EXHIBIT 99.1
Phase III Trial of Active Immunotherapy (FavId®, Id/KLH) Following Treatment with Rituximab: Clinical Responses in Patients with Follicular Non-Hodgkin’s Lymphoma (fNHL)
Arnold Freedman, MD1, Paul Hamlin, MD2, Sattva Neelapu, MD3, Craig Nichols, MD4, Michael Robertson, MD5, Benjamin Djulbegovic, MD6, Jane Winter, MD7, Jonathan Polikoff, MD8, Thomas Lin, MD, PhD9, Brad Pohlman, MD10, Vanessa Esquibel11, Terri Melink, NP11, John F Bender, PharmD11
1Dana Farber Cancer Institute, Boston, MA, 02115; 2Memorial Sloan-Kettering Cancer Center, New York, NY, 10021; 3MD Anderson Cancer Center, Houston, TX, 77030; 4Oregon Health Sciences University, Portland, OR, 97201; 6H. Lee Moffitt Cancer Center, Tampa, FL, 33612; 7Northwestern Univ., Chicago, IL, 60611; 8Kaiser Foundation Hospital, San Diego, CA, 92120; 9The Ohio State Univ., Columbus, OH 43210; 10Cleveland Clinic Foundation, Cleveland, OH 44195; 11Favrille, Inc., San Diego, CA, 92121
Study Objectives
Primary Objective: To compare time to progression (TTP) in subjects receiving FavId/GM-CSF to the TTP in subjects receiving placebo/GM-CSF. Secondary Objectives: Response rate improvement (RRI), overall complete response rate, duration of response (DR), and safety.
Background
A Phase II trial (Koc et al. Blood 2005, 106:11 abstr #772) showing that Id/KLH following rituximab may improve objective response and TTP in fNHL pts formed the basis for this randomized, double-blind, placebo-controlled Phase III trial.
1
Eligibility
Patients with Grade 1-3 fNHL who were treatment naive (TN) or relapsed (R/R) with <2 prior therapies.
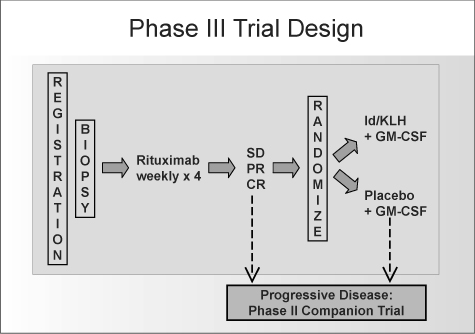
Registration and Randomization
| Registered |
| No Biopsy |
| Off Study* |
| Adequate |
| PD post |
| Randomized |
| |
TN |
| 380 |
| 32 |
| 61 |
| 287 |
| 12 |
| 275 |
|
R/R |
| 115 |
| 9 |
| 28 |
| 78 |
| 5 |
| 73 |
|
Total |
| 495 |
| 41 |
| 89 |
| 365 |
| 17 |
| 348 |
|
* Not eligible (11); Not fNHL (19); No tumor cells (11); No clonality (27); Insufficent quantity (18); withdrew (3)
Production Success
Adequate biopsies |
| FavId successfully manufactured |
| Production success |
|
365 |
| 336 |
| 92% |
|
Treatment
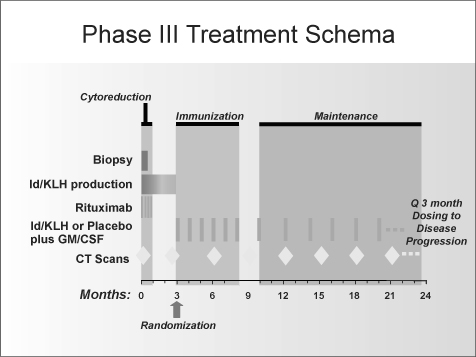
Pts received rituximab (375mg/m2 weekly x 4) during weeks 1-4 and were assessed for response by each investigator at week 11. Pts with SD, PR or CR were randomized to receive either FavId or Placebo (1 mg s.q. monthly x 6) along with GM-CSF (250 mcg, s.q. on days 1-4 of each course). Randomization was stratified by treatment status (TN vs. R/R) and rituximab response
2
(SD vs. PR/CR). Pts continued to receive maintenance injections every 2 months for 6 doses and then every 3 months until disease progression. Enrollment to this trial was completed in January 2006 and follow-up evaluations are ongoing. The trigger for TTP analysis, 248 pts with PD, has not been reached.
3
Results - Planned Interim Analysis
This interim analysis of the secondary endpoint, Response Rate Improvement (RRI), was designed to occur once 12 months of follow-up on at least 200 subjects was available.
Patients Characteristics
Patient Characteristics |
| TN |
| R/R |
|
Patient Distribution |
| 77% |
| 23% |
|
Age: Mean (range) |
| 54 (21 -84) |
| 55(23 -86) |
|
Sex: Male |
| 60% |
| 55% |
|
Histology (WHO) |
|
|
|
|
|
Grade 1 |
| 56% |
| 44% |
|
Grade 2 |
| 39% |
| 43% |
|
Grade 3 |
| 5% |
| 12% |
|
FLIPI Risk Group (# factors)* |
| TN |
| R/R |
| Total |
|
Number of patients |
| 152 |
| 44 |
| 196 |
|
Low (0 - 1) |
| 23% |
| 43% |
| 28% |
|
Intermediate (2) |
| 50% |
| 39% |
| 47% |
|
High (> 3) |
| 27% |
| 18% |
| 25% |
|
* Based on nodal areas from Central Assessment
Response Assessment
Investigator responses to rituximab, based on the International Workshop Response Criteria, were used prospectively to randomize patients. Retrospective central radiology and medical oncology review (2 independent radiologists, 1 adjudicator and 1 oncologist) were used to determine clinical responses and response improvements.
4
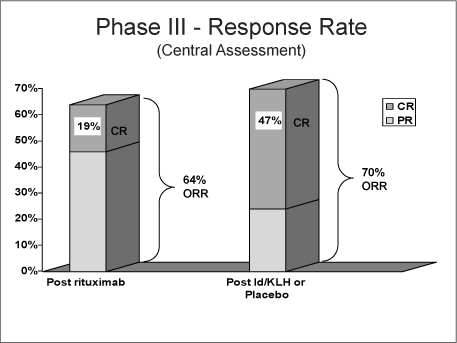
Response to Rituximab: Investigator vs Central Assessment
|
|
| Central Assessment |
| |||||
Response |
| Investigator (N=365) |
| All (N=226)* |
| TN (N=169) |
| Relapsed (N=57) |
|
ORR (CR+PR) |
| 206 (56%) |
| 144 (64%) |
| 107 (63%) |
| 37 (65%) |
|
CR/CRu |
| 24 (7%) |
| 42 (19%) |
| 29 (17%) |
| 13 (23%) |
|
PR |
| 182 (50%) |
| 102 (45%) |
| 78 (46%) |
| 24 (42%) |
|
SD |
| 141 (39%) |
| 80 (35%) |
| 60 (36%) |
| 20 (35%) |
|
PD |
| 17 (5%) |
| 2 (1%) |
| 2 (1%) |
| 0 (0%) |
|
NE |
| 1 (0.3%) |
|
|
|
|
|
|
|
*No. of CTs available for interim assessment.
Response Rate Improvement (RRI)*
RRI |
| All Patients |
| TN |
| R/R |
|
N |
| 182 |
| 138 |
| 44 |
|
SD to PR |
| 9/80 (11%) |
| 7/60 (12%) |
| 2/20 (10%) |
|
SD to CR |
| 6/80 (8%) |
| 5/60 (8%) |
| 1/20 (5%) |
|
PR to CR |
| 59/102 (58%) |
| 49/78 (63%) |
| 10/24 (42%) |
|
Total RRI |
| 74/182 (41%) |
| 61/138 (44%) |
| 13/44 (30%) |
|
*Response Rate Improvent (RRI) represents the percentage of SD+PR patients that convert to a better response after Month 3 (start of FavId or placebo) based on Central Assessment.
At 12 months follow-up, a planned interim analysis of the secondary endpoint RRI did not demonstrate a statistically significant difference between FavId and placebo (alpha = 0.05).
5
Safety
The most frequent adverse event observed to date has been a localized injection site reaction consisting of erythema, induration and pruritis. All but one has been CTC Grade <2. In addition, several patients have now developed generalized allergic / hypersensitivity reactions during the maintenance phase of blinded injection. All patient treatments have remained blinded so attribution to Id/KLH/GMCSF or placebo/GM-CSF can not be determined.
Conclusions
Interim analysis of initial and best objective responses in 226 pts with at least 12 months follow-up showed:
· Response to rituximab (central assessment) is equivalent for TN and R/R pts.
· RRI was seen in 41% of 182 RRI eligible pts with most converting to CR.
· RRI occurred at a higher rate in TN pts.
· Interim analysis of RRI between Id/KLH and placebo did not reach statistical significance at an alpha = 0.05.
· The primary end-point, TTP, is event driven and is likely to occur in Q4 2007 and will allow for the determination of the durability of responses following FavId administration.
Disclosure
In compliance with ACCME policy, ASH requires the following disclosures, ASF, PAH, SN, CN, MR, GB, JNW, JP, TL, and BP are all Investigators in at least one Favrille clinical trial and VE, TM, and JFB are employees of Favrille, Inc.
Unaudited Data
6