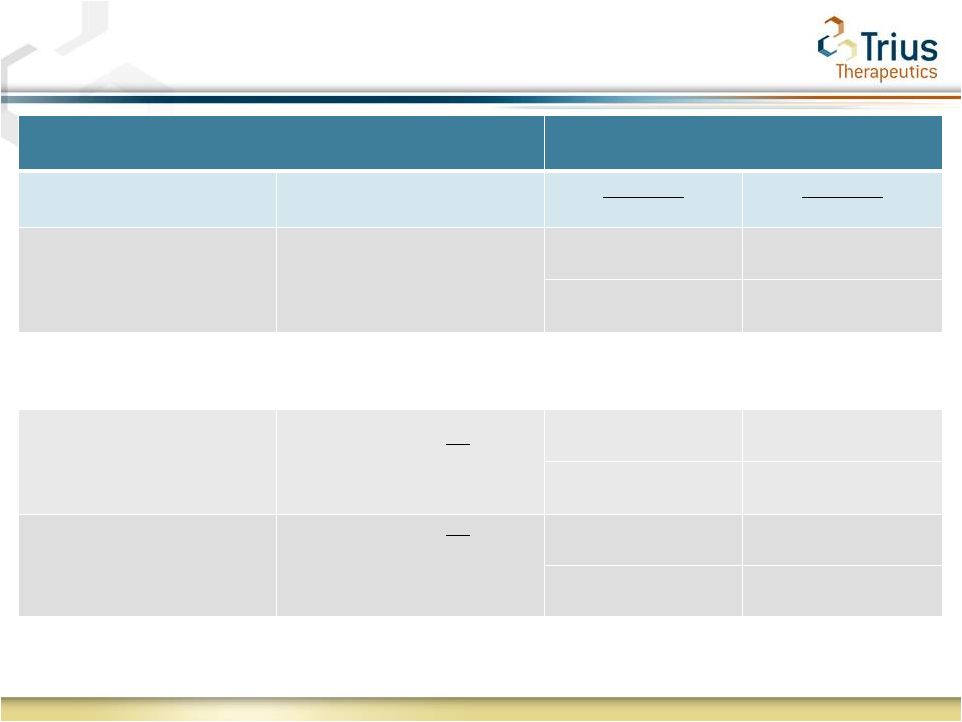
2 Forward Looking Statements Statements made in this presentation regarding matters that are not historical facts are “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995. Because such statements are subject to risks and uncertainties, actual results may differ materially from those expressed or implied by such forward-looking statements, and the results of the 112 study are not necessarily indicative of the results of the 113 study. Such statements include, but are not limited to, statements regarding Trius’ ability to successfully complete its ongoing clinical trials and development programs and the expected timing for reporting of top-line data for the TR701-113 study. Risks that contribute to the uncertain nature of the forward-looking statements include: the success and timing of Trius’ preclinical studies and clinical trials; regulatory developments in the United States and foreign countries; changes in Trius’ plans to develop and commercialize its product candidates; the outcome of final analyses of data from recently-completed clinical trials of tedizolid may vary from Trius’ initial analyses and the FDA may not agree with Trius’ interpretation of such results; additional ongoing or planned clinical trials of tedizolid may produce negative or inconclusive results; Trius may decide, or the FDA may require Trius, to conduct additional clinical trials or to modify Trius’ ongoing clinical trials; Trius may experience delays in the commencement, enrollment, completion or analysis of clinical testing for its product candidates, or significant issues regarding the adequacy of its clinical trial designs or the execution of its clinical trials, which could result in increased costs and delays, or limit Trius’ ability to obtain regulatory approval; the third parties with whom Trius has partnered with for the development of tedizolid and upon whom Trius relies to conduct its clinical trials and manufacture its product candidates may not perform as expected; tedizolid may not receive regulatory approval or be successfully commercialized; unexpected adverse side effects or inadequate therapeutic efficacy of tedizolid could delay or prevent regulatory approval or commercialization; Trius’ ability to obtain and maintain intellectual property protection for its product candidates; the loss of key scientific or management personnel, Trius’ ability to obtain additional financing; and the accuracy of Trius’ estimates regarding expenses, future revenues and capital requirements. These and other risks and uncertainties are described more fully in Trius’ most recent Form 10-K, Forms 10-Q and other documents filed with the United States Securities and Exchange Commission, including those factors discussed under the caption “Risk Factors” in such filings. All forward-looking statements contained in this press release speak only as of the date on which they were made. Trius undertakes no obligation to update such statements to reflect events that occur or circumstances that exist after the date on which they were made. |