SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K/A
[X] ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2006
[ ] TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934
FOR THE TRANSITION PERIOD FROM ______ TO _____
-------------
COMMISSION FILE NUMBER 000-551030
OccuLogix, Inc. (Exact name of Registrant as specified in its charter) |
DELAWARE (State or other jurisdiction of incorporation or organization) | 59 343 4771 (I.R.S. Employer Identification No.) |
2600 Skymark Avenue, Unit 9, Suite 201 Mississauga, Ontario L4W 5B2 (Address of principal executive offices) |
(905) 602-0887
(Registrant’s telephone number, including area code)
SECURITIES REGISTERED PURSUANT TO SECTION 12(b) OF THE ACT:
COMMON STOCK, $0.001 PAR VALUE
(Title of Class)
SECURITIES REGISTERED PURSUANT TO SECTION 12(g) OF THE ACT: NONE
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes [ ] No [X]
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes [ ] No [X]
Indicate by check mark whether the Registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the Registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes [X] No [ ]
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the Registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. [ ]
Indicate by check mark whether the Registrant is an accelerated filer (as defined in Rule 12b-2 of the Exchange Act). Yes [ X] No [ ]
Indicate by check mark if the Registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes [ ] No [X]
The aggregate market value of the voting common stock held by non-affiliates of the Registrant (assuming officers, directors and 10% stockholders are affiliates), based on the last sale price for such stock on June 30, 2006: $42,348,498. The Registrant has no non-voting common stock.
As of March 8, 2007, there were 57,303,895 shares of the Registrant’s Common Stock outstanding.
EXPLANATORY NOTE
This Amendment No. 1 to the Annual Report on Form 10-K filed on March 15, 2007 (the “Original Annual Report”) is being filed in order to replace Exhibits 31.1 and 31.2 to the Original Annual Report.
DOCUMENTS INCORPORATED BY REFERENCE
Portions of the Registrant’s Proxy Statement for the 2007 Annual Meeting of Stockholders of the Registrant to be held June 29, 2007 are incorporated by reference into Part III of this Form 10-K.
The Registrant makes available free of charge on or through its website (http://www.occulogix.com) its Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K and any amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. The material is made available through the Registrant’s website as soon as reasonably practicable after the material is electronically filed with or furnished to the U.S. Securities and Exchange Commission, or SEC. All of the Registrant’s filings may be read or copied at the SEC’s Public Reference Room at 100F Street, N.E., Room 1580, Washington D.C. 20549. Information on the hours of operation of the SEC’s Public Reference Room can be obtained by calling the SEC at 1-800-SEC-0330. The SEC maintains a website (http://www.sec.gov) that contains reports and proxy and information statements of issuers that file electronically.
PART I
SPECIAL NOTE REGARDING FORWARD LOOKING STATEMENTS
This Annual Report on Form 10-K contains forward-looking statements relating to future events and our future performance within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. In some cases, you can identify forward-looking statements by terms such as “may”, “will”, “should”, “could”, “would”, “expects”, “plans”, “intends”, “anticipates”, “believes”, “estimates”, “projects”, “predicts”, “potential” and similar expressions intended to identify forward-looking statements. These statements involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performances, time frames or achievements expressed or implied by the forward-looking statements.
Given these risks, uncertainties and other factors, you should not place undue reliance on these forward-looking statements. Information regarding market and industry statistics contained in this Annual Report on Form 10-K is included based on information available to us that we believe is accurate. It is generally based on academic and other publications that are not produced for purposes of securities offerings or economic analysis. We have not reviewed or included data from all sources and cannot assure you of the accuracy of the market and industry data we have included.
Unless the context indicates or requires otherwise, in this Annual Report on Form 10-K, references to the “Company” shall mean OccuLogix, Inc. and its subsidiaries. References to “$” or “dollars” shall mean U.S. dollars unless otherwise indicated. References to “C$” shall mean Canadian dollars.
ITEM 1. BUSINESS.
Overview
We are an ophthalmic therapeutic company in the business of commercializing innovative treatments for age-related eye diseases, including age-related macular degeneration, or AMD. AMD is the leading cause of late onset visual impairment and legal blindness in people over the age of 50 in the United States and other Western industrialized societies. We believe that Dry AMD, the most common form of the disease, afflicts approximately 13.0 to 13.5 million people in the United States, representing approximately 85% to 90% of all AMD cases. Although the exact cause of AMD is not known, researchers have identified several factors that are associated with AMD, including poor microcirculation and the gradual build-up of cellular waste material in the retina. We believe that improved microcirculation increases the supply of oxygen and nutrients to the compromised retina and facilitates the removal of cellular waste material from the retina. We believe that a treatment that improves microcirculation in the retina can help to enhance the metabolic efficiency of the retina and the removal of waste material and thereby aid in the treatment of Dry AMD. We believe there is a significant opportunity for such a treatment.
Our product for Dry AMD, the RHEO™ System, is designed to improve microcirculation in the eye by filtering high molecular weight proteins and other macromolecules from the patient’s plasma. The RHEO™ System is used to perform the Rheopheresis™ procedure, which we refer to under our trade name RHEO™ Therapy. The Rheopheresis™ procedure is a blood filtration process that selectively removes molecules from plasma. The RHEO™ System consists of the OctoNova Pump and a disposable treatment set, containing two filters, through which the patient’s blood circulates. We believe that the RHEO™ System is the only Dry AMD treatment to target what we believe to be the underlying cause of AMD rather than its symptoms and that, based on early data, appeared to demonstrate improved vision in some patients. The only currently accepted treatment option for persons with advanced cases of Dry AMD are over-the-counter vitamins, antioxidants and zinc supplements that can reduce the five-year risk of conversion to Wet AMD, the other form of the disease, by approximately 25%.
We conducted a pivotal clinical trial, called MIRA-1, or Multicenter Investigation of Rheopheresis for AMD, which, if successful, was expected to support our application to the U.S. Food and Drug Administration, or FDA, to obtain approval to market the RHEO™ System in the United States. On February 3, 2006, we announced that, based on a preliminary analysis of the data from MIRA-1, MIRA-1 did not meet its primary efficacy endpoint as it did not demonstrate a statistically significant difference in the mean change of Best Spectacle-Corrected Visual Acuity applying the Early Treatment Diabetic Retinopathy Scale, or ETDRS BCVA, between the treated and placebo groups in MIRA-1 at 12 months post-baseline. As expected, the treated group demonstrated a positive result. An anomalous response of the control group is the principal reason why the primary efficacy endpoint was not met. There were subgroups that did demonstrate statistical significance in their mean change of ETDRS BCVA versus control.
The MIRA-1 protocol required us to obtain a minimum of 150 complete clinical data sets. To that end, we had enrolled a total of 185 patients in MIRA-1 as of December 31, 2004. On November 17, 2005, we announced that we had collected complete 12-month post-treatment data sets for 169 of these patients. As of December 31, 2004, we had also submitted to the FDA the first three of four modules of the Pre-market Approval Application, or PMA, filing, the non-clinical portion. The non-clinical portion of the PMA consisted of technical data relating to components of the RHEO™ System. In late 2001, with the permission of the FDA, we submitted an interim analysis of 36 complete data sets from the first 43 patients enrolled. The remaining seven patients did not complete all of the required follow-up and thus their results did not qualify as a complete data set. Of the 36 data sets analyzed, 11 were from placebo patients. Fifty-eight percent of, or 11 of 19, patients in the MIRA-1 interim analysis entering the clinical trial with worse than legal driving vision, which is defined as best corrected visual acuity, or BCVA, of worse than 20/40, improved to meet or exceed the requirements to regain a driver’s license. Although we had intended to submit the fourth module, which consists of the follow-up clinical data, in two components, following discussions with the FDA, we subsequently elected to file only one PMA clinical module following completion of our 12-month data on at least 150 data sets.
Subsequent to the February 3, 2006 announcement, the Company completed an in-depth analysis of the MIRA-1 study data identifying subjects that were included in the intent-to-treat, or ITT, population but who deviated from the MIRA-1 protocol as well as those patients who had documented losses or gains in vision for reasons not related to retinal disease such as cataracts. Those subjects in the ITT population who met the protocol requirements, and who did not exhibit ophthalmic changes unrelated to retinal disease, comprised the modified per-protocol population.
In the modified per-protocol analysis, eyes treated with RHEO™ Therapy demonstrated a mean vision gain of 0.8 lines of ETDRS BCVA at 12 months post-baseline, compared to a mean vision loss of 0.1 lines of ETDRS BCVA in the eyes in the placebo group. The result was statistically significant (repeated measure p value = 0.0147). The following table presents a summary of the ETDRS BCVA changes observed 12 months post-baseline in the modified per-protocol analysis of MIRA-1:
| | Treatment Group (n=69) | Placebo Group (n=46) |
| Vision improvement greater or equal to: | | |
| 1 line | 46.4% | 19.6% |
| 2 lines | 27.5% | 8.7% |
3 lines | 8.7% | 2.2% |
| Vision loss greater or equal to: | | |
| 1 line | 11.6% | 23.9% |
| 2 lines | 5.8% | 6.5% |
3 lines | 2.9% | 2.2% |
Within the modified per-protocol population with pre-treatment vision worse than 20/40, 50.0% of RHEO™ Therapy-treated eyes improved, after treatment, to 20/40 or better and would be able to qualify for a driver’s license 12 months post-baseline, compared to 20.0% of placebo eyes.
MIRA-1 data support historical clinical and commercial experience with respect to the safety of RHEO™ Therapy, with observed treatment side effects generally being mild, transient and self-limiting.
In light of the MIRA-1 study results, we re-evaluated our PMA submission strategy and then met with representatives of the FDA on June 8, 2006 in order to discuss the impact on our PMA submission strategy of the MIRA-1 study results and the fact of the per-protocol population being fewer than 150. As expected, in light of MIRA-1’s failure to meet its primary efficacy endpoint, the FDA advised us that it would require an additional study of the RHEO™ System to be performed.
At that meeting, the FDA confirmed its willingness to allow the substitution, in the new study, of the new polysulfone Rheofilter™ filter for the older cellulose acetate filter which currently forms part of the RHEO™ System. The immediate replacement of the filter avoids the regulatory uncertainties that would arise, were the replacement to take place following receipt of FDA approval. Furthermore, due to manufacturing constraints on the number of cellulose acetate filters that can be produced by their manufacturer, Asahi Kasei Medical Co., Ltd. (formerly Asahi Medical Co., Ltd.), or Asahi Medical, the replacement of the filter in the new trial eliminates the need to continue to build and maintain adequate inventories of the older cellulose acetate filter that the Company had been building and maintaining in preparation for commercial launch.
On January 29, 2007, the Company announced that it had obtained Investigational Device Exemption clearance from the FDA to commence the new pivotal clinical trial of the RHEO™ System, called RHEO-AMD, or Safety and Effectiveness in a Multi-center, Randomized, Sham-controlled Investigation for Dry, Non-exudative Age-Related Macular Degeneration (AMD) Using Rheopheresis. The company has been actively preparing a protocol, putting in place the required resources and obtaining clinical trial site commitments for RHEO-AMD.
We cannot begin commercialization in the United States until we receive FDA approval. Until we commence patient enrollment in RHEO-AMD and gain a clear understanding of the progress of that clinical trial, we will not be able to anticipate when, if ever, we will receive FDA approval for the RHEO™ System. Accordingly, at this time, we do not know when we can expect to begin to generate revenues in the United States from the commercialization of the RHEO™ System.
In 2003, we received licenses from Health Canada for the components of the RHEO™ System. These licenses allow us to market the RHEO™ System in Canada for use in the treatment of patients suffering from dysproteinemia due, for example, to abnormal plasma viscosity and/or macular disease. Upon receiving our licenses, we began limited commercialization of the RHEO™ System through sales of OctoNova pumps and disposable treatment sets to three clinics in Canada. In September 2004, we signed an agreement with a private Canadian company called Rheo Therapeutics Inc. (now Veris Health Services Inc., or Veris), a provider of RHEO™ Therapy and the Company’s sole commercial customer, which agreed to purchase approximately 8,000 treatment sets and 20 OctoNova pumps by the end of 2005, with an option to purchase up to an additional 2,000 treatment sets, subject to availability. However, due to delays in its plans to open a number of commercial treatment centers in various Canadian cities where RHEO™ Therapy would be performed, Veris no longer required the contracted-for number of treatment sets for such period. We agreed to keep the original pricing for a reduced number of treatment sets. In December 2005, by letter agreement, we agreed to the volume and other terms for the purchase and sale of treatment sets and pumps for the period ending February 28, 2006. During 2006, the Company continued to sell treatment sets to Veris at the discounted price of $200 per treatment set, which is lower than the Company’s cost. On November 6, 2006, the Company amended its agreement with Veris and forgave a certain amount receivable which had been owing to the Company for the sale of treatment sets and pumps, and the provision of related services, to Veris during the period from September 14, 2005 to December 31, 2005. In consideration of the forgiveness of this debt, Veris agreed that the Company did not owe Veris certain specified amounts. In January 2007, the Company further agreed to forgive an amount receivable owing by Veris for the purchase of 348 treatment sets which had been delivered to Veris in November 2006. We have been notified that Veris has initiated restructuring proceedings under the Bankruptcy and Insolvency Act (Canada) but that it is continuing to carry on its operations in the normal course during its restructuring proceedings.
We have exclusive rights to commercialize the RHEO™ System for ophthalmic uses in North America, certain countries in the Caribbean, Australia, New Zealand, Colombia and Venezuela. We have a non-exclusive right to commercialize the RHEO™ System for ophthalmic uses in Italy. In order to sell or export a medical device in the European community, a Conformité Européene or CE Mark, is required. The Rheopheresis™ procedure for the selective removal of molecules from plasma received CE Mark approval in 1998.
Until our announcement on February 3, 2006 of the preliminary analysis of the data from MIRA-1, our primary activities included commercialization of the RHEO™ System in Canada, working to obtain FDA regulatory approval for the RHEO™ System and building an operating infrastructure to support potential U.S. sales following approval by the FDA. Since February 3, 2006, in addition to conducting a full analysis of the MIRA-1 study data, our primary activities have included negotiating the parameters of RHEO-AMD with the FDA, designing the protocol for RHEO-AMD, recruiting clinical trial sites and otherwise preparing for the launch of RHEO-AMD.
In anticipation of the delay in commercialization of the RHEO™ System in the United States, the Company accelerated its diversification plans by acquiring Solx, Inc., or SOLX, a Boston University Photonics Center-incubated company that has developed a system for the treatment of glaucoma, called the SOLX Glaucoma System, and by acquiring 50.1% of the capital stock, on a fully diluted basis, of OcuSense, Inc., or OcuSense, a San Diego-based company that is in the process of developing technologies that will enable eye care practitioners to test, at the point-of-care, for highly sensitive and specific biomarkers using nanoliters of tear film.
The SOLX Glaucoma System is a next-generation glaucoma treatment platform designed to reduce intra-ocular pressure, or IOP, without a bleb (which is a surgically created flap that serves as a drainage pocket underneath the surface of the eye), thus avoiding its related complications. The SOLX Glaucoma System consists of the SOLX 790 Laser, a titanium sapphire laser used in laser trabeculoplasty procedures, and the SOLX Gold Shunt, a 24-karat gold, ultra-thin drainage device designed to bridge the anterior chamber and the suprachoroidal space in the eye, using the pressure differential that exists naturally in the eye in order to reduce IOP.
The SOLX 790 Laser received CE Mark approval in December 2004, and the SOLX Gold Shunt received CE Mark approval in October 2005. The SOLX 790 Laser has a Health Canada license, and we will be seeking the corresponding approval for the SOLX Gold Shunt.
We are in the process of actively training and certifying physicians in the use of the SOLX Gold Shunt, for commercial purposes, in various European and Asian jurisdictions, including Spain, Italy, Germany, Poland, France, the United Kingdom and Thailand. In addition, in order to establish and maintain a reliable distribution network for SOLX’s products, we are continuing to maintain our relationships with distributors in France, Germany, Spain, the United Kingdom and Canada and are engaged actively in pursuing relationships with other distributors in Europe.
Both the SOLX 790 Laser and the SOLX Gold Shunt are currently the subject of randomized, multi-center clinical trials, the purposes of which are to demonstrate equivalency to the argon laser, in the case of the SOLX 790 Laser, and to the Ahmed Glaucoma Valve manufactured by New World Medical, Inc., in the case of the SOLX Gold Shunt. The results of these clinical trials will be used in support of applications to the FDA for a 510(k) clearance for each of the SOLX 790 Laser and the SOLX Gold Shunt, the receipt of which, if any, will enable the Company to market and sell these products in the United States.
OcuSense’s first product, which is currently under development, is a hand-held tear film osmolarity test for the diagnosis and management of dry eye syndrome, or DES, known as the TearLab™ test for DES. The anticipated innovation of the TearLab™ test for DES will be its ability to measure precisely and rapidly certain biomarkers in nanoliter volumes of tear samples, using inexpensive hardware. Historically, eye care researchers have relied on expensive instruments to perform tear biomarker analysis. In addition to their cost, these conventional systems are slow, highly variable in their measurement readings and not waived by the FDA under the Clinical Laboratory Improvement Amendments, or CLIA.
The TearLab™ test for DES will require the development of the following three components: (1) the TearLab™ disposable, which is a single-use microfluidic cartridge; (2) the TearLab™ pen, which is a hand-held interface with the TearLab™ disposable; and (3) the TearLab™ reader, which is a physical housing for the TearLab™ pen connections and measurement circuitry. OcuSense is currently engaged actively in industrial, electrical and software design efforts for the three components of the TearLab™ test for DES and, to these ends, is working with two expert partners, both based in Melbourne, Australia, one of which is a leader in biomedical instrument development and the other of which is a leader in customized microfluidics.
OcuSense’s intention is to seek a 510(k) clearance and a CLIA waiver from the FDA for the TearLab™ test for DES, following its development and subsequent clinical trials.
Our History and Major Relationships
Shortly after our inception, we began commercialization of therapeutic apheresis by opening a therapeutic apheresis center in Florida. This site generated revenues of $900,200 and $1,277,800 for the years ended June 30, 1999 and 1998, respectively. The therapeutic apheresis center was closed in 1999 pursuant to a directive issued by the FDA. After obtaining an FDA investigational device exemption in 1999, we initiated the MIRA-1 pivotal clinical trial to support an application to the FDA for approval to market the RHEO™ System and completed this trial in 2005.
Relationship with TLC Vision Corporation
TLC Vision Corporation, or TLC Vision, beneficially owns approximately 36.08% of our outstanding common stock, or 33.79% on a fully diluted basis. Elias Vamvakas, a director of TLC Vision (and formerly its Chairman and CEO), became our Chairman in 2003 and is now also our CEO. In addition, two of our other directors, Thomas N. Davidson and Richard L. Lindstrom, are also directors of TLC Vision. Mr. Vamvakas beneficially owns 2,827,589 common shares of TLC Vision, representing approximately 4.09% of TLC Vision’s outstanding shares. Mr. Davidson beneficially owns 71,954 common shares of TLC Vision, representing approximately 0.10% of TLC Vision’s outstanding shares, and Dr. Lindstrom does not beneficially own any common shares of TLC Vision.
On December 8, 2004, we purchased TLC Vision’s 50% interest in OccuLogix, L.P. in exchange for which we issued 19,070,234 shares of our common stock to TLC Vision. This resulted in OccuLogix, L.P. becoming our wholly-owned subsidiary. Accordingly, 100% of the results of OccuLogix, L.P.’s operations are included in the consolidated financial statements since that date. We licensed to OccuLogix, L.P. all of the distribution and marketing rights for the RHEO™ System for ophthalmic indications to which we are entitled. Prior to the acquisition, our only profit stream had come from our share of OccuLogix, L.P.’s earnings. Our acquisition of TLC Vision’s 50% ownership interest in OccuLogix, L.P. transferred the earnings potential for sales of the RHEO™ System entirely to us.
As part of the formation of OccuLogix, L.P. in July 2002, we licensed certain patent rights, trademark rights and know-how rights to OccuLogix, L.P. We also provided OccuLogix, L.P. with licenses to our in-house software as well as sublicensing software that we have licensed from TLC Vision. TLC Vision agreed to provide OccuLogix, L.P., upon request, with $200,000 in funding at an annual interest rate equal to the Bank of America prime rate of interest on the date the loan is made, plus two percent. As at December 8, 2004, Occulogix, L.P. had not requested funding from TLC Vision.
On December 31, 2005, OccuLogix, L.P. transferred all of its assets and liabilities, including the licensed patent, trademark and know-how rights and the licensed distribution and marketing rights for the RHEO™ System, to our then newly incorporated subsidiary, OccuLogix Canada Corp. We completed the wind-up of OccuLogix, L.P. on February 6, 2006. We believe that going forward, our value with respect to the RHEO™ System resides solely in OccuLogix Canada Corp.
Until June 2005, one of Occulogix, L.P.’s primary customers was RHEO Clinic Inc., or RHEO Clinic, a subsidiary of TLC Vision, for which Occulogix, L.P. has reported revenues of $81,593, $401,236, $459,730 and nil for the years ended December 31, 2005, 2004, 2003 and 2002, respectively. RHEO Clinic used the RHEO™ System to treat patients, for which it charged its customers (the patients) a per-treatment fee. RHEO Clinic has advised us that all of its revenues, in Canadian dollars, of $192,430, $595,275, $836,696 and nil for the years ended December 31, 2005, 2004, 2003 and 2002, respectively, are derived from sales to unrelated third parties. The revenues reported from RHEO Clinic are unaudited and have not been independently verified by us. However, management believes the amounts to be accurate.
Since it has ceased the treatment of commercial patients in 2005, RHEO Clinic has not been a source of revenue for us, nor will it be a source of revenue for us in the future. On July 29, 2005, the Company entered into an agreement with RHEO Clinic to purchase fixed assets and intellectual property valued at C$61,812 to be used for the Company’s clinical trial activities and other purposes. The Company agreed to share equally in losses incurred by RHEO Clinic, to a maximum of C$28,952, for assets that RHEO Clinic is not able to dispose of as at December 31, 2005. In addition, the Company reimbursed RHEO Clinic C$281,581, which amount represented that proportion of the costs incurred by RHEO Clinic deemed applicable to our clinical trial activities from October 1, 2004 to June 30, 2005. On May 1, 2006, the Company paid RHEO Clinic C$31,859, which amount included the amount owing for losses incurred for assets that RHEO Clinic was not able to dispose of as at December 31, 2005.
Other Major Relationships
The components of the RHEO™ System were developed by our suppliers, Diamed Medizintechnik GmbH, or Diamed, and Asahi Kasei Medical Co., Ltd., or Asahi Medical.
In 2002, Apheresis Technologies, Inc., or Apheresis Technologies, which is managed by John Cornish, one of our stockholders, our Vice President of Operations and one of our directors from April 1997 to September 2004, was spun off from us. The purpose of the spin-off was to allow us to focus on our clinical trials. This spin-off was accomplished by our transferring all the assets we had in connection with our plasma filter distribution business to our then wholly-owned subsidiary, Apheresis Technologies. In consideration for the transfer of those assets, Apheresis Technologies agreed to pay us $25,000. The full amount of this consideration was applied to amounts owing by us to Apheresis Technologies. Following this transfer, we distributed the stock we owned in Apheresis Technologies to our stockholders, such that the identity and relative ownership of our stockholders and Apheresis Technologies’ stockholders were the same. We did not assume any liabilities in connection with this transfer. Shortly after the spin-off, we entered into a distribution services agreement with Apheresis Technologies to provide us with logistical support, including warehousing, order fulfillment, shipping and billing services. We had the right to terminate this agreement at any time and terminated it on March 28, 2005.
In June 2003, we entered into a reimbursement agreement with Apheresis Technologies whereby we reimbursed it for the applicable percentage of time that its employees provided services to us. One of these employees was John Cornish, our Vice President of Operations. Effective April 1, 2005, the Company terminated its reimbursement agreement with Apheresis Technologies such that the Company no longer compensates Apheresis Technologies in respect of any salary paid to, or benefits provided to, Mr. Cornish by Apheresis Technologies. On April 1, 2005, the Company and Mr. Cornish entered into an employment agreement, which has been amended several times. Effective April 13, 2006, Mr. Cornish is paid an annual base salary of $68,660, representing compensation to him for devoting 50% of his time to the business and affairs of the Company. Mr. Cornish participates in the Company’s bonus plan and is entitled to receive, and has received, stock options pursuant to the Company’s stock option plan.
Prior to the Company’s acquisition of SOLX, Doug P. Adams served as the President and Chief Executive Officer of SOLX and was a significant stockholder of SOLX. As of September 1, 2006, the closing date of the acquisition, Mr. Adams became an executive officer of the Company. The Company paid Mr. Adams a total of $1,005,791 and issued to him 1,309,329 shares of our common stock in consideration of his proportionate share of the purchase price of SOLX. Mr. Adams is owed an additional amount of up to $2,663,084 by the Company in consideration of his proportionate share of the outstanding balance of the purchase price of SOLX.
In addition, the Company paid Peter M. Adams, Doug P. Adams’ brother, a total of $229,967 and issued to him and his spouse an aggregate of 300,452 shares of our common stock in consideration of his proportionate share of the purchase price of SOLX. The Company owes Mr. Adams an additional amount of up to $615,983 in consideration of his proportionate share of the outstanding balance of the purchase price of SOLX.
On November 30, 2006, Mr. Vamvakas agreed to provide the Company with a standby commitment to purchase convertible debentures of the Company in an aggregate maximum principal amount of up to $8 million. When the Company raised gross proceeds in the amount of $10,016,000 on February 6, 2007 in a private placement of shares of its common stock and warrants, the commitment amount under Mr. Vamvakas’ standby commitment was reduced to zero, thus effectively terminating the standby commitment. No portion of the standby commitment was ever drawn down by the Company, and the Company has paid Mr. Vamvakas a total of $29,808 in commitment fees.
Industry (Retina)
Overview of the Human Eye
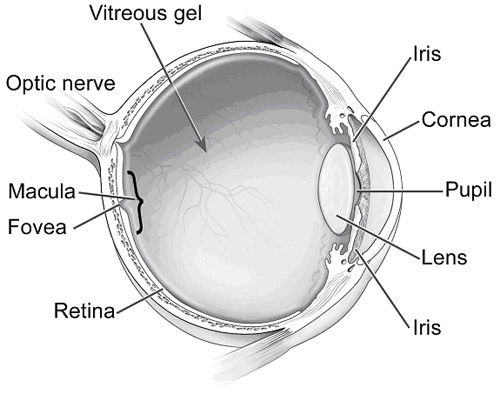
The human eye is composed of focusing elements in the front, the cornea and lens, and a light-sensing element in the back, the retina. Light falls on the photoreceptors that are part of the retina and is converted into electrical energy, which travels via the optic nerve to the brain. The brain processes the complex signals sent from the retina into vision. The central 5% of the area of the retina is the macula, the region responsible for seeing color and for the central vision necessary for activities such as reading, face recognition, watching television and driving. Due to its extremely small size, any damage to the macula can result in significant visual impairment, including legal blindness. In the Western World, the major diseases that usually result in blindness in adults are those affecting the retina, including AMD.
Age-Related Macular Degeneration (AMD)
AMD is a chronic, progressive disease of the macula that results in the loss of central vision. The most common symptoms include central distortion, loss of contrast sensitivity and loss of color vision, none of which can be corrected by refractive means, including glasses, contact lenses or laser eye surgery. Peripheral vision usually remains unaffected so that patients are often forced to look to the side of objects to see them but are still unable to see detail. AMD typically affects people initially in one eye, with a high probability of occurrence in the second eye over time. People with AMD often have difficulty living independently and performing routine daily activities.
We believe that approximately 15 million people in the United States suffer from AMD. According to a ten-year study published in Ophthalmology in October 2002, the prevalence of AMD among a selected sample of U.S. residents increased sharply with age, from 28.2% among people 65 to 74 years of age to 46.2% among people 75 years and older. A study by Duke University published in 2003 reported that the prevalence of AMD among a selected sample of U.S. residents aged 65 and older was 27% in 1999. According to the U.S. Census Bureau, the number of people in the United States aged 50 or older is approximately 80 million and is expected to increase by approximately 40% over the next two decades. We expect that this increase in the number of elderly people will result in a significant increase in the number of cases of AMD in the United States.
AMD occurs in two forms — a non-exudative ‘‘dry’’ form and an exudative ‘‘wet’’ form.
Dry AMD. Dry AMD is the most common form of the disease. We believe that Dry AMD affects approximately 13.0 to 13.5 million people in the United States, or approximately 85% to 90% of all AMD cases. Dry AMD is characterized by a gradual decrease of visual acuity, by pigment abnormalities on the macula and by the build-up of protein and lipid deposits, called drusen. This build-up of macromolecules affects the microcirculation in the eye. Research suggests that the retinal cells, overwhelmed by the lack of oxygen and nutrients and the build-up of debris, enter into a dysfunctional state of dormancy. Without treatment, the retinal cells ultimately die and do not regenerate, leading to irreversible vision loss either through the progression of Dry AMD or conversion to Wet AMD. Patients with Dry AMD are classified at the time of diagnosis into four categories of worsening severity. The higher the category, the greater the risk of progression, or conversion, to Wet AMD within five years.
The following table contains the principal characteristics of each category as described by the Age Related Eye Disease Report, or AREDS Report, No. 8:
Category | Risk of Wet AMD in Five Years | | Key Characteristics |
Category 1 | No Risk | | • no pigment changes and less than five small drusen |
| | | | • BCVA better than 20/32 in each eye |
| | | | • neither eye with Wet AMD |
| Category 2 | Low Risk (Less than 2%) | | • any combination of multiple small drusen, one isolated intermediate drusen or mild pigment abnormalities in one or both eyes |
| | | | • BCVA better than 20/32 in each eye |
| | | | • neither eye with Wet AMD |
Category 3(1) | Moderate Risk (18%) | | • any combination of at least one large drusen, extensive intermediate drusen or geographic atrophy not involving the central macula |
| | | | • neither eye with Wet AMD |
| | | | • BCVA better than 20/32 in at least one eye |
Category 4(1) | High Risk | | • one eye with no signs of Wet AMD |
| | (42%) | | • other eye with either Wet AMD or BCVA worse than 20/32 due to Dry AMD |
__________
(1) Categories 3 and 4 are commonly referred to as “Advanced Dry AMD”.
Wet AMD. We believe that Wet AMD affects approximately 1.5 to 2.0 million people in the United States, representing approximately 10% to 15% of all cases of AMD in the United States. Wet AMD occurs when new blood vessels grow into the macular tissues of the eye. This abnormal blood vessel growth generally is known as neovascularization. These new blood vessels tend to be fragile and often bleed, leaking fluid into the macula, resulting in loss of vision. Untreated, this blood vessel growth and leakage can lead to scarring, atrophy and, eventually, macular cell death. Wet AMD patients experience vision loss more rapidly than Dry AMD patients, usually within months of diagnosis. If treatment is not received in this small window of time, the damage is usually irreversible. As a result, the number of people who have Wet AMD that are considered ‘‘potentially treatable’’, or hoping for significant, positive visual outcomes, will stay relatively small each year as opposed to the number of people who have Dry AMD.
Treatment Alternatives for Wet and Dry AMD
Wet AMD
There is currently no cure for Wet AMD. However, retinal specialists may treat the symptoms in an attempt to reduce blood vessel growth and leakage, using one of a few approved therapies currently available — thermal laser treatment, photodynamic therapy and drug therapies. In addition, there are currently more than 30 therapies being evaluated in U.S. clinical studies for the treatment of Wet AMD. These treatments may slow the progression of the disease but do not prevent the reoccurrence of abnormal blood vessel growth and do not restore lost vision.
| · | Thermal Laser Treatment and Photodynamic Therapy. Thermal laser treatment of Wet AMD entails the use of a high-energy laser to destroy the abnormal blood vessels that are growing and leaking in the macula. This is a surgical procedure involving a medical device that was approved more than two decades ago by the FDA. Because the laser-treated portions of the retina are irreversibly destroyed due to collateral damage from intense heat, thermal laser treatment generally is now used only for the minority of Wet AMD patients whose abnormal blood vessel growth and vessel leakage occur away from the center of the macula. A more targeted approach, photodynamic therapy, involves the use of a light-activated drug named Visudyne, which was developed by QLT Inc. This therapy involves a two-step process in which the drug is administered systemically by intravenous infusion, after which a dose of low energy light is delivered to the target site to activate the drug and destroy the newly grown abnormal blood vessels. |
| · | Drug Therapies. Rather than attempting to destroy abnormal blood vessels, many drug therapies are designed to slow or stop the proliferation of abnormal blood vessels before they can further damage the retina. Genentech, Inc.’s Lucentis received FDA approval in June 2006 and appears to be gaining significant momentum in the ophthalmic community. Lucentis, as well as other drug therapies in clinical trials for Wet AMD, including ones sponsored by Regeneron Pharmaceuticals, Inc., Sirna Therapeutics, Inc. and Acuity Pharmaceuticals, is believed to block the effect of vascular endothelial growth factor, or VEGF, a natural protein that stimulates the production and growth of blood vessels, using different mechanisms of action. Avastin, a cancer drug of Genentech, Inc. which is molecularly similar to Lucentis, is reported to be the subject of much off-label use by physicians for the treatment of Wet AMD. Alcon Laboratories, Inc.’s Retaane is a modified steroid targeting enzymes produced by stimulated blood vessels by blocking the effects of multiple growth factors. Eyetech Pharmaceuticals, Inc.’s Macugen is a pegylated anti-VEGF aptamer, which binds to VEGF. Eyetech Pharmaceuticals, Inc. is owned by OSI Pharmaceuticals, Inc. Sirna Therapeutics, Inc. was recently acquired by Merck & Co., Inc. |
Dry AMD
Dry AMD is not a well understood disease, and there is no medical consensus regarding its underlying cause. As a result, there have been few resources devoted to developing a therapy for Dry AMD. However, there is some research that suggests a vascular component to the disease. This ‘‘vascular model’’ suggests that Dry AMD results from a disorder of the vascular microcirculation in the retina which leads to a reduction in the amount of oxygen and nutrients that reach the retina. This disorder also results in the accumulation of debris between the cellular layers of the retina and the subsequent formation of drusen. In addition, studies have shown that AMD progression may be related to the presence of elevated blood levels of certain macromolecules. Current research has identified a number of high molecular weight blood components that may have a detrimental effect on normal cellular functions and microcirculation.
There is currently no FDA-approved therapy for Dry AMD. Dry AMD is diagnosed and monitored by a primary eye care doctor, such as an optometrist or ophthalmologist, through a routine retinal exam. The AREDS Report provides evidence that vitamin, antioxidant and zinc supplements only reduce the five-year risk of conversion into Wet AMD by up to 25% for Category 3 and Category 4 Dry AMD cases. Regardless of the supplement treatments, Dry AMD may ultimately lead to irreversible vision loss, whether or not it converts into Wet AMD.
Potential Causes of AMD
The precise cause of AMD is not known. However, researchers have identified certain factors that are associated with AMD:
| · | Reduced Metabolic Efficiency of Retina. The macula must be able to function at an extremely high rate of metabolic efficiency to provide sharp vision. The macula, therefore, has an unusually high nutrient and oxygen requirement. Intact cell transport mechanisms are required to supply the necessary nutrients and oxygen. In addition to blood vessels in the retina, the macula receives its blood supply from a tiny meshwork of blood vessels, called the choroid, which lies underneath the retina. The blood supply in this network decreases in older people but even more so in some AMD patients. It has been proposed that the decreased blood flow in the retina of AMD patients reduces the metabolism in the retina, resulting in significant degradation of visual function. |
| · | Poor Waste Material Disposal. Conversion of light in the retina into electrical energy is a photochemical process which produces a large quantity of cellular waste materials. Some researchers believe that life-long environmental, oxidative and chemical stresses progressively injure eye tissues, making it more difficult to clear away the waste material generated by the vision-producing cells. This may explain why waste products like drusen are often seen in the retinas of AMD patients and why their presence is associated with an increased risk of progressive vision loss. |
We believe that a treatment that improves microcirculation in the retina can help to enhance the metabolic efficiency of the retina and the removal of waste material and thereby aid in the treatment of Dry AMD. We continue to believe there is a significant market opportunity for such a treatment.
Our Solution (Retina)
The RHEO™ System, which consists of a pump and a disposable treatment set, containing two filters, is designed to filter high molecular weight proteins and macromolecules from the patient’s plasma, leading to improved microcirculatory function. Researchers involved in MIRA-1 believe that blood filtered with the RHEO™ System is able to flow more easily through the tiny capillaries of the eye and that the resulting improved microcirculation more effectively supplies the macular cells with oxygen and nutrients which facilitates removal of cellular waste materials. The RHEO™ System represents a fundamentally new approach to the treatment of Dry AMD and offers the following potential benefits:
| · | Addresses a large AMD patient population with limited current treatment options. Current Wet AMD treatments are effective only on patients who are newly diagnosed with Wet AMD, of which there are approximately 200,000 in the United States each year. RHEO™ Therapy, however, is a treatment for most patients in the Category 3 and Category 4 Dry AMD populations, which, according to the AREDS Report, represent approximately 54% of the total U.S. AMD patients, or currently approximately 8 million people. RHEO™ Therapy is not appropriate for everyone in the Category 3 and Category 4 Dry AMD population. For example, RHEO™ Therapy would not be appropriate for potential patients who may have existing ailments that would make it unsafe for them to receive any blood transfusion type procedure. |
| · | Preserves or improves vision of Dry AMD patients. Success in treating AMD is generally measured by the ability to slow or halt progression of the disease. We believe that RHEO™ Therapy is currently the only Dry AMD therapy that, based on an interim analysis of 36 complete data sets from the first 43 patients enrolled in MIRA-1 and the modified per-protocol analysis of the final MIRA-1 study data, appears to demonstrate improved vision in some patients. However, MIRA-1 did not meet its primary efficacy endpoint as it did not demonstrate a statistically significant difference in the mean change of ETDRS BCVA between the treated and placebo groups in MIRA-1 at 12 months post-baseline. |
| · | Patient-friendly procedure. RHEO™ Therapy is a form of therapeutic apheresis, a procedure that selectively removes molecules from the plasma. Apheresis has been used safely for more than twenty years in the United States and Europe to treat various diseases, including leukemia, rheumatoid arthritis, sickle cell disease and several other medical conditions. Although RHEO™ Therapy is a patient-friendly procedure, it is time consuming, with an initial course of RHEO™ Therapy requiring eight procedures over a 10- to 12-week period, with each procedure lasting between two and four hours depending on patient weight and height. Patients recline in a comfortable chair and typically listen to music or otherwise relax during the procedure. As with any medical procedure, there are potential side effects associated with RHEO™ Therapy, which are all temporary and generally mild, including drops in blood pressure, abnormal heart rate, nausea, chills and localized bleeding, swelling, pain and numbness in the area of the arms where the needles are inserted. |
| · | Limited barriers to adoption for eye care professionals and health care service providers. We believe that the RHEO™ System requires lower capital expenditures and less physical space than equipment used in many other procedures performed by eye care professionals, including laser vision correction and cataract surgery. The RHEO™ System requires no special installation and minimal maintenance costs. We believe that RHEO™ Therapy, which can be administered by a nurse, can be easily integrated into our potential customers’ workflow and offers an attractive source of additional revenues for both facilities and providers. However, our success is dependent upon achieving widespread acceptance of RHEO™ Therapy among ophthalmologists and optometrists who may be reluctant to accept RHEO™ Therapy. |
| · | Cost-effective procedure. The initial course of RHEO™ Therapy is initially expected to cost between $16,000 and $25,600. We believe that Medicare and third-party payors will determine that the benefits of RHEO Therapy™ will justify the cost of reimbursement. However, should Medicare and third-party payors decline to provide coverage of RHEO™ Therapy or set broad restrictions on patient coverage or on treatment settings in which RHEO™ Therapy is covered, our potential revenues may be significantly limited, particularly if potential patients deem our treatment to be too expensive. Nonetheless, we believe that to the extent that RHEO™ Therapy is not reimbursed by the government or private third-party payors, some patients with the economic means to do so will be willing to pay for RHEO™ Therapy themselves in order to avoid the consequences of uncorrectable impaired vision, including, but not limited to, the inability to drive. |
Our Strategy (Retina)
Our goal is to establish RHEO™ Therapy as the leading treatment for Dry AMD in North America. To date, key elements of our strategy have included creating a plan to develop market awareness of RHEO™ Therapy by educating eye care professionals and patients, establishing third-party reimbursement for RHEO™ Therapy, securing relationships with key multi-facility health care service providers, ensuring sufficient manufacturing capacity and inventory to support our commercialization plan and maintaining our intellectual property portfolio and other barriers to entry. However, as a result of our discussions with the FDA following the full analysis of the MIRA-1 study results and the FDA’s requirement that a follow-up clinical trial of the RHEO™ System be conducted, the timetable for the achievement of our goal to establish RHEO™ Therapy as the leading treatment for Dry AMD in North America and the implementation of our strategy for achieving this goal have been delayed for at least the duration of the RHEO-AMD clinical trial. To the extent that it makes sound business sense, we will continue to lay the foundation for the achievement of our goal and the implementation of our strategy.
Our Product (Retina)
The RHEO™ System
The RHEO™ System employs a double filtration apheresis process, whereby a pair of single-use blood and plasma filters sequentially separate and partially remove the targeted plasma components. The system removes macromolecules greater than a specified size from the plasma. The RHEO™ System consists of two primary components:
| · | OctoNova Pump. The OctoNova pump is a microprocessor-controlled device used to circulate blood and plasma from the patient, through the filter and back to the patient. The OctoNova pump is complemented by single-use sterilized tubing which creates a closed-loop system. Blood is pumped through the tubing with small gear-like sprockets that create a peristaltic action in the tube similar to that which occurs in our intestines. The smooth-edged teeth of the sprockets press against the outside surface of the tube pushing the blood along the length of the tube as the wheels turn all at the same rate and direction. No blood ever leaves the closed-loop system. The OctoNova pump was developed in the 1990s by Diamed and licensed to us in 2002. We will be seeking FDA approval of the OctoNova pump as part of the RHEO™ System PMA. |
| · | Disposable Treatment Sets. Disposable treatment sets consist of the tubing and two filters, the Plasmaflo filter and the Rheofilter filter. One treatment set is used for each treatment undertaken by the patient. The Plasmaflo filter performs the initial function of separating the blood cells from the plasma. The Rheofilter filter is a single-use, hollow-fiber nanopore membrane, which is used to filter specific high molecular weight proteins and other macromolecules from the plasma. Following this, the filtered plasma is reconstituted with the blood cells and returned into the patient. The tubing and the filters are easily disposed of after each patient procedure by the administering nurse, providing us with a recurring source of revenue. The Rheofilter filter was developed in the early 1980s by Asahi Medical. We will be seeking FDA approval of the tubing and two filters as part of the RHEO™ System PMA and will be working with Asahi Medical on preparing the PMA following the completion of RHEO-AMD. Upon FDA approval of the PMA, we have an agreement to transfer this FDA approval to a special purpose corporation which will be owned as to 51% by Asahi Medical and as to 49% by us. In that same agreement, Asahi Medical agreed to us being the exclusive distributor of the Plasmaflo filter and the Rheofilter filter in North America, certain countries in the Caribbean, Australia, New Zealand, Colombia and Venezuela and a non-exclusive distributor in Italy. With respect to the United States, subject to early termination under certain circumstances, this agreement has a term which will end ten years following the date on which the FDA approval is received, if ever, and contemplates successive one-year renewal terms thereafter. The Rheofilter filter is currently made of a cellulose acetate filter material. We had been working with Asahi Medical to develop a new filter made of polysulfone to replace the older cellulose acetate filter, and the FDA has confirmed its willingness to allow the substitution, in RHEO-AMD, of the new polysulfone filter. |
The disposable treatment sets received Health Canada regulatory approval in 2002. The OctoNova pump received a Health Canada license in 2003. The RHEO™ System components have also been granted a CE Mark in Europe, where, other than in Italy, the commercialization rights for the Rheopheresis™ procedure are exclusively held by Diamed, one of our principal stockholders and suppliers. Although we had been conducting clinical studies with the goal of obtaining FDA approval as well as with a view to gaining widespread physician acceptance of RHEO™ Therapy, our clinical study efforts with respect to the RHEO™ System, in the near future, will be focused principally on RHEO-AMD.
The RHEO™ Procedure
Each RHEO™ Therapy procedure typically takes between two and four hours to complete and begins by placing one intravenous line in each forearm of the patient. Blood is pumped from a large vein in one arm and circulated through the filtration system where the whole blood is separated from the plasma by the Plasmaflo filter. The plasma is filtered through the Rheofilter filter, which filters high molecular weight proteins and other macromolecules from the patient’s plasma. The plasma is then remixed with the blood and is returned to the patient intravenously. Only approximately 1.25 pints of blood are outside the patient’s body, and, at all times, blood remains in a sterile closed circuit. Throughout the RHEO™ Therapy procedure, the attending nurse monitors the blood pressure, heart rate, oxygen saturation, cardiac rhythm and activated clotting time of the patient. The attending nurse also gauges the flow rates, temperature and pressures of the filters. No blood products or medications are added, other than a small amount of heparin to prevent clotting in the tubing system. We believe the initial course of eight procedures of RHEO™ Therapy given over a 10- to 12-week period provides the best results for patients with Dry AMD. Typically, one or two booster procedures are given each 12 to 18 months thereafter to maintain the clinical benefits derived from the initial course of RHEO™ Therapy. The referring physician monitors post-procedure follow-up. The following graphic shows the RHEO™ Therapy process:

Background of Rheopheresis™
Researchers discovered Rheopheresis™ for AMD during the search for a blood treatment for elevated cholesterol levels in the mid-1980s. Asahi Medical developed a filter aimed at selectively removing the low-density lipid, or LDL, macromolecules known as the ‘‘bad’’ cholesterol in an apheresis procedure. Although the filter successfully removed LDL, it also removed several other large molecules, including von Willebrand’s factor, fibrinogen, lipoprotein A and C reactive protein. Researchers have confirmed that apheresis, a plasma filtering or exchange procedure, is a relatively safe procedure and that there do not appear to be negative consequences to also filtering out these large molecules. At approximately the same time, however, the first statin drug was proven to be effective in lowering LDL levels in the blood, thereby eliminating the need for an apheresis procedure to remove LDL. Shortly thereafter, Asahi Medical ceased its efforts to develop and commercialize apheresis treatment for elevated LDL levels.
In the late 1980s, researchers at the University of Cologne in Germany were searching for a treatment for a small group of patients referred to the university with a condition known as refractory uveitis, a chronic inflammatory eye condition that was not responding to conventional therapy. Having learned that the Asahi Medical filters had the ability to remove large molecules from the blood and that the eye condition was related to significant levels of many of the same molecules, the researchers performed a small pilot study. The filtration procedure was effective for uveitis but also showed preliminary success in improving the vision of two patients in the study who also had AMD. This led the researchers to conduct several years of clinical research to develop apheresis for AMD in Germany. The research suggested that eight procedures over a 10- to 12-week period was the optimal treatment regime.
Clinical Studies (Retina)
In 2006, we completed our FDA clinical trial, MIRA-1, or Multicenter Investigation of Rheopheresis for AMD, which, if successful, was expected to support our application to the FDA to obtain approval to market the RHEO™ System in the United States. On February 3, 2006, we announced that, based on a preliminary analysis of the data from MIRA-1, MIRA-1 did not meet its primary efficacy endpoint. Two other clinical trials have been conducted by third parties: MAC-1, which was conducted in Germany from 1995 to 1998; and the Rheopheresis™ pilot study which was conducted by the University of Utah from 1997 to 1998. While the protocols of these three clinical trials were not identical, the interim results of MIRA-1 and results of each of these other two studies have been generally consistent.
As expected, in light of MIRA-1’s failure to meet its primary efficacy endpoint, the FDA advised us that it would require an additional study of the RHEO™ System to be performed. On January 29, 2007, the Company announced that it had obtained Investigational Device Exemption clearance from the FDA to commence the new pivotal clinical trial of the RHEO™ System, called RHEO-AMD, or Safety and Effectiveness in a Multi-center, Randomized, Sham-controlled Investigation for Dry, Non-exudative Age-Related Macular Degeneration (AMD) Using Rheopheresis.
RHEO-AMD
RHEO-AMD is a randomized, placebo-controlled trial designed to evaluate the safety and efficacy of RHEO™ Therapy in patients with intermediate-to-late stage, or Category 3 and Category 4 (as defined in the AREDS Report), Dry AMD.
To be included in RHEO-AMD, a patient’s study eye must demonstrate intermediate-to-late stage Dry AMD, with three or more large, or ten or more intermediate, drusen. Primary eyes in the study must show no signs of active Wet AMD and must have demonstrated best corrected visual acuity, or BCVA, between 20/40 and 20/100 inclusive. Potential study subjects who demonstrate central geographic atrophy within 250 µm of the fovea of the eye being studied or with any serious pigment epithelium detachments within 500 µm of the fovea will not qualify to participate in the study. Other ophthalmic exclusion criteria of RHEO-AMD, among others, will require potential study subjects to not require cataract surgery, to not have had cataract surgery during the three-month period prior to screening and to not have had a Yag capsulotomy during the six-week period prior to screening. RHEO-AMD will not apply any inclusion or exclusion criteria based on blood factors.
Two out of every three patients will be treated in the trial, while the third will be a placebo, or control, patient. Patients will receive eye exams prior to treatment and at three-, six-, nine- and 12-month follow-up intervals. Patients in the placebo-control group will be made to believe that they are receiving RHEO™ Therapy. All subjects, including those randomized to the placebo group, will be shrouded from the neck down to prevent them from observing their treatment and will receive actual needle sticks in both arms. Additionally, a partition will be positioned in front of the OctoNova pump in order to prevent the patient from seeing the system. The machine will be activated so that patients can hear the background noise of the machine, but those patients in the placebo group will not be connected to the tubing circuit. In addition, all subjects, including those randomized to the placebo group, will be required to take the same dose of antioxidant vitamins that are commonly recommended for Dry AMD patients as a possible inhibitor of conversion into Wet AMD.
Vision research typically uses a “standard measurement” called the “change in BCVA”, which is measured using the Early Treatment Diabetic Retinopathy Study, or ETDRS, eye chart, which is the standard eye chart used in ophthalmic trials and which provides five letters per line of decreasing size or increasing difficulty. Each letter has a relative value of 0.2 or 20% of the entire line. A patient entering the study who gains two lines of vision will be able to read ten additional letters or two complete lines of vision. “Mean change” is the cumulative averaging of all patient results in a specific category. For example, a patient entering the study with 20/40 vision and gaining 1.4 lines following treatment would have improved to 20/32 plus two letters on the 20/25 line. This number, 1.4, would be included in the calculation with all other individual patient results when calculating the cumulative average.
RHEO-AMD’s primary endpoint will be the mean change in BCVA and will be achieved if there is demonstrated a statistically significant difference in the proportion of study eyes achieving a ten-letter (two-line) or greater improvement in BCVA between actively treated patients and placebo patients. RHEO-AMD will have various secondary and tertiary endpoints.
The protocol for RHEO-AMD was designed with the input of members of our Scientific Advisory Board, and the protocol design specifically takes into account the learnings derived from MIRA-1.
It is anticipated that up to 25 ophthalmic clinical trial sites and up to 20 apheresis clinical trial sites will participate in RHEO-AMD. The initiation of clinical trial sites has been underway and is continuing.
MIRA-1
MIRA-1 was a randomized, placebo-controlled trial designed to evaluate the safety and efficacy of RHEO™ Therapy in patients with intermediate-to-late stage, or Category 3 and Category 4 (as defined in the AREDS Report), Dry AMD.
In September 1999, we received an Investigational Device Exemption from the FDA to begin MIRA-1. Between early 2000 and August 2001, we enrolled 98 patients in MIRA-1. In August 2001, due to financial constraints, we temporarily suspended the new enrollment of patients but continued to pursue follow-up with the remaining patients in MIRA-1. In late 2001, with the permission of the FDA, we submitted the data sets of the 43 patients who had reached their full 12-month follow-up in MIRA-1 for independent third-party analysis. Over the course of the next several months, the FDA addressed a number of matters relating to MIRA-1. First, the FDA allowed us to submit the PMA in modules. Second, it acknowledged that MIRA-1 is intended to be the pivotal trial for obtaining FDA approval for RHEO™ Therapy. Third, the FDA allowed us to treat the patients in the placebo group with RHEO™ Therapy free of charge once their full 12-month follow-up data had been obtained. Fourth, it confirmed that we would be required to submit at least 150 full data sets from the 180 patients that were to be enrolled in the trial. Following disclosure of the interim results of MIRA-1 and these changes to the MIRA-1 protocol, we were able to obtain new financing. As a result of the new financing, in October 2003, we began screening additional patients for enrollment in MIRA-1 and then opened five additional MIRA-1 sites and, at the completion of the study, were operating 12 MIRA-1 sites.
As of December 31, 2004, we had enrolled a total of 185 patients in MIRA-1 and had also submitted to the FDA the first three of four modules of the PMA filing, the non-clinical portion. These first three modules contained non-clinical results of bench tests and quality assurance and document manufacturing processes on the components of the RHEO™ System. Although we had intended to submit the fourth module, which consists of the follow-up clinical data, in two components, following discussions with the FDA, we elected to file only one PMA clinical module following completion of our 12-month data on at least 150 data sets.
To be included in MIRA-1, a patient’s eyes must have demonstrated intermediate-to-late stage Dry AMD, with ten or more intermediate or large drusen. Additionally, patients must have shown elevated serum levels of at least two out of three macromolecules associated in previous studies that suggested the best positive treatment outcomes. Primary eyes in the study must have shown no signs of Wet AMD and must have demonstrated BCVA between 20/32 and 20/125 inclusive.
Two out of every three patients were treated in the trial, while the third was a placebo or control patient. Patients received eye exams prior to treatment and at three-, six-, nine-, and 12-month follow-up intervals. Each patient received either eight RHEO™ Therapy or eight placebo procedures over ten weeks. Patients in the placebo-control group were made to believe that they were receiving RHEO™ Therapy. All subjects, including those randomized to the placebo group, were shrouded from the neck down to prevent them from observing their treatment and received actual needle sticks in both arms. Additionally, a partition was positioned in front of the OctoNova pump so that the patient could not see the system. The machine was activated so that the patients could hear the background noise of the machine, but those patients in the placebo group were not connected to the tubing circuit. In addition, all subjects, including those randomized to the placebo group, were required to take the same dose of antioxidant vitamins that are commonly recommended for Dry AMD patients as a possible inhibitor of conversion into Wet AMD.
The study’s primary endpoint was the mean change in BCVA. In this trial, visual acuity was measured as the number of letters that the patient can read on the ETDRS eye chart. Secondary and tertiary endpoints included:
| · | the ability to pass a vision test in order to regain a driver’s license; |
| · | the Pepper Visual Skills for Reading Test, which is a measure of reading ability; |
| · | the National Eye Institute visual functioning questionnaire; and |
| · | progression to legal blindness. |
On February 3, 2006, we announced that, based on a preliminary analysis of the data from MIRA-1, MIRA-1 did not meet its primary efficacy endpoint as it did not demonstrate a statistically significant difference in the mean change of ETDRS BCVA between the treated and placebo groups in MIRA-1 at 12 months post-baseline. As expected, the treated group demonstrated a positive result. An anomalous response of the control group is the principal reason why the primary efficacy endpoint was not met. There were subgroups that did demonstrate statistical significance in their mean change of ETDRS BCVA versus control.
Subsequent to the February 3, 2006 announcement, the Company completed an in-depth analysis of the MIRA-1 study data identifying subjects that were included in the intent-to-treat, or ITT, population but who deviated from the MIRA-1 protocol as well as those patients who had documented losses or gains in vision for reasons not related to retinal disease such as cataracts. Those subjects in the ITT population who met the protocol requirements, and who did not exhibit ophthalmic changes unrelated to retinal disease, comprised the modified per-protocol population.
In the modified per-protocol analysis, eyes treated with RHEO™ Therapy demonstrated a mean vision gain of 0.8 lines of ETDRS BCVA at 12 months post-baseline, compared to a mean vision loss of 0.1 lines of ETDRS BCVA in the eyes in the placebo group. The result was statistically significant (repeated measure p value = 0.0147). The following table presents a summary of the ETDRS BCVA changes observed 12 months post-baseline in the modified per-protocol analysis of MIRA-1:
| | Treatment Group (n=69) | Placebo Group (n=46) |
| Vision improvement greater or equal to: | | |
| 1 line | 46.4% | 19.6% |
| 2 lines | 27.5% | 8.7% |
3 lines | 8.7% | 2.2% |
| Vision loss greater or equal to: | | |
| 1 line | 11.6% | 23.9% |
| 2 lines | 5.8% | 6.5% |
3 lines | 2.9% | 2.2% |
Within the modified per-protocol population with pre-treatment vision worse than 20/40, 50.0% of RHEO™ Therapy-treated eyes improved, after treatment, to 20/40 or better and would be able to qualify for a driver’s license 12 months post-baseline, compared to 20.0% of placebo eyes.
MIRA-1 data support historical clinical and commercial experience with respect to the safety of RHEO™ Therapy, with observed treatment side effects generally being mild, transient and self-limiting.
LEARN Studies
On February 28, 2005, we announced that the FDA had completed a review of the Long-term Efficacy in AMD from Rheopheresis in North America, or LEARN, protocols submitted to it by us on January 21, 2005 and had given us permission to initiate two studies.
LEARN-1 is an open-label multi-center study that enrolled 50 subjects who were treated in the MIRA-1 study. At nine investigational sites, subjects were randomized in a 1:1 fashion to receive either two or four RHEO™ Therapy “booster” procedures. The results between the groups will be compared after three, six, nine and 12 months of follow-up from baseline.
LEARN-2 is an open-label multi-center study that enrolled 29 subjects who had been placebo patients in the MIRA-1 study. At nine investigational sites, subjects received eight RHEO Therapy procedures and will have a three-, six-, nine- and 12-month follow-up from baseline evaluation.
The screening and treatment phases of both LEARN-1 and LEARN-2 have been completed, and both clinical trials are currently in the follow-up phase.
OMER
We have been conducting a small clinical study, called OMER, or Objective Measurement of the Effect of Rheopheresis, with the assistance of Columbia University, New York Presbyterian Hospital and New York Blood Center. OMER is an open-label study of ten patients, the objective of which is to evaluate any change, from baseline evaluation to post-treatment, in the multi-focal electrophysiological activity of their macula and in the thickness of their retina. Each patient participating in the study has been, or will be, receiving a series of eight RHEO™ Therapy treatments over a ten- to 12-week period, and clinical data will be collected at three-, six- and 12-month intervals following the baseline evaluation. Although a number of patients have already been treated or will commence treatment shortly, OMER is still in the enrollment phase and is recruiting patients.
Among other things, it is hypothesized that an increase in electrophysiological activity in the macula may be indicative of an improvement in the functioning of the macula.
MAC-1
The MAC-1 trial was a 40-patient study conducted in Germany by the University of Cologne from 1995 to 1998 and resulted in the Rheopheresis™ procedure for Dry AMD achieving the CE Mark. The patients were randomized into two groups, a treatment group and a placebo-control group. The treatment group was treated ten times over a period of 21 weeks.
Unlike in RHEO-AMD and MIRA-1, the investigators and each patient knew whether that patient was in the treatment group or the control group, because the 20 patients in the control group did not receive placebo treatments but were simply examined at the designated follow-up intervals. The MAC-1 study also included patients with signs of Wet AMD and included patients with significant soft drusen. 18 of the patients in the study had signs of Wet AMD and would be excluded from RHEO-AMD under the RHEO-AMD protocol and also would have been excluded from MIRA-1 under the MIRA-1 protocol.
The main parameter of the study was BCVA. Electrical activity in the eye was also recorded. Plasma and whole-blood speed and volume in the macular region were also measured. The results of MAC-1 were similar to the interim results that have been seen in MIRA-1: statistically significant relative improvement of 1.6 lines of BCVA immediately following the course of treatment, with the same level of benefit seen at 12 months. For patients with soft drusen, the average difference was 2.3 lines (p<0.01); for patients without soft drusen, the difference was only 0.64 lines (p=0.43). In the treated group, improvement in electrical activity was statistically significant, indicating that the cells of the retina were functioning more efficiently. The speed and volume of blood flow in the choridial arteries which supply blood to the retina were found to be decreased by 37% and 33%, respectively, in patients with AMD. Following treatment of those patients, blood flow increased by 22%. There were no serious adverse events noted.
Rheopheresis™ Pilot Study
The study was conducted from 1997 to 1998 by physicians at the University of Utah Health Sciences Center in Salt Lake City, Utah, under an Investigational Device Exemption from the FDA. The University of Utah’s Institutional Review Board also provided approval for human experimentation prior to enrollment. The study involved 30 patients. The trial measured electrical activity in the cells of the macula before and after treatment. The results of this study were used to support the application for the Investigational Device Exemption to conduct MIRA-1.
PERC Study
In April 2004, RHEO Clinic, a subsidiary of TLC Vision, received Institutional Review Board approval for and launched a new study called the Prospective Evaluation of Rheopheresis in Canada, or PERC.
PERC is a single center study in Canada designed to examine the effect of RHEO™ Therapy on 60 patients with Dry AMD to gain a greater understanding of the treatment’s method of action. Although at the outset PERC was contemplated to study the outcome variables for 60 patients, it was subsequently decided to limit the enrollment in PERC to 20 patients. Each patient received a series of eight RHEO™ Therapy treatments over a 10- to 12-week period. Clinical data were collected at three-month intervals for one year following the initial treatments.
One objective of the study is to develop a complete description of the physiological changes produced by RHEO™ Therapy. This will be done using structural and functional objective tests and subjective measures of vision in its broad context. This includes measurements of the size and shape of the retina, retinal electrical activity and vascular function as well as general visual performance using standard measurements of acuity, reading speed, and color and contrast sensitivity. Subjective vision assessments using the National Eye Institute Visual Functioning Questionnaire 25 were evaluated to gain understanding about general quality of life and AMD-specific visual symptoms.
Early analysis of the visual acuity of the 20 patients in PERC showed findings similar to those shown by the interim analysis conducted on 36 complete data sets from the first 43 patients enrolled in MIRA-1. Further analysis of the other parameters measured in the PERC study will continue to be conducted.
RHEONET Registry
The RHEONET Registry is a collaborative effort between the Apheresis Research Institute in Cologne, Germany, and us. The registry contains a database of Rheopheresis™ procedures from centers and clinics performing the Rheopheresis™ procedure commercially in Germany, using systems sold by Diamed or provided by Diamed for some local research projects and, in Canada, using systems sold by us. In January 2007, a total of 6,726 Rheopheresis™ procedures (5,272 in Germany and 1,454 in Canada) on 1,005 patients were registered, including 739 patients with AMD. Ophthalmological data of 322 eyes of 218 patients with Dry AMD could be analyzed from the registry as of January 2007. Results of RHEONET Registry analyses will be presented, when available, at scientific meetings in 2007.
Supplier Relationships (Retina)
We have three key supplier arrangements — with Asahi Medical, who manufactures the treatment sets, including the Rheofilter filter and the Plasmaflo filter, and with Diamed and MeSys GmbH, or MeSys, the designer and the manufacturer, respectively, of the OctoNova pump. The Rheofilter filter, the Plasmaflo filter and the OctoNova pump are all key components of the RHEO™ System.
Rheofilter Filter and Plasmaflo Filter. We purchase the Rheofilter filter and the Plasmaflo filter from Asahi Medical. We make these purchases pursuant to a distribution agreement which appoints us Asahi Medical’s exclusive distributor of the Rheofilter filter and the Plasmaflo filter for use in treating AMD in North America, certain countries in the Caribbean, Australia, New Zealand, Colombia and Venezuela, subject to us obtaining necessary regulatory approvals in those agreed countries where we choose to sell the filters. The distribution agreement appoints us a non-exclusive distributor of the Rheofilter filter and the Plasmaflo filter in Italy where we are also obligated to obtain regulatory approval therefor. Under this agreement:
| · | we may not market or sell any product that is similar to or competitive with the filters; |
| · | we must use our best efforts to support providers in their efforts to secure reimbursement from public and private health insurers, in those territories where we have exclusive distribution rights, on behalf of patients whose Dry AMD treatment involves utilization of these filters; |
| · | for the United States and the Caribbean, we must purchase a minimum of 9,000 filter sets during the one-year period commencing six months following FDA approval, 15,000 filter sets in the succeeding one-year period and 22,500 filter sets in the next succeeding one-year period. If we fail to meet our minimum purchase requirements under our agreement with Asahi Medical, our agreement may be terminated or rendered non-exclusive at the sole discretion of Asahi Medical; |
| · | for Canada, we must purchase a minimum of 900 filter sets during the one-year period commencing upon the earlier to occur of the sale of our current inventory of Rheofilter filters or their expiry, 1,500 filter sets in the succeeding one-year period and 2,250 filter sets in the next succeeding one-year period; |
| · | for Australia, New Zealand, Colombia, Venezuela and Italy, we have committed to purchase an aggregate of 300 filter sets and 500 filter sets in 2009 and 2010, respectively; |
| · | we must transfer the whole ownership of the FDA approval, if obtained and upon receipt, to a special purpose corporation which will be owned as to 51% by Asahi Medical and as to 49% by us; |
| · | regulatory approvals obtained, if any, in Australia, New Zealand, Colombia, Venezuela or Italy will be held by Asahi Medical; |
| · | the clinical trial data from RHEO-AMD will be jointly owned by Asahi Medical and us, and we will have the ability to use such clinical trial data in those territories where we have exclusive distribution rights; and |
| · | provided that certain conditions are met, Asahi Medical will be obligated to contribute $3,000,000 toward the cost of RHEO-AMD. |
Although we have an obligation to purchase a minimum annual quantity of filters, Asahi Medical has the right to reject any order but may not unreasonably reject any order placed by us in order to satisfy our minimum purchase requirements. Under the agreement, Asahi Medical can cease to supply Rheofilter filters and Plasmaflo filters to us, after a 12-month notice period, in the event that: (1) Asahi Medical cannot economically supply the product; (2) due to special circumstances, such as patent infringement liability or product liability issues, Asahi Medical cannot supply the product; or (3) Asahi Medical develops an improved product, in which case, we have a right of first refusal to become the exclusive distributor of that new product in the same territories where we are the exclusive distributor of the filters on terms and conditions satisfactory to Asahi Medical and to us.
With respect to the United States, this agreement has a term of ten years from our obtaining FDA approval to use the filters to treat AMD and is automatically renewable for one-year terms unless terminated upon six months’ notice. In addition, Asahi Medical may terminate our agreement in certain circumstances, including:
| · | if we become insolvent or are petitioned into bankruptcy; |
| · | if we transfer all or an important part of our business to a third party; |
| · | if we are unable to obtain FDA approval and other necessary approvals in the territories for which we have distribution rights by the respective deadlines provided for in the agreement which, in the case of FDA approval, is December 31, 2010; |
| · | if we breach the agreement and do not remedy the default within 30 days of Asahi Medical notifying us that we are in default; or |
| · | if a competitor of Asahi Medical acquires a majority of the voting stock of the Company or substantially takes control of the Company’s management which, in either case, would adversely affect the sale of filters in the territories in which we have distribution rights. |
Our distribution agreement with Asahi covers the newer polysulfone Rheofilter filter which will form part of the RHEO™ System that will be the subject of RHEO-AMD and which will replace the older cellulose acetate filter which currently forms part of the RHEO™ System.
OctoNova Pump. We purchase the OctoNova pump pursuant to a marketing and distribution agreement with Diamed, the developer of the OctoNova pump, and a distribution agreement with MeSys, the company that manufactures the pumps for Diamed.
Under the agreement with Diamed, we have been appointed Diamed’s exclusive distributor of the OctoNova pump in the United States, Canada, Mexico and certain countries in the Caribbean. Under this agreement:
| · | we have committed to use our best efforts in promoting the sale and use of, and securing orders and developing the market for, the OctoNova pump in the territories for which we have distribution rights; and |
| · | we are obligated to use our best efforts in promoting public and private medical insurance reimbursement for the treatment of hemo-rheological disorders in microcirculation in the United States. |
This agreement has a term of ten years from FDA approval of the RHEO™ System and is automatically renewable for one-year terms unless terminated upon six months’ notice. In addition, Diamed may terminate this agreement in certain circumstances, including:
| · | if we become insolvent or are petitioned into bankruptcy; |
| · | if the whole or an important part of our business is transferred to a third party and such transfer would adversely affect the sale of the OctoNova pump; |
| · | if we breach the agreement and do not remedy the default within 30 days of Diamed notifying us that we are in default; |
| · | if any essential changes in our management or our share ownership would adversely affect the sale of the OctoNova pump; |
| · | if our distribution agreement with MeSys is terminated; or |
| · | if we are unable to obtain FDA approval and other necessary approvals in the territories for which we have distribution rights. |
Under this agreement, we have an obligation to purchase a minimum quantity of 1,000 OctoNova pumps before the fifth anniversary of FDA approval. If we fail to meet our minimum purchase requirements under our agreement with Diamed, our agreement may be terminated or rendered non-exclusive at the sole discretion of Diamed. Subsequent minimum purchase orders will be as mutually agreed.
Under our agreement with MeSys, MeSys agrees to manufacture and sell to us the OctoNova pump. Under this agreement, we have an obligation to purchase a minimum annual quantity of OctoNova pumps. This agreement expires on the third anniversary of our obtaining FDA approval to use the OctoNova pump to treat AMD. In addition, MeSys may terminate our agreement in certain circumstances, including:
| · | if we become insolvent or are petitioned into bankruptcy; |
| · | if we breach the agreement and do not remedy the default within 60 days of MeSys notifying us that we are in default; |
| · | if Diamed’s manufacturing agreement with MeSys is terminated; or |
| · | if our marketing agreement with Diamed is terminated. |
Sales and Marketing (Retina)
We currently have limited sales and marketing capabilities and no distribution capabilities. We had been seeking to develop our own sales and marketing infrastructure to commercialize the RHEO™ System, and we were intending to recruit our domestic ophthalmic sales force in order to have an established sales and marketing capability if and when we receive FDA approval to market the RHEO™ System in the United States. However, as a result of our discussions with the FDA following the full analysis of the MIRA-1 study results and the FDA’s requirement for a follow-up clinical trial, we have limited all sales and marketing activities that were being conducted in anticipation of commercialization in the United States. However, we will continue to develop and execute our conference and podium strategy to ensure the visibility, and the “evidence-based” positioning, of the RHEO™ System within the ophthalmic community.
In Canada, we previously had been marketing and selling the RHEO™ System through a small, dedicated Canadian sales force. During 2006, our sales and marketing activities in Canada diminished significantly. Currently, Veris is the Company’s sole commercial customer for the RHEO™ System. During 2006, the Company sold treatment sets to Veris at the discounted price of $200 per treatment set, which is lower than the Company’s cost. On November 6, 2006, the Company amended its agreement with Veris and forgave a certain amount receivable which had been owing to the Company for the sale of treatment sets and pumps, and the provision of related services, to Veris during the period from September 14, 2005 to December 31, 2005. In consideration of the forgiveness of this debt, Veris agreed that the Company did not owe Veris certain specified amounts. In January 2007, the Company further agreed to forgive an amount receivable owing by Veris for the purchase of 348 treatment sets which had been delivered to Veris in November 2006. We have been notified that Veris has initiated restructuring proceedings under the Bankruptcy and Insolvency Act (Canada) but that it is continuing to carry on its operations in the normal course during its restructuring proceedings. Our agreement with Veris continues to remain in place.
Competition (Retina)
The pharmaceutical, biotechnology and medical industries are intensely competitive. The RHEO™ System specifically targets people afflicted with Dry AMD. While we are aware that a number of companies have developed, or are in the process of developing, treatments for Wet AMD, including Eyetech Pharmaceuticals, Inc./Pfizer Inc., Genentech, Inc./Novartis Ophthalmics, Regeneron Pharmaceuticals, Inc./Bayer HealthCare, Sirna Therapeutics, Inc., Acuity Pharmaceuticals, Alcon Laboratories, Inc., Iridex Corporation and QLT Inc., we are not aware of any companies developing treatments specifically for Dry AMD, other than Acuity Medical, Inc. which, we understand, is pursuing an electrical stimulation technology to treat Dry AMD. This puts us in a strong competitive position. However, some of these companies may develop new treatments for Dry AMD or may develop modifications to their treatments for Wet AMD that may be effective for Dry AMD as well. In addition, other companies also may be involved in competitive activities of which we are not aware.
While there are other suppliers who manufacture a pump that could be used in the RHEO™ Therapy, there are no other suppliers of Asahi Medical’s Rheofilter filter, and, consequently, we believe that a third party could not readily make a system similar to the RHEO™ System. Furthermore, if a third party were to be successful in making a system similar to the RHEO™ System, it would be required to have that system approved for marketing in the United States by the FDA.
Patents and Proprietary Rights (Retina)
Our success depends in part on our ability to develop a competitive intellectual property advantage over potential competitors for the treatment of Dry AMD. There is currently no FDA-approved therapy for Dry AMD, and, to date, we are not aware of any other treatment in clinical development in North America. We own or have licenses to certain patents, and we have exclusive arrangements with certain suppliers that we believe will help us develop this competitive advantage. We also rely on know-how, continuing technological innovation and in-licensing opportunities to further develop our proprietary position. Our ability and the ability of our licensors to obtain intellectual property protection for the RHEO™ System and related processes, and our ability to operate without infringing the intellectual property rights of others and to prevent others from infringing our intellectual property rights, will be an important factor to our success. Our strategy is to seek to protect our proprietary position by, among other methods, filing U.S. patent applications related to our technology, inventions and improvements that are important to the development of our business.
One aspect of the RHEO™ System is a treatment method described in an issued U.S. patent which expires in 2017. This patent, issued under U.S. patent number 6,245,038 and entitled “Method for Treatment of Ophthalmological Diseases”, is directed to a process for treating ocular diseases using apheresis. We license this patent from the two co-owners of the patent under a separate license agreement with each owner. Under the license agreements, we have the exclusive right to use the claimed treatment method in the U.S. during the term of the patent. As part of those agreements, we are required to make royalty payments in the aggregate of 2% of the sales for the OctoNova pumps and filters, subject to minimum required payments in the aggregate amount of $25,000 during each calendar quarter.
We expect that we will request re-issuance of the patent licensed to us at the U.S. Patent and Trademark Office, and we believe that a more detailed claim set will be issued. Subsequent to entering into these license agreements, we determined that certain prior art publications may not have been considered by the Examiner during prosecution of this patent and that these references may warrant the submission of new claims. We therefore intend to request re-issuance in order to have the issued claims in this patent considered in view of these publications. During the re-issuance proceeding, we also intend to submit additional claims, which are narrower in scope than the issued claims, and are limited to the use of plasma filtration processes for treatment of ophthalmological diseases. The timing of the submission of our re-issuance application has not yet been determined and will depend, to some degree, on our future estimate of when we will be in a position to begin commercializing the RHEO™ System in the United States.
In addition, we own one issued patent in the United States, which expires in 2019. This patent, issued under U.S. patent number 6,551,266 and entitled ‘‘Rheological Treatment Methods and Related Apheresis Systems’’, is directed to methods of screening and identifying patient candidates for RHEO™ Therapy. We also have three additional pending patent applications in the United States, Europe and Japan relating to the 6,551,266 patent.
The patent position of companies like ours is generally uncertain and involves complex legal and factual questions. Our ability to maintain and solidify a proprietary position for our technology will depend on our success in obtaining effective claims and enforcing those claims once granted. We do not know whether any part of our patent applications will result in the issuance of any patents. Our issued patents and those that may issue in the future, or those licensed to us, may be challenged, invalidated or circumvented, which could limit our ability to stop competitors from marketing our product or the length of term of patent protection that we may have for our processes. The request for re-issuance of patent 6,245,038 may result in the patent being rejected and no claims of commercial value being issued or it may result in competitors acquiring intervening rights. In addition, the rights granted under any issued patents may not provide us with proprietary protection or competitive advantages against competitors with similar technology. Because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before any of our products can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent.
In addition to patent protection, we have registered the following U.S. trademarks:
We also have the right to use the following registered trademarks from Asahi Medical: Rheofilter and Plasmaflo.
We may rely, in some circumstances, on trade secrets to protect our technology. However, trade secrets are difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements with our employees, consultants, scientific advisors and other contractors. These agreements may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our employees, consultants or contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Industry (Glaucoma)
Glaucoma
Glaucoma is a leading cause of irreversible blindness and is largely related to aging. Approximately 2.2 million people in the United States aged 40 and older suffer from this disease, and the U.S. National Eye Institute estimates that at least 3.3 million people in the United States will suffer from this disease by the year 2020. The worldwide incidence of glaucoma is approximately 67 million people. Although largely related to aging, glaucoma still afflicts people of all ages. For example, it is estimated that one out of every 10,000 babies born in the United States will suffer from glaucoma.
Although the mechanisms of glaucoma are not completely understood, it is known that abnormally high pressure in the eye eventually leads to optic nerve damage and that the resultant vision loss is never regained. Glaucoma is a disease in which the aqueous humor, the watery fluid that fills the eye, builds up. In a healthy eye, aqueous humor is secreted by the ciliary processes and passes through the angle between the cornea and the iris. There, a spongy, self-cleaning filter known as the trabecular meshwork is designed to filter fluid into a passageway known as Schlemm’s canal, where it is reabsorbed into the venous system. Glaucoma is thought to be the result of the trabecular meshwork clogging, by the exfoliation of cells or other debris, or by its deformation for other reasons. When the fluid doesn’t drain properly from the clogged meshwork, it builds up in the eye and causes increased pressure. Medical treatment strategies for glaucoma focus on keeping intra-ocular pressure, or IOP, down for the remainder of a glaucoma patient’s life in an effort to preserve his or her remaining vision.
Treatment Alternatives for Glaucoma
Pharmaceuticals have become the front-line therapy for glaucoma. All of them are designed to lower IOP through a variety of mechanisms. Prostaglandin agonists do so by increasing the outflow of fluid. Examples of such prostaglandin agonists include Pfizer Inc.’s Xalatan, Alcon, Inc.’s Travatan and Allergan, Inc.’s Lumigan. Beta blockers, such as Merck & Co., Inc.’s Timoptic, and alpha adrenergic receptor agonists, such as Allergan, Inc.’s Alphagan, lower pressure by decreasing the production of aqueous humor. Another class of drugs, the muscarinic agonists, increase outflow by contracting the pupil and stimulating ciliary muscles.
Although glaucoma drugs operate in a $2 billion market today, they are plagued by low patient compliance and limited long-term efficacy. Drugs may cause unacceptable side effects in many patients or fail to control IOP adequately. Drugs also often eventually stop working as the disease progresses, and patients ultimately find themselves on daily regimens of as many as three drugs in eyedrop form that must be administered three times a day. As a result, lack of compliance is a big problem. The high cost of chronic medications (approximately $700 to $3,000 per year) exacerbates this problem further.
Device interventions for glaucoma today are focused on improving drainage, or compensating for blockages, in the trabecular meshwork. There are two types of non-invasive laser procedures—argon laser trabeculoplasty, or ALT, and a newer, gentler approach known as selective laser trabeculoplasty, or SLT. In ALT, the cornea is anesthetized, and burns are applied to the junction of the non-pigmented and pigmented trabeculum. Although the mechanism of effect is unknown, it appears to cause coagulative damage that results in collagen shrinkage and scarring in the trabecular meshwork. The laser may also be inducing macrophages to clear debris from the trabecular meshwork. SLT uses an (ND):YAG laser to target pigmented trabecular meshwork cells selectively, without producing collateral thermal damage to non-pigmented cells or structures. Coherent Inc., HGM Medical Lasers Inc., Nidek Incorporated, Iridex Corporation and Lightmed Corporation are among the companies competing in the argon laser space. Laserex Technologies Pty. Ltd., Lumenis Ltd. and Coherent Inc. are among the companies competing in the selective laser space.
If laser trabeculoplasty fails, a glaucoma patient could have an incisional or filtering surgery, the most common of which is trabeculectomy. Trabeculectomy has a 20% chance of failure at five years and high morbidity. Known negative effects of trabeculectomy include dangerously low pressure, or hypotony, inflammation, scarring and a foreign body sensation in the eye. In trabeculectomy, a piece of tissue in the drainage angle of the eye is removed, thus creating an opening. The opening is partially covered with a flap of tissue from the sclera (the white part of the eye) and the conjunctiva (its clear covering) to protect it from infection from the outside. The surgically created flap is known as a “bleb”, and it serves as a drainage pocket underneath the surface of the eye. As an adjunct or an alternative, surgeons may implant a tube or a shunt that connects the anterior chamber to the sclera or, rather, the inside of the eye to the outside of the eye. The bleb causes many complications. Manufacturers of shunts used in trabeculectomy include Optonol Ltd., Advanced Medical Optics, Inc., Pfizer Inc., New World Medical, Inc., Staar Surgical Company and Molteno Ophthalmic Limited.
Our Product (Glaucoma)
The SOLX Glaucoma System is a next-generation glaucoma treatment platform designed to reduce IOP without a bleb, thus avoiding its related complications. The SOLX Glaucoma System consists of the SOLX 790 Laser, a titanium sapphire laser used in laser trabeculoplasty, and the SOLX Gold Shunt, a 24-karat gold, ultra-thin drainage device designed to bridge the anterior chamber and the suprachoroidal space in the eye, using the pressure differential that exists naturally in the eye in order to reduce IOP.
We believe that the SOLX 790 Laser achieves deeper penetration than other lasers that are currently being used in trabeculoplasty and causes less thermal damage to the trabecular meshwork than other lasers. We believe that the SOLX Gold Shunt’s main comparative advantages are its ability to reduce IOP without a bleb, the relative ease and speed with which surgeries using the SOLX Gold Shunt can be performed and the low incidence of post-surgical complications relative to incisional surgeries and surgeries involving the implantation of other shunts.
Clinical Trials (Glaucoma)
Both the SOLX 790 Laser and the SOLX Gold Shunt are currently the subject of randomized, multi-center clinical trials, the purposes of which are to demonstrate equivalency to the argon laser, in the case of the SOLX 790 Laser, and to the Ahmed Glaucoma Valve manufactured by New World Medical, Inc., in the case of the SOLX Gold Shunt.
Both clinical trials are currently in enrollment phase and involve the participation of clinical trial sites in the United States, Canada, Spain and Israel. Our objectives are to complete enrollment in the SOLX 790 Laser clinical trial by the end of the second quarter of 2007 and to complete enrollment in the SOLX Gold Shunt clinical trial by the end of 2007. The results of these clinical trials will be used in support of applications to the FDA for a 510(k) clearance for each of the SOLX 790 Laser and the SOLX Gold Shunt, the receipt of which, if any, will enable the Company to market and sell these products in the United States. Currently, our intention is to file the application for a 510(k) clearance for the SOLX 790 Laser by the end of 2007 and to file the application for a 510(k) clearance for the SOLX Gold Shunt by the end of the second quarter of 2008.
Sales and Marketing (Glaucoma)
The SOLX 790 Laser received CE Mark approval in December 2004, and the SOLX Gold Shunt received CE Mark approval in October 2005. We are in the process of actively training and certifying physicians in the use of the SOLX Gold Shunt, for commercial purposes, in various European and Asian jurisdictions, including Spain, Italy, Germany, Poland, the United Kingdom and Thailand. In addition, we are engaged actively in pursuing relationships with distributors in Europe in order to establish a reliable distribution network for these products.
In order to ensure the visibility and awareness of the SOLX Glaucoma System within the ophthalmic community, we have been arranging to have a presence at most, if not all, of the major ophthalmology conferences in North America and Europe. We will continue to develop and execute our conference and podium strategy in order to ensure continued visibility and awareness of the SOLX Glaucoma System.
Supplier and Distribution Relationships (Glaucoma)
Cynosure, Inc., or Cynosure, a former stockholder of SOLX, is currently the exclusive manufacturer and supplier of the SOLX 790 Laser. In accordance with the terms of our agreement with Cynosure, Cynosure has designed and developed the SOLX 790 Laser in accordance with SOLX’s specifications and is supplying lasers to SOLX.
To date, we have relied on a single source of supply and manufacturing for the SOLX Gold Shunt which produces it in accordance with our specifications. Although it is our current intention to continue working with our existing supply and manufacturing partners, we are concurrently seeking to establish relationships with other manufacturers, both to ensure ourselves a cost-effective, scalable manufacturing capability and a back-up supply of any new products that we may develop in the future.
In order to establish and maintain a reliable distribution network for the SOLX Glaucoma System, we are continuing to maintain our relationships with our distributors in France, Germany, Spain and Canada and are engaged actively in pursuing relationships with other distributors in Europe.
Competition (Glaucoma)
The pharmaceutical, biotechnology and medical industries are intensely competitive. At the present time, there are many treatment alternatives for glaucoma. Numerous companies are engaged in the development, manufacture and marketing of drugs and devices for the treatment of glaucoma that are competitive with our products. Although we believe that the SOLX Glaucoma System offers notable improvements in connection with trabeculoplasty procedures and invasive glaucoma surgery, many of our competitors in this space have much greater resources than we have, thus enabling them, among other things, to make greater research and development investments, and to make much more significant investments in marketing, promotion and sales, than we are capable of at the present time or will be capable of during the foreseeable future.
Patents and Proprietary Rights (Glaucoma)
We own or have licenses to four U.S. patents relating to the SOLX Glaucoma System and related processes and have applied for a number of other patents in the United States, Canada, Europe, Australia and Japan.
We will rely on know-how, continuing technological innovation and in-licensing opportunities to develop further our proprietary position. Our ability to obtain intellectual property protection for the SOLX Glaucoma System and related processes, and our ability to operate without infringing the intellectual property rights of others and to prevent others from infringing our intellectual property rights, will have a substantial impact on our ability to succeed in the glaucoma business. We will seek to protect our proprietary position by, among other methods, continuing to file patent applications related to our technology, inventions and improvements that are important to the development of our glaucoma business. However, the patent position of companies like ours is generally uncertain and involves complex legal and factual questions. Our ability to maintain and solidify a proprietary position for our technology will depend on our success in obtaining effective claims and enforcing those claims once granted. We do not know whether any part of our patent applications will result in the issuance of any patents. Our issued patents or those that may issue in the future, or those licensed to us, may be challenged, invalidated or circumvented, which could limit our ability to stop competitors from marketing our glaucoma products or the lengths of term of patent protection that we may have for our glaucoma products and processes.
In addition to patent protection, we have registered the following U.S. trademarks:
| · | THE MODERN SYMBOL OF GLAUCOMA THERAPY |
OcuSense, Inc.
In anticipation of the delay in commercialization of the RHEO™ System in the United States, the Company accelerated its diversification plans which, in addition to the Company’s acquisition of SOLX, resulted in the Company’s acquisition of 50.1% of the capital stock, on a fully diluted basis, of OcuSense, Inc., or OcuSense. OcuSense is a San Diego-based company that is in the process of developing technologies that will enable eye care practitioners to test, at the point-of-care, for highly sensitive and specific biomarkers in tears using nanoliters of tear film.
OcuSense’s first product, which is currently under development, is a hand-held tear film osmolarity test for the diagnosis and management of dry eye syndrome, or DES, known as the TearLab™ test for DES. It is estimated that over 90 million people in the United States suffer from DES. The anticipated innovation of the TearLab™ test for DES will be its ability to measure precisely and rapidly certain biomarkers in nanoliter volumes of tear samples, using inexpensive hardware. Historically, eye care researchers have relied on expensive instruments to perform tear biomarker analysis. In addition to their cost, these conventional systems are slow, highly variable in their measurement readings and not CLIA-waived by the FDA.
The TearLab™ test for DES will require the development of the following three components: (1) the TearLab™ disposable, which is a single-use microfluidic cartridge; (2) the TearLab™ pen, which is a hand-held interface with the TearLab™ disposable; and (3) the TearLab™ reader, which is a physical housing for the TearLab™ pen connections and measurement circuitry. OcuSense is currently engaged actively in industrial, electrical and software design efforts for the three components of the TearLab™ test for DES and, to these ends, is working with two expert partners, both based in Melbourne, Australia, one of which is a leader in biomedical instrument development and the other of which is a leader of customized microfluidics.
OcuSense’s objective is to complete product development of the TearLab™ test for DES by the end of 2007. Following the completion of product development and subsequent clinical trials, OcuSense intends to seek a 510(k) clearance and a CLIA waiver from the FDA for the TearLab™ test for DES.
Government Regulation
Government authorities in the United States and other countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, promotion, advertising, distribution and marketing of the Company’s products, all of which are medical devices. In the United States, the FDA regulates medical devices under the Federal Food, Drug, and Cosmetic Act and implementing regulations. Failure to comply with the applicable FDA requirements, both before and after approval, may subject us to administrative and judicial sanctions, such as a delay in approving or refusal by the FDA to approve pending applications, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, administrative fines and/or criminal prosecution.
Unless exempted by regulation, medical devices may not be commercially distributed in the United States unless they have been cleared or approved by the FDA. Medical devices are classified into one of the three classes, Class I, II or III, on the basis of the controls necessary to reasonably assure their safety and effectiveness. Class I devices are subject to general controls, such as labeling, pre-market notification and adherence to good manufacturing practices. Class II devices are subject to general and specific controls, such as performance standards, pre-market notification, patient registries and FDA guidelines. Generally, Class III devices are those which must receive approval of a PMA by the FDA to provide reasonable assurance of their safety and effectiveness. For example, life-sustaining, life-supporting and implantable devices, or new devices which have not been found substantially equivalent to legally marketed devices, generally require approval of a PMA by the FDA.
There are two review procedures by which medical devices can receive clearance or approval. Some products may qualify for clearance under a Section 510(k) procedure, in which the manufacturer provides a pre-market notification that it intends to begin marketing the product, and shows that the product is substantially equivalent to another legally marketed product, that is that it has the same intended use and is as safe and effective as a legally marketed device and does not raise different questions of safety and effectiveness than does a legally marketed device. In some cases, the submission must include data from human clinical studies. Marketing may commence when the FDA issues a clearance letter finding substantial equivalence.
By statute and regulation, the FDA is required to clear, deny or request additional information on a 510(k) pre-market notification within 90 days of its submission. However, as a practical matter, 510(k) clearance often takes significantly longer. The FDA may require additional information, including clinical data, to make a determination regarding substantial equivalence. In addition, after a device receives 510(k) clearance, any modification to the device that could significantly affect its safety or effectiveness, or that would constitute a major change in its intended use, would require a new 510(k) clearance or an approval of a PMA. Although the FDA requires the manufacturer to make the initial determination regarding the effect of a modification to the device that is subject to 510(k) clearance, the FDA can review the manufacturer’s determination at any time and require the manufacturer to seek another 510(k) clearance or an approval of a PMA.
The components of the SOLX Glaucoma System and the TearLab™ test for DES are all Class II devices and qualify for the 510(k) procedure.
The Clinical Laboratory Improvement Amendments, or CLIA, is intended to ensure the quality and reliability of clinical laboratories in the United States by mandating specific standards in the areas of personnel qualifications, administration, participation in proficiency testing, patient test management, quality control, quality assurance and inspections. The regulations promulgated under CLIA establish three levels of in vitro diagnostic tests: (1) waiver; (2) moderately complex; and (3) highly complex. The standards applicable to a clinical laboratory depend on the level of diagnostic tests it performs. A CLIA waiver is available to clinical laboratory test systems if they meet certain requirements established by the statute. Waived tests are simple laboratory examinations and procedures employing methodologies that are so simple and accurate as to render the likelihood of erroneous results negligible or to pose no reasonable risk of harm to patients if the examinations or procedures are performed incorrectly. These tests are waived from regulatory oversight.
We cannot be sure of when, or whether, we will be successful in obtaining a 510(k) clearance for the components of the SOLX Glaucoma System, nor can we be sure of when, or whether, OcuSense will be successful in obtaining a 510(k) clearance or a CLIA waiver for the TearLab™ test for DES.
If the medical device does not qualify for the 510(k) procedure, either because it is not substantially equivalent to a legally marketed device or because it is a Class III device required to have an approved PMA, then the FDA must approve a submitted PMA before marketing can begin. A PMA must demonstrate, among other matters, that the medical device is safe and effective. A PMA is typically a complex submission, usually including the results of preclinical and clinical studies, and preparing an application is a detailed and time-consuming process. The PMA must be accompanied by the payment of user fees which currently exceed $200,000 for most submissions. When modular submissions are used, the entire fee is due when the first module is submitted to the FDA. Once a PMA has been submitted, the FDA’s review may be lengthy and may include requests for additional data. The FDA usually inspects device manufacturers before approval of a PMA, and the FDA will not approve the PMA unless the manufacturer’s compliance with the quality systems regulation is satisfactory.
The RHEO™ System is a Class III device and will require approval of a PMA. Until we commence patient enrollment in RHEO-AMD and gain a clear understanding of the progress of that clinical trial, we will not be able to anticipate the timing of our PMA submission. Once it is submitted, we cannot be sure when the FDA’s review will be complete or that the FDA will approve a PMA for our product in a timely fashion, or at all. FDA requests for additional studies during the review period are not uncommon and can significantly delay approvals. Even if we were able to obtain approval of a PMA of a product for one indication, changes to the product, its indication or its labeling can require additional clearances or approvals.
To obtain approval of a PMA, clinical studies demonstrating the safety and effectiveness of the medical device must be conducted. Prior to beginning such studies, an Investigational Device Exemption, or IDE, for the study must become effective. The IDE will automatically become effective 30 days after its receipt by the FDA, unless the FDA raises concerns or questions about the conduct of the study. In that case, the concerns and questions must be resolved before the study can begin. Even after an IDE becomes effective, the FDA may suspend it at any time on various grounds, including a finding that patients are being exposed to an unacceptable health risk. The RHEO™ System is the subject of an effective IDE, but we cannot be sure that the FDA will not suspend it, which would prevent us from commencing RHEO-AMD or completing our ongoing studies using the RHEO™ System.
Regardless of whether a medical device requires FDA clearance or approval, a number of other FDA requirements apply to the device, its manufacturer and those who distribute it. Device manufacturers must be registered and their products listed with the FDA, and certain adverse events and product malfunctions must be reported to the FDA. The FDA also regulates the product labeling, promotion and, in some cases, advertising, of medical devices. In addition, manufacturers and their suppliers must comply with the FDA’s quality system regulation which establishes extensive requirements for quality and manufacturing procedures. Thus, suppliers, manufacturers and distributors must continue to spend time, money and effort to maintain compliance, and failure to comply can lead to enforcement action. The FDA periodically inspects facilities to ascertain compliance with these and other requirements.
Employees
As of December 31, 2006, we had 34 full-time employees. Of our full-time workforce, 16 employees are engaged in clinical trial activities and 18 are engaged in business development, finance and administration. We also retain outside consultants. None of our employees are covered by collective bargaining arrangements, and our management considers its relationships with our employees to be good. To date, our strategy has been to limit the size of our full-time workforce and to outsource several of our key operating functions. Although we are, and will continue to be, actively engaged in the oversight of RHEO-AMD and the clinical trials of the components of the SOLX Glaucoma System, their day-to-day management has been outsourced to our contract research organization, The Emmes Corporation. We also rely on the resources of one of our major stockholders, TLC Vision, to provide us with infrastructure support.
Risk Factors
Risks Relating to Our Business
Our financial condition and history of losses have caused our auditors to express doubt as to whether we will be able to continue as a going concern.
We have prepared our consolidated financial statements on the basis that we will continue as a going concern. However, the Company has sustained substantial losses for each of the years ended December 31, 2006, 2005 and 2004. The Company’s working capital at December 31, 2006 is $13,539,026, which represents a $30,875,921 reduction of its working capital of $44,414,947 at December 31, 2005. As indicated in their audit report dated March 2, 2007, our auditors have expressed substantial doubt as to whether we will be able to continue as a going concern because of the losses that we have sustained during the past three years and our current cash position.
Although the Company realized gross proceeds of $10,016,000 (less transaction costs of approximately $750,000) on February 6, 2007 from the private placement of shares of its common stock and warrants, management believes that these proceeds, together with the Company’s existing cash, will be only be sufficient to cover its operating activity and other demands until early 2008.
Our consolidated financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary if the Company were not able to continue as a going concern.
We have incurred losses since inception and anticipate that we will incur continued losses for the foreseeable future.
We have incurred losses in each year since our inception in 1996. Our net loss for the fiscal years ended December 31, 2006, 2005, 2004, 2003 and 2002 was $82.2 million, $163.0 million, $21.8 million, $2.5 million and $2.9 million, respectively. The losses in 2006 and 2005 include a charge for impairment of goodwill of $65.9 million and $147.5 million, respectively. As of December 31, 2006, we had an accumulated deficit of $293.2 million. These losses, among other things, have had and will continue to have an adverse effect on our stockholders’ equity and working capital. We expect our clinical and regulatory expenses to increase significantly in connection with RHEO-AMD, the clinical trials of the components of the SOLX Glaucoma System and other clinical trials that we may initiate. In connection with the acquisition of SOLX on September 1, 2006, we remain indebted to the former stockholders of SOLX in an aggregate amount of up to $13 million for the outstanding portion of the purchase price of SOLX. We also remain indebted to OcuSense in an aggregate amount of up to $4 million for the outstanding portion of the purchase price of the capital stock of OcuSense that we acquired on November 30, 2006. Furthermore, we are legally committed to make an additional equity investment of $3 million upon receipt, if any, from the FDA of a 510(k) clearance for the TearLab™ test for DES and another additional equity investment of $3 million upon receipt, if any, from the FDA of a CLIA waiver for the TearLab™ test for DES. In addition, subject to FDA approval of any of the RHEO™ System and the components of the SOLX Glaucoma System, we expect to incur significant sales, marketing and procurement expenses. As a result, we expect to continue to incur significant and increasing operating losses for the next several years. Because of the numerous risks and uncertainties associated with developing new medical therapies, we are unable to predict the extent of any future losses or when we will become profitable, if ever.
Our business may not generate the cash necessary to fund our operations.
Since inception, we have funded our operations through early private placements of our equity and debt securities, early stage revenues, a successful initial public offering, or IPO, and, most recently on February 6, 2007, a private placement of shares of our common stock and warrants. Prior to the IPO, our cash resources were limited. We will need additional capital in the future, and our prospects for obtaining it are uncertain. We expect that the funding requirements for our operating activities will continue to increase substantially in the future, especially in view of the requirement to conduct RHEO-AMD, in support of our PMA application to the FDA, and to support the clinical trials of the SOLX Glaucoma System, in support of our applications to the FDA for 510(k) clearance, and as a consequence of our planned subsequent commercialization of the RHEO™ System and the components of the SOLX Glaucoma System. Sources of additional funds may include product licensing, joint development and other financing arrangements, or a combination of these sources. In addition, we may issue debt or additional equity securities. Future financings could result in significant dilution of existing stockholders. Additional capital may not be available on terms favorable to us, or at all. If adequate capital is unavailable, and if our operations do not generate cash, our commercialization of the RHEO™ System and/or the SOLX Glaucoma System will be delayed and we may be unable to continue our operations. See “Risk Factors—Risks Relating to Our Business—Our financial condition and history of losses have caused our auditors to express doubt as to whether we will be able to continue as a going concern.”
We do not know whether we will be able to increase our revenues or become profitable in the future.
We were founded in 1996, but the principal focus of our operations since 2000 has been directed towards our pivotal trials for the RHEO™ System—MIRA-1 initially and now RHEO-AMD. Prior to 2000, our focus had been on commercializing and performing therapeutic apheresis, or blood filtering. We generated revenues of approximately $900,200 and $1,277,800 for the years ended June 30, 1999 and 1998, respectively, all of which were earned in the United States. For the year ended December 31, 2006, we had revenues of $205,884, of which $174,259 was derived from sales of the RHEO™ System and $31,625 was derived from sales of components of the SOLX Glaucoma System. For the years ended December 31, 2005, 2004 and 2003, we had revenues of $1,840,289, $969,357 and $390,479, respectively, of which $81,593, $731,757 and $390,479, respectively, were derived from sales of the RHEO™ System to OccuLogix, L.P., a related party, which then sold the RHEO™ System to three clinics in Canada, one of which is a related party, RHEO Clinic Inc., a subsidiary of TLC Vision. For the period from July 2002 to December 8, 2004, our only customer was OccuLogix, L.P., a related party. Subsequent to December 8, 2004, OccuLogix, L.P. became wholly owned by us. Our ability to increase our revenues and to earn revenues in the United States is dependent on a number of factors, including:
| | | obtaining FDA approvals to market the RHEO™ System and the components of the SOLX Glaucoma System in the United States which will require their respective clinical trials to have successful outcomes; |
| | • | successfully building the infrastructure and manufacturing capacity to market and sell the RHEO™ System and the components of the SOLX Glaucoma System; |
| | • | achieving widespread acceptance of RHEO™ Therapy among physicians and patients, as well as the widespread acceptance by physicians and patients of the components of the SOLX Glaucoma System; and |
| | • | agreement of governmental and third-party payors to reimburse for RHEO™ Therapy and for procedures involving the components of the SOLX Glaucoma System. |
We cannot begin commercialization in the United States until we receive FDA approval. At this time, we do not know when we can expect to begin to generate revenues in the United States. If we do not obtain FDA approvals and are required to focus our efforts on marketing the RHEO™ System to clinics solely in Canada and on marketing the components of the SOLX Glaucoma System in Canada and Europe, or if we are unable to generate significant revenues in the United States, we may not become profitable and we may be unable to continue our operations.
The business prospects and financial condition of our sole commercial customer, Veris, is uncertain.
We have been notified that Veris has initiated restructuring proceedings under the Bankruptcy and Insolvency Act (Canada) but that it is continuing to carry on its operations in the normal course during its restructuring proceedings. A failure on the part of Veris to restructure its business successfully may adversely affect our ability to generate any revenues since, at this time, there is no other commercial provider of RHEO™ Therapy in any jurisdiction in which we have distribution and marketing rights to the RHEO™ System.
During the year ended December 31, 2005, the Company had recorded an allowance for doubtful accounts of $1,047,622 against the amount due from Veris. In June 2006, Veris returned four OctoNova pumps which had been sold to it in December 2005. Accordingly, during fiscal year 2006, amounts receivable, net and the allowance for doubtful account recorded against the amount due from Veris were reduced by $143,520, the invoiced amount for the four pumps that were returned to the Company in June 2006. In addition, in November 2006, the Company forgave an amount receivable of $904,101 (for which an allowance for doubtful account had been recorded previously) which had been owing to the Company for the sale of treatment sets and pumps, and the provision of related services, to Veris during the period from September 14, 2005 to December 31, 2005.
In November 2006, the Company sold 348 treatment sets to Veris for $73,776, including applicable taxes, the revenues for which were not recognized by the Company for the year ended December 31, 2006 as the Company believed that Veris would not be able to meet its financial obligations to the Company. In January 2007, the Company agreed to forgive this outstanding amount receivable of $73,776, and the Company has recorded an inventory loss of $60,987 in the year ended December 31, 2006 for the sale of these 348 treatment sets since these treatment sets had been delivered to Veris already.
As of April 2006, the Company has been selling treatment sets to Veris at the negotiated discounted price of $200 per treatment set, which is lower than the Company’s cost. That price continues to remain in effect at the present time.
MIRA-1 did not meet its primary efficacy endpoint, and we are required to conduct a follow-up clinical trial of the RHEO™ System, RHEO-AMD, to support our PMA application.
We are required to obtain FDA approval to market the RHEO™ System in the United States. To support an application for FDA approval, we conducted, at our own expense, MIRA-1 to evaluate the safety and efficacy of RHEO™ Therapy in humans. MIRA-1 did not meet its primary efficacy endpoint, and we are required to conduct a follow-up clinical trial to support our PMA application, RHEO-AMD. The outcome of RHEO-AMD is uncertain. Clinical testing is expensive and can take many years. Failure can occur at any stage of the testing. We may encounter numerous factors during, or as a result of, RHEO-AMD that could delay or prevent us altogether from completing it and receiving FDA approval for a number of reasons, including:
· we may be unable to obtain the complete number of data sets required by the protocol for RHEO-AMD;
· the costs of RHEO-AMD may be greater than we anticipate;
· we, or the regulators, may suspend or terminate RHEO-AMD if the participating patients are being exposed to unacceptable health risks; and
· negative or inconclusive results may arise, and we may decide, or regulators may require us, to conduct additional clinical and/or preclinical testing.
The clinical trials of the components of the SOLX Glaucoma System may not succeed.
We are required to obtain FDA approval to market the components of the SOLX Glaucoma System in the United States. In order to support our 510(k) clearance pre-market notifications, we are conducting, at our own expense, two randomized, multi-center clinical trials to demonstrate substantial equivalency to the argon laser, in the case of the SOLX 790 Laser, and to the Ahmed Glaucoma Valve, in the case of the SOLX Gold Shunt. The outcomes of these clinical trials is uncertain. Clinical testing is expensive and can take many years. Failure can occur at any stage of the testing. We may encounter numerous factors during, or as a result of, these clinical trials that could delay us or prevent us altogether from completing them and receiving the sought-after FDA approvals, including:
| · | we may be unable to obtain the complete number of data sets required by the respective protocols for these clinical trials; |
| · | the costs of these clinical trials may be greater than we anticipate; |
| · | we, or the regulators, may suspend or terminate either or both of these clinical trials if the participating patients are being exposed to unacceptable health risks; |
| · | negative or inconclusive results may arise, and we may decide, or regulators may require us, to conduct additional clinical and/or preclinical testing; and |
| · | one or both of these clinical trials may fail to demonstrate the substantial equivalency, to its predicate device, of the device being tested. |
We may not receive the necessary FDA approvals to market, in the United States, the RHEO™ System or the components of the SOLX Glaucoma System.
We may not receive the necessary FDA approvals to market, in the United States, the RHEO™ System or the components of the SOLX Glaucoma System. Obtaining FDA approval is a lengthy and expensive process, and approval is uncertain. We may be delayed in receiving, or may never receive, the necessary FDA approvals for the RHEO™ System or the components of the SOLX Glaucoma System. One or more delays in obtaining or failure to obtain such FDA approvals would delay or prevent the successful commercialization of the RHEO™ System and/or one or both components of the SOLX Glaucoma System, diminish our competitive advantage and/or defer or decrease our receipt of revenues.
Even if we eventually obtain FDA approval for the RHEO™ System, this approval may only be for a limited or narrow class of Dry AMD patients, thereby diminishing the size of the class of prospective patients for whose use the RHEO™ System can be promoted.
In addition, changes to the RHEO™ System or the components of the SOLX Glaucoma System can require additional FDA approvals.
Our purchase commitments may adversely affect our liquidity.
We currently have commitments to purchase approximately $21.9 million of OctoNova pumps (based on current exchange rates) within five years after FDA approval, $13.3 million of Rheofilter filters and Plasmaflo filters over a three-year period beginning six months after FDA approval with respect to the United States, $1.3 million of Rheofilter filters and Plasmaflo filters over a three-year period commencing upon the earlier to occur of the sale of our current inventory of Rheofilter filters or their expiry with respect to Canada and $0.4 million of Rheofilter filters and Plasmaflo filters in 2009 and 2010 with respect to Australia, New Zealand, Colombia, Venezuela and Italy. We expect to fund our purchase commitments with cash generated from operations following receipt of the FDA approvals being sought or, in the event we do not have sufficient cash from operations, other financing sources. Should these sources be insufficient to fund our purchase commitments, our liquidity may be adversely affected.
We currently depend on single sources for key components of the RHEO™ System and the components of the SOLX Glaucoma System. The loss of any of these sources could delay our clinical trials or prevent or delay commercialization of the RHEO™ System or the components of the SOLX Glaucoma System.
We currently depend on single sources for the filters and the OctoNova pump used in the RHEO™ System. We have entered into a supply agreement for the filters with Asahi Medical and for the OctoNova pump with Diamed, which designed the OctoNova pump, and MeSys, which manufactures the pumps for Diamed. We currently have commitments to purchase approximately $21.9 million of OctoNova pumps (based on current exchange rates) within five years after FDA approval, $13.3 million of Rheofilter filters and Plasmaflo filters over a three-year period beginning six months after FDA approval with respect to the United States, $1.3 million of Rheofilter filters and Plasmaflo filters over a three-year period commencing upon the earlier to occur of the sale of our current inventory of Rheofilter filters or their expiry with respect to Canada and $0.4 million of Rheofilter filters and Plasmaflo filters in 2009 and 2010 with respect to Australia, New Zealand, Colombia, Venezuela and Italy. If we fail to meet our minimum purchase requirements under our agreements with Diamed or Asahi Medical, those agreements may be terminated or rendered non-exclusive at the sole discretion of the supplier. If any of these suppliers ceases to supply components to us or does not supply an adequate number of components, our sales and growth could be restricted, potentially materially. If we do not achieve FDA approval and other necessary approvals in certain of the territories for which we have distribution rights by the applicable deadlines (which, under our agreement with Asahi Medical, are February 28, 2009 for Canada and December 31, 2010 for the United States and all of the other territories where we have distribution rights under this agreement), Asahi Medical can terminate the supply agreement for the filters and Diamed can terminate the supply agreement for the pumps. Our agreement with Asahi Medical as it relates to the United States and our agreement with Diamed each has a term ending ten years after the date of FDA approval and is automatically renewable for one-year terms unless terminated upon six months’ notice. In addition, Diamed may terminate its agreement upon the termination of our manufacturing agreement with MeSys, which has a term of three years following FDA approval. We believe that establishing additional or replacement suppliers for these components may not be possible as these suppliers have trade secrets, patents and other intellectual property that may prevent a third party from manufacturing a suitable replacement product. Even if we switch to replacement suppliers and the supplier can manufacture the necessary components without violating any third-party intellectual property rights, we may face additional regulatory delays and the distribution of the RHEO™ System could be interrupted for an extended period of time, which may delay or slow down the commercialization of RHEO™ Therapy and adversely impact our financial condition and results of operations.
We currently depend on single sources for the manufacture and supply of the SOLX 790 Laser and the SOLX Gold Shunt. If any of our suppliers ceases to supply products to us or does not supply them in adequate quantities, the clinical trials of the components of the SOLX Glaucoma System could be delayed and our sales and growth in the glaucoma business could be restricted, potentially materially. The establishment of a cost-effective, scalable manufacturing capability for the SOLX Gold Shunt will be critical to the success of our glaucoma business. Although, at the present time, we are not aware of any reason why additional or replacement suppliers of the components of the SOLX Glaucoma System cannot be found, including the SOLX Gold Shunt, there is a risk that trade secrets, patents and other intellectual property of our current suppliers may prevent a third party from manufacturing suitable replacement products. Even if no such intellectual property barriers exist, we may face additional regulatory delays and the distribution of the components of the SOLX Glaucoma System could be interrupted for an extended period of time, which may delay or slow down their commercialization and adversely impact our financial condition and results of operations.
Our supply agreement with Asahi Medical requires us to transfer the FDA approval of the RHEO™ System, upon its receipt, to a special purpose corporation, to be majority-owned by Asahi Medical, which will limit our control of the FDA approval.
Under our supply agreement with Asahi Medical for the filters that are used in the RHEO™ System, we agreed to obtain FDA approval of the RHEO™ System and to transfer it upon receipt, if any, to a special purpose corporation which will be owned as to 51% by Asahi Medical and as to 49% by us. This transfer of the FDA approval to this special purpose corporation may limit our flexibility to make changes in the FDA approval, such as the addition of alternate suppliers of RHEO™ System components, without Asahi Medical’s consent, or limit our ability to prevent changes to the FDA approval that we might consider detrimental, such as the addition of labeling changes or the substitution of alternate component suppliers. Regulatory approvals obtained for the RHEO™ System, if any, in Canada, Australia, New Zealand, Colombia, Venezuela or Italy will be held by Asahi Medical.
If we or our suppliers fail to comply with the extensive regulatory requirements to which we and our products are subject, the RHEO™ System and the components of the SOLX Glaucoma System could be subject to restrictions or withdrawals from the market and we could be subject to penalties.
We, our suppliers and all of our products are subject to numerous FDA requirements covering the design, testing, manufacturing, quality control, labeling, advertising, promotion and export of our products and other matters. Failure to comply with statutes and regulations administered by the FDA could result in, among other things, any of the following actions:
• warning letters;
• fines and other civil penalties;
• unanticipated expenditures;
• withdrawal of FDA approval;
• delays in approving or refusal to approve our products;
• product recall or seizure;
• interruption of production;
• operating restrictions;
• border stops;
• injunctions; and
• criminal prosecution.
We and our suppliers are subject to numerous federal, state and local laws relating to such matters as safe working conditions, manufacturing practices, environmental protection, fire hazard control and disposal of hazardous or potentially hazardous substances. In addition, advertising and promotional materials relating to medical devices are, in certain instances, subject to regulation by the Federal Trade Commission. We and our suppliers may be required to incur significant costs to comply with such laws and regulations in the future, and such laws and regulations may materially harm our business. Unanticipated changes in existing regulatory requirements, the failure by us or our manufacturers to comply with such requirements or the adoption of new requirements could materially harm our business.
We may be unable to commercialize the RHEO™ System or the components of the SOLX Glaucoma System successfully in the United States.
Even if we successfully obtain FDA approval for the RHEO™ System, our success depends on our ability to market and sell the RHEO™ System. Successful commercialization of the RHEO™ System depends on a number of factors, including:
• achieving widespread acceptance of RHEO™ Therapy among physicians and patients;
• agreement of governmental and third-party payors to provide reimbursement for RHEO™ Therapy;
• maintaining our relationships with our single source suppliers;
• obtaining sufficient quantities of components for the RHEO™ System;
• establishing adequate sales and marketing capabilities;
• obtaining sufficient facility space;
• our ability to identify and sell the RHEO™ System to key multi-facility health care providers as well as to private eye care professional practices;
• our ability to successfully sell the RHEO™ System at our projected selling price;
• whether there are adverse side effects or unfavorable publicity concerning the RHEO™ System; and
• whether there is competition for the RHEO™ System from new or existing products, which may prove to be safer, more efficacious or more cost-effective than the RHEO™
System.
Other than the ability to identify and make sales to key multi-facility health care providers, all of the above-listed factors (substituting the above-noted references to the RHEO™ System and RHEO™ Therapy with references to the SOLX Glaucoma System and procedures using the SOLX Glaucoma System, respectively) will be equally influential in the success or failure of our efforts to commercialize the SOLX Glaucoma System in the United States. In addition to such factors, the establishment of a cost-effective, scalable manufacturing capability for the SOLX Gold Shunt will be critical to the success of our glaucoma business.
RHEO™ Therapy is based on a model that has not achieved widespread acceptance and may be proven incorrect. If we are unsuccessful in achieving widespread acceptance of RHEO™ Therapy among physicians and patients, our business may not succeed.
AMD is not a well understood disease and its underlying cause is not known. RHEO™ Therapy is based on a disease model that has not achieved widespread acceptance with eye care professionals. Unlike traditional therapeutic treatments for eye diseases, RHEO™ Therapy is a systemic approach for the treatment of Dry AMD, rather than a localized approach. Our success is dependent upon achieving widespread acceptance of RHEO™ Therapy among ophthalmologists and optometrists. Eye care professionals and health care service providers may not be willing to integrate RHEO™ Therapy into their workflow. In addition, because RHEO™ Therapy can be performed by health care providers other than eye care professionals, eye care professionals may be reluctant to endorse RHEO™ Therapy. The fact that MIRA-1 did not meet its primary efficacy endpoint may strengthen opposition to RHEO™ Therapy or impede its acceptance.
Even if we are successful in achieving widespread acceptance of RHEO™ Therapy among physicians, we may be unable to achieve widespread acceptance among potential patients. An initial course of RHEO™ Therapy is time consuming, requiring eight procedures over a 10- to 12-week period, with each procedure lasting between two and four hours. Some patients may be reluctant to undergo RHEO™ Therapy because of the time commitment. In addition, RHEO™ Therapy providers may not be easily accessible to all patients and some patients may be unwilling or unable to travel to receive RHEO™ Therapy. If we are unable to achieve widespread acceptance, our financial condition and results of operations will be adversely affected.
In August 1997, our predecessor opened its sole client facility, the Rheotherapy Center, in Tampa, Florida, to perform therapeutic apheresis commercially. In 1999, the FDA’s Office of Compliance issued a directive notifying our predecessor that further conducting of therapeutic apheresis would need to be conducted under the authority of an Investigational Device Exemption filed with the FDA. In a related action, our predecessor, on behalf of one of our founders, Dr. Richard C. Davis, made a payment in the amount of $10,000 to cover legal expenses incurred by the Florida Board of Medicine in prosecuting our predecessor’s unauthorized advertising of new medical therapies. Our predecessor closed the Rheotherapy Center in 1999, after which time we received an Investigational Device Exemption and subsequently completed MIRA-1. Dr. Davis was our Chief Executive Officer from January to June 2003 and was our Chief Science Officer from July 2003 to April 2004 and since then has served as a consultant to us, although he no longer serves us in any capacity. Dr. Davis is also a former director of ours. We believe that the activities of the Rheotherapy Center engendered opposition in certain segments of the eye care community to RHEO™ Therapy and if this opposition continues, acceptance of RHEO™ Therapy among eye care professionals and patients may be difficult to achieve.
If RHEO™ Therapy and the procedures involving the components of the SOLX Glaucoma System are not reimbursed by governmental and other third-party payors, or are only reimbursed on a limited basis, our business may not succeed.
Undergoing RHEO™ Therapy is expensive, with an initial course of treatment expected to initially cost between $16,000 and $25,600 in the United States. The cost of procedures involving the components of the SOLX Glaucoma System are not anticipated to be insubstantial either. Continuing efforts of governmental and third-party payors to contain or reduce the costs of health care could negatively affect the sale of the RHEO™ System and the components of the SOLX Glaucoma System. Our ability to commercialize our products successfully will depend in substantial part on favorable determinations by governmental payors, most prominently Medicare, private health insurers and state-funded health care coverage programs. Without the establishment of timely, favorable coverage and reimbursement policies, we may be unable to set or maintain price levels sufficient to realize an appropriate return on our investment in product development. Other significant insurance coverage limitations, such as narrow restrictions on patient coverage criteria and restrictions on treatment settings in which RHEO™ Therapy is covered, may also limit our potential revenues.
Our patents may not be valid and we may not be able to obtain and enforce patents to protect our proprietary rights from use by competitors.
Our owned and licensed patents may not be valid, and we may not be able to obtain and enforce patents and to maintain trade secret protection for our technology. The extent to which we are unable to do so could materially harm our business.
We have applied for and will continue to apply for patents for certain processes used in the RHEO™ System and for patents important to the development of our glaucoma business. Such applications may not result in the issuance of any patents, and any patents now held or that may be issued may not provide us with adequate protection from competition. In addition, we expect that we will seek to have the patent licensed to us re-issued at the U.S. Patent and Trademark Office, and we believe that a more detailed claim set will be issued. The timing of the submission of our re-issuance application has not yet been determined and will depend, to some degree, on our future estimate of when we will be in a position to begin commercializing the RHEO™ System in the United States. The application for re-issuance of this patent may result in the patent being rejected or no claims of commercial value being issued or it may result in competitors acquiring intervening rights. Furthermore, it is possible that patents issued or licensed to us may be challenged successfully. In that event, if we have a preferred competitive position because of such patents, any preferred position held by us would be lost. If we are unable to secure or to continue to maintain a preferred position, the components of the RHEO™ System could become subject to competition from the sale of generic products.
Patents issued or licensed to us may be infringed by the products or processes of others. The cost of enforcing our patent rights against infringers, if such enforcement is required, could be significant, and the time demands could interfere with our normal operations. There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical, biotechnology and medical technology industries. We may become a party to patent litigation and other proceedings. The cost to us of any patent litigation, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of such litigation more effectively than we can because of their substantially greater financial resources. Litigation may also absorb significant management time.
Unpatented trade secrets, improvements, confidential know-how and continuing technological innovation are important to our scientific and commercial success. Although we attempt to, and will continue to attempt to, protect our proprietary information through reliance on trade secret laws and the use of confidentiality agreements with our corporate partners, collaborators, employees and consultants and other appropriate means, these measures may not effectively prevent disclosure of our proprietary information, and, in any event, others may develop independently, or obtain access to, the same or similar information.
Certain of our patent rights are licensed to us by third parties. If we fail to comply with the terms of these license agreements, our rights to those patents may be terminated, and we will be unable to conduct our business.
Patents of other companies could require us to stop using or pay to use required technology.
It is possible that a court may find us to be infringing upon validly issued patents of third parties. In that event, in addition to the cost of defending the underlying suit for infringement, we may have to pay license fees and/or damages and we may be enjoined from conducting certain activities. Obtaining licenses under third-party patents can be costly, and such licenses may not be available at all. Under such circumstances, we may need to materially alter our products or processes and we may be unable to do so successfully.
If we are unable to establish adequate sales and marketing capabilities, we may not be able to generate significant revenue and may not become profitable.
While our management team has some experience in marketing medical technology, we do not have a sales organization and have limited experience as a company in the sales, marketing and distribution of ophthalmic therapy products. In order to commercialize our products, we must develop our sales, marketing and distribution capabilities or make arrangements with a third party to perform these functions. If and when marketing of our products is eventually approved by the FDA, our plan will be to establish our own sales force to market them in the United States. Developing a sales force is expensive and time consuming, and we may not be able to develop this capacity. If we are unable to establish adequate sales, marketing and distribution capabilities, independently or with others, we may not be able to generate significant revenue and may not become profitable.
Our suppliers may not have sufficient manufacturing capacity and inventory to support our commercialization plans.
Our success requires that our suppliers have adequate manufacturing capacity and inventory in order to facilitate a rapid rollout of our products. In particular, the establishment of a cost-effective, scalable manufacturing capability for the SOLX Gold Shunt will be critical to the success of our glaucoma business. We have not achieved such capability.
Our ability to conduct our clinical trials and commercialize our products depends, in large part, on our ability to have components manufactured at a competitive cost and in accordance with FDA and other regulatory requirements. We do not control the manufacturing processes of our suppliers. If current manufacturing processes are modified, or the source or location of our product supply is changed, voluntarily or involuntarily, the FDA will require us to demonstrate that the material produced from the modified or new process or facility is equivalent to the material used in the clinical trials or products previously approved. Any such modifications to the manufacturing process or supply may not achieve or maintain compliance with the applicable regulatory requirements. In many cases, prior approval by regulatory authorities may be required before any changes can be made, which may adversely affect our business.
To date, we have used $4.8 million to stockpile an inventory of the older cellulose acetate Rheofilter filters that currently form part of the RHEO™ System in anticipation of manufacturing constraints to which such filters are subject. With the FDA’s confirmation of its willingness to allow the substitution of the older Rheofilter filter with the new polysulfone Rheofilter filter in RHEO-AMD, such manufacturing constraints are no longer of immediate concern. However, each of the older Rheofilter filters that we’ve accumulated in inventory has a shelf life of approximately three years, and it is possible that some or all of these filters will expire before they are used. Holding inventory in this manner decreases our short term liquidity.
Our success in the commercialization of the RHEO™ System depends upon our ability to sell to key multi-facility health care providers as well as private eye care professional practices.
In order to facilitate a rapid rollout of the RHEO™ System if and when we receive FDA approval, we will need to establish relationships with key organized groups of multi-facility health care service providers, including hospitals, dialysis clinics and ambulatory surgery centers, as well as private practices. We may be unsuccessful in establishing these relationships, which could limit our ability to commercialize the RHEO™ System.
We anticipate that RHEO™ Therapy will be prescribed by physicians and administered by nurses, and therefore our service provider customers will need the support of an adequate supply of trained nurses. Training nurses to administer RHEO™ Therapy may be costly, and our customers may experience shortages of nurses from time to time. If there is a shortage of trained nurses to work in our customers’ facilities, our commercialization of RHEO™ Therapy may be unsuccessful.
RHEO™ Therapy and the procedures involving the components of the SOLX Glaucoma System may produce adverse side effects in patients that prevent its adoption or that necessitate withdrawal from the market.
RHEO™ Therapy may produce undesirable, unexpected and unintended side effects not previously observed during clinical trials. These side effects in patients may prevent or limit its commercial adoption and use. Side effects that have been observed in MIRA-1 were all temporary and generally mild and included temporary drops in blood pressure, abnormal heart rate, nausea, chills and localized bleeding, pain, numbness and swelling in the area of the arms where the needles were inserted. The procedures involving the components of the SOLX Glaucoma System may also produce undesirable, unexpected and unintended side effects not previously observed during clinical trials. Should that occur, the commercial adoption and use of the components of the SOLX Glaucoma System may be limited or may be prevented altogether.
Even after approval by the FDA and other regulatory authorities, the RHEO™ System and/or the components of the SOLX Glaucoma System may later be found to produce adverse side effects that prevent widespread use or necessitate withdrawal from the market. The manifestation of such side effects could cause our business to suffer. In some cases, regulatory authorities may require additional disclosure to patients that could add warnings or restrict usage based on unexpected side effects seen after marketing a medical treatment.
We may face future product liability claims that may result from the use of our products.
The testing, manufacturing, marketing and sale of therapeutic products entails significant inherent risks of allegations of product liability. Our use of such products in clinical trials and the commercial sale of our products may expose us to liability claims. These claims might be made directly by patients, health care providers or others selling our products. We carry clinical trials and product liability insurance to cover certain claims that could arise during our clinical trials or during the commercial use of our products. We currently maintain clinical trials and product liability insurance with coverage limits of $5,000,000 in the aggregate annually. We also maintain some separate clinical trials insurance for clinical trial activities in Spain and Israel. Such coverage, and any coverage obtained in the future, may be inadequate to protect us in the event of a successful product liability claim, and we may not be able to increase the amount of such insurance coverage or even renew it. A successful product liability claim could materially harm our business. In addition, substantial, complex or extended litigation could cause us to incur large expenditures and divert significant resources.
In the medium or long term, we will need to increase the size of our organization, and we may experience difficulties in managing our growth.
In order to commercialize the RHEO™ System and the components of the SOLX Glaucoma System, we will need to expand our employee base for management of operational, sales and marketing, financial and other resources. It is not clear when we will be able to commercially launch our products in the United States, if ever. Future growth will impose significant additional responsibilities on members of management, including the need to identify, recruit, maintain and integrate additional employees. Our future financial performance and our ability to commercialize our products and to compete effectively will depend, in part, on our ability to manage any future growth effectively. To that end, we must be able to:
| | • | integrate additional management, administrative, distribution and sales and marketing personnel; |
| | • | develop our administrative, accounting and management information systems and controls; and |
• hire and train additional qualified personnel.
We may not be able to accomplish these tasks, and our failure to accomplish any of them could prevent us from achieving or maintaining profitability.
We may face competition and may not be successful in addressing it.
The pharmaceutical, biotechnology and medical technology industries are characterized by rapidly changing technology and intense competition. AMD is not a well understood disease and researchers are continuing to investigate different theories of the cause of AMD. If the cause of AMD is determined, competitors could potentially develop a treatment for Dry AMD that would replace RHEO™ Therapy. In addition, competitors may develop alternative treatments for Dry AMD that prove to be superior to, or more cost-effective than, RHEO™ Therapy. Some of these competitors may include companies which have access to financial, technical and marketing resources significantly greater than ours and substantially greater experience in developing, manufacturing and distributing products, conducting preclinical and clinical testing and obtaining regulatory approvals.
We are aware of a number of companies which have developed or are in the process of developing treatments for Wet AMD, including Eyetech Pharmaceuticals, Inc./Pfizer Inc., Genentech, Inc./Novartis Ophthalmics, Alcon Laboratories, Inc., Regeneron Pharmaceuticals, Inc./Bayer HealthCare, Sirna Therapeutics, Inc., Acuity Pharmaceuticals, Iridex Corporation and QLT Inc. Some of these treatments are in late-stage clinical development or have been approved by the FDA already. Some of these companies may develop new treatments for Dry AMD or may develop modifications to their treatments for Wet AMD that may be effective for Dry AMD as well. We are aware that Acuity Medical, Inc. is pursuing an electrical stimulation technology to treat Dry AMD. In addition, other companies also may be involved in competitive activities of which we are not aware.
At the present time, there are many treatment alternatives for glaucoma. Numerous companies are engaged in the development, manufacture and marketing of drugs and devices for the treatment of glaucoma, including, but not limited to, Optonol Ltd., Advanced Medical Optics, Inc., Pfizer Inc., New World Medical, Inc., Allergan, Inc. and Alcon, Inc. Although we believe that the SOLX Glaucoma System offers notable improvements in connection with trabeculoplasty procedures and glaucoma surgery, many of our competitors in this space have much greater resources than we have, thus enabling them, among other things, to make greater research and development investments, and to make much more significant investments in marketing, promotion and sales, than we are capable of at the present time or may ever become capable of in the future.
We may be unable to attract and retain key personnel which may adversely affect our business.
Our success depends on the continued contributions of our executive officers and scientific personnel. Many of our key responsibilities have been assigned to a relatively small number of individuals. We will be required to hire eyecare specialists as well as personnel with skill sets in apheresis, nursing, training, equipment maintenance, finance, distribution, logistics, warehousing, sales and service, and possibly other areas of expertise, to meet our personnel needs. There is competition for qualified personnel, and the failure to secure the services of key personnel or loss of services of key personnel could adversely affect our business.
The additional uncertainty regarding our business prospects that has been created by MIRA-1’s failure to reach its primary efficacy endpoint may impede our ability to attract and retain key personnel.
For as long as TLC Vision owns a substantial portion of our common stock, our other stockholders may be effectively unable to affect the outcome of stockholder voting.
TLC Vision beneficially owns approximately 36.08% of our outstanding common stock, or 33.79% on a fully diluted basis. Accordingly, TLC Vision, in conjunction with other stockholders, could possess an effective controlling vote on matters submitted to a vote of the holders of our common stock.
While it owns a substantial portion of our common stock, TLC Vision could effectively control decisions with respect to:
• our business direction and policies, including the election and removal of our directors;
• mergers or other business combinations involving us;
• the acquisition or disposition of assets by us;
• our financing; and
• amendments to our certificate of incorporation and bylaws.
Furthermore, TLC Vision may be able to cause or prevent a change of control of the Company, and this concentration of ownership may have the effect of discouraging others from pursuing transactions involving a potential change of control of the Company, in either case regardless of whether a premium is offered over then-current market prices.
Conflicts of interest may arise between us and TLC Vision, which has three directors on our board and for which our Chief Executive Officer and Chairman served as Chairman until June 2006.
TLC Vision beneficially owns approximately 36.08% of our outstanding common stock, or 33.79% on a fully diluted basis. Our directors, Elias Vamvakas, Thomas Davidson and Richard Lindstrom, are also directors of TLC Vision. Mr. Vamvakas beneficially owns 2,827,589 common shares of TLC Vision, representing approximately 4.09% of TLC Vision’s outstanding shares. Mr. Davidson beneficially owns 71,954 common shares of TLC Vision, representing approximately 0.10% of TLC Vision’s outstanding shares, and Dr. Lindstrom does not beneficially own any common shares of TLC Vision. Because Messrs. Vamvakas and Davidson and Dr. Lindstrom are directors of TLC Vision, a conflict of interest could arise. Conflicts may arise between TLC Vision and us as a result of our ongoing agreements. We may not be able to resolve all potential conflicts with TLC Vision, and even if we do, the resolution may be less favorable to us than if we were dealing with an unaffiliated third party.
We have entered into a number of related party transactions with suppliers, creditors, stockholders, officers and other parties, each of which may have interests which conflict with those of our public stockholders.
We have entered into several related party transactions with our suppliers, creditors, stockholders, officers and other parties, each of which may have interests which conflict with those of our public stockholders.
Certain of our directors and management team members have been with us for only a short time.
Nozhait Chaudry-Rao, our Vice President, Clinical Research, and Stephen Parks, our Vice President, Sales, and our directors, Adrienne Graves and Gilbert Omenn, have all served as members of our management team for less than two years. This poses a number of risks, including the risk that these persons may:
• have limited familiarity with our past practices;
• lack experience in communicating effectively within the team and with other employees;
• lack settled areas of responsibility; and
• lack an established track record in managing our business strategy, including clinical trials.
In December 2004, we moved from our previous headquarters which we subleased from TLC Vision to our current headquarters, which are also in Mississauga. Until January 31, 2006, we subleased our current headquarters from Echo Online Internet, Inc. and, as of February 1, 2006, have been leasing them from Penyork Properties III Inc. The facility presently consists of approximately 6,600 square feet of office space utilized for corporate finance and clinical trial management personnel. Our current arrangement expires on July 31, 2007. Our current monthly lease obligation for rent for this facility is C$11,512. The future minimum obligation under this lease is C$80,581 for 2007. TLC Vision has advised us that it does not have any ownership interest in our current headquarters.
We also lease space in a facility in Palm Harbor, Florida consisting of 5,020 square feet of space used for warehousing the RHEO™ System components and providing office space for certain members of our clinical trial personnel and John Cornish, who is our Vice President, Operations, and records. The facility consists of office and working space and an approximately 1,700 square foot warehouse in the back. Our lease on this property expired on December 31, 2006 and has been renewed until December 31, 2007. Our current monthly lease obligation for rent for this facility is approximately $2,168. The landlord under this lease is Cornish Properties, which is owned by Mr. Cornish. Mr. Cornish was also one of our directors from April 1997 to September 2004.
In addition, Solx, Inc. and OcuSense, Inc. lease office space in facilities owned by parties unrelated to us. The total future minimum obligation under these leases is $30,000 for 2007.
We believe that if our existing facilities are not adequate to meet our business requirements for the near-term, additional space will be available on commercially reasonable terms.
| ITEM 3. | LEGAL PROCEEDINGS. |
We are not aware of any material litigation involving us that is outstanding, threatened or pending.
| ITEM 4. | SUBMISSION OF MATTERS TO A VOTE OF SECURITY HOLDERS. |
No matter was submitted during the fourth quarter of the Company’s 2006 fiscal year to a vote of security holders, through the solicitation of proxies or otherwise.
PART II
| ITEM 5. | MARKET FOR THE REGISTRANT’S COMMON EQUITY AND RELATED STOCKHOLDER MATTERS. |
Market for Common Equity
Our Common Stock trades on the NASDAQ Global Market (the “NASDAQ”) under the symbol “OCCX” and the Toronto Stock Exchange (the “TSX”) under the symbol “OC”.
The following table sets forth the range of high and low sales prices per share of our Common Stock on both the NASDAQ and the TSX for the fiscal periods indicated.
| | Common Stock Prices |
| | Fiscal 2006 | Fiscal 2005 |
| | High | Low | High | Low |
| NASDAQ | | | | |
| First Quarter | $12.85 | $3.25 | $10.68 | $7.06 |
| Second Quarter | 3.70 | 1.86 | 9.35 | 5.92 |
| Third Quarter | 2.90 | 1.56 | 9.78 | 6.05 |
Fourth Quarter | 2.68 | 1.55 | 8.78 | 5.88 |
TSX | | | | |
| First Quarter | C$14.99 | C$3.76 | C$12.90 | C$8.75 |
| Second Quarter | 4.33 | 2.12 | 11.17 | 7.57 |
| Third Quarter | 3.00 | 1.69 | 11.70 | 7.00 |
| Fourth Quarter | 3.00 | 1.80 | 10.49 | 6.94 |
The closing share price for our Common Stock on March 14, 2007 as reported by the NASDAQ, was $1.66. The closing share price for our Common Stock on March 14, 2007, as reported by the TSX was C$2.05.
As of March 12, 2007, there were approximately 117 stockholders of record of our Common Stock.
We have never declared or paid any cash dividends on shares of our capital stock. We currently intend to retain all available funds to support operations and to finance the growth and development of our business. Any determination related to payments of future dividends will be at the discretion of our board of directors after taking into account various factors that our board of directors deems relevant, including our financial condition, operating results, current and anticipated cash needs, plans for expansion and debt restrictions, if any.
Unregistered Issuances of Capital Stock
On October 6, 2006, we issued an aggregate of 10,000 shares of Common Stock to Irving Siegel as a result of the exercise of options to purchase common shares at an exercise price per share of $0.99 in consideration for cash.
On November 2, 2006, we issued an aggregate of 10,000 shares of Common Stock to Irving Siegel as a result of the exercise of options to purchase common shares at an exercise price per share of $0.99 in consideration for cash.
Each of the above issuances was exempt from registration under the Securities Act of 1933, as amended (the “Securities Act”), pursuant to Rule 701 thereunder.
| ITEM 6. | SELECTED FINANCIAL DATA. |
The following tables set forth our selected historical consolidated financial data for the years ended December 31, 2006, 2005, 2004, 2003 and 2002 which have been derived from our consolidated financial statements included elsewhere in this Annual Report on Form 10-K and our consolidated financial statements included on Form S-1 for the years ended December 31, 2003 and 2002. The following tables should be read in conjunction with our financial statements, the related notes thereto and the information contained in “Item 7 - Management’s Discussion and Analysis of Financial Condition and Results of Operations”.
| | Year Ended December 31, |
| | 2002 | 2003 | 2004 | 2005 | 2006 |
| | (in thousands except per share amounts) |
Consolidated Statements of Operations Data: | | | | | |
| Revenue | | | | | |
| Retina | | | | | |
| Revenue from related parties | $94 | $390 | $732 | $81 | $— |
| Revenue from unrelated parties | — | — | 238 | 1,759 | 174 |
| Glaucoma | — | — | — | — | 32 |
| Total revenue | 94 | 390 | 970 | 1,840 | 206 |
| Cost of goods sold | | | | | |
| Retina | | | | | |
| Cost of goods sold to related parties | 81 | 373 | 689 | 43 | — |
| Cost of goods sold to unrelated parties | — | — | 134 | 3,251 | 3,429 |
| Royalty costs | 78 | 109 | 135 | 100 | 100 |
| Glaucoma | — | — | — | — | 19 |
| Gross margin (loss) | (65) | (92) | 12 | (1,554) | (3,342) |
| Operating expenses | | | | | |
General and administrative | 449 | 1,565 | 17,530 | 8,729 | 9,831 |
| Clinical and regulatory | 1,447 | 731 | 3,995 | 5,251 | 5,711 |
| Sales and marketing | — | — | 220 | 2,165 | 1,970 |
| Impairment of goodwill | — | — | — | 147,452 | 65,946 |
| Restructuring charges | — | — | — | — | 820 |
| | 1,896 | 2,296 | 21,745 | 163,597 | 84,278 |
| Other (expenses) income | (921) | (82) | (110) | 1,536 | 1,271 |
Loss before income taxes and cumulative effect of a change in accounting principle | (2,882) | (2,470) | (21,843) | (163,615) | (86,349) |
| Recovery of income taxes | — | — | 24 | 643 | 4,070 |
Loss before cumulative effect of a change in accounting principle | (2,882) | (2,470) | (21,819) | (162,972) | (82,279) |
Cumulative effect of a change in accounting principle | — | — | — | — | 107 |
| Net loss for the year | $(2,882) | $(2,470) | $(21,819) | $(162,972) | $(82,172) |
Per Share Data: | | | | | |
Loss before cumulative effect of a change in accounting principle per share — basic and diluted | $(0.77) | $(0.62) | $(2.96) | $(3.89) | $(1.83) |
Cumulative effect of a change in accounting principle per share — basic and diluted | — | — | — | — | — |
Net loss per share — basic and diluted | $(0.77) | $(0.62) | $(2.96) | $(3.89) | $(1.83) |
Weighted average number of shares used in per share calculations — basic and diluted | 3,735 | 3,977 | 7,370 | 41,931 | 44,980 |
| | As at December 31, |
| | |
| | 2002 | 2003 | 2004 | 2005 | 2006 |
| | (in thousands) |
Consolidated Balance Sheet Data: | | | | | |
| Cash and cash equivalents | $602 | $1,237 | $17,531 | $9,600 | $5,741 |
| Short-term investments | — | — | 42,500 | 31,663 | 9,785 |
Working capital (deficiency) | (1,780) | (2,538) | 58,073 | 44,415 | 13,539 |
| Total assets | 1,038 | 1,868 | 301,601 | 137,806 | 90,404 |
Long-term debt (including current portion due to stockholders) | 1,507 | 3,694 | 517 | 158 | 152 |
Other long-term obligations (including amount classified as current portion of other liability) | ― | ― | ― | ― | 6,421 |
| Total liabilities | 2,693 | 4,134 | 13,502 | 11,765 | 27,999 |
| Minority interest | ― | ― | ― | ― | 1,185 |
| Common stock | 4 | 5 | 42 | 42 | 51 |
Series A Convertible Preferred Stock | 2 | 2 | ― | ― | ― |
Series B Convertible Preferred Stock | 1 | 1 | ― | ― | ― |
Additional paid-in capital | 22,057 | 23,915 | 336,064 | 336,978 | 354,320 |
| Accumulated deficit | (23,718) | (26,188) | (48,007) | (210,979) | (293,151) |
Total stockholders’ equity (deficiency) | (1,655) | (2,266) | 288,098 | 126,041 | 61,220 |
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our consolidated financial statements and related notes, included in Item 8 of this Form 10-K. Unless otherwise specified, all dollar amounts are U.S. dollars.
Overview
We are an ophthalmic therapeutic company in the business of commercializing innovative treatments for age-related eye diseases, including age-related macular degeneration, or AMD, and glaucoma. We also hold a majority interest in a company that is in the process of developing ocular diagnostic technologies. Our core purpose is to improve life through evidence-based medical therapies.
Our product for Dry AMD, the RHEO™ System, is used to perform the Rheopheresis™ procedure, which we refer to under our trade name RHEO™ Therapy. The Rheopheresis™ procedure is a blood filtration procedure that selectively removes molecules from plasma, which is designed to treat Dry AMD, the most common form of the disease.
We conducted a pivotal clinical trial, called MIRA-1, or Multicenter Investigation of Rheopheresis for AMD, which, if successful, was expected to support our application to the U.S. Food and Drug Administration, or FDA, to obtain approval to market the RHEO™ System in the United States. On February 3, 2006, we announced that, based on a preliminary analysis of the data from MIRA-1, MIRA-1 did not meet its primary efficacy endpoint as it did not demonstrate a statistically significant difference in the mean change of Best Spectacle-Corrected Visual Acuity applying the Early Treatment Diabetic Retinopathy Scale, or ETDRS BCVA, between the treated and placebo groups in MIRA-1 at 12 months post-baseline. As expected, the treated group demonstrated a positive result. An anomalous response of the control group is the principal reason why the primary efficacy endpoint was not met. There were subgroups that did demonstrate statistical significance in their mean change of ETDRS BCVA.
Subsequent to the February 3, 2006 announcement, the Company completed an in-depth analysis of the MIRA-1 study data identifying subjects that were included in the intent-to-treat, or ITT, population but who deviated from the MIRA-1 protocol as well as those patients who had documented losses or gains in vision for reasons not related to retinal disease such as cataracts. Those subjects in the ITT population who met the protocol requirements, and who did not exhibit ophthalmic changes unrelated to retinal disease, comprised the modified per-protocol population. In the modified per-protocol analysis, eyes treated with RHEO™ Therapy demonstrated a mean vision gain of 0.8 lines of ETDRS BCVA at 12 months post-baseline, compared to a mean vision loss of 0.1 lines of ETDRS BCVA in the eyes in the placebo group. The result was statistically significant (repeated measure p value = 0.0147). The following table presents a summary of the ETDRS BCVA changes observed 12 months post-baseline in the modified per-protocol analysis of MIRA-1:
| | Treatment Group (n=69) | Placebo Group (n=46) |
| Vision improvement greater or equal to: | | |
| 1 line | 46.4% | 19.6% |
| 2 lines | 27.5% | 8.7% |
3 lines | 8.7% | 2.2% |
| Vision loss greater or equal to: | | |
| 1 line | 11.6% | 23.9% |
| 2 lines | 5.8% | 6.5% |
| 3 lines | 2.9% | 2.2% |
Within the modified per-protocol population with pre-treatment vision worse than 20/40, 50% of RHEO™ Therapy-treated eyes improved, after treatment, to 20/40 or better (which is the required visual acuity to qualify for a driver’s license) 12 months post-baseline, compared to 20% of placebo eyes.
MIRA-1 data support historical clinical and commercial experience with respect to the safety of RHEO™ Therapy, with observed treatment side effects generally being mild, transient and self-limiting.
In light of the MIRA-1 study results, we re-evaluated our Pre-market Approval Application, or PMA, submission strategy and then met with representatives of the FDA on June 8, 2006 in order to discuss the impact the MIRA-1 results would have on our PMA to market the RHEO™ System in the United States. As expected, in light of MIRA-1’s failure to meet its primary efficacy endpoint, the FDA advised us that it will require an additional study of the RHEO™ System to be performed. At that meeting, the FDA confirmed its willingness to allow the substitution, in the new study, of the new polysulfone Rheofilter™ filter for the older cellulose acetate filter which currently forms part of the RHEO™ System. The immediate replacement of the filter avoids the regulatory uncertainties that would arise, were the replacement to take place following receipt of FDA approval. Furthermore, due to manufacturing constraints on the number of cellulose acetate filters that can be produced by their manufacturer, Asahi Kasei Medical Co., Ltd. (formerly Asahi Medical Co., Ltd.), or Asahi Medical, the replacement of the filter in the new trial eliminates the need to continue to build and maintain adequate inventories of the older cellulose acetate filter that the Company had been building and maintaining in preparation for commercial launch. On January 29, 2007, the Company announced that it had obtained Investigational Device Exemption clearance from the FDA to commence the new pivotal clinical trial of the RHEO™ System, called RHEO-AMD, or Safety and Effectiveness in a Multi-center, Randomized, Sham-controlled Investigation for Dry, Non-exudative Age-Related Macular Degeneration (AMD) Using Rheopheresis.
As a result of the announcement on February 3, 2006, the per share price of our common stock as traded on the NASDAQ National Market, or NASDAQ, decreased from $12.75 on February 2, 2006 to close at $4.10 on February 3, 2006. The 10-day average price of the stock immediately following the announcement was $3.65 and reflected a decrease in our market capitalization from $536.6 million on February 2, 2006 to $153.6 million based on the 10-day average share price subsequent to the announcement. On June 12, 2006, we announced that the FDA will require us to perform an additional study of the RHEO™ System. In addition, on June 30, 2006, we announced that we had terminated negotiations with Sowood Capital Management LP (“Sowood”) in connection with a proposed private purchase of approximately $30,000,000 of zero-coupon convertible notes of the Company. The per share price of our common stock decreased subsequent to the June 12, 2006 announcement and again after the June 30, 2006 announcement. Based on the result of the analysis of the data from MIRA-1 and the events that occurred during the second quarter of fiscal 2006, we concluded that there were sufficient indicators of impairment leading to an analysis of our intangible assets and goodwill and resulting in our reporting an impairment charge to goodwill of $65,945,686 and $147,451,758 in the second quarter of 2006 and in the fourth quarter of 2005, respectively.
On September 29, 2004, we signed a product purchase agreement with Veris Health Sciences Inc. (formerly RHEO Therapeutics, Inc.), or Veris, for the purchase and sale of 8,004 treatment sets over the period from October 2004 to December 2005, a transaction valued at $6,003,000, after introductory rebates. However, due to delays in opening its planned number of clinics throughout Canada, Veris no longer required the contracted-for number of treatment sets in the period. We agreed to the original pricing for the reduced number of treatment sets required in the period. In December 2005, by letter agreement, we agreed to the volume and other terms for the purchase and sale of treatment sets and pumps for the period ending February 28, 2006. As at December 31, 2005, the Company had received a total of $1,779,566 from Veris. Included in amounts receivable, net as at December 31, 2005 was $1,049,297 due from Veris for the purchase of additional pumps and treatment sets.
We believed that the announcement on February 3, 2006 made it unlikely that we would be able to collect on amounts outstanding from Veris as at December 31, 2005. This resulted in a provision for bad debts of $1,049,297 during the year ended December 31, 2005, of which $518,852 related to revenue recognized prior to December 31, 2005 and $530,445 related to goods shipped to Veris in December 2005, for which revenue was not recognized. We also recognized an inventory loss of $252,071 during the year ended December 31, 2005, representing the cost of goods shipped to Veris in December 2005 which we do not anticipate will be returned by Veris. During the year ended December 31, 2005, we also fully expensed the C$195,000 advance paid to Veris in connection with clinical trial services to be provided by Veris for MIRA-PS, one of our clinical trials which we have suspended. In addition, we evaluated our ending inventories as at December 31, 2005 on the basis that Veris may not be able to increase its commercial activities in Canada in line with our initial expectations. Accordingly, we set up a provision for obsolescence of $1,990,830 during the year ended December 31, 2005 for treatment sets that will unlikely be utilized prior to their expiration dates.
During the year ended December 31, 2006, we sold a number of treatment sets, with a negotiated discount, to Veris at a price lower than our cost. Accordingly, the price which we charged to Veris, net of a negotiated discount, represents the current net realizable value; therefore, we wrote down the value of our treatment sets by $1,625,000 to reflect their current net realizable value as at December 31, 2006. We also set up an additional provision for obsolescence of $1,679,124 during the year ended December 31, 2006 for treatment sets that will unlikely be utilized prior to their expiration dates.
As at December 31, 2006 and 2005, we had combined inventory reserves of $5,101,394 and $1,990,830, respectively.
In June 2006, Veris returned four pumps which had been sold to it in December 2005. In fiscal 2005, we did not recognize revenue on sales made to Veris in December 2005 and had recorded an inventory loss associated with all sales made to Veris in December 2005. Accordingly, as at December 31, 2006, amounts receivable and the allowance for doubtful account recorded against the amount due from Veris have been reduced by the invoiced amount for the four pumps of $143,520. In addition, the cost of the four pumps returned by Veris, valued at $85,058, was used to reduce the cost of sales in the period.
On November 6, 2006, we amended the product purchase agreement with Veris and agreed to forgive the outstanding amount receivable of $904,101 from Veris which had been owing for the purchase of treatment sets and pumps and for related services delivered or provided to Veris from September 14, 2005 to December 31, 2006. In consideration of the forgiveness of this debt, Veris agreed that we do not owe any amounts whatsoever in connection with (i) our use of the leasehold premises located at 5280 Solar Drive in Mississauga, Ontario or (ii) legal fees and expenses incurred by Veris prior to February 14, 2006 with respect to those trademarks of Veris that were assigned to us on February 14, 2006.
In November 2006, we sold a total of 348 treatment sets to Veris for $73,776, including applicable taxes, payment for which was not received by the Company within the agreed credit period. The sale of these treatment sets was not recognized as revenue during the year ended December 31, 2006 as we believe that Veris would not be able to meet its financial obligations to the Company. In January 2007, we met with the management of Veris and agreed to forgive the outstanding amount receivable of $73,776 which was owing for the purchase of the 348 treatment sets delivered to Veris in November 2006. We also recognized an inventory loss of $60,987 during the year ended December 31, 2006, representing the cost of the 348 treatment sets shipped to Veris in November 2006.
We entered into a new distributorship agreement (the “2006 Distributorship Agreement”), effective October 20, 2006, with Asahi Medical. The 2006 Distributorship Agreement replaced the 2001 distributorship agreement between Asahi Medical and us, as supplemented and amended by the 2003, 2004 and 2005 Memoranda. Pursuant to the 2006 Distributorship Agreement, we have distributorship rights to Asahi Medical's Plasmaflo filter and Asahi Medical's second generation polysulfone Rheofilter filter on an exclusive basis in the United States, Mexico and certain Caribbean countries (collectively, “Territory 1-a”), on an exclusive basis in Canada, on an exclusive basis in Colombia, Venezuela, New Zealand, Australia (collectively, “Territory 2”) and on a non-exclusive basis in Italy.
Pursuant to the 2006 Distributorship Agreement, we will be responsible for obtaining regulatory approvals for the Plasmaflo filter and Rheofilter filter for use in the treatment of AMD in Territory 1-a, Territory 2 and Italy by December 31, 2010 and in Canada by February 28, 2009. With the exception of the FDA approval of the RHEO™ System in the United States, all of such regulatory approvals, when and if obtained, will be held in Asahi Medical’s name. The FDA approval of the RHEO™ System will be held by a special purpose corporation, to be owned as to 51% by Asahi Medical and as to 49% by the Company. Under the 2006 Distributorship Agreement, the Company will be responsible for covering costs relating to the pursuit of regulatory approvals in Territory 1-a, Canada and Territory 2, and the Company and Asahi Medical will share the costs relating to the pursuit of regulatory approval in Italy. In addition, provided that certain conditions are met, Asahi Medical will be obligated to contribute $3,000,000 toward the cost of RHEO-AMD, our new pivotal clinical trial of the RHEO™ System which is intended to support our PMA.
With respect to the United States, subject to early termination under certain circumstances, the 2006 Distributorship Agreement has a term which will end 10 years following the date on which FDA approval to market the RHEO™ System in the United States is received and contemplates successive one-year renewal terms thereafter.
We are subject to certain minimum purchase requirements in each of the territories covered by the 2006 Distributorship Agreement.
On September 1, 2006, we completed the acquisition of Solx, Inc. (“SOLX”) for a total purchase price of $29,068,443 which includes acquisition-related transaction costs of $851,279. SOLX is a Boston University Photonics Center-incubated company that has developed a system for the treatment of glaucoma called the SOLX Glaucoma System. The results of SOLX’s operations have been included in our consolidated financial statements since September 1, 2006. The SOLX Glaucoma Treatment System is a next-generation treatment platform designed to reduce intra-ocular pressure, or IOP, without a bleb, thus avoiding its related complications. The SOLX Glaucoma System consists of the SOLX 790 Laser, a titanium sapphire laser used in laser trabeculoplasty procedures, and the SOLX Gold Shunt, a 24-karat gold, ultra-thin drainage device designed to bridge the anterior chamber and the suprachoroidal space in the eye, using the pressure differential that exists naturally in the eye in order to reduce IOP.
Both the SOLX 790 Laser and the SOLX Gold Shunt are currently the subject of randomized, multi-center clinical trials. The results of these clinical trials will be used in support of applications to the FDA for a 510(k) clearance for each of the SOLX 790 Laser and the SOLX Gold Shunt, the receipt of which, if any, will enable the Company to market and sell these products in the United States. Currently, our intention is to file the application for a 510(k) clearance for the SOLX 790 Laser by the end of 2007 and to file the application for a 510(k) clearance for the SOLX Gold Shunt by the end of the second quarter of 2008.
The acquisition of SOLX represents an expansion of the Company’s ophthalmic product portfolio beyond the RHEO™ procedure for Dry AMD. This expansion or diversification has become a corporate objective, and in light of the delay in the U.S. commercial launch of the RHEO™ System, we accelerated these plans. Our focus is on age-related eye diseases like AMD and glaucoma as they are expected to be the fastest growing segments of eye care over the next 10 years.
On November 30, 2006, we acquired 50.1% of the capital stock of OcuSense, Inc., or OcuSense, measured on a fully diluted basis, for a total purchase price of $4,171,098 which includes acquisition-related transaction costs of $171,098. The Company will make additional payments totaling $4,000,000 upon the attainment of two pre-defined milestones by OcuSense prior to May 1, 2009, The contingent payments totaling $4,000,000 were not included in the determination of the purchase price or recorded as a liability as the achievement of the two pre-defined milestones prior to May 1, 2009 is not guaranteed.
OcuSense is a San Diego-based company that is in the process of developing technologies that will enable eye care practitioners to test, at the point-of-care, for highly sensitive and specific biomarkers using nanoliters of tear film. The results of OcuSense’s operations have been included in our consolidated financial statements since November 30, 2006. OcuSense’s first product, which is currently under development, is a hand-held tear film osmolarity test for the diagnosis and management of dry eye syndrome, or DES, known as the TearLab™ test for DES. It is estimated that over 90 million people in the United States suffer from DES. The anticipated innovation of the TearLab™ test for DES will be its ability to measure precisely and rapidly certain biomarkers in nanoliter volumes of tear samples, using inexpensive hardware. Historically, eye care researchers have relied on expensive instruments to perform tear biomarker analysis. In addition to their cost, these conventional systems are slow, highly variable in their measurement readings and not waived by the FDA under the Clinical Laboratory Improvement Amendments, or CLIA.
The TearLab™ test for DES will require the development of the following three components: (1) the TearLab™ disposable, which is a single-use microfluidic cartridge; (2) the TearLab™ pen, which is a hand-held interface with the TearLab™ disposable; and (3) the TearLab™ reader, which is a physical housing for the TearLab™ pen connections and measurement circuitry. OcuSense is currently engaged actively in industrial, electrical and software design efforts for the three components of the TearLab™ test for DES and, to these ends, is working with two expert partners, both based in Melbourne, Australia, one of which is a leader in biomedical instrument development and the other of which is a leader of customized microfluidics.
OcuSense’s objective is to complete product development of the TearLab™ test for DES by the end of 2007. Following the completion of product development and subsequent clinical trials, OcuSense intends to seek a 510(k) clearance and a CLIA waiver from the FDA for the TearLab™ test for DES.
On November 30, 2006, we announced that Elias Vamvakas, our Chairman and Chief Executive Officer, had agreed to provide us with a standby commitment to purchase convertible debentures of the Company (“Convertible Debentures”) in an aggregate maximum amount of $8,000,000 (the “Total Commitment Amount”). Pursuant to the Summary of Terms and Conditions, executed and delivered as of November 30, 2006 by the Company and Mr. Vamvakas, during the 12-month commitment term commencing on November 30, 2006, upon no less than 45 days’ written notice by the Company to Mr. Vamvakas, Mr. Vamvakas was obligated to purchase Convertible Debentures in the aggregate principal amount specified in such written notice. A commitment fee of 200 basis points was payable by the Company on the undrawn portion of the total $8,000,000 commitment amount. Any Convertible Debentures purchased by Mr. Vamvakas would have carried an interest rate of 10% per annum and would have been convertible, at Mr. Vamvakas’ option, into shares of the Company’s common stock at a conversion price of $2.70 per share. The Summary of Terms and Conditions of the standby commitment further provided that if the Company closed a financing with a third party, whether by way of debt, equity or otherwise and there are no Convertible Debentures outstanding, then, the Total Commitment Amount was to be reduced automatically upon the closing of the financing by the lesser of: (i) the Total Commitment Amount; and (ii) the net proceeds of the financing. On February 6, 2007, the Company raised gross proceeds in the amount of $10,016,000 in a private placement of shares of its common stock and warrants. The Total Commitment Amount was therefore reduced to zero, thus effectively terminating Mr. Vamvakas’ standby commitment. No portion of the standby commitment was ever drawn down by the Company, and the Company paid Mr. Vamvakas a total of $29,808 in commitment fees in February 2007.
Our results of operations for the year ended December 31, 2006 were impacted by our adoption of Statement of Financial Accounting Standards (“SFAS”) No. 123R (revised 2004), “Share-Based Payments” (“SFAS No. 123R”), which requires us to recognize a non-cash expense related to the fair value of our stock-based compensation awards. We elected to use the modified prospective transition method of adoption requiring us to include this stock-based compensation charge in our results of operations beginning on January 1, 2006 without restating prior periods to include stock-based compensation expense. Of the $2,221,133 stock-based compensation expense recognized during the year ended December 31, 2006, $1,442,023 is included in general and administrative expenses, $237,567 in clinical and regulatory expenses and $541,543 in sales and marketing expenses. This method also required us to estimate forfeitures as of the effective date of adoption of SFAS No. 123R and to eliminate any compensation cost previously recognized in income for periods before the effective date of adoption. This compensation cost previously recognized in income should be recognized as the cumulative effect of a change in accounting principle as of the required effective date. We also recognized $107,045 as the cumulative effect of a change in accounting principle reflecting the impact of our estimated forfeitures of outstanding awards as of January 1, 2006.
At the annual meeting of stockholders of the Company held on June 23, 2006, our stockholders approved the re-pricing of all then out-of-the-money stock options of the Company. Consequently, the exercise price of all outstanding stock options that, on June 23, 2006, was greater than $2.05, being the weighted average trading price of our common stock on NASDAQ during the five-trading day period immediately preceding June 23, 2006, was adjusted downward to $2.05. 2,585,000 of the outstanding stock options with a weighted average exercise price of $8.42 were affected by the re-pricing. SFAS No. 123R treats the re-pricing of equity awards as a modification of the original award and provides that such a modification is an exchange of the original award for a new award. SFAS No. 123R considers the modification to be the repurchase of the old award for a new award of equal or greater value, incurring additional compensation cost for any incremental value. This incremental difference in value is measured as the excess, if any, of the fair value of the modified award determined in accordance with the provisions of SFAS No. 123R over the fair value of the original award immediately before its terms are modified, measured based on the share price and other pertinent factors at that date. SFAS No. 123R provides that this incremental fair value, plus the remaining unrecognized compensation cost from the original measurement of the fair value of the old option, must be recognized over the remaining vesting period. Of the 2,585,000 options affected by the re-pricing, 1,401,073 were vested as at December 31, 2006. Therefore, additional compensation cost of $423,338 for the 1,401,073 options was recognized and is included in the stock-based compensation expense for the year ended December 31, 2006. The remaining unrecognized incremental fair value of $169,057 plus the compensation cost from the original measurement of the fair value of the old options of $2,607,496, which totaled $2,776,553 in unrecognized compensation expense as at December 31, 2006, is expected to be amortized over a weighted average vesting period of 2.3 years.
In accordance with SFAS No. 123R, we also recorded a compensation expense of $3,363 in the second quarter of fiscal 2006 as our board of directors approved accelerating the vesting of 1,250 unvested stock options granted to a terminated employee on April 28, 2006. SFAS No. 123R treats such a modification as a cancellation of the original unvested award and the grant of a new fully vested award as of that date.
Prior to the adoption of SFAS No. 123R, we applied the provisions of SFAS No. 123, “Accounting for Stock-Based Compensation” (“SFAS No. 123”), which allowed companies either to expense the estimated fair value of employee stock options or to follow the intrinsic value method set forth in Accounting Principles Board Opinion No. 25, “Accounting for Stock Issued to Employees” (“APB No. 25”), but required companies to disclose the pro forma effects on net loss as if the fair value of the options had been expensed. We elected to apply APB No. 25 in accounting for employee stock options. As required by SFAS No. 123, prior to the adoption of SFAS No. 123R, we provided pro forma net loss and pro forma net loss per share disclosures for stock-based awards as if the fair value of the options had been expensed.
As at December 31, 2006, $3,978,530 of total unrecognized compensation cost related to stock options is expected to be recognized over a weighted-average period of 2.53 years.
Recent Development
On February 1, 2007, we entered into a Securities Purchase Agreement (the “Securities Purchase Agreement”) with certain institutional investors, pursuant to which we agreed to issue to the investors an aggregate of 6,677,333 shares of our common stock (the “Shares”) and five-year warrants exercisable into an aggregate of 2,670,933 shares of our common stock (the “Warrants”). The per share purchase price of the Shares is $1.50, and the per share exercise price of the Warrants is $2.20, subject to adjustment. The Warrants will become exercisable on August 6, 2007. Pursuant to the Securities Purchase Agreement, on February 6, 2007, we issued the Shares and the Warrants. The gross proceeds of sale of the Shares totaled $10,016,000 (less transaction costs of approximately $750,000). On February 6, 2007, we also issued to Cowen and Company, LLC a warrant exercisable into an aggregate of 93,483 shares of our common stock (the “Cowen Warrant”) in part payment of the placement fee payable to Cowen and Company, LLC for the services it had rendered as the placement agent in connection with the sale of the Shares and the Warrants. All of the terms and conditions of the Cowen Warrant (other than the number of shares of our common stock into which the Cowen Warrant is exercisable) are identical to those of the Warrants.
RESULTS OF OPERATIONS
Revenues, Cost of Sales and Gross Margin
For the years ended December 31,
(in thousands)
| | | 2006 | | Change | | 2005 | | Change | | 2004 |
| | | | | | | | | | | |
| Retina revenue | | | | | | | | | | |
| Sales to related parties | $ | — | | N/M* | $ | 81 | | (89)% | $ | 732 |
| Sales to unrelated parties | | 174 | | (90)% | | 1,759 | | 639% | | 238 |
| Glaucoma revenue | | 32 | | N/M* | | — | | — | | — |
| Total revenues | $ | 206 | | (89)% | $ | 1,840 | | 90% | $ | 970 |
| | | | | | | | | | | |
| Retina cost of sales | | | | | | | | | | |
| Cost of sales to related parties | $ | — | | N/M* | $ | 43 | | (94)% | $ | 689 |
| Cost of sales to unrelated parties | | 3,429 | | 5% | | 3,251 | | 2,326% | | 134 |
| Royalty costs | | 100 | | — | | 100 | | (26)% | | 135 |
| Glaucoma cost of sales | | 19 | | N/M* | | — | | — | | — |
| Total cost of sales | $ | 3,548 | | 5% | $ | 3,394 | | 254% | $ | 958 |
| | | | | | | | | | | |
| Retina gross margin (loss) | $ | (3,355) | | (116)% | $ | (1,554) | | (13,050)% | $ | 12 |
| Percentage of retina revenue | | (1,928)% | | (1,844) pts | | (84)% | | (85) pts | | 1% |
| Glaucoma gross margin | | 13 | | N/M* | | — | | — | | — |
| Percentage of glaucoma revenue | | 41% | | N/M* | | — | | — | | — |
| Total gross margin | | (3,342) | | (115)% | | (1,554) | | (13,050)% | | 12 |
| Percentage of total revenue | | (1,622)% | | (1,538) pts | | (84)% | | (85) pts | | 1% |
| *N/M - Not meaningful | | | | | | | | | | |
Revenues
Retina Revenue
Retina revenue consists of revenue generated from the sale of components of the RHEO™ System which consists of the OctoNova pump and the disposable treatment sets, which include two disposable filters and applicable tubing.
Subsequent to the Company’s February 3, 2006 announcement that MIRA-1 had not met its primary efficacy endpoint, our sole customer, Veris, halted purchases while the Company completed its in-depth analysis of the MIRA-1 study data. The modified per-protocol analysis of the MIRA-1 data support historical clinical and commercial experience with respect to the safety of RHEO™ Therapy, with observed treatment side effects generally being mild, transient and self-limiting. Based on the results of the analysis and in line with our continued support of Veris in Canada, we agreed that all sales of treatment sets made to Veris in fiscal 2006 will be sold at a negotiated discounted price of $200 per treatment set, which is lower than our cost. We also agreed that payment for the treatment sets sold subsequent to June 2006 must be received by the Company within 60 days of shipment. In November 2006, we sold a total of 348 treatment sets to Veris, payment for which was not received by the Company within the agreed credit period. The sale of these treatment sets has not been included in revenue for the year ended December 31, 2006 as we do not expect to receive payment from Veris for these treatment sets.
During fiscal 2006, as compared with fiscal 2005, revenues decreased significantly primarily due to reduced sales of components of the RHEO™ System to Veris as a result of our February 3, 2006 announcement that MIRA-1 did not meet its primary efficacy endpoint. In addition, included in revenues in fiscal 2005 are sales made to RHEO Clinic Inc., a subsidiary of TLC Vision Corporation (“TLC Vision”) and a related party, for which we reported revenues of $81,593 in the period. RHEO Clinic Inc. has since ceased the treatment of commercial patients and is therefore no longer a source of revenue for us.
During fiscal 2005, as compared with fiscal 2004, revenues increased, due primarily to increased sales of treatment sets, pumps and services to Veris, and reflect the impact of the acquisition of TLC Vision’s 50% ownership interest in OccuLogix, L.P.
Glaucoma Revenue
Glaucoma revenue consists of revenue generated from the sale of components of the SOLX Glaucoma System.
On September 1, 2006, the Company completed the acquisition of SOLX, and the results of SOLX’s operations have been included in our consolidated financial statements since that date. Revenue therefore includes the sale of SOLX Gold Shunts from September 1, 2006. There was no comparative revenue during the years ended December 31, 2005 and 2004.
Cost of Sales
Cost of sales includes costs of goods sold and royalty costs. Our cost of goods sold consists primarily of costs for the manufacture of the RHEO™ System and the SOLX Glaucoma System, including the costs we incur for the purchase of component parts from our suppliers, applicable freight and shipping costs, fees related to warehousing, logistics inventory management and recurring regulatory costs associated with conducting business and ISO certification.
Retina Cost of Sales
During fiscal 2006, we sold a total of 1,207 treatment sets to Veris at a price, net of negotiated discounts, which was lower than our cost. As Veris is currently our sole customer for the RHEO™ System treatment sets, the price at which we sold the treatment sets to Veris represents our inventory’s current net realizable value, and therefore, we have written down the value of the treatment sets to reflect this net realizable value. Included in cost of sales is $1,625,000 which reflects the write-down of the treatment sets to their net realizable value. In addition, we evaluated our ending inventories as at December 31, 2006 and 2005 on the basis that Veris may not be able to increase its commercial activities in Canada in line with our initial expectations. Accordingly, we set up a provision for obsolescence of $1,990,830 during the year ended December 31, 2005, and an additional provision for obsolescence of $1,679,124 during the year ended December 31, 2006 for treatment sets that will unlikely be utilized prior to their expiration dates.
During the year ended December 31, 2006, as compared with the corresponding period in fiscal 2005, cost of sales increased due primarily to the charge of $1,625,000 which reflects the write-down of our inventory of treatment sets to its net realizable value. There was no comparative expense in fiscal 2005. Cost of sales for the years ended December 31, 2006 and 2005 includes a provision for obsolescence of $1,679,124 and 1,990,830, respectively, for treatment sets that will unlikely be utilized prior to their expiration dates.
Cost of sales increased during the year ended December 31, 2005, as compared with the corresponding period in 2004, as a result of the increase in sales from the prior period and the impact of the acquisition of TLC Vision’s 50% ownership interest in OccuLogix, L.P. Based on our evaluation of our ending inventories as at December 31, 2005 we set up a provision for obsolescence of $1,990,830 for treatment sets that are unlikely to be utilized prior to their expiration dates. Also included in cost of sales expenses for the year ended December 31, 2005 is $252,071 which reflects the inventory loss associated with treatment sets and pumps shipped to Veris in December 2005 for which revenue was not recognized due to the likelihood that the customer will not return the products shipped and would not be able to pay for the amounts invoiced. There was no comparative expense in fiscal 2004.
Glaucoma Cost of Sales
Cost of sales includes the cost of SOLX Gold Shunts sold during the four-month period ended December 31, 2006. There was no comparative expense during the years ended December 31, 2005 and 2004 as we completed the acquisition of SOLX on September 1, 2006.
Gross Margin
Retina Gross Margin
During fiscal 2006 as compared with fiscal 2005, our retina gross margin decreased 1,844 percentage points due to reduced sales in fiscal 2006 and increased cost of sales due to the inventory write-down and the provision for obsolescence recorded in the period.
Retina gross margin on sales for the year ended December 31, 2005, as compared with the corresponding period in 2004, decreased by 85 percentage points due primarily to the impact of the provision for inventory obsolescence of $1,990,830 and the inventory loss of $252,071 recorded during the year. Gross margin on sales was 1% for the year ended December 31, 2004.
Glaucoma Gross Margin
Gross margin on the sale of SOLX Gold Shunts was 40% during the four months ended December 31, 2006.
Operating Expenses
For the years ended December 31,
(in thousands)
| | | 2006 | | Change | | 2005 | | Change | | 2004 |
| | | | | | | | | | | |
| General and administrative | $ | 9,831 | | 13% | $ | 8,729 | | (50)% | $ | 17,530 |
| Clinical and regulatory | | 5,711 | | 9% | | 5,251 | | 31% | | 3,995 |
| Sales and marketing | | 1,970 | | (9)% | | 2,165 | | 884% | | 220 |
| Impairment of goodwill | | 65,946 | | (55)% | | 147,452 | | N/M* | | — |
| Restructuring charges | | 820 | | N/M* | | — | | — | | — |
| Total operating expenses | $ | 84,278 | | (48)% | $ | 163,597 | | 652% | $ | 21,745 |
| *N/M - Not meaningful | | | | | | | | |
General and Administrative Expenses
General and administrative expenses increased by $1,101,996 during the year ended December 31, 2006, as compared with the corresponding period of fiscal 2005, due to an increase of $1,212,385 in stock-based compensation expense associated with the adoption of SFAS No. 123R beginning January 1, 2006 which requires us to recognize a non-cash expense related to the fair value of our stock-based compensation awards. General and administrative expenses also include a charge for $1,032,545 which represents the amortization of the intangible assets acquired during fiscal 2006 upon the acquisition of SOLX and OcuSense. These increases were partially offset by the decrease in employee and travel costs of $366,472 due in part to the grant of options to an employee in lieu of salary. Professional fees and fees associated with compliance with Section 404 of the Sarbanes-Oxley Act of 2002 also decreased by $819,468 while directors’ fees decreased by $50,207 due to the grant of options to directors in lieu of their annual fees payable for board and committee memberships.
General and administrative expenses decreased by $8,800,563 during the year ended December 31, 2005, as compared with the corresponding period of fiscal 2004. This decrease is due primarily to the requirement to expense the intrinsic value of options granted in December 2003 over the vesting period of these options. All of these options became fully vested upon the Company’s initial public offering, and, therefore, $15,392,323, reflecting the remaining unamortized balance of stock-based compensation charges as of December 31, 2003, was expensed in the year ended December 31, 2004. This decrease was partially offset by increased professional fees to establish agreements, to review and amend existing contracts, the fees associated with compliance with Section 404 of the Sarbanes-Oxley Act of 2002 as well as other public company costs, the cost of several key executives and employees hired part way through the third quarter of 2004 and amortization expense of the intangible asset acquired on the purchase of TLC Vision’s 50% interest in OccuLogix, L.P.
We are continuing to focus our efforts on achieving additional operating efficiencies by reviewing and improving upon our existing business processes and cost structure.
Clinical and Regulatory Expenses
Clinical and regulatory expenses increased by $460,338 during the year ended December 31, 2006, as compared with the corresponding prior year period, due to an increase in clinical trial expenses associated with the LEARN trials, or Long-term Efficacy in AMD from Rheopheresis in North America trials, other clinical trials and the cost of SOLX’s clinical and regulatory expenses during the period. Stock-based compensation expense also increased by $100,924 during the year ended December 31, 2006 as a result of the adoption of SFAS No. 123R beginning January 1, 2006.
During the year ended December 31, 2005, clinical and regulatory expenses increased by $1,255,525, as compared with the corresponding period in fiscal 2004, as a result of increased activities associated with the MIRA-1, LEARN and other clinical trials. Also included in clinical trial expenses for the year ended December 31, 2005 is the advance payment of C$195,000 or $165,661 made to Veris for the provision of clinical trial services in connection with our MIRA-PS trial which we have suspended. This unrecoverable amount has been fully expensed in the year ended December 31, 2005. No other adjustments were made as a result of the announcement.
Our goal is to establish the RHEO™ Therapy as the leading treatment for Dry AMD in North America and to establish the SOLX Glaucoma System as a unique, new surgery of choice for glaucoma. Accordingly, we expect clinical and regulatory expenses to increase in the future as we are required to conduct RHEO-AMD, an additional study of the RHEO™ System in order to support our PMA application to the FDA. We also have to complete ongoing studies of the SOLX 790 Laser and the SOLX Gold Shunt in order to obtain 510(k) approval to market them in the United States. In addition, we have to complete product development of OcuSense’s TearLab™ test for DES. Following the completion of product development, OcuSense will have to conduct clinical trials in order to seek a 510(k) clearance and a CLIA waiver from the FDA for the TearLab™ test for DES.
Sales and Marketing Expenses
Sales and marketing expenses decreased by $195,699 during the year ended December 31, 2006, as compared with the prior period in fiscal 2005, due to reduced employee and travel costs during the period of $371,976 and a decrease in marketing expenses of $344,067 due to reduced marketing efforts in the year following the announcement of MIRA-1 results. Bad debt expense also decreased during the year ended December 31, 2006 by $510,913 as the Company only recognized revenue on sale of treatment sets sold to its sole customer, Veris, on receipt of payment. These decreases in costs were offset by increased stock-based compensation expense of $541,043 associated with the adoption of SFAS No. 123R beginning January 1, 2006 and increased fees and expenses of the Company’s Scientific Advisory Board members of $210,456. Sales and marketing expenses also include SOLX’s sales and marketing expenses of $269,114 for trade shows and other marketing public relations during the four-month period ended December 31, 2006. There were no comparable expenses in the prior year period.
Sales and marketing expenses increased by $1,945,781 during the year ended December 31, 2005, as compared with the corresponding prior year period, since virtually all sales and marketing expenses had been incurred by OccuLogix, L.P. prior to our acquisition of TLC Vision’s 50% interest in OccuLogix, L.P. In the third and fourth quarters of 2004, we hired three new employees to begin establishing sales and marketing efforts to promote the use of the RHEO™ System in Canada and, upon FDA approval, in the United States. Sales and marketing expenses consist primarily of costs of establishing sales and marketing efforts to promote the use of the RHEO™ System in Canada and, upon FDA approval, in the United States. Sales and marketing expenses for the year ended December 31, 2005 include bad debt expense of $518,852 and reflect the allowance for doubtful amounts due to us from Veris for the purchase of treatment sets, pumps and other services.
The cornerstone of our sales and marketing strategy to date has been to increase awareness of our products among eye care professionals and, in particular, the key opinion leaders in the eye care professions. We will continue to develop and execute our conference and podium strategy to ensure visibility and evidence-based positioning of the RHEO™ System, the SOLX Glaucoma System and the TearLab™ test for DES among eye care professionals.
Impairment of Goodwill
The decrease in our stock price subsequent to the February 3, 2006 announcement of the MIRA-1 trial's failure to meet its primary efficacy endpoint, the June 12, 2006 announcement of the outcome of our meeting with the FDA and the June 30, 2006 announcement of the termination of negotiations with Sowood were identified as indicators of impairment which led to an analysis of our intangible assets and goodwill which, in turn, resulted in the reporting of an impairment charge of $65,946,686 and $147,451,758 during the years ended December 31, 2006 and 2005, respectively. The impairment of goodwill charge represents the write-down of the value of goodwill acquired on the purchase of TLC Vision's 50% interest in OccuLogix, L.P. on December 8, 2004 to nil as at December 31, 2006. There was no comparable charge in the year ended December 31, 2004.
Restructuring Charges
In accordance with SFAS No. 146, “Accounting for Costs Associated with Exit or Disposal Activities” (“SFAS No. 146”), we recognized a total of $819,642 in restructuring charges during the year ended December 31, 2006. The Company implemented a number of structural and management changes designed both to support the continued development of the RHEO™ System and to execute the Company’s accelerated diversification strategy within ophthalmology. The restructuring charge of $819,642, recorded in the year ended December 31, 2006, consists solely of severance and benefit costs related to the termination of a total of 12 employees at both the Company’s Mississauga and Palm Harbor offices. The severance and benefit costs were fully paid by December 31, 2006. There was no comparable expense in the years ended December 31, 2005 and 2004.
Other Income, Net
For the years ended December 31,
(in thousands)
| | | 2006 | | Change | | 2005 | | Change | | 2004 |
| | | | | | | | | | | |
| Interest income | $ | 1,370 | | (27)% | $ | 1,593 | | 2,555% | $ | 60 |
| Interest and amortization of discount on future payment expense | | (288) | | N/M* | | — | | N/M* | | (24) |
| Other income (expense) | | 31 | | 154% | | (57) | | 61% | | (146) |
| Minority interests | | 158 | | N/M* | | — | | — | | — |
| | $ | 1,271 | | (17)% | $ | 1,536 | | 1,496% | $ | (110) |
| * N/M - Not meaningful | | | | | | | | |
Interest Income
Interest income consists of interest income earned in the current period and the corresponding prior periods as a result of the Company’s cash and short-term investment position following the raising of capital in the Company’s initial public offering in December 2004.
Interest and Amortization of Discount on Future Payment Expense
In connection with the acquisition of SOLX on September 1, 2006, we remain indebted to the former stockholders of SOLX in an aggregate amount of up to $13,000,000 for the outstanding portion of the purchase price of SOLX. $5,000,000 of this amount is payable in cash on the second anniversary of the September 1, 2006 closing. The $5,000,000 has been recorded as a long-term liability at its present value, discounted at the incremental borrowing rate of the Company as at August 1, 2006. The difference between the discounted value and the $5,000,000 payable is being amortized using the effective yield method over the two-year period with the monthly expense being charged as an interest expense in the Company’s consolidated statement of operations. Interest and amortization of discount on future payment expense for the year ended December 31, 2006 consists primarily of the amortization expense for the four months from September to December 2006. There was no comparable expense in the years ended December 31, 2005 and 2004.
Interest and amortization of discount on future payment expense for the year ended December 31, 2004 consists of interest expense on certain debt prior to its repayment by the Company on December 22, 2004.
Other Income (Expense)
Other income for the year ended December 31, 2006 consists primarily of foreign exchange gain of $37,316 due to exchange rate fluctuations on the Company’s foreign currency transactions. This gain was offset by miscellaneous tax expense of $6,449 during the year ended December 31, 2006. Other expense was $57,025 for the year ended December 31, 2005 and consists of a provision for subscription receivable of $34,927 and miscellaneous tax expense of $23,021.
Other expense was $145,925 for the year ended December 31, 2004 due to the expense of the $100,000 owed to Apheresis Technologies, Inc. in accordance with the amended distribution services agreement. The agreement has since been terminated. Also included in other expense for the year ended December 31, 2004 is foreign exchange loss of $43,548.
Minority Interest
Minority interest is from our acquisition of 50.1% of the capital stock of OcuSense, on a fully diluted basis, on November 30, 2006. The results of OcuSense’s operations have been included in our consolidated financial statements since that date. Income from minority interest of $157,624 for the year ended December 31, 2006 relates to the loss reported by OcuSense in which the Company has a shared interest with minority partners.
Recovery of Income Taxes
For the years ended December 31,
(in thousands)
| | | 2006 | | Change | | | 2005 | | Change | | 2004 |
| | | | | | | | | | | | |
| Recovery of income taxes | $ | 4,070 | | 534% | | $ | 642 | | 2,575% | $ | 24 |
Recovery of Income Taxes
Recovery of income taxes increased by $3,427,966 during the year ended December 31, 2006, as compared with the prior period in 2005. This increase is due primarily to a deferred tax recovery amount of $2,784,000 associated with the recognition of a deferred tax asset from the availability of 2006 net operating losses in the United States which may be utilized to reduce taxes in future years.
Recovery of income taxes for the years ended December 31, 2006, 2005 and 2004 also includes the amortization of the deferred tax liability which was recorded based on the difference between the fair value of intangible assets acquired and their tax bases. The increase in the amount recorded during the year ended December 31, 2006 as compared with the corresponding period in fiscal 2005 is due to the additional deferred tax liability recorded upon the acquisition of SOLX and OcuSense, being the difference between the fair value of the intangible assets acquired by the Company upon its acquisition of SOLX and OcuSense and their tax bases. The deferred tax liability totaling $23,462,064 is being amortized over an average period of 11.98 years, the estimated weighted-average useful life of the intangible assets.
Cumulative Effect of a Change in Accounting Principle
For the years ended December 31,
(in thousands)
| | | 2006 | | Change | | | 2005 | | Change | | | 2004 |
| | | | | | | | | | | | | |
| Cumulative effect of a change in accounting principle | $ | 107 | | N/M* | | $ | — | | — | | $ | — |
| *N/M - Not meaningful | | | | | | | | | | |
Cumulative Effect of a Change in Accounting Principle
The cumulative effect of a change in accounting principle reflects the impact of our estimated forfeitures of outstanding stock option awards as of January 1, 2006. On January 1, 2006, the effective date of adopting SFAS No. 123R, we were required to estimate the number of forfeitures of our outstanding awards as of the effective date. Consolidated balance sheet amounts related to any compensation cost for these estimated forfeitures previously recognized in prior periods before the adoption of SFAS No. 123R have to be eliminated and recognized in income as the cumulative effect of a change in accounting principle as of the effective date. The compensation cost previously recognized in prior periods before the adoption of SFAS No. 123R relates to compensation expense associated with non-employee stock options.
LIQUIDITY AND CAPITAL RESOURCES
As at December 31,
(in thousands)
| | | 2006 | | 2005 | | Change |
| | | | | | | |
| Cash and cash equivalents | $ | 5,741 | $ | 9,600 | $ | (3,859) |
| Short-term investments | | 9,785 | | 31,663 | | (21,878) |
| Total cash and cash equivalents and short-term investments | $ | 15,526 | $ | 41,263 | $ | (25,737) |
| | | | | | | |
| Percentage of total assets | | 17% | | 30% | | (13) pts |
| Working capital | $ | 13,539 | $ | 44,415 | $ | (30,876) |
In December 2004, the Company raised $67,200,000 of gross cash proceeds (less issuance costs of $7,858,789) in an initial public offering of shares of its common stock. Immediately prior to the offering, the primary source of the Company’s liquidity was cash raised through the issuance of debentures.
On February 6, 2007, the Company raised gross proceeds in the amount of $10,016,000 (less issuance costs of approximately $750,000) in a private placement of shares of its common stock and warrants.
To date, cash has been primarily utilized to finance increased infrastructure costs, to accumulate inventory and to fund costs of the MIRA-1, LEARN, RHEO-AMD and other clinical trials and, more recently to acquire SOLX and OcuSense in line with our diversification strategy. We expect that, in the future, we will use our cash resources to continue to fund our diversification strategy, the development of our infrastructure and to conduct RHEO-AMD and complete on-going clinical trials. In addition, we will use our cash resources to fund the ongoing clinical trials of the SOLX Glaucoma System, the completion of product development of OcuSense’s TearLab™ test for DES and clinical trials that will be required for the TearLab™ test for DES. In addition, in connection with the acquisition of SOLX on September 1, 2006, we remain indebted to the former stockholders of SOLX in an aggregate amount of up to $13,000,000 for the outstanding portion of the purchase price of SOLX. We also remain indebted to OcuSense in an aggregate amount of up to $4,000,000 for the outstanding portion of the purchase price of the capital stock of OcuSense that we acquired on November 30, 2006. Furthermore, we are legally committed to make an additional equity investment of $3,000,000 upon receipt, if any, from the FDA of a 510(k) clearance for the TearLab™ test for DES and another additional equity investment of $3,000,000 upon receipt, if any, from the FDA of a CLIA waiver for the TearLab™ test for DES.
Changes in Cash Flows
Years ended December 31,
(in thousands)
| | | 2006 | | Change | | | 2005 | | Change | | | 2004 | |
| | | | | | | | | | | | | | |
| Cash used in operating activities | $ | (14,548) | $ | 4,162 | | $ | (18,710) | $ | (13,328) | | $ | (5,382) | |
| Cash provided by (used in) investing activities | | 10,418 | | (33) | | | 10,451 | | 53,879 | | | (43,428) | |
| Cash provided by financing activities | | 271 | | (57) | | | 328 | | (64,776) | | | 65,104 | |
| Net (decrease) increase in cash and cash equivalents during the year | $ | (3,859) | $ | 4,072 | | $ | (7,931) | $ | (24,225) | | $ | 16,294 | |
Cash Used in Operating Activities
Net cash used to fund our operating activities during the year ended December 31, 2006 was $14,548,344. Net loss during the year was $82,171,548. The non-cash charges which comprise a portion of the net loss during that period consisted primarily of the goodwill impairment of $65,945,686 and the amortization of intangible assets, fixed assets, patents and trademarks, discounts on future cash payments and premium/discounts on investments of $3,277,488 netted by applicable deferred income taxes of $4,065,962 and minority interest of $157,624. Additional non-cash charges consist of $2,221,133 in stock-based compensation charges netted by the cumulative effect of a change in accounting principle of $107,045.
The net change in non-cash working capital balances related to operations for the years ended December 31, 2006, 2005 and 2004 consists of the following:
| | Years ended December 31, |
| | 2006 $ | 2005 $ | 2004 $ |
| | | | |
| Due to related party | (5,065) | 13,291 | 110,749 |
| Amounts receivable | 390,634 | (82,810) | (222,218) |
| Inventory | 2,250,554 | (3,431,743) | (136,527) |
| Prepaid expenses | 247,361 | (322,455) | (324,353) |
| Deposit | (5,551) | 4,105 | (8,996) |
| Accounts payable | (1,225,575) | 301,457 | 26,548 |
| Accrued liabilities | (1,155,335) | (563,925) | 2,511,897 |
| Deferred revenue and rent inducement | — | (485,047) | (152,153) |
| Due to stockholders | (5,827) | (358,523) | (931,652) |
| Other current assets | 18,332 | — | — |
| | 509,528 | (4,925,650) | 873,295 |
| · | Amounts receivable decreased due primarily to the receipt of accrued interest receivable on investments and the refund of sales taxes received in 2006. |
| · | Decrease in inventory balance reflects the write-down of inventory and the provision for obsolescence offset by the purchase of additional OctoNova pumps during the first quarter of fiscal 2006 to complete outstanding purchase obligations in line with supplier expectations. |
| · | Decrease in prepaid expenses is primarily due to the utilization of advances paid to various organizations involved in the MIRA-1 and related clinical trials. |
| · | Accounts payable and accrued liabilities decreased as payments are being made for costs associated with the Company’s activities. |
| · | The decrease in amounts due to stockholders is due to payments made to TLC Vision during the year ended December 31, 2006. |
Cash Provided by (Used in) Investing Activities
Net cash provided by (used in) investing activities for the year ended December 31, 2006 was $10,418,156 and resulted from cash provided from the net sale of short-term investments of $21,841,860. Cash used in investing activities during the period consists of $255,886 used to acquire fixed assets and $105,217 used to protect and maintain patents and trademarks. Additional cash used in investing activities includes cash of $7,906,968 paid by the Company, including costs of acquisition, to acquire SOLX net of cash acquired from SOLX of $34,719. In addition, the Company advanced a total of $2,434,537 to SOLX to support its operations prior to the acquisition. The Company also invested $2,076,312 to acquire 50.1% of the capital stock of OcuSense, on a fully diluted basis, including acquisition costs of $76,312. Cash acquired upon the acquisition of OcuSense was $1,320,497. The $2,000,000 invested by the Company in OcuSense is being utilized to fund the operations of OcuSense.
Cash provided by (used in) investing activities was $10,451,255 and ($43,428,156) for the years ended December 31, 2005 and 2004, respectively. Net cash provided by investing activities for the year ended December 31, 2005 was from the net sale of short-term investments of $10,689,818 (2004 - $42,500,000) offset by cash used to protect and maintain patents and trademarks in the amount of $36,290 (2004 - $28,990) and the purchase of fixed assets of $202,273 (2004 - $192,281).
Cash Provided by Financing Activities
Net cash provided by financing activities for the year ended December 31, 2006 was $270,935 and reflects cash received from the exercise of options to purchase shares of common stock of the Company.
Cash provided by financing activities was $328,463 for the year ended December 31, 2005 and relates to the exercise of stock options for cash proceeds of $231,235 and the receipt of $186,734 as part of the balance due from stockholders from the exercise of warrants in 2004. This was offset by additional share issue costs of $88,714 in the year ended December 31, 2005 in relation to our initial public offering. Net cash provided by financing activities was $65,104,005 in the year ended December 31, 2004 and primarily reflects the issuances of common stock and convertible debentures as well as the issuance of convertible preferred stock.
Borrowings
On November 30, 2006, we announced that Elias Vamvakas, our Chairman and Chief Executive Officer, had agreed to provide us with a standby commitment to purchase convertible debentures of the Company (“Convertible Debentures”) in an aggregate maximum amount of $8,000,000 (the “Total Commitment Amount”). Pursuant to the Summary of Terms and Conditions, executed and delivered as of November 30, 2006 by the Company and Mr. Vamvakas, during the 12-month commitment term commencing on November 30, 2006, upon no less than 45 days’ written notice by the Company to Mr. Vamvakas, Mr. Vamvakas was obligated to purchase Convertible Debentures in the aggregate principal amount specified in such written notice. A commitment fee of 200 basis points was payable by the Company on the undrawn portion of the total $8,000,000 commitment amount. Any Convertible Debentures purchased by Mr. Vamvakas would have carried an interest rate of 10% per annum and would have been convertible, at Mr. Vamvakas’ option, into shares of the Company’s common stock at a conversion price of $2.70 per share. The Summary of Terms and Conditions of the standby commitment further provided that if the Company closed a financing with a third party, whether by way of debt, equity or otherwise and there are no Convertible Debentures outstanding, then, the Total Commitment Amount was to be reduced automatically upon the closing of the financing by the lesser of: (i) the Total Commitment Amount; and (ii) the net proceeds of the financing. On February 6, 2007, the Company raised gross proceeds in the amount of $10,016,000 in a private placement of shares of its common stock and warrants. The Total Commitment Amount was therefore reduced to zero, thus effectively terminating Mr. Vamvakas’ standby commitment. No portion of the standby commitment was ever drawn down by the Company, and the Company paid Mr. Vamvakas a total of $29,808 in commitment fees in February 2007.
Contractual Obligations and Contingencies
The following table summarizes our contractual commitments as of December 31, 2006 and the effect those commitments are expected to have on liquidity and cash flow in future periods.
| | Payments Due by Period |
Contractual Commitments | Total | Less than 1 year | 1 to 3 years | More than 3 years |
| | $ | $ | $ | $ |
| | | | | |
Operating leases | 142,379 | 130,418 | 11,961 | — |
Royalty payments | 1,325,000 | 125,000 | 375,000 | 825,000 |
Consulting and non-competition agreements | 1,058,991 | 630,180 | 428,811 | — |
On November 30, 2006, pursuant to the Series A Preferred Stock Purchase Agreement between us and OcuSense, we purchased 1,744,223 shares of OcuSense’s Series A Preferred Stock representing 50.1% of OcuSense’s capital stock on a fully diluted basis for an aggregate purchase price of up to $8,000,000 (the “Purchase Price”). On the closing of the purchase which took place on November 30, 2006, we paid $2,000,000 of the Purchase Price. We paid another $2,000,000 installment of the Purchase Price on January 3, 2007. We will pay the third $2,000,000 installment of the Purchase Price upon the attainment by OcuSense of the first of two pre-defined milestones and the last $2,000,000 installment of the Purchase Price upon the attainment by OcuSense of the second of such milestones, provided that both milestones are achieved prior to May 1, 2009. The Series A Preferred Stock Purchase Agreement also makes provision for an ability on our part to increase our ownership interest in OcuSense for nominal consideration if OcuSense fails to meet certain milestones by specified dates. In addition, pursuant to the Series A Preferred Stock Purchase Agreement, we have agreed to purchase $3,000,000 of shares of OcuSense’s Series B Preferred Stock, which shall constitute 10% of OcuSense’s capital stock on a fully diluted basis at the time of purchase, upon OcuSense’s receipt from the FDA of 510(k) clearance for the DES Test and to purchase another $3,000,000 of shares of OcuSense’s Series B Preferred Stock, which shall constitute an additional 10% of OcuSense’s capital stock on a fully diluted basis at the time of purchase, upon OcuSense’s receipt from the FDA of CLIA waiver for the DES Test.
Pursuant to the terms of our distribution agreement with MeSys GmbH, or MeSys, dated January 1, 2002, we undertook a minimum purchase commitment of 25 OctoNova pumps per year beginning after FDA approval of the RHEO™ System, representing an annual commitment after FDA approval of €405,000, or approximately $534,900. The marketing and distributorship agreement with Diamed provides for a minimum purchase of 1,000 OctoNova pumps during the period from the date of the agreement until the end of the five-year period following receipt of FDA approval, representing an aggregate commitment of €16,219,000, or approximately $21,397,727, based on exchange rates as of December 31, 2006.
Pursuant to the terms of the 2006 Distribution Agreement with Asahi Medical, dated October 20, 2006, we undertook a commitment to purchase a minimum of 9,000, 15,000, and 22,500 of each of the Plasmaflo filters and the Rheofilter filters in years 1, 2 and 3, respectively, beginning six months after FDA approval of the RHEO™ System. Minimum purchase orders for the fourth year shall be determined immediately after the term of the first year by mutual consent but shall not be less than that of the previous year. This same method shall be used in subsequent years to determine future minimum purchase quantities such that minimum purchase quantities are always fixed for three years. Future minimum annual commitments in respect of United States, Mexico and certain Caribbean countries, after FDA approval, are approximately as follows:
Year 1 | ............................................................................................................................................................................ | $2,565,000 |
Year 2 | ............................................................................................................................................................................ | $4,275,000 |
Year 3 | ............................................................................................................................................................................ | $6,412,500 |
Pursuant to the terms of the 2006 Distribution Agreement with Asahi Medical, in respect of Canada, we undertook a commitment to purchase a minimum of 900, 1,500 and 2,250 of each of the Plasmaflo filters and the Rheofilter filters in years 1, 2 and 3, respectively, beginning upon the earlier to occur of (a) the sale by the Company of its current inventory of Rheofilter filters and (b) the expiration of the Company’s current inventory of Rheofilter filters. Minimum purchase orders for the fourth year shall be determined immediately after the term of the first year by mutual consent but shall not be less than that of the previous year. This same method shall be used in subsequent years to determine future minimum purchase quantities such that minimum purchase quantities are always fixed for three years. Future minimum annual commitments in respect of Canada are approximately as follows:
Year 1 | ............................................................................................................................................................................... | $256,500 |
Year 2 | ............................................................................................................................................................................... | $427,500 |
Year 3 | ............................................................................................................................................................................... | $641,250 |
In respect of Colombia, Venezuela, New Zealand and Australia, we undertook a commitment to purchase a minimum of 300 and 500 of each of the Plasmaflo filters and the Rheofilter filters in 2009 and 2010, respectively. In respect of Italy, we undertook a commitment to purchase a minimum of 200 and 500 of each of the Plasmaflo filters and the Rheofilter filters in 2007 and 2008, respectively. Minimum purchase orders for the years 2009 and 2010 shall be discussed and determined at the beginning of the year 2008 by mutual consent.
Future minimum annual commitments, in respect of Colombia, Venezuela, New Zealand, Australia and Italy, are approximately as follows:
2007 | ............................................................................................................................................................................... | $57,000 |
2008 | ............................................................................................................................................................................... | $142,500 |
2009 | ............................................................................................................................................................................... | $85,500 |
2010 | ............................................................................................................................................................................... | $142,500 |
As of September 1, 2006, SOLX granted a security interest in all of its intellectual property to Doug P. Adams, John Sullivan and Peter M. Adams, in their capacity as members of the Stockholder Representative Committee acting on behalf of the former stockholders of SOLX, in order to secure SOLX’s obligations under the Guaranty, dated as of September 1, 2006, by SOLX in favor of Doug P. Adams, John Sullivan and Peter M. Adams, in their capacity as members of the Stockholder Representative Committee (the “Guaranty”). Pursuant to the Guaranty, SOLX guaranteed the Company’s obligation to pay the Stockholder Representative Committee, acting on behalf of the former stockholders of SOLX, an aggregate amount of up to $13,000,000, being the maximum aggregate amount of the purchase price remaining payable to the former stockholders of SOLX.
Off-Balance-Sheet Arrangements
As of December 31, 2006, we did not have any significant off-balance-sheet arrangements as defined in Item 303(a)(4)(ii) of SEC Regulation S-K.
Financial Condition
Management believes that the existing cash and cash equivalents and short-term investments, together with funds expected to be generated from operations and funds generated from the private placement of the Company’s shares, will be sufficient to fund the Company’s anticipated level of operations and other demands and commitments until early 2008.
Our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties. Actual results could vary as a result of a number of factors. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect. Our future funding requirements will depend on many factors, including but not limited to:
| · | the costs of operating the businesses that have been acquired in the implementation of the Company’s diversification strategy; |
| · | the cost and results RHEO-AMD; |
| · | the rate of progress, cost and results of the LEARN and other clinical trials of the RHEO™ System; |
| · | our ability to obtain FDA approval to market and sell the RHEO™ System in the United States and the timing of such approval, if any; |
| · | our ability to continue to sell the RHEO™ System in Canada; |
| · | the cost and results, and the rate of progress, of the clinical trials of the components of the SOLX Glaucoma System to support SOLX’s application to obtain 510(k) approval from the FDA to market and sell the components of the SOLX Glaucoma System in the United States; |
| · | SOLX’s ability to obtain 510(k) approval to market and sell the components of the SOLX Glaucoma System in the United States and the timing of such approval, if any; |
| · | the cost and results of the product development of OcuSense’s TearLab™ test for DES; |
| · | the cost and results of the clinical trials that will be required of the TearLab™ test for DES that will support OcuSense’s application to obtain 510(k) clearance and a CLIA waiver from the FDA to market and sell the TearLab™ test for DES in the United States; |
| · | OcuSense’s ability to obtain 510(k) approval to market and sell the TearLab™ test for DES in the United States and the timing of such approval, if any; |
| · | whether government and third-party payors agree to reimburse treatments using the RHEO™ System and the components of the SOLX Glaucoma System; |
| · | the costs and timing of building the infrastructure to market and sell the RHEO™ System and the components of the SOLX Glaucoma System; |
| · | the costs of filing, prosecuting, defending and enforcing any patent claims and other intellectual property rights; and |
| · | the effect of competing technological and market developments. |
We cannot begin commercialization of the RHEO™ System, the components of the SOLX Glaucoma System and the TearLab™ test for DES in the United States until we receive FDA approval. At this time, we do not know when we can expect to begin to generate revenues from the RHEO™ System, the components of the SOLX Glaucoma System or the TearLab™ test for DES in the United States. We expect that the funding requirements of our operating activities will continue to increase substantially in the future. Until we can generate a sufficient amount of revenue, we expect to finance future cash needs through public or private equity offerings, debt financings, corporate collaboration or licensing or other arrangements. We cannot be certain that additional funding will be available on acceptable terms, or at all. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience significant dilution. In addition, future debt financing, if available, may involve restrictive covenants. To the extent that we raise additional funds through collaboration and licensing arrangements, it may be necessary to relinquish some rights to our technologies, or grant licenses on terms that are not favorable to us. If adequate funds are not available, we may be required to delay or reduce the scope of, or eliminate, some of our commercialization efforts, or we may even be unable to continue our operations.
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations is based upon our audited consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amount of assets, liabilities, sales and expenses, and related disclosure of contingent assets and liabilities. On an ongoing basis, we evaluate our estimates, including those related to our intangible assets, uncollectible receivables, inventories, goodwill and stock-based compensation. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Because this can vary in each situation, actual results may differ from these estimates under different assumptions or conditions.
We believe the following critical accounting policies affect our more significant judgments and estimates used in the preparation of our audited consolidated financial statements.
Revenue Recognition
We recognize revenue from the sale of the RHEO™ System which is comprised of OctoNova pumps and the related disposable treatment sets and from the sale of the components of the SOLX Glaucoma System which is comprised of the SOLX 790 Laser and the SOLX Gold Shunt. We receive a signed binding purchase order from our customers. The pricing is a negotiated amount between our customers and us. The Company sells the components of the SOLX Glaucoma System directly to physicians and also through distributors. Revenue is reported net of distributors’ commissions.
We have the obligation to train our customers and to calibrate the OctoNova pumps delivered to them. Only upon the completion of these services, do we recognize revenue for the pumps. We are also responsible for providing a one-year warranty on the OctoNova pumps, and the estimated cost of providing this service is accrued at the time revenue is recognized. The treatment sets and the components of the SOLX Glaucoma System do not require any additional servicing and revenue is recognized upon passage of title. However, our revenue recognition policy requires an assessment as to whether collectibility is reasonably assured, which requires us to evaluate the credit worthiness of our customers. The result of our assessment could materially impact the timing of revenue recognition.
Bad Debt Reserves
We evaluate the collectibility of our accounts receivable based on a combination of factors. In cases where we are aware of circumstances that may impair a specific customer’s ability to meet its financial obligations to us, a specific allowance against amounts due to us is recorded which reduces the net recognized receivable to the amount we reasonably believe will be collected. For all other customers, we recognize allowances for doubtful accounts based on the length of time the receivables are past due, the current business environment and historical experience. As at December 31, 2006 and 2005, we had bad debt reserves of nil and $1,049,297, respectively. We expensed amounts related to bad debt reserves of nil, $518,852 and nil during the years ended December 31, 2006, 2005 and 2004, respectively, and set up a provision for $530,445 representing invoices for product shipped to customers in December 2005 for which revenue was not recognized due to the likelihood that the customer would not be able to pay for the amounts invoiced.
Inventory Valuation
Inventory is recorded at the lower of cost and net realizable value and consists of finished goods. Cost is accounted for on a first-in, first-out basis. Deferred cost of sales (included in finished goods) consists of products shipped but not recognized as revenue because they did not meet the revenue recognition criteria.
Management must make estimates about future customer demand for our products when establishing the appropriate provisions for inventory and also determine whether market demand has had any impact on our inventory’s net realizable value. When making these estimates, we consider general economic conditions and growth prospects, including the impact of us receiving FDA approval for the RHEO™ System.
We received free inventory from Asahi Medical for the purpose of the MIRA-1 and related clinical trials. We accounted for this inventory at a value equivalent to the cost we pay for the same filters purchased for commercial sales to third parties.
With respect to our provisioning policy, in general, we fully reserve for surplus inventory in excess of our demand forecast, taking into consideration the expiry date of our inventory. In addition, we assess whether recent transactions provide indicators as to whether the net realizable value of our inventory is below our cost.
As at December 31, 2006 and 2005, we had inventory reserves of $5,101,394 and $1,990,830, respectively. During the years ended December 31, 2006, 2005 and 2004 we recognized a provision related to inventory of $3,304,124, $1,990,830 and nil, respectively, based on the above analysis.
Impairment of long-lived assets
We review our fixed assets and intangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset might not be recoverable. When such an event occurs, management estimates the future undiscounted cash flows expected to result from the use of the asset and its eventual disposition. In the event the undiscounted cash flows are less than the carrying amount of the asset, a further analysis is required to estimate the fair value of the asset using the discounted cash flow method and then an impairment loss equal to the excess of the carrying amount over the fair value is charged to operations.
Our intangible assets consist of the value of the exclusive distribution agreements we have with Asahi Medical and MeSys and other acquisition-related intangibles arising from our acquisition of SOLX and OcuSense during fiscal 2006. The distribution agreements and other acquisition-related intangible assets are amortized using the straight-line method over an estimated useful life of 15 and 10 years, respectively.
As a result of the analysis of the data from MIRA-1 and the result of our subsequent meeting with the FDA, we concluded that there were sufficient indications that the carrying value of the distribution intangible may no longer be recoverable, thus requiring us to assess whether the Company’s distribution intangible was impaired as of June 30, 2006. Based on management’s estimates of forecasted undiscounted cash flows as of June 30, 2006, we concluded that there is no indication of an impairment of the Company’s distribution intangible. Accordingly, we did not perform the second step of this analysis which would have required us to compare the carrying value of our intangible asset to the present value of future cash flows of the Company.
We determined that, as at December 31, 2006, there have been no significant events which may affect the carrying value of our distribution intangible and no significant changes in the methods used to determine the fair market value of other acquisition-related intangible assets. However, our prior history of losses and losses incurred during the current fiscal year reflects a potential indication of impairment, thus requiring us to assess whether the Company’s intangible assets were impaired as of December 31, 2006. Based on management’s estimates of forecasted undiscounted cash flows as of December 31, 2006, we concluded that there is no indication of an impairment of the Company’s intangible assets. Therefore, no impairment charge was recorded during the year ended December 31, 2006.
Impairment of Goodwill
Effective January 1, 2002, goodwill is no longer amortized and is subject to an annual impairment test. Goodwill impairment is evaluated between annual tests upon the occurrence of certain events or circumstances. Goodwill impairment is assessed based on a comparison of the fair value of the reporting unit to the underlying carrying value of the reporting unit’s net assets, including goodwill. When the carrying amount of the reporting unit exceeds its fair value, the fair value of the reporting unit’s goodwill is compared with its carrying amount to measure the amount of impairment loss, if any.
Prior to the acquisition of SOLX and OcuSense during the second half of fiscal 2006, the Company was a single reporting unit. Therefore, management determined the fair value of the Company’s goodwill using the Company’s market capitalization as opposed to the fair value of its assets and liabilities. As a result of the announcement on February 3, 2006, the per share price of our common stock as traded on NASDAQ decreased from $12.75 on February 2, 2006 to close at $4.10 on February 3, 2006. The 10-day average price of the stock immediately following the announcement was $3.65 and reflected a decrease in our market capitalization from $536.6 million on February 2, 2006 to $153.6 million based on the 10-day average share price subsequent to the announcement. On June 12, 2006, we announced that the FDA will require us to perform an additional study of the RHEO™ System. In addition, on June 30, 2006, we announced that we had terminated negotiations with Sowood in connection with a proposed private purchase of approximately $30,000,000 of zero-coupon convertible notes of the Company. The per share price of our common stock decreased subsequent to the June 12, 2006 announcement and again after the June 30, 2006 announcement. Based on the result of the preliminary analysis of the data from MIRA-1 and the events that occurred during the second quarter of fiscal 2006, we concluded that there were sufficient indicators of impairment leading to an analysis of our intangible assets and goodwill and resulting in our reporting an impairment charge to goodwill of $65,945,686 and $147,451,758 during the years ended December 31, 2006 and 2005, respectively.
Subsequent to the acquisition of SOLX and OcuSense, the Company will determine the fair value of its acquired goodwill based on a comparison of the fair value of the reporting unit to the underlying carrying value of the reporting unit’s net assets, including goodwill.
The Company determined that, as at December 31, 2006, no significant event or circumstance has occurred that may lead to an impairment of its acquired goodwill. The Company further determined that there have not been any significant changes in the methods used to value the net tangible and intangible assets acquired. Therefore, no impairment charge was recorded during the year ended December 31, 2006.
Stock-based Compensation
We account for stock-based compensation in accordance with the provisions of SFAS 123R. Under the fair value recognition provision of SFAS 123R, stock-based compensation cost is estimated at the grant date based on the fair value of the award and is recognized as an expense ratably over the requisite service period of the award. We have selected the Black-Scholes option-pricing model as our method of determining the fair value for all our awards and will recognize compensation cost on a straight-line basis over the awards’ vesting periods.
At the annual meeting of stockholders of the Company held on June 23, 2006, our stockholders approved the re-pricing of all then out-of-the-money stock options of the Company. Consequently, the exercise price of all outstanding stock options that, on June 23, 2006, was greater than $2.05, being the weighted average trading price of our common stock on NASDAQ during the five-trading day period immediately preceding June 23, 2006, was adjusted downward to $2.05. 2,585,000 of the outstanding stock options with a weighted average exercise price of $8.42 were affected by the re-pricing. SFAS No. 123R treats the re-pricing of equity awards as a modification of the original award and provides that such a modification is an exchange of the original award for a new award. SFAS No. 123R considers the modification to be the repurchase of the old award for a new award of equal or greater value, incurring additional compensation cost for any incremental value. This incremental difference in value is measured as the excess, if any, of the fair value of the modified award determined in accordance with the provisions of SFAS No. 123R over the fair value of the original award immediately before its terms are modified, measured based on the share price and other pertinent factors at that date. SFAS No. 123R provides that this incremental fair value, plus the remaining unrecognized compensation cost from the original measurement of the fair value of the old option, must be recognized over the remaining vesting period. Of the 2,585,000 options affected by the re-pricing, 1,401,073 were vested as at December 31, 2006. Therefore, additional compensation cost of $423,338 for the 1,401,073 options was recognized and is included in the stock-based compensation expense for the year ended December 31, 2006. The remaining unrecognized incremental fair value of $169,057 plus the compensation cost from the original measurement of the fair value of the old options of $2,607,496, which totaled $2,776,553 in unrecognized compensation expense as at December 31, 2006, is expected to be amortized over a weighted average vesting period of 2.3 years.
In accordance with SFAS No. 123R, we also recorded a compensation expense of $3,363 in the second quarter of fiscal 2006 as our board of directors approved accelerating the vesting of 1,250 unvested stock options granted to a terminated employee on April 28, 2006. SFAS No. 123R treats such a modification as a cancellation of the original unvested award and the grant of a new fully vested award as of that date.
Effective Corporate Tax Rate
Income Taxes
As of December 31, 2006, we had net operating loss carry forwards for federal income taxes of $24 million. Our utilization of the net operating loss and tax credit carry forwards may be subject to annual limitations pursuant to Section 382 of the Internal Revenue Code, and similar state provisions, as a result of changes in our ownership structure. The annual limitations may result in the expiration of net operating losses and credits prior to utilization.
At December 31, 2006, we had recorded a deferred tax liability due to the difference between the fair value of our intangible assets and their tax bases. We also recorded a deferred tax asset, netted off against the deferred tax liability, from the availability of 2006 net operating losses in the United States which may be utilized to reduce taxes in future years. In addition, we also had additional deferred tax asset representing the benefit of net operating loss carry forwards and certain stock issuance costs capitalized for tax purposes. We did not record a benefit for this deferred tax asset because realization of the benefit was uncertain, and, accordingly, a valuation allowance is provided to offset the deferred tax asset.
The Company and its subsidiaries have current and prior year losses available to reduce taxable income and taxes payable in future years which, if not utilized, will expire as follows:
| | $ |
| | |
| 2012 | 3,455,029 |
| 2018 | 4,500,401 |
| 2019 | 2,420,681 |
| 2020 | 5,241,917 |
| 2021 | 3,855,009 |
| 2022 | 3,313,031 |
| 2023 | 3,188,708 |
| 2024 | 7,849,643 |
| 2025 | 15,690,473 |
| 2026 | 13,877,166 |
Recent Accounting Pronouncements
In December 2004, the FASB issued SFAS No. 123R which revised SFAS No. 123 and supersedes APB No. 25. SFAS No. 123R requires all share-based payments to employees, including grants of employee stock options to be recognized in the consolidated financial statements based on their fair values. The pro forma disclosure previously permitted under SFAS No. 123 is no longer an alternative to financial statement recognition. SFAS No. 123R is effective at the beginning of the first annual period beginning after June 15, 2005. Accordingly, we adopted SFAS No. 123R beginning January 1, 2006. We have selected the Black-Scholes option-pricing model as our method of determining the fair value for our awards and will recognize compensation cost on a straight-line basis over the awards’ vesting periods.
The adoption of the following recent accounting pronouncements in fiscal 2006 did not have a material impact on our results of operations and financial condition:
| · | SFAS No. 151, “Inventory Costs — An Amendment of ARB No. 43, Chapter 4”; |
| · | SFAS No. 153, “Exchanges of Nonmonetary Assets — An Amendment of APB Opinion No. 29”; |
| · | SFAS No. 154, “Accounting Changes and Error Corrections”, which replaces APB No. 20, “Accounting Changes”; |
| · | SFAS No. 3, “Reporting Accounting Changes in Interim Financial Statements — An Amendment of APB Opinion No. 28”; and |
| · | FASB Staff Position FAS 115-1 and FAS 124-1, “The Meaning of Other-Than-Temporary Impairment and Its Application to Certain Investments”. |
In February 2006, the FASB issued SFAS No. 155, “Accounting for Certain Hybrid Financial Instruments — An Amendment of FASB Statements No. 133 and 140”. SFAS No. 155 simplifies accounting for certain hybrid instruments currently governed by SFAS No. 133, “Accounting for Derivative Instruments and Hedging Activities”, by allowing fair value re-measurement of hybrid instruments that contain an embedded derivative that otherwise would require bifurcation. SFAS No. 155 also eliminates the guidance in SFAS No. 133 Implementation Issue No. D1, “Application of Statement 133 to Beneficial Interests in Securitized Financial Assets”, which provides that such beneficial interests are not subject to SFAS No. 133. SFAS No. 155 amends SFAS No. 140, “Accounting for Transfers and Servicing of Financial Assets and Extinguishments of Liabilities — A Replacement of FASB Statement No. 125”, by eliminating the restriction on passive derivative instruments that a qualifying special-purpose entity may hold. SFAS No. 155 is effective for financial instruments acquired or issued after the beginning of fiscal years beginning after September 15, 2006. We do not expect the adoption of this statement to have a material impact on our results of operations and financial condition.
In March 2006, the FASB issued SFAS No. 156, “Accounting for Servicing of Financial Assets — An Amendment of FASB Statement No. 140”. SFAS No.156 requires an entity to recognize a servicing asset or servicing liability each time it undertakes an obligation to service a financial asset by entering into a servicing contract in specific situations. Additionally, the servicing asset or servicing liability shall be initially measured at fair value; however, an entity may elect the “amortization method” or “fair value method” for subsequent balance sheet reporting periods. SFAS No.156 is effective for fiscal years beginning after September 15, 2006. We do not expect the adoption of this statement to have a material impact on our results of operations and financial condition.
In June 2006, the FASB issued FASB Interpretation (“FIN”) No. 48, “Accounting for Uncertainty in Income Taxes — An Interpretation of FASB Statement No. 109”, which clarifies the accounting for uncertainty in tax positions. FIN No. 48 requires that we recognize, in our financial statements, the impact of a tax position, if that position is more likely than not of being sustained on audit, based on the technical merits of the position. FIN No. 48 is effective for fiscal years beginning after December 15, 2006. We do not expect the adoption of FIN No. 48 to have a material effect on our consolidated results of operations and financial position.
In September 2006, the SEC released Staff Accounting Bulletin (“SAB”) No. 108, “Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current Year Financial Statements”. SAB No. 108 provides interpretive guidance on the SEC’s views regarding the process of quantifying materiality of financial statement misstatements. SAB No. 108 is effective for fiscal years ending after November 15, 2006, and early application for the first interim period of the same fiscal year is encouraged. The application of SAB No. 108 in fiscal 2006 did not have a material effect on our results of operations or financial position.
In September 2006, the FASB issued SFAS No. 157, “Fair Value Measurements”. SFAS No. 157 defines fair value, establishes a framework and gives guidance regarding the methods used for measuring fair value, and expands disclosures about fair value measurements. SFAS No. 157 is effective for financial statements issued for fiscal years beginning after November 15, 2007, and interim periods within those fiscal years. We are currently evaluating the impact the adoption of SFAS No. 157 would have on our consolidated financial statements.
In September 2006, the FASB issued SFAS No. 158, “Employers’ Accounting for Defined Benefit Pension and Other Post-retirement Plans, an amendment of FASB Statements No. 87, 88, 106, and 132(R)”. SFAS No.158 requires companies to recognize the overfunded or underfunded status of a defined benefit post-retirement plan as an asset or liability on their balance sheet and to recognize changes in that funded status in the year in which the changes occur through comprehensive income, effective for fiscal years ending after December 15, 2006. SFAS No. 158 also requires companies to measure the funded status of the plan as of the date of its fiscal year-end, with limited exceptions, effective for fiscal years ending after December 15, 2008. The adoption of SFAS No. 158 did not have a material effect on our consolidated results of operations and financial position.
| ITEM 7A. | QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK |
Quantitative and Qualitative Disclosure of Market Risk
Currency Fluctuations and Exchange Risk
All of our sales are in U.S. dollars or are linked to the U.S. dollar, while a portion of our expenses are in Canadian dollars and Euros. We cannot predict any future trends in the exchange rate of the Canadian dollar or Euro against the U.S. dollar. Any strengthening of the Canadian dollar or Euro in relation to the U.S. dollar would increase the U.S. dollar cost of our operations, and affect our U.S. dollar measured results of operations. We do not engage in any hedging or other transactions intended to manage these risks. In the future, we may undertake hedging or other similar transactions or invest in market risk sensitive instruments if we determine that is advisable to offset these risks.
Interest Rate Risk
The primary objective of our investment activity is to preserve principal while maximizing interest income we receive from our investments, without increasing risk. We believe this will minimize our market risk.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA.
Consolidated Financial Statements
OccuLogix, Inc.
December 31, 2006 and 2005
REPORT OF INDEPENDENT REGISTERED
PUBLIC ACCOUNTING FIRM
To the Board of Directors and Shareholders of OccuLogix, Inc.
We have audited the accompanying consolidated balance sheets of OccuLogix, Inc. as of December 31, 2006 and 2005, and the related consolidated statements of operations, changes in stockholders’ equity (deficiency) and cash flows for each of the years in the three-year period ended December 31, 2006. Our audits also included the financial statement schedule listed in the in the index at Item 15(a). These financial statements are the responsibility of OccuLogix, Inc.’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the consolidated financial position of OccuLogix, Inc. at December 31, 2006 and 2005, and the consolidated results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2006, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the financial statements taken as a whole, presents fairly, in all material respects, the information set forth therein.
The accompanying consolidated financial statements have been prepared assuming that OccuLogix, Inc. will continue as a going concern. As more fully described in Note 1 to the consolidated financial statements, OccuLogix, Inc. has incurred recurring operating losses and its working capital at December 31, 2006 is $13,539,026, which represents a $30,875,921 reduction of its working capital during 2006. OccuLogix, Inc.’s history of losses and financial condition raises substantial doubt about its ability to continue as a going concern, such as those described in Note 1 to the accompanying consolidated financial statements. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
We have also audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the effectiveness of OccuLogix, Inc.’s internal control over financial reporting as of December 31, 2006, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission, and our report dated March 2, 2007 expressed an unqualified opinion thereon.
As described in Notes 2 & 14 to these consolidated financial statements, OccuLogix, Inc. changed its accounting policy in regards to the fair value recognition provisions of accounting for stock-based compensation.
Toronto, Canada, /s/ Ernst & Young LLP
March 2, 2007. &# 160; Chartered Accountants
OccuLogix, Inc.
CONSOLIDATED BALANCE SHEETS
(expressed in U.S. dollars)
(Going Concern Uncertainty - See Note 1)
| | As of December 31, |
| | 2006 $ | 2005 $ |
ASSETS | | |
Current | | |
| Cash and cash equivalents | 5,740,697 | 9,599,950 |
| Short-term investments | 9,785,000 | 31,662,845 |
Amounts receivable, net of bad debt reserves of nil in 2006 and $518,852 in 2005 (note 10(f)) | 166,209 | 554,966 |
| Inventory, net of provision for inventory obsolescence of $5,101,394 in 2006 and $1,990,830 in 2005 | 2,715,737 | 4,701,464 |
| Prepaid expenses | 680,476 | 803,268 |
| Deposit | 10,442 | 4,891 |
| Other current assets | 79,200 | — |
Total current assets | 19,177,761 | 47,327,384 |
Fixed assets, net (note 5) | 860,717 | 470,561 |
Patents and trademarks, net (note 6) | 234,841 | 135,232 |
Intangible assets, net (note 7) | 55,683,399 | 23,927,195 |
Goodwill (note 4) | 14,446,977 | 65,945,686 |
| | 90,403,695 | 137,806,058 |
| | | |
LIABILITIES AND STOCKHOLDERS’ EQUITY | | |
Current | | |
Accounts payable (note 10(f)) | 395,392 | 522,520 |
Accrued liabilities (notes 10(f) and 12) | 2,090,937 | 2,226,619 |
Due to related party (note 10) | — | 5,065 |
Due to stockholders (note 9) | 152,406 | 158,233 |
Current portion of other long-term liability (note 3) | 3,000,000 | — |
Total current liabilities | 5,638,735 | 2,912,437 |
Deferred tax liability, net (note 11) | 18,939,417 | 8,853,062 |
Other long-term liability (note 3) | 3,420,609 | — |
Total liabilities | 27,998,761 | 11,765,499 |
Commitments and contingencies (notes 10 and 13) | | |
Minority interest | 1,184,844 | — |
Stockholders’ equity | | |
Capital stock (note 14) | | |
Common stock | 50,627 | 42,086 |
Par value of $0.001 per share; | | |
Authorized: 75,000,000; Issued and outstanding: | | |
December 31, 2006 - 50,626,562; December 31, 2005 - 42,085,853 | | |
| Additional paid-in capital | 354,320,116 | 336,977,578 |
| Accumulated deficit | (293,150,653) | (210,979,105) |
Total stockholders’ equity | 61,220,090 | 126,040,559 |
| | 90,403,695 | 137,806,058 |
See accompanying notes | | |
OccuLogix, Inc.
CONSOLIDATED STATEMENTS OF OPERATIONS
(expressed in U.S. dollars except number of shares)
| | Years ended December 31, |
| | 2006 | 2005 | 2004 |
| | $ | $ | $ |
Revenue | | | |
Retina | | | |
Sales to related parties (note 10) | — | 81,593 | 731,757 |
Sales to unrelated parties | 174,259 | 1,758,696 | 237,600 |
Glaucoma | 31,625 | — | — |
Total revenue | 205,884 | 1,840,289 | 969,357 |
Cost of goods sold | | | |
Retina | | | |
Cost of goods sold to related parties (note 10) | — | 43,236 | 688,102 |
Cost of goods sold to unrelated parties | 3,428,951 | 3,250,866 | 133,710 |
Royalty costs (note 10) | 100,000 | 100,000 | 135,457 |
Glaucoma | | | |
Cost of goods sold | 11,053 | — | — |
Royalty costs (note 10) | 8,332 | — | — |
Total cost of goods sold | 3,548,336 | 3,394,102 | 957,269 |
Gross (loss) profit | (3,342,452) | (1,553,813) | 12,088 |
Operating expenses | | | |
General and administrative (notes 9, 10 and 14) | 9,831,452 | 8,729,456 | 17,530,019 |
Clinical and regulatory (notes 10 and 14) | 5,710,830 | 5,250,492 | 3,994,967 |
Sales and marketing (notes 10 and 14) | 1,969,638 | 2,165,337 | 219,556 |
Impairment of goodwill (note 4) | 65,945,686 | 147,451,758 | — |
Restructuring charges (note 8) | 819,642 | — | — |
| | 84,277,248 | 163,597,043 | 21,744,542 |
| Loss from operations | (87,619,700) | (165,150,856) | (21,732,454) |
Other income (expenses) | | | |
| Interest income | 1,370,208 | 1,593,366 | 60,227 |
| Interest and amortization of discount on future payment expense | (288,088) | — | (24,492) |
| Other | 30,868 | (57,025) | (145,925) |
| Minority interest | 157,624 | — | — |
| | 1,270,612 | 1,536,341 | (110,190) |
| Loss before income taxes and cumulative effect of a change in accounting principle | (86,349,088) | (163,614,515) | (21,842,644) |
Recovery of income taxes (note 11) | 4,070,495 | 642,529 | 23,771 |
| Loss before cumulative effect of a change in accounting principle | (82,278,593) | (162,971,986) | (21,818,873) |
| Cumulative effect of a change in accounting principle | 107,045 | — | — |
Net loss for the year | (82,171,548) | (162,971,986) | (21,818,873) |
Weighted average number of shares outstanding - basic and diluted | 44,979,692 | 41,931,240 | 7,369,827 |
| Loss before cumulative effect of a change in accounting principle per share - basic and diluted | $(1.83) | $(3.89) | $(2.96) |
| Cumulative effect of a change in accounting principle per share - basic and diluted | — | — | — |
Net loss per share - basic and diluted | $(1.83) | $(3.89) | $(2.96) |
See accompanying notes | | | |
OccuLogix, Inc.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY (DEFICIENCY)
(expressed in U.S. dollars)
| | Voting common stock at par value | Series A convertible preferred stock at par value | Series B convertible preferred stock at par value | Additional paid-in capital | Accumulated deficit | Stockholders’ equity (deficiency) |
| | shares issued | shares issued | shares issued | | | |
| | # | $ | # | $ | # | $ | $ | $ | $ |
| | | | | | | | | | |
Balance, December 31, 2003 | 5,032,906 | 5,033 | 1,767,740 | 1,768 | 620,112 | 620 | 23,915,012 | (26,188,246) | (2,265,813) |
Stock-based compensation (note 14(e)) | — | — | — | — | — | — | 15,439,960 | — | 15,439,960 |
Stock issued on exercise of options (note 14(e)) | 272,200 | 273 | — | — | — | — | 129,147 | — | 129,420 |
Stock issued on exercise of warrants (note 14(f)) | 102,369 | 102 | 379,284 | 379 | — | — | 1,415,840 | — | 1,416,321 |
Subscription receivable (note 14(f)) | — | — | — | — | — | — | (221,661) | — | (221,661) |
Contribution of inventory from related party (note 10) | — | — | — | — | — | — | 146,905 | — | 146,905 |
Conversion of Series A convertible preferred stock into common stock (note 14(b)) | 3,603,350 | 3,603 | (2,147,024) | (2,147) | — | — | (1,456) | — | — |
Conversion of Series B convertible preferred stock into common stock (note 14(b) | 1,019,255 | 1,019 | — | — | (620,112) | (620) | (399) | — | — |
Conversion of convertible grid debentures into common stock (note 14(b)) | 7,106,454 | 7,107 | — | — | — | — | 6,992,893 | — | 7,000,000 |
Fractional payout of converted shares due to preferred stockholders | — | — | — | — | — | — | (747) | — | (747) |
Shares issued on acquisition of OccuLogix, L.P. (notes 3 and 14(b)) | 19,070,234 | 19,070 | — | — | — | — | 228,823,738 | — | 228,842,808 |
Initial public offering, net of issue costs (note 14(d)) | 5,600,000 | 5,600 | — | — | — | — | 59,424,325 | — | 59,429,925 |
| Net loss for the year | — | — | — | — | — | — | — | (21,818,873) | (21,818,873) |
Balance, December 31, 2004 | 41,806,768 | 41,807 | — | — | — | — | 336,063,557 | (48,007,119) | 288,098,245 |
Stock-based compensation (note 14(e)) | — | — | — | — | — | — | 366,781 | — | 366,781 |
Stock issued on exercise of options (note 14(e)) | 279,085 | 279 | — | — | — | — | 230,956 | — | 231,235 |
Subscription receivable (note 14(f)) | — | — | — | — | — | — | 221,661 | — | 221,661 |
Contribution of inventory from related party (note 10) | — | — | — | — | — | — | 167,730 | — | 167,730 |
| Contribution of inventory from unrelated party | — | — | — | — | — | — | 15,652 | — | 15,652 |
Fractional payout of converted shares due to preferred stockholders | — | — | — | — | — | — | (45) | — | (45) |
Additional share issue costs related to initial public offering (note 14(d)) | — | — | — | — | — | — | (88,714) | — | (88,714) |
| Net loss for the year | — | — | — | — | — | — | — | (162,971,986) | (162,971,986) |
Balance, December 31, 2005 | 42,085,853 | 42,086 | — | — | — | — | 336,977,578 | (210,979,105) | 126,040,559 |
OccuLogix, Inc.
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY (DEFICIENCY) continued
(expressed in U.S. dollars)
| | Voting common stock at par value | Series A convertible preferred stock at par value | Series B convertible preferred stock at par value | Additional paid-in capital | Accumulated deficit | Stockholders’ equity (deficiency) |
| | shares issued | shares issued | shares issued | | | |
| | # | $ | # | $ | # | $ | $ | $ | $ |
Balance, December 31, 2005 (balance forward) | 42,085,853 | 42,086 | — | — | — | — | 336,977,578 | (210,979,105) | 126,040,559 |
Stock-based compensation (note 14(e)) | — | — | — | — | — | — | 2,098,526 | — | 2,098,526 |
Stock issued on exercise of options (note 14(e)) | 140,726 | 141 | — | — | — | — | 270,794 | — | 270,935 |
Free inventory returned to related party (note 10) | — | — | — | — | — | — | (60,000) | — | (60,000) |
| Contribution of inventory from unrelated party | — | — | — | — | — | — | 11,994 | — | 11,994 |
Shares issued on acquisition of Solx, Inc. (notes 3 and 14(d)) | 8,399,983 | 8,400 | — | — | — | — | 15,027,570 | — | 15,035,970 |
| Shares issue costs | — | — | — | — | — | — | (21,908) | — | (21,908) |
Change in OcuSense, Inc.’s stockholders’ equity, stock-based compensation | — | — | — | — | — | — | 15,562 | — | 15,562 |
| Net loss for the year | — | — | — | — | — | — | — | (82,171,548) | (82,171,548) |
Balance, December 31, 2006 | 50,626,562 | 50,627 | — | — | — | — | 354,320,116 | (293,150,653) | 61,220,090 |
See accompanying notes | | | | | | | | | |
OccuLogix, Inc.
CONSOLIDATED STATEMENTS OF CASH FLOWS
(expressed in U.S. dollars)
| | Years ended December 31, |
| | 2006 $ | 2005 $ | 2004 $ |
OPERATING ACTIVITIES | | | |
| Net loss for the year | (82,171,548) | (162,971,986) | (21,818,873) |
| Adjustments to reconcile net loss to cash used in operating activities: | | | |
Stock-based compensation (note 14(e)) | 2,221,133 | 366,781 | 15,439,960 |
Amortization of fixed assets | 213,488 | 99,301 | 42,956 |
Amortization of patents and trademarks | 5,608 | 5,712 | 5,480 |
Amortization of intangible assets | 2,749,212 | 1,716,667 | 106,138 |
Impairment of goodwill (note 4) | 65,945,686 | 147,451,758 | — |
Amortization of discount on future cash payments (note 3) | 273,195 | — | — |
Amortization of premiums/discounts on short-term investments | 35,985 | 147,337 | — |
Subscription receivable - provision for doubtful amount (note 14(f)) | — | 34,927 | — |
Deferred income taxes (note 11) | (4,065,962) | (635,167) | (39,271) |
Gain on sale of fixed assets | — | — | (6,000) |
Impairment of fixed assets | — | — | 13,850 |
| Cumulative effect of a change in accounting principle | (107,045) | — | — |
Minority interests | (157,624) | — | — |
Net change in non-cash working capital balances related to operations (note 15) | 509,528 | (4,925,650) | 873,295 |
Cash used in operating activities | (14,548,344) | (18,710,320) | (5,382,465) |
INVESTING ACTIVITIES | | | |
| Proceeds on sale of fixed assets | — | — | 6,000 |
| Sale of (purchase of) short-term investments | 21,841,860 | 10,689,818 | (42,500,000) |
| Additions to fixed assets | (255,886) | (202,273) | (192,281) |
| Additions to patents and trademarks | (105,217) | (36,290) | (28,990) |
Acquisition costs (note 3) | (949,499) | — | (768,808) |
| Advance to Solx, Inc., pre-acquisition | (2,434,537) | — | — |
Payments for acquisitions, net of cash acquired (note 3) | (7,678,565) | — | 55,923 |
Cash provided by (used in) investing activities | 10,418,156 | 10,451,255 | (43,428,156) |
FINANCING ACTIVITIES | | | |
Increase in long-term convertible debentures (note 10) | — | — | 4,350,000 |
| Share issuance costs | — | (88,714) | (7,770,075) |
Proceeds from exercise of common stock options and warrants (notes 14(e) and 14(f)) | 270,935 | 231,235 | 263,900 |
Proceeds from exercise of Series A convertible preferred stock warrants (note 14(f)) | — | 186,734 | 1,060,180 |
Fractional payout of converted shares due to preferred stockholders | — | (792) | — |
Proceeds from issuance of common stock (note 14(d)) | — | — | 67,200,000 |
Cash provided by financing activities | 270,935 | 328,463 | 65,104,005 |
Net (decrease) increase in cash and cash equivalents during the year | (3,859,253) | (7,930,602) | 16,293,384 |
| Cash and cash equivalents, beginning of year | 9,599,950 | 17,530,552 | 1,237,168 |
Cash and cash equivalents, end of year | 5,740,697 | 9,599,950 | 17,530,552 |
See accompanying notes | | | |
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
1. NATURE OF OPERATIONS AND GOING CONCERN UNCERTAINTY
Nature of Operations
OccuLogix, Inc. (the “Company”) is an ophthalmic therapeutic company in the business of commercializing innovative treatments for age-related eye diseases, including age-related macular degeneration, or AMD, and glaucoma. The Company also holds a majority interest in a company that is in the process of developing ocular diagnostic technologies. The Company’s core purpose is to improve life through evidence-based medical therapies.
The Company’s product for Dry AMD, the RHEO™ System contains a pump that circulates blood through two filters and is used to perform the Rheopheresis™ procedure, which is referred to under the Company’s trade name as RHEO™ Therapy. The Rheopheresis™ procedure is a blood filtration procedure that selectively removes molecules from plasma, which is designed to treat Dry AMD, the most common form of the disease.
The Company owns and/or has licensed certain patents relating to the RHEO™ System and has the exclusive right to develop and sell the equipment which comprises the RHEO™ System in the North American markets.
The Company conducted a clinical trial, called MIRA-1, or Multicenter Investigation of Rheopheresis for AMD, which, if successful, was expected to support its application with the U.S. Food and Drug Administration (the “FDA”) to obtain approval to market the RHEO™ System in the United States. On February 3, 2006, the Company announced that, based on a preliminary analysis of the data from MIRA-1, MIRA-1 did not meet its primary efficacy endpoint as it did not demonstrate a statistically significant difference in the mean change of Best Spectacle-Corrected Visual Acuity applying the Early Treatment Diabetic Retinopathy Scale, or ETDRS BCVA, between the treated and placebo groups in MIRA-1 at 12 months post-baseline. As expected, the treated group demonstrated a positive result. An anomalous response of the control group is the principal reason why the primary efficacy endpoint was not met.
On June 8, 2006, the Company met with the FDA to discuss the results of MIRA-1 and the impact the results will have on our application to market the RHEO™ System in the United States. In light of MIRA-1’s failure to meet its primary efficacy endpoint, the FDA advised that it will require an additional study of the RHEO™ System to be performed. On December 28, 2006, the Company submitted its Investigational Device Exemption, or IDE, package to the FDA together with the proposed protocol for the additional study of the RHEO™ System called RHEO-AMD, or Safety and Effectiveness in a Multi-Center, Randomized, Sham-Controlled Investigation for Dry Non-exudative Age-Related Macular Degeneration (AMD) using Rheopheresis. On January 26, 2007, the FDA issued an IDE number for RHEO-AMD allowing patient enrollment to commence within the first quarter of 2007.
During the second half of fiscal 2006, the Company completed the acquisition of Solx, Inc (“SOLX”) and acquired 50.1% of the capital stock of OcuSense, Inc. (“OcuSense”) on a fully diluted basis. The acquisition of SOLX and the significant investment in OcuSense represent an expansion of the Company’s ophthalmic product portfolio beyond the RHEO™ procedure for Dry AMD (note 3).
Going concern uncertainty
The consolidated financial statements have been prepared on the basis that the Company will continue as a going concern. However, the Company has sustained substantial losses for each of the years ended December 31, 2006, 2005 and 2004. The Company’s working capital at December 31, 2006 is $13,539,026, which represents a $30,875,921 reduction of its working capital of $44,414,947 at December 31, 2005. As a result of the Company’s history of losses and financial condition, there is substantial doubt about the ability of the Company to continue as a going concern.
Although the Company realized gross proceeds of $10,016,000 (less transaction costs of approximately $750,000) on February 7, 2007 from the private placement of shares of its common stock and warrants, management believes that these proceeds, together with the Company’s existing cash, will be only sufficient to cover its operating activity and other demands until early 2008.
The consolidated financial statements do not include any adjustments relating to the recoverability and classification of recorded asset amounts or the amounts and classification of liabilities that might be necessary if the Company were not able to continue in existence as a going concern.
Historically, the Company has obtained financing from its initial public offering of shares of its common stock, the private placement of its shares of common stock and from certain of its significant stockholders.
2. SIGNIFICANT ACCOUNTING POLICIES
The consolidated financial statements have been prepared by management in conformity with accounting principles generally accepted in the United States (“U.S. GAAP”).
Basis of consolidation
The consolidated financial statements include the accounts of the Company and its subsidiaries. All significant intercompany transactions and balances have been eliminated on consolidation. The Company’s consolidated financial statements also include the results of the companies acquired from the date of each acquisition. The Company completed the acquisition of SOLX on September 1, 2006 and acquired 50.1% of the capital stock of OcuSense, on a fully diluted basis, on November 30, 2006 (note 3).
Use of estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expenses during the reporting periods. Some of the Company’s more significant estimates include those related to uncollectible receivables, inventory, goodwill, stock-based compensation and its intangible assets. Actual results could differ from those estimates.
Revenue recognition
The Company recognizes revenue from the sale of the RHEO™ System which is comprised of OctoNova pumps and the related disposable treatment sets and from the sale of the components of the SOLX Glaucoma System which includes the SOLX 790 Titanium Sapphire Laser (“SOLX 790 Laser”) and the SOLX Gold Shunt. The Company receives a signed binding purchase order from its customers. The pricing is a negotiated amount between the Company and its customers. The Company sells the components of the SOLX Glaucoma System directly to physicians and also through distributors. Revenue is reported net of distributors’ commissions.
The Company has the obligation to train its customers and to calibrate the OctoNova pumps delivered to them. Only upon the completion of these services, does the Company recognize revenue for the pumps. The Company is also responsible for providing a one-year warranty on the OctoNova pumps, and the estimated cost of providing this service is accrued at the time revenue is recognized. The treatment sets and the components of the SOLX Glaucoma System do not require any additional servicing and revenue is recognized upon passage of title. However, the Company’s revenue recognition policy requires an assessment as to whether collectibility is reasonably assured, which requires the Company to evaluate the creditworthiness of its customers. The result of the assessment could materially impact the timing of revenue recognition.
During the years ended December 31, 2006, 2005 and 2004, the Company did not recognize as revenue $69,600, $530,445 and nil, respectively, in sales of products that were shipped during the last quarter of each of the fiscal years due to collectibility not being reasonably assured.
Cost of goods sold
Cost of sales includes costs of goods sold and royalty costs. The Company’s cost of goods sold consists primarily of costs for the manufacture of the RHEO™ System and the SOLX Glaucoma System, including the costs the Company incurs for the purchase of component parts from its suppliers, applicable freight and shipping costs, fees related to warehousing, logistics inventory management and recurring regulatory costs associated with conducting business and ISO certification. In addition to these direct costs, included in the cost of goods sold are licensing costs associated with distributing the RHEO™ System in Canada and minimum royalty payments due to Mr. Hans Stock and Dr. Richard Brunner that are only recoverable based on sufficient volume (notes 9 and 10).
Cash and cash equivalents
Cash and cash equivalents comprise cash on hand and highly liquid short-term investments with original maturities of 90 days or less at the date of purchase.
Short-term investments
Short-term investments consist of investments in auction rate securities, which are available to support the Company’s current operations. These investments are classified as available-for-sale securities and are recorded at fair value with unrealized gains or losses reported in other comprehensive income. Due to the short time period between the reset dates of the interest rates, there are no unrealized gains or losses associated with these securities. All of the auction rate securities have contractual maturities of more than three years.
Bad debt reserves
The Company evaluates the collectibility of its accounts receivable based on a combination of factors. In cases where management is aware of circumstances that may impair a specific customer’s ability to meet its financial obligations to the Company, a specific allowance against amounts due to the Company is recorded, which reduces the net recognized receivable to the amount management reasonably believes will be collected. For all other customers, the Company recognizes allowances for doubtful accounts based on the length of time the receivables are past due, the current business environment and historical experience. As at December 31, 2006 and 2005, the Company had bad debt reserves of nil and $1,049,297, respectively. The Company expensed amounts related to bad debt reserves of nil, $518,852 and nil during the years ended December 31, 2006, 2005 and 2004, respectively, and set up a provision for $530,445 representing invoices for products shipped, plus related taxes, to a customer in December 2005 for which revenue was not recognized due to the likelihood that the customer would not be able to pay for the amounts invoiced.
Inventory
Inventory is recorded at the lower of cost and net realizable value and consists of finished goods. Cost is accounted for on a first-in, first-out basis. Deferred cost of sales (included in finished goods) consists of products shipped but not recognized as revenue because they did not meet the revenue recognition criteria.
Management must make estimates about future customer demand for the Company’s products when establishing the appropriate provisions for inventory and also determine whether market demand has had any impact on the Company’s inventory’s net realizable value. When making these estimates, management considers general economic conditions and growth prospects, including the impact of the Company receiving FDA approval for the RHEO™ System.
The Company received free inventory from Asahi Kasai Medical Co., Ltd. (“Asahi Medical”) (formerly Asahi Medical Co., Ltd.) for the purpose of the MIRA-1 and related clinical trials. The free inventory received from Asahi Medical was accounted for at a value equivalent to the cost the Company pays for the same filters purchased for commercial sales to third parties. Of the total inventory value of $2,642,249 and $4,701,464 as at December 31, 2006 and 2005, respectively, $85,590 and $264,615 relate to free inventory received by the Company from Asahi Medical. In addition, the Company received free vitamins from a certain supplier for the purpose of the MIRA-1 and related clinical trials. Included in inventory as at December 31, 2006 and 2005 are $7,316 and $6,397, respectively, which relate to free vitamins received by the Company.
With respect to the Company’s provisioning policy, in general, the Company fully reserves for surplus inventory in excess of its demand forecast, taking into consideration the expiry date of its inventory. In addition, the Company assesses whether recent transactions provide indicators as to whether the net realizable value of its inventory is below its recorded cost.
As at December 31, 2006 and 2005, the Company had inventory reserves of $5,101,394 and $1,990,830, respectively. During the years ended December 31, 2006, 2005 and 2004, the Company recognized a provision related to inventory of $3,304,124, $1,990,830 and nil, respectively, based on the above analysis.
Fair value of financial instruments
Fair value of a financial instrument is defined as the amount at which the instrument could be exchanged in a current transaction between willing parties. The estimated fair values of cash and cash equivalents, short-term investments, amounts receivable, due to related party, accounts payable, accrued liabilities, due to stockholders and other long-term liability approximate their carrying values due to the short-term maturities of these instruments.
Fixed assets
Fixed assets are recorded at cost less accumulated amortization. Amortization is calculated using the straight-line method, commencing when the assets become available for productive use, based on the following estimated useful lives:
| Furniture and office equipment | 2 - 7 years | |
| Computer equipment and software | 3 years | |
| Medical equipment | 1 - 5 years | |
Impairment of long-lived assets
The Company reviews its fixed assets and intangible assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset might not be recoverable. When such an event occurs, management estimates the future undiscounted cash flows expected to result from the use of the asset and its eventual disposition. In the event the undiscounted cash flows are less than the carrying amount of the asset, an impairment loss equal to the excess of the carrying amount over the fair value is charged to operations.
The Company’s intangible assets are comprised of the value of the exclusive distribution agreements the Company has with Asahi Medical, the manufacturer of the Rheofilter filters and the Plasmaflo filters, and Diamed Medizintechnik GmbH (“Diamed”) and MeSys GmbH, the designer and manufacturer, respectively, of the OctoNova pumps and other acquisition-related intangibles. The distribution agreements and other acquisition-related intangible assets are amortized using the straight-line method over an estimated useful life of 15 and 10 years, respectively.
Patents and trademarks
Patents and trademarks have been recorded at historical cost and are amortized using the straight-line method over their estimated useful lives, not to exceed 15 years.
Goodwill
Goodwill is not amortized and instead is subject to an annual impairment test. The Company’s annual impairment test is conducted effective October 1 and is evaluated between annual tests upon the occurrence of certain events or circumstances. Goodwill impairment is assessed based on a comparison of the fair value of the reporting unit to the underlying carrying value of the reporting unit’s net assets, including goodwill. When the carrying amount of the reporting unit exceeds its fair value, the fair value of the reporting unit’s goodwill is compared with its carrying amount to measure the amount of impairment loss, if any. Prior to the acquisition of SOLX and OcuSense during the second half of fiscal 2006, the Company was a single reporting unit. Therefore, management determined the fair value of its goodwill using the Company’s market capitalization as compared to the fair value of its assets and liabilities.
Foreign currency translation
The Company’s functional and reporting currency is the U.S. dollar. The assets and liabilities of the Company’s Canadian operations are maintained in U.S. dollars. Monetary assets and liabilities denominated in foreign currencies are translated into U.S. dollars at exchange rates in effect at the consolidated balance sheet dates, and non-monetary assets and liabilities are translated at exchange rates in effect on the date of the transaction. Revenue and expenses are translated into U.S. dollars at average exchange rates prevailing during the year. Resulting exchange gains and losses are included in net loss for the year and are not material in any of the years presented.
Clinical and regulatory costs
Clinical and regulatory costs attributable to the performance of contract services are recognized as the services are performed. Non-refundable, up-front fees paid in connection with these contracted services are deferred and recognized as an expense on a straight-line basis over the estimated term of the related contract.
Income taxes
The Company uses the liability method of accounting for income taxes. Deferred tax assets and liabilities are recorded based on the differences between the income tax bases of assets and liabilities and their carrying amounts for financial reporting purposes at the applicable enacted statutory tax rates. Deferred tax assets are reduced by a valuation allowance if, based on the weight of available evidence, it is more likely than not that some portion or all of the deferred tax assets will not be realized.
Stock-based compensation
The Company accounts for stock-based compensation in accordance with the provisions of Statement of Financial Accounting Standard (“SFAS”) No. 123R (revised 2004), “Stock-Based Compensation” (“SFAS No. 123R”). Under the fair value recognition provision of SFAS 123R, stock-based compensation cost is estimated at the grant date based on the fair value of the award and is recognized as an expense ratably over the requisite service period of the award. The Company has selected the Black-Scholes option-pricing model as its method of determining the fair value for all its awards and will recognize compensation cost on a straight-line basis over the awards’ vesting periods (note 14(e)).
Net loss per share
The Company follows SFAS No. 128, “Earnings Per Share” (“SFAS No. 128”). In accordance with SFAS No. 128, companies that are publicly held or have complex capital structures are required to present basic and diluted earnings per share (“EPS”) on the face of the statement of income. Basic EPS excludes dilution and is computed by dividing net loss available to common stockholders by the weighted average number of shares of common stock outstanding for the year. Diluted EPS reflects the potential dilution that could occur if securities or other contracts to issue common stock were exercised or converted and the resulting additional shares are dilutive because their inclusion decreases the amount of EPS.
The following table presents the potentially dilutive effects of outstanding securities:
| | Years ended December 31, |
| | 2006 # | 2005 # | 2004 # |
| | | | |
Weighted average number of shares outstanding - basic | 44,979,692 | 41,931,240 | 7,369,827 |
| Effect of dilutive securities: | | | |
| Stock options | 934,591 | 1,246,809 | 1,498,950 |
Weighted average number of shares outstanding - diluted | 45,914,283 | 43,178,049 | 8,868,777 |
Potentially dilutive securities have not been used in the calculation of diluted loss per share as they are anti-dilutive.
Comprehensive income
The Company follows SFAS No. 130, “Reporting Comprehensive Income” (“SFAS No. 130”). SFAS No. 130 establishes standards for reporting and the presentation of comprehensive income and its components in a full set of financial statements. SFAS No. 130 requires only additional disclosures in the financial statements and does not affect the Company’s financial position or results of operations. For each of the periods presented, comprehensive income (loss) is consistent with the reported loss.
Recent accounting pronouncements
In December 2004, the Financial Accounting Standards Board (“FASB”) issued SFAS No. 123R which revised SFAS No. 123 and supersedes Accounting Principles Board Opinion (“APB”) No. 25. SFAS No. 123R requires all share-based payments to employees, including grants of employee stock options to be recognized in the financial statements based on their fair values. The pro forma disclosure previously permitted under SFAS No. 123 is no longer an alternative to financial statement recognition. SFAS No. 123R is effective at the beginning of the first annual period beginning after June 15, 2005. Accordingly, the Company adopted SFAS No. 123R beginning January 1, 2006. The Company has selected the Black-Scholes option-pricing model as the method of determining the fair value for its awards and will recognize compensation cost on a straight-line basis over the awards’ vesting periods.
The adoption of the following recent accounting pronouncements in fiscal 2006 did not have a material impact on the Company’s results of operations and financial condition:
| · | SFAS No. 151, “Inventory Costs - An Amendment of ARB No. 43, Chapter 4”; |
| · | SFAS No. 153, “Exchanges of Nonmonetary Assets - An Amendment of APB Opinion No. 29”; |
| · | SFAS No. 154, “Accounting Changes and Error Corrections” which replaces APB No. 20, “Accounting Changes”; |
| · | SFAS No. 3, “Reporting Accounting Changes in Interim Financial Statements - An Amendment of APB Opinion No. 28”; and |
| · | FASB Staff Position FAS 115-1 and FAS 124-1, “The Meaning of Other-Than-Temporary Impairment and Its Application to Certain Investments” |
In February 2006, FASB issued SFAS No. 155, "Accounting for Certain Hybrid Financial Instruments - An Amendment of FASB Statements No. 133 and 140" (“SFAS No. 155”). SFAS No. 155 simplifies accounting for certain hybrid instruments currently governed by SFAS No. 133, "Accounting for Derivative Instruments and Hedging Activities" (“SFAS No. 133”), by allowing fair value re-measurement of hybrid instruments that contain an embedded derivative that otherwise would require bifurcation. SFAS No. 155 also eliminates the guidance in SFAS No. 133 Implementation Issue No. D1, "Application of Statement 133 to Beneficial Interests in Securitized Financial Assets", which provides that such beneficial interests are not subject to SFAS No. 133. SFAS No. 155 amends SFAS No. 140, "Accounting for Transfers and Servicing of Financial Assets and Extinguishments of Liabilities - A Replacement of FASB Statement No. 125", by eliminating the restriction on passive derivative instruments that a qualifying special-purpose entity may hold. SFAS No. 155 is effective for financial instruments acquired or issued for fiscal years beginning after September 15, 2006. The Company does not expect the adoption of this statement to have a material impact on its results of operations and financial condition.
In March 2006, FASB issued SFAS No. 156, "Accounting for Servicing of Financial Assets - An Amendment of FASB Statement No. 140" (“SFAS No. 156”). SFAS No. 156 requires an entity to recognize a servicing asset or servicing liability each time it undertakes an obligation to service a financial asset by entering into a servicing contract in specific situations. Additionally, the servicing asset or servicing liability shall be initially measured at fair value; however, an entity may elect the "amortization method" or "fair value method" for subsequent balance sheet reporting periods. SFAS No. 156 is effective for fiscal years beginning after September 15, 2006. The Company does not expect the adoption of this statement to have a material impact on its results of operations and financial condition.
In June 2006, FASB issued FASB Interpretation (“FIN”) No. 48, "Accounting for Uncertainty in Income Taxes - An Interpretation of FASB Statement No. 109" (“FIN No. 48”), which clarifies the accounting for uncertainty in tax positions. FIN No. 48 requires that we recognize, in our financial statements, the impact of a tax position, if that position is more likely than not of being sustained on audit, based on the technical merits of the position. FIN No. 48 is effective for fiscal years beginning after December 15, 2006. The Company does not expect the adoption of FIN No. 48 to have a material effect on the Company’s results of operations and financial position.
In September 2006, the U.S. Securities and Exchange Commission (the “SEC”) released Staff Accounting Bulletin No. 108, “Considering the Effects of Prior Year Misstatements when Quantifying Misstatements in Current Year Financial Statements” (“SAB No. 108”). SAB No. 108 provides interpretive guidance on the SEC’s views regarding the process of quantifying materiality of financial statement misstatements. SAB No. 108 is effective for fiscal years ending after November 15, 2006, and early application for the first interim period of the same fiscal year is encouraged. The application of SAB No. 108 in fiscal 2006 did not have a material effect on the Company’s results of operations and financial position.
In September 2006, FASB issued Statement No. 157, “Fair Value Measurements” (“SFAS No. 157”). SFAS No. 157 defines fair value, establishes a framework and gives guidance regarding the methods used for measuring fair value, and expands disclosures about fair value measurements. SFAS No. 157 is effective for financial statements issued for fiscal years beginning after November 15, 2007 and for interim periods within those fiscal years. The Company is currently evaluating the impact the adoption of SFAS No. 157 would have on its results of operations and financial position.
In September 2006, FASB issued Statement No. 158, “Employers’ Accounting for Defined Benefit Pension and Other Postretirement Plans, an amendment of FASB Statements Nos. 87, 88, 106 and 132(R)” (“SFAS No. 158”). SFAS No. 158 requires companies to recognize the overfunded or underfunded status of a defined benefit post-retirement plan as an asset or liability in their balance sheet and to recognize changes in that funded status in the year in which the changes occur through comprehensive income, effective for fiscal years ending after December 15, 2006. SFAS No. 158 also requires companies to measure the funded status of the plan as of the date of its fiscal year end, with limited exceptions, effective for fiscal years ending after December 15, 2008. The adoption of SFAS No. 158 did not have a material effect on the Company’s results of operations and financial position.
3. ACQUISITIONS
During the two fiscal years ended December 31, 2006 and 2004, the Company completed three acquisitions. The results of each purchase acquisition are included in the Company’s consolidated statements of operations from the date of each acquisition. There were no acquisitions made during fiscal 2005.
The Company’s acquisitions during fiscal 2006 are described below.
SOLX
On September 1, 2006, the Company acquired SOLX, a privately held company that has developed a system for the treatment of glaucoma. The SOLX Glaucoma System developed by SOLX includes the SOLX 790 Laser and the SOLX Gold Shunt which can be used separately or together to provide physicians with multiple options to manage intraocular pressure. The acquisition of SOLX represents an expansion of the Company’s ophthalmic product portfolio beyond the RHEO™ procedure for Dry AMD. The results of SOLX’s operations have been included in the Company’s consolidated financial statements since September 1, 2006.
The Company acquired SOLX by way of a merger, in connection with which the Company issued an aggregate of 8,399,983 shares of its common stock and paid $7,000,000 in cash to the stockholders of SOLX. The Company will make additional payments of $3,000,000 and $5,000,000 in cash on the first and second anniversaries of the September 1, 2006 closing, respectively. In addition, if SOLX receives final FDA approval for the marketing and sale of the SOLX Gold Shunt on or prior to December 31, 2007, the Company will pay an additional $5,000,000 in cash to the former stockholders of SOLX. The stock consideration was valued based on a per share price of $1.79, being the weighted-average closing sale price of the Company’s common stock as traded on the NASDAQ Global Market (“NASDAQ”) over the two-day trading period before and after August 1, 2006, being the date the terms of the acquisition of SOLX were agreed to and announced. The Company has recorded the cash payment expected to be paid on the first anniversary of the closing date as a current liability as of December 31, 2006. The $5,000,000 due on the second anniversary of the closing date has been recorded as a long-term liability at its present value, discounted at the incremental borrowing rate of the Company as at August 1, 2006. The difference between the discounted value and the $5,000,000 payable is being amortized using the effective yield method over the two-year period with the monthly expense being charged as an interest expense in the Company’s consolidated statement of operations. In accordance with SFAS No. 141, “Business Combinations”, the contingent payment of $5,000,000 was not included in the determination of the purchase price or recorded as a liability as the receipt of FDA approval for the marketing and sale of the SOLX Gold Shunt on or prior to December 31, 2007 is subject to many variables, the outcome of which is not determinable beyond reasonable doubt.
The total purchase price of $29,068,443, which includes acquisition-related transaction costs of $851,279, has been allocated as follows:
| | | | $ |
| | | | |
| Net tangible assets | | | (2,908,384) |
| Deferred tax liability | | | (12,270,150) |
| Intangible assets: | | | |
| Shunt and laser technology | | | 27,000,000 |
| Regulatory and other | | | 2,800,000 |
| | | | 14,621,466 |
| Goodwill | | | 14,446,977 |
| | | | 29,068,443 |
Acquisition-related transaction costs include investment banking, legal and accounting fees and other third party costs directly related to the acquisition.
In estimating the fair value of the intangible assets acquired, the Company considered a number of factors, including the valuation performed by an independent third-party valuator that used the income approach to value SOLX’s shunt and laser technology (consisting of the SOLX Gold Shunt and the SOLX 790 Laser) and the cost approach to value the regulatory and other intangible assets acquired (note 7).
The unaudited financial information below summarizes the consolidated results of operations of the Company and SOLX, on a pro forma basis, as though the acquisition of SOLX had been completed on January 1, 2005. The unaudited pro forma information presented below is for information purposes only and may not be indicative of the results of operations as they would have been if the acquisition had occurred on January 1, 2005, nor is it necessarily indicative of future results of operations.
| | | | December 31, |
| | | | 2006 | | 2005 |
| | | | $ | | $ |
| | | | | | | | |
| Revenue | | | 251,763 | | | 1,931,590 | |
| Loss before cumulative effect of a change in accounting principle | | | (86,412,413) | | | (169,117,548) | |
| Net loss for the year | | | (86,305,368) | | | (169,117,548) | |
| Net loss per share - basic and diluted | | | (1.71) | | | (3.36) | |
OcuSense
On November 30, 2006, the Company acquired 50.1% of the capital stock of OcuSense, measured on a fully diluted basis. OcuSense’s first product under development is a hand-held tear film osmolarity test for the diagnosis and management of dry eye syndrome, or DES, known as the TearLab™ test for DES. The results of OcuSense’s operations have been included in the Company’s consolidated financial statements since November 30, 2006.
Pursuant to the terms of the Series A Preferred Stock Purchase Agreement (the “Series A Preferred Stock Purchase Agreement”), dated as of November 30, 2006, between OcuSense and the Company, the Company purchased 1,744,223 shares of OcuSense’s Series A Preferred Stock, par value $0.001 per share, representing 50.1% of OcuSense’s capital stock on a fully diluted basis for an aggregate purchase price of up to $8,000,000 (the “Purchase Price”). On the closing of the purchase which took place on November 30, 2006, the Company paid $2,000,000 of the Purchase Price. The Company paid another $2,000,000 installment of the Purchase Price on January 3, 2007. The Company will pay the third $2,000,000 installment of the Purchase Price upon the attainment by OcuSense of the first of two pre-defined milestones and the last $2,000,000 installment of the Purchase Price upon the attainment by OcuSense of the second of such milestones, provided that both milestones are achieved prior to May 1, 2009. The contingent payments totaling $4,000,000 were not included in the determination of the purchase price or recorded as a liability as the attainment by OcuSense of the two pre-determined milestones prior to May 1, 2009 is subject to many variables, the outcome of which is not determinable beyond reasonable doubt.
The Series A Preferred Stock Purchase Agreement also makes provision for an ability on the part of the Company to increase its ownership interest in OcuSense for nominal consideration if OcuSense fails to meet certain other milestones by specified dates. In addition, pursuant to the Series A Preferred Stock Purchase Agreement, the Company has agreed to purchase $3,000,000 of shares of OcuSense’s Series B Preferred Stock, which shall constitute 10% of OcuSense’s capital stock on a fully diluted basis at the time of purchase, upon OcuSense’s receipt from the FDA of 510(k) clearance for the TearLab™ test for DES and to purchase another $3,000,000 of shares of OcuSense’s Series B Preferred Stock, which shall constitute an additional 10% of OcuSense’s capital stock on a fully diluted basis at the time of purchase, upon OcuSense’s receipt from the FDA of Clinical Laboratory Improvement Amendments, or CLIA, waiver for the TearLab™ test for DES.
The total purchase price of $4,171,098 includes acquisition-related transaction costs of $171,098. Acquisition-related transaction costs include legal fees and other third party costs directly related to the acquisition.
The purchase price of $4,171,098 has been allocated as follows:
| | | | $ |
| | | | |
| Net tangible assets | | | 1,347,848 |
| Deferred tax liability | | | (1,882,166) |
| Intangible asset | | | 4,705,416 |
| | | | 4,171,098 |
In estimating the fair value of the intangible assets acquired, the Company considered a number of factors, including the valuation performed by an independent third-party valuator that used the income approach to value OcuSense’s TearLab™ technology (note 7).
If the Company’s acquisition of 50.1% of the capital stock of OcuSense, measured on a fully diluted basis, had been completed on January 1, 2005, the effect on the pro forma statements of operations would have been to increase net loss by $1,320,036 and $378,224 for the years ended December 31, 2006 and 2005, respectively. Net loss per share would have increased by $0.03 and $0.01 for the years ended December 31, 2006 and 2005, respectively. There is no pro forma effect on the Company’s revenue for each of the years ended December 31, 2006 and 2005.
The unaudited pro forma information is presented for information purposes only and may not be indicative of the results of operations if the acquisition had occurred on January 1, 2005, nor is it necessarily indicative of future results of operations.
The Company’s acquisition during fiscal 2004 is described below.
OccuLogix, L.P.
On December 8, 2004, as part of the Company’s reorganization transactions (note 14(b)), the Company acquired 50% interest in OccuLogix, L.P. (the “Partnership”) then held by TLC Vision Corporation (“TLC Vision”) in exchange for the issuance to TLC Vision of 19,070,234 shares of the Company’s common stock. The stock consideration was valued based on the Company’s initial offering share price of $12.00 per share. The results of the Partnership’s operations have been included in the consolidated financial statements since that date.
The purchase price of the acquisition consisted of 19,070,234 shares of the Company’s common stock valued at $228,842,808, plus acquisition costs of $768,808 for a total acquisition cost of $229,611,616. The purchase price was allocated as follows:
| | | $ |
| | | |
| Net tangible assets | | (8,328) |
| Deferred tax liability | | (9,527,500) |
| Intangible asset | | 25,750,000 |
| | | 16,214,172 |
| Goodwill | | 213,397,444 |
| | | 229,611,616 |
If the acquisition of TLC Vision’s 50% share of the Partnership had been completed by January 1, 2004, the unaudited pro forma effects on the consolidated statements of operations for the year ended December 31, 2004 would have been to decrease revenue by $166,634 and to increase net loss by $1,008,371. As a result of the impact of the above pro forma change to net loss, combined with the dilutive effect from the increased number of shares, the unaudited net loss per share for the year ended December 31, 2004 would have been reduced by $2.06. There is no pro forma effect on the consolidated statements of operations and the net loss per share for the years ended December 31, 2006 and 2005 as the results of the Partnership’s operations are included in the Company’s consolidated financial statements.
The unaudited pro forma information is presented for information purposes only and may not be indicative of the results of operations if the acquisition had occurred on January 1, 2004, nor is it necessarily indicative of future results of operations.
On December 31, 2005, as part of the Company’s continued reorganization, the Partnership transferred all of its assets and liabilities, including the licensed patent, trademark and know-how rights for the RHEO™ System, to the Company’s then newly incorporated subsidiary, OccuLogix Canada Corp. The Company completed the wind-up of the Partnership on February 6, 2006 (note 14(b)).
4. GOODWILL
The Company follows the provisions of SFAS No. 142, “Goodwill and Other Intangible Assets” (“SFAS No. 142”), which requires that goodwill not be amortized but instead be tested for impairment at least annually and more frequently if circumstances indicate possible impairment.
The Company’s goodwill amount by reporting unit is as follows:
| | | Retina | | Glaucoma | | Total | |
| | | $ | | $ | | $ | |
| | | | | | | | | | | |
| Balance, December 31, 2004 | | | 213,397,444 | | | — | | | 213,397,444 | |
| Impairment loss recognized | | | (147,451,758 | ) | | — | | | (147,451,758 | ) |
| Balance, December 31, 2005 | | | 65,945,686 | | | — | | | 65,945,686 | |
| Acquired during the period | | | — | | | 14,446,977 | | | 14,446,977 | |
| Impairment loss recognized | | | (65,945,686 | ) | | — | | | (65,945,686 | ) |
| Balance, December 31, 2006 | | | — | | | 14,446,977 | | | 14,446,977 | |
The Company performs its annual goodwill impairment analysis on its acquired goodwill on October 1 of each year and evaluates the carrying value of its goodwill between annual tests upon the occurrence of certain events and circumstances.
Retina
The Company conducted a pivotal clinical trial, called MIRA-1, which, if successful, was expected to support its application to the FDA to obtain approval to market the RHEO™ System in the United States. On February 3, 2006, the Company announced that, based on a preliminary analysis of the data from MIRA-1, MIRA-1 did not meet its primary efficacy endpoint as it did not demonstrate a statistically significant difference in the mean change of ETDRS BCVA between the treated and placebo groups in MIRA-1 at 12 months post-baseline. As a result of the announcement on February 3, 2006, the per share price of the Company’s common stock as traded on NASDAQ decreased from $12.75 on February 2, 2006 to close at $4.10 on February 3, 2006. The 10-day average price of the stock immediately following the announcement was $3.65 and reflected a decrease in the Company’s market capitalization from $536.6 million on February 2, 2006 to $153.6 million based on the 10-day average share price subsequent to the announcement. Based on this, the Company concluded that there were sufficient indicators to require management to re-assess whether the Company’s recorded goodwill was impaired as of December 31, 2005. Prior to the acquisition of SOLX and OcuSense during the second half of fiscal 2006, the Company was a single reporting unit. Therefore, management determined the fair value of the Company's goodwill using the Company’s market capitalization as opposed to the fair value of its assets and liabilities. The Company recorded a goodwill impairment charge of $147,451,758 during the year ended December 31, 2005 as a result of a goodwill impairment re-assessment performed subsequent to the February 3, 2006 announcement.
On June 12, 2006, the Company announced that it met with the FDA to discuss the results of MIRA-1 and confirmed that the FDA will require the Company to perform an additional study of the RHEO™ System to obtain approval to market the RHEO™ System in the United States. In addition, on June 30, 2006, the Company announced that it had terminated negotiations with Sowood Capital Management LP in connection with a proposed private purchase of approximately $30,000,000 of zero-coupon convertible notes of the Company. In accordance with SFAS No. 142, the Company concluded that, based on the price of the Company’s common stock subsequent to the June 12, 2006 announcement and again after the June 30, 2006 announcement, there were sufficient indicators to require management to re-assess whether the Company’s recorded goodwill was impaired as at June 30, 2006. Based on the goodwill impairment analysis performed, the Company concluded that a further goodwill impairment charge of $65,945,686 should be recorded during the second quarter of 2006.
Glaucoma
On September 1, 2006, the Company acquired SOLX by way of a merger for a total purchase price of $29,068,443 (note 3). Of this amount, $14,446,977 has been allocated to goodwill. The Company determined that, as of December 31, 2006, no significant event or circumstance has occurred that may lead to an impairment of its acquired goodwill. The Company further determined that there have not been any significant changes in the methods used to value the net tangible and intangible assets acquired. Therefore, no impairment charge was required to be recorded during the year ended December 31, 2006.
5. FIXED ASSETS
| | 2006 | 2005 |
| | Cost | Accumulated Amortization | Cost | Accumulated Amortization |
| | $ | $ | $ | $ |
| | | | | |
| Furniture and office equipment | 119,776 | 49,566 | 52,077 | 23,924 |
| Computer equipment and software | 268,955 | 145,001 | 155,194 | 53,345 |
| Medical equipment | 1,805,228 | 1,138,675 | 846,555 | 505,996 |
| | 2,193,959 | 1,333,242 | 1,053,826 | 583,265 |
| Less accumulated amortization | 1,333,242 | | 583,265 | |
| | 860,717 | | 470,561 | |
Amortization expense was $213,488, $99,301 and $42,956 during the years ended December 31, 2006, 2005 and 2004, respectively.
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
6. PATENTS AND TRADEMARKS
| | 2006 | 2005 |
| | Cost | Accumulated Amortization | Cost | Accumulated Amortization |
| | $ | $ | $ | $ |
| | | | | |
| Patents | 139,461 | 14,909 | 95,289 | 10,843 |
| Trademarks | 117,513 | 7,224 | 56,468 | 5,682 |
| | 256,974 | 22,133 | 151,757 | 16,525 |
| Less accumulated amortization | 22,133 | | 16,525 | |
| | 234,841 | | 135,232 | |
Estimated amortization expense for patents and trademarks for each of the next five years is as follows:
| | Patents $ | Trademarks $ | Total $ |
| | | | |
| 2007 | 4,066 | 3,760 | 7,826 |
| 2008 | 4,066 | 3,760 | 7,826 |
| 2009 | 4,066 | 3,760 | 7,826 |
| 2010 | 4,066 | 3,760 | 7,826 |
| 2011 | 4,066 | 3,760 | 7,826 |
| | 20,330 | 18,800 | 39,130 |
7. INTANGIBLE ASSETS
The Company’s intangible assets consist of the value of the exclusive distribution agreements that the Company has with its major suppliers and other acquisition-related intangibles. The Company has no indefinite-lived intangible assets. The distribution agreements and other acquisition-related intangible assets are amortized using the straight-line method over an estimated useful life of 15 and 10 years, respectively. Amortization expense for the years ended December 31, 2006, 2005 and 2004 was $2,749,212, $1,716,667 and $106,138, respectively.
As of December 31, 2006, the remaining weighted average amortization period for the distribution agreements is 13 years and 9.7 years for the other acquisition-related intangible assets.
Intangible assets subject to amortization consist of the following:
| | | December 31, | |
| | | 2006 | | 2005 | |
| | | Gross Carrying Amount | | Accumulated Amortization | | Gross Carrying Amount | | Accumulated Amortization | |
| | | | $ | | | $ | | | $ | | | $ | |
| | | | | | | | | | | | | | |
| Distribution agreements | | | 25,750,000 | | | 3,539,472 | | | 25,750,000 | | | 1,822,805 | |
| Shunt and laser technology | | | 27,000,000 | | | 900,000 | | | — | | | — | |
| Regulatory and other | | | 2,800,000 | | | 93,333 | | | — | | | — | |
| TearLab™ technology | | | 4,705,416 | | | 39,212 | | | — | | | — | |
| | | | 60,255,416 | | | 4,572,017 | | | 25,750,000 | | | 1,822,805 | |
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
Estimated amortization expense for the intangible assets for each of the below-listed fiscal years ending December 31 is as follows:
| | | | $ |
| | | | |
| 2007 | | | 5,167,208 |
| 2008 | | | 5,167,208 |
| 2009 | | | 5,167,208 |
| 2010 | | | 5,167,208 |
| 2011 | | | 5,167,208 |
| | | | 29,847,359 |
As a result of the analysis of the data from MIRA-1 and the result of the meeting held with the FDA in June 2006, the Company concluded that there were sufficient indications that the carrying value of the distribution intangible may no longer be recoverable, thus requiring management to assess whether the Company’s distribution intangible was impaired as of June 30, 2006. Based on management’s estimates of forecasted undiscounted cash flows as of June 30, 2006, the Company concluded that there is no impairment of the Company’s distribution intangible.
The Company determined that, as at December 31, 2006, there have been no significant events which may affect the carrying value of its distribution intangible and no significant changes in the methods used to determine the fair market value of other acquisition-related intangible assets. However, the Company’s prior history of losses and losses incurred during the current fiscal year reflects a potential indication of impairment, thus requiring management to assess whether the Company’s intangible assets were impaired as of December 31, 2006. Based on management’s estimates of forecasted undiscounted cash flows as of December 31, 2006, the Company concluded that there is no indication of an impairment of the Company’s intangible assets. Therefore, no impairment charge was recorded during the year ended December 31, 2006.
8. RESTRUCTURING CHARGES
In March 2006, the Company implemented a number of structural and management changes designed to support both the continued development of its RHEO™ System and to execute its accelerated diversification strategy within ophthalmology. In accordance with SFAS No. 146, “Accounting for Costs Associated with Exit or Disposal Activities”, the Company recognized a total of $819,642 in restructuring charges in the year ended December 31, 2006.
The restructuring charges of $819,642 recorded in the year ended December 31, 2006 consist solely of severance and benefit costs related to the termination of a total of 12 employees at both the Company’s Mississauga, Ontario and Palm Harbor, Florida offices. All severance and benefit costs have been fully paid as at December 31, 2006.
9. DUE TO STOCKHOLDERS
| | December 31, |
| | 2006 $ | 2005 $ |
| | | |
Due to | | |
TLC Vision Corporation (note 10) | 91,884 | 116,070 |
Other stockholders (note 10) | 60,522 | 42,163 |
| | 152,406 | 158,233 |
The balance owing to TLC Vision of $91,884 and $116,070 as of December 31, 2006 and 2005, respectively, is related to computer and administrative support provided by TLC Vision, all of which has been expensed during the years ended December 31, 2006 and 2005, respectively, and included in general and administrative expenses.
10. RELATED PARTY TRANSACTIONS
The following are the Company’s related party transactions in addition to those disclosed in notes 9 and 13.
(a)RHEO Clinic Inc.
| | | December 31, |
| | | 2006 | | 2005 |
| | | $ | | $ |
| | | | | | | |
Due to | | | | | | |
| RHEO Clinic Inc. | | | — | | | 5,065 |
One of the Company’s primary customers had been RHEO Clinic Inc., a subsidiary of TLC Vision. RHEO Clinic Inc. used the RHEO™ System to treat patients for which it charged its customers (the patients) a per-treatment fee. During the third quarter of 2005, RHEO Clinic Inc. determined that it will no longer treat patients and subsequently sold certain of its assets to the Company at a purchase price of C$61,812, including all applicable taxes. In connection with that sale, the Company agreed to share equally in losses incurred by RHEO Clinic Inc., to a maximum of C$28,952, for assets that RHEO Clinic Inc. is not able to dispose of as at the agreed date, being December 31, 2005. On May 1, 2006, the Company paid RHEO Clinic Inc. C$31,859 which included the amount owing for losses incurred for assets that RHEO Clinic Inc. was not able to dispose of as at the agreed date. Included in the balance due to RHEO Clinic Inc. as at December 31, 2005 is the amount owing to the Company for the purchase of the components of the RHEO™ System, net of the amount owing to RHEO Clinic Inc. for clinical and administrative support provided by RHEO Clinic Inc. for the Company’s MIRA-1 and related clinical trials.
(b)TLC Vision and Diamed
On June 25, 2003, the Company entered into agreements with TLC Vision and Diamed to issue grid debentures in the maximum aggregate principal amount of $12,000,000 in connection with the funding of the Company’s MIRA-1 and related clinical trials. $7,000,000 of the aggregate principal amount was convertible into shares of the Company’s common stock at a price of $0.98502 per share, and $5,000,000 of the aggregate principal amount was non-convertible.
The $5,000,000 portion of the $12,000,000 commitment which was not convertible into the Company’s common stock was not advanced and the commitment was terminated prior to the completion of the Company’s initial public offering of shares of its common stock. During the years ended December 31, 2004 and 2003, the Company issued an aggregate of $4,350,000 and $2,650,000 to TLC Vision and Diamed, respectively, under the convertible portion of the grid debentures. On December 8, 2004, as part of the corporate reorganization relating to the Company’s initial public offering, the Company issued 7,106,454 shares of its common stock to TLC Vision and Diamed, upon conversion of $7,000,000 of aggregate principal amount of convertible debentures at a conversion price of $0.98502 per share. Collectively, as at December 31, 2006, the two companies have a combined 48.6% equity interest in the Company on a fully diluted basis.
If and when the Company receives FDA approval to market the RHEO™ System in the United States, it will be economically dependent on Diamed to control the supply of the OctoNova pumps used in the RHEO™ System. The Company believes that the OctoNova pump is a critical component of the RHEO™ System.
(c)Asahi Medical
Since 2001, the Company has been party to a distributorship agreement with Asahi Medical pursuant to which Asahi Medical supplies the filter products used in the RHEO™ System.
The Company is economically dependent on Asahi Medical to continuously provide filters and believes that the filter products provided by Asahi Medical are a critical component in the RHEO™ System.
The Company entered into a new distributorship agreement (the “2006 Distributorship Agreement”), effective October 20, 2006, with Asahi Medical. The 2006 Distributorship Agreement replaced the 2001 distributorship agreement between the Company and Asahi Medical, as supplemented and amended by the 2003, 2004 and 2005 Memoranda. Pursuant to the 2006 Distributorship Agreement, the Company has distributorship rights to Asahi Medical's Plasmaflo filter and Asahi Medical's second generation polysulfone Rheofilter filter on an exclusive basis in the United States, Mexico and certain Caribbean countries (collectively, “Territory 1-a”), on an exclusive basis in Canada, on an exclusive basis in Colombia, Venezuela, New Zealand and Australia (collectively, “Territory 2”) and on a non-exclusive basis in Italy.
Pursuant to the 2006 Distributorship Agreement, the Company will be responsible for obtaining regulatory approvals for the Rheofilter filters and Plasmaflo filters for use in the treatment of AMD in Territory 1-a, Territory 2 and Italy by December 31, 2010 and in Canada by February 28, 2009. With the exception of the FDA approval of the RHEO™ System, all of such regulatory approvals, when and if obtained, will be held in Asahi Medical’s name. The FDA approval of the RHEO™ System will be held by a special purpose corporation, to be owned as to 51% by Asahi Medical and as to 49% by the Company. Under the 2006 Distributorship Agreement, the Company will be responsible for covering costs relating to the pursuit of regulatory approvals in Territory 1-a, Canada and Territory 2, and the Company and Asahi Medical will share the costs relating to the pursuit of regulatory approval in Italy. In addition, provided that certain conditions are met, Asahi Medical will be obligated to contribute $3,000,000 toward the cost of RHEO-AMD, the Company's new pivotal clinical trial of the RHEO™ System which is intended to support the Company's Pre-Market Approval application to the FDA.
With respect to the United States, subject to early termination under certain circumstances, the 2006 Distributorship Agreement has a term which will end ten years following the date on which FDA approval to market the RHEO™ System in the United States is received and contemplates successive one-year renewal terms thereafter.
The Company is subject to certain minimum purchase requirements in each of the territories covered by the 2006 Distributorship Agreement.
The Company received free inventory from Asahi Medical for the purpose of the MIRA-1, LEARN or Long-term Efficacy in AMD from Rheopheresis in North America and related clinical studies. The Company has accounted for this inventory at a value equivalent to the cost the Company has paid for the same filters purchased from Asahi Medical for purposes of commercial sales to the Company’s customers. During fiscal 2006, the Company returned 300 filters, valued at $60,000, to Asahi Medical as the filters were labeled for commercial use and cannot be utilized for clinical trials. The value of the free inventory received from Asahi Medical was nil and $167,730 for the years ended December 31, 2006 and 2005, respectively.
(d)Mr. Hans Stock (note 9)
On February 21, 2002, the Company entered into an agreement with Mr. Stock as a result of his assistance in procuring a distributor agreement for the filter products used in the RHEO™ System from Asahi Medical. Mr. Stock agreed to further assist the Company in procuring new product lines from Asahi Medical for marketing and distribution by the Company. The agreement will remain effective for a term consistent with the term of the distributorship agreement with Asahi Medical, and Mr. Stock will receive a 5% royalty payment on the purchase of the filters from Asahi Medical. During the years ended December 31, 2006 and 2005, the Company paid Mr. Stock nil and $240,657, respectively, as royalty fees. Included in due to stockholders as at December 31, 2006 and 2005 is $48,022 and $29,663, respectively, due to Mr. Stock.
On June 25, 2002, the Company entered into a consulting agreement with Mr. Stock for the purpose of procuring a patent license for the extracorporeal applications in ophthalmic diseases for that period of time in which the patent was effective. Mr. Stock was entitled to 1.0% of total net revenue from the Company’s commercial sales of products sold in reliance and dependence upon the validity of the patent’s claims and rights in the United States. The Company agreed to make advance consulting payments to Mr. Stock of $50,000 annually, payable on a quarterly basis, to be credited against any and all future consulting payments payable in accordance with this agreement. Due to the uncertainty of future royalty payment requirements, all required payments to date have been expensed.
On August 6, 2004, the Company entered into a patent license and royalty agreement with Mr. Stock to obtain an exclusive license to U.S. Patent No. 6,245,038. The Company is required to make royalty payments totaling 1.5% of product sales to Mr. Stock, subject to minimum advance royalty payments of $12,500 per quarter. The advance payments are credited against future royalty payments to be made in accordance with the agreement. This agreement replaces the June 25, 2002 consulting agreement with Mr. Stock which provided for a royalty payment of 1% of product sales. In each of the years ended December 31, 2006 and 2005, the Company paid $50,000 to Mr. Stock as royalty fees. Included in due to stockholders as at December 31, 2006 and 2005 is $12,500 and $12,500, respectively, due to Mr. Stock.
(e)Apheresis Technologies, Inc.
On May 1, 2002, the Company entered into an exclusive distribution services agreement with Apheresis Technologies, Inc. (“ATI”), a company controlled by certain stockholders of the Company pursuant to which the Company paid ATI 5% of the Company’s cost of components of the RHEO™ System. Under this agreement, ATI was the exclusive provider of warehousing, order fulfillment, shipping, billing services and customer service related to shipping and billing to the Company.
On July 30, 2004, the Company amended its distribution services agreement with ATI such that the Company would have the sole discretion as to when the agreement would terminate. In consideration of this amendment, the Company agreed to pay ATI $100,000 on the successful completion of the Company’s initial public offering. On January 18, 2005, the Company paid ATI $100,000 as provided for in the amended distribution services agreement. On March 28, 2005, the Company terminated its distribution services agreement with ATI.
(f)Other
On June 25, 2003, the Company entered into a reimbursement agreement with ATI, pursuant to which employees of ATI, including John Cornish, one of the Company’s stockholders and its Vice President, Operations, provide services to the Company and ATI is reimbursed for the applicable percentage of time the employees spend working for the Company. Effective April 1, 2005, the Company terminated its reimbursement agreement with ATI such that the Company no longer compensates ATI in respect of any salary paid to, or benefits provided to, Mr. Cornish by ATI. Until April 1, 2005, Mr. Cornish did not have an employment contract with the Company and received no direct compensation from the Company. On April 1, 2005, Mr. Cornish entered into an employment agreement with the Company under which he received an annual base salary of $106,450, representing compensation to him for devoting 80% of his time to the business and affairs of the Company. Effective June 1, 2005, the Company amended its employment agreement with Mr. Cornish such that he began to receive an annual base salary of $116,723, representing compensation to him for devoting 85% of his time to the business and affairs of the Company. Effective April 13, 2006, the Company further amended its employment agreement with Mr. Cornish such that his annual base salary was decreased to $68,660 in consideration of his devoting 50% of his time to the business and affairs of the Company. Mr. Cornish continues to participate in the Company’s bonus plan and is entitled to receive, and has received, stock options pursuant to the Stock Option Plan.
Effective January 1, 2004, the Company entered into a rental agreement with Cornish Properties Corporation, a company owned and managed by Mr. Cornish, pursuant to which the Company leases space from Cornish Properties Corporation at $2,745 per month. The original term of the lease extended to December 31, 2005. On November 8, 2005, as provided for in the rental agreement, the Company extended the term of the rental agreement with Cornish Properties Corporation for another year, ending December 31, 2006. In each of the years ended December 31, 2006 and 2005, the Company paid Cornish Properties Corporation an amount of $32,940 as rent. On December 19, 2006, the Company extended the term of the rental agreement with Cornish Properties Corporation for another year, ending December 31, 2007, at a lease payment of $2,168 per month.
Effective June 25, 2003, Elias Vamvakas, then the Chairman of TLC Vision, became the Chairman and Secretary of the Company. 500,000 stock options issued to Mr. Vamvakas in December 2003 were accounted for in accordance with APB No. 25. The Company estimated the intrinsic value of these options granted to Mr. Vamvakas to be approximately $5,880,000. Management estimated the fair value of the underlying common stock based on management’s estimate of the Company’s value. The intrinsic value of the options is being amortized over the vesting period. However, upon the successful completion of the Company’s initial public offering in December 2004, the options vested immediately, and therefore, any unvested compensation expense was expensed immediately.
On November 30, 2006, the Company announced that Mr. Vamvakas had agreed to provide the Company with a standby commitment to purchase convertible debentures of the Company (“Convertible Debentures”) in an aggregate maximum amount of $8,000,000 (the “Total Commitment Amount”). Pursuant to the Summary of Terms and Conditions, executed and delivered as of November 30, 2006 by the Company and Mr. Vamvakas, during the 12-month commitment term commencing on November 30, 2006, upon no less than 45 days’ written notice by the Company to Mr. Vamvakas, Mr. Vamvakas was obligated to purchase Convertible Debentures in the aggregate principal amount specified in such written notice. A commitment fee of 200 basis points was payable by the Company on the undrawn portion of the total $8,000,000 commitment amount. Any Convertible Debentures purchased by Mr. Vamvakas would have carried an interest rate of 10% per annum and would have been convertible, at Mr. Vamvakas’ option, into shares of the Company’s common stock at a conversion price of $2.70 per share. The Summary of Terms and Conditions of the standby commitment further provided that if the Company closes a financing with a third party, whether by way of debt, equity or otherwise and there are no Convertible Debentures outstanding, then, the Total Commitment Amount was to be reduced automatically upon the closing of the financing by the lesser of: (i) the Total Commitment Amount; and (ii) the net proceeds of the financing. On February 6, 2007, the Company raised gross proceeds in the amount of $10,016,000 in a private placement of shares of its common stock and warrants. The Total Commitment Amount was therefore reduced to zero, thus effectively terminating Mr. Vamvakas’ standby commitment. No portion of the standby commitment was ever drawn down by the Company, and the Company paid Mr. Vamvakas a total of $29,808 in commitment fees in February 2007. (note 18).
The Company entered into a consultancy and non-competition agreement on July 1, 2003 with the Center for Clinical Research (“CCR”), then a significant shareholder of the Company, which requires the Company to pay a fee of $5,000 per month. For the year ended December 31, 2003, CCR agreed to forego the payment of $75,250 due to it in exchange for options to purchase 20,926 shares of the Company’s common stock at an exercise price of $0.13 per share. In addition, CCR agreed to the repayment of the balance of $150,500 due to it at a rate of $7,500 per month beginning in July 2003. On August 22, 2005, the Company amended the consultancy and non-competition agreement with CCR such that the fee payable to it was increased from $5,000 to $15,000 per month effective January 1, 2005. The monthly fee is fixed regardless of actual time incurred by CCR in performance of the services rendered to the Company. The agreement allows either party to convert the payment arrangement to a fee of $2,500 daily. In the event of such conversion, CCR shall provide services on a daily basis as required by the Company and will invoice the Company for the total number of days that services were provided in that month. The amended consultancy and non-competition agreement provides for the payment of a one-time bonus of $200,000 upon receipt by the Company of FDA approval of the RHEO™ System and the grant of 60,000 options to CCR at an exercise price of $7.15 per share. The stockholders of the Company approved the adjustment of the exercise price of these options to $2.05 per share on June 23, 2006 (note 14(e)). These options were scheduled to vest as to 100% when and if the Company receives FDA approval of the RHEO™ System on or before November 30, 2006, as to 80% when and if the Company receives FDA approval after November 30, 2006 but on or before January 31, 2007 and as to 60% when and if the Company receives FDA approval after January 31, 2007. In August 2006, by letter agreement between the Company and CCR, it was agreed that the monthly fee of $15,000 would be suspended at the end of August 2006 until CCR’s services will be required by the Company in the future. This resulted in a combined consulting expense, included within clinical and regulatory expense for the years ended December 31, 2006 and 2005, of $147,857 and $249,831, respectively.
On September 29, 2004, the Company signed a product purchase agreement with Veris Health Sciences Inc. (“Veris”) (formerly RHEO Therapeutics, Inc.) for its purchase from the Company of 8,004 treatment sets over the period from October 2004 to December 2005, a transaction valued at $6,003,000, after introductory rebates. However, due to delays in opening its planned number of clinics throughout Canada, Veris no longer required the contracted-for number of treatment sets in the period. The Company agreed to the original pricing for the reduced number of treatment sets required in the period. Dr. Jeffrey Machat, who is an investor in, and one of the directors of, Veris, was a co-founder and former director of TLC Vision. In December 2005, by letter agreement, the Company agreed to the volume and other terms for the purchase and sale of treatment sets and pumps for the period ending February 28, 2006. As at December 31, 2005, the Company had received a total of $1,779,566 from Veris. Included in amounts receivable, net as at December 31, 2005, was $1,047,622 due from Veris for the purchase of additional pumps and treatment sets. Veris agreed to the payment of interest at the rate of 8% per annum on all amounts outstanding for more than 45 days up to March 31, 2006, the expected date of final payment. In January 2006, the Company received from Veris an interest payment of $4,495 on amounts outstanding for more than 45 days to December 31, 2005. On February 3, 2006, the Company announced that the MIRA-1 clinical trial had not met its primary efficacy endpoint and that it would be more likely than not that the Company will be required to conduct a follow-up clinical trial of the RHEO™ System in order to support its Pre-Market Approval application to the FDA. Because of this delay in being able to pursue commercialization of the RHEO™ System in the United States and the resulting market reaction to this news and based on discussions with Veris, the Company believed that Veris would not be able to meet its financial obligations to the Company. Therefore, during the year ended December 31, 2005, the Company recorded an allowance for doubtful accounts of $1,047,622 against the amount due from Veris and did not accrue additional interest on the amount outstanding during the year ended December 31, 2006.
In April 2006, the Company agreed to sell a total of 1,000 treatment sets, with a negotiated discount, to Veris at a price of $200 per treatment set, which is lower than the Company’s cost. It was also agreed that payment for the treatment sets must be received by the Company in advance of shipment. In July 2006, Veris negotiated new payment terms with the Company, and it was agreed that payment for treatment sets shipped subsequent to June 2006 must be received within 60 days of shipment. The Company also agreed that all sales of treatment sets made to Veris to the end of 2006 will remain at the discounted price of $200 per treatment set. During the year ended December 31, 2006, the Company received a total of $171,800 from Veris for the purchase of 1,207 treatment sets. The sale of the treatment sets is included in revenue for the year ended December 31, 2006 as all the treatment sets have been delivered to Veris. In November 2006, the Company sold 348 treatment sets to Veris for $73,776, including applicable taxes, payment for which was not received by the Company within the agreed 60-day credit period. The sale of these treatment sets was not recognized as revenue for the year ended December 31, 2006 as the Company believed that Veris would not be able to meet its financial obligations to the Company. In January 2007, the Company met with the management of Veris and agreed to forgive the outstanding amount receivable of $73,776 for the purchase of 348 treatment sets delivered to Veris in November 2006. This amount is therefore not included in amounts receivable as of December 31, 2006. In addition, the Company has recorded an inventory loss of $60,987 in the year ended December 31, 2006 for the sale of these 348 treatment sets since these treatment sets have been delivered to Veris already.
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
In June 2006, Veris returned four pumps which had been sold to it in December 2005. In fiscal 2005, the Company had recorded an inventory loss associated with all sales made to Veris in December 2005 and did not recognize revenue due to the Company’s anticipation that Veris may not return the products shipped to it and would not be able to pay for the amounts invoiced. Accordingly, during fiscal 2006, amounts receivable, net and the allowance for doubtful account recorded against the amount due from Veris have been reduced by the invoiced amount for the four pumps of $143,520. In addition, the cost of the four pumps returned by Veris, valued at $85,058, was used to reduce the cost of goods sold in the period.
On November 6, 2006, the Company amended its product purchase agreement with Veris and agreed to forgive the outstanding amount receivable of $904,101 from Veris which had been owing for the purchase of treatment sets and pumps and for related services delivered or provided to Veris during the period from September 14, 2005 to December 31, 2005. In consideration of the forgiveness of this debt, Veris agreed that the Company did not owe Veris any amounts whatsoever in connection with (i) the use by the Company of the leasehold premises located at 5280 Solar Drive in Mississauga, Ontario or (ii) legal fees and expenses incurred by Veris prior to February 14, 2006 with respect to certain of Veris’ trademarks that had been assigned to the Company, and licensed back to Veris, on February 14, 2006.
The Company also entered into a clinical trial agreement on November 22, 2005 with Veris which required Veris to provide certain clinical trial services to the Company. The agreement provided for an advance payment of C$195,000 to Veris which represents 30% of the total value of the contract. The Company paid Veris C$195,000 on November 22, 2005 as provided for in the clinical trial agreement. This amount has been expensed during the year ended December 31, 2005 as the Company has suspended the clinical trial in question.
In October 2003, the Company’s stockholders created a new company called Rheogenx BioSciences Corporation (“Rheogenx”) to further develop the use of the current components of the RHEO™ System for non-ophthalmic uses. On March 28, 2005, the Company entered into a supply and co-marketing agreement with Rheogenx for the supply of pumps and disposable treatment sets to Rheogenx and its affiliates, including PhereSys Therapeutics Corporation (“PhereSys”), Rheogenx’s wholly-owned subsidiary. Under this agreement, the Company will provide marketing support for PhereSys’s mobile apheresis business upon obtaining FDA approval to market the RHEO™ System in the United States. In connection with entering into this agreement, the Company also entered into an asset purchase agreement with Rheogenx on March 28, 2005 to effectively terminate the patent, know-how and trademark rights to non-ophthalmic indications for the RHEO™ System in North America which the Company had previously licensed to Rheogenx. The purchase price of the assets under the asset purchase agreement was $10 and has been included within accrued liabilities.
During the fourth quarter of 2004, the Company began a business relationship with Innovasium Inc. Innovasium Inc. designed and built some of the Company’s websites and also created some of the sales and marketing materials to reflect the look of the Company’s websites. Daniel Hageman, who is the President and one of the owners of Innovasium Inc., is the spouse of an officer of the Company. During the years ended December 31, 2006 and 2005, the Company paid Innovasium Inc. C$44,219 and C$123,967, respectively. Included in accounts payable and accrued liabilities as at December 31, 2006 and 2005 is nil and C$15,798, respectively, due to Innovasium Inc. These amounts are expensed in the period incurred and paid when due.
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
11. INCOME TAXES
Significant components of the Company’s deferred tax assets and liabilities are as follows:
| | December 31, |
| | 2006 $ | 2005 $ |
| | | |
Deferred tax assets | | |
| Intangibles | 1,547,214 | ― |
| Fixed assets | 3,457 | (52,219) |
| Stock options | 4,845,559 | 4,191,762 |
| Accruals and other | 2,244,941 | 1,332,602 |
| Research tax credit | 215,719 | ― |
| Net operating loss carryforwards | 23,355,282 | 15,251,744 |
| | 32,212,172 | 20,723,889 |
| Valuation allowance | (29,428,172) | (20,723,889) |
Deferred tax asset | 2,784,000 | ― |
| | | |
Deferred tax liability | | |
| Intangible assets (other than goodwill) | (21,723,417) | (8,853,062) |
Deferred tax liability | (21,723,417) | (8,853,062) |
| | | |
Deferred tax liability, net | (18,939,417) | (8,853,062) |
The following is a reconciliation of the recovery of income taxes between those that are expected, based on substantively enacted tax rates and laws, to those currently reported:
| | December 31, |
| | 2006 $ | 2005 $ | 2004 $ |
| | | | |
Loss for the year before income taxes | (86,242,043) | (163,614,515) | (21,803,373) |
| | | | |
| Expected recovery of income taxes | (31,189,248) | (60,537,371) | (8,067,250) |
| Goodwill impairment (permanent difference basis) | 23,740,447 | 54,557,150 | — |
| Stock-based compensation | 55,117 | 38,628 | 312,292 |
| Rate change | 322,321 | 12,923 | ― |
| Tax free income | (864) | (46,979) | — |
| Return to provision | (180,455) | 1,252,842 | ― |
| Non-deductible expenses | 89,360 | 19,656 | 3,700 |
| Change in valuation allowance | 3,092,827 | 4,060,622 | 7,727,487 |
Recovery of income taxes | (4,070,495) | (642,529) | (23,771) |
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
The Company and its subsidiaries have current and prior year losses available to reduce taxable income and taxes payable in future years, and, if these losses are not utilized, they will expire as follows:
| | $ |
| | |
| 2012 | 3,455,029 |
| 2018 | 4,500,401 |
| 2019 | 2,420,681 |
| 2020 | 5,241,917 |
| 2021 | 3,855,009 |
| 2022 | 3,313,031 |
| 2023 | 3,188,708 |
| 2024 | 7,849,643 |
| 2025 | 15,690,473 |
| 2026 | 13,877,166 |
12. ACCRUED LIABILITIES
| | December 31, |
| | 2006 $ | 2005 $ |
Due to professionals | 709,047 | 348,044 |
| Due to clinical trial sites | 195,074 | 32,936 |
| Due to clinical trial specialists | 206,642 | 227,009 |
| Product development costs | 124,312 | — |
| Due to ATI | — | 7,490 |
| Due to employees and directors | 464,146 | 993,177 |
| Sales tax and capital tax payable | 12,394 | 155,604 |
| Due to MeSys GmbH for pumps | — | 191,692 |
| Corporate compliance | 227,475 | 141,667 |
| Interest payable | 10,758 | — |
| Miscellaneous | 141,089 | 129,000 |
| | 2,090,937 | 2,226,619 |
13. COMMITMENTS AND CONTINGENCIES
Commitments
The Company leases office space from a related party under a lease agreement expiring December 31, 2007 (note 10). The Company may terminate the lease with three months’ notice. The future minimum obligation under the lease is $26,016 for 2007. Rent paid amounted to $32,940 for each of the years ended December 31, 2006, 2005 and 2004, respectively.
The Company also has commitments relating to operating leases for rental of office space and equipment from unrelated parties. The total future minimum obligation under the various leases is $104,402 for 2007. Rent paid under these leases was $80,329, $60,207 and $10,188 for the years ended December 31, 2006, 2005 and 2004, respectively. All Canadian dollar amounts have been converted at the respective year-end exchange rate.
In May and June 2002, the Company entered into two separate agreements with Dr. Richard Brunner and Mr. Stock, respectively, to obtain the exclusive license to U.S. Patent No. 6,245,038. The Company is required to make royalty payments totaling 1.5% of product sales. The Company is required to make minimum advance quarterly royalty payments of $25,000 and amounts credited against future royalty payments to be made in accordance with the agreements. These agreements may be terminated by the Company upon the first to occur of:
(a) all patents of the patent rights expiring, which is June 2017;
(b) all patent claims of the patent rights being invalidated; or
(c) the introduction of a similar competing technology deployed in the United States which could not be deterred by enforcement of the patent.
On August 6, 2004, the Company entered into a patent license and royalty agreement with Mr. Stock to obtain an exclusive license to U.S. Patent No. 6,245,038. The Company is required to make royalty payments totaling 1.5% of product sales to Mr. Stock, subject to minimum advance royalty payments of $12,500 per quarter. The advance payments are credited against future royalty payments to be made in accordance with the agreement. This agreement replaces the June 2002 consulting agreement with Mr. Stock, which provided for a royalty payment of 1% of product sales. This agreement effectively increases the total royalty payments required to be made in respect of U.S. Patent No. 6,245,038 to 2% of product sales (note 10).
Future minimum royalty payments under the agreements as at December 31, 2006 are approximately as follows:
| | $ |
| | |
| 2007 | 100,000 |
| 2008 | 100,000 |
| 2009 | 100,000 |
| 2010 | 100,000 |
| 2011 and thereafter | 650,000 |
| | 1,050,000 |
In June 2000, SOLX entered into a Patent License Agreement with Candela Corporation (“Candela”) to obtain the exclusive license to U.S. Patent No. 08/781,504 and No. 6,059,772. In accordance with the terms of this agreement, the Company is required to make royalty payments to Candela of 2% of SOLX 790 Laser sales worldwide and an annual royalty payment of $25,000 and to make a royalty payment to Dr. Shlomo Melamed of 1.5% of SOLX 790 Laser sales in the United States. The annual royalty payment to Candela is payable by January 31 of each year. The term of the agreement is for the legal life of the patents which will end in June 2017.
Future minimum royalty payments under the agreement as at December 31, 2006 are approximately as follows:
| | $ |
| | |
| 2007 | 25,000 |
| 2008 | 25,000 |
| 2009 | 25,000 |
| 2010 | 25,000 |
| 2011 and thereafter | 175,000 |
| | 275,000 |
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
In addition, the Company entered into a consultancy and non-competition agreement on July 1, 2003 with CCR (note 10), which requires the Company to pay a fee of $5,000 per month. On August 22, 2005, the Company amended the consultancy and non-competition agreement with CCR such that the fee payable was increased from $5,000 to $15,000 per month effective January 1, 2005. The monthly fee is fixed regardless of actual time incurred by CCR in performance of the services rendered to the Company. The agreement allows either party to convert the payment arrangement to a fee of $2,500 daily. In the event of such conversion, CCR shall provide services on a daily basis as required by the Company and will invoice the Company for the total number of days that services were provided in that month. The amended consultancy and non-competition agreement provides for the payment of a one-time bonus of $200,000 upon receipt by the Company of FDA approval of the RHEO™ System and the grant of 60,000 options to CCR at an exercise price of $7.15 per share. The stockholders of the Company approved the adjustment of the exercise price of these options to $2.05 per share on June 23, 2006 (note 14(e)). These options were scheduled to vest as to 100% when and if the Company receives FDA approval of the RHEO™ System on or before November 30, 2006, as to 80% when and if the Company receives FDA approval after November 30, 2006 but on or before January 31, 2007 and as to 60% when and if the Company receives FDA approval after January 31, 2007. In August 2006, by letter agreement between the Company and CCR, it was agreed that the monthly fee of $15,000 would be suspended at the end of August 2006 until CCR’s services will be required by the Company in the future. The future minimum obligation under the consultancy and non-competition agreement for 2007 is therefore nil.
The Company entered into consulting agreements with individual members of its Scientific Advisory Board (“SAB”). The SAB was established in fiscal 2005 to advise the Company on its continuing research and development activities. The future minimum obligation under the various consulting agreements is $258,930 for 2007. Consulting fees paid amounted to $244,165 for the year ended December 31, 2006 and nil for each of the years ended December 31, 2005 and 2004.
SOLX entered into consulting agreements with various physicians, hospitals and principal investigators for the purposes of its clinical trials and other medical consulting services. The future minimum obligation under the various consulting agreements is $191,250 for 2007.
On January 1, 2006, OcuSense entered into a consulting agreement with Benjamin David Sullivan which requires the Company to pay a consulting fee of $15,000 per month. The future minimum obligation under the consulting agreement is $180,000 for 2007.
On November 30, 2006, pursuant to the Series A Preferred Stock Purchase Agreement between the Company and OcuSense, the Company purchased 1,744,223 shares of OcuSense’s Series A Preferred Stock representing 50.1% of OcuSense’s capital stock on a fully diluted basis for an aggregate purchase price of up to $8,000,000 (the “Purchase Price”). On the closing of the purchase which took place on November 30, 2006, we paid $2,000,000 of the Purchase Price. We paid another $2,000,000 installment of the Purchase Price on January 3, 2007. We will pay the third $2,000,000 installment of the Purchase Price upon the attainment by OcuSense of the first of two pre-defined milestones and the last $2,000,000 installment of the Purchase Price upon the attainment by OcuSense of the second of such milestones, provided that both milestones are achieved prior to May 1, 2009.. The Series A Preferred Stock Purchase Agreement also makes provision for an ability on the part of the Company to increase its ownership interest in OcuSense for nominal consideration if OcuSense fails to meet certain milestones by specified dates. In addition, pursuant to the Series A Preferred Stock Purchase Agreement, the Company has agreed to purchase $3,000,000 of shares of OcuSense’s Series B Preferred Stock, which shall constitute 10% of OcuSense’s capital stock on a fully diluted basis at the time of purchase, upon OcuSense’s receipt from the FDA of 510(k) clearance for the DES Test and to purchase another $3,000,000 of shares of OcuSense’s Series B Preferred Stock, which shall constitute an additional 10% of OcuSense’s capital stock on a fully diluted basis at the time of purchase, upon OcuSense’s receipt from the FDA of CLIA waiver for the DES Test (note 3).
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
Guaranty
As of September 1, 2006, SOLX granted a security interest in all of its intellectual property to Doug P. Adams, John Sullivan and Peter M. Adams, in their capacity as members of the Stockholder Representative Committee acting on behalf of the former stockholders of SOLX, in order to secure SOLX’s obligations under the Guaranty, dated as of September 1, 2006, by SOLX in favor of Doug P. Adams, John Sullivan and Peter M. Adams, in their capacity as members of the Stockholder Representative Committee (the “Guaranty”). Pursuant to the Guaranty, SOLX guaranteed the Company’s obligation to pay the Stockholder Representative Committee, acting on behalf of the former stockholders of SOLX, an aggregate amount of up to $13,000,000, being the maximum aggregate amount of the purchase price remaining payable to the former stockholders of SOLX.
Contingencies
During the ordinary course of business activities, the Company may be contingently liable for litigation and a party to claims. Management believes that adequate provisions have been made in the accounts where required. Although it is not possible to estimate the extent of potential costs and losses, if any, management believes that the ultimate resolution of any such contingencies will not have a material adverse effect on the financial position and results of operations of the Company.
Pursuant to the terms of the distribution agreement with MeSys GmbH, dated January 1, 2002, the Company undertook a commitment to purchase a minimum of 25 OctoNova pumps yearly, beginning after receipt of FDA approval of the RHEO™ System, representing an annual commitment of approximately $534,900. The marketing and distributorship agreement with Diamed provides for a minimum purchase of 1,000 OctoNova pumps during the period from the date of the agreement until the end of the five-year period following receipt of FDA approval, representing an aggregate commitment of €16,219,000, or approximately $21,397,727, based on exchange rates as of December 31, 2006.
Pursuant to the terms of the 2006 Distributorship Agreement with Asahi Medical, dated October 20, 2006, the Company undertook a commitment to purchase a minimum of 9,000, 15,000, and 22,500 of each of the Plasmaflo filters and the Rheofilter filters in years 1, 2 and 3, respectively, beginning six months after receipt of FDA approval of the RHEO™ System. Minimum purchase orders for the fourth year shall be determined immediately after the term of the first year by mutual consent but shall not be less than that of the previous year. This same method shall be used in subsequent years to determine future minimum purchase quantities such that minimum purchase quantities are always fixed for three years. Future minimum annual commitments in respect of Territory 1-a, after receipt of FDA approval, are approximately as follows:
| | $ |
| | |
| Year 1 | 2,565,000 |
| Year 2 | 4,275,000 |
| Year 3 | 6,412,500 |
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
In respect of Canada, the Company undertook a commitment to purchase a minimum of 900, 1,500 and 2,250 of each of the Plasmaflo filters and the Rheofilter filters in years 1, 2 and 3, respectively, beginning upon the earlier to occur of (a) the sale by the Company of its current inventory of Rheofilter filters and (b) the expiration of the Company’s current inventory of Rheofilter filters. Minimum purchase orders for the fourth year shall be determined immediately after the term of the first year by mutual consent but shall not be less than that of the previous year. This same method shall be used in subsequent years to determine future minimum purchase quantities such that minimum purchase quantities are always fixed for three years. Future minimum annual commitments, in respect of Canada, are approximately as follows:
| | $ |
| | |
| Year 1 | 256,500 |
| Year 2 | 427,500 |
| Year 3 | 641,250 |
In respect of Territory 2, the Company undertook a commitment to purchase a minimum of 300 and 500 of each of the Plasmaflo filters and the Rheofilter filters in 2009 and 2010, respectively. In respect of Italy, the Company undertook a commitment to purchase a minimum of 200 and 500 of each of the Plasmaflo filters and the Rheofilter filters in 2007 and 2008, respectively. Minimum purchase orders for the years 2009 and 2010 shall be discussed and determined at the beginning of calendar year 2008 by mutual consent. Future minimum annual commitments, in respect of Territory 2 and Italy, are approximately as follows:
| | $ |
| | |
| 2007 | 57,000 |
| 2008 | 142,500 |
| 2009 | 85,500 |
| 2010 | 142,500 |
14. CAPITAL STOCK
(a) Authorized share capital
The total number of authorized shares of common stock of the Company is 75,000,000. Each share of common stock has a par value of $0.001 per share. The total number of authorized shares of preferred stock of the Company is 10,000,000. Each share of preferred stock has a par value of $0.001 per share.
(i)On July 18, 2002, the Company’s former parent company, OccuLogix Corp. (“Old OccuLogix”), merged with the Company, which was then a wholly-owned subsidiary of Old OccuLogix, to form OccuLogix, Inc. Pursuant to the merger, the Company effected a one-for-four stock split of its common and convertible preferred stock pursuant to which each share of Old OccuLogix common stock outstanding immediately prior to the merger was converted into one-fourth of one fully paid and non-assessable share of the Company’s common stock. Each outstanding share of Old OccuLogix Series A preferred stock was converted into one-fourth of one fully paid and non-assessable share of the Company’s Series A convertible preferred stock.
At the effective time of the merger, each outstanding warrant and option to purchase common stock of Old OccuLogix was assumed by the Company and converted into a warrant or option to purchase common stock of the Company, with appropriate adjustments to the exercise price and number of shares for which such warrants or options were exercisable.
(ii)On December 8, 2004, the Company consummated certain reorganization transactions, which are collectively referred to as the “Reorganization” and which consisted of the following:
| · | 4,622,605 shares of common stock issued upon the automatic conversion of all outstanding shares of Series A and Series B convertible preferred stock; |
| · | 7,106,454 shares of common stock issued to TLC Vision and Diamed upon conversion of $7,000,000 aggregate principal amount of convertible grid debentures held by them, the conversion price was $0.98502 per share; and |
| · | 19,070,234 shares of common stock issued to TLC Vision in connection with the purchase by the Company of TLC Vision’s 50% interest in the Partnership, this amount included 1,281,858 shares of common stock which were issued upon the exchange of shares of OccuLogix ExchangeCo ULC, one of the Company’s Canadian subsidiaries, issued for tax purposes to TLC Vision in connection with the Company’s purchase of TLC Vision’s interest in the Partnership. |
Following the Reorganization, the Partnership’s U.S. business was carried on, and will continue to be carried on, by OccuLogix LLC, a Delaware limited liability company that is the Company’s wholly-owned, indirect subsidiary. The Partnership carried on the Canadian business until December 31, 2005.
The Company had licensed to the Partnership all of the distribution and marketing rights for the RHEO™ System for ophthalmic indications to which it is entitled. Prior to the Reorganization, the Company’s only profit stream came from its share of the Partnership’s earnings. The Company’s acquisition of TLC Vision’s 50% ownership interest in the Partnership, achieved through the Reorganization, moved the earnings potential for sales of the RHEO™ System to the Company.
(iii)On December 31, 2005, the Partnership transferred all of its assets and liabilities, and assigned its right to develop and sell the RHEO™ System, to OccuLogix Canada Corp., a wholly-owned subsidiary of the Company. Following the transfer, the Partnership’s Canadian business will be carried on by OccuLogix Canada Corp. The Partnership and its general partner have subsequently been wound up.
(c) Convertible preferred stock
Convertible preferred stockholders were entitled to one vote per share, on an “as-converted to common stock” basis. Each share of Series A and Series B Convertible Preferred Stock was entitled to receive a non-cumulative dividend of $0.411216 and $0.34698, respectively, prior to the payment of any dividend on common stock. Each share of Series A and Series B Convertible Preferred Stock was entitled to a liquidation preference of $4.836 and $3.5183, respectively, plus any declared but unpaid dividend before any payment could be made to holders of common stock.
After giving effect to the anti-dilution adjustment resulting from the issuance of the June 25, 2003 related party secured grid debentures (note 10), each share of Series A and Series B Convertible Preferred Stock was convertible into 1.678323 and 1.643683 shares of common stock, respectively, at the option of the holder. Each share of Series A and Series B Convertible Preferred Stock would automatically convert into shares of common stock at the conversion rate previously described if the Company obtained a firm underwriting commitment for an initial public offering. The conversion rate would be adjusted for stock dividends, stock splits and other dilutive events. Shares of Series A and Series B Convertible Preferred Stock would automatically convert in the event of sale of all or substantially all of the assets or capital stock of the Company.
(d) Common stock
In December 2004, 5,600,000 shares of common stock of the Company at $12.00 per share were issued in connection with the initial public offering for gross cash proceeds of $67,200,000 (less issuance costs of $7,858,789).
On September 1, 2006, the Company issued 8,399,983 shares of its common stock to the former stockholders of SOLX in connection with the acquisition of SOLX. The stock consideration was valued based on a per share price of $1.79, being the weighted-average closing sale price of the Company’s common stock as traded on NASDAQ over the two-day trading period before and after August 1, 2006, being the date the terms of the acquisition of SOLX were agreed to and announced (note 3).
As at December 31, 2006, the number of shares of common stock of the Company reserved for issuance upon the exercise of stock options is as follows:
Range of exercise prices $ | Expiry date | # |
| | | |
| 2.05 | 2008 | 25,000 |
| 2.00 - 2.05 | 2009 | 167,625 |
| 2.00 - 2.05 | 2010 | 119,375 |
| 0.80 - 2.00 | 2012 | 96,090 |
| 0.99 - 1.30 | 2013 | 1,082,048 |
| 2.05 | 2014 | 675,000 |
| 2.05 | 2015 | 1,183,333 |
| 1.77 - 2.14 | 2016 | 888,750 |
| | | 4,237,221 |
The Company has a stock option plan, the 2002 Stock Option Plan (the “Stock Option Plan”). Under the Stock Option Plan, up to 4,456,000 options are available for grant to employees, directors and consultants. Options granted under the Stock Option Plan may be either incentive stock options or non-statutory stock options. Under the terms of the Stock Option Plan, the exercise price per share for an incentive stock option shall not be less than the fair market value of a share of stock on the effective date of grant and the exercise price per share for non-statutory stock options shall not be less than 85% of the fair market value of a share of stock on the date of grant. No option granted to a holder of more than 10% of the Company’s common stock shall have an exercise price per share less than 110% of the fair market value of a share of stock on the effective date of grant.
Options granted may be time-based or performance-based options. The vesting of performance-based options is contingent upon meeting company-wide goals, including obtaining FDA approval of the Company’s RHEO™ System and the achievement of a minimum amount of sales over a specified period. Generally, options expire 10 years after the date of grant. No incentive stock options granted to a 10% owner optionee shall be exercisable after the expiration of five years after the effective date of grant of such option, no option granted to a prospective employee, prospective consultant or prospective director may become exercisable prior to the date on which such person commences service, and with the exception of an option granted to an officer, director or a consultant, no option shall become exercisable at a rate less than 20% per annum over a period of five years from the effective date of grant of such option unless otherwise approved by the board of directors of the Company (the “Board of Directors”).
The Company has also issued options outside of the Stock Option Plan. These options were issued before the establishment of the Stock Option Plan, when the authorized limit of the Stock Option Plan was exceeded or as permitted under stock exchange rules when the Company was recruiting executives. In addition, options issued to companies for the purpose of settling amounts owing were issued outside of the Stock Option Plan, as the Stock Option Plan prohibited the granting of options to companies. The issuance of such options was approved by the Board of Directors, and such options were granted on terms and conditions similar to those options issued under the Stock Option Plan.
On January 1, 2006, the Company adopted the provisions of SFAS No. 123R, requiring the recognition of expense related to the fair value of its stock-based compensation awards. The Company elected to use the modified prospective transition method as permitted by SFAS No. 123R and therefore has not restated its financial results for prior periods. Under this transition method, the stock-based compensation expense for the year ended December 31, 2006 includes compensation expense for all stock-based compensation awards granted prior to, but not yet vested as of, January 1, 2006 based on the grant date fair value estimated in accordance with the original provisions of SFAS No. 123. Stock-based compensation expense for all stock-based compensation awards granted subsequent to January 1, 2006 is based on the grant date fair value estimated in accordance with the provisions of SFAS No. 123R. The Company recognizes compensation expense for stock option awards on a straight-line basis over the requisite service period of the award.
The following table sets forth the total stock-based compensation expense resulting from stock options included in the Company’s consolidated statements of operations:
| | | December 31, |
| | | 2006 (i) | | 2005 (ii) | | 2004 (iii) |
| | | $ | | $ | | $ |
| | | | | | | |
| General and administrative | | | 1,442,023 | | | 229,638 | | | 15,403,242 |
| Clinical and regulatory | | | 237,567 | | | 136,643 | | | 36,718 |
| Sales and marketing | | | 541,543 | | | 500 | | | — |
Stock-based compensation expense before income taxes (iv) | | | 2,221,133 | | | 366,781 | | | 15,439,960 |
(i) At the annual meeting of stockholders of the Company held on June 23, 2006, the stockholders of the Company approved the re-pricing of all then out-of-the-money stock options of the Company. Consequently, the exercise price of all outstanding stock options of the Company that, on June 23, 2006, was greater than $2.05, being the weighted average trading price of the Company’s common stock on NASDAQ during the five-trading day period immediately preceding June 23, 2006, was adjusted downward to $2.05. 2,585,000 of the Company’s outstanding stock options with a weighted average exercise price of $8.42 were affected by the re-pricing. SFAS No. 123R requires the re-pricing of equity awards to be treated as a modification of the original award and provides that such a modification is an exchange of the original award for a new award. SFAS No. 123R considers the modification to be the repurchase of the old award for a new award of equal or greater value, incurring additional compensation cost for any incremental value. This incremental difference in value is measured as the excess, if any, of the fair value of the modified award determined in accordance with the provisions of SFAS No. 123R over the fair value of the original award immediately before its terms are modified, measured based on the share price and other pertinent factors at that date. SFAS No. 123R provides that this incremental fair value, plus the remaining unrecognized compensation cost from the original measurement of the fair value of the old option, must be recognized over the remaining vesting period. Of the 2,585,000 options affected by the re-pricing, 1,401,073 were vested as at December 31, 2006. Therefore, additional compensation cost of $423,338 for the 1,401,073 stock options that were vested has been recognized and is included in the stock-based compensation expense for the year ended December 31, 2006. The remaining unrecognized incremental fair value of $169,057 plus the compensation cost from the original measurement of the fair value of the old options of $2,607,496, which totaled $2,776,553 in unrecognized compensation expense as at December 31, 2006, is expected to be amortized over a weighted average vesting period of 2.3 years.
In accordance with SFAS No. 123R, the Company also recorded a compensation expense of $3,363 in the year ended December 31, 2006 as the Board of Directors approved accelerating the vesting of 1,250 unvested stock options granted to a terminated employee on April 28, 2006. SFAS No. 123R treats such a modification as a cancellation of the original unvested award and the grant of a new fully vested award as of that date.
| (ii) | Stock-based compensation expense for the year ended December 31, 2005 relates primarily to compensation expense associated with non-employee stock options. The fair value of these options was determined using the Black-Scholes option-pricing model and was recorded in the Company’s consolidated statements of operations in accordance with the provisions of SFAS No. 123. |
On December 11, 2005, the Board of Directors approved accelerating the vesting of unvested stock options granted prior to December 31, 2004 to employees, officers and directors. As a result of the vesting acceleration, options to purchase 438,561 shares of the Company’s common stock became exercisable immediately, including 308,611 held by executive officers, 48,958 held by non-employee directors and 80,992 held by other employees. These accelerated stock options represent approximately 30% of the total employee stock options of the Company that would not have been vested as at December 31, 2005. The weighted average exercise price of the options that were accelerated was $11.78. The purpose of the acceleration was to enable the Company to avoid recognizing compensation expense associated with these options of $1,532,203 and $1,466,253 during the years ending December 31, 2006 and 2007, respectively, in its consolidated statements of operations as a result of the adoption of SFAS No. 123R on January 1, 2006. In accordance with APB No. 25, the Company recorded a compensation expense of $53,295 for the year ended December 31, 2005 as 9,033 of the total options, of which the vesting was accelerated, were “in-the-money” as at the date of the accelerated vesting. With respect to SFAS No. 123, the Company recognized, for purposes of pro forma disclosures, the incremental increase in fair value and the remaining balance of unrecognized compensation cost for the affected options at the time of acceleration.
In accordance with APB No. 25, the Company also recorded a compensation expense of $4,431 for the year ended December 31, 2005 as certain performance-based options granted to an employee and two directors were “in-the-money” as at December 31, 2005.
| (iii) | In December 2003, the Company granted a total of 1,352,500 stock options to its employees, directors and certain executives. The Company estimated the intrinsic value of these options to be $15,905,400. Management estimated the fair value of the underlying common stock based on management’s estimate of the Company’s value. The intrinsic value of the options was being amortized over the vesting period. However, upon the successful completion of the Company’s initial public offering in December 2004, the options vested immediately, and therefore the remaining $15,392,323 of stock-based compensation expense as at December 31, 2003 was expensed during the year ended December 31, 2004. |
| (iv) | The tax benefit associated with the Company’s stock-based compensation expense for the year ended December 31, 2006 was $781,527. This amount has not been recognized in the Company’s consolidated financial statements for the year ended December 31, 2006 as it is more likely than not that the Company will not realize this benefit. |
Net cash proceeds from the exercise of common stock options were $270,935, $231,235 and $129,420 for the years ended December 31, 2006, 2005 and 2004, respectively. No income tax benefit was realized from stock option exercises during the years ended December 31, 2006, 2005 and 2004. In accordance with SFAS No. 123R, the Company presents excess tax benefits from the exercise of stock options, if any, as financing cash flows rather than operating cash flows.
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
As a result of adopting SFAS No. 123R on January 1, 2006, the Company’s net loss for the year ended December 31, 2006 was $1,547,926 higher than if it had continued to account for share-based compensation under APB No. 25. Basic and diluted loss per share for the year ended December 31, 2006 increased by $0.04 with the adoption of SFAS No. 123R.
The Company did not estimate forfeitures, resulting from the failure to satisfy performance conditions, on its outstanding awards prior to the adoption of SFAS No. 123R. Under SFAS No. 123, the Company could assume all awards will vest and reverse recognized compensation cost or adjust its disclosure for forfeited awards when the awards are actually forfeited. SFAS No. 123R requires a company to estimate the number of awards that are expected to vest and revise the estimate as actual forfeitures differ from the estimate. On January 1, 2006, the effective date of adopting SFAS No. 123R, the Company was required to estimate the number of forfeitures of its outstanding awards as of the effective date. Consolidated balance sheet amounts related to any compensation cost for these estimated forfeitures previously recognized in prior periods before the adoption of SFAS No. 123R have to be eliminated and recognized in income as the cumulative effect of a change in accounting principle as of the effective date. During the year ended December 31, 2006, the Company recognized $107,045 as the cumulative effect of a change in accounting principle resulting from the requirement to estimate forfeitures on its outstanding awards as at January 1, 2006. The compensation cost previously recognized in prior periods before the adoption of SFAS No. 123R relates to compensation expense associated with non-employee stock options.
Prior to the adoption of SFAS No. 123R, the Company applied the provisions of SFAS No. 123, which allowed companies either to expense the estimated fair value of employee stock options or to follow the intrinsic value method as set forth in APB No. 25 but required companies to disclose the pro forma effects on net loss as if the fair value of the options had been expensed. The Company elected to apply APB No. 25 in accounting for employee stock options. Therefore, as required by SFAS No. 123, prior to the adoption of SFAS No. 123R, the Company provided pro forma net loss and pro forma net loss per share disclosures for stock-based awards as if the fair value of the options had been expensed.
The following table illustrates the pro forma net loss and net loss per share of common stock as if the fair value method had been applied to all awards during the years ended December 31, 2005 and 2004:
| | | | Years ended December 31, |
| | | | 2005 | | | 2004 | |
| | | | $ | | | $ | |
| | | | | | | | |
| Net loss, as reported | | | (162,971,986 | ) | | (21,818,873 | ) |
| Adjustment for APB No. 25 | | | 57,726 | | | 15,392,323 | |
| Adjustment for SFAS No. 123 | | | (6,664,395 | ) | | (15,673,031 | ) |
| Pro forma net loss | | | (169,578,655 | ) | | (22,099,581 | ) |
| Pro forma net loss per share - basic and diluted | | | (4.04 | ) | | (3.00) | |
The weighted average fair value of stock options granted during the years ended December 31, 2006, 2005 and 2004 was $1.77, $3.54 and $6.96, respectively. The estimated fair value was determined using the Black-Scholes option-pricing model with the following weighted-average assumptions:
| | | Years ended December 31, |
| | | 2006 | | 2005 | 2004 |
| | | | | | |
| Volatility | | | 0.901 | | | 0.728 | | 0.891 |
| Expected life of options | | | 5.56 years | | | 2.33 years | | 3 years |
| Risk-free interest rate | | | 4.83% | | | 3.87% | | 3.21% |
| Dividend yield | | | 0% | | | 0% | | 0% |
The Company’s computation of expected volatility for the years ended December 31, 2006, 2005 and 2004 is based on a comparable company’s historical stock prices as the Company did not have sufficient historical data. The Company’s computation of expected life has been estimated using the “short-cut approach” as provided in SAB No. 107 as options granted by the Company meet the criteria of “plain vanilla” options as defined in SAB 107. Under this approach, estimated life is calculated to be the mid-point between the vesting date and the end of the contractual period. The risk-free interest rate for an award is based on the U.S. Treasury yield curve with a term equal to the expected life of the award on the date of grant.
A summary of the options issued during the year ended December 31, 2006 and the total number of options outstanding as of that date and changes since December 31, 2003 are set forth below:
| | Number of Options Outstanding | Weighted-Average Exercise Price (i) $ | Weighted-Average Remaining Contractual Life (years) | Aggregate Intrinsic Value $ |
| | | | | |
| Outstanding, December 31, 2003 | 2,389,961 | 1.45 | | |
| Granted | 828,000 | 12.00 | | |
| Exercised | (272,200) | 0.48 | | |
| Forfeited | (196,562) | 2.48 | | |
| Outstanding, December 31, 2004 | 2,749,199 | 4.64 | 8.31 | 934,777 |
| Granted | 1,823,750 | 8.10 | | |
| Exercised | (279,085) | 0.83 | | |
| Forfeited | (186,250) | 9.99 | | |
Outstanding, December 31, 2005 (i) | 4,107,614 | 1.75 | 8.20 | 724,812 |
| Granted | 890,000 | 1.99 | | |
| Exercised | (140,726) | 1.93 | | |
| Forfeited | (619,667) | 2.05 | | |
Outstanding, December 31, 2006 | 4,237,221 | 1.75 | 7.61 | 653,307 |
| | | | | |
Vested or expected to vest, December 31, 2006 | 3,005,956 | 1.63 | 7.17 | 653,307 |
| | | | | |
Exercisable, December 31, 2006 | 2,581,804 | 1.56 | 6.95 | 653,307 |
(i) At the annual meeting of stockholders of the Company held on June 23, 2006, the stockholders of the Company approved the re-pricing of all then out-of-the-money stock options of the Company. Consequently, the exercise price of all outstanding stock options of the Company that, on June 23, 2006, was greater than $2.05, being the weighted average trading price of the Company’s common stock on NASDAQ during the five-trading day period immediately preceding June 23, 2006, was adjusted downward to $2.05.
The aggregate intrinsic value in the table above represents the total pre-tax intrinsic value (i.e., the difference between the Company’s closing stock price on the last trading day of fiscal 2006 of $1.57 and the exercise price, multiplied by the number of shares that would have been received by the option holders if the options had been exercised on December 31, 2006). This amount changes according to the fair market value of the Company’s stock.
As at December 31, 2006, $3,978,530 of total unrecognized compensation cost related to stock options is expected to be recognized over a weighted-average period of 2.53 years.
(f) Warrants
Purchasers of Series A convertible preferred stock received warrants to purchase shares of common stock at an exercise price of $1.00 per share. The warrants were exercisable for the purchase of one share of common stock for each share of Series A convertible preferred stock owned. In February 1998, an additional voluntary warrant was granted to each Series A convertible preferred stockholder to purchase an equal number of shares of common stock at an exercise price of $2.00 per share. Additionally, warrants to purchase 50,000 shares of common stock at an exercise price of $1.00 per share were granted to an officer and certain directors and stockholders of the Company in exchange for providing certain private credit guarantees.
| | | Weighted average exercise price |
Common stock warrants | # | $ |
| | | |
Outstanding, December 31, 2003 (i) | 150,000 | 2.83 |
Exercised (ii) | (102,369) | 2.29 |
| Expired | (47,631) | 4.00 |
Outstanding, December 31, 2004, 2005 and 2006 | — | — |
| | | Weighted average exercise price |
Series A convertible preferred stock warrants | # | $ |
| | | |
Outstanding, December 31, 2003 (i) | 482,710 | 6.80 |
Exercised (ii) | (379,284) | 6.73 |
| Expired | (103,426) | 7.04 |
Outstanding, December 31, 2004, 2005 and 2006 | — | — |
(i) As a result of the issuance of Series B convertible preferred stock on July 25, 2002 at a price lower than the exercise price of the Series A convertible preferred stock warrants, anti-dilution adjustments were applied to reduce the exercise price of the Series A convertible preferred stock warrants and to increase the number of shares issuable upon the exercise of the Series A convertible preferred stock warrants.
As a result of the TLC Vision and Diamed convertible grid note debenture agreements entered into on June 25, 2003 at a conversion price lower than the exercise price of the Series A convertible preferred stock warrants, further anti-dilution adjustments were applied to reduce the exercise price of the Series A convertible preferred stock warrants and to increase the number of shares issuable upon the exercise of the Series A convertible preferred stock warrants.
Of the 102,369 warrants exercised to purchase shares of common stock, 24,999 shares of common stock were issued on a cashless basis (note 15). The remaining 77,370 shares of common stock were issued for total cash proceeds of $134,480.
(ii) Of the 379,284 warrants exercised to purchase shares of Series A convertible preferred stock, 165,189 shares of Series A convertible preferred stock were issued on a cashless basis (note 15). The remaining 214,095 shares of Series A convertible preferred stock were issued for total cash proceeds of $1,281,841 of which $34,927 has yet to be received as at December 31, 2006. This amount is included as a subscription receivable within paid-in capital and has been fully provided for.
All warrants to purchase shares of common stock and Series A convertible preferred stock at exercise prices between $1.20 per share and $7.83 per share expired on July 17, 2004, other than 379,284 warrants to purchase shares of Series A convertible preferred stock and 102,369 warrants to purchase shares of common stock which were exercised prior to the expiration of the warrants. As at December 31, 2006, 2005 and 2004, no common stock warrants and no Series A convertible preferred stock warrants remained outstanding.
15. CONSOLIDATED STATEMENTS OF CASH FLOWS
The net change in non-cash working capital balances related to operations consists of the following:
| | Years ended December 31, |
| | 2006 $ | 2005 $ | 2004 $ |
| | | | |
| Due to related party | (5,065) | 13,291 | 110,749 |
| Amounts receivable | 390,634 | (82,810) | (222,218) |
| Inventory | 2,250,554 | (3,431,743) | (136,527) |
| Prepaid expenses | 247,361 | (322,455) | (324,353) |
| Deposit | (5,551) | 4,105 | (8,996) |
| Accounts payable | (1,225,575) | 301,457 | 26,548 |
| Accrued liabilities | (1,155,335) | (563,925) | 2,511,897 |
| Deferred revenue and rent inducement | — | (485,047) | (152,153) |
| Due to stockholders | (5,827) | (358,523) | (931,652) |
| Other current assets | 18,332 | — | — |
| | 509,528 | (4,925,650) | 873,295 |
The following table lists those items that have been excluded from the consolidated statements of cash flows as they relate to non-cash transactions and additional cash flow information:
| | Years ended December 31, |
| | 2006 $ | 2005 $ | 2004 $ |
| Non-cash investing and financing activities | | | |
| Conversion of debentures | — | ― | 7,000,000 |
Cashless exercise of warrants to purchase shares of Series A convertible preferred stock | — | ― | 1,269,845 |
| Cashless exercise of warrants to purchase shares of common stock | — | ― | 99,996 |
| Free inventory | (48,006) | 183,382 | 146,905 |
| Common stock issued on acquisition | 15,035,969 | ― | 228,842,808 |
| | | | |
| Additional cash flow information | | | |
| Interest paid | — | ― | (26,575) |
| Income taxes recovered (paid), net | 4,533 | (8,138) | ― |
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
16. FINANCIAL INSTRUMENTS
Currency risk
The Company’s activities which result in exposure to fluctuations in foreign currency exchange rates consist of the purchase of equipment from suppliers billing in foreign currencies. The Company does not use derivative financial instruments to reduce its currency risk.
Credit risk
The Company’s financial instruments that are exposed to concentration of credit risk consist primarily of cash and cash equivalents and amounts receivable. The Company maintains its accounts for cash with large low credit risk financial institutions in the United States and Canada in order to reduce its exposure.
During fiscal 2006, the Company derived all of its revenue from the sale of the components of the RHEO™ System and the SOLX Glaucoma System. The Company sold components of the RHEO™ System to only one customer in the year, Veris. As previously discussed in note 10, the Company fully provided for the balance due from Veris. Accordingly, no trade receivables due from Veris have been recognized as at December 31, 2006. There were no trade receivables outstanding from the sale of the components of the SOLX Glaucoma System as at December 31, 2006 as all amounts were fully paid as of that date.
17. SEGMENTED INFORMATION
As a result of the acquisition of SOLX and OcuSense (note 3), the Company has two reportable segments: retina and glaucoma. The retina segment is in the business of commercializing the RHEO™ System which is used to perform the Rheopheresis™ procedure, a procedure that selectively removes molecules from plasma, which is designed to treat Dry AMD. The Company began limited commercialization of the RHEO™ System in Canada in 2003 and continues to support its sole customer, Veris, in its commercial activities in Canada. The Company has recently obtained investigational device exemption clearance from the FDA to commence RHEO-AMD, its new clinical study of the RHEO™ System. The glaucoma segment is in the business of providing treatment for glaucoma with the use of the components of the SOLX Glaucoma System which are used to provide physicians with multiple options to manage intraocular pressure. The Company is seeking to obtain 510(k) approval to market the components of the SOLX Glaucoma System in the United States. The Company acquired the glaucoma segment in the acquisition of SOLX on September 1, 2006; therefore, no amounts are shown for the segment in periods prior to September 1, 2006. Other is made up of the TearLab™ business which is currently developing technologies that enable eye care practitioners to test, at the point-of-care, for highly sensitive and specific biomarkers in tears using nanoliters of tear film. The Company acquired the TearLab™ business in the acquisition of 50.1% of the capital stock of OcuSense, on a fully diluted basis, on November 30, 2006; therefore, no amounts are shown in periods prior to November 30, 2006. The TearLab™ business does not meet the quantitative criteria to be disclosed separately as a reportable segment.
The accounting policies of the segments are the same as those described in significant accounting policies (note 2). Intersegment sales and transfers are minimal and are accounted for at current market prices, as if the sales or transfers were to third parties.
The Company’s reportable units are strategic business units that offer different products and services. They are managed separately, because each business unit requires different technology and marketing strategies. The Company’s business units were acquired or developed as a unit, and in the case of SOLX and OcuSense, their respective management was retained at the time of acquisition.
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
The Company’s business units were as follows:
| | | Retina | Glaucoma | Other | Total | |
| | | $ | | $ | | $ | | $ | |
Year ended December 31, 2006 | | | | | | | | | |
| Revenue | | 174,259 | | 31,625 | | — | | 205,884 | |
| Expenses: | | | | | | | | | |
| Cost of goods sold | | 3,528,951 | | 19,385 | | — | | 3,548,336 | |
| Operating | | 12,507,953 | | 1,723,265 | | 312,394 | | 14,543,612 | |
| Depreciation and amortization | | 1,860,849 | | 1,067,943 | | 39,516 | | 2,968,308 | |
| Impairment of goodwill | | 65,945,686 | | — | | — | | 65,945,686 | |
| Restructuring charges | | 819,642 | | — | | — | | 819,642 | |
| Loss from operations | | (84,488,822) | | (2,778,968) | | (351,910) | | (87,619,700) | |
| Interest income | | 1,370,205 | | 3 | | — | | 1,370,208 | |
| Interest expense | | (286,784) | | — | | (1,304) | | (288,088) | |
| Other income (expense), net | | 31,108 | | (67) | | (173) | | 30,868 | |
| Minority interest | | — | | — | | 157,624 | | 157,624 | |
| Recovery of income taxes | | 2,819,805 | | 1,182,005 | | 68,685 | | 4,070,495 | |
| Cumulative effect of a change in accounting principle | | 107,045 | | — | | — | | 107,045 | |
| Net loss | | (80,447,443) | | (1,597,027) | | (127,078) | | (82,171,548) | |
| | | | | | | | | | |
| Total assets | | 40,762,771 | | 44,158,205 | | 5,482,719 | | 90,403,695 | |
| | | | | | | | | | |
Year ended December 31, 2005 | | | | | | | | | |
| Revenue | | 1,840,289 | | — | | — | | 1,840,289 | |
| Expenses: | | | | | | | | | |
| Cost of goods sold | | 3,394,102 | | — | | — | | 3,394,102 | |
| Operating | | 14,323,605 | | — | | — | | 14,323,605 | |
| Depreciation and amortization | | 1,821,680 | | — | | — | | 1,821,680 | |
| Impairment of goodwill | | 147,451,758 | | — | | — | | 147,451,758 | |
| Loss from operations | | (165,150,856) | | — | | — | | (165,150,856) | |
| Interest income | | 1,593,366 | | — | | — | | 1,593,366 | |
| Other expense, net | | (57,025) | | — | | — | | (57,025) | |
| Recovery of income taxes | | 642,529 | | — | | — | | 642,529 | |
| Net loss | | (162,971,986) | | — | | — | | (162,971,986) | |
| | | | | | | | | | |
| Total assets | | 137,806,058 | | — | | — | | 137,806,058 | |
| | | | | | | | | | |
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
| | | Retina | Glaucoma | Other | Total | |
| | | $ | | $ | | $ | | $ | |
Year ended December 31, 2004 | | | | | | | | | |
| Revenue | | 969,357 | | — | | — | | 969,357 | |
| Expenses: | | | | | | | | | |
| Cost of goods sold | | 957,269 | | — | | — | | 957,269 | |
| Operating | | 21,589,968 | | — | | — | | 21,589,968 | |
| Depreciation and amortization | | 154,574 | | — | | — | | 154,574 | |
| Loss from operations | | (21,732,454) | | — | | — | | (21,732,454) | |
| Interest income | | 60,227 | | — | | — | | 60,227 | |
| Interest expense | | (24,492) | | — | | — | | (24,492) | |
| Other expense, net | | (145,925) | | — | | — | | (145,925) | |
| Recovery of income taxes | | 23,771 | | — | | — | | 23,771 | |
| Net loss | | (21,818,873) | | — | | — | | (21,818,873) | |
| | | | | | | | | | |
| Total assets | | 301,600,631 | | — | | — | | 301,600,631 | |
| | | | | | | | | | |
The Company’s geographic segments are as follows:
| | | United States | | Canada | | Europe | | Israel | Total |
| | | $ | | $ | | $ | | $ | | $ |
Year ended December 31, 2006 | | | | | | | | | | |
| Revenues | | — | | 174,384 | | 31,500 | | — | | 205,884 |
| Fixed assets and intangibles | | 70,932,850 | | 186,987 | | 63,484 | | 42,613 | | 71,225,934 |
| | | | | | | | | | | |
Year ended December 31, 2005 | | | | | | | | | | |
| Revenues | | — | | 1,840,289 | | — | | — | | 1,840,289 |
| Fixed assets and intangibles | | 90,340,988 | | 137,686 | | — | | — | | 90,478,674 |
| | | | | | | | | | | |
Year ended December 31, 2004 | | | | | | | | | | |
| Revenues | | — | | 969,357 | | — | | — | | 969,357 |
| Fixed assets and intangibles | | 239,446,055 | | 67,494 | | — | | — | | 239,513,549 |
Revenues from Veris, of the Company’s retina segment, accounted for approximately 85%, 96% and 25% of the Company’s revenue for the years ended December 31, 2006, 2005 and 2004, respectively.
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
18. SUBSEQUENT EVENT
On February 1, 2007, the Company entered into a Securities Purchase Agreement (the “Securities Purchase Agreement”) with certain institutional investors, pursuant to which the Company agreed to issue to those investors an aggregate of 6,677,333 shares of the Company’s common stock (the “Shares”) and five-year warrants exercisable into an aggregate of 2,670,933 shares of the Company’s common stock (the “Warrants”). The per share purchase price of the Shares is $1.50, and the per share exercise price of the Warrants is $2.20, subject to adjustment. The Warrants will become exercisable on August 6, 2007. Pursuant to the Securities Purchase Agreement, on February 6, 2007, the Company issued the Shares and the Warrants. The gross proceeds of sale of the Shares totaled $10,016,000 (less transaction costs of approximately $750,000). On February 6, 2007, the Company also issued to Cowen and Company, LLC a warrant exercisable into an aggregate of 93,483 shares of the Company’s common stock (the “Cowen Warrant”) in part payment of the placement fee payable to Cowen and Company, LLC for the services it had rendered as the placement agent in connection with the sale of the Shares and the Warrants. All of the terms and conditions of the Cowen Warrant (other than the number of shares of the Company's common stock into which the Cowen Warrant is exercisable) are identical to those of the Warrants.
19. QUARTERLY FINANCIAL DATA (UNAUDITED)
The following tables contain selected unaudited consolidated statement of operations data for each quarter of fiscal 2006 and 2005:
| | | | Fiscal 2006 Quarter Ended | |
| | | March 31 | | June 30 | | September 30 | | December 31 | |
| | | $ | | $ | | $ | | $ | |
| | | | | | | | | | | | | | |
| Revenues | | | — | | | 82,715 | | | 85,444 | | | 37,725 | |
Gross profit (loss) (i) | | | (1,650,000 | ) | | 78,398 | | | (31,961 | ) | | (1,738,889 | ) |
Loss from operations (ii) | | | (6,367,600 | ) | | (70,557,888 | ) | | (3,922,773 | ) | | (6,771,439 | ) |
Net loss (iii) | | | (5,731,952 | ) | | (69,995,592 | ) | | (3,583,808 | ) | | (2,860,196 | ) |
| Weighted average number of shares outstanding - basic and diluted | | | 42,166,561 | | | 42,186,579 | | | 44,911,018 | | | 50,622,496 | |
Net loss per share - basic and diluted (iv) | | | (0.14 | ) | | (1.66 | ) | | (0.08 | ) | | (0.06 | ) |
| | | | Fiscal 2005 Quarter Ended | |
| | | March 31 | | June 30 | | September 30 | | December 31 | |
| | | $ | | $ | | $ | | $ | |
| | | | | | | | | | | | | | |
| Revenue | | | 403,739 | | | 597,841 | | | 632,330 | | | 206,379 | |
Gross profit (loss) (i) | | | 103,805 | | | 193,986 | | | 295,120 | | | (2,146,724 | ) |
Loss from operations (ii) | | | (3,806,780 | ) | | (3,695,436 | ) | | (3,394,985 | ) | | (154,253,655 | ) |
| Net loss | | | (3,281,364 | ) | | (3,159,720 | ) | | (2,853,914 | ) | | (153,676,988 | ) |
| Weighted average number of shares outstanding - basic and diluted | | | 41,810,679 | | | 41,860,288 | | | 41,982,057 | | | 42,070,457 | |
Net loss per share - basic and diluted (iv) | | | (0.08 | ) | | (0.08 | ) | | (0.07 | ) | | (3.65 | ) |
| (i) | Gross profit (loss) for the three months ended December 31, 2006, March 31, 2006 and December 31, 2005 includes the expense of amounts related to inventory reserves of $1,679,124, $1,625,000 and $1,990,830, respectively. |
| (ii) | Loss from operations for the three months ended June 30, 2006 and December 31, 2005 includes a goodwill impairment charge of $65,945,686 and $147,451,758, respectively. |
| (iii) | Net loss for the three months ended December 31, 2006 includes a deferred tax recovery of $2,784,000 associated with the recognition of a deferred tax asset due to the availability of 2006 net operating losses which may be utilized to reduce taxes in future years. |
| (iv) | Net loss per share - basic and diluted are computed independently for the quarters presented. Therefore, the sum of the quarterly per share information may not be equal to the annual per share information. |
OCCULOGIX, INC.
Notes to Consolidated Financial Statements
(expressed in U.S. dollars except as otherwise noted)
| ITEM 9. | CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE. |
Not applicable.
ITEM 9A. CONTROLS AND PROCEDURES.
The Company maintains disclosure controls and procedures that are designed to ensure that information required to be disclosed in the Company’s reports under the Securities Exchange Act of 1934, as amended (the “Exchange Act”), is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms, and that such information is accumulated and communicated to the Company’s management, including its Chief Executive Officer and Chief Financial Officer, as appropriate, to allow timely decisions regarding required disclosure. In designing and evaluating the disclosure controls and procedures, management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving the desired control objectives, and management necessarily is required to apply its judgment in evaluating the cost-benefit relationship of possible controls and procedures. The Company’s disclosure controls and procedures are designed to provide reasonable assurance of achieving their desired objectives, and the Company’s Chief Executive Officer and Chief Financial Officer have concluded that the Company’s disclosure controls and procedures are effective to provide that reasonable assurance.
As of the end of the period covered by the report, the Company carried out an evaluation, under the supervision and with the participation of the Company’s management, including the Company’s Chief Executive Officer and Chief Financial Officer, of the effectiveness of the design and operation of the Company’s disclosure controls and procedures (as defined in Rule 13a-15(e) of the Exchange Act). Based on that evaluation, the Company’s Chief Executive Officer and Chief Financial Officer concluded that the Company’s disclosure controls and procedures (as defined in Rule 13a-15(e) of the Exchange Act) were effective to ensure that information required to be disclosed in the reports the Company files and submits under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the Securities and Exchange Commission’s rules and forms.
There have been no significant changes in the Company’s internal control over financial reporting that occurred during the year ended December 31, 2006, that have materially affected, or are reasonably likely to materially affect, the Company’s internal control over financial reporting.
MANAGEMENT’S REPORT ON INTERNAL CONTROL OVER FINANCIAL REPORTING
Management of the Company is responsible for establishing and maintaining effective internal control over financial reporting as defined in Rule 13a-15(f) under the Securities Exchange Act of 1934. The Company’s internal control over financial reporting is designed to provide reasonable assurance to the Company’s management and board of directors regarding the preparation and fair presentation of published financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Therefore, even those systems determined to be effective can provide only reasonable assurance with respect to financial statement preparation and presentation.
Management assessed the effectiveness of the Company’s internal control over financial reporting as of December 31, 2006. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (COSO) in Internal Control - Integrated Framework. Based on our assessment, we believe that, as of December 31, 2006, the Company’s internal control over financial reporting is effective based on those criteria.
Management’s assessment of the effectiveness of internal control over financial reporting as of December 31, 2006, has been audited by Ernst & Young LLP, an independent registered public accounting firm who also audited the Company’s consolidated financial statements. Ernst & Young’s attestation report on management’s assessment of the Company’s internal control over financial reporting is included elsewhere herein.
REPORT OF INDEPENDENT REGISTERED
PUBLIC ACCOUNTING FIRM
The Board of Directors and Shareholders of OccuLogix, Inc.
We have audited management’s assessment, included in the accompanying Management’s Report on Internal Control over Financial Reporting, that OccuLogix, Inc. maintained effective internal control over financial reporting as of December 31, 2006, based on criteria established in Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the “COSO criteria”). OccuLogix Inc.’s management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting. Our responsibility is to express an opinion on management’s assessment and an opinion on the effectiveness of the Company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, evaluating management’s assessment, testing and evaluating the design and operating effectiveness of internal control, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, management’s assessment that OccuLogix, Inc. maintained effective internal control over financial reporting as at December 31, 2006, is fairly stated, in all material respects, based on the COSO criteria. Also, in our opinion, OccuLogix, Inc. maintained, in all material respects, effective internal control over financial reporting as at December 31, 2006, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the consolidated balance sheets of OccuLogix, Inc. as at December 31, 2006 and 2005, and the related consolidated statements of operations, changes in stockholders’ equity (deficiency) and cash flows for each of the three years in the period ended December 31, 2006 and our report dated March 2, 2007 expressed an unqualified opinion thereon. Our audits also included the financial statement schedule listed in the index at Item 15(a).
| Toronto, Canada, | /s/ Ernst & Young LLP |
| March 2, 2007. | Chartered Accountants |
ITEM 9B. OTHER INFORMATION.
None.
PART III
| ITEM 10. | DIRECTORS AND EXECUTIVE OFFICERS OF THE REGISTRANT. |
The information required with respect to directors is incorporated herein by reference to the information contained in the General Proxy Information for our 2007 Annual Meeting of Stockholders (the “Proxy Statement”). The information with respect to our audit committee financial expert is incorporated herein by reference to the information contained in the sections captioned “Appointment of Auditors” and “Audit Committee Report” of the Proxy Statement.
Information about our Code of Ethics appears under the heading “Code of Business Conduct and Ethics” in the Proxy Statement. That portion of the Proxy Statement is incorporated by reference into this report.
Information about compliance with Section 16(a) of the Exchange Act appears under the heading “Section 16(a) Beneficial Ownership Reporting Compliance” in the Proxy Statement. That portion of the Proxy Statement is incorporated by reference into this report.
| ITEM 11. | EXECUTIVE COMPENSATION. |
Information about compensation of our named executive officers appears under the headings “Executive Officers” and “Information on Executive Compensation” in the Proxy Statement. Information about compensation of our directors appears under the heading “Compensation of Directors” in the Proxy Statement. These portions of the Proxy Statement are incorporated by reference into this report.
| ITEM 12. | SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS. |
Information about security ownership of certain beneficial owners and management and information regarding securities authorized for issuance under equity compensation plans appears under the headings “Information on Executive Compensation”, “Employee Benefit Plans” and “Principal Stockholders” in the Proxy Statement. These portions of the Proxy Statement are incorporated by reference to this report.
| ITEM 13. | CERTAIN RELATIONSHIPS AND RELATED PARTY TRANSACTIONS. |
Information about certain relationships and related transactions appears under the heading “Certain Relationships and Related Party Transactions” in the Proxy Statement. That portion of the Proxy Statement is incorporated by reference into this report.
| ITEM 14. | PRINCIPAL ACCOUNTANT FEES AND SERVICES. |
Information about the principal accountant fees and services as well as related pre-approval policies and procedures appears under the headings “Appointment of Auditors” and “Audit Committee Report” in the Proxy Statement. These portions of the Proxy Statement are incorporated by reference into this report.
PART IV
| ITEM 15. | EXHIBITS, FINANCIAL STATEMENT SCHEDULES. |
| (a) | The following documents are filed as part of the report: |
| | (1) Financial Statements included in PART II of this report: |
| Included in PART II of this report: | Page |
| Report of Independent Auditors | 82 |
| Consolidated Balance Sheets as at December 31, 2006 and December 31, 2005 | 84 |
| Consolidated Statements of Operations for the three years ended December 31, 2006 | 85 |
Consolidated Statements of Changes in Stockholders’ Equity (Deficiency) for the three years ended December 31, 2006 | 86 |
| Consolidated Statements of Cash Flows for the three years ended December 31, 2006 | 88 |
| Notes to Consolidated Financial Statements | 89 |
| (2) | Financial Statement Schedules: |
Schedule II - Valuation and Qualifying Accounts and Reserves
Except as noted above, all financial statement schedules for which provisions have been made in the applicable accounting regulations of the Commission have been omitted because they are inapplicable, not required by the instructions or because the required information is either incorporated herein by reference or included in the financial statements or notes thereto included in this report.
(3) Exhibits:
The exhibits required to be filed as part of this Annual Report on Form 10-K are listed in the attached Index to Exhibits. Items 10.5, 10.7 to 10.11 inclusive, 10.13, 10.14, 10.20 to 10.28 inclusive, 10.30 to 10.37 inclusive, 10.41 and 10.47 in the attached Index to Exhibits are management contracts or compensatory plans or arrangements.
(b) Exhibits
The exhibits required to be filed as part of this Annual Report on Form 10-K are listed in the attached Index to Exhibits.
* * *
Copies of the exhibits filed with this Annual Report on Form 10-K or incorporated by reference herein do not accompany copies hereof for distribution to stockholders of the Registrant. The Registrant will furnish a copy of any of such exhibits to any stockholder requesting the same for a nominal charge to cover duplicating costs.
EXPERTS
Valuation analyses of the fair value of certain of our net assets included in this Annual Report on Form 10-K have been performed by Peter Ott & Associates Inc., an independent appraiser, and have been included in reliance upon such company’s authority as an expert in business valuation.
POWER OF ATTORNEY
The registrant and each person whose signature appears below hereby appoint Elias Vamvakas and William G. Dumencu as attorney-in-fact with full power of substitution, severally, to execute in the name and on behalf of the registrant and each such person, individually and in each capacity stated below, one or more amendments to this Annual Report on Form 10-K, which amendments may make such changes in this Annual Report as the attorney-in-fact acting in the premises deems appropriate and to file any such amendments to this Annual Report on Form 10-K with the Securities and Exchange Commission.
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this Annual Report on Form 10-K to be signed on its behalf by the undersigned thereunto duly authorized.
Dated: March 15, 2007 | | OCCULOGIX, INC. |
By: /s/ Elias Vamvakas
____________________________________________
Elias Vamvakas
Chief Executive Officer
Pursuant to the requirements of the Securities Exchange Act of 1934, this Annual Report on Form 10-K has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
Dated: March 15, 2007 | | By: | /s/ Elias Vamvakas |
| | Elias Vamvakas Chief Executive Officer and Chairman of Board of Directors |
| | | | |
Dated: March 15, 2007 | | By: | /s/ William G. Dumencu |
| | William G. Dumencu Chief Financial Officer and Treasurer |
| | | | |
Dated: March 15, 2007 | | By: | /s/ Jay T. Holmes |
| | Jay T. Holmes Director |
| | | | |
Dated: March 15, 2007 | | By: | /s/ Thomas N. Davidson |
| | Thomas N. Davidson Director |
| | | | |
Dated: March 15, 2007 | | By: | /s/ Richard L. Lindstrom |
| | Richard L. Lindstrom, M.D. Director |
| | | | |
Dated: March 15, 2007 | | By: | /s/ Georges Noël |
| | Director |
Dated: March 15, 2007 | | By: | /s/ Adrienne L. Graves |
| | Adrienne L. Graves Director |
Dated: March 15, 2007 | | By: | /s/ Gilbert S. Omenn |
| | |
SCHEDULE II
VALUATION AND QUALIFYING ACCOUNTS AND RESERVES
| | | Balance at beginning of period | | Charged to costs and expenses | | Charged to other accounts | | Deductions | | Balance at end of period | |
| | | $ | | $ | | $ | | $ | | $ | |
| | | | | | | | | | | | |
| Fiscal 2004 | | — | | — | | — | | — | | — | |
| | | | | | | | | | | | |
| Fiscal 2005 | | | | | | | | | | | |
| Bad debt reserves | | — | | 518,852 | | — | | — | | 518,852 | |
| Inventory reserves | | — | | 1,990,830 | | — | | — | | 1,990,830 | |
| | | | | | | | | | | | |
Fiscal 2006 | | | | | | | | | | | |
Bad debt reserves | | 518,852 | | — | | — | | (518,852) | 1 | — | |
Inventory reserves | | 1,990,830 | | 3,304,124 | | — | | (193,560) | 2 | 5,101,394 | |
| 1. | During fiscal 2006, OccuLogix, Inc. (“the Company”) agreed to forgive the amount receivable from Veris Health Services Inc. (“Veris”) which had been owing for products and related services delivered or provided to Veris during the period from September 14, 2005 to December 31, 2005. |
| 2. | During fiscal 2006, the Company utilized inventory that had previously been provided for. |
2.1 | Form of Plan of Reorganization (incorporated by reference to Exhibit 2.1 to the Registrant’s Registration Statement on Form S-1/A No. 4, filed with the Commission on December 6, 2004 (file no. 333-118024)). |
3.1 | Amended and Restated Certificate of Incorporation of the Registrant as currently in effect (incorporated by reference to Exhibit 10.4 to the Registrant’s Registration Statement on Form S-1/A No. 3, filed with the Commission on November 16, 2004 (file no. 333-118024)). |
3.2 | Amended and Restated By-Laws of the Registrant as currently in effect (incorporated by reference to Exhibit 10.4 to the Registrant’s Registration Statement on Form S-1/A No. 3, filed with the Commission on November 16, 2004 (file no. 333-118024)). |
10.1 | 2004 Memorandum dated July 18, 2004, by and between Asahi Medical Co., Ltd. and the Registrant (incorporated by reference to Exhibit 10.4 to the Registrant’s Registration Statement on Form S-1/A No. 1, filed with the Commission on October 7, 2004 (file no. 333-118024)). |
10.2 | Amended and Restated Marketing and Distribution Agreement dated October 25, 2004 between Diamed Medizintechnik GmbH and the Registrant (incorporated by reference to Exhibit 10.6 to the Registrant’s Registration Statement on Form S-1/A No. 1, filed with the Commission on October 7, 2004 (file no. 333-118024)). |
10.3 | Amended and Restated Patent License and Royalty Agreement dated October 25, 2004 between the Registrant and Dr. Richard Brunner (incorporated by reference to Exhibit 10.8 to the Registrant’s Registration Statement on Form S-1/A No. 1, filed with the Commission on October 7, 2004 (file no. 333-118024)). |
10.4 | Amendment to the Distribution Services Agreement dated July 30, 2004 between the Registrant and Apheresis Technologies, Inc. (incorporated by reference to Exhibit 10.9 to the Registrant’s Registration Statement on Form S-1/A No. 1, filed with the Commission on October 7, 2004 (file no. 333-118024)). |
10.5 | 2002 Stock Option Plan (incorporated by reference to Exhibit 10.22 to the Registrant’s Registration Statement on Form S-1/A No. 3, filed with the Commission on November 16, 2004 (file no. 333-118024)). |
10.6 | Amended and Restated Patent License and Royalty Agreement dated October 25, 2004 between the Registrant and Hans Stock (incorporated by reference to Exhibit 10.12 to the Registrant’s Registration Statement on Form S-1/A No. 1, filed with the Commission on October 7, 2004 (file no. 333-118024)). |
10.7 | Employment Agreement between the Registrant and Elias Vamvakas dated September 1, 2004 (incorporated by reference to Exhibit 10.13 to the Registrant’s Registration Statement on Form S-1/A No. 1, filed with the Commission on October 7, 2004 (file no. 333-118024)). |
10.8 | Employment Agreement between the Registrant and Thomas P. Reeves dated August 1, 2004 (incorporated by reference to Exhibit 10.14 to the Registrant’s Registration Statement on Form S-1/A No. 1, filed with the Commission on October 7, 2004 (file no. 333-118024)). |
10.9 | Employment Agreement between the Registrant and Stephen Kilmer dated July 30, 2004 (incorporated by reference to Exhibit 10.17 to the Registrant’s Registration Statement on Form S-1/A No. 1, filed with the Commission on October 7, 2004 (file no. 333-118024)). |
10.10 | Employment Agreement between the Registrant and Julie Fotheringham dated September 1, 2004 (incorporated by reference to Exhibit 10.18 to the Registrant’s Registration Statement on Form S-1/A No. 1, filed with the Commission on October 7, 2004 (file no. 333-118024)). |
10.11 | Employment Agreement between the Registrant and Zayed (Joe) Zawaideh dated September 7, 2004 (incorporated by reference to Exhibit 10.19 to the Registrant’s Registration Statement on Form S-1/A No. 1, filed with the Commission on October 7, 2004 (file no. 333-118024)). |
10.12 | Product Purchase Agreement dated September 29, 2004 between the Registrant and Promedica International (incorporated by reference to Exhibit 10.20 to the Registrant’s Registration Statement on Form S-1/A No. 2, filed with the Commission on November 2, 2004(file no. 333-118024)). |
10.13 | Employment Agreement between the Registrant and Dr. David Eldridge dated November 9, 2004 ((incorporated by reference to Exhibit 10.23 to the Registrant’s Registration Statement on Form S-1/A No. 3, filed with the Commission on November 16, 2004 (file no. 333-118024)). |
10.14 | Consulting Agreement between the Registrant and Richard Davis dated May 1, 2004 (incorporated by reference to Exhibit 10.24 to the Registrant’s Registration Statement on Form S-1/A No. 4, filed with the Commission on December 6, 2004 (file no. 333-118024)). |
10.15 | Rental Agreement between the Registrant and Cornish Properties Corporation dated January 1, 2004 (incorporated by reference to Exhibit 10.27 to the Registrant’s Registration Statement on Form S-1/A No. 4, filed with the Commission on December 6, 2004 (file no. 333-118024)). |
10.16 | Sub-sublease between Echo Online Internet, Inc. and the Registrant dated September 29, 2004 (incorporated by reference to Exhibit 10.28 to the Registrant’s Registration Statement on Form S-1/A No. 4, filed with the Commission on December 6, 2004 (file no. 333-118024)). |
10.17 | Asset Purchase Agreement between Rheogenx Biosciences Corporation and the Registrant dated as of March 28, 2005 (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on May 6, 2005 (file no. 000-51030)). |
10.18 | Agreement between the Registrant and Rheogenx Biosciences Corporation dated March 28, 2005 (incorporated by reference to Exhibit 10.2 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on May 6, 2005 (file no. 000-51030)). |
10.19 | Termination Agreement between the Registrant and Apheresis Technologies, Inc. dated as of March 28, 2005 (incorporated by reference to Exhibit 10.3 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on May 6, 2005 (file no. 000-51030)). |
10.20 | Employment Agreement between the Registrant and John Cornish dated as of April 1, 2005 (incorporated by reference to Exhibit 10.4 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on May 6, 2005 (file no. 000-51030)). |
10.21 | Settlement Agreement among the Registrant, David Craig Eldridge and David C. Eldridge O.D., P.C. dated as of May 20, 2005 (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on August 8, 2005 (file no. 000-51030)). |
10.22 | Employment Agreement between John Caloz and the Registrant dated as of May 18, 2005 incorporated by reference to Exhibit 10.2 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on August 8, 2005 (file no. 000-51030)). |
10.23 | Amending Agreement between the Registrant and John Cornish, dated as of June 1, 2005, amending the Employment Agreement between the Registrant and John Cornish dated as of April 1, 2005 (incorporated by reference to Exhibit 10.3 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on August 8, 2005 (file no. 000-51030)). |
10.24 | Amending Agreement between the Registrant and Thomas P. Reeves, dated as of July 1, 2005, amending the Employment Agreement between the Registrant and Thomas P. Reeves dated August 2004 (incorporated by reference to Exhibit 10.4 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on August 8, 2005 (file no. 000-51030)). |
10.25 | Amending Agreement between the Registrant and Irving Siegel, dated as of September 1, 2005, amending the Employment Agreement between the Registrant and Irving Siegel dated as of August 1, 2003 (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on November 10, 2005 (file no. 000-51030)). |
10.26 | Consulting Agreement among the Registrant, AMD Medical Services Inc. and Irving Siegel dated as of September 1, 2005 (incorporated by reference to Exhibit 10.2 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on November 10, 2005 (file no. 000-51030)). |
10.27 | Employment Agreement between Steve Parks and the Registrant dated as of October 4, 2005 (incorporated by reference to Exhibit 10.3 to the Registrant’s Quarterly Report on Form 10-Q filed with the Commission on November 10, 2005 (file no. 000-51030)). |
10.28 | Option Agreement between Steve Parks and the Registrant dated as of October 4, 2005 (incorporated by reference to Exhibit 10.4 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on November 10, 2005 (file no. 000-51030)). |
10.29 | 2005 Memorandum between Asahi Kasei Medical Co., Ltd. and the Registrant dated October 17, 2005 (incorporated by reference to Exhibit 10.29 to the Registrant’s Annual Report on Form 10-K filed with the Commission on March 16, 2006 (file no. 0000-51030)). |
10.30 | Release Agreement between Zayed (Joe) Zawaideh and the Registrant, dated as of November 22, 2005, terminating the Employment Agreement between the Registrant and Zayed (Joe) Zawaideh dated September 7, 2004 (incorporated by reference to Exhibit 10.30 to the Registrant’s Annual Report on Form 10-K filed with the Commission on March 16, 2006 (file no. 0000-51030)). |
10.31 | Employment Agreement between Nozhat Choudry and the Registrant dated as of February 10, 2006 (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on May 10, 2006 (file no. 000-51030)). |
10.32 | Release Agreement between John Caloz and the Registrant, dated as of April 13, 2006, terminating the Employment Agreement between the Registrant and John Caloz dated May 18, 2006 (incorporated by reference to Exhibit 10.2 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on May 10, 2006 (file no. 000-51030)). |
10.33 | Release Agreement between Irving Siegel and the Registrant, dated as of April 13, 2006, terminating the Employment Agreement between the Registrant and Irving Siegel dated as of August 3, 2003, as amended by the Amending Agreement between the Registrant and Irving Siegel dated as of September 1, 2005 (incorporated by reference to Exhibit 10.3 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on May 10, 2006 (file no. 000-51030)). |
10.34 | Termination Agreement among the Registrant, AMD Medical Services Inc., Irving Siegel, OccuLogix Canada Corp., Rheo Clinic Inc. and TLC Vision Corporation, dated as of April 13, 2006, terminating, among other things, the Consulting Agreement among the Registrant, AMD Medical Services Inc. and Irving Siegel dated September 1, 2005 (incorporated by reference to Exhibit 10.4 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on May 10, 2006 (file no. 000-51030)). |
10.35 | Amending Agreement between the Registrant and William G. Dumencu, dated as of April 14, 2006, amending the Employment Agreement between the Registrant and William G. Dumencu dated as of August 1, 2003, as amended by the Amendment between the Registrant and William G. Dumencu dated August 1, 2003 and effective September 30, 2003 (incorporated by reference to Exhibit 10.5 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on May 10, 2006 (file no. 000-51030)). |
10.36 | Amending Agreement between the Registrant and Nozhat Choudry, dated as of April 1, 2006, amending the Employment Agreement between the Registrant and Nozhat Choudry dated as of February 10, 2006 (incorporated by reference to Exhibit 10.6 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on May 10, 2006 (file no. 000-51030)). |
10.37 | Amending Agreement between the Registrant and John Cornish, dated as of April 13, 2006, amending the Employment Agreement between the Registrant and John Cornish dated as of April 1, 2005, as amended by the Amending Agreement between the Registrant and John Cornish dated as of June 1, 2005 (incorporated by reference to Exhibit 10.7 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on May 10, 2006 (file no. 000-51030)). |
10.38 | Convertible Unsecured Promissory Note of Solx, Inc., dated April 1, 2006, in the principal amount of $2,000,000 (incorporated by reference to Exhibit 10.8 to the Registrant’s Quarterly Report on Form 10-Q/A, filed with the Commission on May 25, 2006 (file no. 000-51030)). |
10.39 | Agreement and Plan of Merger, dated as of August 1, 2006, by and among the Registrant, OccuLogix Mergeco, Inc., Solx, Inc. and Doug P. Adams, John Sullivan and Peter M. Adams, acting, in each case, in his capacity as a member of the Stockholder Representative Committee referred to therein (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on August 9, 2006 (file no. 000-51030)). |
10.40 | Convertible Unsecured Promissory Note of Solx, Inc., dated August 1, 2006, in the principal amount of $240,000 (incorporated by reference to Exhibit 10.2 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on August 9, 2006 (file no. 000-51030)). |
10.41 | Employment Agreement between the Registrant and Doug P. Adams dated as of September 1, 2006. |
10.42 | Registration Rights Agreement, dated as of September 1, 2006, among the Registrant, Doug P. Adams, John Sullivan and Peter M. Adams, acting, in each case, in his capacity as a member of the Stockholder Representative Committee referred to in the Agreement and Plan of Merger, dated as of August 1, 2006, by and among the Registrant, OccuLogix Mergeco, Inc., Solx, Inc. and Doug P. Adams, John Sullivan and Peter M. Adams, acting, in each case, in his capacity as a member of the Stockholder Representative Committee referred to therein. |
10.43 | 2006 Distributorship Agreement between Asahi Kasei Medical Co., Ltd. and the Registrant dated October 20, 2006 (incorporated by reference to Exhibit 10.1 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on November 9, 2006 (file no. 000-51030)). |
10.44 | Summary of Terms and Conditions between the Registrant and Elias Vamvakas dated November 30, 2006. |
10.45 | Series A Stock Purchase Agreement by and among OcuSense, Inc. and the Registrant dated as of November 30, 2006. (Exhibits have been omitted pursuant to Item 601(b)(2) of Regulation S-K and will be provided to the Securities and Exchange Commission upon request.) |
10.46 | Securities Purchase Agreement, dated as of February 1, 2007, by and among the Registrant and the investors listed on the Schedule of Investors attached thereto as Exhibit A (incorporated by reference to Exhibit 10.1 to the Registrant’s Current Report on Form 8-K, filed with the Commission on February 6, 2007 (file no. 000-51030)). |
10.47 | Employment Agreement between the Registrant and Suh Kim dated as of March 12, 2007. |
10.48 | License Agreement between OcuSense, Inc. and The Regents of the University of California dated March 12, 2003. (Portions of this exhibit have been omitted pursuant to a request for confidential treatment.) |
10.49 | Amendment No. 1, dated June 9, 2003, to the License Agreement between OcuSense, Inc. and The Regents of the University of California dated March 12, 2003. |
10.50 | Amendment No. 2, dated September 5, 2005, to the License Agreement between OcuSense, Inc. and The Regents of the University of California dated March 12, 2003. (Portions of this exhibit have been omitted pursuant to a request for confidential treatment.) |
10.51 | Amendment No. 3, dated July 7, 2006, to the License Agreement between OcuSense, Inc. and The Regents of the University of California dated March 12, 2003. |
10.52 | Amendment No. 4, dated October 9, 2006, to the License Agreement between OcuSense, Inc. and The Regents of the University of California dated March 12, 2003. |
14.1 | Code of Conduct of the Registrant (incorporated by reference to Exhibit 14.1 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on November 10, 2005 (file no. 000-51030)). |
14.2 | Complaint and Reporting Procedures of the Registrant (incorporated by reference to Exhibit 14.2 to the Registrant’s Quarterly Report on Form 10-Q, filed with the Commission on August 8, 2005 (file no. 000-51030)). |
21.1 | Subsidiaries of Registrant (incorporated by reference to Exhibit 21.1 to the Registrant’s Registration Statement on Form S-1/A No. 1, filed with the Commission on October 7, 2004 (file no. 333-118024)). |
23.1 | Consent of Ernst & Young LLP. |
23.2 | Consent of Peter Ott & Associates Inc. |
24.1 | Power of Attorney (included on signature page). |
31.1 | CEO’s Certification required by Rule 13A-14(a) of the Securities Exchange Act of 1934. |
31.2 | CFO’s Certification required by Rule 13A-14(a) of the Securities Exchange Act of 1934. |
32.1 | CEO’s Certification of periodic financial reports pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, U.S.C. Section 1350. |
32.2 | CFO’s Certification of periodic financial reports pursuant to Section 906 of the Sarbanes-Oxley Act of 2002, U.S.C. Section 1350. |