UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
| x | ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
For the Fiscal Year Ended December 31, 2012
or
| ¨ | TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
Commission File Number 001-33103
CADENCE PHARMACEUTICALS, INC.
(Exact name of registrant as specified in its charter)
| | |
| Delaware | | 41-2142317 |
(State or other jurisdiction of incorporation or organization) | | (I.R.S. Employer Identification No.) |
12481 High Bluff Drive, Suite 200
San Diego, California 92130
(858) 436-1400
(Address, including zip code, and telephone number, including area code, of principal executive offices)
Securities registered pursuant to Section 12(b) of the Act:
| | |
| Common Stock, $0.0001 par value per share | | NASDAQ Global Market |
| (Title of class) | | (Name of exchange on which registered) |
Securities registered pursuant to Section 12(g) of the Act: None
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes ¨ No x
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes ¨ No x
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes x No ¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§ 232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yes x No ¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of the registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. x
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer or a non-accelerated filer. See the definitions of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| | | | | | |
Large accelerated filer ¨ | | Accelerated filer x | | Non-accelerated filer ¨ | | Smaller reporting company ¨ |
| | | | (Do not check if a smaller reporting company) | | |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act). Yes ¨ No x
The aggregate market value of the voting common stock held by non-affiliates of the registrant, based upon the closing sale price of the common stock on June 29, 2012, the last business day of the Registrant’s second fiscal quarter, reported on the NASDAQ Global Market, was approximately $115,000,000. Shares of common stock held by each executive officer and director and by each person who owns 10% or more of the Registrant’s outstanding common stock have been excluded from this computation. The determination of affiliate status for this purpose is not necessarily a conclusive determination for other purposes. The Registrant does not have any non-voting common equity securities.
As of February 28, 2013, there were 85,668,668 shares of the Registrant’s common stock outstanding.
DOCUMENTS INCORPORATED BY REFERENCE
Part III incorporates certain information by reference from the Registrant’s Definitive Proxy Statement to be filed with the Commission pursuant to Regulation 14A in connection with the Registrant’s 2013 Annual Meeting of Stockholders, which is scheduled to be held on June 12, 2013. Such Definitive Proxy Statement will be filed with the Commission not later than 120 days after the conclusion of the Registrant’s fiscal year ended December 31, 2012.
Forward-Looking Statements
You should read the following together with the more detailed information regarding our company, our common stock and our financial statements and notes to those statements appearing elsewhere in this document or incorporated by reference. The Securities and Exchange Commission, or SEC, allows us to “incorporate by reference” information that we file with the SEC, which means that we can disclose important information to you by referring you to those documents. The information incorporated by reference is considered to be part of this report.
This report and the information incorporated herein by reference contain forward-looking statements that involve a number of risks and uncertainties. Although our forward-looking statements reflect the good faith judgment of our management, these statements can only be based on facts and factors currently known by us. Consequently, these forward-looking statements are inherently subject to risks and uncertainties, and actual results and outcomes may differ materially from results and outcomes discussed in the forward-looking statements.
Forward-looking statements can be identified by the use of forward-looking words such as “believes,” “expects,” “hopes,” “may,” “will,” “plan,” “intends,” “estimates,” “could,” “should,” “would,” “continue,” “seeks,” “pro forma,” or “anticipates,” or other similar words (including their use in the negative), or by discussions of future matters such as the development of new products, technology enhancements, possible changes in legislation or the regulations that impact our business and other statements that are not historical. These statements include but are not limited to statements under the captions “Business,” “Risk Factors” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” as well as other sections in this report. You should be aware that the occurrence of any of the events discussed under the caption “Risk Factors” and elsewhere in this report could substantially harm our business, results of operations and financial condition and that if any of these events occurs, the trading price of our common stock could decline and you could lose all or a part of the value of your shares of our common stock.
The cautionary statements made in this report are intended to be applicable to all related forward-looking statements wherever they may appear in this report. We urge you not to place undue reliance on these forward-looking statements, which speak only as of the date of this report. Except as required by law, we assume no obligation to update our forward-looking statements, even if new information becomes available in the future.
CADENCE PHARMACEUTICALS, INC.
Annual Report on Form 10-K
For the Fiscal Year Ended December 31, 2012
Table of Contents
PART I
Company Overview
We are a biopharmaceutical company focused on acquiring, in-licensing, developing and commercializing proprietary products principally for use in the hospital setting. We currently have rights to one product, OFIRMEV® (acetaminophen) injection, a proprietary intravenous, or IV, formulation of acetaminophen. We in-licensed the exclusive United States, or U.S., and Canadian rights to OFIRMEV from Bristol-Myers Squibb Company, or BMS, which sells intravenous acetaminophen in Europe and other markets for the treatment of acute pain and fever under the brand name Perfalgan®. In November 2010, the U.S. Food and Drug Administration, or FDA, granted marketing approval for OFIRMEV, which is indicated for the management of mild to moderate pain, the management of moderate to severe pain with adjunctive opioid analgesics and the reduction of fever in adults and children two years of age and older. We launched commercial sales of OFIRMEV in the U.S. in January 2011.
We believe that OFIRMEV fills significant unmet medical needs and that the hospital pharmaceuticals market is both concentrated and underserved. We have established a hospital-focused sales force to promote OFIRMEV to this market, along with any other products we may acquire in the future. We intend to build a leading franchise in the hospital setting, continuing to focus on differentiated products with significant unmet commercial potential that are complementary to OFIRMEV and enable us to effectively leverage our commercial infrastructure.
We were incorporated under the laws of the State of Delaware in May 2004. Our principal executive offices are located at 12481 High Bluff Drive, Suite 200, San Diego, California 92130 and our telephone number is (858) 436-1400. Information about the company is also available at our website at www.cadencepharm.com, which includes links to reports we have filed with the Securities and Exchange Commission, or SEC, which are available free of charge. The contents of our website are not incorporated by reference in this Annual Report on Form 10-K. The public may read and copy any materials that we file with the SEC at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. Information on the operation of the Public Reference Room is available by calling the SEC at 1-800-SEC-0330. These reports may also be accessed free of charge via the SEC’s website at www.sec.gov.
We own or have rights to various trademarks, copyrights and tradenames used in our business, including the following: Cadence®, OFIRMEV® and the OFIRMEV logo. This report also contains trademarks of others, including Caldolor®, EXPAREL®, IONSYSTM, Percocet®, Perfalgan®, Toradol®, Tylenol®, Tylenol Codeine #3 McNeil®, Ultram®, and Vicodin®.
Our Business Strategy
Our goal is to become a leading biopharmaceutical company focused on the acquisition, in-licensing, development and commercialization of proprietary products principally for use in the hospital setting. Our near-term strategy is to continue to work directly with physicians and hospitals to increase demand for OFIRMEV. Longer-term, our strategy is to acquire, in-license, develop and commercialize differentiated products with significant unmet commercial potential that are complementary to OFIRMEV and enable us to effectively leverage our commercial infrastructure. Specifically, we intend to:
| | • | | Successfully expand the sales of OFIRMEV.We continue to work to accelerate demand for OFIRMEV by seeking to increase the number of new customers for the product, as well as the frequency and size of our customers’ orders, and ensuring that OFIRMEV is readily available at the point of care. Our sales force is well equipped with promotional materials that are designed to expand the utilization of |
1
| | the product as part of a multimodal approach to safe and effective pain management in an increasing variety of surgical procedures. Our medical science liaisons inform and educate hospital-based physicians and other healthcare professionals on the importance of improving the quality of pain management. |
| | • | | Focus our highly leverageable sales organization on high-potential hospitals.In the near term, our promotional efforts are focused on surgeons, physician assistants, nurse practitioners and other healthcare professionals at the approximately 1,800 to 1,900 strategic hospitals and integrated delivery networks we have targeted throughout the U.S. that account for a substantial portion of prescribing activity. We believe the concentrated nature of this market creates the opportunity for significant marketing synergies, and we intend to ultimately leverage our sales force with multiple products across multiple therapeutic categories in the hospital. Outside the U.S., we intend to establish strategic partnerships for the commercialization of any products we may acquire or in-license in the future in areas where we have commercialization rights. |
| | • | | Expand our product portfolio by acquiring or in-licensing additional commercial hospital-focused products with well-understood risk profiles.We continue to seek opportunities to acquire or in-license products to continue to exploit our commercial and development capabilities. We believe that our focus on the hospital market enables us to evaluate a broad range of products across multiple therapeutic areas for possible acquisition. To reduce the time to market and the risks and costs of clinical development, we will continue to focus on differentiated products with significant unmet commercial potential that are complementary to OFIRMEV and enable us to effectively leverage our commercial infrastructure. In addition, we will also consider strategically attractive opportunities to co-promote commercial hospital products that would be complementary to our existing business or that otherwise have attractive potential. |
| | • | | Pursue additional indications and commercial opportunities for OFIRMEV and future products. We will seek to maximize the value of OFIRMEV and any other products we may acquire, in-license or develop. These activities may include pursuing additional indications and commercial opportunities for OFIRMEV and any other products we may acquire. |
The U.S. Hospital Market
Large, multinational pharmaceutical companies have generally decreased marketing efforts focused on hospital-use drugs, instead focusing on drugs that can be marketed in the larger outpatient setting. We believe this reduced emphasis on the hospital marketplace presents us with an excellent opportunity to market products that address unmet medical needs in the hospital setting. We believe the concentrated nature of the hospital marketplace will allow for our expansion into other therapeutic areas without substantial investment in additional commercial infrastructure.
U.S. hospitals accounted for approximately $56 billion or 14% of U.S. pharmaceutical sales in 2012, according to Symphony Health Solutions, an independent marketing research firm. Furthermore, we believe pharmaceutical sales to acute care hospitals are highly concentrated among a relatively small number of large institutions. For example, 1,800 to 1,900 of the approximately 7,000 acute care hospitals in the U.S. represent approximately 80% of hospital injectable analgesic sales. The concentration of high-prescribing institutions enables effective promotion of pharmaceuticals utilizing a relatively small, dedicated sales and marketing organization. We believe the relative lack of promotional efforts directed toward the highly concentrated hospital marketplace makes it an underserved and compelling opportunity, especially for a biopharmaceutical company commercializing its products directly through its own dedicated sales force.
We believe a typical sales representative focused on office-based physicians can generally promote only two to three products effectively; whereas, a typical hospital-focused sales representative can effectively promote five to six products. Furthermore, we believe a typical sales representative focused on office-based physicians can effectively reach five to seven physicians per day; whereas, a typical hospital-focused sales representative can
2
reach many more physicians, nurses and pharmacy directors within a given institution. Notably, a hospital-focused sales representative also faces significantly less travel time between sales calls and less wait time in physician offices as a large number of prescribers can be found in a single location. Thus, a single sales representative can effectively promote products from multiple therapeutic categories to multiple prescribers within the institution. Furthermore, drug sampling generally does not occur in hospitals, which represents a significant cost advantage versus marketing to office-based physicians.
Intravenous Acetaminophen and U.S. Market Opportunity
Prior to our commercial launch of OFIRMEV in January 2011, the U.S. IV analgesic therapy market consisted of opioids, such as morphine, meperidine, hydromorphone and fentanyl, and two NSAIDs. These two NSAIDs, Toradol (ketorolac tromethamine), a generic form of which is also available in the U.S. from a number of manufacturers, and Caldolor (ibuprofen), represented the only non-opioid IV analgesics available for treating acute pain in adults in the U.S. prior to OFIRMEV. According to Symphony Health Solutions, approximately 240 million vials of injectable analgesics were sold in the U.S. in 2012. The price of ketorolac in the U.S. in 1997, prior to the entry of generic competitors, was approximately $7.00 (U.S. dollars) per vial, according to the American Journal of Health-System Pharmacy. The price of Caldolor in the U.S. was $10.50 (U.S. dollars) per 800 mg vial in 2010. The list price, or wholesale acquisition cost, of OFIRMEV as of January 3, 2013, was $12.43 per vial. We have signed agreements with all of the major group purchasing organizations to provide services and discounted pricing. Our pricing strategy is intended to allow hospitals to access OFIRMEV at a fair price while facilitating prompt formulary adoption at many institutions.
We believe that the key product attributes that will drive the adoption of OFIRMEV in the U.S. include the efficacy and safety profile of the product, as demonstrated in multiple clinical studies, the established safety profile and familiarity physicians have with oral acetaminophen, alone and in combination with opioids, the potential for reducing concomitant use of morphine and other opioids, improved patient satisfaction, and the desire for a dosage form for patients unable to take medication orally.
Marketed Product
OFIRMEV Product Overview
The FDA approved OFIRMEV (acetaminophen) injection in November 2010 for the management of mild to moderate pain, the management of moderate to severe pain with adjunctive opioid analgesics and the reduction of fever in adults and children two years of age and older.
In its oral form, acetaminophen is the most widely used drug for the treatment of pain and fever in the U.S. Acetaminophen was discovered in the late 19th century and was made available for sale in 1955, when it was introduced in the U.S. under the brand name Tylenol. Acetaminophen is currently available in over 600 combination and single-ingredient prescription and over-the-counter medicines, including tablet, caplet, orally-dosed liquid suspension, powder and suppository forms for both adults and children. Despite the broad usage of acetaminophen, prior to the commercial launch of OFIRMEV in January 2011, there was no intravenous formulation available in the U.S. for patients who have pain or fever warranting an intravenous formulation, were unable to take medications by other routes, required faster onset of pain relief or fever reduction, or for whom it was otherwise more convenient to receive an injectable analgesic.
Since 2002, our licensor, BMS, has marketed this proprietary intravenous formulation of acetaminophen for the treatment of acute pain and fever in Europe and several other markets outside the U.S., where it is known as paracetamol and marketed under the brand name Perfalgan. We in-licensed the exclusive U.S. and Canadian rights to OFIRMEV from BMS in March 2006.
3
Pain Management
Despite major improvements in surgical techniques and the introduction of novel drugs, the overall treatment of post-operative pain has not substantially improved over the last 20 years. According to the industry research group Datamonitor, up to 75% of patients report inadequate pain relief after surgery. Inadequate treatment of pain may lead to a variety of symptoms, including anxiety, depression, insomnia, fatigue, decreased appetite, nausea and vomiting. Decreased mobilization may also result from the inadequate treatment of pain, which may increase the risk of deep venous thrombosis, reduced lung tidal volume, and partial collapse or incomplete inflation of the lungs, as well as potentially prolonging hospital stays. All of these factors have the potential to significantly impact patient care and create additional costs for hospitals.
Drugs used to treat pain are collectively known as analgesics. Injectable formulations of analgesics are typically used when patients are unable to take medications by mouth, would benefit from a faster onset or more potent forms of analgesia, when other administration routes are contraindicated, or when it is more convenient to administer drugs in injectable form. Hospitalized patients may be unable to take medications by mouth for a variety of reasons, including gastric or intestinal dysfunction, pre-operative or pre-procedural restrictions, sedation, mental status changes or neurological conditions that increase the risk of aspiration, nausea or vomiting, or as a result of conditions that make swallowing painful, such as oral or esophageal infections, inflammation or ulceration. Additionally, absorption of oral analgesics may be compromised following surgery due to factors such as delays in gastric function and opioid-related increased pyloric tone. As a result, published clinical studies have shown that dosing with IV acetaminophen provides higher plasma and cerebrospinal fluid concentrations of the drug than oral administration. Prior to the approval of OFIRMEV, only two classes of injectable analgesics, opioids and non-steroidal anti-inflammatory drugs, or NSAIDs, were available in the U.S. for the treatment of pain.
Opioids have been used as analgesics for over 2,000 years and continue to be the mainstay of post-operative pain management. Opioids interact with certain receptors in the central and peripheral nervous system to produce beneficial effects, which include analgesia, sedation and euphoria. A range of naturally occurring, semi-synthetic and synthetic opioids are available for intravenous use, including morphine, fentanyl, hydromorphone, meperidine, sufentanil, and alfentanil. Opioids, however, may also be associated with a variety of unwanted side effects when used to treat acute pain, including respiratory depression, excessive sedation, nausea, vomiting, constipation, urinary retention, itchiness, chest wall rigidity, cognitive impairment, and seizures. Respiratory depression may lead to death if not monitored closely. Side effects from opioids have been demonstrated to reduce patients’ quality of life. Opioid use may prolong a patient’s stay in the post-anesthesia care unit or ambulatory surgical facility, as well as a patient’s overall length of stay in the hospital, as a result of opioid side effects and the need to administer additional medications or treatments to resolve opioid side effects. Studies have demonstrated that hospital costs may be increased by opioid use, not only due to additional personnel time required to handle and dispose of these controlled substances, but also as a result of costs associated with treating opioid-related side effects, including the potential need for the patient to remain in the hospital for an extended period of time.
Other than OFIRMEV, the only non-opioid intravenous analgesics currently available in the U.S. are the NSAIDs Toradol (ketorolac tromethamine), a generic form of which is also available in the U.S. from a number of manufacturers, and Caldolor (ibuprofen), which was approved by the FDA in mid-2009 for the treatment of mild to moderate pain in adults, and moderate to severe pain in adults as an adjunct to opioid therapy, and reduction of fever. Caldolor is not approved for use in children under 17 years of age, and ketorolac is only approved for use as a single dose in children greater than two years of age. NSAIDs act as non-selective inhibitors of the enzyme cyclooxygenase, inhibiting both the cyclooxygenase-1, or COX-1, and cyclooxygenase-2, or COX-2, enzymes. Since NSAIDs do not produce respiratory depression or impair gastrointestinal motility, they are considered to be useful alternatives or adjuncts to opioids for the relief of acute pain.
However, the use of NSAIDs is limited in the post-operative period due to their potential to cause adverse effects. NSAIDs such as ketorolac and ibuprofen exert a direct inhibitory effect on platelet aggregation, which
4
could result in increased bleeding susceptibility in the post-operative setting. NSAIDs are often avoided in surgical patients because they may be associated with renal toxicity, particularly in the elderly and patients with compromised renal function or hypoperfusion. NSAIDs may also be associated with gastric irritation and gastric bleeding, and an increased incidence of cardiovascular adverse events has been found to be associated with postoperative use of certain NSAIDs. All NSAIDs carry a boxed warning for a number of side effects. A boxed warning is the strongest type of warning that the FDA can require for a drug and is generally reserved for situations where prescribers should be aware of the potential for adverse drug reactions that can cause serious injury or death.
Multimodal Pain Management
Multimodal analgesia is the use of two or more analgesic agents that act by different mechanisms to provide superior analgesic efficacy with equivalent or reduced adverse effects. The Practice Guidelines for Acute Pain Management in the Perioperative Setting from the American Society of Anesthesiologists, or ASA, recommend that multimodal pain management therapy should be employed whenever possible. The ASA guidelines recommend that all surgical patients receive an around-the-clock regimen of acetaminophen, NSAIDs, or COX-2 inhibitors, and that dosing regimens should be administered to optimize efficacy while minimizing the risk of adverse events. The only intravenous NSAIDs approved in the U.S., Caldolor (ibuprofen), Toradol (ketorolac tromethamine), and generic ketorolac, all carry a boxed warning for the risk of bleeding, renal dysfunction, and other adverse effects.
In August 2012, The Joint Commission, an independent, not-for-profit organization that accredits and certifies more than 20,000 health care organizations and programs in the U.S., issued Sentinel Event Alert No. 49, on the safe use of opioids in hospitals. This publication advised hospitals to take specific pharmacologic and non-pharmacologic measures to reduce the incidence of serious complications that are often associated with opioid use. Specifically, this publication endorsed a multimodal approach to pain management, and recommended the use of non-narcotic analgesics, such as acetaminophen, NSAIDs, antidepressants, anticonvulsants and muscle relaxants, to reduce opioid use, particularly in patients at higher risk for over sedation and respiratory depression.
The concept of using acetaminophen for multimodal management of pain to improve pain relief and reduce opioid consumption is not new to physicians. In fact, oral acetaminophen-opioid combination products are very commonly prescribed for the treatment of acute pain, including post-operative pain. Such products include Vicodin (hydrocodone plus acetaminophen), Percocet (oxycodone plus acetaminophen), Tylenol Codeine #3 McNeil (codeine plus acetaminophen), and Ultram (tramadol plus acetaminophen). Approximately 73% of the 14.4 billion doses of oral opioids sold in the U.S. in 2008 were combination products that included acetaminophen. As the only IV formulation of acetaminophen available in the U.S., OFIRMEV provides the only option to extend this common multimodal approach to the perioperative setting when patients are unable to take oral medications.
Fever Reduction
Fever is an increase in internal body temperature above its average normal value due to an increased temperature regulatory set-point. A significant fever is usually defined as an oral temperature of greater than 100.4 degrees Fahrenheit (38 degrees Centigrade). Fever is typically a sign of the body’s response to an underlying infection, disease process or allergic reaction. Very high fevers may cause hallucinations, confusion, irritability, convulsions or death.
Hospitalized patients are at especially high risk for developing fever due to the prevalence of infections, whether community- or hospital-acquired, and as a result of invasive procedures and treatments that may cause fevers. Surgery is the most common predisposing factor for fever in the hospital setting, with published incidence rates ranging from 14% to 91% of post-operative patients. Aside from the body’s reaction to surgical trauma,
5
infections such as surgical wound infections, urinary tract infections, and pneumonia are the most common causes of post-operative fevers. However, deep venous thrombosis, pulmonary emboli, myocardial infarction, transfusions of blood products, and medications are also important potential causes of post-operative fever. Many patients also enter hospitals and emergency rooms with fevers that are caused by infections or complications from an underlying disease or medical condition. While the origin of a fever is often unknown, treatment to reduce fever will typically be given even if the cause cannot be determined.
Fever is also the most common reason parents bring their children to hospital emergency rooms. Pediatric fever is particularly worrisome, as approximately 4% of children under age five and nearly one in five children born prematurely experience fever-induced seizures, or febrile seizures. The signs of febrile seizures, which occur when a child’s temperature rises or falls rapidly, include loss of consciousness and convulsions.
Acetaminophen, ibuprofen and aspirin are the most commonly used oral medications to treat fever. Caldolor (intravenous ibuprofen) is not approved for treating fever or pain in children under 17 years of age. Aspirin has been reported to be associated with Reye’s syndrome, a potentially fatal disease, in children and teenagers with viral infections.
Treating fever in a hospitalized patient with oral medication may be difficult or infeasible due to the severe nausea and vomiting that often accompany a high fever, or because the patient is unconscious, sedated, fasting or experiencing gastrointestinal dysfunction. Oral medications are also precluded in patients on a restricted oral intake regimen due to a concomitant medical condition or upcoming medical procedure. In the U.S., OFIRMEV is the only available intravenous form of acetaminophen, and aspirin is currently not available in intravenous dosage forms. While rectal delivery of these medications is sometimes possible, drug absorption using this method is highly variable, resulting in the potential for subtherapeutic dosing. Rectal delivery is further complicated if the drug is expelled with a bowel movement, which leads to difficulty determining the amount of medication delivered.
Therapeutic drug levels often may be achieved more rapidly when a drug is administered intravenously compared to oral or rectal administration, offering the potential advantage of a more rapid onset of action. This may be particularly desirable in patients with high fever, or in whom fever is causing undesirable symptoms or complications such as febrile seizures. It may also be more convenient to administer medications in an intravenous dosage form, particularly for patients who currently have an intravenous line in place. We believe that the availability of OFIRMEV in the U.S. offers a significant treatment option for hospitalized patients with fever and addresses unmet medical needs, particularly with respect to the management of fever in children two years of age and older.
Clinical Development
In November 2010, the FDA granted marketing approval for OFIRMEV. OFIRMEV is indicated for the management of mild to moderate pain, the management of moderate to severe pain with adjunctive opioid analgesics and the reduction of fever in adults and children two years of age and older. We submitted our new drug application, or NDA, for OFIRMEV under Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act. This approach allows at least some of the information required for approval to come from studies not conducted by or for the applicant, and for which the applicant has not obtained a right of reference. Supportive information may also include scientific literature and publicly available information contained in the labeling of other medications. Accordingly, the NDA we submitted for OFIRMEV included data from our own clinical trials in the U.S., trials of IV acetaminophen previously completed by BMS in the U.S. and Europe, and other studies published in the scientific and medical literature.
The approval of OFIRMEV was supported by the results of 20 clinical trials involving 1,375 patients. Procedure types included, but were not limited to, orthopedic surgery (including total hip or knee replacement), gynecologic surgery, general surgery, ear, nose, and throat surgery, and cardiothoracic surgery. Across this
6
clinical data set, IV acetaminophen showed a significant and reproducible benefit in analgesia as measured by a variety of endpoints relating to pain relief or reduction in pain intensity. Importantly, several studies demonstrated that including IV acetaminophen in the analgesic regimen resulted in significant reductions in opioid consumption. The clinical benefit of reduced opioid consumption was not demonstrated. There are three pivotal clinical trials that supported our NDA for OFIRMEV and are currently included in the OFIRMEV prescribing information.
Adult Pain Study 1, RC 210 3 002 / Sinatra Study (BMS)
This was a phase III, randomized, double-blind, placebo-controlled, multicenter study that evaluated the analgesic efficacy and safety of single and repeated doses of OFIRMEV 1 g in comparison with placebo in 101 patients experiencing moderate to severe pain following total hip or knee replacement. Patients were allowed rescue medication with patient-controlled analgesia, or PCA, morphine.
| | • | | Pain Relief and Pain Intensity.In a 6-hour, single-dose evaluation period, OFIRMEV 1 g + PCA morphine demonstrated superior pain relief vs. placebo + PCA morphine (15 minutes through 6 hours, P<0.05). In a repeated-dose evaluation period, OFIRMEV 1 g delivered Q6h for 24 hours showed a greater reduction in pain intensity over 24 hours (SPID24) compared to placebo (P<0.001). |
| | • | | Morphine Consumption.OFIRMEV 1 g + PCA morphine significantly reduced morphine consumption vs. placebo + PCA morphine alone (–46% after first dose over 6 hours, P<0.01; –33% over 24 hours, P<0.01). Median time to first rescue medication was significantly longer with OFIRMEV 1 g compared with placebo (3 hours vs. 0.8 hours, P=0.0001). The clinical benefit of reduced opioid consumption was not demonstrated. |
| | • | | Patient Satisfaction.Patients’ global evaluation of study treatment (excellent plus good scores) significantly favored the OFIRMEV group over PCA morphine alone (40.8% vs. 23.1%, P=0.004). There were no differences between OFIRMEV and placebo groups in incidence of adverse events. No serious hepatic events were related to treatment with OFIRMEV 1 g. The most common adverse reactions in patients treated with OFIRMEV were nausea, vomiting, headache, and insomnia in adult patients. |
Adult Pain Study 2, Cadence Study 304
This was a phase III, randomized, double-blind, placebo-controlled, multicenter, parallel-group, repeated-dose study of the analgesic efficacy and safety of OFIRMEV vs. placebo for the treatment of postoperative pain after abdominal laparoscopic surgery. A total of 244 patients received OFIRMEV 1 g or placebo Q6h, or OFIRMEV 650 mg or placebo Q4h. Opioid rescue medication was available to all patients.
| | • | | Pain Intensity.A significantly greater reduction in pain intensity differences from baseline was seen with OFIRMEV 1 g compared to the combined placebo group over the 24-hour period (P=0.0068). Time to meaningful pain relief after the first dose was significantly shorter in subjects who received OFIRMEV 1 g compared to the matched placebo group, with median values of 24.9 minutes and 53.9 minutes, respectively (P=0.0028). Similarly, there was a significant difference in pain intensity differences from baseline seen with OFIRMEV 650 mg compared with the combined placebo group over 24 hours (P=0.0183). |
| | • | | Morphine Consumption.No statistical differences were found between OFIRMEV 1 g or 650 mg and the combined placebo groups in total rescue medication consumption or in the first time to rescue medication. |
| | • | | Patient Satisfaction.Patient global evaluation of study treatment (excellent plus good scores) significantly favored OFIRMEV 1 g over the control group (P=0.0004). |
7
Adult Fever Study 1, Cadence Study 302
This was a phase III, randomized, double-blind, placebo-controlled, single-center study that evaluated the antipyretic efficacy and safety of a single dose of OFIRMEV 1 g compared with placebo in 60 healthy adult males who developed fever induced by a standard dose of endotoxin.
| | • | | Antipyretic Efficacy.OFIRMEV 1 g was shown to be effective in blunting the peak temperature produced by endotoxin and reducing the fever it produced for a period of up to 6 hours. The weighted sum of temperature differences over 6 hours (primary endpoint) was significantly better for OFIRMEV 1 g vs. placebo (P=0.0001). Importantly, OFIRMEV 1 g demonstrated a rapid onset of action and showed statistically significant temperature differences from baseline vs. placebo at T30 minutes (15 minutes after completing the infusion) (P=0.0085). Statistically significant reductions in temperature at each time point from 30 minutes through 5.5 hours were also observed for subjects who received OFIRMEV 1 g vs. placebo. |
Post-Approval Commitments
In accordance with a Pediatric Research Equity Act requirement included in the NDA approval of OFIRMEV, we began enrolling patients in the third quarter of 2012 in a post-marketing efficacy study of OFIRMEV in infants and neonates. In addition, we plan to use the data from this study to satisfy a formal written request from the FDA under Section 505A of the U.S. Food, Drug and Cosmetic Act that was made as part of the approval process for OFIRMEV. The FDA has agreed to a due date for completion of this study in August 2015. Upon timely completion and the acceptance by the FDA of the data from this study, OFIRMEV will be eligible for an additional six months of marketing exclusivity in the U.S.
Competition
The pharmaceutical industry is subject to intense competition and characterized by extensive research efforts and rapid technological progress. Competition in our industry occurs on a variety of fronts, including developing and bringing new products to market before others, developing new technologies to improve existing products, developing new products to provide the same benefits as existing products at lower cost and developing new products to provide benefits superior to those of existing products. There are many companies, including generic manufacturers as well as large pharmaceutical companies, that have significantly greater financial and other resources than we do, as well as academic and other research institutions that are engaged in research and development efforts for the indications targeted by our product.
A variety of competitive products from two main drug classes, opioids and NSAIDs, are currently available in the market for treatment of pain and fever in hospitalized patients, including:
Injectable Opioids
| | • | | morphine, the leading product for the treatment of acute post-operative pain, a generic version of which is available from several manufacturers; and |
| | • | | other injectable opioids, including fentanyl, meperidine and hydromorphone, each of which is available generically from several manufacturers. |
Injectable NSAIDs
| | • | | Toradol (ketorolac tromethamine), an injectable NSAID, a generic version of which is available from several manufacturers; and |
| | • | | Caldolor (ibuprofen), another injectable NSAID. |
8
In addition, Exparel, a long-acting, controlled release, local analgesic formulation of bupivacaine, was approved by the FDA in October 2011 as a single intraoperative injection given at the close of surgery. Exparel’s manufacturer, Pacira Pharmaceuticals, Inc., has indicated that it is exploring additional indications for this product, which include regional anesthetic techniques.
Product Candidates
Competitors may seek to develop alternative formulations of intravenous acetaminophen for our targeted indications that do not directly infringe on our in-licensed patent rights. We are aware of several third-party U.S. and Canadian patents and patent applications directed to various potential injectable formulations of acetaminophen, including intravenous formulations, as well as methods of making and using these potential formulations. We are also aware of a number of product candidates in development to treat acute pain, including injectable NSAIDs, novel opioids, new formulations of currently available opioids, long-acting local anesthetics and new chemical entities as well as alternative delivery forms of various opioids, COX2 inhibitors, and NSAIDs. A variety of pharmaceutical and biotechnology companies are developing these new product candidates, including but not limited to Acusphere, Inc., Cara Therapeutics, Inc., Durect Corporation, Hospira, Inc., NeurogesX, Inc., Pacira Pharmaceuticals, Inc., Paion AG, QRx Pharma Limited, Relmada Therapeutics, Inc., Teva Pharmaceutical Industries Ltd., and The Medicines Company.
Generic IV Acetaminophen
In connection with a settlement and license agreements entered into in November 2012, Perrigo Company, or Perrigo, has been granted the exclusive right of first refusal to negotiate an agreement with us to market an authorized generic version of OFIRMEV in the U.S., in the event that we elect to launch an authorized generic version of the product. The license agreement also provides that, if we enter into an agreement for Perrigo to market an authorized generic version of OFIRMEV, during the license period, Perrigo would purchase the product exclusively from us. We would receive product costs plus an administrative fee, as well as a royalty payment based on the net profits achieved by Perrigo from the sale of the authorized generic product. Additionally, we have granted Perrigo the non-exclusive right to market a generic intravenous acetaminophen product in the U.S. under Perrigo’s Abbreviated New Drug Application, or ANDA, after December 6, 2020, or earlier under certain circumstances. The Federal Trade Commission, or FTC, or the Department of Justice, or DOJ, could seek to challenge our settlement with Perrigo, or a competitor, customer or other third-party could initiate a private action under antitrust or other laws challenging our settlement with Perrigo. Additional information about the intellectual property for OFIRMEV, including the Perrigo settlement agreement and ongoing intellectual property litigation, is set forth below under the heading “Business – Intellectual Property – OFIRMEV and Pending Litigation.”
Sales and Marketing
We have established a sales force of hospital sales specialists that is supported by an experienced commercial management, marketing and sales operations team. Additionally, our field-based medical science liaisons inform and educate hospital-based physicians regarding the appropriate uses of OFIRMEV.
The primary target audience for OFIRMEV includes anesthesiologists and surgeons. Other targets include certified registered nurse anesthetists, emergency medicine physicians, intensivists, internists, hospitalists, obstetricians and other physicians throughout the hospital, as well as hospital-based pharmacists. Our commercial sales force is focused on reaching the top 1,800 to 1,900 U.S. hospitals, which we believe represent approximately 80% of the market opportunity for OFIRMEV.
We believe that our sales force is differentiated by its level of experience and background in the industry. Our sales management team has an average of more than 15 years of pharmaceutical industry experience, and an average of more than eight years of hospital sales management experience. We require that our sales
9
representatives complete a comprehensive training program focused on our product, therapeutic area, competitive products, sales techniques and compliance with applicable laws and regulations. This training program includes field-based learning to provide our representatives with a comprehensive understanding and perspective on the unmet medical needs in the management of pain and fever in adults and children and how OFIRMEV addresses those needs.
Field-based regional business directors and district sales managers provide oversight for our hospital sales specialists and direct our efforts to provide hospital customers with the information needed to obtain formulary approval for, and increase utilization of, OFIRMEV. Because our clinical studies of OFIRMEV have been conducted across a wide range of surgical procedures, we believe that providing access to this data and the unique characteristics of OFIRMEV assists physicians in using OFIRMEV safely and effectively. In addition to our hospital sales specialists, we also implement a variety of marketing programs to educate customers, including direct-to-physician promotional materials, peer-to-peer educational programs, medical journal advertising, and participation in targeted medical convention programs.
Business Relationships
Licensing Agreement with Bristol-Myers Squibb Company
In March 2006, we in-licensed from BMS the patents and the exclusive development and commercialization rights to OFIRMEV in the U.S. and Canada. BMS has sublicensed these rights to us under a license agreement with SCR Pharmatop S.A., or Pharmatop.
As consideration for the license, we paid a $25.0 million up-front fee and an additional $15.0 million fee in November 2010 after approval of the product. In addition, we may be required to make future milestone payments totaling up to $25.0 million upon the achievement of various milestones related to achievement of certain net sales levels of OFIRMEV. We are also obligated to pay a royalty on net sales of the product. We have the right to grant sublicenses to our affiliates.
The term of the OFIRMEV agreement generally extends on a country-by-country basis until the last licensed patent expires, which is expected to occur in the U.S. in 2021. Either party may terminate the OFIRMEV agreement upon delivery of written notice if the other party commits a material breach of its obligations and fails to remedy the breach within a specified period or upon the occurrence of specified bankruptcy, reorganization, liquidation or receivership proceedings. In addition, BMS may terminate the OFIRMEV agreement if we breach, in our capacity as a sublicensee, any provision of the agreement between BMS and Pharmatop. The OFIRMEV agreement will automatically terminate in the event of a termination of the license agreement between BMS and Pharmatop. We may terminate the OFIRMEV agreement at any time upon specified written notice to BMS after the occurrence of events of default that relate to our territory and would entitle BMS to terminate the Pharmatop license agreement. The events of default include Pharmatop’s inability to maintain specified claims under listed patents, the marketing by a third party of a parenterally-administered product containing acetaminophen, subject to certain conditions, or a successful third party action that deprives Pharmatop of its rights to specified patents. We may also terminate the OFIRMEV agreement upon specified written notice after an uncured failure by Pharmatop to perform any of its material obligations under the Pharmatop license agreement with respect to our territory that would permit BMS to terminate the Pharmatop license agreement.
Either BMS or Pharmatop may terminate the license agreement between them upon delivery of written notice after an uncured failure by the other party to perform any of its material obligations under the license agreement. BMS may generally terminate the agreement upon written notice to Pharmatop within a specified period so long as all payments due under the agreement to Pharmatop are current. Pharmatop may terminate the agreement upon specified written notice if BMS opposes any of the listed patent applications or challenges the validity or enforceability of any of the listed licensed patents. BMS is also entitled to terminate the Pharmatop agreement upon the occurrence of events of default that relate to the territory described above.
10
Amended and Restated Supply Agreement with Lawrence Laboratories, Inc.
In February 2013, we entered into an amended and restated supply agreement with Lawrence Laboratories, an operating division of Swords Laboratories, and a member of the BMS group of companies, which amended and restated our original agreement from December 2010, for the manufacture of commercial supplies of the finished drug product for OFIRMEV packaged in vials, for sale and distribution by us in the United States and Canada. Bristol-Myers Squibb Srl, or BMS Anagni, an indirect subsidiary of BMS located in Anagni, Italy, manufactures the product on behalf of Lawrence Laboratories. BMS Anagni is currently our sole supplier of OFIRMEV.
Pursuant to the terms of the this Agreement, we pay Lawrence Laboratories a set price for each unit of product purchased, based upon the aggregate quantity of product that we have specified that we intend to order during a calendar year, and whether Lawrence Laboratories has implemented certain agreed-upon manufacturing capacity increase improvements. We are obligated to purchase a minimum number of units each year, or pay Lawrence Laboratories an amount equal to the shortfall between the minimum purchase requirement and the number of units of product actually ordered during such year, multiplied by a pre-set amount that also varies depending upon whether Lawrence Laboratories has implemented certain agreed-upon manufacturing capacity increase improvements. We are obligated to purchase at least 75% of our annual product requirements from Lawrence Laboratories each contract year. The agreement also requires us to pay Lawrence Laboratories for additional services requested by us at a specified hourly rate and for any validation batches that we may require, not to exceed a specified rate. All amounts payable under the agreement will be paid in U.S. dollars.
The term of this agreement extends through December 31, 2018, unless extended by the mutual agreement of us and Lawrence Laboratories, unless the agreement is terminated sooner: (1) by the mutual agreement of the parties, (2) by either party for convenience following 24 months’ prior written notice of termination to the other party, (3) upon the termination of our license agreement for the product with BMS, or (4) upon our dissolution or termination, other than in connection with or following the assignment of this agreement. In addition, either party may terminate the agreement: (a) within 60 days, after written notice in the event of a material uncured breach of the agreement by the other party, or (b) immediately, if the other party becomes insolvent or admits in writing its inability to pay its debts as they become due, files a petition for bankruptcy, makes an assignment for the benefit of its creditors or has a receiver or other court officer appointed for its properties or assets.
If the agreement is terminated by us for our convenience or by Lawrence Laboratories due to our material breach of the agreement, we will reimburse Lawrence Laboratories for: (1) any product ordered under a firm order and received by us, and (2) any inventory of materials used to manufacture the product that are specific to the product and that Lawrence Laboratories is unable to reasonably utilize. Additionally, our minimum purchase requirement for the year in which the termination takes effect will be reduced proportionally, and we will not be required to fulfill the minimum purchase requirement for any subsequent contract year. If the agreement is terminated for any reason other than by us for our convenience or by Lawrence Laboratories due to our material breach of the agreement, we will not be required to reimburse Lawrence Laboratories for any inventory of materials used to manufacture the product, and we will have no obligation to purchase the minimum purchase requirement for the year in which the termination takes effect, or for any subsequent contract year.
Manufacturing and Supply Agreement with Laboratorios Grifols, S.A.
In March 2013, we entered into an agreement with Laboratorios Grifols, S.A., or Grifols, a division of Grifols, S.A., a global healthcare company headquartered in Barcelona, Spain, for the development, manufacture and supply of commercial quantities of OFIRMEV in flexible IV bags. Grifols has supplied IV acetaminophen in flexible plastic bags to BMS for distribution in certain markets outside of the U.S. and Canada since 2010. We plan to submit a supplemental NDA to the FDA in the second half of 2013 seeking approval of the product to be manufactured by Grifols.
11
Pursuant to the terms of the agreement, we will pay Grifols a set price for the OFIRMEV we purchase, which price may be adjusted annually by Grifols, subject to specified limitations. In addition, we will be obligated to pay Grifols a reservation fee, in lieu of any minimum purchase commitment, calculated by multiplying the shortfall between the annual production capacity we have reserved with Grifols and the amount of product actually ordered during that year by a fixed amount. Pending review and subsequent approval of the submission by the FDA, the agreement will terminate on the sixth anniversary of the approval by the FDA of the product manufactured by Grifols, unless it is terminated sooner by Cadence upon the termination of its license agreement for the product with BMS, or after 60 days written notice following the discontinuation of the distribution of the product by Cadence. In addition, either party may terminate the agreement after 60 days written notice in the event of a material uncured breach of the agreement by the other party (or 30 days in the case of a payment default), or immediately upon an insolvency event.
Settlement and Termination Agreement with Baxter Healthcare Corporation
In January 2011, we entered into an amended and restated development and supply agreement with Baxter Healthcare Corporation, or Baxter, for the manufacture of OFIRMEV for commercial distribution by us in the U.S. In February 2012, we temporarily suspended production of OFIRMEV by Baxter and that suspension has remained in effect through December 31, 2012, pending an investigation into unidentified particulate matter observed during routine product stability testing and pursuant to two recalls of the product during 2012.
In March 2013, we and Baxter mutually agreed to terminate the amended and restated development and supply agreement. Under the termination agreement, we are required to remove our manufacturing equipment from Baxter’s facility within 180 days, and pay Baxter for anticipated costs or expenses related to such removal, but we are not required to restore Baxter’s manufacturing facility to its condition prior to the installation of OFIRMEV-related improvements. In connection with the product, we are not required to reimburse Baxter for any remaining materials purchased by Baxter in connection with its manufacture of OFIRMEV. The termination agreement also contains customary mutual releases.
Distribution and Wholesaler Agreements
We distribute OFIRMEV primarily to drug wholesalers, who in turn distribute the product to hospital pharmacies and other institutional customers. We have retained third-party service providers to perform a variety of functions related to the distribution of OFIRMEV, including warehousing, customer service, order-taking, invoicing, collections, shipment and returns processing. We have entered into agreements with the major pharmaceutical wholesalers for distribution management services and data reporting in exchange for a fee.
Intellectual Property
We are the exclusive licensee of two U.S. patents and two Canadian patents from Pharmatop, under BMS’s license to these patents from Pharmatop. U.S. Patent No. 6,028,222 (Canadian patent 2,233,924), or the ‘222 patent, covers the formulation of OFIRMEV and formulations made by that process and expires in August 2017. U.S. Patent No. 6,992,218 (Canadian patent 2,415,403), or the ‘218 patent, covers the process used to manufacture OFIRMEV and expires in June 2021. We plan to complete a pediatric clinical trial by August 2015 and, upon timely completion and the acceptance by the FDA of the data from this study, OFIRMEV will be eligible for an additional six months of marketing exclusivity.
OFIRMEV and Pending Litigation
In August 2011, we and Pharmatop filed suit in the United States District Court for the District of Delaware against Paddock Laboratories, Inc., Perrigo Company and Paddock Laboratories, LLC, collectively referred to herein as Perrigo, and against Exela Pharma Sciences, LLC, Exela PharmaSci, Inc. and Exela Holdings, Inc., collectively referred to herein as Exela. The lawsuit follows the notices that we received in July 2011 from each
12
of Perrigo and Exela concerning their filings of Abbreviated New Drug Applications, or ANDAs, containing a “Paragraph IV” patent certification with the FDA for a generic version of OFIRMEV. In the lawsuit, we allege that Perrigo and Exela have each infringed the ‘222 patent and the ‘218 patent by filing their respective ANDAs seeking approval from the FDA to market a generic version of OFIRMEV prior to the expiration of these patents. The ‘222 and the ‘218 patents are listed in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. The patent infringement lawsuit was filed within 45 days of receipt of the pertinent notice letters, thereby triggering a stay of FDA approval of the Perrigo ANDA and the Exela ANDA until the earlier of the expiration of a 30-month period, the expiration of the ‘222 and ‘218 patents, the entry of a settlement order or consent decree stating that the ‘222 and ‘218 patents are invalid or not infringed, a decision in the case concerning infringement or validity that is favorable to Perrigo or Exela, or such shorter or longer period as the Court may order. Each of Perrigo and Exela has filed an answer in the case that asserts, among other things, non-infringement and invalidity of the asserted patents, and has also filed counterclaims. A date for the bench trial in the case with Exela has been scheduled for May 2013.
In November 2012, we settled the litigation with Perrigo by entering into settlement and license agreements. Under these agreements, Perrigo was granted the exclusive right of first refusal to negotiate an agreement with us to market an authorized generic version of OFIRMEV in the U.S. in the event that we elect to launch an authorized generic version of the product. The license agreement also provides that, if we enter into an agreement for Perrigo to market an authorized generic version of OFIRMEV during the license period, Perrigo would purchase the product exclusively from us. We would receive product costs plus an administrative fee, as well as a royalty payment based on the net profits achieved by Perrigo from the sale of the authorized generic product. Additionally, Perrigo was granted the non-exclusive right to market a generic intravenous acetaminophen product in the U.S. under Perrigo’s ANDA after December 6, 2020, or earlier under certain circumstances. The FTC or the DOJ could seek to challenge our settlement with Perrigo, or a competitor, customer or other third-party could initiate a private action under antitrust or other laws challenging the settlement.
In September 2012, an unidentified third party filed with the U.S. Patent and Trademark Office, or USPTO, a Request for Ex Parte Reexamination of the ‘222 patent. In December 2012, we received notice that the USPTO had granted the Request for Reexamination. The reexamination process is provided for by law and requires the USPTO to consider the scope and validity of the patent based on substantial new questions of patentability raised by a third party or the USPTO. Because we and Pharmatop believe that the scope and validity of the patent claims in this patent are appropriate and that the USPTO’s prior issuance of the patent was correct, we, in conjunction with Pharmatop, will vigorously defend this patent. We cannot predict whether we and Pharmatop ultimately will succeed in maintaining the scope and validity of the claims of this patent during reexamination. If the patent claims in this patent ultimately are narrowed during prosecution before the USPTO, the extent of the patent coverage afforded to OFIRMEV could be impaired, which could potentially harm our business and operating results.
In April 2012, Exela filed suit against David J. Kappos and the USPTO in the United States District Court for the Eastern District of Virginia for declaratory judgment seeking a reversal of the USPTO’s decision not to act on a petition by Exela to vacate the USPTO’s April 2003 order reviving the international application for the ‘218 patent. The lawsuit followed the USPTO’s rejection of Exela’s petition to the USPTO filed in November 2011, which sought to vacate the April 23, 2003 order granting Pharmatop’s petition to revive the ‘218 patent. The USPTO determined that Exela lacks standing to seek such relief. Exela also seeks declaratory judgment that the USPTO’s rules and regulations that allow for revival of abandoned, international patent applications under the “intentional” standard are invalid, and similar relief in connection with one or more counterclaims it has filed in the Delaware litigation. Our motion to intervene in this lawsuit was granted in October 2012. In December 2012, the district court dismissed the case with prejudice as barred by the applicable statute of limitations. In February 2013, Exela appealed the district court’s decision to the Court of Appeals for the Federal Circuit. A decision by the Court of Appeals in favor of Exela could result in the invalidation of the ‘218 patent.
13
In January 2013, we and Pharmatop filed suit in the United States District Court for the Southern District of California and the Northern District of Illinois against Fresenius Kabi USA, LLC, or Fresenius. The lawsuits follow a December 2012 notice by Fresenius concerning its filing of a New Drug Application, or NDA, containing a Paragraph IV patent certification with the FDA for a generic version of OFIRMEV. In the lawsuits, we allege that Fresenius has infringed the ‘222 patent and the ‘218 patent by filing its NDA seeking approval from the FDA to market a generic version of OFIRMEV prior to the expiration of these patents. Fresenius has filed an answer in the Southern District of California that asserts, among other things, non-infringement and invalidity of the asserted patents, and has also filed counterclaims.
In February 2013, we and Pharmatop filed suit in the United States District Court for the Southern District of California and the District of New Jersey against Sandoz, Inc., or Sandoz. The lawsuits follow a December 2012 notice by Sandoz concerning its filing of an ANDA containing a Paragraph IV patent certification with the FDA for a generic version of OFIRMEV. In the lawsuits, we allege that Sandoz has infringed the ‘222 patent and the ‘218 patent by filing its ANDA seeking approval from the FDA to market a generic version of OFIRMEV prior to the expiration of these patents. Sandoz has not yet filed an answer in either District.
Both the Fresenius and Sandoz lawsuits were filed within 45 days of receipt of the respective notice letters, thereby triggering a stay of FDA approval of the Fresenius NDA and the Sandoz ANDA until the earlier of the expiration of a 30-month period, the expiration of the ‘222 and ‘218 patents, the entry of a settlement order or consent decree stating that the ‘222 and ‘218 patents are invalid or not infringed, a decision in the case concerning infringement or validity that is favorable to Fresenius and/or Sandoz, or such shorter or longer period as the courts may order.
Regardless of the outcome of any litigation, no NDA or ANDA can receive final approval from the FDA before expiration of the regulatory exclusivity period for OFIRMEV. Specifically, the FDA has granted OFIRMEV three years of regulatory exclusivity, which expires November 2, 2013. We intend to vigorously enforce our intellectual property rights relating to OFIRMEV to prevent the marketing of infringing generic products prior to the expiration of our patents. The ‘222 patent expires August 5, 2017 (or February 5, 2018 if pediatric exclusivity is granted) and the ‘218 patent expires June 6, 2021 (or December 6, 2021 if pediatric exclusivity is granted). However, given the unpredictability inherent in litigation, we cannot predict the outcome of these matters or any other litigation. Regardless of how these matters are ultimately resolved, these matters may be costly, time-consuming and distracting to our management, which could have a material adverse effect on our business.
Research and Development
Our research and development expenses were $6.5 million in 2012, $8.9 million in 2011 and $13.8 million in 2010. Our historical research and development expenses relate predominantly to OFIRMEV and our discontinued omiganan pentahydrochloride product candidate. Our research and development expenses consist primarily of salaries and related employee benefits for our research and development team, license fees paid to our licensors prior to approval of our drug candidates, pre-commercialization manufacturing development activities, costs associated with clinical trials, and costs associated with non-clinical activities, such as expenses related to regulatory submissions. License fees are paid to the patent holders of our product candidates that give us the exclusive licenses to the patent rights and know-how for selected indications and territories. Manufacturing development activities include the costs to develop facilities for the commercial production of our drug products prior to approval, the production of supply and validation lots, and other manufacturing support activities related to the requirements for submitting NDAs for our product candidates. The clinical trial expenses include payments to vendors such as clinical research organizations and investigator sites, clinical suppliers and related consultants.
We expect to continue to incur research and development expenses related to OFIRMEV, however, it is difficult to anticipate the scope and magnitude of our future research and development expenses. For example,
14
we began enrollment in our FDA-required post-approval clinical trial for OFIRMEV in pediatric patients under two years of age during the third quarter of 2012, which we plan to complete by August 2015. We may also conduct clinical studies to expand the indications for OFIRMEV. In addition, in July 2012, we filed a New Drug Submission for OFIRMEV with Health Canada which was accepted for review in August 2012. We are currently evaluating the commercial prospects and partnering opportunities for the product in Canada and anticipate that the product would not be approved by Canadian regulatory authorities for at least 18 months after this submission, if at all. We expect to incur costs associated with seeking approval for OFIRMEV in Canada and any marketing activities that may take place if approval is obtained. Moreover, any product candidates we may in-license or acquire in the future would likely require significant research and development resources. Therefore, we are unable to estimate with any certainty the costs we will incur in completing our development efforts for OFIRMEV or any other product candidate we might acquire or in-license.
Government Regulation
Governmental authorities in the U.S. and other countries extensively regulate the testing, manufacturing, labeling, storage, record-keeping, advertising, promotion, export, marketing and distribution, among other things, of pharmaceutical products. In the U.S., the FDA, under the Federal Food, Drug and Cosmetic Act and other federal statutes and regulations, subjects pharmaceutical products to rigorous review. If we do not comply with applicable requirements, we may be fined, the government may refuse to approve our marketing applications or allow us to manufacture or market our products, and we may be criminally prosecuted.
FDA Approval Process
To obtain approval of a new product from the FDA, we must, among other requirements, submit data supporting safety and efficacy as well as detailed information on the manufacture and composition of the product and proposed labeling. The testing and collection of data and the preparation of necessary applications are expensive and time-consuming. The FDA may not act quickly or favorably in reviewing these applications, and we may encounter significant difficulties or costs in our efforts to obtain FDA approvals that could delay or preclude us from marketing our products.
The process required by the FDA before a new drug may be marketed in the U.S. generally involves the following: completion of preclinical laboratory and animal testing in compliance with FDA regulations, submission of an investigational new drug application, or IND, which must become effective before human clinical trials may begin, performance of adequate and well-controlled human clinical trials to establish the safety and efficacy of the proposed drug for its intended use, and submission and approval of an NDA by the FDA. The sponsor typically conducts human clinical trials in three sequential phases, but the phases may overlap. In Phase I clinical trials, the product is tested in a small number of patients or healthy volunteers, primarily for safety at one or more dosages. In Phase II clinical trials, in addition to safety, the sponsor evaluates the efficacy of the product on targeted indications, and identifies possible adverse effects and safety risks in a patient population. Phase III clinical trials typically involve testing for safety and clinical efficacy in an expanded population at geographically-dispersed test sites.
Clinical trials must be conducted in accordance with the FDA’s good clinical practices requirements. The FDA may order the partial, temporary or permanent discontinuation of a clinical trial at any time or impose other sanctions if it believes that the clinical trial is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The institutional review board, or IRB, generally must approve the clinical trial design and patient informed consent at each clinical site and may also require the clinical trial at that site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements, or may impose other conditions.
The applicant must submit to the FDA the results of the preclinical and clinical trials, together with, among other things, detailed information on the manufacture and composition of the product and proposed labeling, in
15
the form of an NDA, including payment of a user fee. The FDA reviews all NDAs submitted before it accepts them for filing and may request additional information rather than accepting an NDA for filing. Once the submission is accepted for filing, the FDA begins an in-depth review of the NDA. Under the policies agreed to by the FDA under the Prescription Drug User Fee Act, or PDUFA, the FDA has 10 months from the filing date in which to complete its initial review of a standard NDA and respond to the applicant. The review process and the PDUFA goal date may be extended by three months if the FDA requests or the NDA sponsor otherwise provides additional information or clarification regarding information already provided in the submission within the last three months of the PDUFA goal date. If the FDA’s evaluations of the NDA and the clinical and manufacturing procedures and facilities are favorable, the FDA will issue an approval letter. If the FDA’s evaluation of the NDA submission and the clinical and manufacturing procedures and facilities is not favorable, the FDA may refuse to approve the NDA and issue a Complete Response Letter, or CRL. The CRL usually describes the specific deficiencies in the NDA identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the CRL typically contains the conditions that must be met in order to secure final approval of the NDA. If a CRL is issued, the applicant may either resubmit the NDA, addressing all of the deficiencies identified in the letter, or withdraw the application. If and when those conditions have been met to the FDA’s satisfaction, the FDA will issue an approval letter, authorizing commercial marketing of the drug for certain indications.
Section 505(b)(2) New Drug Applications
As an alternate path to FDA approval for new indications or improved formulations of previously-approved products, a company may file a Section 505(b)(2) NDA, instead of a “stand-alone” or “full” NDA. Section 505(b)(2) of the Federal Food, Drug and Cosmetic Act was enacted as part of the Drug Price Competition and Patent Term Restoration Act of 1984, otherwise known as the Hatch-Waxman Act. Section 505(b)(2) permits the submission of an NDA where at least some of the information required for approval comes from studies not conducted by or for the applicant and for which the applicant has not obtained a right of reference. For example, the Hatch-Waxman Act permits the applicant to rely upon the FDA’s findings of safety and effectiveness for an approved product. The FDA may also require companies to perform additional studies or measurements to support the change from the approved product. The FDA may then approve the new formulation for all or some of the label indications for which the referenced product has been approved, or the new indication sought by the Section 505(b)(2) applicant.
To the extent that the Section 505(b)(2) applicant is relying on the FDA’s findings for an already-approved product, the applicant is required to certify to the FDA concerning any patents listed for the approved product in the FDA’s Orange Book publication. Specifically, the applicant must certify that: (1) the required patent information has not been filed; (2) the listed patent has expired; (3) the listed patent has not expired, but will expire on a particular date and approval is sought after patent expiration; or (4) the listed patent is invalid or will not be infringed by the manufacture, use or sale of the new product. A certification that the new product will not infringe the already approved product’s Orange Book-listed patents or that such patents are invalid is called a paragraph IV certification. If the applicant does not challenge the listed patents, the Section 505(b)(2) application will not be approved until all the listed patents claiming the referenced product have expired. The Section 505(b)(2) application may also not be approved until any non-patent exclusivity, such as exclusivity for obtaining approval of a new chemical entity, listed in the Orange Book for the referenced product, has expired.
If the applicant has provided a paragraph IV certification to the FDA, the applicant must also send notice of the paragraph IV certification to the NDA and patent holders once the NDA has been accepted for filing by the FDA. The NDA and patent holders may then initiate a legal challenge to the paragraph IV certification. The filing of a patent infringement lawsuit within 45 days of their receipt of a paragraph IV certification automatically prevents the FDA from approving the Section 505(b)(2) NDA until the earlier of the expiration of a 30-month period, the expiration of the patent, the entry of a settlement order or consent decree stating that the patent are invalid or not infringed, a decision in the case concerning infringement or validity that is favorable to the Section 505(b)(2) applicant. For drugs with five-year exclusivity, if an action for patent infringement is
16
initiated after year four of that exclusivity period, then the 30-month stay period is extended by such amount of time so that 7.5 years has elapsed since the approval of the NDA with five-year exclusivity. This period could be extended by six months if the NDA sponsor obtains pediatric exclusivity. Thus, the Section 505(b)(2) applicant may invest a significant amount of time and expense in the development of its products only to be subject to significant delay and patent litigation before its products may be commercialized. Alternatively, if the listed patent holder does not file a patent infringement lawsuit within the required 45-day period, the applicant’s NDA will not be subject to the 30-month stay.
Notwithstanding the approval of many products by the FDA pursuant to Section 505(b)(2), over the last few years, certain brand-name pharmaceutical companies and others have objected to the FDA’s interpretation of Section 505(b)(2) and one pharmaceutical company has sued the FDA on the matter. Although the issues in that litigation are specific to the products involved, if the FDA does not prevail, it may be required to change its interpretation of Section 505(b)(2), which could delay or even prevent the FDA from approving any Section 505(b)(2) NDA that we submit.
Fast Track Designation
A drug designated as a fast track product by the FDA must be intended for the treatment of a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the condition. Fast track designation does not apply to a product alone, but applies to a combination of the product and specific indication for which it is being studied. A sponsor may submit a request for fast track designation at the time of original submission of its IND, or at any time thereafter prior to receiving marketing approval of its NDA. Fast track status enables the sponsor to have more frequent and timely communication and meetings with the FDA regarding the product development plans. If fast track designation is obtained, the FDA may initiate review of sections of an NDA before the application is complete. This “rolling review” is available if the applicant provides and the FDA approves a schedule for the remaining information. In some cases, a fast track product may be eligible for accelerated approval, pursuant to which the product is approved on the basis of either a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Fast track status may also result in eligibility for NDA priority review, under which the PDUFA review goal for the NDA is six months rather than ten months.
The Hatch-Waxman Act
Under the Hatch-Waxman Act, newly-approved drugs and indications benefit from a statutory period of non-patent marketing exclusivity. The Hatch-Waxman Act provides five-year marketing exclusivity to the first applicant to gain approval of an NDA for a new chemical entity, meaning that the FDA has not previously approved any other new drug containing the same active moiety. The Hatch-Waxman Act prohibits the submission of an ANDA or a Section 505(b)(2) NDA for another version of such drug during the five-year exclusive period; however, as explained above, submission of an ANDA or Section 505(b)(2) NDA containing a paragraph IV certification is permitted after four years, which may trigger a 30-month stay of approval of the ANDA or Section 505(b)(2) NDA. Protection under the Hatch-Waxman Act will not prevent the submission or approval of another full NDA; however, the applicant would be required to conduct its own preclinical and adequate and well-controlled clinical trials to demonstrate safety and effectiveness. The Hatch-Waxman Act also provides three years of marketing exclusivity for the approval of new and supplemental NDAs, including Section 505(b)(2) NDAs such as we filed for OFIRMEV, for, among other things, new indications, dosages or strengths of an existing drug, if new clinical investigations that were conducted or sponsored by the applicant are essential to the approval of the application. Additionally, six months of marketing exclusivity is available under Section 505A of the FDCA if, in response to a written request from the FDA, a sponsor submits and the agency accepts certain requested information relating to the use of the approved drug in the pediatric population.
17
Other Regulatory Requirements
FDA Post-Approval Regulatory Requirements
We may also be subject to a number of post-approval regulatory requirements. For example, in accordance with a Pediatric Research Equity Act requirement included in the NDA approval of OFIRMEV, we began enrolling patients in the third quarter of 2012 in a post-marketing efficacy study of OFIRMEV in infants and neonates. In addition, we plan to use the data from this study to satisfy a formal written request from the FDA under Section 505A of the U.S. Food, Drug and Cosmetic Act that was made as part of the approval process for OFIRMEV. The FDA has agreed to a due date for completion of this study in August 2015. If we seek to make certain changes to an approved product, such as promoting or labeling a product for a new indication, making certain manufacturing changes or product enhancements or adding labeling claims, we will need FDA review and approval before the change can be implemented. While physicians may use products for indications that have not been approved by the FDA, we may not label or promote the product for an indication that has not been approved. Securing FDA approval for new indications or product enhancements and, in some cases, for manufacturing and labeling claims, is generally a time-consuming and expensive process that may require us to conduct clinical trials under the FDA’s IND regulations. Even if such studies are conducted, the FDA may not approve any change in a timely fashion, or at all. In addition, it has been reported that the current presidential administration may be seeking to curb practices that could result in the extension of the term of patent protection for pharmaceuticals, which may include applications for new indications or product enhancements.
Adverse experiences associated with use of the products must be reported to the FDA, and FDA rules govern how we can label, advertise or otherwise commercialize our products. There are current post-marketing safety surveillance requirements that we will need to meet to continue to market an approved product. The FDA also may, in its discretion, require post-marketing testing and surveillance to monitor the effects of approved products or place conditions on any approvals that could restrict the commercial applications of these products.
In addition, we and the manufacturers on which we rely for the manufacture of our products are subject to requirements that drugs be manufactured, packaged and labeled in conformity with current good manufacturing practice. To comply with current good manufacturing practice requirements, manufacturers must continue to spend time, money and effort to meet requirements relating to personnel, facilities, equipment, production and process, labeling and packaging, quality control, record-keeping and other requirements. The FDA periodically inspects drug manufacturing facilities to evaluate compliance with current good manufacturing practice requirements.
Also, as part of the sales and marketing process, pharmaceutical companies frequently provide samples of approved drugs to physicians. This practice is regulated by the FDA and other governmental authorities, including, in particular, requirements concerning record-keeping and control procedures.
Outside of the U.S., our ability to market our products will also depend on receiving marketing authorizations from the appropriate regulatory authorities. The foreign regulatory approval process includes all of the risks associated with the FDA approval process described above. The requirements governing the conduct of clinical trials and marketing authorization vary widely from country to country.
Other Government Regulatory Requirements
In addition to FDA restrictions on marketing of pharmaceutical products, we are subject to various state and federal fraud and abuse laws, including the anti-kickback statute and false claims laws and regulations. The federal anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any health care item or service reimbursable under Medicare, Medicaid or other federally financed health care programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other.
18
Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. There are also federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. Recently, several pharmaceutical and other health care companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn were used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws. The majority of states also have statutes or regulations similar to the federal anti-kickback statute and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor.
Further, the recently enacted Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act, or collectively, the PPACA, among other things, amends the intent requirement of the federal anti-kickback and criminal healthcare fraud statutes. A person or entity can now be found guilty of fraud or false claims under PPACA without actual knowledge of the statute or specific intent to violate it. In addition, PPACA provides that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the false claims statutes. Possible sanctions for violation of these anti-kickback laws include monetary fines, civil and criminal penalties, exclusion from Medicare, Medicaid and other government programs and forfeiture of amounts collected in violation of such prohibitions. Any violations of these laws, or any action against us for violation of these laws, even if we successfully defend against it, could result in a material adverse effect on our reputation, business, results of operations and financial condition.
The PPACA also imposes new reporting requirements on device and pharmaceutical manufacturers to make annual public disclosures of payments and other transfers of value to healthcare providers and ownership of their stock by certain healthcare professionals. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (or up to an aggregate of $1 million per year for “knowing failures”), for all covered payments, transfers of value or ownership or investment interests that are not reported in the annual submission. Under the PPACA, we will be required to begin data collection regarding reportable payments, transfers of value and ownership of investment interests on August 1, 2013 and report such data to the Centers for Medicare & Medicaid Services, or CMS, by March 31, 2014. CMS will publish this data on a public website by September 31, 2014.
Additionally, if not preempted by this federal law, several states require pharmaceutical companies to report expenses relating to the marketing and promotion of pharmaceutical products and to report gifts and payments to individual physicians in the states. Other states prohibit providing various other marketing related activities. Still other states require the posting of information relating to clinical studies and their outcomes. In addition, certain states require pharmaceutical companies to implement compliance programs or marketing codes. Currently, several additional states are considering similar proposals. Companies must also be registered or licensed by the federal and state governments prior to manufacturing or distributing prescription drug products. Meanwhile, California has enacted legislation that requires development of an electronic pedigree to track and trace each prescription drug at the saleable unit level through the distribution system. California’s electronic pedigree requirement is scheduled to take effect in January 2015.
We also make our products available to authorized users of the Federal Supply Schedule, or FSS, of the General Services Administration. As a result of the Veterans Health Care Act of 1992, or the VHC Act, federal
19
law requires that we offer deeply discounted FSS contract pricing for purchases by the Department of Veterans Affairs, the Department of Defense, the Coast Guard and the Public Health Service, including the Indian Health Service, in order for federal funding to be available for these four federal agencies and certain federal grantees to purchase our products. FSS pricing to these four federal agencies must be equal to or less than the Federal Ceiling Price, or FCP, which is 24% below the Non-Federal Average Manufacturer Price, or Non-FAMP, for the prior fiscal year. The accuracy of the pricing and other information we report may be audited by the government under applicable federal procurement laws and the terms of our FSS contract. Among the remedies available to the government for inaccuracies in our pricing information is recoupment of any overcharges resulting from such inaccuracies and civil monetary penalties of $100,000 per item that is incorrect.
We and our contract manufacturers and clinical research organizations may also be subject to regulations under other federal, state and local laws, including the Occupational Safety and Health Act, the Environmental Protection Act, the Clean Air Act and import, export and customs regulations as well as the laws and regulations, of other countries.
Additionally, the U.S. Foreign Corrupt Practices Act, or FCPA, prohibits U.S. corporations and their representatives from offering, promising, authorizing or making payments to any foreign government official, government staff member, political party or political candidate in an attempt to obtain or retain business abroad. The scope of the FCPA would include interactions with certain healthcare professionals in many countries. Our present and future business has been and will continue to be subject to various other U.S. and foreign laws, rules and/or regulations.
Third-Party Reimbursement and Pricing Controls
In the U.S. and elsewhere, sales of pharmaceutical products depend in significant part on the availability of coverage and reimbursement to customers from third-party payors, such as government and private insurance plans. Third-party payors are increasingly challenging the prices charged for medical products and services. Our products may not be considered cost effective, and coverage and reimbursement received by our customers may not be sufficient to allow us to sell our products on a competitive and profitable basis.
In many foreign markets, including Canada, pricing of pharmaceutical products is subject to governmental control. In the U.S., there have been, and we expect that there will continue to be, a number of federal and state proposals to implement similar governmental pricing control. In March 2010, the PPACA became law and made extensive changes to the delivery of health care in the U.S. The PPACA includes numerous provisions that affect pharmaceutical companies, some of which became effective immediately and others of which will be taking effect over the next several years. The PPACA seeks to expand health care coverage to the uninsured through private health insurance reforms and an expansion of Medicaid. The PPACA also imposes substantial costs on pharmaceutical manufacturers, such as an increase in liability for rebates paid to Medicaid for certain outpatient drugs, new drug discounts that must be offered to certain enrollees in the Medicare prescription drug benefit, an annual fee imposed on all manufacturers of brand prescription drugs in the U.S., and an expansion of an existing program requiring pharmaceutical discounts to certain types of hospitals and federally subsidized clinics. The PPACA also promotes programs that increase the federal government’s comparative effectiveness research, which may be used to evaluate the selection of medical services by clinicians and others. In addition, PPACA implements payment system reforms such as a national pilot program on payment bundling to encourage hospitals, physicians and other providers to improve the coordination, quality and efficiency of certain healthcare services through bundled payment models, and creates an independent payment advisory board that will submit recommendations to reduce Medicare spending if projections of such spending exceed a specified growth rate.
Other legislative changes have been proposed and adopted since the PPACA was enacted. On August 2, 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the
20
legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. On January 2, 2013, the President signed into law the American Taxpayer Relief Act of 2012, or the ATRA, which, among other things, delayed for another two months the budget cuts mandated by these sequestration provisions of the Budget Control Act of 2011. The ATRA also reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws, as well as legislative and regulatory proposals that may be adopted from time to time in the future, may result in additional reductions in Medicare and other health care funding, which could have a material adverse effect on our customers and accordingly, our financial operations.
Employees
As of March 1, 2013, we had approximately 206 employees.
21
The following information sets forth risk factors that could cause our actual results to differ materially from those contained in forward-looking statements we have made in this Annual Report on Form 10-K and those we may make from time to time. You should carefully consider the risks described below, in addition to the other information contained in this report, before making an investment decision. Our business, financial condition or results of operations could be harmed by any of these risks. The risks and uncertainties described below are not the only ones we face. Additional risks not presently known to us or other factors not perceived by us to present significant risks to our business at this time also may impair our business operations.
Risks Related to Our Business and Industry
Our success depends on the commercial success of our only product, OFIRMEV®.
Our success depends on the continued success of our efforts to commercialize our only product, OFIRMEV, which was approved by the FDA in November 2010 for the management of mild to moderate pain, the management of moderate to severe pain with adjunctive opioid analgesics and the reduction of fever in adults and children two years of age and older.
We launched OFIRMEV in January 2011, but our ability to maintain and increase revenues from sales of OFIRMEV will depend on several factors, including:
| | • | | our ability to increase market demand for OFIRMEV through our own marketing and sales activities, and any other arrangements to promote this product we may later establish; |
| | • | | our ability to maintain and defend our patent protection and regulatory exclusivity for OFIRMEV; |
| | • | | our ability to continue to procure a supply of OFIRMEV from our sole source third-party manufacturer in sufficient quantities and at acceptable quality and pricing levels in order to meet commercial demand; |
| | • | | the performance of our third-party manufacturer and our ability to ensure that our supply chain for OFIRMEV efficiently and consistently delivers OFIRMEV to our customers; |
| | • | | our ability to continue to deploy and support a qualified sales force; |
| | • | | our ability to maintain fees and discounts payable to the wholesalers and distributors who distribute OFIRMEV, as well as to group purchasing organizations, at commercially reasonable levels; |
| | • | | whether the FTC, DOJ or third parties seek to challenge and are successful in challenging our settlement agreement with Perrigo; |
| | • | | the occurrence of adverse side effects or inadequate therapeutic efficacy of OFIRMEV, and any resulting product liability claims or product recalls; and |
| | • | | our ability to achieve hospital formulary acceptance for OFIRMEV, and to the extent third-party payors separately cover and reimburse for OFIRMEV, the availability of adequate levels of reimbursement for OFIRMEV from third-party payors. |
Any disruption in our ability to generate revenues from the sale of OFIRMEV or lack of success in its commercialization will have a substantial adverse impact on our results of operations.
The continued success of our commercialization of OFIRMEV is subject to many internal and external challenges and if we cannot overcome these challenges in a timely manner, our revenues and profits could be materially and adversely impacted.
OFIRMEV was launched in January 2011. Since that time, we have continued to expend significant time and resources to provide effective promotional materials to our sales force and medical affairs staff for their use in communicating about OFIRMEV with physicians, nurses, hospitals and other customers, and to ensure that a
22
consistent and appropriate message about OFIRMEV is being delivered to our potential customers. The effectiveness of our promotional and medical communication materials about OFIRMEV is critically important to our efforts to inform and educate potential customers about the benefits and risks of OFIRMEV and its proper administration, and the continued success of our commercialization activities for the product.
In addition to extensive internal efforts, the continued successful commercialization of OFIRMEV requires many third parties, over whom we have no control, to decide to utilize OFIRMEV and to make it readily available at the point of care throughout their hospitals. These third parties include physicians, pharmacists, and hospital pharmacy and therapeutics committees, which are commonly referred to as P&T committees. Generally, before we can attempt to sell OFIRMEV in a hospital, OFIRMEV must be approved for addition to that hospital’s list of approved drugs, or formulary list, by the hospital’s P&T committee. A hospital’s P&T committee typically governs all matters pertaining to the use of medications within the institution, including review of medication formulary data and recommendations for the appropriate use of drugs within the institution to the medical staff. The frequency of P&T committee meetings at various hospitals varies considerably, and P&T committees often require additional information to aide in their decision-making process, so we may experience substantial delays in obtaining formulary approvals. Additionally, hospital pharmacists may be concerned that the cost of acquiring OFIRMEV for use in their institutions will adversely impact their overall pharmacy budgets, which could cause pharmacists to resist efforts to add OFIRMEV to the formulary, or to implement restrictions on the usage of the drug in order to control costs, either initially or later, when the increasing use of OFIRMEV within their institution begins to significantly impact their budgets. We cannot guarantee that we will be successful in getting the approvals we need from enough P&T committees and overcoming any financial objections raised by hospital pharmacists quickly enough to maintain and grow hospital sales of OFIRMEV.
We have no manufacturing capabilities and depend entirely upon our sole source contract manufacturer to produce OFIRMEV. If our contract manufacturer fails to meet our requirements for OFIRMEV, or fails to fully comply with cGMP regulations, we may be unable to meet market demand, and may lose potential revenues.
The manufacture of pharmaceutical products requires significant expertise and capital investment, including the development of advanced manufacturing techniques and process controls, and the use of specialized processing equipment. We have no such manufacturing capabilities, so we have relied upon contract manufacturers as our source for OFIRMEV.
In February 2013, we amended our supply agreement with Lawrence Laboratories, an operating division of Swords Laboratories and a member of the BMS group of companies, under which BMS Anagni, an indirect subsidiary of BMS located in Anagni, Italy, manufactures OFIRMEV for us on behalf of Lawrence Laboratories. BMS Anagni, which is currently our sole source for OFIRMEV, has manufactured the product for more than ten years for sale and distribution by BMS and its affiliates in a number of countries outside of the U.S. and Canada.
In March 2013, we entered into an agreement with Grifols for the development, manufacture and supply of commercial quantities of OFIRMEV in flexible plastic bags. We plan to submit a supplemental NDA to the FDA in the second half of 2013 seeking approval of the product to be manufactured by Grifols, but Grifols will not be able to supply us with OFIRMEV until FDA approval is granted, if ever.
Our contract manufacturers must comply with strictly enforced federal, state and foreign regulations, including GMP regulations. The FDA will inspect our contract manufacturers’ facilities from time to time and, in the event that any such inspection reveals that the facility is not in compliance with applicable regulations, the FDA may issue fines and civil penalties, suspend production, suspend or delay any subsequent product approvals, seize or recall our products, or withdraw our product approval, which would limit the availability of OFIRMEV. Any manufacturing defect or error discovered after products have been produced and distributed could result in even more significant consequences, including costly recall procedures, re-stocking costs, damage to our reputation and our relationships with our customers, product liability claims and litigation.
23
We also currently rely upon a single source for the manufacture of the active pharmaceutical ingredient, or API, for OFIRMEV, as well as for other critical components of OFIRMEV. We have entered into a supply agreement for the commercial supply of the API. If our supplier becomes unable to meet our demand for the API, the process of changing or adding a new API manufacturer may require additional testing and prior FDA approval and may be expensive and time-consuming. If we were unable to manage such changes effectively, we could face supply disruptions that could result in significant costs and delays, damage to our reputation or commercial prospects and cause us to lose potential revenues.
Although we actively manage these third party relationships to ensure continuity and quality, some events beyond our control could result in the complete or partial failure of these goods and services. Any such failure could have a material adverse effect on our financial condition and operations. In addition, as OFIRMEV is a relatively new product in the U.S., the effect of any delay or failure to deliver could be magnified due to the short sales track record for OFIRMEV.
For example, in February 2012, we announced a voluntary recall of a single lot of OFIRMEV that was manufactured by our previous contract manufacturer, Baxter, and in July 2012, we announced a second voluntary recall of product manufactured at Baxter’s facility, due to the presence of unidentified, visible particles in a limited number of vials of the product, which were detected during routine stability testing. Although we received no adverse event reports associated with the particulate matter or product complaints involving similar particulate matter, as a precautionary measure we suspended production by Baxter in connection with the initial recall and decided to recall all remaining lots of OFIRMEV manufactured by Baxter in connection with the second recall. As a result of the first recall, during the first quarter of 2012, some of our customers experienced short-term supply delays due to the temporary suspension of shipments from Baxter before we were able to expedite sufficient shipments of OFIRMEV from BMS Anagni. In addition, during that time we incurred higher freight costs to expedite shipments of OFIRMEV from BMS Anagni in order to meet demand for the product following the temporary suspension at Baxter’s facility. We also continued to incur unabsorbed manufacturing costs due to fixed costs that continued to accrue under our supply agreement with Baxter during the time that Baxter’s manufacturing of the product was suspended.
As a result of the second recall, we decided to destroy the Baxter-manufactured finished product inventory that we previously placed on indefinite hold. We recorded charges of $5.8 million in relation to this product due to uncertainty as to the amount of time that would be required to complete the investigation and whether the product would have sufficient remaining shelf life or otherwise be saleable after the investigation was completed. In March 2013, we and Baxter mutually agreed to terminate our supply agreement for OFIRMEV. Under the termination agreement, we are required to remove our manufacturing equipment from Baxter’s facility within 180 days and pay Baxter for any pre-approved costs or expenses related to such removal. We incurred impairment charges of $7.7 million and a loss on the sale of equipment of $0.9 million during the fourth quarter of 2012 in relation to certain manufacturing assets involved with the manufacture of OFIRMEV under the terminated development and supply agreement with Baxter.
Although we have not completed our investigation into the cause of the particulate matter discovered in the product manufactured by Baxter, our review of data for product manufactured by BMS Anagni has confirmed that no similar particulate material has been observed in any product manufactured there. However, any future recalls of OFIRMEV could negatively affect customer perceptions and reduce revenue from OFIRMEV, and could also result in unexpected costs for replacement product, investigational costs and the write down of inventory and equipment. Additionally, any termination or disruption of our relationship with BMS Anagni, our sole source for OFIRMEV, may materially harm our business and financial condition and adversely impact our commercialization and sales efforts with respect to the product.
24
If OFIRMEV does not achieve broad market acceptance, the revenues that we generate from its sales will be limited.
The commercial success of OFIRMEV will depend upon its acceptance by the medical community and our ability to ensure that the drug is included in hospital formularies, and coverage and reimbursement for OFIRMEV by third-party payors, including government payors. The degree of market acceptance of OFIRMEV, or any other product or product candidate we may license or acquire, will depend on a number of factors, including:
| | • | | limitations or warnings contained in the product’s FDA-approved labeling; |
| | • | | changes in the standard of care for the targeted indications for our product candidates, which could reduce the marketing impact of any superiority claims that we could make following FDA approval; and |
| | • | | potential advantages over, and availability of, alternative treatments, including, in the case of OFIRMEV, a number of products already used to treat pain or fever in the hospital setting. |
Our ability to effectively promote and sell OFIRMEV and any other product or product candidate we may license or acquire in the hospital marketplace will also depend on pricing and cost effectiveness, including our ability to produce a product at a reasonable cost and achieve hospital formulary acceptance for the product and sell the product at a competitive price, as well as our ability to obtain sufficient third-party coverage or reimbursement. Since many hospitals are members of group purchasing organizations, which leverage the purchasing power of a group of entities to obtain discounts based on the collective buying power of the group, our ability to attract customers in the hospital marketplace will also depend on our ability to effectively promote OFIRMEV and any other product to hospitals that are members of group purchasing organizations. We will also need to demonstrate acceptable evidence of safety and efficacy, as well as relative convenience and ease of administration. Market acceptance could be further limited depending on the prevalence and severity of any expected or unexpected adverse side effects associated with OFIRMEV and any other product or product candidates we may license or acquire. If OFIRMEV, or any other product or product candidate that is approved, does not achieve an adequate level of acceptance by physicians, health care payors and patients, we may not generate sufficient revenue from these products, and we may not become or remain profitable. In addition, our efforts to educate the medical community and third-party payors on the benefits and risks of OFIRMEV or any other product or product candidate may require significant resources and may never be successful.
We rely on third parties to perform many essential services for OFIRMEV and any other products that we commercialize, including services related to warehousing and inventory control, distribution, customer service, accounts receivable management, cash collection and adverse event reporting, and if such third parties fail to perform as expected or to comply with legal and regulatory requirements, our efforts to commercialize OFIRMEV or any other products may be significantly impacted and we may be subject to regulatory sanctions.
We rely on third-party service providers to perform a variety of functions related to the sale and distribution of OFIRMEV, key aspects of which are out of our direct control. The services provided by these third parties include warehousing and inventory control, distribution, customer service, accounts receivable management and cash collection. As a result, most of our inventory is stored at a single warehouse maintained by one such service provider. If these third-party service providers fail to comply with applicable laws and regulations, fail to meet expected deadlines, or otherwise do not carry out their contractual duties to us, or if our products encounter physical or natural damage at their facilities, our ability to deliver product to meet commercial demand would be significantly impaired. In addition, we have engaged third parties to perform various other services for us relating to adverse event reporting, safety database management, fulfillment of requests for medical information regarding OFIRMEV and related services. If the quality or accuracy of the data maintained or services performed by these third parties is insufficient, we could be subject to regulatory sanctions.
25
We face potential product liability exposure, and if successful claims are brought against us, we may incur substantial liability for OFIRMEV or other products or product candidates we may license or acquire and may have to limit their commercialization.
The use of OFIRMEV and any other products or product candidates we may license or acquire in clinical trials and the sale of any products for which we obtain marketing approval expose us to the risk of product liability claims. Product liability claims might be brought against us by consumers, health care providers or others using, administering or selling our products. If we cannot successfully defend ourselves against these claims, we will incur substantial liabilities. Regardless of merit or eventual outcome, liability claims may result in:
| | • | | decreased demand for OFIRMEV or other products or product candidates; |
| | • | | impairment of our business reputation; |
| | • | | costs of related litigation; |
| | • | | substantial monetary awards to patients or other claimants; |
| | • | | withdrawal of clinical trial participants; |
| | • | | significant distraction of our scientific and management personnel who may be involved in our efforts to defend against such claims; and |
| | • | | the inability or lack of commercial rationale to continue commercialization of OFIRMEV or any other products or product candidates. |
Although we currently have commercial product liability coverage for OFIRMEV, which includes coverage for any clinical trials we may perform, insurance coverage is becoming increasingly expensive and we may be unable to obtain commercially reasonable product liability insurance coverage at a reasonable cost or in sufficient amounts to protect us against losses due to liability. On occasion, large judgments have been awarded in class action lawsuits based on drugs that had unanticipated side effects. Our commercial product liability insurance coverage may not reimburse us or may not be sufficient to reimburse us for any expenses or losses we may suffer. A successful product liability claim or series of claims brought against us could cause our stock price to fall and, if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
We expect intense competition for OFIRMEV, and new products may emerge that provide different or better therapeutic alternatives for our targeted indications.
The biotechnology and pharmaceutical industries are subject to rapid and intense technological change. We will continue to face competition in our efforts to market and sell OFIRMEV from other biotechnology and pharmaceutical companies. There can be no assurance that developments by others will not render OFIRMEV obsolete or noncompetitive. Furthermore, new developments, including the development of other drug technologies and methods of preventing the incidence of disease, occur in the pharmaceutical industry at a rapid pace. These developments may render OFIRMEV obsolete or noncompetitive.
OFIRMEV will compete with well-established products with similar indications. Competing injectable products available for the treatment of pain include opioids such as morphine, fentanyl, meperidine and hydromorphone, each of which is available generically from several manufacturers, and several of which are available as proprietary products using novel delivery systems. Ketorolac, an injectable non-steroidal anti-inflammatory drug, or NSAID, is also available generically in the U.S. from several manufacturers, and Caldolor (ibuprofen for injection), an NSAID, is available for the treatment of pain and fever in adults and children 17 years of age and older. Competing products available for the treatment of fever in the hospital setting include acetaminophen administered orally and rectally, aspirin and NSAIDs, which may be administered orally,
26
topically or intravenously. Additional products may be developed for the treatment of acute pain, including new injectable NSAIDs, novel opioids, new formulations of currently available opioids and NSAIDS, long-acting local anesthetics and new chemical entities as well as alternative delivery forms of various opioids and NSAIDs.
Competitors may seek to develop alternative formulations of intravenous acetaminophen for our targeted indications that do not directly infringe our in-licensed patent rights. The commercial opportunity for OFIRMEV could be significantly harmed if competitors are able to develop alternative formulations outside the scope of our in-licensed patents.
Compared to us, many of our potential competitors have substantially greater:
| | • | | research development resources, including personnel and technology; |
| | • | | clinical trial experience; |
| | • | | expertise in prosecution of intellectual property rights; and |
| | • | | manufacturing, distribution, and sales and marketing experience. |
As a result of these factors, our competitors may be able to obtain patent protection or other intellectual property rights that limit our ability to commercialize OFIRMEV. Our competitors may also develop drugs that are more effective, useful and less costly than ours and may be more successful than us in manufacturing and marketing their products. We expect to face similar competition in our efforts to identify appropriate collaborators or partners to help commercialize OFIRMEV in Canada.
We may require substantial additional funding and may be unable to raise capital when needed, which would force us to delay, reduce or eliminate some or all of our planned activities.
We began generating revenue from the launch of OFIRMEV in January 2011, however, we expect our negative cash flow from operations to continue until we are able to generate significant revenues from sales of OFIRMEV. As a result, we may need to raise additional capital to:
| | • | | fund our operations as we implement our marketing strategies, maintain our sales force and commercial infrastructure and commercialize OFIRMEV; |
| | • | | purchase sufficient quantities of OFIRMEV from our contract manufacturers to meet customer demand or our minimum purchase obligations; |
| | • | | continue to fund the expansion of our contract manufacturers’ capacity to produce OFIRMEV in order to meet future demand for this product; |
| | • | | complete our ongoing efficacy, pharmacokinetic and pharmacodynamic study of OFIRMEV in pediatric patients under two years of age, as required to comply with our post-commercialization commitment to the FDA; or |
| | • | | acquire or in-license additional products, businesses or technologies that we believe are a strategic fit. |
Our funding requirements related to the commercialization of OFIRMEV may exceed our current projections as a result of many factors, including, but not limited to:
| | • | | our sales of OFIRMEV may be lower than expected; |
| | • | | the costs associated with our efforts to sell, market and distribute OFIRMEV, including costs associated with maintaining our sales force and commercial infrastructure, may be greater than anticipated; |
27
| | • | | we may incur unexpected costs in order to ensure a sufficient supply of OFIRMEV from our contract manufacturers in order to meet customer demand, including any replacement of product or write down of inventory related to any product recall or other quality issue, or we may be required to pay fees based on minimum purchase obligations; and |
| | • | | we may be required to file lawsuits to defend our patent rights or regulatory exclusivities from challenges by companies seeking to market generic versions of intravenous acetaminophen, such as our intellectual property litigation, including any such costs we may be required to expend if our licensors are unwilling or unable to do so. |
Until we can generate a sufficient amount of revenue from sales of OFIRMEV, if ever, we expect to finance future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements, as well as through interest income earned on cash and investment balances. We have engaged in various financing activities in the past. In May 2011, for example, we established a universal shelf registration statement to permit us, from time to time, to offer and sell up to $150.0 million of equity or debt securities. In November 2011, we undertook a public offering of common stock using our universal shelf registration statement that raised net proceeds of approximately $77.3 million. In addition, we have refinanced our $30.0 million secured credit facility with Oxford Finance LLC, Silicon Valley Bank and General Electric Capital Corporation on various occasions, including most recently in December 2012. However, there can be no assurance in the future that we would be able to enter into similar financing arrangements or complete any securities offerings, including under our universal shelf registration statement, and to the extent that we raise additional capital by issuing equity securities, our existing stockholders’ ownership will be diluted.
We believe we have sufficient financial resources to fund our projected operating requirements, at a minimum, for the next twelve months. This estimate does not reflect any participation in strategic transactions. We have based this estimate on assumptions that may prove to be wrong and we could spend our available financial resources faster than we currently expect. We cannot be certain that additional funding will be available on acceptable terms, or at all. If adequate funds are not available, we may be required to reduce the scope of or eliminate some or all of our sales, marketing and commercialization efforts for OFIRMEV, or we may not be able to adequately fund our intellectual property litigation, which could decrease sales of this product and have a material adverse effect on our financial condition, stock price and operations.
Although OFIRMEV has received regulatory approval from the FDA, it remains subject to substantial, ongoing regulatory requirements.
OFIRMEV remains subject to ongoing FDA requirements with respect to manufacturing, labeling, packaging, storage, distribution, advertising, promotion, record-keeping and submission of safety and other post-market information on the drug. The FDA has the authority to regulate the claims we make in marketing OFIRMEV to ensure that such claims are true, not misleading, supported by scientific evidence and consistent with the approved label for the drug. In addition, the discovery of previously unknown problems with OFIRMEV, such as adverse events of unanticipated severity or frequency, or problems with the facility where the product is manufactured, may result in the imposition of additional restrictions, including withdrawal of the product from the market.
For example, as a condition of the approval of OFIRMEV, we are required to complete an efficacy, pharmacokinetic and pharmacodynamic study of OFIRMEV in pediatric patients under two years of age, and to submit the final results of this clinical trial to the FDA. Depending on the outcome of this study, we may be unable to expand the indications for OFIRMEV or we may be required to include specific warnings or limitations on dosing this product, which could negatively impact our sales of OFIRMEV. Enrollment in this study began in the third quarter of 2012.
We have implemented a comprehensive compliance program and related infrastructure, but we cannot provide absolute assurance that we are or will be in compliance with all potentially applicable laws and
28
regulations. If our operations in relation to OFIRMEV fail to comply with applicable regulatory requirements, the FDA or other regulatory agencies may:
| | • | | issue warning letters or untitled letters; |
| | • | | impose consent decrees, which may include the imposition of various fines, reimbursements for inspection costs, required due dates for specific actions and penalties for noncompliance; |
| | • | | impose fines other civil or criminal penalties; |
| | • | | suspend regulatory approval; |
| | • | | suspend any ongoing clinical trials; |
| | • | | refuse to approve pending applications or supplements to approved applications filed by us; |
| | • | | impose restrictions on operations, including costly new manufacturing requirements; |
| | • | | exclude us from participating in U.S. federal healthcare programs, including Medicaid or Medicare; or |
| | • | | seize or detain products or require a product recall. |
In addition to FDA restrictions, numerous other federal, state and local laws and regulations apply to the promotion and sale of pharmaceutical products, such as the federal anti-kickback statute and false claims laws and regulations. The federal anti-kickback statute prohibits, among other things, knowingly and willfully offering, paying, soliciting or receiving remuneration to induce or in return for purchasing, leasing, ordering or arranging for the purchase, lease or order of any healthcare item or service reimbursable under Medicare, Medicaid or other federally financed healthcare programs. This statute has been interpreted to apply to arrangements between pharmaceutical manufacturers on the one hand and prescribers, purchasers and formulary managers on the other. Although there are a number of statutory exemptions and regulatory safe harbors protecting certain common activities from prosecution or other regulatory sanctions, the exemptions and safe harbors are drawn narrowly, and practices that involve remuneration intended to induce prescribing, purchases or recommendations may be subject to scrutiny if they do not qualify for an exemption or safe harbor. Our practices may not in all cases meet all of the criteria for safe harbor protection from anti-kickback liability.
Federal false claims laws prohibit any person from knowingly presenting, or causing to be presented, a false claim for payment to the federal government, or knowingly making, or causing to be made, a false statement to have a false claim paid. There are also federal criminal laws that prohibit executing a scheme to defraud any healthcare benefit program or making false statements relating to healthcare matters. Recently, several pharmaceutical and other healthcare companies have been prosecuted under these laws for allegedly inflating drug prices they report to pricing services, which in turn are used by the government to set Medicare and Medicaid reimbursement rates, and for allegedly providing free product to customers with the expectation that the customers would bill federal programs for the product. In addition, certain marketing practices, including off-label promotion, may also violate false claims laws. The majority of states also have statutes or regulations similar to the federal anti-kickback statute and false claims laws, which apply to items and services reimbursed under Medicaid and other state programs, or, in several states, apply regardless of the payor. Sanctions under these federal and state laws may include civil monetary penalties, exclusion of a manufacturer’s products from reimbursement under government programs, criminal fines and imprisonment.
Because of the breadth of these laws and the narrowness of the safe harbors, it is possible that some of our business activities could be subject to challenge under one or more of such laws. Such a challenge could have a material adverse effect on our business, financial condition, results of operations and growth prospects. Further, the recently enacted PPACA, among other things, amends the intent requirement of the federal anti-kickback and criminal healthcare fraud statutes. A person or entity can now be found guilty of fraud or false claims under PPACA without actual knowledge of the statute or specific intent to violate it. In addition, PPACA provides that the government may assert that a claim including items or services resulting from a violation of the federal anti-kickback statute constitutes a false or fraudulent claim for purposes of the false claims statutes. Possible
29
sanctions for violation of these anti-kickback laws include monetary fines, civil and criminal penalties, exclusion from Medicare, Medicaid and other government programs and forfeiture of amounts collected in violation of such prohibitions. Any violations of these laws, or any action against us for violation of these laws, even if we successfully defend against it, could result in a material adverse effect on our reputation, business, results of operations and financial condition.
The PPACA also imposes new reporting and disclosure requirements on device and drug manufacturers for any “transfer of value” made or distributed to prescribers and other healthcare providers. In addition, device and drug manufacturers will also be required to report and disclose any investment interests held by physicians and their immediate family members during the preceding calendar year. Failure to submit required information may result in civil monetary penalties of up to an aggregate of $150,000 per year (and up to an aggregate of $1 million per year for “knowing failures”), for all payments, transfers of value or ownership or investment interests not reported in an annual submission. Manufacturers will be required to begin data collection on August 1, 2013 and report such data to CMS by March 31, 2014 and on the 90th day of every calendar year for the reporting period of the previous year.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians for marketing. Some states, such as California, Massachusetts and Vermont, mandate implementation of compliance programs, along with the tracking and reporting of gifts, compensation and other remuneration to physicians. The shifting commercial compliance environment and the need to build and maintain robust and expandable systems to comply with different compliance and/or reporting requirements in multiple jurisdictions increase the possibility that a healthcare company may run afoul of one or more of the requirements.
The scope and enforcement of these laws is uncertain and subject to change in the current environment of healthcare reform, especially in light of the lack of applicable precedent and regulations. We cannot predict the impact on our business of any changes in these laws. Federal or state regulatory authorities may challenge our current or future activities under these laws. Any such challenge could have a material adverse effect on our reputation, business, results of operations, and financial condition. Any state or federal regulatory review of us, regardless of the outcome, would be costly and time-consuming.
Regulatory approval for any approved product is limited by the FDA to those specific indications and conditions for which clinical safety and efficacy have been demonstrated.
Any regulatory approval is limited to those specific diseases and indications for which a product is deemed to be safe and effective by the FDA. In addition to the FDA approval required for new formulations, any new indication for an approved product also requires FDA approval. If we are not able to obtain FDA approval for any desired future indications for our products, our ability to effectively market and sell our products may be reduced and our business may be adversely affected.
While physicians may choose to prescribe drugs for uses that are not described in the product’s labeling and for uses that differ from those tested in clinical studies and approved by the regulatory authorities, our ability to promote the products is limited to those indications that are specifically approved by the FDA. Regulatory authorities in the U.S. generally do not regulate the behavior of physicians in their choice of treatments, and such off-label uses by healthcare professionals are common. Regulatory authorities do, however, restrict communications by pharmaceutical companies on the subject of off-label use. If our promotional activities fail to comply with these regulations or guidelines, we may be subject to warnings from, or enforcement action by, these authorities. In addition, our failure to follow FDA rules and guidelines relating to promotion and advertising may cause the FDA to suspend or withdraw an approved product from the market, require a recall or institute fines, or could result in disgorgement of money, operating restrictions, injunctions or criminal prosecution, any of which could harm our business.
30
We are subject to new legislation, regulatory proposals and healthcare payor initiatives that may increase our costs of compliance and adversely affect our ability to market our products, obtain collaborators and raise capital.
In March 2010, the PPACA became law and made extensive changes to the delivery of health care in the U.S. The PPACA includes numerous provisions that affect pharmaceutical companies, some of which became effective immediately and others of which will be taking effect over the next several years. The PPACA seeks to expand health care coverage to the uninsured through private health insurance reforms and an expansion of Medicaid. The PPACA also imposes substantial costs on pharmaceutical manufacturers, such as an increase in liability for rebates paid to Medicaid, new drug discounts that must be offered to certain enrollees in the Medicare prescription drug benefit, an annual fee imposed on all manufacturers of brand prescription drugs in the U.S., and an expansion of an existing program requiring pharmaceutical discounts to certain types of hospitals and federally subsidized clinics. The PPACA also promotes programs that increase the federal government’s comparative effectiveness research, which may be used to evaluate the selection of medical services by clinicians and others. In addition, PPACA implements payment system reforms such as a national pilot program on payment bundling to encourage hospitals, physicians and other providers to improve the coordination, quality and efficiency of certain healthcare services through bundled payment models, and creates an independent payment advisory board that will submit recommendations to reduce Medicare spending if projections of such spending exceed a specified growth rate.
Other legislative changes have been proposed and adopted since the PPACA was enacted. On August 2, 2011, the Budget Control Act of 2011, among other things, created measures for spending reductions by Congress. A Joint Select Committee on Deficit Reduction, tasked with recommending a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, was unable to reach required goals, thereby triggering the legislation’s automatic reduction to several government programs. This includes aggregate reductions to Medicare payments to providers of up to 2% per fiscal year, starting in 2013. On January 2, 2013, President Obama signed into law the American Taxpayer Relief Act of 2012, or the ATRA, which, among other things, delayed for another two months the budget cuts mandated by these sequestration provisions of the Budget Control Act of 2011. The ATRA also reduced Medicare payments to several providers, including hospitals, imaging centers and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years. These new laws, as well as legislative and regulatory proposals that may be adopted from time to time in the future, may result in additional reductions in Medicare and other health care funding, which could have a material adverse effect on our customers and accordingly, our financial operations. The continuing efforts of the government, insurance companies, managed care organizations and other payors of health care services to contain or reduce costs of health care could result in decreased net revenues from our pharmaceutical products and decrease potential returns from our development efforts. Although there is some uncertainty as to the exact extent of the requirements and definitive guidance has not yet been provided by the government, it is currently anticipated that data collection requirements will begin no earlier than January 1, 2013. In addition, pharmaceutical and device manufacturers will also be required to report and disclose investment interests held by physicians and their immediate family members during the preceding calendar year. Failure to submit required information may result in civil monetary penalties for payments, transfers of value or ownership or investment interests not reported in an annual submission. Compliance with the PPACA and state laws with similar provisions is difficult and time consuming, and companies that do not comply with these state laws face civil penalties. Because of the breadth of these laws and the narrowness of the safe harbors, it is possible that some of our business activities could be subject to challenge under one or more of such laws. Such a challenge could have a material adverse effect on our business, financial condition, results of operations and growth prospects.
In addition, there have been a number of other legislative and regulatory proposals aimed at changing the pharmaceutical industry. In particular, California has enacted legislation that requires development of an electronic pedigree to track and trace each prescription drug at the saleable unit level through the distribution system. California’s electronic pedigree requirement is scheduled to take effect in January 2015. Compliance with California and future federal or state electronic pedigree requirements may increase our operational expenses and impose significant administrative burdens. As a result of these and other new proposals, we may determine to change our current manner of operation, provide additional benefits or change our contract
31
arrangements, any of which could have a material adverse effect on our business, financial condition and results of operations. Managed care organizations are increasingly challenging the prices charged for medical products and services and, in some cases, imposing restrictions on the coverage of particular drugs. Many managed care organizations negotiate the price of medical services and products and develop formularies which establish pricing and reimbursement levels. Exclusion of a product from a formulary can lead to its sharply reduced usage in the managed care organization’s patient population. The process for obtaining coverage can be lengthy and costly, and we expect that it could take several months before a particular payor initially reviews our product and makes a decision with respect to coverage. For example, third-party payors may require cost-benefit analysis data from us in order to demonstrate the cost-effectiveness of OFIRMEV or any other product we might bring to market. For any individual third-party payor, we may not be able to provide data sufficient to gain reimbursement on a similar or preferred basis to competitive products, or at all.
We may never receive approval outside of the U.S. to commercialize OFIRMEV or any other products or product candidates we may acquire.
Our rights to OFIRMEV include Canada, as well as the U.S. In order to market OFIRMEV, and any other products or product candidates we may acquire, in Canada or other jurisdictions outside of the U.S., we must comply with numerous and varying regulatory requirements of other countries regarding non-clinical testing, manufacturing, clinical safety and efficacy. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval. For example, in July 2012, we filed a New Drug Submission for OFIRMEV with Health Canada which was accepted for review in August 2012. We are currently evaluating the commercial prospects and partnering opportunities for the product in Canada and anticipate that the product would not be approved by Canadian regulatory authorities for at least 18 months after this submission, if at all. The regulatory approval process in other countries may include all of the risks detailed above as well as other risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval could have the same adverse effects detailed above regarding FDA approval in the U.S. As described above, such effects include the risks that OFIRMEV and any other products may not be approved for all indications requested, which could limit the uses of our products and have an adverse effect on product sales and potential royalties, and that any regulatory approvals we may obtain may be subject to limitations on the indicated uses for which our products may be marketed or require us to perform costly, post-marketing follow-up studies.
Public concern regarding the safety of drug products such as OFIRMEV could result in new requirements from regulatory agencies to include unfavorable information in our labeling, or require us to undertake other activities that may entail additional costs.
In light of widely publicized events concerning the safety risk of certain drug products, the FDA, members of Congress, the Government Accountability Office, medical professionals and the general public have raised concerns about potential drug safety issues. These events have resulted in the withdrawal of drug products, revisions to drug labeling that further limit use of the drug products and the establishment of risk management programs that may, for example, restrict distribution of drug products after approval. For example, in January 2011, the FDA issued a press release and posted on its website a drug safety communication asking manufacturers of prescription drug products containing combinations of acetaminophen and opioid medications to limit the amount of acetaminophen to no more than 325 milligrams (mg) in each dosage unit (i.e. each tablet or caplet). In the announcement, the FDA also requested manufacturers to update labels for such products to include a boxed warning highlighting the potential for severe acetaminophen-induced liver injury and a warning highlighting the potential for allergic reactions. The boxed warning required for affected products reaffirms previous statements made by the FDA that most cases of liver injury are associated with acetaminophen doses that exceed 4,000 mg per day. While the FDA has indicated that this communication does not apply to
32
intravenous acetaminophen, it is possible that the FDA may apply similar labeling requirements to OFIRMEV in the future. We reaffirmed our dosing recommendations for OFIRMEV in July 2011 following a news release by a major manufacturer of over-the-counter acetaminophen products announcing its plan to lower the recommended maximum daily dose of some oral acetaminophen products in an effort to reduce the risk of accidental acetaminophen overdose among its customers in the over-the-counter setting.
Also, the California “State’s Experts” acting under Proposition 65 have recommended a high priority for a review of acetaminophen by the Office of Environmental Health Hazard Assessment, which, depending on subsequent research and findings, could lead to the requirement for a warning statement to be added to the label for over-the-counter acetaminophen products that such products contain chemicals known to the State of California to cause cancer. We believe that OFIRMEV, like other prescription products, would be exempt from this additional labeling requirement. However, any perception or concern that acetaminophen is unsafe could harm our ability to successfully commercialize and sell OFIRMEV, which could have a material adverse effect on our business, financial condition and results of operations.
In addition, the Food and Drug Administration Amendments Act of 2007, or FDAAA, granted significant expanded authority to the FDA, much of which is aimed at improving the safety of drug products before and after approval. In particular, the law authorizes the FDA to, among other things, require post-approval studies and clinical trials, mandate changes to drug labeling to reflect new safety information and require risk evaluation and mitigation strategies for certain drugs, including certain currently approved drugs. It also significantly expands the federal government’s clinical trial registry and results databank, which we expect will result in significantly increased government oversight of clinical trials. Under the FDAAA, companies that violate these and other provisions of that law are subject to substantial civil monetary penalties, among other regulatory, civil and criminal penalties. Data from clinical trials may receive greater scrutiny, particularly with respect to safety, which may make the FDA or other regulatory authorities more likely to require additional preclinical studies or clinical trials.
If our hospital customers fail to receive adequate reimbursement from the government or third-party payors for OFIRMEV or any future products we may license or acquire, if any, or if hospitals choose to use therapies that are less expensive, our revenue and prospects for profitability will be limited.
In both domestic and foreign markets, our anticipated sales of OFIRMEV or any future products will depend in part upon the reimbursement rates our customers receive for OFIRMEV. In particular, many U.S. hospitals receive a fixed reimbursement amount per procedure for certain surgeries and other treatment therapies they perform. Because this amount may not be based on the actual expenses the hospital incurs, hospitals may choose to use therapies which are less expensive when compared to our products. In addition, some third-party payors, including government health programs such as Medicare, managed care providers and commercial payors, are emphasizing the substitution of branded pharmaceuticals with less expensive generic equivalents. An increase in the sales of generic pharmaceutical products could result in a decrease in revenues of branded pharmaceuticals. While there are no generic equivalents competing with OFIRMEV at this time, in the future we could face generic competition.
OFIRMEV or any other products or product candidates that we may in-license or acquire, if approved, will face competition from other therapies and drugs, as well as other routes of administration of acetaminophen, for these limited hospital financial resources. We may need to conduct post-marketing studies in order to demonstrate the cost-effectiveness of any future products to the satisfaction of hospitals, other target customers and their third-party payors. Such studies might require us to commit a significant amount of management time and financial and other resources. Our future products might not ultimately be considered cost-effective and accordingly, we may be unable to maintain price levels sufficient to realize an appropriate return on investment in product development.
Governments continue to propose and pass legislation designed to reduce the cost of healthcare. In some foreign markets, such as Canada, the government controls the pricing of prescription pharmaceuticals. In these
33
countries, pricing negotiated with governmental authorities can take six to twelve months or longer after the receipt of regulatory marketing approval for a product. In the U.S., we expect that there will be an increase in federal and state proposals to implement pricing controls for prescription drugs, and new legislation and regulations affecting the pricing of pharmaceuticals might change before our product candidates are approved for marketing or after our marketed products have been approved. For example, the U.S. Congress is considering a number of legislative and regulatory proposals with an objective of ultimately reducing healthcare costs. Legislative and regulatory actions under consideration in the U.S. include health care reform initiatives that could significantly alter the market for pharmaceuticals (such as private health insurance expansion, the creation of competing public health insurance plans, a variety of proposals that would reduce government expenditures for prescription drugs to help finance healthcare reform, or the eventual transition of the U.S. multiple payer system to a single payer system). Other actions under consideration include proposals for government intervention in pharmaceutical pricing, changes in government reimbursement, an accelerated approval process for “follow-on” biologics, legalization of commercial drug importation into the U.S., and involuntary approval of medicines for over-the-counter use. Such legislation could result in the exclusion of OFIRMEV and any other products or product candidates we may license or acquire from hospital formularies, or lower the prices we would receive for our products or product candidates. Our revenues from the sale of OFIRMEV or any other approved products could be significantly reduced as a result of these cost containment measures and reforms, which would negatively impact our profitability.
If we breach any of the agreements under which we license rights to OFIRMEV from others, we could lose the ability to sell OFIRMEV.
In March 2006, we entered into an exclusive license agreement with BMS relating to OFIRMEV for the U.S. and Canada. Because we have in-licensed the rights to this product from a third party, if there is any dispute between us and our licensor regarding our rights under our license agreement, our ability to continue to sell this product may be adversely affected. Any uncured, material breach under our license agreement could result in our loss of exclusive rights to OFIRMEV and may lead to a complete termination of our related commercial efforts.
If BMS breaches the underlying agreement under which we sublicense the rights to OFIRMEV, we could lose the ability to sell OFIRMEV.
Our license for OFIRMEV is subject to the terms and conditions of a license from Pharmatop to BMS, under which BMS originally licensed the intellectual property rights covering OFIRMEV. If BMS materially breaches the terms or conditions of this underlying license from Pharmatop, and neither BMS nor we adequately cure that breach, or BMS and Pharmatop otherwise become involved in a dispute, the breach by BMS or disputes with Pharmatop could result in a loss of, or other material adverse impact on, our rights under our license agreement with BMS. While we would expect to exercise all reasonable rights and remedies available to us, including seeking to cure any breach by BMS, and otherwise seek to preserve our rights under the patents licensed by Pharmatop, we may not be able to do so in a timely manner, at an acceptable cost or at all. Any uncured, material breach under the license from Pharmatop to BMS could result in our loss of exclusive rights to OFIRMEV and may lead to a complete termination of our commercial efforts for OFIRMEV.
We may experience difficulties in managing the growth of our organization.
As of March 1, 2013, we had approximately 206 employees. The commercial launch of OFIRMEV in January 2011 required us to substantially expand our managerial, commercial, financial and other personnel resources, particularly in sales and marketing positions. Additionally, beginning in November 2011, we implemented a reduction in force of 17 employees, or approximately 7% of our total work force at that time, primarily in our development and general and administrative areas. This action was taken in order to focus our resources on commercialization activities for OFIRMEV and to reduce programmatic costs not directly associated with such efforts. Despite these efforts, our management, personnel, systems and facilities currently in place may not be adequate to support our commercially-focused organization, and we may not be able to retain or
34
recruit qualified personnel in the future due to competition for personnel among pharmaceutical businesses. The failure to do so could have a significant negative impact on our future product revenues and business results.
Our need to effectively manage our operations, growth and various projects requires that we:
| | • | | effectively train and manage our employees, and establish appropriate systems, policies and infrastructure to support our organization; |
| | • | | ensure that our consultants and other service providers successfully carry out their contractual obligations, provide high quality results, and meet expected deadlines; |
| | • | | continue to carry out our own contractual obligations to our licensors and other third parties; and |
| | • | | continue to improve our operational, financial and management controls, reporting systems and procedures. |
We may be unable to successfully implement these tasks on a larger scale and, accordingly, may not achieve our commercialization goals.
We may not be able to manage our business effectively if we are unable to attract and retain key personnel.
We may not be able to attract or retain qualified management and commercial, scientific and clinical personnel in the future due to the intense competition for qualified personnel among biotechnology, pharmaceutical and other businesses, particularly in the San Diego, California area. If we are not able to attract and retain necessary personnel to accomplish our business objectives, we may experience constraints that will significantly impede the achievement of our development objectives, our ability to raise additional capital and our ability to implement our business strategy.
Our industry has experienced a high rate of turnover of management personnel in recent years. We are highly dependent on the expertise of our senior management, particularly Theodore R. Schroeder, our President and Chief Executive Officer, William R. LaRue, our Senior Vice President, Chief Financial Officer, Treasurer and Assistant Secretary, and Scott A. Byrd, our Senior Vice President and Chief Commercial Officer. If we lose one or more of these key employees, our ability to successfully implement our business strategy could be seriously harmed. Replacing key employees may be difficult and may take an extended period of time because of the limited number of individuals in our industry with the breadth of skills and experience required to commercialize products successfully. Competition to hire from this limited pool is intense, and we may be unable to hire, train, retain or motivate these additional key personnel. Although we have employment agreements with Mr. Schroeder, Mr. LaRue and Mr. Byrd, these agreements are terminable at will at any time with or without notice and, therefore, we may not be able to retain their services as expected. During the third quarter of 2012, our former Executive Vice President and Chief Medical Officer departed in connection with our current focus on commercialization, rather than new product development. Any attempt to develop new products in the future could be limited unless we were able to hire a suitable replacement.
In addition, we have scientific and clinical advisors who assist us in product development and clinical strategies. These advisors are not our employees and may have commitments to, or consulting or advisory contracts with, other entities that may limit their availability to us, or may have arrangements with other companies to assist in the development of products that may compete with ours.
Our business and operations would suffer in the event of system failures.
Despite the implementation of security measures, our internal computer systems are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. Any system failure, accident or security breach that causes interruptions in our operations could result in a material disruption of our drug development programs. To the extent that any disruption or security breach results in a loss or damage to our data or applications, or inappropriate disclosure of confidential or proprietary information, we may incur liability and our operations may be set back.
35
Our future growth depends on our ability to identify and acquire or in-license products and if we do not successfully identify and acquire or in-license related products or product candidates or integrate them into our operations, we may have limited growth opportunities.
An important part of our business strategy is to continue to develop a pipeline of products and product candidates by acquiring or in-licensing products, businesses or technologies that we believe are a strategic fit with our focus on the hospital marketplace. As part of our efforts to acquire businesses or to in-license products, we conduct technical, business and legal due diligence with the goal of identifying and evaluating material risks involved in such transactions, which may include:
| | • | | exposure to unknown liabilities; |
| | • | | disruption of our business and diversion of our management’s time and attention to develop acquired products or technologies; |
| | • | | difficulty or inability to secure financing to fund development activities for such acquired or in-licensed technologies in the current economic environment; |
| | • | | incurrence of substantial debt or dilutive issuances of securities to pay for acquisitions; |
| | • | | higher than expected acquisition and integration costs; |
| | • | | increased amortization expenses; |
| | • | | difficulty and cost in combining the operations and personnel of any acquired businesses with our operations and personnel; |
| | • | | effectiveness of the acquired business’s internal controls and procedures; |
| | • | | impairment of relationships with key suppliers or customers of any acquired businesses due to changes in management and ownership; and |
| | • | | inability to retain key employees of any acquired businesses. |
Additionally, in connection with any such acquisition or in-licensing transaction, we must estimate the value of the transaction by making certain assumptions about, among other things, likelihood of regulatory approval for unapproved products and the market potential for marketed products and/or product candidates. Ultimately, our assumptions may prove to be incorrect, which could cause us to fail to realize the anticipated benefits of a transaction. Despite our efforts, we ultimately may be unsuccessful in ascertaining or evaluating all such risks and, as a result, we might not realize the intended advantages of the acquisition or in-licensing transaction. If we fail to realize the expected benefits from the transactions we have consummated or may consummate in the future, the results of our operations and financial condition could be adversely affected.
It cannot be assured that, following an acquisition, we will achieve revenues, specific net income or loss levels that justify the acquisition or that the acquisition will result in increased earnings, or reduced losses, for the combined company in any future period. Moreover, we may need to raise additional funds through public or private debt or equity financings to acquire any businesses, which would result in dilution for stockholders or the incurrence of indebtedness. We may not be able to operate acquired businesses profitably or otherwise implement our growth strategy successfully.
We have limited resources to identify and execute the acquisition or in-licensing of third-party products, businesses and technologies and integrate them into our current infrastructure. In particular, we may compete with larger pharmaceutical companies and other competitors in our efforts to establish new collaborations and in-licensing opportunities. These competitors likely will have access to greater financial resources than us and may have greater expertise in identifying and evaluating new opportunities. Moreover, we may devote resources to potential acquisitions or in-licensing opportunities that are never completed, or we may fail to realize the anticipated benefits of such efforts.
36
Our business involves the use of hazardous materials and we and our third-party manufacturer must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
Our third-party manufacturer’s activities and, to a lesser extent, our own activities involve the controlled storage, use and disposal of hazardous materials, including the components of OFIRMEV and other hazardous compounds. We and our manufacturer are subject to federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of these hazardous materials. Although we believe that the safety procedures for handling and disposing of these materials comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental contamination or injury from these materials. In the event of an accident, state or federal authorities may curtail our use of these materials and interrupt our business operations.
Risks Related to Intellectual Property
The patent rights that we have in-licensed covering OFIRMEV are limited to a specific intravenous formulation of acetaminophen. As a result, our market opportunity for this product may be limited by the lack of patent protection for the active ingredient itself and other formulations of intravenous acetaminophen may be developed by competitors.
The active ingredient in OFIRMEV is acetaminophen. Patent protection is not available for the acetaminophen molecule itself in the territories licensed to us, which include the U.S. and Canada. As a result, competitors who obtain the requisite regulatory approval can offer products with the same active ingredient as OFIRMEV so long as the competitors do not infringe any process or formulation patents that we have in-licensed from BMS and its licensor, Pharmatop. We are the exclusive licensee of two U.S. patents and two issued Canadian patents owned by Pharmatop, under BMS’s license to these patents from Pharmatop. U.S. Patent No. 6,028,222, or the ‘222 patent (Canadian patent number 2,233,924), covers the formulation of OFIRMEV, and this patent expires in August 2017. U.S. Patent No. 6,992,218, or the ‘218 patent (Canadian patent number 2,415,403), covers the process used to manufacture OFIRMEV, and this patent expires in June 2021. We plan to complete a pediatric clinical trial of OFIRMEV by August 2015 and, upon timely completion and the acceptance by the FDA of the data from this study, OFIRMEV will be eligible for an additional six months of marketing exclusivity in the U.S.
We are also aware of several U.S. and Canadian patents and patent applications directed to various potential injectable formulations of acetaminophen as well as methods of making and using these potential formulations. For example, Injectapap, a liquid formulation of acetaminophen for intramuscular injection, was approved by the FDA for the reduction of fever in adults in March 1986, although it was subsequently withdrawn from the market by McNeil Pharmaceutical in July 1986. The number of patents and patent applications directed to products in the same field as OFIRMEV indicates that competitors have sought to develop and may seek to market competing formulations that may not be covered by our licensed patents and patent applications. The commercial opportunity for OFIRMEV could be significantly harmed if competitors are able to develop alternative formulations of acetaminophen outside the scope of our in-licensed patents. We are also aware of a number of third-party patents in the U.S. that claim methods of making acetaminophen. If a supplier of the API for OFIRMEV is found to infringe any of these method patents covering acetaminophen, our supply of the API could be delayed and we may be required to locate an alternative supplier.
Four third-parties have challenged, and additional third parties may challenge, the patents covering OFIRMEV, which could result in the invalidation or unenforceability of some or all of the relevant patent claims. If a third party files an ANDA for a generic drug product containing acetaminophen and relies in whole or in part on studies conducted by or for us, the third party will be required to certify to the FDA that, in the opinion of that third party, the patent listed in the Orange Book for a branded product is invalid, unenforceable, or will not be infringed by the manufacture, use or sale of the third party’s generic drug product. A third party certification that the new product will not infringe the Orange Book-listed patents for OFIRMEV, or that such patents are invalid, is called a Paragraph IV patent certification. If the third party submits a Paragraph IV patent certification to the
37
FDA, a notice of the Paragraph IV patent certification must also be sent to us once the third-party’s ANDA is accepted for filing by the FDA. A lawsuit may then be initiated to defend the patents identified in the notice. The filing of a patent infringement lawsuit within 45 days of the receipt of notice of a Paragraph IV patent certification automatically prevents the FDA from approving the ANDA until the earlier of the expiration of a 30-month period, the expiration of the patents, the entry of a settlement order stating that the patents are invalid or not infringed, a decision in the infringement case that is favorable to the ANDA applicant, or such shorter or longer period as the court may order. If a patent infringement lawsuit is not initiated within the required 45-day period, the third-party’s ANDA will not be subject to the 30-month stay. Regardless of the outcome of any litigation, no ANDA or NDA can receive final approval from the FDA before expiration of the regulatory exclusivity period for OFIRMEV in November 2013.
In August 2011, we and Pharmatop filed suit in the United States District Court for the District of Delaware against Perrigo and Exela alleging that each has infringed the ‘222 and ‘218 patents, which are listed in the Orange Book for OFIRMEV, by filing their respective ANDA seeking approval from the FDA to market a generic version of OFIRMEV prior to the expiration of these patents. The lawsuit was filed within 45 days of receipt of the pertinent notice letters, thereby triggering a stay of FDA approval of the Perrigo ANDA and the Exela ANDA until the earlier of the expiration of a 30-month period, the expiration of the ‘222 and ‘218 patents, the entry of a settlement order or consent decree stating that the ‘222 and ‘218 patents are invalid or not infringed, a decision in the case concerning infringement or validity that is favorable to Exela and/or Perrigo, or such shorter or longer period as the Court may order. Each of Perrigo and Exela filed an answer in the case that asserts, among other things, non-infringement and invalidity of the asserted patents, and has also filed counterclaims. A date for the bench trial in this case has been scheduled for May 2013.
We settled with Perrigo and the case against Perrigo was dismissed in November 2012. Under the settlement and license agreements with Perrigo, Perrigo has been granted the exclusive right of first refusal to negotiate an agreement with us to market an authorized generic version of OFIRMEV in the U.S. in the event that we elect to launch an authorized generic version of the product. Additionally, we granted Perrigo the non-exclusive right to market a generic intravenous acetaminophen product in the U.S. under Perrigo’s ANDA after December 6, 2020, or earlier under certain circumstances. The FTC, or the DOJ could seek to challenge our settlement with Perrigo, or a competitor, customer or other third-party could initiate a private action under antitrust or other laws challenging our settlement with Perrigo. Any such challenge could be both expensive and time consuming and may render the settlement agreement unenforceable.
In January 2013, we and Pharmatop filed suit in the United States District Court for the Southern District of California and the Northern District of Illinois against Fresenius Kabi USA, LLC, or Fresenius. In February 2013, we filed suit in the United States District Court for the Southern District of California and the District of New Jersey against Sandoz, Inc., or Sandoz. In these cases, we have alleged that each of Fresenius and Sandoz has each infringed the ‘222 and ‘218 patents by filing their respective NDA, in the case of Fresenius, or ANDA, in the case of Sandoz, seeking approval from the FDA to market a generic version of OFIRMEV prior to the expiration of our patents for OFIRMEV. Both the Fresenius and Sandoz lawsuits were filed within 45 days of receipt of the respective notice letters, thereby triggering a stay of FDA approval of the Fresenius NDA and the Sandoz ANDA until the earlier of the expiration of a 30-month period, the expiration of the ‘222 and ‘218 patents, the entry of a settlement order or consent decree stating that the ‘222 and ‘218 patents are invalid or not infringed, a decision in the case concerning infringement or validity that is favorable to Fresenius and/or Sandoz, or such shorter or longer period as the courts may order. Fresenius has filed an answer in the Southern District of California that asserts, among other things, non-infringement and invalidity of the asserted patents, and has also filed counterclaims. Sandoz has not yet filed an answer in either District.
Litigation or other proceedings to enforce or defend intellectual property rights are often very complex in nature, may be very expensive and time-consuming, may divert our management’s attention from our core business, and may result in unfavorable results that could adversely impact our ability to prevent third parties from competing with our products. Any adverse outcome of such litigation could result in one or more generic
38
versions of OFIRMEV being launched before the expiration of the patents we have in-licensed from BMS and its licensor, Pharmatop, which could adversely affect our ability to successfully execute our business strategy to increase sales of OFIRMEV and negatively impact our financial condition and results of operations. We intend to vigorously enforce our intellectual property rights relating to OFIRMEV to prevent the marketing of infringing generic products prior to the expiration of our patents. However, given the unpredictability inherent in litigation, we cannot predict or guarantee the outcome of these matters or any other litigation. Regardless of how these matters are ultimately resolved, these matters may be costly, time-consuming and distracting to our management, which could have a material adverse effect on our business.
The protection of our intellectual property rights is critical to our success and any failure on our part to adequately secure such rights would materially affect our business.
Our commercial success depends on maintaining patent protection and trade secret protection for OFIRMEV, as well as for any other products or product candidates that we may license or acquire, and successfully defending these patents and trade secrets against third-party challenges. We will only be able to protect our technologies from unauthorized use by third parties to the extent that valid and enforceable patents or trade secrets cover them.
For example, in April 2012, Exela filed suit against David J. Kappos and the USPTO in the United States District Court for the Eastern District of Virginia for declaratory judgment seeking a reversal of the USPTO’s decision not to act on a petition by Exela to vacate the USPTO’s April 2003 order reviving the international application for the ‘218 patent. The lawsuit followed the USPTO’s rejection of Exela’s petition to the USPTO filed in November 2011, which sought to vacate the April 23, 2003 order granting Pharmatop’s petition to revive the ‘218 patent. The USPTO determined that Exela lacks standing to seek such relief. Exela also seeks declaratory judgment that the USPTO’s rules and regulations that allow for revival of abandoned, international patent applications under the “intentional” standard are invalid, and similar relief in connection with one or more counterclaims it has filed in the Delaware litigation. Our motion to intervene in this lawsuit was granted in October 2012. In December 2012, the district court dismissed the case with prejudice as barred by the applicable statute of limitations. In February 2013, Exela appealed the court’s decision to the Court of Appeals for the Federal Circuit. A decision by the Court of Appeals in favor of Exela could result in the invalidation of the ‘218 patent.
Additionally, in September 2012, an unidentified third party filed with the USPTO a Request for Ex Parte Reexamination of the ‘222 patent. In December 2012, we received notice that the USPTO had granted the Request for Reexamination. The reexamination process is provided for by law and requires the USPTO to consider the scope and validity of the patent based on substantial new questions of patentability raised by a third party or the USPTO. Because we and Pharmatop believe that the scope and validity of the patent claims in this patent are appropriate and that the USPTO’s prior issuance of the patent was correct, we, in conjunction with Pharmatop, will vigorously defend this patent. We cannot predict whether we and Pharmatop ultimately will succeed in maintaining the scope and validity of the claims of this patent during reexamination. If the patent claims in this patent ultimately are narrowed during prosecution before the USPTO, the extent of the patent coverage afforded to OFIRMEV could be impaired, which could potentially harm our business and operating results.
The patent positions of pharmaceutical and biotechnology companies can be highly uncertain and involve complex legal and factual questions for which important legal principles remain unresolved. No consistent policy regarding the breadth of claims allowed in pharmaceutical or biotechnology patents has emerged to date in the U.S. The patent situation outside the U.S. is even more uncertain. Changes in either the patent laws or in interpretations of patent laws in the U.S. and other countries may diminish the value of our intellectual property. Accordingly, we cannot predict the breadth of claims that may be allowed or enforced in our patents or in third-party patents.
39
The degree of future protection for our proprietary rights is uncertain, because legal means afford only limited protection and may not adequately protect our rights or permit us to gain or keep our competitive advantage. For example:
| | • | | our licensors might not have been the first to make the inventions covered by each of our pending patent applications and issued patents; |
| | • | | our licensors might not have been the first to file patent applications for these inventions; |
| | • | | others may independently develop similar or alternative technologies or duplicate any of our products, product candidates or technologies; |
| | • | | it is possible that none of the pending patent applications licensed to us will result in issued patents; |
| | • | | the issued patents covering our products or product candidates may not provide a basis for commercially viable active products, may not provide us with any competitive advantages, or may be challenged by third parties; |
| | • | | we may not develop additional proprietary technologies that are patentable; or |
| | • | | patents of others may have an adverse effect on our business. |
Patent applications in the U.S. are maintained in confidence for at least 18 months after their earliest effective filing date. Consequently, we cannot be certain that our licensors were the first to invent or the first to file patent applications on our products or product candidates. In the event that a third party has also filed a U.S. patent application relating to our products or product candidates or a similar invention, we may have to participate in interference proceedings declared by the USPTO to determine priority of invention in the U.S. The costs of these proceedings could be substantial and it is possible that our efforts would be unsuccessful, resulting in a material adverse effect on our U.S. patent position. Furthermore, we may not have identified all U.S. and foreign patents or published applications that affect our business either by blocking our ability to commercialize our drugs or by covering similar technologies that affect our drug market.
In addition, some countries, including many in Europe, do not grant patent claims directed to methods of treating humans, and in these countries patent protection may not be available at all to protect our products or product candidates. Even if patents are issued, we cannot guarantee that the claims of those patents will be valid and enforceable or provide us with any significant protection against competitive products, or otherwise be commercially valuable to us.
We also rely on trade secrets to protect our technology, particularly where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. While we use reasonable efforts to protect our trade secrets, our licensors, employees, consultants, contractors, outside scientific collaborators and other advisors may unintentionally or willfully disclose our information to competitors. Enforcing a claim that a third party illegally obtained and is using our trade secrets is expensive and time consuming, and the outcome is unpredictable. In addition, courts outside the U.S. are sometimes less willing to protect trade secrets. Moreover, our competitors may independently develop equivalent knowledge, methods and know-how.
If our licensors or we fail to obtain or maintain patent protection or trade secret protection for OFIRMEV or any other product or product candidate we may license or acquire, third parties could use our proprietary information, which could impair our ability to compete in the market and adversely affect our ability to generate revenues and achieve profitability.
40
We depend on our licensors for the maintenance and enforcement of our intellectual property and have limited control, if any, over the amount or timing of resources that our licensors devote on our behalf, or whether any financial difficulties experienced by our licensors could result in their unwillingness or inability to secure, maintain and enforce patents protecting our intellectual property.
We depend on our licensor, BMS, and its licensor, Pharmatop, to protect the proprietary rights covering OFIRMEV and we have limited, if any, control over the amount or timing of resources that BMS or Pharmatop devote on our behalf, or the priority they place on maintaining and enforcing our patent rights, and prosecuting patent applications to our advantage.
Pharmatop is under a contractual obligation to BMS to maintain the issued OFIRMEV patents in the U.S., and to diligently prosecute the patent applications and maintain any issued patents related to OFIRMEV in Canada. BMS has the opportunity to consult, review and comment on any patent office communications. We may not receive any patent from the applications in Canada, or if patents are issued they may be inadequate to protect our OFIRMEV product from competition.
For a third-party challenge to the validity or enforceability of the OFIRMEV patents, we will have some ability to participate in either Pharmatop’s or BMS’ defense thereof. In the event that neither Pharmatop nor BMS elects to defend the third-party challenge, we may have the opportunity to defend it. BMS has the first right to prosecute a third-party infringement of the OFIRMEV patents relating to OFIRMEV, and Pharmatop has the second right. We may not have the ability to cooperate with BMS or Pharmatop in any such third-party infringement suits. In certain instances, we may be allowed to pursue a third-party infringement claim ourselves.
It is possible that Pharmatop or BMS could take some action or fail to take some action that could harm the patents related to OFIRMEV. For example, if Pharmatop decides it no longer wants to maintain the OFIRMEV patents, to prosecute the patent applications related to OFIRMEV in Canada, or if Pharmatop or BMS decide not to defend the patents against third party challenges, we risk losing the benefit of all or some of those patent rights. Moreover, Pharmatop or BMS may experience serious difficulties related to their respective businesses or financial stability, and may be unwilling or unable to continue to expend the financial resources required to maintain and prosecute these patents and patent applications, or to defend the patents against third party challenges.
Our success will depend in part on our ability to obtain and maintain patent protection for OFIRMEV, both in the U.S. and Canada. While we intend to take actions reasonably necessary to enforce our patent rights, we depend on our licensors to protect a substantial portion of our proprietary rights. We may have limited, if any, control or involvement over the defense of these claims, and our licensors could be subject to injunctions and temporary or permanent exclusionary orders in the U.S. or other countries.
We or our licensors may also be notified of alleged infringement and be sued for infringement of third-party patents or other proprietary rights. Our licensors are not obligated to defend or assist in our defense against third-party claims of infringement. We have limited, if any, control over the amount or timing of resources, if any, that our licensors devote on our behalf or the priority they place on defense of such third-party claims of infringement. Because of the uncertainty inherent in any patent or other litigation involving proprietary rights, we or our licensors may not be successful in defending claims of intellectual property infringement by third parties, which could have a material adverse effect on our results of operations. Regardless of the outcome of any litigation, defending the litigation may be expensive, time-consuming and distracting to management.
If we are sued for infringing intellectual property rights of third parties, it will be costly and time consuming, and an unfavorable outcome in any litigation would harm our business.
Our ability to develop, manufacture, market and sell OFIRMEV or any other products or product candidates that we may license or acquire depends upon our ability to avoid infringing the proprietary rights of third parties. Numerous U.S. and foreign issued patents and pending patent applications, which are owned by third parties,
41
exist in the general fields of pain treatment and cover the use of numerous compounds and formulations in our targeted markets. Because of the uncertainty inherent in any patent or other litigation involving proprietary rights, we and our licensors may not be successful in defending intellectual property claims by third parties, which could have a material adverse effect on our results of operations. Regardless of the outcome of any litigation, defending the litigation may be expensive, time-consuming and distracting to management. In addition, because patent applications can take many years to issue, there may be currently pending applications, unknown to us, which may later result in issued patents that OFIRMEV may infringe. There could also be existing patents of which we are not aware that OFIRMEV may inadvertently infringe.
There is a substantial amount of litigation involving patent and other intellectual property rights in the biotechnology and biopharmaceutical industries generally. If a third party claims that we infringe on their products or technology, we could face a number of issues, including:
| | • | | infringement and other intellectual property claims which, with or without merit, can be expensive and time consuming to litigate and can divert management’s attention from our core business; |
| | • | | substantial damages for past infringement which we may have to pay if a court decides that our product infringes on a competitor’s patent; |
| | • | | a court prohibiting us from selling or licensing our product unless the patent holder licenses the patent to us, which it would not be required to do; |
| | • | | if a license is available from a patent holder, we may have to pay substantial royalties or grant cross licenses to our patents; and |
| | • | | redesigning our processes so they do not infringe, which may not be possible or could require substantial funds and time. |
We may be subject to claims that our employees have wrongfully used or disclosed alleged trade secrets of their former employers.
As is common in the biotechnology and pharmaceutical industry, we employ individuals who were previously employed at other biotechnology or pharmaceutical companies, including our competitors or potential competitors. Although no claims against us are currently pending, we may be subject to claims that these employees or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers. Litigation may be necessary to defend against these claims. Even if we are successful in defending against these claims, litigation could result in substantial costs and be a distraction to management.
Risks Related to Our Finances and Capital Requirements
We have incurred significant operating losses since our inception and anticipate that we will incur continued losses for the foreseeable future.
We began generating revenues from the commercialization of OFIRMEV in January 2011. Prior to that time, we focused primarily on in-licensing and developing OFIRMEV and our former product candidate, omiganan pentahydrochloride, with the goal of supporting regulatory approval for these product candidates. We have incurred losses in each year since our inception in May 2004, including net losses of $81.0 million, $93.0 million and $56.6 million for the years ended December 31, 2012, 2011 and 2010, respectively. As of December 31, 2012, we had an accumulated deficit of $447.7 million. These losses, among other things, have had and will continue to have an adverse effect on our stockholders’ equity and our working capital. For example, while our development expenses have decreased since 2010 due to the completion of our clinical development program for OFIRMEV, we have incurred increased commercialization and marketing expenses since that time in connection with our launch of OFIRMEV. Further, since the launch of OFIRMEV, we have also incurred significant increased sales, marketing and outsourced manufacturing expenses. In addition, we are required to pay a minimum annual royalty under our license agreement for OFIRMEV and we have minimum
42
purchase obligations under our supply agreements with our contract manufacturers for OFIRMEV. If our sales of OFIRMEV are insufficient to meet our minimum annual royalty obligations, we will be required to make larger royalty payments than would have otherwise been required based on sales of OFIRMEV alone. As a result, we expect to continue to incur significant operating losses for the foreseeable future. Because of the numerous risks and uncertainties associated with developing pharmaceutical products, we are unable to predict the extent of any future losses or when we will become profitable, if at all.
We currently have a limited history of revenue and may never be profitable.
Our ability to become profitable depends upon our ability to generate revenue. We began to market OFIRMEV in January 2011, and we had not generated any revenue prior to that time. Our ability to generate revenue depends on a number of factors, including, but not limited to, our ability to:
| | • | | effectively commercialize OFIRMEV; |
| | • | | manufacture commercial quantities of OFIRMEV at acceptable cost levels; |
| | • | | successfully manage our commercial organization and the supporting infrastructure required to successfully market and sell OFIRMEV; and |
| | • | | obtain regulatory approval for any other product or product candidates that we may license or acquire. |
We have incurred and anticipate continuing to incur significant costs associated with our efforts to commercialize, market and sell OFIRMEV. We also do not anticipate that we will achieve profitability for a period of time after generating material revenues, if ever. If we are unable to generate sufficient revenues, we will not become profitable and may be unable to continue operations without continued funding.
Our short operating history makes it difficult to evaluate our business and prospects.
We were incorporated in May 2004 and have only been conducting operations with respect to OFIRMEV since March 2006. Prior to 2011, our operations were limited to organizing and staffing our company, in-licensing and conducting product development activities, including clinical trials and manufacturing development activities, and preparing to commercialize OFIRMEV. In January 2011, we launched OFIRMEV and began generating revenues. The revenues we have generated from OFIRMEV have changed significantly since launch, and we anticipate that they will continue to change. Consequently, any predictions about our future performance may not be as accurate as they could be if we had a longer history of marketing OFIRMEV or other pharmaceutical products.
Our quarterly operating results may fluctuate significantly.
We expect our operating results to be subject to quarterly fluctuations. Our net loss and other operating results will be affected by numerous factors, including:
| | • | | our ability to successfully market and sell OFIRMEV; |
| | • | | our capacity to manage our commercial infrastructure and related expenses, including our hired sales and marketing personnel and costs incurred under our agreements with third parties for warehousing, distribution, cash collection and related commercial activities; |
| | • | | our execution of acquisition, in-licensing, co-promotion or similar agreements for new products and the timing of payments we may make or receive under these arrangements; |
| | • | | variations in the level of expenses related to our development programs for any future product candidates and any further development costs associated with OFIRMEV, including our ongoing pediatric clinical trial; |
| | • | | costs associated with our ongoing intellectual property infringement lawsuits related to OFIRMEV, and any product liability or other litigation in which we may become involved; |
43
| | • | | our ability to successfully defend the patents for the OFIRMEV and maintain our market exclusivity; |
| | • | | costs associated with any product recall or investigation into quality concerns; |
| | • | | our ability to successfully procure sufficient quantities of OFIRMEV and maintain adequate supply levels; |
| | • | | regulatory developments affecting OFIRMEV or the products or product candidates of our competitors; and |
| | • | | the level of underlying hospital demand for OFIRMEV and wholesalers’ buying patterns. |
If our quarterly or annual operating results fall below the expectations of investors or securities analysts, the price of our common stock could decline substantially. Furthermore, any quarterly or annual fluctuations in our operating results may, in turn, cause the price of our stock to fluctuate substantially. We believe that quarterly comparisons of our financial results are not necessarily meaningful and should not be relied upon as an indication of our future performance.
Raising additional funds by issuing securities may cause dilution to existing stockholders and raising funds through lending and licensing arrangements may restrict our operations or require us to relinquish proprietary rights.
To the extent that we raise additional capital by issuing equity securities, our existing stockholders’ ownership will be diluted. For example, we undertook a public offering of our common stock in November 2010 through which we issued a total of 12.5 million shares of common stock and raised net proceeds of $93.6 million, and in November 2011 we issued a total of 21.8 million shares of common stock in a public offering and raised net proceeds of $77.3 million. If we raise additional funds through alternative means such as licensing arrangements, it may be necessary to relinquish potentially valuable rights to our potential products or proprietary technologies, or grant licenses on terms that are not favorable to us.
Any debt financing we enter into may involve covenants that restrict our operations. These restrictive covenants may include limitations on additional borrowing and specific restrictions on the use of our assets as well as prohibitions on our ability to create liens, pay dividends, redeem our stock or make investments. For example, we have refinanced our $30.0 million secured credit facility with Oxford Finance LLC, Silicon Valley Bank and General Electric Capital Corporation on various occasions, including most recently in December 2012. This secured credit facility contains a variety of affirmative and negative covenants, including minimum quarterly product revenue requirements, required financial reporting, limitations on the disposition of assets other than in the ordinary course of business, limitations on the incurrence of additional debt and other requirements. To secure our performance of our obligations under our current loan and security agreement, we pledged substantially all of our assets other than intellectual property assets, to the lenders. Our failure to comply with the covenants in the current loan and security agreement could result in an event of default that, if not cured or waived, could result in the acceleration of all or a substantial portion of our debt and potential foreclosure on the assets pledged to secure the debt.
We will continue to incur significant increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives.
As a public company, we incur significant legal, accounting and other expenses under the Sarbanes-Oxley Act of 2002, as well as rules subsequently implemented by the SEC and The NASDAQ Stock Market LLC, or NASDAQ. These rules impose various requirements on public companies, including requiring establishment and maintenance of effective disclosure and financial controls and appropriate corporate governance practices. Our management and other personnel have devoted and will continue to devote a substantial amount of time to these compliance initiatives. Moreover, these rules and regulations increase our legal and financial compliance costs and make some activities more time-consuming and costly. For example, these rules and regulations make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be
44
required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified persons to serve on our board of directors, our board committees or as executive officers.
The Sarbanes-Oxley Act of 2002 requires, among other things, that we maintain effective internal controls for financial reporting and disclosure controls and procedures. As a result, we are required to periodically perform an evaluation of our internal controls over financial reporting to allow management to report on the effectiveness of those controls, as required by Section 404 of the Sarbanes-Oxley Act. Additionally, our independent auditors are required to perform a similar evaluation and report on the effectiveness of our internal controls over financial reporting. These efforts to comply with Section 404 and related regulations have required, and continue to require, the commitment of significant financial and managerial resources. While we anticipate maintaining the integrity of our internal controls over financial reporting and all other aspects of Section 404, we cannot be certain that a material weakness will not be identified when we test the effectiveness of our control systems in the future. If a material weakness is identified, we could be subject to sanctions or investigations by NASDAQ, the SEC or other regulatory authorities, which would require additional financial and management resources, costly litigation or a loss of public confidence in our internal controls, which could have an adverse effect on the market price of our stock.
In addition, in July 2010, the Dodd-Frank Wall Street Reform and Protection Act, or the Dodd-Frank Act, was enacted. There are significant corporate governance and executive compensation-related provisions in the Dodd-Frank Act that require the SEC to adopt additional rules and regulations in these areas such as “say on pay” and proxy access, and the SEC has since issued final rules implementing “say on pay” measures. Our efforts to comply with corporate governance and related requirements have resulted in, and are likely to continue to result in, an increase in expenses and a diversion of management’s time from other business activities.
The use of our net operating loss carryforwards and research tax credits may be limited.
Our net operating loss carryforwards and research and development tax credits may expire and not be used. As of December 31, 2012, we had generated federal and state net operating loss carryforwards of approximately $369.7 million and $374.2 million, respectively. We also had federal and state research and development tax credit carryforwards of approximately $4.8 million and $3.2 million, respectively.
Our net operating loss carryforwards will begin expiring in 2024 for federal purposes and 2014 for state purposes if we have not used them prior to that time. Our federal tax credits will begin expiring in 2025 unless previously used and our state tax credits carryforward indefinitely. Additionally, under Internal Revenue Code Sections 382 and 383, the annual use of our net operating loss carryforwards and research tax credits will be limited in the event a cumulative change in our ownership occurs within a three-year period. We expect to complete an analysis as to whether such a change of ownership has occurred in the next three months, and in such an event, we may be limited to the amount of net operating loss carryforwards and research tax credits that could be utilized annually in the future to offset taxable income. Any such annual limitation may significantly reduce the utilization of the net operating loss carryforwards and research tax credits before they expire. In addition, certain states have suspended the use of net operating loss carryforwards for certain taxable years, and other states are considering similar measures. Currently, California allows companies to utilize their net operating losses, however, new legislation could suspend the use of those losses in the future. As a result, we may incur higher state income tax expense in the future. Depending on our future tax position, continued suspension of our ability to use net operating loss carryforwards in states in which we are subject to income tax could have an adverse impact on our operating results and financial condition.
Our results of operations and liquidity needs could be materially negatively affected by market fluctuations and economic downturn.
Our results of operations could be materially negatively affected by economic conditions generally, both in the U.S. and elsewhere around the world. Continuing concerns over inflation, energy costs, geopolitical issues,
45
the availability and cost of credit, the U.S. mortgage market and a difficult residential real estate market in the U.S. have contributed to increased volatility and shifting expectations for the economy and the markets going forward. These factors, combined with volatile oil prices, fluctuating business and consumer confidence and continued unemployment concerns, have precipitated significant economic uncertainty. Domestic and international equity markets continue to experience heightened volatility and turmoil. These events and the continuing market changes may have an adverse effect on us. In the event of continuing market turbulence, our results of operations could be adversely affected by those factors in many ways, including making it more difficult for us to raise funds, if necessary, and our stock price may decline.
Risks Relating to Securities Markets and Investment in Our Stock
Our stock may be subject to substantial price and volume fluctuations due to a number of factors, many of which are beyond our control and may prevent our stockholders from reselling our common stock at a profit.
The market prices for securities of biotechnology and pharmaceutical companies have historically been highly volatile, and the market has recently experienced significant price and volume fluctuations that are unrelated to the operating performance of particular companies. Continued volatility in the overall capital markets could reduce the market price of our common stock in spite of our operating performance. Further, high stock price volatility could result in higher stock-based compensation expense.
The trading prices for our common stock during the 52 weeks ending December 31, 2012 ranged from a high of $5.00 to a low of $2.56. The market price of our common stock is likely to continue to be highly volatile and may fluctuate substantially due to many factors, including:
| | • | | announcements concerning our operating results and the hospital formulary acceptance of OFIRMEV; |
| | • | | market conditions in the pharmaceutical and biotechnology sectors or the economy as a whole; |
| | • | | price and volume fluctuations in the overall stock market; |
| | • | | the failure of OFIRMEV to achieve commercial success; |
| | • | | announcements of the introduction of new products by us or our competitors; |
| | • | | developments pertaining to the intellectual property lawsuits relating to OFIRMEV, including any future lawsuits, and any other challenges to our patents and other intellectual property rights; |
| | • | | developments concerning product development results or intellectual property rights of others; |
| | • | | product recalls, quality concerns or manufacturing difficulties; |
| | • | | litigation or public concern about the safety of our potential products; |
| | • | | actual fluctuations in our quarterly operating results, and concerns by investors that such fluctuations may occur in the future; |
| | • | | deviations in our operating results from the estimates of securities analysts or other analyst comments; |
| | • | | additions or departures of key personnel; |
| | • | | health care reform legislation, including measures directed at controlling the pricing of pharmaceutical products and the amount of reimbursement received by our customers; |
| | • | | developments concerning current or future strategic collaborations; and |
| | • | | discussion of us or our stock price by the financial and scientific press and in online investor communities. |
The realization of any of the risks described in these “Risk Factors” could have a dramatic and material adverse impact on the market price of our common stock. In addition, class action litigation has often been
46
instituted against companies whose securities have experienced periods of volatility in market price. Any such litigation brought against us could result in substantial costs and a diversion of our management’s attention and resources, which could hurt our business, operating results and financial condition.
Future sales of our common stock may cause our stock price to decline.
Persons who were our stockholders prior to the sale of shares in our initial public offering continue to hold a substantial number of shares of our common stock that they may now be able to sell in the public market. Significant portions of these shares are held by a small number of stockholders. Sales by our current stockholders of a substantial number of shares, or the expectation that such sales may occur, could significantly reduce the market price of our common stock. Moreover, the holders of a substantial number of shares of common stock may have rights, subject to certain conditions, to require us to file registration statements to permit the resale of their shares in the public market or to include their shares in registration statements that we may file for ourselves or other stockholders.
For example, we undertook public offerings of our common stock through which we issued totals of 21.8 million shares of common stock in November 2011 and 12.5 million shares of common stock in November 2010, and in May 2009, we completed the registration of approximately 18.1 million shares of our common stock in connection with a financing transaction completed in February 2009. As a result, all of the shares currently outstanding may generally be freely sold in the public market, subject to volume and other limitations applicable to our affiliates. We have also registered all common stock that we may issue under our employee benefits plans. As a result, these shares can be freely sold in the public market upon issuance, subject to restrictions under the securities laws.
Furthermore, any future equity financing we may undertake, or the expectation of such financing, could reduce the market price of our common stock over dilution concerns. In addition, certain of our officers have established, and other of our directors and executive officers may in the future establish, programmed selling plans under Rule 10b5-1 of the Securities Exchange Act of 1934, as amended, for the purpose of effecting sales of our common stock. If any of these events cause a large number of our shares to be sold in the public market, the sales could reduce the trading price of our common stock and impede our ability to raise future capital.
Our executive officers and directors and their affiliates may exercise control over stockholder voting matters in a manner that may not be in the best interests of all of our stockholders.
As of December 31, 2012, our executive officers and directors and their affiliates together controlled approximately 30% of our outstanding common stock. As a result, these stockholders will collectively be able to significantly influence all matters requiring approval of our stockholders, including the election of directors and approval of significant corporate transactions. The concentration of ownership may delay, prevent or deter a change in control of our company even when such a change may be in the best interests of all stockholders, could deprive our stockholders of an opportunity to receive a premium for their common stock as part of a sale of our company or our assets, and might affect the prevailing market price of our common stock.
Anti-takeover provisions under our charter documents and Delaware law could delay or prevent a change of control which could limit the market price of our common stock and may prevent or frustrate attempts by our stockholders to replace or remove our current management.
Our amended and restated certificate of incorporation and amended and restated bylaws contain provisions that could delay or prevent a change of control of our company or changes in our board of directors that our stockholders might consider favorable. Some of these provisions include:
| | • | | a board of directors divided into three classes serving staggered three-year terms, such that not all members of the board will be elected at one time; |
| | • | | a prohibition on stockholder action through written consent; |
47
| | • | | a requirement that special meetings of stockholders be called only by the chairman of the board of directors, the president or by a majority of the total number of directors; |
| | • | | advance notice requirements for stockholder proposals and nominations; |
| | • | | a requirement of approval of not less than 66-2/3% of all outstanding shares of our capital stock entitled to vote to amend any bylaws by stockholder action, or to amend specific provisions of our certificate of incorporation; and |
| | • | | the authority of the board of directors to issue preferred stock on terms determined by the board of directors without stockholder approval. |
In addition, we are governed by the provisions of Section 203 of the Delaware General Corporate Law, which may prohibit certain business combinations with stockholders owning 15% or more of our outstanding voting stock. These and other provisions in our amended and restated certificate of incorporation, amended and restated bylaws and Delaware law could make it more difficult for stockholders or potential acquirers to obtain control of our board of directors or initiate actions that are opposed by the then-current board of directors, including to delay or impede a merger, tender offer or proxy contest involving our company. Any delay or prevention of a change of control transaction or changes in our board of directors could cause the market price of our common stock to decline.
We have never paid dividends on our capital stock, and we do not anticipate paying any cash dividends in the foreseeable future.
We have paid no cash dividends on any of our classes of capital stock to date and we currently intend to retain our future earnings, if any, to fund the development and growth of our business. We do not anticipate paying any cash dividends on our common stock in the foreseeable future. Furthermore, our current loan and security agreement with Oxford Finance LLC, Silicon Valley Bank and General Electric Capital Corporation restricts our ability to pay dividends. As a result, capital appreciation, if any, of our common stock will be your sole source of gain for the foreseeable future.
We may become involved in securities class action litigation that could divert management’s attention and harm our business.
The stock markets have from time to time experienced significant price and volume fluctuations that have affected the market prices for the common stock of biotechnology and pharmaceutical companies. These broad market fluctuations may cause the market price of our common stock to decline. In the past, securities class action litigation has often been brought against a company following a decline in the market price of its securities. This risk is especially relevant for us because biotechnology and biopharmaceutical companies have experienced significant stock price volatility in recent years. We may become involved in this type of litigation in the future. Litigation often is expensive and diverts management’s attention and resources, which could adversely affect our business.
| Item 1B. | Unresolved Staff Comments |
Not applicable.
We currently lease approximately 16,600 square feet of space in our headquarters in San Diego, California under a lease that expires in December 2013. We have no laboratory, research, manufacturing or warehouse facilities; however we do own manufacturing equipment located at a third-party contractor. We currently do not plan to purchase or lease facilities for manufacturing, packaging or warehousing as such services are provided to us by third-party contractors. We believe that our current facilities are adequate for our needs for the immediate future and that, should it be needed, suitable additional space will be available to accommodate expansion of our operations on commercially reasonable terms.
48
In August 2011, we and Pharmatop filed suit in the United States District Court for the District of Delaware against Paddock Laboratories, Inc., Perrigo Company and Paddock Laboratories, LLC, collectively referred to herein as Perrigo, and against Exela Pharma Sciences, LLC, Exela PharmaSci, Inc. and Exela Holdings, Inc., collectively referred to herein as Exela. The lawsuit follows the notices that we received in July 2011 from each of Perrigo and Exela concerning their filings of Abbreviated New Drug Applications, or ANDAs, containing a “Paragraph IV” patent certification with the FDA for a generic version of OFIRMEV. In the lawsuit, we allege that Perrigo and Exela have each infringed the ‘222 patent and the ‘218 patent by filing their respective ANDAs seeking approval from the FDA to market a generic version of OFIRMEV prior to the expiration of these patents. The ‘222 and the ‘218 patents are listed in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. The patent infringement lawsuit was filed within 45 days of receipt of the pertinent notice letters, thereby triggering a stay of FDA approval of the Perrigo ANDA and the Exela ANDA until the earlier of the expiration of a 30-month period, the expiration of the ‘222 and ‘218 patents, the entry of a settlement order or consent decree stating that the ‘222 and ‘218 patents are invalid or not infringed, a decision in the case concerning infringement or validity that is favorable to Perrigo or Exela, or such shorter or longer period as the Court may order. Each of Perrigo and Exela has filed an answer in the case that asserts, among other things, non-infringement and invalidity of the asserted patents, and has also filed counterclaims. A date for the bench trial in the case with Exela has been scheduled for May 2013.
We settled with Perrigo and the case against Perrigo was dismissed on November 30, 2012. In connection with a settlement and license agreements entered into in November 2012, Perrigo has been granted the exclusive right of first refusal to negotiate an agreement with us to market an authorized generic version of OFIRMEV in the U.S. in the event that we elect to launch an authorized generic version of the product. The license agreement also provides that, if we enter into an agreement for Perrigo to market an authorized generic version of OFIRMEV, during the license period, Perrigo would purchase the product exclusively from us. We would receive product costs plus an administrative fee, as well as a royalty payment based on the net profits achieved by Perrigo from the sale of the authorized generic product. Additionally, we have granted Perrigo the non-exclusive right to market a generic intravenous acetaminophen product in the U.S. under Perrigo’s ANDA after December 6, 2020, or earlier under certain circumstances. The Federal Trade Commission, or FTC, or the Department of Justice, or DOJ, could seek to challenge our settlement with Perrigo, or a competitor, customer or other third-party could initiate a private action under antitrust or other laws challenging our settlement with Perrigo.
In September 2012, an unidentified third party filed with the USPTO a Request for Ex Parte Reexamination of the ‘222 patent. In December 2012, we received notice that the USPTO had granted the Request for Reexamination. The reexamination process is provided for by law and requires the USPTO to consider the scope and validity of the patent based on substantial new questions of patentability raised by a third party or the USPTO. Because we and Pharmatop believe that the scope and validity of the patent claims in this patent are appropriate and that the USPTO’s prior issuance of the patent was correct, we, in conjunction with Pharmatop, will vigorously defend this patent. We cannot predict whether we and Pharmatop ultimately will succeed in maintaining the scope and validity of the claims of this patent during reexamination. If the patent claims in this patent ultimately are narrowed during prosecution before the USPTO, the extent of the patent coverage afforded to OFIRMEV could be impaired, which could potentially harm our business and operating results.
In April 2012, Exela filed suit against David J. Kappos and the USPTO in the United States District Court for the Eastern District of Virginia for declaratory judgment seeking a reversal of the USPTO’s decision not to act on a petition by Exela to vacate the USPTO’s April 2003 order reviving the international application for the ‘218 patent. The lawsuit followed the USPTO’s rejection of Exela’s petition to the USPTO filed in November 2011, which sought to vacate the April 23, 2003 order granting Pharmatop’s petition to revive the ‘218 patent. The USPTO determined that Exela lacks standing to seek such relief. Exela also seeks declaratory judgment that the USPTO’s rules and regulations that allow for revival of abandoned, international patent applications under the “intentional” standard are invalid, and similar relief in connection with one or more counterclaims it has filed in the Delaware litigation. Our motion to intervene in this lawsuit was granted in
49
October 2012. In December 2012, the district court dismissed the case with prejudice as barred by the applicable statute of limitations. In February 2013, Exela appealed the district court’s decision to the Court of Appeals for the Federal Circuit. A decision by the Court of Appeals in favor of Exela could result in the invalidation of the ‘218 patent.
In January 2013, we filed suit in the United States District Court for the Southern District of California and the Northern District of Illinois against Fresenius Kabi USA, LLC, or Fresenius. The lawsuits follow a December 2012 notice by Fresenius concerning its filing of a New Drug Application, or NDA, containing a Paragraph IV patent certification with the FDA for a generic version of OFIRMEV. In the lawsuits, we allege that Fresenius has infringed the ‘222 patent and the ‘218 patent by filing its NDA seeking approval from the FDA to market a generic version of OFIRMEV prior to the expiration of these patents. Fresenius has filed an answer in the Southern District of California that asserts, among other things, non-infringement and invalidity of the asserted patents, and has also filed counterclaims.
In February 2013, we filed suit in the United States District Court for the Southern District of California and the District of New Jersey against Sandoz, Inc., or Sandoz. The lawsuits follow a December 2012 notice by Sandoz concerning its filing of an ANDA containing a Paragraph IV patent certification with the FDA for a generic version of OFIRMEV. In the lawsuits, we allege that Sandoz has infringed the ‘222 patent and the ‘218 patent by filing its ANDA seeking approval from the FDA to market a generic version of OFIRMEV prior to the expiration of these patents. Sandoz has not yet filed an answer in either District.
Both the Fresenius and Sandoz lawsuits were filed within 45 days of receipt of the respective notice letters, thereby triggering a stay of FDA approval of the Fresenius NDA and the Sandoz ANDA until the earlier of the expiration of a 30-month period, the expiration of the ‘222 and ‘218 patents, the entry of a settlement order or consent decree stating that the ‘222 and ‘218 patents are invalid or not infringed, a decision in the case concerning infringement or validity that is favorable to Fresenius and/or Sandoz, or such shorter or longer period as the courts may order.
Regardless of the outcome of any litigation, no NDA or ANDA can receive final approval from the FDA before expiration of the regulatory exclusivity period for OFIRMEV. Specifically, the FDA has granted OFIRMEV three years of regulatory exclusivity, which expires November 2, 2013. We intend to vigorously enforce our intellectual property rights relating to OFIRMEV to prevent the marketing of infringing generic products prior to the expiration of our patents. The ‘222 patent expires August 5, 2017 (or February 5, 2018 if pediatric exclusivity is granted) and the ‘218 patent expires June 6, 2021 (or December 6, 2021 if pediatric exclusivity is granted). However, given the unpredictability inherent in litigation, we cannot predict the outcome of these matters or any other litigation. Regardless of how these matters are ultimately resolved, these matters may be costly, time-consuming and distracting to our management, which could have a material adverse effect on our business. At this time, we are unable to estimate possible losses or ranges of losses for current litigation, and we have not accrued any amounts for current litigation other than ongoing attorney’s fees.
| Item 4. | Mine Safety Disclosures |
Not applicable.
50
PART II
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities |
Market Information
Our common stock is traded on the NASDAQ Global Market under the symbol “CADX.” As of February 28, 2013, there were 85,668,668 shares of common stock outstanding held by approximately 20 stockholders of record. Many stockholders hold their shares in street name and we believe that there are more than 4,000 beneficial owners of our common stock. The closing price of our common stock on the NASDAQ Global Market on December 31, 2012, the last trading day in 2012, was $4.79 per share. The following table sets forth the high and low sales prices for our common stock as reported on the NASDAQ Global Market for the periods indicated:
| | | | | | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2012 | | | 2011 | |
| | | High | | | Low | | | High | | | Low | |
Period: | | | | | | | | | | | | | | | | |
First Quarter | | $ | 4.37 | | | $ | 3.44 | | | $ | 9.21 | | | $ | 7.05 | |
Second Quarter | | $ | 3.70 | | | $ | 2.56 | | | $ | 10.00 | | | $ | 6.94 | |
Third Quarter | | $ | 4.87 | | | $ | 3.49 | | | $ | 9.47 | | | $ | 5.80 | |
Fourth Quarter | | $ | 5.00 | | | $ | 2.88 | | | $ | 6.99 | | | $ | 3.41 | |
Securities Authorized for Issuance under Equity Compensation Plans
The following table provides information as of December 31, 2012, about our common stock that may be issued upon the exercise of stock options and the vesting of restricted stock units granted to employees and members of our board of directors under all existing equity compensation plans, including our 2006 Equity Incentive Award Plan and our 2004 Equity Incentive Award Plan. The 2006 Equity Incentive Award Plan was adopted at the time of our initial public offering in October 2006, which coincided with the discontinuance of the 2004 Equity Incentive Award Plan. We amended and restated this plan in April 2010, which became effective in June 2010 upon the approval of our stockholders. See Note 11 to the Notes to Financial Statements in Item 8 of this Annual Report on Form 10-K for further discussion of our equity plans.
Equity Compensation Plan Information
| | | | | | | | | | | | |
| | | Number of securities to
be issued upon exercise
of outstanding options,
restricted stock units,
warrants and rights | | | Weighted average
exercise price of
outstanding options,
warrants and rights | | | Number of securities
remaining available for
future issuance under
equity compensation
plans (excluding
securities reflected in
column (a)) | |
| | | (a) | | | (b) | | | (c) | |
Plan Category: | | | | | | | | | | | | |
Equity compensation plans approved by security holders | | | 10,038,922 | (1) | | $ | 6.81 | (2) | | | 2,126,842 | (3) |
Equity compensation plans not approved by security holders. | | | — | | | | — | | | | — | |
| | | | | | | | | | | | |
Total | | | 10,038,922 | | | $ | 6.81 | (2) | | | 2,126,842 | (3) |
| | | | | | | | | | | | |
| (1) | Of these shares of common stock, 9,136,444 shares were subject to outstanding options under the 2006 Equity Incentive Award Plan and 901,540 shares were subject to outstanding options under the 2004 Equity Incentive Award Plan. In addition, 938 of the shares were subject to outstanding restricted stock units under the 2006 Equity Incentive Award Plan. |
51
| (2) | As restricted stock units do not have an exercise price, the weighted average exercise price does not take into account the 938 restricted stock units outstanding under the 2006 Equity Incentive Award Plan. |
| (3) | The 2006 Equity Incentive Award Plan contains an “evergreen” provision which allows for annual increases in the number of shares available for future issuance on January 1 of each year through January 1, 2016. The annual increase in the number of shares shall be equal to the lesser of (1) 4% of our outstanding common stock on the applicable January 1 or (2) such lesser amount determined by our board of directors. At January 1, 2012, 2011, 2010, 2009 and 2008, the board of directors approved the amount of shares authorized for future issuance under the 2006 Plan to be increased by 3,334,952 shares, 1,893,220 shares, 1,766,960 shares, 1,269,576 shares and 1,018,939 shares, respectively, under this provision. Effective January 1, 2013, the board of directors authorized an additional 2,570,060 shares for future issuance under the 2006 Plan pursuant to the provision. |
Performance Graph
The following stock performance graph illustrates a comparison of the total cumulative stockholder return on our common stock to two indices; the NASDAQ Composite Index and the NASDAQ Biotechnology Index. The graph assumes an initial investment of $100 at the close of business on December 31, 2007, and that all dividends, if any, were reinvested. No cash dividends have been declared or paid on our common stock. Stockholder returns over the indicated period should not be considered indicative of future stockholder returns.
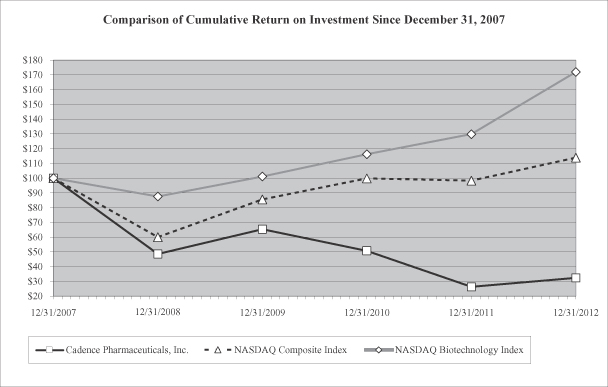
Dividend Policy
We have never declared or paid any cash dividends on our capital stock and we do not currently intend to pay any cash dividends on our common stock. We expect to retain future earnings, if any, to fund the development and growth of our business. The payment of dividends by us on our common stock is limited by our loan and security agreement with Oxford Finance LLC, Silicon Valley Bank and General Electric Capital Corporation. Any future determination to pay dividends on our common stock will be at the discretion of our board of directors and will depend upon, among other factors, our results of operations, financial condition, capital requirements and contractual restrictions.
52
Issuer Repurchases of Equity Securities
Not applicable.
| Item 6. | Selected Financial Data |
The selected financial data set forth below is derived from our audited financial statements and may not be indicative of future operating results. Audited balance sheets at December 31, 2012 and 2011 and the related audited statements of operations and of cash flows for each of the three years in the period ended December 31, 2012 and notes thereto appear elsewhere in this Annual Report on Form 10-K. Audited balance sheets at December 31, 2010, 2009 and 2008 and the related audited statements of operations and of cash flows for 2009 and 2008 are not included elsewhere in this Annual Report on Form 10-K.
The following selected financial data should be read in conjunction with the financial statements, related notes and other financial information appearing elsewhere in this Annual Report on Form 10-K.
| | | | | | | | | | | | | | | | | | | | |
| | | Year Ended December 31, | |
(in thousands, except share and per share data) | | 2012 | | | 2011 | | | 2010 | | | 2009 | | | 2008 | |
Statement of Operations Data: | | | | | | | | | | | | | | | | | | | | |
Product revenue, net | | $ | 50,066 | | | $ | 11,486 | | | $ | — | | | $ | — | | | $ | — | |
License revenue | | | 118 | | | | 5,210 | | | | — | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | |
Total net revenue | | | 50,184 | | | | 16,696 | | | | — | | | | — | | | | — | |
Cost of product sales | | | 23,256 | | | | 12,406 | | | | — | | | | — | | | | — | |
Amortization of patent license | | | 1,343 | | | | 1,567 | | | | — | | | | — | | | | — | |
Research and development | | | 6,519 | | | | 8,885 | | | | 13,757 | | | | 19,464 | | | | 40,018 | |
Selling, general and administrative | | | 86,843 | | | | 81,504 | | | | 39,347 | | | | 24,620 | | | | 14,131 | |
Impairment (adjustment to impairment) of long-lived assets | | | 7,723 | | | | — | | | | 1,522 | | | | (181 | ) | | | 2,353 | |
Other | | | 1,174 | | | | 1,076 | | | | 291 | | | | 594 | | | | 31 | |
| | | | | | | | | | | | | | | | | | | | |
Loss from operations | | | (76,674 | ) | | | (88,742 | ) | | | (54,917 | ) | | | (44,497 | ) | | | (56,533 | ) |
Interest income | | | 123 | | | | 136 | | | | 106 | | | | 182 | | | | 1,530 | |
Interest expense | | | (4,449 | ) | | | (4,424 | ) | | | (2,144 | ) | | | (1,137 | ) | | | (1,916 | ) |
Other income (expense) | | | 27 | | | | 9 | | | | 312 | | | | (39 | ) | | | (180 | ) |
| | | | | | | | | | | | | | | | | | | | |
Net loss | | $ | (80,973 | ) | | $ | (93,021 | ) | | $ | (56,643 | ) | | $ | (45,491 | ) | | $ | (57,099 | ) |
| | | | | | | | | | | | | | | | | | | | |
Basic and diluted net loss per share(1) | | $ | (0.95 | ) | | $ | (1.41 | ) | | $ | (1.09 | ) | | $ | (0.93 | ) | | $ | (1.55 | ) |
| | | | | | | | | | | | | | | | | | | | |
| (1) | There is a lack of comparability in the per share amounts between the periods presented as a result of the issuance of 21,800,000 shares of common stock pursuant to a public offering in the fourth quarter of 2011, 12,500,000 shares of common stock pursuant to a public offering in the fourth quarter of 2010, 12,040,000 shares of common stock pursuant to a private placement in the first quarter of 2009 and 9,240,000 shares of common stock pursuant to a registered direct offering in the first quarter of 2008. |
53
| | | | | | | | | | | | | | | | | | | | |
| | | As of December 31, | |
(in thousands) | | 2012 | | | 2011 | | | 2010 | | | 2009 | | | 2008 | |
Balance Sheet Data: | | | | | | | | | | | | | | | | | | | | |
Cash, cash equivalents and marketable securities | | $ | 62,072 | | | $ | 127,227 | | | $ | 134,141 | | | $ | 82,006 | | | $ | 47,627 | |
Accounts receivable, net | | | 6,152 | | | | 2,208 | | | | — | | | | — | | | | — | |
Inventory | | | 6,498 | | | | 1,388 | | | | 485 | | | | — | | | | — | |
Working capital | | | 55,517 | | | | 116,892 | | | | 121,319 | | | | 67,193 | | | | 28,385 | |
Total assets | | | 97,679 | | | | 163,665 | | | | 163,786 | | | | 92,563 | | | | 55,148 | |
Long-term debt, less current portion and discount | | | 28,818 | | | | 28,696 | | | | 24,654 | | | | — | | | | 6,098 | |
Accumulated deficit | | | (447,656 | ) | | | (366,683 | ) | | | (273,662 | ) | | | (217,019 | ) | | | (171,528 | ) |
Total stockholders’ equity | | | 47,811 | | | | 119,310 | | | | 123,960 | | | | 75,063 | | | | 26,440 | |
54
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results of Operations |
You should read the following discussion and analysis together with “Item 6 — Selected Financial Data” and the financial statements and related notes included in “Item 8 — Financial Statements and Supplementary Data” in this Annual Report on Form 10-K. This discussion may contain forward-looking statements that involve risks and uncertainties. For a complete discussion of forward-looking statements, see the section above entitled “Forward-Looking Statements.” Our actual results could differ materially from those anticipated in any forward-looking statements as a result of many factors, including those set forth in the section above entitled “Risk Factors.”
Overview
We are a biopharmaceutical company focused on acquiring, in-licensing, developing and commercializing proprietary products principally for use in the hospital setting. We intend to build a leading franchise in the hospital setting, continuing to focus on differentiated products with significant unmet commercial potential that are complementary to our current product, OFIRMEV® (acetaminophen) injection, and enable us to effectively leverage our commercial infrastructure.
In 2006, we in-licensed the exclusive U.S. and Canadian rights to OFIRMEV an intravenous formulation of acetaminophen, from BMS, which currently markets the product in Europe and several other markets under the brand name Perfalgan®. In November 2010, OFIRMEV was approved by the FDA, and we commercially launched OFIRMEV in the U.S. in January 2011. Our initial focus during the launch of OFIRMEV was to ensure formulary adoption, which we believe was an important first step to broad market acceptance, and by the end of 2012, OFIRMEV had been successfully placed on formulary at approximately 2,150 institutions. Our current focus is educating doctors, pharmacists and other healthcare professionals on the appropriate use of OFIRMEV and effective approaches to utilizing multimodal analgesia. We believe we are beginning to see the dividends of this strategy as our revenue has consistently grown each quarter since launch. More specifically, during the year ended December 31, 2012, we recorded net product revenue of $50.1 million, an increase of $38.6 million, or more than four times, from the $11.5 million reported for 2011.
In executing our business strategy, we have incurred significant net losses since our inception and have financed our operations primarily through the sale of equity securities in both public and private offerings. Most recently, we sold 21.8 million shares in a public offering in the fourth quarter of 2011 and received aggregate net proceeds of approximately $77.3 million (after underwriting discounts and offering costs). From inception through December 31, 2012, we have received total net proceeds of approximately $444.1 million from the sale of our preferred stock, common stock and warrants to purchase common stock. Additionally, we have entered into multiple loan and security agreements with Oxford Finance LLC, Silicon Valley Bank and General Electric Capital Corporation to provide us with growth capital. As of December 31, 2012, the principal balance outstanding on our current facility with this loan syndicate was $30.0 million.
Operations
Revenue
Our primary source of revenue is from the sale of OFIRMEV to hospitals and other end-user customers. Additionally, we have licensed certain data, and are providing on-going consulting support, to Terumo Corporation, or Terumo, for their use in seeking regulatory approval and commercializing the same intravenous formulation of acetaminophen in Japan. Further detail of these sources of revenue is provided below.
Product Revenue
In January 2011, we launched commercial sales of OFIRMEV and began shipping product to independent wholesalers, which sell OFIRMEV to hospitals and other end-user customers. Our initial focus for revenue growth was to promote rapid hospital formulary adoption of the product. During the second half
55
of 2011, our sales force placed additional emphasis on generating pull-through hospital sales of OFIRMEV from these institutions. During 2012, we have continued our focus on generating pull-through sales by actively promoting the product through a variety of marketing programs to inform customers about OFIRMEV.
As a result of these campaigns, over 3,750 unique accounts had ordered OFIRMEV as of December 31, 2012, which represents an increase of approximately 66% from the approximately 2,200 unique accounts at December 31, 2011. Further, these customers have increased the frequency with which they have placed orders for OFIRMEV as well as the average size of these orders. Specifically, the average frequency of customer orders was more than 16% higher for the fourth quarter of 2012 than for the fourth quarter of 2011, and the average size of customer orders for the fourth quarter of 2012 was approximately 40% higher than for the fourth quarter of 2011.
The impact of the continued growth in these metrics is evident in our net product revenue trends. Specifically, our net product revenue for the year ended December 31, 2012 was $50.1 million, more than four times the $11.5 million reported for the year ended December 31, 2011. No similar revenue was recognized during the year ended December 31, 2010.
We intend to continue our marketing strategies to promote OFIRMEV for the foreseeable future and we believe that there are substantial growth opportunities through continued promotion of the product.
License Revenue
In November 2010, we entered into a data license agreement with Terumo and Pharmatop. As part of the data license agreement, we provided to Terumo certain data and information resulting from our clinical development program for OFIRMEV for Terumo’s use in obtaining regulatory approval for and commercializing the same intravenous formulation of acetaminophen in Japan. Further, under the agreement we were to provide to Terumo, without charge, up to 500 hours of technical assistance and consulting services through November 2012 in relation to the licensed technical information, data and know-how in order to assist Terumo in obtaining regulatory approval and manufacturing capacity for the product candidate. In April 2011, we received an upfront payment of $5.3 million under the terms of the data license agreement and during the year ended December 31, 2011, we recognized $5.2 million of licensing revenue under the data license agreement with Terumo for the data provided and consulting hours incurred. The remaining payment balance of $0.1 million was recognized during the year ended December 31, 2012 and our obligation to provide up to 500 hours of technical assistance and consulting services has expired. No similar revenue was recognized during the year ended December 31, 2010.
If Terumo is successful in its efforts to obtain regulatory approval for and commercialize the product in Japan, we may be entitled to an additional lump-sum payment upon the first commercial sale of the product candidate and royalty payments upon any commercial sales of the product in Japan.
Cost of Sales
Our cost of sales consists primarily of our third-party manufacturing costs, indirect and personnel overhead costs, freight, excess or obsolete inventory adjustment charges and, if applicable, internal manufacturing overhead and the cost of purchasing acetaminophen, which is the active pharmaceutical ingredient for OFIRMEV. Further, cost of sales includes the royalties due under our license agreement with BMS, which range from the mid-teens to the mid-twenties, depending on the aggregate amount of net sales we record per contract year. The cost of sales we report for the quarterly and annual periods are primarily driven by sales volume, however, they are also impacted by production volumes of our product, manufacturing efficiencies during the production of our product, price variances of our manufacturing input costs and any inventory adjustment charges we may record.
The finished product, OFIRMEV, is currently supplied to us by BMS Anagni. Previously, Baxter Healthcare Corporation, or Baxter, also manufactured OFIRMEV for us, however, in February 2012, we temporarily
56
suspended production of OFIRMEV by Baxter and that suspension has remained in effect through December 31, 2012, pending an investigation into unidentified particulate matter observed during routine product stability testing and pursuant to two recalls of the product in 2012. In March 2013, we and Baxter, mutually agreed to terminate our supply agreement for OFIRMEV. We continued to incur certain manufacturing costs related to Baxter during this suspension, which are included in cost of sales for the year ended December 31, 2012. We placed certain inventory produced by Baxter on indefinite hold in February 2012 as a result of our February 2012 voluntary recall of product manufactured by Baxter and recorded the write-off of this inventory in costs of sales. We have decided to destroy this product and have accrued for the destruction costs as of December 31, 2012. Moreover, due to the termination of the supply agreement with Baxter, we reduced the carrying value of our manufacturing assets and manufacturing equipment and facility construction assets in process to their current estimated fair value. Further, we fully impaired our asset retirement obligation associated with the supply agreement because of our obligation to remove our equipment. See Critical Accounting Policies and Estimates— Long-Lived Assets for further discussion of this impairment.
During the suspension of production by Baxter, we transitioned our supply of OFIRMEV to BMS Anagni, which is presently acting as our sole supplier for the product. As a result of this transition, and in an effort to minimize any potential short-term supply disruption, we incurred expedited freight costs on certain shipments of OFIRMEV during the first half of 2012. These expedited freight costs have been recognized through the sale of the related inventory and we do not anticipate further impact from these shipments on our costs of sales in future periods. No further supply shortages are anticipated as a result of the termination of the Baxter agreement, as we continue to distribute product manufactured by BMS Anagni.
License Fees and Patent Amortization
As a result of the FDA’s approval of OFIRMEV, we paid a $15.0 million license fee in the fourth quarter of 2010 pursuant to the term of our license agreement with BMS. This payment was capitalized on our balance sheets as an intangible asset and we are amortizing the balance on a straight-line basis based upon the estimated life of the underlying patent assets. We may be required to make two additional milestone payments totaling up to $25.0 million based upon the achievement of certain levels of net sales of OFIRMEV, which will be recognized as license fees in the period they are incurred, as appropriate. However, as these payments are dependent upon future levels of net sales, we are unable to estimate with any certainty the timing of when these charges may be incurred.
Research and Development Expenses
Our research and development expenses relate predominantly to the development of product candidates, including OFIRMEV. These expenses have consisted of salaries and related employee benefits for our research and development team, license fees paid to our licensors prior to approval of our drug candidates, pre-commercialization manufacturing development activities, costs associated with clinical trials, and costs associated with non-clinical activities, such as expenses related to regulatory submissions. We have expensed these charges as the costs were incurred in developing, testing and seeking marketing approval of our product candidates. We received marketing approval for OFIRMEV from the FDA in November 2010 and with our OFIRMEV program continuing to progress, we implemented a restructuring of our workforce in November 2011 to focus our resources on the commercialization of OFIRMEV and reduce program costs that were not directly related to such efforts. This action resulted in a reduction in force of twelve employees in research and development in the fourth quarter of 2011.
As a result of these changes, our research and development expenses during the years ended December 31, 2012 and 2011 were significantly lower than during 2010 as fewer resources were being utilized by the development program since we received approval for OFIRMEV in November 2010. However, we expect to incur additional research and development expenses related to OFIRMEV in future periods, although it is difficult to anticipate the scope and magnitude of our future research and development expenses. For example, we began enrolling patients in an FDA-required post-approval clinical trial for OFIRMEV in pediatric patients
57
under two years of age during the third quarter of 2012. We may also conduct clinical studies to expand the indications for OFIRMEV. Moreover, any product candidates we may in-license or acquire in the future would likely require significant research and development resources. Therefore, we are unable to estimate with any certainty the costs we will incur in completing our development efforts for OFIRMEV or any other product candidate we might acquire or in-license.
Selling, General and Administrative Expenses
Our selling, general and administrative expenses consist primarily of salaries and related employee benefits for our sales and marketing staff; advertising, marketing and other promotional costs for OFIRMEV; selling expenses for our sales representatives, including travel-related costs; salaries and related employee benefits for our administrative, finance, human resources, legal, business development and internal systems support functions; costs incurred in relation to our medical affairs programs, including salaries, related employee benefits and costs incurred by our medical science liaisons; as well as the related professional fees for these functions, insurance and facility costs.
Our selling, general and administrative costs increased significantly following the approval of OFIRMEV in November 2010 as we hired our sales force and related personnel to support the commercial efforts for OFIRMEV and we continue to incur these costs. Further, we have incurred additional legal costs in 2012 related to our intellectual property litigation and we will continue to incur these costs as we enforce our intellectual property rights. Therefore, we expect to continue to incur significant selling, general and administrative expenses as we continue to execute our marketing and sales strategies for OFIRMEV, enforce our intellectual property rights and operate our business.
Interest and Other Income and Expense
Our interest income consists primarily of interest earned on our cash, cash equivalents and short-term investments. Interest expense consists of the interest we incur under our loan and security agreements and the amortization of debt issuance costs. Other income and expense includes federal grants we have received, gains or losses recognized on transactions denominated in foreign currencies and other transactions not related to our operations.
Our current loan and security agreement had a principal balance of $30.0 million as of December 31, 2012 and we are currently making interest-only payments on the outstanding balance of this facility, which will continue through December 2013. In January 2014, we will begin making equal monthly principal and interest payments to fully amortize the balance over a 30-month term. This facility has a fixed interest rate of 10.9545% and, as we begin making principal payments, we anticipate that our interest expense will decline.
Income Taxes
We assess income tax positions and record tax benefits for all years subject to examination based upon our evaluation of the facts, circumstances and information available at the reporting date. For those tax positions where there is a greater than 50% likelihood that a tax benefit will be sustained, we have recorded the largest amount of tax benefit that may potentially be realized upon ultimate settlement with a taxing authority that has full knowledge of all relevant information. For those income tax positions where there is less than 50% likelihood that a tax benefit will be sustained, no tax benefit has been recognized in the financial statements.
As of December 31, 2012, we had federal and state net operating loss carryforwards of approximately $369.7 million and $374.2 million, respectively. If not utilized, the net operating loss carryforwards will begin expiring in 2024 for federal purposes and 2014 for state purposes. Additionally, we had both federal and state research and development tax credit carryforwards of approximately $4.8 million and $3.2 million, respectively. The federal tax credits will begin expiring in 2025 unless previously utilized and the state tax credits carryforward indefinitely.
58
Under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, or the Internal Revenue Code, substantial changes in our ownership may limit the amount of net operating loss carryforwards and development tax credit carryforwards that could be utilized annually in the future to offset taxable income. Any such annual limitation may significantly reduce the utilization of the net operating losses and tax credits before they expire. We have not completed a Section 382/383 study at this time to determine the impact ownership changes have had on our carryforwards but expect to complete the analysis within the next three months and, as a result, we may have a change in the unrecognized tax benefits that are recorded. In each period since our inception, we have recorded a valuation allowance for the full amount of our deferred tax asset, as the realization of the deferred tax asset is uncertain. As a result, we have not recognized any federal or state income tax benefit in our statement of operations and, due to the existence of the valuation allowance, future changes in our unrecognized tax benefits will not impact our effective tax rate.
Critical Accounting Policies and Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the U.S., or GAAP, requires us to make certain estimates, judgments and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements, and the reported amounts of expenses during the reporting period. Some of our accounting policies require us to make difficult and subjective judgments, often as a result of the need to make estimates of matters that are inherently uncertain. The following accounting policies involve critical accounting estimates because they are particularly dependent on estimates and assumptions made by management about matters that are highly uncertain at the time the accounting estimates are made. In addition, while we have used our best estimates based on facts and circumstances available to us at the time, different estimates reasonably could have been used. Changes in the accounting estimates we use are reasonably likely to occur from time to time, which may have a material impact on the presentation of our financial condition and results of operations.
Our most critical accounting estimates include the recognition of revenue; the valuation of our inventory, which impacts gross margin; stock-based compensation which impacts operating expenses; and the assessment of recoverability of long-lived assets, which primarily impacts operating expenses when we impair assets or accelerate depreciation. We review our estimates, judgments, and assumptions used in our accounting practices periodically and reflect the effects of revisions in the period in which they are deemed to be necessary. We believe that these estimates are reasonable, however, our actual results may differ from these estimates.
Revenue Recognition
We sell OFIRMEV to wholesalers and directly to hospitals and other end-user customers. Our primary distribution channel for OFIRMEV involves our third party logistics distributor, which distributes the product to independent wholesalers, which in turn distribute the product directly to hospitals and other end-user customers. We also sell the product directly to end-user customers, and we have contracted with group purchasing organizations.
Our wholesaler agreements provide selling prices that are fixed on the date of sale, although we offer certain discounts to group purchasing organizations, certain end-user hospitals and governmental programs. The wholesalers take title to the product, bear the risk of loss of ownership and have economic substance to the inventory. Further, we have no significant obligations for future performance to generate pull-through sales, however we do allow our wholesalers to return product that is damaged or received in error. Additionally, we allow for product to be returned beginning six months prior to, and ending twelve months following, product expiration. OFIRMEV is our first and only commercially available product, and we have had limited product return data since the commercial launch of OFIRMEV in January 2011. As a result, we have not had sufficient data to reasonably estimate product returns from our wholesaler distribution channel and therefore we have deferred the recognition of revenue until the time that product has been sold by a wholesaler to a hospital or other end-user customer. Shipments of product that have not been recognized as revenue are treated as deferred
59
revenue until evidence exists to confirm that pull-through sales to hospitals or other end-user customers has occurred. However, we have begun to accumulate a reasonable amount of product return data, which we continue to gather, that we believe will allow us to reasonably estimate returns on product sold in the future.
We record certain fees, sales reserves and allowances as a reduction to gross revenue and deferred revenue, as applicable. These reserves and allowances include distribution service fees, a prompt payment reserve, a group purchasing discount and administrative service fee, and discounts to certain end-user hospitals and governmental programs, as applicable. Distribution service fees arise from contractual agreements between us and certain wholesalers for distribution services they provide with respect to OFIRMEV. These fees are generally a fixed percentage of the price of the product purchased by these wholesalers. The prompt payment reserve is based upon cash discounts we offer certain wholesalers as an incentive to meet certain payment terms. We account for these cash discounts at the time the sale is made to the wholesalers. The group purchasing discount and administrative service fee is based upon contracted discounts we provide to members of certain purchasing groups. We estimate the sales through our wholesalers to the group purchasing organization members and accrue for the chargebacks we anticipate from such sales based on the difference between the current retail price and the reduced price paid by the group purchasing organization members. A group purchasing organization administrative fee that we incur in exchange for administrative services provided by the group purchasing organizations for these transactions is also accrued at the time of sale. We also provide governmental programs and certain customers a predetermined discount that is recorded at the time of sale.
Revenue from our data license agreement with Terumo is recognized upon delivery of the goods and services provided, based upon the consideration allocated to each deliverable. We allocated the consideration to each deliverable based upon our review of the agreement pursuant to multiple-element arrangement guidance. We determined both the data license and consulting service deliverables were separate units of accounting, each having value on a standalone basis, and estimated the fair value of each item. The value of the data license was based upon similar proposals from third parties and internal costs we incurred in developing the data and obtaining similar rights. The value of the consulting services was based on contracts we had engaged with third parties for similar services. These values were consolidated and adjusted based upon the relative fair value of the consideration received pursuant to the agreement and there is no right of return or similar refund provisions in the data license agreement. Consideration allocated to the data license was recognized as revenue upon delivery of the data in 2011. Consideration allocated to the consulting services has been recognized as revenue as such services were rendered, or upon the termination of the requisite service period. As of December 31, 2012, we had fully recognized the license revenue received from the agreement and our consulting services obligation pursuant to the terms of the agreement had expired.
Inventories
We state our inventories at the lower of cost or market. We use a combination of standard and actual costing methodologies to determine the cost basis for our inventories. These methodologies approximate actual costs on a first-in, first-out basis. In addition to stating inventory at the lower of cost or market, we also evaluate our inventories each period for excess quantities and obsolescence. This evaluation includes identifying those items specifically identified as obsolete and analyzing forecasted demand versus quantities on hand so that this inventory can be valued appropriately.
Our inventory costs consist primarily of our third-party manufacturing fees, indirect and personnel overhead costs, freight-in, and, if applicable, internal manufacturing overhead, and other direct costs, if any. Fixed production overheads are allocated to the unit production costs based upon normal production capacity. Unallocated overhead costs incurred during periods of abnormally low production or unplanned facility downtime are recognized as expense in the period in which they are incurred.
In February 2012, we placed certain inventory produced by Baxter on indefinite hold and temporarily suspended production of OFIRMEV by Baxter pending the outcome of an investigation into unidentified particulate matter observed during routine product stability testing. Production by Baxter remained suspended
60
through December 31, 2012, and in March 2013, we and Baxter mutually agreed to terminate our supply agreement for OFIRMEV. We recorded charges of $5.6 million for the fourth quarter of 2011 and $0.2 million for the first quarter of 2012 in cost of sales to fully write-down the value of this inventory. Further, we decided to destroy this product as a result of a second voluntary recall of product manufactured by Baxter and have accrued for those costs as of December 31, 2012. During the suspension, we transitioned the supply of OFIRMEV to BMS Anagni, which is presently acting as our sole supplier for the product. No supply shortages are anticipated as a result of the suspension of production at Baxter and the termination of our supply agreement as we continue to distribute product manufactured by BMS Anagni.
Stock-Based Compensation
We account for stock-based compensation by calculating the fair value of the award on the date of grant for equity awards or, in the case of liability classified awards, we revalue the awards each reporting period until the awards are subsequently classified as equity awards, or otherwise vest. We calculate the fair value of stock options using the Black-Scholes pricing model, which requires a number of estimates, including the expected lives of awards, interest rates, stock volatility and other assumptions. Restricted stock units, or RSUs, are measured based on the fair market values of the underlying stock on the date of grant. We apply a forfeiture rate to estimate the number of grants that will ultimately vest. If the awards are performance based, we also assess the likelihood of the vesting conditions occurring and apply an appropriate factor in recognizing the expense.
Long-Lived Assets
A substantial portion of our capital assets are associated with our manufacturing equipment at Baxter, our initial third-party manufacturer. In building these assets and creating additional capacity, we have entered into agreements whereby we fund specified improvements to the facilities and the construction of the manufacturing equipment to be used for the production of OFIRMEV. During the build-out of the facility and construction of our equipment, we have accrued for costs incurred based on factors such as estimates of work performed, milestones achieved and experience with similar contracts. As actual costs become known, we adjust our accruals accordingly.
We evaluate these long-lived assets for impairment of their carrying value when events or circumstances indicate that the carrying value may not be recoverable. Factors we consider in deciding when to perform an impairment review include significant negative industry or economic trends, significant changes or planned changes in our use of the assets, technological obsolescence, or other changes in circumstances which indicate the carrying value of the assets may not be recoverable. If such an event occurs, we evaluate whether the sum of the estimated undiscounted cash flows attributable to the assets in question is less than their carrying value. If this is the case, we recognize an impairment loss to the extent that carrying value exceeds fair value. Fair value is determined based on market prices or discounted cash flow analysis, depending on the nature and planned use of the asset and the availability of market data. Any estimate of future cash flows is inherently uncertain. The factors we take into consideration in making estimates of future cash flows include product life cycles, pricing trends, future capital needs, cost trends, product development costs, competitive factors and technology trends as they each affect cash inflows and outflows. If an asset is written down to fair value, that value becomes the asset’s new carrying value and is depreciated over the remaining useful life of the asset.
In February 2012, we suspended production of OFIRMEV by Baxter and that suspension has remained in effect through December 31, 2012, pending an investigation into unidentified particulate matter in the product, which was observed during routine product stability testing and pursuant to two recalls of the product during 2012. In March 2013, we and Baxter mutually terminated our supply agreement for OFIRMEV. As a result, we reduced the carrying value of our manufacturing assets and manufacturing equipment and facility construction assets in process to their current estimated fair value, resulting in an impairment charge of $7.0 million for the year ended December 31, 2012. Moreover, we fully impaired the asset retirement obligation associated with the supply agreement, resulting in a charge of $0.7 million as of December 31, 2012. The carrying value on our
61
balance sheet of the manufacturing assets located at Baxter at December 31, 2012 was $1.0 million, and the value of the manufacturing equipment and facility construction assets in process was $0.4 million. The carrying value of our asset retirement obligation, included was $0.7 million.
Results of Operations
Years ended December 31, 2012 and 2011
Revenue
During the year ended December 31, 2012, we recognized $50.1 million of net product revenue from the sale of OFIRMEV to hospitals and other end-users, an increase of $38.6 million, or 336%, from the $11.5 million reported for the year ended December 31, 2011. Additionally, we recognized $5.2 million of revenue during 2011 related to the data license agreement with Terumo, for which data was provided and consulting hours were incurred pursuant to the terms of a data license agreement. During 2012, we recognized $0.1 million for the remaining consulting hours required under the agreement.
The increase in product revenue during 2012 as compared to 2011, our initial launch year for OFIRMEV, was primarily due to the continued expansion of our end-user customer base and the penetration of their use of OFIRMEV in a variety of surgical settings. For example, unique end-user accounts increased from approximately 2,200 at December 31, 2011 to over 3,750 at December 31, 2012. Further, this increasingly broad customer base has increased the frequency at which they are ordering the product, as well as their average order size. For example, the average order size for end-user customers for the fourth quarter of 2012 increased more than 40%, as compared to the average order size for the fourth quarter of 2011. Moreover, the frequency with which these customers ordered the product increased by 16% in the fourth quarter of 2012 compared to the fourth quarter of 2011. In addition, we implemented a price increase during 2012, which resulted in our average net selling price for 2012 increasing 3% from the average net selling price for 2011.
Costs and Expenses
Cost of Product Sales.Our cost of product sales for the year ended December 31, 2012, was $23.3 million, or 47% of net product revenue, compared to $12.4 million, or 108% of net product revenue, for the comparable period in 2011. The improvement in our costs of sales as a percentage of net product revenue in 2012 was primarily due to a reduction in inventory losses we recorded during the 2012 period as compared to 2011. For example, we recorded a $5.6 million charge in 2011 for inventory losses, compared to a similar charge of $0.2 million in 2012. These losses relate to inventory that was placed on indefinite hold due to the investigation into the cause of unidentified particulate matter observed in the product during routine product stability testing at Baxter’s manufacturing facility.
Additional improvements in our cost of sales as a percentage of net product revenue for 2012 include economies of scale we realized on increased sales volume during the 2012 period. However, these efficiencies realized in 2012 were partially offset by higher freight costs related to a supply disruption during the first quarter of the year and unabsorbed manufacturing costs we incurred on our idle manufacturing site. These excess costs were mostly related to our suspension of production by Baxter in connection with the aforementioned investigation. More specifically, we incurred expedited freight costs on certain shipments of OFIRMEV from BMS Anagni in order to meet demand for OFIRMEV following the suspension of manufacturing at Baxter’s facility in February 2012. Additionally, we continued to incur certain fixed manufacturing costs at the Baxter manufacturing facility during 2012, which were recognized as a cost of product sales during the period in which they were incurred.
Patent Amortization. For the year ended December 31, 2012, we incurred $1.3 million of non-cash expense related to the amortization of the $15.0 million license payment made to BMS following the approval of our NDA for OFIRMEV in November 2010. We are amortizing the balance of the payment on a straight-line basis, based upon the estimated life of the underlying patent assets. For the year ended December 31, 2011, we incurred $1.6 million of patent amortization expense.
62
Research and Development Expenses. Research and development expenses decreased $2.4 million for the year ended December 31, 2012, to $6.5 million, compared to $8.9 million for 2011. This decrease was primarily due to our transition to a commercially-focused organization whereby we implemented a restructuring of our workforce in November 2011, resulting in the reduction of twelve employees in our research and development organization. As a result, our personnel-related costs and the corporate overhead cost allocations which are based on headcount, decreased significantly in 2012 as compared to 2011. However, in 2012 we began establishing sites and enrolling patients in our FDA-required post-approval clinical trial for OFIRMEV in pediatric patients under two years of age and began to incur costs for this trial in 2012, which we will continue to incur in the near future until the trial and associated activities are complete.
Selling, General and Administrative Expenses. Selling, general and administrative expenses increased $5.3 million, or 6.5%, for the year ended December 31, 2012, to $86.8 million, compared to $81.5 million for 2011. This increase was mostly attributable to higher legal expenses incurred during 2012 related to our intellectual property litigation and increased commissions earned by our hospital sales force, which was partially offset by reductions in other personnel-related costs.
Impairment of Long-Lived Assets. As of December 31, 2012, we recorded impairment charges totaling $7.0 million to reduce the value of our manufacturing assets at Baxter to their estimated net realizable value. The production of OFIRMEV at Baxter had been suspended since February 2012, and in March 2013, we and Baxter mutually agreed to terminate our supply agreement. In addition, we impaired the anticipated asset retirement obligation associated with the terminated supply agreement as of December 31, 2012, pursuant to the terms of the terminated supply agreement and the Settlement and Termination Agreement, resulting in a charge of $0.7 million for the year ended December 31, 2012. No similar impairment charges were recorded during the year ended December 31, 2011.
Other Expenses. In December 2012, we recorded a loss of $0.9 million upon the disposal of a manufacturing asset being constructed. Further, we accrued for the anticipated destruction costs for the recalled inventory we wrote-off during the year, resulting in a charge of $0.3 million for the year ended December 31, 2012. During the year ended December 31, 2011, we recorded one-time employee termination costs of $1.1 million as part of the restructuring of our workforce in November 2011. The restructuring was due to our transition to a commercially-focused organization, which resulted in a reduction to our research and development expenses for 2012.
Other Income and Expenses, Net
Net other expense for the year ended December 31, 2012, was $4.3 million, which was consistent with 2011. Moreover, our interest expense of $4.4 million for 2012 was consistent with our interest expense for 2011 as the outstanding principal balance on our debt remained at $30.0 million throughout 2012. In December 2012, we amended and restated our existing loan facility whereby we delayed the commencement of principal payments by one year, from January 1, 2013 to January 1, 2014. The 2012 amendment did not provide for additional capital, but the stated interest rate was reduced from 10.99% to 10.9545%. As part of the 2012 amendment, we issued warrants to purchase an aggregate of 154,638 shares of our common stock at an exercise price of $3.88 per share and made a term loan final payment in December 2012 in accordance with the terms of the prior facility, which we had been accruing during the term of the facility. We are obligated to pay a similar final payment in accordance with the terms of the 2012 amendment. Our effective interest rate under the 2012 amendment is 15.31%.
Years ended December 31, 2011 and 2010
Revenue
During the year ended December 31, 2011, our initial launch year for OFIRMEV, we recognized $11.5 million of net product revenue from the sale of OFIRMEV to hospitals and other end-users. Additionally, we
63
recognized $5.2 million of revenue during 2011 related to the data license agreement with Terumo, for which data was provided and consulting hours were incurred pursuant to the terms of the agreement. No similar revenue was recognized during the year ended December 31, 2010.
Costs and Expenses
Cost of Product Sales.Our cost of product sales for the year ended December 31, 2011, was $12.4 million and includes a charge recorded in the fourth quarter of 2011 of $5.6 million. Further, our cost of sales for 2011 includes higher per-unit costs as we commenced the manufacturing of OFIRMEV, resulting in inefficiencies and higher allocated overhead costs. Additionally, we incurred certain periods of idle manufacturing capacity, resulting in unabsorbed manufacturing costs, mostly attributable to depreciation expense and labor-related costs. These costs were recognized as a cost of product sales during the period in which they were incurred.
Patent Amortization. For the year ended December 31, 2011, we incurred $1.6 million of non-cash expense related to the amortization of the $15.0 million license payment made to BMS following the approval of our NDA for OFIRMEV in November 2010. No such amortization was incurred during the year ended December 31, 2010.
Research and Development Expenses. Research and development expenses decreased $4.9 million for the year ended December 31, 2011, to $8.9 million, compared to $13.8 million for 2010. This decrease was due to the completion of our development program for OFIRMEV, and transition to a commercially-focused organization. Specifically, in 2010, we incurred manufacturing development expenses in preparation for the commercialization of OFIRMEV that were not incurred in 2011. Further, during 2011, certain quality assurance expenses incurred in the manufacturing process for OFIRMEV were allocated to manufacturing, which were not allocated in 2010. In November 2011, we continued this transition by implementing a restructuring of our workforce, resulting in the reduction of twelve employees in our research and development organization.
Selling, General and Administrative Expenses. Selling, general and administrative expenses increased by $42.2 million for the year ended December 31, 2011, to $81.5 million, compared to $39.3 million for 2010. This increase was primarily related to costs associated with our sales representatives, who were hired following the FDA approval of OFIRMEV in November 2010. These additional costs include the labor-related costs for the sales representatives and the related support staff. Other added costs include the travel expenses incurred by our sale representatives and other selling and education related costs. Further, we incurred additional marketing and promotion expenses, medical affairs costs, legal fees and other general corporate expenses in support of our commercial operations during 2011, as compared to 2010.
Impairment of Long-Lived Assets. In 2010, we recorded a charge of $1.5 million related to the partial cancellation of an equipment order resulting from a modification in the design of our planned second production line for OFIRMEV. The charge recorded impaired the costs that had been accumulated in construction-in-progress for the equipment. No similar charges were recorded for the year ended December 31, 2011.
Other Expenses. In November 2011, we implemented a restructuring of our workforce, which resulted in the termination of 17 employees, or approximately 7% of our total workforce at that time. The reduction was primarily in our research and development and general and administrative areas. As a result, we recorded one-time employee termination costs of $1.1 million, including severance and other benefits. These costs were partially offset by a $0.3 million recovery of previously accrued labor related charges, which was recorded at the time of the termination and is included in research and development expenses. For the year ended December 31, 2010, we incurred a charge of $0.3 million related to a reduction in force of six individuals during the second quarter of the year.
Other Income and Expenses, Net
Net other expense increased $2.6 million for the year ended December 31, 2011, to $4.3 million, compared to $1.7 million in 2010. This increase was primarily due to the additional interest expense incurred under our
64
loan and security agreement and the amendments thereto, and a higher outstanding principal balance during the year ended December 31, 2011, as compared to the same period in 2010. Further, in 2010, we received a $0.2 million federal government grant from the Qualifying Therapeutic Discovery Project program under section 48D of the Internal Revenue Code. No similar grants were received in 2011.
Liquidity and Capital Resources
As a biopharmaceutical company focused on acquiring, in-licensing, developing and commercializing proprietary products principally for use in the hospital setting, we enter into agreements to acquire commercial products and the right to develop and commercialize product candidates, which requires a significant amount of resources. Further, these agreements and related development programs may not result in commercially successful products that generate significant revenue and, for product candidates, even if a commercial product is developed, it could take a substantial amount of time to recover the investment in the program, if at all. For example, we obtained the exclusive patent rights and know-how for OFIRMEV, which is currently our only product, for the U.S. and Canada pursuant to our license agreement with BMS. Under this agreement, we have paid a total of $40.0 million in up-front fees and milestone payments, and we may be required to make two future milestone payments totaling up to $25.0 million upon the achievement of certain levels of net sales of the product in addition to royalties on the net sales of OFIRMEV. Further, in developing OFIRMEV, we have incurred over $44.5 million in research and development costs through December 31, 2012 specific to the product. However, our total investment in the OFIRMEV program is significantly more as these costs exclude a substantial portion of our internal costs, such as salaries and related personnel costs, which are not tracked on a project basis. In January 2011, we commenced sales of OFIRMEV however we have yet to recover our investment in the drug product and development program. For example, as of December 31, 2012, we had realized less than $26.0 million in gross profit on sales of OFIRMEV and we continued to operate at a loss.
OFIRMEV is currently our only product and we have no ongoing development programs for other product candidates and if we acquire, in-license or develop other drug products or drug candidates, it will likely require substantial capital resources. We previously entered into an option agreement with Incline Therapeutics, Inc., or Incline, whereby we had the option to acquire Incline. However, in December 2012, we entered into a Waiver, Consent and Option Termination Agreement, or option waiver agreement, with Incline pursuant to which we agreed to the buy-out and termination of our option. In January 2013, under the terms of the waiver agreement, we relinquished our option for consideration of $13.1 million in cash. Additionally, we received $1.5 million for the shares of Incline stock we sold as part of the transaction.
Since inception, our operations have been financed primarily through the sale of equity securities, in both public and private offerings. From our inception through December 31, 2012, we have received net proceeds of approximately $444.1 million from the sale of our preferred stock, common stock and warrants to purchase common stock. Through December 31, 2012, the sales of shares of our preferred stock, common stock and warrants were as follows:
| | • | | from July 2004 to December 2012 (excluding our initial public offering, our February 2008 registered direct offering, our February 2009 private placement and our 2010 and 2011 public offerings), we issued and sold a total of 3,217,883 shares of common stock to our founders, employees, directors and consultants for aggregate net proceeds of $3.1 million; |
| | • | | from July 2004 to August 2004, we issued and sold a total of 8,085,108 shares of Series A-1 preferred stock for aggregate net proceeds of $7.5 million; |
| | • | | from June 2005 to September 2005, we issued and sold a total of 17,675,347 shares of Series A-2 preferred stock for aggregate net proceeds of $17.6 million; |
| | • | | in March 2006, we issued and sold a total of 53,870,000 shares of Series A-3 preferred stock for aggregate net proceeds of $53.8 million; |
| | • | | in the fourth quarter of 2006, we completed our initial public offering in which we issued and sold a total of 6,900,000 shares of our common stock for aggregate net proceeds of $55.9 million; |
65
| | • | | in February 2008, we completed a registered direct offering pursuant to an effective shelf registration in which we issued and sold a total of 9,240,307 shares of our common stock for aggregate net proceeds of $49.1 million; |
| | • | | in February 2009, we raised aggregate net proceeds of approximately $86.2 million through a private placement transaction in which we issued 12,039,794 shares of common stock and warrants to purchase up to 6,019,897 additional shares of common stock at a price of $7.84, all of which remain outstanding at December 31, 2011; |
| | • | | in November and December 2010, we completed a public offering in which we issued and sold a total of 12,500,000 shares of our common stock for aggregate net proceeds of $93.6 million; and |
| | • | | in November 2011, we completed a public offering in which we issued and sold a total of 21,800,000 shares of our common stock for aggregate net proceeds of $77.3 million. |
Additionally, we have obtained growth capital through loans with Oxford Finance LLC, Silicon Valley Bank and General Electric Capital Corporation. As of December 31, 2012, the current secured credit facility with this syndicate had an outstanding principal balance of $30.0 million and we had no further available credit. We are currently making interest-only payments on the outstanding balance of this facility, which will continue through December 2013. In January 2014, we will begin making equal monthly principal and interest payments to fully amortize the balance over a 30-month term. In connection with the establishment of our loan agreements, we have issued warrants to the lenders to purchase shares of our stock. As of December 31, 2012, 63,079 shares of common stock had been issued from the exercise of such warrants. Warrants to purchase an additional 50,331 common shares at $12.67 per share, 254,793 common shares at $7.0645 per share, 158,311 common shares at $3.79 per share and 154,638 common shares at $3.88 per share, remain outstanding from our loan agreements.
Liquidity
As of December 31, 2012, we had $58.3 million in cash and cash equivalents, compared to $82.6 million at December 31, 2011. This $24.3 million decrease in our cash and cash equivalent balance during 2012 was primarily due to our use of cash in our operations of $64.3 million, $40.9 million of which was funded through net maturities of our marketable securities. Correspondingly, our marketable securities balance at December 31, 2012 was $3.7 million, as compared to $44.6 million at December 31, 2011. Other uses of cash during 2012 included $1.7 million for the purchase of property and equipment. However, our cash outflows in 2012 were partially offset by the proceeds we received from stock option exercises of $0.5 million and the sale of property and equipment of $0.4 million. In December 2012, we entered into an agreement to waive our option to purchase Incline in order to allow Incline to be sold to a third party. Upon the sale of Incline in January 2013, we received $13.1 million in cash in consideration for our waiver plus an additional $1.5 million for the shares of Incline stock we held. The proceeds received from this transaction are not included in our financial statements at December 31, 2012.
The $64.3 million of cash used in operations during 2012, represents a decrease of $13.9 million from the $78.2 million of cash used in operations during 2011. This reduced utilization of cash was mostly due to our reduced operating loss in 2012 as compared to 2011, driven by an increase in our revenue. During 2012 we used approximately $3.7 million to support our working capital requirements, an increase of $0.1 million from the $3.6 million used for working capital in 2011. The increase in our working capital for 2012 was primarily related to increases in our inventory balances to support our increased demand, and an increase in our accounts receivable as a result of our increased revenue. More specifically, as of December 31, 2012, our accounts receivable balance was $6.2 million, an increase of $4.0 million from the $2.2 million at December 31, 2011. Despite this increase in our accounts receivable balance, however, our days sales outstanding at December 31, 2012, as calculated by wholesaler shipments, remained relatively constant at approximately 35 days. Further, through December 31, 2012 we had incurred less than $0.1 million in bad debt expense to reserve or write-off our receivable balances.
66
Our net property and equipment balance at December 31, 2012, decreased $8.6 million to $2.0 million from $10.6 million as of December 31, 2011. This reduction was primarily due to an impairment charge of $7.0 million recorded in the fourth quarter of 2012 to reduce the value of our manufacturing assets, including construction-in-process assets, to their estimated net realizable value as a result of the protracted investigation and uncertainty regarding the resolution of manufacturing concerns at Baxter. In addition, we sold a construction-in-process asset in December 2012, resulting in a loss on the transaction of $0.9 million. Under our settlement and termination agreement with Baxter, which was executed in March 2013, we have no future commitments for facility improvements. However, we are responsible for the removal costs of our equipment which we have estimated at $0.7 million and we fully recognized this liability as of December 31, 2012.
We made no principal payments under our debt agreements during 2012 and our outstanding principal balance remained at $30.0 million as of December 31, 2012. Further, in December 2012, we amended our credit facility to delay the commencement of principal payments by one year, from January 1, 2013 to January 1, 2014. Under the amended agreement, we are required to maintain minimum quarterly product revenue of $12.5 million and we are subject to various other financial and non-financial covenants. We believe we were in compliance will all such covenants under the agreement as of December 31, 2012.
Capital Resources
Our cash, cash equivalents and short-term investment balances are our primary source of liquidity and currently the only sources available to us. However, in January 2013, we consummated our option waiver agreement with Incline whereby we received $13.1 million as consideration for relinquishing our option to acquire Incline. Further, we received an additional payment of $1.5 million as consideration for the 500,000 shares of Incline Series A preferred stock we held.
We believe we have sufficient financial resources to fund our operations, at a minimum, for the next twelve months. However, our future funding requirements will depend on many factors, including, but not limited to:
| | • | | our ability to successfully market and sell OFIRMEV; |
| | • | | our capacity to manage our commercial infrastructure and related expenses, including our hired sales and marketing personnel, and costs incurred under our agreements with third parties for warehousing, distribution, cash collection and related commercial activities; |
| | • | | our execution of acquisition, in-licensing, co-promotion, or similar agreements for new products, and the timing of payments we may make or receive under these arrangements; |
| | • | | variations in the level of expenses related to our development programs for any future product candidates and any further development costs associated with OFIRMEV, including our ongoing pediatric clinical trial; |
| | • | | costs associated with our ongoing intellectual property infringement lawsuits related to OFIRMEV, and any product liability or other litigation in which we may become involved; |
| | • | | our ability to successfully defend the patents for the OFIRMEV and maintain our market exclusivity; |
| | • | | costs associated with any product recall or investigation into quality concerns; |
| | • | | our ability to successfully procure sufficient quantities of OFIRMEV and maintain adequate supply levels; |
| | • | | regulatory developments affecting OFIRMEV or the products of our competitors; and |
| | • | | the level of underlying hospital demand for OFIRMEV and our wholesalers’ buying patterns. |
Until we can generate significant cash from our operations, we expect to continue to fund our operations with our available financial resources, generated from the proceeds of offerings of our equity securities and our existing borrowings under our loan and security agreement. These financial resources may not be adequate to
67
sustain our operations until we are able to generate significant positive cash flow from our operations and we may be required to finance future cash needs through the sale of additional equity securities, strategic collaboration agreements or debt financing. However, we cannot be certain that additional financing will be available when needed or that, if available, financing will be obtained on terms favorable to us or our stockholders. The capital markets have experienced volatility in recent years and the availability of credit has been adversely affected by illiquid credit markets and wide credit spreads. Further, concern about the stability of the markets in general, and the strength of counterparties specifically, has led many lenders and institutional investors to reduce, and in some cases, cease to provide funding to borrowers. Additional turbulence in the U.S. and international markets and economies may adversely affect our ability to obtain additional financing on terms acceptable to us, or at all, which may limit our ability to timely replace maturing liabilities and to access the capital markets to meet liquidity needs. Having insufficient funds may require us to delay, scale-back or eliminate some or all of our programs or renegotiate less favorable terms than we would otherwise choose. Failure to obtain adequate financing also may adversely affect our ability to operate as a going concern. Additionally, if we raise funds by issuing equity securities, dilution to existing stockholders would result; and if we raise funds by incurring additional debt financing, the terms of the debt may involve significant cash payment obligations as well as covenants and specific financial ratios that may restrict our ability to operate our business.
Other Significant Cash and Contractual Obligations
In operating our business, we enter into contracts and agreements that require capital resources to be consumed in future periods. The following table summarizes our scheduled contractual obligations and commitments that will affect our future liquidity as of December 31, 2012 (in thousands):
| | | | | | | | | | | | | | | | | | | | |
| | | | | | Payments By Period | |
| | | Total | | | Less than
1 year | | | 1-3 years | | | 3-5 years | | | More than
5 years | |
Long-term debt obligations, including interest | | $ | 39,480 | | | $ | 3,250 | | | $ | 27,544 | | | $ | 8,686 | | | $ | — | |
Third-party manufacturing obligations(1) | | | 73,350 | | | | 12,225 | | | | 24,450 | | | | 24,450 | | | | 12,225 | |
Operating leases(2) | | | 892 | | | | 810 | | | | 65 | | | | 17 | | | | — | |
Other purchase obligations(3) | | | 226 | | | | 136 | | | | 90 | | | | — | | | | — | |
License obligations(4) | | | — | | | | — | | | | — | | | | — | | | | — | |
| | | | | | | | | | | | | | | | | | | | |
Total(5) | | $ | 113,948 | | | $ | 16,421 | | | $ | 52,149 | | | $ | 33,153 | | | $ | 12,225 | |
| | | | | | | | | | | | | | | | | | | | |
| (1) | We have contracted with third-party manufacturers for the commercial supply of OFIRMEV. Under these agreements, we are required to purchase a certain minimum number of vials each year during the terms of the contracts. The amounts presented represent our estimates of the minimum required expenditures under our supply agreement with Lawrence Laboratories; however, the ultimate liability for these obligations may be reduced if our supplier fails or declines to supply a sufficient quantity of OFIRMEV in accordance with our purchase orders. The amounts presented do not include obligations under our previous supply agreement with Baxter as this agreement was terminated in March 2013. Further, the amounts presented do not include obligations under our Manufacturing and Supply Agreement with Laboratorios Grifols, S.A., as the initial contract year does not commence until the FDA has approved the product and manufacturing at this facility, and would not commence if such approval was not received. |
| (2) | The amounts presented represent commitments for minimum lease payments related to leases of office space and certain equipment under non-cancelable operating leases. |
| (3) | Includes purchase commitments for capital expenditures and other purchase obligations for services at fixed minimum costs. |
| (4) | Under our license agreement with BMS, we may be required to make additional future payments up to a total of $25.0 million upon the achievement of certain levels of net sales of OFIRMEV. We are also required to pay royalties on any net sales of OFIRMEV under the agreement and are subject to annual minimum royalty obligations. License payments may be increased based on the timing of various milestones and the extent to which the licensed technologies are pursued for other indications. These milestone |
68
| | payments under our license agreements are not included in the table above because at this time we cannot determine when, or if, the related milestones will be achieved or the events triggering the commencement of payment obligations will occur. Further, our minimum royalty obligations are not included in the table as we cannot determine the extent, if any, we will be required to pay as our obligation may be offset by payments from other parties. |
| (5) | We also enter into unconditional purchase obligations with various vendors and suppliers of goods and services in the normal course of operations through purchase orders or other documentation, or that are undocumented except for an invoice. Such unconditional purchase obligations are generally outstanding for periods less than a year and are settled by cash payments upon delivery of goods and services and are not reflected in this line item. |
Off-Balance Sheet Arrangements
We had no off-balance sheet arrangements as of December 31, 2012.
Recent Accounting Pronouncements
See Note 2 to the Notes to Financial Statements in Item 8 below for further discussion of recent accounting pronouncements.
| Item 7A. | Quantitative and Qualitative Disclosures About Market Risk |
Cash Equivalents and Investments
As of December 31, 2012, our cash equivalents and short-term investment holdings consisted of investments in money market funds, debt obligations of municipalities, commercial paper and certificates of deposit. These investments were made in accordance with an investment policy approved by our board of directors which specifies the categories, allocations, and ratings of securities we may consider for investment. The primary objective of our investment activities is to preserve principal while at the same time maximizing the income we receive without significantly increasing risk. Some of the financial instruments that we invest in could be subject to market risk. This means that a change in prevailing interest rates may cause the value of the instruments to fluctuate. For example, if we purchase a security that was issued with a fixed interest rate and the prevailing interest rate later rises, the value of that security will probably decline. To minimize this risk, we intend to maintain a portfolio which may include cash, cash equivalents and investment securities available-for-sale in a variety of securities which may include money market funds, government debt securities and commercial paper, all with various maturity dates. Based on our current investment portfolio, we do not believe that our results of operations would be materially impacted by an immediate change of 10% in interest rates.
We do not hold or issue derivatives, derivative commodity instruments or other financial instruments for speculative trading purposes. Further, we do not believe our cash, cash equivalents and investment securities have significant risk of default or illiquidity. We made this determination based on discussions with our investment advisors and a review of our holdings. While we believe our cash, cash equivalents and investment securities do not contain excessive risk, we cannot provide absolute assurance that in the future our investments will not be subject to adverse changes in market value. All of our cash, cash equivalents and investment securities are held at fair value. The following table shows the fair value of our cash equivalents and investment securities as of December 31, 2012 (in thousands):
| | | | | | | | |
| | | Amortized
Cost Basis | | | Fair Value | |
Cash equivalents | | $ | 55,736 | | | $ | 55,736 | |
Available-for-sale marketable securities | | $ | 3,745 | | | $ | 3,745 | |
69
Debt
Our current loan and security agreement has a fixed interest rate. Consequently, we do not have significant interest rate cash flow exposure on our debt. The outstanding principal balance of the loan and security agreement at December 31, 2012 was $30.0 million, and is collateralized by substantially all of our assets (excluding intellectual property). Under the terms of our current agreement, we must maintain minimum quarterly product revenue of at least $12.5 million, are precluded from entering into certain financing and other transactions, including disposing of certain assets and paying dividends, and are subject to prepayment penalties and various non-financial covenants. We believe we were in compliance with all such covenants under the agreement as of December 31, 2012.
70
| Item 8. | Financial Statements and Supplementary Data |
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
The Board of Directors and
Stockholders of Cadence Pharmaceuticals, Inc.
We have audited the accompanying balance sheets of Cadence Pharmaceuticals, Inc. as of December 31, 2012 and 2011, and the related statements of operations, comprehensive income, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2012. Our audits also included the financial statement schedule listed in the Index at Item 15(a)2. These financial statements and schedule are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements and schedule based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Cadence Pharmaceuticals, Inc. at December 31, 2012 and 2011, and the results of its operations and its cash flows for each of the three years in the period ended December 31, 2012, in conformity with U.S. generally accepted accounting principles. Also, in our opinion, the related financial statement schedule, when considered in relation to the basic financial statements taken as a whole, presents fairly in all material respects the information set forth therein.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), Cadence Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2012, based on criteria established in Internal Control-Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission and our report dated March 8, 2013 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
San Diego, California
March 8, 2013
71
CADENCE PHARMACEUTICALS, INC.
BALANCE SHEETS
(in thousands, except share and per share data)
| | | | | | | | |
| | | December 31, | |
| | 2012 | | | 2011 | |
| Assets | | | | | | | | |
Current assets: | | | | | | | | |
Cash and cash equivalents | | $ | 58,327 | | | $ | 82,609 | |
Investments in marketable securities | | | 3,745 | | | | 44,618 | |
Restricted cash | | | 640 | | | | 450 | |
Accounts receivable, net | | | 6,152 | | | | 2,208 | |
Inventory | | | 6,498 | | | | 1,388 | |
Prepaid expenses | | | 1,064 | | | | 1,071 | |
Other current assets | | | 90 | | | | 90 | |
| | | | | | | | |
Total current assets | | | 76,516 | | | | 132,434 | |
Property and equipment, net | | | 1,967 | | | | 10,569 | |
Intangible assets, net | | | 12,090 | | | | 13,433 | |
Restricted cash | | | — | | | | 190 | |
Other assets | | | 7,106 | | | | 7,039 | |
| | | | | | | | |
Total assets | | $ | 97,679 | | | $ | 163,665 | |
| | | | | | | | |
| Liabilities and Stockholders’ Equity | | | | | | | | |
Current liabilities: | | | | | | | | |
Accounts payable | | $ | 5,796 | | | $ | 3,801 | |
Accrued liabilities | | | 12,969 | | | | 10,450 | |
Deferred revenue | | | 2,234 | | | | 1,291 | |
| | | | | | | | |
Total current liabilities | | | 20,999 | | | | 15,542 | |
Long-term debt, less discount of $1,182 and $1,304, respectively | | | 28,818 | | | | 28,696 | |
Other long-term liabilities | | | 51 | | | | 117 | |
| | | | | | | | |
Total liabilities | | | 49,868 | | | | 44,355 | |
Commitments and contingencies (Note 8) | | | | | | | | |
Stockholders’ equity: | | | | | | | | |
Preferred stock, $0.0001 par value; 10,000,000 shares authorized, no shares issued and outstanding at December 31, 2012 and 2011, respectively | | | — | | | | — | |
Common stock, $0.0001 par value; 200,000,000 shares authorized and 85,668,668 shares issued and outstanding at December 31, 2012 and 100,000,000 shares authorized and 85,511,607 shares issued and outstanding at December 31, 2011 | | | 9 | | | | 9 | |
Additional paid-in capital | | | 495,458 | | | | 485,982 | |
Accumulated other comprehensive income | | | — | | | | 2 | |
Accumulated deficit | | | (447,656 | ) | | | (366,683 | ) |
| | | | | | | | |
Total stockholders’ equity | | | 47,811 | | | | 119,310 | |
| | | | | | | | |
Total liabilities and stockholders’ equity | | $ | 97,679 | | | $ | 163,665 | |
| | | | | | | | |
The accompanying notes are an integral part of these financial statements.
72
CADENCE PHARMACEUTICALS, INC.
STATEMENTS OF OPERATIONS
(in thousands, except per share data)
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
Revenues: | | | | | | | | | | | | |
Product revenue, net | | $ | 50,066 | | | $ | 11,486 | | | $ | — | |
License revenue | | | 118 | | | | 5,210 | | | | — | |
| | | | | | | | | | | | |
Total net revenues | | | 50,184 | | | | 16,696 | | | | — | |
| | | | | | | | | | | | |
Costs and expenses: | | | | | | | | | | | | |
Cost of product sales | | | 23,256 | | | | 12,406 | | | | — | |
Amortization of patent license | | | 1,343 | | | | 1,567 | | | | — | |
Research and development | | | 6,519 | | | | 8,885 | | | | 13,757 | |
Selling, general and administrative | | | 86,843 | | | | 81,504 | | | | 39,347 | |
Impairment of long-lived assets | | | 7,723 | | | | — | | | | 1,522 | |
Other | | | 1,174 | | | | 1,076 | | | | 291 | |
| | | | | | | | | | | | |
Total costs and expenses | | | 126,858 | | | | 105,438 | | | | 54,917 | |
| | | | | | | | | | | | |
Loss from operations | | | (76,674 | ) | | | (88,742 | ) | | | (54,917 | ) |
Other (expense) income: | | | | | | | | | | | | |
Interest income | | | 123 | | | | 136 | | | | 106 | |
Interest expense | | | (4,449 | ) | | | (4,424 | ) | | | (2,144 | ) |
Other income | | | 27 | | | | 9 | | | | 312 | |
| | | | | | | | | | | | |
Total other expense, net | | | (4,299 | ) | | | (4,279 | ) | | | (1,726 | ) |
| | | | | | | | | | | | |
Loss before income tax | | | (80,973 | ) | | | (93,021 | ) | | | (56,643 | ) |
| | | | | | | | | | | | |
Net loss | | $ | (80,973 | ) | | $ | (93,021 | ) | | $ | (56,643 | ) |
| | | | | | | | | | | | |
Basic and diluted net loss per share(1) | | $ | (0.95 | ) | | $ | (1.41 | ) | | $ | (1.09 | ) |
| | | | | | | | | | | | |
Shares used to compute basic and diluted net loss per share(1) | | | 85,556 | | | | 66,075 | | | | 52,042 | |
| | | | | | | | | | | | |
| (1) | As a result of the issuance of common stock pursuant to public offerings in the fourth quarter of 2011 and 2010, there is a lack of comparability in the per share amounts between the periods presented. Please see Note 2 of the Notes to Financial Statements for further discussion. |
The accompanying notes are an integral part of these financial statements.
73
CADENCE PHARMACEUTICALS, INC.
STATEMENTS OF COMPREHENSIVE INCOME
(in thousands)
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
Net loss | | $ | (80,973 | ) | | $ | (93,021 | ) | | $ | (56,643 | ) |
Other comprehensive income (loss) gain: | | | | | | | | | | | | |
Net unrealized (loss) gain on securities available for sale | | | (2 | ) | | | 2 | | | | — | |
| | | | | | | | | | | | |
Other comprehensive income (loss) gain | | | (2 | ) | | | 2 | | | | — | |
| | | | | | | | | | | | |
Comprehensive loss | | $ | (80,975 | ) | | $ | (93,019 | ) | | $ | (56,643 | ) |
| | | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements.
74
CADENCE PHARMACEUTICALS, INC.
STATEMENTS OF STOCKHOLDERS’ EQUITY
(in thousands, except per share data)
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | Common Stock | | | Additional
Paid-in
Capital | | | Accumulated
Other
Comprehensive
Income | | | Accumulated
Deficit | | | Total
Stockholders’
Equity | |
| | | Shares | | | Amount | | | | | |
Balance at December 31, 2009 | | | 50,485 | | | $ | 5 | | | $ | 292,077 | | | $ | — | | | $ | (217,019 | ) | | $ | 75,063 | |
Issuance of warrants in June to purchase 255 shares of common stock at $7.0645 per share | | | — | | | | — | | | | 1,237 | | | | — | | | | — | | | | 1,237 | |
Public offering of common stock, net of $6,445 offering costs, in November and December at $8.00 per share | | | 12,500 | | | | 1 | | | | 93,554 | | | | — | | | | — | | | | 93,555 | |
Issuance of common stock from option exercises and lapse of restricted stock units under equity compensation plans | | | 122 | | | | — | | | | 235 | | | | — | | | | — | | | | 235 | |
Stock-based compensation | | | — | | | | — | | | | 10,513 | | | | — | | | | — | | | | 10,513 | |
Net Loss | | | — | | | | — | | | | — | | | | — | | | | (56,643 | ) | | | (56,643 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2010 | | | 63,107 | | | | 6 | | | | 397,616 | | | | — | | | | (273,662 | ) | | | 123,960 | |
Public offering of common stock, net of $4,448 offering costs, in November at $3.75 per share | | | 21,800 | | | | 2 | | | | 77,300 | | | | — | | | | — | | | | 77,302 | |
Issuance of warrants in December to purchase 158 shares of common stock at $3.79 per share | | | — | | | | — | | | | 390 | | | | — | | | | — | | | | 390 | |
Issuance of common stock from option exercises and lapse of restricted stock units under equity compensation plans | | | 605 | | | | 1 | | | | 1,443 | | | | — | | | | — | | | | 1,444 | |
Stock-based compensation | | | — | | | | — | | | | 9,233 | | | | — | | | | — | | | | 9,233 | |
Unrealized gain on marketable securities, net | | | — | | | | — | | | | — | | | | 2 | | | | — | | | | 2 | |
Net Loss | | | — | | | | — | | | | — | | | | — | | | | (93,021 | ) | | | (93,021 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2011 | | | 85,512 | | | | 9 | | | | 485,982 | | | | 2 | | | | (366,683 | ) | | | 119,310 | |
Issuance of warrants in December to purchase 155 shares of common stock at $3.88 per share | | | — | | | | — | | | | 416 | | | | — | | | | — | | | | 416 | |
Issuance of common stock from option exercises and lapse of restricted stock units under equity compensation plans | | | 157 | | | | — | | | | 451 | | | | — | | | | — | | | | 451 | |
Stock-based compensation | | | — | | | | — | | | | 8,609 | | | | — | | | | — | | | | 8,609 | |
Unrealized loss on marketable securities, net | | | — | | | | — | | | | — | | | | (2 | ) | | | — | | | | (2 | ) |
Net Loss | | | — | | | | — | | | | — | | | | — | | | | (80,973 | ) | | | (80,973 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Balance at December 31, 2012 | | | 85,669 | | | $ | 9 | | | $ | 495,458 | | | $ | — | | | $ | (447,656 | ) | | $ | 47,811 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements.
75
CADENCE PHARMACEUTICALS, INC.
STATEMENTS OF CASH FLOWS
(in thousands)
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
Operating activities | | | | | | | | | | | | |
Net loss | | $ | (80,973 | ) | | $ | (93,021 | ) | | $ | (56,643 | ) |
Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | | | | | |
Depreciation | | | 1,560 | | | | 1,670 | | | | 758 | |
Loss (gain) on disposal of assets | | | 873 | | | | (66 | ) | | | 55 | |
Impairment of long-lived assets | | | 7,723 | | | | — | | | | 1,522 | |
Inventory write-down | | | 163 | | | | 5,574 | | | | — | |
Stock-based compensation | | | 8,609 | | | | 9,233 | | | | 10,513 | |
Non-cash interest expense | | | 27 | | | | 49 | | | | 32 | |
Amortization of intangible assets | | | 1,343 | | | | 1,567 | | | | — | |
Amortization of discount on note payable | | | 122 | | | | 409 | | | | 373 | |
Accretion of discounts on available-for-sale securities, net of accretion of premiums | | | (16 | ) | | | 5 | | | | 29 | |
Changes in operating assets and liabilities: | | | | | | | | | | | | |
Accounts receivable | | | (3,944 | ) | | | (2,208 | ) | | | — | |
Prepaid expenses and other assets | | | (78 | ) | | | 104 | | | | (585 | ) |
Intangible assets | | | — | | | | — | | | | (15,000 | ) |
Inventory | | | (5,273 | ) | | | (6,477 | ) | | | (485 | ) |
Accounts payable | | | 2,140 | | | | 360 | | | | 653 | |
Deferred revenue | | | 943 | | | | 1,291 | | | | — | |
Accrued liabilities and other liabilities | | | 2,482 | | | | 3,358 | | | | (82 | ) |
| | | | | | | | | | | | |
Net cash used in operating activities | | | (64,299 | ) | | | (78,152 | ) | | | (58,860 | ) |
| | | | | | | | | | | | |
| | | |
Investing activities | | | | | | | | | | | | |
Purchases of marketable securities | | | (1,397 | ) | | | (82,681 | ) | | | (24,201 | ) |
Maturities and sales of marketable securities | | | 42,275 | | | | 60,006 | | | | 8,250 | |
Payment for option purchase right | | | — | | | | (3,500 | ) | | | (3,500 | ) |
Restricted cash | | | — | | | | (300 | ) | | | 1,348 | |
Purchases of property and equipment | | | (1,705 | ) | | | (2,733 | ) | | | (3,754 | ) |
Proceeds from the sale of property and equipment | | | 393 | | | | 66 | | | | 3 | |
| | | | | | | | | | | | |
Net cash provided by (used in) investing activities | | | 39,566 | | | | (29,142 | ) | | | (21,854 | ) |
| | | | | | | | | | | | |
| | | |
Financing activities | | | | | | | | | | | | |
Proceeds from issuance of common stock | | | 451 | | | | 78,746 | | | | 93,790 | |
Borrowings under debt agreements, net of fees | | | — | | | | 3,434 | | | | 29,591 | |
Principal payments under debt agreements | | | — | | | | (4,452 | ) | | | (6,351 | ) |
| | | | | | | | | | | | |
Net cash provided by (used in) financing activities | | | 451 | | | | 77,728 | | | | 117,030 | |
| | | | | | | | | | | | |
Net (decrease) increase in cash and cash equivalents | | | (24,282 | ) | | | (29,566 | ) | | | 36,316 | |
Cash and cash equivalents at beginning of period | | | 82,609 | | | | 112,175 | | | | 75,859 | |
| | | | | | | | | | | | |
Cash and cash equivalents at end of period | | $ | 58,327 | | | $ | 82,609 | | | $ | 112,175 | |
| | | | | | | | | | | | |
| | | |
Supplemental disclosures | | | | | | | | | | | | |
Issuance of warrants in connection with loan and security agreement | | $ | 416 | | | $ | 390 | | | $ | 1,237 | |
Shares in unconsolidated entity acquired in option purchase agreement | | $ | — | | | $ | — | | | $ | 500 | |
Property and equipment purchases in accounts payable and accrued expenses | | $ | 338 | | | $ | 891 | | | $ | 371 | |
Cash paid for interest and fees | | $ | 3,865 | | | $ | 4,311 | | | $ | 1,986 | |
The accompanying notes are an integral part of these financial statements.
76
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS
Cadence Pharmaceuticals, Inc. (the “Company”) was incorporated in the state of Delaware in May 2004. The Company is a biopharmaceutical company focused on acquiring, in-licensing, developing and commercializing proprietary products principally for use in the hospital setting. In March 2006, the Company in-licensed the exclusive U.S. and Canadian rights to OFIRMEV® (acetaminophen) injection, an intravenous formulation of acetaminophen, from Bristol-Myers Squibb Company (“BMS”). In November 2010, the Food and Drug Administration (“FDA”) approved the Company’s New Drug Application (“NDA”) for OFIRMEV for the management of mild to moderate pain, moderate to severe pain with adjunctive opioid analgesics, and the reduction of fever in adults and children two years of age and older. In January 2011, the Company commenced commercial sales of the product in the U.S.
| 2. | | Summary of Significant Accounting Policies |
Management Estimates
The preparation of financial statements in accordance with accounting principles generally accepted in the United States of America (“GAAP”) requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. The Company bases its estimates on historical experience and on various other assumptions that are believed to be reasonable under the circumstances. Examples of such estimates include, but are not limited to, the fair value of property and equipment, inventory obsolescence and valuation, restructuring liabilities, stock-based compensation, and commitments and contingencies. On a regular basis, the Company reviews its estimates to ensure the estimates appropriately reflect changes in its business or as new information becomes available. Management believes that these estimates are reasonable, however, actual results could materially differ from these estimates.
Reclassifications
The Company has reclassified certain prior period amounts to conform to the current period presentation. Specifically, it has classified its accrued wholesaler fees payable from accrued liabilities to an offset of its accounts receivable and its accrued group purchasing administrative service fees from accounts receivable to accrued liabilities. As of December 31, 2011, these reclassifications reduced the Company’s current and total assets and current and total liabilities by $495,000, respectively. The reclassification had no impact on the Company’s net loss from operations or stockholders’ equity as previously reported.
Revenue Recognition
The Company recognizes revenue when there is persuasive evidence that an arrangement exists, title has passed, collection is reasonably assured and the price is fixed or determinable. It sells OFIRMEV mostly to wholesalers who in-turn sell the product to hospitals and other end-user customers. Sales to wholesalers provide for selling prices that are fixed on the date of sale, although the Company offers certain discounts to group purchasing organizations, certain end-user hospitals and governmental programs. The wholesalers take title to the product, bear the risk of loss of ownership, and have economic substance to the inventory. Further, the Company has no significant obligations for future performance to generate pull-through sales, however it does allow wholesalers to return product that is damaged or received in error. In addition, the Company allows for product to be returned beginning six months prior to, and ending twelve months following, product expiration. OFIRMEV is the Company’s first and only commercially available product, and the Company had limited product return
77
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
data since the commercial launch of OFIRMEV in January 2011. As a result, the Company has not had sufficient data to reasonably estimate product returns from its wholesaler distribution channel and therefore it has deferred the recognition of revenue until the time that product has been sold by a wholesaler to a hospital or other end-user customer. Shipments of product that have not been recognized as revenue are treated as deferred revenue until evidence existed to confirm that pull-through sales to hospitals or other end-user customers has occurred. However, the Company has begun to accumulate a reasonable amount of product return data, which it continues to gather, and the Company believes that this data will allow it to reasonably estimate returns on product sold in the future.
The Company records certain sales reserves and allowances as a reduction to gross revenue and deferred revenue, as applicable. These reserves and allowances include distribution service fees, a prompt payment reserve, a group purchasing discount and chargeback reserve, discounts to governmental programs and other contractual discounts, as applicable. Distribution service fees arise from contractual agreements the Company has with certain wholesalers for distribution services they provide with respect to OFIRMEV. These fees are generally a fixed percentage of the price of the product purchased by these wholesalers. The prompt payment reserve is based upon cash discounts the Company offers certain wholesalers as an incentive to meet certain payment terms. It accounts for these cash discounts at the time the sale is made to the wholesalers and reduces its accounts receivable accordingly. The group purchasing discount and chargeback reserve is based upon contracted discounts the Company provides to members of certain purchasing groups. The Company estimates the sales through its wholesalers to these group purchasing organizations and accrues for the chargebacks it anticipates from its wholesalers for the difference between the current retail price and the reduced price paid by the members of the group purchasing organizations. A group purchasing organization fee the Company incurs for these transactions is also accrued at the time of sale. The Company also provides governmental programs and certain customers a predetermined discount that is recorded at the time of sale.
Revenue from the Company’s data license agreement has been recognized upon delivery of the goods and services provided, based upon the consideration allocated to each deliverable, or the termination of the service period. The Company allocated the consideration to each deliverable based upon its review of the agreement pursuant to multiple-element arrangement guidance. See Note 9 for further discussion.
Accounts Receivable
The Company extends credit to its customers in the normal course of business based upon an evaluation of the customer’s credit history, financial condition and other factors. Trade accounts receivable are recorded on gross sales to wholesalers, net of allowances for prompt payment and other discounts, wholesaler fees, chargebacks and doubtful accounts. Estimates of allowances for doubtful accounts are determined by evaluating individual customer circumstances, historical payment patterns, length of time past due and economic and other factors. During the year ended December 31, 2012, the Company recorded bad debt expense of $56,000 to reserve for past due accounts. As of December 31, 2012, this balance remained as a reserve against gross receivables. No similar charges were recorded for the years ended December 31, 2011 and 2010 and the Company did not have a reserve balance at December 31, 2011.
Fair Value Reporting
The Company’s financial instruments consist of cash and cash equivalents, marketable securities, restricted cash, trade receivables and payables, an option purchase right, equity securities of an unconsolidated privately-held entity, accrued liabilities and long-term debt. Fair value estimates of these instruments are made at a specific point in time, based on relevant market information. These estimates may be subjective in nature and involve uncertainties and matters of significant judgment and therefore cannot be determined with precision. The
78
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
carrying amount of cash and cash equivalents, restricted cash, trade receivables and payables and accrued liabilities are generally considered to be representative of their respective fair values because of the short-term nature of those instruments. Further, based upon the borrowing rates currently available to the Company for loans with similar terms, the Company believes the fair value of long-term debt approximates its carrying value. The Company’s option purchase right and equity securities of an unconsolidated privately-held entity have been initially valued based upon the transaction price under the cost method of accounting. These assets are subject to fair value adjustments in certain circumstances, such as when there is evidence of impairment. The fair value of marketable securities is based upon market prices quoted on the last day of the fiscal period.
Current accounting guidance defines fair value, establishes a framework for measuring fair value in accordance with GAAP, and requires certain disclosures about fair value measurements. The valuation techniques included in the guidance are based on observable and unobservable inputs. Observable inputs reflect readily obtainable data from independent sources, while unobservable inputs reflect market assumptions and are classified into the following fair value hierarchy:
| | | | | | |
Level 1 Inputs | | | – | | | Quoted prices for identical instruments in active markets. |
| | |
Level 2 Inputs | | | – | | | Quoted prices for similar instruments in active markets; quoted prices for identical or similar instruments in markets that are not active; and model-derived valuations in which all significant inputs and significant value drivers are observable. |
| | |
Level 3 Inputs | | | – | | | Valuation derived from valuation techniques in which one or more significant inputs or significant value drivers are unobservable. |
The following tables present further detail of the financial instruments carried at fair value on the Company’s balance sheet as of December 31, 2012 and 2011. The tables do not include assets and liabilities which are measured at historical cost or on any basis other than fair value (in thousands):
| | | | | | | | | | | | | | | | |
Description | | Balance at
December 31, 2012 | | | Fair Value Measurements
as of December 31, 2012 | |
| | | Level 1 | | | Level 2 | | | Level 3 | |
Assets: | | | | | | | | | | | | | | | | |
Cash equivalents: | | | | | | | | | | | | | | | | |
Money market funds | | $ | 55,736 | | | $ | 55,736 | | | $ | — | | | $ | — | |
Investments in marketable securities—short-term: | | | | | | | | | | | | | | | | |
Debt instruments—Corporate debt obligations | | | 1,398 | | | | — | | | | 1,398 | | | | — | |
Debt instruments—Municipal debt obligations | | | 1,347 | | | | — | | | | 1,347 | | | | — | |
Certificates of deposit | | | 1,000 | | | | — | | | | 1,000 | | | | — | |
| | | | | | | | | | | | | | | | |
Assets at fair value | | $ | 59,481 | | | $ | 55,736 | | | $ | 3,745 | | | $ | — | |
| | | | | | | | | | | | | | | | |
| | |
Description | | Balance at
December 31, 2011 | | | Fair Value Measurements
as of December 31, 2011 | |
| | | | | | | | | | | | | |
| | | Level 1 | | | Level 2 | | | Level 3 | |
Assets: | | | | | | | | | | | | | | | | |
Cash equivalents: | | | | | | | | | | | | | | | | |
Money market funds | | $ | 74,389 | | | $ | 74,389 | | | $ | — | | | $ | — | |
Debt instruments—Corporate debt obligations | | | 6,346 | | | | — | | | | 6,346 | | | | — | |
Investments in marketable securities—short-term: | | | | | | | | | | | | | | | | |
Debt instruments—Corporate debt obligations | | | 37,394 | | | | — | | | | 37,394 | | | | — | |
Debt instruments—Municipal debt obligations | | | 6,224 | | | | — | | | | 6,224 | | | | — | |
Certificates of deposit | | | 1,000 | | | | — | | | | 1,000 | | | | — | |
| | | | | | | | | | | | | | | | |
Assets at fair value | | $ | 125,353 | | | $ | 74,389 | | | $ | 50,964 | | | $ | — | |
| | | | | | | | | | | | | | | | |
79
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
The Company’s Level 2 financial instruments are valued using market prices on less active markets and model-derived valuations with observable valuation inputs such as interest rates and yield curves. The Company obtains the fair value of Level 2 financial instruments from a third-party pricing service, which the Company validates through independent valuation testing and review of portfolio valuations provided by the Company’s investment managers.
Cash Equivalents
The Company considers all highly liquid investments purchased with maturities of three months or less from the date of purchase to be cash equivalents. These investments may include money market funds, U.S. Government agencies, corporate debt securities and commercial paper. As of December 31, 2012 and 2011, the Company’s cash equivalents were $55,736,000 and $80,735,000, respectively.
Marketable Securities
The Company determines the appropriate classification of its investments at the time of acquisition and reevaluates such determination at each balance sheet date. The Company has classified its investment holdings as available-for-sale, as the sale of such securities may be required prior to maturity to implement management strategies. The Company’s investment policy set minimum credit quality criteria and maximum maturity limits on its investments to provide for safety of principle, liquidity and a reasonable rate of return. Available-for-sale securities are recorded at fair value, based on current market valuations. Unrealized holding gains and losses on available-for-sale securities are excluded from earnings and are reported as a separate component of other comprehensive income (loss) until realized. Realized gains and losses are included in non-operating other income (expense) on the statement of operations and are derived using the specific identification method for determining the cost of the securities sold. During the years ended December 31, 2012, 2011 and 2010, no realized gains or losses were recorded on the sale or maturity of the Company’s marketable securities. Further, no impairments to reduce the value of an available-for-sale equity security were taken during the years ended December 31, 2012, 2011 and 2010. See Note 3 for further discussion.
Concentration Risk
Credit Risk.Financial instruments that potentially subject the Company to a significant concentration of credit risk consist primarily of cash, cash equivalents, restricted cash and marketable securities. The Company maintains deposits in federally insured financial institutions in excess of federally insured limits. However, management believes the Company is not exposed to significant credit risk due to the financial position of the depository institutions in which those deposits are held. Additionally, the Company has established guidelines regarding diversification of its investments and their maturities, which are designed to maintain safety and liquidity. To date, the Company has not experienced any material realized losses on its cash, cash equivalents, restricted cash and marketable securities.
Manufacturing.The Company depends on an outsourced manufacturing strategy for its products. It has contracts in place with one third-party manufacturer that is approved for the production of OFIRMEV and one third-party manufacturer for which approval will be sought. Further, the Company relies upon a single source for the active pharmaceutical ingredient for OFIRMEV.
Customers.The Company has entered into distribution agreements with major pharmaceutical wholesalers to supply OFIRMEV across the U.S. through their distribution centers, and a majority of the Company’s sales are to these customers. The Company’s three primary wholesaler customers represented approximately 93% of the Company’s product revenue for 2012 and approximately 94% of the Company’s accounts receivable balance at December 31, 2012. See Note 12 for further detail of the Company’s significant customers.
80
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
Inventories
The Company states its inventories at the lower of cost or market. The Company uses a combination of standard and actual costing methodologies to determine its cost basis for its inventories. These methodologies approximate actual costs on a first-in, first-out basis. In addition to stating inventory at the lower of cost or market, the Company also evaluates inventory each period for excess quantities and obsolescence. This evaluation includes identifying those items specifically identified as obsolete and reserving them, analyzing forecasted demand versus quantities on hand and reserving for the excess, and identifying other specific reserves. During the years ended December 31, 2012 and 2011, the Company recorded charges for inventory losses of $163,000 and $5,574,000, respectively, in cost of sales to write-down certain inventory manufactured to its estimated net realizable value. The product in question had been placed on indefinite hold pending the outcome of an investigation into unidentified particulate matter observed during routine product stability testing however, the Company has decided to destroy this product and has recorded a charge of $290,000 in Other operating expenses for the year ended December 31, 2012 for the destruction costs. See “Supply Agreements” in Note 8 below for further information.
Royalty and License Payments
Pursuant to the terms of its license agreement with BMS, the Company is required to make royalty payments based upon net sales of OFIRMEV, subject to annual minimums, that range from the mid-teens to the mid-twenties, depending on the aggregate amount of net sales. The Company accrues for these payments as the product is sold, or otherwise deemed obligated. Additionally, the Company paid $15,000,000 under the license agreement upon the NDA approval of OFIRMEV in November 2010 and may be required to make future milestone payments of up to $25,000,000 based on the achievement of certain levels of net sales. The Company has capitalized the $15,000,000 payment as an intangible asset on its balance sheet and is amortizing this balance over the estimated useful life of the licensed patents. As of December 31, 2012, the Company had amortized an aggregate $2,910,000 of the payment and the estimated aggregate amortization expense of the payment for each of the five succeeding fiscal years is approximately $1,343,000. With respect to future milestone payments, at December 31, 2012, the Company had not yet achieved the levels of net sales necessary to require it to make payments under these milestone obligations, and therefore had not accrued for such potential payments in its financial statements. The Company will accrue for future milestone payments as they are anticipated and amortize the payments over the period in which the milestone is achieved. See Note 9 for further discussion.
Advertising Expense
The Company records the cost of its advertising efforts when services are performed or goods are delivered. The Company incurred advertising costs of approximately $1,594,000, $2,181,000 and $828,000, respectively, for the years ended December 31, 2012, 2011 and 2010. As of December 31, 2011, the Company had capitalized $24,000 of advertising costs in prepaid expenses. No similar balance remained at December 31, 2012.
Shipping and Handling Costs
The costs incurred by the Company for shipping and handling are classified as cost of product sales. The Company does not charge its customers shipping and handling costs.
Property and Equipment
Property and equipment, including leasehold improvements, are stated at cost or, if the assets are impaired, at fair value. Depreciation is calculated using the straight-line method over the estimated useful lives of the
81
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
assets, which are generally as follows: seven years for manufacturing equipment; five years for furniture and equipment; and three years for computer equipment and software. Leasehold improvements are amortized over the shorter of their useful lives or the terms of the related leases. Asset lives are reviewed periodically to determine if appropriate and adjustments are made as necessary. Depreciation begins at the time the asset is placed in service. Maintenance and repairs are expensed as incurred.
For the years ended December 31, 2012, 2011 and 2010, the Company recorded depreciation expense of $1,560,000, $1,670,000 and $758,000, respectively.
Impairment of Long-Lived Assets
Long-lived assets such as property and equipment are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset may not be recoverable. Recoverability of assets to be held and used is measured by a comparison of the carrying amount of an asset to estimated undiscounted future cash flows expected to be generated by the asset. If the carrying amount of an asset exceeds its estimated future cash flows, an impairment charge is recognized by the amount by which the carrying amount of the asset exceeds the fair value of the asset. Assets to be disposed of would be separately presented in the balance sheet and reported at the lower of the carrying amount or the fair value less costs to sell, and are no longer depreciated. The assets and liabilities of a disposed group classified as held for sale would be presented separately in the appropriate asset and liability sections of the balance sheet.
As of December 31, 2012, the Company recorded a charge of $6,973,000 to impair the value of its manufacturing assets and certain construction-in-process to their estimated fair value. The charge was due to the termination of a supply agreement with one of its third-party manufacturers. Additionally, the Company fully impaired its asset retirement obligation related to the removal of the equipment located at that manufacturer’s facility, resulting in an additional charge of $750,000. During 2010, the Company recorded a charge of $1,522,000 due to the modification of the design of the planned second production line for OFIRMEV, resulting in the partial cancellation of a capital equipment order for which the Company had identified an alternative supplier. No similar charge was incurred during the year ended December 31, 2011. See Note 6 and Note 8 for further discussion.
Research and Development
The Company’s research and development expenses consist primarily of salaries and related employee benefits, costs associated with clinical trials managed by the Company’s contract research organizations (“CROs”), and costs associated with non-clinical activities, such as regulatory and pre-commercialization manufacturing expenses. The Company uses external service providers and vendors to conduct clinical trials, to manufacture product candidates to be used in clinical trials and to provide various other research and development related products and services. The Company accounts for research and development expenditures as incurred and accrues expenses based upon estimates of work performed, patient enrollment and experience with similar contracts.
Income Taxes
Income taxes are accounted for under the asset and liability method. Under this method, deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carryforwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that
82
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
includes the enactment date. The Company provides a valuation allowance against net deferred tax assets unless, based upon the available evidence, it is more likely than not that the deferred tax assets will be realized. A valuation allowance is recorded when it is more likely than not that some of the deferred tax assets will not be realized. In determining the need for valuation allowances the Company considers projected future taxable income and the availability of tax planning strategies. If in the future the Company determines that it would not be able to realize its recorded deferred tax assets, an increase in the valuation allowance would be recorded, decreasing earnings in the period in which such determination is made.
The Company assesses its income tax positions and record tax benefits for all years subject to examination based upon its evaluation of the facts, circumstances and information available at the reporting date. For those tax positions where there is a greater than 50% likelihood that a tax benefit will be sustained, the Company has recorded the largest amount of tax benefit that may potentially be realized upon ultimate settlement with a taxing authority that has full knowledge of all relevant information. For those income tax positions where there is less than 50% likelihood that a tax benefit will be sustained, no tax benefit has been recognized in the financial statements.
Stock-Based Compensation
The Company has stock-based compensation plans, which are described in Note 11. As of December 31, 2012, the Company had issued both stock option awards and restricted stock units under its stock-based compensation plans. During the year ended December 31, 2012, the Company issued stock option awards which were initially classified as liability awards as the issuance of any common stock under these options was subject to the Company’s receipt of stockholder approval for an amendment to the Company’s Amended and Restated Certificate of Incorporation to increase the number of authorized shares of common stock. These awards were subsequently classified as equity awards during the year following receipt of the requisite stockholder approval. The Company revalued the awards on the approval date and is amortizing this value throughout the vesting period of these awards. As of December 31, 2012, all stock-based compensation awards were classified as equity awards.
Stock option awards.Stock options are valued using the Black-Scholes option pricing model. The Company values option awards on the date of grant or, if the awards are classified as liability awards, it revalues the awards each reporting period using this model until the awards are subsequently classified as equity awards, or otherwise vest. The Black-Scholes option pricing model involves a number of estimates, including the expected lives of stock options, the Company’s anticipated stock volatility and interest rates. The following table summarizes the average estimates the Company used in the Black-Scholes option pricing model for the years ended December 31, 2012, 2011 and 2010, to determine the fair value of stock options granted during each period. The estimates listed for the year ended December 31, 2012 represent the estimates applied to equity classified awards on the date of grant or, in the case of the awards initially classified as liability awards, the estimates applied to the awards following their classification change to equity awards:
| | | | | | |
| | | Year Ended December 31, |
| | | 2012 | | 2011 | | 2010 |
Risk free interest rates | | 0.9% | | 2.2% | | 2.7% |
Expected life in years | | 5.7 years | | 6.2 years | | 5.9 years |
Expected dividend yield | | 0.0% | | 0.0% | | 0.0% |
Expected volatility | | 72.0% | | 73.9% | | 76.3% |
The Company determines its risk-free interest rate assumption based on the U.S. Treasury yield for obligations with contractual lives similar to the expected lives of the Company’s share-based payment awards being valued. The weighted-average expected life of options has historically been calculated using the simplified
83
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
method, as prescribed by the Securities and Exchange Commission (“SEC”), due to the lack of relevant historical exercise data. In addition, due to the Company’s limited historical stock price volatility data, the estimated volatility has historically been calculated by incorporating the historical volatility of comparable companies. In 2011, the Company began to incorporate the historical stock price volatility and the implied volatility of its exchanged traded options in determining the expected volatility. The assumed dividend yield was based on the Company’s expectation of not paying dividends in the foreseeable future. Forfeitures are estimated based upon the historical and anticipated future experience.
Based upon these assumptions, the Company has estimated the per share weighted-average grant date fair value of its options granted for the years ended December 31, 2012, 2011 and 2010 at $1.86, $5.67 and $5.99, respectively.
Restricted stock unit awards. Restricted stock units (“RSUs”) are valued based on the fair market value of the Company’s stock on the date of grant and the Company recognizes expense for RSUs if vesting is considered probable. The weighted-average grant date fair value of the RSUs granted in 2010 was $10.38. There were no RSUs granted in 2012 or 2011.
Compensation expense for all stock-based payment awards is recognized using the straight-line method. Stock-based compensation expense recognized during the period is based on the value of the portion of awards that is ultimately expected to vest. Hence, the gross expense is reduced for estimated forfeitures and adjusted for the probability of achieving performance criteria. If awards are forfeited prior to vesting, all previous expense recognized is recovered during the period in which the forfeiture occurs.
The table below summarizes the total stock-based compensation expense included in the Company’s statements of operations for the periods presented (in thousands):
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
Cost of product sales | | $ | 343 | | | $ | 297 | | | $ | — | |
Research and development | | | 1,651 | | | | 2,308 | | | | 3,058 | |
Selling, general and administrative | | | 6,615 | | | | 6,628 | | | | 7,455 | |
| | | | | | | | | | | | |
Total stock-based compensation expense included in loss from operations | | $ | 8,609 | | | $ | 9,233 | | | $ | 10,513 | |
| | | | | | | | | | | | |
The compensation expense related to unvested stock options and RSUs not yet recognized was approximately $11,578,000 at December 31, 2012. This expense is expected to be recognized over a weighted-average period of approximately 32 months. The total fair value of shares vested during the years ended December 31, 2012, 2011 and 2010 was $9,212,000, $9,852,000 and $9,273,000, respectively.
Comprehensive Income (Loss)
Comprehensive income (loss) is defined as the change in equity during a period from transactions and other events and circumstances from non-owner sources. Components of comprehensive income (loss) includes, among other items, unrealized gains and losses on the changes in fair value of investments. These components are added, net of their related tax effect, to the reported net income (loss) to arrive at comprehensive income (loss). The balance of accumulated other comprehensive income at December 31, 2011 was comprised of the net unrealized net holding gains on the Company’s investments in marketable securities. There was no similar accumulated other comprehensive income or loss at December 31, 2012 and 2010. See Note 3 for further detail of the unrealized holdings gains and losses on the Company’s investments in marketable securities.
84
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
Net Loss Per Share
Net loss per share is presented as basic and diluted net loss per share. Basic net loss per share is calculated by dividing the net loss by the weighted average number of common shares outstanding for the period, without consideration for common stock equivalents. Diluted net loss per share is computed by dividing the net loss by the weighted average number of common share equivalents outstanding for the period determined using the treasury-stock method. For purposes of this calculation, stock options, restricted stock units and warrants are considered to be common stock equivalents and are not included in the calculations of diluted net loss per share as their effect is anti-dilutive. Additionally, the restricted stock units outstanding during 2012, 2011 and 2010 were excluded from the basic net loss calculation as these units do not include dividend rights and therefore are not considered to be participating securities.
The actual net loss per share amounts for the years ended December 31, 2012, 2011 and 2010 were computed based on the weighted average shares of common stock outstanding during the respective periods. The net loss per share for the years presented include the effect of the (1) 21,800,000 common shares issued pursuant to a public offering in the fourth quarter of 2011; and (2) 12,500,000 common shares issued pursuant to a public offering in the fourth quarter of 2010. As a result of the issuance of these common shares, there is a lack of comparability in the basic and diluted net loss per share amounts for the periods presented.
The following is a reconciliation of the basic and diluted shares for the periods presented (in thousands):
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
Shares for basic and dilutive net loss per share: | | | | | | | | | | | | |
Weighted average common shares outstanding | | | 85,556 | | | | 66,075 | | | | 52,043 | |
Weighted average unvested common shares subject to repurchase | | | — | | | | — | | | | (1 | ) |
| | | | | | | | | | | | |
Denominator for basic and diluted earnings per share | | | 85,556 | | | | 66,075 | | | | 52,042 | |
| | | | | | | | | | | | |
At December 31, 2012, 2011 and 2010, options, restricted stock units and warrants totaling approximately 16,677,000 shares, 14,457,000 shares and 13,460,000 shares, respectively, were excluded from the calculation as their effect would have been anti-dilutive.
Recent Accounting Pronouncements
In January 2013, the Financial Accounting Standards Board (“FASB”) issued Accounting Standards Update (“ASU”) No. 2013-01,Clarifying the Scope of Disclosures About Offsetting Assets and Liabilities. ASU 2013-01 clarifies which instruments and transactions are subject to the offsetting disclosures requirements established by ASU 2011-11,Disclosures About Offsetting Assets and Liabilities. Under ASC 2013-01, instruments or transactions subject to the disclosures include recognized derivative instruments accounted for in accordance with ASC 815,Derivative Hedging, including bifurcated embedded derivatives, repurchase agreements and reverse repurchase agreements, and securities borrowing and securities lending transactions, to the extent they are offset in the financial statements or subject to an enforceable master netting agreement or similar agreement. Further, the disclosures are required irrespective of whether the transactions are offset in the statement of financial position. The Company applies the offsetting principle to certain assets and liabilities, however it does not hold instruments, and has not had transactions involving such instruments, which would be subject to the disclosure requirements. The guidance is effective for annual reporting periods beginning on or after January 1, 2013, and interim period within those annual periods. The adoption of this standard is not expected to have an impact on the Company’s financial results or disclosures.
85
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
In February 2013, the FASB issued ASU No. 2013-02, Comprehensive Income (Topic 220): Reporting of Amounts Reclassified Out of Accumulated Other Comprehensive Income. Under ASU 2013-02, an entity is required to provide information about the amounts reclassified out of accumulated other comprehensive income (“AOCI”) by component. In addition, an entity is required to present, either on the face of the financial statements or in the notes, significant amounts reclassified out of AOCI by the respective line items of net income, but only if the amount reclassified is required to be reclassified in its entirety in the same reporting period. For amounts that are not required to be reclassified in their entirety to net income, an entity is required to cross-reference to other disclosures that provide additional details about those amounts. ASU 2013-02 does not change the current requirements for reporting net income or other comprehensive income in the financial statements. The guidance is effective for annual reporting periods beginning on or after January 1, 2013, and interim periods within those annual periods. The adoption of this standard is not expected to have an impact on the Company’s financial results or disclosures.
| 3. | | Investments in Marketable Securities |
In accordance with the Company’s investment policy, it has invested funds in marketable securities. The cost, gross unrealized holding gains, gross unrealized holding losses and fair value of these investments by types and classes of security at December 31, 2012 and December 31, 2011 consisted of the following (in thousands):
| | | | | | | | | | | | | | | | | | | | |
At December 31, 2012 | | Amortized
Cost Basis | | | Other-than-
temporary
Impairments | | | Gross
Unrealized
Holding Gains | | | Gross
Unrealized
Holding Losses | | | Fair Value | |
Available-for-sale: | | | | | | | | | | | | | | | | | | | | |
Debt instruments—Corporate debt obligations | | $ | 1,398 | | | $ | — | | | $ | — | | | $ | — | | | $ | 1,398 | |
Debt instruments—Municipal debt obligations | | | 1,347 | | | | — | | | | — | | | | — | | | | 1,347 | |
Certificates of deposit | | | 1,000 | | | | — | | | | — | | | | — | | | | 1,000 | |
| | | | | | | | | | | | | | | | | | | | |
| | $ | 3,745 | | | $ | — | | | $ | — | | | $ | — | | | $ | 3,745 | |
| | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | |
At December 31, 2011 | | Amortized
Cost Basis | | | Other-than-
temporary
Impairments | | | Gross
Unrealized
Holding Gains | | | Gross
Unrealized
Holding Losses | | | Fair Value | |
Available-for-sale: | | | | | | | | | | | | | | | | | | | | |
Debt instruments—Corporate debt obligations | | $ | 37,392 | | | $ | — | | | $ | 3 | | | $ | (1 | ) | | $ | 37,394 | |
Debt instruments—Municipal debt obligations | | | 6,224 | | | | — | | | | — | | | | — | | | | 6,224 | |
Certificates of deposit | | | 1,000 | | | | — | | | | — | | | | — | | | | 1,000 | |
| | | | | | | | | | | | | | | | | | | | |
| | $ | 44,616 | | | $ | — | | | $ | 3 | | | $ | (1 | ) | | $ | 44,618 | |
| | | | | | | | | | | | | | | | | | | | |
Investments by contractual maturity are as follows (in thousands):
| | | | | | | | | | | | | | | | |
| | | December 31, 2012 | | | December 31, 2011 | |
| | | Cost | | | Fair Value | | | Cost | | | Fair Value | |
Due or callable in one year or less | | $ | 3,745 | | | $ | 3,745 | | | $ | 44,616 | | | $ | 44,618 | |
Due after one year | | $ | — | | | $ | — | | | $ | — | | | $ | — | |
86
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
As of December 31, 2012, there were no investments in unrealized loss positions. As of December 31, 2011, the Company held two investments in an unrealized loss position, all of which had been in such a position for less than 12 months. During the year ended December 31, 2012, these positions matured and no gains or losses were recognized.
| 4. | | Selected Financial Statement Data |
| | | | | | | | |
| | | As of December 31, | |
| | | 2012 | | | 2011 | |
Accounts receivable (in thousands): | | | | | | | | |
Trade accounts receivable | | $ | 6,208 | | | $ | 2,208 | |
Allowance for doubtful accounts | | | (56 | ) | | | — | |
| | | | | | | | |
| | $ | 6,152 | | | $ | 2,208 | |
| | | | | | | | |
Inventory (in thousands): | | | | | | | | |
Raw materials | | $ | 83 | | | $ | 96 | |
Finished goods | | | 6,415 | | | | 1,292 | |
| | | | | | | | |
| | $ | 6,498 | | | $ | 1,388 | |
| | | | | | | | |
Property and equipment (in thousands): | | | | | | | | |
Manufacturing equipment | | $ | 2,999 | | | $ | 6,925 | |
Leasehold improvements | | | 1,639 | | | | 1,610 | |
Computer equipment and software | | | 1,489 | | | | 1,629 | |
Furniture and fixtures | | | 478 | | | | 458 | |
Construction-in-process | | | 724 | | | | 3,965 | |
| | | | | | | | |
| | | 7,329 | | | | 14,587 | |
Less accumulated depreciation | | | (5,362 | ) | | | (4,018 | ) |
| | | | | | | | |
Total | | $ | 1,967 | | | $ | 10,569 | |
| | | | | | | | |
Accrued liabilities (in thousands): | | | | | | | | |
Accrued personnel costs | | $ | 6,560 | | | $ | 6,896 | |
Accrued royalties payable | | | 2,652 | | | | 917 | |
Accrued manufacturing costs and equipment purchases | | | 564 | | | | 1,613 | |
Accrued asset retirement obligation | | | 750 | | | | — | |
Other accrued liabilities | | | 2,443 | | | | 1,024 | |
| | | | | | | | |
Total | | $ | 12,969 | | | $ | 10,450 | |
| | | | | | | | |
On June 21, 2010, the Company entered into an option agreement (the “Option Agreement”) with Incline Therapeutics, Inc. (“Incline”), a privately held specialty pharmaceutical company, pursuant to which the Company obtained an exclusive, irrevocable option (the “Option”) to acquire Incline, which is developing IONSYS™ (fentanyl iontophoretic transdermal system), an investigational product candidate intended to provide patient-controlled analgesia for adult inpatients requiring opioids following surgery. As consideration for the Option, the Company paid Incline a $3,500,000 upfront option fee in June 2010 and made a second payment of $3,500,000 in September 2011. Additionally, in consideration of the Company’s expenditure of funds in connection with conducting its initial due diligence on IONSYS, the Company received $500,000 of Incline Series A preferred stock, or 500,000 shares, on terms generally consistent with Incline’s other Series A preferred stock investors.
87
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
In December 2012, the Company and Incline entered into a Waiver, Consent and Option Termination Agreement (the “Waiver Agreement”) pursuant to which the Company agreed to the buy-out and termination of its Option, contingent upon the closing of a separate agreement and plan of merger between Incline and The Medicines Company whereby The Medicines Company agreed to acquire Incline (the “Incline Acquisition”). In January 2013, The Medicines Company completed its acquisition of Incline. As consideration for entering into the Waiver Agreement and relinquishing its Option, the Company received a payment of $13,125,000 upon the closing of the Incline Acquisition. The Company also received an additional payment of $1,529,000 as consideration for the 500,000 shares of Incline Series A preferred stock held by the Company. The Company could also receive future milestone payments related to potential future licensing, regulatory approval and sales of the product candidate. Such milestones, if any, will be recorded as they are earned.
At the time the Option Agreement was entered, the Company determined that Incline was a variable interest entity (“VIE”). However, because it would not absorb a disproportionate amount of Incline’s expected losses or receive a disproportionate amount of Incline’s expected residual returns, the Company was not the primary beneficiary of this entity. Further, the Company did not have oversight of the day-to-day operations of Incline, nor did it have sufficient rights or voting representation to influence the operating or financial decisions of Incline, and the Company was not a founder of Incline and had no additional equity or funding requirements in future financings or otherwise. As such, the Company did not consolidate Incline into its financial statements. Alternatively, it valued its investment in the option, and the shares received from the due diligence, using the cost method and classified these investments as Level 3 in the fair value hierarchy with a carrying value of $7,000,000. No adjustments were recorded to the carrying value of these assets during the years ended December 31, 2012, 2011 and 2010, as no impairment indicators were present. Further, as the Company did not initially elect the fair value option to value of these assets, it did not record an adjustment to the carrying value as a result of the Waiver Agreement. The Company will record the $14,654,000 consideration received at the time of the closing of the Incline Acquisition in January 2013, and a corresponding gain on the investment of $7,654,000 as of that time.
The $7,000,000 carrying value of the Company’s Incline investments were recorded as other long-term assets on the Company’s balance sheets at December 31, 2012 and 2011, respectively. See Note 16 for further discussion.
| 6. | | Restructuring and Impairment Charges |
In February 2012, the Company observed particulate matter during routine product stability testing of OFIRMEV that was manufactured at one of its third-party manufacturers, Baxter Healthcare Corporation (“Baxter”). As a result, the Company decided to temporarily suspend further production by Baxter until the investigation had been completed and any necessary corrective and preventative actions had been implemented. In March 2013, the Company and Baxter mutually agreed to terminate the supply agreement for OFIRMEV. As a result, the Company reduced the carrying value of its manufacturing assets and its manufacturing equipment and facility construction assets in process to their current estimated fair value as of December 31, 2012, resulting in an impairment charge of $6,973,000 during the year. The fair value of these assets was determined through a third-party valuation assessment based upon research of market prices for similar equipment and the Company’s prior experience with asset disposals. The determination of the fair value of the manufacturing assets was considered a Level 3 measurement. The Company also fully impaired the asset retirement obligation related to the removal of the equipment as of December 31, 2012, resulting in a charge of $750,000 during the year. No such obligation had been recorded as of December 31, 2011. During 2010, the Company recorded a charge of $1,522,000 due to the modification of the design of the planned second production line for OFIRMEV, resulting in the partial cancellation of a capital equipment order for which the Company had identified an alternative supplier. See further discussion of the Baxter agreement in Note 8.
88
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
In November 2011, the Company commenced a restructuring of its workforce to focus its resources on the commercialization of OFIRMEV and reduce program costs not directly associated with such efforts. As a result of the 2011 restructuring, the Company recorded one-time employee termination charges of $1,142,000 in connection with the termination of 17 employees. The following table details the restructuring charges for severance-related costs and termination of contractual obligations for periods presented (in thousands):
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
Beginning restructuring liability | | $ | 931 | | | $ | — | | | $ | — | |
Severance and termination charges incurred | | | — | | | | 1,142 | | | | — | |
Adjustments to severance and termination charges | | | — | | | | — | | | | — | |
Severance and termination disbursements | | | (931 | ) | | | (211 | ) | | | — | |
| | | | | | | | | | | | |
Ending restructuring liability | | $ | — | | | $ | 931 | | | $ | — | |
| | | | | | | | | | | | |
| 7. | | Loan and Security Agreement |
In December 2012, the Company entered into a First Amendment to Second Amended and Restated Loan and Security Agreement (the “2012 Amendment”) with Oxford Finance LLC, Silicon Valley Bank and General Electric Capital Corporation (collectively, the “Lenders”). The 2012 Amendment amends and restates the Company’s previous Second Amended and Restated Loan and Security Agreement entered into in December 2011 (the “2011 Amendment”) with the Lenders, which had amended and restated the Company’s previous Amended and Restated Loan and Security Agreement entered in June 2010 (the “2010 Amendment”). The 2010 Amendment established the Company’s current $30,000,000 credit facility by extinguishing the Company’s previous $15,000,000 credit facility established in 2007 (the “2007 Facility”).
The 2012 Amendment delayed the commencement of principal payments by one year, from January 1, 2013 to January 1, 2014, but did not increase the amount of principal available under the facility. At the time of closing the 2012 Amendment, the Company made a term loan final payment of $752,000 in accordance with the terms of the 2011 Amendment, which had been amortized over the term of the 2011 Amendment. The Company also paid customary closing fees and expenses of $18,000 in connection with the closing of the 2012 Amendment. The stated interest rate under the 2012 Amendment is 10.9545% and the Company will be required to make a final payment of 6% of the total advance at the termination of the loan.
Pursuant to the terms of the 2012 Amendment, the Company will make interest only payments through December 2013, and in January 2014, will begin to make equal monthly principal and interest payments to fully amortize the balance over the remaining 30-month term.
The Company issued warrants to purchase 154,638 shares of the Company’s common stock, as detailed below, to the Lenders in connection with the 2012 Amendment at an exercise price $3.88 per share. The warrants are immediately exercisable, and excluding certain mergers or acquisitions, will expire on the seven-year anniversary of the date of issuance. The Company determined the relative fair value of these warrants, as detailed below, and has classified the warrants as equity, recognizing the cost as a discount on the loan issuance. The credit facility contains customary default and acceleration provisions and is secured by the Company’s assets, excluding intellectual property. Further, the Company was required to make a negative pledge of its intellectual property, which generally prohibits the Company from granting liens on its intellectual property. Under the terms of the 2012 Amendment, the Company may be precluded from entering into certain financing and other transactions, including disposing of certain assets and paying dividends, and is subject to prepayment penalties and certain financial and non-financial covenants, including the maintenance of minimum quarterly product
89
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
revenue of at least $12,500,000. Upon the occurrence of an event of default, including a Material Adverse Change (as defined in 2011 Amendment), the lenders may declare all outstanding amounts due and payable under the 2012 Amendment. As of December 31, 2012, the Company was in compliance will all covenants under the 2012 Amendment.
The Company determined that the terms of the 2012 Amendment were not substantially different than the 2011 Amendment and has therefore accounted for the transaction as a loan modification. As such, the fair value of the warrants issued in connection with the 2012 Amendment and the carrying value of the issuance costs and discount related to the 2011 Amendment were aggregated and are being amortized to interest expense throughout the life of the 2012 Amendment using an effective interest rate of 15.30%. Similarly, in connection with the 2011 Amendment, the Company determined that the terms were not substantially different than the 2010 Amendment and therefore accounted for the transaction as a loan modification of the 2010 Amendment. The 2011 Amendment provided the Company with $3,434,000 of additional net capital after deducting a $954,000 term loan final payment paid under the 2010 Amendment and customary closing fees and expenses of $63,000 paid in connection with the closing of the 2011 Amendment. As part of the 2011 Amendment, the Company issued warrants to purchase an aggregate of 158,311 shares of the Company’s common stock to the Lenders, as detailed below, and classified the warrants as equity, recognizing the fair value as a discount on the loan issuance. The fair value of the warrants was aggregated with the carrying value of the issuance costs and discount related to the 2010 Amendment, and was being amortized over the term of the 2011 Amendment using an effective interest rate of 15.31% prior to the 2012 Amendment.
In connection with the 2010 Amendment, the outstanding balance of the Company’s 2007 Facility was paid in full, including accrued interest, and a $375,000 term loan final payment. This transaction was accounted for as a loan extinguishment and upon the repayment of the $15,000,000 facility, and the Company recorded a charge of approximately $145,000 in the second quarter of 2010 to (1) fully amortize the balance of the loan discount and related issuance costs and (2) fully accrue the term loan final payment. The warrants to purchase an aggregate of 254,793 shares of the Company’s common stock issued, as detailed below, and the upfront fees paid in connection with the 2010 Amendment were recognized as a discount on the loan issuance while the legal and related expenses were recognized as debt issuance costs on the Company’s balance sheets. At the time of the 2012 Amendment, the carrying values of these costs were subsequently aggregated with the relative fair value of the warrants issued in connection with the 2011 Amendment and amortized accordingly.
As of December 31, 2012 and 2011, the aggregate outstanding principal balance of the loans included on the Company’s balance sheets for each period was $30,000,000. Future maturities and interest payments under the Company’s 2012 Amendment as of December 31, 2012 were as follows (in thousands):
| | | | |
2013 | | $ | 3,250 | |
2014 | | | 13,772 | |
2015 | | | 13,772 | |
2016 | | | 8,686 | |
2017 | | | — | |
| | | | |
Total future payments | | | 39,480 | |
Less amount representing interest and fees | | | (9,480 | ) |
| | | | |
Gross balance of long-term debt | | | 30,000 | |
Less unamortized discount | | | (1,182 | ) |
| | | | |
Total present value of long-term debt | | $ | 28,818 | |
| | | | |
90
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
Warrants
In connection with the establishment of the Company’s credit facilities and related amendments, the Company has issued warrants to the Lenders to purchase shares of the Company’s common stock. Most recently, in December 2012, the Company issued four fully exercisable warrants to purchase an aggregate of 154,638 shares of the Company’s common stock at an exercise price of $3.88 per share, expiring December 5, 2019, in connection with its 2012 Amendment. The Company classified the warrants from the 2012 Amendment as equity and determined their relative fair value to be $416,000, using the Black-Scholes valuation model. The value of the warrants was recorded as a discount to the note payable, and is being amortized to interest expense over the expected term of the loan agreement. The warrants were valued using the following assumptions: risk-free interest rate of 1.02%; dividend yield of 0.0%; expected volatility of 70.17%; and a contractual term of seven years.
Additionally, in December 2011, the Company issued four fully exercisable warrants to the Lenders to purchase an aggregate of 158,311 shares of the Company’s common stock at an exercise price of $3.79 per share, expiring December 22, 2018, in connection with its 2011 Amendment. In June 2010, the Company issued three fully exercisable warrants to the Lenders to purchase an aggregate of 254,793 shares of the Company’s common stock at an exercise price of $7.0645 per share, expiring June 18, 2017, in connection with its 2010 Amendment, and in June 2007, the Company issued six fully exercisable warrants to the Lenders to purchase an aggregate of 50,331 shares of the Company’s common stock at an exercise price of $12.67 per share, expiring November 30, 2014, in connection with the 2007 Facility. The Company has classified each of the warrants issued as equity and determined their relative fair value using the Black-Scholes valuation model. More specifically, the warrants issued in December 2011 were determined to have a relative fair value of $390,000, using the assumptions of a 1.4% risk-free interest rate, a dividend yield of 0.0%, an expected volatility of 72.4% and a contractual term of seven years. The warrants issued in June 2010 were determined to have a relative fair value of $1,237,000, using the assumptions of a 2.7% risk-free interest rate, a dividend yield of 0.0%, an expected volatility of 76.5% and a contractual term of seven years. Finally, the warrants issued in November 2007 were determined to have a relative fair value of $474,000, using the assumptions of a 3.64% risk-free interest rate, a dividend yield of 0.0%, an expected volatility of 70.0% and a contractual term of seven years.
As of December 31, 2012, the aforementioned warrants to purchase 618,073 shares of the Company’s common stock were outstanding.
| 8. | | Commitments and Contingencies |
Leases
In May 2006, the Company entered into a six-year operating lease for corporate office space. In December 2011, the Company amended the lease to reduce the monthly rent charge, extend the lease term and terminate a portion of the lease, returning space to the lessor. Pursuant to the terms of the amended agreement, the basic monthly per square foot fee was reduced commencing in April 2012 and the Company returned a portion of the leased space in September 2012. The lease will expire in December 2013 with no option to extend the term.
As security for the initial lease, a letter of credit in the initial amount of $1,581,000 was required by the landlord. The letter of credit is collateralized by a certificate of deposit in the same amount that is classified as restricted cash in the Company’s balance sheet. The required amount subject to the letter of credit and corresponding certificate of deposit was eligible to be reduced by 22% on each of the first four anniversaries of the commencement of the lease and as of December 31, 2012, the letter of credit had been reduced by $1,391,000 in accordance with the agreement and the related restricted cash had been adjusted by a like amount. The value of the letter of credit and corresponding certificate of deposit, classified as restricted cash on the Company’s balance sheet at December 31, 2012 and 2011, was $190,000.
91
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
The Company also leases certain office equipment under capital and operating leases. Its current capital lease has a term of four years and expires in 2016. As of December 31, 2012, the assets under its current capital lease had a gross value of $56,000 and as of December 31, 2012, the Company had recorded amortization expense of $1,000 related to the assets. The obligation under its capital lease is recorded on the Company’s balance sheet in Accrued Expenses at $12,000 and Other Long-Term Liabilities at $43,000. No assets were recorded under capital leases as of December 31, 2011.
As of December 31, 2012, the total future minimum payments under its operating and capital leases, including rent and office equipment, were as follows (in thousands):
| | | | |
2013 | | $ | 810 | |
2014 | | | 43 | |
2015 | | | 22 | |
2016 | | | 17 | |
2017 | | | — | |
Thereafter | | | — | |
| | | | |
Total | | $ | 892 | |
| | | | |
Rent expense for operating leases is recorded on a straight-line basis over the life of the lease term. If a lease has a fixed and determinable escalation clause, the difference between the rent expense and rent paid is recorded as deferred rent. Rent expense under the Company’s facility and equipment leases for the years ended December 31, 2012, 2011 and 2010 was $927,000, $928,000 and $919,000, respectively.
Corporate Credit Card
In 2009, the Company entered into a pledge agreement pursuant to the establishment of a corporate credit card program whereby the Company pledged $150,000 in a certificate of deposit as collateral. During 2011, the Company increased its pledged amount by $300,000 related to an increase in its credit limit. At December 31, 2012, the Company maintained the pledge agreement and the funds under the agreement are classified as restricted cash on the Company’s balance sheet at December 31, 2012 and 2011, respectively.
Supply Agreements
Baxter Healthcare Corporation
In July 2007, the Company entered into a development and supply agreement (the “Supply Agreement”) with Baxter for the completion of pre-commercialization manufacturing development activities and the manufacture of commercial supplies of the finished drug product for OFIRMEV with an initial term of five years. In January 2011, the Company amended and restated the Supply Agreement (the “Amended Supply Agreement”) in connection with a plan to expand the manufacturing capacity for OFIRMEV at Baxter.
In February 2012, the Company announced a voluntary recall of a single lot of OFIRMEV that was manufactured at Baxter’s facility due to the presence of an unidentified, visible particle in that lot during routine stability testing. The Company also placed certain finished product inventory of OFIRMEV manufactured by Baxter on indefinite hold pending the outcome of the Company’s investigation into unidentified particulate matter observed during routine product stability testing and decided to temporarily suspend further production by Baxter until the investigation had been completed and any necessary corrective and preventative actions had been implemented. In July 2012, the Company announced a second voluntary recall of the remaining 41 unexpired lots of OFIRMEV manufactured at Baxter’s facility due to the presence of unidentified, visible particles in a limited
92
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
number of vials from one lot of the product, which were detected during routine stability testing. Although the Company received no adverse event reports associated with the particulate matter, and no product complaints involving similar particulate matter have been received, the Company decided to recall the remaining lots of OFIRMEV manufactured by Baxter as a precautionary measure. All of the 41 recalled lots, which were manufactured between January and March 2011, had expired by December 31, 2012. In March 2013, the Company and Baxter mutually agreed to terminate the supply agreement for OFIRMEV. As part of the settlement and termination with Baxter, the Company agreed that it would be responsible for the removal of the equipment, which the Company has estimated will cost approximately $750,000. Accordingly, it has recorded this retirement obligation on its balance sheet at December 31, 2012 as the conditions existed under the terms of the Amended Supply Agreement at that time. Further, as of December 31, 2012, the Company fully impaired this retirement obligation and recognized a charge of $750,000 in its statement of operations for the year ended December 31, 2012.
As a result of the initial recall, the Company recorded charges of $5,574,000 for the fourth quarter of 2011 and $163,000 for the first quarter of 2012 to fully write-down the value of the inventory placed on hold due to uncertainty as to the amount of time that would be required to complete the investigation and whether the product would have sufficient remaining shelf life or otherwise be saleable after the investigation is completed. As a result of the second recall, the Company decided to destroy the product that was previously placed on hold and has estimated the destruction charges at $290,000, which the Company recorded as a charge in “Other” operating expenses for the year ended December 31, 2012. In addition, the Company has incurred costs associated with these recalls, including administration costs of the recalls, of approximately $300,000 through December 31, 2012, and the Company will continue to incur storage fees for the quarantined product until the time of its destruction. Through December 31, 2012, approximately 5,000 vials have been returned as a result of these recalls, and the Company believes that the potential number of additional vials that will be returned is minimal. The costs related to the recalls are being recognized as selling, general and administrative expenses on the Company’s statement of operations as they are incurred. The charge to reduce the value of the inventory was recorded as a cost of product sales on the Company’s statement of operations during the period in which the impairment was taken.
Additionally, due to the termination of the supply agreement with Baxter, the Company reduced the carrying value of its manufacturing assets and its manufacturing equipment and facility construction assets in process to their current estimated fair value, resulting in an impairment charge of $6,973,000 for the year ended December 31, 2012. The fair value of these assets was determined through a third-party valuation assessment and market prices for similar assets. Additionally, in December 2012, the Company sold a construction-in-process asset resulting in a loss on the disposal of $858,000. As a result, the carrying value of the manufacturing assets on the Company’s balance sheet at December 31, 2012 was $975,000 and the manufacturing equipment and facility construction assets in process was $357,000. These assets were classified as held and used at December 31, 2012 as a formal plan to sell the assets, or otherwise dispose of them, had not been implemented at that time. See Note 6 for further discussion of the impairment.
No reimbursements under the Amended Supply Agreement were made during the year ended December 31, 2012. However, for the year ended December 31, 2011, the Company had reimbursed Baxter approximately $262,000 for facility improvements pursuant to the Amended Supply Agreement.
Lawrence Laboratories
In February 2013, the Company entered into an Amended and Restated Supply Agreement (the “Supply Agreement”) with Lawrence Laboratories, an operating division of Swords Laboratories, and a member of the BMS group of companies, which amended and restated the original agreement entered into between the parties in
93
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
December 2010, for the manufacture of commercial supplies of the finished drug product for OFIRMEV packaged in vials (the “Product”), for sale and distribution by the Company in the United States and Canada. Bristol-Myers Squibb Srl (“BMS Anagni”), an indirect subsidiary of BMS located in Anagni, Italy, manufactures the Product on behalf of Lawrence Laboratories. BMS Anagni is currently the Company’s sole supplier of OFIRMEV.
Pursuant to the terms of the Supply Agreement, the Company will pay Lawrence Laboratories a set price for each unit of Product purchased, based upon the aggregate quantity of Product the Company has specified that it intends to order during a calendar year, and whether Lawrence Laboratories has implemented certain agreed-upon manufacturing capacity increase improvements. The Company is obligated to purchase a minimum number of units each year, or pay Lawrence Laboratories an amount equal to the shortfall between the minimum purchase requirement and the number of units of Product actually ordered during such year, multiplied by a pre-set amount that also varies depending upon whether Lawrence Laboratories has implemented certain agreed-upon manufacturing capacity increase improvements. The Company is obligated to purchase at least 75% of its annual Product requirements from Lawrence Laboratories each contract year. The Supply Agreement also requires the Company to pay Lawrence Laboratories for additional services requested by the Company at a specified hourly rate and for any validation batches that may be required by the Company, not to exceed a specified rate. All amounts payable under the Supply Agreement will be paid in U.S. dollars.
The term of the Supply Agreement extends through December 31, 2018, unless extended by mutual agreement of the Company and Lawrence Laboratories, unless the Supply Agreement is terminated sooner: (1) by the mutual agreement of the parties, (2) by either party for convenience following 24 months’ prior written notice of termination to the other party, (3) upon the termination of the Company’s license agreement for the Product with BMS, or (4) upon the dissolution or termination of the Company, other than in connection with or following the assignment of the Supply Agreement. In addition, either party may terminate the Supply Agreement: (a) within 60 days, after written notice in the event of a material uncured breach of the Supply Agreement by the other party, or (b) immediately, if the other party becomes insolvent or admits in writing its inability to pay its debts as they become due, files a petition for bankruptcy, makes an assignment for the benefit of its creditors or has a receiver or other court officer appointed for its properties or assets.
If the Supply Agreement is terminated by the Company for its convenience or by Lawrence Laboratories due to the Company’s material breach of the Supply Agreement, the Company will reimburse Lawrence Laboratories for: (1) any Product ordered under a firm order and received by the Company, and (2) any inventory of materials used to manufacture the Product that are specific to the Product and that Lawrence Laboratories is unable to reasonably utilize. Additionally, the Company’s minimum purchase requirement for the year in which the termination takes effect will be reduced proportionally, and the Company will not be required to fulfill the minimum purchase requirement for any subsequent contract year. If the Supply Agreement is terminated for any reason other than by the Company’s for its convenience or by Lawrence Laboratories due to the Company’s material breach of the Supply Agreement, the Company will not be required to reimburse Lawrence Laboratories for any inventory of materials used to manufacture the Product, and will have no obligation to purchase the minimum purchase requirement for the year in which the termination takes effect, or for any subsequent contract year.
94
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
The minimum purchase requirements under the Company’s Supply Agreement with Lawrence Laboratories as of December 31, 2012 were as follows (in thousands):
| | | | |
2013 | | $ | 12,225 | |
2014 | | | 12,225 | |
2015 | | | 12,225 | |
2016 | | | 12,225 | |
2017 | | | 12,225 | |
Thereafter | | | 12,225 | |
| | | | |
Total | | $ | 73,350 | |
| | | | |
| 9. | | License Agreements and Acquired Development and Commercialization Rights |
In March 2006, the Company in-licensed the technology and the exclusive development and commercialization rights to OFIRMEV in the U.S. and Canada from BMS. BMS sublicensed these rights to the Company under a license agreement with Pharmatop and the Company has the right to grant sublicenses to third parties. As consideration for the license, the Company paid a $25,000,000 up-front fee in March 2006 and, as a result of the approval of the Company’s NDA for OFIRMEV in the fourth quarter of 2010, the Company paid an additional milestone payment of $15,000,000 in the fourth quarter of 2010. The Company may be required to make future milestone payments totaling up to $25,000,000 upon the achievement of certain levels of net sales. In addition, the Company is obligated to pay a royalty on net sales of the licensed products which range from the mid-teens to the mid-twenties, depending on the aggregate amount of net sales, and is subject to annual minimum royalty obligations. The $25,000,000 up-front fee was recognized as research and development expense at the time the payment was made. The $15,000,000 milestone payment was recorded as an intangible asset on the Company’s balance sheets and is being amortized over the estimated useful life of the licensed patents. Royalty liabilities are recognized at the time the product is sold or, for minimum royalty obligations that are not anticipated to be met, over the period in which the minimum liability is incurred.
In November 2010, the Company entered into a data license agreement among Terumo Corporation (“Terumo”), the Company and SCR Pharmatop S.A. (“Pharmatop”). Under the data license agreement, the Company provided to Terumo certain data and information resulting from the Company’s clinical development program for OFIRMEV for Terumo’s use in obtaining regulatory approval for and commercializing the same intravenous formulation of acetaminophen in Japan. Further, the Company was to provide to Terumo, without charge, up to 500 hours of technical assistance and consulting services regarding the licensed technical information, data and know-how, as reasonably necessary to assist Terumo in obtaining regulatory approval and manufacturing capacity for the product candidate. In April 2011, the Company received an upfront payment of $5,329,000 under the terms of the data license agreement. If Terumo is successful in obtaining regulatory approval for and commercializing the product in Japan, the Company may also be entitled to an additional lump-sum payment upon the first commercial sale of the product candidate and royalty payments on any commercial sales of the product in Japan.
In accordance with multiple-element arrangement guidance, the Company determined both the data license and consulting service deliverables were separate units of accounting, each having value on a standalone basis. The Company estimated the fair value of the data license based upon similar proposals from third parties and internal costs incurred in developing the data and obtaining similar rights. The value of the consulting services was based on contracts the Company had engaged with third parties for similar services. The Company allocated the value of the payment received on a relative fair value basis and will recognize the consideration allocated to the data license upon delivery and the consideration allocated to the consulting services as such services are rendered. There is no right of return or similar refund provisions in the data license agreement. During 2011, the
95
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
Company transferred the data and related information to Terumo and provided a portion of the consulting hours. For the year ended December 31, 2011, the Company recognized $5,210,000 of licensing revenue pursuant to the agreement for the data transfer and consulting hours provided. During the year ended December 31, 2012, the Company recognized the remaining $118,000 balance as licensing revenue during the period. No similar revenue was recognized during the year ended December 31, 2010. Any milestones or royalties the Company could receive from the agreement will be recognized as revenue in the period earned. No such revenue was recognized during the years ended December 31, 2012, 2011 or 2010.
In August 2011, the Company and Pharmatop filed suit in the United States District Court for the District of Delaware against Paddock Laboratories, Inc., Perrigo Company and Paddock Laboratories, LLC, collectively referred to herein as Perrigo, and against Exela Pharma Sciences, LLC, Exela PharmaSci, Inc. and Exela Holdings, Inc., collectively referred to herein as Exela. The lawsuit follows the notices that the Company received in July 2011 from each of Perrigo and Exela concerning their filings of Abbreviated New Drug Applications, or ANDAs, containing a “Paragraph IV” patent certification with the FDA for a generic version of OFIRMEV. In the lawsuit, the Company alleges that Perrigo and Exela have each infringed the ‘222 patent and the ‘218 patent by filing their respective ANDAs seeking approval from the FDA to market a generic version of OFIRMEV prior to the expiration of these patents. The ‘222 and the ‘218 patents are listed in the FDA’s Approved Drug Products with Therapeutic Equivalence Evaluations, commonly known as the Orange Book. The patent infringement lawsuit was filed within 45 days of receipt of the pertinent notice letters, thereby triggering a stay of FDA approval of the Perrigo ANDA and the Exela ANDA until the earlier of the expiration of a 30-month period, the expiration of the ‘222 and ‘218 patents, the entry of a settlement order or consent decree stating that the ‘222 and ‘218 patents are invalid or not infringed, a decision in the case concerning infringement or validity that is favorable to Perrigo or Exela, or such shorter or longer period as the Court may order. Each of Perrigo and Exela has filed an answer in the case that asserts, among other things, non-infringement and invalidity of the asserted patents, and has also filed counterclaims. A date for the bench trial in the case with Exela has been scheduled for May 2013.
The Company settled with Perrigo and the case against Perrigo was dismissed on November 30, 2012. In connection with a settlement and license agreements entered into in November 2012, Perrigo Company, or Perrigo, has been granted the exclusive right of first refusal to negotiate an agreement with us to market an authorized generic version of OFIRMEV in the U.S. in the event that the Company elects to launch an authorized generic version of the product. The license agreement also provides that, if the Company enters into an agreement for Perrigo to market an authorized generic version of OFIRMEV during the license period, Perrigo would purchase the product exclusively from us. The Company would receive product costs plus an administrative fee, as well as a royalty payment based on the net profits achieved by Perrigo from the sale of the authorized generic product. Additionally, the Company has granted Perrigo the non-exclusive right to market a generic intravenous acetaminophen product in the U.S. under Perrigo’s ANDA after December 6, 2020, or earlier under certain circumstances. The Federal Trade Commission, or FTC, or the Department of Justice, or DOJ, could seek to challenge the Company’s settlement with Perrigo, or a competitor, customer or other third-party could initiate a private action under antitrust or other laws challenging the Company’s settlement with Perrigo.
In September 2012, an unidentified third party filed with the USPTO a Request for Ex Parte Reexamination of the ‘222 patent. In December 2012, the Company received notice that the USPTO had granted the Request for Reexamination. The reexamination process is provided for by law and requires the USPTO to consider the scope and validity of the patent based on substantial new questions of patentability raised by a third party or the USPTO. Because the Company and Pharmatop believe that the scope and validity of the patent claims in this patent are appropriate and that the USPTO’s prior issuance of the patent was correct, the Company, in
96
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
conjunction with Pharmatop, will vigorously defend this patent. The Company cannot predict whether it and Pharmatop ultimately will succeed in maintaining the scope and validity of the claims of this patent during reexamination. If the patent claims in this patent ultimately are narrowed during prosecution before the USPTO, the extent of the patent coverage afforded to OFIRMEV could be impaired, which could potentially harm the Company’s business and operating results.
In April 2012, Exela filed suit against David J. Kappos and the USPTO in the United States District Court for the Eastern District of Virginia for declaratory judgment seeking a reversal of the USPTO’s decision not to act on a petition by Exela to vacate the USPTO’s April 2003 order reviving the international application for the ‘218 patent. The lawsuit followed the USPTO’s rejection of Exela’s petition to the USPTO filed in November 2011, which sought to vacate the April 23, 2003 order granting Pharmatop’s petition to revive the ‘218 patent. The USPTO determined that Exela lacks standing to seek such relief. Exela also seeks declaratory judgment that the USPTO’s rules and regulations that allow for revival of abandoned, international patent applications under the “intentional” standard are invalid, and similar relief in connection with one or more counterclaims it has filed in the Delaware litigation. The Company’s motion to intervene in this lawsuit was granted in October 2012. In December 2012, the district court dismissed the case with prejudice as barred by the applicable statute of limitations. In February 2013, Exela appealed the district court’s decision to the Court of Appeals for the Federal Circuit. A decision by the Court of Appeals in favor of Exela could result in the invalidation of the ‘218 patent.
In January 2013, the Company filed suit in the United States District Court for the Southern District of California and the Northern District of Illinois against Fresenius Kabi USA, LLC, or Fresenius. The lawsuits follow a December 2012 notice by Fresenius concerning its filing of a New Drug Application, or NDA, containing a Paragraph IV patent certification with the FDA for a generic version of OFIRMEV. In the lawsuits, the Company alleges that Fresenius has infringed the ‘222 patent and the ‘218 patent by filing its NDA seeking approval from the FDA to market a generic version of OFIRMEV prior to the expiration of these patents. Fresenius has filed an answer in the Southern District of California that asserts, among other things, non-infringement and invalidity of the asserted patents, and has also filed counterclaims.
In February 2013, the Company filed suit in the United States District Court for the Southern District of California and the District of New Jersey against Sandoz, Inc., or Sandoz. The lawsuits follow a December 2012 notice by Sandoz concerning its filing of an ANDA containing a Paragraph IV patent certification with the FDA for a generic version of OFIRMEV. In the lawsuits, the Company alleges that Sandoz has infringed the ‘222 patent and the ‘218 patent by filing its ANDA seeking approval from the FDA to market a generic version of OFIRMEV prior to the expiration of these patents. Sandoz has not yet filed an answer in either District.
Both the Fresenius and Sandoz lawsuits were filed within 45 days of receipt of the respective notice letters, thereby triggering a stay of FDA approval of the Fresenius NDA and the Sandoz ANDA until the earlier of the expiration of a 30-month period, the expiration of the ‘222 and ‘218 patents, the entry of a settlement order or consent decree stating that the ‘222 and ‘218 patents are invalid or not infringed, a decision in the case concerning infringement or validity that is favorable to Fresenius and/or Sandoz, or such shorter or longer period as the courts may order.
Regardless of the outcome of any litigation, no NDA or ANDA can receive final approval from the FDA before expiration of the regulatory exclusivity period for OFIRMEV. Specifically, the FDA has granted OFIRMEV three years of regulatory exclusivity, which expires November 2, 2013. The Company intends to vigorously enforce its intellectual property rights relating to OFIRMEV to prevent the marketing of infringing generic products prior to the expiration of its patents. The ‘222 patent expires August 5, 2017 (or February 5, 2018 if pediatric exclusivity is granted) and the ‘218 patent expires June 6, 2021 (or December 6, 2021 if
97
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
pediatric exclusivity is granted). However, given the unpredictability inherent in litigation, the Company cannot predict the outcome of these matters or any other litigation. At this time, the Company is unable to estimate possible losses or ranges of losses for current litigation, and it has not accrued any amounts for current litigation other than ongoing attorney’s fees.
Authorized Shares
In June 2012, following approval by the Company’s stockholders, the Company filed a Certificate of Amendment of Amended and Restated Certificate of Incorporation with the Secretary of State of the State of Delaware, which increased the number of authorized shares of common stock of the Company from 100,000,000 to 200,000,000.
Public Offerings
In November 2011, the Company issued an aggregate of 21,800,000 shares of its common stock at a purchase price of $3.75 per share pursuant to a public offering. In November and December 2010, the Company issued an aggregate of 12,500,000 shares of its common stock at a purchase price of $8.00 per share pursuant to a public offering. The 2011 and 2010 offerings raised proceeds, net of offering costs and underwriting discounts and commissions, of $77,302,000 and $93,555,000, respectively.
Private Placement
In February 2009, the Company issued 12,039,794 shares of its common stock at a purchase price of $7.13 per share pursuant to a private placement. In addition to the shares of the Company’s common stock, warrants to purchase up to 6,019,897 additional shares of the Company’s common stock were also issued as part of the transaction at a price of $0.125 per warrant. Each warrant is immediately exercisable and has a five-year term. The warrants may be exercised through either cash or net exercise for one share of common stock at a price of $7.84 and have been accounted for as permanent equity. As of December 31, 2012, all warrants related to the private placement were outstanding.
The private placement raised proceeds, net of offering costs, of $86,243,000. The purchasers in the offering consisted of new investors and existing stockholders of the Company, including six funds affiliated with three directors of the Company. In March 2009, the Company filed a registration statement covering the resale of the shares of common stock acquired by the investors in this offering, which was declared effective by the SEC in May 2009. The Company is required to maintain the effectiveness of the registration statement and may be subject to liquidated damages of one percent per month of the aggregate purchase price of the common shares then held by the investor that are registrable securities, subject to an aggregate cap of eight percent per calendar year. The Company has not recorded a liability for the potential damages associated with these liquidated damages provisions as it does not currently believe that the transfer of consideration is probable under the agreement.
Equity Awards
In 2006, the Company adopted the 2006 Equity Incentive Award Plan (the “2006 Plan”) in connection with the Company’s initial public offering which became effective on October 24, 2006. Upon adoption of the 2006 Plan, the Company restricted future grants from its 2004 Equity Incentive Award Plan (the “2004 Plan”). The 2006 Plan was amended and restated in 2010 to preserve the ability to deduct compensation associated with future performance-based awards made under the plan to certain executives. The term of the 2006 Plan was also extended under the 2010 amendment to 2020.
98
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
The 2006 Plan initially reserved 2,100,000 shares of common stock for future issuance and allowed for the initial number of reserved shares to be increased by (1) the 90,772 shares of common stock that remained available for issuance under the 2004 Plan as of the effective date of the 2006 Plan and (2) the number of shares under the 2004 Plan that are repurchased, forfeited, expired or cancelled on or after the effective date of the 2006 Plan. As of December 31, 2012, options to purchase 75,816 shares issued under the 2004 Plan have been repurchased, forfeited and/or cancelled since the effective date of the 2006 Plan, increasing the number of shares reserved for issuance under the 2006 Plan accordingly.
Beginning on January 1, 2008, the 2006 Plan allows for an annual increase in the number of shares available for issuance under the 2006 Plan by the lesser of (1) 4% of the outstanding common stock on January 1 and (2) a lesser amount determined by the board of directors, subject to an aggregate of 20,000,000 shares of common stock that may be issued through January 1, 2016. Through December 31, 2012, the board of directors approved the amount of shares authorized for future issuance under the 2006 Plan to be increased by an aggregate 9,283,647 shares under this provision.
As of December 31, 2012, the Company had issued both stock options and restricted stock units (“RSUs”) under the 2006 Plan and only stock options under the 2004 Plan. The following table presents shares authorized, available for future grant and outstanding under each of the Company’s plans at December 31, 2012:
| | | | | | | | | | | | |
| | | Authorized | | | Available | | | Outstanding | |
2004 Equity Incentive Plan | | | 2,708,412 | | | | — | | | | 901,540 | |
2006 Equity Incentive Plan | | | 11,550,235 | | | | 2,126,842 | | | | 9,137,382 | |
| | | | | | | | | | | | |
| | | 14,258,647 | | | | 2,126,842 | | | | 10,038,922 | |
| | | | | | | | | | | | |
The Company issues new shares of common stock upon the exercise of stock options and vesting of RSUs. RSUs that are tendered or withheld to satisfy the exercise price or tax withholding obligation pursuant to the award are returned to the pool of available shares for future grant.
Stock Options
Stock options granted under the 2006 Plan expire no later than 10 years from the date of grant and generally vest over a four-year period. Vesting generally occurs at the rate of 25% at the end of the first year, and thereafter in 36 equal monthly installments, however certain grants to the Company’s executive officers have been made in lieu of their annual bonus awards and vest over a term of generally less than one-year. In addition, annual grants to the Company’s board members vest over a period of one-year. The exercise price of the Company’s stock options shall not be less than 100% of the fair value of the Company’s common stock on the date of grant. Further, the exercise price of any option granted to a 10% stockholder may not be less than 110% of the fair value of the Company’s common stock on the date of grant.
99
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
The following table summarizes the Company’s stock option activity as of December 31, 2012, and changes for the year then ended:
| | | | | | | | | | | | | | | | |
| | | Shares | | | Weighted-
Average
Exercise
Price | | | Weighted-
Average
Remaining
Contractual
Life - Years | | | Aggregate
Intrinsic Value | |
Options outstanding at beginning of period | | | 7,969,233 | | | $ | 8.21 | | | | | | | | | |
Granted | | | 3,177,008 | | | $ | 3.49 | | | | | | | | | |
Exercised | | | (155,612 | ) | | $ | 2.91 | | | | | | | | | |
Cancelled | | | (952,645 | ) | | $ | 8.05 | | | | | | | | | |
| | | | | | | | | | | | | | | | |
Options outstanding at end of period | | | 10,037,984 | | | $ | 6.81 | | | | 6.88 | | | $ | 6,234,000 | |
| | | | | | | | | | | | | | | | |
Options exercisable at end of period | | | 6,184,112 | | | $ | 7.60 | | | | 5.75 | | | $ | 3,212,000 | |
| | | | | | | | | | | | | | | | |
The aggregate intrinsic value of options exercised during 2012, 2011 and 2010 was $175,000, $2,774,000 and $792,000, respectively. During 2012, the Company received $452,000 upon the exercise of stock options in satisfaction of the exercise price.
Restricted Stock Units
The Company has granted a limited number of RSUs with vesting schedules based upon performance criteria, service conditions or a combination of both performance criteria and service conditions. In August 2009, the Company granted 150,250 RSUs to certain officers and employees that were scheduled to vest upon the first anniversary of the approval by the FDA of the NDA for OFIRMEV, or November 2, 2011, of which 29,125 RSUs had been cancelled prior to vesting. An additional 13,500 RSUs were granted in 2010, of which a total of 3,407 RSUs have been cancelled prior to vesting. As of December 31, 2012, 938 RSUs remain outstanding.
The following table summarizes the Company’s RSU activity as of December 31, 2012, and changes for the year then ended:
| | | | | | | | | | | | |
| | | Shares | | | Weighted-Average
Grant Date Fair
Value per Share | | | Aggregate
Intrinsic Value | |
Restricted stock units outstanding at beginning of period | | | 4,376 | | | $ | 10.38 | | | | | |
Granted | | | — | | | | — | | | | | |
Vested | | | (1,563 | ) | | $ | 10.38 | | | | | |
Cancelled | | | (1,875 | ) | | $ | 10.38 | | | | | |
| | | | | | | | | | | | |
Restricted stock units outstanding at end of period | | | 938 | | | $ | 10.38 | | | $ | 4,000 | |
| | | | | | | | | | | | |
The aggregate intrinsic value of RSUs vested during 2012, 2011 and 2010 was $6,000, $716,000 and $33,000, respectively. During 2012, a total of 114 vested shares were withheld from distribution in satisfaction of statutory minimum tax obligations and the Company used less than $1,000 to satisfy such tax obligations.
Operating segments are identified as components of an enterprise which separate discrete financial information is available for evaluation by the chief operating decision-maker, or decision-making group, in making decisions regarding resource allocation and assessing performance and the Company operates and manages its business as principally one segment. It sells its only product, OFIRMEV, primarily to established wholesale distributors in the pharmaceutical industry, including the nation’s three leading wholesale pharmaceutical distributors: Cardinal Health, Inc., AmerisourceBergen Corporation and McKesson Corporation.
100
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
Shipments to wholesalers representing approximately 10% or more of total product revenue for the periods presented were as follows (as a percentage of total gross product revenue):
| | | | | | | | | | | | |
| | | Year Ended December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
Cardinal Health, Inc. | | | 33 | % | | | 37 | % | | | — | |
AmerisourceBergen Corporation | | | 33 | % | | | 33 | % | | | — | |
McKesson Corporation | | | 27 | % | | | 23 | % | | | — | |
Related receivables from customers representing approximately 10% or more of total product revenue for each period are as follows (as a percentage of total gross trade receivables):
| | | | | | | | | | | | |
| | | As of December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
Cardinal Health, Inc. | | | 31 | % | | | 35 | % | | | — | |
AmerisourceBergen Corporation | | | 32 | % | | | 36 | % | | | — | |
McKesson Corporation | | | 31 | % | | | 21 | % | | | — | |
The Company is subject to taxation in the U.S. and various state jurisdictions. The Company’s tax years for 2004 and forward are subject to examination by the Federal and state tax authorities due to the carryforward of unutilized net operating losses and research and development credits. The Company’s practice is to recognize interest and/or penalties related to income tax matters in income tax expense. The Company had no accrued interest and/or penalties related to income tax matters in the Company’s balance sheets at December 31, 2012 and 2011, and has recognized no interest and/or penalties in the Company’s statement of operations for the years ended December 31, 2012, 2011 and 2010. Further, as of December 31, 2012, the Company had not recorded any unrecognized tax benefits.
Pursuant to Internal Revenue Code (“IRC”) Sections 382 and 383, annual use of the Company’s net operating loss and research and development credit carryforwards may be limited in the event of a cumulative change in ownership of more than 50% within a three-year period. The Company has not completed this analysis regarding the limitation and therefore has removed the (1) deferred tax assets for net operating losses of approximately $149,071,000 and (2) research and development credits of approximately $6,809,000 generated through 2012 from its deferred tax asset schedule. Further, the Company has recorded a corresponding decrease to its valuation allowance. When this analysis is finalized, the Company plans to update its deferred tax asset and valuation allowance accordingly. The Company expects to complete this analysis within the next three months and, as a result, the Company may have a change in the unrecognized tax benefits that are recorded. Due to the existence of the valuation allowance, future changes in the Company’s unrecognized tax benefits will not impact the Company’s effective tax rate.
101
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
A valuation allowance has been established as realization of such deferred tax assets has not met the more likely than not threshold requirement. Other significant components of the Company’s net deferred tax assets for federal and state income taxes at December 31, 2012 and 2011 are shown below (in thousands):
| | | | | | | | |
| | | As of December 31, | |
| | | 2012 | | | 2011 | |
Deferred tax assets: | | | | | | | | |
Net operating loss carryforwards | | $ | — | | | $ | — | |
Tax credit carryforwards | | | — | | | | — | |
Stock-based compensation | | | 12,876 | | | | 10,394 | |
Capitalized research and development | | | 5,348 | | | | 6,360 | |
Other, net | | | 5,954 | | | | 2,144 | |
| | | | | | | | |
| | | 24,178 | | | | 18,898 | |
Valuation allowance for deferred tax assets | | | (23,272 | ) | | | (18,405 | ) |
| | | | | | | | |
Net deferred tax assets | | $ | 906 | | | $ | 493 | |
| | | | | | | | |
Deferred tax liabilities: | | | | | | | | |
Deferred tax liabilities | | | (906 | ) | | | (493 | ) |
| | | | | | | | |
Net deferred tax liabilities | | $ | (906 | ) | | $ | (493 | ) |
| | | | | | | | |
Net deferred tax assets | | $ | — | | | $ | — | |
| | | | | | | | |
A reconciliation of the Company’s effective tax rate and federal statutory tax rate is as follows:
| | | | | | | | | | | | |
| | | As of December 31, | |
| | | 2012 | | | 2011 | | | 2010 | |
Federal income taxes | | | 35.0 | % | | | 35.0 | % | | | 35.0 | % |
State income taxes | | | 4.7 | % | | | 4.3 | % | | | 5.8 | % |
Research and development credits | | | 0.3 | % | | | 0.9 | % | | | 1.0 | % |
Stock-based compensation | | | (1.0 | )% | | | (0.7 | )% | | | (1.7 | )% |
Change in valuation allowance | | | (5.6 | )% | | | 0.0 | % | | | (4.3 | )% |
Prior year true-up | | | 0.6 | % | | | (0.0 | )% | | | 0.3 | % |
Removal of net operating loss and research and development tax credits | | | (32.4 | )% | | | (37.8 | )% | | | (35.7 | )% |
Other, net | | | (1.6 | )% | | | (1.7 | )% | | | (0.4 | )% |
| | | | | | | | | | | | |
| | | 0.0 | % | | | 0.0 | % | | | 0.0 | % |
| | | | | | | | | | | | |
At December 31, 2012, the Company had federal and state net operating loss carryforwards of approximately $369,696,000 and $374,249,000, respectively. The federal and state tax loss carryforwards will begin to expire in 2024 and 2014, respectively, unless previously utilized. The Company also had federal research and development tax credit carryforwards of approximately $4,752,000 which will begin expiring in 2025 unless previously utilized, and state research and development tax credit carryforwards of approximately $3,164,000 which carryforward indefinitely.
Included in the net operating loss carryforwards is approximately $975,000 of losses attributable to excess stock option deductions. Under current accounting guidance concerning when tax benefits related to excess stock option deductions can be credited to paid in capital, the related valuation allowance cannot be reversed, even if the facts and circumstances indicate that it is more likely than not that the deferred tax asset can be realized. The valuation allowance will only be reversed as the related deferred tax asset is applied to reduce taxes payable.
102
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
| 14. | | Employee Benefit Plan |
The Company has a qualified retirement plan under the provisions of Section 401(k) of the Internal Revenue Code covering substantially all employees. Employees may contribute up to 100% of their annual compensation up to the maximum annual amount prescribed by the Internal Revenue Service. The Company may elect to make a discretionary contribution or match a discretionary percentage of employee contributions. During 2012, 2011 and 2010, the Company elected not to make any contributions to the plan.
| 15. | | Summarized Quarterly Data (Unaudited) |
The following financial information reflects all normal recurring adjustments, which are, in the opinion of management, necessary for a fair statement of the results of the interim periods. Summarized quarterly data for the years ended December 31, 2012 and 2011 are as follows (in thousands, except share and per share data):
| | | | | | | | | | | | | | | | | | | | |
| | | Fiscal Year 2012 Quarters | | | | |
| | | 1st(4) | | | 2nd(5) | | | 3rd | | | 4th(5)(6) | | | Total | |
Revenues | | $ | 8,004 | | | $ | 11,108 | | | $ | 13,898 | | | $ | 17,174 | | | $ | 50,184 | |
Gross profit (loss)(1) | | $ | 3,758 | | | $ | 5,352 | | | $ | 7,822 | | | $ | 9,996 | | | $ | 26,928 | |
Net loss | | $ | (22,673 | ) | | $ | (20,989 | ) | | $ | (15,890 | ) | | $ | (21,421 | ) | | $ | (80,973 | ) |
Basic and diluted net loss per share(2)(3) | | $ | (0.27 | ) | | $ | (0.25 | ) | | $ | (0.19 | ) | | $ | (0.25 | ) | | $ | (0.95 | ) |
| | | | | | | | | | | | | | | | | | | | |
| | | Fiscal Year 2011 Quarters | | | | |
| | | 1st | | | 2nd(5) | | | 3rd | | | 4th(4)(7) | | | Total | |
Revenues | | $ | 350 | | | $ | 6,916 | | | $ | 3,541 | | | $ | 5,889 | | | $ | 16,696 | |
Gross profit (loss)(1) | | $ | 61 | | | $ | 5,935 | | | $ | 1,223 | | | $ | (2,929 | ) | | $ | 4,290 | |
Net loss | | $ | (24,372 | ) | | $ | (19,214 | ) | | $ | (21,829 | ) | | $ | (27,606 | ) | | $ | (93,021 | ) |
Basic and diluted net loss per share(2)(3) | | $ | (0.39 | ) | | $ | (0.30 | ) | | $ | (0.34 | ) | | $ | (0.37 | ) | | $ | (1.41 | ) |
| (1) | Determined by subtracting cost of sales from net revenue. |
| (2) | Loss per share is computed independently for each of the quarters presented. Therefore, the sum of the quarterly net loss per share may not necessarily equal the total for the year. |
| (3) | In the fourth quarter of 2011, the Company issued 21,800,000 shares of common stock. As a result, there is a lack of comparability in the basic and diluted net loss per share amounts for the periods presented. |
| (4) | During the fourth quarter of 2011, the Company recorded a charge of $5,574 to write-down the value of certain inventory. During the first quarter of 2012, the Company recorded an additional charge of $163 to write-down additional inventory. |
| (5) | During the second quarter of 2011, the Company recognized $5,210 in license revenue under a data license agreement with Terumo Corporation, primarily related to the one-time transfer of data and related information. During the second and fourth quarters of 2012, the Company recognized additional license revenue of $33 and $85 pursuant to the license agreement. |
| (6) | During the fourth quarter of 2012, the Company recorded charges of $6,973 to impair certain manufacturing equipment and construction-in-process to their fair values and, a related asset retirement obligation impairment charge of $750 for the removal of the equipment. Additionally, the Company recorded a loss on the sale of one of its construction-in-process assets of $858 and a charge of $290 to accrue for inventory destruction costs. |
| (7) | During the fourth quarter of 2011, the Company recorded one-time employee charges of $1,142 related to a reduction in force of 17 employees. Relatedly, $251 of previously accrued labor-related costs for these individuals of was reversed at the time of the termination. |
103
CADENCE PHARMACEUTICALS, INC.
NOTES TO FINANCIAL STATEMENTS—Continued
In January 2013, The Medicines Company completed its acquisition of Incline. As a result, the Option Agreement between the Company and Incline terminated and the Company waived and relinquished its right to exercise its exclusive, irrevocable option to acquire Incline, pursuant to the Waiver Agreement entered on December 11, 2012. At the closing of the Incline Acquisition, the Company received total consideration of $14,654,000 for the waiver of its option and the 500,000 shares of Incline stock held by the Company. The aggregate value of the Incline option purchase right and the shares of Incline stock on the Company’s balance sheet at December 31, 2012 was $7,000,000. See Note 5 for further discussion.
In February 2013, the Company entered into an Amended and Restated Supply Agreement with Lawrence Laboratories, an operating division of Swords Laboratories, and a member of the BMS group of companies, which amended and restated the original agreement entered into between the parties in December 2010, for the manufacture of commercial supplies of the finished drug product for OFIRMEV packaged in vials, for sale and distribution by the Company in the United States and Canada. BMS Anagni, an indirect subsidiary of BMS located in Anagni, Italy, manufactures the product on behalf of Lawrence Laboratories. See Note 8 for further discussion.
In March 2013, the Company and Baxter mutually agreed to terminate the Amended Development and Supply Agreement between the two parties. Under the termination agreement, the Company is responsible to remove its equipment located at Baxter’s facility, which the Company has estimated will cost approximately $750,000. As of December 31, 2012, the Company recorded impairment charges of $6,973,000 to reduce the carrying value of its manufacturing assets and manufacturing assets in process related to this agreement to their current estimated fair value. Additionally, the Company fully impaired the retirement obligation as of December 31, 2012, resulting in a charge of $750,000. See Note 6 and Note 8 for further discussion.
In March 2013, the Company entered into an agreement with Laboratorios Grifols, S.A., or Grifols, a division of Grifols, S.A., a global healthcare company headquartered in Barcelona, Spain, for the development, manufacture and supply of commercial quantities of OFIRMEV in flexible IV bags. Grifols has supplied IV acetaminophen in flexible plastic bags to BMS for distribution in certain markets outside of the U.S. and Canada since 2010. Cadence plans to submit a supplemental NDA to the FDA in the second half of 2013 seeking approval of the product to be manufactured by Grifols.
Pursuant to the terms of the agreement, the Company will pay Grifols a set price for the OFIRMEV it purchases, which price may be adjusted annually by Grifols, subject to specified limitations. In addition, the Company will be obligated to pay Grifols a reservation fee, in lieu of any minimum purchase commitment, calculated by multiplying the shortfall between the annual production capacity it has reserved with Grifols and the amount of product actually ordered during that year by a fixed amount. Pending review and subsequent approval of the submission by the FDA, the agreement will terminate on the sixth anniversary of the approval by the FDA of the product manufactured by Grifols, unless it is terminated sooner by the Company upon the termination of its license agreement for the product with BMS, or after 60 days written notice following the discontinuation of the distribution of the product by the Company. In addition, either party may terminate the agreement after 60 days written notice in the event of a material uncured breach of the agreement by the other party (or 30 days in the case of a payment default), or immediately upon an insolvency event.
104
Schedule II
CADENCE PHARMACEUTICALS, INC.
Valuation and Qualifying Accounts
For the Years ended December 31, 2012, 2011 and 2010
(in thousands)
| | | | |
| | | Allowance
for
Doubtful
Accounts | |
| |
Balance at December 31, 2009 | | $ | — | |
Deductions charged to costs and expenses | | | — | |
Write-offs | | | — | |
| | | | |
Balance at December 31, 2010 | | | — | |
Deductions charged to costs and expenses | | | — | |
Write-offs | | | — | |
| | | | |
Balance at December 31, 2011 | | | — | |
Deductions charged to costs and expenses | | | 56 | |
Write-offs | | | — | |
| | | | |
Balance at December 31, 2012 | | $ | 56 | |
| | | | |
105
| Item 9. | Changes in and Disagreements with Accountants on Accounting and Financial Disclosure |
Not applicable.
| Item 9A. | Controls and Procedures |
Our management evaluated, with the participation of our Chief Executive Officer and our Chief Financial Officer, the effectiveness of our disclosure controls and procedures as of the end of the period covered by this Annual Report on Form 10-K. Based on this evaluation, our Chief Executive Officer and our Chief Financial Officer have concluded that our disclosure controls and procedures are effective to ensure that information we are required to disclose in reports that we file or submit under the Securities Exchange Act of 1934, as amended, (the “Exchange Act”) is accumulated and communicated to our management, including our principal executive and principal financial officers, as appropriate to allow timely decisions regarding required disclosure, and that such information is recorded, processed, summarized and reported within the time periods specified in Securities and Exchange Commission rules and forms. Management has determined that there were no significant changes to our internal control over financial reporting during the year or quarter ended December 31, 2012 that has materially affected, or is reasonably likely to materially affect, our internal control over financial reporting.
Management’s Report on Internal Control Over Financial Reporting
Management is responsible for establishing and maintaining an adequate system of internal control over financial reporting pursuant to Section 404 of the Sarbanes-Oxley Act of 2002 and as implemented in Rule 13a-15(f) under the Exchange Act. Our internal control over financial reporting is designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles general accepted in the U.S. All internal control systems, no matter how well designed, have inherent limitations. Internal control over financial reporting includes those policies and procedures that:
| | • | | pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; |
| | • | | provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and |
| | • | | provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of the company’s assets that could have a material effect on the company’s financial statements. |
Management has adopted the Committee of Sponsoring Organizations of the Treadway Commission framework to evaluate the effectiveness of our internal control over financial reporting. Management’s evaluation of the results of testing included consideration of susceptibility to loss or fraud, subjectivity, complexity, the extent of judgment, the amount and volume of the transactions exposed to the deficiency, the existence of mitigating controls, the cause of detected exceptions, how the exception was detected, the pervasiveness of the exception, the significance of the deviation from policy and the frequency of exceptions relative to the frequency of operation.
Indicators of deficiencies that may be material weaknesses and are at least significant include restatement, material misstatement in the current period, ineffective Audit Committee oversight, ineffective internal audit function, identification of fraud of any magnitude by management, significant deficiencies that remain uncorrected for some period of time, ineffective control environment, and the aggregate effect of all deficiencies.
As of December 31, 2012, management assessed the effectiveness of our internal control over financial reporting, and concluded that such control over financial reporting was effective and there were no material weaknesses in our internal control over financial reporting that have been identified by management. Our independent registered public accounting firm, Ernst & Young LLP, has issued an audit report on the effectiveness of our internal control over financial reporting as of December 31, 2012 and is included below.
106
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM ON
INTERNAL CONTROL OVER FINANCIAL REPORTING
The Board of Directors and
Stockholders of Cadence Pharmaceuticals, Inc.
We have audited Cadence Pharmaceuticals, Inc.’s internal control over financial reporting as of December 31, 2012, based on criteria established in Internal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (the COSO criteria). Cadence Pharmaceuticals, Inc.’s management is responsible for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting included in the accompanying Management’s Report on Internal Control Over Financial Reporting. Our responsibility is to express an opinion on the company’s internal control over financial reporting based on our audit.
We conducted our audit in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects. Our audit included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we considered necessary in the circumstances. We believe that our audit provides a reasonable basis for our opinion.
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, Cadence Pharmaceuticals, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2012, based on the COSO criteria.
We also have audited, in accordance with the standards of the Public Company Accounting Oversight Board (United States), the balance sheets of Cadence Pharmaceuticals, Inc. as of December 31, 2012 and 2011, and the related statements of operations and comprehensive loss, stockholders’ equity and cash flows for each of the three years in the period ended December 31, 2012 of Cadence Pharmaceuticals, Inc. and our report dated March 8, 2013 expressed an unqualified opinion thereon.
/s/ Ernst & Young LLP
San Diego, California
March 8, 2013
| Item 9B. | Other Information |
Not applicable.
107
PART III
| Item 10. | Directors, Executive Officers and Corporate Governance |
Information required by this item will be included under the captionsElection of Directors, Information Regarding the Board of Directors and Corporate Governance, Executive Compensation and Other Information, and Section 16(a) Beneficial Ownership Reporting Compliance contained in our definitive Proxy Statement to be filed with the Commission within 120 days after the conclusion of our year ended December 31, 2012 pursuant to General Instruction G(3) of Form 10-K and is incorporated herein by reference.
| Item 11. | Executive Compensation |
We maintain employee compensation programs and benefit plans in which our executive officers are participants. Copies of these plans and programs are set forth or incorporated by reference as Exhibits to this report. The information required by this item will be included in our definitive Proxy Statement under the captionExecutive Compensation and Other Information to be filed with the Commission within 120 days after the conclusion of our year ended December 31, 2012 pursuant to General Instruction G(3) of Form 10-K and is incorporated herein by reference.
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters |
Information required by this item will be included under the captionSecurity Ownership of Certain Beneficial Owners and Management contained in our definitive Proxy Statement to be filed with the Commission within 120 days after the conclusion of our year ended December 31, 2012 pursuant to General Instruction G(3) of Form 10-K and is incorporated herein by reference. The information required by this item regarding our equity compensation plan is included in the section above entitled “Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities—Securities Authorized for Issuance under Equity Compensation Plans.”
| Item 13. | Certain Relationships and Related Transactions, and Director Independence |
Information required by this item will be included under the captionsCertain Relationships and Related Transactions andInformation Regarding the Board of Directors and Corporate Governancecontained in our definitive Proxy Statement to be filed with the Commission within 120 days after the conclusion of our year ended December 31, 2012 pursuant to General Instruction G(3) of Form 10-K and is incorporated herein by reference.
| Item 14. | Principal Accounting Fees and Services |
Information required by this item will be included under the captionRatification of Selection of Independent Registered Public Accounting Firm contained in our definitive Proxy Statement to be filed with the Commission within 120 days after the conclusion of our year ended December 31, 2012 pursuant to General Instruction G(3) of Form 10-K and is incorporated herein by reference.
108
PART IV
| Item 15. | Exhibits, Financial Statement Schedules |
(a) Documents filed as part of this report:
(1) Financial Statements.The following financial statements of Cadence Pharmaceuticals, Inc., together with the report thereon of Ernst & Young LLP, required to be filed pursuant to Part II, Item 8 of this Annual Report on Form 10-K, are included on pages 71 through 104, as follows:
(2) Financial Statements Schedules.The following financial statement schedule of Cadence Pharmaceuticals, Inc., required to be filed pursuant to Part IV, Item 15 of this Annual Report on Form 10-K, is included on page 105, as follows:
| | | | |
| | | Page | |
Schedule II –Valuation and Qualifying Accounts | | | 105 | |
(3) List of exhibits required by Item 601 of Regulation S-K. See part (b) below.
(b) Exhibits.
| | |
Exhibit Number | | Description of Exhibit |
| |
| 3.1 | | Amended and Restated Certificate of Incorporation of the Company, incorporated herein by reference to Exhibit 3.1 to the Company’s Quarterly Report on Form 10-Q (File No. 001-33103) for the period ended September 30, 2006 as filed with the SEC on November 30, 2006 |
| |
| 3.2 | | Certificate of Amendment of Amended and Restated Certificate of Incorporation of the Company, incorporated herein by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on June 15, 2012 |
| |
| 3.3 | | Amended and Restated Bylaws of the Company, incorporated herein by reference to Exhibit 3.2 to the Company’s Quarterly Report on Form 10-Q (File No. 001-33103) for the period ended September 30, 2006 as filed with the SEC on November 30, 2006 |
| |
| 3.4 | | Amendment of Amended and Restated Bylaws of the Company, incorporated herein by reference to Exhibit 3.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on December 17, 2007 |
| |
| 4.1 | | Form of the Company’s Common Stock Certificate, incorporated herein by reference to Exhibit 4.1 to the Company’s Quarterly Report on Form 10-Q (File No. 001-33103) for the period ended September 30, 2006 as filed with the SEC on November 30, 2006 |
| |
| 4.2 | | Amended and Restated Investor Rights Agreement dated February 21, 2006, incorporated herein by reference to Exhibit 4.2 to the Company’s Registration Statement on Form S-1 (File No. 333-135821) as filed with the SEC on July 17, 2006 |
109
| | |
Exhibit Number | | Description of Exhibit |
| |
| 4.3 | | Registration Rights Waiver and Amendment dated November 29, 2007, incorporated herein by reference to Exhibit 4.5 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on December 3, 2007 |
| |
| 4.4 | | Form of Warrant to Purchase Stock issued to Silicon Valley Bank on November 30, 2007, incorporated herein by reference to Exhibit 4.6 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on December 3, 2007 |
| |
| 4.5 | | Form of Warrant to Purchase Stock issued to Oxford Finance Corporation on November 30, 2007, incorporated herein by reference to Exhibit 4.7 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on December 3, 2007 |
| |
| 4.6 | | Form of Warrant to Purchase Stock issued to GE Business Financial Services Inc. (formerly known as Merrill Lynch Business Financial Services Inc.), on November 30, 2007, incorporated herein by reference to Exhibit 4.8 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on December 3, 2007 |
| |
| 4.7 | | Form of Warrant to Purchase Stock issued on February 18, 2009, incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on February 20, 2009 |
| |
| 4.8 | | Form of Warrant to Purchase Stock issued on June 18, 2010, incorporated herein by reference to Exhibit 4.10 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on June 21, 2010 |
| |
| 4.9 | | Form of Warrant to Purchase Stock issued on December 22, 2011, incorporated herein by reference to Exhibit 4.10 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on December 27, 2011 |
| |
| 4.10 | | Form of Warrant to Purchase Stock issued on December 5, 2012, incorporated herein by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on December 6, 2012 |
| |
| 10.1# | | Form of Director and Executive Officer Indemnification Agreement, incorporated herein by reference to Exhibit 10.1 to Amendment No. 1 of the Company’s Registration Statement on Form S-1 (File No. 333-135821) as filed with the SEC on August 30, 2006 |
| |
| 10.2# | | Amended and Restated Cadence Pharmaceuticals, Inc. Director Compensation Policy, effective September 1, 2011, incorporated herein by reference to Exhibit 10.1 to the Company’s Quarterly Report on Form 10-Q (File No. 001-33103) for the period ended September 30, 2011 as filed with the SEC on November 4, 2011 |
| |
| 10.3# | | Form of Second Amended and Restated Employment Agreement, incorporated herein by reference to Exhibit 10.31 to the Company’s Annual Report on Form 10-K (File No. 001-33103) for the year ended December 31, 2008 as filed with the SEC on March 16, 2009 |
| |
| 10.4# | | Employment Agreement between the Company and Scott A. Byrd, incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on June 22, 2009 |
| |
| 10.5# | | 2004 Equity Incentive Award Plan and forms of Option Agreements thereunder, incorporated herein by reference to Exhibit 10.3 to the Company’s Registration Statement on Form S-8 (File No. 333-138226) as filed with the SEC on October 26, 2006 |
| |
| 10.6# | | 2010 Amendment and Restatement of the 2006 Equity Incentive Award Plan, incorporated herein by reference to Exhibit 10.5 to the Company’s Registration Statement on Form S-8 (File No. 333-171396) as filed with the SEC on December 23, 2010 |
110
| | |
Exhibit Number | | Description of Exhibit |
| |
| 10.7 | | Forms of Option and Restricted Stock Agreements under the 2006 Equity Incentive Award Plan, incorporated herein by reference to Exhibit 10.5 to the Company’s Registration Statement on Form S-8 (File No. 333-138226) as filed with the SEC on October 26, 2006 |
| |
| 10.8# | | Form of Deferral Restricted Stock Unit Agreement under the 2006 Equity Incentive Award Plan, incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on September 2, 2009 |
| |
| 10.9# | | Form of Non-Deferral Restricted Stock Unit Agreement under the 2006 Equity Incentive Award Plan, incorporated herein by reference to Exhibit 10.2 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on September 2, 2009 |
| |
| 10.10# | | 2012 Corporate Bonus Plan, incorporated herein by reference to Exhibit 10.11 to the Company’s Annual Report on Form 10-K (File No. 001-33103) for the year ended December 31, 2011 as filed with the SEC on March 13, 2012 |
| |
| 10.11±# | | 2013 Corporate Bonus Plan |
| |
| 10.12±# | | Executive Severance Plan |
| |
| 10.13 | | Form of Amended and Restated Restricted Common Stock Purchase Agreement, incorporated herein by reference to Exhibit 10.6 to Amendment No. 1 of the Company’s Registration Statement on Form S-1 (File No. 333-135821) as filed with the SEC on August 30, 2006 |
| |
| 10.14 | | Form of Common Stock Purchase Agreement dated February 14, 2008, incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on February 15, 2008 |
| |
| 10.15 | | Securities Purchase Agreement, dated February 13, 2009, incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on February 20, 2009 |
| |
| 10.16 | | Lease dated May 12, 2006 by and between the Company and Prentiss/Collins Del Mar Heights LLC, incorporated herein by reference to Exhibit 10.9 to the Company’s Registration Statement on Form S-1 (File No. 333-135821) as filed with the SEC on July 17, 2006 |
| |
| 10.17 | | First Amendment to Lease dated September 29, 2006 by and between the Company and Prentiss/Collins Del Mar Heights LLC, incorporated herein by reference to Exhibit 10.16 to the Company’s Annual Report on Form 10-K (File No. 001-33103) for the year ended December 31, 2011 as filed with the SEC on March 13, 2012 |
| |
| 10.18 | | Second Amendment to Lease dated December 8, 2011 by and among the Company and PRII High Bluffs LLC and Collins Corporate Center Partners, LLC, incorporated herein by reference to Exhibit 10.17 to the Company’s Annual Report on Form 10-K (File No. 001-33103) for the year ended December 31, 2011 as filed with the SEC on March 13, 2012 |
| |
| 10.19† | | IV APAP Agreement (U.S. and Canada) dated February 21, 2006 by and between the Company and Bristol-Myers Squibb Company, incorporated herein by reference to Exhibit 10.11 to Amendment No. 2 of the Company’s Registration Statement on Form S-1 (File No. 333-135821) as filed with the SEC on September 25, 2006 |
| |
| 10.20† | | License Agreement dated December 23, 2002 by and among SCR Pharmatop and Bristol-Myers Squibb Company, incorporated herein by reference to Exhibit 10.12 to Amendment No. 2 of the Company’s Registration Statement on Form S-1 (File No. 333-135821) as filed with the SEC on September 25, 2006 |
| |
| 10.21† | | Supply Agreement dated December 1, 2010 by and between the Company and Lawrence Laboratories, incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on December 2, 2010 |
111
| | |
Exhibit Number | | Description of Exhibit |
| |
| 10.22† | | Amended and Restated Supply Agreement dated February 22, 2013 by and between the Company and Lawrence Laboratories, incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on February 28, 2013. |
| |
| 10.23† | | Amended and Restated Development and Supply Agreement dated January 28, 2011 by and between the Company and Baxter Healthcare Corporation, incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on February 2, 2011 |
| |
| 10.24† | | Amended and Restated Loan and Security Agreement dated June 18, 2010 by and among the Company and Oxford Finance Corporation, Silicon Valley Bank and GE Business Financial Services, Inc., incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on June 21, 2010 |
| |
| 10.25 | | First Amendment to Amended and Restated Loan and Security Agreement dated November 22, 2010 by and among the Company and Oxford Finance Corporation, Silicon Valley Bank and GE Business Financial Services, Inc., incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on November 26, 2010 |
| |
| 10.26 | | Second Amended and Restated Loan and Security Agreement dated December 22, 2011 by and among the Company and Oxford Finance LLC, Silicon Valley Bank and General Electric Capital Corporation, incorporated herein by reference to Exhibit 10.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on December 27, 2011 |
| |
| 10.27 | | First Amendment to Second Amended and Restated Loan and Security Agreement, dated December 5, 2012, by and among the Company and Oxford Finance LLC, Silicon Valley Bank and General Electric Capital Corporation, incorporated herein by reference to Exhibit 4.1 to the Company’s Current Report on Form 8-K (File No. 001-33103) as filed with the SEC on December 6, 2012 |
| |
| 10.28† | | Option Agreement dated June 21, 2010 by and among the Company and Incline Therapeutics, Inc., incorporated herein by reference to Exhibit 10.7 to the Company’s Quarterly Report on Form 10-Q (File No. 001-33103) for the period ended June 30, 2010 as filed with the SEC on August 6, 2010 |
| |
| 10.29± | | Waiver, Consent and Option Termination Agreement dated December 11, 2012 by and between the Company and Incline Therapeutics, Inc. |
| |
| 10.30±† | | Settlement Agreement dated November 27, 2012 by and between the Company and SCR Pharmatop and Paddock Laboratories, LLC and Perrigo Company |
| |
| 10.31±† | | License Agreement dated November 27, 2012 by and between the Company and SCR Pharmatop and Paddock Laboratories, LLC and Perrigo Company |
| |
| 23.1± | | Consent of Independent Registered Public Accounting Firm |
| |
| 31.1± | | Certification of Chief Executive Officer pursuant to Rule 13a – 14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| |
| 31.2± | | Certification of Chief Financial Officer pursuant to Rule 13a – 14(a) and 15d-14(a), as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 |
| |
| 32.1± | | Certification of Chief Executive Officer and Chief Financial Officer pursuant to 18.U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes Oxley Act of 2002 |
| |
| 101.INS± | | XBRL Instance Document |
| |
| 101.SCH± | | XBRL Taxonomy Extension Schema Document |
112
| | |
Exhibit Number | | Description of Exhibit |
| |
| 101.CAL± | | XBRL Taxonomy Extension Calculation Linkbase Document |
| |
| 101.DEF± | | XBRL Taxonomy Extension Definition Linkbase Document |
| |
| 101.LAB± | | XBRL Taxonomy Extension Label Linkbase Document |
| |
| 101.PRE± | | XBRL Taxonomy Extension Presentation Linkbase Document |
| ± | Included in this Report. |
| # | Indicates management contract or compensatory plan. |
| † | Confidential treatment has been requested as to certain portions, which portions have been omitted and filed separately with the Securities and Exchange Commission. |
113
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, as amended, the registrant has duly caused this Annual Report on Form 10-K to be signed on its behalf by the undersigned, thereunto duly authorized.
| | |
| CADENCE PHARMACEUTICALS, INC. |
| |
| By: | | /s/ THEODORE R. SCHROEDER |
| | Theodore R. Schroeder |
| | President, Chief Executive Officer and Director |
| | (Principal Executive Officer) |
Dated: March 8, 2013
Pursuant to the requirements of the Securities Exchange Act of 1934, as amended, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | |
Signature | | Title | | Date |
| | |
/s/ THEODORE R. SCHROEDER Theodore R. Schroeder | | President, Chief Executive Officer and Director (Principal Executive Officer) | | March 8, 2013 |
| | |
/s/ WILLIAM R. LARUE William R. LaRue | | Senior Vice President, Chief Financial Officer, Treasurer and Assistant Secretary (Principal Financial and Accounting Officer) | | March 8, 2013 |
| | |
/s/ CAM L. GARNER Cam L. Garner | | Chairman of the Board of Directors | | March 8, 2013 |
| | |
/s/ BRIAN G. ATWOOD Brian G. Atwood | | Director | | March 8, 2013 |
| | |
/s/ SAMUEL L. BARKER, PH.D. Samuel L. Barker, Ph.D. | | Director | | March 8, 2013 |
| | |
/s/ MICHAEL A. BERMAN, M.D. Michael A. Berman, M.D. | | Director | | March 8, 2013 |
| | |
/s/ JAMES C. BLAIR, PH.D. James C. Blair, Ph.D. | | Director | | March 8, 2013 |
| | |
/s/ ALAN D. FRAZIER Alan D. Frazier | | Director | | March 8, 2013 |
| | |
/s/ MICHAEL L. EAGLE Michael L. Eagle | | Director | | March 8, 2013 |
| | |
/s/ TODD W. RICH Todd W. Rich | | Director | | March 8, 2013 |
| | |
/s/ CHRISTOPHER J. TWOMEY Christopher J. Twomey | | Director | | March 8, 2013 |
114