QuickLinks -- Click here to rapidly navigate through this document
As filed with the Securities and Exchange Commission on March 1, 2007
File 333-139293
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
AMENDMENT NO. 4 TO
FORM F-1
REGISTRATION STATEMENT
UNDER THE
SECURITIES ACT OF 1933
ONCOGENEX TECHNOLOGIES INC.
(Name of Registrant as specified in its Charter)
| Canada State or other Jurisdiction of Incorporation or Organization | 2834 Primary Standard Industrial Classification Code Number | N/A I.R.S Employer Identification No | ||
1001 West Broadway, Suite 400 Vancouver, British Columbia Canada V6H 4B1 (604) 736-3678 (Address, including zip code, and telephone number, including area code, of registrant's principal executive offices) | ||||
DL Services Inc. 1420 Fifth Avenue, Suite 3400 Seattle, Washington 98101 (206) 903-8800 (Name, Address, Including Zip Code, and Telephone Number, Including Area Code, of Agent for Service) | ||||
Copies of Communications to: | |||
| Randal R. Jones Christopher L. Doerksen Dorsey & Whitney LLP 1420 Fifth Avenue, Suite 3400 Seattle, Washington 98101 (206) 903-8800 | J. Douglas Seppala DuMoulin Black LLP 10th Floor — 595 Howe Street Vancouver, BC Canada V6C 2T5 (604) 687-1224 | R. Hector MacKay-Dunn, Q.C. Farris, Vaughan, Wills & Murphy LLP 25th Floor, 700 West Georgia Street Vancouver, BC Canada V7Y 1B3 (604) 684-9151 | Christopher J. Cummings Shearman & Sterling LLP Suite 4405 P.O. Box 247 Toronto, Ontario Canada M5L 1E8 (416) 360-8484 |
Approximate date of commencement of proposed sale to the public:
As soon as practicable after this Registration Statement is declared effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. o
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration number of the earlier effective registration statement for the same offering. o
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Commission, acting pursuant to said Section 8(a), may determine.
The information in this prospectus is not complete and may be changed. These securities may not be sold until the registration statement filed with the United States Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and is not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
Subject to Completion
Preliminary Prospectus dated February 28, 2007
5,000,000 Shares

OncoGenex Technologies Inc.
Common Shares
This is our initial public offering. We are selling 5,000,000 of our common shares.
We expect the public offering price to be between $7.50 and $8.50 per common share. Currently, no public market exists for our common shares. The Nasdaq Global Market has approved the listing of our common shares under the symbol "OGXI" and the Toronto Stock Exchange has conditionally approved the listing of our common shares under the symbol "OGX".
Investing in our common shares involves risks that are described in the "Risk Factors" section beginning on page 9 of this prospectus.
| | Price to Public | Underwriting Discount | Net Proceeds to Us | ||||||
|---|---|---|---|---|---|---|---|---|---|
| Per common share | $ | $ | $ | ||||||
| Total | $ | $ | $ | ||||||
The underwriters may also purchase up to an additional 750,000 common shares from us, at the public offering price, less the underwriting discount, within 30 days from the date of this prospectus to cover overallotments.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The common shares will be ready for delivery on or about , 2007.
RBC CAPITAL MARKETS
| NEEDHAM & COMPANY, LLC | LAZARD CAPITAL MARKETS | |||||||
CANACCORD ADAMS INC. | SUSQUEHANNA FINANCIAL GROUP, LLLP | |||||||
The date of this prospectus is , 2007.
| | Page | |
|---|---|---|
| Prospectus Summary | 1 | |
| The Offering | 6 | |
| Summary Consolidated Financial Data | 7 | |
| Risk Factors | 9 | |
| Special Note Regarding Forward-Looking Statements | 34 | |
| Use of Proceeds | 35 | |
| Dividend Policy | 36 | |
| Consolidated Capitalization | 37 | |
| Dilution | 38 | |
| Selected Consolidated Financial Data | 40 | |
| Management's Discussion and Analysis of Financial Condition and Results of Operation | 42 | |
| Business | 53 | |
| Management | 94 | |
| Principal Shareholders | 116 | |
| Certain Relationships and Related Party Transactions | 120 | |
| Description of Capital Stock | 122 | |
| Shares Eligible for Future Sale | 131 | |
| Material United States and Canadian Income Tax Consequences | 135 | |
| Underwriting | 147 | |
| Legal Matters | 152 | |
| Experts | 152 | |
| Relationship between Our Company and Certain Underwriters | 152 | |
| Expenses of the Offering | 153 | |
| Where You Can Find More Information | 153 | |
| Prior Sales | 154 | |
| Reorganization | 154 | |
| Material Contracts | 154 | |
| Promoters | 155 | |
| Eligibility for Investment | 155 | |
| Purchasers' Statutory Rights | 156 | |
| Index to Consolidated Financial Statements | F-1 |
You should rely only on the information contained in this prospectus. We have not, and the underwriters have not, authorized any other person to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely on it. We are not, and the underwriters are not, making an offer to sell these securities in any jurisdiction where the offer or sale is not permitted. You should assume that the information appearing in this prospectus is accurate only as of the date on the front cover of this prospectus. Our business, financial condition, results of operations and prospects may have changed since that date.
In this prospectus, unless otherwise specified, all monetary amounts are in United States dollars. To the extent such monetary amounts have been derived from our consolidated financial statements, they have been translated into United States dollars in accordance with our accounting policies as described therein. Unless otherwise indicated, all other Canadian dollar, or C$, amounts have been translated into United States dollars at the February 23, 2007 noon buying rate published by the Federal Reserve Bank of New York, being US$1.00 = C$1.1586.
This summary does not contain all of the information you should consider before buying our common shares. You should read the entire prospectus carefully, especially the "Risk Factors" section, the "Special Note Regarding Forward-Looking Statements" section and our consolidated financial statements and the related notes appearing at the end of this prospectus, before deciding to invest in our common shares.
OncoGenex Technologies Inc.
Our Company
We are a biopharmaceutical company committed to the development and commercialization of new cancer therapies that address treatment resistance in cancer patients. Our product candidates are being developed to block the production of specific proteins associated with the development of treatment resistance, which we believe will increase survival time and improve the quality of life for cancer patients.
In response to many cancer therapies (including hormone ablation therapy, chemotherapy and radiation therapy), tumor cells become stressed and increase production of certain proteins. These proteins cause tumor cells to become resistant to the cancer therapies and promote the survival of tumor cells. Therefore, in spite of the initial effectiveness of cancer therapies, many patients develop treatment resistance and the patients die due to the lack of an effective therapy.
We currently have three product candidates in development: OGX-011, OGX-427 and OGX-225. These product candidates are designed to selectively inhibit the production of proteins that are associated with treatment resistance and that are over-produced in response to a variety of cancer treatments. Our aim in targeting these particular proteins is to disable the tumor cell's adaptive defenses, render the tumor cells susceptible to attack with a variety of cancer therapies, including chemotherapy, and facilitate tumor cell death.
Cancer is a group of diseases characterized by the uncontrolled growth and spread of abnormal cells. Cancer growth can cause tissue damage, organ failure and, ultimately, death. In North America, cancer is expected to strike one in two men and one in three women in their lifetimes and has recently surpassed heart disease as the leading cause of death in the United States. Our product candidates are currently being developed for use in the treatment of prostate, non-small cell lung, breast, ovarian and bladder cancers and multiple myeloma. Collectively, these cancers account for approximately 44 percent of the cancer deaths in the United States each year.
Product Development Programs
While we are encouraged by the results of our clinical trials to-date, we will need to conduct additional clinical trials for each of our product candidates in order to generate the safety and efficacy data needed to support an application with the FDA and regulatory agencies in other countries. Successful early clinical trials do not ensure that later clinical trials will also be successful.
Our Lead Product Candidate, OGX-011
The development program for our lead product candidate, OGX-011, is focused on reducing clusterin production to enhance treatment sensitivity and delay tumor progression in patients who
1
have not fully developed treatment resistance and to restore treatment sensitivity in patients who have developed treatment resistance. Clusterin is a cell survival protein that is over-produced in several cancer indications and in response to many cancer treatments, including hormone ablation therapy, chemotherapy and radiation therapy. Increased clusterin production is observed in many human cancers, including prostate, non-small cell lung, breast, ovarian, bladder, renal, pancreatic, anaplastic large cell lymphoma and colon cancers and melanoma. Increased clusterin production is linked to faster rates of cancer progression, treatment resistance and shorter survival duration.
A broad range of pre-clinical studies conducted by the Prostate Centre at Vancouver General Hospital, or the Prostate Centre, and others have shown that reducing clusterin production facilitates tumor cell death by sensitizing human prostate, non-small cell lung, breast, ovarian, bladder, renal, and melanoma tumor cells to chemotherapy. Pre-clinical studies conducted by the Prostate Centre and others also indicate that reducing clusterin production sensitizes prostate tumor cells to hormone ablation therapy and sensitizes prostate and non-small cell lung tumor cells to radiation therapy.
Three phase 1 clinical trials, involving a total of 75 patients, have been completed with OGX-011. In all of these clinical trials, OGX-011 was well tolerated by the patients, some of whom have experienced various adverse events, the majority of which are associated with other treatments in the protocol and the disease. The majority of adverse events were mild and the most common adverse events consisted of flu-like symptoms.
In the first phase 1 clinical trial, OGX-011 was intravenously administered once per week in combination with hormone ablation therapy to 25 patients with localized prostate cancer in advance of surgery to remove the prostate gland to evaluate safety and to determine drug concentration and clusterin inhibition in the prostate gland. Six different doses of OGX-011 were evaluated ranging from 40 mg per patient per dose to 640 mg per patient per dose. This clinical trial showed that OGX-011 reduced clusterin mRNA levels in both prostate cancer tissue and lymph node tissue in a dose dependent manner which was statistically significant (PTrend = 0.008 and PTrend<0.001, respectively) in each case. This clinical trial also showed that OGX-011 increased prostate tumor cell death in a dose dependent manner which was statistically significant (PTrend<0.001). A PTrend value of less than 0.05 was deemed statistically significant. At the 640 mg dose, OGX-011 reduced clusterin mRNA by approximately 92 percent in prostate cancer tissue and approximately 98 percent in lymph node tissue, and more than doubled the number of prostate tumor cells undergoing cell death compared to hormone ablation therapy alone. OGX-011 was well-tolerated and there were no serious adverse events reported. The most common adverse events were a mild suppression of white blood cell counts, flu-like symptoms and mild elevations in liver enzyme levels.
In the second phase 1 clinical trial, OGX-011 was intravenously administered once per week in combination with docetaxel chemotherapy to 40 patients with solid tumors known to express clusterin. Six different doses of OGX-011 were evaluated ranging from 40 mg per patient per dose to 640 mg per patient per dose. This clinical trial showed that serum clusterin levels dropped in patients while on treatment with 640 mg OGX-011 in combination with docetaxel. A statistical analysis was not performed. Ten patients experienced 13 serious adverse events. Six of the 13 serious adverse events were attributed to the combination of OGX-011 and the chemotherapy.
In the third phase 1 clinical trial, OGX-011 was intravenously administered once per week in combination with two commonly used chemotherapeutic agents to 10 patients with advanced non-small cell lung cancer. Two different doses of OGX-011 were evaluated ranging from 480 mg
2
per patient per dose to 640 mg per patient per dose. The one-year survival rate for all ten patients was 60 percent and for patients that received at least one dose of OGX-011 in combination with these chemotherapies was approximately 67 percent. This compares with results from prior published phase 3 clinical trials which reported one-year survival rates of 33 to 43 percent for patients that received chemotherapy treatment alone. Statistical analysis for treatment response was not performed. Five serious adverse events were reported in four of the 10 patients evaluated. All of the serious adverse events were attributed to the chemotherapy or disease, except for one case of elevated creatinine and one case of fever with a reduced number of cells of a specific white blood cell type which were both attributed to the combination of OGX-011 and the chemotherapeutic agents.
OGX-011 is currently in five phase 2 clinical trials investigating the potential to improve treatment outcomes for patients with prostate cancer, non-small cell lung cancer and breast cancer. We are conducting these clinical trials in parallel, rather than sequentially, to accelerate our evaluation of OGX-011 in several cancer indications and in combination with several treatment regimes, which will accelerate our assessment of these indications and combinations for further development. We plan to evaluate the results of our ongoing phase 2 clinical trials to determine which cancer indications and which treatment combinations demonstrate promise and we will design our phase 3 clinical trials accordingly. We have completed enrollment of four of our five phase 2 clinical trials and we expect that data from all five phase 2 clinical trials will be available by the end of 2007.
OGX-011 is being developed to work in combination with therapies that are broadly used by clinicians and considered highly effective in the treatment of each cancer indication that we are targeting with the intent of delaying treatment resistance to those therapies. Since production of clusterin and the resulting treatment resistance occurs in an array of cancer indications and in response to a variety of cancer treatments, we believe that our development options for OGX-011 are numerous.
Our Second Product Candidate, OGX-427
The development program for our second product candidate, OGX-427, is focused on reducing heat shock protein 27 (Hsp27) production to enhance treatment sensitivity and delay tumor progression in patients who have not fully developed treatment resistance and to restore treatment sensitivity in patients who have developed treatment resistance. Hsp27 is a cell survival protein that is over-produced in response to many cancer treatments, including hormone ablation therapy, chemotherapy and radiation therapy. Increased Hsp27 production is observed in many human cancers, including prostate, non-small cell lung, breast, ovarian, bladder, renal, pancreatic, multiple myeloma and liver cancers. Increased Hsp27 production is linked to faster rates of cancer progression, treatment resistance and shorter survival duration.
A number of pre-clinical studies conducted by the Prostate Centre and others have shown that inhibiting the production of Hsp27 in human prostate, breast, ovarian, pancreatic and bladder tumor cells sensitizes the cells to chemotherapy. The Prostate Centre has also conducted pre-clinical studies that indicate that reducing Hsp27 production sensitizes prostate tumor cells to hormone ablation therapy. Pre-clinical studies conducted by the Prostate Centre and others have shown that reducing Hsp27 production induces tumor cell death in prostate, breast, non-small cell lung, bladder and pancreatic cancers.
3
We intend to file our investigational new drug application for OGX-427 in early 2007 and begin dosing patients in our initial phase 1 clinical trial by mid-2007.
Our Third Product Candidate, OGX-225
The development program for our third product candidate, OGX-225, is focused on reducing the production of both insulin-like growth factor binding protein-2 (IGFBP-2) and insulin-like growth factor binding protein-5 (IGFBP-5) with a single product to enhance treatment sensitivity and delay tumor progression in patients who are resistant to hormone ablation therapy. In the treatment of cancers that require hormones for growth, clinicians use hormone ablation therapy to inhibit the production of the primary hormone (e.g., testosterone or estrogen) required for tumor growth. While tumors often regress initially following hormone ablation therapy, IGFBP-2 or IGFBP-5 make an alternate hormone, insulin-like growth factor-1, or IGF-1, available to the tumor that facilitates continued tumor growth. By inhibiting the production of IGFBP-2 and IGFBP-5, the tumor's access to IGF-1 is reduced and tumor growth is delayed.
Increased IGFBP-2 or IGFBP-5 production is observed in several cancers, including prostate, non-small cell lung, breast, ovarian, bladder, pancreatic and colon cancers, acute myeloid leukemia, acute lymphoblastic leukemia, neuroblastoma, glioma and melanoma. Increased IGFBP-2 or IGFBP-5 production is linked to faster rates of cancer progression, treatment resistance and shorter survival duration.
Pre-clinical studies conducted by the Prostate Centre and others in human prostate, bladder, glioma and breast cancer, have shown that reducing IGFBP-2 and IGFBP-5 production with OGX-225 induced tumor cell death or sensitized all of these tumor types to chemotherapy. We intend to continue to evaluate OGX-225 in pre-clinical studies to assess its ability to inhibit or delay progression of hormone-dependent and other tumors.
Our Approach to Treatment Resistance
We are focused on inhibiting the processes by which cancers develop treatment resistance. Our current product candidates are designed to target and selectively inhibit the production of proteins that facilitate the survival and growth of tumor cells. A protein's influence in the body can be inhibited by either interfering with its structure and function, or by disrupting its production; however, the structure of certain proteins, such as clusterin and Hsp27, have not been solved and hence are difficult to inhibit by interfering with their structure or function. For this reason, we are focusing on disrupting the production of these proteins. We have chosen to disrupt the production of these proteins using "second-generation" antisense technology because we believe that it is the most effective means of disrupting production of these proteins. To facilitate this approach, our protein production inhibitors have been combined with Isis Pharmaceuticals, Inc.'s second- generation antisense chemistry to overcome the limitations of first-generation antisense technology.
Our Strategy
- •
- Focus on developing and commercializing new cancer therapies to inhibit treatment resistance in cancer patients.
- •
- Focus on enhancing the effectiveness of broadly used cancer therapeutics through combination with OGX-011.
4
- •
- Accelerate the assessment of OGX-011 by conducting concurrent clinical trials in multiple indications.
- •
- Advance our product pipeline by conducting clinical trials across multiple cancer indications for each of OGX-427 and OGX-225.
- •
- Optimize the development of our product candidates through use of outsourcing and internal expertise.
- •
- Selectively establish collaborations that expand capabilities and accelerate development and marketing of our product candidates.
Risk Factors
Investing in our common shares involves substantial risk. In executing our business strategy we face significant risks and uncertainties, which are highlighted in the section entitled "Risk Factors". These risks include the following:
- •
- We are not profitable and have incurred significant net losses in each year since our inception. As of September 30, 2006, we had incurred cumulative losses before taxes of $19.9 million, and we expect to incur losses for the foreseeable future.
- •
- In order to successfully implement our strategy, we will need to raise substantial additional capital to support our operations for a number of years while we continue our research activities and pursue the development of, and regulatory approval for, our product candidates.
- •
- All of our product candidates are subject to regulatory approval by the U.S. Food and Drug Administration and comparable agencies in other countries. None of our product candidates have completed the expensive and lengthy clinical trials required for obtaining regulatory approval.
- •
- If we are unable to develop, receive regulatory approval for and successfully commercialize any of our product candidates, we will be unable to generate significant revenues, and we may never become profitable.
Company Information
We were incorporated under the Canada Business Corporations Act, or CBCA, on May 26, 2000 as 3766284 Canada Inc. We changed our named to OncoGenex Technologies Inc. on July 6, 2000. We have one wholly-owned subsidiary: OncoGenex, Inc., a corporation incorporated under the laws of the State of Washington. Unless the context otherwise requires, any reference to "OncoGenex", "the Company", "we", "our" and "us" in this prospectus refers to OncoGenex Technologies Inc. and our subsidiary. Our principal place of business is at 1001 West Broadway, Suite 400, Vancouver, British Columbia, V6H 4B1. Our telephone number is (604) 736-3678 and our facsimile number is (604) 736-3687. We also maintain a web site atwww.oncogenex.ca. The information contained in, or that can be accessed through, our web site is not a part of this prospectus.
5
| Common shares we are offering | 5,000,000 shares | |
Common shares to be outstanding after this offering | 16,079,738 shares | |
Use of proceeds | We estimate that our net proceeds from this offering will be $35.2 million at an assumed initial public offering price of $8.00 per share, after deducting underwriting discounts and commissions and estimated offering expenses. We plan to use the net proceeds from this offering to fund the further development and expansion of our product candidates and other working capital and general corporate purposes. See "Use of Proceeds". | |
Nasdaq Global Market symbol | "OGXI" | |
Toronto Stock Exchange symbol | "OGX" |
The number of our common shares to be outstanding immediately after this offering is based on 11,079,738 common shares outstanding as of February 28, 2007 after giving effect to the conversion of all of our outstanding classes and series of preferred shares as described under the heading "Reorganization", and excludes:
- •
- 1,423,147 common shares issuable upon the exercise of stock options outstanding, with a weighted average exercise price of C$0.92 per share; and
- •
- 482,410 common shares reserved for future stock option grants under our 2006 Stock Incentive Plan.
Except as otherwise noted, all of the information presented in this prospectus is based on the assumption that the Reorganization has been implemented and that the underwriters will not have exercised their overallotment option. In addition, all historical share numbers have been adjusted retroactively to give effect to the one-for-five share consolidation we completed on September 23, 2003. See "Description of Capital Stock".
6
SUMMARY CONSOLIDATED FINANCIAL DATA
In the table below we provide a summary of our historical financial data. We have prepared this data using our audited consolidated financial statements for the years ended December 31, 2001, 2002, 2003, 2004 and 2005 and our unaudited interim consolidated financial statements for the nine months ended September 30, 2005 and 2006 and for the period from May 26, 2000 (inception) to September 30, 2006. You should read this data together with our consolidated financial statements, including the related notes, and "Management's Discussion and Analysis of Financial Condition and Results of Operations" included elsewhere in this prospectus. Our consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States. The summary consolidated balance sheet data is presented on an actual basis and on an as adjusted basis to reflect the sale of the 5,000,000 common shares offered at an assumed initial public offering price of $8.00 per share, after deducting underwriting discounts and commissions and estimated offering expenses and the conversion of all our series preferred shares into an aggregate of 9,794,238 common shares pursuant to the Reorganization.
| | | | | | | Nine Months Ended September 30, | Period from May 26, 2000 (Inception) to September 30 2006 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Year Ended December 31, | |||||||||||||||||||||||||
| | 2005 | 2004 | 2003 | 2002 | 2001 | 2006 | 2005 | |||||||||||||||||||
| | (In thousands, except share and per share amounts) | |||||||||||||||||||||||||
| Consolidated Statements of Loss Data | ||||||||||||||||||||||||||
| Operating expenses: | ||||||||||||||||||||||||||
| Research and development | $ | 3,143 | $ | 2,778 | $ | 1,381 | $ | 1,124 | $ | 253 | $ | 6,822 | $ | 1,857 | $ | 15,502 | ||||||||||
| General and administrative (1) | 1,523 | 930 | 487 | 249 | 65 | 1,553 | 1,128 | 4,813 | ||||||||||||||||||
| Total expenses | 4,666 | 3,708 | 1,868 | 1,373 | 318 | 8,375 | 2,985 | 20,315 | ||||||||||||||||||
| Other income (expense): | ||||||||||||||||||||||||||
| Interest income | 313 | 199 | 41 | 17 | — | 357 | 160 | 928 | ||||||||||||||||||
| Other | (144 | ) | (156 | ) | (99 | ) | — | — | (111 | ) | (139 | ) | (510 | ) | ||||||||||||
| Total other income (expense) | 169 | 43 | (58 | ) | 17 | — | 246 | 21 | 418 | |||||||||||||||||
| Loss for the period before taxes | $ | 4,497 | $ | 3,665 | $ | 1,926 | $ | 1,356 | $ | 318 | $ | 8,129 | $ | 2,964 | $ | 19,897 | ||||||||||
| Tax Expense | $ | 432 | $ | 346 | $ | — | $ | — | $ | — | $ | 473 | $ | 274 | $ | 1,251 | ||||||||||
| Net Loss | $ | 4,929 | $ | 4,011 | $ | 1,926 | $ | 1,356 | $ | 318 | $ | 8,602 | $ | 3,238 | $ | 21,148 | ||||||||||
| Redeemable convertible preferred share accretion | $ | 1,843 | $ | 1,248 | $ | 385 | $ | 129 | $ | 3 | $ | 1,912 | $ | 1,202 | $ | 5,520 | ||||||||||
| Loss attributable to common shareholders | $ | 6,772 | $ | 5,259 | $ | 2,311 | $ | 1,485 | $ | 321 | $ | 10,514 | $ | 4,440 | $ | 26,668 | ||||||||||
| Basic and diluted loss per common share | $ | 5.43 | $ | 4.55 | $ | 2.01 | $ | 1.46 | $ | 0.35 | $ | 8.18 | $ | 3.59 | $ | 25.19 | ||||||||||
| Weighted average number of common shares | 1,248,158 | 1,155,500 | 1,148,925 | 1,018,958 | 906,050 | 1,285,500 | 1,235,573 | 1,058,755 | ||||||||||||||||||
| Pro forma basic and diluted loss per common share (2) | $ | 0.81 | $ | 0.83 | $ | 0.87 | $ | 0.93 | $ | 0.35 | $ | 0.95 | $ | 0.60 | $ | 5.95 | ||||||||||
| Pro forma weighted average number of common shares (2) | 8,365,079 | 6,374,239 | 2,648,968 | 1,589,776 | 919,066 | 11,079,738 | 7,450,248 | 4,485,061 | ||||||||||||||||||
| (1) Includes stock-based compensation expense as follows: | ||||||||||||||||||||||||||
$ | 55 | $ | 40 | $ | 15 | $ | 5 | $ | — | $ | 144 | $ | 40 | $ | 260 | |||||||||||
(2) Pro forma figures retroactively give effect to the Reorganization. See "Reorganization". | ||||||||||||||||||||||||||
7
| | As of December 31, 2005 | As of September 30, 2006 | |||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Actual | Pro Forma As Adjusted(4) | Actual | Pro Forma As Adjusted(4) | |||||||||
| | (In thousands) | (In thousands) | |||||||||||
| Consolidated Balance Sheet Data | |||||||||||||
| Cash, cash equivalents, short-term investments and long-term investments (3) | $ | 18,369 | $ | 53,589 | $ | 11,040 | $ | 46,260 | |||||
| Working capital | 14,037 | 49,257 | 9,994 | 45,214 | |||||||||
| Total assets | 19,750 | 54,970 | 13,533 | 48,753 | |||||||||
| Net assets | 17,998 | 53,218 | 10,174 | 45,394 | |||||||||
| Series preferred shares | 31,825 | — | 33,737 | — | |||||||||
| Common shares | 399 | 67,444 | 399 | 69,356 | |||||||||
| Deficit accumulated during the development stage | (16,154 | ) | (16,154 | ) | (26,668 | ) | (26,668 | ) | |||||
| Total shareholders' equity (deficiency) | (13,827 | ) | 21,393 | (23,563 | ) | 11,657 | |||||||
- (3)
- Investments with maturities greater than one year are classified as long-term investments.
- (4)
- Assumes completion of the offering and the Reorganization. See "Reorganization".
8
Investing in our common shares involves a high degree of risk. You should consider carefully the risks and uncertainties described below, together with all of the other information contained in this prospectus, before deciding to invest in our common shares. If any of the following risks materialize, our business, financial condition, results of operation and future prospects will likely be materially and adversely affected. In that event, the market price of our common shares could decline and you could lose all or part of your investment.
Risks Related to Our Business
We have a limited operating history, have incurred losses since inception and anticipate that we will continue to incur losses for the foreseeable future. We have never had any products available for commercial sale and we may never achieve or sustain profitability.
We are a clinical-stage biopharmaceutical company with a limited operating history. We are not profitable and have incurred losses in each year since our inception in 2000. We have never had any products available for commercial sale and we have not generated any revenue from product sales. We do not anticipate that we will generate revenue from the sale of products for the foreseeable future. We have not yet submitted any products for approval by regulatory authorities. We continue to incur research and development and general and administrative expenses related to our operations. Our loss before taxes for the years ended December 31, 2003, 2004 and 2005 was $1.9 million, $3.7 million and $4.5 million, respectively. As of September 30, 2006, we had incurred cumulative losses before taxes of $19.9 million. We expect to continue to incur losses for the foreseeable future, and we expect these losses to increase as we continue our research activities and conduct development of, and seek regulatory approvals for, our product candidates, and prepare for and begin to commercialize any approved products. If our product candidates fail in clinical trials or do not gain regulatory approval, or if our product candidates do not achieve market acceptance, we may never become profitable. Even if we achieve profitability in the future, we may not be able to sustain profitability in subsequent periods.
We are highly dependent on the success of our lead product candidate, OGX-011, and we cannot give any assurance that it or any of our other product candidates will receive regulatory approval or be successfully commercialized.
OGX-011 is in several phase 2 clinical trials, which we expect will be completed by the end of 2007. In order to market OGX-011, we will have to conduct additional clinical trials, including phase 3 clinical trials, to demonstrate safety and efficacy. We have not initiated any phase 3 clinical trials with any of our product candidates. If our ongoing phase 2 clinical trials generate safety concerns or lack of efficacy, or competitive products developed by third parties show significant benefit in the cancer indications in which we are developing our product candidates, any planned phase 3 clinical trial may be delayed, altered or not initiated and OGX-011 may never receive regulatory approval or be successfully commercialized. Our other product candidates, OGX-427 and OGX-225, have not yet been tested in humans. Our pre-clinical testing of either of these product candidates may not be successful and we may be unable to initiate clinical evaluation of them. Our clinical development programs for OGX-011, OGX-427 and OGX-225 may not receive regulatory approval either if we fail to demonstrate that they are safe and effective in clinical trials and consequently fail to obtain necessary approvals from the U.S. Food and Drug Administration, or the FDA, or similar non-U.S. regulatory agencies, or if we have
9
inadequate financial or other resources to advance these product candidates through the clinical trial process. Even if our product candidates receive regulatory approval, we may not be successful in marketing them for a number of reasons, including the introduction by our competitors of more clinically-effective or cost-effective alternatives or failure in our sales and marketing efforts. Any failure to obtain approval of OGX-011 or our other product candidates, and successfully commercialize them, would have a material and adverse impact on our business.
Clinical trials may not demonstrate a clinical benefit of our product candidates.
Positive results from pre-clinical studies and early clinical trials should not be relied upon as evidence that later-stage or large-scale clinical trials will succeed. We will be required to demonstrate with substantial evidence through well-controlled clinical trials that our product candidates are safe and effective for use in a diverse population before we can seek regulatory approvals for their commercial sale. Success in early clinical trials does not mean that future clinical trials will be successful because product candidates in later-stage clinical trials may fail to demonstrate sufficient safety and efficacy to the satisfaction of the FDA and other non-U.S. regulatory authorities despite having progressed through initial clinical trials.
Even after the completion of phase 3 clinical trials, the FDA or other non-U.S. regulatory authorities may disagree with our clinical trial design and our interpretation of data, and may require us to conduct additional clinical trials to demonstrate the efficacy of our product candidates.
Failure to recruit and enroll patients for clinical trials may cause the development of our product candidates to be delayed.
We may encounter delays or rejections if we are unable to recruit and enroll enough patients to complete clinical trials. In the past, we have experienced delays enrolling patients in our clinical trials due to slower than expected approvals by ethics review boards and patient availability. We have been enrolling patients in our ongoing phase 2 clinical trials for OGX-011 as follows:
- •
- phase 2 clinical trial for hormone refractory prostate cancer patients in combination with initial chemotherapy;
- •
- phase 2 clinical trial for hormone refractory prostate cancer patients that have failed initial chemotherapy;
- •
- phase 2 clinical trial for advanced non-small cell lung cancer patients in combination with chemotherapy;
- •
- phase 2 clinical trial for localized prostate cancer in combination with hormone ablation therapy; and
- •
- phase 2 clinical trial for advanced breast cancer patients in combination with chemotherapy.
As of February 28, 2007, we have enrolled 100%, 95%, 100%, 100% and 100% of our planned number of patients, respectively, in these trials. Patient enrollment depends on many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites and the eligibility criteria for the clinical trial. Any delays in planned patient enrollment may result in delays to our product development and increased development costs, which could harm our ability to develop products.
10
Our drug development activities could be delayed or stopped.
We do not know whether any of our ongoing or planned clinical trials for OGX-011, or pre-clinical studies or clinical trials for our other product candidates, will proceed or be completed on schedule, or at all. The commencement of our planned clinical trials could be substantially delayed or prevented by several factors, including:
- •
- limited number of, and competition for, suitable patients with the particular types of cancer required for enrollment in our clinical trials;
- •
- limited number of, and competition for, suitable sites to conduct our clinical trials;
- •
- delay or failure to obtain FDA or non-U.S. regulatory agencies' approval or agreement to commence a clinical trial;
- •
- delay or failure to obtain sufficient supplies of the product candidate for our clinical trials;
- •
- delay or failure to reach agreement on acceptable clinical trial agreement terms or clinical trial protocols with prospective sites or investigators; and
- •
- delay or failure to obtain institutional review board, or IRB, approval to conduct a clinical trial at a prospective site.
The completion of our current clinical trials could also be substantially delayed or prevented by several factors, including:
- •
- slower than expected rates of patient recruitment and enrollment;
- •
- failure of patients to complete the clinical trial;
- •
- unforeseen safety issues;
- •
- lack of efficacy evidenced during clinical trials;
- •
- termination of our clinical trials by one or more clinical trial sites;
- •
- inability or unwillingness of patients or medical investigators to follow our clinical trial protocols;
- •
- inability to monitor patients adequately during or after treatment; and
- •
- introduction of competitive products that may impede our ability to retain patients in our clinical trials.
Our clinical trials may be suspended or terminated at any time by the FDA, other regulatory authorities, the IRB overseeing the clinical trial at issue, any of our clinical trial sites with respect to that site, or us. Any failure or significant delay in completing clinical trials for our product candidates could materially harm our financial results and the commercial prospects for our product candidates.
Our product candidates may cause undesirable and potentially serious side effects during clinical trials that could delay or prevent their regulatory approval or commercialization.
Three phase 1 clinical trials, involving a total of 75 patients, have been completed with OGX-011. Some of the patients experienced various adverse events, the majority of which are associated with other treatments in the protocol and the disease. The majority of adverse events were mild and the most common adverse events consisted of flu-like symptoms. Since patients in
11
our clinical trials have advanced stages of cancer, we expect additional adverse events, including serious adverse events, will occur.
Undesirable side effects caused by any of our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or non-U.S. regulatory authorities for any or all targeted indications. This, in turn, could prevent us from commercializing our product candidates and generating revenues from their sale. In addition, if our product candidates receive marketing approval and we or others later identify undesirable side effects caused by the product:
- •
- regulatory authorities may withdraw their approval of the product;
- •
- we may be required to recall the product, change the way the product is administered, conduct additional clinical trials or change the labeling of the product;
- •
- a product may become less competitive and product sales may decrease; or
- •
- our reputation may suffer.
Any one or a combination of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase the costs and expenses of commercializing the product, which in turn could delay or prevent us from generating significant revenues from the sale of the product.
Recent events have raised questions about the safety of marketed drugs and may result in increased cautiousness by the FDA in reviewing new drugs based on safety, efficacy or other regulatory considerations and may result in significant delays in obtaining regulatory approvals, additional clinical trials being required, or more stringent product labeling requirements. Any delay in obtaining, or inability to obtain, applicable regulatory approvals, would prevent us from commercializing our product candidates.
The success of OGX-011 is influenced by our collaboration with Isis Pharmaceuticals, Inc., or Isis, and involves a complex sharing of decisions, responsibilities, costs and benefits. The loss of Isis as a partner, or any adverse developments in the collaboration, would materially harm our business.
In November 2001, we entered into a co-development and license agreement with Isis to co-develop OGX-011, the material terms of which are described under the heading, "License and Collaboration Agreements" within the Business section of this prospectus.
We are subject to a number of risks associated with our collaboration with Isis, including:
- •
- we and Isis could disagree as to development plans, including clinical trials or regulatory approval strategy, or as to which additional indications for OGX-011 should be pursued. Disputes regarding the collaboration agreement that delay or terminate the development, commercialization or receipt of regulatory approvals of OGX-011 would harm our business and could result in significant litigation or arbitration;
- •
- Isis could fail to devote sufficient resources to the development, approval, or partnering of OGX-011;
- •
- Isis could refuse to manufacture OGX-011 or facilitate transfer of manufacturing knowledge to an alternate drug substance manufacturer;
12
- •
- Isis could fail to effectively manage its manufacturing relationships with raw materials suppliers necessary for the manufacture of OGX-011. Raw materials used in the manufacture of OGX-011 are available from a limited number of suppliers and Isis has established and maintains these relationships for OGX-011 and themselves; and
- •
- Isis may terminate our collaboration agreement upon our material breach of the collaboration agreement or our bankruptcy.
In addition, we do not currently have the resources necessary to develop and market OGX-011 on our own. Isis is currently funding 35 percent of the development costs of OGX-011. If either we or Isis do not perform our respective obligations under, or devote sufficient resources to, our collaboration, or if we and Isis do not work effectively together, OGX-011 may not be successfully commercialized, or its development may be delayed. If our collaboration were to be terminated, we may need to secure an alternative source of funding and may not be able to do so on acceptable terms, or at all.
If we fail to obtain additional financing, we may be unable to complete the development and commercialization of OGX-011 and our other product candidates, or continue our research and development programs.
Our operations have consumed substantial amounts of cash since inception. We expect to continue to spend substantial amounts to:
- •
- continue and complete the clinical development of OGX-011:
- •
- initiate and complete the clinical development of our other product candidates;
- •
- develop, license or acquire additional product candidates;
- •
- launch and commercialize any product candidates for which we receive regulatory approval; and
- •
- continue our research and development programs.
We expect that our existing capital resources, together with the proceeds from this offering, will be sufficient to fund our operations through at least early 2009. We anticipate that we will need to raise additional capital, through private placements or public offerings of our equity or debt securities, in addition to the capital provided by this offering to complete the development and commercialization of our current product candidates.
We anticipate that the cost of our first phase 3 clinical trial, including any proceeds of this offering that are used to plan and initiate our first phase 3 clinical trial, will be approximately $20 million to $40 million. The actual cost of our first phase 3 clinical trial will vary depending on a number of factors, including the cancer indication and stage of disease for which the clinical trial is undertaken, the number of patients included in the clinical trial, and the number and geographic distribution of clinical trial sites.
Many factors will affect our ability to develop our product candidates as anticipated. We may be subject to unanticipated costs or delays that would accelerate our need for additional capital or increase the costs of individual clinical trials. If we are unable to raise additional capital when required or on acceptable terms, we may have to significantly delay, scale back or discontinue the
13
development and/or commercialization of one or more of our product candidates. We also may be required to:
- •
- seek collaborators for our product candidates at an earlier stage than otherwise would be desirable and on terms that are less favorable than might otherwise be available; and
- •
- relinquish or license on unfavorable terms our rights to technologies or product candidates that we otherwise would seek to develop or commercialize ourselves.
If we raise additional financing, the terms of such transactions may cause dilution to existing shareholders or contain terms that are not favorable to us.
To date, our sources of cash have been limited primarily to proceeds from the private placement of our securities. In the future, we may seek to raise additional financing through private placements or public offerings of our equity or debt securities. We cannot be certain that additional funding will be available on acceptable terms, or at all. To the extent that we raise additional funds by issuing equity securities, our shareholders may experience significant dilution. Any debt financing, if available, may involve restrictive covenants, such as limitations on our ability to incur additional indebtedness, limitations on our ability to acquire or license intellectual property rights and other operating restrictions that could adversely impact our ability to conduct our business.
If our competitors develop and market products that are more effective, safer or less expensive than our future product candidates, our clinical trials and commercial opportunities will be negatively impacted.
The life sciences industry is highly competitive, and we face significant competition from many pharmaceutical, biopharmaceutical and biotechnology companies that are researching and marketing products designed to address cancer indications for which we are currently developing products or for which we may develop products in the future. We are aware of several other companies, including Genta Incorporated, Conforma Therapeutics Corporation, Eli Lilly and Company, Aegera Therapeutics Inc. and Gemin X Biotechnologies Inc., which are developing therapeutics that seek to promote tumor cell death by inhibiting cell survival proteins. Any products we may develop in the future are also likely to face competition from other drugs and therapies. Many of our competitors have significantly greater financial, manufacturing, marketing and drug development resources than we do. Large pharmaceutical companies, in particular, have extensive experience in clinical testing and in obtaining regulatory approvals for drugs. These companies also have significantly greater research and marketing capabilities than we do. In addition, many universities and private and public research institutes are, or may become, active in cancer research, the products of which may be in direct competition with us. If our competitors market products that are more effective, safer or less expensive than our future product candidates, if any, or that reach the market sooner than our future product candidates, if any, we may not achieve commercial success.
14
If new therapies become broadly used we may need to conduct clinical trials of our product candidates in combination with these new therapies to demonstrate safety and efficacy of the combination. Additional trials will delay the development of our product candidates and increase our costs. The failure of our product candidates to work in combination with these new therapies would have an adverse effect on our business.
Our intention is to combine our product candidates with therapies that are broadly used by clinicians and considered highly effective. As new therapies are developed we will need to assess these therapies to determine whether to conduct clinical trials of our product candidates in combination with them to demonstrate safety and efficacy of the combination. If we determine to conduct additional clinical trials of our product candidates in combination with these new therapies, the development of our product candidates will be delayed and our costs increased. If these clinical trials generate safety concerns or lack of efficacy, our business would be adversely affected.
If our product candidates become approved in combination with a specific therapy that is broadly used and that therapy becomes displaced by another product, the market for our product candidate may decrease.
If product liability lawsuits are successfully brought against us, we may incur substantial liabilities and may be required to limit commercialization of our product candidates.
We face an inherent risk of product liability lawsuits related to the testing of our product candidates, and will face an even greater risk if product candidates are introduced commercially. An individual may bring a liability claim against us if one of our product candidates causes, or merely appears to have caused, an injury. Because we conduct clinical trials in humans, we face the risk that the use of our product candidates will result in adverse side effects. We cannot predict the possible harms or side effects that may result from our clinical trials. Although we have clinical trial liability insurance for up to C$10 million in aggregate per annum, our insurance may be insufficient to cover any such events. We do not know whether we will be able to continue to obtain clinical trial coverage on acceptable terms, or at all. We may not have sufficient resources to pay for any liabilities resulting from a claim excluded from, or beyond the limit of, our insurance coverage. There is also a risk that third parties that we have agreed to indemnify, such as the University of British Columbia, or UBC, could incur liability. If we cannot successfully defend ourselves against a product liability claim, we may incur substantial liabilities. Regardless of merit or eventual outcome, liability claims either during clinical trials or following commercial introduction may result in:
- •
- decreased demand for our product candidates;
- •
- injury to our reputation;
- •
- withdrawal of clinical trial participants;
- •
- significant litigation costs;
- •
- substantial monetary awards to or costly settlement with patients;
- •
- product recalls;
- •
- loss of revenue; and
- •
- the inability to commercialize our product candidates.
15
We could also be adversely affected if any of our product candidates or any similar products distributed by other companies prove to be, or are asserted to be, harmful to consumers.
If we fail to acquire and develop products or product candidates at all or on commercially reasonable terms, we may be unable to grow our business.
We currently neither have nor intend to establish internal discovery capabilities and are dependent upon pharmaceutical and biotechnology companies and other researchers to sell or license products or product candidates to us. To date, our product candidates have been derived from technologies discovered by the Prostate Centre at Vancouver General Hospital, or the Prostate Centre, and licensed to us by UBC. We intend to continue to rely on the Prostate Centre, UBC and other research institutions as sources of product candidates. We cannot guarantee that the Prostate Centre or UBC will continue to develop new product candidate opportunities, that we will continue to have access to such opportunities or that we will be able to purchase or license these product candidates on commercially reasonable terms, or at all. If we are unable to purchase or license new product candidates from the Prostate Center or UBC, we will be required to identify alternative sources of product candidates.
The success of our product pipeline strategy depends upon our ability to identify, select and acquire pharmaceutical product candidates. Proposing, negotiating and implementing an economically viable product acquisition or license is a lengthy and complex process. We compete for partnering arrangements and license agreements with pharmaceutical and biotechnology companies and academic research institutions. Our competitors may have stronger relationships with third parties with whom we are interested in collaborating and/or may have more established histories of developing and commercializing products. As a result, our competitors may have a competitive advantage in entering into partnering arrangements with such third parties. In addition, even if we find promising product candidates, and generate interest in a partnering or strategic arrangement to acquire such product candidates, we may not be able to acquire rights to additional product candidates or approved products on terms that we find acceptable, or at all. If we fail to acquire and develop product candidates from UBC, or otherwise, we may be unable to grow our business.
We expect that any product candidate to which we acquire rights will require additional development efforts prior to commercial sale, including extensive clinical testing and approval by the FDA and non-U.S. regulatory authorities. All product candidates are subject to the risks of failure inherent in pharmaceutical product development, including the possibility that the product candidate will not be shown to be sufficiently safe and effective for approval by regulatory authorities. Even if the product candidates are approved, we cannot be sure that they would be capable of economically feasible production or commercial success.
If we fail to attract and retain key management and scientific personnel, we may be unable to successfully develop or commercialize our product candidates.
We will need to expand and effectively manage our managerial, operational, financial, development and other resources in order to successfully pursue our research, development and commercialization efforts for our existing and future product candidates. Our success depends on our continued ability to attract, retain and motivate highly qualified management, pre-clinical and clinical personnel, including our key management personnel, Scott Cormack, Martin Gleave, Cindy Jacobs and Stephen Anderson. The loss of the services of any of our senior management could
16
delay or prevent the commercialization of our product candidates. Although we have entered into employment agreements with each of Mr. Cormack, Dr. Jacobs and Mr. Anderson for an indefinite term, such agreements permit the executive to terminate his or her employment with us at any time, subject to providing us with advance written notice. Our existing consulting agreement with Dr. Gleave is for a fixed term expiring on December 31, 2008. We maintain "key man" insurance policies on the lives of Mr. Cormack and Dr. Gleave. We will need to hire additional personnel as we continue to expand our development activities.
We have scientific and clinical advisors who assist us in formulating our development and clinical strategies. These advisors are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us. In addition, our advisors may have arrangements with other companies to assist those companies in developing products or technologies that may compete with ours.
We may not be able to attract or retain qualified management and scientific personnel in the future due to the intense competition for qualified personnel among biotechnology, pharmaceutical and other businesses. If we are not able to attract and retain the necessary personnel to accomplish our business objectives, we may experience constraints that will impede significantly the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy. In particular, if we lose any members of our senior management team, we may not be able to find suitable replacements in a timely fashion or at all and our business may be harmed as a result.
We may encounter difficulties in managing our expected growth and in expanding our operations successfully.
As we advance our product candidates through development and clinical trials, we will need to develop or expand our development, regulatory, manufacturing, marketing and sales capabilities or contract with third parties to provide these capabilities for us. Maintaining additional relationships and managing our future growth will impose significant added responsibilities on members of our management. We must be able to: manage our development efforts effectively; manage our clinical trials effectively; hire, train and integrate additional management, development, administrative and sales and marketing personnel; improve our managerial, development, operational and finance systems; and expand our facilities, all of which may impose a strain on our administrative and operational infrastructure.
Furthermore, we may acquire additional businesses, products or product candidates that complement or augment our existing business. To date, we have no experience in acquiring and integrating other businesses. Integrating any newly acquired business, product or product candidate could be expensive and time-consuming. We may not be able to integrate any acquired business, product or product candidate successfully or operate any acquired business profitably. Our future financial performance will depend, in part, on our ability to manage any future growth effectively and our ability to integrate any acquired businesses. We may not be able to accomplish these tasks, and our failure to accomplish any of them could prevent us from successfully growing our company.
17
We rely, in part, on third parties to conduct clinical trials for our product candidates and plan to rely on third parties to conduct future clinical trials. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be unable to obtain regulatory approval for or commercialize our current and future product candidates.
To implement our product development strategies, we rely on third parties, such as contract research organizations, medical institutions, clinical investigators and contract laboratories, to conduct our clinical trials of our product candidates. Although we rely on these third parties to conduct our clinical trials, we are responsible for ensuring that each of our clinical trials is conducted in accordance with its investigational plan and protocol. Moreover, the FDA and non-U.S. regulatory authorities require us to comply with regulations and standards, commonly referred to as Good Clinical Practices, or GCPs, for conducting, monitoring, recording and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate and that the clinical trial subjects are adequately informed of the potential risks of participating in clinical trials. Our reliance on third parties does not relieve us of these responsibilities and requirements. If the third parties conducting our clinical trials do not perform their contractual duties or obligations, do not meet expected deadlines or need to be replaced, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to GCPs or for any other reason, we may need to enter into new arrangements with alternative third parties and our clinical trials may be extended, delayed or terminated. In addition, a failure by such third parties to perform their obligations in compliance with GCPs may cause our clinical trials to fail to meet regulatory requirements, which may require us to repeat our clinical trials.
We rely on third parties to manufacture and supply our product candidates.
We do not own or operate manufacturing facilities, and we depend on third-party contract manufacturers for production of our product candidates. We have no experience in drug formulation or manufacturing, and we lack the resources and the capability to manufacture any of our product candidates. To date, our product candidates have been manufactured in limited quantities for pre-clinical studies and clinical trials. All active pharmaceutical ingredient for OGX-011 has been manufactured for us by Isis and all drug product has been manufactured for us by Formatech, Inc. and Pyramid Laboratories, Inc., in each case pursuant to a purchase order or short-term contract that has been fulfilled. Although we have sufficient quantities of OGX-011 to complete our current phase 2 clinical trials and initiate our first phase 3 clinical trial, we will need to obtain additional quantities to complete our first phase 3 clinical trial. All active pharmaceutical ingredient for OGX-427 for IND-enabling toxicology studies and initial clinical trials has been manufactured for us by Avecia Biotechnology Inc. and all drug product has been manufactured for us by Laureate Pharma, Inc., in each case pursuant to a purchase order or short-term contract that has been fulfilled. If, in the future, one of our product candidates is approved for commercial sale, we will need to manufacture that product candidate in commercial quantities. We cannot assure you that the third-party manufacturers with which we have contracted in the past will have sufficient capacity to satisfy our future manufacturing needs, or that we will be able to negotiate additional purchases of active pharmaceutical ingredient or drug product from these or alternative manufacturers on terms favorable to us, or at all.
Third party manufacturers may fail to perform under their contractual obligations, or may fail to deliver the required commercial quantities of bulk drug substance or finished product on a timely basis and at commercially reasonable prices. Any performance failure on the part of our
18
contract manufacturers could delay clinical development or regulatory approval of our product candidates or commercialization of our future product candidates, depriving us of potential product revenue and resulting in additional losses. If we are required to identify and qualify an alternate manufacturer, we may be forced to delay or suspend our clinical trials, regulatory submissions, required approvals or commercialization of our product candidates, which may cause us to incur higher costs and could prevent us from commercializing our product candidates successfully. If we are unable to find one or more replacement manufacturers capable of production at a reasonably favorable cost, in adequate volumes, of adequate quality, and on a timely basis, we would likely be unable to meet demand for our product candidates and our clinical trials could be delayed or we could lose potential revenue. Our ability to replace an existing active pharmaceutical ingredient manufacturer may be difficult because the number of potential manufacturers is limited to approximately four manufacturers, and the FDA must approve any replacement manufacturer before it can begin manufacturing our product candidates. Such approval would require new testing and compliance inspections. It may be difficult or impossible for us to identify and engage a replacement manufacturer on acceptable terms in a timely manner, or at all. We expect to continue to depend on third-party contract manufacturers for the foreseeable future.
Our product candidates require precise, high quality manufacturing. Any of our contract manufacturers will be subject to ongoing periodic unannounced inspection by the FDA and non-U.S. regulatory authorities to ensure strict compliance with current Good Manufacturing Practice, or cGMP, and other applicable government regulations and corresponding standards. If our contract manufacturers fail to achieve and maintain high manufacturing standards in compliance with cGMP regulations, we may experience manufacturing errors resulting in patient injury or death, product recalls or withdrawals, delays or interruptions of production or failures in product testing or delivery, delay or prevention of filing or approval of marketing applications for our product candidates, cost overruns or other problems that could seriously harm our business.
Significant scale-up of manufacturing may require additional validation studies, which the FDA must review and approve. Additionally, any third party manufacturers we retain to manufacture our product candidates on a commercial scale must pass an FDA pre-approval inspection for conformance to the cGMPs before we can obtain approval of our product candidates. If we are unable to successfully increase the manufacturing capacity for a product candidate in conformance with cGMPs, the regulatory approval or commercial launch of any related products may be delayed or there may be a shortage in supply.
If we are unable to develop our sales and marketing and distribution capability on our own or through collaborations with marketing partners, we will not be successful in commercializing our product candidates. We currently do not have a marketing staff nor a sales or distribution organization.
We currently do not have marketing, sales or distribution capabilities. If our product candidates are approved, we may establish a sales and marketing organization with technical expertise and supporting distribution capabilities to commercialize our product candidates, which will be expensive and time consuming. Any failure or delay in the development of internal sales, marketing and distribution capabilities would adversely impact the commercialization of these product candidates. We may choose to collaborate with third parties that have direct sales forces and established distribution systems, either to augment our own sales force and distribution systems or in lieu of our own sales force and distribution systems. To the extent that we enter into
19
co-promotion or other licensing arrangements, our product revenue is likely to be lower than if we directly marketed or sold our products, when and if we have any. In addition, any revenue we receive will depend in whole or in part upon the efforts of such third parties, which may not be successful and will generally not be within our control. If we are unable to enter into such arrangements on acceptable terms or at all, we may not be able to successfully commercialize our existing and future product candidates. If we are not successful in commercializing our existing and future product candidates, either on our own or through collaborations with one or more third parties, our future product revenue will suffer and we may incur significant additional losses.
Risks Related to Our Intellectual Property
Our proprietary rights may not adequately protect our technologies and product candidates.
Our commercial success will depend on our ability to obtain patents and/or regulatory exclusivity and maintain adequate protection for our technologies and product candidates in Canada, the United States and other countries. As of February 28, 2007, we owned or had exclusive rights to approximately ten issued U.S. and foreign patents and approximately 89 pending U.S. and foreign patent applications. We will be able to protect our proprietary rights from unauthorized use by third parties only to the extent that our proprietary technologies and future product candidates are covered by valid and enforceable patents or are effectively maintained as trade secrets.
We apply for patents covering both our technologies and product candidates, as we deem appropriate. However, we may fail to apply for patents on important technologies or product candidates in a timely fashion, or at all. Our existing patents and any future patents we obtain may not be sufficiently broad to prevent others from practicing our technologies or from developing competing products and technologies. In addition, we do not always control the patent prosecution of subject matter that we license from others. Accordingly, we are sometimes unable to exercise the same degree of control over this intellectual property as we would over our own. Moreover, the patent positions of biopharmaceutical companies are highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. As a result, the validity and enforceability of our patents cannot be predicted with certainty. In addition, we cannot guarantee that:
- •
- we or our licensors were the first to make the inventions covered by each of our issued patents and pending patent applications;
- •
- we or our licensors were the first to file patent applications for these inventions;
- •
- others will not independently develop similar or alternative technologies or duplicate any of our technologies;
- •
- any of our or our licensors' pending patent applications will result in issued patents;
- •
- any of our or our licensors' patents will be valid or enforceable;
- •
- any patents issued to us or our licensors and collaboration partners will provide us with any competitive advantages, or will not be challenged by third parties;
- •
- we will develop additional proprietary technologies that are patentable; or
- •
- the patents of others will not have an adverse effect on our business.
20
The actual protection afforded by a patent varies on a product-by-product basis, from country to country and depends upon many factors, including the type of patent, the scope of its coverage, the availability of regulatory related extensions, the availability of legal remedies in a particular country and the validity and enforceability of the patents. Our ability to maintain and solidify our proprietary position for our product candidates will depend on our success in obtaining effective claims and enforcing those claims once granted. Our issued patents and those that may issue in the future, or those licensed to us, may be challenged, invalidated or circumvented, and the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar products. Due to the extensive amount of time required for the development, testing and regulatory review of a potential product, it is possible that, before any of our product candidates can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
Protection afforded by U.S. patents may be adversely affected by proposed changes to patent related U.S. statutes and to U.S. Patent and Trademark Office, or U.S.PTO, rules, especially changes to rules concerning the filing of continuation applications. If implemented, the rules may require that second or subsequent continuing application filings be supported by a showing as to why the new amendments or claims, argument or evidence presented could not have been previously submitted. Other rules, if implemented, may limit consideration by the U.S.PTO of up to only ten claims per application. It is common practice to file multiple patent applications with many claims in an effort to maximize patent protection. If the first set of proposed U.S.PTO rules are implemented, they may limit our ability to file continuing applications directed to our product candidates and methods and related competing products and methods. In addition, if the second set of U.S.PTO rules are implemented, they may limit our ability to patent a number of claims sufficient to cover our product candidates and methods and related competing products and methods. Other changes to the patent statutes may adversely affect the protection afforded by U.S. patents and/or open U.S. patents up to third party attack in non-litigation settings.
We also rely on trade secrets to protect some of our technology, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to maintain. While we use reasonable efforts to protect our trade secrets, our or our collaboration partners' employees, consultants, contractors or scientific and other advisors may unintentionally or willfully disclose our proprietary information to competitors. Enforcement of claims that a third party has illegally obtained and is using trade secrets is expensive, time consuming and uncertain. In addition, non-U.S. courts are sometimes less willing than U.S. courts to protect trade secrets. If our competitors independently develop equivalent knowledge, methods and know-how, we would not be able to assert our trade secrets against them and our business could be harmed.
The intellectual property protection for our product candidates is dependent on third parties.
With respect to OGX-011, OGX-427 and OGX-225, we have exclusively licensed from UBC certain issued patents and pending patent applications covering the respective antisense sequences underlying these product candidates and their commercialization and use. These patents and pending patent applications do not cover all potential antisense sequences for the corresponding molecular targets of these product candidates nor all potential uses. We direct patent prosecution and are responsible for all fees and costs related to the preparation, filing, prosecution, enforcement and maintenance of the patent rights that we have licensed from UBC.
21
Similarly, we have licensed from Isis certain issued patents and pending patent applications directed to chemical modification of our product candidates for commercialization, use and the manufacturing thereof, as well as some alternative antisense sequences. We have also received a sublicense from Isis under certain third party patent portfolios directed to such modifications. These patents and pending patent applications do not cover all potential modifications of our product candidates. We do not have and have not had any control over the filing, prosecution or enforcement of these patents or patent applications. We cannot be certain that such prosecution efforts have been or will be conducted in compliance with applicable laws and regulations or will result in valid and enforceable patents. We also cannot be assured that Isis or its licensors will agree to enforce any such patent rights at our request or devote sufficient efforts to attain a desirable result. Any failure by Isis, or any of their licensing partners, to properly protect the intellectual property rights relating to our product candidates could have a material adverse effect on our financial condition and results of operation.
We may not be able to protect our intellectual property rights throughout the world.
Filing, prosecuting and defending patents on all of our product candidates and products, when and if we have any, in every jurisdiction would be prohibitively expensive. Competitors may use our technologies in jurisdictions where we or our licensors have not obtained patent protection to develop their own products. These products may compete with our products, when and if we have any, and may not be covered by any of our or our licensors' patent claims or other intellectual property rights.
The laws of some non-U.S. countries do not protect intellectual property rights to the same extent as the laws of the United States, and many companies have encountered significant problems in protecting and defending such rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to biotechnology and/or pharmaceuticals, which could make it difficult for us to stop the infringement of our patents. Proceedings to enforce our patent rights in foreign jurisdictions could result in substantial cost and divert our efforts and attention from other aspects of our business.
The patent protection for our product candidates or products may expire before we are able to maximize their commercial value which may subject us to increased competition and reduce or eliminate our opportunity to generate product revenue.
The patents for our product candidates have varying expiration dates and, when these patents expire, we may be subject to increased competition and we may not be able to recover our development costs. For example, the first granted U.S. patent directed to OGX-011 and licensed from UBC is due to expire in 2021. A U.S. patent issuing from one of the pending patent families directed to OGX-427 and OGX-225 and licensed from UBC would expire as early as 2023 and 2020, respectively. Generally, we anticipate that the patents we license from Isis will expire prior to the patents we license from UBC. In some of the larger economic territories, such as the United States and Europe, patent term extension/restoration may be available to compensate for time taken during aspects of the product candidate's regulatory review. However, we cannot be certain that an extension will be granted, or if granted, what the applicable time period or the scope of patent protection afforded during any extended period will be. In addition, even though some regulatory agencies may provide some other exclusivity for a product candidate under its
22
own laws and regulations, we may not be able to qualify the product candidate or obtain the exclusive time period.
If we are unable to obtain patent term extension/restoration or some other exclusivity, we could be subject to increased competition and our opportunity to establish or maintain product revenue could be substantially reduced or eliminated. Furthermore, we may not have sufficient time to recover our development costs prior to the expiration of our U.S. and non-U.S. patents.
We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights and we may be unable to protect our rights to, or use of, our technology.
If we choose to go to court to stop someone else from using the inventions claimed in our patents or our licensed patents, that individual or company has the right to ask the court to rule that these patents are invalid and/or should not be enforced against that third party. These lawsuits are expensive and would consume time and other resources even if we were successful in stopping the infringement of these patents. In addition, there is a risk that the court will decide that these patents are invalid or unenforceable and that we do not have the right to stop the other party from using the inventions. There is also the risk that, even if the validity of these patents is upheld, the court will refuse to stop the other party on the ground that such other party's activities do not infringe our rights.
If we wish to use the technology or compound claimed in issued and unexpired patents owned by others, we will need to obtain a license from the owner, enter into litigation to challenge the validity or enforceability of the patents or incur the risk of litigation in the event that the owner asserts that we infringed its patents. The failure to obtain a license to technology or the failure to challenge an issued patent that we may require to discover, develop or commercialize our product candidates may have a material adverse impact on us.
If a third party asserts that we infringed its patents or other proprietary rights, we could face a number of risks that could seriously harm our results of operations, financial condition and competitive position, including:
- •
- patent infringement and other intellectual property claims, which would be costly and time consuming to defend, whether or not the claims have merit, and which could delay the regulatory approval process and divert management's attention from our business;
- •
- substantial damages for past infringement, which we may have to pay if a court determines that our product candidates or technologies infringe a competitor's patent or other proprietary rights;
- •
- a court prohibiting us from selling or licensing our technologies or future drugs unless the third party licenses its patents or other proprietary rights to us on commercially reasonable terms, which it is not required to do; and
- •
- if a license is available from a third party, we may have to pay substantial royalties or lump sum payments or grant cross licenses to our patents or other proprietary rights to obtain that license.
The biotechnology industry has produced a proliferation of patents, and it is not always clear to industry participants, including us, which patents cover various types of products or methods of use. The coverage of patents is subject to interpretation by the courts, and the interpretation is
23
not always uniform. If we are sued for patent infringement, we would need to demonstrate that our product candidates or methods of use either do not infringe the patent claims of the relevant patent and/or that the patent claims are invalid, and we may not be able to do this. Proving invalidity, in particular, is difficult since it requires a showing of clear and convincing evidence to overcome the presumption of validity enjoyed by issued patents.
U.S. patent laws as well as the laws of some foreign jurisdictions provide for provisional rights in published patent applications beginning on the date of publication, including the right to obtain reasonable royalties, if a patent subsequently issues and certain other conditions are met.
We are aware of an issued U.S. patent and corresponding foreign counterparts containing claims relating to antisense sequences that inhibit IGFBP-2. Certain of these claims may be broad enough in scope such that, if we choose to commercialize OGX-225 in the U.S. or in any foreign jurisdiction in which a corresponding patent has issued, we may infringe such claims. While we believe that there may be multiple grounds on which to challenge the validity of the U.S. patent and the foreign counterparts, we cannot predict the outcome of any invalidity challenge. Alternatively, it is possible that we may determine it prudent to seek a license from the patent holder to avoid potential litigation and other potential disputes. We cannot be sure that a license would be available to us on acceptable terms, or at all.
Because some patent applications in the United States may be maintained in secrecy until the patents are issued, because patent applications in the United States and many foreign jurisdictions are typically not published until 18 months after filing, and because publications in the scientific literature often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for technology covered by our licensors' issued patents or our pending applications or our licensors' pending applications, or that we or our licensors were the first to invent the technology.
Patent applications filed by third parties that cover technology similar to ours may have priority over our or our licensors' patent applications and could further require us to obtain rights to issued patents covering such technologies. If another party files a United States patent application on an invention similar to ours, we may elect to participate in or be drawn into an interference proceeding declared by the U.S.PTO to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful, resulting in a loss of our United States patent position with respect to such inventions.
Some of our competitors may be able to sustain the costs of complex patent litigation more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations. We cannot predict whether third parties will assert these claims against us or against the licensors of technology licensed to us, or whether those claims will harm our business. If we are forced to defend against these claims, whether they are with or without any merit, whether they are resolved in favor of or against us or our licensors, we may face costly litigation and diversion of management's attention and resources. As a result of these disputes, we may have to develop costly non-infringing technology, or enter into licensing agreements. These agreements, if necessary, may be unavailable on terms acceptable to us, if at all, which could seriously harm our business or financial condition.
24
If we breach any of the agreements under which we license rights to our product candidates or technology from third parties, we could lose license rights that are important to our business. Certain of our license agreements may not provide an adequate remedy for their breach by the licensor.
We license the development and commercialization rights for each of our product candidates, and we expect to enter into similar licenses in the future. For instance, we licensed exclusive worldwide rights from UBC that enable us to use, manufacture, distribute and sell the antisense sequences corresponding to OGX-011, OGX-427 and OGX-225. Under these licenses we are subject to various obligations, including royalty and milestone payments, annual maintenance fees, limits on sublicensing, insurance obligations and the obligation to use commercially reasonable best efforts to develop and exploit the licensed technology. If we fail to comply with any of these obligations or otherwise breach these agreements, our licensors may have the right to terminate the license in whole or in part or to terminate the exclusive nature of the license. Loss of any of these licenses or the exclusivity rights provided therein could harm our financial condition and operating results. In addition, our license agreements with UBC eliminate our ability to obtain money damages in respect of certain claims against UBC.
We may be subject to damages resulting from claims that we, or our employees or consultants, have wrongfully used or disclosed alleged trade secrets of third parties.
Many of our employees were previously employed, and certain of our consultants are currently employed, at universities or biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although we have not received any claim to date, we may be subject to claims that these employees or consultants or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of these current or former employers. Litigation may be necessary to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel. We may be subject to claims that employees of our partners or licensors of technology licensed by us have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. We may become involved in litigation to defend against these claims. If we fail in defending such claims, in addition to paying monetary damages, we may lose valuable intellectual property rights or personnel.
Risks Related to Our Industry
The regulatory approval process is expensive, time consuming and uncertain and may prevent us from obtaining approvals for the commercialization of some or all of our product candidates.
The research, testing, manufacturing, labeling, approval, selling, marketing and distribution of drug products are subject to extensive regulation by the FDA and non-U.S. regulatory authorities, which regulations differ from country to country. We are not permitted to market our product candidates in the United States until we receive approval of a New Drug Application, or NDA, from the FDA. We have not submitted an application for or received marketing approval for any of our product candidates. Obtaining approval of an NDA can be a lengthy, expensive and uncertain process. In addition, failure to comply with FDA, non-U.S. regulatory authorities' or other applicable United States and non-U.S. regulatory requirements may, either before or after
25
product approval, if any, subject our company to administrative or judicially imposed sanctions, including:
- •
- restrictions on the products, manufacturers or manufacturing process;
- •
- warning letters;
- •
- civil and criminal penalties;
- •
- injunctions;
- •
- suspension or withdrawal of regulatory approvals;
- •
- product seizures, detentions or import bans;
- •
- voluntary or mandatory product recalls and publicity requirements;
- •
- total or partial suspension of production;
- •
- imposition of restrictions on operations, including costly new manufacturing requirements; and
- •
- refusal to approve pending NDAs or supplements to approved NDAs.
Regulatory approval of an NDA or NDA supplement is not guaranteed, and the approval process is expensive and may take several years. The FDA also has substantial discretion in the drug approval process. Despite the time and expense exerted, failure can occur at any stage, and we could encounter problems that cause us to abandon clinical trials or to repeat or perform additional pre-clinical studies and clinical trials. The number of pre-clinical studies and clinical trials that will be required for FDA approval varies depending on the drug candidate, the disease or condition that the drug candidate is designed to address, and the regulations applicable to any particular drug candidate. The FDA can delay, limit or deny approval of a drug candidate for many reasons, including:
- •
- a drug candidate may not be deemed safe or effective;
- •
- the FDA may not find the data from pre-clinical studies and clinical trials sufficient;
- •
- the FDA might not approve our third-party manufacturer's processes or facilities;
- •
- the FDA may change its approval policies or adopt new regulations; or
- •
- third party products may enter the market and change approval requirements.
Even if we obtain regulatory approvals for our product candidates, the terms of approvals and ongoing regulation of our product candidates may limit how we manufacture and market our product candidates, which could materially impair our ability to generate revenue.
Once regulatory approval has been granted, the approved product and its manufacturer are subject to continual review. Any regulatory approval that we receive for a product candidate is likely to be subject to limitations on the indicated uses for which the end product may be marketed, or include requirements for potentially costly post-approval follow-up clinical trials. In addition, if the FDA and/or non-U.S. regulatory authorities approve any of our product candidates, the labeling, packaging, adverse event reporting, storage, advertising and promotion for the end product will be subject to extensive regulatory requirements. We and the manufacturers of our products, when and if we have any, will also be required to comply with cGMP regulations,
26
which include requirements relating to quality control and quality assurance as well as the corresponding maintenance of records and documentation. Further, regulatory agencies must approve these manufacturing facilities before they can be used to manufacture our products, when and if we have any, and these facilities are subject to ongoing regulatory inspection. If we fail to comply with the regulatory requirements of the FDA and other non-U.S. regulatory authorities, or if previously unknown problems with our products, when and if we have any, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions, including:
- •
- restrictions on the products, manufacturers or manufacturing process;
- •
- warning letters;
- •
- civil or criminal penalties or fines;
- •
- injunctions;
- •
- product seizures, detentions or import bans;
- •
- voluntary or mandatory product recalls and publicity requirements;
- •
- suspension or withdrawal of regulatory approvals;
- •
- total or partial suspension of production;
- •
- imposition of restrictions on operations, including costly new manufacturing requirements; and
- •
- refusal to approve pending NDAs or supplements to approved NDAs.
In addition, the FDA and non-U.S. regulatory authorities may change their policies and additional regulations may be enacted that could prevent or delay regulatory approval of our product candidates. We cannot predict the likelihood, nature or extent of government regulation that may arise from future legislation or administrative action, either in the United States, Canada or abroad. If we are not able to maintain regulatory compliance, we would likely not be permitted to market our future product candidates and we may not achieve or sustain profitability.
Even if we receive regulatory approval to market our product candidates, the market may not be receptive to our products.
Even if our product candidates obtain regulatory approval, resulting products may not gain market acceptance among physicians, patients, health care payors and/or the medical community. We believe that the degree of market acceptance will depend on a number of factors, including:
- •
- timing of market introduction of competitive products;
- •
- safety and efficacy of our products;
- •
- prevalence and severity of any side effects;
- •
- potential advantages or disadvantages over alternative treatments;
- •
- strength of marketing and distribution support;
- •
- price of our products, both in absolute terms and relative to alternative treatments; and
- •
- availability of coverage and reimbursement from government and other third-party payors.
27
If our future product candidates fail to achieve market acceptance, we may not be able to generate significant revenue or achieve or sustain profitability.
There is a high risk that our drug development activities will not result in commercial products.
Our product candidates are in various stages of development and are prone to the risks of failure inherent in drug development. We will need to complete significant additional clinical trials before we can demonstrate that our product candidates are safe and effective to the satisfaction of the FDA and non-U.S. regulatory authorities. Clinical trials are expensive and uncertain processes that take years to complete. Failure can occur at any stage of the process, and successful early clinical trials do not ensure that later clinical trials will be successful. Product candidates in later-stage clinical trials may fail to show desired efficacy and safety traits despite having progressed through initial clinical trials. A number of companies in the pharmaceutical industry have suffered significant setbacks in advanced clinical trials, even after obtaining promising results in earlier clinical trials. In addition, a clinical trial may prove successful with respect to a secondary objective, but fail to demonstrate clinically significant benefits with respect to a primary objective. Failure to satisfy a primary objective in a phase 3 clinical trial (registration trial) would generally mean that a product candidate would not receive regulatory approval.
If government and third-party payors fail to provide coverage and adequate reimbursement rates for our product candidates, our revenues and potential for profitability will be reduced.
In the United States and elsewhere, our product revenues will depend principally upon the reimbursement rates established by third-party payors, including government health administration authorities, managed-care providers, public health insurers, private health insurers and other organizations. These third-party payors are increasingly challenging the price, and examining the cost effectiveness, of medical products and services. In addition, significant uncertainty exists as to the reimbursement status, if any, of newly approved drugs, pharmaceutical products or product indications. We may need to conduct post-marketing clinical trials in order to demonstrate the cost-effectiveness of products. Such clinical trials may require us to commit a significant amount of management time and financial and other resources. If reimbursement of such product is unavailable or limited in scope or amount or if pricing is set at unsatisfactory levels, our revenues could be reduced.
In some countries other than the United States, particularly the countries of the European Union and Canada, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, obtaining pricing approval from governmental authorities can take six to twelve months or longer after the receipt of regulatory marketing approval of a product for an indication. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of one of our product candidates to other available therapies. If reimbursement of such product candidate is unavailable or limited in scope or amount or if pricing is set at unsatisfactory levels, our revenues could be reduced.
Domestic and foreign governments continue to propose and pass legislation designed to reduce the cost of healthcare, including drugs. In the United States, there have been, and we expect that there will continue to be, federal and state proposals to implement similar governmental control. In addition, increasing emphasis on managed care in the United States will continue to put pressure on the pricing of pharmaceutical products. For example, the Medicare Prescription Drug Improvement and Modernization Act of 2003 reforms the way Medicare will
28
cover and reimburse for pharmaceutical products. The legislation expands Medicare coverage for drug purchases by the elderly and eventually will introduce a new reimbursement methodology based on average sales prices for certain drugs. In addition, the new legislation provides authority for limiting the number of outpatient drugs that will be covered in any therapeutic class. As a result of the new legislation and the expansion of federal coverage of drug products, we expect that there will be additional pressure to contain and reduce costs. The Medicaid program and state healthcare laws and regulations may also be modified to change the scope of covered products and/or reimbursement methodology. Cost control initiatives could decrease the established reimbursement rates that we receive for any products in the future, which would limit our revenues and profitability. Legislation and regulations affecting the pricing of pharmaceutical products, including OGX-011, may change at any time, which could further limit or eliminate reimbursement rates for OGX-011 or other product candidates.
Failure to obtain regulatory approval outside the United States would prevent us from marketing our product candidates abroad.
We intend to market certain of our existing and future product candidates in non-North American markets. In order to market our existing and future product candidates in the European Union and many other non-North American jurisdictions, we must obtain separate regulatory approvals. We have had no interactions with non-North American regulatory authorities, and the approval procedures vary among countries and can involve additional testing, and the time required to obtain approval may differ from that required to obtain FDA approval. Approval by the FDA or other regulatory authorities does not ensure approval by regulatory authorities in other countries, and approval by one or more non-North American regulatory authorities does not ensure approval by regulatory authorities in other countries or by the FDA. The non-North American regulatory approval process may include all of the risks associated with obtaining FDA approval. We may not obtain non-North American regulatory approvals on a timely basis, if at all. We may not be able to file for non-North American regulatory approvals and may not receive necessary approvals to commercialize our existing and future product candidates in any market.
Risks Related to this Offering
There has been no active trading market for our common shares, our common share price will likely be highly volatile, and your investment could decline in value.
Prior to this offering, there has been no public market for our common shares. We have received approval to list the securities distributed under this prospectus on the Nasdaq Global Market. Additionally, the Toronto Stock Exchange has conditionally approved the listing of our common shares, subject to us fulfilling all the listing requirements of the exchange. However, an active public market for our common shares may not develop or be sustained after this offering. Following this offering, the market price of our common shares is likely to be highly volatile and may fluctuate significantly in response to various factors and events, many of which we cannot control. The stock market in general, and the market for biopharmaceutical and biotechnology company stocks in particular, have historically experienced significant price and volume fluctuations. Volatility in the market price for a particular company's shares has often been unrelated or disproportionate to the operating performance of that company. Market and industry factors may depress the market price of our common shares, regardless of our operating performance.
29
If you purchase our common shares in this offering, you will incur immediate and substantial dilution of your investment.
The assumed initial public offering price of our common shares is $8.00 per share. This amount is substantially higher than the pro forma net tangible book value that our outstanding common shares will have immediately after this offering. Accordingly, if you purchase our common shares at the assumed initial public offering price, you will incur immediate and substantial dilution of $5.18 per share. If the holders of outstanding options exercise those options, you will suffer further dilution. Investors who purchase common shares in this offering will contribute 58% of the total amount of contributed capital but will own only 31% of the outstanding share capital and 31% of the voting rights.
Future sales of our currently outstanding shares could cause the market price of our common shares to decrease significantly, even if our business is doing well.
Our current shareholders hold a substantial number of common shares that they will be able to sell in the future. Sales of a significant number of our common shares, or the perception that these sales could occur, particularly with respect to sales by affiliates, directors, executive officers or other insiders, could materially and adversely affect the market price of our common shares and impair our ability to raise capital through the sale of additional equity securities.
After this offering and assuming completion of the Reorganization, 16,079,738 common shares will be outstanding based on the number of shares outstanding as of February 28, 2007. This includes the 5,000,000 common shares that we are selling in this offering. 11,058,702 common shares, or approximately 69 percent of the common shares outstanding after this offering, are currently restricted as a result of securities laws or lock-up agreements. However, the underwriters can waive the provisions of the lock-up agreements and allow the shareholders bound by the lock-up agreements to sell their common shares at any time, subject to applicable securities laws. Taking into account the lock-up agreements and assuming completion of the Reorganization, the number of shares that will be available for sale in the public market under the provisions of Rule 144, 144(k) and 701 and Regulation S will be as follows:
| Days after Date of this Prospectus | Number of Shares Eligible for Sale in Public Market | Comment | ||
|---|---|---|---|---|
| Upon Effectiveness | 5,021,036 | Shares sold in this offering and shares not subject to lock-up | ||
180 Days after our common shares first commence trading on the Toronto Stock Exchange | 11,058,702 | Lock-up period expires; 7,813,138 shares eligible for sale without Securities Act restrictions and 3,245,564 shares eligible for sale under Rules 144 and 701 and Regulation S | ||
Thereafter | Nil | Restricted securities held for one year or less |
In Canada, holders of common shares sold in this offering will generally be able to freely sell those shares following a receipt being issued for the final prospectus filed with Canadian provincial securities regulatory authorities, which is expected to occur concurrently with the effectiveness of the U.S. registration statement. Taking into account the lock-up agreements and the securities laws of the provinces of Canada, holders of any of the remaining 7,813,138 common shares outstanding will generally be able to freely sell those shares in Canada 180 days after our
30
common shares first commence trading on the Toronto Stock Exchange, when the lock-up is released.
Upon the expiry of six months from the closing of this offering, holders of a significant percentage of our previously-issued common shares will have the right, subject to some conditions, to require us to file registration statements under the Securities Act of 1933, as amended, or the Securities Act, covering their shares or to include their shares in registration statements that we may file under the Securities Act for ourselves or other shareholders. We also intend to register with the United States Securities and Exchange Commission, or SEC, all common shares that we may issue upon exercise of the stock options we have granted under our Stock Option Plan and all common shares reserved for issuance under our 2006 Stock Incentive Plan. Once we register these common shares, they can be freely sold in the public market upon issuance, subject to the lock-up agreements discussed above and the restrictions imposed on our affiliates under Rule 144.
We will have broad discretion in the use of the net proceeds of this offering and may not use them to effectively manage our business.
We will have broad discretion over the use of the net proceeds from this offering. Because of the number and variability of factors that will determine our use of such proceeds, our ultimate use might vary substantially from our planned use. You may not agree with how we allocate or spend the proceeds from this offering. We may pursue collaborations, clinical trials or other activities that do not result in an increase in the market value of our common shares and may increase our losses.
After this offering our executive officers, directors and major shareholders will continue to have substantial control over us and will maintain the ability to control all matters submitted to shareholders for approval.
As of February 28, 2007, our directors, executive officers and principal shareholders, together with their affiliates, beneficially owned approximately 86 percent of our common shares, including shares subject to outstanding stock options. We expect that following the completion of this offering, this same group will continue to hold at least a majority of our outstanding common shares. These shareholders, acting together, will exercise significant influence over all matters requiring shareholder approval, including the election of directors and any amendment of our notice of articles or by-laws. This concentration of ownership could also have the effect of delaying or preventing a change in our control.
Certain Canadian laws could delay or deter a change of control.
Limitations on the ability to acquire and hold our common shares may be imposed by the Competition Act (Canada). This legislation permits the Commissioner of Competition of Canada to review any acquisition of a significant interest in us. This legislation grants the Commissioner jurisdiction to challenge such an acquisition before the Canadian Competition Tribunal if the Commissioner believes that it would, or would be likely to, result in a substantial lessening or prevention of competition in any market in Canada.
The Investment Canada Act (Canada) subjects an acquisition of control of a company by a non-Canadian to government review if the value of our assets as calculated pursuant to the legislation exceeds a threshold amount. A reviewable acquisition may not proceed unless the relevant minister is satisfied that the investment is likely to be a net benefit to Canada.
31
Any of the foregoing could prevent or delay a change of control and may deprive or limit strategic opportunities for our shareholders to sell their shares.
We believe that we were a passive foreign investment company, or PFIC, for U.S. federal income tax purposes for previous taxable years and expect that we will be a PFIC for the current taxable year, which may negatively affect U.S. investors.
For U.S. federal income taxation purposes, we will be a passive foreign investment company, or PFIC, if in any taxable year either: (a) 75 percent or more of our gross income consists of passive income; or (b) 50 percent or more of the value of our assets is attributable to assets that produce, or are held for the production of, passive income. Because we are a clinical-stage biopharmaceutical company which has not yet recognized significant operating income, and as a result, our gross income consisted mostly of interest, we have been a PFIC in prior taxable years. We may also be a PFIC in the current taxable year as well as future taxable years. If you are a U.S. investor and we are considered a PFIC in any taxable year in which you hold common shares, we will be considered a PFIC with respect to such common shares held by you in subsequent taxable years even if we have operating income and are otherwise not a PFIC in such subsequent taxable years. Gain realized by a U.S. investor from the sale of PFIC shares is taxed as ordinary income, as opposed to capital gain, and subject to an interest charge. You may avoid these adverse tax consequences by timely making one of the tax elections described in the section titled "United States Federal Income Tax Consequences".
The PFIC rules are extremely complex. A U.S. person is encouraged to consult his or her U.S. tax advisor before making an investment in our shares.
As a foreign private issuer, we are subject to different U.S. securities laws and rules than a domestic U.S. issuer, which may limit the information publicly available to our shareholders.
As a foreign private issuer, we are exempt from the proxy disclosure requirements of the Securities Exchange Act of 1934, or Exchange Act, and we are not required to comply with all the periodic disclosure requirements of the Exchange Act. Therefore, there may be less publicly available information about us than if we were a U.S. domestic issuer. In addition, our officers, directors, and principal shareholders are exempt from the reporting and "short-swing" profit recovery provisions of Section 16 of the Exchange Act and the rules thereunder. Therefore, our shareholders may not know on a timely basis when our officers, directors and principal shareholders purchase or sell our common shares.
You may be unable to enforce actions against us, certain of our directors and officers, or the experts named in this prospectus under U.S. federal securities laws.
We are a corporation organized under the laws of Canada. A majority of our directors and officers, and certain of the experts named in this prospectus reside in Canada. Because all or a substantial portion of our assets and the assets of these persons are located outside the U.S., it may not be possible for you to effect service of process within the United States upon us or those persons. Furthermore it may not be possible for you to enforce against us or them in the United States, judgments obtained in U.S. courts based upon the civil liability provisions of the U.S. federal securities laws or other laws of the U.S. There is doubt as to the enforceability, in original actions in Canadian courts, of liabilities based upon the U.S. federal securities laws and as to the enforceability in Canadian courts of judgments of U.S. courts obtained in actions based upon the
32
civil liability provisions of the U.S. federal securities laws. Therefore, it may not be possible to enforce those actions against us, certain of our directors and officers or the experts named in this prospectus.
An increase in the market price of our common shares, which is uncertain and unpredictable, may be your sole source of gain from an investment in our common shares. An investment in our common shares may not be appropriate for investors who require dividend income.
We have never declared or paid cash dividends on our capital stock. We do not anticipate paying any cash dividends on our capital stock in the foreseeable future. We currently intend to retain all available funds and any future earnings to fund the development and growth of our business. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future. Accordingly, an investment in our common shares may not be appropriate for investors who require dividend income.
We are at risk of securities class action litigation.
In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies have experienced significant stock price volatility in recent years. If we face such litigation, it could result in substantial costs and a diversion of management's attention and resources, which could harm our business.
33
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus includes forward-looking statements. Statements regarding future events, expectations and beliefs of management and other statements that do not express historical facts, are forward-looking statements. In this prospectus, the words "believe", "may", "will", "estimate", "continue", "anticipate", "intend", "expect", "plan", "predict", "potential" and similar expressions, as they relate to OncoGenex, our business and our management, are intended to identify forward-looking statements. We have based these forward-looking statements largely on our current expectations and projections about future events and financial trends affecting the financial condition of our business. Forward-looking statements should not be read as a guarantee of future performance or results, and will not necessarily be accurate indications of the times at, or by, which such performance or results will be achieved. Forward-looking statements are based on information available at the time those statements are made and/or management's good faith belief as of that time with respect to future events, and are subject to risks and uncertainties that could cause actual performance or results to differ materially from those expressed in or suggested by the forward-looking statements. Important factors that could cause such differences include, but are not limited to:
- •
- delays or unfavorable results from our current and planned clinical trials;
- •
- our ability to establish and maintain intellectual property protection for our product candidates;
- •
- our access to additional capital;
- •
- our ability to enter into and maintain relationships with third parties, such as licensors, manufacturers, suppliers and those who conduct clinical trials for us;
- •
- our ability to enroll patients for our clinical trials;
- •
- our ability to implement and manage our sales and commercialization initiatives;
- •
- the impact of competition and technological change;
- •
- the timing of necessary regulatory clearances;
- •
- coverage and reimbursement policies of governmental and private third-party payors, including the Medicare and Medicaid programs;
- •
- general economic and business conditions, both nationally and in our markets;
- •
- our ability to attract and retain key management and scientific personnel;
- •
- existing and future regulations that affect our business; and
- •
- other risk factors included under "Risk Factors" in this prospectus.
In light of these risks and uncertainties, the forward-looking events and circumstances discussed in this prospectus may not occur and actual results could differ materially from those anticipated or implied in the forward-looking statements.
Forward-looking statements speak only as of the date the statements are made. You should not put undue reliance on any forward-looking statements. We assume no obligation to update forward-looking statements to reflect actual results, changes in assumptions or changes in other factors affecting forward-looking information, except to the extent required by applicable securities laws. If we do update one or more forward-looking statements, no inference should be drawn that we will make additional updates with respect to those or other forward-looking statements.
34
We estimate the net proceeds to us from the sale of the 5,000,000 common shares in this offering will be approximately $35.2 million, at an assumed initial public offering price of $8.00 per share and after deducting underwriting discounts and commissions and estimated offering expenses. If the underwriters' overallotment option is exercised in full, we estimate the net proceeds to us will be approximately $40.8 million.
We anticipate using the net proceeds from this offering together with our available cash resources and revenues, if any, as follows:
- •
- to complete our five phase 2 clinical trials of OGX-011 — approximately $1.0 million;
- •
- to complete pre-clinical studies and two phase 1 clinical trials of OGX-427 — approximately $1.5 million;
- •
- to fund additional capital equipment required by a third party contract manufacturing company approximately $1.0 million;
- •
- to manufacture clinical trial drug supplies, initiate manufacturing method validation and plan and initiate our first phase 3 clinical trial of OGX-011 — approximately $23.5 million; and
- •
- the balance of the net proceeds not used for the specific purposes listed above will be used for general corporate purposes and working capital.
As of January 31, 2007, we had cash, cash equivalents, short-term and long-term investments of approximately $7.2 million in the aggregate. We expect our existing capital resources and the net proceeds from this offering to be sufficient to enable us to maintain currently planned operations through at least early 2009.
We will need to raise substantial additional capital to fund our operations and to complete development of our product candidates. We may obtain this funding through a variety of means in the future, including, among other means, corporate partnering arrangements and collaborations and through debt and equity financings.
The actual costs and timing of clinical trials are highly uncertain, subject to risk and may change depending upon the clinical indication targeted, the development strategy pursued and the results of pre-clinical studies and earlier clinical trials. The amounts and timing and the allocation of resources among our existing clinical development programs, as well as our other expenditures, will depend upon numerous factors, including the status of our product development and commercialization efforts, the amount of proceeds actually raised in this offering, competition, any strategic partnership arrangements we may enter into and any additional product candidates we may in-license. As a result, our management will have broad discretion to allocate the net proceeds from this offering.
Net proceeds allocated to general corporate purposes and working capital may be used for, among other things: furthering our currently planned clinical trials, planning and initiating additional clinical trials, capital expenditures or general and administrative expenses. In addition, we may use a portion of such net proceeds for the acquisition of, or investment in, companies, technologies, products or assets that complement our business. We have not yet determined the manner in which we will allocate this portion of the net proceeds due to the fact that disease
35
indication, trial size and overall cost of our initial phase 3 clinical trial can only be determined upon completion of our ongoing phase 2 clinical trials.
Pending the use of the net proceeds of this offering, we intend to invest the net proceeds of this offering in short-term, investment-grade interest-bearing securities or guaranteed obligations of the Canadian or U.S. government.
We have never declared or paid any dividends on our capital stock. We currently intend to retain any future earnings to finance operations and the expansion of our business and do not intend to declare or pay cash dividends on our capital stock in the foreseeable future. Any future determination to pay dividends will be at the discretion of our board of directors and will depend upon our results of operations, financial condition, current and anticipated cash needs, contractual restrictions, restrictions imposed by applicable law and other factors that our board of directors deem relevant.
36
The following table sets forth our capitalization as of September 30, 2006:
- •
- on an actual basis; and
- •
- on an as adjusted basis to reflect the sale of the 5,000,000 common shares offered at an assumed initial public offering price of $8.00 per share, after deducting underwriting discounts and commissions and estimated offering expenses and the conversion of all our series preferred shares into an aggregate of 9,794,238 common shares pursuant to the Reorganization.
You should read this information together with our consolidated financial statements, including the related notes, and "Management's Discussion and Analysis of Financial Condition and Results of Operations" and "Reorganization" included elsewhere in this prospectus.
| | Actual as at December 31, 2005 | Actual as at September 30, 2006 | As Adjusted as at September 30, 2006 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | (In thousands, except share data) | ||||||||||
| Series 1 Class A preferred shares without par value, 760,000 designated as authorized, 513,394 issued and outstanding, actual; no shares designated as authorized, no shares issued and outstanding, as adjusted | $ | 1,966 | $ | 2,130 | $ | — | |||||
| Series 2 Class A preferred shares without par value, 420,000 designated as authorized, 335,411 issued and outstanding, actual; no shares designated as authorized, no shares issued and outstanding, as adjusted | 1,509 | 1,629 | — | ||||||||
| Series 1 Class B preferred shares without par value, 4,543,553 designated as authorized, 4,543,553 issued and outstanding, actual; no shares designated as authorized, no shares issued and outstanding, as adjusted | 14,475 | 15,348 | — | ||||||||
| Series 2 Class B preferred shares without par value, 5,058,084 designated as authorized, 4,401,895 issued and outstanding, actual; no shares designated as authorized, no shares issued and outstanding, as adjusted | 13,875 | 14,630 | — | ||||||||
| Shareholder equity (deficiency): | |||||||||||
| Common shares, without par value, unlimited shares authorized, 1,285,500 shares issued and outstanding, actual; unlimited shares authorized, 16,079,738 shares issued and outstanding, as adjusted | 399 | 399 | 69,356 | ||||||||
| Additional paid-in capital | 122 | 266 | 266 | ||||||||
| Deficit accumulated during the development stage | (16,154 | ) | (26,668 | ) | (26,668 | ) | |||||
| Accumulated other comprehensive income (loss) | 1,806 | 2,440 | 2,440 | ||||||||
| Total capitalization | $ | 17,998 | $ | 10,174 | $ | 45,394 | |||||
Actual and as adjusted shares excludes:
- •
- 1,424,147 common shares issuable upon the exercise of stock options outstanding, with a weighted average exercise price of C$0.92 per share; and
- •
- 481,410 common shares reserved for future stock option grants under our 2006 Stock Incentive Plan.
37
Our historical net tangible book value as of September 30, 2006 was approximately $(23.6) million, or $(18.33) per common share, based on 1,285,500 common shares outstanding. Historical net tangible book value per share is determined by dividing our total tangible assets less total liabilities and series preferred shares, by the actual number of our outstanding common shares. Our pro forma net tangible book value as of September 30, 2006 was approximately $10.2 million, or $0.92 per common share, based on 11,079,738 common shares outstanding after giving effect to the conversion of all outstanding series preferred shares into 9,794,238 common shares. Pro forma net tangible book value per share represents our total tangible assets less our total liabilities, divided by the pro forma number of common shares outstanding before giving effect to this offering.
After giving effect to the issuance and sale of 5,000,000 common shares in this offering at an assumed initial public offering price of $8.00 per share and after deducting underwriting discounts and commissions and estimated offering expenses, our pro forma net tangible book value as of September 30, 2006 would have been $45.4 million or $2.82 per share. This represents an immediate increase in pro forma net tangible book value to existing shareholders of $1.90 per share and an immediate dilution in pro forma net tangible book value of $5.18 per share to new investors purchasing our common shares in the offering at an initial public offering price of $8.00 per share. Dilution per share to new investors is determined by subtracting pro forma net tangible book value per share after this offering from the initial public offering price per share paid by a new investor. The following table illustrates the per share dilution without giving effect to the overallotment option granted to the underwriters:
| Initial public offering price per share | $ | 8.00 | |||||
| Historical net tangible book value per share as of September 30, 2006 | $ | (18.33 | ) | ||||
| Increase attributable to the conversion of series preferred shares | 19.25 | ||||||
| Pro forma net tangible book value per share as of September 30, 2006 | 0.92 | ||||||
| Increase per share attributable to new investors in this offering | 1.90 | ||||||
| Pro forma net tangible book value per share after this offering | 2.82 | ||||||
| Dilution of net tangible book value per share to new investors | $ | 5.18 | |||||
The following table sets forth, as of September 30, 2006, on the pro forma basis discussed above, the number of common shares purchased from us, the total consideration paid to us and the average price per share paid to us by existing shareholders and to be paid by new investors purchasing common shares in this offering. The table reflects an assumed initial public offering price of $8.00 per share, before deducting underwriting discounts and commissions and estimated offering expenses:
| | Shares Purchased | Total Consideration | | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Average Price Per Share | |||||||||||
| | Number | Percent | Amount | Percent | ||||||||
| Existing shareholders | 11,079,738 | 69 | % | $ | 28,606,000 | 42 | % | $ | 2.58 | |||
| New investors | 5,000,000 | 31 | % | $ | 40,000,000 | 58 | % | $ | 8.00 | |||
| Total | 16,079,738 | 100 | % | $ | 68,606,000 | 100 | % | $ | 4.27 | |||
38
The above discussion and tables exclude:
- •
- 1,424,147 common shares issuable upon the exercise of stock options outstanding, with a weighted average exercise price of C$0.92 per share; and
- •
- 481,410 common shares reserved for future stock option grants under our 2006 Stock Incentive Plan.
If the underwriters exercise their overallotment option in full to purchase 750,000 additional common shares in this offering, the pro forma net tangible book value per share after the offering would be $3.03 per share, the increase in pro forma net tangible book value per share to existing shareholders would be $2.11 per share and the dilution to new investors purchasing shares in this offering would be $4.97 per share.
39
SELECTED CONSOLIDATED FINANCIAL DATA
We have derived the selected consolidated statement of loss data for the years ended December 31, 2003, 2004 and 2005, for the nine months ended September 30, 2006 and for the period from May 26, 2000 (inception) to September 30, 2006 and the selected consolidated balance sheet data as of December 31, 2004 and 2005 and September 30, 2006, from our audited consolidated financial statements and our unaudited interim consolidated financial statements that are included elsewhere in this prospectus. We have derived the selected consolidated statements of operations data for the years ended December 31, 2001 and 2002 and the selected consolidated balance sheet data as of December 31, 2001, 2002 and 2003 from our audited consolidated financial statements that are not included in this prospectus. Our audited consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States. Historical results are not necessarily indicative of the results to be expected in the future periods.
You should read the following selected consolidated financial data together with our consolidated financial statements, including the related notes, and "Management's Discussion and Analysis of Financial Condition and Results of Operations" included elsewhere in this prospectus.
| | | | | | | Nine Months Ended September 30, | Period from May 26, 2000 (Inception) to September 30 2006 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Year Ended December 31, | |||||||||||||||||||||||||
| | 2005 | 2004 | 2003 | 2002 | 2001 | 2006 | 2005 | |||||||||||||||||||
| | (In thousands, except share and per share amounts) | |||||||||||||||||||||||||
| Consolidated Statements of Loss Data | ||||||||||||||||||||||||||
| Operating expenses: | ||||||||||||||||||||||||||
| Research and development | $ | 3,143 | $ | 2,778 | $ | 1,381 | $ | 1,124 | $ | 253 | $ | 6,822 | $ | 1,857 | $ | 15,502 | ||||||||||
| General and administrative (1) | 1,523 | 930 | 487 | 249 | 65 | 1,553 | 1,128 | 4,813 | ||||||||||||||||||
| Total expenses | 4,666 | 3,708 | 1,868 | 1,373 | 318 | 8,375 | 2,985 | 20,315 | ||||||||||||||||||
| Other income (expense): | ||||||||||||||||||||||||||
| Interest income | 313 | 199 | 41 | 17 | — | 357 | 160 | 928 | ||||||||||||||||||
| Other | (144 | ) | (156 | ) | (99 | ) | — | — | (111 | ) | (139 | ) | (510 | ) | ||||||||||||
| Total other income (expense) | 169 | 43 | (58 | ) | 17 | — | 246 | 21 | 418 | |||||||||||||||||
| Loss for the period before taxes | $ | 4,497 | $ | 3,665 | $ | 1,926 | $ | 1,356 | $ | 318 | $ | 8,129 | $ | 2,964 | $ | 19,897 | ||||||||||
| Tax expense | $ | 432 | $ | 346 | $ | — | $ | — | $ | — | $ | 473 | $ | 274 | $ | 1,251 | ||||||||||
| Net loss | $ | 4,929 | $ | 4,011 | $ | 1,926 | $ | 1,356 | $ | 318 | $ | 8,602 | $ | 3,238 | $ | 21,148 | ||||||||||
| Redeemable convertible preferred share accretion | $ | 1,843 | $ | 1,248 | $ | 385 | $ | 129 | $ | 3 | $ | 1,912 | $ | 1,202 | $ | 5,520 | ||||||||||
| Loss attributable to common shareholders | $ | 6,772 | $ | 5,259 | $ | 2,311 | $ | 1,485 | $ | 321 | $ | 10,514 | $ | 4,440 | $ | 26,668 | ||||||||||
| Basic and diluted loss per common share | $ | 5.43 | $ | 4.55 | $ | 2.01 | $ | 1.46 | $ | 0.35 | $ | 8.18 | $ | 3.59 | $ | 25.19 | ||||||||||
| Weighted average number of common shares | 1,248,158 | 1,155,500 | 1,148,925 | 1,018,958 | 906,050 | 1,285,500 | 1,235,573 | 1,058,755 | ||||||||||||||||||
| Pro forma basic and diluted loss per common share (2) | $ | 0.81 | $ | 0.83 | $ | 0.87 | $ | 0.93 | $ | 0.35 | $ | 0.95 | $ | 0.60 | $ | 5.95 | ||||||||||
| Pro forma weighted average number of common shares (2) | 8,365,079 | 6,374,239 | 2,648,968 | 1,589,776 | 919,066 | 11,079,738 | 7,450,248 | 4,485,061 | ||||||||||||||||||
- (1)
- Includes stock-based compensation expense as follows:
| $ | 55 | $ | 40 | $ | 15 | $ | 5 | $ | — | $ | 144 | $ | 40 | $ | 260 | |||||||||
- (2)
- Pro forma figures retroactively give effect to the Reorganization. See "Reorganization".
40
| | | December 31, | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | September 30, 2006 | ||||||||||||||||||
| | 2005 | 2004 | 2003 | 2002 | 2001 | ||||||||||||||
| | (In thousands) | ||||||||||||||||||
| Consolidated Balance Sheet Data | |||||||||||||||||||
| Cash, cash equivalents and short-term investments and long-term investments (1) | $ | 11,040 | $ | 18,369 | $ | 10,420 | $ | 6,233 | $ | 1,484 | $ | 1,088 | |||||||
| Working capital | 9,994 | 14,037 | 6,488 | 6,116 | 1,201 | 1,245 | |||||||||||||
| Total assets | 13,533 | 19,750 | 11,397 | 7,025 | 1,733 | 1,597 | |||||||||||||
| Net assets | 10,174 | 17,998 | 8,802 | 6,187 | 1,215 | 1,245 | |||||||||||||
| Redeemable convertible series preferred shares | 33,737 | 31,825 | 16,531 | 9,465 | 2,620 | 1,270 | |||||||||||||
| Common shares | 399 | 399 | 378 | 378 | 378 | 294 | |||||||||||||
| Deficit accumulated during the development stage | (26,668 | ) | (16,154 | ) | (9,382 | ) | (4,123 | ) | (1,812 | ) | (327 | ) | |||||||
| Total shareholders' equity (deficiency) | (23,563 | ) | (13,827 | ) | (7,729 | ) | (3,278 | ) | (1,405 | ) | (25 | ) | |||||||
- (1)
- Investments with maturities greater than one year are classified as long-term investments.
41
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION
AND RESULTS OF OPERATIONS
You should read the following discussion of our financial condition and results of operations in conjunction with the financial statements and the notes thereto included elsewhere in this prospectus. The following discussion contains forward-looking statements that reflect our plans, estimates and beliefs. Our actual results could differ materially from those discussed in the forward-looking statements. Factors that could cause or contribute to these differences include those discussed below and elsewhere in this prospectus, particularly in the "Risk Factors".
Overview
We are a biopharmaceutical company committed to the development and commercialization of new cancer therapies that address treatment resistance in cancer patients. Our product candidates are being developed to block the production of specific proteins associated with the development of treatment resistance which we believe will increase survival time and improve the quality of life for cancer patients. We currently have three product candidates in development: OGX-011, OGX-427 and OGX-225. Our lead drug candidate, OGX-011, is currently in five phase 2 clinical trials investigating the potential to improve treatment outcomes for patients with prostate cancer, non-small cell lung cancer and breast cancer. OGX-427, our second drug candidate, is currently being developed to improve treatment outcomes for patients with various solid and hematological cancers. OGX-427 is expected to enter clinical investigation in mid 2007. Our third drug candidate, OGX-225, is in pre-clinical development to assess its ability to inhibit or delay progression of hormone dependent and other tumors.
We incorporated in May 2000 and continue to be a development stage company. We have devoted substantially all of our resources to the development of our product candidates. To date we have funded our operations primarily through the private placement of equity securities. We have never been profitable and we incurred a loss before taxes for the nine months ended September 30, 2006 of $8.1 million and a cumulative loss before taxes of $19.9 million since our inception in 2000 through September 30, 2006. We expect our net losses to increase primarily due to our anticipated clinical trial activities. Clinical trials are costly, and as we continue to advance our product candidates through development, we expect our research and development expenses to increase significantly, especially as we initiate our phase 3 clinical trials of OGX-011. As compared to phase 1 and phase 2 clinical trials, phase 3 clinical trials are typically more expensive as they involve a greater number of patients, are conducted at multiple sites and sometimes in several countries, are conducted over a longer period of time and require greater quantities of drug product. In addition, we plan to expand our infrastructure and facilities and hire additional personnel, including clinical development, administrative, and marketing personnel. We are unable to predict when, if ever, we will be able to commence the sale of any of our product candidates.
Revenues
We have not generated any revenues to date, and we do not expect to generate any revenues from licensing or product sales until we execute a partnership or collaboration arrangement or are able to commercialize our product candidates ourselves.
42
Research and Development Expenses
Research and development expenses consist primarily of costs for: personnel, including salaries and benefits; regulatory activities; pre-clinical studies; clinical trials; materials and supplies; licensing and intellectual property; and allocations of other research and development-related costs. External research and development expenses include fees paid to universities, hospitals and other entities that conduct certain research and development activities and that manufacture our product candidates for use in our clinical trials. We expect our research and development expenses to increase significantly in the future as we continue to develop our product candidates. Currently, we manage our clinical trials through independent medical investigators at their sites and at hospitals.
A majority of our expenditures to date have been related to the development of OGX-011 and OGX-427. From inception in 2000 through September 30, 2006, we have incurred an aggregate of $15.5 million of research and development expenses. Of this amount, approximately $2.4 million was spent on pre-clinical activities required to advance the development of our drug candidates to the initiation of clinical trials, approximately $0.9 million was spent on conducting our phase 1 and phase 2 clinical trials, approximately $5.4 million was spent on manufacturing and supplies for our clinical trials and approximately $2.6 million was spent on licenses and intellectual property. The remaining amount of $4.2 million was expended primarily on employee costs, consultants, supplies, and other research and development administrative expenses.
OGX-011 is being co-developed with Isis. We share research and development expenses for OGX-011 on the basis of 65 percent OncoGenex and 35 percent Isis. Please see "Business" for a detailed description of our relationship with Isis. Several of our prostate cancer clinical trials have been supported by grant funding which was received directly by the hospitals and/or clinical investigators conducting the clinical trials allowing us to complete these clinical trials with minimal expense. From inception to September 30, 2006 we have spent approximately $880,000 on OGX-011 clinical trials as follows: prostate cancer, $190,000; non-small cell lung cancer, $625,000; and breast cancer, $65,000.
We expect to incur further research and development expenses of approximately $900,000 to complete our five ongoing phase 2 clinical trials of OGX-011.
We have completed enrollment of four of our five ongoing phase 2 clinical trials of OGX-011 and anticipate completing enrollment of all five clinical trials by early 2007. We then plan to evaluate the results of our ongoing clinical trials and determine which cancer indications and which treatment combinations demonstrate promise. Therefore, our initial phase 3 clinical trial may be in any of prostate, non-small cell lung or breast cancer. We anticipate that phase 3 clinical trials will be required for each cancer indication we decide to pursue. In addition to the clinical trials currently in progress, we may elect to pursue additional clinical trials for OGX-011 in other indications.
From inception to September 30, 2006 we have spent approximately $3.8 million on OGX-427 for pre-clinical expenses and manufacturing supplies. We expect to incur further research and development expenses of approximately $1.6 million to complete our pre-clinical development and two phase 1 clinical trials of OGX-427.
Since our drug candidates are in the early stage of development, we cannot estimate completion dates for development activities or when we might receive material net cash inflows from our research and development projects.
43
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related costs for our personnel in executive, business development, human resources, external communications, finance and other administrative functions, as well as consulting costs, including market research and business consulting. Other costs include professional fees for legal and accounting services, insurance and facility costs. We anticipate that general and administrative expenses will increase significantly in the future as we continue to expand our operating activities and as a result of costs associated with being a public company. From inception through September 30, 2006, we have incurred an aggregate of $4.8 million of general and administrative expenses.
Critical Accounting Policies
Our management's discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with United States generally accepted accounting principles. The preparation of these financial statements requires us to make judgments, assumptions, and estimates that affect the amounts reported in the financial statements and accompanying notes. Actual results could differ from those estimates.
While our significant accounting policies are described in more detail in Note 2 to our consolidated financial statements included elsewhere in this prospectus, we believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our financial statements.
Stock-Based Compensation
Effective January 1, 2006, we adopted the fair value recognition provisions of the Financial Accounting Standards Board Statement No. 123(R) (or SFAS 123(R)), "Share-Based Payment," using the modified prospective method with respect to options granted to employees and directors. Under this transition method, compensation cost is recognized in the financial statements beginning with the effective date for all share-based granted after January 1, 2006 and for all awards granted prior to but not yet vested as of January 1, 2006. The expense is amortized on a straight-line basis over the vesting period. Accordingly, prior period amounts have not been restated.
Since inception we have accounted for stock options issued to non employees using the fair value method and recognized the compensation on our statement of loss.
Stock-based compensation expense, which is a non-cash charge, results in part from estimating the fair value of stock options granted using the Black Scholes option pricing model. The Black Scholes option pricing model requires the input of several subjective assumptions including the expected life of the option and the expected volatility at the time the option is granted as well as the input of the fair value of our shares at the date of grant of the stock options. The estimated fair value is amortized over the vesting period, which is generally three years.
Given the absence of an active market for our common shares, our board of directors is required to estimate the fair value of our common stock. The board performed a contemporaneous analysis of fair value at each grant date and did not use an unrelated valuation specialist as the board determined that the benefits of using such a specialist were not warranted given the stage of the company's development and limited resources. Our board considered
44
numerous objective and subjective factors in determining the value of our common shares at each option grant date, including the factors described below:
- •
- the option grants involved illiquid securities in a private company;
- •
- prices for our series preferred shares, which we have sold to outside investors in arms-length transactions, and the rights, preferences and privileges of the series preferred shares over the common shares, especially with respect to liquidation;
- •
- progress and milestones achieved in our business; and
- •
- the likelihood of achieving a liquidity event for the common shares underlying these options, such as an initial public offering or sale of the Company, given prevailing market conditions.
In January 2007 management performed a retrospective analysis of the fair value of our common stock covering the period from December 2003 through March 2006. We used the market approach valuation methodology under which we measured our enterprise value through comparisons to recent valuations of private and publicly-traded North American biopharmaceutical companies at a similar stage and with a similar focus. We applied a 40% discount to the public companies to account for the typically higher valuations recognized by public biopharmaceutical companies over similar private companies. We then applied the option-pricing method to allocate enterprise value between common and preferred shares, taking into account the liquidation preferences of the preferred shareholders. Estimating the volatility of the share price of a private company is complex because there is no readily available market for the shares. We estimated the annualized volatility of our shares for the purposes of this analysis based on the historical volatility of our preferred share financings from December 2001 through August 2005. Had we used different estimates of volatility, the allocations between preferred and common shares would have been different. Based on this valuation analysis, we concluded that the estimated values of common shares of C$0.90 for the period from December 2003 to August 2005 and C$0.95 for the period from August 2005 to March 2006 used for stock option valuation at those dates were reasonable.
We recorded total stock-based compensation expense for non-employees of $15,000, $40,000 and $55,000 for the years ended December 31, 2003, 2004 and 2005 respectively. For the nine months ended September 30, 2006 we recorded total stock-based compensation expense for both non-employees and employees of $144,000.
We expect our stock-based compensation charges to increase as we expand our operations and hire new employees. These charges will increase our expenses and may increase our losses for the foreseeable future. As stock-based compensation is a non-cash charge, it will not have any effect upon our liquidity or capital resources.
Research and Development Costs
Research and development expenditures are charged to operations as incurred, pursuant to SFAS No. 2,Accounting for Research and Development Costs. Costs to acquire technologies to be used in research and development, but which have not reached technological feasibility and have no alternative future use are expensed when incurred. Payments to licensors that relate to the achievement of pre-approval development milestones are recorded as research and development expense when incurred. Research and development costs for activities conducted through third
45
parties with whom we contract are expensed as the costs are incurred. To the extent we make a payment to a third party vendor representing a refundable deposit, such payment is recorded as a prepaid expense. These third party vendors may include contract research organizations, third-party manufacturers of drug material and clinical supplies and other vendors. Investigator costs related to patient enrollment are accrued as patients enter the clinical trial. We monitor patient enrollment levels and related activities to the extent possible through internal reviews and correspondence and discussions with external vendors in order to estimate our incurred expenses. Due to the possibility of incomplete or inaccurate information, we may underestimate or overestimate activity levels and related expenses associated with any of our clinical trials at a given point in time. In such an event, we would record adjustments to research and development expenses in future periods when the actual activity level becomes known. In the past, we have not had to make any material adjustments to research and development expenses due to deviations between the estimates used in determining our accruals for clinical trial expenses and the actual clinical trial expenses incurred. Additionally, we do not expect material adjustments to research and development expenses to result from changes in the nature and level of clinical trial activity and related expenses that are currently subject to estimation. In the future, as we expand our clinical trial activities for our product candidates, we expect to have increased levels of research and development costs that will be subject to estimation. Our processes pertaining to those estimates may need to be enhanced in order to continue to adequately determine our accruals for those costs.
We have received funding in the form of investment tax credits (ITC) through the Government of Canada's Scientific Research and Development (SR&ED) program since 2002. We do not record the funds as income, but apply them against the costs of the qualified expenditures; generally research and development expenses. Generally, a Canadian-controlled private corporation (CCPC) can earn a refundable ITC of 35 percent up to the first Cdn $2 million of qualified expenditures for SR&ED carried out in Canada, and 20 percent on any excess amount. After a public offering, we will no longer qualify as a CCPC, but we will be eligible to earn an ITC of 20 percent of qualified expenditures for SR&ED carried out in Canada. The ITC earned by a Canadian corporation that is not a CCPC is non-refundable, but may be used to reduce any taxes payable. Unused ITC's may be carried forward indefinitely.
Income Taxes
We use the liability method for accounting for income taxes. Under this method, deferred tax assets and liabilities are determined based on differences between financial reporting and tax reporting bases of assets and liabilities and are measured using enacted tax rates and laws that are expected to be in effect when the differences are expected to reverse. Currently, there is no provision for income taxes, as we have incurred net losses to date. We have not recorded a benefit from our net operating loss carry-forwards because we believe that it is uncertain that we will have sufficient income from future operations to realize the carry-forwards prior to their expiration. Accordingly, we have established a valuation allowance against the deferred tax asset arising from the carry-forwards. In the event that we were to determine that we would be able to realize our deferred tax assets in the future, an adjustment to the valuation allowance would increase net income in the period such determination was made. We believe that the most significant uncertainty that will impact the determination of our valuation allowance will be our estimation of the extent and timing of future net income, if any.
46
Results of Operations
We have a limited operating history. We present below a comparison of our results of operations for the nine months ended September 30, 2006 compared to the nine months ended September 30, 2005, the year ended December 31, 2005 compared to the year ended December 31, 2004 and for the year ended December 31, 2004 compared to the year ended December 31, 2003.
Nine Months Ended September 30, 2006 Compared to Nine Months Ended September 30, 2005
Research and development expenses for the nine months ended September 30, 2006 were $6.8 million compared to $1.9 million for the nine months ended September 30, 2005, an increase of $4.9 million due mainly to pre-clinical and manufacturing expenses for the development of OGX-427, manufacturing expenses for clinical trial supplies of OGX-011 and the addition of eight new employees including the Chief Medical Officer, the V.P. Regulatory Affairs, and the Senior Director of Medical Affairs and Clinical Research who were hired in the fourth quarter of 2005. The increase in development expenses was offset by a decrease in the upfront license fees for OGX-427 incurred in the second quarter of 2005.
General and administrative expenses for the nine months ended September 30, 2006 were $1.6 million compared to $1.1 million for the nine months ended September 30, 2005, an increase of $0.5 million due mainly to higher spending on salaries and related expenses required to expand the business.
Interest income for the nine months ended September 30, 2006 was $0.36 million compared to $0.16 million for the nine months ended September 30, 2005, an increase of $0.20 million. The increase is due to the increase in cash balances available to be invested and higher yields realized on those investments.
Year Ended December 31, 2005 Compared to Year Ended December 31, 2004
Research and development expenses for the year ended December 31, 2005 were $3.1 million compared to $2.8 million for the year ended December 31, 2004, an increase of $0.3 million which is primarily due to the net impact of $1.5 million lower manufacturing expenses for OGX-011 and OGX-427, $0.8 million higher intellectual property expenses related to the acquisition of a license for OGX-427; $0.2 million higher clinical development expenses; and $0.7 million higher employee and administration expenses.
General and administrative expenses for the year ended December 31, 2005 were $1.5 million compared to $0.9 million for the year ended December 31, 2004. The $0.6 million increase in general and administrative expenses was due primarily to the addition of staff positions in the areas of business development, intellectual property and contract management, and senior accounting. The office costs and other related expenses also increased with the expansion of office space and the addition of the new positions.
Interest income for the year ended December 31, 2005 was $0.3 million compared to $0.2 million for the year ended December 31, 2004. The $0.1 million 2005 increase resulted primarily from the increase in cash balances available to be invested and higher yields realized on those investments.
47
Year Ended December 31, 2004 Compared to Year Ended December 31, 2003
Research and development expenses for the year ended December 31, 2004 were $2.8 million compared to $1.4 million for the year ended December 31, 2003, an increase of $1.4 million which is primarily due to the net impact of $2.0 million higher manufacturing expenses of OGX-011, $0.8 million lower intellectual property and pre-clinical expenses related to the acquisition of a license for OGX-225 in 2003, and $0.2 million higher employee and administration expenses due to the increase in staffing of the clinical team.
General and administrative expenses for the year ended December 31, 2004 were $0.9 million compared to $0.5 million for the year ended December 31, 2003. The $0.4 million increase in general and administrative expenses was due primarily to the increase in staffing and the increase in consulting fees and marketing expenses.
Interest income for the year ended December 31, 2004 was $0.20 million compared to $0.04 million for the year ended December 31, 2003. The $0.16 million increase resulted primarily from the increase in cash balances available to be invested and higher yields realized on those investments.
Liquidity and Capital Resources
We have incurred cumulative losses attributable to common shareholders of $26.7 million since inception through September 30, 2006. We do not expect to generate revenue from product candidates for several years. Since inception, we have funded our operations primarily through the private placement of our preferred shares. We raised net proceeds of $1.4 million through the sale of our Series A preferred shares in 2001/2002, $1.2 million through the sale of our Series A preferred shares in 2002, $5.7 million through the sale of our Series 1 Class B preferred shares in 2003, $5.8 million through the sale of our Series 1 Class B preferred shares in 2004 and $12.7 million through the sale of our Series 2 Class B preferred shares in August 2005.
As at September 30, 2006, we had cash, cash equivalents, short-term and long-term investments of $11.0 million in the aggregate. At December 31, 2005, our aggregate cash, cash equivalents, short-term and long-term investments were $18.4 million as compared to $10.4 million at December 31, 2004 and $6.2 million at December 31, 2003.
We do not have any borrowing or credit facilities available to us.
Cash Flows
Cash Used in Operations
For the nine months ended September 30, 2006 cash used in operations of $7.6 million was attributable primarily to our loss offset by an increase in accounts payable primarily related to the manufacturing of the first batch of OGX-427 of $0.6 million and an increase in taxes payable due to Part VI.1 tax of $0.5 million.
For the year ended December 31, 2005, cash used in operations of $5.3 million was attributable primarily to our loss less items not involving cash of $3.6 million and a decrease in accounts payable due mainly for payments made for clinical trial supplies expensed in 2004 of $1.0 million and an increase in taxes payable due to Part VI.1 tax of $0.5 million.
48
For the year ended December 31, 2004, cash used in operations of $2.2 million was attributable primarily to our loss less items not involving cash of $3.6 million, partially offset by an increase in accounts payable primarily for invoices received for clinical trial supplies paid in early 2005 of $1.4 million and an increase in taxes payable due to Part VI.1 tax of $0.4 million.
For the year ended December 31, 2003 cash used in operations of $1.3 million was attributable primarily to our loss less items not involving cash of $1.1 million, partially offset by an increase in accounts payable of $0.3 million, and an increase in investment tax credits and other receivables for clinical trial reimbursement costs of $0.5 million.
Cash Provided by Financing Activities
Net cash used by financing activities for the nine months ended September 30, 2006 was for deferred share issue costs related to the initial public offering.
Net cash provided by financing activities for the nine months ended September 30, 2005 was $12.7 million and the years ended December 31, 2005, 2004 and 2003 was $12.7 million, $5.8 million and $5.7 million, respectively. Net cash provided by financing activities in all three years was related to the sale of preferred shares.
Cash Used /Provided by Investing Activities
Net cash provided by investing activities was $7.1 million for the nine months ended September 30, 2006 due primarily to maturities of short-term and long-term investments. Net cash used in investing activities was $8.1 million for the year ended December 31, 2005 due primarily to the purchases of both short-term and long-term investments of $8.0 million (net of maturities) and the purchases of equipment of $0.1 million. Net cash used by investing activities was $3.4 million for the year ended December 31, 2004 due primarily to the purchase of short-term and long-term investments of $3.2 million (net of maturities) and the purchases of equipment of $0.2 million. Net cash used in investing activities was $2.9 million for the year ended December 31, 2003 due primarily to net purchases of short-term investments.
Operating Capital and Capital Expenditure Requirements
We believe that the net proceeds from this offering, together with our cash, cash equivalents, short-term investments and long-term investments, will be sufficient to fund our currently planned operations through at least early 2009, including:
- •
- completion of our ongoing phase 2 clinical trials of OGX-011;
- •
- completion of our pre-clinical studies and two phase 1 clinical trials of OGX-427;
- •
- planning and initiating our first phase 3 clinical trial of OGX-011; and
- •
- working capital, capital expenditures and general corporate purposes.
We anticipate the need to raise additional capital or incur indebtedness to continue to fund our operations in the future.
We expect to incur losses from operations in the future. We expect to incur increasing research and development expenses, including expenses related to clinical trials and additional personnel. We expect that our general and administrative expenses will increase in the future as
49
we expand our staff, add infrastructure and incur additional expenses related to being a public company, including directors' and officers' insurance, investor relations and increased professional fees.
Contractual Obligations and Commitments
We have no capital leases or debt obligations. Our operating lease and purchase obligations as of December 31, 2005 were as follows:
| | Payments Due By Period | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Total | Less than 1 Year | 1-3 Years | 3-5 Years | More than 5 Years | ||||||||||
| Vancouver operating lease | $ | 463,000 | $ | 121,000 | $ | 342,000 | $ | — | $ | — | |||||
| Seattle operating lease | 174,000 | 65,000 | 109,000 | — | — | ||||||||||
| Purchase obligations(1) | 106,000 | 94,000 | 12,000 | — | — | ||||||||||
| Total | $ | 743,000 | $ | 280,000 | $ | 463,000 | $ | — | $ | — | |||||
- (1)
- Consists of agreements to purchase goods and services. In addition, we are required to make certain payments to UBC and Isis under the terms of our license and collaboration agreements, upon the occurrence of certain clinical milestones and upon the generation of revenue from our product candidates. Due to the uncertainty of the timing and occurrence of these milestones and the generation of royalty-generating revenue, these payments have not been classified as purchase obligations and are not estimated in the foregoing table. See "Business" for a detailed description of our milestone and royalty obligations.
Income Taxes
We have established a wholly-owned subsidiary, OncoGenex Inc., a United States based company which employs eight members of our clinical and regulatory team. We are required to file separate income tax returns for both OncoGenex and the subsidiary. Canadian income tax rules require us to treat the United States based operations as an arms length company and require the services provided by the United States subsidiary to be charged to us at fair market value. All profit and losses are eliminated upon consolidation.
Canadian Operations
We have incurred net operating losses on a consolidated basis for the years ended December 31, 2005, 2004 and 2003 and, accordingly, we did not pay or record any federal taxes. As of December 31, 2005, we had non-capital loss carry forwards of $8.3 million which expire over various periods to the year 2015. We also had unclaimed tax deductions of approximately $2.7 million related to scientific research and experimental development expenditures available to carry forward indefinitely to reduce taxable income of future years.
As of December 31, 2005 we had deferred tax assets of $5.2 million, of which $0.8 million relate to the tax basis in excess of book value of assets, $0.9 million of research and development deductions and credits available indefinitely, $2.8 million in non-capital loss carry-forwards and $0.6 million for taxes accrued on the redeemable convertible preferred share accretion as these
50
taxes are available to reduce future income taxes payable. A valuation allowance is provided to offset the deferred tax assets because the realization of the benefit does not meet the more likely than not criteria. In the event that we determine that we will be able to utilize our deferred tax assets in the future, an adjustment to the valuation allowance would increase net income in the period such a determination is made.
United States Operations
Our United States operations were minimal for the year ended December 31, 2005.
Other taxes
In the event that holders of Class A and Class B preferred shares are paid the cumulative preferred return adjustment, the Company would become liable for payment of taxes under Part VI.1 of the Income Tax Act (Canada) which is calculated at 25% of the amount paid in excess of Cdn $500,000. On the payment of this tax, the Company will be entitled to claim a deduction equal to nine-fourths times the amount of any Part VI.1 taxes actually paid. The Company has accrued for the Part VI.1 tax liability in its financial statements.
Inflation
We do not believe that inflation has had a material impact on our business and operating results during the periods presented.
Foreign Currency Fluctuations
For a description of the effect on us of foreign currency fluctuations, see "Quantitative and Qualitative Disclosure of Market Risks".
Related Party Transactions
For a description of our related party transactions, see "Certain Relationships and Related Party Transactions".
Off-Balance Sheet Arrangements
Since inception, we have not engaged in any off-balance sheet activities, including the use of structured finance, special purpose entities or variable interest entities.
Quantitative and Qualitative Disclosure of Market Risks
Our concentration of credit risk consists principally of cash, cash equivalents, and short-term investments. Our exposure to market risk is limited primarily to interest income sensitivity, which is affected by changes in the general level of Canadian and United States interest rates, particularly because the majority of our investments are in short-term debt securities.
Our investment policy restricts investments to high-quality investments and limits the amounts invested with any one issuer, industry, or geographic area. The goals of our investment policy are as follows: preservation of capital; assurance of liquidity needs; best available return on invested capital; and minimization of capital taxation and a reduction of impact on Scientific Research and Experimental Development refundable tax credits under theIncome Tax Act (Canada). Some of
51
the securities in which we invest may be subject to market risk. This means that a change in prevailing interest rates may cause the principal amount of the investment to fluctuate. For example, if we hold a security that was issued with an interest rate fixed at the then-prevailing rate and the prevailing interest rate later rises, the principal amount of our investment will probably decline. To minimize this risk, in accordance with our investment policy, we maintain our portfolio of cash equivalents, short-term marketable securities and restricted cash in a variety of securities, including money market mutual funds, T-bills, GICs, and commercial papers. The risk associated with fluctuating interest rates is limited to our investment portfolio. Due to the short term nature of our investment portfolio we believe we have minimal interest rate risk arising from our investments.
As a Canadian company with our executive offices and a significant portion of our operations located in Canada, we are also subject to currency risk. Many of our expenditures in Canada, including payroll, are in Canadian dollars. In addition, the exercise prices of stock options we grant under our stock compensation plans have historically been expressed in Canadian dollars. We are therefore subject to currency risk from changes in the United States dollar / Canadian dollar exchange rate. We do not currently hedge our exposures to currency risk. A hypothetical 10 percent change in the value of the Canadian dollar relative to the U.S. dollar during the fiscal year ended December 31, 2005 would have had a $400,000 impact on our total expenditures for the fiscal year ended December 31, 2005.
Recent Accounting Pronouncements
In July 2006, the Financial Accounting Standards Board (or FASB) issued Interpretation No. 48, "Accounting for Uncertainty in Income Taxes — an interpretation of FASB Statement No. 109", or FIN 48. This interpretation clarifies the accounting for uncertainty in income taxes recognized in financial statements in accordance with FASB Statement No. 109, "Accounting for Income Taxes." FIN 48 will require companies to determine whether it is more-likely-than-not that a tax position taken or expected to be taken in a tax return will be sustained upon examination, including resolution of any related appeals or litigation processes, based on the technical merits of the position. If a tax position meets the more-likely-than-not recognition threshold, it is measured to determine the amount of benefit to recognize in the financial statements based on guidance in the interpretation. FIN 48 is effective for fiscal years beginning after December 15, 2006. We have not determined the effect, if any, that the adoption of FIN 48 will have on our consolidated financial position or results of operations.
52
Overview
We are a biopharmaceutical company committed to the development and commercialization of new cancer therapies that address treatment resistance in cancer patients. Our product candidates are being developed to block the production of specific proteins that are associated with the development of treatment resistance, which we believe will increase survival time and improve the quality of life for cancer patients.
In response to many cancer therapies (including hormone ablation therapy, chemotherapy and radiation therapy), tumor cells become stressed and increase production of certain proteins. These proteins cause tumor cells to become resistant to the cancer therapies and promote the survival of tumor cells. Therefore, in spite of the initial effectiveness of cancer therapies, many patients develop treatment resistance and the patients die due to the lack of an effective therapy.
We currently have three product candidates in development: OGX-011, OGX-427 and OGX-225. These product candidates are designed to selectively inhibit the production of proteins that are associated with treatment resistance and that are over-produced in response to a variety of cancer treatments. Our aim in targeting these particular proteins is to disable the tumor cell's adaptive defenses, render the tumor cells susceptible to attack with a variety of cancer therapies, including chemotherapy, and facilitate tumor cell death.
Product Candidate Development Summary
| Product Candidate | Cancer Indication and Study | Treatment Combination | Development Phase/Status | Status of Patient Enrollment1 | Results/Status2 | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| OGX-011 | Localized Prostate Cancer (OGX-011-01) | OGX-011 with hormone ablation therapy | Phase 1 Completed | Fully enrolled, 25 patients | • Well tolerated by patients • No dose-limiting toxicity observed • Over 92% inhibition of clusterin mRNA in prostate cancer compared to hormone therapy alone • Double the number of tumor cells undergoing cell death compared to hormone therapy alone • 98% inhibition of clusterin mRNA in lymph nodes compared to hormone therapy alone | |||||
Solid Tumors (prostate, breast, NSCL, ovarian, renal, bladder, peritoneum) (OGX-011-02) | OGX-011 with chemotherapy (docetaxel) | Phase 1 Completed | Fully enrolled, 40 patients | • Well tolerated by patients across cancer indications evaluated • Maximum tolerated dose not reached | ||||||
53
Advanced Non-Small Cell Lung Cancer (OGX-011-05) | OGX-011 with initial chemotherapy (gemcitabine/cisplatin) | Phase 1 Completed | Fully enrolled, 10 patients | • Well tolerated by patients • No dose-limiting toxicity observed • 1-year survival = 67% (compared to published data3 of 33 to 43%)4 • Median survival = 17.1 months (compared to published data3 for median survival of 8 to 10.8 months) | ||||||
Hormone Refractory Prostate Cancer (OGX-011-03) | Initial chemotherapy (docetaxel/prednisone) with and without OGX-011 | Randomized Phase 2 Ongoing | Fully enrolled, 82 patients | • Primary endpoint data expected by the end of 2007 | ||||||
Hormone Refractory Prostate Cancer (OGX-011-07) | OGX-011 with secondary chemotherapy (docetaxel/prednisone or mitoxantrone/prednisone) | Randomized non-comparative Phase 2 Ongoing | 95% enrolled, 38 of 40 patients | • Accrual completion expected by early 2007 • Primary endpoint data expected by mid-2007 | ||||||
Advanced Non-Small Cell Lung Cancer (OGX-011-05) | OGX-011 with initial chemotherapy (gemcitabine/cisplatin) | Phase 2 Ongoing | Fully enrolled, 75 patients | • Primary endpoint data expected by the end of 2007 | ||||||
Localized Prostate Cancer (OGX-011-04) | OGX-011 with hormone ablation therapy | Phase 2 Ongoing | Fully enrolled, 23 patients | • Primary endpoint data expected by the end of 2007 | ||||||
Advanced Breast Cancer (OGX-011-06) | OGX-011 with secondary chemotherapy (docetaxel) | Phase 2 Ongoing | Fully enrolled, 15 patients | • Data from this trial is expected by mid-2007 | ||||||
OGX-427 | Solid Tumors | OGX-427 with and without chemotherapy | Pre-clinical Ongoing | NA | • Lead compound identified • Pre-clinical pharmacology completed • Clinical manufacturing completed • Investigational new drug filing expected by early 2007 • Patient treatment expected by mid 2007 | |||||
OGX-225 | Solid Tumors | OGX-225 with and without chemotherapy | Pre-clinical Ongoing | NA | • Lead compound identified • Pre-clinical pharmacology ongoing |
- (1)
- As of February 28, 2007.
- (2)
- See "Completed Phase 1 Clinical Trials" and "Ongoing Phase 2 Clinical Trials".
- (3)
- Prior published results from randomized clinical trials totalling 1,260 patients using gemcitabine and platinum-based chemotherapy.
- (4)
- For patients treated with at least one dose of OGX-011 in combination with chemotherapy. The one-year survival rate for all patients was 60 percent and included one patient that died before receiving any doses of OGX-011 in combination with chemotherapy.
54
While we are encouraged by the results of our clinical trials to-date, we will need to conduct additional clinical trials for each of our product candidates in order to generate the safety and efficacy data needed to support an application with the FDA. Successful early clinical trials do not ensure that later clinical trials will also be successful.
Our Lead Product Candidate, OGX-011
The development program for our lead product candidate, OGX-011, is focused on reducing clusterin production to enhance treatment sensitivity and delay tumor progression in patients who have not fully developed treatment resistance and to restore treatment sensitivity in patients who have developed treatment resistance. Clusterin is a cell survival protein that is over-produced in several cancer indications and in response to many cancer treatments, including hormone ablation therapy, chemotherapy and radiation therapy. Increased clusterin production is observed in many human cancers, including prostate, non-small cell lung, breast, ovarian, bladder, renal, pancreatic, anaplastic large cell lymphoma and colon cancers and melanoma. Increased clusterin production is linked to faster rates of cancer progression, treatment resistance and shorter survival duration.
A broad range of pre-clinical studies conducted by the Prostate Centre and others have shown that reducing clusterin production facilitates tumor cell death by sensitizing human prostate, non-small cell lung, breast, ovarian, bladder, renal, and melanoma tumor cells to chemotherapy. Pre-clinical studies conducted by the Prostate Centre and others also indicate that reducing clusterin production sensitizes prostate tumor cells to hormone ablation therapy and sensitizes prostate and non-small cell lung tumor cells to radiation therapy.
Three phase 1 clinical trials, involving a total of 75 patients, have been completed with OGX-011. In all of these clinical trials, OGX-011 was well tolerated by the patients.
In the first phase 1 clinical trial, OGX-011 was intravenously administered once per week in combination with hormone ablation therapy to patients with localized prostate cancer in advance of surgery to remove the prostate gland. This clinical trial showed that once weekly administration of OGX-011 reduced clusterin mRNA levels by approximately 92 percent in prostate cancer tissue and approximately 98 percent in lymph node tissue, and more than doubled the rate of prostate tumor cell death compared to hormone ablation therapy alone. Patients who received hormone ablation therapy alone, and who were used for comparison, had prostate cancer that was of similar stage to those patients treated with OGX-011.
In the second phase 1 clinical trial, OGX-011 was intravenously administered once per week in combination with docetaxel chemotherapy to patients with solid tumors known to express clusterin. This clinical trial showed that serum clusterin levels dropped in patients while on treatment with 640 mg OGX-011 in combination with docetaxel.
In the third phase 1 clinical trial, OGX-011 was intravenously administered once per week in combination with two commonly used chemotherapeutic agents to patients with advanced non-small cell lung cancer. The one-year survival rate for all ten patients was 60 percent and for patients that received at least one dose of OGX-011 in combination with these chemotherapies was approximately 67 percent. This compares with results from prior published randomized clinical trials which reported one-year survival rates of 33 to 43 percent for patients that received chemotherapy treatment alone. Patients in these published clinical trials who received chemotherapy alone, and who were used for comparison, had non-small cell lung cancer of similar stage to those patients treated with OGX-011.
55
OGX-011 is currently in five phase 2 clinical trials investigating the potential to improve treatment outcomes for patients with prostate cancer, non-small cell lung cancer and breast cancer. We are conducting these clinical trials in parallel, rather than sequentially, to accelerate our evaluation of OGX-011 in several cancer indications and in combination with several treatment regimes, which will accelerate our assessment of these indications and combinations for further development. We plan to evaluate the results of our ongoing phase 2 clinical trials to determine which cancer indications and which treatment combinations demonstrate promise and we will design our phase 3 clinical trials accordingly. We have completed enrollment of four of our five phase 2 clinical trials and we expect that data from these phase 2 clinical trials will be available by the end of 2007.
OGX-011 is being developed to work in combination with therapies that are broadly used by clinicians and considered highly effective in the treatment of each cancer indication that we are targeting with the intent of delaying treatment resistance to those therapies. Since production of clusterin and the resulting treatment resistance occurs in an array of cancer indications and in response to a variety of cancer treatments, we believe that our development options for OGX-011 are numerous.
Our Second Product Candidate, OGX-427
The development program for our second product candidate, OGX-427, is focused on reducing heat shock protein 27 (Hsp27) production to enhance treatment sensitivity and delay tumor progression in patients who have not fully developed treatment resistance and to restore treatment sensitivity in patients who have developed treatment resistance. Hsp27 is a cell survival protein that is over-produced in response to many cancer treatments, including hormone ablation therapy, chemotherapy and radiation therapy. Increased Hsp27 production is observed in many human cancers, including prostate, non-small cell lung, breast, ovarian, bladder, renal, pancreatic, multiple myeloma and liver cancers. Increased Hsp27 production is linked to faster rates of cancer progression, treatment resistance and shorter survival duration.
A number of pre-clinical studies conducted by the Prostate Centre and others have shown that inhibiting the production of Hsp27 in human prostate, breast, ovarian, pancreatic and bladder tumor cells sensitizes the cells to chemotherapy. The Prostate Centre has also conducted pre-clinical studies that indicate that reducing Hsp27 production sensitizes prostate tumor cells to hormone ablation therapy. Pre-clinical studies conducted by the Prostate Centre and others have shown that reducing Hsp27 production induces tumor cell death in prostate, breast, non-small cell lung, bladder and pancreatic cancers.
We intend to file our investigational new drug application for OGX-427 in early 2007 and begin dosing patients in our initial phase 1 clinical trial by mid-2007.
Our Third Product Candidate, OGX-225
The development program for our third product candidate, OGX-225, is focused on reducing the production of both insulin-like growth factor binding protein-2 (IGFBP-2) and insulin-like growth factor binding protein-5 (IGFBP-5) with a single product to enhance treatment sensitivity and delay tumor progression in patients who are resistant to hormone ablation therapy. In the treatment of cancers that require hormones for growth, clinicians use hormone ablation therapy to inhibit the production of the primary hormone (e.g. testosterone or estrogen) required for tumor growth. While tumors often regress initially following hormone ablation therapy,
56
IGFBP-2 or IGFBP-5 make an alternate hormone, insulin-like growth factor-1, or IGF-1, available to the tumor that facilitates continued tumor growth. By inhibiting the production of IGFBP-2 and IGFBP-5, the tumor's access to IGF-1 is reduced and tumor growth is delayed.
Increased IGFBP-2 or IGFBP-5 production is observed in several cancers, including prostate, non-small cell lung, breast, ovarian, bladder, pancreatic and colon cancers, acute myeloid leukemia, acute lymphoblastic leukemia, neuroblastoma, glioma and melanoma. Increased IGFBP-2 or IGFBP-5 production is linked to faster rates of cancer progression, treatment resistance and shorter survival duration.
Pre-clinical studies conducted by the Prostate Centre and others in human prostate, bladder, glioma and breast cancer, have shown that reducing IGFBP-2 and IGFBP-5 production with OGX-225 induced tumor cell death or sensitized all of these tumor types to chemotherapy. We intend to continue to evaluate OGX-225 in pre-clinical studies to assess its ability to inhibit or delay progression of hormone-dependent and other tumors.
Our Therapeutic Approach
We are focused on inhibiting the processes by which cancers develop treatment resistance. Since treatment resistance may be caused by several different biological processes, our choice of therapeutic approach will depend on what we believe is best suited to inhibit the specific treatment resistance target, taking into consideration its biology, including structure and function, and the cost and intended clinical use of the therapy.
Our current product candidates are designed to target and selectively inhibit the production of proteins that facilitate the survival and growth of tumor cells. A protein's influence in the body can be inhibited by either interfering with its structure and function, or by disrupting its production; however, the structure of certain proteins, such as clusterin and Hsp27, have not been determined and hence are difficult to inhibit by interfering with their structure or function. For this reason, we are focusing on disrupting the production of these proteins. We have chosen to disrupt the production of these proteins using "second-generation" antisense technology because we believe that it is the most effective means of disrupting protein production. To facilitate this approach, our protein production inhibitors have been combined with Isis' second-generation antisense chemistry to overcome the limitations of first-generation antisense technology.
The majority of antisense therapeutics that have been evaluated in late-stage human clinical trials are based on "first-generation" antisense chemistry. First-generation antisense therapeutics are susceptible to rapid degradation and loss of functionality when introduced to the human body. Thus these first-generation antisense therapeutics typically require daily dosing by continuous or frequent infusion to achieve sufficient concentrations to be effective.
In contrast, therapies that are based on second-generation antisense chemistry have increased target binding affinity, improved resistance to degradation and therefore better tissue distribution. These improvements result in slower clearance of the therapies from the body, allowing for less frequent dosing and thereby making treatment easier on patients and decreasing the associated cost.
Clinical data from our phase 1 clinical trial in prostate cancer patients demonstrated that weekly intravenous administration of OGX-011 resulted in drug distribution to prostate cancer tissue and over 92 percent inhibition of its target, clusterin mRNA, in prostate tumor cells in these patients. These data demonstrate that our second-generation antisense product candidate
57
can be systemically administered and results in the product candidate entering tumor cells and effectively inhibiting clusterin production.
Our Collaborators
Isis Pharmaceuticals
We have established a co-development and license agreement with Isis in which we jointly direct the development activities for OGX-011. This strategic relationship provides us with access to Isis' proprietary second-generation antisense chemistry for use in OGX-011. We have been responsible for developing and managing the clinical trials for OGX-011 and for establishing the pre-clinical pharmacology, while Isis has been responsible for product manufacturing and completing the pre-clinical toxicology. We share with Isis, on a proportionate share basis of 65 percent OncoGenex and 35 percent Isis, the costs and revenues related to the development and commercialization of OGX-011.
We have also entered into license and collaboration agreements with Isis for OGX-427 and OGX-225. These agreements provide us with access to Isis' proprietary second-generation antisense chemistry which has been incorporated into OGX-427 and OGX-225. Following our joint discovery and lead optimization efforts with Isis, we are solely responsible for all product development activities for each of these product candidates. Under these agreements, product development milestones and product sales royalties may be payable to Isis.
Prostate Centre at Vancouver General Hospital/University of British Columbia
OncoGenex was created to further develop and commercialize certain product candidates which were discovered by the Prostate Centre and licensed to us by UBC in 2001. UBC is responsible for licensing activities related to inventions derived through the Prostate Centre.
The Prostate Centre is a research facility dedicated primarily to understanding the causes of prostate disease progression. Its resources for discovery and research include an established human tumor tissue bank that provides the basis for identifying targets associated with treatment resistance. The Prostate Centre has assembled a trans-disciplinary team of more than 120 basic and clinical researchers and support staff involved in activities from discovery to clinical trials. The strategy that the Prostate Centre employs is to conduct its initial research in the area of prostate cancer and then seek application in other cancer indications. Our other product candidates, OGX-427 and OGX-225, were also derived from technologies discovered by the Prostate Centre, and were licensed to us by UBC between 2001 and 2005. The initial discovery and research activities undertaken by the Prostate Centre and UBC with respect to our product candidates have resulted in significant cost efficiencies to us. Under our license agreements with UBC, product development milestones and product sales royalties may be payable to UBC.
Our Strategy
- •
- Focus on developing and commercializing new cancer therapies to inhibit treatment resistance in cancer patients.
We are focused on inhibiting the processes by which cancers develop treatment resistance. Since treatment resistance may be caused by several different biological processes, our choice of therapeutic approach will depend on what we believe is best suited to inhibit the specific
58
treatment resistance target, taking into consideration its biology, including structure and function, and the cost and intended clinical use of the therapy.
Our current product candidates are designed to target and selectively inhibit the production of proteins that facilitate the survival and growth of tumor cells. Our aim in targeting these particular proteins is to disable the tumor cell's adaptive defenses, render the tumor cells susceptible to attack with a variety of cancer therapies, including chemotherapy, and facilitate tumor cell death.
- •
- Focus on enhancing the effectiveness of broadly used cancer therapeutics through combination with OGX-011.
We are developing OGX-011 in combination with therapies that are broadly used by clinicians and considered highly effective in the treatment of each cancer indication with the intent of delaying treatment resistance to those therapies. Since production of clusterin and the resulting treatment resistance occurs in an array of cancer indications and in response to a variety of cancer treatments, we have assessed and will continue to assess OGX-011 in combination with various existing and future treatments.
- •
- Accelerate the assessment of OGX-011 by conducting concurrent clinical trials in multiple indications.
Since clusterin expression is associated with treatment resistance in a broad spectrum of cancer indications and treatment combinations, OGX-011 is being evaluated in five parallel phase 2 clinical trials. This will accelerate our assessment of OGX-011 for these indications and treatment combinations for further development. If our ongoing phase 2 clinical trials indicate efficacy in more than one indication and treatment combination, we may elect to initiate more than one phase 3 clinical trial of OGX-011.
- •
- Advance our product pipeline by conducting clinical trials across multiple cancer indications for each of OGX-427 and OGX-225.
Concurrent with our programs for OGX-011, we are continuing to develop our other two product candidates, OGX-427 and OGX-225. Consistent with the strategy we are following for OGX-011, we intend to conduct parallel clinical trials to evaluate these product candidates in several cancer indications and treatment combinations to accelerate our assessment of these product candidates for further development.
We intend to maintain and develop our relationships with the Prostate Centre and other research institutions in order to identify and source additional product candidates.
- •
- Optimize the development of our product candidates through use of outsourcing and internal expertise.
We consider various factors, including cost, available external expertise, capacity and quality assurance in determining which activities will be conducted internally versus those that will be carried out externally. The product development functions that we have chosen to establish within the company include clinical trial management and regulatory affairs.
We have chosen to acquire our product candidates through our relationships with leading research institutions. This strategy has allowed us to avoid the significant expense and risk that we believe is associated with internal discovery programs. To date, we have relied on the Prostate Centre for discovery capabilities that would be difficult or impossible for us to recreate. We
59
anticipate continuing to source additional product candidates through our relationships with the Prostate Centre and other research institutions.
We have also chosen to outsource pre-clinical and manufacturing activities. With respect to pre-clinical activities, we have established relationships with laboratories internationally which we believe enable us to complete requisite pre-clinical testing faster and more efficiently than if we performed these activities ourselves. With respect to manufacturing activities, our manufacturing needs are currently sporadic and therefore we have not internalized this function.
- •
- Selectively establish collaborations that expand capabilities and accelerate development and marketing of our product candidates.
We may establish an agreement with a pharmaceutical or biotechnology company that has substantial drug development and sales and marketing capabilities in oncology to augment our development activities and to market our products. This strategy is intended to maximize our returns from our product candidates by enabling us to utilize the partner's marketing capabilities and, if the partner's funding is sufficient, to further accelerate development of our product candidates in more than one cancer indication simultaneously.
Cancer — Treatment and Resistance
Cancer is a group of diseases characterized by the uncontrolled growth and spread of abnormal cells. As cancer progresses, the tumor cells may invade other tissues throughout the body producing additional tumors, called metastases. Cancer growth can cause tissue damage, organ failure and, ultimately, death.
In North America, cancer is expected to strike one in two men and one in three women in their lifetimes and has recently surpassed heart disease as the leading cause of death in the United States. The American Cancer Society estimates that in 2006 approximately 1,399,790 new patients in the United States will be diagnosed with cancer and that there will be approximately 564,830 patient deaths attributable to these cancers.
Typically, cancer treatment is given sequentially and can include surgery, radiation therapy, chemotherapy and hormone therapy. Although a particular therapy may initially be effective, tumor cells often react to therapeutic treatment by increasing the production of proteins that afford them a survival advantage, which enable them to become resistant to therapy, multiply and spread to additional organs. As a result, many patients progress rapidly through all available therapies and ultimately die.
The progression cycle of cancer is often described as a four-stage process, where:
- •
- Stage I usually means a tumor is relatively small and contained within the organ in which it started;
- •
- Stage II usually means the cancer is localized, but the tumor is larger than in Stage I. In some cases, Stage II means there are nearby lymph nodes containing tumor cells;
- •
- Stage III usually means the tumor is larger and there are tumor cells in the lymph nodes in the area; and
- •
- Stage IV means that the cancer has spread from where it started to another body organ, such as the liver, bone or lungs.
60
- •
- For many cancers, these stages are further subdivided into an "A" or "B" stage.
Our Approach to Treatment Resistance
We are focused on inhibiting the processes by which cancers develop treatment resistance. Since treatment resistance may be caused by several different biological processes, our choice of therapeutic approach will depend on what we believe is best suited to inhibit the specific treatment resistance target, taking into consideration its biology, including structure and function, and the cost and intended clinical use of the therapy.
Inhibiting the Production of Proteins that are Associated with Treatment Resistance
Our current product candidates are designed to target and selectively inhibit the production of proteins that facilitate the survival and growth of tumor cells. A protein's influence in the body can be inhibited by either interfering with its structure and function or disrupting its production; however, the structure of certain proteins, such as clusterin and Hsp27, have not been solved and hence are difficult to inhibit by interfering with their structure or function. For this reason, we are focusing on disrupting the production of these proteins. We have chosen to disrupt the production of these proteins using second-generation antisense technology because we believe that it is the most effective means of disrupting protein production. To facilitate this approach, our protein production inhibitors have been combined with Isis' second-generation antisense chemistry to overcome the limitations of first-generation antisense technology.
Background on Antisense Drug Chemistry
Proteins are the functional workhorses of a cell. They are complex molecules assembled from simpler building blocks (twenty distinct amino acids). Amino acids are linked together in series in a single chain to generate a protein. The order in which amino acids are linked determines the identity of a protein (i.e., a protein may be described by its particular sequence of amino acids). The order of amino acids in a given protein chain is in turn dictated by a corresponding gene.
Genes are composed of DNA and are located on chromosomes in the nucleus of a cell. The nucleus of a human cell contains 23 pairs of chromosomes and a collection of approximately 30,000-40,000 genes. Genes are the master templates for protein production. Each gene contains information on the amino acid sequence its corresponding protein will have, as well as when, in what cell type, and in what quantity its corresponding protein will be produced. Genes act within a cell's nucleus to drive the production of corresponding proteins at particular times and at particular levels appropriate to the cell.
Genes sit at the top of a unidirectional flow of information in a cell, summarized as follows: gene (DNA) gives rise to mRNA through a process called transcription, and mRNA gives rise to protein through a process called translation. mRNA is a second type of nucleic acid (RNA) that bridges the gap between genes and proteins. mRNA takes protein sequence information held by a gene and translates it into appropriate protein production. An mRNA molecule, like a protein, is composed of a single chain of serially linked building blocks (four distinct ribonucleotides, rather than amino acids) to which ribosomes bind for protein assembly. The order in which ribonucleotides are linked in a particular mRNA molecule (the mRNA sequence) is dictated by the protein information held by a gene. The mRNA sequence in turn dictates (encodes) the order in which amino acids are to be linked in a particular protein chain, and thus
61
the type of protein that is to be made. Thus, a gene directs production of a corresponding mRNA, and an mRNA directs assembly of a corresponding protein.
As the flow of information is unidirectional, with synthesis of a protein dependent upon production of an mRNA by a gene, one can intervene at the level of DNA or mRNA to disrupt the production of the protein.
Our current product candidates are designed to selectively bind to mRNA molecules encoding the tumor cell survival proteins we are targeting. The structure of an mRNA molecule is referred to as "sense", and therapeutic candidates that are engineered to be complementary binding partners for the particular mRNAs (i.e., particular "sense" molecules) that encode the targeted proteins are referred to as "antisense" therapeutics. Complementary binding is achieved through the specific sequence design of the antisense therapeutics, based on knowledge of the mRNAs encoding the targeted proteins. When an antisense molecule binds to its target mRNA, the mRNA is then degraded and therefore it is not translated by the ribosome into a functional protein.
Mechanism of Protein Production

- (1)
- In the nucleus of the cell, mRNA, which is a copy of the DNA's genetic information, is made through a process called transcription
- (2)
- The mRNA ("sense") strand travels out of the nucleus
- (3)
- The ribosomes attach to the single stranded mRNA and then move along the mRNA strand while translating the genetic information of the mRNA to assemble amino acids in a specified sequence to form a specific protein
62
Disrupting Protein Production Using Antisense Technology

- (1)
- In the nucleus of the cell, mRNA, which is a copy of the DNA's genetic information, is made through a process called transcription
- (2)
- The mRNA ("sense") strand travels out of the nucleus
- (3)
- An antisense strand binds to the mRNA strand to create a double stranded sense/antisense molecule
- (4)
- When an antisense molecule binds to its target mRNA, the mRNA is then degraded and therefore it is not translated by the ribosome into a functional protein
Second Generation Antisense Technology Overcomes Challenges of Early Antisense Science
The majority of antisense therapeutics that have been evaluated in late-stage human clinical trials are based on first-generation antisense chemistry. First-generation antisense therapeutics are susceptible to rapid degradation and loss of functionality when introduced to the human body. Thus these first-generation antisense therapeutics typically require daily dosing by continuous infusion to achieve sufficient concentrations to be effective.
In contrast, therapies that are based on second-generation antisense chemistry have increased target binding affinity, improved resistance to degradation and therefore better tissue distribution. These improvements result in slower clearance of the therapies from the body, allowing for less frequent dosing and thereby making treatment easier on patients and decreasing the associated cost.
Clinical data from our phase 1 clinical trial in prostate cancer patients demonstrated that weekly intravenous administration of OGX-011 resulted in drug distribution to prostate cancer tissue and over 92 percent inhibition of its target, clusterin mRNA, in prostate tumor cells in these patients. These data demonstrate that our second-generation antisense product candidate can be systemically administered and results in it entering tumor cells and effectively inhibiting clusterin production.
Cancer Indications For Which Our Product Candidates Are Currently Being Developed
Our product candidates are currently being developed for use in the treatment of prostate, non-small cell lung, breast, ovarian and bladder cancers and multiple myeloma. Collectively, these cancers account for approximately 44 percent of the cancer deaths in the United States each year.
63
The American Cancer Society estimates that the incidence of new diagnosis and deaths resulting from these cancers during 2006 will be as follows:
| Cancer Type | Estimated U.S. Incidence | Estimated Annual U.S. Deaths | ||
|---|---|---|---|---|
| Prostate Cancer | 234,460 | 27,350 | ||
| Non-Small Cell Lung Cancer | 151,789 | 141,340 | ||
| Breast Cancer(1) | 212,920 | 40,970 | ||
| Ovarian Cancer | 20,180 | 15,310 | ||
| Bladder Cancer | 61,420 | 13,060 | ||
| Multiple Myeloma | 16,570 | 11,310 |
- (1)
- Refers to incidence of breast cancer in females only.
Prostate Cancer
Prostate cancer is a leading cause of cancer death in men. The American Cancer Society estimates that in 2006, approximately 234,460 new cases of prostate cancer will be diagnosed in the United States and 27,350 men will die from prostate cancer in the United States. Primary treatment of prostate cancer that is localized to the prostate gland is usually either surgical removal or ablation through radiation of the prostate. For recurrent prostate cancer, hormone ablation therapy is often prescribed to retard the growth of the cancer. Hormone ablation therapy is generally effective for 18 to 24 months of treatment. When hormone ablation therapy ceases to be effective, the patient is considered to have hormone refractory prostate cancer, or HRPC. At this stage, patients become eligible for initial chemotherapy with docetaxel. The mortality of prostate cancer patients is primarily associated with those at the hormone refractory prostate cancer-stage of the disease, as HRPC is uniformly fatal.
Lung Cancer
Lung cancer accounts for 25 percent of all cancer deaths in women and more than 30 percent of cancer deaths in men. The American Cancer Society estimates that in 2006, approximately 174,470 new cases of lung cancer will be diagnosed in the United States and 162,460 will die from lung cancer in the United States. It is one of the most common cancers in industrialized nations and an increasing problem in developing countries. According to the American Cancer Society, non-small cell lung cancer, or NSCLC, accounts for 87 percent of lung cancers. Because less than 50 percent of patients live for more than one year after diagnosis and the overall five-year survival rate is approximately 15 percent, lung cancer is characterized by an unmet need for more effective therapies that can extend survival. Current treatment options are determined by the type and stage of the cancer, and include surgery, radiation therapy, chemotherapy and targeted biological therapies.
Breast Cancer
Breast cancer is the most common cancer in women and the second-most common cause of cancer death in women worldwide. The American Cancer Society estimates that in 2006, approximately 212,920 new cases of breast cancer will be diagnosed in women in the United States and 40,970 women will die from breast cancer in the United States. Lumpectomy, mastectomy, radiation therapy, chemotherapy, or hormone therapy, often in combination, are current
64
treatments. Current therapies have limited benefit in patients with regional or distant metastases, and five-year survival rates are approximately 81 and 26 percent, respectively.
Ovarian Cancer
Ovarian cancer ranks second among gynecologic cancers. The American Cancer Society estimates that in 2006, approximately 20,180 new cases of ovarian cancer will be diagnosed and 15,310 women will die this year from ovarian cancer in the United States. Treatment of ovarian cancer is primarily managed by surgery and chemotherapy, while radiotherapy is used as a palliative treatment for patients with inoperable cancer to relieve symptoms. If the cancer is diagnosed at the localized stage, the five-year survival rate is 94 percent; however, only about 19 percent of ovarian cancer is detected this early. For women with regional and distant disease, survival rates of five years are 68 and 29 percent, respectively.
Bladder Cancer
The American Cancer Society estimates that in 2006, approximately 61,420 new cases of urinary bladder cancer will be diagnosed in the United States, and 13,060 people will die this year from bladder cancer in the United States. Currently, the five-year survival rate for regional stage bladder cancer is 48 percent, but for distant stage bladder cancer it is only six percent.
Multiple Myeloma
Multiple myeloma is a hematological cancer with high morbidity and mortality that is caused by uncontrolled growth of plasma cells in bone tissue. The American Cancer Society estimates that in 2006, approximately 16,570 new cases of multiple myeloma will be diagnosed in the United States and approximately 11,310 people will die this year from multiple myeloma in the United States. The goal of therapy in multiple myeloma is to relieve symptoms (bone pain, immune compromise) and bring the disease into remission. When relapse occurs, therapy in these late-stage patients will involve chemotherapy. The five-year survival rate for all stages of multiple myeloma is approximately 32 percent.
Product Development Programs
Our Lead Product Candidate, OGX-011
The development program for our lead product candidate, OGX-011, is focused on reducing clusterin production to enhance treatment sensitivity and delay tumor progression in patients who have not fully developed treatment resistance and to restore treatment sensitivity in patients who have developed treatment resistance. Clusterin is a cell survival protein that is over-produced in several cancer indications and in response to many cancer treatments, including hormone ablation therapy, chemotherapy and radiation therapy. Increased clusterin production is observed in many human cancers, including prostate, non-small cell lung, breast, ovarian, bladder, renal, pancreatic, anaplastic large cell lymphoma and colon cancers and melanoma. Increased clusterin production is linked to faster rates of cancer progression, treatment resistance and shorter survival duration.
A broad range of pre-clinical studies conducted by the Prostate Centre and others have shown that reducing clusterin production facilitates tumor cell death by sensitizing human prostate, non-small cell lung, breast, ovarian, bladder, renal and melanoma tumor cells to chemotherapy. Pre-clinical studies conducted by the Prostate Centre and others also indicate that reducing
65
clusterin production sensitizes prostate tumor cells to hormone ablation therapy and prostate and non-small cell lung tumor cells to radiation therapy.
OGX-011 Market Opportunity
OncoGenex is initially developing OGX-011 to enhance the effects of chemotherapy in prostate, non-small cell lung and breast cancer. We believe that the results of our pre-clinical studies and clinical trials demonstrate that OGX-011 could also be used in combination with hormone ablation therapy, radiation or certain other treatments that induce tumor cell death. Because clusterin is over-produced and associated with treatment resistance in a variety of cancers, we believe that OGX-011 may have broad market potential to address many cancer indications. Since localized (non-metastatic) disease of the target cancers is most often treated with surgery, OGX-011 is intended primarily for use by patients with non-local (metastatic) or recurrent disease, which represents the stage of disease with the largest unmet need.
Potential Advantages of OGX-011
Based on the results of our pre-clinical studies and our phase 1 clinical trials, we believe that administering OGX-011 to cancer patients inhibits the production of clusterin, and therefore, when used in combination with other therapies, may offer clinically meaningful anti-cancer activity, including:
- •
- improved overall survival in multiple cancer indications;
- •
- enhanced effectiveness of multiple chemotherapy agents when used in combination with OGX-011; and
- •
- multiple mechanisms of action for synergistic tumor cell death.
In our phase 2 clinical trials, we are seeking to estimate the survival and safety benefits of OGX-011 when used in addition to various chemotherapy regimens in prostate, non-small cell lung and breast cancers.
Development Status and Strategy — Completed Phase 1 Clinical Trials
OGX-011 in Combination with Hormone Ablation Therapy in Patients with Localized Prostate Cancer (Clinical Trial OGX-011-01)
For men with localized prostate cancer, hormone ablation therapy with surgery remains the initial standard of care. Hormone ablation causes death of prostate tumor cells.
The purpose of this clinical trial was to:
- •
- determine safety and tolerability and define the recommended phase 2 dose of OGX-011 when given in combination with hormone ablation therapy;
- •
- determine the concentration and elimination profile of OGX-011 in serum and in the prostate when administered in combination with hormone ablation therapy;
- •
- measure clusterin mRNA and protein levels in the prostate and other tissues and correlate with dose of OGX-011;
- •
- measure serum clusterin levels and correlate with dose of OGX-011; and
- •
- attempt to establish possible correlations between plasma and/or prostate concentrations of OGX-011 with patient response or toxicity measures.
66
In order to determine these objectives, OGX-011 was administered in combination with hormone ablation therapy to patients with localized prostate cancer in advance of a surgical procedure to remove the prostate gland.
There were a total of 25 patients enrolled into treatment groups who received increasing doses of OGX-011 from 40 milligrams (mg) up to 640 mg. There was no dose limiting toxicity observed during OGX-011 dose escalation and adverse events were mild to moderate (grade 1 or 2). In summary, the results of the phase 1 clinical trial of OGX-011 in combination with hormone ablation therapy were that:
- •
- OGX-011 achieved high drug concentrations in the prostate and lymph nodes through once weekly intravenous administration;
- •
- clusterin mRNA was inhibited in prostate tumor cells by more than 92 percent in patients receiving the highest dose of OGX-011 (640 mg);
- •
- approximately double the number of tumor cells undergoing cell death occurred in the prostate of patients receiving the highest dose of OGX-011 (640 mg) compared to hormone ablation therapy alone;
- •
- OGX-011 was well-tolerated, with no dose limiting toxicity observed; and
- •
- the recommended phase 2 dose of OGX-011 was established at 640 mg.
Figure 1: Concentration of OGX-011 in Prostate Tissue

A trend test was used to test the statistical significance of the association between increasing the dose level of OGX-011 and the parameter being measured (PTrend). A PTrend value of less than 0.05 was deemed statistically significant. OGX-011 concentration in prostate tissue increased with
67
higher doses (PTrend < 0.001), as shown in Figure 1 above. Dose-dependent decreases in clusterin mRNA in prostate tissue were also observed (PTrend = 0.008). At the highest dose (640 mg) of OGX-011, clusterin mRNA was decreased by more than 92 percent in prostate tissue when compared to the lowest dose level and other historical controls (patients with no prior hormone ablation therapy and patients with less than two months of hormone ablation therapy), as shown in Figure 2 below. Similarly, clusterin mRNA was decreased by approximately 98 percent in lymph node tissue when compared to the lowest dose level (data not shown) (PTrend < 0.001).
Figure 2: OGX-011 Inhibits Clusterin mRNA Expression in Prostate Tissue
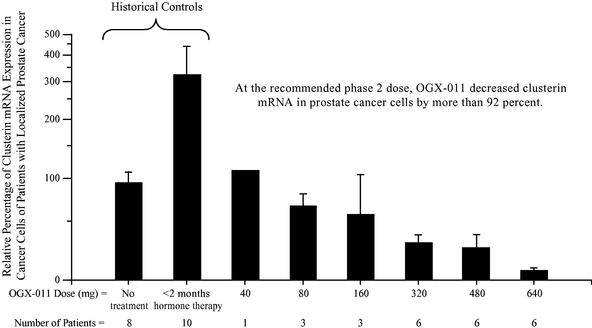
To determine whether suppression of clusterin levels by OGX-011 treatment could increase tumor cell death in prostate cancer tissue, the percentage of dying tumor cells per high powered field of the microscope were counted as shown in Figure 3 below. At the phase 2 dose (640 mg) the addition of OGX-011 more than doubled the percentage of dying tumor cells compared to hormone ablation therapy alone. The percentage of tumor cell death in prostate tissue increased proportionally with the dose of OGX-011 (PTrend < 0.001).
68
Figure 3: OGX-011 Increases the Apoptotic Index in Prostatectomy Specimens

These data demonstrate that OGX-011 was able to:
- •
- be delivered to prostate tumors and lymph nodes;
- •
- get inside tumor cells where it is required in order to potentially have a therapeutic effect;
- •
- significantly decrease clusterin production; and
- •
- increase tumor cell death when combined with hormone ablation therapy.
Based on achieving high drug concentration in prostate tissue and over 92 percent inhibition of clusterin mRNA in prostate tumor cells and lymph nodes, and a favorable safety profile, 640 mg was selected as the optimal dose for phase 2 clinical trials of OGX-011.
Patients in this clinical trial experienced various "adverse events", the majority of which are known to be associated with the other treatments in the protocol (hormone ablation therapy or prostate surgery). An adverse event is any unfavorable and unintended clinical laboratory value or symptom which is encountered during or after the use of an investigational product (i.e. OGX-011). These events may or may not be related to the patient's disease, other therapies administered to the patient or the investigational product.
None of the events that occurred resulted in patients discontinuing OGX-011 and the majority of the adverse events were considered mild. The adverse events thought to be possibly related to OGX-011 occurred mainly within the first week and decreased with continued dosing. These events included mild suppression of white blood cell counts; flu-like symptoms that included fever, fatigue, and rigors; and mild elevations in liver enzyme levels. Most symptoms went away after one to three weeks despite continued administration of OGX-011. Suppression of cells involved in blood clotting (thrombocytopenia) and incidence of fever and rigors correlated statistically with increasing the dose of OGX-011 (P=0.04, P=.001 and P<0.001, respectively).
There were no "serious adverse events" reported during this clinical trial. Serious adverse event means any adverse experience that results in any of the following outcomes: death, a
69
life-threatening experience, inpatient hospitalization or prolongation of existing hospitalization, a persistent or significant disability/incapacity, or a congenital anomaly/birth defect.
OGX-011 in Combination with Docetaxel Chemotherapy in the Treatment of Solid Tumors (Clinical Trial OGX-011-02)
OGX-011 treatment in animal models has been shown to enhance chemosensitivity and to reverse chemotherapy resistance to docetaxel, a chemotherapeutic agent used in a number of cancers including lung, breast, ovarian, bladder, renal cell and hormone refractory prostate cancer. This clinical trial was undertaken to evaluate the safety profile of OGX-011 in combination with docetaxel chemotherapy.
The purpose of this clinical trial was to:
- •
- determine safety and tolerability and define the recommended phase 2 dose of OGX-011 when given in combination with docetaxel chemotherapy given either weekly or every three weeks;
- •
- determine the concentration of OGX-011 and docetaxel in serum when administered in combination;
- •
- measure evidence of the effect of OGX-011 on serum clusterin levels; and
- •
- document any objective tumor responses.
In order to determine these objectives, OGX-011 was administered in combination with docetaxel chemotherapy to patients with cancers known to over-produce clusterin (prostate, non-small cell lung, breast, ovary, bladder and renal cell).
There were a total of 40 patients enrolled into treatment groups who received increasing doses of OGX-011 from 40 mg up to 640 mg. In addition to OGX-011 treatment, patients also received docetaxel chemotherapy given either as a weekly or every three week schedule as one cycle. The clinical trial was conducted at multiple clinical sites.
Although this was a group of patients with various cancer types, responses and stable disease were documented. Since this clinical trial did not include patients who did not receive OGX-011, statistical analysis for treatment response was not performed. OGX-011 treatment resulted in a trend towards dose dependent decreases in serum clusterin levels with the 640 mg dose level having the greatest change from baseline. There was no evidence that administration of OGX-011 affected the metabolism of docetaxel chemotherapy or that docetaxel chemotherapy affected the metabolism of OGX-011.
The recommended OGX-011 dose for treatment in combination with docetaxel chemotherapy was determined as 640 mg, based on acceptable safety results, documented responses/stable disease, and lowering of serum clusterin levels observed from this phase 1 clinical trial.
Patients in this clinical trial experienced various adverse events, the majority of which are known to be associated with the other treatment in the protocol (docetaxel chemotherapy). Adverse events associated with docetaxel chemotherapy include fatigue, diarrhea, vomiting, fluid retention, taste disturbance, infection, reduced appetite, nausea, hair loss and decrease in blood counts.
Adverse events were primarily mild to moderate. The incidence of reduced appetite, nausea and hair loss increased as the OGX-011 dose levels increased. The decrease in white blood cell
70
counts was consistent with other trials using docetaxel and did not decrease further with increased doses of OGX-011. Five of the 40 patients experienced dose-limiting toxicity. Four of these events were attributed to the docetaxel chemotherapy and one, fatigue, was attributed to the combination of OGX-011, docetaxel and the disease. Ten patients experienced 13 serious adverse events. Six of the 13 serious adverse events were attributed to the combination of OGX-011 and the chemotherapy.
OGX-011 in Combination with Gemcitabine and Cisplatin Chemotherapy in Patients with Advanced Non-Small Cell Lung Cancer (Clinical Trial OGX-011-05)
Our phase 1 clinical trial evaluating OGX-011 in combination with docetaxel chemotherapy established a favorable safety profile and a phase 2 dose of OGX-011 for this combination. In the treatment of advanced non-small cell lung cancer, two chemotherapy agents (doublet chemotherapy) are routinely used. This clinical trial was undertaken to evaluate the safety profile of OGX-011 in combination with the doublet chemotherapy of gemcitabine/cisplatin.
The purpose of this clinical trial was to:
- •
- determine safety and tolerability and define the recommended phase 2 dose of OGX-011 when given in combination with the doublet chemotherapy regime of gemcitabine/cisplatin;
- •
- estimate objective tumor response rate;
- •
- determine the profile of OGX-011 in serum when administered in combination with gemcitabine/cisplatin;
- •
- estimate progression-free survival and overall survival of the treated patients; and
- •
- measure evidence of the effect of OGX-011 on serum clusterin levels.
In order to determine these objectives, OGX-011 was administered in combination with gemcitabine/cisplatin chemotherapy to patients with advanced non-small cell lung cancer.
There were a total of 10 patients with metastatic or locally advanced non-small cell lung cancer enrolled into two treatment groups that received either 480 mg or 640 mg of OGX-011, of which 90 percent of the patients had Stage IV disease. The clinical trial was conducted at multiple clinical sites.
Ten patients were treated in Phase I. Nine of the ten patients received at least one dose of OGX-011 in combination with chemotherapy and one patient died on day 19 having been removed from further protocol therapy for disease progression prior to initiating any chemotherapy. All but one of the nine patients treated with OGX-011 and chemotherapy showed either a partial response, defined by a greater than or equal to 30 percent tumor size reduction, or stable disease, defined by less than 30 percent tumor size reduction. Of the 55 percent of patients reported as stable disease, tumor reduction ranged from 10.3 percent to 29.7 percent.
The median time for survival (n = 10) was 17.1 months. The one-year survival rate for all ten patients was 60% and for patients that received at least one dose of OGX-011 in combination with these chemotherapies was approximately 67 percent. As at December 19, 2006, three patients survive at 25.8, 24.1 and 20.1 months. Prior published results from randomized clinical trials using gemcitabine/cisplatin or carboplatin (another platinum-based chemotherapy agent) have reported one-year survival rates of 33 to 43 percent and median survival times of 8 to 10.8 months. Since treatment response of patients treated only with chemotherapy (without OGX-011) was derived from historical controls of third party clinical trials and not from this clinical trial, and this clinical
71
trial involves few patients and patient follow up is ongoing, statistical analysis for treatment response was not performed. There was no evidence that administration of OGX-011 affected the metabolism of gemcitabine or cisplatin chemotherapy or that either of these chemotherapeutic agents affected the metabolism of OGX-011.
Patients in this clinical trial experienced various adverse events, the majority of which are known to be associated with the other treatments in the protocol (gemcitabine and cisplatin chemotherapy) and the disease. The majority of the events were mild. The most common events were constipation, change in taste, fatigue and chills. Decreased white blood cell counts and sodium levels were also observed. Five serious adverse events were observed in four of the ten patients evaluated in this clinical trial. All of the serious adverse events were attributed to the chemotherapy or disease, except for one case of elevated creatinine and one case of fever with a reduced number of cells of a specific white blood cell type which were both attributed to the combination of OGX-011 and the chemotherapeutic agents.
Development Status and Strategy — Ongoing Phase 2 Clinical Trials
Our phase 1 clinical trials evaluated the safety and established the recommended phase 2 dose of OGX-011 in combination with either docetaxel chemotherapy (two different schedules), gemcitabine/cisplatin chemotherapy or hormone ablation therapy. In all clinical trials, 640 mg was established as the phase 2 dose.
We are currently conducting five phase 2 clinical trials of OGX-011 to further evaluate the safety and efficacy of OGX-011 in combination with various cancer therapies for prostate, non-small cell lung and breast cancer, as further described below. Four of the five phase 2 clinical trials have completed patient enrollment.
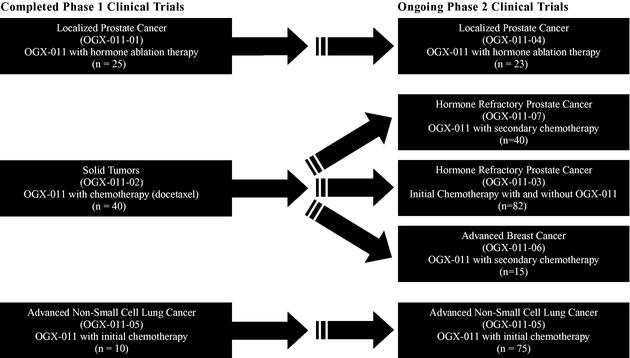
72
OGX-011 in Combination with Chemotherapy in Patients with Metastatic Hormone Refractory Prostate Cancer (Clinical Trial OGX-011-03)
Our ongoing phase 2 clinical trial in patients with metastatic hormone refractory prostate cancer is designed to estimate the efficacy and safety benefits resulting from the addition of OGX-011 to the standard chemotherapy regimen of docetaxel and prednisone. This clinical trial is a randomized, open-label, multi-center trial in which we have enrolled 82 patients, or 41 patients per treatment arm, at centers in Canada and the United States. This clinical trial will compare the weekly dosing regimen of OGX-011 in combination with docetaxel (dosed every three weeks) and daily prednisone to docetaxel (dosed every three weeks) and daily prednisone alone. It is anticipated that the response rate data from this clinical trial will be available by the end of 2007. We completed accrual to this trial in December 2006 with a total of 82 patients.
The primary objective of this phase 2 clinical trial is to measure prostate specific antigen, or PSA, response, a biomarker of prostate cancer. Secondary objectives of the clinical trial include: (a) measuring objective response and duration, (b) determining tolerability and toxicity of OGX-011 in combination with docetaxel chemotherapy and prednisone, (c) measuring evidence of OGX-011 and docetaxel and prednisone effect on serum clusterin levels and (d) describing time to progression and overall patient survival for both treatment groups. We are describing time to progression and overall survival because we intend to use these endpoints to form the basis of subsequent registration clinical trials, if any. It is our belief that an improvement in overall survival is recognized as the most meaningful outcome in hormone refractory prostate cancer, as evidenced by its use as the basis for approval by the FDA in 2004 of docetaxel, which is the current chemotherapeutic standard of care for initial chemotherapy treatment of hormone refractory prostate cancer.
OGX-011 in Combination with Chemotherapy in Patients with Prostate Cancer that are Refractory to Hormone Ablation Therapy and Primary Chemotherapy (Clinical Trial OGX-011-07)
Our ongoing phase 2 clinical trial in patients with metastatic hormone refractory prostate cancer who have progressed while on or within six months of docetaxel chemotherapy treatment and require additional chemotherapy is designed to evaluate the feasibility of treating this patient group and ability of OGX-011 treatment in combination with either mitoxantrone (20 patients) or docetaxel (20 patients) to overcome chemotherapy resistance and enhance further chemotherapy in these patients. We believe that at least two three-week cycles of OGX-011 therapy will be required to reduce clusterin production levels sufficiently to impact chemotherapy resistance. However, disease progression or death occurs rapidly in these patients which may result in termination of the existing chemotherapy prior to the patients receiving six weeks of OGX-011 treatment. If only a small number of patients receive six weeks of chemotherapy without disease progression or death, analysis of the effectiveness of OGX-011 may not be relevant, in which case it may not be feasible to conduct a phase 3 clinical trial in this group of patients. The trial was initiated in June 2006. As of February 28, 2007, we have accrued 38 patients to this trial. We expect to enroll a total of 40 patients at centers in Canada.
In addition to the above objective, other objectives of this clinical trial include assessing safety, PSA response and time delay in pain progression. A further objective of this clinical trial is to explore relationships between serum clusterin levels and change in serum PSA levels to progression-free survival. It is anticipated that data from this trial related to the feasibility of treating this patient group will be available by mid-2007.
73
OGX-011 in Combination with Chemotherapy in Patients with Advanced Non-Small Cell Lung Cancer (Clinical Trial OGX-011-05)
Our ongoing phase 2 clinical trial in patients with metastatic or locally advanced non-small cell lung cancer is designed to estimate objective response rates. This clinical trial is a single-arm, multi-center trial in which we have enrolled 75 patients at centers in Canada and the United States. It is anticipated that the data from this clinical trial will be available by the end of 2007. We completed accrual to this trial in October 2006 with a total of 75 patients.
The primary objective of this phase 2 clinical trial is to estimate objective response rates and further describe the safety profile of OGX-011 in combination with a gemcitabine/platinum-based regimen in patients with metastatic or locally advanced non-small cell lung cancer. Secondary objectives include: (a) measuring progression-free survival and overall survival of patients treated with OGX-011 in combination gemcitabine/platinum-based regimen, (b) measuring the effect of OGX-011 on serum clusterin levels and, whenever possible, within accessible tumors, and (c) assessing the influence of initial clusterin levels on time-to-disease progression and response rates for NSCLC patients.
OGX-011 in Combination with Hormone Ablation Therapy in Patients with Localized Prostate Cancer (Clinical Trial OGX-011-04)
Our ongoing phase 2 clinical trial in patients with localized prostate cancer is designed to assess the effects of combined hormone ablation therapy and weekly treatment with OGX-011 for three months prior to surgical removal of the prostate gland. This clinical trial is a single-arm, single center trial in which we have enrolled 23 patients. It is anticipated that the data from this clinical trial will be available by the end of 2007. We completed accrual to this trial in December 2006 with a total of 23 patients.
The primary objective of this phase 2 clinical trial is to assess for complete response rates as determined by microscopic analysis of the removed prostate gland in patients with high-risk localized prostate cancer. Secondary objectives include: (a) quantifying changes in clusterin expression in residual prostate tumor cells after treatment with hormone ablation therapy and OGX-011, (b) measuring levels of OGX-011 in prostate tissues after weekly treatment for three months, (c) assessing the safety and tolerability of the treatment regime, (d) measuring evidence of OGX-011 effect on clusterin expression in blood cells, (e) measuring evidence of OGX-011 effect on serum clusterin levels, (f) assessing the effects of treatment with hormone ablation therapy and OGX-011 on time to lowest PSA levels, and (g) determining PSA recurrence rates after treatment with hormone ablation therapy and OGX-011.
OGX-011 in Combination with Secondary Chemotherapy in Patients with Advanced Breast Cancer (Clinical Trial OGX-011-06)
Our ongoing phase 2 clinical trial is designed to estimate objective response rates, or tumor shrinkage, in patients with advanced breast cancer when treated with OGX-011 in combination with docetaxel chemotherapy. Secondary objectives include: (a) determining the tolerability and toxicity of OGX-011 in combination with docetaxel chemotherapy, (b) describing time to progression, (c) describing overall survival, and (d) measuring evidence of OGX-011 effect on serum clusterin levels. This clinical trial is a single arm, multi-center trial that is being conducted at centers in Canada in 15 patients. A further 27 patients were contemplated in the event that six or more of the first 15 patients achieved tumor shrinkage equal to or greater than 30 percent from
74
baseline tumor size. Since fewer than six of the 15 patients achieved tumor shrinkage greater than 30 percent, this study has been limited to accrual for the first-stage of the trial and is pending assessment of the influence of the treatment on these patients' serum clusterin levels and survival. These data are consistent with our pre-clinical data with secondary chemotherapies and preliminary data from our phase 1 clinical trials which indicate that OGX-011 may increase the number of patients with disease stabilization and may improve survival without necessarily increasing the number of patients with greater than 30 percent tumor shrinkage. It is anticipated that data from the 15 patients in this clinical trial will be available by mid-2007.
Safety Data
As of November 2006, OGX-011 has been administered to 243 patients in seven clinical trials. See "Completed Phase 1 Studies" for summaries of safety results from our completed phase 1 clinical trials. Analysis of adverse event data for each of the ongoing phase 2 clinical trials will be available following completion of each of these trials.
Patients in our completed and ongoing clinical trials have experienced various "adverse events", the majority of which are known to be associated with the other treatments in the protocol (hormone ablation therapy, chemotherapy or prostate surgery). An adverse event is any unfavorable and unintended clinical laboratory value or symptom which is encountered during or after the use of an investigational product (i.e. OGX-011). Serious adverse event means any adverse experience that results in any of the following outcomes: death, a life-threatening experience, inpatient hospitalization or prolongation of existing hospitalization, a persistent or significant disability/incapacity, or a congenital anomaly/birth defect. These events may or may not be related to the patient's disease, other therapies administered to the patient or the investigational product.
Through November 2006, 54 of the 243 patients treated with OGX-011 experienced 73 serious adverse events. The majority of these serious adverse events were in trials where OGX-011 was administered in combination with chemotherapy. Most of the serious adverse events that occurred are known to be associated with chemotherapy or hormone ablation therapy, or are commonly associated with the patients' disease. The serious adverse events experienced by 31 of the 54 patients were considered by the clinical investigator to be related to the combination of OGX-011 and chemotherapy.
Future Phase 3 Clinical Trials
We plan to evaluate the results of our ongoing phase 2 clinical trials to determine which cancer indications and which treatment combinations demonstrate promise. Therefore, our initial phase 3 clinical trial may be in any of prostate, non-small cell lung or breast cancer. Our ongoing phase 2 clinical trials may indicate efficacy across several indications and treatment combinations, in which case we may elect to initiate more than one phase 3 clinical trial. In determining the cancer indications for which we may conduct future phase 3 clinical trials, we will consider, among others, the following factors:
- •
- our phase 2 clinical trial results;
- •
- patient availability;
- •
- estimated time to regulatory approval;
75
- •
- estimated trial size required to assess endpoint;
- •
- the size of the market for each cancer indication; and
- •
- the availability of commercial products and products in development.
Our Second Product Candidate, OGX-427
The development program for our second product candidate, OGX-427, is focused on reducing heat shock protein 27 (Hsp27) production to enhance treatment sensitivity and delay tumor progression in patients who have not fully developed treatment resistance and to restore treatment sensitivity in patients who have developed treatment resistance. Hsp27 is a cell survival protein that is over-produced in response to many cancer treatments, including hormone ablation therapy, chemotherapy and radiation therapy. Increased Hsp27 production is observed in many human cancers, including prostate, non-small cell lung, breast, ovarian, bladder, renal, pancreatic, multiple myeloma and liver cancers. Increased Hsp27 production is linked to faster rates of cancer progression, treatment resistance and shorter survival duration.
A number of pre-clinical studies conducted by the Prostate Centre and others have shown that inhibiting the production of Hsp27 in human prostate, breast, ovarian, pancreatic and bladder tumor cells sensitizes the cells to chemotherapy. Pre-clinical studies conducted by the Prostate Centre and others have shown that reducing Hsp27 production induced tumor cell death in prostate, breast, non-small cell lung, bladder and pancreatic cancers. The Prostate Centre has also conducted pre-clinical studies that indicate that reducing Hsp27 production sensitizes prostate tumor cells to hormone ablation therapy.
OGX-427 works via antisense mechanism to inhibit the production of Hsp27, thereby impairing the tumor cells' survival mechanism and facilitating tumor cell death when used alone or in combination with other cancer therapies. OGX-427 employs second-generation antisense chemistry and therefore overcomes issues of stability, affinity and dosage scheduling.
OGX-427 Market Opportunity
Since Hsp27 is over-produced in response to many cancer treatments, enhances tumor cell survival and is associated with treatment resistance in a variety of cancers, we believe that OGX-427 may have broad market potential to treat many cancer indications and disease stages.
OncoGenex is initially developing OGX-427 to enhance the effects of chemotherapy in a variety of cancers. We believe that the results of our pre-clinical studies demonstrate that OGX-427 could also be used in combination with hormone ablation therapy, radiation therapy and certain other treatments that induce tumor cell death. Since localized (non-metastatic) disease of the target cancers is most often treated with surgery, OGX-427 is intended primarily for use by patients with non-local (metastatic) or recurrent disease, which represents the stage of disease with the largest unmet need.
We believe that additional opportunities exist for OGX-427 to inhibit treatment resistance to Velcade® or to restore treatment sensitivity in patients who have already developed treatment resistance to Velcade®. Velcade® is a drug approved for the treatment of relapsed refractory multiple myeloma. Since Hsp27 levels are significantly elevated in patients that have failed Velcade® therapy, OncoGenex may pursue a combination treatment with Velcade® to enhance
76
treatment sensitivity in patients who may develop treatment resistance and to restore treatment sensitivity in patients who have developed treatment resistance.
Potential Advantages of OGX-427
Pre-clinical studies conducted by the Prostate Centre and others suggest that inhibiting the production of Hsp27 can induce tumor cell death on its own in prostate, breast, non-small cell lung, bladder and pancreatic cancers and can enhance the efficacy of chemotherapy in ovarian, prostate, breast, non-small cell lung and bladder cancers. Based on these pre-clinical studies, we believe that OGX-427 may offer clinically meaningful anti-cancer activity, including:
- •
- improved overall survival in multiple cancer indications;
- •
- enhanced anti-cancer activity with multiple chemotherapy agents; and
- •
- multiple mechanisms of action providing for synergistic tumor cell death.
Development Status and Strategy
We have completed manufacturing of OGX-427 for our initial human clinical trials and are concluding drug distribution and toxicology studies required before requesting regulatory approval to initiate phase 1 clinical trials. We will seek to evaluate safety and define our phase 2 dose in two phase 1 clinical trials. In our phase 1 clinical trials, we intend to evaluate OGX-427 alone, as well as in combination with other cancer therapies. In contrast to only evaluating OGX-427 in combination with other cancer therapies, evaluating OGX-427 alone will enable attribution of clinical activity and side effects to the use of OGX-427. Clinical trials evaluating OGX-427 in combination with other cancer therapies represent the likely clinical strategy for future phase 2 and phase 3 clinical trials, since patients with treatment resistant cancer are most often treated with combinations of drugs.
Our Third Product Candidate, OGX-225
The development program for our third product candidate, OGX-225, is focused on reducing the production of both insulin-like growth factor binding protein-2 (IGFBP-2) and insulin-like growth factor binding protein-5 (IGFBP-5) with a single product to enhance treatment sensitivity and delay tumor progression in patients who are resistant to hormone ablation therapy. In the treatment of tumors that require hormones for growth, clinicians use hormone ablation therapy to inhibit the production of the primary hormone (e.g. testosterone or estrogen) required for tumor growth. While tumors often regress initially following hormone ablation therapy, IGFBP-2 or IGFBP-5 make an alternate hormone, insulin-like growth factor-1, or IGF-1, available to the tumor that facilitates continued tumor growth. By inhibiting the production of IGFBP-2 and IGFBP-5, the tumor's access to IGF-1 is reduced and tumor growth is delayed.
Increased IGFBP-2 or IGFBP-5 production is observed in several cancers including prostate, breast, non-small cell lung, bladder, ovarian, pancreatic and colon cancers, acute myeloid leukemia, acute lymphoblastic leukemia, neuroblastoma, glioma, and melanoma. Increased IGFBP-2 or IGFBP-5 production is linked to faster rates of cancer progression, treatment resistance and shorter survival duration. Since IGFBP-2 and IGFBP-5 serve similar functions, inhibiting the production of both proteins may be superior to inhibiting the production of either protein on its own. Employing OGX-225 as a single product to simultaneously inhibit the
77
production of both IGFBP-2 and IGFBP-5 has the potential to delay disease progression in cancers dependent upon IGF-1.
Pre-clinical studies conducted by the Prostate Centre and others in human prostate, bladder, glioma and breast cancer, have shown that reducing IGFBP-2 and IGFBP-5 production with OGX-225 induced tumor cell death or sensitized all of these tumor types to chemotherapy.
We intend to continue to evaluate OGX-225 in pre-clinical studies to assess its ability to inhibit or delay progression of hormone-dependent and other tumors.
OGX-225 Market Opportunity
Since IGFBP-2 and IGFBP-5 are over-expressed in a variety of cancers, OGX-225 may have broad market potential to treat many cancer indications. We believe that the initial opportunity for OGX-225 would be in breast and prostate cancer patients early in the course of their recurrence after failed hormone ablation therapy.
Development Status and Strategy
We have completed numerous pre-clinical studies with OGX-225 indicating that it delays progression to hormone independence in prostate and breast cancer model systems. We have not defined when we will initiate the pre-clinical studies required to request initiation of phase 1 clinical trials.
License and Collaboration Agreements
Isis Pharmaceuticals, Inc.
OGX-011
In November 2001, we entered into an agreement with Isis to jointly develop and commercialize OGX-011. This strategic relationship provides us with access to Isis' proprietary position in second-generation antisense chemistry for use in OGX-011, Isis' expertise in developing antisense therapeutics, including their manufacturing expertise, and has allowed us to develop OGX-011 cost efficiently. We share with Isis, on a proportionate share basis of 65 percent OncoGenex and 35 percent Isis, the costs and revenues resulting from the development and commercialization of OGX-011. In 2001, we paid Isis $500,000 for a specified amount of OGX-011 to support the initial IND-directed toxicology and drug distribution (pharmacokinetic) studies. Under the agreement, neither of us can pursue the development or commercialization of any antisense compound for clusterin outside of the collaboration. This exclusive arrangement will continue until OGX-011 is no longer being developed or commercialized or unless the agreement is terminated by one party due to the other's insolvency.
The agreement establishes that both parties will agree to the development and commercialization activities with respect to OGX-011. We are currently obligated to conduct clinical and regulatory development of OGX-011 pursuant to a project plan approved by the parties. For our ongoing phase 2 clinical trials, we are responsible for conducting (or having conducted) the clinical trials, preparing all regulatory filings in connection with these clinical trials and analyzing the clinical trial data. Any new development and commercialization activities will require the approval of both parties.
78
Under our agreement with Isis, we and Isis acknowledge our respective obligations to pay certain third parties royalties on net sales of OGX-011. The amount of the royalties is dependent on whether we or Isis owe royalty payments to third parties pursuant to our respective license agreements with the third parties. In the event that the patents held by these third parties expire, our royalty obligations to them will be reduced accordingly. We do not anticipate making any royalty payments under the terms of the agreement in 2007. Each of Isis and OncoGenex will receive its proportionate share of the licensing revenue generated by sales of OGX-011 after payments are made to the third parties. In the event that OGX-011 is being marketed by OncoGenex or Isis, the company marketing OGX-011 will subtract expenses from revenues received from OGX-011, pay or distribute third party payments and distribute the remaining revenue to the parties according to their proportionate share. In the event that expenses for marketing OGX-011 are greater than revenues received, the parties will divide such expenses that are in excess of revenues, according to their proportionate share. See "Summary of Milestone, Royalty and Revenue/Cost-Sharing Obligations by Product".
OncoGenex has agreed to indemnify Isis and certain individuals affiliated with Isis against liabilities caused by our and our licensees' and sublicensees' gross negligence or willful misconduct, or our material breach of the collaboration and license agreement. If the agreement is materially breached by OncoGenex, Isis can independently develop or commercialize OGX-011.
OGX-427
In January 2005, we entered into a collaboration and license agreement with Isis to jointly identify antisense compounds designed to inhibit the production of proteins encoded by specified gene targets. We are solely responsible for all product development activities for antisense compounds under this collaboration. This relationship provides us with access to Isis' proprietary position in second-generation antisense chemistry for use in specified products. We were permitted to designate up to two collaboration gene targets for collaborative research, development and commercialization. In April, 2005, Hsp27 was confirmed as a collaboration gene target. This gene target combined with Isis' technology is our product candidate OGX-427. Our right to designate a second collaboration gene target expired on January 5, 2007.
Under the terms of the agreement, in the event that we abandon OGX-427, Isis may elect to unilaterally continue development of OGX-427, in which case we must provide Isis with a worldwide license or sublicense (as the case may be) of our relevant technology solely to develop and commercialize OGX-427 in exchange for a royalty on Isis' sales of OGX-427.
In consideration for the grant of rights related to OGX-427, we issued Isis a promissory note which was converted into 244,300 of our Series 2 Class B preferred shares (which upon completion of the Reorganization and this Offering will convert into 244,300 common shares). Under the terms of the agreement, we may be obligated to make certain milestone payments to Isis contingent upon the occurrence of certain clinical development and regulatory events related to OGX-427. We are also obligated to pay to Isis certain milestone payments as well as certain royalties on net sales for OGX-427, with the amount of royalties depending on whether third party royalty payments are owed. We do not anticipate making any milestone or royalty payments to Isis under the terms of the agreement in 2007. See "Summary of Milestone, Royalty and Revenue/Cost-Sharing Obligations by Product".
We have agreed to indemnify Isis and certain individuals affiliated with Isis against liabilities caused by our and our licensees' and sublicensees' gross negligence or willful misconduct, our
79
material breach of the collaboration and license agreement, and the manufacture, use, handling, storage, sale or other disposition of OGX-427 that is sold by us or our affiliates, agents or sublicensees.
The term of the agreement will continue for each product until the later of 10 years after the date of the first commercial sale of OGX-427, or the expiration of the last to expire of any patents required to be licensed in order to use or sell OGX-427, unless we abandon OGX-427 and Isis does not elect to unilaterally continue development of OGX-427.
OGX-225
In August 2003, we entered into a collaboration and license agreement with Isis to jointly identify antisense compounds related to OGX-225 targeted to inhibit the production of IGFBP-2 and IGFBP-5. We are solely responsible for all product development activities for OGX-225. This relationship provides us with access to Isis' proprietary position in second-generation antisense chemistry for use in OGX-225. We will owe Isis payments upon completion of product development milestones and royalties on product sales.
Under the agreement, neither of us can pursue the development or commercialization of any antisense compound that inhibits the production of either IGFBP-5 or IGFBP-2 outside of the collaboration. Under the terms of the agreement, in the event that we abandon all products developed under this agreement, including OGX-225, Isis may elect to unilaterally continue development of any or all of such abandoned product(s), in which case we must provide Isis with a worldwide license or sublicense (as the case may be) of our relevant technology solely to develop and commercialize the abandoned product(s) in exchange for a royalty on Isis' sales of such abandoned product(s).
In connection with entering into this agreement, we issued 272,232 Series 1 Class B preferred shares to Isis (which upon completion of the Reorganization and this Offering will convert into 272,232 common shares). Under the terms of the agreement, we may be obligated to make certain milestone payments to Isis contingent upon the occurrence of certain clinical development and regulatory events related to OGX-225. We are also obligated to pay to Isis certain royalty payments on net sales of OGX-225, with the amount depending on whether Isis owes royalty payments to third parties pursuant to license agreements between Isis and those third parties. We do not anticipate making any milestone or royalty payments to Isis under the terms of the agreement in 2007. See "Summary of Milestone, Royalty and Revenue/Cost-Sharing Obligations by Product".
Isis has the first right to manufacture OGX-225. If Isis is unable or unwilling to manufacture OGX-225 or the parties cannot reach mutually acceptable terms, OncoGenex may have OGX-225 manufactured by a manufacturer licensed under Isis' proprietary manufacturing and analytical technology or have OGX-225 manufactured using a process not covered by Isis' proprietary manufacturing and analytical technology.
We have agreed to indemnify Isis and certain individuals affiliated with Isis in respect of liabilities caused by our and our licensees' and sublicensees' gross negligence or willful misconduct, our material breach of the collaboration and license agreement, or the manufacture, use, handling, storage, sale or other disposition of OGX-225 that is sold by us or our affiliates, agents or sublicensees.
80
The term of this agreement will continue for so long as any product is being developed or commercialized, unless the agreement is earlier terminated by our abandoning all product(s) developed under this agreement, including OGX-225, and Isis does not elect to unilaterally continue development of any such product(s), or unless the agreement is earlier terminated by one party due to the other's insolvency.
University of British Columbia
OGX-011
Under an agreement made in November 2001, as amended, UBC granted to us an exclusive, worldwide license to commercialize its existing intellectual property and any improvements related to clusterin. This technology combined with Isis' second-generation antisense chemistry is our product candidate, OGX-011. In connection with entering into this license agreement, we issued 70,000 common shares to UBC. We agreed to pay to UBC certain royalties on the revenue from sales of OGX-011. See "Summary of Milestone, Royalty and Revenue/Cost-Sharing Obligations by Product". We do not anticipate making any royalty or other payments to UBC under the terms of the agreement in 2007, except for a C$2,000 annual maintenance fee.
We agreed to use our commercially reasonable efforts to develop and exploit the licensed technology and any improvements. We also agreed to promote, market and sell any resulting products and to cause the market demand for such products to be satisfied. We are permitted to sublicense the technology, subject to certain consent and other requirements. We direct patent prosecution and are responsible for all fees and costs related to the preparation, filing, prosecution and maintenance of the patent rights underlying the agreement. We indemnify UBC and certain of its affiliates against liability arising out of the exercise of any rights granted pursuant to the agreement. The term of this agreement will expire on the later of 20 years from its effective date or the expiry of the last patent licensed under the agreement. Subject to patent term extensions, the current granted patent for OGX-011 expires in the United States in 2021 and would expire in all other jurisdictions by 2020. We have additional patent applications pending which, if issued and not invalidated, may extend the expiration date of the last-to-expire patents. We may also file additional patent applications related to clusterin that could potentially extend the expiration date of the last to expire patent in this area.
OGX-427
Under an agreement made in April 2005, as amended, UBC granted to us an exclusive, worldwide license to commercialize its existing intellectual property and any improvements related to Hsp27. This technology combined with Isis' second-generation antisense chemistry is our product candidate, OGX-427. In connection with entering into this license agreement, we issued 30,000 common shares to UBC. We agreed to pay to UBC certain royalties on the revenue from sales of OGX-427, which royalty rate may be reduced in the event that we must pay additional royalties under patent licenses entered into with third parties in order to manufacture, use or sell OGX-427. We may be obligated to make milestone payments to UBC contingent upon the occurrence of certain clinical development and regulatory events related to OGX-427. See "Summary of Milestone, Royalty and Revenue/Cost-Sharing Obligations by Product". We do not anticipate making any milestone or royalty payments to UBC under the terms of the agreement in 2007, except for a C$2,000 annual maintenance fee.
81
Subject to certain exceptions, we agreed to use our commercially reasonable efforts to (i) develop and exploit the licensed technology and any improvements, and (ii) promote, market and sell any resulting products. We are permitted to sublicense the technology, subject to certain consent and other requirements. We direct patent prosecution and are responsible for all fees and costs related to the preparation, filing, prosecution and maintenance of the patent rights underlying the agreement. We indemnify UBC and certain of its affiliates against liability arising out of the exercise of any rights granted pursuant to the agreement. The term of this agreement will expire on the later of 20 years from its effective date or the expiry of the last patent licensed under the agreement. Depending on the outcome of the pending patent applications in the licensed patent family, and subject to any applicable patent term extensions, a patent issuing from this family would expire in all jurisdictions by 2023. We may also file additional patent applications related to Hsp27 that could potentially extend the expiration date of the last to expire patent in this area.
OGX-225
Under a series of agreements made between November 2001 and October 2005, as amended, UBC granted to us exclusive, worldwide licenses to commercialize its existing intellectual property and any improvements related to IGFBP-2 and IGFBP-5. This technology combined with Isis' second-generation antisense chemistry is our product candidate, OGX-225. In connection with entering into these license agreements, we issued 62,000 common shares to UBC. We agreed to pay to UBC certain royalties on the revenue from sales of OGX-225, which royalty rate may be reduced in the event that we must pay additional royalties under patent licenses entered into with third parties in order to manufacture, use or sell OGX-225. We may be obligated to make milestone payments to UBC contingent upon the occurrence of certain clinical development and regulatory events related to OGX-225. See "Summary of Milestone, Royalty and Revenue/Cost-Sharing Obligations by Product". We do not anticipate making any milestone or royalty payments to UBC under the terms of the agreement in 2007, except for a C$4,000 annual maintenance fee.
Subject to certain exceptions, we agreed to use our commercially reasonable efforts to (i) develop and exploit the licensed technology and any improvements, and (ii) promote, market and sell any resulting products and cause the market demand for such products to be satisfied. We are permitted to sublicense the technology, subject to certain consent and other requirements. We direct patent prosecution and are responsible for all fees and costs related to the preparation, filing, prosecution and maintenance of the patent rights underlying the agreement. We indemnify UBC and certain of its affiliates against liability arising out of the exercise of any rights granted pursuant to the agreement. The term of this agreement will expire on the later of 20 years from its effective date or the expiry of the last patent licensed under the agreement. The patent estate for OGX-225 comprises three patent families: inhibitors of IGFBP-2 production, IGFBP-5 production and single product candidates that simultaneously inhibit both IGFBP-2 and IGFBP-5 production. OGX-225 is a single product which inhibits the production of both IGFBP-2 and IGFBP-5. Patent protection for OGX-225 may rely on one or more of these patent families. Depending on the outcome of the pending patent applications within these families, and subject to any applicable patent term extensions, the patents issuing from these families would expire in all jurisdictions between 2020 and 2024. We may also file additional patent applications related to IGFBP-2 and/or IGFBP-5 that could potentially extend the expiration date of the last to expire patent in this area.
82
Summary of Milestone, Royalty and Revenue/Cost-Sharing Obligations by Product
The table below sets forth, by product candidate, the estimated milestone payments, royalty payments, revenue-sharing and cost-sharing arrangements to which we are subject under the license and collaboration agreements described above. Except for C$8,000 in annual maintenance fees payable to UBC, we do not anticipate making any milestone or royalty payments with respect to any of our product candidates until at least 2008.
| | Total Payable | ||||
|---|---|---|---|---|---|
| OGX-011 | |||||
| Milestone Payments to Third Parties | Nil | ||||
| Royalties to Third Parties(1)(2) | 1.0 - 3.5 | % | |||
| Revenue (net of royalties to third parties) and development costs are shared 65% OncoGenex and 35% Isis | |||||
OGX-427 | |||||
| Milestone Payments to Third Parties(3)(4) | |||||
| Start Phase 2 Clinical Trial | $ | 840,000 | |||
| Start Phase 3 Clinical Trial | $ | 1,474,000 | |||
| 1st Major Market Approval | $ | 1,948,000 | |||
| 2nd Major Market Approval | $ | 1,500,000 | |||
| Royalties to Third Parties(2)(5) | 3.25 - 5.75 | % | |||
OGX-225 | |||||
| Milestone Payments to Third Parties(4) | |||||
| Start Phase 2 Clinical Trial | $ | 590,000 | |||
| Start Phase 3 Clinical Trial | $ | 1,224,000 | |||
| 1st Major Market Approval | $ | 1,448,000 | |||
| 2nd Major Market Approval | $ | 1,000,000 | |||
Royalties to Third Parties(2)(5) | 2.88 - 5.25 | % | |||
- (1)
- Royalties to third parties are paid prior to any distribution to OncoGenex or Isis.
- (2)
- Minimum royalty rates assume certain third party royalty rates are not payable at the time that the product candidate is marketed due to expiration of patents held by such third parties. Maximum royalty rates assume all third party royalty rates currently in effect continue in effect at the time that the product candidate is marketed and are net of anti-stacking provisions specified in our agreements.
83
- (3)
- Additional milestone payments may be required in respect of OGX-427 for product approvals outside the field of oncology.
- (4)
- Certain milestone payments are payable in Canadian dollars, which are translated based on the September 30, 2006 exchange rate of US$1.00 = C$1.1151, and rounded to the nearest $1,000.
- (5)
- Includes royalties payable to UBC, a portion of which is distributable to Martin Gleave, and other co-inventors of the technology in accordance with UBC's patents and licensing policy.
Contract Research Agreements
Consistent with our strategy to outsource certain activities, we have established contract research agreements for pre-clinical, manufacturing and data management services. We choose which business or institution to use for these services based on their expertise, capacity and reputation and the cost of the service.
We also provide quantities of our product candidates to academic research institutions to investigate the mechanism of action and evaluate novel combinations of our product candidates with other cancer therapies in various cancer indications. These collaborations expand our research activities for our product candidates with modest contribution from OncoGenex.
Manufacturing
We do not own facilities for the manufacture of materials for clinical or commercial use. We rely and expect to continue to rely on contract manufacturers to manufacture our product candidates in accordance with current cGMP for use in clinical trials. We will ultimately depend on contract manufacturers for the manufacture of our products, when and if we have any, for commercial sale, as well as for process development.
To date, all active pharmaceutical ingredient, or API, for OGX-011 has been manufactured by Isis on a purchase order basis, under standards of Good Manufacturing Practices. Drug product manufactured from API has been performed by Formatech, Inc. and Pyramid Laboratories Inc. in separate manufacturing campaigns, pursuant to purchase orders or short-term contracts with us or our licensors, each of which has been fulfilled. For OGX-427, all API has been manufactured for us by Avecia Biotechnology Inc. and all drug product has been manufactured for us by Laureate Pharma, Inc., in each case pursuant to a purchase order or short-term contract that has been fulfilled. Contract manufacturing for commercial product is being evaluated and may or may not be performed at the current manufacturers. Larger contract manufacturers that can meet higher commercial drug quantities may be required and contracted to manufacture our products for commercial sale, when and if we have any.
Intellectual Property
Our success depends in part on our ability to obtain and maintain proprietary protection for our product candidates, technology and know-how, as well as operate without infringing on the proprietary rights of others and to prevent others from infringing the proprietary rights for our product candidates.
For each of OGX-011, OGX-427 and OGX-225, our intellectual property results from our licenses with UBC and Isis. As of February 28, 2007, we had exclusive rights through our license
84
with UBC to approximately ten issued U.S. and foreign patents and approximately 89 pending U.S. and foreign patent applications. These include one issued U.S. and associated pending foreign patent applications related to intellectual property that was jointly invented by employees of Isis and UBC.
We are aware of an issued U.S. patent and corresponding foreign counterparts containing claims relating to antisense sequences that inhibit IGFBP-2. Certain of these claims may be broad enough in scope such that, if we choose to commercialize OGX-225 in the U.S. or in any foreign jurisdiction in which a corresponding patent has issued, we may infringe such claims. We believe that there may be multiple grounds on which to challenge the validity of the U.S. patent and possibly the foreign counterparts, and we may determine to make such a challenge. Alternatively, it is possible that we may determine it prudent to seek a license from the patent holder to avoid potential extended litigation and other potential disputes.
For intellectual property under license from UBC, we direct patent prosecution and are responsible for all fees and costs related to the preparation, filing, prosecution and maintenance of the patent rights underlying the agreement. For this intellectual property, we file patent applications in the United States (U.S.), Canada, Europe (through the European Patent Office), Japan, and numerous other jurisdictions. Where necessary or preferable, we also rely on trade secrets and know-how. We pursue proprietary protection that we consider important to our business by filing patent applications on compositions of matter and their methods of use.
We have been granted rights to all intellectual property owned, licensed or otherwise controlled by Isis at the date of our agreements with them that relate to second-generation antisense chemistry and that are required for our product candidates. Isis directs patent prosecution and is responsible for all fees and costs related to the preparation, filing, prosecution and maintenance of their patent rights. Isis also pursues proprietary protection in numerous jurisdictions.
Individual patents have terms of protection depending on the laws of the countries in which the applications are made. Generally, patents issued in the U.S. are effective for 20 years from the earliest non-provisional filing date, if the application from which the patent issues was filed on or after June 8, 1995 (otherwise the term is the longer of 17 years from the issue date or 20 years from the earliest non-provisional filing date). The duration of patent terms for non-U.S. patents is typically 20 years from the earliest corresponding national or international filing date.
Patent term extensions, specifically to make up for regulatory delays, are available in the U.S., Europe, and Japan, and are under review in some other jurisdictions. Although we believe that our product candidates will meet the criteria for patent term extensions, there can be no assurance that we will obtain such extensions. Our licensed UBC patent estate, based on those patents and applications existing now and expected by us to issue, will expire in years ranging from 2020 to 2024, without the benefit of extensions.
Competition
The development and commercialization of new drugs is highly competitive. Our major competitors are large pharmaceutical, specialty pharmaceutical and biotechnology companies, in Canada, the United States and abroad. Many oncology drugs in clinical trials are being developed for the four primary cancer indications: lung, breast, colorectal, and prostate cancer. Certain of these drugs are, like ours, designed to interfere with treatment resistance. If new treatment
85
resistance drugs are approved for sale for the indications that OncoGenex is targeting in advance of our two lead product candidates, OGX-011 and OGX-427, or even after their commercialization, it may reduce the market's interest in our product candidates. We are aware of several other companies developing therapeutics, whether antisense or otherwise, that seek to promote tumor cell death by inhibiting cell survival proteins. Our competitors may seek to identify gene sequences, protein targets or antisense chemistry different from ours, and outside the scope of our intellectual property protection, to develop antisense therapeutics that serve the same function as our product candidates. Our competitors may also seek to use mechanisms other than antisense to inhibit the proteins that our product candidates are designed to inhibit the production of.
Many of our existing and potential competitors have substantially greater financial resources and expertise in manufacturing, developing products, conducting clinical trials, obtaining regulatory approvals, and marketing than OncoGenex. These entities also compete with us in recruiting and retaining qualified scientific and management personnel, as well as in acquiring products and technologies complementary to our programs.
Standard treatments vary considerably by cancer indication, and new drugs may be more effective in treating one cancer indication than another. In addition, it must be recognized that cancer is a difficult disease to treat and it is likely that no one therapeutic will replace all other therapies in any particular indication. Therapeutic strategies for treating cancer are increasingly focused on combining a number of drugs in order to yield the best results. Since OGX-011 and OGX-427 are intended to be used in multiple cancer indications and target the tumors' adaptive survival mechanisms, these drugs will potentially be synergistic with many new and currently marketed therapies.
Our ability to compete successfully will depend largely on our ability to:
- •
- advance the development of our lead programs, including the enrollment of patients for our clinical trials;
- •
- gain regulatory approval for our product candidates in their respective first indications as well as expand into additional indications;
- •
- commercialize our lead product candidates successfully, including convincing physicians, insurers and other third-party payors of the advantages of our products, when and if we have any, over current therapies;
- •
- obtain intellectual property protection and protect the exclusivity for our product candidates and products, when and if we have any; and
- •
- acquire other product candidates to expand our pipeline.
Government Regulation
The FDA and comparable regulatory agencies in state and local jurisdictions and in foreign countries impose substantial requirements upon the clinical development, premarket approval, manufacture, marketing and distribution of pharmaceutical and biological products. These agencies and other federal, state and local entities regulate research and development activities and the testing, approval, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, advertising and promotion of our product candidates. Failure to comply with applicable
86
FDA or other requirements may result in civil or criminal penalties, recall or seizure of products, partial or total suspension of production or withdrawal of a product from the market.
In the United States, the FDA regulates drug products under the Federal Food, Drug, and Cosmetic Act, or FD&C Act. The process required by the FDA before our drug and biologic product candidates may be marketed in the United States generally involves the following steps:
- •
- completion of extensive pre-clinical laboratory tests, pre-clinical animal studies and formulation studies primarily performed in accordance with FDA's current Good Laboratory Practice, or cGLP, regulations;
- •
- submission to the FDA's Center for Drug Evaluation and Research, or CDER, of an investigational new drug application, or IND, which must become effective before human clinical trials may begin. The IND contains the plan for the clinical trial. CDER specialists carefully review the IND application to determine whether there are any flaws in the initial studies and whether the overall development plan is feasible;
- •
- performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the product candidate for each proposed indication, as approved by an institutional review board, or IRB;
- •
- submission to the FDA of an NDA;
- •
- satisfactory completion of an FDA pre-approval inspection of the manufacturing facilities at which the product is produced to assess compliance with current Good Manufacturing Practice, or cGMP, regulations; and
- •
- FDA review and approval of the NDA prior to any commercial marketing, sale or shipment of the drug.
The testing and approval process requires substantial time, effort and financial resources, and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all.
Pre-clinical tests include laboratory evaluation of product chemistry, formulation and stability, as well as studies to evaluate toxicity in animals. The results of pre-clinical tests, together with manufacturing information and analytical data, are submitted as part of an IND to the FDA. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, raises concerns or questions about the conduct of the clinical trial, including concerns that human research subjects will be exposed to unreasonable health risks. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. A separate submission to an existing IND must also be made for each successive clinical trial conducted during product development.
Also, an IRB for each medical center proposing to conduct the clinical trial must review and approve the plan for any clinical trial before it commences at that center and it must monitor the clinical trial until completed. An IRB is a separate board of scientists, physicians, and nurses who are not associated with the clinical trial. Once approved by the board, the clinical trial is closely monitored by the IRB and given a formal review each year, or other interval, depending on the length of the clinical trial.
The FDA, the IRB, or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable
87
health risk. Clinical testing also must satisfy extensive good clinical practices, or GCPs, regulations and regulations for informed consent.
Clinical Trials
For purposes of NDA submission and approval, human clinical trials are typically conducted in the following four sequential phases, which may overlap:
- •
- Phase 1 Clinical Trials. Trials are initially conducted in a limited population to test the product candidate for safety, dose tolerance, absorption, metabolism, distribution and excretion in healthy humans or, on occasion, in patients, such as cancer patients.
- •
- Phase 2 Clinical Trials. Trials are generally conducted in the intended patient population to identify possible adverse effects and safety risks, to determine the efficacy of the product candidate for the specific targeted indications and to determine dose tolerance and optimal dosage.
- •
- Phase 3 Clinical Trials. These are commonly referred to as pivotal or registration studies or pivotal or registration trials, when designed to provide the principal basis for approval. When phase 2 clinical trials demonstrate that a dose range of the product candidate is effective and has an acceptable safety profile, phase 3 clinical trials are undertaken in large patient populations to further evaluate dosage, to provide substantial evidence of clinical efficacy and to further test for safety in an expanded and diverse patient population at multiple, geographically-dispersed clinical trial sites. If side effects become too severe, the clinical trial may be cancelled.
- •
- Phase 4 Clinical Trials. In some cases, the FDA may condition approval of an NDA for a product candidate on the sponsor's agreement to conduct additional clinical trials to further assess the drug's safety and effectiveness after NDA approval. Such post approval trials are typically referred to as phase 4 clinical trials.
New Drug Applications
The results of product development, pre-clinical studies and clinical trials are submitted to the FDA as part of an NDA. NDAs also must contain extensive manufacturing information. Once the submission has been accepted for filing, the FDA targets 10 months to review the application and respond to the applicant. This 10 month review time from the date of the receipt of the application is in accordance with the Prescription Drug User Fee Act. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the application to an advisory committee for review, evaluation and recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations. The FDA may deny approval of an NDA if the applicable regulatory criteria are not satisfied, or it may require additional clinical data and/or an additional pivotal phase 3 clinical trial. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data from clinical trials are not always conclusive and the FDA may interpret data differently than we or our collaborators interpret data. Once issued, the FDA may withdraw product approval if ongoing regulatory requirements are not met or if safety problems occur after the end product reaches the market. In addition, the FDA may require testing, including phase 4 clinical trials, and surveillance programs to monitor the safety effects of approved products which have been commercialized, and
88
the FDA has the power to prevent or limit further marketing of a product based on the results of these post-marketing programs or other information.
Fast Track
Fast track products are those products intended for the treatment of a serious or life-threatening condition and which demonstrate the potential to address unmet medical needs for such conditions. If fast track designation is obtained, FDA may initiate review of sections of an NDA before the application is complete. This "rolling review" is available if the applicant provides and the FDA approves a schedule for the remaining information. In some cases, a fast track product may be approved on the basis of either a clinical objective or a surrogate objective that is reasonably likely to predict clinical benefit under the FDA's accelerated approval regulations. Approvals of this kind typically include requirements for appropriate post-approval phase 4 clinical trials to validate the surrogate objective or otherwise confirm the effect of the clinical objective. As part of the fast track process, our product candidates may be eligible for "priority review," or review within a six month timeframe from the date a complete NDA is accepted for filing, if we show that our product candidate provides a significant improvement compared to marketed drugs. Because we are studying our product candidates for the treatment of serious and life-threatening conditions, we regularly assess the potential for using this program. For example, we have applied for fast track designation for OGX-011 for use in hormone refractory prostate cancer in combination with chemotherapy and intend to pursue this application with data from our ongoing phase 2 clinical trial in this patient population. However, there can be no assurance that any of our product candidates in development will receive designation as fast track products or that our product candidates will be reviewed or approved more expeditiously than would otherwise have been the case.
Special Protocol Assessment and Agreement
In the United States, certain protocols can be submitted to the FDA for Special Protocol Assessment (SPA). Under a SPA, the company and the FDA can reach an agreement on the design and size of the clinical trial. This agreement can be in writing and cannot be changed after the clinical trial begins except: (i) with written agreement of the company and the FDA; or (ii) if the director of the FDA reviewing division determines that "a substantial scientific issue essential to determining the safety or effectiveness of the drug" was identified after testing began. This, however, will not apply to approvals outside the U.S. We may submit one or more of our protocols to the FDA for Special Protocol Assessment in the future.
Other Regulatory Requirements
Any end products manufactured or distributed by us or our collaborators pursuant to FDA approvals are subject to continuing regulation by the FDA, including recordkeeping and reporting requirements. Adverse event experience with the product must be reported to the FDA in a timely fashion and pharmacovigilance programs to proactively look for these adverse events may be mandated by the FDA. Drug manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with ongoing regulatory requirements, including cGMPs, which impose certain procedural and documentation requirements upon us and our third-party manufacturers. Failure to comply with the statutory and regulatory requirements can subject a manufacturer to possible legal or regulatory action, such as warning
89
letters, suspension of manufacturing, seizure of product, injunctive action or possible civil penalties.
The FDA closely regulates the post-approval marketing and promotion of drugs, including standards and regulations for direct-to-consumer advertising, off-label promotion, industry-sponsored scientific and educational activities and promotional activities involving the Internet. Drugs and biologics may be marketed only for the approved indications and in accordance with the provisions of the approved label. Further, if there are any modifications to the drug, including changes in indications, labeling, or manufacturing processes or facilities, we may be required to submit and obtain FDA approval of a new or supplemental NDA, which may require us to develop additional data or conduct additional pre-clinical studies and clinical trials. Physicians may prescribe legally available drugs for uses that are not described in the product's labeling and that differ from those tested by us and approved by the FDA. Such off-label uses are common across medical specialties. Physicians may believe that such off-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, impose stringent restrictions on manufacturers' communications regarding off-label use.
International Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our future product candidates. Whether or not we obtain FDA approval for a product candidate, we must obtain approval of a product candidate by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product candidate in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Under European Union regulatory systems, marketing authorizations may be submitted either under a centralized or mutual recognition procedure. The centralized procedure provides for the grant of a single marking authorization that is valid for all European Union member states. The mutual recognition procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marking authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessment report, each member state must decide whether to recognize approval.
In Canada, applications for marketing authorizations are submitted to Health Canada, which is a centralized regulatory body overseeing prescription drug approvals for all of Canada. At present, Health Canada targets 355 days for application review and approvals. Once approved, the sponsor has the right to sell the drug in Canada; however, placement on the reimbursement formularies in the various Canadian provinces may take an unspecified amount of time.
In addition to regulations in the United States, Europe and Canada, we will be subject to a variety of foreign regulations governing clinical trials and commercial distribution of our future product candidates.
90
Third-Party Reimbursement
Sales of pharmaceutical products depend in significant part on the availability of coverage and adequate reimbursement from government and other third-party payors, including the Medicare and Medicaid programs. Third-party payors are increasingly challenging the pricing of pharmaceutical products and may not consider our future product candidates cost-effective or may not provide coverage of and adequate reimbursement for our future product candidates, in whole or in part. In the United States, there have been and we expect there will continue to be a number of legislative and regulatory proposals to change the health care system in ways that could significantly affect our business. The Medicare Prescription Drug, Improvement and Modernization Act of 2003 (MMA) establishes, among other things, a new prescription drug benefit beginning January 1, 2006 and changes reimbursement for certain oncology drugs under existing benefits.
United States Anti-Kickback and False Claims Laws
In the United States, we are subject to various federal and state laws pertaining to healthcare "fraud and abuse," including anti-kickback and false claims laws. The federal Anti-Kickback Law makes it illegal for any person, including a prescription drug manufacturer (or a party acting on its behalf) to knowingly and willfully solicit, offer, receive or pay any remuneration, directly or indirectly, in exchange for, or to induce, the referral of business, including the purchase, order or prescription of a particular drug, for which payment may be made under federal healthcare programs such as Medicare and Medicaid. The federal government has issued regulations, commonly known as safe harbors, that set forth certain provisions which, if fully met, will assure healthcare providers and other parties that they will not be prosecuted under the federal Anti-Kickback Law. Although full compliance with these provisions ensures against prosecution under the federal Anti-Kickback Law, the failure of a transaction or arrangement to fit within a specific safe harbour does not necessarily mean that the transaction or arrangement is illegal or that prosecution under the federal Anti-Kickback Law will be pursued. Violations of the law are punishable by up to five years in prison, criminal fines, administrative civil money penalties, and exclusion from participation in federal healthcare programs. In addition, many states have adopted laws similar to the federal Anti-Kickback Law. Some of these state prohibitions apply to referral of patients for healthcare services reimbursed by any source, not only the Medicare and Medicaid programs. Due to the breadth of these laws, and the potential for additional legal or regulatory change addressing some of our practices, it is possible that our sales and marketing practices or our relationships with physicians might be challenged under anti-kickback laws, which could harm us. In anticipation of commercializing a product or products which may be reimbursed under a federal healthcare program and other governmental healthcare programs, we are in the process of developing a comprehensive compliance program that will seek to establish internal controls to facilitate adherence to the rules and program requirements to which we may be or may become subject.
False claims laws prohibit anyone from knowingly presenting, or causing to be presented, for payment to third-party payors (including Medicare and Medicaid) claims for reimbursed items or services, including drugs, that are false or fraudulent, claims for items or services not provided as claimed, or claims for medically unnecessary items or services. Our future activities relating to the reporting of wholesaler or estimated retail prices for our products, when and if we have any, the reporting of Medicaid rebate information and other information affecting federal, state and third-party reimbursement of such products, and the sale and marketing of such products, are subject to
91
scrutiny under these laws. In addition, pharmaceutical companies have been prosecuted under the federal False Claims Act in connection with their off-label promotion of drugs. Suits filed under the False Claims Act, known as "qui tam" actions, can be brought by any individual on behalf of the government and such individuals (known as "relators" or, more commonly, as "whistleblowers") may share in the amounts paid by the entity to the government in fines or settlement. Penalties for a violation include three times the actual damages sustained by the government, plus mandatory civil penalties of between $5,500 and $11,000 for each separate false claim. In addition, certain states have enacted laws modeled after the federal False Claims Act. If the government were to allege that we were or our partners were, or convict us or our partners of, violating these false claims laws, we could be harmed, be subject to a substantial fine and suffer a decline in our stock price.
Insurance
We maintain director and officer insurance, key person insurance for certain individuals, general liability insurance for each of our facilities, shipping and storage insurance for OGX-011, and liability insurance for our clinical trials. We intend to expand our insurance coverage to include the sale of commercial products if marketing approval is obtained for any of our product candidates.
Trademarks
We own two approved Canadian trade-marks: OncoGenex™ and Bringing Hope to Life™.
Employees
We have a total of 25 employees; 21 full-time and four part-time. In our Vancouver office, we have 15 full-time employees and two part-time employees, eight of whom are engaged in clinical, regulatory affairs and business development and nine of whom are engaged in administration, accounting and finance. In our Seattle office, we have six full-time employees and two part-time employees, seven of whom are engaged in clinical and regulatory affairs and one who is engaged in administration. The following table sets forth information regarding our employees as at December 31, 2006, 2005, 2004 and 2003:
| | Current | December 31, 2006(1) | December 31, 2005(1) | December 31, 2004 | December 31, 2003 | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| Total Employees (full time (FT)/part time (PT)) | 25 (21/4) | 25 (21/4) | 17 (15/2) | 11 (11/0) | 5 (5/1) | ||||||
| Clinical, regulatory affairs and business development (FT/PT) | 15 (14/1) | 15 (14/1) | 11 (10/1) | 6 (6/0) | 2 (1/1) | ||||||
| Administration, accounting and finance (FT/PT) | 10 (7/3) | 10 (7/3) | 6 (5/1) | 5 (5/0) | 3 (3/0) | ||||||
- (1)
- December 31, 2006 figures include eight employees (6FT/2PT) in our Seattle office and December 31, 2005 figures include seven employees (6FT/1PT). In both years, all but one of the employees were engaged in clinical and regulatory affairs. We did not have any employees in Seattle as at December 31, 2004 and 2003.
92
All of our employees have entered into non-disclosure agreements with us regarding our intellectual property, trade secrets and other confidential information. None of our employees are represented by a labor union or covered by a collective bargaining agreement, nor have we experienced any work stoppages. We believe that we maintain satisfactory relations with our employees.
From time to time, we also use outside consultants to provide advice on our clinical development plans, research programs and potential acquisitions of new technologies.
Facilities
We have business offices located in both Vancouver, British Columbia and Seattle, Washington. In our Vancouver office, we lease approximately 4,857 square feet, currently at a rent of approximately $121,000 per annum. This lease expires in September 2009. We have an option to renew the lease for a further term of five years. In our Seattle office, we lease approximately 3,687 square feet, currently at a rent of approximately $65,000 per annum. This lease expires in November 2008, with an option to renew the lease for a further term of three years. We are in good standing, and not in default, under the leases for our Vancouver and Seattle premises.
Legal Proceedings
From time to time, we may be involved in litigation relating to claims arising out of our operations. We are not currently involved in any material legal proceedings.
Company Information
We were incorporated under the laws of Canada on May 26, 2000 as 3766284 Canada Inc. We changed our named to OncoGenex Technologies Inc. on July 6, 2000. We were extra-provincially registered in British Columbia on July 27, 2000. We have one wholly-owned subsidiary, OncoGenex, Inc., incorporated under the laws of the State of Washington on August 19, 2005, through which we conduct specified clinical services.
Our principal place of business is at 400 - 1001 West Broadway, Vancouver, British Columbia, V6H 4B1. Our telephone number is (604) 736-3678 and our facsimile number is (604) 736-3687. Our registered office is at c/o DuMoulin Black LLP, 10th floor, 595 Howe Street, Vancouver, British Columbia, V6C 2T5. The phone number for our registered office is (604) 687-1224 and the facsimile number is (604) 687-3635. Our agent for service of process in the United States is DL Services Inc., 1420 5th Avenue, Suite 3400, Seattle, Washington, 98101-4010, (206) 903-8800. We also maintain a website at www.oncogenex.ca. The information contained in, or that can be assessed through, our website is not a part of this prospectus.
Our principal legal advisor in Canada is DuMoulin Black LLP, 10th floor, 595 Howe Street, Vancouver, British Columbia, V6C 2T5, and our principal legal advisor in the United States is Dorsey & Whitney LLP, 1420 Fifth Avenue, Suite 3400, Seattle, Washington, 98101. Since our inception, our auditors have been Ernst & Young LLP, 2300 - 700 West Georgia Street, Vancouver, British Columbia, V7Y 1C7.
93
Directors, Executive Officers and Key Employees
Our directors, executive officers and key employees as of the date of this prospectus are as follows:
| Name & Municipality of Residence | Age | Position | |||
|---|---|---|---|---|---|
| Executive Officers and Key Employees | |||||
| Scott Cormack Richmond, British Columbia | 42 | Chief Executive Officer, President, Director and co-founder | |||
| Martin Gleave Vancouver, British Columbia | 48 | Chief Science Officer, Director and co-founder | |||
| Cindy Jacobs East King County, Washington | 49 | Executive Vice President, Chief Medical Officer | |||
| Stephen Anderson West Vancouver, British Columbia | 44 | Chief Financial Officer and Secretary | |||
| Monica Krieger Seattle, Washington | 57 | Vice President, Regulatory Affairs | |||
| Patricia Stewart Seattle, Washington | 64 | Senior Director, Clinical Research and Medical Affairs | |||
| Susan Tees Vancouver, British Columbia | 43 | Senior Director, Intellectual Property and Contracts | |||
| Directors | |||||
| Scott Cormack Vancouver, British Columbia | 42 | Director | |||
| Martin Gleave Vancouver, British Columbia | 48 | Director | |||
| James Shepard(1)(2)(3) Vancouver, British Columbia | 68 | Chairman of the Board | |||
| Aaron Davidson(2)(3)(4) Caledon, Ontario | 39 | Director | |||
| Neil Clendeninn(3)(4) Hanalei, Hawaii | 57 | Director | |||
| Christopher Laird(1)(2)(4) North Vancouver, British Columbia | 32 | Director | |||
| K. Thomas Bailey(1)(2) Mercer Island, Washington | 38 | Director | |||
Except as indicated in the following biographical summaries, the business address of each officer and director is c/o OncoGenex Technologies Inc., 400 - 1001 West Broadway, Vancouver, British Columbia, V6H 4B1.
- (1)
- Member of the audit committee.
- (2)
- Member of the finance committee.
- (3)
- Member of the nominating and governance committee.
- (4)
- Member of the compensation committee.
94
Executive Officers
Scott Cormack is a co-founder of OncoGenex and has been our President since May 2000 and our Chief Executive Officer since February 2002 and has served as a member of our board of directors since May 2000. Mr. Cormack served as interim President, CEO and Chairman of the Board of Salpep Biotechnology Inc., an asthma and inflammation biotechnology company, from 2000 to 2001. From 1998 to 2001, Mr. Cormack served as Vice President of Milestone Medica Corporation, a seed venture capital firm investing in life sciences opportunities. From 1995 to 1998, Mr. Cormack served as Chief Operating Officer of NeuroSpheres Ltd, a neural stem cell biotechnology company. Mr. Cormack was President and founder of For Tomorrow, a sole proprietorship engaged in business consulting, from 1991 to 1999. From 1986 to 1988, Mr. Cormack served as Territory Manager of Vetrepharm Inc. (now Bioniche Life Sciences, Inc.), a biopharmaceutical company developing products for human and animal health markets, and from 1988 to 1991 served as Technology Manager, Immunomodulators of Vetrepharm Inc. Since October 2005, Mr. Cormack has served as Director of the British Columbia Biotechnology Alliance Society. Mr. Cormack holds a Bachelor of Science degree from the University of Alberta.
Martin Gleave, M.D., is a co-founder of OncoGenex, the principal inventor of our product candidates, and has been our Chief Science Officer and a member of our board of directors since May 2000. In addition to his part-time duties at OncoGenex, Dr. Gleave is a Distinguished Professor and Research Director in the Department of Urologic Sciences at the University of British Columbia since 2000, and Director of the Prostate Centre at Vancouver General Hospital since 1993. He has also been a Senior Research Scientist at the Prostate Research Lab at Vancouver General Hospital and the Department of Cancer Endocrinology at BC Cancer Agency since 1992. Dr. Gleave has been a consultant Urologist for the Department of Urology at the University of Washington since 1997. Dr. Gleave received his M.D. from the University of British Columbia in 1984, his Fellow at the Royal College of Surgeons of Canada (FRCSC) in 1989, his Urologic Oncology Fellowship, University of Texas MD Anderson Cancer Center in 1992 and Fellow of the American College of Surgeons (FACS) in 1998.
Cindy Jacobs, Ph.D., M.D., has served as our Executive Vice President, Chief Medical Officer since September 2005. From 1999 to 2005, Dr. Jacobs served as Chief Medical Officer and Senior Vice President, Clinical Development of Corixa Corporation, a biopharmaceutical company (now a subsidiary of Glaxo-Smith-Kline). Prior to 1998, Dr. Jacobs held Vice President, Clinical Research positions at two other biopharmaceutical companies. Dr. Jacobs received her Ph.D. in Veterinary Pathology/Microbiology from Washington State University and M.D. from the University of Washington Medical School.
Stephen Anderson has served as our Chief Financial Officer since January 2006 and our Secretary since August 2006. From 2005 to 2006, Mr. Anderson served as Chief Financial Officer at Perceptronix Medical Inc., a medical diagnostic company. From 2003 to 2005, he served as President of his consulting company, SLA Enterprises Ltd. From 1998 to 2003, he served in various positions at Westport Innovations Inc., an alternative energy company, including Secretary, Controller, Vice President, Finance and Chief Financial Officer. Mr. Anderson holds Bachelor of Arts and Master of Business Administration degrees from the University of British Columbia. He is a Chartered Accountant.
95
Key Employees
Monica Krieger, Ph.D. has served as our Vice President, Regulatory Affairs since October 2005. From 1999 to 2005, Dr. Krieger served as Vice President, Regulatory Affairs at Corixa Corporation, a biopharmaceutical company. Dr. Krieger holds an M.B.A. from the Darden School at the University of Virginia and received her Ph.D. in Zoology from Rutgers University.
Patricia Stewart, M.D. has served as our Senior Medical Director Clinical Research and Medical Affairs since October 2005. From 2001 to 2005, Dr. Stewart served as Medical Director and Senior Medical Director, Medical Affairs and Clinical Research at Corixa Corporation, a biopharmaceutical company. Prior to 2001, Dr. Stewart was the In-Patient Medical Director, Fred Hutchinson Cancer Research Center and a member of the Seattle Bone Marrow Transplant Team for over 20 years.
Susan Tees, B.Sc. has served as our Senior Director, Intellectual Property and Contracts since August 2005. From 2003 to 2005, Ms. Tees was an associate at the intellectual property law firm of Smart & Biggar, Fetherstonhaugh. From 1997 to 2003, Ms. Tees was Senior Director, Contracts and Intellectual Property, at Kinetek Pharmaceuticals, Inc. Ms. Tees holds a B.Sc. in Microbiology from UBC and became a registered U.S. patent agent in 1999 and a Canadian patent agent in 2004.
With the exception of Dr. Gleave, all of our officers work full time for us.
Directors
Scott Cormack is a co-founder of OncoGenex and has been our President since May 2000 and our Chief Executive Officer since February 2002 and has served as a member of our board of directors since May 2000. See "Executive Officers" above.
Martin Gleave, M.D., is a co-founder of OncoGenex, a co-inventor of our product candidates, and has been our Chief Science Officer and a member of our board of directors since May 2000. See "Executive Officers" above.
James Shepard has served as Chairman of our board of directors since March 2002. Mr. Shepard served as Chief Executive Officer of Finning International Inc., a distributor of Caterpillar products and support services, from 1991 until his retirement in 2000. He has served as Chairman of the Board of Finning International Inc., Chairman of the Board of MacDonald Dettweiler and Associates, Vice-Chairman of the Conference Board of Canada, Vice-Chairman of the Business Council on National Issues, Director of the ABN-AMRO Bank of Canada, BC Rail and the Vancouver Symphony Orchestra, honorary Chairman of Leadership Vancouver, and was the founding Chairman of the Business Summit of B.C. Mr. Shepard is currently a Director of Imperial Oil Limited. Mr. Shepard holds a Bachelor of Applied Science in Civil Engineering from the University of British Columbia.
Aaron Davidson has served as a member of our board of directors since September 2003. Mr. Davidson is a Managing Director of H.I.G. Ventures, a venture capital firm since 2003. Prior to joining H.I.G. Ventures, he was a Vice President with Ventures West Management Inc., a venture capital company, from 2002 to 2003. From 2001 to 2002 Mr. Davidson served as Vice President, Corporate Development of Synx Pharma Inc., a biopharmaceutical company. From 1991 to 2001 he served in various management roles at Eli Lilly and Company, a pharmaceutical company. Mr. Davidson currently serves on the boards of Gemin X Biotechnologies Inc.,
96
HyperBranch Medical Technology, NeurAxon Inc., Novadaq Technologies Inc., Tranzyme Pharma, and Alder Biopharmaceuticals Inc., and serves as a board observer of Avera Pharmaceuticals Inc. Mr. Davidson has an M.B.A. from Harvard Business School and a Bachelor of Commerce degree from McGill University. Mr. Davidson's business address is c/o H.I.G. Ventures, 1001 Brickell Bay Drive, Floor 27, Miami, Florida, 33131.
Neil Clendeninn, M.D., Ph.D., has served as a member of our board of directors since September 2004. Dr. Clendeninn served as Corporate Vice President, Head of Clinical Affairs of Agouron Pharmaceuticals, Inc., a subsidiary of Pfizer Inc., from 1993 until his retirement in 2001. Dr. Clendeninn holds a B.A. Biology/Chemistry from Wesleyan University, and a Ph.D. Microbiology/Pharmacology and M.D. from New York University.
Christopher Laird has served as a member of our board of directors since December 2005. Mr. Laird is Vice President, biotechnology of Ventures West Management Inc., a venture capital firm, and has been with Ventures West since 2000. Mr. Laird currently serves as a director of Alder Biopharmaceuticals and an observer of Celator Pharmaceuticals. Mr. Laird holds a Bachelor of Science in microbiology from the University of British Columbia. Mr. Laird's business address is c/o Ventures West Management Inc., 2500 - 1066 West Hastings Street, Vancouver, British Columbia, V6E 3X1.
K. Thomas Bailey has served as a member of our board of directors since January 2006. Mr. Bailey is the Chief Financial Officer at Angiotech Pharmaceuticals, Inc., a biotechnology company. Prior to joining Angiotech in 2003, he worked as an investment banker from 1997 to 2002 at Credit Suisse First Boston. Mr. Bailey holds a Bachelor of Arts in Economics from Harvard University and an M.B.A. from Harvard Business School in 1995. Mr. Bailey's business address is c/o Angiotech Pharmaceuticals, Inc., 1618 Station Street, Vancouver, British Columbia, V6A 1B6.
Board Composition
Our directors are elected at each annual general meeting of shareholders and serve until their successors are elected or appointed, unless they resign or are removed earlier. The next annual general meeting at which directors will be elected will be in 2007. Our bylaws permit our shareholders to establish by resolution the authorized number of directors, and seven directors are currently authorized.
Board Committees
Our board of directors currently has four committees: the audit committee, the compensation committee, the governance and nomination committee and the finance committee.
Audit Committee
The members of our audit committee are Mr. Bailey, Mr. Shepard and Mr. Laird, each of whom is a non-employee member of our board of directors. Mr. Bailey chairs the committee and is our audit committee financial expert (as is currently defined under the SEC rules implementing Section 407 of the Sarbanes Oxley Act of 2002). Our board of directors has determined that each member of our audit committee is independent under the audit committee requirements of the Nasdaq Global Market and the rules and regulations of the SEC and Canadian provincial securities regulatory authorities.
97
Our audit committee is responsible for overseeing our financial reporting processes and control systems on behalf of our board of directors. Our independent auditors report directly to our audit committee. The duties of our audit committee are set forth in a written audit committee charter, which will become effective upon the closing of this offering. Specific responsibilities of our audit committee include:
- •
- Evaluating the performance, and assessing the qualifications, of our independent auditors and recommending to our board of directors the appointment of our independent auditors for the purpose of preparing or issuing an auditor's report or performing other audit, review or attest services;
- •
- Subject to the appointment of our independent auditors by our shareholders, determining and approving the engagement of, and compensation to be paid to, our independent auditors;
- •
- Determining and approving the engagement, prior to the commencement of such engagement, of, and compensation for, our independent auditors to perform any proposed permissible non-audit services;
- •
- Reviewing our financial statements and management's discussion and analysis of financial condition and results of operations and recommending to our board of directors whether or not such financial statements and management's discussion and analysis of financial condition and results of operations should be approved by our board of directors;
- •
- Conferring with our independent auditors and with our management regarding the scope, adequacy and effectiveness of internal financial reporting controls in effect;
- •
- Reviewing and discussing with our management that appropriate procedures exist for the receipt, retention and treatment of complaints received by us regarding accounting, internal accounting controls or auditing matters and the confidential and anonymous submission by our employees of concerns regarding questionable accounting or auditing matters;
- •
- Reviewing and assessing our risk management policies and procedures with regard to identification of our principal risks annually, including the adequacy of the implementation of appropriate systems to mitigate and manage these risks, and to report periodically to the board of directors;
- •
- Reviewing with our management the timeliness and accuracy of our filings with regulatory authorities; and
- •
- Reviewing all related party transactions and development with our management of policies and procedures related to those transactions.
Compensation Committee
The members of our compensation committee are Mr. Davidson, Dr. Clendeninn and Mr. Laird. Mr. Davidson chairs the committee. Our board of directors has determined that each member of our compensation committee is an independent director for purposes of the compensation committee requirements of the Nasdaq Global Market and as defined in the rules and regulations of the Canadian provincial securities regulatory authorities.
98
The duties of our compensation committee are set forth in a written compensation committee charter, which will become effective upon the closing of this offering. Specific responsibilities of our compensation committee include:
- •
- Reviewing and making recommendations to our board of directors for the compensation of our chief executive officer and other executive officers, including salary, incentives, benefits and other perquisites;
- •
- Reviewing and making recommendations to our board of directors regarding general compensation philosophy and guidelines for all executive officers, including our CEO, including incentive plan design and other remuneration;
- •
- Preparing any executive compensation report to be included in our periodic filings or proxy statement; and
- •
- Acting as administrator of our stock option plan (and other equity based plans established from time to time, if and to the extent that such administration is delegated to the Compensation Committee by our board of directors).
Compensation Committee Interlocks and Insider Participation
None of the members of our compensation committee has, at any time during the prior three years, been an officer or employee of ours. None of our directors has, at any time during the prior three years, been employed as one of our executive officers while concurrently serving as a director or member of the compensation committee (or other board committee performing equivalent functions or, in the absence of any such committee, the entire board of directors) of another issuer, when such issuer has had any of its executive officers serving on our compensation committee (or other board committee performing equivalent functions or, in the absence of any such committee, our entire board of directors) or as one of our directors. None of our directors or officers has at any time during the prior three years been indebted to us or, except as otherwise disclosed in this prospectus, had an interest in a material transaction involving the Company.
Governance and Nomination Committee
The members of our governance and nomination committee are Mr. Shepard, Dr. Clendeninn and Mr. Davidson. Mr. Shepard chairs the committee. Our board of directors has determined that each member of our governance and nomination committee is an independent director for purposes of the nominating and corporate governance committee requirements of the Nasdaq Global Market and as defined in the rules and regulations of the Canadian provincial securities regulatory authorities.
The duties of our governance and nomination committee are set forth in a written governance and nomination committee charter, which will become effective upon the closing of this offering. Specific responsibilities of our governance and nomination committee include:
- •
- Reviewing the appropriateness of our board structure, composition and practices, and making recommendations on these matters to our board of directors;
- •
- Reviewing and recommending to our board of directors candidates for appointment to the office of Chair of our board of directors or to the office of CEO;
99
- •
- Reviewing and recommending to our board of directors appropriate corporate governance policies, including our framework for delegating authority from our board to management;
- •
- Making recommendations to our board of directors to fill any vacancies on the board or its committees;
- •
- Reviewing, approving and reporting to our board on the performance evaluation of the Chair of the board and each of its committees;
- •
- Reviewing, approving and reporting to our board and to the Compensation Committee on the performance evaluation of the CEO, including performance against our corporate objectives; and
- •
- Overseeing compliance with our timely disclosure, confidentiality, and insider trading policies, and with our code of business conduct and ethics, and authorizing any waiver granted in connection with such policies and code.
Finance Committee
The members of our finance committee are Mr. Shepard, Mr. Davidson, Mr. Laird and Mr. Bailey. The mandate of the committee is to oversee our finance strategy. The committee's mandate will terminate upon completion of this offering.
Compensation of Directors
From inception to September 16, 2004, we did not pay any of our directors cash compensation for serving as a director.
On October 26, 2006, our board of directors approved a policy regarding compensation for each of our directors that is neither management nor a contractually appointed investor representative. The policy provides that each such director will receive:
- •
- an annual retainer of $5,000 and $750 for each board or committee meeting attended in person ($375 for meetings attended by telephone conference);
- •
- upon his or her first election to our board, an initial stock option grant to purchase 9,000 of our common shares, which shall vest over eight equal quarterly periods commencing after the completion of the first quarter following the date of grant, as well as, following each annual general meeting of our shareholders at which such director is re-elected to our board, a stock option grant to purchase 5,000 of our common shares, which shall vest over four equal quarterly periods commencing after the completion of the first quarter following the annual general meeting of our shareholders;
- •
- upon his or her first appointment to a committee of our board, an initial stock option grant to purchase 1,000 of our common shares, which shall vest over four equal quarterly periods commencing after the completion of the first quarter following the date of grant, as well as, following each annual general meeting of our shareholders at which such director is re-elected to our board and is subsequently appointed by our board as a member of the audit committee, the compensation committee or the governance and nomination committee, a stock option grant to purchase 1,000 of our common shares for each committee of which he or she is a member, which shall vest over four equal quarterly
100
- •
- following each annual general meeting of our shareholders at which such director is elected to our board and is subsequently appointed as the chairperson of our board, the audit committee, the compensation committee or the governance and nomination committee, a stock option grant to purchase 1,500 of our common shares for each chairperson role, which shall vest over four equal quarterly periods commencing after the completion of the first quarter following the director's appointment as a chairperson.
periods commencing after the completion of the first quarter following the director's appointment as a committee member; and
Options were last granted to these directors in March 2006 as compensation for past services during the 2005 calendar year. These directors have agreed to defer their current entitlement to further options under the policy from June 29, 2006, the date of our last annual general meeting, until the closing of this offering. The options will be granted after the offering at an exercise price at least equal to the fair market value of our common shares at the time of grant and will have an accelerated vesting schedule.
For the year ended December 31, 2005, James Shepard and Neil Clendeninn received stock options in lieu of their cash compensation as follows:
| Name | No. of Shares Subject to Options | Exercise Price | Expiry Date | ||||
|---|---|---|---|---|---|---|---|
| James Shepard | 2,768 | C$ | 0.95 | March 23, 2013 | |||
| Neil Clendeninn | 2,359 | C$ | 0.95 | March 23, 2013 | |||
In addition, effective August 3, 2005, all outstanding stock options granted at an exercise price of C$4.00 or C$5.00 were repriced to C$0.90 per share. The repriced options included the following options held by our non-employee directors:
| Name | No. of Shares Subject to Repriced Options | Expiry Date | ||
|---|---|---|---|---|
| Martin Gleave | 20,000 | March 1, 2009 | ||
| James Shepard | 3,200 | March 25, 2009 | ||
| James Shepard | 1,700 | April 14, 2010 |
All of our directors are reimbursed for out-of pocket expenses incurred in attending board and committee meetings.
In February, 2005, we entered into a consulting agreement with CANAID, Inc., a company controlled by Dr. Clendeninn, under which CANAID, Inc. agreed to provide advice and counsel on the design, implementation and analysis of clinical trials. The agreement terminated on February 15, 2006. We paid CANAID, Inc. $6,000 during 2005 and no amount during 2006 in respect of this agreement.
For more information regarding Martin Gleave's services agreement, see "Management — Executive Compensation — Employment and Consulting Agreements".
101
Executive Compensation
Summary Compensation Table
The following table presents compensation information for our fiscal years ended December 31, 2006, 2005, 2004 and 2003 paid to or accrued for our Chief Executive Officer, our Chief Financial Officer, and our other executive officer whose total cash compensation exceeded $100,000 during the year ended December 31, 2006, who we refer to as our named executive officers.
| | | | | | Long-Term Compensation | | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Annual Compensation | | ||||||||||
| | Awards Shares Underlying Number of Options Granted (#) | | ||||||||||
| Name and Principal Position(s) | Year | Salary ($) | Bonus ($) | Other Annual Compensation ($) | All Other Compensation ($) | |||||||
| Scott Cormack(1) President and Chief Executive Officer and Director | 2006 2005 2004 2003 | 246,914 196,451 175,156 157,053 | 88,889 59,530 42,037 71,388 | — — — — | — 212,000 99,000 94,000 | — — — — | ||||||
Stephen Anderson(1)(2) Chief Financial Officer | 2006 2005 2004 2003 | 155,678 — — — | 36,584 — — — | — — — — | 100,000 — — — | — — — | ||||||
Sherry Tryssenaar(1)(3) Chief Financial Officer | 2006 2005 2004 2003 | — 109,575 132,135 117,790 | — 20,636 21,802 35,694 | — — — — | — — 31,500 62,500 | — 156,913 — — | ||||||
Cindy Jacobs, M.D.(4) Executive Vice President, Chief Medical Officer | 2006 2005 2004 2003 | 320,000 93,330 — — | 90,240 10,634 — — | — — — — | — 125,000 — — | — — — — | ||||||
- (1)
- Mr. Cormack, Mr. Anderson and Ms. Tryssenaar have received their compensation in Canadian dollars. Annual amounts for these executives have been translated from Canadian dollars for each year based on the applicable yearly average exchange rate, except where otherwise noted.
- (2)
- Mr. Anderson commenced employment with us as our Chief Financial Officer on January 9, 2006 at an annual salary of C$180,000, which translates into $158,730 based on the 2006 yearly average noon buying rate published by the Federal Reserve Bank of New York, being US$1.00 = C$1.1340.
- (3)
- Ms. Tryssenaar was a consultant with us from August 6, 2002 to January 31, 2003 and was our Chief Financial Officer from February 1, 2003 to September 30, 2005. Amount under "All Other Compensation" represents severance payment as at September 30, 2005.
102
- (4)
- Cindy Jacobs commenced employment with us on September 12, 2005 and was appointed Chief Medical Officer.
Option Grants in Fiscal Year 2006
The following table sets forth information regarding options granted to each of our named executive officers during the fiscal year ended December 31, 2006. The exercise prices of the options we granted were the fair market value of our common stock on the date of grant, as determined by our board of directors. Options generally vest either in three equal amounts on the first, second and third anniversary of the date of grant or in four equal amounts beginning on the end of a probationary period and thereafter on the first, second and third anniversary of the optionee's commencement of employment with us.
| | Individual Grants | | | |||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Potential Realizable Value at Assumed Annual Rates of Share Price Appreciation for Option Term(3) | |||||||||||||||
| | | | | Market Value of Shares Underlying Options on the Date of Grant in C$(2) | | |||||||||||
| | Number of Securities Underlying Options Granted | Percent of Total Options Granted to Employees in 2006(1) | Exercise Price Per Share in C$(2) | | ||||||||||||
| Name | Expiration Date | |||||||||||||||
| 5% | 10% | |||||||||||||||
| Scott Cormack | — | — | — | — | — | — | — | |||||||||
| Stephen Anderson | 100,000 | (4) | 99.0 | $ | 0.95 | $ | 0.95 | March 23, 2013 | 1,125,680 | 1,558,973 | ||||||
| Sherry Tryssenaar | — | — | — | — | — | — | — | |||||||||
| Cindy Jacobs | — | — | — | — | — | — | — | |||||||||
- (1)
- The figures representing percentages of total options granted to employees in 2006 are based on a total of 101,000 common shares underlying stock options granted to our employees during fiscal year 2006.
- (2)
- The exercise price of each stock option granted was equal to the fair market value of our common shares as determined by our board of directors on the date of grant. The exercise price must be paid in cash or by bank draft or certified check, or by using such consideration as our compensation committee may permit.
- (3)
- The amounts shown in the table above as potential realizable value represent hypothetical gains that could be achieved for the respective stock options if exercised at the end of the option term. These amounts represent assumed rates of appreciation in the value of our common shares from the fair market value on the date of grant. Potential realizable values in the table above are calculated by:
- •
- Multiplying the number of common shares subject to the stock option by the assumed initial public offering price of $8.00 per share;
- •
- Assuming that the aggregate share value derived from that calculation compounds at the annual 5 percent or 10 percent rates shown in the table for the balance of the term of the option; and
- •
- Subtracting from that result the total option exercise price. The 5 percent and 10 percent assumed rates of appreciation are suggested by the rules of the SEC and do not represent our estimate or projection of the future common share price. Actual gains, if any, on stock option exercises will be dependent on the future performance of our common shares.
- (4)
- The stock options vest in four equal amounts on the end of a probationary period and on the first, second and third anniversary of the commencement of employment with us.
Option Grants in Fiscal Year 2005
The following table sets forth information regarding options granted to each of our named executive officers during the fiscal year ended December 31, 2005. The exercise prices of the options we granted were the fair market value of our common stock on the date of grant, as determined by our board of directors. Options generally vest either in three equal amounts on the first, second and third anniversary of the date of grant or in four equal amounts beginning on the
103
end of a probationary period and thereafter on the first, second and third anniversary of the optionee's commencement of employment with us.
| | | | | | | Potential Realizable Value at Assumed Annual Rates of Share Price Appreciation for Option Term(3) | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Individual Grants | |||||||||||||
| | | | | Market Value of Shares Underlying Options on the Date of Grant in C$(2) | | |||||||||
| Name | Number of Securities Underlying Options Granted | Percent of Total Options Granted to Employees in 2005(1) | Exercise Price Per Share in C$(2) | Expiration Date | ||||||||||
| 5% | 10% | |||||||||||||
| Scott Cormack | 212,000 | (4) | 48.6 | 0.95 | 0.95 | August 8, 2012 | 2,386,442 | 3,305,023 | ||||||
| Stephen Anderson | — | — | — | — | — | — | — | |||||||
| Sherry Tryssenaar | — | — | — | — | — | — | — | |||||||
| Cindy Jacobs | 125,000 | (5) | 28.6 | 0.95 | 0.95 | September 12, 2012 | 1,407,100 | 1,948,716 | ||||||
- (1)
- The figures representing percentages of total options granted to employees in 2005 are based on a total of 436,500 common shares underlying stock options granted to our employees during fiscal year 2005.
- (2)
- The exercise price of each stock option granted was equal to the fair market value of our common shares as determined by our board of directors on the date of grant. The exercise price must be paid in cash or by bank draft or certified check, or by using such consideration as our compensation committee may permit.
- (3)
- The amounts shown in the table above as potential realizable value represent hypothetical gains that could be achieved for the respective stock options if exercised at the end of the option term. These amounts represent assumed rates of appreciation in the value of our common shares from the fair market value on the date of grant. Potential realizable values in the table above are calculated by:
- •
- Multiplying the number of common shares subject to the stock option by the assumed initial public offering price of $8.00 per share;
- •
- Assuming that the aggregate share value derived from that calculation compounds at the annual 5 percent or 10 percent rates shown in the table for the balance of the term of the option; and
- •
- Subtracting from that result the total option exercise price. The 5 percent and 10 percent assumed rates of appreciation are suggested by the rules of the SEC and do not represent our estimate or projection of the future common share price. Actual gains, if any, on stock option exercises will be dependent on the future performance of our common shares.
- (4)
- The stock options vest in three equal amounts on the first, second and third anniversary of the date of grant.
- (5)
- The stock options vest in four equal amounts on the end of a probationary period and on the first, second and third anniversary of the commencement of employment with us.
In addition, effective August 3, 2005, all outstanding stock options granted at an exercise price of C$4.00 or C$5.00 were repriced to C$0.90 per share. The repriced options included the following options held by our named executive officers:
| Name | Date of Repricing | Securities Under Options Repriced or Amended (#) | Market Price of Securities at Time of Repricing (C$/Security)(1) | Exercise Price at Time of Repricing or Amendment (C$/Security) | New Exercise Price (C$/Security) | Expiry Date | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Scott Cormack | August 3, 2005 | 114,000 | 0.90 | 4.00 | 0.90 | January 1, 2009 | ||||||
| Sherry Tryssenaar | August 3, 2005 | 4,000 | 0.90 | 4.00 | 0.90 | August 1, 2009 | ||||||
| Sherry Tryssenaar | August 3, 2005 | 32,500 | 0.90 | 5.00 | 0.90 | February 1, 2010 |
- (1)
- The new exercise price of each stock is equal to the fair market value of our common shares at the date of repricing as determined by our board of directors on the date of repricing.
104
Fiscal Year 2006 Option Values
The following table sets forth the number of common shares covered by stock options outstanding as of December 31, 2006 and the value of stock options as of December 31, 2006 for the named executive officers. No stock options were exercised by our named executive officers during 2006.
| | Number of Common Shares Underlying Unexercised Options at December 31, 2006 | Value of Unexercised in-the-Money Options at December 31, 2006(1) | ||||||
|---|---|---|---|---|---|---|---|---|
| Name | ||||||||
| Exercisable | Unexercisable | Exercisable | Unexercisable | |||||
| Scott Cormack | 344,667 | 174,333 | (2) | 2,763,343 | 1,250,073 | |||
| Stephen Anderson | 25,000 | 75,000 | 179,056 | 537,169 | ||||
| Sherry Tryssenaar | 98,000 | — | 706,222 | — | ||||
| Cindy Jacobs | 62,500 | 62,500 | 447,641 | 447,641 | ||||
- (1)
- Amounts presented under the caption "Value of Unexercised in-the-Money Options at December 31, 2006" are based on the assumed initial public offering price of $8.00 per share minus the exercise price, multiplied by the number of shares subject to the stock option, without taking into account any taxes that might be payable in connection with this transaction.
- (2)
- Three quarters of Mr. Cormack's unvested options automatically vest on the date that we become a reporting issuer.
Fiscal Year 2005 Option Values
The following table sets forth the number of common shares covered by stock options outstanding as of December 31, 2005 and the value of stock options as of December 31, 2005 for the named executive officers. No stock options were exercised by our named executive officers during 2005.
| | Number of Common Shares Underlying Unexercised Options at December 31, 2005 | Value of Unexercised in-the-Money Options at December 31, 2005(1) | ||||||
|---|---|---|---|---|---|---|---|---|
| Name | ||||||||
| Exercisable | Unexercisable | Exercisable | Unexercisable | |||||
| Scott Cormack | 209,667 | 309,333 | (2) | 1,515,445 | 2,226,723 | |||
| Stephen Anderson | — | — | — | — | ||||
| Sherry Tryssenaar | 98,000 | — | 708,331 | — | ||||
| Cindy Jacobs | 31,250 | 93,750 | 224,530 | 673,591 | ||||
- (1)
- Amounts presented under the caption "Value of Unexercised in-the-Money Options at December 31, 2005" are based on the assumed initial public offering price of $8.00 per share minus the exercise price, multiplied by the number of shares subject to the stock option, without taking into account any taxes that might be payable in connection with this transaction.
- (2)
- Three quarters of Mr. Cormack's unvested options will automatically vest on the completion of this offering.
105
Employment and Consulting Agreements
Scott Cormack
In December 2001, we entered into an employment agreement with Mr. Cormack, our President and Chief Executive Officer. This agreement was amended in August 2005. Mr. Cormack currently receives an annual base salary of C$300,000, subject to increases at the discretion of our board of directors. Mr. Cormack is also eligible for a performance bonus of up to 40 percent of his annual base salary based on mutually agreed upon objectives as established by the board of directors and Mr. Cormack. We agreed to advance Mr. Cormack an interest-free housing relocation loan of up to C$150,000 repayable over ten years, or within 60 days of Mr. Cormack's resignation or termination for just cause; however, this loan was never drawn.
Upon occurrence of a change of control, one-half of any options Mr. Cormack has which have not yet vested at the time of the change of control shall automatically vest upon the date of the change of control. In addition, if Mr. Cormack is terminated without just cause after such change of control, all remaining options he has which have not yet vested shall automatically vest. Mr. Cormack shall be entitled to exercise the options that vest as provided above together with any options that had previously vested but had not yet been exercised at the time of the change of control.
If we complete this offering, three quarters of the options Mr. Cormack has that have not yet vested shall automatically vest. He will be entitled to exercise such options so vested together with any options which had previously vested but had not yet been exercised at the time of this offering.
Under the agreement, we may terminate his employment at any time, and Mr. Cormack may terminate his employment with us by providing us with two months written notice. If we terminate Mr. Cormack's employment without just cause, or if Mr. Cormack resigns following any material adverse change, without Mr. Cormack prior written consent, in his title, status, position, job function, compensation or reporting responsibilities (referred to in this section as a constructive dismissal), we are obligated to pay to him a lump sum or continuance of salary of 12 months of his compensation, calculated based on the average of the base salary plus bonus paid to him in the prior two year period. In addition, if Mr. Cormack is terminated without just cause or is constructively dismissed, any stock options granted to Mr. Cormack prior to July 13, 2005 but not yet vested shall automatically vest and be exercisable and Mr. Cormack shall be entitled to exercise such options until the date that is seven years after the grant date of such options, and any stock options granted to Mr. Cormack after July 13, 2005 shall, to the extent already vested, be exercisable until the expiry date of such options or the 365th day after his termination or constructive dismissal, whichever is earlier. Subject to certain limitations, we are also obligated to maintain Mr. Cormack's benefits for up to 12 months or until he becomes employed elsewhere, whichever date comes first.
During the term of his employment agreement and within 12 months following the termination thereof, Mr. Cormack agrees that, without our written consent, he will not, within Canada or the United States, own or have any interest directly in, except for an interest of less than 5 percent in a publicly traded company, act as an officer, director, agent, employee or consultant of, or assist in any way or in any capacity any person that is engaged in a business that is substantially similar to our business or that competes with us. Mr. Cormack also agrees, for a period of twelve months following the termination of his employment, not to, directly or indirectly,
106
contact for the purpose of solicitation or actually solicit any person who is or was a customer of us on or at any time within the two years before the date of termination of his employment agreement or who was scheduled to become a customer of us within 12 months prior to the date of termination of his employment agreement; induce or attempt to induce any person who was our employee at the date of termination or has been our employee during the twelve months before the date of termination or solicit, divert or take away any of our staff or business or otherwise compete for our accounts or personnel which become known to him through his relationship with us; or influence or attempt to influence any of our customers, suppliers, resellers or personnel not to do business with us or take any action which may be reasonably foreseen to result in harm to us.
Martin Gleave
In December 2001, we entered into a services agreement with Dr. Gleave, under which Dr. Gleave agreed to perform all functions normally associated with the position of Chief Scientific Officer, as amended from time to time. We paid Dr. Gleave $73,000, $74,000 and $78,000 for the year ended December 31, 2003, 2004 and 2005, respectively. Dr. Gleave currently receives an annual consulting fee of C$100,000 for serving as our Chief Scientific Officer as well as C$2,000 per day for any travel outside of British Columbia. Dr. Gleave is also eligible for a bonus of up to 40 percent of his annual consulting fee. Under the agreement, we may terminate the agreement at any time without cause, in which case we are required to pay Dr. Gleave an amount equal to the lesser of C$100,000 or the aggregate remaining consulting fees payable to the end of the December 31, 2008. In addition, if Dr. Gleave is terminated without cause, any stock options granted to him after July 13, 2005 shall, to the extent already vested, be exercisable until the expiry date of such options or the 365th day after his termination, whichever is earlier. We may also terminate the agreement if Dr. Gleave defaults on his obligation and fails to correct such default within 30 days of our written notice to him of his default or with five days written notice if Dr. Gleave ceases to beneficially own or control any of our shares. The agreement is for a fixed term and will automatically expire on December 31, 2008, if not terminated earlier or extended by mutual agreement. Dr. Gleave is required to be available to render his services in good faith as is reasonably requested by us.
During the term of the agreement and for two years after termination thereof, Dr. Gleave agrees not to engage, directly or indirectly, with any other person or in any other capacity participate in any business competitive with our business unless we otherwise agree, except for his holding a less than five percent interest in a publicly traded company. During the same period, Dr. Gleave also agrees not to induce or solicit, or attempt to induce or solicit or assist any third party in inducing, soliciting or interfering with any of our employees, consultants, customers, or strategic partners.
Cindy Jacobs
In September 2005, we entered into an employment agreement with Dr. Jacobs, our Executive Vice President, Chief Medical Officer. Dr. Jacobs currently receives an annual base salary of $340,000, subject to increases at the discretion of our board of directors. Dr. Jacobs is also eligible for a discretionary performance bonus of up to 30 percent of her annual base salary, based on annual corporate objectives established by the board of directors and annual personal objectives established by Dr. Jacobs' supervisor. Under the agreement, we may terminate her employment at any time and Dr. Jacobs shall provide the Company with 30 days written notice. If we terminate
107
Dr. Jacob's employment without cause, we are obligated to pay her severance pay equal to 12 months of her then current base salary.
During the term of the agreement, Dr. Jacobs agrees not, within Canada, the United States or Europe, without our written consent, to own or have any interest directly in, act as an officer, director, agent, employee or consultant of, or assist in any way or in any capacity any person or entity that is engaged in a business that is substantially similar to or competes with our business, except for owning an interest of less than five percent in a publicly traded company. Dr. Jacobs further agrees that during the first year after termination of her employment with us, she will not, within Canada or the State of Washington, without our written consent, act as an officer, director, agent, employee or consultant of, or assist in any way or in any capacity any person or entity that is engaged in a business that is substantially similar to or competes with our business. In addition, Dr. Jacobs agrees not, during the term of this agreement and twelve months thereafter, to, directly or indirectly, solicit or attempt to solicit any person or entity that is or was our customer on or at any time within twelve months before the date of termination, any of our employees or those who have been our employees during the twelve months before the date of termination, or any of our suppliers not to do business with us or take any action that may be reasonably foreseen to result in harm to us.
Stephen Anderson
In January 2006 we entered into an employment agreement with Stephen Anderson, our Chief Financial Officer. Mr. Anderson currently receives an annual base salary of C$184,000. Mr. Anderson is also eligible for a performance bonus of up to 25 percent of his annual base salary, based on annual corporate objectives established by the board of directors and annual personal objectives established by Mr. Anderson's supervisor. Under the agreement, we may terminate his employment for just cause at any time. We may terminate Mr. Anderson's employment without just cause, or we may constructively dismiss him by making a material adverse change, without his prior written consent, in his title, position, job function or compensation, by providing him with four weeks notice plus an additional two weeks for each full year of employment to a maximum of 26 weeks, or paying to Mr. Anderson in lieu of notice an amount determined by multiplying his average weekly earnings (inclusive of base salary and bonus) by the number of weeks in the severance period. Subject to certain limitations, we are also obligated to continue benefit entitlement during the severance period or until he becomes employed elsewhere, whichever date comes first.
During the term of the agreement and six months thereafter, Mr. Anderson agrees not, within Canada, the United States or Europe, without our written consent, to own or have any interest directly in, act as an officer, director, agent, employee or consultant of, or assist in any way or in any capacity any person or entity that is engaged in a business that is substantially similar to or competes with our business, except for owning an interest of less than 5 percent in a publicly traded company. In addition, Mr. Anderson agrees not, during the term of this agreement and six months thereafter, to, directly or indirectly, solicit or attempt to solicit any person or entity that is or was our customer on or at any time within twelve months before the date of termination, any of our employees or those who have been our employees during the twelve months before the date of termination, or any of our suppliers not to do business with us or take any action that may be reasonably foreseen to result in harm to us.
108
Employee Benefit Plans
In October, 2001, our board of directors adopted a Stock Option Plan that was subsequently approved by our shareholders. The Stock Option Plan was amended in September 2003, December 2004 and June 2005. Under the Stock Option Plan, as of February 28, 2007, options have been granted to directors, officers, employees and consultants to purchase up to 1,423,147 common shares.
In anticipation of this offering, our board of directors has approved the adoption of a new 2006 Stock Incentive Plan, which was subsequently approved by our shareholders. An amendment and restatement of the 2006 Stock Incentive Plan was subsequently approved by our board of directors and our shareholders. The 2006 Stock Incentive Plan will cease to be effective if we do not complete this offering by December 31, 2007.
Options which are outstanding under our existing Stock Option Plan will continue to be exercisable and will continue to be governed by and be subject to the terms of our existing Stock Option Plan and the stock option agreements evidencing their existence.
The board of directors has determined that no options or awards will be granted under the 2006 Stock Incentive Plan prior to the completion of this offering. The board has also determined that if this offering is completed, no further options will be granted under our existing Stock Option Plan.
The total number of common shares issuable under our existing Stock Option Plan and the 2006 Stock Incentive Plan together is the greater of 1,905,557 common shares and 10 percent of our issued and outstanding common shares from time to time.
2006 Stock Incentive Plan
Purpose. The purpose of the 2006 Stock Incentive Plan is to promote the interests of our company and our shareholders by aiding us in attracting and retaining employees, officers, consultants, independent contractors and directors capable of assuring our future success, to offer such persons incentives to put forth maximum efforts for the success of our business and to afford such persons an opportunity to acquire a proprietary interest in us.
To provide us with flexibility, the 2006 Stock Incentive Plan allows for the issuance of various forms of securities to eligible persons including, without limitation, stock options, stock appreciation rights, restricted stock and restricted stock units. Any such issuance of securities under the 2006 Stock Incentive Plan is referred to as an "Award".
Persons Eligible. Any employee, officer, consultant, independent contractor or director of, or providing services to us or any parent, affiliate, or subsidiary of us will be eligible to participate in the 2006 Stock Incentive Plan.
Administration. The 2006 Stock Incentive Plan is administered by our compensation committee, or such other committee as the board may assign administrative responsibility. The board of directors may, without any further action of the administering committee, or Committee, exercise the powers and duties of the Committee under the plan. The Committee will have the power to: (i) designate plan participants; (ii) determine the type or types of Awards to be granted to participants under the 2006 Stock Incentive Plan; (iii) determine the number of shares to be covered by each Award; (iv) determine the terms and conditions of any Award or Award agreement; (v) amend the terms and conditions of any Award or Award agreement and accelerate
109
the exercisability of any option or waive any restrictions relating to any Award; (vi) determine whether and to what extent, and under what circumstances, Awards may be exercised in cash, shares of us, promissory notes (which do not conflict with the provisions of applicable legislation), other securities, other Awards, or other property, or may be canceled, forfeited, or suspended; (vii) interpret and administer the 2006 Stock Incentive Plan and any instrument or agreement, including Award agreements, relating to the plan; (viii) establish, amend, suspend, or waive such rules and regulations and appoint such agents as it deems appropriate to administer the 2006 Stock Incentive Plan; and (ix) make any other determination or take any other action that the Committee deems necessary or desirable to the administration of the 2006 Stock Incentive Plan.
Shares Available under the 2006 Stock Incentive Plan. Subject to adjustment as provided in the 2006 Stock Incentive Plan, the aggregate number of common shares that may be issued under the 2006 Stock Incentive Plan, including shares issuable under the existing Stock Option Plan, shall be the greater of 1,905,557 common shares and 10 percent of our issued and outstanding common shares from time to time.
Limitations on Awards to Insiders. Awards in any form to our insiders, as defined in Part VI of the Company Manual of the Toronto Stock Exchange (generally, directors, officers, and 10 percent shareholders), will be limited under the 2006 Stock Incentive Plan. No Award shall be granted under the 2006 Stock Incentive Plan which may result in the aggregate number of our common shares:
- •
- issued to our insiders within any one year period, and
- •
- issuable to our insiders at any time,
under the 2006 Stock Incentive Plan, or when combined with all of our other security based compensation arrangements, exceeding 10 percent of our common shares issued and outstanding at the relevant time.
Options. The Plan will allow for the issuance of both "incentive stock options" (as defined in section 422 of the United States Internal Revenue Code of 1986, as amended) and options which do not qualify as "incentive stock options".
The exercise price of options granted under the 2006 Stock Incentive Plan will be determined by the Committee and will be in accordance with all applicable regulatory requirements. If our shares are listed on the Toronto Stock Exchange, the exercise price of options granted under the 2006 Stock Incentive Plan will not be less than the volume weighted average trading price, or VWAP, of one share for the five trading days immediately preceding the grant of the option.
The term of each option shall be fixed by the Committee at the time of the grant, but may not exceed ten (10) years. The Committee also determines the vesting schedule of options, the method in which payment of the exercise price may be made and may also provide that if an option would otherwise expire during or immediately after a self-imposed black-out period it will be automatically extended until 10 business days following the expiration of the black-out period, provided that no option term shall be extended beyond 10 years from the date of grant.
Incentive stock options granted under the 2006 Stock Incentive Plan that are exercisable for the first time by any participant during any calendar year may not exceed $100,000 in aggregate fair market value. All incentive stock options must be granted within ten (10) years from the date on which the 2006 Stock Incentive Plan was adopted by the board of directors. Incentive stock
110
options granted under the 2006 Stock Incentive Plan must terminate no later than ten (10) years after the date of grant, except incentive stock options granted to 10 percent shareholders must terminate five (5) years after the date of grant. Incentive stock options must have an exercise price of at least 100 percent of the VWAP, but incentive stock options granted to 10 percent shareholders will have an exercise price of at least 110 percent of the VWAP.
Stock Appreciation Rights, or SARs. The Committee will be authorized to grant SARs to eligible persons subject to the terms of the 2006 Stock Incentive Plan. Each SAR granted under the 2006 Stock Incentive Plan shall confer on the holder upon exercise the right to receive, as determined by the Committee, cash or a number of common shares equal to the excess of (a) the VWAP of one common share on the date of exercise (or, if the Committee shall so determine, at any time during a specified period before or after the date of exercise) over (b) the grant price of the SAR as determined by the Committee, which grant price shall not be less than 100 percent of the VWAP of one share on the date of grant of the SAR.
Restricted Stock and Restricted Stock Units. The Committee is authorized to grant Awards consisting of common shares, or Restricted Stock, or consisting of the right to receive common shares in the future, or Restricted Stock Units, to eligible persons under the 2006 Stock Incentive Plan. Shares of Restricted Stock shall be subject to such restrictions as the Committee may impose (including, without limitation, a restriction on or prohibition against the right to receive any dividend or other right or property with respect thereto), which restrictions may lapse separately or in combination at such time or times, in such installments or otherwise as the Committee may deem appropriate. Restricted Stock Units shall be subject to a Restricted Stock Unit award agreement containing such terms and conditions, not inconsistent with the provisions of the 2006 Stock Incentive Plan, as the Committee shall determine. If a Restricted Stock or Restricted Stock Unit holder terminates his or her employment with us during the applicable restricted period, all Restricted Stock and Restricted Stock Units subject to restriction shall be subject to forfeiture and reacquisition by us at our discretion.
Performance Awards. The Committee is authorized to grant Performance Awards to eligible persons subject to the terms of the 2006 Stock Incentive Plan. A Performance Award granted under the 2006 Stock Incentive Plan (i) may be denominated or payable in cash, shares (including, without limitation, Restricted Stock and Restricted Stock Units), other securities, other Awards or other property, and (ii) shall confer on the holder thereof the right to receive payments, in whole or in part, upon the achievement of such performance goals during such performance periods as the Committee shall establish. Subject to the terms of the 2006 Stock Incentive Plan, the performance goals to be achieved during any performance period, the length of any performance period, the amount of any Performance Award granted, the amount of any payment or transfer to be made pursuant to any Performance Award and any other terms and conditions of any Performance Award shall be determined by the Committee.
Other Stock Grants. The Committee is authorized, subject to the terms of the 2006 Stock Incentive Plan, to grant to eligible persons shares without restrictions thereon as are deemed by the Committee to be consistent with the purpose of the 2006 Stock Incentive Plan.
Other Stock-Based Awards. The Committee is authorized to grant to eligible persons, subject to the terms of the 2006 Stock Incentive Plan, such other Awards that are denominated or payable in, valued in whole or in part by reference to, or otherwise based on or related to, shares
111
(including, without limitation, securities convertible into shares), as are deemed by the Committee to be consistent with the purpose of the 2006 Stock Incentive Plan.
Term of Awards. Subject to the terms of the 2006 Stock Incentive Plan, the term of each Award shall be for such period as may be determined by the Committee. Whether options or Restricted Stock Units terminate on termination of employment is determined by the Committee.
Additional Conditions in Connection with Awards Granted to Participants Employed in Canada. Certain additional terms, conditions and restrictions apply to Awards granted to participants employed in Canada, as specified in the plan.
Amendment of the 2006 Stock Incentive Plan. Subject to any required regulatory approvals, the board of directors may amend the terms of the 2006 Stock Incentive Plan or any outstanding Award, or discontinue the 2006 Stock Incentive Plan at any time; provided, however, that the board must obtain the approval of our shareholders to make any of the following amendments:
- •
- the increase of: (i) limitations on grants of options to insiders; (ii) the term of any Award held by an insider; or (iii) the number of shares that may be reserved for issuance to insiders;
- •
- the increase of the maximum number of shares reserved for issuance upon exercise of Awards granted under the 2006 Stock Incentive Plan; or
- •
- the increase of the maximum number of shares reserved for issuance of options available under the existing Stock Option Plan; and
provided, further, that no amendment to the 2006 Stock Incentive Plan adversely affecting the rights of the holder of an outstanding Award may be made without such holder's consent, unless such holder is also provided with additional similar rights or other compensation of equal or greater value.
Existing Stock Option Plan
Expiration of Stock Options. An option will expire on the date determined by the board of directors and specified in the option agreement pursuant to which such option is granted, which date shall not be later than the seventh anniversary of the date of grant, or such earlier date as may be required by applicable, law, rules or regulations, including those of any exchange or market on which the common shares are listed or traded. If an optionee's status as a non-employee director or non-employee officer terminates for any reasons other than death, disability or termination for cause, the option will expire on the expiry date of the option. If an optionee's status as an employee director or an employee officer terminates for any reason other than death, disability or termination for cause, the option will expire on the earlier of the expiry date of the option or one year from the optionee ceasing to be an employee. If the optionee's status as an employee terminates for any reason other than death, disability or termination for cause, the option will expire on the earlier of the expiry date of the option or 60 days from the optionee ceasing to be an employee. If the optionee's status as a director, officer, employee or consultant is terminated for cause, the option shall terminate immediately. In the event that the optionee ceases to be a director, officer, employee or consultant due to death or disability, the option will expire on the earlier of the expiry date of the option or six months after the optionee ceases to be a director, officer, employee or consultant.
112
Take-Over Bid. In the event of a bona fide offer by a third party to either (i) the optionee, (ii) a class of shareholders that includes the optionee or (iii) all of our shareholders and such offer would result in a change of control if accepted, then all option shares shall become vested and the option may be exercised in order for the optionee to tender the option shares to the offeror. The option will be reinstated, and the exercise price refunded if the offer is not completed within a specified time or if the offeror does not take up and pay for all of the option shares, in which case only the options underlying the unpurchased shares will be reinstated.
Change in Control. If a change of control occurs, 50 percent (or such larger percentage as may be determined by the board of directors) of all option shares which have not yet vested will become vested, whereupon such option may be exercised in whole or in part by the optionee.
Transferability. Options granted under our existing Stock Option Plan are not transferable or assignable except where such transfer or assignment does not involve a change of beneficial ownership of the options and is not otherwise prohibited by applicable laws.
California Residents. The Stock Option Plan contains additional provisions for options that are granted to a resident of California, which shall remain in place as long as they are required by California law. Under these provisions, such options are subject to a minimum exercise price, a minimum vesting schedule, additional transfer restrictions, the right to receive financial statements, and a shareholder approval requirement for the plan.
Amendments or Termination. Our existing Stock Option Plan will terminate on September 16, 2013. Our board of directors has the right at any time to suspend, amend or terminate the plan subject to certain exceptions.
401(k) Plan
Our wholly-owned subsidiary, OncoGenex, Inc., maintains a 401(k) Plan that is a defined contribution plan intended to qualify under Section 401(a) of the Internal Revenue Code of 1986, as amended. United States employees who are 21 years of age or older, have been employed by OncoGenex, Inc. and have met the applicable service requirements are eligible to participate. Non-resident aliens and employees covered by collectively bargained agreements are not eligible to participate. Our 401(k) Plan is a discretionary contribution plan, whereby participants may voluntarily make pre-tax contributions to the 401(k) plan of up to a maximum statutory limit, which for most employees was $15,000 in 2006. Under the 401(k) Plan, each employee is fully vested in his or her deferred salary contributions. Employee contributions are held and invested by the 401(k) Plan's trustee. Each participant's contributions, and the corresponding investment earnings, are generally not taxable until withdrawn. Individual participants may direct the trustee to invest their accounts in authorized investment alternatives.
Limitations of Liability and Indemnification
Under the CBCA, the corporate statute governing us, we may indemnify an individual who:
- •
- is or was our director or officer; or
- •
- at our request, is or was a director or officer of, or acted in a similar capacity for, another entity,
113
against all costs, charges and expenses, including an amount paid to settle an action or satisfy a judgment reasonably incurred by the individual in respect of any civil, criminal, administrative, investigative or other proceeding in which that individual is involved because of that association with us or other entity.
However, we are prohibited from indemnifying an individual under the CBCA unless:
- •
- such individual acted honestly and in good faith with a view to our best interests (or to the best interests of the other entity for which the individual acted as a director or officer or in a similar capacity at our request, as the case may be); and
- •
- in the case of a criminal or administrative action or proceeding that is enforced by a monetary penalty, such individual had reasonable grounds for believing that such individual's conduct was lawful.
The CBCA allows us to advance moneys to the individual for the costs, charges and expenses of any civil, criminal, administrative, investigative or other proceeding, but the individual must repay the moneys if the individual does not fulfill the conditions listed above.
We may not indemnify or pay the expenses of the individual in respect of an action brought against the individual by or on behalf of us unless such indemnity or payment has been approved by the appropriate court.
We or the individual or other entity may apply to the appropriate court for an order approving an indemnity under section 124 of the CBCA and the court may so order or make any further order that it sees fit. An applicant in such court application must give the Director under the CBCA notice of the application and the Director is entitled to appear and be heard in person or by counsel.
The CBCA provides that we may purchase and maintain insurance for the benefit of the individual against any liability incurred by the individual in their capacity as one of our directors or officers or as a director or officer, or someone a similar capacity, of another entity, if they act or acted in that capacity at our request. Our bylaws provide that, subject to the limitations of the CBCA, we may purchase and maintain such insurance for the benefit of our directors and officers as our board of directors may determine.
Our by-laws provide that, subject to the CBCA, we may indemnify our directors and officers, our former directors or officers or another individual who acts or acted at our request as a director or officer, or an individual acting in a similar capacity, of another entity, against all costs, charges and expenses, including an amount paid to settle an action or satisfy a judgment, reasonably incurred by the individual in respect of any civil, criminal, administrative, investigative or other proceeding in which the individual is involved because of that association with us or other entity if:
- •
- the individual acted honestly and in good faith with a view to our best interest, or as the case may be, to the best interests of the other entity for which the individual acted as director or officer or in a similar capacity at our request; and
- •
- in the case of a criminal or administrative action or proceeding that is enforced by a monetary penalty, the individual had reasonable grounds for believing that the individual's conduct was lawful.
114
We maintain liability insurance which insures our directors and officers against certain losses and which insures us against our obligations to indemnify our directors and officers.
We have entered into indemnity agreements with Scott Cormack, Stephen Anderson, Cindy Jacobs, Patricia Stewart and Monica Krieger which provide, among other things, that we will indemnify him or her against certain expenses incurred in respect of a proceeding in which such person is or may be joined as a party by reason of his or her corporate status. Such persons will be indemnified against all expenses, judgments, penalties, fines and amounts paid in settlement, provided that we will not indemnify such person if he or she did not act honestly and in good faith with a view to our best interests and, in the case of a criminal proceeding, the person did not have reasonable grounds for believing that his or her conduct in respect of which the proceeding was brought was lawful.
In the event a claim is brought against an indemnified person by us or in our right, if applicable law so provides, we will not indemnify such person if he or she is finally adjudged to be liable to us, unless and to the extent that a court of competent jurisdiction determines that such indemnification will be made, and in such event we will indemnify such person in respect of certain legal expenses, which will not include any amounts to be paid in respect of any judgments, penalties, fines or amounts paid in settlement.
Notwithstanding the foregoing, in the event that the indemnified person is wholly or partially successful in a proceeding, we will indemnify such person in respect of certain legal expenses actually and reasonably incurred by him or her up to the maximum extent permitted by law for such expenses incurred in connection with each successfully resolved claim, issue or matter. Additionally, we will indemnify such persons against certain expenses, judgments, penalties, fines and amounts paid in settlement actually and reasonably incurred including, without limitation, in respect of liability arising out of the negligence or active or passive wrongdoing of such indemnified person, provided that such payment may be limited as prescribed by law.
We have entered into additional indemnity agreements with each of our directors, including Scott Cormack, which provide, among other things, that we will indemnify him against any and all charges and claims brought or made by any person or organization, and against expenses, penalties or fines incurred by such person in connection with the execution of his duties as a director, irrespective of whether they are incurred by reason of his negligence or breach of duty.
Notwithstanding the foregoing, we will not indemnify such person if, among other things, a court of competent jurisdiction finds that such person did not act honestly and in good faith with a view to our best interests, in the case of a criminal proceeding enforced by a monetary penalty, the person did not have reasonable grounds for believing that his or her conduct in respect of which the proceeding was brought was lawful, in the case of any act, error or omission of such person, such person acted fraudulently or maliciously, or for any liabilities for which such person is entitled to indemnity pursuant to any valid or collectible policy of insurance obtained and maintained by us, to the extent of the amounts actually collected by such indemnified person under such policy.
At present, we are not aware of any pending or threatened litigation or proceeding involving any of our directors, officers, employees or agents in which indemnification would be required or permitted.
115
The following table sets forth information regarding beneficial ownership of our common shares as of February 28, 2007 by:
- •
- each person known by us to be the beneficial owner of more than 5 percent of our common shares;
- •
- our executive officers;
- •
- our directors; and
- •
- all of our executive officers and directors as a group.
Beneficial ownership is determined in accordance with the rules of the SEC and generally includes voting or investment power with respect to securities. Unless otherwise indicated below, to our knowledge, the persons and entities named in the table have sole voting and sole investment power with respect to all shares beneficially owned, subject to community property laws where applicable.
As of February 28, 2007, there are two U.S. record holders holding 4.79 percent of the Class A preferred shares and three U.S. record holders holding 8.59 percent of the Class B preferred shares.
The table below lists the applicable percentage ownership based on 11,079,738 shares outstanding as of February 28, 2007, after giving effect to the Reorganization. The number of common shares to be outstanding after the closing of this offering is based on the foregoing, plus 5,000,000 common shares to be sold in this offering. Unless otherwise indicated, the address of each beneficial owner is c/o OncoGenex Technologies Inc., 400 - 1001 West Broadway, Vancouver, British Columbia, Canada, V6H 4B1.
116
| | Beneficial Ownership Prior to the Offering | | | | | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | | Options Exercisable Within 60 Days of February 28, 2007 | Percentage of Shares Outstanding(1) | Percentage of Shares Outstanding (Fully Diluted Basis)(1) | |||||||||
| Name and Address of Beneficial Owner | Shares Beneficially Owned(2) | Before the Offering | After the Offering(2) | Before the Offering | After the Offering(2) | ||||||||
| 5% Shareholders | |||||||||||||
Ventures West 7 Limited Partnership(3) 2500 - West Hastings Street Vancouver, BC, V6E 3X1 | 2,816,681 | — | 25.42 | % | 17.52 | % | 22.52 | % | 16.09 | % | |||
H.I.G. Horizon Corp.(4) Worthing Corporate Centre Worthing Main Road Christ Church Barbados | 2,230,365 | — | 20.13 | 13.87 | 17.84 | 12,74 | |||||||
Working Opportunity Fund (EVCC) Ltd.(5) 2600 - 1055 West Georgia Street Vancouver, BC, V6E 3R5 | 1,546,212 | — | 13.96 | 9.62 | 12.36 | 8.83 | |||||||
BDC Capital Inc.(6) 444 7th Ave SW, Suite 110 Calgary, Alberta, T2P 0X8 | 1,357,934 | — | 12.26 | 8.45 | 10.86 | 7.76 | |||||||
Milestone Medica Corporation(7) 101 College Street - Suite 230 Toronto, ON, M5G 1L7 | 779,734 | — | 7.04 | 4.85 | 6.24 | 4.45 | |||||||
Executive Officers and Directors | |||||||||||||
Scott Cormack(8) | 10,664 | 581,667 | 5.08 | 3.56 | 4.74 | 3.38 | |||||||
| Martin Gleave(9) | 738,000 | 189,204 | 8.23 | 5.70 | 7.41 | 5.30 | |||||||
| Cindy Jacobs | — | 62,500 | * | * | |||||||||
| Stephen Anderson | — | 50,000 | * | * | |||||||||
| James Shepard | 9,328 | 67,543 | * | * | |||||||||
| Aaron Davidson(10) | — | — | — | — | |||||||||
| Neil Clendeninn | — | 34,734 | * | * | |||||||||
| Christopher Laird(11) | — | — | — | — | |||||||||
| K. Thomas Bailey | — | 6,000 | * | * | |||||||||
| All executive officers and directors as a group (9 persons)(12) | 757,992 | 991,647 | 14.49 | 10.25 | 13.99 | 10.00 | |||||||
- *
- Represents beneficial ownership of less than one percent (1 percent) of our outstanding common shares.
- (1)
- Percentage of shares outstanding assumes, for each person or group, that the options exercisable within 60 days of February 28, 2007 and held by such person or group have been exercised, and that no other person has exercised any outstanding options.
- (2)
- Upon the completion of this offering and giving effect to the Reorganization, our existing shareholders will own 11,079,738 shares (excluding any shares they may purchase in the offering), representing 68.90 percent of our outstanding common shares on a non-diluted basis and 63.30 percent of our outstanding common shares on a fully-diluted basis.
117
- (3)
- Includes 2,569,977 shares owned by Ventures West 7 Limited Partnership and 246,704 shares owned by Ventures West 7 U.S. Limited Partnership. Investment decisions on behalf of Ventures West 7 Limited Partnership are made by its general partner, Ventures West 7 Management Ltd. Investment decisions on behalf of Ventures West 7 U.S. Limited Partnership are made by its manager, Ventures West 7 Management (International) Inc. The officers of each of Ventures West 7 Management Ltd. and Ventures West 7 Management (International) Inc. are Robin Louis, Sam Znaimer, David Berkowitz, Barry Gekiere, Edward Anderson and Howard Riback. Each such individual disclaims beneficial ownership of the common shares held by Ventures West 7 Limited Partnership and Ventures West 7 U.S. Limited Partnership. Christopher Laird, one of our directors, is an employee of Ventures West Management Inc., which is the manager of Ventures West 7 Limited Partnership. Ventures West Capital Ltd. is the parent company of each of Ventures West 7 Management Ltd., Ventures West 7 Management (International) Inc. and Ventures West Management Inc.
- (4)
- Investment decisions on behalf of H.I.G. Horizon Corp. are made by its board of directors, which consists of Tony Tamer, Michael Weetch, and David Correa. Each such individual disclaims beneficial ownership of the common shares held by H.I.G. Horizon Corp.
- (5)
- Working Opportunity Fund (EVCC) Ltd. is managed by GrowthWorks Capital Ltd., which is a wholly-owned subsidiary of GrowthWorks Ltd. As at October 31, 2006, Working Enterprise Ltd. owned an approximate 40% interest in GrowthWorks Ltd. Mr. David Levi is the President and Chief Executive Officer of each of the foregoing entities, and as at October 31, 2006, Mr. Levi owned an approximate 21.6% interest in GrowthWorks Ltd. Mr. Levi disclaims beneficial ownership of the common shares held by Working Opportunity Fund (EVCC) Ltd., except to the extent of his pecuniary interest in GrowthWorks Ltd. Working Enterprise Ltd. is owned by the British Columbia Federation of Unions and six British Columbia labour unions.
- (6)
- Based on information available to us, we believe that BDC Capital Inc. is a wholly-owned subsidiary of the Business Development Bank of Canada, a Crown corporation.
- (7)
- Based on information available to us, we believe that: (i) RBC Technologies Ventures Inc., a wholly-owned subsidiary of Royal Bank of Canada, the indirect parent company of RBC Dominion Securities Inc. and RBC Capital Markets Corporation, holds a 52.9% interest in Milestone Medica Corporation; and (ii) the other 47.1% interest in Milestone Medica Corporation is held by Research Technologies Corporation, a company that has no affiliation with Royal Bank of Canada.
- (8)
- Includes 10,664 shares held by Trycor Investment Trust, a trust of which a majority of the beneficiaries are Mr. Cormack, his spouse, and his and his spouse's family members, 106,000 shares issuable pursuant to stock options that will vest automatically upon the closing of this offering and 98,000 shares issuable pursuant to stock options held by Sherry Tryssenaar, Mr. Cormack's spouse, within 60 days of February 28, 2007.
- (9)
- Includes 338,000 shares owned by Martin Gleave and 400,000 shares owned by 603356 B.C. Ltd., a company controlled by Martin Gleave.
- (10)
- Excludes 2,230,365 shares beneficially owned by H.I.G. Horizon Corp. H.I.G. Horizon Corp. is an affiliate of H.I.G. Ventures. Aaron Davidson is an employee of H.I.G. Ventures. Mr. Davidson disclaims control over or ownership of shares held by H.I.G. Horizon Corp.
- (11)
- Excludes 2,816,681 shares held by Ventures West 7 Limited Partnership and Ventures West 7 U.S. Limited Partnership. Christopher Laird disclaims control over or beneficial ownership of shares held by Ventures West 7 Limited Partnership and Ventures West 7 U.S. Limited Partnership. See footnote 3 above.
- (12)
- Includes shares held by persons or entities affiliated with Scott Cormack and Martin Gleave. See footnotes 8 and 9.
118
Shares and Options Held by Directors and Officers
| Name of Executive Officer and Director | Shares Beneficially Owned | Number of Securities Underlying Options Granted | Exercise Price in C$ | Expiration Date | Total of Shares and Options Beneficially Owned(1) | |||||
|---|---|---|---|---|---|---|---|---|---|---|
| Scott Cormack(2) | 10,664 | 114,000 | 0.90 | Jan.1, 2009 | 627,664 | |||||
| 4,000 | 0.90 | Aug. 1, 2009 | ||||||||
| 32,500 | 0.90 | Feb. 1, 2010 | ||||||||
| 124,000 | 0.90 | Dec. 16, 2010 | ||||||||
| 130,500 | 0.90 | Jan. 14, 2011 | ||||||||
| 212,000 | 0.95 | Aug. 8, 2012 | ||||||||
Martin Gleave | 738,000 | 20,000 | 0.90 | March 1, 2009 | 997,870 | |||||
| 133,870 | 0.90 | Sept. 24, 2010 | ||||||||
| 106,000 | 0.95 | Aug. 8, 2012 | ||||||||
Cindy Jacobs | — | 125,000 | 0.95 | Sept. 12, 2012 | 125,000 | |||||
Stephen Anderson | — | 100,000 | 0.95 | March 23, 2013 | 100,000 | |||||
James Shepard | 9,328 | 3,200 | 0.90 | March 25, 2009 | 76,871 | |||||
| 1,700 | 0.90 | April 14, 2010 | ||||||||
| 10,000 | 0.90 | Dec. 16, 2010 | ||||||||
| 12,000 | 0.90 | Jan. 14, 2011 | ||||||||
| 4,375 | 0.90 | Dec. 20, 2011 | ||||||||
| 33,500 | 0.95 | Aug. 8, 2012 | ||||||||
| 2,768 | 0.95 | March 23, 2013 | ||||||||
Aaron Davidson(3) | — | 0 | — | — | — | |||||
Neil Clendeninn | — | 11,000 | 0.90 | Sept. 16, 2011 | 34,734 | |||||
| 4,375 | 0.90 | Dec. 20, 2011 | ||||||||
| 17,000 | 0.95 | Aug. 8, 2012 | ||||||||
| 2,359 | 0.95 | March 23, 2013 | ||||||||
Christopher Laird(4) | — | 0 | — | — | — | |||||
K. Thomas Bailey | — | 10,500 | 0.95 | March 23, 2013 | 10,500 |
- (1)
- Includes unvested stock options.
- (2)
- Includes 10,664 shares held by Trycor Investment Trust, a trust of which a majority of beneficiaries are Mr. Cormack and his and his spouse's family members, and 98,000 shares issuable pursuant to stock options held by Sherry Tryssenaar, Mr. Cormack's spouse.
- (3)
- Excludes 2,230,365 shares beneficially owned by H.I.G. Horizon Corp. H.I.G. Horizon Corp. is an affiliate of H.I.G. Venture. Aaron Davidson is an employee of H.I.G. Ventures. Mr. Davidson disclaims control over or ownership of shares held by H.I.G. Horizon Corp.
- (4)
- Excludes 2,816,681 shares held by Ventures West 7 Limited Partnership. Christopher Laird disclaims control over or beneficial ownership of shares held by Ventures West 7 Limited Partnership and Ventures West 7 U.S. Limited Partnership.
119
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
We have entered into the following transactions with our executive officers, directors and certain other related parties since January 1, 2003.
Share Sales
On September 24, 2003, we sold 1,324,738 Series 1 Class B preferred shares to Ventures West 7 Limited Partnership at a price of $2.755 per share for gross proceeds of $3,649,653; and 127,168 Series 1 Class B preferred shares to Ventures West 7 U.S. Limited Partnership at a price of $2.755 per share for gross proceeds of $350,348. Upon completion of the Reorganization, these shares will convert into a total of 1,451,906 common shares. Mr. Laird, one of our directors, is an employee of Ventures West Management Inc., the managing partner of Ventures West 7 Limited Partnership and Ventures West 7 U.S. Limited Partnership.
On September 24, 2003, we sold 1,088,930 Series 1 Class B preferred shares to H.I.G. Horizon Corp. at a price of $2.755 per share for gross proceeds of $3,000,002. Upon completion of the Reorganization, these shares will convert into a total of 1,088,930 common shares. Mr. Davidson, one of our directors, is an employee of H.I.G. Ventures. H.I.G. Horizon Corp. is an affiliate of H.I.G. Ventures.
On September 24, 2003, we sold 725,954 Series 1 Class B preferred shares to Working Opportunity Fund (EVCC) Ltd. at a price of $2.755 per share for gross proceeds of $2,000,003. Upon completion of the Reorganization, these shares will convert into a total of 725,954 common shares.
On September 24, 2003, we sold 635,208 Series 1 Class B preferred shares to Business Development Bank of Canada at a price of $2.755 per share for gross proceeds of $1,749,998. These shares were subsequently transferred to BDC Capital Inc. Upon completion of the Reorganization, these shares will convert into a total of 635,208 common shares.
On September 24, 2003, we sold 261,342 Series 1 Class B preferred shares to Milestone Medica Corporation at a price of $2.755 per share for gross proceeds of $719,997. Upon completion of the Reorganization, these shares will convert into a total of 261,342 common shares.
On August 10, 2005, we sold 1,245,239 Series 2 Class B preferred shares to Ventures West 7 Limited Partnership at a price of $3.07 per share for gross proceeds of $3,822,884 and 119,536 Series 2 Class B preferred shares to Ventures West 7 U.S. Limited Partnership at a price of $3.07 per share for gross proceeds of $366,976. Upon completion of the Reorganization, these shares will convert into a total of 1,364,775 common shares. Mr. Laird, one of our directors, is an employee of Ventures West Management Inc., the managing partner of Ventures West 7 Limited Partnership and Ventures West 7 U.S. Limited Partnership.
On August 10, 2005, we sold 1,141,435 Series 2 Class B preferred shares to H.I.G. Horizon Corp. at a price of $3.07 per share for gross proceeds of $3,504,205. Upon completion of the Reorganization, these shares will convert into a total of 1,141,435 common shares. Mr. Davidson, one of our directors, is an employee of H.I.G. Ventures. H.I.G. Horizon Corp. is an affiliate of H.I.G. Ventures.
On August 10, 2005, we sold 820,258 Series 2 Class B preferred shares to Working Opportunity Fund (EVCC) Ltd. at a price of $3.07 per share for gross proceeds of $2,518,192.
120
Upon completion of the Reorganization, these shares will convert into a total of 820,258 common shares.
On August 10, 2005, we sold 407,166 Series 2 Class B preferred shares to Business Development Bank of Canada at a price of $3.07 per share for gross proceeds of $1,250,000. These shares were subsequently transferred to BDC Capital Inc. Upon completion of the Reorganization, these shares will convert into a total of 407,166 common shares.
Shareholders' Agreement
On August 10, 2005, we entered into a further amended and restated shareholders' agreement with Ventures West 7 Limited Partnership, Ventures West 7 U.S. Limited Partnership, H.I.G. Horizon Corp., Working Opportunity Fund (EVCC) Ltd., Business Development Bank of Canada, Milestone Medica Corporation, Martin Gleave, 603356 B.C. Ltd., Scott Cormack and WHI Morula Fund, LLC to establish certain rights and obligations in respect of the conduct of the affairs of the Company and among other things, the holding and sale of the parties respective securities. The shareholders' agreement was further amended on September 7, 2006. The shareholders' agreement, as amended, will automatically terminate upon the completion of this offering, except that certain of the shareholders will continue to hold registration rights as further described under "Description of Capital Stock".
EIA Agreement
On September 24, 2003, we entered into an EIA Agreement with Working Opportunity Fund (EVCC) Ltd., or WOF. Under the terms of the EIA Agreement, we agreed to provide WOF, upon request, with a certificate as to the continuing eligibility under the Employee Investment Act (British Columbia) of WOF's investments in our company. We also agreed to use all funds that WOF invests in us in accordance with such Act. Any funds received from WOF in this offering will be subject to the terms of the EIA Agreement. We believe that compliance with the terms of the EIA Agreement will have no material effect on our use of proceeds from this offering.
Employment and Consulting Agreements
We have entered into employment or consulting agreements with our executive officers and certain of our directors. For more information regarding these agreements, see "Management — Executive Compensation — Employment and Consulting Agreements" and "Compensation of Directors".
Indemnification of Directors and Officers
Our corporate statute and by-laws contain provisions limiting the liability of directors and officers. In addition, we have entered into indemnification agreements with each of our directors and officers. For further information, see "Management — Limitations of Liability and Indemnification".
121
The following is a summary of the rights of our common shares and preferred shares as set forth in our articles and certain sections of the CBCA.
Our authorized capital stock consists of an unlimited number of common shares, each without par value, and an unlimited number of Class A, B and C preferred shares issuable in series, of which 760,000 are designated Series 1 Class A preferred shares, 420,000 are designated Series 2 Class A preferred shares, 4,543,553 Series 1 Class B preferred shares and 5,058,084 Series 2 Class B preferred shares, each without par value.
As of February 28, 2007, we have issued and outstanding 1,285,500 common shares, 513,394 Series 1 Class A preferred shares, 335,411 Series 2 Class A preferred shares, 4,543,553 Series 1 Class B preferred shares, 4,401,895 Series 2 Class B preferred shares, without giving effect to the Reorganization or this offering. As of February 28, 2007 we also had outstanding options to purchase 1,423,147 common shares, without giving effect to this Reorganization.
On September 23, 2003, we effected a one-for-five share consolidation of our common shares and our preferred shares, which we refer to in this prospectus as the Share Consolidation, whereby the holders of our common shares received one common share for every five common shares held immediately prior to the Share Consolidation, and the holders of each series of our preferred shares received one share of the applicable series of preferred shares for every five shares of such series of preferred shares held immediately prior to the Share Consolidation. All share numbers in this prospectus are presented on a post Share Consolidation basis.
Upon completion of the Reorganization and this offering, all of the 9,794,253 outstanding series A and B preferred shares will convert into 9,794,238 common shares and the preferred shares so converted will be automatically cancelled and will not be reissuable. The number of common shares resulting from the conversion is 15 shares lower than the outstanding preferred shares as fractions of preferred shares will be eliminated upon completion of the Reorganization. As a result, upon completion of the Reorganization and this offering, based upon shares outstanding as of February 28, 2007, we will have issued and outstanding 16,079,738 common shares and no preferred shares. Upon completion of the Reorganization and this offering our authorized capital will consist of an unlimited number of common shares.
Share Capital — Common Shares
The holders of our common shares are entitled to one vote for each share held at any meeting of the shareholders. The holders of our common shares are entitled to receive dividends as and when declared by our board of directors. In the event of our liquidation, dissolution or winding-up or other distribution of our assets among our shareholders, the holders of our common shares are entitled to share pro rata in the distribution of our assets. Upon completion of the Reorganization, our common shares will be our only class of shares.
Key Provisions of our Articles and Bylaws and the CBCA
The following is a summary of certain key provisions of our articles and bylaws and certain related sections of the CBCA. Please note that this is only a summary and is not intended to be exhaustive. For further information please refer to the full version of our articles and bylaws which are included as an exhibit to the registration statement of which this prospectus is a part.
122
Stated Objects or Purposes
Our articles and bylaws do not contain stated objects or purposes and do not place any limitations on the business that we may carry on.
Directors
Power to Vote on Matters in Which a Director is Materially Interested. The CBCA states that a director shall disclose to us the nature and extent of any interest (a disclosable interest) that he or she has in a material contract or material transaction, whether made or proposed, with us, if the director is: (a) is a party to the contract or transaction; (b) is a director or an officer, or an individual acting in a similar capacity, of a party to the contract or transaction; or (c) has a material interest in a party to the contract or transaction. Furthermore, any director that has a disclosable interest is not entitled to vote on any directors' resolution to approve that contract or transaction, unless the contract or transaction relates primarily to his or her remuneration as a director, officer, employee or agent of our company or an affiliate of our company; is for indemnity or insurance under section 124 of the CBCA; or is with an affiliate of our company.
Under the CBCA, in the event that the board of directors enters into a material contract or transaction in which a director has a disclosable interest, the contract or transaction is not invalid, and the director is not accountable to us or our shareholders for any profit realized from the contract or transaction, because of the director's interest in the contract or transaction or because the director was present or was counted to determine whether a quorum existed at the meeting of directors or committee of directors that considered the contract or transaction, if disclosure of the interest was made in accordance with the provisions of the CBCA; the directors approved the contract or transaction; and the contract or transaction was reasonable and fair to us when it was approved. Even if such disclosure was not provided, the director with a disclosable interest, if he or she acted honestly and in good faith, is not accountable to us or to our shareholders for any profit realized from a contract or transaction for which disclosure is required under the CBCA, and the contract or transaction is not invalid by reason only of the interest of the director in the contract or transaction, if the contract or transaction is approved or confirmed by special resolution at a meeting of the shareholders; disclosure of the interest was made to the shareholders in a manner sufficient to indicate its nature before the contract or transaction was approved or confirmed; and the contract or transaction was reasonable and fair to us when it was approved or confirmed.
Directors' Power to Determine the Compensation of Directors. Our bylaws provide that our directors shall be paid such remuneration for their services as our board of directors may, from time to time, determine. Our bylaws also provide that our directors shall also be entitled to be reimbursed for traveling and other expenses properly incurred by them in attending meetings of our board of directors or any committee thereof. Nothing in the bylaws precludes any of our directors from serving us in any other capacity and receiving remuneration for such services.
Borrowing Powers Exercisable by the Board of Directors. Our articles and bylaws provide that our board of directors may, borrow money on our credit; issue, reissue, sell or pledge bonds, debentures, notes or other evidence of our indebtedness or guarantee; subject to the CBCA, issue a guarantee on our behalf to secure performance of an obligation of any person; and charge, mortgage, hypothecate, pledge or otherwise create a security interest in all or any of our owned or subsequently acquired property and undertaking, to secure any present or future indebtedness or
123
liability of ours. The board of directors may delegate these borrowing powers to one or more directors or officers of our company.
The borrowing powers exercisable by the board of directors may be limited by amending our bylaws, which would require an ordinary resolution of the board of directors followed by ratification by shareholders at the next scheduled shareholders meeting; by amending our articles, which would require a special resolution of shareholders at a shareholders meeting; or by all our shareholders entering into a unanimous shareholders' agreement containing such limitation.
Retirement or Non-Retirement of Directors Under an Age Limit Requirement. Our articles and bylaws do not impose any mandatory age-related retirement or non-retirement requirement for our directors.
Number of Shares Required to be Owned by a Director. Our bylaws provide that a director is not required to be a shareholder of the company.
Action Necessary to Change the Rights of Holders of Our Shares
Our shareholders can authorize the alteration of our articles to create or vary the special rights or restrictions attached to any of our shares by passing a special resolution. Such a special resolution will not be effective until articles of amendment are filed with Industry Canada.
Upon completion of the Reorganization we will only have one class of shares, being our common shares. Any rights attached to our common shares may not be prejudiced or interfered with unless the holders of our common shares consent to such action by a special resolution.
Shareholder Meetings
We must hold an annual general meeting of our shareholders at such time in each year that is not more than 15 months after the preceding annual general meeting and no later than six months after the end of our preceding financial year and at such place as our board of directors, the chairperson of the board or our president may, from time to time, determine.
A meeting of our shareholders may be held at our registered office or elsewhere in the municipality in which the registered office is situated (currently being Vancouver, British Columbia) or, if the board of directors shall so determine, at some other place in Canada or, if all the shareholders entitled to vote at the meeting so agree, at some place outside Canada.
Under our bylaws, our board of directors, the chairperson of the board or our president may, at any time, call a special meeting of our shareholders. Under the CBCA, shareholders holding not less than five percent of our issued voting shares may also cause our directors to call a shareholders meeting.
Upon completion of the offering, we will be a "distributing corporation" under the CBCA by virtue of having filed this prospectus with each province under applicable securities legislation. As a distributing corporation, a notice convening a general meeting, specifying the date, time, and location of the meeting, and, where a meeting is to consider special business, the general nature of the special business, must be given to shareholders not less than 21, and not more than 60, days prior to the meeting. Shareholders entitled to notice of a meeting may waive or reduce the period of notice for such meeting and shareholder attendance at a meeting is deemed a waiver of such notice unless the shareholder attends a meeting for the express purpose of objecting to the transaction of any business on the grounds that the meeting is not lawfully called.
124
A quorum for general meetings is two persons present and being, or representing by proxy, shareholders holding in the aggregate not less than five percent of the outstanding shares entitled to be voted at the meeting. If within half an hour from the time set for a general meeting, a quorum is not present, the shareholders present or represented by proxy may adjourn the meeting to a fixed time and place but may not transact any other business, provided however that if no provision for adjournment is made at any such meeting or adjourned meeting at which a quorum is not present, the meeting shall be dissolved.
Holders of our common shares are entitled to attend shareholder meetings. Our directors and our auditors are also entitled to be present at such meetings, but are not entitled to vote, except to the extent that they are shareholders. In addition, others who, although not entitled to vote, are entitled or required under any provision of the CBCA, our articles or our bylaws, to be present at such meetings, may attend, but not vote at, such meetings.
Limitations on the Right to Own Securities
Neither our articles nor our bylaws provide for any limitations on the rights to own our securities. See also "Description of Capital Stock — Competition Act, Exchange Controls and Investment Canada Act".
Change of Control
Neither our articles nor our bylaws contain any change of control limitations with respect to a merger, acquisition or corporate restructuring that involves us.
Shareholder Ownership Disclosure
Although U.S. and Canadian securities laws regarding shareholder ownership by certain persons require certain disclosure, neither our articles nor our bylaws provide for any ownership threshold above which shareholder ownership must be disclosed.
Options
As of December 31, 2005, there were outstanding options to purchase 1,332,720 common shares at a weighted average exercise price of C$0.91 per share.
125
As of February 28, 2007, there were outstanding options to purchase 1,423,147 common shares at a weighted average exercise price of C$0.92 per share, as set forth in the following table:
| Category | Common Shares under Options Granted | Exercise Price in C$ | Expiration Date | ||||
|---|---|---|---|---|---|---|---|
| All current and past executive officers | 114,000 | $ | 0.90 | January 1, 2009 | |||
| 4,000 | 0.90 | August 1, 2009 | |||||
| 32,500 | 0.90 | February 1, 2010 | |||||
| 124,000 | 0.90 | December 16, 2010 | |||||
| 130,500 | 0.90 | January 14, 2011 | |||||
| 212,000 | 0.95 | August 8, 2012 | |||||
| 125,000 | 0.95 | September 12, 2012 | |||||
| 100,000 | 0.95 | March 23, 2013 | |||||
All current and past directors | 20,000 | 0.30 | October 30, 2008 | ||||
| 12,300 | 0.90 | March 25, 2009 | |||||
| 6,300 | 0.90 | April 14, 2010 | |||||
| 10,000 | 0.90 | December 16, 2010 | |||||
| 12,000 | 0.90 | January 14, 2011 | |||||
| 11,000 | 0.90 | September 16, 2011 | |||||
| 8,750 | 0.90 | December 20, 2011 | |||||
| 50,500 | 0.95 | August 8, 2012 | |||||
| 15,627 | 0.95 | March 23, 2013 | |||||
All other current and past employees | 500 | 0.90 | August 19, 2009 | ||||
| 25,000 | 0.90 | December 8, 2010 | |||||
| 25,000 | 0.90 | December 15, 2010 | |||||
| 500 | 0.90 | December 16, 2010 | |||||
| 16,000 | 0.90 | May 10, 2011 | |||||
| 500 | 0.90 | April 15, 2011 | |||||
| 500 | 0.90 | April 26, 2011 | |||||
| 10,000 | 0.95 | September 12, 2012 | |||||
| 16,000 | 0.95 | October 3, 2012 | |||||
| 50,000 | 0.95 | October 17, 2012 | |||||
| 10,000 | 0.95 | October 17, 2012 | |||||
| 11,500 | 0.95 | December 14, 2012 | |||||
| 1,000 | 0.95 | March 23, 2013 | |||||
All consultants | 20,000 | 0.90 | March 1, 2009 | ||||
| 133,870 | 0.90 | September 24, 2010 | |||||
| 1,000 | 0.90 | March 10, 2011 | |||||
| 2,000 | 0.90 | February 28, 2012 | |||||
| 1,000 | 0.90 | April 4, 2012 | |||||
| 1,000 | 0.90 | June 15, 2012 | |||||
| 106,000 | 0.95 | August 8, 2012 | |||||
| 2,500 | 0.95 | December 14, 2012 | |||||
| 800 | 0.95 | March 23, 2013 | |||||
| Total | 1,423,147 | ||||||
126
Registration Rights
Upon the expiry of six months from the closing of this offering, any party to our shareholders' agreement that either:
- •
- is an "affiliate" of us within the meaning of SEC Rule 144, and holds more than one percent of our outstanding shares; or
- •
- is a person that holds or has the right to receive securities of us that are "restricted securities" within the meaning of SEC Rule 144, and that cannot be sold under Rule 144 within a single three-month period,
will be entitled to registration rights under the terms of our shareholders' agreement. Subject to limitations specified in the shareholders' agreement, these registration rights include the following:
Demand Registration Rights. After September 15, 2008, if we receive a written request of one or more of the "Major Investors", as defined in the shareholders' agreement, seeking to register securities for sale to the public for gross proceeds of at least $25,000,000, we will use our reasonable best efforts to register such securities for resale under the Securities Act and under such state securities laws as may be reasonably requested by such investors. Notwithstanding the foregoing, if we determine that it would be seriously detrimental to us and our shareholders to file such registration, we may defer such filing for not more than 180 days, not more than once in any 24-month period. We are not obligated to effect more than one registration pursuant to this demand registration right.
Piggyback Registration Rights. If we register any of our securities under the Securities Act for sale to the public (other than on Forms S-8, S-4 or F-4 or their successors, or any other form not available for registering the registrable securities), we must give notice to all parties to the shareholders' agreement with registration rights of our intention to do so, at least 20 days prior to filing the registration statement. Upon the written request of any party to the shareholders' agreement with registration rights, we must use our reasonable best efforts to include in such registration all securities which we have been requested to register. In the case of an underwritten offering, the lead underwriter shall be entitled to reduce the amount of the selling shareholder's shares being registered by any amount it deems appropriate, and require that each selling shareholder accept the terms of the underwriting, assuming usual and customary underwriting terms.
Form S-3 Registration Rights. If we receive a written request of one or more "Major Investors", as defined in the shareholders' agreement, seeking to register shares for sale to the public for gross proceeds of at least $5,000,000 at a time when we are eligible to register such shares under the Securities Act on Forms S-3 or F-3, we must use our reasonable best efforts to register such shares for resale under the Securities Act on one of such forms.
The registration rights granted under the shareholders' agreement cannot be exercised in connection with this offering or within the six-month period immediately following this offering. Thereafter, the registration rights may be exercised by qualifying shareholders, but we will not be required to keep any registration statement effective for a period of more than 180 days. We will bear all registration expenses other than underwriting discounts and commissions. The registration rights will terminate as to any shareholder when such shareholder ceases to hold or have the right to receive any restricted securities (including securities subject to Rule 144 due to the holder's status as our affiliate), or such shareholder is eligible to sell all restricted securities it holds or has
127
the right to receive pursuant to Rule 144 within a three-month period. On the fifth anniversary of this offering, all registration rights under the shareholders' agreement will terminate.
Transfer Agent and Registrar
Upon the closing of this offering, the transfer agent and registrar for the common shares in the United States will be Computershare Investor Services Inc. in Golden, Colorado and in Canada will be Computershare Investor Services Inc. in Vancouver, British Columbia and Toronto, Ontario.
Nasdaq Global Market and Toronto Stock Exchange
The Nasdaq Global Market has approved the listing of our common shares under the symbol "OGXI". In addition, the Toronto Stock Exchange has conditionally approved the listing of our common shares under the symbol "OGX". Listing is subject to us fulfilling all of the requirements of the Toronto Stock Exchange on or before April 4, 2007, including the distribution of our shares to a minimum number of public shareholders.
Statement of Nasdaq Corporate Governance Differences
Rule 4350(a) of the NASD Manual permits a foreign private issuer, such as our company, to follow its home country practice in lieu of certain of the requirements of Rule 4350, provided that the company discloses in its initial registration statement filed with the SEC and in its annual reports filed with the SEC each requirement of Rule 4350 that it does not follow and describes the home country practice that it follows in lieu of such requirements.
Rule 4350(f) requires that the quorum for a meeting of the common shareholders of a Nasdaq-listed company be not less than 331/3% of the company's outstanding common shares. As permitted by the laws of Canada and the rules of the Toronto Stock Exchange (which defer to the laws of our jurisdiction of incorporation in respect of this matter), the quorum for a meeting of our shareholders is two persons present in person or represented by proxy holding in the aggregate not less than 5% of the outstanding shares entitled to vote at the meeting.
Rule 4350(i) specifies certain types of transactions in which a Nasdaq-listed company must obtain shareholder approval in advance of an issuance of securities, including a change of control transaction, the issuance of shares representing more than 20% of the company's total voting power, certain acquisitions in which officers, directors or substantial shareholders have an economic interest, and the creation or material amendment of an equity compensation plan available to the company's officers, directors, employees or consultants. Rule 4350(i) differs in certain ways from the laws of Canada and the rules of the Toronto Stock Exchange relating to shareholder approval. For example, the Toronto Stock Exchange does not require shareholder approval for a private placement of shares representing more than 20% of the company's total voting power, but will generally require shareholder approval of a private placement if it would materially affect the control of the issuer or: (a) would provide consideration to insiders in aggregate of 10% or greater of the market capitalization of the company and has not been negotiated at arm's length; (b) that is made at below-market price and would involve the issuance of more than 25% of the outstanding listed shares; or (c) that is made to insiders which would, when aggregated with all placements made to insiders during any six month period, involve greater than 10% of the number of securities of the company which are outstanding on a non-diluted basis, prior to the date of closing of the first private placement to an insider during the six month
128
period. A further example is that, in certain circumstances, the Toronto Stock Exchange will not require shareholder approval of an amendment to an equity-based compensation arrangement, including where the arrangement contains amendment provisions that specify shareholder approval is not required for the amendment in question. In the event that we enter into a transaction that is treated differently under the Nasdaq and Toronto Stock Exchange rules, we intend to comply with the shareholder approval requirements of the Toronto Stock Exchange.
Competition Act, Exchange Controls and Investment Canada Act
Limitations on the ability to acquire and hold our common shares may be imposed by the Competition Act (Canada). This legislation permits the Commissioner of Competition of Canada, or Commissioner, to review any acquisition of a significant interest in us. This legislation grants the Commissioner jurisdiction for up to three years, to challenge this type of acquisition before the Canadian Competition Tribunal if the Commissioner believes that it would, or would be likely to, substantially reduce or prevent competition in any market in Canada.
This legislation also requires any person who intends to acquire our common shares to file a notification with the Canadian Competition Bureau if certain financial thresholds are exceeded, and that person would hold more than 20 percent of our common shares. If a person already owns 20 percent or more of our common shares, a notification must be filed when the acquisition would bring that person's holdings to over 50 percent. Where a notification is required, the legislation prohibits completion of the acquisition until the expiration of a statutory waiting period, unless the Commissioner provides written notice that he or she does not intend to challenge the acquisition.
There is no law, governmental decree, or regulation in Canada that restricts the export or import of capital, or which would affect the remittance of dividends or other payments by us to non-resident holders of our common shares, other than withholding tax requirements.
There is no limitation imposed by Canadian law or our by-laws or articles on the right of non-residents to hold or vote our common shares, other than those imposed by the Investment Canada Act (Canada), or Investment Act.
The Investment Act requires each individual, government or agency thereof, corporation, partnership, trust or joint venture that is not a "Canadian" as defined in the Investment Act, referred to in this discussion as a "non-Canadian" who commences a new business activity in Canada or acquires control of an existing Canadian business, where the establishment or acquisition of control is not a reviewable transaction, to file a notification with Industry Canada. The Investment Act generally prohibits the implementation of a reviewable transaction by a non-Canadian unless after review the minister responsible for the Investment Act is satisfied that the investment is likely to be of net benefit to Canada. An investment in our common shares by a non-Canadian would be reviewable under the Investment Act if it were an investment to acquire control of us and the value of our assets were C$5.0 million or more. The Investment Act provides for special review thresholds for World Trade Organization, or WTO, member country investors, including United States investors. Under the Investment Act, an investment in our common shares by a non-Canadian who is a "WTO investor" (as defined in the Investment Act) would be reviewable only if it were an investment to acquire control of us and the value of our assets was equal to or greater than a specified amount, which increases in stages. The specified amount is C$265 million in 2006. The threshold amount is subject to an annual adjustment on the basis of a prescribed formula in the Investment Act to reflect inflation and real growth within Canada.
129
The acquisition of a majority of the voting interests of an entity or of a majority of the undivided ownership interests in the voting shares of an entity that is a corporation is deemed to be acquisition of control of that entity. The acquisition of less than a majority but one-third or more of the voting shares of a corporation or of an equivalent undivided ownership interest in the voting shares of the corporation is presumed to be acquisition of control of that corporation unless it can be established that, on the acquisition, the corporation is not controlled in fact by the acquirer through the ownership of voting shares. The acquisition of less than one-third of the voting shares of a corporation or of an equivalent undivided ownership interest in the voting shares of the corporation is deemed not to be acquisition of control of that corporation. Certain transactions in relation to our common shares would be exempt from review from the Investment Act, including:
- •
- acquisition of our common shares by a person in the ordinary course of that person's business as a trader or dealer in securities;
- •
- acquisition or control of us in connection with the realization of security granted for a loan or other financial assistance and not for any purpose related to the provisions of the Investment Act; and
- •
- acquisition or control of us by reason of an amalgamation, merger, consolidation or corporate reorganization following which the ultimate direct or indirect control in fact of us, through the ownership of voting interests, remains unchanged.
130
SHARES ELIGIBLE FOR FUTURE SALE
Prior to this offering, there has been no market for our common shares and a significant public market for our common shares may not develop or be sustained after this offering. Future sales of substantial amounts of our common shares in the public market could adversely affect prevailing market prices from time to time. Furthermore, since only a limited number of shares will be available for sale shortly after this offering because of certain contractual and legal restrictions on resale described below, sales of substantial amounts of our common shares in the public market after the restrictions lapse could adversely affect the prevailing market price and our ability to raise equity capital in the future.
United States Resale Restrictions
Based on shares outstanding as of February 28, 2007, upon completion of the Reorganization and the closing of this offering, 16,079,738 common shares will be outstanding, assuming no exercise of the underwriters' overallotment option and no exercise of outstanding options. Of these outstanding shares, the 5,000,000 shares sold in this offering and 7,813,138 shares previously sold by us outside the United States pursuant to Regulation S under the Securities Act, to the extent not subject to lock-up agreements, will be freely tradable without restrictions or further registration under the Securities Act, unless the shares are held by our affiliates, as that term is defined in Rule 144 under the Securities Act.
The remaining 3,245,564 common shares are restricted securities as defined under Rule 144. Restricted securities may be sold in the public market only if registered, if sold outside the United States pursuant to Regulation S under the Securities Act, or if they qualify for an exemption from registration under Rules 144, 144(k) or 701 of the Securities Act, which are summarized below. Taking into account the lock-up agreements described below, the number of shares that will be available for sale in the public market under the provisions of Rules 144, 144(k) and 701 and Regulation S will be as follows:
| Days after Date of this Prospectus | Number of Shares Eligible for Sale in Public Market | Comment | ||
|---|---|---|---|---|
| Upon Effectiveness | 5,021,036 | Shares sold in this offering and shares not subject to lock-up | ||
| 180 Days after our common shares first commence trading on the Toronto Stock Exchange | 11,058,702 | Lock-up period expires; 7,813,138 shares eligible for sale without Securities Act restrictions and 3,245,564 shares eligible for sale under Rules 144 and 701 and Regulation S | ||
| Thereafter | Nil | Restricted securities held for one year or less |
Additionally, of the 1,423,147 shares issuable upon exercise of options to purchase our common shares outstanding as of February 28, 2007, approximately 1,040,000 shares will be vested and eligible for sale upon release of the lock-up.
131
Regulation S
We are a foreign private issuer as defined in Regulation S. As a foreign private issuer, securities that we sell outside the United States pursuant to Regulation S are not considered to be restricted securities under the Securities Act, and are freely tradable without registration or restrictions under the Securities Act, unless the securities are held by our affiliates. Therefore, except to the extent such shares are held by our affiliates or are subject to lock-up agreements, the 7,813,138 shares previously sold by us outside the United States pursuant to Regulation S, and any additional shares issued by us upon exercise of stock options or otherwise outside the United States pursuant to Regulation S, may be immediately resold in the public market without registration or restriction under the Securities Act.
In addition, provided that we remain a foreign private issuer, and subject to any applicable lock-up agreements, the holders of our outstanding restricted securities may immediately resell such securities outside the United States without registration under the Securities Act pursuant to Regulation S. Generally, subject to certain limitations, holders of our restricted shares who are not our affiliates or who are our affiliates solely by virtue of their status as an officer or director of us may, under Regulation S, resell their restricted shares in an "offshore transaction" (which would include a sale through the Toronto Stock Exchange that is not pre-arranged with a U.S. buyer) if neither the seller nor any person acting on the seller's behalf engages in directed selling efforts in the United States and, in the case of a sale of our restricted shares by an officer or director who is an affiliate of us solely by virtue of holding such position, no selling commission, fee or other remuneration is paid in connection with the offer or sale other than a usual and customary broker's commission. Additional restrictions are applicable to a holder of our restricted shares who will be an affiliate of us other than by virtue of his or her status as an officer or director of us.
Rule 144
In general, under Rule 144 as currently in effect, beginning 90 days after the effective date of this offering, a person who has beneficially owned restricted securities for at least one year, including the holding period of any prior owner other than one of our affiliates, is entitled to sell a number of restricted shares within any three-month period that does not exceed the greater of:
- •
- one percent of the number of common shares then outstanding, which will equal shares immediately after this offering; or
- •
- the average weekly trading volume of our common shares on the Nasdaq Global Market during the four calendar weeks preceding the filing of a notice on Form 144 with respect to the sale.
Sales of restricted shares under Rule 144 are also subject to requirements regarding the manner of sale, notice, and the availability of current public information about us. Rule 144 also provides that affiliates that sell our common shares in the U.S. public market that are not restricted shares must nonetheless comply with the same restrictions applicable to restricted shares, other than the holding period requirement.
Rule 144(k)
Under Rule 144(k), a person who is not deemed to have been our affiliate at any time during the 90 days preceding a sale of restricted shares and who has beneficially owned the shares proposed to be sold for at least two years, including the holding period of any prior owner other
132
than an affiliate, may sell those shares without complying with the manner-of-sale, public information, volume limitation or notice provisions of Rule 144.
Rule 701
Restricted shares acquired upon the exercise of stock options granted under our Stock Option Plan or our 2006 Stock Incentive Plan in the United States pursuant to Rule 701 may be resold, beginning 90 days after the date of this prospectus, to the extent not subject to lock-up agreements, by:
- •
- persons other than affiliates, subject only to the manner-of-sale provisions of Rule 144; and
- •
- our affiliates, subject to the manner-of-sale, current public information, and filing requirements of Rule 144, in each case, without compliance with the one-year holding period requirement of Rule 144.
As of February 28, 2007, an aggregate of 1,905,557 common shares were authorized for issuance under our existing stock option plan. As of September 30, 2006, options to purchase a total of 1,424,147 common shares were outstanding, including 259,443 granted pursuant to Rule 701 (of which approximately 128,559 were vested), and 481,410 shares were available for future grants. Of the total number of common shares issuable under these Rule 701 options, substantially all are subject to contractual lock-up agreements with the underwriters.
Form S-8 Registration Statements
We intend to file one or more registration statements on Form S-8 under the Securities Act following this offering to register the common shares that are issuable upon exercise of outstanding stock options granted under our Stock Option Plan or our 2006 Stock Incentive Plan. These registration statements are expected to become effective upon filing. Shares covered by these registration statements will thereupon be eligible for sale in the public market, upon the expiration or release from the terms of the lock-up agreements, to the extent applicable, or subject in certain cases to vesting of such shares and to Rule 144 limitations applicable to affiliates.
Canadian Resale Restrictions
Under the securities laws of the provinces of Canada, a person who owns our common shares, or a security convertible into our common shares (other than options granted to our directors, officers and employees), distributed by us more than four months prior to the date of this prospectus will generally be able to freely sell those common shares, or the common shares issued upon the conversion of that convertible security, in Canada following the date of this prospectus. To the extent that such common shares or convertible securities were distributed by us during the four months preceding the date of this prospectus, those common shares, or the common shares issued upon the conversion of those convertible securities, may not be resold, except under a prospectus or an exemption from the prospectus requirement, until four months have passed since the date of distribution of those securities by us, at which time such a person will generally be able to freely sell those common shares in Canada. Any of our directors, officers and employees who purchase common shares from us pursuant to the exercise of options granted at any time prior to the date of this prospectus will generally be able to freely resell those common shares in Canada following the date of this prospectus. Any sales of our common shares in Canada will be subject to the terms of applicable lock-up agreements.
133
Lock-up Agreements
Each of our directors and officers, and the holders of substantially all of our outstanding shares and options to acquire our shares have agreed not to sell or otherwise dispose of, directly or indirectly (for example, through a hedging or monetization transaction), any of our common shares (or any security convertible into or exchangeable or exercisable for our common shares), whether now owned or later acquired, without the prior written consent of RBC Dominion Securities Inc. and RBC Capital Markets Corporation (referred to together as RBC) for a period of 180 days from the date our common shares first commence trading on the Toronto Stock Exchange.
Shareholders subject to the lock-up agreements may nevertheless transfer securities without the prior written consent of RBC:
- •
- pursuant to a bona fide third party take-over bid made to all holders of our common shares or similar acquisition transaction;
- •
- by way of pledge or security interest provided that the pledgee or beneficiary of the security interest agrees in writing with RBC to be bound by the lock-up agreement; or
- •
- to certain types of trusts, corporations or other entities established for the benefit of the shareholder, or its immediate family or affiliates, provided that the transferee agrees in writing with RBC to be bound by the lock-up agreement.
In addition, the lock-up agreements do not apply to any common shares purchased by our shareholders in this offering or through the facilities of the Toronto Stock Exchange or Nasdaq.
134
MATERIAL UNITED STATES AND CANADIAN INCOME TAX CONSEQUENCES
Material Canadian Income Tax Consequences
The following summarizes the principal Canadian federal income tax consequences under theIncome Tax Act (Canada), or the Tax Act, generally applicable to the acquisition, holding and disposition of common shares by holders who acquire common shares pursuant to this prospectus. This summary is applicable only to holders who, at all relevant times, for the purposes of the Tax Act, are resident in Canada, deal at arm's length and are not affiliated with us, all within the meaning of the Tax Act, and acquire and hold their common shares as capital property (a Resident Shareholder), and to holders of common shares who, at all relevant times, for the purposes of the Tax Act, are not resident in Canada, deal at arm's length with us, acquire and hold their common shares as capital property, do not use and are not deemed to use or hold their common shares in the course of carrying on, or otherwise in connection with, a business in Canada and who, at all relevant times, for the purposes of the Canada-United States Income Tax Convention 1980, as amended, or the Treaty, are resident in the United States, have never been resident in Canada, and have not held or used (and do not hold or use) common shares in connection with a permanent establishment or fixed base in Canada (a US Shareholder). Generally, common shares will be considered to be capital property to a holder thereof provided that the holder does not use or hold the common shares in the course of carrying on a business or in one or more transactions considered to be an adventure or concern in the nature of trade. Certain Resident Shareholders may, in certain circumstances, treat common shares as capital property by making an irrevocable election permitted by subsection 39(4) of the Tax Act. This summary does not deal with special situations, such as the particular circumstances of traders or dealers in securities, limited liability companies, tax-exempt entities, insurers, "authorized foreign banks", "specified financial institutions" and "financial institutions" as defined in the Tax Act (including those to which the mark-to-market provisions of the Tax Act apply), holders of an interest which is a "tax shelter investment" for the purposes of the Tax Act, or otherwise.
This summary is based on the current provisions of the Tax Act, the regulations under the Tax Act, or the Regulations, all proposals to amend the Tax Act publicly announced by the Minister of Finance (Canada) prior to the date hereof, or the Proposed Amendments, and our understanding of the current published administrative and assessing practices and policies of the Canada Revenue Agency, or the CRA which have been made publicly available prior to the date hereof. This summary assumes that the Proposed Amendments will be enacted in the form proposed and does not take into account, or anticipate any other changes in law, whether by way of judicial, legislative, regulatory, administrative or governmental decision or action, or changes in the administrative practices of the CRA, is not exhaustive of all Canadian federal income tax consequences, nor does it take into account provincial, territorial or foreign income tax legislation or considerations, which may differ from the Canadian federal income tax consequences discussed herein. No assurance can be given that any of the Proposed Amendments will be enacted into law as proposed or that judicial, legislative, regulatory, and administrative or government changes will not modify or change the statements expressed herein.
This summary is not exhaustive of all possible Canadian federal income tax consequences. It is not intended as legal or tax advice to any prospective holder of common shares and should not be construed as such. No representations with respect to the income tax consequences to any such holder are made. The tax consequences to any prospective holder of common shares will vary according to the status of that holder as an individual, trust, corporation or member of a
135
partnership, the jurisdictions in which that holder is subject to taxation and, generally, according the holder's own particular circumstances. Each holder should consult the holder's own tax advisor with respect to the tax consequences applicable to the holder's own particular circumstances including the application and effect of the income and other tax law of any country, province, state or local tax authority.
All amounts relevant in computing the liability of a US Shareholder under the Tax Act are to be reported in Canadian currency at the rate of exchange prevailing at the relevant time.
Taxation of Resident Shareholder
Adjusted Cost Base of Common Shares
The adjusted cost base to a Resident Shareholder of the common shares acquired pursuant to this prospectus will be determined by averaging the cost of the common shares so acquired with the adjusted cost base to the Resident Shareholder of any other common shares of the Company that are held by the Resident Shareholder at the time of the acquisition.
Dividends on Common Shares
A Resident Shareholder of common shares that is an individual (including a trust) will be subject to the normal treatment under the Tax Act applicable to dividends received from a corporation resident in Canada as defined in the Tax Act. That is, a Resident Shareholder who is an individual will be subject to the usual gross-up and dividend tax credit rules normally applicable in respect of taxable dividends received from corporations resident in Canada.
A new dividend regime for "eligible dividends" has been brought into effect by Bill C-28,A second act to implement certain provisions of the budget tabled in parliament on May 2, 2006, which received royal assent on February 21, 2007. The effect of these new rules is to reduce the total tax paid by individuals on eligible dividends from corporations resident in Canada. The reduction in tax results from both an increase in the dividend gross-up (from 25% to 45% of dividends received) and a decrease in the dividend tax credit (from2/3 to11/18 of the gross up). To be eligible, dividends paid must be designated as such by the corporation at the time of payment.
Taxable dividends received by an individual (other than certain specified trusts) will be relevant in computing possible liability for alternative minimum tax.
A Resident Shareholder that is a corporation will include dividends received or deemed received on the common shares in computing its income for tax purposes and generally will be entitled to deduct the amount of such dividends in computing its taxable income, with the result that no tax will be payable by it in respect of such dividends. Certain corporations, including private corporations or subject corporations (as such terms are defined in the Tax Act) and certain other corporations controlled by or for the benefit of an individual (other than a trust) or related group of individuals (other than trusts) may be liable to pay a refundable tax under Part IV of the Tax Act at the rate of 331/3 percent of the dividends received or deemed to be received on the common shares to the extent such dividends are deductible in computing taxable income. This tax generally will be refunded to the corporation at a rate of $1 for every $3 of taxable dividends paid while it is a private corporation.
136
Disposition of Common Shares
In general, a disposition or deemed disposition of a common share by a Resident Shareholder will give rise to a capital gain (or capital loss) equal to the amount by which the proceeds of disposition of such common share, net of any reasonable costs of disposition, exceed (or are less than) the adjusted cost base of such common share to that Resident Shareholder immediately before the disposition.
One-half of any capital gain realized by a Resident Shareholder on the disposition or deemed disposition of a common share in a taxation year must be included in computing the Resident Shareholder's income for that year as a taxable capital gain. Similarly, one-half of any capital loss realized by a Resident Shareholder on the disposition or deemed disposition of a common share in a taxation year is an allowable capital loss that may (subject to detailed rules in the Tax Act) be deducted from the Resident Shareholder's capital gains for that year or the three preceding taxation years or any subsequent taxation year.
In general, in the case of a Resident Shareholder that is a corporation, the amount of any capital loss otherwise determined arising from a disposition or deemed disposition of common shares may be reduced by the amount of dividends received thereon, or deemed received thereon, to the extent and under circumstances prescribed in the Tax Act. Analogous rules apply where a corporation is, directly or through a trust or partnership, a member of a partnership or a beneficiary of a trust which owns common shares.
A Resident Shareholder that is throughout the relevant taxation year a "Canadian-controlled private corporation" (as defined in the Tax Act) may be liable to pay, in addition to the tax otherwise payable under the Tax Act, a refundable tax of 62/3 percent on its "aggregate investment income" for the year which is defined to include taxable capital gains.
Capital gains realized by a Resident Shareholder that is an individual may give rise to a liability for alternative minimum tax.
Taxation of US Shareholder
Disposition of Common Shares
A US Shareholder will not be subject to tax under the Tax Act in respect of any capital gain on a disposition of our common shares unless such shares constitute "taxable Canadian property", as defined in the Tax Act, of the US Shareholder and the US Shareholder is not entitled for relief under the Treaty. Provided the common shares are then listed on a prescribed stock exchange, which currently includes the Toronto Stock Exchange and the Nasdaq Global Market, the common shares generally will not constitute taxable Canadian property of a US Shareholder, unless at any time during the 60-month period immediately preceding the disposition of the common shares, the US Shareholder, persons with whom the US Shareholder did not deal at arm's length, or the US Shareholder together with all such persons, own 25 percent or more of the issued shares of any class of shares of our capital stock. In this case, the common shares will constitute taxable Canadian property to the US Shareholder. A US Shareholder whose common shares are taxable Canadian property should consult their own advisors. In addition, the Treaty generally will exempt a US Shareholder who would otherwise be liable to pay Canadian income tax in respect of any capital gain realized by the US Shareholder on the disposition of our common shares, from such liability provided that the value of our common shares is not derived principally from real property
137
situated in Canada. The Treaty may not be available to a US Shareholder that is a U.S. limited liability company which is not subject to tax in the U.S.
Dividends on Common Shares
Amounts in respect of our common shares paid or credited or deemed to be paid or credited as, on account or in lieu of payment of, or in satisfaction of, dividends to a US Shareholder will generally be subject to Canadian non-resident withholding tax at the rate of 25 percent. Currently, under the Treaty the rate of Canadian non-resident withholding tax will generally be reduced to:
- •
- 5 percent of the gross amount of dividends if the beneficial owner is a company that is resident in the U.S. and that owns at least 10 percent of our voting shares; or
- •
- 15 percent of the gross amount of dividends in all other cases.
In addition, under the Treaty, dividends may be exempt from Canadian non-resident withholding tax if paid to certain U.S. Residents that are qualifying religious, scientific, literary, educational or charitable tax-exempt organizations and qualifying trusts, companies, organizations or arrangements operated exclusively to administer or provide pension, retirement or employee benefits that are exempt from tax in the United States and that have complied with specific administrative procedures.
United States Federal Income Tax Consequences
The following is a summary of the anticipated material U.S. federal income tax consequences to a U.S. Holder (as defined below) arising from and relating to the acquisition, ownership, and disposition of our common shares.
This summary is for general information purposes only and does not purport to be a complete analysis or listing of all potential U.S. federal income tax consequences that may apply to a U.S. Holder as a result of the acquisition, ownership, and disposition of our common shares. In addition, this summary does not take into account the individual facts and circumstances of any particular U.S. Holder that may affect the U.S. federal income tax consequences of the acquisition, ownership, and disposition of common shares.
This summary is not intended to be, and should not be construed as, legal or U.S. federal income tax advice with respect to any U.S. Holder. Each U.S. Holder should consult its own tax advisor regarding the U.S. federal income, U.S. state and local, and foreign tax consequences of the acquisition, ownership, and disposition of our common shares.
Scope of this Summary
Authorities
This summary is based on the Internal Revenue Code of 1986, as amended (the Code), Treasury Regulations (whether final, temporary, or proposed), published rulings of the Internal Revenue Service (the IRS), published administrative positions of the IRS, the Treaty and U.S. court decisions that are applicable and, in each case, as in effect and available, as of the date of this prospectus. Any of the authorities on which this summary is based could be changed in a material and adverse manner at any time, and any such change could be applied on a retroactive basis. This summary does not discuss the potential effects, whether adverse or beneficial, of any proposed legislation that, if enacted, could be applied on a retroactive basis.
138
U.S. Holders
For purposes of this summary, a "U.S. Holder" is a beneficial owner of our common shares that, for U.S. federal income tax purposes, is (a) an individual who is a citizen or resident of the U.S., (b) a corporation, or any other entity classified as a corporation for U.S. federal income tax purposes, that is created or organized in or under the laws of the U.S., any state in the U.S., or the District of Columbia, (c) an estate if the income of such estate is subject to U.S. federal income tax regardless of the source of such income, or (d) a trust if (i) such trust has validly elected to be treated as a U.S. person for U.S. federal income tax purposes or (ii) a U.S. court is able to exercise primary supervision over the administration of such trust and one or more U.S. persons have the authority to control all substantial decisions of such trust.
Non-U.S. Holders
For purposes of this summary, a "non-U.S. Holder" is a beneficial owner of common shares other than a U.S. Holder. This summary does not address the U.S. federal income tax consequences of the acquisition, ownership, and disposition of our common shares to non-U.S. Holders. Accordingly, a non-U.S. Holder should consult its own tax advisor regarding the U.S. federal income, U.S. state and local, and foreign tax consequences (including the potential application of and operation of any income tax treaties) of the acquisition, ownership, and disposition of our common shares.
U.S. Holders Subject to Special U.S. Federal Income Tax Rules Not Addressed
This summary does not address the U.S. federal income tax consequences of the acquisition, ownership, and disposition of our common shares to U.S. Holders that are subject to special provisions under the Code, including the following U.S. Holders: (a) U.S. Holders that are tax-exempt organizations, qualified retirement plans, individual retirement accounts, or other tax-deferred accounts; (b) U.S. Holders that are financial institutions, insurance companies, real estate investment trusts, or regulated investment companies; (c) U.S. Holders that are dealers in securities or currencies or U.S. Holders that are traders in securities that elect to apply a mark-to-market accounting method; (d) U.S. Holders that have a "functional currency" other than the U.S. dollar; (e) U.S. Holders that are liable for the alternative minimum tax under the Code; (f) U.S. Holders that own our common shares as part of a straddle, hedging transaction, conversion transaction, constructive sale, or other arrangement involving more than one position; (g) U.S. Holders that acquired our common shares in connection with the exercise of employee stock options or otherwise as compensation for services; (h) U.S. Holders that hold our common shares other than as a capital asset within the meaning of Section 1221 of the Code; or (i) U.S. Holders that own (directly, indirectly, or constructively) 10 percent or more of the total combined voting power of all classes of our shares entitled to vote. U.S. Holders that are subject to special provisions under the Code, including U.S. Holders described immediately above, should consult their own tax advisors regarding the U.S. federal income tax consequences of the acquisition, ownership, and disposition of our common shares.
If an entity that is classified as a partnership for U.S. federal income tax purposes holds our common shares, the U.S. federal income tax consequences of the acquisition, ownership, and disposition of our common shares to such partnership and the partners of such partnership generally will depend on the activities of the partnership and the status of such partners. Partners of entities that are classified as partnerships for U.S. federal income tax purposes should consult
139
their own tax advisors regarding the U.S. federal income tax consequences of the acquisition, ownership, and disposition of our common shares.
Tax Consequences Other than U.S. Federal Income Tax Consequences Not Addressed
This summary does not address the U.S. state and local, U.S. federal estate and gift, or foreign tax consequences to U.S. Holders of the acquisition, ownership, and disposition of our common shares. Each U.S. Holder should consult its own tax advisor regarding the U.S. state and local, U.S. federal estate and gift, and foreign tax consequences of the acquisition, ownership, and disposition of our common shares. (See, however, "Material Canadian Income Tax Consequences — Taxation of US Shareholder" above).
U.S. Federal Income Tax Consequences of the Acquisition, Ownership, and Disposition of Common Shares
Distributions on Common Shares
General Taxation of Distributions
Subject to the "passive foreign investment company", or PFIC, rules discussed below, a U.S. Holder that receives a distribution, including a constructive distribution, with respect to our common shares will be required to include the amount of such distribution in gross income as a dividend (without reduction for any Canadian income tax withheld from such distribution) to the extent of our current or accumulated "earnings and profits." To the extent that a distribution exceeds our current and accumulated "earnings and profits," such distribution will be treated (a) first, as a tax-free return of capital to the extent of a U.S. Holder's tax basis in the common shares and, (b) thereafter, as gain from the sale or exchange of such common shares. (See "Disposition of common shares" below). Dividends received on the common shares generally will not be eligible for the "dividends received deduction".
Reduced Tax Rates for Certain Dividends
For taxable years beginning after December 31, 2002 and before January 1, 2011, a dividend paid by us generally will be taxed at the preferential tax rates applicable to long-term capital gains if (a) we are a "qualified foreign corporation" (as defined below), (b) the U.S. Holder receiving such dividend is an individual, estate, or trust, and (c) such dividend is paid on common shares that have been held by such U.S. Holder for at least 61 days during the 121-day period beginning 60 days before the "ex-dividend date."
We generally will be a "qualified foreign corporation" under Section 1(h)(11) of the Code (a QFC) if (a) we were incorporated in a possession of the U.S., (b) we are eligible for the benefits of the Treaty, or (c) the common shares are readily tradable on an established securities market in the U.S. However, even if we satisfy one or more of such requirements, we will not be treated as a QFC if we are a PFIC for the taxable year during which we pay a dividend or for the preceding taxable year. In 2003, the U.S. Department of the Treasury (the Treasury) and the IRS announced that they intended to issue Treasury Regulations providing procedures for a foreign corporation to certify that it is a QFC. Although these Treasury Regulations have not yet been issued, the Treasury and the IRS have confirmed their intention to issue these Treasury Regulations. It is expected that these Treasury Regulations will obligate persons required to file information returns to report a dividend paid by a foreign corporation as a dividend from a QFC if the foreign
140
corporation has, among other things, certified under penalties of perjury that the foreign corporation was not a PFIC for the taxable year during which the foreign corporation paid the dividend or for the preceding taxable year.
As discussed below, we believe that we were a PFIC for previous taxable years, and expect that we will be a PFIC for the current taxable year. (See "Passive Foreign Investment Company" below). Accordingly, we do not expect to be a QFC for the current or subsequent taxable year.
Distributions Paid in Foreign Currency
The amount of a distribution received on the common shares in foreign currency generally will be equal to the U.S. dollar value of such distribution based on the exchange rate applicable on the date of receipt. A U.S. Holder that does not convert foreign currency received as a distribution into U.S. dollars on the date of receipt generally will have a tax basis in such foreign currency equal to the U.S. dollar value of such foreign currency on the date of receipt. Such a U.S. Holder generally will recognize ordinary income or loss on the subsequent sale or other taxable disposition of such foreign currency (including an exchange for U.S. dollars).
Disposition of Common Shares
A U.S. Holder will recognize gain or loss on the sale or other taxable disposition of our common shares in an amount equal to the difference, if any, between (a) the amount of cash plus the fair market value of any property received and (b) such U.S. Holder's adjusted tax basis in the common shares sold or otherwise disposed of. Subject to the PFIC rules discussed below, any such gain or loss generally will be capital gain or loss, which will be long-term capital gain or loss if the common shares are held for more than one year.
Preferential tax rates apply to long-term capital gains of a U.S. Holder that is an individual, estate, or trust. Deductions for capital losses are subject to significant limitations under the Code.
Foreign Tax Deduction or Credit
A U.S. Holder that pays (whether directly or through withholding) Canadian income tax with respect to dividends received on our common shares generally will be entitled, at the election of such U.S. Holder, to receive either a deduction or a credit for such Canadian income tax paid. Generally, a credit will reduce a U.S. Holder's U.S. federal income tax liability on a dollar-for-dollar basis, whereas a deduction will reduce a U.S. Holder's income subject to U.S. federal income tax. This election is made on a year-by-year basis and applies to all foreign taxes paid (whether directly or through withholding) by a U.S. Holder during a taxable year.
Complex limitations apply to the foreign tax credit, including the general limitation that the credit cannot exceed the proportionate share of a U.S. Holder's U.S. federal income tax liability that such U.S. Holder's "foreign source" taxable income bears to such U.S. Holder's worldwide taxable income. In applying this limitation, a U.S. Holder's various items of income and deduction must be classified, under complex rules, as either "foreign source" or "U.S. source." In addition, this limitation is calculated separately with respect to specific categories of income (including "passive income," "high withholding tax interest," "financial services income," "general income," and certain other categories of income). Gain or loss recognized by a U.S. Holder on the sale or other taxable disposition of common shares and foreign currency gains generally will be treated as "U.S. source" for purposes of applying the foreign tax credit rules.
141
Dividends received on the common shares generally will be treated as "foreign source" and generally will be categorized as "passive income" or, in the case of certain U.S. Holders, "financial services income" for purposes of applying the foreign tax credit rules. However, for taxable years beginning after December 31, 2006, the foreign tax credit limitation categories are reduced to "passive category income" and "general category income" (and the other categories of income, including "financial services income," are eliminated). The foreign tax credit rules are complex, and each U.S. Holder should consult its own tax advisor regarding the foreign tax credit rules.
Information Reporting; Backup Withholding Tax
Payments made within the United States or by a U.S. payor or U.S. middleman, of dividends on, or proceeds arising from the sale or other taxable disposition of, our common shares generally will be subject to information reporting and backup withholding tax, at the rate of 28 percent, if a U.S. Holder (a) fails to furnish such U.S. Holder's correct U.S. taxpayer identification number (generally on Form W-9), (b) furnishes an incorrect U.S. taxpayer identification number, (c) is notified by the IRS that such U.S. Holder has previously failed to properly report items subject to backup withholding tax, or (d) fails to certify, under penalty of perjury, that such U.S. Holder has furnished its correct U.S. taxpayer identification number and that the IRS has not notified such U.S. Holder that it is subject to backup withholding tax. However, U.S. Holders that are corporations generally are excluded from these information reporting and backup withholding tax rules. Any amounts withheld under the U.S. backup withholding tax rules will be allowed as a credit against a U.S. Holder's U.S. federal income tax liability, if any, or will be refunded, if such U.S. Holder furnishes required information to the IRS. Each U.S. Holder should consult its own tax advisor regarding the information reporting and backup withholding tax rules.
Passive Foreign Investment Company
We generally will be a PFIC under Section 1297(a) of the Code if, for a taxable year, (a) 75 percent or more of our gross income for such taxable year is passive income or (b) on average, 50 percent or more of the assets held by us either produce passive income or are held for the production of passive income, based on the fair market value of such assets (or on the adjusted tax basis of such assets, if we are not publicly traded and make an election). "Passive income" includes, for example, dividends, interest, certain rents and royalties, certain gains from the sale of stock and securities, and certain gains from commodities transactions.
For purposes of the PFIC income test and asset test described above, if we own, directly or indirectly, 25 percent or more of the total value of the outstanding shares of another foreign corporation, we will be treated as if we (a) held a proportionate share of the assets of such other foreign corporation and (b) received directly a proportionate share of the income of such other foreign corporation. In addition, for purposes of the PFIC income test and asset test described above, "passive income" does not include any interest, dividends, rents, or royalties that are received or accrued by us from a "related person" (as defined in Section 954(d)(3) of the Code), to the extent such items are properly allocable to the income of such related person that is not passive income.
Because we are a clinical-stage biopharmaceutical company which has not yet recognized significant operating income and our gross income consists mostly of interest, we have been a PFIC for previous taxable years. We may also be a PFIC in the current taxable year as well as future taxable years until we generate significant operating income. A U.S. Holder can avoid the
142
adverse U.S. federal income tax consequences of holding shares in a PFIC by making a QEF Election (see "QEF Election", below). Under a QEF Election, generally, an electing U.S. Holder will be required each taxable year in which we are a PFIC to recognize, as ordinary income, a pro rata share of our earnings, and to recognize, as capital gain, a pro rata share of our net capital gain. Accordingly, because we expect that we only will be a PFIC in taxable years in which we do not generate any net income, an electing U.S. Holder would not have any income inclusions as a result of the QEF Election. Furthermore, in any taxable year in which we generate significant operating income, we may cease to be a PFIC and the QEF Election will not be applicable.
The determination of whether we were, or will be, a PFIC for a taxable year depends, in part, on the application of complex U.S. federal income tax rules, which are subject to various interpretations. In addition, whether we will be a PFIC for the current taxable year and each subsequent taxable year depends on our assets and income over the course of each such taxable year and, as a result, cannot be predicted with certainty as of the date of this prospectus. Accordingly, there can be no assurance that the IRS will not challenge the determination made by us concerning our PFIC status or that we were not, or will not be, a PFIC for any taxable year.
Default PFIC Rules Under Section 1291 of the Code
If we are a PFIC, the U.S. federal income tax consequences to a U.S. Holder of the acquisition, ownership, and disposition of common shares will depend on whether such U.S. Holder makes an election to treat us as a "qualified electing fund" or "QEF" under Section 1295 of the Code (a QEF Election) or a mark-to-market election under Section 1296 of the Code (a Mark-to-Market Election). A U.S. Holder that does not make either a QEF Election or a Mark-to-Market Election will be referred to in this summary as a "Non-Electing U.S. Holder."
A Non-Electing U.S. Holder will be subject to the rules of Section 1291 of the Code with respect to (a) any gain recognized on the sale or other taxable disposition of common shares and (b) any excess distribution received on the common shares. A distribution generally will be an "excess distribution" to the extent that such distribution (together with all other distributions received in the current taxable year) exceeds 125 percent of the average distributions received during the three preceding taxable years (or during a U.S. Holder's holding period for the common shares, if shorter).
Under Section 1291 of the Code, any gain recognized on the sale or other taxable disposition of our common shares, and any excess distribution received on our common shares, must be ratably allocated to each day in a Non-Electing U.S. Holder's holding period for such common shares. The amount of any such gain or excess distribution allocated to prior years of such Non-Electing U.S. Holder's holding period for the common shares (other than years prior to our first taxable year beginning after December 31, 1986 for which we were not a PFIC) will be subject to U.S. federal income tax at the highest tax rate applicable to ordinary income in each such prior year. A Non-Electing U.S. Holder will be required to pay interest on the resulting tax liability for each such prior year, calculated as if such tax liability had been due in each such prior year. Such a Non-Electing U.S. Holder that is not a corporation must treat any such interest paid as "personal interest," which is not deductible. The amount of any such gain or excess distribution allocated to the current year of such Non-Electing U.S. Holder's holding period for the common shares will be treated as ordinary income in the current year, and no interest charge will be incurred with respect to the resulting tax liability for the current year.
143
If we are a PFIC for any taxable year during which a Non-Electing U.S. Holder holds our common shares, we will continue to be treated as a PFIC with respect to such Non-Electing U.S. Holder, regardless of whether we cease to be a PFIC in one or more subsequent taxable years. A Non-Electing U.S. Holder may terminate this deemed PFIC status by electing to recognize gain (which will be taxed under the rules of Section 1291 of the Code discussed above) as if such common shares were sold on the last day of the last taxable year for which we were a PFIC.
In addition, if we are a PFIC and own shares of another foreign corporation that also is a PFIC, under certain indirect ownership rules, a disposition by us of the shares of such other foreign corporation or a distribution received by us from such other foreign corporation generally will be treated as an indirect disposition by a U.S. Holder or an indirect distribution received by a U.S. Holder, subject to the rules of Section 1291 of the Code discussed above. To the extent that gain recognized on the actual disposition by a U.S. Holder of our common shares or income recognized by a U.S. Holder on an actual distribution received on our common shares was previously subject to U.S. federal income tax under these indirect ownership rules, such amount generally should not be subject to U.S. federal income tax.
QEF Election
A U.S. Holder that makes a timely QEF Election generally will not be subject to the rules of Section 1291 of the Code as discussed above. A U.S. Holder that makes a timely QEF Election generally will recognize capital gain or loss on the sale or other taxable disposition of our common shares. Furthermore, for each taxable year in which we are a PFIC, an electing U.S. Holder will recognize, for U.S. federal income tax purposes, such U.S. Holder's pro rata share of (a) our net capital gain, which will be taxed as long-term capital gain to such U.S. Holder, and (b) and our ordinary earnings, which will be taxed as ordinary income to such U.S. Holder. Accordingly, an electing U.S. Holder would not have any income inclusions as a result of the QEF Election so long as we do not generate any net income. Generally, "net capital gain" is the excess of (a) net long-term capital gain over (b) net short-term capital loss, and "ordinary earnings" are the excess of (a) "earnings and profits" over (b) net capital gain. A U.S. Holder that makes a QEF Election will include such U.S. Holder's pro rata share of our net capital gain and ordinary earnings for each taxable year in which we are a PFIC, even though such amounts may not be distributed to such U.S. Holder by us. A U.S. Holder that makes a QEF Election may, subject to certain limitations, elect to defer payment of current U.S. federal income tax on such amounts, subject to an interest charge. If such U.S. Holder is not a corporation, any such interest paid will be treated as "personal interest," which is not deductible.
A U.S. Holder that makes a QEF Election generally (a) may receive a tax-free distribution from us to the extent that such distribution represents our "earnings and profits" that were previously included in income by the U.S. Holder because of such QEF Election and (b) will adjust such U.S. Holder's tax basis in our common shares to reflect the amount included in income or allowed as a tax-free distribution because of such QEF Election.
The procedure for making a QEF Election, and the U.S. federal income tax consequences of making a QEF Election, will depend on whether such QEF Election is timely. A QEF Election generally will be "timely" if it is made for the first year in a U.S. Holder's holding period for our common shares in which we are a PFIC. In this case, a U.S. Holder may make a timely QEF Election by filing the appropriate QEF Election documents with such U.S. Holder's U.S. federal income tax return for such first year. However, if we were a PFIC in a prior year in a U.S.
144
Holder's holding period for the common shares, then in order to be treated as making a "timely" QEF Election, such U.S. Holder must elect to recognize gain (which will be taxed under the rules of Section 1291 of the Code discussed above) as if the common shares were sold on the qualification date for an amount equal to the fair market value of the common shares on the qualification date. The "qualification date" is the first day of the first taxable year in which we were a QEF with respect to such U.S. Holder. In addition, under very limited circumstances, a U.S. Holder may make a retroactive QEF Election if such U.S. Holder failed to file the QEF Election documents in a timely manner.
A QEF Election will apply to the taxable year for which such QEF Election is made and to all subsequent taxable years, unless such QEF Election is invalidated or terminated or the IRS consents to revocation of such QEF Election. If a U.S. Holder makes a QEF Election and, in a subsequent taxable year, we cease to be a PFIC, the QEF Election will remain in effect (although it will not be applicable) during those taxable years in which we are not a PFIC. Accordingly, if we become a PFIC in another subsequent taxable year, the QEF Election will be effective and the U.S. Holder will be subject to the QEF rules described above during any such subsequent taxable year in which we qualify as a PFIC. In addition, the QEF Election will remain in effect (although it will not be applicable) with respect to a U.S. Holder even after such U.S. Holder disposes of all of such U.S. Holder's direct and indirect interest in our common shares. Accordingly, if such U.S. Holder reacquires an interest in OncoGenex, such U.S. Holder will be subject to the QEF rules described above for each taxable year in which we were a PFIC.
In the event we are a PFIC, we will satisfy record keeping requirements that apply to a QEF and supply U.S. Holders with information that such U.S. Holders require to report under the QEF rules. Each U.S. Holder should consult its own tax advisor regarding the availability of, and procedure for making, a QEF Election.
Mark-to-Market Election
A U.S. Holder may make a Mark-to-Market Election only if our common shares are marketable stock. Our common shares generally will be "marketable stock" if the common shares are regularly traded on a qualified exchange or other market. For this purpose, a "qualified exchange or other market" includes (a) a national securities exchange that is registered with the U.S. Securities and Exchange Commission, (b) the national market system established pursuant to section 11A of the Securities and Exchange Act of 1934, or (c) a foreign securities exchange that is regulated or supervised by a governmental authority of the country in which the market is located, provided that (i) such foreign exchange has trading volume, listing, financial disclosure, surveillance, and other requirements designed to prevent fraudulent and manipulative acts and practices, remove impediments to and perfect the mechanism of a free, open, fair, and orderly market, and protect investors (and the laws of the country in which the foreign exchange is located and the rules of the foreign exchange ensure that such requirements are actually enforced) and (ii) the rules of such foreign exchange effectively promote active trading of listed stocks. If the common shares are traded on such a qualified exchange or other market, the common shares generally will be "regularly traded" for any calendar year during which the common shares are traded, other than inde minimis quantities, on at least 15 days during each calendar quarter.
A Mark-to-Market Election applies to the taxable year in which such Mark-to-Market Election is made and to each subsequent taxable year, unless our common shares cease to be "marketable stock" or the IRS consents to revocation of such election. Each U.S. Holder should
145
consult its own tax advisor regarding the availability of, and procedure for making, a Mark-to-Market Election.
A U.S. Holder that makes a Mark-to-Market Election generally will not be subject to the rules of Section 1291 of the Code discussed above. However, if a U.S. Holder makes a Mark-to-Market Election after the beginning of such U.S. Holder's holding period for the common shares and such U.S. Holder has not made a timely QEF Election, the rules of Section 1291 of the Code discussed above will apply to certain dispositions of, and distributions on, our common shares.
A U.S. Holder that makes a Mark-to-Market Election will include in ordinary income, for each taxable year in which we are a PFIC, an amount equal to the excess, if any, of (a) the fair market value of the common shares as of the close of such taxable year over (b) such U.S. Holder's adjusted tax basis in such common shares. A U.S. Holder that makes a Mark-to-Market Election will be allowed a deduction in an amount equal to the lesser of (a) the excess, if any, of (i) such U.S. Holder's adjusted tax basis in the common shares over (ii) the fair market value of such common shares as of the close of such taxable year or (b) the excess, if any, of (i) the amount included in ordinary income because of such Mark-to-Market Election for prior taxable years over (ii) the amount allowed as a deduction because of such Mark-to-Market Election for prior taxable years.
A U.S. Holder that makes a Mark-to-Market Election generally will adjust such U.S. Holder's tax basis in the common shares to reflect the amount included in gross income or allowed as a deduction because of such Mark-to-Market Election. In addition, upon a sale or other taxable disposition of our common shares, a U.S. Holder that makes a Mark-to-Market Election will recognize ordinary income or loss (not to exceed the excess, if any, of (a) the amount included in ordinary income because of such Mark-to-Market Election for prior taxable years over (b) the amount allowed as a deduction because of such Mark-to-Market Election for prior taxable years).
The PFIC rules are complex, and each U.S. Holder should consult its own tax advisor regarding the PFIC rules and how the PFIC rules may affect the U.S. federal income tax consequences of the acquisition, ownership, and disposition of common shares.
146
This offering is being made concurrently in the United States and the provinces of Canada. Our common shares are being offered in the United States by RBC Capital Markets Corporation, Needham & Company, LLC, Lazard Capital Markets LLC, Canaccord Adams Inc. and Susquehanna Financial Group, LLLP, and such other registered dealers as may be designated by the underwriters. Our common shares are being offered in Canada by RBC Dominion Securities Inc. and Canaccord Capital Corporation. Subject to applicable law, the underwriters may offer the common shares outside of the United States and Canada.
RBC Dominion Securities Inc. is acting as representative for the underwriters named above. Subject to the terms and conditions described in an underwriting agreement among us and the underwriters, we have agreed to sell to the underwriters, and the underwriters severally have agreed to purchase from us, the number of common shares listed opposite their names below.
| Underwriters | Number of Shares | ||
|---|---|---|---|
| RBC Capital Markets Corporation | |||
| Needham & Company, LLC | |||
| Lazard Capital Markets LLC | |||
| Canaccord Adams Inc. | |||
| Susquehanna Financial Group, LLLP | |||
| RBC Dominion Securities Inc. | |||
| Canaccord Capital Corporation | |||
| Total shares | 5,000,000 | ||
The underwriters have agreed to purchase all of the common shares sold under the underwriting agreement if any of these common shares are purchased. If an underwriter defaults, the underwriting agreement provides that the purchase commitments of the non-defaulting underwriters may be increased or the underwriting agreement may be terminated. The public offering price for our common shares offered in the United States and elsewhere outside Canada is payable in U.S. dollars and the public offering price for our common shares offered in Canada is payable in Canadian dollars, except as we and the underwriters may otherwise agree. The Canadian dollar amount is the approximate equivalent of the U.S. dollar price of the common shares being offered hereby determined based on the prevailing Canadian-U.S. dollar exchange rate on the date of the underwriting agreement.
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act and applicable securities laws of the provinces of Canada, or to contribute to payments the underwriters may be required to make in respect of those liabilities.
The underwriters are offering the common shares, subject to prior sale, when, as and if issued to and accepted by them, subject to approval of legal matters by their counsel, including the validity of the common shares, and other conditions contained in the underwriting agreement, such as the receipt by the underwriters of officer's certificates and legal opinions. The underwriters reserve the right to withdraw, cancel or modify offers to the public and to reject orders in whole or in part.
147
Commissions and Discounts
The underwriters have advised us that the underwriters propose initially to offer the common shares to the public at the initial public offering price on the cover page of this prospectus and to dealers at that price less a concession in the United States and elsewhere outside Canada not in excess of $ (or C$ in the case of the Canadian offering) per share. The underwriters may allow, and the dealers may re-allow, a discount not in excess of $ per share to other dealers. After the initial public offering, the public offering price, concession and discount may be changed. In Canada, after the underwriters have made a bona fide effort to sell all of the common shares offered under this prospectus at the initial public offering price fixed in this prospectus, the offering price may be decreased and thereafter further changed, from time to time, to an amount not greater than the initial public offering price. In this event, the compensation realized by the underwriters will be decreased by the amount that the aggregate price paid by purchasers for the common shares is less than the gross proceeds paid by the underwriters to us.
The following table shows the assumed public offering price, underwriting discount and proceeds before expenses to OncoGenex. The information assumes either no exercise or full exercise by the underwriters of their overallotment options.
| | Per Share | Without Option | With Option | |||
|---|---|---|---|---|---|---|
| Public offering price | ||||||
| Underwriting discount | ||||||
| Proceeds, before expenses, to OncoGenex |
The expenses of the offering, not including the underwriting discount, are estimated at $1,980,000 and are payable by us. We have agreed to reimburse the underwriters for their out-of-pocket expenses and taxes thereon, not to exceed $500,000. The National Association of Securities Dealers, Inc. has deemed any such reimbursement to be compensation in connection with this offering.
Lazard Frères & Co. LLC referred this transaction to Lazard Capital Markets LLC and will receive a referral fee from Lazard Capital Markets LLC in connection therewith.
Overallotment Option
We have granted an option to the underwriters to purchase up to 750,000 additional common shares at the public offering price less the underwriting discount. The underwriters may exercise this option for 30 days from the date of the underwriting agreement solely to cover any overallotments. If the underwriters exercise this option, each will be obligated, subject to conditions contained in the underwriting agreement, to purchase a number of additional common shares proportionate to that underwriter's initial amount reflected in the above table.
Reserved Shares
At our request the underwriters have reserved for sale, at the initial public offering price, up to shares offered by this prospectus for sale to some of our directors, officers and employees, and other persons having relationships with us. If these persons purchase reserved shares, this will reduce the number of shares available for sale to the general public. Any reserved shares which are not orally confirmed for purchase within one day of the pricing of the offering
148
will be offered by the underwriters to the general public on the same terms as the other shares offered by this prospectus.
No Sales of Similar Securities
We have agreed, with exceptions, not to sell any of our common shares for 180 days after the date our common shares first commence trading on the Toronto Stock Exchange without first obtaining the written consent of RBC. Specifically, we have agreed, subject to exceptions, not to directly or indirectly:
- •
- offer, pledge, sell or contract to sell any of our common shares;
- •
- sell any option or contract to purchase any of our common shares;
- •
- purchase any option or contract to sell any of our common shares;
- •
- grant any option, right or warrant for the sale of any of our common shares;
- •
- lend or otherwise dispose of or transfer any of our common shares;
- •
- file a registration statement related to our common shares; or
- •
- enter into any swap or other agreement that transfers, in whole or in part, the economic consequence of ownership of any of our common shares whether any such swap or transaction is to be settled by delivery of shares or other securities, in cash or otherwise.
This lock-up provision applies to our common shares and to securities convertible into or exchangeable or exercisable for or repayable with our common shares.
In addition, each of our directors and officers, and the holders of substantially all of our outstanding shares and options to acquire our shares have agreed, subject to certain exceptions, not to sell or otherwise dispose of, directly or indirectly (for example, through a hedging or monetization transaction), any of our common shares (or any security convertible into or exchangeable or exercisable for our common shares), whether now owned or later acquired (other than common shares purchased in this offering or through the facilities of the Toronto Stock Exchange or Nasdaq Global Market), without the prior written consent of RBC for a period of 180 days from the date our common shares first commence trading on the Toronto Stock Exchange.
Listing on the Nasdaq Global Market and the Toronto Stock Exchange
The Nasdaq Global Market has approved the listing of our common shares under the symbol "OGXI". In addition, the Toronto Stock Exchange has conditionally approved the listing of our common shares under the symbol "OGX". Listing is subject to us fulfilling all of the requirements of the Toronto Stock Exchange on or before April 4, 2007, including the distribution of our shares to a minimum number of public shareholders. In order to meet the requirements for listing on the Toronto Stock Exchange, the underwriters have undertaken to sell a minimum number of common shares to a minimum number of beneficial owners.
Before this offering, there has been no public market for our common shares. The initial public offering price was determined through negotiations among us and the underwriters. In
149
addition to prevailing market conditions, the factors considered in determining the initial public offering price were:
- •
- the valuation multiples of publicly traded companies that the underwriters believe to be comparable to us;
- •
- our financial information;
- •
- the history of, and the prospects for, our company and the industry in which we compete;
- •
- an assessment of our management, its past and present operations, and the prospects for, and timing of, our future revenues;
- •
- the present state of our development; and
- •
- the above factors in relation to market values and various valuation measures of other companies engaged in activities similar to ours.
An active trading market for the common shares may not develop. It is also possible that after the offering the common shares will not trade in the public market at or above the initial public offering price.
The underwriters do not expect to sell more than five percent of the common shares in the aggregate to accounts over which they exercise discretionary authority.
Price Stabilization, Short Positions and Penalty Bids
Until the distribution of the common shares is completed, SEC and applicable Canadian and stock exchange rules may limit underwriters and selling group members from bidding for and purchasing our common shares. However, the underwriters may engage in transactions that stabilize the price of our common shares, such as bids or purchases to peg, fix or maintain that price, subject to certain conditions described below.
In accordance with policy statements of the Ontario Securities Commission and the Authorité des marches financiers, the underwriters may not throughout the period of distribution, bid for or purchase the shares. Exceptions, however, exist where the bid or purchase is not made to create the appearance of active trading in, or rising prices of, the common shares. These exceptions include a bid or purchase permitted under the by-laws and rules of applicable regulatory authorities and the Toronto Stock Exchange relating to market stabilization and passive market making activities and a bid or purchase made for and on behalf of a customer where the order was not solicited during the period of distribution. Subject to the foregoing and applicable laws, in connection with the offering and pursuant to the first exception mentioned above, the underwriters may overallot or effect transactions that stabilize or maintain the market price of the shares at levels other than those which might otherwise prevail on the open market. Any of these activities may have the effect of preventing or retarding a decline in the market price of the common shares. They may also cause the price of the common shares to be higher than the price that would otherwise exist in the open market in the absence of these transactions. The underwriters may conduct these transactions on the Nasdaq Global Market, the Toronto Stock Exchange or in the over-the-counter market, or otherwise. If the underwriters commence any of these transactions, they may discontinue them at any time.
If the underwriters create a short position in our common shares in connection with the offering, i.e., if they sell more common shares than are listed on the cover of this prospectus, the
150
underwriters may reduce that short position by purchasing shares in the open market. "Covered" short sales are sales of shares made in an amount up to the number of shares represented by the underwriters' overallotment option. In determining the source of shares to close out the covered short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through the overallotment option. Transactions to close out the covered short involve either purchases of the common stock in the open market after the distribution has been completed or the exercise of the overallotment option. The underwriters may also make "naked" short sales of shares in excess of overallotment option. The underwriters must close out any naked short position by purchasing shares of the common stock in the open market. A naked short position is more likely to be created if the underwriters are concerned that there may be downward pressure on the price of the shares in the open market after pricing that could adversely affect investors who purchase in the offering. Purchases of our common shares to stabilize their price or to reduce a short position may cause the price of our common shares to be higher than it might be in the absence of such purchases.
RBC Dominion Securities Inc. may also impose a penalty bid on underwriters and selling group members. This means that if RBC Dominion Securities Inc. purchases common shares in the open market to reduce the underwriter's short position or to stabilize the price of such shares, they may reclaim the amount of the selling concession from the underwriters and selling group members who sold those shares. The imposition of a penalty bid may also affect the price of the common shares in that it discourages resales of those shares.
Neither we nor any of the underwriters makes any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of our common shares. In addition, neither we nor any of the underwriters makes any representation that the underwriters will engage in these transactions or that these transactions, once commenced, will not be discontinued without notice.
No Public Offering Outside the United States and Canada
No action has been or will be taken in any jurisdiction (except in the United States and Canada) that would permit a public offering of our common shares, or the possession, circulation or distribution of this prospectus or any other material relating to our company or our common shares in any jurisdiction where action for that purpose is required. Accordingly, our common shares may not be offered or sold, directly or indirectly, and neither this prospectus nor any other offering material or advertisements in connection with the common shares may be distributed or published, in or from any country or jurisdiction except in compliance with any applicable rules and regulations of any such country or jurisdiction.
Purchasers of the common shares offered by this prospectus may be required to pay stamp taxes and other charges in accordance with the laws and practices of the country of purchase in addition to the offering price on the cover page of this prospectus.
Electronic Distribution
RBC Dominion Securities Inc. will be facilitating Internet distribution for this offering to certain of its Internet subscription customers. Its representatives intend to allocate a limited number of shares for sale to their online brokerage customers. An electronic prospectus is available on the web sites maintained by RBC Dominion Securities Inc. Other than the electronic
151
prospectus, the information on the RBC Dominion Securities Inc. web site is not part of this prospectus.
Legal matters relating to Canadian law, the offering and the validity of the common shares offered in this offering are being passed upon for us by DuMoulin Black LLP, Vancouver, British Columbia. Legal matters relating to U.S. law and the offering are being passed upon for us by Dorsey & Whitney LLP, Seattle, Washington and San Francisco, California. Legal matters relating to Canadian law and the offering are being passed upon for the underwriters by Farris, Vaughan, Wills & Murphy LLP, Vancouver, British Columbia. The underwriters have been advised by Shearman & Sterling LLP with respect to certain matters involving U.S. law.
Ernst & Young LLP, 700 Georgia Street P.O Box 10101, Vancouver, British Columbia, V7Y 1C7, independent registered public accounting firm, has audited our consolidated balance sheets as at December 31, 2004 and 2005 and the related consolidated statements of operations, shareholders' deficiency and cash flows for each of the years ended December 31, 2003, 2004 and 2005, as set forth in their report thereon appearing elsewhere herein. We have included our consolidated financial statements in the prospectus in reliance on Ernst & Young LLP's report, given on their authority as experts in accounting and auditing.
As of the date hereof, none of the partners and associates of DuMoulin Black LLP, Farris, Vaughan, Wills & Murphy LLP or Ernst & Young LLP as a group beneficially own, directly or indirectly, any of our outstanding equity securities.
RELATIONSHIP BETWEEN OUR COMPANY AND CERTAIN UNDERWRITERS
RBC Technologies Ventures Inc., a wholly owned subsidiary of Royal Bank of Canada, is a controlling shareholder of one of our principal shareholders, Milestone Medica Corporation. Royal Bank of Canada is the indirect parent company of both RBC Capital Markets Corporation and RBC Dominion Securities Inc., the representative for the underwriting syndicate. Milestone Medica Corporation currently holds 7.04% of our outstanding voting shares and will hold 4.85% of our outstanding common shares after giving effect to the offering and the Reorganization. See "Principal Shareholders". Accordingly, we may be considered to be a connected issuer of RBC Dominion Securities Inc. and RBC Capital Markets Corporation under Canadian securities legislation. As the representative for the underwriting syndicate, RBC Dominion Securities Inc. negotiated the offering price of the common shares with us and managed the due diligence of our company and the relationship with underwriters' legal counsel prior to filing this prospectus. Other than as described in this prospectus, RBC Dominion Securities Inc. and RBC Capital Markets Corporation will not receive any proceeds of the offering. Milestone Medica Corporation will not receive any proceeds of the offering.
152
We estimate that we will pay approximately $1,980,000 of expenses for this offering, consisting of the following:
| U.S. and Canadian filing fees | $ | 40,000 | |
| Nasdaq Global Market and Toronto Stock Exchange listing fees | $ | 240,000 | |
| Accounting fees and expenses | $ | 300,000 | |
| Legal fees and expenses | $ | 1,200,000 | |
| Printing, mailing and administrative fees | $ | 120,000 | |
| Transfer agent and registrar fees | $ | 10,000 | |
| Miscellaneous | $ | 70,000 |
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC, Washington, D.C. 20549, a registration statement on Form F-1 under the Securities Act, with respect to our common shares offered hereby. This prospectus, which forms part of the registration statement, does not contain all of the information set forth in the registration statement and the exhibits and schedules to the registration statement. Some items are omitted in accordance with the rules and regulations of the SEC. For further information about us and our common shares, we refer you to the registration statement and the exhibits and schedules to the registration statement filed as part of the registration statement. Statements contained in this prospectus as to the contents of any contract or other document filed as an exhibit are qualified in all respects by reference to the actual text of the exhibit. You may read and copy the registration statement, including the exhibits and schedules to the registration statement, at the SEC's Public Reference Room at 450 Fifth Street, N.W., Washington, D.C. 20549. You can obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. In addition, the SEC maintains a web site atwww.sec.gov, from which you can electronically access the registration statement, including the exhibits and schedules to the registration statement.
We are a "foreign private issuer" as defined under Rule 3b-4 under of the Exchange Act. As a result, although upon completion of the offering we will become subject to the informational requirements of the Exchange Act, as a foreign private issuer, we will be exempt from certain informational requirements of the Exchange Act which domestic issuers are subject to, including the proxy rules under Section 14 of the Exchange Act, the insider reporting and short-profit provisions under Section 16 of the Exchange Act. Although we would be permitted to file periodic reports with the SEC under the Exchange Act on Form 20-F or 40-F, and to make submissions on Form 6-K, we intend to voluntarily file periodic reports with the SEC on domestic Forms 10-K, 10-Q and 8-K. We will also be subject to the full informational requirements of the securities commissions in all provinces of Canada. You are invited to read and copy any reports, statements or other information, other than confidential filings, that we intend file with the Canadian provincial securities commissions. These filings are electronically available from the Canadian System for Electronic Document Analysis and Retrieval (SEDAR) (http://www.sedar.com), the Canadian equivalent of the SEC's electronic document gathering and retrieval system. We also intend to furnish our shareholders with annual reports containing consolidated financial statements audited by an independent registered public accounting firm.
153
On August 10, 2005, we issued 4,401,895 Series 2 Class B preferred shares at an issue price per share of $3.07. These shares will be converted into 4,401,895 common shares on completion of the Reorganization and this offering.
Prior to closing of this offering, and as a condition of the closing of this offering, our articles will be amended to change our share capital through the conversion of all of the issued and outstanding preferred shares (encompassing all classes and series) into common shares on the basis of one common share for each preferred share. Specifically, each series preferred share will become one common share and fractions of preferred shares will be eliminated. The articles of the Company will also be amended to, among other things, delete all of the classes and series of preferred shares from our authorized share capital. Consequently, at the time of the closing of this offering, the Company will be authorized to issue an unlimited number of common shares. The implementation of the foregoing is referred to in this prospectus as the "Reorganization". As of February 28, 2007, after giving effect to the Reorganization, there are 11,079,738 common shares outstanding. Except as otherwise noted, the information in this prospectus, including all information about our outstanding share capital dated within 30 days of this prospectus, assumes the completion of the Reorganization.
The following are the only material contracts, other than those contracts entered into in the ordinary course of business, which we have entered into during the two years before the date of this prospectus.
- 1.
- License agreement effective November 1, 2001 between UBC and OncoGenex relating to clusterin, as amended as of August 30, 2006 referred to under "Business-License and Collaboration Agreements";
- 2.
- License agreement effective November 1, 2001 between UBC and OncoGenex relating to IGFBP-5, as amended as of November 14, 2002, November 23, 2002 and August 30, 2006 referred to under "Business-License and Collaboration Agreements";
- 3.
- License agreement effective September 1, 2002 between UBC and OncoGenex relating to IGFBP-2 and IGFBP-5, as amended as of September 12, 2002, November 14, 2002 and August 30, 2006 referred to under "Business-License and Collaboration Agreements";
- 4.
- License agreement effective April 5, 2005 between UBC and OncoGenex relating to Hsp27, as amended as of August 30, 2006 referred to under "Business-License and Collaboration Agreements";
- 5.
- Investment agreement dated August 10, 2005 among OncoGenex, Ventures West 7 Limited Partnership, Ventures West 7 U.S. Limited Partnership, H.I.G. Horizon Corp., Working Opportunity Fund (EVCC) Ltd., Business Development Bank of Canada and WHI Morula Fund, LLC, pursuant to which we issued 4,157,595 Series 2 Class B preferred shares referred to under "Prior Sales";
- 6.
- Further Amended and Restated Shareholders' Agreement dated August 10, 2005 as amended September 7, 2006 among OncoGenex Technologies Inc. and Ventures West
154
- 7.
- Underwriting agreement dated , 2007 among OncoGenex and the underwriters, referred to under "Underwriting".
7 Limited Partnership, Ventures West 7 U.S. Limited Partnership, H.I.G. Horizon Corp., Working Opportunity Fund (EVCC) Ltd., Business Development Bank of Canada, Milestone Medica Corporation, Martin Gleave, 603356 B.C. Ltd., Scott Cormack and WHI Morula Fund, LLC. Referred to under "Certain Relationships and Related Party Transactions"; and
Copies of the above may be inspected during ordinary office business hours at our registered office, located at 10th Floor, 595 Howe Street, Vancouver, British Columbia, Canada during the period of distribution of our common shares or may be viewed at the website maintained by the SEC at http://www.sec.gov or the website maintained by the Canadian Securities Administrators at http://www.sedar.com. Certain information in the agreements with Isis and UBC available for inspection has been redacted for reasons of confidentiality.
Martin Gleave and Scott Cormack took the initiative in founding and organizing our business and, as a result, each of them may be considered to be a "promoter" under the securities legislation in certain of the provinces of Canada. A description of the nature of the relationship between us and such persons is described under "Certain Relationships and Related Party Transactions".
Subject to compliance with the prudent investment standards and general investment provisions and restrictions of the statutes referred to below (and, where applicable, the regulations made under those statutes) and, in certain cases, subject to the satisfaction of additional requirements relating to investment or lending policies, standards, procedures and goals and, in certain cases, subject to the filing of such policies, standards, procedures or goals, the common shares offered under this prospectus would not, if the date hereof was the closing date, be precluded as investments under the following statutes:
Cooperative Credit Associations Act(Canada);
Insurance Companies Act (Canada);
Pension Benefits Standards Act, 1985 (Canada);
Trust and Loan Companies Act (Canada);
Alberta Heritage Savings Trust Fund Act (Alberta);
Employment Pension Plans Act (Alberta);
Insurance Act (Alberta);
Loan and Trust Corporations Act (Alberta);
Financial Institutions Act (British Columbia);
Pension Benefits Standards Act (British Columbia);
The Insurance Act (Manitoba);
The Pension Benefits Act (Manitoba);
The Trustee Act (Manitoba);
Pension Benefits Act (Nova Scotia);
Trustee Act (Nova Scotia);
Loan and Trust Corporations Act (Ontario);
Pension Benefits Act (Ontario);
Trustee Act (Ontario);
An Act respecting insurance (Québec) (for an insurer, as defined therein, incorporated under the laws of the Province of Québec, other than a guarantee fund);
An Act respecting trust companies and savings companies (Québec) (for a trust company, as defined therein, which invests its own funds and funds received as deposits and a savings company, as defined therein, investing its funds);
Supplemental Pension Plans Act (Québec); and
The Pension Benefits Act, 1992 (Saskatchewan).
155
DuMoulin Black LLP, our Canadian counsel, and Farris, Vaughan, Wills & Murphy LLP, Canadian counsel to the underwriters, are each of the opinion that our common shares, if, as and when they are listed for trading on a prescribed stock exchange within the meaning of the Tax Act, will be qualified investments under the Tax Act for trusts governed by registered retirement savings plans, registered retirement income funds, deferred profit sharing plans and registered education savings plans, each as defined in the Tax Act. For these purposes, each of the Toronto Stock Exchange and the Nasdaq Global Market is a prescribed stock exchange.Investors are advised to consult their own tax advisors.
Securities legislation in certain of the provinces of Canada provides purchasers with the right to withdraw from an agreement to purchase securities. This right may be exercised within two business days after receipt, or deemed receipt, of a prospectus and any amendment. In several of the provinces, the securities legislation further provides a purchaser with remedies for rescission or, in some provinces, damages if the prospectus and any amendment contains a misrepresentation or is not delivered to the purchaser, provided that the remedies for rescission or damages are exercised by the purchaser within the time limit prescribed by the securities legislation of the purchaser's province. The purchaser should refer to any applicable provisions of the securities legislation of the purchaser's province for the particulars of these rights or consult with a legal advisor.
156
ONCOGENEX TECHNOLOGIES INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| Report of Independent Registered Public Accounting Firm | F-2 | |
Consolidated Balance Sheets as of September 30, 2006 (unaudited) and December 31, 2004 and 2005 | F-3 | |
Consolidated Statements of Loss for the nine months ended September 30, 2005 and 2006 (unaudited), the years ended December 31, 2003, 2004 and 2005 and for the period from May 26, 2000 (Date of Inception) to September 30, 2006 (unaudited) | F-4 | |
Consolidated Statements of Shareholders' Deficiency for the nine months ended September 30, 2006 (unaudited) and the years ended December 31, 2003, 2004 and 2005 | F-5 | |
Consolidated Statements of Cash Flows for the nine months ended September 30, 2005 and 2006 (unaudited), the years ended December 31, 2003, 2004 and 2005 and for the period from May 26, 2000 (Date of Inception) to September 30, 2006 (unaudited) | F-7 | |
Notes to Consolidated Financial Statements | F-8 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors
OncoGenex Technologies Inc.
(a development stage enterprise)
We have audited the accompanying consolidated balance sheets ofOncoGenex Technologies Inc. (a development stage enterprise) as of December 31, 2005 and 2004, and the related consolidated statements of loss, shareholders' deficiency, and cash flows for each of the three years in the period ended December 31, 2005. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company's internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the consolidated financial position ofOncoGenex Technologies Inc. (a development stage enterprise) at December 31, 2005 and 2004, and the consolidated results of its operations and its cash flows for each of the three years in the period ended December 31, 2005, in conformity with U.S. generally accepted accounting principles.
As discussed in Note 2(a) to the financial statements, the Company has restated its financial statements to accrue for the preferred return payable upon redemption of its convertible redeemable preferred shares.
| Ernst & Young LLP | ||
Vancouver, Canada, | ||
| February 24, 2006, except for notes 2a, 10, 11 and 12, as to which the date is February 1, 2007 | Chartered Accountants |
F-2
OncoGenex Technologies Inc.
(a development stage enterprise)
(Incorporated under the laws of Canada)
CONSOLIDATED BALANCE SHEETS AS RESTATED — NOTE 2(A)
(In thousands of U.S. dollars)
| | | December 31, | ||||||
|---|---|---|---|---|---|---|---|---|
| | September 30, 2006 | |||||||
| | 2005 | 2004 | ||||||
| | $ | $ | $ | |||||
| | (Unaudited) | | | |||||
| ASSETS | ||||||||
| Current | ||||||||
| Cash and cash equivalents[note 4] | 452 | 1,282 | 1,947 | |||||
| Short-term investments[note 5] | 10,097 | 12,503 | 5,968 | |||||
| Amounts receivable[note 13] | 497 | 384 | 198 | |||||
| Investment tax credit recoverable | 704 | 558 | 478 | |||||
| Prepaid expenses | 249 | 227 | 118 | |||||
| Total current assets | 11,999 | 14,954 | 8,709 | |||||
| Long-term investments[note 6] | 491 | 4,584 | 2,505 | |||||
| Property and equipment[note 9] | 164 | 201 | 172 | |||||
| Deferred share issue costs[note 8] | 867 | — | — | |||||
| Other assets[note 7] | 12 | 11 | 11 | |||||
| Total assets | 13,533 | 19,750 | 11,397 | |||||
LIABILITIES AND SHAREHOLDERS' DEFICIENCY | ||||||||
| Current | ||||||||
| Accounts payable and accrued liabilities[notes 15 and 18] | 2,005 | 917 | 2,004 | |||||
| Funding advances | — | — | 217 | |||||
| Total current liabilities | 2,005 | 917 | 2,221 | |||||
| Tax Payable | 1,354 | 835 | 374 | |||||
| Total liabilities | 3,359 | 1,752 | 2,595 | |||||
Commitments and contingencies[notes 13 and 17] | ||||||||
Class A redeemable convertible preferred shares: no par value; unlimited number authorized; 848,805 shares issued and outstanding at September 30, 2006 and December 31, 2005 and 2004 (aggregate retraction amount of $4,605 at September 30, 2006, $4,157 at December 31, 2005 and $3,728 at December 31, 2004)[note 10] | 3,759 | 3,475 | 3,146 | |||||
| Class B redeemable convertible preferred shares: no par value; unlimited number authorized; 8,945,448 shares issued and outstanding at September 30, 2006 and December 31, 2005 and 4,543,553 at December 31, 2004 (aggregate retraction amount of $30,377 at September 30, 2006, $28,662 at December 31, 2005 and $13,634 at December 31, 2004)[note 10] | 29,978 | 28,350 | 13,385 | |||||
Shareholders' deficiency: | ||||||||
| Common shares: no par value; unlimited number authorized; 1,285,500 shares issued and outstanding at September 30, 2006 and December 31, 2005 and 1,255,500 at December 31, 2004[note 11[b]] | 399 | 399 | 378 | |||||
| Additional paid-in capital[note 11 [d]] | 266 | 122 | 67 | |||||
| Deficit accumulated during the development stage | (26,668 | ) | (16,154 | ) | (9,382 | ) | ||
| Accumulated other comprehensive income | 2,440 | 1,806 | 1,208 | |||||
| Total shareholders' deficiency | (23,563 | ) | (13,827 | ) | (7,729 | ) | ||
| Total liabilities and shareholders' deficiency | 13,533 | 19,750 | 11,397 | |||||
See accompanying notes
F-3
OncoGenex Technologies Inc.
(a development stage enterprise)
CONSOLIDATED STATEMENTS OF LOSS AS RESTATED — NOTE 2(A)
(In thousands of U.S. dollars,
except share and per share amounts)
| | Nine months ended September 30, | Years ended December 31, | | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Period from May 26, 2000 (inception) to September 30, 2006 | ||||||||||||
| | 2006 | 2005 | 2005 | 2004 | 2003 | ||||||||
| | $ | $ | $ | $ | $ | $ | |||||||
| | (Unaudited) | | | | (Unaudited) | ||||||||
| EXPENSES | |||||||||||||
| Research and development[note 14] | 6,822 | 1,857 | 3,143 | 2,778 | 1,381 | 15,502 | |||||||
| General and administrative | 1,553 | 1,128 | 1,523 | 930 | 487 | 4,813 | |||||||
| Total expenses | 8,375 | 2,985 | 4,666 | 3,708 | 1,868 | 20,315 | |||||||
OTHER INCOME (EXPENSE) | |||||||||||||
| Interest income | 357 | 160 | 313 | 199 | 41 | 928 | |||||||
| Other | (111 | ) | (139 | ) | (144 | ) | (156 | ) | (99 | ) | (510 | ) | |
| Total other income (expense) | 246 | 21 | 169 | 43 | (58 | ) | 418 | ||||||
| Loss for the period before taxes | (8,129 | ) | (2,964 | ) | (4,497 | ) | (3,665 | ) | (1,926 | ) | (19,897 | ) | |
| Tax Expense | 473 | 274 | 432 | 346 | 0 | 1,251 | |||||||
| Net Loss | (8,602 | ) | (3,238 | ) | (4,929 | ) | (4,011 | ) | (1,926 | ) | (21,148 | ) | |
| Redeemable convertible preferred share accretion | 1,912 | 1,202 | 1,843 | 1,248 | 385 | 5,520 | |||||||
| Loss attributable to common shareholders | (10,514 | ) | (4,440 | ) | (6,772 | ) | (5,259 | ) | (2,311 | ) | (26,668 | ) | |
Basic and diluted loss per common share[note 11[f]] | $(8.18 | ) | $(3.59 | ) | $(5.43 | ) | $(4.55 | ) | $(2.01 | ) | |||
| Weighted average number of common shares[note 11[f]] | 1,285,500 | 1,235,573 | 1,248,158 | 1,155,500 | 1,148,925 | ||||||||
| Pro forma basic and diluted loss per common share[note 2] | $(0.95 | ) | $(0.60 | ) | $(0.81 | ) | $(0.83 | ) | $(0.87 | ) | |||
| Pro forma weighted average number of common shares[note 2] | 11,079,738 | 7,450,248 | 8,365,079 | 6,374,239 | 2,648,968 | ||||||||
See accompanying notes
F-4
OncoGenex Technologies Inc.
(a development stage enterprise)
CONSOLIDATED STATEMENTS OF SHAREHOLDERS' DEFICIENCY
AS RESTATED — NOTE 2(A)
Period from May 26, 2000 (inception) to September 30, 2006
(Information for the nine months ended September 30, 2006 is unaudited)
| | Common Shares | | Accumulated Other Comprehensive Income (Loss) | | | | |||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Additional Paid-In Capital | Other Comprehensive Income (Loss) | Deficit Accumulated During the Development Stage | Total Shareholders' Deficiency | |||||||||||||||||
| | Shares | Amount | |||||||||||||||||||
| | (in thousands of U.S. dollars, except share amounts) | ||||||||||||||||||||
| Shares issued for cash | 266,000 | $ | 51 | $ | — | $ | — | $ | — | $ | — | $ | 51 | ||||||||
| Shares issued for research and development expenses | 820,000 | — | |||||||||||||||||||
| Cumulative translation adjustment from application of US dollar reporting | (1 | ) | (1 | ) | (1 | ) | |||||||||||||||
| Loss for the period | (6 | ) | (6 | ) | (6 | ) | |||||||||||||||
| Comprehensive loss for the period | (7 | ) | |||||||||||||||||||
| Balance, December 31, 2000 | 1,086,000 | 51 | (1 | ) | (6 | ) | 44 | ||||||||||||||
| Shares issued for cash | 60,000 | 94 | 94 | ||||||||||||||||||
| Shares issued for acquisition of licenses | 100,000 | 156 | 156 | ||||||||||||||||||
| Shares cancelled under terms of issuance | (28,500 | ) | (7 | ) | 7 | ||||||||||||||||
| Cumulative translation adjustment from application of US dollar reporting | 2 | 2 | 2 | ||||||||||||||||||
| Redeemable convertible preferred share accretion | (3 | ) | (3 | ) | |||||||||||||||||
| Loss for the year | (318 | ) | (318 | ) | (318 | ) | |||||||||||||||
| Comprehensive loss for the year | (316 | ) | |||||||||||||||||||
| Balance, December 31, 2001 | 1,217,500 | 294 | 7 | 1 | (327 | ) | (25 | ) | |||||||||||||
| Shares issued for acquisition of licenses | 32,000 | 83 | 83 | ||||||||||||||||||
| Shares issued on exercise of options | 6,000 | 1 | 1 | ||||||||||||||||||
| Cumulative translation adjustment from application of US dollar reporting | 9 | 9 | 9 | ||||||||||||||||||
| Stock-based compensation expense | 5 | 5 | |||||||||||||||||||
| Unrealized gain on marketable securities | 7 | 7 | 7 | ||||||||||||||||||
| Redeemable convertible preferred share accretion | (129 | ) | (129 | ) | |||||||||||||||||
| Loss for the year | (1,356 | ) | (1,356 | ) | (1,356 | ) | |||||||||||||||
| Comprehensive loss for the year | (1,340 | ) | |||||||||||||||||||
| Balance, December 31, 2002 | 1,255,500 | 378 | 12 | 17 | (1,812 | ) | (1,405 | ) | |||||||||||||
| Cumulative translation adjustment from application of US dollar reporting | 417 | 417 | 417 | ||||||||||||||||||
| Stock-based compensation expense | 15 | 15 | |||||||||||||||||||
| Reclassification of unrealized gain on marketable securities | (7 | ) | (7 | ) | (7 | ) | |||||||||||||||
| Unrealized gain on marketable securities | 13 | 13 | 13 | ||||||||||||||||||
| Redeemable convertible preferred share accretion | (385 | ) | (385 | ) | |||||||||||||||||
| Loss for the year | (1,926 | ) | (1,926 | ) | (1,926 | ) | |||||||||||||||
| Comprehensive loss for the year | (1,503 | ) | |||||||||||||||||||
| Balance, December 31, 2003 | 1,255,500 | 378 | 27 | 440 | (4,123 | ) | (3,278 | ) | |||||||||||||
F-5
OncoGenex Technologies Inc.
(a development stage enterprise)
CONSOLIDATED STATEMENTS OF SHAREHOLDERS' DEFICIENCY
AS RESTATED — NOTE 2(A) (Continued)
Period from May 26, 2000 (inception) to September 30, 2006
(Information for the nine months ended September 30, 2006 is unaudited)
| | Common Shares | | Accumulated Other Comprehensive Income (Loss) | | | | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Additional Paid-In Capital | Other Comprehensive Income (Loss) | Deficit Accumulated During the Development Stage | Total Shareholders' Deficiency | |||||||||||
| | Shares | Amount | |||||||||||||
| | (in thousands of U.S. dollars, except share amounts) | ||||||||||||||
| Cumulative translation adjustment from application of US dollar reporting | 723 | 723 | 723 | ||||||||||||
| Stock-based compensation expense | 40 | 40 | |||||||||||||
| Reclassification of unrealized gain on marketable securities | (13 | ) | (13 | ) | (13 | ) | |||||||||
| Unrealized gain on marketable securities | 58 | 58 | 58 | ||||||||||||
| Redeemable convertible preferred share accretion | (1,248 | ) | (1,248 | ) | |||||||||||
| Loss for the year | (4,011 | ) | (4,011 | ) | (4,011 | ) | |||||||||
| Comprehensive loss for the year | (3,243 | ) | |||||||||||||
| Balance, December 31, 2004 | 1,255,500 | 378 | 67 | 1,208 | (9,382 | ) | (7,729 | ) | |||||||
| Shares issued for acquisition of licenses | 30,000 | 21 | 21 | ||||||||||||
| Cumulative translation adjustment from application of US dollar reporting | 619 | 619 | 619 | ||||||||||||
| Stock-based compensation expense | 55 | 55 | |||||||||||||
| Reclassification of unrealized gain on marketable securities | (58 | ) | (58 | ) | (58 | ) | |||||||||
| Unrealized gain on marketable securities | 37 | 37 | 37 | ||||||||||||
| Redeemable convertible preferred share accretion | (1,843 | ) | (1,843 | ) | |||||||||||
| Loss for the year | (4,929 | ) | (4,929 | ) | (4,929 | ) | |||||||||
| Comprehensive loss for the year | (4,331 | ) | |||||||||||||
| Balance, December 31, 2005 | 1,285,500 | 399 | 122 | 1,806 | (16,154 | ) | (13,827 | ) | |||||||
| Stock-based compensation expense | 144 | 144 | |||||||||||||
| Cumulative translation adjustment from application of US dollar reporting | 680 | 680 | 680 | ||||||||||||
| Reclassification of unrealized gain on marketable securities | (37 | ) | (37 | ) | (37 | ) | |||||||||
| Unrealized loss on marketable securities | (9 | ) | (9 | ) | (9 | ) | |||||||||
| Redeemable convertible preferred share accretion | (1,912 | ) | (1,912 | ) | |||||||||||
| Loss for the period | (8,602 | ) | (8,602 | ) | (8,602 | ) | |||||||||
| Comprehensive loss for the period | (7,968 | ) | |||||||||||||
| Balance, September 30, 2006 | 1,285,500 | 399 | 266 | 2,440 | (26,668 | ) | (23,563 | ) | |||||||
See accompanying notes
F-6
OncoGenex Technologies Inc.
(a development stage enterprise)
CONSOLIDATED STATEMENTS OF CASH FLOWS AS RESTATED — NOTE 2(A)
(In thousands of U.S. dollars)
| | Nine months ended September 30, | | | | Period from May 26, 2000 (inception) to September 30, 2006 | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Years ended December 31, | |||||||||||||
| | 2006 | 2005 | 2005 | 2004 | 2003 | |||||||||
| | $ | $ | $ | $ | $ | $ | ||||||||
| | (Unaudited) | | | | (Unaudited) | |||||||||
| OPERATING ACTIVITIES | ||||||||||||||
| Loss for the period | (8,602 | ) | (3,238 | ) | (4,929 | ) | (4,011 | ) | (1,926 | ) | (21,148 | ) | ||
| Add items not involving cash | ||||||||||||||
| Amortization | 72 | 51 | 73 | 67 | 14 | 230 | ||||||||
| Stock-based collaboration expense | — | 771 | 771 | — | 748 | 1,758 | ||||||||
| Stock-based compensation[note 11[d]] | 144 | 40 | 55 | 40 | 15 | 260 | ||||||||
| Changes in non-cash working capital items | ||||||||||||||
| Amounts receivable | (112 | ) | (156 | ) | (186 | ) | 3 | (191 | ) | (497 | ) | |||
| Investment tax credit recoverable | (146 | ) | (260 | ) | (80 | ) | (6 | ) | (320 | ) | (704 | ) | ||
| Prepaid expenses | (22 | ) | (659 | ) | (109 | ) | (71 | ) | 26 | (250 | ) | |||
| Other assets | — | — | — | (11 | ) | — | (12 | ) | ||||||
| Accounts payable and accrued liabilities | 563 | (852 | ) | (1,087 | ) | 1,386 | 328 | 1,479 | ||||||
| Funding advances | — | (217 | ) | (217 | ) | (5 | ) | (8 | ) | — | ||||
| Changes in taxes payable | 519 | 303 | 461 | 374 | — | 1,355 | ||||||||
| Cash used in operating activities | (7,584 | ) | (4,217 | ) | (5,248 | ) | (2,234 | ) | (1,313 | ) | (17,529 | ) | ||
FINANCING ACTIVITIES | ||||||||||||||
| Issuance of preferred shares, net of share issue costs | — | 12,701 | 12,701 | 5,818 | 5,712 | 26,719 | ||||||||
| Issuance of common shares, net of share issue costs | — | — | — | — | — | 146 | ||||||||
| Deferred share issue costs | (342 | ) | — | — | — | — | (342 | ) | ||||||
| Cash provided by (used in) financing activities | (342 | ) | 12,701 | 12,701 | 5,818 | 5,712 | 26,523 | |||||||
INVESTING ACTIVITIES | ||||||||||||||
| Purchase of investments | (14,413 | ) | (20,172 | ) | (27,088 | ) | (19,545 | ) | (10,508 | ) | (73,734 | ) | ||
| Proceeds from investments | 21,528 | 11,319 | 19,043 | 16,288 | 7,674 | 65,492 | ||||||||
| Purchase of property and equipment | (27 | ) | (36 | ) | (96 | ) | (156 | ) | (64 | ) | (359 | ) | ||
| Cash provided by (used in) investing activities | 7,088 | (8,889 | ) | (8,141 | ) | (3,413 | ) | (2,898 | ) | (8,601 | ) | |||
Effect of exchange rate changes on cash | 8 | (63 | ) | 23 | 112 | (100 | ) | 59 | ||||||
| Increase (decrease) in cash and cash equivalents during the period | (830 | ) | (468 | ) | (665 | ) | 283 | 1,401 | 452 | |||||
| Cash and cash equivalents, beginning of the period | 1,282 | 1,947 | 1,947 | 1,664 | 263 | — | ||||||||
| Cash and cash equivalents, end of the period | 452 | 1,479 | 1,282 | 1,947 | 1,664 | 452 | ||||||||
Supplemental cash flow information [note 11[b]].
See accompanying notes
F-7
OncoGenex Technologies Inc.
(a development stage enterprise)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
AS RESTATED — NOTE 2(A)
(in U.S. dollars)
Information as at September 30, 2006 and 2005 and for the nine months
ended September 30, 2006 and 2005 is unaudited.
1. NATURE OF BUSINESS AND BASIS OF PRESENTATION
OncoGenex Technologies Inc. (the "Company") is a development stage enterprise incorporated on May 26, 2000 under the Canada Business Corporations Act and is registered as an extraprovincial company in the province of British Columbia. The Company's principal business activities include the development and commercialization of the Company's technologies for the treatment of cancer.
The Company has financed, and expects to continue to finance, its cash requirements primarily from share issuances. The Company's ability to realize the carrying value of its assets is dependent on successfully bringing its technologies to market and achieving future profitable operations, the outcome of which cannot be predicted at this time. The Company's lead drug candidate, OGX-011, is being co-developed with the Company's partner, Isis Pharmaceuticals Inc. ("Isis") [note 13]. Substantially all of the Company's research and development activities to date have focused on OGX-011.
These consolidated financial statements include the accounts of the Company and its wholly owned subsidiary, OncoGenex Inc., which was incorporated on August 19, 2005. Inter-company accounts and transactions have been eliminated.
The accompanying interim consolidated balance sheet as at September 30, 2006, the consolidated statements of loss and cash flows for the nine month periods ended September 30, 2006 and 2005 and for the period from May 26, 2000 (inception) to September 30, 2006 and the consolidated statement of shareholders' deficiency for the nine month period ended September 30, 2006 are unaudited. These unaudited interim consolidated financial statements have been prepared in accordance with United States generally accepted accounting principles. In the opinion of the Company's management, the unaudited interim consolidated financial statements have been prepared on the same basis as the audited consolidated financial statements and include all adjustments (which include reclassifications and normal recurring adjustments) necessary for the fair presentation of the Company's financial position, results of operations and cash flows as at September 30, 2006 and for the nine month periods ended September 30, 2006 and 2005 and for the period from May 26, 2000 (inception) to September 30, 2006. The results for the nine month period ended September 30, 2006 are not necessarily indicative of the results to be expected for the year ending December 31, 2006.
F-8
2. RESTATEMENT AND ACCOUNTING POLICIES
a) Restatement
The Company has restated its financial statements to accrue for the preferred return payable upon redemption of its convertible redeemable preferred stock. In its previously issued financial statements, the Company did not accrue the preferred return payable upon redemption over the period from issuance to the redemption date. The financial statements have been restated to reflect the accretion of the preferred return over the period from issuance to the redemption date. The accretion of the preferred return is charged to the deficit and increases the net loss attributable to common stockholders for purposes of calculating net loss per share. In Canada, the preferred return is subject to Part VI.1 tax. This tax expense increases net loss.
The impact of this change on previously issued financial statements in thousands of U.S. dollars, except per share amounts is as follows:
| | September 30, | December 31, | |||||||
|---|---|---|---|---|---|---|---|---|---|
| | 2006 | 2005 | 2005 | 2004 | |||||
| | (Unaudited) | (Unaudited) | | | |||||
| Convertible redeemable preferred shares | 5,520 | 3,608 | 1,765 | ||||||
| Deficit | (6,771 | ) | (4,386 | ) | (2,111 | ) | |||
| | Nine Months ended September 30, | Years ended December 31, | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2006 | 2005 | 2005 | 2004 | 2003 | |||||||||||
| | (Unaudited) | (Unaudited) | | | | |||||||||||
| Net loss | (473 | ) | (274 | ) | (432 | ) | (346 | ) | — | |||||||
| Loss attributable to common shareholders | (2,385 | ) | (1,476 | ) | (2,275 | ) | (1,594 | ) | (385 | ) | ||||||
| Basic and diluted loss per common share | $ | (1.86 | ) | $ | (1.19 | ) | $ | (1.83 | ) | $ | (1.38 | ) | $ | (0.33 | ) | |
| Pro forma basic and diluted loss per common share | $ | (0.22 | ) | $ | (0.20 | ) | $ | (0.27 | ) | $ | (0.26 | ) | $ | (0.14 | ) | |
b) Accounting Policies
The accompanying consolidated financial statements have been prepared in accordance with United States generally accepted accounting principles and are expressed in U.S. dollars unless otherwise noted. The following is a summary of significant accounting policies used in the preparation of these consolidated financial statements.
F-9
Use of estimates
The preparation of consolidated financial statements in conformity with United States generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the consolidated financial statements and notes thereto. Actual results could differ from those estimates.
Cash and cash equivalents
The Company considers all highly liquid investments with an original maturity of three months or less to be cash equivalents, which are carried at market value with unrealized gains and losses, if any, reported as accumulated other comprehensive income or loss, which is a separate component of shareholders' deficiency.
Short-term investments
Short-term investments consist of financial instruments purchased with an original maturity of greater than three months and less than one year. The Company considers its short-term investments as available-for-sale and they are carried at market value with unrealized gains and losses, if any, reported as accumulated other comprehensive income or loss, which is a separate component of shareholders' deficiency. Realized gains and losses on the sale of these securities are recognized in net income or loss. The cost of investments sold is based on the specific identification method.
Long-term investments
Long-term investments consist of financial instruments purchased with an original maturity of greater than one year. The Company considers its long-term investments as available-for-sale and they are carried at market value with unrealized gains and losses, if any, reported as accumulated other comprehensive income or loss, which is a separate component of shareholders' deficiency. Realized gains and losses on the sale of these securities are recognized in net income or loss. The cost of investments sold is based on the specific identification method.
F-10
Property and equipment
Property and equipment assets are recorded at cost less accumulated amortization. Amortization is provided on a straight-line basis over the following periods:
| Computer equipment | 3 years | |
| Computer software | 3 years | |
| Furniture and fixtures | 5 years | |
| Leasehold improvements | Over the term of the lease |
Reporting currency and foreign currency translation
The Company follows the temporal method for the translation of foreign currency amounts including those of its foreign integrated subsidiary, into Canadian dollars. Under this method, monetary assets and liabilities denominated in foreign currencies are translated into Canadian dollars using exchange rates in effect at the balance sheet date. Revenue and expense items are translated at the exchange rate in effect on the date of the transaction. Foreign exchange gains and losses are included in the determination of loss for the period.
The consolidated financial statements are based on a Canadian dollar functional currency and have been translated into the U.S. reporting currency using the current rate method as follows: assets and liabilities using the rate of exchange prevailing at the balance sheet date; shareholders' deficiency using the applicable historic rate; and revenue and expenses at the average rate of exchange for the respective periods. Translation gains and losses have been included as part of the cumulative translation adjustment which is reported as a component of accumulated other comprehensive loss.
Funding advances
Funds received in advance for specific projects are classified as funding advances. Costs related to these projects are charged against the funding advances as they are incurred and shown as a reduction of research and development costs.
Income taxes
Income taxes are accounted for under the liability method. Deferred tax assets and liabilities are recognized for the differences between the carrying values of assets and liabilities and their respective income tax bases and for operating losses and tax credit carry forwards. A valuation
F-11
allowance is provided for the portion of deferred tax assets that is more likely than not to be unrealized. Deferred tax assets and liabilities are measured using the enacted tax rates and laws.
Investment tax credits
The benefits of investment tax credits for scientific research and development expenditures are recognized in the year the qualifying expenditure is made providing there is reasonable assurance of recoverability. The investment tax credits recorded are based on management's estimates of amounts expected to be recovered and are subject to audit by taxation authorities. The investment tax credit reduces the carrying cost of expenditures for research and development expenses to which it relates.
Research and development costs
Research and development costs are expensed in the year incurred.
Stock-based compensation
Effective January 1, 2006, the Company adopted the fair value recognition provisions of the Financial Accounting Standards Board Statement No. 123(R) (or SFAS 123(R)), "Share-Based Payment", using the modified prospective method with respect to options granted to employees and directors. Under this transition method, compensation cost is recognized in the financial statements beginning with the effective date for all share-based payments granted after January 1, 2006 and for all awards granted prior to but not yet vested as of January 1, 2006. The expense is amortized on a straight-line basis over the vesting period. Accordingly, prior period amounts have not been restated.
As a result of adopting SFAS 123(R) on January 1, 2006, the Company's loss and basic and diluted loss per common share for the nine months ended September 30, 2006 is $118,000 and $0.09 higher, respectively than if it had continued to account for employee and director share based compensation under APB Opinion No. 25.
Prior to January 1, 2006, the Company accounted for stock options granted to employees and directors under the recognition and measurement provisions of APB Opinion No. 25 (or APB 25), "Accounting for Stock Issued to Employees", as amended by SFAS No. 148, using the intrinsic value method, as permitted by Statement of Financial Accounting Standards No. 123 (or SFAS 123), "Accounting for Stock-Based Compensation". As the exercise price of the Company's employee stock options equals the estimated fair value of the underlying stock on the date of grant, no compensation expense has been recognized under APB 25.
F-12
The Company accounts for stock-based awards issued to non-employees prior to January 1, 2006 in accordance with Statement of Financial Accounting Standards No. 123, Accounting for Stock-Based Compensation, as amended by SFAS No. 148. Stock-based awards for non-employees are measured at the fair value of the equity instruments issued using the Black-Scholes option pricing model. The fair value of stock options granted is amortized to the consolidated statement of operations over the vesting period.
The Company discloses the proforma effects to the loss for periods prior to the adoption of FAS 123(R) as if the fair value method had been used for awards to employees and directors granted, modified or settled prior to December 31, 2005 (see Note 11(d)).
Comprehensive income (loss)
Comprehensive income (loss) is comprised of net income (loss) and other comprehensive income (loss). Other comprehensive income (loss) consists of translation adjustments from the application of U.S. dollar reporting and unrealized gains and losses on the Company's available-for-sale marketable securities. The Company has reported the components of comprehensive loss in the statement of shareholders' deficiency.
Loss per common share
Basic loss per common share is computed using the weighted average number of common shares outstanding during the period, excluding contingently issuable shares, if any. Diluted net loss per common share is computed in accordance with the treasury stock method which uses the weighted average number of common shares outstanding during the period and includes the dilutive effect of potentially issuable common shares from outstanding stock options and convertible preferred shares. Diluted loss per common share is equivalent to basic loss per common share for all periods presented as the outstanding stock options and convertible preferred shares are anti-dilutive.
The Company has filed a prospectus/registration statement with Canadian and United States regulatory authorities to sell common shares to the public. If the initial public offering contemplated by this prospectus is consummated, all of the 9,794,253 convertible redeemable preferred shares outstanding as at September 30, 2006, will automatically convert on a one-for-one basis into 9,794,238 common shares (after adjusting for fractional shares). The pro forma basic and diluted loss per common share calculations assume the conversion of the convertible redeemable preferred shares into common shares using the as-if-converted method as at date of issue.
F-13
Recent accounting pronouncements
In July 2006, the Financial Accounting Standards Board (orFASB) issued Interpretation No. 48, "Accounting for Uncertainty in Income Taxes — an interpretation of FASB Statement No. 109" (orFIN 48). This interpretation clarifies the accounting for uncertainty in income taxes recognized in financial statements in accordance with FASB Statement No. 109, "Accounting for Income Taxes". FIN 48 will require companies to determine whether it is more-likely-than-not that a tax position taken or expected to be taken in a tax return will be sustained upon examination, including resolution of any related appeals or litigation processes, based on the technical merits of the position. If a tax position meets the more-likely-than-not recognition threshold, it is measured to determine the amount of benefit to recognize in the financial statements based on guidance in the interpretation. FIN 48 is effective for fiscal years beginning after December 15, 2006. The Company has not determined the effect, if any, that the adoption of FIN 48 will have on the Company's consolidated financial position or results of operations.
3. FINANCIAL INSTRUMENTS AND RISK
For certain of the Company's financial instruments including cash and cash equivalents, amounts receivable and accounts payable, the carrying values approximate fair value due to their short-term nature. The Company's short-term investments and long-term investments are recorded at fair value.
Financial risk is the risk to the Company's results of operations that arises from fluctuations in interest rates and foreign exchange rates and the degree of volatility of these rates as well as credit risk associated with the financial stability of the issuers of the financial instruments. The Company purchases the majority of its goods and services in Canadian dollars and maintains the majority of its cash, cash equivalents, short-term investments and long-term investments in Canadian dollars. Accordingly, the Company has minimal exposure to foreign exchange risk. The Company's cash and cash equivalents, short-term investments and long-term investments are invested in fixed rate securities.
4. CASH AND CASH EQUIVALENTS
Cash equivalents include treasury bills and commercial paper. The balance as of September 30, 2006 was $241,000 [December 31, 2005 — $890,000; 2004 — $1,141,000] with average interest rates of 4.62% at September 30, 2006, 2.11% at December 31, 2005 and 1.58% at December 31, 2004.
F-14
5. SHORT-TERM INVESTMENTS
Short-term investments are comprised of treasury bills and commercial paper with an average interest rate of 4.34% [December 31, 2005 — 2.55%; 2004 — 2.11%] and maturities to March 2007. At September 30, 2006, the short-term investments with a cost of $10,098,000 [December 31, 2005 — $12,454,000; 2004 — $5,913,000] are recorded at fair value of $10,097,000 [December 31, 2005 — $12,503,000; 2004 — $5,968,000], with a corresponding net unrealized loss of $1,000 [December 31, 2005 — gain of $49,000; 2004 — gain of $55,000] based on quoted market prices as follows:
| | Cost | Gross Unrealized Gains | Gross Unrealized Losses | Fair Value | ||||
|---|---|---|---|---|---|---|---|---|
| | $ | $ | $ | $ | ||||
| | (In thousands) | |||||||
| September 30, 2006 | 10,098 | — | 1 | 10,097 | ||||
| December 31, 2005 | 12,454 | 49 | — | 12,503 | ||||
| December 31, 2004 | 5,913 | 55 | — | 5,968 | ||||
6. LONG-TERM INVESTMENTS
Long-term investments are comprised of commercial paper with an average interest rate of 2.63% [December 31, 2005 — 2.97%; 2004 — 5.95%] and maturities to November 2006. At September 30, 2006 the long-term investments with a cost of $498,000 [December 31, 2005 — $4,596,000; 2004 — $2,502,000] are recorded at fair value of $491,000 [December 31, 2005 — $4,584,000; 2004 — $2,505,000], with a corresponding net unrealized loss of $7,000 [December 31, 2005 — loss of $12,000; 2004 — gain of $3,000] based on quoted market prices as follows:
| | Cost | Gross Unrealized Gains | Gross Unrealized Losses | Fair Value | ||||
|---|---|---|---|---|---|---|---|---|
| | $ | $ | $ | $ | ||||
| | (In thousands) | |||||||
| September 30, 2006 | 498 | — | 7 | 491 | ||||
| December 31, 2005 | 4,596 | 28 | 40 | 4,584 | ||||
| December 31, 2004 | 2,502 | 6 | 3 | 2,505 | ||||
7. OTHER ASSETS
Other assets include a deposit paid for the Canadian office space as per the operating lease agreement which expires in September 2009.
F-15
8. DEFERRED SHARE ISSUE COSTS
Deferred share issue costs comprise of costs associated with the issuance of share capital. Share issue costs incurred prior to the issuance of share capital are deferred and applied against the proceeds when the shares are issued. In the event the issuance is not successful, such costs are expensed.
9. PROPERTY AND EQUIPMENT
| | Cost | Accumulated Amortization | Net Book Value | |||
|---|---|---|---|---|---|---|
| | $ | $ | $ | |||
| | (In thousands) | |||||
| September 30 2006 | ||||||
| Computer equipment | 166 | 105 | 61 | |||
| Computer software | 103 | 76 | 27 | |||
| Furniture and fixtures | 102 | 33 | 69 | |||
| Leasehold improvements | 46 | 39 | 7 | |||
| 417 | 253 | 164 | ||||
December 31 2005 | ||||||
| Computer equipment | 140 | 70 | 70 | |||
| Computer software | 97 | 50 | 47 | |||
| Furniture and fixtures | 92 | 17 | 75 | |||
| Leasehold improvements | 44 | 35 | 9 | |||
| 373 | 172 | 201 | ||||
December 31 2004 | ||||||
| Computer equipment | 99 | 33 | 66 | |||
| Computer software | 87 | 20 | 67 | |||
| Furniture and fixtures | 45 | 6 | 39 | |||
| Leasehold improvements | 33 | 33 | — | |||
| 264 | 92 | 172 | ||||
F-16
10. REDEEMABLE CONVERTIBLE PREFERRED SHARES
[a] Authorized
Unlimited number of Class A preferred voting shares, issuable in series, no par value
Unlimited number of Class B preferred voting shares, issuable in series, no par value
From December 2001 through October 2002, the Company issued 848,805 Class A Series 1 and 2 Redeemable Convertible Preferred Shares for net proceeds of $2,488,000. From September 2003 through August 2005, the Company issued 8,945,448 Class B Series 1 and 2 Redeemable Convertible Preferred Shares for net proceeds of $25,729,000, consisting of cash of $24,231,000 and payment of collaboration expenses of $1,498,000.
The Class A preferred shares, Series 1 and Series 2, and the Class B preferred shares, Series 1 and Series 2, are convertible at any time at the option of the holder into common shares. Pursuant to the share rights of these shares (see note 19[b] all preferred shares will automatically convert into common shares if the Company completes an initial public offering of the Company's common shares at an offering price per Common Share of not less than $9.21 per share resulting in not less than $25 million net proceeds (including treasury and secondary shares) and which results in the Common Shares being listed and posted for trading on the Toronto Stock Exchange, New York Stock Exchange or quoted on the NASDAQ National Market. All preferred shares will convert, initially on a one for one basis and adjusted thereafter for capital alterations [see note 11[e][i]].
The Class B preferred shares, Series 1 and Series 2 are retractable, subject to theCanada Business Corporations Act, at any time after August 10, 2010 and on not less than 120 days notice by holders of not less than 50% of the outstanding respective Class B preferred shares, Series 1 or Series 2. In the event of any liquidation, dissolution, or winding up of the Company, the Class B preferred shareholders have a liquidation preference senior to the Class A shareholders and Common shareholders and are entitled to receive $2.755 per share for Class B Series 1 shares and $3.07 per share for Class B Series 2 shares.
The Class A preferred shares, Series 1 and Series 2, are retractable, subject to theCanada Business Corporations Act, at the option of the holder behind the Class B preferred shares with such right becoming effective after August 10, 2010 and on not less than 120 days notice by holders of not less than 50% of the outstanding respective Class A preferred shares, Series 1 or Series 2, and provided that no Class B preferred shares, Series 1 and Series 2, are then outstanding. In the event of any liquidation, dissolution, or winding up of the Company, the Class A preferred shareholders have a liquidation preference senior to the Common shareholders
F-17
and are entitled to receive CAD$4.42 per share for Class A Series 1 shares and CAD$5.36 per share for Class A Series 2 shares.
Any remaining assets available for distribution, subsequent to the liquidation payments to Class A and Class B preferred shareholders, shall be distributed ratably among the holders of all share classes as if all shares had been converted to common shares.
The retraction price for the Class A preferred shares, Series 1 and Series 2, and the Class B preferred shares, Series 1 and Series 2 is equal to the issue price for such shares plus a preferred return adjustment (being an amount required to generate an 8% annual cumulative return for the holder of such shares).
If dividends are declared on common shares, Class A and Class B preferred shareholders are entitled to receive a dividend based upon the number of common shares they would receive if they elected to convert their preferred shares into common shares.
In the event that holders of Class A and Class B preferred shares are paid the cumulative preferred return adjustment referred to above, the Company would become liable for payment of taxes under Part VI.1 of the Income Tax Act (Canada) which is calculated at 25% of the amount paid in excess of Cdn $500,000. On the payment of this tax, the Company will be entitled to claim a deduction equal to nine-fourths times the amount of any Part VI.1 taxes actually paid.
For accounting purposes, the Preferred Shares are presented as mezzanine equity in the consolidated financial statements as the shares are redeemable at the option of the holders.
[b] Issued and outstanding
A summary of the preferred share transactions is as follows:
| | Shares | Amount | ||
|---|---|---|---|---|
| | # | $ | ||
| | | (In thousands of U.S. dollars, except share amounts) | ||
| Class A preferred — Series 1 | ||||
| Balance, December 31, 2000 | — | — | ||
| Issued for cash, net of issue costs of $58 | 475,113 | 1,267 | ||
| Accretion of redeemable convertible preferred shares | 3 | |||
| Balance, December 31, 2001 | 475,113 | 1,270 | ||
| Issued for cash, net of issue costs of $14 | 38,281 | 92 | ||
F-18
| Accretion of redeemable convertible preferred shares | 112 | |||
| Balance, December 31, 2002 | 513,394 | 1,474 | ||
| Accretion of redeemable convertible preferred shares | 140 | |||
| Balance, December 31, 2003 | 513,394 | 1,614 | ||
| Accretion of redeemable convertible preferred shares | 163 | |||
| Balance, December 31, 2004 | 513,394 | 1,777 | ||
| Accretion of redeemable convertible preferred shares | 189 | |||
| Balance, December 31, 2005 | 513,394 | 1,966 | ||
| Accretion of redeemable convertible preferred shares | 164 | |||
| Balance, September 30, 2006 | 513,394 | 2,130 | ||
Class A preferred — Series 2 | ||||
| Balance, December 31, 2001 | — | — | ||
| Issued for cash, net of issue costs of $17 | 335,411 | 1,129 | ||
| Accretion of redeemable convertible preferred shares | 17 | |||
| Balance, December 31, 2002 | 335,411 | 1,146 | ||
| Accretion of redeemable convertible preferred shares | 102 | |||
| Balance, December 31, 2003 | 335,411 | 1,248 | ||
| Accretion of redeemable convertible preferred shares | 121 | |||
| Balance, December 31, 2004 | 335,411 | 1,369 | ||
| Accretion of redeemable convertible preferred shares | 140 | |||
| Balance, December 31, 2005 | 335,411 | 1,509 | ||
| Accretion of redeemable convertible preferred shares | 120 | |||
| Balance, September 30, 2006 | 335,411 | 1,629 | ||
F-19
Class B preferred — Series 1 | ||||
| Balance, December 31, 2002 | — | — | ||
| Issued for cash, net of issue costs of $193 | 2,153,354 | 5,712 | ||
| Issued pursuant to collaboration agreement | 272,232 | 748 | ||
| Accretion of redeemable convertible preferred shares | 143 | |||
| Balance, December 31, 2003 | 2,425,586 | 6,603 | ||
| Issued for cash, net of issue costs of $9 | 2,117,967 | 5,818 | ||
| Accretion of redeemable convertible preferred shares | 964 | |||
| Balance, December 31, 2004 | 4,543,553 | 13,385 | ||
| Accretion of redeemable convertible preferred shares | 1,090 | |||
| Balance, December 31, 2005 | 4,543,553 | 14,475 | ||
| Accretion of redeemable convertible preferred shares | 873 | |||
| Balance, September 30, 2006 | 4,543,553 | 15,348 | ||
Class B preferred — Series 2 | ||||
| Balance, December 31, 2004 | — | — | ||
| Issued for cash, net of issue costs of $66 | 4,157,595 | 12,701 | ||
| Issued pursuant to collaboration agreement | 244,300 | 750 | ||
| Accretion of redeemable convertible preferred shares | 424 | |||
| Balance, December 31, 2005 | 4,401,895 | 13,875 | ||
| Accretion of redeemable convertible preferred shares | 755 | |||
| Balance, September 30, 2006 | 4,401,895 | 14,630 | ||
F-20
| Totals, December 31, 2001 | 475,113 | 1,270 | ||
| Totals, December 31, 2002 | 848,805 | 2,620 | ||
| Totals, December 31, 2003 | 3,274,391 | 9,465 | ||
| Totals, December 31, 2004 | 5,392,358 | 16,531 | ||
| Totals, December 31, 2005 | 9,794,253 | 31,835 | ||
| Total September 30, 2006 | 9,794,253 | 33,737 | ||
11. COMMON SHARES
[a] Authorized
Unlimited number of common voting shares, no par value
[b] Issued and outstanding shares
During the year ended December 31, 2005, the Company obtained a license to certain technologies from the University of British Columbia. Under the terms of the agreement, the Company issued a total of 30,000 Common shares at a value of $21,000. This amount has been included in the research and development expenses for the year ended December 31, 2005.
As at December 31, 2005 nil [2004 and 2003 - 100,000] common shares of the Company were held in escrow, the release of which was subject to the achievement of certain milestones stipulated within a service agreement.
[c] Stock options(All options are exercisable in Canadian Dollars)
In September 2003, the Board of Directors approved an amended stock option plan, which was an amendment of the stock option plan first established in October 2001. Under such plan, the Company may grant options to purchase common shares in the Company to employees, directors, officers, and consultants of the Company. The exercise price of the options is determined by the Board but generally will be at least equal to the fair value of the shares at the
F-21
grant date. In December 2006, the shareholders approved the 2006 Stock Incentive Plan ("2006 Plan") which provides, among other matters, the granting of options to acquire common shares equal to the greater of 1,905,557 common shares and 10% of the issued and outstanding common shares. The 2006 Plan is only effective upon the completion of an initial public offering on or before December 31, 2007 and ceases to be in force if an initial public offering is not completed by that date. Accordingly, as of September 30, 2006, no options have been granted pursuant to this plan.
The options vest in accordance with terms as determined by the Board, typically over three years. The expiry date for each option is set by the Board with a maximum expiry date of seven years and a minimum expiry of five years from the date of grant.
As at September 30, 2006 the Company has reserved, pursuant to the 2001 plan, 1,905,557 [December 31, 2005 - 1,905,557] common shares for issuance of stock options to employees, directors, officers and consultants of the Company of which 481,410 [December 31, 2005 - 572,837] are available for future issuance.
During the year ended December 31, 2005 the Company repriced 189,600 options originally granted in 2002 and 2003 from exercise prices of $4.00 to $5.00 to $0.90 in order to be consistent with the option pricing model used to price options granted subsequently. The impact of the repricing was not significant.
F-22
Stock option transactions and the number of share options outstanding are summarized below:
| Exercisable in Canadian Dollars | Number of Optioned Common Shares | Weighted Average Exercise Price | ||
|---|---|---|---|---|
| | # | $ | ||
| Balance, December 31, 2002 | 224,800 | 0.85 | ||
| Options granted | 357,170 | 0.90 | ||
| Balance, December 31, 2003 | 581,970 | 0.88 | ||
| Options granted | 257,250 | 0.90 | ||
| Options forfeited | (54,000 | ) | 0.90 | |
| Balance, December 31, 2004 | 785,220 | 0.88 | ||
| Options granted | 599,500 | 0.95 | ||
| Options forfeited | (52,000 | ) | 0.90 | |
| Balance, December 31, 2005 | 1,332,720 | 0.91 | ||
| Options granted | 117,427 | 0.95 | ||
| Options forfeited | (26,000 | ) | 0.90 | |
| Balance, September 30, 2006 | 1,424,147 | 0.92 | ||
The following table summarizes information about options outstanding at September 30, 2006:
| | Option Outstanding | Option Exercisable | ||||||
|---|---|---|---|---|---|---|---|---|
| Exercise Price | Issuable | Remaining Contractual Life (years) | Exercisable | Exercise Price | ||||
| $ | # | | # | $ | ||||
| Exercisable in Canadian dollars | | | | | ||||
| 0.30 | 20,000 | 2.1 | 20,000 | 0.30 | ||||
| 0.90 | 692,220 | 4.0 | 610,971 | 0.90 | ||||
| 0.95 | 711,927 | 6.2 | 286,177 | 0.95 | ||||
| 0.92 | 1,424,147 | 5.1 | 917,148 | 0.89 | ||||
F-23
[d] Stock-based compensation(exercisable in Canadian dollars)
The estimated fair value of options granted to consultants since inception, is amortized to expense over the vesting period of the stock options resulting in compensation expense of $15,000, $40,000 and $55,000 for the years ended December 31, 2003, 2004 and 2005 respectively. For the nine months ended September 30, 2006 total stock-based compensation expense for both consultants and employees and directors amounted to $144,000.
The weighted average fair value of stock options granted during the nine month period ended September 30, 2006 was $0.72 per share [December 31, 2005 — $0.62; 2004 — $0.74 and 2003 — $0.78]. The estimated fair value of stock options granted in the respective periods was determined using the Black-Scholes option pricing model using the following weighted average assumptions:
| | | December 31 | ||||||
|---|---|---|---|---|---|---|---|---|
| | September 30, 2006 | |||||||
| | 2005 | 2004 | 2003 | |||||
| Annualized volatility | 100% | 100% | 100% | 100% | ||||
| Risk-free interest rate | 4.09% | 3.61% | 4.37% | 4.53% | ||||
| Expected life | 5 years | 5 years | 7 years | 7 years | ||||
| Dividend yield | 0.0% | 0.0% | 0.0% | 0.0% | ||||
As at September 30, 2006, the total unrecognized compensation expense related to stock options granted is $214,000, which is expected to be recognized into expense over a period of approximately three years.
Pro-forma disclosure is required to reflect the impact on the Company had it elected to adopt the fair value method of accounting for options granted to employees and directors since inception. If the computed fair values of stock options granted since inception to December 31,
F-24
2005 had been amortized to expense over their vesting periods, the loss and basic and diluted loss per common share would have been:
| | Nine Month period ended September 30, 2005 | Years ended December 31 | |||||||
|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2004 | 2003 | ||||||
| | $ | $ | $ | $ | |||||
| | (In thousands except for share amounts) | ||||||||
| Loss attributable to common shareholders as reported | (4,440 | ) | (6,772 | ) | (5,259 | ) | (2,311 | ) | |
| Add stock based compensation expense related to consultants included in loss | 40 | 55 | 40 | 15 | |||||
| Deduct stock-based compensation for all awards | (195 | ) | (262 | ) | (152 | ) | (57 | ) | |
| Pro forma loss for the period | (4,595 | ) | (6,979 | ) | (5,371 | ) | (2,353 | ) | |
| Basic and diluted loss per common share | |||||||||
| As reported | (3.59 | ) | (5.43 | ) | (4.55 | ) | (2.01 | ) | |
| Pro forma | (3.72 | ) | (5.59 | ) | (4.65 | ) | (2.05 | ) | |
[e] Anti-dilution
- [i]
- Pursuant to the Company's share provisions, if the Company issues common shares, securities to purchase or acquire common shares, or securities convertible, exercisable into or exchangeable for common shares at a price less than the then current conversion price of the Class A preferred shares, Series 1 and 2 or of the Class B preferred shares, Series 1 and 2, the then-existing conversion price is reduced to a lower amount calculated in accordance with a formula defined in the share provisions. The current conversion price of the Class A preferred shares, Series 1 and 2 and the Class B preferred shares, Series 1, is $2.755. The current conversion price of the Class B preferred shares, Series 2, is $3.07.
- [ii]
- One investor, being a registered venture capital corporation under the Small Business Venture Capital Act (British Columbia) ("SBVCA"), has a put clause allowing it to require the Company to repurchase all of its Class A preferred shares (Series 1: 40,724; Series 2: 22,388) only if the Company no longer qualifies as an investment under the SBVCA and the Administrator of the SBVCA requires the investor to sell its shares in the Company. If the
F-25
put is exercised, the repurchase price will be the greater of the issue price and the fair market value for such shares.
[f] Loss per common share
| | Nine months ended September 30 | Years ended December 31 | ||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2006 | 2005 | 2005 | 2004 | 2003 | |||||||||||
| Numerator | ||||||||||||||||
| Loss attributable to common shareholders as reported (in thousands) | $ | (10,514 | ) | $ | (4,440 | ) | $ | (6,772 | ) | $ | (5,259 | ) | $ | (2,311 | ) | |
| Denominator | ||||||||||||||||
| Weighted average number of common shares outstanding including escrowed shares | 1,285,500 | 1,270,005 | 1,273,911 | 1,255,500 | 1,255,500 | |||||||||||
| Less: weighted average number of escrowed shares | — | (34,432 | ) | (25,753 | ) | (100,000 | ) | (106,575 | ) | |||||||
| Weighted average number of common shares outstanding | 1,285,500 | 1,235,573 | 1,248,158 | 1,155,500 | 1,148,925 | |||||||||||
| Basic and diluted loss per common share | $ | (8.18 | ) | $ | (3.59 | ) | $ | (5.43 | ) | $ | (4.55 | ) | $ | (2.01 | ) | |
12. INCOME TAXES
At December 31, 2005, the Company has investment tax credits of $5,000 [2004 — $5,000 and 2003 — $3,000] available to reduce future income taxes otherwise payable. The Company also has non-capital loss carry forwards of $8,332,000 [2004 — $5,325,000 and 2003 — $1,727,000] available
F-26
to offset future taxable income in Canada. The investment tax credits and non-capital losses for income tax purposes expire as follows:
| | Investment Tax Credits | Non-capital Losses | ||
|---|---|---|---|---|
| | $ | $ | ||
| | (In thousands) | |||
| 2009 | — | 1,049 | ||
| 2010 | — | 866 | ||
| 2011 | — | — | ||
| 2012 | 2 | — | ||
| 2013 | 2 | — | ||
| 2014 | 1 | 3,582 | ||
| 2015 | — | 2,835 | ||
| 5 | 8,332 | |||
In addition, the Company has unclaimed tax deductions of approximately $2,706,000 related to scientific research and experimental development expenditures available to carry forward indefinitely to reduce taxable income of future years.
Significant components of the Company's future tax assets as of December 31 are shown below.
| | 2005 | 2004 | ||||
|---|---|---|---|---|---|---|
| | $ | $ | ||||
| | (In thousands) | |||||
| Future tax assets: | ||||||
| Tax basis in excess of book value of assets | 805 | 484 | ||||
| Non-capital loss carryforwards | 2,843 | 1,817 | ||||
| Research and development deductions and credits | 923 | 422 | ||||
| Part VI.1 deduction | 642 | 287 | ||||
| Share issue costs | 54 | 58 | ||||
| Total future tax assets | 5,267 | 3,068 | ||||
| Valuation allowance | (5,267 | ) | (3,068 | ) | ||
| — | — | |||||
F-27
The potential income tax benefits relating to these future tax assets have not been recognized in the accounts as their realization did not meet the requirements of "more likely than not" under the liability method of tax allocation. Accordingly, a valuation allowance has been recorded and no future tax assets have been recognized as at December 31, 2005, 2004 and 2003.
The reconciliation of income tax attributable to operations computed at the Canadian statutory tax rate to income tax expense, using a statutory tax rate of 34.86%, 35.62% and 37.62% for the years ended December 31, 2005, 2004 and 2003 respectively is as follows:
| | 2005 | 2004 | 2003 | ||||
|---|---|---|---|---|---|---|---|
| | $ | $ | $ | ||||
| | (In thousands) | ||||||
| Income tax recovery at statutory rates | (1,557 | ) | (1,305 | ) | (725 | ) | |
| Benefit of future tax assets not recognized | 1,301 | 1,067 | 602 | ||||
| Investment tax credits | 214 | 214 | 99 | ||||
| Other | 42 | 24 | 24 | ||||
| Part VI.1 tax on redeemable Convertible Preferred Shares | 432 | 346 | — | ||||
| 432 | 346 | — | |||||
13. AGREEMENTS AND COMMITMENTS
Pursuant to a collaboration and co-development agreement between Isis and the Company, each of Isis and the Company are obligated to share development costs for OGX-011 on a specified basis. In return, any future revenues received in respect of OGX-011 will be shared between Isis and the Company on a specified basis, after payments are made to certain third party licensors. Included in amounts receivable at September 30, 2006, is an amount due from Isis of approximately $178,000 [December 31, 2005 — $196,000; 2004 — $78,000].
Pursuant to license agreements the Company has with the University of British Columbia ("UBC") and Isis, the Company is obligated to pay royalties on future product sales and milestone payments of up to $10.0 million upon the achievement of specified product development milestones. In addition, the Company is obligated to pay to UBC certain patent costs and annual license maintenance fees of C$8,000.
The UBC agreements have effective dates ranging from November 1, 2001 to April 5, 2005 and each agreement expires upon the later of 20 years from its effective date or the expiry of the last patent licensed thereunder, unless otherwise terminated.
F-28
Unless otherwise terminated, the Isis agreements generally expire upon the expiration of the last issued patent related to the technologies.
The Company utilizes contract research organizations to perform services related to the conduct of human clinical studies with OGX-011. The Company has also entered into contracts with clinical sites for the conduct of clinical studies. Pursuant to these agreements with the contract research organizations and clinical sites, the Company has the right to terminate the agreements.
The Company has an operating lease agreement for Canadian office space which expires in September 2009, with an option for the Company to terminate the lease at any point after September 2007, subject to a declining termination fee which is limited to a maximum of $34,000, and with an option to renew through 2014 at the then fair market value.
In addition, the Company has an operating lease agreement for US office space which expires in November 2008, with an option for the Company to renew the lease for an additional three years at the then fair market value.
Future minimum annual lease payments under these are as follows:
| | $ | |
|---|---|---|
| | (In thousands) | |
| 2006 (remainder of year) | 35 | |
| 2007 | 165 | |
| 2008 | 198 | |
| 2009 | 105 | |
| 503 | ||
Rent expense for the nine months ended September 30, 2006 and 2005 was $140,000 and $78,000 respectively and for the years ended December 31, 2005, 2004 and 2003 was $119,000, $47,000, and $20,000 respectively.
F-29
14. RESEARCH AND DEVELOPMENT
| | Nine Month period ended September 30 | Years ended December 31 | | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2006 | 2005 | 2005 $ | 2004 $ | 2003 $ | Inception to date $ | |||||||
| | $ | $ | $ | $ | $ | $ | |||||||
| | (In thousands) | ||||||||||||
| Research and development expenditures | 6,941 | 2,220 | 3,973 | 3,434 | 1,665 | 17,552 | |||||||
| Less: investment tax credit | (119 | ) | (148 | ) | (615 | ) | (601 | ) | (264 | ) | (1,752 | ) | |
| Less: funding advances | — | (215 | ) | (215 | ) | (55 | ) | (20 | ) | (298 | ) | ||
| 6,822 | 1,857 | 3,143 | 2,778 | 1,381 | 15,502 | ||||||||
15. RELATED PARTY TRANSACTIONS
Upon incorporation of the Company, a director and shareholder assigned certain intellectual property to the Company in exchange for 820,000 common shares. These common shares were recorded at a nominal amount representing the director's original cost of the intellectual property.
The Company incurred consulting fees of $70,000 and $42,000 for the nine months ended September 30, 2006 and 2005 respectively and $73,000, $74,000 and $84,000 for the years ended December 31, 2003, 2004 and 2005, respectively payable to two directors. Included in accounts payable and accrued liabilities are $9,000 as at September 30, 2006 [December 31, 2005 — $33,000; 2004 — $32,000] payable under normal trade terms. All transactions were recorded at their exchange amounts.
16. SEGMENTED INFORMATION
The Company operates primarily in one business segment with substantially all its assets located in Canada and operations located in Canada and the United States.
17. GUARANTEES
Occasionally, the Company enters into agreements with third parties in the ordinary course of business that include indemnification provisions that are customary in the industry. Those indemnifications generally require the Company to compensate the other party for certain damages and costs incurred as a result of third party claims or damages arising from these transactions. These indemnification provisions may survive termination of the underlying agreement. The nature of the indemnification obligation prevents the Company from making a
F-30
reasonable estimate of the maximum potential amount it could be required to pay. Historically, the Company has not made any indemnification payments under such agreements and no amount has been accrued in the accompanying consolidated financial statements with respect to these indemnification obligations.
18. ACCOUNTS PAYABLE AND ACCRUED LIABILITIES
| | September 30 2006 | December 31 2005 | December 31 2004 | |||
|---|---|---|---|---|---|---|
| | $ | $ | $ | |||
| | (In thousands) | |||||
| Trade accounts payable | 839 | 585 | 1,677 | |||
| Employee related accruals | 347 | 233 | 133 | |||
| Accrued research and development expenses | 341 | 15 | 134 | |||
| Other | 478 | 84 | 60 | |||
| 2,005 | 917 | 2,004 | ||||
19. Subsequent Events
- [a]
- Subsequent to September 30, 2006 there was an amendment to the agreement referred to in note 11[e] such that the put clause shall automatically cease and be of no further force or effect upon completion of an initial public offering by the Company of its securities.
- [b]
- On September 1, 2006, the shareholders passed a special resolution in accordance with the Canada Business Corporations Act to amend the articles to eliminate all authorized classes of shares (except common shares), to convert all of the issued and outstanding preferred shares into common shares on a fixed one-for-one basis and to authorize the directors to file such amendment to give effect to the amendment immediately prior to the completion of an initial public offering at a price of $9.21 per share or such lower price that is approved by Major Investors Approval (as defined in the existing articles). The amendment does not become effective until filed with the appropriate regulatory authority.
F-31
Through and including , 2007 (the 25th day after the date of this prospectus), all dealers effecting transactions in these securities, whether or not participating in this offering, may be required to deliver a prospectus. This is in addition to the dealers' obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscription.
5,000,000 Shares
OncoGenex Technologies Inc.
Common Shares
RBC CAPITAL MARKETS
| NEEDHAM & COMPANY, LLC | LAZARD CAPITAL MARKETS | |||||||
CANACCORD ADAMS INC. | SUSQUEHANNA FINANCIAL GROUP, LLLP | |||||||
The date of this prospectus is , 2007.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
ITEM 6. INDEMNIFICATION OF DIRECTORS AND OFFICERS
Included in the prospectus, under the heading, "Management — Limitations of Liability and Indemnification".
ITEM 7. RECENT SALES OF UNREGISTERED SECURITIES
The following is a summary of the securities sold by the Registrant in the past three years which were not registered under the Securities Act.
(a) On April 25, 2005, the Registrant issued 30,000 common shares to the University of British Columbia, or UBC, in consideration for intellectual property rights granted pursuant to a License Agreement dated April 25, 2005 between the Registrant and UBC.
(b) On May 5, 2005, the Registrant issued Isis Pharmaceuticals, Inc. a $750,000 convertible promissory note in consideration for intellectual property rights granted under a License and Collaboration Agreement dated January 5, 2005 between the Registrant and Isis. On August 10, 2005, the note converted into 244,300 Series 2 Class B preferred shares of the Registrant.
(c) On August 10, 2005, the Registrant entered into an Investment Agreement pursuant to which it sold 3,896,696 shares of Series 2 Class B preferred shares at a price of $3.07 per share for gross proceeds of $11,962,857. With respect to each investor, the Registrant sold:
- •
- 1,245,239 Series 2 Class B preferred shares to Ventures West 7 Limited Partnership for gross proceeds of $3,822,884;
- •
- 119,536 Series 2 Class B preferred shares to Ventures West 7 U.S. Limited Partnership for gross proceeds of $366,976;
- •
- 1,141,435 Series 2 Class B preferred shares to H.I.G. Horizon Corp. for gross proceeds of $3,504,205;
- •
- 820,258 Series 2 Class B preferred shares to Working Opportunity Fund (EVCC) Ltd. for gross proceeds of $2,518,192;
- •
- 407,166 Series 2 Class B preferred shares to Business Development Bank of Canada for gross proceeds of $1,250,000. These shares were subsequently transferred to BDC Capital Inc.; and
- •
- 163,062 Series 2 Class B preferred shares to WHI Morula Fund, LLC for gross proceeds of $500,600.
(d) On August 10, 2005, the Registrant entered into an Investment Agreement pursuant to which it sold 16,306 Series 2 Class B preferred shares to Cape Family Fund L.P. at a price of $3.07 per share for gross proceeds of $50,059.
(e) On August 10, 2005, the Registrant entered into an Investment Agreement pursuant to which it sold 244,593 Series 2 Class B preferred shares to BC Advantage Funds (VCC) Ltd. at a price of $3.07 per share for gross proceeds of $750,901.
II-1
(f) The Company has granted a total of 1,209,547 options to purchase common stock of the Registrant to its officers, directors, employees and consultants.
In the case of the sales and issuances of securities described in paragraphs (b), (c) and (d) above and made to purchasers in the United States or that were U.S. persons, the Registrant claimed exclusion from registration under the Securities Act pursuant to Rule 506 of Regulation D and Section 4(2) of the Securities Act, in that the Registrant did not engage in any general solicitation or general advertising and the purchasers represented that they were accredited investors, were sophisticated, were purchasing for investment and had adequate information or access to information regarding the Registrant. With respect to all other sales and issuances of securities described in paragraphs (a), (c), (d) and (e), such sales and issuances were made to purchasers outside the United States that were not U.S. persons, in reliance upon the exclusion from Securities Act registration provided by Rule 903 of Regulation S thereunder, in that no offers or directed selling efforts were made in the United States and at the time the buy order was originated, the purchaser was, or the Registrant reasonably believed that the purchaser was, outside the United States.
In the case of the option grants described in paragraph (f) above and made to optionees resident in the United States, the Registrant claimed exclusion from registration under the Securities Act pursuant to Rule 701 under the Securities Act, in that the optionees were officers, directors, employees or qualified consultants of the Registrant or its subsidiary under Rule 701, and such grants were compensatory in nature, were made pursuant to a written stock option plan and option agreements, and were otherwise made pursuant to Rule 701. With respect to all other option grants described in paragraph (k), such grants were made to optionees outside the United States that were not U.S. persons, in reliance upon the exclusion from Securities Act registration provided by Rule 903 of Regulation S thereunder, in that no offers or directed selling efforts were made in the United States and at the time the buy order was originated, the purchaser was, or the Registrant reasonably believed that the purchaser was, outside the United States.
ITEM 8. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
- (a)
- Exhibits
The following exhibits are filed as part of this Registration Statement:
| Exhibit Number | Description | |
|---|---|---|
| 1.1 | (3) | Form of underwriting agreement |
| 3.1 | (1) | Form of restated articles of the Registrant |
| 3.2 | (1) | By-Law No. 1 of the Registrant dated February 8, 2002 |
| 4.1 | (2) | Specimen certificate evidencing common shares of the Registrant |
| 4.2 | (2) | Further amended and restated shareholders' agreement between the Registrant and the shareholders named therein dated as of August 10, 2005, and further amendment dated September 7, 2006 |
| 4.3 | EIA agreement between the Registrant and Working Opportunity Fund (EVCC) Ltd. dated September 24, 2003 | |
| 5.1 | (2) | Legal opinion of DuMoulin Black LLP |
II-2
| 10.1 | (1) | Amended and restated stock option plan, as further amended through June 15, 2005 (pre-existing plan) |
| 10.2 | (1) | 2006 stock incentive plan (new plan) |
| 10.3 | (1) | Employment agreement between the Registrant and Scott D. Cormack dated December 21, 2001, and amendment dated as of August 10, 2005 |
| 10.4 | (2) | Services agreement between the Registrant and Dr. Martin Gleave dated December 21, 2001, and amendments dated January 1, 2002, September 24, 2003 and August 10, 2005* |
| 10.5 | (2) | Employment agreement between the Registrant and Stephen Anderson dated January 9, 2006* |
| 10.6 | (2) | Employment agreement between OncoGenex, Inc. and Cindy Jacobs dated September 12, 2005* |
| 10.7 | (1) | Form of indemnification agreement between the Registrant and each of Scott Cormack, Stephen Anderson and Cindy Jacobs, as executive officers of the Registrant |
| 10.8 | (1) | Form of indemnification agreement between the Registrant and each director of the Registrant |
| 10.9 | (1) | Consulting agreement between the Registrant and CANAID, Inc. (Neil Clendeninn) dated February 15, 2005 |
| 10.10 | (2) | Collaboration and co-development agreement between the Registrant and Isis Pharmaceuticals, Inc. dated as of November 16, 2001 (OGX-011)* |
| 10.11 | (2) | Collaboration and co-development agreement between the Registrant and Isis Pharmaceuticals, Inc. dated as of January 5, 2005 (OGX-427)* |
| 10.12 | (2) | Collaboration and co-development agreement between the Registrant and Isis Pharmaceuticals, Inc. dated as of August 31, 2003 (OGX-225)* |
| 10.13 | (2) | License agreement between the Registrant and the University of British Columbia effective as of November 1, 2001, and amending agreement dated as of August 30, 2006 (OGX-011)* |
| 10.14 | (2) | License agreement between the Registrant and the University of British Columbia effective as of April 5, 2005, and amending agreement dated as of August 30, 2006 (OGX-427)* |
| 10.15 | (2) | License agreement between the Registrant and the University of British Columbia effective as of November 1, 2001 (BP5)* |
| 10.16 | (2) | License agreement between the Registrant and the University of British Columbia dated November 22, 2002 but effective as of September 1, 2002, and amendment dated October 12, 2005 but effective as of September 12, 2002 (BP2 and OGX-225)* |
| 10.17 | (1) | Letter agreement between the Registrant and the University of British Columbia dated November 14, 2002 (BP2, BP5 and OGX-225) |
| 10.18 | (2) | Amending agreement between the Registrant and the University of British Columbia dated October 12, 2005 but effective as of November 23, 2002 (BP5)* |
II-3
| 10.19 | (1) | Second amending agreement between the Registrant and the University of British Columbia dated as of August 30, 2006 (BP5) |
| 10.20 | (2) | Second amending agreement between the Registrant and the University of British Columbia dated as of August 30, 2006 (BP2 and OGX-225)* |
| 10.21 | (1) | Indenture between the Registrant and Rosebud Properties No. 3 Ltd. dated October 1, 2004 (Vancouver office lease) |
| 10.22 | (1) | Agreement of lease between the OncoGenex, Inc. and First & Yesler, L.L.C. dated September 15, 2005, and amendment dated September 16, 2005 (Seattle office lease) |
| 14.1 | (1) | Code of conduct for directors, officers and employees |
| 21.1 | List of subsidiaries (included in the prospectus, under the heading, "Business — Company Information") | |
| 23.1 | Consent of Ernst & Young LLP | |
| 23.2 | (2) | Consent of DuMoulin Black LLP (included in Exhibit 5.1) |
| 24.1 | (1) | Power of attorney (included on signature page) |
| 99.1 | (1) | Audit committee charter |
| 99.2 | (1) | Compensation committee charter |
| 99.3 | (1) | Governance and nomination committee charter |
| 99.4 | (2) | Request for waiver |
- *
- Confidential portions of this exhibit have been omitted and filed separately with the Commission pursuant to an application for Confidential Treatment under Rule 406 promulgated under the Securities Act of 1933, as amended.
- (1)
- Previously filed as an exhibit to the Registrant's Form F-1 filed on December 13, 2006.
- (2)
- Previously filed as an exhibit to the Registrant's Form F-1 Amendment No. 1 filed on January 29, 2007.
- (3)
- Previously filed as an exhibit to the Registrant's Form F-1 Amendment No. 3 filed on February 23, 2007.
- (b)
- Financial Statement Schedules
All schedules have been omitted because the information required to be set forth therein is not applicable or has been included in the Consolidated Financial Statements and notes thereto.
The undersigned Registrant hereby undertakes to provide to the underwriter at the closing specified in the underwriting agreements certificates in such denominations and registered in such names as required by the underwriter to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the Securities and
II-4
Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer or controlling person of the Registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
The undersigned Registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-5
Pursuant to the requirements of the Securities Act of 1933, the Registrant certifies that reasonable grounds to believe that it meets all of the requirements for filing on Form F-1 and has duly caused this amendment to the registration statement to be signed on our behalf by the undersigned, thereunto duly authorized, in the City of Vancouver, Province of British Columbia, Canada, on February 28, 2007.
| ONCOGENEX TECHNOLOGIES INC. | |||
By: | /s/ SCOTT CORMACK Scott Cormack President, Chief Executive Officer and Director | ||
Pursuant to the requirements of the Securities Act of 1933, this amendment to the registration statement has been signed by the following persons in the capacities and on the dates indicated.
| By: | /s/ SCOTT CORMACK Scott Cormack | President, Chief Executive Officer and Director (principal execuitve officer) | February 28, 2007 | ||
By: | * Stephen Anderson | Chief Financial Officer and Secretary (principal financial officer and principal accounting officer) | February 28, 2007 | ||
By: | * Martin Gleave | Chief Science Officer and Director | February 28, 2007 | ||
By: | * James Shepard | Director | February 28, 2007 | ||
By: | * Aaron Davidson | Director | February 28, 2007 | ||
By: | * Neil Clendeninn | Director | February 28, 2007 | ||
By: | * Christopher Laird | Director | February 28, 2007 | ||
By: | * K. Thomas Bailey | Director | February 28, 2007 |
*By: | /s/ SCOTT CORMACK Attorney-in-Fact |
II-6
The undersigned has signed this amendment to the registration statement solely in the capacity of the duly authorized representative of OncoGenex Technologies Inc. in the United States on February 28, 2007.
| ONCOGENEX, INC., a Washington corporation | |||
By: | /s/ SCOTT CORMACK Scott Cormack President, Treasurer and Director | ||
II-7
TABLE OF CONTENTS
PROSPECTUS SUMMARY
RISK FACTORS
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
USE OF PROCEEDS
DIVIDEND POLICY
CONSOLIDATED CAPITALIZATION
DILUTION
SELECTED CONSOLIDATED FINANCIAL DATA
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
BUSINESS
Disrupting Protein Production Using Antisense Technology
Figure 1: Concentration of OGX-011 in Prostate Tissue
Figure 2: OGX-011 Inhibits Clusterin mRNA Expression in Prostate Tissue
Figure 3: OGX-011 Increases the Apoptotic Index in Prostatectomy Specimens
MANAGEMENT
PRINCIPAL SHAREHOLDERS
CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS
DESCRIPTION OF CAPITAL STOCK
SHARES ELIGIBLE FOR FUTURE SALE
MATERIAL UNITED STATES AND CANADIAN INCOME TAX CONSEQUENCES
UNDERWRITING
LEGAL MATTERS
EXPERTS
RELATIONSHIP BETWEEN OUR COMPANY AND CERTAIN UNDERWRITERS
EXPENSES OF THE OFFERING
WHERE YOU CAN FIND MORE INFORMATION
PRIOR SALES
REORGANIZATION
MATERIAL CONTRACTS
PROMOTERS
ELIGIBILITY FOR INVESTMENT
PURCHASERS' STATUTORY RIGHTS
ONCOGENEX TECHNOLOGIES INC. INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
OncoGenex Technologies Inc. (a development stage enterprise) (Incorporated under the laws of Canada) CONSOLIDATED BALANCE SHEETS AS RESTATED — NOTE 2(A) (In thousands of U.S. dollars)
OncoGenex Technologies Inc. (a development stage enterprise) CONSOLIDATED STATEMENTS OF LOSS AS RESTATED — NOTE 2(A) (In thousands of U.S. dollars, except share and per share amounts)
OncoGenex Technologies Inc. (a development stage enterprise) CONSOLIDATED STATEMENTS OF SHAREHOLDERS' DEFICIENCY AS RESTATED — NOTE 2(A) Period from May 26, 2000 (inception) to September 30, 2006 (Information for the nine months ended September 30, 2006 is unaudited)
OncoGenex Technologies Inc. (a development stage enterprise) CONSOLIDATED STATEMENTS OF SHAREHOLDERS' DEFICIENCY AS RESTATED — NOTE 2(A) (Continued) Period from May 26, 2000 (inception) to September 30, 2006 (Information for the nine months ended September 30, 2006 is unaudited)
OncoGenex Technologies Inc. (a development stage enterprise) CONSOLIDATED STATEMENTS OF CASH FLOWS AS RESTATED — NOTE 2(A) (In thousands of U.S. dollars)
OncoGenex Technologies Inc. (a development stage enterprise) NOTES TO CONSOLIDATED FINANCIAL STATEMENTS AS RESTATED — NOTE 2(A) (in U.S. dollars) Information as at September 30, 2006 and 2005 and for the nine months ended September 30, 2006 and 2005 is unaudited.
PART II INFORMATION NOT REQUIRED IN PROSPECTUS
- ITEM 6. INDEMNIFICATION OF DIRECTORS AND OFFICERS
ITEM 7. RECENT SALES OF UNREGISTERED SECURITIES
ITEM 8. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
ITEM 9. UNDERTAKINGS
AUTHORIZED REPRESENTATIVE