| Item 2. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
You should read the following discussion and analysis of our financial condition and results of operations together with the consolidated financial statements and related notes that are included elsewhere in this Quarterly Report on Form 10-Q and our Annual Report on Form 10-K for the fiscal year ended December 31, 2020 filed with the U.S. Securities and Exchange Commission, or the SEC, on March 1, 2021 (“2020 Form 10-K”). This discussion contains forward-looking statements based upon current plans, expectations and beliefs that involve risks and uncertainties. Our actual results may differ materially from those anticipated in these forward-looking statements as a result of various factors, including, but not limited to, those discussed in the section entitled “Risk Factors” and elsewhere in this Quarterly Report on Form 10-Q. In preparing this MD&A, we presume that readers have access to and have read the MD&A in our 2020 Form 10-K, pursuant to Instruction 2 to paragraph of Item 303 of Regulation S-K. Unless stated otherwise, references in this Quarterly Report on Form 10-Q to “us,” “we,” “our,” or our “Company” and similar terms refer to Rocket Pharmaceuticals, Inc.
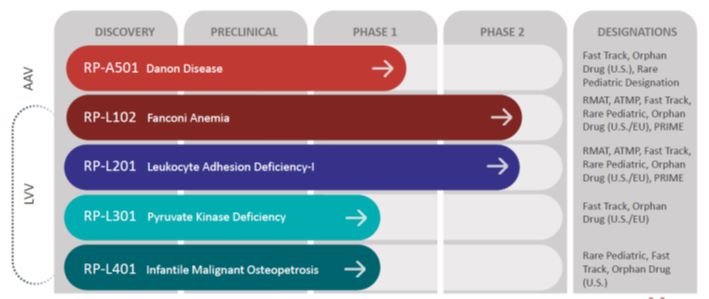
We are a clinical-stage, multi-platform biotechnology company focused on the development of first, only and best-in-class gene therapies, with direct on-target mechanism of action and clear clinical endpoints, for rare and devastating diseases. We have four clinical-stage ex vivo lentiviral vector (“LVV”). These include programs for Fanconi Anemia (“FA”), a genetic defect in the bone marrow that reduces production of blood cells or promotes the production of faulty blood cells, Leukocyte Adhesion Deficiency-I (“LAD-I”), a genetic disorder that causes the immune system to malfunction, Pyruvate Kinase Deficiency (“PKD”), a rare red blood cell autosomal recessive disorder that results in chronic non-spherocytic hemolytic anemia and Infantile Malignant Osteopetrosis (“IMO”), a genetic disorder characterized by increased bone density and bone mass secondary to impaired bone resorption. Of these, both the Phase 2 FA program and the Phase 1/2 LAD-I program are in registration-enabling studies in the United States (“U.S.”) and Europe (“EU”). In addition, in the U.S., we have a clinical stage in vivo adeno-associated virus (“AAV”) program for Danon disease, a multi-organ lysosomal-associated disorder leading to early death due to heart failure. Additional discovery efforts on a gene therapy program for the less common FA subtypes C and G is ongoing. We have global commercialization and development rights to all of these product candidates under royalty-bearing license agreements.
Recent Developments
On April 26, 2021, we redeemed in full our 2022 Convertible Notes prior to the redemption date. Holders of approximately $38.4 million remaining principal amount of the 2022 Convertible Notes converted such notes into approximately 1.3 million shares of the Company’s common stock and cash in lieu of fractional shares. On August 2, 2021, holders of $5.15 million of the 2021 Convertible Notes converted the $5.15 million remaining balance of the 2021 Convertible Notes into 160,614 shares. As of September 30, 2021, none of the 2021 Convertible Notes or 2022 Convertible Notes were outstanding.
On May 10, 2021, we announced that the RP-A501 Danon Disease program was placed on clinical hold by the FDA. No new drug-related safety events were observed in the low- or high-dose adult cohorts of the Phase 1 trial. On August 16, 2021, the FDA lifted the clinical hold on our Danon Disease program allowing patient enrollment in our Phase 1 clinical trial of RP-A501 for the treatment of Danon Disease to resume. The hold was removed after we addressed the FDA’s requests to modify the trial protocol and other supporting documents with revised guidelines for patient selection and management. We initiated steps to resume the program as soon as possible and commenced dosing in the low-dose (6.7e13 vg/kg) pediatric patient cohort in the third quarter of 2021. We anticipate reporting updated clinical data results in the fourth quarter of 2021.
On August 2, 2021, holders of the 2021 Convertible Notes converted the $5.15 million remaining balance of the 2021 Convertible Notes into 160,614 shares of common stock. In accordance with ASC 470-Debt, the conversion of the 2021 Convertible Notes was accounted for as a conversion since the 2021 Convertible Notes did not include a beneficial conversion feature and the carrying amount of the 2021 Convertible Notes, including any unamortized premium or discount, was credited to additional paid in capital upon conversion to reflect the common stock issued and no gain or loss was recognized. As of September 30, 2021, there were no 2021 Convertible Notes outstanding.
On August 27, 2021, we entered into a Securities Purchase Agreement (the “Purchase Agreement”) with a fund affiliated with RTW Investments, LP, our largest shareholder (the “Purchaser”), pursuant to which we agreed to sell and issue to the Purchaser, in a private placement (the “Private Placement”), 812,516 shares of our common stock at a purchase price of $32.48 per share for aggregate net proceeds of approximately $26.4 million before deducting estimated offering expenses payable. The Private Placement closed on August 31, 2021.
Gene Therapy Overview
Genes are composed of sequences of deoxyribonucleic acid (“DNA”), which code for proteins that perform a broad range of physiologic functions in all living organisms. Although genes are passed on from generation to generation, genetic changes, also known as mutations, can occur in this process. These changes can result in the lack of production of proteins or the production of altered proteins with reduced or abnormal function, which can in turn result in disease.
Gene therapy is a therapeutic approach in which an isolated gene sequence or segment of DNA is administered to a patient, most commonly for the purpose of treating a genetic disease that is caused by genetic mutations. Currently available therapies for many genetic diseases focus on administration of large proteins or enzymes and typically address only the symptoms of the disease. Gene therapy aims to address the disease-causing effects of absent or dysfunctional genes by delivering functional copies of the gene sequence directly into the patient’s cells, offering the potential for curing the genetic disease, rather than simply addressing symptoms.
We are using modified non-pathogenic viruses for the development of our gene therapy treatments. Viruses are particularly well suited as delivery vehicles because they are adept at penetrating cells and delivering genetic material inside a cell. In creating our viral delivery vehicles, the viral (pathogenic) genes are removed and are replaced with a functional form of the missing or mutant gene that is the cause of the patient’s genetic disease. The functional form of a missing or mutant gene is called a therapeutic gene, or the “transgene.” The process of inserting the transgene is called “transduction.” Once a virus is modified by replacement of the viral genes with a transgene, the modified virus is called a “viral vector.” The viral vector delivers the transgene into the targeted tissue or organ (such as the cells inside a patient’s bone marrow). We have two types of viral vectors in development, LVV and AAV. We believe that our LVV and AAV-based programs have the potential to offer a significant therapeutic benefit to patients that is durable (long-lasting).
The gene therapies can be delivered either (1) ex vivo (outside the body), in which case the patient’s cells are extracted and the vector is delivered to these cells in a controlled, safe laboratory setting, with the modified cells then being reinserted into the patient, or (2) in vivo (inside the body), in which case the vector is injected directly into the patient, either intravenously (“IV”) or directly into a specific tissue at a targeted site, with the aim of the vector delivering the transgene to the targeted cells.
We believe that scientific advances, clinical progress, and the greater regulatory acceptance of gene therapy have created a promising environment to advance gene therapy products as these products are being designed to restore cell function and improve clinical outcomes, which in many cases include prevention of death at an early age. The FDA approval of several gene therapies in recent years is supportive of a regulatory pathway forward for gene therapy products.
Pipeline Overview
The chart below shows the current phases of development of Rocket’s programs and product candidates:
AAV Program:
Danon Disease:
Danon disease is a multi-organ lysosomal-associated disorder leading to early death due to heart failure. Danon disease is caused by mutations in the gene encoding lysosome-associated membrane protein 2 (“LAMP-2”), a mediator of autophagy. This mutation results in the accumulation of autophagic vacuoles, predominantly in cardiac and skeletal muscle. Male patients often require heart transplantation and typically die in their teens or twenties from progressive heart failure. Along with severe cardiomyopathy, other Danon disease symptoms can include skeletal muscle weakness, liver disease, and intellectual impairment. There are no specific therapies available for the treatment of Danon disease. RP-A501 is in clinical trials as an in vivo therapy for Danon disease, which is estimated to have a prevalence of 15,000 to 30,000 patients in the U.S. and the EU.
Danon disease is an autosomal dominant, rare inherited disorder characterized by progressive cardiomyopathy which is almost universally fatal in males even in settings where cardiac transplantation is available. Danon disease predominantly affects males early in life and is characterized by absence of LAMP2B expression in the heart and other tissues. Pre-clinical models of Danon disease have demonstrated that AAV-mediated transduction of the heart results in reconstitution of LAMP2B expression and improvement in cardiac function.
We have treated five patients in the RP-A501 Phase 1 clinical trial, completed the first cohort of the study evaluating an initial low-dose in male patients aged 15 or greater and initiated dosing in the pediatric cohort at the low dose level (6.7e13 vg/kg). The preliminary data announced in December 2020 for the low dose cohort included safety and clinical activity results from the three patients treated with the low dose of 6.7×1013 genome copies (gc)/kilogram (kg) and early safety information from the two patients treated with the higher dose of 1.1×1014 gc/kg as of the cutoff date of November 2020.
In the three patients treated in the low dose cohort, RP-A501 showed manageable safety results. No unexpected and serious drug product-related adverse events or severe adverse events were observed in this low dose cohort. The most common adverse events were predominantly mild, not associated with clinical symptoms and were related to elevated transaminases post treatment. Elevation in transaminases was observed in all three low-dose patients and returned to baseline levels within the first one to two months post-treatment. There was also a transient and reversible decline in platelets observed in these three patients. These changes were largely responsive to corticosteroids and other immunosuppressive therapies. All patients were given oral steroids to prevent or minimize potential immune-related events.
At the higher dose administered (1.1×1014 gc/kg), additional immunosuppressive therapies were stipulated and administered to mitigate the immune response associated with RP-A501. One of the two treated patients, who received the higher absolute AAV9 dose and had some degree of pre-existing anti-AAV9 immunity, experienced a non-persistent, immune-related event that was classified as a drug product-related serious adverse event. This thrombotic microangiopathy (“TMA”) event (which was later reclassified as a Sudden Unexpected Serious Adverse Reaction (“SUSAR”)) was believed to be likely due to immune-mediated complement activation, resulting in reversible thrombocytopenia and acute kidney injury requiring eculizumab and transient hemodialysis. This patient regained normal kidney function within three weeks.
From the perspective of gene expression results, all three low dose patients demonstrated evidence of cardiac LAMP2B expression by Western blot and/or immunohistochemistry. In two of the three patients in the low dose cohort who had closely monitored compliance with the immunosuppressive regimen, high levels of cardiac LAMP2B expression were observed along with clinical biomarker improvements. In cardiac biopsies of the low dose patients, LAMP2B gene expression was observed in 67.8% of cells compared to normal as determined by immunohistochemistry at 12 months in one patient, and at 92.4% of cells compared to normal at month 9 in the other patient. In this latter patient, Western blot assessment showed 61% of normal LAMP2B protein expression at month 9. The 12-month Western blot data was still pending for all three patients as of the data cutoff.
At least two of the three low dose patients demonstrated key clinical biomarker improvements consistent with improved cardiac function. Notably, photographic evidence for all three patients showed improvements (i.e., decrease) in autophagic vacuoles, a hallmark of Danon disease pathology, as assessed by electron microscopy of cardiac tissue via endomyocardial biopsy. Additionally, two of the three low dose patients with closely monitored immunosuppressive regimen compliance demonstrated improvement in cardiac output and brain natriuretic peptide (“BNP”) and also upgraded from Class II to I in the New York Heart Association Functional Classification, which is the most commonly used heart failure classification system. Class II is where a patient exhibits a slight limitation of physical activity, are comfortable at rest, and ordinary physical activity results in fatigue, palpitation and/or dyspnea. Class I is where a patient exhibits no limitation of physical activity and ordinary physical activity does not cause undue fatigue, palpitation and/or dyspnea. In these two patients, we also observed substantial improvement of a key marker of heart failure, BNP, which decreased from a pretreatment baseline by 75 percent in one patient and 79 percent in the other as well as improvement in cardiac output by 35 percent in one patient and 62 percent in the other as measured by invasive hemodynamics. The third patient has demonstrated stabilization of NYHA class and BNP.
As disclosed in December 2020, one patient in the high dose cohort, who was the heaviest treated to date and had highly advanced disease developed complement-mediated TMA which resolved fully with transient hemodialysis (this event was later reclassified as a SUSAR). This patient continued to have progressive disease considered unrelated to gene therapy by the trial investigator as well as his transplant cardiologist and successfully received a heart transplant. The patient is currently doing well clinically and reports resolution of his baseline myopathy that was present prior to treatment. Analysis of the explanted heart demonstrated fibrosis that was consistent with end-stage Danon disease. Of note, this patient had advanced features of heart failure upon treatment; the updated trial protocol will exclude enrollment of Danon patients with end-stage disease. Data collection from this patient and the other high dose patient is ongoing with full presentation of the data from the high dose as well as low dose level expected to be presented in the fourth quarter.
Given the activity observed in the low dose cohort and to mitigate TMA and associated safety concerns observed in the high dose cohort, in agreement with FDA we will focus on the low dose moving forward (6.7e13) and will no longer administer the higher doses (1.1e14 or higher) in this trial. Additional safety measures have been implemented and are reflected in the updated trial protocol. The FDA lifted the clinical hold on August 16, 2021 and dosing of the pediatric cohort was initiated in September 2021.
In September 2021, previously disclosed data for the ongoing Phase 1 trial of RP-A501 was presented at the Heart Failure Society of America (“HFSA”) 2021 Annual Scientific Meeting. This academic presentation included data from the low dose (6.7e13) cohort which demonstrated the investigational gene therapy RP-A501 was well tolerated and showed progressive and durable clinical benefit. We expect to report updated data for the study in the fourth quarter of 2021 including: long-term clinical data for the low-dose cohort (6.7e13), safety and efficacy data for the two patients dosed at the high dose (1.1e14) and updates on the pediatric cohort.
Fanconi Anemia Complementation Group A (FANCA):
FA, a rare and life-threatening DNA-repair disorder, generally arises from a mutation in a single FA gene. An estimated 60 to 70% of cases arise from mutations in the Fanconi-A (“FANCA”) gene, which is the focus of our program. FA results in bone marrow failure, developmental abnormalities, myeloid leukemia, and other malignancies, often during the early years and decades of life. Bone marrow aplasia, which is bone marrow that no longer produces any or very few red and white blood cells and platelets leading to infections and bleeding, is the most frequent cause of early morbidity and mortality in FA, with a median onset before 10 years of age. Leukemia is the next most common cause of mortality, ultimately occurring in about 20% of patients later in life. Solid organ malignancies, such as head and neck cancers, can also occur, although at lower rates during the first two to three decades of life.
Although improvements in allogeneic (donor-mediated) hematopoietic stem cell transplant (“HSCT”), currently the most frequently utilized therapy for FA, have resulted in more frequent hematologic correction of the disorder, HSCT is associated with both acute and long-term risks, including transplant-related mortality, graft versus host disease (“GVHD”), a sometimes fatal side effect of allogeneic transplant characterized by painful ulcers in the GI tract, liver toxicity and skin rashes, as well as increased risk of subsequent cancers. Our gene therapy program in FA is designed to enable a minimally toxic hematologic correction using a patient’s own stem cells during the early years of life. We believe that the development of a broadly applicable autologous gene therapy can be transformative for these patients.
In December 2020, we presented updated interim data from our FA at the 62nd American Society of Hematology (“ASH”) Annual Meeting. The FA data presented at the ASH Annual Meeting were from seven of the nine patients treated as of October 2020 in both the U.S. Phase 1 and global Phase 2 studies of RP-L102 for FA. Patients in these studies received a single intravenous infusion of “Process B” RP-L102 which incorporates a modified stem cell enrichment process, transduction enhancers, as well as commercial-grade vector. Preliminary data from these studies support “Process B” as a consistent and reproducible improvement over “Process A” which was used in earlier academic FA studies.
Seven patients had follow-up data of at least two-months and three of the seven patients had been followed for twelve-months or longer. As patients are treated with gene therapy product without the use of a conditioning regimen, the data indicated that RP-L102 was generally well-tolerated with no significant safety issues reported with infusion or post-treatment. One drug related serious adverse event of Grade 2 transient infusion-related reaction was observed. In five out of the seven patients for whom there was follow-up data, evidence of preliminary engraftment was observed, with bone marrow (“BM”) vector copy numbers (“VCNs”) from 0.16 to 0.22 (long-term follow-up only) and peripheral VCNs ranging from 0.01 (2-month follow-up) to 0.11 (long-term follow-up). Further, two of the three patients with greater than 12-months follow-up showed evidence of increasing engraftment, mitomycin-C (“MMC”) resistance and stable blood counts, which suggests a halt in the progression of bone marrow failure. The third patient with greater than 12-month follow-up contracted Influenza B nine months post-treatment resulting in progressive BM failure, for which, such patient received a successful bone marrow transplant at 18 months post-treatment.
In May 2021, we presented positive clinical data at the 24th Annual Meeting of the American Society of Gene and Cell Therapy (“ASGCT”). The preliminary results from the Phase 1/2 trials presented in a poster at ASGCT are from nine pediatric patients and showed increasing evidence of engraftment in at least six of the nine patients, including two patients with at least 15-months of follow-up and four patients with at least 6-months of follow-up. RP-L102 demonstrated a highly favorable safety profile with all subjects being treated without conditioning and with no sign of dysplasia. One patient experienced a Grade 2 transient infusion-related reaction.
We expect to report longer-term follow up on these patients in the fourth quarter of 2021.
Leukocyte Adhesion Deficiency-I (LAD-I):
LAD-I is a rare autosomal recessive disorder of white blood cell adhesion and migration, resulting from mutations in the ITGB2 gene encoding for the Beta-2 Integrin component, CD18. Deficiencies in CD18 result in an impaired ability for neutrophils (a subset of infection-fighting white blood cells) to leave blood vessels and enter tissues where these cells are needed to combat infections. As is the case with many rare diseases, accurate estimates of incidence are difficult to confirm; however, several hundred cases have been reported to date.
Most LAD-I patients are believed to have the severe form of the disease. Severe LAD-I is notable for recurrent, life-threatening infections and substantial infant mortality in patients who do not receive an allogeneic HSCT. Mortality for severe LAD-I has been reported as 60 to 75% by age two in the absence of allogeneic HCST.
We currently have one ex-vivo program targeting LAD-I, RP-L201. RP-L201 is a clinical program that we in-licensed from CIEMAT. We have partnered with UCLA to lead U.S. clinical development efforts for the LAD-I program. UCLA and its Eli and Edythe Broad Center of Regenerative Medicine and Stem Cell Research is serving as the lead U.S. clinical research center for the registrational clinical trial for LAD-I, and HNJ and GOSH serving as the lead clinical sites in Spain and London, respectively. This study has received a $6.5 million CLIN2 grant award from the California Institute for Regenerative Medicine (“CIRM”) to support the clinical development of gene therapy for LAD-I.
The ongoing open-label, single-arm, Phase 1/2 registration enabling clinical trial of RP-L201 intended to assess the safety and tolerability of RP-L201 in severe LAD-I patients. The first patient was treated at UCLA with RP-L201 in the third quarter of 2019.
In April 2021, we presented positive clinical updates from RP-L201 at the Clinical Immunology Society (CIS) Annual Meeting. The Phase 1/2 data presented in a poster at CIS 2021 are from four pediatric patients with severe LAD-I. RP-L201 was well tolerated with no safety issues reported with treatment or post-treatment. All four patients achieved hematopoietic reconstitution within 5-weeks. The first patient with 18-months of follow up demonstrated durable CD18 expression of approximately 40%, peripheral blood vector copy number (VCN) levels of 1.2 at 12-months post-treatment and resolution of skin lesions with no new lesions. The second patient with 9-months of follow up demonstrated CD18 expression of approximately 28% and peripheral blood VCN levels of 0.75 at 6-months post-treatment with kinetics consistent with those of the first patient. The third and fourth patients demonstrated high CD18 expression of approximately 70% and approximately 51%, respectively at 3-months post treatment, and peripheral blood VCN kinetics consistent with those of the first two patients.
The LAD-I program received Regenerative Medicine Advanced Therapy designation from the FDA and Priority Medicines designation from the European Medicines Agency (“EMA”), completing the full complement of all U.S. and EU accelerated regulatory designations for the program.
In May 2021, we presented the Phase 1/2 clinical data at the 24th Annual Meeting of ASGCT in an oral presentation from four pediatric patients with severe LAD-I. RP-L201 that showed preliminary efficacy in all four patients, including one patient with 18-months of follow-up and one patient with 9-months of follow-up. All four patients demonstrated CD18 expression substantially exceeding the 4-10% threshold associated with survival into adulthood and consistent with the reversal of severe LAD-I phenotype. RP-L201 was well tolerated with no safety issues reported with treatment or post-treatment.
In October 2021, we presented phase 1/2 clinical data of the first seven of nine patients with at least 3 months of follow-up data at the 28th Annual Congress of ESGCT. The safety profile of RP-L201 appears favorable, with all infusions well tolerated and no drug product-related serious adverse events. Preliminary efficacy was evident in all seven patients, including two patients with at least 12 months of follow-up. All seven patients demonstrated durable neutrophil CD18 expression that exceeded the 4-10% threshold associated with survival into adulthood and consistent with reversal of the severe LAD-I phenotype. Peripheral blood vector copy number (VCN) levels have been stable and in the 0.5 – 2.5 copy per genome range. No patients have had LAD-I related infections requiring hospitalization subsequent to hematopoietic reconstitution post RP-L201. All 9 LAD-I patients have been treated and enrollment in the study is complete.
We expect to announce additional clinical data for our LAD-I program in the fourth quarter of 2021.
Pyruvate Kinase Deficiency (PKD):
Red blood cell PKD is a rare autosomal recessive disorder resulting from mutations in the pyruvate kinase L/R (“PKLR”) gene encoding for a component of the red blood cell (“RBC”) glycolytic pathway. PKD is characterized by chronic non-spherocytic hemolytic anemia, a disorder in which RBCs do not assume a normal spherical shape and are broken down, leading to decreased ability to carry oxygen to cells, with anemia severity that can range from mild (asymptomatic) to severe forms that may result in childhood mortality or a requirement for frequent, lifelong RBC transfusions. The pediatric population is the most commonly and severely affected subgroup of patients with PKD, and PKD often results in splenomegaly (abnormal enlargement of the spleen), jaundice and chronic iron overload which is likely the result of both chronic hemolysis and the RBC transfusions used to treat the disease. The variability in anemia severity is believed to arise in part from the large number of diverse mutations that may affect the PKLR gene. Estimates of disease incidence have ranged between 3.2 and 51 cases per million in the white U.S. and EU population. Industry estimates suggest at least 2,500 cases in the U.S. and EU have already been diagnosed despite the lack of FDA-approved molecularly targeted therapies. Market research indicates the application of gene therapy to broader populations could increase the market opportunity from approximately 250 to 500 patients per year.
We currently have one ex-vivo LVV-based program targeting PKD, RP-L301. RP-L301 is a clinical stage program that we in-licensed from CIEMAT. The IND for RP-L301 to initiate the global Phase 1 study cleared in October 2019. This program has been granted US and EMA orphan drug disease designation.
This global Phase 1 open-label, single-arm, clinical trial is expected to enroll six adult and pediatric PKD patients in the U.S. and Europe. The trial will be comprised of three cohorts to assess RP-L301 in young pediatric (age 8-11), older pediatric (age 12-17) and adult populations. The trial is designed to assess the safety, tolerability, and preliminary activity of RP-L301, and initial safety evaluation will occur in the adult cohort before evaluation in pediatric patients. Stanford will serve as the lead site in the U.S. for adult and pediatric patients, HNJ will serve as the lead site in Europe for pediatrics, and Hospital Universitario Fundación Jiménez Díaz will serve as the lead site in Europe for adult patients. In July 2020, we treated the first patient in our clinical trial of RP-L301.
The data presented at the 2020 ASH Annual Meeting were from two adult PKD patients with significant anemia and transfusion requirements. Patient L301-006-1001 was treated with RP-L301. Preliminary data from this first patient supported initial tolerability of RP-L301, hemoglobin improvement to a normal range at 3-months post treatment and additional normalization of hemolysis markers. The patient was 31-years of age at the time of enrollment and had been followed for 3-months post treatment.
Patient L301-006-1001 received a cell dose of 3.9x106 cells/kilogram (“kg”) with a drug product mean VCN of 2.73. For this patient, hematopoietic reconstitution was observed in less than two weeks. Furthermore, the patient attained peripheral blood VCN of 2.21 at 1-month and 1.55 at 3-months and normalized hemoglobin (“Hgb”) and hemolysis markers at 3-months post-treatment. In particular at baseline, the patient had Hb of approximately 7.4 grams (“g”)/deciliter (“dL”) to Hb of 14.3 g/dL at 3-months post treatment with RP-L301. In the two years prior to enrollment, the patient underwent approximately 14 transfusion episodes; subsequent to engraftment from RP-L301 treatment, the patient to date has not required any red blood cell transfusions. The patient also exhibited normalization of bilirubin, lactate dehydrogenase and erythropoietin levels at 3-months post treatment, each of which had been substantially elevated prior to study enrollment. The patient also had an increase in hepcidin and a decrease in reticulocytes at 3-months post treatment.
The data from Patient L301-006-1001 indicated that RP-L301 was generally well-tolerated and there were no serious safety issues or infusion-related complications observed 3-months post treatment. The patient experienced Grade 3 treatment-emergent adverse events of neutropenia, stomatitis, increased liver transaminase levels (AST and ALT) and a Grade 4 treatment-emergent adverse event of hypertriglyceridemia; the investigator did not consider these adverse events related to RP-L301.
Patient L301-006-1001 also experienced Grade 2 serious adverse events of chest pain, dyspnea, and nausea during the apheresis collection. The investigator considered these events related to apheresis, hyperleukocytosis and the mobilizing agents. They resolved with supportive care and without sequelae. Other events included Grade 2 bone pain and Grade 3 leukocytosis.
A second patient L301-006-1002, was 47 years old at the time of enrollment and had been recently treated with RP-L301, receiving a cell dose of 2.4x106 cells/kg with a mean drug product VCN of 2.08.
In March 2021, we announced updated positive preliminary clinical data from the two adult patients in the Phase 1 trial of RP-L301 for the treatment of PKD which showed sustained safety and tolerability 6- and 3-months after treatment, respectively. The two patients demonstrated durable normalization of hemoglobin levels of 13.9 g/dL at 6-months post treatment in the first patient and 13.8 g/dL at 3-months post treatment in the second patient. Both patients also demonstrated significant improvements in bilirubin 6- and 3-months after treatment, which had been substantially elevated prior to study enrollment.
In May 2021, we presented positive clinical data in an oral presentation at the 24th Annual Meeting of ASGCT. Updated preliminary Phase 1 data from the first two patients with significant anemia and transfusion requirements that showed sustained safety and tolerability. Preliminary efficacy, measured by peripheral blood VCN levels, was evident in both patients during the initial 9-months and 3-months post-treatment, respectively. The two patients continued to demonstrate durable normalization of hemoglobin levels of 13.1 g/dL at 9-months post treatment in the first patient and 14.4 g/dL at 6-months post treatment in the second patient.
The Phase 1 trial continues to enroll patients with longer-term data on track for the fourth quarter of 2021.
Infantile Malignant Osteopetrosis (IMO):
IMO is a genetic disorder characterized by increased bone density and bone mass secondary to impaired bone resorption. During normal growth and development small areas of bone are constantly being broken down by special cells called osteoclasts, then made again by cells called osteoblasts. In IMO, the cells that break down bone (osteoclasts) do not work properly, which leads to the bones becoming thicker and not as healthy. Untreated IMO patients may suffer from a compression of the bone-marrow space, which results in bone marrow failure, anemia, and increased infection risk due to the lack of production of white blood cells. Untreated IMO patients may also suffer from a compression of cranial nerves, which transmit signals between vital organs and the brain, resulting in blindness, hearing loss and other neurologic deficits.
IMO represents the autosomal recessive, severe variants of a group of disorders characterized by increased bone density and bone mass secondary to impaired bone resorption. IMO typically presents in the first year of life and is associated with severe bone and hematologic manifestations leading to death within the first decade of life in the absence of allogeneic HSCT, although HSCT results have been limited to-date and notable for frequent graft failure, GVHD and other severe complications including pneumonitis and pulmonary hypertension.
Approximately 50% of IMO results from mutations in the TCIRG1 gene, resulting in cellular defects that prevent osteoclast bone resorption. As a result of this defect, bone growth is markedly abnormal. It is estimated that IMO occurs in 1 out of 250,000-300,000 within the general global population, although incidence is higher in specific geographic regions including Costa Rica, parts of the Middle East, the Chuvash Republic of Russia, and the Vasterbotten Province of Northern Sweden.
We currently have one LVV-based program targeting IMO, RP-L401. RP-L401 is a preclinical program that we in-licensed from Lund University, Sweden. This program has been granted ODD and Rare Pediatric Disease designation from the FDA. We have partnered with UCLA to lead U.S. clinical development efforts for the IMO program and UCLA will serve as the lead U.S. clinical site for IMO. The IND for RP-L401 to initiate a global Phase 1 study was cleared by the FDA in June 2020. The non-randomized, open-label Phase 1 clinical trial will enroll two pediatric patients, one month of age or older. The trial is designed to assess safety and tolerability of RP-L401, as well as preliminary efficacy, including potential improvements in bone abnormalities/density, hematologic status, and endocrine abnormalities.
In October 2020, we presented pre-clinical data from our LVV-based program targeting IMO, RP-L401, at the ESID 2020 Meeting. Preclinical data on IMO indicate that a modest level of engraftment can correct the disease phenotype in vivo, with increased long-term survival, tooth eruption, weight gain and normalized bone resorption. A comprehensive review of pre-clinical gene therapy investigations in TCIRG1-mediated osteopetrosis published in December 2020 supports acceleration into clinical development for RP-L401.
A clinical trial for RP-L401 was initiated in the fourth quarter of 2020.
The first patient treated with RP-L401 received investigational therapy during the second quarter of 2021. This was a 6-year-old child with severe IMO-related anemia and bone abnormalities. Although a drug product was successfully manufactured and infused without immediate complications, this patient died during the initial weeks after therapy of pulmonary complications, most likely pulmonary hemorrhage related to thrombocytopenia following conditioning therapy, and also related to underlying osteopetrosis. Corroborated by autopsy findings, this patient death was not considered to be RP-L401 related by the study investigators. Importantly, pulmonary hemorrhage is a rare but documented complication of HSCT, and pulmonary complications (including life threatening and fatal complications) have been observed to occur with high frequency in osteopetrosis patients undergoing allogenic HSCT procedures. Nonetheless in accordance with protocol-stipulated stopping rules, we paused enrollment to enable a comprehensive evaluation in collaboration with the Independent Data Monitoring Committee (“IDMC”). A thorough review was conducted by the IDMC, supplemented by additional clinical and scientific advisors, including evaluation of the patient’s clinical course, autopsy findings, additional scientific studies on autopsy tissue and on IMO-patient derived cells transduced with the relevant vector. The IDMC concluded that this event was not related to RP-L401 and endorsed further investigation by means of a revised protocol including modified eligibility, patient-monitoring and supportive care elements.
We expect to report clinical updates for our IMO program in the fourth quarter of 2021.
Strategy
We seek to bring hope and relief to patients with devastating, undertreated, rare pediatric diseases through the development and commercialization of potentially curative first-in-class gene therapies. To achieve these objectives, we intend to develop into a fully-integrated biotechnology company. In the near- and medium-term, we intend to develop our first-in-class product candidates, which are targeting devastating diseases with substantial unmet need, develop proprietary in-house analytics and manufacturing capabilities and continue to commence registration trials for our currently planned programs. In the medium and long-term, we expect to submit our first biologics license applications (“BLAs”) and establish our gene therapy platform and expand our pipeline to target additional indications that we believe to be potentially compatible with our gene therapy technologies. In addition, during that time, we believe that our currently planned programs will become eligible for priority review vouchers from the FDA that provide for expedited review. We have assembled a leadership and research team with expertise in cell and gene therapy, rare disease drug development and product approval.
We believe that our competitive advantage lies in our disease-based selection approach, a rigorous process with defined criteria to identify target diseases. We believe that this approach to asset development differentiates us as a gene therapy company and potentially provides us with a first-mover advantage.
Financial Overview
Since our inception, we have devoted substantially all our resources to organizing and staffing, business planning, raising capital, acquiring, or discovering product candidates and securing related intellectual property rights, conducting discovery, research and development activities for the programs and planning for potential commercialization. We do not have any products approved for sale and have not generated revenue from product sales. Since our inception in 2015 through September 30, 2021, we received net cash proceeds of approximately $680.5 million from investors through both equity and convertible debt financing to fund operating activities. As of September 30, 2021, we had cash, cash equivalents and investments of $421.5 million.
Since inception, we have incurred significant operating losses. Our ability to generate product revenue sufficient to achieve profitability will depend heavily on the successful development and eventual commercialization of one or more of the current or future product candidates and programs. We had net losses of $124.8 million for the nine months ended September 30, 2021, and $139.7 million for the year ended December 31, 2020. As of September 30, 2021, we had an accumulated deficit of $447.7 million. We expect to continue to incur significant expenses and higher operating losses for the foreseeable future as we advance our current product candidates from discovery through preclinical development and clinical trials and seek regulatory approval of our product candidates. In addition, if we obtain marketing approval for any of our product candidates, we expect to incur significant commercialization expenses related to product manufacturing, marketing, sales, and distribution. Furthermore, we expect to incur additional costs as a public company having transitioned out of emerging growth company status. Accordingly, we will need additional financing to support continuing operations and potential acquisitions of licensing or other rights for product candidates.
Until such a time as we can generate significant revenue from product sales, if ever, we will seek to fund our operations through public or private equity or debt financings or other sources, which may include collaborations with third parties and government programs or grants. Adequate additional financing may not be available to us on acceptable terms, or at all. We can make no assurances that we will be able to raise the cash needed to fund our operations and, if we fail to raise capital when needed, we may have to significantly delay, scale back or discontinue the development and commercialization of one or more product candidates or delay pursuit of potential in-licenses or acquisitions.
Because of the numerous risks and uncertainties associated with product development, we are unable to predict the timing or amount of increased expenses or when or if we will be able to achieve or maintain profitability. Even if we are able to generate product sales, we may not become profitable. If we fail to become profitable or are unable to sustain profitability on a continuing basis, then we may be unable to continue our operations at planned levels and be forced to reduce or terminate our operations.
Revenue
To date, we have not generated any revenue from any sources, including from product sales, and we do not expect to generate any revenue from the sale of products in the near future. If our development efforts for product candidates are successful and result in regulatory approval or license agreements with third parties, we may generate revenue in the future from product sales.
Operating Expenses
Research and Development Expenses
Our R&D program expenses consist primarily of external costs incurred for the development of our product candidates. These expenses include:
| • | expenses incurred under agreements with research institutions and consultants that conduct R&D activities including, process development, preclinical, and clinical activities on our behalf; |
| • | costs related to process development, production of preclinical and clinical materials, including fees paid to contract manufacturers and manufacturing input costs for use in internal manufacturing processes; |
| • | consultants supporting process development and regulatory activities; |
| • | costs related to in-licensing of rights to develop and commercialize our product candidate portfolio. |
We recognize external development costs based on contractual payment schedules aligned with program activities, invoices for work incurred, and milestones which correspond with costs incurred by third parties. Nonrefundable advance payments for goods or services to be received in the future for use in R&D activities are recorded as prepaid expenses.
Our direct R&D expenses are tracked on a program-by-program basis for product candidates and consist primarily of external costs, such as research collaborations and third-party manufacturing agreements associated with our preclinical research, process development, manufacturing, and clinical development activities. Our direct R&D expenses by program also include fees incurred under license agreements. Our personnel, non-program and unallocated program expenses include costs associated with activities performed by our internal R&D organization and generally benefit multiple programs. These costs are not separately allocated by product candidate and consist primarily of:
| • | salaries and personnel-related costs, including benefits, travel, and stock-based compensation, for our scientific personnel performing R&D activities; |
| • | facilities and other expenses, which include expenses for rent and maintenance of facilities, and depreciation expense; and |
| • | laboratory supplies and equipment used for internal R&D activities. |
Our direct R&D expenses consist principally of external costs, such as fees paid to investigators, consultants, laboratories and CROs in connection with our clinical studies, and costs related to acquiring and manufacturing clinical study materials. We allocate salary and benefit costs directly related to specific programs. We do not allocate personnel-related discretionary bonus or stock-based compensation costs, costs associated with our general discovery platform improvements, depreciation or other indirect costs that are deployed across multiple projects under development and, as such, the costs are separately classified as other R&D expenses.
| | Three Months Ended September 30, | | | Nine Months Ended September 30, | |
| | | 2021 | | | 2020 | | | 2021 | | | 2020 | |
| Direct Expenses: | | | | | | | | | | | | |
| Danon Disease (AAV) | | $ | 3,497 | | | $ | 5,131 | | | $ | 11,804 | | | $ | 13,575 | |
| Leukocyte Adhesion Deficiency (LVV) | | | 7,896 | | | | 1,568 | | | | 18,167 | | | | 3,784 | |
| Fanconi Anemia (LVV) | | | 6,379 | | | | 4,661 | | | | 11,986 | | | | 11,318 | |
| Pyruvate Kinase Deficiency (LVV) | | | 1,109 | | | | 1,606 | | | | 3,530 | | | | 3,406 | |
| Infantile Malignant Osteopetrosis (LVV) | | | 729 | | | | 704 | | | | 1,975 | | | | 1,882 | |
| Other product candidates | | | 2,127 | | | | 262 | | | | 3,998 | | | | 349 | |
| Total direct expenses | | | 21,738 | | | | 13,932 | | | | 51,460 | | | | 34,314 | |
| Unallocated Expenses | | | | | | | | | | | | | | | | |
| Employee compensation | | | 5,524 | | | | 3,380 | | | | 15,265 | | | | 9,662 | |
| Share based compensation expense | | | 3,084 | | | | 1,780 | | | | 9,148 | | | | 4,960 | |
| Depreciation and amortization expense | | | 1,258 | | | | 755 | | | | 3,627 | | | | 1,633 | |
| Laboratory and related expenses | | | 651 | | | | 726 | | | | 2,133 | | | | 1,770 | |
| Legal and patent fees | | | 386 | | | | 328 | | | | 937 | | | | 751 | |
| Professional Fees | | | 400 | | | | 357 | | | | 1,217 | | | | 977 | |
| Other expenses | | | 6,934 | | | | 399 | | | | 9,526 | | | | 1,278 | |
| Total other research and development expenses | | | 18,236 | | | | 7,725 | | | | 41,854 | | | | 21,031 | |
| Total research and development expense | | $ | 39,975 | | | $ | 21,657 | | | $ | 93,315 | | | $ | 55,345 | |
For the three and nine months ended September 30, 2021, LAD-1 costs of $7.9 million or 36% of direct expenses and $18.2 million or 35% of direct expenses primarily relate to foundational investments in the company’s LVV platform, pre-commercial investment in manufacturing and BLA filing preparation.
Our R&D activities are central to our business model. Product candidates in later stages of clinical development generally have higher development costs than those in earlier stages of clinical development. As a result, we expect that R&D expenses will increase substantially over the next several years as we increase personnel costs, including stock-based compensation, support ongoing clinical studies, seek to achieve proof-of-concept in additional product candidates, advance preclinical programs to clinical programs, and prepare regulatory filings for product candidates.
We cannot determine with certainty the duration and costs to complete current or future clinical studies of product candidates or if, when, or to what extent we will generate revenues from the commercialization and sale of any of its product candidates that obtain regulatory approval. We may never succeed in achieving regulatory approval for any of our product candidates. The duration, costs, and timing of clinical studies and development of product candidates will depend on a variety of factors, including:
| • | the scope, rate of progress, and expense of ongoing as well as any clinical studies and other R&D activities that we undertake; |
| • | future clinical study results; |
| • | uncertainties in clinical study enrollment rates; |
| • | changing standards for regulatory approval; and |
| • | the timing and receipt of any regulatory approvals. |
We expect R&D expenses to increase for the foreseeable future as we continue to invest in R&D activities related to developing product candidates, including investments in manufacturing, as our programs advance into later stages of development and as we conduct additional clinical trials. The process of conducting the necessary clinical research to obtain regulatory approval is costly and time-consuming, and the successful development of product candidates is highly uncertain. As a result, we are unable to determine the duration and completion costs of R&D projects or when and to what extent we will generate revenue from the commercialization and sale of any of our product candidates.
Our future R&D expenses will depend on the clinical success of our product candidates, as well as ongoing assessments of the commercial potential of such product candidates. In addition, we cannot forecast with any degree of certainty which product candidates may be subject to future collaborations, when such arrangements will be secured, if at all, and to what degree such arrangements would affect our development plans and capital requirements.
The successful development and commercialization of our product candidates is highly uncertain. This is due to the numerous risks and uncertainties associated with product development and commercialization, including the uncertainty of:
| • | the scope, progress, outcome and costs of our clinical trials and other R&D activities; |
| • | the efficacy and potential advantages of our product candidates compared to alternative treatments, including any standard of care; |
| • | the market acceptance of our product candidates; |
| • | obtaining, maintaining, defending, and enforcing patent claims and other intellectual property rights; |
| • | significant and changing government regulation; and |
| • | the timing, receipt, and terms of any marketing approvals. |
A change in the outcome of any of these variables with respect to the development of our product candidates that we may develop could mean a significant change in the costs and timing associated with the development of our product candidates. For example, if the FDA or another regulatory authority were to require us to conduct clinical trials or other testing beyond those that we currently contemplate for the completion of clinical development of any of our product candidates that we may develop or if we experience significant delays in enrollment in any of our clinical trials, we could be required to expend significant additional financial resources and time on the completion of clinical development of that product candidate.
General and Administrative Expenses
General and administrative expenses consist primarily of salaries and related benefit costs for personnel, including stock-based compensation and travel expenses for our employees in executive, operational, finance, legal, business development, and human resource functions. In addition, other significant general and administrative expenses include professional fees for legal, consulting, investor and public relations, auditing, and tax services as well as other expenses for rent and maintenance of facilities, insurance and other supplies used in general and administrative activities. We expect general and administrative expenses to increase for the foreseeable future due to anticipated increases in headcount to support the continued advancement of our product candidates. We also anticipate that as we continue to operate as a public company with increasing complexity, we will continue to incur increased accounting, audit, legal, regulatory, compliance and director and officer insurance costs as well as investor and public relations expenses.
Interest Expense
Interest expense for the three and nine months ended September 30, 2021, is related to the 2021 Convertible Notes, which were converted into common stock on August 2, 2021, the 2022 Convertible Notes, which were redeemed and converted into common stock in April 2021, and our financing lease obligation for the Cranbury, NJ facility.
Interest Income
Interest income is related to interest earned from investments and cash equivalents.
Critical Accounting Policies and Significant Judgments and Estimates
Our consolidated financial statements are prepared in accordance with generally accepted accounting principles in the U.S. The preparation of our financial statements and related disclosures requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, costs and expenses, and the disclosure of contingent assets and liabilities in our financial statements. We base our estimates on historical experience, known trends and events and various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. We evaluate estimates and assumptions on an ongoing basis. Actual results may differ from these estimates under different assumptions or conditions.
Our significant accounting policies are described in more detail in our 2020 Form 10-K.
Results of Operations
Comparison of the Three Months Ended September 30, 2021 and 2020
The following table summarizes the results of operations for the three months ended September 30, 2021 and 2020 ($ in thousands):
| | | Three Months Ended September 30, | |
| | | 2021 | | | 2020 | | | Change | |
| Operating expenses: | | | | | | | | | |
| Research and development | | $ | 39,975 | | | $ | 21,657 | | | $ | 18,318 | |
| General and administrative | | | 9,671 | | | | 5,730 | | | | 3,941 | |
| Total operating expenses | | | 49,646 | | | | 27,387 | | | | 22,259 | |
| Loss from operations | | | (49,646 | ) | | | (27,387 | ) | | | (22,259 | ) |
| Research and development incentives | | | - | | | | - | | | | - | |
| Interest expense | | | (534 | ) | | | (1,967 | ) | | | 1,433 | |
| Interest and other income net | | | 806 | | | | 518 | | | | 288 | |
| Amortization of premium on investments - net | | | (744 | ) | | | (244 | ) | | | (500 | ) |
| Total other expense, net | | | (472 | ) | | | (1,693 | ) | | | 1,221 | |
| Net loss | | $ | (50,118 | ) | | $ | (29,080 | ) | | $ | (21,038 | ) |
Research and Development Expenses
R&D expenses increased $18.3 million to $40.0 million for the three months ended September 30, 2021 compared to the three months ended September 30, 2020. The increase was related to the increase in manufacturing and development costs of $5.4 million, an increase in new research agreements of $7.6 million in non-cash expenses due to the issuance of a warrant in August 2021, increases in compensation and benefits of $2.1 million due to increased R&D headcount, and an increase in non-cash stock compensation expense of $1.3 million,
General and Administrative Expenses
G&A expenses increased $3.9 million to $9.7 million for the three months ended September 30, 2021 compared to the three months ended September 30, 2020. The increases in G&A expenses were primarily driven by an increase in non-cash stock compensation expense of $1.6 million, an increase in compensation and benefits of $0.8 million due to increased G&A headcount and an increase in commercial preparation expenses of $0.8 million.
Other Expense, Net
Other expense, net was $0.5 million for the three months ended September 30, 2021 compared to $1.7 million for the three months ended September 30, 2020. The change was primarily due to reduced interest expense associated with the 2022 Convertible Notes that were redeemed in April 2021 and the 2021 Convertible Notes that were converted in August 2021, as well as a decrease of $0.5 million in accretion income related to our investments, due to lower interest rates in 2021 as compared to the same period in 2020.
Comparison of the Nine Months Ended September 30, 2021 and 2020
The following table summarizes the results of operations for the nine months ended September 30, 2021 and 2020 ($ in thousands):
| | | Nine Months Ended September 30, | |
| | | 2021 | | | 2020 | | | Change | |
| Operating expenses: | | | | | | | | | |
| Research and development | | $ | 93,315 | | | $ | 55,345 | | | $ | 37,970 | |
| General and administrative | | | 29,600 | | | | 19,720 | | | | 9,880 | |
| Total operating expenses | | | 122,915 | | | | 75,065 | | | | 47,850 | |
| Loss from operations | | | (122,915 | ) | | | (75,065 | ) | | | (47,850 | ) |
| Research and development incentives | | | 500 | | | | - | | | | 500 | |
| Interest expense | | | (2,514 | ) | | | (5,326 | ) | | | 2,812 | |
| Interest and other income net | | | 2,218 | | | | 1,913 | | | | 305 | |
| Amortization of premium on investments - net | | | (2,111 | ) | | | (306 | ) | | | (1,805 | ) |
| Total other expense, net | | | (1,907 | ) | | | (3,719 | ) | | | 1,812 | |
| Net loss | | $ | (124,822 | ) | | $ | (78,784 | ) | | $ | (46,038 | ) |
Research and Development Expenses
R&D expenses increased $38.0 million to $93.3 million for the nine months ended September 30, 2021 compared to the nine months ended September 30, 2020. The increase was primarily due to an increase in manufacturing and development costs of $13.9 million, an increase in new research agreements of $7.6 million in non-cash expenses due to the issuance of a warrant in August 2021, an increase in compensation and benefits of $5.6 million due to increased R&D headcount, an increase in non-cash stock compensation expense of $4.2 million, an increase in lab supplies and office expense of $2.1 million, an increase in depreciation and amortization of $2.1 million, and an increase in clinical trial expenses of $1.3 million.
General and Administrative Expenses
G&A expenses increased $9.9 million to $29.6 million for the nine months ended September 30, 2021 compared to the nine months ended September 30, 2020. The increases in G&A expenses were primarily driven by an increase in non-cash stock compensation expense of $5.5 million, an increase in compensation and benefits of $2.2 million due to increased G&A headcount, an increase in office and administrative costs of $1.9 million, an increase in accounting and consulting expenses of $0.9 million, an increase in commercial preparation expenses of $0.8 million, offset by a decrease in debt conversion expense recorded for the nine months ended September 30, 2020 of $2.0 million due to the refinancing of the 2021 Convertible Notes in February 2020. There were no debt conversion expenses recorded for the nine months ended September 30, 2021.
Other Expense, Net
Other expense, net was $1.9 million for the nine months ended September 30, 2021 compared to $3.7 million for the nine months ended September 30, 2020. The change was primarily due to reduced interest expense associated with the 2022 Convertible Notes which were fully redeemed in April 2021 and the 2021 Convertible Notes which were converted in August 2021, as well as a decrease of $1.8 million in accretion income related to our investments, due to lower interest rates for the nine months ended September 30, 2021 as compared to the nine months ended September 30, 2020.
Liquidity, Capital Resources and Plan of Operations
Since inception, we have not generated any revenue from any sources, including from product sales, and have incurred significant operating losses and negative cash flows from our operations. We have funded operations to date primarily with proceeds from the sale of preferred shares, common stock, and the issuance of convertible notes.
Cash Flows
The following table summarizes our cash flows for each of the periods presented:
| | | Nine Months Ended September 30, | |
| | | 2021 | | | 2020 | |
| Cash used in operating activities | | $ | (90,222 | ) | | $ | (61,531 | ) |
| Cash provided by (used in) investing activities | | | 1,989 | | | | (42,129 | ) |
| Cash provided provided by financing activities | | | 36,545 | | | | 546 | |
| Net change in cash, cash equivalents and restricted cash | | $ | (51,688 | ) | | $ | (103,114 | ) |
Operating Activities
During the nine months ended September 30, 2021, operating activities used $90.2 million of cash, primarily resulting from our net loss of $124.8 million offset by net non-cash charges of $34.8 million, including non-cash stock-based compensation expense of $22.2 million, warrant issuance of $7.6 million, accretion of discount on investments of $2.1 million, and depreciation and amortization expense of $2.2 million. Changes in our operating assets and liabilities for the nine months ended September 30, 2021 consisted of an increase in accounts payable and accrued expenses for $2.8 million and a decrease in our prepaid expenses of $1.0 million.
During the nine months ended September 30, 2020, operating activities used $61.5 million of cash, primarily resulting from our net loss of $78.8 million offset by net non-cash charges of $15.5 million, including non-cash stock-based compensation expense of $12.5 million and accretion of the discount on convertible notes of $2.1 million. Changes in our operating assets and liabilities for the nine months ended September 30, 2020 consisted of increases in prepaid expenses and other assets of $1.4 million, and a decrease in accounts payable and accrued expenses of $1.8 million.
Investing Activities
During the nine months ended September 30, 2021, net cash provided by investing activities was $2.0 million, consisting of proceeds of $234.1 million from the maturities of investments, offset by purchases of investments of $226.5 million, purchases of property and equipment of $4.9 million, and purchases of internal use software of $0.7 million.
During the nine months ended September 30, 2020, net cash used by investing activities was $42.1 million, consisting of proceeds of $104.1 million from the maturities of investments, offset by purchases of investments of $132.1 million, purchases of property and equipment of $7.0 million, payments made to acquire a right of use asset of $6.5 million, and purchases of internal use software of $0.6 million.
Financing Activities
During the nine months ended September 30, 2021, net cash provided by financing activities was $36.5 million, consisting of issuance of common stock related to the August 2021 Private Placement and issuance of common stock, pursuant to exercises of stock options.
During the nine months ended September 30, 2020, net cash provided by financing activities was $0.5 million, consisting primarily of issuance of common stock of $0.9 million pursuant to exercise of stock options, offset by payments of withholding tax on option exercises.
Funding Requirements
We expect expenses to increase substantially in connection with our ongoing activities, particularly as we advance our preclinical activities, and initiate additional clinical trials and manufacturing of our product candidates. In addition, we expect to incur additional costs associated with operating as a public company having transitioned from being an emerging growth company. Our expenses will also increase as we:
| • | leverage our programs to advance other product candidates into preclinical and clinical development; |
| • | seek regulatory agreements to initiate clinical trials in Europe, the U.S. and ROW; |
| • | establish a sales, marketing, medical affairs, and distribution infrastructure to commercialize any product candidates for which Rocket may obtain marketing approval and intend to commercialize on its own or jointly; |
| • | hire additional preclinical, clinical, regulatory, quality, and scientific personnel; |
| • | expand our operational, financial and management systems and increase personnel, including personnel to support our clinical development, manufacturing and commercialization efforts and our operations as a public company; |
| • | maintain, expand, and protect our intellectual property portfolio; and |
| • | acquire or in-license other product candidates and technologies. |
As of September 30, 2021, we had cash, cash equivalents and investments of $421.5 million. We expect such resources would be sufficient to fund our operating expenses and capital expenditure requirements into the second half of 2023.
Because of the numerous risks and uncertainties associated with research, development, and commercialization of pharmaceutical product candidates, we are unable to estimate the exact amount of working capital requirements. Our future funding requirements will depend on, and could increase significantly as a result of, many factors, including:
| • | the scope, progress, results, and costs of researching and developing our product candidates, and conducting preclinical studies and clinical trials; |
| • | the costs, timing, and outcome of regulatory review of our product candidates; |
| • | the costs of future activities, including product sales, medical affairs, marketing, manufacturing, and distribution, for any of our product candidates for which we receive marketing approval; |
| • | the costs of manufacturing commercial-grade product to support commercial launch; |
| • | the ability to receive additional non-dilutive funding, including grants from organizations and foundations; |
| • | the revenue, if any, received from commercial sale of its products, should any of its product candidates receive marketing approval; |
| • | the costs of preparing, filing and prosecuting patent applications, maintaining and enforcing our intellectual property rights and defending intellectual property-related claims; |
| • | our ability to establish and maintain collaborations on favorable terms, if at all; |
| • | the extent to which we acquire or in-license other product candidates and technologies; and |
| • | the timing, receipt, and amount of sales of, or milestone payments related to our royalties on, current or future product candidates, if any. |
Until such time, if ever, as we can generate substantial product revenue, we expect to finance our cash needs through a combination of public or private equity offerings, debt financings, collaborations, strategic partnerships or marketing, distribution, or licensing arrangements with third parties. To the extent that we raise additional capital through the sale of equity or convertible debt securities, our ownership interest may be materially diluted, and the terms of such securities could include liquidation or other preferences that adversely affect the rights of our common stockholders. Debt financing and preferred equity financing, if available, may involve agreements that include restrictive covenants that limit our ability to take specified actions, such as incurring additional debt, making capital expenditures, or declaring dividends. In addition, additional debt financing would result in increased fixed payment obligations.
If we raise funds through governmental funding, collaborations, strategic partnerships or marketing, distribution, or licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates or grant licenses on terms that may not be favorable to us. If we are unable to raise additional funds through equity or debt financings when needed, we may be required to delay, reduce, or eliminate our product development or future commercialization efforts or grant rights to develop and market product candidates that it would otherwise prefer to develop and market themselves.
Contractual Obligations and Commitments
There were no material changes outside the ordinary course of our business to the contractual obligations specified in the table of contractual obligations included in “Management’s Discussion and Analysis of Financial Condition and Results of Operations” in our 2020 Form 10-K. Information regarding contractual obligations and commitments may be found in Note 11 of our Consolidated Unaudited Financial Statements in this Form 10-Q.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and do not currently have, any off-balance sheet arrangements, as defined in the rules and regulations of the Securities and Exchange Commission.
Recently Issued Accounting Pronouncements
A description of recently issued accounting pronouncements that may potentially impact our financial position and results of operations is disclosed in Note 3 of our “Consolidated Unaudited Financial Statements,” in this Quarterly Report on Form 10-Q.
| Item 3 | Quantitative and Qualitative Disclosures About Market Risk |
We are exposed to market risks in the ordinary course of our business. These market risks are principally limited to interest rate fluctuations. We had cash, cash equivalents and investments of $421.5 million at September 30, 2021, consisting primarily of funds in a money market account, corporate and municipal bonds, and United States Treasury securities. The primary objective of our investment activities is to preserve principal and liquidity while maximizing income without significantly increasing risk. We do not enter into investments for trading or speculative purposes. Due to the short-term nature of our investment portfolio, we do not believe an immediate 1.0% increase in interest rates would have a material effect on the fair market value of our portfolio, and accordingly we do not expect a sudden change in market interest rates to affect materially our operating results or cash flows.
Our 2021 Convertible Notes bore interest at a fixed rate and therefore a change in interest rates would not have impacted the amount of interest we would have had to pay on this indebtedness, while it was outstanding.
| Item 4 | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
Our management, with the participation of our principal executive officer and our principal financial officer, evaluated, as of the end of the period covered by this Form 10-Q, the effectiveness of our disclosure controls and procedures. Based on that evaluation of our disclosure controls and procedures as of September 30, 2021, our principal executive officer and principal financial officer concluded that our disclosure controls and procedures as of such date are effective at the reasonable assurance level. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act are recorded, processed, summarized, and reported within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by us in the reports we file or submit under the Exchange Act is accumulated and communicated to our management, including our principal executive officer and principal financial officer, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and our management necessarily applies its judgment in evaluating the cost-benefit relationship of possible controls and procedures.
Inherent Limitations of Internal Controls
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation. Projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting (as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act) during the nine months ended September 30, 2021, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
PART II – OTHER INFORMATION
From time to time, we may be subject to various legal proceedings and claims that arise in the ordinary course of our business activities. Although the results of litigation and claims cannot be predicted with certainty, we do not believe we are party to any other claim or litigation the outcome of which, if determined adversely to us, would individually or in the aggregate be reasonably expected to have a material adverse effect on our business. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Our material risk factors are disclosed in Item 1A of our Annual Report on Form 10-K for the year ended December 31, 2020 filed with the Securities and Exchange Commission on March 1, 2021. There have been no material changes from the risk factors previously disclosed in such filing.
| Item 2. | Unregistered Sales of Equity Securities and Use of Proceeds |
None.
| Item 3. | Defaults Upon Senior Securities |
None.
| Item 4. | Mine Safety Disclosures |
Not applicable.
None
| Exhibit | | |
| Number | | Description of Exhibit |
| | Agreement and Plan of Merger and Reorganization, dated as of September 12, 2017, by and among Inotek Pharmaceuticals Corporation, Rocket Pharmaceuticals, Ltd. and Rome Merger Sub (incorporated by reference to Exhibit 2.1 to the Company’s Current Report on Form 8-K (001-36829), filed with the SEC on September 13, 2017) |
| | Seventh Amended and Restated Certificate of Incorporation of Rocket Pharmaceuticals, Inc., effective as of February 23, 2015(incorporated by reference to Exhibit 3.1 to the Company’s Annual Report on Form 10-K (001-36829), filed with the SEC on March 31, 2015) |
| | Certificate of Amendment (Reverse Stock Split) to the Seventh Amended and Restated Certificate of Incorporation of the Registrant, effective as of January 4, 2018 (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K (001-36829), filed with the SEC on January 5, 2018) |
| | Certificate of Amendment (Name Change) to the Seventh Amended and Restated Certificate of Incorporation of the Registrant, effective January 4, 2018 (incorporated by reference to Exhibit 3.2 to the Company’s Current Report on Form 8-K (001-36829), filed with the SEC on January 5, 2018) |
| | Certificate of Amendment to the Seventh Amended and Restated Certificate of Incorporation of the Registrant, effective as of June 25, 2018 (incorporated by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K (001-36829), filed with the SEC on June 25, 2019 |
| | Amended and Restated By-Laws of Rocket Pharmaceuticals, Inc., effective as of March 29, 2018 (incorporated by reference to Exhibit3.2 to the Company’s Current Report on Form 8-K (001-36829), filed with the SEC on April 4, 2018) |
| | Securities Purchase Agreement, dated as of August 27, 2021, between the Registrant and the purchaser party thereto (incorporated by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (001-36829), filed with the SEC on August 30, 2021) |
| | Registration Rights Agreement, dated as of August 27, 2021, between the Registrant and the purchaser party thereto (incorporated by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K (001-36829), filed with the SEC on August 30, 2021) |
| | Certification of Principal Executive Officer pursuant to Rule 13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| | Certification of Principal Financial Officer pursuant to Rule 13a-14(a) or Rule 15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| | Certification of Principal Executive Officer and Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 |
| 101.INS | | Inline XBRL Instance Document. |
| 101.SCH | | Inline XBRL Taxonomy Extension Schema Document. |
| 101.CAL | | Inline XBRL Taxonomy Extension Calculation Document. |
| 101.DEF | | Inline XBRL Taxonomy Extension Definition Linkbase Document. |
| 101.LAB | | Inline XBRL Taxonomy Extension Labels Linkbase Document. |
| 101.PRE | | Inline XBRL Taxonomy Extension Presentation Link Document. |
| 104 | | Cover Page Interactive Data File (the cover page XBRL tags are embedded within the Inline XBRL document) |
Pursuant to the requirements of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| | ROCKET PHARMACEUTICALS, INC. |
| | | |
| November 5, 2021 | By: | /s/ Gaurav Shah, MD |
| | | Gaurav Shah, MD |
| | | Chief Executive Officer and Director |
| | | (Principal Executive Officer) |
| | | |
| November 5, 2021 | By: | /s/ Carlos Garcia-Parada |
| | | Carlos Garcia-Parada |
| | | Chief Financial Officer |
| | | (Principal Financial Officer) |