As filed with the Securities and Exchange Commission on April 18, 2006
Registration Statement No. 333-
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM F-10
REGISTRATION STATEMENT
Under
THE SECURITIES ACT OF 1933
LABOPHARM INC.
(Exact name of Registrant as specified in its charter)
| Québec, Canada | 2834 | NOT APPLICABLE | ||
| (Province or other jurisdiction of company or organization) | (Primary Standard Industrial Classification Code Number) | (I.R.S. Employer Identification Number) |
480 Armand-Frappier Blvd. Laval, Québec, Canada H7V 4B4 (450) 686-1017
(Address and telephone number of Registrant’s principal executive offices)
CT Corporation System
101 Federal Street
Boston, MA 02110
(617) 757-6400
(Name, address (including zip code) and telephone number (including area code)
of agent for service in the United States)
Copies to:
| James R. Howard-Tripp Labopharm inc. 480 Armand-Frappier Blvd. Laval, Québec, Canada H7V 4B4 (450) 686-1017 | Lawrence S. Wittenberg Goodwin Procter, LLP 53 State Street Boston, MA 02109 (617) 570-1000 | David E. Redlick Stuart M. Falber Wilmer Cutler Pickering Hale and Dorr LLP 60 State Street Boston, MA 02109 (617) 526-6000 | Louis-François Hogue Frédéric Boucher Fasken Martineau Dumoulin LLP Stock Exchange Tower Suite 3400, P.O. Box 242 800 Place Victoria Montreal, Québec, Canada H4Z 1E9 (514) 397-7400 |
Approximate date of commencement of proposed sale of the securities to the public: As soon as practicable after the effective date of this Registration Statement.
Province of Québec, Canada
(Principal jurisdiction regulating this offering)
It is proposed that this filing shall become effective (check appropriate box):
| A. | o | upon filing with the Commission, pursuant to Rule 467(a) (if in connection with an offering being made contemporaneously in the United States and Canada). | |
| B. | x | at some future date (check the appropriate box below): |
| 1. | o | pursuant to Rule 467(b) on (date) at (time) (designate a time not sooner than 7 calendar days after filing). |
| 2. | o | pursuant to Rule 467(b) on (date) at (time) (designate a time 7 calendar days or sooner after filing) because the securities regulatory authority in the review jurisdiction has issued a receipt or notification of clearance on (date). | ||
| 3. | o | pursuant to Rule 467(b) as soon as practicable after notification of the Commission by the Registrant or the Canadian securities regulatory authority of the review jurisdiction that a receipt or notification of clearance has been issued with respect hereto. | ||
| 4. | x | after the filing of the next amendment to this Form (if preliminary material is being filed). |
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to the home jurisdiction’s shelf prospectus offering procedures, check the following box.o
CALCULATION OF REGISTRATION FEE
| Title of each | Proposed maximum | Proposed maximum | ||||||||||||||
| class of securities | Amount to be | offering price | aggregate offering | Amount of | ||||||||||||
| to be registered | registered* | per unit** | price** | registration fee | ||||||||||||
| Common Shares | 11,500,000 | US$7.64 | US$87,860,000 | US$9,401.02 | ||||||||||||
| * | Includes 1,500,000 shares that the underwriters have an option to purchase to cover over-allotments, if any. | |
| ** | Estimated solely for the purpose of calculating the amount of the registration fee pursuant to Rule 457(c), based on the average of the high and low prices of the Registrant’s common shares on the Toronto Stock Exchange on April 13, 2006 converted into U.S. dollars at the inverse of the noon buying rate in New York City for cable transfers in Canadian dollars by the Federal Reserve Bank of New York on April 13, 2006. |
The Registrant hereby amends this registration statement on such date or dates as may be necessary to delay its effective date until the registration statement shall become effective as provided in Rule 467 under the Securities Act of 1933 or on such date as the Commission, acting pursuant to Section 8(a) of the Act, may determine.
PART I
INFORMATION REQUIRED TO BE
DELIVERED TO OFFEREES OR PURCHASERS
I-1
| Information contained herein is subject to completion or amendment. A registration statement relating to these securities has been filed with the Securities and Exchange Commission. These securities may not be sold nor may offers to buy be accepted prior to the time the registration statement becomes effective. This prospectus shall not constitute an offer to sell or the solicitation of an offer to buy nor shall there be any sale of these securities in any State in which such offer, solicitation or sale would be unlawful prior to registration or qualification under the securities laws of any such State. |
SUBJECT TO COMPLETION,
PRELIMINARY PROSPECTUS DATED APRIL 18, 2006
PROSPECTUS
10,000,000 Shares

Common Shares
Labopharm inc. is offering 10,000,000 common shares. Our common shares are listed on the Toronto Stock Exchange under the symbol “DDS.” This is our initial public offering in the United States, and no public market currently exists in the United States for our shares. The closing price of our common shares on the Toronto Stock Exchange on April 17, 2006 was C$8.42 per share.
We have applied to list our common shares on the Nasdaq National Market under the symbol “DDSS.”
Investing in our common shares involves certain risks. See “Risk Factors” beginning on page 5.
| Per Share | Total | |||
| Offering price | US$ | US$ | ||
| Underwriting discounts and commissions | US$ | US$ | ||
| Offering proceeds to Labopharm inc., before expenses | US$ | US$ | ||
This offering is made by a foreign private issuer that is permitted, under a multijurisdictional disclosure system adopted by the United States, to prepare this prospectus in accordance with the disclosure requirements of its home country, Canada. Prospective investors should be aware that such requirements are different from those of the United States. Financial statements included or incorporated herein, if any, have been prepared in accordance with Canadian generally accepted accounting principles, and may be subject to foreign auditing and auditor independence standards, and thus may not be comparable to financial statements of United States companies that are prepared in accordance with United States generally accepted accounting principles.
You should be aware that the acquisition of the securities described herein may have tax consequences both in the United States and in Canada. Such consequences for investors who are residents in, or citizens of, the United States may not be fully described herein. You should read the tax discussion contained in this prospectus and consult your own tax advisor.
The enforcement by investors of civil liabilities under U.S. federal securities laws may be affected adversely by the fact that we are organized under the laws of the Province of Québec, Canada, that most of our officers and directors are residents of Canada, that some of the underwriters or experts named in this prospectus are residents of Canada, and that a substantial portion of our assets and the assets of those officers, directors, underwriters and experts are located outside of the United States.
THESE SECURITIES HAVE NOT BEEN APPROVED OR DISAPPROVED BY THE SECURITIES AND EXCHANGE COMMISSION NOR HAS THE COMMISSION PASSED UPON THE ACCURACY OR ADEQUACY OF THIS PROSPECTUS. ANY REPRESENTATION TO THE CONTRARY IS A CRIMINAL OFFENSE.
The public offering price for our common shares offered in Canada is payable in Canadian dollars, and the public offering price for our common shares offered outside Canada is payable in Canadian or U.S. dollars. The U.S. dollar price is the equivalent of the Canadian dollar price for the common shares based on the prevailing exchange rate on the date of this prospectus.
We have granted the underwriters the right to purchase up to 1,500,000 additional common shares at the public offering price, less underwriting discounts and commissions, to cover any over-allotments. The underwriters can exercise this right at any time within thirty days after the offering. The underwriters expect to deliver the common shares to investors on or about , 2006.
Joint Book-Running Managers
| Merrill Lynch & Co. | Banc of America Securities LLC |
| Canaccord Capital Corporation | Leerink Swann & Company |
| Orion Securities Inc. | Dundee Securities Corporation | Westwind Partners Inc. |
The date of this prospectus is , 2006
TABLE OF CONTENTS
| Prospectus Summary | 1 | |||
| Risk Factors | 7 | |||
| Special Note Regarding Forward-Looking Statements | 25 | |||
| Enforceability of Civil Liabilities | 26 | |||
| Exchange Rate Information | 26 | |||
| Use of Proceeds | 27 | |||
| Price Range of Common Shares | 28 | |||
| Dividend Policy | 28 | |||
| Capitalization | 29 | |||
| Dilution | 30 | |||
| Selected Consolidated Financial Data | 31 | |||
| Management’s Discussion and Analysis of Financial Condition and Results of Operations | 34 | |||
| Business | 44 | |||
| Management | 68 | |||
| Related Party Transactions | 72 | |||
| Principal Shareholders | 73 | |||
| Description of Share Capital | 74 | |||
| Certain Material Income Tax Considerations | 75 | |||
| Underwriting | 81 | |||
| Transfer Agent and Registrar | 84 | |||
| Legal Matters | 84 | |||
| Experts | 85 | |||
| Documents Incorporated by Reference | 85 | |||
| Where You Can Find More Information | 86 | |||
| Documents Filed as Part of the Registration Statement | 86 | |||
| Index to Consolidated Financial Statements | F-1 |
i
You should rely only on the information contained or incorporated by reference in this prospectus. We have not, and the underwriters have not, authorized anyone to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely on it. We are not making an offer to sell or seeking an offer to buy these securities in any jurisdiction where the offer or sale is not permitted. You should assume that the information contained in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or of any sale of the common shares. Our business, financial condition, results of operations and prospects may have changed since that date.
Our estimates of market share and market size in this prospectus were based on, in certain cases, public disclosure, industry and trade publications and reports prepared by third parties, which we believe to be reliable.
All dollar amounts in this prospectus are expressed in Canadian dollars, except where otherwise indicated. References to “$” or “C$” are to Canadian dollars and references to “US$” are to U.S. dollars.
Our consolidated financial statements and certain other financial information of ours contained or incorporated by reference in this prospectus are, except where otherwise noted, reported in Canadian dollars and have been prepared in accordance with Canadian generally accepted accounting principles, or Canadian GAAP. To the extent applicable to our consolidated financial statements included elsewhere in this prospectus, these principles conform in all material respects with United States generally accepted accounting principles, or U.S. GAAP, except as described in note 23 to our consolidated financial statements found elsewhere in this prospectus.
This prospectus contains a translation of some Canadian dollar amounts into U.S. dollars solely for your convenience. See “Exchange Rate Information.”
The names “Labopharm”® and “Contramid”® appearing in this prospectus are our registered trademarks. The name “Polymeric Nano-Delivery System”TM is our trademark. Other trademarks and service marks appearing in this prospectus are the property of their respective holders.
ii
PROSPECTUS SUMMARY
This summary highlights selected information contained elsewhere in this prospectus or in the documents incorporated by reference. This summary does not contain all of the information that you should consider before investing in our common shares. You should carefully read the entire prospectus, including the section titled “Risk Factors” and the documents and financial statements incorporated by reference into this prospectus, before making an investment decision.
In this prospectus, references to “Labopharm,” “we,” “our” and “us” refer to Labopharm inc. and, unless the context otherwise states, its subsidiaries.
Our Business
We are an international, specialty pharmaceutical company focused on improving existing drugs by incorporating our proprietary, advanced controlled-release technologies. Our lead product, a once-daily formulation of the pain-killer tramadol designed to address the worldwide market for moderate to severe pain, has been approved in Europe and is currently the subject of a New Drug Application, or NDA, under review by the United States Food and Drug Administration, or FDA, with a Prescription Drug User Fee Act, or PDUFA, date of September 28, 2006. We are developing additional product candidates using our drug delivery technologies and formulation expertise and have three product candidates in the clinical stage of development including a once-daily formulation of trazodone. In addition we are currently pursuing tramadol product line extensions, including combination products using our Contramid technology.
Pain Market
The treatment of pain represents a significant commercial opportunity and we believe that this market will continue to grow. Pain can be classified by duration (acute or chronic) and severity (mild, moderate or severe). The World Health Organization, or WHO, established a three-step analgesic ladder for the treatment of chronic pain states. In the case of mild pain, Step 1 suggests prompt treatment with a non-opioid such as acetaminophen, aspirin or NSAIDs. For moderate pain, Step 2 recommends introducing a mild opioid such as tramadol or codeine. For severe pain, Step 3 suggests treatment with morphine or other strong opioids until the patient is pain free. Effective management of moderate to severe chronic pain requires treatment that can be tolerated over a long period of time with relatively few side effects and that can be taken in a dosing regimen that facilitates patient compliance and reduces the risk of breakthrough pain. We believe that our once-daily tramadol product addresses these issues.
Tramadol
Tramadol is a proven analgesic that has been on the market for nearly three decades. It has been marketed in approximately 100 countries worldwide. Studies have shown that tramadol provides relief from both acute and chronic pain conditions, including osteoarthritis pain, low back pain, cancer pain, post-operative pain and dental pain. Tramadol avoids the potentially serious side effects such as gastrointestinal bleeding that may be caused by NSAIDs and the potential cardiovascular events recently attributed to COX-2 inhibitors. Scheduled opioids, such as morphine and morphine-derived drugs, are sub-optimal chronic medications due to their gastrointestinal, respiratory and addictive side-effects, which can compromise patient compliance. Compared to stronger opioids, tramadol has, in clinical studies, demonstrated a lower incidence of major side effects, such as respiratory depression. When used over the long-term, tramadol has a significantly lower incidence of analgesic tolerance, withdrawal symptoms and addiction as compared to stronger opioids. However, tramadol has been available primarily as formulations requiring more than one dose per day.
Our Once-Daily Tramadol Product
We have overcome these limitations by developing a tramadol formulation that patients can take once a day and that has been shown clinically to provide pain relief lasting a full 24 hours, thereby maintaining the patient’s pain under control. Furthermore, our formulation provides for an early phase of tramadol release,
1
with a rate comparable to that of the immediate release formulations. We are positioned to address the worldwide market for moderate to severe pain through our established global distribution network. Our once-daily tramadol product has completed six Phase III clinical trials in the United States and Europe, including two long-term safety studies, as well as 13 pharmacokinetic studies. In total, more than 2,600 subjects have received our once-daily tramadol product in various dosage forms. Our once-daily tramadol product consists of a dual matrix delivery system designed to provide both rapid and sustained drug release to maintain blood levels within the therapeutic range. By achieving this, we believe our product confers the following advantages:
| • | early phase tramadol release rate comparable to immediate release formulations; | |
| • | pain relief lasting a full 24 hours; and | |
| • | lower incidence of major side effects. |
We launched our once-daily tramadol product in Germany in November 2005. In September 2005, we received regulatory approval for 22 European countries, under the Mutual Recognition Procedure, or MRP, and have now obtained marketing authorizations, or MAs, for our once-daily tramadol product in 15 countries. We continue to seek MA in the remaining seven countries. We intend to market our once-daily tramadol product primarily through a series of marketing and distribution arrangements in the United States, Europe and the rest of the world. To date, we have entered into agreements for the distribution of our once-daily tramadol product in the United States, in 21 European countries and in 20 Latin American and Caribbean countries. Our marketing and distribution collaborators include Purdue Pharma Products L.P., or Purdue, HEXAL AG, Laboratoire Aventis, or Aventis, Grünenthal GmbH, Recordati Ireland Limited, or Recordati, Azienda Chimiche Rionite Angelini Francesco S.P.A., or Gruppo Angelini, CSC Pharmaceuticals SA, or CSC Pharma, Laboratorios del Dr. Esteve S.A., or Esteve S.A., and Glaxo Group Limited, or GSK. Although the structures of these agreements are different, each of these agreements is designed specifically by territory and to provide us with overall economic participation in sales of our once-daily tramadol product that is proportionate to the royalties that Purdue has agreed to pay us on sales of our once-daily tramadol product.
Under our licensing and distribution arrangement with Purdue for the United States, we received a licensing fee of US$20 million and are eligible to receive up to another US$150 million in milestone payments subject to receiving regulatory approval and meeting specific sales targets in the United States, as well as royalties ranging from 20% to 25% on net product sales by Purdue. We have retained co-promotion rights in the United States to market our once-daily tramadol product to medical specialties that focus on the central nervous system and pain. Purdue has agreed to assist us in building and training our sales force, as well as to compensate us for our co-promotion efforts on a fee-for-service basis that we believe will offset the majority of our sales force costs.
Our Once-Daily Trazodone
Pursuant to a feasibility and formulation agreement with Gruppo Angelini, we are developing a formulation of our once-daily trazodone product that would be bioequivalent in terms of drug absorption to a currently marketed immediate release product administered three times daily or twice-daily product. Trazodone is indicated and prescribed for depression with or without anxiety. Trazodone continues to be widely prescribed in the United States, with more than 14 million prescriptions written in 2005 and having a compounded annual growth rate of 5% over the past five years. Trazodone is currently available in the United States in both branded and generic immediate release formulations that typically require three doses per day. In Europe, trazodone is available in both immediate release and twice-daily formulations. We are currently developing clinical protocols with the intent of advancing this program into Phase II clinical trials in 2006. We expect that any NDA for this product candidate would be submitted under Section 505(b)(2) of the U.S. Federal Food, Drug, and Cosmetic Act.
2
Our Drug Delivery Technologies
Our once-daily formulation of tramadol and trazodone, as well as our other product candidates in clinical development, are based on our patented technology, Contramid. Contramid is a controlled-release drug delivery technology based on cross-linked, high amylose starch, for oral administration of solid dosage medications. We have developed the expertise to formulate novel products that combine our Contramid technology with existing drugs to optimize their release profile. Our success in formulating our once-daily tramadol product illustrates our ability to employ our Contramid technology, formulate and execute a clinical plan for a complex small, highly water soluble molecule and control its release over a 24-hour period with the desired pharmacokinetic profile.
Our patented polymeric nano-delivery systems technology is a complementary platform and provides us with the capabilities to address the delivery challenges associated with water insoluble compounds, as well as proteins, peptides and nucleic acids. We believe this technology may have the potential to improve the delivery of currently administered intravenous products, as well as to increase bioavailability for potential oral delivery.
Our Strategy
Our goal is to become a fully integrated, international specialty pharmaceutical company, with the expertise and infrastructure to develop and commercialize proprietary therapeutics by taking them from the formulation stage through clinical development, regulatory approval, marketing and sales. Key elements of our strategy are to:
| • | maximize the global commercial opportunity for our once-daily tramadol product; | |
| • | develop a robust pipeline of additional products; | |
| • | develop a specialty sales and marketing infrastructure in the United States; and | |
| • | leverage global relationships for future opportunities. |
Corporate Information
We amalgamated under Part IA of the Companies Act (Quebec) in June 1996. Our principal offices
are located at 480 Armand-Frappier Blvd, Laval, Québec, H7V 4B4, and our telephone number is (450) 686-1017. Our website address is www.labopharm.com. Information contained on our website, or which can be accessed through our website, is not a part of, and is not incorporated by reference in, this prospectus.
3
THE OFFERING
| Common shares offered | 10,000,000 shares | |
| Common shares to be outstanding after the offering | 53,901,863 shares | |
| Over-allotment option | The underwriters have an option to purchase up to an additional 1,500,000 common shares from us at the public offering price, less underwriting discounts and commissions, to cover over-allotments, if any. This option is exercisable in whole or in part for a period of 30 days from the date of the closing of the offering. | |
| Use of proceeds | We expect to use the net proceeds from the offering: | |
| • to support the commercialization of our once-daily tramadol product; | ||
| • to advance development of existing and new product candidates within our product pipeline; and | ||
| • for working capital and other general corporate purposes. | ||
| See “Use of Proceeds.” | ||
| Toronto Stock Exchange symbol | DDS | |
| Proposed Nasdaq National Market symbol | DDSS | |
| Risk factors | An investment in our common shares involves certain risks that you should carefully consider. See “Risk Factors” and other information included in this prospectus. |
The number of common shares that will be outstanding immediately after the completion of the offering is based on the number of common shares outstanding as of April 13, 2006 and excludes:
| • | 3,384,875 common shares issuable upon the exercise of stock options then outstanding at a weighted average exercise price of $5.76 (approximately US$5.00) per share; | |
| • | 164,504 common shares reserved for future issuance under our stock option plan; and | |
| • | up to 1,500,000 common shares, if any, issuable pursuant to the underwriters’ over-allotment option as described under “Underwriting”. |
Except as otherwise indicated, information in this prospectus assumes no exercise of the underwriters’ over-allotment option for up to an additional 1,500,000 common shares.
4
Summary Consolidated Financial Data
The following summary consolidated financial data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our audited consolidated financial statements and related notes included elsewhere in this prospectus. We have derived the consolidated statement of operations data for the year ended December 31, 2004 and the year ended December 31, 2005 and the consolidated balance sheet data as at December 31, 2005 from our consolidated financial statements that have been audited by Ernst & Young LLP, which are included elsewhere in this prospectus. Our audited consolidated financial statements have been prepared in Canadian dollars in accordance with Canadian GAAP. We have included a reconciliation of the significant differences between Canadian GAAP and U.S. GAAP of our net loss, balance sheet and cash flow items in note 23 to our audited consolidated financial statements. For your convenience, the following table presents certain of our operations data in both U.S. dollars and Canadian dollars. The U.S. dollar amounts have been converted from Canadian dollars at the rate of U.S.$0.8254 per $1.00 Canadian, which is the average exchange rate for the year ended December 31, 2005, as quoted by the Bank of Canada. In addition, note 1 to our audited consolidated financial statements provides a description of our business and details on our going concern uncertainty. Our historical results from any prior period are not necessarily indicative of results to be expected for any future period.
| Year Ended December 31, | |||||||||||||
| 2005 | 2005 | 2004 | |||||||||||
| US$ | C$ | C$ | |||||||||||
| (in thousands, except share and per share data) | |||||||||||||
Consolidated Statement of Operations Data: | |||||||||||||
Canadian GAAP | |||||||||||||
| Revenues: | |||||||||||||
| Product sales | US$ | 1,047 | $ | 1,269 | $ | — | |||||||
| Cost of goods sold | 626 | 758 | — | ||||||||||
| Gross profit on product sales | 421 | 511 | — | ||||||||||
| Licensing | 1,575 | 1,908 | 106 | ||||||||||
| Research and development contracts | 50 | 61 | 1,288 | ||||||||||
| 2,046 | 2,480 | 1,394 | |||||||||||
| Expenses: | |||||||||||||
| Research and development expenses, net | 16,292 | 19,738 | 15,214 | ||||||||||
| Selling, general and administrative expenses | 10,060 | 12,188 | 9,878 | ||||||||||
| Financial expenses | 1,599 | 1,937 | 864 | ||||||||||
| Depreciation and amortization | 1,373 | 1,664 | 1,662 | ||||||||||
| Interest income | (460 | ) | (557 | ) | (538 | ) | |||||||
| Foreign exchange gain | (293 | ) | (355 | ) | (57 | ) | |||||||
| Expenses related to an incomplete financing initiative | — | — | 1,509 | ||||||||||
| 28,571 | 34,615 | 28,532 | |||||||||||
| Loss before income taxes | (26,525 | ) | (32,135 | ) | (27,138 | ) | |||||||
| Income taxes: | |||||||||||||
| Current | 990 | 1,199 | 41 | ||||||||||
| Net loss for the period | US$ | (27,515 | ) | $ | (33,334 | ) | $ | (27,179 | ) | ||||
| Net loss per share — basic and diluted | US$ | (0.64 | ) | $ | (0.78 | ) | $ | (0.68 | ) | ||||
| Weighted average number of shares outstanding | 42,922,741 | 42,922,741 | 39,954,488 | ||||||||||
U.S. GAAP | |||||||||||||
| Net loss for the period | US$ | (27,541 | ) | $ | (33,367 | ) | (27,441 | ) | |||||
| Net loss per share — basic and diluted | US$ | (0.64 | ) | $ | (0.78 | ) | $ | (0.69 | ) | ||||
| Weighted average number of shares outstanding | 42,922,741 | 42,922,741 | 39,954,488 | ||||||||||
5
The as adjusted consolidated balance sheet data below reflects our sale of 10,000,000 common shares in this offering at an assumed public offering price of US$7.31 per share and our receipt of estimated net proceeds of US$67.2 million, after deducting estimated underwriting discounts and commissions and offering expenses payable by us. For your convenience, the following table is presented in both U.S. dollars and Canadian dollars. The U.S. dollar amounts have been converted from Canadian dollars at the rate of US$0.8577 per $1.00 Canadian, which is the noon exchange rate for December 31, 2005, as quoted by the Bank of Canada, except for the estimated net proceeds of the offering which we have assumed will be received in U.S. dollars, and will be converted to Canadian dollars at the noon exchange rate of US$0.8681 per $1.00 Canadian as quoted by the Bank of Canada on April 13, 2006.
| As of December 31, 2005 | |||||||||||||||||
| Actual | As Adjusted | Actual | As Adjusted | ||||||||||||||
| US$ | US$ | C$ | C$ | ||||||||||||||
| (unaudited) | (unaudited) | ||||||||||||||||
| (in thousands) | |||||||||||||||||
Consolidated Balance Sheet Data: | |||||||||||||||||
Canadian GAAP | |||||||||||||||||
| Cash, cash equivalents and short-term investments | US$ | 29,928 | US$ | 97,142 | $ | 34,893 | $ | 112,320 | |||||||||
| Working capital | 14,361 | 81,575 | 16,743 | 94,170 | |||||||||||||
| Total assets | 46,755 | 113,969 | 54,512 | 131,939 | |||||||||||||
| Obligations under capital leases — long-term portion | 5,009 | 5,009 | 5,840 | 5,840 | |||||||||||||
| Long-term debt | 9,607 | 9,607 | 11,201 | 11,201 | |||||||||||||
| Total liabilities | 48,987 | 48,987 | 57,115 | 57,115 | |||||||||||||
| Total shareholders’ equity (deficiency) | (2,233 | ) | 64,981 | (2,603 | ) | 74,824 | |||||||||||
U.S. GAAP | |||||||||||||||||
| Total assets | 44,980 | 112,194 | 52,443 | 129,870 | |||||||||||||
| Total shareholders’ equity (deficiency) | (4,007 | ) | 63,207 | (4,672 | ) | 72,755 | |||||||||||
6
RISK FACTORS
Investing in our common shares involves a high degree of risk. You should carefully consider the following risks as well as the other information contained in this prospectus before investing in our common shares. If any of the following risks occur, our business, results of operations or financial condition could be materially adversely affected. In that case, the trading price of our common shares could decline, and you may lose all or part of your investment. The risks set out below are not the only risks we face. You should also refer to the other information set forth in this prospectus, including our consolidated financial statements and the related notes.
Risks Related to Our Business
| We have not generated significant revenues to date and expect to continue to experience losses for at least the next year. |
For the year ended December 31, 2005, we had a net loss of $33.3 million. As at December 31, 2005, we had an accumulated deficit of approximately $144.6 million. We have incurred losses in each year of our operations and we expect to continue to incur operating losses for at least the next year.
We expect our operating expenses to continue to be significant. The process of developing our product candidates requires significant laboratory testing, manufacturing development, validation and clinical trials to support regulatory approvals. In addition, we must establish sales, marketing and manufacturing capabilities, either through internal hiring or through contractual relationships with others, to commercialize our product candidates, which we expect will require us to increase our selling, general and administrative expenses over the next several years. As a result, we will need to generate significant revenues to achieve profitability.
We believe our future profitability will depend on a number of factors, including:
| • | sales of our once-daily tramadol product; | |
| • | payments from Purdue under our U.S. collaboration with Purdue, including milestone payments; | |
| • | our selling, general and administrative expenses; and | |
| • | our development of other products. |
We may never achieve profitability. Even if we achieve profitability, we may not be able to maintain profitability on an annual or quarterly basis. Our failure to become and remain profitable would depress the market price of our common shares and could impair our ability to raise capital, expand our business, expand our product pipeline or continue our operations.
| We depend heavily on the success of our lead product candidate, our once-daily tramadol, and if our NDA for our once-daily tramadol product is not approved by the FDA on a timely basis or at all, it would have a material adverse effect on our business. |
We have invested a significant portion of our financial resources in the development of our lead product candidate, our once-daily tramadol. We anticipate that in the near term our ability to generate significant revenues will depend primarily on the successful development and commercialization of this product, especially in the United States. Although we have several other products under development, they are at an earlier stage of development.
Regulatory approval of our once-daily tramadol product has been obtained in certain European countries but not the United States. In November 2005, we submitted an NDA for our once-daily tramadol product to the FDA under Section 505(b)(2) of the U.S. Federal Food, Drug, and Cosmetic Act. The NDA was accepted for review and filed by the FDA in January 2006.
7
The FDA has confirmed to us a PDUFA action date of September 28, 2006, which is the date by which we expect to receive an action letter from the FDA on our NDA. However, the FDA may extend the PDUFA date or fail to provide us with an action letter by such date. We have agreed to submit to the FDA the data from our Phase III clinical trial, MDT3-005, of our once-daily tramadol product. The FDA has stated that it will accept and review the data if we submit the data to the FDA on or prior to June 28, 2006. If we do not submit the data to the FDA by June 28, 2006, the FDA could extend the PDUFA date. Other responses to additional requests for information from the FDA may also delay the action date. If the NDA for our once-daily tramadol product is not approved on a timely basis the milestone payment due to us from Purdue upon NDA approval could be reduced and our business could be adversely affected.
The FDA could take action on our NDA by the PDUFA date but not approve the NDA or require us to conduct additional clinical trials. In addition, Section 505(b)(2) permits us to rely for approval on studies that we have not conducted and for which we have not obtained a right of reference. In the case of our once-daily tramadol product, we relied in part on the FDA’s previous findings of safety for Ultram, the immediate release brand of tramadol sold by Ortho-McNeil, Inc., or OMI.
| Our products, including our once-daily tramadol product, if approved for marketing, may fail to achieve market acceptance. |
Any products we successfully develop, including our once-daily tramadol product, if approved for marketing, may not achieve market acceptance and, as a result, may not generate significant revenues. Market acceptance of our products by hospitals, physicians or patients will depend on a number of factors, including:
| • | our ability to provide acceptable evidence of safety and efficacy; | |
| • | relative convenience and ease of administration; | |
| • | the prevalence and severity of any adverse side effects; | |
| • | pricing and cost effectiveness, which may be subject to regulatory control; | |
| • | availability, cost and effectiveness of alternative treatments; | |
| • | availability and cost of competing versions of our products, including OMI’s once-daily tramadol product which was launched in the United States in February 2006, once-daily and twice-daily tramadol products being sold in Europe and generic immediate release tramadol products that are available worldwide; | |
| • | effectiveness of our collaborators’ and distributors’ sales and marketing strategy; and | |
| • | our ability to obtain sufficient third-party insurance coverage or reimbursement. |
If any product that we commercialize does not provide a treatment regimen that is as beneficial as the current standard of care or otherwise does not provide patient benefit, that product likely will not achieve market acceptance. This may result in a shortfall in revenues and an inability to achieve or maintain profitability.
| Competition in the pharmaceutical industry is intense, and if we fail to compete effectively, our business, financial condition and results of operations will suffer. |
We are engaged in a business characterized by extensive research efforts, rapid technological developments and intense competition. Any product that we develop may not compete successfully. Given that our business is reformulating existing drugs, our product candidates will face competition from generic and branded formulations of the existing drugs we reformulate. For instance, our once-daily tramadol product faces competition in the United States from OMI, which launched its own once-daily formulation of tramadol under license from Biovail Corporation, or Biovail, in February 2006 and may benefit from its first to market position. We may also face competition from Cipher Pharmaceuticals Ltd., or Cipher, which has announced its intention to submit an NDA to the FDA in 2006 for its once-daily formulation of tramadol. In Europe, our once-daily tramadol product faces competition from the multiple tramadol formulations that are widely
8
available, including extended release tramadol formulations for once-daily dosing that have been and are marketed in certain European countries, including the United Kingdom, Belgium, France, Germany and Spain. Similarly, generic immediate release tramadol products are available worldwide. Our once-daily tramadol product will also face competition from non-narcotic analgesics, other opioids and NSAIDs, including COX-2 inhibitors, which are used for the treatment of moderate to severe pain. We expect that successful competition will depend, among other things, on product efficacy, safety, reliability, availability, timing and scope of regulatory approval, costs of supply, marketing and sales capabilities, reimbursement, patent positions and price.
We expect competition to increase as technological advances are made and commercial applications broaden. In addition, research and development activities by competitors may render our products obsolete or uneconomical. In commercializing our initial product candidates, and any additional product candidate we develop using our drug delivery technologies, we will face substantial competition from large pharmaceutical, biotechnology and other companies, universities and research institutions. To the extent that we develop or market products incorporating drugs that are off-patent, or are being developed by multiple companies, we will face competition from other companies developing and marketing similar products.
Relative to us, we believe that most of our competitors have substantially greater capital resources, research and development staff, facilities and experience in conducting clinical trials and obtaining regulatory approvals, as well as in manufacturing and marketing pharmaceutical products. Many of our competitors may achieve product commercialization or patent protection earlier than we will. Finally, our competitors may use different technologies for, or approaches to, the development of products similar to the products we are seeking to develop.
| We may require additional funding and may not be able to raise additional capital in which case we will be unable to complete planned clinical trials, obtain regulatory approvals or commercialize our product candidates. |
As of December 31, 2005, we had cash, cash equivalents and short-term investments of $34.9 million. Unless this offering is completed, we expect that our committed sources of funds and our cash and cash equivalents and short-term investments on hand will not be sufficient to meet our committed cash obligations and expected level of expenses through the fourth quarter of 2006. As a result, there is significant uncertainty about our ability to continue as a going concern. We will need to successfully complete this offering or successfully execute alternative financing and operational plans to help alleviate this uncertainty.
Even if we complete this offering, we will require substantial future capital in order to continue to conduct the research and development, clinical and regulatory activities necessary to bring our product candidates to market and to establish commercial manufacturing, marketing and sales capabilities. We believe that the net proceeds from this offering, together with our existing cash and cash equivalents and short-term investments and anticipated revenues from our once-daily tramadol product, will be sufficient to enable us to fund our operating expenses and capital expenditures for at least the next two years. Our future capital requirements will depend on many factors, including the following factors:
| • | time and costs involved in performing clinical trials and obtaining regulatory approvals; | |
| • | costs of manufacturing and supply; | |
| • | costs of establishing or contracting for sales and marketing capabilities; | |
| • | progress of clinical and formulation development; | |
| • | establishment of agreements with collaborators and distributors and activities required for product commercialization; | |
| • | number of product candidates we pursue; | |
| • | revenues from sales of our once-daily tramadol product or from our collaborators and distributors in connection with our once-daily tramadol product; |
9
| • | costs involved in filing and prosecuting patent applications and enforcing or defending patent claims; and | |
| • | existence, availability and the rate of refundable tax credits attributable to scientific research and development expenditures carried out in Québec, Canada. |
We intend to seek additional funding from a variety of sources, including additional public or private financings and collaborative relationships, to fund all or a part of particular programs. Our ability to obtain funding will depend in part upon prevailing capital market conditions and our business performance. Any additional equity financings may significantly dilute existing shareholders.
If we cannot obtain adequate funds or funds on reasonable terms, we may need to:
| • | terminate or delay clinical trials for one or more of our product candidates; | |
| • | delay our establishment of sales or marketing capabilities; | |
| • | curtail significant product development programs that are designed to identify new product candidates; and | |
| • | sell or assign rights to our technologies or product candidates. |
| We may not achieve our projected development goals in the time frames we announce and expect. |
We set goals for and make public statements regarding our expected timing of meeting the objectives material to our success, such as the commencement and completion of clinical trials, anticipated regulatory approval and product launch dates. The actual timing of these forward looking events can vary dramatically due to factors such as delays or failures in our clinical trials, the need to develop additional data required by regulators as a condition of approval, the uncertainties inherent in the regulatory approval process and delays in achieving manufacturing or marketing arrangements necessary to commercialize our product candidates.
| If our clinical trials do not produce successful results, we will not be able to commercialize our product candidates. |
No product can receive regulatory approval unless human clinical trials show safety and efficacy for each target indication or bioequivalence to an approved drug in accordance with applicable regulatory requirements and standards. Success in pre-clinical studies and early clinical trials does not ensure that later clinical trials will be successful and interim results from clinical trials do not necessarily predict the final results of such trials. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks from their inability to satisfy these requirements in late-stage clinical trials, even after achieving promising results in early-stage clinical studies. Data obtained from pre-clinical and clinical activities are subject to varying interpretations by regulatory agencies that could delay, limit or prevent regulatory agency approval. Clinical trials for our once-daily tramadol product may not successfully address the concerns of regulatory authorities. For instance, the results of the clinical trials for our once-daily tramadol product may not be interpreted by the FDA as establishing the safety and efficacy of our once-daily tramadol product sufficiently to obtain marketing approval.
The timing and success of clinical trials depend on various factors, including:
| • | sufficient patient enrollment which may be affected by the incidence of the disease studied, the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites, the eligibility criteria for a patient to participate in the study and the rate of patient drop-out; | |
| • | regulatory agency policies regarding requirements for approval of a drug; | |
| • | our capacity to produce or have produced clinical trial material in sufficient quantities and of sufficient quality to meet the schedule for our planned clinical trials; and |
10
| • | performance by third parties, such as clinical research organizations, on whom we rely to support our clinical trials. |
A failure of one or more of our clinical trials can occur at any stage of testing. If clinical trials do not show any potential product to be safe and effective, if we are required to conduct additional clinical trials or other testing of our products in development beyond those that we currently contemplate or if we are unable to successfully complete our clinical trials or other testing, we may:
| • | be delayed in obtaining marketing approval for our products; | |
| • | not be able to obtain marketing approval for our products; or | |
| • | obtain approval with limitations or restrictions, such as limitations on the indications for use. |
Our product development costs may also increase if we experience delays in testing or approvals or have to conduct additional testing. In addition, significant clinical trial and regulatory delays also could allow our competitors to bring products to market before we do and impair our ability to commercialize our products.
| If we fail to obtain regulatory approvals for our product candidates under development, we will not be able to commercialize our products. |
We must receive regulatory approval of a product candidate before it can be commercialized. The research, development, testing and manufacturing of any product candidate we develop independently or in collaboration with third parties, as well as the distribution, marketing, promotion, advertising, and record keeping of such product candidate, are regulated by numerous federal, state, provincial and local governmental authorities, principally the FDA in the United States, and other similar agencies in other countries. The approval procedures in the United States vary depending on such factors as the novelty of the drug and its intended use and also vary among countries. Changes in regulatory approval policies during the development period, changes in or the enactment of additional statutes or regulations or changes in regulatory review for each submitted product application, may cause delays in the approval or rejection of an application. The FDA has substantial discretion in the approval process and may refuse to accept any application or may decide that our data are insufficient for approval and require additional preclinical, clinical or other studies. The development and regulatory approval process in each jurisdiction takes many years, requires the expenditure of substantial resources, is uncertain and subject to delays. In addition, approval by a regulatory authority of one country does not ensure the approval by regulatory authorities of other countries.
Failure to obtain regulatory approval, any delay or setback in obtaining regulatory approval, any limitation on drug use required as a condition of approval could:
| • | adversely affect our ability to market any drugs we develop independently or with collaborators; | |
| • | affect our ability to negotiate collaboration or distribution agreements or continue current agreements with certain collaborators or distributors; | |
| • | impose additional costs and diminish any competitive advantages that we may attain; or | |
| • | adversely affect our ability to generate product sales and/or royalties based on these sales. |
11
| Even if we obtain marketing approval, there may be limits on the approval and our products will be subject to ongoing regulatory review and regulatory requirements. If we fail to comply with these requirements, we could lose marketing approval and sales of any approved commercial products could be suspended. |
Even if regulatory approval of a product is granted, the approval may be subject to limitations on the uses for which the product may be marketed or to conditions of approval, which could affect the marketability of the product. In addition, the terms of approval may contain requirements for costly post-market follow-up studies or post-market surveillance to monitor the safety or efficacy of the product, which could reduce our revenues, increase our expenses or render the approved product not commercially viable. For example, the FDA has indicated that if our once-daily tramadol product is approved, it would likely require us to implement a risk management program in order to monitor the potential abuse, misuse, or diversion of our product.
Also, an NDA is required to contain adequate data to assess the safety and efficacy of the drug for the claimed indication in all relevant pediatric subpopulations. We have requested from the FDA a waiver of this requirement for children under 12 years of age, and a deferral for children 12 years and older. If the FDA grants our waiver and deferral requests, we expect to satisfy the FDA’s pediatric pharmacokinetic requirements by conducting studies on a post-approval basis in adolescents who are 12 to 16 years old.
If we receive regulatory approval to market a particular product, we will be subject to extensive ongoing regulatory requirements, including requirements relating to registration, manufacturing, labeling, advertising, promotion, adverse event reporting, packaging, distribution, storage, and record keeping. In addition, the manufacturing facilities for such product will be subject to continual review and periodic inspections by regulatory authorities. If we fail to comply with the regulatory requirements of the FDA and other applicable United States and foreign regulatory authorities, or if previously unknown problems with any approved commercial products, manufacturers or manufacturing processes are discovered, we could be subject to administrative or judicially imposed sanctions or other setbacks, including:
| • | restrictions on the products, manufacturers or manufacturing processes; | |
| • | warning letters; | |
| • | civil or criminal penalties; | |
| • | fines; | |
| • | injunctions; | |
| • | product seizures or detentions; | |
| • | import or export bans or restrictions; | |
| • | product recalls and related publicity requirements; | |
| • | suspension or withdrawal of regulatory approvals; | |
| • | total or partial suspension of production; and | |
| • | refusal to approve pending applications for marketing approval of new products or supplements to approved applications. |
In addition, as clinical experience with a drug expands after approval because it is typically used by a greater number and more diverse group of patients after approval than during clinical trials, side effects and other problems may be observed after approval that were not seen or anticipated during pre-approval clinical trials or other studies. Any adverse effect observed after the approval and marketing of a product candidate could result in limitations on the use of or withdrawal of marketing approval for the product.
Also, the United States or other countries’ statutes, regulations, or policies may change and additional statutes or government regulations or policies may be enacted which could prevent, or impose additional restrictions on the continued marketing of drug products. We cannot predict the likelihood, nature
12
or extent of adverse governmental regulation that might arise from future legislative or administrative action, either in the United States or abroad. For example, the Drug Enforcement Administration, or the DEA, currently does not regulate tramadol as a controlled substance. However, tramadol has been under review by the DEA since July 2004 because of the potential for abuse and because of reports received of actual abuse of tramadol products. As a result, the DEA may determine that tramadol should be regulated as a controlled substance in the future. DEA regulation would impose significant additional requirements on our business. We may be slow to adapt, or we may never adapt, to changes in existing requirements or adoption of new requirements or policies. If we are slow to adapt, or are unable to adapt, to changes in existing regulatory requirements or adoption of new regulatory requirements or policies, we may lose marketing approval for our products when and if any of them are approved, resulting in decreased revenue from milestones, product sales or royalties.
| Claims by other companies that we infringe their intellectual property rights may result in liability for damages or stop our development and commercialization efforts, including with respect to our once-daily tramadol product. |
We are aware of issued United States, Canadian and European patents that contain claims that might be infringed by our once-daily tramadol product. If our once-daily tramadol product or our other product candidates infringe or were claimed to infringe a valid claim of a third-party patent, including these patents, we may choose or may be required to obtain a license under such patent or seek a court judgment that such patent claims are invalid. Any patent litigation in which we are involved, whether brought against us or brought by us to invalidate a patent would be costly and could divert our technical and management personnel from their normal responsibilities, and we may not have sufficient financial resources to conduct such litigation. Any adverse outcome of such litigation or settlement of such a dispute could subject us to significant liabilities and could require us to stop developing our product candidates, stop selling our products or enter into royalty or licensing agreements which may or may not be available on terms acceptable to us, if at all. Even if we were able to obtain a license, the rights may be non-exclusive, which could result in our competitors gaining access to the same intellectual property. If we do not obtain any required licenses or sublicenses, we could encounter delays in product introductions while we attempt to design around these patents, or could find that the development, manufacture or sale of products requiring these licenses is foreclosed to us.
If we obtain a license to patents from a third party, as we have obtained under our agreement with Purdue, we will need to comply with the terms of the license or risk termination of the license and the rights to practice the technology covered by such license.
A number of patent applications may be currently pending or may be filed by third parties in the future for technologies generally related to our business, including patent applications that remain confidential after filing. Due to these factors and the inherent difficulty in conducting patent searches and interpreting their results, there can be no guarantee that we will not violate third-party patent rights that we have not yet identified.
Many of our competitors have obtained or may be in the process of obtaining patents covering products and processes generally related to some of our products and processes, and they may assert their rights in these patents against us. Moreover, these competitors may have sought or may seek additional patents that could cover aspects of our technology. As a result, there is a greater likelihood of a patent dispute than would be expected if our competitors were pursuing unrelated products or technologies.
In addition, a competitor may claim misappropriation of a trade secret by an employee hired from that competitor. Any patent infringement or trade secret misappropriation action could cause us to incur substantial costs defending the lawsuit and could distract our management from our business, even if the allegations of infringement or misappropriation are unwarranted. The defense of multiple claims could have a disproportionately greater impact. Furthermore, a party making this type of claim could secure a judgment that requires us to pay substantial damages. A judgment could also include an injunction or other court order that could prevent us from making, using, selling, offering for sale or importing our products or prevent our
13
customers from using our products. If a court determined or if we independently discovered, that any of our products or manufacturing processes violated third-party proprietary rights, we may not be able to reengineer the product or processes to avoid those rights, or to obtain a license under those rights on commercially reasonable terms, if at all. As a result, we could be prevented from commercializing our product candidates.
| We may become involved in lawsuits to protect or enforce our intellectual property rights that would be expensive and time consuming. |
In order to protect or enforce our intellectual property rights, we may initiate litigation against third parties. In addition, we may become subject to interference or opposition proceedings conducted in patent and trademark offices to determine the priority of inventions. The defense of intellectual property rights, including patent rights, through lawsuits, interference or opposition proceedings, and other legal and administrative proceedings, would be costly and could divert our technical and management personnel from their normal responsibilities and we may not have sufficient financial or other resources to conduct such defence. Any adverse outcome of such litigation, proceedings or settlement of such dispute may require us to enter into royalty or licensing agreements which may or may not be available on terms acceptable to us, if at all, and could put one or more of our patents at risk of being invalidated or interpreted narrowly and could put our patent applications at risk of not issuing.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. For example, confidential information may be inadvertently disclosed in the form of documents or testimony in connection with discovery requests, depositions or trial testimony. This disclosure would provide our competitors with access to our proprietary information and may harm our competitive position.
| Rapid technological change could make our products or drug delivery technologies obsolete. |
Pharmaceutical technologies are subject to rapid and significant technological change. We expect our competitors will develop new technologies and products that may render our products and drug delivery technologies uncompetitive or obsolete. The products and drug delivery technologies of our competitors may be more effective than the products and drug delivery technologies developed by us. As a result, our products may become obsolete before we recover expenses incurred in connection with their development or realize revenues from any commercialized product.
| We have received regulatory approval for only one product that uses any of our drug delivery technologies. |
Our drug delivery technologies can be quite complex, with many different components. The development required to take a technology from its earliest stages to its incorporation in a product that is sold commercially can take many years and cost a substantial amount of money. Significant technical challenges are common as products incorporating our technologies progress through development, particularly in the first product candidate incorporating a new technology. No product employing our Contramid technology has received regulatory approval, other than our once-daily tramadol product, which has been approved in certain European countries. We have not received regulatory approval in any country for any product that uses our polymeric nano-delivery systems technologies. In addition, any particular technology may not perform in the same manner when used with different therapeutic agents and therefore these technologies may not prove to be as useful or valuable as originally thought, resulting in additional development work. If our efforts do not repeatedly lead to successful development of product candidates, we may not be able to grow our pipeline or to enter into agreements with distributors or collaborators who are willing to distribute or develop our product candidates. Delays or unanticipated increases in costs of development at any stage, or failure to solve a technical challenge, could adversely affect our operating results.
14
| If we are unable to protect our intellectual property rights, our competitors may develop and market products with similar features to our products which may reduce demand for our products and inhibit their effective commercialization. |
We will be able to protect our intellectual property rights from unauthorized use by third parties only to the extent that our intellectual property rights are covered by valid and enforceable patents or are effectively maintained as trade secrets. We try to protect our intellectual property position by filing patent applications related to our proprietary technology, inventions and improvements that are important to the development of our business. Because the patent position of pharmaceutical companies involves complex legal and factual questions, the issuance, scope and enforceability of patents cannot be predicted with certainty. Patents, if issued, may be challenged, invalidated or circumvented. If our patents are invalidated, we will lose the ability to exclude others from making, using or selling the invention claimed. Moreover, an issued patent does not guarantee us the right to practice the patented technology or commercialize a product using that technology. Third parties may have blocking patents that could be used to prevent us from developing our product candidates, selling our products or commercializing our patented technology. Thus, any patents that we own or license from others may not allow us to exploit the rights conferred by our intellectual property protection. Our future and pending patent applications may not result in patents being issued. Even if issued, they may not be issued with claims sufficiently broad to protect our products and technologies or may not provide us with a competitive advantage against competitors with similar products or technologies. Furthermore, others may independently develop products or technologies similar to those that we have developed or discover our trade secrets. In addition, the laws of many countries do not protect our intellectual property rights to the same extent as the laws of the United States, and those countries may also lack adequate rules and procedures for defending our intellectual property rights.
We also rely on trade secrets, know-how and technology, which are not protected by patents, to maintain our competitive position. We try to protect this information by entering into confidentiality undertakings with parties that have access to it, such as our corporate and prospective distributors, collaborators, employees and consultants. Any of these parties may breach the undertakings and disclose our confidential information or our competitors might learn of the information in some other way. Enforcing a claim that a third party illegally obtained and is using our trade secrets is expensive and time consuming and the outcome is unpredictable. In addition, courts outside the United States may be less willing to or may not protect trade secrets. If any trade secret, know-how or other technology not protected by a patent were to be disclosed to or independently developed by a competitor, our competitive position would be harmed.
| Disputes may arise regarding the ownership or inventorship of our products and technologies. |
From time to time we may become involved in disputes relating to the ownership or inventorship of our products and technologies. We have not yet completed the filing of the assignments of patents from the original inventors to us for certain of our patents and we have not yet obtained executed assignments of patents from all of the inventors for this subset of our patents. If we are unsuccessful in obtaining these assignments of patents or are otherwise not able to establish our ownership of the invention covered by the patents, we may face additional expense in perfecting our title to these patents and our business may be adversely affected.
| In the past we have entered into agreements that may require us to make royalty payments, which would adversely affect our operating results and financial condition. |
We have entered into purchase and development agreements that may obligate us to make royalty payments. In 1994, concurrently with the purchase of a controlled-release technology from Université de Montréal and Université du Québec à Montréal, we acquired a right of first refusal with respect to any improvements and additions to the purchased technology for which we agreed to pay royalties of 4% on net revenues that we receive from the commercialization of the technology purchased in 1994. On February 7, 2005, Université de Montréal and Université du Québec à Montréal served us with a motion to institute legal proceedings in the Superior Court of Québec. The motion seeks payment of royalties allegedly owed by us with respect to the commercialization of the purchased technology and any improvements and additions
15
thereto, and its use in various of our product candidates, including our once-daily tramadol product. It is our position that we are not commercializing that particular technology that had been purchased in 1994 or any improvements or additions thereto and that we do not use it in our once-daily tramadol product or any of our product candidates. Consequently, we consider that no amounts are owing and we are vigorously contesting the motion. This matter is in the examination for discovery phase of the proceedings.
However, despite our views regarding the merits of legal proceedings, in each case, if a court were to determine that we are obligated to make royalty or other payments, our operating results and financial condition may be adversely affected.
| We currently have a single source of supply for our Contramid cross-linked high amylose starch. |
We have an exclusive supply agreement with Cargill Inc., or Cargill, to purchase from Cargill all of our requirements for cross-linked high amylose starch comprising our bulk Contramid. This agreement expires on the later of November 4, 2018 or the expiry date of the last to expire patent covering the Contramid technology. It may be difficult to find another manufacturer if Cargill is unable to supply us with a sufficient quantity of our Contramid cross-linked high amylose starch or if we are forced for any reason to find another manufacturer. Cargill may terminate the manufacturing agreement if we fail to make payments that are due or become insolvent. If Cargill decides to cease manufacturing Contramid, Cargill may also terminate the agreement, subject to Cargill using reasonable efforts to notify us at least two years in advance of such termination We believe that it would take any other company at least a year to develop and certify the necessary processes to manufacture our Contramid technology on terms acceptable to us. There may not be third-party manufacturers that are able to meet our volume or quality requirements at a price that is as favorable to us as the price that we currently have with Cargill. Any financial, operational, production or quality assurance difficulties experienced by these third-party manufacturers that result in a reduction or interruption in supply to us could significantly delay the development and manufacture of our products.
| If third-party manufacturers of our products fail to devote sufficient time and resources to our concerns, or if their performance is substandard, the commercialization of our products could be delayed or prevented, and this may result in higher costs or deprive us of potential product revenues. |
We have limited manufacturing facilities and have not previously commercially manufactured drugs. Although we manufacture clinical trial supplies in-house, we rely on third parties for commercial scale manufacturing. We may also rely on third parties for the manufacturing of certain clinical trial materials. Our reliance on contract manufacturers will expose us to the following risks, any of which could delay or prevent the commercialization of our products, result in higher costs, or deprive us of potential product revenues:
| • | Contract manufacturers can encounter difficulties in achieving volume production, quality control and quality assurance, as well as with shortages of qualified personnel. Accordingly, a manufacturer might not be able to manufacture sufficient quantities to meet our clinical trial needs or to commercialize our products. | |
| • | Contract manufacturers are required to undergo a satisfactory current good manufacturing practices, or GMPs, inspection prior to regulatory approval and are obliged to operate in accordance with FDA, European and other nationally mandated GMPs, which govern manufacturing processes, stability testing, record keeping and quality standards. Any failure of these contract manufacturers to establish and follow GMPs and to document their adherence to such practices may lead to significant delays in the availability of material for clinical studies, may delay or prevent filing or approval of marketing applications for our products or result in sanctions being imposed on us. | |
| • | For each of our current product candidates we will initially rely on a limited number of contract manufacturers. Changing these or future manufacturers may be difficult and the number of potential manufacturers is limited. Changing manufacturers generally requires re-validation of the manufacturing processes and procedures in accordance with FDA, European and other nationally mandated GMPs and may require prior regulatory approval. It |
16
| may be difficult or impossible for us to quickly find replacement manufacturers on acceptable terms, if at all. Such re-validation may be costly and time-consuming and we could suffer delays in advancing our product candidates in clinical trials or in supplying the commercial market with our products. | ||
| • | With respect to our once-daily tramadol product, our ability to reach full commercial scale manufacturing depends upon the ability of our commercial scale contract manufacturer to be approved under such GMPs. Reaching full commercial scale has a direct impact on our overall costs of goods, which, in turn, directly affects our operating margins. Any delay in obtaining GMP approval beyond the time we anticipate may have a negative impact on our operating margins and other financial results, as well as our ability to adequately supply the market with our product. | |
| • | Our contract manufacturers may not perform as agreed or may not remain in the contract manufacturing business for the time required to produce, store and distribute our products successfully. | |
| • | Our contract manufacturers may terminate or not renew our agreements based on their own priorities at a time that is costly or inconvenient for us. |
Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA, and corresponding state and foreign agencies, including European agencies and their designees, to ensure strict compliance with GMPs and other government regulations. While we are obligated to audit the performance of third-party contractors, we do not have complete control over our third-party manufacturers’ compliance with these regulations and standards. Failure by either our third-party manufacturers or by us to comply with applicable regulations could result in sanctions being imposed on us, including fines, injunctions, civil penalties, failure of the government to grant review of submissions or market approval of drugs, delays, suspension or withdrawal of approvals, product seizures or recalls, operating restrictions, facility closures and criminal prosecutions, any of which could harm our business.
Under our collaboration and marketing and distribution arrangements, we have committed to supply these third parties with product. In the event that we are unable to fulfill such obligations as a result of a failure of our contract manufacturers, we may be in breach of our obligations under those arrangements.
| We rely on third parties to conduct, supervise and monitor our clinical trials, and those third parties may not perform satisfactorily. |
We rely on third parties such as contract research organizations, medical institutions and clinical investigators to enroll qualified patients and conduct, supervise and monitor our clinical trials. Our reliance on these third parties for clinical development activities reduces our control over these activities. Our reliance on these third parties, however, does not relieve us of our regulatory responsibilities, including ensuring that our clinical trials are conducted in accordance with good clinical practice regulations, or GCPs, and the investigational plan and protocols contained in an investigational new drug application, or IND, or comparable foreign submission. Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. In addition, they may not complete activities on schedule, or may not conduct our preclinical studies or clinical trials in accordance with regulatory requirements or our trial design. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, our efforts to obtain regulatory approvals for, and commercialize, our product candidates may be delayed or prevented.
| We have no experience in selling, marketing or distributing our products, and we have no internal capability to do so yet. |
If we receive regulatory approval to commence commercial sales of any of our product candidates, we will then be required to sell, market and distribute our product candidates. As we currently do not have the capabilities to market our products directly, we must develop our own marketing and sales force with technical expertise and supporting distribution capability. For example, in our licensing and distribution
17
agreement with Purdue, we retained the right to co-promote our once-daily tramadol product to certain medical specialties that focus on the central nervous system and pain. We anticipate building both internal sales and marketing management infrastructure, including the hiring of employees, as well as a sales force to co-promote our once-daily tramadol product should the product receive regulatory approval in the United States. However, we may not successfully establish sales and distribution capabilities on our own.
| Our agreements relating to the development and distribution of products may expose us to |
a number of risks.
We currently have nine collaboration or distribution agreements relating to the marketing and distribution of our once-daily tramadol product in Europe, the United States, Latin America and the Caribbean. We rely on these agreements because we do not have our own capabilities. We intend to secure additional agreements relating to the marketing and distribution of our once-daily tramadol product and any other product for which we may receive regulatory approval. If we are unable to reach agreements with suitable collaborators and distributors, we may fail to meet our business objectives for the affected product or program. We face, and will continue to face significant competition in seeking appropriate collaborators and distributors. Moreover, collaboration and distribution arrangements are complex and time consuming to negotiate, document and implement. We may not be successful in our efforts to establish and implement collaboration or marketing and distribution arrangements upon satisfactory terms or at all. Reliance on these agreements exposes us to a number of risks, including the following:
| • | our collaborators and distributors may not devote sufficient resources to our products or product candidates; | |
| • | disputes may arise with respect to payments that we believe are due under such distribution and collaboration agreements; | |
| • | we may face unwillingness on the part of collaborators and distributors to keep us informed regarding the progress of its development, commercialization or marketing activities, or to permit public disclosure of these activities; | |
| • | our collaborators and distributors may terminate the relationship; | |
| • | disputes may arise in the future with respect to the ownership of rights to technology developed with collaborators; | |
| • | disagreements with collaborators and distributors could delay or terminate the research and development, regulatory approval, commercialization or marketing of product candidates, or result in litigation or arbitration; | |
| • | our collaborators may elect to pursue the development of any additional product candidates and pursue technologies or products either on their own or in collaboration with other parties, including our competitors which technologies or products may be competitive with ours; | |
| • | our collaborators and distributors may pursue higher priority programs or change the focus of their programs, which could affect the collaborator’s and distributor’s commitment to the United States; and | |
| • | our collaborators and distributors may develop or distribute products that compete with our products. |
The occurrence of any of these or other events may delay product development or impair commercialization of our products.
| If we are unable to retain key personnel and hire additional qualified personnel, we may not be able to successfully achieve our goals. |
We depend on the principal members of our scientific and management staff including our President and Chief Executive Officer, Mr. James Howard-Tripp. The loss of any of these individuals’ services might
18
significantly delay or prevent our achievement of research, development or business objectives. Our employment with Mr. Howard-Tripp is for an indefinite term and can be terminated by him at any time. Except for Mr. Howard-Tripp, we do not maintain key person life insurance on any of these individuals.
Our success depends, in large part, on our ability to continue to attract and retain qualified scientific and management personnel. We face intense competition for such personnel and consultants. We may not be able to attract and retain qualified management and scientific personnel in the future. Also, we must provide training for our growing employee base due to the highly specialized nature of pharmaceutical products.
Further, we expect that our potential expansion into areas and activities requiring additional expertise, such as further clinical trials, governmental approvals, manufacturing, sales and marketing, will place additional requirements on our management, operational and financial resources. We expect these demands will require an increase in the number of management and scientific personnel and the development of additional expertise by existing management personnel. The failure to attract and retain such personnel, or to develop such expertise, could materially adversely affect prospects for our success.
Our current personnel may be inadequate and we may fail to assimilate and train new employees. Highly skilled employees with the education and training that we require, especially employees with significant experience and expertise in drug delivery systems, are in high demand. Once trained, our employees may be hired by our competitors.
| We have international operations that expose us to additional business risks. |
We have expanded our operations outside of Canada, primarily in Europe, in order to market and distribute our products and may expand further in the future. Any expansion in international markets requires additional resources and management attention and subjects us to new business risks, including the following:
| • | different regulatory requirements for approval of our product candidates; | |
| • | dependence on local distributors; | |
| • | longer payment cycles and problems in collecting accounts receivable; | |
| • | adverse changes in trade and tax regulations; | |
| • | the absence or significant lack of legal protection for intellectual property rights; | |
| • | difficulty in managing widespread operations; | |
| • | unfavourable labour regulations; | |
| • | political and economic instability; and | |
| • | currency risks. |
The occurrence of any of these or other factors may cause our international operations not to be successful or otherwise have an adverse effect on our operating results.
| We may not be able to successfully acquire and integrate complementary technologies or businesses needed for the development of our business and any acquisitions we make could disrupt our business and harm our financial condition. |
We may pursue product or business acquisitions that could complement or expand our business. However, we may not be able to identify appropriate acquisition candidates in the future. If an acquisition candidate is identified, we may not be able to successfully negotiate the terms of any such acquisition or finance such acquisition. Any such acquisition could result in unanticipated costs or liabilities, diversion of management’s attention from our core business, the expenditure of resources and the potential loss of key employees, particularly those of the acquired organizations. In addition, we may not be able to successfully integrate any businesses, products, technologies or personnel that we might acquire in the future, which may harm our business.
19
To the extent we issue common shares or other rights to finance any acquisition, existing shareholders may be diluted and earnings per share may decrease. They may also result in goodwill and other long-lived assets that are subject to impairment tests, which could result in future impairment charges.
| We may incur losses associated with foreign currency fluctuations. |
We anticipate that the majority of the revenue from commercialization of our product candidates may be in currencies other than Canadian dollars. Fluctuation in the exchange rate of the Canadian dollar relative to these other currencies could result in us realizing a lower profit margin on sales of our product candidates than we anticipate at the time of entering into commercial agreements. Our financial results will be subject to the impact of currency transactions and translation gains and losses. As we commercialize our products, the functional currency of certain of our subsidiaries may change from the Canadian dollar to the subsidiaries local currency. This could have an impact on our exposure to foreign currency fluctuations. Adverse movements in exchange rates could have a material adverse effect on our financial condition and results of operations.
Risks Relating to Our Industry
| Generic drug manufacturers will increase competition for certain products and may reduce our royalties. |
Because our product development strategy involves the reformulation of existing drugs with active ingredients that are off-patent, our products are likely to face competition from generic versions of such drugs. Regulatory approval for generic drugs may be obtained without investing in costly and time-consuming clinical trials. Because of substantially reduced development costs, manufacturers of generic drugs are often able to charge much lower prices for their products than the original developer of a new product. If we face competition from manufacturers of generic drugs on products we may commercialize such as our once-daily tramadol product, the prices at which such products are sold and the revenues we receive may be reduced.
| Market acceptance of our products will be limited if users of our products are unable to obtain adequate reimbursement from third-party payors. |
Government health administration authorities, private health insurers and other organizations generally provide reimbursement for products like ours, and our commercial success will depend in part on whether appropriate reimbursement levels for the cost of our products and related treatments are obtained from government authorities, private health insurers and other organizations, such as health maintenance organizations and managed care organizations. Even if we succeed in bringing any of our products to market, third-party payors may not provide reimbursement in whole or in part for their use.
Significant uncertainty exists as to the reimbursement status of newly approved health care products. Each of our product candidates is intended to replace or alter existing therapies or procedures. These third-party payors may conclude that our products are less safe, less effective or less economical than those existing therapies or procedures. Therefore, third-party payors may not approve our products for reimbursement. We may be required to make substantial pricing concessions in order to gain access to formularies of large managed-care organizations. If third-party payors do not approve our products for reimbursement or fail to reimburse them adequately, sales will suffer as some physicians or their patients may opt for a competing product that is approved for reimbursement or is adequately reimbursed. Even if third-party payors make reimbursement available, these payors’ reimbursement policies may adversely affect our ability and our potential distributors’ and collaborators’ ability to sell our products on a profitable basis.
| If we are unable to obtain adequate reimbursement from governments or third-party payors for any product that we may develop or to obtain acceptable prices for such product, our revenues and prospects for profitability will suffer. |
Our revenues and profits will depend heavily upon the availability of adequate reimbursement for the use of our approved product candidates from governmental and other third-party payors, both in the United
20
States and in other markets. Reimbursement by a third-party payor may depend upon a number of factors, including the third-party payor’s determination that use of a product is:
| • | a covered benefit under its health plan; | |
| • | safe, effective and medically necessary; | |
| • | appropriate for the specific patient; | |
| • | cost-effective; and | |
| • | neither experimental nor investigational. |
Obtaining reimbursement approval for a product from each government or other third-party payor is a time consuming and costly process that could require us to provide supporting scientific, clinical and cost-effectiveness data for the use of our products to each payor. We may not be able to provide data sufficient to gain acceptance with respect to reimbursement. Even when a payor determines that a product is eligible for reimbursement, the payor may impose coverage limitations that preclude payment for some uses that are approved by the FDA or comparable authorities. In addition, there is a risk that full reimbursement may not be available for high priced products. Moreover, eligibility for coverage does not imply that any product will be reimbursed in all cases or at a rate that allows us to make a profit or even cover our costs. Interim payments for new products, if applicable, may also not be sufficient to cover our costs and may not be made permanent. A trend in the United States healthcare industry and elsewhere is cost containment. We expect recent changes in the Medicare program and increasing emphasis on managed care to continue to put pressure on pharmaceutical product pricing.
| Governments outside the United States tend to impose strict price controls, which may adversely affect our revenues, if any. |
In some countries, particularly the countries of the European Economic Area, or EEA, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time and delay the placing on the market of a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies. If reimbursement of our product is unavailable or limited in scope or amount, or if pricing is set at unsatisfactory levels, our business could be adversely affected.
| We are subject to the risk of product liability claims, for which we may not have or will not be able to obtain adequate insurance coverage. |
Drugs involve the risk of product liability claims and associated adverse publicity. For products on the market, such as once-daily tramadol, claims might be made directly by patients, healthcare providers or pharmaceutical companies or others selling our products. There are also risks related to participants in our clinical trials, who may suffer unintended consequences. We may not have or be able to obtain or maintain sufficient and affordable insurance coverage. Also, our warranty recourses, if any, against third party manufacturers of our products may be limited or unavailable. Furthermore, product liability claims, regardless of their merits, could be costly and divert our management’s attention from other business concerns, or adversely affect our reputation and the demand for our products.
| Our products involve the use of hazardous materials, and as a result we are exposed to potential liability claims and to costs associated with complying with laws regulating hazardous waste. |
Our research and development activities involve the use of hazardous materials, including chemicals and biological materials, and are subject to Canadian federal, provincial and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous materials and waste products. However, accidental injury or contamination from these materials may occur. In the event of an accident, we
21
could be held liable for any damages, which could exceed our available financial resources. In addition, we may be required to incur significant costs to comply with environmental laws and regulations in the future.
Risks Related to this Offering
| Our share price has been volatile, and an investment in our common shares could suffer a decline in value. |
Our valuation and share price since the beginning of trading after our initial public offering in Canada have had no meaningful relationship to current or historical financial results, asset values, book value or many other criteria based on conventional measures of the value of common shares. For example, over the last two years, the trading price of our common shares on the Toronto Stock Exchange varied between $2.28 and $9.75. The market price of our common shares will fluctuate due to various factors including:
| • | clinical and regulatory developments regarding our product candidates; | |
| • | developments regarding current, potential or future third-party distributors or collaborators; | |
| • | announcements by us or our competitors regarding technological, product development or other matters; | |
| • | government regulatory action affecting our product candidates and our competitors’ products in the United States, Europe and other countries; | |
| • | additions or departures of key personnel; | |
| • | developments or disputes concerning our or third-party intellectual proprietary rights; | |
| • | actual or anticipated fluctuations in our revenues or expenses; | |
| • | deviations in our operating results from the estimates of securities analysts; | |
| • | timing and receipt of milestone payments from distributors or collaborators; | |
| • | general market conditions and fluctuations in the biotechnology or pharmaceutical industry market sectors; and | |
| • | economic conditions and political factors in the United States, Europe, Canada or abroad. |
In the past, when the market price of a stock has been volatile, holders of that stock have often instituted securities class action litigation against that company. If any of our shareholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit. The lawsuit could also divert the time and attention of our management.
| Our common shares have no prior trading history in the United States, and an active market may not develop. |
Our common shares are currently listed on the Toronto Stock Exchange but are not listed on any U.S. stock exchange or quoted on any U.S. quotation system. Accordingly, prior to this offering, there has been no public market in the United States for our common shares. The initial U.S. public offering price for our common shares may bear no relationship to the price at which our common shares will trade upon the completion of this offering. The price of our common shares may be lower than the price of the shares sold in this offering. In addition, because the liquidity and trading patterns of securities listed on the Toronto Stock Exchange may be substantially different from those of securities quoted on the Nasdaq National Market, historical trading prices may not be indicative of the prices at which our shares will trade in the future. Although we have applied to have our common shares approved for quotation on the Nasdaq National Market, an active trading market for our shares may never develop or be sustained in the United States following this offering. If an active market for our common shares does not develop, it may be difficult for U.S. residents to sell the shares they purchase in this offering without depressing the market price for the shares or at all.
22
| Future issuances of common shares by us or sales by our existing shareholders may cause our stock price to fall. |
The market price of our common shares could decline as a result of issuances by us or sales by our existing shareholders of common shares in the market after this offering, or the perception that these sales could occur. Sales by shareholders might also make it more difficult for us to sell equity securities at a time and price that we deem appropriate. Upon completion of this offering, and assuming the over-allotment option is exercised in full, we will have 55,401,863 common shares issued and outstanding, all of which will be freely tradable on either the Toronto Stock Exchange or the Nasdaq National Market. All of the shares offered pursuant to this offering, plus any shares sold pursuant to the exercise of the underwriters’ over-allotment option, will be freely tradable without restriction under the United States Securities Act of 1933, as amended, or the Securities Act, unless purchased by our affiliates.
| We have never paid dividends on our common shares, and we do not anticipate paying any cash dividends in the foreseeable future. |
We have paid no cash dividends on any of our common shares to date and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. In addition, the terms of any future debt or credit facility may preclude us from paying these dividends.
| Investors in this offering will pay a much higher price than the book value of our common shares. |
If you purchase common shares in this offering, you may pay more for your shares than the amounts paid by existing shareholders for their shares. As a result, you will incur immediate and substantial dilution. In the past, we issued options to acquire common shares at prices significantly below the public offering price. To the extent these outstanding options are ultimately exercised, you will incur further dilution. See “Dilution.”
| We have broad discretion in the use of the net proceeds from this offering and may not use them effectively. |
Our management will have broad discretion in the application of the net proceeds from this offering and could spend the proceeds in ways that do not improve our results of operations or enhance the value of our common shares. The failure by our management to apply these funds effectively could result in financial losses that could have a material adverse effect on our business, cause the price of our common shares to decline and delay the development of our product candidates. Pending their use, we may invest the net proceeds from this offering in a manner that does not produce income or that loses value.
| Because we are a Canadian company, certain civil liabilities and judgments may not be enforceable against us. |
We are incorporated under the laws of Quebec, Canada. Most of our directors and officers and certain of the experts named elsewhere in this prospectus are residents of Canada. All or a substantial portion of our assets and the assets of these persons are located outside of the United States. As a result, it may be difficult for a shareholder to initiate a lawsuit within the United States against these non-U.S. residents, or to enforce in the United States judgments that are obtained in a U.S. court against us or these persons. It may also be difficult for shareholders to enforce a U.S. judgment in Canada, or to succeed in a lawsuit in Canada, based solely on violations of U.S. securities laws.
| Our articles and certain Canadian laws could delay or deter a change of control. |
Our authorized preferred shares are available for issuance from time to time at the discretion of our board of directors, without shareholder approval. Our articles grant our board of directors the authority, subject to the corporate law of Quebec, to determine or alter the special rights and restrictions granted to or imposed on any wholly unissued series of preferred shares, and such rights may be superior to those of our common shares.
23
The Investment Canada Act (Canada) subjects an acquisition of control of a company by a non-Canadian to government review if the value of our assets as calculated pursuant to the legislation exceeds a threshold amount. A reviewable acquisition may not proceed unless the relevant minister is satisfied that the investment is likely to be a net benefit to Canada.
Any of the foregoing could prevent or delay a change of control and may deprive or limit strategic opportunities for our shareholders to sell their shares.
| We may be a passive foreign investment company for U.S. tax purposes which may negatively affect U.S. investors. |
For U.S. federal income taxation purposes, we will be a passive foreign investment company, or PFIC, if in any taxable year either: (a) 75% or more of our gross income consists of passive income; or (b) 50% or more of the value of our assets is attributable to assets that produce, or are held for the production of, passive income. If we meet either test, our shares held by a U.S. person in that year will be PFIC shares for that year and all subsequent years in which they are held by that person. It is possible that we will be considered a PFIC for 2006 and subsequent years due to the nature of our income and assets and the manner in which the relevant provisions of the U.S. Internal Revenue Code of 1986, as amended, may be applied to our situation. Gain realized by a U.S. investor from the sale of PFIC shares is taxed as ordinary income, as opposed to capital gain, and subject to an interest charge unless the U.S. person has timely made one of the tax elections described in the section titled “Certain Material Income Tax Considerations — U.S. Federal Income Tax Considerations.”
The PFIC rules are extremely complex. A U.S. person is encouraged to consult his or her U.S. tax advisor before making an investment in our shares.
| As a foreign private issuer, we are subject to different U.S. securities laws and rules than a domestic U.S. issuer, which may limit the information publicly available to our shareholders. |
As a foreign private issuer we are not required to comply with all the periodic disclosure requirements of the Securities Exchange Act of 1934 and therefore there may be less publicly available information about us than if we were a U.S. domestic issuer. In addition, our officers, directors, and principal shareholders are exempt from the reporting and “short swing” profit recovery provisions of Section 16 of the Securities Exchange Act of 1934 and the rules thereunder. Therefore, our shareholders may not know on a timely basis when our officers, directors and principal shareholders purchase or sell our common shares.
24
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements. The forward-looking statements are principally contained in the sections entitled “Summary”, “Risk Factors”, “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and “Business.” These statements involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by the forward-looking statements. Forward-looking statements include, but are not limited to statements about:
| • | our plans to develop and commercialize product candidates and the timing of these development programs; | |
| • | whether we will receive, and the timing and costs of obtaining, regulatory approvals; | |
| • | clinical development of our product candidates, including the results of current and future clinical trials; | |
| • | the benefits of our drug delivery technologies and product candidates as compared to others; | |
| • | our ability to maintain and establish intellectual property rights in our drug delivery technologies and product candidates; | |
| • | our need for additional financing and our estimates regarding our capital requirements and future revenues and profitability; | |
| • | our estimates of the size of the potential markets for our product candidates; | |
| • | our selection and licensing of product candidates; | |
| • | our ability to attract distributors and collaborators with acceptable development, regulatory and commercialization expertise and the benefits to be derived from such collaborative efforts; | |
| • | sources of revenues and anticipated revenues, including contributions from distributors and collaborators, product sales, license agreements and other collaborative efforts for the development and commercialization of product candidates; | |
| • | our ability to create an effective direct sales and marketing infrastructure for products we elect to market and sell directly; | |
| • | the rate and degree of market acceptance of our products; | |
| • | the timing and amount of reimbursement for our products; | |
| • | the success and pricing of other competing therapies that may become available; | |
| • | our ability to retain and hire qualified employees; and | |
| • | the manufacturing capacity of third-party manufacturers for our product candidates. |
In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “could,” “would,” “expect,” “plan,” “anticipate,” “believe,” “estimate,” “project,” “predict,” “intend,” “potential,” “continue” and similar expressions intended to identify forward-looking statements. Forward-looking statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. Given these risks and uncertainties, the forward-looking events and circumstances discussed in this prospectus may not transpire, and you should not place undue reliance on these forward-looking statements. We discuss many of these risks in greater detail under the heading “Risk Factors.” Also, these forward-looking statements represent our estimates and assumptions only as of the date of this prospectus. We undertake no obligation and do not intend to update or revise these forward-looking statements, unless required by law. We qualify all of the information presented in this prospectus, and particularly our forward-looking statements, with these cautionary statements.
25
ENFORCEABILITY OF CIVIL LIABILITIES
We are incorporated and organized under the laws of the Province of Québec, Canada. Most of our directors and officers, as well as some of the experts named in this prospectus, are residents of Canada, and a substantial portion of their assets and our assets are located outside of the United States. As a result, it may be difficult for shareholders to effect service of process within the United States upon us or those directors, officers and experts who are not residents of the United States or to enforce against us or them judgments obtained in the courts of the United States based upon the civil liability provisions of the federal securities laws or other laws of the United States. There is doubt as to the enforceability in Canada, or elsewhere, against us or against any of our directors, officers or experts who are not residents of the United States, in original actions or in actions for enforcement of judgments of United States courts, of liabilities based solely upon the civil liability provisions of the U.S. federal securities laws. Therefore, it may not be possible for U.S. shareholders to enforce those actions against us, our directors and officers or the experts named in this prospectus.
EXCHANGE RATE INFORMATION
The following table sets forth for each period indicated: (i) the noon exchange rates in effect at the end of the period; (ii) the high and low noon exchange rates during such period; and (iii) the average noon exchange rates for such period, for one Canadian dollar, expressed in U.S. dollars, as quoted by the Bank of Canada.
| Year Ended | Year Ended | |||||||
| December 31, | December 31, | |||||||
| 2005 | 2004 | |||||||
| Period End | U | S$0.8577 | U | S$0.8308 | ||||
| High | U | S$0.8690 | U | S$0.8493 | ||||
| Low | U | S$0.7872 | U | S$0.7159 | ||||
| Average | U | S$0.8254 | U | S$0.7683 | ||||
The average exchange rate is calculated based on the last business day of each month for the applicable period. Unless otherwise indicated, all U.S. dollar amounts referred to in this prospectus which have been converted into U.S. dollars from Canadian dollars have been so converted using the noon exchange rate of US$0.8681 per one Canadian dollar, as quoted by the Bank of Canada on April 13, 2006.
26
USE OF PROCEEDS
We estimate that the net proceeds to us from the sale of 10,000,000 of our common shares in this offering, after deducting underwriting discounts and commissions and the estimated expenses of the offering, will be approximately US$67.2 million, based on an assumed public offering price of US$7.31. If the over-allotment option is exercised in full, we estimate that our net proceeds will be approximately US$77.5 million.
We plan to use the net proceeds from this offering:
| • | to support the commercialization of our once-daily tramadol product; | |
| • | to advance development of existing and new product candidates within our product pipeline; and | |
| • | for working capital and other general corporate purposes. |
Pending such uses, we plan to invest the net proceeds of the offering in short-term, interest bearing investment grade securities.
A US$1.00 increase (decrease) in the assumed public offering price of US$7.31 per share would increase (decrease) our net proceeds from this offering by approximately US$9.4 million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions.
27
PRICE RANGE OF COMMON SHARES
Our common shares are traded on the Toronto Stock Exchange under the symbol “DDS.” We have applied to list our common shares on the Nasdaq National Market under the symbol “DDSS.” The following table sets forth the high and low closing sale prices for our common shares for the periods indicated, as reported on the Toronto Stock Exchange. However, you should not view this presentation as an indication that the market price of our common shares will continue at such levels.
| High | Low | |||||||
Year ended December 31, 2003: | ||||||||
| First Quarter | $ | 4.80 | $ | 3.24 | ||||
| Second Quarter | 5.50 | 3.35 | ||||||
| Third Quarter | 7.56 | 4.40 | ||||||
| Fourth Quarter | 9.27 | 6.92 | ||||||
Year ended December 31, 2004: | ||||||||
| First Quarter | $ | 8.97 | $ | 5.49 | ||||
| Second Quarter | 6.58 | 4.00 | ||||||
| Third Quarter | 4.40 | 2.48 | ||||||
| Fourth Quarter | 4.15 | 2.52 | ||||||
Year ended December 31, 2005: | ||||||||
| First Quarter | $ | 4.20 | $ | 3.16 | ||||
| Second Quarter | 3.49 | 2.60 | ||||||
| Third Quarter | 5.23 | 2.80 | ||||||
| Fourth Quarter | 7.03 | 3.54 | ||||||
Year ended December 31, 2006: | ||||||||
| First Quarter | $ | 9.35 | $ | 6.92 | ||||
On April 17, 2006, the closing price of our common shares as reported on the Toronto Stock Exchange was $8.42.
DIVIDEND POLICY
We have never declared or paid cash dividends on our common shares and do not anticipate paying any cash dividends on our common shares in the foreseeable future. We presently intend to retain future earnings, if any, to finance the expansion and growth of our business. Any future determination to pay dividends will be at the discretion of our board of directors and will depend on our financial condition, results of operations, capital requirements and other factors the board of directors deems relevant. In addition, the terms of any future debt or credit facility may preclude us from paying dividends.
28
CAPITALIZATION
The following table describes our cash, cash equivalents and short-term investments and our capitalization as at December 31, 2005:
| • | on an actual basis; and | |
| • | on an as adjusted basis to give effect to our sale of 10,000,000 common shares in the offering at an assumed public offering price of US$7.31 per share and our receipt of estimated net proceeds of US$67.2 million, after deducting estimated underwriting discounts and commissions and offering expenses payable by us and assuming no exercise of the underwriters’ over-allotment option. |
You should read the information below with our consolidated financial statements and related notes and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this prospectus. For your convenience, the following table is presented in both U.S. dollars and Canadian dollars. The U.S. dollar amounts have been converted from Canadian dollars at the rate of U.S.$0.8577 per $1.00 Canadian, which is the noon exchange rate for December 31, 2005, as quoted by the Bank of Canada, except for the estimated net proceeds of the offering which we have assumed will be received in U.S. dollars, and will be converted to Canadian dollars at the noon exchange rate of US$0.8681 per $1.00 Canadian as quoted by the Bank of Canada on April 13, 2006.
| As at December 31, 2005 | ||||||||||||||||||
| Actual | As Adjusted | Actual | As Adjusted | |||||||||||||||
| US$ | US$ | C$ | C$ | |||||||||||||||
| (unaudited) | (unaudited) | |||||||||||||||||
| (in thousands, except share data) | ||||||||||||||||||
| Cash, cash equivalents and short-term investments(1) | US$ | 29,928 | US$ | 97,142 | $ | 34,893 | $ | 112,320 | ||||||||||
| Long-term debt, including current portion | US$ | 9,607 | US$ | 9,607 | $ | 11,201 | $ | 11,201 | ||||||||||
| Obligations under capital leases, including current portion | 5,080 | 5,080 | 5,923 | 5,923 | ||||||||||||||
| Shareholders’ equity (deficiency): | ||||||||||||||||||
| Common shares, without par value; unlimited number of shares authorized; 43,673,863 shares issued and outstanding, actual and 53,673,863 shares issued and outstanding, as adjusted | 116,331 | 183,545 | 135,631 | 213,058 | ||||||||||||||
| Contributed surplus | 5,446 | 5,446 | 6,350 | 6,350 | ||||||||||||||
| Deficit | (124,010 | ) | (124,010 | ) | (144,584 | ) | (144,584 | ) | ||||||||||
| Total shareholders’ equity (deficiency)(1) | (2,233 | ) | 64,981 | (2,603 | ) | 74,824 | ||||||||||||
| Total capitalization(1) | US$ | 12,454 | US$ | 79,668 | $ | 14,521 | $ | 91,948 | ||||||||||
| (1) | A US$1.00 increase (decrease) in the assumed public offering price of US$7.31 per share would increase (decrease) each of cash, cash equivalents and short-term investments, total shareholders’ equity (deficiency) and total capitalization by approximately US$9.4 million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting estimated underwriting discounts and commissions. |
The above table excludes:
| • | 228,000 common shares issued upon the exercise of stock options for cash proceeds of $887,530 in the period January 1, 2006 to April 13, 2006; | |
| • | 3,384,875 common shares issuable upon the exercise of stock options outstanding as of April 13, 2006 at a weighted average exercise price of $5.76 (approximately US$5.00) per share; and | |
| • | 164,504 common shares reserved for future issuance under our stock option plan as of April 13, 2006. |
29
DILUTION
Our negative net tangible book value as at December 31, 2005 was approximately $8.0 million (approximately US$7.0 million), or $0.18 (approximately US$0.16) per common share. Net tangible book value per share represents the total amount of our assets, including assets under capital leases, less total liabilities, intangible assets, deferred financing costs and leasehold and building improvements, divided by the number of common shares outstanding. Dilution in net tangible book value per share represents the difference between the amount per share that you pay in this offering and the net tangible book value per share after this offering.
After giving effect to the issuance and sale by us of 10,000,000 common shares in the offering at an assumed public offering price of $8.42 per share (US$7.31 per share) and after deducting estimated underwriting discounts and commissions and offering expenses of $6.8 million (approximately US$5.9 million) payable by us, our pro forma net tangible book value as at December 31, 2005 is $69.4 million (approximately US$60.2 million), or $1.29 per share (approximately US$1.12 per share). This represents an immediate increase in the net tangible book value of $1.47 per share (approximately US$1.28 per share) to existing shareholders and an immediate dilution of $7.13 per share (approximately US$6.19 per share) to new investors. This dilution is illustrated by the following table:
| (US$) | |||||||||||||||||
| ) | |||||||||||||||||
| ($ | |||||||||||||||||
| Assumed public offering price per share | U | S$7.31 | $8.42 | ||||||||||||||
| Net tangible book value per share as at December 31, 2005 | U | S$(0.16 | ) | $(0.18 | ) | ||||||||||||
| Increase per share attributable to new investors | 1.28 | 1.47 | |||||||||||||||
| Pro forma net tangible book value per share after this offering | 1.12 | 1.29 | |||||||||||||||
| Dilution per share to new investors | U | S$6.19 | $7.13 | ||||||||||||||
Assuming the exercise in full of the underwriters’ over-allotment option, our pro forma net tangible book value as at December 31, 2005 would have been $81.3 million (approximately US$70.6 million), or $1.47 per share (approximately US$1.28 per share). This represents an immediate increase in the net tangible book value of $1.65 per share (approximately US$1.44 per share) to existing shareholders and an immediate dilution of $6.95 per share (approximately US$6.03 per share) to new investors.
The foregoing discussion and tables assume no exercise of any stock options and no issuance of common shares reserved for future issuance under our stock option plan. As at December 31, 2005, there were options outstanding to purchase 3,560,875 common shares at a weighted average exercise price of $5.59 (approximately US$4.85) per share. In addition, we may grant more options in the future.
For your convenience, the foregoing discussion and tables are presented in both U.S. dollars and Canadian dollars. The U.S. dollar amounts have been converted from Canadian dollars at the rate of US$0.8577 per $1.00 Canadian, which is the noon exchange rate for December 31, 2005, as quoted by the Bank of Canada, except for the assumed public offering price and the net proceeds of the offering which we have assumed will be received in U.S. dollars, and will be converted to Canadian dollars at the noon exchange rate of US$0.8681 per $1.00 Canadian as quoted by the Bank of Canada on April 13, 2006.
30
SELECTED CONSOLIDATED FINANCIAL DATA
The following selected consolidated financial data should be read in conjunction with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our audited consolidated financial statements and related notes included elsewhere in this prospectus. The consolidated statement of operations data for the year ended December 31, 2004 and the year ended December 31, 2005 and the consolidated balance sheet data as at December 31, 2004 and 2005 have been derived from our consolidated financial statements that have been audited by Ernst & Young LLP, and which are included elsewhere in this prospectus. Our audited consolidated financial statements have been prepared in accordance with Canadian GAAP. We have included a reconciliation of significant differences between Canadian GAAP and U.S. GAAP of our net loss, balance sheet and cash flow items is presented in note 23 to our audited consolidated financial statements. In addition, note 1 to our audited consolidated financial statements provides a description of our business and details on our going concern uncertainty. Our historical results from any prior period are not necessarily indicative of results to be expected for any future period.
For your convenience, the following table presents certain of our operations data in both U.S. dollars and Canadian dollars. The U.S. dollar amounts have been converted from Canadian dollars at the rate of US$0.8254 per $1.00 Canadian, which is the average exchange rate for the year ended December 31, 2005, as quoted by the Bank of Canada.
31
| Year Ended December 31, | |||||||||||||
| 2005 | 2005 | 2004 | |||||||||||
| US$ | C$ | C$ | |||||||||||
| (in thousands, except share and per share data) | |||||||||||||
Consolidated Statement of Operations Data: | |||||||||||||
Canadian GAAP | |||||||||||||
| Revenues: | |||||||||||||
| Product sales | US$ | 1,047 | $ | 1,269 | $ | — | |||||||
| Cost of goods sold | 626 | 758 | — | ||||||||||
| Gross profit on product sales | 421 | 511 | — | ||||||||||
| Licensing | 1,575 | 1,908 | 106 | ||||||||||
| Research and development contracts | 50 | 61 | 1,288 | ||||||||||
| 2,046 | 2,480 | 1,394 | |||||||||||
| Expenses: | |||||||||||||
| Research and development expenses, net | 16,292 | 19,738 | 15,214 | ||||||||||
| Selling, general and administrative expenses | 10,060 | 12,188 | 9,878 | ||||||||||
| Financial expenses | 1,599 | 1,937 | 864 | ||||||||||
| Depreciation and amortization | 1,373 | 1,664 | 1,662 | ||||||||||
| Interest income | (460 | ) | (557 | ) | (538 | ) | |||||||
| Foreign exchange gain | (293 | ) | (355 | ) | (57 | ) | |||||||
| Expenses related to an incomplete financing initiative | — | — | 1,509 | ||||||||||
| 28,571 | 34,615 | 28,532 | |||||||||||
| Loss before income taxes | (26,525 | ) | (32,135 | ) | (27,138 | ) | |||||||
| Income taxes: | |||||||||||||
| Current | 990 | 1,199 | 41 | ||||||||||
| Net loss for the period | US$ | (27,515 | ) | $ | (33,334 | ) | $ | (27,179 | ) | ||||
| Net loss per share — basic and diluted | US$ | (0.64 | ) | $ | (0.78 | ) | $ | (0.68 | ) | ||||
| Weighted average number of shares outstanding | 42,922,741 | 42,922,741 | 39,954,488 | ||||||||||
U.S. GAAP | |||||||||||||
| Net loss for the period | US$ | (27,541 | ) | $ | (33,367 | ) | $ | (27,441 | ) | ||||
| Net loss per share — basic and diluted | US$ | (0.64 | ) | $ | (0.78 | ) | $ | (0.69 | ) | ||||
| Weighted average number of shares outstanding | 42,922,741 | 42,922,741 | 39,954,488 | ||||||||||
32
The as adjusted consolidated balance sheet data below reflects our sale of 10,000,000 common shares in the offering at an assumed public offering price of US$7.31 per share and our receipt of estimated net proceeds of US$67.2 million, after deducting estimated underwriting discounts and commissions and offering expenses payable by us.
For your convenience, the following table is presented in both U.S. dollars and Canadian dollars. The U.S. dollar amounts have been converted from Canadian dollars at the rate of US$0.8577 per $1.00 Canadian, which is the noon exchange rate for December 31, 2005, as quoted by the Bank of Canada, except for the estimated net proceeds of the offering which we have assumed will be received in U.S. dollars, and will be converted to Canadian dollars at the noon exchange rate of US$0.8681 per $1.00 Canadian as quoted by the Bank of Canada on April 13, 2006.
| As of | ||||||||||||||||||||
| December 31, | ||||||||||||||||||||
| As of December 31, 2005 | 2004 | |||||||||||||||||||
| Actual | As Adjusted | Actual | As Adjusted | Actual | ||||||||||||||||
| US$ | US$ | C$ | C$ | C$ | ||||||||||||||||
| (unaudited) | ||||||||||||||||||||
| (unaudited) | ||||||||||||||||||||
| ($ in thousands) | ||||||||||||||||||||
Consolidated Balance Sheet Data: | ||||||||||||||||||||
Canadian GAAP | ||||||||||||||||||||
| Cash, cash equivalents and short-term investments | US$ | 29,928 | US$ | 97,142 | $ | 34,893 | $ | 112,320 | $ | 23,623 | ||||||||||
| Working capital | 14,361 | 81,575 | 16,743 | 94,170 | 19,307 | |||||||||||||||
| Total assets | 46,755 | 113,969 | 54,512 | 131,939 | 39,916 | |||||||||||||||
| Obligations under capital leases — long-term portion | 5,009 | 5,009 | 5,840 | 5,840 | 5,923 | |||||||||||||||
| Long-term debt | 9,607 | 9,607 | 11,201 | 11,201 | — | |||||||||||||||
| Total liabilities | 48,987 | 48,987 | 57,115 | 57,115 | 13,763 | |||||||||||||||
| Total shareholders’ equity (deficiency) | (2,233 | ) | 64,981 | (2,603 | ) | 74,824 | 26,153 | |||||||||||||
U.S. GAAP | ||||||||||||||||||||
| Total assets | 44,980 | 112,194 | 52,443 | 129,870 | 37,880 | |||||||||||||||
| Total shareholders’ equity (deficiency) | (4,007 | ) | 63,207 | (4,672 | ) | 72,755 | 24,117 | |||||||||||||
33
MANAGEMENT’S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
The following information should be read in conjunction with the section entitled “Selected Consolidated Financial Data” and our audited consolidated financial statements and related notes thereto included elsewhere in this prospectus. Our audited consolidated financial statements have been prepared in accordance with Canadian GAAP. A reconciliation of the significant differences between Canadian GAAP and U.S. GAAP of our net loss, balance sheet and cash flow items is presented in note 23 to our audited consolidated financial statements. To the extent any statements made in this prospectus contain information that is not historical, these statements are forward-looking and are subject to risks and uncertainties, as described in the section entitled “Risk Factors” and elsewhere in this prospectus. Actual results, levels of activity, performance, or achievements could differ materially from those projected in this prospectus and depend on a number of factors, including the successful and timely completion of clinical studies, the uncertainties related to the regulatory process, and the commercialization of product candidates thereafter. See “Forward-Looking Information.”
Overview
| Liquidity and Going Concern Uncertainty |
We have incurred substantial operating losses since our inception due in large part to expenditures for our research and development activities. As at December 31, 2005, we had an accumulated deficit of $144.6 million. We expect our operating losses to decrease going forward as we generate revenue through sales of our once-daily tramadol product while continuing to advance our other product candidates towards commercialization and expand our development pipeline. However, our committed sources of funds and our cash and cash equivalents and short-term investments on hand will not be sufficient to meet our committed cash obligations and expected level of expenses through the fourth quarter of 2006. Our ability to continue as a going concern is dependent upon receiving funds from this offering, through product licensing agreements or collaborative research contracts, raising additional financing through borrowings or equity financing, or achieving future profitable operations. The outcome of these matters is dependent on a number of items outside of our control. As a result there is significant uncertainty as to whether we will have the ability to continue as a going concern.
| Revenue |
During the fourth quarter of 2005, we achieved a significant milestone with the commercial launch of our first product, once-daily tramadol, which precipitated our first ever product sale to our marketing and distribution distributor for Germany, HEXAL AG. Revenue from product sales should in the future be a key driver in evaluating our performance towards achieving profitability, as we continue to launch our once-daily tramadol product during 2006 and thereafter, in various markets. The selling price of our once-daily tramadol product will vary for each licensee depending on specific market conditions in the prevailing jurisdictions including competing products and regulatory pricing policies.
Revenue to date has been generated primarily under our licensing and distribution agreements, and in prior periods by our research collaboration agreements. Since 2002, we have secured nine such agreements for marketing and distribution of our once-daily tramadol product, that cover 42 countries. To date we have received approximately $32 million of licensing payments from our once-daily tramadol product licensees, including US$20 million from Purdue. We will also receive additional licensing payments from Purdue upon achieving various milestones, including up to US$40 million upon the regulatory approval of our once-daily tramadol product in the U.S., and up to US$110 million upon meeting specified sales targets. In addition, we will receive from Purdue royalty rates ranging from 20% to 25% of net product sales.
| Research and Development Expenses |
Our research and development expenses consist primarily of fees paid to outside parties that we use to conduct clinical studies and manufacturing process validation, salaries and related personnel expenses,
34
laboratory supplies and costs for facilities and equipment. In 2005, research and development expenses increased in comparison to the year ended December 31, 2004, primarily due to the timing and progress of expenditures related to our clinical trial program for our once-daily tramadol product, the MRP submission costs and the validation of the commercial manufacturing process of tramadol at a second manufacturer.
Change in Accounting Policy
In June 2003, the Canadian Institute of Chartered Accountants, or CICA, issued Accounting Guideline AcG-15,Consolidation of Variable Interest Entities which requires consolidation of variable interest entities, or VIE, for fiscal years beginning on or after November 1, 2004. A VIE is any legal structure used to conduct activities or hold assets which is not controlled by voting interests but rather by contractual or other interests that change with that entity’s underlying net asset value. AcG-15 requires the consolidation of a VIE by its primary beneficiary, i.e., the party that receives the majority of the expected residual returns and/or absorbs the majority of the entity’s expected losses. The adoption of AcG-15 did not result in any change to the consolidated financial statements.
Critical Accounting Policies and Estimates
In preparing our consolidated financial statements, we are required to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expenses during the reporting periods. We have identified the following accounting policies that we believe require application of management’s most subjective judgments, often requiring the need to make estimates about the effect of matters that are inherently uncertain and may change in subsequent periods. Our actual results could differ from these estimates and such differences could be material.
| Revenue Recognition |
We recognize revenue from research and development contracts, and licensing and distribution arrangements, which may include multiple elements. Revenue arrangements with multiple elements are reviewed in order to determine whether the multiple elements can be divided into separate units of accounting, if certain criteria are met, and as such requires judgement. If separable, the consideration received is allocated among the separate units of accounting based on their respective fair values, and the applicable revenue recognition criteria are applied to each of the separate units. Otherwise, the applicable revenue recognition criteria are applied to combined elements as a single unit of accounting.
Licensing revenues. Up-front non-refundable licensing revenue is deferred and recognized on a straight-line basis over the term which we maintain substantive contractual obligations, and as such require estimates of the applicable period of performance. Licensing revenue received upon the achievement of milestones is recognized when the underlying condition is met if it has stand-alone value to the customer, we have no further obligations in relation to that milestone and collectibility is reasonably assured. Otherwise, it is recognized over the remaining term of the underlying agreement. Amounts received in advance of recognition are included in deferred revenue, as are amounts which are refundable if underlying conditions are not met.
In August 2005, we granted Purdue the exclusive right to market, sell and distribute our once-daily formulation of the analgesic tramadol in the United States, its territories and possessions. Under the terms of the agreement, we received a non-refundable up-front licensing fee of US$20 million. We will also receive additional licensing payments from Purdue upon achieving various milestones, including up to US$40 million upon the regulatory approval of once-daily tramadol in the U.S. and up to US$110 million upon meeting specified sales targets, and we will also receive reimbursements for products manufactured. We currently are recognizing the up-front payment of US$20 million over 33 months until August 2008, which is the estimated term over which we will be continuing research and development activities and supplying products. This period may be revised if circumstances change. When and if received, the milestone of up to US$40 million upon regulatory approval of our once-daily tramadol product in the U.S. will be recognized over
35
approximately 18 months representing the period for which we are responsible to supply products. This period is also an estimate and is subject to change if circumstances require. We plan to recognize the milestones of up to US$110 million upon meeting specified sales targets when the underlying conditions are met.
As at December 31, 2005, we had entered into seven licensing and distribution agreements for once-daily tramadol for commercialization in Europe, Latin America and the Caribbean. Under the terms of these agreements, we have received $8,836,000 of up-front payments as of December 31, 2005. Payments which are non-refundable are recognized over the term of the agreement and $536,000 was recognized as licensing revenue in the year ended December 31, 2005 (2004 — $106,000), and the remainder was recorded as deferred revenue and will be recognized over the term of the respective agreements ranging between five and 19 years. Additionally, we are entitled to receive between $7,076,000 (US$750,000 and Euro 4,490,000) and $8,667,000 (US$1,700,000 and Euro 4,840,000) upon the achievement of various additional milestones including the price approval in territories covered by the agreements, product launches in specific territories, or attaining specified sales targets. These future payments will be recognized over the remaining term of the respective agreements unless they relate to the attainment of sales targets, in which case they will be recognized immediately.
Product sales. Revenue is recognized when the product is shipped to our customers, provided we have not retained any significant risks of ownership or future obligations with respect to the product shipped and collection is reasonably assured. Our products are indirectly subject to pricing regulations in certain markets.
Research and development contracts. We recognize revenues from various research agreements as the contracted services are performed or when milestones are achieved, in accordance with the terms of the specific agreements. Up-front payments for the use of technology where further services are to be provided or fees received on the signing of research agreements are recognized over the period of performance of the related activities. Amounts received in advance of recognition are included in deferred revenue.
| Impairment of Long-lived Assets |
Property, plant and equipment and other long-lived assets including intellectual property are regularly reviewed for impairment whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. Impairment is assessed by comparing the carrying amount of an asset to be held and used with the sum of the undiscounted cash flows expected from its use and disposal, and as such requires us to make significant estimates on expected revenues from the commercialization of our products and services and the related expenses. If such assets are considered impaired, the impairment loss to be recognized is measured by the amount by which the carrying amount of the assets exceeds its fair value generally determined on a discounted cash flow basis. Any impairment results in a write-down of the asset and a charge to income during the year. Despite our going concern uncertainty, we did not deem necessary to write-down any of our property, plant and equipment and other long-lived assets. We believe we should be able to obtain additional financing to allow us to pursue our activities until profitability is achieved and positive cash flow is generated. However, there can be no assurance that we will be able to raise such capital on favorable terms or that commercial agreements will be concluded or that we will receive payments under existing agreements.
| Refundable Investment Tax Credits |
We incur research and development expenditures which are eligible for refundable investment tax credits from the Province of Québec. The investment tax credits recorded are based on our estimates of amounts we expect to be recovered and are subject to audit by the taxation authorities and, accordingly, these amounts may vary. The amount of research and development tax credit receivable recorded as at December 31, 2005 is $875,000.
36
| Valuation Allowance on Future Tax Assets |
We recorded a valuation allowance on future tax assets related primarily to operating losses and research and development expenses carryforwards. We have concluded that the related tax benefits are more likely than not, not to be realized based on our historical results and estimated future taxable income and tax planning strategies in the related jurisdictions. The implementation of tax planning strategies or the generation of future taxable income in these jurisdictions could result in the recognition of some portion or all of these carryforwards, which could result in a material increase in our results of operations through the recovery of future income taxes.
| Stock-Based Compensation |
We account for our stock option plan for our directors, executives and employees, for stock option awards granted after March 1, 2002, using the fair value method. The fair value of stock options at the grant date is determined using the Black-Scholes option pricing model and expensed over the vesting period of the options. Assumptions that affect our application of the fair value method include the determination of volatility factors and the expected life of the options issued.
Results of Operations
Our results of operations have fluctuated significantly from period to period in the past and are likely to do so in the future. We anticipate that our quarterly and annual results of operations will be impacted for the foreseeable future by several factors, including the timing and amount of payments received pursuant to our current and future collaborations, and the progress and timing of expenditures related to our research, development and commercialization efforts particularly for our once-daily tramadol product. Due to these fluctuations, we believe that the period-to-period comparisons of our operating results are not a good indication of our future performance.
| Year Ended December 31, 2005 Compared to Year Ended December 31, 2004 |
| Revenue |
For the year ended December 31, 2005, total revenue amounted to $3,238,000 compared to $1,394,000 for the year ended December 31, 2004.
During fiscal 2005, we recognized licensing revenue of $1,908,000, representing a portion of the licensing payments received from Purdue, HEXAL AG, Gruppo Angelini, and Esteve S.A, under our agreements for our once-daily tramadol product. Licensing revenue for the year ended December 31, 2004 was $106,000 and represented a portion of the up-front payments received from the above mentioned distributors or collaborators excluding Purdue. Over the next several quarters, we anticipate receiving additional milestone payments as provided for in the current agreements for our once-daily tramadol product as we receive market and/or price approval or launch the product in the various countries. We expect to recognize these licensing payments, for which the deliverable has no stand-alone value to our customers, rateably over the term which we maintain substantive contractual obligations, as provided for in our revenue recognition policy.
For the year ended December 31, 2005, product sales were $1,269,000 and were attributable to the first sales of our once-daily tramadol product to HEXAL AG in Germany, and included the sale of samples for the initial promotion of the product. The launch in Germany was the first of what we expect will be a series of launches in key markets in Europe and globally.
Revenue generated from research and development contracts for the year ended December 31, 2005 was $61,000 and was derived from our co-development agreement with Gruppo Angelini under which we are formulating a once-daily version of the anti-depressant trazodone. For the previous year, revenue generated from research and development contracts was $1,288,000 and was derived from the aforementioned agreement with Gruppo Angelini, as well as from our agreement with Debiopharm, under which we were conducting
37
research on the potential oral delivery of a current intravenous cancer drug using our proprietary polymeric nano-delivery systems technology (previously referred to as micelles technology).
| Cost of Goods Sold |
For the year ended December 31, 2005, cost of goods sold was $758,000 and consisted primarily of raw materials, third-party bulk tablet manufacturing and third-party packaging costs for our once-daily tramadol product. Our cost of goods sold is subject to variability due primarily to currency fluctuation and size of packaging runs. Gross margin as a percentage of product sales revenue for the period was 40% and will be subject in the future to variability due to our cost of goods sold structure and to our respective selling prices in the various territories.
| Research and Development Expenses |
Research and development expenses (before tax credits) for the year ended December 31, 2005 were $22,451,000 compared with $16,037,000 for the year ended December 31, 2004. The increase was primarily the result of the following factors: (i) the timing and progress of our clinical trial program for our once-daily tramadol product; (ii) costs related to our NDA filing to the FDA; (iii) costs related to optimizing the commercial manufacturing process at our first third-party manufacturer for launch in Europe; (iv) the validation of the commercial manufacturing process for tramadol at a second third-party manufacturer; and (v) our efforts to increase our research and development capacities. In addition, during the year ended December 31, 2005, we incurred costs for pharmacokinetic studies related to our once-daily formulation of betahistine and our once-daily formulation of trazodone while in the previous year, we incurred costs for pharmacokinetic studies related to our controlled-release formulation of gabapentin and a once-daily formulation of oxybutynin. We have since de-emphasized development efforts for the two latter programs.
Research and development tax credits for the year ended December 31, 2005 were $2,713,000 compared to $823,000 in the previous year. The increase is primarily due to our recognition of previously unrecorded federal research and development tax credits, which were used to offset federal income tax payable generated as a result of certain tax planning strategies, and due to a favourable ruling received in 2005 for previous taxation years.
For the year ended December 31, 2005, research and development costs, net of tax credits, allocated to our once-daily tramadol product program amounted to approximately $16.3 million compared to $10.5 million for the year ended December 31, 2004.
| Selling, General and Administrative Expenses |
Selling, general and administrative expenses for the year ended December 31, 2005 were $12,188,000 compared to $9,878,000 for the year ended December 31, 2004, an increase of $2,310,000 or 23%. The increase is primarily due to additional headcount required for the launch and commercialization of our once-daily tramadol product globally, increased executive variable compensation expense as a result of a year of significant achievements, and increased professional and consulting fees associated with our review of our long-term strategy.
| Financial Expenses |
Financial expenses for the year ended December 31, 2005 were $1,937,000 compared with $864,000 for year ended December 31, 2004. The increase is primarily due to the financial expenses related to the term loan agreement that we entered into in June 2005.
| Foreign Exchange Gain |
Net loss for the year ended December 31, 2005 included a foreign exchange gain of $355,000, primarily corresponding to the favourable effect of the currency fluctuation on the term loan denominated in U.S. currency, partially offset by the loss incurred on the cash held in the same currency.
38
| Income Taxes |
For the year ended December 31, 2005, the income tax expense amounted to $1,199,000 compared to $41,000 for the preceding fiscal year. For the current year, we are generating taxable income at the Canadian federal level and since we have utilized all our accumulated tax losses, we have chosen to utilize our non refundable federal research and development tax credits which have a limited carryforward period to offset these taxes instead of using the research and development expenditure pool, which has an unlimited carryforward period.
| Net Loss |
Net loss for the year ended December 31, 2005 was $33,334,000, or $0.78 per share, compared with $27,179,000, or $0.68 per share, for the year ended December 31, 2004. The increase in net loss is the result of higher expenses related to the development and preparation for commercial launch of our once-daily tramadol product, as well as higher financial expenses partially offset by an increase in revenue in 2005.
Quarterly Information
The following selected financial information is derived from our unaudited quarterly financial statements for each of the last eight quarters, all of which cover periods of three months.
| Three Months Ended | ||||||||||||||||||||||||||||||||
| Dec. 31, | Sept. 30, | June 30, | March 31, | Dec. 31, | Sept. 30, | June 30, | March 31, | |||||||||||||||||||||||||
| 2005 | 2005 | 2005 | 2005 | 2004 | 2004 | 2004 | 2004 | |||||||||||||||||||||||||
| (C$ in thousands, except per share data) | ||||||||||||||||||||||||||||||||
| Revenue(1) | 2,415 | 72 | 11 | 740 | 970 | 238 | 93 | 93 | ||||||||||||||||||||||||
| Net loss | (11,067 | ) | (7,549 | ) | (8,789 | ) | (5,929 | ) | (6,631 | ) | (5,895 | ) | (7,084 | ) | (7,569 | ) | ||||||||||||||||
| Basic and diluted net loss per share | (0.26 | ) | (0.18 | ) | (0.21 | ) | (0.14 | ) | (0.16 | ) | (0.14 | ) | (0.18 | ) | (0.21 | ) | ||||||||||||||||
| (1) | The comparative figures for revenue were reclassified to conform with the presentation in the current period. |
Liquidity and Capital Resources
Cash and cash equivalents, short-term and restricted long-term investments and accrued interest on investments (included in accounts receivable) as at December 31, 2005 were $36,248,000. On June 28, 2005 we entered into a term loan agreement which generated gross proceeds of $12,317,000. As part of the transaction we issued 543,104 warrants to purchase common shares. In addition, throughout 2005 we received a US$20 million up-front licensing fee from Purdue and other licensing payments from HEXAL AG, Aventis and Recordati, totalling $6,902,000. Despite the abovementioned proceeds, our committed sources of funds and our cash and cash equivalents and short-term investments on hand will not be sufficient to meet our committed cash obligations and expected level of expenses through the fourth quarter of 2006. Our ability to continue as a going concern is dependent upon receiving additional funds through product licensing agreements or collaborative research contracts, raising additional financing through borrowings or equity financing, or generating significant revenue from product sales and ultimately achieving future profitable operations. The outcome of these matters is dependent on a number of items outside of our control. As a result, there is significant uncertainty as to whether we will have the ability to continue as a going concern. However, we expect to raise additional funds using one or a combination of alternatives including the receipt of licensing payments in relation to existing and additional product distribution and licensing agreements, the obtaining of credit facilities or through equity financing. In addition, we expect to generate increased revenue from product sales as we progress with the launch of our once-daily tramadol product. However, there can be no assurance that we will be able to raise such capital on favourable terms, that further commercial agreements will be concluded or that we will receive payments under existing agreements.
39
Funds used in operating activities amounted to $1,040,000 for the year ended December 31, 2005 as compared to $23,082,000 for the corresponding period last year. The decrease in funds used in the operating activities is a direct result of licensing payments of $30,033,000 received during the year for our once-daily tramadol product, of which $1,908,000 was recognized as revenue and the remaining recorded as deferred revenue. The funds used in our operating activities were used primarily to develop our in-house product portfolio, principally our once-daily tramadol product, and for general operating purposes.
Funds provided from investing activities for the year ended December 31, 2005 amounted to $5,187,000 compared to funds used in investing activities of $3,275,000 for the previous fiscal year. Capital expenditures for the current year were $1,016,000 compared to $1,187,000 for the year ended December 31, 2004. Capital expenditures for the year were principally related to acquisition of laboratory and plant equipment and information technology infrastructure. Investment activities also include the purchase and disposal of marketable securities, as we invest our excess funds generated from previous financings or cash received from our distributors and collaborators according to our investment policy.
For the year ended December 31, 2005, funds provided by financing activities amounted to $13,802,000 as compared to $28,446,000 for the preceding fiscal year. On June 28, 2005, we entered into a term loan agreement which generated gross proceeds of $12,317,000, of which $11,586,000 was attributed to the term loan and $731,000 to the 543,104 warrants issued as part of the agreement. Related financing costs paid during the period totalled $474,000. In the year ending December 31, 2004, net proceeds of $27,804,000 were generated from the equity financing completed in May 2004. Proceeds of $2,093,000 were obtained from the exercise of stock options during the current year, compared to $813,000 for the exercise of stock options and warrants for the previous year.
As at December 31, 2005, our working capital was $16,743,000. Accounts receivable totalled $532,000 as at December 31, 2005 and included primarily amounts receivable for sales tax, trade receivables, as well as accrued interest on investments. Research and development tax credits receivable totalled $875,000 and included the estimated tax credits for the year ended December 31, 2005. In preparation for commercial launch of our product in other European countries, we have accumulated $2,188,000 of inventories consisting of raw materials and intermediate finished product (bulk tablets). Inventory levels are likely to increase over the next several quarters as we prepare to sell to additional marketing and distribution collaborators. Accounts payable and accrued liabilities increased from $5,930,000 at December 31, 2004 to $10,090,000 at December 31, 2005, due to the timing of the payments of expenses related to the U.S. Phase III study associated with our once-daily tramadol product and to the amendment to the supply agreement with Cargill, Inc. (formerly Cerestar) for the purchase by us of Contramid patent rights and Contramid inventory. Deferred revenue totalled $29,901,000 as at December 31, 2005 and included the unrecognized portion of the licensing payments received from the various licensees of our once-daily tramadol product. These licensing fees will be recognized as revenue generally over the term which we maintain substantive contractual obligations. Approximately $4,501,000 of the licensing fees included in deferred revenue are subject to payback provisions if certain future conditions are not met. Obligations under capital leases decreased in 2005 by $134,000 to $5,923,000 as at December 31, 2005, as a result of payments made since December 31, 2004.
Cash and cash equivalents, short-term and restricted long-term investments and accrued interest on investments (included in accounts receivable) totalled $36,248,000 as at December 31, 2005 compared to $25,101,000 as at December 31, 2004, an increase of $11,147,000, primarily as a result of the milestone payments received during the year and the proceeds from the financing completed in June 2005, net of the loss incurred during the year. Our investment policy regulates our investment activities relating to cash resources. We invest in liquid, high-grade investment securities with varying terms to maturity, selected with regard to the expected timing of expenditures for continuing operations. As at December 31, 2005, our short-term investments included commercial paper from major Canadian corporations and bonds issued by governments in amounts ranging from $993,000 to $2,405,000.
Even if we complete the offering, we will require substantial future capital in order to continue to conduct the research and development, clinical and regulatory activities necessary to bring our product candidates to market and to establish commercial manufacturing, marketing and sales capabilities. We believe
40
that the net proceeds from the offering, together with our existing cash, cash equivalents and short-term investments and anticipated revenues from our once-daily tramadol product, will be sufficient to enable us to fund our operating expenses and capital expenditures for at least the next two years. Our future capital requirements will depend on many factors, including the following factors:
| • | time and costs involved in performing clinical trials and obtaining regulatory approvals; | |
| • | costs of manufacturing and supply; | |
| • | costs of establishing or contracting for sales and marketing capabilities; | |
| • | progress of clinical and formulation development; | |
| • | establishment of agreements with collaborators and distributors and activities required for product commercialization; | |
| • | number of product candidates we pursue; | |
| • | revenues from sales of our once-daily tramadol product or from our collaborators and distributors in connection with our once-daily tramadol product; | |
| • | costs involved in filing and prosecuting patent applications and enforcing or defending patent claims; and | |
| • | existence, availability and the rate of refundable tax credits attributable to scientific research and development expenditures carried out in Québec, Canada. |
Related Party Transactions
We have entered into a consulting services agreement with ACPharma Ltd., a company controlled by Anthony C. Playle, who is a director of Labopharm and Managing Director and member of the Board of Directors of our wholly-owned subsidiary, Labopharm Europe Limited. The fees paid by us to ACPharma Ltd. during 2005 and 2004 were $324,768 and $318,840 respectively. The amounts were recorded at their exchange amounts and are subject to normal trade terms.
Contractual Obligations
In the normal course of our operations, we have entered into several contracts providing for the following payments over the next fiscal years:
| Payments due by year | ||||||||||||||||||||
| Less than | 1 - 3 | 4 - 5 | After | |||||||||||||||||
| Total | 1 year | years | years | 5 years | ||||||||||||||||
| (in thousands of Canadian dollars) | ||||||||||||||||||||
| Capital lease obligations | $ | 12,875 | $ | 889 | $ | 1,864 | $ | 2,002 | $ | 8,120 | ||||||||||
| Long-term debt obligations | 14,098 | 5,097 | 9,001 | — | — | |||||||||||||||
| Operating leases | 452 | 82 | 124 | 116 | 130 | |||||||||||||||
| Purchase obligations | 28,155 | 4,630 | 23,525 | — | — | |||||||||||||||
| Total contractual obligations | $ | 55,580 | $ | 10,698 | $ | 34,514 | $ | 2,118 | $ | 8,250 | ||||||||||
Capital and operating lease obligations pertain primarily to our facilities in Canada and Europe and the related amounts shown in the above table are the payments due by us in connection with such agreements. The purchase obligations in the table above include the minimum amounts we would be required to pay with regard to certain long-term supply agreements with third-party manufacturers for bulk tablets of our once-daily tramadol product. These agreements provide for the purchase of minimum quantities over the term of the agreements.
41
Off-Balance Sheet Arrangements
To date, we have not had any relationships with unconsolidated entities or financial partnerships, such as entities referred to as structured finance or special purpose entities, which are established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes.
We periodically enter into research agreements or strategic alliances with third parties that include indemnification provisions that are customary in the industry. These guarantees are discussed in more detail in note 13 to our audited consolidated financial statements.
Quantitative and Qualitative Disclosures of Market Risk
| Foreign Currency Risk |
We operate internationally, and a substantial portion of our expense activities and capital expenditures are in Canadian dollars, whereas our revenue (current and potential) from research contracts and licensing and distribution agreements is, and will be, primarily in U.S. dollars or Euros. In addition, in June 2005 we contracted a $10 million term loan denominated in U.S. currency. A significant adverse change in foreign currency exchange rates between the Canadian dollar relative to the U.S. dollar and Euro, could have a material effect on our consolidated results of operations, financial position or cash flows. We have not hedged exposures denominated in foreign currencies.
| Interest Rate Risk |
We invest our excess cash in investment-grade, interest-bearing securities. The primary objective of our investment activities is to preserve principal while at the same time maximizing yields without significantly increasing risk. To achieve this objective, we invest in highly liquid and high quality debt instruments or commercial paper of major corporations, government agencies and financial institutions with maturities generally of less than two years. A significant change in interest rates could have a material effect on the fair value of our investments if these investments were not held to maturity.
Recent Accounting Pronouncements
| Comprehensive Income and Equity |
In January 2005, the CICA released new Handbook Section 1530, Comprehensive Income, and Section 3251, Equity, effective for annual and interim periods beginning on or after October 1, 2006. Section 1530 establishes standards for reporting comprehensive income. The section does not address issues of recognition or measurement for comprehensive income and its components. Section 3251 establishes standards for the presentation of equity and changes in equity during the reporting period. The requirements in this section are in addition to Section 1530. We have not yet determined the impact of the adoption of this standard on the presentation of the consolidated results of operations or financial position.
| Financial Instruments — Recognition and Measurement |
In January 2005, the CICA released new Handbook Section 3855, Financial Instruments — Recognition and Measurement, effective for annual and interim periods beginning on or after October 1, 2006. This new section prescribes when a financial instrument is to be recognized on the balance sheet and at what amount, sometimes using fair value and other times using cost-based measures. It also specifies how financial instrument gains and losses are to be presented and defines financial instruments to include accounts receivable and payable, loans, investments in debt and equity securities, and derivative contracts. We have not yet determined the impact of the adoption of this standard on the consolidated results of operations or financial position.
42
| Non-Monetary Transactions |
In June 2005, the CICA released new Handbook Section 3831, Non-monetary Transactions, effective for fiscal periods beginning on or after January 1, 2006. This standard requires all non-monetary transactions to be measured at fair value unless they meet one of four very specific criteria. Commercial substance replaces culmination of the earnings process as the test for fair value measurement. A transaction has commercial substance if it causes an identifiable and measurable change in the economic circumstances of the entity. Commercial substance is a function of the cash flows expected by the reporting entity. We have not yet determined the impact of the adoption of this standard on the consolidated results of operations or financial position.
| Hedges |
In April 2005, the CICA issued Section 3865 of the CICA Handbook entitled “Hedges”, effective for years beginning on or after October 1, 2006. This Section establishes standards for when and how hedge accounting may be applied. Hedging is an activity designed to modify an entity’s exposure to one or more risks. Hedge accounting modifies the normal basis for recognizing the gains, losses, revenues and expenses associated with a hedged item or a hedging item in an entity’s income statement. It ensures that counterbalancing gains, losses, revenues and expenses are recognized in the same period. We have not yet determined the impact of the adoption of this standard on the consolidated results of operations or financial position.
Effectiveness of Internal Disclosure Controls
The Chief Executive Officer and the Chief Financial Officer have evaluated the effectiveness of the Company’s disclosure controls and procedures as of December 31, 2005 and have concluded that the Company’s disclosure controls and procedures provide reasonable assurance that material information relating to the Company, including its consolidated subsidiaries, would be made known to them by others within those entities, particularly during the period in which this report was being prepared.
43
BUSINESS
Overview
We are an international, specialty pharmaceutical company focused on improving existing drugs by incorporating our proprietary, advanced controlled-release technologies. Our lead product, a once-daily formulation of the pain-killer tramadol designed to address the worldwide market for moderate to severe pain, has been approved in Europe and is currently the subject of an NDA under review by the FDA with a PDUFA date of September 28, 2006. We currently have three additional product candidates in the clinical stage of development and several product candidates in earlier stages of research and development.
We are developing our once-daily tramadol product to treat patients with moderate to severe pain who want the convenience of once-a-day dosing. Our once-daily tramadol product has completed six Phase III clinical trials in the United States and Europe, including two long-term safety studies, as well as 13 pharmacokinetic studies. In total, more than 2,600 subjects have received our once-daily tramadol product in various dosage forms. We believe that our once-daily tramadol product has generally exhibited a favourable safety and efficacy profile. Specifically, our once-daily tramadol product consists of a dual matrix delivery system designed to provide both rapid and sustained drug release to maintain blood levels within the therapeutic range providing a full 24 hours of pain relief. We believe that maintaining drug concentrations within the therapeutic range has the advantage of fewer and less severe side effects while maintaining efficacy.
We launched our once-daily tramadol product in Germany in November 2005. In September 2005, we received regulatory approval for 22 European countries, under the MRP, and have now obtained MAs for our once-daily tramadol product in 15 countries. We continue to seek marketing authorizations in the remaining seven countries. We plan to launch the product in those countries for which we obtained MAs, but in certain countries, such as France and Spain, we cannot launch the product until we receive pricing and reimbursement approval. We intend to market our once-daily tramadol product primarily through a series of marketing and distribution arrangements in the United States, Europe and the rest of the world. To date, we have entered into agreements for the distribution of our once-daily tramadol product in the United States, in 21 European countries and in 20 Latin American and Caribbean countries. Our marketing and distribution collaborators include Purdue, HEXAL AG, Aventis, Grünenthal GmbH, Recordati, Gruppo Angelini, CSC Pharma, Esteve S.A. and GSK. Although the structures of these agreements are different, they are all designed to provide us with a similar economic return.
Under our licensing and distribution arrangement with Purdue, we received a licensing fee of US$20 million and are eligible to receive up to another US$150 million in milestone payments subject to receiving regulatory approval and meeting specific sales targets in the United States, as well as royalties ranging from 20% to 25% on net product sales by Purdue. We have retained co-promotion rights in the United States to market our once-daily tramadol product to medical specialties that focus on the nervous system and pain. Purdue has agreed to assist us in building and training our sales force, as well as to compensate us for our co-promotion efforts on a fee-for-service basis that we believe will offset the majority of our sales force costs.
Our once-daily formulation of tramadol, as well as our product candidates in clinical development, are based on our proprietary technology, Contramid. Contramid is a versatile, safe, patented and cost-effective technology that we use to provide the benefits of controlled-release drug delivery to existing drugs that have proven safety and efficacy. With tramadol, we successfully demonstrated our technology’s ability to contain a small, highly water soluble molecule and control its release over a 24-hour period with the desired pharmacokinetic profile. We are also developing novel polymeric nano-delivery systems, including polymeric micelles, for delivery of water-insoluble and poorly bio-available drugs, and have developed product candidates based on this technology, both in intravenous and oral formulations.
We are developing additional product candidates using our drug delivery technologies and formulation expertise. Our most advanced product candidates in clinical development include a once-daily formulation of trazodone, a once-daily formulation of betahistine and DDS-2001. We plan to advance our
44
once-daily trazodone into Phase II clinical trials in 2006. We believe that a safe and effective once-daily trazodone could substantially improve compliance, and therefore, performance, of a well-known drug to treat depression. In addition we are currently pursuing tramadol product line extensions, including combination products using our Contramid technology. Our product pipeline includes predominantly proprietary and a few collaborative programs, with products in both clinical trials and pre-clinical development.
Our Strategy
Our goal is to become a fully integrated, international specialty pharmaceutical company, with the expertise and infrastructure to develop and commercialize proprietary therapeutics by taking them from the formulation stage through clinical development, regulatory approval, marketing and sales. Key elements of our strategy are to:
| • | Maximize the global commercial opportunity for our once-daily tramadol product. |
| • | In Europe, we have MRP approval in 22 countries and marketing and distribution collaborations covering 21 countries. We launched in Germany with HEXAL AG in November 2005 and plan to continue launching throughout 2006 in the other countries in which our once-daily tramadol product has been approved, following receipt of MAs and any required pricing and reimbursement approvals. | |
| • | In the United States the FDA has accepted our NDA for review and we have a PDUFA action date of September 28, 2006. We believe that the addition of our recently announced MDT3-005 data to our NDA will enhance our NDA and improve its potential for approval without delaying the timing of our action date. At the same time, we are working with Purdue to launch as soon as possible after approval. | |
| • | In the rest of the world, we are leveraging our European approval to obtain additional approvals. We have received approval in Mexico and have entered into a marketing and distribution arrangement with GSK with the goal of launching the product in Latin America and the Caribbean in 2006 and 2007. |
| • | Develop a robust pipeline of additional products.In addition to our once-daily tramadol product, we have three product candidates in clinical development, several additional product candidates in pre-clinical development and we continue to expand our technological capabilities. Among the product candidates in clinical development our once-daily trazodone has the highest priority, tying into our approach to bring additional central nervous system opportunities to support the further development of our specialty sales force. Simultaneously, we are developing line-extensions, such as combination products, to our once-daily tramadol product. New technology applications for Contramid, for example, abuse resistant formulations, may allow us to develop additional, innovative products, and advances in our polymeric nano delivery systems are encouraging, with potential products in development for intravenous and oral applications. In addition, we may selectively acquire marketed products, development stage compounds, drug delivery technologies or companies that augment or accelerate our strategy. |
| • | Develop a specialty sales and marketing infrastructure in the United States.As part of our strategy to become an integrated specialty pharmaceutical company, we plan to build a U.S. specialty sales force comprising between 50 and 75 sales representatives and an appropriate sales and marketing management infrastructure. Under our agreement with Purdue, the costs of initially building and training this sales force will be borne by them and they will compensate us for our co-promotion efforts on a fee-for-service basis that we believe will offset the majority of our sales force costs. The co-promotion rights for our once-daily tramadol product in the United States will allow us to market to certain medical specialties that focus on the central nervous system and pain. We anticipate leveraging our sales force to market other products that we may develop or acquire. We intend to commercialize ourselves, or obtain co-promotion rights for, products targeted at certain markets that can be addressed with a smaller sales force or leveraged through additional co-promotion |
45
| opportunities. We believe this strategy allows us to maximize the value we retain from the commercialization of our product candidates. | |
| • | Leverage global relationships for future opportunities.We have established a global distribution network for our once-daily tramadol product which we believe can be leveraged for new opportunities. We form strong alliances and relationships which may provide us with access to these markets for our future products. In addition, the existing product pipelines of our collaborators provide an opportunity to identify other products for reformulation, which can be developed and marketed in territories where individual collaborators may not have the required capabilities. For example, after establishing our alliance with Gruppo Angelini, we entered into a feasibility agreement to develop a once-daily formulation of trazodone. |
Our Once-Daily Tramadol
| Overview |
We are developing a once-daily formulation of tramadol. Tramadol is a non-scheduled, centrally acting analgesic for the treatment of pain. It is currently available in the United States for the treatment of moderate to moderately severe pain as an immediate release formulation, typically requiring four to six doses per day, and as a recently launched once-daily formulation. In Europe, tramadol is currently available as immediate release, twice-daily and once-daily formulations for the treatment of moderate to severe pain. Our once-daily tramadol product consists of a dual matrix delivery system designed to provide both rapid and sustained drug release to maintain blood levels within the therapeutic range providing a full 24 hours of pain relief. We believe that maintaining drug concentrations within the therapeutic range has the advantage of fewer and less severe side effects while maintaining efficacy.
The figure below is a representative data plot from our MDTI-011 clinical trial of mean tramadol plasma concentrations over a 24-hour dosing interval following a single oral dose of 200 mg of our once-daily tramadol product and what we believe to be the optimal therapeutic range.
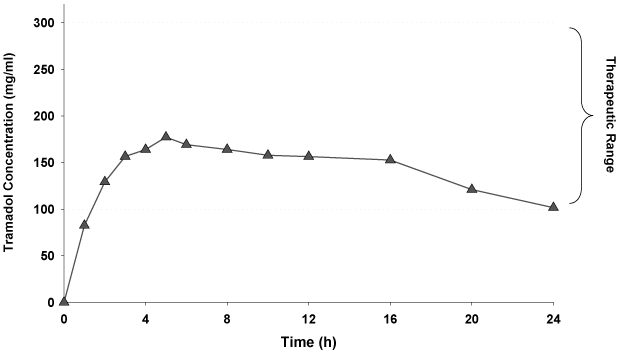
46
| Pain Market |
The treatment of pain represents a significant commercial opportunity and we believe continued growth of this market will be fueled by a number of factors including:
| • | a rapidly aging demographic with longer life expectancy; | |
| • | physicians’ recognition of pain as a health problem and their increased willingness to treat the condition; | |
| • | increasing number of pain sufferers seeking help and demanding effective treatment; and | |
| • | new reformulations of existing products. |
Pain can be classified by duration (acute or chronic) and severity (mild, moderate or severe). Acute pain has a well-defined point of onset, is typically associated with a known trauma or surgery, and usually lasts for less than one month, but may last for up to three months. Chronic pain has a less defined temporal onset and cause, and persists for more than three months. Examples of chronic pain include chronic low back pain, cancer pain and various arthritic conditions.
To help practitioners deal with this significant health issue, the WHO established a three-step analgesic ladder for the treatment of chronic pain states. In the case of mild pain, Step 1 suggests prompt treatment with a non-opioid such as acetaminophen, aspirin or NSAIDs. For moderate pain, Step 2 recommends introducing a mild opioid such as tramadol or codeine. For severe pain, Step 3 suggests treatment with morphine or other strong opioids until the patient is pain free. The WHO further recommends that to maintain freedom from pain, drugs should be given “by the clock”, that is every three to six hours for immediate release formulations, rather than “on demand”. However, patient compliance is one of the leading challenges to effective treatment. In an aging population, more likely taking multiple medications, it is increasingly difficult for patients to manage their medication regimen and take the next dose at the appropriate time.
Scheduled opioids, such as morphine and morphine-derived drugs, are sub-optimal chronic medications due to their gastrointestinal, respiratory and addictive side-effects, which can compromise patient compliance. Effective management of moderate to severe chronic pain requires treatment that can be tolerated over a long period of time with relatively few side effects and that can be taken in a dosing regimen that facilitates patient compliance and reduces the risk of breakthrough pain. We believe that our once-daily tramadol product addresses these issues.
| Tramadol |
Tramadol is a proven analgesic that has been on the market for nearly three decades. It has been marketed in approximately 100 countries worldwide. Studies have shown that tramadol provides relief from both acute and chronic pain conditions, including osteoarthritis pain, low back pain, cancer pain, post-operative pain and dental pain. Tramadol avoids the potentially serious side effects such as gastrointestinal bleeding that may be caused by NSAIDs and the potential cardiovascular events recently attributed to COX-2 inhibitors. Compared to stronger opioids, tramadol has, in clinical studies, demonstrated a lower incidence of major side effects, such as respiratory depression. When used over the long-term, tramadol has a significantly lower incidence of analgesic tolerance, withdrawal symptoms and addiction as compared to stronger opioids. However, tramadol has been available primarily as formulations requiring more than one dose per day. We have overcome these limitations by developing a tramadol formulation that patients can take once a day and that has been shown clinically to provide pain relief lasting a full 24 hours, thereby maintaining the patient’s pain under control. Furthermore, our formulation provides for an early phase of tramadol release, with a rate comparable to that of the immediate release formulations.
| Commercialization of our once-daily tramadol product in the United States |
In the United States the prescription pain product market exceeded US$15 billion in 2005 on more than 333 million prescriptions. The United States is the world’s single biggest market for tramadol products.
47
In 2005, the number of prescriptions of tramadol products surpassed 19 million, more than doubling over the last five years, with a growth of 12% from the previous year.
The table below sets out the retail sales and prescriptions of selected major classes of pain products in the United States in 2005 as reported by Verispan.
| Prescriptions | Sales | |||||||
| Class | (in millions) | (in US$billions) | ||||||
Opioids | 217.9 | 8.3 | ||||||
| Tramadols (e.g., Ultram, Ultracet) | 19.1 | 0.7 | ||||||
| Hydrocodones (e.g., Vicoden, Vicoprofen, Norco) | 106.6 | 1.7 | ||||||
| Propoxyphenes (e.g., Darvon, Darvocet) | 24.0 | 0.3 | ||||||
| Other | 68.2 | 5.6 | ||||||
Non-opioids | 115.5 | 7.5 | ||||||
| NSAIDs (e.g., Mobic, Naprosyn) | 76.2 | 2.4 | ||||||
| COX-2 (e.g., Celebrex) | 12.4 | 1.4 | ||||||
| Other | 26.9 | 3.7 | ||||||
Tramadol is part of a sub-class of opioids used to treat moderate to moderately severe pain. This sub-class, which also includes the hydrocodone combinations and propoxyphenes, generated approximately 150 million prescriptions in the United States in 2005. As all of these products required multiple daily dosing in 2005, patients may benefit from an effective once-daily pain reliever indicated for the same pain severity.
Osteoarthritis sufferers had been well served by the introduction of the once-daily COX-2 inhibitors, including Celebrex, Vioxx and Bextra, in addition to the opioid analgesics. As a consequence of the recent safety issues regarding increased cardiovascular risk and the subsequent withdrawals from the market of Vioxx and Bextra, sales and prescriptions of the COX-2 class dropped by approximately 70% in 2005. We believe that some of these lost prescriptions were re-addressed within the NSAID class, and in particular, to Mobic, a once-daily NSAID. However, for those patients unwilling to take or unable to tolerate NSAIDs, few treatment options are available with the benefits of once-daily dosing.
In January 2003, we initiated two double-blind, multi-center, randomized Phase III clinical studies in the United States designed to evaluate the safety and efficacy of our once-daily tramadol product. These studies compared our once-daily tramadol product to placebo. The two studies, MDT3-002 and MDT3-003, were similar in design and size, each enrolling more than 500 patients diagnosed with moderate to moderately severe pain associated with osteoarthritis of the knee. The patients were treated over a 12-week period. We completed the patient treatment stage of these trials in September 2003 and announced the results in January 2004.
The pivotal MDT3-003 trial achieved statistical significance for the three co-primary endpoints at the 200 mg and 300 mg doses, demonstrating an improvement as measured by the change in the WOMAC Osteoarthritis Index pain subscale, the WOMAC Osteoarthritis Index physical function subscale and the patient’s global assessment of drug effectiveness. The WOMAC Osteoarthritis Index is a validated tri-dimensional measure used to assess changes in patients with osteoarthritis using pain, stiffness and physical function subscales. We included a 100 mg dose to determine the lowest effective dose and this dose is intended to be used as a titration dose. The MDT3-002 trial, partly due to, we believe, an unusually high response to placebo, did not achieve statistical significance for all of the co-primary endpoints. However, in both trials, our once-daily tramadol product provided pain relief for a full 24-hour period and the incidence of adverse events was lower than that reported in the literature for the immediate-release formulation of tramadol currently available in the United States requiring typically four to six doses per day.
In October 2004, we initiated a third double-blind, randomized Phase III study, MDT3-005, to compare the safety and efficacy of our once-daily formulation of tramadol to placebo over a 12-week period in patients with moderate to severe pain associated with osteoarthritis of the knee. The study was conducted
48
at more than 100 centers and enrolled more than 1,000 patients. The study design differed from that of the previous U.S. Phase III trials by allowing patients to titrate to optimum dose. In addition, the study was conducted under a Special Protocol Assessment, or SPA. An SPA is designed to facilitate the FDA’s review and approval of drug products by allowing the agency to evaluate the proposed design and size of clinical trials that are intended to form the primary basis for determining a drug product’s efficacy. If agreement is reached with the FDA, an SPA documents the terms and conditions under which the design and planned analysis of the subject trial will be adequate to address the study’s objectives in support of an NDA.
We completed the patient treatment stage in January 2006 and announced top line results in April 2006. The primary endpoint of the study was to compare baseline pain intensity with pain intensity at the end of the study period as measured by the 11-point Pain Intensity Numerical Rating Score (PI-NRS) in the once-daily tramadol group versus the placebo group. Statistical significance was achieved for the primary endpoint (p value of 0.0157) and was maintained under additional methods of analysis. The drop-out rate was comparable to that of our European Phase III trial (MDT3-001). The incidence of adverse events was lower than in study MDT3-001, and comparable to the incidence in our MDT3-002 and MDT3-003 trials.
We have also completed two long-term safety and tolerability studies using once-daily tramadol. In MDT3-004, patients were treated on a 300 mg daily dose for up to 12 months and in MDT3-001-EIA1, an extension of the double-blind MDT3-001, patients were maintained on either 200 mg, 300 mg or 400 mg for up to 12 months. A total of 630 patients entered the two studies, of which 493 completed at least six months and 243 completed 12 months on our once-daily tramadol product. The incidence of the most common adverse events in these long-term studies was comparable to that observed in our 12-week efficacy studies.
The following chart summarizes our Phase III development program for our once-daily tramadol product.
| Number of | Endpoints | |||||||||||
| Study | Duration | Subjects in Trial | Primary Endpoints | Met | ||||||||
| MDT3-001 | 12 weeks | 431 | WOMAC pain (non-inferiority vs twice-daily) | ü | ||||||||
| MDT3-001-E1A1 | Up to 12 months | 238 | Long-term safety and tolerability | ü | ||||||||
| MDT3-002 | 12 weeks | 565 | 3 co-primary endpoints (vs. placebo) | |||||||||
| i) WOMAC pain | ||||||||||||
| ii) WOMAC function | ||||||||||||
| iii) patient global assessment | ü | (*) | ||||||||||
| MDT3-003 | 12 weeks | 552 | 3 co-primary endpoints (vs. placebo) | |||||||||
| i) WOMAC pain | ü | |||||||||||
| ii) WOMAC function | ü | |||||||||||
| iii) patient global assessment | ü | |||||||||||
| MDT3-004 | Up to 12 months | 392 | Long-term safety and tolerability | ü | ||||||||
| MDT3-005 | 12 weeks | 1,028 | Pain intensity score (vs. placebo) | ü | ||||||||
| (*) | Statistical significance only achieved at the 300 mg dose. |
49
We submitted an NDA to the FDA in November 2005 under Section 505(b)(2) of the United States Federal Food, Drug, and Cosmetic Act and relied on the agency’s previous findings of safety and efficacy for Ultram. The submission included 11 pharmacokinetic studies and five Phase III studies, including two long-term safety studies. Approximately 1,600 subjects were exposed to the drug in these studies. The NDA was accepted for review and filed by the FDA in January 2006, with an action date under the PDUFA of September 28, 2006.
In the course of discussions with the FDA in 2005, they advised us that a single positive, well controlled pivotal Phase III study would be sufficient for an NDA submission and that MDT3-005 would no longer be required. This position was an evolution from the FDA’s previous requirement of two positive Phase III studies versus placebo. However, as announced in April 2006, we plan to submit the results of clinical trial MDT3-005 to the FDA no later than June 28, 2006 as a positive, well-controlled clinical study in support of our NDA. The FDA has agreed to accept the MDT3-005 report and review the data in that report, if received by June 28, 2006, within the original 10-month review cycle ending September 28, 2006.
In August 2005, we entered into a license agreement with Purdue pursuant to which we granted to Purdue the exclusive right to manufacture, package, distribute, market and sell our once-daily tramadol product in the United States. We have retained co-promotion rights in the United States to market our once-daily tramadol product to medical specialties that focus on the central nervous system and pain.
Under the license agreement, we are responsible for continuing the development of our once-daily tramadol product and conducting the necessary clinical trials in order to obtain regulatory approval from the FDA. Purdue has agreed to assist us in building and training our sales force, as well as to compensate us for our co-promotion efforts on a fee-for-service basis that we believe will offset the majority of our sales force costs.
Purdue has paid us a non-refundable, non-creditable, up-front fee of US$20 million. Purdue has also agreed to pay us additional non-refundable, non-creditable development milestone payments of up to a maximum of US$40 million depending upon the time of launch of our once-daily tramadol product and sales success milestone payments of up to a maximum of US$110 million based on aggregate net sales of our once-daily tramadol product. We are also entitled to receive a royalty at rates ranging from 20% to 25% of net sales of our once-daily tramadol product sold by Purdue in the United States.
In addition to the license agreement, we entered into a supply agreement with Purdue in October 2005 to supply Purdue with our once-daily tramadol product until Purdue assumes responsibility for manufacturing, packaging, testing, and supplying such product or until 18 months following the launch of our once-daily tramadol product in the United States, whichever is earlier. However, if we are not able to supply Purdue with sufficient quantities of our once-daily tramadol product to meet Purdue’s launch requirements, we have agreed to extend the 18-month deadline. The term of the license agreement lasts until the end of the commercial life of our once-daily tramadol product or of any generic product which may be distributed by Purdue under the license agreement. If Purdue terminates the agreement due to an uncured material breach by us, then, under the agreement, we are obligated to transfer our rights to our once-daily tramadol product in the United States to Purdue, and Purdue will be obligated to pay us a specified royalty on net sales from that point forward. If we terminate the agreement due to an uncured material breach by Purdue, then, under the agreement, Purdue is obligated to transfer its rights to our once-daily tramadol product in the United States to us, and we will retain the license to commercialize the once-daily tramadol product under the Purdue intellectual property rights in the United States without any royalty obligation to Purdue.
In November 2005, Purdue, with our consent, granted a license to Biovail’s licensee, OMI, under certain patents owned or controlled by Purdue covering Biovail’s extended release tramadol product to make, use and sell such product in the United States. This license would allow OMI to list those Purdue patents in the FDA’s Orange Book of Approved Drug Products with Therapeutic Equivalence Evaluations, thus providing a reference. In exchange, we secured a waiver from OMI, as an agent of Biovail, of the three-year non-patent exclusivity granted to Biovail under the Hatch-Waxman Act for Biovail’s tramadol product, allowing the FDA to grant approval of our once-daily tramadol product based on its merits. We also received a right of reference from OMI authorizing the FDA to rely upon any data contained in any regulatory filing
50
held by OMI for drug products containing tramadol in support of our regulatory filing for our once-daily tramadol product.
| Commercialization of our once-daily tramadol product in Europe |
As tramadol was first launched in Germany in 1977, the European market is the most mature of the global tramadol markets. Europe accounts for about half of the 4.5 billion tramadol standard units sold in the world in 2005. As defined by IMS Health, standard unit is determined by dividing the number of units sold by the smallest common dose of a product representation such as one tablet or capsule for solid oral formulations. Last year, sales of tramadol products in Europe exceeded US$638 million and have increased approximately 16% on a compounded annual basis over the past five years.
In early 2003, we completed the pivotal Phase III clinical trial MDT3-001 in Europe for our once-daily tramadol product. The double-blind, randomized, parallel group study enrolled 431 patients and was designed to compare the efficacy and safety of our once-daily tramadol product to that of an extended release, twice-daily formulation of tramadol currently marketed in Europe. The primary endpoint of the study was pain relief as measured by the WOMAC Osteoarthritis Index pain subscale. We reported positive results from this trial in January 2003. Our product candidate met all primary and secondary endpoints and exhibited a statistically and clinically significant reduction in pain associated with osteoarthritis of the knee. The adverse events profile of our once-daily tramadol product showed a clinical advantage with respect to the incidence of dizziness/vertigo, vomiting and headache compared to twice-daily tramadol. All of these adverse events occurred less frequently and were less severe in our once-daily tramadol product group than in the active comparator group. Our once-daily tramadol product also provided sustained pain relief for a full 24-hour period, similar to the performance of the twice-daily European formulation taken every 12 hours.
In January 2005, we received the MA in France for our once-daily tramadol product. Under the MRP, France, acting as the Reference Member State, or RMS, requested that all Concerned Member States in the EU recognize the MA granted in France as the RMS. Pursuant to this request, we received MRP approval in September 2005 in the following additional 21 European countries: Germany, the United Kingdom, Italy, Spain, Portugal, Austria, Belgium, Cyprus, the Czech Republic, Denmark, Estonia, Iceland, Latvia, Lithuania, Luxembourg, Malta, the Netherlands, Poland, Slovakia, Slovenia and Sweden. In the European Economic Area, a medicinal product may only be placed on the market when an MA has been issued by a competent authority. As such, we have since received MAs in 15 of the countries in which we obtained MRP approval and anticipate receiving the remaining MAs during 2006. In those countries that do not require pricing approval or reimbursement, such as Germany and the United Kingdom, upon receipt of the MA, we will be able to market our once-daily tramadol product through distributors to whom we have granted the right to market and sell our once-daily formulation, whereas in other countries, such as Spain and France, we are required to obtain government approval of pricing and reimbursement prior to product launch. In addition, we intend to submit national registration dossiers in other strategic European tramadol markets not party to the MRP, as well as in countries that were withdrawn from the MRP due to labeling issues.
Following receipt of the MA in Germany, our distributor HEXAL AG launched our once-daily tramadol product into the German market in November 2005, under the tradename Tramadolor einmal täglich, supported by a sales force of more than 400 sales representatives. The product is being marketed with the common label approved under the MRP, which designates it as a once-daily product for the treatment of moderate to severe pain, including both acute and chronic conditions, for use in a dose range from 100 to 400 mg per day. It is available in three dosage strengths (100 mg, 200 mg and 300 mg) and three pack sizes (20 tablets, 50 tablets and 100 tablets).
To date, we have executed agreements with seven pharmaceutical companies covering 21 European countries and some of their overseas territories. We have signed licensing and distribution agreements with HEXAL AG for Germany, Aventis and Grünenthal GmbH for France and its overseas territories, Esteve S.A. for Spain and Portugal, Gruppo Angelini for Italy, Recordati for the United Kingdom and CSC Pharma for Austria, Russia and 13 Eastern European countries. Through our agreements, we have established a distribution network in territories accounting for 87% of total European tramadol sales. Our distribution
51
relationships in Europe and the rest of the world are designed specifically by territory and aim to achieve similar overall economic participation on sales to our Purdue agreement.
Pursuant to these relationships, we are responsible for supplying finished packaged goods to the distributors. Under these relationships, we received up-front payments with additional payments due upon achieving certain milestones. We will also receive revenue from the sale of finished packaged product to our distributors. This product revenue will include revenue from sales of product for inventory stocking and product samples, as well as product for commercial sale. With anticipated launches in multiple countries throughout 2006, we anticipate that it will take a period of time for product revenues to normalize and correspond more directly to future retail sales. We are actively engaged in discussions with additional prospective marketing and distribution collaborators for other European countries.
| Commercialization of our once-daily tramadol product in the rest of the world |
Tramadol has been marketed in approximately 100 countries worldwide. Global sales in countries other than the United States and Europe in 2005 were more than US$160 million. Compounded annual sales growth over the last five years approximates 20%, outpacing growth in both the European and U.S. markets. We believe that Latin America, Canada, Southeast Asia and Australia each represent sizeable and expanding commercial opportunities for us.
The Latin American analgesic market has experienced accelerated growth in recent years, surpassing sales of US$1.9 billion in 2005. In 2005 we entered into a licensing and distribution agreement for our once-daily tramadol product with GSK. The agreement covers 20 Latin American and Caribbean countries.
Under the terms of the licensing and distribution agreement with GSK for the 20 Latin American and Caribbean countries, we have agreed to supply GSK with product and GSK has agreed to register and distribute the product throughout the licensed territory. We are entitled to receive a transfer price on tablet supply based on a percentage of the selling price, resulting in an effective royalty rate commensurate with that of our agreement with Purdue. We are also in discussions for marketing and distribution agreements for other countries.
We are pursuing regulatory approval for our once-daily tramadol product in other countries around the world. In most cases, we can use the regulatory dossier approved in Europe as a basis for the registration of the product in additional markets. For example, we received regulatory approval in Mexico in October 2005 following the submission of a marketing application based on the approved European dossier to the Mexican regulatory authority, General Directorate for the Control of Medicinal Products and Technologies Related to Health, in March 2005. Other regulatory dossiers are in preparation for key tramadol markets.
Product Pipeline
We are developing additional product candidates using our drug delivery technologies and formulation expertise. Our most advanced product candidates in clinical development include a once-daily formulation of trazodone, a once-daily formulation of betahistine and DDS-2001. In addition we are currently pursuing tramadol product line extensions, including combination products, using our Contramid technology. Our product pipeline includes predominantly proprietary and a few collaborative programs, with products in both clinical trials and pre-clinical development.
52
The following table sets forth our product candidates and their current development status.

| Our Once-Daily Trazodone |
Trazodone is an antidepressant currently available in the United States in both branded and generic immediate release formulations that typically require three doses per day. In Europe, trazodone is available in both immediate release and twice-daily formulations. Trazodone is indicated and prescribed for depression with or without anxiety.
Depression is among the most prevalent central nervous system disorders, affecting at least 121 million people worldwide according to the WHO. Depression is characterized by depressed mood, loss of interest or pleasure, weight or appetite changes, and insomnia or excessive sleep or fatigue. In the United States, sales of antidepressants approximated US$13 billion in 2005. Trazodone continues to be widely prescribed in the United States, with more than 14 million prescriptions written in 2005 and having a compounded annual growth rate of 5% over the past five years.
Pursuant to a feasibility and formulation agreement with Gruppo Angelini, we are developing a formulation of our once-daily trazodone product that would be bioequivalent in terms of drug absorption to a currently marketed immediate release product administered three times daily or twice-daily product. We have developed once-daily trazodone formulations in dosage strengths that have demonstrated controlled release over a 24-hour period and dose proportionality in pharmacokinetic trials. We are currently developing clinical protocols with the intent of advancing this program into Phase II clinical trials in 2006. We expect that any NDA for this product candidate would be submitted under Section 505(b)(2) of the U.S. Federal Food, Drug, and Cosmetic Act. We are discussing the terms and conditions of an agreement with Gruppo Angelini with regard to the potential clinical development and commercialization of our formulation of once-daily trazodone.
53
| Once-Daily Betahistine |
We are developing a once-daily formulation of betahistine, an oral histamine analogue, for the treatment of the symptoms related to Ménière’s disease, a disorder characterized by recurrent vertigo and hearing loss. Currently marketed versions of betahistine require two or three doses per day resulting in significant fluctuations in the plasma concentrations of the drug. Betahistine is thought to treat vertigo by normalizing the flow of impulses from the vestibular organs to the brain and improving blood flow in the inner ear.
Ménière’s disease affects approximately one in 500 people throughout the world. Betahistine products are marketed in more than 80 countries, including most of Europe and Latin America, but not in the United States. Worldwide sales of betahistine products approximated US$220 million for the 12 months ended March 31, 2004.
We formulated two dosage strengths of our once-daily betahistine product, manufactured clinical batches of the highest dosage strength internally and tested the highest dosage strength in pilot comparative pharmacokinetic studies. The results demonstrated controlled release of betahistine consistent with once-daily dosing of the drug. We are exploring the possibility of out-licensing the formulation as a novel once-daily betahistine product for development by a third party. We do not plan to conduct any further development of this product candidate until we determine whether to out-license the product.
| DDS-2001 |
Together with MedPointe Inc., we are developing a novel sustained-release product, which we call DDS-2001. The details of this product have not been disclosed publicly, but an immediate release formulation of this product is currently marketed by MedPointe.
We have completed the initial formulation phase of this product, scaled-up the manufacturing process for clinical trial batch manufacture and tested prototype formulations in a pilot pharmacokinetic study. Subsequently, we proposed an agreement and a development plan to MedPointe which has not been finalized. In the event that we do not conclude an agreement with MedPointe, we do not plan to move forward with the development of this product.
Drug Delivery Technologies
Our current product candidates make use of one of our following drug delivery technologies:
| • | Contramid, for oral administration of soluble to sparingly soluble drugs; and | |
| • | Polymeric nano-delivery systems technologies, for delivery of water insoluble or poorly bioavailable drugs via a variety of administration routes. |
We believe controlled-release drug delivery has the ability to improve the performance of a wide variety of existing drugs. The therapeutic effect of a drug will typically vary according to its concentration in the bloodstream. When the concentration is too low, efficacy may be reduced and when it is too high, side effects may increase. For any drug, there is a range of concentrations, the therapeutic range, in which the balance between efficacy and side effects is optimized. In traditional tablets without controlled-release, blood levels of the drug tend to rise quickly after administration, reach a peak and then drop fairly rapidly, thus requiring frequent dosing. Side effects may also occur at the high peak blood levels experienced. Controlled-release allows a drug’s concentration to be maintained within the therapeutic range, extending its therapeutic effect over a prolonged period and often improving its safety profile. This added convenience may increase patient compliance and therefore improve medical outcomes.
| Contramid |
Contramid is a patented, controlled-release drug delivery technology based on cross-linked, high amylose starch, for oral administration of solid dosage medications. Following ingestion, gastric fluids transform the surface of a Contramid-based product into a semi-permeable membrane that stabilizes rapidly.
54
This self-forming membrane regulates the release of the drug. We have developed the expertise to formulate novel products that combine our Contramid technology with existing drugs to optimize their release profile.
We believe that our Contramid technology possesses advantages that make it an attractive drug delivery technology, including:
| • | Compatibility.Our Contramid technology may be combined with numerous drugs, including those requiring high drug loading or possessing varying degrees of solubility. Typically, our Contramid technology is best suited for highly soluble to sparingly soluble drugs. | |
| • | Versatility.Our Contramid technology may be used to achieve multiple release profiles, including delayed-released, dual-release and controlled-release in conjunction with gastric retention. | |
| • | Cost Effectiveness.Together with Cerestar USA Inc. and Cerestar France S.A., affiliates of Cargill Inc., we believe we have developed a reliable, reproducible and economical manufacturing process for our Contramid technology on a commercial scale. As it is a compressible, versatile excipient, we believe our Contramid technology allows for simple and efficient manufacturing of the final pharmaceutical product using standard equipment and processes. We expect these manufacturing synergies, together with our Contramid technology’s relatively low cost, to result in minimal incremental cost to the final marketed product. | |
| • | Manufacturing Robustness.Our Contramid technology has demonstrated relative insensitivity to broad variations in production parameters such as compression pressure, compression time, type of tabletting equipment and batch size. This results in simplified process development and technology transfer to a commercial manufacturing site. We believe this also allows for robust and reproducible batch-to-batch consistency. | |
| • | Safety and Regulatory Acceptance.Our Contramid technology conforms to the specifications for modified starch in the U.S. National Formulary and the Food Chemicals Codex and as such is considered a food additive by the FDA and European regulatory authorities. As modified starches have a long history for food use under these regulations, our Contramid technology may be used without limit to quantity with orally administered drugs. Regulatory authorities in 22 European countries and in Mexico have recently approved the use of our Contramid technology in our once-daily tramadol product. | |
| • | Intellectual Property.Our Contramid drug delivery technology is covered by one patent that was issued in the United States, which is scheduled to expire in 2020. A counterpart patent application was allowed in Europe in November 2005 and additional related applications have been filed and are either issued or pending in various jurisdictions outside the United States and Europe, including Canada. In addition, we own or have rights to six issued U.S. patents and foreign counterparts thereof which relate to modified starches in drug delivery technologies. |
Our once-daily formulations of tramadol and trazodone, as well as our other product candidates in clinical development, are based on our patented technology, Contramid. Our success in formulating our once-daily tramadol product illustrates our ability to employ our Contramid technology, formulate and execute a clinical plan for a complex small, highly water soluble molecule and control its release over a 24-hour period with the desired pharmacokinetic profile. As an extension of our core technology, we are developing new presentations of Contramid that will potentially broaden its applications, including Contramid hard and soft gels, Contramid films and abuse resistant formulations employing our Contramid technology.
| Polymeric Nano-Delivery Systems Technology |
Our polymeric nano-delivery systems technology is a complementary platform and provides us with the capabilities to address the delivery challenges associated with water insoluble compounds, as well as proteins, peptides and nucleic acids. We believe this technology may have the potential to improve the
55
delivery of currently administered intravenous products, as well as to increase bioavailability for potential oral delivery.
We are currently developing a series of novel polymeric nano-delivery technologies, including polymeric micelles, for effective delivery of poorly water-soluble or poorly bioavailable drugs via a variety of routes including oral and intravenous administration. The ability to deliver poorly soluble, often highly toxic, drugs safely and effectively is a continuing challenge to the pharmaceutical industry. Consequently, we believe many potentially valuable drug candidates fail to reach commercialization, and, lacking an alternative, those that do may by necessity be formulated using agents often associated with dose-limiting side effects. The polymeric nano-delivery system we are developing is intended to offer safer, more efficient and lower cost alternatives to those agents currently used. They comprise block copolymers that self-assemble in water to form micelles and similar nanostructures with high solubilizing and loading capacities. We believe that we have developed an efficient drug loading process for these systems that is scalable and suitable for commercial production. We expect this technology to have applications in many fields, including oncology and immunology, and to facilitate delivery of proteins, peptides and nucleic acids.
Completed proof of concept studies confirm our belief that our polymeric nano-delivery systems technology has the potential to improve the deliverability of current intravenous products like Propofol, a commonly used anaesthetic. Using our polymeric nano-delivery systems, we have developed a freeze-dried formulation of Propofol that in pre-clinical models has comparable efficacy to that of a currently marketed branded formulation, but has additional benefits in that it alone does not support bacterial growth, is clear and highly stable and can be resuspended in a variety of infusion solutions for greater ease of use. We have also completed proof of concept studies which suggest that our oral platform has the potential to increase bioavailability and may allow for targeted delivery. This could result in the substantial improvement of a wide variety of therapeutics, particularly in the field of oncology. We will work towards completing further proof of concept studies involving this technology during 2006.
Intellectual Property
Our success depends in part on our ability to obtain patents, protect trade secrets, operate without infringing the proprietary rights of others and prevent others from infringing our proprietary rights. Our policy is to seek to protect our proprietary position by, among other methods, filing in the United States and other countries patent applications related to our proprietary drug delivery technologies, inventions and improvements that are important to the development of our business.
Our patent portfolio currently protects our products and technologies as follows:
| • | Once-Daily Tramadol Product.We have filed patent applications in the United States and abroad relating to our once-daily tramadol product. Our patent applications in the United States contain claims directed to once-daily tramadol formulations which provide for specified analgesic effects, pharmacokinetic profiles, in vitro dissolution profiles, specified clinical profiles and methods and kits for titrating tramadol dosages. In addition to our tramadol applications, because our once-daily tramadol product contains our Contramid drug delivery technology, our once-daily tramadol product is protected by our issued U.S. patent and a corresponding patent application that has been allowed by the European Patent Office relating to our Contramid drug delivery technology. We also have a license from Purdue to certain U.S. patents owned by Purdue directed to the controlled-release formulation of tramadol. | |
| • | Contramid Drug Delivery Technology.We own an issued U.S. product-by-process patent pertaining to our Contramid drug delivery technology. This patent is scheduled to expire in 2020. Counterpart patent applications have been filed and are either pending or issued in jurisdictions outside the United States, including an allowance received in November 2005 from the European Patent Office. We own or have rights to six other issued U.S. patents and foreign counterparts thereof which relate to modified starches as drug delivery technologies. |
56
| • | Polymeric Nano-Delivery Systems Technology.We own or have exclusive rights, including by license, to five issued U.S. patents, certain U.S. pending patent applications and foreign counterpart patents and applications that relate generally to polymeric nano-delivery systems technology. | |
| • | Other Products.Because our other products utilize our proprietary drug delivery technologies we believe that they may also benefit from the protection provided by our issued U.S. patents and their counterparts relating to our Contramid technology and polymeric nano-delivery systems technology. Nevertheless, we endeavour to file patent applications on our potential product development candidates. For example, we have filed applications pertaining to our once-daily tramadol product and once-daily trazodone formulations. |
The patent positions of companies like ours are generally uncertain and involve complex legal and factual questions. Our ability to maintain and solidify our proprietary position for our technology will depend on our success in obtaining effective claims and enforcing those claims once granted. We do not know whether any of our patent applications will result in the issuance of any patents. Even if issued, they may not provide us with a competitive advantage against competitors with similar technologies. Furthermore, others may design around our patents and develop similar technologies or duplicate any technologies that we have developed. The laws of many countries do not protect our intellectual property to the same extent as the laws of the United States.
We are aware of issued United States, Canadian and European patents that contain claims that might be infringed by our once-daily tramadol product. If our once-daily tramadol product or our other product candidates infringe or are claimed to infringe a valid claim of a third party patent, including these patents, we may choose or may be required to obtain a license under such patent or seek a court judgment that such patent claims are invalid. In the United States, we have entered into a license agreement with Purdue, pursuant to which we obtained rights to all U.S. patents owned by Purdue relating to the manufacturing, marketing and distribution of our once-daily tramadol product. Claims of issued patents are presumed to be valid, and any finding of invalidity would come, if at all, only after litigation that could prove to be lengthy, costly and unsuccessful. There are numerous issued patents and pending patent applications owned by others in the U.S. and other countries directed to the technological areas in which we are developing products, including our lead product candidate, our once-daily tramadol product. These patents and patent applications could materially affect our ability to develop our product candidates or sell our products, including preventing us from making, using or selling our products. Because patent applications can take many years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that our product candidates or technologies may infringe. These patent applications may have priority over patent applications filed by us.
Any patent litigation in which we are involved, whether brought against us or brought by us to invalidate a patent would be costly and could divert our technical and management personnel from their normal responsibilities and we may not have sufficient financial or other resources to conduct a successful litigation. Any adverse outcome of such litigation or settlement of such a dispute could subject us to significant liabilities and could require us to stop developing our product candidates, stop selling our products or enter into royalty or licensing agreements which may or may not be available on terms acceptable to us, if at all. Even if we were able to obtain a license, the rights may be non-exclusive, which could result in our competitors gaining access to the same intellectual property. If we do not obtain any required licenses or sublicenses, we could encounter delays in product introductions while we attempt to design around these patents, or could find that the development, manufacture or sale of products requiring these licenses is foreclosed.
We may rely, in some circumstances, on trade secrets to protect our drug delivery technologies. However, trade secrets are difficult to protect. We seek to protect our proprietary drug delivery technologies and processes, in part, by agreements with our employees, consultants and contractors. These agreements may be breached and we may not have adequate remedies for any breach. Also, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our employees, consultants
57
or contractors use intellectual property owned by others in their work for us, disputes may also arise as to the rights in related or resulting know-how and inventions.
We may also be obligated to make royalty payments pursuant to several purchase and development agreements that we have entered into, including pursuant to certain research contracts pertaining to our Contramid technology with Université de Montréal and Université du Québec à Montréal. In 1994, concurrently with the purchase of a controlled-release technology from Université de Montréal and Université du Québec à Montréal, we acquired a right of first refusal with respect to any improvements and additions to the purchased technology for which we agreed to pay royalties of 4% on net revenues that we receive from the commercialization of the purchased technology. On February 7, 2005, Université de Montréal and Université du Québec à Montréal served us with a motion to institute legal proceedings in the Superior Court of Québec. The motion seeks payment of royalties allegedly owed by us with respect to the commercialization of the technology purchased in 1994 and any improvements and additions thereto and its use in various of our product candidates, including our once-daily tramadol product. It is our position that we are not commercializing the 1994 purchased technology or any improvements or additions thereto and that we do not use it in our once-daily tramadol product or any of our product candidates. Consequently, we consider that no amounts are owing and we are vigorously contesting the motion. This matter is in the examination for discovery phase of the proceedings.
We have acquired and licensed our polymeric nano-delivery systems technology from Université de Montréal pursuant to an agreement under which we have agreed to pay royalties of three to six percent of (i) the sale price of the technology; (ii) our revenues arising from the commercialization of the technology; or (iii) our sales of any product using the technology.
Manufacturing
We currently outsource all commercial manufacturing for our once-daily tramadol product and intend to continue this strategy for our pipeline products. We now perform manufacturing of clinical trial materials in our GMP pilot plant which allows for a more cost-effective and timely initiation of clinical studies.
We have an exclusive supply agreement with Cargill to purchase from Cargill all of our requirements for cross-linked, high amylose starch comprising Contramid. Cargill is a leader in the production of starch and starch-based products in both the European and North American markets. Cargill has submitted a Drug Master File for our Contramid technology in the United States. Together with Cargill, we have produced several multi-ton lots of Contramid and demonstrated the ability to achieve reproducible results at commercial scale.
We initially executed the supply agreement in 1998 for a period of ten years with Cerestar USA Inc. and Cerestar France S.A., which became wholly-owned subsidiaries of Cargill. In 2001, we entered into a joint technology license agreement with Cerestar pursuant to which Cerestar granted to us an exclusive license to Cerestar’s rights in U.S. patent No. 6607748 covering the Contramid technology. Our supply agreement with Cerestar was amended in May 2001 and November 2005. As part of the November 2005 amendment, Cargill, through its Cerestar subsidiaries, assigned to us all right, title and interest in U.S. patent No. 6607748, and the joint technology license agreement was terminated. In exchange for such patent assignment, we are obligated to pay Cargill US$1 million.
The supply agreement will expire on the later of November 4, 2018 or the expiry date of the last-to-expire patent covering the Contramid technology. Cargill may terminate the supply agreement if we fail to make payments that are due or we become insolvent. If Cargill decides to cease manufacturing Contramid, Cargill may also terminate the supply agreement, subject to Cargill using reasonable efforts to notify us at least two years in advance of such termination.
We are currently in the process of negotiating with Cargill a new, exclusive long-term supply agreement extending throughout the expected life of our patent protection, which agreement will supersede and replace the 1998 supply agreement. Upon execution of a new supply agreement, Cargill shall have an
58
option to negotiate with us an exclusive license to exploit our Contramid technology in the food and flavours industry, not including nutraceuticals and dietary supplements. This option expires on November 17, 2010.
As our product development candidates are reformulations of off-patent drugs, there are numerous sources of supply for active pharmaceutical ingredients, or APIs. We have a secure source of supply for API for our once-daily tramadol product and have identified several alternative suppliers. We have agreements with two contract manufacturers for the production of our once-daily tramadol product. We also have a contract manufacturing arrangement with another third-party for the packaging of our once-daily tramadol product into finished product.
Competition
We are engaged in a business characterized by extensive research efforts, rapid technological developments and intensive competition. Our competitors include large pharmaceutical, biotechnology and other companies, universities and research institutions. All of these competitors currently engage in, have engaged in or may engage in the future in the development, manufacturing, marketing and commercialization of new pharmaceuticals and existing pharmaceuticals, some of which may compete with our present or future product candidates. We expect that successful competition will depend, among other things, on product efficacy, safety, reliability, availability, timing and scope of regulatory approval and price.
Given that our business is reformulating existing drugs, our product candidates will face competition from generic and branded formulations of the existing drugs we reformulate. Our drug delivery technologies will compete with existing drug delivery technologies, as well as new drug delivery technologies that may be developed or commercialized in the future. Any of these drugs and drug delivery technologies may receive government approval or gain market acceptance more rapidly than our product candidates, may offer therapeutic or cost advantages over our product candidates or may cure our targeted diseases or their underlying causes completely. As a result, our product candidates may become non-competitive or obsolete.
Competition currently exists for our once-daily tramadol product from pain medications marketed by various pharmaceutical companies. In the United States, in addition to branded and generic immediate release formulations of tramadol, Biovail through its marketing partner OMI, has launched its once-daily tramadol product and Cipher recently announced its plan to submit an NDA to the FDA in 2006 for its once-daily tramadol product. Other competitors may be developing formulations for tramadol as well. In addition, extended-release tramadol formulations for once-daily dosing have been approved and are and will be marketed in certain European countries, including the United Kingdom, Belgium, France, Germany and Spain. Our once-daily tramadol product will also face competition from non-narcotic analgesics, other opioids and NSAIDs, including COX-2 inhibitors, which are used for the treatment of moderate to severe pain.
We anticipate that our other product candidates will also experience competition. For example, our once-daily trazodone product will compete with numerous proprietary and generic antidepressants.
We believe that our ability to successfully compete will depend on, among other things:
| • | the efficacy, safety and reliability of our product candidates; | |
| • | the timing and scope of regulatory approval; | |
| • | the speed at which we develop product candidates; | |
| • | our ability to manufacture and sell commercial quantities of a product to the market; | |
| • | product acceptance by physicians and other health care providers; | |
| • | the quality and breadth of our technology; | |
| • | the skills of our employees and our ability to recruit and retain skilled employees; | |
| • | the protection of our intellectual property; and |
59
| • | the availability of substantial capital resources to fund development and commercialization activities. |
Drug Development Process
Our drug development process involves applying our drug delivery technologies and development expertise to reformulate existing drug compounds which have proven efficacy and safety. We reintroduce them to the market as branded products with improved deliverability and significant commercial potential. In contrast to the drug development process for new chemical entities, or NCEs, which generally lasts 12 years and costs hundreds of millions of dollars, we believe the development process for our newly formulated versions of existing drugs can be shorter and less costly. Our products typically require fewer clinical trials to generate the safety and efficacy data needed to achieve regulatory approval than drug compounds that have not been previously marketed. In addition, we may be able to refer to existing safety data in our regulatory applications and therefore, may be able to avoid having to conduct the extensive pre-clinical safety and toxicity studies usually required for new drugs. We may also be able to take advantage of existing pharmacokinetic and clinical data to accelerate our development and regulatory process and reduce the risk as compared to traditional pharmaceutical and biotechnology companies. As a result of these advantages, we have been able to take our once-daily tramadol product from formulation to regulatory filing for marketing approval in Europe in approximately four years.
Our drug development and testing process consists of three phases:
| • | Formulation.This phase involves laboratory work to select an appropriate delivery technology for a compound, define the parameters of drug release in vitro, define product specifications which are expected to meet clinical and market requirements and develop formulations to be tested in human clinical trials. | |
| • | Pilot Pharmacokinetics.In this phase, a new formulation which has been produced under GMP is tested in Phase I trials in healthy human volunteers to assess its safety and to determine the pharmacokinetics of the drug. If the results do not meet clinical or market targets, the product is reformulated and retested. A formulation that emerges from this phase may be advanced into the final stage of development. | |
| • | Pharmacokinetics (Phase I) and Clinical Testing for demonstrating Efficacy and Safety (Phases II and III).The type of studies conducted in this phase are dependent on the regulatory requirements for each product candidate. For product candidates which are equivalent to existing marketed products, pharmacokinetic data demonstrating bioequivalence or similar bioavailability may be sufficient for obtaining regulatory approval. For novel formulations, new routes of administration or new clinical uses, we may need to conduct Phase II and/or Phase III clinical trials designed to demonstrate safety and efficacy prior to submitting an application for regulatory approval. See “ — Government Regulation.” |
Government Regulation
Drug delivery and drug companies must comply with the regulatory framework applicable to the pharmaceutical industry. Although the approval process is similar in many countries, pharmaceutical companies are generally required to submit to the regulatory authorities extensive data and information prior to marketing the pharmaceutical product in a country.
Government authorities in all of our target markets, including Europe, the United States and other countries, extensively regulate, among other things, the research, development, testing, manufacture, packaging, labeling, promotion, advertising, distribution, marketing and export and import of drugs such as those we are developing.
In the United States, pharmaceutical products, such as ours, are regulated under federal laws and regulations administered by the FDA and state laws and regulations administered by state health agencies, while in the European Union, these products are regulated under laws and regulations administered by the
60
Enterprise Directorate General, Pharmaceuticals and Cosmetics Unit, of the European Commission in Brussels, Belgium, with the assistance of the European Medicines Agency, or EMEA. We expect the United States and the European Union to be major markets for our products. A summary of the United States and European Union regulatory process follows below.
| United States |
In the United States, drugs are subject to rigorous regulation by the FDA. The U.S. Federal Food, Drug, and Cosmetic Act, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, record keeping, packaging, labeling, adverse event reporting, advertising, promotion, marketing, distribution, and import and export of pharmaceutical products. Failure to comply with applicable regulatory requirements may subject a company to a variety of administrative or judicially-imposed sanctions and/or the inability to obtain or maintain required approvals or to market drugs.
The steps ordinarily required before a new pharmaceutical product may be marketed in the United States include:
| • | preclinical laboratory tests, animal studies and formulation studies under FDA’s good laboratory practices regulations or GLPs; | |
| • | the submission to the FDA of an IND, which must become effective before human clinical trials may begin; | |
| • | the execution of adequate and well-controlled clinical trials according to GCPs to establish the safety and efficacy of the product for each indication for which approval is sought; | |
| • | submission to the FDA of an NDA; | |
| • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with GMPs to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and | |
| • | FDA review and approval of the NDA. |
Preclinical tests include laboratory and animal evaluation of product pharmacology and toxicology. The conduct of the preclinical tests and formulation of compounds for testing must comply with federal regulations and requirements for GLPs and GMPs. Violation of GLPs may, in some cases, lead to invalidation of the studies, requiring the studies to be repeated. The results of preclinical testing and formulation development are submitted to the FDA as part of an IND.
The IND must become effective before clinical testing may commence. A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has not commented on or questioned the IND within this 30-day period, clinical trials may begin. If the FDA has comments or questions, the questions must be answered to the satisfaction of the FDA before clinical testing can begin.
The FDA may refuse to accept the IND for review if applicable regulatory requirements are not met. Moreover, the FDA may delay or prevent the start of clinical studies if the manufacturing of the test drugs fails to meet GMP requirements or the clinical studies are not adequately designed. Such government regulation may delay or prevent the study and marketing of potential products for a considerable period of time and may impose costly procedures upon a manufacturer’s activities. In addition, the FDA may, at any time, impose a clinical hold on ongoing clinical trials. If the FDA imposes a clinical hold, clinical trials cannot continue without FDA authorization and then only under terms authorized by the FDA.
Success in early stage clinical trials does not assure success in later stage clinical trials. Data obtained from clinical activities are not always conclusive and may be susceptible to varying interpretations that could delay, limit or prevent regulatory approval. Even if a product receives regulatory approval, later
61
discovery of previously unknown problems with a product may result in restrictions on the product or even withdrawal of the marketing approval for the product.
Clinical Trials
Clinical trials involve the administration of the drug to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted in compliance with federal, state, and local regulations and requirements, under protocols detailing, for example, the objectives of the trial, the parameters to be used in monitoring safety and the efficacy criteria to be evaluated. Each protocol involving testing on human subjects must be submitted to the FDA as part of the IND prior to starting the trial. In addition, the study protocol and informed consent information for patients in clinical trials must be submitted for approval to institutional review boards at each local institution involved in the studies. Clinical trials to support NDAs for marketing approval are typically conducted in three sequential phases, but the phases may overlap. In Phase I, the drug is tested, generally in healthy volunteers, to assess metabolism, pharmacokinetics and safety, including side effects associated with increasing doses. Absorption, metabolism, distribution and excretion studies are generally performed at this stage.
Phase II usually involves trials in a limited patient population to determine optimum dosage, identify possible adverse effects and provide preliminary support for the efficacy of the drug for any new indication being studied.
If a compound demonstrates evidence of effectiveness and an acceptable safety profile in Phase I and II studies, Phase III trials are undertaken to further evaluate clinical efficacy and to further test for safety within an expanded patient population, typically at geographically dispersed clinical trial sites, in order to assess the overall benefit-risk relationship of the drug and provide an adequate basis for physician labeling. Phase I, Phase II or Phase III testing of any product candidate may not be completed successfully within any specified time period, if at all. In the case of drugs that are copies of previously approved drugs, which are also known as generic drugs, an applicant may submit an abbreviated new drug application, or ANDA, that demonstrates that the proposed generic product is “bioequivalent” to the previously approved product.
| NDAs and ANDAs |
After successful completion of the required clinical testing, generally an NDA, or an ANDA for generic drugs, is prepared and submitted to the FDA. In certain cases, an application for marketing approval may include information regarding safety and efficacy of a proposed drug that comes from studies not conducted by or for the applicant and for which the applicant has not obtained a specific right to reference those studies. Such applications, known as a 505(b)(2) NDA, are permitted for new drug products that incorporate previously approved active ingredients, even if the proposed new drug incorporates an approved active ingredient in a novel formulation or for a new indication. Section 505(b)(2) also permits the FDA to rely for such approvals on literature or on a finding by the FDA of safety and/or efficacy for a previously approved drug product. In addition, a 505(b)(2) NDA for changes to a previously approved drug product may rely on the FDA’s finding of safety and efficacy of the previously approved product coupled with new clinical information needed by FDA to support the change. FDA approval of the NDA or ANDA is required before marketing of the product may begin in the United States. The NDA must include the results of any clinical and other testing and a compilation of data relating to the product’s pharmacology, toxicology chemistry, manufacture and manufacturing controls. The cost of preparing and submitting an NDA may be substantial. Under U.S. federal law, the submission of NDAs, including 505(b)(2) NDAs, is generally subject to substantial application user fees, and the manufacturer and/or sponsor under an approved NDA are also subject to annual product and establishment user fees. These fees are typically increased annually. Currently, there are no fees assessed for ANDAs.
The FDA has 60 days from its receipt of an NDA to determine whether the application will be accepted for filing based on the FDA threshold determination that the NDA is sufficiently complete to permit substantive review. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under U.S. federal law, the FDA has agreed to certain performance goals in the review of NDAs. Most
62
such applications for non-priority drug products are to be reviewed within ten months. The review process may be significantly extended by FDA requests for additional information or clarification. The FDA may also refer applications for novel drug products or novel uses of drug products or drug products which present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved. The FDA is not bound by the recommendation of an advisory committee.
Before approving an application, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will not approve the application unless GMP compliance is satisfactory. If the FDA determines that the NDA or ANDA, manufacturing process and manufacturing facilities are acceptable, the FDA may issue an approval letter, or, in some cases, an approvable letter followed by an approval letter. An approvable letter generally contains a statement of specific conditions that must be met in order to secure final approval of the application. If and when those conditions have been met to the FDA’s satisfaction, the FDA will typically issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications. As a condition of approval, the FDA may require post approval testing and surveillance to monitor the drug’s safety or efficacy and may impose other conditions, including labeling restrictions on the use of the drug which can materially impact its potential market and profitability. Once granted, product approvals may be withdrawn if compliance with regulatory standards for manufacturing and quality control are not maintained or if additional safety problems are identified following initial marketing.
If the FDA’s evaluation of the NDA submission or manufacturing processes and facilities is not favorable, the FDA may refuse to approve the NDA or ANDA and may issue a not approvable letter. The not approvable letter outlines major deficiencies in the submission and often requires substantial additional testing or information in order for the FDA to reconsider the application. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The Pediatric Research Equity Act, or PREA, requires NDAs (or NDA supplements) for a new active ingredient, new indication, new dosage form, new dosing regimen, or new route of administration to contain data assessing the safety and efficacy for the claimed indication in all relevant pediatric subpopulations. Data to support dosing and administration also must be provided for each pediatric subpopulation for which the drug is safe and effective. FDA may grant deferrals for the submission of data, or full or partial waivers from the PREA requirements.
Once the NDA or ANDA is approved, the sponsor of the product will be subject to certain post-approval requirements, including requirements for adverse event reporting and submission of periodic reports. Also, some types of changes to the approved product, such as manufacturing changes and labeling claims, are subject to further FDA review and approval. Additionally, the FDA strictly regulates the promotional claims that may be made about prescription drug products. In particular, the FDA requires substantiation of any claims of superiority of one product over another including, in many cases, requirements that such claims be proven by adequate and well controlled head-to-head clinical trials. To the extent that market acceptance of our products may depend on their superiority over existing therapies, any restriction on our ability to advertise or otherwise promote claims of superiority, or requirements to conduct additional expensive clinical trials to provide proof of such claims, could negatively affect the sales of our products and our costs.
Once an NDA, including a 505(b)(2) NDA, is approved, the product covered thereby becomes a “listed drug” which can, in turn, be cited by potential competitors in support of approval of an ANDA. Specifically, a generic drug that is the subject of an ANDA must be bioequivalent and have the same active ingredient(s), route of administration, dosage form, and strength, as well as the same labeling, with certain exceptions, as the listed drug. If the FDA deems that any of these requirements are not met, additional data may be necessary to seek approval.
ANDA applicants do not have to conduct extensive clinical trials to prove the safety or efficacy of the drug product; rather, they are required to conduct less rigorous bioequivalence testing. Drugs approved in
63
this way are commonly referred to as “generic equivalents” to the listed drug, are listed as such by the FDA, and can often be substituted by pharmacists under prescriptions written for the original listed drug.
With respect to NDAs, U.S. federal law provides for a period of three years of exclusivity following approval of a listed drug that contains previously approved active ingredients but is approved in a new dosage, dosage form, route of administration or combination, or for a new use, the approval of which was required to be supported by new clinical trials, other than bioavailability studies, conducted by or for the sponsor. During this three-year period the FDA cannot grant effective approval of an ANDA or a 505(b)(2) NDA for the same conditions of approval under which the NDA was approved.
U.S. federal law also provides a period of five years following approval of a new chemical entity, that is a drug containing no previously approved active ingredients, during which ANDAs for generic versions of those drugs, as well as 505(b)(2) NDAs, cannot be submitted unless the submission contains a certification that the listed patent is invalid or will not be infringed, in which case the submission may be made four years following the original product approval. If an ANDA or 505(b)(2) NDA applicant certifies that it believes one or more listed patents are invalid or not infringed, it is required to provide notice of its filing to the NDA sponsor and the patent holder. If the patent holder or exclusive patent licensee then initiates a suit for patent infringement against the ANDA or 505(b)(2) NDA sponsor within 45 days of receipt of the notice, the FDA cannot grant effective approval of the ANDA or 505(b)(2) NDA until either 30 months has passed or there has been a court decision holding that the patents in question are invalid or not infringed. If an infringement action is not brought within 45 days, the ANDA or 505(b)(2) NDA applicant may bring a declaratory judgement action to determine patent issues prior to marketing. If the ANDA or 505(b)(2) NDA applicant certifies as to the date on which the listed patents will expire, then the FDA cannot grant effective approval of the ANDA or 505(b)(2) NDA until those patents expire. The first ANDA(s) submitting substantially complete application(s) certifying that listed patents for a particular product are invalid or not infringed may qualify for a period of 180 days of marketing exclusivity, starting from the date of the first commercial marketing of the drug by the applicant and during which subsequently submitted ANDAs cannot be granted effective approval. The first ANDA applicant can forfeit its exclusivity, under certain circumstances, for example if it fails to market its product or meet other regulatory requirements within specified time periods.
From time to time, including presently, legislation is drafted and introduced in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of drug products. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our product candidates. It is impossible to predict whether legislative changes will be enacted, or FDA regulations, guidance or interpretations changed, or what the impact of such changes, if any, may be.
| European Economic Area |
A medicinal product may only be placed on the market in the European Economic Area, or EEA, composed of the 25 European Union Member States, plus Norway, Iceland and Lichtenstein, when a marketing authorization, referred to as a national authorization, has been issued by the competent authority of a member state, or EEA country, for its own territory or when an MA, referred to as a Community authorization, has been granted in accordance with Regulation (EC) No. 726/2004 for the entire EEA. In 1995, when the European system for authorizing medicinal products was introduced, the EMEA began its activities. The EMEA’s main responsibility is the protection and promotion of public health, through the evaluation and supervision of medicines for human and veterinary use. The EMEA works as a network, bringing together the scientific resources of the European Union Member States for the evaluation and supervision of medicines in Europe. The EMEA also undertakes inspections of production facilities to assure compliance with GMP, GCP, Good Laboratory Practice, or GLP, and other quality assurance systems.
The type and extent of data to be submitted to support an MA application in the European Union depends on the amount of information already publicly available for a particular medicinal product or its active ingredient for a specific treatment. There are three principal forms of MA applications:
64
| • | Complete Applications.These are applications for products containing drugs not previously approved within the European Union and must contain complete results of physico-chemical, biological or microbiological tests, full information on all pharmacological and toxicological tests, as well as appropriate clinical trials of safety and efficacy. | |
| • | Mixed Data Applications.These are applications for products that develop different pharmaceutical forms of drugs previously approved within the European Union. In these applications, published scientific literature is presented together with original results of tests and trials. It is possible to replace results of pharmacological and toxicological tests or clinical trials by detailed references to published scientific literature if it can be demonstrated that the constituent(s) of a medicinal product have a well established medicinal use, with recognized efficacy and an acceptable level of safety. However, if the drug contained in the product is to be used for an entirely new therapeutic use, it is not possible to refer solely to the drug’s prior use; instead, additional data on the new therapeutic indication together with appropriate safety data would need to be provided. | |
| • | Abridged Applications.These are applications for products essentially similar to ones already on the market in the member country and which applications do not require the provision of results of pharmacological and toxicological tests or the results of clinical trials. This applies if the product candidate is essentially similar to an authorized product in the member state and either consent has been obtained from the company responsible for the marketing of the original product to reference the pharmacological, toxicological or clinical references or if the product candidate is essentially similar to a product authorized within the European Union for not less than eight to ten years and is marketed within the member state for which the application is made. Abridged MA applications must demonstrate the safety and efficacy of the medicinal product and justify the content of the application. |
With the MRP, an MA application for drugs is made in a single member state (i.e., a national authorization) and marketing authorization may then be granted in other states of the European Union. With the centralized procedure, a community authorization is obtained which is valid for the entire EEA. A Community authorization is mandatory for medicines manufactured using certain biotechnological processes and is optional for innovative medicinal products, e.g., new drugs and medicinal products administered by means of new delivery systems, which, in the opinion of the EMEA, constitute a significant innovation or for medicinal products presented for an entirely new indication which, in the opinion of the EMEA, are of significant therapeutic interest.
The centralized application procedure involves making an application directly to the EMEA which is then assessed by the Committee for Medicinal Products for Human Use, or CHMP, during an evaluation period that may last up to 210 days. Upon completion of the evaluation, the EMEA has 15 days to submit the CHMP’s opinion to the Commission in Brussels which has 15 days to prepare a draft decision. This draft decision is then sent to a committee composed of representatives of the member states for additional comments. Ultimately, if successful, an MA is issued. MAs obtained by means of this centralized application procedure are valid for an initial five-year period, renewable on the basis of a re-evaluation by the EMEA.
The MRP involves making an application to an individual member state, known as the RMS, whose approval is then recognized in other member states. Under this process, the RMS has 90 days after receipt of a valid application to provide an assessment report, together with the approved Summary of Product Characteristics, or SmPC, to the member states and the MA holder. The other member states then have 90 days to recognize the decision of the RMS and the SmPC as approved by it. If the member states fail to reach an agreement because one of them considers that there are grounds for supposing that the authorization of the medicinal product may present a potential serious risk to public health, the disagreement may be submitted to the appropriate EMEA scientific committee for arbitration. The decision that ultimately results from this process is binding on all concerned member states and the MA holder. Other member states not directly concerned at the time of the decision are also bound as soon as they receive an MA application for the same product.
65
| Other Countries |
In addition to regulations in the United States and the European Union, we are subject to a variety of other foreign regulations governing clinical trials and commercial sales and distribution of drugs in other countries. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
| Related Matters |
From time to time, legislation is drafted, introduced and passed in governmental bodies that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA or EMEA and other regulatory bodies to which we are subject. In addition, regulations and guidance are often revised or reinterpreted by the national agency in ways that may significantly affect our business and our product candidates. It is impossible to predict whether such legislative changes will be enacted, or FDA or EMEA regulations, guidance or interpretations changed, or what the impact of such changes, if any, may be. We may need to adapt our business and product candidates and products to these changes that occur in the future.
Pharmaceutical Pricing and Reimbursement
In the United States and markets in other countries, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third-party payors. Third-party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. These third-party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare product candidates. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. Our product candidates may not be considered cost-effective. Adequate third-party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
In 2003, the United States government enacted legislation providing a partial prescription drug benefit for Medicare recipients, beginning in 2006. Government payment for some of the costs of prescription drugs may increase demand for any products for which we receive FDA marketing approval. However, to obtain payments under this program, we would be required to sell products to Medicare recipients through drug procurement organizations operating pursuant to this legislation. These organizations would negotiate prices for our products, which are likely to be lower than we might otherwise obtain. Federal, state and local governments in the United States continue to consider legislation to limit the growth of healthcare costs, including the cost of prescription drugs. Future legislation could limit payments for pharmaceuticals including our once-daily tramadol product and other drug candidates that we are developing.
In addition, an increasing emphasis on managed care in the United States has increased and will continue to increase the pressure on pharmaceutical pricing. The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third-party payors fail to provide adequate coverage and reimbursement.
In some countries, particularly the countries of the EEA, the pricing of prescription pharmaceuticals is subject to governmental control. In these countries, pricing negotiations with governmental authorities can take considerable time and delay the placing on the market of a product. To obtain reimbursement or pricing approval in some countries, we may be required to conduct a clinical trial that compares the cost-effectiveness of our product candidate to other available therapies.
66
Employees
As at March 1, 2006, we had 98 full-time employees, 71 of whom work in our research and development and regulatory and clinical development groups and 27 in business development and administration. 12 of our employees hold doctorate degrees, 23 hold masters degrees, two are M.D. graduates and an additional 32 have bachelor degrees.
We are not a party to any collective bargaining agreements. We consider our relations with our employees to be good and our operations have never been interrupted as the result of a labour dispute.
Facilities
In April 2003, we moved to our new corporate headquarters in the Parc scientifique et de haute technologie in Laval, Québec, which we now occupy under a 15-year lease. This 48,180 square-foot facility now houses both our corporate offices as well as our laboratory and pilot plant for producing GMP-grade clinical supplies. Our new facility has consolidated our employees from three separate locations and will allow us to develop an expanded number of products more efficiently and economically.
Labopharm Europe Limited, our wholly-owned European subsidiary, leases administrative facilities of 1,711 square feet in Dublin, Ireland.
Legal Proceedings
On February 7, 2005, Université de Montréal and Université du Québec à Montréal commenced an action against us claiming non payment of outstanding royalties owed by us with respect to the commercialization of a technology we purchased in 1994 and any improvements and additions thereto. A description of this litigation is set forth in “ — Intellectual Property.” Other than this litigation, we are not involved in any legal or arbitration proceedings that are material to our operations nor do we know of any material threatened or contemplated proceedings against us.
Corporate Information
We were incorporated under the Part IA of the Companies Act (Québec) on October 26, 1990 under the name Centre de recherche appliquée pharmaceutique CRAP inc. On September 13, 1994, we changed our name to Labopharm inc. and amalgamated with 9033-3394 Québec inc. pursuant to a certificate of amalgamation dated June 11, 1996 issued under Part IA of the Companies Act (Québec). On July 11, 1997, our articles were amended to change the location of the judicial district of our head office from Terrebonne, Québec to Laval, Québec.
Our head office, registered office and laboratory facilities are located at 480 Armand-Frappier Blvd., Laval, Québec, Canada, H7V 4B4. Our telephone number is (450) 686-1017 (or toll free at 1-888-686-1017). Our website is www.labopharm.com. The information contained on our website is not part of this prospectus. Our website address is included in this prospectus as an inactive textual reference only.
Labopharm inc. has four wholly-owned subsidiaries:
| • | Labopharm USA, Inc., a Delaware Corporation; | |
| • | Labopharm Europe Limited, an Ireland corporation; | |
| • | Labopharm (Barbados) Limited, a Barbados corporation; and | |
| • | Labopharm Cyprus Limited, a Cyprus corporation. |
67
MANAGEMENT
Executive Officers and Directors
The following table sets forth information about our executive officers and directors.
| Name | Age | Position | ||||
| Richard J. MacKay(1) | 71 | Interim Chairman of the Board and Director | ||||
| James R. Howard-Tripp | 56 | President, Chief Executive Officer and Director | ||||
| Warren Whitehead | 54 | Chief Financial Officer | ||||
| Sylvie Bouchard | 50 | Vice-President, Clinical Development | ||||
| Lynda Covello | 47 | General Counsel and Corporate Secretary | ||||
| Allan Mandelzys | 41 | Vice-President, Business Development | ||||
| Damon Smith | 43 | Vice-President, Research and Development | ||||
| Anthony C. Playle | 61 | Director and Managing Director of Labopharm Europe Limited | ||||
| Frédéric Porte(2) | 54 | Director | ||||
| Robert Raich(1) | 53 | Director | ||||
| Jacques L. Roy(1)(2) | 54 | Director | ||||
| James S. Scibetta(2) | 41 | Director | ||||
| Santo J. Costa | 60 | Director | ||||
| (1) | Member of the human resources and corporate governance committee. |
| (2) | Member of the audit committee. |
Richard J. MacKayhas been one of our directors since 1990. Mr. MacKay has been President and Chief Executive Officer of Stiefel Canada Inc. since 1976. He served as a director of both the Non-Prescription Drug Manufacturers Association of Canada from 1992 to 2002 and has been a director of the Canadian Dermatology Foundation since 1984. He was on the board of the Pharmaceutical Manufacturers Association of Canada (now Canada’s Research-Based Pharmaceutical Companies) from 1978 to 1986, serving as the chairman from 1982 to 1983. Mr. MacKay holds a B.A. from Sir George Williams University and an Advanced Pharmaceutical Marketing Diploma from Dartmouth College and completed the Program for Management Development (PMD) from Harvard University.
James R. Howard-Tripphas been our President and Chief Executive Officer since July 2000 and has been one of our directors since 1999. Prior to joining us, Mr. Howard-Tripp was Senior Vice President, Operations with Allelix Biopharmaceuticals, Inc. (now NPS Allelix Corp.) and President and Chief Executive Officer of its subsidiary, Allelix Neuroscience Inc. from 1998 to 2000. Mr. Howard-Tripp is a director of ChondroGene Ltd. He is a founding member and continues to serve as the originating chairman of the Canadian Healthcare Licensing Association.
Warren Whiteheadhas been our Chief Financial Officer since August 2000. Mr. Whitehead was also Chief Financial Officer of Resolution Pharmaceuticals, Inc. from October 1998 to February 2001 and one of their directors from 2000 to 2001. Prior to that, he was Manager of Business Development at Glaxo Wellcome Inc. from August 1995 to September 1998. Mr. Whitehead is a Certified Management Accountant and holds a M.B.A. and a B.Comm. from the University of Windsor and a B.A. from the University of Western Ontario.
Dr. Sylvie Bouchard, M.D., Ph.D., has been our Vice President, Clinical Development since February 2001. Prior to joining us, Dr. Bouchard held several medical and clinical positions at Sandoz Ltd. (now Novartis A.G.) from September 1981 to August 1994, most recently as Vice-President and Medical Director at Sandoz Canada Inc. During her tenure as Medical Director, Dr. Bouchard was involved in Sandoz’s national and international research projects working in France, Switzerland, the United States and Canada. Dr. Bouchard, concurrently with her position with us, works as an emergency physician at the Lakeshore
68
General Hospital in Montreal. She holds a M.D. from Université Laval with residency training at Université de Montréal. She also obtained a B.Sc. and Ph.D. in sciences (neurochemistry) from Université Laval.
Lynda Covellohas been our General Counsel and Corporate Secretary since November 2005. Prior to joining us, Ms. Covello gathered more than 15 years experience in legal practice, including as a partner with Margolis Covello, LLP from March 2000 to December 2004, and merged her practice with the law firm of Sim, Hughes, Ashton & McKay LLP (now Sim, Lowman, Ashton & McKay) in January 2005, where she practised law until November 2005. For more than a decade, she has been an active member of the Licensing Executives Society (LES) in both Canada and the U.S. and LES International, serving in a variety of executive roles. Ms. Covello received her B.A. in International Relations from the University of Toronto, a LL.B. and a LL.M from York University and Osgoode Hall, and was called to the Ontario Bar in 1987.
Allan Mandelzys, Ph.D.,has been our Vice President, Business Development since October 2000. Dr. Mandelzys was Director of Business Development at Allelix Biopharmaceuticals, Inc. (now NPS Allelix Corp.) from April 1999 to September 2000 and Business Development Specialist from November 1997 to March 1999. Prior to that, Dr. Mandelzys held several scientific positions within Allelix Biopharmaceuticals, including the management of pharmaceutical alliances. He holds a Ph.D. in physiology, an M.B.A. from McGill University and a B.Sc. from the University of Toronto. In addition, Dr. Mandelzys received post-doctoral training at the Roche Institute of Molecular Biology.
Damon Smith, Ph.D.,has been our Vice President, Research and Development since May 2002. Prior to joining us, Dr. Smith was Director of Research at ConjuChem Inc. from February 2000 to April 2002. Prior to that, he was Director of Development at Protherics Ltd. (formerly Therapeutic Antibodies Inc.) from June 1996 to December 1999. Dr. Smith holds a B.Sc. and a Ph.D. in biological chemistry from the University of Essex in the United Kingdom.
Anthony C. Playlehas been one of our directors since 2001 and has been the Managing Director of Labopharm Limited Europe since April 2002. Mr. Playle has been President and Chief Executive Officer of ACpharma Ltd., a consulting firm specializing in the pharmaceutical industry, since June 2001. Prior to that, Mr. Playle held various positions with Aventis Pharma SA, including Vice President of European Coordination for Commercial Operations from October 1999 to May 2001 and Vice President, Business Development and Strategic Planning, Europe from 1998 to October 1999. He has served as a director for Phoqus Ltd., a drug delivery company, since June 2003. Mr. Playle holds a B. Pharm. from University of London and he is a member of the Royal Pharmaceutical Society.
Frédéric Portehas been one of our directors since 1998. Mr. Porte is the founder and has been the President of Medipress Management Inc., a company offering strategic and financial planning in the health care sector since 1982. Mr. Porte has also served as the President of Angelus Investment Corporation, a venture capital organization, since 2003. In 1981, he founded L’Actualité Médicale Inc., a publishing company which was sold in 1985. In 1987, he founded and was President of Clinidata Inc., a medical and pharmaceutical software company, which was purchased by Hoechst Marion Roussel (now Aventis Pharma SA) in 1994. He holds a Diplôme d’études approfondies (D.E.A.) in Social and Economical Information from L’Université Paris-Sorbonne in France. He also holds a master’s Degree in Finance and Administration from L’École Supérieure de Commerce in Lyon, France.
Robert Raichhas been one of our directors since 2004. Mr. Raich was a senior partner at the law firm Spiegel Sohmer, where he has served as Managing Partner since 1985. Mr. Raich holds a B.C.L. from McGill University.
Jacques L. Royhas been one of our directors since 2001. Mr. Roy has been Vice President, Finance and Corporate Development of Omega Laboratories Limited, a privately held pharmaceutical company, since July 2005. From May 2002 to April 2005, Mr. Roy held various positions with Fonds de solidarité des travailleurs du Québec (F.T.Q.), a development capital fund in the Province of Québec. Prior to joining Fonds de solidarité, Mr. Roy worked with ABB (Canada) as Vice President, Business Development, Mergers and Acquisitions between February 1998 and December 2001. Mr. Roy is a director of LBL SkySystems Corporation, a public company previously engaged in the manufacture and sale of high-rise curtain-wall
69
systems which was declared bankrupt on September 27, 2005. Mr. Roy holds a B. Comm. from McGill University.
James S. Scibettahas been one of our directors since 2001. Since September 2001, Mr. Scibetta has been Executive Vice President and Chief Financial Officer of Merrimack Pharmaceuticals, Inc. of Cambridge, Massachusetts, and he served on the board of directors of Merrimack Pharmaceuticals, Inc. from 1998 to March 2004. Prior to joining Merrimack, he was a senior investment banker for Shattuck Hammond Partners, a healthcare investment boutique in New York, from 1997 to 2001, and for the Healthcare Finance Group of PaineWebber from 1988 to 1997. Mr. Scibetta holds a B.S. in Physics from Wake Forest University, and a M.B.A. in Finance from the University of Michigan.
Santo J. Costahas been one of our directors since March 2006. Since June 2001, Mr. Costa has been of counsel with the law firm Maupin Taylor P.A. Prior to that Mr. Costa was President of Quintiles Transnational Corporation, an international provider of pharmaceutical product development and commercialization activities, from April 1994 to November 1999, and Vice Chairman from November 1999 to May 2001. Mr. Costa also held the positions of General Counsel and Senior Vice-President Administration with Glaxo Inc. from 1986 to 1993, US Area Counsel with Merrell Dow Pharmaceuticals from 1977 to 1986 and Counsel, Food & Drug with Norwich Eaton Pharmaceuticals from 1971 to 1977. Mr. Costa holds a B.S. in Pharmacy and a J.D. from St. John’s University. He is a member of the North Carolina Bar and the New York and Ohio Bars.
Term of Our Executive Officers
Each of our executive officers has been elected by our board of directors and serves until his or her successor is duly elected and qualified.
Term of Our Board of Directors
Our directors are elected at the annual meeting of shareholders for one-year terms and serve until the close of the next annual meeting of shareholders, unless they resign or are removed earlier. Our articles of incorporation provide that the board of directors must have a minimum of three and a maximum of 25 directors. Our board of directors currently consists of 8 directors.
Committees of Our Board of Directors
We have in place an audit committee and a human resources and corporate governance committee. Because we are a foreign private issuer, we are not required to comply with all of the corporate governance requirements set forth in Nasdaq Rule 4350 as they apply to U.S. domestic companies. Our corporate governance measures differ in the following significant ways. First, we have not appointed an independent nominations committee or adopted a board resolution addressing the nomination process. Second, we have not appointed a compensation committee. Our human resources and corporate governance committee performs similar functions to that of a compensation committee, and each of the three members of the committee are non-executive independent directors. Since 2004, at each of our regularly scheduled board meetings, we hold an executive session at which only non-management and independent directors, except Anthony C. Playle, may attend. During the year ended December 31, 2005, 14 such executive sessions were held.
In respect of the practices noted above, our practices conform with Multilateral Instrument 52-110 —Audit Committees and National Policy 58-201 —Corporate Governance Practices in lieu of Nasdaq Rule 4350.
70
| Audit Committee |
The mandate of the audit committee includes, among others:
| • | to review with representatives of management and representatives of the independent auditors our interim quarterly and annual audited financial statements prior to their filing and the related press releases and to report thereon to our board of directors; | |
| • | to review and satisfy itself, on behalf of the board of directors, that the information delivered to shareholders referred to above is accurate; | |
| • | to ensure the integrity of our internal control over financial reporting and monitor the annual review and evaluation by management of internal control over financial reporting; | |
| • | to ensure our compliance with, and process for monitoring compliance with, legal and regulatory requirements; | |
| • | to ensure the independent auditors’ qualifications and independence; and | |
| • | to engage and review the performance of the independent auditors. |
The current members of the audit committee are Messrs Frederic Porte, Jacques L. Roy and James S. Scibetta, each of whom meets the independence and audit committee composition requirements promulgated by all governmental or regulatory bodies exercising authority over us, including the Autorité des marchés financiers (Québec), the Securities and Exchange Commission, or SEC, the National Association of Securities Dealers and the rules of all stock exchanges on which our shares are listed, or Regulatory Bodies.
| Human Resources and Corporate Governance Committee |
The mandate of the human resources and corporate governance committee includes, among others:
| • | to develop and monitor the effectiveness of our system of corporate governance, including our Code of Ethics and Business Conduct; | |
| • | to establish procedures for the identification of new nominees to our board of directors and lead the candidate selection process; | |
| • | to develop and implement orientation procedures for new directors; | |
| • | to assess the effectiveness of individual directors, the board of directors and the various committees of the board; | |
| • | to ensure appropriate corporate governance and proper delineation of the roles, duties and responsibilities of management, the board of directors and its committees; | |
| • | to ensure the recruitment and ongoing long-term development of high caliber senior management; | |
| • | to establish levels of salary, bonus, benefits and incentives for our executive and senior officers; and | |
| • | to assist the board of directors in setting the objectives for our chief executive officer and to evaluate his performance. |
The current members of the human resources and corporate governance committee are Messrs. Richard J. MacKay, Robert Raich and Jacques L. Roy, each of whom meets the independence requirements promulgated by the Regulatory Bodies.
71
Employment Agreements
Each of our executive officers has entered into an employment agreement with us or, in the case of Mr. Playle, with Labopharm Europe Limited. Each employment agreement determines the annual salary of the named executive officer subject to an annual review by the human resources and corporate governance committee. In addition, each named executive officer is entitled to be reimbursed for his/her business expenses and a performance bonus, and, except for Mr. Playle, to benefits under our corporate medical, dental and insurance plan, as well as an automobile allowance. Each employment agreement has an indefinite term. If an executive officer is terminated other than for cause or if we enter into a transaction resulting in a change in control, the executive officer is entitled to 18 months severance and his/her performance bonus, except for Mr. Howard-Tripp who is entitled to 24 months severance and his performance bonus. Upon such termination of employment or change in control, we will also continue to provide the same insurance and health care benefits throughout the severance period and all options that were granted to such executive officer pursuant to our stock option plan shall become immediately vested and will remain exercisable throughout the severance period.
RELATED PARTY TRANSACTIONS
Except as described below, we are not aware that any of our directors, officers, other insiders or any persons associated with or otherwise related to any of the foregoing has had an interest in any material transaction carried out since the beginning of our most recently completed financial year or in any proposed transaction which has materially affected or is likely to materially affect us or any of our subsidiaries.
In May 2001, we entered into a shareholders’ agreement with Fonds de solidarité des travailleurs du Québec (F.T.Q.), or FSTQ. In accordance with this agreement:
| • | so long as FSTQ holds 5% or more of our issued and outstanding common shares, FSTQ has the right to designate two nominees to serve as a director on our board of directors; and | |
| • | so long as FSTQ holds less than 5% but at least 1% of our issued and outstanding common shares, FSTQ has the right to designate one nominee to serve as a director on our board of directors. |
The agreement terminates when FSTQ ceases to hold at least 1% of our issued and outstanding shares. As at March 1, 2006, to our knowledge, FSTQ held 4,544,671 common shares, representing approximately 10.4% of our common shares then issued and outstanding. FSTQ nominated our current director, Mr. Jacques L. Roy.
We have entered into a consulting services agreement with ACPharma Ltd., a company controlled by Anthony C. Playle, who is a member of our board of directors and Managing Director of Labopharm Europe Limited. The fees paid by us to ACPharma Ltd. were $324,768, $318,840 and $244,714 in 2005, 2004 and 2003 respectively.
72
PRINCIPAL SHAREHOLDERS
The following table shows information with respect to the beneficial ownership of our common shares as of March 1, 2006 by:
| • | each of our named executive officers; | |
| • | each of our directors; and | |
| • | all of our directors and executive officers as a group. |
Beneficial ownership is determined in accordance with the rules of the SEC. The information does not necessarily indicate beneficial ownership for any other purpose. Under these rules, the number of common shares deemed outstanding includes shares issuable upon exercise of options held by the respective person or group which may be exercised or converted within 60 days after March 1, 2006. Percentage of beneficial ownership before the offering is based on 43,816,913 common shares outstanding as of March 1, 2006.
For purposes of calculating percentages of beneficial ownership, stock options exercisable within 60 days after March 1, 2006 are included for that person or group but not the stock options of any other person or group.
All of the 13 persons mentioned below beneficially owned, as of March 1, 2006, an aggregate of 2,462,621 common shares representing approximately 6% of our then outstanding common shares. Other than FSTQ which held 4,544,671 common shares as at March 1, 2006, representing approximately 10.4% of our common shares then issued and outstanding, no person or group or affiliated persons known by us is the beneficial owner of more than 10% of our common shares. Unless otherwise indicated and subject to applicable community property laws, to our knowledge, each shareholder named in the following table possesses sole voting and investment power over the shares listed, except for those jointly owned with that person’s spouse.
| Number of | ||||
| Common Shares | ||||
| Name of Beneficial Owner | Beneficially Owned | |||
Directors and Named Executive Officers | ||||
| James R. Howard-Tripp | 723,100 | |||
| Allan Mandelzys | 350,800 | |||
| Sylvie Bouchard | 295,000 | |||
| Lynda Covello | — | |||
| Richard J. MacKay | 229,071 | |||
| Warren Whitehead | 174,700 | |||
| Anthony C. Playle | 226,600 | |||
| Frédéric Porte | 120,000 | |||
| Damon Smith | 113,300 | |||
| James S. Scibetta | 87,500 | |||
| Jacques L. Roy(1) | 61,250 | |||
| Santo J. Costa | 50,000 | |||
| Robert Raich | 31,300 | |||
| All directors and executive officers as a group (13 persons) | 2,462,621 | |||
| (1) | Mr. Roy is FSTQ’s nominee pursuant to the FSTQ Agreement. FSTQ held 4,544,671 common shares as at March 1, 2006. |
73
DESCRIPTION OF SHARE CAPITAL
General
Our authorized share capital currently consists of an unlimited number of common shares, without par value, and an unlimited number of preferred shares, no par value and issuable in series, of which 43,816,913 common shares and no preferred shares were outstanding as of March 1, 2006.
The following is a brief description of our common shares and preferred shares.
Common Shares
The common shares rank junior to the preferred shares with respect to the payment of dividends, return of capital and distribution of assets in the event of our liquidation, dissolution or winding-up. The holders of outstanding common shares are entitled to receive dividends on a share-for-share basis out of the assets legally available for that purpose at such times and in such amounts as our board of directors may determine. The common shares carry one vote per share. There is no cumulative voting. The holders of common shares are entitled to receive notice of any of the meetings of shareholders and to attend and vote at all such meetings as a single class on all matters to be voted on by the shareholders, except a meeting where only the holders of shares of a class or of a particular series are entitled to vote separately. The common shares are not redeemable nor retractable. The holders of common shares have no preemptive rights. Upon our liquidation, dissolution or winding-up, the holders of common shares shall be entitled to participate equally, on a share-for-share basis, in the remaining property and assets available for distribution to such holders.
Preferred Shares
The preferred shares are issuable, from time to time, in one or more series, as determined by our board of directors. The preferred shares, if issued, rank prior to the common shares with respect to the payment of dividends and the distribution of assets in the event of our dissolution or liquidation or the distribution of all or part of our assets among the shareholders for an amount equal to the value of the consideration paid in respect of such outstanding shares, as credited to our issued and paid-up capital account, grossed up by the declared and unpaid dividends. Subject to the provisions of the Companies Act (Québec), the preferred shares do not carry voting rights.
74
CERTAIN MATERIAL INCOME TAX CONSIDERATIONS
Certain Canadian Material Income Tax Considerations
The following summarizes the principal Canadian federal income tax considerations under the Income Tax Act (Canada), or the Tax Act, generally applicable to holders of common shares who, at all relevant times, for purposes of the Tax Act, are resident in Canada, deal at arm’s length and are not affiliated with Labopharm, all within the meaning of the Tax Act, and acquire and hold their common shares as capital property, or the Resident Shareholders, and to holders of common shares who, at all relevant times, for purposes of the Tax Act, are not resident in Canada, deal at arm’s length with Labopharm, acquire and hold their common shares as capital property, do not use and are not deemed to use or hold their common shares in the course of carrying on, or otherwise in connection with, a business in Canada and who, at all relevant times, for the purposes of the Canada-United States Income Tax Convention 1980, as amended, or the Treaty, are resident in the United States, have never been resident in Canada, and have not held or used (and do not hold or use) common shares in connection with a permanent establishment or fixed base in Canada, or the U.S. Shareholders. Two related persons (such as a corporation and a person that controls the corporation) are deemed not to deal with each other at arm’s length and it is a question of fact whether persons not related to each other are dealing with each other at arm’s length. Generally, common shares will be considered to be capital property to a holder thereof provided that the holder does not use the common shares in the course of carrying on a business and has not acquired them in one or more transactions considered to be an adventure in the nature of trade. Certain Resident Shareholders may, in certain circumstances, treat common shares as capital property by making an irrevocable election permitted by subsection 39(4) of the Tax Act. This summary does not deal with special situations, such as the particular circumstances of traders or dealers in securities, limited liability companies, tax-exempt entities, insurers, “specified financial institutions” and “financial institutions” as defined in the Tax Act (including those to which the mark-to-market provisions of the Tax Act apply), holders of an interest in which is a “tax shelter investment” for the purposes of the Tax Act, or otherwise.
This summary is based on the current provisions of the Tax Act and the regulations thereunder, or the Regulations, all specific proposals to amend the Tax Act and Regulations publicly announced by the Minister of Finance (Canada) prior to the date hereof, or the Proposed Amendments, the current provisions of the Treaty and counsel’s understanding of the current administrative policy and practices of the Canada Revenue Agency, or the Administrative Practices. It has been assumed that all Proposed Amendments will be enacted substantially as proposed and that there will be no other relevant changes in any governing law, the Treaty or Administrative Practices, although no assurances can be given in these respects. This summary does not take into account provincial, territorial, United States or other foreign income tax considerations, which may differ significantly from those discussed herein.
This summary is not exhaustive of all possible Canadian federal income tax consequences. It is not intended as legal or tax advice to any prospective holder of common shares and should not be construed as such. No representations with respect to the income tax consequences to any such holder are made. The tax consequences to any prospective holder of common shares will vary according to the status of that holder as an individual, trust, corporation or member of a partnership, the jurisdictions in which that holder is subject to taxation and, generally, according to that holder’s particular circumstances. Each holder should consult the holder’s own tax advisor with respect to the tax consequences applicable to the holder’s own particular circumstances including the application and effect of the income and other tax law of any country, province, state or local tax authority.
All amounts relevant in computing the liability of a U.S. Shareholder under the Tax Act are to be reported in Canadian Currency at the rate of exchange prevailing at the relevant time.
| Taxation of Resident Shareholders |
In the case of a Resident Shareholder who is an individual, any dividends received on the common shares will be included in computing his income and will be subject to gross-up and dividend tax credit rules normally applicable to taxable dividends paid by taxable Canadian corporations.
75
In the case of a Resident Shareholder that is a corporation, dividends received on the common shares will be included in computing the corporation’s income and will generally be deductible in computing its taxable income. A Resident Shareholder that is a corporation may be liable to pay refundable tax under Part IV of the Tax Act on dividends received on the common shares to the extent that such dividends are deductible in computing the corporation’s taxable income. However, a public corporation which is not controlled, whether because of a beneficial interest in one or more trusts or otherwise, by or for the benefit of an individual (other than a trust) or a related group of individuals (other than trusts) will not be liable to pay refundable tax under Part IV of the Tax Act.
A disposition, or a deemed disposition, of a common share (other than a disposition or a deemed disposition to Labopharm, unless purchased by Labopharm in the open market in the manner in which shares are normally purchased in the open market) will give rise to a capital gain (or a capital loss) equal to the amount by which the proceeds of disposition of the common share, net of any costs of disposition, exceed (or are less than) the adjusted cost base of the common shares to the Resident Shareholder. For this purpose, the adjusted cost base to a Resident Shareholder of a common share will be determined by averaging the cost of that common share with the adjusted cost base of all common shares held at that time by the Resident Shareholder.
One-half of any capital gain realized by a Resident Shareholder will be included in computing the Resident Shareholder’s income as a taxable capital gain. One-half of any capital loss realized by a Resident Shareholder may generally be deducted against taxable capital gains realized in the year of realization of such loss or the three preceding taxation years or any subsequent taxation year, subject to detailed rules contained in the Tax Act in this regard. A capital loss realized by a Resident Shareholder that is a corporation, a partnership of which a corporation is a member or a trust of which a corporation is a beneficiary will be reduced by the amount of dividends received in certain circumstances.
Capital gains realized by an individual may give rise to a liability for alternative minimum tax.
| Taxation of U.S. Shareholders |
Amounts in respect of common shares paid or credited or deemed to be paid or credited as, on account or in lieu of payment of, or in satisfaction of, dividends to a U.S. Shareholder by Labopharm are subject to Canadian withholding tax at a rate of 25%. Under the Treaty, the rate of withholding tax on dividends beneficially owned by a U.S. Shareholder is generally limited to 15% of the gross amount of the dividend (or 5% in the case of corporate U.S. Shareholders owing at least 10% of the voting shares of Labopharm).
A U.S. Shareholder is generally not subject to tax under the Tax Act in respect of a capital gain realized on the disposition of a common share unless such share is “taxable Canadian property,” as defined in the Tax Act, to the holder thereof and the U.S. Shareholder is not entitled to relief from taxation in Canada under the Treaty with respect to such disposition.
A common share will generally not be taxable Canadian property to a U.S. Shareholder at the time of a disposition provided that the common shares are listed on a prescribed stock exchange (which includes the TSX) and that at no time during the 60 month period ending at the time of disposition of the common share did the U.S. Shareholder, persons with whom the U.S. Shareholder did not deal at arm’s length, or the U.S. Shareholder together with such persons, own 25% of more of the issued shares of any class or series of Labopharm. In the case of the U.S. Shareholder to whom common shares constitute, or are deemed to constitute, taxable Canadian property, no tax under the Tax Act will generally be payable on a capital gain realized on the disposition of such shares by virtue of the relieving provisions of the Treaty unless, at the time of disposition, the value of such shares is derived principally from real property situated in Canada. We believe that, at the date of this prospectus, the value of the common shares is not derived principally from real property situated in Canada.
76
U.S. Federal Income Tax Considerations
The following discussion summarizes certain of the material U.S. federal income tax consequences to you if you are a U.S. Holder, as defined below, of the acquisition, ownership and disposition of our common shares.
This discussion is not a comprehensive description of all of the tax considerations that may be relevant to each holder’s decision to purchase common shares. Except where noted, this summary deals only with common shares that are held as capital assets by U.S. Holders and are acquired in this offering. This discussion does not address all aspects of U.S. federal income taxation that may be relevant to a U.S. Holder based on such holder’s particular situation. This discussion does not address the U.S. federal income tax consequences to U.S. Holders that are subject to special treatment under the U.S. federal income tax laws, including, but not limited to:
| • | banks; | |
| • | dealers in securities or currencies; | |
| • | financial institutions; | |
| • | real estate investment trusts; | |
| • | insurance companies; | |
| • | tax-exempt organizations; | |
| • | persons holding common shares as part of a hedging, integrated or conversion transaction, constructive sale or straddle; | |
| • | traders in securities that have elected the mark-to-market method of accounting; | |
| • | persons liable for the alternative minimum tax; | |
| • | individual retirement and other tax-deferred accounts; | |
| • | persons who own directly, indirectly or by attribution at least 10% of our voting power; or | |
| • | U.S. persons whose “functional currency” is not the U.S. dollar. |
This summary is based upon the provisions of the Internal Revenue Code of 1986, as amended, or the Code, and U.S. Treasury regulations, rulings and judicial decisions thereunder as of the date hereof, and such authorities may be replaced, revoked or modified, possibly on a retroactive basis, so as to result in U.S. federal income tax consequences different from those discussed below.
This discussion does not address any aspect of U.S. federal gift or estate taxes, or of state, local or non-U.S. tax laws. Additionally, this discussion does not consider the tax treatment of partnerships or persons who hold common shares through a partnership or other pass-through entity.
If you are considering the purchase, ownership or disposition of our common shares, you are urged to consult your tax advisor concerning the tax consequences in light of your particular circumstances.
A U.S. Holder is a beneficial owner of common shares that is a U.S. person. A U.S. person is:
| • | a citizen or resident of the United States for U.S. federal income tax purposes; | |
| • | a corporation (or other entity taxable as a corporation) that is created or organized in or under the laws of the United States, any state thereof, or the District of Columbia; | |
| • | an estate the income of which is subject to U.S. federal income taxation, regardless of its source; or | |
| • | a trust if it is subject to the primary supervision of a court within the United States and one or more U.S. persons have the authority to control all substantial decisions of the trust or if it has |
77
| a valid election in effect under applicable U.S. Treasury regulations to be treated as a U.S. person. |
If a partnership holds common shares, the tax treatment of a partner will generally depend on the status of the partner and the activities of the partnership. If you are a U.S. Holder that is a partner of a partnership holding common shares, you are urged to consult your tax advisor.
| Distributions on Common Shares |
Subject to the discussion under “— Passive Foreign Investment Company Rules” below, the gross amount of any cash distributions on the common shares (before reduction for Canadian withholding taxes) will be taxable to you as dividends to the extent of our current and accumulated earnings and profits, as determined under U.S. federal income tax principles. Subject to certain limitations, dividends paid to noncorporate U.S. Holders, including individuals, may be eligible for a reduced rate of taxation if we are deemed to be a “qualified foreign corporation” for U.S. federal income tax purposes. A qualified foreign corporation includes a foreign corporation that is eligible for the benefits of a comprehensive income tax treaty with the United States that includes an exchange of information program and that the U.S. Treasury Department has determined to be satisfactory for purposes of the qualified dividend income provisions of the Code. Distributions from foreign corporations may also qualify for the reduced tax rate if the distributions are received with respect to stock that is “readily tradable on an established securities market in the United States.” Dividends received from an otherwise qualified foreign corporation that is a passive foreign investment company are not eligible for the reduced rate of taxation.
The U.S. Treasury Department has determined that the Treaty is satisfactory for purposes of the qualified dividend provisions of the Code. We believe that we are eligible for the benefits of the Treaty but may be a passive foreign investment company and, therefore, not a qualified foreign corporation. Our status as a qualified foreign corporation may change.
Dividends will be includable in your gross income on the date actually or constructively received by you. These dividends will not be eligible for the dividends-received deduction generally allowed to U.S. corporations in respect of dividends received from other U.S. corporations.
To the extent that the amount of any cash distribution exceeds our current and accumulated earnings and profits, the distribution will first be treated as a tax-free return of capital, causing a reduction in the adjusted basis of the common shares (thereby increasing the amount of gain, or decreasing the amount of loss, you would recognize on a subsequent disposition of the common shares), and the balance in excess of adjusted basis will be subject to tax as capital gain.
To the extent we pay dividends on the common shares in Canadian dollars, the U.S. dollar value of such dividends should be calculated by reference to the exchange rate prevailing on the date of actual or constructive receipt of the dividend, regardless of whether the Canadian dollars are converted into U.S. dollars at that time. If Canadian dollars are converted into U.S. dollars on the date of actual or constructive receipt of such dividends, your tax basis in such Canadian dollars will be equal to their U.S. dollar value on that date and, as a result, you generally should not be required to recognize any foreign currency exchange gain or loss. Any gain or loss recognized on a subsequent conversion or other disposition of the Canadian dollars generally will be treated as U.S. source ordinary income or loss.
You may be entitled to deduct, or claim a U.S. foreign tax credit for, Canadian taxes that are withheld on dividends received by you, subject to applicable limitations in the Code. Dividends paid on the common shares generally will constitute “passive income” or, in the case of certain U.S. Holders, “financial services income” and will be treated as income from sources without the United States for U.S. foreign tax credit limitation purposes. The amount of foreign income taxes that may be claimed as a credit in any year is subject to complex limitations and restrictions, which must be determined on an individual basis by each holder. You are urged to consult your tax advisor regarding the availability of the U.S. foreign tax credit in your particular circumstances.
78
| Sale, Exchange or Other Disposition of Common Shares |
Subject to the discussion under “— Passive Foreign Investment Company Rules” below, upon the sale, exchange or other disposition of common shares, you generally will recognize capital gain or loss equal to the difference between the amount realized upon the sale, exchange or other disposition and your adjusted tax basis in the common shares. Your tax basis in a common share will be, in general, the price you paid for that common share, subject to adjustments for return of capital distributions, as described above. The capital gain or loss generally will be long-term capital gain or loss if, at the time of sale, exchange or other disposition, you have held the common share for more than one year. Net long-term capital gains of noncorporate U.S. Holders, including individuals, are eligible for reduced rates of taxation. The deductibility of capital losses is subject to limitations. Any gain or loss that you recognize generally will be treated as gain or loss from sources within the United States for U.S. foreign tax credit limitation purposes.
| Passive Foreign Investment Company Rules |
In general, a foreign corporation will be classified as a passive foreign investment company for U.S. federal income tax purposes if:
| • | 75% or more of its gross income in a taxable year falls within specific categories of passive income; or | |
| • | the average percentage of its assets in a taxable year (ordinarily determined based on their market value as long as a corporation is not a controlled foreign corporation, as defined for U.S. federal income tax purposes) which produce passive income or are held for the production of passive income is at least 50%. |
If we were classified as a passive foreign investment company, and you did not make a qualifying election either to treat us as a “qualified electing fund” or to mark our common shares to market:
| • | Excess distributions by us to you would be taxed in a special way. “Excess distributions” are amounts received by you with respect to our common shares in any taxable year that exceed 125% of the average distributions received by you from us in the shorter of either the three previous years or your holding period for the common shares before the current taxable year. Excess distributions must be allocated ratably to each day that you have held our common shares. You would be required to include in your gross income amounts allocated to the current taxable year and years before we became a passive foreign investment company as ordinary income. In addition, amounts allocated to each taxable year beginning with the year we first became a passive foreign investment company would be taxed at the highest rate in effect for that year on ordinary income and the tax would be subject to an interest charge at the rate applicable to deficiencies for income tax. | |
| • | The entire amount of gain that is realized by you upon the sale or other disposition of common shares would also be considered an excess distribution and would be subject to tax as described above. |
It is possible that we will be considered a passive foreign investment company for 2006 and subsequent years due to the nature of our income and assets and the manner in which the relevant provisions of the Code dealing with attribution of income and assets of subsidiaires to a parent corporation may be applied to our situation. You are strongly urged to consult your tax advisor about the passive foreign investment company rules.
If we were classified as a passive foreign investment company, you could, subject to certain conditions, elect to treat our common shares as stock in a “qualified electing fund,” in which case you would be required to include in current U.S. taxable income a proportionate share of our ordinary earnings and net capital gain in years in which we are classified as a passive foreign investment company, but under certain conditions any gain subsequently recognized upon the sale of our common shares by you generally may be taxed as capital gain. If we determine that we are, or may be, a passive foreign investment company for 2006
79
or a subsequent year, we will provide shareholders with the necessary information to make a “qualified electing fund” election upon request. Alternatively, you could make a “mark-to-market” election pursuant to which you would recognize an amount of ordinary income or loss each year in an amount equal to the difference between the fair market value of your common shares and your adjusted tax basis therein. Losses would be allowed only to the extent of income, net of losses, previously recognized pursuant to the “mark-to-market” election. You should consult with your own tax advisors regarding the availability, consequences, advisability, and manner of making either the “qualified electing fund” or “mark-to-market” election if we are treated as a passive foreign investment company.
If you hold common shares during a period when we are a passive foreign investment company, you will be subject to the preceding rules, even if we cease to be a passive foreign investment company, subject to exceptions if you made a “qualified electing fund” election or “mark-to-market” election.
Information Reporting and Backup Withholding
In general, unless you belong to a category of certain exempt recipients (such as corporations), information reporting requirements will apply to distributions on common shares made within the United States and to the proceeds of sales of common shares that are effected through the U.S. office of a broker or the non-U.S. office of a broker that has certain connections with the United States. Backup withholding at a current rate of 28% may apply to these payments if you fail to: (i) provide a correct taxpayer identification number and certify that such number is correct; (ii) certify that you are not subject to backup withholding; or (iii) otherwise comply with applicable certification requirements.
Any amounts withheld under the backup withholding rules will be allowed as a credit against your U.S. federal income tax, provided you furnish the required information to the Internal Revenue Service in a timely manner.
80
UNDERWRITING
We are offering the common shares described in this prospectus through a number of underwriters. We have entered into a purchase agreement with the underwriters. The representatives of the underwriters are Merrill Lynch, Pierce, Fenner & Smith Incorporated and Banc of America Securities LLC. Subject to the terms and conditions of the purchase agreement, we have agreed to sell to the underwriters, and each underwriter has severally agreed to purchase from us, the number of common shares listed next to its name in the following table:
| Number | ||||
| Underwriter | of Shares | |||
| Merrill Lynch, Pierce, Fenner & Smith Incorporated | ||||
| Banc of America Securities LLC | ||||
| Canaccord Capital Corporation | ||||
| Leerink Swann & Co., Inc. | ||||
| Orion Securities Inc. | ||||
| Dundee Securities Corporation | ||||
| Westwind Partners Inc. | ||||
| Total | ||||
The purchase agreement is subject to a number of terms and conditions and provides that the underwriters must buy all of the shares if they buy any of them. The underwriters will sell the shares to the public when and if the underwriters buy the shares from us.
We will indemnify the underwriters against some liabilities, including liabilities under the Securities Act and Canadian provincial securities legislation. If we are unable to provide this indemnification, we will contribute to payments the underwriters may be required to make in respect of those liabilities.
The offering is being made concurrently in the United States and Canada pursuant to the multi-jurisdictional disclosure system implemented by the securities regulatory authorities in the United States and Canada. The common shares will be offered in the United States through those underwriters who are registered to offer the common shares for sale in the United States and such other registered dealers as may be designated by the underwriters. The common shares will be offered in all provinces of Canada through Merrill Lynch Canada Inc., the Canadian affiliate of Merrill Lynch, Pierce, Fenner & Smith Incorporated, Banc of America Securities Canada Co., the Canadian affiliate of Banc of America Securities LLC, Canaccord Capital Corporation, Orion Securities Inc., Dundee Securities Corporation and Westwind Partners Inc. and such other registered dealers as may be designated by the underwriters. Subject to applicable law, the underwriters may offer the common shares outside the United States and Canada pursuant to prospectus exemptions.
Discounts and Commissions.The underwriters initially will offer the shares to the public at the price specified on the cover page of this prospectus. The underwriters may allow a concession of not more than $ per share to selected dealers. The underwriters may also allow, and those dealers may re-allow, a concession of not more than $ per share to some other dealers. If all the shares are not sold at the public offering price, the underwriters may change the public offering price and the other selling terms. The common shares are offered subject to a number of conditions, including:
| • | receipt and acceptance of the common shares by the underwriters; and | |
| • | the underwriters’ right to reject orders in whole or in part. |
81
The following table shows the public offering price, total underwriting discounts and commissions to be paid to the underwriters by us and proceeds, before expenses, to us. These amounts are shown assuming no exercise and full exercise of the underwriters’ option to purchase additional shares.
| Paid by Us | |||||||||
| No Exercise | Full Exercise | ||||||||
| Per Share | US$ | US$ | |||||||
| Underwriting Discounts and Commissions | US$ | US$ | |||||||
| Total | US$ | US$ | |||||||
The expenses of the offering, not including the underwriting discounts and commissions, are estimated to be approximately US$1.5 million and will be paid by us.
Over-Allotment Option.We have granted the underwriters an over-allotment option to buy up to 1,500,000 additional common shares at the same price per share as they are paying for the shares shown in the table above. These additional shares would cover sales of shares by the underwriters which exceed the total number of shares shown in the table above. The underwriters may exercise this option at any time within 30 days after the date of this prospectus. To the extent that the underwriters exercise this option, each underwriter will purchase additional shares from us in approximately the same proportion as it purchased the shares shown in the table above. If purchased, the additional shares will be sold by the underwriters on the same terms as those on which the other shares are sold. We will pay the expenses associated with the exercise of this option.
No Sales of Similar Securities.We and our directors and executive officers have agreed, with exceptions, not to sell or transfer any common shares for 90 days after the date of this prospectus without first obtaining the written consent of Merrill Lynch, Pierce, Fenner & Smith Incorporated and Banc of America Securities LLC on behalf of the underwriters. Specifically, we and these other persons have agreed not to directly or indirectly:
| • | offer, pledge, sell or contract to sell any common shares; | |
| • | sell or grant any option or contract to purchase any common shares; | |
| • | purchase any option or contract to sell any common shares; | |
| • | grant any option, right or warrant for the sale of any common share (except pursuant to our employee stock option plan); | |
| • | otherwise dispose of or transfer any common shares; | |
| • | request or demand that we file a registration statement related to their common shares; or | |
| • | enter into any swap or other agreement that transfers in whole or in part the economic consequence of ownership of any common shares whether any such swap or transaction is to be settled by delivery of common shares or other securities, in cash or otherwise. |
Notwithstanding the foregoing, if: (1) during the last 17 days of the 90-day lock-up period, we issue an earnings release or publicly announce material news or a material event relating to us occurs; or (2) prior to the expiration of the 90-day lock-up period, we announce that we will release earnings results or we become aware that material news or a material event will occur during the 16-day period beginning on the last day of the 90-day lock-up period, the restrictions imposed by the lock-up provision shall continue to apply until the expiration of the 18-day period beginning on the issuance of the earnings release or the public announcement of the material news or the occurrence of the material event, as applicable, unless each of Merrill Lynch, Pierce, Fenner & Smith Incorporated and Banc of America Securities LLC waive, in writing, such extension.
This lock-up provision applies to common shares and to securities convertible into or exchangeable or exercisable for or repayable with common shares. It also applies to common shares owned now or acquired
82
by the person executing the agreement or for which the person executing the agreement later acquires the power of disposition.
Listing.Our common shares are listed on the Toronto Stock Exchange under the symbol “DDS.” We expect our common shares to be approved for quotation on the Nasdaq National Market under the symbol “DDSS.”
Price Stabilization and Short Positions.In connection with this offering, the underwriters may engage in activities that stabilize, maintain or otherwise affect the price of our common shares, including:
| • | stabilizing transactions; | |
| • | short sales; | |
| • | syndicate covering transactions; | |
| • | imposition of penalty bids; and | |
| • | purchases to cover positions created by short sales. |
Stabilizing transactions consist of bids or purchases made for the purpose of preventing or retarding a decline in the market price of our common shares while this offering is in progress. Stabilizing transactions may include making short sales of our common shares, which involves the sale by the underwriters of a greater number of common shares than they are required to purchase in this offering, and purchasing common shares from us or on the open market to cover positions created by short sales. Short sales may be “covered” shorts, which are short positions in an amount not greater than the underwriters’ over-allotment option referred to above, or may be “naked” shorts, which are short positions in excess of that amount. Syndicate covering transactions involve purchases of our common shares in the open market after the distribution has been completed in order to cover syndicate short positions.
The underwriters may close out any covered short position either by exercising their over-allotment option, in whole or in part, or by purchasing shares in the open market. In making this determination, the underwriters will consider, among other things, the price of shares available for purchase in the open market compared to the price at which the underwriters may purchase through the over-allotment option.
A naked short position is more likely to be created if the underwriters are concerned that there may be downward pressure on the price of the common shares in the open market that could adversely affect investors who purchased in this offering. To the extent that the underwriters create a naked short position, they will purchase shares in the open market to cover the position.
The representatives of the underwriters also may impose a penalty bid on underwriters and dealers participating in the offering. This means that the representatives may reclaim from any syndicate members or other dealers participating in the offering the underwriting discounts and commissions on shares sold by it and purchased by the representatives in stabilizing or short covering transactions.
These activities may have the effect of raising or maintaining the market price of our common shares or preventing or retarding a decline in the market price of our common shares. As a result of these activities, the price of our common shares may be higher than the price that otherwise might exist in the open market. If the underwriters commence the activities, they may discontinue them at any time. The underwriters may carry out these transactions on the Nasdaq National Market, the Toronto Stock Exchange, in the over-the-counter market or otherwise.
In accordance with policy statements of the Autorité des marchés financiers (Québec) and the Ontario Securities Commission, the underwriters in Canada may not, throughout the period of distribution, bid for or purchase common shares. Exceptions, however, exist where the bid or purchase is not made to create the appearance of active trading in, or raising prices of, the common shares. These exceptions include a bid or purchase permitted under the by-laws and rules of the Toronto Stock Exchange relating to market stabilization and passive market making activities and a bid or purchase made for and on behalf of a customer where the order was not solicited during the period of distribution. We have been advised that in
83
connection with this offering and pursuant to the first exception mentioned above, the underwriters may over-allot or effect transactions which stabilize or maintain the market price of the common shares at levels other than those which might otherwise prevail on the open market. These transactions, if commenced, may be discontinued at any time.
Market Making.In connection with this offering, some underwriters and any selling group members who are qualified market makers on the Nasdaq National Market may engage in passive market making transactions in our common shares on the Nasdaq National Market. Passive market making is allowed during the period when the SEC’s rules would otherwise prohibit market activity by the underwriters and dealers who are participating in this offering. Passive market making may occur during the business day before the pricing of this offering, before the commencement of offers or sales of the common shares. A passive market maker must comply with applicable volume and price limitations and must be identified as a passive market maker. In general, a passive market maker must display its bid at a price not in excess of the highest independent bid for our common shares; but if all independent bids are lowered below the passive market maker’s bid, the passive market maker must also lower its bid once it exceeds specified purchase limits. Net purchases by a passive market maker on each day are limited to a specified percentage of the passive market maker’s average daily trading volume in our common shares during the specified period and must be discontinued when that limit is reached. Passive market making may cause the price of our common shares to be higher than the price that otherwise would exist in the open market in the absence of those transactions. The underwriters and dealers are not required to engage in a passive market making and may end passive market making activities at any time.
Internet Offering.A prospectus in electronic format may be made available on the web sites maintained by one or more of the underwriters participating in this offering. Other than the prospectus in electronic format, the information on any such web site, or accessible through any such web site, is not part of the prospectus. The representatives may agree to allocate a number of shares to underwriters for sale to their online brokerage account holders. Internet distributions will be allocated by the underwriters that will make internet distributions on the same basis as other allocations. In addition, shares may be sold by the underwriters to securities dealers who resell shares to online brokerage account holders.
Other Relationships.Some of the underwriters and their affiliates have provided, and may in the future provide, various investment banking, commercial banking and other financial services for us and our affiliates for which services they have received, and may in the future receive, customary fees.
TRANSFER AGENT AND REGISTRAR
The transfer agent and registrar for our common shares in Canada is National Bank Trust, at its principal offices in Montreal and Toronto and in the United States is Computershare Trust Company, Inc. at its principal office in Golden, Colorado.
LEGAL MATTERS
Legal matters relating to this offering as to Canadian law and the validity of the common shares offered by this prospectus are being passed upon for us by Fasken Martineau DuMoulin LLP, Montreal, Québec. Legal matters relating to this offering as to United States law will be passed upon for us by Goodwin Procter LLP, Boston, Massachusetts. Legal matters relating to this offering as to Canadian law are being passed upon for the underwriters by McCarthy Tétrault LLP, Montreal, Québec. Legal matters relating to this offering as to United States law are being passed upon for the underwriters by Wilmer Cutler Pickering Hale and Dorr LLP, Boston, Massachusetts.
As of April 17, 2006, the partners and associates of these firms beneficially owned, directly or indirectly, less than 1% of our issued and outstanding securities.
84
EXPERTS
Our consolidated financial statements as at December 31, 2004 and 2005, and for the years ended December 31, 2004 and December 31, 2005, appearing and incorporated by reference in this prospectus and registration statement have been audited by Ernst & Young LLP, independent auditors, as set forth in their report appearing elsewhere in this prospectus and incorporated by reference in this prospectus and registration statement, and are included and incorporated by reference in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
DOCUMENTS INCORPORATED BY REFERENCE
Information has been incorporated by reference in this prospectus from documents filed with securities commissions or similar authorities in Canada (including the permanent information record in the Province of Québec). Copies of documents incorporated by reference in this prospectus and not delivered with this prospectus may be obtained upon request without charge from our Secretary at 480 Armand-Frappier Blvd., Laval, Québec, H7V4B4, telephone (450) 686-1017 or by accessing the disclosure documents available through the Internet on the Canadian System for Electronic Document Analysis and Retrieval, or SEDAR, which can be accessed at http://www.sedar.com. For the purpose of the Province of Québec, this simplified prospectus contains information to be completed by consulting the permanent information record. A copy of the permanent information record may be obtained from our Secretary at the above-mentioned address and telephone number. The following documents, filed with the various securities commissions or similar authorities in each of the provinces of Canada, are specifically incorporated by reference and form an integral part of this prospectus:
| • | our Annual Information Form dated March 30, 2006 for the fiscal year ended December 31, 2005; | |
| • | our Management’s Discussion and Analysis contained in our Annual Report for the fiscal year ended December 31, 2005; | |
| • | our Audited Comparative Consolidated Financial Statements, including the notes thereto, for the fiscal year ended December 31, 2005, together with the auditors’ report thereon as refiled on April 18, 2006, prepared in accordance with Canadian GAAP and including a reconciliation of the significant differences between Canadian GAAP and U.S. GAAP; and | |
| • | our Management Proxy Circular dated March 20, 2006 in connection with the annual meeting of shareholders of the Company to be held on May 4, 2006. |
Any documents of the type referred to in the preceding paragraph and any material change reports (excluding confidential material change reports) we filed with a securities commission or any similar authority in Canada or the SEC after the date of this prospectus and prior to the termination of the offering shall be deemed to be incorporated by reference into this prospectus.
Any statement contained in a document incorporated or deemed to be incorporated by reference in this prospectus shall be deemed to be modified or superseded for the purposes of this prospectus to the extent that a statement contained in this prospectus or in any subsequently filed document which also is or is deemed to be incorporated by reference in this prospectus modifies or supersedes that statement. The modifying or superseding statement need not state that it has modified or superseded a prior statement or include any other information set forth in the document that it modifies or supersedes. The making of a modifying or superseding statement shall not be deemed an admission for any purposes that the modified or superseded statement, when made, constituted a misrepresentation, an untrue statement of a material fact or an omission to state a material fact that is required to be stated or that is necessary to make a statement not misleading in light of the circumstances in which it was made. Any statement so modified or superseded shall not be deemed, except as so modified or superseded, to constitute a part of this prospectus.
85
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form F-10 under the Securities Act with respect to the common shares offered by this prospectus. This prospectus, which forms part of the registration statement, does not contain all the information set forth in the registration statement. For further information with respect to us and the common shares to be sold in this offering, you should refer to the registration statement and its exhibits.
Upon completion of this offering, we will become subject to the reporting and information requirements of the United States Exchange Act of 1934, as amended, or the Exchange Act, and, as a result, will file periodic and current reports, proxy statements, and other information with the SEC. Under a multi-jurisdictional disclosure system adopted by Canada and the United States, such reports and other information may be prepared in accordance with the disclosure requirements of Canada, which requirements are different from those of the United States. As a “foreign private issuer” under the Exchange Act, we intend to provide to our shareholders proxy statements and annual reports prepared in accordance with applicable Canadian law. Our annual reports will be available within 140 days of the end of each fiscal year and will contain our audited consolidated financial statements. We will also make available quarterly reports containing unaudited summary consolidated financial information for each of the first three fiscal quarters. We intend to prepare these financial statements in accordance with Canadian GAAP and to include a reconciliation of the significant differences between Canadian GAAP and U.S. GAAP in the notes to our annual consolidated financial statements. We are exempt from provisions of the Exchange Act which require us to provide proxy statements in prescribed form to shareholders and which relate to short swing profit reporting and liability.
You may read and copy this information and a copy of the registration statement at the Public Reference Room of the SEC located at 100 F Street, N.E., Washington, D.C. 20549. Please call the SEC at 1-800-SEC-0330 for further information on the operation of the Public Reference Room. Copies of all or any part of the registration statement may be obtained from the SEC’s offices upon payment of fees prescribed by the SEC. The SEC maintains an Internet site that contains periodic and current reports, proxy and information statements, and other information regarding issuers that file electronically with the SEC. The address of the SEC’s website is http://www.sec.gov.
In addition, we are subject to the filing requirements prescribed by the securities legislation of all Canadian provinces. You are invited to read and copy any reports, statements or other information that we file with the Canadian provincial securities commissions or other similar regulatory authorities at their respective public reference rooms. These filings are also electronically available from SEDAR at http://www.sedar.com. Reports and other information about us should also be available for inspection at the offices of the Toronto Stock Exchange and the Nasdaq National Market.
DOCUMENTS FILED AS PART OF THE REGISTRATION STATEMENT
The following documents have been filed with the SEC as part of the registration statement of which this prospectus forms a part:
| • | the documents referred to under “Documents Incorporated by Reference”; | |
| • | the form of purchase agreement referred to under “Underwriting”; | |
| • | the consent of Ernst & Young LLP; and | |
| • | the powers of attorney pursuant to which amendments to the registration statement may be signed. |
86
LABOPHARM INC.
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
Auditors’ Report | F-2 | |||
Consolidated Financial Statements | ||||
| Consolidated Balance Sheets | F-3 | |||
| Consolidated Statements of Operations | F-4 | |||
| Consolidated Statements of Deficit | F-5 | |||
| Consolidated Statements of Cash Flows | F-6 | |||
| Notes to Consolidated Financial Statements | F-7 |
These consolidated financial statements are presented in Canadian dollars.
F-1
AUDITORS’ REPORT
To the Directors of
Labopharm inc.
We have audited the consolidated balance sheets of Labopharm inc. as at December 31, 2005 and 2004 and the consolidated statements of loss, deficit and cash flows for the years ended December 31, 2005 and December 31, 2004. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with Canadian generally accepted auditing standards. Those standards require that we plan and perform an audit to obtain reasonable assurance whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation.
In our opinion, these consolidated financial statements present fairly, in all material respects, the financial position of the Company as at December 31, 2005 and 2004 and the results of its operations and its cash flows for the years ended December 31, 2005 and December 31, 2004 in accordance with Canadian generally accepted accounting principles.
| Montréal, Canada | (signed) Ernst & Young LLP. |
| February 2, 2006 | Chartered Accountants. |
(except note 23, which is as at April 17, 2006).
COMMENTS BY AUDITORS FOR U.S. READERS ON
CANADA — U.S. REPORTING DIFFERENCE
In the United States, reporting standards for auditors require the addition of an explanatory paragraph (following the opinion paragraph) when the financial statements are affected by conditions and events that cast substantial doubt on the Company’s ability to continue as a going concern, such as those described in note 1 to the consolidated financial statements. Our report to the directors dated February 2, 2006 (except note 23, which is as at April 17, 2006) is expressed in accordance with Canadian reporting standards which do not permit a reference to such events and conditions in the auditors’ report when these are adequately disclosed in the financial statements.
| Montréal, Canada | (signed) Ernst & Young LLP. |
| February 2, 2006 | Chartered Accountants. |
(except note 23, which is as at April 17, 2006).
F-2
LABOPHARM INC.
CONSOLIDATED BALANCE SHEETS(note 1)
| As at December 31, | ||||||||
| 2005 | 2004 | |||||||
| (thousands of | ||||||||
| Canadian dollars) | ||||||||
ASSETS(note 11) | ||||||||
Current | ||||||||
| Cash and cash equivalents | $ | 20,282 | $ | 2,809 | ||||
Short-term investments(note 4) | 14,611 | 20,814 | ||||||
Accounts receivable(note 5) | 532 | 967 | ||||||
Research and development tax credits receivable(note 15) | 875 | 800 | ||||||
| Income tax receivable | 426 | — | ||||||
Inventories(note 6) | 2,188 | — | ||||||
| Prepaids and other assets | 452 | 247 | ||||||
Total current assets | 39,366 | 25,637 | ||||||
Restricted long-term investments(note 7) | 1,271 | 1,282 | ||||||
Property, plant and equipment(note 8) | 10,280 | 10,961 | ||||||
Intangible assets(note 9) | 3,231 | 2,036 | ||||||
| Deferred financing costs | 364 | — | ||||||
| $ | 54,512 | $ | 39,916 | |||||
LIABILITIES | ||||||||
Current | ||||||||
| Accounts payable and accrued liabilities | $ | 10,090 | $ | 5,930 | ||||
| Current portion of deferred revenue | 9,067 | 266 | ||||||
Current portion of obligations under capital leases(note 10) | 83 | 134 | ||||||
Current portion of long-term debt(note 11) | 3,383 | — | ||||||
| 22,623 | 6,330 | |||||||
| Deferred revenue | 20,834 | 1,510 | ||||||
Obligations under capital leases(note 10) | 5,840 | 5,923 | ||||||
Long-term debt(note 11) | 7,818 | — | ||||||
| 57,115 | 13,763 | |||||||
Shareholders’ equity (deficiency) | ||||||||
Common shares, no par value, unlimited authorized shares 43,673,863 and 42,510,630 issued as at December 31, 2005 and 2004, respectively(note 12) | 135,631 | 132,658 | ||||||
| Contributed surplus | 6,350 | 4,745 | ||||||
| Deficit | (144,584 | ) | (111,250 | ) | ||||
Total shareholders’ equity (deficiency) | (2,603 | ) | 26,153 | |||||
| $ | 54,512 | $ | 39,916 | |||||
Commitments, Guarantees and Contingency(notes 13 and 14)
On behalf of the Board:
| (Signed) Jacques L. Roy Director | (Signed) James S. Scibetta Director |
See accompanying notes
F-3
LABOPHARM INC.
CONSOLIDATED STATEMENTS OF OPERATIONS(note 1)
| For the year ended | |||||||||
| December 31, | |||||||||
| 2005 | 2004 | ||||||||
| (thousands of Canadian | |||||||||
| dollars, except share and | |||||||||
| per share amounts) | |||||||||
REVENUE | |||||||||
| Product sales | $ | 1,269 | $ | — | |||||
| Cost of goods sold | 758 | — | |||||||
| Gross profit on product sales | 511 | — | |||||||
Other revenue | |||||||||
| Licensing | 1,908 | 106 | |||||||
| Research and development contracts | 61 | 1,288 | |||||||
| 2,480 | 1,394 | ||||||||
EXPENSES | |||||||||
| Research and development expenses | 22,451 | 16,037 | |||||||
Government assistance(note 15) | (2,713 | ) | (823 | ) | |||||
| 19,738 | 15,214 | ||||||||
| Selling, general and administrative expenses | 12,188 | 9,878 | |||||||
Financial expenses(note 16) | 1,937 | 864 | |||||||
| Depreciation and amortization | 1,664 | 1,662 | |||||||
| Interest income | (557 | ) | (538 | ) | |||||
| Foreign exchange gain | (355 | ) | (57 | ) | |||||
| Expenses related to an incomplete financing initiative | — | 1,509 | |||||||
| 34,615 | 28,532 | ||||||||
| Loss before income taxes | (32,135 | ) | (27,138 | ) | |||||
Income taxes(note 14): | |||||||||
| Current | 1,199 | 41 | |||||||
Net loss | $ | (33,334 | ) | $ | (27,179 | ) | |||
| �� | |||||||||
Net loss per share — basic and diluted | $ | (0.78 | ) | $ | (0.68 | ) | |||
Weighted average number of shares outstanding | 42,922,741 | 39,954,488 | |||||||
See accompanying notes
F-4
LABOPHARM INC.
CONSOLIDATED STATEMENTS OF DEFICIT(note 1)
| For the year ended | ||||||||
| December 31, | ||||||||
| 2005 | 2004 | |||||||
| (thousands of | ||||||||
| Canadian dollars) | ||||||||
Balance, beginning of year (as previously reported) | $ | (111,250 | ) | $ | (81,095 | ) | ||
Adjustment for change in accounting policy(note 3) | — | (2,976 | ) | |||||
Balance, beginning of year (adjusted) | (111,250 | ) | (84,071 | ) | ||||
| Net loss | (33,334 | ) | (27,179 | ) | ||||
Balance, end of year | $ | (144,584 | ) | $ | (111,250 | ) | ||
See accompanying notes
F-5
LABOPHARM INC.
CONSOLIDATED STATEMENTS OF CASH FLOWS(note 1)
| For the year ended | |||||||||
| December 31, | |||||||||
| 2005 | 2004 | ||||||||
| (thousands of | |||||||||
| Canadian dollars) | |||||||||
OPERATING ACTIVITIES | |||||||||
| Net loss for the year | $ | (33,334 | ) | $ | (27,179 | ) | |||
| Items not affecting cash: | |||||||||
| Depreciation of property, plant and equipment | 1,524 | 1,520 | |||||||
| Amortization of intangible assets | 140 | 142 | |||||||
| Financial expenses | 271 | — | |||||||
| Amortization of deferred financing costs | 110 | — | |||||||
| Unrealized foreign exchange gain | (232 | ) | — | ||||||
| Stock-based compensation | 1,754 | 1,775 | |||||||
| (29,767 | ) | (23,742 | ) | ||||||
Net change in non-cash operating items(note 17) | 28,727 | 660 | |||||||
| (1,040 | ) | (23,082 | ) | ||||||
INVESTING ACTIVITIES | |||||||||
| Acquisition of investments | (20,440 | ) | (43,547 | ) | |||||
| Disposals of investments | 958 | 4,353 | |||||||
| Maturities of investments | 25,685 | 37,106 | |||||||
| Acquisition of property, plant and equipment | (843 | ) | (1,000 | ) | |||||
| Acquisition of intangible assets | (173 | ) | (187 | ) | |||||
| 5,187 | (3,275 | ) | |||||||
FINANCING ACTIVITIES | |||||||||
| Repayment of capital lease obligations | (134 | ) | (171 | ) | |||||
Proceeds from issuance of capital stock(note 12) | 2,093 | 30,813 | |||||||
Issuance costs of capital stock(note 12) | — | (2,196 | ) | ||||||
| Proceeds from issuance of long-term debt | 11,586 | — | |||||||
| Proceeds from issuance of warrants | 731 | — | |||||||
| Deferred financing costs | (474 | ) | — | ||||||
| 13,802 | 28,446 | ||||||||
| Foreign exchange loss on cash held in foreign currencies | (476 | ) | — | ||||||
Increase in cash and cash equivalents during the year | 17,473 | 2,089 | |||||||
| Cash and cash equivalents, beginning of year | 2,809 | 720 | |||||||
Cash and cash equivalents, end of year | $ | 20,282 | $ | 2,809 | |||||
Cash flows include the following items: | |||||||||
| Interest paid | $ | 1,434 | $ | 863 | |||||
| Income taxes paid (received) | $ | 427 | $ | (18 | ) | ||||
See accompanying notes
F-6
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
| 1. | Description of Business and Going Concern Uncertainty |
The Company, incorporated under theCompanies Act (Québec)is specialized in the development of drugs using advanced controlled-release technologies and the development of pharmaceutical products incorporating its proprietary technologies.
The Company’s strategy is to develop products internally in order to form strategic alliances or enter into licensing agreements with national or international pharmaceutical companies that have the necessary resources and distribution networks to market and sell the pharmaceutical products incorporating the Company’s proprietary technologies. To date, the Company has financed its cash requirements primarily through share issuances, a term loan, investment tax credits, collaborative research contracts, licensing and distribution agreements, interest income and product sales. The future profitability of the Company is dependent upon such factors as the success of the clinical trials, the approval by regulatory authorities of products developed by the Company, the ability of the Company to successfully market, sell and distribute its products and the ability of the Company to obtain the necessary financing to complete its projects.
The Company has incurred significant operating losses and cash outflows from operations and has incurred an accumulated deficit of $144,584. As at December 31, 2005, the Company’s committed cash obligations and expected level of expenses for the upcoming twelve months exceed the committed sources of funds and the Company’s cash and cash equivalents on hand. The ability of the Company to continue as a going concern is dependent upon raising additional financing through borrowings or equity financing, receiving funds through collaborative research contracts or product licensing agreements, obtaining regulatory approval in various jurisdictions, and achieving future profitable operations. The outcome of these matters is dependent on a number of items outside of the Company’s control. As a result, there is significant uncertainty as to whether the Company will have the ability to continue as a going concern.
The consolidated financial statements have been prepared on a going concern basis, which assumes the Company will continue in operation for the foreseeable future and will be able to realize its assets and discharge its liabilities and commitments in the ordinary course of business. These financial statements do not include any adjustments to the amounts and classification for assets and liabilities that may be necessary should the Company not be successful in its effort to obtain additional financing, to receive significant funds on signing collaborative research contracts or by out licensing its products or making significant product sales.
| 2. | Significant Accounting Policies |
These financial statements have been prepared by management in accordance with Canadian generally accepted accounting principles. The most significant accounting policies are summarized below. A reconciliation of significant differences with generally accepted accounting principles in the United States is presented in note 23.
Use of estimates
The preparation of financial statements in conformity with generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting periods. Actual results could differ from those estimates and such differences could be material.
F-7
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
| 2. | Significant Accounting Policies — (Continued) |
Principles of consolidation
The consolidated financial statements include the accounts of the Company and those of its wholly owned subsidiaries, Labopharm Europe Limited, Labopharm (Barbados) Limited, Labopharm Cyprus Limited and Labopharm USA, Inc., incorporated in 2005. All significant inter-company transactions and balances have been eliminated upon consolidation.
Revenue recognition
The Company recognizes revenue from research and development contracts, and licensing arrangements, which may include multiple elements. Revenue arrangements with multiple elements are reviewed in order to determine whether the multiple elements can be divided into separate units of accounting, if certain criteria are met. If separable, the consideration received is allocated among the separate units of accounting based on their respective fair values, and the applicable revenue recognition criteria are applied to each of the separate units. Otherwise, the applicable revenue recognition criteria are applied to combined elements as a single unit of accounting.
Licensing revenues — Up-front non-refundable licensing revenue is deferred and recognized on a straight-line basis over the term which the Company maintains substantive contractual obligations. Licensing revenue received upon the achievement of milestones is recognized when the underlying condition is met if it has stand-alone value to the customer, the Company has no further obligations in relation to that milestone and collectibility is reasonably assured. Otherwise, it is recognized over the remaining term of the underlying agreement. Amounts received in advance of recognition are included in deferred revenue, as are amounts which are refundable if underlying conditions are not met.
Product sales — Revenue is recognized when the product is shipped to the Company’s customers, provided the Company has not retained any significant risks of ownership or future obligations with respect to the product shipped and collection is reasonably assured. The Company’s products are indirectly subject to pricing regulations in certain markets.
Research and development contracts — The Company recognizes revenues from various research agreements as the contracted services are performed or when milestones are achieved, in accordance with the terms of the specific agreements. Up-front payments for the use of technology where further services are to be provided or fees received on the signing of research agreements are recognized over the period of performance of the related activities. Amounts received in advance of recognition are included in deferred revenue.
Cash and cash equivalents
Cash and cash equivalents consist of cash and all highly liquid short-term investments that are readily convertible to known amounts of cash and which are subject to an insignificant risk of change in value. The Company considers these highly liquid short-term investments with a maturity on acquisition of less than three months to be cash equivalents.
F-8
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
| 2. | Significant Accounting Policies — (Continued) |
Short-term and long-term investments
Short-term investments are recorded at the lower of amortized cost or fair market value on a portfolio basis. Long-term investments are accounted for at amortized cost. Investments are written down to their fair market value based on quoted market prices, when a decline in value is other than temporary.
Inventories
Inventories are valued at the lower of cost, which is determined on an average cost basis, and net realizable value for finished goods and work-in-progress and replacement cost for raw materials.
Property, plant and equipment
Property, plant and equipment are carried at cost less related investment tax credits for research and development equipment.
Assets acquired under capital leases are carried at cost, being the present value of the minimum lease payments after deduction of executory costs.
Depreciation of property, plant and equipment and assets acquired under capital leases are calculated over their estimated useful life using the following methods and rates:
| Laboratory and plant equipment | Diminishing balance | 20% to 30% | ||
| Computer hardware and software | Diminishing balance | 30% | ||
| Furniture and office equipment | Straight-line | 5 to 10 years | ||
| Leasehold improvements | Straight-line over the term of the lease up to 10 years | |||
| Building and building improvements | Straight-line over the term of the lease up to 15 years | |||
Intangible assets
Intangible assets consist of patent and trademark costs and intellectual property rights which consist of fees paid to license technology or to acquire patents. The patent costs include legal fees to obtain patents and patent application fees.
Amortization of intangible assets is calculated over their estimated useful life, which cannot exceed the life of the underlying patents using the following methods and rates:
| Patents and trademarks | Straight-line up to 20 years | |||
| Intellectual property rights | Straight-line up to 20 years |
Deferred financing costs
Financing costs associated with the issuance of debt are deferred and amortized over the term of the related debt using the effective yield method.
F-9
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
| 2. | Significant Accounting Policies — (Continued) |
Impairment of long-lived assets
Property, plant and equipment and other long-lived assets are regularly reviewed for impairment whenever events or changes in circumstances indicate that the carrying value of an asset may not be recoverable. Impairment is assessed by comparing the carrying amount of an asset to be held and used with the sum of the undiscounted cash flows expected from its use and disposal. If such assets are considered impaired, the impairment loss to be recognized is measured by the amount by which the carrying amount of the assets exceeds its fair value generally determined on a discounted cash flow basis. Any impairment results in a write-down of the asset and a charge to income during the year.
Research and development expenses
Research expenses are charged to income in the year of expenditure less related tax credits. Development costs net of related tax credits are charged to income as incurred unless a development project meets generally accepted accounting principles in Canada for deferral and amortization. The Company has not deferred any such development costs to date.
Government assistance
Grant amounts resulting from government assistance programs, including investment tax credits on research and development expenses, are reflected as reductions to the cost of the assets or to the expenses to which they relate at the time the assistance becomes receivable.
Foreign currency translation
The Company’s foreign subsidiaries are considered to be integrated foreign entities and are accounted for in accordance with the temporal method, as are transactions in foreign currencies entered into by the Company. Monetary assets and liabilities denominated in foreign currencies are translated into Canadian dollars at the year-end exchange rate, non-monetary assets and liabilities are translated at the historical exchange rate, and revenue and expense items are translated in Canadian dollars at rates of exchange in effect at the related transaction dates. Exchange gains and losses arising from these transactions are included as a charge to income during the year.
Stock-based compensation plan
The Company has a stock-based compensation plan and has applied the fair value based method to expense all options awarded to employees and directors since March 1, 2002. The fair value of stock options granted is determined at the date of grant using the Black-Scholes option pricing model, and expensed over the vesting period of the options.
Net loss per share
The basic net loss per share is calculated using the weighted average number of common shares outstanding during the year.
The diluted net loss per share is calculated based on the weighted average number of common shares outstanding during the year, plus the effects of dilutive common share equivalents such as options. This
F-10
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
| 2. | Significant Accounting Policies — (Continued) |
method requires that the diluted net loss per share be calculated using the treasury stock method, as if all common share equivalents had been exercised at the beginning of the reporting period, or period of issuance, as the case may be, and that the funds obtained thereby were used to purchase common shares of the Company at the volume weighted average trading price of the common shares during the period.
Diluted loss per share is equal to the basic loss per share since the effect of exercising 1,574,425 options (2004 — 2,055,946) would be antidilutive for all periods presented.
Income taxes
The Company follows the liability method of accounting for income taxes. Under this method, future income tax assets and liabilities are determined based on the differences between the financial reporting and tax bases of assets and liabilities and are measured using substantively enacted tax rates and laws that are expected to be in effect in the periods in which the future tax assets or liabilities are expected to be realized or settled. A valuation allowance is provided to the extent that it is more likely than not that future income tax assets will not be realized.
Issuance costs of capital stock
The Company records issuance costs of capital stock against capital stock.
Recent accounting pronouncements
Comprehensive Income and Equity
In January 2005, the CICA released new Handbook Section 1530, Comprehensive Income, and Section 3251, Equity, effective for the annual and interim period of the Company beginning on January 1, 2007. Section 1530 establishes standards for reporting comprehensive income. The section does not address issues of recognition or measurement for comprehensive income and its components. Section 3251 establishes standards for the presentation of equity and changes in equity during the reporting period. The requirements in this section are in addition to Section 1530. We have not yet determined the impact of the adoption of this standard on the presentation of the consolidated results of operations or financial position.
Financial Instruments — Recognition and Measurement
In January 2005, the CICA released new Handbook Section 3855, Financial Instruments — Recognition and Measurement, effective for the annual and interim period of the Company beginning on January 1, 2007. This new section prescribes when a financial instrument is to be recognized on the balance sheet and at what amount, sometimes using fair value and other times using cost-based measures. It also specifies how financial instrument gains and losses are to be presented and defines financial instruments to include accounts receivable and payable, loans, investments in debt and equity securities, and derivative contracts. We have not yet determined the impact of the adoption of this standard on the consolidated results of operations or financial position.
F-11
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
| 2. | Significant Accounting Policies — (Continued) |
Non-Monetary Transactions
In June 2005, the CICA released new Handbook Section 3831, Non-monetary Transactions, effective for the fiscal year beginning on January 1, 2006. This standard requires all non-monetary transactions to be measured at fair value unless they meet one of four very specific criteria. Commercial substance replaces culmination of the earnings process as the test for fair value measurement. A transaction has commercial substance if it causes an identifiable and measurable change in the economic circumstances of the entity. Commercial substance is a function of the cash flows expected by the reporting entity. The Company believes that the adoption of this standard will not have a material impact on the Company’s consolidated results of operations or financial position.
Hedges
In April 2005, the CICA issued Section 3865 of the CICA Handbook entitled “Hedges”, effective for the fiscal year beginning on January 1, 2007. This Section establishes standards for when and how hedge accounting may be applied. Hedging is an activity designed to modify an entity’s exposure to one or more risks. Hedge accounting modifies the normal basis for recognizing the gains, losses, revenues and expenses associated with a hedged item or a hedging item in an entity’s income statement. It ensures that counterbalancing gains, losses, revenues and expenses are recognized in the same period. The Company believes that the adoption of this standard will not have a material impact on the Company’s consolidated results of operations or financial position.
3. Changes in Accounting Policy
In 2003, the Canadian Institute of Chartered Accountants (“CICA”) amended its pronouncement relating to stock-based compensation requiring companies to measure and expense all equity instruments awarded to employees and directors starting in fiscal years beginning on or after January 1, 2004 in accordance with the fair value method. The fair value of stock options to employees and directors is determined at the date of grant using the Black-Scholes option pricing model, and expensed over the vesting period of the options. The transitional provisions provide for a prospective treatment to this change in accounting policy for those companies who adopt this new pronouncement in fiscal years beginning before January 1, 2004 and a retroactive treatment for those companies adopting on or after January 1, 2004.
Effective January 1, 2004, the Company adopted the retroactive treatment without restatement, for options granted since March 1, 2002. Consequently, the opening deficit and contributed surplus balances as at January 1, 2004 increased by $2,976. The compensation expense for the year ended December 31, 2005 was $1,754 (2004 — $1,775) of which $685 (2004 — $600) was included in research and development expenses and $1,069 (2004 — $1,175) was included in selling, general and administrative expenses. The counterpart has been recorded as contributed surplus. Prior to January 1, 2004, no compensation expense was recognized when stock options were granted to employees and directors, however the Company provided pro forma information as if the fair value method had been applied.
F-12
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
4. Short-term Investments
Short-term investments are comprised of 9 investment grade instruments (21 as at December 31, 2004). These investments have a maturity of less than one year and have an average weighted yield of 3.05% (December 31, 2004 — 2.42%). The market value of short-term investments held as at December 31, 2005 and 2004 approximates their carrying value. Short-term investments include the following:
| 2005 | 2004 | |||||||
| Commercial paper | $ | 12,209 | $ | 6,872 | ||||
| Corporate bonds | 2,402 | 6,719 | ||||||
| Government bonds | — | 7,223 | ||||||
| $ | 14,611 | $ | 20,814 | |||||
5. Accounts Receivable
| 2005 | 2004 | |||||||
| Trade | $ | 42 | $ | 329 | ||||
| Sales taxes | 386 | 436 | ||||||
| Interest on investments | 104 | 196 | ||||||
| Other | — | 6 | ||||||
| $ | 532 | $ | 967 | |||||
6. Inventories
As at December 31, 2005, the Company has accumulated inventories comprised of raw materials totalling $1,710 and intermediate finished goods (bulk tablets) totalling $478.
7. Restricted Long-Term Investments
Long-term investments are comprised of two debt instruments (two as at December 31, 2004) maturing in 2006 with an average weighted yield of 2.9% (December 31, 2004 — 2.0%). The market value of the long-term investments held as at December 31, 2005 approximates their carrying value. These two investments collateralize letters of credit issued with regard to lease obligations on behalf of the Company by its bankers(note 13). Although these investments mature in 2006, they will be renewed on an ongoing basis to maintain the letters of credit.
F-13
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
8. Property, Plant and Equipment
| Net | ||||||||||||
| Accumulated | carrying | |||||||||||
| Cost | depreciation | value | ||||||||||
2005 | ||||||||||||
| Building and building improvements | $ | 8,044 | $ | 1,409 | $ | 6,635 | ||||||
| Laboratory and plant equipment | 4,290 | 2,389 | 1,901 | |||||||||
| Computer hardware and software | 2,110 | 1,019 | 1,091 | |||||||||
| Furniture and office equipment | 1,090 | 518 | 572 | |||||||||
| Leasehold improvements | 106 | 25 | 81 | |||||||||
| $ | 15,640 | $ | 5,360 | $ | 10,280 | |||||||
2004 | ||||||||||||
| Building and building improvements | $ | 8,030 | $ | 871 | $ | 7,159 | ||||||
| Laboratory equipment | 3,862 | 1,945 | 1,917 | |||||||||
| Computer hardware and software | 1,872 | 726 | 1,146 | |||||||||
| Furniture and office equipment | 1,062 | 415 | 647 | |||||||||
| Leasehold improvements | 106 | 14 | 92 | |||||||||
| $ | 14,932 | $ | 3,971 | $ | 10,961 | |||||||
Property, plant and equipment include the following assets under capital leases:
| Net | ||||||||||||
| Accumulated | carrying | |||||||||||
| Cost | depreciation | value | ||||||||||
2005 | ||||||||||||
| Building | $ | 5,970 | $ | 1,061 | $ | 4,909 | ||||||
| Laboratory equipment | 243 | 130 | 113 | |||||||||
| Office equipment | 229 | 103 | 126 | |||||||||
| $ | 6,442 | $ | 1,294 | $ | 5,148 | |||||||
2004 | ||||||||||||
| Building | $ | 5,970 | $ | 663 | $ | 5,307 | ||||||
| Laboratory equipment | 243 | 101 | 142 | |||||||||
| Office equipment | 229 | 57 | 172 | |||||||||
| $ | 6,442 | $ | 821 | $ | 5,621 | |||||||
Amortization expense of assets under capital leases of $473 (2004 — $481) is included with depreciation of property, plant and equipment.
The right to use the building and the building improvements reverts to its owners in 2018 unless the Company renews or extends the lease.
F-14
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
9. Intangible Assets
| Net | ||||||||||||
| Accumulated | carrying | |||||||||||
| Cost | amortization | value | ||||||||||
2005 | ||||||||||||
| Intellectual property rights | $ | 500 | $ | 271 | $ | 229 | ||||||
| Patents and trademarks | 3,720 | 718 | 3,002 | |||||||||
| $ | 4,220 | $ | 989 | $ | 3,231 | |||||||
2004 | ||||||||||||
| Intellectual property rights | $ | 500 | $ | 246 | $ | 254 | ||||||
| Patents and trademarks | 2,385 | 603 | 1,782 | |||||||||
| $ | 2,885 | $ | 849 | $ | 2,036 | |||||||
| 10. | Capital Lease Obligations |
| 2005 | 2004 | |||||||
| Building, repayable in monthly instalments of $71 until May 2008, $83 from June 2008 to May 2013, and $96 from June 2013 to April 2018, including interest calculated at 14.6% | $ | 12,765 | $ | 13,615 | ||||
| Various leases for office equipment, repayable in monthly instalments totalling $3 including interest ranging from 8.0% to 9.9%, with maturity from February 2008 to March 2009 | 110 | 210 | ||||||
| 12,875 | 13,825 | |||||||
| Interest included in instalments | 6,952 | 7,768 | ||||||
| 5,923 | 6,057 | |||||||
| Current portion | 83 | 134 | ||||||
| $ | 5,840 | $ | 5,923 | |||||
Minimum lease payments under capital leases for the next five years are as follows:
| 2006 | $ | 889 | ||
| 2007 | 889 | |||
| 2008 | 975 | |||
| 2009 | 1,002 | |||
| 2010 | 1,000 | |||
| Thereafter | 8,120 |
During 2005 no property, plant and equipment were financed through capital leases (2004 — $13).
F-15
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
| 11. | Long-Term Debt |
| 2005 | ||||
| Term loan, maturing on July 1, 2008 with interest payable monthly and repayable in 30 monthly payments of $452 including interest, commencing on February 1, 2006 | $ | 11,660 | ||
| Less adjustment for the termination fee and the portion of the warrants not yet recognized as financial expenses | (459 | ) | ||
| Carrying value of long-term debt | $ | 11,201 | ||
| Current portion of long-term debt | 3,383 | |||
| $ | 7,818 | |||
On June 28, 2005, the Company entered into a US$10,000 term loan agreement bearing interest at 11.95%, resulting in gross proceeds of $12,317. As part of the agreement, the Company issued 543,104 warrants to purchase common shares (note 12). Proceeds were allocated to the long-term debt for $11,586 and the warrants for $731. As a result of the value assigned to the warrants and the loan termination fee of $408 (US$350) due on repayment, the effective interest rate of the term loan is approximately 17%.
The financing costs related to the term loan agreement amounted to $474 and were recorded as deferred financing costs.
The term loan is collaterized by all of the Company’s assets except for its intellectual property.
Minimum annual principal repayments of the carrying value of the long-term debt during the next twelve-month periods ending December 31, are as follows:
| 2006 | $ | 3,383 | ||
| 2007 | 4,423 | |||
| 2008 | 3,395 |
12. Capital Stock
Authorized
Unlimited number of preferred shares, non-participating, non-voting, without par value
Unlimited number of common shares, voting, without par value
Issued
| 2005 | 2004 | |||||||
| 43,673,863 Common shares (December 31, 2004 — 42,510,630) | $ | 135,631 | $ | 132,658 | ||||
Capital stock transactions
During 2005, 834,600 (2004 — 212,600) options were exercised for a total cash consideration of $2,093 (2004 — $453). In addition, capital stock was increased by $149 (2004 — $6) and contributed surplus reduced by the same amount to record options exercised which were granted after March 1, 2002. In addition, 328,633 shares were issued on a cashless conversion of 543,104 warrants resulting in a transfer of $731 from contributed surplus to share capital.
F-16
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
12. Capital Stock — (Continued)
On May 26, 2004, the Company issued 6,122,449 common shares for consideration of $30,000. Share issue expenses amounted to $2,196.
Warrants
On June 29, 2005, as part of the term loan agreement described in note 11, the Company issued 543,104 warrants to purchase one common share per warrant, having an exercise price of $2.71 each and expiring on June 29, 2010. The Company valued the warrants using the Black-Scholes option pricing model with the following weighted average assumptions:
| Expected volatility | 0.63 | |
| Expected life | 5.0 years | |
| Risk-free interest rate | 3.16% | |
| Dividend yield | Nil |
Proceeds from the term loan agreement were allocated on a pro-rata basis to the long-term debt and the warrants. The fair value attributed to the warrants was $731.
During 2005, the 543,104 warrants were exercised on a cashless basis for a total of 328,633 shares. As at December 31, 2005, no warrants were outstanding (2004 — nil).
During the year ended February 28, 2000, the Company issued 200,000 warrants to purchase one common share per warrant, to the former supplier of Contramid®. These warrants had an exercise price of $2.00 and an expiry date of May 2004. During the year ended February 28, 2002, 10,000 warrants were exercised for a total of 10,000 shares for a cash consideration of $20. During 2004, 180,000 warrants were exercised for a total of 180,000 shares for a total cash consideration of $360 and 10,000 warrants expired unexercised.
Stock option plan
The Company established a stock option plan for directors, executive officers, employees and consultants of the Company. On April 20, 2005, the Company’s stock option plan was amended in order to express the maximum number of securities issuable thereunder as a percentage of the Company’s issued and outstanding shares rather than a fixed number. The maximum number of common shares that are issuable under the plan shall not exceed 9.9% of the Company’s total issued and outstanding shares at any time.
The maximum number of common shares that may be optioned in favour of any individual will not exceed 5% of the number of outstanding common shares. Under the stock option plan, the price at which the common shares may be purchased will not be lower than the closing price of the common shares on the Toronto Stock Exchange on the grant date. Any options issued are non-transferable.
All of the options that may be granted under the plan are exercisable according to a schedule up to a maximum period of ten years following the grant date thereof. The outstanding options as at December 31, 2005, may be exercised no later than December 2010. Options generally vest over a three-year period except for options to Board of Directors which vest immediately.
As of December 31, 2005, 4,323,712 securities are issuable under the plan, and 193,937 options are available for grant.
F-17
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
12. Capital Stock — (Continued)
The changes in the number of stock options granted by the Company, and their weighted average exercise prices are as follows:
| 2005 | 2004 | |||||||||||||||
| # | # | |||||||||||||||
| Balance, beginning of year | 3,363,475 | $ | 5.01 | 3,415,025 | $ | 4.87 | ||||||||||
| Granted | 1,071,400 | 4.92 | 190,800 | 4.66 | ||||||||||||
| Exercised | (834,600 | ) | 2.51 | (212,600 | ) | 2.13 | ||||||||||
| Expired | (39,000 | ) | 2.59 | — | — | |||||||||||
| Forfeited | (400 | ) | 7.92 | (29,750 | ) | 7.36 | ||||||||||
| Balance, end of year | 3,560,875 | $ | 5.59 | 3,363,475 | $ | 5.01 | ||||||||||
| Options eligible to be exercised | 2,887,975 | $ | 5.70 | 3,047,375 | $ | 4.76 | ||||||||||
Additional information concerning stock options as at December 31, 2005 is as follows:
| Options outstanding | Options exercisable | |||||||||||||||||||
| Weighted- | ||||||||||||||||||||
| average | Weighted- | Weighted- | ||||||||||||||||||
| remaining | average | average | ||||||||||||||||||
| Number of | contractual | exercise | Number of | exercise | ||||||||||||||||
| Range of exercise price | options | life | price | options | price | |||||||||||||||
| $2.16 to $3.94 | 1,476,025 | 2.70 years | $ | 3.07 | 1,167,625 | $ | 2.99 | |||||||||||||
| $4.10 to $6.61 | 986,700 | 3.58 years | $ | 5.79 | 645,600 | $ | 5.35 | |||||||||||||
| $7.00 to $10.29 | 1,098,150 | 1.88 years | $ | 8.83 | 1,074,750 | $ | 8.86 | |||||||||||||
| 3,560,875 | 2,887,975 | |||||||||||||||||||
A compensation expense of $1,754 (2004 — $1,775) has been recognized during the year ended December 31, 2005 for stock options granted to employees and directors since March 1, 2002.
The fair value of options granted during the year was estimated at the date of grant using the Black-Scholes option pricing model with the following weighted average assumptions:
| For the year ended December 31, | 2005 | 2004 | ||||||
| Expected volatility | 0.67 | 0.72 | ||||||
| Expected life | 4 years | 4 years | ||||||
| Risk-free interest rate | 3.67% | 3.65% | ||||||
| Dividend yield | Nil | Nil | ||||||
The weighted average grant date fair value of stock options granted during the year ended December 31, 2005 using the above assumptions amounted to $2.65 (2004 — $2.63).
F-18
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
13. Commitments, Guarantees and Contingency
(a) Commitments
The Company occupies certain facilities under operating lease arrangements and leases certain equipment. Estimated future minimum payments under these operating leases for the years ending December 31 are as follows:
| 2006 | $ | 82 | ||
| 2007 | 64 | |||
| 2008 | 60 | |||
| 2009 | 58 | |||
| 2010 | 58 | |||
| Thereafter | 130 | |||
| $ | 452 | |||
Rental expenses for facilities and equipment under operating leases totalled $128 in 2005 (2004 — $84).
Letters of credit amounting to $1,271 were issued to the lessors of the Company’s facilities as a collateral for the Company’s performance of obligations under the leases. These letters of credit are collateralized by specific investments totalling $1,271 which have been classified as long-term.
The Company has entered into long-term supply agreements with third-party manufacturers in anticipation of the commercialization of its products. These agreements include clauses under which the Company would have to reimburse certain equipment and set-up costs if minimum quantities are not purchased or not purchased within the prescribed delay, and clauses requiring the purchase of minimum quantities of product. Management has estimated that the minimum commitments related to these agreements would be approximately $267 for equipment and set-up costs, and approximately $27,888 for the purchase of bulk tablets.
(b) Guarantees
The Company periodically enters into research, licensing, distribution or supply agreements with third parties that include indemnification provisions that are customary in the industry. These guarantees generally require the Company to compensate the other party for certain damages and costs incurred as a result of third-party intellectual property claims or damages arising from these transactions. In some cases, the maximum potential amount of future payments that could be required under these indemnification provisions is unlimited. These indemnification provisions generally survive termination of the underlying agreement. The nature of the intellectual property indemnification obligations prevents the Company from making a reasonable estimate of the maximum potential amount it could be required to pay. Historically, the Company has not made any indemnification payments under such agreements and no amount has been accrued in the accompanying consolidated financial statements with respect to these indemnification obligations.
F-19
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
13. Commitments, Guarantees and Contingency — (Continued)
(c) Contingency
In 1994, concurrently with the purchase of a controlled-release technology, the Company acquired a right of first refusal with respect to an improved technology for which it agreed to pay royalties of 4% on net revenues generated from the commercialization of the 1994 purchased technology. On February 7, 2005, the Company was served with a motion to institute legal proceedings in the Québec Superior Court. The motion seeks payment of an unspecified amount of royalties said to be outstanding since 1999. The Company considers that, as at December 31, 2005, no amounts are owing.
14. Income Taxes
Income tax expense on loss from operations as presented differs from the amount calculated by applying the statutory income tax rate to loss before income taxes.
The reasons for this difference and the effect on income tax expense are as follows:
| 2005 | |||||||||||||||||
| 2004 | |||||||||||||||||
| Loss before income taxes: | |||||||||||||||||
| Canadian operations | $ | (1,007 | ) | $ | (3,284 | ) | |||||||||||
| Foreign operations | (31,128 | ) | (23,854 | ) | |||||||||||||
| (32,135 | ) | (27,138 | ) | ||||||||||||||
| Tax recovery at the Canadian statutory rate | 9,961 | 31.0 % | 8,413 | 31.0 % | |||||||||||||
| Change in income taxes arising from the following: | |||||||||||||||||
| Effect of foreign tax rate differential | (6,217 | ) | (19.3) | (2,974 | ) | (11.0) | |||||||||||
| Non-deductible items | (497 | ) | (1.5) | (960 | ) | (3.5) | |||||||||||
| Unrecognized tax benefits of losses carried forward and other differences | (5,044 | ) | (15.7) | (4,479 | ) | (16.5) | |||||||||||
| Impact of changes in tax rates | 970 | 3.0 | — | — | |||||||||||||
| Other | (372 | ) | (1.2) | (41 | ) | (0.2) | |||||||||||
Tax expense | $ | (1,199 | ) | (3.7)% | $ | (41 | ) | (0.2)% | |||||||||
The Company created a Canadian Federal tax expense and liability of $1,199 in order to utilize a corresponding amount of non-refundable tax credits related to research and development expenditures not previously recognized in the financial statements and which would eventually expire. The corresponding amount was recognized as government assistance and a reduction of research and development expenses(note 15)and offsets the Canadian Federal income tax payable in the year.
F-20
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
14. Income Taxes — (Continued)
The tax effect of temporary differences and net operating losses that give rise to future income tax assets are as follows:
| 2005 | 2004 | |||||||
Future income tax liabilities | ||||||||
| Investment tax credits | $ | (384 | ) | $ | — | |||
| Total future income tax liabilities | (384 | ) | — | |||||
Future income tax assets | ||||||||
| Tax basis of capital assets in excess of carrying values | 2,595 | 2,722 | ||||||
| Net operating losses carried forward of foreign entities | 9,145 | 6,708 | ||||||
| Net operating losses carried forward of the Canadian entity | — | 665 | ||||||
| Research and development expenditures | 10,293 | 9,111 | ||||||
| Share issue costs | 642 | 995 | ||||||
| Total future income tax assets | 22,675 | 20,201 | ||||||
| Valuation allowance | (22,291 | ) | (20,201 | ) | ||||
| Net future income tax assets | — | — | ||||||
| Net future income tax liability | $ | — | $ | — | ||||
During the year ended December 31, 2005, the valuation allowance increased by $2,090.
The Company has accumulated loss carryforwards in foreign jurisdictions which are available to reduce future taxable income, the benefits of which have not been recognized in these financial statements. These loss carryforwards expire as follows:
| Barbados | ||||
| 2012 | $ | 6,244 | ||
| 2013 | 4,847 | |||
| 2014 | 3,633 | |||
| $ | 14,724 | |||
In addition, the Company has $70,155 and $76 of loss carryforwards in Ireland and Cyprus respectively that are available to reduce taxable income in future years and have an unlimited carryforward period, the benefit of which has not been reflected in these financial statements.
The Company has approximately $30,818 of research and development expenditures for Canadian Federal tax purposes and $29,259 for Québec purposes that are available to reduce taxable income in future years and have an unlimited carryforward period, the benefit of which has not been reflected in these financial statements. Research and development expenditures are subject to audit by the taxation authorities and accordingly, these amounts may vary.
The Company also has accumulated share issue costs which have not been deducted for income tax purposes amounting to approximately $1,986. The benefits of these expenses have not been recorded in the financial statements.
F-21
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
14. Income Taxes — (Continued)
During 2005, the Company was audited by the Canada Revenue Agency (CRA) with respect to the 2002 sale, to its foreign subsidiaries, of an undivided interest in certain of its intellectual property rights. Subsequent to year end the Company received a proposed adjustment from the CRA of the amount for which the assets were sold. The proposed adjustment would increase significantly the taxable income of the Company for 2002. The Company believes that the proposed adjustment is largely unfounded and it intends to vigorously oppose the proposal and any subsequent assessment, should one arise.
The Company believes it has taken adequate reserves to address this issue through a reduction in future tax assets and a corresponding reduction in the 2005 valuation allowance of $2,072. The ultimate resolution of this matter could result in material adjustments to the amounts provided in the accounts.
15. Government Assistance
The Company incurred research and development expenditures which are eligible for investment tax credits. The investment tax credits recorded are based on management’s estimates of amounts expected to be recovered and are subject to audit by the taxation authorities and, accordingly, these amounts may vary. These amounts have been recorded as a reduction of research and development expenditures as follows:
| 2005 | 2004 | ||||||||
| Research and development tax credits | |||||||||
| Non-refundable | $ | 1,199 | $ | — | |||||
| Refundable | 1,514 | 823 | |||||||
| $ | 2,713 | $ | 823 | ||||||
The Company has non-refundable tax credits available amounting to $6,010 related to research and development expenditures which may be utilized to reduce Canadian Federal income taxes payable in the future years and expire as follows:
| 2010 | $ | 19 | ||
| 2011 | 529 | |||
| 2012 | 791 | |||
| 2013 | 1,404 | |||
| 2014 | 1,494 | |||
| 2015 | 1,773 | |||
| $ | 6,010 | |||
The benefits of these non-refundable investment tax credits have not been recognized in the financial statements.
F-22
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
16. Financial Expenses
| 2005 | 2004 | |||||||
| Interest on long-term debt | $ | 1,010 | $ | — | ||||
| Interest on capital lease obligations | 817 | 864 | ||||||
| Amortization of deferred financing costs | 110 | — | ||||||
| $ | 1,937 | $ | 864 | |||||
17. Net Change in Non-Cash Operating Items
| 2005 | 2004 | |||||||
| Accounts receivable | $ | 355 | $ | 328 | ||||
| Research and development tax credits receivable | (75 | ) | 100 | |||||
| Income tax receivable | (425 | ) | — | |||||
| Inventories | (2,188 | ) | — | |||||
| Prepaids and other assets | (205 | ) | 90 | |||||
| Accounts payable and accrued liabilities | 3,140 | (1,157 | ) | |||||
| Deferred revenues | 28,125 | 1,299 | ||||||
| $ | 28,727 | $ | 660 | |||||
18. Employee Benefit Plan
The Company has two defined contribution plans, a contributory Canadian plan effective July 1, 2005 and a non-contributory European plan effective December 2003. The Company funded and charged to expense contributions of $108 and $43 in 2005 and 2004, respectively.
19. Economic Dependence
The Company is dependent on two key raw material suppliers, two contract manufacturers and two packagers. The loss of any of these suppliers may have a materially adverse effect on the Company’s financial position and results of operations.
20. Financial Instruments
Fair value
Given their short-term maturity, the fair value of cash and cash equivalents, accounts receivable, research and development tax credits receivable, income tax receivable and current accounts payable approximate the carrying value.
The fair value of the obligations under capital leases, calculated at the present value of future contractual payments of principal and interest and discounted at the current market rate of interest available to the Company for debt instruments with similar terms and maturity, approximates their carrying values.
The fair value of the long-term debt, including current portion approximates its carrying value. The Company believes that the interest rates used to determine the carrying value are similar to the rates which it could have obtained as at the balance sheet date for loans with similar terms, conditions and maturity dates.
F-23
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
20. Financial Instruments — (Continued)
Credit risk
Financial instruments that potentially subject the Company to significant concentrations of credit risk consist principally of short-term and long-term investments. The Company has investment policies that monitor the safety and preservation of principal and investments are limited to $2,500 with a single issuer.
Also, the Company provides credit to its clients in the normal course of operations. It carries out on a continuing basis, credit evaluations of its customers. As at December 31, 2005, 100% (December 31, 2004 — 100%) of trade accounts receivable is due from one customer (December 31, 2004 — one customer). For the year ended December 31, 2005, 100% (December 31, 2004 — 100%) of revenue from research and development contracts was earned from one customer (December 31, 2004 — two customers), located in Europe.
Currency risk
A substantial portion of the Company’s revenues from research contracts and licensing and distribution agreements are denominated in U.S. dollars or Euros. This results in financial risk due to fluctuations in the value of the Canadian dollar relative to the U.S. dollar and Euro. The Company does not use derivative financial instruments to reduce its foreign exchange exposure, however, the Company has a natural hedge for a portion of this risk, in that many of its expenditures are in U.S. dollars and Euros. Fluctuations in payments made for the Company’s services could cause unanticipated fluctuations in the Company’s operating results. The significant balances in foreign currencies at December 31, 2005 are as follows:
| U.S. Dollars | Euros | |||||||||||||||
| 2005 | 2004 | 2005 | 2004 | |||||||||||||
| Cash and cash equivalents | $ | 9,614 | $ | 118 | € | 5,851 | € | 881 | ||||||||
| Accounts receivables | 143 | — | 30 | 213 | ||||||||||||
| Restricted long-term investment | — | — | 51 | 50 | ||||||||||||
| Accounts payables and accrued liabilities | (2,704 | ) | (219 | ) | (616 | ) | (275 | ) | ||||||||
| Long-term debt | (10,000 | ) | — | — | — | |||||||||||
| $ | (2,947 | ) | $ | (101 | ) | € | 5,316 | € | 869 | |||||||
21. Segment Disclosure
The Company carries on business in Canada, Barbados, the United States, and Ireland and substantially all of the Company’s tangible assets are located in Canada. All licensing and product revenue has been derived from the business carried on in Ireland, by the Company’s subsidiary Labopharm Europe Limited, and all other revenue has been derived from business carried on in Canada. The intangible assets are jointly owned
F-24
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
21. Segment Disclosure — (Continued)
by the Company and its foreign subsidiaries. The following table presents revenue by the customers’ domicile and property, plant and equipment by location.
| Total revenue | ||||||||
| 2005 | 2004 | |||||||
| Europe | $ | 1,878 | $ | 1,394 | ||||
| United States | 1,360 | — | ||||||
| $ | 3,238 | $ | 1,394 | |||||
| Property, plant and | ||||||||
| equipment | ||||||||
| 2005 | 2004 | |||||||
| Canada | $ | 9,912 | $ | 10,762 | ||||
| Europe | 368 | 199 | ||||||
| $ | 10,280 | $ | 10,961 | |||||
22. Related Party Transaction
During 2005, in the normal course of business, the Company paid consulting fees of $325 (2004 — $319) to a company related to a director and recorded these as selling, general and administrative expenses as incurred. These transactions are measured at their exchange amount.
| 23. | Reconciliation of Significant Differences between Accounting Principles Generally Accepted in Canada and the United States |
These financial statements were prepared in accordance with accounting principles generally accepted in Canada (“Canadian GAAP”) and conform in all material respects with U.S. generally accepted accounting principles (“U.S. GAAP”) except as follows:
(a) Stock-based compensation
Under Canadian GAAP, the Company accounts for stock-based compensation to employees and directors as described in note 2. Under U.S. GAAP, the Company has similarly elected to follow the fair value method in accordance with FAS 123,Accounting for Stock-Based Compensationas of May 1995. As such, all compensation expense is recorded as a charge to income and a credit to contributed surplus. However, compensation expense under U.S. GAAP takes into account options and warrants granted prior to the effective date of the Canadian GAAP requirements.
During the year ended February 28, 2001, the Company granted 196,725 compensation options to its agent in connection with the Company’s issuance of common shares. The options had an exercise period of twenty-four months from August 31, 2000, vested immediately and entitled the holder to purchase common shares at an exercise price of $3.40 per share. Under Canadian GAAP no compensation cost was recorded. For purposes of reconciliation to U.S. GAAP, these options were accounted for under the fair value method on the date of grant using the Black-Scholes option pricing model, and an expense of $356 was recorded as a
F-25
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
| 23. | Reconciliation of Significant Differences between Accounting Principles Generally Accepted in Canada and the United States — (Continued) |
share issue cost against share capital, with a corresponding credit to contributed surplus. During the year ended February 28, 2002, 100,000 options were exercised resulting in a transfer of $181 from contributed surplus to share capital, and the remaining 96,725 options expired unexercised in August 2002.
(b) Warrants
During the year ended February 28, 2000, the Company issued 200,000 warrants to purchase one common share per warrant, to the former supplier of Contramid® in partial settlement for the termination of an agreement. No expense was recorded for the fair value of these warrants under Canadian GAAP. For purposes of reconciliation to U.S. GAAP, these warrants were accounted for under the fair value method on the date of grant using the Black-Scholes option pricing model, and an expense of $224 was recorded as a charge to operations in fiscal 2000, with a corresponding credit to contributed surplus. In 2002 and 2004, 190,000 warrants were exercised resulting in a total transfer of $213 from contributed surplus to capital stock.
(c) Patent and intellectual property costs
Under Canadian GAAP, the Company capitalizes certain patent costs and costs of acquiring intellectual property as further described in note 2. Under U.S. GAAP, such costs are classified as in-process research and development and expensed as incurred until such time as the underlying product is approved for commercialization. Amounts included in the reconciliation of consolidated net loss and comprehensive loss reflect an increase in the net loss for amounts capitalized under Canadian GAAP but expensed under U.S. GAAP, offset by a decrease to the net loss for the related amortization expense.
On November 29, 2005, the Company acquired certain patents which relate to a product approved for sale in Europe and capitalized the purchase cost of $1,162 for U.S. and Canadian GAAP purposes.
(d) Research and development expenses
Under U.S. GAAP, research and development costs are expensed as incurred and include salaries and benefits, costs paid to third-party contractors to perform research, develop and manufacture drug material, and a portion of facilities cost. Clinical trial costs are a significant component of research and development expenses and include costs associated with third-party contractors. Under Canadian GAAP certain development costs may be deferred.
(e) Available for sale investment
Under U.S. GAAP, short-term investments are classified as available-for-sale and carried at market values with unrealized gains or losses reflected as a component of accumulated other comprehensive income until the investments are sold, at which time the realized gains and losses are included in the results of operations. The reconciling differences were insignificant in 2005 and 2004. Under Canadian GAAP short-term investments are recorded at amortized cost unless there is a decline in fair value which is other than temporary.
F-26
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
| 23. | Reconciliation of Significant Differences between Accounting Principles Generally Accepted in Canada and the United States — (Continued) |
(f) Comprehensive loss
Comprehensive loss includes all changes in equity (deficiency) during the periods presented except shareholder transactions. For the purpose of reporting under U.S. GAAP, the components of comprehensive loss and total comprehensive loss are reported in the consolidated statements of changes in shareholders’ equity (deficiency), and below net loss in the consolidated statements of operations and deficit. For the periods presented, accumulated other comprehensive loss equals net loss.
The effect of the above on the Company’s consolidated financial statements is set out below:
Reconciliation of Consolidated Net Loss and Comprehensive Loss
| Year ended | Year ended | |||||||
| December 31, | December 31, | |||||||
| 2005 | 2004 | |||||||
Net loss under Canadian GAAP | $ | (33,334 | ) | $ | (27,179 | ) | ||
| Adjustment for: | ||||||||
| Stock based compensation (a) | — | (217 | ) | |||||
| Patent and intellectual property costs (c)(d) | (33 | ) | (45 | ) | ||||
Net loss and comprehensive loss under U.S. GAAP | $ | (33,367 | ) | $ | (27,441 | ) | ||
Net loss per share under U.S. GAAP — basic and diluted | $ | (0.78 | ) | $ | (0.69 | ) | ||
The weighted average number of common shares outstanding for purposes of determining basic and diluted loss per share are the same as those used for Canadian GAAP purposes.
The effects of any permanent or temporary timing differences for tax purposes are not significant and therefore have not been reflected in the reconciliation.
Reconciliation of Reported Amounts on Consolidated Balance Sheets
Material variations in selected balance sheet accounts under U.S. GAAP are as follows:
| Canadian | U.S. | |||||||||||
| GAAP | Adjustments | GAAP | ||||||||||
2005 | ||||||||||||
| Intangible assets (c)(d) | $ | 3,231 | $ | (2,069 | ) | $ | 1,162 | |||||
| Capital stock (a)(b) | 135,631 | 2,939 | 138,570 | |||||||||
| Contributed surplus (a)(b) | 6,350 | 7,381 | 13,731 | |||||||||
| Deficit | (144,584 | ) | (12,389 | ) | (156,973 | ) | ||||||
2004 | ||||||||||||
| Intangible assets (c)(d) | 2,036 | (2,036 | ) | — | ||||||||
| Capital stock (a) (b) | 132,658 | 1,706 | 134,364 | |||||||||
| Contributed surplus (a)(b) | 4,745 | 8,614 | 13,359 | |||||||||
| Deficit | (111,250 | ) | (12,356 | ) | (123,606 | ) | ||||||
F-27
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
| 23. | Reconciliation of Significant Differences between Accounting Principles Generally Accepted in Canada and the United States — (Continued) |
Reconciliation of Consolidated Cash Flow Captions
| Year ended | Year ended | |||||||||||||||
| December 31, 2005 | December 31, 2004 | |||||||||||||||
| Canadian | U.S. | Canadian | U.S. | |||||||||||||
| GAAP | GAAP | GAAP | GAAP | |||||||||||||
| Operating activities (c)(d) | $ | (1,040 | ) | $ | (1,213 | ) | $ | (23,082 | ) | $ | (23,269 | ) | ||||
| Investing activities (c)(d) | 5,187 | 5,360 | (3,275 | ) | (3,088 | ) | ||||||||||
As at December 31, 2005 and 2004, cash and cash equivalents are comprised of cash.
Additional disclosures required under U.S. GAAP are as follows:
(i) Consolidated statement of changes in shareholders’ equity (deficiency) in accordance with U.S. GAAP:
| �� | ||||||||||||||||||||
| Outstanding common | ||||||||||||||||||||
| shares | ||||||||||||||||||||
| Contributed | ||||||||||||||||||||
| Number | Surplus | Deficit | Total | |||||||||||||||||
Balance, December 31, 2003 | 35,995,581 | $ | 105,297 | $ | 11,817 | $ | (96,165 | ) | $ | 20,949 | ||||||||||
| Share issuance | 6,122,449 | 27,804 | — | — | 27,804 | |||||||||||||||
| Issued on the exercise of stock options | 212,600 | 701 | (248 | ) | — | 453 | ||||||||||||||
| Issued on the exercise of warrants | 180,000 | 562 | (202 | ) | — | 360 | ||||||||||||||
| Stock-based compensation | — | — | 1,992 | — | 1,992 | |||||||||||||||
| Net loss | — | — | — | (27,441 | ) | (27,441 | ) | |||||||||||||
Balance, December 31, 2004 | 42,510,630 | $ | 134,364 | $ | 13,359 | $ | (123,606 | ) | $ | 24,117 | ||||||||||
| Issued on the exercise of stock options | 834,600 | 3,475 | (1,382 | ) | — | 2,093 | ||||||||||||||
| Grant of warrants | — | — | 731 | — | 731 | |||||||||||||||
| Issued on the exercise of warrants | 328,633 | 731 | (731 | ) | — | — | ||||||||||||||
| Stock-based compensation | — | — | 1,754 | — | 1,754 | |||||||||||||||
| Net loss | — | — | — | (33,367 | ) | (33,367 | ) | |||||||||||||
Balance, December 31, 2005 | 43,673,863 | $ | 138,570 | $ | 13,731 | $ | (156,973 | ) | $ | (4,672 | ) | |||||||||
(ii) Accounts payable and accrued liabilities consist of the following:
| 2005 | 2004 | |||||||
| Trade payables | $ | 7,850 | $ | 4,162 | ||||
| Accrued payroll and related expenses | 1,742 | 1,451 | ||||||
| Other | 498 | 317 | ||||||
| $ | 10,090 | $ | 5,930 | |||||
F-28
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
| 23. | Reconciliation of Significant Differences between Accounting Principles Generally Accepted in Canada and the United States — (Continued) |
(iii) Intangible assets
Under U.S. GAAP, the estimated amortization expense for each of the next five succeeding fiscal years for the capitalized costs of acquiring patents will amount to $81.
(iv) Allowance for doubtful accounts
The Company maintains an allowance for doubtful accounts related to its accounts receivable. The Company reviews its accounts receivable on a regular basis to determine if any receivables have a high risk of being uncollectible, and include these in its allowance. Based on the information available, management believes the allowance for doubtful accounts is appropriate; however, actual write-offs might exceed the recorded allowance. The Company did not have an allowance for doubtful accounts as of December 31, 2005 and 2004.
(v) Recent accounting pronouncements under U.S. GAAP
In December 2004, the Financial Accounting Standards Board issued SFAS 123(R)Share-Based Payment, a revision to SFAS 123Accounting for Stock Based Compensation. SFAS 123(R) requires all share-based payments to be recognized in the financial statements based on their fair values using either a modified-prospective or modified-retrospective transition method. Accordingly, from the date of adoption of the revised standard, the Company will be required to recognize compensation expense for all share-based payments based on grant-date fair value, including those granted, modified or settled prior to December 1, 2002. Under U.S. GAAP, the Company had already elected to follow the fair value method in accordance with SFAS 123, and as such, SFAS 123(R) does not represent a significant change in accounting policy for the Company.
In November 2004, the FASB issued SFAS No. 151,Inventory Costs, an amendment of ARB No. 43, Chapter 4. This statement clarifies that abnormal inventory costs such as costs of idle facilities, excess freight and handling costs, and wasted material (spoilage) are required to be recognized as current period charges. In addition, the statement requires that allocation of fixed production overhead to the costs of conversion be based on the normal capacity of the production facilities. The provisions of this statement became effective for inventory costs incurred during fiscal years beginning after June 15, 2005. The Company believes that the adoption of this statement will not have a significant impact on the Company’s consolidated results of operations or financial condition.
In December 2004, the FASB issued SFAS No. 153,Exchanges of Non monetary Assets, an amendment of APB Opinion No. 29. The amendments made by SFAS 153 are based on the principle that exchanges of non monetary assets should be measured based on the fair value of the assets exchanged. Further, the amendments broaden the exception for exchanges of non monetary assets that do not have commercial substance. SFAS 153 is effective for non monetary asset exchanges occurring in fiscal periods beginning after June 15, 2005. The Company believes that the adoption of SFAS 153 will not have a material impact on the Company’s consolidated results of operations or financial position.
In May 2005, the FASB issued SFAS 154Accounting Changes and Error Corrections. The statement replaces APB Opinion No. 20, Accounting Changes and SFAS No. 3, Reporting Accounting Changes in Interim Financial Statements. SFAS 154 requires companies to apply voluntary changes in principles retrospectively
F-29
LABOPHARM INC.
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS — (Continued)
December 31, 2005
(thousands of Canadian dollars, except share and per share amounts)
| 23. | Reconciliation of Significant Differences between Accounting Principles Generally Accepted in Canada and the United States — (Continued) |
whenever practicable. SFAS 154 is effective for accounting changes and correction of errors made in fiscal years beginning after December 15, 2005. The Company presently does not believe that the adoption of the provisions of SFAS 154 will have a material impact on its financial statements.
24. Comparative Figures
Certain comparative figures have been reclassified to conform to the presentation adopted in the current period.
F-30
Until , 2006 (25 days after the commencement of this offering), all dealers that buy, sell or trade common shares may be required to deliver a prospectus regardless of whether they are participating in this offering. This is in addition to the dealers’ obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
10,000,000 Shares
Common Shares
PROSPECTUS
, 2006
PART II
INFORMATION NOT REQUIRED TO BE
DELIVERED TO OFFEREES OR PURCHASERS
INDEMNIFICATION
Under the Companies Act (Québec) (the “Act”), except in respect of an action by or on behalf of the Registrant to procure a judgment in its favor, the Registrant shall assume the defense of its mandatary (which covers directors and officers) prosecuted by a third person for an act done in the exercise of his duties and shall pay damages, if any, resulting from that act, unless such mandatary has committed a grievous offence or a personal offence separable from the exercise of his duties. However, in a penal or criminal proceeding, the Registrant shall assume only the payment of the expenses of its mandatary if he had reasonable grounds to believe that his conduct was in conformity with the law, or the payment of the expenses of its mandatary if he has been freed or acquitted. The Registrant shall assume the expenses of its mandatary if, having prosecuted him for an act done in the exercise of his duties, it loses its case and the court so decides. If the Registrant wins its case only in part, the court may determine the amount of the expenses it shall assume.
In addition, the By-laws of the Registrant provide in effect for the indemnification by the Registrant of each director and officer of the Registrant to the fullest extent permitted by applicable law.
The Registrant has purchased insurance for the benefit of all directors and officers of the Registrant and its subsidiaries against liability incurred by them in such capacity.
The Underwriting Agreement provides that the Underwriters are obligated, under certain circumstances, to indemnify directors, officers and controlling persons of the Company against certain liabilities, including liabilities under the Securities Act of 1933, as amended (the “Securities Act”) and liabilities under Canadian provincial securities laws. Reference is made to the form of Underwriting Agreement filed as Exhibit 3.1 hereto.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling the Registrant pursuant to the foregoing provisions, the Registrant has been informed that in the opinion of the U.S. Securities and Exchange Commission (the “Commission”) such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
II-1
EXHIBITS
The following exhibits have been filed as part of the Registration Statement:
| Exhibit | ||
| Number | Description | |
| 3.1* | Form of underwriting agreement | |
| 4.1 | Annual Information Form of the Registrant for the year ended December 31, 2005, dated March 30, 2006 | |
| 4.2 | Management Proxy Circular dated March 20, 2006 in connection with the annual meeting of shareholders of the Registrant to be held on May 4, 2006 | |
| 4.3 | Audited Comparative Consolidated Financial Statements of the Registrant, including the notes thereto, for the fiscal year ended December 31, 2005 together with the Auditor’s Report thereon as refiled on April 18, 2006, prepared in accordance with Canadian GAAP and including a reconciliation of the significant differences between Canadian GAAP and U.S. GAAP | |
| 4.4 | Management’s Discussion and Analysis contained in our Annual Report for the fiscal year ended December 31, 2005 | |
| 5.1 | Consent of Ernst & Young LLP | |
| 6.1 | Powers of Attorney (included on page III-3 of this Registration Statement) |
| * | To be filed by Amendment. |
II-2
PART III
UNDERTAKING AND CONSENT TO SERVICE OF PROCESS
Item 1. Undertaking.
The Registrant undertakes to make available, in person or by telephone, representatives to respond to inquiries made by the Commission staff, and to furnish promptly, when requested to do so by the Commission staff, information relating to the securities registered pursuant to Form F-10 or to transactions in such securities.
Item 2. Consent to Service of Process.
Concurrently with the filing of this Registration Statement on Form F-10, the Registrant is filing with the Commission a written irrevocable consent and power of attorney on Form F-X.
III-1
SIGNATURES
Pursuant to the requirements of the Securities Act, the Registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form F-10 and has duly caused this Registration Statement to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Montreal, Province of Québec, Canada on April 18, 2006.
| Labopharm inc. | ||
| By: | /s/ James R. Howard-Tripp Name: James R. Howard-Tripp Title: President and Chief Executive Officer | |
III-2
POWER OF ATTORNEY
Each person whose signature appears below constitutes and appoints each of James R. Howard-Tripp and Warren Whitehead with full power to act without the other, his true and lawful attorneys-in-fact and agents, with full and several power of substitution, for him and in his name, place and stead, in any and all capacities, to sign any or all amendments, including post-effective amendments, and supplements to this Registration Statement on Form F-10 (or to any other registration statement for the same offering which may be filed pursuant to Rule 429 under the Securities Act), and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Commission, granting unto said attorneys-in-fact and agents full power and authority to do and perform each and every act and thing requisite and necessary to be done in and about the premises, as fully to all intents and purposes as they or he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents or his or their substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act, this Registration Statement has been signed below by or on behalf of the following persons in the capacities indicated on April 18, 2006.
| /s/ James R. Howard-Tripp James R. Howard-Tripp | President and Chief Executive Officer and Director (Principal Executive Officer) | |
| /s/ Santo J. Costa Santo J. Costa | Director | |
| /s/ Frédéric Porte Frédéric Porte | Director |
III-3
| /s/ Jacques L. Roy Jacques L. Roy | Director | |
| /s/ James S. Scibetta James S. Scibetta | Director | |
| /s/ Warren Whitehead Warren Whitehead | Chief Financial Officer (Principal Financial Officer and Principal Accounting Officer) |
III-4
AUTHORIZED REPRESENTATIVE
Pursuant to the requirements of Section 6(a) of the Securities Act, the Authorized Representative certifies that it is the duly authorized United States representative of Labopharm inc. and has duly caused this Registration Statement to be signed on its behalf by the undersigned, solely in its capacity as the duly authorized representative of Labopharm inc. in the United States, in the City of Boston, Commonwealth of Massachusetts, United States of America on the 18th day of April, 2006.
| | ||
| By: | /s/ James S. Scibetta Name: James S. Scibetta Title: Director, Labopharm inc. | |
III-5
| Exhibit | ||
| Number | Description | |
| 3.1* | Form of underwriting agreement | |
| 4.1 | Annual Information Form of the Registrant for the year ended December 31, 2005, dated March 30, 2006 | |
| 4.2 | Management Proxy Circular dated March 20, 2006 in connection with the annual meeting of shareholders of the Registrant to be held on May 4, 2006 | |
| 4.3 | Audited Comparative Consolidated Financial Statements of the Registrant, including the notes thereto, for the fiscal year ended December 31, 2005 together with the Auditor’s Report thereon as refiled on April 18, 2006, prepared in accordance with Canadian GAAP and including a reconciliation of the significant differences between Canadian GAAP and U.S. GAAP | |
| 4.4 | Management’s Discussion and Analysis contained in our Annual Report for the fiscal year ended December 31, 2005 | |
| 5.1 | Consent of Ernst & Young LLP | |
| 6.1 | Powers of Attorney (included on page III-3 of this Registration Statement) |
| * | To be filed by Amendment. |