As filed with the Securities and Exchange Commission on July 10, 2008
Registration No. 333-144804
UNITED STATES SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Post-Effective
Amendment No. 1 to
Form SB-2
on
Form S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
OSTEOLOGIX, INC.
(Name of small business issuer in its charter)
| | | | | |
| Delaware | | 2834 | | 32-0104570 |
(State or jurisdiction of
incorporation or organization) | | (Primary Standard Industrial
Classification Code Number) | | (I.R.S. Employer
Identification Number) |
4415 Cox Road
Glen Allen, Virginia 23060
(804) 747-6025
(Address, including zip code and telephone number,
including area code, of Registrant’s principal executive offices)
Phillip J. Young
President and Chief Executive Officer
4415 Cox Road
Glen Allen, Virginia 23060
(Name, address, including zip code and telephone number,
including are code, of agent for service)
Copy to:
Stephen B. Thau, Esq.
Morrison & Foerster LLP
755 Page Mill Road
Palo Alto, CA 94304
650-813-5640
Approximate date of proposed sale to the public: From time to time after this Registration Statement becomes effective.
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, check the following box. þ
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act of 1933, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act of 1933, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act of 1933, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer,” and “smaller reporting company” in Rule 12b-2 of the Securities and Exchange Act.
| | | | | | | |
Large accelerated filer o | | Accelerated filer o | | Non-accelerated filer o | | Smaller reporting company þ |
| | | (Do not check if a smaller reporting company) |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act of 1933 or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to said Section 8(a), may determine.
EXPLANATORY NOTE
Thispost-effective amendment is being filed for the purpose of bringing current the information (including audited financial statements) and other information, as required by Section 10(a)(3) of the Securities Act of 1933, as amended, which appeared in the Registrant’s registration statement onForm SB-2 declared effective by the Securities and Exchange Commission on August 14, 2007. A secondary purpose of thispost-effective amendment is to convert such registration statement previously filed onForm SB-2 to a registration statement filed onForm S-1 in accordance with Securities Exchange Commission ReleaseNo. 33-8876. The Registrant is not registering any additional securities. All filing fees payable in connection with the registration of securities were previously paid in connection with the filing of theForm SB-2.
Osteologix, Inc.
1,988,631 Shares of Common Stock
This prospectus relates to the resale from time to time by the selling stockholders identified in this prospectus for their own account of up to a total of 1,988,631 shares of our common stock, including up to an aggregate of 662,877 shares of our common stock issuable upon the exercise of warrants with an exercise price of $1.20 per share subject to adjustment under certain circumstances. The selling stockholders acquired their shares in a private placement of shares completed on April 17, 2008.
We will not receive any of the proceeds from the sale of shares by the selling stockholders. However, we will receive the proceeds from the exercise of the warrants by the selling stockholders, if any, to the extent that the warrants are not exercised on a cashless basis. See “Use of Proceeds” beginning on page 17 of this prospectus.
Our common stock trades on the OTC Bulletin Board, or OTCBB, under the symbol “OLGX.” The last reported sales price per share of our common stock as reported by the OTCBB on July 7, 2008 was $0.92.
INVESTING IN OUR COMMON STOCK INVOLVES A HIGH DEGREE OF RISK. BEFORE BUYING ANY SHARES, YOU SHOULD CAREFULLY READ THE DISCUSSION OF MATERIAL RISKS OF INVESTING IN OUR COMMON STOCK IN “RISK FACTORS” BEGINNING ON PAGE 4 OF THIS PROSPECTUS.
NEITHER THE SECURITIES AND EXCHANGE COMMISSION NOR ANY STATE SECURITIES COMMISSION HAS APPROVED OR DISAPPROVED THESE SECURITIES, OR DETERMINED IF THIS PROSPECTUS IS TRUTHFUL OR COMPLETE. ANY REPRESENTATION TO THE CONTRARY IS A CRIMINAL OFFENSE.
The date of this prospectus is July 10, 2008.
TABLE OF CONTENTS
About this Prospectus
You should rely only on the information contained in this prospectus. We have not authorized anyone to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely on it. You should assume that the information contained in this prospectus is accurate only as of the date of this prospectus, and that information contained in any document included in this prospectus is accurate only as of the date of that document, regardless of the time of delivery of this prospectus or any sale of shares of our common stock. Our business, financial condition, results of operations and prospects may have changed since such dates.
Market data and certain industry forecasts used in this prospectus and the documents included in this prospectus in this prospectus were obtained from market research, publicly available information and industry publications. We believe that these sources are generally reliable, but the accuracy and completeness of such information is not guaranteed. We have not independently verified this information, and we do not make any representation as to the accuracy of such information.
In this prospectus, unless the context otherwise requires, references to “we”, “us”, “our” or similar terms, as well as references to “Osteologix” or the “Company”, refer to Osteologix, Inc., either alone or together with our subsidiaries.
The name Osteologix is our trademark. Other trademarks, product names and company names appearing in this prospectus and documents included in this prospectus or incorporated by reference in this prospectus are the property of their respective owners.
PROSPECTUS SUMMARY
This summary highlights key aspects of the information contained elsewhere in this prospectus. This summary does not contain all the information you should consider before investing in our securities. You should read this entire prospectus carefully, especially the risks of investing in our securities discussed under “Risk Factors” beginning on page 4 of this prospectus and our consolidated financial statements and the notes to those consolidated financial statements beginning onpage F-1, before making an investment decision.
Our Business
We are in the business of developing pharmaceuticals for the treatment and prevention of diseases of bone and joint tissues. Our lead product candidate, NB S101 (strontium malonate) is a strontium salt for treatment of metabolic bone diseases. Currently, our primary goal is to obtain approval for NB S101 for the treatment and prevention of osteoporosis. No product currently approved (or, to our knowledge, under investigation) for the treatment of osteoporosis in the U.S. has demonstrated the ability to increase bone formation and decrease resorption.
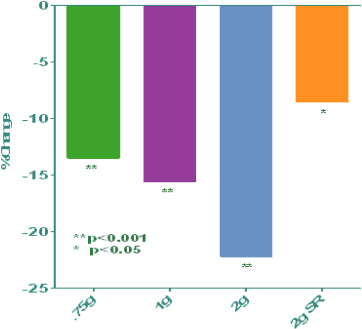
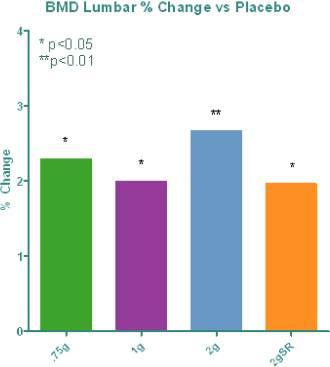
We have completed phase I and II clinical trials with NB S101, and our Investigational New Drug Application, or IND, for NB S101 was recently approved by the U.S. Food and Drug Administration, or FDA, for the treatment and prevention of osteoporosis. We are currently planning to meet with the FDA and European Medicines Agency, or EMEA, to gain approval to conduct larger phase III clinical studies. Our phase I study demonstrated that at a significantly lower dose, our tablet formulation of strontium has shown bioequivalent levels of strontium to a marketed sachet product that has been proven safe and effective in osteoporotic patients in Europe. More importantly, the recent results of our phase II study demonstrated that NB S101 decreased an established biomarker of bone resorption, CTX-1, in a dose-dependent manner by an amount statistically equivalent to or superior to the product approved in Europe. The phase II results also showed that NB S101 significantly increased bone mineral density at the lumbar spine and hip with only 12 weeks of treatment, and no significant side effects were noted in the trial.
Servier, SA, a French pharmaceutical company, has developed their own formulation of a strontium salt, strontium ranelate, and now sells strontium renelate as a drug for treatment of osteoporosis throughout Europe. Their clinical studies of strontium ranelate include data from more than 7,000 subjects, and revealed clinical efficacy with adverse event rates comparable to placebo. Most notably, their product also showed few gastrointestinal side effects, a problem associated with the most common current treatment for osteoporosis, an antiresorptive class of drugs called bisphosphonates. In addition, strontium ranelate did not exhibit side effects associated with other classes of approved osteoporosis drugs, such as estrogens, selective estrogen receptor modulators, and parathyroid hormone. We believe that our investigational drug, strontium malonate (produced in tablet form as NB S101), which is a new salt and improved formulation and dosage form of strontium, represents a more commercially attractive product than strontium ranelate and the treatments which are approved for osteoporosis.
We are currently in the process of discussing development and marketing collaborations with larger pharmaceutical and biotech companies. Successfully completing larger phase III clinical trials will be necessary to receive regulatory approval to commercialize NB S101.
Risks Factors
You should carefully consider the matters discussed in the section “Risk Factors” beginning on page 4 of this prospectus, including the following, before you invest in our common stock:
| | |
| | • | We expect to need additional financing in the near term and may be unable to raise funding when needed, which could force us to delay, curtail or discontinue development of our products or operations |
| |
| | • | We have only one product that is currently being evaluated for commercial development, and even if our continued development of this product is successful, it will be several years before it can reach market |
1
| | |
| | • | Obtaining and maintaining the necessary U.S. or worldwide regulatory approvals for our product candidates will be time consuming, difficult and costly. If we fail to do so, we will be unable to commercialize our product candidates. |
Company Information
We were initially incorporated in Copenhagen, Denmark in 2003 under the name Nordic Bone A/S, before changing our name to Osteologix A/S in 2004. On May 24, 2006, Osteologix A/S merged with Castle & Morgan Holdings, Inc., a U.S. public “shell” company with no operations. Concurrent with the merger, we also raised $10 million in a private placement transaction. As a result of the merger agreement, Osteologix A/S became a wholly-owned subsidiary of Castle & Morgan Holdings, Inc., which subsequently changed its name to Osteologix, Inc. Our principal executive offices are located at 4415 Cox Road Glen Allen, Virginia 23060, and our telephone number is(804) 747-6025. Our website address iswww.osteologix.com. The information on our website does not constitute part of this prospectus.
THE OFFERING
| | |
| Securities Offered | | Up to 1,988,631 shares of our common stock, $0.0001 par value, by certain selling stockholders, of which 662,877 are issuable upon the exercise of warrants. |
| |
| Manner of Offering | | The selling stockholders, either directly or through underwriters, broker-dealers or agents, may offer and sell their securities on a continuous or delayed basis in the future. These sales may be conducted in the open market or in privately negotiated transactions and at prevailing market prices, fixed prices or negotiated prices. See “Plan of Distribution” beginning on page 56 of this prospectus. |
| |
| Use of proceeds | | We will not receive any proceeds from the sale of the shares by the selling stockholders. Nevertheless, we may receive proceeds from the exercise of the warrants, if any, to the extent the warrants are not exercised on a cashless basis. See “Use of Proceeds” beginning on page of this prospectus. |
| |
Trading symbol for our common stock | | Our common stock is traded on the OTCBB under the symbol “OLGX.” |
Unless specifically stated otherwise, the information in this prospectus assumes no exercise no outstanding options or warrants.
2
SUMMARY FINANCIAL DATA
The following table provides a summary of our consolidated financial data for the periods indicated. The summary consolidated financial data for each of the three fiscal years ended December 31, 2007, has been derived from our audited consolidated financial statements and the notes thereto included elsewhere in this prospectus, which were prepared in accordance with United States generally accepted accounting principals (“GAAP”). The summary consolidated financial data for the three months ended March 31, 2008 has been derived from our unaudited consolidated financial statements included elsewhere in this prospectus. These unaudited consolidated financial statements have been prepared on the same basis as the audited consolidated financial statements and, in our opinion, contain all adjustments, consisting only of normal recurring adjustments, necessary for a fair statement of our financial position and results of operations. Operating results for the three months ended March 31, 2008 are not necessarily indicative of the results that may be expected for the fiscal year ending December 31, 2008.
The information presented below should be read in conjunction with “Use of Proceeds,” “Capitalization,” “Selected Consolidated Financial Data,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and the audited consolidated financial statements and notes thereto included elsewhere in this prospectus.
| | | | | | | | | | | | | | | | | | | | | |
| | | For the Years Ended
| | | For the Three Months
| |
| | | December 31, | | | Ended March 31, | |
Statement of Operations Data | | 2005 | | | 2006 | | | 2007 | | | 2007 | | | 2008 | |
| | | (In thousands, except per share data) | |
| |
| Revenue | | $ | 750 | | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| | | | | | | | | | | | | | | | | | | | | |
| Operating expenses: | | | | | | | | | | | | | | | | | | | | |
| Research and development | | | 1,966 | | | | 2,159 | | | | 4,594 | | | | 1,194 | | | | 672 | |
| General and administrative | | | 1,996 | | | | 2,929 | | | | 3,745 | | | | 849 | | | | 951 | |
| | | | | | | | | | | | | | | | | | | | | |
| Total operating expenses | | | 3,962 | | | | 5,088 | | | | 8,339 | | | | 2,043 | | | | 1,623 | |
| | | | | | | | | | | | | | | | | | | | | |
| Loss from operations | | | (3,212 | ) | | | (5,088 | ) | | | (8,339 | ) | | | (2,043 | ) | | | (1,623 | ) |
| Interest income | | | 16 | | | | 230 | | | | 302 | | | | 74 | | | | 28 | |
| | | | | | | | | | | | | | | | | | | | | |
| Net loss | | $ | (3,196 | ) | | $ | (4,858 | ) | | $ | (8,037 | ) | | $ | (1,969 | ) | | $ | (1,595 | ) |
| | | | | | | | | | | | | | | | | | | | | |
| Net loss per share | | $ | (0.34 | ) | | $ | (0.29 | ) | | $ | (0.35 | ) | | $ | (0.09 | ) | | $ | (0.06 | ) |
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | |
| | | As of December 31, | | | As of March 31,
| |
Balance Sheet Data | | 2006 | | | 2007 | | | 2008 | |
| | | (In thousands) | |
| |
| Cash, cash equivalents and short-term investments | | $ | 6,200 | | | $ | 4,145 | | | $ | 2,237 | |
| Working capital | | | 5,788 | | | | 2,999 | | | | 1,476 | |
| Total assets | | | 6,542 | | | | 4,345 | | | | 2,403 | |
| Stockholders’ equity | | | 5,797 | | | | 3,008 | | | | 1,485 | |
3
RISK FACTORS
Investing in our common stock involves a high degree of risk. You should carefully consider the risks described below, together with all other information contained in this prospectus, including the financial statements and the related notes appearing at the end of this prospectus, before deciding to invest in our common stock. If any of the following risks actually occur, our business, financial condition and results of operations could suffer. In these circumstances, the market price of our common stock could decline, and you may lose all or part of your investment in our securities.
Risks Related to Our Financial Condition
We expect to need additional financing in the near term and may be unable to raise funding when needed, which could force us to delay, curtail or discontinue development of our products or operations.
Developing drugs, conducting clinical trials, and commercializing products is expensive. Our future funding requirements will depend on many factors, including, but not limited to:
| | |
| | • | the progress and cost of our clinical trials and other research and development activities; |
| |
| | • | the costs and timing of obtaining regulatory approvals; |
| |
| | • | the terms and timing of any collaborative, licensing, acquisition or other arrangements that we may establish; |
| |
| | • | the costs of filing, prosecuting, defending and enforcing any patent applications, claims, patents and other intellectual property rights; |
| |
| | • | the cost and timing of securing manufacturing capabilities for our clinical product candidates and commercial products, if any; |
| |
| | • | the costs of lawsuits involving us or our product candidates; and |
| |
| | • | the costs of establishing sales, marketing and distribution capabilities. |
We will not receive any proceeds from the sale of the shares by the selling stockholders pursuant to this prospectus. As a result, we believe that our existing cash will only be sufficient to support our current operating plan through approximately the second quarter of 2009. This expectation is based on our current operating plan, which could change as a result of the above-described or other factors, and we may need additional funding sooner than expected. We will need to raise additional funds to support our future development programs, including completion of the clinical trials required to market our investigational drug if we do not enter into an agreement with a larger company to conduct these trials. Our funding requirements may change as a result of many factors, including delays in development activities, underestimates of budget items, unanticipated cash requirements, limitation of development of new potential products, future product opportunities with collaborators, future licensing opportunities and future business combinations. Consequently, we will need to seek additional sources of financing, which may not be available on a timely basis or terms favorable to us, if at all.
If we do not succeed in raising additional funds on a timely basis or on terms favorable to us, we may be unable to complete planned development of NB S101 or obtain approval of our product candidates from the FDAand/or other regulatory authorities. In addition, insufficient funds may force us to discontinue product development, curtail operations, forego sales and marketing efforts and lose attractive business opportunities, which would harm our business, financial condition and results of operation.
Raising additional funds may cause dilution to existing stockholders or require us to relinquish valuable rights.
We may seek to raise additional financing through public or private equity offerings, debt financings, or additional corporate collaboration and licensing arrangements. We cannot be certain that additional funding will be available on a timely basis, on terms favorable to us, or at all. To the extent we raise additional capital by issuing equity securities, our stockholders may experience dilution. To the extent that we raise additional capital by issuing debt securities, we would incur substantial interest obligations, may be required to pledge assets as security for the debt and may be constrained by restrictive financialand/or operational covenants that could limit our flexibility in
4
conducting future business activities. Debt financing would also be senior to our stockholders’ interests in bankruptcy or liquidation. To the extent we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies or product candidates, or grant licenses on terms unfavorable to us.
We have incurred significant net operating losses since our inception and may not achieve profitability in future periods.
We have incurred significant net operating losses since our inception in 2003. To date, we have devoted significant financial resources to the research and development of our investigational drug NB S101. The only revenue we have received since our inception was from a related party in 2005. As a result, we recorded losses of $8.0 million for the year ended December 31, 2007 and $1.6 million for the three months ended March 31, 2008, and our consolidated balance sheet had an accumulated deficit of approximately $19.4 million at March 31, 2008. We do not expect to generate any revenue from the sale of our NB S101 in the near term, and we expect to continue to have significant losses for the foreseeable future as we continue clinical development, seek to advance our product candidates closer to market, seek to expand our pipeline of research and development projects, implement additional internal systems and build our infrastructure.
To become profitable, we must develop and obtain regulatory approval for our product candidates and effectively manufacture, market and sell these product candidates. Accordingly, we may never generate significant revenues and, even if we do generate significant revenues, we may never achieve profitability in future periods.
Risks Related to Our Business
We have only one product that is currently being evaluated for commercial development, and even if our continued development of this product is successful, it will be several years before it can reach market.
Our current product candidate NB S101 is the only pharmaceutical product we are testing in humans, and it may never be successfully marketed or manufactured. To date, this product candidate has only been tested on a limited number of humans. The additional clinical trials required by the FDA and other regulatory authorities to obtain approval to market the product are long and complex and will take a number of years to complete. Our preferred single-tablet dosage may not be accepted by the FDA or supported by future clinical trial data. The FDA and other regulatory authorities also may disagree with our current clinical and pre-clinical research plans and require us to conduct more extensive studies than we currently anticipate before considering our investigational drug for marketing approval.
Most of our future planned studies of NB S101 involve drug exposures for durations that are significantly longer than we have tested thus far and may go out to three years of exposure on the drug as compared to our longest treatment period thus far of only 12 weeks. The longer-term studies could reveal safety or other issues that could adversely affect marketing approval. We need to commit substantial time and additional resources in order to conduct further clinical trials before we can submit a NDA with respect to any of these product candidates. We cannot predict with any certainty when we might submit any NDA for regulatory approval for any of our product candidates, if at all.
Obtaining and maintaining the necessary U.S. or worldwide regulatory approvals for our product candidates will be time consuming, difficult and costly. If we fail to do so, we will be unable to commercialize our product candidates.
Government regulations in the U.S. and other countries have a significant impact on our business and affect research and development, manufacture and marketing of our product candidates. We will require FDA approval to commercialize our product candidates in the U.S. and approvals from similar foreign regulatory authorities to commercialize our product candidates outside the U.S. In order to obtain FDA approval of a product candidate, we must submit to the FDA an NDA demonstrating that the product candidate is safe for humans and effective for its intended use. This demonstration requires significant research and animal tests, which are referred to as pre-clinical studies, as well as human tests, which are referred to as clinical trials. These studies will be time consuming, difficult and costly, and we cannot predict whether our efforts will result in any drugs that the FDA or other regulatory authorities consider safe and effective for humans. The FDA has substantial discretion in the drug
5
approval process, and may refuse to accept our application or may deny approval. If the FDA does not accept or approve our application, it may require us to conduct additional pre-clinical testing or manufacturing studies and submit that data before it will reconsider our application, or require us to perform post-marketing studies. The approval process may also be delayed by changes in government regulation, future legislation, administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals will increase our operating expenses and delay the commercialization of our product candidates and our ability to derive product revenues from them.
Even if we comply with all FDA requests, the FDA may ultimately determine, under its statutory authority, to deny our requests for approval of our drug candidates. We cannot be certain that we will ever obtain regulatory clearance for any product candidate. Failure to obtain FDA approval of NB S101 or any other product candidates will severely undermine our business by reducing our number of salable products and, therefore, corresponding product revenues.
In foreign jurisdictions, we must also receive approval from the appropriate regulatory, pricing and reimbursement authorities before we can commercialize and market our drugs. These processes generally include all of the risks associated with FDA procedures. Pursuing foreign approvals will be time-consuming and expensive. Regulations vary among countries, and foreign authorities may require different or additional clinical trials than we conduct in our attempts to obtain FDA approval. We may never receive any of the approvals necessary to commercialize our product candidates.
We currently plan to commercialize NB S101 and other potential products internationally through collaborative relationships with pharmaceutical companies that have foreign sales capabilities. Future collaborative relationships are important to the success of our product candidates internationally because we do not have regulatory, clinical and commercial resources necessary to obtain approval for and sell our product candidate throughout the world. We may not be able to enter into collaboration agreements with appropriate other companies for important foreign markets on acceptable terms, or at all. Such collaborations may not be effective or profitable for us.
In addition, even if we get to the point where our product candidates are marketed, the products and our manufacturers are still subject to continual review by applicable regulatory authorities, including FDA adverse event reporting requirements and FDA requirements governing product distribution, advertising, and promotion. At any stage of development or commercialization, the discovery of previously unknown problems with our product candidates, our own manufacturing or the manufacture by third parties may result in restrictions on our product candidates or in their manufacture, including withdrawal of the product from the market.
Clinical trials are time-consuming, difficult and costly to design and implement.
Human clinical trials are expensive and difficult to design and implement, in part because the science behind them is complex and they are therefore subject to rigorous regulatory requirements. Further, the medical, regulatory and commercial environment for pharmaceutical products changes quickly and often in ways we may not be able to accurately predict. The clinical trial process is also time-consuming. We estimate that clinical trials of our product candidates will take at least several more years to complete. Furthermore, as failure can occur at any stage, we could encounter problems that cause us to abandon or repeat clinical trials. The commencement and completion of clinical trials may be delayed by, among other things, changes in regulatory requirements, unforeseen safety issues, determination of dosing issues, lack of effectiveness in clinical trials, slower than expected patient recruitment, inability to monitor patients adequately during or after treatment, inability or unwillingness of medical investigators to follow our clinical protocols, inability to maintain a sufficient supply of the investigational drug to support the trials, suspension or termination of clinical trials for noncompliance with regulatory requirements and changes in clinical care protocols and standards of care within the institutions in which our trials take place.
In addition, we or the FDA may suspend our clinical trials at any time if it appears we are exposing participants to unacceptable health risks or if the FDA finds deficiencies in our IND submissions or the conduct of these trials.
6
The results of our clinical trials may not support our product candidate claims or may result in the discovery of adverse side effects.
Even if our clinical trials are completed as planned, we cannot be certain that the results of those trials will support our product candidate claims or that the FDA or foreign authorities will agree with our conclusions regarding the results. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will be successful, and we cannot be sure that the later trials will replicate the results of prior trials and pre-clinical testing. The clinical trial process may fail to demonstrate that our product candidates are safe for humans and effective for indicated uses, which could cause us to abandon a product candidate and may delay or cancel development of others. Any delay of our clinical trials will delay the filing of our NDAs and, ultimately, our ability to commercialize our product candidates and generate revenues. Any cancellation of our clinical trials will eliminate our ability to file an NDA for that product and eliminate our ability to generate any revenue from that product, unless we are later able to conduct a different trial that would satisfy the regulatory authorities.
It is possible that patients enrolled in clinical trials will experience adverse side effects that are not currently part of our product candidate’s profile. In addition, our clinical trials involve a comparatively small patient population. Because of the small sample size, their results may not be indicative of future results. Companies typically collect the most reliable information on side effects during the large phase III studies, when significant numbers of patients are tested. We have not reached this development stage yet, and we cannot accurately predict if phase III studies with our investigational drug, strontium malonate, in tablet form as NB S101, will reveal unexpected side effects. Occurrence of any side effect could delay or terminate further development and hamper or prevent regulatory approval or marketing of our lead product candidate.
The strontium salt that is approved for treating osteoporosis in Europe has exhibited a new potential side effect not seen in clinical trials.
NB S101 contains a salt form of strontium as its active ingredient. There is one product approved in Europe and countries around the world that contains a different salt form of strontium, which is marketed by Servier SA as Protelos®. Protelos was studied in clinical trials involving approximately 7,000 patients prior to its approval in late 2004. In November 2007, the EMEA identified a potential side effect and recommended a label change for Protelos. The side effect “drug rash with eosinophilia and systemic symptoms,” or DRESS, was noted in 16 patients following 570,000 patient-years of worldwide exposure (approximately one case per 35,000 patient years), and two of the cases were fatal. The EMEA advised physicians and patients to stop treatment with Protelos should a rash occur (generally after three to six weeks of treatment), and to seek further medical advice. For these patients Protelos should not be reintroduced.
We currently do not know whether DRESS was caused by the strontium, the different synthetic salt used by Servier, a combination of Protelos with other drugs, or other factors. The infrequency of the cases of DRESS identified by post marketing observation will make it very difficult to ever detect in human clinical trials. It is possible that the FDA or other regulatory authorities will require us to conduct additional clinical or preclinical work on DRESS, which would increase our costsand/or increase the expected time it may take us to receive approval for NB S101. It is also possible that we will be required to note DRESS as a potential side effect in our label if we receive approval for NB S101, even if we do not observe any symptoms in our clinical trials.
Delays in patient enrollment for clinical trials could increase costs and delay regulatory approvals.
We may face substantial competition in seeking to enroll qualified patients into our future clinical trials. Competition for patients has delayed clinical trials of other biotechnology and drug development companies. In addition, recent improvements in existing drug therapy, particularly for medical products for prevention and treatment of osteoporosis, may make it more difficult for us to enroll patients in our clinical trials as the patient population may choose to enroll in clinical trials sponsored by other companies or choose alternative therapies. Delays in patient enrollment can result in increased development costs and delays in regulatory approvals.
The FDA or other regulatory authorities may also change the requirements for testing new drugs for osteoporosis. For example, the current guidelines require placebo-controlled clinical trials conducted in osteoporotic patients. There are ethical concerns regarding treating osteoporotic patients with placebo, and the FDA or other regulatory authorities may at some time require osteoporosis drugs in the future to include active
7
controls (such as existing osteoporosis treatments) instead of placebo. If the FDA makes these changes, we may need to enroll more patients, conduct longer clinical trials, or otherwise change our planned clinical trial designs, potentially and substantially increasing the costsand/or decreasing our chances of demonstrating the efficacy needed to obtain approval.
Physicians and patients may not accept and use our drugs.
Even if the FDA approves one or more of our drug candidates, physicians and patients may not accept and use them. Post-approval market acceptance and use of our drugs will depend upon a number of factors, including perceptions by the health care community, including physicians, about their safety and effectiveness, their cost-effectiveness relative to competing products, the availability of reimbursement for our products from government or other healthcare payors, such as major insurance companies, and the effectiveness of marketing and distribution efforts by us, our licensees and distributors. The failure of NB S101, which we expect to generate substantially all of our product revenues for the foreseeable future, or any of our other product candidates to find post-approval market acceptance would harm to our business, financial condition and results of operations.
Patients may not be able to obtain adequate reimbursement for our drugs.
Our ability to commercialize our drugs, alone or with collaborators, will depend in part on the extent to which reimbursement will be available from government and health administration authorities, private health maintenance organizations, health insurers, and other payors. Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Healthcare payors, including Medicare, routinely challenge prices charged for drugs. Government and other healthcare payors limit both coverage and reimbursement. Even if our product candidates are approved by the FDA, insurance coverage may not be available and reimbursement may be inadequate. If payors do not provide adequate coverage and reimbursement levels, the post-approval market acceptance of our products could be diminished, which would harm our business, financial condition and results of operations.
Our drug-development programs depend upon third-party researchers who are outside our control.
We depend upon independent investigators and collaborators, such as medical institutions, clinical research organizations, and universities, to conduct our pre-clinical and clinical trials and, in certain cases, to develop a product for its target indication. We contract with these collaborators, but they are not our employees, and we cannot control the amount or timing of resources they devote to our programs. They may not assign as great a priority to our programs or pursue them as diligently as we would if we were undertaking the programs ourselves. If outside collaborators fail to devote sufficient time and resources to our programs, or if their performance is substandard, the approval of our FDA applications and our introduction of new drugs, if any, will be delayed, which would harm our business, financial condition and results of operations. These collaborators may also have relationships with other commercial entities, some of whom may compete with us. If our collaborators were to assist our competitors at our expense, our competitive position would be harmed.
We rely exclusively on third parties to manufacture our product candidates.
We rely exclusively on a limited number of vendors to supply raw materials and finished goods necessary to manufacture our product candidates, and the loss of any one of these vendors could harm our business. The FDA and regulatory agencies in other countries also periodically inspect manufacturing facilities, including third parties who manufacture our product candidates or our active ingredients. The FDA may take the position that our chosen manufacturers do not have enough experience manufacturing the dosage forms we have contracted them to produce and may subject those manufacturers to increased scrutiny. Pharmaceutical manufacturing facilities must comply with applicable good manufacturing practice (GMP) standards, and manufacturers typically must invest substantial funds, time and effort to ensure full compliance with these standards and make quality products. We do not have control over our manufacturers’ compliance with these requirements. If our third party manufacturers fail to comply with regulatory requirements, it can result in denial of approval for our product candidate, a requirement to repeat clinical trials, sanctions, fines, delays, suspensions of approvals, seizures or recalls of products, operating restrictions, manufacturing interruptions, corrective actions, injunctions, adverse publicity against us and our product candidatesand/or criminal prosecutions, any of which would harm our business.
8
If we are unable to obtain sufficient supplies of raw materials or if there is a significant increase in the price of raw materials, our business would be harmed. If any of our product candidates receives FDA approval, we expect to rely on one or more third-party contractors to supply our drugs. If our current or future third-party suppliers cease to supply drugs in the quantity and quality we need to manufacture our drug candidates, or if they are unable to comply with GMP standards and other government regulations, the qualification of other suppliers could be a lengthy process, and there may not be adequate alternatives to meet our needs. As a result, we may not be able to obtain the necessary ingredients used in our products in the future on a timely basis, if at all. This would negatively affect our business.
We currently rely on a single source for our supply of the active pharmaceutical ingredient in NB S101 and a different single source for the finished dosage form manufacturing. If either of these sole suppliers fail to provide us with sufficient quantities and with the acceptable level of quality, we may not be able to obtain an alternative supply on a timely or commercially acceptable basis. Any such interruption would disrupt our ability to manufacture NB S101 and could harm our business.
Competition for qualified personnel is intense in our industry, and we may not be able to recruit and retain qualified personnel.
Recruiting and retaining qualified personnel will be critical to our success. We need to hire additional qualified personnel with expertise in pre-clinical testing, clinical research and testing, government regulation, formulation, manufacturing, finance and accounting, and sales and marketing. Competition for such individuals is intense in our industry. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions, some of which are more established than we are and have the ability to pay more cash compensation than we do. As a result, we cannot be certain that we will be able to recruit and retain qualified new hires or retain existing highly skilled employees, which could harm our business, financial condition and operating results.
Developments by competitors may render our products or technologies obsolete or noncompetitive.
Companies that currently sell osteoporosis products include Merck, Procter & Gamble, GlaxoSmithKline, Eli Lilly, Wyeth Ayerst, Novartis, Hoffmann-LaRoche and Sanofi-Aventis. Alternative products and technologies are being developed by these companies and others to improve upon or replace current products for the treatment of osteoporosis, several of which are awaiting FDA approval or are in late-stage clinical trials. Other companies pursuing similar therapeutic areas also represent substantial competition. Many of these competitors have substantially greater financial resources and operate larger research and development organizations than we do. They may also have greater experience in drug development and in obtaining FDA and other regulatory approvals, as well as greater experience in launching, marketing and selling drugs. The greater size and experience of our competitors allows them to develop and market competing products more rapidly and more effectively than we can. If we fail to compete successfully with these competitors, our business, financial condition and operating results would be harmed.
If we fail to protect our intellectual property rights, or to secure rights to patents of others, our competitive position could be harmed or we could be required to incur significant expenses to enforce our rights.
Our success, competitive position and future revenues will depend in part on our ability, and the ability of our licensors, to obtain and maintain patent protection for our product candidates, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties.
We currently have one patent issued in Europe and 25 patent applications pending worldwide. We anticipate filing additional patent applications both in the U.S. and in other countries, as appropriate. These patent applications may not result in the issuance of any additional patents. Moreover, our patent, or any of the patents that may be issued to us in the future, could be challenged, invalidated, circumvented or may otherwise not provide a competitive advantage to us. As a result, others may independently develop similar products or design around our patents and other intellectual property rights.
9
In July 2007, Intellectual Property Services filed an objection to our one issued patent with the European Patent Office. Although we have submitted a response and believe the objection is without merit, we cannot predict the outcome of this opposition, which is likely to take several years to complete. In January 2008, the U.S. Patent and Trademark Office sent us a rejection notice on our application entitled “Water soluble strontium salts for use in the treatment of cartilageand/or bone conditions.” Although we believe we have addressed the issues raised by the examiner by filing additional data in April 2008 and requesting a narrowing of the patient claims, this patent may never issue.
Our issued patents and those that may be issued in the future may be challenged, invalidated or circumvented, which could limit our ability to prevent competitors from marketing related product candidates or could limit the length of the term of patent protection of our product candidates. In addition, the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar technology. Furthermore, our competitors may independently develop similar technologies. In addition, because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before any of our product candidates can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
In addition, legal standards relating to the validity, enforceability and scope of protection of intellectual property rights in the U.S. and other countries are uncertain and may afford inadequate protection of our intellectual property. Consequently, we may be unable to prevent our intellectual property from being exploited by others in the U.S. or abroad, which could require costly efforts to protect our intellectual property. Policing the unauthorized use of our intellectual property is expensive, difficult and, in some instances, impracticable. Litigation may be necessary in the future to enforce or defend our intellectual property rights. Such litigation could result in substantial costs and diversion of management resources, either of which could harm our business.
If we infringe the rights of third parties, we could be prevented from selling products, forced to pay damages, and compelled to defend against litigation.
An issued patent does not guarantee us the right to practice the patented technology or commercialize the patented product. Third parties may have blocking patents that could be used to prevent us from commercializing our patented products and practicing our patented technology. There are patents held by others for the use of strontium. Within the class of strontium-based pharmaceutical products, Servier has several patents. These include U.S. Patent No. 5,128,367, which is directed to strontium ranelate and other metal ranelates, as well as methods and compositions for treating osteoporosis. In addition, Servier’s U.S. Patent No. 4,939,164 is directed to a specific strontium salt of pentanedioic acid, as well as methods and compositions for treating osseous diseases using this salt. Also, Servier’s U.S. Patent No. 5,075,336 describes specific types of carboxylic acid salts, including strontium salts as well as methods and compositions for treating osseous diseases using these salts. Servier’s U.S. Patent No. 5,856,356 (European equivalent: EP813869) describes the use of strontium salts, including strontium ranelate, for treatment of arthrosis. Servier also has three additional U.S. patents directed to methods of manufacturing strontium salts. Although we do not believe that we infringe any patents held by others, there can be no assurance that we will not be accused of infringement, which could lead to expensive and time-consuming litigation or that our product candidates will be held not to infringe valid third party patent claims.
If our product candidates, methods, processes and other technologies infringe proprietary rights of other parties, we could incur substantial costs, and we may have to obtain licenses, which may not be available on commercially reasonable terms, if at all. We may be required to redesign our product candidates or processes, stop using the subject matter claimed in the asserted patents, pay damages, or defend litigation or administrative proceedings, which may be costly whether we win or lose. All these could result in a substantial diversion of valuable management and scientific resources. Resolving intellectual property issues could result in lengthy and costly legal proceedings, the outcome of which cannot be predicted.
We may be exposed to liability claims associated with using hazardous materials and chemicals.
Our research and development activities involve the controlled use of hazardous materials and chemicals. Although we believe our safety procedures for using, storing, handling and disposing of these materials comply with applicable laws and regulations, we cannot eliminate the risk of accidental injury or contamination from these materials. In the event of such an accident, we could be held liable for resulting damages, which could materially
10
adversely affect our business, financial condition and results of operations. In addition, laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous or radioactive materials and waste products may require us to incur substantial compliance costs that could materially adversely affect our business, financial condition and results of operations.
We may incur substantial liabilities and be required to limit commercialization of our product candidates in response to product liability lawsuits.
Testing and marketing medical products entails an inherent risk of product liability. Although side effects from our phase I and phase II clinical studies have been mild and similar to placebo, we may be held liable if serious adverse reactions from the use of our product candidates occur either in clinical trials or in subsequent marketing. If we cannot successfully defend ourselves against product liability claims, we may incur substantial liabilities or be required to limit commercialization of our product candidates. Our inability to obtain sufficient product liability insurance at acceptable cost against claims could prevent or inhibit the commercialization of pharmaceutical products we develop, alone or with collaborators. We carry clinical trial insurance for our trials but do not carry product liability insurance. We, or any collaborators, may not be able to obtain insurance at reasonable cost, if at all. Agreements with future collaborators entitling us to indemnification against losses may not be adequate if claims arise.
Risk Related to Management
We rely on key executive officers and scientific and medical advisors. Their knowledge of our business and technical expertise would be difficult to replace.
We are highly dependent on our limited number of employees to provide services to us, including our president and chief executive officer, Philip J. Young. We do not have “key person” life insurance policies for any of our officers. The loss of the technical knowledge and management and industry expertise of any of our key personnel could result in delays in product development, inability to obtain financing for our business, and diversion of management resources, which could adversely affect our operating results, our ability to obtain approval for our product candidates, or even our ability to continue operations as planned.
In addition, we rely on members of our scientific advisory board and clinical advisors to assist us in formulating research and development strategy. Our scientific advisory board members and clinical advisors generally have other full-time employment and other commitments, and may be subject to non-disclosure obligations that may limit their availability to work with us.
On March 20, 2008, we reached an understanding with our chief financial officer, Matthew M. Loar, regarding his resignation which became effective May 1, 2008. On April 15, 2008, we entered into an agreement with Mr. Loar whereby he agreed to provide services as a consultant with us for up to ten hours per month through September 1, 2008. We cannot be certain that we will be able to extend the terms of the letter agreement beyond September 1, 2008. If we are unable extend the terms of the letter agreement or hire a chief financial officer, or the equivalent, on or before September 1, 2008, it will have a material adverse affect on our internal control over financial reporting in the future. Additionally, we may be forced to implement other temporary remediation measures, such as engaging outside financial consultants to serve as an interim solution, which could be costly. Any such costs would adversely affect our business, financial condition and results of operations.
We may not successfully manage our growth.
Our success will depend upon the effective management of the development of NB S101 and the effective management of our growth, which will place a significant strain on our management and administrative, operational, and financial resources. To manage this anticipated growth, we must consolidate and potentially expand our facilities, augment our operational, financial and management systems, and hire and train additional qualified personnel. We recently moved our operations to Virginia. Such a move may adversely affect the research and development of our product candidates and disrupt our operations. If we are unable to manage our growth effectively, our business would be harmed.
11
The concentrated ownership of our common stock may have the effect of delaying or preventing mergers, acquisitions or other significant corporate transactions.
Nordic Biotech K/S, a venture capital firm and limited partnership in Denmark, and its affiliates together own approximately 62% of our outstanding common stock. Two of the seven members of our board of directors (Florian Schönharting and Christian Hansen) are employed by the Nordic Biotech Advisors ApS, which is the sole advisor of Nordic Biotech K/S. Because of its concentrated ownership of our stock and their positions on our board, Nordic Biotech is able to control all matters requiring stockholder approval and is able to exercise significant influence over all matters requiring board approval, including the election of directors and approval of mergers, acquisitions and other significant corporate transactions. Nordic Biotech is also a shareholder and investor in a number of other biotechnology companies, and there may be conflicts between our business interests and Nordic Biotech’s other investments. In addition, if Nordic Biotech elects to sell any of our shares of common stock which they own in the open market, our stock price is likely to decrease.
Risks Related to Our Common Stock
We cannot assure you that our common stock will become liquid or that it will become listed on a national securities exchange.
We plan to list our common stock on the Nasdaq Global Market, the Nasdaq Capital Market or the American Stock Exchange as soon as practical after we meet initial listing requirements. However, we may never be able to meet the initial listing standards of any stock exchange, or if we do we may not be able to maintain any such listing. Until our common stock is listed on an exchange, we expect that it will continue to be eligible to be quoted on the OTC Bulletin Board, another over-the-counter quotation system, or in the “pink sheets.” In these venues, however, investors and potential investors may find it difficult to obtain accurate stock price quotationsand/or may have limitations imposed on their ability to purchase our common stock. In addition, various restrictions may be imposed by law on broker-dealers who sell our securities to persons other than established customers and accredited investors. Consequently, such regulations may deter broker-dealers from recommending or selling the common stock, which may further affect its liquidity. These restrictions also make it more difficult for us to raise additional capital.
A significant number of shares of our common stock are subject to issuance upon exercise of outstanding warrants, which upon such exercise would result in dilution to our security holders.
As of June 30, 2008, we have issued outstanding warrants to purchase an aggregate of 4,842,753 shares of our common stock, 2,030,152 of which have an exercise price of $1.32 per share, 1,912,877 of which have an exercise price of $1.20 per share and 899,724 of which have an exercise price of $1.03 per share. Although we cannot determine at this time which of these warrants will ultimately be exercised, it is reasonable to assume that such warrants will be exercised only if the exercise price is below the market price of our common stock. To the extent the warrants are exercised, additional shares of our common stock will be issued that will be eligible for resale in the public market, which will result in dilution to our security holders. The issuance of additional securities could also have an adverse effect on the market price of our common stock.
An investor will only be able to sell shares of common stock registered for resale under this prospectus if those shares been registered or qualified or are deemed exempt under the securities laws of the state of residence of the holder of the shares.
The resale of the shares of common stock registered for resale under this prospectus are only being registered for resale under the federal securities laws. Nevertheless, in order for the shares of common stock to be resold by the holder, the shares must either be registered for resale under applicable state securities laws or an exemption from registration is available. We plan to seek to have our securities registered for resale on a national securities exchange as soon as we meet and satisfy the applicable listing requirements. If and when our securities are listed on a national securities exchange, that listing would provide an exemption from registration in every state in the United States. However, until and unless our securities are listed for trading on a national securities exchange, absent an available exemption, you will not be able to sell the shares of common stock registered for resale under this prospectus. We do not plan to register the shares of resale under the securities laws of the individual states.
12
There may be issuances of shares of preferred stock in the future.
Although we currently do not have preferred shares outstanding, the board of directors could authorize the issuance of a series of preferred stock that would grant holders preferred rights to our assets upon liquidation, the right to receive dividends before dividends would be declared to common stockholders, and the right to the redemption of such shares, possibly together with a premium, prior to the redemption of the common stock. To the extent that we do issue preferred stock, the rights of holders of common stock could be impaired thereby, including, without limitation, with respect to liquidation.
Our compliance with the Sarbanes-Oxley Act and SEC rules concerning internal controls may be time consuming, difficult and costly.
Under current requirements, our auditors will be required to audit the effectiveness of our internal controls for the first time when we file our 2008 Annual Report onForm 10-K. It may be time consuming, difficult and costly for us to develop and implement the internal controls and reporting procedures required by Sarbanes-Oxley in a manner adequate to withstand the rigors of an external audit, and due to our small size the proportion of costs we need to devote to comply with these requirements may be substantially greater than it is for other companies. We will likely need to hire additional financial reporting, internal control and other finance staff in order to develop and implement appropriate internal controls and reporting procedures, particularly as we expand our planned operations and consolidate our facilities to a new location. If we are unable to maintain compliance with Sarbanes-Oxley’s internal controls requirements, or the audit of the effectiveness of our internal controls identifies new issues when the audit requirements become effective for us, we may not be able to obtain the independent accountant certifications that Sarbanes-Oxley Act requires publicly-traded companies to obtain.
Our common stock is considered a “penny stock.”
The SEC has adopted regulations which generally define “penny stock” to be an equity security that has a market price of less than $5.00 per share, subject to specific exemptions. The market price of our common stock is less than $5.00 per share and therefore is a “penny stock.” Brokers and dealers effecting transactions in “penny stock” must disclose certain information concerning the transaction, obtain a written agreement from the purchaser and determine that the purchaser is reasonably suitable to purchase the securities. These rules may restrict the ability of brokers or dealers to sell our common stock and may affect your ability to sell shares.
Anti-takeover provisions in our charter and by-laws and under Delaware law may make an acquisition of us or a change in our management more difficult, even if an acquisition or a management change would be beneficial to our stockholders.
Provisions in our charter and by-laws may delay or prevent an acquisition of us or a change in our management. Some of these provisions divide our board into three classes with only a portion of our directors subject to election at each annual meeting of stockholders, allow us to issue preferred stock without any vote or further action by the stockholders, provide that a special meeting of stockholders may be called only by resolutions adopted by our board of directors and prohibit stockholders from acting by written consent without a meeting. These provisions may prevent or delay a change in our board of directors or our management, which is appointed by our board of directors. In addition, because we are incorporated in Delaware, we will become subject to the provisions of Section 203 of the Delaware General Corporation Law upon the completion of this offering. Section 203 may prohibit large stockholders, in particular those owning 15% or more of our outstanding voting stock, from merging or combining with us. These provisions in our charter, by-laws and under Delaware law could reduce the price that investors might be willing to pay for shares of our common stock in the future and result in the market price being lower than it would be without these provisions.
13
CAUTIONARY NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements within the “safe harbor” provisions of the Private Securities Litigation Reform Act of 1995. All statements included in this prospectus, other than statements that are purely historical, are forward-looking statements. Words such as “anticipate,” “contemplate,” “expect,” “intend,” “plan,” “believe,” “seek,” “estimate,” “will,” “will continue to be,” or the negative of foregoing and similar expressions regarding beliefs, plans expectations or intentions regarding the future also identify forward-looking statements. Forward-looking statements in this prospectus include, without limitation:
| | |
| | • | The statements in “Prospectus Summary” regarding our strategies, beliefs, plans, expectations, anticipations and intentions including (1) the commercial attractiveness of strontium malonate compared to other treatments for osteoporosis; |
| |
| | • | The statements in “Use of Proceeds” regarding our strategies, beliefs, plans, expectations, anticipations and intentions including (1) that we will use any funds from the exercise of warrants to fund clinical trials; |
| |
| | • | The statements in “Dividend Policy” regarding our strategies, beliefs, plans, expectations, anticipations and intentions including (1) that we do not anticipate declaring dividends in the foreseeable future; and (2) that we anticipate that all of our available cash will be needed for operations; |
| |
| | • | The statements in “Management’s Discussion and Analysis” regarding our strategies, beliefs, plans, expectations, anticipations and intentions including (1) that we do not anticipate declaring dividends in the foreseeable future; (2) that we plan to continue developing NB S101 in 2008; (3) that we plan to consolidate our operations to a single location and further build our infrastructure to support our operations as a public company; (4) that we plan to continue to acquire and develop products and to either develop resources to market those products or out-license marketing rights to larger pharmaceutical companies; (5) our belief that our currently existing resources will provide liquidity to fund our planned operations through approximately the second quarter of 2009; (6) that we plan to follow the recent phase II clinical trial with a larger phase IIand/or phase III clinical trials estimated to be at least two years to complete; and (7) that we plan to seek approval for NB S101 throughout the world if the results of the clinical trials are positive; |
| |
| | • | The statements in “Business” regarding our strategies, beliefs, plans, expectations, anticipations and intentions including (1) the commercial attractiveness of strontium malonate compared to other treatments for osteoporosis; (2) meeting with the FDA and EMEA to gain approval to conduct larger phase III clinical studies in connection with NB S101; (3) exploring the possibility of developing a strontium product for non-osteoporosis indications; (4) our belief that dual action may stimulate formation and in- growth of new bone metabolism; (5) the submission of a full safety and toxicology package for NB S101; (6) the improved bioavailabilty of our product candidates is due to the different anion to the strontium; (7) the benefits of NB S101 compared to the current therapies and its potential to be a leading treatment for osteoporosis; (8) the strontium class of pharmaceuticals provides a valuable alternative to existing prophylactic and therapeutic treatment options for osteoporosis; (9) the success of NB S101 will depend in part on our ability to achieve market share at the expense of existing, established products, to leverage favorably with future products in development and to grow new or existing markets; (10) our product candidate is in a superior dosage form and that it may have a superior side-effect profile compared to Protelos, while having similar or better efficacy on the skeletal system; (11) expanding our current product pipeline by internal development, acquisitionsand/or in licensing of additional product candidates; (12) future phase III human clinical trials will include measuring the effect of NB S101 on bone fracture reduction; (13) using the preclinical package, phase I studies, phase II studies and phase III studies for registration in both the U.S. and Europe; (14) conducting more preclinical studies for one or more non-osteoporosis indications in the near future; (15) the external performance of clinical studies in collaboration with suitable CROs and academic research institutions; (16) the primary objective of a phase III study will be to evaluate the ability of NB S101 to prevent incident vertebral fracture; (17) the types of data on we will collect during various studies; (18) continuing analysis of all data available on strontium, NB S101 specifically, and potentially competing drugs in development will increase our knowledge in the field, and may lead to our modifying our current development strategy; (19) carrying out an additional phase I study addressing and quantifying the effect of |
14
| | |
| | | food and calcium intake on absorption of NB S101; (20) discussing with U.S. and European regulators whether the significant increases in BMD that we observed in the phase II trial will be sufficient to allow us or a collaborator to immediately enter phase III testing, or whether a longer duration BMD study would be required; (21) the increase in osteoporosis prevalence in the Western population ages in the coming decades; (22) better treatments will lead to a higher percentage of patients on medication; (23) the continued growth of the osteoporosis treatments as the population grows older and lives longer; (24) the future impact on the market due to patients with osteoporosis not complying with their prescribed treatments due to the adverse side effects; (25) HRT’s role in the treatment and prevention of osteoporosis in the future; (26) approval of our NDA will require a full development program complying with FDA guidelines; (27) collaborating with a major global pharmaceutical company with experience marketing products to physicians that commonly prescribe treatments for osteoporosis; (28) retaining the internal core competency required for design, planning, supervision and interpretation of the clinical studies; (29) granting licenses to large, regional pharmaceutical companies that have capability to market the product adequately in their respective territories; (30) our ability to develop our strontium salts for the treatment of metabolic bone diseases such as osteoporosis; (31) the likelihood that Aditech will not develop products that are licensed from us; (32) submitting the preclinical test results, together with manufacturing information and analytical data, to the FDA as part of an IND in the near future; and (33) our belief that we will be able to renew our Glen Allen lease upon its expiration or find alternative suitable office space which is readily available should we chose to relocate. |
| | |
| | • | The statements in “Market for Stock and Related Matters” regarding our strategies, beliefs, plans, expectations, anticipations and intentions including (1) that we intend to retain any earnings for use in the operation and expansion of our business and do not anticipate paying cash dividends in the foreseeable future; |
| |
| | • | The statements in “Plan of Distribution” regarding our strategies, beliefs, plans, expectations, anticipations and intentions including (1) that we intend to make copies of this prospectus available to the selling stockholders; |
| |
| | • | The statements in “Certain U.S. Federal Income Tax Consequences” regarding our strategies, beliefs, plans, expectations, anticipations and intentions including (1) that we do not expect to declare or pay any dividends on our common stock in the foreseeable future; and (2) that we do not expect our stock to constitute a United States real property interest; |
| |
| | • | The statements in the Financial Statements regarding our strategies, beliefs, plans, expectations, anticipations and intentions including (1) that management believes that its current cash and short-term investments will be enable it to continue planned operations only through the second quarter of 2009; (2) that management plans to raise additional capital in order to fund its operations; (3) our belief that we have not taken any uncertain income tax positions that would impact our consolidated financial statements as of December 31, 2007; (4) our belief that no financial statement accounts will be impacted by SFAS 157; and (5) our belief that the fair value of our obligations under our indemnification commitments is minimal. |
Our expectations, beliefs, objectives, intentions and strategies regarding the future, including, without limitation, those concerning expected operating results, revenues and earnings and current and potential litigation are not guarantees of future performance and are subject to risks and uncertainties that could cause actual results to differ materially from results contemplated by the forward-looking statements. These risks and uncertainties include, but are not limited to: uncertainties associated with the research and registration process; our inability to meet government regulation standards; market demand for our product candidates; the availability and terms for licensing, capital raising and partnership opportunities; changes in the cost of administration and clinical studies; advances in health in the aging population; growth of alternative medicine and techniques; our inability to retain employees; fluctuations in the real estate market; speed of development of our product candidates; increased success and introduction of competing products; effectiveness of our efforts to protect our intellectual property rights; and decisions made by holders of third party patent rights; listing standards of national stock exchanges; general economic conditions; changes in government regulations and administrative procedures; changes in our business strategy; fluctuations of interest rates and credit markets.
15
The forward-looking statements in prospectus are subject to additional risks and uncertainties set forth “Risk Factors” beginning on page 4 of this prospectus, and are based on information available to us on the date hereof. We assume no obligation to update any forward-looking statements. Readers are cautioned not to place undue reliance on forward-looking statements, which speak only as of the date of the filing of this prospectus. Readers should also review carefully the cautionary statements and risk factors listed in our Annual Report onForm 10-K for the year ended December 31, 2007, and in our other filings with the SEC, including ourForms 10-Q and8-K and our Annual Report to Stockholders.
16
USE OF PROCEEDS
We will not receive any proceeds from the sales by the selling stockholders of the shares covered by this prospectus. The selling stockholders identified in this prospectus will receive the proceeds from such sale of shares. If the selling shareholders exercise, on a cash basis, all of the warrants underlying the shares being registered, we will receive approximately $795,000. We will use such funds, if any, to fund continued development of NB S101, for working capital and general corporate purposes. We will not receive any proceeds from the exercise of warrants on a cashless basis.
DIVIDEND POLICY
We have not declared any cash dividends on our common stock, and we do not anticipate declaring dividends for the foreseeable future, as we anticipate that all of our available cash will be needed for operations. We are not subject to any legal restrictions respecting the payment of dividends, except that we may not pay dividends if the payment would render us insolvent. Any future determination as to the payment of dividends will be at our board of directors’ discretion and will depend on our financial condition, operating results, capital requirements, plans for expansion and other factors that our board of directors considers to be relevant.
17
CAPITALIZATION
The following table sets forth our cash, cash equivalents and capitalization as of March 31, 2008. You should read this information together with the sections of this prospectus entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and the consolidated financial statements and related notes appearing elsewhere in this prospectus.
| | | | | |
| | | As of March 31, 2008 | |
| | | ($ in thousands) | |
| |
| Cash, cash equivalents and short-term investments | | $ | 2,237 | |
| Long-term debt | | | — | |
| Capital lease obligations | | | — | |
| Stockholders’ equity (deficit): | | | | |
| Preferred Stock; par value $0.0001 per share; 1,000,000 voting, participating shares authorized, none issued and outstanding | | | — | |
| Common stock; par value $0.0001 per share; 100,000,000 shares authorized and 25,011,898 shares issued and outstanding | | | 3 | |
| Additional paid-in capital | | | 21,025 | |
| Accumulated other comprehensive loss | | | (140 | ) |
| Accumulated deficit | | | (19,403 | ) |
| Total stockholders’ equity | | | 1,485 | |
| Total capitalization | | | 1,485 | |
18
MANAGEMENT’S DISCUSSION AND ANALYSIS
We are in the business of developing pharmaceuticals for the treatment and prevention of diseases of bone and joint tissues. Our lead product candidate, NB S101 (strontium malonate), is in clinical development for treatment of osteoporosis, and in late 2007 we completed a human phase II clinical trial of this investigational drug which demonstrated a positive effect of NB S101 on biomarkers of bone loss and on bone mineral density after 12 weeks of treatment. We are currently in process of discussing development and marketing collaborations with larger pharmaceutical and biotech companies. Successfully completing larger phase III clinical trials will be necessary to receive regulatory approval to commercialize NB S101. We are publicly traded in the United States on the OTC Bulletin Board under the stock ticker symbol “OLGX.”
Currently, our primary goal is to obtain approval for NB S101 for the treatment and prevention of osteoporosis. We believe, based on preclinical and clinical data, that NB S101 simultaneously decreases resorption (loss) of existing bone tissue while also increasing formation of new bone tissue. No product currently approved (or, to our knowledge, under investigation) for the treatment of osteoporosis in the U.S. has demonstrated the ability to increase bone formation and decrease resorption. Our phase I study of the pharmacokinetic, or PK, properties of NB S101 revealed that a one gram tablet dose of NB S101 resulted in approximately the same level of strontium in human serum as a European company’s approved product containing two grams of strontium ranelate in sachet formulation, which must be mixed with water before ingestion. Thus, at a significantly lower dose our tablet formulation of strontium has shown bioequivalent levels of strontium to a marketed sachet product that has been proven safe and effective in osteoporotic patients in Europe. More importantly, the recent results of our phase II study demonstrated that NB S101 decreased an established biomarker of bone resorption, CTX-1, in a dose-dependent manner by an amount statistically equivalent to or superior to the product approved in Europe. The phase II results also showed that NB S101 significantly increased bone mineral density at the lumbar spine and hip with only 12 weeks of treatment, and no significant side effects were noted in the trial.
Critical Accounting Policies and Use of Estimates
The preparation of consolidated financial statements requires us to make certain estimates and assumptions that affect the reported amounts of assets and liabilities, the reported amounts and classification of expense, and the disclosure of contingent assets and liabilities. We evaluate our estimates and assumptions on an ongoing basis. We base our estimates on historical experience and various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions. The following items in our consolidated financial statements require significant estimates and judgments:
Accounting for stock options. Beginning on January 1, 2006, we began accounting for stock options under the provisions of SFAS 123R, which requires the recognition of the fair value of stock-based compensation. The fair value of stock options was estimated using a Black-Scholes option valuation model. This model requires the input of subjective assumptions in implementing SFAS 123R, including expected stock price volatility, expected life and estimated forfeitures of each award. The fair value of equity-based awards is amortized over the vesting period of each award, and we have elected to use the straight-line method of amortization. Due to the limited amount of historical data available to us, particularly with respect to stock-price volatility, employee exercise patterns and forfeitures, actual results could differ from our assumptions.
Prior to the implementation of SFAS 123R, we accounted for stock options under the provisions of Accounting Principles Board Opinion No. 25,“Accounting for Stock Issued to Employees”and made pro forma footnote disclosures as required by SFAS No. 148,“Accounting For Stock-Based Compensation — Transition and Disclosure,”which amended SFAS No. 123,“Accounting For Stock-Based Compensation.”Pro forma net loss and pro forma net loss per share disclosed in the footnotes to the consolidated financial statements were estimated using a Black-Scholes option valuation model.
We estimate the fair value of options granted using the Black-Scholes option valuation model. As allowed by Staff Accounting Bulletin (SAB) No. 107,Share-Based Payment,we have opted to use the simplified method for estimating our expected term equal to the midpoint between the vesting period and the contractual term of our stock
19
options. We estimate the volatility of our common stock at the date of grant based on the historical volatility of our common stock and, due to our limited time as an operating public company, that of other small public companies in the healthcare industry, consistent with provisions of SFAS 123R and SAB 107. We base the risk-free interest rate that we use in the Black-Scholes option valuation model on the implied yield in effect at the time of option grant on U.S. Treasury zero-coupon issues with equivalent remaining terms. We have never paid any cash dividends on our common stock and we do not anticipate paying any cash dividends in the foreseeable future. Consequently, we use an expected dividend yield of zero in the Black-Scholes option valuation model. SFAS 123R requires us to estimate forfeitures at the time of grant and revise those estimates in subsequent periods if actual forfeitures differ from those estimates. We use historical data to estimate pre-vesting option forfeitures and record stock-based compensation expense only for those awards that are expected to vest. For options granted on or after January 1, 2006, we amortize the fair value on a straight-line basis. All options are amortized over the requisite service periods of the awards, which are generally the vesting periods. We may elect to use different assumptions under the Black-Scholes option valuation model in the future, which could materially affect our net income or loss and net income or loss per share.
We account for equity instruments issued to non-employees in accordance with the provisions of SFAS No. 123 and Emerging Issues Task Force (“EITF”) IssueNo. 96-18,“Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling Goods, or Services.”As a result, the non-cash charge to operations for non-employee options with vesting or other performance criteria is affected each reporting period by changes in the estimated fair value of our common stock. The two factors which most affect these changes are the fair value of the common stock underlying stock options for which stock-based compensation is recorded and the volatility of such fair value. If our estimates of the fair value of these equity instruments change, it would have the effect of changing compensation expenses.
Research and Development Costs. All costs incurred for research and development are expensed as incurred. Research and development costs include costs for clinical trials, which are incurred from planning through patient enrollment tofollow-up visits and reporting of the underlying data. We estimate expenses incurred for clinical trials that are in process based on patient screening, enrollment and physician visits as reported by each of the various sites involved in the trial, but these estimates can vary based on estimates of total patient numbers, patients that do not meet trial enrollment criteria, the number of visits missed by the patients, and the patients that drop out of the trial or chose to discontinue treatment.
Recently Adopted or Issued Accounting Pronouncements
On January 1, 2008, we adopted Statement of Financial Accounting Standards No. 157, “Fair Value Measurements,” or SFAS 157, which clarifies and prioritizes methods for measuring fair value under generally accepted accounting principles. SFAS 157 generally increases the level of disclosure required for fair value measurements, although it does not impact the valuations or disclosures required under Statement of Financial Accounting Standards No. 123R, “Share-based Payment.” The adoption of SFAS 157 did not have a material impact on our fair value measurement.
Important Developments
During 2007, there were two key company events impacting our business of developing pharmaceutical products to treat human disease — a financing transaction and the completion of our phase II clinical trial. On June 7, we announced that we had sold 3,825,754 shares of common stock and warrants to purchase 1,912,877 shares of common stock to certain institutional investors for gross proceeds of $5,050,000. On November 5, 2007, we announced positive top-line results of an international Phase II clinical trial of our lead investigational drug, NB S101. This large, well-controlled clinical trial of 289 postmenopausal women with low bone mineral density met its primary endpoint by significantly decreasing CTX-1, a well-validated biomarker for measuring bone resorption activity, at all dose levels compared with the control group. Results of the trial also showed that NB S101 significantly increased lumbar spine bone mineral density at all doses tested.
On April 17, 2008, the Company sold 4,030,304 shares of the Company’s common stock and 2,015,152 common stock purchase warrants with an exercise price of $1.32 per share for gross proceeds of $5,320,000. The shares and warrants were offered and sold only to accredited investors, and have not been registered under the
20
Securities Act of 1933, as amended, or any state securities laws and may not be offered or sold in the United States absent registration with the SEC or an applicable exemption from SEC registration requirements. An affiliate of Nordic Biotech K/S, which owned approximately 60% of the Company’s outstanding common stock prior to the transaction, purchased 3,030,304 shares of common stock and warrants to purchase 1,515,152 shares of common stock in the April 17, 2008 transaction, for gross proceeds of approximately $4,000,000, increasing the Nordic Biotech K/S funds’ beneficial ownership percentage to approximately 62%. Punk Ziegel + Co., L.P., acted as placement agent in connection with this transaction. For its services, we issued to Punk Ziegel warrants to purchase 15,000 shares of common stock upon the same terms as the warrants issued to the investors, as well as a cash fee and reimbursement for certain expenses equal to $55,024 in the aggregate.
On May 7, 2008, we announced that our IND application for NB S101 for the treatment and prevention of osteoporosis was accepted by the U.S. Food and Drug Administration.
Comparison of Three Months Ended March 31, 2008 and 2007
Revenues
Osteologix’s products are currently in various stages of research and development, and there were no revenues recorded in the three month periods ended March 31, 2008 and 2007.
Research and Development Expenses
Expenditures relating to research and development are expensed as incurred. Research and development expenses include clinical trial costs, preclinical studies, development and manufacturing costs for medicinal products under investigation, payments to contract research organizations, compensation expenses for research and development personnel, supplies and related consulting and advisor costs.
Research and development expenses, and the changes compared to the previous year, were as follows:
| | | | | | | | | | | | | |
| Three Months Ended March 31, | | | Decrease from Period in Prior Year | |
| 2008 | | 2007 | | | Dollars | | | Percent | |
| |
| $672,000 | | $ | 1,194,000 | | | $ | (522,000 | ) | | | (44 | )% |
Research and development expenses decreased $522,000 for the first three months of 2008 compared to the first three months of 2007 primarily because during the 2008 period, we did not incur costs for the human phase II clinical trial that we completed in November 2007. While the phase II clinical trial was underway substantially all of our research and development costs were related to this study. For the 2008 period, our research and development costs consisted primarily of costs paid to outside medical advisors and consultants assisting us with formatting the preclinical and clinical trial documentation in a manner that is designed to meet the quality requirements of the FDA, completion of documentation regarding drug manufacturing and quality specifications for NB S101, manufacturing additional investigational drug product, and costs related to the preparation and filing of an IND filing with the FDA.
We expect research and development expenses for the balance of 2008 to be lower than for the comparable periods in 2007 because we completed our phase II clinical trial in late 2007. We expect that our costs for 2008 will include planning for a phase III clinical trial, additional manufacturing and quality work on the drug product, consultants and medical advisors, and potentially smaller human and animal studies of NB S101 as we continue preparing for a larger phase III trial.
General and Administrative Expenses
General and administrative expenses include compensation expense for personnel not directly involved in research and development activities, management and other administrative personnel costs, insurance, accounting, legal and patent expenses.
21
General and administrative expenses, and the changes compared the previous year, were as follows:
| | | | | | | | | | | | | |
| Three Months Ended March 31, | | | Increase from Period in Prior Year | |
| 2008 | | 2007 | | | Dollars | | | Percent | |
| |
| $951,000 | | $ | 849,000 | | | $ | 102,000 | | | | 12 | % |
General and administrative expenses increased by $102,000 for the three months ended March 31, 2008, compared to the three months ended March 31, 2007, primarily because of higher patent costs for the intellectual property protecting NB S101.
We expect general and administrative expenses for the second quarter of 2008 to be lower than for the second quarter of 2007 due to the additional charge recorded in the second quarter of 2007 related to the resignation of our prior chief executive officer and president. Excluding this item, for the balance of 2008, we expect general and administrative expenses to increase modestly compared to the amounts incurred during 2007.
Interest Income
Interest income decreased by $46,000, to $28,000 for the first three months of 2008 compared to the first three months of 2007 because of lower average interest rates earned on our cash and short-term investment balances and lower average cash and short-term investments balances.
Comparison of Fiscal Years Ended December 31, 2007, 2006 and 2005
Revenues
Our product candidates are currently in various stages of research and development. Since we were established in 2003, there has only been one source of revenue, which was $750,000 received from an agreement signed in 2005 for the license of intellectual property rights to a related party. We recognized the full amount in the statement of operations because we have no on-going obligations under the agreement. Because we are still developing our investigational drug for the treatment of osteoporosis, we do not expect significant revenues for several years, if at all.
Research and Development Expenses
Expenditures relating to research and development are expensed as incurred. Research and development expenses include clinical trial costs, preclinical studies, payments to contract research organizations, compensation expenses for research and development personnel, development and manufacturing costs for investigational drugs, supplies, and related consulting and advisor costs.
Research and development expenses, and the changes compared to the previous year, were as follows:
| | | | | | | | | | | | | | | | | |
| For the Years Ended December 31, | | | Increase Compared to Previous Year | |
| 2007 | | 2006 | | | 2005 | | | 2007 | | | 2006 | |
| |
| $4,594,000 | | $ | 2,159,000 | | | $ | 1,966,000 | | | $ | 2,435,000 | | | $ | 193,000 | |
| | | | | | | | | | | | +113 | % | | | +10 | % |
Research and development expenses more than doubled in 2007 compared to 2006. The increase was primarily related to our human phase II clinical trial of NB S101 which was started in late 2006 and continued throughout most of 2007 until we reported the results in November 2007. The phase II clinical trial costs include expenditures incurred by the physicians treating the patients being evaluated for and enrolled into the trial, expenditures by a contract research organization that is assisting us with the oversight of the trial, and costs to acquire, manufacture and package the drug products that are being used. Conducting the trial and subsequently analyzing the results occupied substantially all of our research and development resources during 2007.
Research and development expenses increased $193,000 in 2006 compared to 2005 primarily due to ourstart-up of a human phase II clinical trial in late 2006. Largely offsetting the increase in research and development expenses related to the phase II clinical trial was a decrease in costs in 2006 as a result of a smaller phase I clinical trial that was both started and completed in 2005. Because later-state clinical trials enroll more patients and treat
22
them for a longer period of time, our incremental costs for the portion of the phase II trial that occurred in 2006 were greater than the costs for the full phase I trial incurred in 2005.
Although in 2008 we will not have any costs for the phase II clinical trial, we do plan to continue developing NB S101 and to have increases in costs relating to additional studies of the drug, manufacturing and productionscale-up costs, regulatory costs as we present the results to authorities in the U.S. and in Europe and complete the planning for the next human clinical trial.
General and Administrative Expenses
General and administrative expenses include compensation expense for personnel not directly involved in research and development activities, management and other administrative personnel costs, insurance and accounting, legal and patent expenses.
General and administrative expenses, and the changes compared the previous year, were as follows:
| | | | | | | | | | | | | | | | | |
| For the Years Ended December 31, | | | Increase Compared to Previous Year | |
| 2007 | | 2006 | | | 2005 | | | 2007 | | | 2006 | |
| |
| $3,745,000 | | $ | 2,929,000 | | | $ | 1,996,000 | | | $ | 816,000 | | | $ | 933,000 | |
| | | | | | | | | | | | +28 | % | | | +47 | % |
General and administrative expenses increased by $816,000 in 2007 compared to 2006. The largest component of the increase was $514,000 for severance following the resignation of our previous Chief Executive Officer. Excluding this event, our general and administrative expenses increased by $302,000, or approximately 10%. The higher costs in the 2007 period were largely related to increased costs of operating as a public company for the full 2007 period as compared to only a portion of the 2006 period, including increased staffing, and external legal and accounting costs. Partially offsetting the increased costs in the 2007 period were lower costs for stock-based compensation following a 2006 modification to outstanding equity awards.
General and administrative expenses increased by $933,000 in 2006 compared to 2005. The largest component of the increase was $508,000 in costs recorded for the value of stock options granted to our officers, directors and employees following our implementation of SFAS 123R on January 1, 2006 and the modification of the awards on the date of the merger transaction. In addition, we incurred increased expenses, including legal and accounting costs, of operating as a public company since our merger transaction was completed in May 2006.
We expect general and administrative expenses for 2008 to be higher than for the comparable periods in 2007 because we plan to consolidate our operations to a single location and further build our infrastructure to support our operations as a public company.
Interest Income
Interest income was $302,000 in 2007, an increase of $72,000 from 2006. The increase was a result of higher average cash and short-term investment balances for 2007 as compared to 2006, partially offset by lower interest rates received in 2007. Interest income increased $214,000 in 2006 compared to 2005 due to higher average cash and short-term investments balances following our private placement and merger transaction in May 2006.
Liquidity and Capital Resources
We measure our liquidity primarily by the cash, cash equivalents and short-term investments, as well as the working capital, available to fund our operations. Since we were founded in 2003, we have applied the majority of our resources to research and development programs, primarily the development of NB S101, and the administrative expenses to support the our pharmaceutical development operations. We have operated at a loss since inception and expect to continue to incur losses in the future as a result of our ongoing research and development efforts. To date we have funded our operations primarily through the sale of our common stock
23
In evaluating our liquidity, we do not distinguish between cash and short-term investments because when we hold short-term investments all of our short-term investments are available for sale and we believe they can readily be converted into cash when needed. We do not hold any auction rate securities in our portfolio. The following table summarizes our cash, cash equivalents and short-term investments, and our working capital available to fund our operations:
| | | | | | | | | |
| | | March 31,
| | | December 31,
| |
| | | 2008 | | | 2007 | |
| |
| Cash, cash equivalents and short-term investments | | $ | 2,237,000 | | | $ | 4,145,000 | |
| Working capital | | | 1,476,000 | | | | 2,999,000 | |
The decrease in our cash, cash equivalents and short-term investments of $1,908,000 during the first three months of 2008 funded the continuing development of NB S101, our investigational drug for osteoporosis, including manufacturing and quality specifications, preparation and completion of an IND with the FDA, and payment of 2007 clinical trial expenses which became due during 2008. The decrease in cash, cash equivalents and short-term investments also included the general and administrative expenses of operating a public company. We have experienced net losses from our inception through March 31, 2008, and we expect losses to continue as we further our research and development programs.
After March 31, 2008, we completed a sale of common stock and warrants to purchase common stock for gross proceeds of $5,320,000. We expect these proceeds, together with our cash and cash equivalents as of March 31, 2008, to fund our planned operations through approximately the second quarter of 2009. We may consume available resources more rapidly than currently anticipated, resulting in the need for funding at an earlier date. If adequate funds are not available, we may be required to significantly reduce, refocus or cease our operations, or to obtain funds through arrangements that may require us to relinquish rights to NB S101, which could have a material adverse affect on the price of our common stock.
Plan of Operation
Our business strategy is to develop drug candidates that target major medical needs and can be rapidly commercialized. We are currently focusing on the treatment and prevention of diseases of bone and joint tissues. NB S101 for the treatment of osteoporosis fits this strategy because the osteoporosis market is large, with over $7 billion in annual product sales, and preclinical, phase I and one phase II human clinical studies have been completed. In addition, a drug which contains a different strontium compound as its active ingredient (as compared to NB S101) has been proven safe and effective and is approved for treatment of osteoporosis in Europe, which we believe reduces the development risk for NB S101 when compared to other investigational drugs in completely unproven chemical classes. In executing our business strategy, our management oversees the human clinical trials necessary to establish preliminary evidence of efficacy, and we are seeking to establish collaborative agreements with pharmaceutical and biotechnology companies for their late-stage development and marketing of our product candidatesand/or our obtaining rights to develop and market certain of their product candidates. We plan to continue to acquire and develop products, and plan to either develop the resources to market these products in selected world regions or out-license marketing rights to larger pharmaceutical companies.
We believe that our currently available existing resources will provide liquidity to fund our planned operations through approximately the second quarter of 2009. Because the business of developing pharmaceutical products is time-consuming and expensive, and NB S101, our most advanced investigational drug, still must complete pivotal human clinical trials before it can be marketed, we will require additional financial resources to carry out our business strategy. We plan to follow the recent phase II clinical trial with a larger phase IIand/or phase III clinical trial that we estimate will take at least two years to complete. After or possibly concurrent with the next clinical trial, either one or two similar or potentially larger phase III trials will be required. We may license the rights to NB S101 to a larger company before any of the clinical trials are complete, or we may choose to retain all rights ourselves without entering into a license or collaborative agreement. If the results of the clinical trials are positive, we plan to seek approval for NB S101 throughout the world, either on our own or in collaboration with a larger pharmaceutical company that has more financial resources than we do.
24
Because we are developing NB S101 and are also seeking to expand our line of investigational drugs by acquiring rights to additional drugs in development, we will require additional financial resources to carry out our plans. We are exploring various options to obtain additional capital, including the licensing of certain rights to NB S101, the sale of equity or debt securities and other alternatives. Our ability to successfully raise additional funds for our operations will be based, in part, on overall market conditions, conditions in the biotechnology and pharmaceutical industries, the status and results of our clinical trials (including interpretations of the data and the market potential, which can be subjective), and other parties’ opinions about NB S101 and the osteoporosis market, among other factors. Additional funding may not be available on acceptable terms, if at all. The actual amounts we ultimately need for our future operating requirements will vary depending on a number of other factors, including:
| | |
| | • | the requirements of regulatory agencies; |
| |
| | • | our ability to establish collaborative relationships and the terms of these relationships; |
| |
| | • | the progress we make in our clinical development programs and the results of our clinical trials; |
| |
| | • | competing technological and market developments; and |
| |
| | • | our acquisition or licensing of new drug candidates. |
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements that have or are reasonably likely to have a material current or future effect on our financial condition, results of operations, liquidity or capital resources.
25
BUSINESS
Overview
We are in the business of developing pharmaceuticals for the treatment and prevention of diseases of bone and joint tissues. Our lead product candidate, NB S101 (strontium malonate), is in clinical development for treatment of osteoporosis, and in late 2007 we completed a human phase II clinical trial of this investigational drug that demonstrated a positive effect of NB S101 on biomarkers of bone loss and on bone mineral density after 12 weeks of treatment. We are currently in the process of discussing development and marketing collaborations with larger pharmaceutical and biotech companies. Successfully completing larger phase III clinical trials will be necessary to receive regulatory approval to commercialize NB S101. We are publicly traded in the United States on the OTC Bulletin Board under the stock ticker symbol “OLGX.”
NB S101 is a strontium salt for treatment of metabolic bone diseases, and we are currently studying its application for the prevention and treatment of osteoporosis. Strontium salts exhibit pharmacologic effects on skeletal tissue that set this class of compounds apart from other existing treatments for osteoporosis. These other treatments either reduce the regular resorption (loss) of bone tissue and are called anti-resorptives, or increase the regular formation of bone tissue and are called anabolic agents. Most notably for strontium products, unlike other approved treatments, both preclinical and clinical studies indicate that strontium simultaneously decreases resorption (loss) of bone while also increasing formation of new bone. The French pharmaceutical company Servier, SA has developed their own formulation of a strontium salt, strontium ranelate (“Protelos”), and now sells this as a drug for treatment of osteoporosis throughout Europe. Their clinical studies of strontium ranelate include data from more than 7,000 subjects, and revealed clinical efficacy with adverse event rates comparable to placebo. Most notably, their product also showed few gastrointestinal side effects, a problem associated with the most common current treatment for osteoporosis, an antiresorptive class of drugs called bisphosphonates. In addition, strontium ranelate did not exhibit side effects associated with other classes of approved osteoporosis drugs, such as estrogens, selective estrogen receptor modulators, and parathyroid hormone. We believe that our investigational drug, strontium malonate (produced in tablet form as NB S101), which is a new salt and improved formulation and dosage form of strontium, represents a more commercially attractive product than strontium ranelate and the treatments which are approved for osteoporosis. We have completed phase I and II clinical trials with NB S101, and our IND for NB S101 was recently approved by the FDA for the treatment and prevention of osteoporosis. We are currently planning to meet with the FDA and EMEA to gain approval to conduct larger phase III clinical studies.
Currently, our primary goal is to obtain approval for NB S101 for the treatment and prevention of osteoporosis. No product currently approved (or, to our knowledge, under investigation) for the treatment of osteoporosis in the U.S. has demonstrated the ability to increase bone formation and decrease resorption. Our phase I study of the pharmacokinetic, or PK, properties of NB S101 revealed that a one gram tablet dose of NB S101 resulted in approximately the same level of strontium in human serum as the European company’s approved product containing two grams of strontium ranelate in sachet formulation, which must be mixed with water before ingestion. Thus, at a significantly lower dose our tablet formulation of strontium has shown bioequivalent levels of strontium to a marketed sachet product that has been proven safe and effective in osteoporotic patients in Europe. More importantly, the recent results of our phase II study demonstrated that NB S101 decreased an established biomarker of bone resorption, CTX-1, in a dose-dependent manner by an amount statistically equivalent to or superior to the product approved in Europe. The phase II results also showed that NB S101 significantly increased bone mineral density at the lumbar spine and hip with only 12 weeks of treatment, and no significant side effects were noted in the trial.
Company History
After our initial formation in 2003, we began operating activities in early 2004 with chemistry andin vitrostudies for the selection and development of NB S101, our lead product candidate for the treatment of osteoporosis. Later in 2004 we established a United States presence. In 2005 we expanded our operational capabilities to enter human clinical studies and completed our first phase I clinical trial. In 2006 we completed a merger transaction which provided us with additional financing and a public stock listing. Later in 2006 we initiated a phase II clinical trial of NB S101, which we completed in the second half of 2007. On November 5, 2007 we announced that the
26
results of this study were positive and that the study met its primary endpoint by showing a statistically significant reduction in a validated biomarker of bone resorption, CTX-1.
We were initially incorporated in Copenhagen, Denmark in 2003 under the name Nordic Bone A/S, before changing our name to Osteologix A/S in 2004. On May 24, 2006, Osteologix A/S merged with Castle & Morgan Holdings, Inc., a U.S. public “shell” company with no operations. Concurrent with the merger, we also raised $10 million in a private placement transaction. As a result of the merger agreement, OsteologixA/S became a wholly-owned subsidiary of Castle & Morgan Holdings, Inc., which subsequently changed its name to Osteologix, Inc. As a result of the May 24, 2006 transactions, the previously outstanding stock of Osteologix A/S exchanged into a 50.0% ownership of Osteologix, Inc., the private placement purchasers acquired 36.4% of Osteologix, Inc., and the previous stockholders of Castle & Morgan Holdings, Inc. owned 13.6% of Osteologix, Inc.
Our principal executive offices are located at 4415 Cox Road, Glen Allen, Virginia 23060, and our corporate telephone number is(804) 747-6025. We also maintain an office in California at 425 Market Street, Suite 2200, San Francisco, California 94105 and in Denmark at Østergade 5, 3, 1100 Copenhagen, Denmark. Our website is located athttp://www.osteologix.com. The information on our website is not part of this prospectus.
Development of NB S101 for Osteoporosis
We have conducted various studies of NB S101 in animal species and have conducted phase I and II clinical trials in humans. The phase I pharmacokinetic and bioequivalence study was conducted in healthy human volunteers in order to determine the blood levels of strontium at various points in time following administration of a single dose of NB S101. The phase II dose-ranging and efficacy study was conducted in postmenopausal women with low bone mineral density in order to determine the drug’s impact on bone resorption and bone mineral density, and to determine the blood levels of strontium over longer periods of time. In addition to these human clinical trials, we have completed long term toxicology and safety studies in rodents and non-rodents.
Development of NB S101 for Other Indications
We intend to explore the possibility of developing a strontium product for non-osteoporosis indications in which the uncoupling action of strontium on bone metabolism would be expected to provide substantial therapeutic benefit. The pharmacodynamic action of strontium on bone simultaneously provides an anabolic and an antiresorptive effect. We believe this dual action may stimulate formation and in-growth of new bone, while at the same time allowing normal bone remodeling and removal of damaged, or necrotic, bone from specific skeletal sites that need bone rejuvenation. Thus, we expect that new clinical indications may include orthopedics, secondary osteoporosis (including glucocorticoid-induced osteoporosis), osteonecrosis, and osteoarthritis.
Commercial products for these indications could be developed as different indications for NB S101, or, alternatively, we could develop other strontium product(s) covered by our intellectual property portfolio for these other indications. We are currently conducting feasibility studies and evaluations to assess commercial potential, clinical opportunity and possible regulatory and development strategies. We plan to conduct more preclinical studies for one or more of these opportunities in the near future. Additionally, we may enlist strategic partners to develop NB S101 or other products in our portfolio for indications that we choose not to pursue ourselves. Development plans for the orthopedic and other indications are still under consideration. We also intend to expand our current product pipeline by internal development, acquisitionsand/or in licensing of additional product candidates, which may include pharmaceutical projects within the field of bone and joint-related disorders. We may look to major pharmaceutical companies as prospective partners to assist in the developing and eventual marketing of certain of our product candidates.
Osteoporosis Overview
Osteoporosis is a disease characterized by low bone mass and structural deterioration of bone tissue, leading to bone fragility and increased susceptibility to fractures. In initial stages, the disease predominantly affects trabecular bone, and skeletal sites (such as spine, hip and wrist) that are rich in trabecular bone are particularly subject to increased risk of osteoporotic fracture. Osteoporosis is defined according to bone mineral density, or BMD, measurements, and a woman is diagnosed with osteoporosis when BMD is at least 21/2 standard deviations below the
27
healthy premenopausal mean. A woman is diagnosed with osteopenia, a precursor to osteoporosis, when her BMD is between one and 21/2 standard deviations below the healthy premenopausal mean.
Osteoporosis is often called the “silent disease” because bone loss occurs without symptoms. People may not know that they have osteoporosis until their bones become so weak that a sudden strain, bump or fall causes a fracture, or a vertebra to collapse. Collapsed vertebrae may initially be felt or seen in the form of severe back pain, loss of height, or spinal deformities such as kyphosis or stooped posture. Assessment methods for evaluating skeletal strength, integrity and turnover rate are key in diagnosing and treating osteoporosis. BMD measurement is the currently-recommended primary clinical diagnosis tool. Reliable equipment exists for quantitative assessment of BMD at various anatomical sites, the most important of which is the lower part of the spine, which is very susceptible to the deleterious effects of osteoporosis because it is made up almost exclusively of trabecular bone. In the last decade, other tools, including imaging techniques such as MRI, ultrasound and very specific biochemical markers of bone resorption and formation have been used to accurately quantify bone status and rapidly demonstrate therapeutic effects of medical interventions on bone metabolism.
According to the National Osteoporosis Foundation, or NOF, osteoporosis is a major public health threat for an estimated 44 million Americans, or 55% of people 50 and older. In the U.S. today, 10 million people are estimated to already have osteoporosis and almost 34 million more are estimated to have osteopenia, which places them at increased risk for osteoporosis. Approximately 80% of those with osteoporosis in the U.S. are women. Osteoporosis is similarly widespread in Europe, and epidemiological studies in China and India point to similar prevalence there. Extensive scientific literature documents osteoporosis prevalence in both sexes. Depending on the skeletal sites and populations studied, most studies show an osteoporosis prevalence of 15% to 25% among postmenopausal women; prevalence of osteopenia is substantially higher. Although the prevalence of osteoporosis is lower among men, studies show that by age 70, most men lose bone at a rate equivalent to women. Studies in both the U.S. and in Europe estimate that approximately five percent of men over 50 have osteoporosis.
The most severe consequence of osteoporosis is skeletal fracture. According to the NOF, one in two women and one in four men over age 50 can be expected to have an osteoporosis-related fracture at some time. Osteoporosis is responsible for more than 1.5 million fractures in the U.S. annually, including more than 300,000 hip fractures, approximately 700,000 fractures of vertebrae, 250,000 of wrist, and 300,000 at other skeletal sites. While hip fractures are two to three times more frequent in women than men, the one-year mortality is nearly twice as high for men, indicating the seriousness of osteoporosis in men.
Also according to the NOF, currently an average of only 20% of people with osteoporosis are diagnosed, and of those diagnosed, less than half are treated with medication to prevent further bone loss or to increase bone density. In men particularly, osteoporosis awareness is very low. Attempts to close this gap are expected to be a major driver for future growth of the osteoporosis market. As Western populations age in the coming decades, we and others believe that osteoporosis prevalence will increase substantially. We also believe that better treatments will lead to a higher percentage of patients on medication.
28
Current Osteoporosis Treatments
Several classes of compounds are currently used in routine clinical practice for treatment and prevention of osteoporosis, including bisphosphonates, Selective Estrogen Receptor Modulators, or SERMs, Hormone Replacement Therapy, or HRT, and parathyroid hormone, or PTH. Calcitonin products are also used in osteoporosis. There is one strontium compound approved in Europe, Protelos®, which is from the same class of pharmaceuticals as our investigational drug NB S101. A summary of the reported magnitude of skeletal effects and commonly reported side effects associated with each class is briefly outlined below.
| | | | | | | | | |
Pharmaceutical
| | Magnitude of
| | | | | | |
Class | | Fracture Prevention | | Market Leader(s) | | Known Side Effects | | Mechanism of Action |
| |
| Bisphosphonates | | 41 to 49% overall reduction in vertebral fracture risk after 3 years. | | Fosamax (Alendronate), Merck. Actonel (Risedronate), Procter & Gamble. Boniva, GlaxoSmithKline and Roche. | | Gastrointestinal side effects such as esophagitis, esophageal ulcers and esophageal erosions. Possible association with osteonecrosis of the jaw, atrial fibrillation and bone/muscle pain. | | Antiresorptive (potent) |
| SERMs | | 35% overall risk reduction in vertebral fracture after 3 years. | | Evista (Raloxifene), Eli Lilly | | Certain estrogen-related effects such as hot flashes and leg cramps. | | Antiresorptive |
| HRT | | Varying reports of 25 to 50% risk reduction. | | Premarin, Wyeth. | | Increased risk of certain cancers and possibly cardiovascular disease. | | Antiresorptive |
| PTH | | 65% reduction in relative risk of vertebral fracture after 19 months. | | Forteo, Eli Lilly. Preos, NPS pharmaceuticals (Europe only). | | Few and minor in clinical trials (dizziness, leg cramps). Osteosarcoma seen in animals. | | Anabolic hormone |
| Strontium | | 49% reduction in vertebral fracture risk after 12 months. 41% overall reduction after 3 years. | | Protelos (strontium ranelate), Servier. | | Diarrhea (usually transient). Slight increase in venous thromboembolism. | | Both an antiresorptive and an anabolic effect |
| Calcitonin | | Limited clinical documentation for significant fracture reduction. | | Miacalcin, (salmon Calcitonin) Novartis. Fortical (Unigene Laboratories). | | Nasal irritation. More rare: Bloody urine, breathing difficulty, dizziness | | Antiresorptive. Calcium regulating hormone |
Calcium and vitamin D are common and widespread interventions for promoting bone health, and both play a vital role in the maintenance of skeletal integrity. Although they also serve a useful role in correcting dietary deficiencies, calcium and vitamin D treatment does not alter current bone turnover or reverse established osteoporosis. Calcium and vitamin D, however, are useful supplements to other osteoporosis therapies.
29
Development History of NB S101 and Overview of Development Strategy
Early in the development of NB S101, during 2003 and 2004, we performed studies of chemical and physical properties of a selected range of physiologically acceptable strontium salts. Based on the outcome of these studies we selected certain salts for furtherin vivopharmacokinetic and pharmacodynamic testing. Based on these studies, we chose strontium malonate as our lead product candidate for further clinical development. We also refer to the pharmaceutical formulation of strontium malonate that we are developing as NB S101.
The clinical development of NB S101 was initiated in 2004 with preclinical safety, toxicology and pharmacodynamic studies, in addition to work on formulation. During 2005, we conducted human phase I studies to obtain pharmacokinetic data. From 2006 to 2007 we conducted a human phase II clinical trial to obtain evidence of the efficacy by measuring BMD and biochemical markers of bone resorption and bone formation. Our preclinical and clinical studies have been conducted according to International Conference on Harmonization, or ICH, guidelines. Future clinical trials are anticipated to include at least one phase III human clinical trial, measuring the effect of NB S101 on bone fracture reduction.
Our clinical development plan for NB S101 contemplates pursuing multiple registration pathways and clinical indications with the main emphasis being on filing a New Drug Application, or NDA, with the FDA for NB S101 for prevention and treatment of osteoporosis. We anticipate that approval of our NDA will require a full development program complying with FDA guidelines, including its “Clinical evaluation of agents used in the prevention and treatment of postmenopausal osteoporosis,” published in 1994. We also believe we will need to submit a full safety and toxicology package for NB S101. The European Agency for Evaluation of Medicinal Products, or EMEA, has published guidelines similar to the FDA’s for development of products for osteoporosis, and we have designed our program to also comply with these requirements. We plan to use the preclinical package, phase I studies, phase II studies and phase III studies for registration in both the U.S. and Europe. Because strontium ranelate has been approved by EMEA, we were able to include strontium ranelate treatment for comparison purposes in clinical trials performed in Europe. We may also be able to use efficacy and safety data from published studies on strontium ranelate to a somewhat lesser extent, although this will have to be discussed with the regulatory authorities. We may also use other published literature on strontium toxicology to a limited extent.
As we continue development of NB S101, we plan to retain the internal core competency required for design, planning, supervision and interpretation of the studies, although we expect all the clinical studies will be performed externally in collaboration with suitable Clinical Research Organizations, or CROs, and that preclinical studies will be conducted in collaboration with academic research institutions and CROs. We have outsourced all manufacturing of the active pharmaceutical ingredient, or API, and the finished medical product, which is also called the investigational medical product, or IMP, to contract manufacturers who produce, package and supply the required materials according to our specifications.
Preclinical Experience with NB S101
We have designed our preclinical program to comply with regulatory requirements and relevant ICH guidelines. In compliance with the requirements, we carried out toxicological studies in rodents and in non-rodent species. We are continuing additional safety and toxicology work as NB S101 advances in clinical development and we expect to continue to accrue additional data throughout the development timeline of NB S101. To assess the effects of NB S101 on skeletal metabolism and bone strength, we have also conducted various pharmacodynamic studies, including pre-clinical efficacy studies in an animal model of osteoporosis.
In addition to the preclinical studies with NB S101 we carried out, comprehensive documentation on the safety and toxicological properties of strontium salts is available from reference literature. A number of studies to assess possible health effects of various strontium salts suggest strontium itself is considered safe. Based primarily on rat studies, the U.S. Department of Human and Health Services has established a lowest observed adverse effects level of 550 mg/kg/day. For comparison purposes, the highest strontium malonate dose in our recent phase II clinical was approximately 40 mg/kg/day, corresponding to about 20 mg/kg/day ionic strontium.
30
Phase I Studies
In September 2005, we completed a phase I randomized, pharmacokinetic and bioequivalence study of NB S101 in 60 healthy male volunteers divided into five different groups. The study’s primary objective was to obtain pharmacokinetic data (Area under the curve, or AUC, and plasma concentration at the peak, or Cmax) on strontium uptake and elimination from oral administration of NB S101. Secondary objectives included identification of the dose of NB S101 bioequivalent to 2.0 grams Protelos® (the dose approved in Europe), obtaining data on markers of calcium balance and assessment of the dynamic effects of NB S101 on biochemical markers of bone turnover.
From the phase I clinical trial, we calculated that 0.99 grams of NB S101 was bioequivalent to 2.0 grams of Protelos®, and based on this finding we included a 1.0 gram dose of NB S101 in our phase II study, as well as a higher dose (2.0 grams) and a lower dose (0.75 grams), each administered in a tablet once a day. For comparison, our 1.0 gram tablet contains 465 mg of strontium, as compared to 680 mg of strontium contained in the approved dosage of Protelos as a result of our product candidate’s improved bioavailability. We believe the improved bioavailability is due to the different anion (ranelate versus malonate) to the strontium, since ranelate (used in Protelos) appears to bind to strontium stronger than malonate (used in NB S101), thus impairing the uptake of free strontium from the intestinal lumen.
At a later point we plan to carry out an additional phase I study addressing and quantifying the effect of food and calcium intake on absorption of NB S101. Calcium is known to affect strontium absorption, probably because strontium and calcium are taken up in the intestine by the same transport mechanisms, resulting in reduced absorption of strontium when calcium is present. Our study plan currently calls for the food and calcium interaction study to be carried out as a controlled, randomized, cross-over study with four interventions, comprising single dose administrations. We estimate that the study population will comprise approximately 30 postmenopausal women. While we believe this study does not need to be completed prior to initiating our next efficacy-demonstrating clinical trial, we believe it needs to be completed prior to our submitting an NDA to the FDA.
Our complete phase I program and the package of preclinical safety and toxicology experiments has been designed to be compatible with pursuit of clinical development of NB S101 in osteoporosis and in other clinical indications.
Phase II Studies
In November 2007, we completed a phase II double-blind, placebo-controlled randomized study of NB S101 in 289 post-menopausal women with low bone mineral density in order to assess the efficacy of our investigational drug. The trial participants were divided into five groups, with three of these groups receiving different doses of NB S101 (0.75 grams, 1.0 grams, and 2.0 grams), one group receiving placebo and one group receiving the approved 2.0 gram dose of Protelos® in sachet formulation (because Protelos is not in tablet formulation, the Protelos arm was not blinded to the treatment received). The patients in the trial were treated for 12 weeks andfollowed-up for four to eight weeks following completion of the treatment portion of the trial.
The primary endpoint in the study was the change in patients’ bone resorption, as measured by the biochemical marker CTX-1. We also captured data regarding the effect of NB S101 on bone formation, bone mineral density and a marker of cartilage degradation as well as the levels of strontium in patients’ serum and the side effects. The goal of our phase II study was to document the efficacy of NB S101 and to establish an effective and tolerable dose of NB S101 that will enable us or a pharmaceutical collaborator to initiate phase III clinical trials that would be designed to obtain approval of NB S101.
31
We met the primary endpoint for all three doses of NB S101 evaluated in the trial, showing a statistically significant reduction in CTX-1 as compared to placebo, in a dose dependent manner. The reductions in serumCTX-1 compared to placebo were 13.5 percent in the group receiving the lowest dose of NB S101 (0.75 grams), 15.5 percent in the group receiving the middle dose of NB S101 (1.0 gram), and 22.2 percent in the group receiving the highest dose of NB S101 (2.0 gram) (p<0.001 for all doses vs. placebo). These reductions in the marker for bone resorption were also numerically superior compared to the approved dose of Protelos (Protelos reduced the marker for bone resorption by 8.5 percent), although only the highest dose of NB S101 was statistically significant compared to Protelos. Data from the primary endpoint of the trial are shown in the chart below.
Primary endpoint — percent change in CTX-1 from baseline to week 12
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | Strontium Malonate | | | | |
| | | Placebo
| | | 0.75 g
| | | 1 g
| | | 2 g
| | | Protelos®
| |
| | | N = 56 | | | N = 57 | | | N = 57 | | | N = 58 | | | N = 56 | |
| |
| Mean change from baseline relative to placebo, in percent | | | N/A | | | | (13.48 | ) | | | (15.54 | ) | | | (22.17 | ) | | | (8.50 | ) |
| p-value versus placebo | | | N/A | | | | <0.001 | | | | <0.001 | | | | <0.001 | | | | 0.030 | |
32
In addition to meeting the primary endpoint, we also saw increases in BMD among the patients receiving NB S101 in comparison to patients on placebo, as shown in the chart of lumbar spine BMD below.
Lumbar spine — percent change in BMD from screening to week 12
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | Strontium Malonate | | | | |
| | | Placebo
| | | 0.75 mg
| | | 1 g
| | | 2 g
| | | Protelos®
| |
| | | N = 56 | | | N = 56 | | | N = 51 | | | N = 56 | | | N = 55 | |
| |
| Mean change from screening relative to placebo | | | N/A | | | | 2.29 | | | | 1.99 | | | | 2.66 | | | | 1.96 | |
| p-value versus placebo | | | N/A | | | | 0.020 | | | | 0.049 | | | | 0.008 | | | | 0.048 | |
In our phase II clinical trial, NB S101 was well-tolerated and side effects were generally mild in all dose groups. There were no significant differences in the side effect profiles between the different treatment groups. It is our belief that the trial clearly demonstrates the beneficial effect of NB S101 on bone resorption and on patients’ bone mineral density after only a short treatment period and that more advanced clinical trials should be conducted to support the approval of NB S101.
Phase III Studies
We are designing our development plan for NB S101 for osteoporosis to comply with both U.S. and European guidelines. We plan to discuss with U.S. and European regulators whether the significant increases in BMD that we observed in the phase II trial will be sufficient to allow us or a collaborator to immediately enter phase III testing, or whether a longer duration BMD study would be required.
The FDA and EMEA require positive results from phase III studies before they will approve an NDA. In order to obtain approval for a new drug for the treatment of osteoporosis, current regulatory guidelines require placebo-controlled double-blind studies with fracture reduction as the primary endpoint. An adequate fracture-prevention trial represents a significant time and resource investment, as the treatment duration in the required clinical studies can be up to three years, although recent trials have been conducted with a two-year treatment duration and more recently an advisory group has recommended that these trials be reduced to 18 to 24 months in duration. These trials raise ethical questions concerning the use of placebo groups when approved drugs are available to treat
33
osteoporosis, and both European and U.S. regulatory authorities are currently debating whether continued use of such trials can be recommended. If future guidelines prevent or limit the use of placebo control in such studies, we may plan to alternatively conduct a non-inferiority study against an established osteoporosis treatment or otherwise modify our plans based on any new guidelines established for approval of drugs to treat or prevent osteoporosis.
Based on the current FDA and EMEA guidelines and anticipated changes, we anticipate the primary objective of a phase III study will be to evaluate the ability of NB S101 to prevent incident vertebral fracture. We also anticipate that we would collect data on non-vertebral fractures, spine, hip and forearm BMD, and biochemical markers of bone and cartilage turnover. We estimate that the phase III study may enroll up to 3,000 osteoporotic women, comparing one dose of NB S101 to placebo over a two-to three-year treatment period, with a possible two-year extension. Based on continuing analysis of the phase II data and discussions with regulatory agencies, we are continuing statistical assessments before we finalize any phase III clinical trial design and determine the specific number of patients needed.
Our clinical development plans are based on the current profile of NB S101, our experience with preclinical and clinical testing of NB S101, our understanding of current FDA and EMEA guidelines, and our review of publicly available data on potentially competing products. We expect that continuing analysis of all data available on strontium, NB S101 specifically, and potentially competing drugs in development will increase our knowledge in the field, and may lead to our modifying our current development strategy. In addition, changes in market conditions or regulatory requirements may also require us to modify our development plans.
Osteoporosis Market
We believe that osteoporosis and other diseases of skeletal deterioration remain underserved medical needs, despite recent medical advances in the area and the progress of other drugs in development. For this reason, the American Society for Bone and Mineral Research, or ASBMR, and the International Osteoporosis Foundation, or IOF, have actively promoted osteoporosis awareness among medical professionals, legislators and the public.
The worldwide market for prescription osteoporosis products is estimated by Datamonitor at $7.9 billion in product sales in 2008. We believe that the market for osteoporosis treatments will continue to grow as the population grows older and lives longer. We also believe that the market potential is even greater because many patients with osteoporosis do not comply with their prescribed treatments due to the adverse side effects. We believe that our investigational drug NB S101 offers benefits compared to the current therapies and thus has the potential to be a leading treatment for osteoporosis, if approved.
Our Strategy
Our goal is to add value to clinical development products in areas where we have expertise such as bone disease and women’s health. We aim to develop and commercialize new medications that offer advantages over existing treatments. Our development program for NB S101 is an example of our progress in executing upon this goal. We are also seeking to develop or acquire other new treatments for bone disease or women’s health. Our strategy is to:
| | |
| | • | Focus on unmet, underserved or high value markets; |
| |
| | • | Efficiently select product candidates to minimize development risk and maximize commercialization opportunities; |
| |
| | • | Structure attractive co-development and commercialization agreements to maximize up-front guaranteed and milestone payments; and |
| |
| | • | Identify business opportunities and product candidates for attractive indications. |
We plan to use external collaborators, contract and clinical research organizations, and scientific and business contacts and consultants during our product evaluation and development efforts in order to access the most relevant expertise and identify the most appropriate potential development programs and partners.
34
Sales and Marketing
To commercialize NB S101, we intend to collaborate with a major global pharmaceutical company with experience marketing products to physicians that commonly prescribe treatments for osteoporosis, such as general practitioners, with especially broad sales, marketing, and distribution capabilities. However, we also plan to evaluate granting licenses to large, regional pharmaceutical companies (such as one license for North America, another for Europe and another for Japan and Asia) that have capability to market the product adequately in their respective territories.
Competition
The success of NB S101 will depend in part on our ability to achieve market share at the expense of existing, established products, to leverage favorably with future products in development and to grow new or existing markets. Currently, there are five classes of compounds approved for sale in the U.S. which we see as competition for the treatment and prevention of osteoporosis: bisphosphonates, parathyroid hormone, or PTH, Selective Estrogen Receptor Modulators, or SERMs, Hormone replacement therapy, or HRT, and Calcitonin. The following is a summary of these treatments:
| | |
| | • | Bisphosphonates are potent anti-resorptive agents with well established fracture reduction efficacy. Bisphosphonates are the leading treatments for osteoporosis, dominated by alendronate (Fosamax, Merck), and risedronate (Actonel, Procter&Gamble). Newer bisphosphonates have been introduced by Roche (ibandronate, Bonviva) and Novartis (zoledronate, Zometa). Bisphosphonates may be taken by men, unlike HRT and SERMs. New formulations for parenteral administration (i.e., intravenous or subcutaneous) are also being introduced. Major side effects include irritation of the gastrointestinal system, which require the patients to stand upright for a period after administration. Furthermore, the poor bioavailability of bisphosphonates limit oral intake (they are generally taken in the morning, and require fasting before and after intake). Osteonecrosis of the jaw bone is being reported in some bisphosphonate users. The FDA is looking into whether bisphosphonates increase the incidence of atrial fibrillation. More recently, the FDA issued an alert that advises physicians that the FDA believes many cases of bone and muscle pain are attributable to bisphosphonates. |
| |
| | • | PTH takes a central role in the body’s regulation of calcium homeostasis and bone turnover. It has a potent anabolic effect on bone tissues, stimulating the formation and maturation of bone-forming osteoblasts which in turn causes significant increases in new bone formation. To date, only one PTH product, Forteo (Eli Lilly), is marketed in the United States. PTH products, given as daily injections, are approved for severe osteoporosis, wherebuild-up of new bone is key in preventing further deterioration. However, PTH is not recommended for prophylactic or longer term therapy. Typically, treatment duration is limited to two years because of the risk of osteosarcoma revealed in preclinical testing. Moreover, treatment with PTH is inconvenient because it is administered by daily injections. |
| |
| | • | SERMs were developed to preserve beneficial effects of estrogen while minimizing potential side effects. Currently, only one SERM is approved (raloxifene, Evista, by Eli Lilly), but additional SERMs are currently in development or being evaluated by the FDA for approval. SERM action on bone is mainly anti-resorptive, with less potency than the bisphosphonates, but SERMs still significantly reduce fractures in large clinical trials. A primary concern is the SERMs potential ability to affect tissues other than bone, due to the widespread occurrence and pleitrophic effects of sex steroid receptors. Endometrial safety and CNS related effects (e.g. hot flash occurrence) also remain issues with SERMs. |
| |
| | • | HRT was a common therapy among postmenopausal women for many years, as estrogen offers relief of many menopausal inconveniences in addition to its bone protecting effect. However, studies such as the Women’s Health Initiative, show that HRT use is associated with increased risk of breast cancer, cardiovascular disorders and other side effects. Since these studies have been published, there have been significant declines in the usage of HRT, and we believe it will play a much smaller role in the treatment and prevention of osteoporosis in the future. The class is dominated by Premarin (Wyeth). |
35
| | |
| | • | Calcitonin is a natural hormone involved in the physiological maintenance of calcium homeostasis and regulation of skeletal metabolism. It decreases release of calcium by down-regulating the activity of the bone resorbing osteoclasts. Calcitonin is available in a nasal spray, but it is currently used in a relatively small proportion of the osteoporotic population. Although the effect of calcitonin on BMD is lower than those seen with HRT, SERMs, PTH and bisphosphonates, calcitonin has been reported to have some analgesic effects, which may be useful in patients with painful symptoms. Calcitonin has one of the best side effect profiles among currently approved treatments for osteoporosis, which probably is among the key rationale for its use in osteoporosis therapy. Widespread use in osteoporosis is hampered by the lack of clinical proof for fracture prevention. |
One osteoporosis product containing strontium has been developed by the French pharmaceutical company Servier, SA. Their product contains strontium ranelate as the active ingredient and is sold under the trade name Protelos® in Europe. Protelos was approved for sale by the Committee for Medicinal Products for Human Use, or CHMP, of the EMEA in late 2004, and Servier began marketing Protelos in most European countries during 2005. Protelos is formulated in daily 2.0 gram sachets, which are packets of powdered drug substance that must be reconstituted in water prior to ingestion. The phase III program conducted by Servier prior to receiving approval of Protelos consisted of two double-blind placebo controlled clinical trials, the Spinal Osteoporosis Therapeutic Intervention, or SOTI, trial for assessing the ability of strontium ranelate to reduce the risk of vertebral (spinal) fractures and the Treatment Of Peripheral Osteoporosis study, or TROPOS, for assessing the ability of Protelos to reduce the risk of peripheral (non-spinal) fractures. Almost 7,000 osteoporotic women in total completed these two three-year studies. In these trials, strontium ranelate demonstrated statistically significant reductions in both vertebral and non-vertebral fractures. Strontium ranelate treatment also increased patients’ BMD and showed concomitant decreases in bone resorption and increases in bone formation. One possible adverse effect of the use of strontium ranelate was an increase in venous thromboembolism (25 cases among 3,352 strontium ranelate treated women compared to 14 cases among 3,317 placebo treated women). In addition, transient mild diarrhea was also seen among some of the strontium-ranelate treated women. Other side-effects in the clinical trials were generally few. In November 2007, the EMEA noted that approximately 16 cases of drug rash with eosinophilia and systemic symptoms, or DRESS, had occurred in patients treated with Protelos after approximately 570,000 patient-years of worldwide exposure.
To our knowledge, Protelos® is currently the only prescription pharmaceutical product containing ionic non-radioactive strontium, and thus is a relevant competitor for NB S101. We believe that NB S101 is in a superior dosage form (tablet vs. sachet) and that it may have a superior side-effect profile compared to Protelos, while having similar or better efficacy on the skeletal system. Since its introduction, quarterly sales of Protelos have increased to over $60 million (fourth quarter of 2007), reaching an annual level of over $200 million in 2007, an increase of over 100% year over year (2007 compared to 2006, as reported by IMS, an organization that tracks sales of prescription pharmaceutical products).
Most major pharmaceutical companies have established an osteoporosis franchise or are in late-stage clinical development with osteoporosis products and several biotechnology companies are pursuing new osteoporosis drugs. One new approach is based on Osteoprotegrin, or OPG, a circulating ’decoy-receptor’ for the RANK-L hormone (receptor activator of nuclear factor-kappaB-ligand). RANK-L up-regulates formation of osteoclasts and thus increases bone degradation, and a RANK-L-specific monoclonal antibody (AMG-162, or denosumab, Amgen) that inhibits osteoclast activity is currently in late stage clinical development. Other drug targets currently being investigated include: matrix metallo-proteinases, or MMPs, such as cathepsin K; ion channels of importance for osteoclast function such as CLC-7 (Nordic Bioscience); and other endocrine factors involved in physiological regulation of bone turnover (e.g. Calcitonin, marketed by Novartis andGLP-2 developed by Sanos Bioscience). An important goal for future therapies is to evoke a long-lasting skeletal benefit to enable abuild-up of lost bone mass. We believe that one possible way to achieve net bone growth is by positively uncoupling bone resorption and formation as has been observed in long-duration strontium therapy. Thus, we anticipate that the strontium class of pharmaceuticals provides a valuable alternative to existing prophylactic and therapeutic treatment options for osteoporosis.
36
Intellectual Property
Our goal is to (a) obtain, maintain, and enforce patent protection for our product candidates, formulations, processes, methods, and other proprietary technologies, (b) preserve our trade secrets, and (c) operate without infringing on the proprietary rights of other parties, worldwide. We seek, where appropriate, the broadest intellectual property protection for product candidates, proprietary information, and proprietary technology through a combination of contractual arrangements and patents.
We have filed more than 25 individual patent applications that are pending worldwide. Three international patent applications filed on May 6, 2004, form the foundation of our patent portfolio, and the first of these patents was issued in Europe on October 18, 2006. These three patent applications claim water soluble strontium salts and methods of treatment using these salts, controlled release strontium salt compositions, and combinations of strontium salts and other agents. The three key patent applications and their primary claims are as follows:
| | |
| | • | PCT/DK2004/000328: “Water Soluble Strontium Salts for Use in the Treatment of Cartilageand/or Bone Conditions.” Claims are generally directed to water soluble strontium salts and the use of such formulations to treat metabolic bone diseases. The most notable clinical indication claimed in this application is osteoporosis, but several other metabolic bone diseases are also claimed. The application has entered the national/regional phase in the U.S., Japan, Australia, Canada and before the European Patent Organization, or EPO. The European application was granted as a patent on October 18, 2006. A divisional application in Europe containing broader claims is still pending. Acontinuation-in-part, or CIP, application based on PCT/DK2004/000328 was filed in the U.S. This CIP application contains additional data and is generally directed to compositions and methods for treating bone diseases using strontium salts including NB S101. The U.S. Patent Office issued an initial office action rejecting the claims in the CIP application, and we have submitted a response to the office action requesting more limited claims. In Australia and Japan a request for examination has been filed. In Canada a request for examination is due in May 2009. |
| |
| | • | PCT/DK2004/000327: “Combination Treatment with Strontium for the Prophylaxisand/or Treatment of Cartilageand/or Bone Conditions.” This application is generally directed to compositions containing strontium salts and one or more other active substances for the treatment of boneand/or cartilage disorders such as osteoporosis. The application has entered the national/regional phases in the U.S., Japan, Australia, Canada and before the EPO. In Australia and Japan a request for examination has been filed. In Canada a request for examination is due in May 2009. In Europe the examination has started and an official communication has issued to which we are preparing a response. |
| |
| | • | PCT/DK2004/000326: “Controlled Release Composition Containing a Strontium Salt.” This application is generally directed to controlled release formulations of strontium salts and uses for treating a broad variety of cartilageand/or bone diseases, including osteoporosis. The application has entered the national/regional phases in the U.S., Japan, Australia, Canada and before the EPO. In Australia and Japan a request for examination has been filed. In Canada a request for examination is due in May 2009. In Europe the examination has started and an official communication has issued to which we have filed a response. |
We have received international search reports (performed by the European Patent Office) for the patent applications listed above. The International Searching Authorities of the European Patent Organization identified some prior art, which might have potential impact on the patentabilityand/or allowable scope of the claims of these applications. As is typical in patent prosecution, certain limitations to the claims may have to be introduced during the prosecution of the patent applications before their actual grant and there can be no assurance that any of our pending patent applications will result in issued patent claims. However, it is difficult to predict the ultimate scope of the patent claims, if any, we will be granted by the patent authorities.
In July 2007, Intellectual Property Services filed an objection to the issued patent with the European Patent Office corresponding to PCT/DK2004/000328. We responded to the European Patent Office in March 2008. We cannot predict the outcome of this opposition, which is likely to take several years to complete. However, regardless of the outcome of this opposition, the manufacture and use of our investigational drug NB S101 is the subject of other pending patent applications which would provide additional patent protection, if received.
37
In addition to the above, we have filed a number of additional applications in order to protect relevant new developments and scientific and clinical knowledge that we have gained. These additional patent applications are for specific methods of manufacturing strontium salts and for other clinical indications, such as treatment of osteonecrosis. All international patent applications which we have filed designate all countries which are party to the Patent Cooperation Treaty, or PCT, which is effectively all major industrialized countries. Our current strategy is to file applications as PCT applications seeking worldwide coverage, and then file applications nationally in the U.S., Europe, Canada, Japan, and Australia.
In addition to the protection that may be provided by issued U.S. patents, under the Drug Price Competition and Patent Term Restoration Act of 1984, which is also known as the Hatch-Waxman Act, a portion of a product’s patent term that was lost during clinical development and application review by the FDA may be restored. The Hatch-Waxman Amendments also provide for a statutory protection, known as nonpatent market exclusivity, against the FDA’s acceptance or approval of certain competitor NDAs. Patent term restoration can compensate for patent life lost during product development and the regulatory review process by returning up to five years of patent life for a patent that covers a new product or its use. This period is generally one-half the time between the effective date of an IND (falling after issuance of the patent) and the submission date of a NDA, plus the time between the submission date of a NDA and the approval of that application. Patent term restorations, however, are subject to a maximum extension of five years, and the patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years. The application for patent term extension is subject to approval by the United States Patent and Trademark Office in conjunction with the FDA. It takes at least six months to obtain approval of the application for patent term extension. Up to five years of interim one year extensions are available if a product is still undergoing development or FDA review at the time of its expiration.
Third Party Patent Rights
We believe we will have freedom to develop our strontium salts for the treatment of metabolic bone diseases such as osteoporosis. However, there are other patents for the use of strontium. Within the class of strontium-based pharmaceutical products, Servier has several patents. These include U.S. Patent No. 5,128,367 (European equivalent EP-B-0 415 850), which is directed to strontium ranelate and other metal ranelates, as well as methods and compositions for treating osteoporosis. In addition, Servier’s U.S. Patent No. 4,939,164 (European equivalent EB-P-0 349 432) is directed to a specific strontium salt of pentanedioic acid, as well as methods and compositions for treating osseous diseases using this salt. Also, Servier’s U.S. Patent No. 5,075,336 (European equivalent EB-P-0 445 025) describes specific types of carboxylic acid salts, including strontium salts as well as methods and compositions for treating osseous diseases using these salts. Servier’s U.S. Patent No. 5,856,356 (European equivalent EB-P-0 813 869) describes the use of various strontium salts, including strontium ranelate and strontium malonate, for treatment of arthrosis. Due to prior art restrictions, Servier’s European patent EB-P-0 813 869 has been limited to one claim, the use of strontium ranelate for the treatment of osteoarthritis. Servier has also obtained three additional U.S. patents, Nos. 7,091,364 (European equivalent EP-A-1 403 265), 7,105,683 (European equivalent EP-B-1 403 264 and 7,214,805 (European equivalent EP-A-1 403 266), which are directed to methods of manufacturing strontium salts and chemical intermediates used to prepare such salts.
In addition to the patents held by Servier, there are other issued patents describing the use of strontium salts for treatment of human subjects. U.S. Patent No. 5,851,556 issued to the French company L’Oreal describes the use of various alkaline earth metal salts such as strontium salts for the treatment of skin conditions, bronchopulmonary conditions, pain, gastrointestinal conditions, central nervous system disorders, disorders associated with the release of TNF alpha and disorders associated with the release of substance P, although these claims were not allowed in Europe. The Norwegian company Santosolve has one issued patent — EP 1429792, which generally describes strontium salts, and compositions and methods using such salts for treating sub-dermal soft tissue pain and herpetic infections, and one U.S. patent, No. 7,241,460, with claims directed to compositions and methods of treating sub-dermal soft tissue pain by topically applying a strontium composition. Santosolve also has one published patent application, WO 04084920 (European equivalent EP-A-1 605 955 and US 2007053994), which is generally directed to methods for treating inflammation with a strontium compound. There may be other patents or patent applications of which we are not aware that may represent a more dominant position compared to ours.
38
In November 2005, we entered into a patent license agreement with Aditech AB, or Aditech. Aditech is 100% owned by Nordic Biotech, and at the time of the agreement Nordic Biotech owned 100% of us. The agreement provides Aditech with rights to develop pharmaceutical products for certain non-osteoporosis indications contained in our patent portfolio, in exchange for Aditech’s obligation to pay us a royalty on product revenues generated from products developed under the agreement. Also under the agreement, Aditech has an exclusive worldwide license to our patents containing certain compounds other than strontium compounds, which are outside the core focus of our business, for which we are also entitled to receive a royalty on product revenues. We, in turn, have an exclusive worldwide license to Aditech’s patents for strontium compounds. In exchange for the rights granted, Aditech paid us $750,000 in February 2006. We are also entitled to a 2.5% royalty on future net sales of products developed by Aditech under the agreement. Aditech is entitled to a 1.5% royalty on future net sales of products containing strontium compounds that we develop. We currently believe that it is unlikely that Aditech will develop products that are licensed from us.
To supplement our patent portfolio, we also depend on the skills, knowledge, and experience of our scientific and technical personnel, as well as that of our advisors, consultants, and other contractors. To help protect our patentable proprietary know-how, and for inventions for which patents may be difficult to enforce, we rely on trade secret protection and confidentiality agreements to protect our interests. We require all employees, consultants, advisors and other contractors to enter into confidentiality agreements that prohibit disclosure of confidential information and, where applicable, require disclosure and assignment to us of ideas, developments, discoveries and inventions important to our business.
Government Regulation
The FDA, comparable foreign regulators and state and local pharmacy regulators impose substantial requirements upon clinical development, manufacture and marketing of pharmaceutical products. These and other entities regulate research and development and the testing, manufacture, quality control, safety, effectiveness, labeling, storage, record keeping, approval, advertising, and promotion of our product candidates.
The drug approval process required by the FDA under the Food, Drug, and Cosmetic Act generally involves:
| | |
| | • | Preclinical laboratory and animal tests; |
| |
| | • | Submission of an IND prior to commencing human clinical trials; |
| |
| | • | Adequate and well-controlled human clinical trials to establish safety and efficacy for intended use; |
| |
| | • | Submission to the FDA of a New Drug Application, or NDA; and |
| |
| | • | FDA review and approval of a NDA. |
The testing and approval process requires substantial time, effort, and financial resources, and we cannot be certain that any approval will be granted on a timely basis, if at all.
Preclinical tests include laboratory evaluation of the product candidate, its chemistry, formulation and stability, and animal studies to assess potential safety and efficacy. Certain preclinical tests must be conducted in compliance with good laboratory practice regulations. Violations of these regulations can, in some cases, lead to invalidation of the studies, requiring them to be replicated. In some cases, long-term preclinical studies are conducted concurrently with clinical studies.
We plan to submit the preclinical test results, together with manufacturing information and analytical data, to the FDA as part of an IND in the near future. This IND must become effective before we begin human clinical trials in the United States. The IND automatically becomes effective 30 days after filing, unless the FDA raises questions about conduct of the trials outlined in the IND and imposes a clinical hold, in which case, we must resolve the matters with the FDA before beginning clinical trials. It is possible that our submission may not result in FDA authorization to commence clinical trials.
Clinical trials must be supervised by a qualified investigator in accordance with good clinical practice regulations, which include informed consent requirements. An independent Institutional Review Board, or IRB, at each medical center reviews and approves and monitors the study, and is periodically informed of the study’s
39
progress, adverse events and changes in research. Progress reports are submitted annually to the FDA, and more frequently if adverse events occur.
Human clinical trials typically have three sequential phases that may overlap:
| | |
| | • | Phase I: The drug is initially tested in healthy human subjects or patients for safety, dosage tolerance, absorption, metabolism, distribution, and excretion. |
| |
| | • | Phase II: The drug is studied in a limited patient population to identify possible adverse effects and safety risks, determine efficacy for specific diseases and establish dosage tolerance and optimal dosage. |
| |
| | • | Phase III: When phase II evaluations demonstrate that a dosage range is effective with an acceptable safety profile, phase III trials to further evaluate dosage, clinical efficacy and safety, are undertaken in an expanded patient population, often at geographically dispersed sites. |
We cannot be certain that we will successfully complete any additional testing of our product candidates within any specific time period, if at all. Furthermore, the FDA, an IRB or the IND sponsor may suspend clinical trials at any time for various reasons, including a finding that subjects or patients are exposed to an unacceptable health risk.
Concurrent with these trials and studies, we also develop chemistry and physical characteristics data and finalize a manufacturing process in accordance with good manufacturing practice, or GMP, requirements. The manufacturing process must conform to consistency and quality standards, and we must develop methods for testing the quality, purity, and potency of the final products. Appropriate packaging is selected and tested, and chemistry stability studies are conducted to demonstrate that the product does not undergo unacceptable deterioration over its shelf-life. While we do not own manufacturing facilities, we contract with qualified third parties who must comply with manufacturing practices and procedures established by the FDA for the manufacture of bulk active pharmaceutical ingredients and finished products.
Results of preclinical and clinical trials, as well as manufacturing specifications, are submitted to the FDA, along with other data on the product, as part of a NDA for approval. Once the FDA accepts an NDA for filing, it begins an in-depth review. The FDA has substantial discretion in the approval process and may disagree with our interpretation of the data submitted. The process may be significantly extended by requests for additional information or clarification regarding information already provided. As part of this review, the FDA may also refer the application to an appropriate advisory committee, which is typically a panel of clinicians. Manufacturing establishments often are inspected prior to NDA approval to assure compliance with GMP requirements and with manufacturing commitments made in the application.
Submission of an NDA to the FDA typically requires payment of a fee, which we estimate may exceed $1 million at the time an application for NB S101 may be ready, if at all. Currently, the FDA generally assigns a goal of ten months for issuing its “complete response,” in which the FDA may approve the NDA, deny the NDA, or require additional information before coming to a decision on approval or denial of the NDA. The FDA can always decide the NDA does not warrant approval. If the FDA does approve the NDA, the product can become available for physicians to prescribe to patients. However, product approval may be withdrawn at any time if regulatory compliance is not maintained or safety problems occur. The FDA may require post-marketing studies, also known as phase IV studies, as a condition of approval, and may also require surveillance programs to monitor products that have been approved. The FDA has the power to require changes in product labeling or prohibit further marketing based on its discretion.
Satisfaction of the FDA’s regulatory requirements typically takes a number of years, but the actual time required may vary substantially based upon the type, complexity and novelty of the product. Government regulation may delay or prevent marketing of potential products for a considerable period of time and impose costly procedures on our activities. We cannot be certain that the FDA or other regulatory agencies will approve any of our product candidates on a timely basis, if at all. Success in earlier trials does not assure success in subsequent clinical trials. Data obtained from pre-clinical and clinical activities are not always conclusive and may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. Even if one of our product candidates receives regulatory approval, the approval may be significantly limited to specific indications or uses. Also, after regulatory approval is obtained, later discovery of previously unknown problems with a product may result in
40
restrictions on the product or even complete withdrawal of the product from the market. Delays in obtaining, or failures to obtain regulatory approvals would have a material adverse effect on our business.
Any products manufactured or distributed by us pursuant to FDA approvals are subject to pervasive and continuing FDA regulation, including record-keeping requirements, reporting of adverse experiences, submitting periodic reports, drug sampling and distribution requirements, manufacturing or labeling changes, record-keeping requirements, and compliance with FDA promotion and advertising requirements. Drug manufacturers and their subcontractors are required to register their facilities with the FDA and state agencies and are subject to periodic unannounced inspections for GMP compliance, imposing procedural and documentation requirements upon us and third-party manufacturers. Failure to comply with these regulations could result, among other things, in suspension of regulatory approval, recalls, suspension of production or injunctions, seizures, or civil or criminal sanctions. We cannot be certain that we or our present or future subcontractors will be able to comply with these regulations.
The FDA regulates drug labeling and promotion activities. We and our product candidates are also subject to a variety of state laws and regulations which may hinder our ability to market our product candidates. Whether or not FDA approval has been obtained, approval by foreign regulatory authorities must be obtained prior to commencing clinical trials, sales and marketing efforts in those countries. These approval procedures vary in complexity from country to country, and the processes may be longer or shorter than that required for FDA approval. We will likely incur significant costs to comply with these laws and regulations now or in the future.
In addition, the FDA’s policies may change, and additional government regulations may be enacted which could prevent or delay regulatory approval of our potential products. Increased attention to the containment of health care costs worldwide could result in new government regulations materially adverse to our business. We cannot predict the likelihood, nature or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the U.S. or abroad.
The U.S. Federal Trade Commission and the Office of the Inspector General of the U.S. Department of Health and Human Services, or HHS, also regulate certain pharmaceutical marketing practices. Government reimbursement practices and policies with respect to our product candidates are also important to our success.
We are subject to numerous federal, state and local laws relating to safe working conditions, manufacturing practices, environmental protection, fire hazard control, and disposal of hazardous or potentially hazardous substances. We may incur significant costs to comply with these laws and regulations. The regulatory framework under which we operate will inevitably change in light of scientific, economic, demographic and policy developments, and such changes may have a material adverse effect on our business.
Similar to requirements in the U.S., in Europe prior regulatory approval is required for phase I clinical trials. Thereafter, data from successful phase I studies are submitted to regulatory authorities to support applications for phase II studies. European authorities typically have one to three months, which may be extended in their discretion, to raise objections to proposed studies. One or more independent ethics committees also review relevant ethical issues, similar to reviews preformed by IRBs.
For European marketing approval, we submit to the relevant authority for review a Marketing Authorization Application, or MAA, providing information on the quality of the chemistry, manufacturing and pharmaceutical aspects of the product as well as non-clinical and clinical data.
The European Union provides two different, elective authorization routes for approval: centralized and decentralized. For NB S101 we have selected the centralized route, which, if successful, leads to approval for the entire European Union. Under this procedure our application will be reviewed by members of the Committee for Proprietary Medicinal Products, or CPMP, on behalf of EMEA. Based on that review, the CPMP will provide an opinion on safety, quality and efficacy to the European Commission, which makes the decision to grant or refuse authorization.
Approval in Europe can take several months to several years, and can be denied. Regulatory authorities conduct facilities inspections and review manufacturing procedures, operating systems and personnel qualifications. In many cases, each drug manufacturing facility must be approved, and further inspections may occur over the product’s life. The regulatory agency may require additional studies prior to approval and may also require post-
41
marketing studies or additional product surveillance to monitor for adverse effects. Further clinical studies are usually necessary for approval of additional indications. As in the U.S., the terms of any approval, including labeling content, may be more restrictive than expected and could affect the marketability of a product.
Failure to comply with ongoing requirements can result in suspension of regulatory approval and civil and criminal sanctions. European regulatory authorities have the authority to revoke, suspend or withdraw approvals, prevent companies and individuals from participating in the drug approval process, require product recalls, seize products, obtain injunctions to close non-compliant manufacturing plants and stop shipments of products.
Pricing Controls
Pricing for products that are approved is also subject to regulation. Requirements vary widely between countries and can be implemented differently in each country. Even if our drug candidates are approved, we may not be able to obtain favorable pricing arrangements for our products.
Third-Party Reimbursements
In the U.S., the European Union and elsewhere, pharmaceutical sales are dependent in part on the availability and adequacy of reimbursement from third party payers such as governments and private insurance plans. Third party payers are increasingly challenging established prices, and new products may have difficulty finding ready acceptance unless there is a clear therapeutic benefit.
Employees
As of June 30, 2008, we had three employees, all of whom were full time. Our Chief Executive Officer and two other employees are based in our Virginia office. On March 20, 2008, we reached an understanding with Matthew M. Loar, our Chief Financial Officer, regarding his intention to resign as our chief financial officer, effective May 1, 2008, in connection with our anticipated relocation to Virginia. On April 15, 2008, we entered into an agreement with Mr. Loar whereby he agreed to provide services as a consultant with us for up to ten hours per month and serve as our Principal Financial and Accounting Officer through September 1, 2008. There were no disagreements between Mr. Loar and us on any matter relating to our financial practices or policies that resulted in his resignation.
Facilities
We have office locations in Glen Allen, Virginia; San Francisco, California; and Copenhagen, Denmark. Research and development activities are generally contracted to others under our supervision, and accordingly we do not have our own research and development facilities. The Glen Allen, Virginia location comprises approximately 1,500 square feet of office space, and the operating lease requires monthly payments of approximately $2,500, expiring on December 31, 2008. The San Francisco location comprises approximately 250 square feet and requires monthly payments of approximately $3,600, also expiring on December 31, 2008. We recently reduced the size of its facilities in Copenhagen, Denmark and relocated its Danish operations to a shared location with an affiliate that does not require monthly lease payments or other consideration. During 2008, we plan to consolidate the financial and administrative functions currently performed at our San Francisco location to our Glen Allen facility and we do not plan to renew the San Francisco lease upon its expiration. We believe that we will be able to renew our Glen Allen lease upon its expiration or find alternative suitable office space which is readily available should we chose to relocate.
Legal Proceedings
We are not a party to any material legal proceedings nor are we aware of any circumstance that may reasonably lead a third party to initiate legal proceedings against us.
42
MANAGEMENT
Directors and Executive Officers
Set forth below are the names, ages and certain biographical information relating to our directors and executive officers, as of June 30, 2008.
| | | | | | | |
Name | | Age | | Position |
| |
| Jeremy Curnock Cook | | | 58 | | | Director |
| Klaus Eldrup-Jørgensen, M.D. | | | 49 | | | Chairman and Director |
| Christian Hansen, Ph.D. | | | 41 | | | Director |
| Bobby W. Sandage, Jr., Ph.D. | | | 54 | | | Director |
| Florian Schönharting, M.Sc. | | | 39 | | | Director |
| Christopher B. Wood, M.D. | | | 62 | | | Director |
| Philip J. Young | | | 50 | | | President, Chief Executive Officer and Director |
| Matthew M. Loar | | | 45 | | | Principal Financial and Accounting Officer |
There are no agreements or understandings for any of our directors or executive officers to resign at the request of another person and none of our directors or executive officers is acting on behalf of or will act at the direction of any other person.
Our executive officers are appointed at the discretion of our board of directors with no fixed term. There are no family relationships between or among any of our directors or executive officers.
Jeremy Curnock Cook, Executive Chairman, Bioscience Managers Limited
Jeremy Curnock Cook has been Executive Chairman of Bioscience Managers limited since August 2000. Previously, he was a Director of Rothschild Asset Management Limited and was responsible for the Rothschild Bioscience Unit. Earlier, he founded the International Biochemicals Group, which was later sold to Royal Dutch Shell, and Mr. Curnock Cook continued to serve as Managing Director for a period of time after the sale. Mr. Curnock Cook is currently Chairman of both Targeted Genetics Inc. and Inflazyme Pharmaceuticals Ltd. and is also a director of Biocompatibles International, plc and Silence Therapeutics plc.
Klaus Eldrup-Jørgensen, M.D., President and Chief Executive Officer, ISG A/S; Chairman of the Board of Directors
Klaus Eldrup-Jørgensen, M.D., has been our Chairman since November 2003. From May 2003 through October 2003 he was also our interim Chief Executive Officer. Dr. Eldrup-Jørgensen is currently one of the founders and the President and Chief Executive Officer of the high-tech company ISG A/S. Previously he held senior positions in a number of companies, including Novo Nordisk A/S, Dako A/S and Nordic Bioscience A/S. Dr. Klaus Eldrup Jørgensen presently serves on the board of two biotech venture funds, Seed Capital Denmark A/S and Symbion Capital A/S.
Christian Hansen, Ph.D., Co-Founder and Partner, Nordic Biotech K/S
Christian Hansen, Ph.D. has been a partner of Nordic Biotech K/S since he co-founded it in June 2001. Previously, from 2000 to 2001, Dr. Hansen was co-President of the Protein Pharmaceuticals Division of Maxygen, Inc. From 1999 to 2000, Dr. Hansen was the founder and co-CEO of ProFound Pharma A/S, a company focused on developing second generation protein pharmaceuticals. From 1992 to 1999, Dr. Hansen worked in various positions at Novo Nordisk A/S, including as Director of Intellectual Property Strategy. Dr. Hansen holds an M.Sc. in chemical engineering from the Technical University of Denmark and a Ph.D. in molecular biology/biochemistry undertaken at the Institut Pasteur, Paris, and the University of Salamanca, Spain, as well as a MBA from the Edinburgh Business School. He is currently a director of Curalogic A/S.
43
Florian Schönharting, M.Sc., Co-Founder and Partner, Nordic Biotech K/S
Florian Schönharting has been a partner of Nordic Biotech K/S since he co-founded it in June 2001. Previously, Mr. Schönharting was with Bank Invest Asset Management, a Danish private & public equity group, for approximately ten years, most recently as managing director in charge of biotech investments. During his tenure with BankInvest, Mr. Schönharting executed some 20 investments, including Biomarin, Genmab, Profound Pharma (Maxygen Inc.), Bavarian Nordic, Pharmexa, Pyrosequencing and Glyko Biomedical. Mr. Schönharting earned a master’s degree in business economics (finance) from the Copenhagen Business School, and was given the McKinsey & Co award for best-of-year student. Florian Schönharting is currently a member of the board of directors of the Danish Venture Capital Association and the Danish Corporate Governance Association.
Bobby W. Sandage, Jr., Ph.D., Executive Vice President, Research and Development and Chief Scientific Officer, Indevus Pharmaceuticals, Inc.
Bobby W. Sandage Jr., Ph.D. has been Executive V.P., Research and Development and Chief Scientific Officer of Indevus Pharmaceuticals, Inc. since 1995. He joined Indevus in 1991 following senior development positions within the cardiovascular R&D division of DuPont Merck Pharmaceutical Company. Dr. Sandage was previously affiliated with the Medical Department of DuPont Critical Care, most recently as associate medical director. Dr. Sandage is adjunct professor in the Department of Pharmacology at the Massachusetts College of Pharmacy. Dr. Sandage received his Ph.D. in Clinical Pharmacy from Purdue University and a B.S. in Pharmacy from the University of Arkansas.
Christopher B. Wood, M.D., Chairman, BioMed (UK), Ltd.
Christopher B. Wood, M.D. has been Executive Chairman of BioMed (UK), Ltd. since November 2007. From January 1999 through October 2007, Dr. Wood was Chairman and Chief Executive Officer of Bioenvision, Inc. Previously Dr. Wood was chairman of the board of Eurobiotech, Inc., and before then served as a specialist surgeon in the National Health Service in the United Kingdom. For approximately 12 years Dr. Wood was a specialist surgeon at The Royal Postgraduate Medical School, London, England. Dr. Wood holds an M.D. from the University of Wales School of Medicine and is a fellow of the Royal College of Surgeons of Edinburgh.
Philip J. Young, President and Chief Executive Officer
Mr. Young has been our President and Chief Executive Officer since May 1, 2007. He was elected to our board of directors on June 7, 2007. Prior to joining us, Mr. Young served as Executive Vice President of Commercial Operations and Chief Business Officer of Insmed, Inc. from April 2004 until March 2007. Previously, Mr. Young served as President and Chief Operations Officer for AGY Therapeutics and as Chief Executive Officer of GanTech International from 2000 until 2004. From 1998 until 2000, Mr. Young was Vice President and General Manager of Neurex Pharmaceuticals. Before joining Neurex, Mr. Young was Business Director and General Manager of the Peptide Hormones Division at Pharmacia (a division of Pfizer). Mr. Young earlier served for seven years at Genentech, where he was the Product Manager of Growth Hormone Products. Mr. Young has a B.S. in Sociology from James Madison University.
Matthew M. Loar, Principal Financial and Accounting Officer
Mr. Loar has been a consultant acting as Principal Financial and Accounting Officer beginning May 1, 2008, after serving us as Chief Financial Officer from September 1, 2006 to April 30, 2008. Prior to joining us, Mr. Loar was Chief Financial Officer of Genelabs Technologies, Inc. He was with Genelabs for approximately eleven years, and CFO for his last five years. For the five years prior to joining Genelabs he held finance positions with an international manufacturing company. Previously he was an audit manager with a major public accounting firm. Mr. Loar is a Certified Public Accountant in California and holds a BA degree from University of California, Berkeley. Pursuant to the scheduled move of our principal executive office, Mr. Loar’s employment with us as Chief Financial Officer ended on May 1, 2008. On April 15, 2008, we entered into an agreement with Mr. Loar whereby he agreed to provide services as a consultant with us for up to ten hours per month from May 1, 2008 to September 1, 2008, to serve the role of our principal financial and accounting officer while assisting with an orderly transition of his previous responsibilities
44
Board and Committee Meetings
Our board of directors held five meetings during fiscal year ended December 31, 2007. No director attended fewer than 75% of the meetings of the board and any committee of which the director was a member.
Our board of directors has an Audit Committee and a Compensation Committee. We do not have a policy with regard to board members’ attendance at annual meetings of stockholders.
Compensation Committee
Our board of directors has appointed Klaus Eldrup-Jørgensen, Christian Hansen and Christopher B. Wood to our compensation committee. Our compensation committee has the responsibility to make recommendations to our board of directors with respect to all forms of compensation paid to our executive officers, members of our board of directors, to such other officers as directed by our board of directors, and any other compensation matters as from time to time directed by our board of directors. Our compensation committee met two times in 2007.
Audit Committee
Our audit committee assists our board of directors by overseeing the performance of the independent auditors and the quality and integrity of our internal accounting, auditing and financial reporting practices. Our board of directors had appointed Christopher B. Wood, Klaus Eldrup-Jørgensen and Bobby W. Sandage, Jr. to our audit committee. Each of the members meets the independence requirements and standards currently established by the SEC. Our board of directors has designated the chairman of our audit committee, Christopher B. Wood, as an “audit committee financial expert” within the meaning of the rules and regulations of the SEC because he has considerable experience overseeing the chief financial officer at a U.S. public biotechnology company.
Our audit committee is responsible for retaining and, as necessary, terminating the independent auditors; reviewing, on an annual basis, the qualifications, performance and independence of the independent auditors; and pre-approving audit and non-audit services to be performed by the auditors and related fees. In performing all of these functions, our audit committee relies on the work and assurances of our management, which has the primary responsibility for financial statements and reports, and the independent auditors, who, in their report, express an opinion on the conformity of the our annual financial statements to generally accepted accounting principles. Our audit committee met four times in 2007.
Review of our Audited Financial Statements for the Fiscal Year Ended December 31, 2007
In discharging its oversight responsibility as to the audit process, our audit committee has reviewed and discussed our audited financial statements for the fiscal year ended December 31, 2007 with management and the independent registered public accounting firm Weinberg & Company, P.A.
Our audit committee discussed with the independent auditors all matters required to be discussed under Statement on Auditing Standards No. 61 (“Communication with Audit Committees”). Our audit committee also received and discussed with the independent auditors their annual written report on their independence from us and our management, which is made under Independence Standards Board Standard No. 1 (“Independence Discussions with Audit Committees”), and considered with the independent auditors whether the provision of non-audit services during the fiscal year ended December 31, 2007 was compatible with the independent auditors’ independence.
Based on these reviews and discussions, our audit committee recommended to our board of directors that the audited financial statements be included in our Annual Report onForm 10-K for the year ended December 31, 2007.
AUDIT COMMITTEE
Christopher B. Wood, Chairman
Klaus Eldrup-Jørgensen
Bobby W. Sandage, Jr.
45
Process for Sending Communications to Our Board of Directors
Our board of directors maintains a process for stockholders to communicate with the board. Stockholders wishing to communicate with our board of directors or any individual director must mail a communication addressed to the board or the individual director to the Board of Directors at Osteologix, Inc., 4415 Cox Road, Glen Allen, VA 23060. Any such communication must state the number of shares of common stock beneficially owned by the stockholder making the communication. All of such communications will be forwarded to the full board of directors or to any individual director or directors to whom the communication is directed unless the communication is clearly of a marketing nature or is unduly hostile, threatening, illegal, or similarly inappropriate, in which case we have the authority to discard the communication or take appropriate legal action regarding the communication.
Nomination by Stockholders
Our board of directors accepts director nominations made by stockholders. Our board of directors may consider those factors it deems appropriate in evaluating director nominees, including judgment, skill, diversity, strength of character, experience with businesses and organizations comparable in size or scope to us, experience and skill relative to other board members, and specialized knowledge or experience. Depending upon the current needs of our board of directors, certain factors may be weighed more or less heavily. In considering candidates for our board of directors, they evaluate the entirety of each candidate’s credentials and do not have any specific minimum qualifications that must be met by a nominee. They will consider candidates from any reasonable source, including current board members, stockholders, professional search firms or other persons. They will not evaluate candidates differently based on who has made the recommendation.
Our amended and restated by-laws include a provision that permits a stockholder of record that beneficially owned more than five percent of our voting stock for at least one year as of the date of the recommendation to submit to us the name of any person whom the stockholder wishes to nominate as a candidate for election to the board of directors. In general, such a submission must be received by our corporate secretary at our principal office 30 days prior to the filing of our proxy statement before the annual stockholder meeting, and must contain all information about the candidate that would be required to be disclosed in a proxy statement prepared and filed under federal and state law, as well as the proposed nominee’s consent to be named as a nominee and to serve if elected. The stockholder must also provide information about his or her identity and the number of shares owned. If the nomination is made by a stockholder holding shares in “street name,” then the identity and ownership information must be furnished about the beneficial owner of the shares. A candidate submitted by a stockholder as a nominee need not be nominated by the independent directors.
We are required to include in our future proxy statements information about a recommended stockholder nominee, but only when the following criteria are met:
| | |
| | • | The proposed nomination is received by a date not later than the 120th day before the date (i.e., the month and day) of our proxy statement released to stockholders in connection with the prior year’s annual meeting. |
| |
| | • | The stockholder or stockholder group making the proposal has beneficially owned more than 5% of our voting stock for at least a year. |
If those criteria are met, and provided that we have written consent from the proposed candidate and from the stockholder or stockholder group, we would be obliged to identify in our proxy statement the name of the candidate and the stockholder or stockholder group making the nomination, and to disclose our position regarding the nomination.
46
EXECUTIVE COMPENSATION
Summary Compensation Table
The following table sets forth certain information concerning compensation by all persons who served as the our principal executive officer during 2007 and 2006, our two other most highly compensated executive officers during 2007 and 2006, and an individual who would have been one of the two most highly compensated executive officers during 2007 but for the fact that the individual was not one of our executive officers of as of December 31, 2007 (collectively, the “Named Executive Officers”):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | Warrant and
| | Non-Equity
| | | | |
| | | | | | | | | Option
| | Incentive Plan
| | | | |
| | | | | | | | | Awards
| | Compensation
| | All Other
| | |
Name | | | | Salary | | Bonus | | (1) | | (2) | | Compensation | | Total |
| |
| Philip J. Young(3) | | | 2007 | | | $ | 233,333 | | | $ | 69,417 | | | $ | 76,855 | | | | — | | | $ | 17,944 | (4) | | $ | 397,549 | |
| President and Chief Executive Officer | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Charles J. Casamento(5) | | | 2007 | | | $ | 108,333 | | | | — | | | $ | 76,073 | | | | — | | | $ | 564,582 | (6) | | $ | 748,987 | |
| Former Chief Executive Officer and President | | | 2006 | | | $ | 420,000 | | | | — | | | $ | 170,309 | | | $ | 35,590 | | | $ | 81,444 | (7) | | $ | 707,343 | |
| Matthew M. Loar(8) | | | 2007 | | | $ | 250,000 | | | $ | 77,778 | | | $ | 82,979 | | | | — | | | | — | | | $ | 410,757 | |
| Chief Financial Officer | | | 2006 | | | | 83,333 | | | | — | | | $ | 27,482 | | | | — | | | | — | | | $ | 110,815 | |
| Stephan Christgau(9) | | | 2007 | | | $ | 114,160 | | | | — | | | $ | 32,924 | | | | — | | | $ | 73,690 | (10) | | $ | 220,774 | |
| Former Chief Operating Officer | | | 2006 | | | | 152,993 | | | | — | | | $ | 102,571 | | | $ | 13,133 | | | | — | | | $ | 268,697 | |
| | |
| (1) | | Warrant and option awards are valued based on a Black-Scholes option pricing model as used in our consolidated financial statements pursuant to Statement of Accounting Standards No. 123R, “Share-based Payment.” The Black-Scholes option pricing model reflects certain assumptions regarding variable factors such stock price volatility and the term of the option. Stock options have value to the recipient only as a result of appreciation in the price of our common stock. For the purposes of establishing the value shown in the table, the model assumed a dividend yield of zero, risk-free interest rate of 4.5% to 5.0%, volatility factor of 70%, and an expected life of the options of three to six years. For financial reporting purposes, in 2007 we used an estimated forfeiture rate of 15% for the stock options granted. This estimated forfeiture rate has not been included in the balances in the table because the forfeiture rate is an overall estimate for the organization as a whole and cannot be attributed to the anticipated forfeiture of the options granted to specifically to any of our executive officers. |
| |
| (2) | | Non-equity incentive plan compensation represents amounts paid as an incentive bonus following the financing and merger transactions of May 24, 2006. |
| |
| (3) | | Mr. Young was hired as President and Chief Executive Officer effective May 1, 2007 with a 2007 annual salary of $350,000. |
| |
| (4) | | We paid $1,275 per month for Mr. Young in the form of an auto lease payment allowance and reimbursed all auto maintenance and operating expenses, which together with the lease payment allowance aggregated $11,925. We paid insurance premiums aggregating $6,019 on life insurance and disability insurance policies. These amounts were paid on behalf of Mr. Young under the terms of his Employment Agreement dated April 3, 2007. |
| |
| (5) | | Mr. Casamento resigned as Chief Executive Officer and President on April 3, 2007. Following his resignation, warrants to purchase 79,583 shares of common stock remained unvested and were canceled. In addition, at the time of his resignation Mr. Casamento held options to purchase 350,000 shares of common stock; of these, options to purchase 291,667 shares of common stock were unvested and canceled. The options to purchase the remaining 58,333 shares expired unexercised three months following termination of his service to us. |
| |
| (6) | | In accordance with Mr. Casamento’s Employment Agreement dated October 18, 2004, a Preliminary Binding Agreement dated as of April 3, 2007, and a Separation and Mutual General Release dated as of April 3, 2007, we paid made cash payments to Mr. Casamento of $420,000. We also paid Mr. Casamento’s accrued vacation |
47
| | |
| | balance as of the date of resignation, which aggregated $32,307. In addition, in accordance with the agreements, for each month of 2007, we paid Mr. Casamento $1,275 per month towards an auto lease plus all of his auto maintenance and operating expenses, which together with the lease payments aggregated $35,573. In addition to medical insurance coverage provided while Mr. Casamento was an employee, we also paid for the COBRA medical premiums following Mr. Casamento’s resignation, and paid for all 2007 family medical expenses, which together with the COBRA payments aggregated $31,637 in 2007. We also paid premiums of $44,565 for life and disability insurance policies. Finally, following his resignation, we provided Mr. Casamento with a computer valued at $500. |
| |
| (7) | | In accordance with Mr. Casamento’s Employment Agreement dated October 18, 2004, during 2006 we paid $1,275 per month for Mr. Casamento’s auto lease and paid all auto maintenance and operating expenses, which together with lease payments aggregated $24,513. In addition to medical insurance coverage which is provided to all employees, we paid for all family medical expenses, which amounted to $14,952 in excess of the medical insurance. We paid premiums of $37,104 on life insurance and disability insurance policies. We also provided Mr. Casamento with a parking space in the building that houses our corporate headquarters at a cost of $4,875. |
| |
| (8) | | Mr. Loar was hired on September 1, 2006 with an annual salary of $250,000. |
| |
| (9) | | Dr. Christgau is a resident of Copenhagen Denmark, and was paid in Danish Kroner by Osteologix ApS prior to his resignation effective September 7, 2007. His 2007 salary paid through date of termination was 618,750 DKK, converted into U.S. dollars at the weighted average exchange rate as used in our consolidated statement of operations of 0.1845 DKK per U.S. dollar. His 2006 salary and non-equity incentive plan compensation were 908,626 DKK and 78,000 DKK, respectively, both converted into U.S. dollars at the weighted average exchange rate as used in our consolidated statement of operations of 0.1684 DKK per U.S. dollar. |
| |
| (10) | | Other compensation paid to Dr. Christgau represents payment of 160,543 DKK for unused vacation time (converted into $29,620), $3,750 cash for consulting services and 28,000 shares of common stock, which was valued at $40,320 as of the date the stock was issued. |
Additional information regarding the fair value of warrant and option awards in the Summary Compensation Table is contained in the table below:
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | Number of
| | | | | | | | | | |
| | | | | | Securities
| | | Exercise
| | | Grant Date
| | | | |
| | | | | | Underlying
| | | Price of
| | | Fair Value of
| | | Fair Value
| |
| | | | | | Warrants or
| | | Warrants or
| | | Warrants or
| | | Recognized under
| |
| | | | | | Options
| | | Options
| | | Options
| | | SFAS 123R
| |
Name | | Grant Date | | | (#) | | | ($/share) | | | ($) | | | ($) | |
| |
| Philip J. Young | | | 5/1/2007 | | | | 1,000,000 | (1) | | $ | 1.20 | | | $ | 613,017 | | | | 2007 | | | $ | 76,855 | |
| Charles J. Casamento | | | 5/24/2006 | | | | 509,240 | (2) | | $ | 1.03 | | | $ | 371,745 | | | | 2006 | | | $ | 158,707 | |
| | | | | | | | | | | | | | | | | | | | 2007 | | | $ | 41,237 | |
| Charles J. Casamento | | | 10/5/2006 | | | | 350,000 | (3) | | $ | 1.20 | | | $ | 278,453 | | | | 2006 | | | $ | 11,602 | |
| | | | | | | | | | | | | | | | | | | | 2007 | | | $ | 34,836 | |
| Matthew M. Loar | | | 9/1/2006 | | | | 400,000 | (4) | | $ | 1.50 | | | $ | 331,600 | | | | 2006 | | | $ | 27,482 | |
| | | | | | | | | | | | | | | | | | | | 2007 | | | $ | 82,979 | |
| Stephan Christgau | | | 5/24/2006 | | | | 244,827 | (2) | | $ | 1.03 | | | $ | 178,724 | | | | 2006 | | | $ | 97,599 | |
| | | | | | | | | | | | | | | | | | | | 2007 | | | | — | |
| Stephan Christgau | | | 10/5/2006 | | | | 150,000 | (3) | | $ | 1.20 | | | $ | 119,337 | | | | 2006 | | | $ | 4,972 | |
| | | | | | | | | | | | | | | | | | | | 2007 | | | $ | 32,924 | |
| | |
| (1) | | The options granted to Mr. Young vest over a period of four years and seven months, with options to purchase 125,000 shares vesting on December 1, 2007, and the remaining 875,000 shares vesting monthly thereafter over the next 48 months (approximately 18,229 shares vest per month). |
| |
| (2) | | The warrants to purchase shares of our common stock that were granted to Mr. Casamento and Dr. Christgau on May 24, 2006 replaced warrants to purchase stock of Osteologix A/S, a Danish company that was merged into our predecessor company. Mr. Casamento and Dr. Christgau received the same exchange ratio for their warrants that the stockholder of Osteologix A/S did for its stock in the exchange of shares our shares. The grant date fair |
48
| | |
| | value of warrants shown in the column represents the incremental fair value of the warrants as of the exchange date. The fair value recognized under SFAS 123R for 2006 represents the amount recognized in the statement of operations both prior to and subsequent to the exchange date. |
| |
| (3) | | The options granted to Mr. Casamento and Dr. Christgau vest over a period of four years, beginning six months from the date of grant. Mr. Casamento’s vesting terminated on July 3, 2007 and Dr. Christgau’s vesting terminated on December 31, 2007. |
| |
| (4) | | The options granted to Mr. Loar vest over a period of four years, beginning one year from the date of grant. |
Outstanding Equity Awards at Fiscal Year-End
The following table provides certain information with respect to outstanding stock warrants and options as of December 31, 2007:
| | | | | | | | | | | | | | | | | |
| | | Warrant and Option Awards | |
| | | Number of
| | | Number of
| | | | | | | |
| | | Securities
| | | Securities
| | | | | | | |
| | | Underlying
| | | Underlying
| | | | | | | |
| | | Unexercised
| | | Unexercised
| | | Warrant or
| | | | |
| | | Warrants and
| | | Warrants and
| | | Option
| | | Warrant or
| |
| | | Options
| | | Options
| | | Exercise
| | | Option
| |
| | | (#)
| | | (#)
| | | Price
| | | Expiration
| |
Name | | Exercisable | | | Unexercisable | | | ($/share) | | | Date | |
| |
| Philip J. Young | | | 125,000 | | | | 875,000 | | | $ | 1.20 | | | | 4/30/2017 | |
| Charles J. Casamento | | | 429,657 | | | | — | | | $ | 1.03 | | | | 10/15/2014 | |
| Matthew M. Loar | | | 125,000 | | | | 275,000 | | | $ | 1.50 | | | | 8/31/2016 | |
| Stephan Christgau | | | 244,827 | | | | — | | | $ | 1.03 | | | | 6/30/2013 | |
| Stephan Christgau | | | 43,750 | | | | — | | | $ | 1.20 | | | | 3/31/2008 | |
Director Compensation
The following table shows the compensation received by the non-employee members of our board of directors for the year ended December 31, 2007:
| | | | | | | | | | | | | | | | | |
| | | Fees
| | | | | | Warrant and
| | | | |
| | | Earned or
| | | Stock
| | | Option
| | | | |
Name | | Paid in Cash | | | Awards(1) | | | Awards(2) | | | Total | |
| |
| Klaus Eldrup-Jørgensen, Chairman | | | — | | | $ | 41,500 | | | $ | 4,977 | | | $ | 46,477 | |
| Jeremy Curnock Cook | | | — | | | $ | 22,000 | | | $ | 4,150 | | | $ | 26,150 | |
| Christian Hansen(3) | | | — | | | | — | | | | — | | | | — | |
| Bobby W. Sandage, Jr. | | | — | | | $ | 28,000 | | | $ | 16,950 | | | $ | 44,950 | |
| Florian Schönharting(3) | | | — | | | | — | | | | — | | | | — | |
| Christopher B. Wood | | | — | | | $ | 31,500 | | | $ | 4,977 | | | $ | 36,477 | |
| | |
| (1) | | Members of our board of directors are currently entitled to receive their base compensation either as a cash payment at the end of each quarter or as newly issued, unregistered shares of common stock, valued as of the last day of the quarter. All board members that have not waived their right to receive compensation have elected to receive shares of our common stock in lieu of cash. The directors can elect to revoke their election to receive stock instead of cash at any time, although to date none have done so. |
| |
| (2) | | Warrant and option awards are valued based on a Black-Scholes option pricing model as used in our consolidated financial statements pursuant to Statement of Accounting Standards No. 123R, “Share-based Payment.” The Black-Scholes option pricing model reflects certain assumptions regarding variable factors such stock price volatility and the term of the option. Stock options have value to the recipient only as a result of appreciation in the price of our common stock. For the purposes of establishing the value shown in the table, the model assumed a dividend yield of zero, risk-free interest rate of 4.5% to 5.0%, volatility factor of 70%, and an expected life of the options of three to six years. |
| |
| (3) | | Dr. Hansen and Mr. Schönharting have elected to waive receipt of compensation as directors. |
49
Additional information regarding the fair value of warrant and option awards in the Director Compensation Table is contained in the table below:
| | | | | | | | | | | | | | | | | | | | | |
| | | | | | Number of
| | | Exercise
| | | | | | | |
| | | | | | Securities
| | | Price
| | | Grant Date
| | | Fair Value
| |
| | | | | | Underlying
| | | of Warrants
| | | Fair Value of
| | | Recognized
| |
| | | | | | Warrants or
| | | or
| | | Warrants or
| | | under
| |
| | | | | | Options(1)
| | | Options
| | | Options
| | | SFAS 123R
| |
Name | | Grant Date | | | (#) | | | ($/share) | | | ($) | | | ($) | |
| |
| Klaus Eldrup-Jørgensen | | | 10/5/2006 | | | | 25,000(1 | ) | | $ | 1.20 | | | $ | 19,889 | | | $ | 4,977 | |
| Jeremy Curnock Cook | | | 11/9/2006 | | | | 25,000(1 | ) | | $ | 1.00 | | | $ | 16,585 | | | $ | 4,150 | |
| Bobby W. Sandage, Jr. | | | 5/24/2006 | | | | 48,965(2 | ) | | $ | 1.03 | | | $ | 35,744 | | | $ | 11,973 | |
| Bobby W. Sandage, Jr. | | | 10/5/2006 | | | | 25,000(1 | ) | | $ | 1.20 | | | $ | 19,889 | | | $ | 4,977 | |
| Christopher B. Wood | | | 10/5/2006 | | | | 25,000(1 | ) | | $ | 1.20 | | | $ | 19,889 | | | $ | 4,977 | |
| | |
| (1) | | The options vest over four years beginning at the end of the month following the six month anniversary of the grant date. After the six month anniversary of the grant date, the options vest monthly for the following 42 months. |
| |
| (2) | | The warrants to purchase shares of our common stock that were granted to Dr. Sandage on May 24, 2006 replaced warrants to purchase stock of Osteologix A/S, a Danish company that was merged into our predecessor company. Dr. Sandage received the same exchange ratio for his warrants that the stockholder of Osteologix A/S did for its stock in the exchange of shares for shares of our common stock. Vesting of the warrant to purchase common stock was the same as when the warrant was initially granted. The grant date fair value of warrants shown in the column represents the incremental fair value of the warrants as of the exchange date. |
Board members are paid quarterly in arrears. Our compensation committee adopted a schedule for compensation of members of our board of directors as follows, which was unchanged throughout 2007:
| | | | | |
| Annual retainer | | $ | 10,000 | |
| Additional retainer for Chairman | | $ | 12,000 | |
| Fee per each regularly scheduled meeting | | $ | 3,000 | |
| Additional annual retainer for Audit Committee chairman | | $ | 8,000 | |
| Additional annual retainer for Audit Committee member | | $ | 6,000 | |
| Additional annual retainer for Compensation Committee | | $ | 1,500 | |
Directors are also reimbursed for any out-of-pocket expenses incurred in attending board meetings or other company business.
LIMITS ON DIRECTOR LIABILITY AND INDEMNIFICATION
Our Certificate of Incorporation limits the liability of our directors for monetary damages arising from a breach of their fiduciary duty as directors, except to the extent otherwise required by the General Corporation Law of Delaware. Such limitation of liability does not affect the availability of equitable remedies such as injunctive relief or rescission.
Our amended and restated by-laws provide that we shall indemnify our directors and officers to the fullest extent permitted by Delaware law, including in circumstances in which indemnification is otherwise discretionary under Delaware law. We have entered into indemnification agreements with our officers and directors containing provisions that may require us, among other things, to indemnify such officers and directors against certain liabilities that may arise by reason of their status or service as directors or officers (other than liabilities arising from willful misconduct of a culpable nature), to advance their expenses incurred as a result of any proceeding against them as to which they could be indemnified, and to obtain directors’ and officers’ insurance if available on reasonable terms.
50
CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS
Parties are considered to be related if one party has the ability, directly or indirectly, to control the other party or exercise significant influence over the other party in making financial and operational decisions. Parties are also considered to be related if they are subject to common control or common significant influence.
From January 1, 2007 to the present, there have been two transactions in which the amount involved exceeded one percent (1%) average of our total assets at year end for our past three completed fiscal years to which Osteologix was a party and in which any executive officer, director, 5% beneficial owner of common stock or member of the immediate family of any of the foregoing persons had or has a direct or indirect material interest other than compensation agreements and other arrangements, that are described where required under “Executive Compensation.” Further information on these two transactions is as follows:
On June 4, 2007, we entered into a securities purchase agreement with certain investors for the sale of an aggregate of 1,912,877 units, each unit consisting of two shares of our common stock and one common stock purchase warrant with an exercise price equal to $1.20 per share and an expiration date of August 31, 2008. The purchase price of each unit was $2.64 per unit, for an aggregate purchase price of $5,050,000. Nordic Biotech K/S purchased 1,250,000 units in the transaction, for total consideration of $3,300,000. Pursuant to a registration rights agreement, also entered into on June 4, 2007, Nordic has the right at an time that is the later of (i) one hundred eighty days after the Closing Date and (ii) thirty days after the date of the effectiveness of the registration statement described above to request that we prepare and file a registration statement covering the resale of the registrable securities held by Nordic. To date, Nordic has not requested us to prepare and file a registration statement covering the resale of their registrable securities. This transaction was approved by a committee of our board of directors consisting only of disinterested directors.
On April 17, 2008, we closed the private placement of 4,030,304 shares of common stock to the selling stockholders and an additional investor, Nordic Biotech Opportunity Fund K/S and warrants to purchase 2,015,152 shares. The aggregate purchase price was approximately $5,320,000. Nordic Biotech Opportunity Fund K/S, an affiliate of Nordic Biotech K/S, our largest shareholder, purchased a total of 3,030,304 shares of common stock in the private placement and warrants to purchase 1,515,152 shares, for an aggregate purchase price of approximately $4,000,000. Pursuant to a registration rights agreement, dated March 27, 2008, entered into in connection with the private placement, we agreed to file with the SEC, no later than 30 days after the closing date of the transaction, a registration statement registering the shares of common stock purchased by the selling stockholders for resale. Nordic Biotech Opportunity Fund K/S has the right at any time after the earlier of (i) six months after the closing date of the transaction and (ii) 30 days after the date of the effectiveness of the registration statement described above to cause us to prepare and file a registration statement covering the resale of the shares of common stock purchased by it, subject to certain limitations. This transaction was approved by a committee of our board of directors consisting only of disinterested directors. Punk Ziegel + Co., L.P., acted as placement agent in connection with this transaction. For its services, we issued to Punk Ziegel warrants to purchase 15,000 shares of common stock upon the same terms as the warrants issued to the investors, as well as a cash fee and reimbursement for certain expenses equal to $55,024 in the aggregate.
51
SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT
The following table sets forth as June 30, 2008, the number of shares of our common stock beneficially owned by (i) each person who is known by us to be the beneficial owner of more than five percent of the our common stock; (ii) each director and nominee for election to our board of directors; (iii) each of the named executive officers in the Summary Compensation Table; and (iv) all directors and executive officers as a group. Unless otherwise indicated, the stockholders listed in the table have sole voting and investment power with respect to the shares indicated, and have an address atc/o Osteologix, Inc., 4415 Cox Road, Glen Allen, Virginia, 23060.
| | | | | | | | | | | | | | | | | |
| | | | | | Number of
| | | | | | | |
| | | Number of
| | | Shares with
| | | Total Shares
| | | | |
| | | Shares
| | | Right to
| | | Beneficially
| | | Percent of
| |
Name and Address of Beneficial Owner | | Owned | | | Acquire | | | Owned(1) | | | Class | |
| |
| Christian Hansen, Ph.D.(2) | | | 18,252,265 | | | | 2,765,152 | | | | 21,017,417 | | | | 66.0 | % |
| Florian Schönharting, M.Sc.(3) | | | 18,183,365 | | | | 2,765,152 | | | | 20,948,517 | | | | 65.8 | % |
Nordic Biotech K/S
Oestergade 5,3
DK-1100, Copenhagen, Denmark(4) | | | 18,163,365 | | | | 2,765,152 | | | | 20,928,517 | | | | 65.7 | % |
| Jeremy Curnock Cook(5) | | | 1,941,482 | | | | 16,395 | | | | 1,957,877 | | | | 6.7 | % |
BML Healthcare I, LP
243 Knightsbridge
London, United Kingdom SW7 IDN | | | 1,914,000 | | | | — | | | | 1,914,000 | | | | 6.6 | % |
| Charles J. Casamento(6) | | | 55,320 | | | | 429,657 | | | | 484,977 | | | | 1.6 | % |
| Klaus Eldrup-Jørgensen, M.D. | | | 47,566 | | | | 219,111 | | | | 266,677 | | | | * | |
| Stephan Christgau(7) | | | 11,484 | | | | 244,827 | | | | 256,311 | | | | * | |
| Philip J. Young | | | — | | | | 270,833 | | | | 270,833 | | | | * | |
| Matthew M. Loar | | | — | | | | 191,667 | | | | 191,667 | | | | * | |
| Bobby W. Sandage, Jr., Ph.D. | | | 50.613 | | | | 117,511 | | | | 168,124 | | | | * | |
| Christopher B. Wood, M.D. | | | 46,605 | | | | 69,497 | | | | 116,102 | | | | * | |
| All Directors and Officers (10 people) | | | 20,425,335 | | | | 4,324,650 | | | | 24,749,825 | | | | 74.1 | % |
| | |
| * | | Less than one percent. |
| |
| (1) | | Beneficial ownership is determined in accordance with the rules of the Securities and Exchange Commission, the SEC, and generally includes voting or investment power with respect to the shares shown. Except as indicated and subject to community property laws where applicable, to our knowledge, the stockholders named in the table have sole voting and investment power with respect to all common stock shares shown as beneficially owned by them. A person is deemed to be the beneficial owner of securities that can be acquired by such person within 60 days upon the exercise of options, warrants or convertible securities (in any case, the “Currently Exercisable Options”). Each beneficial owner’s percentage ownership is determined by assuming that the Currently Exercisable Options that are held by such person (but not those held by any other person) have been exercised and converted. Except as indicated in this table or the footnotes to this table and pursuant to applicable community property laws, the persons named in the table have sole voting and investment power with respect to all shares of common stock. |
| |
| (2) | | Christian Hansen is a general partner of Nordic Biotech K/S and holds voting and dispositive power for the 15,133,061 shares owned by Nordic Biotech K/S and the 3,030,304 shares held by Nordic Biotech Opportunity Fund K/S. Dr. Hansen disclaims beneficial ownership of these shares, except to the extent of his pecuniary interest therein. In addition, Dr. Hansen is the sole owner of CKH Invest ApS, which owns 88,900 shares. |
| |
| (3) | | Florian Schönharting is a general partner of Nordic Biotech K/S and holds voting and dispositive power for the 15,133,061 shares owned by Nordic Biotech K/S and the 3,030,304 shares held by Nordic Biotech Opportunity Fund K/S. Mr. Schönharting disclaims beneficial ownership of these shares, except to the extent of his pecuniary interest therein. |
| |
| (4) | | Stock held by Nordic Biotech K/S includes 15,133,061 shares held directly by Nordic Biotech K/S and 3,030,304 shares held by Nordic Biotech Opportunity Fund K/S. |
52
| | |
| (5) | | Jeremy Curnock Cook is Executive Director of Bioscience Managers Limited, which has voting and dispositive power for the 1,914,000 shares owned by BML Healthcare I, LP. Mr. Cook disclaims beneficial ownership of these shares, except to the extent of his pecuniary interest therein. |
| |
| (6) | | Charles Casamento resigned on April 3, 2007 and his holdings represent the most recent data available to us, which we believe to be correct. |
| |
| (7) | | Dr. Stephen Christgau resigned on September 7, 2007 and his holdings represent the most recent data available to us which, we believe to be correct. The “number of shares owned” and the “total shares beneficially owned” both include 5,742 shares held in the name of Janelyn Mangulad-Christgau, Dr. Christgau’s wife. |
SELLING STOCKHOLDERS
The following table sets forth as of June 30, 2008 information regarding the current beneficial ownership of our common stock by the persons identified, based on information provided to us by them, which we have not independently verified. Although we have assumed for purposes of the table that the selling stockholders will sell all of the shares offered by this prospectus, because they may from time to time offer all or some of their shares under this prospectus or in another manner, no assurance can be given as to the actual number of shares that will be resold by the selling stockholders (or any of them), or that will be held after completion of the resales. In addition, a selling stockholder may have sold or otherwise disposed of shares in transactions exempt from the registration requirements of the Securities Act or otherwise since the date he or she provided information to us. The selling stockholders are not making any representation that the shares covered by this prospectus will be offered for sale.
Except as set forth below, no selling stockholder has held any position nor had any material relationship with us or our affiliates during the past three years.
| | | | | | | | | | | | | | | | | |
| | | Shares
| | | Maximum
| | | Shares
| | | | |
| | | Beneficially
| | | Number of
| | | Beneficially
| | | Percentage
| |
| | | Owned Prior to
| | | Shares to be
| | | Owned After
| | | Ownership After
| |
Name of Selling Stockholder | | Offering | | | Sold | | | Offering | | | Offering | |
| |
| Goldman Sachs Intl(1) | | | 852,270 | (2) | | | 852,270 | (2) | | | 0 | | | | 0 | |
| BVF Investments, LLC(3) | | | 623,307 | (4) | | | 623,307 | (4) | | | 0 | | | | 0 | |
| Biotech Value Fund I, LP(3) | | | 259,050 | (5) | | | 259,050 | (5) | | | 0 | | | | 0 | |
| Biotech Value Fund II, LP(3) | | | 177,792 | (6) | | | 177,792 | (6) | | | 0 | | | | 0 | |
| Investment 10, LLC(3) | | | 76,212 | (7) | | | 76,212 | (7) | | | 0 | | | | 0 | |
| | | | | | | | | | | | | | | | | |
| Total | | | 1,988,631 | | | | 1,988,631 | | | | 0 | | | | 0 | |
| | |
| (1) | | Mr. Edward Lees has voting and investment control over the securities held by the stockholder. Mr. Lees disclaims beneficial ownership of the shares. The stockholder’s address is Peterborough Court, 133 Fleet Street, London, United Kingdom EC4A 2BB. |
| |
| (2) | | Includes 284,090 shares of Common Stock issuable upon exercise of warrants to purchase outstanding warrants to purchase shares of Common Stock at a purchase price of $1.20 per share. |
| |
| (3) | | Mr. Mark Lambert, President of BVF, Inc., general partner of BVF Partners L.P., which is manager of the stockholder has voting and investment control over the securities held by the stockholder. Mr. Lambert disclaims beneficial ownership of the shares. The stockholder’s address is One Sansome Street, 31st Floor, San Francisco, CA 94104. |
| |
| (4) | | Includes 207,769 shares of Common Stock issuable upon exercise of warrants to purchase outstanding warrants to purchase shares of Common Stock at a purchase price of $1.20 per share. |
| |
| (5) | | Includes 86,769 shares of Common Stock issuable upon exercise of warrants to purchase outstanding warrants to purchase shares of Common Stock at a purchase price of $1.20 per share. |
| |
| (6) | | Includes 59,264 shares of Common Stock issuable upon exercise of warrants to purchase outstanding warrants to purchase shares of Common Stock at a purchase price of $1.20 per share. |
| |
| (7) | | Includes 25,404 shares of Common Stock issuable upon exercise of warrants to purchase outstanding warrants to purchase shares of Common Stock at a purchase price of $1.20 per share. |
53
DESCRIPTION OF CAPITAL STOCK
The following description of our capital stock does not purport to be complete and is subject to and qualified in its entirety by our certificate of incorporation and amended and restated by-laws, which are included as exhibits to the registration statement of which this prospectus forms a part, and by the applicable provisions of Delaware law.
General
Our current authorized capital stock consists of 100,000,000 shares of common stock, par value $0.0001 per share, of which 29,067,825 shares were issued and outstanding as of June 30, 2008, and 1,000,000 shares of preferred stock, par value $0.0001 per share, of which no shares are issued or outstanding.
Common Stock
Each common share entitles the holder to one vote on all matters submitted to a vote of the Company’s stockholders. When a dividend is declared by the Board, all stockholders are entitled to receive a fixed dividend. To date, no dividends have been declared. All shares issued in the company are of the same class, and have equal liquidation, preference, and adjustment rights.
Preferred Stock
We are authorized, subject to limitations prescribed by Delaware law, to issue preferred stock and to fix the rights, preferences and privileges of the preferred stock. The rights of holders of common stock will be subject to, and may be adversely affected by, the rights of holders of any preferred stock. To date, no shares of preferred stock have been issued.
Warrants
As of March 31, 2008, we have outstanding warrants to purchase an aggregate of 4,842,753 shares of our common stock. The exercise price for warrants to purchase 2,030,152 shares of our common stock is $1.32. The exercise price for warrants to purchase 1,912,877 shares of our common stock is $1.20 per share. The exercise price for warrants to purchase 899,724 shares of our common stock is $1.03 per share.
Registration Rights
In connection with a private placement of our common stock, which closed on April 17, 2008, we entered into a registration rights agreement, dated as of March 27, 2008, with the selling stockholders and Nordic Biotech Opportunity Fund K/S. Pursuant to the registration rights agreement, we agreed to file with the SEC, no later than 30 days after the closing date of the transaction, a registration statement registering the shares of common stock purchased by the selling stockholders for resale. Nordic Biotech Opportunity Fund K/S has the right at any time after the earlier of (i) six months after the closing date of the transaction and (ii) 30 days after the date of the effectiveness of the registration statement described above to cause us to prepare and file a registration statement covering the resale of the shares of common stock purchased by it, subject to certain limitations. This transaction was approved by a committee of our board of directors consisting only of disinterested directors.
Effects of Certain Provisions of Our Certificate of Incorporation and Amended and Restated By-Laws and the Delaware Anti-Takeover Statute
Certificate of Incorporation and Amended and Restated By-Laws
Some provisions of Delaware law and our certificate of incorporation and amended and restated by-laws contain provisions that could make the following transactions more difficult:
| | |
| | • | acquisition of us by means of a tender offer; |
| |
| | • | acquisition of us by means of a proxy contest or otherwise; or |
| |
| | • | removal of our incumbent officers and directors. |
54
These provisions, summarized below, are expected to discourage coercive takeover practices and inadequate takeover bids and to promote stability in our management. These provisions are also designed to encourage persons seeking to acquire control of us to first negotiate with our board of directors.
| | |
| | • | Undesignated preferred stock. The ability to authorize undesignated preferred stock makes it possible for our board of directors to issue one or more series of preferred stock with voting or other rights or preferences that could impede the success of any attempt to change control of us. These and other provisions may have the effect of deferring hostile takeovers or delaying changes in control or management of our company. |
| |
| | • | Stockholder meetings. Our amended and restated by-laws provide that a special meeting of stockholders may be called only by resolution adopted by our board of directors. |
| |
| | • | Elimination of stockholder action by written consent. Our amended and restated certificate of incorporation eliminates the right of stockholders to act by written consent without a meeting. |
| |
| | • | Election and removal of directors. Our amended and restated by-laws will provide for the division of our board of directors into three classes, as nearly equal in number as possible, with the directors in each class serving for three-year terms and one class being elected each year by our stockholders. In addition, our directors will be removable only for cause and, subject to certain exceptions, any vacancies of the board of directors shall be filled only by the affirmative vote of a majority of the directors then in office. Because this system of election, appointing, removing and replacing directors generally makes it more difficult for stockholders to replace a majority of the board of directors, it may discourage a third party from making a tender offer or otherwise attempting to gain control of us and may maintain the incumbency of the board of directors. |
Delaware Anti-Takeover Statute
Upon the completion of this offering, we will be subject to Section 203 of the Delaware General Corporation Law. This law prohibits a publicly-held Delaware corporation from engaging in any business combination with any interested stockholder for a period of three years following the date that the stockholder became an interested stockholder unless:
| | |
| | • | prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction, which resulted in the stockholder becoming an interested stockholder; |
| |
| | • | upon consummation of the transaction, which resulted in the stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the number of shares outstanding those shares owned by persons who are directors and also officers and by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or |
| |
| | • | on or subsequent to the date of the transaction, the business combination is approved by the board of directors and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least two-thirds of the outstanding voting stock, which is not owned by the interested stockholder. |
Section 203 defines “business combination” to include:
| | |
| | • | any merger or consolidation involving the corporation and the interested stockholder; |
| |
| | • | any sale, transfer, pledge or other disposition of 10% or more of our assets involving the interested stockholder; |
| |
| | • | in general, any transaction that results in the issuance or transfer by us of any of our stock to the interested stockholder; or |
| |
| | • | the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation. |
55
In general, Section 203 defines an “interested stockholder” as an entity or person beneficially owning 15% or more of the outstanding voting stock of the corporation and any entity or person affiliated with or controlling or controlled by the entity or person.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is Continental Stock Transfer & Trust Company, 17 Battery Place, 8th Floor, New York, New York 10004, telephone number212-509-4000.
MARKET FOR COMMON STOCK AND RELATED MATTERS
Our Common Stock trades on the OTC Bulletin Board under the symbol “OLGX”. Some quotation systems require the symbol to be entered as “OLGX.OB”. Prior to May 24, 2006, our predecessor was a non-operating shell company that traded under the symbol “CSMH” on the OTC Bulletin Board. No shares of our Common Stock traded between January 1, 2005 and June 30, 2006. The following table sets forth, for the periods indicated, the quarterly high and low trading prices of our common stock as reported on the OTC Bulletin Board for the past two fiscal years. The trading prices reflect the prices prior to and after the merger transaction on May 24, 2006. The trading prices prior to April 13, 2006, have been adjusted to reflect a one-for-three reverse split which occurred on April 13, 2006.
| | | | | | | | | | | | | |
| | | High | | | Low | | | | |
| |
| Second Quarter 2008 | | $ | 1.20 | | | $ | 0.91 | | | | | |
| First Quarter 2008 | | $ | 1.60 | | | $ | 1.10 | | | | | |
| Fourth Quarter 2007 | | $ | 2.60 | | | $ | 0.90 | | | | | |
| Third Quarter 2007 | | $ | 1.27 | | | $ | 0.88 | | | | | |
| Second Quarter 2007 | | $ | 1.25 | | | $ | 0.80 | | | | | |
| First Quarter 2007 | | $ | 1.30 | | | $ | 0.90 | | | | | |
| Fourth Quarter 2006 | | $ | 1.45 | | | $ | 0.85 | | | | | |
| Third Quarter 2006 | | $ | 1.60 | | | $ | 1.20 | | | | | |
| Second Quarter 2006 | | $ | 1.05 | * | | $ | 1.05 | * | | | | |
| First Quarter 2006 | | $ | 1.05 | * | | $ | 1.05 | * | | | | |
| | |
| * | | These prices represent the most recent trade, which was for 5,000 shares at $0.35 per share on December 31, 2004 (before the Castle & Morgan reverse split, after the reverse split the trade would be adjusted to 1,667 shares at $1.05). |
As of June 30, 2008, there were approximately 75 holders of record of our Common Stock.
We have never declared or paid any cash dividends on our capital stock. We currently intend to retain any earnings for use in the operation and expansion of our business and do not anticipate paying any cash dividends in the foreseeable future.
PLAN OF DISTRIBUTION
The selling stockholders, which as used herein includes donees, pledgees, transferees or othersuccessors-in-interest selling shares of common stock or interests in shares of common stock received after the date of this prospectus from a selling stockholder as a gift, pledge, partnership distribution or other transfer, may, from time to time, sell, transfer or otherwise dispose of any or all of their shares of common stock or interests in shares of common stock on any stock exchange, market or trading facility on which the shares are traded or in private transactions. These dispositions may be at fixed prices, at prevailing market prices at the time of sale, at prices related to the prevailing market price, at varying prices determined at the time of sale, or at negotiated prices.
56
The selling stockholders may use any one or more of the following methods when disposing of shares or interests therein:
| | |
| | • | ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; |
| |
| | • | block trades in which the broker-dealer will attempt to sell the shares as agent, but may position and resell a portion of the block as principal to facilitate the transaction; |
| |
| | • | purchases by a broker-dealer as principal and resale by the broker-dealer for its account; |
| |
| | • | an exchange distribution in accordance with the rules of the applicable exchange; |
| |
| | • | privately negotiated transactions; |
| |
| | • | short sales effected after the date the registration statement of which this Prospectus is a part is declared effective by the SEC; |
| |
| | • | through the writing or settlement of options or other hedging transactions, whether through an options exchange or otherwise; |
| |
| | • | broker-dealers may agree with the selling stockholders to sell a specified number of such shares at a stipulated price per share; and |
| |
| | • | a combination of any such methods of sale. |
The selling stockholders may, from time to time, pledge or grant a security interest in some or all of the shares of common stock owned by them and, if they default in the performance of their secured obligations, the pledgees or secured parties may offer and sell the shares of common stock, from time to time, under this prospectus, or under an amendment or supplement to this prospectus under Rule 424(b)(3) or other applicable provision of the Securities Act amending the list of selling stockholders to include the pledgee, transferee or other successors in interest as selling stockholders under this prospectus. The selling stockholders also may transfer the shares of common stock in other circumstances, in which case the transferees, pledgees or other successors in interest will be the selling beneficial owners for purposes of this prospectus.
In connection with the sale of our common stock or interests therein, the selling stockholders may enter into hedging transactions with broker-dealers or other financial institutions, which may in turn engage in short sales of the common stock in the course of hedging the positions they assume. The selling stockholders may also sell shares of our common stock short and deliver these securities to close out their short positions, or loan or pledge the common stock to broker-dealers that in turn may sell these securities. The selling stockholders may also enter into option or other transactions with broker-dealers or other financial institutions or the creation of one or more derivative securities which require the delivery to such broker-dealer or other financial institution of shares offered by this prospectus, which shares such broker-dealer or other financial institution may resell pursuant to this prospectus (as supplemented or amended to reflect such transaction).
The aggregate proceeds to the selling stockholders from the sale of the common stock offered by them will be the purchase price of the common stock less discounts or commissions, if any. Each of the selling stockholders reserves the right to accept and, together with their agents from time to time, to reject, in whole or in part, any proposed purchase of common stock to be made directly or through agents. We will not receive any of the proceeds from this offering.
The selling stockholders also may resell all or a portion of the shares in open market transactions in reliance upon Rule 144 under the Securities Act of 1933, provided that they meet the criteria and conform to the requirements of that rule.
The selling stockholders and any underwriters, broker-dealers or agents that participate in the sale of the common stock or interests therein may be “underwriters” within the meaning of Section 2(11) of the Securities Act. Any discounts, commissions, concessions or profit they earn on any resale of the shares may be underwriting discounts and commissions under the Securities Act. Selling stockholders who are “underwriters” within the meaning of Section 2(11) of the Securities Act will be subject to the prospectus delivery requirements of the Securities Act.
57
To the extent required, the shares of our common stock to be sold, the names of the selling stockholders, the respective purchase prices and public offering prices, the names of any agents, dealer or underwriter, any applicable commissions or discounts with respect to a particular offer will be set forth in an accompanying prospectus supplement or, if appropriate, a post-effective amendment to the registration statement that includes this prospectus.
In order to comply with the securities laws of some states, if applicable, the common stock may be sold in these jurisdictions only through registered or licensed brokers or dealers. In addition, in some states the common stock may not be sold unless it has been registered or qualified for sale or an exemption from registration or qualification requirements is available and is complied with.
We have advised the selling stockholders that the anti-manipulation rules of Regulation M under the Exchange Act may apply to sales of shares in the market and to the activities of the selling stockholders and their affiliates. In addition, we will make copies of this prospectus (as it may be supplemented or amended from time to time) available to the selling stockholders for the purpose of satisfying the prospectus delivery requirements of the Securities Act. The selling stockholders may indemnify any broker-dealer that participates in transactions involving the sale of the shares against certain liabilities, including liabilities arising under the Securities Act.
We have agreed to indemnify the selling stockholders against liabilities, including liabilities under the Securities Act and state securities laws, relating to the registration of the shares offered by this prospectus.
We have agreed with the selling stockholders to keep the registration statement of which this prospectus constitutes a part effective until the earlier of (1) such time as all of the shares covered by this prospectus have been disposed of pursuant to and in accordance with the registration statement or (2) the date on which the shares may be sold pursuant to Rule 144(k) of the Securities Act.
CERTAIN U.S. FEDERAL INCOME TAX CONSEQUENCES
The following is a summary of the material United States federal income and estate tax considerations relating to the purchase, ownership and disposition of shares of our common stock, but does not purport to be a complete analysis of all the potential tax considerations relating thereto. This summary is based upon the Internal Revenue Code and United States Treasury Regulations, administrative rulings and court decisions thereunder now in effect, all of which are subject to change, possibly on a retroactive basis.
This summary addresses only holders that will hold shares of our common stock as “capital assets” (generally, property held for investment) and does not address tax considerations applicable to investors that may be subject to special tax rules, including financial institutions, tax-exempt organizations, insurance companies, tax-qualified retirement plans, dealers in securities or currencies, traders in securities that elect to use a mark-to-market method of accounting for their securities holdings, persons that will hold the shares of our common stock as a position in a hedging transaction, “straddle” or “conversion transaction” for tax purposes, regulated investment companies, real estate investment trusts, United States holders that have a functional currency other than the U.S. dollar, certain United States expatriates, controlled foreign corporations, passive foreign investment companies, corporations that accumulate earnings to avoid United States federal income tax or partnerships or other pass-through entities or holders of an interest in such entities. Moreover, this summary does not discuss alternative minimum tax consequences, if any, or any state, local or foreign tax consequences to holders of the shares of our common stock. We have not sought any ruling from the Internal Revenue Service (the “IRS”) with respect to the statements made and the conclusions reached in the following summary, and there can be no assurance that the IRS will agree with such statements and conclusions.
Investors considering the purchase of shares of our common stock should consult their own tax advisors with respect to the application of the United States federal income and estate tax laws to their particular situations as well as any tax consequences arising under the laws of any state, local or foreign taxing jurisdiction or under any applicable tax treaty.
As used in this discussion, a “United States holder” is a beneficial owner of shares of our common stock that for United States federal income tax purposes is:
| | |
| | • | an individual who is a citizen or resident of the United States; |
58
| | |
| | • | a corporation or other entity taxable as a corporation for United States federal income tax purposes, that was created or organized in or under the laws of the United States, any state thereof or the District of Columbia; |
| |
| | • | an estate whose income is subject to United States federal income taxation regardless of its source; and |
| |
| | • | a trust (i) if it is subject to the supervision of a court within the United States and one or more United States persons have the authority to control all substantial decisions of the trust, or (ii) if it has a valid election in effect under applicable United States Treasury Regulations to be treated as a United States person. |
As used in this summary, the term“non-United States holder” means a beneficial owner of common stock who is not a United States holder.
If a partnership (or an entity taxable as a partnership for United States federal income tax purposes) holds the shares of our common stock, the United States federal income tax treatment of a partner will generally depend upon the status of the partner and the activities of the partnership. Partners of a partnership (including an entity treated as a partnership for United States federal income tax purposes) holding shares of our common stock should consult their own tax advisor.
United States Holders
Dividends
We do not expect to declare or pay any dividends on our common stock in the foreseeable future. However, if we do pay dividends on our common stock, such distributions will constitute dividends for United States federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under United States federal income tax principles. To the extent that distributions to you constitute dividends for United States federal income tax purposes, they will be included in your gross income as ordinary income. For individuals, amounts treated as dividends may be eligible for the reduced tax rate (generally 15%) applicable to “qualified dividend income” of non-corporate United States holders for tax years beginning before January 1, 2011, provided certain holding period requirements are met. For corporations, amount treated as dividends generally will be eligible for the dividends received deduction allowed to United States corporations.
Distributions in excess of earnings and profits will constitute a return of capital that is applied against and reduces (but not below zero) your adjusted tax basis in the common stock. Any remaining excess will be treated as gain realized on the sale or other disposition of the common stock and will be treated as described under “United States holders — Disposition of common stock” below.
Disposition of Common Stock
Upon the sale, exchange or other disposition of common stock, you will recognize gain or loss in an amount equal to the difference between your adjusted tax basis in such common stock and the amount realized on the sale, exchange or other disposition. Such gain or loss will be capital gain or loss, and generally will be long-term capital gain or loss if your holding period for the shares of common stock exceeds one year. The deductibility of capital losses is subject to limitations.
Information Reporting and Backup Withholding
Dividends paid to you and proceeds from the sale or disposition of shares of our common stock may be subject to backup withholding unless you (a) are a corporation; (b) provide a valid taxpayer identification number; or (c) establish qualification for another exemption.
Backup withholding is not an additional tax. Rather, amounts withheld under the backup withholding rules may be credited against your United States federal income tax liability. Furthermore, you may obtain a refund of any excess amounts withheld by filing an appropriate claim for refund with the IRS and furnishing any required information in a timely manner.
59
Non-United States Holders
Dividends
We do not expect to declare or pay any dividends on shares of our common stock in the foreseeable future. However, if we do pay dividends on shares of our common stock, such distributions will constitute dividends for United States federal income tax purposes to the extent paid from our current or accumulated earnings and profits, as determined under United States federal income tax principles. Distributions in excess of earnings and profits will constitute a return of capital that is applied against and reduces (but not below zero) your adjusted tax basis in shares of our common stock. Any remaining excess will be treated as gain realized on the sale or other disposition of shares of our common stock and will be treated as described under“Non-United States holders — Disposition of common stock” below. Any dividend paid to you ordinarily will be subject to withholding of United States federal income tax at a rate of 30%, or such lower rate as may be specified under an applicable income tax treaty. In order to receive a reduced treaty rate, you must provide an IRSForm W-8BEN or other appropriate version ofForm W-8 certifying eligibility for the reduced rate.
Generally, dividends paid to you that are effectively connected with a trade or business you conduct in the United States (and, if an income tax treaty applies, are attributable to a permanent establishment you maintain in the United States) will be exempt from the withholding tax described above and instead will be subject to United States federal income tax on a net income basis at the regular graduated United States federal income tax rates in much the same manner as if you were a United States holder. In such cases, we will not have to withhold United States federal income tax if you comply with applicable certification and disclosure requirements. In order to obtain this exemption from withholding tax, you must provide an IRSForm W-8ECI properly certifying eligibility for such exemption. If you are a corporate holder, dividends you receive that are effectively connected with your conduct of a trade or business in the United States may also be subject to an additional branch profits tax at a rate of 30% or such lower rate specified by an applicable income tax treaty.
Disposition of Common Stock
Generally, you will not be subject to United States federal income tax on any gain realized on your disposition of shares of our common stock, unless (i) the gain is effectively connected with your conduct of a trade or business in the United States (and, in the case of an applicable tax treaty, is attributable to a permanent establishment you maintain in the United States), (ii) if you are an individual, you are present in the United States for 183 or more days in the taxable year of the sale or other disposition and certain other conditions are met, or (iii) you are subject to tax pursuant to the provisions of the Internal Revenue Code regarding the taxation of United States expatriates. In addition, you may be subject to United States federal income and withholding tax upon a disposition of our common stock if our stock were considered to be a United States real property interest. We do not expect our stock to constitute a United States real property interest.
Federal Estate Taxes
Shares of our common stock owned or treated as being owned by you at the time of your death will be included in your gross estate for United States federal estate tax purposes, and may be subject to United States federal estate tax, unless an applicable estate tax treaty provides otherwise.
Information Reporting and Backup Withholding
Generally, the amount of dividends paid, the name and address of the recipient, and the amount, if any, of tax withheld must be reported annually to the IRS. A similar report is sent to you. Copies of the information returns reporting those dividends and amounts withheld may also be made available to the tax authorities in the country in which you reside pursuant to the provisions of an applicable tax treaty or exchange of information treaty.
In general, backup withholding at the applicable rate (currently 28%) will not apply to dividends on shares of our common stock paid to you if you have provided the required certification and neither we nor our paying agent has actual knowledge or reason to know that you are a United States person.
60
Information reporting and backup withholding generally will not apply to a payment of the proceeds of a sale of shares of our common stock effected outside the United States by a foreign office of a foreign broker. However, information reporting requirements will apply to a payment of the proceeds of a sale of shares of our common stock effected outside the United States by a foreign office of a broker that (i) is a United States person, (ii) derives 50% or more of its gross income for certain periods from the conduct of a trade or business in the United States, (iii) is a “controlled foreign corporation” as to the United States, or (iv) is a foreign partnership that, at any time during its taxable year, is more than 50% (by income or capital interests) owned by United States persons or is engaged in the conduct of a trade or business in the United States, unless in any such case the broker has documentary evidence in its records that you are anon-United States holder and certain other conditions are met, or you otherwise establish an exemption. Payment of the proceeds of a sale of shares of our common stock by a United States office of a broker will be subject to both information reporting and backup withholding unless you certify yournon-United States holder status under penalties of perjury and the broker does not have actual knowledge or reason to know that you are a United States person, or you otherwise establish an exemption.
Backup withholding is not an additional tax. Rather, amounts withheld under the backup withholding rules may be credited against your United States federal income tax liability. Furthermore, you may obtain a refund of any excess amounts withheld by filing an appropriate claim for refund with the IRS and furnishing any required information in a timely manner.
You should consult your own tax advisor with respect to the application of the above rules to your ownership and disposition of shares of our common stock.
LEGAL MATTERS
The validity of the securities offered hereby has been passed upon for us by Morrison & Foerster LLP, Palo Alto, California.
EXPERTS
Weinberg & Company, P.A., an independent registered public accounting firm, has audited our consolidated balance sheets as of December 31, 2007 and 2006, and the related consolidated statements of operations, stockholders’ equity and comprehensive loss, and cash flows for each of the years in the three-year period ended December 31, 2007, and for the period from June 16, 2003 (inception) through December 31, 2007, which are included in this prospectus. Our consolidated financial statements are included in reliance on the report of Weinberg & Company, P.A. given their authority as experts in accounting and auditing.
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We are a public company and file annual, quarterly and special reports, proxy statements and other information with the SEC. You may read and copy any document we file at the SEC’s public reference room at 100 F Street, N.E., Washington, D.C. 20549. You can request copies of these documents by writing to the SEC and paying a fee for the copying cost. Please call the SEC at1-800-SEC-0330 for more information about the operation of the public reference room. Our SEC filings are also available, at no charge, to the public at the SEC’s web site athttp://www.sec.gov.
61
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| | | | | |
| | | F-2 | |
| | | F-3 | |
| | | F-4 | |
| | | F-5 | |
| | | F-6 | |
| | | F-7 | |
| | | F-19 | |
| | | F-20 | |
| | | F-21 | |
| | | F-22 | |
| | | F-23 | |
F-1
Report of Independent Registered Public Accounting Firm
To the Board of Directors and Stockholders of:
Osteologix, Inc.
We have audited the accompanying consolidated balance sheet of Osteologix, Inc. (the “Company”), a development stage company, as of December 31, 2007 and 2006, and the related consolidated statements of operations, stockholders’ equity and comprehensive loss, and cash flows for each of the years in the three-year period ended December 31, 2007, and for the period from June 16, 2003 (inception) through December 31, 2007. These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these consolidated financial statements based on our audits.
We conducted our audits in accordance with standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the consolidated financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
We were not engaged to examine management’s assertion about the effectiveness of Osteologix, Inc.’s internal control over financial reporting as of December 31, 2007 included in the Company’s Item 9A(T) “Controls and Procedures” in the Annual Report onForm 10-K and, accordingly, we do not express an opinion thereon.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Osteologix, Inc. as of December 31, 2007 and 2006 and the consolidated results of their operations and their cash flows for each of the years in the three-year period ended December 31, 2007, and for the period from June 16, 2003 (inception) through December 31, 2007, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 1 to the consolidated financial statements, the Company has limited revenues and has experienced net losses and negative cash flows from operations since its inception through December 31, 2007 and expects such losses to continue as research and development programs continue. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Weinberg & Company, P.A.
Boca Raton, Florida
March 7, 2008
F-2
| | | | | | | | | |
| | | December 31,
| | | December 31,
| |
| | | 2007 | | | 2006 | |
| | | (In thousands, except per share amounts) | |
| |
ASSETS |
| Current assets: | | | | | | | | |
| Cash and cash equivalents | | $ | 2,872 | | | $ | 481 | |
| Short-term investments | | | 1,273 | | | | 5,719 | |
| Prepaid expenses and other assets | | | 191 | | | | 333 | |
| | | | | | | | | |
| Total current assets | | | 4,336 | | | | 6,533 | |
| Equipment, net | | | 9 | | | | 9 | |
| | | | | | | | | |
| Total assets | | $ | 4,345 | | | $ | 6,542 | |
| | | | | | | | | |
| |
LIABILITIES AND STOCKHOLDERS’ EQUITY |
| Current liabilities: | | | | | | | | |
| Accounts payable | | $ | 1,059 | | | $ | 600 | |
| Accrued liabilities | | | 278 | | | | 145 | |
| | | | | | | | | |
| Total current liabilities | | | 1,337 | | | | 745 | |
| | | | | | | | | |
| Commitments and contingencies | | | | | | | | |
| Stockholders’ Equity | | | | | | | | |
| Preferred stock, $0.0001 par value, 1,000 shares authorized; none issued or outstanding at December 31, 2007 and 2006 | | | — | | | | — | |
| Common stock, $0.0001 par value, 100,000 shares authorized; 24,989 shares issued and outstanding at December 31, 2007, 21,021 shares issued and outstanding at December 31, 2006 | | | 2 | | | | 2 | |
| Additional paid-in capital | | | 20,946 | | | | 15,622 | |
| Accumulated other comprehensive loss | | | (132 | ) | | | (56 | ) |
| Deficit accumulated during development stage | | | (17,808 | ) | | | (9,771 | ) |
| | | | | | | | | |
| Total stockholders’ equity | | | 3,008 | | | | 5,797 | |
| | | | | | | | | |
| Total liabilities and stockholders’ equity | | $ | 4,345 | | | $ | 6,542 | |
| | | | | | | | | |
See accompanying notes to consolidated financial statements.
F-3
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | For the Period
| |
| | | | | | | | | | | | from June 16,
| |
| | | | | | | | | | | | 2003 (Inception)
| |
| | | | | | | | | | | | through
| |
| | | For the Years Ended December 31, | | | December 31,
| |
| | | 2007 | | | 2006 | | | 2005 | | | 2007 | |
| | | (In thousands, except per share amounts) | |
| |
| Contract revenue from related party | | $ | — | | | $ | — | | | $ | 750 | | | $ | 750 | |
| | | | | | | | | | | | | | | | | |
| Operating expenses: | | | | | | | | | | | | | | | | |
| Research and development | | | 4,594 | | | | 2,159 | | | | 1,966 | | | | 9,678 | |
| General and administrative | | | 3,745 | | | | 2,929 | | | | 1,996 | | | | 9,442 | |
| | | | | | | | | | | | | | | | | |
| Total operating expenses | | | 8,339 | | | | 5,088 | | | | 3,962 | | | | 19,120 | |
| | | | | | | | | | | | | | | | | |
| Loss from operations | | | (8,339 | ) | | | (5,088 | ) | | | (3,212 | ) | | | (18,370 | ) |
| Interest income, net | | | 302 | | | | 230 | | | | 16 | | | | 562 | |
| | | | | | | | | | | | | | | | | |
| Net loss | | $ | (8,037 | ) | | $ | (4,858 | ) | | $ | (3,196 | ) | | $ | (17,808 | ) |
| | | | | | | | | | | | | | | | | |
| Net loss per common share, basic and diluted | | $ | (0.35 | ) | | $ | (0.29 | ) | | $ | (0.34 | ) | | | | |
| | | | | | | | | | | | | | | | | |
| Weighted average number of common shares outstanding — basic and diluted | | | 23,275 | | | | 16,872 | | | | 9,415 | | | | | |
| | | | | | | | | | | | | | | | | |
See accompanying notes to consolidated financial statements.
F-4
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | Deficit
| | | | |
| | | | | | | | | | | | Accumulated
| | | Accumulated
| | | | |
| | | | | | | | | Additional
| | | Other
| | | During the
| | | Total
| |
| | | Common Stock | | | Paid-in
| | | Comprehensive
| | | Development
| | | Stockholders’
| |
| | | Shares | | | Amount | | | Capital | | | Loss | | | Stage | | | Equity | |
| | | (In thousands, except per share amounts) | |
| |
| Issuance of common stock to founders for cash of $0.02 per share in June 2003 | | | 4,897 | | | $ | 1 | | | $ | 84 | | | $ | — | | | $ | — | | | $ | 85 | |
| Issuance of common stock for cash of $0.95 per share in October 2003 | | | 979 | | | | — | | | | 930 | | | | — | | | | — | | | | 930 | |
| Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss for the period | | | — | | | | — | | | | — | | | | — | | | | (246 | ) | | | (246 | ) |
| Foreign currency translations | | | — | | | | — | | | | — | | | | (21 | ) | | | — | | | | (21 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (267 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2003 | | | 5,876 | | | | 1 | | | | 1,014 | | | | (21 | ) | | | (246 | ) | | | 748 | |
| Issuance of common stock for cash of $1.01 per share in May 2004 | | | 979 | | | | — | | | | 988 | | | | — | | | | — | | | | 988 | |
| Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss for the period | | | — | | | | — | | | | — | | | | — | | | | (1,471 | ) | | | (1,471 | ) |
| Foreign currency translations | | | — | | | | — | | | | — | | | | (148 | ) | | | — | | | | (148 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (1,619 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2004 | | | 6,855 | | | | 1 | | | | 2,002 | | | | (169 | ) | | | (1,717 | ) | | | 117 | |
| Issuance of common stock for cash of $0.83 per share in January 2005, net of issuance costs | | | 1,430 | | | | — | | | | 1,158 | | | | — | | | | — | | | | 1,158 | |
| Issuance of common stock for cash of $1.24 per share in June 2005, net of issuance costs | | | 2,220 | | | | — | | | | 2,699 | | | | — | | | | — | | | | 2,699 | |
| Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss for the period | | | — | | | | — | | | | — | | | | — | | | | (3,196 | ) | | | (3,196 | ) |
| Foreign currency translations | | | — | | | | — | | | | — | | | | 165 | | | | — | | | | 165 | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (3,031 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2005 | | | 10,505 | | | | 1 | | | | 5,859 | | | | (4 | ) | | | (4,913 | ) | | | 943 | |
| Common stock transferred in merger with Castle & Morgan Holdings, Inc. | | | 2,860 | | | | — | | | | — | | | | — | | | | — | | | | — | |
| Issuance of common stock for cash of $1.31 per share in private placement in May 2006, net of issuance costs | | | 7,656 | | | | 1 | | | | 9,255 | | | | — | | | | — | | | | 9,256 | |
| Stock based compensation expense | | | — | | | | — | | | | 508 | | | | — | | | | — | | | | 508 | |
| Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss for the period | | | — | | | | — | | | | — | | | | — | | | | (4,858 | ) | | | (4,858 | ) |
| Unrealized gain on short-term investments | | | — | | | | — | | | | — | | | | 2 | | | | — | | | | 2 | |
| Foreign currency translations | | | — | | | | — | | | | — | | | | (54 | ) | | | — | | | | (54 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (4,910 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2006 | | | 21,021 | | | | 2 | | | | 15,622 | | | | (56 | ) | | | (9,771 | ) | | | 5,797 | |
| Issuance of common stock and warrants for cash of $1.32 per share in private placement in June 2007, net of issuance costs | | | 3,825 | | | | — | | | | 4,970 | | | | — | | | | — | | | | 4,970 | |
| Stock issued in lieu of cash for services | | | 143 | | | | — | | | | 170 | | | | — | | | | — | | | | 170 | |
| Stock based compensation expense | | | — | | | | — | | | | 184 | | | | — | | | | — | | | | 184 | |
| Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss for the period | | | — | | | | — | | | | — | | | | — | | | | (8,037 | ) | | | (8,037 | ) |
| Unrealized loss on short-term investments | | | — | | | | — | | | | — | | | | (2 | ) | | | — | | | | (2 | ) |
| Foreign currency translations | | | — | | | | — | | | | — | | | | (74 | ) | | | — | | | | (74 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (8,113 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2007 | | | 24,989 | | | $ | 2 | | | $ | 20,946 | | | $ | (132 | ) | | $ | (17,808 | ) | | $ | 3,008 | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
See accompanying notes to consolidated financial statements.
F-5
| | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | For the Period
| |
| | | | | | | | | | | | from June 16,
| |
| | | | | | | | | | | | 2003 (Inception)
| |
| | | | | | | | | | | | through
| |
| | | For the Years Ended December 31, | | | December 31,
| |
| | | 2007 | | | 2006 | | | 2005 | | | 2007 | |
| | | (In thousands) | |
| |
Operating activities | | | | | | | | | | | | | | | | |
| Net loss | | $ | (8,037 | ) | | $ | (4,858 | ) | | $ | (3,196 | ) | | $ | (17,808 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | | | | | |
| Depreciation | | | 8 | | | | 5 | | | | 3 | | | | 19 | |
| Loss on disposal of equipment | | | 2 | | | | — | | | | — | | | | 2 | |
| Issuance of stock in lieu of cash for services | | | 170 | | | | — | | | | — | | | | 170 | |
| Stock based compensation | | | 184 | | | | 508 | | | | — | | | | 692 | |
| Changes in operating assets and liabilities: | | | | | | | | | | | | | | | | |
| Receivable from related party | | | — | | | | 750 | | | | (750 | ) | | | — | |
| Prepaid expenses and other current assets | | | 142 | | | | (37 | ) | | | (216 | ) | | | (191 | ) |
| Accounts payable | | | 459 | | | | 119 | | | | 337 | | | | 1,059 | |
| Accrued liabilities | | | 133 | | | | (55 | ) | | | 122 | | | | 278 | |
| | | | | | | | | | | | | | | | | |
| Net cash used in operating activities | | | (6,939 | ) | | | (3,568 | ) | | | (3,700 | ) | | | (15,779 | ) |
| | | | | | | | | | | | | | | | | |
Investing activities | | | | | | | | | | | | | | | | |
| Purchases of short-term investments | | | (6,873 | ) | | | (6,547 | ) | | | — | | | | (13,420 | ) |
| Sales and maturities of short-term investments | | | 11,317 | | | | 830 | | | | — | | | | 12,147 | |
| Purchases of equipment | | | (10 | ) | | | (7 | ) | | | — | | | | (30 | ) |
| | | | | | | | | | | | | | | | | |
| Net cash provided by (used in) investing activities | | | 4,434 | | | | (5,724 | ) | | | — | | | | (1,303 | ) |
| | | | | | | | | | | | | | | | | |
Financing activities | | | | | | | | | | | | | | | | |
| Net proceeds from issuance of common stock | | | 4,970 | | | | 9,256 | | | | 3,857 | | | | 20,086 | |
| | | | | | | | | | | | | | | | | |
| Effect of exchange rates on cash and cash equivalents | | | (74 | ) | | | (54 | ) | | | 165 | | | | (132 | ) |
| | | | | | | | | | | | | | | | | |
| Net increase/(decrease) in cash and cash equivalents | | | 2,391 | | | | (90 | ) | | | 322 | | | | 2,872 | |
| Cash and cash equivalents at beginning of period | | | 481 | | | | 571 | | | | 249 | | | | — | |
| | | | | | | | | | | | | | | | | |
| Cash and cash equivalents at end of period | | $ | 2,872 | | | $ | 481 | | | $ | 571 | | | $ | 2,872 | |
| | | | | | | | | | | | | | | | | |
See accompanying notes to consolidated financial statements.
F-6
Osteologix, Inc.
(a development stage company)
Notes to Consolidated Financial Statements
December 31, 2007
(Tabular amounts in thousands, except per share amounts)
| |
| 1. | Basis of Presentation and Summary of Significant Accounting Policies |
Business Description
Osteologix, Inc. and its subsidiary (“Osteologix” or the “Company”) are in the business of developing pharmaceuticals for the treatment and prevention of diseases of bone and joint tissues. The Company’s lead product candidate, NB S101, is in clinical development for the treatment of osteoporosis. Osteologix has not yet generated substantial revenues from its operations and, accordingly, the Company is in the development stage.
Basis of Presentation
The consolidated financial statements include the accounts of the Company and its wholly owned Danish subsidiary, Osteologix ApS, which was previously incorporated as Osteologix A/S. All intercompany accounts and transactions have been eliminated. Osteologix operates in one business segment, the development of pharmaceutical products.
The consolidated financial statements have been prepared assuming the Company will continue as a going concern. Osteologix has experienced net losses and negative cash flows from operations since its inception and expects its losses to continue as the Company furthers its research and development programs. The Company’s management believes that its current cash and short-term investments will enable it to continue planned operations only into the fourth quarter of 2008. These conditions raise substantial doubt about the Company’s ability to continue as a going concern. Management plans to raise additional capital in order to fund is operations, and therefore believes it is appropriate for the consolidated financial statements to be prepared on a going concern basis. The consolidated financial statements do not contain any adjustments that may be required if Osteologix is unable to continue as a going concern.
Background of Company Organization and Description of 2006 Merger Transaction
Osteologix was initially formed in Denmark on June 16, 2003 under the name Nordic Bone A/S, and in 2004 changed its name to Osteologix A/S. All of the issued and outstanding shares of Osteologix A/S were owned by Nordic Biotech K/S, a Danish venture capital fund, prior to a merger transaction that occurred on May 24, 2006. On May 24, 2006, Osteologix A/S completed a “reverse merger” transaction with Castle & Morgan Holdings, Inc., a U.S. public company incorporated in Delaware and traded on the Over-the-Counter Bulletin Board. In the merger, Castle and Morgan Holdings, Inc. issued new shares of common stock in exchange for all of the issued and outstanding common stock of Osteologix A/S. Also on May 24, 2006, a subsidiary of Osteologix A/S completed a private placement transaction, raising $10 million in gross proceeds by issuing new shares of its common stock. Immediately thereafter, Castle & Morgan Holdings, Inc. changed its name to Osteologix, Inc. and the newly issued shares in the subsidiary of Osteologix A/S converted into Osteologix Inc. shares. As a result of these transactions, Nordic Biotech K/S’s previous ownership of 100% of the outstanding stock of Osteologix A/S exchanged into a 50.0% ownership position in Osteologix, Inc. The stock in the subsidiary of Osteologix A/S that was sold in the private placement exchanged into a 36.4% ownership position in Osteologix, Inc. The previously outstanding stock in Castle & Morgan Holdings represented 13.6% ownership position in Osteologix, Inc. After Nordic Biotech K/S participation in the private placement financing, they owned 60.1% of Osteologix, Inc.
Although Osteologix A/S became a wholly owned subsidiary of Castle & Morgan Holdings Inc. (subsequently renamed Osteologix, Inc.) in the merger, for financial reporting purposes Osteologix A/S is treated as the acquirer because its’ previous shareholder continued to own a majority of the surviving company. Accordingly, the historical consolidated financial statements prior to the date of the merger that are included in these consolidated financial statements for comparative purposes are the consolidated financial statements of Osteologix A/S and its subsidiary.
F-7
Osteologix, Inc.
(a development stage company)
Notes to Consolidated Financial Statements — (Continued)
The number of shares of common stock, the par value and additional paid-in capital reported prior to May 24, 2006 in the consolidated financial statements have been amended to reflect the impact of the merger with Castle & Morgan Holdings, Inc. Following the merger, the functional currency of the Company became the United States Dollar whereas it was previously the Danish Krone.
Use of Estimates
The preparation of financial statements in conformity with U.S. generally accepted accounting principles requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual amounts may differ from those estimates.
Concentration of Credit and Other Risks and Uncertainties
Financial instruments which potentially subject the Company to concentration of credit risk consist primarily of cash, cash equivalents and short-term investments. The Company’s cash and cash equivalents are generally invested in deposit accounts or money market accounts with U.S and Danish banks, and deposits may exceed the amount covered by insurance for loss. As of December 31, 2007 and 2006, the Company’s uninsured cash totaled $174,000 and $324,000, respectively, and the Company’s uninsured money market accounts and cash equivalents totaled $2,621,000 at December 31, 2007. The Company’s money market accounts, cash equivalents and short-term investments are invested in high quality government or corporate debt securities, and, other than U.S. Government securities, the Company limits its exposure to any single corporation to no more than 5% of its debt security portfolio.
The Company’s lead product candidate, NB S101, is in clinical development for the treatment of osteoporosis and is currently the Company’s only pharmaceutical product being tested in humans. Development of new pharmaceutical products involves a high degree of risk, and failure can occur at any point in a product’s development. Accordingly, NB S101 may never be successfully marketed. The business risk as a result of the Company concentrating its efforts in a single product under development is significant.
Fair Value of Financial Instruments
For financial instruments consisting of cash and cash equivalents, short-term investments, prepaid expenses and other assets, accounts payable and accrued liabilities included in the consolidated financial statements, the carrying amounts are reasonable estimates of the fair value due to their short maturities. The fair value of other short-term and long-term obligations is estimated based on current interest rates available for debt instruments with similar terms, degrees of risk and remaining maturities. The carrying values of these obligations approximate their fair values.
Foreign Currency Translation
These financial statements are presented in U.S. dollars for all periods presented. Translation of balance sheet accounts denominated in foreign currencies is made at the exchange rate in effect on the balance sheet date. Translation of amounts reported in the statement of operations and statement of cash flows is made at the average exchange rate for the periods reported. Translation gains and losses are recognized within “Other Comprehensive Loss.”
Revenue Recognition
Revenue recognized under corporate license agreements and collaborations is recognized as earned based on the performance requirements of the contracts. Revenue from non-refundable license fees where the Company continues involvement is recognized on a straight-line basis over the period of the Company’s continued
F-8
Osteologix, Inc.
(a development stage company)
Notes to Consolidated Financial Statements — (Continued)
involvement. Revenue from non-refundable license fees for which no further performance obligations exist and for which the Company has no continuing involvement is recognized either when the payments are received or collection is assured.
Cash and Cash Equivalents
The Company considers all highly liquid investments with a maturity period of three months or less at the time of acquisition to be cash equivalents.
Equipment
Equipment is stated at cost and depreciated on a straight-line basis over the estimated useful lives of the related assets, which is generally three years. Upon sale or retirement of the assets, the costs and related accumulated depreciation are removed from the accounts and the resulting gain or loss is reflected in the statement of operations. Repair and maintenance expenses are charged to the statement of operations as they are incurred.
Impairment of Long-Lived Assets
The Company reviews long-lived assets for impairment whenever events or changes in business circumstances indicate that the carrying amount of the assets may not be fully recoverable. An impairment loss would be recognized when future estimated undiscounted cash flows expected to result from use of the asset, and its eventual disposition, are less than the carrying amount of the asset. The impairment loss would be based on the excess of the carrying value over its respective fair value. Through December 31, 2007, the Company has not recorded any impairment losses.
Research and Development Expenses
The Company’s research and development costs are expensed as incurred. Research and development costs include clinical trial costs, preclinical studies, payments to contract research organizations, compensation expenses for research and development personnel, development and manufacturing costs for investigational drugs, supplies, and related consulting and advisor costs.
General and Administrative Expenses
The Company’s general and administrative expenses include compensation expense for personnel not directly involved in research and development activities, management and other administrative personnel costs, insurance, accounting, legal and patent expenses.
Stock-based Compensation
Stock options issued to employees are accounted for using an estimate of the fair value of the stock option on the date it is granted. The estimated fair value on the grant date is recognized in the statement of operations on a straight-line basis over the vesting period of the underlying stock options.
Income Taxes
The Company uses the liability method of accounting for income taxes, and determines deferred tax assets and liabilities based on differences between the financial reporting and tax reporting basis of assets and liabilities. The Company measures these assets and liabilities using enacted tax rates and laws that are scheduled to be in effect when the differences are expected to reverse. Because the realization of deferred tax assets is dependent on future earnings, if any, and the Company’s future earnings are uncertain, all of the Company’s net deferred tax assets have been fully offset by a valuation allowance.
F-9
Osteologix, Inc.
(a development stage company)
Notes to Consolidated Financial Statements — (Continued)
Net Loss per Share and Anti-dilutive Securities
Net loss per share has been computed using the weighted average number of shares of common stock outstanding during the period. During the years ended December 31, 2007, 2006 and 2005, potentially dilutive options and warrants to purchase common stock aggregating 4,656,000, 2,129,000 and 979,000 shares, respectively, were outstanding and not considered in the loss per share computation because their effect would have been antidilutive.
Recent Accounting Pronouncement Issued and Adopted by the Company
In June 2006, the Financial Accounting Standards Board (“FASB”) issued FIN 48, “Accounting for Uncertainty in Income Taxes — an interpretation of FASB Statement No. 109” (“FIN 48”), which seeks to reduce the diversity in practice associated with the accounting and reporting for uncertainty in income tax positions. FIN 48 prescribes a model for the financial statement recognition, measurement, presentation and disclosure of uncertain tax positions taken or expected to be taken in an income tax return and was effective for fiscal years beginning after December 15, 2006. Accordingly, the Company adopted the provisions of FIN 48 on January 1, 2007. Under FIN 48, the Company may recognize the tax benefit from an uncertain tax position only if it is more-likely-than-not (greater than 50% likely) that the tax position will be upheld on examination by the taxing authority, based on the technical merits of the tax position. FIN 48 also provides guidance on income tax recognition and classification, and requires additional financial statement disclosures, including information about interest and penalties on income taxes and accounting for income taxes in interim periods.
The Company believes that it has not taken any uncertain income tax positions that would impact its consolidated financial statements as of December 31, 2007. As of January 1, 2007, the date Osteologix adopted FIN 48, the Company had deferred tax assets of $2,869,000, which were not recognized in the financial statements because they were fully reserved by a valuation allowance of the same amount. Also as of the date of adoption, and as of December 31, 2007, Osteologix does not have a liability for unrecognized tax benefits. The Company’s policy is to record interest and penalties on uncertain tax positions as income tax expense. As of December 31, 2007, the company has no accrued interest or penalties related to uncertain income tax positions.
Recent Accounting Pronouncements Issued and Not Yet Adopted by the Company
In September 2006, the FASB issued Statement of Financial Accounting Standards No. 157, “Fair Value Measurements” (“SFAS 157”), which clarifies and prioritizes methods for measuring fair value under generally accepted accounting principles. SFAS 157 generally increases the level of disclosure required for fair value measurements, although it does not impact the valuations or disclosures required under SFAS 123R. For Osteologix, implementation of SFAS 157 will be required on January 1, 2008 and the Company is completing its evaluation of the impact of SFAS 157 on its consolidated financial statements and disclosures. Osteologix currently does not believe any financial statement accounts will be impacted by SFAS 157, but footnote disclosures may be expanded.
| |
| 2. | Short-Term Investments |
The Company invests funds that are not required for immediate operating needs primarily in a diversified portfolio of debt securities which are classified as short-term investments on the balance sheet. Management determines the appropriate classification of these marketable debt securities at the time of purchase and reevaluates such designation as of each balance sheet date. As of December 31, 2007, all marketable securities are classified as available-for-sale. These securities are stated at their estimated fair value based upon market quotes. Unrealized gains and losses are included in accumulated other comprehensive loss. Amortization of premiums and discounts and realized gains and losses are included in interest income. The cost of securities sold is based on the specific identification method. The Company has not experienced any significant losses on its investments.
F-10
Osteologix, Inc.
(a development stage company)
Notes to Consolidated Financial Statements — (Continued)
As of December 31, 2007 all of the Company’s short-term investments had maturity dates of less than one year. The components of short-term investments were as follows:
| | | | | | | | | |
| | | December 31,
| | | December 31,
| |
| | | 2007 | | | 2006 | |
| |
| Corporate securities | | $ | 968 | | | $ | 3,982 | |
| Municipal obligations | | | 305 | | | | 1,112 | |
| U.S. government securities | | | — | | | | 474 | |
| Asset-backed and other securities | | | — | | | | 151 | |
| | | | | | | | | |
| | | $ | 1,273 | | | $ | 5,719 | |
| | | | | | | | | |
The fair value of corporate securities included immaterial unrealized gains. There were no unrealized losses in the Company’s short-term investments. Realized gains and losses on sales of the Company’s short-term investments have not been material.
| |
| 3. | Prepaid Expenses and Other Assets |
The components of prepaid expenses and other assets are as follows:
| | | | | | | | | |
| | | December 31,
| | | December 31,
| |
| | | 2007 | | | 2006 | |
| |
| Danish value-added tax (VAT) receivable | | $ | 86 | | | $ | 232 | |
| Prepaid expenses and deposits | | | 105 | | | | 101 | |
| | | | | | | | | |
| | | $ | 191 | | | $ | 333 | |
| | | | | | | | | |
Equipment primarily consists of computer equipment and an ultra-cold freezer. As of December 31, 2007 and 2006, accumulated depreciation on equipment aggregated $19,000 and $11,000, respectively.
| |
| 5. | Commitments and Contingencies |
The Company has two noncancelable operating leases with initial terms in excess of 12 months for the San Francisco, California and Glen Allen, Virginia corporate offices. These leases both expire on December 31, 2008. At December 31, 2007, future minimum payments required under the leases aggregate $73,000, which is all due in 2008. Total rental expense was $124,000, $81,000 and $55,000 for the years ended December 31, 2007, 2006 and 2005, respectively.
The Company has an employment agreement in place with its President and Chief Executive Officer which in addition to salary requires the Company to pay the premium on certain life insurance benefits, an automobile allowance and automobile operating expenses and disability insurance coverage based on salary. If the CEO’s employment is terminated under certain circumstances as specified in his employment agreement he may be entitled to nine months continued salary and benefits.
The Company has an employment agreement in place with its Chief Financial Officer. If the CFO’s employment is terminated under certain circumstances as specified in his employment agreement he may be entitled to six months continued salary and benefits.
The Company, as permitted under Delaware law and in accordance with its by-laws, has agreed to pay certain expenses and indemnify its officers and directors, subject to certain limits, if the officer or director becomes involved in a lawsuit or other proceeding arising from his service to the Company. The maximum amount of
F-11
Osteologix, Inc.
(a development stage company)
Notes to Consolidated Financial Statements — (Continued)
potential future indemnification is unlimited. The Company has a director and officer insurance policy that may enable the Company to recover a portion of any future amounts paid under the Company’s indemnity obligations. The Company believes that the fair value of its obligations under its indemnification commitments is minimal and at present no claims are being asserted against the Company for indemnification under these obligations. Accordingly, the Company has not recognized any liabilities relating to these obligations as of December 31, 2007.
Preferred Stock
The Company is authorized, subject to limitations prescribed by Delaware law, to issue preferred stock and to fix the rights, preferences and privileges of the preferred stock. As of December 31, 2007, no shares of preferred stock have been issued.
Rights of Common Stockholders
The Company has only common stock issued and outstanding. Accordingly, all outstanding shares are of the same class and have equal liquidation, preference and adjustment rights. Each share of common stock entitles the holder to one vote on all matters submitted to a vote of the Company’s stockholders.
Issuances of Common Stock
In June 2007, Osteologix completed the sale of 3,825,000 shares of its common stock to a group of investors, including Nordic Biotech K/S, at a price of $1.32 per share, for gross proceeds of $5,050,000. Net proceeds were $4,970,000 after issuance costs. As part of the transaction, the Company issued each investor a warrant to purchase one share of common stock for every two shares of common stock purchased in the placement, and an aggregate of 1,913,000 warrants were issued. The warrants have an exercise price of $1.20 per share and expire on August 31, 2008.
In May 2006, Osteologix completed the sale of 7,656,000 shares of its common stock to a group of investors, including Nordic Biotech K/S, at a price of $1.31 per share, for gross proceeds of $10,000,000. Net proceeds were $9,256,000 after issuance costs.
In June 2005, Osteologix completed the sale of 2,220,000 shares of its common stock to Nordic Biotech K/S at a price of $1.24 per share, for gross proceeds of $2,747,000. Net proceeds were $2,699,000 after issuance costs.
In January 2005, Osteologix completed the sale of 1,430,000 shares of its common stock to Nordic BiotechK/S at a price of $0.83 per share, for gross proceeds of $1,180,000. Net proceeds were $1,158,000 after issuance costs.
In May 2004, Osteologix completed the sale of 979,000 shares of its common stock to Nordic Biotech K/S at a price of $1.01 per share, for gross and net proceeds of $988,000.
In October 2003, Osteologix completed the sale of 979,000 shares of its common stock to Nordic Biotech K/S at a price of $0.95 per share for gross and net proceeds of $930,000.
In June 2003, at the Company’s formation, Osteologix completed the sale of 4,897,000 shares of its common stock to its founder, Nordic Biotech K/S, at a price of $0.02 per share, for gross and net proceeds of $85,000.
Securities Exercisable into Common Stock
At December 31, 2007, in addition to the warrants and options outstanding under the Company’s stock-based compensation programs, there were warrants to purchase 1,913,000 shares of common stock outstanding. The warrants are exercisable at a price of $1.20 per share and expire on August 31, 2008.
F-12
Osteologix, Inc.
(a development stage company)
Notes to Consolidated Financial Statements — (Continued)
| |
| 7. | Stock-Based Compensation |
Osteologix’s Stock-based Compensation Plans
In 2003, the Osteologix A/S board of directors adopted an Equity Incentive Plan (the “Osteologix A/S Equity Plan”) which provided for the issuance of warrants to purchase common stock at fair market value on the date of the grant. As a part of the May 24, 2006 merger transaction, all outstanding warrants to purchase shares of Osteologix A/S under this plan were canceled, and warrants to purchase 979,000 shares of Osteologix, Inc. common stock were issued in their place under similar terms. The Osteologix A/S Equity Plan was then canceled and no further warrants have been issued.
In 2006, the Castle & Morgan Holdings, Inc. board of directors adopted the 2006 Equity Incentive Plan (the “2006 Stock Option Plan”) which provides for the issuance of incentive and nonqualified stock options to employees and independent consultants at or above the fair market value on the date of grant. The 2006 Stock Option Plan was ratified by the Company’s stockholders in 2007. A total of 2,400,000 shares have been reserved for issuance under the 2006 Stock Option Plan. As of December 31, 2007, there were 806,000 shares available for future issuance under the 2006 Stock Option Plan.
Under the Company’s director compensation program, the Company has issued nonqualified stock options to its directors with exercise prices equal to the stock’s fair market value on the date of grant. Although no shares are reserved for future issuance under this program, the board of directors retains the ability to issue additional options to purchase shares of common stock under this program at its discretion.
As of December 31, 2007, the outstanding stock options and warrants were comprised as follows:
| | | | | |
| Warrants converted from Osteologix A/S Equity Plan | | | 900 | |
| Options granted under the 2006 Stock Option Plan | | | 1,594 | |
| Options granted under the director compensation program | | | 250 | |
| | | | | |
| Total outstanding options and warrants for which stock-based compensation expense has been recognized | | | 2,744 | |
| | | | | |
Stock-based Compensation Expense
Since adopting Statement of Financial Accounting Standards No. 123R, “Share-Based Payment” (“SFAS 123R”) on January 1, 2006, the Company has elected to use the Black-Scholes option-pricing model (the “Black-Scholes model”) as its method of valuing stock-based compensation. The Black-Scholes model requires assumptions to be made regarding expected volatility, expected term, risk-free interest rate and dividend yield of the security into which the options can be converted.
Since the date of the merger transaction in 2006, Osteologix has issued options to purchase its common stock to officers, employees and directors. The following were the weighted average assumptions used to value the Company’s stock options and warrants:
| | | | | | | | | |
| | | Options Granted During the
| |
| | | Years Ended December 31 | |
| | | 2007 | | | 2006 | |
| |
| Expected volatility | | | 70 | % | | | 70 | % |
| Expected term | | | 6.1 | years | | | 6.1 | years |
| Risk-free interest rate | | | 4.6 | % | | | 4.7 | % |
| Dividend yield | | | 0.0 | % | | | 0.0 | % |
| | | | | | | | | |
| Weighted-average fair value of options | | $ | 0.64 | | | $ | 0.80 | |
| | | | | | | | | |
F-13
Osteologix, Inc.
(a development stage company)
Notes to Consolidated Financial Statements — (Continued)
The expected volatility was determined by the Company based on the historical volatility of Osteologix and comparable companies. The expected term was determined as the mean of the weighted average term to vesting and the expiration date of the option. The risk-fee interest rate was based upon the U.S. Treasury yield for expected life of the Company’s stock options on the date of grant. The dividend rate was based on the Company’s projections that show it will not be able to pay dividends for the foreseeable future. As share-based compensation expense for stock options is based on awards ultimately expected to vest, the share-based compensation expense related to stock options issued has been reduced for estimated forfeitures. Forfeitures are estimated at the time of grant and revised in subsequent periods if actual forfeitures differ from those estimated. In 2007 the Company estimated the expected forfeiture rate at 15%.
When the merger transaction occurred in 2006 Osteologix exchanged warrants to purchase common stock of Osteologix A/S for warrants to purchase common stock of Osteologix, Inc. at the same exercise price and exchange ratio as received by the shareholder of Osteologix A/S. The Company recorded an expense for the modification to the stock awards based on the excess of the fair value of the replacement warrant over the fair value of the cancelled warrant. The incremental stock-based compensation expense on the date of the merger for the increased fair value of the modified warrants aggregated $372,000, of which $275,000 was immediately recognized in the consolidated statement of operations for the warrants that were vested at that time. The remaining $97,000 is being recognized over the remaining vesting period of the warrants ending in 2008. The estimated per share fair value of the modified warrants was $0.73, with the following assumptions: expected volatility of 70%; expected term of 3.0 years; annualized risk-free interest rate of 5.0%; and dividend yield of zero. From the date of adoption of SFAS 123R until the merger transaction, no options or warrants were granted.
In addition to the assumptions used to estimate the value of stock-based compensation, the actual stock price and the option exercise price at the time of grant will also have a significant impact on the valuation of the stock options. The stock options granted during 2007 all had exercise prices of $1.20 per share (with stock prices at the time of grant ranging from $0.98 to $1.18), vesting periods of four to approximately four and one-half years, and expiration dates ten years from the date of grant. The stock options granted in 2006 included exercise prices ranging from $1.00 to $1.50 per share (with stock prices at the time of grant ranging from $1.00 to $1.30), vesting periods of up to four years and expiration dates ten years from the date of grant.
Stock option transactions for the years 2005 through 2007 are summarized as follows:
| | | | | | | | | |
| | | | | | Weighted
| |
| | | | | | Average
| |
| | | Number of
| | | Exercise
| |
| | | Shares | | | Price | |
| |
| Outstanding at December 31, 2004 | | | 392 | | | $ | 1.03 | |
| Granted | | | 587 | | | | 1.03 | |
| | | | | | | | | |
| Outstanding at December 31, 2005 | | | 979 | | | | 1.03 | |
| Granted or modified | | | 2,279 | | | | 1.18 | |
| Canceled | | | (1,129 | ) | | | 1.05 | |
| | | | | | | | | |
| Outstanding at December 31, 2006 | | | 2,129 | | | | 1.18 | |
| Granted | | | 1,150 | | | | 1.20 | |
| Forfeited | | | (477 | ) | | | 1.17 | |
| Expired | | | (58 | ) | | | 1.20 | |
| | | | | | | | | |
| Outstanding at December 31, 2007 | | | 2,744 | | | $ | 1.19 | |
| | | | | | | | | |
F-14
Osteologix, Inc.
(a development stage company)
Notes to Consolidated Financial Statements — (Continued)
The following table summarizes information about stock options and warrants outstanding and exercisable as of December 31, 2007:
| | | | | | | | | | | | | | | | | |
| | | | | Weighted
| | | | Weighted
|
| | | | | Average
| | | | Average
|
| | | Number
| | Remaining
| | | | Remaining
|
| | | of Options
| | Contractual
| | Number
| | Contractual
|
Exercise Price | | Outstanding | | Life (Years) | | Exercisable | | Life (Years) |
| |
| $1.00 - $1.03 | | | 925 | | | | 6.5 | | | | 895 | | | | 6.4 | |
| $1.20 | | | 1,419 | | | | 9.2 | | | | 347 | | | | 9.0 | |
| $1.50 | | | 400 | | | | 8.6 | | | | 125 | | | | 8.6 | |
| | | | | | | | | | | | | | | | | |
| | | | 2,744 | | | | 8.2 | | | | 1,367 | | | | 7.3 | |
| | | | | | | | | | | | | | | | | |
The weighted average exercise price for options that were exercisable at December 31, 2007 was $1.12. There were options for 958,000 and 527,000 shares exercisable at December 31, 2006 and 2005, respectively. The weighted average exercise price for the 1,377,000 unvested options at December 31, 2007 was $1.26 per share, with an average grant-date fair value of $0.68. The weighted average exercise price for the 1,171,000 unvested options at December 31, 2006 was $1.27 per share, with an average grant date fair value of $0.77.
The aggregate intrinsic value of the options and warrants outstanding as of December 31, 2007 was $556,000, and the aggregate intrinsic value of the options and warrants that were exercisable was $364,000, based on a closing stock price of $1.37.
The 2007 stock-based compensation expense of $184,000 includes $92,000 related to stock options granted during 2007 and $92,000 related to stock options and warrants granted prior to 2007. The 2006 stock-based compensation expense of $508,000 includes $480,000 related to stock options and warrants modified or granted during 2006 and stock-based compensation expense of $28,000 related to stock warrants granted prior to 2006. As of December 31, 2007, the total unrecognized expense for unvested stock warrants and options is $920,000, which will be expensed over the remaining weighted-average vesting period of 3.5 years. The Company recorded no income tax benefits for share-based compensation arrangements for the year ended December 31, 2007 because the Company has cumulative operating losses for which a valuation allowance has been established.
Pro-Forma Information under SFAS 123
Prior to adopting SFAS 123R on January 1, 2006, the Company applied APB Opinion No. 25 “Accounting for Stock Issued to Employees” in accounting for its stock-based compensation. Because the Company granted warrants to purchase its common stock with an exercise price that was equal to the fair market value of the stock on the date of grant, Osteologix accordingly recognized no compensation expense for the warrants. The Company followed the disclosure-only provisions of SFAS 123, as amended by SFAS No. 148, and for purposes of pro forma disclosures the estimated fair value of the warrants was amortized to expense over the vesting period of the warrants
F-15
Osteologix, Inc.
(a development stage company)
Notes to Consolidated Financial Statements — (Continued)
using the straight-line method. The following table presents information showing the effects to the reported net loss and net loss per share as if Osteologix had accounted for stock-based compensation using the fair-value method:
| | | | | |
| | | For the Year Ended
| |
| | | December 31, 2005 | |
| |
| Net loss applicable to common stockholders, as reported | | $ | (3,196 | ) |
| Less: total stock-based employee compensation determined under fair value based method for all awards | | | (120 | ) |
| | | | | |
| Net loss applicable to common stockholders, pro forma | | $ | (3,316 | ) |
| | | | | |
| Net loss per common share, basic and diluted: | | | | |
| As reported | | $ | (0.34 | ) |
| | | | | |
| Pro forma | | $ | (0.35 | ) |
| | | | | |
The fair value of warrants granted prior to the adoption of SFAS 123R was estimated on the date of grant using the Black-Scholes option valuation model under the minimum value method, which assumes a volatility of 0%, expected term of five years for employee grants and ten years for non-employee director grants, expected dividend of zero, and a risk free rate for periods related to the expected life of the warrants based on the U.S. Treasury yield curve in effect at the time of grant.
General Stock Option Accounting Information
The Company believes it is important for investors to be aware that there is a high degree of subjectivity involved in estimating the value of stock-based compensation, including under the requirements of SFAS 123R, and that changes in input assumptions, particularly the estimated volatility and estimated term, can materially affect the resulting estimates of the fair values of the options and warrants granted. The expenses recorded for stock-based compensation in the Company’s consolidated financial statements may differ significantly from the actual value realized by the recipients of the stock awards. The stock awards may expire worthless or otherwise result in zero intrinsic value to the recipient, or value may be realized from these instruments that are significantly in excess of the fair values reported in consolidated financial statements. Under SFAS 123R, the expenses recorded in the consolidated financial statements are not adjusted to the actual amounts realized by the stock option recipients. The expenses recognized under SFAS 123R will not result in any payment of cash by the Company.
Stock-based compensation arrangements for non-employees are accounted for using a fair value approach, and the compensation costs under such arrangements are subject to re-measurement over their vesting terms, as earned.
The Company is subject to income taxes in the United States and in Denmark based on its operations in each country. The losses for each country are as follows:
| | | | | | | | | | | | | |
| | | For the Years Ended December 31, | |
| | | 2007 | | | 2006 | | | 2005 | |
| |
| United States | | $ | (3,160 | ) | | $ | (1,719 | ) | | $ | (474 | ) |
| Denmark | | | (4,877 | ) | | | (3,139 | ) | | | (2,722 | ) |
| | | | | | | | | | | | | |
| | | $ | (8,037 | ) | | $ | (4,858 | ) | | $ | (3,196 | ) |
| | | | | | | | | | | | | |
There is no provision for income taxes because the Company has incurred operating losses. Realization of deferred tax assets is dependent upon future earnings, if any, the timing and amount of which are uncertain;
F-16
Osteologix, Inc.
(a development stage company)
Notes to Consolidated Financial Statements — (Continued)
therefore the net deferred tax assets have been fully offset by a valuation allowance. For 2007, 2006 and 2005, the valuation allowance increased by $2,479,000, $1,727,000 and $849,000, respectively.
The significant components of the Company’s deferred tax assets are as follows:
| | | | | | | | | |
| | | December 31,
| | | December 31,
| |
| | | 2007 | | | 2006 | |
| |
| Net operating loss carryforwards | | $ | 4,854 | | | $ | 2,450 | |
Capitalizedstart-up costs | | | 196 | | | | 196 | |
| Stock-based compensation | | | 284 | | | | 209 | |
| Accrued expenses & depreciation | | | 14 | | | | 14 | |
| | | | | | | | | |
| | | | 5,348 | | | | 2,869 | |
| Valuation allowance | | | (5,348 | ) | | | (2,869 | ) |
| | | | | | | | | |
| | | $ | — | | | $ | — | |
| | | | | | | | | |
At December 31, 2007, the Company’s federal net operating loss carrryforwards were approximately $4,000,000, which expire in the years2026-2027. The Company’s state net operating loss carryforwards were approximately $7,200,000 which expire in the years2016-2017. At December 31, 2007, the Company’s Danish net operating loss carryforwards, which do not expire, were approximately 66,000,000 DKK, or US$12,900,000. The availability of the Company’s U.S and Danish net operating loss carryforwards may be subject to limitations based on ownership changes as defined in the respective tax codes, which could prevent the Company from realizing some or all of its net operating loss carryforwards.
A reconciliation of income taxes at the statutory federal income tax rate to income taxes included in the consolidated statements of operations is as follows:
| | | | | | | | | | | | | |
| | | For the Years Ended December 31 | |
| | | 2007 | | | 2006 | | | 2005 | |
| |
| United States federal tax rate | | | 35 | % | | | 35 | % | | | 35 | % |
| State taxes, net of federal benefit | | | 6 | | | | 6 | | | | 6 | |
| Difference in tax rate for foreign earnings | | | (9 | ) | | | (5 | ) | | | (6 | ) |
| Non-deductible items | | | 1 | | | | 1 | | | | 1 | |
| Effect of graduated tax rates & other | | | (2 | ) | | | (1 | ) | | | (9 | ) |
| Change in valuation allowance | | | (31 | ) | | | (36 | ) | | | (27 | ) |
| | | | | | | | | | | | | |
F-17
Osteologix, Inc.
(a development stage company)
Notes to Consolidated Financial Statements — (Continued)
| |
| 9. | Related Party Transactions |
In November 2005, Osteologix entered into a patent license agreement with Aditech AB (“Aditech”), a Swedish company. At the time of the agreement both Osteologix and Aditech were 100% owned by Nordic Biotech K/S. The agreement provided Aditech with an exclusive worldwide license to the Company’s patents containing certain compounds other than strontium compounds. In return, Aditech paid the Company $750,000 and agreed to pay the Company a 2.5% royalty on future net sales of products Aditech develops under the agreement. Also in the agreement, Aditech granted Osteologix an exclusive worldwide license to Aditech’s patents for strontium compounds. Under the terms of the agreement Aditech is entitled to a 1.5% royalty on all future net sales of products containing strontium compounds which are developed by Osteologix.
During 2007, the Company issued 143,000 shares of common stock for services received from related parties. In all cases, the stock was valued at the closing price on the date immediately prior to the date of issuance, at prices ranging from $0.97 per share to $1.44 per share. The stock was issued to related parties as follows:
| | | | | | | | | |
| | | Number of
| | | | |
| | | Shares | | | Value | |
| |
| Stock issued to members of the board of directors for service on the board instead of cash payments (at election of each board member) | | | 115 | | | $ | 130 | |
| Stock issued to former Chief Operating Officer for consulting services following term of employment | | | 28 | | | | 40 | |
| | | | | | | | | |
| | | | 143 | | | $ | 170 | |
| | | | | | | | | |
F-18
| | | | | | | | | |
| | | March 31,
| | | December 31,
| |
| | | 2008 | | | 2007 | |
| | | (Unaudited) | | | | |
| | | (In thousands, except per share amounts) | |
| |
ASSETS |
| Current assets: | | | | | | | | |
| Cash and cash equivalents | | $ | 2,237 | | | $ | 2,872 | |
| Short-term investments | | | — | | | | 1,273 | |
| Prepaid expenses and other assets | | | 157 | | | | 191 | |
| | | | | | | | | |
| Total current assets | | | 2,394 | | | | 4,336 | |
| Equipment, net | | | 9 | | | | 9 | |
| | | | | | | | | |
| Total assets | | $ | 2,403 | | | $ | 4,345 | |
| | | | | | | | | |
| |
LIABILITIES AND STOCKHOLDERS’ EQUITY |
| Current liabilities: | | | | | | | | |
| Accounts payable | | $ | 749 | | | $ | 1,059 | |
| Accrued liabilities | | | 169 | | | | 278 | |
| | | | | | | | | |
| Total current liabilities | | | 918 | | | | 1,337 | |
| | | | | | | | | |
| Commitments and Contingencies | | | | | | | | |
| Stockholders’ Equity Preferred stock, $0.0001 par value, 1,000 shares authorized, none issued or outstanding | | | — | | | | — | |
| Common stock, $0.0001 par value, 100,000 shares authorized, 25,012 and 24,989 shares issued and outstanding at March 31, 2008 and December 31, 2007, respectively | | | 3 | | | | 2 | |
Additionalpaid-in-capital | | | 21,025 | | | | 20,946 | |
| Accumulated other comprehensive loss | | | (140 | ) | | | (132 | ) |
| Deficit accumulated during development stage | | | (19,403 | ) | | | (17,808 | ) |
| | | | | | | | | |
| Total stockholders’ equity | | | 1,485 | | | | 3,008 | |
| | | | | | | | | |
| Total liabilities and stockholders’ equity | | $ | 2,403 | | | $ | 4,345 | |
| | | | | | | | | |
See accompanying notes to the condensed consolidated financial statements.
F-19
| | | | | | | | | | | | | |
| | | | | | | | | For the Period
| |
| | | | | | | | | from June 16,
| |
| | | For the Three Months Ended March 31, | | | 2003 (Inception) to
| |
| | | 2008 | | | 2007 | | | March 31, 2008 | |
| | | (In thousands, except per share amounts)
| |
| | | (Unaudited) | |
| |
| Revenues | | $ | — | | | $ | — | | | $ | 750 | |
| | | | | | | | | | | | | |
| Operating expenses: | | | | | | | | | | | | |
| Research and development | | | 672 | | | | 1,194 | | | | 10,350 | |
| General and administrative | | | 951 | | | | 849 | | | | 10,393 | |
| | | | | | | | | | | | | |
| Total operating expenses | | | 1,623 | | | | 2,043 | | | | 20,743 | |
| | | | | | | | | | | | | |
| Loss from operations | | | (1,623 | ) | | | (2,043 | ) | | | (19,993 | ) |
| Interest income, net | | | 28 | | | | 74 | | | | 590 | |
| | | | | | | | | | | | | |
| Net loss | | $ | (1,595 | ) | | $ | (1,969 | ) | | $ | (19,403 | ) |
| | | | | | | | | | | | | |
| Net loss per share, basic and diluted | | $ | (0.06 | ) | | $ | (0.09 | ) | | | | |
| | | | | | | | | | | | | |
| Weighted average number of shares outstanding basic and diluted | | | 25,011 | | | | 21,051 | | | | | |
| | | | | | | | | | | | | |
See accompanying notes to the condensed consolidated financial statements.
F-20
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | Deficit
| | | | |
| | | | | | | | | | | | Accumulated
| | | Accumulated
| | | | |
| | | | | | | | | Additional
| | | Other
| | | During the
| | | Total
| |
| | | Common Stock | | | Paid-In
| | | Comprehensive
| | | Development
| | | Stockholders’
| |
| | | Shares | | | Amount | | | Capital | | | Loss | | | Stage | | | Equity | |
| | | (In thousands, except per share amounts) | |
| |
| Issuance of common stock to founders for cash of $0.02 per share in June 2003 | | | 4,897 | | | $ | 1 | | | $ | 84 | | | $ | — | | | $ | — | | | $ | 85 | |
| Issuance of common stock for cash of $0.95 per share in October 2003 | | | 979 | | | | — | | | | 930 | | | | — | | | | — | | | | 930 | |
| Issuance of common stock for cash of $1.01 per share in May 2004 | | | 979 | | | | — | | | | 988 | | | | — | | | | — | | | | 988 | |
| Issuance of common stock for cash of $0.83 per share in January 2005, net of issuance costs | | | 1,430 | | | | — | | | | 1,158 | | | | — | | | | — | | | | 1,158 | |
| Issuance of common stock for cash of $1.24 per share in June 2005, net of issuance costs | | | 2,220 | | | | — | | | | 2,699 | | | | — | | | | — | | | | 2,699 | |
| Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss for the period | | | — | | | | — | | | | — | | | | — | | | | (4,913 | ) | | | (4,913 | ) |
| Foreign currency translations | | | — | | | | — | | | | — | | | | (4 | ) | | | — | | | | (4 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (4,917 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2005 | | | 10,505 | | | | 1 | | | | 5,859 | | | | (4 | ) | | | (4,913 | ) | | | 943 | |
| Common stock transferred in merger with Castle & Morgan Holdings, Inc. | | | 2,860 | | | | — | | | | — | | | | — | | | | — | | | | — | |
| Issuance of common stock for cash of $1.31 per share in private placement in May 2006, net of issuance costs | | | 7,656 | | | | 1 | | | | 9,255 | | | | — | | | | — | | | | 9,256 | |
| Stock based compensation expense | | | — | | | | — | | | | 508 | | | | — | | | | — | | | | 508 | |
| Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss for the period | | | — | | | | — | | | | — | | | | — | | | | (4,858 | ) | | | (4,858 | ) |
| Unrealized gain on short-term investments | | | — | | | | — | | | | — | | | | 2 | | | | — | | | | 2 | |
| Foreign currency translations | | | — | | | | — | | | | — | | | | (54 | ) | | | — | | | | (54 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (4,910 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2006 | | | 21,021 | | | | 2 | | | | 15,622 | | | | (56 | ) | | | (9,771 | ) | | | 5,797 | |
| Issuance of common stock and warrants for cash of $1.32 per share in private placement in June 2007, net of issuance costs | | | 3,825 | | | | — | | | | 4,970 | | | | — | | | | — | | | | 4,970 | |
| Stock issued in lieu of cash for services | | | 143 | | | | — | | | | 170 | | | | — | | | | — | | | | 170 | |
| Stock based compensation expense | | | — | | | | — | | | | 184 | | | | — | | | | — | | | | 184 | |
| Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss for the period | | | — | | | | — | | | | — | | | | — | | | | (8,037 | ) | | | (8,037 | ) |
| Unrealized loss on short-term investments | | | — | | | | — | | | | — | | | | (2 | ) | | | — | | | | (2 | ) |
| Foreign currency translations | | | — | | | | — | | | | — | | | | (74 | ) | | | — | | | | (74 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (8,113 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2007 | | | 24,989 | | | $ | 2 | | | $ | 20,946 | | | $ | (132 | ) | | $ | (17,808 | ) | | $ | 3,008 | |
| Stock issued in lieu of cash for services | | | 23 | | | | 1 | | | | 30 | | | | — | | | | — | | | | 31 | |
| Stock based compensation expense | | | — | | | | — | | | | 49 | | | | — | | | | — | | | | 49 | |
| Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | |
| Net loss for the period | | | — | | | | — | | | | — | | | | — | | | | (1,595 | ) | | | (1,595 | ) |
| Foreign currency translations | | | — | | | | — | | | | — | | | | (8 | ) | | | — | | | | (8 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Total comprehensive loss | | | | | | | | | | | | | | | | | | | | | | | (1,603 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | |
Balance at March 31, 2008 | | | 25,012 | | | $ | 3 | | | $ | 21,025 | | | $ | (140 | ) | | $ | (19,403 | ) | | $ | 1,485 | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
See accompanying notes to the condensed consolidated financial statements.
F-21
| | | | | | | | | | | | | |
| | | | | | | | | For the Period
| |
| | | | | | | | | from June 16,
| |
| | | | | | | | | 2003
| |
| | | For the Three Months Ended March 31, | | | (Inception) to
| |
| | | 2008 | | | 2007 | | | March 31, 2008 | |
| | | (In thousands, except per share amounts)
| |
| | | (Unaudited) | |
| |
Operating activities | | | | | | | | | | | | |
| Net loss | | $ | (1,595 | ) | | $ | (1,969 | ) | | $ | (19,403 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
| Depreciation | | | — | | | | 1 | | | | 19 | |
| Loss on disposal of equipment | | | — | | | | — | | | | 2 | |
| Issuance of stock in lieu of cash for services | | | 31 | | | | 38 | | | | 201 | |
| Stock based compensation | | | 49 | | | | 70 | | | | 741 | |
| Changes in operating assets and liabilities: | | | | | | | | | | | | |
| Prepaid expenses and other current assets | | | 34 | | | | 136 | | | | (157 | ) |
| Accounts payable | | | (310 | ) | | | 473 | | | | 749 | |
| Accrued liabilities | | | (109 | ) | | | 74 | | | | 169 | |
| | | | | | | | | | | | | |
| Net cash used in operating activities | | | (1,900 | ) | | | (1,177 | ) | | | (17,679 | ) |
| | | | | | | | | | | | | |
Investing activities | | | | | | | | | | | | |
| Purchase of short-term investments | | | — | | | | (775 | ) | | | (13,420 | ) |
| Sales and maturities of short-term investments | | | 1,273 | | | | 2,378 | | | | 13,420 | |
| Purchase of equipment | | | — | | | | — | | | | (30 | ) |
| | | | | | | | | | | | | |
| Net cash provided by (used in) investing activities | | | 1,273 | | | | 1,603 | | | | (30 | ) |
| | | | | | | | | | | | | |
Financing activities — | | | | | | | | | | | | |
| Net proceeds from issuance of common stock | | | — | | | | — | | | | 20,086 | |
| | | | | | | | | | | | | |
| Effect of exchange rates on cash and cash equivalents | | | (8 | ) | | | (9 | ) | | | (140 | ) |
| | | | | | | | | | | | | |
| Net increase (decrease) in cash and cash equivalents | | | (635 | ) | | | 417 | | | | 2,237 | |
| Cash and cash equivalents at beginning of period | | | 2,872 | | | | 481 | | | | — | |
| | | | | | | | | | | | | |
| Cash and cash equivalents at end of period | | $ | 2,237 | | | $ | 898 | | | $ | 2,237 | |
| | | | | | | | | | | | | |
See accompanying notes to the condensed consolidated financial statements.
F-22
| |
| 1. | Basis of Presentation and Summary of Significant Accounting Policies |
Business Description
Osteologix, Inc. (“Osteologix” or the “Company”) is in the business of developing pharmaceuticals for the treatment and prevention of diseases of bone and joint tissues. The Company’s lead product candidate, NB S101, is in clinical development for the treatment of osteoporosis. Osteologix has not yet generated substantial revenues from its operations and, accordingly, the Company is in the development stage.
Basis of Presentation
The unaudited condensed consolidated financial statements include the accounts of the Company and its wholly owned Danish subsidiary, Osteologix ApS. All intercompany accounts and transactions have been eliminated. Osteologix operates in one business segment, the development of pharmaceutical products.
The preparation of financial statements in conformity with U.S. generally accepted accounting principles (“GAAP”) requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Actual amounts may differ from those estimates. These condensed consolidated financial statements have been prepared in accordance with GAAP for interim financial information and with the instructions toForm 10-Q and Article 10 ofRegulation S-X. Accordingly, they do not include all of the information and footnotes required for complete financial statements. In the opinion of management, all adjustments (consisting of normal recurring adjustments) considered necessary for a fair presentation have been included. Operating results for the three month period ended March 31, 2008, are not necessarily indicative of results that may be expected for the year ending December 31, 2008. The condensed consolidated balance sheet information as of December 31, 2007 was derived from the audited consolidated financial statements included in the Company’s Annual Report onForm 10-K. These interim financial statements should be read in conjunction with that report.
The audited consolidated financial statements as of December 31, 2007 included a “going concern” audit opinion from the Company’s independent registered public accounting firm because Osteologix has experienced net losses since its inception and the year-end cash and short-term investments balance was insufficient to fund the Company’s operations through the end of 2008. Osteologix has raised additional capital since these audited financial statements were issued (see Note 6) and management believes its capital is sufficient to fund planned operations until approximately mid-2009. Therefore, the condensed consolidated financial statements do not contain any adjustments that may be required if Osteologix is unable to continue as a going concern.
The Company has been in the development stage since its formation. All losses accumulated since the inception of Osteologix have been considered as part of the Company’s development stage activities.
Fair Value of Financial Instruments
For financial instruments consisting of cash and cash equivalents, short-term investments, prepaid expenses and other assets, accounts payable and accrued liabilities included in the condensed consolidated financial statements, the carrying amounts are reasonable estimates of the fair value due to their short maturities. The fair value of other short-term and long-term obligations is estimated based on current interest rates available for debt instruments with similar terms, degrees of risk and remaining maturities. The carrying values of these obligations approximate their fair values.
| |
| 2. | Cash Equivalents and Short-Term Investments |
The Company considers all highly liquid investments with a maturity period of three months or less at the time of acquisition to be cash equivalents.
The Company generally invests funds that are not required for immediate operating needs in a diversified portfolio of debt securities. Based on the term and liquidity of the investments, these securities are classified as
F-23
Osteologix, Inc.
(a development stage company)
Notes to Condensed Consolidated Financial Statements — (Continued)
either cash equivalents or short-term investments on the condensed consolidated balance sheets. Management determines the appropriate classification of these marketable securities at the time of purchase and reevaluates such designation as of each balance sheet date. As of March 31, 2008, all of the Company’s debt securities were considered cash equivalents. The Company does not hold any auction rate securities.
| |
| 3. | Stock-Based Compensation |
The Company uses the Black-Scholes option-pricing model as its method for valuing stock-based compensation, which consists of options and warrants to purchase common stock at or above its fair value at the time of grant. During the three month period ended March 31, 2008, the Company granted to its directors options to purchase a total of 100,000 shares of common stock. These options have an exercise price of $1.45 per share, vest over four years, and expire 10 years from the date of grant; the Company has estimated the fair value of these options at $0.93 per share using a volatility factor of 70%, expected life of 6.1 years, risk-free interest rate of 3.3% and no payment of dividends. The Company did not grant any options to purchase common stock during the three months ended March 31, 2007. Total expense for stock-based compensation was $49,000 and $70,000 for the three months ended March 31, 2008 and 2007, respectively. As of March 31, 2008, the total unrecognized expense for unvested stock warrants and options was $965,000, which will be expensed over the remaining vesting period of 3.8 years. The aggregate intrinsic value of the options and warrants outstanding and exercisable as of March 31, 2008 was $153,000 based on a closing stock price of $1.20.
On January 2, 2008, the Company issued 22,443 shares of its common stock to members of its board of directors for their services on the board during the last three months of 2007. The common stock was issued in place of cash payments, and was valued at $1.37 per share, the closing price on December 31, 2007.
The net loss per share has been computed using the weighted-average number common shares outstanding during the period. During the three months ended March 31, 2008 and 2007, potentially dilutive options and warrants to purchase common stock aggregating 4,678,000 and 2,129,000 shares, respectively, were outstanding and not considered because their effect would have been antidilutive.
| |
| 5. | Recently Adopted Accounting Pronouncement |
In September 2006, the Financial Accounting Standards Board (“FASB”) issued SFAS No. 157, “Fair Value Measurements” (“SFAS 157”), which defines fair value, establishes a framework for measuring fair value in accordance with generally accepted accounting principles and expands disclosures about fair value measurements. SFAS No. 157 became effective for the Company on January 1, 2008, and accordingly Osteologix has adopted this pronouncement. In February 2008, the FASB issued Staff PositionNo. 157-2(“FSP 157-2”) which delays the effective date of SFAS 157 one year for all nonfinancial assets and nonfinancial liabilities, except those recognized or disclosed at fair value in the financial statements on a recurring basis.
The assets and liabilities measured at fair value under SFAS 157 in the first quarter of 2008 did not have a material impact on our condensed consolidated financial statements. In accordance withFSP 157-2, the Company will evaluate the remaining assets and liabilities no later than the first quarter of 2009.
On April 17, 2008, the Company closed a private placement transaction under which Osteologix issued 4,030,000 shares of common stock and warrants to purchase 2,030,000 shares of common stock for gross proceeds of $5,320,000. The Company has agreed to file a registration statement covering the resale of the shares.
F-24
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
| |
| Item 13. | Other Expenses of Issuance and Distribution |
The estimated expenses payable by the Registrant in connection with the issuance and distribution of the securities being registered are as follows:
| | | | | |
| SEC Registration Fee | | $ | 60 | |
| Printing Expenses | | $ | 5,000 | |
| Legal Fees and Expenses | | $ | 35,000 | |
| Accounting Fees and Expenses | | $ | 10,000 | |
| | | | | |
| Total | | $ | 50,060 | |
| | | | | |
| |
| Item 14. | Indemnification of Directors and Officers |
Our certificate of incorporation provides that to the fullest extent permitted by the Delaware General Corporation Law, directors of the registrant shall not be liable to it or its stockholders for monetary damages for breach of fiduciary duty as a director. The Company is also subject to Section 145 of the Delaware General Corporation Law, set forth below.
“Section 145. Indemnification of officers, directors, employees and agents; insurance.
“(a) A corporation shall have power to indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of the corporation) by reason of the fact that the person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by the person in connection with such action, suit or proceeding if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe the person’s conduct was unlawful. The termination of any action, suit or proceeding by judgment, order, settlement, conviction, or upon a plea of nolo contendere or its equivalent, shall not, of itself, create a presumption that the person did not act in good faith and in a manner which the person reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had reasonable cause to believe that the person’s conduct was unlawful.
“(b) A corporation shall have power to indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the corporation to procure a judgment in its favor by reason of the fact that the person is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against expenses (including attorneys’ fees) actually and reasonably incurred by the person in connection with the defense or settlement of such action or suit if the person acted in good faith and in a manner the person reasonably believed to be in or not opposed to the best interests of the corporation and except that no indemnification shall be made in respect of any claim, issue or matter as to which such person shall have been adjudged to be liable to the corporation unless and only to the extent that the Court of Chancery or the court in which such action or suit was brought shall determine upon application that, despite the adjudication of liability but in view of all the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which the Court of Chancery or such other court shall deem proper.
“(c) To the extent that a present or former director or officer of a corporation has been successful on the merits or otherwise in defense of any action, suit or proceeding referred to in subsections (a) and (b) of this section, or in
II-1
defense of any claim, issue or matter therein, such person shall be indemnified against expenses (including attorneys’ fees) actually and reasonably incurred by such person in connection therewith.
“(d) Any indemnification under subsections (a) and (b) of this section (unless ordered by a court) shall be made by the corporation only as authorized in the specific case upon a determination that indemnification of the present or former director, officer, employee or agent is proper in the circumstances because the person has met the applicable standard of conduct set forth in subsections (a) and (b) of this section. Such determination shall be made, with respect to a person who is a director or officer at the time of such determination, (1) by a majority vote of the directors who are not parties to such action, suit or proceeding, even though less than a quorum, or (2) by a committee of such directors designated by majority vote of such directors, even though less than a quorum, or (3) if there are no such directors, or if such directors so direct, by independent legal counsel in a written opinion, or (4) by the stockholders.
“(e) Expenses (including attorneys’ fees) incurred by an officer or director in defending any civil, criminal, administrative or investigative action, suit or proceeding may be paid by the corporation in advance of the final disposition of such action, suit or proceeding upon receipt of an undertaking by or on behalf of such director or officer to repay such amount if it shall ultimately be determined that such person is not entitled to be indemnified by the corporation as authorized in this section. Such expenses (including attorneys’ fees) incurred by former directors and officers or other employees and agents may be so paid upon such terms and conditions, if any, as the corporation deems appropriate.
“(f) The indemnification and advancement of expenses provided by, or granted pursuant to, the other subsections of this section shall not be deemed exclusive of any other rights to which those seeking indemnification or advancement of expenses may be entitled under any bylaw, agreement, vote of stockholders or disinterested directors or otherwise, both as to action in such person’s official capacity and as to action in another capacity while holding such office.
“(g) A corporation shall have power to purchase and maintain insurance on behalf of any person who is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise against any liability asserted against such person and incurred by such person in any such capacity, or arising out of such person’s status as such, whether or not the corporation would have the power to indemnify such person against such liability under this section.
“(h) For purposes of this section, references to (the corporation) shall include, in addition to the resulting corporation, any constituent corporation (including any constituent of a constituent) absorbed in a consolidation or merger which, if its separate existence had continued, would have had power and authority to indemnify its directors, officers, and employees or agents, so that any person who is or was a director, officer, employee or agent of such constituent corporation, or is or was serving at the request of such constituent corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, shall stand in the same position under this section with respect to the resulting or surviving corporation as such person would have with respect to such constituent corporation if its separate existence had continued.
“(i) For purposes of this section, references to (other enterprises) shall include employee benefit plans; references to “fines” shall include any excise taxes assessed on a person with respect to any employee benefit plan; and references to “serving at the request of the corporation” shall include any service as a director, officer, employee or agent of the corporation which imposes duties on, or involves services by, such director, officer, employee or agent with respect to an employee benefit plan, its participants or beneficiaries; and a person who acted in good faith and in a manner such person reasonably believed to be in the interest of the participants and beneficiaries of an employee benefit plan shall be deemed to have acted in a manner “not opposed to the best interests of the corporation” as referred to in this section.
“(j) The indemnification and advancement of expenses provided by, or granted pursuant to, this section shall, unless otherwise provided when authorized or ratified, continue as to a person who has ceased to be a director, officer, employee or agent and shall inure to the benefit of the heirs, executors and administrators of such a person.
II-2
“(k) The Court of Chancery is hereby vested with exclusive jurisdiction to hear and determine all actions for advancement of expenses or indemnification brought under this section or under any bylaw, agreement, vote of stockholders or disinterested directors, or otherwise. The Court of Chancery may summarily determine a corporation’s obligation to advance expenses (including attorneys’ fees).”
| |
| Item 15. | Recent Sales of Unregistered Securities |
During the last three years, we have issued the following securities that were not registered under the Securities Act of 1933.
On April 17, 2008, we sold an aggregate of 4,030,304 shares of the Company’s common stock and 2,015,152 common stock purchase warrants with an exercise price of $1.32 per share for gross proceeds of $5,320,000 in a private placement exempt from registration pursuant to the exemption for transactions by an issuer not involving any public offering under Section 4(2) of the Securities Act of 1933, and Regulation D thereunder. The warrants expire on September 30, 2009. Punk Ziegel + Co., L.P., acted as placement agent in connection with this transaction. For its services, we issued to Punk Ziegel warrants to purchase 15,000 shares of common stock upon the same terms as the warrants issued to the investors.
On June 7, 2007, we sold an aggregate of 5,738,631 shares of our common stock to a group of accredited investors for an aggregate offering price of approximately $5,050,000 in a private placement exempt from registration pursuant to the exemption for transactions by an issuer not involving any public offering under Section 4(2) of the Securities Act of 1933, and Regulation D thereunder. Of the 5,738,631 shares of common stock sold, 1,912,877 are issuable upon the exercise of warrants to purchase common stock at an exercise price equal to $1.20 per share. The warrants expire on August 31, 2008.
On May 24, 2006, we sold an aggregate of 7,656,000 shares of our common stock to certain accredited investors for aggregate gross proceeds of $10,000,000 in a series of transactions exempt from registration either pursuant to the exemption for transactions by an issuer not involving any public offering under Section 4(2) of the Securities Act of 1933, and Regulation D thereunder, or the exemption for offers and sales outside the United States under Regulation S.
| |
| Item 16. | Exhibits and Financial Statement Schedules. |
(a) Exhibits. The following exhibits are included herein or incorporated herein by reference:
| | | | | |
Exhibit
| | |
Number | | Description |
| |
| | 2 | .1 | | Share and Warrant Exchange Agreement, dated as of May 24, 2006, by and among Castle & Morgan Holdings, Inc., various Warrantholders, Nordic Biotech K/S, Charles J. Casamento as attorney-in-fact for various investors, and Osteologix A/S (incorporated herein by reference to Exhibit 2.1 to the Registrant’s Current Report onForm 8-K filed on May 31, 2006 (the “May 31, 2006Form 8-K”)). |
| | 3 | .1 | | Articles of Incorporation, as amended (incorporated herein by reference to Exhibit 3.1 to the Registrant’s Annual Report onForm 10-KSB filed on March 27, 2007 (the “2007Form 10-K”)). |
| | 3 | .2 | | Amended and Restated By-Laws of Osteologix, Inc. (incorporated herein by reference to Exhibit 3.1 to the Registrant’s Quarterly Report onForm 10-QSB filed for the quarter ended September 30, 2006). |
| | 5 | .1 | | Opinion of Morrison & Foerster LLP. |
| | 10 | .1 | | Subscription Agreement, dated as of May 24, 2006 (incorporated herein by reference to Exhibit 10.2 to the May 31, 2006Form 8-K). |
| | 10 | .2 | | Registration Rights Agreement, dated as of May 24, 2006 (incorporated herein by reference to Exhibit 10.3 to the May 31, 2006Form 8-K). |
| | 10 | .3 | | Securities Purchase Agreement dated as of March 27, 2008, by and among Osteologix, Inc., and the purchasers named on the signature page thereto (incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K filed on April 2, 2008 (the “April 2, 2008Form 8-K”)). |
| | 10 | .4 | | Registration Rights Agreement entered into on March 27, 2008 by and among Osteologix, Inc., and the Holders named therein (incorporated herein by reference to Exhibit 10.2 to the April 2, 2008Form 8-K). |
II-3
| | | | | |
Exhibit
| | |
Number | | Description |
| |
| | 10 | .5 | | Form of Warrant issued in connection with the Securities Purchase Agreement listed as Exhibit 4.2 above (incorporated herein by reference to Exhibit 10.3 to the April 2, 2008Form 8-K). |
| | 10 | .6 | | Patent License Agreement between Osteologix A/S and Aditech Pharma AB dated November 30, 2005 (incorporated herein by reference to Exhibit 10.3 to the 2007Form 10-K). |
| | 10 | .7 | | Osteologix, Inc. Equity Incentive Plan (incorporated herein by reference to Exhibit 10.5 to the 2007Form 10-K).* |
| | 10 | .8 | | Employment Agreement between Osteologix, Inc. and Philip J. Young dated as of April 3, 2007 (incorporated herein by reference to Exhibit 10.2 to the Registrant’s Current Report onForm 8-K filed on April 3, 2007 (the “April 3, 2007Form 8-K”)).* |
| | 10 | .9 | | Amendment to Employment Agreement between Osteologix, Inc. and Philip J. Young dated as of December 12, 2007 (incorporated herein by reference to Exhibit 10.6 to the Registrant’s Annual Report onForm 10-K filed on March 25, 2008 (the “2008Form 10-K”)).* |
| | 10 | .10 | | Amendment to Employment Agreement between Osteologix, Inc. and Philip J. Young dated as of June 5, 2008 (incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K filed on June 11, 2008).* |
| | 10 | .11 | | Securities Purchase Agreement, dated as of June 4, 2007, by and between the Registrant and the subscribers identified on the signature pages thereto (incorporated herein by reference to Exhibit 10.1 to the June 7, 2007Form 8-K). |
| | 10 | .12 | | Registration Rights Agreement, dated as of June 4, 2007, by and between the Registrant and the subscribers identified on the signature pages thereto (incorporated herein by reference to Exhibit 10.2 to the June 7, 2007Form 8-K). |
| | 10 | .13 | | Letter Agreement, dated as of April 15, 2008, by and between the Registrant and Matthew M. Loar, the Registrant’s former Chief Financial Officer (incorporated herein by reference to Exhibit 10.1 to the Registrant’s Current Report onForm 8-K filed on April 18, 2008).* |
| | 21 | .1 | | Subsidiary List (incorporated herein by reference to Exhibit 21.1 to the 2008Form 10-K). |
| | 23 | .1 | | Consent of Independent Registered Public Accounting Firm. |
| | 23 | .2 | | Consent of Morrison & Foerster LLP (included in Exhibit 5.1). |
| | |
| * | | management contract or compensatory plan or arrangement |
(b) Financial Statement Schedules. All financial statements supporting schedules are omitted because the information is inapplicable or presented in the Notes to Consolidated Financial Statements.
The undersigned Company hereby undertakes to:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this registration statement:
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the SEC pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20 percent change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement, and
II-4
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the SEC such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
II-5
SIGNATURES
In accordance with the requirements of the Exchange Act, the registrant caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
OSTEOLOGIX, INC.
Principal Executive Officer:
Name: Philip J. Young
| | |
| | Title: | President and Chief Executive Officer |
Date: July 10, 2008
Principal Financial and Accounting Officer:
Name: Matthew M. Loar
| | |
| | Title: | Principal Financial and Accounting Officer |
Date: July 10, 2008
POWER OF ATTORNEY
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Philip J. Young as his true and lawful attorney-in-fact, with full power of substitution and resubstitution for him and in his name, place and stead, in any and all capacities to sign any and all amendments to this registration statement, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, hereby ratifying and confirming all that said attorney-in-fact or his substitute may lawfully do or cause to be done by virtue thereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | | | | |
Name | | Title | | Date |
| |
| | | | | |
/s/ Klaus Eldrup-Jørgensen
Klaus Eldrup-Jørgensen | | Chairman of the Board of Directors | | July 10, 2008 |
| | | | | |
/s/ Philip J. Young
Philip J. Young | | President, Chief Executive Officer and Director | | July 10, 2008 |
| | | | | |
/s/ Matthew M. Loar
Matthew M. Loar | | Principal Financial and Accounting Officer | | July 10, 2008 |
II-6
| | | | | | | |
Name | | Title | | Date |
| |
| | | | | |
/s/ Jeremy Curnock Cook
Jeremy Curnock Cook | | Director | | July 10, 2008 |
| | | | | |
/s/ Christian Hansen
Christian Hansen | | Director | | July 10, 2008 |
| | | | | |
/s/ Bobby W. Sandage, Jr.
Bobby W. Sandage, Jr. | | Director | | July 10, 2008 |
| | | | | |
/s/ Florian Schönharting
Florian Schönharting | | Director | | July 10, 2008 |
| | | | | |
/s/ Christopher B. Wood
Christopher B. Wood | | Director | | July 10, 2008 |
II-7