QuickLinks -- Click here to rapidly navigate through this document
As filed with the Securities and Exchange Commission on August 2, 2005
Registration No. 333-124396
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Post-Effective
Amendment No. 1
to
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
Advanced Life Sciences Holdings, Inc.
(Exact Name of Registrant as Specified in Its Charter)
| Delaware (State or other jurisdiction of incorporation or organization) | 2854 (Primary Standard Industrial Classification Code Number) | 30-0296543 (I.R.S. Employer Identification Number) |
1440 Davey Road
Woodridge, Illinois 60517
(630) 739-6744
(Address, including zip code, and telephone number, including area code, of registrant's principal executive offices)
Michael T. Flavin, Ph.D.
Chairman and Chief Executive Officer
Advanced Life Sciences Holdings, Inc.
1440 Davey Road
Woodridge, Illinois 60517
(630) 739-6744
(Name, address, including zip code, and telephone number, including area code, of agent for service)
| Copies to: | ||
| R. Cabell Morris, Jr. Winston & Strawn LLP 35 West Wacker Drive Chicago, Illinois 60601 (312) 558-5600 | Paul M. Kinsella Ropes & Gray LLP One International Place Boston, Massachusetts 02110 (617) 951-7000 | |
Approximate date of commencement of proposed sale to the public:
As soon as practicable after the effective date of this Registration Statement.
If the only securities being registered on this Form are being offered pursuant to dividend or interest reinvestment plans, please check the following box. o
If any of the securities being registered on this Form are being offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, as amended check the following box. o
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. o
If delivery of the prospectus is expected to be made pursuant to Rule 434, please check the following box. o
Subject to Completion or Amendment
Preliminary Prospectus dated August 2, 2005
PRELIMINARY PROSPECTUS

6,000,000
Shares of Common Stock
We are offering 5,571,429 shares of our common stock through the underwriters. We are also offering 428,571 shares of our common stock in a concurrent offering directly to a current stockholder in exchange for a $3 million reduction of a milestone payment obligation under our license agreement with the stockholder. The consummation of the initial public offering and the concurrent offering are conditioned upon each other.
We expect the initial public offering price to be between $6.00 and $8.00 per share. The number of shares we are offering in the concurrent offering assumes an initial public offering price of $7.00 per share. The total public offering price, underwriting discount and the net proceeds to us shown in the table below reflect only shares of common stock we are offering through the underwriters.
No public market currently exists for our common stock. Our common stock has been approved for listing on the Nasdaq National Market under the symbol "ADLS."
Investment in our common stock involves risks.
See "Risk Factors" beginning on page 6 of this prospectus.
| | Per Share | Total | ||||
|---|---|---|---|---|---|---|
| Public offering price | $ | $ | ||||
| Underwriting discount | $ | $ | ||||
| Proceeds, before expenses, to us | $ | $ | ||||
The underwriters may also purchase up to an additional 835,714 shares from us at the public offering price less the underwriting discount within 30 days from the date of this prospectus to cover over-allotments of shares.
The underwriters expect to deliver the shares in New York, New York on or about , 2005.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or passed upon the accuracy or adequacy of this prospectus. Any representation to the contrary is a criminal offense.
| C.E. UNTERBERG, TOWBIN | THINKEQUITY PARTNERS LLC | |
MERRIMAN CURHAN FORD & CO. | ||
The date of this prospectus is , 2005.
| | Page | |
|---|---|---|
| Prospectus Summary | 1 | |
| Risk Factors | 6 | |
| Special Note Regarding Forward-Looking Statements | 22 | |
| Concurrent Offering | 23 | |
| Use of Proceeds | 24 | |
| Dividend Policy | 25 | |
| Capitalization | 26 | |
| Dilution | 27 | |
| Selected Financial Data | 29 | |
| Management's Discussion and Analysis of Financial Condition and Results of Operations | 30 | |
| Business | 40 | |
| Management | 69 | |
| Related Party Transactions | 81 | |
| Corporate Reorganization | 82 | |
| Principal Stockholders | 83 | |
| Description of Capital Stock | 85 | |
| Shares Eligible for Future Sale | 88 | |
| Underwriting | 90 | |
| Material U.S. Federal Income Tax Consequences to Non-U.S. Holders | 94 | |
| Legal Matters | 97 | |
| Experts | 97 | |
| Where You Can Find More Information | 97 | |
| Index to Consolidated Financial Statements | F-1 |
If it is against the law in any state to make an offer to sell these shares, or to solicit an offer from someone to buy these shares, then this prospectus does not apply to any person in that state, and no offer or solicitation is made by this prospectus to any such person.
You should rely only on the information contained in this prospectus. We have not authorized any person to provide you with different information. If anyone provides you with different or inconsistent information, you should not rely on it. We are not making an offering to distribute or sell these securities in any jurisdiction where the distribution or sale is not permitted. You should assume that the information appearing in this prospectus is accurate only as of the date on the front cover of this prospectus. Our business, financial condition, results of operations and prospects may have changed since that date.
i
This summary highlights information contained elsewhere in this prospectus. This summary does not contain all of the information you should consider before investing in our common stock. You should read this entire prospectus carefully, including the section entitled "Risk Factors," and our financial statements and related notes included elsewhere in this prospectus, before making an investment decision. References in this prospectus to our amended and restated certificate of incorporation and bylaws refer to the certificate of incorporation and bylaws as the same shall be in effect upon completion of the offerings. Unless otherwise specified or the context otherwise requires, references in this prospectus to "we," "our" and "us" refer to Advanced Life Sciences Holdings, Inc. and its wholly-owned operating subsidiary, Advanced Life Sciences, Inc.
Our Business
We are a biopharmaceutical company focused on the discovery, development and commercialization of novel drugs in the areas of infectious disease, inflammation and oncology. Using our internal discovery capabilities and our network of pharmaceutical and academic partners, we have assembled a promising pipeline of clinical and preclinical product candidates for the treatment of respiratory tract infections, human immunodeficiency virus (HIV), acute respiratory distress syndrome (ARDS) and cancer. We currently have four product candidates that are either in clinical development or approved to begin clinical development, and six additional product candidates that are in preclinical development.
We have an exclusive worldwide license (excluding Japan) from Abbott Laboratories to develop and commercialize cethromycin, a novel ketolide antibiotic in Phase III clinical development for the treatment of respiratory tract infections. Ketolides, a new class of antibiotic to treat respiratory tract infections, have demonstrated strong activity against bacteria that are resistant to many currently marketed antibiotics. Following the offerings, we intend to conduct two pivotal Phase III clinical trials of cethromycin for the treatment of mild-to-moderate community acquired pneumonia.
Through our 50/50 joint venture, Sarawak MediChem Pharmaceuticals, Inc., we have managed the development of Calanolide A, a potential treatment for HIV. We completed a series of Phase I clinical trials for Calanolide A and, subject to reaching a satisfactory agreement with our joint venture partner regarding our current business arrangement and our respective funding obligations for the next phase of development, we plan to enroll HIV-infected patients for a Phase IIa clinical trial of Calanolide A.
ALS-886 is a novel therapeutic entering clinical development for the treatment of inflammation-related tissue damage, including tissue damage associated with ARDS. We have established an open investigational new drug, or IND, application for ALS-886 with the FDA. We expect that the first Phase I clinical trial will involve approximately 40 patients to determine the safety profile of the compound in human subjects.
ALS-357 is a compound that has shown anti-tumor activity against malignant melanoma in preclinical trials. We have established an open IND application for ALS-357 with the FDA. The expected Phase I protocol contemplates using a topical cream to treat 16 patients with melanoma that has spread beyond the primary growth site.
In addition to the compounds summarized above, we have additional product candidates in preclinical development. These include compounds derived from natural products to treat infectious diseases, a novel class of synthetic chemotherapeutics to treat various cancers and small molecule inhibitors that may lead to a treatment for Alzheimer's disease.
1
Our Opportunity
We believe that there are attractive opportunities in the discovery, development and commercialization of product candidates in the areas of infectious disease, inflammation and oncology. Although these areas are highly competitive, we believe that the markets for the indications we intend to pursue are either too small or too fragmented for many large pharmaceutical companies. Many large pharmaceutical companies are exiting certain therapeutic areas and looking to out-license developmental products that fail to meet revenue thresholds. Our goal is to take advantage of in-licensing opportunities as they arise while also building on our foundation of internal drug discovery and development. We expect to compete effectively because of the proficiency of our experienced management team and the utility of our integrated chemistry and biology drug discovery platform.
Our Strategy
Our objective is to become a fully integrated pharmaceutical company that discovers and develops small molecule therapeutics to treat life-threatening diseases in the areas of infectious disease, inflammation and cancer, and then markets these products directly to physicians and hospitals. We plan to sustain our drug development pipeline through our internal drug discovery capabilities and by in-licensing promising compounds that fit into our areas of focus. Specific key aspects of our strategy include the following:
- •
- maximize the commercial potential of cethromycin;
- •
- advance the development of our other infectious disease, inflammation and oncology product candidates;
- •
- leverage our drug discovery and development capabilities;
- •
- continue to develop strategic collaborations; and
- •
- build commercial infrastructure to market our products.
None of our product candidates has received FDA marketing approval, and our revenues received to date consist of very limited amounts earned through management fees, grant programs and royalties. Our ability to generate any significant revenues in the near-term will depend solely on the successful development and commercialization of cethromycin, our most advanced product candidate, and our ability to market cethromycin to consumers effectively.
Our Collaborations
An integral part of our strategy is to establish strategic collaborations with leading pharmaceutical and biotechnology companies, governmental institutions and academic laboratories. We have entered into a collaboration with Abbott Laboratories to in-license cethromycin. We have established a 50/50 joint venture with the State Government of Sarawak, Malaysia for the development of Calanolide A. We have also licensed drug compounds from the National Institutes of Health, the University of Illinois at Chicago, Argonne National Laboratory and Baxter International in exchange for royalty and/or milestone obligations. In each of our collaborations, we have assumed complete responsibility to manage the development of the related product candidate.
Drug Discovery and Development Capabilities
We intend to expand our product portfolio by exploiting and enhancing our internal drug discovery and development capabilities. Our drug discovery efforts are focused on natural products, structural biology, medicinal chemistry and targeted biological screening. The integration of chemistry and biology gives us the capability to design, optimize and evaluate high potential drug candidates quickly and cost-effectively. Our chemistry capabilities include a strong emphasis on medicinal chemistry, the
2
discipline that seeks to identify optimal drug candidates through consideration of the biological effects of newly synthesized compounds. We then use our biological screening capabilities to obtain feedback regarding the therapeutic potential of the compounds synthesized by our chemists.
Risks Affecting Us
We are subject to a number of risks that you should be aware of before you decide to buy our common stock. These risks are discussed more fully in "Risk Factors." Our cumulative net loss and working capital deficit as of June 30, 2005 were $41.9 million and $16.4 million, respectively. We anticipate that our cumulative net losses will increase after the offerings. The opinion that we received from our independent registered public accounting firm regarding our 2004 financial statements contains an explanatory paragraph as to our ability to continue as a going concern. We have not received regulatory approval for any of our product candidates, and we do not anticipate generating any significant revenues for at least the next several years, if ever. It is possible that we may never successfully commercialize any of our product candidates.
Recent Development
On August 1, 2005, we agreed with Abbott Laboratories to reduce our royalty payment obligation to Abbott on the net sales of cethromycin if it is approved for commercialization. Under the new terms, we will owe Abbott royalty payments of 19.0% on the first $100 million of aggregate net sales of cethromycin, 18.0% on net sales once aggregate net sales exceed $100 million but are less than $200 million, and 17.0% on all net sales once aggregate net sales exceed $200 million. In addition, we agreed to amend our license agreement with Abbott in order to terminate our in-license of ABT-210, a second-generation ketolide antibiotic product candidate in preclinical development.
We were incorporated as a holding company in Delaware in 2004. Our wholly-owned operating subsidiary, Advanced Life Sciences, Inc., was incorporated in Illinois in 1999. Our principal executive offices are located at 1440 Davey Road, Woodridge, Illinois 60517. Our telephone number is (630) 739-6744. Our web site is http://www.advancedlifesciences.com. We do not intend the information found on our web site to be part of this prospectus and our web site may not contain all of the information that is important to you.
"Advancing Discoveries For Health" and the Advanced Life Sciences logo are trademarks of Advanced Life Sciences, Inc.
References in this prospectus to the offerings are to both the 5,571,429 shares of common stock we are offering through the underwriters in the initial public offering and the 428,571 shares of common stock we are offering in a concurrent offering directly to one of our stockholders.
References to data compiled by IMS Health are reprinted with permission, IMS Health, IMS National Prescription Audit, Copyright 2005, all rights reserved.
3
The Offerings
| Common stock we are offering in our initial public offering | 5,571,429 shares | |
| Common stock we are offering in the concurrent offering | 428,571 shares | |
Common stock to be outstanding after the offerings | 16,826,801 shares | |
Nasdaq National Market symbol | ADLS | |
Use of proceeds | Of the net proceeds from the initial public offering, we intend to use approximately $28.5 million to continue the clinical development of cethromycin and to make milestone and license payments to Abbott Laboratories and the remainder for general corporate purposes, including potential further development of our other product candidates and working capital. See "Use of Proceeds" section of this prospectus. |
The number of shares to be outstanding immediately after this offering is based on the number of shares outstanding as of June 30, 2005, excluding:
- •
- 686,837 shares of common stock issuable upon the exercise of options outstanding under our 1999 Stock Incentive Plan at a weighted average exercise price of $0.93 per share;
- •
- 2,072,455 additional shares of common stock reserved for future issuances under our 2005 Stock Incentive Plan; and
- •
- 14,887 shares of common stock issuable upon the exercise of warrants outstanding at an exercise price of $8.02 per share.
Except as otherwise indicated, information in this prospectus:
- •
- gives effect to a 3.97-for-one stock split effective on June 29, 2005; and
- •
- assumes the underwriters have not exercised their option to purchase 835,714 shares to cover over-allotments.
4
The following table summarizes our consolidated financial data. The summary financial data as of December 31, 2003 and 2004 and for the years ended December 31, 2002, 2003 and 2004 are derived from our audited financial statements included elsewhere in this prospectus. The balance sheet data as of December 31, 2000, 2001 and 2002, and the statement of operations data for the years ended December 31, 2000 and 2001, have been derived from our audited financial statements not included in this prospectus. The balance sheet data as of June 30, 2005 and the statement of operations data for each of the six months ended June 30, 2004 and 2005 have been derived from our unaudited financial statements, which include, in the opinion of management, all adjustments necessary to present fairly the data for such periods. You should read this data together with our financial statements and related notes included elsewhere in this prospectus and the information under "Selected Financial Data" and "Management's Discussion and Analysis of Financial Condition and Results of Operations."
| | Years ended December 31, | Six months ended June 30, | Period from Inception (January 1, 1999) to June 30, 2005 | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2000 | 2001 | 2002 | 2003 | 2004 | 2004 | 2005 | |||||||||||||||||||
| Statement of Operations Data: | ||||||||||||||||||||||||||
| Revenue | $ | 447,525 | $ | 225,218 | $ | 496,207 | $ | 180,427 | $ | 319,780 | $ | 182,973 | $ | 44,379 | $ | 2,125,129 | ||||||||||
| Operating expenses: | ||||||||||||||||||||||||||
| Research and development(1) | 3,159,383 | 2,771,517 | 924,799 | 1,362,255 | 25,661,868 | 570,986 | 724,347 | 37,516,266 | ||||||||||||||||||
| Selling, general and administrative | 344,607 | 461,001 | 668,622 | 1,198,722 | 1,649,953 | 639,085 | 780,161 | 5,177,282 | ||||||||||||||||||
| Loss from operations | (3,056,465 | ) | (3,007,300 | ) | (1,097,214 | ) | (2,380,550 | ) | (26,992,041 | ) | (1,027,098 | ) | (1,460,129 | ) | (40,568,419 | ) | ||||||||||
Interest expense, net | 231,930 | 377,029 | 164,337 | 170,938 | 194,877 | 96,389 | 204,249 | 1,370,252 | ||||||||||||||||||
| Net loss | (3,288,395 | ) | (3,384,329 | ) | (1,261,551 | ) | (2,551,488 | ) | (27,186,918 | ) | (1,123,487 | ) | (1,664,378 | ) | (41,938,671 | ) | ||||||||||
| Less accumulating preferred dividends for the period | 175,000 | 175,000 | 175,000 | 175,000 | 175,000 | 87,500 | 87,500 | 1,057,292 | ||||||||||||||||||
| Net loss available to common shareholders | $ | (3,463,395 | ) | $ | (3,559,329 | ) | $ | (1,436,551 | ) | $ | (2,726,488 | ) | $ | (27,361,918 | ) | $ | (1,210,987 | ) | $ | (1,751,878 | ) | $ | (42,995,963 | ) | ||
Basic and diluted loss per common share | $(2.18 | ) | $(2.24 | ) | $(0.90 | ) | $(1.72 | ) | $(13.27 | ) | $(0.76 | ) | $(0.16 | ) | ||||||||||||
Weighted average shares outstanding—basic | 1,588,000 | 1,588,000 | 1,588,000 | 1,588,000 | 2,062,351 | 1,588,134 | 10,780,148 | |||||||||||||||||||
| | As of December 31, | | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
As of June 30, 2005 | |||||||||||||||||||
| | 2000 | 2001 | 2002 | 2003 | 2004 | ||||||||||||||
| Balance Sheet Data: | |||||||||||||||||||
| Cash | $ | 94,757 | $ | 7,846 | $ | 18,739 | $ | 61,203 | $ | 194,555 | $ | 39,951 | |||||||
| Total assets | 251,687 | 160,467 | 146,917 | 255,127 | 1,015,932 | 1,769,858 | |||||||||||||
| Long-term debt and lease payable, less current portion | 5,002,992 | 2,000,000 | 2,000,000 | 2,000,000 | 2,027,279 | 2,020,498 | |||||||||||||
| Total liabilities(2) | 5,876,597 | 2,252,835 | 2,339,836 | 2,661,806 | 19,289,733 | 21,102,004 | |||||||||||||
| Deficit accumulated during the development stage | (5,874,910 | ) | (9,274,336 | ) | (10,535,887 | ) | (13,087,375 | ) | (40,274,293 | ) | (41,938,671 | ) | |||||||
| Total stockholders' deficit | (5,624,910 | ) | (2,092,368 | ) | (2,192,919 | ) | (2,406,679 | ) | (18,273,801 | ) | (19,332,146 | ) | |||||||
- (1)
- Amount for 2004 includes approximately $24.5 million of in-process research and development expense related to compounds licensed from Abbott Laboratories in December 2004.
- (2)
- Amount for 2004 includes $14.0 million in payments due in 2005 under our license agreement with Abbott Laboratories executed in December 2004. See Note 11 to our consolidated financial statements. As a result of the concurrent offering, the amount of license payments due to Abbott will be reduced by $3.0 million. See "Concurrent Offering" and "Capitalization."
5
An investment in our common stock involves a high degree of risk. You should carefully consider the following risk factors and other information in this prospectus, including our consolidated financial statements and the notes thereto, before deciding whether to purchase our common stock. Additional risks and uncertainties not presently known to us or that we currently deem immaterial may also impair our business, results of operations and your investment. If any of the events or developments described below actually occurs, our business, financial condition and results of operations may suffer. In that case, the value of our common stock may decline, and you could lose all or part of your investment.
Risks Related to Our Industry and Business
We are at an early stage of development and may never attain product sales.
We have not received FDA approval for any of our product candidates. Any compounds that we discover or in-license will require extensive and costly development, preclinical testing and/or clinical trials prior to seeking regulatory approval for commercial sales. Our most advanced product candidate, cethromycin, and any other compounds we discover, develop or in-license, may never be approved for commercial sale. The time required to attain product sales and profitability is lengthy and highly uncertain, and we cannot assure you that we will be able to achieve or maintain product sales.
We expect our net operating losses to continue for at least several years, and we are unable to predict the extent of future losses or when we will become profitable, if ever.
We have incurred significant net losses since our formation in 1999. Our cumulative net loss was $41.9 million as of June 30, 2005, and we anticipate that our cumulative net losses will increase after the offerings. Our operating losses are due in large part to the significant research and development costs required to identify, validate and license potential product candidates, conduct preclinical studies and conduct clinical trials of our more advanced product candidates. To date, we have generated only limited revenues, consisting of management fees and one-time or limited payments associated with our collaborations or grants, and we do not anticipate generating any significant revenues for at least the next several years, if ever. We expect to increase our operating expenses over the next several years as we plan to:
- •
- expand the clinical trial program for cethromycin;
- •
- continue the preclinical development and commence the clinical development of our other product candidates, such as ALS-886 and ALS-357;
- •
- expand our research and development activities;
- •
- acquire or in-license new technologies and product candidates; and
- •
- increase our required corporate infrastructure and overhead.
As a result, we expect to continue to incur significant and increasing operating losses for the foreseeable future. Because of the numerous risks and uncertainties associated with our research and product development efforts, we are unable to predict the extent of any future losses or when we will become profitable, if ever. Even if we do achieve profitability, we may not be able to sustain or increase profitability on an ongoing basis.
We will require additional funding to satisfy our future capital needs, and future financing strategies may adversely affect holders of our common stock.
Our operations will require significant additional funding in large part due to our research and development expenses, future preclinical and clinical testing costs, the possibility of expanding our
6
facilities, and the absence of any meaningful revenues during the foreseeable future. We do not know whether additional financing will be available to us on favorable terms or at all. If we cannot raise additional funds, we may be required to reduce our capital expenditures, scale back product development or programs, reduce our workforce and license to others products or technologies that we may otherwise be able to commercialize. We believe that the net proceeds from the initial public offering will be sufficient to meet our anticipated operating requirements through 2006; however, we have based this estimate on assumptions that may prove to be incorrect.
To the extent we raise additional capital by issuing equity securities, our stockholders could experience substantial dilution. Any additional equity securities we issue may have rights, preferences or privileges senior to those of existing holders of stock, including purchasers of our common stock in the offerings. To the extent that we raise additional funds through collaboration and licensing arrangements, we may be required to relinquish some rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us.
Our financial statements include a "going concern" limitation, and there is no guarantee that we will be able to operate our business or generate revenues.
Our ability to continue as a going concern is dependent on our ability to raise additional capital, including from the offerings. We do not currently have sufficient capital to fund our operations through 2005. The opinion that we received from our independent registered public accounting firm regarding our 2004 financial statements contains an explanatory paragraph as to our ability to continue as a going concern. If the proceeds from the initial public offering are inadequate to fund our operations through 2006, we may also receive a going concern modification on our 2005 financial statements. If doubts are raised about our ability to continue as a going concern following the offerings, our stock price could drop and our ability to raise additional funds may be adversely affected. Any of these outcomes would be detrimental to our operations.
Following the offerings, we intend to conduct two pivotal Phase III clinical trials of cethromycin for the treatment of mild-to-moderate community acquired pneumonia, and our business would be materially harmed if these trials are unsuccessful.
We anticipate that our ability to generate any significant product revenues in the near future will depend solely on the successful development and commercialization of cethromycin, our most advanced product candidate. Following the offerings, we intend to conduct two pivotal Phase III clinical trials of cethromycin for the treatment of mild-to-moderate community acquired pneumonia. Prior to licensing the compound to us, Abbott Laboratories conducted an initial Phase III clinical trial of cethromycin for the treatment of community acquired pneumonia. Although the results of this study met the primary endpoints for efficacy, this is no assurance that our pivotal Phase III clinical trials will be successful.
The Abbott study used cethromycin dosing levels of 150 mg once-daily and 150 mg twice-daily. Our proposed pivotal Phase III protocol uses a dosing level of 300 mg once-daily, even though Abbott was able to demonstrate efficacy at 150 mg once-daily. We do not expect that the different dosing regimen will reduce the effectiveness of cethromycin in our pivotal Phase III clinical trials, but we could be incorrect. Clinical trials at the higher dosage level are also likely to increase the frequency of adverse side effects. The Abbott study did not include a standard of care comparator arm. Antibiotic products currently in the marketplace are generally effective and, for this reason, the required threshold to demonstrate non-inferiority for new antibiotic compounds is often very high. There can be no assurance that our pivotal Phase III clinical trials of cethromycin will establish non-inferiority when a comparator arm is applied.
The success of our pivotal Phase III clinical trials in treating mild-to-moderate community acquired pneumonia may depend on many factors that are beyond our control and unrelated to the safety or
7
efficacy of cethromycin. For instance, clinical studies may demonstrate lower than expected cure rates because of the lack of patient compliance with prescribed dosing regimens. Cethromycin is our most advanced product candidate, and, as a company, we do not have any prior experience in conducting Phase III clinical trials. If we are unable to complete these trials successfully or ultimately receive FDA marketing approval for cethromycin, our ability to generate revenues and our business will be materially harmed.
Prior clinical trials of cethromycin have shown evidence of side effects that may diminish its prospects for commercialization and wide market acceptance.
The gastrointestinal adverse events reported in prior clinical trials of cethromycin were generally at frequencies consistent with Ketek and other antibiotic treatments. These gastrointestinal side effects included nausea, diarrhea, vomiting, headache and abdominal pain. Taste perversion was the adverse event most frequently observed in prior clinical trials of cethromycin, occurring in 17% of subjects across once-daily dosing regimens of 150 mg, 300 mg and 600 mg. According to available data, taste perversion has not occurred at any significant level for other antibiotic treatments. We do not believe that, by itself, taste perversion presents a safety risk for patients. However, taste perversion may lead to higher levels of patient non-compliance, which could have the effect of reducing overall cure rates in our planned pivotal Phase III clinical trials of cethromycin. If overall cure rates in our pivotal Phase III clinical trials are not sufficient to establish non-inferiority, we may not receive FDA approval for cethromycin and our business will be materially harmed.
Prior pivotal Phase III comparator trials of cethromycin in the treatment of bronchitis and pharyngitis failed to establish non-inferiority, and our inability to expand cethromycin into these indications would harm our ability to generate additional revenues in the future.
Abbott Laboratories conducted four pivotal Phase III comparator trials for cethromycin in treating bronchitis and pharyngitis at a dosing level of 150 mg once-daily. Each of these trials failed to establish non-inferiority against comparator antibiotics. While we believe that the negative outcomes of the Abbott comparator trials were related to dosing levels, we may be incorrect. The failure to meet primary endpoints in the Abbott trials may not have been dose related, as we believe, but rather a result of the compound's lack of sufficient clinical efficacy. Clinical trials using a 300 mg once-daily regimen are also likely to increase the occurrence of adverse side effects. Even if we receive FDA approval to market cethromycin for the treatment of community acquired pneumonia, our failure to expand cethromycin into other indications would harm our ability to generate additional revenues in the future.
Because we are heavily dependent on our license agreement with Abbott Laboratories and our collaborations with other third parties, our product development programs may be delayed or terminated by factors beyond our control.
In December 2004, we entered into a license agreement with Abbott Laboratories for certain patent applications, patents and proprietary technology relating to cethromycin. We have also entered into a number of license agreements for intellectual property and other rights needed to develop our product candidates that are in earlier stages of development. Our primary collaborators other than Abbott include the National Institutes of Health, the University of Illinois at Chicago, Argonne National Laboratory and Baxter International. A description of our agreements with collaborators can be found under the headings "Business—Abbott Laboratories Collaboration" and "Business—Other Collaborations and License Agreements." Our collaborations generally present additional risks to our business, such as the risk that our collaborators encounter conflicts of interest to their arrangements with us, inadequately defend our intellectual property rights or develop other products that compete with us. Our ability to generate any significant product revenues in the near future will depend solely
8
on the successful commercialization of cethromycin. If for any reason we are unable to realize the expected benefits of our license agreement with Abbott, or under any of our other collaborations, then our business and financial condition may be materially harmed.
If we are unable to restructure the debt and joint venture relationship with the State Government of Sarawak, Malaysia, then our development program for Calanolide A may be delayed or abandoned.
The intellectual property for Calanolide A is held by Sarawak MediChem Pharmaceuticals, Inc., our 50/50 joint venture with the State Government of Sarawak, Malaysia. The original intent of our business relationship was that we would contribute intellectual property and management to the joint venture and our partner would provide funding. To advance Calanolide A, the Sarawak Government funded $9 million by purchasing its 50% equity position and loaned an additional $12 million to the joint venture. According to the terms of the loan agreement with the Sarawak Government, $9 million of this debt is past due and $0.5 million and $2.5 million are due in the third and fourth quarters of 2005, respectively. As a result of this default in payment by the joint venture, the Sarawak Government has the option under the loan agreement to take over the control and management of the Calanolide A project or the affairs of the joint venture until such time as the loan is repaid in full. The Sarawak Government recently asserted its rights to assume control of the management of the joint venture. We have initiated discussions with the Sarawak Government to determine the form in which their representative would assume control of the affairs and management of the joint venture. The Sarawak Government also expressly reserved all contractual and legal remedies available in light of the default and indicated its intent to fully exercise its rights as a creditor of the joint venture. Ultimately, we believe that in order for the joint venture to continue, the terms of the debt must be amended to extend its maturity to a date after commercialization of Calanolide A.
Further development will require that we and the Sarawak Government agree to restructure and extend the debt and determine the source of financing for Phase IIa clinical trials of Calanolide A. We have proposed to the Sarawak Government that, for the first time, we would jointly fund the next development phase of Calanolide A through future advances of debt or purchases of equity. We currently estimate that it would cost approximately $2 million to conduct a Phase IIa clinical trial of Calanolide A. No agreement has yet been reached, and we do not intend to expend any meaningful funds advancing the development of Calanolide A unless and until we restructure the joint venture's debt and reach a satisfactory agreement with the Sarawak Government to amend our joint venture relationship and establish our respective obligations regarding the future funding of the next phase of developing Calanolide A. As a result, we are unable to predict when, or if at all, we will proceed with our plans to commence Phase IIa clinical trials for Calanolide A.
Further, our joint venture agreement with the Sarawak Government provides that, upon termination of the joint venture, any liabilities of the joint venture will be settled by the parties equally and all patents or licenses of the joint venture shall be reassigned back to us, including rights to clinical trials. If we and the Sarawak Government are unable to restructure the debt of the joint venture or if any dispute arises with the Sarawak Government, then our ability to continue the development of Calanolide A may be adversely affected, and we may cease future participation in the program. See "Business—Other Collaborations and License Agreements—Sarawak MediChem Pharmaceuticals, Inc."
Because the results of preclinical studies for our preclinical product candidates are not necessarily predictive of future results, our product candidates may not have favorable results in later clinical trials or ultimately receive regulatory approval.
Only two product candidates in our development pipeline, cethromycin and Calanolide A, have been tested in clinical trials. Our other product candidates have only been through preclinical studies. Positive results from preclinical studies, particularlyin vitro studies, are no assurance that later clinical trials will succeed. Preclinical trials are not designed to establish the efficacy of our preclinical product
9
candidates. We will be required to demonstrate through clinical trials that these product candidates are safe and effective for use before we can seek regulatory approvals for their commercial sale. There is typically an extremely high rate of failure as product candidates proceed through clinical trials. If our product candidates fail to demonstrate sufficient safety and efficacy in any clinical trial, we would experience potentially significant delays in, or be required to abandon, development of that product candidate. This would adversely affect our ability to generate revenues and may damage our reputation in the industry and in the investment community.
The future clinical testing of our product candidates could be delayed, resulting in increased costs to us and a delay in our ability to generate revenues.
Our product candidates will require preclinical testing and extensive clinical trials prior to submitting a regulatory application for commercial sales. We do not know whether planned clinical trials will begin on time, if at all. Delays in the commencement of clinical testing could significantly increase our product development costs and delay product commercialization. In addition, many of the factors that may cause, or lead to, a delay in the commencement of clinical trials may also ultimately lead to denial of regulatory approval of a product candidate. Each of these results would adversely affect our ability to generate revenues.
The commencement of clinical trials can be delayed for a variety of reasons, including delays in:
- •
- demonstrating sufficient safety to obtain regulatory approval to commence a clinical trial;
- •
- reaching agreement on acceptable terms with prospective contract research organizations and trial sites;
- •
- manufacturing sufficient quantities of a product candidate; and
- •
- obtaining institutional review board approvals to conduct clinical trials at prospective sites.
In addition, the commencement of clinical trials may be delayed due to insufficient patient enrollment, which is a function of many factors, including the size of the patient population, the nature of the protocol, the proximity of patients to clinical sites, the availability of effective treatments for the relevant disease, and the eligibility criteria for the clinical trial. Because community acquired pneumonia is generally seasonal in nature, achieving sufficient patient enrollment for our pivotal Phase III clinical trials of cethromycin may be particularly difficult. We have not yet received approval from the FDA for our proposed pivotal Phase III protocol in treating mild-to-moderate community acquired pneumonia, and we are not able to begin our pivotal Phase III clinical trials of cethromycin until after we receive this approval.
The FDA required one of our competitors, Aventis Pharmaceuticals, to undertake a 24,000 patient safety study prior to approving Ketek® for commercialization. We do not expect the FDA to require a similarly sized trial for cethromycin because Ketek was the first ketolide introduced and no previous safety testing had been conducted for this class of antibiotics. Since that time, Ketek has been administered to over ten million patients and, to our knowledge, has not demonstrated any severe safety risks. If the FDA does require a large safety trial for cethromycin, however, it would significantly increase our costs and extend the time required to receive regulatory approval.
We will depend on outside parties to conduct our clinical trials, and any failure of those parties to fulfill their obligations could result in costs and delays beyond our control that prevent us from obtaining regulatory approval or successfully commercializing product candidates.
Although we do not currently have any agreements with contract research organizations, we intend to engage contract research organizations to perform data collection and analysis and other aspects of our clinical trials of cethromycin and our other product candidates. We do not have the physical and
10
human resources to conduct our clinical trials independently. The contract research organizations we engage will interact with clinical investigators and medical institutions to enroll patients in our clinical trials. As a result, we will depend on these clinical investigators, medical institutions and contract research organizations to perform the studies and trials properly. If these parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, or if the quality, completeness or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols or for other reasons, our clinical trials may be extended, delayed or terminated. Many of these factors will be outside of our oversight and beyond our control. We may not be able to enter into replacement arrangements without undue delays or considerable expenditures. If there are delays in testing or regulatory approvals as a result of the failure to perform by third parties, our drug discovery and development costs will increase, and we may not be able to obtain regulatory approval for our product candidates. In addition, we may not be able to establish or maintain relationships with these parties on favorable terms, if at all.
Even if we successfully complete clinical trials of cethromycin or any other product candidate, we may fail to obtain FDA approval of a new drug application.
The FDA may not approve in a timely manner, or at all, any NDA we submit. If we are unable to submit an NDA for cethromycin or any other product candidate, or if any NDA we submit is not approved by the FDA, we will be unable to commercialize that product in the United States. The FDA can and does reject NDAs, and often requires additional clinical trials, even when product candidates performed well or achieved favorable results in large-scale Phase III clinical trials. The FDA imposes substantial requirements on the introduction of pharmaceutical products through lengthy and detailed laboratory and clinical testing procedures, sampling activities and other costly and time-consuming procedures. Satisfaction of these requirements typically takes several years and may vary substantially based upon the type and complexity of the pharmaceutical product. A number of our product candidates are novel compounds, which may further increase the period of time required for satisfactory testing procedures.
Data obtained from preclinical and clinical activities are susceptible to varying interpretations, which could delay, limit or prevent regulatory approval. In addition, delays or rejections may be encountered based on changes in, or additions to, regulatory policies for drug approval during the period of product development and regulatory review. The effect of government regulation may be to delay or prevent the commencement of clinical trials or marketing of our product candidates for a considerable period of time, to impose costly procedures upon our activities and to provide an advantage to our competitors that have greater financial resources or are more experienced in regulatory affairs. The FDA may not approve our product candidates for clinical trials or marketing on a timely basis or at all. Delays in obtaining or failure to obtain such approvals would adversely affect the marketing of our product candidates and our liquidity and capital resources.
Drug products and their manufacturers are subject to continual regulatory review after the product receives FDA approval. Later discovery of previously unknown problems with a product or manufacturer may result in additional clinical testing requirements or restrictions on such product or manufacturer, including withdrawal of the product from the market. Failure to comply with applicable regulatory requirements can, among other things, result in fines, injunctions and civil penalties, suspensions or withdrawals of regulatory approvals, product recalls, operating restrictions or shutdown and criminal prosecution. We may lack sufficient resources and expertise to address these and other regulatory issues as they arise.
11
If we fail to obtain regulatory approvals in other countries for our product candidates under development, we will not be able to generate revenues in such countries.
In order to market our products outside of the United States, we must comply with numerous and varying regulatory requirements of other countries. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval in the United States. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may negatively impact the regulatory process in others. The risks involved in the non-U.S. regulatory approval process, as well as the consequences for failing to comply with applicable regulatory requirements, generally include the same considerations as in the United States. A description of U.S. regulatory considerations can be found under the section entitled "—Even if we successfully complete clinical trials of cethromycin or any other product candidate, we may fail to obtain FDA approval of a new drug application."
Even if we successfully develop and obtain approval for cethromycin or any of our other product candidates, our business will not be profitable if those products do not achieve and maintain market acceptance.
Even if any of our product candidates are approved for commercial sale by the FDA or other regulatory authorities, the degree of market acceptance of any approved product candidate by physicians, healthcare professionals, patients and third-party payors, and our resulting profitability and growth, will depend on a number of factors, including:
- •
- our ability to provide acceptable evidence of safety and efficacy;
- •
- relative convenience and ease of administration;
- •
- the prevalence and severity of any adverse side effects;
- •
- the availability of alternative treatments;
- •
- the details of FDA labeling requirements, including the scope of approved indications and any safety warnings;
- •
- pricing and cost effectiveness;
- •
- the effectiveness of our or our collaborators' sales and marketing strategy;
- •
- our ability to obtain sufficient third-party insurance coverage or reimbursement; and
- •
- our ability to have the product listed on insurance company formularies.
If any of our product candidates achieve market acceptance, we may not be able to maintain that market acceptance over time if new products or technologies are introduced that are received more favorably or are more cost effective. Complications may also arise, such as antibiotic or viral resistance, that render our products obsolete. We rely on the favorable resistance profile of cethromycin exhibited in clinical trials to be a potential competitive distinction from currently marketed compounds. Even if we receive FDA approval to market cethromycin, antibiotic resistance may emerge that will substantially harm our ability to generate revenues from its sale.
We are initially seeking FDA approval for cethromycin as a 7-10 day treatment regimen. There are currently a number of antibiotic products that are marketed as 5-day therapies. In the event that the marketplace considers this to be a significant competitive distinction, it is uncertain whether we will be able to make cethromycin available for a lower dosing period. In addition, we expect that cethromycin, if approved for sale, would be used primarily in the outpatient setting.
12
Our most advanced product candidate, cethromycin, will face significant competition in the marketplace if it receives marketing approval from the FDA.
Initially, we plan to conduct pivotal Phase III clinical trials of cethromycin for the treatment of mild-to-moderate community acquired pneumonia. We also intend to pursue opportunities for cethromycin in the treatment of other respiratory tract infections such as bronchitis, sinusitis and pharyngitis. There are several classes of antibiotics that are primary competitors for the treatment of one or more of these indications, including:
- •
- macrolides such as Biaxin® (clarithromycin), a product of Abbott Laboratories; and Zithromax® (azithromycin), a product of Pfizer Inc.;
- •
- one other ketolide antibiotic, Ketek® (telithromycin), recently launched in the United States and Europe by Aventis Pharmaceuticals;
- •
- semi-synthetic penicillins such as Augmentin® (amoxicillin and clavulanate potassium), a product of GlaxoSmithKline; and
- •
- fluoroquinolones such as Levaquin® (levofloxacin), a product of Ortho-McNeil Pharmaceutical, Inc.; Tequin® (gatifloxacin), a product of Bristol-Myers Squibb Company; FACTIVE® (gemifloxacin mesylate) tablets, a product of Oscient Pharmaceuticals; and Cipro® (ciprofloxacin) and Avelox® (moxifloxacin), both products of Bayer Corporation.
If cethromycin is approved by the FDA, it will not be the first ketolide antibiotic introduced to the marketplace. Ketek has been available for sale in Europe since 2002 and in the United States since August 2004. There are several additional ketolide product candidates in preclinical development or entering clinical development, including ABT-210, a second-generation ketolide antibiotic product candidate that we transferred back to Abbott Laboratories in July 2005. If ultimately approved by the FDA, these product candidates may have improved efficacy, ease of administration or side effect profiles when compared to cethromycin. The availability of additional ketolide antibiotics may have an adverse affect on our ability to generate product revenues and achieve profitability.
The availability of generic equivalents may adversely affect our ability to generate product revenues from cethromycin.
Many generic antibiotics are currently prescribed to treat respiratory tract infections. As competitive products lose patent protection, makers of generic drugs will likely begin to market additional competing products. Companies that produce generic equivalents are generally able to offer their products at lower prices. Ketek may lose patent protection as early as 2015, which would enable generic drug manufacturers to sell generic ketolide antibiotics at a lower cost than cethromycin. Generic equivalents of Biaxin and Zithromax, two macrolide antibiotic products, may be available as early as 2005. Cethromycin, if approved for commerical sale, may be at a competitive disadvantage because of its higher cost relative to generic products. This may have an adverse effect on our ability to generate product revenues from cethromycin.
We will face significant competition from other biotechnology and pharmaceutical companies, and our operating results will suffer if we fail to compete effectively.
We are a development stage company with 20 employees. Many of our competitors, such as Pfizer, GlaxoSmithKline and Bayer, are large pharmaceutical companies with substantially greater financial, technical and human resources than we have. The biotechnology and pharmaceutical industries are intensely competitive and subject to rapid and significant technological change. Many of the drugs that we are attempting to discover or develop will compete with existing therapies if we receive marketing approval. Because of their significant resources, our competitors may be able to use discovery technologies and techniques, or partnerships with collaborators, in order to develop competing products
13
that are more effective or less costly than the product candidates we develop. This may render our technology or product candidates obsolete and noncompetitive. Academic institutions, government agencies, and other public and private research organizations may seek patent protection with respect to potentially competitive products or technologies and may establish exclusive collaborative or licensing relationships with our competitors.
As a company, we do not have any prior experience in conducting Phase III clinical trials. Our competitors may succeed in obtaining FDA or other regulatory approvals for product candidates more rapidly than us. Companies that complete clinical trials, obtain required regulatory agency approvals and commence commercial sale of their drugs before we do may achieve a significant competitive advantage, including certain FDA marketing exclusivity rights that would delay or prevent our ability to market certain products. Any approved drugs resulting from our research and development efforts, or from our joint efforts with our existing or future collaborative partners, might not be able to compete successfully with our competitors' existing or future products.
Our collaborators and third party manufacturers may not be able to manufacture our product candidates in larger quantities, which would prevent us from commercializing our product candidates.
To date, each of our product candidates has been manufactured in small quantities by our collaborators and third party manufacturers for preclinical and clinical trials. We do not currently have any agreements with third party manufacturers. If any of our product candidates is approved by the FDA or other regulatory agencies for commercial sale, we will need to enter into agreements with third parties to manufacture the product in larger quantities. Due to factors beyond our control, our collaborators and third party manufacturers may not be able to increase their manufacturing capacity for any of our product candidates in a timely or economic manner, or at all. Significant scale-up of manufacturing may require additional validation studies, which the FDA must review and approve. If we are unable to increase the manufacturing capacity for a product candidate successfully, the regulatory approval or commercial launch of that product candidate may be delayed or there may be a shortage in the supply of the product candidate. Our product candidates require precise, high-quality manufacturing. The failure of our collaborators and third party manufacturers to achieve and maintain these high manufacturing standards, including the incidence of manufacturing errors, could result in patient injury or death, product recalls or withdrawals, delays or failures in product testing or delivery, cost overruns or other problems that could seriously harm our business.
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to sell and market any products we may develop, we may be unable to generate revenues.
We do not currently have product sales and marketing capabilities. If we receive regulatory approval to commence commercial sales of any of our product candidates, we will have to establish a sales and marketing organization with appropriate technical expertise and distribution capabilities or make arrangements with third parties to perform these services. If we receive approval to commercialize cethromycin for the treatment of community acquired pneumonia, we intend to market the product directly in the United States. Our ability to generate any significant revenues in the near-term is dependent entirely on the successful commercialization and market acceptance of cethromycin. Factors that may inhibit our efforts to commercialize cethromycin or other product candidates without strategic partners or licensees include:
- •
- difficulty recruiting and retaining adequate numbers of effective sales and marketing personnel;
- •
- the inability of sales personnel to obtain access to, or persuade adequate numbers of, physicians to prescribe our products;
- •
- the lack of complementary products to be offered by sales personnel, which may put us at a competitive disadvantage against companies with broader product lines; and
14
- •
- unforeseen costs associated with creating an independent sales and marketing organization.
As an alternative to establishing our own sales and marketing organization, we may engage other pharmaceutical or health care companies with existing distribution systems and direct sales organizations to assist us in North America and abroad. We may not be able to negotiate favorable distribution partnering arrangements, if at all. To the extent we enter co-promotion or other licensing arrangements, any revenues we receive will depend on the efforts of third parties and will not be under our control. If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, our ability to generate product revenues, and become profitable, would be severely limited.
Off-label promotion of our products could result in substantial penalties.
If any of our product candidates receives marketing approval, we will only be permitted to promote the product for the uses indicated on the label cleared by the FDA. Our pivotal Phase III clinical trials of cethromycin are for the treatment of mild-to-moderate community acquired pneumonia, although we believe that cethromycin may have other applications in bronchitis, pharyngitis and sinusitis. If we request additional label indications for cethromycin or our other product candidates, the FDA may deny those requests outright, require extensive clinical data to support any additional indications or impose limitations on the intended use of any approved products as a condition of approval. U.S. Attorneys' offices and other regulators, in addition to the FDA, have recently focused substantial attention on off-label promotional activities and have initiated civil and criminal investigations related to such practices. If it is determined by these or other regulators that we have promoted our products for off-label use, we could be subject to fines, legal proceedings, injunctions or other penalties.
If our efforts to obtain rights to new products or product candidates from third parties are not successful, we may not generate product revenues or achieve profitability.
Our long-term ability to earn product revenues depends on our ability to identify, through internal research programs, potential product candidates that may be developed into new pharmaceutical products and/or obtain new products or product candidates through licenses from third parties. If our internal research programs do not generate sufficient product candidates, we will need to obtain rights to new products or product candidates from third parties. We may be unable to obtain suitable products or product candidates from third parties for a number of reasons, including:
- •
- we may be unable to purchase or license products or product candidates on terms that would allow us to make an appropriate return from resulting products;
- •
- competitors may be unwilling to assign or license product or product candidate rights to us;
- •
- we may not have access to the capital necessary to purchase or license products or product candidates; or
- •
- we may be unable to locate suitable products or product candidates within, or complementary to, our areas of interest.
If we are unable to obtain rights to new products or product candidates from third parties, our ability to generate product revenues and achieve profitability may suffer.
Because our product candidates and development and collaboration efforts depend on our intellectual property rights, adverse events affecting our intellectual property rights will harm our ability to commercialize products.
Our success will depend to a large degree on our own, our licensees' and our licensors' ability to obtain and defend patents for each party's respective technologies and the compounds and other
15
products, if any, resulting from the application of such technologies. The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions. No consistent policy regarding the breadth of claims allowed in biotechnology patents has emerged to date. Accordingly, we cannot predict the breadth of claims that will be allowed or maintained, after challenge, in our or other companies' patents.
The degree of future protection for our proprietary rights is uncertain, and we cannot ensure that:
- •
- we were the first to make the inventions covered by each of our pending patent applications;
- •
- we were the first to file patent applications for these inventions;
- •
- others will not independently develop similar or alternative technologies or duplicate any of our technologies;
- •
- any of our pending patent applications will result in issued patents;
- •
- any patents issued to us or our collaborators will provide a basis for commercially viable products, will provide us with any competitive advantages or will not be challenged by third parties;
- •
- we will develop additional proprietary technologies that are patentable; or
- •
- the patents of others will not have a negative effect on our ability to do business.
We are a party to certain in-license agreements that are important to our business, and we generally do not control the prosecution of in-licensed technology. Accordingly, we are unable to exercise the same degree of control over this intellectual property as we exercise over our internally developed technology. Moreover, some of our academic institution licensors, research collaborators and scientific advisors have rights to publish data and information in which we have rights. If we cannot maintain the confidentiality of our technology and other confidential information in connection with our collaborations, then our ability to receive patent protection or protect our proprietary information will be impaired. In addition, some of the technology we have licensed relies on patented inventions developed using U.S. government resources. Under applicable law, the U.S. government has the right to require us to grant a nonexclusive, partially exclusive, or exclusive license for such technology to a responsible applicant or applicants, upon terms that are reasonable under the circumstances, if the government determines that such action is necessary.
Confidentiality agreements with employees and others may not adequately prevent disclosure of trade secrets and other proprietary information and may not adequately protect our intellectual property.
We rely on trade secrets to protect our technology, particularly when we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. In order to protect our proprietary technology and processes, we rely in part on confidentiality and intellectual property assignment agreements with our corporate partners, employees, consultants, outside scientific collaborators and sponsored researchers and other advisors. These agreements may not effectively prevent disclosure of confidential information nor result in the effective assignment to us of intellectual property, and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information or other breaches of the agreements. In addition, others may independently discover our trade secrets and proprietary information, and in such case we could not assert any trade secret rights against such party. Enforcing a claim that a party illegally obtained and is using our trade secrets is difficult, expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the United States may be less willing to protect trade secrets. Costly and time-consuming litigation could be necessary to seek to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
16
Market acceptance and sales of our product candidates will be severely limited if we cannot arrange for favorable reimbursement policies.
Our ability to commercialize any product candidates successfully will depend in part on the extent to which governmental authorities, private health insurers and other organizations establish reimbursement levels for the cost of our products and related treatments. Third-party payors are increasingly challenging the prices charged for medical products and services. Also, the trend toward managed health care in the United States, as well as legislative proposals to reform health care, control pharmaceutical prices or reduce government insurance programs, may also result in exclusion of our product candidates from reimbursement programs. Because many generic antibiotics are available for the treatment of respiratory tract infections, our ability to list cethromycin on insurance company formularies will depend on its effectiveness compared to lower-cost products. The cost containment measures that health care payors and providers are instituting, and the effect of any health care reform, could materially and adversely affect our ability to earn revenues from the sales of cethromycin and our other product candidates.
The recent Medicare prescription drug coverage legislation and future legislative or regulatory reform of the healthcare system could limit future revenues from our product candidates.
In the United States, there have been a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to market and sell our product candidates profitably. In particular, in December 2003, President Bush signed into law new Medicare prescription drug coverage legislation. Under this legislation, the Centers for Medicare and Medicaid Services, or CMS, the agency within the Department of Health and Human Services that administers Medicare and will be responsible for reimbursement of the cost of drugs, has asserted the authority of Medicare to elect not to cover particular drugs if CMS determines that the drugs are not "reasonable and necessary" for Medicare beneficiaries or to elect to cover a drug at a lower rate similar to that of drugs that CMS considers to be "therapeutically comparable." Changes in reimbursement policies or health care cost containment initiatives that limit or restrict reimbursement for our products may cause our revenues to decline.
Another development that may affect the pricing of drugs is regulatory action regarding drug reimportation into the United States. The Medicare Prescription Drug, Improvement and Modernization Act of 2003, which became law in December 2003, requires the Secretary of the U.S. Department of Health and Human Services to promulgate regulations allowing drug reimportation from Canada into the United States under certain circumstances. These provisions will become effective only if the Secretary certifies that such imports will pose no additional risk to the public's health and safety and result in significant cost savings to consumers. To date, the Secretary has made no such finding, but he could do so in the future. Proponents of drug reimportation may also attempt to pass legislation that would remove the requirement for the Secretary's certification or allow reimportation under circumstances beyond those anticipated under current law. If legislation is enacted, or regulations issued, allowing the reimportation of drugs, it could decrease the reimbursement we receive for any products that we may commercialize, negatively affecting our anticipated revenues and prospects for profitability.
We will need to increase the size of our organization, and we may encounter difficulties managing our growth, which could adversely affect our results of operations.
We are currently a development stage company with 20 employees. We will need to expand and effectively manage our managerial, operational, financial and other resources in order to successfully pursue our research, development and commercialization effort. To manage any growth, we will be required to continue to improve our operational, financial and management controls, reporting systems and procedures and to attract and retain sufficient numbers of talented employees. We may be unable
17
to successfully manage the expansion of our operations or operate on a larger scale and, accordingly, may not achieve our research, development and commercialization goals.
If we are unable to attract and retain qualified scientific, technical and key management personnel, or if any of our key executives, Michael T. Flavin, Ph.D., Ze-Qi Xu, Ph.D., or John L. Flavin, discontinues his employment with us, it may delay our research and development efforts.
We are highly dependent upon the efforts of our senior management team and scientific staff. The loss of the services of one or more members of the senior management team might impede the achievement of our development objectives. In particular, we are highly dependent upon and our business would be significantly harmed if we lost the services of Michael T. Flavin, Ph.D., our founder and Chairman and Chief Executive Officer, Ze-Qi Xu, Ph.D., our Executive Vice President and Chief Scientific Officer, or John L. Flavin, our President. We do not currently have any key man life insurance policies. We have entered into employment agreements with members of our senior management team, but this does not ensure that we will retain their services for any period of time in the future. Our research and drug discovery programs also depend on our ability to attract and retain highly skilled chemists, biologists and preclinical and clinical personnel. We may not be able to attract or retain qualified scientific personnel in the future due to intense competition among biotechnology and pharmaceutical businesses, particularly in the Chicago area. If we are not able to attract and retain the necessary personnel to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our research and development objectives and our ability to meet the demands of our collaborators in a timely fashion.
Our business will expose us to potential product liability risks and there can be no assurance that we will be able to acquire and maintain sufficient insurance to provide adequate coverage against potential liabilities.
Our business will expose us to potential product liability risks that are inherent in the testing, manufacturing and marketing of pharmaceutical products. The use of our product candidates in clinical trials also exposes us to the possibility of product liability claims and possible adverse publicity. These risks will increase to the extent our product candidates receive regulatory approval and are commercialized. We do not currently have any product liability insurance, although we plan to obtain product liability insurance in connection with future clinical trials of our product candidates. There can be no assurance that we will be able to obtain or maintain any such insurance on acceptable terms. Moreover, our product liability insurance may not provide adequate coverage against potential liabilities. On occasion, juries have awarded large judgments in class action lawsuits based on drugs that had unanticipated side effects. A successful product liability claim or series of claims brought against us would decrease our cash reserves and could cause our stock price to fall significantly.
We face regulation and risks related to hazardous materials and environmental laws, violations of which may subject us to claims for damages or fines that could materially affect our business, financial condition and results of operations.
Our research and development activities involve the controlled use of hazardous materials and chemicals. The risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of an accident, we could be held liable for any damages or fines that result, and the liability could have a material adverse effect on our business, financial condition and results of operations. We are also subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous materials and waste products. If we fail to comply with these laws and regulations or with the conditions attached to our operating licenses, the licenses could be revoked, and we could be subjected to criminal sanctions and substantial liability or be required to suspend or modify our operations. In addition, we may have to incur significant costs to
18
comply with future environmental laws and regulations. We do not currently have a pollution and remediation insurance policy.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Our drug discovery and preclinical testing systems are highly technical and proprietary. Any system failure, accident or security breach that causes interruptions in our operations could result in a material disruption of our drug discovery programs. To the extent that any disruption or security breach results in a loss or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we may incur liability as a result, our drug discovery programs may be adversely affected and the further development of our product candidates may be delayed. In addition, we may incur additional costs to remedy the damages caused by these disruptions or security breaches.
Risks Related to the Offerings
An active trading market for our common stock may not develop, and we expect that our stock price will fluctuate significantly due to events and developments unique to our business or the life sciences industry generally.
Prior to the offerings, you could not buy or sell our common stock publicly. Our common stock has been approved for listing on the Nasdaq National Market, but an active trading market for our shares may never develop or be sustained following the offerings. The initial public offering price for our common stock will be determined through negotiations with the underwriters. This initial public offering price may vary from the market price of the common stock after the offerings and you may not be able to sell your common stock at or above the initial public offering price. The stock market has recently experienced significant volatility, particularly for pharmaceutical and biotechnology stocks. Factors that could cause this volatility in the market price of our common stock include:
- •
- the timing of and the results from our clinical trial programs, including the initiation and completion of Phase I clinical trials for ALS-886 and ALS-357, Phase IIa clinical trials for Calanolide A and pivotal Phase III clinical trials for cethromycin;
- •
- the in-licensing or acquisition of additional product candidates;
- •
- the loss of licenses or proprietary rights to technologies and products;
- •
- FDA or international regulatory actions;
- •
- failure of any of our product candidates, if approved, to achieve commercial success;
- •
- announcements of new products by our competitors;
- •
- market conditions in the pharmaceutical and biotechnology sectors;
- •
- litigation or public concern about the safety of our potential products;
- •
- comments by securities analysts;
- •
- actual and anticipated fluctuations in our quarterly operating results;
- •
- deviations in our operating results from the estimates of securities analysts;
- •
- rumors relating to us or our competitors;
- •
- public concern as to the efficacy or safety of new technologies;
19
- •
- third party reimbursement policies;
- •
- developments concerning current or future collaborations, including disputes or termination events and the achievement, timing and accounting treatment of milestone payments; and
- •
- the addition or termination of research programs or funding support.
These and other factors may cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent investors from readily selling their shares of common stock and may otherwise negatively affect the liquidity of our common stock. In addition, in the past, when the market price of a stock has been volatile, holders of that stock have instituted securities class action litigation against the company that issued the stock. If any of our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit and the time and attention of our management may be diverted.
Future sales of common stock by our existing stockholders may cause our stock price to fall.
The market price of our common stock could decline as a result of sales by our existing stockholders of shares of common stock in the market after the offerings, or the perception that these sales could occur. These sales might also make it more difficult for us to sell equity securities at a time and price that we deem appropriate. The lock-up agreements delivered by our executive officers, directors and primary stockholders provide that C.E. Unterberg, Towbin, LLC may release those parties, at any time or from time to time and without notice, from their obligation not to dispose of shares of common stock for a period of 180 days (which period could be extended by C.E. Unterberg, Towbin, LLC for up to an additional 34 days under certain circumstances) after the date of this prospectus. C.E. Unterberg, Towbin, LLC has no pre-established conditions to waiving the terms of the lock-up agreements, and any decision by it to waive those conditions would depend on a number of factors, which may include market conditions, the performance of the common stock in the market and our financial condition at that time. After the offerings, 1,192,853 shares of our common stock will be subject to registration rights. Please see "Description of Capital Stock."
Investors in the offerings will pay a much higher price than the book value of our common stock and will incur immediate and substantial dilution and may incur dilution in the future.
If you purchase common stock in the initial public offering, you will pay more for your shares than the amounts paid by our existing stockholders for their shares. You will incur immediate and substantial dilution of $5.89 per share, representing the difference between our pro forma net tangible book value per share after giving effect to the offerings and an assumed initial offering price of $7.00 per share. In the past, we have issued options and warrants to acquire common stock at prices significantly below the assumed initial offering price. To the extent these options or warrants are ultimately exercised, you will sustain further dilution. Moreover, we may require additional funds to support our working capital requirements or for other purposes, and may seek to raise additional funds through public or private equity financing. We also may acquire other companies or technologies or finance strategic alliances by issuing equity. Any of these or other capital raising transactions may result in additional dilution to our stockholders.
We will have broad discretion in how we use the proceeds of the initial public offering, and we may not use these proceeds effectively, which could negatively impact our results of operations and cause our stock price to decline.
Our management will have considerable discretion in the application of the proceeds of the initial public offering, and you will not have the opportunity, as part of your investment decision, to assess whether we are using the proceeds appropriately. We currently intend to use these proceeds for payments to Abbott Laboratories under our license agreement, pivotal Phase III clinical trials of
20
cethromycin, the repayment of indebtedness and general corporate purposes, including, potentially, clinical trials of our other product candidates, research and development expenses, general and administrative expenses, and potential acquisitions of products and technologies that complement our business. We may use the proceeds for corporate purposes that do not yield a significant return, or any return at all, for our stockholders.
Our Chairman and Chief Executive Officer will have significant voting control over our company which may delay, prevent or deter corporate actions that may be in the best interest of our stockholders.
After the offerings, Flavin Ventures, LLC will control approximately 57.0% of our outstanding common stock. Dr. Michael Flavin, our founder and Chairman and Chief Executive Officer, has effective voting control for all shares of our common stock held by Flavin Ventures. As a result, Dr. Flavin will be able to exert significant influence for all matters requiring approval of our stockholders, including the election of directors and approval of significant corporate transactions. This concentration of ownership may delay, prevent or deter a change in control of our company even when such a change may be in the best interest of all the stockholders, could deprive stockholders of an opportunity to receive a premium for their common stock as part of a sale of our company or assets and might affect the prevailing market price of our common stock.
Provisions of Delaware law or our charter documents could delay or prevent an acquisition of our company, even if the acquisition would be beneficial to our stockholders, and could make it more difficult for you to change management.
Provisions of our amended and restated certificate of incorporation and bylaws may discourage, delay or prevent a merger, acquisition or other change in control that stockholders may consider favorable, including transactions in which stockholders might otherwise receive a premium for their shares. This is because these provisions may prevent or frustrate attempts by stockholders to replace or remove our current management or members of our board of directors.
These provisions include:
- •
- a classified board of directors under which approximately one third of the directors will be elected each year;
- •
- a requirement that the authorized number of directors be changed only by a resolution of the board of directors;
- •
- authorized and unissued additional shares of our common stock and preferred stock;
- •
- advance notice requirements for proposals that can be acted upon at stockholder meetings; and
- •
- a requirement that only our Chairman or our board of directors, acting by resolution, may call stockholder meetings.
As a result, these provisions and others available under Delaware law could limit the price that investors are willing to pay in the future for shares of our common stock.
21
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements that are based on our management's beliefs and assumptions and on information currently available to our management. The forward-looking statements are contained principally in the sections entitled "Prospectus Summary," "Risk Factors," "Management's Discussion and Analysis of Financial Condition and Results of Operations" and "Business." In some cases, you can identify forward-looking statements by terms such as "anticipates," "believes," "could," "estimates," "expects," "intends," "may," "plans," "potential," "predicts," "projects," "should," "will," "would," and similar expressions intended to identify forward-looking statements. Forward-looking statements include, but are not limited to, statements about:
- •
- the timing and progress of our clinical trials, research and development and drug discovery programs;
- •
- our ability to in-license or acquire new technology or product candidates;
- •
- our ability to receive regulatory approvals to develop and commercialize our product candidates;
- •
- our collaboration and strategic alliance efforts;
- •
- our ability to contract for and maintain third party manufacturing services;
- •
- our ability to protect our intellectual property and avoid infringing upon others' intellectual property;
- •
- our ability to achieve market acceptance for our product candidates and successfully compete in the market;
- •
- our estimates for future performance; and
- •
- our estimates regarding anticipated operating losses, use of proceeds from the initial public offering, future revenues, capital requirements and our needs for additional financing.
Forward-looking statements involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements. We discuss many of these risks in greater detail under the heading "Risk Factors." Given these uncertainties, you should not place undue reliance on these forward-looking statements. Also, forward-looking statements represent our management's beliefs and assumptions only as of the date of this prospectus. You should read this prospectus and the documents that we reference in this prospectus and have filed as exhibits to the registration statement, of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from what we expect. The forward-looking statements contained in this prospectus are excluded from the safe harbor protection provided by the Private Securities Litigation Reform Act of 1995 and Section 27A of the Securities Act of 1933, as amended.
Except as required by law, we assume no obligation to update these forward-looking statements publicly, or to explain the reasons actual results could differ materially from those anticipated in these forward-looking statements, even if new information becomes available in the future.
22
Concurrent with the shares we are offering by this prospectus in the initial public offering, we are also offering 428,571 shares of our common stock to Abbott Laboratories, the beneficial owner of approximately 10.4% of our common stock prior to the offerings. The initial public offering and the concurrent offering are conditioned upon each other. The number of shares that we are offering to Abbott in the concurrent offering is determined by dividing $3 million by the assumed initial public offering price of $7.00 per share. In exchange for receiving shares of our common stock in the concurrent offering, a $5 million milestone obligation under our license agreement with Abbott that is payable on October 31, 2005, or, if earlier, upon our initiation of pivotal Phase III clinical trials for cethromycin, will be reduced by $3 million. The underwriters will not receive a fee in connection with the concurrent offering.
23
We estimate that the net proceeds from the sale of our common stock in the initial public offering will be approximately $35.0 million, based on an assumed initial public offering price of $7.00 per share and after deducting estimated underwriting discounts, commissions and offering expenses. If the underwriters exercise their over-allotment option in full we estimate that our net proceeds will be approximately $40.5 million.
We currently intend to use the net proceeds from the initial public offering as follows:
- •
- $8.0 million for a payment to Abbott Laboratories under our license agreement, which is due and payable on the earlier of September 1, 2005 or five days following the closing of the offerings;
- •
- $2.0 million for a payment to Abbott Laboratories on October 31, 2005 or, if earlier, upon our initiation of pivotal Phase III clinical trials for cethromycin; and
- •
- $18.5 million over approximately the next 18 months to conduct pivotal Phase III clinical trials for cethromycin in the treatment of mild-to-moderate community acquired pneumonia.
We intend to use any remaining net proceeds from the initial public offering for working capital, capital expenditures and general corporate purposes, including, potentially, depending on the amount of proceeds, clinical trials of ALS-886, ALS-357 and, subject to reaching a satisfactory agreement with our joint venture partner, Calanolide A, research and development in the discovery of new compounds, potential acquisitions of product candidates through in-licensing activities and continued laboratory studies of preclinical product candidates.
We believe that the net proceeds from the initial public offering will be sufficient to meet our anticipated operating requirements through 2006. We expect to satisfy our future cash needs through the sale of equity securities, debt financings, corporate collaborations and licensing arrangements and grant funding, as well as through interest income earned on cash balances.
As of the date of this prospectus, we cannot specify with certainty all of the particular uses for the net proceeds to be received upon the completion of the offerings. The amount and timing of our expenditures will depend on several factors, including the progress of our clinical trials and research and development efforts as well as the amount of cash used in our operations. Accordingly, our management will have broad discretion in the application of the net proceeds and investors will be relying on the judgment of our management regarding the application of the proceeds of the initial public offering. We reserve the right to change the use of these proceeds as a result of certain contingencies such as the results of our drug discovery and development activities, competitive developments, opportunities to acquire products, technologies or businesses and other factors.
We have no current agreements or commitments for acquisitions of complementary businesses or technologies. Pending these uses, we will invest the net proceeds of the initial public offering in short-term, investment grade and interest-bearing securities.
Concurrent with the shares we are offering by this prospectus in the initial public offering, we are also offering 428,571 shares of our common stock to Abbott Laboratories in exchange for a $3 million reduction of a $5 million milestone obligation under our license agreement with Abbott that is payable on October 31, 2005, or, if earlier, upon our initiation of pivotal Phase III clinical trials for cethromycin.
24
We have never declared or paid any cash dividends on our capital stock. We presently intend to retain future earnings, if any, to finance the expansion of our business, and we do not expect to pay any cash dividends. Any payment of dividends will be at the discretion of our board of directors and will be dependent upon our financial condition, results of operations, capital requirements, general business conditions and other factors that the board of directors may deem relevant.
25
The following sets forth our capitalization as of June 30, 2005:
- •
- on an actual basis; and
- •
- on an as adjusted basis to give effect to our receipt of net proceeds from the sale of shares of common stock in the initial public offering at an assumed initial public offering price of $7.00 per share, after deducting the underwriting discounts and commissions and estimated offering expenses.
You should read the information in the following table together with "Selected Financial Data," "Management's Discussion and Analysis of Financial Condition and Results of Operations," and the consolidated financial statements and the accompanying notes, included elsewhere in this prospectus.
| | As of June 30, 2005 | ||||||||
|---|---|---|---|---|---|---|---|---|---|
| | Actual | As Adjusted | |||||||
| Long-term debt and lease payable, less current portion | $ | 2,020,498 | $ | 2,020,498 | |||||
Series A preferred stock of Advanced Life Sciences, Inc.: | |||||||||
| 250,000 shares authorized and 250,000 shares issued and outstanding at June 30, 2005 and on an as adjusted basis | — | — | |||||||
Stockholders' equity (deficit): | |||||||||
| Common stock, $0.01 par value, 60,000,000 shares authorized, 10,826,801 shares issued and outstanding at June 30, 2005 and 16,826,801 shares issued and outstanding on an as adjusted basis | 108,268 | 168,268 | |||||||
| Additional paid-in capital | 22,779,215 | 60,739,218 | |||||||
| Deferred stock-based compensation | (280,958 | ) | (280,958 | ) | |||||
| Deficit accumulated during the development stage | (41,938,671 | ) | (41,938,671 | ) | |||||
| Total stockholders' equity (deficit) | (19,332,146 | ) | 18,687,857 | ||||||
| Total capitalization | $ | (17,311,648 | ) | $ | 20,708,355 | ||||
On July 28, 2005, we entered into a commitment letter with our bank to extend the maturity of our line of credit until December 2007 and eliminate the bank's demand payment rights. Consequently, following the offering amounts outstanding under the line of credit will be reclassified as long-term debt and included in our capitalization.
In June 2005, our board of directors adopted and our shareholders approved an amendment to our certificate of incorporation that authorizes us to issue 60,000,000 shares of our common stock and 5,000,000 shares of undesignated preferred stock.
The table above does not include, as of June 30, 2005:
- •
- 686,837 shares of common stock issuable upon the exercise of options outstanding under our 1999 Stock Incentive Plan at a weighted average exercise price of $0.93 per share;
- •
- 2,072,455 additional shares of common stock reserved for future issuances under our 2005 Stock Incentive Plan; and
- •
- 14,887 shares of common stock issuable upon the exercise of warrants outstanding at an exercise price of $8.02 per share.
26
If you invest in our common stock, your interest will be diluted to the extent of the difference between the public offering price per share of our common stock and the as adjusted net tangible book value per share of our common stock after the offerings. As of June 30, 2005, we had a historical net tangible book value of $(19.3) million, or $(1.79) per share of common stock. Net tangible book value per share is equal to our total tangible assets (total assets less intangible assets) less total liabilities, divided by the number of shares of our outstanding common stock. After giving effect to the sale of 5,571,429 shares of common stock in the initial public offering at an assumed initial public offering price of $7.00 per share and 428,571 shares of common stock in the concurrent offering in exchange for a $3 million reduction of our milestone payment obligation, and after deducting the estimated underwriting discounts, commissions and offering expenses, our as adjusted net tangible book value as of June 30, 2005 would have been approximately $18.7 million, or approximately $1.11 per pro forma share of common stock. This represents an immediate increase in pro forma net tangible book value of $2.90 per share to our existing stockholders and an immediate dilution of $5.89 per share to new public investors and the concurrent offering investor in the offerings. The following table illustrates this per share dilution:
| Assumed initial public offering price per share | $ | 7.00 | ||||
| Historical net tangible book value per share as of June 30, 2005 | $ | (1.79 | ) | |||
| Increase per share attributable to the offerings | 2.90 | |||||
| As adjusted net tangible book value per share after the offerings | 1.11 | |||||
| Dilution per share to new public investors and concurrent offering investor | $ | 5.89 | ||||
The following table summarizes, on a pro forma basis as of June 30, 2005, the differences between existing stockholders and the new investors with respect to the number of shares of common stock purchased from us, the total consideration paid and the average price per share paid before deducting the underwriting discounts and commissions and our estimated offering expenses.
| | Shares purchased | Total consideration | | |||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Average price per share | |||||||||||
| | Number | Percent | Amount | Percent | ||||||||
| Existing stockholders | 10,826,801 | 64.34 | % | $ | 22,546,718 | 34.93 | % | $ | 2.08 | |||
| New public investors | 5,571,429 | 33.11 | % | 39,000,003 | 60.42 | % | $ | 7.00 | ||||
| Concurrent offering investor | 428,571 | 2.55 | % | 3,000,000 | 4.65 | % | $ | 7.00 | ||||
| Total | 16,826,801 | 100.00 | % | $ | 64,546,721 | 100.00 | % | |||||
The discussion and tables above assume no exercise of the underwriters' over-allotment option or any outstanding stock options or warrants. If the underwriters' over-allotment option is exercised in full, the number of shares of common stock held by existing stockholders will be reduced to 61.30% of the total number of shares of common stock to be outstanding after the offerings, and the number of shares of common stock held by the new public investors will be increased to 6,407,143 shares or 36.28% of the total number of shares of common stock outstanding after this offering. See "Principal Stockholders" for a description of beneficial ownership after the offerings.
The information in the two tables above excludes, as of June 30, 2005:
- •
- 686,837 shares of common stock issuable upon the exercise of options outstanding under our 1999 Stock Incentive Plan at a weighted average exercise price of $0.93 per share;
27
- •
- 2,072,455 additional shares of common stock reserved for future issuances under our 2005 Stock Incentive Plan; and
- •
- 14,887 shares of common stock issuable upon the exercise of warrants outstanding at an exercise price of $8.02 per share.
To the extent that these options and warrants are exercised, there will be further dilution to new investors.
28
You should read the following selected financial data in conjunction with the consolidated financial statements and the notes to such statements and "Management's Discussion and Analysis of Financial Condition and Results of Operations" included elsewhere in this prospectus. The financial data as of December 31, 2003 and 2004 and for the years ended December 31, 2002, 2003 and 2004 are derived from our audited financial statements included elsewhere in this prospectus. The balance sheet data as of December 31, 2000, 2001 and 2002, and the statements of operations data for the years ended December 31, 2000 and 2001, have been derived from our audited financial statements not included in this prospectus. The balance sheet data as of June 30, 2005 and the statement of operations data for each of the six months ended June 30, 2004 and 2005 have been derived from our unaudited financial statements, which include, in the opinion of management, all adjustments necessary to present fairly the data for such periods. Historical results are not necessarily indicative of the results to be expected in the future.
| | Years ended December 31, | Six months ended June 30, | Period from Inception (January 1, 1999) to June 30, 2005 | |||||||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2000 | 2001 | 2002 | 2003 | 2004 | 2004 | 2005 | |||||||||||||||||||
| Statement of Operations Data: | ||||||||||||||||||||||||||
Revenue | $ | 447,525 | $ | 225,218 | $ | 496,207 | $ | 180,427 | $ | 319,780 | $ | 182,973 | $ | 44,379 | $ | 2,125,129 | ||||||||||
| Operating expenses: | ||||||||||||||||||||||||||
| Research and development(1) | 3,159,383 | 2,771,517 | 924,799 | 1,362,255 | 25,661,868 | 570,986 | 724,347 | 37,516,266 | ||||||||||||||||||
| Selling, general and administrative | 344,607 | 461,001 | 668,622 | 1,198,722 | 1,649,953 | 639,085 | 780,161 | 5,177,282 | ||||||||||||||||||
| Loss from operations | (3,056,465 | ) | (3,007,300 | ) | (1,097,214 | ) | (2,380,550 | ) | (26,992,041 | ) | (1,027,098 | ) | (1,460,129 | ) | (40,568,419 | ) | ||||||||||
Interest expense, net | 231,930 | 377,029 | 164,337 | 170,938 | 194,877 | 96,389 | 204,249 | 1,370,252 | ||||||||||||||||||
| Net loss | (3,288,395 | ) | (3,384,329 | ) | (1,261,551 | ) | (2,551,488 | ) | (27,186,918 | ) | (1,123,487 | ) | (1,664,378 | ) | (41,938,671 | ) | ||||||||||
| Less accumulating preferred dividends for the period | 175,000 | 175,000 | 175,000 | 175,000 | 175,000 | 87,500 | 87,500 | 1,057,292 | ||||||||||||||||||
| Net loss available to common shareholders | $ | (3,463,395 | ) | $ | (3,559,329 | ) | $ | (1,436,551 | ) | $ | (2,726,488 | ) | $ | (27,361,918 | ) | $ | (1,210,987 | ) | $ | (1,751,878 | ) | $ | (42,995,963 | ) | ||
Basic and diluted loss per common share | $(2.18 | ) | $(2.24 | ) | $(0.90 | ) | $(1.72 | ) | $(13.27 | ) | $(0.76 | ) | $(0.16 | ) | ||||||||||||
Weighted average shares outstanding-basic | 1,588,000 | 1,588,000 | 1,588,000 | 1,588,000 | 2,062,351 | 1,588,134 | 10,780,148 | |||||||||||||||||||
| | As of December 31, | | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
As of June 30, 2005 | |||||||||||||||||||
| | 2000 | 2001 | 2002 | 2003 | 2004 | ||||||||||||||
| Balance Sheet Data: | |||||||||||||||||||
| Cash | $ | 94,757 | $ | 7,846 | $ | 18,739 | $ | 61,203 | $ | 194,555 | $ | 39,951 | |||||||
| Total assets | 251,687 | 160,467 | 146,917 | 255,127 | 1,015,932 | 1,769,858 | |||||||||||||
| Long-term debt and lease payable, less current portion | 5,002,992 | 2,000,000 | 2,000,000 | 2,000,000 | 2,027,279 | 2,020,498 | |||||||||||||
| Total liabilities(2) | 5,876,597 | 2,252,835 | 2,339,836 | 2,661,806 | 19,289,733 | 21,102,004 | |||||||||||||
| Deficit accumulated during the development stage | (5,874,910 | ) | (9,274,336 | ) | (10,535,887 | ) | (13,087,375 | ) | (40,274,293 | ) | (41,938,671 | ) | |||||||
| Total stockholders' deficit | (5,624,910 | ) | (2,092,368 | ) | (2,192,919 | ) | (2,406,679 | ) | (18,273,801 | ) | (19,332,146 | ) | |||||||
- (1)
- Amount for 2004 includes approximately $24.5 million of in-process research and development expense related to compounds licensed from Abbott Laboratories in December 2004.
- (2)
- Amount for 2004 includes $14.0 million in payments due in 2005 under our license agreement with Abbott Laboratories executed in December 2004. See Note 11 to our consolidated financial statements. As a result of the concurrent offering, the amount of license payments due to Abbott will be reduced by $3.0 million. See "Concurrent Offering" and "Capitalization."
29
MANAGEMENT'S DISCUSSION AND ANALYSIS
OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion of our financial condition and results of operations in conjunction with our consolidated financial statements and the related notes included elsewhere in this prospectus. This discussion contains forward-looking statements that involve risks and uncertainties. As a result of many factors, including those set forth under the section entitled "Risk Factors" and elsewhere in this prospectus, our actual results may differ materially from those anticipated in these forward-looking statements.
Overview
We are a biopharmaceutical company focused on the discovery, development and commercialization of novel drugs in the areas of infectious disease, inflammation and oncology. Using our internal discovery capabilities and our network of pharmaceutical and academic partners, we have assembled a promising pipeline of clinical and preclinical product candidates. Our most advanced product candidate, cethromycin, is a novel ketolide antibiotic in Phase III clinical development for the treatment of respiratory tract infections. We also have product candidates in earlier stages of development for the treatment of indications including HIV, inflammation-related tissue damage and malignant melanoma.
None of our product candidates has been approved by the FDA or any comparable foreign agencies, and we have not generated any significant revenues to date. Our ability to generate revenues in the future will depend on our ability to meet development or regulatory milestones under any license agreements that trigger payments to us, to enter into new license agreements for other products or territories and to receive regulatory approvals for, and successfully commercialize, our product candidates either directly or through commercial partners.
Since our inception we have incurred net losses each year. Our net losses for the years ended December 31, 2004 and 2003 were $27.2 million and $2.6 million, respectively. Our net loss for the year ended December 31, 2004 resulted principally from $24.5 million of in-process research and development expenses relating to our collaboration with Abbott Laboratories for the development of cethromycin. As of June 30, 2005, we had an accumulated deficit of $41.9 million. Our operations to date have been funded principally through debt and capital contributions made by our founder and controlling stockholder and borrowings under our bank line of credit.
Even if we succeed in developing and commercializing one or more of our product candidates, we may never become profitable. We expect to continue to incur increasing expenses over the next several years as we plan to:
- •
- expand the clinical trial program for cethromycin;
- •
- continue the preclinical development and commence the clinical development of our other product candidates, such as ALS-886 and ALS-357;
- •
- expand our research and development activities;
- •
- acquire or in-license new technologies and product candidates; and
- •
- increase our corporate infrastructure and overhead.
Upon receiving the net proceeds from the initial public offering, we expect to significantly increase the rate of our research and development expenditures as we accelerate our development of cethromycin. Accordingly, our total expenses and net losses will similarly increase. If the clinical testing for our product candidates is delayed, we could incur increased development costs and a delay in our ability to generate revenues.
30
If we receive regulatory approval for cethromycin, we expect to incur significant sales and marketing costs in order to establish a sales and marketing organization with the appropriate technical expertise and distribution capabilities, or make arrangements with third parties to perform these services. We also anticipate that our general and administrative costs will increase as we expand our administrative staff, systems infrastructure and facilities and begin to operate as a public company.
Financial Operations Review
Revenue
Revenue received to date is limited to amounts earned through management fees, grant programs and royalties. Revenue was derived primarily from fees received from Flavin Ventures, LLC, and related companies that are also controlled by our controlling stockholder, in exchange for administrative management services that we provided under a management services agreement. These services included general management, administrative support and financial services. In 2003, revenue was also derived from a similar management agreement with Sarawak MediChem Pharmaceuticals, our 50/50 joint venture with the State Government of Sarawak, Malaysia. We have also received grants through federal government programs and royalty payments from our transfer of certain patented technologies to a related party.
Royalty expense
Upon receipt of marketing approval and commencement of commercial sales, which may not occur for several years, if ever, we will owe royalties to licensors of certain patents. Under a royalty agreement with Abbott Laboratories we are obligated to pay a royalty based on net sales of cethromycin. Royalty payments will also be due to other parties if we receive FDA approval to commercialize ALS-886 and ALS-357.
Research and development expense
Research and development expense consists of development work associated with product candidates, including employee compensation, costs of preclinical studies, clinical trials, clinical supplies and consultant fees. We are responsible for all of the research and development costs related to cethromycin, ALS-886 and ALS-357.
The large majority of our research and development expenses to date relate to our cethromycin program. From our inception in 1999 through June 30, 2005, we incurred $37.5 million in research and development expenses. Of this amount, $24.5 million related to our purchase of in-process research and development for cethromycin and ABT-210 from Abbott Laboratories in 2004. These research and development expenses under the Abbott license are allocated between cethromycin and ABT-210 as $23.1 million and $1.4 million, respectively. In the fourth quarter of 2005, we intend to conduct pivotal Phase III clinical trials of cethromycin for the treatment of mild-to-moderate community acquired pneumonia. ABT-210 is a preclinical product candidate. In July 2005, we transferred back to Abbott all of our license rights to ABT-210 in exchange for a reduction in our future royalty obligations on net sales of cethromycin if it is approved for commercialization.
Even if our pivotal Phase III clinical trials of cethromycin are successful, we do not expect to receive FDA approval for commercialization until 2008 at the earliest. Our cethromycin program will require an additional $40 million in research and development expenses after completion of our pivotal Phase III clinical trials. These expenses represent additional milestone payments to Abbott triggered upon submitting an NDA and receiving FDA approval. Development timelines and related costs are difficult to estimate and may vary significantly from our current estimates. If our cethromycin research and development program does not lead to commercialization, then our business would be materially harmed. You should read the risk factors beginning on page 6 of this prospectus that summarize the
31
risks inherent in our research and development programs for cethromycin and our other product candidates.
We have not begun clinical development of ALS-357 or ALS-886. On a direct cost basis, preclinical development costs for ALS-357 and ALS-886 have amounted to approximately $2.0 million and $1.4 million, respectively, from our inception in 1999 through June 30, 2005. The majority of our other research and development spending to date has been directed to discovery programs in the inflammation and oncology areas. As we commence more extensive development activities for our early stage product candidates, including clinical trials and commercial scale-up, we expect research and development expense to increase substantially. Development timelines and costs are difficult to estimate and may vary significantly for each product candidate and from quarter-to-quarter. The process of seeking regulatory approvals, and the subsequent compliance with applicable regulations, require the expenditure of substantial resources. We anticipate that we will make determinations as to which of our early stage product candidates is best-suited for further development, as well as how much funding to direct to each project, on an on-going basis in response to the scientific and clinical success and commercial potential of each product candidate.
Selling, general and administrative expense
General and administrative expense consists primarily of compensation for employees in executive and operational functions, including finance and accounting, business development and corporate development. Other significant costs include facilities costs and professional fees for accounting and legal services. After completion of the offerings, we anticipate our general and administrative expenses to increase due to increased costs for insurance, professional fees, external reporting requirements, Sarbanes-Oxley compliance and investor relations associated with operating as a publicly-traded company. These increases will also likely include the hiring of additional personnel.
Stock-based compensation
In connection with the grant of stock options to employees, we recorded deferred stock-based compensation as a component of stockholders' deficit. Deferred stock compensation for options granted to employees is equal to the fair value of our common stock on the date such options were granted. We amortize this stock-based compensation as charges to operations over the vesting periods of the options, generally 36 months.
We recorded $6,000 in amortization of deferred stock-based compensation related to options granted to employees during the year ended December 31, 2004. There was a balance of $23,000 of deferred stock-based compensation at December 31, 2004 which we expect to be amortized by 2007.
Interest expense—net
Interest expense—net consists of non-operating items, including interest income and interest expense. Interest income is earned from funds on deposit. Interest expense is incurred from our various financing arrangements.
Results of Operations
Six months ended June 30, 2005 compared to six months ended June 30, 2004
Revenue. Total revenue decreased 76% to $44,000 for the six months ended June 30, 2005 from $183,000 for the six months ended June 30, 2004. Management fee revenue, the largest component of revenue, declined 69% to $44,000 for the six months ended June 30, 2005 from $145,000 for the six months ended June 30, 2004. The decline is attributable to a reduction in revenue derived from our management agreement with Flavin Ventures wherein we provide administrative and management
32
services to a series of companies jointly owned by Flavin Ventures. We believe this reduction in the service level required by Flavin Ventures and its related companies will be permanent and that revenue derived from this agreement will continue at the level recognized in the second quarter of 2005 throughout the remainder of 2005. Grant revenue decreased by 100% to $0 for the six months ended June 30, 2005 from $38,000 for the six months ended June 30, 2004, resulting from the completed funding of a Small Business Innovation Research (SBIR) grant. We regularly submit grant applications to the National Institutes of Health and anticipate being awarded an additional Phase I SBIR grant in the third quarter of 2005. Phase I SBIR grants have a budget of $100,000 and last between six months and one year. The ownership structure of our company after the offerings could make us ineligible to receive SBIR grants.
Research and Development Expense. Research and development expenses increased 27% to $724,000 for the six months ended June 30, 2005 from $571,000 for the six months ended June 30, 2004. Increased research expenditures primarily resulted from additional scientific headcount added subsequent to the second quarter of 2004.
Selling, general and administrative expense. General and administrative expense increased 22% to $780,000 for the six months ended June 30, 2005 from $639,000 for the six months ended June 30, 2004. The increase in general and administrative expenses was attributable to additional headcount in the finance, information technology and human resources functions to enhance our reporting and internal control structures.
Interest expense—net. Interest expense—net increased 113% to $204,000 for the six months ended June 30, 2005 from $96,000 for the six months ended June 30, 2004 as a result of the $3.0 million line of credit secured on December 13, 2004 and the subsequent increase of $1.0 million in May of 2005. Total capacity for borrowing under this line stands at $4.0 million. Total borrowings under the loan were $3.5 million, net of a $9,000 debt discount at June 30, 2005. Interest on the loan is paid monthly and is based upon the prime rate (currently 6.25%) plus 0.75% per annum.
Year ended December 31, 2004 compared to year ended December 31, 2003
Revenue. Total revenue increased 77% to $320,000 in 2004 from $180,000 in 2003. Management fees, the largest component of revenue, increased 129% in 2004 to $272,000 from $119,000 in 2003. During 2004 we entered into a management agreement with Flavin Ventures, a company controlled by our founder and controlling stockholder, to provide administrative and management services to a series of companies jointly owned by Flavin Ventures. Management revenue from our 50/50 joint venture, Sarawak MediChem Pharmaceuticals, is recorded on a cash basis and totaled $0 and $119,000 in 2004 and 2003, respectively. Billed and unrecognized management fee revenue totaled $160,000 and $185,000 for the years ended December 31, 2004 and 2003, respectively. Grant revenue decreased by 38% in 2004 to $38,000 from $62,000 in 2003, resulting from the completed funding of a SBIR grant. Royalties from a related party were $10,000 in 2004 compared to $0 in 2003. Royalty revenue is derived through a license with deCODE genetics and is based upon deCODE's sales of licensed technology.
Research and development expense. Research and development expenses increased $24.3 million from $1.4 million in 2003 to $25.7 million in 2004. Approximately $24.5 million of the 2004 expense incurred relates to our purchase of in-process research and development for cethromycin and ABT-210 from Abbott Laboratories. These purchased in-process expenses represent $1.5 million of payments and $9.0 million of our common stock issued to Abbott by December 31, 2004 and $14 million in payments that are due in 2005 under the terms of the contract. The remainder of our research and development expenditures in 2004, totaling approximately $1.2 million, was expended under a variety of early-stage discovery programs.
33
Selling, general and administrative expense. General and administrative expenses increased 38% to $1.6 million in 2004 from $1.2 million in 2003. The increase in general and administrative expenses was the result of higher legal, audit and travel expenses associated with our financing activities and increased business development efforts in connection with the negotiation and conclusion of our license agreement with Abbott Laboratories. General and administrative expense consists primarily of compensation for employees in executive and operational functions, facilities costs and professional fees for accounting and legal services.
Interest expense—net. Interest expense—net increased 14% to $195,000 in 2004 from $171,000 in 2003. The increase in interest expense is attributable to the $3.0 million note issued to Leaders Bank in December of 2004, which bears interest at the prime rate plus 0.75% per annum. In connection with this note, we issued warrants to purchase 14,887 shares of our common shares at a price of $8.02 per share. These warrants were determined to have a fair value at the time of issuance of approximately $12,000, which is being amortized ratably over the two-year life of the note. Approximately $185,000 and $171,000 of interest expense incurred in 2004 and 2003, respectively, related to the $2.0 million loan that Dr. Michael T. Flavin provided to us in September 2001. The loan accrues interest at the rate of 7.75% per year, compounded annually. The loan is due and payable in December 2007.
Year ended December 31, 2003 compared to year ended December 31, 2002
Revenue. Total revenue decreased 64% to $180,000 in 2003 from $496,000 in 2002. Management fees, the largest component of revenue, decreased 76% in 2003 to $119,000 from $495,000 in 2002 as a result of a change in accounting for revenue derived from our 50/50 joint venture. We began recognizing management fee revenue from our 50/50 joint venture during the second quarter of 2003 on a cash basis. As a result, three months of revenue during the year was recognized. Billed and unrecognized management fees total $185,000 at December 31, 2003. Grant revenue increased to $62,000 in 2003 from $0 in 2002 as a result of an SBIR grant award by the National Institutes of Health. SBIR grants have a budget of $100,000 and last between six months and one year.
Research and development expense. Research and development expenses increased $475,000 from $925,000 in 2002 to $1.4 million in 2003 and resulted from increased scientific headcount and related supplies used in various early stage discovery programs.
Selling, general and administrative expense. General and administrative expenses increased 79% to $1.2 million in 2003 from $669,000 in 2002. The increase in general and administrative expenses was the result of higher legal, audit and travel expenses associated with our financing activities and increased business development efforts in connection with the negotiation of a potential acquisition that was not consummated. General and administrative expense consists primarily of compensation for employees in executive and operational functions, facilities costs and professional fees for accounting and legal services.
Interest expense—net. Interest expense—net increased 4% to $171,000 in 2003 from $164,000 in 2002. Approximately $171,000 and $159,000 of interest expense incurred in 2003 and 2002, respectively, relates to the $2.0 million loan that Dr. Michael T. Flavin provided to us in September 2001. The loan accrues interest at the rate of 7.75% per year, compounded annually. The loan is due and payable in December 2007.
Liquidity and Capital Resources
We have devoted substantially all of our cash resources to research and development and general and administrative expenses. To date, we have not generated any revenues from the sale of products, and we do not expect to generate any such revenues for a number of years, if at all. As a result, we have incurred an accumulated deficit of $41.9 million, $40.3 million and $13.1 million as of June 30,
34
2005, December 31, 2004 and December 31, 2003, respectively, and we expect to incur significant and increasing operating losses for the foreseeable future. Our working capital deficit as of June 30, 2005 amounted to $16.4 million. Cash was $40,000 as of June 30, 2005. Since our inception in 1999, we have financed our operations primarily through debt and capital contributions from our founder and controlling stockholder and borrowings under our bank line of credit.
In September 2001, Dr. Michael T. Flavin made a $2.0 million loan to us. The loan accrues interest at the rate of 7.75%, compounded annually, and is due and payable in December 2007.
On December 21, 2004, we entered into a $3.0 million line of credit with a local financial institution. Interest is based upon the prime rate plus 0.75% and is payable monthly. The principal balance is due upon demand or by December 21, 2006, if no demand is made. The line of credit is secured by substantially all of our assets and is guaranteed by Flavin Ventures, Dr. Flavin and his wife and John L. Flavin. Flavin Ventures secured its guaranty by issuing a mortgage on our principal facilities and pledging the shares it owns in us. In May 2005, we increased our line of credit to $4.0 million. In connection with this increase, Dr. Flavin entered into a subordination agreement with the bank under which he agreed to subordinate his $2.0 million loan to us to the line of credit. On July 28, 2005, we entered into a commitment letter with our bank to extend the maturity of our line of credit until December 2007 and eliminate the bank's demand payment rights.
During the twelve months ended December 31, 2004, cash used in operating activities totaled $2.3 million, primarily due to general operating activities. Cash used in investing activities totaled $91,000 and $105,000 during the years ended December 31, 2004 and 2003, respectively, primarily for the purchase of laboratory and office equipment. Future cash used in investing activities for property and equipment are not expected to be significant. Cash provided from financing activities totaled approximately $2.5 million during the twelve months ended December 31, 2004 and consisted of $2.3 million related to the sales of our common stock and $235,000 from the receipt of proceeds from our loan with Leaders Bank.
During the six months ended June 30, 2005, cash used in operating activities totaled $2.5 million, primarily due to general operating activities. Cash used in investing activities during the same period was negligible. Cash provided from financing activities totaled approximately $2.3 million during the six months ended June 30, 2005 principally as a result of sales of our common stock.
We believe that the net proceeds from the initial public offering will be sufficient to meet our anticipated operating requirements through 2006. We expect that we will need to raise additional capital or incur additional indebtedness to continue to fund our operations for a longer period. Our future capital uses and requirements depend on numerous forward-looking factors. These factors include, but are not limited to, the following:
- •
- progress in our research and development programs, as well as the magnitude of these programs;
- •
- the timing, receipt and amount of milestone and other payments, if any, from present and future collaborators;
- •
- our ability to establish and maintain additional collaborative arrangements;
- •
- the resources, time and costs required to successfully initiate and complete our preclinical and clinical trials, obtain regulatory approvals, protect our intellectual property and obtain and maintain licenses to third-party intellectual property;
- •
- the cost of preparing, filing, prosecuting, maintaining and enforcing patent claims; and
- •
- the timing, receipt and amount of sales and royalties, if any, from our potential products.
35
If, at any time, our prospects for financing our clinical development programs are limited, we may decide to reduce research and development expenses by delaying, discontinuing or reducing our funding of development of one or more product candidates. Alternatively, we might raise funds through public or private financings, strategic relationships or other arrangements. We cannot assure you that any additional financing will be available on attractive terms, or at all. Furthermore, any additional equity financing may be dilutive to stockholders and debt financing, if available, may involve restrictive covenants and increased interest expense. Similarly, financing obtained through future co-development arrangements may require us to forego certain commercial rights to future product candidates. Our failure to raise capital as and when needed could have a negative impact on our financial condition and our ability to pursue our business strategy.
Contractual Obligations
As of December 31, 2004, the annual amounts of future minimum payments under debt obligations, interest, lease obligations and other long term liabilities consisting of research, development, and license agreements are as follows:
| | Payments Due by December 31, | | | ||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2005 | 2006 | 2007 | 2008 | 2009 | Thereafter | Total | ||||||||||||||
| Short-term debt | $ | — | $ | 2,250,000 | $ | — | $ | — | $ | — | $ | — | $ | 2,250,000 | |||||||
| Long-term debt | — | 2,000,000 | — | — | — | — | 2,000,000 | ||||||||||||||
| Interest | — | 1,277,149 | — | — | — | — | 1,277,149 | ||||||||||||||
| Abbott License Payments | 14,000,000 | — | 10,000,000 | 30,000,000 | — | — | 54,000,000 | ||||||||||||||
| Operating Leases | 218,463 | 225,759 | 234,789 | 160,645 | — | — | 839,656 | ||||||||||||||
| Capital Leases | 17,421 | 17,421 | 14,110 | — | — | — | 48,952 | ||||||||||||||
| Total | $ | 14,235,884 | $ | 5,770,329 | $ | 10,248,899 | $ | 30,160,645 | $ | — | $ | — | $ | 60,415,757 | |||||||
Amounts outstanding under our bank line of credit, equal to $2.25 million at December 31, 2004, can be called at any time and have therefore been classified as short-term debt. In the absence of a request for repayment, the outstanding amounts are due in December 2006. We borrowed an additional $750,000 under our bank line of credit in the first quarter of 2005 and $517,000 in the second quarter of 2005. These amounts remain outstanding and can be called at any time. On July 28, 2005, we entered into a commitment letter with our bank to extend the maturity of our line of credit until December 2007 and eliminate the bank's demand payment rights. Under the terms of the line of credit, we will owe a fee in the amount of $30,000 upon the completion of the offerings.
Under our license agreement with Abbott Laboratories, we have paid $1 million to Abbott in 2005 and we are obligated to pay an additional $8 million on September 1, 2005, or, if earlier, five days following the closing of the offerings. This $8 million payment includes our purchase of the remaining cethromycin inventory from Abbott. We are also required to make payments to Abbott if we achieve certain milestones. We owe Abbott $5 million on October 31, 2005, or, if earlier, upon our initiation of pivotal Phase III clinical trials for cethromycin. Concurrent with the shares we are offering by this prospectus in the initial public offering, we are also offering 428,571 shares of our common stock to Abbott Laboratories in exchange for reducing this milestone payment obligation from $5 million to $2 million. We will also owe Abbott $10 million if we submit an NDA for cethromycin and $30 million if and when the FDA approves the NDA, which we estimate will occur in 2007 and 2008, respectively, at the earliest. Thereafter, we would owe Abbott an additional $2.5 million upon reaching $200 million in aggregate net sales of cethromycin and $5 million upon reaching $400 million in aggregate net sales. The periods in which milestone obligations become payable are only estimates due to uncertainties in the regulatory approval process.
36
Our long-term commitments under operating leases shown above consist of payments relating to our facility lease in Woodridge, IL, which expires in September of 2008.
Calanolide A Joint Venture
Our Calanolide A product candidate is held by our 50/50 joint venture with the Sarawak Government. To date, the Sarawak Government has funded substantially all of the joint venture's activities through a $9 million equity investment and $12 million in loans. As of the date of this prospectus, $9 million of this debt is past due. In July 2005, the Sarawak Government exercised certain of its contractual rights under its loan agreement with the joint venture and reserved all rights and remedies available as a creditor. Further development will require that we and the Sarawak Government agree to restructure and extend the joint venture's debt and determine the source of financing for the next stage of clinical trials for Calanolide A. We have proposed to the Sarawak Government that, for the first time, we would jointly fund the next level of development of Calanolide A, which we currently estimate to cost approximately $2 million. No agreement has yet been reached, and we do not intend to expend any meaningful funds advancing the development of Calanolide A unless and until we restructure the joint venture's debt and reach a satisfactory agreement with the the Sarawak Government to amend our joint venture relationship and establish our respective obligations regarding future funding.
Our joint venture could be subject to termination if we are unable to successfully negotiate a restructuring with the Sarawak Government. Under our joint venture agreement, upon termination any liabilities of the joint venture will be settled by the parties equally and all patents or licenses of the joint venture would be reassigned back to us. We believe that this equal settlement of liabilities provision was intended to recognize the 50/50 equity ownership relationship and does not constitute our guaranty of the joint venture's indebtedness to the Sarawak Government. Consequently, if the joint venture is terminated, the joint venture partners would work together to monetize the joint venture's assets for the purpose of satisfying its liabilities. In the event the joint venture's assets are insufficient to settle its liabilities, we do not believe that the equal settlement of liabilities provision obligates us to contribute funds to the joint venture. See "Other Collaborations and License Agreements—Sarawak MediChem Pharmaceuticals, Inc."
Market Risks
Our exposure to interest rate risk is limited to interest income sensitivity, which is affected by changes in the general level of U.S. dollar interest rates, particularly as the majority of our investments are in short-term debt securities and bank deposits. The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive without significantly increasing risk. To minimize risk, we maintain our portfolio of cash and cash equivalents in a variety of interest-bearing instruments, including U.S. government and agency securities, high-grade U.S. corporate bonds, commercial paper and money market funds. Due to the nature of our short-term and restricted investments, we believe that we are not exposed to any material market risk.
We do not have any relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special purpose entities, which would have been established for the purpose of facilitating off-balance sheet arrangements or other contractually narrow or limited purposes. In addition, we do not engage in trading activities involving non-exchange traded contracts. Therefore, we are not materially exposed to any financing, liquidity, market or credit risk that could arise if we had engaged in these relationships.
37
Critical Accounting Policies and Estimates
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses and related disclosure of contingent assets and liabilities. We review our estimates on an ongoing basis. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances. Actual results may differ from these estimates under different assumptions or conditions. While our significant accounting policies are described in more detail in the notes to our financial statements included in this prospectus, we believe the following accounting policies to be critical to the judgments and estimates used in the preparation of our financial statements.
In-Process Research and Development
We recognize in-process research and development in accordance with FASB Interpretation No. 4,Applicability of FASB Statement No. 2 to Business Combinations Accounted for by the Purchase Method. Assets to be used in research and development activities, specifically, compounds that have yet to receive new drug approval from the FDA and would have no alternative use should approval not be given, are immediately charged to income when acquired.
In December 2004, we incurred $24.5 million of in-process research and development expense as a result of entering into our exclusive license with Abbott Laboratories for cethromycin and ABT-210. We determined to immediately expense the purchased in-process research and development costs because the ultimate outcome of clinical trials for cethromycin is unknown and, therefore, FDA approval is uncertain. If cethromycin does not receive FDA approval then it does not have an alternative future use. As part of the consideration for the cethromycin license, we issued to Abbott shares of our common stock. In order to determine the value of the common stock issued to Abbott, we relied on the arms-length nature of the transaction, precedent market data and several internal valuation models and studies. An independent appraisal was also obtained to provide additional support for the valuation methodologies utilized. By their nature, valuations involve judgments of management that rely on estimates regarding the timing and amount of cash flows and discount rates that, if changed, could significantly affect the conclusions as to value.
Variable Interest Entities
We determine consolidation of variable interest entities in accordance with FASB Interpretation 46(R), Consolidation of Variable Interest Entities. Our 50/50 joint venture, Sarawak MediChem Pharmaceuticals, has not been included in our consolidated financial statements because the majority of its financial support in the form of contributed capital and debt has been funded by our joint venture partner.
Revenue Recognition
We recognize revenue relating to our collaboration agreements in accordance with the Securities and Exchange Commission's Staff Accounting Bulletin No. 101,Revenue Recognition in Financial Statements, SAB No. 104,Revenue Recognition, and Emerging Issues Task Force Issue No. 00-21,Revenue Arrangements with Multiple Deliverables. Revenue under collaborative arrangements may include the receipt of non-refundable license fees, milestone payments and research and development payments. SBIR grant revenue is recorded as expenses are incurred and services performed in accordance with the terms of the grant agreements. Royalty revenue is earned from a license of our technology. Under the license, revenue is earned based upon a percentage of reported net sales of MediChem Research, Inc. for services that incorporate the licensed technology. Management fees,
38
which are generated from a shared services agreement with Sarawak MediChem Pharmaceuticals, are recognized on the cash basis as the collectibility of such fees is not reasonably assured. Management fees derived from Flavin Ventures are recorded as services are provided.
Stock-based Compensation
Effective January 1, 2004, we adopted the fair value method, which is considered the preferable accounting method, of recording stock-based employee compensation as contained in Statement of Financial Accounting Standards No. 123 "Accounting for Stock-Based Compensation." As prescribed in SFAS No. 123(R) "Share-Based Payment" we elected to use the "prospective method." The prospective method requires expense to be recognized for all awards granted, modified or settled in the year of adoption. Historically, we applied the intrinsic method as provided in Accounting Principles Board Opinion No. 25 "Accounting for Stock Issued to Employees" and related interpretations and accordingly, no compensation cost had been recognized for stock options issued to employees in prior years. As a result of adopting the fair value method for stock compensation, all future awards will be expensed over the stock options vesting period. We utilized the Black-Scholes option pricing model to estimate the fair value of options granted in 2004. The adoption did not have a significant effect in 2004.
Recent Accounting Pronouncements
In November 2003, the FASB issued Staff Position 150-3,Effective Date, Disclosures, and Transition for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable NonControlling Interests Under FASB 150,deferring the effective date for applying the provisions of SFAS No. 150,Accounting for Certain Financial Instruments with Characteristics of Both Liabilities and Equity. SFAS No. 150 requires that certain instruments be classified as liabilities in the statements of financial position. For nonpublic entities, the classification, measurement and disclosure provisions of SFAS No. 150 have been deferred indefinitely for financial instruments that are not mandatorily redeemable on fixed dates for fixed or determinable amounts. SFAS No. 150 is effective for financial instruments that are mandatorily redeemable on fixed dates for fixed or determinable amounts for fiscal periods beginning after December 15, 2004, and its adoption is not expected to have a material impact on the Company's financial position or results of operations.
39
Overview
We are a biopharmaceutical company focused on the discovery, development and commercialization of novel drugs in the areas of infectious disease, inflammation and oncology. Using our internal discovery capabilities and our network of pharmaceutical and academic partners, we have assembled a promising pipeline of clinical and preclinical product candidates. The following is a summary of each of our primary programs:
- •
- Infectious Disease. We have an exclusive worldwide license (excluding Japan) from Abbott Laboratories to develop and commercialize cethromycin, a novel ketolide antibiotic in Phase III clinical development for the treatment of respiratory tract infections. Following the offerings, we intend to conduct two pivotal Phase III clinical trials for the treatment of mild-to-moderate community acquired pneumonia, the indication for which we initially plan to seek FDA approval. Cethromycin has been tested in approximately 3,800 human subjects during clinical trials. Abbott Laboratories completed six Phase III clinical trials of the compound to test safety and efficacy.
- •
- Inflammation. ALS-886 is a novel therapeutic in preclinical development for the treatment of inflammation-related tissue damage, including tissue damage associated with ARDS. Patients suffering from acute respiratory distress syndrome (ARDS) have a high fatality rate, and there are currently only limited treatment options available. We have established an open IND application for ALS-886 with the FDA. We expect that the first clinical study will involve approximately 40 patients to determine the safety profile of the compound in human subjects.
- •
- Oncology. ALS-357 is a compound that has shown evidence of anti-tumor activity against malignant melanoma in preclinical trials. Currently available therapies have not had significant success at prolonging survival for patients with melanoma that has spread beyond the primary growth site. We believe that ALS-357 has the potential for either a topical or systemic formulation. We established an open IND application for ALS-357 with the FDA. The expected Phase I clinical trial protocol contemplates treating 16 patients with in-transit metastatic melanoma by topical application.
Through our 50/50 joint venture, Sarawak MediChem Pharmaceuticals, we have managed the development of Calanolide A, a non-nucleoside reverse transcriptase inhibitor for the treatment of HIV. We completed a series of Phase I clinical trials for Calanolide A, which showed effectiveness in reducing viral load and that it was well-tolerated by patients. Subject to reaching a satisfactory agreement with our joint venture partner regarding our current business arrangement and our respective funding obligations for the next phase of development, we plan to enroll HIV-infected patients for a Phase IIa clinical trial of Calanolide A.
In addition to the compounds summarized above, we have additional product candidates in preclinical development. These include compounds derived from natural products to treat infectious diseases, a novel class of synthetic chemotherapeutics to treat various cancers and small molecule inhibitors that may lead to a treatment for Alzheimer's disease.
Our Opportunity
We believe that there are attractive opportunities in the discovery, development and commercialization of product candidates in the areas of infectious disease, inflammation and oncology. Although these areas are highly competitive, we believe that the markets for the indications we intend to pursue are either too small or too fragmented for many large pharmaceutical companies. Many large pharmaceutical companies are exiting certain therapeutic areas and looking to out-license developmental products that fail to meet revenue thresholds. Our goal is to take advantage of in-licensing opportunities as they arise while also building on our foundation of internal drug discovery
40
and development. We expect to compete effectively because of the proficiency of our experienced management team and the utility of our integrated chemistry and biology drug discovery platform.
Our Strategy
Our objective is to become a fully integrated pharmaceutical company that discovers and develops small molecule therapeutics to treat life-threatening diseases in the areas of infectious disease, inflammation and cancer, and then markets these products directly to physicians and hospitals. We plan to sustain our drug development pipeline through our internal drug discovery capabilities and by opportunistically in-licensing promising compounds that fit into our areas of focus. Specific key aspects of our strategy include the following:
Maximize the Commercial Potential of Cethromycin
Initially, we intend to focus a significant portion of our business efforts on the further development of cethromycin for the treatment of mild-to-moderate community acquired pneumonia. Abbott Laboratories, which granted us an exclusive worldwide license (except in Japan) to commercialize cethromycin, completed six Phase III clinical trials of the compound to determine the safety and efficacy of 150 mg once-daily and twice-daily regimens. To date, approximately 3,800 human subjects have been dosed with cethromycin in clinical trials. Following the offerings, we plan to conduct two pivotal Phase III clinical trials for the treatment of mild-to-moderate community acquired pneumonia using a 300 mg once-daily dosing regimen.
In addition to treating mild-to-moderate community acquired pneumonia in pivotal Phase III clinical trials, we intend to pursue opportunities for cethromycin in the treatment of other respiratory tract infections such as bronchitis, sinusitis and pharyngitis.
Advance the Development of our other Infectious Disease, Inflammation and Oncology Product Candidates
We intend to leverage our existing clinical trial experience from developing Calanolide A to advance product candidates in our inflammation and oncology programs through clinical trials. For instance, to advance ALS-886 for the treatment of inflammation-related tissue damage, we plan to conduct Phase I clinical trials that test safety and provide preliminary indications regarding efficacy in preventing lung tissue damage in ARDS patients. To develop ALS-357 for the treatment of malignant melanoma, we intend to conduct Phase I clinical trials with a topical formulation to test safety and provide preliminary indications regarding efficacy. We also plan to develop an oral and IV formulation of ALS-357 and initiate related clinical trials. Subject to reaching a satisfactory agreement with our joint venture partner regarding our current business arrangement and our respective obligations for the next phase of development, we plan to enroll HIV-infected patients for a Phase IIa clinical trial of Calanolide A.
Leverage our Drug Discovery and Development Capabilities
We intend to expand our product portfolio by exploiting and enhancing our internal drug discovery and development capabilities using our integrated chemistry and biology skills. We plan to continue utilizing our integrated chemistry and biology drug discovery platform to design, optimize and evaluate high-potential product candidates.
Continue to Develop Strategic Collaborations
We plan to continue developing relationships with key pharmaceutical and biotechnology companies, governmental institutions and academic laboratories in order to in-license promising compounds that are not core to their strategy but fit closely with our corporate strengths. We also intend to identify co-development partners for the out-licensing of certain product candidates. Further,
41
we may choose to establish collaborative partnerships through which certain of our clinical candidates can be marketed and commercialized.
Build Commercial Infrastructure to Market our Products
We plan to retain U.S. marketing and sales rights or co-promotion rights for certain of our products that we believe can be marketed through a focused sales force targeting specialists and high-prescribing physicians. For situations in which a large sales force is required to access the market, and for markets outside the United States, we generally plan to commercialize our product candidates through a variety of collaboration arrangements with leading pharmaceutical companies and contract sales organizations.
Our Lead Product Candidates
We have developed a promising pipeline of clinical and preclinical product candidates from both in-licensing efforts and our own discovery programs in the areas of infectious disease, inflammation and oncology. The following chart provides a summary of our product candidate pipeline and indicates the stage of development that each product candidate is currently in or ready to begin:
Development Pipeline
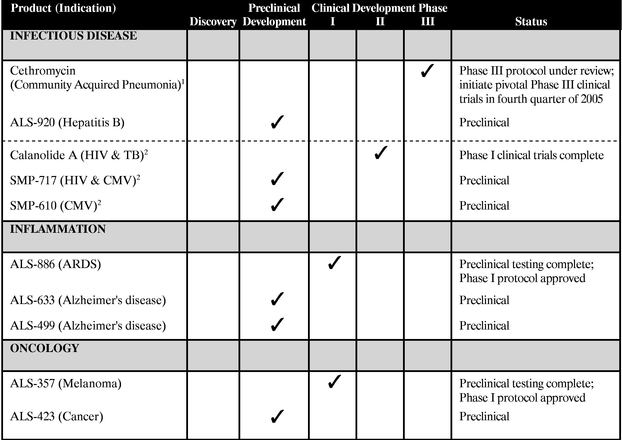
- (1)
- Six Phase III clinical trials have been completed by Abbott Laboratories for the treatment of respiratory tract infections, including community acquired pneumonia, bronchitis, pharyngitis and sinusitis.
- (2)
- Developed through our Sarawak MediChem Pharmaceuticals joint venture.
42
Our Lead Programs
Infectious Disease
Antibiotics
Overview
Our most advanced product candidate, cethromycin, is a novel ketolide antibiotic in Phase III clinical development for the treatment of respiratory tract infections. We are initially developing cethromycin as a treatment for mild-to-moderate community acquired pneumonia. Over the last decade, the rapid rise in severe and fatal infections caused by antibiotic-resistant bacteria has posed a serious threat to public health. There is a need to discover new antibiotics that are effective against resistant bacteria. As a new class of antibiotics, ketolides have shown activity against penicillin-and macrolide-resistant Gram-positive pathogens. Clinical trials indicate that cethromycin has enhanced activity towards drug-resistantStreptococcus pneumoniae andHaemophilus influenzae, two of the pathogens commonly found in community acquired pneumonia, when compared to published data on antibiotics currently on the market. Cethromycin has also shownin vitro evidence of an extended post-antibiotic effect, meaning that the suppression of bacterial growth persists in the absence of measurable antibiotic concentration. Six Phase III clinical trials have been completed for the treatment of respiratory tract infections, and cethromycin has demonstrated a clinical cure rate as high as 93% for subjects with community acquired pneumonia. All clinical studies to date were conducted by Abbott Laboratories. In December 2004, Abbott Laboratories granted us an exclusive worldwide license, except in Japan, to commercialize cethromycin. Following the offerings, in order to advance cethromycin toward marketing approval, we intend to conduct pivotal Phase III clinical trials focused on the clinical and bacteriological cure rates in treating mild-to-moderate community acquired pneumonia.
Market Overview
Bacterial infections occur when bacteria that naturally exist in the body, or that are acquired through inhalation, ingestion or direct penetration, are not controlled by the normal immune defense system. These uncontrolled bacteria can multiply and either excrete toxins or provoke the immune system to mount a response, in either case damaging tissue. Antibiotics work by binding to specific targets in a bacterial pathogen, thereby inhibiting a function that is essential to the pathogen's survival. Many antibiotics were developed and introduced into the market during the 1970s and 1980s and have proven to be effective in treating most bacterial infections. We believe this historic efficacy prompted pharmaceutical companies to shift their resources to other areas of drug discovery and development. As a result, very few antibiotics from new chemical classes have been introduced in the last several years.
Antibiotic resistance has become widely considered a significant threat to public health, and the problem continues to worsen. The Centers for Disease Control, or CDC, continue to report on new strains of bacteria that are resistant to one or more antibiotics currently on the market. The increasing prevalence of drug-resistant bacteria has led to prolonged illnesses and hospitalizations, increased healthcare costs and significantly higher mortality rates. As a result, there is a strong demand for new treatments that are more effective against resistant strains and do not show potential for inducing the rapid development of additional resistant strains. We do not believe that this demand for new antibiotic therapies is being met by large pharmaceutical companies because of a shift in research and development focus toward chronic conditions that require sustained medication over long periods of time.
43
Respiratory tract infections arise when bacterial infections develop in the inhalation passageway, including the nose, throat, sinuses and lungs. The four main types of respiratory tract infections are pneumonia, bronchitis, pharyngitis and sinusitis. Many of these bacterial infections can be life threatening if not treated quickly. Bacterial pneumonia, which involves an infection of the lung itself, is the most severe of the common respiratory tract infections. Pneumonia is often classified as community acquired pneumonia or hospital acquired pneumonia, depending upon the setting in which contraction of the bacterial infection occurred. The Textbook of Primary and Acute Care Medicine estimates that, in the United States alone, there are approximately 5-6 million cases of community acquired pneumonia each year.
Current Treatment Options and Limitations
Until recently, there have been three main classes of antibiotics prescribed for respiratory tract infections such as community acquired pneumonia. These are semi-synthetic penicillins (also known as beta-lactams), such as Augmentin® (amoxicillin and clavulanate potassium) sold by GlaxoSmithKline; macrolides, such as erythromycin or Zithromax® (azithromycin) sold by Pfizer; and fluoroquinolones, such as Cipro® (ciprofloxacin) sold by Bayer. Penicillins, macrolides and fluoroquinolones have all been shown to have certain shortcomings with respect to the treatment of respiratory tract infections. Studies have shown that, in the United States, approximately 26% ofStreptococcus pneumoniaeisolates, the most common pathogens that cause respiratory tract infections such as community acquired pneumonia, are resistant to macrolides. In addition, numerous reports in the medical literature have noted the emergence of penicillin-resistant pneumococci. Because fluoroquinolones have a broad-spectrum activity against both Gram-negative and Gram-positive bacteria, there is a greater potential for side effects. The wide use of fluoroquinolones also increases the potential for development of resistance amongStreptococcus pneumoniae.
Increased bacterial resistance to many of the currently available antibiotics has been caused by certain common medical practices and sociological factors. By necessity, a wide variety of antibiotics is often administered before the specific disease-causing pathogen has been identified. Bacterial resistance is fostered through the erroneous prescription of antibiotics for non-bacterial infections. The lack of full patient compliance with prescribed courses of therapies has further contributed to bacterial resistance against currently marketed antibiotics. Patients will frequently discontinue a prescribed dosing regimen after symptoms subside, but bacteria that are not entirely eradicated may re-emerge in resistant forms.
Ketolides are a new class of antibiotics that show promise to address many of the shortcomings found in the current treatment options for community acquired pneumonia and other respiratory tract infections, such as bronchitis, sinusitis and pharyngitis. Ketolides are effective against penicillin- and macrolide-resistant pathogens known to cause respiratory tract infections. When compared to published data on current fluoroquinolone antibiotic treatments, ketolides have shown a favorable safety profile. The only ketolide available for sale in the United States, Ketek® (telithromycin), was approved by the FDA in April 2004 and launched commercially by Aventis Pharmaceuticals in August 2004. Ketek has been available for sale in Europe since the summer of 2002. According to Aventis' publicly available financial data, global sales of Ketek in 2004 totaled approximately $250 million, with approximately $93 million of sales in the fourth quarter. According to data compiled by IMS Health, an independent pharmaceutical information and consulting firm, sales of Ketek in the United States totaled approximately $70 million in 2004. While Ketek has been determined to be effective against resistant pathogens, the FDA has imposed a labeling requirement for Ketek indicating that it may cause visual disturbances including double-vision, blurred vision and difficulty focusing.
ALS Solution
We are developing cethromycin, a novel ketolide antibiotic licensed from Abbott Laboratories, in response to the emerging antibiotic resistance observed in the treatment of community acquired pneumonia. Cethromycin has been tested in approximately 3,800 human subjects during clinical trials.
44
Following the offerings, we intend to conduct two pivotal Phase III clinical trials of cethromycin for the treatment of mild-to-moderate community acquired pneumonia. We also intend to pursue opportunities for cethromycin in the treatment of other respiratory tract infections such as bronchitis, sinusitis and pharyngitis.
Based on publicly available data regarding current antibiotic compounds, we believe that there is a potential opportunity for further development of cethromycin in the treatment of respiratory tract infections for a number of reasons:
- •
- cethromycin has shown higherin vitro potency and a broader range of activity than macrolides and Ketek against Gram-positive bacteria associated with respiratory tract infections;
- •
- cethromycin appears to be clinically effective against penicillin- and macrolide-resistant bacteria;
- •
- cethromycin has a mechanism of action, unique to ketolides, that may slow the onset of future resistance;
- •
- cethromycin has shown specific activity against Gram-positive pathogens, unlike fluoroquinolones, while leaving normally-present Gram-negative bacteria undisturbed;
- •
- cethromycin has shownin vitro evidence of extended post-antibiotic effects against the pathogens commonly seen in respiratory tract infections; and
- •
- cethromycin, unlike Ketek, has not demonstrated visual disturbance side effects in clinical trials.
We believe that cethromycin, if approved, would build upon the growing market acceptance of ketolide compounds in the antibiotic marketplace.
Mechanism of Action. Ketolides are often considered the next generation of macrolides because of their similar mechanisms of action. Unlike macrolides, which have one binding site on bacterial ribosomes, ketolides such as cethromycin have shown effectiveness in establishing dual binding sites with the target pathogen. We believe that having dual binding sites is an important factor for their activity against antibiotic-resistant bacteria. One way in which bacteria develop resistance to antibiotics is by modifying the site at which the drug binds to the pathogen. Because cethromycin has two binding sites, cethromycin maintains its ability to bind to the bacteria even after the site common to both macrolides and cethromycin has been modified. This allows cethromycin to remain effective against bacterial strains resistant to macrolides. As a ketolide, cethromycin retains the activity of the macrolides against the predominant community acquired respiratory pathogens and, at the same time, expands the spectrum to include penicillin- and macrolide-resistant pathogens. Cethromycin has demonstrated a greater affinity, or tighter binding, for its ribosomal targets than Ketek and macrolides. This high affinity, along with the dual binding sites, results in a slower dissociation time from the ribosomal target and an enhanced post-antibiotic effect.
The majority of drug-resistant bacteria has emerged from the class of bacteria called Gram-positive pathogens. Gram-positive bacteria can be differentiated from Gram-negative bacterial pathogens by the differences in the structure of the bacterial cell membrane and cell wall. Gram-positive bacteria possess a single cellular membrane and a thick cell wall, while Gram-negative bacteria possess a double cellular membrane and a thin cell wall. Cethromycin is active against the Gram-positive pathogens most likely to be responsible for bacterially mediated respiratory disease, as well as a number of the atypical pathogens, without affecting normally present Gram-negative bacteria. This provides an advantage over the fluoroquinolone class of antibiotics, which have activity against both Gram-positive and Gram-negative organisms. By restricting activity to Gram-positive pathogens, cethromycin should be able to retain effectiveness in treating respiratory infections while reducing the incidence of side effects seen when Gram-negative pathogens normally present in the gastrointestinal tract are eradicated.
The mechanism of action, efficacy and safety of cethromycin were studied in preclinical and clinical trials by Abbott Laboratories. Abbott Laboratories granted us an exclusive worldwide license,
45
except in Japan, to commercialize cethromycin in December 2004, and we intend to conduct two pivotal Phase III clinical studies for the treatment of mild-to-moderate community acquired pneumonia beginning in the fourth quarter of 2005. Provided below is a summary of the preclinical and clinical trials and results conducted by Abbott, as well as a description of our future clinical plans for the compound. The results of preclinical and clinical trials to date provide evidence of safety and efficacy, but clinical testing is not complete and future results may vary from those found in earlier clinical trials.
In Vitro Activity. Whilein vitro activity alone does not necessarily indicate clinical significance, thein vitro antibacterial activity of cethromycin has been shown to be more potent than clarithromycin, azithromycin and Ketek against Gram-positive bacteria. Multiple publications in 2001 and 2002 confirmed and expanded the spectrum ofin vitroactivity of cethromycin compared to various antibiotics including Ketek. At comparable concentrations, cethromycin has been shown by various investigators to be 2-4 times more potent than Ketek in killingStreptococcus pneumoniae, including macrolide-resistant strains. Thein vitro activity, together within vivo animal model efficacy studies, provide evidence that cethromycin may be effective against community acquired pneumonia, acute exacerbation of chronic bronchitis, acute otitis media, skin and soft tissue infections, acute bacterial sinusitis and pharyngitis/tonsillitis.
Phase I Clinical Trials. The pharmacokinetics and safety of cethromycin were assessed in Phase I clinical trials of dosing ranges, the effects of food on oral dosage, drug-drug interaction and penetration of cethromycin into pulmonary tissues. The potential for cardiac arrhythmia by cethromycin was evaluated on healthy volunteers in a trial that was specifically designed to assess potential electrocardiographic changes, and no clinically significant events were observed at the anticipated dosing levels. Overall, approximately 1,000 healthy subjects have been exposed to cethromycin in Phase I clinical trials, and the results provide evidence that the compound is well-tolerated.
Phase II Clinical Trials. In Phase II clinical trials, the safety and effectiveness of cethromycin were evaluated in 863 individuals with community acquired pneumonia, bronchitis and sinusitis. The clinical cure rate is defined as the percentage of studied individuals treated with the antibiotic that demonstrated complete resolution of the signs and symptoms of the original infection. For individuals with community acquired pneumonia treated with once-daily doses of 300 mg or 600 mg of cethromycin, results demonstrated clinical cure rates of 92% and 80%, respectively. For subjects with bronchitis treated with once-daily doses of 150 mg, 300 mg or 600 mg, results demonstrated clinical cure rates of 87%, 90% and 90%, respectively. For subjects with sinusitis treated with once-daily doses of 150 mg, 300 mg or 600 mg, results demonstrated clinical cure rates of 89%, 83% and 71%, respectively. We believe that the reduced efficacy findings at higher dosing levels in Phase II clinical trials is attributable to patient compliance factors. As dosing levels increase, administration of the prescribed regimen is more burdensome and patients may be more likely to comply irregularly or discontinue the treatment.
The side effect findings for cethromycin were generally consistent with other current antibiotic treatments. The reported adverse event frequencies for cethromycin were considerably lower at the 150 mg and 300 mg dosing levels compared to the 600 mg dosing level. Taste perversion was the adverse event most frequently observed in the 863 cethromycin subjects evaluated, occurring in 17% of subjects across the three dosing regimens. The other adverse events reported by patients included nausea, diarrhea, vomiting, headache and abdominal pain, gastrointestinal side effect findings common to, and at frequencies consistent with, Ketek and other antibiotic treatments. Because approximately 2% of subjects reported visual disturbances in clinical trials of Ketek, the FDA imposed a labeling requirement informing patients and physicians of this side effect. Subjects have not reported visual disturbance side effects at any significant level in clinical trials for cethromycin, and we do not believe that the FDA will impose a similar labeling requirement. We believe that the lack of visual disturbance
46
side effects is important because patients undergoing an antibiotic regimen for respiratory tract infections are often otherwise able to drive a car and re-enter the workplace.
Phase III Clinical Trials. Cethromycin was evaluated for the treatment of community acquired pneumonia, bronchitis, pharyngitis and sinusitis in Phase III clinical trials. The initial Phase III clinical trial for the treatment of community acquired pneumonia examined the effectiveness of cethromycin at doses of 150 mg once-daily and 150 mg twice-daily. Results from this study demonstrated that both dosing regimens were safe and well-tolerated, and that each dose was effective in resolving or improving clinical signs and symptoms of community acquired pneumonia in adult subjects. The table below presents a summary of the initial Phase III clinical trial results for cethromycin in the treatment of community acquired pneumonia. For comparative purposes, the table also provides clinical results from a Phase II clinical trial of cethromycin and those found in independent studies for Ketek for the treatment of community acquired pneumonia as reported in FDA briefing documents. Clinical testing of cethromycin is not complete and our findings in future clinical trials may vary from previous studies.
| | Cethromycin 300 mg once- daily(1) | Cethromycin 600 mg once- daily(1) | Cethromycin 150 mg once- daily | Cethromycin 150 mg twice- daily | Ketek 800 mg once- daily | Ketek Study Pooled Comparators(2) | ||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Clinical Cure Rate | 92% (71/77) | 80% (56/70) | 93% (224/240) | 87% (224/254) | 92% (1032/1120) | 89% (339/381) | ||||||
| Bacteriological Cure Rate | 92% (54/59) | 82% (46/56) | 93% (140/150) | 89% (131/148) | 91% (312/344) | 93% (84/90) |
- (1)
- Phase II clinical trial
- (2)
- Includes amoxicillin, clarithromycin and trovafloxacin
Trials of cethromycin against comparators for the treatment of community acquired pneumonia have yet to be performed, although we expect to commence pivotal Phase III comparator trials in the fourth quarter of 2005. Four pivotal Phase III comparator trials of cethromycin have been performed for the treatment of bronchitis and pharyngitis, at a dosing level of 150 mg once-daily, but in each case the assessment of clinical cure rates failed to establish non-inferiority of cethromycin relative to the comparator. In a typical antibiotics trial, two endpoints are used to determine non-inferiority. One endpoint is the clinical cure rate, discussed above, and the other endpoint is the bacteriological cure rate, which is defined as the percentage of studied individuals treated with the antibiotic that showed complete eradication of the bacterial pathogen believed to be responsible for the clinical condition. In general, to demonstrate non-inferiority, the studied drug must be statistically non-inferior to the comparator drug for both endpoints. Clinical cure rates in two pivotal Phase III clinical trials for the treatment of bronchitis were 78% for cethromycin against 86% for azithromycin (a macrolide), and 89% for cethromycin against 90% for levofloxacin (a fluoroquinolone). Clinical cure rates in two pivotal Phase III clinical trials for the treatment of pharyngitis were 85% for cethromycin compared to 93% for penicillin, and 90% for cethromycin compared to 95% for penicillin. Results from all trials demonstrated that both cethromycin and the comparators were safe and well-tolerated.
An initial Phase III clinical trial has been performed to examine the effectiveness of cethromycin in the treatment of sinusitis at doses of 150 mg once-daily and 150 mg twice-daily. Results from this study demonstrated that both dosing regimens were safe and well-tolerated, and that each dose was effective in resolving or improving clinical signs and symptoms of sinusitis in adult subjects. Clinical cure rates were 90% for the 150 mg once-daily regimen and 81% for the 150 mg twice-daily dosing regimen.
Pivotal Phase III Clinical Trial Plans for the Treatment of Mild-to-Moderate Community Acquired Pneumonia. In order to complete regulatory requirements for marketing approval, we intend to conduct two pivotal Phase III clinical trials, one focused in the United States and the other in Europe, beginning in the fourth quarter of 2005. Both studies will evaluate a 300 mg once-daily dose of
47
cethromycin over 10 days for the treatment of mild-to-moderate community acquired pneumonia. The U.S. study will evaluate cethromycin against a standard course of levofloxacin (a fluoroquinolone), and the study in Europe will evaluate cethromycin against a standard course of Augmentin (a combinationb-lactam aminopenicillin andb-lactamase inhibitor). Each study will be a double-blinded, multi-center trial with the goal of enrolling 300 subjects in each arm, for a total of 1,200 subjects. The clinical cure rate as well as the bacteriological cure rate will be measured and statistical non-inferiority to the comparator regimen will be evaluated. We anticipate that a worldwide enrollment period of 12 months will be required to complete clinical evaluation.
The dosing regimen selected for pivotal Phase III clinical trials of cethromycin is based on the safety and tolerability findings for the compound when dosed at 300 mg once-daily in earlier studies. While previous data has suggested that cethromycin may be effective in the treatment of community acquired pneumonia at lower doses, earlier comparator trials in other indications using doses of 150 mg once-daily failed to demonstrate non-inferiority. We believe that a 300 mg once-daily dosage level will be effective in establishing non-inferiority for the treatment of community acquired pneumonia, and will be within the safety margins previously defined for cethromycin. We also believe that a 300 mg once-daily dosing regimen may have applicability for other respiratory tract infections such as bronchitis, pharyngitis and sinusitis.
Human Immunodeficiency Virus (HIV)
Overview
We are managing the development of Calanolide A through Sarawak MediChem Pharmaceuticals, Inc. a Delaware corporation in which we and the State Government of Sarawak, Malaysia each own 50% of the outstanding equity. Calanolide A is a natural non-nucleoside reverse transcriptase inhibitor for the treatment of patients with HIV. Currently, HIV-positive patients are usually treated with three or more anti-HIV drugs used in combination. However, some patients do not respond to current drug regimens, and some HIV strains are resistant to current antiviral therapies. Additionally, many current therapies, including Combivir, the top-selling anti-HIV drug, have significant negative side effects, including headache and nausea.In vitro, Calanolide A has shown a broad spectrum of antiviral activity against several different strains of HIV. In Phase I clinical trials at the doses studied, Calanolide A demonstrated a favorable safety profile associated with a reduction of viral load. These results lead us to believe that Calanolide A may be effective for both treatment-naïve HIV patients and for HIV patients who are failing current treatments, known as salvage therapy, in combination with currently available anti-HIV medications. Subject to reaching a satisfactory agreement with our joint venture partner regarding our current business arrangement and our respective funding obligations for the next phase of development, we plan to enroll HIV-infected patients for a Phase IIa clinical trial of Calanolide A. Our intended Phase IIa protocol would use Calanolide A in combination with another commonly prescribed HIV drug in order to attempt to enhance the viral load reduction seen in earlier trials.
Market Overview
HIV is the causative agent of the debilitating disease of AIDS. HIV disease is characterized by a gradual deterioration of immune functions. During the course of infection, crucial immune cells, called CD4+ T-cells, are disabled and killed, and their numbers progressively decline. If HIV goes untreated, AIDS develops in most people within 12 to 13 years after the initial infection. With treatment for HIV, the progression to AIDS may be delayed or prevented.
According to the United Nations AIDS Program, as of the end of 2003, approximately 38 million people worldwide were living with HIV. The majority of these individuals do not receive treatment. According to the CDC, an additional 45 million people could become newly infected by 2010. The primary driver for growth in the anti-HIV pharmaceutical market is expected to be the increasing number of patients who will receive treatment because of greater accessibility to anti-HIV drugs and
48
better and more cost-effective diagnostic procedures. Newer therapies will become available and are expected to command premium prices for those patient groups that can afford the cost.
Current Treatment Options and Limitations
The FDA approved the first anti-HIV drug, AZT, in 1987. Approximately 20 distinct anti-HIV drugs have received marketing approval in the United States since the approval of AZT. In today's treatment approach, three or more anti-HIV drugs are used in combination to create an effective antiviral therapy. Even so, the most potent drug combinations are still unable to eradicate the HIV virus from a patient completely. Another shortfall associated with currently available anti-HIV therapies is the evolution of drug-resistant HIV strains that emerge progressively against even the most highly active HIV therapies. HIV treatments that are currently marketed, or under development, fall into the following five categories:
- •
- nucleoside reverse transcriptase inhibitors (NRTIs);
- •
- non-nucleoside reverse transcriptase inhibitors (NNRTIs);
- •
- protease inhibitors (PIs);
- •
- entry inhibitors; and
- •
- vaccines.
NRTIs mimic the building blocks required by HIV in the early stages of replication and terminate the growth of viral RNA, ultimately preventing the virus from replicating itself. PIs block a protease enzyme that HIV requires in order to make new virus particles, rendering the virus unable to perform the splicing and joining of its protein building blocks. Entry inhibitors prevent entry of HIV into the host cell. Vaccines have only resulted in limited success.
NNRTIs bind directly to viral reverse transcriptase to inhibit enzyme activity. By inhibiting the enzymatic activity necessary for reverse transcription, NNRTIs interfere with one of the steps of the HIV life-cycle and thus prevent the virus from replicating itself. Although NNRTIs are very potent, resistance can develop quickly if the drugs are not taken as prescribed. Once viral resistance develops to one drug in this class, patients often become less responsive to other drugs in the NNRTI class. Because of this, there is an increasing demand for new NNRTIs that differentiate themselves from existing drugs with respect to their resistance profile.
GlaxoSmithKline's Combivir, a formulation combining the NRTIs lamivudine and zidovudine, was the top-selling HIV drug worldwide in 2004, with sales of over $1 billion according to GlaxoSmithKline's publicly available financial information.
ALS Solution
Calanolide A is an NNRTI of HIV that was originally extracted from theCalophyllum lanigerum tree indigenous to the island of Borneo in Malaysia. We developed a proprietary process to synthesize and manufacture Calanolide A in larger quantities.In vitro studies indicate that Calanolide A possesses properties that may overcome many of the shortcomings, especially the drug resistance and negative side effects, of currently marketed and developmental anti-HIV drugs in the NNRTI class. Calanolide A, due to its distinct mechanism of action, has a different resistance profile than currently available anti-HIV NNRTIs. Calanolide A has demonstrated synergistic activityin vitro when used in combination with NRTIs as well as certain PIs. The results of preclinical and clinical trials to date provide evidence of safety and efficacy, but clinical testing of Calanolide A is not complete and future results may vary from those found in earlier studies.
Preclinical Results. Ourin vitro preclinical testing provided evidence that Calanolide A is active against a wide range of HIV strains, including the Y181C mutant (a point mutation that confers
49
resistance to most NNRTIs) and other NNRTI-resistant mutants strains. Studies have also indicated that Calanolide A is synergistic when used in combination with other commonly used HIV drugs.
Phase I Clinical Trial Results. A total of 190 healthy human volunteers and HIV-positive individuals have been safely dosed with Calanolide A in escalating exposure levels up to 800 mg twice-a-day and up to 21 days continuous dosing. We have observed that Calanolide A is rapidly absorbed and distributed with a long half-life, while blood levels rise with increasing doses. Our Phase I clinical studies with Calanolide A have shown the drug to have only mild and transient side effects, including dizziness and an oily aftertaste. In addition, in early efficacy evaluations, HIV RNA concentrations decreased in a dose-dependent manner. This generally means that the HIV viral load decreased with increasing dosage levels of Calanolide A.
Phase IIa Clinical Trial Plans. Subject to reaching a satisfactory agreement with our joint venture partner regarding our current business arrangement and our respective funding obligations for the next phase of development, we plan to conduct a clinical trial utilizing a protocol entitled "A Phase IIa Study of Calanolide A to Evaluate the Safety, Pharmacokinetics and Effects of Calanolide A with Low-Dose Ritonavir on Surrogate Markers in HIV-Positive Patients with No Previous or Limited Experience of Antiretroviral Therapy." An earlier drug-drug interaction study showed that levels of Calanolide A could be increased substantially in healthy subjects by adding another therapeutic agent for HIV at low levels (i.e., non-therapeutic doses). This is called pharmacokinetic enhancement, or PK enhancement. The low-dose drug serves to enhance the pharmacokinetics of Calanolide A by slowing down the rate at which Calanolide A is metabolized. In our Phase IIa clinical trials, PK-enhanced Calanolide A will be given to sixteen HIV-infected subjects and the dosage will be increased to 800 mg and 1000 mg twice-a-day for a period of 14 days. The objectives will be to observe any changes in the side effect profile of Calanolide A and to monitor the antiviral activity of the drug over the study period.
50
The primary objective of the Phase IIa clinical trial will be to determine the potential therapeutic effect of PK-enhanced Calanolide A monotherapy, combined with a low dose of Norvir® (ritonavir), on the viral load levels of subjects. Plasma HIV RNA levels will be measured over fourteen days. The safety and pharmacokinetics of three different regimens will be monitored over the study period. Study subjects must be treatment-naïve prior to enrollment in order to qualify for the Phase IIa clinical trial, meaning that they must not have used, or have had limited use only, of anti-retroviral drugs (limited protease inhibitor and nucleoside reverse transcriptase inhibitor, but not non-nucleoside reverse transcriptase inhibitor use). Upon completion of the study period, all subjects will be offered a standard three-drug regimen, in consultation with their primary healthcare provider, for three to six months post-study.
If we determine to proceed with the Phase IIa clinical trial for Calanolide A, upon its completion we will again evaluate our strategic alternatives for the joint venture relationship and the future advancement of Calanolide A. We may determine to continue the program through our joint venture, independently or under a new joint venture arrangement. It is also possible that we will elect to transfer our rights in the compound to our current joint venture partner or an independent third party. Factors that we will consider in making this decision include the success of the Phase IIa study, the relative financial obligations of us and our joint venture partner and our capabilities at such time to continue development independently. For a more detailed discussion of our Calanolide A joint venture, see "—Other Collaborations and License Agreements—Sarawak MediChem Pharmaceuticals, Inc."
Other Opportunities in Infectious Disease Treatments
We intend to continue our evaluation of Calanolide A for other indications, such as tuberculosis, where the compound has shown initial efficacy inin vitro studies. We also plan to continue our evaluation of SMP-717 and SMP-610 for their antiviral properties, which may provide a treatment for viral indications such as cytomegalovirus, or CMV. In addition, ALS-920 is in preclinical development as a therapy for the treatment of hepatitis B.
Inflammation
Overview
We are developing ALS-886, a novel treatment intended to reduce and prevent the tissue damage associated with diseases such as ARDS. ARDS is a serious medical condition often caused by the inflammation and tissue damage associated with excess free radicals in the lungs. According to an article published inThe New England Journal of Medicine, there are approximately 200,000 cases of ARDS in the United States annually and the mortality rate is approximately 50%. Currently, there are no treatments for ARDS that have been shown to be effective. In preclinical testing, ALS-886 showed evidence of efficacy in reducing harmful increases in lung leakage. We believe the compound may also be effective as a prophylactic therapy against the initial tissue damage that often leads to ARDS. ALS-886 may also have applicability for other conditions in which free radicals cause tissue damage, such as ischemic stroke and myocardial infarction.
Market Overview
Inflammation is a complex process associated with a variety of medical conditions. One outcome of the inflammation process is the excess production of reactive oxygen metabolites or oxygen free radicals. When produced in excess, these free radicals can lead to tissue damage and destruction. The production of free radicals is enhanced in the presence of iron. Thus, therapeutic agents are needed that can eliminate or "scavenge" oxygen free radicals and/or sequester or remove free iron. Several serious clinical conditions, such as ARDS, ischemic stroke and myocardial infarction can attribute some of their tissue-damaging pathology to the overproduction of free radicals.
51
ARDS is sudden, life-threatening lung failure brought about by a variety of conditions including sepsis, severe pneumonia, trauma, burns and drug overdose. ARDS is not caused by one pathogen or trauma, but is brought about by a cascade of events leading to a severe limitation of lung function, usually twenty-four to forty-eight hours after the causative injury or illness. Often, ARDS begins when excess free radicals, brought about by one or more of the conditions described above, damage lung vascular tissue causing leaks through which fluid can enter. As fluid enters the air sacs, or alveoli, they collapse, gas exchange ceases, and the body becomes starved of oxygen, resulting in pulmonary edema, or the excessive retention of fluid in the lungs. If severe enough, this edema can lead to ARDS. ARDS patients require mechanical ventilation or some other form of assisted breathing in order to survive.
Current Treatment Options and Limitations
Current treatment options for ARDS are extremely limited. Most patients are currently only treated in a supportive manner using assisted ventilation. To our knowledge, the only product currently in development for the treatment of ARDS is Surfaxin®, being developed by Discovery Laboratories and Johnson & Johnson. This product mimics the fluids that coat the inside of lung surfaces and is currently in Phase II clinical trials for the treatment of ARDS. Surfaxin is a surfactant that must be applied directly to the lining of the lung, and thus cannot be given either orally or intravenously.
ALS Solution
We are initially developing ALS-886 as a supportive intravenous treatment for individuals with ARDS. ALS-886 is a small molecule therapeutic that, in preclinical studies, has demonstrated efficacy in both scavenging existing free radicals and sequestering free iron. The goal of treatment with ALS-886 for ARDS will be to reduce the tissue damage brought about by the free radicals in the lungs. Previous animal toxicology studies showed evidence that ALS-886 is safe and has a benign side effect profile. This could prove beneficial in the use of the compound in the critical care setting. In addition, the mild safety profile shown in animal studies supports the possibility that ALS-886 could be used as a prophylactic. That is, it could be administered to individuals who are at high risk of developing ARDS, such as patients suffering from sepsis or severe trauma, in order to prevent the underlying condition from leading to ARDS.
Mechanism of Action. The mechanisms of action for ALS-886 include:
- •
- Oxygen radical scavenging. A neutrophil cell is an especially abundant type of granular white blood cell that is highly destructive to microorganisms. In healthy individuals, neutrophils play an important role in the immune system. However, it is believed that neutrophil cells can become sequestered in the lungs among patients at risk for ARDS. Certain free radical byproducts of these activated and sequestered neutrophils can potentially produce the type of tissue damage observed in ARDS. ALS-886 has shown indications of effectiveness as an oxygen radical scavenger that can reduce or eliminate the tissue-damaging radicals created by neutrophils.
- •
- Iron chelation. It has been suggested that the presence of iron in lung surface lining fluid renders the surrounding tissues susceptible to increased oxidative stress. Thus, sequestering excess iron may result in a reduction in tissue damage due to the reduction in free radical production. ALS-886 has been known to be an iron chelator, thereby reducing the availability of free iron.
Preclinical Results. In three different animal models of lung injury, each resulting in damage to the lungs similar to that seen in human ARDS, ALS-886 was effective in reducing the increases in lung leakage commonly seen. Conscious sheep were administered LPS, a bacterial product that results in lung tissue damage. In these sheep, the co-administration of ALS-886 significantly reduced the leakage of the lung vasculature. This leakage was quantified by measuring the concentration of protein present in the lung lymph fluid. Increased protein concentration serves as an indicator of increased lung leakage.
52
Lung lymph protein clearance in conscious sheep administered LPS
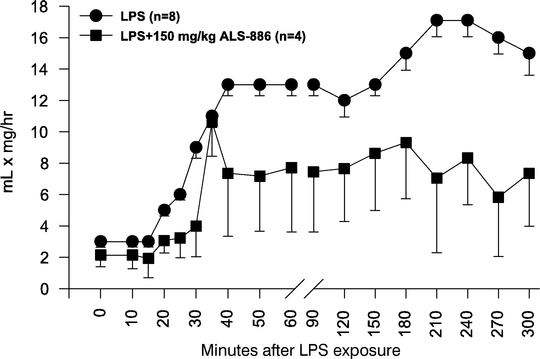
Similarly, in guinea pigs treated with aerosolized LPS, ALS-886 administration significantly diminished the leakage of the lung as determined by measuring the protein concentration of fluid removed from the lungs. ALS-886 was also successful in reducing the vascular permeability changes in a rat model of sepsis, or overwhelming bacterial infection, commonly seen in humans as a precursor to ARDS. Taken as a whole, the results of animal studies showed evidence that ALS-886 exerted a protective effect against the lung tissue damage caused by oxygen radicals. We have not conducted any clinical trials of ALS-886. Future clinical trial results may vary from those found in earlier preclinical studies.
Phase I Clinical Trial Plans. We received approval from the FDA to initiate human clinical trials with ALS-886. The initial protocol will evaluate the safety and tolerability of ALS-886 when administered as a continuous IV infusion in single doses to healthy volunteers. The study will involve four groups of ten healthy subjects each. A single continuous IV infusion of 10 mg/kg, 20 mg/kg and 40 mg/kg of ALS-886, or matching placebo (normal saline), will be administered respectively in the first three single dose cohorts. After completing enrollment for the first three cohorts, a fourth cohort will be enrolled at a dosage level to be determined on the basis of safety and pharmacokinetic results from the initial three single-dose cohorts. Clinical laboratory tests to be completed in connection with this Phase I clinical trial include hematology, blood chemistry and urinalysis as well as physical examination findings, 12-lead ECG results, vital signs and the reporting of adverse events. Serial blood sampling will be done to determine human pharmacokinetic properties of ALS-886.
In addition to studies in the treatment of ARDS, we intend to expand our future preclinical development work for ALS-886 to other indications characterized by tissue damage. For example, we have been awarded grant funding by the National Institute of Neurological Disorders and Stroke to pursue studies investigating the use of ALS-886 in the prevention of tissue damage following ischemic stroke.
Other Opportunities in Inflammation Treatment
We are developing therapies to treat Alzheimer's disease, which is also associated with inflammation. We are currently applying for additional grant funding to continue the preclinical
53
development of ALS-633, a product candidate we believe holds promise in the treatment of Alzheimer's disease and other serious medical conditions. ALS-633 is believed to inhibit the formation of amyloid fibrils, which are protein clusters that have been implicated in many serious diseases such as Alzheimer's disease, Parkinson's disease and amyotrophic lateral sclerosis (Lou Gehrig's disease).
While it is not known that these various diseases are caused by the formation of amyloid fibrils, they are each characterized by the assembly of proteins into amyloid fibrils, which are shown to crisscross the brain in affected patients. ALS-663 has been shown in the laboratory to prevent these proteins from coming together as amyloid fibrils, and we hope that this may prevent or reverse the progress of degenerative diseases such as Alzheimer's disease. We intend to continue to explore the development of ALS-633 and its possible use as a treatment for Alzheimer's disease and other serious medical conditions that may be caused by the presence of amyloid fibrils. In addition to ALS-633, we are also studying ALS-499 as a possible treatment for Alzheimer's disease.
Oncology
Melanoma
Overview
ALS-357 is a natural product we are developing that has demonstrated anti-tumor activity against malignant melanoma. ALS-357 has shown promise in bothin vivo andin vitro preclinical studies. In preclinical animal studies, ALS-357 concentrated in tumors, resulting in rapid tumor regression with no observable toxicity to the animal even at high doses. In addition, preclinical studies showed evidence that direct injection of ALS-357 into grafted human tumors induced apoptosis, or programmed cell death, within the tumors. We have an exclusive worldwide license from the University of Illinois at Chicago, or UIC, to develop and commercialize ALS-357 for the treatment of malignant melanoma. Preclinical development of ALS-357 is complete and we received approval from the FDA to enter Phase I clinical trials in 2005.
Market Overview
Melanoma is estimated to be the sixth most common cancer among new cases of cancer in the United States in 2005. Melanoma occurs when melanocytes (pigment cells) become malignant. Most commonly, melanoma originates in the skin or the eye. The chance of developing melanoma increases with age. Melanoma accounts for about 4% of all skin cancers but causes about 79% of skin cancer deaths.
The American Cancer Society estimates that, in 2004, approximately 55,000 new cases of melanoma were diagnosed in the United States and an estimated 7,910 deaths occurred as a result of melanoma. According to the American Cancer Society, survival is highly correlated to disease stage, with Stage I (tumor still in epidermis) patients having a 90% ten-year survival rate and Stage IV (tumor reaches lower part of dermis) patients having only a 10% ten-year survival rate. If detected early, melanoma can be treated successfully through surgical removal. If surgical removal of the melanoma is not an option or is ineffective, chemotherapeutics, biologic drugs or radiation therapies are often used to treat melanoma patients. The most effective non-surgical treatments currently used include bio-immunotherapy and combination therapy.
Current Treatment Options and Limitations
Surgical removal of early primary melanoma is often sufficient as a long-term cure. However, metastatic melanoma, which occurs after the cancerous cells spread from the primary growth area, remains resistant to most of the currently available forms of therapy and in most instances results in the death of the patient.
54
Numerous combination therapy regimens have been tested in metastatic melanoma. Although some combination therapy regimens, like the Dartmouth regimen (dacarbazine, cisplatin, carmustine and tamoxifen) provide enhanced anti-tumor response, they have yet to demonstrate an effect on prolonging disease-free or overall survival. Radiation therapy and certain drugs currently under development, including vaccines, bio-immunotherapy, monoclonal antibodies, gene-based therapies and cell signaling modifiers may also be used to treat melanoma, but they have not had a demonstrable effect at prolonging survival. There have been no significant improvements in survival rates for patients with advanced melanoma in the past 30 years.
ALS Solution
ALS-357 is a natural product that has shown specific anti-tumor activity in animal models of malignant melanoma. We have an exclusive worldwide license from UIC to develop and commercialize ALS-357 for the treatment of malignant melanoma. Scientists at UIC evaluated the potential anti-tumor activity of several thousand extracts derived from plants collected globally. One of such extracts displayed selective cytotoxicity against cultured human melanoma cells. This cytotoxicity was determined to be mediated through the induction of apoptosis. ALS-357 was isolated from the plant extract and identified through this process as an active anti-cancer compound. ALS-357 is available in abundance from natural sources such as the bark of the common white birch tree.
Preclinical testing of ALS-357 in animal models showed evidence that it has activity against apoptosis-resistant melanoma tumors. When administered by injection into mice bearing human melanoma tumors, ALS-357 concentrated in the tumors, resulting in rapid tumor regression with no observable toxicity to the animal even at high doses. Moreover, the mechanism of action induced cell death within the tumorsin vivo. Preclinical development of ALS-357 is complete, and in January 2005 we received approval from the FDA to enter Phase I clinical trials.
Mechanism of Action. ALS-357 acts by causing the cell to undergo a process called apoptosis. This process is initiated by at least two independent pathways in the cell. One pathway functions through signaling molecules located on the cell surface, while the other functions through changes in the membrane of the mitochondria, a structure inside the cell responsible for energy production. ALS-357 induces these changes in the mitochondria, which result in the activation of the apoptosis pathway. The precise pathway that is activated by ALS-357 functions independently of a number of proteins known to be altered in cancer cells, allowing for activation of the apoptotic program in tumors normally resistant to induction of programmed cell death.
Preclinical Results Showing In Vitro Activity. The cytotoxic potential of ALS-357 was tested against a battery of cell lines derived from a wide variety of human cancers, including multiple melanoma lines established at UIC. In these experiments, ALS-357 demonstrated activity at doses ranging from 1 to 10mg/ml. Further experiments verified that the cytotoxic activity was mediated through the induction of CD95- and p53-independent apoptosis.In vitro results showed evidence that ALS-357 has activity against a variety of melanoma cell lines and is able to induce apoptosis successfully.
Efficacy in Animal Studies. Based on the activity demonstrated by ALS-357 in cell culture, the potential of the agent to serve as an anti-cancer drug was assessed within vivo model systems. In initial studies, mice injected with human melanoma cells were treated with ALS-357 twenty-four hours following tumor injection. ALS-357 demonstrated activity at each dose injected (50, 250 and 500 mg/kg).
In a second series of experiments, mice were again injected with human melanoma cells; however, initiation of treatment of ALS-357 was delayed for two weeks to allow the tumors to grow and establish. ALS-357 was then administered every three days for a total of six treatments, and tumor size was measured twice per week. As illustrated in the graph below entitled "Inhibition of the growth of established tumors by ALS-357 in mice," ALS-357 significantly inhibited the growth of these established
55
tumors relative to an untreated, negative control group. Although dacarbazine, or DTIC, is a known anti-tumor agent for melanoma cells, ALS-357 was a significantly better chemotherapeutic agent against melanoma.
Inhibition of the growth of established tumors by ALS-357 in mice
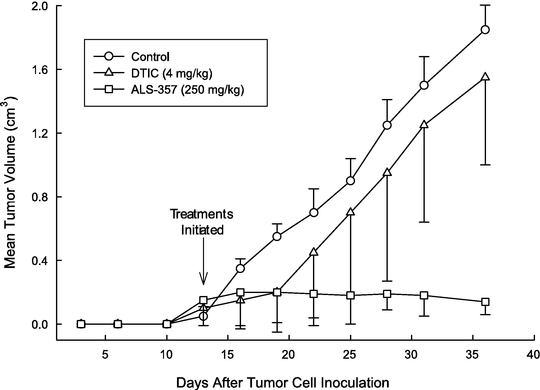
In order to evaluate the effects of ALS-357 on existing tumors, we conducted an additional test with control animals that did not receive any therapeutic agent for the first forty days following the tumor-cell injection. These mice had large tumor volumes by day forty. Following six doses of 50 mg/kg ALS-357 administered between days 41 and 59, the tumor volume was reduced in all mice by approximately 80%, further demonstrating the effectiveness of ALS-357 in reducing established tumors.
Topical Treatment of Human Melanoma Using ALS-357. In experiments designed to determine the efficacy of ALS-357 when administered topically as a cream, human skin was grafted onto mice. Human melanoma cells were then injected into the human skin graft and tumors were allowed to form and establish on the mice. ALS-357 was administered topically as a cream once a day for thirty days. After thirty days, treatment with ALS-357 resulted in complete regression of the melanoma tumors in all treated animals. Results from this experiment led to the preparation and submission of an IND application to the FDA for the topical treatment of melanoma in human subjects. We have not conducted any clinical trials of ALS-886. Future clinical trial results may vary from those found in earlier preclinical studies.
Future Clinical Studies. The FDA has approved a Phase I protocol that would examine the efficacy of topical application of ALS-357 in the treatment of patients with in-transit metastatic melanoma. These individuals have melanoma that has metastasized from the primary tumor site and re-established back to the skin. Often, the affected areas of re-establishment involve entire limbs or the entire trunk, preventing surgical excision. While this type of metastatic disease carries a poor prognosis, ALS-357 will be tested as a palliative treatment in these patients in an attempt to reduce suffering and prolong a reasonable quality of life. In addition, data obtained from both macroscopic and microscopic evaluations of efficacy may be important in demonstrating the effectiveness of ALS-357 in the
56
treatment of melanoma. Sixteen patients will be studied in a dose-escalating protocol design, which we anticipate can be completed over a period of nine months.
Ongoing Laboratory Studies. Laboratory investigations are underway to develop ALS-357 as a systemic treatment for metastatic melanoma. Studies are focused on obtaining a viable clinical IV formulation that could be utilized in the treatment of metastatic disease inaccessible to topical therapy. In addition, treatment of other tumor types of the same cellular origin as melanomas, such as brain malignancies, has been successfulin vitro and in clinical isolates. Further studies are underway to examine the feasibility of expanding the use of ALS-357 for the treatment of these tumor types. Finally, we are pursuing an active ALS-357 analogue design and testing program in an effort to identify novel molecules that may display improved potency and enhanced biopharmaceutical properties.
Additional Opportunities in Oncology
We are currently developing ALS-423, a novel cancer therapeutic, through preclinical testing. This proprietary product belongs to a class of indolocarbazoles and has demonstratedin vitro potency and efficacy. It is a Topoisomerase I (Topo I) inhibitor that may reduce the serious side effects observed with currently marketed Topo I inhibitors. Topo I is a protein that represents a promising target for the development of new cancer chemotherapeutic agents against a number of solid tumors. Development of anti-Topo I agents offers a new approach to the multi-regimental arsenal of therapies currently used in the clinic for the treatment of cancer. The best characterized Topo I inhibitors are camptothecin, or CPT, and its analogues. Topotecan (Hycamtin®) and CPT-11 (Camptosar®) are the only Topo I drugs approved for clinical use, and a significant need exists for new drugs that both improve the potency of the first generation Topo I inhibitors and/or minimize the drug-associated side effects.
Drug Discovery and Development Capabilities
We intend to expand our product portfolio by exploiting and enhancing our internal drug discovery and development capabilities. Our drug discovery efforts are focused on natural products, structural biology, medicinal chemistry and targeted biological screening. The integration of chemistry and biology gives us the capability to design, optimize and evaluate high potential product candidates quickly and cost-effectively. Our chemistry capabilities include a strong emphasis on medicinal chemistry, the discipline that seeks to identify optimal product candidates through consideration of the biological effects of newly synthesized compounds. We then use our biological screening capabilities to obtain feedback regarding the potential of the compounds synthesized by our chemists.
We utilize our specialized biological screening capabilities in the areas of mitochondria-mediated apoptosis and Topo I in oncology and iron chelation and amyloid fibril formation in inflammation. Our apoptosis program is aimed at identifying novel inducers of apoptosis. Certain malignant conditions are especially resistant to the induction of apoptosis, and part of our apoptosis platform involves identifying these often difficult-to-treat conditions by targeting their specific biological idiosyncrasies. Our efforts to design and synthesize a number of promising small molecules aimed at the inhibition of Topo I, a key protein target in the treatment of cancer, have identified several potential therapeutic candidates in a new class of compounds for which we recently received an issued U.S. Patent. We also are leveraging our expertise in protein-protein interactions and medicinal chemistry to design and synthesize a number of promising small molecules aimed at the inhibition of amyloid fibril formation. Amyloid fibrils are implicated in many important diseases such as amyloidosis, Alzheimer's disease and diabetes. We have developed a high throughput screening assay to identify inhibitors of amyloid fibril formation and identified a new class of compounds effective in inhibiting the formation of amyloid fibrils. Medicinal chemistry techniques are being utilized in optimizing these leads into even more potent compounds.
57
Abbott Laboratories Collaboration
In December 2004, we entered into an agreement with Abbott Laboratories under which we acquired from Abbott a license to certain patent applications, patents and proprietary technology relating to cethromycin and ABT-210, a second-generation ketolide antibiotic product candidate in preclinical development. Under the terms of the license agreement, we have an exclusive worldwide license (excluding Japan) to develop and commercialize cethromycin. On August 1, 2005, we agreed to amend our license agreement with Abbott in order to terminate our in-license of ABT-210. The license for cethromycin includes rights under U.S. Patent No. 5,866,549 (for cethromycin) and foreign equivalents other than in Japan, and all related applications and patents that are entitled to benefit from the original filing of applications that issued as U.S. Patent No. 5,866,549 or their foreign equivalents. We intend to conduct and fund the remaining clinical development of cethromycin and commercialize the product for the treatment of mild-to-moderate community acquired pneumonia. In February 2005, Abbott transferred to us all of its interests in Investigational New Drug application No. 57,836 relating to cethromycin. Under the terms of our agreement, Abbott received 1,122,569 shares of our common stock and has the right to receive $11 million in cash. To date we have paid $3 million of this amount, and we are obligated to pay an additional $8 million on September 1, 2005, or, if earlier, five days following the closing of the offerings. The $8 million payment includes our purchase of the remaining cethromycin inventory from Abbott. The license agreement also requires us to make certain milestone payments. We owe Abbott $5 million on October 31, 2005, or, if earlier, upon our intiation of pivotal Phase III clinical trials for cethromycin. Concurrent with the shares we are offering by this prospectus in the initial public offering, we are also offering 428,571 shares of our common stock to Abbott Laboratories in exchange for reducing this milestone payment obligation from $5 million to $2 million. We will also owe Abbott $10 million if we submit an NDA and $30 million if and when the FDA approves the NDA. Pursuant to the August 1, 2005 amendment, we also agreed to pay to Abbott $2.5 million upon cethromycin reaching $200 million in aggregate net sales and $5 million upon it reaching $400 million in aggregate net sales. To date we have made no milestone payments under the license agreement. Finally, we were originally obligated to pay Abbott royalties, generally equal to 19.0% of net sales, for commercialized products on a country-by-country basis. In exchange for our agreement with Abbott on August 1, 2005 to terminate our in-license of ABT-210 and provide the $2.5 million and $5.0 million milestone payments, Abbott agreed to reduce its royalty rights for cethromycin if it is approved for commercialization. Under the new terms, we will owe Abbott royalty payments of 19.0% on the first $100 million of aggregate net sales of cethromycin, 18.0% on net sales once aggregate net sales exceed $100 million but are less than $200 million, and 17.0% on all net sales once aggregate net sales exceed $200 million.
The term of the agreement commenced on December 13, 2004 and continues until the expiration of the last patent licensed under the agreement, unless the agreement is otherwise terminated. The primary patent licensed under the agreement, used by us in connection with cethromycin, expires in the U.S. on September 4, 2016, and in most foreign countries or jurisdictions on September 2, 2017 and October 29, 2018, respectively, all subject to any term restoration that may be granted for the time necessary for regulatory approval in each respective jurisdiction. Upon the expiration of the license agreement, we maintain a non-exclusive, perpetual and irrevocable license to use Abbott's proprietary technology and other types of information directly related or used in connection with cethromycin and its manufacture into pharmaceutical products without any further payment obligations to Abbott, except for those payment obligations accruing prior to such expiration. The agreement may be terminated by either party on 30 days' notice if the other party ceases its business operations or if the other party passes a resolution or a court of competent jurisdiction makes an order for winding up its business. Either party may also terminate the agreement for material breach if not cured within 90 days of notice or if not cured within 30 days of notice if the breach relates to a payment provision.
We have the right to sublicense our rights under the agreement at our discretion. In addition, if we determine to co-market or co-distribute cethromycin, Abbott has a 90-day exclusive right of first negotiation to be the co-marketer, co-distributor or exclusive distributor. This exclusive right begins
58
when we notify Abbott of our marketing and/or distributing plans, and we have no further obligation to Abbott after the 90-day period of time elapses.
Other Collaborations and License Agreements
In addition to our collaboration with Abbott Laboratories, we have entered into a number of license agreements for intellectual property and other rights needed to develop our products.
Sarawak MediChem Pharmaceuticals, Inc. In 1996, we entered into a 50/50 joint venture with Craun Research Sdn. Bhd., acting as agent for the State Government of Sarawak, Malaysia, to finance the development of Calanolide A. This joint venture is a Delaware corporation named Sarawak MediChem Pharmaceuticals, Inc. Upon the formation of the joint venture, the Sarawak Government invested $9 million in equity and MediChem Life Sciences, Inc., which assigned its interest in Sarawak MediChem Pharmaceuticals to us, received its 50% equity interest in exchange for its contribution of intellectual property. Subsequent to the formation of the joint venture, the Sarawak Government issued $12 million in long-term debt to the joint venture. According to the terms of the debt instruments, $9 million of this debt is past due and $0.5 million and $2.5 million are due in the third and fourth quarters of 2005, respectively. As a result of this default in payment by the joint venture, the Sarawak Government has the option under the loan agreement to take over the control and management of the Calanolide A project or the affairs of the joint venture until such time as the loan is repaid in full. To date, the Sarawak Government has not pursued the collection of this debt or sought to control or manage the Calanolide A project. On July 15, 2005, the Sarawak Government exercised certain of its contractual rights by notifying the joint venture of its intention to appoint its representative as an officer of the joint venture for purposes of assuming control of the affairs and management of the joint venture effective immediately. We have initiated discussions with the Sarawak Government to determine the form in which their representative would assume control of the affairs and management of the joint venture and are complying with their request to provide access to the joint venture's accounting, financial and corporate records. The Sarawak Government also expressly reserved all contractual and legal remedies available in light of the default and indicated its intent to fully exercise its rights as a creditor of the joint venture. Ultimately, we believe that for the joint venture to continue, the terms of the debt must be amended to extend its maturity to a date after commercialization of Calanolide A.
We previously commenced discussions with the Sarawak Government to restructure and extend the existing $12 million debt and determine the source of financing for a Phase IIa clinical trial of Calanolide A. We have proposed to the Sarawak Government that, for the first time, we would jointly fund the next development phase of Calanolide A through future advances of debt or purchases of equity. We currently estimate that it would cost approximately $2 million to conduct a Phase IIa clinical trial of Calanolide A. We do not intend to expend any meaningful funds advancing the development of Calanolide A unless and until we restructure the joint venture's debt and reach a satisfactory agreement with the Sarawak Government to amend our joint venture relationship and establish our respective obligations regarding the future funding of the next phase of developing Calanolide A.
If we are able to restructure our joint venture relationship and determine to proceed with the Phase IIa clinical trial for Calanolide A, upon completion of this trial we will evaluate our strategic alternatives for the joint venture relationship and the future advancement of Calanolide A. We may determine to continue the program through our joint venture, independently or under a new joint venture arrangement. It is also possible that we will elect to transfer our rights in the compound to our current joint venture partner or an independent third party. Factors that we will consider in making this decision include the success of the Phase IIa clinical trial, the relative financial obligations of us and our joint venture partner and our capabilities at such time to continue development independently.
The term of the joint venture agreement continues indefinitely unless terminated by the written agreement of both parties not to proceed further with the development of Calanolide A. In the event only one of the parties wishes to discontinue development of Calanolide A, the other party has the
59
right, but not the obligation, to purchase the discontinuing party's equity interest in the joint venture for $9 million. If the other party does not choose to exercise its purchase right within six months of receiving written notice from the discontinuing party, then the joint venture is deemed mutually terminated. Upon such mutual termination, the joint venture agreement provides that any liabilities of the joint venture will be settled by the parties equally and all patents or licenses of the joint venture shall be reassigned back to us, including rights to clinical trials. We believe that the equal settlement of liabilities provision in the joint venture agreement was intended to recognize the 50/50 equity ownership of the joint venture and does not constitute our guaranty of the joint venture's indebtedness to the Sarawak Government. Consequently, if the joint venture is mutually terminated, the joint venture partners would work together to monetize the joint venture's assets for the purpose of satisfying its liabilities, which currently are solely comprised of indebtedness to a 50% partner, the Sarawak Government. In the event the joint venture's assets are insufficient to settle its liabilities, we do not believe that the equal settlement of liabilities provision obligates us to contribute funds to the joint venture.
In addition to the financing provided by our joint venture partner, the joint venture's product portfolio and discovery and preclinical programs have been funded through SBIR grants awarded by the National Institutes of Health. Sarawak MediChem Pharmaceuticals has been successful in being awarded over $1.5 million in SBIR funding to advance the calanolide analogue development pipeline. The joint venture is also building upon its relationship with the Sarawak Government by collaborating with the Sarawak Biodiversity Center in Malaysia to discover future drug leads for treating infectious diseases and cancer. To date, this drug discovery collaboration with the Sarawak Biodiversity Center has not produced any drug leads for future development.
National Institutes of Health. In 1996, our joint venture, Sarawak MediChem Pharmaceuticals, acquired an exclusive worldwide license, under patent rights and know-how controlled by the National Institutes of Health (NIH), to develop, make, use and sell Calanolide A and related compounds to treat HIV and other viral infections. The license was originally entered by MediChem Research, Inc., and MediChem subsequently assigned the license to our joint venture. In consideration of this license, our joint venture is obligated to make up to $350,000 in aggregate milestone payments upon the achievement of various development and commercialization milestones. To date, the joint venture has not been required to make any milestone payments. MediChem made an up-front payment of $20,000 to NIH. Since our inception in 1999, the joint venture has paid NIH approximately $133,000 for the reimbursement of patent maintenance expenses, as required by the license agreement. In addition, the joint venture agreed to pay royalties to NIH based on net sales of licensed products. Even if there are no sales of licensed products, the joint venture is obligated to pay NIH a minimum annual royalty of approximately $6,700. Minimum annual royalties paid by the joint venture to NIH since our inception in 1999 are approximately $33,000. The license from NIH is subject to certain reserved rights for the U.S. Government, including the Government's right to develop, make, use and sell Calanolide A pursuant to any treaties or agreements with foreign governments that the U.S. Government may enter, and the Government's rights to require us to sublicense the rights granted in the interest of public health and safety. This license may be terminated by NIH if NIH determines we have not achieved certain benchmarks for drug development or we cannot reasonably satisfy unmet health and safety needs of the public.
University of Illinois at Chicago. In 1999, we acquired an exclusive worldwide license, under patent rights and know how controlled by UIC, to develop, make, use and sell ALS-357 and related compounds to treat melanoma and other forms of cancer. In consideration for this license, we paid an upfront license fee of $15,000 upon the execution of the agreement. We are also obligated to make up to $135,000 in aggregate milestone payments upon the achievement of various development and commercialization milestones for each of ALS-357 and any other compound developed by us under the licensed technology. To date, we have paid $10,000 in milestone payments. Under the terms of the agreement, we are obligated to reimburse UIC for past patent preparation, filing and prosecution expenses, and have agreed to reimburse UIC for similar expenses related to foreign patents throughout
60
the term of the agreement. To date we have paid UIC $233,000 for patent reimbursement and we are currently obligated to pay an additional $65,000 during the fiscal year ended December 31, 2005. In addition, we agreed to pay royalties to UIC based on net sales of licensed products by us, our affiliates and sublicensees, as well as a percentage of all other income we receive from sublicensees, but in any event, a minimum royalty of $5,000 annually once commercial sales commence.
Argonne National Laboratory. In 2003, we acquired an exclusive license, under patent rights and know-how controlled by Argonne National Laboratory, to develop, make, use and sell in the United States and its territories a class of peptides and related compounds to inhibit the formation of amyloid fibrils. These fibrils are implicated in a number of diseases such as Alzheimer's disease and amyloidosis. In consideration of this license, we agreed to pay royalties to Argonne equal to 4% of net sales of licensed products by us, our affiliates and sublicensees, as well as 10% of any benchmark or milestone fees we receive from sublicensees. We also agreed to reimburse Argonne for the cost of ongoing patent prosecution and maintenance. The license is subject to certain reserved rights for the U.S. Government based on Federal statutes, including the Government's right to develop, make, use, and sell the amyloid inhibiting peptides pursuant to any treaties or agreements with foreign governments that the U.S. Government may enter, and the Government's right to require us to sublicense the rights granted in certain limited circumstances. We are obligated to provide at least one half-time employee to this project until completion of Phase I clinical trials. Argonne has the right to terminate the agreement if due diligence requirements, including the devotion of one half-time employee, are not met.
Baxter International. As part of our spin-off from MediChem Life Sciences in 1999, we obtained the entire right, title and interest to certain patented inventions relating to ALS-886 that were assigned by Baxter International Inc. to Dr. Michael T. Flavin. We have assumed the obligation of Dr. Flavin to pay Baxter, as a payment for the assignment, 3% of net sales of ALS-886. Further, we are also obliged to share with Baxter 50% of the sublicensing fees we collect, other than royalties. In addition, if we sell the ongoing business of making, using or selling ALS-886, we are obligated to pay Baxter 50% of the fair market value of the right or license to manufacture, use or sell ALS-886 that is conveyed as part of the sale.
Intellectual Property
Patents and Trade Secrets
We have assembled a broad intellectual property portfolio encompassing the use, methods of preparation and methods of manufacture for our product candidates in the areas of infectious disease, inflammation and cancer. Together with our joint venture, we currently have exclusive access to 57 issued U.S. and international patents. In addition, 47 U.S. and international patent applications have been filed and are in various stages of processing.
Key U.S. Patents and Expiration Dates
| Drug Candidate | U.S. Patent Number | Expiration Date | ||
|---|---|---|---|---|
| Cethromycin | 5866549 | 9/4/2016 | ||
| Calanolide A | 5591770 | 1/7/2014 | ||
| ALS-886 | 5504111 | 4/2/2013 | ||
| ALS-357 | 5658947 | 8/19/2014 |
The patent positions of companies like ours are generally uncertain and involve complex legal and factual questions. Our ability to maintain and solidify our proprietary position for our technology will depend on our success in obtaining effective claims and enforcing those claims once granted. We do not know whether any of our patent applications or those patent applications that we license will result in the issuance of any patents. Our issued patents and those that may issue in the future, or those licensed to us, may be challenged, invalidated or circumvented, which could limit our ability to stop competitors from marketing related products or the length of term of patent protection that we may have for our products. In addition, there can be no assurance that this patent coverage will be broad
61
enough to prevent third parties from developing or commercializing similar or identical technologies and thus the rights granted under any issued patents may not provide us with any meaningful competitive advantages against our competitors. Furthermore, because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before any of our products can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantage of the patent. There can also be no assurance that our technologies will not be deemed to infringe the IP rights of third parties or that we will be able to acquire licenses to the IP rights of third parties under satisfactory terms or at all.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. We face competition from many different sources, including commercial pharmaceutical and biotechnology enterprises, academic institutions, government agencies and private and public research institutions. We believe that our most significant competitors are Oscient Pharmaceuticals, Aventis, Pfizer, GlaxoSmithKline and Johnson & Johnson.
Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, clinical trials, regulatory approvals and marketing approved products than we do. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. Our commercial opportunity will be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects or are less expensive than any products that we may develop. These third parties compete with us in recruiting and retaining qualified scientific and management personnel, establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies and technology licenses complementary to our programs or advantageous to our business.
We rely upon our collaborators for support in advancing certain of our product candidates and intend to rely on our collaborators for the commercialization of these products. Our collaborators may be conducting multiple product development efforts within the same disease areas that are the subjects of their agreements with us. Generally, our agreements with our collaborators do not preclude them from pursuing development efforts using a different approach from that which is the subject of our agreement with them. Therefore, any of our product candidates may be subject to competition with a product candidate under development by a collaborator.
There are also a number of companies working to develop new drugs and other therapies for these diseases that are undergoing clinical trials. The key competitive factors affecting the success of all of our product candidates are likely to be their efficacy, safety, price and convenience. See "Risk Factors—We will face significant competition from other biotechnology and pharmaceutical companies, and our operating results will suffer if we fail to compete effectively."
Government Regulation
Government authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, labeling, promotion, advertising, distribution, marketing and export and import of pharmaceutical products such as those we are developing. The process of obtaining regulatory approvals and the subsequent substantial compliance with appropriate federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
U.S. Government Regulation
In the United States, the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act and implementing regulations. If we fail to comply with the applicable United States requirements at
62
any time during the product development process, approval process or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include the FDA's refusal to approve pending applications, withdrawal of an approval, clinical holds, warning letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution. Any agency enforcement action could have a material adverse effect on us.
The steps required before a drug may be marketed in the United States include:
- •
- preclinical laboratory tests, animal studies and formulation studies under the FDA's good laboratory practices regulations;
- •
- submission to the FDA of an IND application for human clinical testing, which must become effective before human clinical trials may begin;
- •
- adequate and well-controlled clinical trials in accordance with FDA good clinical practice regulations, to establish the safety and efficacy of the product for each indication;
- •
- submission to the FDA of an NDA;
- •
- satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product is produced to assess compliance with current good manufacturing practices, or cGMP; and
- •
- FDA review and approval of the NDA.
Preclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. The IND must become effective before human clinical trials may begin. An IND will automatically become effective 30 days after receipt by the FDA, unless before that time the FDA raises concerns or questions about issues such as the conduct of the trials as outlined in the IND. In that case, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed. If these issues are unresolved, the FDA may not allow the clinical trials to commence.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators. Clinical trials are conducted under protocols detailing, among other things, the objectives of the study, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol must be submitted to the FDA as part of the IND.
Clinical trials typically are conducted in three sequential phases, but the phases may overlap or be combined. Each trial must be reviewed and approved by an independent Institutional Review Board before it can begin. Phase I clinical trials usually involve the initial introduction of the investigational drug into humans to evaluate the product's safety, dosage tolerance and pharmacodynamics and, if possible, to gain an early indication of its effectiveness.
Phase II clinical trials usually involve controlled trials in a limited patient population to:
- •
- evaluate dosage tolerance and appropriate dosage;
- •
- identify possible adverse effects and safety risks; and
- •
- evaluate preliminarily the efficacy of the drug for specific indications.
Phase III clinical trials usually further evaluate clinical efficacy and test further for safety in an expanded patient population. Phase I, Phase II and Phase III clinical trials may not be completed successfully within any specified period, if at all. Furthermore, the FDA or we may suspend or terminate clinical trials at any time on various grounds, including a finding that the subjects or patients are being exposed to an unacceptable health risk.
Assuming successful completion of the required clinical testing, the results of the preclinical studies and of the clinical studies, together with other detailed information, including information on the manufacture and composition of the product, are submitted to the FDA in the form of an NDA
63
requesting approval to market the product for one or more indications. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and has a favorable risk/benefit profile.
Under the Pediatric Research Equity Act of 2003, or PREA, NDAs or supplements to NDAs must contain data to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the drug is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. In most cases, the NDA must be accompanied by a substantial user fee.
Before approving an application, the FDA will inspect the facility or the facilities where the product is manufactured. The FDA will not approve the product unless cGMP compliance is considered satisfactory. The FDA will issue an approval letter if it determines that the application, manufacturing process and manufacturing facilities are acceptable. If the FDA determines the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all. We may encounter difficulties or unanticipated costs in our efforts to secure necessary governmental approvals, which could delay or preclude us from marketing our products. The FDA may limit the indications for use or place other conditions on any approvals that could restrict the commercial application of the products. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval.
After regulatory approval of a product is obtained, we are required to comply with a number of post-approval requirements. For example, as a condition of approval of an application, the FDA may require post-marketing testing and surveillance to monitor the product's safety or efficacy. In addition, holders of an approved NDA are required to report certain adverse reactions and production problems to the FDA, to provide updated safety and efficacy information and to comply with requirements concerning advertising and promotional labeling for their products. Also, quality control and manufacturing procedures must continue to conform to cGMP after approval. The FDA periodically inspects manufacturing facilities to assess compliance with cGMP, which imposes numerous procedural and documentation requirements. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other regulations.
We rely, and expect to continue to rely, on third parties for the production of clinical and commercial quantities of our product candidates. Future FDA inspections may identify compliance issues at our facilities or at the facilities of our contract manufacturers that may disrupt production or distribution, or require substantial resources to correct. In addition, discovery of problems with a product, or the failure to comply with requirements, may result in restrictions on a product, manufacturer or holder of an approved NDA, including withdrawal or recall of the product from the market or other voluntary or FDA-initiated action that could delay further marketing. Newly discovered or developed safety or efficacy data may require changes to a product's approved labeling, including the addition of new warnings and contraindications. Also, new government requirements may be established that could delay or prevent regulatory approval of our products under development.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sale and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable
64
regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Under European Union regulatory systems, we may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure, which is compulsory for medicines produced by biotechnology and optional for those which are highly innovative, provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessment report, each member state must decide whether to recognize approval. If a member state does not recognize the marketing authorization, the disputed points are eventually referred to the European Commission, whose decision is binding on all member states.
Pharmaceutical Pricing and Reimbursement
In both domestic and foreign markets, sales of any products for which we receive regulatory approval for commercial sale will depend in part on the availability of reimbursement from third party payors. Third party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. These third party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare product candidates. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. Our product candidates may not be considered cost-effective. Adequate third party reimbursement may not be available to enable us to maintain price levels sufficient to realize an appropriate return on our investment in product development.
In 2003, the United States government enacted legislation providing a partial prescription drug benefit for Medicare recipients, beginning in 2006. Government payment for some of the costs of prescription drugs may increase demand for any products for which we receive marketing approval. However, to obtain payments under this program, we would be required to sell products to Medicare recipients through drug procurement organizations operating pursuant to this legislation. These organizations would negotiate prices for our products, which are likely to be lower than we might otherwise obtain. Federal, state, and local governments in the United States continue to consider legislation to limit the growth of healthcare costs, including the cost of prescription drugs. Future legislation could limit payments for pharmaceuticals such as the product candidates that we are developing.
The marketability of any products for which we receive regulatory approval for commercial sale may suffer if the government and third party payors fail to provide adequate coverage and reimbursement. In addition, an increasing emphasis on managed care in the United States has increased and will continue to increase the pressure on pharmaceutical pricing.
Another development that may affect the pricing of drugs is proposed Congressional action regarding drug reimportation into the United States. Proposed legislation would allow the reimportation of approved drugs originally manufactured in the United States back into the United States from other countries where the drugs are sold at a lower price. If legislation or regulations are passed allowing the reimportation of drugs, they could reduce the price we receive for any products that we may develop, negatively affecting our revenues and prospects for profitability. Even without legislation authorizing reimportation, patients have been purchasing prescription drugs from Canadian and other non-United States sources, which has reduced the price received by pharmaceutical companies for their products.
65
Manufacturing
We produce small quantities of our compounds for research purposes. All of our compounds are small molecules and use industry standard processes and readily accessible raw materials. We intend to contract with qualified third parties for the manufacture of larger quantities of any investigational products for clinical trials or approved products for commercial sale. Further, we have identified multiple contract manufacturers for our products to minimize supply constraints and manage cost of goods.
Sales and Marketing
If we receive regulatory approval for our product candidates, we plan to commence commercialization activities by building a focused sales and marketing organization complemented by co-promotion and other arrangements with pharmaceutical or biotechnology collaborators. Our sales and marketing strategy is to:
- •
- Build our own domestic sales force. We believe that we can access high-prescribing physicians and specialists in the United States for a number of the product candidates that we are developing through a relatively small, specialized sales force.
- •
- Use contract sales organizations to sell our products in selected markets. We expect to use select contract sales organizations to help us access the general physician market to sell our products.
- •
- Recruit a marketing organization. We plan to build a marketing and sales management organization to create and implement marketing strategies for any products that we market through our own sales organization and to oversee and support our sales force. The responsibilities of the marketing organization would include developing educational initiatives with respect to approved products and establishing relationships with thought leaders in relevant fields of medicine.
- •
- Establish marketing and sales alliances. We plan to selectively enter into new strategic alliances with leading pharmaceutical and biotechnology companies to assist us in advancing our drug discovery and development programs. We plan to enter into commercialization agreements with partners to access geographic or therapeutic segments of the markets that our focused sales force cannot address. We also plan to retain U.S. marketing and sales rights or co-promotion rights for our product candidates for which we receive marketing approvals in situations in which we believe it is possible to access the market through a focused, specialized sales force.
Scientific and Clinical Advisors
We have established an independent scientific advisory board to obtain outside expertise in our scientific drug programs and scientific operations. We generally compensate our scientific advisors for their services with cash honorariums. The scientists on our advisory board are not employees, and they may have conflicting obligations that limit their involvement with us. Scientific advisory board members include:
Gordon R. Bernard, M.D., is Chief of Pulmonary Service and Professor of Medicine at Vanderbilt University School of Medicine. In addition, he is Medical Director for the Institutional Review Board. Dr. Bernard is a leader in the area of cardiopulmonary medicine and sepsis research and he has published more than 100 peer-reviewed articles on these topics. He chairs the Steering Committee for NHLBI Acute Respiratory Distress Syndrome Clinical Trials Network. Dr. Bernard received his M.D. degree from Louisiana State University. He serves on the editorial boards ofClinical Intensive Care,Journal of Critical Care andSepsis.
66
Robert W. Buckheit, Jr., Ph.D., is the Director of Infectious Disease and Immunology at TherImmune Research Corporation. Previously, he was the Director of the Microbiology Research Department at Southern Research Institute. Dr. Buckheit received a Ph.D. in Microbiology and Immunology from Duke University and completed a postdoctoral fellowship in Virology, Cell and Microbiology at the University of North Carolina. Dr. Buckheit has received numerous honors and awards in the areas of cancer and viral research.
Stan Bukofzer, M.D., is Divisional Vice President and Head of Global Medical Affairs of Abbott Laboratories, where he is responsible for health economics and outcomes, physician development, affiliate medical departments as well as Abbott's internal contract research organization. Dr. Bukofzer has also served as the head of Abbott's Anti-Infectives Venture and as the medical director of clinical research for Abbott International. Prior to joining Abbott, he practiced gastroenterology and was involved in research into molecular aspects of the hepatitis B virus.
Patricia Frank, Ph.D., is President of Patricia Frank & Associates, Inc., a company specializing in assisting the pharmaceutical industry in the management of the drug development process. Her expertise is in the design and implementation of safety studies, preparation of INDs, NDAs and other regulatory documents for submission to the FDA and worldwide regulatory agencies. She has been involved with over 100 drug candidates for 60 clients. Dr. Frank received a Ph.D. in Pharmacology from Northwestern University Medical School. Dr. Frank's publications include over 30 articles and book chapters, and she holds three patents. Her previous experience includes drug development at Lakeside Laboratories, Searle/Monsanto and Pitman-Moore.
James A. Longstreth, Ph.D., is the founder and President of Longstreth & Associates, Inc., a company specializing in assisting the pharmaceutical industry in the management of pharmacokinetic aspects of the drug development and life cycle management processes. His expertise is in the design, implementation and analysis of animal and human trials for the identification of the time course of critical indicators of a drug's presence and its performance. He is also an expert at integrating pharmacokinetic and pharmacodynamic findings into IND, NDA and supplemental New Drug Application filings with the FDA and other international regulatory bodies. Dr. Longstreth received his Ph.D. in Biomedical Engineering from The Johns Hopkins University and is an author on over 30 articles and book chapters. Dr. Longstreth's previous experience includes a faculty position at the University of North Carolina School of Pharmacy and employment at Searle.
Renslow D. Sherer, Jr., M.D., is Clinical Associate at the University of Chicago Hospitals. He was Director of Coordinated HIV Services at the CORE Center, Cook County Hospital. He has extensive experience and expertise in HIV primary care and in model care programs for women, chemical dependency, and the medically indigent. He has been active in HIV clinical research and in the design and implementation of clinical trials, in HIV prevention programs, and in local and federal policy on HIV disease. In September 2003, he became the Director of HIV/STI/TB for Project HOPE and he joined the faculty of the Section of Infectious Diseases at the University of Chicago. Dr. Sherer received an M.D. degree from Rush University and is a Diplomat of the American Board of Internal Medicine. Dr. Sherer has published extensively in the area of HIV and AIDS and has been a member of numerous committees, task forces and panels on AIDS. He is the recipient of the Albert Ebert award presented by the National Pharmacist Honor Society and a member of the Department of Health and Human Services AIDS Treatment Guideline Panel.
Stacy N. Suberg, Ph.D., is the founder of a regulatory consulting group, Research-Based Regulatory, Ltd., providing regulatory services in the areas of human drugs, biologics, medical devices and veterinary drugs. Clients include multi-billion dollar pharmaceutical companies, biotech start-ups, academic institutions, law firms and other life science companies. Dr. Suberg is also actively involved with the biologics group at the FDA with expertise in the area of cellular therapies. Dr. Suberg was
67
employed with Searle and Bristol-Myers prior to starting her own company. She received her Ph.D. in Physiology from the University of California-Davis.
Nicholas J. Vogelzang, M.D., is Director of the Nevada Cancer Institute. He was formerly Director of the University of Chicago Cancer Research Center, Professor of Medicine and Surgery (Urology) and the Buffet Chair in Genitourinary Oncology at the University of Chicago. Dr. Vogelzang is a leading researcher in the area of oncology. He received his M.D. from the University of Illinois at Chicago and completed a fellowship in Medical Oncology at the University of Minnesota. Dr. Vogelzang has served on the board of directors for the American Cancer Society and the American Society of Clinical Oncology, the Editorial Review Board forCancer Research and is a member of the Illinois State Legislative Committee of the American Association for Cancer Research. He has over 250 publications and is the senior editor of theComprehensive Textbook of Genitourinary Oncology.
Mark A. Wainberg, Ph.D., is the Director of Research of the Lady Davis Institute for Medical Research at the Jewish General Hospital in Montreal, Canada. He received a B.S. degree from McGill University and a Ph.D. degree from Columbia University. Dr. Wainberg has received numerous awards and honors and has published extensively in the areas of HIV and cancer. The antiviral activity of the HIV drug 3TC was first identified in Dr. Wainberg's laboratory. Dr. Wainberg was the President of the International AIDS Society between 1998-2000. Additionally, he is Professor and Director of the McGill AIDS Centre and Professor of Microbiology and Immunology at McGill University.
Herbert P. Wiedemann, M.D., is Chairman of the Department of Pulmonary, Allergy & Critical Care Medicine at The Cleveland Clinic. Dr. Wiedemann received his M.D. from Cornell University and completed a postdoctoral fellowship in pulmonary and critical care medicine at Yale University. Dr. Wiedemann has conducted extensive research in the area of ARDS and has been principal investigator for several clinical studies sponsored by major pharmaceutical companies. He serves on the editorial boards forCleveland Clinic Journal of Medicine,Clinical Pulmonary Medicine andEvidenced- Based On-Call.
Employees
We had 20 employees as of June 30, 2005, including 8 Ph.D.-level scientists. None of our employees are represented by labor unions or covered by collective bargaining agreements. We consider our relationship with our employees to be good.
Facilities
In September of 2003, we relocated our operations to our new Advanced Life Sciences Product Development Center in Woodridge, Illinois, which houses our corporate offices and chemistry and biology laboratories. The Product Development Center consists of approximately 15,000 square feet of laboratory and office space. We believe that our current facilities are adequate to meet our needs for the foreseeable future. We believe that suitable additional or alternative space will be available in the future on commercially reasonable terms as needed.
Legal Proceedings
We are not currently a party to any material legal proceedings.
68
Set forth below is the name, age, position and a brief account of the business experience of each of our executive officers, directors and key employees as of June 30, 2005.
| Name | Age | Position | ||
|---|---|---|---|---|
| Michael T. Flavin, Ph.D. | 49 | Chief Executive Officer and Chairman of the Board of Directors | ||
| John L. Flavin | 36 | President and Director | ||
| R. Richard Wieland II | 60 | Executive Vice President and Chief Financial Officer | ||
| Ze-Qi Xu, Ph.D. | 43 | Executive Vice President and Chief Scientific Officer | ||
| Suseelan Pookote, Ph.D. | 52 | Executive Vice President of Corporate Development | ||
| Patrick W. Flavin, J.D. | 30 | Chief Legal Counsel | ||
| David A. Eiznhamer, Ph.D. | 42 | Vice President of Biological Sciences | ||
| Terry W. Osborn, Ph.D.(2)(3) | 62 | Director | ||
| Israel Rubinstein, M.D.(1)(2) | 53 | Director | ||
| Rosalie Sagraves, Pharm.D.(1)(3) | 59 | Director | ||
| Thomas V. Thornton(2)(3) | 39 | Director |
- (1)
- Member of the compensation committee
- (2)
- Member of the audit committee
- (3)
- Member of the nominating and corporate governance committee
Michael T. Flavin, Ph.D., Chief Executive Officer and Chairman, founded Advanced Life Sciences, Inc. in 1999. Prior to this date, he was the founder, Chief Executive Officer and Chairman of MediChem Life Sciences, Inc., a drug discovery technology and services company. Dr. Flavin took MediChem from start-up in 1987 through many stages of development including the completion of MediChem's private placement, the acquisition and integration of ThermoGen and Emerald Biostructures as MediChem subsidiaries and MediChem's initial public offering in October 2000. MediChem was acquired in March 2002 by deCODE genetics. Dr. Flavin was the Chief Executive Officer of MediChem, as a subsidiary of deCODE, through May 2002. Dr. Flavin received a B.S. in Chemistry from the University of Notre Dame, a Ph.D. in Medicinal Chemistry from the University of Illinois at Chicago and he completed a postdoctoral fellowship at Harvard University. Dr. Flavin is an Adjunct Professor for the Department of Medicinal Chemistry & Pharmacognosy at the University of Illinois at Chicago. Dr. Flavin is the brother of John L. Flavin and Patrick W. Flavin.
John L. Flavin, President and Director, joined us in June 2002 as Executive Vice President and Chief Financial Officer. He was elected to our board of directors in 2003 and promoted to President in 2004. From May 2000 through May 2002, Mr. Flavin was the Chief Operating Officer and a director of MediChem Life Sciences, Inc. At MediChem, Mr. Flavin was responsible for developing and managing the business and scientific operations and was the Chief Executive Officer of its protein engineering and structural proteomics subsidiaries, Emerald BioStructures, ThermoGen and AXAS. Mr. Flavin holds a B.S. in Business Administration from Marquette University and an M.B.A. from Lewis University. Mr. Flavin is the brother of Michael T. Flavin, Ph.D. and Patrick W. Flavin.
R. Richard Wieland II, Executive Vice President and Chief Financial Officer, was appointed as our Executive Vice President and Chief Financial Officer in June 2004. From May 2000 to April 2002, Mr. Wieland served as Chief Financial Officer of MediChem Life Sciences, Inc., where he was
69
responsible for overseeing the financial aspects of acquiring ThermoGen and Emerald Biostructures as MediChem subsidiaries and assisting in the financial effort to complete MediChem's initial public offering. Mr. Wieland obtained his B.A. in Accounting and Economics from Monmouth College and his M.B.A. from Washington University.
Ze-Qi Xu, Ph.D., Executive Vice President and Chief Scientific Officer, joined us in October 2002 from MediChem Life Sciences, Inc. From January 2000 until September 2002, Dr. Xu was the Vice President of Strategic Drug Development at MediChem and oversaw the discovery, preclinical and clinical development of Calanolide A as well as the preclinical development of ALS-886 and ALS-357. Dr. Xu has published 52 articles and holds 19 patents in the fields of infectious disease and cancer. Dr. Xu earned a B.S. degree in Chemistry from Jiangxi Normal University (China), a M.S. degree in Organic Chemistry from Shanghai Medical University and a Ph.D. in Organic Chemistry from the Shanghai Institute of Organic Chemistry. He performed his postdoctoral fellowships at Clemson University and the Michigan Cancer Foundation.
Suseelan Pookote, Ph.D., Executive Vice President of Corporate Development, joined us in March 2001. Prior to joining us, Dr. Pookote worked at Monsanto from 1980 to 2000, where he had responsibility for technology and business development in a variety of industry segments such as food, nutrition, biotechnology and specialty chemicals. At Monsanto, Dr. Pookote concentrated on technology evaluation, licensing and corporate transactions, including the negotiation of technology licensing deals with academic institutions. Dr. Pookote received his Ph.D. in Chemical Engineering from Northwestern University.
Patrick W. Flavin, J.D., Chief Legal Counsel, joined us in November 2002 as legal counsel. He was promoted to Chief Legal Counsel in 2004. Prior to joining us, Mr. Flavin served as a counsel to the Illinois Speaker of the House from January 2002 to November 2002. Prior to this position, he was the Director of Legal Affairs for MediChem Life Sciences from May 2000 to September 2001, where he managed the in-house legal staff and outside legal counsel. Mr. Flavin obtained his B.A. from Providence College and received his J.D. from DePaul University College of Law. Mr. Flavin is the brother of Michael T. Flavin, Ph.D. and John L. Flavin.
David A. Eiznhamer, Ph.D., Vice President of Biological Sciences, joined us as Director of Biological Sciences in 2003 from MediChem Life Sciences, Inc., where he was the Assistant Director of Pharmacology and ADME from January 2002 to May 2003. Dr. Eiznhamer was promoted to Vice President of Biological Sciences in May 2004. In his role at MediChem, Dr. Eiznhamer was responsible for the design and development of pharmacology and ADME research programs. Prior to that, he served as the Manager of Regulatory Affairs for MediChem Life Sciences from December 1999 to January 2002. Dr. Eiznhamer also worked as a clinical coordinator for the Division of Gastroenterology at Loyola University Medical Center in Maywood, Illinois. He has authored seven publications and made 19 presentations at national and international meetings. Dr. Eiznhamer received his Ph.D. in Molecular Biology from Loyola University of Chicago.
Terry W. Osborn, Ph.D., Director, joined our board of directors in July 2001. Dr. Osborn is a pharmaceutical executive with significant experience in establishing strategic direction to develop sales and enhance profitability in both startup and turnaround environments. He is the President and CEO of Gene Express, Inc., a genomics company in Toledo, Ohio. From 1999 until July 2002 Dr. Osborn was the Chief Executive Officer of Pharmaceutical Development Center, a contract formulation, development and cGMP manufacturer of biopharmaceutical and pharmaceutical drugs. He was also the founder, President and Chief Executive Officer of Health Advance Institute, a National Clinical Research Organization providing clinical research services to pharmaceutical and biotechnology companies. Dr. Osborn received his Ph.D. in Biochemistry from the University of California at Riverside and his M.B.A. from Pepperdine University.
70
Israel Rubinstein, M.D., Director, joined our board of directors in May 2001. Dr. Rubinstein is Professor of Medicine and Biopharmaceutical Sciences in the Colleges of Medicine and Pharmacy at the University of Illinois at Chicago, where he has worked since 1993. Dr. Rubinstein is a leading physician and research scientist in respiratory medicine and oncology. He has served on the Editorial Boards of several medical journals and is grant reviewer for the NIH. Dr. Rubinstein received an M.D. degree from Hebrew University-Hadassah School of Medicine and completed fellowships in Respirology at the University of Toronto and in Cardiovascular Research at the University of California San Francisco.
Rosalie Sagraves, Pharm.D., Director, joined our board of directors in May 2001. Dr. Sagraves is Dean and Professor at the University of Illinois at Chicago, College of Pharmacy, where she has worked since 1995. Her practice area is pediatric critical care with educational/research interests in pediatric pharmacotherapeutics and maternal-child health. She has authored more than 60 journal articles and book chapters. She is a speaker on pediatric pharmacotherapy and women's health. She has served as Council of Deans chair for the American Association of Colleges of Pharmacy and is a member of AACP board of directors, the American Pharmaceutical Association, and the APhA Board of Trustees. She has also served as president of the Academy of Pharmaceutical Research and Science and as a member of the editorial board ofPharmacy Today. She is a fellow of APhA and of the American College of Clinical Pharmacy. She serves on the National Institutes of Health Office of Research on Women's Health Advisory Board. She has received awards for outstanding teaching and leadership. She graduated from the Ohio State University and the Philadelphia College of Pharmacy and Science.
Thomas V. Thornton, Director, joined our board of directors in June 2001. Mr. Thornton is a successful early-stage venture capital investor and recognized leader in the development of public/private technology development initiatives. From July 2001 to March 2002, Mr. Thornton was the Senior Vice President—Midwest Region for Convergent Technology Group, a mergers and acquisitions advisory services firm. From October 1999 to May 2001, Mr. Thornton was the Managing Partner for divine interVentures, Inc., a service and software company, and led seed- and early-stage venture investing teams that managed over $120 million and contributed to divine interVenture's initial public offering. Presently, Mr. Thornton is the president of the Illinois Technology Development Alliance, a public/private partnership established to strengthen Illinois' economy through science and technology. Mr. Thornton received a B.A. degree from the University of Wisconsin-Madison.
Board Composition and Election Of Directors
Our by-laws provide that the number of our directors shall be fixed from time to time by a resolution of the majority of our board of directors. We are authorized to have nine directors and we currently have six directors. Director vacancies and newly created directorships may be filled by the vote of a majority of the directors then in office. In accordance with the terms of our amended and restated certificate of incorporation and bylaws, the terms of office of the directors are divided into three classes:
- •
- Class I, whose term will expire at the annual meeting of stockholders to be held in 2006. The Class I directors are Drs. Terry W. Osborn and Israel Rubinstein.
- •
- Class II, whose term will expire at the annual meeting of stockholders to be held in 2007. The Class II directors are Dr. Rosalie Sagraves and Mr. John L. Flavin.
- •
- Class III, whose term will expire at the annual meeting of stockholders to be held in 2008. The Class III directors are Mr. Thomas V. Thornton and Dr. Michael T. Flavin.
At each annual meeting of stockholders after the initial classification, or special meeting in lieu thereof, the successors to directors whose terms will then expire will be elected to serve from the time
71
of election and qualification until the third annual meeting following election, or special meeting held in lieu thereof. Any additional directorships resulting from an increase in the number of directors will be distributed among the three classes so that, as nearly as possible, each class will consist of one third of the total number of directors. This classification of the board of directors may have the effect of delaying or preventing changes in the control or management of our company.
Board Committees
Our board of directors has an audit committee, a compensation committee and a nominating and corporate governance committee, each of which has the composition and responsibilities described below. The board may also establish other committees from time to time to assist in the discharge of its duties. We believe that our board of directors and the composition of its committees will meet the standards for independence under the current applicable requirements of the Sarbanes-Oxley Act of 2002, the Nasdaq National Market and SEC rules and regulations.
Audit Committee
The audit committee of the board of directors reviews and monitors our corporate financial reporting, our external audits, the results and scope of the annual audit, other services provided by our independent auditors and our compliance with legal matters that have a significant impact on our financial reports. The audit committee also consults with management and our independent auditors before the presentation of financial statements to stockholders and, as appropriate, initiates inquiries into aspects of our financial affairs. In addition, the audit committee has the responsibility to consider and recommend the appointment of, and to review fee arrangements with, our independent auditors. The current members of the audit committee are Dr. Israel Rubinstein, Dr. Terry W. Osborn and Mr. Thomas V. Thornton, each of whom is an independent director. Mr. Thornton is our audit committee financial expert under the SEC rule implementing Section 407 of the Sarbanes-Oxley Act of 2002. We believe that the functioning of our audit committee will comply with the applicable requirements of the Sarbanes-Oxley Act of 2002, the Nasdaq National Market and SEC rules and regulations.
Compensation Committee
The compensation committee of the board of directors reviews and makes recommendations to the board regarding all forms of compensation provided to our executive officers and directors, including stock compensation. In addition, the compensation committee reviews and makes recommendations on bonus and stock compensation arrangements for all of our employees. As part of these responsibilities, the compensation committee also administers our stock incentive plans. The current members of the compensation committee are Drs. Rosalie Sagraves and Israel Rubinstein, each of whom is an independent director.
Nominating and Corporate Governance Committee
Our nominating and corporate governance committee intends to develop and recommend to the board of directors a set of corporate governance principles and procedures applicable to us, identify individuals qualified to become board members and recommend the director nominees for each annual meeting of our stockholders. The current members of our nominating and corporate governance committee are Dr. Terry W. Osborn, Dr. Rosalie Sagraves and Mr. Thomas V. Thornton, each of whom is an independent director.
72
Compensation Committee Interlocks And Insider Participation
Our compensation committee currently consists of Drs. Israel Rubinstein and Rosalie Sagraves. No member of the compensation committee has been an officer or employee of ours at any time. None of our executive officers serves as a member of the board of directors or compensation committee of any other company that has one or more executive officers serving as a member of our board of directors or compensation committee. Prior to the formation of the compensation committee in 2004, the board of directors as a whole made decisions relating to compensation of our executive officers.
Director Compensation
Our directors who are also our employees do not receive any additonal compensation for their services as our directors. Following the offerings, we will pay each of our non-employee directors an annual fee of $40,000 for serving on our board. We pay an additional $15,000 annual fee to the chairman of our audit committee, and $10,000 to the chairpersons of our compensation and nominating and corporate governance committees. As of June 30, 2005, each non-employee, non-significant stockholder director had been issued 4,069 options under our 1999 Stock Incentive Plan. All of our directors who are not employees of the company are eligible to receive additional equity incentives in the form of stock option grants under our 2005 Stock Incentive Plan. Each new non-employee director that joins our board of directors will receive 15,000 options that vest over a three-year period under our 2005 Stock Incentive Plan. In addition, each non-employee director will annually receive 5,600 options with immediate vesting.
Certain of our directors have, from time to time, received compensation for consulting services provided to us in connection with our product development programs that are within such directors' areas of expertise. In the year ended December 31, 2004, we paid $4,375 to Dr. Rubinstein and $413 to Dr. Osborn in exchange for these services. Drs. Rubinstein and Osborn provided these consulting services prior to their appointment as members of our board committees and while we were a privately held corporation. Members of our audit committee and nominating and corporate governance committee are no longer eligible to receive fees for consulting or other advisory services beyond their director compensation. Our non-employee directors are also reimbursed for out-of-pocket expenses incurred in attending board and committee meetings.
Limitation of Liability and Indemnification of Officers and Directors
Our amended and restated certificate of incorporation and our bylaws provide that our directors and officers shall be indemnified by us to the fullest extent authorized by Delaware law, as it now exists or may in the future be amended, against all expenses and liabilities reasonably incurred in connection with their service for or on our behalf. In addition, our amended and restated certificate of incorporation provides that our directors will not be personally liable for monetary damages to us for breaches of their fiduciary duty as directors, unless they violated their duty of loyalty to us or to our stockholders, acted in bad faith, knowingly or intentionally violated the law, authorized illegal dividends or redemptions or derived an improper personal benefit from their action as directors. We have entered, and intend to continue to enter, into separate indemnification agreements with our directors and executive officers, in addition to the indemnification provided for in our amended and restated certificate of incorporation and our bylaws. These agreements, among other things, provide that we will indemnify our directors and executive officers for certain expenses, including attorneys' fees, judgments, fines and settlement amounts incurred by a director or executive officer in any action or proceeding arising out of their services as one of our directors or executive officers, or any of our subsidiaries or any other company or enterprise to which the person provides services at our request.
We have obtained insurance which insures our directors and officers against specified losses and which insures us against specific obligations to indemnify our directors and officers. At present, there is no pending litigation or proceeding involving any of our directors or executive officers as to which indemnification is required or permitted, and we are not aware of any threatened litigation or proceeding that may result in a claim for indemnification.
73
Executive Compensation
Summary Compensation Table
The following table sets forth the total compensation paid to or earned by our Chief Executive Officer and each of our four other most highly compensated executive officers for the fiscal year ended December 31, 2004, each of whom was serving as an executive officer on December 31, 2004. We refer to these individuals elsewhere in this prospectus as our "named executive officers."
| | | | | Long-Term Compensation Awards | | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | | Annual Compensation | | ||||||||||
| Name and principal position | | All Other Compensation(1) | |||||||||||
| Year | Salary | Bonus | Option Shares | ||||||||||
| Michael T. Flavin Chairman and Chief Executive Officer | 2004 2003 2002 | $ | 180,000 180,000 70,615 | $ | — 30,000 — | — — — | $ | 5,400 6,000 1,661 | |||||
John L. Flavin President | 2004 2003 2002 | 157,500 157,500 87,841 | — — — | 27,790 20,842 4,962 | 1,575 4,725 1,454 | ||||||||
R. Richard Wieland II(2) Executive Vice President and Chief Financial Officer | 2004 | 80,167 | — | 23,026 | 1,352 | ||||||||
Suseelan Pookote Executive Vice President of Corporate Development | 2004 2003 2002 | 150,000 150,000 148,944 | — — — | 3,970 — 41,685 | 4,500 4,500 4,500 | ||||||||
Ze-Qi Xu Chief Scientific Officer | 2004 2003 2002 | 130,000 130,000 27,500 | — 2,500 — | 3,970 — 9,925 | 3,899 3,825 — | ||||||||
- (1)
- Except for other compensation reported for Mr. Wieland, other compensation consists of matching payments made under our 401(k) employee savings plan.
- (2)
- Mr. Wieland began his employment with us in June 2004. Amounts reported as other compensation for Mr. Wieland consist primarily of reimbursed health insurance premiums and health club membership expenses.
Stock Option Grants in 2004
The following table sets forth information concerning stock options granted to each of the named executive officers during the fiscal year ended December 31, 2004. Each option represents the right to purchase one share of our common stock. We granted these options at an exercise price equal to the estimated fair value of the common stock on the date of grant as determined by our board of directors. There was no public market for our common stock on the grant dates of these options. Accordingly, we calculated the potential realizable value by multiplying the number of shares of our common stock subject to a given option by the initial public offering price of $7.00 per share. The potential realizable value at assumed annual rates of stock price appreciation for the option term represents hypothetical gains that could be achieved for the respective options if exercised at the end of the option term. The 5% and 10% assumed annual rates of compounded stock price appreciation are required by rules of the SEC and do not represent our estimate or projection of our future common stock prices. These amounts represent assumed rates of appreciation in the value of our common stock from the exercise price per share. Actual gains, if any, on stock option exercises are dependent on the future
74
performance of our common stock and overall stock market conditions. The amounts reflected in the table may not necessarily be achieved.
| | | | | | Potential Realizable Value at Assumed Annual Rates of Stock Price Appreciation for Option Term | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Number of Securities Underlying Options Granted | Percent of Total Options Granted to Employees in Fiscal Year | | | |||||||||||
| Name | Exercise Price Per Share | Expiration Date | |||||||||||||
| 5% | 10% | ||||||||||||||
| Michael T. Flavin | — | — | % | $ | — | — | — | — | |||||||
John L. Flavin | 27,790 | 25.2 | 0.157 | 6/1/2014 | $ | 243,912 | $ | 308,929 | |||||||
R. Richard Wieland II | 23,026 | 20.9 | 0.157 | 6/1/2014 | 202,099 | 255,970 | |||||||||
Suseelan Pookote | 3,970 | 3.6 | 0.157 | 6/1/2014 | 34,845 | 44,133 | |||||||||
Ze-Qi Xu | 3,970 | 3.6 | 0.157 | 6/1/2014 | 34,845 | 44,133 | |||||||||
Fiscal Year-End Option Values
The following table shows information concerning the number and value of unexercised options held by each of the named executive officers as of December 31, 2004. There were no options exercised by the named executive officers in 2004. There was no public trading market for the common stock as of December 31, 2004. Accordingly, the value of unexercised in-the-money options listed below has been calculated on the basis of an assumed initial public offering price of $7.00 per share, less the applicable exercise price per share, multiplied by the number of shares underlying such options.
| | Number of Shares of Common Stock Underlying Unexercised Options at Year End | Value of Unexercised In-the-Money Options at Year End | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|
| Name | ||||||||||
| Exercisable | Unexercisable | Exercisable | Unexercisable | |||||||
| Michael T. Flavin | — | — | — | — | ||||||
John L. Flavin | 40,085 | 29,389 | $ | 274,302 | $ | 201,109 | ||||
R. Richard Wieland II | 46,059 | 18,651 | 315,182 | 127,629 | ||||||
Suseelan Pookote | 42,439 | 3,215 | 290,410 | 22,000 | ||||||
Ze-Qi Xu | 42,439 | 3,215 | 290,410 | 22,000 | ||||||
Employment Contracts, Termination of Employment and Change-In-Control Arrangements
We have entered into employment agreements with each of our executive officers. Under the employment agreements, each executive officer has agreed not to compete with us and not to solicit our employees and customers during his employment and for one year after his employment terminates for any reason. Some agreements provide for the payment of salary for a maximum of 12 months after the termination of employment if the executive fails to attain other employment as a result of the non-compete provisions. Each of the executive officers has further agreed not to disclose confidential information or use such information for personal benefit, either during or after employment with us. The agreements give us full rights to any inventions of the executive officers made during or within 120 days after employment with us, unless the particular invention does not relate directly to our business and was not produced during company time or with company resources.
The employment agreement with Dr. Michael Flavin, our Chairman and Chief Executive Officer, provides that Dr. Flavin or we can terminate his employment at any time, with or without cause. The agreement also provides that if we terminate his employment without cause (as defined in the
75
agreement), Dr. Flavin is entitled to continuation of his then-current base salary and continuation of employee benefit coverage for 24 months following the termination of his employment. Dr. Flavin's stock options would also be exercisable for 24 months following the termination date. Alternatively, if within 24 months of the date on which we experience a change in control (as defined in the agreement) we terminate Dr. Flavin other than for cause, he will be entitled to a lump sum payment equal to 2 years of his base salary and we will continue employee benefit coverage for a period of 2 years. Upon a change of control, Dr. Flavin is also entitled to bonus compensation in accordance with the terms of our Annual Bonus Plan. In the event that Dr. Flavin is terminated or we experience a change in control, all debt owed by us to Dr. Flavin would come due.
The employment agreement with John L. Flavin, our President, provides that Mr. Flavin or we can terminate his employment at any time, with or without cause. The agreement also provides that in the event that we terminate his employment without cause, Mr. Flavin is entitled to continuation of his then-current base salary and continuation of employee benefits coverage for 9 months following the termination of his employment. If his employment is terminated without cause during the 24-month period after a change of control, Mr. Flavin is entitled to receive a lump sum payment generally equal to two times the sum of his base salary and target bonus for that fiscal year. During his term of employment, Mr. Flavin is eligible for bonus compensation under our Annual Bonus Plan and stock option and other equity-based incentive compensation under our 2005 Stock Incentive Plan.
The employment agreement with Suseelan Pookote, Ph.D., our Executive Vice President of Corporate Development, provides that Dr. Pookote or we can terminate his employment at any time, with or without cause. The agreement also provides that in the event that we terminate his employment without cause, Dr. Pookote is entitled to continuation of his then-current base salary and continuation of employee benefits coverage for 9 months following the termination of his employment. If his employment is terminated without cause during the 24-month period after a change of control, Dr. Pookote is entitled to receive a lump sum payment generally equal to two times the sum of his base salary and target bonus for that fiscal year. During his term of employment, Dr. Pookote is eligible for bonus compensation under our Annual Bonus Plan and stock option and other equity-based incentive compensation under our 2005 Stock Incentive Plan.
The employment agreement with Ze-Qi Xu, Ph.D., our Chief Scientific Officer, provides that Dr. Xu or we can terminate his employment at any time, with or without cause. The agreement also provides that in the event that we terminate his employment without cause, Dr. Xu is entitled to continuation of his then-current base salary and continuation of employee benefits coverage for 6 months following the termination of his employment. If his employment is terminated without cause during the 24-month period after a change of control, Dr. Xu is entitled to receive a lump sum payment generally equal to two times the sum of his base salary and target bonus for that fiscal year. During his term of employment, Dr. Xu is also eligible for bonus compensation under our Annual Bonus Plan and stock option and other equity-based incentive compensation under our 2005 Stock Incentive Plan.
The employment agreement with R. Richard Wieland II, our Executive Vice President and Chief Financial Officer, provides that Mr. Wieland or we can terminate his employment at any time, with or without cause. The agreement also provides that in the event that we terminate his employment without cause, Mr. Wieland is entitled to continuation of his then-current base salary and continuation of employee benefits coverage for 6 months following the termination of his employment. If his employment is terminated without cause during the 24-month period after a change of control, Mr. Wieland is entitled to receive a lump sum payment generally equal to two times the sum of his base salary and target bonus for that fiscal year. During his term of employment, Mr. Wieland is eligible for bonus compensation under our Annual Bonus Plan and stock option and other equity-based incentive compensation under our 2005 Stock Incentive Plan.
76
Stock Option and Other Compensation Plans
1999 Stock Incentive Plan
In December 2004, in connection with our recapitalization, our board of directors adopted the 1999 Stock Incentive Plan of Advanced Life Sciences, Inc. and assumed all options granted thereunder. The compensation committee of our board of directors administers the plan. As of June 30, 2005, we had 686,837 options to purchase shares of our common stock outstanding to employees, directors and consultants under the plan, at a weighted average exercise price of $0.93 per share. We issued 110,364 of these options in the fiscal year ended December 31, 2004, at an exercise price of $0.157 per share. We issued 67,290 options under the plan in the first quarter of 2005, at an exercise price of $8.02 per share. According to the terms of the grant agreements for awards under the 1999 Stock Incentive Plan, all unvested options currently outstanding will vest automatically upon the closing of the offerings. There will be no further options issued under the 1999 Stock Incentive Plan.
2005 Stock Incentive Plan
In April 2005, our board of directors adopted the 2005 Stock Incentive Plan. The 2005 Stock Inventive Plan permits awards of nonqualified stock options, incentive stock options, stock appreciation rights, restricted stock, restricted stock units, and performance shares. In addition, the plan provides an opportunity for the deferral of salary and bonuses into restricted stock units and the deferral of gains or payments due upon the vesting or exercise of awards under our stock incentive plan.
Term of plan. Awards may be made under our 2005 Stock Incentive Plan until the earliest of:
- •
- the date when all of the shares reserved for issuance under the plan have been exhausted;
- •
- April 21, 2015, which is the tenth anniversary of the date on which our board of directors adopted the plan; and
- •
- the date as of which our compensation committee terminates the plan.
Shares subject to the plan. Subject to the adjustment provisions discussed below, the maximum number of shares of common stock that may be subject to awards under our stock incentive plan will be 2,072,455. Any shares subject to an award under our 1999 Stock Incentive Plan or 2005 Stock Incentive Plan that are forfeited, canceled, settled or otherwise terminated without a distribution of shares, or withheld by us in connection with the exercise of an option, or in payment of any required income tax withholding, will be deemed to be available for award under our 2005 Stock Incentive Plan. If there is a change in our capitalization, our compensation committee may make appropriate adjustments to the number and class of shares that may be delivered under the 2005 Stock Incentive Plan, to the number, class or price of shares subject to outstanding awards, and to the limits on individual awards under the plan to prevent dilution or enlargement of rights.
Limitations. The maximum number of shares and share equivalent units that may be granted during any calendar year to any one participant under all types of awards is 100,000. In addition, the maximum number of shares that may be issued or transferred to participants under our 2005 Stock Incentive Plan as incentive stock options is 200,000 and as restricted stock is 200,000. Each of these limits is subject to adjustment, as discussed above.
Section 162(m) of the Internal Revenue Code places limits on the deductibility for federal income tax purposes of compensation paid to certain executive officers. Our compensation committee may adjust targets set for pre-established performance objectives, but awards designed to qualify for the performance-based exception under Internal Revenue Code Section 162(m) may not be adjusted upward, except to reflect accounting changes or other events. Under certain circumstances, our
77
compensation committee will have discretion to change the performance objectives without obtaining stockholder approval.
Administration. Our 2005 Stock Incentive Plan will be administered by our compensation committee, which will consist of at least two directors who satisfy the "nonemployee director" requirements of Rule 16b-3 under Securities Exchange Act of 1934, the independence rules of the Nasdaq National Market and the "outside director" provisions of Internal Revenue Code Section 162(m). The compensation committee may select award recipients, determine the size, types and terms of awards, interpret the plan, and prescribe, amend and rescind rules and make all other determinations necessary or desirable for the administration of the plan.
Eligibility. Our employees, directors and consultants and those of our affiliates are eligible to participate in our 2005 Stock Incentive Plan. An individual becomes a plan participant when he or she is chosen by our compensation committee to receive an award.
Stock options. Our compensation committee may grant either incentive stock options that are intended to qualify under Section 422 of the Internal Revenue Code or nonqualified stock options. Each stock option will be evidenced by an award agreement that describes the terms and conditions of the grant. Our compensation committee determines the exercise price of an option at the time it is granted, but that exercise price must equal at least 100% of the fair market value of our common stock on the grant date. The term of a stock option may not exceed ten years.
Exercise of options. An option vests and becomes exercisable according to the terms specified in the stock option award agreement. The stock option award agreement will specify whether and under what circumstances an option may be exercised after the participant's death, retirement, disability or other termination of employment. A participant must pay the full exercise price when exercising a vested option.
Stock appreciation rights (SARs). Our compensation committee may grant freestanding SARs, tandem SARs, and/or any combination of these forms of SARs under the plan. A tandem SAR is an SAR issued in connection with a stock option. A tandem SAR may be exercised only as to the shares for which its related option is then exercisable and upon the surrender of the right to exercise the equivalent portion of the related option. The term of a SAR may not exceed ten years. When exercised, an SAR entitles the participant to a payment based on the excess of the fair market value of a share of common stock on the exercise date over the grant price of the SAR. The grant price of a freestanding SAR will equal the fair market value of our common stock on the date of grant, and the grant price of a tandem SAR will equal the exercise price of the related option.
Restricted stock and restricted stock units. Selected participants may defer a portion of their annual bonus and/or base salary in exchange for restricted stock units. Each participant who elects to make a deferral will be credited with a number of restricted stock units equal to the amount deferred divided by the fair market value of the common stock on the date designated by the compensation committee.
The number of restricted stock units granted to a participant may be increased. Restricted stock units must be settled in shares, except that fractional shares are settled in cash. During the restriction period, participants holding restricted stock may exercise full voting rights with respect to the underlying common stock. In addition, a participant may be entitled to receive regular cash dividends or dividend equivalents that are paid with respect to the underlying shares or share equivalent units during the restriction period.
Performance shares. Our compensation committee may grant performance shares under the plan. Each performance share must have an initial value equal to the fair market value of our common stock on the date of grant. Our compensation committee will set performance periods and performance objectives that will determine the number or value, or both, of the performance shares that will be paid
78
out to the participant. Earned performance shares may be paid in cash, shares or a combination of cash and shares.
Transferability of plan awards. Awards granted under the plan are generally not transferable. However, nonqualified stock options may, if no consideration is paid, be transferred or assigned to a participant's spouse or children or to one or more trusts for the exclusive benefit of a participant's spouse or children. Only the participant or the participant's guardian or legal representative may exercise awards during his or her lifetime.
Amendment, modification and termination. Our compensation committee may amend, modify or terminate the plan without stockholder approval, except as to certain matters. Our compensation committee may also modify, extend or renew awards that have been granted, as long as no termination, amendment, or modification of an award adversely affects in any material way these awards without the written consent of the participant holding the affected award. Our compensation committee may not increase the number of shares that may be issued or transferred to participants under the plan, or modify any outstanding option so as to specify a lower exercise price, without stockholder approval. The plan will automatically terminate in 2015.
The compensation committee may make adjustments in the terms and conditions of awards in recognition of unusual or nonrecurring events affecting us or of changes in applicable laws, regulations or accounting principles, if the compensation committee determines these adjustments are appropriate to prevent dilution or enlargement of the benefits or potential benefits intended to be made available under our stock incentive plan.
Our board of directors granted to our non-employee directors, effective upon the date that shares of our common stock are first quoted on the Nasdaq National Market, options to purchase shares of our common stock at an exercise price equal to the initial public offering price of $7.00 under our 2005 Stock Incentive Plan. These options vest immediately upon grant and are allocated among our non-employee directors as follows:
| Director | Number of options | |
|---|---|---|
| Terry W. Osborn | 10,931 | |
| Israel Rubinstein | 10,931 | |
| Rosalie Sagraves | 10,931 | |
| Thomas V. Thornton | 10,931 |
Annual Bonus Plan
In April 2005, our board of directors adopted the Annual Bonus Plan. The Annual Bonus Plan is a performance-based incentive bonus plan under which participants may receive cash bonuses based on meeting performance targets established by the compensation committee that may be based on the performance of the company as a whole, specific business units or divisions, or individual performance.
Only our full-time and part-time employees and the full-time and part-time employees of our affiliates are eligible to participate in the Annual Bonus Plan. The compensation committee designates those eligible employees who will become participants in the Annual Bonus Plan.
Performance targets for the Annual Bonus Plan will be established by our compensation committee at the beginning of each year. These targets may be based on any of the following criteria: net earnings; operating earnings or income; earnings growth; net income (absolute or competitive growth rates comparative); net income applicable to common stock; gross revenue or revenue by pre-defined business segment (absolute or competitive growth rates comparative); revenue backlog; margins realized on delivered services; cash flow, including operating cash flow, free cash flow, discounted cash flow return on investment, and cash flow in excess of cost of capital; earnings per share of common stock;
79
return on stockholders equity (absolute or peer-group comparative); stock price (absolute or peer-group comparative); absolute and/or relative return on common stockholders equity; absolute and/or relative return on capital; absolute and/or relative return on assets; economic value added (income in excess of cost of capital); customer satisfaction; expense reduction; ratio of operating expenses to operating revenues; product development milestones; and capital raising objectives.
Bonus payouts will be paid in cash in an amount determined by the compensation committee at the end of the year. The amount of each participant's bonus is based upon the company's success in meeting the performance targets, and the participant's contribution toward achieving these targets. If, after a change of control of the company, any participant's employment is terminated without cause, the participant shall receive payment of his or her performance bonus as if the performance targets had been met in full and the participant was employed for the entire performance period. Our compensation committee administers the Annual Bonus Plan and has the authority to construe, interpret, and implement the plan, and prescribe, amend, and rescind rules and regulations relating to the plan. The determination of the compensation committee on all matters relating to the Annual Bonus Plan or any award agreement is final, binding, and conclusive.
401(k) Retirement Plan
We maintain a 401(k) retirement plan which is intended to be a tax-qualified defined contribution plan under Section 401(k) of the Internal Revenue Code. In general, all of our employees are eligible to participate, subject to a 60-day waiting period. The 401(k) plan includes a salary deferral arrangement pursuant to which participants may elect to reduce their current compensation by up to the statutorily prescribed limit, equal to $13,000 in 2004, and have the amount of the reduction contributed to the 401(k) plan. We are permitted to match employees' 401(k) plan contributions. For the year ended December 31, 2004, we elected to match 50% of eligible employees' contributions to the 401(k) plan.
80
Loan from Dr. Michael T. Flavin
In September 2001, Dr. Michael T. Flavin, our founder and Chairman and Chief Executive Officer, made a $2.0 million loan to us. The loan accrues interest at the rate of 7.75% per year, compounded annually. As of December 31, 2004 the principal and accrued interest on the loan totaled approximately $2.6 million. On July 28, 2005, Dr. Flavin agreed to extend the maturity on the loan until December 31, 2007.
Loan from Flavin Ventures
In anticipation of increasing our line of credit from $3.0 million to $4.0 million, on May 17, 2005, Flavin Ventures loaned to us $91,000 pursuant to a thirty-day bridge loan. The increase to the line of credit went into effect as of May 31, 2005, and the bridge loan was repaid in full on June 3, 2005.
Line of Credit Guarantees
Our $4.0 million bank line of credit has been guaranteed by Flavin Ventures, Dr. Flavin and his wife, Karen A. Flavin, and John L. Flavin. Flavin Ventures secured its guaranty by issuing a mortgage on our principal facilities and pledging the shares it owns in us. Dr. Flavin also entered into a subordination agreement with the bank under which he agreed to subordinate his $2.0 million loan to us to the line of credit.
Management Services Agreement with Sarawak MediChem Pharmaceuticals, Inc.
In 2001, we entered into a management services agreement with Sarawak MediChem Pharmaceuticals, Inc., our 50/50 joint venture with the State Government of Sarawak, Malaysia. Under this agreement, Sarawak MediChem Pharmaceuticals owes us a flat monthly service fee in exchange for us providing Sarawak MediChem Pharmaceuticals with administrative management services relating to our development program for Calanolide A. We did not have any management fees recognized as revenue in 2004. Billed and unrecognized revenue in 2004 totaled approximately $160,000.
Facility Lease with BioStart Property Group, LLC
We lease real property facilities from BioStart Property Group, LLC, a wholly-owned subsidiary of Flavin Ventures, LLC. Flavin Ventures is the controlling member of ALS Ventures, LLC, our primary stockholder. Michael T. Flavin and John L. Flavin are the members of Flavin Ventures, LLC. The lease lasts for five years and commenced on October 1, 2003. It may be renewed in writing 60 days prior to the lease termination date. The lease provides us with 15,000 square feet at $14 per square foot. The rental rate increases by 3.0% for the second and third years and by 4.0% in the final two years of the lease term reaching approximately $16 per square foot in 2007. This rate is comparable with surrounding prices for office and laboratory space, which range from $13 per square foot in Lemont, Illinois to $25 per square foot in Chicago, Illinois. Our rental expense payable to BioStart Property Group in 2004 totaled approximately $243,000.
Management Services Agreement with Flavin Ventures
In January 2004, we entered into a management services agreement with Flavin Ventures under which we provide administrative management services to Flavin Ventures and its wholly-owned subsidiaries Shamrock Structures, BioStart Property Group and Molecular Formulations. The agreement also requires Flavin Ventures and its related subsidiaries to reimburse us for rent and related facility costs based upon the actual square footage occupied by each of the companies. Revenue recognized under this agreement was approximately $272,000 in 2004.
Concurrent Offering
Concurrent with the shares we are offering by this prospectus in the initial public offering, we are also offering 428,571 shares of our common stock to Abbott Laboratories, the beneficial owner of approximately 10.4% of our common stock prior to the offerings. The initial public offering and the concurrent offering are conditioned upon each other. The number of shares that we are offering to Abbott in the concurrent offering is determined by dividing $3 million by the assumed initial public offering price of $7.00 per share. In exchange for receiving shares of our common stock in the concurrent offering, a $5 million milestone obligation under our license agreement with Abbott that is payable on October 31, 2005, or, if earlier, upon our initiation of pivotal Phase III clinical trials for cethromycin, will be reduced by $3 million.
81
On December 13, 2004, Advanced Life Sciences, Inc. ("ALS Inc.") executed a plan of recapitalization and reorganization in order to simplify its capital structure and give effect to capital contributions made by Dr. Flavin, our founder, Chairman and Chief Executive Officer. In connection with the recapitalization, Advanced Life Sciences Holdings, Inc. ("ALS Holdings") was formed as a Delaware corporation. Common stockholders of ALS Inc. exchanged their 1,629,685 shares of common stock in ALS Inc. for 1,629,685 shares of common stock in ALS Holdings. Flavin Ventures, LLC, a Delaware limited liability company, exchanged its 40% interest in Advanced Life Sciences General Partnership, an Illinois general partnership between Flavin Ventures and ALS Inc. ("ALS GP"), for 7,852,330 shares of common stock in ALS Holdings. After the exchanges were completed, ALS GP was dissolved and its assets and liabilities were assumed by ALS Inc. According to the plan of recapitalization and reorganization, ALS Ventures, LLC was formed as a Delaware limited liability company, and Dr. Flavin and Flavin Ventures simultaneously contributed to ALS Ventures all of their shares of common stock in ALS Holdings. The result of the reorganization and recapitalization was for ALS Inc. to become a wholly-owned subsidiary of ALS Holdings. ALS Holdings is offering shares of its common stock pursuant to this prospectus.
82
The following table sets forth certain information regarding beneficial ownership of shares of our common stock as of the completion of the offerings for:
- •
- each of our directors;
- •
- each of our officers named in the summary compensation table;
- •
- all directors and executive officers as a group; and
- •
- each person known to us to be the beneficial owner of more than 5% of our outstanding shares of common stock.
Beneficial ownership is determined in accordance with the rules and regulations of the Securities and Exchange Commission. In computing the number of shares beneficially owned by a person and the percentage ownership of that person, shares of common stock subject to options or warrants held by that person that are currently exercisable or exercisable within 60 days of the date hereof are deemed outstanding. Such shares, however, are not deemed outstanding for the purposes of computing the percentage ownership of any other person. Except as indicated in the footnotes to this table and subject to community property laws where applicable, the persons named in the table have sole voting and investment power with respect to all shares of our common stock shown opposite such stockholder's name. The percentage of beneficial ownership is based on 10,826,801 shares of common stock outstanding prior to the offerings and 16,826,801 shares of our common stock outstanding immediately after the offerings. Concurrent with the shares we are offering by this prospectus in the initial public offering, we are also offering 428,571 shares of our common stock to Abbott Laboratories in exchange for a $3 million reduction of a $5 million milestone obligation under our license agreement with Abbott that is payable on October 31, 2005, or, if earlier, upon our initiation of pivotal Phase III clinical trials for cethromycin.
| | | Percentage of Common Stock Outstanding | ||||||
|---|---|---|---|---|---|---|---|---|
| Beneficial Owner(1) | Number of Shares | Before Offerings | After Offerings | |||||
| Five percent stockholders: | ||||||||
| Flavin Ventures, LLC(2) | 9,591,866 | 88.6 | % | 57.0 | % | |||
| Abbott Laboratories(3)(4) | 1,122,569 | 10.4 | 9.2 | |||||
| Directors and Executive Officers | ||||||||
| Michael T. Flavin(5) | 9,591,866 | 88.6 | 57.0 | |||||
| John L. Flavin(6) | 9,661,341 | 88.7 | 57.2 | |||||
| R. Richard Wieland II(7) | 64,711 | * | * | |||||
| Suseelan Pookote(7) | 45,655 | * | * | |||||
| Ze-Qi Xu(7) | 45,655 | * | * | |||||
| Terry W. Osborn(7) | 4,069 | * | * | |||||
| Israel Rubinstein(7) | 4,069 | * | * | |||||
| Rosalie Sagraves(7) | 4,069 | * | * | |||||
| Thomas V. Thornton(7) | 4,069 | * | * | |||||
| All executive officers and directors as a group (11 persons) | 9,879,293 | 88.9 | 57.7 | |||||
- *
- Less than 1%
- (1)
- Unless otherwise indicated, the address for each five percent stockholder, director and executive officer is c/o Advanced Life Sciences Holdings, Inc., 1440 Davey Road, Woodridge, Illinois 60517.
83
- (2)
- Represents 151,534 shares held directly and 9,440,330 shares held by ALS Ventures, LLC. Flavin Ventures, LLC is the sole voting member of ALS Ventures, LLC, and in such capacity it may be deemed to have beneficial ownership of all 9,440,330 shares held by ALS Ventures, LLC. Flavin Ventures, LLC disclaims beneficial ownership of the shares held by ALS Ventures, LLC, except to the extent of its proportionate pecuniary interest therein. Dr. Michael Flavin and Mr. John Flavin are members and managers of Flavin Ventures, LLC.
- (3)
- The address for Abbott Laboratories is 100 Abbott Park Road, Department 309, Building AP30, Abbott Park, Illinois 60064-3537.
- (4)
- Percentage of common stock outstanding after the offerings includes 428,571 shares of our common stock offered to Abbott in the concurrent offering, which will bring Abbott's total holdings to 1,551,140 shares.
- (5)
- Dr. Michael Flavin is a member and a manager of Flavin Ventures, LLC, which is the sole voting member of ALS Ventures, LLC. In such capacity he may be deemed to have shared voting and investment power with respect to 9,440,330 shares held by ALS Ventures, LLC and 151,534 shares held by Flavin Ventures, LLC. Dr. Flavin disclaims beneficial ownership of the shares held by ALS Ventures, LLC and Flavin Ventures, LLC, except to the extent of his proportionate pecuniary interest therein.
- (6)
- Mr. John Flavin is a member and a manager of Flavin Ventures, LLC, which is the sole voting member of ALS Ventures, LLC. In such capacity he may be deemed to have shared voting and investment power with respect to 9,440,330 shares held by ALS Ventures, LLC and 151,534 shares held by Flavin Ventures, LLC. Mr. Flavin disclaims beneficial ownership of the shares held by ALS Ventures, LLC and Flavin Ventures, LLC, except to the extent of his proportionate pecuniary interest therein. Includes 69,475 shares issuable upon the exercise of stock options that are currently exercisable or exercisable within 60 days.
- (7)
- Represents shares issuable upon the exercise of stock options that are currently exercisable or exercisable within 60 days. According to the terms of the grant agreements for awards under the 1999 Stock Incentive Plan, all unvested options currently outstanding will vest automatically upon the closing of the offerings.
84
The following information describes our common stock, as well as options and warrants to purchase our common stock, and provisions of our amended and restated certificate of incorporation and our bylaws, all as will be in effect upon the closing of the offerings. This description is only a summary and is qualified by reference to our certificate of incorporation and bylaws. Our certificate of incorporation and bylaws have been filed with the SEC as exhibits to our registration statement of which this prospectus forms a part. The description of the common stock, as well as options and warrants to purchase our common stock, reflect changes to our capital structure that will occur upon the closing of the offerings in accordance with the terms of the certificate.
Upon completion of the offerings, our authorized capital stock will consist of 60,000,000 shares of our common stock, par value $0.01 per share, and 5,000,000 shares of preferred stock that may be issued in one or more series.
Common Stock
As of June 30, 2005, there were 10,826,801 shares of our common stock outstanding and held of record by 12 stockholders. There will be 16,826,801 shares of common stock outstanding upon the closing of the offerings, which gives effect to the issuance of 6,000,000 shares of common stock offered by us under this prospectus.
Each share of common stock will have identical rights and privileges in every respect upon the closing of the offerings. The holders of our common stock are entitled to vote upon all matters submitted to a vote of our stockholders and are entitled to one vote for each share of common stock held. The holders of common stock are entitled to receive such dividends, payable in cash, stock or otherwise, as may be declared by our board out of any funds legally available for the payment of dividends. If we voluntarily or involuntarily liquidate, dissolve or wind-up, the holders of common stock will be entitled to receive after distribution in full of the preferential amounts, if any, all of the remaining assets available for distribution ratably in proportion to the number of shares of common stock held by them. Holders of common stock have no preferences or any preemptive, conversion or exchange rights.
Preferred Stock
Under our amended and restated certificate of incorporation, our board of directors, without any further action by our stockholders, will be authorized to issue shares of our undesignated preferred stock in one or more classes or series. The board may fix the rights, preferences and privileges of the preferred stock, along with any limitations or restrictions, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences of each class or series of preferred stock. The preferred stock could have voting or conversion rights that could adversely affect the voting power or other rights of holders of our common stock. The issuance of preferred stock could also have the effect, under certain circumstances, of delaying, deferring or preventing a change of control of our company. We currently have no plans to issue any shares of preferred stock.
In connection with our spin-off from MediChem Life Sciences, Inc. in 1999, Advanced Life Sciences, Inc., our current operating subsidiary, issued 250,000 shares of its Series A preferred stock to MediChem Life Sciences. The preferred stock accumulates cash dividends at a rate of 7.0% per annum on the face liquidation amount of $2,500,000. Dividends are only payable when, as and if declared by the board of directors of Advanced Life Sciences, Inc. The preferred stock is not convertible into our common stock and only has voting rights to the extent necessary to protect the powers, preferences or rights of the shares of preferred stock. The preferred stock has a liquidation preference upon the dissolution or winding up of Advanced Life Sciences, Inc., not including a sale of all its assets or a merger or consolidation. If there were to be a dissolution or winding up of Advanced Life Sciences, Inc., the preferred stock would be entitled to a liquidation preference, prior to any
85
distribution on the common stock, in the amount of $2,500,000 plus accrued and unpaid dividends at the time of liquidation. The total liquidation preference as of June 30, 2005 was approximately $3.6 million, including approximately $1.1 million of accrued dividends. We do not intend ever to declare dividends on the outstanding preferred stock.
Stock Options
As of June 30, 2005, options to purchase a total of 686,837 shares of our common stock were outstanding at a weighted average exercise price of $0.93 per share. Options to purchase an additional total of 2,072,455 shares of common stock may be granted under our 2005 Stock Incentive Plan. Please see "Management—Stock Option and Other Compensation Plans."
Warrants
In December 2004, we issued warrants to purchase 14,887 shares of our common stock to Leaders Bank in connection with the line of credit provided by Leaders Bank. The warrants expire in December 2009 and have an exercise price equal to the lower of $8.02 per share or the initial public offering price per share. The warrants contain anti-dilution protection upon the occurrence of certain events, including the issuance of shares in the offerings, any recapitalization, reclassification, stock dividend, stock split, stock combination or similar transaction. All of the warrants are currently exercisable.
Registration Rights
Under the terms of registration rights agreements with holders of 1,192,853 shares of our common stock, we have granted to these holders rights to register such shares under the Securities Act. If we propose to register any of our securities under the Securities Act, either for our own account or for the account of other stockholders, the holders of these shares are entitled, under certain circumstances, to include in the registration statement, at our expense, their shares of common stock. In addition, the holders of these shares may require us, at our expense and on not more than two occasions at any time beginning approximately six months from the date of the closing of the offerings, to file a registration statement under the Securities Act with respect to their shares of common stock, and we will be required to use our best efforts to effect the registration. Further, the holders of these shares may require us, at our expense, to register their shares on Form S-3 when we can avail ourselves of this form. These registration rights are subject to certain conditions and limitations, including the right of the underwriters to limit the number of shares included in any such registration under certain circumstances. Each holder of our common stock that has registration rights waived such rights to the extent the rights arise in connection with the offerings.
Anti-Takeover Effects of Provisions of Delaware Law and our Certificate of Incorporation and By-laws
Delaware Anti-Takeover Law
We are subject to Section 203 of the Delaware General Corporation Law. Section 203 generally prohibits a public Delaware corporation from engaging in a "business combination" with an "interested stockholder" for a period of three years after the date that the stockholder became an interested stockholder, unless:
- •
- prior to the date of the transaction, the board of directors of the corporation approved either the business combination or the transaction which resulted in the stockholder becoming an interested stockholder;
- •
- upon the completion of the transaction that resulted in the interested stockholder becoming an interested stockholder, the interested stockholder owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding for purposes of determining the number of shares outstanding (a) shares owned by persons who are directors
86
- •
- on or subsequent to the date of the transaction, the business combination is approved by the board and authorized at an annual or special meeting of stockholders, and not by written consent, by the affirmative vote of at least 662/3% of the outstanding voting stock that is not owned by the interested stockholder.
and also officers, and (b) shares owned by employee stock plans in which employee participants do not have the right to determine confidentially whether shares held subject to the plan will be tendered in a tender or exchange offer; or
Section 203 defines a business combination to include:
- •
- any merger or consolidation involving the corporation and the interested stockholder;
- •
- any sale, transfer, pledge or other disposition involving the interested stockholder of 10% or more of the assets of the corporation;
- •
- subject to exceptions, any transaction that results in the issuance or transfer by the corporation of any stock of the corporation to the interested stockholder;
- •
- subject to exceptions, any transaction involving the corporation that has the effect of increasing the interested stockholder's proportionate share of the stock of any class or series of the corporation; or
- •
- the receipt by the interested stockholder of the benefit of any loans, advances, guarantees, pledges or other financial benefits provided by or through the corporation.
In general, Section 203 defines an interested stockholder as any entity or person that, together with affiliates and associates, owns, or within three years prior to the determination of interested stockholder status, did own 15% or more of the outstanding voting stock of the corporation.
Amended and Restated Certificate of Incorporation and Bylaws
Provisions of our amended and restated certificate of incorporation and bylaws may delay or discourage transactions involving an actual or potential change in our control or change in our management, including transactions in which stockholders might otherwise receive a premium for their shares, or transactions that our stockholders might otherwise deem to be in their best interests. Therefore, these provisions could adversely affect the price of our common stock. Among other things, our amended and restated certificate of incorporation and bylaws:
- •
- permit our board of directors to issue up to 5,000,000 shares of preferred stock, with any rights, preferences and privileges as they may designate (including the right to approve an acquisition or other change in control);
- •
- provide that the authorized number of directors may be changed only by resolution of the board of directors;
- •
- divide our board of directors into three staggered classes;
- •
- provide that stockholders seeking to present proposals before a meeting of stockholders or to nominate candidates for election as directors at a meeting of stockholders must provide timely notice in writing, and also must comply with specified requirements as to the form and content of a stockholder's notice;
- •
- do not provide for cumulative voting rights (therefore allowing the holders of a majority of the shares of common stock entitled to vote in any election of directors to elect all of the directors standing for election, if they should so choose); and
- •
- provide that special meetings of our stockholders may be called only by our Chairman or by our board of directors.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is LaSalle Bank National Association. The transfer agent's address is 135 South LaSalle Street, Chicago, Illinois.
87
SHARES ELIGIBLE FOR FUTURE SALE
Prior to the offerings, there has been no public market for our common stock. Future sales of substantial amounts of our common stock in the public market could adversely affect prevailing market prices. Furthermore, since no shares will be available for sale shortly after the offerings because of contractual and legal restrictions on resale as described below, sales of substantial amounts of our common stock in the public market after these restrictions lapse could adversely affect the prevailing market price and our ability to raise equity capital in the future.
Upon completion of the offerings, we will have outstanding an aggregate of 16,826,801 shares of common stock, assuming no exercise of outstanding options after June 30, 2005. Of these shares, all of the shares sold in the offerings will be freely tradable without restriction or further registration under the Securities Act, unless these shares are purchased by our "affiliates" as that term is defined in Rule 144 under the Securities Act. The remaining 10,826,801 shares of common stock held by existing stockholders are "restricted securities" under Rule 144. Restricted securities may be sold in the public market only if registered or if they qualify for an exemption from registration described below under Rules 144, 144(k) or 701 promulgated under the Securities Act.
As a result of the contractual restrictions described below and the provisions of Rules 144, 144(k) and 701, the restricted shares will be available for sale in the public market as follows:
- •
- no shares will be eligible for sale upon completion of the offerings; and
- •
- 11,255,372 shares will be eligible for sale upon the expiration of the lock-up agreements, described below, beginning 180 days after the date of this prospectus, subject to Rule 144 volume limitations.
Lock-Up Agreements
Our directors, officers and substantially all of our stockholders, who combined hold approximately 99% of our common stock, have entered into lock-up agreements with the underwriters of the initial public offering generally providing that they will not offer, sell, contract to sell or grant any option to purchase or otherwise dispose of our shares of common stock or any securities exercisable for or convertible into our common stock owned by them prior to the offerings for a period of 180 days after the date of this prospectus (which period could be extended by those underwriters for up to an additional 34 days under certain circumstances) without the prior written consent of C.E. Unterberg, Towbin, LLC on behalf of our underwriters. C.E. Unterberg, Towbin, LLC has advised us that it has no current intention to shorten or release any of the shares subject to the lock-up agreements prior to the expiration of the lock-up period.
Rule 144
In general, under Rule 144 as currently in effect, beginning 90 days after the date of this prospectus a person who has beneficially owned restricted securities for at least one year, including the holding period of any prior owner except an affiliate, would be entitled to sell within any three-month period a number of shares that does not exceed the greater of:
- •
- 1% of the number of shares of our common stock then outstanding, which will equal approximately 168,268 shares immediately after the offerings; or
- •
- the average weekly trading volume of our common stock on the Nasdaq National Market during the four calendar weeks preceding the filing of a notice on Form 144 with respect to the sale.
Sales under Rule 144 are also subject to matter of sale provisions and notice requirements and to the availability of current public information about us.
88
Rule 144(k)
Under Rule 144(k), a person who is not deemed to have been one of our affiliates at any time during the 3 months preceding a sale, and who has beneficially owned the shares proposed to be sold for at least two years, including the holding period of any prior owner except an affiliate, is entitled to sell these shares without complying with the manner of sale, public information, volume limitation or notice provisions of Rule 144. No shares of our common stock will be eligible for sale under 144(k) prior to October 26, 2006.
Rule 701
In general, under Rule 701 of the Securities Act as currently in effect, any of our employees, consultants or advisors, other than affiliates, who purchases or receives shares from us in connection with a compensatory stock purchase plan or option plan or other written agreement is eligible to resell their shares beginning 90 days after the effective date of the offerings under Rule 144, without having to comply with its holding period, public information, volume limitation or notice provisions, and by affiliates under Rule 144 without having to comply with its holding period requirements.
89
Under the terms and subject to the conditions contained in an underwriting agreement dated , 2005, we have agreed to sell to the underwriters named below, for whom C.E. Unterberg, Towbin, LLC, ThinkEquity Partners LLC and Merriman Curhan Ford & Co. are acting as representatives, the following respective numbers of shares of common stock:
| Underwriters | Number of Shares | |
|---|---|---|
| C.E. Unterberg, Towbin, LLC | ||
| ThinkEquity Partners LLC | ||
| Merriman Curhan Ford & Co. | ||
Total | 5,571,429 |
C.E. Unterberg, Towbin, LLC is acting as book-running manager for the initial public offering.
The underwriters have agreed to purchase all of the shares shown in the above table if any of those shares are purchased. If an underwriter defaults, the underwriting agreement provides that the purchase commitments of the non-defaulting underwriters may be increased or the underwriting agreement may be terminated.
The shares of common stock are offered by the underwriters, subject to prior sale, when, as and if issued to and accepted by them, subject to advice on legal matters from counsel for the underwriters and other conditions contained in the underwriting agreement, such as the receipt by the underwriters of officers' certificates and legal opinions.
The underwriters have advised us that they do not expect sales to accounts over which they exercise discretionary authority to exceed 5% of the shares of common stock being offered.
Concurrent Offering
The closing of our concurrent offering is conditioned upon the closing of our initial public offering. The price of our shares to be sold in our concurrent offering is equal to the initial public offering price. The underwriters will not receive a fee in connection with the concurrent offering.
Public Offering Price and Dealers Concession
The underwriters propose initially to offer the shares of common stock offered by this prospectus directly to the public at the offering price per share set forth on the cover page of this prospectus, and to certain broker/dealers at that price less a concession not in excess of $ per share. The underwriters may allow, and these broker/dealers may re-allow, a discount not in excess of $ per share on sales to certain other broker/dealers. After commencement of the initial public offering, the offering price, discount and re-allowance may be changed by the underwriters.
Over-Allotment Option
We have granted to the underwriters an option, exercisable during the 30-day period after the date of this prospectus, to purchase up to a total of 835,714 additional shares of common stock at the public offering price per share less the underwriting discount per share shown on the cover page of this prospectus. To the extent that the underwriters exercise this option, each of the underwriters will have a firm commitment, subject to conditions, to purchase approximately the same percentage of the additional shares that the number of shares of common stock to be purchased by that underwriter as shown in the above table represents as a percentage of the total number of shares shown in that table.
90
Underwriting Compensation
The underwriting discount is equal to the public offering price per share of common stock less the amount paid by the underwriters to us per share of common stock. The following table summarizes the compensation to be paid to the underwriters by us in connection with the initial public offering. The following amounts are shown assuming both no exercise and full exercise of the underwriters' option to purchase additional shares:
| | Per Share | Total | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Without Over-allotment | With Over-allotment | Without Over-allotment | With Over-allotment | ||||||||
| Underwriting discount paid by us | $ | $ | $ | $ | ||||||||
| Expenses payable by us | $ | $ | $ | $ | ||||||||
We estimate that the total expenses of the offerings, excluding the underwriting discount, will be approximately $1.25 million.
Indemnification of Underwriter
We have agreed to indemnify the underwriters against certain civil liabilities, including liabilities under the Securities Act, and, where such indemnification is unavailable, contribute to payments the underwriters may be required to make in connection with any such liabilities.
Lock-Up Arrangements
We have agreed, subject to certain exceptions, that we will not offer, sell, contract to sell, pledge or otherwise dispose of, directly or indirectly, or file with the SEC a registration statement under the Securities Act relating to, any shares of our common stock or securities convertible into or exchangeable or exercisable for any shares of our common stock, or publicly disclose the intention to make any offer, sale, pledge, disposition or filing, without the prior written consent of C.E. Unterberg, Towbin, LLC for a period of 180 days after the date of this prospectus. However, in the event that either (1) during the last 17 days of the "lock-up" period, we release earnings results or material news or a material event relating to us occurs or (2) prior to the expiration of the "lock-up" period, we announce that we will release earnings results during the 16-day period beginning on the last day of the "lock-up" period, then in either case the expiration of the "lockup" will be extended until the expiration of the 18-day period beginning on the date of the release of the earnings results or the occurrence of the material news or event, as applicable, unless C.E. Unterberg, Towbin, LLC waives, in writing, such an extension.
Our officers, directors and security holders, who combined hold approximately 99% of our stock, have agreed that they will not offer, sell, contract to sell, pledge or otherwise dispose of, directly or indirectly, any shares of our common stock or securities convertible into or exchangeable or exercisable for any shares of our common stock, enter into a transaction that would have the same effect, or enter into any swap, hedge or other arrangement that transfers, in whole or in part, any of the economic consequences of ownership of our common stock, whether any of these transactions are to be settled by delivery of our common stock or other securities, in cash or otherwise, or publicly disclose the intention to make any offer, sale, pledge or disposition, or to enter into any transaction, swap, hedge or other arrangement, without, in each case, the prior written consent of C.E. Unterberg, Towbin, LLC for a period of 180 days after the date of this prospectus. However, in the event that either (1) during the last 17 days of the "lock-up" period, we release earnings results or material news or a material event relating to us occurs or (2) prior to the expiration of the "lock-up" period, we announce that we will release earnings results during the 16-day period beginning on the last day of the "lock-up" period, then in either case the expiration of the "lock-up" will be extended until the expiration of the 18-day
91
period beginning on the date of the release of the earnings results or the occurrence of the material news or event, as applicable, unless C.E. Unterberg, Towbin, LLC waives, in writing, such an extension.
Quotation on the Nasdaq National Market
Our common stock has been approved for listing on the Nasdaq National Market under the symbol "ADLS."
Pricing of the Initial Public Offering
Prior to the offerings, there has been no public market for our common stock. The initial public offering price will be negotiated among the company and the underwriters. Among the factors to be considered in determining the initial public offering price of the shares, in addition to prevailing market conditions, will be the company's historical performance, estimates of the business potential and earnings prospects of the company, an assessment of the company's management and the consideration of the above factors in relation to market valuation of companies in related businesses. We cannot be sure that the initial public offering price will correspond to the price at which the common stock will trade in the public market following the offerings or that an active trading market for the common stock will develop and continue after the offerings.
Stabilization and Other Transactions
In connection with the offerings, the underwriters may engage in transactions that stabilize, maintain or otherwise affect the market price of our common stock. These transactions may include stabilization transactions effected in accordance with Rule 104 of Regulation M under the Securities Exchange Act, pursuant to which the underwriters may make a bid for, or purchase, common stock for the purpose of stabilizing the market price. The underwriters also may create a short position by selling more common stock in connection with the initial public offering than the underwriters are committed to purchase from us, and in such case may purchase common stock in the open market following completion of the offerings to cover all or a portion of such short position. In addition, the underwriters may impose "penalty bids" whereby the underwriters may reclaim from a dealer participating in the initial public offering the selling concession with respect to the common stock that the underwriters distributed in the initial public offering, but which was subsequently purchased for the accounts of the underwriters in the open market. Any of the transactions described in this paragraph may result in the maintenance of the price of the common stock at a level above that which might otherwise prevail in the open market. None of the transactions described in this paragraph is required and, if they are undertaken, they may be discontinued at any time.
Electronic Prospectus
In connection with the initial public offering, certain of the underwriters or securities dealers may distribute this prospectus electronically.
Disclosure Directed to Offers in the United Kingdom
Each underwriter has represented, warranted and agreed that: (a) it has not offered or sold and, prior to the expiry of a period of six months from the closing date, will not offer or sell any shares to persons in the U.K. except to persons whose ordinary activities involve them in acquiring, holding, managing or disposing of investments (as principal or agent) for the purposes of their businesses or otherwise in circumstances which have not resulted and will not result in an offer to the public in the U.K. within the meaning of the Public Offers of Securities Regulations 1995; (b) it has only communicated or caused to be communicated and will only communicate or cause to be communicated any invitation or inducement to engage in investment activity (within the meaning of Section 21 of the
92
Financial Services and Markets Act 2000 ("FSMA")) received by it in connection with the issue or sale of any shares in circumstances in which Section 21(1) of the FSMA does not apply to the issuer; and (c) it has complied and will comply with all applicable provisions of the FSMA with respect to anything done by it in relation to the shares in, from or otherwise involving the U.K.
93
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES TO NON-U.S. HOLDERS
The following summary describes certain material United States federal income tax consequences of the ownership and disposition of common stock by a non-U.S. holder as of the date of this prospectus. This discussion does not address all aspects of United States federal income taxes and does not deal with estate, gift, foreign, state and local tax consequences that may be relevant to such non-U.S. holders in light of their personal circumstances. Special rules may apply to certain non-U.S. holders, such as "controlled foreign corporations," "passive foreign investment companies," corporations that accumulate earnings to avoid U.S. federal income tax, investors in pass-through entities, dealers in securities, holders of securities held as part of a "straddle," "hedge," "conversion transaction" or other risk reduction transaction, and certain former citizens or long-term residents of the United States that are subject to special treatment under the Internal Revenue Code of 1986, as amended (the "Code"). Such entities and persons should consult their own tax advisors to determine the U.S. federal, state, local and other tax consequences that may be relevant to them. Furthermore, the discussion below is based upon the provisions of the Code, and regulations, rulings and judicial decisions thereunder as of the date hereof, and such authorities may be repealed, revoked or modified with or without retroactive effect so as to result in United States federal income tax consequences different from those discussed below.
If a partnership holds the common stock, the tax treatment of a partner will generally depend on the status of the partner and the activities of the partnership. Persons who are partners in partnerships or other pass-through entities holding the common stock should consult their tax advisors.
The authorities on which this summary is based are subject to various interpretations, and any views expressed within this summary are not binding on the Internal Revenue Service or the courts. No ruling from the IRS or opinion of counsel has been, or will be, requested in connection with the issuance of our stock. No assurance can be given that the IRS or the courts will agree with the tax consequences described herein.
For the purposes of the discussion in this prospectus, a "non-U.S. holder" means a beneficial owner of our common stock that is not any of the following for U.S. federal income tax purposes:
- •
- a citizen or resident of the United States;
- •
- a corporation, or other entity treated as a corporation for United States federal income tax purposes, created or organized in or under the laws of the United States or any political subdivision thereof;
- •
- an estate the income of which is subject to United States federal income taxation regardless of its source; or
- •
- a trust (i) which is subject to primary supervision by a court situated within the United States and as to which one or more United States persons have the authority to control all substantial decisions of the trust, as described in section 7701(a)(30) of the Code, or (ii) that has a valid election in effect under applicable U.S. Treasury regulations to be treated as a United States person.
For purposes of this discussion, a "non-U.S. holder" does not include an individual who is present in the United States for 183 days or more in the taxable year of disposition and is not otherwise a resident of the United States for U.S. federal income tax purposes. Such an individual is urged to consult his or her own tax advisor regarding the U.S. federal income tax consequences of the sale, exchange or other disposition of common stock.
Prospective purchasers are urged to consult their own tax advisors regarding the U.S. federal income tax consequences, as well as other U.S. federal, state, and local income and estate tax consequences, and non-U.S. tax consequences, to them of acquiring, owning, and disposing of our common stock.
94
Dividends
If we make distributions on our common stock, such distributions paid to a non-U.S. holder will generally constitute dividends for U.S. federal income tax purposes to the extent such distributions are paid from our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. If a distribution exceeds our current and accumulated earnings and profits, the excess will be treated as a tax-free return of the non-U.S. holder's investment to the extent of the non-U.S. holder's adjusted basis in our common stock. Any remaining excess will be treated as capital gain.
Dividends paid to a non-U.S. holder of common stock generally will be subject to withholding of United States federal income tax at a 30% rate or such lower rate as may be specified by an applicable income tax treaty. A non-U.S. holder of common stock who wishes to claim the benefit of an applicable treaty rate for dividends, will be required to (a) complete IRS Form W-8BEN (or appropriate substitute form) and certify, under penalty of perjury, that such holder is not a U.S. person and is eligible for the benefits with respect to dividends allowed by such treaty or (b) hold common stock through certain foreign intermediaries and satisfy the certification requirements of applicable Treasury regulations. Special certification requirements apply to certain non-U.S. holders that are "pass-through" entities rather than individuals. A non-U.S. holder of common stock eligible for a reduced rate of United States withholding tax pursuant to an income tax treaty may obtain a refund of any excess amounts withheld by timely filing an appropriate claim for refund with the IRS.
The withholding tax may not apply to dividends that are effectively connected with the conduct of a trade or business by the non-U.S. holder within the United States. The withholding tax may also not apply to dividends paid to a non-U.S. holder where the dividends are attributable to a United States permanent establishment of the non-U.S. holder and a tax treaty provides that withholding taxes will not be applied to such dividends. Dividends effectively connected with the conduct of a trade or business, as well as those attributable to a United States permanent establishment of the non-U.S. holder under an applicable treaty, are subject to United States federal income tax on a net income basis in generally the same manner as if the non-U.S. holder were a U.S. person, as defined under the Code. Certain certification and disclosure requirements must be complied with in order for effectively connected income to be exempt from withholding. Any such effectively connected dividends received by a foreign corporation may, under certain circumstances, be subject to an additional "branch profits tax" at a 30% rate or such lower rate as may be specified by an applicable income tax treaty.
Gain on Disposition of Common Stock
A non-U.S. holder generally will not be subject to United States federal income tax with respect to gain recognized on a sale or other disposition of common stock unless:
- •
- the gain is effectively connected with a trade or business of the non-U.S. holder in the United States and, where a tax treaty applies, is attributable to a United States permanent establishment of the non-U.S. holder;
- •
- the non-U.S. holder is an individual who holds his or her common stock as a capital asset (generally, an asset held for investment purposes) and who is present in the United States for a period or periods aggregating 183 days or more during the calendar year in which the sale or disposition occurs and certain other conditions are met; or
- •
- we are or have been a "U.S. real property holding corporation" within the meaning of Section 897(c)(2) of the Code (referred to in this discussion as a "USRPHC") for United States federal income tax purposes at any time within the five year period preceding the disposition (or, if shorter, the non-U.S. holder's holding period).
Gain recognized on the sale or other disposition of common stock and effectively connected with a United States trade or business, or attributable to a United States permanent establishment of the
95
non-U.S. holder under an applicable treaty, is subject to United States federal income tax on a net income basis in generally the same manner as if the non-U.S. holder were a U.S. person, as defined under the Code. Any such effectively connected gain from the sale or disposition of common stock received by a foreign corporation may, under certain circumstance, be subject to an additional "branch profits tax" at a 30% rate or such lower rate as may be specified by an applicable income tax treaty.
Based on audited financial statements and other data, we believe that we currently are not a USRPHC. In addition, based on these financial statements and current expectations regarding the value and nature of our assets and other relevant date, we do not anticipate becoming a USRPHC.
If we become a USRPHC, a non-U.S. holder of common stock will nevertheless not be subject to United States federal income tax if our common stock is regularly traded on an established securities market, within the meaning of applicable Treasury regulations, and the non-U.S. holder holds no more than five percent of our outstanding common stock, directly or indirectly, during the testing period identified in the third bullet point immediately above. Our common stock has been approved for listing on the Nasdaq National Market, and it may be regularly traded on an established securities market in the United States so long as it is listed.
Information Reporting and Backup Withholding
We must report annually to the IRS and to each non-U.S. holder the amount of dividends paid to such holder and the tax withheld with respect to such dividends, regardless of whether withholding was required. Copies of the information returns reporting such dividends and withholding may also be made available to the tax authorities in the country in which the non-U.S. holder resides under the provisions of an applicable income tax treaty.
The United States imposes a backup withholding tax on dividends and certain other types of payments to United States persons (currently at a rate of 28%) of the gross amount. Dividends paid to a non-U.S. holder will not be subject to backup withholding if proper certification of foreign status (usually on an IRS Form W-8BEN) is provided, and the payor does not have actual knowledge or reason to know that the beneficial owner is a United States person, or the holder is a corporation or one of several types of entities and organizations that qualify for exemption, which we refer to as an "exempt recipient."
Information reporting and backup withholding generally are not required with respect to the amount of any proceeds from the sale of shares of common stock by a non-U.S. holder outside the United States through a foreign office of a foreign broker that does not have certain specified connections to the United States. However, if a non-U.S. holder sells shares of common stock through the U.S. office of a United States or foreign broker, the broker will be required to report the amount of proceeds paid to such holder to the IRS and to apply the backup withholding tax (currently at a rate of 28%) to the amount of such proceeds unless appropriate certification (usually on an IRS Form W-8BEN) is provided to the broker of the holder's status as either an exempt recipient or a non-U.S. person, and the payor does not have actual knowledge or reason to know that the beneficial owner is a United States person. Information reporting also applies if a non-U.S. holder sells its shares of common stock through the foreign office of a broker deriving more than a specified percentage of its income from United States sources or having certain other connections to the United States and the foreign broker does not have certain documentary evidence in its files of the non-U.S. holder's foreign status.
Any amounts withheld under the backup withholding rules may be allowed as a refund or a credit against such holder's U.S. federal income tax liability provided the required information is timely furnished to the IRS.
96
The validity of the shares of common stock offered hereby will be passed upon for us by Winston & Strawn LLP, Chicago, Illinois. On February 8, 2005, we issued and sold 7,940 shares of our common stock to Thomas Fitzgerald of Winston & Strawn LLP at a cash purchase price of $8.02 per share. Certain legal matters related to the sale of the shares of common stock offered hereby will be passed upon for the underwriters by Ropes & Gray LLP, Boston, Massachusetts.
The consolidated financial statements of Advanced Life Sciences Holdings, Inc. (the "Company"), as of December 31, 2003 and 2004 and for each of the three years in the period ended December 31, 2004, have been audited by Deloitte & Touche LLP, an independent registered public accounting firm, as stated in their report appearing herein, which report expresses an unqualified opinion on the consolidated financial statements and includes an explanatory paragraph referring to the uncertainty about the Company's ability to continue as a going concern, and have been so included in reliance upon the report of such firm given upon their authority as experts in accounting and auditing.
WHERE YOU CAN FIND MORE INFORMATION
We have filed with the SEC a registration statement on Form S-1 (including exhibits, schedules and amendments) under the Securities Act of 1933 with respect to the shares of common stock to be sold in the offerings. This prospectus does not contain all the information set forth in the registration statement. For further information with respect to us and the shares of common stock to be sold in the offerings, reference is made to the registration statement. Statements contained in this prospectus as to the contents of any contract, agreement or other document referred to are not necessarily complete. Whenever a reference is made in this prospectus to any contract or other document of ours, the reference may not be complete, and you should refer to the exhibits that are a part of the registration statement for a copy of the contract or document.
You may read and copy all or any portion of the registration statement or any other information Advanced Life Sciences, Inc. files at the SEC's public reference room at 100 F Street, N.E., Room 1580, Washington, D.C. 20549. You can request copies of these documents, upon payment of a duplicating fee, by writing to the SEC. Please call the SEC at 1-800-SEC-0330 for further information on the operation of the public reference rooms. The SEC also maintains a web site (http://www.sec.gov) that contains reports, proxy and information statements and other information regarding issuers that file electronically with the SEC.
As a result of the offerings, we will become subject to the information and reporting requirements of the Securities Exchange Act, and, in accordance with those requirements, will file periodic reports, proxy statements and other information with the SEC.
This prospectus includes statistical data that were obtained from industry publications. These industry publications generally indicate that the authors of these publications have obtained information from sources believed to be reliable, but do not guarantee the accuracy and completeness of their information. While we believe these industry publications to be reliable, we have not independently verified their data.
97
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
| | Page | |
|---|---|---|
| Report of Independent Registered Public Accounting Firm | F-2 | |
Consolidated Balance Sheets | F-3 | |
Consolidated Statements of Operations | F-4 | |
Consolidated Statements of Stockholders' Deficit | F-5 | |
Consolidated Statements of Cash Flows | F-6 | |
Notes to Consolidated Financial Statements | F-7 |
F-1
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Stockholders of
Advanced Life Sciences Holdings, Inc.:
We have audited the accompanying consolidated balance sheets of Advanced Life Sciences Holdings, Inc. (a Delaware corporation in the development stage) and its subsidiary Advanced Life Sciences Inc. as of December 31, 2004 and 2003, and the related consolidated statements of operations, stockholders' deficit and cash flows for each of the three years in the period ended December 31, 2004 and for the period from January 1, 1999 (inception) through December 31, 2004. These financial statements are the responsibility of the Company's management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financials statements are free of material misstatement. An audit includes consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company's internal control over financial reporting. Accordingly, we express no such opinion. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, such consolidated financial statements present fairly, in all material respects, the financial position of Advanced Life Sciences Holdings, Inc. and its subsidiary as of December 31, 2004 and 2003, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2004, and for the period from January 1, 1999 (inception) through December 31, 2004, in conformity with accounting principles generally accepted in the United States of America.
The accompanying consolidated financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Notes 1 and 10 to the consolidated financial statements, the Company's recurring losses from operations, stockholders' capital deficiency and substantial payments due in 2005 raise substantial doubt about its ability to continue as a going concern. Management's plans concerning these matters are also described in Note 10. The consolidated financial statements do not include any adjustments that might result from the outcome of this uncertainty.
/s/ Deloitte & Touche LLP
Chicago, Illinois
April 25, 2005 (June 29, 2005 as to Note 12 and July 15, 2005 as to Note 3)
F-2
ADVANCED LIFE SCIENCES HOLDINGS, INC. AND SUBSIDIARY
(A Development Stage Company)
CONSOLIDATED BALANCE SHEETS
| | As of Year ended December 31, | | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | As of June 30, 2005 | |||||||||||
| | 2003 | 2004 | ||||||||||
| | | | (unaudited) | |||||||||
| ASSETS | ||||||||||||
CURRENT ASSETS: | ||||||||||||
| Cash | $ | 61,203 | $ | 194,555 | $ | 39,951 | ||||||
| Clinical trials supplies | — | 533,333 | 533,333 | |||||||||
| Cost of proceeds | — | — | 948,785 | |||||||||
| Accounts receivable—related party | — | 24,868 | 11,096 | |||||||||
| Prepaid expenses and other | 29,833 | 25,806 | 17,745 | |||||||||
| Total current assets | 91,036 | 778,562 | 1,550,910 | |||||||||
| FURNITURE AND EQUIPMENT: | ||||||||||||
| Furniture and fixtures | 131,627 | 139,005 | 140,849 | |||||||||
| Laboratory equipment | 56,513 | 142,928 | 142,928 | |||||||||
| Computer equipment | 52,698 | 79,146 | 81,466 | |||||||||
| Leasehold improvements | 28,672 | 40,646 | 40,646 | |||||||||
| Total furniture and equipment—at cost | 269,510 | 401,725 | 405,889 | |||||||||
| Less accumulated depreciation | (112,473 | ) | (180,807 | ) | (217,184 | ) | ||||||
| Furniture and equipment—net | 157,037 | 220,918 | 188,705 | |||||||||
| OTHER LONG-TERM ASSETS: | ||||||||||||
| Deferred financing costs | — | 15,000 | 25,461 | |||||||||
| Other assets | 7,054 | 1,452 | 4,782 | |||||||||
| Total other assets | 7,054 | 16,452 | 30,243 | |||||||||
TOTAL ASSETS | $ | 255,127 | $ | 1,015,932 | $ | 1,769,858 | ||||||
| LIABILITIES AND STOCKHOLDERS' DEFICIT | ||||||||||||
CURRENT LIABILITIES: | ||||||||||||
| Accounts payable | $ | 210,571 | $ | 255,880 | $ | 982,645 | ||||||
| Accrued expenses | 73,551 | 185,348 | 392,822 | |||||||||
| Accrued interest payable | — | 4,319 | 3,724 | |||||||||
| Licenses payable | — | 14,000,000 | 13,000,000 | |||||||||
| Notes payable—net of $11,766 debt discount in 2004 and $8,796 in 2005 | — | 2,238,234 | 3,523,204 | |||||||||
| Short-term lease payable | — | 12,070 | 13,052 | |||||||||
| Total current liabilities | 284,122 | 16,695,851 | 17,915,447 | |||||||||
Long-term lease payable | — | 27,279 | 20,498 | |||||||||
| Accrued interest payable—related party | 377,684 | 566,603 | 666,059 | |||||||||
| Grant payable | — | — | 500,000 | |||||||||
| Notes payable—related party | 2,000,000 | 2,000,000 | 2,000,000 | |||||||||
| Total liabilities | 2,661,806 | 19,289,733 | 21,102,004 | |||||||||
STOCKHOLDERS' DEFICIT: | ||||||||||||
| Common stock, $0.01 par value—June 30, 2005: 60,000,000 shares authorized; 10,826,801 issued and outstanding (unaudited); December 31, 2004: 10,604,584 shares issued and outstanding; December 31, 2003: no par value; 3,970,000 shares authorized, 1,588,000 issued and outstanding | 250,000 | 106,046 | 108,268 | |||||||||
| Series A preferred stock of Advanced Life Sciences, Inc., no par value—250,000 shares authorized; 250,000 shares issued and outstanding | — | — | — | |||||||||
| Deferred compensation | — | (23,219 | ) | (280,958 | ) | |||||||
| Additional paid-in capital | 10,430,696 | 21,917,665 | 22,779,215 | |||||||||
| Deficit accumulated during the development stage | (13,087,375 | ) | (40,274,293 | ) | (41,938,671 | ) | ||||||
| Total stockholders' deficit | (2,406,679 | ) | (18,273,801 | ) | (19,332,146 | ) | ||||||
| TOTAL LIABILITIES AND STOCKHOLDERS' DEFICIT | $ | 255,127 | $ | 1,015,932 | $ | 1,769,858 | ||||||
See notes to consolidated financial statements.
F-3
ADVANCED LIFE SCIENCES HOLDINGS, INC. AND SUBSIDIARY
(A Development Stage Company)
CONSOLIDATED STATEMENTS OF OPERATIONS
| | | | | Period From Inception (January 1, 1999) Through December 31, 2004 | | | | |||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | | | | | | Period From Inception (January 1, 1999) Through June 30, 2005 | ||||||||||||||||||
| | Years ended December 31, | Six months ended June 30, | ||||||||||||||||||||||
| | 2002 | 2003 | 2004 | 2004 | 2005 | |||||||||||||||||||
| | | | | | (Unaudited) | (Unaudited) | ||||||||||||||||||
| Revenue: | ||||||||||||||||||||||||
| Management fees | $ | 495,252 | $ | 118,563 | $ | 271,644 | $ | 1,099,941 | $ | 144,837 | $ | 44,379 | $ | 1,144,320 | ||||||||||
| Grant | — | 61,864 | 38,136 | 935,571 | 38,136 | — | 935,571 | |||||||||||||||||
| Royalty—related party | 955 | — | 10,000 | 45,238 | — | — | 45,238 | |||||||||||||||||
| Total revenue | 496,207 | 180,427 | 319,780 | 2,080,750 | 182,973 | 44,379 | 2,125,129 | |||||||||||||||||
| Expenses: | ||||||||||||||||||||||||
| Research and development | 902,550 | 1,362,255 | 25,661,868 | 28,811,620 | 570,986 | 724,347 | 29,535,967 | |||||||||||||||||
| Contracted research and development—related party | 22,249 | — | — | 7,980,299 | — | — | 7,980,299 | |||||||||||||||||
| Selling, general and administrative | 668,622 | 1,198,722 | 1,649,953 | 4,397,121 | 639,085 | 780,161 | 5,177,282 | |||||||||||||||||
| Total expenses | 1,593,421 | 2,560,977 | 27,311,821 | 41,189,040 | 1,210,071 | 1,504,508 | 42,693,548 | |||||||||||||||||
| Loss from operations | (1,097,214 | ) | (2,380,550 | ) | (26,992,041 | ) | (39,108,290 | ) | (1,027,098 | ) | (1,460,129 | ) | (40,568,419 | ) | ||||||||||
| Interest expense—net | 164,337 | 170,938 | 194,877 | 1,166,003 | 96,389 | 204,249 | 1,370,252 | |||||||||||||||||
| Net loss | (1,261,551 | ) | (2,551,488 | ) | (27,186,918 | ) | (40,274,293 | ) | (1,123,487 | ) | (1,664,378 | ) | (41,938,671 | ) | ||||||||||
| Less accumulating preferred dividends for the period | 175,000 | 175,000 | 175,000 | 969,792 | 87,500 | 87,500 | 1,057,292 | |||||||||||||||||
| Net loss available to common shareholders | $ | (1,436,551 | ) | $ | (2,726,488 | ) | $ | (27,361,918 | ) | $ | (41,244,085 | ) | $ | (1,210,987 | ) | $ | (1,751,878 | ) | $ | (42,995,963 | ) | |||
| Basic net loss per share available to common shareholders | $(0.90 | ) | $(1.72 | ) | $(13.27 | ) | $(0.76 | ) | $(0.16 | ) | ||||||||||||||
| Weighted average number common shares outstanding—basic | 1,588,000 | 1,588,000 | 2,062,351 | 1,588,134 | 10,780,148 | |||||||||||||||||||
See notes to consolidated financial statements.
F-4
ADVANCED LIFE SCIENCES HOLDINGS, INC. AND SUBSIDIARY
(A Development Stage Company)
CONSOLIDATED STATEMENTS OF STOCKHOLDERS' DEFICIT
FOR THE PERIOD FROM INCEPTION (JANUARY 1, 1999) THROUGH JUNE 30, 2005
| | | | | | | | Deficit Accumulated During the Development Stage | | ||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Common Stock | Preferred Stock | | | | |||||||||||||||||||
| | Additional Paid-in Capital | Deferred Compensation | | |||||||||||||||||||||
| | Shares | Amount | Shares | Amount | Total | |||||||||||||||||||
| BALANCE—January 1, 1999 (inception) | — | $ | — | — | $ | — | $ | — | $ | — | $ | — | $ | — | ||||||||||
Issuance of common stock in exchange for contributed intellectual property in January 1999 | 1,588,000 | — | — | — | — | — | — | — | ||||||||||||||||
| Exchange of shares under recapitalization and spin-off from MediChem Life Sciences, Inc. in June 1999, at historical cost | (1,588,000 | ) | — | 250,000 | — | — | — | — | — | |||||||||||||||
| Issuance of common stock in June 1999, at $0.157 per share | 1,588,000 | 250,000 | — | — | — | — | �� | — | 250,000 | |||||||||||||||
| Compensation expense related to stock options since inception | — | — | — | — | 15,097 | — | — | 15,097 | ||||||||||||||||
| Conversion of loans to stockholder into equity, August 2001 | — | — | — | — | 5,751,753 | — | — | 5,751,753 | ||||||||||||||||
| Capital contributions | — | — | — | — | 1,165,118 | — | — | 1,165,118 | ||||||||||||||||
| Net loss since inception | — | — | — | — | — | — | (9,274,336 | ) | (9,274,336 | ) | ||||||||||||||
BALANCE—December 31, 2001 | 1,588,000 | 250,000 | 250,000 | — | 6,931,968 | — | (9,274,336 | ) | (2,092,368 | ) | ||||||||||||||
Capital contributions | — | — | — | — | 1,161,000 | — | — | 1,161,000 | ||||||||||||||||
| Net loss | — | — | — | — | — | — | (1,261,551 | ) | (1,261,551 | ) | ||||||||||||||
BALANCE—December 31, 2002 | 1,588,000 | 250,000 | 250,000 | — | 8,092,968 | — | (10,535,887 | ) | (2,192,919 | ) | ||||||||||||||
Capital contributions | — | — | — | — | 2,337,728 | — | — | 2,337,728 | ||||||||||||||||
| Net loss | — | — | — | — | — | — | (2,551,488 | ) | (2,551,488 | ) | ||||||||||||||
BALANCE—December 31, 2003 | 1,588,000 | 250,000 | 250,000 | — | 10,430,696 | — | (13,087,375 | ) | (2,406,679 | ) | ||||||||||||||
Issuance of common stock at $0.157 per share | 41,685 | 6,563 | — | — | — | — | — | 6,563 | ||||||||||||||||
| Compensation expense related to stock options | — | — | — | — | 28,823 | (23,219 | ) | — | 5,604 | |||||||||||||||
| Exchange of shares under recapitalization | (1,629,685 | ) | (256,563 | ) | — | — | — | — | — | (256,563 | ) | |||||||||||||
| Issuance of shares under recapitalization | 9,482,015 | 94,820 | — | — | 161,743 | — | — | 256,563 | ||||||||||||||||
| Capital contributions | — | — | — | — | 2,295,731 | — | — | 2,295,731 | ||||||||||||||||
| Issuance of 14,887 warrants | — | — | — | — | 11,898 | — | — | 11,898 | ||||||||||||||||
| Issuance of common stock in exchange for licenses | 1,122,569 | 11,226 | — | — | 8,988,774 | — | — | 9,000,000 | ||||||||||||||||
| Net loss | — | — | — | — | — | — | (27,186,918 | ) | (27,186,918 | ) | ||||||||||||||
BALANCE—December 31, 2004 | 10,604,584 | 106,046 | 250,000 | — | 21,917,665 | (23,219 | ) | (40,274,293 | ) | (18,273,801 | ) | |||||||||||||
Issuance of common stock (unaudited) | 222,217 | 2,222 | — | — | 561,501 | — | — | 563,723 | ||||||||||||||||
| Compensation expense related to stock options (unaudited) | — | — | — | — | 300,049 | (257,739 | ) | — | 42,310 | |||||||||||||||
| Net loss (unaudited) | — | — | — | — | — | — | (1,664,378 | ) | (1,664,378 | ) | ||||||||||||||
BALANCE—June 30, 2005 (unaudited) | 10,826,801 | $ | 108,268 | 250,000 | $ | — | $ | 22,779,215 | $ | (280,958 | ) | $ | (41,938,671 | ) | $ | (19,332,146 | ) | |||||||
See notes to consolidated financial statements.
F-5
ADVANCED LIFE SCIENCES HOLDINGS, INC. AND SUBSIDIARY
(A Development Stage Company)
CONSOLIDATED STATEMENTS OF CASH FLOWS
| | | | | Period From Inception (January 1, 1999) Through December 31, 2004 | | | Period From Inception (January 1, 1999) Through June 30, 2005 | |||||||||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| | Years ended December 31, | Six months ended June 30, | ||||||||||||||||||||||||
| | 2002 | 2003 | 2004 | 2004 | 2005 | |||||||||||||||||||||
| | | | | | (Unaudited) | (Unaudited) | ||||||||||||||||||||
| CASH FLOWS FROM OPERATING ACTIVITIES: | ||||||||||||||||||||||||||
| Net loss | $ | (1,261,551 | ) | $ | (2,551,488 | ) | $ | (27,186,918 | ) | $ | (40,274,293 | ) | $ | (1,123,487 | ) | $ | (1,664,378 | ) | $ | (41,938,671 | ) | |||||
| Adjustments to reconcile net loss to net cash flows used in operating activities: | ||||||||||||||||||||||||||
| Depreciation and amortization | 40,508 | 49,620 | 68,334 | 198,340 | 29,668 | 36,377 | 234,717 | |||||||||||||||||||
| Non-cash interest expense | — | — | 132 | 132 | — | 7,509 | 7,641 | |||||||||||||||||||
| Amortization of deferred compensation | — | — | 5,604 | 20,701 | 801 | 42,310 | 63,011 | |||||||||||||||||||
| Non-cash research and development | — | — | 24,466,667 | 24,466,667 | — | — | 24,466,667 | |||||||||||||||||||
| Loss on disposal | — | 11,533 | — | 11,533 | — | — | 11,533 | |||||||||||||||||||
| Changes in operating assets and liabilities: | ||||||||||||||||||||||||||
| Accounts receivable | 500 | — | (24,868 | ) | (24,868 | ) | (26,770 | ) | 13,772 | (11,096 | ) | |||||||||||||||
| Prepaid expenses and other current assets | — | (29,833 | ) | 4,027 | (25,806 | ) | 19,726 | 8,061 | (17,745 | ) | ||||||||||||||||
| Other assets | — | 8,343 | 5,602 | (1,452 | ) | 427 | (3,330 | ) | (4,782 | ) | ||||||||||||||||
| Accounts payable | (81,478 | ) | 149,131 | 45,309 | 255,880 | (98,076 | ) | 127,980 | 383,860 | |||||||||||||||||
| Accrued expenses | 40,471 | 1,822 | 111,797 | 185,348 | 44,797 | (142,526 | ) | 42,822 | ||||||||||||||||||
| Licenses payable | — | — | — | — | — | (1,000,000 | ) | (1,000,000 | ) | |||||||||||||||||
| Accrued interest on debt | 155,000 | 171,017 | 193,239 | 1,148,985 | 96,616 | 98,861 | 1,247,846 | |||||||||||||||||||
| Net cash flows used in operating activities | (1,106,550 | ) | (2,189,855 | ) | (2,311,075 | ) | (14,038,833 | ) | (1,056,298 | ) | (2,475,364 | ) | (16,514,197 | ) | ||||||||||||
| CASH FLOWS FROM INVESTING ACTIVITIES: | ||||||||||||||||||||||||||
| Capital expenditures | (16,565 | ) | (105,409 | ) | (90,777 | ) | (296,233 | ) | (67,675 | ) | (4,164 | ) | (300,397 | ) | ||||||||||||
| Net cash flows used in investing activities | (16,565 | ) | (105,409 | ) | (90,777 | ) | (296,233 | ) | (67,675 | ) | (4,164 | ) | (300,397 | ) | ||||||||||||
| CASH FLOWS FROM FINANCING ACTIVITIES: | ||||||||||||||||||||||||||
| Proceeds from issuance of common stock and capital contributions | 1,161,000 | 2,337,728 | 2,295,731 | 7,209,575 | 1,080,780 | 563,660 | 7,773,235 | |||||||||||||||||||
| Proceeds from issuance of notes payable | — | — | 235,000 | 7,408,691 | — | 1,267,000 | 8,675,691 | |||||||||||||||||||
| Proceeds from grants | 500,000 | 500,000 | ||||||||||||||||||||||||
| Proceeds from stock options exercised | — | — | 6,563 | 6,563 | 63 | 63 | 6,626 | |||||||||||||||||||
| Payments on capital leases | (26,992 | ) | — | (2,090 | ) | (95,208 | ) | — | (5,799 | ) | (101,007 | ) | ||||||||||||||
| Net cash flows provided by financing activities | 1,134,008 | 2,337,728 | 2,535,204 | 14,529,621 | 1,080,843 | 2,324,924 | 16,854,545 | |||||||||||||||||||
| NET INCREASE IN CASH | 10,893 | 42,464 | 133,352 | 194,555 | (43,130 | ) | (154,604 | ) | 39,951 | |||||||||||||||||
| CASH—Beginning of period | 7,846 | 18,739 | 61,203 | — | 61,203 | 194,555 | — | |||||||||||||||||||
| CASH—End of period | $ | 18,739 | $ | 61,203 | $ | 194,555 | $ | 194,555 | $ | 18,073 | $ | 39,951 | $ | 39,951 | ||||||||||||
| SUPPLEMENTAL DISCLOSURE OF CASH FLOW INFORMATION: | ||||||||||||||||||||||||||
| Cash paid during the period for interest | $ | 7,352 | $ | 3,958 | $ | 1,511 | $ | 16,231 | $ | — | $ | 97,645 | $ | 113,876 | ||||||||||||
| SUPPLEMENTAL DISCLOSURE OF NONCASH TRANSACTIONS: | ||||||||||||||||||||||||||
| Noncash investment activity—purchases of fixed assets under capital leases | $ | — | $ | — | $ | 41,438 | $ | 134,556 | $ | — | $ | — | $ | 134,556 | ||||||||||||
| Noncash financing activity—issuance of common stock for licenses | — | — | 9,000,000 | 9,000,000 | — | — | 9,000,000 | |||||||||||||||||||
| Offering costs | — | — | — | — | — | 948,785 | 948,785 | |||||||||||||||||||
| Clinical supplies | — | — | 533,333 | 533,333 | — | — | 533,333 | |||||||||||||||||||
| Debt discount | — | — | 11,898 | 11,898 | — | — | 11,898 | |||||||||||||||||||
| Deferred financing costs | $ | — | $ | — | $ | 15,000 | $ | 15,000 | $ | — | $ | 15,000 | $ | 30,000 | ||||||||||||
See notes to consolidated financial statements.
F-6
ADVANCED LIFE SCIENCES HOLDINGS, INC. AND SUBSIDIARY
(A Development Stage Company)
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
YEARS ENDED DECEMBER 31, 2004, 2003 AND 2002
1. Nature of Business
On January 1, 1999, MediChem Life Sciences, Inc. ("MediChem") contributed all of the net assets of its proprietary drug development business, including MediChem's 50% interest in Sarawak MediChem Pharmaceuticals, Inc. ("SMP") to a wholly owned subsidiary, Advanced Life Sciences, Inc. ("ALS Inc.") in exchange for 1,588,000 shares of common stock.
On June 14, 1999, MediChem exchanged its investment in 100% of the outstanding common stock of ALS, Inc. for nonvoting preferred stock issued by ALS Inc. affecting a spin-off of ALS Inc. No gain or loss was recognized in MediChem's consolidated statements of operations upon the spin-off of ALS Inc.
Prior to the spin-off, Dr. Michael Flavin, the sole stockholder, owned 100% of MediChem and ALS Inc., then a wholly owned subsidiary of MediChem. As a result of the spin-off, Dr. Flavin became the sole common stockholder of ALS Inc. MediChem holds 100% of the preferred stock of ALS Inc., which was issued in exchange for common stock held at the June 14, 1999 spin-off.
On August 24, 2001, ALS Inc. formed a general partnership, Advanced Life Sciences General Partnership ("ALS GP"), with Dr. Flavin, ALS Inc.'s chief executive officer. ALS Inc. contributed all of its assets and liabilities to ALS GP, with the exception of debt due to Dr. Flavin. In 2002, Dr. Flavin transferred his interest in ALS GP to Flavin Ventures, LLC ("Flavin Ventures"), which he controls.
In March 2002, MediChem was acquired by deCODE genetics, Inc. ("deCODE"). As a result of the merger, Dr. Flavin's ownership in the merged entity was reduced to a noncontrolling interest with Dr. Flavin holding approximately 1% of the common shares of deCODE genetics, Inc. After the merger was complete, Dr. Flavin resigned his position as chief executive officer of MediChem to focus on his role as chief executive officer of ALS Inc.
On December 13, 2004, ALS Inc. executed a plan of reorganization. Advanced Life Sciences Holdings, Inc. ("the Company"), a Delaware corporation, was formed with Dr. Flavin as its chief executive officer. Common shareholders of ALS, Inc. exchanged their 1,629,685 common shares for 1,629,685 common stock of the Company. Under the terms of the reorganization, Flavin Ventures exchanged its 40% ownership of ALS GP for 7,852,330 shares of the Company's common stock. After the exchange was completed, ALS GP was dissolved, its assets and liabilities assumed by ALS Inc.
On December 13, 2004, according to the plan of reorganization, ALS Ventures, LLC ("Ventures"), a Delaware Limited Liability Company was formed. After its formation, Dr. Flavin and Flavin Ventures simultaneously contributed all of their common shares, 1,588,000 and 7,852,330, respectively, to Ventures. ALS Ventures is not included in the consolidated financial statements of the Company.
The Company's principal business activity is to conduct new drug research and development in the fields of infectious disease, oncology and inflammation. Since inception, the Company has devoted substantially all of its efforts to activities such as financial planning, capital raising and product development, and has not derived significant revenues from its primary business activity. Accordingly, the Company is in the development stage, as defined by Statement of Financial Accounting Standards ("SFAS") No. 7,Accounting and Reporting by Development Stage Enterprises.
Business and Credit Risks—The Company is subject to risks and uncertainties common to drug discovery companies, including technological change, potential infringement on intellectual property of
F-7
third parties, new product development, regulatory approval and market acceptance of its products, activities of competitors and its limited operating history.
The Company accomplishes its primary business objectives through focus on the development of new drugs in the fields of infectious disease, inflammation and oncology. In December 2004, the Company entered into an exclusive license agreement with Abbott Laboratories ("Abbott") to in-license Cethromycin, a novel ketolide antibiotic currently in Phase III clinical development for the treatment of resistant respiratory infections. The Company's rights under this agreement give it an exclusive worldwide license to commercialize Cethromycin, except in Japan. Cethromycin has been shown to be active against upper and lower respiratory tract pathogens, including penicillin- and macrolide-resistant Gram-positive bacteria. In addition, the license agreement provides the Company with the exclusive worldwide rights to ABT-210, a novel second-generation ketolide antibiotic in preclinical development.
The Company currently has two compounds entering Phase I human clinical trials. Both compounds, an anti-inflammatory drug and a melanoma treatment are scheduled to begin Phase I clinical trials in 2005.
Should the Company's research and development efforts fail, the Company would likely be unable to reach profitable operations.
In addition to these compounds, the Company has a promising pipeline of product candidates in preclinical development. These include multiple products derived from natural products to treat infectious diseases, a novel class of synthetic chemotherapeutics to treat various cancers and small molecule inhibitors of amyloid fibrils that characterize Alzheimer's disease.
The Company and ALS Inc. have incurred losses since their inception in January 1999, and, prior to their inception, the business activities of ALS Inc. as a segment of MediChem incurred losses. The Company has funded its operations to date primarily from debt financings and capital contributions from its controlling stockholder. The Company will not be generating revenues or realizing cash flows from operations in the near term, and will require additional equity or debt financing to meet working capital needs and to fund operating losses. The Company is currently seeking additional debt or equity financing to fund clinical trials. Although management believes the Company could obtain such financing, there can be no assurances that such financing will be available in the future at terms acceptable to the Company, if at all. The accompanying financial statements do not include any adjustments that might result from the outcome of these uncertainties.
The Company has derived its revenues to date from grant programs sponsored by the U.S. government, management services provided to the SMP joint venture and Flavin Ventures and royalties received from technology licenses. Such revenue is dependent upon the continuance of these programs and licenses.
2. Summary of Significant Accounting Policies
Basis of Presentation—The consolidated financial statements include the accounts of the Company and its wholly-owned subsidiary ALS Inc. All significant intercompany balances and transactions have been eliminated.
F-8
Cash and Cash Equivalents—The Company considers all highly liquid investments with original maturities of three months or less when purchased to be cash equivalents.
Furniture and Equipment—Furniture and equipment are recorded at cost and depreciated using the straight-line method over the estimated useful life of three to five years. Assets under capital leases were recorded at the present value of the minimum lease payments and are amortized over the shorter of their estimated useful lives or the term of the respective leases using the straight-line method. Maintenance and repair costs are expensed as incurred. Amounts capitalized as fixed assets under the capital lease amounted to $41,000 and $0, net of accumulated depreciation of $2,300 and $0, as of December 31, 2004 and 2003, respectively. Depreciation expense was approximately $68,000, $50,000 and $41,000 for 2004, 2003 and 2002, respectively.
Clinical trial supplies—Clinical trials supplies consist of clinical trial raw materials purchased from Abbott and is valued at the lower of cost or market. The Company tests the supplies every two years to determine its purity and compliance with FDA prescribed clinical protocols. When processed into tablets for clinical trial consumption, supplies will be specifically identified and expensed to research and development costs as consumed in clinical trials.
Revenue Recognition—Small Business Innovation Research grant revenue is recorded as expenses are incurred and services performed in accordance with the terms of the grant agreements. Royalty revenue is earned from a license of the Company's chemical process technology. Under the license, revenue is earned based upon a percentage of reported net sales from MediChem Life Sciences, a subsidiary of deCODE, for services that incorporate the licensed technology (see Note 4). Management fees, which are generated from a shared services agreement with SMP, are recognized on the cash basis as the collectibility of such fees is not reasonably assured. Management fees derived from Flavin Ventures, LLC are recorded as services are provided.
Fair Value of Financial Instruments—The carrying amount of the Company's financial instruments, which include cash equivalents, accounts payable, accrued expenses and long-term obligations, approximate their fair values.
Research and Development Expenses—Research and development expenses consist of internal research costs and fees paid for contract research in conjunction with the research and development of anti-infective, anti-cancer and anti-inflammatory therapeutic agents. All such costs are expensed as incurred.
Income Taxes—Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. A valuation allowance is provided when it is more likely than not that some portion or all of a deferred tax asset will not be realized.
Milestone and Intellectual Property Costs—Milestone and intellectual property costs consist of milestone payments made for agreed-upon achievements in the course development of the compounds the Company licenses. Milestone payments are expensed when the milestone is achieved.
F-9
Stock-Based Compensation—In December 2004, the FASB issued Statement 123(R) that will require compensation cost related to share-based payment transactions to be recognized in the financial statements. Compensation cost will be measured based on the grant-date fair value of the equity or liability instruments issued. Compensation expense will be recognized over the period that an employee provides service in exchange for the award.
Effective January 1, 2004, the Company adopted the fair value method, which is considered the preferable accounting method, of recording stock-based employee compensation as contained in SFAS No. 123,Accounting for Stock-Based Compensation. As prescribed in SFAS No. 123(R),Share-Based Payment, the Company elected to use the "prospective method." The prospective method requires expense to be recognized for all awards granted, modified or settled in the year of adoption. Historically, the Company applied the intrinsic method as provided in Accounting Principles Board Opinion No. 25,Accounting for Stock Issued to Employees and related interpretations and accordingly, no compensation cost had been recognized for stock options issued to employees in prior years. As a result of adopting the fair value method for stock compensation, all future awards will be expensed over the stock options vesting period. The Company utilizes the Black-Scholes option pricing model to estimate the fair value of options granted.
The following illustrates the pro forma effect on net loss and loss per share as if the Company had applied the fair value recognition provisions of SFAS No. 123(R) in each period:
| | Pro Forma | ||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | 2002 | 2003 | 2004 | ||||||||
| Net loss available to common shareholders | $ | (1,436,551 | ) | $ | (2,726,488 | ) | $ | (27,361,918 | ) | ||
| Stock-based employee compensation: | |||||||||||
| Less fair value-based expense | 12,904 | 16,750 | — | ||||||||
| Pro forma net loss available to common shareholders | $ | (1,449,455 | ) | $ | (2,743,238 | ) | $ | (27,361,918 | ) | ||
| Loss per share: | |||||||||||
| Basic—as reported | $(0.90 | ) | $(1.72 | ) | $(13.27 | ) | |||||
| Basic—pro forma | (0.91 | ) | (1.73 | ) | (13.27 | ) | |||||
Disclosure About Segments of an Enterprise—Operating segments are identified as components of an enterprise about which separate discrete financial information is available for evaluation by the chief operating decision-maker, or decision-making group, in making decisions regarding resource allocation and assessing performance. To date, the Company has viewed its operations and manages its business as principally one segment.
Use of Estimates—The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements. Estimates also affect the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
F-10
Recent Accounting Pronouncements—In November 2003, the FASB issued Staff Position 150-3,Effective Date, Disclosures, and Transition for Mandatorily Redeemable Financial Instruments of Certain Nonpublic Entities and Certain Mandatorily Redeemable Noncontrolling Interests Under FASB 150,deferring the effective date for applying the provisions of SFAS No. 150,Accounting for Certain Financial Instruments with Characteristics of Both Liabilities and Equity. SFAS No. 150 requires that certain instruments be classified as liabilities in the statements of financial position. For nonpublic entities, the classification, measurement and disclosure provisions of SFAS No. 150 have been deferred indefinitely for financial instruments that are not mandatorily redeemable on fixed dates for fixed or determinable amounts. SFAS No. 150 is effective for financial instruments that are mandatorily redeemable on fixed dates for fixed or determinable amounts for fiscal periods beginning after December 15, 2004, and its adoption is not expected to have a material impact on the Company's financial position or results of operations.
Reclassifications—Certain reclassifications have been made to the prior years' financial statements to conform to the 2004 classifications.
3. Joint Venture
SMP is a joint venture between the State Government of Sarawak, Malaysia (the "Sarawak Government") and the Company. The Sarawak Government, through Craun Sdn Bhd, and ALS Inc. each maintain a 50% share in SMP.
Under the joint venture agreement (the "Agreement"), the Sarawak Government has contributed $9,000,000 to SMP through stock subscription payments based on the attainment of certain milestone achievements outlined in the Agreement. The Company has contributed certain intellectual property and has committed to make scientists available to undertake the research and clinical trials, to train scientists nominated by the Sarawak Government and to permit such scientists to participate in the clinical trials, manufacture, marketing and distribution of any commercial product resulting from the research and development effort. Additionally, the Sarawak Government has provided long-term loans to SMP of $12,000,000 through December 31, 2004. Accrued interest on such loans was approximately $4,690,000 as of December 31, 2004.
The Company accounts for its investment in SMP under the modified equity method of accounting, which provides that the Company record its proportional share of earnings once the accumulated deficit incurred by SMP has been recovered. Accordingly, as of December 31, 2004 and 2003, the Company's investment in SMP had a carrying value of zero.
According to the terms of the loan agreement with the Sarawak Government, $9 million of SMP's debt is past due and $0.5 million and $2.5 million are due in the third and fourth quarters of 2005, respectively. As a result of this default in payment by SMP, the Sarawak Government has the option under the loan agreement to take over the control and management of the Calanolide A project or the affairs of the joint venture until such time as the loan is repaid in full. To date, the Sarawak Government has not pursued collection of this debt or sought to control or manage the Calanolide A project. On July 15, 2005, the Sarawak Government exercised certain of its contractual rights by notifying SMP of its intention to appoint its representative as an officer of SMP for purposes of
F-11
assuming control of the affairs and management of SMP effective immediately. The Sarawak Government also expressly reserved all contractual and legal remedies available in light of the default. Ultimately, the Company believes that in order for the joint venture to continue, the terms of the debt must be amended to extend its maturity to a date after commercialization of Calanolide A.
The Company previously commenced discussions with the Sarawak Government to restructure and extend the debt and determine the source of financing for Phase IIa clinical trials of Calanolide A. The Company has proposed to the Sarawak Government that, for the first time, the Company and the Sarawak Government would jointly fund the next development phase of Calanolide A through future advances of debt or issuances of equity. The Company currently estimates that it would cost approximately $2 million to conduct a Phase IIa clinical trial of Calanolide A. The Company does not intend to expend any meaningful funds advancing the development of Calanolide A unless and until the joint venture's debt is restructured and the Company reaches a satisfactory agreement with the Sarawak Government to amend the joint venture relationship and establish the parties' respective obligations regarding the future funding of the next phase of developing Calanolide A. As a result, the Company is unable to predict when, or if at all, the Company will proceed with its plans to commence Phase IIa clinical trials for Calanolide A.
The term of the joint venture agreement continues indefinitely unless terminated by the written agreement of both parties not to proceed further with the development of Calanolide A. In the event only one of the parties wishes to discontinue development of Calanolide A, the other party has the right, but not the obligation, to purchase the discontinuing party's equity interest in the joint venture for $9 million. If the other party does not choose to exercise its purchase right within six months of receiving written notice from the discontinuing party, then the joint venture is deemed mutually terminated. Upon such mutual termination, the joint venture agreement provides that any liabilities of the joint venture will be settled by the parties equally and all patents or licenses of the joint venture shall be reassigned back to the Company, including rights to clinical trials. We believe that the equal settlement of liabilities provision in the joint venture agreement was intended to recognize the 50/50 equity ownership of the joint venture and does not constitute the Company's guaranty of the joint venture's indebtedness to the Sarawak Government. Consequently, if the joint venture is mutually terminated, the joint venture partners would work together to monetize the joint venture's assets for the purpose of satisfying its liabilities, which currently are solely comprised of indebtedness to a 50% partner, the Sarawak Government. In the event the joint venture's assets are insufficient to settle its liabilities, the Company does not believe that the equal settlement of liabilities provision obligates it to contribute funds to the joint venture.
F-12
A summary of financial information for SMP as of December 31, 2002, 2003 and 2004, and for the years then ended follows:
| | 2002 | 2003 | 2004 | ||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|
| | | | (unaudited) | ||||||||
| Balance sheet information: | |||||||||||
| Total assets | $ | 129,175 | $ | 25,025 | $ | 11,052 | |||||
| Total liabilities | 14,181,286 | 16,258,783 | 17,822,053 | ||||||||
| Accumulated deficit | (23,162,945 | ) | (25,356,535 | ) | (26,933,777 | ) | |||||
| Stockholders' deficit | (14,052,111 | ) | (16,209,324 | ) | (17,811,000 | ) | |||||
| Results of operations information—net loss | $ | (2,537,453 | ) | $ | (2,169,156 | ) | $ | (1,577,243 | ) | ||
4. Related Party Transactions
Subsequent to the spin-off from MediChem on June 14, 1999, the Company and MediChem entered into a master laboratory services agreement under which the Company is provided services. The Company incurred no costs for these services in 2004 and 2003.
The Company did not make any payments in 2004, 2003 and 2002 to MediChem for its achievement of certain milestones in accordance with the terms of research contracts between MediChem and the Company. From inception, the Company has paid $2,350,000 in milestone payments to MediChem. In addition, the Company paid MediChem $400,000 for the purchase of a patent in 1999. These expenses are recorded as contracted research and development-related party expenses in the accompanying consolidated statements of operations.
In March 2002, the existing license agreements between the Company and MediChem were renegotiated at the time of MediChem's merger with deCODE. All of the terms and obligations under the previous licenses were terminated, and a new agreement was formed between the Company and deCODE/MediChem. Under the new agreement, the Company assigned and relinquished all of its rights, title and interest in certain patented technologies in exchange for a 50% royalty on any and all income received by deCODE/MediChem resulting from the licensing, cross licensing or sale to a third party of the patented technologies. In 2004, 2003 and 2002 the Company received $10,000, $0 and $0, respectively, in royalty payments relating to these technologies. With the exception of the master laboratory services agreement, all other agreements between the Company and MediChem were terminated at the time of MediChem's merger with deCODE.
In 2001, the Company initiated a management services agreement with SMP whereby the Company provides administrative management services to SMP. In return, the Company charges SMP a flat monthly service fee. Management fees recognized as revenue under this agreement totaled $0 in 2004, $118,563 in 2003 and $495,252 in 2002. During 2003, management deemed that the collectibility of the monthly management fee was not reasonably assured. Consequently, management concluded that fee revenue will be recognized on the cash basis. As only three months of fees were paid, only three months of revenue was recognized during 2003. The remaining nine months have been billed, and the
F-13
Company continues to provide management services to SMP. Billed and unrecognized revenue in 2004 and 2003 totaled approximately $160,000 and $185,000, respectively.
In January 2004, the Company initiated a management services agreement with Flavin Ventures whereby the Company provides administrative management services to Flavin Ventures and its wholly-owned subsidiaries Shamrock Structures, BioStart Property Group and Molecular Formulations. The agreement also requires Flavin Ventures and its related subsidiaries to reimburse the Company for rent and related facility costs based upon the actual square footage occupied by each of the companies. Revenue recognized related to this agreement was approximately $272,000 in 2004.
The Company's controlling stockholder has loaned to the Company $2.0 million (see Note 5).
The Company's line of credit has been guaranteed by Flavin Ventures, the controlling stockholder and his wife and the Company's President (see Note 5).
The Company leases facilities from Flavin Ventures (see Note 10).
5. Long Term Obligations
On September 1, 2001, the Company entered into a $2.0 million promissory note with the controlling stockholder of the Company which bears interest at 7.75%. Accrued but unpaid interest is added to the outstanding principal annually. Principal plus accrued interest is due in a lump sum on September 1, 2006. As of December 31, 2004 and 2003, the Company had $2,566,603 and $2,377,684, respectively, outstanding under the note.
On December 21, 2004, the Company entered into a $3.0 million line of credit with a local financial institution. Interest is based upon the prime rate plus 0.75% and is payable monthly. The principal balance is due upon demand or by December 21, 2006, if no demand is made. The financial institution also received a warrant to purchase 14,887 of the Company's common stock at an exercise price of $8.02 per share, subject to certain adjustments. The warrant was exercisable at the time the note was issued and will terminate, if not exercised, on December 21, 2009. The warrant was valued, using the Black-Scholes option pricing model, at approximately $11,900. The amount was recorded as a discount to long-term obligations and is being amortized to interest expense over the term of the note. The note is secured by the assets of the Company and guaranteed by Flavin Ventures, the controlling stockholder and his wife and the Company's President.
6. Stockholders' Deficit
Authorized Capital—As of December 31, 2004, the authorized capital stock of the Company consists of 15,880,000 shares of voting common stock authorized for issuance with a par value of $0.01 per share. ALS Inc., our operating subsidiary, has issued and outstanding 250,000 shares of Series A Cumulative Preferred Stock ("Series A Preferred Stock"), which is held by MediChem. The Series A Preferred Stock is nonvoting except as to certain matters including the issuance of any class of stock ranking senior to the shares of Series A Preferred Stock, or the merger or consolidation of ALS Inc. if such merger would adversely affect the powers, preferences or rights of the holder of Series A
F-14
Preferred Stock. The Series A Preferred stockholder is entitled to receive dividends payable in cash at the rate of 7.0% per annum per share on the amount of liquidation preference of the shares.
MediChem received the Series A Preferred Stock on June 14, 1999 in connection with a recapitalization of ALS Inc. MediChem and ALS Inc. effectuated the recapitalization by exchanging the 1,588,000 voting common shares originally issued to MediChem for 250,000 shares of the Series A Preferred Stock. Concurrently, the Company issued the 1,588,000 shares of voting common stock to the controlling stockholder of MediChem for $250,000.
Liquidation Preferences—Upon dissolution or winding up of ALS Inc., whether voluntary or involuntary, the holders of the shares of the Series A Preferred Stock are entitled to receive a liquidation preference of $10 per share, plus all accrued and unpaid dividends. The total liquidation preference values as of December 31, 2004 and 2003 are approximately $3,470,000 and $3,295,000 respectively, including approximately $970,000 and $795,000 of unpaid dividends.
In August 2001, in connection with the formation of ALS GP, the controlling stockholder converted approximately $5.8 million from debt to equity. The conversion was accounted for as a capital contribution, and no common or preferred shares were issued as a result of the conversion.
7. Incentive Stock Plan
Effective December 22, 1999, the Company's Board of Directors and stockholders adopted and approved the Company's Incentive Compensation Plan (the "Plan"). The Plan provides for the grant of stock options and the award of restricted stock to selected officers and consultants of the Company. Options in the amount of 269,960 shares were granted at an exercise price of $0.157 per share and expire 10 years from the date of grant. The Plan provides for graduated vesting whereby options to purchase1/36th of the shares granted vest at the end of each month following the date of grant. Awards of restricted stock are subject to certain terms, conditions, restrictions and limitations as determined by the Board of Directors.
In April 2002, the Company granted an additional 221,526 options to employees and nonemployees. These options were granted at an exercise price of $0.157 per share and expire 10 years from the grant date. These options vest ratably over three years, similar to the 1999 options. However, these options were granted in certain cases for prior service. The vesting period for these options begins on the dates specified in the stock option agreement rather than the grant date.
On January 1, 2003, the Company granted 66,497 options to employees. These options were also granted at an exercise price of $0.157 per share and expire 10 years from the grant date. These options vest ratably over three years. The vesting period for all options granted begins on the date of issuance.
On June 1, 2004, the Company granted 110,364 options to employees. These options were granted at an exercise price of $0.157 per share and expire 10 years from the grant date. The vesting period for all options granted begins on the date of grant.
F-15
The following schedule details the grants under the Company's Plan for the years ended December 31, 2002, 2003 and 2004:
| Options | Shares | Exercise Price | |||
|---|---|---|---|---|---|
| Outstanding at January 1, 2002 | 269,960 | $ | 0.157 | ||
| Granted | 221,526 | 0.157 | |||
| Exercised | — | — | |||
| Terminated | 2,084 | 0.157 | |||
| Outstanding at December 31, 2002 | 489,402 | $ | 0.157 | ||
| Granted | 66,497 | 0.157 | |||
| Exercised | |||||
| Terminated | 1,599 | 0.157 | |||
| Outstanding at December 31, 2003 | 554,300 | $ | 0.157 | ||
| Granted | 110,366 | $ | 0.157 | ||
| Exercised | 41,685 | 0.157 | |||
| Terminated | 3,033 | 0.157 | |||
| Outstanding at December 31, 2004 | 619,948 | $ | 0.157 | ||
| Options exercisable at December 31, 2004 | 513,686 | $ | 0.157 | ||
The fair value of options granted in 2004 was estimated to be approximately $29,000 on the date of grant using the Black-Scholes option pricing model with the following assumptions: no dividend yield, volatility of 54%, risk-free interest rate of 3.5% and expected lives of 3 years. Volatility was determined from the average of similar NASDAQ firms. The fair value of the Company's common stock was estimated based on contemporaneous valuations performed by management with requisite experience. The following table summarizes stock options outstanding at December 31, 2004:
| Options Outstanding | Options Exercisable | ||||||||||||
|---|---|---|---|---|---|---|---|---|---|---|---|---|---|
| Exercise Price | Number Outstanding | Weighted Average Remaining Contractual Life | Weighted Average Exercise Price | | | ||||||||
| $ | 0.157 | 619,948 | 7 years | $ | 0.157 | 513,686 | $ | 0.157 | |||||
F-16
During the 12-month period ended June 30, 2004, the Company granted stock options with exercise prices as follows:
| Grants Made During Quarter Ended | Number of Options Granted | Weighted- Average Exercise Price | Weighted- Average Fair Value per Share | |||||
|---|---|---|---|---|---|---|---|---|
| September 30, 2003 | — | — | — | |||||
| December 31, 2003 | — | — | — | |||||
| March, 31 2004 | — | — | — | |||||
| June 30, 2004 | 110,366 | $ | 0.157 | $ | 4.33 | |||
The fair value per share is being recognized as compensation expense over the applicable vesting period (which also equals the service period).
8. Income Taxes
Net deferred tax assets consist primarily of net operating loss ("NOL") carryforwards. Realization of future tax benefits related to deferred tax assets is dependent on many factors, including the Company's ability to generate future taxable income in the near term. Due to the uncertainty of future earnings, management is unable to predict whether these net deferred tax assets will be realized and, accordingly, has recorded a full valuation allowance against these assets.
NOL carryforwards of approximately $8.8 million for income tax purposes are available to offset future taxable income. If not utilized, these carryforwards will expire in varying amounts beginning in the year 2014.
The following is a summary of the significant components of the Company's deferred tax assets as of December 31, 2003 and 2004:
| | 2003 | 2004 | ||||||
|---|---|---|---|---|---|---|---|---|
| Deferred tax assets: | ||||||||
| Net operating losses | $ | 2,908,000 | $ | 3,464,000 | ||||
| Partnership loss allocation | 1,263,000 | — | ||||||
| In-process research and development | — | 9,503,000 | ||||||
| Less valuation allowance | (4,171,000 | ) | (12,967,000 | ) | ||||
| Deferred tax asset—net | $ | — | $ | — | ||||
9. Employee Benefit Plan
The Company has established a 401(k) plan that covers all employees who meet minimum eligibility requirements. Eligible persons can defer a portion of their annual compensation into the 401(k) plan subject to certain limitations imposed by the Internal Revenue Code. Employees' elective deferrals are immediately vested upon contribution to the 401(k) plan. The Company matches employee contributions at a rate of 50% up to 6% of the employee's annual salary. Company
F-17
contributions vest 25% per year after the first year of service has been completed. Approximately $32,000, $32,000 and $18,000 was recognized as expense in 2004, 2003 and 2002, respectively.
10. Commitments
Lease Obligations—The Company leases facilities under an operating lease with Flavin Ventures. The lease has a five-year term commencing on September 1, 2003, and may be renewed in writing 60 days prior to the lease termination date. The minimum monthly rental payment is $18,000 with an additional amount based on the Company's proportionate share of the variable building costs. Additionally, the Company leases capital equipment of approximately $41,000 under a capital lease.
Minimum annual payments under operating leases and the present value of minimum capital lease payments as of December 31, 2004 are as follows:
| | Operating Leases | Capital lease Obligations | ||||
|---|---|---|---|---|---|---|
| Year ending December 31, | ||||||
| 2005 | $ | 218,463 | $ | 17,421 | ||
| 2006 | 225,759 | 17,421 | ||||
| 2007 | 234,789 | 14,110 | ||||
| 2008 | 160,645 | — | ||||
| 2009 | — | — | ||||
| Total | $ | 839,656 | 48,952 | |||
| Less amount representing interest | 9,604 | |||||
| Present value of minimum lease payments | $ | 39,348 | ||||
Rental expense relating to the Flavin Ventures lease totaled $242,946, $74,785 and $0 in 2004, 2003 and 2002, respectively.
Employment Contracts—The Company has employment contracts with certain executive officers. In the event of termination without cause, the contracts provide for severance benefits ranging from six to 24 months of salary and benefit continuation.
Milestones and Royalties—In December 2004, the Company exercised its option to acquire a license to develop two compounds owned by Abbott. Under the terms of the agreement, certain payments become due over the next 12 months (see Note 11).
The Company has been funded since inception by capital contributions from its principal shareholder and through the issuance of debt. The Company plans to meet its future commitments as outlined in the previous table and those that arise from the Abbott transaction as discussed in Note 11, through additional sales of its common stock and the issuance of debt (see Note 12).
F-18
11. Research and Development Alliances
Abbott Laboratories—In October 2004, the Company executed an option to acquire two antibiotic compounds from Abbott. Under the terms of the option agreement, the Company paid Abbott a nonrefundable $1 million option fee in exchange for the right to acquire an exclusive world-wide license, with the exception of Japan, to develop, manufacture and commercialize each of the compounds.
In December 2004, the Company exercised its option to acquire the rights for both antibiotic compounds. The license agreement requires payments totaling $9 million and issuance of common stock with an estimated value of $9 million in exchange for an exclusive world-wide license to develop the compounds, with the exception of Japan, and purchase Abbott's existing supply of the compounds. Non-milestone based payments and stock issuances under the contract total $25 million, of which $24.5 million and $0.5 million have been classified as in-process research and development expense and clinical trial supplies, respectively. In-process research and development costs are expensed as the outcome of clinical trials is unknown and therefore FDA approval is uncertain and the Company believes there is no alternate use of the compounds should FDA approval not be achieved. Milestone payments totaling $40 million do not become payable under the contract until future milestones are achieved. Additionally, the Company will pay Abbott a royalty on global net sales of the products. As of December 31, 2004, the Company had made payments under its license agreement equal to $1 million. In 2005, the Company made additional payments of $1 million. Payments totaling $8 million that are due by September 1, 2005, or if earlier, five days following the closing of an initial public offering of the Company's common stock, have been classified as a current liability, as has a $5 million payment that is due by October 31, 2005.
University of Illinois at Chicago—In December 1999, the Company entered into a license agreement with the University of Illinois at Chicago ("UIC") to license an anti-melanoma compound. Under the terms of the agreement, the Company was granted an exclusive world-wide license in exchange for certain upfront, milestone and royalty payments. Upon execution, the Company paid UIC a $15,000 non-refundable license fee and $10,000 patent reimbursement fee both of which were charged to research and development expense. The Company is obligated under the contract to make four additional annual payments of $10,000 in reimbursement of previous incurred patent protection costs, which were made as of December 31, 2003. The Company is responsible for reimbursing UIC for all patent costs incurred subsequent to the license date. Patent reimbursement costs totaled approximately $72,000, $91,000 and $44,000 in 2004, 2003 and 2002, respectively. In December 2004, the Company filed an IND with the FDA for the anti-melanoma compound which triggered a $10,000 milestone payment to UIC. The milestone payment was classified as a research and development expense.
12. Subsequent Events
In January 2005, the Company made a payment of $750,000 to Abbott under the terms of the license agreement.
In February 2005, the Company issued 70,284 of its common shares to private investors for $563,660. Additionally, the Company issued 151,534 shares of common stock to Flavin Ventures in relation to $1,215,000 in capital contributions made to ALS GP between July and December 2004.
F-19
In February 2005, the Company issued 67,290 options to purchase shares of its common stock to employees and consultants of the Company, at an exercise price of $8.02 per share. The fair value of options granted in February 2005 was estimated to be approximately $300,000 on the date of grant using the Black-Scholes option pricing model and will be expensed ratably over the three-year vesting period of the options.
In March 2005, the Company was awarded a $500,000 government grant. Under the terms of the grant, proceeds are to be used to construct additional laboratory and office space at the Woodridge facility. In the event the Company does not create and retain 100 full-time positions by January 31, 2007, all issued grant funds become due to the grantor on a pro-rata basis, based on the percentage of the deficiency between the required number of created full-time jobs and the actual number of created full-time jobs at the facility. The grant funds were received by the Company in April 2005.
In April 2005, the Company made a payment of $250,000 to Abbott under the terms of the license agreement.
In May 2005, the Company increased its line of credit to $4.0 million. In connection with this increase, the Company's controlling stockholder entered into a subordination agreement with the bank under which he agreed to subordinate his $2.0 million loan to the Company to the line of credit. In anticipation of increasing the line of credit, on May 17, 2005, Flavin Ventures loaned to the Company $91,000 pursuant to a thirty-day bridge loan. The bridge loan was repaid in full on June 3, 2005.
On June 29, 2005, the Company amended and restated its certificate of incorporation to increase the number of authorized common shares to 60,000,000 and additionally authorized the Company to issue 5,000,000 preferred shares. No preferred shares have been issued to date.
The accompanying financial statements reflect a 3.97-for-one split of the Company's common stock, which became effective as of June 29, 2005. All share and per share information herein has been retroactively restated to reflect this split.
******
F-20
Common Stock
PROSPECTUS
| C.E. UNTERBERG, TOWBIN | THINKEQUITY PARTNERS LLC | |
MERRIMAN CURHAN FORD & CO. | ||
, 2005
Until , 2005, 25 days after the commencement of the offerings, all dealers that effect transactions in our common stock, whether or not participating in the offerings, may be required to deliver a prospectus. This is in addition to the dealers' obligation to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 13. Other Expenses of Issuance and Distribution
The following table sets forth fees payable by us in connection with the issuance and distribution of our common stock in the offerings. All such fees and expenses, except the Securities and Exchange Commission registration fee, are estimated:
| Securities and Exchange Commission Registration Fee | $ | 10,152 | |
| NASD Filing Fee | 9,125 | ||
| Nasdaq National Market Filing Fee | 100,000 | ||
| Transfer Agent Fees and Expenses | 15,000 | ||
| Printing Engraving Fees and Expenses | 100,000 | ||
| Legal Fees and Expenses | 500,000 | ||
| Accounting Fees and Expenses | 350,000 | ||
| Miscellaneous | 165,723 | ||
| TOTAL | $ | 1,250,000 | |
Advanced Life Sciences Holdings, Inc. will bear all of the expenses shown above.
Item 14. Indemnification of directors and officers
Subsection (a) of Section 145 of the Delaware General Corporation Law, or DGCL, provides that a corporation may indemnify any person who was or is a party or is threatened to be made a party to any threatened, pending or completed action, suit or proceeding, whether civil, criminal, administrative or investigative (other than an action by or in the right of the corporation) by reason of the fact that he is or was a director, officer, employee or agent of the corporation, or is or was serving at the request of the corporation as a director, officer, employee or agent of another corporation, partnership, joint venture, trust or other enterprise, against expenses (including attorneys' fees), judgments, fines and amounts paid in settlement actually and reasonably incurred by him in connection with such action, suit or proceeding if he acted in good faith and in a manner he reasonably believed to be in or not opposed to the best interests of the corporation, and, with respect to any criminal action or proceeding, had no reasonable cause to believe his conduct was unlawful. Section 145 further provides that a corporation similarly may indemnify any such person serving in any such capacity who was or is a party or is threatened to be made a party to any threatened, pending or completed action or suit by or in the right of the corporation to procure a judgment in its favor, against expenses (including attorneys' fees) actually and reasonably incurred in connection with the defense or settlement of such action or suit if he acted in good faith and in a manner he reasonably believed to be in or not opposed to the best interests of the corporation and except that no indemnification shall be made in respect of any claim, issue or matter as to which such person shall have been adjudged to be liable to the corporation unless and only to the extent that the Delaware Court of Chancery or such other court in which such action or suit was brought shall determine upon application that, despite the adjudication of liability but in view of all of the circumstances of the case, such person is fairly and reasonably entitled to indemnity for such expenses which the Court of Chancery or such other court shall deem proper.
Our amended and restated certificate of incorporation and by-laws provide that indemnification shall be to the fullest extent permitted by the DGCL for all current or former directors or officers of the Company.
As permitted by the DGCL, our amended and restated certificate of incorporation provides that directors of the Company shall have no personal liability to the Company or its stockholders for
II-1
monetary damages for breach of fiduciary duty as a director, except (i) for any breach of a director's duty of loyalty to the Company or its stockholders, (ii) for acts or omissions not in good faith or which involve intentional misconduct or knowing violations of law, (iii) under Section 174 of the DGCL or (iv) for any transaction in which a director derives an improper personal benefit.
Item 15. Recent Sales of Unregistered Securities
The following list sets forth information regarding all securities sold by us since January 1, 2002:
- (1)
- On December 13, 2004, in connection with our recapitalization, we issued (i) 7,852,330 shares of common stock to Flavin Ventures, LLC at a purchase price of $1.46 per share, giving effect to unallocated contributions of capital made by Flavin Ventures, LLC; (ii) 1,588,000 shares of common stock to the Michael T. Flavin Revocable Trust, in exchange for 1,588,000 shares of common stock of Advanced Life Sciences, Inc., our current operating subsidiary and (iii) 41,685 shares of common stock to Karen Stec, in exchange for 41,685 shares of common stock of Advanced Life Sciences, Inc.
- (2)
- On December 13, 2004, in connection with and as consideration for our license of cethromycin and ABT-210 from Abbott Laboratories, we issued 1,122,569 shares of our common stock to Abbott Laboratories.
- (3)
- On December 13, 2004, we issued a warrant to purchase up to 14,887 shares of our common stock at an exercise price of $8.02 per share to Leaders Bank, in connection with entering our bank line of credit with Leaders Bank.
- (4)
- On February 8, 2005, we issued 151,534 shares of common stock to Flavin Ventures, LLC at a purchase price of $8.02 per share, giving effect to unallocated contributions of capital made by Flavin Ventures, LLC.
- (5)
- On February 8, 2005, we issued 70,284 shares of common stock to accredited investors at a purchase price of $8.02 per share.
- (6)
- Since the adoption of our 1999 Stock Incentive Plan, we have granted options to purchase 735,641 shares of common stock to employees, directors and consultants at exercise prices ranging from $0.157 to $8.02 per share. Of the 735,641 options granted, 686,837 remain outstanding, 6,717 were terminated and 42,082 shares of common stock have been purchased pursuant to the exercises of stock options for the aggregate consideration of $6,625.
The offers, sales, and issuances of the securities described in paragraphs 1, 2, 3, 4 and 5 were deemed to be exempt from registration under the Securities Act in reliance on Section 4(2) of the Securities Act in that the issuance of securities to the recipients did not involve a public offering.
The offers, sales and issuances of the options and common stock described in paragraph 6 were deemed to be exempt from registration under the Securities Act in reliance on Rule 701 in that the transactions were under compensatory benefit plans and contracts relating to compensation as provided under such rule. The recipients of such options and common stock were our employees, directors or bona fide consultants and received the securities under our 1999 Stock Incentive Plan. Appropriate legends were affixed to the securities issued in these transactions. Each of the recipients of securities in these transactions had adequate access, through employment, business or other relationships, to information about us.
II-2
Item 16. Exhibits and Financial Statement Schedules
The following exhibits are filed as part of this Registration Statement:
| Exhibit No. | Description | |
|---|---|---|
1.1* | Form of Underwriting Agreement | |
3.1* | Amended and Restated Certificate of Incorporation of Advanced Life Sciences Holdings, Inc. | |
3.2* | Articles of Amendment of Articles of Incorporation of Advanced Life Sciences, Inc. designating its Series A Preferred Stock | |
3.3* | By-laws of Advanced Life Sciences Holdings, Inc. | |
4.1* | Specimen Common Stock Certificate | |
4.2* | Warrant to Purchase Shares of Common Stock of Advanced Life Sciences Holdings, Inc., dated December 21, 2004, issued to Leaders Bank | |
4.3* | Registration Rights Agreement, dated December 13, 2004, by and between Abbott Laboratories and Advanced Life Sciences Holdings, Inc. | |
4.4* | Stock Purchase Agreement, dated December 13, 2004, by and between Abbott Laboratories and Advanced Life Sciences Holdings, Inc. | |
4.5* | Partnership Exchange Agreement, dated December 13, 2004, by and between Advanced Life Sciences Holdings, Inc. and Flavin Ventures, LLC | |
4.6* | ALS Exchange Agreement, dated December 13, 2004, by and between Advanced Life Sciences Holdings, Inc., the Michael T. Flavin Revocable Trust and Karen Stec | |
4.7* | Partnership Dissolution Agreement, dated December 13, 2004, by and between Advanced Life Sciences Holdings, Inc. and Advanced Life Sciences, Inc. | |
4.8* | Contribution Agreement, dated as of December 13, 2004, by and between Flavin Ventures, LLC and the Michael T. Flavin Revocable Trust | |
4.9* | Ventures Contribution Agreement, dated as of December 13, 2004, by and between ALS Ventures, LLC and Flavin Ventures, LLC | |
5.1* | Opinion of Winston & Strawn LLP | |
10.1* | Stock Incentive Plan Assignment, Assumption and Amendment Agreement, dated December 13, 2004, by and between Advanced Life Sciences, Inc. and Advanced Life Sciences Holdings, Inc. | |
10.2* | Employment Agreement June 1, 2002, by and between Advanced Life Sciences, Inc. and Michael T. Flavin | |
10.3* | Employment Agreement, dated April 11, 2005, by and between Advanced Life Sciences, Inc. and John L. Flavin | |
10.4* | Employment Agreement, dated April 11, 2005, by and between Advanced Life Sciences, Inc. and R. Richard Wieland II | |
10.5* | Employment Agreement, dated April 11, 2005, by and between Advanced Life Sciences, Inc. and Suseelan Pookote | |
II-3
10.6* | Employment Agreement, dated April 11, 2005, by and between Advanced Life Sciences, Inc. and Ze-Qi Xu | |
10.7* | Loan Agreements, dated between November 25, 1999 and October 29, 2001, between the Government of the State of Sarawak and Sarawak MediChem Pharmaceuticals Inc. | |
10.8* | Promissory Note, dated September 1, 2001, from Advanced Life Sciences, Inc. in favor of Michael T. Flavin | |
10.9* | Business Loan Agreement, dated May 31, 2005, between Advanced Life Sciences Holdings, Inc. and The Leaders Bank | |
10.10 | License Agreement, dated December 13, 2004, by and between Abbott Laboratories and Advanced Life Sciences Holdings, Inc. (as amended on April 27, 2005 and August 1, 2005) | |
10.11* | License Agreement, dated April 28, 2003, by and between The University of Chicago, as Operator of Argonne National Laboratory, and Advanced Life Sciences, Inc. | |
10.12* | Exclusive License Agreement, dated December 2, 1999, by and between the Board of Trustees of the University of Illinois and Advanced Life Sciences, Inc. | |
10.13* | Patent Assignment and License Agreement, dated January 24, 1989, Baxter International Inc. and Michael T. Flavin | |
10.14* | Joint Venture Agreement, dated December 21, 1996, between Craun Research Sdn. Bhd, for and on behalf of the Government of the State of Sarawak, Malaysia, and MediChem Research Inc. | |
10.15* | Patent License Agreement, dated May 18, 1995, between the National Institutes of Health and MediChem Research, Inc. | |
10.16* | License Agreement, dated December 27, 1995, between MediChem Research, Inc. and the Government of Sarawak | |
10.17* | Management Services Agreement, dated January 1, 2004, by and between Advanced Life Sciences, Inc. and Flavin Ventures, LLC | |
10.18* | Advanced Life Sciences, Inc. Annual Bonus Plan | |
10.19* | Advanced Life Sciences Holdings, Inc. 2005 Stock Incentive Plan | |
10.20* | Assignment of Lease by Lessor with Consent of Lessee, dated November 1, 2004, by Flavin Ventures, LLC to Biostart Property Group, LLC | |
10.21* | Form of Indemnification Agreement | |
10.22* | Letter agreement, dated July 28, 2005, from Michael T. Flavin, Ph.D., to Advanced Life Sciences Holdings Inc. regarding the extension of the maturity of his loan | |
10.23* | Commitment Letter, dated July 28, 2005, from Leaders Bank to Advanced Life Sciences Holdings, Inc. | |
21.1* | Subsidiaries of Advanced Life Sciences Holdings, Inc. | |
23.1 | Consent of Deloitte & Touche LLP | |
II-4
23.2* | Consent of Winston & Strawn LLP (contained in their opinion filed as Exhibit 5.1) | |
24.1* | Power of Attorney (included on signature page) |
- *
- Previously filed
The undersigned registrant hereby undertakes to provide to the underwriters at the closing specified in the underwriting agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the registrant pursuant to the foregoing provisions, or otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
The undersigned registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act of 1933 shall be deemed to be part of this registration statement as of the time it was declared effective.
(2) For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
II-5
Pursuant to the requirements of the Securities Act of 1933, as amended, the registrant has duly caused this Post-Effective Amendment No. 1 to Registration Statement (No. 333-124396) to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Chicago, Illinois, on the 2nd day of August, 2005.
| ADVANCED LIFE SCIENCES HOLDINGS, INC. (Registrant) | ||||||
By: | ||||||
| /s/ MICHAEL T. FLAVIN | ||||||
| Name: | Michael T. Flavin | |||||
| Title: | Chairman and Chief Executive Officer | |||||
Pursuant to the requirements of the Securities Act of 1933, as amended, this registration statement has been signed below by the following persons, in the capacities and on the dates indicated.
| Signature | Title | Date | ||
|---|---|---|---|---|
| /s/ MICHAEL T. FLAVIN Michael T. Flavin | Chairman and Chief Executive Officer (Principal Executive Officer) | August 2, 2005 | ||
/s/ JOHN L. FLAVIN* John L. Flavin | President and Director | August 2, 2005 | ||
/s/ R. RICHARD WIELAND II* R. Richard Wieland II | Executive Vice President and Chief Financial Officer (Principal Financial and Accounting Officer) | August 2, 2005 | ||
/s/ TERRY W. OSBORN* Terry W. Osborn | Director | August 2, 2005 | ||
/s/ ISRAEL RUBINSTEIN* Israel Rubinstein | Director | August 2, 2005 | ||
/s/ ROSALIE SAGRAVES* Rosalie Sagraves | Director | August 2, 2005 | ||
/s/ THOMAS V. THORNTON* Thomas V. Thornton | Director | August 2, 2005 |
*By: | /s/ MICHAEL T. FLAVIN Michael T. Flavin Attorney-in-fact |
II-6
| Exhibit No. | Description | |
|---|---|---|
1.1* | Form of Underwriting Agreement | |
3.1* | Amended and Restated Certificate of Incorporation of Advanced Life Sciences Holdings, Inc. | |
3.2* | Articles of Amendment of Articles of Incorporation of Advanced Life Sciences, Inc. designating its Series A Preferred Stock | |
3.3* | By-laws of Advanced Life Sciences Holdings, Inc. | |
4.1* | Specimen Common Stock Certificate | |
4.2* | Warrant to Purchase Shares of Common Stock of Advanced Life Sciences Holdings, Inc., dated December 21, 2004, issued to Leaders Bank | |
4.3* | Registration Rights Agreement, dated December 13, 2004, by and between Abbott Laboratories and Advanced Life Sciences Holdings, Inc. | |
4.4* | Stock Purchase Agreement, dated December 13, 2004, by and between Abbott Laboratories and Advanced Life Sciences Holdings, Inc. | |
4.5* | Partnership Exchange Agreement, dated December 13, 2004, by and between Advanced Life Sciences Holdings, Inc. and Flavin Ventures, LLC | |
4.6* | ALS Exchange Agreement, dated December 13, 2004, by and between Advanced Life Sciences Holdings, Inc., the Michael T. Flavin Revocable Trust and Karen Stec | |
4.7* | Partnership Dissolution Agreement, dated December 13, 2004, by and between Advanced Life Sciences Holdings, Inc. and Advanced Life Sciences, Inc. | |
4.8* | Contribution Agreement, dated as of December 13, 2004, by and between Flavin Ventures, LLC and the Michael T. Flavin Revocable Trust | |
4.9* | Ventures Contribution Agreement, dated as of December 13, 2004, by and between ALS Ventures, LLC and Flavin Ventures, LLC | |
5.1* | Opinion of Winston & Strawn LLP | |
10.1* | Stock Incentive Plan Assignment, Assumption and Amendment Agreement, dated December 13, 2004, by and between Advanced Life Sciences, Inc. and Advanced Life Sciences Holdings, Inc. | |
10.2* | Employment Agreement June 1, 2002, by and between Advanced Life Sciences, Inc. and Michael T. Flavin | |
10.3* | Employment Agreement, dated April 11, 2005, by and between Advanced Life Sciences, Inc. and John L. Flavin | |
10.4* | Employment Agreement, dated April 11, 2005, by and between Advanced Life Sciences, Inc. and R. Richard Wieland II | |
10.5* | Employment Agreement, dated April 11, 2005, by and between Advanced Life Sciences, Inc. and Suseelan Pookote | |
10.6* | Employment Agreement, dated April 11, 2005, by and between Advanced Life Sciences, Inc. and Ze-Qi Xu | |
10.7* | Loan Agreements, dated between November 25, 1999 and October 29, 2001, between the Government of the State of Sarawak and Sarawak MediChem Pharmaceuticals Inc. | |
10.8* | Promissory Note, dated September 1, 2001, from Advanced Life Sciences, Inc. in favor of Michael T. Flavin | |
10.9* | Business Loan Agreement, dated May 31, 2005, between Advanced Life Sciences Holdings, Inc. and The Leaders Bank | |
10.10 | License Agreement, dated December 13, 2004, by and between Abbott Laboratories and Advanced Life Sciences Holdings, Inc. (as amended on April 27, 2005 and August 1, 2005) | |
10.11* | License Agreement, dated April 28, 2003, by and between The University of Chicago, as Operator of Argonne National Laboratory, and Advanced Life Sciences, Inc. | |
10.12* | Exclusive License Agreement, dated December 2, 1999, by and between the Board of Trustees of the University of Illinois and Advanced Life Sciences, Inc. | |
10.13* | Patent Assignment and License Agreement, dated January 24, 1989, Baxter International Inc. and Michael T. Flavin | |
10.14* | Joint Venture Agreement, dated December 21, 1996, between Craun Research Sdn. Bhd, for and on behalf of the Government of the State of Sarawak, Malaysia, and MediChem Research Inc. | |
10.15* | Patent License Agreement, dated May 18, 1995, between the National Institutes of Health and MediChem Research, Inc. | |
10.16* | License Agreement, dated December 27, 1995, between MediChem Research, Inc. and the Government of Sarawak | |
10.17* | Management Services Agreement, dated January 1, 2004, by and between Advanced Life Sciences, Inc. and Flavin Ventures, LLC | |
10.18* | Advanced Life Sciences, Inc. Annual Bonus Plan | |
10.19* | Advanced Life Sciences Holdings, Inc. 2005 Stock Incentive Plan | |
10.20* | Assignment of Lease by Lessor with Consent of Lessee, dated November 1, 2004, by Flavin Ventures, LLC to Biostart Property Group, LLC | |
10.21* | Form of Indemnification Agreement | |
10.22* | Letter agreement, dated July 28, 2005, from Michael T. Flavin, Ph.D., to Advanced Life Sciences Holdings Inc. regarding the extension of the maturity of his loan | |
10.23* | Commitment Letter, dated July 28, 2005, from Leaders Bank to Advanced Life Sciences Holdings, Inc. | |
21.1* | Subsidiaries of Advanced Life Sciences Holdings, Inc. | |
23.1 | Consent of Deloitte & Touche LLP | |
23.2* | Consent of Winston & Strawn LLP (contained in their opinion filed as Exhibit 5.1) | |
24.1* | Power of Attorney (included on signature page) |
- *
- Previously filed
TABLE OF CONTENTS
PROSPECTUS SUMMARY
RISK FACTORS
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
CONCURRENT OFFERING
USE OF PROCEEDS
DIVIDEND POLICY
CAPITALIZATION
DILUTION
SELECTED FINANCIAL DATA
MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
BUSINESS
MANAGEMENT
RELATED PARTY TRANSACTIONS
CORPORATE REORGANIZATION
PRINCIPAL STOCKHOLDERS
DESCRIPTION OF CAPITAL STOCK
SHARES ELIGIBLE FOR FUTURE SALE
UNDERWRITING
MATERIAL U.S. FEDERAL INCOME TAX CONSEQUENCES TO NON-U.S. HOLDERS
LEGAL MATTERS
EXPERTS
WHERE YOU CAN FIND MORE INFORMATION
INDEX TO CONSOLIDATED FINANCIAL STATEMENTS
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
PART II INFORMATION NOT REQUIRED IN PROSPECTUS
- Item 13. Other Expenses of Issuance and Distribution
Item 14. Indemnification of directors and officers
Item 15. Recent Sales of Unregistered Securities
Item 16. Exhibits and Financial Statement Schedules
Item 17. Undertakings
EXHIBIT INDEX