UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 10-K
x ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
FOR THE FISCAL YEAR ENDED DECEMBER 31, 2014
OR
o TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
FOR THE TRANSITION PERIOD FROM TO
COMMISSION FILE NO. 001-36534
IRADIMED CORPORATION
(Exact Name of Registrant As Specified In Its Charter)
Delaware |
| 73-1408526 |
(State or other jurisdiction of |
| (I.R.S. Employer |
1025 Willa Springs Drive Winter Springs, Florida |
| 32708 |
(Address of principal executive offices) |
| (Zip Code) |
Registrant’s telephone number, including area code: (407) 677-8022
SECURITIES REGISTERED PURSUANT TO SECTION 12(b) OF THE ACT:
Title of |
| Name of each exchange |
Each Class |
| on which registered |
Common Stock, $0.0001 par value |
| Nasdaq Stock Market LLC |
|
| (Nasdaq Capital Market) |
SECURITIES REGISTERED PURSUANT TO SECTION 12(g) OF THE ACT:
None.
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes o No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes o No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No o
Indicate by checkmark whether the registrant has submitted electronically and posted on its corporate Website, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§ 229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. Yes o No x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
Large accelerated filer o |
| Accelerated filer o |
|
|
|
Non-accelerated filer o |
| Smaller reporting company x |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes o No x
As of June 30, 2014, the last business day of the registrant’s most recently completed second fiscal quarter, there was no established public market for the registrant’s common stock. The registrant’s common stock began trading on the NASDAQ Capital Market on July 16, 2014.
There were 10,972,150 shares outstanding of the registrant’s common stock, par value $0.0001 per share, as of February 28, 2015. The registrant’s common stock is listed on the Nasdaq Capital Market under the stock symbol “IRMD.”
Documents Incorporated by Reference: Information required by Items 10, 11, 12, 13 and 14 of Part III are incorporated by reference from the Proxy Statement for the registrant’s 2015 Annual Meeting of Stockholders. Except with respect to information specifically incorporated by reference in the Form 10-K, the Proxy Statement is not deemed to be filed as part hereof.
IRADIMED CORPORATION.
TABLE OF CONTENTS TO ANNUAL REPORT ON FORM 10-K
For the Fiscal Year Ended December 31, 2014
CAUTIONARY STATEMENTS REGARDING FORWARD-LOOKING STATEMENTS
This Annual Report on Form 10-K contains “forward-looking statements” that involve substantial risks and uncertainties. The forward-looking statements are contained principally in the sections entitled “Business,” “Risk Factors” and “Management’s Discussion and Analysis and Results of Operations.” In some cases, you can identify forward-looking statements by the following words: “may,” “will,” “could,” “would,” “should,” “expect,” “intend,” “plan,” “anticipate,” “believe,” “estimate,” “predict,” “project,” “potential,” “continue,” “ongoing” or the negative of these terms or other comparable terminology, although not all forward-looking statements contain these words. These statements relate to future events or our future financial performance or condition and involve known and unknown risks, uncertainties and other factors that could cause our actual results, levels of activity, performance or achievement to differ materially from those expressed or implied by these forward-looking statements. These forward-looking statements include, but are not limited to, statements about:
· our ability to receive clearance of our 510(k) submission, resolve various matters identified in the FDA Warning Letter, additional actions by or requests from the FDA (including a request to cease domestic distribution of products) and unanticipated costs or delays associated with the resolution of these matters;
· our ability to resolve a securities class-action lawsuit that has been filed against us in connection with the FDA Warning Letter and shipment stoppage;
· our reliance on a single product;
· our ability to retain the continued service of our key professionals and to identify, hire and retain additional qualified professionals;
· our expectations regarding the sales and marketing of our products and product candidates;
· our expectations regarding the integrity of our supply chain for our products;
· the timing and likelihood of FDA approvals and regulatory actions on our product candidates and product marketing activities;
· the potential for adverse application of environmental, health and safety and other laws and regulations on our operations;
· our expectations for market acceptance of our new products;
· the potential for our marketed products to be withdrawn due to recalls, patient adverse events or deaths;
· our ability to establish and maintain intellectual property on our products and our ability to successfully defend these in cases of infringement;
· the implementation of our business strategies;
· the potential for exposure to product liability claims;
· our financial performance expectations;
· our ability to compete in the development and marketing of our products and product candidates with other competitors in the industry;
· difficulties or delays in the development, production, manufacturing and marketing of new or existing products and services, including difficulties or delays associated with obtaining requisite regulatory approvals or clearances associated with those activities;
· changes in laws and regulations or in the interpretation or application of laws or regulations, as well as possible failures to comply with applicable laws or regulations as a result of possible misinterpretations or misapplications;
· cost-containment efforts of our customers, purchasing groups, third-party payers and governmental organizations;
· costs associated with protecting our trade secrets and enforcing our patent, copyright and trademark rights, and successful challenges to the validity of our patents, copyrights or trademarks;
· actions of regulatory bodies and other government authorities, including the FDA and foreign counterparts, that could delay, limit or suspend product development, manufacturing or sales or result in recalls, seizures, consent decrees, injunctions and monetary sanctions;
· costs or claims resulting from potential errors or defects in our manufacturing that may injure persons or damage property or operations, including costs from remediation efforts or recalls;
· the results, consequences, effects or timing of any commercial disputes, patent infringement claims or other legal proceedings or any government investigations;
· interruption in our ability to manufacture our products or an inability to obtain key components or raw materials or increased costs in such key components or raw materials;
· uncertainties in our industry due to government healthcare reform;
· competitive pressures in the markets in which we operate;
· the loss of, or default by, one or more key customers or suppliers; and
· unfavorable changes to the terms of key customer or supplier relationships.
Forward-looking statements are not guarantees of future performance and are subject to substantial risks and uncertainties that could cause the actual results to differ materially from those that we predicted in the forward-looking statements. Investors should carefully review the information contained under the caption “Risk Factors” contained in Item 1A for a description of risks and uncertainties that could cause actual results to differ from those that we predicted. All forward-looking statements are based on information available to us on the date hereof, and we assume no obligation to update forward-looking statements, except as required by Federal Securities laws.
Overview
IRADIMED CORPORATION (“IRADIMED”, the “Company”, “we”, “us”, “our”) develops, manufactures, markets and distributes magnetic resonance imaging (“MRI”) compatible products, and today, we are the only known provider of non-magnetic intravenous (“IV”) infusion pump systems. We were the first to develop an infusion delivery system that neutralizes the dangers and problems present during MRI procedures. Standard infusion pumps contain magnetic and electronic components which can create radio frequency (“RF”) interference and are dangerous to operate in the presence of the powerful magnet that drives an MRI system. Our MRidium MRI compatible IV infusion pump system uses a patented non-magnetic ultrasonic motor and other uniquely-designed non-ferrous parts that enable accurate, safe and dependable fluid delivery to patients undergoing an MRI procedure.
With the expanding use of MRI procedures, both traditional procedures and new intraoperative and interventional procedures, safe and reliable infusion delivery in an MRI environment is becoming increasingly important to hospitals and other medical providers. Our founder, President and Chief Executive Officer, Roger Susi, is a pioneer in the MRI compatible medical device industry, having invented the first MRI compatible patient monitoring system in 1986 and the first non-magnetic MRI safe infusion system in 2004. Since launching our first generation MRI compatible IV infusion pump system in 2005, we have continued to modify and improve our system, and we have leveraged our development strengths and unique market position to expand our customer base and profitability. We were incorporated in Oklahoma in July 1992 and reincorporated in Delaware in April 2014.
We sell our products primarily to acute care facilities and outpatient imaging centers, both in the United States and internationally. In fiscal year 2012, we undertook a direct sales strategy in the United States. Today, our direct sales force consists of ten sales representatives, supplemented by two clinical support representatives. Our goal is to expand our U.S. sales force to 14 by the end of 2015. We have distribution agreements with 35 independent distributors selling our products internationally.
As of December 31, 2014 we estimate that we had approximately 2,300 MRI compatible IV infusion pump systems installed globally. Each system consists of an MRidium MRI compatible IV infusion pump, mobile stand, and proprietary disposable IV tubing sets and many of these systems contain additional optional upgrade accessories. We generate revenue from the one-time sale of pumps and accessories, ongoing service contracts and the sale of disposable IV tubing used during each scan. Our revenue growth has accelerated since initiating our direct sales effort. In fiscal year 2014, our revenue reached $15.6 million and our operating profit was $3.1 million representing an operating margin of 19.6%. This operating margin reflects the blended results of our IV infusion pumps, pump upgrades and disposable IV tubing sets.
History and Development
Mr. Susi founded Invivo Research Inc. in 1979 where he developed the first MRI compatible patient monitoring system. Mr. Susi served as the President of Invivo Research Inc. from 1979 until 1998, and as its Chairman of the Board of Directors from 1998 until 2000. Under Mr. Susi’s leadership, Invivo Research matured from a start-up medical device company into a leading producer of vital signs monitoring devices during MRI procedures. Invivo Research was acquired by Invivo Corporation in 1992, which began trading on the NASDAQ Stock Exchange in 1994. Mr. Susi served as a Director of Invivo Corporation from 1998 until 2000 and oversaw technical areas from 2000 to 2004. Invivo Corporation was acquired by Intermagnetics General Corporation in 2004 for $152 million. The Invivo system, currently owned by Koninklijke Philips NV (NYSE: PHG), continues to maintain its position as the market-leading MRI compatible vital signs monitor.
Mr. Susi began exploring the market for an MRI compatible IV infusion pump while at Invivo. Invivo subsequently disclaimed any interest in the infusion pump and acknowledged that Mr. Susi was free to pursue the infusion pump development for his own account. Accordingly, after leaving Invivo in January 2004, Mr. Susi began the formal and detailed development of what subsequently has become our MRidium MRI compatible IV infusion pump system. During 2005, he assembled a team of individuals experienced in the medical device industry, many of whom were former employees of Invivo. This first generation MRI compatible IV infusion pump system and its associated proprietary IV tubing sets obtained FDA market clearance in March 2005 after which point we began our sales and marketing efforts.
We initially marketed the product ourselves in the U.S. with limited sales staff, and within one year, commenced international sales through a network of distributors. In 2006, we signed an exclusive distribution agreement with Mallinckrodt/Tyco Healthcare (now part of Medtronic plc (NYSE: MDT) for domestic and Canadian distribution of our products including the MRidium 3850 MRI compatible IV infusion pump system. The exclusive arrangement ended in 2010, allowing us to implement a direct marketing strategy with our own sales force in the U.S. and Canada.
In 2009, we introduced our second generation MRI compatible IV infusion pump system, the MRidium 3860+ which improved upon the previous 3850 version in a number of areas, including the addition of SpO2, blood oxygen saturation monitoring, and remote wireless monitoring capability. An SpO2 monitor can signal when an insufficient level of oxygen is being supplied to the body. Our MRidium 3860+ is the leading MRI compatible IV infusion pump system on the market today. In 2011, we introduced the iMagox product line, a standalone SpO2 patient monitor which was based on our MRI compatible SpO2 monitoring system with a wireless remote control for international distribution only.
Industry
We currently compete in the MRI compatible IV infusion pump systems market.
Need for MRI Compatible IV Infusion Pumps
MRI is a widely-used, non-invasive medical imaging technique to visualize vital organs, body function and to identify blockages, abnormalities and growths. MRI is generally considered safer than other scanning techniques that expose the body to radiation. This is particularly true for children. As such, hospitals and other medical facilities have been increasingly developing and using MRI for new procedures. These procedures include cardiac stress testing, intraoperative MRI and neurology MRI techniques. Our MRidium MRI compatible IV infusion pump offers a way to deliver critical IV fluids safely and accurately, thereby allowing the expanded use of MRI procedures.
While the benefits and utility of interventional MR are known, there are hazards intrinsic to the MR environment which must be respected. These hazards may be attributed to a powerful static magnetic field, pulsed gradient magnetic fields, and pulsed radio frequency fields. The MRI suite is a harsh place for medical devices, and safe and proper patient care requires specialty equipment that is specifically designed and built for the MR environment. Many of the dangers and problems present in the MR environment can be solved through use of non-magnetic equipment that have operational safeguards and that maintain performance standards within a harsh magnetic environment while maintaining patient safety. Designing an IV infusion pump system to operate safely and effectively in the MR environment requires overcoming significant technical hurdles.
Intravenous fluids are needed during MRI procedures for many different reasons. Infusion pumps provide sedation to patients who are not able to lie still during an MRI scan and a continuous flow of critical medications to seriously ill patients. Given the benefits to patient safety, radiology departments performing the scan, anesthesia departments delivering sedation and critical care specialists responsible for delivering critical medications during MRI procedures often initiate requests for an MRI compatible IV infusion pump.
Conventional Infusion Pumps and Other Inadequate Alternatives
For those medical facilities that do not currently own an MRI compatible IV infusion pump, there are five general methods that are used to deal with patients undergoing an MRI who require IV medications during their imaging procedure: (1) use conventional (magnetic) pumps with long IV lines that extend outside the MRI scanner room; (2) do not offer MRI treatment to patients requiring IV medications or sedation; (3) proceed and accept patients for an MRI procedure but stop the flow of IV fluids during the procedure; (4) allow the uncontrolled free drip of IV fluids; and (5) attempt to shield a conventional IV infusion pump. All of these approaches have drawbacks.
Use of multiple lengths of extension tubing can cause inaccuracies, waste and false alarms or, more seriously, delayed alarms for equipment issues such as occlusion, especially when low flow rates are being used. Such makeshift extension sets can also affect the effectiveness of fluid delivery. A clinician’s adjustment of dosage and other settings may take longer to reach the patient due to the over-extended tubing.
Further, there are risks in using a conventional IV infusion pump that is mistakenly believed to be at a safe distance from the MR scanner. The powerful magnetic fields may cause metal objects in the MR environment to be drawn with great force into the bore of the MR system, resulting in potentially deadly projectiles. Moreover, an MR scanner’s gradient magnetic field and RF fields can send currents in cables and other conductive materials that are near the MR system and cause the cables to heat. Hot cables may result in burns if they come into contact with a patient. Other problems include devices malfunctioning if they are not properly designed for use in the harsh MR environment and low-quality images due to artifacts caused by RF interference emitted from ancillary equipment.
To deal with the harsh environment of MR, some manufacturers have offered a “shielded box” solution for use with their conventional IV pumps, but the approach was not widely adopted. The major problem with this approach is that a highly magnetic conventional IV infusion pump is still being introduced into a hazardous MRI environment which can lead to projectile accidents. Moreover, a Faraday cage with the conventional IV infusion pump must be kept approximately 5 to 10 feet from the scanner. By contrast, our product can be safely placed anywhere in the scanner room including next to the scanner. We are not aware of any “shielded box” installations in use in the U.S. or any with a FDA 510(k) clearance and hence, we expect little current competition from this approach in the U.S.
We believe that our MRidium MRI compatible IV infusion pump system is the first and only product to provide an easy-to-operate, non-magnetic, safe and RF-quiet solution and hence a truly MRI compatible product.
Market Opportunities
Exit of Competitor from Market
During 2012, our only direct competitor in the MRI compatible IV infusion pump business, Bayer Radiology (formerly MEDRAD, Inc.), became the subject of an FDA recall with respect to its then market-leading Continuum device. In mid-2012, Bayer Radiology announced that it was discontinuing, until further notice, all new sales of Continuum leaving us as the only known global supplier of MRI compatible IV infusion pumps. During 2013, Bayer Radiology announced its decision to commence removal of its pump systems from the U.S. market, and to discontinue support throughout the world by June 30, 2015 due to ongoing regulatory issues, at which time Bayer Radiology will end its limited supply of its proprietary consumable IV sets that some current customers are receiving.
As a result of Bayer Radiology’s announced exit from the market, we anticipate that many Continuum customers will replace their MRI compatible IV infusion pumps with our MRidium 3860+ system. We are continuing to expand our direct sales force in the U.S. and believe that our pump sales will be favorably impacted by the exodus of Bayer. We intend to market aggressively to this existing user base as well as to new potential users.
Expansion of Inter-Hospital Use of MRI Compatible IV Infusion Pumps
We currently market our MRI compatible infusion pumps primarily to the MRI departments of hospitals. We believe, however, that there is potential for expanded deployment of our MRI compatible IV infusion pumps within the Intensive Care Unit (“ICU”), Emergency Room (“ER”), and other locations within the hospital with a high probability that interventional radiology procedures will need to be performed on patients. Expanded use of our additional MRI compatible IV infusion pumps would serve as a type of transport pump and allow for consistent administration of IV fluids without interruption and easy transfer from the ICU or ER to the MRI scanner room.
It frequently becomes necessary for a patient in the ICU or ER who is connected to an IV infusion pump that is delivering critical medications to be quickly moved to the MRI facility for immediate imaging. The presence of multiple MRI compatible IV infusion pumps or pump channels, for each IV line, enables the orderly and rapid transfer between IV infusion pumps by allowing patients to be prepared for an MR procedure and setup on our MRidium MRI compatible IV infusion pump in the ICU or ER. Seriously ill patients are generally at higher risk when they are away from the resources of the ICU or ER, and efficient IV infusion pump transfer minimizes the time the patient spends away from the ICU or ER.
We believe there is a link between the number of infusions and infusion pumps and the onset of equipment-related adverse events during the intra-facility transport of critically ill patients. We therefore believe that placing our MRI compatible IV infusion pumps in ICU and ER facilities could reduce patient adverse events associated with the transport and pump exchange within the hospital.
Recently some hospitals have begun to use MRI during surgical procedures. Neurosurgical interventions have been at the forefront of this development in image-guided surgery, followed by otolaryngological procedures. As MR-guided intervention during surgery has been deployed, the degree of complexity in supplemental devices has increased markedly. Much of the effort required for successful implementation of intraoperative MRI has been in development and testing of anesthesia equipment, patient monitoring devices, infusion pumps and surgical instruments and accessories, all of which need to be MRI compatible if used near the MRI scanner. Intraoperative MRI is expanding demand for our MRI compatible IV infusion pump system from the MRI suite to the surgical suite of the hospital, again with multiple pump channels for multiple IV lines.
Strategy
Company Objective
Our objective is to become the leader in providing safe and effective care for all patients undergoing MRI procedures through the development and commercialization of a portfolio of MRI compatible products. By increasing the safety parameters of equipment operating within the harsh magnetic environment of the MRI scanner room, we hope to enable hospitals and other healthcare providers to offer the MRI diagnostic procedures patients require. In particular, our goal is to increase the safety of MRI diagnostics for critically ill patients and children by minimizing potential complications with IV infusions and vital signs monitoring.
We seek to grow our business by, among other things:
Driving market awareness of MRI compatible IV infusion pumps and the safety risks associated with using conventional IV pumps with long IV lines.
We believe that the largest potential market for our MRI compatible IV infusion pumps is the segment of the market that is currently using workaround solutions. Such solutions include using conventional pumps outside the MRI scanner room and attaching multiple extension lines of IV tubing sets through the wall or under the door into the MRI scanner room to reach the patient. This practice of makeshift setups is fraught with risks to the patient and unnecessary costs and inefficiencies. These risks and inefficiencies include:
· Infection risk from running lengthy IV tubing sets through the wall or under the door;
· Risk of inaccuracy from using a conventional IV infusion pump with multiple extension lines;
· Potential medication occlusion and lengthy alarm notification delays due to multiple extension lines, posing a great risk to a patient on critical medications;
· Excess medication costs due to the disposal of multiple extension IV tubing sets filled with unused medication at the end of the procedure; and
· Lost productivity and MRI scanning time due to the lengthy set up time required for multiple extension lines.
We believe that increased market awareness and education will be required for potential customers to appreciate the value for patients and the hospital of an efficient and patient-safe MRI environment which includes MRI compatible IV infusion pumps.
Continuing to innovate with MRI compatible patient care products.
Our management team collectively has more than 100 years of experience with MRI compatible products. We have entrenched relationships with many of the industry’s top thought leaders and we have, and will continue to, closely collaborate with them to build upon MRidium’s innovative MRI compatible technologies to create next generation pump systems. We intend to leverage this experience and collaboration to innovate and commercialize other technologically-advanced MRI patient care products, such as a device for assisting resuscitation and a thermal management unit.
Developing a New Resuscitation/Monitoring System.
We currently have under development a new resuscitation device with a separate multi-parameter vital signs monitor that is MRI compatible. We plan to launch the vital signs monitor in 2016. When providing anesthesia care in the MRI environment, the same requirements for safe resuscitation and monitoring that apply in a typical operating room must be satisfied. Our device is being developed using MRI compatible technology to safely deliver therapy and monitor all of the required basic vital signs parameters including electrocardiography, pulse oximetry, non-invasive blood pressure, capnography, and temperature. Our device will be designed to have a monitoring/remote station, with wireless communication capability outside of the scanner room (in the control room).
Acquiring synergistic MRI patient care companies, products or technology licenses to accelerate our product development and leverage our existing direct sales organization in the U.S.
We have an experienced team of engineering and operations managers committed to improving on existing MRI patient care designs through our internal development efforts and the acquisition of technologies and intellectual property of others. We have an effective and growing direct sales organization in the U.S. and a team of experienced international distributors that can effectively go to market with additional MRI patient care products. We intend to actively analyze opportunities to improve our product mix and profitability.
Commercial Strategy
We believe that the MRI compatible IV infusion pump market is still in its early stages of growth given the low rate of market penetration, and we aim to drive increased awareness and adoption of our products by:
Expanding our MRI-focused U.S. direct sales force and our international sales efforts.
We believe the most meaningful aspect of our commercialization strategy in the U.S. is the continued development and expansion of our direct sales force. Since there is no current direct competitor for an MRI compatible IV infusion pump, our focus is on expansion of the market through better education on advantages to patients, clinicians and hospitals of our products and the shortcomings of current workaround solutions. Our challenge in the past has been an understaffed direct sales team and a limited ability to educate our potential customers.
Since 2011, our U.S. sales team has grown from one sales representative to a team of ten direct sales representatives and two clinical support representatives. We intend to continue to grow our specialized, MRI product-focused sales team and to support it with clinical support representatives as the business dictates. We will market to current users of the Continuum system of our former competitor, Bayer Radiology, who will soon be without a viable solution. In addition, we believe that we can significantly increase sales of our MRidium MRI compatible IV infusion pump by also calling on anesthesia and critical care departments, to help influence hospitals’ purchasing decisions. We believe that this strategy will likely expand the number of acute care facilities using our MRI compatible products and increase the average number of MRI compatible IV infusion pumps per acute MRI.
Internationally, our focus in 2015 and beyond is to work with our distributors in key target markets, such as Europe and Japan, to expand the business and augment our market penetration rates. In the future, we plan to expand our internal capacity to serve these high potential markets by adding dedicated regional sales managers located outside the U.S. to oversee our relationships at the local level.
Supporting commercial efforts with evidence-based information.
We have developed a white paper that documents the risks and additional costs associated with using a workaround solution of running long lines from conventional IV pumps outside the MRI scanner room. We believe that this kind of evidence-based documentation will help us to provide widespread education to the clinicians that are driving clinical practice. We also believe that documented evidence will serve to inform the quality and risk management leaders in these organizations, which in turn may help drive the overall adoption of our MRidium MRI compatible IV infusion pump.
Providing best in class customer service and user experience.
We believe that the expectations of our customers for service and a superior user experience have risen with the advancement of technology. Once a customer purchases an MRidium MRI compatible IV infusion pump, it is imperative that they receive first-class clinical education and support to encourage usage of our products. We have devoted a significant amount of time and training to ensure that this educational experience is a success. This training is performed most commonly by our sales staff and is augmented by our clinical support representatives; however, we intend to hire more clinical support specialists to improve our initial training experience and ongoing customer support. We believe that a positive user experience will be critical to driving increased rates of utilization of our products.
Our Products
The MRidium MRI compatible IV infusion pump system is based upon a patented non-magnetic ultrasonic motor and other uniquely-designed non-ferrous parts in order to provide accurate and dependable fluid delivery to patients undergoing an MRI procedure. Our MRidium MRI compatible IV infusion pump system has been designed to offer numerous advantages to hospitals, clinicians and patients. MRidium’s strengths include the following:
· The only non-magnetic MRI compatible IV infusion pump system specifically designed and built for the MRI environment.
· A mobile, rugged, easy-to-operate, and reliable system with a strong safety record.
· Able to operate virtually anywhere in the MRI scanner room; approved for use in the presence of 0.2T to 3T magnets and fully operational up to the 10,000 gauss-line.
· The only non-magnetic MRI compatible IV infusion pump available with a Dose Error Reduction System (“DERS”) to reduce the risk of medication errors and simplify clinician monitoring.
· Available with a wireless remote display/control providing clinicians and technicians control and visibility from outside of the MRI scanner room.
· Available with an add-on channel allowing for the easy addition of a second IV line for patients requiring multiple IV medications at a low incremental cost to the hospital.
· Available with a built-in SpO2 monitor using Masimo SET® technology and a specially designed fiber optic SpO2 sensor allowing one device to monitor oxygen saturation levels while safely providing IV infusion during an MRI procedure.
The following diagram is an aerial view of an MRI scanner room with a top-of-the-line 3T magnet. The gauss-lines illustrate the distance from the magnet where various types of infusion pumps can safely operate. Our MRidium MRI compatible IV infusion pump is the only pump on the market approved to operate safely and reliably near the patient (area shown in blue). All of the other pumps must either be placed at a distance from the MRI scanner or outside of the scanner room entirely.
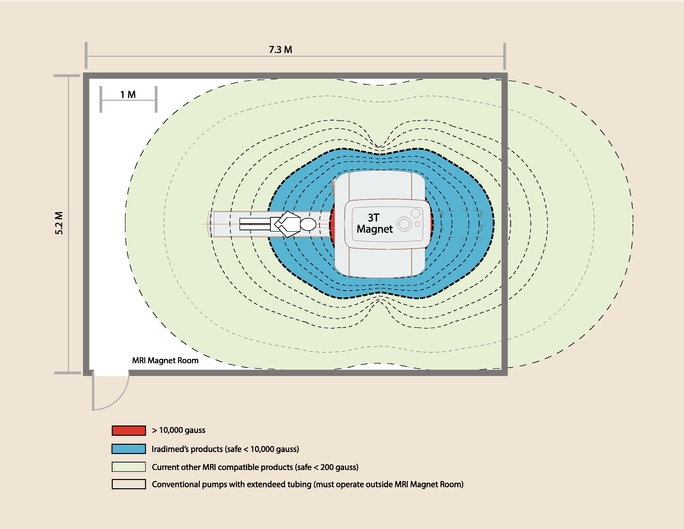
Our MRI compatible IV infusion pump system includes the 3860+ MRI compatible IV infusion pump, proprietary single-use IV tubing sets, a non-magnetic pole and a lithium battery. In addition, we offer optional upgrade systems including the 3865 Remote Display/Control, 3861 Side Car, DERS, and an SpO2 monitor as discussed below.
MRidium 3860+ MRI Compatible IV Infusion Pump
The MRidium 3860+ MRI compatible IV infusion pump system was introduced in 2009 and improved upon the strong performance and features of our first generation MRidium 3850 MRI compatible IV infusion pump system. Our pump systems can operate dependably in the presence of 0.2T to 3T magnets and are fully operational up to the 10,000 gauss-line. This means they are highly versatile and can operate virtually anywhere in the MRI scanner room, including close to the MRI scanner. The MRidium 3860+ MRI compatible IV infusion pump system has a 10-key numeric input keypad making our system easy to accurately program and operate. Our pumping range of 0.1 mL per hour to 1,400 mL per hour provides a broad range of fluid flow control. Our broad range of infusion rates support differing patient needs including low levels for pediatric sedation, mid-levels for continued IV infusion of medications to critically-ill patients and high levels in the event of emergency situations. Our MRidium 3860+ MRI compatible IV infusion pump system offers a dose rate calculator, bolus dose programming, full alarm settings, and a rechargeable battery with a 12-hour battery life.
MRidium 3860+ IV Tubing Sets
The MRidium 3860+ MRI compatible IV infusion pump system utilizes proprietary fluid delivery tubing sets, each known as an “IV tubing set.” Each use of our MRI compatible IV infusion pump requires a disposable IV tubing set. We offer a variety of IV tubing sets for varying MRI scenarios and these include our standard “spike” infusion set, syringe adapter infusion set and extension infusion set. Each of our IV tubing sets is latex-free and DEHP-free.
· MRidium 1056 Standard Infusion Set. Our standard “spike” infusion set features the ability to accurately deliver liquids from either a bottle or IV bag. Our standard infusion set contains two needle-free injection ports and is typically used when starting a new infusion from a bottle or bag.
· MRidium 1057 Syringe Adapter Infusion Set. Our syringe adapter IV set enables users to provide accurate delivery of IV fluids directly from standard syringes. The vented syringe adapter set benefits from a low priming volume of 4 ml, which minimizes inefficient waste of medication. This product is most commonly used for cardiac medications, anesthesia, and pediatric drug delivery.
· MRidium 1058 Extension Infusion Set. Our extension infusion set allows users to transfer a patient on a non-MRI infusion pump to our MRidium MRI compatible IV infusion pump. The user simply disconnects the existing IV tubing at the patient site and connects and primes the MRidium extension set to the existing IV tubing. Once removed from the conventional infusion pump and connected to our MRidium MRI compatible IV infusion pump, the user can program the pump and begin the infusion. The extension set includes one needle-free injection port and is typically used to provide uninterrupted critical medications to a severely ill patient during an MRI procedure.
MR IV Pole
We offer a fully-functional and weighted non-magnetic IV pole that is designed for mobility within the hospital and the MRI scanner room. The IV pole can support two MRidium MRI compatible IV infusion pumps, each with a side car. The IV pole is 66 inches (1.68 meters) high, stabilized with a wide pole radius and mobilized with five casters designed to roll easily during transport. The IV pole is equipped with four hooks for holding fluids.
Optional Features
Our 3860+ MRI compatible IV infusion pump system gives customers the ability to adapt their systems to meet their specific needs. In addition to our standard product features, we also offer system upgrades which include a wireless remote control/display, a modular add-on second IV channel through our “Side Car,” DERS and an imbedded SpO2 monitor. We also offer rechargeable lithium polymer battery packs which have 12-hour battery life when not connected to an electrical outlet.
3865 MRidium Wireless Remote Display/Control
Our wireless remote control units allow for complete control and monitoring of the MRidium MRI compatible IV infusion pump system from the control room (outside of the MRI scan room). The 3865 MRidium Wireless Remote relays all commands and displays information bi-directionally between the MRI compatible IV infusion pump and the remote control unit. Utilizing the same user interface and large bright display as the MRidium pump, our wireless remote control unit permits clinicians to adjust all pump parameters, including SpO2 monitoring parameters, rates, dose, volume, pump run/stop, alarms (adjust or reset), as well as real-time titration. Our remote control unit utilizes a proven MRI compatible 2.4 GHz FH (frequency hopping) spread spectrum radio technology for artifact-free operation that does not disturb the MRI imaging process. Clinicians may also use the remote control unit to adjust a second pump channel when used in combination with our Side Car unit discussed below. Our 3865 MRidium Wireless Remote also functions as a battery charger for our MRidium battery pack.
3861 Side Car Pump Module
Our Side Car Pump Module can be attached to our 3860+ MRidium MRI compatible IV infusion pump to provide a second channel for infusion delivery. This flexible option allows hospitals to convert their single-channel infusion pump into a dual-channel system designed to deliver both large and small volume fluids in the MRI scanner room. The side car is fully functional with our 3865 MRidium Wireless Remote, allowing clinicians the ability to control both channels with one remote control unit outside of the MR scanner room. The additional delivery line has all of the same features and benefits as the 3860+ MRidium MRI compatible IV infusion pump, as described above.
Dose Error Reduction System (“DERS”)
Our DERS for use with our MRidium 3860+ MRI compatible IV infusion pump system incorporates the latest drug safety features for patients. The DERS system enables users to create a unique drug library and establish nominal values and limits for dose and concentration for specified infusion protocols. With DERS, patient safety and user convenience are supported by user-programmed infusion hard limits (maximum and minimum) and soft limits (high and low limits that require user confirmation to exceed). The dose applied via DERS is displayed and can be adjusted directly on the running screen at any time during the infusion. The universal memory card port allows for easy archiving and updating of the drug library.
SpO2 Monitoring with Sensor and Accessories
Our MRidium 3860+ MRI compatible IV infusion pump system also offers state-of-the-art Masimo SET® SpO2TM capability providing a unique ability to have SpO2 monitoring and IV delivery combined in one unit. This feature offers users the ability to start sedations outside of the MRI scanner room, transport to the scanner, and then back to recovery without having to discontinue SpO2 monitoring on the patient. In addition, our fiber optic MRI-SpO2 sensor and accessories provide a safe connection between the patient and our MRI compatible IV infusion pumps. This fiber optic-based SpO2 sensor delivers outstanding performance while avoiding potentially hazardous heating or image artifact during MR scans. The method of patient attachment uses a medical-grade silicone rubber sensor grip that allows easy and convenient attachment to the patient’s hand or foot, and accommodates pediatric, adult, and infant patients with various size grips.
We believe our MRidium 3860+ MRI compatible IV infusion pump system and its customizable features comprehensively and uniquely address the needs of MRI departments within hospitals and other medical facilities.
We also offer two products exclusively to non-U.S. customers. These products consist of the 2460 iMagox MRI SpO2 Monitor and the 2465 iMagox Remote Control.
iMagox 2460 MRI Pulse Oximeter — Available for Use Outside of the U.S.
The iMagox 2460 MRI Pulse Oximeter System uses Masimo SET® Technology and is approved for use in the presence of 0.2T to 3T magnets and operational up to the 10,000 gauss-line. Our digital MRI pulse oximeter simultaneously measures and displays the functional oxygen saturation and pulse rate of adult, pediatric and infant patients. The large display provides digital and waveform data with SpO2 alarms and user messages, which can be easily seen within the MR scanner room. When fully charged, the battery supporting this system will provide up to 24 hours of continuous operation. The unique rear clamp mechanism swivels to allow mounting on either a non-magnetic IV pole, or for mounting to a bed side rail. This portability combined with the system’s extended battery life gives clinicians at medical facilities the freedom to administer continuous patient monitoring before, during and after an MRI scan. Our iMagox system also provides an optional wireless remote and display described below.
iMagox 2465 MRI Oximeter Remote and Display — Available for Use Outside of the U.S.
The iMagox 2465 Wireless Remote and Display allows for monitoring and control of the MRI Pulse Oximeter from outside the MR scanner room. Our remote allows users to adjust all oximeter parameters and reset alarms. The wireless remote, which is designed for plug-and-play use and requires no installation, is fitted with a large display and utilizes the same user interface as the 2460 MRI Pulse Oximeter. The remote also acts as a charger for a backup or spare battery pack for the iMagox 2460 MRI Pulse Oximeter. It utilizes a wireless link at 2.4 GHz for reliable communication with no image artifacts.
Intellectual Property
We protect our proprietary technology through a combination of trade secrets, confidentiality agreements and patents. During the development of our products, our founder, Roger Susi, obtained a number of patents regarding our MRI compatible IV infusion pump and related systems. Mr. Susi has irrevocably assigned these patents to us. We consider our patents important but do not believe our future success is dependent upon patents. We have ten issued U.S. patents and four issued foreign patents. We also have a number of U.S. patent applications pending. These patents and patent applications relate to several of our products, including our MRI compatible IV infusion pump system and its components. We intend to file patent applications with respect to future patentable developments and improvements when we believe that such protection is in our best interest.
We also rely on trade secret, copyright and other laws and on confidentiality agreements to protect our technology, but we believe that neither our patents nor other legal rights will necessarily prevent third parties from developing or using similar or related technology to compete against our products. Moreover, our technology may be viewed as improvements or adaptations of known MRI infusion technology, which might be duplicated or discovered through our patents, reverse engineering or both.
Sales and Marketing
We sell our MRI compatible products through our direct sales force in the U.S. and independent distributors internationally. In the U.S., we sell our products through our ten direct sales representatives and two clinical support representatives. We have distribution agreements for our products with 35 independent distributors selling our products internationally. We have developed an experienced team of distributors that have a strong MRI/radiology product portfolio and focus. Our international distributors are managed by our VP of International Sales, an industry veteran with over 20 years in the IV infusion pump business and nine years as our sales manager.
In fiscal year 2014, 72.6% of our revenue was generated from U.S. sales and 27.4% was generated from international sales.
As of December 31, 2014, our backlog was approximately $19.5 million. We include all purchase orders received from customers in backlog. As part of our commitment to customer service, our goal has been to ship products to meet the customers’ requested shipment dates. Our backlog is occasionally subject to cancellation or rescheduling by the customer on short notice with little or no penalty. Because of the uncertainty of order cancellations or rescheduling, we do not believe our backlog as of any particular date is indicative of actual sales for any future period and, therefore, should not be used as a measure of future revenue.
Selling cycles for our medical devices vary widely but are typically three to six months in duration. To supplement the efforts of our sales and clinical support representatives, we produce and distribute videos that provide users of our MRidium products an easy means for learning clinical applications. These videos guide users through a detailed step-by-step process in using our products, including initial product set-up, selection of infusion sets, loading the infusion pump, programming the pump, managing alarms and alerts and prompts, SpO2 monitoring, and other advanced functions. Users also benefit from our detailed operator manuals and 24/7 technical support via telephone.
The principal customers for our MRI compatible products include hospitals and acute care facilities. A customer’s decision to use our products is typically made by the radiologist and anesthesiologist, or department administrative director. We serve these customers through our sales and service specialists and believe that our specialists are well-positioned to build upon these customer relationships. We communicate with our customers on a regular basis in an attempt to understand potential issues or concerns as well as to improve our products and services in response to their needs. Product orders and inquiries are handled by trained service representatives who communicate with customers after equipment shipments, installations and service repair calls. We have implemented various other programs which enable us to assess our customers’ needs. These programs include regular surveys and visits to customer sites.
As of December 31, 2014, two international customers accounted for approximately 35% of gross accounts receivable. As of December 31, 2013, one international customer accounted for approximately 11% of gross accounts receivable. Revenue for 2013 included sales to an international customer for 129 of our MRI compatible IV infusion pumps, which represented approximately 11% of total revenue for 2013.
We enter into agreements with healthcare supply contracting companies, commonly referred to as GPOs in the U.S., which enable us to sell and distribute our MRidium MRI compatible IV infusion pump systems to their member hospitals. GPOs negotiate volume purchase prices for hospitals, group practices, and other clinics that are members of a GPO. Our agreements with GPOs typically include the following provisions:
· Negotiated pricing for all group members;
· Volume discounts and other preferential terms on their members’ purchases from us;
· Promotion of our products by the GPO to its members; and
· Payment of administrative fees by us to the GPO, based on purchases of our products by group members.
Under these agreements, we are required to pay the GPOs a fee of three percent of the sales of our products to hospitals, group practices, and other acute care facilities that are members of the GPO. We currently have contracts with four major GPOs that effectively give us the ability to sell to more than 95% of all U.S. acute care facilities.
Our MRidium MRI compatible IV infusion pump system received the Frost and Sullivan Technology award in 2005.
Manufacturing and Suppliers
We assemble our products in our facilities in Winter Springs, Florida, from components and sub-assemblies purchased from outside suppliers. We perform final assembly, testing and packaging to control quality and manufacturing efficiency. We purchase components and sub-assemblies from qualified suppliers that are subject to our stringent quality specifications and inspections by us. We conduct quality audits of our key suppliers, several of which are experienced in the supply of components to manufacturers of finished medical devices or disposables for use with these medical devices. Our historical track record of producing MRI compatible IV infusion pump systems has been good; however, there can be no assurance that this trend will continue or that we will be able to produce sufficient units to reach our expected revenue growth rates.
The non-magnetic ultra-sonic motor which drives our MRI compatible IV infusion pump is sole-sourced from a major multinational Japanese manufacturing company with whom we have an excellent long-term relationship. This company has exclusively provided us with these motors since 2005, and we recently renewed our exclusive supply agreement with this company for another five years through 2019. We have never encountered a significant supply interruption from this manufacturer and have received no indications that there might be disruptions of the supply of these motors in the future. We have routinely averaged no more than a two-month supply of these motors in our inventory. The supplier has committed to maintaining our supply and delivering on a timely basis. We have identified two additional suppliers who may be able to produce an ultra-sonic motor essentially identical to our current motor. However, we believe that adding or switching to an alternate supplier would require six months to a year.
We have two alternate suppliers of our IV sets. The first is our own in-house IV set manufacturing capability, and the second is an OEM located in central Florida. We expect our in-house capabilities coupled with production capacity of the OEM should be sufficient to meet expected customer demand for the foreseeable future.
Other sole or limited supply devices and components of our products include the following:
· Force Sensor. This device is used to measure the fluid pressure within the IV tubing set and provide certain alarm functions. This part is supplied by a large multinational U.S.-based manufacturer. We have never experienced a delivery problem of this part, and we typically maintain three to six months inventory in supply. However, we have had an internal development effort underway to create and manufacture this device in-house. In the event of a failure of our current supplier to furnish sufficient force sensors, we believe we could assume the manufacture of this device in three to five months.
· Bubble Detector. This device provides safety and alarm upon detection of air in the IV fluid line. We have never experienced delivery issues with this part, which is supplied by a major multinational U.S.-based company, and we generally maintain a three-month supply on hand. There is one alternate supplier whose part we have tested on a preliminary basis, and we believe we could switch to that source if needed, within approximately three or four months.
· Lithium Polymer Battery Cells. This device is used to power our MRI compatible IV infusion pump. Though we keep several months’ supply on hand and have alternate sources qualified, due to regulatory issues, changing to an alternate source could require three to four months.
· LCD Displays. This component is currently a sole-sourced assembly from a large Chinese supplier who has a history of late delivery. We typically maintain a three-month supply on hand. We have an active ongoing effort to cultivate a second source, which we expect to be finalized by late 2015.
· Various Molded and Cast Components. These custom tooled and molded/cast parts are subject to supply interruption should the tools become damaged. Replacement of these tools could require up to eight months. Our inventory of such custom molded and cast parts is typically on the order of six months.
We place significant emphasis on providing quality products and services to our customers. Quality management and oversight play an essential role in understanding and meeting customer requirements, effectively resolving quality issues and improving our products and services. We have a network of quality systems throughout our facilities that relate to the design, development, assembly, packaging, sterilization, handling, distribution and labeling of our products. To assess and facilitate compliance with applicable requirements, we periodically review our quality systems to determine their effectiveness and identify areas for improvement.
We also conduct compliance training programs for our sales and marketing personnel and perform assessments of our suppliers of raw materials, components and finished goods. In addition, we conduct quality management reviews designed to inform management of key issues that may affect the quality of our products. From time to time, we may determine that products manufactured or marketed by us do not meet our specifications, published standards or regulatory requirements. When a quality issue is identified, we investigate the issue and seek to take appropriate corrective action, such as withdrawal of the product from the market, correction of the product at the customer location, notice to the customer of revised labeling or a combination of these or other corrective actions.
In January 2007, we received ISO 9001 and ISO 13485 certification and met the requirements under the European Medical Device Directive to use the CE Mark, thereby allowing us to continue to market our products in the European Community. These certificates were last renewed on January 17, 2013, and will need renewal again in January 2016.
Competition
We do not believe there is currently any direct competition for our MRI compatible IV infusion pump systems. Our only direct competitor in the MRI compatible IV infusion pump market, Bayer Radiology, formerly MEDRAD, Inc., announced during 2013 its decision to remove its competing Continuum pump system from the market, and to discontinue support throughout the world by June 30, 2015 due to ongoing regulatory issues. As a result, we believe that our MRidium 3860+ MRI compatible IV infusion pump is the only true MRI compatible IV infusion pump available today. Bayer Radiology’s announcement provided that it planned to remove the actual product from customers in the field, and that it would no longer offer the disposable proprietary IV tubing sets after June 30, 2015.
The medical device and IV infusion market is highly regulated and is typically one of the areas that the FDA scrutinizes closely for new market introductions. Because of this, the 510(k) FDA clearance process for new pumps is usually long and requires significant testing and documentation. This long development timeline coupled with the low market penetration to date may discourage new competitors from undertaking a complex project like building an MRI compatible IV infusion pump. However, the medical products industry is generally characterized by intense competition and extensive research and development. We believe, that the market for MRI compatible IV infusion pump products is in relatively early stages of development and may become highly competitive if, and when, the market develops further.
Outside of the U.S., we also compete with manufacturers of “shielded box” solutions that are intended to permit use of conventional IV pumps inside the MR scanner room. The providers of shielded boxes include B. Braun, Fresenius Kabi, MIPM Mammendorfer Institut für Physik und Medizin, and Arcomed. The market for medical products is subject to rapid change due to
an increasingly competitive, cost-conscious environment and to government programs intended to reduce the cost of medical care. Many of these manufacturers and distributors of medical equipment are large, well-established companies whose resources, reputations and ability to leverage existing customer relationships might give them a competitive advantage over us. Our SpO2 products, which measure blood oxygen saturation, also compete indirectly with many other methods currently used to measure blood oxygen levels or the effects of low blood oxygen levels.
Another potential competitor may be CareFusion Corporation (NYSE: CFN). CareFusion is a major medical device manufacturer that has a dominant position in the conventional IV infusion pump market and made an investment in Caesarea Medical Electronics (“CME”) in December 2013. CME manufactured Bayer Radiology’s Continuum Pump System. In addition, B. Braun may seek to obtain FDA clearance for its SpaceStation MRI Trolley, currently only available outside the U.S., which allows traditional B. Braun IV infusion pumps to be used in the MR environment.
Many of our potential customers opt not to purchase our MRI compatible IV infusion pump systems and instead use makeshift workarounds, such as placing conventional IV infusion devices outside of the MR scanning room and utilizing extension tubing to reach the patient. To this extent, we are in competition with conventional IV infusion pump manufacturers and distributors.
There are many manufacturers of conventional IV infusion pump devices, and if any of these manufacturers, or other potential competitors, decide to enter into the MRI compatible IV infusion pump market, they may have competitive advantages over us. Many of these potential competitors have established reputations, customer relationships and marketing, distribution and service networks. In addition, they have substantially longer histories in the medical products industry, larger product lines and greater financial, technical, manufacturing, management, and research and development budgets. Many of these potential competitors may have long-term product supply relationships with our potential customers.
We believe that a company’s reputation for producing accurate, reliable and technologically-advanced products, references from users, features (speed, safety, ease of use, patient convenience and range of applicability), product effectiveness and price are the principal competitive factors in the medical products industry.
Seasonality
Our business is seasonal. Our third quarter sales have typically been lower, compared to other fiscal quarters, principally because the fiscal quarter coincides with the summer vacation season, especially in the U.S., Europe, and Japan.
Research and Development
Our research and development efforts focus on developing innovative products by utilizing our established core competencies in MR compatible technologies and feedback from strategic relationships with hospitals, acute medical facilities and medical equipment manufacturers for new product ideas. Our research and development efforts are driven by the leadership of our founder, Roger Susi, assisted by five engineers and technical professionals with significant experience in product design.
Employees
As of December 31, 2014, we had 54 full-time employees, including 5 in research and development, 24 in manufacturing, 17 in sales and marketing and customer support services and 8 in finance and administration. No employees are represented by a labor union. We have not experienced any work stoppages and consider our relations with our employees to be good.
Regulatory Matters
Governmental Regulation and Other Matters
Our medical device products are subject to extensive, complex and increasing oversight and regulation by the U.S. Food & Drug Administration (“FDA”), and other domestic and foreign governmental authorities. Our manufacturing and other
facilities, and those of our suppliers, are subject to periodic inspections to verify compliance with current FDA and other governmental regulatory requirements. If it were determined that we were not in compliance with these laws and regulations, we could be subject to criminal or civil liability, or both, and other material adverse effects. We have compliance programs in place to support and monitor compliance with these laws and regulations. All of our products and facilities and those of our suppliers are subject to drug and medical device laws and regulations promulgated by the FDA and national and supranational regulatory authorities outside the U.S., including, for example, Health Canada’s Health Products and Foods Branch, the U.K.’s Medicines and Healthcare Products Regulatory Agency, and Australia’s Therapeutic Goods Agency. These authorities regulate a range of activities including, among other matters, manufacturing, post-marketing studies in humans, advertising and promotion, product labeling, post-marketing surveillance and adverse event reporting.
Regulation of Medical Devices in the United States
The development, manufacture, sale and distribution of our medical device products are subject to comprehensive governmental regulation. Most notably, all of our medical devices sold in the United States are subject to the Food, Drug, and Cosmetic Act of 1938, as amended (“FDC Act”), as implemented and enforced by the FDA. The FDA, and in some cases other government agencies, such as the U.S. Federal Communications Commission (“FCC”), administer requirements covering the design, testing, safety, effectiveness, manufacturing, labeling, promotion and advertising, distribution and post-market surveillance of our products.
Unless an exemption applies, each medical device that we market must first receive either premarket notification (by making what is commonly called “a 510(k) submission”) clearance or premarket approval (by filing a premarket approval application (“PMA”) from the FDA pursuant to the FDC Act. In addition, certain modifications made to marketed devices also may require 510(k) clearance or approval of a PMA supplement. The FDA’s 510(k) clearance process usually takes up to twelve months, but it can last longer. The process of obtaining PMA approval is much more costly, lengthy and uncertain. It generally takes from two to three years or even longer. All of our current products that are available in the U.S. were originally cleared through the 510(k) process. However, on September 2, 2014, we received a warning letter from the FDA requesting that we cease commercial distribution of our products and submit a new 510(k). Refer to the section below captioned “FDA Facility Inspection and Warning Letter.” We cannot be sure that future products or modifications of current products, will qualify for the 510(k) pathway or whether 510(k) clearance or PMA approval will be obtained for any future product that we propose to market.
In December 2014, the FDA issued guidance entitled “Infusion Pumps Total Product Life Cycle.” This guidance established substantial additional pre-market requirements for new and modified infusion pumps. Through this guidance, the FDA indicated more data demonstrating product safety will be required for future 510(k) submissions for infusion pumps, including the potential for more clinical and human factors data. The impact of this guidance is likely to result in a more time consuming and costly process to obtain regulatory clearance to market infusion pumps. In addition, new requirements could result in longer delays for the clearance of new products, modification of existing infusion pump products or remediation of existing products in the market. Future delays in the receipt of, or failure to obtain, approvals could result in delayed or no realization of product revenues.
After a device is placed on the market, numerous regulatory requirements continue to apply. Those regulatory requirements include the following: product listing and establishment registration; adherence to the Quality System Regulation (“QSR”), which requires stringent design, testing, control, documentation and other quality assurance procedures; labeling requirements and FDA prohibitions against the promotion of off-label uses or indications; adverse event reporting; post-approval restrictions or conditions, including post-approval study commitments; post-market surveillance requirements; the FDA’s recall authority, whereby it can ask for, or require, the recall of products from the market; and requirements relating to voluntary corrections or removals.
All aspects of our manufacturing and distribution of regulated products and those of our suppliers are subject to substantial governmental oversight. Facilities used for the production, packaging, labeling, storage and distribution of medical devices must be registered with the FDA and other regulatory authorities. All manufacturing activities for these products must be conducted in compliance with current good manufacturing practices (“cGMPs”). Our manufacturing facilities and those of our suppliers are subject to periodic, routine and for-cause inspections to verify compliance with cGMPs. If, upon inspection, the FDA or another regulatory agency finds that a manufacturer has failed to comply with cGMPs, it could institute a wide variety of enforcement actions, ranging from a public warning letter to more severe sanctions, such as product recalls or seizures, monetary sanctions, consent decrees, injunctions to halt manufacturing and distributing products, civil or criminal sanctions, refusal to
grant clearances or approvals or delays in granting such clearances or approvals, import detentions of products made outside of the United States, restrictions on operations or withdrawal or suspension of existing approvals. The FDA also has the authority to request repair, replacement or refund of the cost of any medical device manufactured or distributed by us. These actions could result in, among other things, substantial modifications to our business practices and operations; a total or partial shutdown of production in one or more facilities while we or our suppliers remedy the alleged violation; the inability to obtain future pre-market clearances or approvals; and withdrawals or suspensions of current products from the market. Any of these events could disrupt our business and have a material adverse effect on our revenues, profitability and financial condition.
FDA Facility Inspection and Warning Letter
The FDA conducted a routine inspection of our prior facility between April 7 and April 16, 2014. This was the first FDA inspection of our facility since the voluntary product recall in August 2012 of certain infusion sets and the voluntary recall in July 2013 of our DERS software. The FDA issued a Form 483 on April 16, 2014 that identified eight observations. In general, the observations involved issues related to the 2012 and 2013 product recalls (described in more detail under Product Recalls, below). The majority of the observations related to procedural and documentation issues associated with the design, development, validation testing and documentation of software used in certain of our products. Other observations were related to the design validation of pump labeling, design analysis of tube stretching, procedures for post-market design review, and control and procedures related to handling certain reported complaints. We submitted a response to the Form 483 in May 2014 and June 2014 in which we described our proposed corrective and preventative actions to address each of the FDA’s observations.
On September 2, 2014, we received a warning letter from the FDA relating to this inspection (the “Warning Letter”). The Warning Letter stated that the FDA accepted as adequate several of our responses to Form 483 observations, identified two responses whose accuracy will be determined in the next scheduled inspection of our facility and identified issues for which our response was determined to be inadequate. The issues identified as inadequate concern our procedures for validating device design primarily related to software quality assurance.
Also, the Warning Letter raised a new issue. The Warning Letter stated that modifications made to software on our previously cleared infusion pumps, the MRidium 3860 and MRidium 3850, were “significant” and required submission of new premarket notifications under Section 510(k) (a “510(k) submission”) of the FDC Act. These modifications were made over time. We believe they were insignificant and did not require premarket notification submissions. However, the FDA indicated that the modifications of the software for the MRidium 3860 and the software for the MRidium 3850 were “significant” modifications because they could significantly affect the safety or effectiveness of these devices. As a result, the Warning Letter states that the products being sold by us are “adulterated” and “misbranded” under the FDC Act. The Warning Letter also indicates that the MRidium 3860+ infusion pump requires separate FDA clearance from the MRidium 3860 and MRidium 3850.
The Warning Letter requested that we immediately cease activities that result in the misbranding or adulteration of the MRidium 3860 MRI infusion pump, MRidium 3850 MRI infusion pump, and the MRidium 3860+ MRI infusion pump, including the commercial distribution of the devices. We immediately complied with the Warning Letter and ceased sale and distribution of the identified products in the United States.
On September 4, 2014, we submitted to the FDA our initial response to the Warning Letter and on September 17, 2014 we sent an additional response that included supplemental information related to the Form 483 inspection observations for which the FDA considered our initial responses inadequate.
On November 25, 2014, we announced that we filed the 510(k) submission related to our MRidium 3860+ MRI IV infusion pumps and on December 12, 2014 we were notified that our 510(k) submission had been formally accepted for review by the FDA. On December 22, 2014, under FDA enforcement discretion, we announced that we resumed domestic distribution of our MRI compatible MRidium 3860+ MRI IV infusion pump systems, without the DERS option.
We continue to work with the FDA to fully resolve the Warning Letter and complete the review of the 510(k) submission.
Overall Financial Impact from the Warning Letter. Our financial results for 2014 were significantly impacted by the Warning Letter and resulting stoppage of domestic shipments. We expect to continue to incur higher than normal financial expenses until the issues raised in the Warning Letter are completely resolved.
Product Recalls
Dose Error Reduction System (“DERS”) Software Recall. Some of our MRidium 3860+ MRI compatible IV infusion pumps are equipped with a DERS. Due to a software issue observed on June 19, 2013, the drug dosage calculation indicated an incorrect recommended value for the flow rate when a specific key sequence was used during the infusion setup. As a result, a patient was infused with an incorrect flow rate. No harm to the patient was reported. On July 1, 2013, we issued an urgent medical device recall notice (the “DERS Recall”) and promptly made available to our customers a software update to resolve the error. On July 2, 2013, the subject of the recall was discussed with the FDA by phone. On July 12, 2013, we provided written notification to the FDA of the DERS Recall and submitted a Medical Device Report (MDR) with the FDA describing the incident, the investigative and corrective actions taken, the reason for the DERS Recall and the recall strategy. On September 18, 2013, we notified the FDA that all of the pumps sold with the DERS kits had been successfully upgraded with the software correction and reported that the DERS Recall was completed as of September 16, 2013. We requested that the FDA officially close the DERS Recall. The FDA has not yet responded to our request of September 16, 2013 or more recent requests to close the DERS Recall. It is likely that the FDA wanted to conduct a follow-up inspection prior to closing the DERS Recall. The FDA completed an inspection of our facility on April 16, 2014. Refer to the section above captioned “FDA Facility Inspection and Warning Letter.”
Overall Financial Impact from the Recalls. We believe the financial expenses incurred related to the recall were not significant to our operations or financial results. We expect to incur only minor additional charges over the next several months in connection with the final closeout activities related to the recall.
Corrective Actions from the Recall. We take recalls and related matters seriously and we have responded and will continue to respond fully, and in a timely manner, to the FDA and other governmental regulatory agencies.
We have made substantial investments in quality systems over the past two years. We will continue to make improvements to our products and systems to further reduce potential issues related to patient safety and avoid recalls in the future. Product quality plays a critical role in our success. While we believe that we have made significant improvements to our product quality and overall quality systems, further quality concerns, whether real or perceived, could adversely affect our results. Conversely, improving quality can be a competitive advantage and improve our results. For more information about risks related to these matters, see the section captioned “Defects or failures associated with our products and/or our quality systems could lead to the filing of adverse event reports, recalls or safety alerts and negative publicity and could subject us to regulatory actions” in the “Risk Factors” section.
Healthcare Fraud and Abuse Laws
As a manufacturer and distributor of medical devices to hospitals and other healthcare providers, we and our customers are subject to laws which apply to Medicare, Medicaid, and other federal and state healthcare programs in the U.S. One such law, the Anti-kickback Statute, prohibits the solicitation, offer, payment or receipt of remuneration in return for referral or purchase, or in return for the recommending or arranging for the referral or purchase, of products covered by the programs. The Anti-kickback Statute provides a number of exceptions or “safe harbors” for particular types of transactions. While we generally do not file claims for reimbursement from government payers, the U.S. federal government has asserted theories of liability against manufacturers under the Federal False Claims Act, which prohibits the submission of false claims to Medicare, Medicaid, and other state and federal programs. Many states have similar fraud and abuse laws which may apply to us. Violations of these fraud and abuse-related laws are punishable by criminal or civil sanctions, including substantial fines, imprisonment and exclusion from participation in healthcare programs such as Medicare and Medicaid and health programs outside the United States. We have developed and implemented business practices and processes to train our personnel to perform their duties in compliance with healthcare fraud and abuse laws. While we conduct informal oversight to detect and prevent these types of fraud and abuse, we lack formal written policies and procedures at this time. If we were unable to document and implement the controls and procedures required in a timely manner or otherwise violate such laws, we might suffer adverse regulatory consequences or face criminal sanctions, which could harm our operations, financial reporting or financial results.
Regulation of Medical Devices Outside of the United States
Medical device laws also are in effect in many of the non-U.S. markets in which we do business. These laws range from comprehensive device approval requirements for some or all of our products to requests for product data or certifications.
Inspection of and controls over manufacturing, as well as monitoring of device-related adverse events, also are components of most of these regulatory systems. Most of our business is subject to varying degrees of governmental regulation in the countries in which we operate, and the general trend is toward increasingly stringent regulation balanced with a goal of optimizing international harmonization. For example, the European Union (“EU”), which currently relies on independent third parties, (called “Notified Bodies”) rather than governmental authorities to review and certify medium and high risk medical devices, is moving to more governmental oversight of medical devices. Currently, the regulatory requirements for a broad spectrum of medical devices are covered in three European Medical Device Directives (adopted in the 1990’s) with which manufacturers must comply in order to receive a CE Certificate of Conformity (“CE Mark”) from a Notified Body. Only certified medical devices bearing a CE Mark can be sold in the EU and European Free Trade Association (“EFTA”) countries and Turkey. EFTA includes Iceland, Norway, Principality of Liechtenstein and Switzerland.
In September 2012, the European Commission, (“EC”), proposed significant revisions to the regulatory framework for medical devices in the EU. The proposed changes include more oversight of Notified Bodies by governmental authorities, replacing the three European Medical Service Directives with two regulations and more stringent requirements for clinical evidence while also enhancing alignment with international guidelines to facilitate international trade. It is unknown how the proposed revisions will affect certification of future products or modifications of current products, but it is possible that more clinical data will be needed to support our applications, which would increase the costs and development time involved. We may lose our current quality system certification due to ISO Registrar difficulties as European authorities increase regulatory pressure or increased scrutiny resulting from the EU’s Revised Medical Device Directive. The loss of the quality system certification may prevent product shipments to the EU and to other foreign markets, such as Canada. The EU has enacted legislation restricting the use of hazardous substances in electronic equipment (Directive 2011/65/EU, referred to as RoHS 2), such as our infusion pumps. The application of RoHS 2 to medical devices became effective as of July 22, 2014. Our MRidium 3860+ pumps systems are compliant with RoHS 2. If we are unable to remain compliant with RoHS 2, there may be an interruption of sales to the EU, which could significantly lower our revenues from foreign sales while we take remedial measures.
Anti-Bribery Laws
Our global activities are subject to the U.S. Foreign Corrupt Practices Act, the U.K. Bribery Act, and other countries’ anti-bribery laws that have been enacted in support of the Organization for Economic Cooperation and Development’s Anti-bribery Convention. These laws generally prohibit companies and their intermediaries from making improper payments to non-U.S. government officials with the intent to inappropriately gain a business advantage. They also require companies to maintain accurate books and records and internal financial controls. The U.K. Bribery Act also prohibits commercial bribery and makes it a crime for a company to fail to prevent bribery. Companies have the burden of proving that they have adequate procedures in place to prevent bribery. The enforcement of such laws in the U.S. and elsewhere has increased dramatically in the past few years, and authorities have indicated that the pharmaceutical and medical device industry is a significant focus for enforcement efforts.
Because of the predominance of government-sponsored healthcare systems around the world, many of our customer relationships outside of the United States are with governmental entities. Our policies mandate strict compliance with the anti-bribery laws. We operate in many parts of the world that have experienced governmental corruption to some degree, and in certain circumstances strict compliance with anti-bribery laws may conflict with local customs and practices.
Transparency Laws in the U.S. and Other Countries
There are numerous requirements imposed by states in the U.S. on the interaction of pharmaceutical and medical device companies with physicians. For example, several states and the District of Columbia either require the tracking and reporting of specific types of interactions with healthcare professionals or restrict such interactions. A similar requirement was imposed at the federal level under the “sunshine” provision of Patient Protection and Affordable Care Act, (the “Sunshine Provisions”), to track and report payments and “transfers of value” to U.S. physicians or teaching hospitals by manufacturers of medical products that are available for reimbursement by a federal insurer. It is expected that the Sunshine Provisions will preempt some but not all disclosure requirements under state laws.
Other Laws
We are also subject to a variety of other laws, directives and regulations in and outside of the U.S., including those related to the following:
· environmental laws and regulations;
· the safety and health laws of the U.S. Occupational Safety and Health Act, which sets forth requirements for workplace conditions;
· California’s Proposition 65, which sets forth a list of substances that are deemed by the State of California to pose a risk of carcinogenicity or birth defects; and
· various customs, export control, anti-boycott and trade embargo laws and regulations administered by U.S. and foreign government agencies, including the U.S. Customs and Border Protection, the Bureau of Industry and Security, the Department of Commerce and the Office of Foreign Assets Control Treasury Department, as well as others.
Despite our training and compliance program, our internal control policies and procedures may not always protect us from reckless or criminal acts committed by our employees or agents in violation of any of these laws.
Risks Relating to Our Business and Financial Condition
Our financial performance is currently dependent on a single product, and disruptions in our ability to sell this product may have a material adverse effect on our business.
Our current revenue and profitability is dependent on the sale of the MRidium 3860+ and 3850/R MRI compatible IV infusion pump system and the ongoing sale of disposable tubing sets related to them. Sales of the MRidium 3860+ and 3850/R MRI compatible IV infusion pump systems have historically comprised a substantial majority of our net revenue. Our near-term revenue and profitability will, accordingly, be dependent upon our ability to successfully market and sell this Class II medical device.
On September 2, 2014 we announced we received a Warning Letter from the FDA. The FDA stated in the Warning Letter that modifications made to software on our previously cleared infusion pumps, the MRidium 3860 and MRidium 3850, were “significant” and required submission of new premarket notifications under Section 510(k) (a “510(k) submission”) of the Food, Drug and Cosmetic Act (the “FDCA”). Such updates occurred periodically over time. As stated in the warning letter, the FDA indicated that the modifications of the software related to the MRidium 3860 from version 2.0 to current version 3.5.1 and the modifications of the software related to the MRidium 3850 from version 1.0 to version 585.11.1 as “significant” modifications because they could significantly affect the safety or effectiveness of the devices. As a result, the Warning Letter alleges that the products being sold by us are “adulterated” and “misbranded” under the FDCA. The Warning Letter also indicates that the MRidium 3860+ infusion pump requires separate FDA clearance from the MRidium 3860 and MRidium 3850.
The Warning Letter requested that we immediately cease activities that result in the misbranding or adulteration of the MRidium 3860 MRI infusion pump, MRidium 3850 MRI infusion pump, and the MRidium 3860+ MRI infusion pump, such as the commercial distribution of the devices. We immediately complied with the Warning Letter and ceased sale and distribution of the identified products in the United States. On November 25, 2014, we announced that we filed the 510(k) submission related to our MRidium 3860+ MRI IV infusion pumps and on December 12, 2014 we were notified that our 510(k) submission had been formally accepted for review by the FDA. On December 22, 2014, we announced that we resumed domestic distribution of our MRI compatible MRidium 3860+ MRI IV infusion pump systems, without the Dose Error Reduction System (“DERS”) option. On January 28, 2015, subsequent to our year end, we announced that we resumed domestic distribution of our DERS option.
The disruption in the shipment of our products had a significant adverse effect on our business and our financial condition. We are working with the FDA to resolve this issue and resume commercial distribution of our product. On September 4, 2014, we submitted to the FDA our initial response to the Warning Letter and on September 17, 2014 we sent an additional response that included supplemental information. Although we have resumed commercial distribution of our product, there can be no guarantee that our efforts will be successful in obtaining clearance of our 510(k) submission. The FDA could require us to, again, cease shipment of our products, or notify health professionals and others that the devices present unreasonable risk or substantial harm to public health, order a recall, repair, replacement, or refund of the devices, detain or seize adulterated or misbranded medical devices, or ban the medical devices. The FDA may also issue further warning letters or untitled letters, refuse our request for 510(k) submission or premarket approval, revoke existing 510(k) clearances or premarket approvals previously granted, impose operating restrictions, enjoin and restrain certain violations of applicable law pertaining to medical devices and assess civil or criminal penalties against our officers, employees, or us.
The MRidium 3860+ or 3850/R MRI compatible IV infusion pumps could be rendered obsolete or economically impractical by numerous factors, many of which are beyond our control, including:
· entrance of new competitors into our markets;
· loss of key relationships with suppliers, group purchasing organizations, or end-user customers;
· manufacturing or supply interruptions;
· product liability claims;
· our reputation and product market acceptance; and
· product recalls or safety alerts.
Any major factor adversely affecting the sale of our MRidium 3860+ MRI compatible IV infusion pump would cause our revenues to decline and have a material adverse impact on our business, financial condition and our common stock.
We are currently subject to securities class action litigation and derivative litigation and we may be subject to similar or other litigation in the future.
We and certain of our officers and/or directors are defendants in a lawsuit filed in the United States District Court for the Southern District of Florida, brought on behalf of our stockholders that alleges that the defendants failed to disclose material information concerning our compliance with FDA regulations in violation of Section 10(b) of the Securities Exchange Act of 1934 and Rule 10b-5 thereunder, and that the putative class members suffered damages as a result. The lawsuit additionally alleges “control person” liability against the individual defendants under Section 20(a) of the Securities Exchange Act of 1934. The plaintiffs seek to represent a class comprised of purchasers of our common stock during the period from July 15, 2014 through September 2, 2014 and seeks damages, costs and expenses and such other relief as determined by the Court.
While we believe we have meritorious defenses and intend to defend this lawsuit vigorously, we cannot predict the outcome of this lawsuit. Legal fees and costs incurred in connection with such activities may be significant and we could, in the future, be subject to judgments or enter into settlements of claims for significant monetary damages. With respect to any litigation, our insurance may not reimburse us or may not be sufficient to reimburse us for the expenses or losses we may suffer in contesting and concluding such lawsuits. A decision adverse to our interests on these actions or resulting from these matters could result in the payment of substantial damages and could have a material adverse effect on our business, financial condition and our common stock. Regardless of the outcome, these claims may result in injury to our reputation, significant costs, diversion of management’s attention and resources, and loss of revenue.
Our continued success depends on the integrity of our supply chain, including multiple single-source suppliers, the disruption of which could negatively impact our business.
Many of the component parts of our MRidium MRI compatible IV infusion pumps are obtained through supply agreements with third parties. Some of these parts require our partners to engage in complex manufacturing processes. In light of our dependence on third-party suppliers, several of which are single-source suppliers, we are subject to inherent uncertainties and risks related to their ability to produce parts on a timely basis, to comply with product safety and other regulatory requirements and to provide quality parts to us at a reasonable price.
For example, we are dependent upon a single vendor for the ultrasonic motor at the core of our MRidium MRI compatible IV infusion pump. If this vendor fails to meet our volume requirements, which we anticipate will increase over time, or if the vendor becomes unable or unwilling to continue supplying motors to us, this would impact our ability to supply our pumps to customers until a replacement source is secured. Our executed agreement with this vendor provides that the price at which we purchase products from the vendor is determined by mutual agreement from time to time or should material costs change. Although we have had a long history of stable pricing with this supplier, this provision may make it difficult for us to continue to receive motors from this vendor on favorable terms or at all if we do not agree on pricing in the future. In such event, it could materially and adversely affect our commercial activities, operating results and financial condition.
In the near term, we do not anticipate finding alternative sources for our primary suppliers, including single source suppliers. Therefore, if our primary suppliers become unable or unwilling to manufacture or deliver materials, we could experience protracted delays or interruptions in the supply of materials which would ultimately delay our manufacture of products for commercial sale, which could materially and adversely affect our development programs, commercial activities, operating results and financial condition.
Additionally, any failure by us to forecast demand for, or to maintain an adequate supply of, raw materials or finished products could result in an interruption in the supply of certain products and a decline in our sales.
The manufacture of our products requires strict adherence to regulatory requirements governing medical devices and if we or our suppliers encounter problems our business could suffer.
The manufacture of our pumps and products must comply with strict regulatory requirements governing Class II medical devices in the U.S. and other regulatory requirements in foreign locations. Problems may arise during manufacturing, quality control, storage or distribution of our products for a variety of reasons, including equipment malfunction, failure to follow specific protocols and procedures, manufacturing quality concerns, or problems with raw materials, electromechanical, software and other components, supplier issues, and natural disasters. If problems arise during production of our pump, the batch may have to be discarded. Manufacturing problems or delays could also lead to increased costs, lost sales, damage to customer relations, failure to supply penalties, time and expense spent investigating the cause and, depending on the cause, similar losses with respect to other batches of products. If problems are not discovered before the product is released to the market, voluntary recalls, corrective actions or product liability related costs may also be incurred. Should we encounter difficulties in the manufacture of our products or be subject to a product recall, our business could suffer materially.
We manufacture and store our products at a single facility in Florida.
We manufacture and store our products at a single facility in Winter Springs, Florida. If by reason of fire, hurricane or other natural disaster, or for any other reason, the facility is destroyed or seriously damaged or our access to it is limited, our ability to provide products to our customers would be seriously interrupted or impaired and our operating results and financial condition would be negatively affected.
Our inability to collect on our accounts receivables held by significant customers may have an adverse effect on our business operations and financial condition.
We market our products to end users in the United States and to distributors internationally. Sales to end users in the United States are generally made on open credit terms. Management maintains an allowance for potential credit losses. From time to time, we have had accounts receivables from one or two customers that accounted for 10.0% or more of our gross accounts receivable. As a result, we are exposed to a certain level of concentration of credit risk. If a major customer experiences financial difficulties, the effect on us could be material and have an adverse effect on our business, financial condition and results of operations.
If we fail to maintain relationships with GPOs, sales of our products could decline.
Our ability to sell our products to U.S. acute care facilities and outpatient imaging centers depends in part on our relationships with group purchasing organizations (“GPOs”). Many existing and potential customers for our products are members of GPOs. GPOs negotiate pricing arrangements and contracts, which are sometimes exclusive, with medical supply manufacturers and distributors, and these negotiated prices are made available to a GPO’s affiliated hospitals and other members. We pay the GPOs an administrative fee in the form of a percentage of the volume of products sold to their affiliated hospitals and other members. If we are not an approved provider selected by a GPO, affiliated hospitals and other members may be less likely to purchase our products. Should a GPO negotiate a sole source or bundling contract covering a future competitor’s products, we may be precluded from making sales to members of that GPO for the duration of the contractual arrangement. Our failure to renew contracts with GPOs may cause us to lose market share and could have a material adverse effect on our sales, financial condition and results of operations. In the future, if another competitive supplier emerges, and we fail to keep our relationships and develop new relationships with GPOs, our competitive position would likely suffer.
Cost-containment efforts of our customers and purchasing groups could adversely affect our sales and profitability.
Our MRI compatible IV infusion pumps are considered capital equipment by many potential customers, and hence changes in the budgets of healthcare organizations and the timing of spending under these budgets and conflicting spending priorities can have a significant effect on the demand for our products and related services. Any decrease in expenditures by these healthcare facilities could decrease demand for our products and related services and reduce our revenue.
Any failure in our efforts to educate clinicians, anesthesiologists, radiologists, and hospital administrators regarding the advantages of our products could significantly limit our product sales.
Our future success will require us to educate a sufficient number of clinicians, anesthesiologists, radiologists, hospital administrators and other purchasing decision-makers about our products and the costs and benefits of MRI compatible IV infusion pump systems. If we fail to demonstrate the safety, reliability and economic benefits of our products to hospitals and acute medical facilities, our products may not be adopted and our sales will suffer.
The lengthy sales cycle for the MRidium 3860+ MRI compatible IV infusion pump could delay our sales.
The decision-making process of customers is often complex and time-consuming. Based on our experience, we believe the period between initial discussions concerning the MRidium 3860+ MRI compatible IV infusion pump and a purchase of a unit is three to six months. The process can be delayed as a result of capital budgeting procedures. Moreover, even if one or two units are sold to a hospital, we believe that it will take additional time and experience with the MRidium 3860+ MRI compatible IV infusion pump before other medical professionals routinely use the MRidium 3860+ MRI compatible IV infusion pump for other procedures and in other departments of the hospital. Such time would delay potential sales of additional units and disposable tubing or additional optional accessories to that medical facility or hospital. These delays could have an adverse effect on our business, financial condition and results of operations.
Because we rely on distributors to sell our products outside of the U.S., our revenues could decline if our existing distributors do not continue to purchase products from us or if our relationship with any of these distributors is terminated.
We rely on distributors for all of our sales outside the U.S. and hence do not have direct control over foreign sales activities. These distributors also assist us with regulatory approvals and the education of physicians and government agencies. Our revenues outside the U.S. have historically represented approximately one-quarter to one-third of our net revenues, and we intend to continue our efforts to increase our sales in Europe, Japan and other countries. If our existing international distributors fail to sell our products or sell at lower levels than we anticipate, we could experience a decline in revenues or fail to meet our forecasts. We cannot be certain that we will be able to attract new international distributors nor retain existing ones that market our products effectively or provide timely and cost-effective customer support and service. None of our existing distributors are obligated to continue selling our products.
If we do not successfully develop and commercialize enhanced products or new products that remain competitive, we could lose revenue opportunities and customers, and our ability to achieve growth would be impaired.
The medical device industry is characterized by rapid product development and technological advances, which places our products at risk of obsolescence. Our long-term success depends upon the development and successful commercialization of new products, new or improved technologies and additional applications for non-magnetic infusion technology. The research and development process is time-consuming and costly and may not result in products or applications that we can successfully commercialize. If we do not successfully adapt our technology, products and applications, we could lose revenue opportunities and customers. In addition, we may not be able to improve our products or develop new products or technologies quickly enough to maintain a competitive position in our markets and continue to grow our business.
We are highly dependent on our founder, CEO, President, Director and controlling shareholder, Roger Susi.
Roger Susi developed our MRidium MRI compatible IV infusion pump system, and we believe that he will play a significant role in our continued success and in the development of new products including an MRI compatible device for patient resuscitation. Our current and future operations could be adversely impacted if we were to lose his services. Accordingly, our success will be dependent on appropriately managing the risks related to executing a succession plan for Mr. Susi on a timely basis.
If we fail to attract and retain the talent required for our business, our business could be materially harmed.
Competition for highly skilled personnel is often intense in the medical device industry, and more specifically in the MRI compatible medical device industry. A number of our executives and employees are former employees of Invivo Corporation, where Mr. Susi developed the first MRI compatible patient monitoring system. If our current employees with experience in the MRI compatible device industry leave our company, we may have difficulty finding replacements with an equivalent amount of experience and skill, which could harm our operations. Our future success will also depend in part on our ability to identify, hire and retain additional personnel, including skilled engineers to develop new products, and executives to oversee our marketing, sales, customer support and production staff. We may not be successful in attracting, integrating or retaining qualified personnel to meet our current growth plans or future needs. Our productivity may be adversely affected if we do not integrate and train our new employees quickly and effectively.
Also, to the extent we hire personnel from competitors, we may be subject to allegations that we have improperly solicited, or that they have divulged proprietary or other confidential information, or that their former employers own their inventions or work product.
We may be unable to scale our operations successfully.
Our plan is to grow rapidly. Our growth, if it occurs as planned, will place significant demands on our management and manufacturing capacity, as well as our financial, administrative and other resources. We cannot guarantee that any of the systems, procedures and controls we put in place will be adequate to support the manufacture and distribution of our products. Our operating results will depend substantially on the ability of our officers and key employees to manage changing business conditions and to implement and improve our financial, administrative and other resources. If we are unable to respond to and manage changing business conditions, or the scale of our products, services and operations, then the quality of our services, our ability to retain key personnel and our business could be harmed.
We engage in related party transactions, which result in a conflict of interest involving our management.
We have engaged in the past, and continue to engage, in related party transactions, particularly between our company and Roger Susi and his affiliates. One significant ongoing related party transaction is the lease agreement between our company and Susi, LLC, an affiliate of Roger Susi, with respect to our sole production and headquarters facility in Winter Springs, Florida. Related party transactions present difficult conflicts of interest, could result in disadvantages to our company and may impair investor confidence, which could materially and adversely affect us. Related party transactions could also cause us to become materially dependent on related parties in the ongoing conduct of our business, and related parties may be motivated by personal interests to pursue courses of action that are not necessarily in the best interests of our company and our stockholders.
Any acquisitions of technologies, products and businesses, may be difficult to integrate, could adversely affect our relationships with key customers, and/or could result in significant charges to earnings.
We plan to periodically review potential acquisitions of technologies, products and businesses that are complementary to our products and that could accelerate our growth. However, our company has never completed an acquisition and there can be no assurance that we will be successful in finding any acquisitions in the future. The process of identifying, executing and realizing attractive returns on acquisitions involves a high degree of uncertainty. Acquisitions typically entail many risks and could result in difficulties in integrating operations, personnel, technologies and products. If we are not able to successfully integrate our acquisitions, we may not obtain the advantages and synergies that the acquisitions were intended to create, which may have a material adverse effect on our business, results of operations, financial condition and cash flows, our ability to develop and introduce new products and the market price of our stock.
The environment in which we operate makes it increasingly difficult to forecast our business performance.
Significant changes and volatility in the global financial markets, in the consumer and business environment, and our general competitive landscape may make it increasingly difficult for us to predict our revenues and earnings into the future. Our quarterly sales and profits depend substantially on the volume and timing of orders fulfilled during the quarter, and such orders are difficult to forecast. Product demand is dependent upon the capital spending budgets of our customers and prospects as well as government funding policies, and matters of public policy as well as product and economic cycles that can affect the spending decisions of these entities. As a result, any revenue or earnings guidance or outlook which we have given or might give may turn out to be inaccurate. Though we will endeavor to give reasonable estimates of future revenues and earnings at the time we give such guidance, based on then-current conditions, there is a significant risk that such guidance or outlook will turn out to be incorrect. Historically, companies that have overstated their operating guidance have suffered significant declines in their stock price when such results are announced to the public.
There are inherent uncertainties involved in estimates, judgments and assumptions used in the preparation of financial statements in accordance with United States GAAP. Furthermore, portions of GAAP require the use of fair value models which are variable in application and methodology from appraiser to appraiser. Any changes in estimates, judgments and assumptions used could have a material adverse effect on our business, financial position and operating results.
The preparation of financial statements in conformity with GAAP requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Such assumptions and estimates include those related to revenue recognition, accruals for product returns, valuation of inventory, impairment of intangibles and long-lived assets, accounting for income taxes and stock-based compensation and reserves for potential litigation. We base our estimates on historical experience and on various other assumptions that we believe to be reasonable under the circumstances, as discussed in greater detail in the section titled “Management’s Discussion and Analysis of Financial Condition and Results of Operations.” Our actual operating results may differ and fall below our assumptions and the financial forecasts of securities analysts and investors, resulting in a significant decline in our stock price.
Risks Related to Our Industry
We are subject to substantial government regulation that is subject to change and could force us to make modifications to how we develop, manufacture, market and price our products.
The medical device industry is regulated extensively by governmental authorities, principally the FDA in the U.S. and corresponding state and foreign regulatory agencies. The majority of our manufacturing processes are required to comply with quality systems regulations, including current good manufacturing practice requirements that cover the methods and documentation of the design, testing, production, control, quality assurance, labeling, packaging and shipping of our products. Failure to comply with applicable medical device regulatory requirements could result in, among other things, warning letters, fines, injunctions, civil penalties, repairs, replacements, refunds, recalls or seizures of products, total or partial suspensions of production, refusal of the FDA or other regulatory agencies to grant pre-market clearances or approvals for our products, withdrawals or suspensions of future current clearances or approvals and criminal prosecution.
In addition, our products are subject to pre-approval requirements by the FDA and similar international agencies that govern a wide variety of product activities from design and development to labeling, manufacturing, promotion, sales and distribution. Compliance with these regulations may be time consuming, burdensome and expensive for us. The failure to obtain, or the loss or suspension of any such pre-approval, would negatively affect our ability to sell our products, and harm our anticipated revenues.
Foreign governmental authorities that regulate the manufacture and sale of medical devices have become increasingly stringent and, to the extent we sell our products in foreign countries, we may be subject to rigorous regulation in the future. Regulatory changes could result in restrictions on our ability to carry on or expand our operations, higher than anticipated costs or lower than anticipated revenue.
If we fail to obtain, or experience significant delays in obtaining, FDA clearances or other necessary approvals to commercially distribute new products, our ability to grow will suffer.
Our current products are Class II medical devices and hence require regulatory pre-market approval by the FDA and other federal and state authorities prior to their sale in the U.S. Similar approvals are required by foreign governmental authorities for sale of our products outside of the U.S. We are responsible for obtaining the applicable regulatory approval for the commercial distribution of our products. As part of our growth strategy, we plan to seek approvals for new MRI compatible products. The process of obtaining approvals, particularly from the FDA, can be costly and time consuming, and there can be no assurance that we will obtain the required approvals on a timely basis, or at all. Failure to receive approvals for new products will hurt our ability to grow.
We are subject to risks associated with doing business outside of the U.S.
Sales to customers outside of the U.S. have historically comprised of approximately one-quarter to one-third of our net revenues and we expect that non-U.S. sales will contribute to future growth. A majority of our international sales originate from Europe and Japan, and we also make sales in Canada, Hong Kong, Australia, Mexico and certain parts of the Middle East. The risks associated with operations outside the United States include:
· foreign regulatory and governmental requirements that could change and restrict our ability to manufacture and sell our products;
· possible failure to comply with anti-bribery laws such as the U.S. Foreign Corrupt Practices Act and similar anti-bribery laws in other jurisdictions;
· foreign currency fluctuations that can impact our financial statements when foreign figures are translated into U.S. dollars;
· different local product preferences and product requirements;
· trade protection and restriction measures and import or export licensing requirements;
· difficulty in establishing, staffing and managing non-U.S. operations;
· failure to maintain relationships with distributors, especially those who have assisted with foreign regulatory or government clearances;
· changes in labor, environmental, health and safety laws;
· potentially negative consequences from changes in or interpretations of tax laws;
· political instability and actual or anticipated military or political conflicts;
· economic instability, inflation, deflation, recession or interest rate fluctuations;
· uncertainties regarding judicial systems and procedures; and
· minimal or diminished protection of intellectual property.
These risks, individually or in the aggregate, could have an adverse effect on our results of operations and financial condition.
We may incur product liability losses, or become subject to other lawsuits related to our products, business, and insurance coverage could be inadequate or unavailable to cover these losses.
Our business is subject to potential product liability risks that are inherent in the design, development, manufacture and marketing of our medical devices and consumable products. We carry third party product liability insurance coverage to protect against such risks, but there can be no assurance that our policy is adequate. In the ordinary course of business, we may become the subject of product liability claims and lawsuits alleging that our products have resulted or could result in an unsafe condition or injury to patients. Any product liability claim brought against us, with or without merit, could be costly to defend and could result in settlement payments and adjustments not covered by or in excess of our product liability insurance. We currently have third-party product liability insurance with maximum coverage of $3,000,000; however, such coverage requires a substantial deductible that we must pay before becoming eligible to receive any insurance proceeds. The deductible amount is currently equal to $25,000 per occurrence and $125,000 in the aggregate. We will have to pay for defending product liability or other claims that are not covered by our insurance. These payments could have a material adverse effect on our profitability and financial condition. Product liability claims and lawsuits, safety alerts, recalls or corrective actions, regardless of their ultimate outcome, could have a material adverse effect on our business, financial condition, reputation and on our ability to attract and retain customers. In addition, we may not be able to obtain insurance in the future on terms acceptable to us or at all.
Defects or failures associated with our products and/or our quality control systems could lead to the filing of adverse event reports, recalls or safety alerts and negative publicity and could subject us to regulatory actions.
Safety problems associated with our products could lead to a product recall or the issuance of a safety alert relating to such products and result in significant costs and negative publicity. An adverse event involving one of our products could require us to file an adverse event report with the FDA. Such disclosure could result in reduced market acceptance and demand for all of our products, and could harm our reputation and our ability to market our products in the future. In some circumstances, adverse events arising from or associated with the design, manufacture or marketing of our products could result in the suspension or delay of regulatory reviews of our applications for new product approvals or clearances.
We may also voluntarily undertake a recall of our products or temporarily shut down production lines based on internal safety, quality monitoring and testing data. For example, in August 2012, we initiated a voluntary recall of a particular lot of MRidium Series 1000 MR Infusion Sets, Type 1058 MR IV, an extension set used with our MRidium MRI compatible IV infusion pumps, due to an out-of-specification dimension of one section of the IV set. We retrieved and destroyed all unused infusion sets subject to the recall. In July 2013, the FDA notified us that it had concluded its audit and confirmed that the recall was considered terminated. In July 2013, we issued a voluntary recall of our MRI compatible IV infusion pump systems equipped with MRidium 1145 DERS Drug Library due to their potential risk in providing an incorrect recommended value for the infusion rate during the pump’s initial infusion setup. To avoid future product recalls we have made and continue to invest in our quality systems, processes and procedures. We will continue to make improvements to our products and systems to further reduce issues related to patient safety. However, there can be no assurance our systems will be sufficient. Future quality concerns, whether real or perceived, could adversely affect our operating results.
Our products or product types could be subject to negative publicity, which could have a material adverse effect on our financial position and results of operations and could cause the market value of our common stock to decline.
The market’s perception of our products could be harmed if any of our products or similar products offered by others in our industry become the subject of negative publicity due to a product safety issue, withdrawal, recall, or are proven or are claimed to be harmful to patients. The FDA Warning Letter may harm the market perception of our company and products. The harm to market perception may have a material adverse effect on our business, financial position and results of operations and could cause the market value of our common stock to decline.
Recent U.S. healthcare policy changes, including the Affordable Care Act and PPACA, may have a material adverse effect on our financial condition and results of operations.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act (collectively, the “PPACA”), enacted in March 2010, implemented changes that are expected to significantly impact the medical device industry. Beginning on January 1, 2013, the Affordable Care Act imposed a 2.3% excise tax on sales of products defined as “medical devices” by the regulations of the FDA. We believe that all of our medical products are “medical devices” within the meaning of the FDA regulations. If this tax rate is increased in future years, it would negatively impact our operating results.
Other significant measures contained in the PPACA include research on the comparative clinical effectiveness of different technologies and procedures, initiatives to revise Medicare payment methodologies, such as bundling of payments across the continuum of care by providers and physicians, and initiatives to promote quality indicators in payment methodologies. The PPACA also includes significant new fraud and abuse measures, including required disclosures of financial payments to and arrangements with physician customers, lower thresholds for violations and increasing potential penalties for such violations. In addition, the PPACA established an Independent Payment Advisory Board (“IPAB”), to reduce the per capita rate of growth in Medicare spending. The IPAB has broad discretion to propose policies to reduce health care expenditures, which may have a negative impact on payment rates for services, including treatments and procedures which incorporate use of our products. The IPAB proposals may impact payments for treatments and procedures that use our technology beginning in 2016 and for hospital services beginning in 2020, and may indirectly reduce demand for our products.
The taxes imposed by the new federal legislation and the expansion in government’s effect on the U.S. healthcare industry may result in decreased profits to us, lower reimbursements by payers for our products or reduced medical procedure volumes, all of which may adversely affect our business, financial condition and results of operations.
We are subject to healthcare fraud and abuse regulations that could result in significant liability, require us to change our business practices and restrict our operations in the future.
We and our customers are subject to various U.S. federal, state and local laws targeting fraud and abuse in the healthcare industry, including anti-kickback and false claims laws. Violations of these laws are punishable by criminal or civil sanctions, including substantial fines, imprisonment and exclusion from participation in healthcare programs such as Medicare and Medicaid, and Veterans’ Administration health programs and health programs outside the U.S. These laws and regulations are broad in scope and are subject to evolving interpretations, which could require us to alter one or more of our sales or marketing practices. In addition, violations of these laws, or allegations of such violations, could disrupt our business and result in a material adverse effect on our sales, profitability and financial condition. Furthermore, since many of our customers rely on reimbursement from Medicare, Medicaid and other governmental programs to cover a substantial portion of their expenditures, if we or our customers are excluded from such programs as a result of a violation of these laws, it could have an adverse effect on our results of operations and financial condition. We have developed and implemented business practices and processes to train our personnel to perform their duties in compliance with healthcare fraud and abuse laws and conduct informal oversight to detect and prevent these types of fraud and abuse. However, we lack formal written policies and procedures at this time. If we are unable to formally document and implement the controls and procedures required in a timely manner or we are otherwise found to be in violation of such laws, we might suffer adverse regulatory consequences or face criminal sanctions, which could harm our operations, financial reporting or financial results.
We could be adversely affected by violations of the U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws.
The U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments to non-U.S. officials for the purpose of obtaining or retaining business. We intend to adopt policies for compliance with these anti-bribery laws, which often carry substantial penalties. We cannot assure you that our internal control policies and procedures always will protect us from reckless or other inappropriate acts committed by our affiliates, employees or agents. Violations of these laws, or allegations of such violations, could have a material adverse effect on our business, financial position and results of operations and could cause the market value of our common stock to decline.
We and our suppliers and customers are required to obtain regulatory approvals to comply with FDA regulations applicable to medical devices and infusion pumps, and these approvals could result in delays or increased costs in developing new products.
In December 2014, the FDA issued guidance entitled “Infusion Pumps Total Product Life Cycle.” This guidance established substantial additional pre-market requirements for new and modified infusion pumps. Through this guidance, the FDA indicated more data demonstrating product safety will be required for future 510(k) submissions for infusion pumps, including the potential for more clinical and human factors data. The process for obtaining regulatory approvals to market infusion pumps and related accessories have become more costly and time consuming. The impact of this guidance is likely to result in a more time consuming and costly process to obtain regulatory clearance to market infusion pumps. In addition, new requirements could result in longer delays for the clearance of new products, modification of existing infusion pump products or remediation of existing products in the market. Future delays in the receipt of, or failure to obtain, approvals could result in delayed or no realization of product revenues.
We and our suppliers and customers are required to maintain compliance with FDA regulations applicable to medical devices and infusion pumps, and it could be costly to comply with these regulations and to develop compliant products and processes. Failure to comply with these regulations could subject us to sanctions and could adversely affect our business.
Even if we are able to obtain approval for introducing new products to the market, we and our suppliers may not be able to remain in compliance with applicable FDA and other material regulatory requirements once clearance or approval has been obtained for a product. These requirements include, among other things, regulations regarding manufacturing practices, product labeling, off-label marketing, advertising and post-marketing reporting, adverse event reports and field alerts. Compliance with these FDA requirements is subject to continual review and is monitored through periodic inspections by the FDA. For example, the FDA conducted routine inspections of our facility in Winter Park, Florida in June 2010 and more recently between April 7 and April 16, 2014. The FDA issued a Form 483 on April 16, 2014 that identified eight observations. As described above, the FDA subsequently issued a Warning Letter that has resulted in us ceasing to distribute our primary product in the United States. See the risk factor entitled, “Our financial performance is currently dependent on a single product and the FDA has issued a warning letter requesting that we immediately cease commercial distribution of such product.”
In addition, manufacturing flaws, component failures, design defects, off-label uses or inadequate disclosure of product related information could result in an unsafe condition or the injury or death of a patient. All of these events could harm our sales, margins and profitability in the affected periods and may have a material adverse effect on our business. Any adverse regulatory action or action taken by us to maintain appropriate regulatory compliance, with respect to these laws and regulations could disrupt our business and have a material adverse effect on our sales, profitability and financial condition. Furthermore, an adverse regulatory action with respect to any of our products or operating procedure or to our or our suppliers’ manufacturing facility could materially harm our reputation in the marketplace.
Our operations are subject to environmental laws and regulations, with which compliance is costly and which exposes us to penalties for non-compliance.
Our business, products, and product candidates are subject to federal, state, and local laws and regulations relating to the protection of the environment, worker health and safety and the use, management, storage, and disposal of hazardous substances, waste, and other regulated materials. These environmental laws and regulations could require us to pay for environmental remediation and response costs at third-party locations where we dispose of or recycle hazardous substances. The costs of complying with these various environmental requirements, as they now exist or as may be altered in the future, could adversely affect our financial condition and results of operations.
Risks Relating to our Intellectual Property
Our success depends on our ability to protect our intellectual property.
We intend to rely on a combination of patents, trademarks, trade secrets, know-how, license agreements and contractual provisions to establish and protect our proprietary rights to our technologies and products. We cannot guarantee that the steps we have taken or will take to protect our intellectual property rights will be adequate or that they will deter infringement, misappropriation or violation of our intellectual property. We may fail to secure patents that are important to our business, and we cannot guarantee that any pending U.S. trademark or patent application, if ultimately issued, will provide us some relative competitive advantage. Litigation may be necessary to enforce our intellectual property rights and to determine the validity and scope of our proprietary rights. Any litigation could result in substantial expenses and may not adequately protect our intellectual property rights. In addition, the laws of some of the countries in which our products may in the future be sold may not protect our products and intellectual property to the same extent as U.S. laws, or at all. We may be unable to protect our rights in trade secrets and unpatented proprietary technology in these countries. If our trade secrets become known, we may lose our competitive advantages.
Even if we are able to secure necessary patents in the U.S., we may not be able to secure necessary patents and trademarks in foreign countries in which we sell our products or plan to sell our products. In March 2013, the U.S. transitioned to a “first inventor to file” system for patents in which, assuming the other requirements for patentability are met, the first inventor to file a patent application is entitled to a patent. We may be subject to a third-party pre-issuance submission of prior art to the U.S. Patent and Trademark Office, or become involved in opposition, derivation, reexamination, inter parties review or interference proceedings challenging our patent rights or the patent rights of others. An adverse determination in any such submission, proceeding or litigation could reduce the scope of, or invalidate our patent rights, allow third parties to commercialize our technology or products and compete directly with us, without payment to us, or result in our inability to manufacture or commercialize products without infringing third party patent rights.
Our unpatented trade secrets, know-how, confidential and proprietary information, and technology may be inadequately protected.
We rely on unpatented trade secrets, know-how and technology. This intellectual property is difficult to protect, especially in the medical device industry, where much of the information about a product must be submitted to regulatory authorities during the regulatory approval process. We seek to protect trade secrets, confidential information and proprietary information, in part, by entering into confidentiality and invention assignment agreements with employees, consultants, and others. These parties may breach or terminate these agreements, and we may not have adequate remedies for such breaches. Furthermore, these agreements may not provide meaningful protection for our trade secrets or other confidential or proprietary information or result in the effective assignment to us of intellectual property, and may not provide an adequate remedy in the event of unauthorized use or disclosure of confidential information or other breaches of the agreements. Despite our efforts to protect our trade secrets and our other confidential and proprietary information, we or our collaboration partners, board members, employees, consultants, contractors, or scientific and other advisors may unintentionally or willfully disclose our proprietary information to competitors.
There is a risk that our trade secrets and other confidential and proprietary information could have been, or could, in the future, be shared by any of our former employees with, and be used to the benefit of, any company that competes with us.
If we fail to maintain trade secret protection or fail to protect the confidentiality of our other confidential and proprietary information, our competitive position may be adversely affected. Competitors may also independently discover our trade secrets. Enforcement of claims that a third party has illegally obtained and is using trade secrets is expensive, time consuming and uncertain. If our competitors independently develop equivalent knowledge, methods and know-how, we would not be able to assert our trade secret protections against them, which could have a material adverse effect on our business.
There can be no assurance of timely patent review and approval to minimize competition and generate sufficient revenues.
There can be no assurance that the Patent and Trademark Office will have sufficient resources to review our patent applications in a timely manner. Consequently, even if our patent applications are ultimately successful, our patent applications may be delayed, which would prevent intellectual property protection for our products. If we fail to successfully commercialize our products due to the lack of intellectual property protection, we may be unable to generate sufficient revenues to meet or grow our business according to our expected goals and this may have a materially adverse effect on our profitability, financial condition, and operations.
We may become involved in patent litigation or other intellectual property proceedings relating to our future product approvals, which could result in liability for damages or delay or stop our development and commercialization efforts.
The medical device industry has been characterized by significant litigation and other proceedings regarding patents, patent applications, and other intellectual property rights. The situations in which we may become parties to such litigation or proceedings may include any third parties (which may have substantially greater resources than we have) initiating litigation claiming that our products infringe their patent or other intellectual property rights; in such case, we will need to defend against such proceedings.
The large number of patents, the rapid rate of new patent applications and issuances, the complexities of the technologies involved and the uncertainty of litigation significantly increase the risks related to any patent litigation. Any potential intellectual property litigation also could force us to do one or more of the following:
· stop selling, making, or using products that use the disputed intellectual property;
· obtain a license from the intellectual property owner to continue selling, making, licensing, or using products, which license may require substantial royalty payments and may not be available on reasonable terms, or at all;
· pay substantial damages or royalties to the party whose intellectual property rights we may be found to be infringing;
· pay the attorney fees and costs of litigation to the party whose intellectual property rights we may be found to be infringing; or
· redesign those products that contain the allegedly infringing intellectual property, which could be costly, disruptive and/or infeasible.
If any of the foregoing events occur, we may have to withdraw existing products from the market or may be unable to commercialize one or more of our products, all of which could have a material adverse effect on our business, results of operations and financial condition. As the number of participants in our industry grows, the possibility of intellectual property infringement claims against us increases.
Furthermore, the costs of resolving any patent litigation or other intellectual property proceeding, even if resolved in our favor, could be substantial. Uncertainties resulting from the initiation and continuation of patent litigation or other intellectual property proceedings could have a material adverse effect on our ability to compete in the marketplace. Patent litigation and other intellectual property proceedings may also consume significant management time.
In the event that a competitor infringes upon our patent or other intellectual property rights, enforcing those rights may be costly, difficult, and time-consuming. Even if successful, litigation to enforce our intellectual property rights or to defend our patents against challenge could be expensive and time-consuming and could divert our management’s attention. We may not have sufficient resources to enforce our intellectual property rights or to defend our patent or other intellectual property rights against a challenge. If we are unsuccessful in enforcing and protecting our intellectual property rights and protecting our products, it could materially harm our business.
There may also be situations where we use our business judgment and decide to market and sell products, notwithstanding the fact that allegations of patent infringement(s) have not been finally resolved by the courts (i.e., an “at-risk launch”). The risk involved in doing so can be substantial because the remedies available to the owner of a patent for infringement may include, among other things, damages measured by the profits lost by the patent owner and not necessarily by the profits earned by the infringer. In the case of a willful infringement, the definition of which is subjective, such damages may be increased up to three times. An adverse decision could have a material adverse effect on our business, financial position and results of operations and could cause the market value of our common stock to decline.
In addition, we may indemnify our customers and distributors with respect to infringement by our products of the proprietary rights of third parties. Third parties may assert infringement claims against our customers or distributors. These claims may require us to initiate or defend protracted and costly litigation on behalf of our customers or distributors, regardless of the merits of these claims. If any of these claims succeed, we may be forced to pay damages on behalf of our customers or distributors or may be required to obtain licenses for the products they use. If we cannot obtain all necessary licenses on commercially reasonable terms, our customers may be forced to stop using our products.
We may be subject to claims that we, our board members, employees or consultants have used or disclosed alleged trade secrets or other proprietary information belonging to third parties and any such individuals who are currently affiliated with one of our competitors may disclose our proprietary technology or information.
As is commonplace in the medical device industry, some of our board members, employees and consultants are or have been associated with other medical device companies that compete with us. For example, Mr. Susi and a number of our other employees are former employees of Invivo Corporation. While associated with such other medical device companies, these individuals may have been exposed to research and technology similar to the areas of research and technology in which we are engaged. We may become subject to future claims that we, our employees, board members, or consultants have inadvertently or otherwise used or disclosed alleged trade secrets or other proprietary information of those companies. Litigation may be necessary to defend against such claims.
We have entered into confidentiality agreements with our executives and key consultants. However, we do not have, and are not planning to enter into, any confidentiality agreements with our non-executive directors because they have a fiduciary duty of confidentiality as directors. There is the possibility that any of our former board members, employees, or consultants who are currently employed at, or associated with, one of our competitors may unintentionally or willfully disclose our proprietary technology or information.
Risks Related to Ownership of Our Common Stock
Our common stock price may be subject to significant fluctuations and volatility, and you may be unable to sell your shares at a fair price, or at all.
Our stock could be subject to wide fluctuations in price in response to various factors, including the following:
· a lack of liquidity in the public trading of our common stock;
· the commercial success or failure of our key products;
· delayed or reduced orders from our customers;
· manufacturing or supply interruptions;
· changes or developments in laws or regulations applicable to our product candidates;
· introduction of competitive products or technologies;
· poorly executed acquisitions or acquisitions whose projected potential is not realized;
· actual or anticipated variations in quarterly operating results;
· failure to meet or exceed the estimates and projections of securities analysts or investors;
· varying economic and market conditions in the U.S.;
· negative developments impacting the medical device industry in general and changes in the market valuations of companies deemed similar to us;
· negative developments concerning our sources of manufacturing supply;
· disputes or other developments relating to patents, trademarks or other proprietary rights;
· litigation or investigations involving us, our industry, or both;
· issuances of debt, equity or convertible securities at terms deemed unfavorable by the market;
· major catastrophic events;
· the expiration of contractual lock-up agreements;
· sales of large blocks of our stock;
· exercise of the underwriters’ warrant that may lead to sales that put downward pressure on our stock price;
· changes in our Board of Directors, management or key personnel; or
· the other factors described in this “Risk Factors” section.
Any one of the factors above, or the cumulative effect of some of the factors referred to above, may result in significant fluctuations in our quarterly or annual operating results, fluctuations in our share price and investors’ perception of our business. If we fail to meet or exceed such expectations, our business and stock price could be materially adversely affected.
Future sales of our common stock may cause our stock price to decline.
In July 2014, we sold approximately 2.3 million shares of common stock in our initial public offering, and such shares are registered and freely tradable. If these stockholders sell, or indicate an intention to sell, our common stock in the public market, the trading price of our common stock could decline. In addition, our directors, officers and stockholders who beneficially own approximately 8.4 million shares of common stock, which were subject to lock-up agreements that expired in January 2015. As a result, up to approximately 1.0 million shares became eligible for sale in the public market and approximately 7.4 million shares held by affiliates became eligible for sale subject to volume limitations under Rule 144 under the Securities Act. Moreover, we filed a registration statement under Form S-8 to register all of the shares issuable upon exercise of options outstanding or reserved for future issuance under our equity compensation plans. If these additional shares are sold, or if it is perceived that they will be sold, the trading price of our common stock could decline.
We may need or choose to raise additional capital in the future, which could result in dilution to our stockholders and adversely affect stock price.
While we believe the proceeds from our recent initial public offering will provide us with adequate capital to fund operations for at least the next 12 months, we may need or choose to raise additional funds prior to that time. We may seek to sell additional equity or debt securities or to obtain an additional credit facility, which we may not be able to do on favorable terms, or at all. The sale of additional equity or convertible debt securities could result in additional dilution to our stockholders. If additional funds are raised through the issuance of debt securities or preferred stock, these securities could have rights that are senior to holders of common stock and any debt securities could contain covenants that would restrict our operations. The sale of such securities could hurt demand for our common stock and lead our share price to decline.
Roger Susi, who serves as a director and an executive officer, owns a significant percentage of our stock and will be able to exert significant control over matters subject to stockholder approval.
Roger Susi, our founder, who serves as one of our directors and Chief Executive Officer, and his affiliates beneficially owns a majority of our outstanding common stock. Mr. Susi will be able to influence or control matters requiring approval by our stockholders, including the election of directors and the approval of mergers, acquisitions or other extraordinary transactions. He may also have interests that differ from yours and may vote in a way with which you disagree and which may be adverse to your interests. This concentration of ownership may have the effect of delaying, preventing or deterring a change of control of our company, could deprive our stockholders of an opportunity to receive a premium for their common stock as part of a sale of our company and might ultimately affect the market price of our common stock.
Mr. Susi’s majority ownership also qualifies our company as a “controlled company” and allows us to opt out of compliance with numerous corporate governance listing requirements.
In addition, we qualify for the “controlled company” exemption under the corporate governance rules of the NASDAQ Stock Market until such a time as Mr. Susi does not control a majority of our outstanding common stock. As a “controlled company,” we would be permitted to opt out of compliance with the requirements that a majority of our board of directors consist of independent directors, that our Board of Directors’ compensation committee be comprised solely of independent directors, and that director nominees be selected or recommended to the Board of Directors for selection by independent directors. Notwithstanding the availability of these exemptions, we have elected not to rely upon any of the exemptions afforded to a “controlled company” under NASDAQ rules. A majority of our Board of Directors is comprised of independent directors, our compensation committee is comprised solely of independent directors, and our director nominees are recommended for selection to our Board of Directors by a majority of our independent directors in a vote in which only independent directors may participate. Our compliance is voluntary, however, and there can be no assurance that we will continue to comply with these standards in the future.
We do not intend to pay dividends for the foreseeable future.
The continued expansion of our business will require funding. Accordingly, we do not anticipate that we will pay any cash dividends on shares of our common stock for the foreseeable future. Any determination to pay dividends in the future will be at the discretion of our Board of Directors and will depend upon results of operations, financial condition, contractual restrictions, restrictions imposed by applicable law and other factors our Board of Directors deems relevant. Investors seeking cash dividends should not purchase our common stock. Accordingly, if you purchase shares, realization of a gain on your investment will depend solely on the appreciation of the price of our common stock, which may never occur.
The requirements of being a public company may strain our resources, divert management’s attention and affect our ability to attract and retain executive management and qualified board members.
As a public company, we are subject to the reporting requirements of the Securities Exchange Act of 1934, as amended (“Exchange Act”), the Sarbanes-Oxley Act, the Dodd-Frank Act, the listing requirements of the NASDAQ Stock Market and other applicable securities rules and regulations. Compliance with these rules and regulations will increase our legal and financial compliance costs, make some activities more difficult, time-consuming or costly and increase demand on our systems and resources. The Exchange Act requires, among other things, that we file annual, quarterly and current reports with respect to our business and operating results. As a result, management’s attention may be diverted from other business concerns, which could adversely affect our business and operating results. We may need to hire more employees in the future or engage outside consultants to monitor and advise us regarding compliance, which will increase our costs and expenses.
In addition, changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for public companies, increasing legal and financial compliance costs and making some activities more time consuming. These laws, regulations and standards are subject to varying interpretations, in many cases due to their lack of specificity, and, as a result, their application in practice may evolve over time as new guidance is provided by regulatory and governing bodies. This could result in continuing uncertainty regarding compliance matters and higher costs necessitated by ongoing revisions to disclosure and governance practices. We are investing additional resources to comply with evolving laws, regulations and standards, and this investment may result in increased general and administrative expenses and a diversion of management’s time and attention from revenue-generating activities to compliance activities. If our efforts to comply with new laws, regulations and standards differ from the activities intended by regulatory or governing bodies due to ambiguities related to their application and practice, regulatory authorities may initiate legal proceedings against us, and our business may be adversely affected.
We believe that being a public company and compliant with these new rules and regulations will make it more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced coverage or incur substantially higher costs to obtain coverage. These factors could also make it more difficult for us to attract and retain qualified members of our Board of Directors, particularly to serve on our audit committee and compensation committee, and qualified executive officers.
As a result of becoming a public company, we are obligated to establish and maintain adequate internal controls. Failure to develop and maintain adequate internal controls or to implement new or improved controls could have a material adverse effect on our business, financial position and results of operations and could cause the market value of our common stock to decline.
Ensuring that we have adequate internal financial and accounting controls and procedures in place so that we can produce accurate financial statements on a timely basis is a costly and time-consuming effort. Our internal controls over financial reporting are designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements in accordance with GAAP. We are in the very early stages of the costly and challenging process of compiling the system and processing documentation necessary to perform the evaluation needed to comply with Section 404 of the Sarbanes-Oxley Act of 2002. We may not be able to complete our evaluation, testing and any required remediation in a timely fashion. During the evaluation and testing process, if we identify one or more material weaknesses in our internal controls over financial reporting, we will be unable to assert that our internal controls are effective.
We will be required to disclose changes made in our internal controls and procedures on a quarterly basis. However, our independent registered public accounting firm will not be required to report on the effectiveness of our internal controls over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act until the later of the year following our first annual report required to be filed with the Securities and Exchange Commission (“SEC”), or the date we are no longer an “emerging growth company” as defined in the JOBS Act if we take advantage of the exemptions contained in the JOBS Act. At such time, our independent registered public accounting firm may issue a report that is adverse in the event it is not satisfied with the level at which our controls are documented, designed or operating. Our remediation efforts may not enable us to avoid a material weakness in the future.
We believe that our business practices have become more visible as a public company, and this could impact our competitive environment and our risk of potential litigation.
As a result of disclosure of information in filings required of a public company, our business and financial condition have become more visible potentially exposing us to new competition and threatened or actual litigation, including by competitors and other third parties. New competition could result in reduced sales of our products and adversely impact our profitability. If lawsuits prevail against us, our business and operating results could be adversely affected, and even if the claims do not result in litigation or are resolved in our favor, these claims, and the time and resources necessary to resolve them, could divert the resources of our management and adversely affect our business and operating results.
We may and have become involved in securities class action litigation that could divert management’s attention from our business and adversely affect our business and could subject us to significant liabilities.
The stock markets have from time to time experienced significant price and volume fluctuations that have affected the market prices of small cap medical device companies. These broad market fluctuations as well a broad range of other factors, including the realization of any of the risks described in this “Risk Factors” section, may cause the market price of our common stock to decline. In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. We may become involved in this type of litigation in the future. Litigation is expensive and could divert management’s attention and resources from our primary business, which could adversely affect our operating results. Any adverse determination in any such litigation or any amounts paid to settle any such actual or threatened litigation could require us to make significant payments. Such payment could have a material impact on how investors view our company and result in a decline in our stock price.
We are an “emerging growth company,” and we cannot be certain if the reduced reporting requirements applicable to emerging growth companies will make our common stock less attractive to investors.
We are an “emerging growth company,” as defined in the JOBS Act, and intend to take advantage of certain exemptions from various reporting requirements. We cannot predict if investors will respond negatively to our reliance on these exemptions. If some investors find our common stock less attractive as a result, there may be a less active trading market for our common stock, and our stock price may be more volatile.
As an “emerging growth company” we have also chosen to take advantage of certain provisions of the JOBS Act that allow us to provide you with less information in our public filings than would otherwise be required. As a result it may be more difficult for you to evaluate an investment in our company.
If securities or industry analysts fail to initiate research coverage of our stock, downgrade our stock, or discontinue coverage, our trading volume might never develop and our stock price could decline.
The trading market for our common stock depends, in part, on the research reports that securities or industry analysts publish about our business. If securities or industry analysts do not commence or continue coverage of our company, trading market for our stock may not be robust and the price of our stock could likely be negatively impacted. In the event securities or industry analysts initiate coverage, and later downgrade our stock, our stock price could decline.
Our charter documents and Delaware law have provisions that may discourage an acquisition of us by others and may prevent attempts by our stockholders to replace or remove our current management.
Provisions in our charter documents, as well as provisions of the Delaware General Corporation Law (“DGCL”), could depress the trading price of our common stock by making it more difficult for a third party to acquire us at a price favorable to our shareholders. These provisions include:
· authorizing the issuance of “blank check” preferred stock, the terms of which may be established and shares of which may be issued without stockholder approval to defend against a takeover attempt; and
· establishing advance notice requirements for nominations for election to our Board of Directors or for proposing matters that can be acted upon at stockholder meetings.
In addition, these provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our Board of Directors. We are subject to Section 203 of the DGCL, which generally prohibits a Delaware corporation from engaging in any of a broad range of business combinations with an interested stockholder for a period of three years following the date on which the stockholder became an interested stockholder, unless such transactions are approved by our Board of Directors. This provision could have the effect of delaying or preventing a change of control, whether or not it is desired by or beneficial to our stockholders, which could also affect the price that some investors are willing to pay for our common stock.
ITEM 1B.UNRESOLVED STAFF COMMENTS
Not applicable to smaller reporting companies.
Our principal offices are currently located in a leased facility of approximately 27,000 square feet located in Winter Springs, Florida. This facility has been leased from an entity controlled by our President and CEO, Roger Susi. Pursuant to the terms of our lease, the monthly base rent is $32,583, adjusted annually for changes in the consumer price index. The term of the lease expires on May 31, 2019. The lease will automatically renew for two successive terms of five years each beginning in 2019 and again in 2024, and thereafter, will be renewed for successive terms of one year each.
We do not own any real property that is materially important to our business.
On September 10, 2014, a Civil Action was filed in the U.S. District Court for the Southern District of Florida (“Lam Civil Action”). The Lam Civil Action is a putative class action lawsuit brought against the Company and certain individuals who are officers and / or directors of the Company. The plaintiff is an alleged shareholder of the Company, and seeks relief on behalf of a class of persons who purchased the Company’s common stock during the period from July 15, 2014 through September 2, 2014. The complaint alleges that the defendants failed to disclose material information concerning the Company’s compliance with FDA regulations in violation of Section 10(b) of the Securities Exchange Act of 1934 and Rule 10b-5 thereunder, and that the putative class members suffered damages as a result. The complaint additionally alleges “control person” liability against the individual defendants under Section 20(a) of the Securities Exchange Act of 1934. The Lam Civil Action is presently in the very early stages of litigation. The Company disputes the plaintiff’s allegations and theories of liability, and intends to defend the case vigorously. We have not accrued for any loss related to this matter as we believe that any such loss is not probable or estimable.
In October 2012, Radimed Gesellschaft für Kommunikationsdienstleistungen und Medizintechnik mbH (“Radimed”) brought an action in Düsseldorf Regional Court against our German distributor alleging the name and sign “iRadimed” was confusingly similar to their German trademark “Radimed.” A judgment was rendered against our German distributor preventing use of the name and sign “iRadimed” in Germany. We have however continued to sell products in Germany without any discernible effect by using the name IRI Development. On July 31, 2013, Radimed filed a lawsuit against us and our founder, Roger Susi, in Düsseldorf Regional Court, alleging that we infringed their German and Community trademarks “Radimed” and seeking to prevent our use of the name, sign and domain name “iRadimed” in the European Union. Prior to year end, we began settlement discussions with Radimed and accrued an insignificant amount related to this matter. Subsequent to year end, in March 2015, we settled this matter and paid the amount that had been accrued. Pursuant to this settlement, we may continue to use the name “iRadimed” and our associated signs and domain name in the European Union.
ITEM 4. MINE SAFETY DISCLOSURES
Not applicable.
ITEM 5. MARKET FOR REGISTRANT’S COMMON STOCK, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock trades on the NASDAQ Capital Market under the stock symbol “IRMD.”
Our common stock commenced trading on the NASDAQ Capital Market under the stock symbol “IRMD” on July 16, 2014. Prior to that time, there was no public market for our common stock.
The following table summarizes the highest and lowest sales prices of our common stock during the quarters listed below as reported by the NASDAQ:
Year ended December 31, 2014 |
| Highest |
| Lowest |
| ||
|
|
|
|
|
| ||
Third Quarter |
| $ | 13.60 |
| $ | 6.26 |
|
Fourth Quarter |
| $ | 13.10 |
| $ | 6.87 |
|
The stock market in general has experienced extreme stock price fluctuations in the past few years. In some cases, these fluctuations have been unrelated to the operating performance of the affected companies. Many companies have experienced dramatic volatility in the market prices of their common stock. We believe that a number of factors, both within and outside our control, could cause the price of our common stock to fluctuate, perhaps substantially. Factors such as the following could have a significant adverse impact on the market price of our common stock:
· Our financial position and results of operations;
· Our ability to obtain additional financing and, if available, the terms and conditions of the financing;
· Concern as to, or other evidence of, the reliability and efficiency of our proposed products or our competitors’ products;
· Announcements of innovations or new products by us or our competitors;
· Federal and state governmental regulatory actions and the impact of such requirements on our business;
· The development of litigation against us;
· Period-to-period fluctuations in our operating results;
· Changes in estimates of our performance by any securities analysts;
· The issuance of new equity securities pursuant to a future offering or acquisition;
· poorly executed acquisitions or acquisitions whose projected potential is not realized;
· Changes in interest rates;
· Competitive developments, including announcements by competitors of new products or significant contracts, acquisitions, strategic partnerships, joint ventures or capital commitments;
· sales of large blocks of our stock;
· exercise of the underwriters’ warrant that may lead to sales that put downward pressure on our stock price;
· Investor perceptions of our company; and
· General economic and other national and international conditions.
Stockholders
As of February 28, 2015, we had 16 stockholders of record. This number does not include an indeterminate number of stockholders whose shares are held by brokers in street name.
Dividends
We do not expect to declare or pay any cash dividends on our common stock in the foreseeable future, and we currently intend to retain future earnings, if any, to finance the expansion of our business. The decision whether to pay cash dividends on our common stock will be made by our board of directors, at its discretion, and will depend on our financial condition, operating results, capital requirements and other factors that the board of directors considers significant.
We did not pay cash dividends in the years ended December 31, 2014 or 2013.
Transfer Agent
The transfer agent and registrar for our common stock is Corporate Stock Transfer, Inc.
Equity Compensation Plan Information
Our equity compensation plan information is provided as set forth in Part III, Item 11 herein.
Additional Information
Copies of our annual reports, quarterly reports, current reports, and any amendments to those reports, are available free of charge on the Internet at www.sec.gov. All statements made in any of our filings, including all forward-looking statements, are made as of the date of the document, in which the statement is included, and we do not assume or undertake any obligation to update any of those statements or documents unless we are required to do so by law.
ITEM 6. SELECTED CONSOLIDATED FINANCIAL DATA
Not applicable for a smaller reporting company.
ITEM 7. MANAGEMENT’S DISCUSSION AND ANALYSIS AND RESULTS OF OPERATIONS
You should read this discussion and analysis together with our audited financial statements, the notes to such statements and the other financial information included in this Form 10-K. This discussion contains forward-looking statements that involve risks and uncertainties. As a result of many factors, such as those set forth under the section entitled “Risk Factors” and elsewhere in this Form 10-K, our actual results may differ materially from those anticipated in these forward-looking statements. See “CAUTIONARY STATEMENTS REGARDING FORWARD-LOOKING STATEMENTS” for a discussion of the uncertainties, risks and assumptions associated with these statements.
Our Business
We are the only known provider of non-magnetic intravenous (“IV”) infusion pump systems that are safe for use during magnetic resonance imaging (“MRI”) procedures. Electromechanical medical devices and pumps contain magnetic and electronic parts which generate radio frequency noise, create interference and are dangerous to operate in the presence of the powerful magnet which drives an MRI. Our MRidium (3850/3860+) MRI compatible IV infusion pump systems have been designed with non-ferrous parts, ceramic ultrasonic motors, non-magnetic mobile stand and other special features in order to safely and predictably deliver anesthesia and other IV fluids during various MRI procedures. Our pump solution provides a seamless approach to providing IV fluids before, during and after an MRI scan, which is important to critically-ill patients who cannot be removed from their vital medications, and children and infants must generally be sedated in order to remain immobile during an MRI scan.
As of December 31, 2014 we estimate that we had approximately 2,300 MRI compatible IV infusion pump systems installed globally. Each system consists of an MRidium MRI compatible IV infusion pump, mobile stand, and proprietary disposable IV tubing sets and many of these systems contain additional optional upgrade accessories. We primarily generate revenue from the one-time sale of pumps and accessories, in addition to revenue generated from ongoing service contracts and the sale of proprietary disposable tubing sets used during each patient infusion. The principal customers for our MRI compatible products include hospitals, acute care facilities and outpatient imaging centers.
We sell our MRI compatible products through our direct sales force in the U.S. and independent distributors internationally. In the U.S., we sell our products through our ten direct sales representatives and two clinical support representatives. We have distribution agreements for our products with 35 independent distributors selling our products internationally. Selling cycles for our medical devices vary widely but are typically three to six months in duration. We also enter into agreements with healthcare supply contracting companies in the U.S., which enable us to sell and distribute our MRidium MRI compatible IV infusion pump systems to their member hospitals. Under these agreements, we are required to pay these group purchasing organizations (“GPOs”) a fee of three percent of the sales of our products to their member hospitals. We currently have contracts with four major GPOs that effectively give us the ability to sell to more than 95% of all U.S. acute care facilities.
Financial Highlights and Outlook
Our revenue increased $4.3 million, or 38%, to $15.6 million in 2014, from $11.3 million in 2013. This increase was primarily attributable to an increase in the number of our MRI compatible IV infusion pump systems sold during 2014 compared to 2013 and higher sales of our disposable IV sets. As of December 31, 2014, our estimated installed base of IV infusion pumps increased to approximately 2,300 from approximately 1,771 on December 31, 2013.
During 2013, our largest competitor announced its decision to commence removal of its pump systems from the U.S. market, and to discontinue support throughout the world by June 30, 2015 due to ongoing regulatory issues. As a result, we believe that our MRidium MRI compatible IV infusion pump will be the only known MRI compatible IV infusion pump available to customers beginning in the first half of 2015.
In 2015, we expect our revenues to increase as our expanded U.S. direct sales force continues to expand market awareness of the advantages of patient safety and operating efficiencies provided by our MRI compatible IV infusion pump. We intend to continue focusing our efforts on converting users of our former competitor’s pumps to our systems, in addition to targeting an increased number of hospitals and acute care facilities that have yet to adopt our technology. We expect operating expenses to increase in 2015 due to increased headcount, the costs associated with our new corporate headquarters, costs incurred in addressing the Warning Letter, higher legal expense, higher depreciation expense from additional capital expenditures and the costs of being a public company.
Application of Critical Accounting Policies
We prepare our financial statements in conformity with U.S. generally accepted accounting principles. The preparation of these financial statements requires us to make estimates and use assumptions that affect the reported amounts of assets, liabilities and related disclosures at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results could differ from those estimates.
Our significant accounting policies are more fully described in Note 1 to the financial statements. However, we believe that the following critical accounting policies require the use of significant estimates, assumptions, and judgments. The use of different estimates, assumptions, and judgments could have a material effect on the reported amounts of assets, liabilities and related disclosures as of the date of the financial statements and revenue and expenses during the reporting period.
Revenue Recognition
Revenue is recognized when persuasive evidence of an arrangement exists, the price is fixed or determinable, delivery has occurred and title and risk of loss has transferred and collection of the resulting receivable is reasonably assured. Terms of sale for most domestic sales are FOB destination, reflecting that title and risk of loss are assumed by the purchaser upon delivery. Terms of sales to international distributors are FOB shipping point, reflecting that title and risk of loss are assumed by the distributor at the shipping point.
Under the revenue recognition rules for tangible products, we allocate revenue from arrangements with multiple deliverables to each of the deliverables based upon their relative selling prices as determined by a selling-price hierarchy. A deliverable in an arrangement qualifies as a separate unit of accounting if 1) the delivered item has value to the customer on a stand-alone basis, and 2) the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered items is considered probable and substantially in control of the vendor. The principal deliverables in our multiple deliverable arrangements that qualify as separate units of accounting consist of (i) sales of medical devices and supplies, (ii) installation and training services, and (iii) extended warranty agreements.
We use a hierarchy to determine the selling price to be used for allocating revenue to deliverables as follows: (i) vendor-specific objective evidence of fair value (“VSOE”), (ii) third-party evidence of selling price (“TPE”), and (iii) best estimate of the selling price (“ESP”). VSOE of fair value is defined as the price charged when the same element is sold separately, or if the element has not yet been sold separately, the price for the element established by management having the relevant authority when it is probable that the price will not change before the introduction of the element into the marketplace. VSOE generally exists only when we sell the deliverable separately and is the price actually charged for that deliverable. For certain sales under GPO contracts, we have established VSOE for all of the elements in our multiple element arrangements. This determination is based on the volume of sales to these customers in relation to our total sales and the discount tier in which those sales are made. For all other sales we rely on ESP, reflecting our best estimates of what the selling prices of elements would be if they were sold regularly on a stand-alone basis, to establish the amount of revenue to allocate to the undelivered elements. TPE generally does not exist for our products because of their uniqueness.
For products shipped under FOB shipping point terms, delivery is generally considered to have occurred when shipped. Undelivered elements in our sales arrangements, which are not considered to be essential to the functionality of a product, generally include installation and training services that are performed after the related products have been delivered and extended warranty agreements. Revenue related to undelivered installation and training services is deferred until such time as those services are complete, which is typically within 30 days of the related products being delivered to the customer’s location. Revenue and direct acquisition costs related to undelivered extended warranty agreements are deferred and recognized ratably over the service period, which is between one and four years. Deferred revenue for extended warranty agreements is based on the price charged when the service is sold separately.
Shipping and handling charges billed to customers are included in revenue and shipping and handling related expenses are charged to cost of revenue. Advance payments from customers are recorded as deferred revenue and recognized as revenue as otherwise described above. Most of our sales are subject to 30 to 60 day customer-specified acceptance provisions. These provisions require us to estimate the amount of future returns and recognize revenue net of these potential returns.
In certain states we are required to collect sales taxes from our customers. These amounts are excluded from revenue and recorded as a liability until remitted to the taxing authority.
GPOs negotiate volume purchase prices for hospitals, group practices and other clinics that are members of a GPO. Our agreements with GPOs typically include the following provisions:
· Negotiated pricing for all group members;
· Volume discounts and other preferential terms on their members’ purchases from us;
· Promotion of our products by the GPO to its members;
· Payment of administrative fees by us to the GPO, based on purchases of our products by group members.
We do not sell to GPOs. Hospitals, group practices and other acute care facilities that are members of a GPO purchase products directly from us under the terms negotiated by the GPO. Negotiated pricing and discounts are recognized as a reduction of the selling price of products at the time of the sale. Revenue from sales to members of GPOs is otherwise consistent with revenue recognition policies described above.
Accounts Receivable and Allowance for Doubtful Accounts
Accounts receivable is recorded at the sales price of the related products and services. We assess the sufficiency of the allowance for estimated uncollectible accounts receivable. Estimates are based on historical collection experience and other customer-specific information, such as bankruptcy filings or liquidity problems of our customers. When it is determined that an account receivable is uncollectible, it is written off and relieved from the allowance. Any future determination that the allowance for estimated uncollectible accounts receivable is not properly stated could result in changes in operating expense and results of operations.
Inventory
Inventories are stated at the lower of standard cost, which approximates actual cost on a first-in, first-out basis, or market. We may be exposed to a number of factors that could result in portions of our inventory becoming either obsolete or in excess of anticipated usage. These factors include, but are not limited to, technological changes, competitive pressures in products and prices, and the introduction of new product lines. We regularly evaluate our ability to realize the value of inventory based on a combination of factors, including historical usage rates, forecasted sales, product life cycles, and market acceptance of new products. When inventory that is obsolete or in excess of anticipated usage is identified, it is written down to realizable salvage value or an inventory valuation allowance is established.
The estimates we use in projecting future product demand may prove to be incorrect. Any future determination that our inventory is overvalued could result in increases to our cost of sales and decreases to our operating margins and results of operations.
Stock-based compensation
We apply the fair value recognition provisions of Financial Accounting Standards Board Accounting Standards Codification Topic 718, Compensation — Stock Compensation (“ASC 718”). Determining the amount of stock-based compensation to be recorded requires us to develop estimates of the fair value of stock options as of their grant date. Stock-based compensation expense is recognized ratably over the requisite service period, which is the vesting period of the award. Calculating the fair value of stock-based awards requires that we make highly subjective assumptions. We use the Black-Scholes option pricing model to value our stock option awards. Use of this valuation methodology requires that we make assumptions as to the volatility of our common stock, the expected term of our stock options, the risk-free interest rate for a period that approximates the expected term of our stock options and our expected dividend yield. As we just completed our IPO in July 2014, we utilize the historical stock price volatility from a representative group of public companies to estimate expected stock price volatility. We selected companies from the medical device industry with market capitalizations that are similar to ours. We intend to continue to utilize the historical volatility of the same or similar public companies to estimate expected volatility until a sufficient amount of historical information regarding the price of our publicly traded stock becomes available. We use the simplified method as prescribed by ASC 718 to calculate the expected term of stock options granted to employees as we do not have sufficient historical exercise data to provide a reasonable basis upon which to estimate the expected term of our stock option awards. The risk-free interest rate used for each grant is based on the U.S. Treasury yield curve in effect at the time of the grant for instruments with a similar expected life. We utilize a dividend yield of zero because we have no current intention to pay cash dividends. We estimated the fair value of options granted using a Black-Scholes option pricing model with the following weighted average assumptions:
|
| Years Ended December 31, |
| ||
|
| 2014 |
| 2013 |
|
Volatility |
| 104.3 | % | 113.0 | % |
Expected term (years) |
| 7.0 |
| 7.0 |
|
Risk-free interest rate |
| 2.1 | % | 2.4 | % |
Dividend yield |
| 0.0 | % | 0.0 | % |
Stock-based compensation expense totaled $724,063 and $271,919 for the years ended December 31, 2014 and 2013, respectively. The estimated forfeiture rate used to determine stock-based compensation expense was 3% and 10% for the years ended December 31, 2014 and 2013, respectively. As of December 31, 2014 we had $3,122,686 of total unrecognized stock-based compensation expense, which is expected to be recognized over a weighted-average period of 3.3 years. We expect the future impact of stock-based compensation expense on our financial results to grow due to the potential increases in the value of our common stock, additional stock option grants and increased headcount.
Under ASC 718, we are required to estimate the level of forfeitures expected to occur and record stock-based compensation expense only for those awards that we ultimately expect will vest. We estimate our forfeiture rate based on historical experience and employee class.
As discussed above, one input of the Black-Scholes option pricing model is the fair value of our common stock, which is issued upon exercise of the option. Options to purchase shares of our common stock are intended to be granted with an exercise price per share that is no less than the fair value per share of our common stock on the date of grant, which is based on the information known to us on the date of grant. Prior to our IPO, the fair value of our common stock was assessed on each grant date by our Board. For stock option grants prior to our IPO, the historical valuations of our common stock were determined in accordance with the guidelines outlined in the applicable American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held-Company Equity Securities Issued as Compensation (“AICPA Practice Aid”). In the absence of a public trading market, our Board considered all relevant facts and circumstances known at the time of valuation, made certain assumptions based on future expectations and exercised significant judgment to determine the fair value of our common stock. The factors considered by our Board in determining the fair value include, but are not limited to, the following:
· Contemporaneous third-party valuation of our common stock as of December 31, 2013;
· Our historical financial results and estimated trends and projections of our future operating and financial performance;
· The market performance of comparable, publicly traded companies; and
· The overall economic and industry conditions and outlook.
December 31, 2013 Valuation
Certain members of our Board and management reviewed the contemporaneous third-party valuation of our common stock as of December 31, 2013, discussed the reasonableness of the assumptions, methodologies, analysis and conclusions in this report. After reviewing this report, we determined the fair market value of the Company’s common stock. Our valuation of our common stock was conducted within the guidelines of the applicable AICPA Practice Aid. These guidelines prescribe certain valuation approaches for setting the value of an enterprise, such as the cost, market and income approaches, and various methodologies for allocating the value of an enterprise to common stock, such as the current value method, option pricing method, probability-weighted expected return method and the hybrid method.
Our valuation included the income approach and the market approach. Specifically, the Discounted Future Cash Flow (“DCF”) method was used under the income approach. The Guideline Public Company (“GPC”) method was used under the market approach to substantiate the conclusions derived from the income approach.
The method used to allocate enterprise value was the Probability-Weighted Expected Return Method (“PWERM”). Using the PWERM, the value of our common stock is estimated based upon an analysis of varying values for our common stock assuming the following possible future events for our company: (i) our initial public offering in 2014, (ii) our initial public offering in 2015, and (iii) we continue to operate as a private entity. For each of these possible events, a range of equity values is estimated based on a number of factors, which include income and market valuation approaches that factor in our estimates of future performance and performance of comparable public companies. For each possible future event, we determined the appropriate aggregate value to be allocated to holders of our shares of common stock based on the rights and preferences of each
class of our stock at that time. Next, we estimated the timing of possible future event dates and applied a discount rate, based on our estimated weighted average cost of capital using the venture capital portfolio rates of return as referenced in the AICPA Practice Aid for companies in a similar stage of development as ours. We then multiplied the discounted value of our common stock under each scenario by an estimated probability for each of the possible events, resulting in a probability-weighted value per share of common stock. Finally, we applied a discount for lack of marketability to the weighted value per share to determine a value per common share.
Application of the above described approaches involved the use of estimates, judgments and assumptions, such as future cash flows, selection of comparable publicly traded companies and the selection of discount rates. Changes in our assumptions or the interrelationship of those assumptions could result in changes to our operating results.
Income taxes
We estimate certain components of our provision for income taxes. These estimates include, among other items, depreciation and amortization expense allowable for tax purposes, allowable tax credits, effective rates for state taxes and tax deductibility of certain other items. We adjust our annual effective income tax rate as additional information on outcomes or events becomes available.
We account for income taxes under the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial statements and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. The effect of a change in tax rates on deferred tax assets and liabilities is recognized in income in the period that includes the enactment date.
We record net deferred tax assets to the extent we believe these assets will more likely than not be realized. In making such determination, we consider all available positive and negative evidence, including future reversals of existing taxable temporary differences, projected future taxable income, tax planning strategies and recent financial operations. A valuation allowance is recorded to offset net deferred tax assets if, based upon the available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
We recognize the tax benefit of uncertain tax positions in the financial statements based on the technical merits of the position. When the tax position is deemed more likely than not of being sustained, we recognize the largest amount of tax benefit that is greater than 50 percent likely of being ultimately realized upon settlement. We believe our tax positions are fully supportable
JOBS Act Accounting Election
We are an “emerging growth company” as defined in the Jumpstart Our Business Startups Act (“JOBS Act”), enacted in April 2012. An “emerging growth company” may take advantage of reduced reporting requirements that are otherwise applicable to public companies. For example, we will not have to provide an auditor’s attestation report on our internal controls in future annual reports on Form 10-K as otherwise required by Section 404(b) of the Sarbanes-Oxley Act. The JOBS Act also permits us, as an “emerging growth company,” to take advantage of an extended transition period to comply with new or revised accounting standards applicable to public companies. We chose to “opt out” of this provision and, as a result, we will comply with new or revised accounting standards when they are required to be adopted by issuers. This decision to opt out of the extended transition period under the JOBS Act is irrevocable.
Results of Operations
The following table sets forth for the periods indicated selected statements of operations data as a percentage of total revenue. Our historical operating results are not necessarily indicative of the results for any future period.
|
| Percent of Revenue |
| ||
|
| 2014 |
| 2013 |
|
Revenue |
| 100.0 | % | 100.0 | % |
Cost of revenue |
| 21.7 |
| 25.2 |
|
Gross profit |
| 78.3 |
| 74.8 |
|
Operating expenses: |
|
|
|
|
|
General and administrative |
| 30.8 |
| 21.1 |
|
Sales and marketing |
| 21.1 |
| 20.3 |
|
Research and development |
| 6.8 |
| 8.9 |
|
Total operating expenses |
| 58.7 |
| 50.3 |
|
Income from operations |
| 19.6 |
| 24.6 |
|
Other income, net |
| (0.3 | ) | 0.0 |
|
Income before provision for income taxes |
| 19.3 |
| 24.6 |
|
Provision for income taxes |
| 6.2 |
| 7.5 |
|
Net income |
| 13.1 | % | 17.1 | % |
Years Ended December 31, 2014 and 2013
Revenue
Revenue increased approximately $4.3 million, or 38%, to $15.6 million for the year ended December 31, 2014, compared to $11.3 million for 2013. This increase was primarily attributable to an increase in the number of our MRI compatible IV infusion pump systems sold during 2014 compared to 2013 and higher sales of our disposable IV sets.
Revenue from sales in the U.S. increased approximately $3.3 million, or 40%, to $11.4 million for the year ended December 31, 2014, from $8.1 million for the same period in 2013. Revenue from sales internationally increased approximately $1.1 million, or 33%, to $4.3 million for the year ended December 31, 2014, from $3.2 million for the same period in 2013.
Revenue from sales of devices increased approximately $3.5 million, or 37%, to $12.8 million for the year ended December 31, 2014, from $9.3 million for the same period in 2013. During the year ended December 31, 2014, we sold 536 MRI compatible IV infusion pumps compared to 424 pumps for the same period in 2013. The average selling price of our MRI compatible IV infusion pump systems during the year ended December 31, 2014 was approximately $23,800, compared to $22,000 for the same period in 2013. The increase in our average selling price is the result of higher domestic sales as a percent of revenue during the year ended December 31, 2014 compared to the same period in 2013.
Revenue from sales of our disposable IV sets and services increased approximately $0.8 million, or 42%, to $2.8 million for the year ended December 31, 2014, from $2.0 million for the same period in 2013. We expect revenue from sales of disposables and services to increase relative to the sale of devices as the installed base of our MRI compatible IV infusion pumps systems increases.
Cost of Revenue
Cost of revenue increased approximately $0.5 million, or 19%, to $3.4 million for year ended December 31, 2014, from $2.9 million for the same period in 2013. Gross profit increased approximately $3.7 million, or 44%, to $12.2 million for the year ended December 31, 2014 from $8.5 million for the same period in 2013. Gross profit margin increased to 78.3% for the year ended December 31, 2014, from 74.8% for the same period in 2013 primarily due to favorable materials variances, partially offset by unfavorable overhead and labor utilization rates.
General and Administrative
General and administrative expense increased approximately $2.4 million, or 101%, to $4.8 million for the year ended December 31, 2014, from $2.4 million for the same period last year. This increase is primarily due to higher payroll and employee benefits, legal and professional fees, stock compensation expense, business insurance, administration fees paid to group purchasing organizations, depreciation expense and office rent expense, partially offset by lower bad debt expense.
Sales and Marketing
Sales and marketing expense increased approximately $1.0 million, or 44%, to $3.3 million for the year ended December 31, 2014, from $2.3 million for the same period last year. This is primarily the result of higher salary and travel costs resulting from the increased size of our sales organization, higher sales commissions resulting from higher sales and stock compensation expense.
Research and Development
Research and development expense increased approximately $0.1 million, or 6%, to approximately $1.1 million for the year ended December 31, 2014, from approximately $1.0 million in the same period last year. This is primarily the result of higher salary costs due to increased headcount and stock compensation expense, partially offset by lower outside prototyping and consulting services.
Other Expense, Net
We reported other expense of approximately $(49,000) for the year ended December 31, 2014, compared to other expense of approximately $(3,500) for the same period last year. For the year ended December 31, 2014, we reported approximately $57,000 of foreign currency losses primarily associated with our Japanese Yen cash account, approximately $16,000 of interest expense associated with the purchase of our investments and miscellaneous income of approximately $18,000. For the year ended December 31, 2013, we reported approximately $23,000 of foreign currency losses primarily associated with our Japanese Yen cash account and approximately $20,000 of miscellaneous income.
Income Taxes
We recorded income tax expense of approximately $1.0 million and $0.8 million for the years ended December 31, 2014 and 2013, respectively. The higher income tax expense for the year ended December 31, 2014 was due to higher income before provision for income taxes and an increase in our effective tax rate. Our effective tax rate for the year ended December 31, 2014 was 32.0% compared to 30.4% for the same period in 2013. The increase in our effective tax rate was the result of lower domestic production activities deduction and lower research and development tax.
Liquidity and Capital Resources
Our principal sources of liquidity are our cash and cash equivalents balances, cash flow from operations and access to the financial markets. Our principal uses of cash are operating expenses, working capital requirements and capital expenditures.
As of December 31, 2014, we had cash and cash equivalents and investments of $17.4 million, stockholders’ equity of $20.9 million, and working capital of $19.9 million compared to cash and cash equivalents and investments of $2.7 million, stockholders’ equity of $5.4 million, and working capital of $4.9 million as of December 31, 2013.
In our early stages, our principal stockholder and Chief Executive Officer provided funding for operations in the form of an unsecured interest-free note payable with no specified due date. As of December 31, 2013, approximately $6,000 remained outstanding. In March 2014, we repaid with cash the outstanding balance of the officer note payable. We do not expect to continue to borrow funds from this principal stockholder in the future.
For the years ended December 31, 2014 and 2013, cash provided by operations increased by approximately $1.1 million to $2.6 million in 2014, compared to $1.5 million in 2013. This increase is the result a cash inflow of approximately $313,000 in deferred revenue, a cash inflow of approximately $281,000 in accrued income taxes and a net cash inflow of $183,000 in certain
operating assets and liabilities. Inventory increased by $786,000, or 58.6% in 2014 to $2.1 million, from $1.3 million in 2013. In aggregate, accounts payable and accrued expenses increased by $776,000, or 66.7% in 2014 to $1.9 million, from $1.2 million in 2013. The sum of our net income and certain non-cash expense items, such as stock compensation, depreciation and amortization was $2.9 million in 2014, compared to $2.3 million in 2013.
For the year ended December 31, 2014, cash used in investing activities was approximately $8.3 million, compared to $0.2 million in 2013. During 2014, we used $7.7 million net, of cash to purchase corporate debt securities and $0.6 million to purchase property and equipment. During 2013, we used $0.2 million to purchase property and equipment.
For the year ended December 31, 2014, cash provided by financing activities was approximately $12.7 million primarily resulting from our IPO. Net proceeds from our IPO were $12.4 million. During 2013, we used $0.5 million of payments on the officer note payable. During March 2014, we repaid the officer note payable in full with cash.
Sales to end users in the United States are generally made on open credit terms. Management maintains an allowance for potential credit losses. As of December 31, 2014, two international customers accounted for approximately 35% of gross accounts receivable. As of December 31, 2013, one international customer accounted for approximately 11% of gross accounts receivable.
In July 2014, we completed the move of our manufacturing operations and headquarters facility into a new building that is approximately 27,000 square feet located in Winter Springs, Florida. The new facility has been leased from Susi, LLC, an entity controlled by our President and CEO, Roger Susi. Pursuant to the terms of our lease, the monthly base rent is $32,583, adjusted annually for changes in the consumer price index.
We had an uncommitted revolving credit facility with Bank of America, N.A. that provided for a maximum borrowing capacity of $100,000. This facility was terminated during September 2014 and we no longer have the ability to obtain advances from this revolving credit facility. Prior to the termination of this facility during 2014 and throughout the year ended December 31, 2013, we did not request or obtain any advances from this revolving credit facility.
We believe our sources of liquidity, including cash flow from operations, existing cash, investments, and available financing sources will be sufficient to meet our projected cash requirements for at least the next 12 months. Any equity financing may be dilutive to stockholders and debt financing, if available, may involve restrictive covenants and increase our cost of capital. We will monitor our capital requirements to ensure our needs are in line with available capital resources. From time to time, we may explore additional financing sources to meet our working capital requirements, make continued investment in research and development, expand our business and acquire products or businesses that complement our current business. These actions would likely affect our future capital requirements and the adequacy of our available funds. Our future liquidity and capital requirements will depend on numerous factors, including the:
· Amount and timing of revenue and expenses;
· Extent to which our existing and new products gain market acceptance;
· Extent to which we make acquisitions;
· Cost and timing of product development efforts and the success of these development efforts;
· Cost and timing of selling and marketing activities; and
· Availability of borrowings or other means of financing.
Off-Balance Sheet Arrangements
During the periods presented, we did not have and we do not currently have, any off-balance sheet arrangements, as defined under SEC rules.
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Foreign Currency Exchange Risk
We have foreign currency risks related to our revenue and operating expenses denominated in currencies other than the U.S. dollar, principally the Japanese yen (“Yen”). The volatility of the Yen depends on many factors that we cannot forecast with reliable accuracy. We have experienced and will continue to experience fluctuations in our net income as a result of transaction gains (losses) related to revaluing Yen denominated cash and accounts payable balances that are denominated in Yen. In the event our Yen denominated cash, accounts payable or expenses increase, our operating results may be affected by fluctuations in the Yen exchange rate. If the U.S. Dollar uniformly increased or decreased in strength by 10% relative to the Yen, our net income would have correspondingly increased or decreased by an immaterial amount for the year ended December 31, 2014.
Interest Rate Risk
When able, we invest excess cash in bank money-market funds, corporate debt securities or discrete short-term investments. The fair value of our cash equivalents and short-term investments is sensitive to changes in the general level of interest rates in the U.S., and the fair value of these investments will decline if market interest rates increase. As of December 31, 2014, we had approximately $7.9 million in corporate bonds, with approximately $6.7 million that mature between 1 and 3 years and $1.2 million that mature between 3 and 5 years. These corporate bonds have fixed interest rates and semi-annual interest payment dates. If market interest rates were to change by 100 basis points from levels at December 31, 2014, we expect the corresponding change in fair value of our investments would be approximately $140,000. This is based on sensitivity analyses performed on our financial position as of December 31, 2014. Actual results may differ as our analysis of the effects of changes in interest rates does not account for, among other things, sales of securities prior to maturity and repurchase of replacement securities, the change in mix or quality of the investments in the portfolio, and changes in the relationship between short-term and long-term interest rates.
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
The information required by this Item 8 is incorporated by reference to information beginning on Page F-1 of this Form 10-K.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Disclosure Controls and Procedures
We maintain a set of disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15(e)) designed to ensure that information required to be disclosed by us in reports that we file or submit under the Exchange Act, is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms. In accordance with Rule 13a-15(b) of the Exchange Act, as of the end of the period covered by this Annual Report on Form 10-K, an evaluation was carried out under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, of the effectiveness of our disclosure controls and procedures. Based on that evaluation, our Chief Executive Officer and Chief Financial Officer concluded that our disclosure controls and procedures, as of the end of the period covered by this Annual Report on Form 10-K, were effective to provide reasonable assurance that information required to be disclosed by us in reports that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms and is accumulated and communicated to our management, including the Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
Management’s Report on Internal Control Over Financial Reporting
We are not required to comply with Section 404 of the Sarbanes-Oxley Act under applicable rules for newly public companies and are therefore not required to make an assessment of the effectiveness of our internal control over financial reporting. As a result, our management has not yet performed an evaluation of our internal control over financial reporting. Further, our independent registered public accounting firm is not yet required to, nor have they been engaged to express, nor have they expressed, an opinion on the effectiveness of our internal control over financial reporting.
Changes in Internal Control Over Financial Reporting
There were no changes in our internal control over financial reporting during the fourth quarter ended December 31, 2014 that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Limitations on Controls
Our disclosure controls and procedures and internal control over financial reporting are designed to provide reasonable assurance of achieving their objectives as specified above. Management does not expect, however, that our disclosure controls and procedures or our internal control over financial reporting will prevent or detect all error and fraud. Any control system, no matter how well designed and operated, is based upon certain assumptions and can provide only reasonable, not absolute, assurance that its objectives will be met. Further, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud, if any, within the Company have been detected.
Not applicable.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS, AND CORPORATE GOVERNANCE
The information required by this Item 10 will be included in the Proxy Statement to be filed within 120 days after the fiscal year covered by this annual report on Form 10-K and is incorporated herein by reference.
ITEM 11. EXECUTIVE COMPENSATION
The information required by this Item 11 will be included in the Proxy Statement, and such information is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information required by this Item 12, including Equity Compensation Plan Information, will be included in the Proxy Statement, and such information is incorporated herein by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information required by this Item 13 will be included in the Proxy Statement, and such information is incorporated herein by reference.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information required by this Item 14 will be included in the Proxy Statement, and such information is incorporated herein by reference.
ITEM 15. EXHIBITS AND FINANCIAL STATEMENT SCHEDULES
1. | Financial Statements: See “Index to Financial Statements” in Part II, Item 8 of this annual report on Form 10-K. |
2. | Financial Statement Schedule: Not applicable. |
3. | Exhibits: The exhibits listed in the accompanying “Exhibit Index” are filed or incorporated by reference as part of this Form 10-K. |
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of Winter Springs, State of Florida, on March 23, 2015.
| IRADIMED CORPORATION |
| (Registrant) |
|
|
Dated: March 23, 2015 | /s/ Roger Susi |
| By: Roger Susi |
| Chief Executive Officer and |
| President |
| (Principal Executive Officer) |
KNOW ALL MEN BY THESE PRESENTS, that each person whose signature appears below constitutes and appoints Roger Susi and Chris Scott as his true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this Report on Form 10-K, and to file the same, with all exhibits thereto, and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorney-in-fact and agents full power and authority to do and perform each and every act and thing requisite and necessary to be done in connection therewith, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorney-in-fact and agents, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Company in the capacities and on the dates indicated.
Signature |
| Title |
| Date |
/s/Roger Susi |
| Chief Executive Officer, President, and Director (principal executive officer) |
| March 23, 2015 |
Roger Susi |
|
|
| |
|
|
|
|
|
|
|
|
|
|
/s/ Chris Scott |
| Chief Financial Officer and Secretary (principal financial and accounting officer) |
| March 23, 2015 |
Chris Scott |
|
|
| |
|
|
|
|
|
/s/ James Hawkins |
| Chairman of the Board |
| March 23, 2015 |
James Hawkins |
|
|
| |
|
|
|
|
|
|
|
|
|
|
/s/Serge Novovich |
| Director |
| March 23, 2015 |
Serge Novovich |
|
|
| |
|
|
|
|
|
|
|
|
|
|
/s/Monty Allen |
| Director |
| March 23, 2015 |
Monty Allen |
|
|
|
Exhibit No. |
| Description |
3.1 |
| Certificate of Incorporation (incorporated herein by reference to Exhibit 3.1 to the Company’s Amendment No. 2 to the Registration Statement on Form S-1 (File No. 333-196875), filed on July 10, 2014). |
|
|
|
3.2 |
| Amended and Restated Bylaws (incorporated herein by reference to Exhibit 3.2 to the Company’s Current Report on Form 8-K (File No. 001-36534), filed on December 16, 2014). |
|
|
|
4.1 |
| Specimen common stock certificate (incorporated herein by reference to Exhibit 4.1 to the Company’s Amendment No. 1 to the Registration Statement on Form S-1 (File No. 333-196875), filed on July 9, 2014). |
|
|
|
10.1+ |
| iRadimed Corporation 2005 Incentive Stock Plan adopted February 1, 2005 (incorporated herein by reference to Exhibit 10.1 to the Company’s Registration Statement on Form S-1 (File No. 333-196875), filed on June 18, 2014). |
|
|
|
10.2+ |
| Form of Stock Option Agreement for iRadimed Corp. 2005 Incentive Stock Plan (incorporated herein by reference to Exhibit 10.2 to the Company’s Registration Statement on Form S-1 (File No. 333-196875), filed on June 18, 2014). |
|
|
|
10.3+ |
| Iradimed Corporation 2014 Equity Incentive Plan (incorporated herein by reference to Exhibit 10.3 to the Company’s Registration Statement on Form S-1 (File No. 333-196875), filed on June 18, 2014). |
|
|
|
10.4+ |
| Form of Stock Option Agreement for iRadimed Corporation 2014 Equity Incentive Plan (incorporated herein by reference to Exhibit 10.4 to the Company’s Registration Statement on Form S-1 (File No. 333-196875), filed on June 18, 2014). |
|
|
|
10.5+ |
| Form of Restricted Stock Award Agreement for iRadimed Corporation 2014 Equity Incentive Plan (incorporated herein by reference to Exhibit 4.6 to the Company’s Registration Statement on Form S-8 (File No. 333-198971), filed on September 26, 2014). |
|
|
|
10.6+ |
| Form of Restricted Stock Award Agreement for iRadimed Corporation 2014 Equity Incentive Plan (incorporated herein by reference to Exhibit 4.6 to the Company’s Registration Statement on Form S-8 (File No. 333-198971), filed on September 26, 2014). |
|
|
|
10.7+ |
| Form of Restricted Stock Unit Agreement (Time-Vesting) for iRadimed Corporation 2014 Equity Incentive Plan (incorporated herein by reference to Exhibit 4.7 to the Company’s Registration Statement on Form S-8 (File No. 333-198971), filed on September 26, 2014). |
|
|
|
10.8+ |
| Form of Restricted Stock Unit Agreement (Performance-Vesting) for iRadimed Corporation 2014 Equity Incentive Plan (incorporated herein by reference to Exhibit 4.8 to the Company’s Registration Statement on Form S-8 (File No. 333-198971), filed on September 26, 2014). |
|
|
|
10.9 |
| Lease Agreement regarding 7457 Aloma Avenue dated April 12, 2011 between Roberts Supply Profit Sharing, LLC and the Registrant (incorporated herein by reference to Exhibit 10.5 to the Company’s Registration Statement on Form S-1 (File No. 333-196875), filed on June 18, 2014). |
|
|
|
10.10 |
| Amendment to Lease Agreement regarding 7457 Aloma Avenue dated September 16, 2013 between Roberts Supply Profit Sharing, LLC and the Registrant (incorporated herein by reference to Exhibit 10.6 to the Company’s Registration Statement on Form S-1 (File No. 333-196875), filed on June 18, 2014). |
|
|
|
10.11 |
| Lease Agreement regarding 1025 Willa Springs Dr. dated January 17, 2014 between Susi, LLC and the Registrant (incorporated herein by reference to Exhibit 10.7 to the Company’s Registration Statement on Form S-1 (File No. 333-196875), filed on June 18, 2014). |
10.12+ |
| Employment Agreement between the Registrant and Christopher K. Scott dated December 16, 2013 (incorporated herein by reference to Exhibit 10.8 to the Company’s Registration Statement on Form S-1 (File No. 333-196875), filed on June 18, 2014). |
|
|
|
10.13+ |
| Employment Agreement between the Registrant and Roger Susi dated April 14, 2014 (incorporated herein by reference to Exhibit 10.9 to the Company’s Registration Statement on Form S-1 (File No. 333-196875), filed on June 18, 2014). |
|
|
|
10.14+ |
| Employment Agreement between the Registrant and Brent Johnson dated December 7, 2011 (incorporated herein by reference to Exhibit 10.10 to the Company’s Registration Statement on Form S-1 (File No. 333-196875), filed on June 18, 2014). |
|
|
|
10.15† |
| Supply Agreement between dated January 26, 2014 (incorporated herein by reference to Exhibit 10.11 to the Company’s Registration Statement on Form S-1 (File No. 333-196875), filed on June 18, 2014). |
|
|
|
23.1 |
| Consent of McGladrey LLP, Independent Registered Public Accounting Firm |
|
|
|
24.1 |
| Power of Attorney (included on signature page) |
|
|
|
31.1 |
| Certification of Chief Executive Officer pursuant to Exchange Act Rule, 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
31.2 |
| Certification of Chief Financial Officer pursuant to Exchange Act Rule, 13a-14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
|
|
|
32.1 |
| Certifications of Chief Executive Officer and Chief Financial Officer pursuant to 18 I.S.C Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. |
|
|
|
101.INS |
| XBRL Instance Document |
|
|
|
101.SCH |
| XBRL Taxonomy Extensions Schema Document |
|
|
|
101.CAL |
| XBRL Taxonomy Extension Label Calculation Linkbase Document |
|
|
|
101.DEF |
| XBRL Taxonomy Extension Definition Document |
|
|
|
101.LAB |
| XBRL Taxonomy Extension Label Linkbase Document |
|
|
|
101.PRE |
| XBRL Taxonomy Extension Presentation Linkbase Document |
+ | Indicates a management contract or compensatory plan or arrangement. |
† | Confidential treatment has been granted for portions of this exhibit. These portions have been omitted from the exhibit filed with the Securities and Exchange Commission and submitted separately to the Securities and Exchange Commission. |
IRADIMED CORPORATION FINANCIAL STATEMENTS
| F-2 | |
|
|
|
| F-3 | |
|
|
|
| F-4 | |
|
|
|
| F-5 | |
|
|
|
| F-6 | |
|
|
|
| F-7 |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and Stockholders
IRADIMED CORPORATION
We have audited the accompanying balance sheets of IRADIMED CORPORATION as of December 31, 2014 and 2013, and the related statements of operations and comprehensive income, stockholders’ equity, and cash flows for the years then ended. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of IRADIMED CORPORATION as of December 31, 2014 and 2013, and the results of its operations and its cash flows for the years then ended, in conformity with U.S. generally accepted accounting principles.
/s/ McGladrey LLP
Orlando, Florida
March 23, 2015
IRADIMED CORPORATION
|
| As of December 31, |
| ||||
|
| 2014 |
| 2013 |
| ||
ASSETS |
|
|
|
|
| ||
Current assets: |
|
|
|
|
| ||
Cash and cash equivalents |
| $ | 9,454,150 |
| $ | 2,461,559 |
|
Accounts receivable, net |
| 1,960,214 |
| 1,982,083 |
| ||
Investments |
| 7,913,793 |
| 246,203 |
| ||
Inventory, net |
| 2,125,838 |
| 1,340,331 |
| ||
Prepaid expenses and other current assets |
| 276,540 |
| 119,974 |
| ||
Prepaid income taxes |
| 320,941 |
| 170,496 |
| ||
Deferred income taxes |
| 116,339 |
| 65,961 |
| ||
Total current assets |
| 22,167,815 |
| 6,386,607 |
| ||
Property and equipment, net |
| 794,835 |
| 327,343 |
| ||
Intangible assets, net |
| 250,836 |
| 267,024 |
| ||
Deferred income taxes |
| 76,557 |
| — |
| ||
Other assets |
| 19,676 |
| 5,897 |
| ||
Total assets |
| $ | 23,309,719 |
| $ | 6,986,871 |
|
LIABILITIES AND STOCKHOLDERS’ EQUITY |
|
|
|
|
| ||
Current liabilities: |
|
|
|
|
| ||
Accounts payable |
| $ | 629,167 |
| $ | 427,474 |
|
Accrued payroll and benefits |
| 1,244,898 |
| 655,362 |
| ||
Other accrued taxes |
| 65,790 |
| 80,787 |
| ||
Warranty reserve |
| 27,925 |
| 12,002 |
| ||
Deferred revenue |
| 308,341 |
| 207,395 |
| ||
Officer note payable |
| — |
| 6,333 |
| ||
Accrued income taxes |
| — |
| 62,971 |
| ||
Total current liabilities |
| 2,276,121 |
| 1,452,324 |
| ||
Deferred revenue |
| 142,902 |
| 57,676 |
| ||
Deferred income taxes |
| — |
| 54,087 |
| ||
Total liabilities |
| 2,419,023 |
| 1,564,087 |
| ||
Stockholders’ equity: |
|
|
|
|
| ||
Preferred stock, $0.0001 par value; 10,000,000 shares authorized; none issued and outstanding as of December 31, 2014 and 1,400,000 shares issued and outstanding as of December 31, 2013 |
| — |
| 140 |
| ||
Common stock; $0.0001 par value; 90,000,000 shares authorized; 10,814,650 shares issued and outstanding as of December 31, 2014 and 7,000,000 shares issued and outstanding as of December 31, 2013 |
| 1,082 |
| 700 |
| ||
Additional paid-in capital |
| 15,785,838 |
| 2,346,137 |
| ||
Retained earnings |
| 5,125,249 |
| 3,074,883 |
| ||
Accumulated other comprehensive (loss) income |
| (21,473 | ) | 924 |
| ||
Total stockholders’ equity |
| 20,890,696 |
| 5,422,784 |
| ||
Total liabilities and stockholders’ equity |
| $ | 23,309,719 |
| $ | 6,986,871 |
|
See accompanying notes to financial statements.
IRADIMED CORPORATION
STATEMENTS OF OPERATIONS AND COMPREHENSIVE INCOME
|
| For the Years Ended |
| ||||
|
| 2014 |
| 2013 |
| ||
Revenue |
| $ | 15,653,057 |
| $ | 11,340,097 |
|
Cost of revenue |
| 3,404,400 |
| 2,853,385 |
| ||
Gross profit |
| 12,248,657 |
| 8,486,712 |
| ||
Operating expenses: |
|
|
|
|
| ||
General and administrative |
| 4,816,973 |
| 2,392,305 |
| ||
Sales and marketing |
| 3,297,120 |
| 2,297,309 |
| ||
Research and development |
| 1,068,674 |
| 1,009,872 |
| ||
Total operating expenses |
| 9,182,767 |
| 5,699,486 |
| ||
Income from operations |
| 3,065,890 |
| 2,787,226 |
| ||
Other expense, net |
| (48,549 | ) | (3,458 | ) | ||
Income before provision for income taxes |
| 3,017,341 |
| 2,783,768 |
| ||
Provision for income taxes |
| 966,975 |
| 846,878 |
| ||
Net income |
| $ | 2,050,366 |
| $ | 1,936,890 |
|
Other comprehensive income (loss): |
|
|
|
|
| ||
Change in fair value of available-for-sale securities, net of tax benefit of $13,220 and $3,352 respectively |
| (27,153 | ) | (6,225 | ) | ||
Realized gain on available-for-sale securities reclassified to net income, net of tax expense of $2,560 and $0 |
| (4,756 | ) | — |
| ||
Other comprehensive loss |
|
| (22,397 | ) |
| (6,225 | ) |
Comprehensive income |
| $ | 2,027,969 |
| $ | 1,930,665 |
|
Net income per share: |
|
|
|
|
| ||
Basic |
| $ | 0.23 |
| $ | 0.28 |
|
Diluted |
| $ | 0.20 |
| $ | 0.22 |
|
Weighted average shares outstanding: |
|
|
|
|
| ||
Basic |
| 8,743,461 |
| 7,000,000 |
| ||
Diluted |
| 10,219,143 |
| 8,624,314 |
| ||
See accompanying notes to financial statements.
IRADIMED CORPORATION
STATEMENTS OF STOCKHOLDERS’ EQUITY
|
| Preferred |
| Common |
| Additional |
| Retained |
| Accumulated |
| Stockholders’ |
| ||||||
Balances, December 31, 2012 |
| $ | 140 |
| $ | 700 |
| $ | 2,074,218 |
| $ | 1,137,993 |
| $ | 7,149 |
| $ | 3,220,200 |
|
Net income |
| — |
| — |
| — |
| 1,936,890 |
| — |
| 1,936,890 |
| ||||||
Other comprehensive loss |
| — |
| — |
| — |
| — |
| (6,225 | ) | (6,225 | ) | ||||||
Stock-based compensation |
| — |
| — |
| 271,919 |
| — |
| — |
| 271,919 |
| ||||||
Balances, December 31, 2013 |
| $ | 140 |
| $ | 700 |
| $ | 2,346,137 |
| $ | 3,074,883 |
| $ | 924 |
| $ | 5,422,784 |
|
|
|
|
|
|
|
|
|
|
|
|
|
|
| ||||||
Net income |
| — |
| — |
| — |
| 2,050,366 |
| — |
| 2,050,366 |
| ||||||
Other comprehensive loss |
| — |
| — |
| — |
| — |
| (22,397 | ) | (22,397 | ) | ||||||
Stock-based compensation |
| — |
| — |
| 724,063 |
| — |
| — |
| 724,063 |
| ||||||
Tax benefits credited to equity |
| — |
| — |
| 165,228 |
| — |
| — |
| 165,228 |
| ||||||
Exercise of stock options |
| — |
| 10 |
| 104,990 |
| — |
| — |
| 105,000 |
| ||||||
Issuance of common stock pursuant to initial public offering |
| — |
| 232 |
| 14,489,768 |
| — |
| — |
| 14,490,000 |
| ||||||
Common stock issuance costs and underwriter fees |
| — |
| — |
| (2,044,348 | ) | — |
| — |
| (2,044,348 | ) | ||||||
Conversion of preferred stock |
| (140 | ) | 140 |
| — |
| — |
| — |
| — |
| ||||||
Balances, December 31, 2014 |
| $ | — |
| $ | 1,082 |
| $ | 15,785,838 |
| $ | 5,125,249 |
| $ | (21,473 | ) | $ | 20,890,696 |
|
See accompanying notes to financial statements.
IRADIMED CORPORATION
|
| Years Ended December 31, |
| ||||
|
| 2014 |
| 2013 |
| ||
Operating activities: |
|
|
|
|
| ||
Net income |
| $ | 2,050,366 |
| $ | 1,936,890 |
|
Adjustments to reconcile net income to net cash provided by operating activities: |
|
|
|
|
| ||
Provision for excess and obsolete inventory |
| 62,069 |
| — |
| ||
Depreciation and amortization |
| 149,056 |
| 139,040 |
| ||
Stock-based compensation |
| 724,063 |
| 271,919 |
| ||
Changes in operating assets and liabilities: |
|
|
|
|
| ||
Accounts receivable |
| 21,869 |
| (396,589 | ) | ||
Inventory |
| (847,576 | ) | 45,655 |
| ||
Prepaid expenses and other current assets |
| (154,912 | ) | (40,819 | ) | ||
Other assets |
| (15,433 | ) | (4,594 | ) | ||
Deferred income taxes |
| (167,305 | ) | (48,797 | ) | ||
Accounts payable |
| 201,693 |
| 19,050 |
| ||
Accrued payroll and benefits |
| 589,536 |
| 114,109 |
| ||
Other accrued current liabilities |
| (14,997 | ) | 60,466 |
| ||
Warranty reserve |
| 15,923 |
| (134 | ) | ||
Deferred revenue |
| 186,172 |
| (126,985 | ) | ||
Accrued income taxes, net of prepaid income taxes |
| (213,416 | ) | (494,814 | ) | ||
Other |
| (1,388 | ) | — |
| ||
Net cash provided by operating activities |
| 2,585,720 |
| 1,474,397 |
| ||
Investing activities: |
|
|
|
|
| ||
Purchases of investments |
| (7,951,497 | ) | (4,986 | ) | ||
Proceeds from sale of investments |
| 255,109 |
| — |
| ||
Purchases of property and equipment |
| (583,977 | ) | (163,175 | ) | ||
Capitalized intangible assets |
| (22,311 | ) | (28,586 | ) | ||
Net cash used in investing activities |
| (8,302,676 | ) | (196,747 | ) | ||
Financing activities: |
|
|
|
|
| ||
Proceeds from stock option exercises |
| 105,000 |
| — |
| ||
Income tax benefits credited to equity |
| 165,228 |
| — |
| ||
Repayment of officer note payable |
| (6,333 | ) | (513,397 | ) | ||
Proceeds from the issuance of common stock pursuant to initial public offering |
| 14,490,000 |
| — |
| ||
Payment of initial public offering costs |
| (2,044,348 | ) | — |
| ||
Net cash provided by (used in) financing activities |
| 12,709,547 |
| (513,397 | ) | ||
Net increase in cash and equivalents |
| 6,992,591 |
| 764,253 |
| ||
Cash and cash equivalents, beginning of year |
| 2,461,559 |
| 1,697,306 |
| ||
Cash and cash equivalents, end of year |
| $ | 9,454,150 |
| $ | 2,461,559 |
|
|
|
|
|
|
| ||
Supplemental disclosure of cash flow information: |
|
|
|
|
| ||
Cash paid for income taxes |
| $ | 1,182,430 |
| $ | 1,390,049 |
|
See accompanying notes to financial statements.
IRADIMED CORPORATION
1 — Organization and Significant Accounting Policies
Organization
IRADIMED CORPORATION (“IRADIMED”, the “Company”, “we”, “our”) was incorporated in Oklahoma in July 1992 and reincorporated in Delaware in April 2014. We develop, manufacture, market and distribute Magnetic Resonance Imaging (“MRI”) compatible products, and today, we are the sole known provider of non-magnetic intravenous (“IV”) infusion pump systems. We were the first to develop an infusion delivery system that neutralizes the dangers and problems present during MRI procedures. Our headquarters are in Winter Springs, Florida.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America (U.S. GAAP) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities in the financial statements and the reported amount of revenue and expenses during the reporting period. Such estimates include allowances for potentially uncollectible accounts receivable, valuation of inventory, intangible assets, allocation of revenue arrangement consideration, stock-based compensation, deferred income taxes, reserves for warranty obligations, and the provision for income taxes. Actual results could differ from those estimates.
FDA Warning Letter
On September 2, 2014 we announced we received a Warning Letter from the U.S. Food and Drug Administration (“FDA”) relating to an inspection of our facility that took place in April 2014. At the conclusion of the April inspection, FDA issued a Form 483 that identified eight observations. The majority of the observations related to procedures and documentation associated with the design, development and validation testing of software used in certain of our products. Other observations were related to the design validation of pump labeling, design analysis of tube stretching, procedures for post-market design review, and procedures and processing related to handling certain reported complaints. We submitted responses to the Form 483 in May 2014 and June 2014 in which we described our proposed corrective and preventative actions to address each of the FDA’s concerns.
FDA’s Warning Letter stated that the FDA accepted as adequate several of our responses to Form 483 observations, identified two responses whose accuracy will be determined in the next scheduled inspection of our facility and identified issues for which our response was determined to be inadequate. The issues identified as inadequate concern our procedures for validating device design primarily related to software quality assurance.
Also, the Warning Letter raised a new issue. The Warning Letter stated that modifications made to software on our previously cleared infusion pumps, the MRidium 3860 and MRidium 3850, were “significant” and required submission of new premarket notifications under Section 510(k) (a “510(k) submission”) of the Food, Drug and Cosmetic Act (the “FDC Act”). These modifications were made over time. We believe they were insignificant and did not require premarket notification submissions. However, the FDA indicated that the modifications of the software for the MRidium 3860 and the software for the MRidium 3850 were “significant” modifications because they could significantly affect the safety or effectiveness of these devices. As a result, the Warning Letter states that the products being sold by us are “adulterated” and “misbranded” under the FDC Act. The Warning Letter also indicates that the MRidium 3860+ infusion pump requires separate FDA clearance from the MRidium 3860 and MRidium 3850.
The Warning Letter requested that we immediately cease activities that result in the misbranding or adulteration of the MRidium 3860 MRI infusion pump, MRidium 3850 MRI infusion pump, and the MRidium 3860+ MRI infusion pump, including the commercial distribution of the devices. We immediately complied with the Warning Letter and ceased sale and distribution of the identified products in the United States.
On September 4, 2014, we submitted to the FDA our initial response to the Warning Letter and on September 17, 2014 we sent an additional response that included supplemental information related to the Form 483 inspection observations for which the FDA considered our initial responses inadequate.
On November 25, 2014, we announced that we filed the 510(k) submission related to our MRidium 3860+ MRI IV infusion pumps and on December 12, 2014 we were notified that our 510(k) submission had been formally accepted for review by the FDA. On December 22, 2014, under FDA enforcement discretion, we announced that we resumed domestic distribution of our MRI compatible MRidium 3860+ MRI IV infusion pump systems, without the Dose Error Reduction System (“DERS”) option. On January 28, 2015, subsequent to our year end, we announced that we resumed domestic distribution of our DERS option.
We continue to work with the FDA to fully resolve the Warning Letter and complete the review of the 510(k) submission. See Note 12.
Initial Public Offering
On July 21, 2014, the Company completed an initial public offering (“IPO”) of its common stock and sold 2,318,400 shares of common stock (including 302,400 shares sold upon the underwriters’ exercise of their over-allotment option to purchase additional shares) at a price of $6.25 per share. The IPO generated net proceeds of approximately $12.4 million after deducting underwriting discounts and expenses of approximately $2.0 million. These expenses were recorded against the proceeds received from the IPO. Concurrent with the closing of the IPO, all outstanding preferred stock was automatically converted into common stock on a 1:1 basis.
Associated with our IPO, we issued the underwriters warrants to purchase up to a total of 201,600 shares of our common stock. The grant date aggregate fair value of the warrants was $611,000. The warrants are exercisable, in whole or in part, commencing July 21, 2015 through July 21, 2017. The warrants are exercisable at a per share price equal to $8.13 per share, or 130% of the public offering price per share of our common stock in the IPO. The exercise price and number of warrant shares may be adjusted upon (1) voluntarily at our discretion, or (2) if we undertake a stock split, stock dividend, recapitalization or reorganization of our common stock into a lesser / greater number of shares, the warrant exercise price will be proportionately reduced / increased and the number of warrant shares will be proportionately increased / decreased. The warrants may only be settled through the issuance of our common stock in exchange for cash. We have classified the warrants as equity and incremental direct costs associated with our IPO. Accordingly, the warrants do not impact our financial statements.
Revenue Recognition
Revenue is recognized when persuasive evidence of an arrangement exists, the price is fixed or determinable, delivery has occurred and title and risk of loss has transferred and collection of the resulting receivable is reasonably assured. Terms of sale for most domestic sales are FOB destination, reflecting that title and risk of loss are assumed by the purchaser upon delivery. Terms of sales to international distributors are FOB shipping point, reflecting that title and risk of loss are assumed by the distributor at the shipping point.
Under the revenue recognition rules for tangible products, we allocate revenue from arrangements with multiple deliverables to each of the deliverables based upon their relative selling prices as determined by a selling-price hierarchy. A deliverable in an arrangement qualifies as a separate unit of accounting if 1) the delivered item has value to the customer on a stand-alone basis, and 2) the arrangement includes a general right of return relative to the delivered item, delivery or performance of the undelivered items is considered probable and substantially in control of the vendor. The principal deliverables in our multiple deliverable arrangements that qualify as separate units of accounting consist of (i) sales of medical devices and supplies, (ii) installation and training services, and (iii) separately priced extended warranty agreements.
We use a hierarchy to determine the selling price to be used for allocating revenue to deliverables as follows: (i) vendor-specific objective evidence of fair value (“VSOE”), (ii) third-party evidence of selling price (“TPE”), and (iii) best estimate of the selling price (“ESP”). VSOE of fair value is defined as the price charged when the same element is sold separately, or if the element has not yet been sold separately, the price for the element established by management having the relevant authority when it is probable that the price will not change before the introduction of the element into the marketplace. VSOE generally exists only when we sell the deliverable separately and is the price actually charged for that deliverable. For certain sales under group purchasing organization (“GPO”) contracts, we have established VSOE for all of the elements in our multiple element arrangements. This determination is based on the volume of sales to these customers in relation to our total sales and the discount tier in which those sales are made. For all other sales we rely on ESP, reflecting our best estimates of what the selling prices of elements would be if they were sold regularly on a stand-alone basis, to establish the amount of revenue to allocate to the undelivered elements. TPE generally does not exist for our products because of their uniqueness.
For products shipped under FOB shipping point terms, delivery is considered to have occurred when shipped. Undelivered elements in our sales arrangements, which are not considered to be essential to the functionality of a product, generally include installation and training services that are performed after the related products have been delivered and extended warranty agreements. Revenue related to undelivered installation and training services is deferred until such time as those services are complete, which is typically within 30 days of the related products being delivered to the customer’s location. Revenue and direct
acquisition costs related to undelivered extended warranty agreements are deferred and recognized ratably over the service period, which is between one and four years. Deferred revenue for extended warranty agreements is based on the price charged when the service is sold separately.
Shipping and handling charges billed to customers are included in revenue and shipping and handling related expenses are charged to cost of revenue. Advance payments from customers are recorded as deferred revenue and recognized as revenue as otherwise described above. Most of our sales are subject to 30 to 60 day customer-specified acceptance provisions. These provisions require us to estimate the amount of future returns and recognize revenue net of these potential returns.
In certain States we are required to collect sales taxes from our customers. These amounts are excluded from revenue and recorded as a liability until remitted to the taxing authority.
GPOs negotiate volume purchase prices for hospitals, group practices, and other clinics that are members of a GPO. Our agreements with GPOs typically include the following provisions:
· Negotiated pricing for all group members;
· Volume discounts and other preferential terms on their members’ purchases from us;
· Promotion of our products by the GPO to its members; and
· Payment of administrative fees by us to the GPO, based on purchases of our products by group members.
We do not sell to GPOs. Hospitals, group practices, and other acute care facilities that are members of a GPO purchase products directly from us under the terms negotiated by the GPO. Negotiated pricing and discounts are recognized as a reduction of the selling price of products at the time of the sale. Revenue from sales to members of GPOs is otherwise consistent with revenue recognition policies as previously described.
Cash Equivalents
All highly liquid instruments purchased with an original maturity of three months or less are classified as cash equivalents.
Accounts Receivable and Allowance for Doubtful Accounts
Accounts receivable is recorded at the sales price of the related products and services. We assess the sufficiency of the allowance for estimated uncollectible accounts receivable. Estimates are based on historical collection experience and other customer-specific information, such as bankruptcy filings or liquidity problems of our customers. When it is determined that an account receivable is uncollectible, it is written off and relieved from the allowance. Any future determination that the allowance for estimated uncollectible accounts receivable is not properly stated could result in changes in operating expense and results of operations. As of December 31, 2014 and 2013, our allowance for doubtful accounts was $28,119 and $136,971, respectively.
Investments
Our investments consist of corporate debt securities and are considered available-for-sale. The specific identification method is used to determine the cost basis of investments sold. Our investments are recorded in our balance sheets at fair value. We classify our investments as current based on the nature of the investments and their availability for use in current operations. Unrealized gains and losses on our investments are included in accumulated other comprehensive income, net of tax. Realized gains or losses are recorded in sale of investments and impairment losses that are determined to be other-than-temporary are recorded in investment impairment losses in our statements of operations.
Fair Value Measurements
Fair value is the price that would be received to sell an asset or paid to transfer a liability in the principal or most advantageous market in an orderly transaction between market participants on the measurement date. A three-level valuation hierarchy requires an entity to maximize the use of observable inputs and minimize the use of unobservable inputs when measuring fair value.
The valuation hierarchy is based upon the transparency of inputs to the valuation of an asset or liability on the measurement date. The three levels of inputs are:
· Level 1 — quoted prices (unadjusted) in active markets for an identical asset or liability.
· Level 2 — quoted prices for a similar asset or liability in an active market or model-derived valuations in which all significant inputs are observable for substantially the full term of the asset or liability.
· Level 3 — unobservable and significant to the fair value measurement of the asset or liability.
Financial instruments include cash and cash equivalents, investments, accounts receivable, accounts payable and accrued expenses. Cash and cash equivalents and investments are reported at their respective fair values on the balance sheet dates. The recorded carrying amount of accounts receivable, accounts payable and accrued expenses approximates their fair values due to their short-term maturities.
Inventory
Inventories are stated at the lower of standard cost, which approximates actual cost on a first-in, first-out basis, or market. We may be exposed to a number of factors that could result in portions of our inventory becoming either obsolete or in excess of anticipated usage. These factors include, but are not limited to, technological changes, competitive situations in products and prices, and the introduction of new product lines. We regularly evaluate our ability to realize the value of inventory based on a combination of factors, including historical usage rates, forecasted sales, product life cycles, and market acceptance of new products. When inventory that is obsolete or in excess of anticipated usage is identified, it is written down to realizable salvage value or an inventory valuation allowance is established.
Property and Equipment
Property and equipment are stated at cost less accumulated depreciation. Depreciation expense is computed using the straight-line method over estimated useful lives of the respective assets, which are three to five years for computer software and hardware; five to seven years for furniture, fixtures, machinery and equipment. Leasehold improvements are amortized over the shorter of the lease term or the estimated useful life of the improvements.
Repair and maintenance costs that do not extend the useful life of our property and equipment are expensed as incurred.
Intangible Assets
Intangible assets include application and legal costs incurred to obtain patents. We capitalize these costs when we determine that probable future economic benefits exist. In making this determination, we consider the projected future operating results associated with the patents, industry and economic trends, and the entry of new products in the market. Costs incurred prior to this determination are expensed in the period they are incurred. We amortize capitalized patent costs using the straight-line method over their useful lives, which is typically 17 years. Periodic costs incurred to maintain existing patents are expensed as incurred.
Long-lived Assets
Long-lived assets are tested for impairment whenever changes in circumstances indicate the carrying value of these assets may be impaired. Impairment indicators include, but are not limited to, technological obsolescence, unfavorable court rulings, significant negative industry and economic trends, and significant underperformance relative to historical and projected future operating results. Impairment is considered to have occurred when the estimated undiscounted future cash flows related to the asset groups are less than its carrying value. Estimates of future cash flows involve consideration of many factors including the marketability of new products, product acceptance and lifecycle, competition, appropriate discount rates, and operating margins. An impairment is recognized as the amount by which the carrying value is less than the fair value of the asset or asset group.
Warranty
We provide for the estimated cost of product warranties at the time revenue is recognized. While we engage in product quality programs and processes, including actively monitoring and evaluating the quality of our suppliers, the estimated warranty obligation is affected by ongoing product failure rates, material usage costs and direct labor incurred in correcting a product failure.
Actual product failure rates, material usage costs and the amount of labor required to repair products that differ from estimates result in revisions to the estimated liability. We warrant for a limited period of time that our products will be free from defects in materials and workmanship. We estimate warranty allowances based on historical warranty experience. Historically, warranty expenses have not been material to our financial statements.
Research & Development and Capitalized Software Development Costs
Research and development costs are expensed as incurred. Some of our products include embedded software which is essential to the product’s functionality. Costs incurred in the research and development of new software components and enhancements to existing software components are expensed as incurred until technological feasibility has been established. We capitalize software development costs when the project reaches technological feasibility and cease capitalization when the project is ready for release. Capitalized software development costs are included in intangible assets and are amortized on a straight-line basis over the estimated useful life of the product. Amortization begins when the product is available for general release to the customer.
Advertising and Marketing
For the years ended December 31, 2014 and 2013, these costs were $65,369 and $78,882, respectively. Advertising and marketing costs are expensed as incurred and included in sales and marketing expense.
Medical Device Excise Taxes
Effective January 1, 2013, we became subject to the Medical Device Excise Tax applicable to sales of listed medical devices under the Patient Protection and Affordable Care Act (“ACA”) enacted in 2010. The ACA requires us to pay 2.3% of the taxable sales value of devices sold. Qualifying sales are recorded on a gross basis. For the years ended December 31, 2014 and 2013, we recorded medical device excise taxes of $200,496 and $161,246, respectively. Medical device excise taxes are included as a component of general and administrative expense.
Stock-Based Compensation
We recognize stock-based compensation expense associated with employee stock options on a straight-line basis over the requisite service period for the entire award, which is generally four years. The maximum contractual life of our stock options is ten years from the grant date. We utilize the Black-Scholes option pricing model to estimate the grant date fair value of those awards. The Black-Scholes option pricing model requires the input of certain assumptions including stock price, dividend yield, expected volatility, risk-free interest rate, and expected option life. Changes in these assumptions can materially affect the estimated fair value of our employee stock options.
Prior to our IPO, the grant date stock price was based on third-party valuations that have been performed periodically and consideration of significant events impacting us since the date of the respective valuations; subsequent to our IPO, the grant date stock price was based on our closing stock price on the date of grant; dividend yield was based on our expectation of dividend payments over the expected life of the option; expected volatility was based on a study of comparable, publicly traded companies with similar products and product life cycles; risk-free interest rate was the rate available on zero coupon U.S. government obligations with a term approximating the expected option life; the expected option life was calculated using the simplified method.
Forfeitures of employee stock options are estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from initial estimates. Stock-based compensation expense is recorded net of estimated forfeitures, such that expense is recorded only for those stock-based awards that are expected to vest.
The cash flow resulting from the tax benefits from tax deductions in excess of the compensation cost recognized for those options (excess tax benefits) is classified as a cash inflow from financing activities and a cash outflow from operating activities in our statements of cash flows. We treat tax deductions from certain stock option exercises as being realized when they reduce taxes payable in accordance with relevant tax law. Upon exercise, we issues new shares.
Income Taxes
We account for income taxes under the asset and liability method, which requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been included in the financial statements. Under this method, deferred tax assets and liabilities are determined based on the differences between the financial statements and tax basis of assets and liabilities using enacted tax rates in effect for the year in which the differences are expected to reverse. The effect of a change in tax rates on deferred tax assets and liabilities is recognized in income in the period that includes the enactment date.
We record net deferred tax assets to the extent we believe these assets will more likely than not be realized. In making such determination, we consider all available positive and negative evidence, including future reversals of existing taxable temporary differences, projected future taxable income, tax planning strategies and recent financial operations. A valuation allowance is recorded to offset net deferred tax assets if, based upon the available evidence, it is more likely than not that some or all of the deferred tax assets will not be realized.
We recognize the tax benefit of uncertain tax positions in the financial statements based on the technical merits of the position. When the tax position is deemed more likely than not of being sustained, we recognize the largest amount of tax benefit that is greater than 50 percent likely of being ultimately realized upon settlement.
Foreign Currency
Gains and losses from transactions denominated in currencies other than our functional currency are included in other income and expense. For the years ended December 31, 2014 and 2013, net foreign currency transaction losses were $56,969 and $23,432, respectively. Foreign currency gains and losses result primarily from fluctuations in the exchange rate between the U.S. Dollar and the Japanese Yen.
Comprehensive Income
Comprehensive income includes net income and other comprehensive income items that are excluded from net income under U.S. generally accepted accounting principles. Comprehensive income includes unrealized gains and losses on our investments classified as available for sale.
Basic and Diluted Net Income per Share
Basic net income per share is based upon the weighted average number of common shares outstanding during the period. Diluted net income per share reflects the potential dilution that could occur if securities or other contracts to issue common stock were exercised or converted into common stock. As discussed further in Note 13, the effect of our 1.75:1 stock split and recapitalization is reflected in the number of outstanding shares and per share information in the table below. Preferred stock and stock options granted by us represent the only dilutive effect reflected in diluted weighted-average shares outstanding.
The following table presents the computation of basic and diluted net income per share:
|
| Years Ended December 31, |
| ||||
|
| 2014 |
| 2013 |
| ||
Net income |
| $ | 2,050,366 |
| $ | 1,936,890 |
|
Weighted-average shares outstanding — Basic |
| 8,743,461 |
| 7,000,000 |
| ||
Effect of dilutive securities: |
|
|
|
|
| ||
Preferred stock |
| 751,780 |
| 1,400,000 |
| ||
Stock options |
| 723,902 |
| 224,314 |
| ||
Weighted-average shares outstanding — Diluted |
| 10,219,143 |
| 8,624,314 |
| ||
Basic net income per share |
| $ | 0.23 |
| $ | 0.28 |
|
Diluted net income per share |
| $ | 0.20 |
| $ | 0.22 |
|
Warrants and stock options to purchase shares of our common stock excluded from the calculation of diluted net income per share because the effect would have been anti-dilutive are as follows:
|
| As of December 31, |
| ||
|
| 2014 |
| 2013 |
|
Anti-dilutive warrants and stock options |
| 129,340 |
| 467,109 |
|
Certain Significant Risks and Uncertainties
We market our products to end users in the United States and to distributors internationally. Sales to end users in the United States are generally made on open credit terms. Management maintains an allowance for potential credit losses. As of December 31, 2014, two international customers accounted for approximately 35% of gross accounts receivable. As of December 31, 2013, one international customer accounted for approximately 11% of gross accounts receivable.
Revenue for 2013 included sales to an international customer for 129 of our MRI compatible IV infusion pumps, which represented approximately 11% of total revenue for 2013.
We have deposited our cash and cash equivalents with various financial institutions. Our cash and cash equivalents balances exceed federally insured limits throughout the year. We have not incurred any losses related to these balances.
Our products require clearance from the Food and Drug Administration and international regulatory agencies prior to commercialized sales. The Company’s future products may not receive required approvals. If the Company were denied such approvals, or if such approvals were delayed, it would have a materially adverse impact on the Company’s business, results of operations and financial condition.
Certain key components of our products essential to their functionality are sole-sourced. Any disruption in the availability of these components would have a materially adverse impact on our business, results of operations and financial condition.
Recent Accounting Pronouncements
In May 2014, the FASB issued Accounting Standards Update (“ASU”) 2014-09, Revenue Contracts with Customers (Topic 606). This update provides guidance on the recognition of revenue based upon the entity’s contracts with customers to transfer goods or services at an amount that reflects the consideration the entity expects to receive in exchange for those goods or services. This update also requires additional disclosure about the nature, amount, timing and uncertainty of revenue and cash flows arising from contracts with customers. This update is effective for annual periods beginning after December 15, 2016, including interim periods within that reporting period, which will require us to adopt this update in the first quarter of 2017. Early adoption is not permitted. We are evaluating this guidance and have not yet determined the effect it will have on our financial statements and related disclosures, if any.
2 — Inventory
Inventory consists of:
|
| As of December 31, |
| ||||
|
| 2014 |
| 2013 |
| ||
|
|
|
|
|
| ||
Raw materials |
| $ | 1,541,688 |
| $ | 1,143,495 |
|
Work in process |
| 126,188 |
| 14,337 |
| ||
Finished goods |
| 457,962 |
| 182,499 |
| ||
Total |
| $ | 2,125,838 |
| $ | 1,340,331 |
|
3 — Property and Equipment
Property and equipment consist of:
|
| As of December 31, |
| ||||
|
| 2014 |
| 2013 |
| ||
Computer software and hardware |
| $ | 303,076 |
| $ | 154,709 |
|
Furniture and fixtures |
| 198,253 |
| 87,611 |
| ||
Leasehold improvements |
| 182,105 |
| 47,623 |
| ||
Machinery and equipment |
| 849,852 |
| 721,270 |
| ||
Tooling in-process |
| 42,315 |
| 46,562 |
| ||
|
| 1,575,601 |
| 1,057,775 |
| ||
Accumulated depreciation |
| (780,766 | ) | (730,432 | ) | ||
Total |
| $ | 794,835 |
| $ | 327,343 |
|
Depreciation and amortization expense of property and equipment was $110,557 and $101,449 in the years ended December 31, 2014 and 2013, respectively.
4 — Intangible Assets
The following table summarizes the components of intangible asset balances:
|
| As of December 31, |
| ||||
|
| 2014 |
| 2013 |
| ||
Patents — in use |
| $ | 238,548 |
| $ | 228,430 |
|
Patents — in process |
| 31,358 |
| 19,165 |
| ||
Internally developed software |
| 148,967 |
| 148,967 |
| ||
|
| 418,873 |
| 396,562 |
| ||
Accumulated amortization |
| (168,037 | ) | (129,538 | ) | ||
Total |
| $ | 250,836 |
| $ | 267,024 |
|
Amortization expense of intangible assets was $38,499 and $37,591 in the years ended December 31, 2014 and 2013, respectively.
Expected annual amortization expense for the next five years related to intangible assets is as follows:
2015 |
| $ | 39,446 |
|
2016 |
| $ | 22,144 |
|
2017 |
| $ | 14,665 |
|
2018 |
| $ | 14,665 |
|
2019 |
| $ | 14,665 |
|
5 — Stock-Based Compensation
During the year ended December 31, 2013, we maintained one stock plan known as the iRadimed Corporation Incentive Stock Plan (“Plan”). In April 2014, our Board of Directors adopted and our shareholders approved the 2014 Equity Incentive Plan (“2014 Plan”). Upon adoption and approval of the 2014 Plan, the Plan was terminated and the remaining shares available for future awards were canceled. The 2014 Plan reserves 1,000,000 shares of our common stock for awards of incentive stock options, non-qualified stock option, stock appreciation rights, restricted stock, restricted stock units, performance awards and other stock-based awards and cash awards. As of December 31, 2014, there were 791,750 shares available for future awards under the 2014 Plan.
Stock-based compensation was recognized as follows in the statements of operations:
|
| Years Ended December 31, |
| ||||
|
| 2014 |
| 2013 |
| ||
Cost of revenue |
| $ | 6,529 |
| $ | 13 |
|
General and administrative |
| 266,167 |
| 16,461 |
| ||
Sales and marketing |
| 415,021 |
| 255,096 |
| ||
Research and development |
| 36,346 |
| 349 |
| ||
Total |
| $ | 724,063 |
| $ | 271,919 |
|
As of December 31, 2014, we had $3,122,686 of total unrecognized stock-based compensation expense, which is expected to be recognized over a weighted average period of 3.3 years. The total grant date fair value of stock options that vested during the year ended December 31, 2014 was $752,927.
The fair value of our option grants was estimated using the Black-Scholes model with the following weighted average assumptions:
|
| Years Ended December 31, |
| ||
|
| 2014 |
| 2013 |
|
Volatility |
| 104.3 | % | 113.0 | % |
Expected term (years) |
| 7.0 |
| 7.0 |
|
Risk-free interest rate |
| 2.1 | % | 2.4 | % |
Dividend yield |
| 0.0 | % | 0.0 | % |
The weighted-average grant-date fair value of options granted during the years ended December 31, 2014 and 2013 was $6.47 and $2.99, respectively. The estimated forfeiture rate used to determine stock-based compensation expense was 3% and 10% for the years ended December 31, 2014 and 2013, respectively.
Prior to our IPO, historical valuations of our common stock were determined in accordance with the guidelines outlined in the American Institute of Certified Public Accountants Practice Aid, Valuation of Privately-Held-Company Equity Securities Issued as Compensation. In the absence of a public trading market, we considered all relevant facts and circumstances known at the time of valuation, made certain assumptions based on future expectations and exercised significant judgment to determine the fair value of our common stock. The factors considered in determining the fair value include, but are not limited to, the following:
· Retrospective and contemporaneous third-party valuation of our common stock;
· Our historical financial results and estimated trends and projections of our future operating and financial performance;
· The market performance of comparable, publicly traded companies; and
· The overall economic and industry conditions and outlook.
The following table presents a summary of our stock option activity as of and for the year ended December 31, 2014:
|
| Options |
| Weighted-Average |
| Weighted-Average |
| Aggregate |
| ||
Outstanding beginning of period |
| 1,759,692 |
| $ | 1.18 |
| 7.7 |
| $ | 3,741,190 |
|
|
|
|
|
|
|
|
|
|
| ||
Options granted |
| 281,750 |
| 7.55 |
|
|
|
|
| ||
Options exercised |
| (96,250 | ) | 1.09 |
|
|
|
|
| ||
Options cancelled |
|
|
|
|
|
|
|
|
| ||
Outstanding end of period |
| 1,945,192 |
| $ | 2.11 |
| 7.4 |
| $ | 20,996,495 |
|
Exercisable |
| 957,787 |
| $ | 1.12 |
| 6.0 |
| $ | 11,282,000 |
|
Cash received from option exercises during the year ended December 31, 2014 was $105,000. The total intrinsic value of options exercised during the year ended December 31, 2014 was $592,463.
6 — Investments
As of December 31, 2014, our investments consisted of corporate bonds that we have classified as available-for-sale and are summarized in the following table:
|
| Cost |
| Gross |
| Gross |
| Fair |
| ||||
Corporate bonds: |
|
|
|
|
|
|
|
|
| ||||
U.S. corporations |
| $ | 6,433,286 |
| $ | — |
| $ | 27,067 |
| $ | 6,406,219 |
|
International corporations |
| 1,515,200 |
| — |
| 7,626 |
| 1,507,574 |
| ||||
Total |
| $ | 7,948,486 |
| $ | — |
| $ | 34,693 |
| $ | 7,913,793 |
|
As of December 31, 2014, the scheduled maturities of our investments are as follows:
|
| Cost |
| Fair Value |
| ||
Less than 1 year |
| $ | — |
| $ | — |
|
1 to 3 years |
| 6,723,316 |
| 6,697,241 |
| ||
3 to 5 years |
| 1,225,170 |
| 1,216,552 |
| ||
Total |
| $ | 7,948,486 |
| $ | 7,913,793 |
|
As of December 31, 2013, our investments consisted of two mutual funds classified as available-for-sale and are summarized in the following table:
|
| Cost |
| Gross |
| Gross |
| Fair |
| ||||
December 31, 2013 |
| $ | 244,782 |
| $ | 1,421 |
| $ | — |
| $ | 246,203 |
|
7 — Fair Value Measurements
The fair value of our assets and liabilities subject to recurring fair value measurements are as follows:
|
| Fair Value at December 31, 2014 |
| ||||||||||
|
| Fair |
| Quoted Prices |
| Significant |
| Significant |
| ||||
Corporate bonds: |
|
|
|
|
|
|
|
|
| ||||
U.S. corporations |
| $ | 6,406,219 |
| $ | — |
| $ | 6,406,219 |
| $ | — |
|
International corporations |
| 1,507,574 |
| — |
| 1,507,574 |
| — |
| ||||
Total |
| $ | 7,913,793 |
| $ | — |
| $ | 7,913,793 |
| $ | — |
|
Our corporate bonds are valued by the third-party custodian at closing prices from national exchanges or pricing vendors on the valuation date.
|
| Fair Value at December 31, 2013 |
| ||||||||||
|
| Fair |
| Quoted Prices |
| Significant |
| Significant |
| ||||
Mutual funds |
| $ | 246,203 |
| $ | 246,203 |
| $ | — |
| $ | — |
|
The fair values of our mutual funds are based upon quoted market prices and valuations provided by the third-party custodian of our mutual funds.
There were no transfers into or out of any Levels during the years ended December 31, 2014 or 2013.
8 — Accumulated Other Comprehensive Income
The only component of accumulated other comprehensive income is as follows:
|
| Unrealized Gains |
| |
Balances at December 31, 2012 |
| $ | 7,149 |
|
Gains (losses), net |
| (6,225 | ) | |
Reclassification realized in net earnings |
| — |
| |
|
|
|
| |
Balances at December 31, 2013 |
| $ | 924 |
|
|
|
|
| |
Gains (losses), net |
| (27,153 | ) | |
Reclassification realized in net earnings |
| (4,756 | ) | |
|
|
|
| |
Balances at December 31, 2014 |
| (21,473 | ) | |
9 — Income Taxes
The components of the provision for income taxes are as follows:
|
| Years Ended December 31, |
| ||||
|
| 2014 |
| 2013 |
| ||
Current taxes: |
|
|
|
|
| ||
U.S. federal |
| $ | 1,022,098 |
| $ | 764,452 |
|
State |
| 112,144 |
| 130,784 |
| ||
|
|
|
|
|
| ||
Total current tax expense |
| 1,134,242 |
| 895,236 |
| ||
|
|
|
|
|
| ||
Deferred taxes: |
|
|
|
|
| ||
U.S. federal |
| (149,247 | ) | (14,278 | ) | ||
State |
| (18,020 | ) | (34,080 | ) | ||
|
|
|
|
|
| ||
Total deferred tax benefit |
| (167,267 | ) | (48,358 | ) | ||
|
|
|
|
|
| ||
Income tax expense |
| $ | 966,975 |
| $ | 846,878 |
|
Deferred income taxes reflect the net tax effects of temporary differences between the carrying amounts of assets and liabilities for financial reporting purposes and the amounts used for income tax purposes. Significant components of our deferred taxes are as follows:
|
| Years Ended December 31, |
| ||||
|
| 2014 |
| 2013 |
| ||
Deferred tax assets: |
|
|
|
|
| ||
Current deferred tax assets: |
|
|
|
|
| ||
Reserves and allowances |
| $ | 119,548 |
| $ | 109,177 |
|
|
|
|
|
|
| ||
Total current deferred tax assets |
| $ | 119,548 |
| $ | 109,177 |
|
Noncurrent deferred tax assets: |
|
|
|
|
| ||
Stock compensation |
| $ | 417,956 |
| $ | 152,906 |
|
Other |
| 14,689 |
| — |
| ||
|
|
|
|
|
| ||
Total noncurrent deferred tax assets |
| $ | 432,645 |
| $ | 152,906 |
|
|
|
|
|
|
| ||
Deferred tax liabilities: |
|
|
|
|
| ||
Current deferred tax liabilities: |
|
|
|
|
| ||
Reserves and allowances |
| $ | 3,209 |
| $ | 42,674 |
|
Other |
| — |
| 542 |
| ||
|
|
|
|
|
| ||
Total current deferred tax liabilities |
| $ | 3,209 |
| $ | 43,216 |
|
|
|
|
|
|
| ||
Noncurrent deferred tax liabilities: |
|
|
|
|
| ||
Depreciation and amortization |
| $ | 356,088 |
| $ | 206,993 |
|
|
|
|
|
|
| ||
Total noncurrent deferred tax liabilities |
| 356,088 |
| 206,993 |
| ||
A reconciliation of the statutory U.S. federal tax rate to our effective rate is as follows:
|
| Years Ended December 31, |
| ||
|
| 2014 |
| 2013 |
|
Statutory U.S. federal tax rate |
| 34.0 | % | 34.0 | % |
Domestic production activities deduction |
| (2.1 | ) | (3.7 | ) |
Research and development credits |
| (1.8 | ) | (2.8 | ) |
State taxes, net of federal benefit |
| 2.0 |
| 2.3 |
|
Permanent items |
| (0.1 | ) | 0.6 |
|
|
|
|
|
|
|
Total |
| 32.0 | % | 30.4 | % |
As of December 31, 2014 and December 31, 2013, we have not identified or accrued for any uncertain tax positions. We are currently unaware of any uncertain tax positions that could result in significant payments, accruals or other material deviations in this estimate over the next 12 months.
We file tax returns in the United States Federal jurisdiction and many state jurisdictions. Our returns are not currently under examination by the Internal Revenue Service or other taxing authorities. The Company is subject to income tax examinations for our United States federal and State income taxes for 2008 and subsequent years.
10 — Employee Benefit Plan
We sponsor a 401(k) tax-deferred savings plan under which eligible employees may elect to have a portion of their salary deferred and contributed to the plan. Employer matching contributions are determined by management and are discretionary. Employer matching contributions were $162,581 and $103,755, respectively, for the years ended December 31, 2014 and 2013. Employer contributions vest ratably over five years.
11 — Segment, Customer and Geographic Information
We operate in one reportable segment which is the development, manufacture and sale of MRI compatible products and IV infusion pump systems for use by hospitals and acute care facilities during MRI procedures.
In the U.S., we sell our products through our direct sales force and outside of the U.S. we sell our products through distributors who resell our products to end users.
Revenue information by geographic region is as follows:
|
| Years Ended December 31, |
| ||||
|
| 2014 |
| 2013 |
| ||
United States |
| $ | 11,357,705 |
| $ | 8,100,907 |
|
International |
| 4,295,352 |
| 3,239,190 |
| ||
|
| $ | 15,653,057 |
| $ | 11,340,097 |
|
Revenue information by type is as follows:
|
| Years Ended December 31, |
| ||||
|
| 2014 |
| 2013 |
| ||
Devices |
| $ | 12,812,446 |
| $ | 9,335,753 |
|
Disposable IV Sets and Services |
| 2,840,611 |
| 2,004,344 |
| ||
|
| $ | 15,653,057 |
| $ | 11,340,097 |
|
Revenue for 2013 included two sales to a customer in Saudi Arabia for 129 of our MRI compatible IV infusion pumps. Revenue recorded in 2013 related to these orders represented 11% of total revenue for 2013.
Property and equipment, net information by geographic region is as follows:
|
| Years Ended December 31, |
| ||||
|
| 2014 |
| 2013 |
| ||
United States |
| $ | 728,556 |
| $ | 256,386 |
|
International |
| 66,279 |
| 70,957 |
| ||
|
| $ | 794,835 |
| $ | 327,343 |
|
Long-lived assets held outside of the United States consist principally of tooling, which is a component of property and equipment, net.
12 — Commitments and Contingencies
Leases. We have entered into noncancelable operating leases for our facilities.
In January 2014, we entered into a lease, commencing July 1, 2014, for a new facility in Winter Springs, Florida owned by Susi, LLC, and entity controlled by our president and CEO, Roger Susi. Pursuant to the terms of our lease for this property, the monthly base rent will be $32,583, adjusted annually for changes in the consumer price index. The term of the lease expires on May 31, 2019. The lease will automatically renew for two successive terms of five years each beginning in 2019 and again in 2024, and thereafter, will be renewed for successive terms of one year each.
Rent expense for the years ended December 31, 2014 and 2013 was $284,210 and $116,277, respectively. Minimum lease payments for each of our operating leases are even throughout their respective lease term.
Future minimum lease payments under noncancelable operating leases as of December 31, 2014 are as follows:
|
| Operating |
| |
2015 |
| $ | 390,996 |
|
2016 |
| 390,996 |
| |
2017 |
| 390,996 |
| |
2018 |
| 390,996 |
| |
2019 |
| 162,915 |
| |
Thereafter |
| — |
| |
Total minimum lease payments |
| $ | 1,726,899 |
|
Purchase commitments. We had various purchase orders for goods or services totaling approximately $1,993,801 at December 31, 2014. No amounts related to these purchase orders have been recognized in our balance sheet.
Uncommitted Revolving Credit Facility. We had an uncommitted revolving credit facility with Bank of America, National Association that provided for a maximum borrowing capacity of $100,000. This facility was terminated during September 2014 and we no longer have the ability to obtain advances from this revolving credit facility. Prior to the termination of this facility during the third quarter 2014 and throughout the year ended December 31, 2013, we did not request or obtain any advances from this revolving credit facility.
Indemnifications. Under our amended and restated bylaws, we have agreed to indemnify our officers and directors for certain events or occurrences arising as a result of the officer or director serving in such capacity. We have a director and officer liability insurance policy that limits our exposure under these indemnifications and enables us to recover a portion of any future loss arising out of them.
In addition, in the normal course of business, we enter into contracts that contain indemnification clauses whereby the Company indemnifies our customers against damages associated with product failures. We have determined that these agreements fall within the scope of ASC 460, Guarantees. We have obtained liability insurance providing coverage that limits our exposure for these indemnified matters. We have not incurred costs to defend lawsuits or settle claims related to these indemnities. We believe the estimated fair value of these indemnities is minimal and have not recorded a liability for these agreements as of December 31, 2014.
Legal matters. We may from time to time become a party to various legal proceedings or claims that arise in the ordinary course of business. We do not believe that any current legal or administrative proceedings are likely to have a material effect on our business, financial condition, or results of operations.
On September 10, 2014, a Civil Action was filed in the U.S. District Court for the Southern District of Florida (“Lam Civil Action”). The Lam Civil Action is a putative class action lawsuit brought against the Company and certain individuals who are officers and / or directors of the Company. The plaintiff is an alleged shareholder of the Company, and seeks relief on behalf of a class of persons who purchased the Company’s common stock during the period from July 15, 2014 through September 2, 2014. The complaint alleges that the defendants failed to disclose material information concerning the Company’s compliance with FDA regulations in violation of Section 10(b) of the Securities Exchange Act of 1934 and Rule 10b-5 thereunder, and that the putative class members suffered damages as a result. The complaint additionally alleges “control person” liability against the individual defendants under Section 20(a) of the Securities Exchange Act of 1934. The Lam Civil Action is presently in the very early stages of litigation. The Company disputes the plaintiff’s allegations and theories of liability, and intends to defend the case vigorously. We have not accrued for any loss related to this matter as we believe that any such loss is not probable or estimable.
In October 2012, Radimed Gesellschaft für Kommunikationsdienstleistungen und Medizintechnik mbH (“Radimed”) brought an action in Düsseldorf Regional Court against our German distributor alleging the name and sign “iRadimed” was confusingly similar to their German trademark “Radimed.” A judgment was rendered against our German distributor preventing use of the name and sign “iRadimed” in Germany. We have however continued to sell products in Germany without any discernible effect by using the product name IRI Development. On July 31, 2013, Radimed filed a lawsuit against us and our founder, Roger Susi, in Düsseldorf Regional Court, alleging that we infringed their German and Community trademarks “Radimed” and seeking to prevent our use of the name, sign and domain name “iRadimed” in the European Union. Prior to year end, we began settlement discussions with Radimed and accrued an insignificant amount related to this matter. Subsequent to year end, in March 2015, we settled this matter and paid the amount that had been accrued. Pursuant to this settlement, we may continue to use the name “iRadimed” and our associated signs and domain name in the European Union.
13 — Capital Stock
Reincorporation
Effective April 14, 2014, we reincorporated as a Delaware corporation. As part of this reincorporation, we converted all previously outstanding shares of our Class A Common Stock and Class B Common Stock into a single class of common stock on a 1.75:1 conversion ratio and all previously outstanding shares of our Series A Preferred Stock were split on a 1.75:1 conversion ratio into new Series A Preferred Stock. In accordance with our Certificate of Incorporation, upon the sale of shares pursuant to an initial public offering, which was completed in July 2014, all of our Series A Preferred Stock was automatically converted into common stock on a 1:1 conversion ratio (see Note 1). The table below summarizes the effect of the stock split and conversion on our capital stock that was previously outstanding as of December 31, 2013:
Series A Preferred Stock outstanding — Pre recapitalization |
| 800,000 |
|
Stock split ratio |
| 1.75:1 |
|
Series A Preferred Stock outstanding — Post recapitalization |
| 1,400,000 |
|
Common stock outstanding — Pre recapitalization |
|
|
|
Class A Common Stock |
| 400,000 |
|
Class B Common Stock |
| 3,600,000 |
|
Total |
| 4,000,000 |
|
Stock split ratio |
| 1.75:1 |
|
Common stock outstanding — Post recapitalization |
| 7,000,000 |
|
As of the effective date of the reincorporation, we are now authorized to issue 90,000,000 shares of Common Stock with a par value of $0.0001 per share and 10,000,000 shares of Preferred Stock with a par value of $0.0001.
The effect of this stock split has been retroactively applied to per-share computations, share and option amounts for all periods presented within these financial statements and accompanying notes.
The rights and privileges of our Series A Preferred Stock and Common Stock are as follows:
Series A Preferred Stock
We are authorized to issue 10,000,000 shares of preferred stock, of which 800,000 of these shares shall be designated as Series A Preferred Stock (“Preferred Stock”) with a par value of $0.0001 per share.
Voting and Dividends. The holder of each share of Preferred Stock has the right to one vote for each share of Common Stock into which such Preferred Stock could then be converted. The holders of the Preferred Stock are entitled to receive dividends from legally available assets prior to any declaration or payment of dividends to Common Stock holders. Dividends on each share of Preferred Stock are initially at $0.06429 per year payable when and as declared by the Board and are non-cumulative. After payment of such dividends, any additional dividends or distributions are distributed among all holders of Common Stock and Preferred Stock in proportion to the number of shares of Common Stock that would be held by each holder if all shares of Preferred Stock were converted to Common Stock at the then effective conversion rate. To date, no dividends have been declared.
Liquidation. In the event of any liquidation, dissolution or winding up of our Company, either voluntary or involuntary, the holders of the Preferred Stock are entitled to receive, prior and in preference to any distribution of the proceeds resulting from such liquidation event to holders of the Common Stock, an amount equal to $1.07143 plus declared but unpaid dividends. If, upon occurrence of such liquidation event, the proceeds are insufficient to permit the payment of the aforementioned amount in full, then the entire proceeds shall be distributed ratably among all holders of the Preferred Stock in proportion to the full amount each holder would otherwise receive.
Conversion. Each share of Preferred Stock is convertible at any time, at the option of the holder, into such number of fully paid non-assessable shares of Common Stock as is determined by dividing the original issue price of each share of Preferred Stock by the applicable conversion price. The initial conversion price per share is $1.07143. Adjustments to the initial conversion price may result from a recapitalization event or changes in the number of common shares outstanding. Each share of Preferred Stock automatically converts into shares of fully paid non-assessable shares of Common Stock, at the then applicable conversion rate, upon the date specified by written consent or agreement of the holders of a majority of the then outstanding shares of Preferred Stock, voting as a single class on an as-converted basis.
Redemption. Upon a majority vote of the then outstanding shares of Preferred Stock, we may, at our discretion, redeem or purchase shares of Preferred Stock. We also have a first right of refusal to repurchase shares of the Preferred Stock arising from a holder’s proposal to sell such Preferred Stock.
Common Stock
We are authorized to issue 90,000,000 shares of Common Stock with a par value of $0.0001 per share.
Voting and Dividends. Each outstanding share of Common Stock shall entitle the holder thereof to one vote on each matter properly submitted to the stockholders of the Company for their vote except for matters related to potential amendments to our Certificate of Incorporation or matters that solely relate to the terms of one or more outstanding series of our Preferred Stock. Holders of our Common Stock are entitled to receive, when, as and if declared by the Board, dividends pro rata based on the number of shares of Common Stock held. These dividend rights are junior to those of the Preferred Stock holders’ rights to dividends.
Liquidation. Liquidation preference of the Common Stock holders is junior to that of the Preferred Stock holders.
Redemption. The Common Stock is not redeemable.
14 — Officer Note Payable
In the early stages of the Company, our CEO provided funding for operations in the form of an unsecured interest-free note payable with no specified due date. As of December 31, 2013, $6,333 remained outstanding. In March 2014 we repaid with cash the outstanding balance of the note payable.