UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the fiscal year ended December 31, 2013
OR
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number: 001-35953
REGADO BIOSCIENCES, INC.
(Exact name of registrant as specified in its charter)
| | |
| Delaware | | No. 03-0422069 |
(State or other jurisdiction of incorporation or organization) | | (I.R.S. Employer Identification No.) |
120 Mountain View Boulevard
Basking Ridge, New Jersey 07920
(Address of principal executive offices) (Zip Code)
(908) 580-2100
(Registrant’s telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| | |
Title of Each Class | | Name of Each Exchange on Which Registered |
| Common Stock, $0.001 par value per share | | The NASDAQ Capital Market |
Securities registered pursuant to Section 12(g) of the Act:
None.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act. (Check one):
| | | | | | |
| Large accelerated filer | | ¨ | | Accelerated filer | | ¨ |
| | | |
| Non-accelerated filer | | ¨ (Do not check if a smaller reporting company) | | Smaller reporting company | | x |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes ¨ No x
The aggregate market value of the registrant’s voting and non-voting common stock held by non-affiliates of the registrant (without admitting that any person whose shares are not included in such calculation is an affiliate) computed by reference to the price at which the common stock was last sold on February 28, 2014 was approximately $90.5 million. The registrant has provided this information as of February 28, 2014 because its common stock was not publicly traded as of the last business day of its most recently completed second fiscal quarter.
As of March 10, 2014 there were 25,327,266 shares of the registrant’s common stock, $0.001 par value, outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
None
REGADO BIOSCIENCES, INC.
Annual Report on Form 10-K
Fiscal Year Ended December 31, 2013
Table of Contents
2
PART I
Cautionary Statement Regarding Forward-Looking Statements
This annual report on Form 10-K includes forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended. For this purpose, any statements contained herein, other than statements of historical fact, including statements regarding the progress and timing of our product development programs and related trials; our future opportunities; our strategy, future operations, anticipated financial position, future revenues and projected costs; our management’s prospects, plans and objectives; and any other statements about management’s future expectations, beliefs, goals, plans or prospects constitute forward-looking statements within the meaning of the Private Securities Litigation Reform Act of 1995. We may, in some cases, use words such as “anticipate,” “believe,” “could,” “estimate,” “expect,” “intend,” “may,” “plan,” “project,” “should,” “target,” “will,” “would” or other words that convey uncertainty of future events or outcomes to identify these forward-looking statements. Actual results may differ materially from those indicated by such forward-looking statements as a result of various important factors, including our “critical accounting estimates”; our plans to complete our single, open-label 13,200 subject Phase 3 trial of REG1; our ability to satisfy domestic and international regulatory requirements with respect to REG1 and our other product candidates, many of which are new and still evolving, and the labeling under any approval we may obtain; the performance of contract research organizations who conduct our clinical trials for us; the performance of third-party manufacturers who supply or manufacture our products; our ability to develop commercialization and marketing capabilities or to enter into strategic partnerships to develop and commercialize REG1 or any of our other product candidates; the timing and success of the commercialization of REG1 or any of our other product candidates; the rate and degree of market acceptance of REG1; the size and growth of the potential markets for REG1 and our ability to serve those markets; our plans to expand the indications of REG1; our ability to discover, develop and commercialize novel and innovative therapies using our proprietary technology platform; regulatory developments in the United States and foreign countries; competition from existing antithrombotic drugs or new antithrombotic drugs that may emerge; potential product liability claims; our ability to attract and retain a sufficient number of scientists, clinicians, sales personnel and other key personnel; our ability to obtain, maintain, defend and enforce intellectual property rights protecting our product candidates; the accuracy of our estimates regarding expenses and capital requirements and our ability to adequately support future growth; and the factors listed in this report under “Part I—Item 1A Risk Factors.” All forward-looking statements are expressly qualified in their entirety by this cautionary notice. You are cautioned not to place undue reliance on any forward-looking statements, which speak only as of the date of this report or the date of the document incorporated by reference into this report. We have no obligation, and expressly disclaim any obligation, to update, revise or correct any of the forward-looking statements, whether as a result of new information, future events or otherwise. We have expressed our expectations, beliefs and projections in good faith and we believe they have a reasonable basis. However, we cannot assure you that our expectations, beliefs or projections will result or be achieved or accomplished.
Overview
We are a biopharmaceutical company focused on the discovery and development of novel, first-in-class, actively controllable antithrombotic drug systems for acute and sub-acute cardiovascular indications. We are pioneering the discovery and development of two-component drug systems consisting of a therapeutic aptamer and its specific active control agent. Our actively controllable product candidates have the potential to improve outcomes, enhance the patient experience and reduce overall treatment costs.
Each of our product candidates consists of a two-component system: an aptamer and its specific active control agent. The aptamer is administered first and achieves its therapeutic effect within minutes. When the therapeutic effect of the aptamer is no longer needed, the control agent is administered to rapidly and precisely reduce or eliminate it. The level of reduction is determined by the amount of control agent given compared to the aptamer dose. By contrast and for example, the therapeutic effect of existing antithrombotic drugs is not rapidly and precisely controllable and persists until the drug is metabolized by the patient, a process which varies from patient to patient and can take several hours or more.
Our lead product candidate, REG1, consists of pegnivacogin, a highly potent and selective anticoagulant, and anivamersen, its specific active control agent. We are developing REG1 as an anticoagulant for use in patients with a wide variety of cardiovascular conditions undergoing percutaneous coronary intervention, or PCI, a hospital-based procedure used to mechanically open or widen obstructed coronary arteries. Interventional cardiologists performing PCIs must consider the risk of major bleeding events in determining the level of anticoagulation administered to patients to prevent ischemic events, including death, stroke, myocardial infarction, or MI, or the need for revascularization of the artery. As the anticoagulant effect of existing drugs persists long after administration, intervention cardiologists are forced to make a compromising medical decision because they lack the means to simultaneously reduce the risks of ischemic and major bleeding events. In 2005, we filed an investigational new drug application, or IND, for the use of REG1 in this initial indication. In March 2014, we announced that REG1 has received Fast Track designation from the U.S. Food and Drug Administration, or FDA, for anticoagulant therapy to be used in patients with coronary artery disease during PCI.
3
We believe that REG1 has the potential to become the standard of care for anticoagulation therapy for patients undergoing PCI and other cardiovascular procedures because it gives the physician precise, on-demand control over anticoagulation levels. REG1 is the first and only anticoagulant to demonstrate a reduction in both ischemic and major bleeding events in a clinical trial for PCI. In our clinical trials, REG1 demonstrated a rapid and predictable anticoagulant effect that was precisely modulated or completely reversible in real time. In our randomized, partially blinded, dose-ranging Phase 2b trial involving 640 subjects, or the RADAR trial, when compared to standard of care heparin, REG1 demonstrated both a rapid and predictable anticoagulant effect and ability to precisely modulate or eliminate that effect in real time. REG1 also demonstrated the following important clinical and pharmacoeconomic benefits:
| | • | | an approximate 66.0% reduction in ischemic events; |
| | • | | a reduction of up to 60.0% in major bleeding events; |
| | • | | a substantial reduction in time from catheterization to catheter sheath removal from a median of 3.8 hours to a median of one hour; |
| | • | | a substantial reduction in time of completion of the PCI procedure to catheter sheath removal from a median of three hours to a median of 24 minutes; and |
| | • | | a substantial reduction in the time patients were required to remain still following catheter sheath removal from a median of 5.7 hours to a median of 2.8 hours. |
Based on these clinical results and after discussion with the FDA and the European Medicines Agency, or EMEA, in September of 2013, we initiated a single, open-label, 13,200 subject Phase 3 trial of REG1, or the REGULATE-PCI trial, in patients undergoing PCI procedures other than for the treatment of ST elevation myocardial infarctions. REGULATE-PCI, if successful, will serve as the basis for product registration applications worldwide. We believe that REG1 has potential use in other PCI and interventional cardiovascular procedures, such as open heart surgery, or OHS, PCI as a treatment for ST segment elevation myocardial infarction as well as transcatheter aortic valve replacement or implantation, or TAVI.
Using our proprietary technology platform, we are developing a portfolio of additional clinical candidates in acute and sub-acute cardiovascular and other indications. REG3 is a preclinical stage antiplatelet therapy, consisting of a glycoprotein VI, or GPVI, inhibitor to be evaluated in diabetic vasculopathy and/or other inflammatory diseases. The specific active control agent component of REG3 is designed to permit modulation or elimination, if necessary, of the GPVI inhibition, if necessary, to optimize dosing and minimize unwanted side effects that might result from GPVI inhibition. We intend to file an IND for REG3 upon completion of our remaining pre-IND preclinical and chemistry, manufacturing and controls work, and to initiate a Phase 1 study of REG3 subsequent to that, depending upon availability of financial resources. REG2 is an early clinical stage program evaluating an extended release formulation of pegnivacogin intended to provide a controllable level of anticoagulation for up to two weeks for venous antithrombotic applications such as venous thromboembolism, or VTE, prophylaxis. In REG2, anivamersen would be used as an active control agent if needed. We have completed a single escalating dose Phase 1 clinical trial of REG2 and plan to conduct additional clinical testing in the future. We filed an IND for REG2 in November 2013 which was accepted by FDA and became active in December 2013. Additionally, we are considering potential product candidates against a variety of traditional and novel antiplatelet receptor targets as well as developing aptamer-based antidotes specific to respective oral FXa inhibitors.
4
The following table lists our current product candidates and discovery programs and their respective stages of development:

Our product candidates and proprietary technology platform are protected by a patent estate of 30 issued or allowed patents, including 14 in the United States, covering our composition of matter and methods of use for our product candidates as well as our fundamental controllable aptamer technology. We maintain worldwide commercialization rights to all of our product candidates except in Armenia, Azerbaijan, Belarus, Georgia, Kazakhstan, Kyrgyzstan, Moldova, Russia, Tajikistan, Turkmenistan, Ukraine and Uzbekistan, or the Covered Territory.
PCI Overview
PCIs are hospital-based procedures used to mechanically open or widen obstructed coronary arteries. To treat this condition, a deflated balloon or other device on a catheter is inserted into an artery through an arterial sheath in the groin or wrist and advanced to the site of a blockage. Normalized blood flow is restored by opening or widening the artery. A stent is often placed at the site of blockage to permanently maintain blood flow.
Patients undergoing PCI fall into three treatment categories: emergency, urgent or elective. Patients undergoing PCI as an emergency or urgent procedure suffer from acute coronary syndromes, or ACS, defined as:
| | • | | unstable angina, or UA, a condition in which the heart does not get enough blood flow and oxygen causing chest discomfort even at rest and which may lead to an MI without prompt medical intervention; |
| | • | | Non-ST Elevation Myocardial Infarction, or N-STEMI, which is an MI without electrocardiogram, or ECG, evidence of cell death but where chemical enzyme analysis shows muscle damage has occurred or is occurring; and |
| | • | | ST Elevation Myocardial Infarction, or STEMI,which is an acute MI with ECG evidence of cell death and abnormally high levels of chemical enzymes. |
Patients undergoing PCI as an elective procedure include patients with:
| | • | | chest pain or discomfort that usually occurs with activity or stress, or stable angina, which is due to a coronary arterial blockage; |
| | • | | a significant change or worsening of coronary arterial blood flow on a stress test or echocardiogram; and |
| | • | | previous heart damage with a known coronary arterial blockage who return for additional treatment. |
PCI procedures involve significant medical risks. Blood clots may exist at the blockage or may result from arterial damage at the point of catheter insertion or from the presence of the catheter or other foreign materials in the vasculature. If blood clotting is not eliminated or controlled effectively, ischemic events, including death, stroke, MI or the need for revascularization of the artery, may occur. Because of this risk, powerful anticoagulant drugs are administered prior to and throughout the PCI procedure. However, anticoagulants create a significant risk of major bleeding events, including bleeding into the heart, the brain or other organs, massive internal bleeding that requires a significant transfusion of blood and blood products, and uncontrolled bleeding at the access site. These major bleeding events can result in the need for emergency surgery and increase the risk of MI, stroke, other life-threatening complications and death.
5
Because the therapeutic effect of most existing anticoagulants persists until they are metabolized by the patient, the risk of major bleeding events remains long after the procedure is completed, even when the anticoagulant administration is stopped at the end of the PCI. As a result, patients must wait up to six hours post-procedure before the arterial sheath can be safely removed, increasing patient discomfort and requiring extended monitoring and potential intervention by medical staff. In certain circumstances, a vascular closure device, or VCD, may be used to block or control bleeding at the access site to reduce this waiting time. However, it is not always possible to use a VCD and a VCD may not significantly reduce the risk of a major bleeding event. In addition, the use of a VCD adds significant cost to the procedure and may cause increased risk of vascular complications. As a result, the use of VCDs varies significantly from physician to physician and from hospital to hospital. If a VCD is not utilized, direct compression at the access site by medical staff for up to 30 minutes is typically applied followed by application of a sandbag or other mechanical compression throughout the total wait time to prevent bleeding.
Interventional cardiologists performing a PCI are forced to make a compromising medical decision because they lack the means to simultaneously reduce the risk of ischemic and major bleeding events. We believe that an antithrombotic treatment that can reduce both risks would create a new standard of care.
PCI Market
The PCI market is a large and growing market. According to the American Heart Association, or the AHA, approximately 770,000 PCIs were performed in the United States in 2013. Based on information obtained from publicly available PCI procedure registries, we estimate that in 2013 approximately 540,000 PCIs, excluding STEMI patients but including N-STEMI acute coronary syndrome patients and elective PCI for coronary arterial disease were performed in Europe and at least 1.2 million in the rest of the world.
We expect that the global market for PCIs will continue to grow while the market for PCIs in the United States and Europe will be relatively stable. Based on historical statistics published by the AHA, we believe that the growth in the number of PCIs performed in the United States will correlate to the growth of the population over age 45, which the U.S. Census Bureau estimates will increase at a rate of approximately 1.2% annually for the next decade. PCI growth in emerging countries is expected to be much greater. The National Center for Cardiovascular Diseases China reported that the number of PCIs performed in the People’s Republic of China increased approximately 26% annually from 2005 to 2009 while the National Intervention Council estimated in 2010 that PCI procedures in India would grow at approximately 25% annually. Based on 2008 statistics provided by the Brazilian Public Health System we estimated a projected annual growth rate of approximately 9% in PCI procedures in Brazil. Based on similar information obtained from other countries, we believe that similar increases in the number of PCI procedures are occurring throughout the developing world. This expected worldwide growth in PCI procedures is due to an expected increase in cardiovascular disease and such factors as an aging population, increasing clinical adoption of PCI procedures and increasing PCI survival rates. Based upon the estimated cost per procedure of branded anticoagulants, we believe that this represents a greater than $3.0 billion annual market opportunity for anticoagulants used in PCI procedures.
We intend to seek initial regulatory approval for the use of REG1 in all PCI procedures other than in the treatment of STEMI. Based on 2013 statistics published by the American Heart Association, we believe that approximately 2.5 million PCI procedures were performed worldwide in 2013 in this segment of the market. We anticipate that this segment of the market will grow faster than the overall PCI market as a result of an ongoing clinical shift toward less invasive cardiovascular procedures.
6
Current PCI Anticoagulants and Their Limitations
Anticoagulants are administered throughout a PCI procedure to reduce the chances of unwanted blood clots forming due to arterial damage at the point of catheter insertion or to the presence of the catheter or other foreign materials in the vasculature. The anticoagulants most commonly used in PCI procedures are heparin and bivalirudin.
Heparin
Discovered in 1916, heparin is derived from the entrails of pigs and cows. It is a non-specific, indirect inhibitor of thrombin and blood coagulation Factor Xa, or FXa. There are two forms of heparin: unfractionated heparin, or UFH, and low molecular weight heparin, or LMWH. LMWH is primarily used as an antithrombotic treatment outside of the acute care setting and is used on a limited basis in PCI. When we refer to heparin, we mean UFH unless otherwise indicated. In PCI, heparin is first administered by IV bolus injection followed by a continuous IV infusion during the procedure. Heparin has a number of shortcomings including:
| | • | | Limited effectiveness. As an indirect thrombin inhibitor, heparin is ineffective on clots that have already formed or are in the process of forming. |
| | • | | Major bleeding risk. Heparin requires 20 to 30 minutes to reach its peak anticoagulant effect; consequently physicians may exceed the minimum effective dose to access a compromised artery faster. As a result, patients are exposed to a supratherapeutic level of anticoagulation well after the procedure is completed as the anticoagulant effect of heparin persists at significant levels for up to six hours and the typical PCI procedure only requires approximately 30 to 60 minutes to complete. Female patients as well as patients who are elderly, frail or have kidney disease are particularly at risk for major bleeding events. Also, physicians often use intravenous glycoprotein platelet, or GPIIb/IIIa, inhibitors along with heparin in patients with a high thrombus risk. Recent clinical trials have shown that bleeding risk increases when heparin is used in combination with GPIIb/IIIa inhibitors. |
| | • | | Risk of immunological reaction. Patients receiving heparin can develop heparin-induced thrombocytopenia and thrombosis, or HIT/HITTS, a dangerous and life-threatening immunological reaction that can result in amputation or death. A 2005 article inThrombosis reported HIT/HITTS rates of up to 2.7% in all heparin patients. |
| | • | | Unpredictable pharmacokinetics. Heparin dosing is imprecise because its dose-response relationship is non-linear and it is metabolized at varying rates from patient to patient. In addition, some patients are heparin-resistant. As a result, a patient’s unpredictable response to heparin requires close and continuous monitoring of anticoagulation levels. |
| | • | | No specific active control. Protamine, a drug that reverses the anticoagulant effect of heparin, is rarely used in PCI due to its unpredictable effect and its adverse event profile, which includes allergic reactions, pulmonary hypertension and cross-reactivity with certain types of insulin. As a result, its use is generally limited to OHS, where the high level of heparin used makes a reversal agent medically necessary despite protamine’s shortcomings. If a physician needs to modulate heparin’s anticoagulant effect in a PCI procedure, an extensive transfusion of blood and blood products is the most commonly used method. Additionally, if mechanical complications, such as the inadvertent perforation of an artery occur, extensive intervention including possible emergency surgery is required. |
| | • | | Quality and purity concerns. A substantial portion of the world supply of heparin comes from outside of the United States and the European Union. In the recent past, contaminated heparin has been responsible for a number of deaths and other complications. In addition, variations in product potency are not uncommon. |
Bivalirudin
Bivalirudin is a direct thrombin inhibitor administered by IV bolus injection followed by continuous IV infusion during the PCI procedure. In clinical studies, patients metabolized bivalirudin at a faster rate than heparin. Although bivalirudin has demonstrated a lower rate of major bleeding events compared to heparin in clinical studies, it has a number of shortcomings:
| | • | | Ischemic risk. In clinical trials, patients receiving bivalirudin had a numerically higher rate of ischemic events, including MIs and stent clotting, than patients receiving heparin. As a result, physicians typically use heparin when treating unstable patients and patients at a higher risk of having an ischemic event. |
| | • | | Bleeding risk. Although bivalirudin is metabolized more quickly than heparin, its anticoagulant effect persists at significant levels for up to four hours. As a result, patients are exposed to the risk of a major bleeding event for an extended period. Use of GPIIb/IIIa inhibitors with bivalirudin further exacerbates this risk. |
| | • | | No reversal agent. Because there is no reversal agent for bivalirudin, an extensive transfusion of blood and blood products is the only means to modulate its anticoagulant effect. Due to its mechanism of action, emergency surgical intervention is more likely with bivalirudin than heparin in the event of mechanical complications such as the inadvertent perforation of an artery. |
7
| | • | | Unpredictable dosing. Because bivalirudin is metabolized by the kidneys, dose adjustments are necessary for patients with renal insufficiency. While standard coagulation tests can be performed, the results do not accurately indicate quantitative bivalirudin levels and therefore cannot be used to precisely make dose adjustments. In addition, because the amount of bivalirudin necessary for the treatment of a specific patient depends both on the patient’s weight and the length of the PCI procedure, the amount and cost of bivalirudin used per procedure is difficult to predict. |
| | • | | Risk of immunological reaction. According to its package insert, use of bivalirudin has been associated with severe allergic reactions and publicly available safety data has reported two anaphylaxis deaths. |
The REG1 Anticoagulation System
Overview
REG1 is a two-component system consisting of pegnivacogin, an anticoagulant targeting coagulation Factor IXa, or FIXa, and its specific active control agent, anivamersen, both of which are administered solely by IV bolus injection.
Pegnivacogin is a single-stranded oligonucleotide conjugated to polyethylene glycol, or PEG. Pegnivacogin is a potent anticoagulant that binds to FIXa with high affinity and specificity, thereby preventing blood clot formation and progression. Pegnivacogin is administered prior to the start of a PCI procedure and achieves its maximal anticoagulant effect within five minutes of injection. This anticoagulant effect lasts for 24 hours or more unless modulated or eliminated by the specific active control agent.
In developing pegnivacogin, we targeted FIXa for the following reasons:
| | • | | FIXa controls the rate of thrombin formation so inhibiting it should more effectively slow or prevent clotting than inhibition of FXa or thrombin; |
| | • | | FIXa is seven times more potent than FXa and 60 times more potent than thrombin, so less drug is needed to achieve a desired anticoagulant effect; and |
| | • | | due to the biochemical nature of the interactions of FIXa in the clotting process, aptamers are the only known means to achieve controllable inhibition of FIXa. |
Anivamersen is an oligonucleotide, a biological polymer consisting of a relatively small number of nucleotides chemically bound in a linear sequence that forms a chain-like structure, or strand, that is complementary to a portion of pegnivacogin and has no pharmacologic activity other than to bind to pegnivacogin. When anivamersen binds to pegnivacogin, it changes the shape of pegnivacogin so that it can no longer bind to FIXa, thereby permanently eliminating its anticoagulant activity within minutes. At the end of a PCI procedure, when the antithrombotic effect of the pegnivacogin is no longer needed, anivamersen is administered to rapidly and precisely reduce or eliminate it.
The effect of anivamersen on pegnivacogin is solely dependent on the amount of anivamersen administered relative to the dose of pegnivacogin. As a result, and unlike heparin and bivalirudin, REG1’s anticoagulation reversal is independent of an individual patient’s metabolism or health. By adjusting the dose of anivamersen relative to pegnivacogin, the anticoagulant effect of pegnivacogin can be precisely and rapidly controlled or eliminated.
Competitive Advantages
We believe that REG1 has the potential to become the standard of care for anticoagulation therapy in PCI and other cardiovascular procedures because it gives physicians precise, on-demand control over anticoagulation levels. We believe the key advantages of REG1 over existing therapies are the following:
| | • | | Reduced ischemic events. Because the anticoagulant effect of REG1 can be precisely controlled or eliminated, REG1 allows a higher level of anticoagulation to be used safely during the PCI procedure when needed most. A higher level of anticoagulation will reduce the occurrence of ischemic events such as death, MI, stroke or the need for revascularization. |
| | • | | Reduced major bleeding events. REG1’s anticoagulant effect can be modulated or eliminated at the end of the PCI procedure, when it is no longer necessary or desirable, to reduce the risk of major bleeding events. Once the anticoagulation effect is reduced or eliminated, ordinary clotting can occur. |
| | • | | Precise and predictable dosing. Because REG1’s effect is independent of an individual patient’s metabolism or health, dosing is precise and predictable, thereby eliminating the need for time-consuming and costly patient monitoring during and after PCI. |
| | • | | Broad applicability. REG1’s use in PCI is unrestricted for high risk patients such as those with kidney or liver impairment. |
8
| | • | | Shorter procedure and recovery times. REG1 significantly reduces the time between the end of the PCI procedure and the time the arterial catheter sheath can be pulled safely, as well as the time between the end of the procedure and the time the patient is ambulatory. As a result, we believe that overall procedure times can be significantly reduced, resulting in shorter hospital stays. |
| | • | | Improved patient outcomes and experience. We believe that REG1 will improve patient outcomes by reducing the overall number of ischemic events and the risk of major bleeding events. Also, REG1 can shorten procedure and recovery times and reduce the need for VCDs or other procedure-related follow-up interventions and re-hospitalizations. Because procedure and recovery times will be reduced and patients will experience fewer complications and will be able to leave the hospital more quickly, we believe that the patient experience will also be improved. |
| | • | | Increased staff and facility efficiency. Shorter procedure times will increase staff and facility efficiency by allowing more patients to be treated with reduced staff attention and intervention necessary after the procedure ends. We believe that REG1 will also enhance efficiency by reducing complications that may require emergency intervention or re-hospitalization, such as the need for revascularizations or major blood transfusions. |
| | • | | Reduced overall treatment costs. We believe that REG1 will reduce overall treatment costs by reducing ischemic and major bleeding events, shortening procedure and recovery times, reducing re-hospitalizations due to complications, increasing staff and facility efficiency and improving patient outcomes. |
Early Clinical Trials
REG1 has been studied in three Phase 1 trials involving a total of 174 subjects and one Phase 2a proof-of-concept PCI trial involving 26 subjects. These early clinical trials collectively demonstrated that:
| | • | | pegnivacogin, at the intended PCI dose of 1 mg/kg, provided near complete FIXa inhibition; |
| | • | | a single injection of pegnivacogin reliably produced anticoagulation in humans for more than 24 hours; |
| | • | | a single injection of anivamersen partially or completely reversed pegnivacogin anticoagulation rapidly based on the ratio of the anivamersen dose to the pegnivacogin dose; |
| | • | | the anticoagulation effect of REG1 can be measured with existing standard tests; |
| | • | | pegnivacogin and anivamersen were well-tolerated; |
| | • | | elective PCI could be performed successfully using REG1 therapy; and |
| | • | | arterial catheter sheaths could be removed at the end of PCI procedures. |
9
RADAR Trial
In November 2010, we completed the RADAR trial involving 640 subjects at 69 enrolling sites in the United States, Canada, France, Germany, Poland and the Netherlands. RADAR was an adaptive design, randomized, partially-blinded, dose-ranging trial assessing the safety and efficacy of REG1 in patients with UA and N-STEMI intended for PCI. Standard of care heparin, either UFH or LMWH, was the comparator. Subjects received open-label pegnivacogin at 1mg/kg, followed by a blinded dose of anivamersen to achieve 25%, 50%, 75% or 100% levels of pegnivacogin reversal. GPIIb/IIIa inhibitor use was stipulated for the heparin arm per standard of care guidelines and for provisional use in the REG1 arm for procedural or angiographic complications. Arterial catheter sheaths were to be removed at the end of the catheterization in the REG1 arm and per standard of care in the heparin arm. The demographics and background therapies in RADAR were similar to prior ACS studies with heparin and bivalirudin.
RADAR had several objectives, including the following:
| | • | | determine the clinically acceptable dose range of anivamersen that reliably reverses the anticoagulant effect of pegnivacogin as measured by bleeding events; |
| | • | | estimate the efficacy of REG1 in suppressing ischemic events, defined as death, nonfatal MI and recurrent ischemia in target vessel distribution or urgent target vessel revascularization, or TVR, compared to heparin; |
| | • | | confirm that near-complete FIXa inhibition was achieved with pegnivacogin at the 1 mg/kg dose; and |
| | • | | measure various pharmacoeconomic factors such as the feasibility of early arterial catheter sheath removal, time to sheath removal and time to patient ambulation, compared to standard of care heparin. |
The primary endpoint of RADAR was the composite incidence of major and minor bleeding through day 30. The key secondary endpoint was the proportion of subjects with a composite of ischemic events through day 30. In addition, a predefined analysis of the primary and secondary endpoints was made at 48 hours to simulate hospital discharge. All of these endpoints were adjudicated by an independent clinical events committee.
As part of the adaptive design to determine the effective dose response for anivamersen, RADAR included assessments by an independent data safety and monitoring board, or DSMB, at prescribed enrollment milestones. The DSMB was empowered to eliminate one or more of the anivamersen doses if excessive ischemic event rates were observed or, as was expected for the lowest anivamersen dose, excessive bleeding event rates were observed. If an anivamersen dose was eliminated as part of the adaptive design, the remaining REG1 subjects intended for the discontinued group would be randomized into a continuing anivamersen dose. As contemplated by the adaptive design, the DSMB ended the lowest anivamersen dose of 25% reversal early in the study due to excessive bleeding events.
RADAR demonstrated that REG1 was well-tolerated and, excluding the 25% reversal group, that there was no relevant difference in the overall incidence of treatment-emergent adverse events or serious adverse events between the REG1 arm and the heparin arm or among the remaining REG1 treatment groups.
At day 30, the 100% reversal dose significantly reduced total and major bleeding events as compared to the 25% REG1 reversal dose with a stepwise numerical reduction in major bleeding at doses of anivamersen greater than 50% reversal.
At 48 hours, the 100% reversal dose was statistically superior to both heparin (p=0.045) and the 25% reversal dose (p<0.001) in reducing major bleeding events. At anivamersen doses greater than 50% reversal, a decrease in the number of subjects with major bleeding events was observed. There was a trend towards reduction in total bleeding in the 100% reversal group, as compared to heparin (p=0.07). Among the REG1 groups, there was an anivamersen dose-dependent numerical decrease in the number of subjects with total bleeding from time of randomization to 48 hours. The results of RADAR confirmed that there was a consistency of effect across total and major bleeding events with doses above 50% reversal numerically better than heparin. The 100% reversal dose was statistically superior to the 25% reversal dose in reducing the incidence of total bleeding (p<0.001), which also confirmed the dose response. Based upon the odds ratio, which is a measure of the effect size, the REG1 100% reversal group demonstrated a 60% reduction in major bleeding events as compared to standard of care heparin.
10
The results of major bleeding events reported in RADAR for heparin and for REG1 at 48 hours are summarized in the following table:
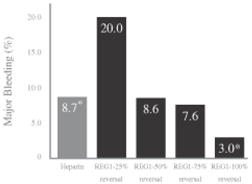
Source: Povsic et al., Controlled anticoagulation in ACS, European Heart Journal, 2012
All REG1 patients received the same pegnivacogin dose. An overall composite ischemic event rate was calculated by combining the composite ischemic events from each REG1 group. In the overall REG1 arm, there was a numerical reduction in the rate of composite ischemic events at day 30 to 3.0% compared to 5.7% for patients in the heparin arm. Overall, 1.9% of the subjects in the REG1 arm had composite ischemic events at 48 hours compared to 5.0% of the subjects in the heparin arm. A consistent effect on ischemic events was seen at 48 hours, 30 days and in those patients undergoing PCI in the REG1 groups, as compared to heparin. For those REG1 subjects undergoing PCI, no evidence of clots on guide wires or catheters was reported. Based upon the odds ratio, which is a measure of the effect size, the REG1 group demonstrated a 66% reduction in ischemic events as compared to standard of care heparin.
Composite ischemic events for the heparin arm and the REG1 arm from time of randomization to 48 hours are shown in the table below:
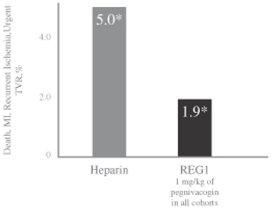
Source: RADAR Clinical Study Report
Measured levels of coagulation corresponded well to targeted levels of reversal after anivamersen dosing. These results indicate the ability to reliably predict reversal with the use of REG1, even when treating physicians are blinded to the level of reversal and the coagulation measurements.
11
REG1 also demonstrated a number of potential pharmacoeconomic effects. Arterial catheter sheaths were removed a median of one hour after catheterization compared to a median of 3.8 hours in the heparin arm. Arterial catheter sheaths were removed a median of 24 minutes after the procedure in the REG1 arm compared to a median of three hours in the heparin arm. Subjects were ambulated approximately a median of 2.8 hours after arterial catheter sheath removal in the REG1 arm compared to a median of 5.7 hours after arterial catheter sheath removal in the heparin arm. These results are summarized below:

Source: RADAR Clinical Study Report
Late in the trial, three REG1 subjects experienced severe allergic events. These events occurred 3 to 20 minutes after administration of pegnivacogin and ranged from a subject with a mild skin reaction to one subject who needed extended hemodynamic support. Based on a blinded assessment of bleeding and ischemic events at that time, it was determined that a sufficient number of endpoint events had occurred to meet the objectives of the trial and the trial was deemed complete. Following the occurrence of the allergic events, a detailed analysis of possible causes was performed. We determined that all three subjects were experiencing activation of their immunological system prior to receiving REG1 treatment and had a history of allergic reactions. We also determined that the REG1 used in these patients conformed to our specifications, was not contaminated, mishandled or stored incorrectly and that no changes to the formulation had occurred. Based on non-human primate studies, we concluded that there was no clinical or biomarker evidence of intravascular immune pathway activation. We also re-examined the complete REG1 clinical database and found no evidence of serious allergic reactions within the remainder of the REG1 development program. Following completion of our investigations and submission of a risk minimization action plan, the FDA and the EMEA agreed that we could proceed to a Phase 3 trial and that no additional exclusion criteria, and no special dosing, pre-treatment or pre-screening requirements were necessary. Additionally, in REGULATE-PCI investigators will receive training on the identification and proper treatment of allergic reactions and blood samples from all subjects will be collected and stored for future reference in the event they are needed for analysis.
REGULATE-PCI Trial
Based on these clinical results and after discussion with the FDA and the EMEA, we commenced the REGULATE-PCI trial September 2013. REGULATE-PCI, if successful, will serve as the basis for product registration applications worldwide.
REGULATE-PCI is a single, open-label Phase 3 trial comparing REG1 to bivalirudin in subjects undergoing PCI as an elective procedure or for the treatment of UA or N-STEMI. The first 1,000 subjects enrolled will be limited to those undergoing elective PCI or PCI as a treatment for UA. REGULATE-PCI will enroll approximately 13,200 subjects at approximately 500 investigational sites worldwide. The number of subjects was determined so that the achievement of the primary efficacy endpoint of a 20% reduction in the occurrence of ischemic events versus bivalirudin would be statistically significant. The primary safety endpoint of REGULATE-PCI is non-inferiority to bivalirudin for the occurrence of major bleeding events. As an adaptive design trial, REGULATE-PCI permits a one-time increase in subject population, if necessary, at the time of 50% enrollment to maintain adequate trial size for statistical significance. Subjects will be randomized once it is determined that they will undergo PCI. REGULATE-PCI has limited exclusion criteria and is designed to be conducted using real life conditions and background therapies recommended by current standards of care. It will allow both femoral and radial approaches to vascular access as determined by the investigator. Investigators will also be allowed to use VCDs.
The primary endpoint of REGULATE-PCI is the efficacy of REG1 compared to bivalirudin for preventing a composite of death, nonfatal MI, nonfatal stroke and urgent target lesion revascularization, or TLR, through day three. The key secondary objective is to determine the safety of REG1 compared to bivalirudin as measured by major bleeding events through day three. Subjects will be followed for 30 days. Subjects will receive on an open-label basis either pegnivacogin at
12
the RADAR dose of 1mg/kg or bivalirudin at the labeled dose. The REG1 subjects will receive 0.5 mg/kg of anivamersen, representing approximately 80% reversal, at the end of the PCI procedure. Bivalirudin subjects will have their drug discontinued at the end of the PCI procedure or otherwise in accordance with standard of care. Investigators will have the discretion to increase the reversal to 100% for REG1 subjects if they determine that the subject has an increased risk of a major bleeding event or in the event of a bleeding complication after the removal of the arterial catheter sheath. Subjects will receive standard guidelines-based background therapy and provisional GPIIb/IIIa inhibitors in both groups. Arterial catheter sheaths will be removed two to ten minutes after the administration of anivamersen in the REG1 arm at the end of the PCI and two to four hours from the end of the PCI for the bivalirudin arm, per standard of care. Pharmacoeconomic endpoints including time in catheterization laboratory, time to sheath removal, time to ambulation and time to hospital discharge will be captured.
REGULATE-PCI includes three interim analyses. The first interim analysis will occur after enrollment of 1,000 subjects and is expected to occur by the beginning of the second quarter of 2014. At this time, the DSMB will evaluate general safety of the subjects enrolled to that point in the REG1 arm and, if the results are satisfactory, will extend trial enrollment to N-STEMI subjects. The second interim analysis will be another general safety assessment by the DSMB after 25% of the subjects are enrolled, which is expected to occur during the third quarter of 2014 with results of the DSMB analysis and deliberation coming6-8 weeks later. At that time, the DSMB will recommend the continuation or discontinuation of REGULATE-PCI based on its evaluation of the safety results in the REG1 arm. The final interim analysis will be performed after 50% of the subjects have been enrolled, which is expected to occur during the fall of 2014 with results of the DSMB analysis and deliberation being announced by the end of 2014. At that time, the DSMB will assess the general safety of REG1 and will perform an analysis of its efficacy. If the DSMB determines that REG1 is not safe or that it is futile to continue the trial because of a lack of efficacy, the trial will be terminated. On the other hand, if the DSMB determines REG1 is safe and its efficacy is overwhelmingly superior to bivalirudin, a positive outcome will be declared and the trial will be considered completed. Also at that time, since REGULATE-PCI is an adaptive design study, the DSMB may recommend an increase of total trial size based on a comparison of actual endpoint event rates to assumed rates to maintain the statistical significance of the trial results. Unless terminated early or extended as described above, we expect to complete REGULATE-PCI by the fourth quarter of 2015 and to have top-line data available by the end of 2015.
We have had extensive discussions with the FDA regarding our REGULATE-PCI trial, including the trial design, the trial protocol, inclusion-exclusion criteria, trial population, endpoint definition, duration of follow-up, statistical analysis plan and interim analyses. Because of the extent of these discussions, and based on the FDA’s view that we would obtain no additional benefit from pursuing a Special Protocol Assessment, or SPA, for the trial, we determined not to seek an SPA from the FDA for the REGULATE-PCI trial.
Other REG1 Indications
We believe that REG1 has potential for use in other PCI and interventional cardiovascular procedures, such as OHS, PCI as a treatment for STEMI, and TAVI, all of which are indications in which bivalirudin is not currently approved for use. These indications are described below.
OHS
We intend to develop REG1 for use in patients undergoing OHS as a replacement for the heparin – protamine combination. Based on information obtained from publicly available OHS procedure registries, we estimate that approximately 1.5 million OHS procedures were performed globally in 2013. If our proof-of-concept human trials are successful, we plan to proceed to pivotal trials for approval of this indication. The EMEA has agreed with our proposed clinical development program and we are currently in discussion with the FDA regarding this program. Because development of this application is at an early stage, we have not filed an IND with the FDA. We intend to file an IND for this application of REG1 after completion of our proof-of-concept human trial.
An extremely high level of anticoagulation is required in OHS to prevent life-threatening clotting, especially when a pump is used to maintain blood flow during the procedure. Heparin is used as the anticoagulant in most OHS procedures. Protamine must be used in OHS patients to reduce heparin’s anticoagulant effect even though protamine is unpredictable and may result in additional complications. OHS patients are subject to intense post-procedure monitoring for major bleeding events. We believe a drug that achieves an optimal level of anticoagulation that can be precisely and predictably controlled or reversed at the end of the procedure and which does not have the significant side effects associated with heparin and protamine would result in improved outcomes for patients undergoing OHS. As a result, REG1 has the potential to become the standard of care anticoagulant for use in OHS.
13
In 2008, we commenced an exploratory Phase 2a feasibility trial of REG1 in patients undergoing off-pump coronary arterial bypass grafting, or CABG, which was intended to enroll ten subjects. The trial was stopped due to clotting in one of three bypass grafts placed in the first study subject so we could assess the cause of clotting, reassess and optimize the protocol, and complete further preclinical development and delineation of optimum methods of use of REG1 in CABG. As a result of our assessments and additional work, future testing of REG1 in CABG patients will be done as a part of the OHS program under a significantly modified protocol.
14
STEMI
We intend to develop REG1 for use in patients undergoing PCI as a treatment for STEMI. Based on information obtained from the GRACE Registry, we believe that approximately 750,000 PCI procedures were performed worldwide in 2013 for the treatment of STEMI. STEMI patients are more at risk if an ischemic event occurs than other patients undergoing PCI treatment because they generally present with significant clotting. As a result, a higher level of anticoagulation is necessary to treat these patients effectively. For these patients, the risk of ischemic events significantly outweighs the risk of major bleeding events. We believe a drug that can safely achieve a high level of anticoagulation would result in improved outcomes for STEMI patients undergoing PCI. As a result, REG1 has the potential to become the standard of care anticoagulant for use in this patient group. Because development of this application is at an early stage, we have not filed an IND with the FDA for this application of REG1.
TAVI
We intend to develop REG1 for use in patients undergoing TAVI. TAVI is an emerging interventional cardiovascular procedure in which myocardial valves are repaired or replaced through a catheter rather than OHS. While TAVI has the potential to be a safer alternative to OHS, TAVI presents many of the same risks applicable to PCI procedures as well as those applicable to OHS. The risk of stroke in TAVI is significantly greater than in OHS and therefore TAVI is not used if a patient can tolerate OHS. Also, the catheters used in TAVI procedures are approximately four to five times larger in diameter than those used in PCI, increasing the potential for arterial damage during the procedure. Because of these factors, in TAVI procedures heparin is dosed in similar amounts to OHS. However, because of the increased stroke risk, protamine is usually not used at the end of the procedure to reverse heparin’s anticoagulant effect, thereby exposing the patient to a high risk of a major bleeding event. In addition, because the stroke risk persists for up to two weeks, a TAVI patient may require sub-acute anticoagulant treatment to prevent a possible stroke. We believe a drug that achieves an optimal level of anticoagulation and that can be precisely and predictably controlled or reversed would result in improved outcomes for patients undergoing TAVI and could dramatically expand the use of TAVI. As a result, REG1 has the potential to become the standard of care anticoagulant for use in TAVI. In addition, REG2 has potential application for use in TAVI patients due to its extended release formulation. Because development of this application is at an early stage, we have not filed an IND with the FDA for this application of REG1.
Our Proprietary Technology Platform
We are pioneering the discovery and development of two-component drug systems consisting of a therapeutic aptamer and its specific active control agent. Our aptamers are single strands of nucleic acids, or oligonucleotides, that are chemically synthesized. Unlike other oligonucleotides, which are designed to control gene expression, an aptamer has a unique geometric shape that binds specifically and tightly to a target protein molecule, leading to inhibition of the target’s activity. Aptamers have been discovered that interact with essentially every class of therapeutic protein target. An aptamer’s pharmacologic activity can be controlled by interaction with a complementary oglionucleotide, which we term a specific active control agent. When the specific active control agent binds to the aptamer, it changes the aptamer’s shape thereby eliminating its therapeutic activity. Our founder co-invented our aptamer control technology to address the unmet medical need for a controllable antithrombotic therapy that could be rapidly modulated or eliminated.
We believe we have the leading position in the development of actively controllable aptamer therapeutic systems, including expertise in aptamer discovery, active control agent design, oligonucleotide chemistry, strong aptamer and active control agent intellectual property rights, including exclusive rights to key patents relating to control of aptamer activity in the body, and disciplined aptamer and active control agent drug discovery and development processes. Central to achieving our goals is the know-how that we have accumulated in aptamer discovery and control agent design and how the specific chemistries we utilize behave in the clinical setting.
We believe that the following aspects of our technology platform give us a competitive advantage:
| | • | | a proven discovery team including the co-inventor of active aptamer control agent technology; |
| | • | | an exclusive license to the patents covering active aptamer control agent technology, which include broad claims directed toward nucleic acid ligands and methods to control aptamer activity by administration of active control agents; |
| | • | | a commercial license to the combinatorial chemistry technology platform, or SELEX™, for aptamer discovery and development; |
| | • | | the know-how to identify, isolate and optimize therapeutic aptamers and their active control agents, which has led to the discovery of multiple product candidates; |
| | • | | a proven pharmaceutical development team with experience in the development of efficient and economical manufacturing processes for aptamers and active control agents; and |
| | • | | an extensive clinical database of systemic aptamer use, which gives us unparalleled expertise in the discovery of new two-component aptamer-based product candidates. |
15
The disciplined approach we take to the discovery and development of controllable aptamer therapeutic systems is as important as the assets assembled to execute on our plans. Every aspect of our development process, including the evaluation of a therapeutic opportunity, the identification of a specific targets, the discovery of a therapeutic aptamer and the optimization of active control agents, is designed to increase our probability of success. Our disciplined approach is summarized in the following four steps:
Step 1–Evaluation of Opportunities
The initiation of our discovery and development efforts is based on rigorous scientific and business criteria, including:
| | • | | clear scientific evidence of the role that a specific target plays in a disease; |
| | • | | scientific evidence to support the belief that the use of an aptamer to inhibit the target protein will provide for a novel mechanism of action compared to existing therapies; |
| | • | | clinical evidence that the ability to control the aptamer’s activity in the treatment of the disease will address an unmet medical need or potentially provide for a superior treatment over existing therapies; and |
| | • | | existence of a significant commercial opportunity. |
Our evaluation criteria potentially apply to a broad range of targets, allowing us to focus on product candidates that we believe have a high probability of clinical and commercial success.
Once we decide to pursue a specific treatment, we use a proprietary methodology to identify and optimize lead aptamer and active control agent pairs for development.
Step 2–Aptamer Identification
We identify aptamers using the SELEX™ process, licensed to us by Archemix Corporation, or Archemix. The SELEX™ process enables us to incubate a large number of nucleic acid sequences with a specific target to more efficiently identify aptamers with the necessary structural and binding properties. Additionally, we use a proprietary process to establish the optimal conditions for performing the SELEX™ process to each target, which we believe provides for a high probability of success in identifying potent aptamers.
Step 3–Control Agent Identification
Once we have identified a potential aptamer candidate, we design potential active control agents based upon rules of Watson-Crick base pairing, the fundamental chemical principles that describe the binding of complementary strands in DNA and RNA molecules to each other. We have developed significant understanding and know-how of the interaction of active control agents with aptamers, which we believe enables us to design and screen potential active control agents with a high probability of success.
Step 4–Aptamer-Control Agent Optimization
The final step in our discovery process is the optimization of the aptamer and active control agent for their pharmacokinetic and pharmacodynamic properties and ease of manufacture. We use our extensive knowledge of oligonucleotide chemistry and clinical performance of controllable aptamer therapeutic systems to:
| | • | | optimize the aptamer size and use of stabilization chemistries within the aptamer to provide for a potent, well-tolerated aptamer with a half-life matched to its intended clinical use that can be manufactured cost effectively; |
| | • | | identify the appropriate carrier for the aptamer, such as PEG, to provide for desirable pharmacokinetic and distribution properties while maintaining the desired potency; and |
| | • | | optimize the active control agent to achieve a suitable half-life, to provide for potent modulation or elimination of the aptamer’s therapeutic activity and to permit cost effective manufacture. |
Aptamer technology has evolved significantly since first generation aptamers were described in 1990. Many of the early aptamers were chemically and metabolically unstable, were potentially immunologically active and were prohibitively expensive to produce. Our product candidates consist of third and fourth generation aptamers that have significantly improved pharmaceutic and pharmacokinetic stability and have not shown immunological activity in a wide range of tests. Due to advances in synthetic techniques, these aptamers can be economically produced using readily available synthesis technology.
16
Our Other Product Candidates
Using our proprietary technology platform, we have developed a pipeline of other product candidates which are described below.
REG3
REG3 consists of a novel, first-in-class GPVI inhibitor and its specific active control agent. Increased levels of GPVI are present in a wide range of platelet-mediated diseases including ACS, stroke, rheumatoid arthritis and diabetic vasculopathy. In recent years, GPVI has emerged as a promising anti-platelet target. REG3 uses aptamer technology to bind to GPVI thereby inhibiting its function. We believe that REG3’s novel mechanism of action will enable it to successfully inhibit GPVI and provide an effective treatment for these conditions. REG3 is being developed as an injectable for sub-acute GPVI inhibition. REG3’s specific active control component is designed to permit modulation or elimination of the GPVI inhibition, if necessary, to optimize dosing and minimize unwanted side effects that might result from GPVI inhibition. We plan to initiate a Phase 1 study of REG3 in the future depending on availability of financial resources.
REG2
The active components of REG2 are identical to that of REG1 except that the pegnivacogin is formulated for subcutaneous depot injection for extended release. REG2 is intended to provide a controllable level of anticoagulation for up to two weeks for sub-acute uses, especially where a patient may be unable to swallow an oral anticoagulant. We are developing REG2 for use in venous thrombosis indications such as VTE prophylaxis in patients undergoing abdominal surgery. REG2 could also be used as a sub-acute anticoagulant treatment for TAVI patients or for bridging patients who are unable to take oral anticoagulants for a period of time before or after a procedure. In REG2, anivamersen is intended for use as an active control agent if needed. We have completed a single escalating dose Phase 1 clinical trial of REG2 and plan to conduct additional clinical testing in the future under the auspices of the newly opened REG2 IND, depending on financial resources.
Antidote to Oral FXa Inhibitors
Oral FXa inhibitors, including rivaroxaban, apixaban and edoxaban, have the potential to replace coumadin in a number of indications including prophylaxis and treatment of deep vein thrombosis, treatment of pulmonary embolism, and prevention of stroke in patients with atrial fibrillation. However, their use is limited by risk of bleeding and lack of antidotes to reverse the drugs activity in the event of bleeding or if discontinuation of therapy is required to conduct medical procedures. Based on this tremendous unmet medical need for specific antidotes to the respective oral FXa inhibitors, we are in the process of discovering high affinity and specificity aptamers to oral FXa agents for use as antidotes. Among the compelling advantages of this approach are: 1) high target specificity and lack of interaction with endogenous blood proteins, 2) ability to design the characteristics of the antidote to match the specific pharmacokinetics and clinical use of a particular FXa inhibitor. Together we believe these advantages will lead to antidotes for which the dosing is determined solely by the concentration of the FXa inhibitor in the blood.
Our Strategy
Our strategy is to discover and develop novel, first-in-class controllable antithrombotic drug systems for use in acute and sub-acute cardiovascular and other indications that we believe are underserved by existing therapies. The key elements of our strategy are:
| | • | | Complete the development and obtain regulatory approval for the first indication of REG1 in the United States, Europe and other countries. We commenced REGULATE-PCI in September 2013. If successfully completed, we intend to seek approval for the first indication of REG1 in the United States, Europe and other countries. We have assembled an experienced team with a successful track record of managing global clinical development activities, obtaining regulatory approvals worldwide for new drugs and maintaining compliance with international regulations governing the development, sales, marketing and distribution of pharmaceutical products. We believe we have the expertise and capacity to manage a global development program without local partners. |
| | • | | Commercialize REG1 in its first indication in the United States, Europe and other countries alone or with a strategic partner. Because REG1 is a hospital-based acute care product, we believe that it is feasible for us to commercialize the first REG1 indication in the United States with a relatively small specialty sales force calling on a targeted group of hospital-based interventional cardiologists. If REG1 is approved, we intend to commercialize REG1 in the United States by building a commercial infrastructure or by utilizing contract reimbursement specialists, sales people and medical education specialists. We may seek to augment our commercial efforts by entering into a collaboration with a pharmaceutical company, if such a collaboration is available on attractive terms. Outside of the United States, we likely will seek to commercialize REG1 through distribution or other collaboration arrangements. |
17
| | • | | Develop and commercialize other applications for REG1. We intend to develop REG1 for use in additional cardiovascular procedures, such as OHS, PCI as a treatment for STEMI, and TAVI. We believe REG1’s two-component anticoagulant system addresses unmet needs in these markets. |
| | • | | Advance the other product candidates in our pipeline. We intend to develop product candidates in our pipeline beyond REG1, including REG3, REG2 and the specific antidotes to oral FXa inhibitors. |
| | • | | Seek joint development agreements for our other product candidates or new treatment targets. We intend to seek partners to assist us in the development of REG3, REG2, and the specific antidotes to oral FXa inhibitors as well as any new product candidates we discover. We also intend to seek collaborations with other life sciences companies that may desire to use our proprietary technology to discover and develop novel treatments in their areas of clinical interest. |
Third-Party Suppliers and Manufacturers
We do not have a manufacturing infrastructure and do not intend to develop one. We have agreements with our third-party manufacturers, or CMOs, to supply bulk drug substances for our product candidates and with third parties to formulate, package and distribute our product candidates. Our employees include professionals with expertise in pharmaceutical manufacturing development who oversee the manufacture and distribution of our product candidates by third-party companies. We may not currently have sufficient amounts of REG1 on hand to complete enrollment in the REGULATE-PCI trial, however the Company is currently contracted to produce sufficient amounts of REG1 in early 2014. The Company may not have enough bivalirudin to complete REGULATE-PCI and may need to secure additional quantities to complete the trial. All of the drug substances used in our product candidates are manufactured by single suppliers. While we have not experienced any supply disruptions, the number of oligonucleotide manufacturers is limited. In the event it is necessary or advisable to acquire supplies from an alternative supplier, we might not be able to obtain them on commercially reasonable terms, if at all. It could also require significant time and expense to redesign our manufacturing processes to work with another company. Formulation and distribution of our finished product candidates are also conducted by a single supplier but we believe that alternative sources for these services are readily available on commercially reasonable terms.
In July 2006, we entered into a supply and service agreement with Agilent Technologies, Inc., or Agilent, which was amended in July 2011, for the manufacture of pegnivacogin and anivamersen bulk drug substance for use in the clinical development of REG1. Drug substances are manufactured pursuant to a good manufacturing practices quality agreement entered into in May 2010. Manufacture of bulk drug substance lots is on a purchase order basis, with no minimum purchase obligation. The supply and service agreement has an indefinite term and expires when all services thereunder have been completed. Agilent may terminate the agreement with or without cause upon 180 days prior written notice to us. We may terminate the agreement if Agilent is unable to timely meet our purchase orders or Agilent’s quoted prices are more than 110% of those available from a third party. Either party may terminate the agreement upon written notice if the other party is in breach of a material provision of the agreement and such breach is not cured within 60 days after receipt of written notice of the breach.
In December 2006, we entered into a license, manufacturing and supply agreement with Nektar Therapeutics, or Nektar, for the supply of PEG used in the manufacture of pegnivacogin. We must provide Nektar with a rolling forecast of our anticipated requirements for the succeeding six quarters, with respect to pre-commercial supply, or eight quarters, with respect to commercial supply, with a required lead time to commit to and guarantee availability of the reagent at an agreed upon pricing structure, which is subject to revision on an annual basis. In addition, we have agreed to pay Nektar specific development and commercial milestones and a specific royalty on product sales. See “License Agreements—Nektar License and Supply Agreement.”
In March 2012, we entered into a clinical supply agreement with Althea Technologies, Inc., for the formulation and packaging of pegnivacogin and anivamersen for use in our clinical trials. The formulation and packaging is conducted pursuant to a quality agreement entered into in January 2012. Formulation and packaging services are provided on a purchase order basis, with no minimum purchase obligation. The clinical supply agreement has an indefinite term and expires when all services there under have been completed. The agreement may be terminated by us with or without cause on 30 days written notice, immediately by either party upon the occurrence of certain bankruptcy events related to the other party, upon 30 days written notice by either party if the parties fail to agree on a change in the scope of the agreement, or upon written notice by one party in the event of a material breach by the other party that is not cured within 30 days or such additional time that may be reasonably necessary to cure such breach if commenced within such 30-day period and diligently pursued to completion.
18
In November 2007, we entered into a development and supply agreement for REG3 bulk drug substance with Avecia Biotechnology, Inc., which expired in November 2009. We anticipate entering into extension of this agreement or a new agreement to manufacture REG3 for our planned Phase I study. Manufacture of bulk drug substance lots is on a purchase order basis, with no minimum purchase obligation.
Except for the agreement with Nektar, all of the agreements described above relate solely to the clinical development of our product candidates. In the event that we obtain marketing approval for a product candidate, we will be required to enter into separate agreements for the commercial production and distribution and manufacturing of that product.
Intellectual Property
We believe that we have a strong patent portfolio and substantial know-how relating to REG1, our other product candidates and our proprietary technology platform. Our patent portfolio, described more fully below, includes claims directed to aptamers, pharmaceutical compositions containing aptamers, aptamer formulations, methods for altering the affinity of an aptamer for a target molecule and for treating disease and disorders, as well as methods of manufacturing aptamers and aptamer formulations. As of December 31, 2013, we are the owner of record of five issued or allowed U.S. patents and six issued or allowed non-U.S. patents, as well as the licensee of at least ten issued or allowed U.S. patents and at least 11 issued or allowed non-U.S. patents. We are actively pursuing an additional nine U.S. patent applications, all of which are non-provisional applications, three international patent application and 50 non-U.S. patent applications in twelve jurisdictions as the owner of record, in addition to at least two U.S. patent applications and 13 non-U.S. patent applications under license.
We strive to protect the proprietary technology that we believe is important to our business, including our proprietary technology platform, our product candidates and our processes. We seek patent protection in the United States and internationally for our products, their methods of use and processes of manufacture, and any other technology to which we have rights, where available and when appropriate. We also rely on trade secrets that may be important to the development of our business.
Our success will depend on the ability to obtain and maintain patent and other proprietary rights in commercially important technology, inventions and know-how related to our business, the validity and enforceability of our patents, the continued confidentiality of our trade secrets as well as our ability to operate without infringing the valid and enforceable patents and proprietary rights of third parties. We also rely on continuing technological innovation and in-licensing opportunities to develop and maintain our proprietary position.
We cannot be sure that patents will be granted with respect to any of our pending patent applications or with respect to any patent applications we may own or license in the future, nor can we be sure that any of our existing patents or any patents we may own or license in the future will be useful in protecting our technology. For this and more comprehensive risks related to our intellectual property, please see “Risk Factors—Risks Relating to Our Intellectual Property.”
The term of individual patents depends upon the legal term of the patents in the countries in which they are obtained. In most countries in which we file, the patent term is 20 years from the date of filing the non-provisional priority application. In the United States, a patent’s term may be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the U.S. Patent and Trademark Office, or PTO, in granting a patent, or may be shortened if a patent is terminally disclaimed over an earlier-filed patent.
The term of a U.S. patent that covers an FDA-approved drug may also be eligible for patent term extension, which permits patent term restoration as compensation for the patent term lost during the FDA regulatory review process. The Hatch-Waxman Amendments permit a patent term extension of up to five years beyond the expiration of the patent. The length of the patent term extension is related to the length of time the drug is under regulatory review. A patent term extension cannot extend the remaining term of a patent beyond a total of 14 years from the date of product approval and only one patent applicable to an approved drug may be extended. Similar provisions are available in Europe and other foreign jurisdictions to extend the term of a patent that covers an approved drug. When possible, depending upon the length of clinical trials and other factors involved in the filing of a new drug application, or NDA, we expect to apply for patent term extensions for patents covering our product candidates and their methods of use.
19
The patent portfolios for our proprietary technology platform and our three most advanced product candidates as of December 31, 2013 are summarized below.
Our Proprietary Technology Platform
Our active aptamer control agent technology was co-discovered by Dr. Christopher P. Rusconi, our chief scientific officer, in his prior role at Duke University, or Duke. Those discoveries are disclosed and claimed in a patent portfolio owned by Duke, or the Duke portfolio, and exclusively licensed to us, on terms described more fully below. The Duke portfolio includes broad claims directed to aptamers and pharmaceutical compositions containing aptamers, as well as methods for modulating coagulation. By stage and geographic focus, the Duke portfolio includes issued U.S. patents, such as U.S. Patent Nos. 7,312,325; 7,776,837; 7,300,922; 8,143,233 and 8,283,330, pending U.S. patent applications, such as U.S. Patent Application Nos. 11/789,992 and 13/428,352, and corresponding issued and foreign national or regional counterpart patents or applications. The issued and pending applications include broad subject matter directed to modulation of aptamer function in the body by a number of different methods. The most significant issued patent within the Duke portfolio, U.S. Patent No. 7,300,922, is expected to expire in 2023, as a result of patent term adjustments. If issued, the last to expire pending patent application within this portfolio would expire in 2022, excluding any patent term adjustments or extensions.
Archemix owns or controls a patent portfolio, or the Archemix portfolio, and related know-how directed to the proprietary SELEX™ method for identifying nucleic acid ligands and directed broadly to nucleic acid ligand compositions, licensed to us under terms described more fully below. By stage and geographic focus, the portfolio includes issued U.S. patents and corresponding foreign national or regional counterpart patents or applications. The most significant issued patent within the Archemix portfolio, U.S. Patent No. 6,011,020 relating to pegylated aptamers, is expected to expire in January 2017. We have a license from Archemix under which we possess rights in certain patent rights and know-how owned or controlled by Archemix, as described more fully below, including an exclusive commercial license and a non-exclusive research license to the SELEX™ processes for identifying aptamers for use in products that contain an aptamer and an active control agent for use in the treatment of certain coagulation-related diseases or conditions in humans.
REG1
The patent portfolio for REG1 includes wholly owned patents and patent applications directed to aptamers, pharmaceutical compositions containing aptamers, aptamer formulations, methods for modulating coagulation and treating coagulation-related diseases utilizing aptamers, and methods of manufacturing aptamers. It includes issued U.S. Patent Nos. 7,304,041; 7,723,315, 8,389,489 and 7,531,524, pending U.S. patent applications, and corresponding foreign national or regional counterpart patents or applications. The most significant issued patents within this portfolio are the issued U.S. patents referenced above, all of which are expected to expire in 2025, excluding any additional term for patent term extensions. If issued, the last to expire pending patent application within this portfolio would expire in 2033, excluding any additional term for patent term adjustments or patent term extensions.
REG3
The patent portfolio for REG3 includes wholly owned patents and patent applications. It includes claims directed to aptamers, pharmaceutical compositions containing aptamers, aptamer formulations, methods of treating a platelet-mediated disorder and methods of modulating platelet function. It includes issued U.S. Patent No. 8,318,923, pending U.S. patent applications, and corresponding foreign national or regional counterpart applications. The last to expire issued patent is expected to expire in 2030, excluding any patent term adjustments or extensions. If issued, the last to expire pending patent applications are expected to expire in 2033, excluding any patent term adjustments or extensions.
REG2
The REG2 patent portfolio includes the same patents and patent applications discussed above with respect to REG1.
Antidotes to Oral FXa Inhibitors
The patent portfolio for this program includes wholly owned provisional patent applications. It includes individual patent applications for specific oral FXa inhibitors with claims directed to aptamers, pharmaceutical compositions containing aptamers, aptamer formulations, methods of discovering such aptamers, and methods of using aptamer antidotes to reverse the pharmacologic activity of the targeted oral FXa inhibitor. It also includes a patent application with broad claims directed to aptamers, pharmaceutical compositions containing aptamers, and methods of discovering aptamers that could be used as antidotes for a wide range of individual pharmaceuticals and classes of pharmaceutical agents.
20
Trade Secrets
In addition to patents, we rely on trade secrets and know-how to develop and maintain our competitive position. For example, significant aspects of our proprietary technology platform are based on unpatented trade secrets and know-how. Trade secrets and know-how can be difficult to protect. We seek to protect our proprietary technology and processes, in part, by confidentiality agreements and invention assignment agreements with our employees, consultants, scientific advisors, contractors and commercial partners. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant us ownership of technologies that are developed through a relationship with a third party. We also seek to preserve the integrity and confidentiality of our data and trade secrets by maintaining physical security of our premises and physical and electronic security of our information technology systems. While we have confidence in these individuals, organizations and systems, agreements or security measures may be breached, and we may not have adequate remedies for any breach. In addition, our trade secrets may otherwise become known or be independently discovered by competitors. To the extent that our contractors use intellectual property owned by others in their work for us, disputes may arise as to the rights in related or resulting know-how and inventions.
Trademarks
We seek trademark protection in the United States and outside of the United States where available and when appropriate. We have registered trademark rights in the TAKING SCIENCE TO HEART mark in the United States and Israel, to the WE TAKE SCIENCE TO HEART mark in the United States, to the REGADO BIOSCIENCES mark in the United States, Canada and Israel, and to the REGADO mark in the United States and Israel. We have registered trademark rights in REGADO BIOSCIENCES THE ACTIVE CONTROL AGENT CONTROL COMPANY in Israel. We use these registered marks in connection with our pharmaceutical research and development as well as our product candidates. We have submitted trademark applications in the United States or REVOLIXYS, SPECIFIX and ENACTIX. We use these marks in connection with our pharmaceutical research and development, as well as our product candidates.
License Agreements
Duke License Agreement
We entered into a license agreement with Duke in November 2004, which was amended in July 2005. Under the amended agreement, we obtained an exclusive worldwide license to make, have made, use, lease, offer for sale, sell, distribute and export license products, including the right to sublicense, under certain Duke patent rights relating to aptamers with an active control agent and to genus claims to anti-FIXa aptamers and a non-exclusive license to related Duke know-how. Duke retains the right to practice the patent rights licensed under the agreement for its educational, research, publication and clinical purposes, and to provide materials covered by such patent rights to certain third parties for non-commercial purposes.
We issued 11,452 shares of our common stock to Duke as consideration for the license agreement. In addition, we may be required to pay to Duke an aggregate of $1.75 million per product upon the achievement of specified development and regulatory approval milestones. Such milestones include $500,000 payable upon the commencement of the REGULATE-PCI trial, which occurred in September 2013. Accordingly, we made a milestone payment of $500,000 to Duke, which was charged to research and development expense in the accompanying consolidated statement of comprehensive loss for the year ended December 31, 2013. All of our product candidates are subject to the terms of the Duke license.
We also are required to pay Duke royalties based on a percentage of net sales for products and services sold by us or our sublicensees that utilize, incorporate or practice any of the licensed patent rights. The percentage royalty rate is in the low single digits. These royalties may be reduced if we are required to obtain a third-party license to practice the Duke patent rights.
The agreement requires us to use reasonable best efforts to commercialize products and services based on the licensed technology and to continue active and diligent marketing efforts of any commercialized products and services for the life of the agreement.
We may terminate the agreement at any time upon three months written notice. Either party may terminate the agreement upon a judgment or conviction holding that the other party has committed fraud, willful misconduct or illegal conduct with respect to the subject matter of the agreement or upon a material breach of the agreement by the other party, subject to the breaching party’s limited right to avoid termination by curing the material breach in certain circumstances. Absent any early termination, the term of the agreement continues until the last of the licensed patent rights has expired or become abandoned. The longest lived patent rights licensed to us under the agreement are expected to expire in 2023 as a result of patent term adjustments.
21
Archemix License Agreement
In October 2003, we entered into a license agreement with Archemix pursuant to which we obtained an exclusive worldwide license to develop, manufacture, use, sell, offer for sale, have sold and import licensed products under certain patent rights and know-how owned or controlled by Archemix for products that contain an aptamer and active control agent for use in the treatment of diseases or conditions in humans caused or characterized by factors involved in, and the modulation of, fibrin deposition, platelet adhesion and/or platelet aggregation. The agreement excludes conditions or diseases of the eye, the orbit and its contents, the eyelids or the lachrymal system, as well as the diagnosis of any diseases or conditions and any uses relating to the handling or storage of blood or blood products.
Under the agreement, we also obtained a non-exclusive research license to the SELEX™ process for identifying and developing aptamers for use solely as part of the licensed products described above.
The agreement provides for a non-exclusive license back to Archemix to use and sublicense our improvements related to the SELEX™ process, as well as a nonexclusive license to use, for internal research solely within Archemix, other improvements we make to the intellectual property licensed from Archemix.
We issued 6,736 shares of our common stock to Archemix as consideration for the license agreement. In addition, we may be required to pay to Archemix an aggregate of $5.5 million upon the achievement of specified development and regulatory approval milestones. Such milestones include $1.0 million payable upon the commencement of the REGULATE-PCI trial, which occurred in September 2013. Accordingly, $1.0 million is included in accrued expenses and in research and development expense in the accompanying consolidated balance sheet and in the consolidated statement of comprehensive loss as of and for the year ended December 31, 2013, respectively. All of our product candidates are subject to the terms of the Archemix license.
We also are required to pay Archemix royalties based on the net sales of licensed products by us and our affiliates. The royalty rate is in the mid-single digits. The longest lived patent rights licensed to us under the agreement are expected to expire in 2017.
To the extent that any intellectual property that we license from Archemix, other than intellectual property relating to the SELEX™ process, is held by a third party licensor of Archemix, we may be responsible for paying incremental costs that Archemix is required to pay to such third-party licensor on account of the license Archemix has granted to us.
The agreement requires us to use commercially reasonable efforts to proceed with the development, manufacture and sale of licensed products. We may enter into collaboration agreements with third parties with respect to part or all of the development or commercialization of one or more licensed products, subject to Archemix’s right of first refusal with respect to any such proposed collaboration agreement in certain limited circumstances. We also have the right to grant sublicenses to the intellectual property licensed from Archemix subject to the obligation to pay Archemix royalties based on amounts received pursuant to any such sublicenses.
We may terminate the agreement at any time upon 60 days’ written notice. Either party may terminate the agreement if the other party commits a material breach of the agreement and fails to cure the breach within 60 days after written notice. Absent any early termination, the term of the agreement continues until the last valid claim within the licensed patent rights expires. The agreement provides that upon expiration, we will have a paid up, exclusive, worldwide licensed under the know-how rights licensed from Archemix in the field licensed to us under the agreement.
Nektar License and Supply Agreement
In December 2006, we entered into a license, manufacturing and supply agreement with Nektar pursuant to which we obtained an exclusive, worldwide license to certain Nektar-controlled patent rights and know-how pertaining to the PEG and other pegylation technology used for pegnivacogin, solely to make, have made, import, use, offer for sale, sell and otherwise exploit pegnivacogin for the treatment, prevention or diagnosis of human diseases or conditions, other than Hemophilia A. Nektar retains the right to practice the licensed patent rights and know-how for research and development of products that it is developing itself or with others and to perform obligations to third parties set forth in agreements existing as of the effective date of our agreement.
We have the right to grant sublicenses to third parties, provided that any sublicensee of the Nektar-controlled patent rights and know-how at the time is not a competitor of Nektar.
We may be required to pay to Nektar an aggregate of $7.75 million upon the achievement of specified development and regulatory approval milestones. Since December 2006, we made payments under this license agreement totaling $1.25 million.
22
We also are required to pay Nektar royalties based on a percentage of net sales of our REG1 product by us or our sublicensees. The percentage royalty rates are in the mid-single digits, and are subject to reduction with respect to sales in a country if none of the patents and patent applications that we license under the agreement cover REG1 or the PEG in such country. These royalties also may be reduced if we are required to pay royalties to a third party due to infringement relating to the PEG. The duration of our royalty obligation expires on a country-by-country basis on the later of ten years after first commercial sale of REG1 in a country and the expiration of the last-to-expire of the licensed patent rights in such country.
If we enter into a transaction where we grant a third party any rights to distribute, promote, market or sell REG1, or we sublicense any of the rights licensed to us under the agreement, then we are required to pay Nektar a specified percentage of the first payment we receive in such transaction, up to a specified maximum payment to Nektar.
We are required to purchase 100% of our and our sublicensees’ requirements of the PEG for the REG1 product from Nektar, at a purchase price defined in the agreement, which purchase price is subject to annual increases based on inflation index changes.
We may terminate the agreement at any time upon 60 days’ written notice, subject to our obligation to pay an early termination fee to Nektar. Our right to terminate the agreement early is further subject to our obligation to continue to make certain minimum purchases from Nektar of the PEG and to continue to pay Nektar royalties for net sales of REG1 that incorporates such PEG purchased from Nektar. Either party may terminate the agreement if the other party fails to comply with the material terms of the agreement and does not correct such failure within 30 days for failures to make timely payment or 90 days for other failures. Nektar also may terminate the agreement if we or a sublicensee challenges the validity, scope or enforceability of or otherwise opposes any of the patent rights licensed to us under the agreement. Absent any early termination, the term of the agreement continues until all royalty obligations under the agreement expire.
NovaMedica Agreements
In connection with our Series E Preferred Stock financing, in December 2012, we entered into a Technology Transfer Agreement, or the Tech Transfer Agreement, with Domain Russia Investments Limited, or DRI, an affiliate of Domain Partners VIII, L.P. Domain Partners VIII, L.P. and Domain Partners VI, L.P., a significant stockholder of our company, are both managed by Domain Associates, L.L.C. Pursuant to the Tech Transfer Agreement, in exchange for a nominal payment, we assigned to DRI certain patents and patents applications in Armenia, Azerbaijan, Belarus, Georgia, Kazakhstan, Kyrgyzstan, Moldova, Russia, Tajikistan, Turkmenistan, Ukraine and Uzbekistan, or the Covered Territory, and granted to DRI an exclusive, fully paid-up, royalty-free, irrevocable and assignable license under our non-patent intellectual property to develop and commercialize REG1 and our other product candidates in the Covered Territory. Immediately thereafter, we, together with DRI and NovaMedica, LLC, or NovaMedica, executed an Assignment and Assumption Agreement, pursuant to which all of DRI’s rights and obligations under the Tech Transfer Agreement were transferred to NovaMedica. DRI also has a right of first negotiation if we desire to partner with a third party to develop or commercialize future product candidates in the Covered Territory, which was assigned to NovaMedica. We agreed to take all action required to register or record the patent transfers to DRI in each country in the Covered Territory and to ensure the assignment of DRI’s rights under the Tech Transfer Agreement to NovaMedica. NovaMedica is jointly owned by Rusnano Medinvest LLC, or Rusnano Medinvest, and DRI. RMI Investments, S.á.r.l, or RMII, a significant stockholder of ours, is a wholly-owned subsidiary of Rusnano Medinvest. In connection with the second tranche closing of our Series E Preferred Stock financing, we agreed to file certain patent transfer applications and to take certain other related actions which have been completed. Under the terms of the Tech Transfer Agreement, upon request we have agreed to provide certain development support to NovaMedica and to use commercially reasonable efforts to assist NovaMedica to establish a manufacturing relationship with our CMOs. We also have agreed to provide NovaMedica with certain manufacturing know-how and support, including making our manufacturing employees available to provide scientific and technical explanations, advice and on-site support that may be reasonably requested by NovaMedica. NovaMedica is required to reimburse us for any out-of-pocket expenses incurred by us in providing this assistance, including travel-related expenses. We estimate that the aggregate out-of-pocket expense of providing such services will be approximately $250,000. In addition, prior to the first commercial sale of a product candidate, we have agreed to sell to NovaMedica sufficient quantities of each product candidate and related compounds to enable NovaMedica to conduct clinical trials of such product candidate in the Covered Territory at cost plus a mark-up in the low double digits so long as any sale does not reasonably interfere with our own development and commercialization activities.
Concurrently with the signing of the Tech Transfer Agreement, we also entered into a letter agreement with DRI pursuant to which we are obligated to pay DRI a make-whole payment up to a maximum amount of $1.2 million in the event that an independent appraiser’s valuation of certain patent applications assigned to DRI under the Tech Transfer Agreement is less than $1.2 million. The letter agreement provides that such payment will be refunded to us if DRI receives certain capital contribution credits with respect to such patent applications in connection with its investment in NovaMedica. DRI has advised us that the independent appraiser valued the assigned patent applications at more than $1.2 million. As a result, DRI is not entitled to any make-whole payment under the terms of the letter agreement. In addition, we have agreed to indemnify DRI against any claims brought by NovaMedica arising out of or resulting from any breach of specified
23
representations and warranties which we made in the Tech Transfer Agreement up to a maximum amount of $1.2 million, less any payments made to DRI in connection with the valuation of the assigned intellectual property. Our indemnification obligation will expire two years following the first commercial sale of REG1 or our other product candidates in the Covered Territory or six years after the date of the letter agreement if no such commercial sales have occurred.
The Tech Transfer Agreement also provides that we will enter into a Clinical Development and Collaboration Agreement, a Supply Agreement and certain related agreements with NovaMedica to implement the terms of the Tech Transfer Agreement. In connection with the second tranche of the initial closing of our Series E Preferred Stock financing, we agreed to use commercially reasonably efforts to negotiate, execute and deliver the Clinical Development and Collaboration Agreement on or before May 31, 2013. The Tech Transfer Agreement provides that the Supply Agreement will cover the commercial supply of product candidates and related drug compounds to NovaMedica at cost plus a mark-up in the low double digits.
In accordance with the terms of the Tech Transfer Agreement, in May 2013 we entered into a Clinical Development and Collaboration Agreement, or the Collaboration Agreement, with NovaMedica pursuant to which we agreed to assist NovaMedica in the development and commercialization of our product candidates in the Covered Territory. The Collaboration Agreement requires the formation of several committees consisting of our representatives and NovaMedica representatives to oversee the general development, day-to-day development work and commercialization of our product candidates for the intended field of use in the Covered Territory. All decisions of these committees must be made by unanimous vote, subject to a dispute resolution process. Under the terms of the Collaboration Agreement, the joint committees will determine a development plan for REG1 for its initial indication and any additional significant commercial indications for REG1, as well as for additional product candidates. NovaMedica will have sole responsibility for the costs and expenses of obtaining regulatory approval for our product candidates and for commercializing any approved products in the Covered Territory and will have the right to conduct its own clinical studies in the Covered Territory at its sole expense. NovaMedica also has the right to file applications for approval of our product candidates in the Covered Territory, subject to committee oversight. We have agreed, among other things, to provide NovaMedica with clinical data necessary for it to obtain necessary approvals in the Covered Territory, information relating to applications for regulatory approval of our product candidates, certain commercialization information and to assist NovaMedica in conducting any clinical trials necessary for regulatory approval of our product candidates in the Covered Territory. We also have agreed to provide NovaMedica with certain development know-how and support, including making our clinical development personnel available to provide scientific and technical explanations, advice and on-site support that may be reasonably requested by NovaMedica.
NovaMedica is required to reimburse us for any out-of-pocket expenses incurred by us in providing this assistance, including travel-related expenses. We estimate that the aggregate out-of-pocket expense of providing such services will be approximately $500,000. Pursuant to the Collaboration Agreement, we have agreed to use commercially reasonable efforts to include sites in the Covered Territory in our clinical trial programs for our product candidates at our sole expense. Under the Collaboration Agreement, prior to the first commercial sale of a product candidate in the Covered Territory, NovaMedica will have the right to purchase product candidates and related compounds from us or through us as are reasonable and necessary for it to conduct clinical trials in the Covered Territory at our cost plus a mark-up in the low double digits pursuant to a clinical supply agreement to be entered into within 120 days of the date of the Collaboration Agreement. NovaMedica has agreed to supervise and maintain sales representatives for the commercialization of any product candidates approved for sale in the Covered Territory. Within 90 days of receipt of FDA approval for the use of any product candidate, we are obligated to enter into a commercial supply agreement with NovaMedica for the supply of such candidate on terms to be negotiated by the parties. In the Collaboration Agreement, the parties also agreed to customary terms and conditions, including the ownership and use of intellectual property, rights to information, prosecution of patent rights, rights under third-party agreements, confidentiality and indemnification obligations and mechanisms for the resolution of disputes. The Collaboration Agreement expires on the earlier of three years following the first commercial sale of a product candidate in the Covered Territory or nine years from the date of effectiveness and terminates upon the termination of the Tech Transfer Agreement. NovaMedica also has the right to terminate the Collaboration Agreement at any time at its convenience upon 90 days prior written notice.
Sales and Marketing
Because REG1 is a hospital-based acute care product, we believe that it is feasible for us to commercialize REG1 with a relatively small specialty sales force in the United States calling on a targeted group of hospital-based interventional cardiologists. If REG1 is approved, our current plan is to commercialize REG1 in the United States by building a commercial infrastructure or utilizing contract reimbursement specialists, sales people and medical education specialists. We may seek to augment our commercial efforts by entering into a collaboration with a pharmaceutical company, if such a collaboration is available on attractive terms. Outside of the United States, we likely will seek to commercialize REG1 through distribution or other collaboration arrangements.
24
Competition
The biopharmaceutical industry is characterized by intense competition and rapid innovation. Our potential competitors include large pharmaceutical and biotechnology companies, specialty pharmaceutical companies and generic drug companies. We believe the key competitive factors that will affect the development and commercial success of our product candidates are efficacy, safety and tolerability profile, mechanism of action, control and predictability, convenience of dosing and price and reimbursement.
Our most advanced product candidate, REG1, is being developed for use in patients undergoing PCI for a wide variety of cardiovascular conditions. If approved for this indication, REG1 would compete with a number of currently-marketed anticoagulants, including bivalirudin, currently marketed and sold by The Medicines Company under the brand name Angiomax® in the United States, and UFH and LMWH, both of which are available as biosimilars and which are currently manufactured and sold by a number of companies.
We intend to develop REG1 for use in additional cardiovascular procedures, such as OHS, PCI as a treatment for STEMI, and TAVI. If approved for these indications, REG1 would compete primarily with UFH and LMWH. We believe that our product candidates offer key potential advantages over these competitive products that could enable our product candidates, if approved, to capture meaningful market share from our competitors. However, many of our potential competitors have substantially greater financial, technical and human resources than we do, as well as greater experience in the discovery and development of product candidates, obtaining FDA and other regulatory approvals of products and the commercialization of those products. Accordingly, our competitors may be more successful than us in obtaining regulatory approval for drugs and achieving widespread market acceptance. Our competitors’ drugs may be more effective, or more effectively marketed and sold, than any product candidate we may commercialize and may render our product candidates obsolete or non-competitive before we can recover the expenses of their development and commercialization. We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available. Finally, the development of new treatment methods for the diseases we are targeting could render our product candidates non-competitive or obsolete.
Government Regulation and Product Approval
Government authorities in the United States at the federal, state and local level extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, marketing, export and import of products such as those we are developing. Our product candidates must receive final approval from the FDA before they may legally be marketed in the United States.
U.S. Drug Development Process
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act, or FDCA, and implementing regulations. The process of obtaining regulatory approvals and ensuring compliance with appropriate federal, state, local and foreign statutes and regulations requires the expenditure of substantial time and financial resources. Failure to comply with the applicable U.S. requirements at any time during the product development process, approval process, or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, withdrawal of an approval, a clinical hold, warning letters, product seizures, product detention, total or partial suspension of production or distribution, injunctions, fines, refusals of government contracts, restitution, disgorgement or civil or criminal penalties. The process required by the FDA before a drug may be marketed in the United States generally involves the following:
| | • | | completion of preclinical laboratory tests and animal studies according to Good Laboratory Practices or other regulations, as well as formulation studies; |
| | • | | submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may begin; |
| | • | | performance of adequate and well-controlled human clinical trials according to Good Clinical Practices, or GCP, to establish the safety and efficacy of the proposed drug for its intended use; |
| | • | | submission to the FDA of an NDA for a new drug; |
| | • | | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with current Good Manufacturing Practices, or cGMP, to assure that the facilities, methods and controls are adequate to preserve the drug’s identity, strength, quality and purity; and |
| | • | | FDA review and approval of the NDA. |
The testing and approval process require substantial time, effort and financial resources and we cannot be certain that any approvals for our product candidates will be granted on a timely basis, if at all.
25
Once a pharmaceutical product candidate is identified for development, it enters the preclinical testing stage. The preclinical testing stage includes laboratory evaluations of product chemistry, toxicity, formulation and stability, as well as animal studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information, analytical data and any available clinical data or literature, to the FDA as part of the IND. The sponsor must also include a protocol detailing, among other things, the objectives of the initial clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated if the initial clinical trial lends itself to an efficacy evaluation. Preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA places the clinical trial on a clinical hold within that 30-day time period. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. Clinical holds also may be imposed by the FDA at any time before or during trials due to safety concerns or non-compliance.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with GCP regulations. These regulations include the requirement that all research subjects provide informed consent. Further, an institutional review board, or IRB, must review and approve the plan for any clinical trial before it commences at any institution. An IRB considers, among other things, whether the risks to individuals participating in the trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the information regarding the clinical trial and the consent form that must be provided to each clinical trial subject or his or her legal representative and must monitor the clinical trial until completed. Once an IND is in effect, each new clinical protocol and any amendments to the protocol must be submitted to the IND for FDA review, and to the IRBs for approval.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| | • | | Phase 1. The product candidate is initially introduced into healthy human subjects and tested for tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing may be conducted in patients having the specific disease. |
| | • | | Phase 2. Phase 2 trials involve investigations in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product candidate for specific targeted diseases and to determine optimal dosage and schedule. |
| | • | | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in a larger patient population at geographically dispersed clinical trial sites. These trials are intended to establish the overall risk/benefit ratio of the product candidate and provide an adequate basis for regulatory approval and product labeling. |
Post-approval studies, also called Phase 4 trials, may be conducted after initial marketing approvals. These studies are used to obtain additional experience from the treatment of patients in the intended therapeutic indication and may be required by the FDA as part of the approval process.
Progress reports detailing the results of the clinical trials must be submitted annually to the FDA and safety reports must be submitted to the FDA and the investigators for serious and unexpected side effects. Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within any specified period, if at all. The FDA or the sponsor may suspend or terminate a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the product and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
26
United States Review and Approval Processes
The results of product development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the drug, proposed labeling and other relevant information, are submitted to the FDA as part of an NDA, requesting approval to market the product. The submission of an NDA is subject to the payment of substantial user fees which may be waived under certain limited circumstances.
FDA Review of New Drug Applications
The FDA reviews all NDAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. The FDA may request additional information rather than accept an NDA for filing. In this event, the NDA must be re-submitted with the additional information. The re-submitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. Under the goals and policies agreed to by the FDA under the Prescription Drug User Fee Act, or PDUFA, the FDA has ten months in which to complete the initial review of a standard NDA and respond to the applicant and six months for a priority NDA. The FDA does not always meet its PDUFA goal dates for standard and priority NDAs. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and whether the chemistry, manufacturing and control documentation is adequate to assure and preserve the product’s identity, strength, quality and purity. Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. The FDA may refer the NDA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions. An advisory committee is a panel of independent experts who provide advice and recommendations when requested by the FDA on matters of importance that come before the agency. The FDA is not bound by the recommendation of an advisory committee.
The approval process is lengthy and difficult and the FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical data or other data and information.
Even if such data and information is submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA will issue a complete response letter if the agency decides not to approve the NDA in its present form. The complete response letter usually describes all of the specific deficiencies that the FDA identified in the NDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to conform the application to a condition suitable for approval. If a complete response letter is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, withdraw the application, or request an opportunity for a hearing.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may require Phase 4 testing which involves clinical trials designed to further assess a drug’s safety and effectiveness after NDA approval and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized.
Expedited Development and Review Programs
The FDA has a Fast Track development program that is intended to expedite or facilitate the process for reviewing new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address an unmet medical need for the condition. Fast Track designation applies to the combination of the product and the specific indication for which it is being studied. A benefit to Fast Track designation is that the FDA may consider for review sections of the NDA on a rolling basis before the complete application is submitted, if the sponsor provides a schedule for the submission of the sections of the NDA, the FDA agrees to accept sections of the NDA and determines that the schedule is acceptable, and the sponsor pays any required user fees upon submission of the first section of the NDA. If FDA accepts a portion of an application, this does not necessarily mean that review will commence or proceed before FDA receives the complete application. Actual commencement and scheduling of review depends on many factors, including FDA staffing, workload, competing priorities, timeline for completion of applications, and the perceived efficiency of commencing review before receipt of the complete application.
Any product submitted to the FDA for marketing approval, including a Fast Track program, may also be eligible for other types of FDA programs intended to expedite development and review, such as priority review and accelerated approval. A product is eligible for priority review if it treats a serious condition and, if approved, has the potential to provide safe and effective therapy where no satisfactory alternative therapy exists or a significant improvement in the treatment, diagnosis or prevention of a disease compared to marketed products. The FDA will attempt to direct additional resources to the evaluation of an application for a new drug or biological product designated for priority review in an effort to facilitate the review. Additionally, a product may be eligible for accelerated approval. Drug or biological products studied for their safety and effectiveness in treating serious or life-threatening illnesses and that provide meaningful therapeutic benefit over existing treatments may receive accelerated approval, which means that they may be approved on the basis of adequate and well-controlled clinical trials establishing that the product has an effect on a surrogate endpoint that is reasonably likely to predict a clinical benefit, or on the basis of an effect on a clinical endpoint other than survival or irreversible morbidity that is reasonable likely to predict an effect on survival, irreversible morbidity or other clinical benefit. As a condition of approval, the FDA may require that a sponsor of a drug or biological product receiving accelerated approval perform adequate and well-controlled post-marketing clinical trials to establish safety and efficacy for the approved indication. In addition, the FDA currently requires as a condition for accelerated approval pre-approval of promotional materials, which could adversely impact the timing of the commercial launch of the product. Fast Track designation, priority review and accelerated approval do not change the standards for approval but may expedite the development or approval process.
Patent Term Restoration and Marketing Exclusivity
Depending upon the timing, duration and specifics of FDA marketing approval of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, commonly referred to as the Hatch-Waxman Amendments. The Hatch-Waxman Amendments permit a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product’s approval date. The patent term restoration period is generally one-half the time between (a) the effective date of an IND and the submission date of an NDA plus (b) the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension and the application for the extension must be submitted prior to the expiration of the patent and within 60 days of approval of the drug. The PTO, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration.
Market exclusivity provisions under the FDCA can also delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the United States to the first applicant to gain approval of an NDA for a new chemical entity. A drug is a new chemical entity if the FDA has not previously approved any other new drug containing the same active pharmaceutical ingredient, or active moiety, which is the molecule or ion responsible for the action of the drug substance. During the exclusivity period, the FDA may not accept for review an
27
abbreviated new drug application, or ANDA, or a Section 505(b)(2) NDA submitted by another company for another version of such drug where the applicant does not own or have a legal right of reference to all the data required for approval. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from clinical trials not conducted by or for the applicant and for which the applicant has not obtained a right of reference. However, the FDCA will not prevent the submission or approval of another full Section 505(b)(1) NDA, but such an NDA applicant would be required to conduct its own preclinical and adequate, well-controlled clinical trials to demonstrate safety and effectiveness. Further, a Section 505(b)(2) application may be submitted after four years if it contains a Paragraph IV certification claiming that the patents covering the drug are either invalid or not infringed by the drug described in the 505(b)(2) application. The FDCA also provides three years of marketing exclusivity for an NDA, Section 505(b)(2) NDA or supplement to an existing NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application. Such clinical trials may, for example, support new indications, dosages, routes of administration or strengths of an existing drug, or for a new use, if new clinical investigations that were conducted or sponsored by the applicant are determined by the FDA to be essential to the approval of the application. This exclusivity, which is sometimes referred to as clinical investigation exclusivity, prevents the FDA from approving an application under Section 505(b)(2) for the same conditions of use associated with the new clinical investigations before the expiration of three years from the date of approval. Such three-year exclusivity, however, would not prevent the approval of another application if the applicant submits a Section 505(b)(1) NDA and has conducted its own adequate, well-controlled clinical trials demonstrating safety and efficacy, nor would it prevent approval of a generic product or Section 505(b)(2) product that did not incorporate the exclusivity-protected changes of the approved drug product. The FDCA, FDA regulations and other applicable regulations and policies provide incentives to manufacturers to create modified, non-infringing versions of a drug to facilitate the approval of an ANDA or other application for generic substitutes.
Post-Approval Requirements
Any drugs for which we receive FDA approval are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse effects with the product, providing the FDA with updated safety and efficacy information, product sampling and distribution requirements, complying with certain electronic records and signature requirements and complying with FDA promotion and advertising requirements. In September 2007, the Food and Drug Administration Amendments Act of 2007 was enacted, giving the FDA enhanced post-marketing authority, including the authority to require post-marketing studies and clinical trials, labeling changes based on new safety information, and compliance with risk evaluations and mitigation strategies approved by the FDA. The FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. Further, manufacturers of drugs must continue to comply with cGMP requirements, which are extensive and require considerable time, resources and ongoing investment to ensure compliance. In addition, certain changes to the manufacturing process generally require prior FDA approval before being implemented and other types of changes to the approved product, such as adding new indications and additional labeling claims, are also subject to further FDA review and approval.
Drug manufacturers and other entities involved in the manufacturing and distribution of approved drugs are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP and other laws. The cGMP requirements apply to all stages of the manufacturing process, including the production, processing, sterilization, packaging, labeling, storage and shipment of the drug. Manufacturers must establish validated systems to ensure that products meet specifications and regulatory standards, and test each product batch or lot prior to its release.
The FDA may restrict market availability or withdraw a product approval if compliance with regulatory standards is not maintained or if problems occur after the product reaches the market. Discovery of previously unknown problems with a product subsequent to its approval may result in restrictions on the product or even complete withdrawal of the product from the market. Further, the failure to maintain compliance with regulatory requirements may result in administrative or judicial actions, such as fines, warning letters, holds on clinical trials, product seizures, product detention or refusal to permit the import or export of products, refusal to approve pending applications or supplements, restrictions on marketing or manufacturing, injunctions or civil or criminal penalties.
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. For example, in July 2012, the Food and Drug Administration Safety and Innovation Act was enacted, expanding drug supply chain requirements and strengthening FDA’s response to drug shortages, among other things. In addition to new legislation, the FDA regulations and policies are often revised or reinterpreted by the agency in ways that may significantly affect our business and our product candidates. It is impossible to predict whether further legislative or FDA regulation or policy changes will be enacted or implemented and what the impact of such changes, if any, may be.
28
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our product candidates to the extent we choose to clinically evaluate or sell any products outside of the United States. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. As in the United States, post-approval regulatory requirements, such as those regarding product manufacture, marketing, or distribution would apply to any product that is approved outside the United States.
Third-Party Payor Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any of our drug candidates for which we obtain regulatory approval. In both the United States and foreign markets, our ability to commercialize our product candidates successfully, and to attract commercialization partners for our product candidates, depends in significant part on the availability of adequate financial coverage and reimbursement from third-party payors, including, in the United States, governmental payors such as the Medicare and Medicaid programs, managed care organizations, and private health insurers. Medicare is a federally funded program managed by the Centers for Medicare and Medicaid Services, or CMS, through local fiscal intermediaries and carriers that administer coverage and reimbursement for certain healthcare items and services furnished to the elderly and disabled. Medicaid is an insurance program for certain categories of patients whose income and assets fall below state defined levels and who are otherwise uninsured that is both federally and state funded and managed by each state. The federal government sets general guidelines for Medicaid and each state creates specific regulations that govern its individual program. Each payor has its own process and standards for determining whether it will cover and reimburse a procedure or particular product. Private payors often rely on the lead of the governmental payors in rendering coverage and reimbursement determinations. Therefore, achieving favorable CMS coverage and reimbursement is usually a significant gating issue for successful introduction of a new product. The competitive position of some of our products will depend, in part, upon the extent of coverage and adequate reimbursement for such products and for the procedures in which such products are used. Prices at which we or our customers seek reimbursement for our product candidates can be subject to challenge, reduction or denial by the government and other payors.
The U.S. Congress and state legislatures may, from time to time, propose and adopt initiatives aimed at cost containment, which could impact our ability to sell our product candidates profitably. For example, in March 2010, President Obama signed into law the Patient Protection and Affordable Care Act and the associated reconciliation bill, which we refer to collectively as the Health Care Reform Law, a sweeping law intended to broaden access to health insurance, reduce or constrain the growth of healthcare spending, enhance remedies against fraud and abuse, add new transparency requirements for healthcare and health insurance industries, impose new taxes and fees on the health industry and impose additional health policy reforms. Effective October 1, 2010, the Health Care Reform Law revised the definition of “average manufacturer price” for reporting purposes, which could increase the amount of Medicaid drug rebates to states once the provision is effective. Further, the law imposes a significant annual fee on companies that manufacture or import branded prescription drug products. Substantial new provisions affecting compliance have also been enacted, which may require us to modify our business practices with healthcare practitioners. We will not know the full effects of the Health Care Reform Law until applicable federal and state agencies issue regulations or guidance under the new law. Although it is too early to determine the effect of the Health Care Reform Law, the new law appears likely to continue the pressure on pharmaceutical pricing, especially under the Medicare program, and may also increase our regulatory burdens and operating costs. Moreover, in the coming years, additional changes could be made to governmental healthcare programs that could significantly impact the success of our product candidates.
The cost of pharmaceuticals continues to generate substantial governmental and third-party payor interest. We expect that the pharmaceutical industry will experience pricing pressures due to the trend toward managed healthcare, the increasing influence of managed care organizations and additional legislative proposals. Our results of operations could be adversely affected by current and future healthcare reforms.
Some third-party payors also require pre-approval of coverage for new or innovative devices or drug therapies before they will reimburse healthcare providers that use such therapies. While we cannot predict whether any proposed cost-containment measures will be adopted or otherwise implemented in the future, the announcement or adoption of these proposals could have a material adverse effect on our ability to obtain adequate prices for our product candidates and operate profitably.
29
Other Healthcare Laws and Compliance Requirements
In the United States, our activities are potentially subject to regulation by various federal, state and local authorities in addition to the FDA, including the CMS, other divisions of the United States Department of Health and Human Services (e.g., the Office of Inspector General), the United States Department of Justice and individual United States Attorney offices within the Department of Justice, and state and local governments. These regulations include:
| | • | | the federal healthcare program anti-kickback law which prohibits, among other things, persons from soliciting, receiving or providing remuneration, directly or indirectly, to induce either the referral of an individual, for an item or service or the purchasing or ordering of a good or service, for which payment may be made under federal healthcare programs such as the Medicare and Medicaid programs; |
| | • | | federal false claims laws which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment from Medicare, Medicaid, or other government reimbursement programs that are false or fraudulent, and which may apply to entities like us which provide coding and billing advice to customers; |
| | • | | the federal Health Insurance Portability and Accountability Act of 1996 which prohibits executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters and which also imposes certain requirements relating to the privacy, security and transmission of individually identifiable health information; |
| | • | | the federal transparency requirements under the Health Care Reform Law requires manufacturers of drugs, devices, biologics, and medical supplies to report to the Department of Health and Human Services information related to physician payments and other transfers of value and physician ownership and investment interests; |
| | • | | the FDCA which, among other things, strictly regulates drug product marketing, prohibits manufacturers from marketing drug products for off-label use and regulates the distribution of drug samples; and |
| | • | | state law equivalents of each of the above federal laws, such as anti-kickback and false claims laws, which may apply to items or services reimbursed by any third party payor, including commercial insurers, and state laws governing the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and often are not preempted by federal laws, thus complicating compliance efforts. |
Employees
As of December 31, 2013, we had a total of 28 employees, 13 of whom work in North Carolina and 15 of whom work in New Jersey. Fourteen of our employees hold graduate degrees including ten doctorate degrees and 15 are engaged in full-time research and development activities. None of our employees are represented by a labor union and we consider our employee relations to be good.
Corporate Information
We were incorporated in Delaware under the name Quartet Biosciences, Inc. in December 2001 and changed our name to Regado Biosciences, Inc. in March 2003. Our principal executive offices are located at 120 Mountain View Boulevard, Basking Ridge, New Jersey 07920, and our telephone number is (908) 580-2100. Our website address is www.regadobio.com. On our website, investors can obtain, free of charge, a copy of our Annual Report on Form 10-K, Quarterly Reports on Form 10-Q, Current Reports on Form 8-K, and other reports filed or furnished pursuant to Section 13(a) or 15(d) of the Exchange Act of 1934, as amended, as soon as reasonably practicable after we file such material electronically with, or furnish it to, the Securities and Exchange Commission. None of the information posted on our website is incorporated by reference into this Annual Report.
Item 1A. Risk Factors
Except for the historical information contained herein or incorporated by reference, this Annual Report and the information incorporated by reference contains forward-looking statements that involve risks and uncertainties. These statements include projections about our accounting and finances, plans and objectives for the future, future operating and economic performance and other statements regarding future performance. These statements are not guarantees of future performance or events. Our actual results may differ materially from those discussed here. Factors that could cause or contribute to differences in our actual results include those discussed in the following section, as well as those discussed in Part II, Item 7 entitled “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and
30
elsewhere throughout this Annual Report and in any other documents incorporated by reference into this Annual Report. You should consider carefully the following risk factors, together with all of the other information included or incorporated in this Annual Report. Each of these risk factors, either alone or taken together, could adversely affect our business, operating results and financial condition, as well as adversely affect the value of an investment in our common stock. There may be additional risks that we do not presently know of or that we currently believe are immaterial which could also impair our business and financial position.
Risks Relating to Our Financial Position and Need for Additional Capital
We have never been profitable. Currently, we have no products approved for commercial sale, and to date we have not generated any revenue from product sales. As a result, our ability to curtail our losses and reach profitability is unproven, and we may never achieve or sustain profitability.
We have never been profitable and do not expect to be profitable in the foreseeable future. We have incurred net losses in each year since our inception, including net losses of approximately $34.4 million for the year ended December 31, 2013 and an accumulated deficit of approximately $145.0 million since inception as of December 31, 2013. Subsequent to December 31, 2013, we raised net proceeds of approximately $18.7 million from the sale of shares of our common stock in a private placement. We have devoted most of our financial resources to research and development, including our preclinical development activities and clinical trials. We have not completed development of any product candidate and we have therefore not generated any revenues from product sales. Because of the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve or maintain profitability. We expect to incur significant increased expenses as we continue our single, open-label 13,200 subject Phase 3 trial of REG1 in patients undergoing PCI (excluding ST elevated myocardial infarction, or STEMI), or the REGULATE-PCI trial, which commenced in September 2013. We also expect an increase in our expenses associated with creating additional infrastructure to support operations as a public company. As a result, we expect to continue to incur net losses and negative cash flows for the foreseeable future. These net losses and negative cash flows have had, and will continue to have, an adverse effect on our stockholders’ equity and working capital.
The amount of our future net losses will depend, in part, on the rate of future growth of our expenses and our ability to generate revenues. In addition, we may not be able to enter into any collaboration that will generate significant cash. If we are unable to develop and commercialize one or more of our product candidates either alone or with collaborators, or if revenues from any product candidate that receives marketing approval are insufficient, we will not achieve profitability. Even if we do achieve profitability, we may not be able to sustain or increase profitability.
We will need to raise additional capital to complete the REGULATE-PCI trial and commercialize REG1. If we are unable to raise sufficient capital, we would be forced to delay, reduce or eliminate our product development programs.
Developing pharmaceutical products, including conducting preclinical studies and clinical trials, is expensive. We expect our research and development expenses to increase in connection with our ongoing activities, particularly as we commence our REGULATE-PCI trial, undertake additional clinical trials of our other product candidates and continue to work on our other research programs. We believe that our existing cash and cash equivalents will be sufficient for us to fund the REGULATE-PCI trial through the second interim analysis, which we expect will occur in the third quarter of 2014. We will need to raise substantial additional capital to fund our operations and complete the development and commercialization of REG1, and to repay our debt with Comerica bank. If the U.S. Food and Drug Administration, or the FDA, or other regulators require that we perform additional studies beyond those we currently expect, or if there are any delays in completing our clinical trials or the development of any of our product candidates, our expenses could increase beyond what we currently anticipate and the timing of any potential product approval may be delayed. We have no commitments or arrangements for any additional financing to fund our research and development programs. We also will need to raise substantial additional capital in the future to complete the development and commercialization of REG1 for additional indications and for our other product candidates. Because successful development of our product candidates is uncertain, we are unable to estimate the actual funds required to complete research and development and commercialize our products under development.
31
Until we can generate a sufficient amount of revenue from our product candidates, if ever, we expect to finance future cash needs through public or private equity offerings, debt financings or corporate collaborations and licensing arrangements. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate one or more of our research or development programs. To the extent that we raise additional funds by issuing equity securities, our stockholders will experience additional dilution, and debt financing, if available, may involve restrictive covenants. To the extent that we raise additional funds through collaborations and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our product candidates or grant licenses on terms that may not be favorable to us. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time.
Our future funding requirements, both near and long-term, will depend on many factors, including, but not limited to:
| | • | | the initiation, progress, timing, costs and results of preclinical studies and clinical trials for our product candidates and potential product candidates, including our REGULATE-PCI trial and the continued development of our other product candidates; |
| | • | | the number and characteristics of product candidates that we pursue; |
| | • | | the terms and timing of any future collaboration, licensing or other arrangements that we may establish; |
| | • | | the outcome, timing and cost of regulatory approvals; |
| | • | | the cost of obtaining, maintaining, defending and enforcing intellectual property rights, including patent rights; |
| | • | | the effect of competing technological and market developments; |
| | • | | the cost and timing of completing commercial-scale outsourced manufacturing activities; |
| | • | | market acceptance of any product candidates for which we may receive regulatory approval; |
| | • | | the cost of establishing sales, marketing and distribution capabilities for any product candidates for which we may receive regulatory approval; and |
| | • | | the extent to which we acquire, license or invest in businesses, products or technologies. |
We have a limited operating history and we expect a number of factors to cause our operating results to fluctuate on a quarterly and annual basis, which may make it difficult to predict our future performance.
We are a development stage biopharmaceutical company with a limited operating history. Our operations to date have been primarily limited to developing our technology and undertaking preclinical studies and clinical trials of REG1 and our other product candidates. We have not yet obtained regulatory approvals for REG1 or any of our other product candidates. Consequently, any predictions made about our future success or viability may not be as accurate as they could be if we had a longer operating history or commercialized products. Our financial condition and operating results have varied significantly in the past and will continue to fluctuate from quarter-to-quarter or year-to-year due to a variety of factors, many of which are beyond our control. Factors relating to our business that may contribute to these fluctuations include other factors described elsewhere in this report and also include:
| | • | | our ability to obtain additional funding to complete development of REG1 and to develop our other product candidates; |
| | • | | delays in the commencement, enrollment and timing of clinical trials; |
| | • | | the success of our clinical trials through all phases of clinical development, including our REGULATE-PCI trial; |
| | • | | any delays in regulatory review and approval of product candidates in clinical development; |
| | • | | our ability to obtain and maintain regulatory approval for REG1 or any of our other product candidates in the United States and foreign jurisdictions; |
| | • | | potential side effects of our product candidates that could delay or prevent commercialization, limit the indications for any approved drug, require the establishment of risk evaluation and mitigation strategies, or REMS, or cause an approved drug to be taken off the market; |
| | • | | our dependence on third-party manufacturers, or CMOs, to supply or manufacture our products; |
| | • | | our dependence on clinical research organizations, or CROs, to conduct our clinical trials; |
32
| | • | | our ability to establish or maintain collaborations, licensing or other arrangements; |
| | • | | market acceptance of our product candidates; |
| | • | | our ability to establish and maintain an effective sales and marketing infrastructure, either through the creation of a commercial infrastructure or through strategic collaborations; |
| | • | | competition from existing products or new products that may emerge; |
| | • | | the ability of patients or healthcare providers to obtain coverage of or sufficient reimbursement for our products; |
| | • | | our ability to leverage our proprietary technology platform to discover and develop additional product candidates; |
| | • | | our ability and our licensors’ abilities to successfully obtain, maintain, defend and enforce intellectual property rights important to our business; |
| | • | | our ability to attract and retain key personnel to manage our business effectively; |
| | • | | our ability to build our finance infrastructure and improve our accounting systems and controls; |
| | • | | potential product liability claims; |
| | • | | potential liabilities associated with hazardous materials; and |
| | • | | our ability to obtain and maintain adequate insurance policies. |
Accordingly, the results of any quarterly or annual periods should not be relied upon as indications of future operating performance.
The audit opinion on our financial statements contains a going concern modification.
Based on our cash balances, recurring losses, net capital deficiency and debt outstanding as of December 31, 2013 and our projected spending in 2014, which raise substantial doubt about our ability to continue as a going concern, the audit opinion on our audited financial statements as of and for the year ended December 31, 2013 contains a going concern modification. We believe that our existing cash and cash equivalents will be sufficient to fund our projected operating requirements and the REGULATE-PCI trial through the second interim analysis, which we expect will occur during the third quarter of 2014. However, if we are unable to continue as a going concern, we might have to liquidate our assets and the values we receive for our assets in liquidation or dissolution could be significantly lower than the values reflected in our financial statements. Amounts due under our loan with Comerica Bank, or Comerica, may become immediately due and payable upon the occurrence of a material adverse change, as defined under the loan agreement. Under the terms of the Comerica loan agreement, we are subject to operational covenants, including limitations on our ability to incur liens or additional debt, pay dividends, redeem stock, make specified investments and engage in merger, consolidation or asset sale transactions, among other restrictions. In addition, the inclusion of a going concern statement by our auditors, our lack of cash resources and our potential inability to continue as a going concern may materially adversely affect our share price and our ability to raise new capital or to enter into critical contractual relations with third parties.
Risks Relating to the Development and Regulatory Approval of Our Product Candidates
Clinical failure can occur at any stage of clinical development. Because the results of earlier clinical trials are not necessarily predictive of future results, any product candidate we advance through clinical trials may not have favorable results in later clinical trials or receive regulatory approval.
Clinical failure can occur at any stage of clinical development. Clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional clinical or preclinical trials. In addition, data obtained from trials are susceptible to varying interpretations, and regulators may not interpret our data as favorably as we do, which may delay, limit or prevent regulatory approval. Success in preclinical testing and early clinical trials does not ensure that later clinical trials will generate the same results or otherwise provide adequate data to demonstrate the efficacy and safety of a product candidate. Frequently, product candidates that have shown promising results in early clinical trials have subsequently suffered significant setbacks in later clinical trials. In addition, the design of a clinical trial can determine whether its results will support approval of a product and flaws in the design of a clinical trial may not become apparent until the clinical trial is well advanced. While members of our management team have experience in designing clinical trials, our company has limited experience in designing clinical trials and we may be unable to design and execute a clinical trial to support regulatory approval. Further, clinical trials of potential products often reveal that it is not practical or feasible to
33
continue development efforts. For example, if the results of our REGULATE-PCI trial do not achieve the primary efficacy endpoints or demonstrate expected safety, the prospects for approval of REG1 would be materially and adversely affected. If REG1 or our other product candidates are found to be unsafe or lack efficacy, we will not be able to obtain regulatory approval for them and our business would be harmed.
REGULATE-PCI includes three interim analyses of REG1 by the Data Safety Monitoring Board, or the DSMB. The first interim analysis will be a general safety analysis after enrollment of 1,000 subjects, which is expected to occur by the beginning of the second quarter of 2014. The second interim analysis will be another general safety analysis after 25% of the subjects are enrolled, which is expected to occur during the third quarter of 2014. The final interim analysis will be an analysis of the general safety and efficacies of REG1 after 50% of the subjects are enrolled, which is expected to occur during the fall of 2014. If, as a result of any of those interim analyses, we or the DSMB determine that REG1 is not safe or that it is futile to continue the trial because of a lack of efficacy, the trial will be terminated. If the results of any one of these analyses is unfavorable, our business would be harmed.
We cannot be certain that REG1 or any of our other product candidates will receive regulatory approval, and without regulatory approval we will not be able to market our product candidates. Any delay in the regulatory review or approval of REG1 or any of our other product candidates will materially or adversely harm our business.
We have invested a significant portion of our efforts and financial resources in the development of REG1, our most advanced product candidate. Our ability to generate revenue related to product sales, which we do not expect will occur for at least the next several years, if ever, will depend on the successful development and regulatory approval of our product candidates. We commenced our REGULATE-PCI trial in September 2013. We may conduct our REGULATE-PCI trial only to learn that REG1 is not a safe or effective treatment, in which case the REGULATE-PCI trial may not lead to regulatory approval for REG1. Similarly, our clinical development programs for our other product candidates may not lead to regulatory approval from the FDA and similar foreign regulatory agencies. This failure to obtain regulatory approvals would prevent our product candidates from being marketed and would have a material and adverse effect on our business.
All of our product candidates require regulatory review and approval prior to commercialization. Any delays in the regulatory review or approval of our product candidates would delay market launch, increase our cash requirements and result in additional operating losses.
The process of obtaining FDA and other required regulatory approvals, including foreign approvals, often takes many years and can vary substantially based upon the type, complexity and novelty of the products involved. Furthermore, this approval process is extremely complex, expensive and uncertain. We may be unable to submit any new drug application, or an NDA, in the United States or any marketing approval application in foreign jurisdictions for any of our products. If we submit an NDA including any amended NDA or supplemental NDA, to the FDA seeking marketing approval for any of our product candidates, the FDA must decide whether to accept or reject the submission for filing. We cannot be certain that any of these submissions will be accepted for filing and reviewed by the FDA, or that the marketing approval application submissions to any other regulatory authorities will be accepted for filing and review by those authorities. We cannot be certain that we will be able to respond to any regulatory requests during the review period in a timely manner, or at all, without delaying potential regulatory action. We also cannot be certain that any of our product candidates will receive favorable recommendations from any FDA advisory committee or foreign regulatory bodies or be approved for marketing by the FDA or foreign regulatory authorities. In addition, delays in approvals or rejections of marketing applications may be based upon many factors, including regulatory requests for additional analyses, reports, data and studies, regulatory questions regarding data and results, changes in regulatory policy during the period of product development and the emergence of new information regarding REG1 or our other product candidates.
Data obtained from preclinical studies and clinical trials are subject to different interpretations, which could delay, limit or prevent regulatory review or approval of any of our product candidates. Furthermore, regulatory attitudes towards the data and results required to demonstrate safety and efficacy can change over time and can be affected by many factors, such as the emergence of new information, including on other products, policy changes and agency funding, staffing and leadership. We do not know whether future changes to the regulatory environment will be favorable or unfavorable to our business prospects.
In addition, the environment in which our regulatory submissions may be reviewed changes over time. For example, average review times at the FDA for NDAs have fluctuated over the last ten years, and we cannot predict the review time for any of our submissions with any regulatory authorities. Review times can be affected by a variety of factors, including budget and funding levels and statutory, regulatory and policy changes. Moreover, in light of widely publicized events concerning the safety risk of certain drug products, regulatory authorities, members of the U.S. Government Accountability Office,
34
medical professionals and the general public have raised concerns about potential drug safety issues. These events have resulted in the withdrawal of drug products, revisions to drug labeling that further limit use of the drug products and establishment of REMS measures that may, for instance, restrict distribution of drug products. The increased attention to drug safety issues may result in a more cautious approach by the FDA to clinical trials. Data from clinical trials may receive greater scrutiny with respect to safety, which may make the FDA or other regulatory authorities more likely to terminate clinical trials before completion, or require longer or additional clinical trials that may result in substantial additional expense and a delay or failure in obtaining approval or may result in approval for a more limited indication than originally sought.
Delays in the commencement, enrollment and completion of our clinical trials could result in increased costs to us and delay or limit our ability to obtain regulatory approval for REG1 and our other product candidates.
Delays in the commencement, enrollment and completion of clinical trials could increase our product development costs or limit the regulatory approval of our product candidates. We commenced our REGULATE-PCI trial in September 2013; however, this clinical trial may not be completed on schedule, if at all. In addition, we do not know whether planned clinical trials of REG1 in additional indications and of our other product candidates will begin on time or will be completed on schedule or at all. The commencement, enrollment and completion of our REGULATE-PCI trial or other clinical trials can be delayed for a variety of reasons, including:
| | • | | inability to reach agreements on acceptable terms with prospective CROs and trial sites, the terms of which can be subject to extensive negotiation and may vary significantly among different CROs and trial sites; |
| | • | | inability to maintain necessary supplies of study drug and comparator to maintain predicted enrollment rates at clinical trial sites |
| | • | | regulatory objections to commencing a clinical trial; |
| | • | | inability to identify and maintain a sufficient number of trial sites, many of which may already be engaged in other clinical trial programs, including some that may be for the same indication as our product candidates; |
| | • | | withdrawal of clinical trial sites from our clinical trials as a result of changing standards of care or the ineligibility of a site to participate in our clinical trials; |
| | • | | inability to obtain institutional review board approval to conduct a clinical trial; |
| | • | | difficulty recruiting and enrolling subjects to participate in clinical trials for a variety of reasons, including meeting the enrollment criteria for our study and competition from other clinical trial programs for the same indication as our product candidates; |
| | • | | inability to retain subjects in clinical trials due to the treatment protocol, personal issues, side effects from the therapy or lack of efficacy; and |
| | • | | difficulty in importing and exporting clinical trial materials and study samples. |
In addition, our REGULATE-PCI trial or any of our other clinical trials may be suspended or terminated by us, the FDA or other regulatory authorities due to a number of factors, including:
| | • | | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| | • | | failure to pass inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities; |
| | • | | failure of any CMOs that we use to comply with current Good Manufacturing Practices, or cGMP; |
| | • | | unforeseen safety issues or any determination that a clinical trial presents unacceptable health risks; |
| | • | | changes in the regulatory requirement and guidance; or |
| | • | | lack of adequate funding to continue the clinical trial due to unforeseen costs resulting from enrollment delays, requirements to conduct additional trials and studies, increased expenses associated with the services of our CROs and other third parties or other reasons. |
If we are required to conduct additional clinical trials or other testing of REG1 or our other product candidates beyond those currently contemplated, we may be delayed in obtaining, or may not be able to obtain, marketing approval for these product candidates.
35
We have never conducted a Phase 3 clinical trial or submitted an NDA before, and may be unable to do so for REG1 and other product candidates we are developing.
We commenced our REGULATE-PCI trial in September 2013. The conduct of Phase 3 clinical trials and the submission of a successful NDA is a complicated process. Although members of our management team have extensive industry experience, including in the development, clinical testing and commercialization of drug candidates, our company has never conducted a Phase 3 clinical trial before, has limited experience in preparing, submitting and prosecuting regulatory filings, and has not submitted an NDA before. Consequently, we may be unable to successfully and efficiently execute and complete these planned clinical trials in a way that leads to NDA submission and approval of REG1 and other product candidates we are developing. We may require more time and incur greater costs than our competitors and may not succeed in obtaining regulatory approvals of product candidates that we develop. Failure to commence or complete, or delays in, our planned clinical trials would prevent or delay commercialization of REG1 and other product candidates we are developing.
We have never performed a clinical trial comparing the safety or efficacy of REG1 to bivalirudin. Because our clinical trials used heparin as a comparator, the risk that our REGULATE-PCI trial does not achieve one or more of its primary endpoints may be increased.
We have never performed a clinical trial directly comparing the safety or efficacy of REG1 to bivalirudin. Our randomized, partially blinded, dose-ranging Phase 2b trial involving 640 subjects, or the RADAR trial, used standard of care heparin as the comparator and, as a result, we have no clinical trial data directly comparing REG1 and bivalirudin. The primary efficacy endpoint of our REGULATE-PCI trial is a 20.0% reduction in the occurrence of ischemic events using REG1 compared to bivalirudin and the primary safety endpoint of the trial is non-inferiority of REG1 compared to bivalirudin with respect to major bleeding events. Because we have no clinical trial data directly comparing REG1 to bivalirudin, the prediction of Phase 3 success based on Phase 2 results is complicated and the risk that REGULATE-PCI does not achieve one or more of these endpoints may be increased.
Our product candidates may cause serious adverse events or undesirable side effects which may delay or prevent marketing approval, or, if approval is received, require them to be taken off the market, require them to include safety warnings or otherwise limit their sales.
Serious adverse events or undesirable side effects from REG1 or any of our other product candidates could arise either during clinical development or, if approved, after the approved product has been marketed. For example, three severe allergic events occurred in our RADAR trial. In addition, in 2008 we terminated an exploratory Phase 2a trial of REG1 in subjects undergoing off-pump coronary arterial bypass grafting when the first enrolled subject experienced clotting in one of three bypass grafts. The results of future clinical trials, including REGULATE-PCI, may show that our product candidates cause serious adverse events or undesirable side effects, which could interrupt, delay or halt clinical trials, resulting in delay of, or failure to obtain, marketing approval from the FDA and other regulatory authorities.
If REG1 or any of our other product candidates cause serious adverse events or undesirable side effects:
| | • | | regulatory authorities may impose a clinical hold which could result in substantial delays and adversely impact our ability to continue development of the product; |
| | • | | regulatory authorities may require the addition of labeling statements, specific warnings, a contraindication or field alerts to physicians and pharmacies; |
| | • | | we may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
| | • | | we may be required to implement a risk minimization action plan, which could result in substantial cost increases and have a negative impact on our ability to commercialize the product; |
| | • | | we may be required to limit the patients who can receive the product; |
| | • | | we may be subject to limitations on how we promote the product; |
| | • | | sales of the product may decrease significantly; |
| | • | | regulatory authorities may require us to take our approved product off the market; |
| | • | | we may be subject to litigation or product liability claims; and |
| | • | | our reputation may suffer. |
36
Any of these events could prevent us from achieving or maintaining market acceptance of the affected product or could substantially increase commercialization costs and expenses, which in turn could delay or prevent us from generating significant revenues from the sale of our products.
We have limited experience manufacturing the oligonucleotides comprising our product candidates at commercial scale and there are no established standards for their manufacture. As a result, delays in regulatory approval of our product candidates may occur. Also, manufacturing issues may arise that could cause delays or increase costs.
We have limited experience manufacturing the oligonucleotides comprising our product candidates at commercial scale. We, together with our CMOs, have developed manufacturing processes that have never been tested in commercial production. Our manufacturing process will be subject to approval by regulators before we can commence the manufacture and sale of an approved product. The standards of the International Conference on Harmonization of Technical Requirements for Registration of Pharmaceuticals for Human Use, which establishes basic guidelines and standards for drug development in the United States, the European Union, Japan and other countries, do not apply to oligonucleotides, including our product candidates. As a result, there is no established generally accepted manufacturing or quality standard for the production of our product candidates. Even though the FDA has agreed to the quality standards for the REG1 to be used in our REGULATE-PCI trial, the FDA has the ability to modify those standards at any time and foreign regulatory agencies may impose differing quality standards and quality control on the manufacture of our drug candidates. The lack of uniform manufacturing and quality standards among regulatory agencies may delay regulatory approval of our product candidates. Also, as we scale-up manufacturing of any approved product, our CMOs may encounter unexpected issues relating to the manufacturing process or the quality, purity and stability of the product and we may be required to refine or alter our manufacturing processes to address these issues. Resolving these issues could result in significant delays and may result in significantly increased costs. If we experience significant delays or other obstacles in producing any approved product for commercial scale, our ability to market and sell any approved products may be adversely affected and our business could suffer.
REG1 and our other product candidates employ novel mechanisms of action and may never be approved or accepted by their intended markets.
Our activities have focused on the discovery and development of therapeutic aptamers and their specific active control agents. Our future success depends on our ability to complete the REGULATE-PCI trial of REG1 successfully, obtain market approval for and successfully commercialize REG1, as well as our ability to develop and market other product candidates that use our proprietary technology platform. We believe only one therapeutic aptamer has been approved for commercial sale and no product candidate consisting of a therapeutic aptamer and its specific active control agent has ever received regulatory approval. The scientific discoveries that form the basis of our proprietary technology platform and our product candidates are relatively new. We are not aware of any other antithrombotic drugs that have the same mechanism of action as our product candidates and there can be no assurance that, even if approved, physicians will be willing to use them. If we do not successfully develop and commercialize product candidates based upon our technological approach, we may not become profitable and the value of our common stock may decline.
In addition, regulatory approval of novel product candidates such as REG1 and our other product candidates manufactured using novel manufacturing processes such as ours can be more expensive and take longer than for other, more well-known or extensively studied pharmaceutical or biopharmaceutical products, due to our and regulatory agencies’ lack of experience with them. We believe that the FDA has only approved one aptamer product to date. This lack of experience may lengthen the regulatory review process, require us to conduct additional studies or clinical trials, increase our development costs, lead to changes in regulatory positions and interpretations, delay or prevent approval and commercialization of these product candidates or lead to significant post-approval limitations or restrictions.
The novel nature of REG1 and our other product candidates also means that fewer people are trained in or experienced with product candidates of this type, which may make it difficult to find, hire and retain capable personnel for research, development and manufacturing positions.
Further, our focus solely on controllable aptamer technology for developing drugs as opposed to multiple, more proven technologies for drug development increases the risks associated with the ownership of our common stock. If we do not obtain regulatory approval for REG1 and our other product candidates and achieve market acceptance for our approved products, we may be required to change the scope and direction of our product development activities. In that case, we may not be able to identify and implement successfully an alternative product development strategy.
37
Risks Relating to the Commercialization of Our Product Candidates
If any of our product candidates for which we receive regulatory approval do not achieve broad market acceptance, the revenues that are generated from their sales will be limited.
The commercial success of REG1 and our other product candidates, if approved, will depend upon the acceptance of these products among physicians, healthcare payors and patients. The degree of market acceptance of our product candidates will depend on a number of factors, including:
| | • | | limitations or warnings contained in a product’s FDA-approved labeling; |
| | • | | changes in the standard of care or the availability of alternative therapies for the targeted indications for any of our product candidates; |
| | • | | limitations in the approved indications for our product candidates; |
| | • | | demonstrated clinical safety and efficacy compared to other products; |
| | • | | lack of significant adverse side effects; |
| | • | | education, sales, marketing and distribution support; |
| | • | | availability and degree of reimbursement from third-party payors; |
| | • | | timing of market introduction and perceived effectiveness of competitive products; |
| | • | | availability of alternative therapies at similar or lower cost, including generics, biosimilar and over-the-counter products; |
| | • | | adverse publicity about our product candidates or favorable publicity about competitive products; |
| | • | | convenience and ease of administration of our products; |
| | • | | potential product liability claims; and |
| | • | | government-imposed pricing restrictions. |
If our product candidates are approved, but do not achieve an adequate level of acceptance by physicians, healthcare payors and patients, sufficient revenue may not be generated from these products, and we may not become or remain profitable. In addition, efforts to educate the medical community and third-party payors on the benefits of our product candidates may require significant resources and may never be successful.
REG1 and each of our other product candidates consist of a therapeutic aptamer and its specific active control agent. These two components are administered at different times and in different strengths and the failure to administer the components correctly may expose a patient to significant risk. Physicians will need to be educated as to our products’ novel mechanisms of action and trained as to the proper use of our products. Physicians may be unwilling to devote the time necessary to learn how to use our product candidates properly and may continue using other competing products even if our products are safer and more effective. As a result, the commercialization of any approved product may be slower than we expect and any approved product may not achieve the level of acceptance we anticipate. If physicians are unwilling to use our products as a result of their novel mechanisms of action or the need to be trained on their use, our business may suffer.
We do not have the capability to sell, distribute and market our product candidates. If we are unable to establish an effective sales force and marketing infrastructure, or enter into acceptable third-party sales and marketing or licensing arrangements, we may not be able to commercialize our product candidates successfully.
We do not have the capability to sell, distribute and market our product candidates. We will need to build a commercial organization or secure a strategic partner to commercialize REG1 or any other product candidates. If we are unable to build a commercial infrastructure or secure a strategic collaboration, our business and results of operations will be materially and adversely affected. Development of an internal commercial organization will require substantial resources and will be time consuming. These costs may be incurred in advance of any approval of our product candidates. In addition, we may not be able to hire a sales force in the United States that is sufficient in size or has adequate expertise in the medical markets that we intend to target. If we are unable to establish a sales and marketing capability, our operating results may be adversely affected. If we seek to enter into sales and marketing or licensing arrangements with third parties for the marketing and sale of any approved products, we may be unable to enter into any such arrangements on acceptable terms, or at all.
38
Fast Track designation does not guarantee approval, or expedited approval, of REG1 and there is no guarantee that REG1 will maintain Fast Track designation.
In March 2014, we announced that the FDA granted Fast Track designation to REG1 for anticoagulant therapy to be used in patients with coronary artery disease during PCI. Under the FDA Modernization Act of 1997, Fast Track designation is designed to facilitate the development and expedite the review of new drugs that are intended to treat serious or life-threatening conditions. Compounds selected must demonstrate the potential to address an unmet medical need for such a condition. Mechanisms intended to facilitate development include opportunities for frequent dialogue with FDA reviewers and for timely review of submitted protocols. However, the designation does not guarantee approval or expedited approval of any application for the product. Furthermore, the FDA may revoke Fast Track designation from a product candidate at any time if it determines that the criteria are no longer met.
Even if our product candidates receive regulatory approval, we may still face future development and regulatory difficulties.
Even if regulatory approval is obtained for any of our product candidates, regulatory authorities may still impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies. Given the number of high profile adverse safety events with certain drug products, regulatory authorities may require, as a condition of approval, costly REMS, which may include safety surveillance, restricted distribution and use, patient education, enhanced labeling, expedited reporting of certain adverse events, pre-approval of promotional materials and restrictions on direct-to-consumer advertising. For example, any labeling approved for any of our product candidates may include a restriction on the term of its use, or it may not include one or more of our intended indications. Furthermore, any new legislation addressing drug safety issues could result in delays or increased costs during the period of product development, clinical trials and regulatory review and approval, as well as increased costs to assure compliance with any new post-approval regulatory requirements.
Our product candidates will also be subject to ongoing regulatory requirements for the labeling, packaging, storage, advertising, promotion, record-keeping and submission of safety and other post-market information. In addition, sellers of approved products, manufacturers and manufacturers’ facilities are required to comply with extensive FDA requirements, including ensuring that quality control and manufacturing procedures conform to cGMP. As such, we and our CMOs are subject to continual review and periodic inspections to assess compliance with cGMP. Accordingly, we and others with whom we work must continue to expend time, money, and effort in all areas of regulatory compliance, including manufacturing, production, and quality control. We will also be required to report certain adverse reactions and production problems, if any, to the FDA, and to comply with certain requirements concerning advertising and promotion for our products. Promotional communications with respect to prescription drugs are subject to a variety of legal and regulatory restrictions and must be consistent with the information in the product’s approved label. As such, we may not promote our products for indications or uses for which they do not have approval.
If a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, or disagrees with the promotion, marketing, or labeling of a product, it may impose restrictions on that product or us, including requiring withdrawal of the product from the market. If our product candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
| | • | | issue warning or other letters; |
| | • | | mandate modifications to promotional materials or require us to provide corrective information to healthcare practitioners; |
| | • | | require us to enter into a consent decree or permanent injunction, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; |
| | • | | impose other civil or criminal penalties; |
| | • | | suspend or withdraw regulatory approval; |
| | • | | suspend any ongoing clinical trials; |
| | • | | refuse to approve pending applications or supplements to approved applications filed by us; |
| | • | | impose restrictions on operations, including costly new manufacturing requirements; or |
| | • | | seize or detain products or require a product recall. |
We expect that our existing and future product candidates will face competition and most of our competitors have significantly greater resources than we do.
The biopharmaceutical industry is characterized by intense competition and rapid innovation. Our potential competitors include large pharmaceutical and biotechnology companies, specialty pharmaceutical companies and generic or biosimilar drug companies. We believe the key competitive factors that will affect the development and commercial success of our product candidates are efficacy, safety and tolerability profile, mechanism of action, control and predictability, convenience of dosing and pricing and reimbursement. Our most advanced product candidate, REG1, is being developed for use in patients undergoing percutaneous coronary intervention for a wide variety of cardiovascular conditions. If approved for this indication, REG1 would compete with a number of currently-marketed anticoagulants, including bivalirudin, currently marketed and sold by The Medicines Company under the brand name Angiomax® in the United States, and heparin, or UFH, and low molecular weight heparin, or LMWH, both of which are available as biosimilars and currently manufactured and sold by multiple manufacturers. If REG1 is approved for this initial indication, we intend to seek approval for the use of REG1 in other cardiovascular indications. If approved for these additional indications, REG1 would potentially compete with the same treatments described above.
39
Many of our potential competitors have substantially greater:
| | • | | resources, including capital, personnel and technology; |
| | • | | research and development capability; |
| | • | | clinical trial expertise; |
| | • | | intellectual property rights, including patent rights; |
| | • | | expertise in obtaining, maintaining, defending and enforcing intellectual property rights, including patent rights; |
| | • | | manufacturing and distribution expertise; and |
| | • | | sales and marketing expertise. |
Accordingly, our competitors may be more successful than us in obtaining regulatory approval for drugs and achieving widespread market acceptance. Our competitors’ drugs may be more effective, or more effectively marketed and sold, than any product candidate we may commercialize and may render our product candidates obsolete or non-competitive before we can recover the expenses of their development and commercialization. We anticipate that we will face intense and increasing competition as new drugs enter the market and advanced technologies become available. Finally, the development of new treatment methods for the diseases we are targeting could render our product candidates non-competitive or obsolete.
Reimbursement decisions by third-party payors may have an adverse effect on pricing and market acceptance of REG1 or any of our other product candidates. If there is not sufficient reimbursement for our products, it is less likely that our products will be widely used.
Market acceptance and sales of REG1 or any other product candidates that we develop will depend on reimbursement policies and may be affected by future healthcare reform measures. Government authorities and third-party payors, such as private health insurers and health maintenance organizations, decide which drugs they will cover and establish payment levels. We cannot be certain that reimbursement will be available for REG1 or any other product candidates that we develop. Also, we cannot be certain that reimbursement policies will not reduce the demand for, or the price paid for, our products. If reimbursement is not available or is available on a limited basis, we may not be able to successfully commercialize REG1 or any other product candidates that we develop.
In the United States, the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, also called the Medicare Modernization Act, or MMA, changed the way Medicare covers and pays for pharmaceutical products. The legislation established Medicare Part D, which expanded Medicare coverage for outpatient prescription drug purchases by the elderly but provided authority for limiting the number of drugs that will be covered in any therapeutic class. The MMA also introduced a new reimbursement methodology based on average sales prices for physician-administered drugs.
The United States and several foreign jurisdictions are considering, or have already enacted, a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and expanding access to healthcare. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives. We expect to experience pricing pressures in connection with the sale of REG1 and any other products that we develop, due to the trend toward managed healthcare, the increasing influence of health maintenance organizations and additional legislative proposals.
In March 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively, the Affordable Care Act, became law in the United States. The goal of the Affordable Care Act is to reduce the cost of healthcare and substantially change the way healthcare is financed by both governmental and private insurers. While we cannot predict what impact on federal reimbursement policies this legislation will have in general or on our business specifically, the Affordable Care Act may result in downward pressure on pharmaceutical reimbursement, which could negatively affect market acceptance of REG1 or any future product candidates.
40
Members of the U.S. Congress and some state legislatures are seeking to overturn at least portions of the legislation and we expect they will continue to review and assess this legislation and possibly alternative healthcare reform proposals. We cannot predict whether new proposals will be made or adopted, when they may be adopted or what impact they may have on us if they are adopted.
If we obtain approval to commercialize any approved products outside of the United States, a variety of risks associated with international operations could materially adversely affect our business.
If REG1 or any of our other product candidates are approved for commercialization outside of the United States, we intend to enter into agreements with third parties to market them on a worldwide basis or in more limited geographical regions, excluding the Covered Territory. We expect that we will be subject to additional risks related to entering into international business relationships, including:
| | • | | different regulatory requirements for drug approvals; |
| | • | | reduced protection for intellectual property rights, including trade secret and patent rights; |
| | • | | unexpected changes in tariffs, trade barriers and regulatory requirements; |
| | • | | economic weakness, including inflation, or political instability in particular foreign economies and markets; |
| | • | | compliance with tax, employment, immigration and labor laws for employees living or traveling abroad; |
| | • | | foreign taxes, including withholding of payroll taxes; |
| | • | | foreign currency fluctuations, which could result in increased operating expenses and reduced revenues, and other obligations incident to doing business in another country; |
| | • | | workforce uncertainty in countries where labor unrest is more common than in the United States; |
| | • | | production shortages resulting from any events affecting raw material supply or manufacturing capabilities abroad; |
| | • | | business interruptions resulting from geopolitical actions, including war and terrorism, or natural disasters including earthquakes, hurricanes, floods and fires; and |
| | • | | difficulty in importing and exporting clinical trial materials and study samples. |
Risks Relating to Our Dependence on Third Parties
We may not succeed in establishing and maintaining collaborative relationships, which may significantly limit our ability to develop and commercialize our product candidates successfully, if at all.
We intend to seek collaborative relationships for the development and commercialization of our product candidates, including REG1. Failure to obtain a collaborative relationship for REG1, particularly in the European Union and for other markets requiring extensive sales efforts, may significantly impair the potential for this product candidate. We will also need to enter into collaborative relationships to provide funding to support our other research and development programs. The process of establishing and maintaining collaborative relationships is difficult, resource intensive and involves significant uncertainty, including:
| | • | | a collaboration partner may shift its priorities and resources away from our product candidates due to a change in business strategies, or a merger, acquisition, sale or downsizing; |
| | • | | a collaboration partner may seek to renegotiate or terminate their relationships with us due to unsatisfactory clinical results, manufacturing issues, a change in business strategy, a change of control or other reasons; |
| | • | | a collaboration partner may cease development in therapeutic areas which are the subject of our strategic collaboration; |
| | • | | a collaboration partner may not devote sufficient capital or resources towards our product candidates; |
| | • | | a collaboration partner may change the success criteria for a product candidate thereby delaying or ceasing development of such candidate; |
| | • | | a significant delay in initiation of certain development activities by a collaboration partner will also delay payment of milestones tied to such activities, thereby impacting our ability to fund our own activities; |
| | • | | a collaboration partner could develop a product that competes, either directly or indirectly, with our product candidate; |
41
| | • | | a collaboration partner with commercialization obligations may not commit sufficient financial or human resources to the marketing, distribution or sale of a product; |
| | • | | a collaboration partner with manufacturing responsibilities may encounter regulatory, resource or quality issues and be unable to meet demand requirements; |
| | • | | a partner may exercise a contractual right to terminate a strategic alliance; |
| | • | | a dispute may arise between us and a partner concerning the research, development or commercialization of a product candidate resulting in a delay in milestones, royalty payments or termination of an alliance and possibly resulting in costly litigation or arbitration which may divert management attention and resources; and |
| | • | | a partner may use our products or technology in such a way as to invite litigation from a third party. |
If any collaborator fails to fulfill its responsibilities in a timely manner, or at all, our research, clinical development, manufacturing, or commercialization efforts related to that collaboration could be delayed or terminated, or it may be necessary for us to assume responsibility for expenses or activities that would otherwise have been the responsibility of our collaborator. If we are unable to establish and maintain collaborative relationships on acceptable terms or to successfully transition terminated collaborative agreements, we may have to delay or discontinue further development of one or more of our product candidates, undertake development and commercialization activities at our own expense or find alternative sources of capital.
We rely on third parties to conduct, supervise and monitor our clinical trials, and if those third parties perform in an unsatisfactory manner, it may harm our business.
We rely on CROs and clinical trial sites to ensure the proper and timely conduct of our clinical trials. While we have agreements governing their activities, we will have limited influence over their actual performance. We will control only certain aspects of our CROs’ activities. Nevertheless, we will be responsible for ensuring that our clinical trials are conducted in accordance with the applicable protocol, legal, regulatory and scientific standards and our reliance on the CROs does not relieve us of our regulatory responsibilities.
We and our CROs are required to comply with the FDA’s current good clinical practices requirements, or cGCP, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the rights, integrity and confidentiality of clinical trial participants are protected. The FDA enforces these cGCPs through periodic inspections of trial sponsors, principal investigators and clinical trial sites. If we or our CROs fail to comply with applicable cGCPs, the clinical data generated in our clinical trials may be deemed unreliable and the FDA may require us to perform additional clinical trials before approving any marketing applications. Upon inspection, the FDA may determine that our clinical trials did not comply with cGCPs. In addition, our clinical trials, including our REGULATE-PCI trial, will require a sufficiently large number of test subjects to evaluate the safety and effectiveness of a product candidate. Accordingly, if our CROs fail to comply with these regulations or fail to recruit a sufficient number of patients, our clinical trials may be delayed or we may be required to repeat such clinical trials, which would delay the regulatory approval process.
Our CROs are not our employees, and we are not able to control whether or not they devote sufficient time and resources to our clinical trials. These CROs may also have relationships with other commercial entities, including our competitors, for whom they may also be conducting clinical trials, or other drug development activities which could harm our competitive position. If our CROs do not successfully carry out their contractual duties or obligations, fail to meet expected deadlines, or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or regulatory requirements, or for any other reasons, our clinical trials may be extended, delayed or terminated, and we may not be able to obtain regulatory approval for, or successfully commercialize our product candidates. As a result, our financial results and the commercial prospects for such product candidates would be harmed, our costs could increase, and our ability to generate revenues could be delayed.
We also rely on other third parties to store and distribute drug products for our clinical trials. Any performance failure on the part of our distributors could delay clinical development or marketing approval of our product candidates or commercialization of our products, if approved, producing additional losses and depriving us of potential product revenue.
42
We do not have multiple sources of supply for the components used in REG1 and our other product candidates. If we were to lose a supplier, it could have a material adverse effect on our ability to complete the development of REG1 or our other product candidates or, if we obtain regulatory approval for REG1 or our other product candidates, to commercialize them.
We do not have multiple sources of supply for the components used in REG1 and our other product candidates. We also do not have long-term supply agreements with any of our suppliers. If for any reason we are unable to obtain drug compounds from a supplier, we would have to seek to obtain it from another oligonucleotide manufacturer. We may not be able to establish additional sources of supply for our product candidates, or may be unable to do so on acceptable terms. Such suppliers are subject to regulatory requirements, covering manufacturing, testing, quality control and record keeping relating to our product candidates and subject to ongoing inspections by the regulatory agencies. Failure by any of our suppliers to comply with applicable regulations may result in long delays and interruptions.
The number of oligonucleotide suppliers is limited. In the event it is necessary or desirable to acquire supplies from an alternative supplier, we might not be able to obtain them on commercially reasonable terms, if at all. It could also require significant time and expense to redesign our manufacturing processes to work with another company.
As part of any marketing approval, a manufacturer and its processes are required to be qualified by the FDA prior to commercialization. If supply from the approved supplier is interrupted, there could be a significant disruption in commercial supply. An alternative vendor would need to be qualified through an NDA supplement which could result in further delay. The FDA or other regulatory agencies outside of the United States may also require additional studies if a new supplier is relied upon for commercial production. Switching vendors may involve substantial costs and is likely to result in a delay in our desired clinical and commercial timelines.
If we are unable to obtain the supplies we need at a reasonable price or on a timely basis, it could have a material adverse effect on our ability to complete the development of REG1 and our other product candidates or, if we obtain regulatory approval for REG1 or our other product candidates, to commercialize them.
We rely on third-party manufacturers to produce our product candidates. If we experience problems with any of these suppliers, the manufacturing of our product candidates or products could be delayed.
We do not have the capability to manufacture our product candidates and do not intend to develop that capability. As a result, we rely on CMOs to produce our product candidates. If REG1 or our other product candidates are approved for sale, we expect to enter into contracts with CMOs for the commercial scale production of the approved product. Reliance on CMOs entails risks, including:
| | • | | the inability to meet our product specifications and quality requirements consistently; |
| | • | | inability to access production facilities on a timely basis; |
| | • | | inability or delay in increasing manufacturing capacity; |
| | • | | manufacturing and product quality issues related to scale-up of manufacturing; |
| | • | | costs and validation of new equipment and facilities required for commercial level activity; |
| | • | | a failure to satisfy the FDA’s cGMP requirements and similar foreign standards on a consistent basis; |
| | • | | the inability to negotiate manufacturing agreements with third parties under commercially reasonable terms; |
| | • | | termination or nonrenewal of manufacturing agreements with third parties in a manner or at a time that is costly or damaging to us; |
| | • | | the reliance on a single sources of supply which, if unavailable, would delay our ability to complete our clinical trials or to sell any product for which we have received marketing approval; |
| | • | | the lack of qualified backup suppliers for supplies that are currently purchased from a single source supplier; |
| | • | | operations of our CMOs or suppliers could be disrupted by conditions unrelated to our business or operations, including the bankruptcy of the CMO or supplier; |
| | • | | carrier disruptions or increased costs that are beyond our control; and |
| | • | | the failure to deliver products under specified storage conditions and in a timely manner. |
43
Any of these risks could cause the delay of clinical trials, regulatory submissions, required approvals or commercialization of our products, cause us to incur higher costs and prevent us from commercializing our product candidates successfully. Manufacturing of our product candidates and any approved products could be disrupted or halted if our CMOs do not comply with cGMP or foreign manufacturing standards, even if the compliance failure does not relate to our product candidates or approved products. Furthermore, if any of our product candidates are approved and our CMOs fail to deliver the required commercial quantities of finished product on a timely basis and at commercially reasonable prices, and we are unable to find one or more replacement manufacturers capable of production at a substantially equivalent cost, in substantially equivalent volumes and quality and on a timely basis, we would likely be unable to meet demand for our products and could lose potential revenue. It may take several years to establish an alternative source of supply for our product candidates and to have any such new source approved by the FDA or a foreign regulator.
Risks Relating to Our Intellectual Property
It is difficult and costly to protect our proprietary rights, and we may not be able to ensure their protection.
Our commercial success will depend in part on obtaining and maintaining proprietary rights important to our business, as well as successfully defending and enforcing those proprietary rights if challenged. The procurement, defense and enforcement of intellectual property rights involve complex legal and factual questions. Changes in either the patent laws or in interpretations of patent laws in the United States and foreign jurisdictions may diminish the value of our intellectual property. Laws relating to patent rights continue to evolve in the United States and foreign jurisdictions, as does their interpretation by national patent offices and judicial systems, creating some uncertainty for patent applicants, patent owners and licensees.
Our ability to stop third parties from using our technology or making, using, selling, offering to sell or importing our products is dependent upon the extent to which we have rights under valid and enforceable patents or trade secrets that cover these activities. If any patent we currently or in the future may own or license is deemed invalid or unenforceable, it could impact our commercial success. We cannot predict the breadth of claims that may be issued from any patent applications we currently or may in the future own or license from third parties.
The degree of future protection our proprietary rights may afford is uncertain because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| | • | | others may be able to make, use, sell, offer to sell or import products that are similar to our product candidates but that are not covered by the claims of our patents; |
| | • | | we might not have been the first to make the inventions covered by our patent portfolio; |
| | • | | we might not have been the first to file patent applications for these inventions; |
| | • | | others may independently develop similar or alternative technologies or duplicate any of our technologies in a manner that does not violate our trade secrets; |
| | • | | our proprietary rights may not provide us with any competitive advantages; |
| | • | | we may not develop additional technologies or products that are patentable or suitable to maintain as trade secrets; or |
| | • | | the proprietary rights of others may have an adverse effect on our business. |
As of December 31, 2013, we are the owner of record of five issued or allowed U.S. patents and six issued or allowed non-U.S. patents, as well as the licensee of at least ten issued or allowed U.S. patents and at least eleven issued or allowed non-U.S. patents. We are actively pursuing an additional nine U.S. patent applications, all of which are non-provisional applications, three international patent applications and 50 non-U.S. patent applications in twelve jurisdictions as the owner of record, in addition to at least two U.S. patent applications and 13 non-U.S. patent applications under license.
We also may rely on trade secrets to protect our technology, especially where we do not believe patent protection is appropriate or obtainable. Our ability to stop third parties from making, using, selling, offering to sell or importing our products or practicing our technology is dependent in part upon the extent to which we have rights in enforceable trade secrets that cover these activities. Trade secret rights can be lost through disclosure to third parties. Although we use reasonable efforts to protect our trade secrets, our employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our trade secrets to third parties, resulting in loss of trade secret protection. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how, which would not constitute a violation of our trade secret rights. Enforcing a claim that a third party is engaged in the unlawful use of our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, recognition of rights in trade secrets and a willingness to enforce trade secrets may differ in certain jurisdictions.
44
Intellectual property disputes are expensive and would consume time and resources and divert the attention of managerial and scientific personnel. We may incur substantial costs as a result of litigation or other proceedings relating to patent and other intellectual property rights and we may be unable to enforce or protect our rights to, or use, our technology.
If we choose to go to court to attempt to stop another party from using our intellectual property without authorization, or any licensor of our intellectual property chooses to do the same, rights in our intellectual property may be lost. More specifically, rights in trade secrets we have or obtain may be lost as the result of disclosure associated with of our efforts to stop their unauthorized use. Rights in any patents we have or obtain may be lost as a result of our efforts to stop their unauthorized use, as the party charged with patent infringement has the right to ask the court to rule that such patents are invalid or should not be enforced against that third party. There is also the risk that, even if the validity of such patents is upheld, the court will refuse to stop the other party on the ground that such other party’s activities do not infringe our rights to such patents. Apart from litigation, adversarial procedures are available in the patent offices of many countries, including the United States, that permit interested third parties to dispute the validity of issued patents or to otherwise impact the course of prosecution of pending patent applications. Intellectual property disputes are expensive and would consume time and resources and divert the attention of managerial and scientific personnel even if we were successful in stopping the unauthorized use of our intellectual property rights. Moreover, the patent laws in the United States and internationally continue to evolve, creating uncertainty as to the likelihood that we will be able to obtain patents and increase the likelihood of challenge to any patents we obtain or license.
Furthermore, a third party may claim that we or our manufacturing partners are engaged in unauthorized use of intellectual property owned by the third party, including patent rights, and may go to court to stop us from engaging in our normal operations and activities, including making or selling our product candidates. These lawsuits are costly and could affect our results of operations and divert the attention of managerial and scientific personnel. There is a risk that a court would decide that we or our CMOs are engaged in unauthorized use of the third party’s valid and enforceable intellectual property, including patent rights, and would order us or our CMOs to stop the activities protected by these rights. In that event, we may not have a viable alternative to the unauthorized use and may need to halt commercialization of the relevant product. In addition, there is a risk that a court will order us or our CMOs to pay the other party damages for having used the other party’s intellectual property in an unauthorized manner. In the future, we may agree to indemnify our CMOs against certain intellectual property claims brought by third parties. Patent rights involve complex factual and legal issues; as a result, it is not always clear to industry participants, including us, whether activities or products are covered by patent rights, or by which patent rights. The breadth of patent claims is subject to interpretation by the courts, and the interpretation is not always uniform. If we are sued for patent infringement, we would need to demonstrate that our products or methods either do not infringe the claims of the relevant patents or that the patent claims are invalid, and we may not be able to do this. Proving invalidity is difficult. For example, in the United States, proving invalidity requires the alleged infringer to overcome the presumption of validity enjoyed by issued patents.
Because some patent applications may be maintained in secrecy until the patents are issued, publication of patent applications is delayed, and publications in the scientific literature often lag behind actual discoveries, we cannot be certain that others have not filed patent applications for technology covered by our pending applications, or that we were the first to invent the technology. Our competitors may have filed, and may in the future file, patent applications covering technology important to our business. Any such patent application may have priority over our patent applications, which could further require us to obtain rights to issued patents covering such technologies and serve as a bar to patentability of our own patent filings. If another party has filed a U.S. patent application on inventions similar to ours, we may have to participate in an interference proceeding declared by the U.S. Patent and Trademark Office to determine priority of invention in the United States. The costs of these proceedings could be substantial, and it is possible that such efforts would be unsuccessful if, unbeknownst to us, the other party had independently arrived at the same or similar invention prior to our own invention, resulting in a loss of our U.S. patent position with respect to such invention.
Some of our competitors may be able to sustain the costs of patent-related disputes, including patent litigation, more effectively than we can because they have substantially greater resources. In addition, any uncertainties resulting from the initiation and continuation of any litigation could have a material adverse effect on our ability to raise the funds necessary to continue our operations.
45
Risks Related to Employee Matters and Managing Growth
We will need to expand our operations and increase the size of our company, and we may experience difficulties in managing growth.
As we advance our product candidates through preclinical studies and clinical trials and develop new product candidates, we will need to increase our product development, scientific and administrative headcount to manage these programs. In addition, to meet our obligations as a public company, we will need to increase our general and administrative capabilities. Our management, personnel and systems currently in place may not be adequate to support this future growth. Our need to effectively manage our operations, growth and various projects requires that we:
| | • | | successfully attract and recruit new employees with the expertise and experience we will require; |
| | • | | manage our clinical programs effectively, which we anticipate being conducted at numerous clinical sites; |
| | • | | develop a marketing, distribution and sales infrastructure if we seek to market our products directly; and |
| | • | | continue to improve our operational, manufacturing, financial and management controls, reporting systems and procedures. |
If we are unable to successfully manage this growth and increased complexity of operations, our business may be adversely affected.
We may not be able to manage our business effectively if we are unable to attract and retain key personnel.
We may not be able to attract or retain qualified management, finance, scientific and clinical personnel in the future due to the intense competition for qualified personnel among biotechnology, pharmaceutical and other businesses. If we are not able to attract and retain necessary personnel to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
Our industry has experienced a high rate of turnover of management personnel in recent years. We are highly dependent on the development, regulatory, commercialization and business development expertise of our executive officers and key employees. If we lose one or more of our executive officers or key personnel, our ability to implement our business strategy successfully could be seriously harmed. Any of our executive officers or key employees may terminate their employment at any time. We have entered into change of control and severance agreements with certain of our officers as part of our retention efforts. Replacing executive officers and key employees may be difficult, will be costly and may take an extended period of time because of the limited number of individuals in our industry with the mix of skills and experience required to develop, gain regulatory approval of and commercialize products successfully. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these additional key personnel. Our failure to attract and retain key personnel could materially harm our business.
Failure to build our finance infrastructure and improve our accounting systems and controls could impair our ability to comply with the financial reporting and internal controls requirements for publicly traded companies.
As a public company, we operate in an increasingly demanding regulatory environment, which requires us to comply with applicable provisions of the Sarbanes-Oxley Act of 2002, or Sarbanes-Oxley Act, and the related rules and regulations of the Securities and Exchange Commission, expanded disclosure requirements, accelerated reporting requirements and more complex accounting rules. Company responsibilities required by the Sarbanes-Oxley Act include establishing corporate oversight and adequate internal control over financial reporting and disclosure controls and procedures. Effective internal controls are necessary for us to produce reliable financial reports and are important to help prevent financial fraud.
We rely on consultants to perform certain of our accounting and financial reporting functions. We will need to hire additional finance personnel and build our financial infrastructure as we transition to operating as a public company, including complying with the applicable requirements of Section 404 of the Sarbanes-Oxley Act. We may be unable to do so on a timely basis. Until we are able to expand our finance and administrative capabilities and establish necessary financial reporting infrastructure, we may not be able to prepare and disclose, in a timely manner, our financial statements and other required disclosures or comply with the applicable provisions of the Sarbanes-Oxley Act or existing or new reporting requirements. If we cannot provide reliable financial reports or prevent fraud, our business and results of operations could be harmed and investors could lose confidence in our reported financial information.
46
We have had a material weakness in our internal control over financial reporting.
Prior to our IPO in August 2013, we had not been a public reporting company and we had limited accounting personnel and systems to adequately execute accounting processes and limited other supervisory resources with which to address internal control over financial reporting. We and our independent registered public accounting firm identified a material weakness in internal control over financial reporting for the years ended December 31, 2012 and 2011. Under standards established by the Public Company Accounting Oversight Board, a deficiency in internal control over financial reporting exists when the design or operation of a control does not allow management or personnel, in the normal course of performing their assigned functions, to prevent or detect misstatements on a timely basis. A material weakness is a deficiency or combination of deficiencies in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements will not be prevented or detected and corrected on a timely basis. We are in the process of remediating the material weakness identified by us and our independent registered public accounting firm; however, we cannot assure that there will not be additional material weaknesses and significant deficiencies that our independent registered public accounting firm or we will identify. If we identify such issues or if we are unable to produce accurate and timely financial statements, our stock price may be adversely affected and we may be unable to maintain compliance with applicable securities laws and listing requirements
Our employees may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements and insider trading.
We are exposed to the risk of employee fraud or other misconduct. Misconduct by employees could include intentional failures to comply with the regulations of the FDA and non-U.S. regulators, provide accurate information to the FDA and non-U.S. regulators, comply with healthcare fraud and abuse laws and regulations in the United States and abroad, report financial information or data accurately or disclose unauthorized activities to us. In particular, sales, marketing and business arrangements in the healthcare industry are subject to extensive laws and regulations intended to prevent fraud, misconduct, kickbacks, self-dealing and other abusive practices. These laws and regulations may restrict or prohibit a wide range of pricing, discounting, marketing and promotion, sales commission, customer incentive programs and other business arrangements. Employee misconduct could also involve the improper use of information obtained in the course of clinical trials, which could result in regulatory sanctions and cause serious harm to our reputation. We have adopted a code of conduct, but it is not always possible to identify and deter employee misconduct, and the precautions we take to detect and prevent this activity may not be effective in controlling unknown or unmanaged risks or losses or in protecting us from governmental investigations or other actions or lawsuits stemming from a failure to comply with these laws or regulations. If any such actions are instituted against us, and we are not successful in defending ourselves or asserting our rights, those actions could have a significant impact on our business, including the imposition of significant fines or other sanctions.
Other Risks Relating to Our Business
We may use our financial and human resources to pursue a particular research program or product candidate and fail to capitalize on programs or product candidates that may be more profitable or for which there is a greater likelihood of success.
Because we have limited financial and human resources, we intend to focus on the regulatory approval of REG1, including the completion of the REGULATE-PCI trial. As a result, we may forego or delay pursuit of opportunities with other product candidates or for other indications that later prove to have greater commercial potential. Our resource allocation decisions may cause us to fail to capitalize on viable commercial products or profitable market opportunities. Our spending on existing and future product candidates for specific indications may not yield any commercially viable products. If we do not accurately evaluate the commercial potential or target market for a particular product candidate, we may relinquish valuable rights to that product candidate through strategic alliance, licensing or other royalty arrangements in cases in which it would have been more advantageous for us to retain sole development and commercialization rights to such product candidate, or we may allocate internal resources to a product candidate in a therapeutic area in which it would have been more advantageous to enter into a partnering arrangement.
47
We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability and may have to limit development of a product candidate or commercialization of an approved product.
The use of our product candidates in clinical trials and the sale of any products for which we may obtain marketing approval expose us to the risk of product liability claims. Product liability claims may be brought against us or our CMOs by participants enrolled in our clinical trials, patients, healthcare providers or others using, administering or selling our products. If we cannot successfully defend ourselves against any such claims, we would incur substantial liabilities. Regardless of merit or eventual outcome, product liability claims may result in:
| | • | | withdrawal of clinical trial participants; |
| | • | | termination of clinical trial sites or entire trial programs; |
| | • | | costs of related litigation; |
| | • | | substantial monetary awards to patients or other claimants; |
| | • | | decreased demand for an approved product and loss of revenue; |
| | • | | impairment of our business reputation; |
| | • | | diversion of management and scientific resources from our business operations; and |
| | • | | the inability to commercialize an approved product. |
We have obtained limited product liability insurance coverage for our clinical trials domestically and in selected foreign countries where we are conducting clinical trials. Our products liability insurance coverage is currently limited to $10.0 million per occurrence and $10.0 million in the aggregate per year. As such, our insurance coverage may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to product liability. We intend to expand our insurance coverage for products to include the sale of commercial products if we obtain marketing approval for our product candidates, but we may be unable to obtain commercially reasonable product liability insurance for any products approved for marketing. Large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us, particularly if judgments exceed our insurance coverage, could cause our stock price to decline and could adversely affect our results of operations and business.
Our operations involve hazardous materials, which could subject us to significant liabilities.
Our research and development processes involve the controlled use of hazardous materials. Our operations produce hazardous waste products. We cannot eliminate the risk of accidental contamination or discharge or injury from these materials. Federal, state and local laws and regulations govern the use, manufacture, storage, handling and disposal of these materials. We could be subject to civil damages in the event of exposure of individuals to hazardous materials. In addition, claimants may sue us for injury or contamination that results from our use of these materials and our liability may exceed our total assets. We have general liability insurance of up to $1.0 million per occurrence, with an annual aggregate limit of $2.0 million, which excludes pollution liability. This coverage may not be adequate to cover all claims related to our hazardous materials. Furthermore, if we were to be held liable for a claim involving hazardous materials, this liability could exceed our insurance coverage, if any, and our other financial resources. Compliance with environmental and other laws and regulations may be expensive and current or future regulations may impair our research, development or production efforts.
Our insurance policies are expensive and protect us only from some business risks, which will leave us exposed to significant uninsured liabilities.
We do not carry insurance for all categories of risk that our business may encounter. Some of the policies we currently maintain include general liability, employment practices liability, property, auto, workers’ compensation, products liability and directors’ and officers’ insurance. We also expect that operating as a public company will make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified people to serve on our board of directors, our board committees or as executive officers. We do not know, however, if we will be able to maintain existing insurance with adequate levels of coverage. Any significant uninsured liability may require us to pay substantial amounts, which would adversely affect our cash position and results of operations.
48
Risks Relating to Ownership of Our Common Stock
Our executive officers and directors will have the ability to control all matters submitted to our stockholders for approval.
Our executive officers and directors, in the aggregate, beneficially own shares representing 52.3% of our common stock as determined as of March 1, 2014. As a result, if these stockholders were to choose to act together, they would be able to control all matters submitted to our stockholders for approval, as well as our management and affairs. For example, these persons, if they choose to act together, will control the election of directors and approval of any merger, consolidation, sale of all or substantially all of our assets or other business combination or reorganization. This concentration of voting power could delay or prevent an acquisition of us on terms that other stockholders may desire. The interests of this group of stockholders may not always coincide with your interests or the interests of other stockholders and they may act in a manner that advances their best interests and not necessarily those of other stockholders, including seeking a premium value for their common stock, and might affect the prevailing market price for our common stock.
We do not anticipate paying cash dividends on our common stock, and accordingly, stockholders must rely on stock appreciation for any return on their investment.
We have never declared or paid any cash dividend on our common stock and do not anticipate paying cash dividends on our common stock in the future. Our Loan Agreement with Comerica Bank prohibits us from paying cash dividends. As a result, the only return to stockholders will be appreciation in the price of our common stock, which may never occur. Investors seeking cash dividends should not invest in our common stock.
Our stock price may be volatile, and investors in our common stock could incur substantial losses.
Our stock price has fluctuated in the past and may be volatile in the future. The stock market in general and the market for biotechnology companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. The market price of shares of our common stock could be subject to wide fluctuations in response to many risk factors listed in this section, and others beyond our control, including:
| | • | | results and timing of our clinical trials; |
| | • | | results of clinical trials of our competitors’ products; |
| | • | | regulatory actions with respect to our products or our competitors’ products; |
| | • | | actual or anticipated fluctuations in our financial condition and operating results; |
| | • | | actual or anticipated changes in our growth rate relative to our competitors; |
| | • | | actual or anticipated fluctuations in our competitors’ operating results or changes in their growth rate; |
| | • | | competition from existing products or new products that may emerge; |
| | • | | announcements by us or our competitors of significant acquisitions, strategic partnerships, joint ventures, collaborations or capital commitments; |
| | • | | issuance of new or updated research or reports by securities analysts; |
| | • | | fluctuations in the valuation of companies perceived by investors to be comparable to us; |
| | • | | share price and volume fluctuations attributable to inconsistent trading volume levels of our shares; |
| | • | | additions or departures of key management or scientific personnel; |
| | • | | disputes or other developments related to proprietary rights, including patents, litigation matters and our ability to obtain, maintain, defend or enforce proprietary rights relating to our products and technologies; |
| | • | | announcement or expectation of additional financing efforts; |
| | • | | sales of our common stock by us, our insiders or our other stockholders; |
| | • | | market conditions for biopharmaceutical stocks in general; and |
| | • | | general economic and market conditions. |
49
Furthermore, the stock markets have experienced extreme price and volume fluctuations that have affected and continue to affect the market prices of equity securities of many companies. These fluctuations often have been unrelated or disproportionate to the operating performance of those companies. These broad market and industry fluctuations, as well as general economic, political and market conditions such as recessions, interest rate changes or international currency fluctuations, may negatively impact the market price of shares of our common stock. In addition, such fluctuations could subject us to securities class action litigation, which could result in substantial costs and divert our management’s attention from other business concerns, which could potentially harm our business.
We may be subject to securities litigation, which is expensive and could divert management attention.
Our stock price has fluctuated in the past and may be volatile in the future, and in the past, companies that have experienced volatility in the market price of their stock have been subject to an increased incidence of securities class action litigation. We may be the target of this type of litigation in the future. Securities litigation against us could result in substantial costs and divert our management’s attention from other business concerns, which could seriously harm our business.
An active trading market for our common stock may not be maintained
Our stock is currently traded on NASDAQ, but we can provide no assurance that we will be able to maintain an active trading market on NASDAQ or any other exchange in the future. If an active market for our common stock is not maintained, it may be difficult for our stockholders to sell shares without depressing the market price for the shares or at all. An inactive market may also impair our ability to raise capital to continue to fund operations by selling shares and may impair our ability to acquire other companies or technologies by using our shares as consideration.
If securities or industry analysts do not publish research or reports about our business, or publish negative reports about our business, our stock price and trading volume could decline.
The trading market for our common stock will depend in part on the research and reports that securities or industry analysts publish about us or our business. We do not have any control over these analysts. There can be no assurance that analysts will continue to cover us or provide favorable coverage. If one or more of the analysts who cover us downgrade our stock or change their opinion of our stock, our stock price would likely decline. If one or more of these analysts cease coverage of our company or fail to regularly publish reports on us, we could lose visibility in the financial markets, which could cause our stock price or trading volume to decline.
A significant portion of our total outstanding shares of common stock is restricted from immediate resale but may be sold into the market in the near future. This could cause the market price of our common stock to drop significantly, even if our business is doing well.
Sales of a substantial number of shares of our common stock in the public market could occur in the future. These sales, or the perception in the market that the holders of a large number of shares of common stock intend to sell shares, could reduce the market price of our common stock. As of March 1, 2014, we have 25,327,266 outstanding shares of common stock. Of these shares, 7,459,777 may be resold in the public market immediately and the remaining 17,867,489 shares are currently restricted under securities laws or as a result of lock-up agreements, which restrict their transfer for a period of 60 days or 90 days after the. Moreover, holders of shares of our common stock will have rights, subject to some conditions, to require us to file registration statements covering the resale or other disposition of up to 13,396,767 shares of our common stock or to require us to include those shares in registration statements that we may file for ourselves or other stockholders.
You may be diluted by exercises of outstanding options and warrants.
As of March 1, 2014, we had outstanding options to purchase an aggregate of 3,865,948 shares of our common stock at a weighted average exercise price of $5.29 per share and warrants to purchase an aggregate of 11,841 shares of our common stock at a weighted average exercise price of $6.91 per share. The exercise of such outstanding options and warrants will result in dilution of your investment. In addition, as described below, you may experience additional dilution if we issue common stock in the future. As a result of this dilution, you may receive significantly less than the full purchase price you paid for the shares in the event of liquidation.
50
Future sales and issuances of our common stock or rights to purchase common stock, including pursuant to our equity incentive plans, could result in additional dilution of the percentage ownership of our stockholders and could cause our stock price to fall.
We expect that significant additional capital will be needed in the future to continue our planned operations. To the extent we raise additional capital by issuing equity securities; our stockholders may experience substantial dilution. We may sell common stock, convertible securities or other equity securities. If we sell common stock, convertible securities or other equity securities, your investment in our common stock will be diluted. These sales may also result in material dilution to our existing stockholders, and new investors could gain rights superior to our existing stockholders.
We are an “emerging growth company,” and will be able take advantage of reduced disclosure requirements applicable to “emerging growth companies,” which could make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or JOBS Act, and, for as long as we continue to be an “emerging growth company,” we intend to take advantage of certain exemptions from various reporting requirements applicable to other public companies but not to “emerging growth companies,” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We could be an “emerging growth company” for up to five years, or until the earliest of (i) the last day of the first fiscal year in which our annual gross revenues exceed $1 billion, (ii) the date that we become a “large accelerated filer” as defined in Rule 12b-2 under the Exchange Act, which would occur if the market value of our common stock that is held by non-affiliates exceeds $700.0 million as of the last business day of our most recently completed second fiscal quarter, or (iii) the date on which we have issued more than $1 billion in non-convertible debt during the preceding three year period. We cannot predict if investors will find our common stock less attractive if we choose to rely on these exemptions. If some investors find our common stock less attractive as a result of any choices to reduce future disclosure, there may be a less active trading market for our common stock and our stock price may be more volatile.
We will incur significantly increased costs and devote substantial management time as a result of operating as a public company particularly after we are no longer an “emerging growth company.”
As a newly public company, we are incurring significant legal, accounting and other expenses that we did not incur as a private company. For example, we are required to comply with certain of the requirements of the Sarbanes-Oxley Act and the Dodd-Frank Wall Street Reform and Consumer Protection Act, as well as rules and regulations subsequently implemented by the Securities and Exchange Commission, and The NASDAQ Capital Market, our stock exchange, including the establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. We expect that compliance with these requirements will increase our legal and financial compliance costs and will make some activities more time consuming and costly. In addition, we expect that our management and other personnel will need to divert attention from operational and other business matters to devote substantial time to these public company requirements. In particular, we expect to incur significant expenses and devote substantial management effort toward ensuring compliance with the requirements of Section 404 of the Sarbanes-Oxley Act. In that regard, we currently do not have an internal audit function, and we will need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge.
However, for as long as we remain an “emerging growth company” as defined in the JOBS Act, we intend to take advantage of certain exemptions from various reporting requirements that are applicable to other public companies that are not “emerging growth companies” including, but not limited to, not being required to comply with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act, reduced disclosure obligations regarding executive compensation in our periodic reports and proxy statements, and exemptions from the requirements of holding a nonbinding advisory vote on executive compensation and stockholder approval of any golden parachute payments not previously approved. We intend to take advantage of these reporting exemptions until we are no longer an “emerging growth company.”
Under the JOBS Act, “emerging growth companies” can delay adopting new or revised accounting standards until such time as those standards apply to private companies. We have irrevocably elected not to avail ourselves of this exemption from new or revised accounting standards and, therefore, we will be subject to the same new or revised accounting standards as other public companies that are not “emerging growth companies.”
51
After we are no longer an “emerging growth company,” we expect to incur additional management time and cost to comply with the more stringent reporting requirements applicable to companies that are deemed accelerated filers or large accelerated filers, including complying with the auditor attestation requirements of Section 404 of the Sarbanes-Oxley Act.
We cannot predict or estimate the amount of additional costs we may incur as a result of becoming a public company or the timing of such costs.
Anti-takeover provisions in our charter documents and under Delaware law could make an acquisition of our company, which may be beneficial to our stockholders, more difficult and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our amended and restated certificate of incorporation and our amended and restated bylaws may delay or prevent a merger, acquisition or other change of control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions include:
| | • | | classifying our board of directors into three classes; |
| | • | | authorizing the issuance of “blank check” convertible preferred stock, the terms of which may be established and shares of which may be issued without stockholder approval; |
| | • | | limiting the removal of directors by the stockholders; |
| | • | | requiring a supermajority vote of stockholders to amend our certificate of incorporation or bylaws; |
| | • | | prohibiting stockholder action by written consent, thereby requiring all stockholder actions to be taken at a meeting of our stockholders; |
| | • | | eliminating the ability of stockholders to call a special meeting of stockholders; |
| | • | | establishing advance notice requirements for nominations for election to the board of directors or for proposing matters that can be acted upon at stockholder meetings; and |
| | • | | establishing Delaware as the exclusive jurisdiction for certain stockholder litigation against us. |
In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, who are responsible for appointing the members of our management team. In addition, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which prohibits, with some exceptions, stockholders owning in excess of 15% of our outstanding voting stock from merging or combining with us. Although we believe these provisions together provide for an opportunity to receive higher bids by requiring potential acquirers to negotiate with our board of directors, they would apply even if the offer may be considered beneficial by some stockholders.
Item 1B. Unresolved Staff Comments
None.
Our corporate headquarters are located in Basking Ridge, New Jersey, where we lease 6,218 square feet of office space. In November 2008, we entered into a three-year lease agreement for that facility and are currently on a month-to-month basis. We have a lab facility in Durham, North Carolina, where we lease 8,495 square feet of office space. In April 2007, we entered into a five-year lease agreement for that facility; thereafter, we were on a month-to-month basis until December 2013 when we signed a new one year lease for this space. In May 2013, we entered into a three-year lease agreement for 1,657 square feet of administrative office space in Durham, North Carolina.
We believe that our existing facilities are adequate for our current needs. We may seek to negotiate new leases or look for additional or alternate space for our operations. We believe that appropriate alternative space is readily available at similar rents.
52
We are not currently a party to any legal proceedings and we are not aware of any claims or actions pending or threatened against us. In the future, we might from time to time become involved in litigation relating to claims arising from our ordinary course of business.
| Item 4. | Mine Safety Disclosures |
Not applicable
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market Information for Common Stock
Our common stock has been listed on the NASDAQ Capital Market under the symbol “RGDO” since August 22, 2013. Prior to that time, there was no public market for our stock. The following table sets forth for the indicated periods the high and low intra-day sales prices per share for common stock on the NASDAQ Global Market.
| | | | | | | | |
| | | High | | | Low | |
Third Quarter 2013(since August 22, 2013) | | $ | 9.39 | | | $ | 4.27 | |
Fourth Quarter 2013 | | $ | 7.10 | | | $ | 4.41 | |
Holders
As of February 28, 2014, there were approximately 45 shareholders of record of our common stock. On March 10, 2014 the closing price of our common stock, as reported by NASDAQ, was $11.30 per share.
Dividend Policy
We have never declared or paid any cash dividends on our common stock, and currently do not plan to declare cash dividends on shares of our common stock in the foreseeable future. We expect that we will retain all of our available funds and future earnings, if any, for use in the operation and expansion of our business. Our loan agreement with Comerica Bank prohibits us from paying cash dividends on our common stock and the terms of any future loan agreement we enter into or any debt securities we may issue are likely to contain similar restrictions on the payment of dividends. Subject to the foregoing, the payment of cash dividends in the future, if any, will be at the discretion of our board of directors and will depend upon such factors as earnings levels, capital requirements, restrictions imposed by applicable law, our overall financial condition and any other factors deemed relevant by our board of directors.
Stock Options and Common Stock Issuances
Since January 1, 2013, the Company granted stock options under its equity compensation plans to purchase an aggregate of 1,918,860 shares of our common stock, net of cancellations at a weighted-average exercise price of $4.25 per share, to certain employees, consultants and directors.
Since January 1, 2013, the Company issued and sold to certain employees an aggregate of 7,602 shares of our common stock upon the exercise of options under the 2004 Plan at a weighted average exercise price of $4.37 per share.
53
Securities Act Exemptions
We deemed the grants of stock options and issuances of common stock upon exercise of such options described above under “—Stock Options and Common Stock Issuances” to be exempt from registration under the Securities Act in reliance on Rule 701 of the Securities Act as offers and sales of securities under compensatory benefit plans and contracts relating to compensation in compliance with Rule 701. Each of the recipients of securities in any transaction exempt from registration either received or had adequate access, through employment, business or other relationships, to information about us.
All certificates representing the securities issued in the transactions described in this Item 5 included appropriate legends setting forth that the securities had not been offered or sold pursuant to a registration statement and describing the applicable restrictions on transfer of the securities. There were no underwriters employed in connection with any of the transactions set forth in this Item 5.
Equity Compensation Plan Information
The following table provides information as of December 31, 2013 regarding shares of our common stock that may be issued under our existing equity compensation plans, including our 2004 Plan and 2013 Plan.
| | | | | | | | | | | | |
| | | Equity Compensation Plan Information | |
| | | Number of securities
to be issued upon
exercise
of outstanding options
and rights | | | Weighted
Average
exercise price of
outstanding
options
and rights | | | Number of securities
remaining available for
future issuance under
equity
compensation plan
(excluding outstanding
options and rights) | |
Equity compensation plans approved by security holders(1) | | | 3,272,847 | | | $ | 5.38 | | | | 191,222 | (2) |
| (1) | Consists of the 2004 Plan, the 2013 Plan and the ESPP Plan. |
| (2) | Includes shares available for future issuance under the 2013 Plan and the 2013 ESPP Plan. |
54
Use of Proceeds from Initial Public Offering of Common Stock
During the third quarter of 2013, we completed our IPO issuing 11,671,500 shares of common stock, inclusive of the exercise of the underwriters’ overallotment option, at a price of $4.00 per share, resulting in net proceeds to us of approximately $41.1 million, after deducting $3.3 million of underwriting discounts and commissions and $2.3 million of offering-related expenses. The offer and sale of all of the shares in the offering were registered under the Securities Act pursuant to a registration statement on Form S-1, which was declared effective on August 21, 2013 (File No. 333-188209). Cowen and Company, LLC and BMO Capital Markets Corp. acted as book-running managers for the offering and as representatives of the underwriters.
No offering costs were paid directly or indirectly to any of our director or officers or persons owning ten percent or more of any class of our equity securities or to any other affiliates, other than payments in the ordinary course of business to officers for salaries and to non-employee directors as compensation for board or board committee service. There has been no material change in the planned use of proceeds from our IPO as described in the final prospectus dated August 22, 2013 filed with the Securities and Exchange Commission pursuant to Rule 424(b) under the Securities Act on August 22, 2013. The proceeds were used to fund our cash basis losses from operations (approximately $34.9 million) for the year ended December 31, 2013.
| Item 6. | Selected Financial Data |
Per §229.301 of Regulation S-K, the Company, designated a Smaller Reporting Company as defined in Section §229.10(f)(1) of Regulation S-K, is not required to provide the disclosure required by this Item.
55
| ITEM 7. | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
Management’s Discussion and Analysis of Financial Condition and Results of Operations is intended to help the reader understand the results of operations and financial condition of the Company. The Management’s Discussion and Analysis of Financial Condition and Results of Operations should be read in conjunction with our audited consolidated financial statements and notes thereto for the year ended December 31, 2013. In addition to historical information, this Annual Report on Form 10-K contains forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended, and Section 21E of the Securities Exchange Act of 1934, as amended, which are intended to be covered by the safe harbors created thereby. See “Cautionary Note Regarding Forward-Looking Statements” in this report. Our actual results and the timing of events could differ materially from those discussed in our forward-looking statements as a result of many factors, including those set forth under the “Part I – Item 1A Risk Factors” section and elsewhere in this report, as well as, in other reports and documents we file with the Securities and Exchange Commission from time to time. Except as required by law, we undertake no obligation to update any forward-looking statements to reflect events or circumstances occurring after the date of this Annual Report on Form 10-K.
Overview
We are a biopharmaceutical company focused on the discovery and development of novel, first-in-class, actively controllable antithrombotic drug systems for acute and sub-acute cardiovascular indications. We are pioneering the discovery and development of two-component drug systems consisting of a therapeutic aptamer and its specific active control agent. Our actively controllable product candidates have the potential to improve outcomes, enhance the patient experience and reduce overall treatment costs.
Our lead product candidate, REG1, consists of pegnivacogin, a highly potent and selective anticoagulant, and anivamersen, its specific active control agent. We are developing REG1 as an anticoagulant for use in patients with a wide variety of cardiovascular conditions undergoing percutaneous coronary intervention, or PCI, a hospital-based procedure used to mechanically open or widen obstructed coronary arteries. Interventional cardiologists performing PCIs must consider the risk of major bleeding events in determining the level of anticoagulation administered to patients to prevent ischemic events, including death, stroke, myocardial infarction, or MI, or the need for revascularization of the artery. As the anticoagulant effect of existing drugs persists long after administration, intervention cardiologists are forced to make a compromising medical decision because they lack the means to simultaneously reduce the risks of ischemic and major bleeding events. In 2005, we filed an investigational new drug application, or IND, for the use of REG1 in this initial indication. We believe that REG1 has the potential to become the standard of care for anticoagulation therapy for patients undergoing PCI and other cardiovascular procedures because it gives the physician precise, on-demand control over anticoagulation levels. REG1 is the first and only anticoagulant to demonstrate a reduction in both ischemic and major bleeding events in a clinical trial for PCI. In our clinical trials, REG1 demonstrated a rapid and predictable anticoagulant effect that was precisely modulated or completely reversible in real time. In our randomized, partially blinded, dose-ranging Phase 2b trial involving 640 subjects, or the RADAR trial, when compared to standard of care heparin, REG1 demonstrated both a rapid and predictable anticoagulant effect and ability to precisely modulate or eliminate that effect in real time, as well as, other important clinical and pharmacoeconomic benefits. Based on these clinical results and after discussion with the U.S. Food and Drug Administration, or FDA, and the European Medicines Agency, or EMEA, in September of 2013, we initiated a single, open-label, 13,200 subject Phase 3 trial of REG1, or the REGULATE-PCI trial, in patients undergoing PCI procedures other than for the treatment of ST elevation myocardial infarctions. REGULATE-PCI, if successful, will serve as the basis for product registration applications worldwide. We believe that REG1 has potential use in other PCI and interventional cardiovascular procedures, such as open heart surgery, or OHS, PCI as a treatment for ST segment elevation myocardial infarction as well as transcatheter aortic valve replacement or implantation, or TAVI.
We completed our initial public offering (“IPO”) in August 2013. Inclusive of the underwriters’ exercise of the over-allotment option in connection with the IPO in September 2013, we issued 11,671,500 shares of common stock at a price of $4.00 per share, resulting in net proceeds of approximately $41.1 million, after deducting underwriting discounts of $3.3 million and offering costs of $2.3 million. Pursuant to the IPO all shares of convertible preferred stock then outstanding automatically converted into an aggregate of 9,396,767 shares of common stock.
In 2014, we sold 4,000,000 shares of our common stock at a purchase price of $5.00 per share to certain accredited and institutional investors (the “2014 Private Placement”). The net proceeds of approximately $ 18.7 million from this offering will be used for general corporate and working capital purposes, including the ongoing Phase 3 REGULATE-PCI clinical trial.
56
We are not profitable and do not expect to be profitable in the foreseeable future. We have suffered negative cash flows from operating activities of $34.7 million during the year ended December 31, 2013 and a net accumulated deficit of $145.0 million since inception as of December 31, 2013. We have devoted most of our financial resources to research and development, including our preclinical development activities and clinical trials. We have not completed development of any product candidate and we have therefore not generated any revenues from product sales. As a result, we expect to continue to incur net losses and negative cash flows for the foreseeable future. These net losses and negative cash flows have had, and will continue to have, an adverse effect on our stockholders’ equity and working capital. Our recurring losses from operations raise substantial doubt about our ability to continue as a going concern, and as a result, our independent registered public accounting firm included an explanatory paragraph in its report on our financial statements as of and for the year ended December 31, 2013 with respect to this uncertainty. Due to the numerous risks and uncertainties associated with pharmaceutical product development, we are unable to accurately predict the timing or amount of increased expenses or when, or if, we will be able to achieve or maintain profitability.
Financial Operations Overview
Revenue
To date, we have not generated any product revenue. Our ability to generate product revenue, which we do not expect will occur for several years, if ever, will depend heavily on the successful development and eventual commercialization of our lead product candidate, REG1.
Research and Development Expenses
Research and development expenses consist of the costs associated with our research and discovery activities, conducting preclinical studies and clinical trials and activities related to regulatory filings. Our research and development expenses consist of:
| | • | | employee salaries and related expenses, which include all compensation benefits for the personnel involved in our drug discovery and development activities, including stock based compensation; |
| | • | | external research and development expenses incurred under agreements with third party AROs and CROs and investigative sites; |
| | • | | clinical trial supplies when used or upon determination that they have no alternative future use and clinical trial supplies shipped to clinical sites for use in clinical studies; |
| | • | | license fees for and milestone payments related to in-licensed products and technologies; and |
| | • | | overhead costs related to facilities, depreciation, and supplies. |
We expense research and development costs as incurred, with the exception of materials purchased and/or manufactured for use in clinical trials. We capitalize clinical trial supplies, which are comprised of materials that will be used in our clinical trials that also have an alternative future use in either ongoing or future clinical research or development projects. Capitalized clinical trial supplies that are determined to be unsuitable for future use are immediately expensed to research and development; otherwise, clinical trial supplies are expensed to research and development when shipped to clinical sites for use in clinical studies or when used in other research and development projects. Costs for clinical agreements, including ARO and CRO contracts, are recognized based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations, or information provided by vendors on their actual costs incurred. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in our financial statements as prepaid or accrued expenses.
Conducting a significant amount of research and development is central to our business model. Product candidates in late stages of clinical development generally have higher development costs than those in earlier stages of clinical development, primarily due to the increased size and duration of late stage clinical trials. We expect our research and development expenses to increase in future periods for the foreseeable future as we seek to complete development of our lead product candidate, REG1, and to further develop our other product candidates.
57
We incurred aggregate research and development expenses of approximately $26.5 million and $8.0 million for the years ended December 31, 2013 and 2012, respectively, and $108.5 million since inception as of December 31, 2013. We expect to incur increased research and development expenses primarily related to our REGULATE-PCI trial.
We track direct external development expenses and direct personnel expenses on each indication for our product candidates. Substantially all of our research and development expenses for REG1 have related to its initial indication, although we expect certain of the data obtained will support development of additional REG1 indications as well as the development of REG2. Indirect expenses, such as, overhead costs related to facilities, depreciation, and small supplies are not allocated to specific product candidates or indications. The following table is a summary of our research and development expenses for the years ended December 31, 2013 and 2012 and for the period from inception to December 31, 2013 (in thousands):
| | | | | | | | | | | | |
| | | Years ended
December 31 | | | Period from Inception
(December 19, 2001) to
December 31, 2013 | |
| | | 2013 | | | 2012 | | |
REG1 | | $ | 23,800 | | | $ | 5,699 | | | $ | 89,029 | |
REG3 | | | 516 | | | | 367 | | | | 4,694 | |
REG2 | | | 507 | | | | 432 | | | | 6,200 | |
Other | | | 24 | | | | — | | | | 24 | |
| | | | | | | | | | | | |
Total direct expenses | | | 24,847 | | | | 6,498 | | | | 99,947 | |
Indirect expenses | | | 1,695 | | | | 1,508 | | | | 8,556 | |
| | | | | | | | | | | | |
Total research and development expense | | $ | 26,542 | | | $ | 8,006 | | | $ | 108,503 | |
| | | | | | | | | | | | |
The successful development of our clinical and preclinical product candidates is highly uncertain. At this time, we can only reasonably estimate the nature, timing and costs of the efforts that will be necessary to complete the remainder of the development of any of our product candidates or the period, if any, in which material net cash inflows from those product candidates may commence. Our estimates are based on reasonable assumptions, past performance, experience and existing contracts. However, unforeseen changes may occur at any time due to the numerous risks and uncertainties associated with developing drugs, including the uncertainty of:
| | • | | the number of sites included in the trials; |
| | • | | the number of countries included in the trials; |
| | • | | the ability to recruit subjects to participate in the trial; |
| | • | | the per subject trial costs; |
| | • | | the length of time required to enroll suitable subjects, achieve interim milestones and complete clinical trials; and |
| | • | | the cost and timeliness of obtaining clinical trial supplies. |
Development timelines, probability of success and development costs vary widely. As a result of the uncertainties discussed above, we anticipate that we will make determinations as to which product candidates and indications to pursue and how much funding to direct to each product candidate and indication on an ongoing basis. Accordingly, we cannot currently estimate with any degree of certainty the amount of time or money that we will be required to expend in the future on the research and development of our product candidates.
General and Administrative Expenses
General and administrative expenses consist principally of salaries and related benefit costs, including stock-based compensation for administrative personnel. Other general and administrative expenses include facility costs, and professional fees for legal, consulting, auditing and tax services. We anticipate that our general and administrative expenses will increase in future periods to support increases in our research and development activities and as a result of increased headcount, expanded infrastructure, and increased legal, compliance, accounting and investor and public relations expenses associated with being a public company.
58
Interest Income (Expense)
Interest income consisted of fair value adjustments related to our warrant liability, as well as, interest earned on our cash and cash equivalents. We expect our interest income earned on cash and cash equivalents to increase as we invest the net proceeds from our IPO and 2014 Private Placement pending their use in our operations. Interest expense in 2013 primarily related to the Comerica Loan (as described more fully under “Liquidity and Capital Resources”) and to our loan with MidCap Financial SBIC, LP, or MidCap, which was paid in full with proceeds from the Comerica Loan. Interest expense in 2012 related to interest incurred on our MidCap loan and on our convertible notes.
Critical Accounting Policies and Significant Judgments and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenue and expenses and the disclosure of contingent assets and liabilities in our financial statements. We evaluate our estimates and judgments, including those related to accrued expenses and share-based compensation, on an ongoing basis. We base our estimates on historical experience, known trends and events and various other factors that are believed to be reasonable under the circumstances, the results of which form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates.
Our significant accounting policies are described in more detail in the notes to our audited consolidated financial statements included in this report. We believe the following accounting policies to be most critical to the judgments and estimates used in preparation of our financial statements and such policies have been reviewed and discussed with our audit committee.
Accrued Expenses
As part of the process of preparing our financial statements, we are required to estimate accrued expenses. This process involves reviewing open contracts and purchase orders, communicating with applicable vendor personnel to identify services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual cost. The majority of our service providers invoice us monthly in arrears for services performed. We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us. We periodically confirm the accuracy of our estimates with the service providers and make adjustments if necessary. Examples of estimated accrued expenses include:
| | • | | fees paid to CROs in connection with clinical trials; |
| | • | | investigative site costs in connection with clinical trials; |
| | • | | milestone payments; and |
| | • | | unpaid salaries, wages and benefits. |
We accrue our expenses related to clinical trials based on our estimates of the services received and efforts expended pursuant to contracts with multiple research institutions and CROs that conduct and manage clinical trials on our behalf. The financial terms of these agreements are subject to negotiation, vary from contract to contract and may result in uneven payment flows. Payments under some of these contracts depend on factors such as the successful enrollment of patients and the completion of clinical trial milestones. In accruing service fees, we estimate the time period over which services will be performed and the level of effort to be expended in each period. If the actual timing of the performance of services or the level of effort varies from our estimate, we will adjust the accrual accordingly. If we do not identify costs that we have begun to incur or if we underestimate or overestimate the level of services performed or the costs of these services, our actual expenses could differ from our estimates. We do not currently anticipate the future settlement of existing accruals to differ materially from our estimates.
Liability–classified warrants
In connection with the funding of Tranche One from Comerica Bank (the “Comerica Loan”), we issued a warrant to Comerica Bank, or the Comerica Warrant, to purchase 156,250 shares of our Series E Preferred Stock at a price of $0.72 per share, or the Warrant Price, subject to adjustment for stock splits, combinations, reclassifications or exchanges and certain dilutive issuances. After giving effect to our IPO and reverse stock-split, the Comerica Warrant was adjusted to a warrant to purchase 9,356 shares of our common stock at a price of $12.02 per share (the “Adjusted Warrant Price”), respectively.
59
In accounting for the Comerica Loan, the loan was separated into debt and warrant liability components. We utilized the Binomial pricing model to determine the fair value of the warrant liability component (see Note 3). The carrying amount of the debt component was determined by deducting the fair value of the warrant liability component from the par value of the Comerica Loan as a whole. The excess of the principal amount of the Comerica Loan component over its carrying amount, referred to as the debt discount, is amortized to interest expense over the term of the loan. The warrant liability component is re-measured at each reporting date and changes in the fair value of the warrant liability are recorded as interest expense or interest income, as applicable.
Stock-based Compensation
In accordance with FASB ASC Topic 718, Stock Compensation, as modified or supplemented, we measure compensation cost for share-based payment awards granted to employees and non-employee directors at fair value using the Black-Scholes option-pricing model. We recognize compensation expense on a straight-line basis over the service period for awards expected to vest. Share-based compensation cost related to share-based payment awards granted to non-employees is adjusted each reporting period for changes in the fair value of our common stock until the measurement date. The measurement date is generally considered to be the date when all services have been rendered or the date that options are fully vested.
We use the Black-Scholes-Merton option pricing model to determine the fair value of our stock options. The determination of the fair value of stock-based payment awards on the date of grant using an option pricing model is affected by our stock price, as well as assumptions regarding a number of complex and subjective variables. These variables include our expected stock price volatility over the term of the awards, risk-free interest rate, actual employee exercise behaviors and expected dividends.
The following table shows the weighted average assumptions used to value stock options on the date of grant, as follows:
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | Year ended December 31, | | | Period from Inception
(December 19, 2001) to
December 31, 2013 | |
| | | 2013 | | | 2012 | | |
| | | Employee | | | Non-
Employees | | | Employee | | | Non-
Employees | | | Employee | | | Non-
Employees | |
Expected stock price volatility | | | 46.90 | % | | | 45.0 | % | | | 44.00 | % | | | 44.00 | % | | | 44.08 | % | | | 41.13 | % |
| | | | | | |
Risk-free interest rate | | | 1.04 | % | | | 0.37 | % | | | 1.38 | % | | | 1.38 | % | | | 1.64 | % | | | 1.21 | % |
| | | | | | |
Expected life of option (in years) | | | 3.69 | | | | 2.00 | �� | | | 6.25 | | | | 6.25 | | | | 4.44 | | | | 3.38 | |
| | | | | | |
Estimated dividend yield | | | 0.00 | % | | | 0.00 | % | | | 0.00 | % | | | 0.00 | % | | | 0.00 | % | | | 0.00 | % |
| | | | | | |
Weighted-average grant date fair value per share | | | 1.91 | | | | 1.49 | | | | 1.67 | | | | 1.67 | | | | 2.32 | | | | 1.99 | |
Expected stock price volatility was calculated based on the weighted-average of historical information of similar public entities. We will continue to use a weighted-average approach using other similar public entities’ volatility information until our historical volatility is relevant to measure expected volatility for future option grants. The risk-free rate was based on the U.S. Treasury yield curve in effect at the time of grant commensurate with the expected life assumption. The average expected life was determined based on anticipated exercise strategy and cancellation behavior for employees and nonemployees, primarily non-employee directors. Effective in 2013, a forfeiture rate of 1% and 0% was used for employees and nonemployees, respectively. We have not paid and do not anticipate paying cash dividends; therefore, the expected dividend rate was assumed to be 0%.
Total stock-based compensation expense recognized based on the total grant date fair value of options vested and expected to vest was approximately $988,000 and $703,000 for the years ended December 31, 2013 and 2012, respectively, and $5.6 million for the period from inception to December 31, 2013. Due to the valuation allowance against our net deferred tax asset, we have never recognized a tax benefit for stock based compensation.
As of December 31, 2013, approximately $2.9 million of total unrecognized compensation cost related to unvested share options is expected to be recognized over a weighted-average period of 2.8 years.
60
Clinical Trial Supplies
We capitalize materials that will be used in our REG I clinical trials that also have an alternative future use in either ongoing or future clinical research and development projects. Clinical trial supplies may comprise material used to manufacture active pharmaceutical ingredients (“API”) used to develop our product candidates, in-process or completed API, in-process or completed unlabeled finished drug product and labeled finished drug product. Clinical trial supplies are stated at cost, using the first-in, first-out method (“FIFO”), and are reported in the accompanying consolidated balance sheets in other current assets. Clinical trial supplies that are determined to be unsuitable for future use are immediately expensed; otherwise clinical trial supplies are expensed when shipped to clinical sites for use in clinical studies or when used in other research and development projects.
Results of Operations
Years Ended December 31, 2013 and 2012
The following table sets forth certain information concerning our results of operations for the periods shown (in thousands):
| | | | | | | | | | | | |
| | | Years Ended
December 31, | | | Increase
(Decrease) | |
| | | 2013 | | | 2012 | | |
Operating expenses: | | | | | | | | | | | | |
Research and development | | $ | (26,542 | ) | | $ | (8,006 | ) | | $ | 18,536 | |
General and administrative | | | (7,297 | ) | | | (4,157 | ) | | | 3,140 | |
| | | | | | | | | | | | |
Total operating expenses | | | (33,839 | ) | | | (12,163 | ) | | | 21,676 | |
| | | | | | | | | | | | |
Other (expense) income: | | | | | | | | | | | | |
Interest income | | | 80 | | | | 5 | | | | 75 | |
Interest expense | | | (680 | ) | | | (904 | ) | | | (224 | ) |
| | | | | | | | | | | | |
Total other (expense) income | | | (600 | ) | | | (899 | ) | | | (299 | ) |
| | | | | | | | | | | | |
Net loss | | $ | (34,439 | ) | | $ | (13,062 | ) | | $ | 21,377 | |
| | | | | | | | | | | | |
Research and Development Expenses
Research and development expenses increased by $18.5 million for the year ended December 31, 2013 compared to the year ended December 31, 2012 primarily due to the start of the REGULATE-PCI trial, including commencement of services provided by AROs and CROs, and due to $1.5 million in license agreement expense for obligations triggered upon the commencement of our REGULATE-PCI trial in September 2013.
Effective September 26, 2013, we reduced our research and development workforce by eliminating 5 of our 32 full-time employees. The majority of the affected employees worked in drug discovery roles at our laboratory facility in North Carolina. The goal of our reduction in workforce was to enable us to focus management and financial resources on advancing our lead product candidate, REG1, in our REGULATE-PCI trial. In connection with the reduction in workforce, severance expense of approximately $250,000 was charged to research and development expense in the accompanying consolidated statement of comprehensive loss for the year ended December 31, 2013. We believe that the reduction in workforce will result in annualized savings of approximately $625,000 beginning in the second quarter of 2014.
General and Administrative Expenses
General and administrative expenses increased by $3.1 million for the year ended December 31, 2013 compared to the year ended December 31, 2012. The increase was primarily due to increased accounting, legal, insurance and other administrative costs associated with becoming a public company, an increase of $437,000 for employee incentive compensation in 2013 compared to 2012, and to severance expense of $417,000 related to the termination of an executive officer in 2013.
61
Other Income (Expense)
Interest income increased by $75,000 for the year ended December 31, 2013, compared to the year ended December 31, 2012 as a result of fair value adjustments related to our warrant liability and to interest earned on the IPO proceeds.
Interest expense decreased by $224,000 for the year ended December 31, 2013, compared to the year ended December 31, 2012 primarily due to the absence of the MidCap Financial loan final fee payment of $192,000 and interest expense related to convertible notes payable.
Liquidity and Capital Resources
Sources of Liquidity
To date, we have not generated any product revenue. We have funded our operations to date through sales of our equity and debt securities, bank borrowings and government grants. As of December 31, 2013, we had $30.7 million in cash and cash equivalents.
We completed our IPO in August 2013. Inclusive of the underwriters’ exercise of the over-allotment option in connection with the IPO in September 2013, we issued 11,671,500 shares of common stock at a price of $4.00 per share, resulting in net proceeds of approximately $41.1 million, after deducting underwriting discounts of $3.3 million and offering costs of $2.3 million. Upon closing of the IPO, all shares of convertible preferred stock then outstanding automatically converted into an aggregate of 9,396,767 shares of common stock.
From inception through December 31, 2013, we received net cash proceeds of $5.8 million from the sale of our Series A Preferred Stock, $19.9 million from sales of our Series B Preferred Stock, $23.0 million from sales of our Series C Preferred Stock, $51.2 million from sales of our Series D Preferred Stock and $32.3 million from sales of our Series E Preferred Stock. Convertible note proceeds of $15.1 million are included in these totals, as well as convertible notes that have been converted to convertible preferred stock as of December 31, 2013.
In 2014, we sold 4,000,000 shares of our common stock at a purchase price of $5.00 per share to certain accredited and institutional investors (the “2014 Private Placement”). The net proceeds of approximately $18.7 million from this offering will be used for general corporate and working capital purposes, including the ongoing Phase 3 REGULATE-PCI clinical trial.
Comerica Loan
In May 2013, we entered into a Loan and Security Agreement, or the Loan Agreement, with Comerica Bank, or Comerica. Pursuant to the terms of the Loan Agreement, we were initially eligible to borrow $4.5 million in an initial tranche, or Tranche One. Upon Comerica’s receipt of evidence satisfactory to Comerica that (i)��the 1,000 patient interim analysis in the REGULATE-PCI study is successful and performed by April 30, 2014 and (ii) upon our completion of the IPO and receipt of net proceeds of at least $50 million prior to September 30, 2013, we had the option to borrow an additional $4 million in the second tranche, or Tranche Two. Since the Tranche Two conditions were not satisfied, Tranche Two is solely at the discretion of Comerica.
The Comerica loan bears interest at Comerica’s Prime Reference Rate (as defined in the Loan Agreement) subject to a floor of 30 day LIBOR plus 250 basis points plus 4.0%. The Comerica loan is interest-only until September 1, 2014. We must repay the principal amount in nine approximately equal consecutive monthly installments commencing on September 1, 2014. The loan matures on May 10, 2015.
In connection with the funding of Tranche One, we issued a warrant to Comerica, or the Comerica Warrant, to purchase 156,250 shares of our Series E Preferred Stock at a price of $0.72 per share, or the Warrant Price, subject to adjustment for stock splits, combinations, reclassifications or exchanges and certain dilutive issuances. After giving effect to our IPO and reverse stock-split, the Comerica Warrant was adjusted to a warrant to purchase 9,356 shares of our common stock at a price of $12.02 per share (the “Adjusted Warrant Price”). If Comerica, in its sole discretion, permits us to borrow the additional $4 million in Tranche Two, the Comerica Warrant will become exercisable for an additional number of shares of our common stock equal to 5,988 divided by the Adjusted Warrant Price.
The proceeds of Tranche One were used to repay in full amounts outstanding under our loan and security agreement with MidCap Financial SBIC, LP which has been terminated, and for general operating purposes.
62
Under the terms of the Loan Agreement, we granted Comerica a first priority security interest in substantially all of our assets other than our intellectual property. The Loan Agreement does not contain any ongoing financial covenants.
The Loan Agreement provides that upon the occurrence of and during a period of default as defined therein, interest on the loan will accrue at a penalty rate. Upon the occurrence and during the continuance of a default, Comerica may, at its election, make all obligations under the Loan Agreement immediately due and payable, cease advancing money or extending credit, exercise its right of setoff, foreclose on our assets, dispose of collateral at a public or private sale, and exercise any other remedies available to a secured creditor at law or in equity.
63
Cash Flows
Our net cash flow from operating, investing and financing activities for the periods below were as follows (in thousands):
| | | | | | | | |
| | | Years Ended
December 31, | |
| | | 2013 | | | 2012 | |
Net cash provided by (used in): | | | | | | | | |
Operating activities | | $ | (34,736 | ) | | $ | (14,229 | ) |
Investing activities | | | (548 | ) | | | (357 | ) |
Financing activities | | | 51,208 | | | | 20,115 | |
| | | | | | | | |
Net increase in cash and cash equivalents | | $ | 15,924 | | | $ | 5,529 | |
| | | | | | | | |
Operating Activities
Net cash used in operating activities was $34.7 million for the year ended December 31, 2013 and $14.2 million for the year ended December 31, 2012. Net cash used in operating activities for the year ended December 31, 2013 principally resulted from our net loss of $34.4 million incurred primarily due to our higher level of expenditures for the REGULATE-PCI trial. Net cash used in operating activities also resulted from a higher level of payments made for clinical supplies, contract signing fees, and other advance payments related to the REGULATE-PCI trial, partially offset by increases in accounts payable and accrued expenses. Net cash used in operating activities for the year ended December 31, 2012 principally resulted from our net loss of $13.1 million and a higher level of clinical supplies, partially offset by a decrease in prepaid expenses.
Investing Activities
Net cash used in investing activities was $548,000 for the year ended December 31, 2013 and $357,000 for the year ended December 31, 2012. Net cash used in investing activities for the years ended December 31, 2013 and 2012 principally resulted from the acquisition of intellectual property rights.
Financing Activities
Net cash provided by financing activities was $51.2 million for the year ended December 31, 2013 and $20.1 million for the year ended December 31, 2012. Net cash provided by financing activities for the year ended December 31, 2013 resulted primarily from $41.1 million in net proceeds from the IPO, and to $10.2 million in net proceeds from the sale of Series E Preferred Stock. Net cash provided by financing activities for the year ended December 31, 2012 principally resulted from $15.0 million in net proceeds from the sale of preferred stock and the receipt of $6.8 million in proceeds from issuing convertible notes payable, partially offset by $1.5 million in note repayments on the MidCap loan.
Funding Requirements
We have not completed development of any of our product candidates. We expect to continue to incur significant expenses and increasing operating losses for at least the next several years. We anticipate that our expenses will increase substantially as we:
| | • | | continue our REGULATE-PCI trial; |
| | • | | continue the research and development activities for our other product candidates; |
| | • | | seek to discover additional product candidates; |
| | • | | seek regulatory approvals for our product candidates that successfully complete clinical trials; |
| | • | | establish a sales, marketing and distribution infrastructure if we do not secure a strategic partner to commercialize products for which we may obtain regulatory approval; |
| | • | | increase manufacturing capabilities in preparation for commercial launch of any such products; and |
| | • | | add operational, financial and management information systems and personnel, including personnel to support our product development, planned commercialization efforts and our operation as a public company. |
64
We believe that our existing cash and cash equivalents plus the proceeds from the 2014 Private Placement together with our existing working capital, will be sufficient to fund the REGULATE-PCI trial through the second interim analysis, which is expected to occur during the third quarter of 2014. We need to raise additional financing in the near term to operate our business and fund our REGULATE-PCI trial to completion, which financing may not be available on favorable terms, or at all. Our forecast of the period of time through which our financial resources will be adequate to support our operations is a forward-looking statement and involves risks and uncertainties, and actual results could vary as a result of a number of factors, including the factors discussed in the section entitled “Risk Factors” and elsewhere in this report. We have based this estimate on assumptions that may prove to be wrong, and we could utilize our available capital resources sooner than we currently expect.
Our future funding requirements, both near and long-term, will depend on many factors, including, but not limited to:
| | • | | the initiation, progress, timing, costs and results of preclinical studies and clinical trials for our product candidates and potential product candidates, including our REGULATE-PCI trial, and the continued development of our other product candidates; |
| | • | | the number and characteristics of product candidates that we pursue; |
| | • | | the terms and timing of any future collaboration, licensing or other arrangements that we may establish; |
| | • | | the outcome, timing and cost of regulatory approvals; |
| | • | | the cost of obtaining, maintaining, defending and enforcing intellectual property rights, including patent rights; |
| | • | | the effect of competing technological and market developments; |
| | • | | the cost and timing of completing commercial-scale outsourced manufacturing activities; |
| | • | | market acceptance of any product candidates for which we may receive regulatory approval; |
| | • | | the cost of establishing sales, marketing and distribution capabilities for any product candidates for which we may receive regulatory approval; and |
| | • | | the extent to which we acquire, license or invest in businesses, products or technologies. |
Until we can generate a sufficient amount of revenue from our product candidates, if ever, we expect to finance future cash needs through public or private equity offerings, debt financings or corporate collaborations and licensing arrangements. Additional funds may not be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available, we may be required to delay our REGULATE-PCI trial, reduce the scope of or eliminate one or more of our research or development programs or our commercialization efforts. To the extent that we raise additional funds by issuing equity securities, our stockholders may experience additional dilution, and debt financing, if available, may involve restrictive covenants. To the extent that we raise additional funds through collaborations and licensing arrangements, it may be necessary to relinquish some rights to our technologies or our product candidates or grant licenses on terms that may not be favorable to us. We may seek to access the public or private capital markets whenever conditions are favorable, even if we do not have an immediate need for additional capital at that time.
We do not expect REG1 to be commercially available before 2017, if at all. The net proceeds of our IPO and 2014 Private Placement will not be sufficient for us to complete the REGULATE-PCI trial and we will need to raise substantial additional capital to complete the development and commercialization of REG1. We also will need to raise substantial additional capital to complete the development and commercialization of REG1 for additional indications and for our other product candidates. Since successful development of our product candidates is uncertain, we are unable to estimate the actual funds required to complete research and development and commercialize our products under development.
65
Supply and Manufacturing Agreements
In July 2006, we entered into a supply and service agreement with Agilent Technologies, Inc., which was amended in July 2011, for the manufacture of pegnivacogin and anivamersen bulk drug substance for use in the clinical development of REG1. Drug substances are manufactured pursuant to a Good Manufacturing Practices and a Quality Agreement entered into in May 2010. Manufacture of bulk drug substance lots is on a purchase order basis, with no minimum purchase obligation.
In December 2006, we entered into a license, manufacturing and supply agreement with Nektar Therapeutics, or Nektar, for the supply of polyethylene glycol, or PEG, used in the manufacture of pegnivacogin. We must provide Nektar with a rolling forecast of our anticipated requirements for the succeeding six quarters, with respect to precommercial supply, or eight quarters, with respect to commercial supply, with a required lead time to commit to and guarantee availability of the reagent at an agreed upon pricing structure, which is subject to revision on an annual basis.
In March 2012, we entered into a clinical supply agreement with Althea Technologies, Inc., for the formulation and packaging of pegnivacogin and anivamersen drug product for use in our clinical trials. Drug products are manufactured and the packaging is conducted pursuant to a Good Manufacturing Practices and a Quality Agreement entered into in January 2012. Formulation and packaging services are provided on a purchase order basis, with no minimum purchase obligation.
Clinical Agreements
We have various clinical trial agreements with AROs and CROs for planning, management and execution of clinical trials. The financial terms of these agreements are subject to negotiation, vary from contract to contract, and may result in uneven payment flows. These contracts generally provide for termination on notice.
Milestone and Other Obligations
Upon the commencement of our REGULATE-PCI trial which occurred in September 2013, we were obligated to make milestone payments of $500,000 to Duke University, or Duke, and $1.0 million to Archemix Corporation, or Archemix. We have paid $500,000 of such milestone obligations and the $1.0 million remaining balance is included in accrued expenses in the accompanying consolidated balance sheet as of December 31, 2013; accordingly, aggregate milestone obligations of $1.5 million are included in research and development expense in the accompanying statement of comprehensive loss for the year ended December 31, 2013. In addition, upon the filing of a new drug application, we are obligated to make additional milestone payments to Duke, Archemix and Nektar Therapeutics AL, Corporation. Given the uncertainty of the drug development process, we cannot be certain when, if ever, we will be required to make these milestone payments.
As a condition of closing the Series E Preferred Stock financing in December 2012, we assigned all intellectual property (“IP”) rights and titles in Armenia, Azerbaijan, Belarus, Georgia, Kazakhstan, Kyrgyzstan, Moldova, Russia, Tajikistan, Turkmenistan, Ukraine and Uzbekistan ( the “Covered Territory”) to Domain Russia Investments Limited (“DRI”). Additionally, we agreed to assist an affiliate of DRI with certain development support related to the development of the IP. Concurrently with the signing of this agreement, we agreed to make a payment to DRI up to a maximum amount of $1.2 million based on an independent appraiser’s valuation of the IP rights transferred. Such independent appraiser’s valuation was received during 2013, the result of which confirmed that we do not have any further obligation pursuant to this agreement.
License Agreements
In December 2012, in connection with its Series E Preferred Stock financing, we entered into a Technology Transfer Agreement, or the Tech Transfer Agreement, with DRI. In accordance with the terms of the Tech Transfer Agreement, in May 2013 we entered into a Clinical Development and Collaboration Agreement with NovaMedica pursuant to which we agreed to assist NovaMedica in the development and commercialization of our product candidates in the Covered Territory, as defined. NovaMedica is required to reimburse us for any out-of-pocket expenses incurred by us in providing this assistance, including travel-related expenses.
Lease Obligations
As of December 31, 2013, we did not have a long-term lease agreement in effect for our office in New Jersey; we are currently leasing this facility on a month-to-month basis. On May 1, 2013, we entered into a three-year lease for administrative office space in North Carolina. Annual rent under this lease is $39,000 for the first year, $40,000 for the second year and $41,000 for the third year. In December 31, 2013, we entered into a new one-year lease for our laboratory facility in North Carolina with an annual rent of approximately $156,000.
66
Tax Loss Carryforwards
As of December 31, 2013, the Company had estimated federal and state operating loss carryforwards of approximately $137.0 million. These federal and state operating loss carryforwards begin to expire in 2022 and 2017, respectively. The utilization of the federal net operating loss carryforwards may be subject to limitations under the rules regarding a change in stock ownership as determined by the Internal Revenue Code and state laws. Section 382 of the Internal Revenue Code of 1986, as amended, imposes annual limitations on the utilization of net operating loss (“NOL”) carry forwards, other tax carry forwards, and certain built-in losses upon an ownership change as defined by that section. In general terms, an ownership change may result from transactions that increase the aggregate ownership of certain stockholders in the Company stock by more than 50 percentage points over a three year testing period (“Section 382 Ownership Change”). If the Company has undergone a Section 382 Ownership change, an annual limitation would be imposed on certain of the Company’s tax attributes, including NOL and capital loss carry forwards, and certain other losses, credits, deductions of tax basis. As of December 31, 2013, the Company has not performed a formal study to determine whether there are Section 382 limitations that apply.
At December 31, 2013, we evaluated and assessed the expected near-term utilization of net operating loss carryforwards, book and taxable income trends, available tax strategies, and the overall deferred tax position. We believe that it is more likely than not that the benefit related to the deferred tax assets will not be realized, therefore we established the valuation allowance required as of December 31, 2013. If actual results differ from the assumptions made in our evaluation, we may record a change in the valuation allowance through income tax expense in the period such determination is made. We believe that our judgments and estimates are reasonable; however, actual results could differ. Our effective tax rate is zero due to continued taxable losses, which generate deferred tax assets which are offset in their entirety by related valuation allowances, due to the uncertainty in realizing these tax benefits. As such, we do not record quarterly accruals for corporate taxes. Deferred income tax assets and liabilities are calculated and reported at year end.
The Company did not have any accrued interest or penalties associated with any unrecognized tax positions at December 31, 2013, and there were no such interest or penalties recognized during the period since inception through December 31, 2013.
Off-Balance Sheet Arrangements
Since inception, we have not engaged in any off-balance sheet arrangements as defined in Item 303(a)(4) of Regulation S-K.
Recent Accounting Pronouncements
Section 107 of the Jumpstart our Business Startups Act of 2012, or the JOBS Act, provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. However, we are choosing to “opt out” of such extended transition period, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable.
| Item 7A. | Quantitative and Qualitative Disclosures about Market Risk |
Per §229.305 of Regulation S-K, the Company, designated a Smaller Reporting Company as defined in Section §229.10(f)(1) of Regulation S-K, is not required to provide the disclosure required by this Item.
| Item 8. | Financial Statements and Supplementary Data |
Our financial statements, together with the independent registered public accounting firm report thereon, are incorporated by reference from the applicable information set forth in Part IV Item 15, “Exhibits, Financial Statement Schedules” of this Annual Report on Form 10-K.
67
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
None.
| Item 9A. | Controls and Procedures |
Evaluation of Disclosure Controls and Procedures
As of December 31, 2013, our management, with the participation of our principal executive officer and principal financial officer, evaluated the effectiveness of our disclosure controls and procedures. The term “disclosure controls and procedures,” as defined in Rules 13a-15(e) and 15d-15(e) under the Exchange Act, means controls and other procedures of a company that are designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is recorded, processed, summarized and reported, within the time periods specified in the SEC’s rules and forms. Disclosure controls and procedures include, without limitation, controls and procedures designed to ensure that information required to be disclosed by a company in the reports that it files or submits under the Exchange Act is accumulated and communicated to the Company’s management, including its principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure. Management recognizes that any controls and procedures, no matter how well designed and operated, can provide only reasonable assurance of achieving their objectives and management necessarily applies its judgment in evaluating the cost benefit relationship of possible controls and procedures. Based on this evaluation, management concluded that our disclosure controls and procedures were not effective due to a material weakness in our internal control over financial reporting that was identified in connection with the preparation of our consolidated financial statements for the years ended December 31, 2011 and 2012 and is described in the following paragraph.
In connection with the preparation of our consolidated financial statements for the years ended December 31, 2012 and 2011, we identified past accounting errors, which resulted in the restatement of our previously issued financial statements. We and our independent registered public accounting firm identified a material weakness in internal control over financial reporting related to these items which required adjustment, specifically: (i) accounting for the purchase of supplies used in the production of our drug product, and (ii) accounting for purchase orders related to manufacturing services where work was contracted but not yet performed. A material weakness is defined as a deficiency, or a combination of deficiencies, in internal control over financial reporting, such that there is a reasonable possibility that a material misstatement of a Company’s annual or interim financial statements will not be prevented or detected on a timely basis by the Company’s internal controls.
We have implemented and are continuing to implement procedures and controls designed to remediate the material weakness and underlying significant deficiencies. Amongst other actions, we have recently hired two additional employees in accounting and finance; commenced implementation of enhanced review procedures; begun a comprehensive documentation of our internal controls and procedures; and implemented more formal procedures as to the evaluation of non-routine judgments and estimates. Although we believe these controls will be effective, we have not yet determined if we have successfully remediated the material weakness previously identified.
Notwithstanding the identified material weakness in internal controls over financial reporting, management believes the consolidated financial statements included in this Annual Report on Form 10-K fairly represent in all material respects our financial condition, results of operations and cash flows at and for the periods presented in accordance with U.S. GAAP.
Management’s Report on Internal Control over Financial Reporting
This annual report does not include a report of management’s assessment regarding internal control over financial reporting or an attestation report of the Company’s registered public accounting firm due to a transition period established by rules of the Securities and Exchange Commission for newly public companies.
Changes in Internal Control over Financial Reporting
Other than remediation efforts described above, there have been no changes in our internal control over financial reporting during the year ended December 31, 2013, that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
68
| Item 9B. | Other Information |
Not applicable.
PART III
Item 10. Directors, Executive Officers and Corporate Governance
The following table sets forth the name, age, position and tenure of each of our directors and executive officers as of March 1, 2014:
| | | | | | |
Name | | Age | | Position(s) | | Served as an
Officer or Director
Since |
| | | |
David J. Mazzo, Ph.D. | | 57 | | Chief Executive Officer and Director | | 2008 |
Michael A. Metzger | | 42 | | President and Chief Operating Officer | | 2013 |
Christopher E. Courts, CPA | | 44 | | Vice President, Finance | | 2006 |
Steven L. Zelenkofske, D.O., F.A.C.C. | | 55 | | Senior Vice President, Clinical and Medical Affairs and Chief Medical Officer | | 2009 |
| | | |
Alexander R. Giaquinto, Ph.D. | | 70 | | Senior Vice President, Regulatory Affairs and Quality Assurance and Chief Compliance Officer | | 2008 |
Christopher P. Rusconi, Ph.D. | | 45 | | Senior Vice President, Discovery/Preclinical Development and Chief Scientific Officer | | 2003 |
Dennis Podlesak(2)(4) | | 55 | | Chairman of the Board of Directors | | 2007 |
B. Jefferson Clark(1)(2) | | 57 | | Director | | 2004 |
Anton Gopka | | 31 | | Director | | 2012 |
Pierre Legault, MBA, CA, CPA(1) | | 53 | | Director | | 2013 |
Michael E. Mendelsohn, (3)(4) | | 58 | | Director | | 2013 |
P. Sherrill Neff(2) | | 62 | | Director | | 2005 |
Jesse Treu, Ph.D.(3)(4) | | 66 | | Director | | 2005 |
| | | |
Raphaël Wisniewski(1)(3) | | 43 | | Director | | 2009 |
| (1) | Member of the audit committee |
| (2) | Member of the compensation committee |
| (3) | Member of the nominating and corporate governance committee |
| (4) | Member of science committee |
69
Executive Officers
Our executive officers are elected by, and serve at the discretion of, our board of directors. The business experience for the past five years, and in some instances, for prior years, of each of our executive officers and directors is as follows:
David J. Mazzo, Ph.D.has served as our chief executive officer and as a member of our board of directors since August 2008. Dr. Mazzo has almost 30 years of experience in the pharmaceutical industry. Most recently, from March 2007 to April 2008, he was president and chief executive officer of Æterna Zentaris, Inc., a publicly held Canadian biopharmaceutical company. From 2003 until 2007, Dr. Mazzo served as president and chief executive officer of Chugai Pharma USA, LLC, a biopharmaceutical company which was the U.S. subsidiary of Chugai Pharmaceutical Co., Ltd. of Japan. Dr. Mazzo has also held senior management and executive positions in research and development at Schering-Plough Corporation, a publicly held pharmaceutical company that was subsequently acquired by Merck & Co., Inc., Hoechst Marion Roussel, Inc., the US subsidiary of Hoechst AG, which was subsequently acquired by Sanofi, a multinational pharmaceuticals company, and Rhone-Poulenc Rorer, Inc., a subsidiary of Rhone-Poulene SA, a French pharmaceuticals company, which was subsequently acquired by Hoechst AG. He currently serves on the boards of directors of pSivida Corp., a publicly held biotechnology company, in the role of non-executive chairman, and Avanir Pharmaceuticals, Inc., a publicly held biopharmaceutical company. Dr. Mazzo earned a B.A. in the Honors Program (Interdisciplinary Humanities) and a B.S. in Chemistry from Villanova University. In addition, Dr. Mazzo received his M.S. in chemistry and his Ph.D. degree in analytical chemistry from the University of Massachusetts, Amherst. He was also a research fellow at the Ecole Polytechnique Federale de Lausanne, Switzerland. We believe Dr. Mazzo possesses specific attributes that qualify him to serve as a member of our board of directors, including his insight and experience as our chief executive officer, which brings historic knowledge, operational expertise and continuity to our board of directors, his experience as chief executive officer and in other senior management roles at other pharmaceutical and biotechnology companies and his service on other boards of directors in the biopharmaceutical industry.
Michael A. Metzger has served as our president and chief operating officer since December 5, 2013. Prior to joining us, from April 2011 to November 2013, Mr. Metzger was executive vice president and chief operating officer at Mersana Therapeutics, Inc., a privately-held biopharmaceutical company. Prior to Mersana, from September 2006 to March 2011, Mr. Metzger held senior positions within business development and led mergers and acquisitions at Forest Laboratories, Inc., an international pharmaceutical manufacturer and marketer. Prior to Forest, from 2001 to 2006, Mr. Metzger served as vice president of corporate development at Onconova Therapeutics, Inc., a biopharmaceutical company focused in oncology. Prior to Onconova, Mr. Metzger was managing director at MESA Partners, a venture capital firm, from 1997 to 2001. Mr. Metzger served as a member on the board of directors of Response Genetics, Inc., a commercial stage company developing molecular diagnostic tests for oncology, from December 2010 to November 2013. In addition, Mr. Metzger has served as a director of various life sciences companies. Mr. Metzger holds a B.A. from George Washington University and an M.B.A. from the New York University Stern School of Business.
Christopher E. Courts, CPA has served as our vice president, finance and principal financial officer since April 2006. From 1997 to 2006, Mr. Courts served as a finance director with ITC DeltaCom, Inc., a publicly held integrated communications provider, which was subsequently acquired by EarthLink, Inc., where his direct responsibilities included corporate budgeting, cost operations and implementation and compliance with Sarbanes-Oxley. Mr. Courts was a key member of the management team involved in business valuation and corporate transactions, including and culminating with the merger of Business Telecom, Inc. and ITC DeltaCom, Inc. in 2003. Mr. Courts is licensed as a Certified Public Accountant in North Carolina, and he holds a B.A. in Finance and M.B.A. from Marshall University.
Steven L. Zelenkofske, D.O., F.A.C.C.has served as our senior vice president, clinical and medical affairs and chief medical officer since January 2009. Dr. Zelenkofske has extensive experience as a principal investigator on a number of important trials in cardiovascular medicine. From June 2007 until January 2009, Dr. Zelenkofske served as vice president, cardiovascular/thrombosis medical unit at Sanofi-Aventis Pharmaceutical, a U.S. subsidiary of Sanofi, a multinational pharmaceutical company, and, from 2006 until 2007, as vice president of clinical sciences and chief patient safety officer for Boston Scientific Corporation’s cardiac rhythm management group. From 2002 to 2006, he was a senior director with Novartis AG, a publicly held multinational healthcare company, where he held positions of increasing responsibility within its cardiovascular division, most recently serving as senior medical director, U.S. clinical development, hypertension and cardiovascular section head. Dr. Zelenkofske is a practicing electrophysiologist at Lehigh Valley Hospital in Allentown, Pennsylvania, and serves as an associate professor of medicine at the College of Medicine of the Pennsylvania State University/Hershey Medical Center and for the Philadelphia College of Osteopathic Medicine. He earned a B.S. and M.Sc. in biochemistry and immunopharmacology at Emory University. He holds a D.O. from the Philadelphia College of Osteopathic Medicine and has certifications in clinical cardiac electrophysiology, cardiology and internal medicine, including a fellow of American Cardiology College.
Alexander R. Giaquinto, Ph.D. has served as our as senior vice president of regulatory affairs and quality assurance and chief compliance officer since January 2009. Dr. Giaquinto has over 42 years of experience in the pharmaceutical industry, including 30 years at Schering-Plough Corporation, a publicly held pharmaceutical company that was subsequently
70
acquired by Merck & Co., Inc., where, from 1972 to 2004, he held executive management positions in the areas of worldwide regulatory affairs and compliance. From 2004 to 2008, Dr. Giaquinto served as the president of ARG Consulting, LLC, a privately held consulting company. Dr. Giaquinto is a member of the Scientific Advisory Boards of Allozyne, Inc., a privately held biopharmaceutical company, and EndoCeutics, Inc., a privately held biopharmaceutical company. Dr.Giaquinto is a Director on the Board of Biothera Inc., a privately held biopharmaceutical company. He also served on, as well as chaired, the Regulatory Affairs Coordinating Committee of the Pharmaceutical Research and Manufacturers of America. Dr. Giaquinto earned a B.S. in pharmacy from St. John’s University and a Ph.D. in pharmaceutics from the University of Connecticut.
Christopher P. Rusconi, Ph.D. .is a co-founder of our company and has served in several executive positions with us since 2003, most recently as our chief scientific officer. From 2000 to 2003, Dr. Rusconi was director of research for the program in combinatorial therapeutics and assistant research professor of surgery at Duke University Medical Center. Dr. Rusconi has authored or co-authored more than 30 peer-reviewed publications, including works appearing inNature, Circulationand theJournal of Clinical Investigation. He is the co-inventor of our aptamer-control agent technology, the inventor of REG1 and several issued and allowed patents and has led the development of this program since its inception. Dr. Rusconi is a member of the Biological Sciences Advisory Board of the University of Kansas. He earned a B.S. in cellular biology from the University of Kansas, and a Ph.D. in molecular and cellular biology as an National Science Foundation pre-doctoral fellow from the University of Colorado, Boulder. He continued his training as a Jane Coffin Childs postdoctoral fellow at Duke University Medical Center.
Board of Directors
Dennis Podlesak is our chairman and has served as a member of our board of directors since December 2007. Mr. Podlesak has been a partner of Domain Associates, or Domain, since 2007. Domain is an exclusively life science focused venture capital firm. From 2007 to December 2009, Mr. Podlesak founded and served as chief executive officer of Calixa Therapeutics, Inc., a privately held biopharmaceutical company, which was acquired by Cubist Pharmaceuticals, Inc. in December 2009. Additionally, Mr. Podlesak was executive chairman of Corthera, Inc., a privately held biopharmaceutical company, which was acquired by Novartis AG in January 2010. Prior to Domain, from 2005 to 2007, Mr. Podlesak was a founder and chief executive officer of Cerexa, Inc., a privately held biopharmaceutical company, which was acquired by Forest Laboratories, Inc. in 2007. Prior to Cerexa, from 2004 to 2005, Mr. Podlesak served as the chief executive officer of Peninsula Pharmaceuticals, Inc., which was acquired by Johnson & Johnson in 2005. Prior to Peninsula, Mr. Podlesak served as senior vice president and led a North American business unit for Novartis AG, a multinational publicly held healthcare company, and as a member of Novartis’ pharmaceutical executive committee and global leadership team. Earlier in his career, Mr. Podlesak served as vice president and headed the CEC division of Allergan, Inc., a publicly held multi-specialty healthcare company, and as member of Allergan’s North American and global management team. Mr. Podlesak spent the first ten years of his career with SmithKline Beecham (now GlaxoSmithKline plc) where he was promoted to eight positions of increasing responsibility during his tenure with the company. Mr. Podlesak currently serves as chairman of the board of directors of Adynxx, Inc., a privately held pharmaceutical company, and Syndax Pharmaceuticals, Inc., a privately held pharmaceutical company, and is a member of the board of directors of Avanir Pharmaceuticals, Inc., a publicly held biopharmaceutical company, RightCare Solutions, Inc., a privately held healthcare technology company, DRI Holdings Limited, and its wholly owned subsidiary, Domain Russian Investments, Limited. Mr. Podlesak received a B.A. in business administration and an M.B.A. from Pepperdine University and has completed postgraduate studies at the Wharton School, University of Pennsylvania. We believe that Mr. Podlesak possesses specific attributes that qualify him to serve on our board of directors, including his experience as chief executive officer at other companies in the biotechnology industry, his over 20 years of strategic, operational and commercial experience within the pharmaceutical industry and his service on other boards of directors in the biotechnology industry.
B. Jefferson Clarkhas served as a member of our board of directors since 2004. Mr. Clark co-founded The Aurora Funds, Inc., or Aurora in 1994 and currently serves as the managing partner of Aurora. He works primarily with Aurora’s life sciences portfolio. Mr. Clark currently serves on the boards of several of Aurora’s private life science portfolio companies. Prior to forming Aurora, Mr. Clark spent 13 years working in development and external affairs for Duke University. Mr. Clark holds a B.S. in Mechanical Engineering from Duke University and a M.B.A. from the Fuqua School of Business at Duke University. We believe that Mr. Clark possesses specific attributes that qualify him to serve on our board of directors including a significant understanding of corporate governance issues and the manner in which a board should operate due to his experience serving on the board for over a dozen early stage life science companies and member of the audit committee for several companies.
Anton Gopkahas served as a member of our board of directors since December 2012. Mr. Gopka is a managing director of D-Pharma, the management company of RusnanoMedInvest, LLC, or RMI, a Russia-based pharmaceutical venture capital firms, founded by RUSNANO State Corporation. Mr. Gopka currently serves on the boards of several of
71
RMI’s portfolio companies. In December 2010, Mr. Gopka founded ParaClassics, a privately held classical music internet media portal where he serves as managing partner. Previously, from October 2010 to April 2011, Mr. Gopka was a vice president for mergers and acquisitions at Barclays Capital, LLC, a subsidiary of Barclays PLC. Prior to Barclays Capital, from October 2008 to October 2010, he was director of mergers and acquisitions at Sistema JSFC. Previously, from April 2006 to October 2008, Mr. Gopka worked at Dresdner Kleinwort, an investment bank, where he advised on a number of M&A and capital markets transactions. Mr. Gopka received a Master’s degree from the Moscow State University for International Affairs. He also earned a corporate finance course diploma from INSEAD. We believe that Mr. Gopka possesses specific attributes that qualify him to serve on our board of directors including his extensive transactional experience in a wide range of industries, strong healthcare exposure as the managing director of a leading Russian pharmaceutical venture capital firm as well as service on the board of directors for a number of healthcare companies.
Pierre Legault, MBA, CA, CPA has served as a member of our board of directors and chair of the audit committee since November 2013. Mr. Legault currently serves as chief executive officer of Nephrogenex, Inc., a publicly-held biotechnology company focused on the treatment of diabetic kidney disease, a position he has held since October 2013 and as the president and chief executive officer of Stone Management LLC, a firm focused in the areas of business development and board assistance, a position he has held since April 2012. From November 2012 to October 2013, Mr. Legault served as executive chairman of Nephrogenex. From September 2010 to April 2012, Mr. Legault was the chief executive officer of Prosidion Ltd., a UK mid-size biotechnology firm and fully integrated subsidiary of Astellas Pharma, Inc. From January 2009 to September 2010, Mr. Legault served as executive vice president, chief financial officer and treasurer of OSI Pharmaceuticals, Inc., a mid-size biotechnology company, which was acquired by Astellas Pharma. From January 2006 to December 2008, Mr. Legault worked for Rite Aid Corporation and its processor companies, a Fortune 500 publicly traded pharmaceutical retail company, most recently serving as senior vice president and chief administrative officer. Prior to Rite Aid, Mr. Legault held several senior positions over a period exceeding 15 years with Sanofi-Aventis and predecessor companies, most recently serving as worldwide president of a large global Sanofi-Aventis business unit. Mr. Legault currently serves on the board of directors of Forest Laboratories, Inc., a publicly traded pharmaceutical manufacturer and marketer. In addition, within the past five years, Mr. Legault previously served on the board of directors of Cyclacel Pharmaceutical Inc., a publicly traded biotech company. Mr. Legault also has served on several private and nonprofit company boards and audit committees. Mr. Legault studied at the Harvard Business School in an executive master’s program and he received his MBA from McGill University and his BAA from University of Montreal (HEC). He also holds CA and CPA degrees. We believe that Mr. Legault possesses specific attributes that qualify him to serve on our board of directors, including his leadership experience at other publicly traded companies in the biotechnology industry, his over 20 years of strategic, operational and commercial experience within the pharmaceutical industry and his service on other boards of directors and audit committees of publicly traded companies.
Michael E. Mendelsohn, M.D. has served as a member of our board of directors since November 2013, served as senior vice president and global franchise head for cardiovascular diseases at Merck & Co., Inc. from June 2010 through October 2013. Prior to Merck, from 1993 to 2010, Dr. Mendelsohn spent 17 years at Tufts Medical Center, where he served as the first-ever chief scientific officer and the executive director of the center’s Molecular Cardiology Research Institute. Prior to Tufts Medical Center, from 1988 to 1993 Dr. Mendelsohn was a member of the faculty of the cardiology division of Brigham and Women’s Hospital and an assistant professor of medicine at Harvard Medical School. Dr. Mendelsohn received his undergraduate degree in Chemistry and English from Amherst College and his M.D. from Harvard Medical School. We believe that Dr. Mendelsohn possesses specific attributes that qualify him to serve on our board of directors including his over 20 years experience and leadership in the field of cardiovascular disease.
P. Sherrill Neffhas served as a member of our board of directors since July 2005. Mr. Neff founded Quaker Partners Management, L.P., an investment firm, in 2002 and has served as a partner since 2002. From 1994 to 2002, Mr. Neff was president and chief operating officer of Neose Technologies, Inc., a biopharmaceutical company, and a director from 1994 to 2003. From 1993 to 1994, he was senior vice president of corporate development at U.S. Healthcare, a publicly traded health maintenance organization which was acquired by Aetna Life & Casualty Co. Prior to that time, Mr. Neff was a managing director at Alex. Brown & Sons, an investment bank, from 1984 to 1993 and a corporate attorney at Morgan Lewis from 1980 to 1984. Mr. Neff holds a B.A. from Wesleyan University and a J.D. from the University of Michigan Law School. Mr. Neff serves on the boards of directors of Resource Capital Corporation, a publicly held real estate investment trust, and Cempra, Inc., a publicly held clinical stage pharmaceutical company, as well as several privately held organizations. Mr. Neff also served on the board of directors of Amicus Therapeutics, Inc., a publicly held biopharmaceutical company, from 1996 until 2011. We believe that Mr. Neff possesses specific attributes that qualify him to serve on our board of directors including his more than 20 years of relevant experience in the healthcare industry and more than eleven years as a founding partner of a leading, healthcare venture capital firm.
72
Jesse Treu, Ph.D. has served as a member of our board of directors since July 2005. Dr. Treu has been a partner of Domain since its inception in 1985. Dr. Treu currently serves on the boards of directors of several of Domain’s portfolio companies. Recently, Dr. Treu served on the board of directors of SenoRx, Inc., a publicly held biotechnology company, which was subsequently acquired by C.R. Baird, Inc. Dr. Treu began his career with General Electric Company in 1973. Dr. Treu holds a B.S. from Rensselaer Polytechnic Institute, and both an M.A. and Ph.D. from Princeton University. We believe that Dr. Treu possesses specific attributes that qualify him to serve on our board of directors including Dr. Treu’s extensive management and board experience in the healthcare industry.
Raphaël Wisniewskihas served as a member of our board of directors since December 2009. Mr. Wisniewski joined Edmond de Rothschild Investment Partners, or Rothschild, in 2001 and has served as a partner of its life sciences practice since 2006. He is a former member of the managing board of Rothschild. Mr. Wisniewski is a graduate of the Ecole des Hautes Etudes Commerciales in Paris and holds a postgraduate diploma in Economics and Finance from the Institut d’Etudes Politiques, Paris. We believe that Mr. Wisniewski possesses specific attributes that qualify him to serve on our board of directors, including his experience investing in and growing biotechnology companies, his experience in finance, capital markets and investing and his understanding of the operations and issues that affect similarly situated private or public companies.
Family Relationships
There are no family relationships among any of our directors or executive officers.
Board Composition
Our board of directors currently consists of nine members, a majority of who are “independent” as that term is defined in the applicable NASDAQ listing standards. Other than Mr. Gopka, Mr. Podlesak and Dr. Treu, each of our directors is deemed “independent” as that term is defined in the applicable NASDAQ listing standards.
Our board of directors is divided into three classes: class I, class II and class III, with each class serving staggered three-year terms. Upon the expiration of the term of a class of directors, directors in that class will be eligible to be elected for a new three-year term at the annual meeting of stockholders in the year in which their term expires.
Our amended and restated certificate of incorporation and amended and restated bylaws provide that the authorized number of directors may be changed only by resolution of the board of directors. Our amended and restated certificate of incorporation and amended and restated bylaws also provide that our directors may be removed only for cause by the affirmative vote of the holders of at least 75% of the votes that all our stockholders would be entitled to cast in an annual election of directors, and that any vacancy on our board of directors, including a vacancy resulting from an increase in the authorized number of directors, may be filled only by vote of a majority of our directors then in office, even if less than a quorum.
We have no formal policy regarding board diversity. Our priority in selection of board members is identification of members who will further the interests of our stockholders through his or her established record of professional accomplishment, the ability to contribute positively to the collaborative culture among board members, knowledge of our business and understanding of the competitive landscape.
Director Independence
Our common stock is listed on The NASDAQ Capital Market. Under the rules of The NASDAQ Stock Market, independent directors must comprise a majority of a listed company’s board of directors within 12 months of the completion of an initial public offering. In addition, the rules of The NASDAQ Stock Market require that, (i) on the date of the completion of the offering, at least one member of each of a listed company’s audit, compensation and nominating and corporate governance committees be independent, (ii) within 90 days of the date of the completion of the offering, a majority of the members of such committees be independent and (iii) within one year of the date of the completion of the offering, all the members of such committees be independent. Audit committee members must also satisfy the independence criteria set forth in Rule 10A-3 under the Securities Exchange Act of 1934, as amended, or the Exchange Act. Under the rules of The NASDAQ Stock Market, a director will only qualify as an “independent director” if, in the opinion of that company’s board of directors, that person does not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director.
In order to be considered to be independent for purposes of Rule 10A-3, a member of an audit committee of a listed company may not, other than in his or her capacity as a member of the audit committee, the board of directors, or any other board committee: (1) accept, directly or indirectly, any consulting, advisory or other compensatory fee from the listed company or any of its subsidiaries or (2) be an affiliated person of the listed company or any of its subsidiaries.
73
Our board of directors undertook a review of its composition, the composition of its committees and the independence of each director. Based upon information requested from and provided by each director concerning his background, employment and affiliations, including family relationships, our board of directors has determined that Messrs. Clark, Legault, Mendelsohn, Neff and Wisniewski, representing five of our nine directors, do not have a relationship that would interfere with the exercise of independent judgment in carrying out the responsibilities of a director and that each of these directors is “independent” as that term is defined under the rules of The NASDAQ Stock Market.
In making this determination, our board of directors considered the relationships that each non-employee director has with our company and all other facts and circumstances our board of directors deemed relevant in determining their independence, including the beneficial ownership of our capital stock by each non-employee director. We intend to comply with the other independence requirements for committees within the time periods specified above.
Board Committees
Our board of directors has established an audit committee, a compensation committee, a nominating and corporate governance committee and a science committee. Each of these committees operates under a charter that has been approved by our board of directors.
Audit Committee
Our audit committee currently consists of Pierre Legault, as chairman, B. Jefferson Clark and Raphaël Wisniewski, each of whom is “independent” as that term is defined under applicable SEC rules and NASDAQ listing standards. Our board of directors has determined that Mr. Legault qualifies as an audit committee financial expert within the meaning of SEC regulations and The NASDAQ Marketplace Rules. In making this determination, our board has considered the formal education and nature and scope of his previous experience, coupled with past and present service on various audit committees. Our audit committee assists our board of directors in its oversight of our accounting and financial reporting process and the audits of our financial statements.
Our audit committee’s responsibilities include:
| | • | | appointing, evaluating, retaining and, when necessary, terminating the engagement of our independent registered public accounting firm; |
| | • | | overseeing the independence of our independent registered public accounting firm, including obtaining and reviewing reports from the firm; |
| | • | | setting the compensation of our independent registered public accounting firm; |
| | • | | overseeing the work of our independent registered public accounting firm, including receiving and considering reports made by our independent registered public accounting firm regarding accounting policies and procedures, financial reporting and disclosure controls; |
| | • | | reviewing and discussing with management and our independent registered public accounting firm our audited financial statements and related disclosures; |
| | • | | preparing the annual audit committee report required by SEC rules; |
| | • | | coordinating internal control over financial reporting, disclosure controls and procedures and code of conduct; |
| | • | | reviewing our policies with respect to risk assessment and risk management; |
| | • | | establishing procedures related to the receipt, retention and treatment of complaints regarding accounting, internal accounting controls or auditing matters, and the confidential, anonymous submission by employees of concerns regarding accounting or auditing matters; |
| | • | | reviewing our policies and procedures for reviewing and approving or ratifying related person transactions, including our related person transaction policy; and |
| | • | | meeting independently with management and our independent registered public accounting firm. |
All audit services to be provided to us and all non-audit services, other than de minimis non-audit services, to be provided to us by our independent registered public accounting firm must be approved in advance by our audit committee.
74
Compensation Committee
Our compensation committee currently consists of P. Sherrill Neff, as chairman, B Jefferson Clark and Dennis Podlesak, a majority of whom are “independent” as that term is defined under applicable SEC rules and NASDAQ listing standards. Our compensation committee assists our board of directors in the discharge of its responsibilities relating to the compensation of our executive officers. Our compensation committee’s responsibilities include:
| | • | | reviewing and recommending to the board of directors our chief executive officer’s compensation, and approving the compensation of our other executive officers reporting directly to our chief executive officer; |
| | • | | overseeing the evaluation of our senior executives; |
| | • | | overseeing, administering, reviewing and making recommendations to the board of directors with respect to our incentive compensation and equity-based plans; |
| | • | | reviewing and making recommendations to the board of directors with respect to director compensation; |
| | • | | reviewing and discussing with management the compensation discussion and analysis required by SEC rules; and |
| | • | | preparing the annual compensation committee report required by SEC rules. |
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee currently consists of Dr. Treu, as chairman, Michael Mendelsohn and Raphaël Wisniewski, a majority of who are “independent” as that term is defined under applicable NASDAQ listing standards. The nominating and corporate governance committee’s responsibilities include:
| | • | | recommending to the board of directors the persons to be nominated for election as directors or to fill any vacancies on the board of directors, and to be appointed to each of the board’s committees; |
| | • | | developing and recommending to the board of directors corporate governance guidelines; and |
| | • | | overseeing an annual self-evaluation of the board of directors. |
Science Committee
Our science committee currently consists of Michael Mendelsohn, as chairman, Dennis Podlesak and Dr. Treu. Our science committee’s responsibilities include:
| | • | | reviewing the science and clinical and regulatory strategies underlying our significant R&D programs, including publication strategies; |
| | • | | reviewing our significant medical affairs strategies and initiatives; |
| | • | | reviewing the annual R&D budget and allocation of resources to discovery and development programs; |
| | • | | reviewing the capacity and skill set of the R&D organization; |
| | • | | reviewing the implications for the R&D organization of significant business development transactions, including mergers, acquisitions, licensing and collaborative agreements; |
| | • | | reviewing the progress toward achievement of key R&D milestones; and |
| | • | | reviewing the interactions of the R&D organization with health care providers and regulatory bodies, especially as with regard to reporting of adverse events and/or unexpected negative data observed in the preclinical and clinical studies conducted by us. |
Code of Business Conduct and Ethics
We have adopted a written code of business conduct and ethics that applies to our directors, officers and employees, including our principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions. A copy of the code is posted on the corporate governance section of our website, which is located at www.regadobio.com. If we make any substantive amendments to, or grant any waivers from, the code of business conduct and ethics for any officer or director, we will disclose the nature of such amendment or waiver on our website.
75
Board Leadership Structure and Board’s Role in Risk Oversight
The positions of our chairman of the board and chief executive officer are separated. We will consider appointing a lead independent director in the event we have a chairman who is not independent within the meaning of The NASDAQ Marketplace Rules. Separating these positions allows our chief executive officer to focus on our day-to-day business, while allowing the chairman of the board to lead the board of directors in its fundamental role of providing advice to and independent oversight of management. Our board of directors recognizes the time, effort and energy that the chief executive officer must devote to his position in the current business environment, as well as the commitment required to serve as our chairman, particularly as the board of directors’ oversight responsibilities continue to grow. Our board of directors also believes that this structure ensures a greater role for the independent directors in the oversight of our company and active participation of the independent directors in setting agendas and establishing priorities and procedures for the work of our board of directors. Our board of directors believes its administration of its risk oversight function has not affected its leadership structure.
Although our amended and restated bylaws do not require our chairman and chief executive officer positions to be separate, our board of directors believes that having separate positions is the appropriate leadership structure for us at this time and demonstrates our commitment to good corporate governance.
Risk is inherent with every business, and how well a business manages risk can ultimately determine its success. We face a number of risks, including those described under “Risk Factors.” Our board of directors is actively involved in oversight of risks that could affect us. This oversight is conducted primarily by our full board of directors, which has responsibility for general oversight of risks.
Our board of directors satisfies this responsibility through full reports by each committee chair regarding the committee’s considerations and actions, as well as through regular reports directly from officers responsible for oversight of particular risks within our company. Our board of directors believes that full and open communication between management and the board of directors is essential for effective risk management and oversight.
Medical Advisory Board
Our Medical Advisory Board comprises many preeminent scientists and physicians from the cardiovascular field. Our board of directors appoints members to the Medical Advisory Board for a term of one year, renewable at the member’s option. The current Medical Advisory Board includes:
Paul W. Armstrong, M.D. has served on our Medical Advisory Board since April 2009. He is a senior attending cardiologist at the University of Alberta Hospital. He is the founding and immediate past president of the Canadian Academy of Health Sciences. Dr. Armstrong received his medical degree at Queen’s University. After post-graduate training both there and at the University of Toronto, he undertook clinical and research training in cardiology at the Massachusetts General Hospital, Harvard Medical School and St. George’s Hospital, London, UK. He is the author, or co-author, of numerous peer-reviewed publications and a member of a number of editorial boards. He is a member of the American College of Cardiology/American Heart Association ST Elevation Myocardial Infarction Guidelines writing committee, CIHR’s Institute of Circulatory and Respiratory Health’s Advisory Board and former member of the FDA Cardiovascular and Renal Advisory Committee and the Board of the Council of the Canadian Academies.
Richard C. Becker, M.D. has served on our Medical Advisory Board since April 2009. Dr. Becker serves as the director of Cardiovascular Thrombosis Research Center at the Duke Clinical Research Institute, or DCRI. Dr. Becker previously spent over a decade at the University of Massachusetts Medical Center, serving in various capacities including Professor of Medicine, and director of the Cardiovascular Thrombosis Research Center, Coronary Care Unit and Anticoagulation Services. He is a member of numerous professional associations such as the International Atherosclerosis Society, the International Society on Thrombosis and Hemostasis and the Thrombosis Council of the American Heart Association. Dr. Becker has published numerous articles in peer-reviewed journals and authored twelve textbooks. He is the founding editor-in-chief of the Journal of Thrombosis and Thrombolysis. Dr. Becker received his MEd in biochemistry and his M.D. from the University of Cincinnati Graduate School and College of Medicine, respectively, in Ohio. He completed fellowships in Hematology at the Cleveland Clinic and Cardiovascular Medicine at the University of Massachusetts Medical Center. He is an NIH-funded investigator in the field of platelet biology, and as co-director, Biomarkers Program coordinates several multi-dimensional research projects that employ gene expression profile and proteomic platforms to determine signatures of thrombosis.
Robert M. Califf, M.D. has served on our Medical Advisory Board since April 2009. Dr. Califf is currently vice chancellor for Clinical Research, director of the Duke Translational Medicine Institute, and professor of medicine in the Division of Cardiology at the Duke University Medical Center in Durham, North Carolina. He graduated from Duke
76
University, summa cum laude and Phi Beta Kappa, in 1973 and from Duke University Medical School in 1978, where he was selected for Alpha Omega Alpha. He performed his internship and residency at the University of California at San Francisco and his fellowship in cardiology at Duke University. He is board-certified in internal medicine (1984) and cardiology (1986) and is a Master of the American College of Cardiology (2006). For 10 years he was the founding Director of the DCRI. He is the editor-in-chief of Elsevier’s American Heart Journal. He has been author or co-author of numerous peer-reviewed journal articles and a contributing editor for theheart.org, an online information resource for academic and practicing cardiologists. Dr. Califf has served on the Cardiorenal Advisory Panel of the U.S. Food and Drug Administration and the Pharmaceutical Roundtable of the Institute of Medicine. He was the founding director of the Coordinating Center for the Centers for Education & Research on Therapeutics™, a public-private partnership among the Agency for Healthcare Research and Quality, academia, the medical products industry, and consumer groups. He is co-chair of the Clinical Trials Transformation Initiative, a public private partnership with the U.S. Food and Drug Administration, or the FDA, academia, industry and multiple government agencies.
John Eikelboom, M.B.B.S.has served on our Medical Advisory Board since June 2009. Dr. Eikelboom completed training in Internal Medicine and Haematology in Perth, Australia in 1998 and subsequently undertook a fellowship in Thrombosis Medicine and Clinical Epidemiology at McMaster University. He returned to McMaster in 2005 to take up a Tier II Canada Research Chair in Cardiovascular Medicine. Dr. Eikelboom has published numerous articles in peer-reviewed journals.
Michael Gibson, M.S., M.D. has served on our Medical Advisory Board since March 2009. Dr. Gibson received his M.D. from the University of Chicago. He was an intern, resident and chief resident at the Brigham and Women’s Hospital, Harvard Medical School. He received his training as an interventional cardiologist and served as the director of the Coronary Care Unit at Beth Israel Hospital, Harvard Medical School. Dr. Gibson founded PERFUSE, an academic research organization which has served as the angiographic core laboratory and data coordinating center for a wide variety of studies. Dr. Gibson also edited a textbook entitled “Management Strategies in Interventional Cardiology.” He has served as lead author of the section on myocardial perfusion imaging in an imaging textbook which is a companion to Braunwald’s Heart Disease. Dr. Gibson is on the editorial board of Circulation and Editor-in-Chief of Clinical Trial Results where he created the first weekly TV show for Cardiologists “This Week in Cardiology.”
Neal S. Kleiman, M.D. has served on our Medical Advisory Board since April 2009. After receiving his medical degree from Columbia University of Physicians and Surgeons in New York, Dr. Kleiman completed an internship and residency in Internal Medicine at Baylor College of Medicine in Houston. He also completed a fellowship in Cardiology at Baylor College of Medicine. Dr. Kleiman has published numerous manuscripts, book chapters and abstracts. In addition, he is on the editorial board of several journals. Dr. Kleiman is a member of a number of professional organizations including The American Heart Association Council on Clinical Cardiology and Council on Thrombosis, and he is a fellow of The American College of Cardiology and the Society for Coronary Angiography and Interventions.
A. Michael Lincoff, M.D. has served on our Medical Advisory Board since August 2009. Dr. Lincoff is a Professor of Medicine at the Cleveland Clinic Lerner College of Medicine of Case Western Reserve University. He holds the Charles and Charlotte Fowler Endowed Chair for Cardiovascular Research at the Cleveland Clinic. Dr. Lincoff has authored or co-authored numerous peer-reviewed articles. He edited a book entitled Glycoprotein IIb/IIIa Inhibitors in Cardiovascular Disease. He has also authored numerous book chapters and reviews and presented numerous lectures at various medical meetings and courses. He is the consulting editor of the American Journal of Cardiovascular Drugs and serves on editorial boards or as a reviewer for a number of peer-reviewed scientific journals. Dr. Lincoff is currently a member of the Cardiovascular and Renal Drugs Advisory Committee of the United States Food and Drug Administration. Dr. Lincoff is a fellow of the American College of Cardiology and the European Society of Cardiology and a member of the American College of Physicians, scientific councils of the American Heart Association, and the Sigma Xi Scientific Research Society.
Roxana Mehran, M.D. has served on our Medical Advisory Board since April 2009. Dr. Mehran is a practicing interventional cardiologist at New York-Presbyterian Hospital, Columbia University Medical Center. She spent several years as an interventional cardiologist before becoming Director of Clinical Research at the Cardiology Research Foundation at the Washington Hospital Center. Dr. Mehran has written numerous publications on these topics, including a chapter in the book, Principles and Practice of Interventional Cardiology. Dr. Mehran completed her training in internal medicine at the University of Connecticut. She then continued her studies in cardiovascular medicine and interventional cardiology at the Cardiovascular Institute at Mount Sinai Medical Center in New York City.
Philippe Menasché, M.D., Ph.D. has served on our Medical Advisory Board since June 2009. Dr. Menasché obtained his M.D. and Ph.D. degrees from the University of Paris. In addition to his clinical positions, he is currently Director of an INSERM (National Institute of Health and Medical Research) laboratory devoted to cell therapy of cardiovascular diseases.
Lars Wallentin, M.D., Ph.D. has served on our Medical Advisory Board since June 2009. Professor Wallentin is chairman of the Committee for National Guidelines for Treatment of Heart Diseases in Sweden, member of the research council of the Swedish Heart Foundation and expert for the Swedish Government Delegation for the development of Clinical Research. Dr. Wallentin became the first professor of Cardiology at Uppsala University Hospital, Uppsala, Sweden in 1991
77
and was head of the Department of Cardiology from 1991 to 1999. He founded the Cardiothoracic Center at Uppsala, becoming its first chairman in 1994 to 1995, and going on to serve as vice chairman from 1995 to 1999. In 2001, Professor Wallentin founded the Uppsala Clinical Research Center, where he has been the director 2002 to 2008. From 1998 to 1999, Professor Wallentin was president of the Swedish Cardiac Society, and from 2000 to 2002, he founded and served as president of the Swedish Heart Association. Dr. Wallentin has published numerous papers, other publications and book chapters. He is a member of numerous medical societies, task forces, working groups and editorial boards, and is an expert for the Swedish Board of Health and Welfare.
Harvey D. White, DSc has served on our Medical Advisory Board since April 2009. Professor White trained as a cardiologist at Green Lane Hospital and Harvard Medical School. In recognition of his work on end-systolic volume, infarct artery patency and fibrinolysis, he was awarded a Doctorate of Science by the University of Otago, New Zealand in 1995 and the 1998 Prince Mahidol Award for Medicine by the King of Thailand. Professor White is an Honorary Clinical Professor, Department of Medicine at the University of Auckland. He is also a fellow of the Royal Australasian College of Physicians, the American College of Cardiology, the European Society of Cardiology, the American Heart Association, the Cardiac Society of Australia and New Zealand and the Royal Society of New Zealand. He has published numerous peer-reviewed articles, editorials and book chapters. He is also the associate editor of circulation and editorial board member for several international journals. He has served on the steering committees of several international trial groups.
John H. Alexander, M.D., MHS has served on our Medical Advisory Board since June 2011. Dr. Alexander received his medical degree from the University of Pennsylvania, School of Medicine. He completed his residency in internal medicine at the Brigham and Women’s Hospital, Harvard Medical School. He was a fellow in cardiology at Duke University Medical Center, where he received training in echocardiography and clinical research training at the DCRI. He joined the Duke faculty in the Division of Cardiology and at the DCRI in 2000, where he is currently an associate professor of Medicine. He is currently the co-director of cardiovascular research at the Duke Clinical Institute, where he oversees a broad portfolio of cardiovascular clinical trials. He is also the director of site-based clinical research at the Duke Heart Center.
Deepak L. Bhatt MD, MPH has served on our Medical Advisory Board since June 2013. Dr. Bhatt obtained his undergraduate degree from the Massachusetts Institute of Technology and his MD from Cornell University. He also completed a Masters in Public Health with a concentration in clinical effectiveness at Harvard University. His internship and residency in internal medicine were performed at the Hospital of the University of Pennsylvania, and his cardiovascular training was completed at Cleveland Clinic. He also completed fellowships in interventional cardiology and cerebral and peripheral vascular intervention as well as serving as Chief Interventional Fellow at Cleveland Clinic where he went on to spend several years as an interventional cardiologist and an Associate Professor of Medicine. Dr. Bhatt is Chief of Cardiology at VA Boston Healthcare System and Director of the Integrated Interventional Cardiovascular Program at Brigham and Women’s Hospital and VA Boston Healthcare System. He is also a Senior Physician at Brigham and Women’s Hospital and Professor of Medicine at Harvard Medical School. Dr. Bhatt’s research interests include preventive cardiology as well as the optimal management of patients with acute coronary syndromes. He also has research interests in advanced techniques in cardiac, cerebral, and peripheral intervention. He has served as the principal investigator or co-principal investigator for a number of cardiology clinical trials. He has authored or co-authored numerous papers and lectured throughout the world.
78
Section 16(a) Beneficial Ownership Reporting Compliance
Section 16(a) of the Securities Exchange Act of 1934, as amended, requires our directors and executive, officers, and persons who are beneficial owners of more than 10% of a registered class of our equity securities, to file reports of ownership and changes in ownership with the Securities and Exchange Commission (the “SEC”). These persons are required by SEC regulations to furnish us with copies of all Section 16(a) forms they file. To our knowledge, based solely on a review of the copies of such reports furnished to us, and written representations that no other reports were required during the fiscal year ended December 31, 2013, all reports required to be filed under Section 16(a) were filed on a timely basis.
Item 11. Executive Compensation
Summary Compensation Table
The following table shows the compensation awarded to or earned by our principal executive officer, our two other most highly compensated executive officers who were serving as executive officers as of December 31, 2013, and all individuals who served as principal executive officer at any time during the fiscal year ended December 31, 2013. The persons listed in the following table are referred to herein as the “named executive officers.”
| | | | | | | | | | | | | | | | | | | | |
Name and Principal Position | | Salary
($) | | | Bonus
($) | | | Option
Awards
($)(1) | | | All Other
Compensation
($) | | | Total
($) | |
David J. Mazzo
Chief Executive Officer | | | 465,825 | | | | 209,621 | | | | 1,094,101 | | | | 14,092 | (2) | | | 1,783,639 | |
Michael A. Metzger
President and Chief Operating Officer | | | 28,688 | (3) | | | 150,000 | (4) | | | 779,081 | | | | — | | | | 957,769 | |
Steven L. Zelenkofske
Senior Vice President,
Clinical and Medical
Affairs and Chief Medical Officer | | | 375,000 | | | | 131,000 | | | | 442,120 | | | | — | | | | 948,120 | |
79
| (1) | Represents the aggregate grant date fair value for grants made in 2013 computed in accordance with FASB ASC Topic 718. The assumptions we used in valuing options are described in Note 10 to our financial statements included in this Annual Report. |
| (2) | Consists of premiums for supplemental life insurance for the benefit of Dr. Mazzo and the related tax gross up benefit. |
| (3) | Mr. Metzger commenced employment on December 5, 2013. Represents salary paid from December 5, 2013 to December 31, 2013. |
| (4) | Represents a $150,000 sign-on bonus. |
Annual Base Salary
The compensation of our named executive officers is generally determined by our board of directors, based upon recommendations from the compensation committee of our board of the directors. The following table sets forth the base salaries for the fiscal year ended December 31, 2013 for our named executive officers.
| | | | |
Name | | 2013 Base
Salary | |
David J. Mazzo | | $ | 465,825 | |
Michael A. Metzger | | $ | 405,000 | |
Steven L. Zelenkofske | | $ | 375,000 | |
Annual Bonus Opportunity
In addition to base salaries, our named executive officers are eligible to receive annual cash bonuses. The annual performance-based bonus each named executive officer is eligible to receive is based on the individual’s target bonus, as a percentage of base salary, and an assessment of individual and corporate performance by our board of directors. Pursuant to their employment agreements, each named executive officer has a target bonus represented as a percentage of base salary, or a target bonus percentage, each of which is set forth below:
| | | | |
Name | | Target bonus | |
David J. Mazzo | | | 50 | % |
Michael A. Metzger | | | 40 | % |
Steven L. Zelenkofske | | | 35 | % |
Long-Term Incentive Compensation
Our long-term, equity-based incentive awards are designed to align the interests of our named executive officers and our other employees, non-employee directors and consultants with the interests of our stockholders. Because vesting is based on continued service, our equity-based incentives also encourage the retention of our named executive officers through the vesting period of the awards.
We use stock options as the primary incentive for long-term compensation to our named executive officers because they are able to profit from stock options only if our stock price increases relative to the stock option’s exercise price. We generally provide initial grants in connection with the commencement of employment of our named executive officers and annual retention grants at or shortly following the end of each year.
We have granted all stock options pursuant to our 2004 Equity Incentive Plan, or the 2004 Plan, or our 2013 Equity Compensation Plan, or the 2013 Plan, the terms of which are described below under “—Equity Compensation Plans.” Following our initial public offering, all options have been or will be granted under our 2013 Plan. We typically grant options with an exercise price equal to the fair market value of our common stock on the date of grant of each award. We typically grant options with a four-year vesting period. In addition, pursuant to the terms of their respective employment agreements, each of our named executive officers is entitled to 100% accelerated vesting of options upon a “change of control” or upon a “corporate reorganization.” We believe these accelerated vesting provisions will allow our named executive officers to focus on closing a transaction that may be in the best interest of our stockholders even though the transaction may otherwise result in a termination of their employment and, absent such accelerated vesting, a forfeiture of their unvested equity awards. Additional information regarding accelerated vesting provisions for our named executive officers is discussed below under “—Employment Agreements, Severance and Change in Control Agreements.”
80
Health and Welfare Benefits
Our named executive officers are eligible to participate in all of our employee benefit plans, including our medical, dental, vision, group life and disability insurance plans, in each case on the same basis as other employees. Generally, we do not provide perquisites or personal benefits to our named executive officers. We do, however, pay the premiums for supplemental life insurance for the benefit of Dr. Mazzo and the related tax gross up benefit.
Employment Agreements, Severance and Change in Control Arrangements
Our employment agreement, as amended, with Dr. Mazzo provides for, among other things: (i) an initial annual base salary of $400,000, (ii) initial eligibility for a target cash bonus of up to 50% of annual base salary, (iii) reimbursement of up to $8,836 per year for supplemental long-term disability insurance and/or supplemental life insurance for Dr. Mazzo’s benefit, (iv) 100% accelerated vesting of options upon a “change of control” or upon a “corporate reorganization” and (v) in the event of a termination “without cause,” payments equal to the sum of 12 months of salary and the target bonus payable bimonthly in accordance with customary payroll practices and continuation of health insurance benefits for 12 months. The agreement further provides that Dr. Mazzo may serve on the board of directors of up to two other companies so long as such service does not interfere with his duties to us and such other companies are not competitors of ours.
We have entered into an amended and restated employment agreement with Dr. Mazzo, which became effective upon the consummation of the IPO. The amended and restated employment agreement provides for, among other things: (i) an initial annual base salary of $465,825, (ii) initial eligibility for a target cash bonus of up to 50% of annual base salary, (iii) reimbursement of up to $8,836 per year for supplemental long-term disability insurance and/or supplemental life insurance for Dr. Mazzo’s benefit, (iv) in the event we terminate Dr. Mazzo’s employment “without cause,” payments equal to the sum of 12 months of base salary and the target bonus payable bimonthly in accordance with customary payroll practices and continuation of health insurance benefits for 12 months, and (v) in the event we terminate Dr. Mazzo’s employment “without cause” within 12 months following a change in control or Dr. Mazzo resigns “for good reason” within 12 months following a change in control, (a) payments equal to the sum of 18 months of base salary and an amount equal to 150% of the target bonus payable bimonthly in accordance with customary payroll practices and continuation of health insurance benefits for 18 months and (b) 100% accelerated vesting of his options. The amended and restated employment agreement further provides that Dr. Mazzo may serve on the board of directors of up to two other companies so long as such service does not interfere with his duties to us and such other companies are not competitors of ours.
On December 19, 2013, we further amended Dr. Mazzo’s amended and restated employment agreement to provide for eligibility for a cash bonus equal to 100% of annual base salary, in lieu of his annual target bonus of 50% of his annual base salary, if certain corporate objectives specified in the amendment are achieved in 2014 or 2015 and certain revisions to the Section 280G cutback provisions in his employment agreement.
Our employment agreement with Mr. Metzger provides for, among other things: (i) an initial annual base salary of $405,000, (ii) a sign-on bonus of $150,000 payable in two equal installments, the initial installment of $75,000 is payable immediately and the second installment is payable at the time 2013 performance bonuses are paid to Company employees, (iii) eligibility for an annual target cash bonus of up to 40% of annual base salary, based on the achievement of certain individual and corporate as established by the board of directors or compensation committee for each year, but, if certain corporate objectives specified in the employment agreement are achieved in 2014 or 2015, his annual bonus for the year in which those objectives are achieved will be 100% of his annual base salary, (iv) reimbursement of reasonable expenses for travel between his place of residence in New York and our office in Basking Ridge, New Jersey and for lodging in the Basking Ridge area, of up to $25,000 per year, (v) in the event Mr. Metzger resigns “for good reason” within the first twelve months of his employment or we terminate Mr. Metzger’s employment “without cause,” payments equal to the sum of 12 months of base salary and the target annual bonus payable bimonthly in accordance with customary payroll practices and continuation of health insurance benefits for 12 months, and (vi) in the event we terminate Mr. Metzger’s employment “without cause” within 12 months following a change in control or Mr. Metzger resigns “for good reason” within 12 months following a change in control, payments equal to the sum of 12 months of base salary and the target annual bonus payable bimonthly in accordance with customary payroll practices and continuation of health insurance benefits for 12 months. Subject to the approval of the Company’s compensation committee at its next meeting, Mr. Metzger will receive initial option grants for an aggregate of 2.25% of the Company’s common stock outstanding, calculated on a fully-diluted basis, consisting of: an option grant representing 0.4375% of the Company’s common stock outstanding, calculated on a fully-diluted basis, which shall vest immediately on the date of grant; an option grant representing 1.3125% of the Company’s common stock outstanding, calculated on a fully-diluted basis, which shall vest in 36 equal monthly installments; and an option grant representing 0.5% of the Company’s common stock outstanding, calculated on a fully-diluted basis, which shall vest upon the achievement of certain performance milestones. The employment agreement further provides that Mr. Metzger may serve on the board of directors of one other company during his first year of employment and up to two other companies in future years so long as such service does not interfere with his duties to us and such other companies are not competitors of ours.
81
Our employment agreement, as amended, with Dr. Zelenkofske provides for among other things: (i) an initial annual base salary of $310,000, (ii) initial eligibility for a target cash bonus of up to 35% of annual base salary, (iii) 100% accelerated vesting of options upon a “change of control” or upon a “corporate reorganization” and (iv) in the event of a termination “without cause,” payments equal to the sum of 12 months of salary and the target bonus payable bimonthly in accordance with customary payroll practices and continuation of health insurance benefits for 12 months. The agreement further provides that Dr. Zelenkofske may continue to practice medicine two days per month (or more with our written permission) and may remain on any boards of directors or advisory boards as long as those positions do not represent a conflict of interest.
We have entered into an amended and restated employment agreement with Dr. Zelenkofske which became effective upon the consummation of the IPO. The amended and restated employment agreement provides for, among other things: (i) an initial annual base salary of $375,000, (ii) initial eligibility for a target cash bonus of up to 35% of annual base salary, (iii) in the event we terminate Dr. Zelenkofske’s employment “without cause” at any time or Dr. Zelenkofske resigns “for good reason” within 12 months following a change in control, payments equal to the sum of 12 months of base salary and the target bonus payable bimonthly in accordance with customary payroll practices and continuation of health insurance benefits for 12 months, and (iv) in the event that, within 12 months following a change in control, we terminate Dr. Zelenkofske’s employment “without cause” or Dr. Zelenkofske resigns for “good reason,” 100% accelerated vesting of his options. The agreement will further provide that Dr. Zelenkofske may continue to practice medicine two days per month (or more with our written permission) and may remain on any boards of directors or advisory boards as long as those positions do not represent a conflict of interest.
On December 19, 2013, we further amended Dr. Zelenkofske’s amended and restated employment agreement to provide for reimbursement of reasonable expenses for travel between his place of residence in Pennsylvania and our office in Basking Ridge, New Jersey and for lodging in the Basking Ridge area, of up to $25,000 per year and certain revisions to the Section 280G cutback provisions in his employment agreement.
Each of our named executive officers signed a proprietary information, inventions and noncompetition agreement which provides for, among other things, a one year non-compete following termination of employment.
Outstanding Equity Awards at Year End
The following table sets forth certain information, on an award-by-award basis, concerning outstanding equity awards for each named executive officer as of December 31, 2013.
| | | | | | | | | | | | | | |
Name | | Number of Securities
Underlying Unexercised
Options Exercisable
(#) | | | Number of Securities
Underlying Unexercised
Options Unexercisable
(#) | | | Option
Exercise
Price
($) | | | Option
Expiration
Date(1) |
David J. Mazzo | | | — | | | | 558,167 | | | | 4.00 | | | 12/31/2023 |
| | | 28,179 | | | | 15,452 | | | | 3.84 | | | 4/25/2022(2) |
| | | 151,794 | | | | — | | | | 4.51 | | | 4/30/2020 |
| | | 134,730 | | | | — | | | | 12.53 | | | 8/01/2018 |
| | | 10,059 | | | | — | | | | 11.69 | | | 12/08/2018 |
82
| | | | | | | | | | | | | | |
Name | | Number of Securities
Underlying Unexercised
Options Exercisable
(#) | | | Number of Securities
Underlying Unexercised
Options Unexercisable
(#) | | | Option
Exercise
Price
($) | | | Option
Expiration
Date(1) |
| | | | |
Michael A. Metzger | | | 108,502 | | | | 325,506 | | | | 4.81 | | | 12/15/2023(3) |
| | | | |
Steven L. Zelenkofske | | | — | | | | 225,552 | | | | 4.00 | | | 12/31/2023 |
| | | 11,275 | | | | 6,183 | | | | 3.84 | | | 4/25/2022(2) |
| | | 43,158 | | | | — | | | | 4.51 | | | 4/30/2020 |
| | | 41,167 | | | | — | | | | 11.69 | | | 1/15/2019 |
| (1) | Unless otherwise indicated, each option to purchase our common stock vests as to 25% of the shares underlying such option on the first anniversary of the grant date of such option and 2.083% of the shares underlying each option vest each month thereafter. Each of these options has a ten-year term from the date of grant. |
| (2) | Such option vested as to 25% on May 25, 2012 and vests as to 2.083% of the shares in equal monthly installments over the subsequent 36 months. Each of these options has a ten-year term from the date of grant. |
| (3) | Such option vested as to 25% on December 16, 2013 and vests as to 2.083% of the shares in equal monthly installments over the subsequent 36 months. This option has a ten-year term from the date of grant. |
Equity Compensation Plans
2004 Equity Compensation Plan
The following is a summary of the material terms of our 2004 Equity Compensation Plan, as amended, or the 2004 Plan. This description is not complete. For more information, we refer you to the full text of the 2004 Plan, which we have filed as an exhibit to this annual report on Form 10-K.
The 2004 Plan permits us to make grants of nonqualified stock options, incentive stock options, restricted stock, and stock awards. Awards under the 2004 Plan may be granted to employees, officers, non-employee board members, consultants and other service providers of our company and its affiliates.
An aggregate of 2,358,105 shares of common stock are authorized for issuance under the 2004 Plan, subject to adjustment as a result of mergers, consolidations, reorganizations, stock splits, stock dividends and other changes in our common stock. Upon effectiveness of the 2013 Equity Compensation Plan, we ceased making awards under the 2004 Plan, and outstanding stock options to acquire 1,406,910 shares of our common stock were assumed under the 2013 Equity Compensation Plan.
The 2004 Plan is administered by the compensation committee, although our board of directors may act in lieu of the compensation committee. The compensation committee has the authority to interpret, construe and administer the 2004 Plan and to act in all matters pertaining to the grant of an award, including:
| | • | | which individuals shall be granted options and other awards; |
| | • | | the number of shares of our common stock subject to options and other awards; and |
| | • | | the form, terms, conditions and duration of each award. |
Unless otherwise determined by the compensation committee, awards of stock options granted under the 2004 Plan will not be transferable other than by will or by the laws of descent and distribution, or, to the extent permitted by applicable law, pursuant to a qualified domestic relations order, and restricted stock will not be transferable until the applicable restrictions lapse. The compensation committee may in its discretion impose restrictions on the transfer of stock awards.
Each award under the 2004 Plan is subject, in the discretion of the committee, to cancellation if the participant is terminated for cause, or if the participant becomes associated with, employed by, renders services to, or owns any interest in (other than any insubstantial interest) any business that is in competition with the Company or any business in which the Company has a substantial interest or that has a substantial interest in the Company.
The board may amend, alter or discontinue the 2004 Plan in any respect at any time, but no amendment or discontinuance may adversely affect the rights of a participant under any awards previously granted, without his or her consent, except that stockholder approval will be needed, to the extent required by Internal Revenue Code section 422 and/or stock market requirements, for any amendment that would increase the maximum number of shares available for awards, materially increase the benefits accruing to participants under the plan, extend the period during which incentive stock options may be granted, change the class of eligible participants, limit the powers of the compensation committee with respect to the administration of the plan, or change the amendment provisions.
83
2013 Equity Compensation Plan
The following is a summary of the material terms of our 2013 Equity Compensation Plan, or the 2013 Plan, which our board of directors and stockholders adopted in May 2013 and became effective upon the consummation of the IPO. This description is not complete. For more information, we refer you to the full text of the 2013 Plan, which is filed as an exhibit to this Annual Report on Form 10-K. Upon effectiveness of the 2013 Plan, outstanding stock options to acquire 1,406,910 shares of our common stock were assumed under the 2013 Plan.
The 2013 Plan permits us to make grants of nonqualified stock options, incentive stock options, stock appreciation rights, restricted stock, restricted stock units, performance shares, performance units, incentive bonus awards, other cash-based awards and other stock-based awards. Awards under the 2013 Plan may be granted to employees, officers, directors, consultants, advisors, other individual service providers of our Company or any subsidiary, or any persons determined by our compensation committee to be prospective employees, officers, directors, consultants, advisors, or other individual service providers of our Company or any subsidiary.
The number of shares available for issuance under the 2013 Plan shall be five percent of the Company’s outstanding common stock on the IPO Date, plus (i) any reserved shares not issued or subject to outstanding grants under the Company’s 2004 Plan on the IPO Date, (ii) outstanding stock options to acquire 1,406,910 shares of our common stock under the 2004 Plan on the IPO Date granted to employees, officers, directors and consultants, (iii) such additional shares covered by options issued under the 2004 Plan and not assumed under the 2013 Plan that are forfeited or otherwise terminated after the IPO Date, and (iv) shares issued under the 2004 Equity Compensation Plan that are repurchased by the Company at or below the original issue price. The number of shares available for issuance is subject to customary adjustments for stock splits, stock dividends or similar transactions.
In addition, the Plan contains an “evergreen” provision allowing for an annual increase in the number of shares of our common stock available for issuance under the 2013 Plan on January 1 of each year during the period beginning January 1, 2014, and ending on (and including) January 1, 2023. The annual increase in the number of shares shall be equal to 5% of the total number of shares of common stock outstanding on December 31st of the preceding calendar year; provided, however, that our board of directors may act prior to the first day of any calendar year to provide that there shall be no increase such calendar year, or that the increase shall be a lesser number of shares of common stock than would otherwise occur.
The 2013 Plan will be administered by the compensation committee of our board of directors. The compensation committee has the authority to determine:
| | • | | which individuals shall be granted options and other awards; |
| | • | | the number of shares of our common stock subject to options and other awards; |
| | • | | the exercise price of each option; |
| | • | | the schedule upon which options become exercisable, including exercise of unvested nonqualified stock options; |
| | • | | the termination or cancellation provisions applicable to options; |
| | • | | the terms and conditions of other awards, including the base or purchase price, and conditions for repurchase, termination or cancellation; and |
| | • | | all other terms and conditions upon which each award may be granted. |
No participant may receive in any one fiscal year options stock appreciation rights, restricted stock, stock units, performance share awards, incentive bonus awards and other stock-based awards that are denominated in shares of common stock with respect to more than 958,083 shares in the aggregate. The maximum dollar value payable to any participant in any one fiscal year of the Company with respect to stock units, performance units or incentive bonus awards or other stock-based awards that may be settled in cash or other property (other than common stock) is $2 million.
In general, awards may not be transferred other than by will or by the laws of descent and distribution. However, the compensation committee may permit the holder of an option, stock appreciation right or other award to transfer the option, right or other award to immediate family members, to a family trust for estate planning purposes, or by gift to charitable institutions.
The compensation committee may, upon the grant of an award, provide for the effect of a change in control on any award, including:
| | • | | acceleration or extension of the time periods for exercising, vesting in, or realizing gain from any award; |
| | • | | elimination or modification of performance or other conditions of an award; and |
| | • | | provision for the cash settlement of an award for an equivalent cash value. |
84
The compensation committee may also, in its discretion, take one or more of the following actions contingent upon a change in control:
| | • | | cause any or all outstanding options and stock appreciation rights to become immediately exercisable, in whole or in part; |
| | • | | cause any other awards to become non-forfeitable, in whole or in part; |
| | • | | cancel any option or stock appreciation right in exchange for a substitute option; |
| | • | | cancel any award of restricted stock, restricted stock units, performance shares or performance units in exchange for a similar award of the capital stock of any successor corporation; |
| | • | | redeem any restricted stock, restricted stock unit, performance share or performance unit for cash and/or other substitute consideration with a value equal to the fair market value of an unrestricted share of our common stock on the date of the change in control; |
| | • | | cancel any option or stock appreciation right in exchange for cash and/or other substitute consideration based on the value of our common stock on the date of the change in control,and cancel any option or stock appreciation right without any payment if its exercise price exceeds the value of our common stock on the date of the change in control; or |
| | • | | make such other modifications, adjustments or amendments to outstanding awards as it deems necessary or appropriate. |
The compensation committee may amend the terms of awards in any manner not inconsistent with the 2013 Plan, provided that no amendment shall adversely affect the rights of a participant with respect to an outstanding award without the participant’s consent. In addition, our board of directors may at any time amend, suspend, or terminate the 2013 Plan, provided that (i) no such amendment, suspension or termination shall materially and adversely affect the rights of any participant under any outstanding award without the consent of such participant and (ii) to the extent necessary to comply with any applicable law or stock exchange rule the 2013 Plan requires us to obtain stockholder consent. Stockholder approval is required for any plan amendment that increases the number of shares of common stock available for issuance under the 2013 Plan or changes the persons or classes of persons eligible to receive awards.
2013 Employee Stock Purchase Plan
The following is a summary of the material terms of our Employee Stock Purchase Plan, or the ESPP, which our board of directors and stockholders adopted in May 2013, effective upon the consummation of the IPO. As this description is not complete, we refer you to the full text of the ESPP, which is filed as an exhibit to this Annual Report on Form 10-K. Upon effectiveness, up to 96,360 shares of our common stock may be issued pursuant to the ESPP.
Our board of directors may implement the ESPP by designating offering periods during which participating employees may be granted options to purchase our common stock. Offering periods will be approximately six months, or such other length as determined by our board of directors. The board of directors has not designated an initial offering period, and has no current plans to do so.
All employees are eligible to be granted options under the ESPP, except no employee may be granted an option (i) to the extent the employee would own shares of our common stock (or options to purchase shares of our common stock) representing five percent or more of the total combined voting power or value of all classes of our capital or of any subsidiary; (ii) to the extent his or her rights to purchase stock under all of our and our subsidiaries’ employee stock purchase plans exceeds $25,000 for each calendar year during which an option is outstanding; and (iii) if the employee received a hardship withdrawal from our 401(k) plan within the preceding six months.
A participant in the ESPP may purchase shares of our common stock by electing payroll deductions of a percentage of his or her gross compensation during an offering period, excluding severance and non-cash compensation. Payroll deductions will be credited to the participant’s account, and will be used to purchase common stock on the last trading day of each offering period. In general, the purchase price of common stock will be 85% of the fair market value of the common stock on the first trading day or the last trading day of the offering period, whichever is lower. At any time, a participant may withdraw all (but not less than all) of the payroll deductions credited to his or her account and not yet used to exercise his or her option.
Our board of directors may amend or terminate the ESPP at any time and for any reason, subject to stockholders approval to the extent required by law, regulation or stock exchange rule. Except as otherwise provided in the ESPP, no amendment or termination may adversely affect options previously granted.
85
Director Compensation
Non-Employee Director Compensation Policy
Our board of directors has approved a compensation policy for our non-employee directors that became effective upon consummation of the IPO. This policy provides for the following compensation to our non-employee directors:
| | • | | each non-employee director, other than the chairman of our board, will receive an annual base fee of $40,000; |
| | • | | in addition to the $40,000 annual base fee, the chairman of our audit committee will receive an annual fee of $20,000 and other members of our audit committee will receive an annual fee of $10,000; |
| | • | | in addition to the $40,000 annual base fee, the chairman of our compensation committee will receive an annual fee of $15,000 and other members of our compensation committee will receive an annual fee of $7,500; |
| | • | | in addition to the $40,000 annual base fee, the chairman of our nominating and corporate governance committee will receive an annual fee of $10,000 and other members of our nominating and corporate governance committee will receive an annual fee of $5,000; and |
| | • | | the chairman of our board of directors will receive an annual fee of $120,000 and will not receive any additional fees for service on any committees or as chair of any committees. |
All fees under the director compensation policy will be paid on a quarterly basis in arrears and no per meeting fees will be paid. We will also reimburse non-employee directors for reasonable expenses incurred in connection with attending board and committee meetings.
Effective with the consummation of the IPO, each non-employee director was granted an option to purchase 17,964 shares of our common stock. Following the consummation of the IPO each non-employee director on the date of his or her initial election to our board of directors will receive an option award with a Black Scholes value equal to $200,000. In addition, each year on January 1 each non-employee director will receive an option award with a Black-Scholes value equal to $100,000 multiplied by a fraction, the numerator of which is the number of full months’ service as a director in the year to which the award pertains, and the denominator of which is 12. Such initial and annual option grants will have an exercise price per share equal to the fair market value of our common stock on the date of grant. Each initial and annual grant will vest in full on the one-year anniversary of the date of grant.
Director Compensation – 2013
The following table summarizes the annual compensation for our non-employee directors during 2013.
| | | | | | | | | | | | | | | | |
| | | | |
| Name | | Fees
Earned or
Paid in Cash
($) | | | Option
Awards
($)(1) | | | All Other
Compensation
($) | | | Total
($) | |
Dennis Podlesak | | | 120,000 | | | | 548,967 | | | | — | | | | 668,967 | |
B. Jefferson Clark | | | 21,617 | | | | 26,900 | | | | — | | | | 48,517 | |
Anton Gopka | | | 15,571 | | | | 26,900 | | | | — | | | | 42,471 | |
Pierre Legault | | | 7,174 | | | | 78,845 | | | | — | | | | 86,019 | |
Michael E. Mendelsohn | | | 19,833 | | | | 77,291 | | | | — | | | | 97,124 | |
P. Sherrill Neff | | | 20,435 | | | | 26,900 | | | | — | | | | 47,335 | |
Jesse Treu | | | 20,435 | | | | 26,900 | | | | — | | | | 47,335 | |
Raphael Wisniewski | | | 19,321 | | | | 26,900 | | | | — | | | | 46,221 | |
| (1) | Represents the aggregate grant date fair value for grants made in 2013 computed in accordance with FASB ASC Topic 718. The assumptions we used in valuing options are described in note 10 to our financial statements included in this annual report on Form 10-K. |
Limitation of Liability and Indemnification
As permitted by Delaware law, our amended and restated certificate of incorporation, limits or eliminates the personal liability of our directors to the maximum extent permitted by Delaware law. Delaware law provides that directors of a corporation will not be personally liable for monetary damages for breaches of their fiduciary duties as directors, except liability for:
| | • | | any breach of the director’s duty of loyalty to us or our stockholders; |
| | • | | any act or omission not in good faith or that involves intentional misconduct or a knowing violation of law; |
86
| | • | | any unlawful payments related to dividends or unlawful stock repurchases, redemptions or other distributions; or |
| | • | | any transaction from which the director derived an improper personal benefit. |
These limitations do not apply to liabilities arising under federal securities laws and do not affect the availability of equitable remedies, including injunctive relief or rescission. If Delaware law is amended to authorize the further elimination or limiting of director liability, then the liability of our directors will be eliminated or limited to the fullest extent permitted by Delaware law as so amended.
As permitted by Delaware law, our amended and restated bylaws that will become effective upon the completion of this offering also provide that:
| | • | | we will indemnify our directors and officers to the fullest extent permitted by law; |
| | • | | we may, to the extent authorized by the board of directors, indemnify our other employees and other agents to the same extent that we indemnify our officers and directors, unless otherwise determined by the board of directors; and |
| | • | | we will advance expenses to our directors and executive officers in connection with legal proceedings to the fullest extent permitted by law. |
These indemnification provisions are not exclusive.
In addition to the indemnification provided for in our amended and restated bylaws, we have entered into indemnification agreements with each of our directors and executive officers. Each indemnification agreement provides that we will indemnify the director or executive officer to the fullest extent permitted by law for claims arising in his or her capacity as our director, officer, employee or agent, provided that he or she acted in good faith and in a manner that he or she reasonably believed to be in, or not opposed to, our best interests and, with respect to any criminal proceeding, had no reasonable cause to believe that his or her conduct was unlawful. In the event that we do not assume the defense of a claim against a director or executive officer, we are required to advance his or her expenses in connection with his or her defense, provided that he or she undertakes to repay all amounts advanced if it is ultimately determined that he or she is not entitled to be indemnified by us. We believe that these provisions and agreements are necessary to attract and retain qualified persons as directors and executive officers. Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers or persons controlling our company pursuant to the foregoing provisions, the opinion of the SEC is that such indemnification is against public policy as expressed in the Securities Act and is therefore unenforceable.
In addition, we maintain standard policies of insurance under which coverage is provided to our directors and officers against losses arising from claims made by reason of breach of duty or other wrongful act, and to us with respect to payments which may be made by us to such directors and officers pursuant to the above indemnification provisions or otherwise as a matter of law.
401(k) Plan
As of the first day of the month following their hire date, all of our employees become eligible to participate in our 401(k) plan, which is a retirement savings defined contribution plan established in accordance with Section 401(a) of the Code. Pursuant to our 401(k) plan, employees may elect to defer their eligible compensation into the plan on a pre-tax basis, up to the statutorily prescribed annual limit of $17,500 in 2013 (additional salary deferrals not to exceed $5,500 are available to those employees 50 years of age or older) and to have the amount of this reduction contributed to our 401(k) plan. In general, eligible compensation is defined as compensation as reflected in each employee’s Form W-2. Effective December 19, 2013 we elected to match participant elective deferrals in an amount equal to 50% of such elective deferrals up to 3% of each participant’s total compensation for the Plan Year. We also declared a retroactive match for the year ended December 31, 2013. Our aggregate 401K match expense for the year ended December 31, 2013 was $87,813. We may, but are not required, to make matching contributions to the 401(k) plan. The 401(k) plan currently does not offer the ability to invest in our securities.
Rule 10b5-1 Sales Plans
Our directors and executive officers may adopt written plans, known as Rule 10b5-1 plans, in which they will contract with a broker to buy or sell shares of our common stock on a periodic basis. Under a Rule 10b5-1 plan, a broker executes trades pursuant to parameters established by the director or executive officer when entering into the plan, without further direction from the director or executive officer. The director or executive officer may amend or terminate the plan in some circumstances. Our directors and executive officers may also buy or sell additional shares outside of a Rule 10b5-1 plan when they are not in possession of material, nonpublic information. However, pursuant to the terms of the lock-up agreements signed by our directors and executive officers, no Rule 10b5-1 plan may provide for the transfer of common stock prior to the expiration of the applicable lock-up agreements.
87
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
PRINCIPAL STOCKHOLDERS
The following table sets forth the beneficial ownership of our common stock as of March 1, 2014 by:
| | • | | each person, or group of affiliated persons, who we know to beneficially own more than 5% of the outstanding shares of our common stock; |
| | • | | each of our named executive officers; |
| | • | | each of our executive officers; |
| | • | | each of our directors; and |
| | • | | all of our executive officers and directors as a group. |
The percentage ownership information shown in the table is based on an aggregate of 25,327,266 shares of our common stock outstanding as of March 1, 2014.
We have determined beneficial ownership in accordance with the rules of the Securities and Exchange Commission. These rules generally attribute beneficial ownership of securities to persons who possess sole or shared voting power or investment power with respect to those securities. In addition, the rules include shares of common stock issuable pursuant to the exercise of stock options or warrants that are either immediately exercisable or exercisable on or before April 30, 2014 which is 60 days after March 1, 2014. These shares are deemed to be outstanding and beneficially owned by the person holding those options or warrants for the purpose of computing the percentage ownership of that person, but they are not treated as outstanding for the purpose of computing the percentage ownership of any other person. Unless otherwise indicated, the persons or entities identified in this table have sole voting and investment power with respect to all shares shown as beneficially owned by them, subject to applicable community property laws.
88
Except as otherwise noted below, the address for persons listed in the table is c/o Regado Biosciences, Inc., 120 Mountain View Boulevard, Basking Ridge, New Jersey 07920. Beneficial ownership representing less than 1% is denoted with an asterisk (*).
| | | | | | | | |
Name of Beneficial Owner | | Total Number of Shares
Beneficially Owned | | | Percentage of Common
Stock Beneficially Owned | |
5% Stockholders | | | | | | | | |
| | |
BVF Partners (1) | | | 3,659,091 | | | | 14.5 | % |
1 Sansome Street 39th Floor San Francisco, CA 94104 | | | | | | | | |
| | |
Edmond de Rothschild Investment Partners (2) | | | 2,407,271 | | | | 9.5 | % |
47 rue du Faubourg Saint Honore 75008 Paris, France | | | | | | | | |
| | |
Entities affiliated with Domain Partners (3) | | | 2,603,208 | | | | 10.3 | % |
One Palmer Square Princeton, NJ 08542 | | | | | | | | |
| | |
RMI Investments, S.á.r.l.(4) | | | 5,236,517 | | | | 20.7 | % |
125047, bldg. 29/22 1st Brestskaya Street Moscow, Russian Federation | | | | | | | | |
| | |
Robert Kierlin | | | 3,709,955 | | | | 14.7 | % |
2001 Theurer Boulevard Winona, MN 55987 | | | | | | | | |
| | |
Quaker Bioventures, L.P. (5) | | | 1,311,189 | | | | 5.2 | % |
Cira Centre 2929 Arch Street Philadelphia, PA 19104-2868 | | | | | | | | |
| | |
Named Executive Officers, Executive Officers and Directors: | | | | | | | | |
| | |
David J. Mazzo, Ph.D.(6) | | | 329,833 | | | | 1.3 | % |
Michael Metzger (7) | | | 144,667 | | | | * | |
Christopher E. Courts (8) | | | 46,297 | | | | * | |
Alexander R. Giaquinto (7) | | | 12,029 | | | | * | |
Christopher P. Rusconi (9) | | | 321,695 | | | | 1.3 | % |
Steven L. Zelenkofske (7) | | | 97,055 | | | | * | |
Dennis Podlesak (7) | | | 199,645 | | | | 0.8 | % |
B. Jefferson Clark (10) | | | 1,147,302 | | | | 4.5 | % |
Anton Gopka (11) | | | 5,236,517 | | | | 20.7 | % |
Pierre Legault | | | — | | | | * | |
Michael E. Mendelsohn | | | — | | | | * | |
P. Sherrill Neff (12) | | | 1,311,189 | | | | 5.2 | % |
Jesse Treu (13) | | | 2,603,208 | | | | 10.3 | % |
Raphaël Wisniewski (14) | | | 2,407,271 | | | | 9.5 | % |
| | |
All current directors and executive officers as a group | | | 13,844,390 | | | | 52.3 | % |
89
| 1) | Consists of 1,979,615 shares of common stock beneficially owned by Biotechnology Value Fund, LP, or BVF, LP, 1,121,257 shares of common stock beneficially owned by Biotechnology Value Fund II, LP, or BVFII, LP, and 558,219 shares of common stock beneficially owned by Investment 10, LLC. BVF Partners LP is the General Partner of BVF, LP and BVFII, LP, and the investment advisor for Investment 10, LLC. Mark Lampert is the President of BVF Inc., which is the General Partner of BVF Partners LP. BVF Partners LP, as the General Partner of BVF, LP and BVFII, LP, and investment advisor for Investment 10, BVF Inc., as the general partner of BVF Partners LP and Mr. Lampert, as director and officer of BVF Inc. share voting or investment control with respect to the securities directly held by BVF, LP, BVFII, LP and Investment 10, LLC. Each of BVF Inc., BVF Partners LP and Mr. Lampert disclaim beneficial ownership of the securities held by BVF, LP, BVFII, LP and Investment 10, LLC except to the extent of their pecuniary interest therein. |
| 2) | Consists of 2,407,271 shares of common stock held by BioDiscovery 3 FCPREdmond de Rothschild Investment Partners, or Rothschild, is the management company for BioDiscovery 3 FCPR. Pierre-Michel Passy is the president of Rothschild and has voting and investment power with respect to the securities held by BioDiscovery 3 FCPR. Raphael Wisniewski is an employee of Rothschild and a partner of BioDiscovery 3 FCPR. Each of Mr. Wisniewski and Mr. Passy disclaims beneficial ownership of the securities held by BioDiscovery 3 FCPR except to the extent of his pecuniary interest therein, if any. |
| 3) | Consists of 2,588,288 shares of common stock owned by Domain Partners VI, L.P and 14,920 shares of common stock owned by DP VI Associates, L.P. The managing members of One Palmer Square are James Blair, Kathleen Schoemaker, Dr. Treu, a member of our board of directors, Brian Dovey and Nicole Vitullo. Each of James Blair, Kathleen Schoemaker, Dr. Treu, Brian Dovey and Nicole Vitullo share voting and investment power with respect to the securities held by Domain Partners VI and DP VI Associates. The managing members of Domain Associates are James Blair, Kathleen Schoemaker, Dr. Treu, Brian Dovey, Nicole Vitullo, Brian Halak and Kim Kamdar. Each of James Blair, Kathleen Schoemaker, Dr. Treu, Brian Dovey, Nicole Vitullo, Brian Halak and Kim Kamdar share voting and investment power with respect to the securities held by Domain Associates. Dennis Podlesak, a member of our board, is an employee of Domain Associates. Each of James Blair, Kathleen Schoemaker, Dr. Treu, Brian Dovey and Nicole Vitullo disclaims beneficial ownership of the securities held by Domain Partners VI and DP VI Associates except to the extent of his or her pecuniary interest therein, if any. Each of James Blair, Kathleen Schoemaker, Dr. Treu, Brian Dovey, Nicole Vitullo, Brian Halak, Kim Kamdar and Dennis Podlesak disclaims beneficial ownership of the securities held by Domain Associates except to the extent of his or her pecuniary interest therein, if any. |
| 4) | Consists of 5,236,517 shares of common stock held by RMI Investments, S.a.r.l., or RMI. RMI LLC, the parent company of RMI, and D-Pharma, management company for RMI LLC, may be deemed to beneficially own the shares of Common Stock. Anton Gopka is a managing director at D-Pharma and a member of the board of directors of the Issuer and may be deemed to beneficially own the shares of Common Stock. Vladimir Gurdus is Director of D-Pharma and may be deemed to beneficially own the shares of Common Stock. RMI LLC, D-Pharma, Mr. Gopka and Mr. Gurdus disclaim beneficial ownership of these securities, except to the extent of their respective pecuniary interest therein, and this report is not an admission that either RMI LLC, D-Pharma, Mr. Gopka or Mr. Gurdus is the beneficial owner of such securities. |
90
| 5) | Consists of 1,294,021 shares of common stock held by Quaker BioVentures L.P., or Quaker BioVentures, and 17,168 options exercisable on or before April 30, 2014, which Mr. Neff holds as nominee for Quaker Management. The shares directly held by Quaker BioVentures are indirectly held by Quaker BioVentures Capital, L.P., or Quaker Capital LP, as general partner of Quaker BioVentures and Quaker BioVentures Capital, LLC, or Quaker Capital LLC, as general partner of Quaker Capital LP. P. Sherrill Neff is a partner in Quaker Partners Management, L.P., or Quaker Management, and he, Richard Kollender, Ira Lubert and Adele Oliva are members of the investment committee of Quaker Management, which controls the voting and disposition of these shares. Each of Mr. Neff, Richard Kollender, Ira Lubert and Adele Oliva disclaims beneficial ownership of the securities held by Quaker BioVentures except to the extent of his pecuniary interest therein, if any. Mr. Neff disclaims beneficial ownership of the securities held by Quaker BioVentures and as nominee for Quaker Management except to the extent of his pecuniary interest therein, if any. |
| 6) | Consists of 328,398 shares of common stock issued to David J. Mazzo underlying options exercisable on or before April 30, 2014. |
| 7) | Represents shares of common stock underlying options exercisable on or before April 30, 2014. |
| 8) | Consists of 5,405 shares of common stock and 40,892 shares of common stock underlying options exercisable on or before April 30, 2014. |
| 9) | Consists of 22,904 shares of common stock and 298,791 shares of common stock underlying options exercisable on or before April 30, 2014. |
| 10) | Consists of 397,147 shares of common stock and 6,285 options exercisable on or before April 30, 2014 owned by Aurora Ventures IV, L.L.C., and 732,987 shares of common stock owned by Aurora Ventures V, L.L.C. which Mr. Clark may be deemed to share voting and investment power. Mr. Clark disclaims beneficial ownership of the securities held by Aurora IV and Aurora V except to the extent of his pecuniary interest therein, if any. |
| 11) | Consists of securities beneficially owned by RMI Investments as set forth in footnote 5, for which Mr. Gopka may be deemed to share voting and investment power. Mr. Gopka disclaims beneficial ownership of the securities held by RMI Investments except to the extent of his pecuniary interest therein, if any. |
| 12) | Consists of securities beneficially owned by Quaker BioVentures as set forth in footnote 5, for which Mr. Neff may be deemed to share voting and investment power. Mr. Neff disclaims beneficial ownership of the securities held by Quaker BioVentures, except to the extent of his pecuniary interest therein, if any. |
| 13) | Consists of securities beneficially owned by Domain Partners VI, DP VI Associates and Domain Associates as set forth in footnote 3, for which Dr. Treu may be deemed to share voting and investment power. Dr. Treu disclaims beneficial ownership of the securities held by Domain Partners VI, DP VI Associates and Domain Associates except to the extent of his pecuniary interest therein, if any. |
| 14) | Consists of securities beneficially owned by BioDiscovery 3 FCRP as set forth in footnote 2, for which Mr. Wisniewski may be deemed to share voting and investment power. Mr. Wisniewski disclaims beneficial ownership of the securities held by BioDiscovery 3 FCRP except to the extent of his pecuniary interest therein, if any. |
91
Equity Compensation Plan Information
Refer to the Equity Compensation Plan Information herein under Part II, Item 5, for information as of December 31, 2013 regarding shares of our common stock that may be issued under our existing equity compensation plans, including our 2004 Plan, 2013 Plan and 2013 ESPP
Item 13. Certain Relationships, Related Transactions and Director Independence
Other than compensation arrangements for our named executive officers and directors, we describe below each transaction or series of similar transactions, since January 1, 2011, to which we were a party or will be a party, in which:
| | • | | the amounts involved exceeded or will exceed $120,000; and |
| | • | | any of our directors, executive officers or holders of more than 5% of our capital stock, or any member of the immediate family of the foregoing persons, had or will have a direct or indirect material interest. |
Compensation and indemnification arrangements for our named executive officers and directors are described in the section entitled “Executive Compensation.”
Series D Financing
Pursuant to the terms of our Series D Preferred Stock Purchase Agreement, we sold an aggregate of 71,666,667 shares of our Series D Preferred Stock at a purchase price of $0.72 per share. The purchasers included the following holders of more than 5% of our capital stock or entities affiliated with them. The following table presents the number of shares issued to such purchasers.
| | | | | | | | |
Name of Investor | | Shares of Series D Preferred
Stock Purchased | | | Aggregate
Purchase
Price | |
DP VI Associates, L.P. and Domain Partners VI, L.P. | | | 15,055,507 | | | $ | 10,839,965 | |
Quaker BioVentures, L.P. | | | 7,769,715 | | | $ | 5,594,195 | |
Mr. Robert Kierlin | | | 23,261,013 | | | $ | 16,747,929 | |
Aurora Ventures V, LP | | | 5,376,964 | | | $ | 3,871,414 | |
BioDiscovery 3 FCPR | | | 17,066,243 | | | $ | 12,287,695 | |
Jesse Treu, a managing member of Domain Associates, LLC, the management company of Domain Partners VI, L.P., is a member of our board of directors. Dennis Podlesak, a partner of Domain Associates, LLC, is a member of our board of directors. P. Sherrill Neff, the founding partner of Quaker Partners Management, L.P., the management company of Quaker BioVentures, L.P., is a member of our board of directors. B. Jefferson Clark, a founder of Aurora Ventures V, LP, is a member of our board of directors. Edmond de Rothschild Investment Partners is BioDiscovery 3 FCPR’s management company and Raphaël Wisniewski, a partner at Edmond de Rothschild Investment Partners, is a member of our board of directors.
Convertible Debt Financing
On May 3, 2012, we sold an aggregate principal amount of approximately $6.8 million in convertible notes to certain of our existing investors, including certain holders of more than 5% of our capital stock or entities affiliated with them. On December 18, 2012, the convertible notes and accrued interest thereon converted into an aggregate 9,888,288 shares of Series E Preferred Stock, as more fully described below in “—Series E Financing.” The following table presents the convertible notes issued to such investors.
| | | | |
Name of Investor | | Principal Amount of
Convertible Notes | |
Mr. Robert Kierlin | | $ | 2,800,071 | |
Domain Partners VI, L.P. | | $ | 1,780,469 | |
BioDiscovery 3 FCPR | | $ | 1,250,000 | |
Quaker BioVentures, L.P. | | $ | 375,000 | |
Aurora Ventures V, LP | | $ | 375,000 | |
92
Series E Financing
On December 18, 2012, we entered into a Series E Preferred Stock purchase agreement, or the Series E Purchase Agreement, pursuant to which we agreed to sell up to 70,528,086 shares of Series E Preferred Stock at a purchase price of $0.72 per share in an initial closing, or the Initial Closing, consisting of three tranches. The first tranche of the Initial Closing closed on December 18, 2012, at which time we issued 31,437,442 shares of Series E Preferred Stock for net cash proceeds of $15.0 million and conversion of $6.8 million in principal amount of convertible notes and accrued interest thereon. The Series E Purchase Agreement provided for closings of second and third tranches of the Initial Closing on or before March 14, 2013 and on or before January 17, 2014, respectively.
The purchasers of the Series E Preferred Stock include the following holders of more than 5% of our capital stock or entities affiliated with them: RMI Investments, S.á.r.l., or RMII, Mr. Robert Kierlin, Domain Partners VI, L.P., BioDiscovery 3 FCPR, Quaker BioVentures, L.P. and Aurora Ventures V, L.P. Jesse Treu, a managing member of Domain Associates, LLC, the management company of Domain Partners VI, L.P., is a member of our board of directors. Dennis Podlesak, a partner of Domain Associates, LLC, is a member of our board of directors. P. Sherrill Neff, the founding partner of Quaker Partners Management, L.P., the management company of Quaker BioVentures, L.P. is a member of our board of directors. B. Jefferson Clark, a member of our board of directors, is a founder of Aurora Ventures V, LP. Edmond de Rothschild Investment Partners, a holder of more than 5% of our capital stock, is BioDiscovery’s management company and Raphaël Wisniewski, a partner at Edmond de Rothschild Investment Partners, is a member of our board of directors. Anton Gopka, the designee of RMII, joined our board of directors in connection with this Series E financing transaction.
The second tranche of the Initial Closing closed on March 22, 2013, at which time we sold 7,160,084 additional shares of Series E Preferred Stock for gross proceeds of $5,155,260. In connection with the closing of the second tranche, we entered into waiver agreements pursuant to which the investors waived certain closing conditions relating to the date of closing, the accuracy of our representations and warranties as of the closing date and the execution and delivery of the Clinical Development and Collaboration Agreement. RMII’s waiver agreement, or the RMII waiver, also waived its separate condition that we complete certain patent assignments as more fully described below. Pursuant to the terms of the waiver agreements, we agreed to use commercially reasonable efforts to negotiate, execute and deliver the Clinical Development and Collaboration Agreement on or before May 31, 2013 and, in the case of the RMII waiver, to file certain patent transfer applications before April 30, 2013 and to take certain other related actions described therein. In accordance with the terms of the RMII waiver, the $5,155,260 to be invested by RMII and the 7,160,084 shares of Series E Preferred Stock to be purchased by RMII at the second tranche closing were deposited into escrow pursuant to the terms of an escrow agreement which provided that the investment proceeds would be released to us upon satisfaction of the revised closing conditions summarized above. We satisfied the closing conditions specified in the RMII waiver and the escrow has been released.
The tables below present the number of shares of Series E Preferred Stock purchased by such purchasers in the first and second tranches of the Initial Closing.
First Tranche
| | | | | | | | | | | | | | | | |
Name of Investor | | Cancellation
of
Indebtedness
Consideration
(including
Interest) | | | Purchase
Price Paid in
Cash | | | Total Consideration
(including Note
Conversion and Cash
Purchase Price) | | | Shares of Series E
Preferred
Stock Issued | |
RMI Investments, S.á.r.l. | | | — | | | $ | 11,317,479 | | | $ | 11,317,479 | | | | 15,718,721 | |
Mr. Robert Kierlin | | $ | 2,940,075 | | | $ | 1,200,000 | | | $ | 4,140,075 | | | | 5,750,104 | |
Domain Partners VI, L.P. | | $ | 1,869,492 | | | $ | 565,373 | | | $ | 2,434,865 | | | | 3,381,757 | |
BioDiscovery 3 FCPR | | $ | 1,312,500 | | | $ | 443,723 | | | $ | 1,756,223 | | | | 2,439,199 | |
Quaker BioVentures, L.P. | | $ | 393,750 | | | $ | 125,346 | | | $ | 519,096 | | | | 720,967 | |
Aurora Ventures V, L.P. | | $ | 393,750 | | | $ | 125,346 | | | $ | 519,096 | | | | 720,967 | |
Second Tranche
| | | | | | | | |
Name of Investor | | Purchase
Price Paid | | | Total Number of Shares of Series E
Preferred Stock Issued | |
RMI Investments, S.á.r.l. | | $ | 5,155,260 | | | | 7,160,084 | |
Mr. Robert Kierlin | | $ | 3,600,001 | | | | 5,000,001 | |
Domain Partners VI, L.P. | | $ | 281,518 | | | | 390,997 | |
BioDiscovery 3 FCPR | | $ | 283,445 | | | | 393,673 | |
Quaker BioVentures, L.P. | | $ | 62,413 | | | | 86,685 | |
Aurora Ventures V, L.P. | | $ | 62,413 | | | | 86,685 | |
93
The Series E Purchase Agreement also provided for the sale of additional shares of Series E Preferred Stock to take place on or before June 30, 2013 in a second closing, or the Second Closing. Purchasers of the Series E Preferred Stock in the Initial Closing had the right, but not the obligation, to purchase additional shares of Series E Preferred Stock at the Second Closing up to the full amount of their pro rata share of the funds to be invested in the Second Closing. RMII agreed to match the amount invested in any Second Closing up to $6.0 million, subject to certain conditions. In the event that more than $6.0 million of Series E Preferred Stock was sold to other investors at the Second Closing, RMII agreed to use its commercially reasonable efforts to obtain additional financing to match the excess investment.
Pursuant to the terms of a Termination Agreement entered into by the parties to the Series E Purchase Agreement, our obligation to sell additional shares of Series E Preferred Stock to the investors and the obligations of the investors to purchase additional shares of Series E Preferred Stock will terminate immediately prior to the consummation of this offering and no additional shares of Series E Preferred Stock will be sold pursuant thereto.
2014 Private Placement
Pursuant to the terms of the security purchase agreements, in 2014 we sold an aggregate of 4,000,000 shares of our common stock at a purchase price of $5.00 per share. The purchasers included Robert Kierlin, a holder of more than 5% of our capital stock. Mr. Kierlin purchased 400,000 shares of our common stock for an aggregate purchase price of $2.0 million. The purchasers also included Biotechnology Value Fund, LP, or BVF, LP, a holder of more than 5% of our capital stock. BVF, LP purchased 2,000,000 shares of our common stock for an aggregate purchase price of $10.0 million.
NovaMedica Agreements
In connection with our Series E Preferred Stock financing, in December 2012, we entered into a Technology Transfer Agreement, or the Tech Transfer Agreement, with Domain Russia Investments Limited, or DRI, an affiliate of Domain Partners VIII, L.P. Domain Partners VIII, L.P. and Domain Partners VI, L.P., a significant stockholder of our company, are both managed by Domain Associates, L.L.C. Pursuant to the Tech Transfer Agreement, in exchange for a nominal payment, we assigned to DRI certain patents and patent applications in Armenia, Azerbaijan, Belarus, Georgia, Kazakhstan, Kyrgyzstan, Moldova, Russia, Tajikistan, Turkmenistan, Ukraine and Uzbekistan, or the Covered Territory, and granted to DRI an exclusive, fully paid-up, royalty-free, irrevocable and assignable license under our non-patent intellectual property to develop and commercialize REG1 and our other product candidates in the Covered Territory. Immediately thereafter, we, together with DRI and NovaMedica LLC, or NovaMedica, executed an Assignment and Assumption Agreement, pursuant to which all of DRI’s rights and obligations under the NovaMedica Agreement were transferred to NovaMedica. DRI also has a right of first negotiation if we desire to partner with a third party to develop or commercialize future product candidates in the Covered Territory, which was assigned to NovaMedica. We agreed to take all action required to register or record the patent transfers to DRI in each country in the Covered Territory and to ensure the assignment of DRI’s rights under the Tech Transfer Agreement to NovaMedica. NovaMedica is jointly owned by RusnanoMed Invest LLC, or RusnanoMed Invest, and DRI. RMII, a significant stockholder of ours, is a wholly owned subsidiary of RusnanoMed Invest. In connection with the second tranche of the Initial Closing, we agreed to file certain patent transfer applications and to take certain other related actions which have been completed.
Under the terms of the Tech Transfer Agreement, upon request we have agreed to provide certain development support to NovaMedica and to use commercially reasonable efforts to assist NovaMedica to establish a manufacturing relationship with our third-party manufacturers. We also have agreed to provide NovaMedica with certain manufacturing know-how and support, including making our manufacturing employees available to provide scientific and technical explanations, advice and on-site support that may be reasonably requested by NovaMedica. NovaMedica is required to reimburse us for any out-of-pocket expenses incurred by us in providing this assistance, including travel-related expenses. We estimate that the aggregate out-of-pocket expense of providing such services will be approximately $250,000. In addition, prior to the first commercial sale of a product candidate, we have agreed to sell to NovaMedica sufficient quantities of each product candidate and related compounds to enable NovaMedica to conduct clinical trials of such product candidate in the Covered Territory at cost plus a mark-up in the low double digits, so long as any sale does not reasonably interfere with our own development and commercialization activities.
Concurrently with the signing of the Tech Transfer Agreement, we also entered into a letter agreement with DRI pursuant to which we are obligated to pay DRI a make-whole payment up to a maximum amount of $1.2 million in the event that an independent appraiser’s valuation of certain patent applications assigned to DRI under the Tech Transfer Agreement is less than $1.2 million. The letter agreement provides that such payment will be refunded to us if DRI receives certain capital contribution credits with respect to such patent applications in connection with its investment in NovaMedica. DRI
94
has advised us that the independent appraiser valued the assigned patent applications at more than $1.2 million. As a result, DRI is not entitled to any make-whole payment under the terms of the letter agreement. In addition, we have agreed to indemnify DRI against any claims brought by NovaMedica arising out of or resulting from any breach of specified representations and warranties which we made in the Tech Transfer Agreement up to a maximum amount of $1.2 million, less any payments made to DRI in connection with the valuation of the assigned intellectual property. Our indemnification obligation will expire two years following the first commercial sale of REG1 or our other product candidates in the Covered Territory or six years after the date of the letter agreement, if no such commercial sales have occurred.
The Tech Transfer Agreement also provides that we will enter into a Clinical Development and Collaboration Agreement, a Supply Agreement and certain related agreements with NovaMedica to implement the terms of the Tech Transfer Agreement. In connection with the second tranche of the Initial Closing, we agreed to use commercially reasonably efforts to negotiate, execute and deliver the Clinical Development and Collaboration Agreement on or before May 31, 2013. The Tech Transfer Agreement provides that the Supply Agreement will cover the commercial supply of product candidates and related drug compounds to NovaMedica at cost plus a mark-up in the low double digits.
In accordance with the terms of the Tech Transfer Agreement, in May 2013 we entered into a Clinical Development and Collaboration Agreement, or the Collaboration Agreement, with NovaMedica pursuant to which we agreed to assist NovaMedica in the development and commercialization of our product candidates in the Covered Territory. The Collaboration Agreement requires the formation of several committees consisting of our representatives and NovaMedica representatives to oversee the general development, day-to-day development work and commercialization of our product candidates for the intended field of use in the Covered Territory. All decisions of these committees must be made by unanimous vote, subject to a dispute resolution process. Under the terms of the Collaboration Agreement, the joint committees will determine a development plan for REG1 for its initial indication and any additional significant commercial indications for REG1, as well as for additional product candidates. NovaMedica will have sole responsibility for the costs and expenses of obtaining regulatory approval for our product candidates and for commercializing any approved products in the Covered Territory and will have the right to conduct its own clinical studies in the Covered Territory at its sole expense. NovaMedica also has the right to file applications for approval of our product candidates in the Covered Territory, subject to committee oversight. We have agreed, among other things, to provide NovaMedica with clinical data necessary for it to obtain necessary approvals in the Covered Territory, information relating to applications for regulatory approval of our product candidates, certain commercialization information and to assist NovaMedica in conducting any clinical trials necessary for regulatory approval of our product candidates in the Covered Territory. We also have agreed to provide NovaMedica with certain development know-how and support, including making our clinical development personnel available to provide scientific and technical explanations, advice and on-site support that may be reasonably requested by NovaMedica.
NovaMedica is required to reimburse us for any out-of-pocket expenses incurred by us in providing this assistance, including travel-related expenses. We estimate that the aggregate out-of-pocket expense of providing such services will be approximately $500,000. Pursuant to the Collaboration Agreement, we have agreed to use commercially reasonable efforts to include sites in the Covered Territory in our clinical trial programs for our product candidates at our sole expense. Under the Collaboration Agreement, prior to the first commercial sale of a product candidate in the Covered Territory, NovaMedica will have the right to purchase product candidates and related compounds from us or through us as are reasonable and necessary for it to conduct clinical trials in the Covered Territory at our cost plus a mark-up in the low double digits pursuant to a clinical supply agreement to be entered into within 120 days of the date of the Collaboration Agreement. NovaMedica has agreed to supervise and maintain sales representatives for the commercialization of any product candidates approved for sale in the Covered Territory. Within 90 days of receipt of FDA approval for the use of any product candidate, we are obligated to enter into a commercial supply agreement with NovaMedica for the supply of such candidate on terms to be negotiated by the parties. In the Collaboration Agreement, the parties also agreed to customary terms and conditions, including the ownership and use of intellectual property, rights to information, prosecution of patent rights, rights under third-party agreements, confidentiality and indemnification obligations and mechanisms for the resolution of disputes. The Collaboration Agreement expires on the earlier of three years following the first commercial sale of a product candidate in the Covered Territory or nine years from the date of effectiveness and terminates upon the termination of the Tech Transfer Agreement. NovaMedica also has the right to terminate the Collaboration Agreement at any time at its convenience upon 90 days prior written notice.
We neither paid nor received monies pursuant to the Collaboration Agreement during 2013.
95
Director Independence
Refer to Part III, Item 10, Directors, Executive Officers and Corporate Governance, herein, for discussion regarding director independence.
Item 14. Principal Accounting Fees and Services
Principal Accountant Fees and Services
The following table summarizes the fees for professional services rendered by Grant Thornton LLP, our independent registered public accounting firm, for each of the last two fiscal years:
| | | | | | | | |
| Fee Category | | 2013 | | | 2012 | |
| | | (In thousands) | |
| | |
Audit fees | | $ | 343 | | | $ | 314 | |
Tax fees | | | 13 | | | | 11 | |
| | | | | | | | |
Total fees | | $ | 356 | | | $ | 325 | |
| | | | | | | | |
Audit Fees
Represents fees, including out of pocket expenses, for professional services provided in connection with the audit of our annual audited financial statements, the review of our quarterly financial statements included in our Forms 10-Q, accounting consultations or advice on accounting matters necessary for the rendering of an opinion on our financial statements, services provided in connection with the offerings of our common stock and audit services provided in connection with other statutory or regulatory filings.
Tax Fees
Tax fees are associated with tax compliance and tax planning related activities.
The Audit Committee is responsible for appointing, setting compensation and overseeing the work of the independent auditors. The Audit Committee has established a policy regarding pre-approval of all auditing services and the terms thereof and non-audit services (other than non-audit services prohibited under Section 10A(g) of the Exchange Act or the applicable rules of the SEC or the Public Company Accounting Oversight Board) to be provided to us by the independent auditor. However, the pre-approval requirement may be waived with respect to the provision of non-audit services for us if the “de minimus” provisions of Section 10A(i)(1)(B) of the Exchange Act are satisfied.
The Audit Committee has considered whether the provision of Audit-Related Fees, Tax Fees, and all other fees as described above is compatible with maintaining Grant Thornton, LLP’s independence and has determined that such services for fiscal year 2013 were compatible. All such services were approved by the Audit Committee pursuant to Rule 2-01 of Regulation S-X under the Exchange Act to the extent that rule was applicable.
96
The Audit Committee is responsible for reviewing and discussing the audit financial statements with management, discussing with the independent registered public accountants the matters required in Auditing Standards No. 61, receiving written disclosures from the independent registered public accountants required by the applicable requirements of the Public Company Accounting Oversight Board regarding the independent registered public accountants’ communications with the Audit Committee concerning independence and discussing with the independent registered public accountants their independence, and recommending to the Board of Directors that the audit financial statements be included in our annual report on Form 10-K.
Item 15. Exhibits and Financial Statement Schedules
| | |
Exhibit
No. | | Description |
| |
| 3.1 | | Sixth Amended and Restated Certificate of Incorporation of Regado Biosciences, Inc. (incorporated by reference to Exhibit 3.1 of the Current Report on Form 8-K filed by the registrant on September 3, 2013.) |
| |
| 3.2 | | Amended and Restated Bylaws of Regado Biosciences, Inc. (incorporated by reference to Exhibit 3.1 of the Current Report on Form 8-K filed by the registrant on September 3, 2013.) |
| |
| 4.1 | | Specimen Common Stock certificate of Regado Biosciences, Inc. (Incorporated by reference to Exhibit 4.1 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 4.2 | | Stock Purchase Warrant dated as of November 19, 2004 to University Medical Discoveries, Inc. (Incorporated by reference to Exhibit 4.4 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 4.3 | | Form of Series A Warrant. (Incorporated by reference to Exhibit 4.5 to the registrant’s Registration Statement onForm S-1, as amended (File No. 333-188209)). |
| |
| 4.4 | | Stock Purchase Warrant, dated May 10, 2013, issued to Comerica Bank. (Incorporated by reference to Exhibit 4.6 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.1† | | Form of Indemnification Agreement. (Incorporated by reference to Exhibit 10.1 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.2† | | Regado Biosciences, Inc. 2004 Equity Compensation Plan, as amended. (Incorporated by reference to Exhibit 10.2 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.3† | | Form of Incentive Stock Option Agreement pursuant to Regado Biosciences, Inc. 2004 Equity Compensation Plan. (Incorporated by reference to Exhibit 10.3 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.4† | | Form of Nonqualified Stock Option Agreement pursuant to Regado Biosciences, Inc. 2004 Equity Compensation Plan. (Incorporated by reference to Exhibit 10.4 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.5† | | Regado Biosciences, Inc. 2013 Equity Compensation Plan. (Incorporated by reference to Exhibit 10.5 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.6† | | Form of Director Nonqualified Stock Option Agreement pursuant to Regado Biosciences, Inc. 2013 Equity Compensation Plan. (Incorporated by reference to Exhibit 10.6 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.7† | | Form of Employee, Officer and Consultant Nonqualified Stock Option Agreement pursuant to Regado Biosciences, Inc. 2013 Equity Compensation Plan. (Incorporated by reference to Exhibit 10.7 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
97
| | |
| |
| 10.8† | | Form of Restricted Stock Agreement pursuant to Regado Biosciences, Inc. 2013 Equity Compensation Plan. (Incorporated by reference to Exhibit 10.8 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.9† | | Regado Biosciences, Inc. Employee Stock Purchase Plan. (Incorporated by reference to Exhibit 10.9 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.10† | | Employment Agreement with David J. Mazzo dated July 25, 2008. (Incorporated by reference to Exhibit 10.10 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.11† | | Amendment No. 1 dated December 22, 2008 to Employment Agreement with David J. Mazzo. (Incorporated by reference to Exhibit 10.11 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.12† | | Amended and Restated Employment Agreement with David J. Mazzo, dated May 16, 2013. (Incorporated by reference to Exhibit 10.12 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.13† | | First Amendment dated December 23, 2013 to Employment Agreement by and between Regado Biosciences, Inc. and David J. Mazzo, dated May 16, 2013. (Incorporated by reference to Exhibit 10.1 of the Current Report on Form 8-K filed by the registrant on December 23, 2013.) |
| |
| 10.14† | | Employment Agreement with Christopher P. Rusconi dated August 1, 2008. (Incorporated by reference to Exhibit 10.13 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.15† | | Amended and Restated Employment Agreement with Christopher P. Rusconi, dated May 16, 2013. (Incorporated by reference to Exhibit 10.14 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.16† | | First Amendment dated December 23, 2013 to Employment Agreement by and between Regado Biosciences, Inc. and Christopher Rusconi, dated May 16, 2013. (Incorporated by reference to Exhibit 10.3 of the Current Report on Form 8-K filed by the registrant on December 23, 2013.) |
| |
| 10.17† | | Employment Agreement with Steven L. Zelenkofske dated November 19, 2008. (Incorporated by reference to Exhibit 10.15 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.18† | | Amendment No. 1 dated December 22, 2008 to Employment Agreement with Steve L. Zelenkofske. (Incorporated by reference to Exhibit 10.16 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.19† | | Amended and Restated Employment Agreement with Steven L. Zelenkofske, dated May 16, 2013. (Incorporated by reference to Exhibit 10.17 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.20† | | First Amendment dated December 23, 2013 to Employment Agreement by and between Regado Biosciences, Inc. and Steven L. Zelenkofske, dated May 16, 2013. (Incorporated by reference to Exhibit 10.2 of the Current Report onForm 8-K filed by the registrant on December 23, 2013.) |
| |
| 10.21† | | Employment Agreement with Christopher E. Courts dated August 1, 2008. (Incorporated by reference to Exhibit 10.21 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.22† | | Amended and Restated Employment Agreement with Christopher E. Courts, dated May 16, 2013. (Incorporated by reference to Exhibit 10.22 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.23† | | First Amendment dated December 23, 2013 to Employment Agreement by and between Regado Biosciences, Inc. and Christopher E. Courts, dated May 16, 2013. (Incorporated by reference to Exhibit 10.4 of the Current Report on Form 8-K filed by the registrant on December 23, 2013.) |
| |
| 10.24† | | Employment Agreement with Alexander Gianquinto dated November 8, 2008. (Incorporated by reference to Exhibit 10.23 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.25† | | Amended and Restated Employment Agreement with Alexander Gianquinto, dated May 16, 2013. (Incorporated by reference to Exhibit 10.24 to the registrant’s Registration Statement on Form S-1, as amended (FileNo. 333-188209) |
| |
| 10.26† | | First Amendment dated December 23, 2013 to Employment Agreement by and between Regado Biosciences, Inc. and Alexander R. Gianquinto, dated May 16, 2013. (Incorporated by reference to Exhibit 10.5 of the Current Report on Form 8-K filed by the registrant on December 23, 2013.) |
98
| | |
| |
| 10.27† | | Employment Agreement by and between Regado Biosciences, Inc. and Michael A. Metzger, dated December 5, 2013. (Incorporated by reference to Exhibit 10.1 of the Current Report on Form 8-K filed by the registrant on December 6, 2013.) |
| |
| 10.28 | | Series D Preferred Stock Purchase Agreement, dated as of December 17, 2009, by and among Regado Biosciences, Inc. and the purchasers named therein. (Incorporated by reference to Exhibit 10.25 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.29 | | Amendment No. 1 dated as of May 25, 2011 to Series D Preferred Stock Purchase Agreement by and among Regado Biosciences, Inc. and the investors named therein. (Incorporated by reference to Exhibit 10.26 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.30 | | Loan and Security Agreement, dated as of May 25, 2011, by and among MidCap Financial SBIC, LP, as administrative agent, the Lenders named therein and Regado Biosciences, Inc. (Incorporated by reference to Exhibit 10.27 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.31 | | First Amendment dated as of August 1, 2011 to the Loan and Security Agreement by and among MidCap Financial SBIC, LP, as administrative agent, the Lenders named therein and Regado Biosciences, Inc. (Incorporated by reference to Exhibit 10.28 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.32 | | Second Amendment dated as of September 30, 2011 to the Loan and Security Agreement by and among MidCap Financial SBIC, LP, as administrative agent, the Lenders named therein and Regado Biosciences, Inc. (Incorporated by reference to Exhibit 10.29 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.33 | | Third Amendment dated as of May 3, 2012 to the Loan and Security Agreement by and among MidCap Financial SBIC, LP, as administrative agent, the Lenders named therein and Regado Biosciences, Inc. (Incorporated by reference to Exhibit 10.30 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.34 | | Convertible Note Purchase Agreement by and among Regado Biosciences, Inc., dated as of May 3, 2012, and the purchasers named therein. (Incorporated by reference to Exhibit 10.31 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.35 | | Form of 8% Unsecured Convertible Note. (Incorporated by reference to Exhibit 10.32 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.36 | | Series E Preferred Stock Purchase Agreement, dated as of December 18, 2012, by and among Regado Biosciences, Inc. and the purchasers named therein. (Incorporated by reference to Exhibit 10.33 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.37# | | Technology Transfer Agreement, dated as of December 18, 2012, by and between Regado Biosciences, Inc. and Domain Russian Investments Limited. (Incorporated by reference to Exhibit 10.34 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.38 | | Assignment and Assumption Agreement, dated December 18, 2012, by and among Domain Russia Investments Limited, Regado Biosciences, Inc. and NovaMedica LLC. (Incorporated by reference to Exhibit 10.35 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.39 | | Letter Agreement, dated as of December 18, 2012, by and between Regado Biosciences, Inc. and Domain Russian Investments Limited. (Incorporated by reference to Exhibit 10.36 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.40# | | License Agreement, dated as of October 3, 2003, by and between Archemix Corp. and Regado Biosciences, Inc. (Incorporated by reference to Exhibit 10.37 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.41# | | License Agreement, dated as of November 18, 2004, by and between Duke University and Regado Biosciences, Inc. (Incorporated by reference to Exhibit 10.38 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
99
| | |
| |
| 10.42# | | First Amendment, dated as of July 13, 2005, to License Agreement by and between Duke University and Regado Biosciences, Inc. (Incorporated by reference to Exhibit 10.39 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.43# | | License, Manufacturing and Supply Agreement, dated as of December 22, 2006, by and between Nektar Therapeutics AL, Corporation and Regado Biosciences, Inc. (Incorporated by reference to Exhibit 10.40 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.44 | | Waiver of Certain Conditions to Closing of Second Tranche of the Initial Closing and Agreement to Revised Conditions, dated as of March 22, 2013, by and between Regado Biosciences, Inc. and RMI Investments, S.á.r.l. (Incorporated by reference to Exhibit 10.41 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.45 | | Waiver of Certain Conditions to Closing of Second Tranche of the Initial Closing and Agreement to Revised Conditions, dated as of March 22, 2013, by and among Regado Biosciences, Inc. and the investors named therein. (Incorporated by reference to Exhibit 10.42 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.46 | | Supply and Service Agreement, dated July 13, 2006, by and between Regado Biosciences, Inc. and Agilent Technologies, Inc. (Incorporated by reference to Exhibit 10.43 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.47 | | Amendment to Supply and Service Agreement, dated July 22, 2011, by and between Regado Biosciences, Inc. and Agilent Technologies, Inc. (Incorporated by reference to Exhibit 10.44 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.48 | | Clinical Supply Agreement, dated March 28, 2012, by and between Regado Biosciences, Inc. and Althea Technologies, Inc. (Incorporated by reference to Exhibit 10.45 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.49 | | Loan and Security Agreement, dated as of May 10, 2013, by and between Regado Biosciences, Inc. and Comerica Bank. (Incorporated by reference to Exhibit 10.46 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.50# | | Clinical Development and Collaboration Agreement, dated May 14, 2013, by and between NovaMedica, LLC and Regado Biosciences, Inc. (Incorporated by reference to Exhibit 10.47 to the registrant’s Registration Statement onForm S-1, as amended (File No. 333-188209)). |
| |
| 10.51 | | Termination Agreement, dated May 16, 2013, by and among Regado Biosciences, Inc. and the investors party to the Series E Purchase Agreement. (Incorporated by reference to Exhibit 10.48 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.52 | | Office Lease Agreement, dated May 1, 2013, by and between Keystone 430 TT, LLC and Regado Biosciences, Inc. (Incorporated by reference to Exhibit 10.49 to the registrant’s Registration Statement on Form S-1, as amended (File No. 333-188209)). |
| |
| 10.53 | | Form of Securities Purchase Agreement (Incorporated by reference to Exhibit 10.1 of the Current Report on Form 8-K filed by the registrant on January 31, 2014.) |
| |
| 21.1* | | List of Subsidiaries of the Company |
| |
| 23.1* | | Consent of Grant Thornton LLP |
| |
| 31.1* | | Certification of Principal Executive Officer pursuant to Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
| |
| 31.2* | | Certification of Principal Financial Officer pursuant to Rule 13a-14(a) and 15d-14(a) of the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
| |
| 32.1* | | Certification of Principal Executive Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| |
| 32.2* | | Certification of Principal Financial Officer pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
| |
| 101** | | The following materials from the Registrant’s quarterly report on Form 10-Q for the quarter ended September 30, 2013, formatted in XBRL (Extensible Business Reporting Language): (i) the Unaudited Consolidated Balance Sheets, (ii) the Unaudited Consolidated Statements of Comprehensive Income (Loss), (iii) the Unaudited Consolidated Statements of Cash Flows, and (iv) Notes to Unaudited Consolidated Financial Statements. |
| ** | XBRL (Extensible Business Reporting Language) information is furnished and not deemed filed or a part of a registration statement or prospectus for purposes of Sections 11 or 12 of the Securities Act of 1933, as amended, or Section 18 of the Securities Exchange Act of 1934, as amended, and otherwise is not subject to liability under these sections. |
| # | The Registrant has received confidential treatment for certain portions of this exhibit. |
| † | Denotes management compensation plan or contract. |
100
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | | | |
| | | | REGADO BIOSCIENCES, INC. |
| | | |
| | | | By: | | /s/ DAVID J. MAZZO |
| Date | | | | | | David J. Mazzo |
| March 12, 2014 | | | | | | Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | |
Signature | | Title | | Date |
| | |
/s/ DAVID J. MAZZO David J. Mazzo | | Chief Executive Officer and Director (Principal Executive Officer) | | March 12, 2014 |
| | |
/s/ CHRISTOPHER E. COURTS Christopher E. Courts | | Vice President, Finance (Principal Financial and Accounting Officer) | | March 12, 2014 |
| | |
/s/ DENNIS PODLESAK Dennis Podlesak | | Director, Chairman of the Board of Directors | | March 12, 2014 |
| | |
/s/ B. JEFFERSON CLARK B. Jefferson Clark | | Director | | March 12, 2014 |
| | |
/s/ ANTON GOPKA Anton Gopka | | Director | | March 12, 2014 |
| | |
/s/ Pierre Legault Pierre Legault | | Director | | March 12, 2014 |
| | |
/s/ MICHAEL E. MENDELSOHN Michael E. Mendelsohn | | Director | | March 12, 2014 |
| | |
/s/ P. SHERRILL NEFF P. Sherrill Neff | | Director | | March 12, 2014 |
| | |
/s/ JESSE TREU Jesse Treu | | Director | | March 12, 2014 |
| | |
/s/ RAPHAEL WISNIEWSKI Raphaël Wisniewski | | Director | | March 12, 2014 |
101
Index to Consolidated Financial Statements
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
Board of Directors and Shareholders
Regado Biosciences, Inc
We have audited the accompanying consolidated balance sheets of Regado Biosciences, Inc. (a Delaware company operating in the development stage) and its subsidiary (the “Company”) as of December 31, 2013 and 2012, and the related consolidated statements of comprehensive loss, changes in stockholders’ equity, and cash flows for each of the years then ended and for the period from inception (December 19, 2001) to December 31, 2013. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. We were not engaged to perform an audit of the Company’s internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the consolidated financial statements referred to above present fairly, in all material respects, the financial position of Regado Biosciences, Inc. and its subsidiary as of December 31, 2013 and 2012, and the results of their operations and their cash flows for the years then ended and for the period from inception (December 19, 2001) to December 31, 2013, in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming the Company will continue as a going concern. As discussed in Note 2 to the financial statements, the Company has recurring significant cash flow deficits from operations and an accumulated deficit as of December 31, 2013. These conditions, along with other matters as set forth in Note 2, raise substantial doubt about the Company’s ability to continue as a going concern. Management’s plans related to these matters are also described in Note 2. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ GRANT THORNTON LLP
Charlotte, NC
March 12, 2014
F-2
Regado Biosciences, Inc.
(a development stage enterprise)
CONSOLIDATED BALANCE SHEETS
(In thousands, except share and per share data)
| | | | | | | | |
| | | December 31,
2013 | | | December 31,
2012 | |
Assets | | | | | | | | |
Current assets: | | | | | | | | |
Cash and cash equivalents | | $ | 30,688 | | | $ | 14,764 | |
Restricted cash | | | 82 | | | | 82 | |
Prepaid expenses | | | 2,147 | | | | 257 | |
Other assets | | | 6,211 | | | | 4,580 | |
| | | | | | | | |
Total current assets | | | 39,128 | | | | 19,683 | |
| | | | | | | | |
Debt issuance costs, net | | | 36 | | | | 87 | |
Property and equipment, net | | | 108 | | | | 66 | |
Intangible assets, net | | | 1,823 | | | | 1,771 | |
Other non-current assets | | | 4,658 | | | | 196 | |
| | | | | | | | |
Total assets | | $ | 45,753 | | | $ | 21,803 | |
| | | | | | | | |
Liabilities and Stockholders’ Equity | | | | | | | | |
Current liabilities: | | | | | | | | |
Accounts payable | | $ | 1,557 | | | $ | 203 | |
Accrued expenses | | | 5,524 | | | | 786 | |
Warrant liability | | | 19 | | | | — | |
Current portion of long-term debt | | | 2,000 | | | | 2,571 | |
| | | | | | | | |
Total current liabilities | | | 9,100 | | | | 3,560 | |
Long-term debt | | | 2,452 | | | | 1,929 | |
| | | | | | | | |
Total liabilities | | | 11,552 | | | | 5,489 | |
| | | | | | | | |
Commitments (Note 6) | | | | | | | | |
Stockholders’ equity: | | | | | | | | |
Series A convertible preferred stock; $0.001 par value; 0 shares designated, issued and outstanding at December 31, 2013 and 5,798,178 shares designated, issued and outstanding (liquidation preference of $5,798) at December 31, 2012 | | | — | | | | 5,778 | |
Series B convertible preferred stock; $0.001 par value; 0 shares designated, issued and outstanding at December 31, 2013 and 16,666,665 shares designated, issued and outstanding (liquidation preference of $20,000) at December 31, 2012 | | | — | | | | 19,888 | |
Series C convertible preferred stock; $0.001 par value; 0 shares designated, issued and outstanding at December 31, 2013 and 17,037,037 shares designated, issued and outstanding (liquidation preference of $23,000) at December 31, 2012 | | | — | | | | 22,978 | |
Series D convertible preferred stock; $0.001 par value; 0 shares designated, issued and outstanding at December 31, 2013 and 71,666,667 shares designated, issued and outstanding (liquidation preference of $51,600) at December 31, 2012 | | | — | | | | 51,237 | |
Series E convertible preferred stock; $0.001 par value; 0 shares designated, issued and outstanding at December 31, 2013 and 31,437,442 shares designated, issued and outstanding (liquidation preference of $22,635) at December 31, 2012 | | | — | | | | 22,169 | |
Common stock, $0.001 par value; 500,000,000 shares authorized; 21,310,614 and 221,272 shares issued and outstanding at December 31, 2013 and December 31, 2012, respectively | | | 21 | | | | — | |
Additional paid-in-capital | | | 179,159 | | | | 4,804 | |
Deficit accumulated during the development stage | | | (144,979 | ) | | | (110,540 | ) |
| | | | | | | | |
Total stockholders’ equity | | | 34,201 | | | | 16,314 | |
| | | | | | | | |
Total liabilities and stockholders’ equity | | $ | 45,753 | | | $ | 21,803 | |
| | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-3
Regado Biosciences, Inc.
(a development stage enterprise)
CONSOLIDATED STATEMENTS OF COMPREHENSIVE LOSS
(In thousands, except share and per share data)
| | | | | | | | | | | | |
| | | For the years ended
December 31, | | | Period from Inception
(December 19, 2001) to | |
| | | 2013 | | | 2012 | | | December 31, 2013 | |
Revenue: | | | | | | | | | | | | |
Grant revenue | | $ | — | | | $ | — | | | $ | 832 | |
| | | | | | | | | | | | |
Total revenue | | | — | | | | — | | | | 832 | |
| | | | | | | | | | | | |
Operating expenses: | | | | | | | | | | | | |
Research and development | | | 26,542 | | | | 8,006 | | | | 108,503 | |
General and administrative | | | 7,297 | | | | 4,157 | | | | 38,195 | |
| | | | | | | | | | | | |
Total operating expenses | | | 33,839 | | | | 12,163 | | | | 146,698 | |
| | | | | | | | | | | | |
Loss from operations | | | (33,839 | ) | | | (12,163 | ) | | | (145,866 | ) |
| | | | | | | | | | | | |
Other (expense) income: | | | | | | | | | | | | |
Realized gain on investments | | | — | | | | — | | | | 176 | |
Interest income | | | 80 | | | | 5 | | | | 2,878 | |
Interest expense | | | (680 | ) | | | (904 | ) | | | (2,167 | ) |
| | | | | | | | | | | | |
Total other (expense) income | | | (600 | ) | | | (899 | ) | | | 887 | |
| | | | | | | | | | | | |
Net loss | | $ | (34,439 | ) | | $ | (13,062 | ) | | $ | (144,979 | ) |
| | | | | | | | | | | | |
Comprehensive loss | | $ | (34,439 | ) | | $ | (13,062 | ) | | $ | (144,979 | ) |
| | | | | | | | | | | | |
Loss per share – basic and diluted | | $ | (4.59 | ) | | $ | (59.03 | ) | | $ | (179.91 | ) |
| | | | | | | | | | | | |
Weighted-average common shares – basic and diluted | | | 7,499,661 | | | | 221,272 | | | $ | 805,826 | |
| | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements
F-4
REGADO BIOSCIENCES, INC.
(a development stage enterprise)
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY
(In thousands)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
For the
Period from
Inception
(December 19,
2001) to
December 31,
2013 | | Common Stock | | | Additional
Capital | | | Series A
Preferred Stock | | | Series B
Preferred Stock | | | Series C
Preferred Stock | | | Series D
Preferred Stock | | | Series E
Preferred Stock | | | Accumulated
Deficit | | | Total | |
| | Shares | | | Amount | | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | |
| | | | | | | | | | | | | | | |
Balance at inception, December 19, 2001 | | | — | | | $ | — | | | $ | — | | | | — | | | $ | — | | | | — | | | $ | — | | | | — | | | $ | — | | | | — | | | $ | — | | | | — | | | $ | — | | | $ | — | | | $ | — | |
Issuance of common stock during the period from inception to December 31, 2011 | | | 101 | | | | — | | | | 53 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 53 | |
Issuance of preferred stock during the period from inception to December 31, 2011, net of issuance costs of $501 | | | — | | | | — | | | | — | | | | 5,798 | | | | 5,778 | | | | 16,667 | | | | 19,888 | | | | 17,037 | | | | 22,978 | | | | 71,667 | | | | 51,253 | | | | — | | | | — | | | | — | | | | 99,897 | |
Exercise of common stock options and stock warrants during the period from inception to December 31, 2011 | | | 120 | | | | — | | | | 205 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 205 | |
Stock compensation during the period from inception to December 31, 2011 | | | — | | | | — | | | | 3,908 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 3,908 | |
Net loss during the period from inception to December 31, 2011 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (97,478 | ) | | | (97,478 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance, December 31, 2011 | | | 221 | | | | — | | | | 4,166 | | | | 5,798 | | | | 5,778 | | | | 16,667 | | | | 19,888 | | | | 17,037 | | | | 22,978 | | | | 71,667 | | | | 51,253 | | | | — | | | | — | | | | (97,478 | ) | | | 6,585 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Issuance costs of $16 related to Series D Preferred Stock | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (16 | ) | | | — | | | | — | | | | — | | | | (16 | ) |
Issuance of Series E Preferred Stock, net of issuance costs of $466 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 31,437 | | | | 22,169 | | | | — | | | | 22,169 | |
Stock compensation | | | — | | | | — | | | | 703 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 703 | |
Other | | | — | | | | — | | | | (65 | ) | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (65 | ) |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (13,062 | ) | | | (13,062 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance, December 31, 2012 | | | 221 | | | $ | — | | | $ | 4,804 | | | | 5,798 | | | $ | 5,778 | | | | 16,667 | | | $ | 19,888 | | | | 17,037 | | | $ | 22,978 | | | | 71,667 | | | $ | 51,237 | | | | 31,437 | | | $ | 22,169 | | | $ | (110,540 | ) | | $ | 16,314 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
F-5
REGADO BIOSCIENCES, INC.
(a development stage enterprise)
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY—(Continued)
(In thousands)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
For the
Period from
Inception
(December 19,
2001) to
December 31,
2013 | | Common Stock | | | Additional
Capital | | | Series A
Preferred Stock | | | Series B
Preferred Stock | | | Series C
Preferred Stock | | | Series D
Preferred Stock | | | Series E
Preferred Stock | | | Accumulated
Deficit | | | Total | |
| | Shares | | | Amount | | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | Shares | | | Amount | | | |
| | | | | | | | | | | | | | | |
Issuance of Series E Preferred stock, net of issuance costs of $147 | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 14,320 | | | | 10,163 | | | | — | | | | 10,163 | |
Issuance of common stock upon initial public offering, net of underwriting discounts and offering costs of $5,611 | | | 11,672 | | | | 12 | | | | 41,063 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 41,075 | |
Conversion of preferred stock to common stock upon initial public offering | | | 9,397 | | | | 9 | | | | 132,204 | | | | (5,798 | ) | | | (5,778 | ) | | | (16,667 | ) | | | (19,888 | ) | | | (17,037 | ) | | | (22,978 | ) | | | (71,667 | ) | | | (51,237 | ) | | | (45,757 | ) | | | (32,332 | ) | | | — | | | | — | |
Issuance of common stock upon exercise of warrants | | | 13 | | | | — | | | | 2 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 2 | |
Issuance of common stock upon exercise of options | | | 8 | | | | — | | | | 33 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 33 | |
Stock compensation | | | — | | | | — | | | | 988 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 988 | |
Other | | | — | | | | — | | | | 65 | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | 65 | |
Net loss | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | — | | | | (34,439 | ) | | | (34,439 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance, December 31, 2013 | | | 21,311 | | | $ | 21 | | | $ | 179,159 | | | | — | | | $ | 0 | | | | — | | | $ | 0 | | | | — | | | $ | 0 | | | | — | | | $ | 0 | | | | — | | | $ | 0 | | | $ | (144,979 | ) | | $ | 34,201 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-6
Regado Biosciences, Inc.
(a development stage enterprise)
CONSOLIDATED STATEMENTS OF CASH FLOWS
(In thousands)
| | | | | | | | | | | | |
| | | For the Years Ended
December 31, | | | Period from Inception
(December 19, 2001) to | |
| | | 2013 | | | 2012 | | | December 31, 2013 | |
Cash flows used in operating activities: | | | | | | | | | | | | |
Net loss | | $ | (34,439 | ) | | $ | (13,062 | ) | | $ | (144,979 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
Depreciation | | | 61 | | | | 42 | | | | 2,395 | |
Amortization of patents and licenses | | | 20 | | | | 401 | | | | 1,807 | |
Impairment of patents | | | 371 | | | | 22 | | | | 536 | |
Accrued final bank fee | | | 55 | | | | — | | | | 55 | |
Amortization of debt discount | | | 61 | | | | — | | | | 61 | |
Amortization of debt issuance costs | | | 108 | | | | 34 | | | | 156 | |
Change in fair value of warrant liability | | | (60 | ) | | | — | | | | (60 | ) |
Stock-based compensation | | | 988 | | | | 703 | | | | 5,599 | |
Loss on disposal of property and equipment | | | 2 | | | | 2 | | | | 30 | |
Gain on sale of investments | | | — | | | | — | | | | (176 | ) |
Other | | | 65 | | | | (65 | ) | | | — | |
Changes in operating assets and liabilities: | | | | | | | | | | | | |
Prepaid expenses | | | (1,890 | ) | | | (32 | ) | | | (2,147 | ) |
Other assets | | | (1,708 | ) | | | (1,436 | ) | | | (6,211 | ) |
Other non-current assets | | | (4,462 | ) | | | (196 | ) | | | (4,658 | ) |
Accounts payable | | | 1,354 | | | | (117 | ) | | | 1,557 | |
Accrued expenses | | | 4,738 | | | | (525 | ) | | | 5,863 | |
| | | | | | | | | | | | |
Net cash used in operating activities | | | (34,736 | ) | | | (14,229 | ) | | | (140,172 | ) |
| | | | | | | | | | | | |
Cash flows used in investing activities: | | | | | | | | | | | | |
Change in restricted cash | | | — | | | | — | | | | (82 | ) |
Purchase of property and equipment | | | (105 | ) | | | (17 | ) | | | (2,536 | ) |
Proceeds received on disposal of property and equipment | | | — | | | | — | | | | 5 | |
Purchase of investments | | | — | | | | — | | | | (163,683 | ) |
Proceeds from sales of investments | | | — | | | | — | | | | 163,859 | |
Patent and license acquisition costs | | | (543 | ) | | | (340 | ) | | | (4,266 | ) |
Proceeds received from sale of patents | | | 100 | | | | — | | | | 100 | |
| | | | | | | | | | | | |
Net cash used in investing activities | | | (548 | ) | | | (357 | ) | | | (6,603 | ) |
| | | | | | | | | | | | |
Cash flows from financing activities: | | | | | | | | | | | | |
Proceeds from borrowing on convertible notes payable | | | — | | | | 6,781 | | | | 15,079 | |
Proceeds from borrowings on bank loan | | | 4,500 | | | | — | | | | 10,500 | |
Repayment of borrowings on bank loan | | | (4,500 | ) | | | (1,500 | ) | | | (6,000 | ) |
Payment of bank origination fee | | | (85 | ) | | | — | | | | (85 | ) |
Payment of debt issuance costs | | | (57 | ) | | | (46 | ) | | | (152 | ) |
Proceeds from issuance of common stock in IPO, net of underwriting discounts | | | 43,418 | | | | — | | | | 43,418 | |
| | | |
Payment of IPO costs | | | (2,266 | ) | | | (77 | ) | | | (2,343 | ) |
Proceeds from sale of preferred stock, net of issuance costs | | | 10,163 | | | | 14,957 | | | | 116,795 | |
Proceeds from issuance of common stock | | | 33 | | | | — | | | | 224 | |
Proceeds from exercise of warrants | | | 2 | | | | — | | | | 27 | |
| | | | | | | | | | | | |
Net cash provided by financing activities | | | 51,208 | | | | 20,115 | | | | 177,463 | |
| | | | | | | | | | | | |
Net increase in cash and cash equivalents | | $ | 15,924 | | | $ | 5,529 | | | $ | 30,688 | |
Cash and cash equivalents, beginning of period | | | 14,764 | | | | 9,235 | | | | — | |
| | | | | | | | | | | | |
Cash and cash equivalents, end of period | | $ | 30,688 | | | $ | 14,764 | | | $ | 30,688 | |
| | | | | | | | | | | | |
Supplemental disclosures of cash flow information: | | | | | | | | | | | | |
Cash paid for interest | | $ | 547 | | | $ | 572 | | | $ | 1,368 | |
Supplemental disclosure of non-cash investing and financing activities: | | | | | | | | | | | | |
Conversion of preferred stock into common stock | | $ | 132,213 | | | $ | — | | | $ | 132,213 | |
Conversion of note payable and interest into preferred stock | | $ | — | | | $ | 7,120 | | | $ | 15,418 | |
The accompanying notes are an integral part of these consolidated financial statements.
F-7
Regado Biosciences, Inc.
(a development stage enterprise)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
| 1 | Organization and Description of Business |
Regado Biosciences, Inc. (the “Company” or “we” or “our” or “us”) is a development stage enterprise incorporated in the State of Delaware on December 19, 2001 operating primarily in Basking Ridge, New Jersey and Durham, North Carolina. We are focused on the discovery and development of novel, first-in-class, actively controllable antithrombotic drug systems for acute and sub-acute cardiovascular indications. Each of our product candidates consists of a two-component system: an antithrombotic aptamer and its specific active control agent. Our lead product candidate, REG1, is a two-component system consisting of pegnivacogin, an anticoagulant aptamer specifically targeting coagulation Factor IXa, and its complementary oligonucleotide active control agent, anivamersen. REG1 is being developed for use in patients with a wide variety of acute coronary syndromes, or ACS, undergoing a percutaneous coronary intervention, or PCI, a hospital-based procedure used to mechanically open or widen obstructed coronary arteries. Our actively controllable product candidates have the potential to improve patient outcomes, enhance the patient experience and reduce overall treatment costs. In September 2013, we commenced our single, open-label, 13,200 subject Phase 3 trial of REG1 in patients undergoing PCI (excluding ST elevated myocardial infarction, or STEMI), or the REGULATE-PCI trial.
In May 2013, our board of directors and our stockholders approved an amendment to the amended and restated certificate of incorporation to effect a one-for-16.7 reverse split of shares of common stock and to increase the number of authorized shares of common stock to 500,000,000, each of which was effected on May 21, 2013. All issued and outstanding common stock, options, warrants and per share amounts contained in these consolidated financial statements have been retroactively adjusted to reflect the reverse split for all periods presented.
We completed our initial public offering (“IPO”) in August 2013. Inclusive of the underwriters’ exercise of the over-allotment option in connection with the IPO in September 2013, we issued 11,671,500 shares of common stock at a price of $4.00 per share, resulting in net proceeds of approximately $41.1 million, after deducting underwriting discounts of $3.3 million and offering costs of $2.3 million. Upon the closing of the IPO, all shares of convertible preferred stock then outstanding automatically converted into an aggregate of 9,396,767 shares of common stock.
| 2 | Basis of Presentation and Summary of Significant Accounting Policies |
Principles of Consolidation and Basis of Presentation
In March 2013, we incorporated Regado Biosciences Europe Limited, a wholly owned subsidiary registered in England and Wales, in order to establish a legal presence in the European Union (EU) for the purpose of conducting clinical trials in the EU. Regado Biosciences Europe Limited had no operations during the year ended December 31, 2013.
The accompanying consolidated financial statements include the accounts of Regado Biosciences, Inc. and its wholly owned subsidiary, Regado Biosciences Europe Limited. There were no significant intercompany accounts or transactions that needed to be eliminated in consolidation. The consolidated financial statements have been prepared in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”). All intercompany balances and transactions have been eliminated in consolidation.
Going Concern Uncertainty
Our financial statements have been prepared on a going-concern basis, which contemplates the realization of assets and settlement of liabilities and commitments in the normal course of business. Operations since inception have consisted primarily of developing and acquiring product technologies and securing financing.
The accompanying financial statements have been prepared assuming that we will operate as a going concern. We have suffered negative cash flows from operating activities of $34.7 million during the year ended December 31, 2013 and a net accumulated deficit of $145.0 million since inception as of December 31, 2013. Prior to our IPO, we were funded primarily through the issuance of preferred stock and debt. We will require additional capital until such time that we can generate operating revenue in excess of operating expenditures. Our plans include continued product development and a move toward completion of clinical trials. We will continue to closely monitor and analyze expenses and make adjustments as necessary to prioritize business operations. We believe that the net proceeds from the IPO and from our 2014 Private Placement pursuant to which we sold $20 million of our common stock to new and existing investors (see Note 14 - Subsequent Events), will be sufficient for us to fund the REGULATE-PCI trial through the
F-8
second interim analysis, which we expect will occur in the third quarter of 2014. We will need to raise additional financing in the first half of 2014 to fund projected operations through 2014 and we can provide no assurances that such additional financing will be available on favorable terms, or at all. Actual results may differ from estimates and the financial statements do not include any adjustments that might be necessary if we are unable to fund operations.
Reclassifications
We have reclassified supplies inventory, net to other current assets in the accompanying consolidated balance sheet at December 31, 2012. This reclassification did not have any impact on our loss from operations or net loss for the year ended December 31, 2012 or on total assets as of December 31, 2012.
We have reclassified certain costs associated with research and development activities, including all laboratory and clinical indirect costs, laboratory and clinical personnel stock compensations costs, and patent and product license amortization and patent impairment costs for the year ended December 31, 2012, and for the period from inception through December 31, 2012, from general and administrative expense to research and development expense in the accompanying consolidated statements of comprehensive loss. These reclassifications did not have any impact on our loss from operations or net loss for the year ended December 31, 2012, or for the period from inception through December 31, 2012.
Both of the aforementioned classifications were made to conform to the current year presentation.
Use of Estimates
The preparation of financial statements in conformity with U.S. GAAP requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Fair Value of Financial Instruments
The carrying amount of certain of our financial instruments, including cash and cash equivalents, prepaid expenses, and accounts payable approximate fair value due to the short maturities of those financial instruments. The carrying amount of our debt at December 31, 2013 and 2012 approximated fair value because the interest rates and terms of our debt approximate market terms. In conjunction with the refinancing of our long term debt in May 2013, we held a warrant liability at December 31, 2013 that is required to be measured at fair value on a recurring basis (see Note 3).
Our valuation of financial instruments is based on a three-tiered approach, which requires that fair value measurements be classified and disclosed in one of three tiers. These tiers are: Level 1, defined as quoted prices in active markets for identical assets or liabilities; Level 2, defined as valuations based on observable inputs other than those included in Level 1, such as quoted prices for similar assets and liabilities in active markets, or other inputs that are observable or can be corroborated by observable input data; and Level 3, defined as valuations based on unobservable inputs reflecting our own assumptions, consistent with reasonably available assumptions made by other market participants.
Cash and Cash Equivalents
We consider all interest-bearing investments due on demand and all highly liquid debt instruments purchased with a maturity of three months or less to be cash equivalents. Cash and cash equivalents included cash of $363 and $235 at December 31, 2013 and 2012, respectively. Cash and cash equivalents at December 31, 2013 and 2012 also included investments of $30.3 million and $14.5 million, respectively, in money market funds invested in U.S. Treasury securities with original maturities of less than three months. Cash deposits are held in federally insured financial institutions in the United States of America. We maintain cash in accounts which are in excess of federally insured limits.
Restricted Cash
Restricted cash consists primarily of funds invested in two certificates of deposit. One certificate of deposit serves as collateral for a lease of laboratory space. The other certificate of deposit serves as collateral for the Company’s corporate credit card. These certificates of deposit mature every seven days and renew weekly.
F-9
Credit Risk
Financial instruments which potentially subject the Company to concentrations of credit risk consist principally of cash and equivalents. The Company’s cash and equivalents were primarily invested in money market funds invested exclusively in treasury securities. Total cash and equivalent balances exceed insured balances by the Federal Depository Insurance Company.
Limited Suppliers
We do not have a manufacturing infrastructure and do not intend to develop one for the foreseeable future. We have agreements with third-party contract manufacturing organizations (“CMOs”), to supply bulk drug substances for our product candidates and with third parties to formulate, package and distribute our drug product candidates. Our employees include professionals with expertise in pharmaceutical manufacturing development who oversee the manufacture and distribution of our drug product candidates by third-party companies. We may not currently have sufficient amounts of REG1 on hand to complete enrollment in the REGULATE-PCI trial, however the Company is currently contracted to produce sufficient amounts of REG1 in early 2014. The Company may not have enough bivalirudin to complete REGULATE-PCI and may need to secure additional quantities to complete the trial. All of the drug substances used in our product candidates are manufactured by single suppliers. While we have not experienced any supply disruptions, the number of oligonucleotide manufacturers is limited. In the event it is necessary or advisable to acquire supplies from an alternative supplier, we might not be able to obtain them on commercially reasonable terms, if at all. It could also require significant time and expense to redesign our manufacturing processes to work with another supplier. Formulation and distribution of our finished drug product candidates are also conducted by a single supplier but we believe that alternative sources for these services are readily available on commercially reasonable terms (see Note 6).
Segment and Geographic Information
Operating segments are defined as components of an enterprise engaging in business activities for which discrete financial information is available and regularly reviewed by the chief operating decision maker in deciding how to allocate resources and in assessing performance. The Company operates and manages its business as one operating segment and all of the Company’s operations were in North America during 2013.
Clinical Trial Supplies
We capitalize materials that will be used in our REG I clinical trial that also have an alternative future use in either ongoing or future clinical research and development projects. Clinical trial supplies may comprise material used to manufacture active pharmaceutical ingredients (“API”) used to develop our product candidates, in-process or completed API, in-process or completed unlabeled finished drug product and labeled finished drug product. Clinical trial supplies are stated at cost, using the first-in, first-out method (“FIFO”), and are reported in the accompanying consolidated balance sheets in other current assets. Clinical trial supplies that are determined to be unsuitable for future use are immediately expensed; otherwise clinical trial supplies are expensed when shipped to clinical sites for use in clinical studies or when used in other research and development projects.
We utilize CMOs to produce API and finished drug product for use in clinical trials. As we do not have facilities that meet the requisite regulatory requirements for storage of API or finished drug product produced, we use a third-party facility for storage. Upon release from the manufacturer, API is shipped to a third-party storage facility. For production of finished drug product, API is shipped from the storage facility to the finished drug product manufacturing site. Unlabelled finished drug product is either shipped from the manufacturer back to the third-party storage facility or directly to the third-party labeling site. Labeled finished drug product is held by the third-party labeling site until it is shipped to the clinical sites for trial use.
We do not have multiple sources of supply for the components of our finished drug product. If we are unable to obtain the supplies needed at a reasonable price or on a timely basis, it could have a material adverse effect on our ability to complete the development of our finished drug product.
As of December 31, 2013 clinical trial supplies included in other current assets were $4.6 million, of which $1.6 million and $3.0 million represented API held at the third-party storage facility and drug product located at depots, respectively. As of December 31, 2012, clinical trial supplies included in other current assets were $4.5 million which represented API held at the third-party storage facility.
F-10
Clinical Agreements
We enter into various clinical trial agreements with academic research organizations (“AROs”) and clinical research organizations (“CROs”) for the planning, management and execution of clinical trials. The financial terms of these agreements are subject to negotiation, vary from contract to contract, and may result in uneven payment flows. The majority of our service providers invoice us monthly in arrears for services performed or when contractual milestones are met. Costs for ARO and CRO contracts are recognized based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations, or information provided by vendors on their actual costs incurred; such costs are charged to research and development expense in the accompanying consolidated statements of comprehensive loss. There may be instances in which payments made to our vendors will exceed the level of services provided and result in a prepayment of the research and development expense. Upfront contract signing fees are amortized over the life of the respective contract; unamortized contract signing fees are included in other non-current assets.
In the accompanying consolidated balance sheet as of December 31, 2013, prepaid expenses include $1.1 million related to clinical agreements, and other non-current assets include $4.5 million of upfront payments of which $2.8 million will be applied to final invoices as required under the respective contract, and $1.7 million will be amortized as contract signing costs over the remaining life of the respective contract, or approximately three years.
In general, our ARO and CRO service agreements permit either party to terminate at will, although we would continue to be responsible for payment of all services completed (or pro-rata completed) at the time of notice of termination, plus any non-cancellable expenses that have been entered into by the ARO and CRO on the Company’s behalf. Accordingly, such expenses would be accrued at time of contract termination and any prepaid expenses and unamortized advance payments would be expensed, accordingly.
Value Added Taxes
We are charged value added taxes on purchases, made on the Company’s behalf by a CRO, of certain clinical supplies from manufacturers in foreign jurisdictions. As of December 31, 2013, the Company had recorded $1,206 as a VAT receivable and $708 as a VAT liability in the accompanying consolidated balance sheet within other current assets and accrued expenses, respectively. There was $0 impact on our loss from operations or net loss for the year ended December 31, 2013 related to VAT.
Property and Equipment
Property and equipment consists primarily of laboratory and computer equipment and furniture, which are recorded at cost and depreciated using the straight-line method over their estimated useful lives, ranging from two to five years. Amortization for leasehold improvements is computed using the straight-line method over the estimated useful lives of the assets or over the term of the related leases, whichever is shorter.
Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation are removed from the accounts, and any resulting gain or loss is credited or charged to income. Repairs and maintenance costs are expensed as incurred.
Intangible Assets
The Company’s policy is to file patent application(s) to protect technology, inventions and improvements that are considered important to the development of its business. The patent positions of technology companies, including the Company, are uncertain and involve complex legal and factual questions for which important legal principles are largely unresolved. Upon receipt of a patent grant; respective costs are amortized over the remaining life of the patent.
The Company amortizes license agreements over the stated contractual life.
Impairment of Long-lived Assets
The Company recognizes impairment of long-lived assets in the event the net book value of such assets exceeds the estimated future undiscounted cash flows attributable to such assets. Accordingly, when indicators of impairment are present, the Company evaluates the carrying value of these assets in relation to the operating performance of the assets and the future undiscounted cash flows expected to result from the use of these assets. No such impairments have been recognized for the years ended December 31, 2013 and 2012, and for the period from inception to December 31, 2013, respectively, except for expense recognized for expired and abandoned patents totaling $371,000, $22,000 and $536,000 for the years ended December 31, 2013 and 2012, and for the period from inception to December 31, 2013, respectively. Such expenses are included in research and development expenses in the accompanying statements of comprehensive loss.
F-11
Grant Revenue
We were awarded several grants under the Therapeutic Discovery Project Grant program during 2010, related to research amounts previously expensed and have historically received modest other grant funding. The total amount recognized within Grant Revenue in the consolidated statement of comprehensive loss was $0 for the years ended December 31, 2013 and 2012, and $832 for the period from inception to December 31, 2013.
Research and Development
Research and development (“R&D”) expenses include direct and indirect R&D costs. Direct R&D consists principally of external costs, such as fees paid to investigators, consultants, central laboratories and clinical research organizations, including costs incurred in connection with our clinical trials, and related clinical trial fees and all employee-related expenses for those employees working in research and development functions, including stock-based compensation for R&D personnel. Indirect R&D costs include overhead costs related to facilities, depreciation, insurance, and small supplies are that not allocated to specific product candidates or indications. R&D costs are expensed as incurred.
Stock-based Compensation
In accordance with FASB ASC Topic 718, Stock Compensation, as modified or supplemented, we measure compensation cost for share-based payment awards granted to employees and non-employee directors at fair value using the Black-Scholes option-pricing model. We recognize compensation expense on a straight-line basis over the service period for awards expected to vest. Share-based compensation cost related to share-based payment awards granted to non-employees is adjusted each reporting period for changes in the fair value of our common stock until the measurement date. The measurement date is generally considered to be the date when all services have been rendered or the date that options are fully vested.
Net Loss Per Share
Basic net loss per share is calculated by dividing the net loss by the weighted average number of common shares outstanding for the period, without consideration for common stock equivalents. Diluted net loss per share is calculated by dividing the net loss by the weighted-average number of common stock equivalents outstanding for the period determined using the treasury-stock method. Dilutive common stock equivalents are comprised of convertible preferred stock, options outstanding under our stock option plan and warrants.
Recent Accounting Pronouncements
Section 107 of the Jumpstart our Business Startups Act of 2012, or the JOBS Act, provides that an “emerging growth company” can take advantage of the extended transition period provided in Section 7(a)(2)(B) of the Securities Act of 1933, as amended, for complying with new or revised accounting standards. In other words, an “emerging growth company” can delay the adoption of certain accounting standards until those standards would otherwise apply to private companies. However, we are choosing to “opt out” of such extended transition period, and as a result, we will comply with new or revised accounting standards on the relevant dates on which adoption of such standards is required for non-emerging growth companies. Section 107 of the JOBS Act provides that our decision to opt out of the extended transition period for complying with new or revised accounting standards is irrevocable.
F-12
| 3 | Fair Value of Financial Instruments |
The following table (in thousands) sets forth our assets and liabilities that were measured at fair value on a recurring basis at December 31, 2013 and 2012 by level within the fair value hierarchy. As required by ASC 820-10, assets and liabilities measured at fair value are classified in their entirety based on the lowest level of input that is significant to the fair value measurement. Our assessment of the significance of a particular input to the fair value measurement in its entirety requires judgment and considers factors specific to the asset or liability.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of December 31, 2013 | | | As of December 31, 2012 | |
Assets and Liabilities | | Quoted Prices
In Active
Markets for
Identical
Assets
(Level 1) | | | Significant
Other
Observable
Inputs
(Level 2) | | | Significant
Unobservable
Inputs
(Level 3) | | | Balance as of
December 31,
2013 | | | Quoted Prices
In Active
Markets for
Identical
Assets
(Level 1) | | | Significant
Other
Observable
Inputs
(Level 2) | | | Significant
Unobservable
Inputs
(Level 3) | | | Balance at
December 31,
2012 | |
Assets: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Money market funds | | $ | 30,325 | | | $ | — | | | $ | — | | | $ | 30,325 | | | $ | 14,529 | | | $ | — | | | $ | — | | | $ | 14,529 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Total assets at fair value | | $ | 30,325 | | | $ | — | | | $ | — | | | $ | 30,325 | | | $ | 14,529 | | | $ | — | | | $ | — | | | $ | 14,529 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Liabilities: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Warrant liability | | $ | — | | | $ | — | | | $ | 19 | | | $ | 19 | | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Total liabilities at fair value | | $ | — | | | $ | — | | | $ | 19 | | | $ | 19 | | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
The change in the fair value measurement using significant unobservable inputs (Level 3) is summarized below (in thousands):
| | | | |
Balance at December 31, 2012 | | $ | — | |
| | | | |
Allocation of long-term debt proceeds to warrant | | | 79 | |
| |
Change in fair value recorded as interest income | | | (69 | ) |
Change in fair value recorded as interest expense | | | 9 | |
| | | | |
Balance at December 31, 2013 | | $ | 19 | |
| | | | |
The warrant liability represents our allocation of a portion of the proceeds from the May 2013 Comerica Loan (as defined in Note 8). The allocation of the proceeds from the Comerica Loan was based on the fair value of the warrant liability on the date of grant. We accounted for the warrant liability in accordance with ASC Topic 815, Derivatives and Hedging, which requires that equity contracts not indexed to the issuer’s own stock and not meeting the definition of a derivative should be reported as an asset or liability. We utilized the Binomial pricing model to determine the fair value of the warrant liability. We record changes in the fair value of the warrant liability as interest expense or interest income, as applicable.
We used significant assumptions in estimating the fair value of the warrant liability including the estimated volatility, risk free interest rate, estimated fair value of the preferred shares, and the estimated life of the warrant. These assumptions were used to establish an expected set of cash flows which were probability-weighted and discounted to present value to determine a fair value.
| 4 | Property and Equipment, net |
Property and equipment consist of the following (in thousands):
| | | | | | | | |
| | | As of December 31, | |
| | | 2013 | | | 2012 | |
Leasehold improvements | | $ | 333 | | | $ | 325 | |
Computer equipment and software | | | 184 | | | | 145 | |
Laboratory equipment | | | 1,775 | | | | 1,775 | |
Furniture and equipment | | | 27 | | | | 27 | |
Less – Accumulated depreciation | | | (2,211 | ) | | | (2,207 | ) |
| | | | | | | | |
Property and equipment, net | | $ | 108 | | | $ | 66 | |
| | | | | | | | |
Depreciation expense for the years ended December 31, 2013 and 2012, and for the period from inception to December 31, 2013, was $61, $42, and $2,395, respectively.
F-13
The following information details the carrying amounts and accumulated amortization of our intangible assets subject to amortization (in thousands):
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of December 31, 2013 | | | As of December 31, 2012 | |
| | | Weighted
Average
Useful Life
Remaining | | | Gross Carrying Amount | | | Accumulated Amortization | | | Net Carrying Amount | | | Gross Carrying Amount | | | Accumulated Amortization | | | Net
Carrying
Amount | |
Identifiable intangible assets: | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Patents | | | 8.0 years | | | $ | 1,518 | | | $ | (383 | ) | | $ | 1,135 | | | $ | 1,607 | | | $ | (349 | ) | | $ | 1,258 | |
Product licenses | | | 2.0 years | | | | 1,555 | | | | (1,424 | ) | | | 131 | | | | 1,547 | | | | (1,439 | ) | | | 108 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Total | | | 7.8 years | | | $ | 3,073 | | | $ | (1,807 | ) | | $ | 1,266 | | | $ | 3,154 | | | $ | (1,788 | ) | | $ | 1,366 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Expected future amortization expenses for intangible assets as of December 31, 2013 are as follows:
| | | | |
For the year ending December 31: | | Amount | |
2014 | | $ | 137 | |
2015 | | | 128 | |
2016 | | | 128 | |
2017 | | | 128 | |
2018 | | | 128 | |
Thereafter | | | 617 | |
| | | | |
| | $ | 1,266 | |
| | | | |
We record amortization of patent and product license costs in research and development expense in the accompanying consolidated statements of comprehensive loss.
Patents
Patent costs consist of expenditures incurred for various patent applications. As of December 31, 2013 and 2012, we had $557,000 and $405,000, respectively, of costs related to patents not subject to amortization as they have yet to be granted.
Product Licenses
We have primary license agreements with Duke University, Archemix Corporation, and Nektar Therapeutics AL and all of the licenses are being amortized over the stated contractual life.
Supply and Manufacturing Agreements
We have agreements with CMOs, to supply bulk drug substances for our product candidates and with third parties to formulate, package and distribute our drug product candidates (see Note 2—Limited Suppliers).
In July 2006, we entered into a supply and service agreement with Agilent Technologies, Inc., which was amended in July 2011, for the manufacture of pegnivacogin and anivamersen bulk drug substance for use in the clinical development of REG1. Drug substances are manufactured pursuant to a Good Manufacturing Practices and a Quality Agreement entered into in May 2010. Manufacture of bulk drug substance lots is on a purchase order basis, with no minimum purchase obligation.
F-14
In December 2006, we entered into a license, manufacturing and supply agreement with Nektar Therapeutics, or Nektar, for the supply of polyethylene glycol, or PEG, used in the manufacture of pegnivacogin. We must provide Nektar with a rolling forecast of our anticipated requirements for the succeeding six quarters, with respect to precommercial supply, or eight quarters, with respect to commercial supply, with a required lead time to commit to and guarantee availability of the reagent at an agreed upon pricing structure, which is subject to revision on an annual basis. In addition, we have agreed to pay Nektar specific development and commercial milestones and a specific royalty on product sales (see Milestone and Other Obligations below).
In March 2012, we entered into a clinical supply agreement with Althea Technologies, Inc., for the formulation and primary packaging of pegnivacogin and anivamersen for use in our clinical trials. Drug products are manufactured and the packaging is conducted pursuant to a Good Manufacturing Practices and a Quality Agreement entered into in January 2012. Formulation and packaging services are provided on a purchase order basis, with no minimum purchase obligation.
Clinical Agreements
We have various clinical trial agreements with AROs and CROs for the planning, management and execution of clinical trials. The financial terms of these agreements are subject to negotiation, vary from contract to contract, and may result in uneven payment flows. These contracts generally provide for termination on notice (see Note 2—Clinical Agreements).
Milestone and Other Obligations
Upon the commencement of our REGULATE-PCI trial which occurred in September 2013, we were obligated to make milestone payments of $500,000 to Duke University (“Duke”), and $1.0 million to Archemix Corporation, (“Archemix”). We have paid $500,000 of such milestone obligations and the $1.0 million remaining balance is included in accrued expenses in the accompanying consolidated balance sheet as of December 31, 2013; accordingly, aggregate milestone obligations of $1.5 million are included in research and development expense in the accompanying statement of comprehensive loss for the year ended December 31, 2013. In addition, upon the filing of a new drug application, we are obligated to make additional milestone payments to Duke, Archemix and Nektar Therapeutics AL, Corporation. Given the uncertainty of the drug development process, we cannot be certain when, if ever, we will be required to make these milestone payments.
As a condition of closing the Series E Preferred Stock financing in December 2012, we assigned all intellectual property (“IP”) rights and titles in Armenia, Azerbaijan, Belarus, Georgia, Kazakhstan, Kyrgyzstan, Moldova, Russia, Tajikistan, Turkmenistan, Ukraine and Uzbekistan ( the “Covered Territory”) to Domain Russia Investments Limited (“DRI”). Additionally, we agreed to assist an affiliate of DRI with certain development support related to the development of the IP. Concurrently with the signing of this agreement, we agreed to make a payment to DRI up to a maximum amount of $1.2 million based on an independent appraiser’s valuation of the IP rights transferred. Such independent appraiser’s valuation was received during 2013, the result of which confirmed that we do not have any further obligation pursuant to this agreement.
We may be required to pay to Nektar an aggregate of $7.75 million upon the achievement of specified development and regulatory approval milestones. Since December 2006, we made payments under this license agreement totaling $1.25 million which are included in research and development expense in the accompanying statement of comprehensive loss for the period from inception to December 31, 2013.
We also are required to pay Nektar royalties based on a percentage of net sales of our REG1 product by us or our sublicensees. The percentage royalty rates are in the mid-single digits, and are subject to reduction with respect to sales in a country if none of the patents and patent applications that we license under the agreement cover REG1 or the PEG in such country. These royalties also may be reduced if we are required to pay royalties to a third party due to infringement relating to the PEG. The duration of our royalty obligation expires on a country-by-country basis on the later of ten years after first commercial sale of REG1 in a country and the expiration of the last-to-expire of the licensed patent rights in such country.
License Agreements
In December 2012, in connection with its Series E Preferred Stock financing, we entered into a Technology Transfer Agreement, (the “Tech Transfer Agreement”), with DRI. In accordance with the terms of the Tech Transfer Agreement, in May 2013 we entered into a Clinical Development and Collaboration Agreement with NovaMedica pursuant to which we agreed to assist NovaMedica in the development and commercialization of our product candidates in the Covered Territory, as defined. NovaMedica is required to reimburse us for any out-of-pocket expenses incurred by us in providing this assistance, including travel-related expenses.
F-15
Operating Leases
We maintain three separate office locations. On May 1, 2013, we entered into a lease agreement for 1,657 square feet of administrative office space located in Durham, North Carolina (the “Durham Office Lease”). The lease is for 36 months with annual lease obligations of $39,000, $40,000 and $41,000 for years one through three, respectively. We had rented an administrative office space in Cary, North Carolina under an operating lease agreement effective December 2012, with base monthly payments at $2,000 per month. The six month lease expired in May 2013 and was replaced with the Durham Office Lease. In April 2007, we entered into an operating lease for 8,495 square feet of laboratory space in Durham, North Carolina. The lease expired July 31, 2012 and subsequent thereto we were on a month-to-month basis with base rent of $11,000 per month. In December 2013 the Company signed a new one year lease for such space with base rent of approximately $13,000 per month. An initial letter of credit of $115,000 was given to the lessor for use of the facility, and the letter of credit was reduced to $46,000 on the fourth anniversary date of the lease. In November 2008, an operating lease was entered into for corporate headquarters in Basking Ridge, New Jersey. The lease expired on November 1, 2011 and is currently on a month-to-month basis. Monthly payments are fixed at $16,000 per month, with an additional $1,000 per month due for utilities.
Rent expense related to operating leases was $464,000 and $498,000 for the years ended December 31, 2013 and 2012, respectively, and $2.9 million for the period from inception to December 31, 2013.
Future minimum aggregate payments under non-cancelable lease obligations as of December 31, 2013 are as follows (in thousands):
| | | | |
For the year ending December 31: | | Amount | |
2014 | | $ | 187 | |
2015 | | | 37 | |
2016 | | | 12 | |
| | | | |
Total minimum lease payments | | $ | 236 | |
| | | | |
Severance
Selected executive employees of ours have employment or severance agreements which provide for severance payments of up to two times base salary, bonuses and benefits upon termination, depending on the reasons for the termination. Certain other employees of ours have employment or severance agreements which generally provide for six months of severance, including base salary, bonuses and benefits upon termination, depending on the reasons for the termination. All employees would be required to execute a release and settlement agreement.
Effective September 26, 2013, we reduced our workforce by eliminating 5 of our 32 full-time employees. The majority of the affected employees worked in drug discovery roles at our laboratory facility in North Carolina. The goal of our reduction in workforce was to enable us to focus management and financial resources on advancing our lead product candidate, REG1, in our REGULATE-PCI trial. In connection with the reduction in workforce, severance expense of approximately $250,000 was charged to research and development expense in the accompanying consolidated statement of comprehensive loss for the year ended December 31, 2013.
In connection with the termination of an executive officer effective as of December 31, 2013, accrued severance expense of $417,000 was recorded and charged to general and administrative expense in the accompanying consolidated statement of comprehensive loss for the year ended December 31, 2013.
F-16
The components of accrued expenses are as follows (in thousands):
| | | | | | | | |
| | | As of December 31, | |
| | | 2013 | | | 2012 | |
Accrued license milestones | | $ | 1,000 | | | $ | — | |
Accrued obligations under clinical contracts | | | 1,957 | | | | — | |
Accrued legal and professional services | | | 152 | | | | 14 | |
Accrued VAT expenses | | | 780 | | | | — | |
Accrued interest | | | 24 | | | | 40 | |
Accrued compensation and benefits | | | 1,225 | | | | 478 | |
Accrued expenses, other | | | 386 | | | | 254 | |
| | | | | | | | |
Total accrued expenses | | $ | 5,524 | | | $ | 786 | |
| | | | | | | | |
In May 2011, we entered into a loan and security agreement with MidCap Financial SBIC, LP pursuant to which we borrowed a total of $6.0 million, at the stated rate of LIBOR, at a 2% rate floor, plus 8% spread per annum. The loan was payable in monthly installments beginning September 2012 through August 2014. Our assets (including intellectual property) were collateral for the borrowings, and we were required to pay a 3% final payment of $180,000 regardless of when the loan was paid in full.
On May 13, 2013, we secured a venture debt loan with Comerica Bank (the “Comerica Loan”). We borrowed $4.5 million (“Tranche One”), and the proceeds of the loan were utilized to repay all amounts due to MidCap Financial SBIC, LP. The Comerica Loan bears interest at Comerica’s Prime Reference Rate (as defined in the Loan Agreement) subject to a floor of 30 day LIBOR plus 250 basis points plus 4.0%, or 7.25% as of December 31, 2013. The terms allow for an interest only period of 15 months, and the remaining principal and interest will be repaid starting September 2014 over a nine-month period (24 months in total). Maturities for 2014 and 2015 are $2.0 million and $2.5 million, respectively. Upon (i) Comerica’s receipt of evidence satisfactory to Comerica that the 1,000 patient interim analysis in the REGULATE-PCI study is successful and performed by April 30, 2014 and (ii) our completion of the IPO and receipt of net proceeds of at least $50.0 million prior to June 30, 2013, we had the option to borrow an additional $4.0 million in the second tranche, or (“Tranche Two”). Since the latter of the Tranche Two conditions was not satisfied, Tranche Two is solely at the discretion of Comerica.
Interest expense recorded related to the Comerica Loan was $346,000 and $0 for the years ended December 31, 2013 and 2012, respectively, and $346,000 for the period from inception to December 31, 2013. Interest expense recorded related to the MidCap Loan was $325,000 and $559,000 for the years ended December 31, 2013 and 2012, respectively, and $1.1 million for the period from inception to December 31, 2013
In connection with the funding of Tranche One, we issued to Comerica a warrant to purchase 156,250 shares of the Series E Preferred Stock at a price of $0.72 per share, or the Warrant Price, subject to adjustment for stock splits, combinations, reclassifications or exchanges and certain dilutive issuances. After giving effect to our IPO and reverse stock split, the warrant was adjusted to a warrant to purchase 9,356 shares of our common stock at a price of $12.02 per share (see Note 3).
F-17
If we had borrowed the additional $4.0 million in Tranche Two, the warrant would have become exercisable for an additional number of shares of the Series E Preferred Stock equal to 100,000 divided by the Warrant Price, as defined, subject to adjustment for stock splits, combinations, reclassifications or exchanges and certain dilutive issuances. As previously noted, as we did not complete an IPO by the required milestone date, Tranche Two is solely at the discretion of Comerica.
Scheduled maturities of long-term debt are as follows (in thousands):
| | | | |
| | | December 31,
2013 | |
Year ending December 31: | | | | |
2014 | | $ | 2,000 | |
2015 | | | 2,500 | |
| | | | |
Total | | $ | 4,500 | |
| | | | |
Less: unamortized discount | | | (103 | ) |
Less - current portion | | | (2,000 | ) |
Plus - fees due at closing(1) | | | 55 | |
| | | | |
Long-term debt, net | | $ | 2,452 | |
| | | | |
| (1) | On the date that all of the principal and interest of the Comerica Loan become due and payable, we must pay an end of term fee of $173,000 (the “Final Fee”). The Final Fee is being accreted to interest expense over the term of the Comerica Loan. |
Included in the consolidated balance sheets within long-term debt are $85,000 in loan origination costs that are being amortized to interest expense over the remaining life of the Comerica Loan using the effective interest rate method.
In accounting for the Comerica Loan, the loan was separated into debt and warrant liability components. We utilized the Binomial pricing model to determine the fair value of the warrant liability component (see Note 3). The carrying amount of the debt component was determined by deducting the fair value of the warrant liability component from the par value of the Comerica Loan as a whole. The excess of the principal amount of the Comerica Loan component over its carrying amount, referred to as the debt discount, is amortized to interest expense over the term of the loan. The warrant liability component is re-measured at each reporting date and changes in the fair value of the warrant liability are recorded as interest expense or interest income, as applicable.
In accounting for the transaction costs related to the issuance of the Comerica Loan, we allocated the total costs incurred to the debt and warrant liability components of the Comerica Loan based on their relative values. Transaction costs attributable to the debt component are amortized to interest expense over the term of the Comerica Loan, and transaction costs attributable to the warrant liability component were immediately expensed.
| 9 | Stockholders’ Equity (in thousands except share and per share amounts) |
IPO
We completed our initial public offering (“IPO”) in August 2013. Inclusive of the underwriters’ exercise of the over-allotment option in connection with the IPO in September 2013, we issued 11,671,500 shares of common stock, at a price of $4.00 per share, resulting in net proceeds to us of approximately $41.1 million after deducting underwriting discounts of $3.3 million and offering costs of $2.3 million.
Pre IPO Common and Preferred Stock Authorized Shares
Prior to the IPO the Company was authorized to issue two (2) classes of shares to be designated, respectively, “Pre-IPO Common Stock” and “Pre-IPO Preferred Stock.” The total number of shares of capital stock that the Company was authorized to issue was 698,363,299 shares. The total number of shares of Pre-IPO Common Stock the Company was authorized to issue was 500,000,000 shares, $0.001 par value per share. The total number of shares of Preferred Stock the Company was authorized to issue was 198,363,299 shares, $0.001 par value per share, of which (a) 5,798,178 shares were to be designated Series A Preferred Stock (the “Series A Preferred Stock”); (b) 16,666,665, shares were to be designated Series B Preferred Stock (the “Series B Preferred Stock”); (c) 17,037,037 shares were to be designated Series C Preferred Stock (the “Series C Preferred Stock”), (d) 71,666,667 shares were to be designated Series D Preferred Stock (the “Series D Preferred Stock”), and (e) 87,194,752 shares were to be designated Series E Preferred Stock (the “Series E Preferred Stock,” and together with the Series A Preferred Stock, the Series B Preferred Stock, the Series C Preferred Stock, and the Series D Preferred Stock, the “Pre-IPO Preferred Stock”).
F-18
During the period from inception to December 31, 2011, the Company issued an aggregate of 111,169 shares of Series A, B, C and D Preferred Stock for aggregate proceeds of $99,897, net of issuance costs of $501.
On December 18, 2012, the first tranche of the Series E financing was completed with 31,437,442 preferred shares issued for $22.6 million. This included $15.5 million in cash plus $6.8 million in convertible loan principal and $339,000 in converted loan interest originally secured on May 3, 2012.
Per the Series E Preferred Stock financing agreement executed on December 18, 2012, a second financing tranche of $10.3 million for 14,320,168 shares of Series E Preferred Stock took place on March 22, 2013. On March 22, 2013 a portion of the second tranche of the Series E financing was completed with 7,160,084 preferred shares issued for $5.2 million. The RMI Investments, S.a.r.l (“RMI”) portion of the second tranche totaling $5.2 million was delivered into an escrow account at the time of the second tranche, and the RMI funds and Series E shares relating to the RMI investment were released on April 26, 2013.
The agreement also provided that a third financing tranche of $17.8 million for 24,770,476 shares of Series E preferred stock would take place on or before January 17, 2014. However, pursuant to the terms of a Termination Agreement entered into by the parties to the Series E Purchase Agreement, our obligation to sell additional shares of Series E Preferred Stock to the investors and the obligations of the investors to purchase additional shares of Series E Preferred Stock terminated immediately prior to the consummation of the IPO and no additional shares of Series E Preferred Stock will be sold pursuant thereto.
Reverse Stock Split
In May 2013, we executed an amendment to our Fifth Amended and Restated Certificate of Incorporation instituting a one–for-16.7 reverse split of common stock and an increase in the number of shares of common stock we are authorized to issue to 500,000,000.
Post IPO Common and Preferred Stock Authorized Shares
Pursuant to the IPO all shares of Series Preferred Stock then outstanding automatically converted into an aggregate of 9,396,767 shares of common stock.
Effective with the IPO, pursuant to the Company’s Sixth Amended and Restated Certificate of Incorporation (the “Restated Certificate”) the Company is authorized to issue two (2) classes of shares to be designated, respectively, “Common Stock” and “Preferred Stock.” The total number of shares of capital stock that the Company is authorized to issue is 501,000,000 shares of capital stock, of which (i) 500,000,000 shares are designated as common stock, par value $0.001 per share (the “Common Stock”), and (ii) 1,000,000 shares are class designated as preferred stock, par value $0.001 per share. (the “Preferred Stock”).
Each holder of record of Common Stock shall have one vote for each share of Common Stock which is outstanding in his, her or its name on the books of the Corporation on all matters on which stockholders are entitled to vote generally. Except as otherwise required by law, holders of Common Stock shall not be entitled to vote on any amendment to the Restated Certificate (including any certificate of designation relating to any series of Preferred Stock) that relates solely to the terms of one or more outstanding series of Preferred Stock if the holders of such affected series are entitled, either separately or together with the holders of one or more other such series, to vote thereon pursuant to the Restated Certificate (including any certificate of designation relating to any series of Preferred Stock) or pursuant to the General Corporation Law of the State of Delaware. Except as otherwise required by law, holders of any series of Preferred Stock shall be entitled to only such voting rights, if any, as shall expressly be granted thereto by the Restated Certificate (including any certificate of designation relating to such series of Preferred Stock).
Subject to applicable law and the rights, if any, of the holders of any outstanding series of Preferred Stock or any class or series of stock having a preference over or the right to participate with the Common Stock with respect to the payment of dividends, dividends may be declared and paid or set apart for payment upon the Common Stock out of any assets or funds of the Corporation legally available for the payment of dividends, but only when and as declared by the Board of Directors or any authorized committee thereof.
Upon the dissolution, liquidation or winding up of the Company, after payment or provision for payment of the debts and other liabilities of the Company and subject to the rights, if any, of the holders of any outstanding series of Preferred Stock or any class or series of stock having a preference over or the right to participate with the Common Stock with respect to the distribution of assets of the Company upon such dissolution, liquidation or winding up of the Company, the holders of Common Stock shall be entitled to receive the remaining assets of the Company available for distribution to its stockholders ratably in proportion to the number of shares held by them.
F-19
The Board of Directors is authorized, by resolution or resolutions, to provide, out of the unissued shares of Preferred Stock, for one or more series of Preferred Stock and, with respect to each such series, to fix the number of shares constituting such series and the designation of such series, and the powers (including voting powers, if any), preferences and relative, participating, optional and other special rights, if any, and any qualifications, limitations or restrictions thereof, of the shares of such series of Preferred Stock. The powers, preferences and relative, participating, optional and other special rights of, and the qualifications, limitations or restrictions thereof, of each series of Preferred Stock, if any, may differ from those of any and all other series at any time outstanding.
Warrants
See Notes 3 and 8 regarding our issuance of a warrant for Series E Preferred Stock in connection with obtaining the Comerica Loan. As of December 31, 2013 and 2012, we had 16,332 and 20,449 warrants outstanding, respectively, that were exercisable into common shares at a weighted average price of $6.96 and $0.17 per share, respectively, at the option of the warrant holder. During the year ended December 31, 2013, a warrant for 13,473 shares of common stock was exercised at an exercise price of $0.17.
| 10 | Stock Based Compensation |
Equity Compensation Plans
The 2013 Equity Compensation Plan (the “2013 Plan”) adopted by our Board of Directors in May 2013, became effective upon consummation of the IPO in August 2013. There are 3,342,839 common shares authorized for future issuance under the 2013 Plan of which 94,862 were available as of December 31, 2013. Upon effectiveness of the 2013 Plan, stock options outstanding under the 2004 Equity Compensation Plan (the “2004 Plan”) to acquire 1,406,910 shares of our common stock were assumed under the 2013 Plan, leaving stock options to acquire 34,342 shares of our common stock outstanding under the 2004 Plan. There will be no further awards made under the 2004 Plan.
The 2013 Plan includes an “evergreen provision” that allows for an annual increase in the number of shares of common stock available for issuance under the 2013 Plan. The annual increase will be added on the first day of each fiscal year starting January 1, 2014, inclusive, and will be equal to five percent of the total number of shares of Common Stock outstanding on December 31st of the preceding calendar year as determined by the board of directors (the Board). The Board may act prior to the first day of any calendar year, to provide that there shall be no increase in the share reserve for such calendar year or that the increase in the share reserve for such calendar year shall be a lesser number of shares of Common Stock than would otherwise occur. On January 1, 2014 another 1,065,530 options became available for grant under this evergreen provision.
Stock Options
We use the Black-Scholes-Merton option pricing model to determine the fair value of our stock options. The determination of the fair value of stock-based payment awards on the date of grant using an option pricing model is affected by our stock price, as well as assumptions regarding a number of complex and subjective variables. These variables include our expected stock price volatility over the term of the awards, risk-free interest rate, actual employee exercise behaviors and expected dividends.
The following table shows the weighted average assumptions used to value stock options on the date of grant, as follows:
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | Year ended December 31, | | | | | | | |
| | | 2013 | | | 2012 | | | Period from Inception
(December 19, 2001) to
December 31, 2013 | |
| | | Employee | | | Non-Employees | | | Employee | | | Non-Employees | | | Employee | | | Non-Employees | |
Expected stock price volatility | | | 46.90 | % | | | 45.30 | % | | | 44.00 | % | | | 44.00 | % | | | 44.08 | % | | | 41.13 | % |
| | | | | | |
Risk-free interest rate | | | 1.04 | % | | | 0.37 | % | | | 1.38 | % | | | 1.38 | % | | | 1.64 | % | | | 1.21 | % |
| | | | | | |
Expected life of option (in years) | | | 3.69 | | | | 2.00 | | | | 6.25 | | | | 6.25 | | | | 4.44 | | | | 3.38 | |
| | | | | | |
Estimated dividend yield | | | 0.00 | % | | | 0.00 | % | | | 0.00 | % | | | 0.00 | % | | | 0.00 | % | | | 0.00 | % |
| | | | | | |
Weighted-average grant date fair value per share | | | 1.91 | | | | 1.49 | | | | 1.67 | | | | 1.67 | | | | 2.32 | | | | 1.99 | |
F-20
Expected stock price volatility was calculated based on the weighted-average of historical information of similar public entities. We will continue to use a weighted-average approach using other similar public entities’ volatility information until our historical volatility is relevant to measure expected volatility for future option grants. The risk-free rate was based on the U.S. Treasury yield curve in effect at the time of grant commensurate with the expected life assumption. The average expected life was determined based on anticipated exercise strategy and cancellation behavior for employees and nonemployees, primarily non-employee directors. Effective in 2013, a forfeiture rate of 1% and 0% was used for employees and nonemployees, respectively. We have not paid and do not anticipate paying cash dividends; therefore, the expected dividend rate was assumed to be 0%.
The following table summarizes our aggregate Equity Compensation Plan activity:
| | | | | | | | | | | | | | | | |
| | | Number of
Options | | | Weighted Average
Exercise Price | | | Weighted Average
Contractual Term
(in years) | | | Aggregate
Intrinsic Value(1) | |
Outstanding – December 31, 2012 | | | 1,409,659 | | | | | | | | | | | | | |
Granted | | | 1,965,696 | | | | | | | | | | | | | |
Exercised | | | (7,602 | ) | | | | | | | | | | | | |
Forfeited | | | (56,623 | ) | | | | | | | | | | | | |
Expired | | | (38,283 | ) | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
Outstanding – December 31, 2013 | | | 3,272,847 | | | $ | 5.38 | | | | 7.87 | | | $ | 2,420,963 | |
| | | | | | | | | | | | | | | | |
Exercisable – December 31, 2013 | | | 1,386,653 | | | $ | 6.98 | | | | 5.43 | | | $ | 800,721 | |
| | | | | | | | | | | | | | | | |
Vested and expected to vest at December 31, 2013(2) | | | 3,255,969 | | | $ | 5.39 | | | | 7.86 | | | $ | 2,403,403 | |
| | | | | | | | | | | | | | | | |
| (1) | Intrinsic value is the excess of the fair value of the underlying common shares as of December 31, 2013 over the weighted-average exercise price. |
| (2) | The number of stock options expected to vest takes into account an estimate of expected forfeitures. |
The total intrinsic value of options exercised during 2013 and 2012, and for the period from inception to December 31, 2013, was $11,000, $0 and $82,000, respectively.
The following table summarizes certain information about all options outstanding as of December 31, 2013:
| | | | | | | | | | | | | | | | |
| | | Options Outstanding | | | Options Exercisable | |
Exercise Price | | Options
Outstanding | | | Weighted Average
Remaining
Contractual Life
(Years) | | | Options
Exercisable | | | Weighted Average
Remaining
Contractual Life
(Years) | |
| | | | |
$1.67-$3.84 | | | 334,921 | | | | 5.8 | | | | 254,829 | | | | 5.0 | |
| | | | |
$4.00 | | | 1,366,760 | | | | 9.6 | | | | 4,256 | | | | 1.0 | |
| | | | |
$4.51-$4.81 | | | 951,961 | | | | 7.8 | | | | 626,455 | | | | 6.7 | |
| | | | |
$4.87-$12.53 | | | 619,205 | | | | 5.2 | | | | 501,113 | | | | 4.1 | |
| | | | | | | | | | | | | | | | |
| | | | |
| | | 3,272,847 | | | | 7.9 | | | | 1,386,653 | | | | 5.4 | |
| | | | | | | | | | | | | | | | |
F-21
Stock-Based Compensation Expense
Total stock-based compensation expense recognized based on the total grant date fair value of options vested and expected to vest was approximately $988,000 and $703,000 for the years ended December 31, 2013 and 2012, respectively, and $5.6 million for the period from inception to December 31, 2013. Due to the valuation allowance against our net deferred tax asset, we have never recognized a tax benefit for stock based compensation.
All stock options issued to nonemployees have been recorded at fair value. Options issued to nonemployees in exchange for services have resulted in expenses of $59,000, $38,000, and $156,000 during the years ended December 31, 2013 and 2012 and for the period from inception to December 31, 2013, respectively.
As of December 31, 2013, approximately $2.9 million of total unrecognized compensation cost related to unvested share options is expected to be recognized over a weighted-average period of 2.8 years.
In 2006, the Company adopted the Regado Biosciences 401k Plan (the Plan). The Plan allows eligible employees to defer up to 87% of their compensation, up to allowable Internal Revenue Service limits. Effective December 19, 2013 the Company elected to match participant elective deferrals in an amount equal to 50% of such elective deferrals up to 3% of each participant’s total compensation for the Plan Year. The Company also declared a retroactive match for the year ended December 31, 2013. The Company’s aggregate 401K match expense for the year ended December 31, 2013 was $87,813.
The Company has a Special Incentive Plan that provides for retention bonuses to be granted to certain participants in the event of a change of control.
Effective upon the consummation of the IPO, the Company adopted an Employee Stock Purchase Plan (the “ESPP”). Upon effectiveness, up to 96,360 shares of the Company’s common stock may be issued pursuant to the ESPP. There were no shares outstanding under this ESPP as of December 31, 2013.
Significant components of the Company’s deferred income tax assets and liabilities at December 31, 2013 and 2012, consisted of the following (in thousands):
| | | | | | | | |
| | | 2013 | | | 2012 | |
Deferred income tax assets: | | | | | | | | |
Net operating loss carryforwards | | $ | 53,436 | | | $ | 41,886 | |
Property and equipment | | | 136 | | | | 121 | |
Stock-based compensation expense | | | 661 | | | | 391 | |
Accruals | | | 1,220 | | | | — | |
Other | | | 44 | | | | 221 | |
Valuation allowance for deferred income tax assets | | | (54,999 | ) | | | (41,997 | ) |
| | | | | | | | |
Net deferred income tax assets | | | 498 | | | | 622 | |
Deferred income tax liabilities: | | | | | | | | |
Patent costs | | | (498 | ) | | | (622 | ) |
| | | | | | | | |
Net deferred income taxes | | $ | — | | | $ | — | |
| | | | | | | | |
At December 31, 2013 and 2012, the Company provided a full valuation allowance against its net deferred income tax assets, as management has assessed that the realization of these benefits is not more-likely-than-not. The change in the valuation allowance is primarily attributable to additional net operating loss carryforwards from current year activity.
F-22
The differences between the statutory rate and the effective rate are due to the valuation allowance and other permanent differences, as shown in the table below (dollars in thousands):
| | | | | | | | | | | | | | | | |
| | | 2013 | | | 2012 | |
| | | Tax | | | Rate | | | Tax | | | Rate | |
Rate Reconciliation | | | | | | | | | | | | | | | | |
Tax at statutory rate | | $ | (12,054 | ) | | | 35.0 | % | | $ | (4,572 | ) | | | 35.0 | % |
Incentive stock options | | | 148 | | | | (0.4 | )% | | | 248 | | | | (1.9 | )% |
State taxes | | | (1,378 | ) | | | 4.0 | % | | | (522 | ) | | | 4.0 | % |
Change in valuation allowance | | | 13,002 | | | | (37.8 | )% | | | 4,846 | | | | (37.1 | )% |
Other | | | 282 | | | | (0.8 | )% | | | — | | | | 0.0 | % |
| | | | | | | | | | | | | | | | |
Income tax expense reported | | $ | — | | | | 0.0 | % | | $ | — | | | | 0.0 | % |
As of December 31, 2013, the Company had estimated federal and state operating loss carry forwards of $137.0 million. These federal and state operating loss carry forwards begin to expire in 2022 and 2017, respectively. The utilization of the federal net operating loss carry forwards may be subject to limitations under the rules regarding a change in stock ownership as determined by the Internal Revenue Code, and state laws. Section 382 of the Internal Revenue Code of 1986, as amended, imposes annual limitations on the utilization of net operating loss (“NOL”) carry forwards, other tax carry forwards, and certain built-in losses upon an ownership change as defined by that section. In general terms, an ownership change may result from transactions that increase the aggregate ownership of certain stockholders in the Company stock by more than 50 percentage points over a three year testing period (“Section 382 Ownership Change”). If the Company has undergone a Section 382 Ownership change, an annual limitation would be imposed on certain of the Company’s tax attributes, including NOL and capital loss carry forwards, and certain other losses, credits, deductions of tax basis. As of December 31, 2013, the Company has not performed a formal study to determine whether there are Section 382 limitations that apply.
The Company recognizes the financial statement benefit of a tax position only after determining that the relevant tax authority would more-likely-than-not sustain the position following an audit. For tax positions meeting the more-likely-than-not threshold, the amount recognized in the financial statements is the largest benefit that has a greater than 50% likelihood of being realized upon ultimate settlement with the relevant tax authority. As of the end of 2013 or 2012, the Company did not record a liability for uncertain tax positions because no material positions existed.
The Company did not have any accrued interest or penalties associated with any unrecognized tax positions at December 31, 2013, and there were no such interest or penalties recognized during the period since inception through December 31, 2013.
F-23
The following table presents the computation of basic and diluted net loss per common share (in thousands, except share and per share data):
| | | | | | | | |
| | | Years ended December 31, | |
| | | 2013 | | | 2012 | |
Net loss attributable to common shareholders | | $ | (34,439 | ) | | $ | (13,062 | ) |
Weighted average common shares outstanding, basic and diluted | | | 7,499,661 | | | | 221,272 | |
| | | | | | | | |
Net loss per share attributable to common shareholders, basic and diluted | | $ | (4.59 | ) | | $ | (59.03 | ) |
| | | | | | | | |
For all periods presented, there is no difference in the number of shares used to calculate basic and diluted shares outstanding due to our net loss position. Securities that may potentially dilute earnings per share in the future that have not been included in the calculation of diluted net loss per share because to do so would be anti-dilutive are as follows (in common equivalent shares):
| | | | | | | | |
| | | Years ended December 31, | |
| | | 2013 | | | 2012 | |
Convertible preferred stock | | | — | | | | 8,539,257 | |
Common stock options | | | 3,272,847 | | | | 1,409,659 | |
Warrants | | | 16,332 | | | | 20,449 | |
| | | | | | | | |
Total | | | 3,289,179 | | | | 9,969,365 | |
| | | | | | | | |
In August 2013, all convertible preferred stock converted into common stock in conjunction with the consummation of our IPO. As of December 31, 2013 and 2012, we had 16,332 and 20,449 of warrants outstanding, respectively, that were exercisable into common shares at a weighted average price of $6.96 and $0.17 per share, respectively, at the option of the warrant holder.
2014 Private Placement
In 2014, we sold 4,000,000 shares of our common stock at a purchase price of $5.00 per share to certain accredited and institutional investors (the “2014 Private Placement”). The net proceeds of approximately $ 18.7 million from this offering will be used for general corporate and working capital purposes, including the ongoing Phase 3 REGULATE-PCI clinical trial.
Related Party Transactions
Purchasers of securities pursuant to the 2014 Private Placement included Robert Kierlin and Biotechnology Value Fund, LP, or BVF, LP each a holder of more than 5% of our capital stock. Mr. Kierlin purchased 400,000 shares of our common stock for an aggregate purchase price of $2.0 million. BVF, LP, purchased 2,000,000 shares of our common stock for an aggregate purchase price of $10.0 million.
F-24