EXHIBIT 99.1
BUSINESS
We are a clinical-stage biopharmaceutical company focused on discovering, developing and commercializing novel treatments for patients suffering from diseases that have high unmet medical need, including those related to fibrosis, respiratory, liver and kidney diseases that have high unmet medical need, including those related to fibrosis in respiratory, liver and kidney diseases. We have a deep understanding of certain biological targets and pathways that have been implicated in the fibrotic process, including fatty acid receptors such as free fatty acid receptor 1, or FFAR1 (also known as G-protein-coupled receptor 40, or GPR40), a related receptor
(G-protein-coupled receptor 84, or GPR84) and peroxisome proliferator-activated receptors, or PPARs. In preclinical studies, we observed that targeting these receptors promoted normal tissue regeneration and scar resolution, including preventing the progression of, and reversing established fibrosis. We also have encouraging clinical data that we believe supports the translatability of our preclinical data observations to the clinic. We have leveraged this understanding, as well as our experience with generating small molecules, to build a pipeline of differentiated product candidates. Our research program is focused on inflammatory, fibrotic and metabolic conditions in patients with liver, respiratory or renal disease, with an emphasis on rare or orphan diseases.
Our Lead Product Candidates
| - | Fezagepras for the treatment of fibrosis: |
Our lead small molecule product candidate, fezagepras (also known as PBI-4050), is an anti-inflammatory and anti-fibrotic small molecule designed to modulate the activity of multiple receptors, including FFAR1 and PPAR alpha.
We have generated compelling preclinical data for fezagepras in multiple organ disease models of fibrosis, scleroderma and osteoporosis, that we believe support the evaluation of fezagepras in clinical trials for the treatment of fibrosis. Some of these studies were performed in collaboration with Vanderbilt University, University of Ottawa, Université de Montréal, McMaster University and the Montreal Heart Institute.
To date, fezagepras has been evaluated in over 250 human subjects (including 166 patients) in a combination of three Phase 1 clinical trials, three open-label Phase 2 clinical trials and one placebo-controlled Phase 2 trial at doses up to 1,200 mg daily. We completed a Phase 2 open-label clinical trial of fezagepras in patients with idiopathic pulmonary fibrosis, or IPF, in January 2017 and a Phase 2 open-label clinical trial of fezagepras in patients with Alström syndrome in June 2018. We also completed an open-label Phase 2 trial of fezagepras in patients with Type 2 Diabetes with Metabolic Syndrome, or T2DMS in November 2016. We also commenced a placebo-controlled Phase 2 trial in T2DMS in May 2017, where patients were dosed up to 1,200mg daily for up to twelve weeks, but terminated the trial due to failure to meet recruitment targets. An open-label rollover, single-arm, Phase 2 clinical trial of fezagepras for the treatment Alström syndrome was started in October 2017 and treatment was terminated in May 2020 as a result of the re-deployment of the investigational site clinical staff as a result of the current COVID-19 pandemic. The rollover study was completed after having been extended by the Data Safety Monitoring Board and the UK Medicines and Healthcare Products Regulatory Agency. Nine patients have completed more than two years of treatment with fezagepras.
This previous preclinical and open-label clinical research with fezagepras has led us to two conclusions that we believe will inform our further study of this compound. The first is that the tolerability and pharmacokinetic characteristics of fezagepras should allow for the use of higher doses than those previous studied in our open-label clinical trials, including the potential for multiple daily dosing regimens. Our current plan is to optimize dosing in a Phase 1 multiple ascending dose ranging, or MAD, clinical trial of fezagepras, including aggregate dosage levels higher than the previously studied doses, commencing in the second half of 2020. Our second conclusion is that fezagepras, based on the preclinical and clinical data currently observed, has the potential to impact fibrosis across multiple organ systems. Following a successful conclusion of the Phase 1 MAD study, our intention is to begin randomized controlled clinical trials in certain of the aforementioned organs to establish human proof of concept for fezagepras’ anti fibrotic activity.
In the second half of 2020, we plan to initiate a Phase 1 clinical trial to evaluate multiple ascending doses of fezagepras in healthy volunteers, at higher aggregate dose levels, taken once daily, twice-daily and three times daily than those previously evaluated in our completed Phase 1 and Phase 2 clinical trials. The data from this Phase 1 clinical trial will inform dose selection and regimen for future clinical trials of fezagepras, including randomized, placebo-controlled Phase 2 clinical trials in respiratory, liver or kidney disease indications. We are currently reviewing the most appropriate indications for fezagepras. Future trials are only expected after we have established the optimal dose level based on the results of our planned Phase 1 multiple ascending dose
trial. We anticipate initiating Phase 2 clinical trials subject to the ongoing evaluation of the mechanism of action of fezagepras, the current COVID-19 pandemic and pending results of the multiple ascending dose study.
Our research has focused on the ability of fezagepras to modulate the receptor activities of FFAR1 and PPAR receptor alpha, and regulating the downstream signaling processes. We believe these combined activities contributed to the effects we observed in our open-label Phase 2 clinical trials of fezagepras. In a Phase 2, open-label clinical trial of fezagepras in patients with IPF, we observed the stabilization of forced vital capacity (FVC), a measure of lung function, over 12 weeks. In a Phase 2, open-label trial in Alström syndrome patients, we observed encouraging effects of fezagepras on both liver stiffness (as measured by Fibroscan, a surrogate for liver fibrosis) and cardiac fibrosis. It should be noted that the open-label study design of both these trials means that there was no placebo control group included.
Fezagepras has been granted Orphan Drug Designation by the FDA and the EMA for the treatment of Alström syndrome as well as for the treatment of IPF. Fezagepras has also been granted the Promising Innovative Medicine (PIM) designation by the UK Medicines and Healthcare products Regulatory Agency (MHRA) for the treatment of Alström syndrome and IPF.
| - | Ryplazim® (human) plasminogen replacement therapy: |
Ryplazim® (human) plasminogen, or Ryplazim, is a highly purified glu-plasminogen derived from human plasma that acts as a plasminogen replacement therapy for patients deficient in plasminogen protein. We are currently developing Ryplazim for the treatment of congenital plasminogen deficiency, a rare disorder associated with abnormal accumulation or growth of fibrin-rich pseudomembranous lesions on mucous membranes. Left untreated, these lesions may impair organ function and impact quality of life. Congenital plasminogen deficiency is caused by mutations in PLG, the gene coding for production of the zymogen plasminogen.
Ligneous conjunctivitis, or LC, appears to be the most common clinical manifestation of plasminogen deficiency, and is characterized by inflamed, woody growths on the conjunctival membranes that, if left untreated, can result in visual impairment or blindness. Other disease manifestations may be observed in the central nervous system, ears, nasopharynx, oral cavity, respiratory, gastrointestinal and genitourinary tracts. Abnormal wound healing and infertility have also been reported in patients with plasminogen deficiency. Although most affected patients survive into adulthood, the disease appears to be most severe when diagnosed in infants and children, where manifestations increase morbidity and mortality risk, including blindness, respiratory failure due to bronchial obstruction, or complications from hydrocephalus (excessive fluid in the brain) and death.
In the Phase 2/3 clinical trial, we evaluated the pharmacokinetics (PK), efficacy and safety of Ryplazim in 15 pediatric and adult patients with congenital plasminogen deficiency, which is also referred to as hypoplasminogenemia. We previously submitted a Biologics Licsense Application, or BLA, to the U.S. Food and Drug Administration, or FDA, for Ryplazim that received a complete response letter, or CRL, from the FDA in April 2018, which identified Chemistry, Manufacturing and Controls, (CMC), deficiencies. We plan to resubmit a BLA, to the FDA in the third quarter of 2020 based on the results from our open-label Phase 2/3 clinical trial completed in October 2018. A resubmission, is a submission to a BLA that purports to answer all the deficiencies that need to be addressed. We believe that our BLA resubmission addresses the deficiencies outlined in the CRL issued by FDA to us in April 2018. We believe that the BLA resubmission will be designated as a Class 2 resubmission by FDA which would provide for a PDUFA goal for FDA review and action within six months of the receipt date of the BLA resubmission. It is possible that when we resubmit, we could experience delays in the timing of review and/or our interactions with FDA due to the impacts of COVID-19, for example, absenteeism by governmental employees, inability to conduct planned physical inspections related to regulatory approval, or the diversion of FDA’s efforts and attention to approval of other therapeutics or other activities related to COVID-19, which could delay approval decisions and otherwise delay or limit our ability to make planned regulatory submissions or obtain approvals.
We conducted a pivotal Phase 2/3 clinical trial of Ryplazim for the treatment of congenital plasminogen deficiency, 15 patients (six children and nine adults) were administered 6.6 mg/kg intravenous doses of Ryplazim every two to four days for 48 weeks. The primary efficacy endpoint was clinical success, defined as 50% of the subjects achieving a greater than 50% improvement in lesion number/size or functionality impact from baseline. The primary pharmacokinetic endpoint was an increase in trough plasminogen activity levels by at least an absolute 10% above baseline at 12 weeks. Ryplazim met both its primary efficacy and pharmacokinetic endpoints following the intravenous administration of Ryplazim to fifteen patients for 48 weeks, as we observed a reduction in visible and non-visible lesions of greater than 50% in all patients at 48 weeks and at least a 10% improvement from baseline trough plasminogen levels at 12 weeks. Both the Phase 2/3 clinical trial and the Phase 1 clinical trial were undertaken at two specialized centers: Indiana Hemophilia and Thrombosis Center in Indianapolis, Indiana and Oslo University Hospital in Oslo, Norway.
All evaluable patients achieved target trough plasminogen activity levels (absolute 10% above baseline) across the initial 12-week treatment period.
Observed Trough Plasminogen Activity Levels after Treatment with Ryplazim
Patient | Age (y) | Prescribed infusion interval (d) | Plasminogen activity (%)* | Plasminogen activity trough levels ‡ absolute 10% above baseline |
Screening | Baseline† | Trough levels |
Wk 2 | Wk 4 | Wk 6 | Wk 8 | Wk 10 | Wk 12 | Any occurrence | ‡3 times |
1 | 39 | 3 | 26 | 29 | 57‡ | 58 | 52 | 61 | 30§ | 48 | 5 | Yes |
2 | 35 | 2 | 29 | 43 | 72 | 81 | 78 | 79 | 83 | 63|| | 6 | Yes |
3 | 16 | 4 | 30 | 28 | 55 | 52 | 42 | 53 | 49 | 55 | 6 | Yes |
4 | 24 | 3 | 32 | 28 | 49 | 61 | 62 | 55 | 56 | 53 | 6 | Yes |
5 | 20 | 3 | 18 | 22 | 45 | 44 | 44 | 41 | 39 | 45 | 6 | Yes |
6 | 37 | 2 | <5 | <5 | 50 | 63 | 60 | 64 | 62 | 70 | 6 | Yes |
7 | 24 | 3 | 26 | 31 | 58 | 46 | 58 | 67 | 57 | 62 | 6 | Yes |
8 | 5 | 3 | 23 | 22 | 41 | 51 | 38 | 34 | 43 | 34 | 6 | Yes |
9 | 16 | 3 | 24 | 20 | 55 | 44 | 64 | 63 | 57 | 64 | 6 | Yes |
10 | 11 | 3 | 18 | 17 | 53 | 55 | 51 | 32|| | 50 | 50 | 6 | Yes |
11 | 6 | 3 | 36 | 29 | 39 | 49 | 52 | 59 | 54 | 61 | 6 | Yes |
12 | 33 | 3 | <5 | <5 | 19 | 33 | 46 | 26|| | 44 | 53 | 6 | Yes |
13 | 33 | 4 | 15 | 15 | 38 | 47 | 61 | 38|| | 45 | 30|| | 6 | Yes |
14 | 42 | 2 | 4 | <5 | 14|| | 24 | 38 | 22|| | 44 | 41 | 5 | Yes |
We observed a significant clinical effect on lesions as early as four weeks after commencement of treatment with Ryplazim, and an overall success rate of 100% after 48 weeks of treatment with Ryplazim. The treatment was generally well tolerated with no serious adverse events or adverse events resulting in drug discontinuation in the trial. Two patients had adverse events of severe intensity: one patient experienced severe back pain that resolved three days later and the other patient, with a medical history of anxiety, experienced an episode of anxiety, temporally related to a motor vehicle accident. Both of these severe adverse events resolved after temporary treatment discontinuation, without re-emergence upon re-start of therapy. The most frequent treatment-emergent adverse events, or TEAEs, observed in subjects in this trial were nasopharyngitis and headache. None of the TEAEs resulted in drug discontinuation in the trial.
Ryplazim was granted Orphan Drug Designation and the Rare Pediatric Disease Designation by the FDA for the treatment of congenital plasminogen deficiency.
We also may explore clinical uses or formulations of plasminogen, including for the treatment of acquired plasminogen deficiencies and in critical care and wound treatment settings. We believe that the expansion of our plasminogen development program may enable us to target additional clinical indications with unmet medical need. Plasminogen was granted Orphan Drug Designation by the FDA for the treatment of IPF.
We are also planning to develop an early-stage pipeline of novel compounds targeting clinically validated and novel biological targets for the treatment of respiratory, liver and kidney diseases by leveraging our own drug
discovery platform, collaborating with third parties and in-licensing or acquiring new compounds and technologies.
Our first development program is a selective GPR84 antagonist candidate that we believe, if approved, could be used as monotherapy or in combination with other approved drugs. GPR84 is a pro-inflammatory target primarily expressed on cells associated with the immune system and its expression levels increase significantly during periods of inflammatory stress. Developing a GPR84 antagonist would build on our in-house expertise in GPR84. Our preclinical research indicates a potential role for antagonism of GPR84 in the reduction of fibrosis in several diseases, including kidney disease and NASH. Our GPR84 antagonist program is currently at the pre-clinical stage.
In addition to our internally developed GPR84, we have recently acquired a series of highly potent synthetic antagonists against the receptor for 5-oxo-ETE, believe to be one of the most powerful chemoattractants and activators of eosinophils, major effector cells in the immune system that play a key role in tissue repair and resolution of inflammation, including a novel selective Oxo-eicosanoid receptor 1 (OXER1) antagonist candidate. OXER1 is a G protein-coupled receptor (GPCR) that is highly selective for 5-Oxo-eicosatetraenoic acid (5-oxo-ETE), believed to be one of the most potent human eosinophil chemo-attractants. Migration of eosinophils to body sites including the lungs and intestines is mediated by eosinophil chemo-attractants such as 5-oxo-ETE. Eosinophils play a key role in Type 2 inflammation-driven diseases, including respiratory diseases and gastro-intestinal diseases. Our OXER1 antagonist program is currently at the pre-clinical stage.
Our Team
We have assembled a team of employees with experience across the spectrum of drug discovery and development who have experience developing and commercializing products. Members of our board of directors and management team have extensive experience in the life sciences industry. We are led by Kenneth Galbraith, our Chief Executive Officer, who brings over 30 years of experience acting as an executive, director, investor and advisor to companies in the biotechnology, medical device, pharmaceutical and healthcare sectors.
COVID-19 Business Update
The World Health Organization has declared the outbreak of a novel coronavirus, referred to as COVID-19, as a global pandemic, which continues to spread throughout Canada and around the world. Following the global outbreak of COVID-19, we have placed a priority on the health and safety of our employees. The COVID-19 pandemic has had a mitigated impact on our business operations in the first two quarters due to early implementation of business continuity plans to protect our workforce and the enforcement of travel restrictions. In March 2020, a significant portion of our workforce transitioned to working remotely. In addition, we implemented social-distancing measures in our plasma collection facilities, manufacturing facility and research laboratories. We have also implemented a range of programs to help support our employees, from additional paid leave for those who must care for vulnerable family members, enabling employees to work from home where possible and offering financial support to those who are financially affected by the pandemic. As a result of this slowdown, we experienced delays in our previously anticipated clinical development timelines and data release milestones, with respect to our BLA submission for Ryplazim and any potential subsequent approval. The extent of these delays cannot be predicted with confidence at the time of this filing. We expect to provide updates to our clinical development programs and timelines as we gain more clarity over the coming months. In addition, we have adopted risk assessments and mitigating actions across the Company related to the COVID-19 pandemic including; an internal resource center to maintain the health and safety of our employees and communicate business continuity plans; the assessment of financial and operating impacts and mitigating actions in response; and enterprise risk management and other functional activities. We also continue to evaluate market conditions as they evolve and take precautionary measures to strengthen our financial position.
Our Strategy
We believe that we have a novel, differentiated approach to treating the complex biology of fibrotic disease in multiple organ systems; a unique near-term commercial asset in Ryplazim if FDA approval is granted; a strategy to build a broader portfolio of novel small molecule compounds over time; and the opportunities for collaboration in both our small molecule development programs and for Ryplazim. The key elements of our strategy include:
| • | Advancing the clinical development of our lead product candidates: |
Fezagepras for the treatment of patients with diseases related to fibrosis, including respiratory, liver or kidney metabolic disease indications. We have generated preclinical data that we believe demonstrated proof-of-mechanism of fezagepras for the treatment of respiratory, liver and kidney disorders associated with chronic or severe fibrosis. In the second half of 2020, we plan to initiate a Phase 1 clinical trial to
evaluate multiple ascending doses of fezagepras in healthy volunteers. The data from this Phase 1 clinical trial will help define dose levels and regimen to determine the most appropriate indications to be pursued and inform the design of any such future clinical trials, evaluating fezagepras in fibrosis related diseases, including respiratory, liver or kidney metabolic disease indications, subject to the ongoing evaluation of the mechanism of action of fezagepras, the current COVID-19 pandemic.
| • | Seek marketing approval in the United States for Ryplazim® (plasminogen) for the treatment of congenital plasminogen deficiency. |
We completed our Phase 2/3 clinical trial evaluating Ryplazim in pediatric and adult patients with congenital plasminogen deficiency in October 2018. If we resubmit the BLA in the third quarter of 2020, and if our resubmission is designated as a Class 2 resubmission, we expect a PDUFA date in the first quarter of 2021. If we receive regulatory approval on this timeline, we currently plan to launch Ryplazim in the United States in 2021 ourselves with a small, focused commercial infrastructure while exploring potential strategic collaborations to access patients in certain other selected markets outside of the U.S. in 2021 and 2022. If our BLA for Ryplazim is approved by the FDA, we may be also eligible to receive a Pediatric Review Voucher, or PRV, from the FDA. If we receive regulatory approval on Ryplazim on the currently expected timeline, and if we receive a PRV for Ryplazim in a timely manner thereafter, we anticipate seeking to monetize any such PRV in 2021. Beyond this initial indication, our intention is to conduct additional clinical trials for other applications of plasma-derived plasminogen where there appears to be a scientific rationale for treatment via increasing plasminogen activity levels.
Ryplazim was granted Orphan Drug Designation and Rare Pediatric Disease Designation for the treatment of congenital plasminogen deficiency and Orphan Drug Designation for the treatment of IPF. We expect to file our BLA resubmission for Ryplazim in the third quarter of 2020.
| • | Leveraging our drug discovery platform and knowledge base to develop an early-stage drug portfolio: |
Our early-stage drug development efforts are focused on expanding our R&D pipeline both for the treatment of diseases associated with fibrosis and other inflammatory diseases. Accordingly, we first intend to develop oral, selective GPR84 antagonists to treat fibrosis and develop an early-stage pipeline of novel compounds for the treatment of respiratory, liver (liver fibrosis and NASH) and kidney diseases by leveraging our own drug discovery platform, entering into collaborations with third parties or in-licensing or acquiring new compounds and technologies. GPR84 is a pro-inflammatory target primarily expressed on cells associated with the immune system and its expression levels increase significantly during periods of inflammatory stress. We believe a GPR84 antagonist has the potential to be used in combination with one or more of our other product candidates in development or with therapeutics developed by third parties. Our GPR84 antagonist program is currently at the pre-clinical stage. Pending the outcome of our preclinical research, we plan to nominate a preclinical product candidate for our GPR84 antagonist program in the second half of 2020. In addition to GRP84, we intend to develop a novel selective OXER1 antagonist candidate. OXER1 is a GPCR that is highly selective 5-oxo-ETE, believe to be one of the most potent human eosinophil chemo-attractants. Eosinophils play a key role in Type 2 inflammation-driven diseases, including respiratory diseases and gastrointestinal diseases. Our OXER1 antagonist program is currently at the pre-clinical stage. Pending the outcome of our preclinical research, we plan to nominate a preclinical product candidate for our OXER1 antagonist program in the second half of 2021.
Our Small Molecule Program
Our research program is focused on inflammatory, fibrotic and metabolic conditions in patients with liver, respiratory or renal disease, with an emphasis on rare orphan disease.
Fibrosis
Fibrosis is an aberrant response of the body to tissue injury that may be caused by trauma, inflammation, infection, cell injury or cancer. The normal response to injury involves the activation of cells that produce collagen and other components of the extracellular matrix, or ECM, that are part of the healing process. This healing process helps to fill in tissue voids created by the injury or damage, segregate infections or cancer, and provide strength to the recovering tissue. Under normal circumstances, where the cause of the tissue injury is limited, the scarring process is self-limited and the scar resolves to approximate normal tissue architecture. However, in certain disease states, this process is prolonged and excessively active, resulting in progressive tissue scarring, or fibrosis, which can cause organ dysfunction and failure as well as, in the case of certain cancers, promote tumor progression.
Excess levels of various profibrotic cytokines and growth factors are associated with fibrosis. Innate inflammatory cells increase the abundance of myofibroblasts, a cell type that drives wound healing, and stimulates them to deposit ECM proteins such as collagen at the site of tissue injury. In the case of normal healing of a limited tissue injury, myofibroblasts eventually die by programmed cell death, or apoptosis, and the fibrous scarring process recedes. Accordingly, we believe there is a need for therapies that could effectively target pathophysiological pathways involved in fibrosis.
Fezagepras
We are developing fezagepras, our lead small molecule product candidate, for the treatment of respiratory, kidney and liver fibrosis-related metabolic diseases (such as NASH). Fezagepras is an anti-inflammatory and anti-fibrotic small molecule designed to modulate the activity of multiple receptors, including FFAR1 and PPAR alpha. Both FFAR1 and PPAR alpha have been shown to have an impact on metabolic control and to play a role in the pathogenesis of diseases including NAFLD and NASH. Further, third-party data suggests that FFAR1 plays a role in glucose control, while PPAR alpha appears to modulate lipids. Given their long association with the control of metabolic disease and the association between metabolic and fibrotic processes, the PPAR agonist class has a strong development rationale in fibrotic disease. Despite this rationale, historical efforts to develop PPAR agonists for the treatment of fibrotic disease have yet to yield particular success, owing to either reasons of efficacy or tolerability depending on the PPAR agonist subtype. More recently, however, an enhanced understanding of both the PPAR class and fibrotic disease had led to the development of several next-generation PPAR agonists, which each interact with the target in a differentiated manner from previous efforts, which has renewed interest in the class as potential anti-fibrotic agents.
Our research has focused on the role of these receptors on the pathogenesis of fibrosis in multiple organs. Fezagepras has demonstrated anti-inflammatory and anti-fibrotic activity in animal models of chronic kidney disease, diabetic kidney disease, lung fibrosis, liver fibrosis, heart fibrosis, scleroderma and osteoporosis.
We are exploring the potential of fezagepras in the treatment of fibroproliferative diseases. Fezagepras has been observed to regulate several cell types involved in the fibrotic pathway: macrophages, fibroblasts/myofibroblasts and epithelial cells. We have observed that fezagepras regulates fibrotic and inflammatory markers in rodent and normal human fibroblasts, IPF patient fibroblasts, human epithelial cells and in rodent macrophages. We believe the modulating effects of fezagepras on known aspects of the fibrosis process suggest it has the potential to treat fibrosis-related diseases, for which there is a substantial unmet medical need. We are planning to further investigate its effect on patients with respiratory diseases, liver or kidney fibrosis disease indications. We believe fezagepras has the potential to treat these patient populations, as fibrosis is at the core of organs losing functionality in each of these diseases.
Although we have completed a Phase 1 clinical trial evaluating single ascending doses of fezagepras in healthy volunteers and three open-label Phase 2 trials in patients with IPF, Alström syndrome and T2DMS, respectively, we have decided to escalate the dose of fezagepras beyond the maximum 1,200 mg daily dose tested to date, to potentially increase its activity. We believe results from repeat dose toxicity animal studies of fezagepras supports a maximum daily dose of up to 2,400 mg in humans. Accordingly, we plan to initiate a Phase 1 clinical trial to evaluate multiple ascending doses of fezagepras in healthy volunteers at aggregate dose levels higher than those previously evaluated in our completed clinical trials, where the doses tested were generally well tolerated. The data from this Phase 1 clinical trial will help define dose levels and regimen to inform the most appropriate indications to be pursued for fezagepras and the design of any future clinical trials evaluating fezagepras in Phase 2 placebo-controlled clinical trial of fezagepras. We continue to review the potential broader use of Fezagepras in other areas such as skin fibrosis, ocular fibrosis and treating patients with high triglyceride levels.
We expect to define a more fulsome development program for Fezagepras after the completion of the additional multiple ascending dose study expected to begin in the second half of 2020. We plan to initiate further clinical trials of fezagepras in selected fibrosis indications, including potential Phase 2 clinical trials of a duration between 12 and 26 weeks with multiple doses being studied against placebo and/or an active control arm.
Fezagepras has been evaluated in more than thirty different animal models involving six organ systems (lung models, heart models, kidney models, liver models, pancreas models and skin models), and has consistently shown significant activity in all models. Given the wide-ranging preclinical data, we believe that fezagepras represents a differentiated and unique approach to treating a complex disease such as fibrosis in patients with broad applicability in multiple therapeutic categories, mainly respiratory, liver and renal, with the opportunity for a significant therapeutic effect compared to other anti-fibrotic product candidates currently under development.
Fezagepras has been granted Orphan Drug Designation by the FDA and the European Medicines Agency (EMA)for the treatment of Alström syndrome as well as for the treatment of IPF. Fezagepras has also been granted the
Promising Innovative Medicine (PIM) designation by the UK Medicines and Healthcare products Regulatory Agency (MHRA) for the treatment of Alström syndrome and IPF.
Fezagepras for the Treatment of Fibrosis of the Respiratory System
Idiopathic Pulmonary Fibrosis and other Interstitial Lung Diseases
Interstitial lung disease, or ILD, is a general category that encompasses a group of many different rare pulmonary diseases that share several pathophysiological features, despite their diverse causes. ILDs involve chronic inflammation and fibrosis, leading to a progressive destruction of the lung interstitium and loss of lung function. There are over 100 different types of ILD, of which IPF is one of the major forms. Other major forms include: chronic hypersensitivity pneumonitis, connective tissue disease-associated ILDs, or CTD-ILDs, such as rheumatoid arthritis-associated ILD, or RA-ILD, and systemic sclerosis-associated ILD, or SSc-ILD, and pulmonary sarcoidosis.
Idiopathic pulmonary fibrosis, or IPF, is a chronic lung disease characterized by a progressive and irreversible decline in lung function when lung tissue becomes damaged, stiff, and scarred. As tissue scarring progresses, transfer of oxygen into the bloodstream is increasingly impaired, leading to irreversible loss of lung function, as well as high morbidity and mortality rates.
Patients with IPF experience debilitating symptoms, including shortness of breath and difficulty performing routine functions, such as walking and talking. Other symptoms include chronic dry, hacking cough, fatigue, weakness, discomfort in the chest, loss of appetite and weight loss.
IPF usually occurs in adult individuals between 50 and 70 years of age, particularly in those with a history of cigarette smoking, and affects men more often than women. IPF affects approximately 130,000 people in the United States, with about 48,000 new cases diagnosed annually. Approximately 40,000 people with IPF die each year. The five-year mortality rate for patients with IPF is estimated to range from 50% to 70% of those affected.
Limitations of Current Treatment Options for Idiopathic Pulmonary Fibrosis
Treatment options for patients with IPF are limited. Two products, nintedanib (OFEV® - Boehringer-Ingelheim) and pirfenidone (Esbriet® - Roche), have been approved for the treatment of IPF. However, neither demonstrated the ability to stabilize lung function in clinical trials, but rather were approved based upon their ability to slow the rate of decline in lung function. In addition, the tolerability profile of each drug is problematic for chronic usage as they are both associated with substantial levels of side effect (particularly GI), which have limited their use. Accordingly, we believe there remains a substantial unmet medical need in IPF.
Beyond the IPF market, competition in product candidate development for other ILDs is limited. Nintedanib, which received FDA approval for the treatment of SSc-ILD in September 2019 and FDA approval for treatment of progressive-fibrosis-ILDs in March 2020, is currently the only anti-fibrotic drug approved for the treatment of another ILD beyond IPF. Roche is also working to expand the potential ILD market for pirfenidone, with clinical trials underway in RA-ILD, SSc-ILD and chronic hypersensitivity pneumonitis. We are not aware of any other industry-sponsored clinical trials evaluating a product candidate for the treatment of SSc-ILD or chronic hypersensitivity pneumonitis in the United States or Europe.
Our Solution for Idiopathic Pulmonary Fibrosis: Fezagepras
Completed Open Label Phase 2 Clinical Trial
In January 2017, we completed an open-label, single arm, exploratory, observational Phase 2 trial of fezagepras, both as a monotherapy and in combination with either nintedanib or pirfenidone, in 41 patients with IPF. The primary endpoints of this trial were safety and tolerability and the key secondary endpoints of this trial included change in pulmonary function and change in inflammatory and fibrotic markers. Nine patients received 800 mg doses of fezagepras alone, 16 patients received 800 mg oral doses of fezagepras in combination with nintedanib and 16 patients received 800 mg doses of fezagepras in combination with pirfenidone, with 40 subjects completing the trial as planned (n= 9, 16 and 15) respectively; all administered orally on a daily basis for 12 weeks. The full results of this trial were published in the European Respiratory Journal in 2018.
The results of the trial showed that the mean change from baseline to Week 12 for forced vital capacity, or FVC, which is the total amount of air exhaled during a forced breath, was either slightly increased (+1.9 mL) or slightly decreased (-12.2 mL) for fezagepras in combination with nintedanib and for fezagepras alone, respectively, but was reduced (-102.0 mL) for fezagepras in combination with pirfenidone.
Observed Effect of 800 mg Dose of Fezagepras on the Change from Baseline
(Week 1, Pre-dose) for FVC at Week 12
| | | | |
Pulmonary Function Test | Fezagepras (n = 9) | Fezagepras + Nintedanib (n = 16) | Fezagepras + Pirfenidone (n = 15) | Total (n = 40) |
Percent-predicted FVC (%) |
Mean change (± SD) at Week 12 | -1.11 (4.457) | 0.06 (4.024) | -2.69 (4.113) | -1.23 (4.228) |
95% confidence interval | -4.537, 2.315 | -2.082, 2.207 | -4.964, -0.409 | -2.585, 0.120 |
P-value* | 0.4759 | 0.9513 | 0.0240 | 0.0729 |
FVC (mL) |
Mean change (± SD) at Week 12 | -12.2 (137.09) | 1.875 (127.6) | -102 (137.80) | -40.3 (138.96) |
95% confidence interval | -117.60, 93.157 | -66.121, 69.871 | -178.31, -25.69 | -84.692, 4.192 |
P-value* | 0.7959 | 0.9539 | 0.0124 | 0.0746 |
FVC = forced vital capacity; N = number of subjects; SD = standard deviation.
a P-value based on paired t-test of the mean change from baseline (Week 1, pre-dose) for FVC at Week 12.
The combination of fezagepras and pirfenidone exhibited reduced pharmacokinetics as compared to fezagepras alone, suggesting a possible drug-drug interaction. Fezagepras’ concentration in plasma was found to be sub-therapeutic at 50% of the expected level in patients that received the fezagepras and pirfenidone combination. In pre-clinical studies using the bleomycin model, it was shown that the combination of fezagepras and pirfenidone had a synergistic effect. In the animal studies, higher doses of fezagepras were used compared to the dose used in the clinical trial, with the goal of maintaining a therapeutic dose level in the animals.
Fezagepras (800 mg) was well tolerated alone and in combination with either nintedanib or pirfenidone. One patient (2.4%) in the fezagepras + nintedanib group had a treatment-emergent serious adverse event of pneumonia. The medical monitor assessed the event as more likely arising from immune suppression due to prolonged use of the concomitant medication prednisone and the patient’s underlying IPF.
Importantly, there were no serious adverse events requiring fezagepras’ discontinuation. The most common adverse event observed in patients in this trial was diarrhea (˃15%). Diarrhea rate, however, was lower in the patients treated with fezagepras alone than in the groups receiving fezagepras in combination with either nintedanib or pirfenidone, both of which are associated with GI side effects.
Most TEAEs were mild or moderate in severity. Three patients (7.3%) had severe TEAEs: one patient had a severe TEAE of diarrhea that resolved and was deemed not related to fezagepras, one patient had severe TEAEs of dyspnea and IPF exacerbation that were moderate in severity and deemed not related to fezagepras, and one patient had a severe TEAE of gastrointestinal disorder that resolved and was deemed not related to fezagepras. In addition, there were no TEAEs of hypoglycemia among eight patients with type 2 diabetes (86-week exposure), including five patients requiring oral antidiabetic therapy with or without insulin (50-week exposure).
Preclinical Data
Antifibrotic Activity of Fezagepras Observed in Animal Models of Lung Fibrosis
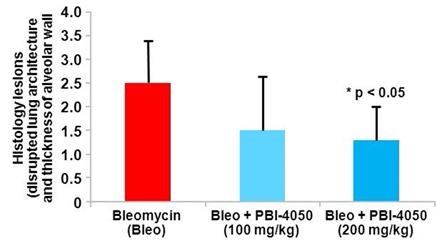
We used a mouse model of bleomycin-induced lung fibrosis to evaluate the effect of fezagepras on histological lesions and inflammatory and fibrotic markers. In this model, female mice were treated with bleomycin (0.025 U) via intratracheal administration on Day 0. On Day 7, 28 mice were randomized into three groups according
to their bleomycin-induced body weight loss. Mice were treated orally with fezagepras (100 mg/kg and 200 mg/kg) or vehicle from Day 7 through Day 20. On day 21, mice were euthanized. We observed a dose-dependent reduction in histological lesions of the lung.
Observed Antifibrotic Activity of Fezagepras in a Mouse Model of Bleomycin-Induced Lung Fibrosis
Parameter | Finding |
Lung lesions | Fezagepras reduced histological lesions of lung fibrosis in a dose-dependent manner |
Inflammatory and fibrotic markers | Fezagepras (200 mg/kg) significantly (p ≤ 0.05) decreased the mRNA expression of CTGF, collagen 1 and IL-23p19. |
CTGF = connective tissue growth factor; GPR = G protein-coupled receptor; IL = interleukin; mRNA = messenger RNA.
Effect of Fezagepras on Bleomycin-Induced Histological Lesions
Fezagepras and Pirfenidone
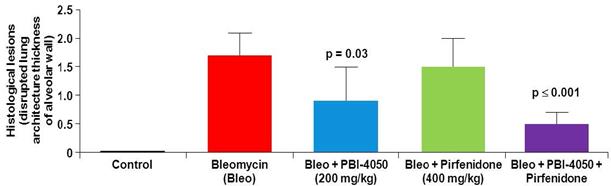
We also used the mouse model of bleomycin-induced lung fibrosis to evaluate the effect of 200 mg/kg doses of fezagepras, 400 mg/kg doses of pirfenidone, and the same doses of fezagepras in combination with pirfenidone on histological lesions and inflammatory and fibrotic markers. In this preclinical study, mice were randomized into three groups according to their bleomycin-induced body weight loss and treated orally with either fezagepras, pirfenidone or a combination of both compounds from Days 7 to 20. Only the animals that recovered their body weight loss by Day 20 were used for data analysis. Mice were euthanized on Day 21. Based on the Ashcroft score, a validated measure of histological lesions of lung fibrosis, fezagepras showed a statistically significant reduction in lung fibrosis compared to the bleomycin control group. We also observed a reduction in the histological lesion score of the mice treated with pirfenidone, but the reduction was not statistically significant compared to the bleomycin control group. Fezagepras also decreased the overexpression of pro-inflammatory and fibrotic markers compared to pirfenidone. We also observed a statistically significant reduction in histological lesions of the lung in the mice treated with fezagepras alone and in the mice treated with fezagepras in combination with pirfenidone.
Observed Antifibrotic Activity of Fezagepras in a Mouse Model of Bleomycin-Induced Lung Fibrosis: Fezagepras and Pirfenidone
Parameter | Finding |
Toxicity | No toxicity was reported with fezagepras. Pirfenidone induced severe vertigo that was reduced in the combination therapy (fezagepras + pirfenidone). |
Bronchoalveolar fluid | All treatments (fezagepras; pirfenidone; and fezagepras + pirfenidone) significantly (p < 0.05) reduced CTGF level. |
Only the combination therapy significantly (p = 0.0424) decreased IL-1β protein level. |
Only the combination therapy significantly (p < 0.05) decreased TNF-α protein level. |
Lung lesions | Only fezagepras and the combination therapy significantly (p < 0.05) reduced histological lesions; pirfenidone had no effect. |
Fezagepras and the combination therapy significantly (p < 0.001) decreased the lesion score determined by the Ashcroft’s score;a pirfenidone alone had a statistically insignificant effect (p = 0.0538). |
All treatments significantly (p ≤ 0.05) reduced the percentage of leukocyte infiltration. |
Inflammatory and fibrotic markers | Fezagepras and the combination therapy significantly (p < 0.05) reduced the percentage of collagen in leukocyte infiltration. |
All treatments significantly (p < 0.05) decreased the overexpression of collagen, fibronectin, IL-23p19, IL-6, and TGF-β. Only fezagepras and the combination therapy significantly (p < 0.05) decreased CTGF and SPARC mRNA expression. Only the combination therapy significantly (p < 0.05) reduced collagen 3 and MMP2 mRNA expression. |
All treatments significantly (p < 0.05) increased IFN-γ protein level in the lung. |
Fezagepras and the combination therapy significantly (p < 0.05) increased IL‑12p40 and IL-1β protein levels in the lung. |
All treatments significantly (p < 0.01) increased the content of TNF-α protein level. |
CTGF = connective tissue growth factor; GPR = G protein-coupled receptor; IFN-γ = interferon gamma; IL = interleukin; MMP = matrix metalloproteinase; mRNA = messenger RNA; SPARC = matricellular protein secreted protein acidic and rich in cysteine; TGF-β = transforming growth factor beta; TNF-α = tumor necrosis factor alpha.
a Ashcroft’s score: Briefly, the entire fields of each lung section were read by a blinded examiner, and each field was visually graded from 0 to 8. Criteria for grading lung fibrosis were as follows: Grade 0= normal lung; Grade 1= minimal fibrous thickening of alveolar or bronchiolar walls; Grade 3= moderate thickening of walls without obvious damage to lung architecture; Grade 5= increased fibrosis with definitive damage to lung structure and formation of fibrous bands or small fibrous masses; Grade 7= severe distortion of structure and large fibrous area; Grade 8= total fibrous obliteration of lung fields.
Observed Effect of Fezagepras and Pirfenidone on Bleomycin-Induced Histological Lesions
Fezagepras and Nintedanib
We used a mouse model of bleomycin-induced pulmonary fibrosis to evaluate the effect of 200 mg/kg doses of fezagepras, 60 mg/kg doses of nintedanib and 200 mg/kg doses of fezagepras in combination with 60 mg/kg doses of nintedanib on inflammatory and fibrotic markers. In this preclinical study, mice were randomized into three groups according to their bleomycin-induced body weight loss and treated orally with either fezagepras, nintedanib or a combination of both compounds from Days 7 to 20. Mice were euthanized on Day 21. We observed a statistically significant reduction in inflammatory and fibrotic markers in lung tissue in the mice treated with fezagepras alone and in the mice treated with fezagepras in combination with nintedanib.
Observed Antifibrotic Activity of fezagepras in a Mouse Model of Bleomycin-Induced Lung Fibrosis: Fezagepras and Nintedanib
| |
Parameter | Finding |
Bronchoalveolar fluid | All treatments (fezagepras; nintedanib; and fezagepras + nintedanib) significantly (p ≤ .05) reduced CTGF. Only fezagepras and the combination therapy significantly (p < 0.05) reduced MCP-1 levels. |
Inflammatory and fibrotic markers | All treatments induced a significant (p < 0.05) decrease of MCP-1 level and iNOS mRNA expression in lung tissue. |
Only fezagepras and the combination therapy significantly (p < 0.05) reduced the percentage of collagen in inflamed lesions and in total lung tissue. |
Only fezagepras significantly (p < 0.05) decreased the mRNA expression of collagen and fibronectin in lung tissue. |
Fezagepras significantly (p < 0.05) reduced TNF-α, IL-6, and CTGF mRNA expression in lung tissue. Nintedanib and the combination therapy induced a significant (p < 0.05) reduction of IL-6 and CTGF. |
CTGF = connective tissue growth factor; IL = interleukin; iNOS = inducible nitric oxide synthase; MCP = monocyte chemotactic protein; mRNA = messenger RNA; TNF-α = tumor necrosis factor alpha.
Fezagepras for the Treatment of Alström Syndrome
Alström Syndrome
Alström syndrome is a rare, inherited, autosomal recessive syndrome resulting from the mutation of the ALMS1 gene that leads to childhood or adolescent obesity, type 2 diabetes with severe insulin resistance, dyslipidemia, hypertension and severe multi-organ fibrosis, involving the heart, liver and kidney. The most common cause of death in patients with Alström syndrome is heart failure with dilated cardiomyopathy due to progressive cardiac fibrosis. Fibrosis leading to liver failure is also responsible for a large number of deaths. Alström syndrome is also characterized by a progressive loss of vision and hearing and short stature. Alström syndrome affects over 800 patients worldwide. There are no therapies approved to treat Alström syndrome.
Completed Open Label Phase 2 Clinical Trial
We completed a single-center, single-arm, open-label Phase 2 trial evaluating fezagepras in 12 subjects with Alström syndrome who were 16 years of age or older. Patients received a total oral daily dose of 800 mg of fezagepras for an initial 24 weeks with continuation for a further 36 or 48 weeks. The primary objective of the trial was to evaluate the safety and tolerability of fezagepras in this patient population. Standard assessments of safety included adverse events, clinical laboratory tests, vital signs, physical examination and electrocardiograms.
Exploratory objectives included assessments of the effect of fezagepras on liver stiffness using transient elastography, measuring fat content and fibrosis burden in the liver using MRI and measuring the effect on cardiac fibrosis and function using MRI. In this Phase 2 open-label safety trial in Alström syndrome patients, we observed encouraging clinical effects of fezagepras on both liver stiffness (as measured by Fibroscan, a surrogate for liver fibrosis) and cardiac fibrosis. Although the data is inconclusive, we believe that fezagepras has the potential to provide benefit to patients with Alström syndrome, since fibrosis is central to disease progression and loss of organ function in this condition. Further controlled studies will be required to further characterize fezagepras' clinical profile.
All patients were recruited from the University Hospitals Birmingham, which is the nationally commissioned specialist service for Alström syndrome in the United Kingdom.
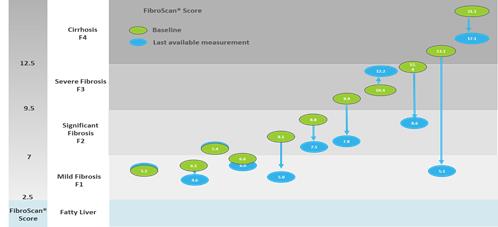
Observed effect on a surrogate of liver fibrosis
Mean transient elastography scores, a measurement of liver stiffness using FibroScan, was observed to decrease from baseline to the last available measurement for 10 out of 11 subjects for whom data was available, with daily 800 mg doses of fezagepras.
Observed Effect of 800 mg Dose of Fezagepras on the Change from Baseline in Liver Stiffness
Using Transient Elastography (FibroScan) –
Observed effect on myocardial fibrosis
Results of cardiac magnetic resonance imaging scans of myocardial fibrosis appeared to yield positive results after treatment with daily doses of 800 mg of fezagepras, when the data from our open-label study subjects was compared to data from a cohort of untreated Alström syndrome patients (shown as the control group in the diagram below).
Observed Effect of 800 mg Dose of Fezagepras on the Change from Baseline in Magnetic Resonance Imaging Assessment of Left Ventricular Function and Myocardial Fibrosis –

Observed tolerability data
Fezagepras was reported to be well tolerated at a daily oral dose of 800 mg for up to 72 weeks in patients 17 to 52 years of age with Alstrom syndrome. No conclusions can be drawn with respect to efficacy as this was a safety study and the sample size was not sufficient to draw any conclusions regarding efficacy. Two patients reported non-treatment related serious adverse events (one with mild dehydration and one with severe cardiac ventricular thrombosis). Neither of these serious adverse events resulted in permanent discontinuation of trial drug. The most common TEAEs observed in patients in this trial were hypoglycemia, rash, headache, and lower respiratory tract infection, urinary tract infection, abdominal discomfort, diarrhea, fatigue, asthma and decreased hemoglobin and the most common treatment-related TEAE observed in patients was hypoglycemia. Most treatment-related TEAEs were mild in severity, and the majority of subjects had mild or moderate TEAEs. None of the TEAEs resulted in permanent discontinuation of the trial drug. There was no severe treatment-related TEAEs.
Rollover Phase 2 Clinical Trial
Following completion of our Phase 2 clinical trial of fezagepras in patients with Alström syndrome, we initiated an open-label, single-arm, multi-center rollover Phase 2 clinical trial evaluating the long-term safety and tolerability of fezagepras in nine patients who completed the end-of-treatment visit for the initial Phase 2 trial. Each patient was administered a total daily dose of 800 mg of fezagepras and undergoes intensive investigation to document the effects of fezagepras on progressive organ fibrosis, including through magnetic resonance imaging of the liver and the heart. Each patient was evaluated against their individual results at trial entry, as well as against their historical disease progression trend, when available. The Data Safety Monitoring Board and the Medicines and Healthcare Products Regulatory Agency have agreed to an extension of this rollover trial. In connection with the COVID-19 pandemic, we recently ended the treatment under this rollover Phase 2 clinical trial. Nine patients have completed more than two years of treatment with fezagepras, and six of which have completed more than three years of treatment with fezagepras.
Fezagepras for the Treatment of Fibrosis Associated with Non-Alcoholic Steatohepatitis
Fibrosis Associated with Non-Alcoholic Steatohepatitis (NASH)
Non-alcoholic fatty liver, or NAFLD, is a condition that is estimated to affect 75 million people in the United States due to the obesity epidemic and is the manifestation of metabolic disease in the liver. NASH is a progressed state of NAFLD, where the chronic injury suffered by the liver due to the excess fatty deposits associated with NAFLD trigger inflammation and fibrosis. Though only a small percentage of NAFLD patients progress to NASH, the sheer number of NAFLD patients has made NASH the most common cause of severe liver disease worldwide. NASH and its associated co-morbidities, such as fibrosis, remain a major unmet medical need with treatment offering little recourse.
Though the biologic mechanisms underlying the pathogenesis of NASH are not fully characterized, the current understanding describes excess lipids present in the liver ultimately leading to hepatoxic injury, followed by inflammation and fibrosis with an associated decline in liver function. Current research implicates multiple pathways for both the initial lipid accumulation and the dysregulated healing response.
There are no currently approved therapies for NAFLD, NASH or associated fibrosis, although one potential therapy (Ocaliva®, developed by Intercept Pharmaceuticals) is currently under FDA review. Current treatment options are limited to diet and lifestyle modifications that may control or reduce the amount of excess fat deposits in the liver.
Observed Antifibrotic Activity of fezagepras Following High Fat Diet (HFD), a Model of Metabolic Syndrome
Obesity and its resulting metabolic disturbances are health threats and a leading cause of a host of diseases, including non-alcoholic fatty liver disease. In preclinical studies, C57BL/6 mice were fed with either a standard diet or a high fat diet, or HFD, for 14 weeks. These mice were then divided into five groups as follows: standard diet, HFD + vehicle, and HFD + fezagepras (200 mg/kg, oral once a day), GPR84-/- + vehicle, and GPR84-/- + fezagepras (200 mg/kg, oral once a day) and treated for an additional six or 10 weeks.
Blood biochemistry, serum insulin level, liver, and white adipose tissue, or WAT, gene expression for markers of inflammation, fibrosis, adipokines, PPARs, fatty acid metabolism, energy expenditure and glucose transporter and histology assessment of these tissues were performed.
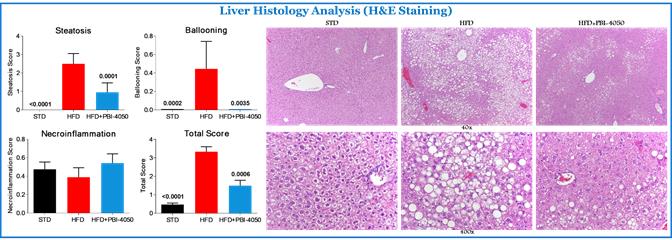
Fezagepras for the Treatment of Fibrosis related Kidney Disease
Chronic Kidney Disease (CKD)
An estimated 37 million American adults (1 in 7 adults; 15% of adults) are estimated to have CKD. Fibrosis is believed to be one of the mechanism via which the condition of CKD patients deteriorates, leading to further loss of renal function, increased cardiovascular complications, and eventually, the need for dialysis treatment while waiting for a kidney transplant. Patients diagnosed with severe CKD stages (3 and 4) often suffer from a gradual and accelerated loss of their renal function (end-stage renal disease or ESRD) leading to the need for hemodialysis. Cardiovascular complications for ESRD patients on hemodialysis are a common cause of death.
Diabetic nephropathy is a complication of long-standing diabetes mellitus, of both Type 1 and Type 2. It is increasing in incidence throughout the world, and in many countries, including the United States and Canada, is the leading cause of end-stage renal disease requiring maintenance dialysis and/or kidney transplantation
Completed Phase 1b clinical trial of fezagepras in patients with stable renal impairment
We conducted a randomized, placebo-controlled Phase 1b clinical trial of fezagepras in patients with stable renal impairment (Phase 3b and 4) with a single 800 mg dose of fezagepras and multiple 800 mg doses administered once daily for 10 days. The objectives of the trial were to evaluate the safety, tolerability and pharmacokinetics of fezagepras in patients with stable renal impairment.
In this trial, we observed that fezagepras was well tolerated in patients with stable renal impairment. None of the TEAEs were severe or serious, and the majority were mild in intensity. Although an imbalance in the frequency of TEAEs and the number of patients who reported TEAEs was observed between the single- and multiple-dose parts and between patients who received fezagepras versus placebo, no trends or consistent pattern were observed. We observed abnormalities with clinical laboratory tests that were considered as TEAEs in three patients.
Based on these results, we concluded that fezagepras did not tend to accumulate following multiple oral dose administrations of 800 mg in patients with stable renal impairment. We observed that the fraction of the drug unbound to plasma proteins was less than 1%.
Observed Effect of Fezagepras in adenine-induced chronic kidney disease
A common histopathological finding in progressive kidney diseases, is the deposition of fibrotic material in the tubulointerstitium, termed tubulointerstitial fibrosis, or TIF. Several studies have concluded that TIF is considered as having the best predictive value for the progression of chronic kidney disease, or CKD. Studying the etiology of TIF in rodents requires adequate models that recapitulate typical CKD progression in humans. To this end, adenine supplementation, causing direct tubule epithelial cell injury, has emerged as a useful murine model of CKD, mimicking several signs of human CKD progression including the development of progressive renal insufficiency, severe anemia, inflammation and TIF. We observed that fezagepras exerted antifibrotic effects in various models of organ fibrosis, including the liver, lungs, heart and kidneys.
A study in which the impact of fezagepras treatment in adenine-induced CKD was undertaken to determine whether this compound could attenuate the renal inflammatory and fibrogenic response. For this, male mice were fed an adenine-supplemented diet for one week, which continued for an additional three weeks alongside daily fezagepras at 200 mg/kg.BW.
| |
| 
|
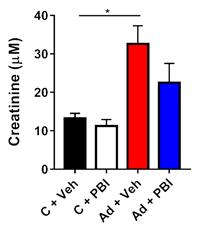
Fig.1 Endpoint plasma creatinine levels and hematocrit. |
After 4 weeks of adenine-feeding, mice from both vehicle (Ad+Veh) and fezagepras (Ad+PBI) groups displayed dramatic weight loss. Adenine led to a significant rise of plasma creatinine in vehicle-treated mice, but not in mice treated for 3 weeks with daily fezagepras at 200 mg/kg, indicative of preserved renal function (Fig.1, left panel).
It is well established that anemia contributes to adverse outcomes in CKD patients. Previously unpublished observations regarding the anti-anemic effects of fezagepras pushed us to investigate this compound’s effects on anemia in this model. Anemia is defined as a decrease in red blood cells and was measured using hematocrit, or Hct. As shown in Fig 1 (right panel), we observed that Hct, declined 5-7% in both Ad-groups after one week. By two-weeks, Hct in the vehicle-treated group decreased by 10%, while PBI-treated mice remained stable. At endpoint, Ad+Veh mice had a 20% drop in Hct while levels in Ad+PBI were unaffected. A sub study revealed that a dose of 200 mg/kg of fezagepras was necessary to prevent the drop in Hct (not shown).
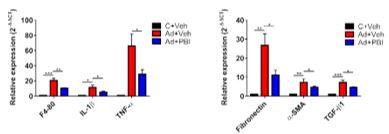
We observed that fezagepras mitigated adenine-induced renal inflammation, fibrosis and histopathological injury. As shown in Fig. 2, we observed that fezagepras treatment for three weeks significantly reduced the deposition of fibrotic material in the kidneys, described as TIF. This was associated with reduced pro-inflammatory (F4/80, IL-1β, and TNF-α) and pro-fibrotic (fibronectin, α-SMA and TGF-β1) gene expression.

|
|
Fig 2. Tubulointerstitial fibrosis and pro-inflammatory / pro-fibrotic gene expression. |
These data are consistent with several preclinical studies highlighting the anti-fibrotic and overall beneficial effects of fezagepras. In this study, fezagepras treatment mitigated important pathogenic mediators of CKD, including ER-stress, TIF, inflammation and anemia suggesting this compound may hold therapeutic potential.
PBI-4547
PBI-4547 is an orally active small molecule that is a GPR84 antagonist, GPR40 (FFAR1)/GPR120 (FFAR4) agonist, and a partial activator of the peroxisome proliferator-activated gamma receptors.
In September 2019, we announced that we had dosed the first patient in a Phase 1 clinical trial of PBI-4547, designed to assess the safety, tolerability and pharmacokinetics of single ascending doses of PBI-4547 in up to 40 healthy volunteers. We voluntarily suspended this clinical trial in the third quarter of 2019. In the second quarter of 2020, following the review of pharmacokinetic data for the first three cohorts obtained, we decided to discontinue the clinical trial pending further assessments and re-evaluation of the development program and not to proceed with the enrolment of the two additional cohorts following . No dose-limiting adverse events have been observed to date.
Our Selective Oxo-eicosanoid receptor 1 (OXER1) Antagonist Program
We are developing an oral, selective OXER1 antagonist candidate. OXER1 is a GPCR that is highly selective for 5-oxo-ETE, believed to be one of the most potent human eosinophil chemo-attractants. Migration of eosinophils to body sites including the lungs and intestines is mediated by eosinophil chemo-attractants such as 5-oxo-ETE. Eosinophils play a key role in Type 2 inflammation-driven diseases, including respiratory diseases and gastro-intestinal diseases.
Eosinophilic-related diseases represent a significant area of unmet need in global health. Several biologics have been approved for the treatment of eosinophil-related diseases, with combined drug sales in 2018 exceeding USD$ 3 Billion. Several of the approved monoclonal antibody treatments for severe eosinophilic asthma are currently in clinical trials aimed at expanding their indications to include other eosinophilic disorders. Compared to approved biologics, small molecule OXE receptor antagonists may offer a promising and potentially more cost-effective option for treatment of eosinophilic-driven disorders. Compared to approved biologics, small molecule OXE receptor antagonists may offer a promising and potentially more cost-effective option for treatment of eosinophilic-driven disorders.
The OXER1 antagonist program is based on the research of Dr. William Powell, Professor Emeritus in the Department of Medicine at McGill University, working in collaboration with Dr. Joshua Rokach of the Florida Institute of Technology.
In addition to our experienced in-house medicinal chemistry group, we have a strong understanding and well-established, in-house expertise in GPCR biology.
Our OXER1 antagonist program is currently at the pre-clinical stage. Pending the outcome of our preclinical research, we plan to nominate a preclinical product candidate for our OXER1 antagonist program in the second half of 2021.
Our Selective G-protein coupled receptor 84 (GPR84) Antagonist Program
We are developing a selective GPR84 antagonist candidate that we believe could be used as monotherapy or in combination with other approved drugs. GPR84 is a pro-inflammatory target primarily expressed on cells associated with the immune system and its expression levels increase significantly during periods of inflammatory stress. Inhibition of GPR84 can inhibit neutrophil and macrophage migration and reduce cytokine release.
However, GPR84 expression is not restricted to cells in the immune system; it is also expressed in tissues such as the brain, heart, muscle, colon, kidney, liver, intestine, adipose and lung. Through its role in inflammation, GPR84 may be a mediator of the relationship between inflammation, obesity and diabetes. Rodent models suggest that GPR84 expression is up-regulated in adipocytes in response to TNF-α released from infiltrating macrophages, and that this in turn can lead to a down-regulation in adiponectin expression in adipocytes. Adiponectin is known to have anti-diabetic, anti-inflammatory, and anti-atherogenic effects, and it also functions as an insulin sensitizer.
In addition to our experienced in-house medicinal chemistry group, we believe we have a strong understanding and well-established, in-house expertise in GPR84 biology. Our prior research has shown that GPR84-deficient mice develop reduced fibrosis in a model of kidney fibrosis (adenine-induced chronic kidney disease). We have also shown that GPR84 mRNA is overexpressed in various acute and chronic kidney models such as 5/6-nephrectomy (Nx)-induced chronic kidney disease, doxorubicin (DOX)-induced nephropathy and in adenine-induced tubulointerstitial injury.
Our GPR84 antagonist program is currently at the pre-clinical stage. Pending the outcome of our preclinical research, we plan to nominate a preclinical product candidate for our GPR84 antagonist program in the second half of 2020. Galapagos Therapeutics GLPG1205 program, currently in Phase 2, is the only other GPR84 antagonist program to our knowledge.
Our Plasma-Derived Therapeutics Program
Patients may be born with the inability to produce sufficient plasminogen naturally, a condition referred to as congenital plasminogen deficiency, or suffer an acute or acquired deficiency following a trauma or an illness. We are initially focused on developing a treatment for congenital plasminogen deficiency, however, we may in the future explore expanding the therapeutic uses Ryplazim, or other plasminogen formulations, to additional indications such as acquired plasminogen deficiency and for use in critical care and wound healing settings. We believe that these indications both represent a near-term opportunity, from a product developed by our plasma-based therapeutics business segment, Prometic. We expect that expansion into new indications, including other indications where low plasminogen activity appears to play a role, would leverage our investment in our proprietary manufacturing process for plasminogen with the potential for different formulations and product presentations suitable for different patient populations.
Ryplazim
We are developing Ryplazim, our lead plasma-derived product candidate, for the treatment of congenital plasminogen deficiency and we expect to resubmit our BLA in the third quarter of 2020
Plasminogen and Congenital Plasminogen Deficiency
Plasminogen is a naturally occurring protein that is synthesized by the liver and circulates in the blood. Activated plasminogen, plasmin, is a fundamental component of the fibrinolytic system and is the main enzyme involved in the lysis of blood clots and clearance of extravasated fibrin. Plasminogen is therefore vital in wound healing, cell migration, tissue remodeling, angiogenesis and embryogenesis. Patients may be born with the inability to produce sufficient plasminogen naturally, a condition referred to as congenital plasminogen deficiency, or suffer an acute or acquired deficiency following a trauma or an illness. Patients with congenital plasminogen deficiency experience an accumulation of fibrin growths or lesions on mucosal surfaces throughout the body. Many cases are first diagnosed in the pediatric population, and if left untreated, disease manifestations may be organ-compromising. Peer-reviewed publications report that the condition may have a prevalence of 1,6 cases per million globally. Proprietary data sources and analyses involving the U.S. population suggest that the number of people potentially affected by plasminogen deficiency in the United States may be greater than these early
epidemiological estimates. Congenital plasminogen deficiency requires lifelong therapy to avoid recurrence of lesions. There are currently no approved therapies for the treatment of congenital plasminogen deficiency.
The most common and visible lesion associated with plasminogen deficiency is ligneous conjunctivitis, or LC, which is characterized by thick, woody (ligneous) growths on the conjunctiva of the eye, and if left untreated, can lead to corneal damage and blindness. Ligneous growths tend to recur after surgical excision, thereby requiring multiple surgeries. While ligneous conjunctivitis is the most common lesion, congenital plasminogen deficiency is a multi-systemic disease that can also affect the ears, tracheobronchial tree, genitourinary tract, and gingiva. Tracheobronchial lesions can result in respiratory failure. Hydrocephalus has also been reported in children with severe hypoplasminogenemia, apparently related to the deposition of fibrin in the cerebral ventricular system. Patients who suffer from plasminogen deficiency also have impaired post-surgical wound healing.
Patients with plasminogen deficiency may be born with the inability to produce sufficient plasminogen naturally, a condition referred to as congenital plasminogen deficiency, or suffer an acute or acquired deficiency following a trauma or an illness. Commercially available diagnostics are used to diagnose patients with plasminogen deficiency and genealogical tracking is also available given the autosomal recessive natures of the disease. We estimate that the prevalence of plasminogen deficiency to be 1.6 cases per million in the United States based on published independent data however our own proprietary research suggests a prevalence of up to twice such published epidemiological estimated data. We are focused on developing a treatment for congenital plasminogen deficiency.
Our Solution for Congenital Plasminogen Deficiency: Ryplazim
Ryplazim is a highly purified glu-plasminogen derived from human plasma that acts as a plasminogen replacement therapy for patients with congenital plasminogen deficiency, a rare inherited disorder caused by a mutation of the PLG gene.
Completed Phase 2/3 Clinical Trial
In a pivotal Phase 2/3 clinical trial of Ryplazim for the treatment of congenital plasminogen deficiency, 15 patients (six children and nine adults) were administered 6.6 mg/kg intravenous doses of Ryplazim every two to four days for 48 weeks. The primary efficacy endpoint was clinical success, defined as 50% of the subjects achieving a greater than 50% improvement in lesion number/size or functionality impact from baseline. The primary pharmacokinetic endpoint was an increase in trough plasminogen activity levels by at least an absolute 10% above baseline at 12 weeks. Ryplazim met both its primary efficacy and pharmacokinetic endpoints following the intravenous administration of Ryplazim to fifteen patients for 48 weeks, as we observed a reduction in visible and non-visible lesions of greater than 50% in all patients at 48 weeks and at least a 10% improvement from baseline trough plasminogen levels at 12 weeks.
We observed that all evaluable patients achieved target trough plasminogen activity levels (absolute 10% above baseline) across the initial 12-week treatment period.
Ryplazim was well tolerated in both children and adults. No serious adverse events were reported and no patient permanently discontinued treatment due to an adverse event. Most adverse events were mild or moderate in severity and deemed by the investigators to be unrelated to the study drug. Two patients had adverse events of severe intensity. One of the patients reported anxiety, nausea, fatigue, arthralgia, back pain, dizziness, paresthesia and flushing after her twentieth infusion, which were resolved after temporary treatment discontinuation without re-emergence with restart of treatment. The other patient had severe back pain that resolved three days later. The most common adverse events observed in patients in this trial were headache and nasopharyngitis. Several patients had adverse events (including epistaxis, hematuria and dysmenorrhea) and/or laboratory abnormalities (including blood in urine and elevated D-dimer levels) consistent with physiologic trial drug activity, specifically increased fibrinolytic capacity with lesion dissolution. Minor bleeding events of hemorrhage (eye, skin, uterine, and vaginal), epistaxis, hematuria, cervical and oral discharge occurred in a few patients in areas near the lesion sites or associated with urinary excretion. No additional analyses were performed on any of these secretions. These findings are consistent with abnormal urinalysis findings of blood and protein in urine, which became macroscopic with urinary tract lesion lysis. Gross hematuria was not persistent or continuous. In addition, there were no clinically significant findings for vital signs or viral testing. Anti-plasminogen antibodies were not detected in any patient.
BLA Submission and Complete Response Letter
In April 2018, we received a CRL from the FDA, following the submission of our BLA for Ryplazim for the treatment of congenital plasminogen deficiency with the FDA and a plant inspection of our Laval, Québec facility
by the FDA. The FDA identified the need for us to make a number of changes related to CMC. These included the implementation and validation of additional analytical assays, such as: developing appropriate reference standards of commercially obtained assays and validating the assays using those standards; increasing the number of assays calculating intermediate data; and establishing the linearity and range of certain assays using analyte in the product matrix versus using standard curves. These also included implementation and validation of “in-process controls” in the manufacturing process of Ryplazim, such as reassessing characterization tests in the process for their utility to control process performance; validating analytical methods; amending the approach to perform assessments of particulates after filtering of a sample to represent the amount of protein aggregation in the product; and establishing the validation of hold times and process times for unit operations for the entire process, as well as the manufacturing of Ryplazim conformance batches to confirm the effectiveness of these process changes.
In October 2018, we announced the completion of a Type C meeting, during which the FDA provided feedback on our proposed action plan for the implementation of additional analytical assays and in-process controls related to the manufacturing process of Ryplazim. As a result of the feedback received during that Type C meeting, we completed the manufacturing process performance qualification protocol and the manufacturing of required Ryplazim conformance lots. We have also engaged external consultants to assist with this process. We plan to resubmit our BLA to the FDA in the third quarter of 2020 based on the results from our open-label Phase 2/3 clinical trial completed in October 2018.
The BLA includes the clinical results on 15 patients with 48 weeks of data. In addition, we plan to continue to supply Ryplazim to those patients enrolled in the original clinical trial under an approved treatment protocol. As of December 31, 2019, all patients in the expanded access studies have rolled over into a treatment protocol (2002C018G). A resubmission is a submission to a BLA that purports to answer all the deficiencies that need to be addressed.
We believe that the BLA resubmission will be designated as a Class 2 resubmission by FDA which would provide for a PDUFA goal for FDA review and action within six months of the receipt date of the BLA resubmission. If we resubmit the BLA in the third quarter of 2020, and if our resubmission is designated as a Class 2 resubmission, we expect a PDUFA date in the first quarter of 2021. If we receive regulatory approval on this timeline, we currently plan to launch Ryplazim ourselves in the United States in 2021 with a small, focused commercial infrastructure while exploring potential marketing collaborations and patient access programs for Ryplazim in selected non-U.S. markets in 2021 and 2022.
If our BLA for Ryplazim is approved by the FDA, we may be also eligible to receive a Pediatric Review Voucher, or PRV, from the FDA. If we receive regulatory approval on Ryplazim on the currently expected timeline, and if we receive a PRV for Ryplazim in a timely manner thereafter, we anticipate seeking to monetize any such PRV in 2021.
We also may explore clinical uses or formulations of plasminogen, including for the treatment of acquired plasminogen deficiencies and in critical care and wound treatment settings and also anticipate initiating additional clinical trials for Ryplazim in the second half of 2021. We believe that the expansion of our plasminogen development program may enable us to target additional clinical indications with unmet medical need.
Plasminogen for critical care indications associated with acquired plasminogen deficiencies
We plan to assess, subject to receiving positive results from pre-clinical research and development initiatives, the potential of Ryplazim, or other presentations or formulations of plasminogen, to address unmet medical needs associated with acute and acquired plasminogen deficiencies.
We have also presented preclinical data showing the benefits of plasminogen administration in reducing lung injury in an animal model of ALI/ARDS associated with acute pancreatitis. Acute lung injury, or ALI, and acute respiratory distress syndrome (ARDS) are life-threatening conditions resulting in respiratory failure in the critically ill patients, including COVID-19 patients.
Plasmin plays a significant role in regulating hemostasis through proteolytic degradation of fibrin. In preclinical studies using mice lacking the plasmin precursor plasminogen (Plg), we have identified additional plasmin functions in inflammation, cell migration, and extracellular matrix degradation that we believe have implications for a variety of pathologic processes. The fibrinolytic activity in bronchoalveolar lavage, or BAL, fluid from patients IPF has been found to be suppressed. The aim of these studies was to investigate the potential effects of plasminogen dosing in two models of ALI: the L-arginine induced, or LPS, model and the chronic bleomycin-induced lung fibrosis model.
Preclinical data suggest that supplementation of plasminogen may offer the potential as a novel therapy, alone or in combination with fezagepras, pirfenidone or nintedanib, for chronic treatment of IPF or in acute exacerbations of IPF.
In December 2017, we received an Orphan Drug Designation from the FDA for the use of plasminogen for the treatment of IPF.
We have also been reviewing some of our preclinical research on other uses of plasminogen and reviewing published data on the use of plasminogen as a potential treatment for patients affected with COVID-19. Any additional preclinical research or potential clinical studies to study plasminogen in COVID-19 infected patients would require additional evaluation and the support of government agencies, collaboration partners and funding agencies.
Plasminogen (sub-cutaneous) for hard-to-treat wounds
We are also exploring the potential to leverage the plasminogen active pharmaceutical ingredient, or API, as an injectable sub-cutaneous formulation to promote the healing of hard-to-treat wounds.
We have previously initiated clinical trials to evaluate Plasminogen (sub-cutaneous) administration in near topical wounds to determine its safety and ability to facilitate the complete healing of otherwise hard-to-treat wounds. Wounds are known to be difficult to heal in certain diabetic patients, and elevated blood sugar level has been shown to greatly reduce the activity of plasminogen. A clinical trial in patients with tympanic membrane perforations, or TMP, was initiated in Sweden in 2018.
This trial was a dose escalation, randomized, placebo-controlled trial designed to investigate the safety, feasibility and initial efficacy of local injections of a novel and proprietary plasminogen formulation for the treatment of chronic tympanic membrane perforation. Nine adult subjects have been enrolled on the completed low dose cohort of the clinical trial. The clinical trial is suspended as a result of a strategic change in focus and pending the development of a novel plasminogen formulation, if ever developed.
Manufacturing, Plasma Collection and Processing
We rely and expect to continue to rely for the foreseeable future, on third-party contract development manufacturing organizations, or CDMOs, to produce our small molecule product candidates for preclinical and clinical testing, as well as for commercial manufacture if our product candidates receive marketing approval. We require that our CDMOs produce bulk drug substances and finished drug products in accordance with current Good Manufacturing Practices, or cGMPs, and all other applicable laws and regulations. We maintain agreements with our manufacturers that include confidentiality and intellectual property provisions to protect our proprietary rights related to our product candidates.
We have engaged CDMOs to manufacture supply of certain of our product candidates for preclinical, clinical and commercial use. Additional CDMOs are used to fill finish, label, package and distribute drug product for preclinical and clinical use. We do not currently have arrangements in place for redundant supply. As our development programs expand and we build new process efficiencies, we expect to continually evaluate this strategy with the objective of satisfying demand for registration trials and, if approved, the manufacture, sale and distribution of commercial products.
Our wholly-owned subsidiary, Prometic Bioproduction Inc., or PBP, operates our plasma processing facility in Laval, Québec, Canada, where we manufacture Ryplazim in accordance with cGMPs to be used in our clinical trials and “compassionate use” programs. “Compassionate Use” is the use outside of a clinical trial of an investigational, or not approved, product candidate when patient enrollment in a clinical trial is not possible, typically due to patient ineligibility or a lack of ongoing clinical trials. We expect to use the Laval facility for commercial manufacture of Ryplazim in bulk drug substance form, if Ryplazim is approved. In May 2015, we entered into a Master Services Agreement with Emergent Biosolutions Inc., or Emergent, pursuant to which Emergent provides additional plasma manufacturing capabilities in Winnipeg, Manitoba, Canada, which we expect to rely on for a larger-scale version of the process implemented in the Laval facility. We continue to evaluate our production needs and plans to be able to support the commercial product launch of Ryplazim, if approved.
We also have a plasma collection center in Winnipeg, Manitoba, Canada, which is an FDA and Health Canada licensed, EMA compliant and International Quality Plasma Program certified plasma collection facility located in close proximity to Emergent’s Winnipeg-based cGMP-compliant manufacturing facility. We believe that the plasma collection center allows for strategic sourcing of plasma for the purification of Ryplazim using our plasma-protein purification platform. Our wholly-owned subsidiary, Prometic Plasma Resources Inc., or PPR, also sells specialty plasma and red blood cells to third parties in the ordinary course. PPR is responsible for supplying and
purchasing our plasma requirements. In 2019, PPR continued to focus on expanding its plasma donor base. In 2020, we filed a BLA with FDA for the approval of our U.S.-based plasma collection center, located in Amherst, New York and received a Clinical Laboratory License from the State of New York. The U.S. collection center is operated by our wholly-owned subsidiary, Prometic Plasma Resources (USA) Inc. We have also leveraged our plasma collection capabilities to commence collecting blood plasma from recovered COVID-19 patients to be potentially used in the manufacture of hyperimmune immunoglobulins by third parties for clinical trial purposes. In July 2020, we joined the CovIg-19 Plasma Alliance, a collaboration among some of the world’s leading plasma-derived therapeutic companies, to contribute to the acceleration of the development of a potential new plasma-derived therapy for COVID-19.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary rights. While we believe that our product candidates, technology, knowledge, experience and scientific resources provide us with competitive advantages, we compete in the highly competitive markets and face significant competition from many sources, including pharmaceutical and biotechnology companies, as well as academic institutions, governmental agencies and private and public research institutions.
We compete in the segments of the biotechnology, pharmaceutical and other related industries that develop and market therapies for the treatment of disorders associated with fibrosis. There are many other companies, including large biotechnology and pharmaceutical companies, that have commercialized and/or are developing therapies for the same therapeutic areas that our product candidates target. There are two approved products available for the treatment of IPF: Boehringer Ingelheim (Ofev / nintedanib) and Roche (Esbriet / pirfenidone). In addition, there are several product candidates in development for IPF including three product candidates already in, or expected to commence, Phase 3 clinical trials: FibroGen Inc. (pamrevlumab), Galapagos NV (GLPG1690), Promedior (recently acquired by Roche), PRM-151. Rhythm Pharmaceuticals, Inc. is developing a product candidate for the treatment of obesity related to Alström syndrome. There are numerous product candidates in development for the treatment of NASH, including one product that is currently under FDA regulatory review (Intercept Pharmaceuticals, Inc. (Ocaliva)), and product candidates that are being evaluated in Phase 3 clinical trials Allergan plc (cenicriviroc), Madrigal Pharmaceuticals, Inc. (resmetirom), Galmed Pharmaceuticals Ltd. (Aramchol), and Galectin Therapeutics Inc., (belapectin). Other PPAR agonists that are approved or in clinical development for the treatment of fibrotic indications include lanifibranor (Inventiva S.A.), Efruxifermin (Akero), pemafibrate (Kowa Pharmaceuticals) and seladelpar (Cymabay Therapeutics, Inc.).
There are currently no approved drugs for the treatment of congenital plasminogen deficiency. Disease management practices include surgical intervention and fresh frozen plasma administration, which have been shown to offer some relief. Third-party reports related to these disease management practices, however, have been part of single cases or small groups of patients, and no standardized treatment protocols or guidelines have been established.
Many of the companies against which we are competing or against which we may compete in the future, either alone or with their strategic collaborators, have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved drugs than we do. Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early-stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies or universities and research institutions. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and enrolling patients for our clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
We could see a reduction or elimination of our commercial opportunity if our competitors develop and commercialize products that are safer, more effective, have fewer or less severe side effects, are more convenient or are less expensive than any products that we may develop. The availability of reimbursement from government and other third-party payors will also significantly affect the pricing and competitiveness of our products. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for ours, which could result in our competitors establishing a strong market position before we are able to enter the market.
License, Manufacturing, Supply and Royalty Agreements
Adsorbent Supply Agreement-
We currently have an adsorbent supply agreement, dated June 18, 2019, with PBL (now Astrea Bioseparations Ltd) for the supply by PBL to us of affinity adsorbents used in the manufacturing activities for plasma-derived products, including our proprietary affinity adsorbents used in the manufacturing of Ryplazim. The price for the adsorbents was detailed in the agreement and is subject to increase in accordance with the agreement, which does not contain any minimum purchase requirements. The agreement has an initial ten-year term with subsequent five-year terms, unless not renewed by us or earlier terminated or not renewed by us in accordance with its terms.
Plasma Purchase Agreements-
We currently have a plasma purchase agreement, dated November 6, 2015, with Interstate Blood Bank, Inc., or IBBI, for the supply by IBBI to us of plasma used in the manufacturing of Ryplazim. The price for the plasma is detailed in the agreement and is subject to increase or decrease in accordance with the agreement. We have agreed to certain annual minimum purchase amounts. The agreement has an initial term until December 31, 2021 with subsequent three-year terms, unless earlier terminated by either party in accordance with its terms.
We also have a plasma purchase agreement, dated June 6, 2017, with Grifols Worldwide Operations Limited, or Grifols, for the supply by Grifols to us of plasma used in the manufacturing of Ryplazim. The price for the plasma is detailed in the agreement and is subject to increase in accordance with the agreement. We have agreed to certain annual minimum purchase amounts. The agreement has an initial term until December 31, 2022, with an option to mutually renew the agreement for additional one year terms, unless earlier terminated by either party in accordance with its terms.
Supply Agreements-
We currently have a master services agreement, or Emergent MSA, dated May 15, 2015, with Cangene Corporation d/b/a Emergent BioSolutions, or Emergent, for the provision of manufacturing services in quantities specified in statement of works. We have agreed to certain annual minimum commitment amounts. The Emergent MSA has an initial fifteen-year term, unless earlier terminated in accordance with its terms.
Royalty Stream Purchase Agreement with Structured Alpha-
In April 2018, we entered into a royalty stream purchase agreement with SALP, our lender under that certain loan agreement dated November 30, 2017. Pursuant to the royalty stream purchase agreement, SALP holds the right to receive a 2% royalty on net sales, licensing revenues, and joint venture revenues attributable to certain of our proprietary intellectual property rights, or royalty income. During a specified period, we have the right to repurchase the royalty income for the fair market value of SALP’s interest under the royalty stream purchase agreement, which is determined on a net present value basis by considering the remaining life of the associated patents (and any relevant extensions or additional exclusivity periods that may be associated with the associated patents or products) and associated cash flows, all as determined pursuant to a valuation performed by a mutually agreed upon third party. The agreement expires upon the earlier of the mutual agreement of the parties and the later of expiration on a country-by-country basis of the last applicable patent right and any applicable data or regulatory exclusivity in such country. The obligation under the royalty stream purchase agreement is secured by all of our assets until the expiry of the last patent anticipated in 2033. Either party may terminate the agreement upon an uncured material breach by the other party.
Assignment Agreement with Innovon-
In October 2001, we and our subsidiary Prometic BioSciences Inc. (now Liminal R&D BioSciences Inc., or LBRD) entered into an assignment agreement with Innovon Pharmaceuticals Inc., or Innovon, and Pierre Laurin pursuant to which Innovon and Pierre Laurin assigned to LBRD and us, all rights, title and interests in certain compounds.
On December 14, 2015, Innovon and Pierre Laurin consented to the assignment and transfer our worldwide intellectual property rights related to the assigned compounds, except for our Canadian rights, to our subsidiary Prometic Pharma SMT Limited (now known as Liminal BioSciences Limited).
In consideration for the assignment by Innovon and Pierre Laurin, we are obligated to pay Innovon a low single digit percentage of any consideration received by us in connection with a license or grant of rights to third parties with respect to such assigned compounds. Additionally, we are obligated to pay a less than one percent
royalty on net sales of such assigned compounds. At any time after a certain period, in the event that (i) we fail to develop or commercialize an assigned compound covered by the assignment agreement, as amended, for an unreasonable period of time, (ii) we elect to cease further development or commercialization of an assigned compound or (iii) of our insolvency, we are obligated to reassign the applicable compound to Innovon and Pierre Laurin subject to certain conditions, including payment by Innovon and Pierre Laurin of a consideration to be negotiated between the parties and payable in the form of royalties.
The assignment agreement expires, on an assigned compound-by-assigned compound basis, on the last sale of the applicable assigned compound, or upon the occurrence of any Event of Default, which includes, subject to applicable laws, insolvency, bankruptcy, an arrangement with creditors, a restructuring, protective administration or liquidation of the Company. Innovon and/or Pierre Laurin may also terminate the agreement upon our breach that remains uncured for a certain number of days.
Amended and Restated License Agreement with The Royal Institution for the Advancement of Learning/ McGill University and Florida Institute of Technology, Inc.
In July 2020, we entered into an amended and restated license agreement with The Royal Institution for the Advancement of Learning/McGill University and Florida Institute of Technology, Inc. (the “Licensors”) effective as of May 16, 2018, pursuant to which the Licensors granted us worldwide, exclusive royalty-bearing right to use and practice certain licensed patents and know-how, with the exclusive right to develop, modify, adapt, improve and customize certain licensed products, and the right to make, have made, commercialize, exploit, reproduce, market, sell, rent, distribute, lease or otherwise transfer, import and export certain licensed products, contingent upon our material compliance with the terms of the Amended and Restated License Agreement. Under the Amended and Restated License Agreement, we are obliged to pay a low single digit royalty to the Licensors based on a percentage of net sales of the licensed products. The Amended and Restated License Agreement terminated upon the last to expire, or become abandoned, licensed patent(s), whether by statute or otherwise, unless it earlier terminates by operation of law or by acts of the parties in accordance with the terms of the Amended and Restated License Agreement.
License Agreement and Contract Manufacturing Agreement with Hematech-
In May 2012, we entered into an amended and restated license agreement with Hematech Biotherapeutics Inc., or Hematech, pursuant to which we granted Hematech an exclusive license under certain of our technology to develop, manufacture and commercialize plasma-derived products in Taiwan and a co-exclusive license under such technology to develop, manufacture and commercialize plasminogen products anywhere in the world other than China as well as a contract manufacturing agreement pursuant to which we have the exclusive right to obtain supply of plasma-derived products from Hematech for use outside of Taiwan. Currently, Hematech is not manufacturing plasma-derived products. Subsequently, in December 2016, we entered into an amendment to the license agreement pursuant to which we re-obtained our exclusive rights to the plasminogen products around the world.
Under the license agreement, we are entitled to receive a low single digit royalty on net sales of plasma-derived products in Taiwan. In connection with our re-obtaining rights to our plasminogen products we issued to Hematech 1,683 common shares and are obligated to pay a mid-single digit royalty on net sales of plasminogen products for congenital deficiency up to December 16, 2020 and subject to both a minimum and cap in the low seven figures.
The license agreement, unless terminated earlier, expires upon the latest to occur of (a) the last patent to expire in Taiwan, (b) thirty years from execution and (c) the date on which Hematech is no longer commercializing a plasma-derived product. Either party may terminate the license agreement upon the insolvency of the other party or an uncured material breach of the license agreement by the other party. Additionally, Hematech has the right to terminate the license agreement for any reason upon prior specified notice. The contract manufacturing agreement expires on the expiration or termination of the license agreement. The contract manufacturing agreement may be terminated upon the mutual agreement of the parties, by either party upon an uncured material breach of the contract manufacturing agreement by the other party, and by Hematech in the event that a governmental entity enjoins the further operation of the production facility or the development, manufacturing, distribution or sale of any product in Taiwan.
Intellectual Property
We own and control the intellectual property utilized in the vast majority of our technologies, products and potential product candidates, giving us the option to develop and eventually commercialize these products in various geographies, to develop new formulations, and to select CDMOs and CROs of our choice. Our intellectual property rights include our trademarks, patents and patent applications, regulatory dossiers, manufacturing, and process know-how. Our intellectual property portfolio has been built in large part from in-house technology
and product research and development over the past 20 years as well as strategic relationships and joint ventures with reputable partners such as American National Red Cross.
Our approach regarding our intellectual property portfolio is to file and/or license patents and patent applications as appropriate and to seek to obtain patent protection in at least the major pharmaceutical markets, including the United States, major European countries and Japan. We also rely on trade secrets, proprietary unpatented information, trademarks, and contractual arrangements to protect our technology and enhance our competitive position. We currently have a patent estate comprised of owned and in-licensed patents and patent applications. Our patent portfolio includes patents and patent applications claiming compounds, pharmaceutical compositions, nutraceuticals, processes, and methods for treating diseases, disorders, or conditions.
Fezagepras
Our fezagepras program is covered by a patent portfolio that we wholly own and that is comprised of issued patents, as well as allowed and pending patent applications, related to fezagepras and related derivative compounds, salt forms and formulations of the same, and methods of making and using the same therapeutically. The portfolio is comprised of patent families granted in Canada, United States, Europe, China, Japan, Russia and other countries, including granted U.S. patents with claims directed to the fezagepras compound and its use in treating various diseases including inflammatory diseases and renal disorders. These patents are expected to expire no sooner than 2030, absent any patent term adjustments or extensions. We also own a patent family with claims directed to methods of treating pulmonary fibrosis (including idiopathic pulmonary fibrosis, or IPF), liver fibrosis, cardiac fibrosis or skin fibrosis with fezagepras or related derivatives. Patents in this family have been granted in Canada, United States, Europe, China, Japan, Russia and other countries and are expected to expire no sooner than 2034, absent any patent term adjustments or extensions.
Plasminogen or Ryplazim
Our Ryplazim program is covered by a wholly-owned patent portfolio comprised of an issued U.S. patent, as well as allowed and pending patent applications, related to the Ryplazim clinical formulation and methods of use. Specifically, one patent family has been granted in the United States, and is pending in Canada, United States, Europe, China, Japan, Russia and other countries with claims directed to the current formulation. This patent and any applications that grant from these applications are expected to expire in 2035, absent any patent term adjustments or extensions. A second patent family is pending in Canada, United States, Europe, China, Japan, Russia and other countries with claims directed to the use of plasminogen in maintaining optimal plasminogen levels in plasminogen-deficient patients. Any patents that may be granted from these patent applications are expected to expire in 2036, absent any patent term adjustments or extensions. We also have wholly-owned pending patent applications related to the use of plasminogen to treat conditions associated with PAI-1 overexpression which are expected to expire in 2038, in-licensed and wholly-owned issued patents and pending patent applications related to the use of plasminogen, including related dosing regimen to treat certain types of wounds, the latter which are expected to expire in 2036.
Our Trademark Portfolio
RYPLAZIM is our registered trademark, in the United States, Canada, Europe, Japan and Turkey used in association with plasminogen for the treatment of plasminogen congenital deficiency in those countries, if approval is received from the relevant regulatory authorities.
LIMINAL BIOSCIENCES is our trademark currently pending registration in Canada and in other countries. This mark is our name and is used in association with several goods and services, pharmaceutical research and development, and pharmaceutical preparations and compositions being developed for the treatment of fibrosis and other indications.
We also have unregistered trademark rights in certain countries in which we operate, where trademark rights arise from use, rather than registration.
Other Intellectual Property Portfolio
Our portfolio of intellectual property contains additional trademarks, pending trademark registrations and domain names associated with our trademarks and pending trademark applications.
Our Policy on Intellectual Property
Our intellectual property practice is to keep all information relating to proprietary compounds, inventions, improvements, trade secrets, know-how and continuing technological innovation confidential and, where practicable, file patent and trademark applications. In particular, as part of our intellectual property protection practice, we, where we deem practicable and commercially reasonable:
| • | file patent applications for any new and patentable invention, development or improvement in the United States and in other countries; |
| • | prosecute pending patent applications in conformity with applicable patent laws and in a manner that covers our activities; |
| • | file trademark applications in countries of interest for our trademarks |
| • | register domain names whose addresses include our trademark names; and |
| • | maintain our intellectual property rights by paying government fees as may be necessary to ensure such rights remain in force. |
Government Regulation
In the United States, pharmaceutical and biological products are subject to extensive regulation by the FDA under the Federal Food, Drug, and Cosmetic Act, or FDCA, and the Public Health Service Act, or PHSA. The FDCA, PHSA, and other federal and state statutes and regulations, govern, among other things, the research, development, testing, manufacture, storage, recordkeeping, approval, labeling, promotion and marketing, distribution, post-approval monitoring and reporting, sampling and import and export of pharmaceutical products. Failure to comply with applicable U.S. requirements may subject a company to a variety of administrative or judicial sanctions, such as FDA refusal to approve pending BLAs, warning or untitled letters, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties and criminal prosecution.
We cannot market a drug or biological product in the United States until the product candidate has received FDA approval. The steps required before a new drug or biologic may be marketed in the United States generally include the following:
| • | completion of extensive nonclinical laboratory tests, animal studies, and formulation studies in accordance with the FDA's Good Laboratory Practice, or GLP, regulations; |
| • | submission to the FDA of an Investigational New Drug, or IND, for human clinical testing, which must become effective before human clinical trials may begin; |
| • | performance of adequate and well-controlled human clinical trials in accordance with Good Clinical Practice, or GCP, requirements to establish the safety and efficacy of the product for each proposed indication; |
| • | submission to the FDA of a BLA, in the case of biological product candidates or a New Drug Application, or NDA in the case of small molecule drug candidates, after completion of all pivotal clinical trials; |
| • | satisfactory completion of an FDA inspection of sites involved in our clinical trials; |
| • | satisfactory completion of an FDA pre-approval inspection of the manufacturing facility or facilities at which the active pharmaceutical ingredient, or API, and finished product are produced and tested to assess compliance with cGMPs; and |
| • | FDA review and approval of the BLA prior to any commercial marketing or sale of the product in the United States. |
Satisfaction of FDA pre-market approval requirements typically takes many years and the actual time required may vary substantially based upon the type, complexity and novelty of the product or disease.
Nonclinical tests include laboratory evaluation of product chemistry, formulation and toxicity, as well as animal trials to assess the characteristics and potential safety and efficacy of the product. The conduct of the nonclinical tests must comply with federal regulations and requirements, including GLP regulations. The results of nonclinical testing are submitted to the FDA as part of an IND along with other information, including information about product chemistry, manufacturing and controls and a proposed clinical trial protocol. Long-term nonclinical tests, such as animal tests of reproductive toxicity and carcinogenicity, may continue after the IND is submitted.
A 30-day waiting period after the submission of each IND is required prior to the commencement of clinical testing in humans. If the FDA has neither commented on nor questioned the IND within this 30-day period, the clinical trial proposed in the IND may begin. If the FDA raises concerns or questions about the conduct of the trial, such as whether human research subjects will be exposed to an unreasonable health risk, the IND sponsor and the FDA must resolve any outstanding FDA concerns or questions before clinical trials can proceed.
Clinical trials involve the administration of the investigational new biologic to healthy volunteers or patients under the supervision of a qualified investigator. Clinical trials must be conducted in compliance with federal regulations, including GCP requirements, as well as under protocols detailing the objectives of the trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. Each protocol and subsequent protocol amendments must be submitted to the FDA as part of the IND.
The FDA may order the temporary, or permanent, discontinuation of a clinical trial at any time, or impose other sanctions, if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. The study protocol and informed consent information for patients in clinical trials must also be submitted to an institutional review board, or IRB, for approval at each site at which the clinical trial will be conducted. An IRB may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB's requirements, or may impose other conditions.
U.S. Biopharmaceutical Products Development Process
Before testing any product candidate, the product candidate enters the nonclinical testing stage. Nonclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies to assess the potential safety and activity of the product candidate. The conduct of the nonclinical tests must comply with federal regulations and requirements including GLPs.
The clinical study sponsor must submit the results of the nonclinical tests, together with manufacturing information, analytical data, any available clinical data or literature and a proposed clinical protocol, to the FDA as part of the IND. Some nonclinical testing typically continues after the IND is submitted. An IND is an exemption from the FDCA that allows an unapproved product to be shipped in interstate commerce for use in an investigational clinical trial and a request for FDA authorization to administer an investigational product to humans. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA requests certain changes to a protocol before the study can begin, or the FDA places the clinical study on a clinical hold within that 30-day time period. The FDA may also impose clinical holds on a product candidate at any time before or during clinical trials due to safety concerns or non-compliance. If the FDA imposes a clinical hold, studies may not recommence without FDA authorization and then only under terms authorized by the FDA. Accordingly, we cannot be sure that submission of an IND will result in the FDA allowing clinical trials to begin, or that, once begun, issues will not arise that suspend or terminate such studies.
Clinical trials involve the administration of the product candidate to healthy volunteers or subjects under the supervision of qualified investigators, generally physicians not employed by or under the study sponsor’s control. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical study, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety, including stopping rules that assure a clinical study will be stopped if certain adverse events should occur. Each protocol and any amendments to the protocol must be submitted to the FDA as part of the IND. Clinical trials must be conducted and monitored in accordance with the FDA’s regulations comprising the GCP requirements, including the requirement that all research subjects provide informed consent. Further, each clinical study must be reviewed and approved by an independent IRB, at or servicing each institution at which the clinical study will be conducted. An IRB is charged with protecting the welfare and rights of study participants and considers such items as whether the risks to individuals participating in the clinical trials are minimized and are reasonable in relation to anticipated benefits. The IRB also approves the form and content of the informed consent that must be signed by each clinical study subject or his or her legal representative and must monitor the clinical study until completed. Additionally, some trials are overseen by an independent group of qualified experts organized by the trial sponsor, known as a data safety monitoring board or committee.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| • | Phase 1. The product is initially introduced into healthy human subjects and tested for safety. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. |
| • | Phase 2. The product is evaluated in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance, optimal dosage and dosing schedule. |
| • | Phase 3. Clinical trials are undertaken to further evaluate dosage, clinical efficacy, potency and safety in an expanded patient population at geographically dispersed clinical trial sites. These clinical trials are |
| | intended to establish the overall risk/benefit ratio of the product and provide an adequate basis for product labeling. |
Post-approval clinical trials, sometimes referred to as Phase 4 clinical trials, may be conducted after initial marketing approval. These clinical trials are used to gain additional experience from the treatment of patients in the intended therapeutic indication, particularly for long-term safety follow-up.
During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data, and clinical trial investigators. Annual progress reports detailing the results of the clinical trials must be submitted to the FDA. Written IND safety reports must be promptly submitted to the FDA and the investigators for serious and unexpected adverse events, any findings from other studies, tests in laboratory animals or in vitro testing that suggest a significant risk for human subjects, or any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigator brochure. The sponsor must submit an IND safety report within 15 calendar days after the sponsor determines that the information qualifies for reporting. The sponsor also must notify the FDA of any unexpected fatal or life-threatening suspected adverse reaction within seven calendar days after the sponsor’s initial receipt of the information. Phase 1, Phase 2 and Phase 3 clinical trials may not be completed successfully within any specified period, if at all. The FDA or the sponsor or its data safety monitoring board may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the biological product has been associated with unexpected serious harm to patients.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the characteristics of the product as well as finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. To help reduce the risk of the introduction of adventitious agents with use of biological products, the PHSA emphasizes the importance of manufacturing control for products whose attributes cannot be precisely defined. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the sponsor must develop methods for testing the identity, strength, quality, potency and purity of the final biological product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
After the completion of clinical trials, FDA approval of a BLA or NDA must be obtained before commercial marketing of the product. The BLA or NDA must include results of product development, laboratory and animal studies, human studies, information on the manufacture and composition of the product, proposed labeling and other relevant information. In addition, under the Pediatric Research Equity Act, or PREA, a BLA or NDA or supplement must contain data to assess the safety and effectiveness of the product for the claimed indications in all relevant pediatric subpopulations and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may grant deferrals for submission of data or full or partial waivers. Unless otherwise required by regulation, PREA does not apply to any biological product for an indication for which orphan designation has been granted. The testing and approval processes require substantial time and effort and there can be no assurance that the FDA will accept the BLA for filing and, even if filed, that any approval will be granted on a timely basis, if at all.
Under the Prescription Drug User Fee Act, as amended, or PDUFA, each BLA or NDA must be accompanied by a significant user fee. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on BLAs or NDAs for product candidates designated as orphan drugs, unless the product candidate also includes a non-orphan indication.
Within 60 days following submission of the application, the FDA reviews the application submitted to determine if it is substantially complete before the agency accepts it for filing. The FDA may refuse to file any application that it deems incomplete or not properly reviewable at the time of submission and may request additional information. In this event, the BLA or NDA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review to determine, among other things, whether the proposed product is safe, potent, and effective, for its intended use, and has an acceptable purity profile, and whether the product is being manufactured in accordance with cGMP to assure and preserve the product’s identity, safety, strength, quality, potency and purity. The FDA may refer applications for novel products that present difficult questions of safety or efficacy to an advisory committee, typically a panel that includes clinicians and other experts, for review, evaluation and a recommendation as to whether the application should be approved
and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions. FDA also will determine whether a Risk Evaluation and Mitigation Strategy, or REMS, is necessary to assure the safe use of the product. If the FDA concludes a REMS is needed, the sponsor must submit a proposed REMS; the FDA will not approve the application without a REMS, if required.
The FDA also offers a number of expedited development and review programs for qualifying product candidates. Priority review designation for BLA is assigned by FDA, if at all, at the time of acceptance of a BLA for a product that treats a serious or life-threatening condition and, if approved, would be a significant improvement in safety or effectiveness relative to available therapies.
Before approving an application, the FDA will inspect the facilities at which the product is manufactured. The FDA will not approve the product unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, the FDA will typically inspect one or more clinical trial sites to assure that the clinical trials were conducted in compliance with IND study requirements and GCP requirements. To assure cGMP and GCP compliance, an applicant must incur significant expenditure of time, money and effort in the areas of training, record keeping, production, and quality control.
Notwithstanding the submission of relevant data and information, the FDA may ultimately decide that the BLA or NDA does not satisfy its regulatory criteria for approval and deny approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. If the agency decides not to approve the BLA or NDA in its present form, the FDA will issue a complete response letter that usually describes all of the specific deficiencies identified by the FDA. The deficiencies identified may be minor, for example, requiring labeling changes, or major, for example, requiring additional clinical trials. Additionally, the complete response letter may include recommended actions that the applicant might take to place the application in a condition for approval. If a complete response letter is issued, the applicant may either resubmit, addressing all of the deficiencies identified in the letter, or withdraw the application.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. The FDA may impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a risk management plan, or otherwise limit the scope of any approval. In addition, the FDA may require post marketing clinical trials, sometimes referred to as Phase 4 clinical trials, designed to further assess a biological product’s safety and effectiveness, and testing and surveillance programs to monitor the safety of approved products that have been commercialized. As a condition for approval, the FDA may also require additional nonclinical testing as a Phase 4 commitment.
One of the performance goals agreed to by the FDA under the PDUFA is to review standard BLAs and NDAs in ten months from filing and priority BLAs and NDAs in six months from filing, whereupon a review decision is to be made. The FDA does not always meet its PDUFA goal dates and its review goals are subject to change from time to time. The review process and the PDUFA goal date may be extended by three months if the FDA requests or the sponsor otherwise provides additional information or clarification regarding information already provided in the submission within the last three months before the PDUFA goal date.
Maintaining substantial compliance with applicable federal, state, and local statutes and regulations requires the expenditure of substantial time and financial resources. Rigorous and extensive FDA regulation of biopharmaceutical products continues after approval, particularly with respect to cGMP. Manufacturers of our products are required to comply with applicable requirements in the cGMP regulations, including quality control and quality assurance and maintenance of records and documentation. Following approval, the manufacturing facilities are subject to biennial inspections and such inspections may result in an issuance of FDA Form 483 deficiency observations or a warning letter, which can lead to plant shutdown and other more serious penalties and fines. Prior to the institution of any manufacturing changes, a determination needs to be made whether FDA approval is required in advance. If not done in accordance with FDA expectations, the FDA may restrict supply and may take further action. Annual product reports are required to be submitted annually. Other post-approval requirements applicable to biological products, include reporting of cGMP deviations that may affect the identity, potency, purity and overall safety of a distributed product, record-keeping requirements, reporting of adverse effects, reporting updated safety and efficacy information, and complying with electronic record and signature requirements. After a BLA is approved, the product also may be subject to official lot release. As part of the manufacturing process, the manufacturer is required to perform certain tests on each lot of the product before it is released for distribution. If the product is subject to official release by the FDA, the manufacturer
submits samples of each lot of product to the FDA together with a release protocol showing a summary of the history of manufacture of the lot and the results of all of the manufacturer’s tests performed on the lot.
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant orphan designation, or ODD, to a drug or biologic intended to treat a rare disease or condition, defined as a disease or condition with either a patient population of fewer than 200,000 individuals in the United States, or a patient population greater of than 200,000 individuals in the United States when there is no reasonable expectation that the cost of developing and making available the drug or biologic in the United States will be recovered from sales in the United States for that drug or biologic. ODD must be requested before submitting a BLA or NDA. After the FDA grants ODD, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA.
If a product that has received ODD and subsequently receives the first FDA approval for a particular active ingredient for the disease for which it has such designation, the product is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications, including a full BLA or NDA, to market the same biologic or drug for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with orphan drug exclusivity or if FDA finds that the holder of the orphan drug exclusivity has not shown that it can assure the availability of sufficient quantities of the orphan drug to meet the needs of patients with the disease or condition for which the drug was designated. Orphan drug exclusivity does not prevent the FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition. Among the other benefits of ODD are tax credits for certain research and a waiver of the application user fee.
A designated orphan drug many not receive orphan drug exclusivity if it is approved for a use that is broader than the indication for which it received ODD. In addition, orphan drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition.
Rare Pediatric Disease Designation
The Rare Pediatric Disease Priority Review Voucher Program, or the PRV Program, is intended to incentivize pharmaceutical companies to develop drugs for rare pediatric diseases. A company that obtains approval of an NDA or a BLA for a designated rare pediatric disease may be eligible for a PRV from the FDA, which may be redeemed to obtain priority review for a subsequent NDA or BLA by the owner of such PRV. A PRV is fully transferable and can be sold to any company, who in turn can redeem the PRV for priority review of a marketing application in six months, compared to the standard timeframe of approximately ten months. A drug that receives a RPDD before October 1, 2020 continues to be eligible for a PRV if the drug is approved before October 1, 2022. Extension beyond these dates will require further Congressional action.
Other Healthcare Laws and Compliance Requirements
Pharmaceutical companies are subject to additional healthcare regulation and enforcement by the federal government and by authorities in the states and foreign jurisdictions in which they conduct their business. Such laws include, without limitation: the U.S. federal Anti-Kickback Statute, which prohibits, among other things, persons and entities from knowingly and willfully soliciting, receiving, offering or paying remuneration, to induce, or in return for, either the referral of an individual, or the purchase or recommendation of an item or service for which payment may be made under any federal healthcare program; federal civil and criminal false claims laws, including the civil False Claims Act, and civil monetary penalty laws, which prohibit, among other things, individuals or entities from knowingly presenting, or causing to be presented, claims for payment to the federal government, including federal healthcare programs, that are false or fraudulent; the federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, which created additional federal criminal statutes which prohibit, among other things, executing a scheme to defraud any healthcare benefit program and making false statements relating to healthcare matters, and which, as amended by the Health Information for Economic and Clinical Health Act of 2009, or HITECH, also imposes certain requirements on HIPAA covered entities and their business associates relating to the privacy, security and transmission of individually identifiable health information; the U.S. federal Physician Payments Sunshine Act, which requires certain manufacturers of drugs, devices, biologics and medical supplies for which payment is available under Medicare, Medicaid or the Children’s Health Insurance Program, with specific exceptions, to annually report to the federal government, information related to payments or other transfers of value made to physicians, as defined by such law, and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members; and U.S. state and foreign law equivalents of each of the above federal laws, which, in some cases, differ from each other in significant ways, and may not have the same effect, thus complicating compliance efforts. If their operations are found to be in violation of any of such laws or any other governmental regulations that apply, they may be subject to significant penalties, including, without limitation, civil, criminal and administrative penalties, damages, fines, exclusion from government-funded healthcare programs, such as Medicare and Medicaid or similar programs in other countries or jurisdictions, integrity oversight and reporting
obligations to resolve allegations of non-compliance, disgorgement, imprisonment, contractual damages, reputational harm, diminished profits and the curtailment or restructuring of our operations.
Coverage and Reimbursement
Significant uncertainty exists as to the coverage and reimbursement status of any pharmaceutical or biological product for which we obtain regulatory approval. Sales of any product depend, in part, on the extent to which such product will be covered by third-party payors, such as federal, state, and foreign government healthcare programs, commercial insurance and managed healthcare organizations, and the level of reimbursement for such product by third-party payors. Decisions regarding the extent of coverage and amount of reimbursement to be provided are made on a plan-by-plan basis. As there is no uniform policy of coverage and reimbursement for drug products among third‑party payors in the United States, coverage and reimbursement policies for drug products can differ significantly from payor to payor. There may be significant delays in obtaining coverage and reimbursement as the process of determining coverage and reimbursement is often time‑consuming and costly which will require us to provide scientific and clinical support for the use of our products to each payor separately, with no assurance that coverage or adequate reimbursement will be obtained. It is difficult to predict at this time what government authorities and third‑party payors will decide with respect to coverage and reimbursement for our drug products. For products administered under the supervision of a physician, obtaining coverage and adequate reimbursement may be particularly difficult because of the higher prices often associated with such drugs. Additionally, separate reimbursement for the product itself or the treatment or procedure in which the product is used may not be available, which may impact physician utilization.
In addition, the U.S. government, state legislatures and foreign governments have continued implementing cost-containment programs, including price controls, restrictions on coverage and reimbursement and requirements for substitution of generic products. Third-party payors are increasingly challenging the prices charged for medical products and services, examining the medical necessity and reviewing the cost effectiveness of pharmaceutical or biological products, medical devices and medical services, in addition to questioning safety and efficacy. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit sales of any product. Decreases in third-party reimbursement for any product or a decision by a third-party payor not to cover a product could reduce physician usage and patient demand for the product.
Healthcare Reform
The United States and some foreign jurisdictions are considering or have enacted a number of reform proposals to change the healthcare system. There is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by federal and state legislative initiatives, including those designed to limit the pricing, coverage, and reimbursement of pharmaceutical and biopharmaceutical products, especially under government-funded health care programs, and increased governmental control of drug pricing.
In March 2010, the Physician Payments Sunshine Act, enacted as part of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or, collectively, ACA, was signed into law, which substantially changed the way healthcare is financed by both governmental and private insurers in the United States, and significantly affected the pharmaceutical industry. The ACA contains a number of other provisions of particular import to the pharmaceutical and biotechnology industries, including, but not limited to, those governing enrollment in federal healthcare programs, a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs that are inhaled, infused, instilled, implanted or injected, and annual fees based on pharmaceutical companies’ share of sales to federal health care programs. Since its enactment, there have been judicial, Congressional, and executive branch challenges to certain aspects of the ACA, and we expect there will be additional challenges and amendments to the ACA in the future. For example, the 2020 federal spending package permanently eliminated, effective January 1, 2020, the ACA-mandated “Cadillac” tax on high-cost employer-sponsored health coverage and medical device tax and, effective January 1, 2021, also eliminates the health insurer tax. In addition, the Tax Act was enacted, which, among other things, removes penalties for not complying with ACA’s individual mandate to carry health insurance. Since the enactment of the Tax Act, there have been additional amendments to certain provisions of the ACA. On December 14, 2018, a U.S. District Court Judge in the Northern District of Texas, ruled that the individual mandate is a critical and inseverable feature of the ACA, and therefore, because it was repealed as part of the Tax Act, the remaining provisions of the ACA are invalid as well. Additionally, on December 18, 2019, the U.S. Court of Appeals for the 5th Circuit upheld the District Court ruling that that the individual mandate was unconstitutional and remanded the case back to the District Court to determine whether the remaining provisions of the ACA are invalid as well. On March 2, 2020, the United States Supreme Court
granted the petitions for writs of certiorari to review this case, and has allotted one hour for oral arguments, which are expected to occur in the fall. It is unclear how such litigation and other efforts to repeal and replace the ACA will impact the ACA.
Other legislative changes have been proposed and adopted since the ACA was enacted, including aggregate reductions of Medicare payments to providers of 2% per fiscal year and reduced payments to several types of Medicare providers, which will remain in effect through 2030 unless additional Congressional action is taken. The Coronavirus Aid, Relief and Economic Security Act, or CARES Act, which was signed into law in March 2020 and is designed to provide financial support and resources to individuals and businesses affected by the COVID-19 pandemic, suspended the 2% Medicare sequester from May 1, 2020 through December 31, 2020, and extended the sequester by one year, through 2030. Moreover, there has recently been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. At the federal level, the Trump administration’s budget proposal for fiscal year 2021 includes a $135 billion allowance to support legislative proposals seeking to reduce drug prices, increase competition, lower out-of-pocket drug costs for patients, and increase patient access to lower-cost generic and biosimilar drugs. On March 10, 2020, the Trump administration sent “principles” for drug pricing to Congress, calling for legislation that would, among other things, cap Medicare Part D beneficiary out-of-pocket pharmacy expenses, provide an option to cap Medicare Part D beneficiary monthly out-of-pocket expenses, and place limits on pharmaceutical price increases. Further, the Trump administration previously released a “Blueprint”, or plan, to lower drug prices and reduce out of pocket costs of drugs that contained proposals to increase drug manufacturer competition, increase the negotiating power of certain federal healthcare programs, incentivize manufacturers to lower the list price of their products, and reduce the out of pocket costs of drug products paid by consumers. The Department of Health and Human Services, or HHS, has solicited feedback on some of these measures and has implemented others under its existing authority. For example, in May 2019, the Centers for Medicare & Medicaid Services, or CMS, issued a final rule to allow Medicare Advantage plans the option to use step therapy for Part B drugs beginning January 1, 2020.This final rule codified CMS’s policy change that was effective January 1, 2019. While some of measures may require additional authorization to become effective, Congress and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs. At the state level, legislatures have increasingly passed legislation and implemented regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. In addition, it is possible that additional governmental action is taken to address the COVID-19 pandemic. For example, the Coronavirus Aid, Relief and Economic Security Act, or CARES Act, which was signed into law in March 2020 and is designed to provide financial support and resources to individuals and businesses affected by the COVID-19 pandemic, suspended the 2% Medicare reduction payment sequester from May 1, 2020 through December 31, 2020, and extended the sequester by one year, through 2030. In addition, on April 18, 2020, CMS announced that qualified health plan issuers under the ACA may suspend activities related to the collection and reporting of quality data that would have otherwise been reported between May and June 2020 given the challenges healthcare providers are facing responding to the COVID-19 virus.
Product Development
We have made significant investments over the last twenty years in the development of our proprietary technologies, our small molecule and plasma-derived therapeutics platforms and the product candidates arising therefrom. These investments and in-house development strategy have allowed us to be flexible in our approaches and adaptive when needed as well as retaining control over intellectual property rights and the potential commercial upside thereon. Furthermore, it allows us to develop the necessary skill sets internally on both the development of manufacturing processes as well as product development (preclinical and clinical) in various disease indications. Notwithstanding the foregoing, we believe that it is important to have a balance between in-house product development and outsourcing same or partnering such activities. Finally, pursuing the development and commercialization phase in partnership with other companies (especially for specific indications and/or geographic regions) is also interesting for us because it provides continuous external validation of our technology and possibilities of short and long term revenues from fees collected at the initiation of the partnership as well as via milestones payments and royalty streams.
Environmental Protection
We produce a certain amount of chemical waste in our R&D and manufacturing activities that is managed in accordance with applicable environmental protection standards by companies that specialize in hazardous waste
management. Our research laboratories generate hazardous waste that is also removed by companies that specialize in hazardous waste management, in accordance with strict internal procedures and applicable regulatory requirements. Compliance with such requirements is not expected to have a significant effect on our competitive position.
Employees
As of June 30, 2020, we had 277 full-time employees, as broken out by function in the table below. None of our employees are represented by collective bargaining agreements. We believe that we maintain good relations with our employees.
| As of
June 30, 2020 |
Function: | |
Research and development | |
| Small molecule therapeutics segment | 32 |
| Plasma-derived therapeutics segment | 149 |
Plasma collection centers | 38 |
General and administrative | 58 |
Total | 277 |
Facilities
We lease our principal executive offices, operational offices, manufacturing plant(s), plasma collection centers and laboratory space, which consists of approximately 9,110 square meters, in Laval, Québec, Canada; Sawston, United Kingdom; Winnipeg, Manitoba, Canada; Rockville, Maryland and Amherst, New York, United States. The lease for our principal executive office in Laval expires on October 31, 2025. The lease for our office in Cambridge, United Kingdom expires on November 12, 2024. The lease for our plasma collection center in Winnipeg expires on February 28, 2023, while the lease for our plasma collection center in Amherst, New York expires on December 1, 2033. The leases for our manufacturing plant and laboratory space, both located in Laval, both expire on October 31, 2025. We own two manufacturing facilities acquired as part of our acquisition of Telesta Therapeutics Inc. in October 2016, located in Pointe-Claire, Quebec, Canada and in Belleville, Ontario, Canada, which are currently not in operation. We believe our current facilities are sufficient to meet our needs. If we need to add new facilities or expand existing facilities as we add employees, we believe that suitable additional space will be available to accommodate any such expansion of our operations.
Legal Proceedings
From time to time, we may become involved in legal proceedings or be subject to claims arising in the ordinary course of our business. Although the results of litigation and claims cannot be predicted with certainty, we currently believe that the final outcome of these ordinary course matters will not have a material adverse effect on our business. Regardless of the outcome, litigation can have an adverse impact on us because of defense and settlement costs, diversion of management resources and other factors.
Liminal Former Employee Dispute
On June 6, 2019, a former employee filed suit and the case is now pending in the United States District Court, District of Connecticut and alleges claims for declaratory judgment, breach of contract, breach of the covenant of good faith and fair dealing, unjust enrichment, and violation of Conn. Gen. Stat. § 31-72 for unpaid wages and benefits, based upon Plaintiff’s Employment Contract and the terms of employee benefit programs. The case has been resolved in principle and the parties are working to finalize the appropriate documentation.