately perform their contractual obligations to us in compliance with regulatory and other legal requirements and our internal policies and procedures. Nevertheless, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific standards, and our reliance on third parties does not relieve us of our regulatory responsibilities. We and our CROs are required to comply with applicable GCP requirements, which are regulations and guidelines enforced by the FDA and foreign regulatory authorities for our product candidates in clinical development, and to implement timely corrective action to address any non-compliance. Regulatory authorities enforce these GCP requirements through periodic inspections of trial sponsors, principal investigators and trial sites, including in connection with the review of marketing applications. If we or any of our CROs fail to comply with applicable GCP requirements, or fail to take any such corrective action in a timely manner or at all, the clinical data generated in our clinical trials may be deemed unreliable and the FDA, EMA, PMDA or other foreign regulatory authorities may require us to take additional action or perform additional clinical trials before approving our marketing applications. We cannot assure you that upon inspection by a given regulatory authority in the future, such regulatory authority will determine that any of our clinical trials comply with GCP requirements.
If our third-party service providers cannot adequately and timely fulfill their obligations to us for any reason, or if the quality and accuracy of our clinical trial data is compromised due to failure by a third party service provider to adhere to our protocols or regulatory requirements or if a third party service provider otherwise fails to meet deadlines, our development plans and/or regulatory reviews for marketing approvals may be delayed or terminated. As a result, our business, prospects, operating results and financial condition would be harmed, and our stock price would likely be negatively impacted.
Risks Related to Managing Our Operations
If we are unable to attract and retain qualified key management and scientists, development, medical and commercial staff, consultants and advisors, our ability to implement our business plan may be adversely affected.
We are highly dependent upon our senior management and our scientific, clinical, sales and medical staff. The loss of the service of any members of our senior management could significantly delay or prevent the achievement of product development and commercialization, and other business objectives, and adversely impact our stock price. Our employment arrangements with our key personnel are terminable without notice. We do not carry key person life insurance on any of our employees.
We have grown our workforce significantly over the past several years and anticipate additional employee growth in the future, and we face intense competition for qualified individuals from numerous pharmaceutical and biotechnology companies, universities, governmental entities and other research institutions, many of which have substantially greater resources to attract and reward qualified individuals than we do. If we are not able to attract and retain highly qualified sales and marketing, research, development, and other professionals, it would negatively impact our ability to commercialize our approved products
and any future products and to support our growing research, development and global commercialization efforts and initiatives, which would have an adverse effect on our ability to implement our future business plans.
We may have difficulty expanding our operations successfully as we continue our evolution from a U.S.- and EU-based company primarily involved in discovery, preclinical testing and clinical development into a global company that develops and commercializes multiple products in multiple geographies.
As we continue the commercial launches of our approved products and increase the number of product candidates we are developing, we will need to continue to expand our operations in the U.S. and further develop operations in the EU and other geographies, including Asia and Latin America. To date, we have received regulatory approval for four products, which we have launched in multiple geographies globally, and we continue to expand the reach of these products with additional regulatory filings and launches.
We have grown our workforce significantly over the last several years and anticipate additional employee growth globally in the future as we focus on the commercialization of our approved products and achieving our Alnylam P5x25 strategy. This growth has placed a strain on our administrative and operational infrastructure and, as a result, we will need to continue to develop additional and/or new infrastructure and capabilities to support our growth and obtain additional space to conduct our global operations in the U.S., EU, Japan, Latin America and other geographies. If we are unable to develop such additional infrastructure or obtain sufficient space to accommodate our growth in a timely manner and on commercially reasonable terms, our business could be negatively impacted. As we continue the commercialization of our approved products, and as the product candidates we develop enter and advance through clinical trials, we will need to continue to expand our global development, regulatory, manufacturing, quality, compliance, and marketing and sales capabilities, or contract with other organizations to provide these capabilities for us. In addition, as our operations continue to expand, we will need to successfully manage additional relationships with various collaborators, suppliers, distributors and other organizations. Our ability to manage our operations and future growth will require us to continue to enhance our operational, financial and management controls and systems, reporting systems and infrastructure, ethics and compliance functions, and policies and procedures. We may not be able to implement enhancements to our management information and control systems in an efficient or timely manner and may discover deficiencies in existing systems and controls.
The use of social media presents risks and challenges.
We use social media to communicate about our clinical development programs and the diseases our investigational RNAi therapeutics are being developed to treat, including in connection with our commercialization efforts for our approved products. We intend to do the same for our future products, if approved. While we believe our social media use is appropriate under current regulatory guidance, social media practices in the biopharmaceutical industry continue to evolve and regulations and regulatory guidance relating to such use are evolving and not always clear. This evolution creates uncertainty and risk of noncompliance with regulations applicable to our business, resulting in potential regulatory actions against us, along with the potential for litigation related to off-label marketing or other prohibited activities. For example, for our clinical-stage candidates, patients may use social media channels to comment on their experience in an ongoing blinded clinical trial or to report an alleged adverse event, or AE. When such disclosures occur, there is a risk that trial enrollment may be adversely impacted, that we may fail to monitor and comply with applicable AE reporting obligations or that we may not be able to defend our business in the face of the political and market pressures generated by social media due to restrictions on what we may say about our investigational products. There is also a risk of inappropriate disclosure of sensitive information or negative or inaccurate posts or comments about us on any online platform, including a blog on the internet, or a post on a website, that can be distributed rapidly and could negatively harm our reputation. If any of these events were to occur or if we otherwise fail to comply with applicable regulations, we could face regulatory actions or incur liability or other harm to our business.
Our business and operations could suffer in the event of system failures or unauthorized or inappropriate use of or access to our systems.
We are increasingly dependent on our information technology systems and infrastructure for our business. We collect, store and transmit sensitive information including intellectual property, proprietary business information, including highly sensitive clinical trial data, and personal information in connection with our business operations. The secure maintenance of this information is critical to our operations and business strategy. Some of this information could be subject to information breaches, unauthorized access, human error, computer viruses, denial-of-service attacks, malicious code, spam attacks, phishing, ransomware or other forms of social engineering and other events that could impact the security, reliability, confidentiality, integrity and availability of our systems, including by third parties with a wide range of motives and expertise, including organized criminal groups, nation-states, “hacktivists,” patient groups, rogue current or former employees and others. Cyber-attacks can be designed to collect sensitive or proprietary information, manipulate, destroy or corrupt data, systems or applications, or accounts, to steal money or extort money through the use of so-called “ransomware”, and to disable the functioning or use of applications or technology assets. Cyber-attacks are of ever-increasing levels of sophistication, and despite our security measures, our information technology and infrastructure may be vulnerable to such attacks or may be breached, including due to employee error or malfeasance. The risk of cyber-attacks is increased with employees working remotely, as remote work increases our vulnerability to cybersecurity-related events such as phishing attacks and other security threats.
Cybersecurity requires ongoing investment and diligence against evolving threats and is subject to federal and state regulation relating to the protection of confidential information. We may be required to expend significant additional resources to modify our protective measures, to investigate and remediate vulnerabilities or other exposures, to make required notifications, to restore our systems and fully recover from a cyber-attack, or to update our technologies and digital properties to comply with industry and regulatory standards, but we may not have adequate personnel, financial or other resources to fully meet these threats and evolving standards. We will also be required to effectively and efficiently govern, manage and ensure timely enhancements to our systems, including in their design, architecture and interconnections as well as their organizational and technical protections.
The pervasiveness of cybersecurity incidents in general and the risks of cyber-crime are complex and continue to evolve. Although we are making significant efforts to maintain the security and integrity of our information systems and are exploring various measures to manage the risk of a security breach or disruption, there can be no assurance that our security efforts and measures will be effective or that attempted security breaches or disruptions would not be successful or damaging. Despite the implementation of security measures, our internal computer systems and those of our contractors, consultants and collaborators are vulnerable to damage or interruption from computer viruses, unauthorized or inappropriate access or use, natural disasters, pandemics or public health emergencies, terrorism, war (including the ongoing conflicts in Ukraine and the Middle East), and telecommunication and electrical failures. Such events could cause interruption of our operations. For example, the loss of preclinical trial data, data from completed or ongoing clinical trials for our product candidates, or manufacturing data could result in delays in our regulatory filings and development efforts, as well as delays in the commercialization of our products, and significantly increase our costs. To the extent that any disruption, security breach or unauthorized or inappropriate use or access to our systems were to result in a loss of or damage to our data, or inappropriate disclosure of confidential or proprietary information, including but not limited to patient, employee or vendor information, we could incur notification obligations to affected individuals and government agencies, face potential lawsuits from patients, collaborators, employees, stockholders or other third parties, and incur liability under foreign, federal and state laws that protect the privacy and security of personal information, and the development and potential commercialization of our products and product candidates could be delayed. Such events may ultimately result in financial losses that are either not insured or are not fully covered through any insurance we maintain.
In addition, our increased use of cloud technologies heightens these third party risks, and any failure by cloud or other technology service providers to adequately safeguard their systems and prevent cyber-attacks could disrupt our operations and result in misappropriation, corruption, or loss of confidential or proprietary information. Although we require certain third party service providers to maintain certain minimum security levels and adopt certain security procedures by policy, we cannot ensure the universal or consistent compliance with these policies across our service providers, or that our policies and procedures will be adequate to address the evolving threat environment and identify and provide controls for all of the risks in a particular service provider’s environment.
Risks Related to Our Industry
Risks Related to Development, Clinical Testing and Regulatory Approval of Our Product Candidates and the Commercialization of Our Approved Products
Any product candidate we or our collaborators develop may fail in development or experience significant delays.
Our business depends upon the successful development and commercialization of our product candidates. These product candidates are in various stages of development and must satisfy rigorous standards of safety and efficacy before they can be approved for sale by the FDA or foreign regulatory authorities. Nonclinical and clinical testing is expensive, difficult to design and implement, can take many years to complete, is uncertain as to outcome, and the historical failure rate for product candidates is high. It is impossible to predict when or if any of our product candidates will prove effective and safe in humans or will receive regulatory approval. Failure to advance our product candidates through clinical development could impair our ability to ultimately commercialize products, which could materially harm our business and long-term prospects.
Clinical trials may fail to demonstrate that our product candidates are safe for humans and effective for indicated uses, and earlier results, both nonclinical and clinical, may not be indicative of future clinical trial results. Many companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in clinical development even after achieving promising results in earlier studies, and any such setbacks in our clinical development could have a material adverse effect on our business, prospects, operating results and financial condition. Moreover, clinical data are often susceptible to varying interpretations, and many companies that have believed their product candidates performed satisfactorily in clinical trials have nonetheless failed to obtain marketing approval of their product candidate. For example, in June 2024, we reported positive results from the HELIOS-B Phase 3 clinical trial of vutrisiran for the treatment of patients with ATTR amyloidosis with cardiomyopathy. While vutrisiran demonstrated positive results in the clinical trial, we cannot be certain that the results from HELIOS-B will support approval of vutrisiran for the treatment of patients with ATTR amyloidosis with cardiomyopathy. Moreover, certain of our product candidates employ novel delivery technologies that have yet to be extensively evaluated in human clinical trials and proven safe and effective.
In addition, from time to time, we report interim, topline, and preliminary data from our clinical trials, which is based on a preliminary analysis of then-available data, and the results and related findings and conclusions are subject to change. Interim or preliminary data from a clinical trial may not be predictive of final results from the clinical trial and are subject to the risk that one or more of the clinical outcomes may materially change as patient enrollment and treatment continues and more patient data become available or as patients from our clinical trials continue other treatments for their disease. Topline data also remain subject to audit and verification procedures that may result in the final data being materially different from the preliminary data we previously published. As a result, topline data should be viewed with caution until the final data are available. If the interim, topline, or preliminary data that we report differ from actual results, or if others, including regulatory authorities, disagree with the conclusions reached, our ability to obtain approval for, and commercialize, our product candidates may be harmed, which could harm our business, operating results, prospects or financial condition.
Additionally, several of our planned and ongoing clinical trials utilize an “open-label” trial design. In an “open-label” clinical trial, both the patient and investigator know whether the patient is receiving the investigational product candidate or, if the trial includes multiple arms, either an existing approved drug or placebo. Most typically, open-label clinical trials test only the investigational product candidate and sometimes may do so at different dose levels. Open-label clinical trials are subject to various limitations that may exaggerate any therapeutic effect as patients in open-label clinical trials are aware when they are receiving treatment. Accordingly, open-label clinical trials may be subject to a “patient bias” where patients perceive their symptoms to have improved merely due to their awareness of receiving an experimental treatment. In addition, open-label clinical trials may be subject to an “investigator bias” where those assessing and reviewing the physiological outcomes of the clinical trials are aware of which patients have received treatment and may interpret the information of the treated group more favorably given this knowledge. The results from an open-label trial may not be predictive of future clinical trial results with any of our product candidates when studied in a blinded, controlled environment with a placebo or active control.
Clinical testing is expensive, is difficult to design and implement, can take many years to complete and is uncertain as to outcome. We cannot guarantee that any clinical trials will be conducted as planned or completed on schedule, or at all. A failure of one or more clinical trials can occur at any stage of testing, which may result from a multitude of factors, including, but not limited to, flaws in trial design, dose selection issues, patient enrollment criteria, operational challenges, site implementation challenges, biostatistical plans and failure to demonstrate favorable safety or efficacy traits. If our product candidates experience any such problems, we may not have the financial resources necessary to continue development of the affected product candidate or any of our other product candidates. We may also lose, or be unable to enter into, collaborative arrangements for the affected product candidate or any of our other product candidates.
A failure of one or more of our clinical trials can occur at any stage of testing. We may experience numerous unforeseen events during, or as a result of, nonclinical testing and the clinical trial process that could extend our clinical development timelines and delay or prevent regulatory approval or our ability to commercialize our product candidates, including:
•our nonclinical tests or clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional nonclinical testing or clinical trials, or we may abandon projects that have the potential to be promising;
•delays in filing IND applications or comparable foreign applications or delays or failure in obtaining the necessary approvals from regulators or IRBs/ethics committees in order to commence a clinical trial at a prospective trial site, or their suspension or termination of a clinical trial once commenced;
•conditions imposed on us by an IRB or ethics committee, or the FDA or comparable foreign regulatory authorities regarding the scope or design of our clinical trials;
•problems in engaging IRBs or ethics committees to oversee clinical trials or problems in obtaining or maintaining IRB or ethics committee approval of clinical trials;
•delays in enrolling patients and volunteers into clinical trials, and variability in the number and types of patients and volunteers available for clinical trials, including as a result of a pandemic or public health emergency or the ongoing conflicts in Ukraine and the Middle East;
•disruptions caused by man-made or natural disasters or pandemics, epidemics or public health emergencies or other business interruptions;
•high drop-out rates for patients and volunteers in clinical trials;
•negative or inconclusive results from our clinical trials or the clinical trials of others for product candidates similar to ours;
•inadequate supply or quality of product candidate materials or other materials necessary for the conduct of our clinical trials or disruption or delays in clinical supply due to a future pandemic or public health emergency;
•serious and unexpected drug-related side effects experienced by patients taking our approved products, participants in our clinical trials or individuals using drugs similar to our products or product candidates;
•poor or disappointing effectiveness of our product candidates during clinical trials;
•the imposition of a clinical hold by regulatory authorities as a result of a serious adverse event or manufacturing concerns or after an inspection of our clinical trial operations or trial sites;
•unfavorable FDA or other regulatory agency inspection and review of a clinical trial site or records of any clinical or nonclinical investigation;
•failure of our third-party contractors or investigators to comply with regulatory requirements, including GLP, GCP and cGMP, or otherwise meet their contractual obligations in a timely manner, or at all;
•governmental or regulatory delays and changes in regulatory requirements, policy and guidelines, including the imposition of additional regulatory oversight around clinical testing generally or with respect to our technology in particular;
•delays in reaching a consensus with regulatory agencies on trial design;
•interpretations of data by the FDA and similar foreign regulatory agencies that differ from ours;
•lack of adequate funding to continue the clinical trial; or
•diminished revenue potential of the applicable program due to competition.
Clinical trials must be conducted in accordance with the legal requirements, regulations or guidelines of the FDA and corresponding regulatory authorities outside the United States. We could encounter delays if a clinical trial is suspended or terminated by us, by the FDA or any other regulatory authority, or if the IRBs of the institutions in which such trials are being conducted suspend or terminate the participation of their clinical investigators and sites subject to their review. Such authorities may suspend or terminate a clinical trial due to a number of factors, including failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols, inspection of the clinical trial operations or trial site by the FDA or other regulatory authorities resulting in the imposition of a clinical hold, unforeseen safety issues or adverse side effects, failure to demonstrate a benefit from using a product candidate, changes in governmental regulations or administrative actions or lack of adequate funding to continue the clinical trial.
Difficulty in enrolling patients could delay or prevent clinical trials of our product candidates, and ultimately delay or prevent regulatory approval.
Clinical trials of a new product candidate require the enrollment of a sufficient number of patients, including patients who are suffering from the disease the product candidate is intended to treat and who meet other eligibility criteria. Our ability to enroll patients in our clinical trials in sufficient numbers and on a timely basis is subject to a number of factors. Clinical trials are expensive and require significant operational resources. Delays in patient enrollment or unforeseen drop-out rates may result in increased costs and longer development times.
Patient enrollment is affected by many factors, including the size of the patient population, the age and condition of the patients, the stage and severity of disease, the availability of clinical trials for other investigational drugs for the same disease or condition, the nature of the protocol, the proximity of patients to clinical sites, the availability of effective treatments for the relevant disease, the eligibility criteria for the clinical trial, the risk that patients enrolled in clinical trials will drop out of the clinical trials before clinical trial completion, and factors we may not be able to control, such as potential pandemics. We or our collaborators may experience difficulty enrolling our clinical trials due to the availability of existing approved treatments, as well as other investigational treatments in development. For example, we may experience delays in recruiting and enrolling patients, and in particular treatment-naive patients, for our planned Phase 3 clinical trial of nucresiran for the treatment of ATTR amyloidosis with cardiomyopathy because there are multiple approved therapies on the market for that indication. Delays or difficulties in patient enrollment, or difficulties retaining trial participants, including as a result of the availability of existing approved treatments or other investigational treatments, safety concerns, or the impact of pandemics or other public health emergencies, can result in increased costs, longer development times or termination of a clinical trial.
If any of our current or future products or product candidates causes undesirable side effects or has other unexpected adverse properties, such side effects or properties could delay or prevent regulatory approval, limit the commercial potential or result in significant negative consequences following any potential marketing approval.
Although our RNAi therapeutics have been generally well-tolerated in our clinical trials to date, new safety findings may emerge. Results of our clinical trials could reveal a high and unacceptable severity and prevalence of side effects or unexpected characteristics. There can be no assurance that our product candidates will not cause undesirable side effects. If any product candidates we develop are associated with unacceptable side effects or deaths, we may need to abandon the development of such product candidates or limit development to certain uses or subpopulations in which the unacceptable side effects or other characteristics are less prevalent, less severe or more acceptable from a risk-benefit perspective, any of which would have a material adverse effect on our business, financial condition, results of operations and prospects. Furthermore, the occurrence of such adverse events could result in the suspension or termination of clinical trials of a product candidate by us, our collaborators, IRBs or ethics committees, or the FDA or a foreign regulatory authority, and may negatively impact the clinical
and/or regulatory timelines of the impacted product candidates. For example, our Phase 1 clinical trial of mivelsiran for Alzheimer’s Disease remains on partial clinical hold in the United States due to findings observed in non-clinical chronic toxicology studies. While our clinical development of mivelsiran has not been impacted by the partial clinical hold because of the dose levels at which it applies, it is possible that the partial clinical hold could have an impact if our development plans change or that future partial or full clinical holds on mivelsiran or our other product candidates could impact our ability to advance the clinical development of such product candidate on our expected timelines or at all.
In addition, the occurrence of serious adverse events could also result in refusal by the FDA or a foreign regulatory authority to approve a particular product candidate for any or all indications of use, or in limitations in the label of any approved product. Even if we are able to demonstrate that any future serious adverse events are not product-related and regulatory authorities do not order us to cease further development of our product candidates, such occurrences could affect patient recruitment or the ability of enrolled patients to complete the trial. Moreover, if we elect, or are required, to delay, suspend or terminate any clinical trial of any product candidate, the commercial prospects of such product candidates may be harmed and our ability to generate product revenues from any of these product candidates may be delayed or eliminated. Any of these occurrences may harm our ability to develop other product candidates, and may harm our business, financial condition and prospects significantly.
We or our collaborators may be unable to obtain U.S. or foreign regulatory approval for our or our collaborated product candidates and, as a result, we or our collaborators may be unable to commercialize such product candidates.
Any product candidates we or our collaborators develop are subject to extensive governmental regulations relating to, among other things, research, testing, development, manufacturing, safety, efficacy, approval, recordkeeping, reporting, labeling, storage, pricing, advertising, promotion and distribution of drugs. Failure to obtain marketing approval for a product candidate we may develop will prevent us from commercializing the product candidate in a given jurisdiction. Securing regulatory approval requires the submission of extensive nonclinical and clinical data and supporting information to the various regulatory authorities for each therapeutic indication to establish the product candidate’s safety and efficacy. Securing regulatory approval also requires the submission of information about the product manufacturing process to, and inspection of manufacturing facilities by, the relevant regulatory authorities. Satisfaction of these and other regulatory requirements is costly, time consuming, uncertain and subject to unanticipated delays. It is possible that the product candidates we and our collaborators are developing will not obtain the regulatory approvals necessary for us or our collaborators to begin selling them, or, in the case of vutrisiran, will not obtain regulatory approval to be sold for an additional, broader indication than the indication for which it is currently approved. It is also possible that the FDA or other regulatory authorities may determine that the data generated in clinical trials for a product candidate is not sufficient to support the approval of an application for regulatory approval. For example, although we reported positive results from the APOLLO-B Phase 3 clinical trial of patisiran in patients with ATTR amyloidosis with cardiomyopathy, in October 2023, the FDA issued a CRL in response to our sNDA for patisiran, indicating the sNDA could not be approved in its present form.
The time required to obtain FDA and other regulatory approvals is unpredictable but typically takes many years following the commencement of clinical trials, depending upon the type, complexity and novelty of the product candidate. The standards that the FDA and its foreign counterparts use when regulating us are not always applied in a predictable or uniform manner and can change over time. Any analysis we perform of data from nonclinical and clinical activities is subject to confirmation and interpretation by regulatory authorities, which could delay, limit or prevent regulatory approval. We or our collaborators may also encounter unexpected delays or increased costs due to new government regulations, for example, from future legislation or administrative action, or from changes in FDA policy during the period of product development, clinical trials and FDA regulatory review. It is impossible to predict whether legislative changes will be enacted, or whether FDA or foreign regulations, guidance or interpretations will be changed, or what the impact of such changes, if any, may be.
Because there may be approved treatments for some of the diseases for which we or our collaborators may seek approval, including vutrisiran for the treatment of ATTR amyloidosis with cardiomyopathy, or treatments in development which will be approved by the time we or our collaborators file for approval, in order to receive regulatory approval, we or they may need to demonstrate through clinical trials that the product candidates we develop to treat these diseases are not only safe and effective, but safer and/or more effective than other products.
Interruption or delays in the operations of the FDA, EMA and comparable foreign regulatory agencies may impact the review, inspection and approval timelines for our or our collaborated product candidates. During the COVID-19 public health emergency, the FDA worked to ensure timely reviews of applications for medical products in line with its user fee performance goals and conducted mission critical domestic and foreign inspections to ensure compliance of manufacturing facilities with FDA quality standards. In addition, during the COVID-19 public health emergency, a number of companies announced receipt of complete response letters due to the FDA’s inability to complete required inspections for their applications. In December 2020, the FDA issued a CRL regarding Novartis’ NDA for inclisiran, stating that the agency could not approve the NDA by the PDUFA action date due to unresolved facility inspection-related conditions. In July 2021, Novartis announced that the resubmission to the FDA of the inclisiran NDA to address the complete response letter was filed, and the FDA approved Leqvio (the trade name under which inclisiran is marketed in the U.S.) in December 2021. This delay in the approval of Leqvio resulted in delayed milestone and royalty revenue to us. Any similar interruption or delay by the FDA, EMA or comparable foreign
regulatory authorities could have a material adverse effect on our or our collaborators’ efforts to obtain regulatory approval for our or our collaborators’ product candidates, which could have a material adverse effect on our business, prospects, operating results or financial condition.
Inadequate funding for the FDA and other government agencies and/or potentially shifting priorities under the new administration could hinder the FDA’s and/or those other government agencies’ ability to hire and retain key leadership and other personnel, prevent new products and services from being developed and/or commercialized in a timely manner, or otherwise prevent those agencies from performing normal business functions on which the operation of our business may rely, which could negatively impact our business. The ability of the FDA to review and approve new products, to provide feedback on clinical trials and development programs, to meet with sponsors and to otherwise review regulatory submissions can be affected by a variety of factors, including government budget and funding levels; the ability to hire and retain key personnel and accept the payment of user fees; and statutory, regulatory, and policy changes, among other factors. Average review times at the agency may fluctuate as a result. Delays at the FDA as a result of these or other factors could impact the FDA’s ability to act on our sNDA submission for vutrisiran by the PDUFA target action date of March 23, 2025, or our other regulatory submissions, including our collaborator Sanofi’s NDA for fitusiran. In addition, government funding of other government agencies or of government programs that provide research funding on which our operations may rely directly or indirectly via third party research and development projects associated with our product development programs, is subject to the political process, which is inherently fluid and unpredictable. The failure for such funding to be furnished or to be furnished in a timely manner could impact our ongoing research and development initiatives.
Any disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies or to otherwise respond to regulatory submissions which would adversely affect our business. For example, the new administration has discussed several changes to the reach and oversight of the FDA, which could affect its relationship with the pharmaceutical industry, transparency in decision making and ultimately the cost and availability of prescription drugs. Additionally, over the last several years, the U.S. government has shut down multiple times and certain regulatory agencies, such as the FDA, have had to furlough critical FDA and other government employees and stop critical activities. For example, the new administration recently announced plans to reduce the number of federal employees by establishing voluntary termination programs, by position eliminations or by involuntary terminations. If funding for the FDA is reduced, if the FDA workforce is reduced, if FDA priorities are changed or if a prolonged government shutdown occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, including our pending sNDA submission for vutrisiran and our collaborator Sanofi’s pending NDA for fitusiran, which could have a material adverse effect on our business.
The FDA or foreign regulatory authorities may request additional clinical or other data or information in connection with the regulatory review of our or our collaborators’ product candidates, including by issuing a complete response letter that may require that we or our collaborators submit additional clinical or other data or impose other conditions that must be met in order to secure final approval of our or our collaborators’ NDA applications, including potentially requiring a facility inspection. Even if such data and information are submitted, or any such inspection is completed, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval.
We may seek a Fast Track, Priority Review, or Breakthrough Therapy designation or similar designations outside the U.S. for some of our product candidates. Product candidates that receive one or more of these designations may be eligible for, among other things, a priority regulatory review. Each of these designations is within the discretion of the FDA. Accordingly, even if we believe one of our product candidates meets the criteria for Fast Track, Priority Review, and/or Breakthrough Therapy designation, the FDA or similar regulatory authorities may disagree and instead determine not to make such designation. The receipt of one or more of these designations for a product candidate does not guarantee a faster development process, review or approval compared to products developed or considered for approval under conventional assessment procedures and does not assure ultimate approval by the FDA or similar regulatory authorities. In addition, even if one or more of our products or product candidates qualifies for Fast Track, Priority Review, and/or Breakthrough Therapy designation, the FDA or similar regulatory authorities may later decide to withdraw such designation if it determines that the product or product candidate no longer meets the conditions for qualification.
Any delay or failure in obtaining required approvals for our product candidates or our collaborated product candidates could have a material adverse effect on our ability to generate revenues from any product candidate for which we or our collaborators may seek approval in the future. For example, as a result of the CRL from the FDA in response to our sNDA for patisiran as a treatment for ATTR amyloidosis with cardiomyopathy, our ability to generate product revenues for patisiran has been negatively impacted.
Furthermore, even if we or our collaborators receive approval of an NDA or foreign marketing application for a product candidate, the FDA or the applicable foreign regulatory agency may grant approval or other marketing authorization contingent on the performance of costly additional clinical trials, including post-market clinical trials. The FDA or the applicable foreign regulatory agency also may approve or authorize for marketing a product candidate for a more limited indication or patient population than we originally request, and the FDA or applicable foreign regulatory agency may not approve or authorize the labeling that we believe is necessary or desirable for the successful commercialization of a product candidate. Any of these
restrictions or commitment could limit an approved product’s market opportunity and have a negative impact on our business, prospects, operating results and financial condition and our stock price. For example, if the FDA or foreign regulatory agency approves the sNDA for vutrisiran for the treatment of ATTR amyloidosis with cardiomyopathy with a more limited label than we are seeking, it could limit the scope of the commercial opportunity for vutrisiran.
In addition, the FDA has the authority to require a Risk Evaluation and Mitigation Strategy, or REMS, as part of its review of an NDA, or after approval, which may impose further requirements or restrictions on the distribution or use of an approved drug, such as limiting prescribing to certain physicians or medical centers that have undergone specialized training, limiting treatment to patients who meet certain safe-use criteria and requiring treated patients to enroll in a registry. In the EU, we or our collaborators could be required to adopt a similar plan, known as a risk management plan, and our products could be subject to specific risk minimization measures, such as restrictions on prescription and supply, the conduct of post-marketing safety or efficacy studies, or the distribution of patient and/or prescriber educational materials. In either instance, these limitations and restrictions may limit the size of the market for our products and affect reimbursement by third-party payors.
We are also subject to numerous foreign regulatory requirements governing, among other things, the conduct of clinical trials, manufacturing and marketing authorization, pricing and third-party reimbursement. The foreign regulatory approval process varies among countries and includes all of the risks associated with FDA approval described above as well as risks attributable to the satisfaction of local regulations in foreign jurisdictions. Approval by the FDA does not ensure approval by any regulatory authority outside the U.S. and vice versa.
Our marketed products and any product candidates for which we obtain approval are subject to extensive and ongoing regulatory oversight. If we or our collaborators fail to comply with continuing U.S. and foreign requirements, our approvals could be limited or withdrawn, we could be subject to other penalties, and in any such case our business would be seriously harmed.
Our five marketed products, including one product that is commercialized by a collaborator, and any product candidates for which we or our collaborators may ultimately receive marketing authorization, subject to ongoing regulatory requirements governing the testing, manufacturing, labeling, packaging, storage, advertising, promotion, sale, distribution, import, export, recordkeeping, and reporting. Ongoing FDA requirements include, among other things, submission of safety and other post-marketing information and reports, registration and listing, continued compliance with GMP requirements relating to manufacturing, quality control, quality assurance and corresponding maintenance of records and documents, requirements regarding the distribution of samples to physicians, and GCP requirements for any clinical trials that we conduct post-approval. In addition, we are conducting, and intend to continue to conduct, clinical trials for our product candidates, and we intend to seek approval to market our product candidates, in jurisdictions outside of the U.S., and therefore will be subject to, and must comply with, regulatory requirements in those jurisdictions.
Our products are subject to continuing regulatory oversight following approval, including the review of adverse drug experiences and clinical results that are reported after our drug products are made commercially available. This includes results from any post-marketing tests or surveillance to monitor the safety and efficacy of our approved products or other products required as a condition of approval or otherwise agreed to by us. Products are more widely used by patients once approval has been obtained and therefore side effects and other problems may be observed after approval that were not seen or anticipated, or were not as prevalent or severe, during pre-approval clinical trials or nonclinical studies. The subsequent discovery of previously unknown or underestimated problems with a product could result in:
•sales of our approved products may be lower than originally anticipated;
•regulatory approvals for our approved products may be restricted or withdrawn;
•we may decide, or be required, to send product warning letters or field alerts to physicians, pharmacists and hospitals;
•additional nonclinical studies or clinical trials, changes in labeling, adoption of a REMS plan, or changes to manufacturing processes, specifications and/or facilities may be required; and/or
•government investigations or lawsuits, including class action suits, may be brought against us.
Any of the above occurrences could reduce or eliminate sales of our approved products, increase our expenses and impair our ability to successfully commercialize one or more of these products.
The CMO and manufacturing facilities we use to make our approved products and certain of our current product candidates, including our Cambridge facility, our Norton facility, as well as facilities at Agilent and other CMOs, will also be subject to periodic review and inspection by the FDA and other regulatory agencies. For example, Agilent and our Cambridge-based facility were subject to regulatory inspection by the FDA and the EMA in connection with the review of our applications for regulatory approval for ONPATTRO and GIVLAARI, and may be subject to similar inspection in connection with any subsequent applications for regulatory approval of one or more of our products filed in other territories. The discovery of any new or previously unknown problems with our or our CMO’s manufacturing processes or facilities, may result in restrictions on the CMO or facility or the products manufactured at such facility, including delay in approval or withdrawal of an approved product from the market. For example, due to a routine inspection by the FDA at a CMO facility that resulted in a pending
inspection classification, we amended our regulatory submission for vutrisiran for the treatment of hATTR-amyloidosis with polyneuropathy in adults, which delayed our PDUFA goal date and AMVUTTRA’s FDA approval. We may not have the ability or capacity to manufacture material at a broader commercial scale in the future. We may manufacture clinical trial materials, or we may contract a third party to manufacture this material for us. Reliance on CMOs entails risks to which we would not be subject if we manufactured products ourselves, including reliance on the applicable CMO for regulatory compliance.
If we or our collaborators, CMOs or service providers fail to comply with applicable continuing regulatory requirements in the U.S. or a foreign jurisdiction in which we seek to market our products, we or they may be subject to, among other things, fines, warning or untitled letters, holds on clinical trials, refusal by the FDA or foreign regulatory authorities to approve pending applications or supplements to approved applications, suspension or withdrawal of regulatory approval, product recalls and seizures, refusal to permit the import or export of products, operating restrictions, fines, injunctions, civil penalties and criminal prosecution.
Any government investigation of alleged violations of law could require us to expend significant time and resources in response and could generate negative publicity. The occurrence of any event or penalty described above may inhibit our ability to commercialize our approved products and any product candidates we may develop and adversely affect our business, financial condition, results of operations and prospects.
The U.S. Supreme Court’s June 2024 decision in Loper Bright Enterprises v. Raimondo overturned the longstanding Chevron doctrine, under which courts were required to give deference to regulatory agencies’ reasonable interpretations of ambiguous federal statutes. The Loper decision could result in additional legal challenges to regulations and guidance issued by federal agencies, including the FDA, on which we rely. Any such legal challenges, if successful, could have a material impact on our business. Additionally, the Loper decision may result in increased regulatory uncertainty, inconsistent judicial interpretations, and other impacts to the agency rule-making process, any of which could adversely impact our business and operations. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action or as a result of legal challenges, either in the United States or abroad. If we are slow or unable to adapt to changes in existing requirements or the adoption of new requirements or policies, or if we are not able to maintain regulatory compliance, our business could be materially harmed.
We may incur significant liability if enforcement authorities allege or determine that we are engaging in commercial activities with respect to our unapproved product candidates or promoting our commercially approved products in a way that violates applicable regulations.
The FDA and other regulatory agencies closely regulate the post-approval marketing and promotion of products to ensure that they are marketed only for the approved indications and in accordance with the provisions of the approved labeling. Although physicians are generally permitted, based on their medical judgment, to prescribe products for indications other than those approved by the applicable regulatory agency, manufacturers are prohibited from promoting their products for such off-label uses. For example, we may not currently promote ONPATTRO or AMVUTTRA in the U.S. for use in any indications other than the treatment of hATTR amyloidosis with polyneuropathy in adults. If a regulatory agency determines that our promotional materials, or other activities constitute off-label promotion, it could request that we modify our promotional materials or other activities, conduct corrective advertising, or subject us to regulatory enforcement actions, such as the issuance of a warning or untitled letter, injunction, seizure, civil fines and criminal penalties. It also is possible that other federal, state, or foreign enforcement authorities might take action if they believe that the alleged improper promotion led to the submission and payment of claims for an off-label use, which could result in significant fines or penalties under other statutory authorities, such as laws prohibiting false claims for reimbursement. Even if it is later determined we were not in violation of these laws, we may be faced with negative publicity, incur significant expenses defending our actions, and have to divert significant management resources from other matters.
Even if we or our collaborators receive regulatory approval to market our product candidates, the market may not be receptive to our product candidates upon their commercial introduction, which could adversely affect our business, prospects, operating results and financial condition.
It may be difficult for us to convince the medical community and third-party payors to accept and use our products, or to provide favorable reimbursement. The factors we believe will materially affect market acceptance of our products include:
•the timing of our receipt of any marketing approvals, the terms of any approvals and the countries in which approvals are obtained;
•the safety and efficacy of our product candidates, as demonstrated in clinical trials and as compared with alternative treatments, if any;
•relative convenience, dosing regimen and ease of administration of our product candidates;
•the willingness of patients to accept potentially new routes of administration or new or different therapeutic approaches and mechanisms of action;
•the success of our physician education programs;
•the availability of adequate government and third-party payor reimbursement;
•the pricing of our products, particularly as compared to alternative treatments, and the market perception of such prices and any price increase that we may implement in the future; and
•availability of alternative effective treatments for the diseases that our product candidates we develop are intended to treat and the relative risks, benefits and costs of those treatments.
For example, ONPATTRO utilizes an intravenous mode of administration with pre-medication that physicians and/or patients may not readily adopt, and which may not compete favorably with other available options for the treatment of hATTR amyloidosis with polyneuropathy in adults, including WAINUA (eplontersen), which is marketed by AstraZeneca and Ionis, and administered subcutaneously, or tafamidis, which is marketed by Pfizer in several countries and administered in pill form. In addition, fitusiran represents a new approach to treating hemophilia, which may not be readily accepted by physicians and patients and their caregivers. Vutrisiran, if approved for the treatment of ATTR amyloidosis with cardiomyopathy, is administered through a subcutaneous injection and could face similar challenges in market acceptance.
We are a multi-product commercial company and expect to continue to invest significant financial and management resources to continue to build our marketing, sales, market access and distribution capabilities and further establish our global infrastructure. If we are not able to continue to develop and scale these capabilities, we may not be able to successfully commercialize our products and product candidates.
We received our first product approval in August 2018 and have established capabilities for marketing, sales, market access and distribution over the last several years. We currently expect to rely on third parties to launch and market certain of our product candidates in certain geographies, if approved. However, we are commercializing ONPATTRO, AMVUTTRA, GIVLAARI and OXLUMO, and intend to commercialize other product candidates, if approved, on our own globally in major markets. Accordingly, we have developed internal marketing, sales, market access and distribution capabilities as part of our core product strategy initially in the U.S., Europe and Japan, with expansion ongoing globally, which has required, and will continue to require, significant financial and management resources. For those products for which we will perform marketing, sales, market access and distribution functions ourselves we could face a number of additional risks, including:
•scaling and retaining our global sales, marketing and administrative infrastructure and capabilities;
•hiring, training, managing and supervising our personnel worldwide;
•the cost of further developing, or leveraging an established, marketing or sales force, which may not be justifiable in light of the revenues generated by any particular product and/or in any specific geographic region; and
•our direct sales and marketing efforts may not be successful.
If we are unable to continue to develop and scale our own global marketing, sales, market access and distribution capabilities for our current and any future products, we will not be able to successfully commercialize our products without reliance on third parties.
The patient populations suffering from hATTR amyloidosis with polyneuropathy, ATTR amyloidosis with cardiomyopathy, AHP and PH1 have not been established with precision. If the actual number of patients suffering from these diseases is smaller than we estimate, or if we fail to raise awareness of these diseases and diagnosis is not improved, our business, prospects, operating results and financial condition may be adversely affected.
Our estimates regarding the potential market size for ONPATTRO, AMVUTTRA, GIVLAARI, OXLUMO or any future products that we may commercialize, may be materially different from the actual market size, including as a result of the indication approved by regulatory authorities, which could result in significant changes in our business plan and may have a material adverse effect on our business, prospects, operating results and financial condition. If we are unable to accurately estimate the number of patients suffering from a disease for which we successfully commercialize a product or we were not able to raise awareness of these diseases and improve diagnosis, it could have a material adverse effect on our business, prospects, operating results or financial condition, and it will be more difficult or impossible to achieve profitability.
Any products we currently market or may develop in the future may become subject to unfavorable pricing regulations, third-party reimbursement practices or healthcare reform initiatives, thereby harming our business, prospects, operating results and financial condition.
The regulations that govern marketing approvals, coverage, pricing and reimbursement for new drugs vary widely from country to country and are subject to change. Some countries require approval of the sale price of a drug before it can be marketed. In many countries, the pricing review period begins after marketing authorization or product licensing approval is granted. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. We are actively monitoring these regulations as we market and sell our approved products and as several of our product candidates move through late stages of development. However, a number of our product candidates
are currently in the earlier stages of development, and we will not be able to assess the impact of such regulations or any changes to such development programs for a number of years. We might also obtain regulatory approval for a product, including one or more of our approved products, in a particular country, but then be subject to price regulations or price controls that delay our commercial launch of the product and/or negatively impact the revenues we are able to generate from the sale of the product in that country and potentially in other countries due to reference pricing.
We believe that the efforts of governments and third-party payors to contain or reduce the cost of healthcare and legislative and regulatory proposals to broaden the availability of healthcare will continue to affect the business and financial condition of pharmaceutical and biopharmaceutical companies.
In the U.S., healthcare and pharmaceutical pricing are subject to both government and public scrutiny and calls for reform, and the U.S. government continues to propose on legislative and regulatory changes designed to control costs. Specifically, there have been several recent U.S. Congressional inquiries into prescription drugs, and proposed and enacted federal and state legislation and regulations designed to, among other things, bring more transparency to drug pricing, reduce the cost and reimbursement of prescription drugs under third-party payor programs, and alter the nature and operation of manufacturer patient support programs. These developments could, directly or indirectly, affect our ability to sell ONPATTRO, AMVUTTRA, GIVLAARI, OXLUMO or future products, if approved, at a favorable price.
At the federal level, for example, the Inflation Reduction Act, or IRA, includes several provisions that will impact our business to varying degrees. For example, the IRA may require us to pay rebates if we increase the net cost of a Medicare Part B or Part D drug faster than the rate of inflation. In addition, our cost-sharing responsibility for any approved product covered by Medicare Part D could be significantly greater under the IRA Part D benefit structure compared to the pre-IRA benefit design. Under the IRA’s Price Negotiation Program, an FDA approval for vutrisiran for treatment of Stargardt Disease would cause us to lose the orphan exemption for AMVUTTRA from Medicare price negotiation. As a result, in October 2022, we announced we would not pursue a Phase 3 clinical trial to study vutrisiran for the treatment of Stargardt Disease. Manufacturers that fail to comply with the IRA may be subject to various penalties, including civil monetary penalties or a potential excise tax. The effect of the IRA on our business and the healthcare industry in general continues to evolve and we may continue to discover additional adverse impacts on our company or our industry. The IRA is anticipated to have significant effects on the pharmaceutical industry and may reduce the prices we can charge and reimbursement we can receive for our products, among other effects.
Furthermore, the new administration has indicated that lowering prescription drug prices is a priority, but the nature and impact of any potential policy changes remain uncertain. While frameworks like the IRA aim to control costs, their implementation under the new administration could introduce further regulatory changes, such as additional price restrictions on products we sell to Medicare or other government purchasers. Any such developments could adversely affect reimbursement, competitive dynamics, and our business. We continue to monitor legislative reforms and assess their potential impact on our operations, but we cannot predict their ultimate effect on our business. Additionally, the new administration may propose policy changes that create additional uncertainty for our business. These may include new price restrictions on products we sell to Medicare or other government purchasers, or other regulatory changes impacting reimbursement or competitive dynamics in multisource markets.
At the state level, governments have become increasingly aggressive in passing legislation and implementing regulations designed to control pharmaceutical product pricing. Some of these measures include upper payment limits on state-regulated payers; regulating product access, copayment assistance, and marketing; imposing drug price, cost, and marketing disclosure and transparency requirements; permitting importation from other countries; and encouraging bulk purchasing. For example, on January 5, 2024, the FDA authorized Florida’s Agency for Health Care Administration’s drug importation proposal, the first step toward Florida facilitating importation of certain prescription drugs from Canada. We cannot predict how further developments of or changes to these policies and rules will affect our business. In addition, regional healthcare authorities and individual hospitals are increasingly using bidding procedures to determine what pharmaceutical products and which suppliers will be included in their prescription drug and other healthcare programs. These measures could reduce the demand for our products, once approved, or put pressure on our product pricing. We cannot predict what healthcare reform initiatives may be adopted in the future in the U.S. or other foreign countries. Further federal, state and foreign legislative and regulatory developments are likely, with expected increased pressure on drug pricing. Such reforms could have a material and adverse effect on our anticipated revenues for one or more of our approved products or other product candidates that we may successfully develop and for which we may obtain regulatory approval and may affect our business, prospects, operating results and financial condition and our ability to develop drug candidates.
Our ability to commercialize our approved products or any future products successfully also will depend in part on the extent to which coverage and reimbursement for these products and related treatments will be available from third-party payors such as government health administration authorities, private health insurers and other organizations. One or more of our approved products and any other products for which we are able to obtain marketing approval may not be considered medically necessary or cost-effective, and the amount reimbursed may be insufficient to allow us to sell such product(s) or any future products on a competitive basis or realize an appropriate return on our investment in product development. There may be significant delays in obtaining coverage for newly approved drugs, and coverage may be more limited than the purposes for
which the drug is approved by the FDA or foreign regulatory authorities. Moreover, eligibility for coverage does not imply that any drug will be reimbursed in all cases or at a rate that covers our costs, including research, development, manufacture, sale and distribution or that covers a particular provider’s cost of acquiring the product. Interim payments for new drugs, if applicable, may also not be sufficient to cover our costs and may not be made permanent. Reimbursement may be based on lower-cost drugs that are already marketed, covered, and reimbursed, may be incorporated into existing payments for other services, and may reflect budgetary constraints or imperfections in data. Net prices for drugs may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors and by any future relaxation of laws that presently restrict imports of drugs from countries where they may be sold at lower prices than in the U.S. In particular, governments in certain markets such as in EU, the U.K., Japan, and China, provide healthcare at low (or zero) direct costs to consumers at the point of care, and thus have significant power as large single payers to regulate prices or impose other cost control mechanisms. In addition, the emphasis on managed care in the U.S. has increased and we expect will continue to exert downward pressure on pharmaceutical pricing. Coverage policies, third-party reimbursement rates and pharmaceutical pricing regulations may change at any time. Even if favorable coverage and reimbursement status is attained for one or more products for which we receive regulatory approval, less favorable coverage policies and reimbursement rates may be implemented in the future.
Increasingly, the third-party payors who reimburse patients or healthcare providers, such as government and private insurance plans, are requiring that drug companies provide them with predetermined discounts from list prices, and are seeking to reduce the prices charged or the amounts reimbursed for drug products. In the U.S., we have entered into value-based agreements, or VBAs, and are negotiating additional VBAs with commercial health insurers. The goal of these agreements is to ensure that we are paid based on the ability of our commercially approved products to deliver results in the real-world setting comparable to those demonstrated in our clinical trials, and the agreements are structured to link the performance of our approved products in real-world use to financial terms. Partnering with payors on these agreements is also intended to provide more confidence regarding the value of our products and help accelerate coverage decisions for patients. If the payment we receive for our products, or the reimbursement provided for such products, is inadequate in light of our significant development and other costs, or if reimbursement is denied, our return on investment could be adversely affected. In addition, we have stated publicly that we intend to grow through continued scientific innovation rather than more substantial price increases. Specifically, we have stated that we will not raise the price of any product for which we receive marketing approval over the rate of inflation, as determined by the consumer price index for urban consumers (approximately 3.5% currently) absent a significant value driver. Our patient access philosophy could also negatively impact the revenues we are able to generate from the sale of one or more of our products in the future.
Insurers are increasingly adopting programs and policies that limit access to medications and increase out-of-pocket costs for patients. In the U.S., to help patients access and afford our approved product(s), we may utilize programs to assist them, including patient assistance programs and co-pay coupon programs for eligible patients. It is possible that changes in insurer policies regarding co-pay coupons (such as co-pay accumulator and maximizer programs) and patient assistance programs (such as alternative funding programs) and/or the introduction and enactment of new legislation or regulatory action could restrict or otherwise negatively affect these co-pay coupon programs and patient support programs, which could result in fewer patients using affected products, and therefore could have a material adverse effect on our sales, business, and financial condition.
We are subject to U.S. and certain foreign export and import controls, sanctions, embargoes, anti-corruption laws, and anti-money laundering laws and regulations. Failure to comply with these legal standards could impair our ability to compete in domestic and international markets. We can face criminal liability and other serious consequences for violations, which can harm our business.
We are subject to export control and import laws and regulations, including the U.S. Export Administration Regulations, U.S. Customs regulations, various economic and trade sanctions regulations administered by the U.S. Treasury Department’s Office of Foreign Assets Control, and anti-corruption laws, including the U.S. Foreign Corrupt Practices Act of 1977, as amended, the U.S. domestic bribery statute contained in 18 U.S.C. § 201, the U.S. Travel Act, the USA PATRIOT Act, the UK Bribery Act 2010, and other applicable anti-bribery and anti-money laundering laws. Anti-corruption laws are interpreted broadly and prohibit companies and their officers, directors, employees, agents, contractors, and other third-party representatives from directly or indirectly authorizing, promising, offering, providing, soliciting, or receiving payments or anything else of value in order to improperly influence the acts or decisions of recipients in the public or private sector or to secure any other improper advantage to obtain or retain business. From time to time, we may engage third parties to conduct clinical trials outside of the U.S., to sell our products abroad, and/or to obtain necessary permits, licenses, patent registrations, and other regulatory approvals. We have direct or indirect interactions with officials and employees of government agencies or government-affiliated hospitals, universities, and other organizations. We can be held liable for the corrupt or other illegal activities of agents, contractors, and third-party representatives acting on our behalf, even if we do not explicitly authorize or have actual knowledge of such activities. Any violations of the laws and regulations described above may result in substantial fines and penalties, reputational harm, and other adverse consequences.
We remain focused on these laws and the activities they regulate and maintain a global compliance program designed to empower our business to operate in compliance with their requirements.
Governments outside the U.S. may impose strict price controls, which may adversely affect our revenues.
The pricing of prescription pharmaceuticals is also subject to governmental control outside the U.S. In these countries, pricing negotiations with governmental authorities can take considerable time after the receipt of regulatory approval for a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidates to other available therapies, which is time-consuming and costly. If reimbursement of our products is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our ability to generate revenues and become profitable could be impaired.
In some countries, including Member States of the EU, or Japan, the pricing of prescription drugs may be subject to governmental control. Additional countries may adopt similar approaches to the pricing of prescription drugs. In such countries, pricing negotiations with governmental authorities can take considerable time after receipt of regulatory approval for a product. In addition, governments and other stakeholders can put considerable pressure on prices and reimbursement levels, including as part of cost containment measures. Moreover, political, economic and regulatory developments may further complicate pricing negotiations, and pricing negotiations may continue after coverage and reimbursement have been obtained. Reference pricing used by various countries and parallel distribution, or arbitrage between low-priced and high-priced countries, can further reduce prices. We cannot be sure that such prices and reimbursement will be acceptable to us or our collaborators. Publication of discounts by third-party payors or authorities may lead to further pressure on the prices or reimbursement levels within the country of publication and other countries. If pricing is set at unsatisfactory levels or if reimbursement of our products is unavailable or limited in scope or amount, our revenues from sales by us or our collaborators and the potential profitability of our approved products or any future products in those countries would be negatively affected. We could also suffer impact from tightening pricing controls on account of greater competition from less expensive generic or biosimilar products once patent or other exclusivity expires. Certain governments have adopted policies to switch prescribed products to generic versions to reduce costs.
If we or our collaborators, CMOs or service providers fail to comply with healthcare laws and regulations, including but not limited to those related to fraud and abuse, we or they could be subject to enforcement actions, which could negatively impact our ability to develop, market and sell our products and may harm our reputation.
Healthcare providers, physicians, and third-party payors play a primary role in the recommendation and prescription of any products for which we obtain marketing approval. Our existing and future arrangements with third-party payors and customers may expose us to broadly applicable fraud and abuse and other healthcare laws and regulations that may constrain our business or financial arrangements and relationships through which we market, sell, and distribute our products. In the United States, these laws include, without limitation, federal and state fraud and abuse laws, transparency laws and patient data privacy and security laws and regulations, including but not limited to the following:
•The U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons or entities from knowingly and willfully soliciting, receiving, offering or paying any remuneration, directly or indirectly, overtly or covertly, in cash or in kind, to induce either the referral of an individual for, or the purchase or ordering of, a good or service for which payment may be made, in whole or in part, under a federal healthcare program, such as the Medicare and Medicaid programs. Violations are subject to civil and criminal fines and penalties, imprisonment, and exclusion from government healthcare programs.
•The U.S. federal false claims laws, including the FCA, which generally prohibit, individuals or entities from knowingly presenting or causing to be presented, claims for payment for good or services by government-funded programs such as Medicare or Medicaid that are false or fraudulent. Manufacturers can be held liable under the FCA even when they do not submit claims directly to government payors if they are deemed to “cause” the submission of false or fraudulent claims. Penalties are three times the amount of the claims in question plus civil monetary penalties.
•The federal civil monetary penalties laws, which generally impose civil fines for, among other things, the offering or transfer of remuneration to a Medicare or Medicaid beneficiary if the person knows or should know it is likely to influence the beneficiary’s selection of a particular provider, practitioner, or supplier of services reimbursable by Medicare or Medicaid. Conduct regulated by the federal civil monetary penalties law often overlaps with other healthcare laws, including the federal Anti-Kickback Statute.
•The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which in addition to privacy and security protections applicable to healthcare providers and other entities, prohibits executing, or attempting to execute, a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters.
•Federal “sunshine” requirements imposed on drug, device, and medical supply manufacturers when payment is available under Medicare, Medicaid or the Children’s Health Insurance Program to report annually to HHS information regarding any “transfer of value” made or distributed to healthcare providers and organizations. Failure to submit timely, accurate and complete information may result in civil monetary penalties.
•Federal price reporting laws, which, among other requirements, require manufacturers to calculate, report, and certify in a timely manner complex pricing and other product data to government programs, where such reported data may be
used in the calculation of reimbursement and/or discounts on approved products; and to pay rebates or offer discounts on pharmaceutical products.
•Federal statutory and regulatory requirements applicable to pricing and sales of products to federal government agencies.
•Federal consumer protection and unfair competition laws, which broadly regulate marketplace activities and activities that potentially harm consumers.
•The Federal Food, Drug, and Cosmetic Act, which among other things, strictly regulates drug product and medical device marketing, prohibits manufacturers from marketing such products prior to approval or for unapproved indications and regulates the distribution of samples.
•State and foreign laws comparable to each of the above federal laws, including in the EU laws prohibiting giving healthcare professionals any gift or benefit in kind as an inducement to prescribe our products and transparency laws requiring the public disclosure of payments made to healthcare professionals and institutions, and data privacy laws, in addition to anti-kickback and false claims laws applicable to commercial insurers and other non-federal payors, requirements for mandatory corporate regulatory compliance programs, and laws relating to price transparency and government reimbursement programs patient data privacy and security.
•European privacy laws including Regulation 2016/679, known as the General Data Protection Regulation, or the EU GDPR, and the EU GDPR as transposed into the laws of the UK, the UK GDPR, collectively referred to as the GDPR, and the e-Privacy Directive (2002/58/EC), and the national laws implementing each of them, as well as the Public and Electronic Communications Regulations 2003 in the UK and the privacy laws of Japan, Brazil and other territories.
•The California Consumer Privacy Act of 2018, as amended by the California Privacy Rights Act of 2020, or, collectively, the CCPA, that, among other provisions, gives California residents rights to their personal information and imposes various privacy and security obligations on regulated businesses. Furthermore, comprehensive privacy laws similar to the CCPA and consumer health data laws have been enacted in more than twelve other states and proposed in nearly one third of all states. Washington’s law regulating consumer health data contains a private right of action. The effects of the CCPA and other state privacy laws, and the creation of new regulatory bodies, such as the California Privacy Protection Agency, increases the cost and complexity of operating our business and our exposure to regulatory investigations, enforcement, fines, and penalties, any of which could negatively impact our business and operations. Failure to comply with these obligations could result in damage to our reputation and legal liability, censures, penalties and fines, disgorgement of profits, restitution to customers, remediation, the issuance of cease-and-desist orders, or injunctive or other equitable relief against us, which individually or in the aggregate could negatively impact our financial results. Depending on the nature of the violation, we may be required to offer restitution or remediation to customers, and the cost of doing so could exceed our loss reserves.
Analogous state and foreign laws and regulations, such as state anti-kickback, anti-bribery and false claims laws, which may apply to healthcare items or services that are reimbursed by non-governmental third-party payors, including private insurers, as well as other state laws that require companies to comply with specific compliance standards, restrict financial interactions between companies and healthcare providers, require companies to report information related to payments to healthcare providers, marketing expenditures or pricing or require the licensing or registration of sales representatives.
If our operations are found to be in violation of any of the aforementioned requirements, we may be subject to penalties, including civil or criminal penalties (including individual imprisonment), criminal prosecution, monetary damages, the curtailment or restructuring of our operations, or exclusion from participation in government contracting, healthcare reimbursement or other government programs, including Medicare and Medicaid, or the imposition of a corporate integrity agreement with the Office of Inspector General of the Department of HHS, or the OIG, any of which could materially and adversely affect our business, prospects, operating results or financial condition. We remain focused on enhancing our global compliance infrastructure as we prepare for the launch of our products in additional countries, assuming regulatory approvals. Although effective compliance programs can mitigate the risk of investigation and prosecution for violations of these laws, these risks cannot be entirely eliminated. For additional information, see the Risk Factor captioned “We may incur significant liability if enforcement authorities allege or determine that we are engaging in commercial activities with respect to our unapproved product candidates or promoting our commercially approved products in a way that violates applicable regulations.” Any action against us for an alleged or suspected violation could cause us to incur significant legal expenses and could divert our management’s attention from the operation of our business, even if our defense is successful. In addition, achieving and sustaining compliance with applicable laws and regulations may be costly to us in terms of money, time and resources.
If we or our collaborators, CMOs or service providers fail to comply with applicable federal, state or foreign laws or regulations, we could be subject to enforcement actions, which could affect our ability to develop, market and sell our approved products, or any future products, successfully and could harm our reputation and lead to reduced acceptance of our products by
the market. These enforcement actions include, among others, civil and criminal penalties, up to and including criminal prosecution resulting in fines, exclusion from healthcare reimbursement programs and imprisonment.
Moreover, federal, state and foreign laws or regulations are subject to change, and while we, our collaborators, CMOs and/or service providers currently may be compliant, we could fall out of compliance due to changes in interpretation, prevailing industry standards or the legal structure.
Activities we undertake related to commercializing our drug products could create risk under laws such as the federal Anti-Kickback Statute and/or the federal False Claims Act, or FCA, with the potential for significant liability, including civil and administrative penalties, criminal sanctions, and potential exclusion from participation in government programs. Third party patient assistance programs that receive financial support from companies have become the subject of enhanced government and regulatory scrutiny, with related legal risk.
We are subject to governmental regulation and other legal obligations related to privacy, data protection and information security, and we are subject to consumer protection laws that regulate our marketing practices and prohibit unfair or deceptive acts or practices. Our actual or perceived failure to comply with such obligations could harm our business.
The GDPR imposes strict requirements on controllers and processors of personal data, including special protections for “special category data,” which includes health, biometric and genetic information of data subjects located in the EEA and UK. Further, GDPR provides a broad right for EEA Member States to create supplemental national laws, such as laws relating to the processing of health, genetic and biometric data, which could further limit our ability to use and share such data or could cause our costs to increase, and harm our business and financial condition.
Failure to comply with the requirements of the GDPR and the related national data protection laws of the EEA Member States and the UK, which may deviate slightly from the GDPR, may result in fines of up to 4% of total global annual revenue, or €20.0 million (₤17.5 million under the UK GDPR), whichever is greater, and in addition to such fines, we may be the subject of litigation and/or adverse publicity, which could have a material adverse effect on our reputation and business. As a result of the implementation of the GDPR, we are required to implement a number of measures to ensure compliance with the data protection regime. The GDPR (i) requires us to inform data subjects of how we process their personal data and how they can exercise their rights, (ii) requires us to ensure we have a valid legal basis to process personal data (if this is consent, the requirements for obtaining consent carries a higher threshold), (iii) requires us to appoint a data protection officer where sensitive personal data (i.e., health data) is processed on a large scale, (iv) introduces mandatory data breach notification requirements throughout the EEA and UK, (v) requires us to maintain records of our processing activities and document data protection impact assessments where there is high risk processing, (vi) imposes additional obligations on us when we are contracting with service providers, requires (vii) appropriate technical and organizational measures to be put in place to safeguard personal data and (viii) requires us to adopt appropriate privacy governance including policies, procedures, training and data audit.
Significantly, the GDPR imposes strict rules on the transfer of personal data out of the EEA and UK to the U.S. or other regions that have not been deemed to offer “adequate” privacy protections. In the past, companies in the U.S. were able to rely upon the EU-U.S., UK-U.S. and the Swiss-U.S. Privacy Shield frameworks as a basis for lawful transfer of personal data from the EU and the UK to the U.S. In July 2020, the Court of Justice of the European Union, or CJEU, in Case C-311/18 (Data Protection Commissioner v Facebook Ireland and Maximillian Schrems, or Schrems II) invalidated the EU-U.S. Privacy Shield on the grounds that the Privacy Shield failed to offer adequate protections to EU personal data transferred to the U.S. The CJEU, in the same decision, deemed that the Standard Contractual Clauses, or SCCs, published by the EC are valid. However, the CJEU ruled that transfers made pursuant to the SCCs need to be assessed on a case-by-case basis to ensure the law in the recipient country provides “essentially equivalent” protections to safeguard the transferred personal data as the EU, and required businesses to adopt supplementary measures if such standard is not met. Subsequent guidance published by the European Data Protection Board, or EDPB, in June 2021 described what such supplementary measures must be, and stated that businesses should avoid or cease transfers of personal data if, in the absence of supplementary measures, equivalent protections cannot be afforded. On June 4, 2021, the EC published new versions of the SCCs, which seek to address the issues identified by the CJEU’s Schrems II decision and provide further details regarding the transfer assessments that the parties are required to conduct when implementing the new SCCs. However, there continue to be concerns about whether the SCCs and other mechanisms will face additional challenges. Similarly, in September 2020, the Swiss data protection authority determined the Swiss-U.S. Privacy Shield framework was no longer a valid mechanism for Swiss-U.S. data transfers and raised questions about the validity of the SCCs as a mechanism for transferring personal data from Switzerland. While SCCs provide an alternative to our Privacy Shield certification for EU-U.S. data flows, the decision (and certain regulatory guidance issued in its wake) casts doubt on the legality of EU-U.S. data flows in general. Any inability to transfer, or burdensome restrictions on the ability to transfer, personal data from the EU to the U.S. in compliance with applicable data protection laws may impede our ability to conduct clinical trials and may adversely affect our business, prospects, operating results and financial condition. The UK is not subject to the EC’s new SCCs but has published its own transfer mechanism, the International Data Transfer Agreement or International Data Transfer Addendum, which enables transfers from the UK. On March 25, 2022, the EC and the U.S. announced a political agreement on a new “Trans-Atlantic Data Privacy Framework” to replace the invalidated Privacy Shield. The framework introduced new binding safeguards to address the concerns raised by the CJEU in Schrems II. On July
10, 2023, the EC announced that it had adopted its adequacy decision for that data privacy framework, labelled the EU-U.S. Data Privacy Framework. The adequacy decision concluded that the U.S. ensures an adequate level of protection for personal data transferred from the EU to US companies under the new framework, and the EC stated that as a result personal data can flow safely from the EU to US companies participating in the framework, without having to put in place additional data protection safeguards. The EU-U.S. Data Privacy Framework is subject to periodic reviews, to be conducted by the EC, together with other European data protection authorities and U.S. authorities, with the first review to take place within a year of adoption of the adequacy decision. A case has been lodged with and remains pending before the EU courts challenging the validity of the EU-U.S. Data Privacy Framework.
EEA Member States have adopted implementing national laws to implement the GDPR which may partially deviate from the GDPR and the competent authorities in the EEA Member States may interpret GDPR obligations slightly differently from country to country, and we do not expect to operate in a uniform legal landscape in the EU. In addition, the UK Government has now introduced a Data Protection and Digital Information Bill, or the UK Bill, into the UK legislative process. The aim of the UK Bill is to reform UK’s data protection regime following Brexit. If passed, the final version of the UK Bill may have the effect of further altering the similarities between the UK and EEA data protection regime. The anticipated UK general election in 2024 could postpone passage of the UK Bill.
We are subject to the supervision of local data protection authorities in those jurisdictions in which we are monitoring the behavior of individuals in the EEA or UK (i.e., undertaking clinical trials). We depend on a number of third parties in relation to the provision of our services, a number of which process personal data of EU and/or UK individuals on our behalf. With each such provider we enter or intend to enter into contractual arrangements under which the provider is contractually obligated to only process personal data according to our instructions, and conduct or intend to conduct diligence to ensure that they have sufficient technical and organizational security measures in place.
We are also subject to evolving European privacy laws on electronic marketing and cookies. The EU is in the process of replacing the e-Privacy Directive (2002/58/EC) with a new set of rules taking the form of a regulation, which will be directly implemented in the laws of each European member state, without the need for further enactment. While the e-Privacy Regulation was originally intended to be adopted on May 25, 2018 (alongside the GDPR), it is still going through the European legislative process. Draft regulations were rejected by the Permanent Representatives Committee of the Council of EU on November 22, 2019; it is not clear when, or even if, new regulations will be adopted. We are also subject to current and evolving privacy laws in other foreign countries, such as Canada.
Compliance with U.S. and international data protection laws and regulations requires that we take on more onerous obligations in our contracts, restrict our ability to collect, use and disclose data, and, in some cases, impacts our ability to operate in certain jurisdictions. Failure to comply with these laws and regulations could result in government enforcement actions (which could include civil, criminal and administrative penalties), private litigation, and/or adverse publicity and could negatively affect our operating results and business. Moreover, clinical trial subjects, employees and other individuals about whom we or our potential collaborators obtain personal information, as well as the providers who share this information with us, may limit our ability to collect, use and disclose the information. Claims that we have violated individuals’ privacy rights, failed to comply with data protection laws, or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
Our ability to obtain services, reimbursement or funding from the federal government may be impacted by possible reductions in federal spending and services, and any inability on our part to effectively adapt to such changes could substantially affect our business, prospects, operating results and financial condition.
Under the Budget Control Act of 2011, the failure of Congress to enact deficit reduction measures of at least $1.2 trillion for the years 2013 through 2021 triggered automatic cuts to most federal programs. These cuts included aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. Certain of these automatic cuts have been implemented resulting in reductions in Medicare payments to physicians, hospitals, and other healthcare providers, among other things. Under federal legislation, including the Bipartisan Budget Act of 2018, these reductions, while temporarily altered due to the COVID-19 pandemic, have resumed as of July 1, 2022 and will stay in effect through 2032 unless additional Congressional action is taken. The American Taxpayer Relief Act of 2012, among other things, reduced Medicare payments to several providers and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. Further, the American Rescue Plan Act of 2021 increased the budget deficit such that additional sequestration was required under the Statutory Pay-As-You-Go Act of 2010, which led to a further payment reduction, up to 4%, that was to take effect in January 2022, although implementation of the reduction was delayed until 2025. These new laws may result in additional reductions in Medicare and other healthcare funding and otherwise affect the prices we may obtain for our approved products or any of our product candidates for which we may obtain regulatory approval, or the frequency with which our products or any future product is prescribed or used. It is uncertain how current and future reforms, including any new legislation enacted during the new administration, in these areas will influence the future of our business operations and financial condition.
Previous actions taken by Congress to reduce spending, disagreements in Congress over government funding levels, high levels of government debt, and the Medicare Trustees’ warnings about the programs’ sustainability as presently structured suggest that uninterrupted/continued growth in funding for relevant programs is not guaranteed. Amounts allocated to federal grants and contracts may be reduced or eliminated. These reductions may also impact the ability of relevant agencies to timely review and approve drug research and development, manufacturing, and marketing activities, which may delay our ability to develop, market and sell our approved products and any other products we may develop.
If we fail to comply with our obligations under the 340B Drug Pricing Program and other U.S. governmental pricing programs, we could be subject to legal consequences, including penalties, sanctions and fines, which could have a material adverse effect on our business, prospects, operating results and financial condition.
We participate in the 340B Drug Pricing Program, Medicaid Drug Rebate Program, and a number of other federal and state government programs in the U.S. Participation in some of these programs is required in order to obtain reimbursement of our drug products under Medicaid or Medicare Part B. These programs generally require that we provide discounts or pay rebates to certain payers when our products are dispensed to beneficiaries of these programs. To support the calculation of these discounts and rebates, these programs may also impose periodic and special price and product data reporting requirements. Changes to our obligations under these government pricing programs occur frequently and program requirements are often ambiguous. Pricing calculations are complex, vary across programs and may be subject to evolving interpretations by legislative and regulatory bodies and the courts. We may be or become subject to penalties for noncompliance, including, but not limited to, civil monetary penalties, exposure under the federal false claims act, and termination from the government program, as a result of our failure to comply with obligations under these programs, including if we fail to provide timely, complete and accurate information to the government, to pay the correct rebates, or to offer the correct discounted pricing. In addition, potential policy changes by the new administration may introduce additional uncertainty for our business. These could include changes to the level of scrutiny applied by the Health Resources and Services Administration to enforce non-compliance with the 340B Drug Pricing Program, new price restrictions on products we sell to Medicaid, Medicare or other government purchasers, or other regulatory changes impacting reimbursement or competitive dynamics in multisource markets. Any such policy shifts could significantly impact our business and operations.
Increasingly, states are enacting legislation requiring manufacturers to report drug pricing information. However, states have not always clearly defined their reporting requirements, resulting in manufacturers failing to properly disclose the required pricing information. Complying with federal and state programs and future changes to these programs can be complex and cost-and resource-intensive, and could have a material adverse effect on our business, prospects, operating results and financial condition.
There is a substantial risk of product liability claims in our business. If we are unable to obtain sufficient insurance, a product liability claim against us could adversely affect our business, prospects, operating results and financial condition.
Our business exposes us to significant potential product liability risks that are inherent in the development, testing, manufacturing and marketing of human therapeutic products. Product liability claims could delay or prevent completion of our clinical development programs. Such claims might not be fully covered by product liability insurance. In addition, product liability claims could result in an FDA investigation of the safety and effectiveness of our approved products, our manufacturing processes and facilities or our marketing programs, and potentially a recall of our products or more serious enforcement action, limitations on the approved indications for which they may be used, or suspension or withdrawal of approvals. Regardless of the merits or eventual outcome, liability claims may also result in decreased demand for our products, injury to our reputation, costs to defend the related litigation, a diversion of management’s time and our resources, substantial monetary awards to trial participants or patients and a decline in our stock price. We currently have product liability insurance that we believe is appropriate for our stage of development, including the marketing and sale of our approved products. Any insurance we have or may obtain may not provide sufficient coverage against potential liabilities. Furthermore, clinical trial and product liability insurance is becoming increasingly expensive. As a result, we may be unable to obtain sufficient insurance at a reasonable cost to protect us against losses caused by product liability claims that could have a material adverse effect on our business.
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements or insider trading violations, which could significantly harm our business, prospects, operating results and financial condition.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with governmental regulations, including healthcare fraud and abuse and anti-kickback laws and regulations in the U.S. and abroad, or failure to report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. As discussed in the Risk Factor above captioned “If we or our collaborators, CMOs or service providers fail to comply with healthcare laws and regulations, or legal obligations related to privacy, data protection and information security, we or they could be subject to enforcement actions, which could negatively impact our ability to develop, market and sell our products and may harm our reputation,” these
laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of, including improper trading based upon, material information obtained in the course of clinical trials or other material non-public information, which could result in regulatory sanctions and serious harm to our reputation. We maintain a global compliance program and remain focused on its evolution and enhancement. Our program includes efforts such as risk assessment and monitoring, fostering a speak-up culture encouraging employees and third parties to raise good faith questions or concerns, and defined processes and systems for reviewing and remediating allegations and identified potential concerns. It is not always possible, however, to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, prospects, operating results and financial condition, including the imposition of significant fines or other sanctions.
If we do not comply with laws regulating the protection of the environment and health and human safety, our business, prospects, operating results and financial condition could be adversely affected.
Our research, development and manufacturing activities involve the use of hazardous materials, chemicals and various radioactive compounds. We maintain quantities of various flammable and toxic chemicals in our facilities in Cambridge and Norton that are required for our research, development and manufacturing activities. We are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. We believe our procedures for storing, handling and disposing these materials in our Cambridge and Norton facilities comply with the relevant guidelines of the City of Cambridge, the town of Norton, the Commonwealth of Massachusetts and the Occupational Safety and Health Administration of the U.S. Department of Labor. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards mandated by applicable regulations, the risk of accidental contamination or injury from these materials cannot be eliminated. If an accident occurs, we could be held liable for resulting damages, which could be substantial. We are also subject to numerous environmental, health and workplace safety laws and regulations, including those governing laboratory procedures, exposure to blood-borne pathogens and the handling of biohazardous materials.
Although we maintain workers’ compensation insurance to cover us for costs and expenses we may incur due to injuries to our employees resulting from the use of these materials, this insurance may not provide adequate coverage against potential liabilities. We do not maintain insurance for environmental liability or toxic tort claims that may be asserted against us in connection with our storage or disposal of biological, hazardous or radioactive materials. Additional federal, state and local laws and regulations affecting our operations may be adopted in the future. We may incur substantial costs to comply with, and substantial fines or penalties if we violate, any of these laws or regulations.
Risks Related to Patents, Licenses and Trade Secrets
If we are not able to obtain and enforce patent protection for our discoveries, our ability to develop and commercialize our product candidates will be harmed.
Our success depends, in part, on our ability to protect proprietary compositions, methods and technologies that we develop under the patent and other intellectual property laws of the U.S. and other countries, so that we can prevent others from unlawfully using our inventions and proprietary information. However, we may not hold proprietary rights to some patents required for us to manufacture and commercialize our current and future products. Because certain U.S. patent applications are confidential until the patents issue, such as applications filed prior to November 29, 2000, or applications filed after such date that will not be filed in foreign countries, third parties may have filed patent applications for subject matter covered by our pending patent applications without our being aware of those applications, and our patent applications may not have priority over those applications. For this and other reasons, we may be unable to secure desired patent rights, thereby losing desired exclusivity. Further, we or our collaborators may be required to obtain licenses under third-party patents to market one or more of our or our collaborator’s approved products, or further develop and commercialize future products, or continue to develop product candidates in our pipeline being developed by us or our collaborators. If licenses are not available to us or not available on reasonable terms or at all, we or our licensees may not be able to market the affected products or conduct the desired activities.
Our strategy depends on our ability to rapidly identify and seek patent protection for our discoveries. In addition, we may rely on third-party collaborators to file patent applications relating to proprietary technology that we develop jointly as part of collaborations. The process of obtaining patent protection is expensive and time-consuming. If we or our collaborators fail to file and prosecute all necessary and desirable patent applications at a reasonable cost and in a timely manner, our business may be adversely affected. Despite our efforts and the efforts of our collaborators to protect our proprietary rights, unauthorized parties may be able to obtain and use information that we regard as proprietary. While issued patents are presumed valid, this does not guarantee that the patent will survive a validity challenge or be held enforceable. Any patents we have obtained, or obtain in the future, may be challenged, invalidated, adjudged unenforceable or circumvented by parties attempting to design
around our intellectual property. Moreover, third parties or the United States Patent and Trademark Office, or USPTO, may commence interference proceedings involving our patents or patent applications. Any challenge to, finding of unenforceability or invalidation or circumvention of, our patents or patent applications, would be costly, would require significant time and attention of our management, could reduce or eliminate milestone and/or royalty payments to us from third party licensors and could have a material adverse effect on our business.
Our pending patent applications may not result in issued patents. The patent position of pharmaceutical or biotechnology companies, including ours, is generally uncertain and involves complex legal and factual considerations. The standards that the USPTO and its foreign counterparts use to grant patents are not always applied predictably or uniformly and can change. Similarly, the ultimate degree of protection that will be afforded to biotechnology inventions, including ours, in the U.S. and foreign countries, remains uncertain and is dependent upon the scope of the protection decided upon by patent offices, courts and lawmakers. Moreover, there are periodic discussions in the U.S. Congress and in international jurisdictions about modifying various aspects of patent law. For example, the America Invents Act, or AIA, included a number of changes to the patent laws of the U.S. If any of the enacted changes do not provide adequate protection for discoveries, including our ability to pursue infringers of our patents for substantial damages, our business could be adversely affected. One major provision of the AIA, which took effect in March 2013, changed U.S. patent practice from a first-to-invent to a first-to-file system. If we fail to file an invention before a competitor files on the same invention, we no longer have the ability to provide proof that we were in possession of the invention prior to the competitor’s filing date, and thus would not be able to obtain patent protection for our invention. There is also no uniform, worldwide policy regarding the subject matter and scope of claims granted or allowable in pharmaceutical or biotechnology patents.
Accordingly, we do not know the degree of future protection for our proprietary rights or the breadth of claims that will be allowed in any patents issued to us or to others. We also rely to a certain extent on trade secrets, know-how and technology, which are not protected by patents, to maintain our competitive position. If any trade secret, know-how or other technology not protected by a patent were to be disclosed to or independently developed by a competitor, our business, prospects, operating results and financial condition could be materially adversely affected.
Failure to obtain and maintain broad patent scope and all available regulatory exclusivities and to maximize patent term restoration or extension on patents covering our products and product candidates may lead to loss of exclusivity and generic entry resulting in a loss of market share and/or revenue.
We license patent rights from third-party owners. If such owners do not properly or successfully obtain, maintain or enforce the patents underlying such licenses, our competitive position and business, prospects, operating results and financial condition may be harmed.
We are a party to a number of licenses that give us rights to third-party intellectual property that is necessary or useful for our business. In particular, we have obtained licenses from, among others, Ionis, Arbutus, and Dicerna. We may also enter into additional licenses to third-party intellectual property in the future.
Our success will depend in part on the ability of our licensors to obtain, maintain and enforce patent protection for our licensed intellectual property, in particular, those patents to which we have secured exclusive rights. Our licensors may not successfully prosecute the patent applications we have licensed. Even if patents issue in respect of these patent applications, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents, or may pursue such litigation less aggressively than we would. Without protection for the intellectual property we license, other companies might be able to offer substantially identical products for sale, which could adversely affect our competitive business position and harm our business, prospects, operating results and financial condition. In addition, we sublicense our rights under various third-party licenses to our collaborators. Any impairment of these sublicensed rights could result in reduced revenues under our collaboration agreements or result in termination of an agreement by one or more of our collaborators.
Other companies or organizations may challenge our patent rights or may assert patent rights that prevent us from developing and commercializing our products.
RNAi is a relatively new and growing scientific field, the commercial exploitation of which has resulted in many different patents and patent applications from organizations and individuals seeking to obtain patent protection in the field. We have obtained grants and issuances of RNAi patents and have licensed many of these patents from third parties on an exclusive basis. The issued patents and pending patent applications in the U.S. and in key markets around the world that we own or license claim many different methods, compositions and processes relating to the discovery, development, manufacture and commercialization of RNAi therapeutics.
Specifically, we have a portfolio of patents, patent applications and other intellectual property covering, among other things: fundamental aspects of the structure and uses of siRNAs, including their use as therapeutics, and RNAi-related mechanisms; chemical modifications to siRNAs that improve their suitability for therapeutic and other uses; siRNAs directed to specific targets as treatments for particular diseases; delivery technologies, such as in the fields of carbohydrate conjugates and cationic liposomes; and all aspects of our specific development candidates.
As the field of RNAi therapeutics is maturing, patent applications are being fully processed by national patent offices around the world. There is uncertainty about which patents will issue, and, if they do, as to when, to whom, and with what claims. It is likely that there will be significant litigation and other proceedings, such as interference, re-examination and opposition proceedings, as well as pre- and post-grant review proceedings in various patent offices relating to patent rights in the RNAi field. In addition, third parties may challenge the validity of our patents. We expect that challenges will be raised relating to patents and patent applications in our portfolio. In many cases, the possibility of appeal exists for either us or our opponents, and it may be years before final, unappealable rulings are made with respect to these patents in certain jurisdictions. The timing and outcome of these and other proceedings is uncertain and may adversely affect our business, prospects, operating results and financial condition if we are not successful in defending the patentability and scope of our pending and issued patent claims. Even if our rights are not directly challenged, disputes could lead to the weakening of our intellectual property rights. Our defense against any attempt by third parties to circumvent or invalidate our intellectual property rights could be costly to us, could require significant time and attention of our management and could have a material adverse effect on our business, prospects, operating results and financial condition and on our ability to successfully compete in the field of RNAi.
There are many issued and pending patents that claim aspects of oligonucleotide chemistry and modifications that we may need for our siRNA products marketed by us or our licensees, our late-stage therapeutic candidates being developed by us or our collaborators, including zilebesiran and fitusiran, as well as our other pipeline products. There are also many issued patents that claim targeting genes or portions of genes that may be relevant for siRNA drugs we wish to develop. In addition, there may be issued and pending patent applications that may be asserted against us in a court proceeding or otherwise based upon the asserting party’s belief that we may need such patents for our siRNA therapeutic candidates or marketed products, or to further develop and commercialize future products, or to continue to develop candidates in our pipeline that are being developed by us or our collaborators. Thus, it is possible that one or more organizations will hold patent rights to which we may need a license, or hold patent rights which could be asserted against us. If those organizations refuse to grant us a license to such patent rights on reasonable terms or at all and/or a court rules that we need such patent rights that have been asserted against us, we may be unable to market our products, including ONPATTRO, AMVUTTRA, GIVLAARI or OXLUMO, or to perform research and development or other activities covered by such patents. For example, on December 12, 2024, the Board of Regents of the University of Texas System, or the University of Texas, filed a lawsuit in the United States District Court for the Western District of Texas, alleging that we infringe one of the University of Texas’ patents by making, using and commercializing ONPATTRO in the U.S.
If we become involved in intellectual property litigation or other proceedings related to a determination of rights, we could incur substantial costs and expenses, and in the case of such litigation or proceedings against us, substantial liability for damages or be required to stop our product development and commercialization efforts.
Third parties may sue us for infringing their patent rights. For example, on December 12, 2024, the Board of Regents of the University of Texas System, or the University of Texas, filed a lawsuit in the United States District Court for the Western District of Texas, alleging that we infringe one of the University of Texas’ patents by making, using and commercializing ONPATTRO in the U.S. A third party may also claim that we have improperly obtained or used its confidential or proprietary information.
Furthermore, third parties may challenge the inventorship of our patents or licensed patents. For example, in March 2011, The University of Utah, or Utah, filed a complaint against us, Max Planck Gesellschaft Zur Foerderung Der Wissenschaften e.V. and Max Planck Innovation, together, Max Planck, Whitehead, MIT and the University of Massachusetts, claiming that a professor of Utah was the sole inventor, or in the alternative, a joint inventor of certain of our in-licensed patents. Utah was seeking correction of inventorship of the Tuschl patents, unspecified damages and other relief. After several years of court proceedings and discovery, the court granted our motions for summary judgment and dismissed Utah’s state law damages claims. During the pendency of this litigation, we incurred significant costs, and in each case, the litigation diverted the attention of our management and other resources that would otherwise have been engaged in other activities.
On July 12, 2024, Acuitas Therapeutics Inc., or Acuitas, filed a declaratory judgment action against us in the U.S. District Court for the District of Delaware, seeking a judgment adding certain Acuitas employees as co-inventors on the patents we have asserted against Pfizer/BioNTech and Moderna in our lawsuits described below. On September 19, 2024, we filed a motion to dismiss arguing that Acuitas did not have standing to sue and failed to state a claim upon which relief could be granted. The court has not yet ruled on our motion.
We may need to resort to litigation to enforce a patent issued or licensed to us or to determine the scope and validity of proprietary rights of others or protect our proprietary information and trade secrets. In March 2022, we announced that we separately filed suit in United States District Court for the District of Delaware against Pfizer and Moderna seeking damages for infringement of U.S. Patent No. 11,246,933, or the ’933 patent in the parties’ manufacture and sale of their messenger RNA, or mRNA, COVID-19 vaccines. Pfizer joined BioNTech SE, or BioNTech, to the suit and filed counterclaims. In July 2022, we filed an additional lawsuit in United States District Court for the District of Delaware against each of Pfizer/BioNTech and Moderna seeking damages for infringing U.S. Patent No. 11,382,979, or the ’979 patent. The court combined the two patents in a single suit for each of Pfizer/BioNTech, or the 2022 Lawsuit, and Moderna with trial dates set for each in November 2024. On May 26, 2023, we filed additional lawsuits against Pfizer and Moderna in Delaware seeking damages for infringing U.S. Patent
No. 11,590,229 in the United States District Court for the District of Delaware. In addition to this patent, we added U.S. Patent Nos. 11,633,479, or the ‘479 patent, and 11,633,480 in the more recently filed suits against both Pfizer and Moderna and of U.S. Patent No. 11,612,657 against Pfizer only. On August 9, 2023, a Markman hearing was held in the U.S. District Court for the District of Delaware to consider the meaning of three disputed terms as used in the ’933 and ’979 patents, and on August 21, 2023, the court issued an order construing two of the three terms, and deferred a ruling on the third term pending an evidentiary hearing, which was held on January 4, 2024 with the final ruling deferred pending the outcome of an additional hearing, which was held on July 12, 2024. Following the August 21, 2023 order, we and Moderna jointly agreed to final judgment of non-infringement of two of our patents, and that judgment was entered by the court on August 30, 2023, and on September 7, 2023, we appealed the claim construction ruling to the Court of Appeals for the Federal Circuit in the 2022 lawsuit against Moderna. The claim construction ruling initially did not affect one of the patents in the lawsuit filed against Moderna on May 26, 2023, but following a Markman hearing held on August 15, 2024, the court entered a ruling on September 10, 2024, in the same manner as in the other earlier case and on October 2, 2024, we and Moderna jointly agreed to final judgment of non-infringement of the ‘479 patent. On October 16, 2024, Moderna filed a motion seeking recovery of fees incurred by them from the time we agreed to a judgment of non-infringement in the first case until the time we agreed to a judgment of non-infringement in the second case, which period runs from approximately September 2023 to October 2024. We opposed the motion in a reply on November 6, 2024. The court has yet to rule on the motion.
The two separate suits against Pfizer/BioNTech are ongoing and in September 2023, we and Pfizer/BioNTech agreed to consolidate the 2022 and 2023 lawsuits into one case, with a trial date set for July 7, 2025. The aforementioned patents relate to our biodegradable cationic lipids that are foundational to the success of the mRNA COVID-19 vaccines.
In protecting our intellectual patent rights through litigation or other means, a third party may claim that we have improperly asserted our rights against them. For example, in August 2017, Dicerna successfully added counterclaims against us in the above-referenced trade secret lawsuit alleging that our lawsuit represented abuse of process and claiming tortious interference with its business. In addition, in August 2017, Dicerna filed a lawsuit against us in the United States District Court of Massachusetts alleging attempted monopolization by us under the Sherman Antitrust Act. As noted above, in April 2018, we and Dicerna settled all claims in the litigation between us.
In addition, in connection with certain license and collaboration agreements, we have agreed to indemnify certain third parties for certain costs incurred in connection with litigation relating to intellectual property rights or the subject matter of the agreements. The cost to us of any litigation or other proceeding relating to such intellectual property rights, even if resolved in our favor, could be substantial, and litigation would divert our management’s efforts. Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. Uncertainties resulting from the initiation and continuation of any litigation or legal proceeding could delay our research, development and commercialization efforts and limit our ability to continue our operations.
If any parties successfully claim that our creation or use of proprietary technologies infringes upon or otherwise violates their intellectual property rights, we might be forced to pay damages, potentially including treble damages, if we are found to have willfully infringed on such parties’ patent rights. In addition to any damages we might have to pay, a court could issue an injunction requiring us to stop the infringing activity or obtain a license from the claimant. Any license required under any patent may not be made available on commercially reasonable terms, or at all. In addition, such licenses are in many instances non-exclusive and, therefore, our competitors may have access to the same technology that is licensed to us. If we fail to obtain a required license and are unable to design around a patent, we may be unable to effectively market some of our technology and products, which could limit our ability to generate revenues or maintain profitability and possibly prevent us from generating revenue sufficient to sustain our operations. Moreover, we expect that a number of our collaboration agreements will provide that royalties payable to us for licenses to our intellectual property may be offset by amounts paid by our collaborators to third parties who have competing or superior intellectual property positions in the relevant fields, which could result in significant reductions in our revenues from products developed through collaborations.
If we fail to comply with our obligations under any licenses or related agreements, we may be required to pay damages and could lose license or other rights that are necessary for developing, commercializing and protecting our RNAi technology, as well as our products and product candidates.
Our current licenses impose, and any future licenses we enter into are likely to impose, various development, commercialization, funding, milestone, royalty, diligence, sublicensing, insurance, patent prosecution and enforcement, and other obligations on us. If we breach any of these obligations, or use the intellectual property licensed to us in an unauthorized manner, we may be required to pay damages and the licensor may have the right to terminate the license or render the license non-exclusive, which could result in us being unable to develop, manufacture, market and sell products that are covered by the licensed technology or enable a competitor to gain access to the licensed technology. Moreover, we could incur significant costs and/or disruption to our business and distraction of our management defending against any breach of such licenses alleged by the licensor. For example, in June 2018, Ionis sent us a notice claiming that it was owed technology access fees, or TAFs, based on rights granted and amounts paid to us in connection with the January 2018 amendment of our collaboration agreement with Sanofi and the related Exclusive TTR License and AT3 License Terms. Following arbitration proceedings, the panel ruled in
favor of each party on certain TAF associated claims and awarded Ionis compensation of $41.2 million for a TAF on certain rights we received in the Sanofi restructuring.
Moreover, our licensors may own or control intellectual property that has not been licensed to us and, as a result, we may be subject to claims, regardless of their merit, that we are infringing or otherwise violating the licensor’s rights. In addition, while we cannot currently determine the amount of the royalty obligations we will be required to pay on sales of each of our approved products or future products, if any, the amounts may be significant. The amount of our future royalty obligations will depend on the technology and intellectual property we use in such products. Therefore, even if we successfully develop and commercialize products, we may be unable to maintain profitability.
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information.
In order to protect our proprietary technology and processes, we rely in part on confidentiality agreements with our collaborators, potential collaborators, employees, consultants, scientific advisors, CMOs, outside scientific collaborators and sponsored researchers, and other advisors. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, other third parties may independently discover trade secrets and proprietary information, and in such cases we could not assert any trade secret rights against such party. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our business, prospects, operating results and financial condition.
Risks Related to Competition
The pharmaceutical market is intensely competitive. If we or our collaborators are unable to compete effectively with existing drugs, new treatment methods and new technologies, we or our collaborators may be unable to commercialize successfully any drugs that we or our collaborators develop.
The pharmaceutical market is intensely competitive and rapidly changing. Many large pharmaceutical and biotechnology companies, academic institutions, governmental agencies and other public and private research organizations are pursuing the development of novel drugs for the same diseases that we are targeting or expect to target. Many of our competitors have:
•substantially greater financial, technical and human resources than we have;
•more extensive experience in preclinical testing, conducting clinical trials, obtaining regulatory approvals, and in manufacturing, marketing and selling drug products;
•product candidates that are based on previously tested or accepted technologies;
•multiple products that have been approved or are in late stages of development; and
•collaborative arrangements in our target markets with leading companies and research institutions.
We face intense competition from drugs that have already been approved and accepted by the medical community for the treatment of the conditions for which we have developed products. In addition, there are a number of drugs currently under development and that may become commercially available in the future, for the treatment of conditions for which we may try to develop product candidates. These drugs may be more effective, safer, less expensive, have more convenient administration or be marketed and sold more effectively, than any products we develop and commercialize.
For example, assuming regulatory approval, vutrisiran, our RNAi therapeutic in development for treatment of ATTR amyloidosis with cardiomyopathy, would compete with VYNDAQEL/VYNDAMAX (tafamidis), which is marketed by Pfizer, and ATTRUBY (acoramidis), which is marketed by BridgeBio, both of which are currently approved to treat ATTR amyloidosis with cardiomyopathy. We are also aware of other product candidates in clinical development for the treatment of ATTR amyloidosis with cardiomyopathy, including WAINUA (eplontersen), which is being developed by Ionis and AstraZeneca plc, or AstraZeneca, and is in Phase 3 clinical development; nexiguran ziclumeran (formerly NTLA-2001), which is being developed by Intellia Therapeutics, Inc. and Regeneron and is in Phase 3 clinical development; ALN-2220 (formerly NI006), which is being developed by Neurimmune AG and Alexion Pharmaceuticals, Inc. (a subsidiary of AstraZeneca) and is in Phase 3 clinical development; coramitug (NNC-6019-0001), which is being developed by Novo Nordisk and is in Phase 2 clinical development; and AT0-2, which is being developed by Attralus, Inc. and is in Phase 2 clinical development. We expect to face competition from any of these and potentially other additional new drugs that enter the market to treat patients with ATTR amyloidosis with cardiomyopathy.
ONPATTRO and AMVUTTRA are approved in certain jurisdictions for the treatment of certain patients with hATTR amyloidosis with polyneuropathy. We are aware of other approved products used to treat this disease, including WAINUA (eplontersen), VYNDAQEL/VYNDAMAX (tafamidis), and TEGSEDI (inotersen), which is marketed by Ionis. There are also product candidates in various stages of clinical development for the treatment of hATTR amyloidosis patients with polyneuropathy, including ATO-02, which is in Phase 2 clinical development, and nexiguran ziclumeran (formerly NTLA-2001), which is in Phase 1 clinical development. While we believe that ONPATTRO and AMVUTTRA have and will
continue to have a competitive product profile for the treatment of patients with hATTR amyloidosis with polyneuropathy, it is possible that ONPATTRO and/or AMVUTTRA may not compete favorably with these products and product candidates, or others, and, as a result, may not achieve commercial success.
We are aware of approved products and product candidates in various stages of clinical development for the treatment of PH1 that would compete with OXLUMO, our RNAi therapeutic approved in the U.S. and EU for the treatment of this disease, including Novo Nordisk’s RIVFLOZA (nedosiran), which was approved for the treatment of PH1 in September 2023 and launched in the U.S. in early 2024. RIVFLOZA is a once-monthly subcutaneous RNAi therapy that was developed by Dicerna. In April 2020, we and Dicerna granted each other a non-exclusive cross-license to our respective intellectual property related to lumasiran and Dicerna’s nedosiran. In addition, several companies have investigational drugs in clinical development for the treatment of PH1, including Biocodex, Inc. in collaboration with M8 Pharmaceuticals, Inc., and YolTech Therapeutics.
If we or our collaborators continue to successfully develop product candidates, and obtain approval for them, we and our collaborators will face competition based on many different factors, including:
•the safety and effectiveness of our or our collaborators’ products relative to alternative therapies, if any;
•the ease with which our or our collaborators’ products can be administered and the extent to which patients accept relatively new routes of administration;
•the timing and scope of regulatory approvals for these products;
•the availability and cost of manufacturing, marketing and sales capabilities;
•the price of our or our collaborators’ products relative to alternative approved therapies;
•reimbursement coverage; and
•patent position.
Our competitors may develop or commercialize products with significant advantages over any products we or our collaborators develop based on any of the factors listed above or on other factors. In addition, our competitors may develop collaborations with or receive funding from larger pharmaceutical or biotechnology companies, providing them with an advantage over us and our collaborators. Our competitors may therefore be more successful in commercializing their products than we or our collaborators are, which could adversely affect our competitive position and business, prospects, operating results and financial condition. Competitive products may make any products we or our collaborators develop obsolete or noncompetitive before we can recover the expenses of developing and commercializing our product candidates. Such competitors could also recruit our employees, which could negatively impact our level of expertise and our ability to execute on our business plan. Furthermore, we and our collaborators also face competition from existing and new treatment methods that reduce or eliminate the need for drugs, such as the use of advanced medical devices. The development of new medical devices or other treatment methods for the diseases we and our collaborators are targeting could make our or our collaborators’ product candidates noncompetitive, obsolete or uneconomical.
We and our collaborators face competition from other companies that are working to develop novel drugs and technology platforms using technology similar to ours, as well as from companies utilizing emerging technologies. If these companies develop drugs more rapidly than we or our collaborators do or their technologies, including delivery technologies, are more effective, our and our collaborators’ ability to successfully commercialize our products may be adversely affected.
In addition to the competition we face from competing drugs in general, we and our collaborators also face competition from other companies working to develop novel drugs using technology that competes more directly with our own. We are aware of several other companies that are working to develop RNAi therapeutic products. Some of these companies are seeking, as we are, to develop chemically synthesized siRNAs as drugs. Others are following a gene therapy approach, with the goal of treating patients with synthetic, exogenously-introduced genes designed to produce siRNA-like molecules within cells. Companies working on chemically synthesized siRNAs include, but are not limited to, Arrowhead and its collaborators, Takeda Pharmaceutical Company Ltd., Janssen Pharmaceutics, Inc., GlaxoSmithKline plc, and Amgen Inc.; Quark Pharmaceuticals, Inc.; Roche; Silence Therapeutics plc and its collaborators, AstraZeneca, Jiangsu Hansoh Pharmaceuticals Group Co., Ltd., and Mallinckrodt plc; Arbutus; Sylentis; Novo Nordisk and its collaborators, Aro Biotherapeutics Company, Boehringer Ingelheim and Eli Lilly and Company; and our collaborators Regeneron, Sanofi and Vir. In addition, we granted licenses or options for licenses to Ionis, Benitec Biopharma Ltd., Arrowhead, Arbutus, Quark, Sylentis and other companies under which these companies may independently develop RNAi therapeutics against a limited number of targets. Any one of these companies may develop its RNAi technology more rapidly and more effectively than we do. In addition, as a result of agreements that we have entered into, Takeda has obtained a non-exclusive license, and Arrowhead, as the assignee of Novartis, has obtained specific exclusive licenses for 30 gene targets, that include access to certain aspects of our technology.
We and our collaborators also compete with companies working to develop antisense-based drugs. Similar to RNAi therapeutics, antisense drugs target mRNAs in order to suppress the activity of specific genes. Akcea Therapeutics, Inc., a wholly owned subsidiary of Ionis, has received marketing approval for an antisense drug, inotersen for the treatment of adult
hATTR amyloidosis patients with stage 1 or stage 2 polyneuropathy. Several antisense drugs developed by Ionis have been approved and are currently marketed, including WAINUA (eplontersen), and Ionis has multiple antisense product candidates in clinical trials. Ionis is also developing antisense drugs using ligand-conjugated GalNAc technology licensed from us, and these drugs have been shown to have increased potency at lower doses in clinical and preclinical studies, compared with antisense drugs that do not use such licensed GalNAc technology. The development of antisense drugs and antisense technology may become the preferred technology for drugs that target mRNAs to silence specific genes.
In addition to competition with respect to RNAi and with respect to specific products, we face substantial competition to discover and develop safe and effective means to deliver siRNAs to the relevant cell and tissue types. If our competitors develop safe and effective means to deliver siRNAs to the relevant cell and tissue types, our ability to successfully commercialize a competitive product would be adversely affected. In addition, third parties are expending substantial resources to discover and develop a safe and effective means of delivering siRNAs into the relevant cell and tissue types, including both private companies and academic laboratories. Some of our competitors have substantially greater resources than we do, and if our competitors negotiate exclusive access to delivery solutions developed by third parties, we may be unable to successfully commercialize our product candidates.
Risks Related to Our Common Stock
Our stock price has been and may in the future be volatile, and an investment in our common stock could suffer a decline in value.
Our stock price has been and may in the future be volatile. From January 1, 2024 to December 31, 2024, our common stock traded between $141.98 and $304.39 per share. The stock market in general and the market for biotechnology companies in particular have experienced extreme price and volume volatility that has often been unrelated to the operating performance of particular companies. The market price of our common stock in the future could be significantly and adversely affected by many factors, including:
•the information contained in our quarterly earnings releases and other public announcements, including updates regarding our or our collaborators’ commercialized products or product candidates, our net product and collaboration revenues and operating expenses for completed periods and financial guidance regarding future periods;
•the success of existing or new competitive products or technologies;
•regulatory actions with respect to our or our collaborators’ products or product candidates;
•announcements by us or our competitors of significant acquisitions, collaborations, joint ventures, collaborations or capital commitments;
•the timing and results of clinical trials of our or our collaborators’ other product candidates;
•commencement or termination of collaborations for our development programs;
•failure or discontinuation of any of our or our collaborators’ development programs;
•results of clinical trials of our competitors’ product candidates;
•regulatory or legal developments in the U.S. and other countries;
•developments or disputes concerning patent applications, issued patents or other proprietary rights;
•the recruitment or departure of key personnel;
•the level of expenses related to any of our product candidates or clinical development programs;
•the results of our or our collaborators’ efforts to develop additional product candidates or products;
•actual or anticipated changes in financial results or development timelines;
•announcement or expectation of additional financing efforts;
•sales of our common stock by us, our insiders or other stockholders;
•variations in our financial results or those of companies that are perceived to be similar to us;
•changes in estimates or recommendations by any of the securities analysts that cover us;
•changes in the structure of healthcare payment systems;
•market conditions in the pharmaceutical and biotechnology sectors;
•general economic, industry and market conditions; and
•the other factors described in this “Risk Factors” section.
In the past, securities class action litigation has often been brought against companies following declines in the market price of their securities. This risk is especially relevant for biopharmaceutical companies, which have experienced significant stock price volatility in recent years. We may be the target of securities litigation that could result in substantial costs and divert our management’s attention and resources, which could cause serious harm to our business, prospects, operating results and financial condition. We maintain liability insurance; however, if any costs or expenses associated with litigation exceed our insurance coverage, we may be forced to bear some or all of these costs and expenses directly, which could be substantial. In addition, we have obligations to indemnify third parties, including our officers and directors, in connection with certain litigation, and those obligations may not be covered by insurance.
Sales of a substantial number of shares of our common stock, including by us, our officers or directors, or our significant stockholders, into the public market could cause the price of our common stock to decline.
A small number of our stockholders beneficially own a substantial amount of our common stock. As of December 31, 2024, our eight largest stockholders beneficially owned in excess of 50% of our outstanding shares of common stock. If we, our officers or directors, or our significant stockholders sell substantial amounts of our common stock in the public market, or there is a perception that such sales may occur, the market price of our common stock could be adversely affected. Sales of common stock by our significant stockholders might make it more difficult for us to raise funds by selling equity or equity-related securities in the future at a time and price that we deem appropriate.
Anti-takeover provisions in our governing documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management or members of our board of directors.
Provisions in our certificate of incorporation and our bylaws may delay or prevent an acquisition of us or a change in the current members of our management or the members of our board of directors. Among other things, these provisions:
•establish a classified board of directors such that all members of our board of directors are not elected at one time;
•establish a prohibition on actions by our stockholders by written consent;
•authorize our board of directors to issue preferred stock without stockholder approval, which could be used to institute a “poison pill” that would work to dilute the stock ownership of a potential hostile acquirer, effectively preventing acquisitions that have not been approved by our board of directors;
•allow the authorized number of our directors to be changed only by resolution of our board of directors.
•limit who may call a special meeting of stockholders;
•require the approval of the holders of at least 75% of the votes that all our stockholders would be entitled to cast to amend or repeal certain provisions of our charter or bylaws;
•limit the manner in which stockholders can remove directors from our board of directors; and
•establish advance notice requirements for election to our board of directors and for proposing matters that can be acted upon at stockholder meetings.
In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits a person who owns in excess of 15% of our outstanding voting stock from merging or combining with us for a period of three years after the date of the transaction in which the person acquired in excess of 15% of our outstanding voting stock, unless the merger or combination is approved in a prescribed manner. These provisions would apply even if the proposed merger or acquisition could be considered beneficial by some stockholders.
We expect that results from our and our collaborators’ clinical development activities and the clinical development activities of our competitors will continue to be released periodically and may result in significant volatility in the price of our common stock.
Any new information regarding our and our collaborators’ products and product candidates or competitive products or potentially competitive product candidates can substantially affect investors’ perceptions regarding our future prospects. We, our collaborators, and our competitors periodically provide updates regarding drug development programs, typically through press releases, conference calls and presentations at medical conferences. These periodic updates often include interim or final results from clinical trials conducted by us or our competitors and/or information about our or our competitors’ expectations regarding regulatory filings and submissions as well as future clinical development of our products or product candidates, competitive products or potentially competitive product candidates. The timing of the release of information by us regarding our drug development programs is often beyond our control and is influenced by the timing of receipt of data from our clinical trials and by the general preference among pharmaceutical companies to disclose clinical data during medical conferences. In addition, the information disclosed about our clinical trials, or our competitors’ clinical trials, may be based on interim rather than final data that may involve interpretation difficulties and may in any event not accurately predict final results. The release of such information may result in volatility in the price of our common stock. For example, in late 2021 our stock price was
negatively impacted following BridgeBio’s public disclosure of the results of Part A of the Phase 3 clinical trial of acoramidis for the treatment of ATTR amyloidosis with cardiomyopathy.
Risks Related to Our Convertible Notes
We may not have sufficient cash flow from our business to pay our indebtedness.
As of December 31, 2024, we had $1.04 billion in total aggregate principal amount of Notes issued and outstanding. The interest rate for the Notes is fixed at 1.00% per annum and is payable semi-annually in arrears on March 15 and September 15 of each year, beginning on March 15, 2023. Our ability to make scheduled payments of the principal of, to pay interest on or to refinance our indebtedness, including the Notes, or to make cash payments in connection with any conversions of Notes, depends on our future performance, which is subject to economic, financial, competitive and other factors beyond our control. Our business may not generate cash flow from operations in the future sufficient to service our debt and make necessary capital expenditures. If we are unable to generate such cash flow, we may be required to adopt one or more alternatives, such as selling assets, restructuring debt or obtaining additional debt financing or equity capital on terms that may be onerous or highly dilutive. Our ability to refinance any future indebtedness will depend on the capital markets and our financial condition at such time. We may not be able to engage in any of these activities or engage in these activities on desirable terms, which could result in a default on our debt obligations.
In addition, our indebtedness, combined with our other financial obligations and contractual commitments, could have other important consequences. For example, it could:
•make us more vulnerable to adverse changes in general U.S. and worldwide economic, industry and competitive conditions and adverse changes in government regulation;
•limit our flexibility in planning for, or reacting to, changes in our business and our industry;
•place us at a disadvantage compared to our competitors who have less debt;
•limit our ability to borrow additional amounts to fund acquisitions, for working capital and for other general corporate purposes; and
•make an acquisition of our company less attractive or more difficult.
Any of these factors could harm our business, prospects, operating results and financial condition. In addition, if we incur additional indebtedness, the risks related to our business and our ability to service or repay our indebtedness would increase.
We may not have the ability to raise the funds necessary to settle for cash conversions of the Notes or to repurchase the Notes for cash upon a fundamental change.
Holders of the Notes have the right to require us to repurchase their Notes upon the occurrence of a fundamental change (as defined in the indenture governing the Notes) at a repurchase price equal to 100% of the principal amount of the Notes to be repurchased, plus accrued and unpaid interest, if any. Upon conversion of the Notes, unless we elect to deliver solely shares of our common stock to settle such conversion (other than paying cash in lieu of delivering any fractional share), we will be required to make cash payments in respect of the Notes being converted. We may not have enough available cash or be able to obtain financing at the time we are required to make repurchases of Notes surrendered or Notes being converted. In addition, our ability to repurchase the Notes or to pay cash upon conversions of the Notes may be limited by law, by regulatory authority or by agreements governing our future indebtedness. Our failure to repurchase Notes at a time when the repurchase is required by the indenture governing such notes or to pay any cash payable on future conversions of the Notes as required by such indenture would constitute a default under such indenture. A default under the indenture governing the Notes or the fundamental change itself could also lead to a default under agreements governing our future indebtedness. If the repayment of the related indebtedness were to be accelerated after any applicable notice or grace periods, we may not have sufficient funds to repay the indebtedness and repurchase the Notes or make cash payments upon conversions.
The conditional conversion feature of the Notes, if triggered, may adversely affect our liquidity.
In the event the conditional conversion feature of the Notes is triggered, holders of the Notes will be entitled to convert the Notes at any time during specified periods at their option. If one or more holders elect to convert their Notes, unless we elect to satisfy our conversion obligation by delivering solely shares of our common stock (other than paying cash in lieu of delivering any fractional share), we would be required to settle a portion or all of our conversion obligation through the payment of cash, which could adversely affect our liquidity. In addition, even if holders do not elect to convert their Notes, we could be required under applicable accounting rules to reclassify all or a portion of the outstanding principal of the Notes as a current liability, rather than long-term liability, which would result in a material reduction of our net working capital.
Transactions relating to the Notes may affect the value of our common stock.
The conversion of some or all of the Notes would dilute the ownership interests of existing stockholders to the extent we satisfy our conversion obligation by delivering shares of our common stock upon any conversion of such Notes. The Notes may become in the future convertible at the option of their holders under certain circumstances. If holders of the Notes elect to
convert their notes, we may settle our conversion obligation by delivering to them a significant number of shares of our common stock, which would cause dilution to our existing stockholders.
In addition, in connection with the issuance of the Notes, we entered into the Capped Calls with certain financial institutions, or the Option Counterparties. The Capped Calls are generally expected to reduce potential dilution to our common stock upon any conversion or settlement of the Notes and/or offset any cash payments we are required to make in excess of the principal amount of converted Notes, with such reduction and/or offset subject to a cap.
In connection with establishing their initial hedges of the Capped Calls, the Option Counterparties or their respective affiliates entered into various derivative transactions with respect to our common stock and/or purchased shares of our common stock concurrently with or shortly after the pricing of the Notes.
From time to time, the Option Counterparties or their respective affiliates may modify their hedge positions by entering into or unwinding various derivative transactions with respect to our common stock and/or purchasing or selling our common stock or other securities of ours in secondary market transactions prior to the maturity of the Notes (and are likely to do so following any conversion of the Notes, any repurchase of the Notes by us on any fundamental change repurchase date, any redemption date, or any other date on which the Notes are retired by us, in each case, if we exercise our option to terminate the relevant portion of the Capped Calls). This activity could cause a decrease and/or increased volatility in the market price of our common stock.
We do not make any representation or prediction as to the direction or magnitude of any potential effect that the transactions described above may have on the price of the Notes or our common stock. In addition, we do not make any representation that the Option Counterparties will engage in these transactions or that these transactions, once commenced, will not be discontinued without notice.
We are subject to counterparty risk with respect to the Capped Calls.
The Option Counterparties are financial institutions, and are subject to the risk that any or all of them might default under the Capped Calls. Our exposure to the credit risk of the Option Counterparties will not be secured by any collateral. Past global economic conditions have resulted in the actual or perceived failure or financial difficulties of many financial institutions. If an Option Counterparty becomes subject to insolvency proceedings, we will become an unsecured creditor in those proceedings with a claim equal to our exposure at that time under the Capped Calls with such Option Counterparty. Our exposure will depend on many factors but, generally, an increase in our exposure will be correlated to an increase in the market price and in the volatility of our common stock. In addition, upon a default by an Option Counterparty, we may suffer adverse tax consequences and more dilution than we currently anticipate with respect to our common stock. We can provide no assurances as to the financial stability or viability of the Option Counterparties.
The accounting method for convertible debt securities that may be settled in cash, such as the Notes, could have a material effect on our reported financial results.
The accounting method for reflecting the Notes on our consolidated balance sheet, accruing interest expense for the Notes and reflecting the underlying shares of our common stock in our reported diluted earnings per share may adversely affect our reported earnings and financial condition.
The Notes are reflected as a liability on our consolidated balance sheets, with the initial carrying amount equal to the principal amount of the Notes, net of issuance costs. The issuance costs were treated as a debt discount for accounting purposes, which is being amortized into interest expense over the term of the Notes. As a result of this amortization, the interest expense that we expect to recognize for the Notes for accounting purposes will be greater than the cash interest payments we will pay on the Notes, which will result in lower reported net income or higher reported net loss, as the case may be.
In addition, the shares of common stock underlying the Notes are reflected in our diluted earnings per share using the “if-converted” method, in accordance with ASU 2020-06. Under this method, diluted earnings per share is generally calculated assuming that all the Notes were converted solely into shares of common stock at the beginning of the reporting period, unless the result would be anti-dilutive. The application of the if-converted method may reduce our reported diluted earnings per share to the extent we are profitable in the future, and accounting standards may change in the future in a manner that may adversely affect our diluted earnings per share.
Furthermore, if any of the conditions to the convertibility of the Notes is satisfied, then we may be required under applicable accounting standards to reclassify the liability carrying value of the Notes as a current, rather than a long-term, liability. This reclassification could be required even if no holders actually convert their Notes and could materially reduce our reported working capital.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not applicable.
ITEM 1C. CYBERSECURITY
Risk Management and Strategy
We have in place a cross-functional, enterprise-wide cybersecurity program that is integrated into our overall risk management process and strategy and has direct involvement from our senior management and board oversight. Our cybersecurity program is based on industry standard CIS critical security controls. The top risks facing our Company, including those related to cybersecurity, are included in our overall enterprise risk management program that is managed by a cross-functional group chaired by our compliance and internal audit functions.
To assess, identify, and manage material risks from cybersecurity threats to our information systems and the associated costs, our cybersecurity program prioritizes vulnerability management and risk reduction, detection and prevention. As part of this program, we conduct continuous monitoring for anomalous behavior using a third-party security operations center. In addition, we conduct an annual cybersecurity risk assessment in conjunction with our third-party consultant that specializes in information security, and we incorporate recommendations from the risk assessment into our cybersecurity strategy, as appropriate. This risk assessment process considers the nature of our business, requirements from our internal and external stakeholders, and industry trends and risks, including new and emerging risks. By continuously assessing the cybersecurity landscape, we develop targeted strategies that identify and address the risks most likely to impact our company. We also conduct at least one cybersecurity incident tabletop exercise each year to test and enhance our incident response plans.
Our cybersecurity program is designed to detect and prevent disruption to critical information systems, minimize the loss or manipulation of sensitive information, efficiently remediate and recover from cybersecurity incidents and ensure compliance with regulations and disclosure requirements. Pursuant to our processes, when a cybersecurity incident occurs, we convene a cross-functional incident response team whose membership is dictated by the severity of the incident but in all instances includes representatives from our information technology, legal and accounting departments. This cross-functional representation allows us to leverage diverse perspectives and expertise when addressing cybersecurity events and to analyze the potential financial, legal, operational, and reputational implications of an incident, thereby enabling us to make informed decisions and take appropriate actions. This incident response framework further enables us to quickly assess the severity of cybersecurity incidents and the materiality of incidents based on pre-defined criteria that considers both quantitative and qualitative factors to determine the appropriate response. Identified incidents are then escalated to the relevant management teams based on their severity, allowing for a swift determination of materiality and an effective mitigation process. If we determine that an incident is not material, we continue to monitor it for subsequent developments.
We also utilize third-party service providers as a normal part of our business operations. These third-party service providers may have access to our systems and/or sensitive information. To address cybersecurity risks arising from our third-party service providers, we assess and monitor risks relating to potential compromises of sensitive information at our third-party service providers and reevaluate these risks periodically. We categorize our third-party service providers by criticality, based on the criticality and sensitivity of our data each third-party service provider has access to, and, based on this, we employ a risk-based approach for review of the security measures implemented by our third-party service providers. In addition, we obtain periodic attestation reports related to data security and privacy from certain third-party service providers to further support compliance with industry-standard cybersecurity protocols.
Additionally, to minimize our enterprise-wide risk and exposure to material cybersecurity incidents, we conduct annual cybersecurity awareness training and education for our employees. By equipping our employees with the necessary knowledge and skills, we intend to cultivate a cybersecurity-conscious culture within our organization.
We maintain insurance to provide coverage for a portion of the losses and damages that may result from a physical attack, cybersecurity attack or a security breach. Such insurance is subject to several exclusions and may not cover the total loss or damage caused by an attack or a breach. Consequently, costs related to incidents may not be covered by insurance.
Our business operations and relationships with customers and suppliers are heavily reliant on technology, and any failure or disruption in our technological systems could have significant negative impacts on our business. For example, we collect, store and transmit sensitive information including intellectual property, proprietary business information, including highly sensitive clinical trial data, and personal information in connection with our business operations. The secure maintenance of this information is critical to our operations and business strategy. If this information was subject to a cybersecurity attack or unauthorized access or use, it could have a material adverse effect on our business and could expose us to potential legal consequences, liabilities, mitigation costs, and damage to our reputation. Managing cybersecurity incidents would also divert management’s attention and resources from regular business operations.
While we have implemented measures to safeguard our operational and technology systems and have established a culture of monitoring and improvement, cybersecurity threats are constantly evolving, becoming more frequent and more sophisticated and are being made by groups of individuals with a wide range of expertise and motives, which increases the difficulty of detecting and successfully defending against them. However, our management has determined that, during the period covered by this Annual Report on Form 10-K, no cybersecurity threats, including as a result of any previous cybersecurity incidents,
have materially affected or are reasonably likely to materially affect our company, including our business strategy, operating results, or financial condition.
Governance
Our board of directors is responsible for cybersecurity risk management and oversight of our cybersecurity program. The nominating and corporate governance committee of the board of directors assists in the oversight of management’s implementation of our information technology policies and monitoring of the risks associated with our information systems, including reviewing and discussing with management our program to identify, assess, manage and monitor cybersecurity risks. The nominating and corporate governance committee regularly reviews technology strategies, physical and cybersecurity threat assessments, emerging issues and related initiatives. In addition, the audit committee of the board of directors coordinates the board of directors’ oversight of our disclosure controls and procedures and ensures that we have in place appropriate internal controls, risk assessment policies and procedures, incident response plans and reporting mechanisms. The nominating and corporate governance committee coordinates with the board of directors and audit committee, as appropriate, on matters related to cybersecurity risk.
Our board of directors delegates execution of our cybersecurity program to our Chief Information Security Officer, or CISO, who is responsible for the day-to-day management of our cybersecurity program. Our CISO has over 25 years of information security and information technology risk expertise in multiple industries, including financial, manufacturing, healthcare and life sciences, and holds industry standard certifications, including CISSP, CRISC and CISM. Our CISO, together with our Chief Information Officer, or CIO, provides at least two presentations each year to our board of directors or nominating and corporate governance committee on cybersecurity incidents, security program updates and ongoing risks, with additional updates being provided on an as-needed basis. Our CISO also meets periodically with our senior leadership team to review metrics on readiness, incidents, mitigations and remediation. In addition, our internal audit team performs periodic audits of our systems and cybersecurity processes, the results of which are reported to the audit committee and our senior management team.
We have established a disclosure committee, which consists of our chief executive officer, chief financial officer, and senior leaders from finance, legal, accounting, corporate communications, and investor relations, including, but not limited to, our Chief Legal Officer, CIO, CISO, chief accounting officer, controller and Chief Corporate Communications Officer. The disclosure committee is actively involved in the review and approval of the Company’s SEC filings and has responsibility for considering the materiality of information for such filings and, on a timely basis, determining the disclosure of that information. The CISO briefs the disclosure committee, as necessary, on cybersecurity related matters, which includes information regarding our detection, prevention, mitigation, and remediation of cybersecurity incidents and monitoring of previously evaluated cybersecurity incidents for subsequent changes that might impact conclusions on materiality, and this information is presented to the nominating and corporate governance committee, as appropriate.
ITEM 2. PROPERTIES
Our operations are based primarily in Cambridge, Massachusetts; Zug, Switzerland; Maidenhead, United Kingdom; Amsterdam, Netherlands; and Tokyo, Japan. A description of certain of the facilities we lease or own as of January 31, 2025 is included in the table below.
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Location | | Primary Use | | Approximate
Square Footage | | Lease
Expiration Date | | Renewal Option |
| 675 West Kendall Street Henri A. Termeer Square Cambridge, Massachusetts | | Corporate headquarters and primary research facility | | 295,000 | | | January 2034 | | Two five-year terms |
300 Third Street
Cambridge, Massachusetts | | Office space and additional research facility | | 129,000 | | | January 2034 | | Two five-year terms |
101 Main Street
Cambridge, Massachusetts | | Office space | | 37,000 | | | June 2026 | | One five-year term |
20 Commerce Way
Norton, Massachusetts | | GMP manufacturing | | 200,000 | | | Not applicable | | Not applicable |
665 Concord Avenue
Cambridge, Massachusetts | | GMP manufacturing | | 15,000 | | | September 2027 | | One five-year term |
Grafenauweg 2 & 4 6300 Zug, Switzerland | | International headquarters | | 15,800 | | | May 2029 | | One five-year term |
Braywick Gate
Braywick Road, Maidenhead
Berkshire, United Kingdom | | Office space | | 21,500 | | | May 2026 | | None |
Wisdom Cross Tower Antonio Vivaldistraat 150
Amsterdam, Netherlands | | Office space | | 12,500 | | | April 2030 | | One five-year term |
Pacific Century Place
1-Chome-11-1 Marunouchi Chiyoda-ku
Tokyo, Japan | | Office space | | 12,700 | | | May 2028 | | None |
_________________________________________
In addition to the locations above, we also occupy small offices in multiple locations in and outside of the U.S. to support our operations and growth.
In the future, we may lease, operate, purchase or construct additional facilities in which to conduct expanded research, development and manufacturing activities and support future commercial operations. We believe that the total space available to us under our current leases will meet our needs for the foreseeable future and that additional space would be available to us on commercially reasonable terms if required.
ITEM 3. LEGAL PROCEEDINGS
For a discussion of material pending legal proceedings, please read the section titled “Legal Matters” within Note 13, Commitments and Contingencies, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K, which is incorporated into this item by reference.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
PART II
ITEM 5. MARKET FOR REGISTRANT’S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock trades on The Nasdaq Global Select Market under the symbol “ALNY.”
Holders of Record
At January 31, 2025, there were 26 holders of record of our common stock. Because many of our shares are held by brokers and other institutions on behalf of stockholders, we are unable to estimate the total number of beneficial holders represented by these record holders.
Stock Performance Graph
The following performance graph and related information shall not be deemed “soliciting material” or to be “filed” with the Securities and Exchange Commission, or SEC, nor shall such information be incorporated by reference into any future filing under the Securities Act of 1933 or Securities Exchange Act of 1934, each as amended, except to the extent that we specifically incorporate it by reference into such filing.
The comparative stock performance graph below compares the five-year cumulative total stockholder return (assuming reinvestment of dividends, if any) from investing $100 on the last trading day of 2019, to the close of the last trading day of 2024, in each of our common stock and the selected indices. The stock price performance reflected in the graph below is not necessarily indicative of future price performance.
Comparison of Five-Year Cumulative Total Return
Among Alnylam Pharmaceuticals, Inc.,
Nasdaq Composite Total Return and Nasdaq Biotechnology Total Return
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| 12/31/2019 | | 12/31/2020 | | 12/31/2021 | | 12/30/2022 | | 12/29/2023 | | 12/31/2024 |
| Alnylam Pharmaceuticals, Inc. | $ | 100.00 | | | $ | 112.85 | | | $ | 147.24 | | | $ | 206.35 | | | $ | 166.20 | | | $ | 204.32 | |
| Nasdaq Composite Total Return | $ | 100.00 | | | $ | 144.92 | | | $ | 177.06 | | | $ | 119.45 | | | $ | 172.77 | | | $ | 223.87 | |
| Nasdaq Biotechnology Total Return | $ | 100.00 | | | $ | 126.42 | | | $ | 126.45 | | | $ | 113.65 | | | $ | 118.87 | | | $ | 118.20 | |
ITEM 6. RESERVED
Not applicable.
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
Overview
We are a global commercial-stage biopharmaceutical company that discovers, develops, manufactures and commercializes novel therapeutics based on RNAi. Our commercial products and broad pipeline of investigational RNAi therapeutics are targeting a broad range of disease areas and indications.
As described in Part I, Item 1. “Business” of this Annual Report on Form 10-K, we currently have five products that have received marketing approval, including one collaborated product, and multiple late-stage investigational programs advancing towards potential commercialization. In Part I, Item 1. “Business” you can also find a summary of key events in 2024 and 2025 to-date related to our marketed products and our clinical development programs.
We have incurred significant losses since we commenced operations in 2002 and as of December 31, 2024, we had an accumulated deficit of $7.29 billion. Historically, we have generated losses principally from costs associated with research and development activities, acquiring, filing and expanding intellectual property rights, and selling, general and administrative costs. As a result of planned expenditures for research and development activities relating to our research platform, our drug development programs, including clinical trial and manufacturing costs, the continued build-out of late-stage clinical and commercial capabilities, including global commercial operations, continued management and growth of our patent portfolio, collaborations and general corporate activities, we may incur additional operating losses. We will require substantial resources over the next several years as we expand our efforts to discover, develop and commercialize RNAi therapeutics, and aim to achieve financial self-sustainability by the end of 2025. We anticipate that our operating results will continue to fluctuate for the foreseeable future, therefore, period-to-period comparisons should not be relied upon as predictive of the results in future periods.
We currently have programs focused on a number of therapeutic areas and, as of December 31, 2024, we generate worldwide product revenues from four commercialized products, ONPATTRO, AMVUTTRA, GIVLAARI and OXLUMO, primarily in the U.S. and Europe. However, our ongoing development and regulatory efforts may not be successful and we may not be able to commence sales of any other products and/or successfully expand the labels of or market and sell our existing commercialized products or any other approved products in the future. A meaningful portion of our total revenues in recent years has been derived from collaboration revenues from collaborations with Roche, Regeneron and Novartis. In addition to revenues from the commercial sales of our approved products and potentially from sales of future products, we expect our sources of potential funding for the next several years to continue to be derived in part from existing and new strategic collaborations. Such collaborations include, or may include in the future, license and other fees, equity investments, funded research and development, milestone payments and royalties on product sales by our licensors, including royalties on sales of Leqvio made by our collaborator Novartis.
Results of Operations
The following data summarizes the results of our operations:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, | | 2024 vs 2023 | | 2023 vs 2022 | | | | | | | | | | | | | | | | |
| (In thousands, except percentages) | | 2024 | | 2023 | | 2022 | | $ Change | | % Change | | $ Change | | % Change | | | | | | | | | | | | | | | | |
| Total revenues | | $ | 2,248,243 | | | $ | 1,828,292 | | | $ | 1,037,418 | | | $ | 419,951 | | | 23 | % | | $ | 790,874 | | | 76 | % | | | | | | | | | | | | | | | | |
| Total operating costs and expenses | | $ | 2,425,128 | | | $ | 2,110,467 | | | $ | 1,822,490 | | | $ | 314,661 | | | 15 | % | | $ | 287,977 | | | 16 | % | | | | | | | | | | | | | | | | |
| Loss from operations | | $ | (176,885) | | | $ | (282,175) | | | $ | (785,072) | | | $ | 105,290 | | | (37) | % | | $ | 502,897 | | | (64) | % | | | | | | | | | | | | | | | | |
| Total other expense, net | | $ | (200,490) | | | $ | (151,342) | | | $ | (341,921) | | | $ | (49,148) | | | 32 | % | | $ | 190,579 | | | (56) | % | | | | | | | | | | | | | | | | |
| Benefit from (provision for) income taxes | | $ | 99,218 | | | $ | (6,725) | | | $ | (4,163) | | | $ | 105,943 | | | * | | $ | (2,562) | | | 62 | % | | | | | | | | | | | | | | | | |
| Net loss | | $ | (278,157) | | | $ | (440,242) | | | $ | (1,131,156) | | | $ | 162,085 | | | (37) | % | | $ | 690,914 | | | (61) | % | | | | | | | | | | | | | | | | |
| * Indicates the percentage change period over period is greater than 500%. | | | | | | | | | | | | | | | | |
For a discussion of our 2023 results and a comparison with 2022 results please refer to “Management’s Discussion and Analysis of Financial Conditions and Results of Operations” in our Annual Report on Form 10-K for the fiscal year ended December 31, 2023, which was filed with the SEC on February 15, 2024.
Discussion of Results of Operations
Revenues
Total revenues consist of the following:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, | | 2024 vs 2023 | | 2023 vs 2022 |
| (In thousands, except percentages) | | 2024 | | 2023 | | 2022 | | $ Change | | % Change | | $ Change | | % Change |
| Net product revenues | | $ | 1,646,228 | | | $ | 1,241,474 | | | $ | 894,329 | | | $ | 404,754 | | | 33 | % | | $ | 347,145 | | | 39 | % |
| Net revenues from collaborations | | 510,221 | | | 546,185 | | | 134,912 | | | (35,964) | | | (7) | % | | 411,273 | | | 305 | % |
| Royalty revenue | | 91,794 | | | 40,633 | | | 8,177 | | | 51,161 | | | 126 | % | | 32,456 | | | 397 | % |
| Total | | $ | 2,248,243 | | | $ | 1,828,292 | | | $ | 1,037,418 | | | $ | 419,951 | | | 23 | % | | $ | 790,874 | | | 76 | % |
|
Net Product Revenues
Net product revenues consist of the following, by product and region:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Years Ended December 31, | | 2024 vs 2023 | | 2023 vs 2022 |
| (In thousands, except percentages) | 2024 | | 2023 | | 2022 | | $ Change | | % Change | | $ Change | | % Change |
| ONPATTRO | | | | | | | | | | | | | |
| United States | $ | 74,787 | | | $ | 97,739 | | | $ | 246,748 | | | $ | (22,952) | | | (23) | % | | $ | (149,009) | | | (60) | % |
| Europe | 134,197 | | | 210,916 | | | 224,063 | | | (76,719) | | | (36) | % | | (13,147) | | | (6) | % |
| Rest of World | 43,873 | | | 45,891 | | | 86,797 | | | (2,018) | | | (4) | % | | (40,906) | | | (47) | % |
| Total | 252,857 | | | 354,546 | | | 557,608 | | | (101,689) | | | (29) | % | | (203,062) | | | (36) | % |
| | | | | | | | | | | | | |
| AMVUTTRA | | | | | | | | | | | | | |
| United States | 630,613 | | | 411,169 | | | 82,521 | | | 219,444 | | | 53 | % | | 328,648 | | | 398 | % |
| Europe | 235,441 | | | 70,898 | | | 4,214 | | | 164,543 | | | 232 | % | | 66,684 | | | * |
| Rest of World | 104,396 | | | 75,771 | | | 7,060 | | | 28,625 | | | 38 | % | | 68,711 | | | * |
| Total | 970,450 | | | 557,838 | | | 93,795 | | | 412,612 | | | 74 | % | | 464,043 | | | 495 | % |
| | | | | | | | | | | | | |
| GIVLAARI | | | | | | | | | | | | | |
| United States | 165,373 | | | 141,954 | | | 115,659 | | | 23,419 | | | 16 | % | | 26,295 | | | 23 | % |
| Europe | 65,906 | | | 57,498 | | | 48,670 | | | 8,408 | | | 15 | % | | 8,828 | | | 18 | % |
| Rest of World | 24,592 | | | 19,799 | | | 8,815 | | | 4,793 | | | 24 | % | | 10,984 | | | 125 | % |
| Total | 255,871 | | | 219,251 | | | 173,144 | | | 36,620 | | | 17 | % | | 46,107 | | | 27 | % |
| | | | | | | | | | | | | |
| OXLUMO | | | | | | | | | | | | | |
| United States | 62,766 | | | 38,159 | | | 27,698 | | | 24,607 | | | 64 | % | | 10,461 | | | 38 | % |
| Europe | 80,753 | | | 60,025 | | | 37,915 | | | 20,728 | | | 35 | % | | 22,110 | | | 58 | % |
| Rest of World | 23,531 | | | 11,655 | | | 4,169 | | | 11,876 | | | 102 | % | | 7,486 | | | 180 | % |
| Total | 167,050 | | | 109,839 | | | 69,782 | | | 57,211 | | | 52 | % | | 40,057 | | | 57 | % |
| | | | | | | | | | | | | |
| Total net product revenues | $ | 1,646,228 | | | $ | 1,241,474 | | | $ | 894,329 | | | $ | 404,754 | | | 33 | % | | $ | 347,145 | | | 39 | % |
| * Indicates the percentage change period over period is greater than 500%. |
Net product revenues increased during the year ended December 31, 2024, compared to the year ended December 31, 2023, primarily due to growth from sales of AMVUTTRA driven by increased patient demand, partially offset by a decrease in sales of ONPATTRO due to patient switches to AMVUTTRA, as well as increased patients on GIVLAARI and OXLUMO therapies.
Please see Note 3, Net Product Revenues, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K for balances and activity in each product revenue allowance and reserve category for the years ended December 31, 2024 and 2023.
Net Revenues from Collaborations and Royalty Revenue
Net revenues from collaborations and royalty revenue consist of the following:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Years Ended December 31, | | 2024 vs 2023 | | 2023 vs 2022 |
| (In thousands, except percentages) | 2024 | | 2023 | | 2022 | | $ Change | | % Change | | $ Change | | % Change |
| Roche | $ | 119,489 | | | $ | 337,802 | | | $ | — | | | $ | (218,313) | | | (65) | % | | $ | 337,802 | | | N/A |
| Regeneron Pharmaceuticals | 302,798 | | | 100,468 | | | 87,844 | | | 202,330 | | | 201 | % | | 12,624 | | | 14 | % |
| Novartis AG | 79,759 | | | 86,727 | | | 43,159 | | | (6,968) | | | (8) | % | | 43,568 | | | 101 | % |
| | | | | | | | | | | | | |
| Other | 8,175 | | | 21,188 | | | 3,909 | | | (13,013) | | | (61) | % | | 17,279 | | | 442 | % |
| Total net revenues from collaborations | $ | 510,221 | | | $ | 546,185 | | | $ | 134,912 | | | $ | (35,964) | | | (7) | % | | $ | 411,273 | | | 305 | % |
| | | | | | | | | | | | | |
| Royalty revenue | $ | 91,794 | | | $ | 40,633 | | | $ | 8,177 | | | $ | 51,161 | | | 126 | % | | $ | 32,456 | | | 397 | % |
Net revenues from collaborations decreased during the year ended December 31, 2024, as compared to the year ended December 31, 2023, primarily driven by:
•a decrease in revenue recognized under our Roche Collaboration in 2024 due to the recognition of $310.0 million of revenue upon the transfer of licenses to Roche during the third quarter of 2023.
Partially offset by:
•revenue of $185.0 million recognized under our Regeneron Collaboration as we modified the collaboration in June 2024 and provided Regeneron with an exclusive license to develop, manufacture and commercialize cemdisiran as a monotherapy; and
•recognition of $65.0 million in revenue under our Roche Collaboration associated with dosing the first patient in the zilebesiran KARDIA-3 clinical trial during 2024.
Royalty revenue increased during the year ended December 31, 2024, as compared to the year ended December 31, 2023, due to increased volume and rate of royalties earned from global net sales of Leqvio by our collaborator, Novartis.
Recognition of our combined net revenues from collaborations and royalty revenue is dependent on a variety of factors, including the level of work reimbursed by collaborators, achievement of milestones under our collaboration agreements, and royalties associated with sales of Leqvio. We expect net revenues from collaborations will increase in 2025, as compared to 2024, primarily driven by higher anticipated revenues under our Roche Collaboration and License Agreement. We expect our royalty revenue will increase in 2025, as compared to 2024, due to the continued growth of royalties earned from global net sales of Leqvio by our collaborator, Novartis.
The amount of revenue from collaborations that we recognize is based, in part, on estimates of total costs to be incurred. These estimates reflect our historical experiences, current contractual requirements, and forecasted plans of development or manufacturing activities. We adjust these estimates for changes in actual costs incurred, contractual terms, and further forecasts. Such changes in estimates could have a significant impact on revenue and earnings in the period of the adjustment.
Operating Costs and Expenses
Operating costs and expenses consist of the following:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Years Ended December 31, | | 2024 vs 2023 | | 2023 vs 2022 |
| (In thousands, except percentages) | 2024 | | 2023 | | 2022 | | $ Change | | % Change | | $ Change | | % Change |
| Cost of goods sold | $ | 306,513 | | | $ | 268,216 | | | $ | 140,174 | | | $ | 38,297 | | | 14 | % | | $ | 128,042 | | | 91 | % |
| | | | | | | | | | | | | |
| Cost of goods sold as a percentage of net product revenues | 18.6 | % | | 21.6 | % | | 15.7 | % | | | | | | | | |
| | | | | | | | | | | | | |
| Cost of collaborations and royalties | 16,857 | | | 42,190 | | | 28,643 | | | (25,333) | | | (60) | % | | 13,547 | | | 47 | % |
| Research and development | 1,126,232 | | | 1,004,415 | | | 883,015 | | | 121,817 | | | 12 | % | | 121,400 | | | 14 | % |
| Selling, general and administrative | 975,526 | | | 795,646 | | | 770,658 | | | 179,880 | | | 23 | % | | 24,988 | | | 3 | % |
| Total | $ | 2,425,128 | | | $ | 2,110,467 | | | $ | 1,822,490 | | | $ | 314,661 | | | 15 | % | | $ | 287,977 | | | 16 | % |
Cost of Goods Sold
Cost of goods sold as a percentage of net product revenues decreased to 18.6% for the year ended December 31, 2024, as compared to 21.6% for the year ended December 31, 2023. Approximately 5.0% of the 21.6% of cost of goods sold as a percentage of net product revenues for the year ended December 31, 2023 was attributable to cancelled manufacturing commitments and the impairment of ONPATTRO inventory that had been manufactured for future demand associated with the use of ONPATTRO for the treatment of patients with ATTR amyloidosis with cardiomyopathy, for which we did not receive regulatory approval in the U.S. These one-time charges in 2023 did not recur in 2024, resulting in the decrease in cost of goods sold as a percentage of net product revenues in 2024, which was partially offset by higher volume and royalty rates payable on net sales of AMVUTTRA in 2024.
We expect our cost of goods sold, including cost of goods sold as a percentage of net product revenues, will increase during 2025, as compared to 2024, primarily as a result of an expected increase in net product revenues and increased royalties on net sales of AMVUTTRA.
Cost of Collaborations and Royalties
Cost of collaborations and royalties decreased during the year ended December 31, 2024, as compared to the year ended December 31, 2023, primarily due to decreased demand for GalNAc material supplied to our collaborators in support of certain
product manufacturing as our collaborators transition to producing the material independently, as well as reduced royalties payable from the expiration of licenses of third-party intellectual property.
We expect our cost of collaborations and royalties will decrease during 2025, as compared to 2024, primarily as a result of our collaborators transitioning to produce GalNAc material independently.
Research and Development
Research and development expenses consist of the following:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, | | 2024 vs 2023 | | 2023 vs 2022 |
| (In thousands, except percentages) | | 2024 | | 2023 | | 2022 | | $ Change | | % Change | | $ Change | | % Change |
| Clinical research and outside services | | $ | 509,129 | | | $ | 485,732 | | | $ | 438,418 | | | $ | 23,397 | | | 5 | % | | $ | 47,314 | | | 11 | % |
| Compensation and related | | 327,929 | | | 260,423 | | | 225,589 | | | 67,506 | | | 26 | % | | 34,834 | | | 15 | % |
Occupancy and all other costs(1) | | 161,425 | | | 160,987 | | | 126,847 | | | 438 | | | — | % | | 34,140 | | | 27 | % |
| Stock-based compensation | | 127,749 | | | 97,273 | | | 92,161 | | | 30,476 | | | 31 | % | | 5,112 | | | 6 | % |
| Total | | $ | 1,126,232 | | | $ | 1,004,415 | | | $ | 883,015 | | | $ | 121,817 | | | 12 | % | | $ | 121,400 | | | 14 | % |
(1) Occupancy and all other costs includes facilities, information technology, depreciation and certain departmental expenses.
Research and development expenses increased during the year ended December 31, 2024, as compared to the year ended December 31, 2023, primarily due to the following:
•increased clinical trial expenses mainly related to the advancement of our KARDIA-3 and cAPPRicorn-1 clinical programs;
•increased costs associated with our preclinical activities as we develop our clinical pipeline of RNAi therapeutics targeting multiple tissue types;
•increased employee compensation and related expenses to support our research and development pipeline and development expenses; and
•increased stock-based compensation expenses primarily due to the accounting for certain performance-based awards.
Partially offset by:
•decreased expenses within other clinical programs, specifically the APOLLO-B Phase 3 clinical trial of patisiran due to the wind down of clinical activities during the open label extension period; and
•decreased costs due to the timing of manufacturing of zilebesiran for clinical activities.
During the years ended December 31, 2024, 2023 and 2022, in connection with advancing activities under our collaboration agreements, we incurred research and development expenses, primarily related to external development and clinical expenses, including the manufacture of clinical product. The following table summarizes research and development expenses incurred, for which we recognize revenue, that are directly attributable to our collaboration agreements, by collaborator:
| | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| (In thousands) | | 2024 | | 2023 | | 2022 |
| Roche | | $ | 92,725 | | | $ | 44,620 | | | $ | — | |
| Regeneron Pharmaceuticals | | 71,659 | | | 77,444 | | | 43,002 | |
| Other | | 8,525 | | | 4,951 | | | 1,172 | |
| Total | | $ | 172,909 | | | $ | 127,015 | | | $ | 44,174 | |
Selling, General and Administrative
Selling, general and administrative expenses consist of the following:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, | | 2024 vs 2023 | | 2023 vs 2022 |
| (In thousands, except percentages) | | 2024 | | 2023 | | 2022 | | $ Change | | % Change | | $ Change | | % Change |
| Compensation and related | | $ | 386,743 | | | $ | 298,888 | | | $ | 273,262 | | | $ | 87,855 | | | 29 | % | | $ | 25,626 | | | 9 | % |
| Consulting and professional services | 274,539 | | | 226,664 | | | 226,941 | | | 47,875 | | | 21 | % | | (277) | | | — | % |
Occupancy and all other costs(1) | | 169,909 | | | 145,687 | | | 131,967 | | | 24,222 | | | 17 | % | | 13,720 | | | 10 | % |
| Stock-based compensation | | 144,335 | | | 124,407 | | | 138,488 | | | 19,928 | | | 16 | % | | (14,081) | | | (10) | % |
| Total | | $ | 975,526 | | | $ | 795,646 | | | $ | 770,658 | | | $ | 179,880 | | | 23 | % | | $ | 24,988 | | | 3 | % |
(1) Occupancy and all other costs includes facilities, information technology, depreciation and certain departmental expenses.
Selling, general and administrative expenses increased during the year ended December 31, 2024, as compared to the year ended December 31, 2023, primarily due to higher costs associated with marketing investments to promote our TTR therapies and prepare for the potential launch of AMVUTTRA for the treatment of ATTR amyloidosis with cardiomyopathy and increased employee compensation expenses.
We expect that research and development expenses combined with selling, general and administrative expenses will increase during 2025, as compared to 2024, as we continue to build out our global commercial and compliance infrastructure, launch our current commercial products into new markets, prepare for future commercial product launches, including the launch of AMVUTTRA in cardiomyopathy, assuming regulatory approvals, advance our product candidates, including collaborated programs, into later-stage development, advance and develop our platform and preclinical pipeline, and prepare regulatory submissions. However, we expect that certain expenses will be variable depending on the timing of manufacturing batches, clinical trial enrollment and results, regulatory review of our product candidates and programs, and stock-based compensation expenses based on our determinations regarding the probability of vesting for performance-based awards.
Other (Expense) Income
Other (expense) income consists of the following:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, | | 2024 vs 2023 | | 2023 vs 2022 |
| (In thousands, except percentages) | 2024 | | 2023 | | 2022 | | $ Change | | % Change | | $ Change | | % Change |
| Interest expense | $ | (141,858) | | | $ | (121,221) | | | $ | (155,968) | | | $ | (20,637) | | | 17 | % | | $ | 34,747 | | | (22) | % |
| Interest income | 121,992 | | | 95,561 | | | 24,808 | | | 26,431 | | | 28 | % | | 70,753 | | | 285 | % |
| Other expense, net | | | | | | | | | | | | | |
| Realized and unrealized losses on marketable equity securities | (3,022) | | | (16,944) | | | (33,312) | | | 13,922 | | | (82) | % | | 16,368 | | | (49) | % |
| Change in fair value of development derivative liability | (170,770) | | | (90,997) | | | (94,659) | | | (79,773) | | | 88 | % | | 3,662 | | | (4) | % |
| Other | (6,832) | | | (17,741) | | | (6,204) | | | 10,909 | | | (61) | % | | (11,537) | | | 186 | % |
| Loss on the extinguishment of debt | — | | | — | | | (76,586) | | | — | | | N/A | | 76,586 | | | (100) | % |
| Total | $ | (200,490) | | | $ | (151,342) | | | $ | (341,921) | | | $ | (49,148) | | | 32 | % | | $ | 190,579 | | | (56) | % |
|
Total other expense, net increased during the year ended December 31, 2024, as compared to the year ended December 31, 2023, primarily due to increased loss associated with the change in fair value of the development derivative liability as a result of valuation updates driven by the positive topline results for the HELIOS-B clinical trial announced in June 2024, partially offset by increased interest income driven by higher market interest rates on our marketable debt securities.
Benefit from (Provision for) Income Taxes
Benefit from (provision for) income taxes was a follows:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Years Ended December 31, | | 2024 vs 2023 | | 2023 vs 2022 |
| (In thousands, except percentages) | 2024 | | 2023 | | 2022 | | $ Change | | % Change | | $ Change | | % Change |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Benefit from (provision for) income taxes | $ | 99,218 | | | $ | (6,725) | | | $ | (4,163) | | | $ | 105,943 | | | * | | $ | (2,562) | | | 62 | % |
| * Indicates the percentage change period over period is greater than 500%. |
We recorded a benefit from income taxes of $99.2 million for the year ended December 31, 2024 and a provision for income taxes of $6.7 million for the year ended December 31, 2023. The benefit from income taxes for the year ended December 31, 2024 primarily relates to the release of the valuation allowance on our certain Switzerland deferred tax assets, which mainly consist of the tax basis of the intangible assets that were transferred to our wholly-owned Switzerland subsidiary in 2020, 2021 and 2023 and net operating loss carryforwards. We maintained a full valuation allowance on our U.S. deferred tax assets as of December 31, 2024.
Liquidity and Capital Resources
The following table summarizes our cash flow activities:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Years Ended December 31, | | $ Change | | |
| (In thousands) | 2024 | | 2023 | | 2022 | | 2024 vs 2023 | | 2023 vs 2022 |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| Net cash (used in) provided by: | | | | | | | | | | | | | |
| Operating activities | $ | (8,312) | | | $ | 104,156 | | | $ | (541,274) | | | $ | (112,468) | | | | | $ | 645,430 | | | |
| Investing activities | $ | (116,840) | | | $ | (336,350) | | | $ | 169,354 | | | $ | 219,510 | | | | | $ | (505,704) | | | |
| Financing activities | $ | 294,159 | | | $ | 172,131 | | | $ | 425,753 | | | $ | 122,028 | | | | | $ | (253,622) | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | | | | | | | | | | | |
Operating Activities
Net cash used in operating activities increased during the year ended December 31, 2024, compared to the year ended December 31, 2023, primarily due to decreased cash received from our collaborators, partially offset by stronger cash receipts from increased product sales.
Investing Activities
Net cash used in investing activities decreased during the year ended December 31, 2024, compared to the year ended December 31, 2023, primarily due to the timing of net investments of cash into our marketable debt securities.
Financing Activities
Net cash provided by financing activities increased during the year ended December 31, 2024, compared to the year ended December 31, 2023, primarily due to increased net proceeds from exercise of stock options.
Additional Capital Requirements
We currently have programs focused in many therapeutic areas and, as of December 31, 2024, have five marketed products, including one product commercialized by a collaborator. However, our ongoing development efforts may not be successful and we may not be able to commence sales of any other products or successfully expand the approved indications for our approved products, including AMVUTTRA, in the future. In addition, we may incur additional operating losses as a result of planned expenditures for research and development activities relating to our research platform, our drug development programs, including clinical trial and manufacturing costs, the continued build-out of late-stage clinical, manufacturing, commercial and compliance capabilities, including global operations, continued management and growth of our intellectual property, including our patent portfolio, collaborations and general corporate activities.
Based on our current operating plan, we believe that our cash, cash equivalents and marketable securities as of December 31, 2024 will be sufficient to satisfy our near-term capital and operating needs for at least 12 months from the filing of this Annual Report on Form 10-K. Recent and expected working and other capital requirements, in addition to the above matters, also include the items described below:
•Amounts related to future lease payments for operating lease obligations as of December 31, 2024 totaled $384.5 million, with $43.4 million expected to be paid within the next 12 months.
•Cash outflows for capital expenditures were $34.3 million in 2024 and $62.2 million in 2023. We expect capital expenditures to increase in 2025 to support the increase in our manufacturing and production capacity needs.
•Amounts related to future long-term debt total $1.02 billion, of which we do not expect to make payments on principal within the next 12 months.
•Payments to Blackstone associated with the liability related to the sale of future royalties were $57.0 million in 2024, with an estimated $131.8 million to be paid within the next 12 months.
•Payments associated with an achieved development milestone due to Blackstone were $21.1 million in 2024, with the same amount to be paid within the next 12 months. Further, we anticipate making an additional $76.5 million of fixed and royalty payments upon regulatory approval of AMVUTTRA for the treatment of ATTR amyloidosis with
cardiomyopathy and the first commercial sale of AMVUTTRA following regulatory approval of AMVUTTRA for the treatment of ATTR amyloidosis with cardiomyopathy, respectively, within the next 12 months.
Since we commenced operations in 2002, we have generated significant losses and as of December 31, 2024, we had an accumulated deficit of $7.29 billion. As of December 31, 2024, we had cash, cash equivalents and marketable securities of $2.69 billion, compared to $2.44 billion as of December 31, 2023.
Due to numerous factors described in more detail under the caption Part I, Item 1A, “Risk Factors” of this Annual Report on Form 10-K, we may require significant additional funds earlier than we currently expect in order to continue to commercialize ONPATTRO, AMVUTTRA, GIVLAARI and OXLUMO, and to develop, conduct clinical trials for, manufacture and, if approved, commercialize additional product candidates.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with GAAP. The preparation of our consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and disclosure of contingent assets and liabilities in our consolidated financial statements. Actual results may differ from these estimates under different assumptions or conditions and could have a material impact on our reported results. While our significant accounting policies are more fully described in the Notes to our consolidated financial statements included elsewhere in this Annual Report on Form 10-K, we believe the following accounting policies to be the most critical in understanding the judgments and estimates we use in preparing our consolidated financial statements:
Net Product Revenues
Our net product revenues are recognized, net of variable consideration related to certain allowances and accruals, at the time the customer obtains control of our product. We record reserves, based on contractual terms, for components related to product sold during the reporting period, as well as our estimate of product that remains in the distribution channel inventory at the end of the reporting period that we expect will be sold to qualified healthcare providers. On a quarterly basis, we update our estimates and record any needed adjustments in the period we identify the adjustments.
The estimates for our product revenue allowances and accruals are most significantly affected by chargebacks, which are contractual commitments with the government and other entities to sell products to qualified healthcare providers at prices lower than the list prices charged to the customer who directly purchases from us, and rebates that represent discount obligations under government programs, including Medicaid in the U.S. and similar programs in certain other countries, including countries in which we are accruing for estimated rebates because final pricing has not yet been negotiated. We are also subject to potential rebates in connection with our value-based agreements, or VBAs, with certain commercial payors.
We use the expected value method, which is the sum of probability-weighted amounts in a range of possible consideration amounts, or the most likely amount method, which is the single most likely amount in a range of possible considerations, to estimate variable consideration related to our product revenues. We use the expected value method to estimate variable consideration for chargebacks, certain rebates, and other incentives and we use the most likely amount method for certain rebates and trade discounts and allowances.
Net Revenues from Collaborations
We earn revenue in connection with collaboration agreements which allow our collaborators to utilize our technology platforms and develop product candidates.
For elements of collaboration arrangements that are accounted for pursuant to Accounting Standards Codification Topic 606, Revenue from Contracts with Customers, or ASC 606, we identify the performance obligations and allocate the total consideration we expect to receive on a relative standalone selling price basis to each performance obligation. Key assumptions to determine the standalone selling price may include forecasted revenues, development timelines, reimbursement rates for personnel costs, the expected number of targets or indications expected to be pursued under each license, discount rates and probabilities of technical and regulatory success. We recognize revenue associated with each performance obligation as the control over the promised goods or services transfer to our collaborator which occurs either at a point in time or over time. If control transfers over time, revenue is recognized by using a method of measuring progress that best depicts the transfer of goods or services, for example based on actual costs incurred relative to total forecasted costs to be incurred over the period the transfer of goods or services occurs. We evaluate the measure of progress and related inputs each reporting period and any resulting adjustments to revenue are recorded on a cumulative catch-up basis. Revenue to be recognized is equal to the total transaction price multiplied by the ratio of actual expense incurred divided by total forecasted expense.
Liability Related to the Sale of Future Royalties
We account for the liability related to the sale of future royalties as a debt financing. Interest on the liability related to the sale of future royalties is recognized using the effective interest rate method over the life of the related royalty stream.
The liability related to the sale of future royalties and the related interest expense are based on our current estimates of future royalties and commercial milestones expected to be paid over the life of the arrangement, which we determine by using third-party data to estimate Leqvio’s global net revenue. We periodically assess the expected payments and to the extent the amount or timing of our future estimated payments is materially different than our previous estimates, we account for any such change by prospectively adjusting the effective interest rate and related non-cash interest expense.
An increase or decrease of 10% to the interest rate would result in an increase or decrease to our liability related to the sale of future royalties of approximately $35.6 million.
Development Derivative Liability
In August 2020, we entered into a co-development agreement, referred to as the Funding Agreement, with BXLS V Bodyguard – PCP L.P. and BXLS Family Investment Partnership V – ESC L.P., collectively referred to as Blackstone Life Sciences, pursuant to which Blackstone Life Sciences will provide up to $150.0 million in funding for the clinical development of vutrisiran and zilebesiran, two of our cardiometabolic programs. As consideration for Blackstone Life Sciences’ funding for certain vutrisiran and zilebesiran clinical development costs, we have agreed to pay Blackstone Life Sciences fixed success-based payments upon achievement of specific milestones for vutrisiran and zilebesiran as well as a 1% royalty on net sales of vutrisiran for ten years.
The development derivative liability is recorded at fair value and represents our current estimate of the expected future payments to Blackstone Life Sciences. The development derivative liability is based on the probability weighted present value of the estimated cash flows pursuant to contractual terms of the Funding Agreement. The most significant assumptions in determining the development derivative liability are the probability of success for the clinical development and regulatory approval of vutrisiran and zilebesiran and our current cost of borrowing. Estimates of the probability of success and our cost of borrowing are based on what we believe to be reasonable and supportable assumptions and require management’s judgment. Actual results could vary materially from these estimates.
Recent Accounting Pronouncements
Please read Note 2, Summary of Significant Accounting Policies, to our consolidated financial statements included in Part II, Item 8, “Financial Statements and Supplementary Data” of this Annual Report on Form 10-K for a description of recent accounting pronouncements.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Interest Rate Risk - Investment Portfolio. We invest a portion of our cash in a number of diversified fixed- and floating-rate securities consisting of cash equivalents and marketable debt securities related to our investment portfolio that are subject to interest rate risk. Changes in the general level of interest rates can affect the fair value of our investment portfolio. If interest rates in the general economy were to rise, our holdings could lose value. As of December 31, 2024 and 2023, a hypothetical increase in interest rates of 100 basis points across the entire yield curve on our holdings would have resulted in an immaterial decrease to the fair value of our holdings.
Foreign Currency Exchange Risk. As a result of our foreign operations, we face exposure to movements in foreign currency exchange rates, primarily the Japanese yen, Euro and British pound against the U.S. Dollar. Fluctuations in the global markets may have a positive or negative effect on our foreign exchange rate exposure. The current exposures arise primarily from net product revenue, operating costs and expenses and balance sheet amounts. Therefore, significant changes in foreign exchange rates of the countries outside the United States where our products are sold, where development expenses are incurred by us or our collaborators, or where we incur operating expenses can impact our operating results and financial condition. As sales outside the United States continue to grow, and as we expand our international operations, we will continue to assess potential steps, including foreign currency hedging and other strategies, to mitigate our foreign exchange risk.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of Alnylam Pharmaceuticals, Inc.
Opinions on the Financial Statements and Internal Control over Financial Reporting
We have audited the accompanying consolidated balance sheets of Alnylam Pharmaceuticals, Inc. and its subsidiaries (the "Company") as of December 31, 2024 and 2023, and the related consolidated statements of operations and comprehensive loss, of stockholders’ equity (deficit) and of cash flows for each of the three years in the period ended December 31, 2024, including the related notes (collectively referred to as the "consolidated financial statements"). We also have audited the Company's internal control over financial reporting as of December 31, 2024, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of the Company as of December 31, 2024 and 2023, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2024 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, the Company maintained, in all material respects, effective internal control over financial reporting as of December 31, 2024, based on criteria established in Internal Control - Integrated Framework (2013) issued by the COSO.
Basis for Opinions
The Company's management is responsible for these consolidated financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in Management’s Annual Report on Internal Control over Financial Reporting appearing under Item 9A. Our responsibility is to express opinions on the Company’s consolidated financial statements and on the Company's internal control over financial reporting based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud, and whether effective internal control over financial reporting was maintained in all material respects.
Our audits of the consolidated financial statements included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
Definition and Limitations of Internal Control over Financial Reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Critical Audit Matters
The critical audit matter communicated below is a matter arising from the current period audit of the consolidated financial statements that was communicated or required to be communicated to the audit committee and that (i) relates to accounts or disclosures that are material to the consolidated financial statements and (ii) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated
financial statements, taken as a whole, and we are not, by communicating the critical audit matter below, providing a separate opinion on the critical audit matter or on the accounts or disclosures to which it relates.
Liability Related to Sale of Future Royalties and Commercial Milestones
As described in Notes 2 and 9 to the consolidated financial statements, the liability related to the sale of future royalties and the related interest expense are based on management’s current estimates of future royalties and commercial milestones expected to be paid over the life of the arrangement. Interest on the liability related to the sale of future royalties will be recognized using the effective interest rate method, resulting in the recognition of interest expense. Management periodically assesses the expected payments and to the extent the amount or timing of the future estimated payments is materially different than the previous estimates, management accounts for any such change by adjusting the liability related to the sale of future royalties and prospectively recognizing the related non-cash interest expense. Management’s estimate of the amount of expected future payments to Blackstone over the life of the arrangement is based on the estimated global net sales of Leqvio. The Company recorded a liability related to the sale of future royalties of $1.45 billion as of December 31, 2024 and recognized interest expense on the liability related to the sale of future royalties of $127.1 million for the year ended December 31, 2024.
The principal considerations for our determination that performing procedures relating to the liability related to the sale of future royalties and commercial milestones is a critical audit matter are the significant judgment by management when developing the estimate of the timing and amount of future royalties and commercial milestones to be paid. This in turn led to a high degree of auditor judgment and effort in performing procedures and in evaluating audit evidence relating to management’s estimate of the expected future royalties and commercial milestones to be paid and the selection of third party data used to estimate global net sales of Leqvio.
Addressing the matter involved performing procedures and evaluating audit evidence in connection with forming our overall opinion on the consolidated financial statements. These procedures included testing the effectiveness of controls relating to the liability for future royalties and commercial milestones, including controls over management’s process for developing the estimate of timing and amount of future royalties and commercial milestones to be paid. These procedures also included, among others (i) testing management’s process for developing the estimate of timing and amount of future royalties and commercial milestones to be paid and (ii) evaluating the reasonableness of significant assumptions used by management when developing the estimate of expected future royalties and commercial milestones to be paid related to the selection of third party data used to estimate global net sales of Leqvio. Evaluating management’s assumption related to the selection of third-party data used to estimate global net sales of Leqvio involved evaluating whether the assumptions used by management were reasonable considering consistency with industry data.
/s/PricewaterhouseCoopers LLP
Boston, Massachusetts
February 13, 2025
We have served as the Company’s auditor since 2003.
ALNYLAM PHARMACEUTICALS, INC.
CONSOLIDATED BALANCE SHEETS
(In thousands, except per share amounts)
| | | | | | | | | | | |
| December 31, |
| 2024 | | 2023 |
| ASSETS | | | |
| Current assets: | | | |
| Cash and cash equivalents | $ | 966,428 | | | $ | 812,688 | |
| Marketable debt securities | 1,719,920 | | | 1,615,516 | |
| Marketable equity securities | 8,156 | | | 11,178 | |
| Accounts receivable, net | 405,308 | | | 327,787 | |
| Inventory | 78,509 | | | 89,146 | |
| Prepaid expenses and other current assets | 116,964 | | | 126,382 | |
| | | |
| Total current assets | 3,295,285 | | | 2,982,697 | |
| Property, plant and equipment, net | 502,784 | | | 526,057 | |
| Operating lease right-of-use assets | 191,148 | | | 199,732 | |
| Deferred tax assets | 116,863 | | | 10,101 | |
| Restricted investments | 68,593 | | | 49,391 | |
| Other assets | 65,310 | | | 61,902 | |
| Total assets | $ | 4,239,983 | | | $ | 3,829,880 | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY (DEFICIT) | | | |
| Current liabilities: | | | |
| Accounts payable | $ | 88,415 | | | $ | 55,519 | |
| Accrued expenses | 887,472 | | | 713,013 | |
| Operating lease liabilities | 41,886 | | | 41,510 | |
| Deferred revenue | 55,481 | | | 102,753 | |
| Liability related to the sale of future royalties | 113,018 | | | 54,991 | |
| Total current liabilities | 1,186,272 | | | 967,786 | |
| Operating lease liabilities, net of current portion | 229,541 | | | 243,101 | |
| Deferred revenue, net of current portion | — | | | 188,175 | |
| Convertible debt | 1,024,621 | | | 1,020,776 | |
| | | |
| Liability related to the sale of future royalties, net of current portion | 1,334,353 | | | 1,322,248 | |
| Other liabilities | 398,108 | | | 308,438 | |
| Total liabilities | 4,172,895 | | | 4,050,524 | |
| Commitments and contingencies (Note 13) | | | |
| Stockholders’ equity (deficit): | | | |
| Preferred stock, $0.01 par value per share, 5,000 shares authorized and no shares issued and outstanding as of December 31, 2024 and December 31, 2023 | — | | | — | |
| Common stock, $0.01 par value per share, 250,000 shares authorized as of December 31, 2024 and December 31, 2023, respectively; 129,294 shares issued and outstanding as of December 31, 2024; 125,794 shares issued and outstanding as of December 31, 2023 | 1,293 | | | 1,259 | |
| Additional paid-in capital | 7,388,061 | | | 6,811,063 | |
| Accumulated other comprehensive loss | (34,518) | | | (23,375) | |
| Accumulated deficit | (7,287,748) | | | (7,009,591) | |
| Total stockholders’ equity (deficit) | 67,088 | | | (220,644) | |
| Total liabilities and stockholders’ equity (deficit) | $ | 4,239,983 | | | $ | 3,829,880 | |
The accompanying notes are an integral part of these consolidated financial statements.
ALNYLAM PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE LOSS
(In thousands, except per share amounts)
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Statements of Operations | | | | | |
| Revenues: | | | | | |
| Net product revenues | $ | 1,646,228 | | | $ | 1,241,474 | | | $ | 894,329 | |
| Net revenues from collaborations | 510,221 | | | 546,185 | | | 134,912 | |
| Royalty revenue | 91,794 | | | 40,633 | | | 8,177 | |
| Total revenues | 2,248,243 | | | 1,828,292 | | | 1,037,418 | |
| Operating costs and expenses: | | | | | |
| Cost of goods sold | 306,513 | | | 268,216 | | | 140,174 | |
| Cost of collaborations and royalties | 16,857 | | | 42,190 | | | 28,643 | |
| Research and development | 1,126,232 | | | 1,004,415 | | | 883,015 | |
| Selling, general and administrative | 975,526 | | | 795,646 | | | 770,658 | |
| Total operating costs and expenses | 2,425,128 | | | 2,110,467 | | | 1,822,490 | |
| Loss from operations | (176,885) | | | (282,175) | | | (785,072) | |
| Other (expense) income: | | | | | |
| Interest expense | (141,858) | | | (121,221) | | | (155,968) | |
| Interest income | 121,992 | | | 95,561 | | | 24,808 | |
| Other expense, net | (180,624) | | | (125,682) | | | (134,175) | |
| Loss on the extinguishment of debt | — | | | — | | | (76,586) | |
| Total other expense, net | (200,490) | | | (151,342) | | | (341,921) | |
| Loss before income taxes | (377,375) | | | (433,517) | | | (1,126,993) | |
| Benefit from (provision for) income taxes | 99,218 | | | (6,725) | | | (4,163) | |
| Net loss | $ | (278,157) | | | $ | (440,242) | | | $ | (1,131,156) | |
| Net loss per common share — basic and diluted | $ | (2.18) | | | $ | (3.52) | | | $ | (9.30) | |
| Weighted-average common shares used to compute basic and diluted net loss per common share | 127,651 | | | 124,906 | | | 121,689 | |
| | | | | |
| Statements of Comprehensive Loss | | | | | |
| Net loss | $ | (278,157) | | | $ | (440,242) | | | $ | (1,131,156) | |
| Other comprehensive income (loss): | | | | | |
| Unrealized (loss) gain on marketable securities | (4) | | | 11,018 | | | (7,840) | |
| Foreign currency translation (loss) gain | (9,643) | | | 11,922 | | | (5,274) | |
| Defined benefit pension plans, net of tax | (1,496) | | | (1,661) | | | 1,719 | |
| Total other comprehensive (loss) income | (11,143) | | | 21,279 | | | (11,395) | |
| Comprehensive loss | $ | (289,300) | | | $ | (418,963) | | | $ | (1,142,551) | |
The accompanying notes are an integral part of these consolidated financial statements.
ALNYLAM PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF STOCKHOLDERS’ EQUITY (DEFICIT)
(In thousands)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Common Stock | | Additional
Paid-in
Capital | | Accumulated Other Comprehensive Loss | | Accumulated
Deficit | | Total Stockholders’ Equity (Deficit) |
| Shares | | Amount | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| | | | | | | | | | | |
| Balance as of December 31, 2021 | 120,182 | | | 1,202 | | | 6,058,453 | | | (33,259) | | | (5,438,193) | | | 588,203 | |
| Exercise of common stock options, net of tax withholdings | 3,103 | | | 32 | | | 263,580 | | | — | | | — | | | 263,612 | |
| Issuance of common stock under equity plans | 640 | | | 6 | | | 13,719 | | | — | | | — | | | 13,725 | |
| Stock-based compensation expense | — | | | — | | | 237,399 | | | — | | | — | | | 237,399 | |
| Purchase of capped calls related to convertible debt | — | | | — | | | (118,611) | | | — | | | — | | | (118,611) | |
| Other comprehensive loss | — | | | — | | | — | | | (11,395) | | | — | | | (11,395) | |
| Net loss | — | | | — | | | — | | | — | | | (1,131,156) | | | (1,131,156) | |
| Balance as of December 31, 2022 | 123,925 | | | 1,240 | | | 6,454,540 | | | (44,654) | | | (6,569,349) | | | (158,223) | |
| Exercise of common stock options, net of tax withholdings | 1,162 | | | 12 | | | 114,237 | | | — | | | — | | | 114,249 | |
| Issuance of common stock under equity plans | 707 | | | 7 | | | 16,421 | | | — | | | — | | | 16,428 | |
| Stock-based compensation expense | — | | | — | | | 225,865 | | | — | | | — | | | 225,865 | |
| Other comprehensive income | — | | | — | | | — | | | 21,279 | | | — | | | 21,279 | |
| Net loss | — | | | — | | | — | | | — | | | (440,242) | | | (440,242) | |
| Balance as of December 31, 2023 | 125,794 | | | 1,259 | | | 6,811,063 | | | (23,375) | | | (7,009,591) | | | (220,644) | |
| Exercise of common stock options, net of tax withholdings | 2,504 | | | 25 | | | 284,256 | | | — | | | — | | | 284,281 | |
| Issuance of common stock under equity plans | 996 | | | 9 | | | 17,425 | | | — | | | — | | | 17,434 | |
| Stock-based compensation expense | — | | | — | | | 275,317 | | | — | | | — | | | 275,317 | |
| Other comprehensive loss | — | | | — | | | — | | | (11,143) | | | — | | | (11,143) | |
| Net loss | — | | | — | | | — | | | — | | | (278,157) | | | (278,157) | |
| Balance as of December 31, 2024 | 129,294 | | | $ | 1,293 | | | $ | 7,388,061 | | | $ | (34,518) | | | $ | (7,287,748) | | | $ | 67,088 | |
The accompanying notes are an integral part of these consolidated financial statements.
ALNYLAM PHARMACEUTICALS, INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Cash flows from operating activities: | | | | | |
| Net loss | $ | (278,157) | | | $ | (440,242) | | | $ | (1,131,156) | |
| Non-cash adjustments to reconcile net loss to net cash (used in) provided by operating activities: | | | | | |
| Depreciation and amortization | 56,670 | | | 54,054 | | | 44,468 | |
| | | | | |
| Non-cash interest expense on liability related to the sale of future royalties | 127,133 | | | 106,554 | | | 104,200 | |
| Stock-based compensation | 272,084 | | | 221,680 | | | 230,649 | |
| Realized and unrealized losses on marketable equity securities | 3,022 | | | 16,944 | | | 33,312 | |
| Loss on extinguishment of debt | — | | | — | | | 76,586 | |
| Change in fair value of development derivative liability | 170,770 | | | 90,997 | | | 94,659 | |
| Deferred income taxes | (106,762) | | | (713) | | | (1,433) | |
| Other | (38,988) | | | (2,379) | | | (177) | |
| Changes in operating assets and liabilities: | | | | | |
| Accounts receivable, net | (86,550) | | | (87,939) | | | (45,597) | |
| Inventory | 13,590 | | | 18,367 | | | (34,136) | |
| Prepaid expenses and other assets | 3,229 | | | (9,029) | | | (38,507) | |
| Accounts payable, accrued expenses and other liabilities | 91,093 | | | 80,840 | | | 191,769 | |
| | | | | |
| Deferred revenue | (235,446) | | | 55,022 | | | (65,911) | |
| Net cash (used in) provided by operating activities | (8,312) | | | 104,156 | | | (541,274) | |
| Cash flows from investing activities: | | | | | |
| Purchases of property, plant and equipment | (34,277) | | | (62,211) | | | (72,059) | |
| Purchases of marketable securities | (1,634,911) | | | (1,823,501) | | | (1,976,961) | |
| Sales and maturities of marketable securities | 1,571,665 | | | 1,553,800 | | | 2,231,568 | |
| Proceeds from maturity of restricted investments | 57,875 | | | 58,475 | | | 89,951 | |
| Purchases of restricted investments | (77,075) | | | (58,475) | | | (98,451) | |
| Other investing activities | (117) | | | (4,438) | | | (4,694) | |
| Net cash (used in) provided by investing activities | (116,840) | | | (336,350) | | | 169,354 | |
| Cash flows from financing activities: | | | | | |
| Proceeds from exercise of stock options and other types of equity, net | 302,951 | | | 147,464 | | | 259,360 | |
| Proceeds from convertible debt, net | — | | | — | | | 1,016,111 | |
| | | | | |
| Purchases of capped calls related to convertible debt | — | | | — | | | (118,611) | |
| | | | | |
| (Repayment of) proceeds from development derivative liability, net | (8,792) | | | 24,667 | | | 31,000 | |
| Repayment of term loan facility | — | | | — | | | (762,107) | |
| | | | | |
| Net cash provided by financing activities | 294,159 | | | 172,131 | | | 425,753 | |
| Effect of exchange rate changes on cash, cash equivalents and restricted cash | (15,239) | | | 6,391 | | | (7,430) | |
| Net increase (decrease) in cash, cash equivalents and restricted cash | 153,768 | | | (53,672) | | | 46,403 | |
| Cash, cash equivalents and restricted cash, beginning of period | 814,884 | | | 868,556 | | | 822,153 | |
| Cash, cash equivalents and restricted cash, end of period | $ | 968,652 | | | $ | 814,884 | | | $ | 868,556 | |
| Supplemental disclosure of cash flows: | | | | | |
| Cash paid for interest | $ | 67,578 | | | $ | 32,118 | | | $ | 45,235 | |
| Cash paid for income taxes | $ | 14,360 | | | $ | 6,021 | | | $ | 2,654 | |
| Operating lease right-of-use assets obtained in exchange for new operating lease liabilities | $ | 9,309 | | | $ | 2,570 | | | $ | 1,013 | |
| Supplemental disclosure of noncash investing activities: | | | | | |
| Capital expenditures included in accounts payable and accrued expenses | $ | 2,324 | | | $ | 3,805 | | | $ | 5,213 | |
The accompanying notes are an integral part of these consolidated financial statements.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
1. NATURE OF BUSINESS
Alnylam Pharmaceuticals, Inc. (also referred to as Alnylam, the Company, we, our or us) commenced operations on June 14, 2002 as a biopharmaceutical company seeking to develop and commercialize novel therapeutics based on ribonucleic acid interference, or RNAi. We are committed to the advancement of our company strategy of building a multi-product, global, commercial biopharmaceutical company with a deep and sustainable clinical pipeline of RNAi therapeutics for future growth and a robust, organic research engine for sustainable innovation and great potential for patient impact. Since inception, we have focused on discovering, developing and commercializing RNAi therapeutics by establishing and maintaining a strong intellectual property position in the RNAi field, establishing strategic collaborations with leading pharmaceutical and life sciences companies, generating revenues through licensing agreements, and ultimately developing and commercializing RNAi therapeutics globally, either independently or with our strategic collaborators. We have devoted substantially all of our efforts to business planning, research, development, manufacturing and commercial efforts, acquiring, filing and expanding intellectual property rights, recruiting management and technical staff, and raising capital.
In early 2021, we launched our Alnylam P5x25 strategy, which focuses on our planned transition to a top-tier biotech company by the end of 2025. With Alnylam P5x25, we aim to deliver transformative therapies across a broad range of disease areas and indications for patients around the world through sustainable innovation, while delivering exceptional financial performance.
As of December 31, 2024, we have five marketed products, including one product commercialized by a collaborator, and multiple late-stage investigational programs advancing towards potential commercialization. We currently generate worldwide product revenues from four commercialized products, ONPATTRO, AMVUTTRA, GIVLAARI and OXLUMO, primarily in the United States, or U.S., and Europe.
2. SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
Basis of Presentation and Principles of Consolidation
The accompanying consolidated financial statements reflect the operations of Alnylam and our wholly owned subsidiaries. All intercompany accounts and transactions have been eliminated. Certain prior period amounts in the consolidated financial statements have been reclassified to conform to the current period presentation.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America, or GAAP, requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities as of the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. In our consolidated financial statements, we use estimates and assumptions related to our inventory valuation and related reserves, clinical accruals, liability related to the sale of future royalties, development derivative liability, income taxes, deferred tax asset valuation allowances, revenue recognition, research and development expenses, and stock-based compensation expense. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable. Actual results could differ from those estimates. Changes in estimates are reflected in reported results in the period in which they become known.
Liquidity
Based on our current operating plan, we believe that our cash, cash equivalents and marketable securities as of December 31, 2024 will be sufficient to satisfy our near-term capital and operating needs for at least the next 12 months from the filing date of this Annual Report on Form 10-K.
Concentrations of Credit Risk and Significant Customers
Financial instruments that potentially expose us to concentrations of credit risk primarily consist of cash, cash equivalents and marketable securities. As of December 31, 2024 and 2023, substantially all of our cash, cash equivalents and marketable securities were invested in money market funds, certificates of deposit, commercial paper, corporate notes, U.S. government-sponsored enterprise securities and U.S. treasury securities through highly rated financial institutions. Corporate notes may also include foreign bonds denominated in U.S. dollars. Investments are restricted, in accordance with our investment policy, to a concentration limit per issuer.
During the years ended December 31, 2024, 2023 and 2022, our revenues were generated primarily from product sales to customers and collaborations with strategic partners. As of December 31, 2024 and 2023, our gross accounts receivable balance was comprised of payments primarily due from customers for product sales and our collaborators.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The following table summarizes customers that represent 10% or greater of our consolidated total gross revenues:
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Distributor A | 29 | % | | 28 | % | | 33 | % |
| Roche | * | | 15 | % | | * |
| Regeneron Pharmaceuticals | 11 | % | | * | | * |
__________________________________________
*Represents less than 10% and/or not a customer in the applicable year
The following table summarizes customers with amounts due that represent 10% or greater of our consolidated gross accounts receivable balance:
| | | | | | | | | | | |
| As of December 31, |
| 2024 | | 2023 |
| Novartis AG | 23 | % | | 18 | % |
| Distributor A | 16 | % | | 16 | % |
| | | |
| | | |
Fair Value Measurements
The fair value is the price that would be received to sell an asset or paid to transfer a liability in an orderly transaction between market participants at the measurement date. In general, fair values determined by Level 1 inputs utilize quoted prices (unadjusted) in active markets for identical assets or liabilities. Fair values determined by Level 2 inputs utilize data points that are observable, such as quoted prices (adjusted), interest rates and yield curves. Fair values determined by Level 3 inputs utilize unobservable data points for the asset or liability, and include situations where there is little, if any, market activity for the asset or liability. The fair value hierarchy level is determined by the lowest level of significant input.
Investments in Marketable Securities and Cash Equivalents
We invest our excess cash balances in marketable debt securities and classify our investments as either trading, held-to-maturity or available-for-sale based on facts and circumstances present at the time we purchased the securities. As of December 31, 2024 and 2023, we classified all of our investments in debt securities as available-for-sale and as current assets on the consolidated balance sheets as they represent the investment of funds available for current operations. We report available-for-sale debt securities at fair value at each balance sheet date, for which fair value measurement data is obtained from independent pricing services, and include any unrealized holding gains and losses (the adjustment to fair value) in accumulated other comprehensive loss. Realized gains and losses are determined using the specific identification method and are included in other expense, net. If any adjustment to fair value reflects a decline in the value of the marketable debt securities, we consider all available evidence to evaluate if an impairment loss exists, and if so, mark the investment to market through a charge to our consolidated statements of operations and comprehensive loss. We did not record any impairment charges related to our marketable debt securities during the years ended December 31, 2024, 2023 or 2022. Our marketable debt securities are classified as cash equivalents if the original maturity, from the date of purchase, is 90 days or less, and as marketable debt securities if the original maturity, from the date of purchase, is in excess of 90 days. Our cash equivalents are generally composed of commercial paper, corporate notes, U.S. government-sponsored enterprise securities, U.S. treasury securities, money market funds and certificates of deposit.
We measure marketable equity investments (except those accounted for under the equity method of accounting or those that result in consolidation of an investee), which have readily available prices, at fair value with changes in fair value recognized in other expense, net on our consolidated statements of operations and comprehensive loss.
Accounts Receivable, Net
We record accounts receivable net of prompt payment discounts and chargebacks based on contractual terms. As of December 31, 2024 and 2023, based on our estimation of expected write-offs, we determined an allowance for doubtful accounts was not material. We have standard payment terms that generally require payment within approximately 30 to 90 days. Accounts receivable, net on our consolidated balance sheets also includes billed and unbilled collaboration receivables and royalty receivables.
Inventory
Inventory is measured at the lower of cost or estimated net realizable value and classified based on the anticipation of when it will be consumed either within our normal operating cycle (short-term) or beyond (long-term). We use a standard cost basis, which approximates cost determined on a first-in, first-out basis. Inventory costs include all raw materials, direct conversion
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
costs and overhead. Raw and intermediate materials that may be used for either research and development or commercial purposes are classified as inventory until the material is consumed or otherwise allocated for research and development. If the material is used for research and development, it is recorded as research and development expenses once that determination is made.
We capitalize inventory costs that are expected to be sold commercially once we determine it is probable that the inventory costs will be recovered through commercial sale based on the review of several factors, including (i) the likelihood that all required regulatory approvals will be received, considering any special filing status, (ii) the expected timing of validation (if not yet completed) of manufacturing processes in the associated facility, (iii) the expected expiration of the inventory, (iv) logistical or commercial constraints that may impede the timely distribution and sale of the product, including transport requirements and reimbursement status, (v) current market factors, including competitive landscape and pricing, (vi) threatened or anticipated litigation challenges, (vii) history of approvals of similar products or formulations, and (viii) FDA (or other appropriate regulatory agencies) correspondence regarding the safety and efficacy of the product. Prior to the capitalization of inventory costs, we record such costs as research and development expenses on our consolidated statements of operations and comprehensive loss.
We reduce our inventory to net realizable value for potentially excess, dated or obsolete inventory based on our quarterly assessment of the recoverability of our capitalized inventory. We periodically review inventory levels to identify what may expire prior to expected sale or has a cost basis in excess of its estimated realizable value and write down such inventories as appropriate.
Property, Plant and Equipment, Net
Property, plant and equipment are stated at cost, net of accumulated depreciation. Depreciation expense is recorded on a straight-line basis over the estimated useful life of the asset. Construction in progress reflects amounts incurred for construction or improvements of property, plant or equipment that have not been placed in service. Costs of construction of certain long-lived assets include capitalized interest, which is amortized over the estimated useful life of the related asset. The cost and accumulated depreciation of assets retired or sold are removed from the respective asset category, and any gain or loss is recognized in our consolidated statements of operations and comprehensive loss. During the years ended December 31, 2024, 2023 and 2022, we recorded $55.1 million, $51.6 million and $39.1 million, respectively, of depreciation expense related to our property, plant and equipment.
The estimated useful lives of property, plant and equipment are as follows:
| | | | | | | | |
| Asset Category | | Useful Life |
| Laboratory equipment | | 5 years |
| Computer equipment and software | | 3-10 years |
| Furniture and fixtures | | 5 years |
| Leasehold improvements | | Shorter of asset life or lease term |
| Manufacturing equipment | | 7-15 years |
| Buildings | | 40 years |
Leases
We determine if an arrangement is a lease at contract inception based on the facts and circumstances present in the arrangement. All of our leases are classified as operating leases. Operating lease right-of-use assets represent our right to use an underlying asset for the lease term and operating lease liabilities represent our obligation to make lease payments arising from the leasing arrangement. Operating lease liabilities are recognized at commencement date based on the present value of lease payments over the lease term. As most of our leases do not provide an implicit rate, in determining the operating lease liabilities, we use an estimate of our incremental borrowing rate based on the information available at commencement. Operating lease right-of-use assets are measured as the lease liability plus initial direct costs and prepaid lease payments less lease incentives. Single lease cost is recognized on a straight-line basis over the lease term.
We do not separate non-lease components from lease components for all classes of underlying assets. Leases with an initial term of 12 months or less are not recorded on the consolidated balance sheets, and the respective lease cost is recognized in the consolidated statements of operations and comprehensive loss on a straight-line basis over the term of the lease.
Clinical Accruals
We record accrued liabilities related to products we have received or services that we have incurred, specifically related to ongoing preclinical studies and clinical trials, for which service providers have not yet billed us, or when billing terms under these contracts do not coincide with the timing of when the work is performed, on our consolidated balance sheets. These costs
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
primarily relate to third-party clinical management costs, laboratory and analysis costs, toxicology studies and investigator fees. The assessment of these costs requires judgment based on our knowledge of the research and development programs, services performed for the period, experience with related activities and the expected duration of the third-party service contract, where applicable.
Revenue Recognition
We recognize revenue when control of promised goods or services is transferred to a customer at an amount that reflects the consideration to which we expect to be entitled in exchange for those goods or services. To determine revenue recognition, we perform the following five steps: (i) identify the contract(s) with a customer; (ii) identify the performance obligation(s) in the contract; (iii) determine the transaction price, including variable consideration, if any; (iv) allocate the transaction price to the performance obligation(s) in the contract; and (v) recognize revenue when (or as) we satisfy the performance obligation(s). We only apply the five-step model to contracts when collectability of the consideration to which we are entitled in exchange for the goods or services we transfer to the customer is determined to be probable.
At contract inception, once the contract is determined to be within the scope of ASC Topic 606, Revenue from Contracts with Customers, or ASC 606, we assess whether the goods or services promised within each contract are distinct and, therefore, represent a separate performance obligation. Goods and services that are determined not to be distinct are combined with other promised goods and services until a distinct bundle is identified. We then allocate the transaction price (the amount of consideration we expect to be entitled to from a customer in exchange for the promised goods or services) to each performance obligation and recognize the associated revenue when (or as) each performance obligation is satisfied. Our estimate of the transaction price for each contract includes all variable consideration to which we expect to be entitled.
Amounts are recorded as accounts receivable when our right to consideration is unconditional. We do not assess whether a contract has a significant financing component if the expectation at contract inception is that the period between payment by the customer and the transfer of the promised goods or services to the customer will be one year or less. We expense incremental costs of obtaining a contract as and when incurred if the expected amortization period of the asset that we would have recognized is one year or less or the amount is immaterial. As of December 31, 2024 and 2023, we had not capitalized any costs to obtain any of our contracts.
Net Product Revenues
Our net product revenues are recognized, net of variable consideration related to certain allowances and accruals, at the time the customer obtains control of our product. We use the expected value method, which is the sum of probability-weighted amounts in a range of possible consideration amounts, or the most likely amount method, which is the single most likely amount in a range of possible considerations, to estimate variable consideration related to our product sales. We use the expected value method to estimate variable consideration for certain rebates, chargebacks, product returns, and other incentives and we use the most likely amount method for certain rebates, trade discounts and allowances.
We record reserves, based on contractual terms, for components related to product sold during the reporting period, as well as our estimate of product that remains in the distribution channel inventory at the end of the reporting period that we expect will be sold to qualified healthcare providers. On a quarterly basis, we update our estimates and record any needed adjustments in the period we identify the adjustments. The following are the components of variable consideration related to product revenues:
Chargebacks: We estimate obligations resulting from contractual commitments with the government and other entities to sell products to qualified healthcare providers at prices lower than the list prices charged to the customer who directly purchases from us. The customer charges us for the difference between what it pays to us for the product and the selling price to the qualified healthcare providers.
Rebates: We are subject to discount obligations under government programs, including Medicaid in the U.S. and similar programs in certain other countries, including countries in which we are accruing for estimated rebates because final pricing has not yet been negotiated. We are also subject to potential rebates in connection with our value-based agreements with certain commercial payors. We record reserves for rebates in the same period the related product revenue is recognized, resulting in a reduction of product revenues and a current liability that is included in accrued expenses on our consolidated balance sheet. Our estimate for rebates is based on statutory discount rates, expected utilization or an estimated number of patients on treatment, as applicable.
Trade discounts and allowances: We provide customary invoice discounts on product sales to our customers for prompt payment and we pay fees for distribution services, such as fees for certain data that customers provide to us. We estimate our customers will earn these discounts and fees, and deduct these discounts and fees in full from gross product revenues and accounts receivable at the time we recognize the related revenues.
Product returns: We offer customers product return rights if products are damaged, defective or expired, with “expired” defined within each customer agreement. We estimate the amount of product that will be returned based on our sales history.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Other incentives: Other incentives include customer discounts, fees, and co-payment assistance we provide to patients with commercial insurance that have coverage and reside in states that allow co-payment assistance. We estimate discounts and fees based on contract terms and other relevant factors, and we estimate the average co-payment assistance amounts for our products based on expected customer demographics. We record any such amounts within accrued expenses on our consolidated balance sheets.
Net Revenues from Collaborations
We earn revenue in connection with collaboration agreements that allow our collaborators to utilize our technology platforms and develop product candidates. Our significant collaboration agreements are detailed in Note 4, Net Revenues from Collaborations. For each collaborator, we discuss our revenue recognition, including our significant performance obligations under each agreement.
At contract inception, we assess whether the collaboration arrangements are within the scope of ASC 606 or ASC Topic 808, Collaborative Arrangements, or ASC 808, to determine whether such arrangements involve joint operating activities performed by parties that are both active participants in the activities and exposed to significant risks and rewards dependent on the commercial success of such activities. This assessment is performed based on the responsibilities of all parties in the arrangement. For collaboration arrangements within the scope of ASC 808 that contain multiple elements, we first determine which elements of the arrangement are within the scope of ASC 808 and which elements are within the scope of ASC 606. For elements of collaboration arrangements that are accounted for pursuant to ASC 808, an appropriate recognition method is determined and applied consistently, either by analogy to authoritative accounting literature or by applying a reasonable and rational policy election.
For elements of collaboration arrangements that are accounted for pursuant to ASC 606, we identify the performance obligations and allocate the total consideration we expect to receive on a relative standalone selling price basis to each performance obligation. Variable consideration, such as performance-based milestones, will be included in the total consideration if we expect to receive such consideration and if it is probable that the inclusion of the variable consideration will not result in a significant reversal in the cumulative amount of revenue recognized under the arrangement. Our estimate of the total consideration we expect to receive under each collaboration arrangement is updated for each reporting period, and any adjustments to revenue are recorded on a cumulative catch-up basis. We exclude sales-based royalty and milestone payments from the total consideration we expect to receive until the underlying sales occur because the license to our intellectual property is deemed to be the predominant item to which the royalties or milestones relate as it is the primary driver of value in our collaboration arrangements.
Key assumptions to determine the standalone selling price may include forecasted revenues, development timelines, reimbursement rates for personnel costs, discount rates and probabilities of technical and regulatory success. We recognize revenue associated with each performance obligation as the control over the promised goods or services transfer to our collaborator which occurs either at a point in time or over time. If control transfers over time, revenue is recognized by using a method of measuring progress that best depicts the transfer of goods or services. We evaluate the measure of progress and related inputs each reporting period and any resulting adjustments to revenue are recorded on a cumulative catch-up basis.
Consideration received that does not meet the requirements to satisfy ASC 808 or ASC 606 revenue recognition criteria is recorded as deferred revenue in the accompanying consolidated balance sheets, classified as either short-term (less than 12 months) or long-term (more than 12 months) deferred revenue based on our best estimate of when such revenue will be recognized.
Cost of Goods Sold
Cost of goods sold includes the cost of producing and distributing inventories that are related to product revenues during the respective period (including salaries and related costs, stock-based compensation expenses and benefits for employees involved with production and distribution, supplies, external services, freight and indirect overhead costs), third-party royalties payable on our net product revenues and amortization of intangible assets associated with the sale of our products. Cost of goods sold may also include costs related to excess or obsolete inventory adjustment charges, abnormal costs, unabsorbed manufacturing and overhead costs, and manufacturing variances.
Cost of Collaborations and Royalties
Cost of collaborations and royalties includes costs we incur in connection with providing commercial drug supplies, such as GalNAc material, to collaborators, in addition to royalties we owe to third parties on the net sales of licensed products.
Income Taxes
We account for income taxes under the asset and liability method. Under this method, deferred tax assets and liabilities are recognized for the estimated future tax consequences attributable to differences between financial statement carrying amounts of existing assets and liabilities and their respective tax basis. Deferred tax assets and liabilities are measured using enacted rates in effect for the year in which these temporary differences are expected to be recovered or settled. Valuation allowances
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
are provided if, based on the weight of available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
Uncertain tax positions, for which management’s assessment is that there is a more than 50% probability of sustaining the position upon challenge by a taxing authority based upon its technical merits, are subject to certain recognition and measurement criteria. The nature of the uncertain tax positions is often very complex and subject to change, and the amounts at issue can be substantial. We develop our cumulative probability assessment of the measurement of uncertain tax positions using internal experience, judgment and assistance from professional advisors. We re-evaluate these uncertain tax positions on a quarterly basis based on a number of factors including, but not limited to, changes in facts or circumstances, changes in tax law, and effectively settled issues under audit and new audit activity. Any change in these factors could result in the recognition of a tax benefit or an additional charge to the tax provision.
Research and Development Expenses
We record research and development expenses as incurred. Included in research and development expenses are salaries and related costs, stock-based compensation expenses, benefits and other operating costs, facilities, supplies, external services, clinical trial and manufacturing costs, certain costs related to our collaboration arrangements, and overhead directly related to our research and development operations, as well as costs to acquire technology licenses.
We have entered into several license agreements for rights to utilize certain technologies. The terms of the licenses may provide for upfront payments, annual maintenance payments, milestone payments based upon certain specified events being achieved and royalties on product sales. We charge costs to acquire and maintain licensed technology that has not reached technological feasibility and does not have alternative future use to research and development expenses as incurred.
Stock-Based Compensation
We recognize stock-based compensation expense for grants under our stock incentive plans and employee stock purchase plan. We account for all stock-based awards granted to employees at their fair value and recognize compensation expense over each employee’s requisite service period, which is generally the vesting period of the award. Determining the amount of stock-based compensation to be recorded requires us to develop estimates of fair values of stock awards as of the grant date. We calculate the grant date fair values of stock options using the Black-Scholes valuation model, which requires us to make assumptions, including but not limited to expected stock price volatility over the term of the awards and the expected term of stock options. The fair value of restricted stock awards granted to employees is based upon the quoted closing market price per share on the date of grant.
We have performance conditions included in certain of our restricted stock awards that are based upon the achievement of pre-specified clinical development, regulatory, commercial and/or financial performance events. As the outcome of each event has inherent risk and uncertainties, and a positive outcome may not be known until the event is achieved, we begin to recognize the grant-date fair value of the performance-based restricted stock awards when we determine the achievement of each performance condition is deemed probable, a determination which requires significant judgment by management. On the date the performance condition is deemed probable, we record a cumulative expense catch-up, with remaining expense amortized over the remaining service period.
Advertising Expenses
We expense the costs of advertising as incurred. Advertising expenses were $29.7 million, $12.4 million and $11.7 million for the years ended December 31, 2024, 2023 and 2022, respectively.
Liability Related to the Sale of Future Royalties
We account for the liability related to the sale of future royalties as a debt financing. Interest on the liability related to the sale of future royalties is recognized using the effective interest rate method over the life of the related royalty stream.
The liability related to the sale of future royalties and the related interest expense are based on our current estimates of future royalties and commercial milestones expected to be paid over the life of the arrangement, which we determine by using third-party data to estimate Leqvio’s global net revenue. We periodically assess the expected payments and to the extent the amount or timing of our future estimated payments is materially different than our previous estimates, we account for any such change by prospectively adjusting the effective interest rate and related non-cash interest expense.
Development Derivative Liability
Development derivative liability is recorded at fair value based on the probability-weighted present value of the estimated cash flows pursuant to contractual terms of the funding agreement within accrued expenses or other liabilities on our consolidated balance sheets, depending on expected timing of our payments. The liability is remeasured quarterly with any change in fair value recorded in other expense, net on the consolidated statements of operations and comprehensive loss.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Comprehensive Loss
Comprehensive loss is comprised of net loss and certain changes in stockholders’ equity (deficit) that are excluded from net loss. Other comprehensive income (loss) includes unrealized holding gains and losses on marketable debt securities classified as available-for-sale, foreign currency translation adjustments (if the functional currency is not the U.S. dollar), and certain changes in the fair value of the plan assets and projected benefit obligations attributed to our defined benefit pension plan.
Net Income (Loss) per Common Share
We compute basic net income (loss) per common share by dividing net income (loss) by the weighted-average number of common shares outstanding. Diluted net income per common share utilizing the treasury stock and if-converted methods is based upon the weighted-average number of common shares and dilutive potential common share equivalents outstanding during the period. For periods in which we have generated a net loss, diluted net loss per common share is the same as basic net loss per common share, as the inclusion of potentially dilutive common shares would be anti-dilutive.
The following table sets forth the potential common shares (prior to consideration of the treasury stock or if-converted methods) excluded from the calculation of diluted net loss per common share because their inclusion would be anti-dilutive:
| | | | | | | | | | | | | | | | | |
| As of December 31, |
| (In thousands) | 2024 | | 2023 | | 2022 |
| Options to purchase common stock, inclusive of performance-based stock options | 5,136 | | | 7,422 | | | 8,424 | |
| Unvested restricted common stock, inclusive of performance-based restricted common stock | 2,679 | | | 2,058 | | | 1,487 | |
| Convertible debt | 3,616 | | | 3,616 | | | 3,616 | |
| Total | 11,431 | | | 13,096 | | | 13,527 | |
Recent Accounting Pronouncements
In November 2024, the Financial Accounting Standards Board, or FASB, issued ASU 2024-04, Induced Conversions of Convertible Debt Instruments, which clarifies the requirements for determining whether certain settlements of convertible debt instruments should be accounted for as an induced conversion or extinguishment of convertible debt. The standard is effective for annual reporting periods beginning after December 15, 2025, and interim periods within those annual periods. We are currently evaluating the impact of the standard on our consolidated financial statements and related disclosures.
In November 2024, the FASB issued ASU 2024-03, Disaggregation of Income Statement Expenses, which is intended to improve disclosures by requiring additional information about specific expense categories in the notes to the financial statements on an annual and interim basis. The standard will be effective for annual reporting periods beginning after December 15, 2026, and interim reporting periods beginning after December 15, 2027, with early adoption permitted. The standard updates may be applied on either a prospective or retrospective basis. We are currently evaluating the disclosure requirements related to this new standard.
In December 2023, the FASB issued ASU 2023-09, Improvements to Income Tax Disclosures, which requires entities to disclose disaggregated information about their effective tax rate reconciliation as well as expanded information on income taxes paid by jurisdiction. The disclosure requirements will be applied on a prospective basis, with the option to apply them retrospectively. The standard is effective for fiscal years beginning after December 15, 2024, with early adoption permitted. We are currently evaluating the disclosure requirements related to this new standard.
In November 2023, the FASB issued ASU 2023-07, Improvements to Reportable Segment Disclosures, which is intended to improve reportable segment disclosure requirements, primarily through additional disclosures about significant segment expenses. The standard became effective for us in the fourth quarter of 2024. Please refer to Note 16, Segment Information, for additional disclosures related to this new standard.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
3. NET PRODUCT REVENUES
Net product revenues, classified based on the geographic region in which the product is sold, consist of the following:
| | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| (In thousands) | | 2024 | | 2023 | | 2022 |
| ONPATTRO | | | | | | |
| United States | | $ | 74,787 | | | $ | 97,739 | | | $ | 246,748 | |
| Europe | | 134,197 | | | 210,916 | | | 224,063 | |
| Rest of World | | 43,873 | | | 45,891 | | | 86,797 | |
| Total | | 252,857 | | | 354,546 | | | 557,608 | |
| | | | | | |
| AMVUTTRA | | | | | | |
| United States | | 630,613 | | | 411,169 | | | 82,521 | |
| Europe | | 235,441 | | | 70,898 | | | 4,214 | |
| Rest of World | | 104,396 | | | 75,771 | | | 7,060 | |
| Total | | 970,450 | | | 557,838 | | | 93,795 | |
| | | | | | |
| GIVLAARI | | | | | | |
| United States | | 165,373 | | | 141,954 | | | 115,659 | |
| Europe | | 65,906 | | | 57,498 | | | 48,670 | |
| Rest of World | | 24,592 | | | 19,799 | | | 8,815 | |
| Total | | 255,871 | | | 219,251 | | | 173,144 | |
| | | | | | |
| OXLUMO | | | | | | |
| United States | | 62,766 | | | 38,159 | | | 27,698 | |
| Europe | | 80,753 | | | 60,025 | | | 37,915 | |
| Rest of World | | 23,531 | | | 11,655 | | | 4,169 | |
| Total | | 167,050 | | | 109,839 | | | 69,782 | |
| | | | | | |
| Total net product revenues | | $ | 1,646,228 | | | $ | 1,241,474 | | | $ | 894,329 | |
As of December 31, 2024 and 2023, net product revenue-related receivables of $269.9 million and $210.1 million, respectively, were included in accounts receivable, net on our consolidated balance sheets.
The following table summarizes balances and activity in each product revenue allowance and reserve category:
| | | | | | | | | | | | | | | | | | | | | | | |
| As of December 31, 2024 |
| (In thousands) | Chargebacks and Rebates | | Trade Discounts and Allowances | | Returns Reserve and Other Incentives | | Total |
| Beginning balance | $ | 325,672 | | | $ | 3,117 | | | $ | 17,152 | | | $ | 345,941 | |
| Provision related to current period sales | 382,892 | | | 19,454 | | | 11,750 | | | 414,096 | |
| Credit or payments made during the period for current year sales | (194,973) | | | (19,696) | | | (15,080) | | | (229,749) | |
| Credit or payments made during the period for prior year sales | (162,683) | | | (1,900) | | | (3,889) | | | (168,472) | |
| Total | $ | 350,908 | | | $ | 975 | | | $ | 9,933 | | | $ | 361,816 | |
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| | | | | | | | | | | | | | | | | | | | | | | |
| As of December 31, 2023 |
| (In thousands) | Chargebacks and Rebates | | Trade Discounts and Allowances | | Returns Reserve and Other Incentives | | Total |
| Beginning balance | $ | 191,772 | | | $ | 2,450 | | | $ | 14,776 | | | $ | 208,998 | |
| Provision related to current period sales | 367,005 | | | 18,500 | | | 19,487 | | | 404,992 | |
| Credit or payments made during the period for current year sales | (147,749) | | | (16,137) | | | (15,134) | | | (179,020) | |
| Credit or payments made during the period for prior period sales | (85,356) | | | (1,696) | | | (1,977) | | | (89,029) | |
| Total | $ | 325,672 | | | $ | 3,117 | | | $ | 17,152 | | | $ | 345,941 | |
4. NET REVENUES FROM COLLABORATIONS
Net revenues from collaborations consist of the following:
| | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| (In thousands) | | 2024 | | 2023 | | 2022 |
| Roche | | $ | 119,489 | | | $ | 337,802 | | | $ | — | |
| Regeneron Pharmaceuticals | | 302,798 | | | 100,468 | | | 87,844 | |
| Novartis AG | | 79,759 | | | 86,727 | | | 43,159 | |
| | | | | | |
| Other | | 8,175 | | | 21,188 | | | 3,909 | |
| Total | | $ | 510,221 | | | $ | 546,185 | | | $ | 134,912 | |
The following table presents the balance of our receivables and contract liabilities related to our collaboration agreements:
| | | | | | | | | | | |
| As of December 31, |
| (In thousands) | 2024 | | 2023 |
| Receivables included in accounts receivable, net | $ | 102,743 | | | $ | 99,576 | |
| Contract liabilities included in deferred revenue and deferred revenue, net of current portion | $ | 55,481 | | | $ | 290,763 | |
We recognized revenue of $266.5 million and $40.5 million in the years ended December 31, 2024 and 2023, respectively, that was included in the contract liability balance at the beginning of the applicable period.
To determine revenue recognized in the period from contract liabilities, we first allocate revenue to the individual contract liability balance outstanding at the beginning of the period until the revenue exceeds that balance. If additional consideration is received on those contracts in subsequent periods, we assume all revenue recognized in the reporting period first applies to the beginning contract liability as opposed to a portion applying to the new consideration for the period.
The following table provides research and development expenses incurred by type, for which we recognize net revenues, that are directly attributable to our collaboration agreements, by collaborator:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2024 | | 2023 | | 2022 |
| (In thousands) | Clinical Trial and Manufacturing | | External Services | | Other | | Clinical Trial and Manufacturing | | External Services | | Other | | Clinical Trial and Manufacturing | | External Services | | Other |
| Roche | $ | 83,454 | | | $ | 7,250 | | | $ | 2,021 | | | $ | 39,320 | | | $ | 2,337 | | | $ | 2,963 | | | $ | — | | | $ | — | | | $ | — | |
| Regeneron | 41,209 | | | 14,366 | | | 16,084 | | | 38,220 | | | 5,836 | | | 33,388 | | | 12,926 | | | 2,141 | | | 27,935 | |
| Other | 7,487 | | | 89 | | | 949 | | | 1,970 | | | 790 | | | 2,191 | | | 156 | | | 679 | | | 337 | |
| Total | $ | 132,150 | | | $ | 21,705 | | | $ | 19,054 | | | $ | 79,510 | | | $ | 8,963 | | | $ | 38,542 | | | $ | 13,082 | | | $ | 2,820 | | | $ | 28,272 | |
The research and development expenses incurred for the agreements included in the table above consist of costs incurred for (i) clinical and manufacturing expenses, including manufacturing of clinical and preclinical product, (ii) external services, including consulting services and lab supplies and services, and (iii) other expenses, including professional services, facilities and overhead allocations, and a reasonable estimate of compensation and related costs as billed to our counterparties, for which
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
we recognize net revenues from collaborations. For the years ended December 31, 2024, 2023 and 2022, we did not incur material selling, general and administrative expenses related to our collaboration agreements.
Product Collaborations
Roche
On July 21, 2023, or the Effective Date, we entered into a Collaboration and License Agreement, or the Roche Agreement, with F. Hoffmann-La Roche Ltd. and Genentech, Inc., or, collectively, Roche, pursuant to which we and Roche established a worldwide, strategic collaboration for the joint development of zilebesiran. Zilebesiran is our investigational small interfering RNA, or siRNA, therapeutic targeting liver-expressed angiotensinogen, which is currently in Phase 2 clinical development for the treatment of hypertension.
Under the Roche Agreement, we granted to Roche (i) co-exclusive rights to develop zilebesiran worldwide and commercialize zilebesiran in the U.S., referred to as the Co-Commercialization Territory, (ii) exclusive rights to commercialize zilebesiran outside of the U.S., referred to as the Roche Territory, and (iii) non-exclusive rights to manufacture zilebesiran for the development and commercialization of zilebesiran in the Roche Territory. In connection with the Roche Agreement, Roche made an upfront, non-refundable payment to us of $310.0 million.
We lead the global clinical development for zilebesiran. We are responsible for forty percent (40%) and Roche is responsible for the remaining sixty percent (60%) of development costs incurred in the conduct of development activities that support regulatory approval of zilebesiran globally. We and Roche share equally (50/50) all costs incurred in connection with development activities that are conducted to support regulatory approval of zilebesiran solely in the Co-Commercialization Territory if incremental development activities are needed. Roche is solely responsible for all costs incurred in the conduct of development activities that primarily support regulatory approval in the Roche Territory. Upon regulatory approval, Roche has the exclusive right to commercialize zilebesiran in the Roche Territory and will pay us tiered, low double-digit royalties based on net sales of zilebesiran on a country-by-country basis during the applicable royalty term. We and Roche will co-commercialize zilebesiran in the Co-Commercialization Territory and share equally (50/50) in profits and losses (including commercialization costs).
Roche has the right to terminate the Roche Agreement for any or no reason at all upon prior written notice; however, if the termination occurs after the achievement of the first development milestone and before the achievement of the third development milestone, Roche is required to pay us a termination fee of $50.0 million. In addition, either party may terminate the Roche Agreement for a material breach by, or insolvency of, the other party, subject to a cure period. Unless earlier terminated pursuant to its terms, the Roche Agreement will remain in effect until expiration on a country-by-country basis (a) in the Roche Territory, upon expiration of the applicable royalty term in the applicable country and (b) in the Co-Commercialization Territory, upon expiration of the term of the co-commercialization efforts.
We evaluated the Roche Agreement and concluded that the Roche Agreement had elements that were within the scope of ASC 606 and ASC 808.
As of the Effective Date, we identified the following promises in the Roche Agreement that were evaluated under the scope of ASC 606: (i) a co-exclusive license to develop zilebesiran worldwide and commercialize zilebesiran within the Co-Commercialization Territory, a non-exclusive license to manufacture zilebesiran in the Roche Territory solely for purposes of developing and commercializing zilebesiran in the Roche Territory, and an exclusive license to commercialize zilebesiran in the Roche Territory, collectively referred to as Roche License Obligation, (ii) development services, including the manufacture of clinical supply, that support regulatory approval of zilebesiran, referred to as the Roche Development Services Obligation, and (iii) a technology transfer of the existing manufacturing process for zilebesiran, referred to as the Roche Technology Transfer Obligation. The three performance obligations under the Roche Agreement are collectively referred to as the Roche Performance Obligations.
We also evaluated whether certain options outlined within the Roche Agreement represented material rights that would give rise to a performance obligation and concluded that none of the options convey a material right to Roche and therefore were not considered separate performance obligations within the Roche Agreement.
We assessed the above promises and determined that the Roche License Obligation, Roche Development Services Obligation and Roche Technology Transfer Obligation were reflective of a vendor-customer relationship and therefore represented performance obligations within the scope of ASC 606. The Roche License Obligation was considered functional intellectual property and distinct from other promises under the contract, as Roche can benefit from the licenses on its own or together with other readily available resources. As the licenses were delivered at the same time, they were considered one performance obligation at contract inception. The Roche Development Services Obligation was considered distinct as Roche could benefit from the development services together with the licenses transferred by us at the inception of the agreement. The development services are not expected to significantly modify or customize the initial intellectual property as zilebesiran was in Phase 2 of clinical development at contract inception. The Roche Technology Transfer Obligation was distinct as Roche can benefit from the manufacturing license transferred by us at the inception of the agreement given the advancements of our RNAi
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
platform and our utilization of third-party contract manufacturing organizations to manufacture zilebesiran. Therefore, each represented a separate performance obligation within the contract with a customer under the scope of ASC 606 at contract inception.
We consider the collaborative activities associated with the co-commercialization of zilebesiran in the U.S. to be a separate unit of account within the scope of ASC 808 as we and Roche are both active participants in the commercialization activities and are exposed to significant risks and rewards that are dependent on the commercial success of the activities in the arrangement.
We determined the transaction price under ASC 606 at the inception of the Roche Agreement was $857.0 million, consisting of the $310.0 million upfront payment and $547.0 million additional variable consideration attributed to cost reimbursement from development and manufacturing services and technology transfer related to the Roche Performance Obligations. We determined that any variable consideration related to development and regulatory milestones was deemed to be fully constrained at inception and therefore excluded from the initial transaction price due to the high degree of uncertainty and risk associated with these potential payments as we determined that we could not assert that it was probable that a significant reversal in the amount of cumulative revenue recognized would not occur. We also determined that royalties and sales milestones relate solely to the licenses of intellectual property and were therefore excluded from the transaction price under the sales- or usage-based royalty exception of ASC 606.
We developed the estimated standalone selling price, at inception, for each of the Roche Performance Obligations with the objective of determining the price at which we would sell such an item if it were to be sold regularly on a standalone basis. We developed the estimated standalone selling price for the Roche License Obligation primarily based on the probability-weighted present value of expected future cash flows associated with each underlying license or activity. In developing such estimates, we applied judgment in determining the forecasted revenues, taking into consideration the applicable market conditions and relevant entity-specific factors, the probability of success, the time needed to develop zilebesiran and the discount rate. We developed the estimated standalone selling price for the services and clinical supply included in the Roche Development Services Obligation and the Roche Technology Transfer Obligation primarily based on the level of efforts necessary to perform the service and the costs for full-time equivalent employees and expected resources to be committed plus a reasonable margin.
We allocated the variable consideration related to the estimated reimbursements for the Roche Development Services Obligation and the Roche Technology Transfer Obligation to each performance obligation as the terms of the variable payment relate specifically to our efforts to satisfy the performance obligation and allocating the variable amount of consideration entirely to the respective performance obligation is consistent with the allocation objective of ASC 606 when considering all of the performance obligations and payment terms in the contract. We allocated the fixed upfront consideration of $310.0 million entirely to the Roche License Obligation as the value of the fixed consideration together with the expected value of the remaining development and regulatory milestones, sales-based milestones, and royalties, all of which are either currently constrained at inception or subject to the sales- or usage-based royalty exception, approximates the standalone selling price of the Roche License Obligation. Therefore, allocating the fixed upfront consideration entirely to the Roche License Obligation is consistent with the allocation objective of ASC 606 when considering all of the performance obligations and payment terms in the contract.
The Roche License Obligation was satisfied at a point in time upon transfer of the license to Roche. Control of the licenses was transferred on the Effective Date and Roche could begin to use and benefit from the licenses. For the Roche Development Services Obligation, we measure proportional performance over time using an input method based on cost incurred relative to the total estimated cost of the obligation, on a quarterly basis, by determining the proportion of effort incurred as a percentage of total effort we expect to expend. This ratio is applied to the transaction price allocated to the obligation. As all costs in the proportional performance model are allowable for reimbursement from Roche, and the assumptions used to determine the total estimated cost of the obligation are consistent with the assumptions used to determine the transaction price allocated to the obligation, the revenue recognized for this obligation will approximate 60% of the actual reimbursable cost incurred. Management has applied significant judgment in the process of developing our estimates. We re-evaluate the transaction price as of the end of each reporting period.
As of December 31, 2024, the total transaction price was determined to be $1.31 billion, an increase of $457.5 million from December 31, 2023. The increase is due to the inclusion of the $65.0 million development milestone for the first patient dosed in the KARDIA-3 Phase 2 clinical trial in the transaction price as this milestone was achieved in 2024, as well as an increase in the level of effort necessary to perform the development services based on an updated development plan approved by the joint steering committee in October 2024 that expanded the size of the clinical trial for the development of zilebesiran.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The following tables provide a summary of the transaction price allocated to each performance obligation, in addition to revenue activity during the period, in thousands:
| | | | | | | | | | | | | | | | | | | | | | | | |
| | Transaction Price Allocated | | | | Revenue Recognized During | | |
| Performance Obligations | | As of December 31, 2024 | | | | Year Ended December 31, 2024 | | Year Ended December 31, 2023 | | |
| Roche License Obligation | | $ | 375,000 | | | | | $ | 65,000 | | | $ | 310,000 | | | |
| Roche Development Services Obligation | | 937,533 | | | | | 45,756 | | | 23,974 | | | |
| Roche Technology Transfer Obligation | | 2,000 | | | | | — | | | — | | | |
| | $ | 1,314,533 | | | | | $ | 110,756 | | | $ | 333,974 | | | |
As of December 31, 2024, the aggregate amount of the transaction price allocated to the Roche Performance Obligations that was unsatisfied was $869.8 million, which is expected to be recognized through the term of the Roche Agreement as the services are performed.
Regeneron Pharmaceuticals, Inc.
Overview
In 2019, we entered into a global, strategic collaboration with Regeneron Pharmaceuticals, Inc., or Regeneron, to discover, develop and commercialize RNAi therapeutics for a broad range of diseases by addressing therapeutic targets expressed in the eye and central nervous system, or CNS, in addition to a select number of targets expressed in the liver, which we refer to as the Regeneron Collaboration. The Regeneron Collaboration is governed by a Master Agreement, referred to as the Regeneron Master Agreement. In connection with the Regeneron Master Agreement, we and Regeneron entered into (i) a co-co collaboration agreement covering the continued development of cemdisiran, our C5 siRNA, currently in development for C5 complement-mediated diseases as a monotherapy, or the C5 Co-Co Collaboration Agreement, and (ii) a license agreement to evaluate anti-C5 antibody-siRNA combinations for C5 complement-mediated diseases including evaluating the combination of Regeneron’s pozelimab and cemdisiran, or the C5 License Agreement. The Master Agreement, the C5 Co-Co Collaboration Agreement and the C5 License Agreement were accounted for as a single arrangement because the agreements were negotiated together.
In November 2022, Regeneron exercised its right under the C5 Co-Co Collaboration Agreement to opt out of the further development and commercialization of cemdisiran monotherapy. As a result of Regeneron’s decision to opt-out, the licenses granted to Regeneron under the C5 Co-Co Collaboration Agreement reverted to us, we had the sole right to continue to develop and commercialize cemdisiran monotherapy, and Regeneron no longer shared in the costs on any monotherapy program. Regeneron remained eligible to receive tiered, double-digit royalties on net sales of cemdisiran as a monotherapy.
In June 2024, we entered into an amended and restated C5 License Agreement, or the Amended C5 License Agreement, which terminated the C5 Co-Co Collaboration Agreement and granted Regeneron a worldwide license to cemdisiran as a monotherapy in addition to the license to cemdisiran in combination with anti-C5 antibodies. Through the Amended C5 License Agreement, Regeneron is now solely responsible for development, manufacturing and commercialization of cemdisiran as a monotherapy and in combination with anti-C5 antibodies. As part of the Amended C5 License Agreement, we will provide manufacturing technology transfer services for cemdisiran to Regeneron. Regeneron provided us with an upfront payment of $10.0 million, and we will receive certain milestone payments upon receipt of regulatory approval for cemdisiran as a monotherapy, and tiered double-digit royalties on net sales. The Amended C5 License Agreement did not change our rights to receive low double-digit royalties and commercial milestones of up to $325.0 million on any potential product sales if cemdisiran is used as part of a combination product.
Under the terms of the Regeneron Collaboration, we are working exclusively with Regeneron to discover RNAi therapeutics for eye and CNS diseases for an initial research period of up to seven years, which we refer to as the Initial Research Term. Regeneron has an option to extend the Initial Research Term (referred to as the Research Term Extension Period, and together with the Initial Research Term, the Research Term) for up to an additional five years, for a research term extension fee of $300.0 million. The Regeneron Collaboration also covers a select number of RNAi therapeutic programs designed to target genes expressed in the liver.
Regeneron leads development and commercialization for all programs targeting eye diseases (subject to limited exceptions), entitling us to certain potential milestone and royalty payments pursuant to the terms of a license agreement, the form of which has been agreed upon by the parties. We and Regeneron are alternating leadership on CNS and liver programs, with the lead party retaining global development and commercial responsibility. For such CNS and liver programs, both we and Regeneron have the option at lead candidate selection to enter into a co-co collaboration agreement, the form of which has been agreed upon by the parties, whereby both companies will share equally all costs of, and profits from, all development and
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
commercialization activities under the program. If the non-lead party elects to not enter into a co-co collaboration agreement with respect to a given CNS or liver program, we and Regeneron will enter into a license agreement with respect to such program and the lead party will be the “Licensee” for the purposes of the license agreement. If the lead party for a CNS or liver program elects to not enter into the co-co collaboration agreement, then we and Regeneron will enter into a license agreement with respect to such program and leadership of the program will transfer to the other party and the former non-lead party will be the “Licensee” for the purposes of the license agreement.
In connection with the Regeneron Master Agreement, we remain eligible to receive an additional $100.0 million milestone payment upon achievement of certain criteria during early clinical development for an eye program. In addition, we and Regeneron are continuing to advance programs nominated during the Initial Research Term, and Regeneron has the right to nominate up to six additional targets per year during this period. For each of these programs, Regeneron will provide us with $2.5 million in funding at program initiation and an additional $2.5 million at lead candidate identification. If Regeneron exercises the option to extend the research term, Regeneron will retain the right to nominate up to six additional targets per year, and we will remain eligible to achieve $2.5 million in funding at each program initiation and an additional $2.5 million at each lead candidate identification during the Research Term Extension Period.
For any license agreement subsequently entered into, the licensee will generally be responsible for its own costs and expenses incurred in connection with the development and commercialization of the collaboration products. The licensee will pay to the licensor certain development and/or commercialization milestone payments totaling up to $150.0 million for each collaboration product. In addition, following the first commercial sale of the applicable collaboration product under a license agreement, the licensee is required to make certain tiered royalty payments, ranging from low double-digits up to 20%, to the licensor based on the aggregate annual net sales of the collaboration product, subject to customary reductions.
For any co-co collaboration agreement subsequently entered into, we and Regeneron will share equally all costs of, and profits from, development and commercialization activities. Reimbursement of our share of costs will be recognized as a reduction to research and development expense in the consolidated statements of operations and comprehensive loss. In the event that a party exercises its opt-out right, the lead party will be responsible for all costs and expenses incurred in connection with the development and commercialization of the collaboration products under the applicable co-co collaboration agreement, subject to continued sharing of costs through defined points. If a party exercises its opt-out right, following the first commercial sale of the applicable collaboration product under a co-co collaboration agreement, the lead party is required to make certain tiered royalty payments, ranging from low double-digits up to 20%, to the other party based on the aggregate annual net sales of the collaboration product and the timing of the exercise of the opt-out right, subject to customary reductions and a reduction for opt-out transition costs.
Contract Modification
In June 2024, we determined the Amended C5 License Agreement does not meet the requirements to account for the contract modification as a separate contract under ASC 606 because the consideration exchanged for the additional distinct goods and services does not reflect the standalone selling price. Therefore, we have accounted for the Amended C5 License Agreement and Regeneron Master Agreement as a single combined contract. The modification date was determined to be the June 2024 effective date of the Amended C5 License Agreement.
Our performance obligations subsequent to the contract modification include: (i) a research license and research services, collectively referred to as the Research Services Obligation; (ii) a worldwide license to cemdisiran for combination therapies, and manufacturing and development service obligations, collectively referred to as the C5 License Obligation; (iii) a worldwide license to cemdisiran for monotherapies, referred to as the C5 Monotherapy Obligation; and (iv) a technology transfer of the existing manufacturing process for cemdisiran, referred to as the Regeneron Technology Transfer Obligation.
The Amended C5 License Agreement did not change the Research Services Obligation or the C5 License Obligation, which were both performance obligations at the inception of our global, strategic collaboration with Regeneron prior to the contract modification. The Amended C5 License Agreement resulted in two additional performance obligations, which are the C5 Monotherapy Obligation and the Regeneron Technology Transfer Obligation. The C5 Monotherapy Obligation is considered functional intellectual property and distinct from other promises as Regeneron can benefit from the cemdisiran monotherapy license on its own or together with other readily available resources and the license is separately identifiable from the other promises in the contract. The Regeneron Technology Transfer Obligation is distinct as Regeneron can benefit from the cemdisiran monotherapy license transferred by us without the technology transfer given cemdisiran is in an advanced stage of clinical development and our utilization of third-party contract manufacturing organizations to manufacture cemdisiran. Therefore, the C5 Monotherapy Obligation and the Regeneron Technology Transfer Obligation each represent a separate performance obligation.
As of the modification date, we established a new transaction price of $329.7 million, which represents the remaining deferred revenue as of the modification date of $260.3 million, variable consideration of $59.4 million, which relates to estimated reimbursements and milestones for the Research Services Obligation, C5 License Obligation and Regeneron Technology Transfer Obligation, and the $10.0 million upfront payment related to the Amended C5 License Agreement. We
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
allocated the $59.4 million of variable consideration to the respective performance obligation as the terms of the variable payments relate specifically to our efforts to satisfy the performance obligations and allocating the variable amount of consideration entirely to the respective performance obligations is consistent with the allocation objective of ASC 606 when considering all of the performance obligations and payment terms in the contract.
We determined that any variable consideration related to regulatory milestones was deemed to be fully constrained and therefore excluded from the transaction price due to the high degree of uncertainty and risk associated with these potential payments and we determined that we could not assert that it was probable that a significant reversal in the amount of cumulative revenue recognized would not occur. We determined that royalties and sales-based milestones relate solely to the license of intellectual property and were therefore excluded from the transaction price under the sales-or usage-based royalty exception of ASC 606.
As of the modification date, the transaction price for each performance obligation is as follows, in thousands:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| Performance Obligations | | Standalone Selling Price | | Fixed Consideration | | Variable Consideration Allocated | | Transaction Price Allocated |
| Research Services Obligation | | $ | 78,820 | | | $ | 45,469 | | | $ | 30,000 | | | $ | 75,469 | |
| C5 License Obligation | | $ | 53,745 | | | 31,004 | | | 25,386 | | | 56,390 | |
| C5 Monotherapy Obligation | | $ | 332,000 | | | 191,520 | | | — | | | 191,520 | |
| Regeneron Technology Transfer Obligation | | $ | 4,000 | | | 2,307 | | | 4,000 | | | 6,307 | |
| | | | $ | 270,300 | | | $ | 59,386 | | | $ | 329,686 | |
The fixed consideration was allocated to the obligations based on the relative estimated standalone selling prices of each obligation, over which management has applied significant judgment. We developed the estimated standalone selling prices for the remaining Research Services Obligation, the remaining C5 License Obligation and the new Regeneron Technology Transfer Obligation primarily based on the levels of effort necessary to perform the services and the costs for full-time equivalent employees and expected resources to be committed plus a reasonable margin. We developed the estimated standalone selling price for the cemdisiran monotherapy license granted under the C5 Monotherapy Obligation using the adjusted market assessment approach based on a discounted cash flow model that establishes the forecasted earnings during the commercial period for cemdisiran as a monotherapy adjusted for probability of success.
The transaction price of $191.5 million allocated to the C5 Monotherapy Obligation performance obligation was recognized immediately as this obligation was satisfied at a point in time upon transfer of the license to Regeneron. Control of the license was transferred in June 2024 and Regeneron could begin to use and benefit from the license as of that time.
A cumulative catch-up adjustment was recognized for the remaining Research Services and the remaining C5 License Obligation as of the contract modification date to reflect the measure of progress and transaction price following the modification. The cumulative catch-up adjustment for the remaining Research Services and the remaining C5 License Obligation was not significant.
For the Research Services Obligation, the C5 License Obligation, and the Regeneron Technology Transfer Obligation, we measure proportional performance over time using an input method based on cost incurred relative to the total estimated costs for each of the identified obligations by determining the proportion of effort incurred as a percentage of total effort we expect to expend. This ratio is applied to the transaction price allocated to each obligation. Management has applied significant judgment in the process of developing our estimates. Any changes to these estimates will be recognized in the period in which they change as a cumulative catch-up. We re-evaluate the transaction price as of the end of each reporting period and as of December 31, 2024, the total transaction price was determined to be $168.6 million. Revenue recognized under this agreement is accounted for as collaboration revenue.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
The following tables provide a summary of the transaction price allocated, deferred revenue as of the balance sheet date, and revenue recognized during the period for the remaining unsatisfied performance obligations in thousands:
| | | | | | | | | | | | | | | | | | | | | | |
| | Transaction Price Allocated | | Deferred Revenue | | |
| Performance Obligations | | As of December 31, 2024 | | As of December 31, 2024 | | As of December 31, 2023 | | |
| Research Services Obligation | | $ | 102,969 | | | $ | 41,155 | | | $ | 63,400 | | | |
| C5 License Obligation | | 59,875 | | | 12,018 | | | 27,500 | | | |
| Regeneron Technology Transfer Obligation | | 5,759 | | | 2,307 | | | — | | | |
| Total | | $ | 168,603 | | | $ | 55,480 | | | $ | 90,900 | | | |
| | | | | | | | | | | | | | | | | | | | | | |
| | Revenue Recognized During | |
| Performance Obligations | | Year Ended December 31, 2024 | | Year Ended December 31, 2023 | | Year Ended December 31, 2022 | | |
| Research Services Obligation | | $ | 39,097 | | | $ | 80,200 | | | $ | 28,600 | | | |
| C5 License Obligation | | 38,341 | | | (15,100) | | | 32,500 | | | |
| Total | | $ | 77,438 | | | $ | 65,100 | | | $ | 61,100 | | | |
As of December 31, 2024, the aggregate amount of the transaction price allocated to the remaining Research Services Obligation, C5 License Obligation, and Regeneron Technology Transfer Obligation that was unsatisfied was $106.6 million, which is expected to be recognized through the term of the Regeneron Collaboration as the services are performed. Deferred revenue related to the Regeneron Collaboration is classified as either current or non-current in the consolidated balance sheets based on the period the revenue is expected to be recognized.
Novartis AG
2013 Collaboration with The Medicines Company
In February 2013, we entered into a license and collaboration agreement with The Medicines Company, or MDCO, pursuant to which we granted to MDCO an exclusive, worldwide license to develop, manufacture and commercialize RNAi therapeutics targeting proprotein convertase subtilisin/kexin type 9 for the treatment of hypercholesterolemia and other human diseases, including inclisiran. We refer to this agreement, as amended through the date hereof, as the MDCO License Agreement. In 2020, Novartis AG, or Novartis, completed its acquisition of MDCO and assumed all of MDCO’s rights and obligations under the MDCO License Agreement.
As of December 31, 2024, we have earned $180.0 million of milestones and we are entitled to royalties ranging from 10% up to 20% based on annual worldwide net sales of licensed products by Novartis, its affiliates and sublicensees, subject to reduction under specified circumstances.
Other
In addition to the collaboration agreements discussed above, we have various other collaboration agreements that are not individually significant to our operating results or financial condition at this time. Pursuant to the terms of those agreements, we may be required to pay, or we may receive, additional amounts contingent upon the occurrence of various future events (e.g., upon the achievement of various development and commercial milestones) which in the aggregate could be significant. We may also incur, or be reimbursed for, significant research and development costs. In addition, if any products related to these collaborations are approved for sale, we may be required to pay, or we may receive, royalties on future sales. The payment or receipt of these amounts, however, is contingent upon the occurrence of various future events. Due to the uncertainty of pharmaceutical development and the high historical failure rates generally associated with drug development and commercialization, it is possible we may not receive any such payments under all of our existing collaboration and license agreements, including the agreements described within this note.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
5. FAIR VALUE MEASUREMENTS
The following tables present information about our financial assets and liabilities that are measured at fair value on a recurring basis and indicate the fair value hierarchy of the valuation techniques we utilized to determine such fair value:
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| (In thousands) | | As of December 31,
2024 | | Quoted Prices in Active Markets
(Level 1) | | Significant Observable Inputs
(Level 2) | | Significant Unobservable Inputs
(Level 3) |
| Financial assets | | | | | | | | |
| Cash equivalents: | | | | | | | | |
| Money market funds | | $ | 190,779 | | | $ | 190,779 | | | $ | — | | | $ | — | |
| U.S. treasury securities | | 36,428 | | | — | | | 36,428 | | | — | |
| Commercial paper | | 22,709 | | | — | | | 22,709 | | | — | |
| U.S. government-sponsored enterprise securities | | 9,952 | | | — | | | 9,952 | | | — | |
| | | | | | | | |
| | | | | | | | |
| Marketable debt securities: | | | | | | | | |
| U.S. treasury securities | | 921,627 | | | — | | | 921,627 | | | — | |
| U.S. government-sponsored enterprise securities | | 396,143 | | | — | | | 396,143 | | | — | |
| Corporate notes | | 361,739 | | | — | | | 361,739 | | | — | |
| Commercial paper | | 35,408 | | | — | | | 35,408 | | | — | |
| | | | | | | | |
| Municipal securities | | 5,003 | | | — | | | 5,003 | | | — | |
| Marketable equity securities | | 8,156 | | | 8,156 | | | — | | | — | |
| Restricted cash (money market funds) | | 910 | | | 910 | | | — | | | — | |
| Total financial assets | | $ | 1,988,854 | | | $ | 199,845 | | | $ | 1,789,009 | | | $ | — | |
| Financial liabilities | | | | | | | | |
| Development derivative liability | | $ | 486,919 | | | $ | — | | | $ | — | | | $ | 486,919 | |
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| | | | | | | | | | | | | | | | | | | | | | | | | | |
| (In thousands) | | As of December 31,
2023 | | Quoted Prices in Active Markets
(Level 1) | | Significant Observable Inputs
(Level 2) | | Significant Unobservable Inputs
(Level 3) |
| Financial assets | | | | | | | | |
| Cash equivalents: | | | | | | | | |
| Money market funds | | $ | 166,059 | | | $ | 166,059 | | | $ | — | | | $ | — | |
| U.S. treasury securities | | 30,712 | | | — | | | 30,712 | | | — | |
| | | | | | | | |
| Commercial paper | | 2,685 | | | — | | | 2,685 | | | — | |
| | | | | | | | |
| Corporate notes | | 762 | | | — | | | 762 | | | — | |
| Marketable debt securities: | | | | | | | | |
| U.S. treasury securities | | 862,022 | | | — | | | 862,022 | | | — | |
| U.S. government-sponsored enterprise securities | | 441,341 | | | — | | | 441,341 | | | — | |
| Corporate notes | | 252,350 | | | — | | | 252,350 | | | — | |
| Commercial paper | | 56,216 | | | — | | | 56,216 | | | — | |
| Certificates of deposit | | 3,587 | | | — | | | 3,587 | | | — | |
| Marketable equity securities | | 11,178 | | | 11,178 | | | — | | | — | |
| Restricted cash (money market funds) | | 1,210 | | | 1,210 | | | — | | | — | |
| Total financial assets | | $ | 1,828,122 | | | $ | 178,447 | | | $ | 1,649,675 | | | $ | — | |
| Financial liabilities | | | | | | | | |
| Development derivative liability | | $ | 324,941 | | | $ | — | | | $ | — | | | $ | 324,941 | |
For the years ended December 31, 2024 and 2023, there were no transfers between Level 1 and Level 2 financial assets or liabilities. The carrying amounts reflected in our consolidated balance sheets for cash, accounts receivable, net, other current assets, accounts payable and accrued expenses approximate fair value due to their short-term maturities.
6. MARKETABLE DEBT SECURITIES
The following tables summarize our marketable debt securities:
| | | | | | | | | | | | | | | | | | | | | | | |
| As of December 31, 2024 |
| (In thousands) | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair Value |
| U.S. treasury securities | $ | 957,145 | | | $ | 1,377 | | | $ | (467) | | | $ | 958,055 | |
| U.S. government-sponsored enterprise securities | 405,890 | | | 575 | | | (370) | | | 406,095 | |
| Corporate notes | 361,311 | | | 769 | | | (341) | | | 361,739 | |
| Commercial paper | 58,117 | | | — | | | — | | | 58,117 | |
| | | | | | | |
| Municipal securities | 5,002 | | | 1 | | | — | | | 5,003 | |
| Total | $ | 1,787,465 | | | $ | 2,722 | | | $ | (1,178) | | | $ | 1,789,009 | |
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| | | | | | | | | | | | | | | | | | | | | | | |
| As of December 31, 2023 |
| (In thousands) | Amortized
Cost | | Gross
Unrealized
Gains | | Gross
Unrealized
Losses | | Fair Value |
| U.S. treasury securities | $ | 892,237 | | | $ | 1,085 | | | $ | (588) | | | $ | 892,734 | |
| U.S. government-sponsored enterprise securities | 440,915 | | | 1,000 | | | (574) | | | 441,341 | |
| Corporate notes | 252,487 | | | 945 | | | (320) | | | 253,112 | |
| Commercial paper | 58,901 | | | — | | | — | | | 58,901 | |
| Certificates of deposit | 3,587 | | | — | | | — | | | 3,587 | |
| Total | $ | 1,648,127 | | | $ | 3,030 | | | $ | (1,482) | | | $ | 1,649,675 | |
The following table summarizes classification of our marketable debt securities in the consolidated balance sheets:
| | | | | | | | | | | |
| As of December 31, |
| (In thousands) | 2024 | | 2023 |
| Cash and cash equivalents | $ | 69,089 | | | $ | 34,159 | |
| Marketable debt securities | 1,719,920 | | | 1,615,516 | |
| Total | $ | 1,789,009 | | | $ | 1,649,675 | |
7. OTHER BALANCE SHEET DETAILS
Inventory
The components of inventory are summarized as follows:
| | | | | | | | | | | |
| As of December 31, |
| (In thousands) | 2024 | | 2023 |
| Raw materials | $ | 23,965 | | | $ | 23,346 | |
| Work in process | 64,978 | | | 76,963 | |
| Finished goods | 26,433 | | | 25,123 | |
| Total inventory | $ | 115,376 | | | $ | 125,432 | |
As of December 31, 2024 and 2023, we had $36.9 million and $36.3 million of long-term inventory, respectively, included within other assets in our consolidated balance sheets as we anticipate it being consumed beyond our normal operating cycle.
Property, Plant and Equipment, Net
Property, plant and equipment, net consist of the following:
| | | | | | | | | | | |
| As of December 31, |
| (In thousands) | 2024 | | 2023 |
| Buildings | $ | 294,508 | | | $ | 271,651 | |
| Leasehold improvements | 236,686 | | | 235,411 | |
| Laboratory equipment | 121,957 | | | 107,147 | |
| Manufacturing equipment | 49,062 | | | 47,976 | |
| Computer equipment and software | 42,534 | | | 35,616 | |
| Furniture and fixtures | 11,647 | | | 12,153 | |
| Land | 9,080 | | | 9,080 | |
| Construction in progress | 7,674 | | | 30,099 | |
| 773,148 | | | 749,133 | |
| Less: accumulated depreciation | (270,364) | | | (223,076) | |
| Total | $ | 502,784 | | | $ | 526,057 | |
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Accrued Expenses
Accrued expenses consist of the following:
| | | | | | | | | | | |
| As of December 31, |
| (In thousands) | 2024 | | 2023 |
| Product rebates and discounts | $ | 361,816 | | | $ | 345,941 | |
| Compensation and related | 214,399 | | | 122,170 | |
| Preclinical, clinical trial and manufacturing | 75,815 | | | 111,503 | |
| Licensing and collaboration agreements | 92,313 | | | 58,282 | |
| Consulting and professional services | 19,974 | | | 21,155 | |
| Other | 123,155 | | | 53,962 | |
| Total | $ | 887,472 | | | $ | 713,013 | |
Cash, Cash Equivalents and Restricted Cash
The following table provides a reconciliation of cash, cash equivalents and restricted cash reported within our consolidated balance sheets to the total of these amounts shown in the consolidated statements of cash flows:
| | | | | | | | | | | | | | | | | |
| As of December 31, |
| (In thousands) | 2024 | | 2023 | | 2022 |
| Cash and cash equivalents | $ | 966,428 | | | $ | 812,688 | | | $ | 866,394 | |
| Total restricted cash included in other assets | 2,224 | | | 2,196 | | | 2,162 | |
| Total cash, cash equivalents, and restricted cash shown in the consolidated statements of cash flows | $ | 968,652 | | | $ | 814,884 | | | $ | 868,556 | |
Accumulated Other Comprehensive (Loss) Income
The following table summarizes the changes in accumulated other comprehensive (loss) income, by component:
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| (In thousands) | Loss on Investment in Joint Venture | | Defined Benefit Pension
Plans, Net of Tax | | Unrealized Gains (Losses) from Debt Securities | | Foreign Currency Translation
Adjustment | | Total Accumulated Other Comprehensive (Loss) Income |
| Balance as of December 31, 2022 | $ | (32,792) | | | $ | (1,092) | | | $ | (9,470) | | | $ | (1,300) | | | $ | (44,654) | |
| Other comprehensive (loss) income before reclassifications | — | | | — | | | (6) | | | 11,922 | | | 11,916 | |
| Amounts reclassified from other comprehensive (loss) income | — | | | (1,661) | | | 11,024 | | | — | | | 9,363 | |
| Net other comprehensive (loss) income | — | | | (1,661) | | | 11,018 | | | 11,922 | | | 21,279 | |
| Balance as of December 31, 2023 | (32,792) | | | (2,753) | | | 1,548 | | | 10,622 | | | (23,375) | |
| Other comprehensive (loss) income before reclassifications | — | | | (1,613) | | | 3 | | | (9,643) | | | (11,253) | |
| Amounts reclassified from other comprehensive (loss) income | — | | | 117 | | | (7) | | | — | | | 110 | |
| Net other comprehensive loss | — | | | (1,496) | | | (4) | | | (9,643) | | | (11,143) | |
| Balance as of December 31, 2024 | $ | (32,792) | | | $ | (4,249) | | | $ | 1,544 | | | $ | 979 | | | $ | (34,518) | |
Amounts reclassified out of accumulated other comprehensive (loss) income relate to settlements of marketable debt securities and amortization of our pension obligation which are recorded as other expense, net in the consolidated statements of operations and comprehensive loss.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
8. CONVERTIBLE DEBT
Convertible Senior Notes Due 2027
In 2022, we commenced a private offering of $900.0 million in aggregate principal amount of 1% Convertible Senior Notes due 2027, or the Initial Notes. On September 13, 2022, the initial purchasers in such offering exercised their option to purchase an additional $135.0 million in aggregate principal amount of our 1% Convertible Senior Notes due 2027, or the Additional Notes, and together with the Initial Notes collectively referred to as the Notes, bringing the total aggregate principal amount of the Notes issued and outstanding to $1.04 billion. The Notes were issued pursuant to an indenture, dated September 15, 2022, or the Indenture. The Indenture includes customary covenants and sets forth certain events of default after which the Notes may be declared immediately due and payable and sets forth certain types of bankruptcy or insolvency events of default involving the Company after which the Notes become automatically due and payable.
The Notes will mature on September 15, 2027, unless earlier converted, redeemed or repurchased. The Notes bear interest at a rate of 1% per year payable semiannually in arrears on March 15 and September 15 of each year, beginning on March 15, 2023. The Notes are convertible at the option of the noteholder on or after June 15, 2027. Prior to June 15, 2027, the Notes are convertible only under the following circumstances: (1) during any calendar quarter commencing after the calendar quarter ending on December 31, 2022 (and only during such calendar quarter), if the last reported sale price of our common stock for at least 20 trading days (whether or not consecutive) during a period of 30 consecutive trading days ending on, and including, the last trading day of the immediately preceding calendar quarter is greater than or equal to 130% of the conversion price on each applicable trading day; (2) during the five business day period after any ten consecutive trading day period in which the trading price per $1,000 principal amount of the Notes for each trading day of that ten consecutive trading day period was less than 98% of the product of the last reported sale price of our common stock and the conversion rate of the Notes on such trading day; (3) if we call any or all of the Notes for redemption; or (4) upon the occurrence of specific corporate events as set forth in the Indenture governing the Notes. We will settle any conversions of Notes by paying or delivering, as applicable, cash, shares of our common stock, or a combination of cash and shares of common stock, at our election.
The conversion rate for the Notes will initially be 3.4941 shares of common stock per $1,000 principal amount of Notes, which is equivalent to an initial conversion price of approximately $286.20 per share of common stock. The initial conversion price of the Notes represents a premium of approximately 35% over the last reported sale price of common stock of $212.00 per share on September 12, 2022. The conversion rate is subject to adjustment under certain circumstances in accordance with the terms of the Indenture.
We may not redeem the Notes prior to September 20, 2025. We may redeem for cash equal to 100% of the principal amount of the Notes being redeemed plus accrued and unpaid interest of all or any portion of the Notes, at our option, on or after September 20, 2025, if the last reported sales price of our common stock has been at least 130% of the conversion price then in effect for at least 20 trading days (whether or not consecutive) during any 30 consecutive trading day period. No sinking fund is provided for the Notes and therefore we are not required to redeem or retire the Notes periodically.
If we undergo a fundamental change, as defined in the Indenture, then subject to certain conditions, holders may require us to repurchase for cash all or any portion of their Notes at a fundamental change repurchase price equal to 100% of the principal amount of the Notes to be repurchased plus accrued and unpaid interest. In addition, if specific corporate events occur prior to the maturity date or if we issue a notice of redemption, we will increase the conversion rate by pre-defined amounts for holders who elect to convert their notes in connection with such corporate event. The conditions allowing holders of the Notes to convert were not met this quarter.
The Notes were issued at par. As of December 31, 2024 and 2023, the Notes are classified as a long-term liability on the consolidated balance sheets and presented net of unamortized issuance costs of $10.4 million and $14.2 million, respectively. The issuance costs are amortized to interest expense over the contractual term of the Notes. As of December 31, 2024 and 2023, the estimated fair value of the Notes was approximately $1.11 billion and $1.02 billion, respectively. The fair value was determined based on the last actively traded price per $100 of the Notes (Level 2) as of December 31, 2024 and 2023, respectively. As of December 31, 2024 and 2023, the effective interest rate of the Notes is 1%.
Capped Call Transactions
In 2022, in connection with the pricing of the Initial Notes and the initial purchasers’ exercise of their option to purchase the Additional Notes, we entered into privately negotiated capped call transactions, or Capped Call Transactions. The Capped Call Transactions initially cover, subject to customary anti-dilution adjustments, the number of shares of common stock that underlie the Notes. The cap price of the Capped Call Transactions is initially $424.00 per share, which represents a premium of 100% over the last reported sale price of common stock of $212.00 per share on September 12, 2022, and is subject to certain adjustments under the terms of the Capped Call Transactions.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
9. LIABILITY RELATED TO THE SALE OF FUTURE ROYALTIES
In April 2020, we entered into a purchase and sale agreement, or Purchase Agreement, with BX Bodyguard Royalties L.P. (an affiliate of The Blackstone Group Inc.), or Blackstone Royalties, pursuant to which Blackstone Royalties acquired a percentage of royalties payable, or the Royalty Interest, initially set at 50% with respect to net sales by MDCO, its affiliates or sublicensees of inclisiran (or the branded drug product, Leqvio) and any other licensed products under the MDCO License Agreement, and 75% of the commercial milestone payments payable under the MDCO License Agreement, together with the Royalty Interest, the Purchased Interest. If Blackstone Royalties does not receive payments in respect to the Royalty Interest by December 31, 2029, equaling at least $1.00 billion, Blackstone Royalties will receive the Royalty Interest at 55% beginning on January 1, 2030. In consideration for the sale of the Purchased Interest, Blackstone Royalties paid us $1.00 billion.
Due to our continuing involvement and an obligation to repay Blackstone Royalties, we record the proceeds from this transaction as a debt, net of closing costs, on our consolidated balance sheets. We account for any royalties and commercial milestones due to us under the MDCO License Agreement as revenue on our consolidated statements of operations and comprehensive loss.
In order to determine the amortization of the liability related to the sale of future royalties, we are required to estimate the total amount of future payments to Blackstone Royalties over the life of the Purchase Agreement. The $1.00 billion liability, recorded at execution of the agreement, will be accreted to the total of these royalty and commercial milestone payments as interest expense over the life of the Purchase Agreement. As of December 31, 2024 and 2023, our estimate of this total interest expense resulted in an effective annual interest rate of 10% and 8%, respectively. These estimates contain assumptions that impact both the amount recorded at execution and the interest expense that will be recognized in future periods.
As payments are made to Blackstone Royalties, the balance of the liability will be effectively repaid over the life of the Purchase Agreement. The exact timing and amount of repayment is likely to change each reporting period. A significant increase or decrease in Leqvio global net revenue will materially impact the liability related to the sale of future royalties, interest expense and the time period for repayment. We will periodically assess the expected payments to Blackstone Royalties and to the extent the amount or timing of such payments is materially different than our initial estimates, we will prospectively adjust the amortization of the liability related to the sale of future royalties and the related interest expense.
As of December 31, 2024 and 2023, the carrying value of the liability related to the sale of future royalties was $1.45 billion and $1.38 billion, net of closing costs of $9.1 million and $10.0 million, respectively. The carrying value of the liability related to the sale of future royalties approximates fair value as of December 31, 2024 and is based on our current estimates of future royalties and commercial milestones expected to be paid to Blackstone Royalties over the life of the arrangement, which are considered Level 3 inputs.
The following table shows the activity with respect to the liability related to the sale of future royalties, in thousands:
| | | | | | | | |
| Carrying value as of December 31, 2022 | | $ | 1,292,304 | |
| Interest expense recognized | | 106,554 | |
| Payments | | (21,619) | |
| Carrying value as of December 31, 2023 | | 1,377,239 | |
| Interest expense recognized | | 127,133 | |
| Payments | | (57,001) | |
| Carrying value as of December 31, 2024 | | $ | 1,447,371 | |
10. DEVELOPMENT DERIVATIVE LIABILITY
In August 2020, we entered into a co-development agreement, referred to as the Funding Agreement, with BXLS V Bodyguard – PCP L.P. and BXLS Family Investment Partnership V – ESC L.P., collectively referred to as Blackstone Life Sciences, pursuant to which Blackstone Life Sciences will provide up to $150.0 million in funding for the clinical development of vutrisiran and zilebesiran, two of our cardiometabolic programs. As of December 31, 2024, Blackstone Life Sciences has provided $70.0 million to fund vutrisiran development costs related to the HELIOS-B Phase 3 clinical trial and $26.0 million to fund Phase 2 clinical trials of zilebesiran. Furthermore, Blackstone Life Sciences has the right, but is not obligated, to fund up to $54.0 million for development costs related to a Phase 3 clinical trial of zilebesiran. The amount of funding ultimately provided by Blackstone Life Sciences for the Phase 3 clinical trial of zilebesiran is dependent on us achieving specified development milestones. As between Blackstone Life Sciences and the Company, we retain sole responsibility for the development and commercialization of both vutrisiran and zilebesiran.
As consideration for Blackstone Life Sciences’ funding for vutrisiran clinical development costs, we have agreed to pay Blackstone Life Sciences a 1% royalty on net sales of AMVUTTRA (vutrisiran) for a 10-year term beginning upon the first
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
commercial sale following regulatory approval of vutrisiran for ATTR-cardiomyopathy, as well as fixed payments of up to 2.5 times their investment over a two-year period upon regulatory approval of vutrisiran for ATTR-cardiomyopathy in specified countries, unless vutrisiran is later withdrawn from the market following a mandatory recall. As consideration for Blackstone Life Sciences’ funding for Phase 2 clinical development costs of zilebesiran, we have agreed to pay Blackstone Life Sciences fixed payments of up to 3.25 times their Phase 2 investment over a four-year period upon the successful completion of the Phase 2 clinical trial of zilebesiran, unless certain regulatory events affecting the continued development of zilebesiran occur. In September 2023, we announced positive topline results from the KARDIA-1 Phase 2 clinical trial of zilebesiran, triggering the achievement of a development milestone of $84.5 million payable to Blackstone Life Sciences in 16 equal quarterly payments over four years. As consideration for Blackstone Life Sciences’ funding for Phase 3 clinical development costs of zilebesiran, we have agreed to pay Blackstone Life Sciences fixed payments of up to 4.5 times their Phase 3 investment over a four-year period upon regulatory approval of zilebesiran in specified countries, unless it is later withdrawn from the market following a mandatory recall.
Our payment obligations under the Funding Agreement will be secured, subject to certain exceptions, by security interests in intellectual property owned by us relating to vutrisiran and zilebesiran, as well as in our bank account in which the funding deposits will be made.
We and Blackstone Life Sciences each have the right to terminate the Funding Agreement in its entirety in the event of the other party’s bankruptcy or similar proceedings. We and Blackstone Life Sciences may each terminate the Funding Agreement in its entirety or with respect to either product in the event of an uncured material breach by the other party, or with respect to a product for certain patient health and safety reasons, or if regulatory approval in specified major market countries is not obtained for the product following the completion of clinical trials for the product. In addition, Blackstone Life Sciences has the right to terminate the Funding Agreement in its entirety upon the occurrence of certain events affecting our ability to make payments under the agreement or to develop or commercialize the products, or upon a change of control of us. Blackstone Life Sciences may also terminate the Funding Agreement with respect to a product if the joint steering committee elects to terminate the development program for that product in its entirety, if certain clinical endpoints are not achieved for that product or, with respect to vutrisiran only, if our right to develop or commercialize vutrisiran is enjoined in a specified major market as a result of an alleged patent infringement. In certain termination circumstances, we will be obligated to pay Blackstone Life Sciences an amount that is equal to, or a multiplier of, the development funding received from Blackstone Life Sciences, and we may remain obligated under certain circumstances to make the payments to Blackstone Life Sciences described above, or the royalty described above in the case of AMVUTTRA, should we obtain regulatory approval for zilebesiran or vutrisiran for ATTR amyloidosis with cardiomyopathy following termination.
We account for the Funding Agreement under ASC 815, Derivatives and Hedging, as a derivative liability, measured at fair value. As of December 31, 2024 and 2023, we recorded $93.8 million and $21.1 million, respectively, within accrued expenses and $393.1 million and $303.8 million, respectively, within other liabilities on our consolidated balance sheets, based on expected timing of our payments to Blackstone Life Sciences. The change in fair value due to the remeasurement of the development derivative liability is recorded within other expense, net on our consolidated statements of operations and comprehensive loss.
As of December 31, 2024 and 2023, the derivative liability is classified as a Level 3 financial liability in the fair value hierarchy. The valuation method incorporates certain unobservable Level 3 key inputs including (i) the probability and timing of achieving stated development milestones to receive payments from Blackstone Life Sciences, (ii) the probability and timing of achieving regulatory approval and payments to Blackstone Life Sciences, (iii) an estimate of the amount and timing of the royalty payable on net sales of AMVUTTRA, assuming regulatory approval for ATTR amyloidosis with cardiomyopathy, (iv) our cost of borrowing (10%), and (v) Blackstone Life Sciences’ cost of borrowing (7%).
The following table presents the activity with respect to the development derivative liability, in thousands:
| | | | | | | | |
| Carrying value as of December 31, 2022 | | $ | 209,277 | |
| Amount received under the Funding Agreement | | 24,667 | |
Loss recorded from change in fair value | | 90,997 | |
| Carrying value as of December 31, 2023 | | 324,941 | |
| Amount received under the Funding Agreement | | 12,333 | |
| Amount paid under the Funding Agreement | | (21,125) | |
Loss recorded from change in fair value | | 170,770 | |
| Carrying value as of December 31, 2024 | | $ | 486,919 | |
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
11. STOCKHOLDERS’ EQUITY (DEFICIT)
Stock-Based Compensation
The following table summarizes stock-based compensation expenses included in operating costs and expenses:
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| (In thousands) | 2024 | | 2023 | | 2022 |
| Research and development | $ | 127,749 | | | $ | 97,273 | | | $ | 92,161 | |
| Selling, general and administrative | 144,335 | | | 124,407 | | | 138,488 | |
| Total | $ | 272,084 | | | $ | 221,680 | | | $ | 230,649 | |
The following table summarizes stock-based compensation expense by type of award:
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| (In thousands) | 2024 | | 2023 | | 2022 |
| | | | | |
| Time-based restricted stock units | $ | 108,501 | | | $ | 55,169 | | | $ | 12,791 | |
| Performance-based restricted stock units | 73,295 | | | 63,879 | | | 102,925 | |
| Time-based stock options | 85,467 | | | 99,165 | | | 114,901 | |
| Other equity programs | 8,054 | | | 7,652 | | | 4,057 | |
| Less: Stock-based compensation expense capitalized to inventory | (3,233) | | | (4,185) | | | (4,025) | |
| Total | $ | 272,084 | | | $ | 221,680 | | | $ | 230,649 | |
The following table summarizes our unrecognized stock-based compensation expense, net of estimated forfeitures, by type of awards, and the weighted-average period over which that expense is expected to be recognized:
| | | | | | | | | | | |
| As of December 31, 2024 |
| Unrecognized Expense, Net of Estimated Forfeitures (in thousands) | | Weighted-average Recognition Period (in years) |
| Type of award: | | | |
| Time-based restricted stock units | $ | 189,577 | | | 1.89 |
| Performance-based restricted stock units * | $ | 8,793 | | | 0.48 |
| Time-based stock options | $ | 66,486 | | | 1.70 |
| Other equity programs | $ | 4,021 | | | 0.51 |
__________________________________________
*Excludes performance-based restricted stock units for which the associated vesting events are not yet determined to be probable.
Time-Based Restricted Stock Units and Awards
The following table summarizes the activity of our time-based restricted stock units and awards, excluding performance-based restricted stock units:
| | | | | | | | | | | |
| Number of Units (in thousands) | | Weighted-average Grant Date Fair Value (per share) |
| Outstanding as of December 31, 2023 | 1,025 | | | $ | 185.24 | |
| Awarded | 1,261 | | | $ | 168.88 | |
| Released | (377) | | | $ | 183.73 | |
| Cancelled | (184) | | | $ | 168.50 | |
| Outstanding as of December 31, 2024 | 1,725 | | | $ | 175.39 | |
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Performance-Based Restricted Stock Units
The following table summarizes the activity of our performance-based restricted stock units:
| | | | | | | | | | | |
| Number of Units (in thousands) | | Weighted-average Grant Date Fair Value (per share) |
| Outstanding as of December 31, 2023 | 1,033 | | | $ | 165.82 | |
| Awarded | 594 | | | $ | 156.43 | |
| Released | (481) | | | $ | 149.67 | |
| Cancelled | (192) | | | $ | 155.65 | |
| Outstanding as of December 31, 2024 | 954 | | | $ | 156.56 | |
The performance-based restricted stock units granted during the years ended December 31, 2024 and 2023 will vest upon the later of the one-year anniversary of the date of grant and the achievement of specific clinical development, regulatory, commercial and/or financial performance events, as approved by our people, culture and compensation committee.
Time-Based Stock Options
The following table summarizes the activity of our time-based stock options, excluding performance-based stock options:
| | | | | | | | | | | | | | | | | | | | | | | |
| Number of Options (in thousands) | | Weighted-average Exercise Price (per share) | | Weighted-average Remaining Contractual Term (in years) | | Aggregate Intrinsic Value (in thousands) |
| Outstanding as of December 31, 2023 | 6,960 | | | $ | 127.11 | | | | | |
| Granted | 463 | | | $ | 154.14 | | | | | |
| Exercised | (2,319) | | | $ | 115.14 | | | | | |
| Cancelled | (241) | | | $ | 159.01 | | | | | |
| Outstanding as of December 31, 2024 | 4,863 | | | $ | 133.80 | | | 5.61 | | $ | 493,677 | |
| Exercisable as of December 31, 2024 | 3,702 | | | $ | 125.19 | | | 4.91 | | $ | 407,707 | |
| Vested or expected to vest as of December 31, 2024 | 4,785 | | | $ | 133.33 | | | 5.57 | | $ | 488,129 | |
The weighted-average fair value of stock options granted was $79.39, $96.53 and $80.65 per share for the years ended December 31, 2024, 2023 and 2022, respectively. The intrinsic value of stock options exercised was $273.2 million, $107.0 million and $289.3 million for the years ended December 31, 2024, 2023 and 2022, respectively. We satisfy stock option exercises with newly issued shares of our common stock.
Performance-Based Stock Options
The following table summarizes the activity of our performance-based stock options granted under our equity plans:
| | | | | | | | | | | | | | | | | | | | | | | |
| Number of Options (in thousands) | | Weighted-average Exercise Price (per share) | | Weighted-average Remaining Contractual Term (in years) | | Aggregate Intrinsic Value (in thousands) |
| Outstanding as of December 31, 2023 | 462 | | | $ | 99.16 | | | | | |
| | | | | | | |
| Exercised | (189) | | | $ | 99.03 | | | | | |
| | | | | | | |
| Outstanding as of December 31, 2024 | 273 | | | $ | 99.24 | | | 2.25 | | $ | 37,205 | |
| Exercisable as of December 31, 2024 | 273 | | | $ | 99.24 | | | 2.25 | | $ | 37,205 | |
The intrinsic value of performance-based stock options exercised was $22.5 million, $9.7 million and $74.4 million for the years ended December 31, 2024, 2023 and 2022, respectively. We satisfy performance-based stock option exercises with newly issued shares of our common stock.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Valuation Assumptions for Stock Options
The grant-date fair value of stock options was estimated using the Black-Scholes option-pricing model. Our expected stock-price volatility assumption is based on the historical volatility of our publicly traded stock. The expected life assumption is based on our historical data. The dividend yield assumption is based on the fact that we have never paid cash dividends and have no present intention to pay cash dividends. The risk-free rate for periods within the expected option life is based on the U.S. Treasury yield curve in effect at the time of grant.
The following table summarizes the Black-Scholes valuation assumption inputs for employee stock options granted:
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Risk-free interest rate | 4.2 - 4.3% | | 3.5 - 4.1% | | 1.3 - 4.2% |
| Expected dividend yield | — | | | — | | | — | |
| Expected option life | 6.0 years | | 5.1 - 7.0 years | | 5.1 - 7.0 years |
| Expected volatility | 47 - 48% | | 49 - 60% | | 50 - 60% |
Stock Plans
In May 2022, our stockholders approved the amendment and restatement of our 2018 Stock Incentive Plan, or, as amended, the Amended and Restated 2018 Plan, which increased the number of shares authorized for issuance thereunder by 6,000,000 shares. The Amended and Restated 2018 Plan provides for the granting of stock options, restricted stock and restricted stock units (together, restricted stock awards), stock appreciation rights and other stock-based awards, and has a fungible share pool. Any award that is not a full value award is counted against the authorized share limits specified as one share for each share of common stock subject to the award, and all full value awards, defined as restricted stock awards or other stock-based awards, are counted as one and a half shares for each one share of common stock subject to such full value award.
As of December 31, 2024, an aggregate of 14,010,827 shares of common stock were reserved for issuance under our stock plans, including outstanding stock options to purchase 5,135,606 shares of common stock, 2,679,429 outstanding restricted stock units, 5,728,086 shares of common stock available for additional equity awards and 467,706 shares of common stock available for future grant under our Amended and Restated 2004 Employee Stock Purchase Plan, as amended, or the Amended and Restated ESPP. Each stock option shall expire within 10 years of grant date. Time-based stock options granted to employees generally vest as to 25% of the shares on the first anniversary of the grant date and 6.25% of the shares at the end of each successive three-month period thereafter until fully vested. Restricted stock units granted to employees generally vest over a three-year period, with one-third of the shares vesting on each of the three successive anniversaries of the grant date. Vesting may be accelerated in certain circumstances, including upon death, disability, retirement or termination without cause in connection with a change of control.
Employee Stock Purchase Plan
In 2004, we adopted the 2004 Employee Stock Purchase Plan and in 2017, our stockholders approved the Amended and Restated ESPP. In 2020, our stockholders approved an amendment to the Amended and Restated ESPP, to increase the number of shares authorized for issuance to 1,965,789 shares. Under the Amended and Restated ESPP, as amended, each offering period is six months, at the end of which employees may purchase shares of common stock through payroll deductions made over the term of the offering. The per-share purchase price at the end of each offering period is equal to the lesser of 85% of the closing price of our common stock at the beginning or end of the offering period. We issued 140,161 and 108,905 shares during the years ended December 31, 2024 and 2023, respectively.
We estimate the fair value of shares to be issued under the Amended and Restated ESPP, as amended, using the Black-Scholes option-pricing model on the date of grant, or first day of the offering period. The following table summarizes the Black-Scholes valuation assumption inputs for stock purchase rights granted under the employee stock purchase plan:
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| 2024 | | 2023 | | 2022 |
| Risk-free interest rate | 4.4% - 5.4% | | 5.1% - 5.4% | | 1.4% - 4.5% |
| Expected dividend yield | — | | | — | | | — | |
| Expected option life | 6 months | | 6 months | | 6 months |
| Expected volatility | 35% - 54% | | 34% - 39% | | 53% - 71% |
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Preferred Stock
We have authorized up to 5,000,000 shares of preferred stock, $0.01 par value per share, for issuance. The preferred stock will have such rights, preferences, privileges and restrictions, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences, as shall be determined by our board of directors upon its issuance. As of December 31, 2024 and 2023, there were no shares of preferred stock outstanding.
12. LEASES
Overview of Significant Leases
We lease three facilities for office and laboratory space in Cambridge, Massachusetts that represent substantially all of our significant lease obligations. An overview of these significant leases are as follows:
675 West Kendall Street
We lease office and laboratory space located at 675 West Kendall Street, Cambridge, Massachusetts for our corporate headquarters from BMR-675 West Kendall Street, LLC under a non-cancelable real property lease. The lease commenced on May 1, 2018 and monthly rent payments became due commencing on February 1, 2019 upon substantial completion of the building improvements, and continue for 15 years, with options to renew for two five-year terms each. We are not reasonably certain to exercise these options, therefore, the periods covered by the options were not included in the lease term as of December 31, 2024 or 2023.
300 Third Street
We lease office and laboratory space located at 300 Third Street, Cambridge, Massachusetts under a non-cancelable real property lease agreement by and between us and ARE-MA Region No. 28, LLC, or ARE-MA, dated as of September 26, 2003, as amended. The term of the lease expires on January 31, 2034 with options to renew for two five-year terms each. We are not reasonably certain to exercise these options, therefore, the periods covered by the options were not included in the lease term as of December 31, 2024 or 2023.
101 Main Street
We lease office space on several floors at 101 Main Street, Cambridge, Massachusetts under non-cancelable real property lease agreements by and between us and RREEF America REIT II CORP. PPP, or RREEF, entered into in 2015. In 2020, we amended our lease agreement, pursuant to which the term of the lease with respect to two floors was extended for an additional five years, through June 2026, with an option to renew for one five-year term. We are not reasonably certain to exercise this option, therefore, the period covered by the option was not included in the lease term as of December 31, 2024 or 2023. In addition, we had a separate lease agreement for an additional floor at 101 Main Street, which expired in March 2024.
Other Lease Disclosures
Our facility leases described above generally contain customary provisions allowing the landlords to terminate the leases if we fail to remedy a breach of any of our obligations under any such lease within specified time periods, or upon our bankruptcy or insolvency.
The following table is a summary of the components of total lease cost for the years ended December 31, 2024, 2023, and 2022:
| | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| (In thousands) | | 2024 | | 2023 | | 2022 |
| Operating lease cost | | $ | 45,986 | | | $ | 46,367 | | | $ | 45,789 | |
| Variable lease cost | | 19,757 | | | 20,278 | | | 17,614 | |
| Total | | $ | 65,743 | | | $ | 66,645 | | | $ | 63,403 | |
Short-term lease costs were not material for the years ended December 31, 2024, 2023 and 2022.
Net cash paid for the amounts included in the measurement of the operating lease liabilities in our consolidated balance sheets and presented within operating activities in our consolidated statements of cash flows was $51.0 million, $46.5 million and $43.1 million for the years ended December 31, 2024, 2023 and 2022, respectively. The weighted-average remaining lease term and weighted-average discount rate for all leases as of December 31, 2024 was eight years and 8%, respectively, and as of December 31, 2023 was nine years and 8%, respectively.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Future lease payments for non-cancellable operating leases and a reconciliation to the carrying amount of the operating lease liabilities presented in the consolidated balance sheet as of December 31, 2024 were as follows, in thousands:
| | | | | |
| Years Ending December 31 | |
| 2025 | $ | 43,384 | |
| 2026 | 46,180 | |
| 2027 | 41,590 | |
| 2028 | 40,525 | |
| 2029 | 40,365 | |
| 2030 and thereafter | 172,445 | |
| Total undiscounted lease liability | 384,489 | |
| Less imputed interest | (113,062) | |
| Total discounted lease liability | $ | 271,427 | |
| |
| Current operating lease liabilities | $ | 41,886 | |
| Non-current operating lease liabilities | 229,541 | |
| Total | $ | 271,427 | |
13. COMMITMENTS AND CONTINGENCIES
Technology License and Other Commitments
We have licensed from third parties the rights to use certain technologies and information in our research processes as well as in any other products we may develop. In accordance with the related license or technology agreements, we are required to make certain fixed payments to the licensor or a designee of the licensor over various agreement terms. Many of these agreement terms are consistent with the remaining lives of the underlying intellectual property that we have licensed. As of December 31, 2024, our commitments over the next five years to make fixed and cancellable payments under existing license agreements were not material.
Legal Matters
From time to time, we may be a party to litigation, arbitration or other legal proceedings in the course of our business, including the matters described below. The claims and legal proceedings in which we could be involved include challenges to the scope, validity or enforceability of patents relating to our products or product candidates, and challenges by us to the scope, validity or enforceability of the patents held by others. These include claims by third parties that we infringe their patents or breach our license or other agreements with such third parties. The outcome of any such legal proceedings, regardless of the merits, is inherently uncertain. In addition, litigation and related matters are costly and may divert the attention of our management and other resources that would otherwise be engaged in other activities. If we were unable to prevail in any such legal proceedings, our business, results of operations, liquidity and financial condition could be adversely affected. Our accounting policy for accrual of legal costs is to recognize such expenses as incurred.
Patent Infringement Lawsuits
In March 2022, we filed separate lawsuits in the U.S. District Court for the District of Delaware against (1) Pfizer, Inc. and its subsidiary Pharmacia & Upjohn Co. LLC, collectively referred to as Pfizer, and (2) Moderna, Inc. and its subsidiaries ModernaTX, Inc., and Moderna US, Inc., collectively referred to as Moderna, seeking damages for infringement of U.S. Patent No. 11,246,933, or ‘933 Patent, in Pfizer’s and Moderna’s manufacture and sale of their messenger RNA, or mRNA, COVID-19 vaccines. The patent relates to the Company’s biodegradable cationic lipids that are foundational to the success of the mRNA COVID-19 vaccines.
We are seeking judgment that each of Pfizer and Moderna is infringing the ‘933 Patent, as well as damages adequate to compensate for the infringement, but in no event less than a reasonable royalty for the unlicensed uses made of our patented lipids by Pfizer and Moderna, together with interest and costs as may be awarded by the court. As stated in the filed complaints, we are not seeking injunctive relief in these lawsuits.
On May 23, 2022, Moderna filed a partial motion to dismiss, asserting an affirmative defense under Section 1498(a). We responded on May 27, 2022, opposing their motion arguing Moderna had significant non-government sales and the government contract ended in April 2022. Moderna responded on June 13, 2022, requesting a partial motion to dismiss those claims for sales under Section 1498(a).
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
On May 27, 2022, Pfizer filed an answer to our complaint, denying the allegations, and asserting invalidity and non-infringement defenses. In addition, Pfizer added BioNTech SE to the suit and added counter-claims seeking a declaratory judgment that our patent is invalid and a second claim alleging that our patent is invalid due to patent misuse. We responded on June 10, 2022, with substantive arguments as to the validity of our claims and the lack of merit of their patent misuse claim.
On July 12, 2022, we filed an additional lawsuit against each of Pfizer and Moderna seeking damages for infringement of U.S. Patent No. 11,382,979, or ‘979 patent, in Pfizer’s and Moderna’s manufacture and sale of their mRNA COVID-19 vaccines. The parties agreed to combine the two patents in one lawsuit, separately against each of Moderna and Pfizer/BioNTech.
On February 8, 2023, we received notification from the U.S. Patent Office that a third patent would issue on February 28, 2023, as U.S. Patent No. 11,590,229, or ‘229 patent, which we also believe Pfizer and Moderna’s COVID-19 vaccines infringe upon. On February 15, 2023, we filed a motion with the court to add this patent to the existing cases against Pfizer and Moderna, and on April 26, 2023, the court held a hearing and denied Moderna’s partial motion to dismiss those claims for sales under Section 1498(a), our motion to add the ‘229 patent to the then current lawsuits as well as a motion filed by Moderna to add certain invalidity arguments made by Pfizer in our case to supplement Moderna’s invalidity arguments previously made.
On May 26, 2023, we filed additional lawsuits against Pfizer and Moderna in Delaware seeking damages for infringing the ‘229 patent. In addition to this patent, we also alleged infringement of U.S. Patent Nos. 11,633,479, or the ‘479 patent, and 11,633,480 against both Pfizer and Moderna and of U.S. Patent No. 11,612,657 against Pfizer only.
On August 9, 2023, a Markman hearing was held in the U.S. District Court for the District of Delaware to consider the meaning of three disputed terms as used in the ’933 and ’979 patents. On August 21, 2023, the court issued an order construing two of the three terms, and deferred a ruling on the third term pending an evidentiary hearing, which was held on January 4, 2024, with the final ruling deferred pending the outcome of an additional evidentiary hearing, which was held on July 12, 2024, followed by an additional evidentiary hearing, which was held on September 19, 2024, at which time the court construed the final claim term narrowly. Following the August 21, 2023 order, we and Moderna jointly agreed to final judgment of non-infringement of two of our patents, and such judgment was entered by the court on August 30, 2023. On September 7, 2023, we appealed the claim construction ruling to the Court of Appeals for the Federal Circuit in the initial lawsuit against Moderna. The claim construction ruling initially did not affect one of the patents in the lawsuit filed against Moderna on May 26, 2023, but following a hearing held on August 15, 2024, on September 10, 2024 the court entered a ruling in the same manner as in the other earlier case and on October 2, 2024, we and Moderna jointly agreed to a final judgment of non-infringement of the ‘479 patent while preserving all rights to appeal. On October 16, 2024, Moderna filed a motion seeking recovery of fees incurred by them from the time we agreed to a judgment of non-infringement in the first case until the time we agreed to a judgment of non-infringement in the second case, which period runs from approximately September 2023 to October 2024. We opposed the motion in a reply filed on November 6, 2024. The court has not yet ruled on the motion. In light of the early stage of this matter, a loss is not probable or reasonably estimable at this time.
The two separate suits against Pfizer are ongoing and in September 2023, we and Pfizer agreed to consolidate the 2022 and 2023 lawsuits into one case with a trial date set for July 7, 2025.
On July 12, 2024, Acuitas Therapeutics, Inc., or Acuitas, filed a declaratory judgment action against us in the U.S. District Court for the District of Delaware, seeking a judgment adding certain Acuitas employees as co-inventors on the patents we have asserted against Pfizer/BioNTech and Moderna in our lawsuits. On September 19, 2024, we filed a motion to dismiss, arguing Acuitas did not have standing to sue and failed to state a claim upon which relief could be granted. The court has not yet ruled on our motion.
On December 12, 2024, The Board of Regents of the University of Texas System filed a lawsuit in the United States District Court for the Western District of Texas, alleging that we infringe U.S. Patent No. 8,895,717 by making, using and commercializing ONPATTRO in the U.S. On February 5, 2025, we filed a motion to dismiss the case for improper venue and an alternative motion to transfer the case to the United States District Court for the District of Massachusetts if the dismissal is not granted. The court has not yet ruled on these motions. In light of the early stage of this matter, a loss is not probable or reasonably estimable at this time.
Indemnifications
In connection with license agreements we may enter with companies to obtain rights to intellectual property, we may be required to indemnify such companies for certain damages arising in connection with the intellectual property rights licensed under the agreements. Under such agreements, we may be responsible for paying the costs of any litigation relating to the license agreements or the underlying intellectual property rights, including the costs associated with certain litigation regarding the licensed intellectual property. We are also a party to a number of agreements entered into in the ordinary course of business, which contain typical provisions that obligate us to indemnify the other parties to such agreements upon the occurrence of certain events, including litigation or other legal proceedings. In addition, we have agreed to indemnify our officers and directors for expenses, judgments, fines, penalties, excise taxes, and settlement amounts paid in connection with any threatened,
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
pending or completed litigation proceedings, in which an officer or director was, is or will be involved as a party, on account of such person’s status as an officer or director, or by reason of any action taken by the officer or director while acting in such capacity, subject to certain limitations. These indemnification costs are charged to selling, general and administrative expense.
Our maximum potential future liability under any such indemnification provisions is uncertain. We have reviewed the estimated aggregate fair value of our potential liabilities under all such indemnification provisions and have not recorded any related liability as of December 31, 2024 or 2023.
14. INCOME TAXES
The domestic and foreign components of loss before income taxes are as follows:
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| (In thousands) | 2024 | | 2023 | | 2022 |
| Domestic | $ | (571,926) | | | $ | (450,311) | | | $ | (1,148,604) | |
| Foreign | 194,551 | | | 16,794 | | | 21,611 | |
| Loss before income taxes | $ | (377,375) | | | $ | (433,517) | | | $ | (1,126,993) | |
During the year ended December 31, 2024, our foreign income of $194.6 million primarily relates to income generated in Switzerland. Our Switzerland foreign income was predominately generated in the second half of 2024 due to growth from sales of AMVUTTRA driven by increased patient demand.
The benefit from (provision for) income taxes consisted of the following:
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| (In thousands) | 2024 | | 2023 | | 2022 |
| Current provision: | | | | | |
| Domestic | $ | (1,600) | | | $ | (4,022) | | | $ | — | |
| Foreign | (5,944) | | | (3,416) | | | (5,596) | |
| Total current provision | (7,544) | | | (7,438) | | | (5,596) | |
| Deferred benefit: | | | | | |
| | | | | |
| Foreign | 106,762 | | | 713 | | | 1,433 | |
| Total deferred benefit | 106,762 | | | 713 | | | 1,433 | |
| Total benefit from (provision for) income taxes | $ | 99,218 | | | $ | (6,725) | | | $ | (4,163) | |
During the year ended December 31, 2024, we recorded a net benefit from income taxes of $99.2 million. This is primarily comprised of $106.8 million of foreign deferred benefit, partially offset by $1.6 million of domestic state current provision and $5.9 million of foreign current provision.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Deferred income taxes reflect the tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting and income tax purposes. We establish a valuation allowance when uncertainty exists as to whether all or a portion of the net deferred tax assets will be realized. Components of the net deferred tax assets are as follows:
| | | | | | | | | | | |
| As of December 31, |
| (In thousands) | 2024 | | 2023 |
| Deferred tax assets: | | | |
| Net operating loss carryforwards | $ | 882,569 | | | $ | 803,221 | |
| Research and development and other credit carryforwards | 391,642 | | | 417,446 | |
| Sale of future royalties | 378,657 | | | 353,974 | |
| Change in fair value of development derivative liability | 107,797 | | | 62,011 | |
| Operating lease liabilities | 60,313 | | | 66,558 | |
| Deferred revenue | 14,515 | | | 74,704 | |
| Deferred compensation | 62,110 | | | 67,150 | |
| Intangible assets | 634,323 | | | 697,784 | |
| Capitalized research and development expenditures | 349,279 | | | 285,411 | |
| Other | 86,425 | | | 69,038 | |
| Total deferred tax assets | 2,967,630 | | | 2,897,297 | |
| Deferred tax liabilities: | | | |
| Property, plant and equipment, net | (18,596) | | | (21,503) | |
| Unrealized gain on marketable securities | (2,042) | | | (2,277) | |
| Operating lease right-of-use assets | (41,196) | | | (46,021) | |
| | | |
| Deferred tax asset valuation allowance | (2,788,933) | | | (2,817,395) | |
| Net deferred tax assets | $ | 116,863 | | | $ | 10,101 | |
Our effective income tax rate differs from the statutory federal income tax rate, as follows:
| | | | | | | | | | | | | | | | | |
| Years Ended December 31, |
| (In thousands) | 2024 | | 2023 | | 2022 |
| At U.S. federal statutory rate | 21.0 | % | | 21.0 | % | | 21.0 | % |
| State taxes, net of federal effect | 10.1 | | | 8.6 | | | 6.0 | |
| Stock-based compensation | 9.6 | | | 3.9 | | | 4.4 | |
| Tax credits | 7.5 | | | 6.7 | | | 2.7 | |
| | | | | |
| Nondeductible compensation | (3.8) | | | (3.0) | | | — | |
| Other permanent items | (1.8) | | | 0.8 | | | (1.5) | |
| Foreign rate differential | 2.7 | | | (0.7) | | | (0.5) | |
| Bermuda tax law enactment | — | | | 85.9 | | | — | |
| Internal reorganization of certain intellectual property rights | (10.5) | | | 12.6 | | | — | |
| Other | (1.2) | | | 1.7 | | | (0.8) | |
| Uncertain tax position reserve | (14.3) | | | — | | | — | |
| Revaluation of deferred taxes due to rate change | (0.6) | | | 5.1 | | | (0.4) | |
| Valuation allowance | 7.6 | | | (144.1) | | | (31.3) | |
| Effective income tax rate | 26.3 | % | | (1.5) | % | | (0.4) | % |
On December 27, 2023, the Government of Bermuda enacted the Corporate Income Tax Act of 2023, or Corporate Income Tax Act, which introduces a corporate income tax regime in Bermuda with a statutory tax rate of 15% effective January 1, 2025. Companies are not subject to income tax in Bermuda prior to this change and with the transition into the Corporate Income Tax Act there is an economic transition adjustment that requires the tax basis of certain Bermudian assets to be established at fair market value. Upon the enactment of the Corporate Income Tax Act, we determined the fair market value of our identifiable intangible assets in Bermuda and recognized a deferred tax asset in our consolidated financial statements. We
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
recorded a full valuation allowance against this deferred tax asset as we have generated historical losses and expect to generate future losses. In the year ended December 31, 2024, we transferred the identifiable intangible assets in Bermuda to Switzerland.
We regularly reassess the valuation allowance on our deferred income tax assets. Valuation allowances require an assessment of both positive and negative evidence when determining whether it is more likely than not that we will be able to recover deferred tax assets. Such assessment is required on a jurisdiction-by-jurisdiction basis. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income during the periods in which those temporary differences become deductible.
We assessed the valuation allowance on our Switzerland deferred tax assets and considered positive evidence, including, among other things, income generated in Switzerland in the current year, three-year cumulative Switzerland income, and expected future profitability in Switzerland. After assessing both the positive evidence and negative evidence, we determined it was more likely than not that certain Switzerland deferred tax assets would be realized in the future and released the associated valuation allowance during the year ended December 31, 2024, which resulted in an income tax benefit of $108.0 million. As of December 31, 2024, we maintained a full valuation allowance against our U.S. deferred tax assets.
The valuation allowance decreased by $28.5 million as of December 31, 2024 compared to December 31, 2023, primarily as a result of the release of the valuation allowance on certain Switzerland deferred tax assets, which primarily consisted of the tax basis in intellectual property transferred from Bermuda and net operating losses, offset by an increase to the valuation allowance due to additional net operating losses in the U.S.
As of December 31, 2024, we had federal and state net operating loss carryforwards, or NOLs, of $3.01 billion and $3.50 billion, respectively, to reduce future taxable income. Federal NOLs of $900.0 million, generated before 2017, will begin expiring in varying amounts through 2037 unless utilized. The remaining federal NOLs of $2.11 billion, generated after 2017, will be carried forward indefinitely and could be used to offset up to 80% of taxable income in all other future tax years. State NOLs will begin expiring in varying amounts through 2044 unless utilized. As of December 31, 2024, we also had foreign NOLs of $443.4 million to reduce future taxable income which will begin expiring in varying amounts through 2030.
As of December 31, 2024, we had federal research and development, including Orphan Drug, carryforwards of $356.4 million, available to reduce future tax liabilities that expire at various dates through 2044. As of December 31, 2024, we had state research and development and investment tax credit carryforwards of $44.6 million, available to reduce future tax liabilities, that expire at various dates through 2039.
We have a full valuation allowance against our U.S. federal and state net operating loss and tax credit carryforwards, as well as a valuation allowance against certain foreign net operating loss carryforwards, as we determined it was more likely than not that we would not realize these assets. Ownership changes, as defined in the Internal Revenue Code and similar state provisions, including those resulting from the issuance of common stock in connection with our public offerings, may limit the amount of federal and state net operating loss and tax credit carryforwards that can be utilized to offset future taxable income or tax liability. The amount of the limitation is determined in accordance with Section 382 of the Internal Revenue Code and similar state provisions. We have performed an analysis of ownership changes through December 31, 2024. Based on this analysis, we do not believe that any of our federal and state tax attributes will expire unutilized due to Section 382 limitations.
Our reserves related to income taxes are based on a determination of whether, and how much of, a tax benefit taken by us in our tax filings or positions is more likely than not to be realized and ultimately sustained upon challenge by a taxing authority based upon its technical merits and subject to certain recognition and measurement criteria.
As of December 31, 2023, we did not have any material gross unrecognized tax benefits related to income tax reserves. As of December 31, 2024, we have certain gross unrecognized tax benefits related to income tax reserves primarily related to federal and state research and orphan drug credit carryforwards of $53.1 million, of which $48.5 million relate to positions taken in prior years and $4.6 million relate to current year positions. We do not have any other material gross unrecognized tax benefits related to income tax reserves. We do not expect any of our unrecognized tax benefits related to income tax reserves, if recognized, to impact our effective tax rate due to full valuation allowance in the U.S. Our policy is to record interest and penalties related to unrecognized tax benefits related to income taxes in our income tax provision. No amounts for interest or penalties related to unrecognized tax benefits have been recognized in our statements of operations and comprehensive loss for the years ended December 31, 2024, 2023 and 2022.
As of December 31, 2024, the unremitted earnings of our foreign subsidiaries are approximately $86.2 million. We have not provided for U.S. income taxes or foreign withholding taxes on these earnings as it is our current intention to permanently reinvest these earnings outside the U.S. Any tax liability on these earnings is not expected to be material. Events that could trigger a tax liability include, but are not limited to, distributions, reorganizations or restructurings and/or tax law changes.
The tax years 2021 through 2024 remain open to examination by the Internal Revenue Service and certain state tax authorities, although net operating loss and tax credit carryforwards generated prior to 2021 may still be adjusted upon examination by the Internal Revenue Service or state tax authorities if they have or will be used in a future period.
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
15. EMPLOYEE BENEFIT PLANS
We maintain a retirement saving plan under Section 401(k) of the Internal Revenue Code, in which eligible U.S. employees may defer compensation for income tax purposes. Contributions made by employees are limited to the maximum allowable for U.S. federal income tax purposes. The plan allows for a discretionary match in an amount up to 100% of each participant’s first 4% of compensation contributed plus 50% of each participant’s next 2% of compensation contributed. The expense related to our 401(k) Savings Plan primarily consists of our matching contributions.
Furthermore, we maintain defined benefit plans for employees in certain countries outside the U.S., including retirement benefit plans required by applicable local law. The benefit obligation corresponds to the projected benefit obligations of which the discounted net present value is calculated based on years of employment, expected salary increases and pension adjustments.
For the years ended December 31, 2024, 2023 and 2022 contributions and net periodic benefit costs to such plans generated a total expense of $22.3 million, $17.5 million and $16.5 million, respectively.
16. SEGMENT INFORMATION
We operate in a single segment dedicated to the discovery, development, manufacturing and commercialization of RNAi therapeutics. RNAi therapeutics are comprised of siRNA that function upstream of conventional medicines by potently silencing messenger RNA, or mRNA, that encode for proteins implicated in the cause or pathway of disease, thus preventing them from being made. To date, our efforts have yielded the approval of five first-in-class RNAi-based medicines, which generate the majority of our consolidated total revenues. Consistent with our operational structure, our Chief Executive Officer, or CEO, as the chief operating decision maker, or CODM, manages and allocates resources on a consolidated basis at the global corporate level. Our global research and development and technical operations and quality organizations are responsible for the discovery, development, and supply of products. Commercial efforts that coordinate the marketing, sales and distribution of these products are organized by geographic region and therapeutic area. All of these activities are supported by global corporate staff functions. Managing and allocating resources at the global corporate level enables our CEO to assess the overall level of resources available and how to best deploy these resources in line with our overarching long-term corporate-wide strategic goals. The determination of a single segment is consistent with the consolidated financial information regularly reviewed by the CODM for the purposes of evaluating performance, forecasting future period financial results, allocating resources and setting incentive targets.
Consistent with our management reporting, results of our operations are reported on a consolidated basis for purposes of segment reporting. The CEO evaluates performance and decides how to allocate resources based on consolidated net income (loss) that is reported on the consolidated statements of operations and comprehensive loss. The measure of segment assets is reported on the consolidated balance sheets as total assets. The CEO uses consolidated net income (loss) to evaluate income generated from the Company’s business activities in deciding how to allocate company resources (such as pursuing clinical development or entering a strategic collaboration), monitoring budget versus actual results, and establishing management’s compensation. Please refer to the consolidated financial statements for further information related to these measures of segment performance. In addition, research and development and selling, general and administrative expenses are significant segment expenses regularly provided to the CEO with the following categories:
Research and Development
| | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| (In thousands) | | 2024 | | 2023 | | 2022 |
| Clinical research and outside services | | $ | 509,129 | | | $ | 485,732 | | | $ | 438,418 | |
| Compensation and related | | 455,678 | | | 357,696 | | | 317,750 | |
Occupancy and all other costs(1) | | 161,425 | | | 160,987 | | | 126,847 | |
| Total research and development expense | | $ | 1,126,232 | | | $ | 1,004,415 | | | $ | 883,015 | |
ALNYLAM PHARMACEUTICALS, INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Selling, General and Administrative
| | | | | | | | | | | | | | | | | | | | |
| | Years Ended December 31, |
| (In thousands) | | 2024 | | 2023 | | 2022 |
| Compensation and related | | $ | 531,078 | | | $ | 423,295 | | | $ | 411,750 | |
| Consulting and professional services | | 274,539 | | | 226,664 | | | 226,941 | |
Occupancy and all other costs(1) | | 169,909 | | | 145,687 | | | 131,967 | |
| Total selling, general and administrative expense | | $ | 975,526 | | | $ | 795,646 | | | $ | 770,658 | |
(1) Occupancy and all other costs includes facilities, information technology, depreciation and certain departmental expenses.
As of December 31, 2024 and 2023, substantially all of our long-lived assets were from U.S. operations. For the years ended December 31, 2024, 2023 and 2022, net revenues from collaborations were attributed to the U.S. Please refer to Note 3, Net Product Revenues, for information regarding our net product revenues by geography.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
Our management, with the participation of our Chief Executive Officer (principal executive officer) and executive vice president, Chief Financial Officer (principal financial officer), evaluated the effectiveness of our disclosure controls and procedures as of December 31, 2024. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to our management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures. Based on the evaluation of our disclosure controls and procedures as of December 31, 2024, our Chief Executive Officer and executive vice president, Chief Financial Officer concluded that, as of such date, our disclosure controls and procedures were effective at the reasonable assurance level.
Management’s Annual Report on Internal Control Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting. Internal control over financial reporting is defined in Rule 13a-15(f) or 15d-15(f) promulgated under the Securities Exchange Act of 1934 as a process designed by, or under the supervision of, our principal executive and principal financial officers and effected by our board of directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles and includes those policies and procedures that:
•Pertain to the maintenance of records that in reasonable detail accurately and fairly reflect the transactions and dispositions of our assets;
•Provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of management and directors; and
•Provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Our management assessed the effectiveness of our internal control over financial reporting as of December 31, 2024. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control-Integrated Framework (2013).
Based on our assessment, our management concluded that, as of December 31, 2024, our internal control over financial reporting is effective based on those criteria.
The effectiveness of our internal control over financial reporting as of December 31, 2024 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in their report, which is included herein.
Changes in Internal Control
There were no changes in our internal control over financial reporting during the quarter ended December 31, 2024 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
Adoption of 10b5-1 Trading Plans by Our Officers and Directors
During our fiscal quarter ended December 31, 2024, one of our directors entered into a written plan for the sale of our securities that is intended to satisfy the conditions specified in Rule 10b5-1(c) under the Exchange Act for an affirmative
defense against liability for trading in securities on the basis of material nonpublic information. We refer to this written plan as a “Rule 10b5-1 trading plan.” We describe the material terms of the Rule 10b5-1 trading plan below.
Michael W. Bonney, Director
On November 12, 2024, Michael W. Bonney, a member of our board of directors, entered into a Rule 10b5-1 trading plan that provides that Mr. Bonney, acting through a broker, may sell up to an aggregate of 22,500 shares of our common stock received upon the exercise of options granted to Mr. Bonney as director compensation, subject to adjustments for stock splits, stock combinations, stock dividends and other similar changes to our common stock. Sales of shares under the plan may only occur if the market price of our common stock is above specified prices from February 12, 2025 to June 3, 2026. The plan is scheduled to terminate on June 3, 2026, subject to earlier termination upon the sale of all shares subject to the plan, upon termination by Mr. Bonney or the broker, or as otherwise provided in the plan.
ITEM 9C. DISCLOSURE REGARDING FOREIGN JURISDICTIONS THAT PREVENT INSPECTIONS
Not applicable.
PART III
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
Incorporated by reference from the information in our Proxy Statement for our 2025 Annual Meeting of Stockholders, or 2025 Proxy Statement, which we will file with the SEC within 120 days of the end of the fiscal year to which this Annual Report on Form 10-K relates.
We have an insider trading policy governing the purchase, sale and other dispositions of our securities that applies to all employees, including directors, officers and other covered persons, as well as the Company. We believe that our insider trading policy is reasonably designed to promote compliance with insider trading laws, rules and regulations, as well as applicable listing standards. A copy of our insider trading policy is filed as Exhibit 19.1 to this Annual Report.
ITEM 11. EXECUTIVE COMPENSATION
The information required under this item is incorporated herein by reference to the information in our 2025 Proxy Statement.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required under this item is incorporated herein by reference to the information in our 2025 Proxy Statement.
Securities Authorized for Issuance Under Equity Compensation Plans
We intend to file our 2025 Proxy Statement not later than 120 days after the close of the fiscal year ended December 31, 2024. The information required by this item relating to our equity compensation plans is incorporated herein by reference to the information contained under the section captioned “Equity Compensation Plan Information” of the Proxy Statement.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required under this item is incorporated herein by reference to the information in our 2025 Proxy Statement.
ITEM 14. PRINCIPAL ACCOUNTANT FEES AND SERVICES
The information required under this item is incorporated herein by reference to the information in our 2025 Proxy Statement.
PART IV
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
(a) (1) Financial Statements
The following consolidated financial statements are filed as part of this report under “Item 8 — Financial Statements and Supplementary Data:”
(a) (2) List of Schedules
All schedules to the consolidated financial statements are omitted as the required information is either inapplicable or presented in the consolidated financial statements.
(a) (3) List of Exhibits
| | | | | | | | |
| Exhibit No. | | Exhibit |
| | |
| | |
| 3.1 | | |
| | |
| 3.2 | | |
| | |
| 4.1 | | |
| | |
| 4.2 | | |
| | |
| 4.3 | | |
| | |
| 4.4 | | |
| | |
| 10.1** | | |
| | |
| 10.2** | | |
| | |
| 10.3** | | |
| | |
| 10.4** | | |
| | | | | | | | |
| Exhibit No. | | Exhibit |
| | |
| | |
| | |
| 10.5** | | |
| | |
| 10.6** | | |
| | |
| 10.7** | | |
| | |
| 10.8** | | |
| | |
| 10.9** | | |
| | |
| 10.10 | | |
| | |
| 10.11** | | |
| | |
| 10.12** | | |
| | |
| 10.13** | | |
| | |
| 10.14** | | |
| | |
| 10.15** | | |
| | |
| 10.16** | | |
| | |
| 10.17** | | |
| | |
| 10.18** | | |
| | |
| 10.19** | | |
| | |
| 10.20** | | |
| | |
| | | | | | | | |
| Exhibit No. | | Exhibit |
| | |
| | |
| 10.21** | | |
| | |
| 10.22** | | |
| | |
| 10.23** | | |
| | |
| 10.24** | | |
| | |
| 10.25 | | |
| | |
| 10.26 | | |
| | |
| 10.27 | | |
| | |
| 10.28 | | |
| | |
| 10.29 | | |
| | |
| 10.30 | | |
| | |
| 10.31 | | |
| | |
| 10.32† | | |
| | |
| 10.33 | | |
| | |
| 10.34 | | |
| | |
| 10.35 | | |
| | |
| | | | | | | | |
| Exhibit No. | | Exhibit |
| | |
| | |
| 10.36 | | |
| | |
| 10.37 | | |
| | |
| 10.38 | | |
| | |
| 10.39 | | |
| | |
| 10.40† | | |
| | |
| 10.41† | | |
| | |
| 10.42† | | |
| | |
| 10.43† | | |
| | |
| 10.44† | | |
| | |
| 10.45† | | |
| | |
| 10.46† | | |
| | |
| 10.47† | | |
| | |
| 10.48*† | | |
| | |
| | | | | | | | |
| Exhibit No. | | Exhibit |
| | |
| | |
| 10.49*† | | |
| | |
| 10.50*† | | |
| | |
| 10.51† | | |
| | |
| 10.52 | | |
| | |
| 10.53† | | |
| | |
| 10.54† | | |
| | |
| 10.55† | | |
| | |
| 10.56† | | |
| | |
| 10.57† | | |
| | |
| 10.58† | | |
| | |
| 10.59† | | |
| | |
| 10.60† | | |
| | |
| 10.61† | | |
| | |
| 10.62† | | |
| | |
| | | | | | | | |
| Exhibit No. | | Exhibit |
| | |
| | |
| 10.63† | | |
| | |
| 10.64† | | |
| | |
| 10.65* | | |
| | |
| 10.66*† | | |
| | |
| 10.67*† | | |
| | |
| 10.68† | | |
| | |
| 10.69† | | |
| | |
| 10.70 | | |
| | |
| 19.1# | | |
| | |
| 21.1# | | |
| | |
| 23.1# | | |
| | |
| 31.1# | | |
| | |
| 31.2# | | |
| | |
| 32.1# | | |
| | |
| 32.2# | | |
| | |
| 97 | | |
| | |
| 101.SCH# | | Inline XBRL Taxonomy Extension Schema Document |
| | |
| 101.CAL# | | Inline XBRL Taxonomy Extension Calculation Linkbase Document |
| | |
| 101.LAB# | | Inline XBRL Taxonomy Extension Label Linkbase Document |
| | |
| 101.PRE# | | Inline XBRL Taxonomy Extension Presentation Linkbase Document |
| | |
| 101.DEF# | | Inline XBRL Taxonomy Extension Definition Linkbase Document |
| | |
| | | | | | | | |
| Exhibit No. | | Exhibit |
| | |
| | |
| 104 | | Cover Page Interactive Data File (formatted as inline XBRL with applicable taxonomy extension information contained in Exhibits 101.) |
| | |
| | | | | | | | |
| * | | Schedules, exhibits and similar supporting attachments or agreements to this exhibit are omitted pursuant to Item 601(b)(2) of Regulation S-K. The Registrant agrees to furnish a supplemental copy of any omitted schedule or similar attachment to the Securities and Exchange Commission upon request. |
| ** | | Management contracts or compensatory plans or arrangements required to be filed as an exhibit hereto pursuant to Item 15(a) of Form 10-K. |
| † | | Portions of this exhibit (indicated by asterisks) have been omitted in accordance with the rules of the Securities and Exchange Commission because such information (i) is not material and (ii) would likely cause competitive harm to the Registrant if publicly disclosed. |
| # | | Filed herewith. |
ITEM 16. FORM 10-K SUMMARY
None.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this Report to be signed on its behalf by the undersigned, thereunto duly authorized, on February 13, 2025.
| | | | | | | | | | | |
| ALNYLAM PHARMACEUTICALS, INC. |
| | | |
| By: | | /s/ Yvonne L. Greenstreet, MBChB |
| | | Yvonne L. Greenstreet, MBChB |
| | | Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, the Report has been signed below by the following persons on behalf of the Registrant and in the capacities indicated as of February 13, 2025.
| | | | | | | | |
| Name | | Title |
| | |
| | |
| /s/ Yvonne L. Greenstreet, MBChB | | Director and Chief Executive Officer |
| Yvonne L. Greenstreet, MBChB | | (Principal Executive Officer) |
| | |
| /s/ Jeffrey V. Poulton | | Executive Vice President, Chief Financial Officer |
| Jeffrey V. Poulton | | (Principal Financial and Accounting Officer) |
| | |
| /s/ Dennis A. Ausiello, M.D. | | Director |
| Dennis A. Ausiello, M.D. | | |
| | |
| /s/ Carolyn R. Bertozzi, Ph.D. | | Director |
| Carolyn R. Bertozzi, Ph.D. | | |
| | |
| /s/ Michael W. Bonney | | Director |
| Michael W. Bonney | | |
| | |
| /s/ Olivier Brandicourt, M.D. | | Director |
| Olivier Brandicourt, M.D. | | |
| | |
| | |
| | |
| | |
| /s/ Margaret A. Hamburg, M.D. | | Director |
| Margaret A. Hamburg, M.D. | | |
| | |
| /s/ Peter N. Kellogg | | Director |
| Peter N. Kellogg | | |
| | |
| /s/ David E.I. Pyott | | Director |
| David E.I. Pyott | | |
| | |
| /s/ Colleen F. Reitan | | Director |
| Colleen F. Reitan | | |
| | |
| /s/ Amy W. Schulman | | Director |
| Amy W. Schulman | | |
| | |
| /s/ Phillip A. Sharp, Ph.D. | | Director |
| Phillip A. Sharp, Ph.D. | | |
| | |
| /s/ Elliott Sigal, M.D., Ph.D. | | Director |
| Elliott Sigal, M.D., Ph.D. | | |