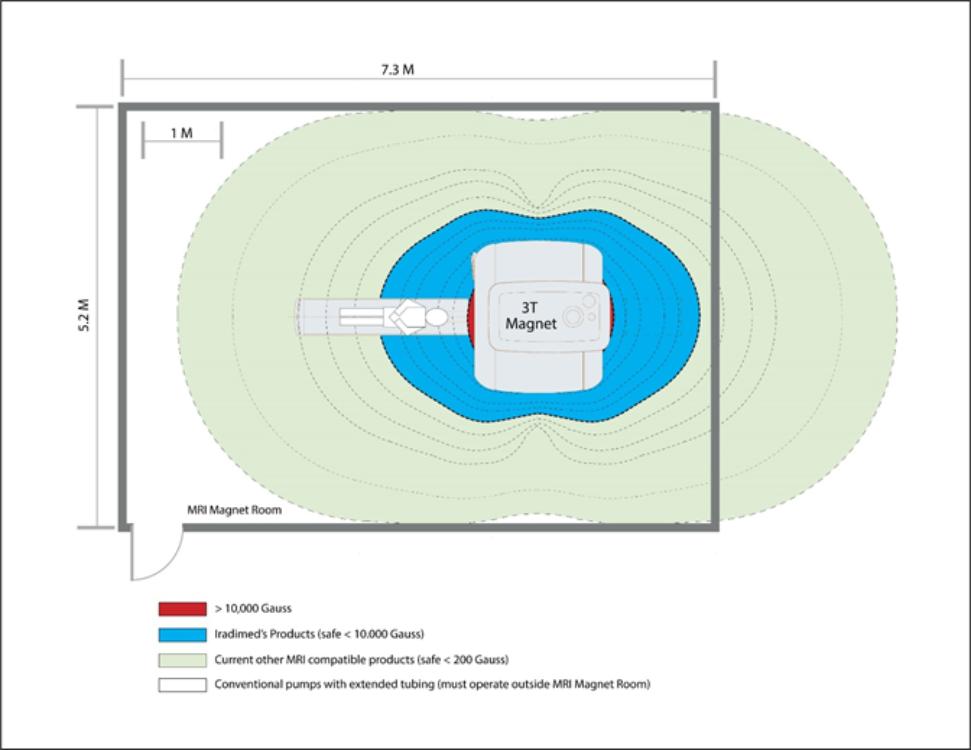
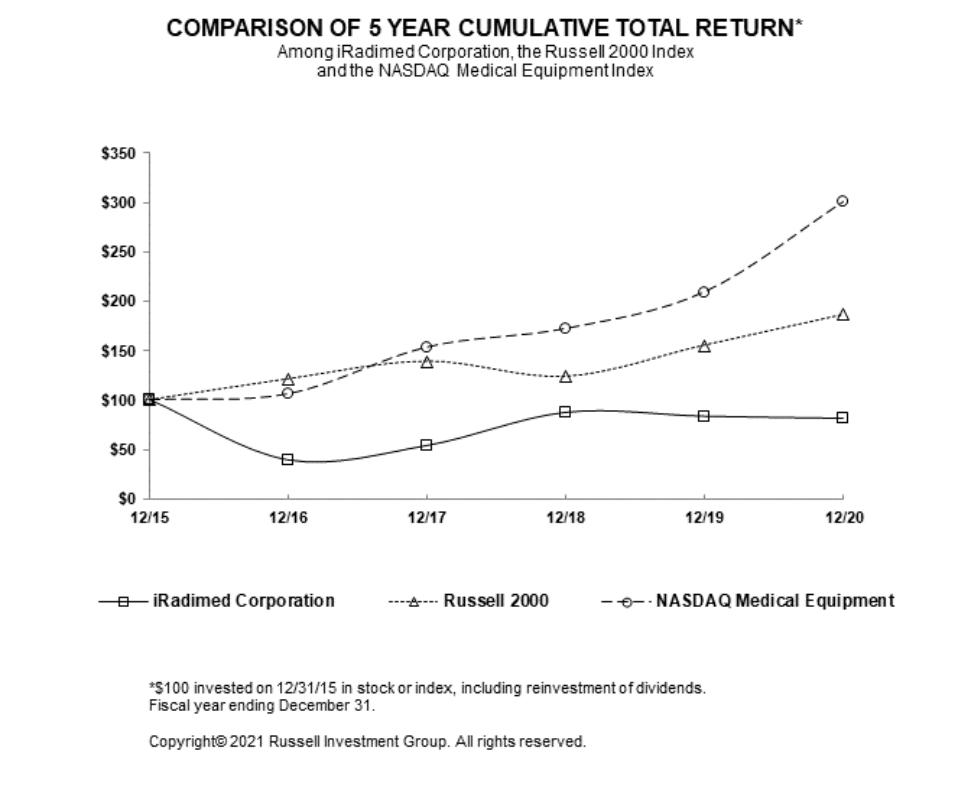
administration and 9 in research and development. No employees are represented by a labor union. We have not experienced any work stoppages and consider our relations with our employees to be good.
Regulatory Matters
Governmental Regulation and Other Matters
Our medical device products are subject to extensive, complex and increasing oversight and regulation by the FDA, and other domestic and foreign governmental authorities. Our manufacturing facility, and those of our suppliers, are subject to periodic inspections to verify compliance with current FDA and other governmental regulatory requirements. If it were determined that we were not in compliance with these laws and regulations, we could be subject to criminal or civil liability, or both, and other material adverse effects. We have compliance programs in place to support and monitor compliance with these laws and regulations. All of our products and facilities and those of our suppliers are subject to drug and medical device laws and regulations promulgated by the FDA and national and supranational regulatory authorities outside the U.S., including, for example, Health Canada’s Health Products and Foods Branch, the U.K.’s Medicines and Healthcare Products Regulatory Agency, and Australia’s Therapeutic Goods Agency. These authorities regulate a range of activities including, among other matters, manufacturing, post-marketing studies in humans, advertising and promotion, product labeling, post-marketing surveillance and adverse event reporting.
Regulation of Medical Devices in the United States
The development, manufacture, sale and distribution of our medical device products are subject to comprehensive governmental regulation. Most notably, all of our medical devices sold in the United States are subject to the Food, Drug, and Cosmetic Act of 1938, as amended (“FDC Act”), as implemented and enforced by the FDA. The FDA, and in some cases other government agencies, such as the U.S. Federal Communications Commission (“FCC”), administer requirements covering the design, testing, safety, effectiveness, manufacturing, labeling, promotion and advertising, distribution and post-market surveillance of our products.
Unless an exemption applies, each medical device that we market must first receive either premarket notification clearance (by making what is commonly called “a 510(k) submission”) or premarket approval (by filing a premarket approval application (“PMA”) from the FDA pursuant to the FDC Act. In addition, certain modifications made to marketed devices also may require 510(k) clearance or approval of a PMA supplement. The FDA’s 510(k) clearance process varies in length and can extend beyond twelve months. The process of obtaining PMA approval is much more costly, lengthy and uncertain. It generally takes from two to three years or even longer. All of our current products that are available in the U.S. were originally cleared through the 510(k) process. We cannot be sure that future products or modifications of current products, will qualify for the 510(k) pathway or whether 510(k) clearance or PMA approval will be obtained for any future product that we propose to market.
In December 2014, the FDA issued guidance entitled “Infusion Pumps Total Product Life Cycle.” This guidance established substantial additional pre-market requirements for new and modified infusion pumps. Through this guidance, the FDA indicated more data demonstrating product safety will be required for future 510(k) submissions for infusion pumps, including the potential for more clinical and human factors data. The impact of this guidance is likely to result in a more time consuming and costly process to obtain regulatory clearance to market infusion pumps. In addition, new requirements beyond the 2014 guidance document could result in longer delays for the clearance of new products, modification of existing infusion pump products or remediation of existing products in the market. Future delays in the receipt of, or failure to obtain, approvals could result in delayed or no realization of product revenues.
After a device is placed on the market, numerous regulatory requirements continue to apply. Those regulatory requirements include the following: product listing and establishment registration; adherence to the Quality System Regulation (“QSR”), which requires stringent design, testing, control, documentation and other quality assurance procedures; labeling requirements and FDA prohibitions against the promotion of off-label uses or indications; adverse event reporting; post-approval restrictions or conditions, including post-approval study commitments; post-market surveillance requirements; the FDA’s recall authority, whereby it can ask for, or require, the recall of products from the market; and requirements relating to voluntary corrections or removals.
All aspects of our manufacturing and distribution of regulated products and those of our suppliers are subject to substantial governmental oversight. Facilities used for the production, packaging, labeling, storage and distribution of