Filed Pursuant to Rule 424(b)(3)
Registration No. 333-258318
PROSPECTUS

7,262,460 shares of Common Stock
This prospectus covers the offer and resale by the selling stockholder identified in this prospectus of up to an aggregate of 7,262,460 shares of our common stock issuable upon the exercise of warrants to purchase 7,262,460 shares of our common stock, or the Warrants. We issued the Warrants to the selling stockholder (i) in exchange for warrants issued by Leading BioSciences, Inc. (“LBS”) pursuant to the Securities Purchase Agreement, dated December 16, 2020, by and between the selling stockholder and LBS which exchange occurred in connection with the Merger described in this prospectus, (ii) in a private placement transaction pursuant to the Securities Purchase Agreement, dated December 16, 2020, by and among us, LBS and the selling stockholder and (iii) in a private placement transaction pursuant to the Waiver and Amendment Agreement, dated July 21, 2021, by and between us and the selling stockholder. We are registering these shares issuable upon exercise of the Warrants on behalf of the selling stockholder, to be offered and sold by them from time to time.
We are not selling any shares of common stock under this prospectus and will not receive any proceeds from the sale by the selling stockholder of such shares. We are paying the cost of registering the shares of common stock covered by this prospectus as well as various related expenses. The selling stockholder is responsible for all selling commissions, transfer taxes and other costs related to the offer and sale of their shares.
Sales of the shares by the selling stockholder may occur at fixed prices, at market prices prevailing at the time of sale, at prices related to prevailing market prices, at negotiated prices and/or at varying prices determined at the time of sale. The selling stockholder may sell shares directly or to or through underwriters, broker-dealers or agents, who may receive compensation in the form of discounts, concessions or commissions from the selling stockholder, the purchasers of the shares, or both. The selling stockholder may sell any, all or none of the securities offered by this prospectus and we do not know when or in what amount the selling stockholder may sell their shares of common stock hereunder following the effective date of the registration statement of which this prospectus forms a part. We provide more information about how the selling stockholder may sell or otherwise dispose of their shares of common stock in the section titled “Plan of Distribution” on page 36.
Our common stock is listed on The Nasdaq Capital Market under the symbol “PALI.” On August 9, the last reported sale price of our common stock was $2.76 per share.
Investing in our common stock involves a high degree of risk. Before making an investment decision, please read the information under "Risk Factors" beginning on page 9 of this prospectus and under similar headings in any amendment or supplement to this prospectus or in any filing with the Securities and Exchange Commission that is incorporated by reference herein.
Neither the SEC nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this prospectus is August 10, 2021
TABLE OF CONTENTS
ABOUT THIS PROSPECTUS
This prospectus is part of a registration statement on Form S-3 that we filed with the Securities and Exchange Commission, or SEC, using a "shelf" registration process. Under this registration statement, the selling stockholder may sell from time to time in one or more offerings the common stock described in this prospectus.
We have not authorized anyone to provide you with information other than the information that we have provided or incorporated by reference in this prospectus and your reliance on any unauthorized information or representation is at your own risk. This prospectus may be used only in jurisdictions where offers and sales of these securities are permitted. You should assume that the information appearing in this prospectus is accurate only as of the date of this prospectus and that any information we have incorporated by reference is accurate only as of the date of the document incorporated by reference, regardless of the time of delivery of this prospectus, or any sale of our common stock. Our business, financial condition and results of operations may have changed since those dates.
Unless otherwise stated, all references in this prospectus to "we," "us," "our," "Palisade," the "Company" and similar designations refer to Palisade Bio, Inc. This prospectus contains references to trademarks belonging to other entities. Solely for convenience, trademarks and trade names referred to in this prospectus, including logos, artwork and other visual displays, may appear without the ® or ™ symbols, but such references are not intended to indicate, in any way, that the applicable licensor will not assert, to the fullest extent under applicable law, its rights to these trademarks and trade names. We do not intend our use or display of other companies' trade names or trademarks to imply a relationship with, or endorsement or sponsorship of us by, any other companies.
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus and any applicable prospectus supplement or free writing prospectus, including the documents that we incorporate by reference herein and therein, contain "forward-looking statements" within the meaning of Section 27A of the Securities Act of 1933, as amended, or the Securities Act, and Section 21E of the Securities Exchange Act of 1934, as amended, or the Exchange Act. These statements relate to future events or to our future operating or financial performance and involve known and unknown risks, uncertainties and other factors which may cause our actual results, performance or achievements to be materially different from any future results, performances or achievements expressed or implied by the forward-looking statements.
In some cases, you can identify forward-looking statements by terms such as “may,” “will,” “should,” “could,” “would,” “expects,” “plans,” “anticipates,” “believes,” “estimates,” “projects,” “predicts,” “potential” and similar expressions intended to identify forward-looking statements. These statements reflect our current views with respect to future events and are based on assumptions and are subject to risks and uncertainties. As such, our actual results may differ significantly from those expressed in any forward-looking statements. Given these uncertainties, you should not place undue reliance on these forward-looking statements.
We discuss many of these risks in greater detail under “Risk Factors” in this prospectus, in the "Business" and "Management's Discussion and Analysis of Financial Condition and Results of Operations" sections incorporated by reference from our most recent Annual Report on Form 10-K and in our Quarterly Reports on Form 10-Q for the quarterly periods ended subsequent to our filing of such Annual Report on Form 10-K, as well as any amendments thereto reflected in subsequent filings with the SEC.
Also, these forward-looking statements represent our estimates and assumptions only as of the date of the document containing the applicable statement. Unless required by law, we undertake no obligation to update or revise any forward-looking statements to reflect new information or future events or developments. Thus, you should not assume that our silence over time means that actual events are bearing out as expressed or implied in such forward-looking statements. You should read this prospectus, any applicable prospectus supplement, together with the documents that we have filed with the SEC that are incorporated by reference and any free writing prospectus we have authorized for use in connection with this offering, completely and with the understanding that our actual future results may be materially different from what we expect. We qualify all of the forward-looking statements in the foregoing documents by these cautionary statements.
PROSPECTUS SUMMARY
This summary highlights certain information about us, this offering and selected information contained elsewhere in or incorporated by reference into this prospectus. This summary is not complete and does not contain all of the information that you should consider before making an investment decision. For a more complete understanding of our company, you should read and consider carefully the more detailed information included or incorporated by reference in this prospectus and any applicable prospectus supplement, including the factors described under the heading “Risk Factors” beginning on page 9 of this prospectus, as well as the information incorporated by reference from our most recent Annual Report on Form 10-K and our most recent Quarterly Report on Form 10-Q, before making an investment decision.
Company Overview
We are a clinical stage biopharmaceutical company focused on discovering, developing, and commercializing innovative oral therapies that target serious diseases associated with the breakdown of the mucosal barrier protecting the gastrointestinal (“GI”) tract. Our goal is to be an industry leader in developing therapies to treat these diseases and to improve the lives of patients suffering from such diseases.
Our approach is founded on the discovery that damage to the intestinal epithelial barrier can result in leakage of digestive enzymes from the GI tract that can damage tissue and promote inflammation, causing a broad array of acute and chronic conditions.
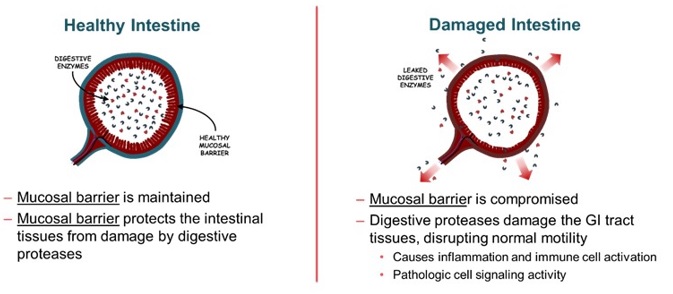
Using our scientific and drug development expertise, we are developing a portfolio of oral product candidates to treat conditions driven by protease (intestinal enzymes) leakage through the intestinal epithelial barrier, including surgical complications and inflammatory conditions.
Our pipeline of product candidates is illustrated in this chart:
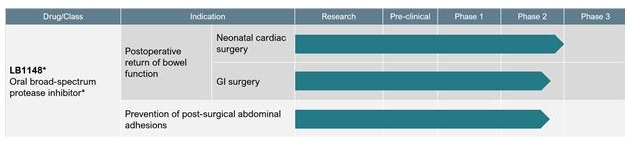
* Commercial right to LB1148 in Greater China (excluding Taiwan) have been out-licensed to Newsoara.
Our lead therapeutic candidate, LB1148, is a novel oral liquid formulation of the well-characterized digestive enzyme inhibitor, tranexamic acid, intended to inhibit digestive enzyme activity and preserve gut integrity during intestinal stress, such as results from reduced blood flow to the intestine, infections, and surgery. Peer reviewed publications of third-party research suggest that digestive enzyme leakage from the GI tract drives GI and organ dysfunction following these events.
We are initially developing LB1148 to be administered to patients prior to major surgeries that risk disrupting the intestinal mucosal barrier. As announced in March of 2020, a randomized, double-blind, parallel, placebo-controlled Phase 2 investigator-sponsored clinical trial of LB1148 in 120 patients undergoing coronary artery bypass grafting and/or heart valve replacement surgery requiring cardiopulmonary bypass was completed. Patients were randomized to receive LB1148 or placebo in conjunction with surgery. The trial’s primary endpoint was time to return of bowel function. Secondary endpoints include Intensive Care Unit (“ICU”) length of stay, hospital length of stay, organ function changes, inflammatory response and glucose control. LB1148 provided an approximately 30% improvement in the time to normal bowel function following cardiovascular (“CV”) surgery (p<0.001) compared to placebo. The treatment group also had an average 1.1-day shorter length of stay in the ICU and an average 1.1-day shorter hospital stay. Generally, treatment with LB1148 was well tolerated. Adverse events (“AEs”) were similar between the treatment groups and not considered unexpected for the subject population. None of the AEs or serious adverse events (“SAEs”) reported were considered drug-related by the sponsor-investigator. One of the primary factors in discharging patients from the hospital following surgery is the return of bowel function. LB1148 has been granted Fast Track designation from the “FDA” for the treatment of postoperative GI dysfunction (which may present as feeding intolerance, ileus, necrotizing enterocolitis (“NEC”), etc.) associated with gut hypoperfusion injury in pediatric patients who have undergone congenital heart disease repair surgery.
We are also currently conducting a randomized, double-blind, placebo-controlled, proof-of-concept Phase 2 clinical trial of LB1148 in patients undergoing elective bowel resection surgery in the Unites States. We expect to have initial data regarding the time to return of GI function from this clinical trial in the second half of 2021. The second part of this trial will evaluate whether patients treated with LB1148 also experience fewer postoperative intra-abdominal adhesions. In the second half of 2021, we are planning to initiate a Phase 2/3 clinical trial of LB1148 in neonatal patients undergoing CV surgery to correct congenital heart defects. We anticipate that this clinical trial will enroll 100 patients and that we will have data from the first 10 patients in late 2021 with final data from the full 100 patients in 2022.
Beyond our lead product candidate, we are continuing to develop additional therapeutic candidates. We believe that protease-based therapeutics hold promise in meeting a number of unmet needs resulting from chronic protease leak, beyond our initial therapeutic focus on GI-related pathology triggered by major surgeries.
Selected Risks Affecting Our Business
| · | The Company’s business depends on the successful clinical development, regulatory approval and commercialization of LB1148. |
| · | Some of the initial indications in which the Company plans to pursue development of LB1148 are indications for which there are no FDA-approved therapies. This makes it difficult to predict the timing and costs of clinical development for LB1148 in these indications, as well as the regulatory approval path. |
| · | The development and commercialization strategy for the Company’s product candidate LB1148 depends, in part, on published scientific literature and the FDA’s prior findings regarding the safety and efficacy of tranexamic acid. If the Company is not able to pursue this strategy, it may be delayed in receiving regulatory authority approval. |
| · | Clinical drug development is very expensive, time-consuming and uncertain. |
| · | The results of previous clinical trials may not be predictive of future results, and the results of the Company’s current and planned clinical trials may not satisfy the requirements of the FDA or non-U.S. regulatory authorities. |
| · | Even if the Company receives marketing approval for LB1148, or any future product candidate, it may not be able to successfully commercialize its product candidates due to unfavorable pricing regulations or third-party coverage and reimbursement policies, which could make it difficult for the Company to sell its product candidates profitably. |
| · | The Company will need to raise additional financing in the future to fund its operations, which may not be available to it on favorable terms or at all. |
| · | The Company currently has no products approved for sale, and it may never obtain regulatory approval to commercialize any of its product candidates. |
| · | The Company’s or third party’s clinical trials may fail to demonstrate the safety and efficacy of its product candidates, or serious adverse or unacceptable side effects may be identified during their development, which could prevent or delay marketing approval and commercialization, increase the Company’s costs or necessitate the abandonment or limitation of the development of the product candidate. |
| · | The Company has expressed substantial doubt about its ability to continue as a going concern. |
| · | The Company may not be able to protect its intellectual property rights throughout the world. |
| · | Our board of directors has broad discretion to issue additional securities, which might dilute the net tangible book value per share of our common stock for existing stockholders. |
Corporate Information
We were originally incorporated in 2001 in the State of Delaware under the name Neuralstem, Inc. In October 2019, we changed our name from Neuralstem, Inc. to Seneca Biopharma, Inc. In April 2021, we effected the Merger (described below). In April 2021, we changed our name from Seneca Biopharma, Inc. to Palisade Bio, Inc. Our principal executive offices are located at 5800 Armada Drive, Suite 210, Carlsbad, California 92008, our telephone number is (858) 7004-4900 and our website address is www.palisadebio.com. The information contained in or accessible through our website does not constitute part of this prospectus.
Implications of Being a Smaller Reporting Company
We are a “smaller reporting company” as defined in Item 10(f)(1) of Regulation S-K. Smaller reporting companies may take advantage of certain reduced disclosure obligations, including, among other things, providing only two years of audited financial statements. We will remain a smaller reporting company until the last day of any fiscal year for so long as either (1) the market value of our shares of common stock held by non-affiliates does not equal or exceed $250.0 million as of the prior June 30th, or (2) our annual revenues did not equal or exceed $100.0 million during such completed fiscal year and the market value of our shares of common stock held by non-affiliates did not equal or exceed $700.0 million as of the prior June 30th. To the extent we take advantage of any reduced disclosure obligations, it may make comparison of our financial statements with other public companies difficult or impossible.
Merger Transaction
On April 27, 2021, pursuant to the Agreement and Plan of Merger (the “Merger Agreement”), dated as of December 16, 2020, by and among Palisade Bio, Inc., formerly known as Seneca Biopharma, Inc. (the “Company”), Leading Biosciences, Inc. (“LBS”) and Townsgate Acquisition Sub 1, Inc., a wholly owned subsidiary of the Company (“Merger Sub”), the Company completed the previously announced merger transaction with LBS, pursuant to which Merger Sub merged with and into LBS, with LBS surviving such merger as a wholly owned subsidiary of the Company (the “Merger”). In connection with the Merger, and immediately prior to the effective time of the Merger (the “Effective Time”), the Company effected a reverse stock split of the Company Common Stock at a ratio of 1-for-6 (the “Reverse Stock Split”). Unless otherwise noted, all references to share and per share amounts in this Prospectus reflect the Reverse Stock Split. Also, in connection with the closing of the Merger (the “Closing”), the Company changed its name from “Seneca Biopharma, Inc.” to “Palisade Bio, Inc.” (the “Name Change”) and the business conducted by the Company became primarily the business conducted by LBS, which is a clinical-stage biopharmaceutical company focused on advancing LBS’s clinical program and developing a therapeutic to combat the interruption of gastrointestinal function following major surgery for which there is currently a significant unmet need for safe and effective therapies.
At the Effective Time:
| | (a) | Each outstanding share of LBS’s common stock, par value $0.001 per share (“LBS Common Stock”), and each outstanding non-voting share of LBS’s Series 1 preferred stock, par value $0.001 per share (“LBS Series 1 Preferred”), issued in the Pre-Merger Financing (as defined below) was converted into the right to receive 0.02719 (the “Exchange Ratio”) shares of Company Common Stock, as set forth in the Merger Agreement. The Exchange Ratio was determined based on the total number of outstanding shares of Company Common Stock and LBS Common Stock, in each case as calculated on an adjusted fully diluted treasury stock method basis, after giving effect to the Pre-Merger Financing, including 50% of the shares subject to the Equity Warrants (as defined below), and taking into account certain adjustments based on the proceeds of the Pre-Merger Financing and the net cash of the Company at the Closing in accordance with the Merger Agreement. |
| | (b) | Each option to purchase shares of LBS Common Stock (each, an “LBS Option”) that was outstanding and unexercised immediately prior to the Effective Time under LBS’s 2013 Equity Incentive Plan (the “LBS Plan”), whether or not vested, was converted into and became an option to purchase shares of Company Common Stock, and the Company assumed the LBS Plan and each such LBS Option in accordance with the terms of the LBS Plan (the “Assumed Options”). The number of shares of Company Common Stock subject to each Assumed Option was determined by multiplying (i) the number of shares of LBS Common Stock that were subject to such Assumed Option, as in effect immediately prior to the Effective Time, by (ii) the Exchange Ratio, and rounding the resulting number down to the nearest whole number of shares of Company Common Stock, and the per share exercise price for the Company Common Stock issuable upon exercise of each Assumed Option was determined by dividing (A) the per share exercise price of such Assumed Option, as in effect immediately prior to the Effective Time, by (B) the Exchange Ratio and rounding the resulting per share exercise price up to the nearest whole cent. |
| | (c) | Each warrant to purchase shares of LBS Common Stock (each, an “LBS Warrant”) outstanding immediately prior to the Effective Time was assumed by the Company and converted into a warrant to purchase shares of Company Common Stock (the “Assumed Warrants”) and thereafter (i) each Assumed Warrant may be exercised solely for shares of Company Common Stock; (ii) the number of shares of Company Common Stock subject to each Assumed Warrant was determined by multiplying (A) the number of shares of LBS Common Stock that were subject to such LBS Warrant, as in effect immediately prior to the Effective Time, by (B) the Exchange Ratio, and rounding the resulting number down to the nearest whole number of shares of Company Common Stock; (iii) the per share exercise price for the Company Common Stock issuable upon exercise of each Assumed Warrant was determined by dividing (A) the exercise price per share of the LBS Common Stock subject to such LBS Warrant, as in effect immediately prior to the Effective Time, by (B) the Exchange Ratio, and rounding the resulting exercise price up to the nearest whole cent. |
Pre-Merger Financing
Securities Purchase Agreement (Bridge Financing)
In connection with signing the Merger Agreement, LBS entered into a securities purchase agreement, dated as of December 16, 2020 (the “Bridge SPA”) with the selling stockholder (the “Investor”), pursuant to which the Investor purchased senior secured promissory notes (the “Bridge Notes”) and warrants to purchase such number of shares of LBS Common Stock equal to the aggregate principal amount of the Bridge Notes issued divided by the initial per share exercise price of $0.4816 (the “Bridge Warrants”), subject to adjustment as disclosed below. The Bridge Warrants have a term of five years from the date all of the shares underlying the Bridge Warrants are registered for resale, and the exercise price shall be subject to price-only full ratchet anti-dilution protection upon the issuance of any shares of LBS Common Stock or securities convertible into LBS Common Stock for a period of two years from the effective date of the registration statement covering such shares. The Bridge Warrants also contain certain participation rights with regard to asset distributions and fundamental transactions. As a result of the Merger, at the Effective Time, each Bridge Warrant was automatically converted into a warrant to purchase that number of shares of the Company’s common stock equal to the number of shares underlying the Bridge Warrants immediately prior to the closing of the Merger multiplied by the Exchange Ratio and the exercise price was proportionately adjusted. At the Effective Time, there were 188,192 shares of the Company’s common stock issuable upon exercise of the Bridge Warrants. The Bridge Warrants were Assumed Warrants pursuant to the Merger Agreement, as described above.
Securities Purchase Agreement (Equity Financing)
In connection with signing the Merger Agreement, on December 16, 2020, LBS, Seneca and the Investor entered into a Securities Purchase Agreement (the “Equity SPA”), pursuant to which, among other things, the Investor agreed to invest $20.0 million in cash and cancel any outstanding principal and interest on the Bridge Notes immediately prior to the closing of the Merger (the aggregate amount of such cash investment and the cancellation of the outstanding principal and interest on the Bridge Notes, the “Purchase Price” and the financing arrangement described herein, the “Pre-Merger Financing”) to fund the combined company following the Merger. In return, LBS issued an amount of shares (the “Initial Shares”) of LBS Series 1 Preferred Stock to the Investor equal to the Purchase Price divided by the per share purchase price of $0.4816 and the Company agreed to issue to the Investor, warrants to purchase shares of the Company’s Common stock (the “Equity Warrants”). The Equity Warrants were issued on the 17th trading day following the closing of the Merger, and had an initial exercise price per share equal to $4.70 and were exercisable for up to 4,995,893 shares of the Company’s common stock, and are immediately exercisable and will have a term of five years from the date of issuance.
The Bridge Warrants and Equity Warrants provide that, until the second anniversary of the date on which all shares of Company’s common stock issued to the Investor (including any shares underlying the Equity Warrants) are registered on one or more registration statements, if the Company publicly announces, issues or sells, enters into a definitive, binding agreement pursuant to which the Company is required to issue or sell or is deemed, pursuant to the provisions of the Bridge Warrants and Equity Warrants, to have issued or sold, any shares of the Company’s common stock for a price per share lower than the exercise price then in effect, subject to certain limited exceptions, then the exercise price of the Bridge Warrants and Equity Warrants shall be reduced to such lower price per share. Further, on preset reset dates, the exercise price of the Bridge Warrants and Equity Warrants was to be adjusted downward (but not increased)(the “Resets”). Further, the Equity Warrants include a provision such that, beginning six months after the closing of the Merger, if the volume weighted average price of Company Common Stock is less than the then-applicable exercise price for five consecutive trading days, the holder of the Equity Warrant shall be entitled to receive 1.0 share of Company Common Stock for each share underlying the Equity Warrants being exercised thereunder in a cashless exercise. The exercise price and the number of shares of Company Common Stock issuable upon exercise of the Bridge Warrants and Equity Warrants will also be subject to adjustment in the event of any stock splits, dividends or distributions or other similar transactions as well as fundamental transactions. Prior to the effectiveness of the Waiver and Amendment Agreement (described below), two Resets occurred and the two Bridge Warrants and the Equity Warrants were exercisable for up to 429,446, 429,446, and 5,303,568 shares, respectively, each at an exercise price of $3.88 per share, and two potential Resets remained.
Waiver and Amendment Agreement
Effective July 21, 2021 (the “Effective Date”), the Investor entered into a Waiver and Amendment Agreement with the Company (the “Waiver Agreement”). Pursuant to the Waiver Agreement, the Investor and the Company agreed to waive certain rights, waive the reset provisions with respect to the exercise price and number of shares subject to the outstanding Bridge Warrants and Equity Warrants, eliminate certain financing restrictions, and accelerate registration rights for the shares underlying the warrants. As consideration for the foregoing, pursuant to the Waiver Agreement, the Company issued to the Investor an additional warrant to purchase up to 1,100,000 shares of the Company’s common stock (the “July Warrant” and together with the Bridge Warrants and the Equity Warrants, the “Warrants”). The July Warrant is exercisable beginning on January 21, 2022 and expires on the later of (x) five years from the date that the underlying shares are registered for resale and (y) January 21, 2027. The per share exercise price for the July Warrant is $3.631, subject to certain adjustments. Pursuant to the Waiver Agreement, Investor agreed to waive the reset provisions in the outstanding Bridge Warrants and Equity Warrants such that the number of shares and exercise price in effect immediately prior to the Effective Time (as described above) shall no longer be subject to price-based resets.
The registration statement of which this prospectus is a part relates to the resale of the shares of common stock that may be issued to the selling stockholder in connection with the exercise of the Warrants issued in the foregoing transactions.
Selected Financial Data of Palisade Bio, Inc. Reflecting Reverse Stock Split
On April 27, 2021, in connection with, and prior to the completion of, the Merger, the Company effected a 1-for-6 reverse stock split of its common stock (the “Reverse Stock Split”). No fractional shares have been issued in the Reverse Stock Split and the remaining fractions were paid out in cash.
Please see below selected financial data presenting selected share and per share data reflecting the effect of the 1-for-6 reverse stock split on all periods previously reported. We derived the selected financial data from our consolidated financial statements included in our Annual Report on Form 10-K filed with the SEC on March 22, 2021, as adjusted to reflect the Reverse Stock Split for all periods presented.
AS REPORTED
| | | Years Ended December 31, |
| (in thousands, except share and per share amounts) | | 2020 | | 2019 |
| Net loss | | $ | (16,267 | ) | | | (8,352 | ) |
| Net loss per share attributable to common stockholders-basic and diluted | | $ | (1.17 | ) | | | (3.80 | ) |
| Weighted-average common shares outstanding, basic and diluted | | | 13,869,272 | | | | 2,197,434 | |
| Common shares outstanding at period end | | | 17,295,703 | | | | 3,866,457 | |
AS ADJUSTED FOR ONE-FOR-SIX REVERSE STOCK SPLIT
| (in thousands, except share and per share amounts) | | Years Ended December 31, |
| | | 2020 | | 2019 |
| | | (unaudited) |
| Net loss | | $ | (16,267 | ) | | $ | (8,352 | ) |
| Net loss per share attributable to common stockholders-basic and diluted | | $ | (7.04 | ) | | $ | (22.80 | ) |
| Weighted-average common shares outstanding, basic and diluted | | | 2,311,545 | | | | 366,239 | |
| Common shares outstanding at period end | | | 2,882,617 | | | | 644,409 | |
The Offering
| Common stock offered by the selling stockholder | | 7,262,460 shares |
| | | |
| Terms of the offering | | Each selling stockholder will determine when and how it will sell the common stock offered in this prospectus, as described in “Plan of Distribution.” |
| | | |
| Use of proceeds | | We will not receive any proceeds from the sale of shares of our common stock by the selling stockholder. |
| | | |
| Risk factors | | See “Risk Factors” beginning on page 9, for a discussion of factors you should carefully consider before deciding to invest in our common stock. |
| | | |
| Nasdaq Capital Market symbol | | PALI |
The selling stockholder named in this prospectus may offer and sell up to 7,262,460 shares of our common stock.
The selling stockholder is prohibited, subject to certain exceptions, from exercising the Warrants to the extent that immediately prior to or after giving effect to such exercise, the selling stockholder, together with its affiliates and other attribution parties, would own more than 4.99% of the total number of shares of the Company’s common stock then issued and outstanding, which percentage may be changed at the selling stockholder’s election to a lower percentage at any time or to a higher percentage not to exceed 9.99% upon 61 days’ notice to the Company.
Our common stock is currently listed on The Nasdaq Capital Market under the symbol “PALI.”
Shares of common stock that may be offered under this prospectus will be fully paid and non-assessable. We will not receive any of the proceeds of sales by the selling stockholder of any of the common stock covered by this prospectus. Throughout this prospectus, when we refer to the shares of our common stock being registered on behalf of the selling stockholder for offer and resale, we are referring to the shares of common stock issued to the selling stockholder in connection with the exercise of warrants issued in the transactions as described above. When we refer to the selling stockholder in this prospectus, we are referring to the selling stockholder identified in this prospectus and, as applicable, their permitted transferees or other successors-in-interest that may be identified in a supplement to this prospectus or, if required, a post-effective amendment to the registration statement of which this prospectus is a part.
RISK FACTORS
Investing in our common stock involves a high degree of risk. Before making an investment decision, you should carefully consider the risks described below and described in the sections entitled "Risk Factors" in our most recent Annual Report on Form 10-K and subsequent Quarterly Reports on Form 10-Q, as filed with the SEC, which are incorporated herein by reference in their entirety, as well any amendment or updates to our risk factors reflected in subsequent filings with the SEC, including any applicable prospectus supplement. Our business, financial condition, results of operations or prospects could be materially adversely affected by any of these risks. The trading price of our securities could decline due to any of these risks, and you may lose all or part of your investment. This prospectus and the documents incorporated herein by reference also contain forward-looking statements that involve risks and uncertainties. Our actual results could differ materially from those anticipated in these forward-looking statements as a result of certain factors, including the risks mentioned elsewhere in this prospectus. For more information, see the section entitled "Where You Can Find Additional Information." Please also read carefully the section entitled "Special Note Regarding Forward-Looking Statements."
Risks Related to the Company’s Development, Commercialization and Regulatory Approval of the Company’s Investigational Therapies
The Company’s business depends on the successful clinical development, regulatory approval and commercialization of LB1148.
The success of the Company’s business, including its ability to finance itself and generate revenue in the future, primarily depends on the successful development, regulatory approval and commercialization of LB1148. The clinical and commercial success of LB1148 depends on a number of factors, including the following:
| | · | timely and successful completion of required clinical trials not yet initiated, which may be significantly slower or costlier than the Company currently anticipates and/or produce results that do not achieve the endpoints of the trials; |
| | · | whether the Company is required by the FDA or similar foreign regulatory agencies to conduct additional studies beyond those planned to support the approval and commercialization of LB1148; |
| | · | achieving and maintaining, and, where applicable, ensuring that the Company’s third-party contractors achieve and maintain compliance with their contractual obligations and with all regulatory requirements applicable to LB1148; |
| | · | ability of third parties with whom the Company contracts to manufacture adequate clinical trial and commercial supplies of LB1148, to remain in good standing with regulatory agencies and to develop, validate and maintain commercially viable manufacturing processes that are compliant with current good manufacturing practices (“cGMP”); |
| | · | a continued acceptable safety profile during clinical development and following approval of LB1148; |
| | | |
| | · | ability to obtain favorable labeling for LB1148 through regulators that allows for successful commercialization, given the drugs may be marketed only to the extent approved by these regulatory authorities (unlike with most other industries); |
| | · | ability to successfully commercialize LB1148 in the United States and internationally, if approved for marketing, sale and distribution in such countries and territories, whether alone or in collaboration others; |
| | · | acceptance by physicians, insurers and payors, and patients of the quality, benefits, safety and efficacy of LB1148, if either is approved, including relative to alternative and competing treatments; |
| | · | existence of a regulatory environment conducive to the success of LB1148; |
| | · | ability to price LB1148 to recover the Company’s development costs and generate a satisfactory profit margin; and |
| | · | The Company’s ability and its partners’ ability to establish and enforce intellectual property rights in and to LB1148. |
If the Company does not achieve one or more of these factors, many of which are beyond its control, in a timely manner or at all, the Company could experience significant delays or an inability to obtain regulatory approvals or commercialize LB1148. Even if regulatory approvals are obtained, the Company may never be able to successfully commercialize LB1148. Accordingly, the Company cannot assure you that it will ever be able to generate sufficient revenue through the sale of LB1148, if approved, to continue its business.
Some of the initial indications in which the Company plans to pursue development of LB1148 are indications for which there are no FDA-approved therapies. This makes it difficult to predict the timing and costs of clinical development for LB1148 in these indications, as well as the regulatory approval path.
There are no FDA-approved therapies for decreasing the time to normal feedings and bowel movement (or preventing necrotizing enterocolitis) in infants after heart surgery. While Entereg is approved to accelerate the time to upper and lower gastrointestinal recovery following surgeries that include partial bowel resection with primary anastomosis, there is no guarantee that regulatory precedence regarding Entereg will apply to the approval of other therapies that may accelerate the time to gastrointestinal recovery following surgery. While there are multiple medical devices approved for the reduction or elimination of postoperative intra-abdominal adhesions, there are no drugs approved to reduce postoperative intra-abdominal adhesions. The regulatory approval process for novel product candidates such as LB1148 can be more expensive and take longer than for other, better known or extensively studied therapeutic approaches.
The development and commercialization strategy for the Company’s product candidate LB1148 depends, in part, on published scientific literature and the FDA’s prior findings regarding the safety and efficacy of tranexamic acid. If the Company is not able to pursue this strategy, it may be delayed in receiving regulatory authority approval.
The Hatch-Waxman Act added Section 505(b)(2) to the U.S. Federal Food, Drug, and Cosmetic Act (“FDCA”). Section 505(b)(2) permits the submission of an NDA or BLA where at least some of the information required for approval comes from investigations that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted. The FDA interprets Section 505(b)(2) of the FDCA, for purposes of approving an NDA/BLA, to permit the applicant to rely, in part, upon published literature and/or the FDA’s previous findings of safety and efficacy for an approved product. The FDA also requires companies to perform additional clinical trials or measurements to support any deviation from the previously approved product and to justify that it is scientifically appropriate to rely on the applicable published literature or referenced product, referred to as bridging. The FDA may then approve the new product candidate for all or some of the indications for which the referenced product has been approved, as well as for any new indication sought by the Section 505(b)(2) applicant, if such approval is supported by study data. The labeling, however, may be required to include all or some of the limitations, contraindications, warnings or precautions or restrictions on use included in the reference product’s labeling, including a boxed warning, or may require additional limitations, contraindications, warnings or precautions or restrictions on use.
The Company currently plans to pursue marketing approval for LB1148, in the United States through a 505(b)(2) NDA and will be completing bridging analyses prior to NDA submissions. If the FDA disagrees with the Company’s conclusions regarding the appropriateness of its reliance on the FDA’s prior findings of safety and efficacy for tranexamic acid (“TXA”) or on published literature, or if the Company is not otherwise able to bridge to the listed drug or published literature to demonstrate that its reliance is scientifically appropriate, the Company could be required to conduct additional clinical trials or other studies to support its NDA, which could lead to unanticipated costs and delays or to the termination of the development program for LB1148. If the Company is unable to obtain approval for LB1148 through the 505(b)(2) NDA process, it may be required to pursue the more expensive and time consuming 505(b)(1) approval process, which consists of full reports of investigations of safety and effectiveness conducted by or for the Company.
Notwithstanding the approval of a number of products by the FDA under Section 505(b)(2), pharmaceutical companies and others have objected to the FDA’s interpretation of Section 505(b)(2). If the FDA’s interpretation of Section 505(b)(2) is successfully challenged, the FDA may be required to change its policies and practices with respect to Section 505(b)(2) regulatory approvals, which could delay or even prevent the FDA from approving any NDA that the Company submits pursuant to the 505(b)(2) process. Even if the Company is allowed to pursue the 505(b)(2) regulatory pathway to FDA approval, it cannot assure you that its product candidates will receive the requisite approvals for commercialization.
The Company may find it difficult to enroll patients in its clinical trials, which could delay or prevent it from proceeding with clinical trials of its product candidates.
Identifying and qualifying subjects to participate in clinical trials of the Company’s product candidates is critical to its success. The timing of clinical trials depends on the Company’s ability to recruit subjects to participate, as well as the completion of required follow-up periods. Patients may be unwilling to participate in clinical trials because of negative publicity from adverse events related to the biotechnology or pharmaceutical fields, competitive clinical trials for similar patient populations, the existence of current treatments or for other reasons. The timeline for recruiting patients, conducting studies and obtaining regulatory approval of the Company’s product candidates may be delayed, which could result in increased costs, delays in advancing its product candidates, delays in testing the effectiveness of its product candidates or termination of the clinical trials altogether.
Patient enrollment and trial completion are affected by numerous additional factors, including the:
| | · | process for identifying patients; |
| | · | design of the trial protocol; |
| | · | eligibility and exclusion criteria; |
| | · | perceived risks and benefits of the product candidate under study; |
| | · | availability of competing therapies and clinical trials; |
| | · | severity of the disease under investigation; |
| | · | proximity and availability of clinical trial sites for prospective patients; |
| | · | ability to obtain and maintain patient consent; |
| | · | risk that enrolled patients will drop out before completion of the trial; |
| | · | patient referral practices of physicians; and |
| | · | ability to monitor patients adequately during and after treatment. |
If the Company has difficulty enrolling a sufficient number of patients to conduct its clinical trials as planned, it may need to delay, limit or terminate ongoing or planned clinical trials, any of which would have an adverse effect on its business, financial condition, results of operations and prospects.
Clinical drug development is very expensive, time-consuming and uncertain.
Clinical development for the Company’s product candidates is very expensive, time-consuming, difficult to design and implement, and the outcomes are inherently uncertain. Most product candidates that commence clinical trials are never approved by regulatory authorities for commercialization and of those that are approved many do not cover their costs of development. In addition, the Company, any partner with which it may in the future collaborate, the FDA, an institutional review board (“IRB”), or other regulatory authorities, including state and local agencies and counterpart agencies in foreign countries, may suspend, delay, require modifications to or terminate the Company’s clinical trials at any time.
The results of previous clinical trials may not be predictive of future results, and the results of the Company’s current and planned clinical trials may not satisfy the requirements of the FDA or non-U.S. regulatory authorities.
The results from the prior preclinical studies and clinical trials for LB1148 discussed elsewhere in this prospectus may not necessarily be predictive of the results of future preclinical studies or clinical trials. Even if the Company is able to complete its planned clinical trials of its product candidates according to its current development timelines, the results from its prior clinical trials of its product candidates may not be replicated in these future trials. Many companies in the pharmaceutical and biotechnology industries (including those with greater resources and experience than the Company) have suffered significant setbacks in late-stage clinical trials after achieving positive results in early stage development, and the Company cannot be certain that it will not face similar setbacks. These setbacks have been caused by, among other things, preclinical findings made while clinical trials were underway or safety or efficacy observations made in clinical trials, including previously unreported adverse events. Moreover, preclinical and clinical data are often susceptible to varying interpretations and analyses, and many companies that believed their product candidates performed satisfactorily in preclinical studies and clinical trials nonetheless have failed to obtain FDA approval. If the Company fails to produce positive results in its clinical trials of any of its product candidates, the development timelines and regulatory approvals and commercialization prospects for its product candidates and its business and financial prospects, would be adversely affected. If the Company fails to produce positive results in its clinical trials of any of its product candidates, the development timelines, regulatory approvals, and commercialization prospects for its product candidates, as well as the Company’s business and financial prospects, would be adversely affected. Further, the Company’s product candidates may not be approved even if they achieve their respective primary endpoints in Phase 3 registration trials. The FDA or non-U.S. regulatory authorities may disagree with the Company’s trial designs or its interpretation of data from preclinical studies and clinical trials. the Company has taken the position that LB1148 has a single active ingredient, TXA. LB1148 also contains polyethylene glycol 3350 (“PEG”). Across different countries and different circumstances, PEG may be regulated as an inactive ingredient, a medical device, or an active ingredient. There is uncertainty on how the FDA and other regulatory agencies will classify the PEG in LB1148. If the FDA determines that LB1148 is a combination product (of TXA and PEG) regulatory approval of this product candidate will require additional clinical trials for which there is not currently a feasible clinical trial design. In addition, any of these regulatory authorities may change requirements for the approval of a product candidate even after reviewing and providing comments or advice on a protocol for a pivotal clinical trial that has the potential to result in approval by the FDA or another regulatory authority. Furthermore, any of these regulatory authorities may also approve the Company’s product candidate for fewer or more limited indications than it requests or may grant approval contingent on the performance of costly post-marketing clinical trials.
If the clinical development of LB1148 is successful, the Company plans to eventually seek regulatory approvals of LB1148 initially in the United States, and may seek approvals in other geographies. Before obtaining regulatory approvals for the commercial sale of any product candidate for any target indication, the Company must demonstrate with substantial evidence gathered in preclinical studies and adequate and well-controlled clinical studies, and, with respect to approval in the United States, to the satisfaction of the FDA, that the product candidate is safe and effective for use for that target indication. The Company cannot assure you that the FDA or non-U.S. regulatory authorities would consider its planned clinical trials to be sufficient to serve as the basis for approval of its product candidates for any indication. The FDA and non-U.S. regulatory authorities retain broad discretion in evaluating the results of the Company’s clinical trials and in determining whether the results demonstrate that its product candidates are safe and effective. If the Company is required to conduct clinical trials of its product candidates in addition to those it has planned prior to approval, the Company will need substantial additional funds, and cannot assure you that the results of any such outcomes trial or other clinical trials will be sufficient for approval.
The Company’s product candidates may cause undesirable side effects or have other unexpected properties that could delay or prevent their regulatory approval, limit the commercial profile of an approved label, or result in post-approval regulatory action.
Unforeseen side effects from LB1148 could arise either during clinical development or, if approved, after it has been marketed. Undesirable side effects could cause the Company, any partners with which the Company may collaborate, or regulatory authorities to interrupt, extend, modify, delay or halt clinical trials and could result in a more restrictive or narrower label or the delay or denial of regulatory approval by the FDA or comparable foreign authorities.
Results of clinical trials could reveal a high and unacceptable severity and prevalence of side effects. In such an event, trials could be suspended or terminated, and the FDA or comparable foreign regulatory authorities could order us to cease further development of or deny approval of a product candidate for any or all targeted indications. The drug-related side effects could affect patient recruitment or the ability of enrolled patients to complete the trial or result in product liability claims. Any of these occurrences may harm the Company’s business, financial condition, operating results and prospects.
Additionally, if the Company or others identify undesirable side effects, or other previously unknown problems, caused by a product after obtaining U.S. or foreign regulatory approval, a number of potentially negative consequences could result, which could prevent the Company or its potential partners from achieving or maintaining market acceptance of the product and could substantially increase the costs of commercializing such product.
The Company may in the future conduct clinical trials for its product candidates outside the United States, and the FDA and applicable foreign regulatory authorities may not accept data from such trials.
The Company, as well as investigator sponsors, have conducted clinical trials, is conducting clinical trials, and may in the future choose to conduct one or more clinical trials outside of the United States. Although the FDA or applicable foreign regulatory authority may accept data from clinical trials conducted outside the United States or the applicable jurisdiction, acceptance of such study data by the FDA or applicable foreign regulatory authority may be subject to certain conditions or exclusion. Where data from foreign clinical trials are intended to serve as the basis for marketing approval in the United States, the FDA will not approve the application on the basis of foreign data alone unless such data are applicable to the U.S. population and U.S. medical practice; the studies were performed by clinical investigators of recognized competence; and the data are considered valid without the need for an on-site inspection by the FDA or, if the FDA considers such an inspection to be necessary, the FDA is able to validate the data through an on-site inspection or other appropriate means. Many foreign regulatory bodies have similar requirements. In addition, such foreign studies would be subject to the applicable local laws of the foreign jurisdictions where the studies are conducted. There can be no assurance the FDA or applicable foreign regulatory authority will accept data from trials conducted outside of the United States or the applicable home country. If the FDA or applicable foreign regulatory authority does not accept such data, it would likely result in the need for additional trials, which would be costly and time-consuming and delay aspects of the Company’s business plan.
The Company expects to rely on third-party CROs and other third parties to conduct and oversee its clinical trials. If these third parties do not meet the Company’s requirements or otherwise conduct the trials as required, the Company may not be able to satisfy its contractual obligations or obtain regulatory approval for, or commercialize, its product candidates.
The Company expects to rely on third-party contract research organizations (“CROs”) to conduct and oversee its LB1148 clinical trials and other aspects of product development. The Company also expects to rely on various medical institutions, clinical investigators and contract laboratories to conduct its trials in accordance with the Company’s clinical protocols and all applicable regulatory requirements, including the FDA’s regulations and good clinical practice (“GCP”) requirements, which are an international standard meant to protect the rights and health of patients and to define the roles of clinical trial sponsors, administrators and monitors, and state regulations governing the handling, storage, security and recordkeeping for drug and biologic products. These CROs and other third parties will play a significant role in the conduct of these trials and the subsequent collection and analysis of data from the clinical trials. the Company will rely heavily on these parties for the execution of its clinical trials and preclinical studies and will control only certain aspects of their activities. The Company and its CROs and other third-party contractors will be required to comply with GCP and good laboratory practice (“GLP”) requirements, which are regulations and guidelines enforced by the FDA and comparable foreign regulatory authorities. Regulatory authorities enforce these GCP and GLP requirements through periodic inspections of trial sponsors, principal investigators and trial sites. If the Company or any of these third parties fail to comply with applicable GCP and GLP requirements, or reveal noncompliance from an audit or inspection, the clinical data generated in the Company’s clinical trials may be deemed unreliable and the FDA or other regulatory authorities may require the Company to perform additional clinical trials before approving the Company’s or the Company’s partners’ marketing applications. the Company cannot assure that upon inspection by a given regulatory authority, such regulatory authority will determine that any of the Company’s clinical or preclinical trials comply with applicable GCP and GLP requirements. In addition, the Company’s clinical trials generally must be conducted with product produced under cGMP regulations. The Company’s failure to comply with these regulations and policies may require it to repeat clinical trials, which would delay the regulatory approval process.
If any of the Company’s CROs or clinical trial sites terminate their involvement in one of its clinical trials for any reason, it may not be able to enter into arrangements with alternative CROs or clinical trial sites or do so on commercially reasonable terms. In addition, if the Company’s relationship with clinical trial sites is terminated, it may experience the loss of follow-up information on patients enrolled in its ongoing clinical trials unless the Company is able to transfer the care of those patients to another qualified clinical trial site. In addition, principal investigators for the Company’s clinical trials may serve as scientific advisors or consultants to it from time to time and could receive cash or equity compensation in connection with such services. If these relationships and any related compensation result in perceived or actual conflicts of interest, the integrity of the data generated at the applicable clinical trial site may be questioned by the FDA.
Even if the Company receives marketing approval for LB1148, or any future product candidate, it may not be able to successfully commercialize its product candidates due to unfavorable pricing regulations or third-party coverage and reimbursement policies, which could make it difficult for the Company to sell its product candidates profitably.
Obtaining coverage and reimbursement approval for a product from a government or other third-party payor is a time consuming and costly process that could require the Company to provide supporting scientific, clinical and cost effectiveness data to the payor. There may be significant delays in obtaining such coverage and reimbursement for newly approved products, and coverage may be more limited than the purposes for which the product is approved by the FDA or comparable foreign regulatory authorities. Moreover, eligibility for coverage and reimbursement does not imply that a product will be paid for in all cases or at a rate that covers costs, including research, development, intellectual property, manufacture, sale and distribution expenses. Interim reimbursement levels for new products, if applicable, may also not be sufficient to cover costs and may not be made permanent. Reimbursement rates may vary according to the use of the product and the clinical setting in which it is used, may be based on reimbursement levels already set for lower cost products and may be incorporated into existing payments for other services. Net prices for products may be reduced by mandatory discounts or rebates required by government healthcare programs or private payors, by any future laws limiting drug prices and by any future relaxation of laws that presently restrict imports of product from countries where they may be sold at lower prices than in the United States.
There is significant uncertainty related to the insurance coverage and reimbursement of newly approved products. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting reimbursement policies, but also have their own methods and approval process apart from Medicare coverage and reimbursement determinations.
Coverage and reimbursement by a third-party payor may depend upon a number of factors, including the third-party payor’s determination that use of a product is:
| | · | a covered benefit under its health plan; |
| | · | safe, effective and medically necessary; |
| | · | appropriate for the specific patient; |
| | · | cost-effective; and |
| | | |
| | · | neither experimental nor investigational. |
The Company cannot be sure that coverage and reimbursement will be available for any product that it commercializes and, if coverage and reimbursement are available, what the level of reimbursement will be. The Company’s inability to promptly obtain coverage and adequate reimbursement rates from both government-funded and private payors for any approved products that the Company develops could have a material adverse effect on its operating results, its ability to raise capital needed to commercialize products and its overall financial condition.
Reimbursement may impact the demand for, and the price of, any product for which the Company obtains marketing approval. Assuming the Company obtains coverage for a given product by a third-party payor, the resulting reimbursement payment rates may not be adequate or may require co-payments that patients find unacceptably high. Patients who are prescribed medications for the treatment of their conditions, and their prescribing physicians, generally rely on third-party payors to reimburse all or part of the costs associated with those medications. Patients are unlikely to use the Company’s products unless coverage is provided and reimbursement is adequate to cover all or a significant portion of the cost of the Company’s products. Therefore, coverage and adequate reimbursement is critical to new product acceptance. Coverage decisions may depend upon clinical and economic standards that disfavor new products when more established or lower cost therapeutic alternatives are already available or subsequently become available.
The Company’s expects to experience pricing pressures in connection with the sale of any of its product candidates due to the trend toward managed healthcare, the increasing influence of health maintenance organizations, and additional legislative changes. The downward pressure on healthcare costs in general, particularly prescription medicines, medical devices and surgical procedures and other treatments, has become very intense. As a result, increasingly high barriers are being erected to the successful commercialization of new products. Further, the adoption and implementation of any future governmental cost containment or other health reform initiative may result in additional downward pressure on the price that the Company may receive for any approved product.
Outside of the United States, many countries require approval of the sale price of a product before it can be marketed and the pricing review period only begins after marketing or product licensing approval is granted. To obtain reimbursement or pricing approval in some of these countries, the Company may be required to conduct a clinical trial that compares the cost-effectiveness of its product candidate to other available therapies. In some foreign markets, prescription pharmaceutical pricing remains subject to continuing governmental control even after initial approval is granted. As a result, the Company might obtain marketing approval for a product candidate in a particular country, but then be subject to price regulations that delay its commercial launch of the product, possibly for lengthy time periods, and negatively impact the revenues, if any, the Company is able to generate from the sale of the product in that country. Adverse pricing limitations may hinder the Company’s ability to recoup its investment in one or more product candidates, even if such product candidates obtain marketing approval.
Even if a product candidate obtains regulatory approval, it may fail to achieve the broad degree of physician and patient adoption and use necessary for commercial success.
The commercial success of both LB1148, if approved, will depend significantly on the broad adoption and use of them by physicians and patients for approved indications, and it may not be commercially successful even though it is shown to be safe and effective. The degree and rate of physician and patient adoption of a product, if approved, will depend on a number of factors, including but not limited to:
| | · | patient demand for approved products that treat the indication for which a product is approved; |
| | · | the effectiveness of the product compared to other available therapies or treatment regimens; |
| | · | the availability of coverage and adequate reimbursement from managed care plans and other healthcare payors; |
| | · | the cost of treatment in relation to alternative treatments and willingness to pay on the part of patients; |
| | | |
| | · | insurers’ willingness to see the applicable indication as a disease worth treating’ |
| | | |
| | · | proper administration; |
| | · | patient satisfaction with the results, administration and overall treatment experience; |
| | · | limitations or contraindications, warnings, precautions or approved indications for use different than those sought by the Company that are contained in the final FDA-approved labeling for the applicable product; |
| | · | any FDA requirement to undertake a risk evaluation and mitigation strategy; |
| | · | the effectiveness of the Company’s sales, marketing, pricing, reimbursement and access, government affairs, and distribution efforts; |
| | · | adverse publicity about a product or favorable publicity about competitive products; |
| | · | new government regulations and programs, including price controls and/or limits or prohibitions on ways to commercialize drugs, such as increased scrutiny on direct-to-consumer advertising of pharmaceuticals; and |
| | · | potential product liability claims or other product-related litigation. |
If LB1148 is approved for use but fails to achieve the broad degree of physician and patient adoption necessary for commercial success, the Company’s operating results and financial condition will be adversely affected, which may delay, prevent or limit its ability to generate revenue and continue its business.
The Company’s product candidates, if approved, will face significant competition and their failure to compete effectively may prevent them from achieving significant market penetration.
The pharmaceutical industry is characterized by rapidly advancing technologies, intense competition, less effective patent terms, and a strong emphasis on developing newer, fast-to-market proprietary therapeutics. Numerous companies are engaged in the development, patenting, manufacturing and marketing of healthcare products competitive with those that the Company is developing, including LB1148. The Company will face competition from a number of sources, such as pharmaceutical companies, generic drug companies, biotechnology companies, medical device companies and academic and research institutions, many of which have greater financial resources, marketing capabilities, sales forces, manufacturing capabilities, research and development capabilities, regulatory expertise, clinical trial expertise, intellectual property portfolios, more international reach, experience in obtaining patents and regulatory approvals for product candidates and other resources than the Company. Some of the companies that offer competing products also have a broad range of other product offerings, large direct sales forces and long-term customer relationships with the Company’s target physicians, which could inhibit the Company’s market penetration efforts.
With respect to the Company’s lead product candidate, LB1148, for the indication of postoperative improvement of bowel function, the Company expects to face competition in the pharmacological therapy space from alvimopan, marketed as a branded product, ENTEREG, by Merck, as well as in generic form. There are no pharmacotherapies for decreasing the time to normal feedings and bowel movement (or preventing necrotizing enterocolitis) in infants after heart surgery or for the reduction or elimination of postoperative intra-abdominal adhesions. However, the Company will face general competition from other medical interventions, namely surgical procedures and adhesion barrier products. Adhesion barrier products approved for abdominal or pelvic surgery in the United States consist of SEPRAFILM, INTERCEED and ADEPT. In addition, several products are used off-label for adhesion prevention in the United States, including EVICEL, SURGIWRAP, COSEAL and PRECLUDE. Adhesion barrier products available outside the United States include HYALOBARRIER, SPRAYSHIELD, PREVADH, and INTERCOAT. Such products are used as adjunctive interventions, have variable efficacy, and are not easily used with laparoscopic procedures, which are becoming increasingly common.
Any adverse developments that occur during any clinical trials conducted by Newsoara may affect the Company’s ability to obtain regulatory approval or commercialize LB1148.
Newsoara Biopharma Co., Ltd. (“Newsoara”) has the rights to develop and commercialize LB1148 in China for return of bowel function, reduction of adhesions, and sepsis. If serious adverse events occur during any clinical trials Newsoara decides to conduct with respect to LB1148, the FDA and other regulatory authorities may delay, limit or deny approval of LB1148 or require the Company to conduct additional clinical trials as a condition to marketing approval, which would increase our costs. If the Company receives FDA approval for LB1148 and a new and serious safety issue is identified in connection with clinical trials conducted by Newsoara, the FDA and other regulatory authorities may withdraw their approval of the product or otherwise restrict the Company’s ability to market and sell the Company’s product. In addition, treating physicians may be less willing to administer the Company’s product due to concerns over such adverse events, which would limit the Company’s ability to commercialize LB1148.
Risks Related to the Company’s Business
The Company has a very limited operating history and has never generated any revenues from product sales.
The Company is an early-stage biotechnology company with a very limited operating history that may make it difficult to evaluate the success of its business to date and to assess its future viability. The Company was initially formed in 2005 and its operations, to date, have been limited to business planning, raising capital, developing the Company’s pipeline assets and other research and development. The Company has not yet demonstrated an ability to successfully complete any clinical trials and has never completed the development of any product candidate, nor has it ever generated any revenue from product sales or otherwise. Consequently, the Company has no meaningful operations upon which to evaluate its business, and predictions about its future success or viability may not be as accurate as they could be if it had a longer operating history or a history of successfully developing and commercializing biopharmaceutical products.
The Company currently has no products approved for sale, and it may never obtain regulatory approval to commercialize any of its product candidates.
The research, testing, manufacturing, safety surveillance, efficacy, quality control, recordkeeping, labeling, packaging, storage, approval, sale, marketing, distribution, import, export and reporting of safety and other post-market information related to its biopharmaceutical products are subject to extensive regulation by the FDA and other regulatory authorities in the United States and in foreign countries, and such regulations differ from country to country and frequently are revised.
Even after the Company achieves U.S. regulatory approval for a product candidate, if any, the Company will be subject to continued regulatory review and compliance obligations. For example, with respect to the Company’s product candidates, the FDA may impose significant restrictions on the approved indicated uses for which the product may be marketed or on the conditions of approval. A product candidate’s approval may contain requirements for potentially costly post-approval studies and surveillance, including Phase 4 clinical trials, to monitor the safety and efficacy of the product. The Company also will be subject to ongoing FDA obligations and continued regulatory review with respect to, among other things, the manufacturing, processing, labeling, packaging, distribution, pharmacovigilance and adverse event reporting, storage, advertising, promotion and recordkeeping for the Company’s product candidates. These requirements include submissions of safety and other post-marketing information and reports, registration, continued compliance with cGMP requirements and with the FDA’s GCP requirements and GLP requirements, which are regulations and guidelines enforced by the FDA for all of the Company’s product candidates in clinical and preclinical development, and for any clinical trials that it conducts post-approval, as well as continued compliance with the FDA’s laws governing commercialization of the approved product, including but not limited to the FDA’s Office of Prescription Drug Promotion (“OPDP”) regulation of promotional activities, fraud and abuse, product sampling, scientific speaker engagements and activities, formulary interactions as well as interactions with healthcare practitioners. To the extent that a product candidate is approved for sale in other countries, the Company may be subject to similar or more onerous (i.e., prohibition on direct-to-consumer advertising that does not exist in the United States) restrictions and requirements imposed by laws and government regulators in those countries.
In addition, manufacturers of drug and biologic products and their facilities are subject to continual review and periodic inspections by the FDA and other regulatory authorities for compliance with cGMP regulations. If the Company or a regulatory agency discovers previously unknown problems with a product, such as adverse events of unanticipated severity or frequency, or problems with the manufacturing, processing, distribution or storage facility where, or processes by which, the product is made, a regulatory agency may impose restrictions on that product or the Company, including requesting that the Company initiate a product recall, or requiring notice to physicians or the public, withdrawal of the product from the market, or suspension of manufacturing.
If the Company, its product candidates or the manufacturing facilities for its product candidates fail to comply with applicable regulatory requirements, a regulatory agency may:
| | · | impose restrictions on the sale, marketing or manufacturing of the product, amend, suspend or withdraw product approvals or revoke necessary licenses; |
| | · | mandate modifications to promotional and other product-specific materials or require the Company to provide corrective information to healthcare practitioners or in its advertising; |
| | · | require the Company or its partners to enter into a consent decree, which can include imposition of various fines, reimbursements for inspection costs, required due dates for specific actions, penalties for noncompliance and, in extreme cases, require an independent compliance monitor to oversee the Company’s activities; |
| | · | issue warning letters, bring enforcement actions, initiate surprise inspections, issue show cause notices or untitled letters describing alleged violations, which may be publicly available; |
| | · | commence criminal investigations and prosecutions; |
| | · | impose injunctions, suspensions or revocations of necessary approvals or other licenses; |
| | · | impose other civil or criminal penalties; |
| | · | suspend any ongoing clinical trials; |
| | · | place restrictions on the kind of promotional activities that can be done; |
| | · | delay or refuse to approve pending applications or supplements to approved applications filed by the Company or its potential partners; |
| | · | refuse to permit drugs or precursor chemicals to be imported or exported to or from the United States; |
| | · | suspend or impose restrictions on operations, including costly new manufacturing requirements; or |
| | · | seize or detain products or require the Company or its partners to initiate a product recall. |
The regulations, policies or guidance of the FDA and other applicable government agencies may change, and new or additional statutes or government regulations may be enacted, including at the state and local levels, which can differ by geography and could prevent or delay regulatory approval of the Company’s product candidates or further restrict or regulate post-approval activities. The Company cannot predict the likelihood, nature or extent of adverse government regulations that may arise from future legislation or administrative action, either in the United States or abroad. If the Company is not able to achieve and maintain regulatory compliance, it may not be permitted to commercialize its product candidates, which would adversely affect its ability to generate revenue and achieve or maintain profitability.
The Company currently has no marketing capabilities and no sales organization. If the Company is unable to establish sales and marketing capabilities on its own or through third parties, the Company will be unable to successfully commercialize its product candidates, if approved, or generate product revenue.
The Company currently has no marketing capabilities and no sales organization. To commercialize the Company’s product candidates, if approved, in the United States and other jurisdictions, the Company must build its marketing, sales, distribution, managerial and other non-technical capabilities or make arrangements with third parties to perform these services, and the Company may not be successful in doing so. Although the Company’s employees, consultants, contractors, and partners have experience in the marketing, sale and distribution of pharmaceutical products, and business development activities involving external alliances, from prior employment at other companies, the Company as a company has no prior experience in the marketing, sale and distribution of pharmaceutical products, and there are significant risks involved in building and managing a sales organization, including its ability to hire, retain and incentivize qualified individuals, generate sufficient sales leads, provide adequate training to sales and marketing personnel, and effectively manage a geographically dispersed sales and marketing team. Any failure or delay in the development of the Company’s internal sales, marketing, distribution and pricing/reimbursement/access capabilities would impact adversely the commercialization of these products.
The Company may face product liability exposure, and if successful claims are brought against it, the Company may incur substantial liability if its insurance coverage for those claims is inadequate.
The Company faces an inherent risk of product liability or similar causes of action as a result of the clinical testing of its product candidates and will face an even greater risk if the Company commercializes any products. This risk exists even if a product is approved for commercial sale by the FDA and manufactured in facilities licensed and regulated by the FDA or an applicable foreign regulatory authority and notwithstanding the Company complying with applicable laws on promotional activity. The Company’s products and product candidates are designed to affect important bodily functions and processes. Any side effects, manufacturing defects, misuse or abuse associated with the Company’s product candidates could result in injury to a patient or potentially even death. The Company cannot offer any assurance that it will not face product liability suits in the future, nor can it assure that its insurance coverage will be sufficient to cover its liability under any such cases.
In addition, a liability claim may be brought against the Company even if its product candidates merely appear to have caused an injury. Product liability claims may be brought against the Company by consumers, healthcare providers, pharmaceutical companies or others selling or otherwise coming into contact with its product candidates, among others, and under some circumstances even government agencies. If the Company cannot successfully defend itself against product liability or similar claims, it will incur substantial liabilities, reputational harm and possibly injunctions and punitive actions. In addition, regardless of merit or eventual outcome, product liability claims may result in:
| | · | withdrawal or delay of recruitment or decreased enrollment rates of clinical trial participants; |
| | · | termination or increased government regulation of clinical trial sites or entire trial programs; |
| | · | the inability to commercialize the Company’s product candidates; |
| | · | decreased demand for the Company’s product candidates; |
| | · | impairment of the Company’s business reputation; |
| | · | product recall or withdrawal from the market or labeling, marketing or promotional restrictions; |
| | · | substantial costs of any related litigation or similar disputes; |
| | · | distraction of management’s attention and other resources from the Company’s primary business; |
| | · | significant delay in product launch; |
| | | |
| | · | substantial monetary awards to patients or other claimants against the Company that may not be covered by insurance; |
| | · | withdrawal of reimbursement or formulary inclusion; or |
The Company intends to obtain product liability insurance coverage for its clinical trials. Large judgments have been awarded in class action or individual lawsuits based on drugs that had unanticipated side effects. The Company’s insurance coverage may not be sufficient to cover all of its product liability-related expenses or losses and may not cover it for any expenses or losses it may suffer. Moreover, insurance coverage is becoming increasingly expensive, restrictive and narrow, and, in the future, the Company may not be able to maintain adequate insurance coverage at a reasonable cost, in sufficient amounts or upon adequate terms to protect it against losses due to product liability or other similar legal actions. The Company will need to increase its product liability coverage if any of its product candidates receive regulatory approval, which will be costly, and it may be unable to obtain this increased product liability insurance on commercially reasonable terms or at all and for all geographies in which the Company wishes to launch. A successful product liability claim or series of claims brought against the Company, if judgments exceed its insurance coverage, could decrease its cash and harm its business, financial condition, operating results and future prospects.
The Company’s employees, independent contractors, principal investigators, other clinical trial staff, consultants, vendors, CROs and any partners with whom the Company may collaborate may engage in misconduct or other improper activities, including noncompliance with regulatory standards and requirements.
The Company is exposed to the risk that its employees, independent contractors, principal investigators, other clinical trial staff, consultants, vendors, CROs and any partners with which the Company may collaborate may engage in fraudulent or other illegal activity. Misconduct by these persons could include intentional, reckless, gross or negligent misconduct or unauthorized activity that violates: laws or regulations, including those laws requiring the reporting of true, complete and accurate information to the FDA or foreign regulatory authorities; manufacturing standards; federal, state and foreign healthcare fraud and abuse laws and data privacy; anticorruption laws, antikickback and Medicare/Medicaid rules, or laws that require the true, complete and accurate reporting of financial information or data, books and records. If any such or similar actions are instituted against the Company and the Company is not successful in defending itself or asserting the Company’s rights, those actions could have a significant impact on the Company’s business, including the imposition of civil, criminal and administrative and punitive penalties, damages, monetary fines, possible exclusion from participation in Medicare, Medicaid and other federal healthcare programs, debarments, contractual damages, reputational harm, diminished profits and future earnings, injunctions, and curtailment or cessation of the Company’s operations, any of which could adversely affect the Company’s ability to operate the Company’s business and the Company’s operating results.
The Company may be subject to risks related to off-label use of its product candidates.
The FDA strictly regulates the advertising and promotion of drug products, and drug products may only be marketed or promoted for their FDA approved uses, consistent with the product’s approved labeling. Advertising and promotion of any product candidate that obtains approval in the United States will be heavily scrutinized by the FDA, the Department of Justice, the Office of Inspector General of the Department of Health and Human Services, state attorneys general, members of Congress and the public. Violations, including promotion of the Company’s products for unapproved or off-label uses, are subject to enforcement letters, inquiries and investigations, and civil, criminal and/or administrative sanctions by the FDA. Additionally, advertising and promotion of any product candidate that obtains approval outside of the United States will be heavily scrutinized by relevant foreign regulatory authorities.
Even if the Company obtains regulatory approval for its product candidates, the FDA or comparable foreign regulatory authorities may require labeling changes or impose significant restrictions on a product’s indicated uses or marketing or impose ongoing requirements for potentially costly post-approval studies or post-market surveillance.
In the United States, engaging in impermissible promotion of the Company’s product candidates for off-label uses can also subject it to false claims litigation under federal and state statutes, which can lead to civil, criminal and/or administrative penalties and fines and agreements, such as a corporate integrity agreement, that materially restrict the manner in which the Company promotes or distributes its product candidates. If the Company does not lawfully promote its products once they have received regulatory approval, the Company may become subject to such litigation and, if it is not successful in defending against such actions, those actions could have a material adverse effect on its business, financial condition and operating results and even result in having an independent compliance monitor assigned to audit the Company’s ongoing operations for a lengthy period of time.
The Company’s or third party’s clinical trials may fail to demonstrate the safety and efficacy of its product candidates, or serious adverse or unacceptable side effects may be identified during their development, which could prevent or delay marketing approval and commercialization, increase the Company’s costs or necessitate the abandonment or limitation of the development of the product candidate.
Before obtaining marketing approvals for the commercial sale of any product candidate, the Company must demonstrate through lengthy, complex and expensive preclinical testing and clinical trials that such product candidate is both safe and effective for use in the applicable indication, and failures can occur at any stage of testing. Clinical trials often fail to demonstrate safety and are associated with side effects or have characteristics that are unexpected. Based on the safety profile seen in clinical testing, the Company may need to abandon development or limit development to more narrow uses in which the side effects or other characteristics are less prevalent, less severe or more tolerable from a risk-benefit perspective. The FDA or an IRB may also require that the Company suspend, discontinue, or limit clinical trials based on safety information. Such findings could further result in regulatory authorities failing to provide marketing authorization for the product candidate. Many pharmaceutical candidates that initially showed promise in early stage testing and which were efficacious have later been found to cause side effects that prevented further development of the drug candidate and, in extreme cases, the side effects were not seen until after the drug was marketed, causing regulators to remove the drug from the market post-approval.
The Company may expend its limited resources to pursue a particular indication and fail to capitalize on indications that may be more profitable or for which there is a greater likelihood of success.
Because the Company has limited financial and managerial resources, it is currently focusing only on development programs that it identifies for specific indications for its product candidates. As a result, the Company may forego or delay pursuit of opportunities for other indications, or with other potential product candidates that later prove to have greater commercial potential. the Company’s resource allocation decisions may cause it to fail to capitalize on viable commercial products or profitable market opportunities. The Company’s spending on current and future research and development programs for specific indications or future product candidates may not yield any commercially viable product. If the Company does not accurately evaluate the commercial potential or target market for its product candidates, it may not gain approval or achieve market acceptance of that candidate, and its business and financial results will be harmed.
The Company may choose not to continue developing or commercializing any of its product candidates, or may choose not to commercialize product candidates in approved indications, at any time during development or after approval, which would reduce or eliminate its potential return on investment for those product candidates.
At any time, the Company may decide to discontinue the development of any of its product candidates for a variety of reasons, including the appearance of new technologies that make its product obsolete, competition from a competing product or changes in or failure to comply with applicable regulatory requirements. If the Company terminates a program in which it has invested significant resources, the Company will not receive any return on its investment and it will have missed the opportunity to have allocated those resources to potentially more productive uses.
Healthcare reform measures could hinder or prevent the commercial success of the Company’s product candidates.
The current presidential administration and certain members of the majority of the U.S. Congress have sought to repeal all or part of the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, “Affordable Care Act”), and implement a replacement program. For example, the so-called “individual mandate” was repealed as part of tax reform legislation adopted in December 2017, such that the shared responsibility payment for individuals who fail to maintain minimum essential coverage under section 5000A of the Code was eliminated beginning in 2019. In addition, litigation may prevent some or all of the Affordable Care Act legislation from taking effect. For example, on December 14, 2018, the U.S. District Court for the Northern District of Texas held that the individual mandate is a critical and inseverable feature of the Affordable Care Act, and therefore, because it was repealed as part of the tax reform legislation, the remaining provisions of the Affordable Care Act are invalid as well. The impact of this ruling is stayed as it is appealed to the Fifth Circuit Court of Appeals. While the ruling will have no immediate effect, it is unclear how this decision, and subsequent appeals, if any, will impact the law. In 2019 and beyond, the Company may face additional uncertainties as a result of likely federal and administrative efforts to repeal, substantially modify or invalidate some or all of the provisions of the Affordable Care Act. There is no assurance that the Affordable Care Act, as amended in the future, will not adversely affect the Company’s business and financial results.
Additionally, in October 2018, the U.S. President proposed to lower Medicare Part B drug prices, in addition to contemplating other measures to lower prescription drug prices. While this proposal has not yet been enacted, the Company expects that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for its product candidates if approved or additional pricing pressures.
There are also calls to ban all direct-to-consumer advertising of pharmaceuticals, which would limit the Company’s ability to market its product candidates. The United States is in a minority of jurisdictions that allow this kind of advertising and its removal could limit the potential reach of a marketing campaign.
The Company may also be subject to stricter healthcare laws, regulation and enforcement, and its failure to comply with those laws could adversely affect its business, operations and financial condition.
Certain federal and state healthcare laws and regulations pertaining to fraud and abuse and patients’ rights are and will be applicable to the Company’s business. The Company is subject to regulation by both the federal government and the states in which it or its partners conduct business. The healthcare laws and regulations that may affect the Company’s ability to operate include: the federal Anti-Kickback Statute; federal civil and criminal false claims laws and civil monetary penalty laws; the federal Health Insurance Portability and Accountability Act of 1996, as amended by the Health Information Technology for Economic and Clinical Health Act; the Prescription Drug Marketing Act (for sampling of drug product among other things); the federal physician sunshine requirements under the Affordable Care Act; the Foreign Corrupt Practices Act as it applies to activities outside of the United States; the new federal Right-to-Try legislation; and state law equivalents of many of the above federal laws.
Because of the breadth of these laws and the narrowness of the statutory exceptions and safe harbors available, it is possible that some of the Company’s business activities could be subject to challenge under one or more of such laws. In addition, recent healthcare reform legislation has strengthened these laws. For example, the recently enacted Affordable Care Act, among other things, amended the intent requirement of the federal Anti-Kickback Statute and certain criminal healthcare fraud statutes. A person or entity no longer needs to have actual knowledge of the statute or specific intent to violate it. In addition, the Affordable Care Act provided that the government may assert that a claim including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal civil False Claims Act.
Achieving and sustaining compliance with these laws may prove costly. In addition, any action against the Company for violation of these laws, even if the Company successfully defends against it, could cause the Company to incur significant legal expenses and divert its management’s attention from the operation of its business and result in reputational damage. If the Company’s operations are found to be in violation of any of the laws described above or any other governmental laws or regulations that apply to the Company, it may be subject to penalties, including administrative, civil and criminal penalties, damages, including punitive damages, fines, disgorgement, the exclusion from participation in federal and state healthcare programs, individual imprisonment or the curtailment or restructuring of its operations, and injunctions, any of which could adversely affect the Company’s ability to operate its business and its financial results.
The Company’s inability to successfully to in-license, acquire, develop and market additional product candidates or approved products would impair its ability to grow its business.
The Company intends to in-license, acquire, develop and market additional products and product candidates. Because the Company’s internal research and development capabilities are limited, it may be dependent on pharmaceutical companies, academic or government scientists and other researchers to sell or license products or technology to it. The success of this strategy depends partly on the Company’s ability to identify and select promising pharmaceutical product candidates and products, negotiate licensing or acquisition agreements with their current owners, and finance these arrangements.
The process of proposing, negotiating and implementing a license or acquisition of a product candidate or approved product is lengthy and complex. Other companies, including some with substantially greater financial, marketing, sales and other resources, may compete with the Company for the license or acquisition of product candidates and approved products. The Company has limited resources to identify and execute the acquisition or in-licensing of third-party products, businesses and technologies and integrate them into its current infrastructure. Moreover, the Company may devote resources to potential acquisitions or licensing opportunities that are never completed, or the Company may fail to realize the anticipated benefits of such efforts. The Company may not be able to acquire the rights to additional product candidates on terms that it finds acceptable or at all.
Further, any product candidate that the Company acquires may require additional development efforts prior to commercial sale, including preclinical or clinical testing and approval by the FDA and applicable foreign regulatory authorities. All product candidates are prone to risks of failure typical of pharmaceutical product development, including the possibility that a product candidate will not be shown to be sufficiently safe and effective for approval by regulatory authorities. In addition, the Company cannot provide assurance that any approved products that it acquires will be manufactured or sold profitably or achieve market acceptance.
The Company may seek to avail itself of mechanisms to expedite the development or approval for product candidates it may pursue in the future, such as fast track or breakthrough designation, but such mechanisms may not actually lead to a faster development or regulatory review or approval process.
LB1148 has received Fast Track designation from the FDA for the treatment of postoperative GI dysfunction (which may present as feeding intolerance, ileus, necrotizing enterocolitis, etc.) associated with gut hypoperfusion injury in pediatric patients who underwent congenital heart disease repair surgery. In addition, the Company may seek fast track designation, breakthrough designation, orphan drug designation, rare pediatric disease designation, priority review, or accelerated approval for product candidates it may pursue in the future. For example, if a drug is intended for the treatment of a serious or life-threatening condition and the drug demonstrates the potential to address unmet medical needs for this condition, the drug sponsor may apply for FDA fast track designation. However, the FDA has broad discretion with regard to these mechanisms, and even if the Company believes a particular product candidate is eligible for any such mechanism, it cannot guarantee that the FDA would decide to grant it. Even if it does obtain fast track or priority review designation or pursue an accelerated approval pathway, the Company may not experience a faster development process, review, or approval compared to conventional FDA procedures. The FDA may withdraw a particular designation if it believes that the designation is no longer supported by data from the Company’s clinical development program.
The Company intends to seek breakthrough designation for LB1148 for the treatment of postoperative GI dysfunction associated with gut hypoperfusion injury in pediatric patients who underwent congenital heart disease repair surgery and for the treatment of postoperative GI dysfunction associated with major surgeries that risk disrupting the intestinal mucosal barrier. A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints. Designation as a breakthrough therapy is within the discretion of the FDA. Accordingly, even if the Company believes a product candidate meets the criteria for designation as a breakthrough therapy, the FDA may disagree and instead determine not to make such designation. The Company cannot be sure that its evaluation of a product candidate as qualifying for breakthrough therapy designation will meet the FDA’s requirements. In any event, the receipt of a breakthrough therapy designation for a product candidate may not result in a faster development process, review, or approval compared to conventional FDA procedures and does not assure ultimate approval by the FDA. In addition, even if one or more product candidates qualifies as a breakthrough therapy, the FDA may later decide that the product candidate no longer meets the conditions for qualification or may decide that the time period for FDA review or approval will not be shortened.
Designation of a drug or biologic as a rare pediatric disease therapy and/or as an orphan drug therapy may also come with accelerated approval rights. In addition, drugs and biologics can also obtain pediatric and/or orphan drug market exclusivity in the United States. Pediatric exclusivity, if granted, adds six months to existing exclusivity periods and patent terms and orphan drug designation may add additional years of exclusivity. However, even if one or more product candidates qualifies for rare pediatric disease designation and/or orphan drug designation, the FDA may later decide that the product candidate no longer meets the conditions for this designation or may decide that the time period for FDA review or approval will not be accelerated.
Risks Related to the Company’s Dependence on Third Parties
The Company expects to rely on collaborations with third parties for the successful development and commercialization of its product candidates.
The Company expects to rely upon the efforts of third parties for the successful development and commercialization of the Company’s current and future product candidates. The clinical and commercial success of the Company’s product candidates may depend upon maintaining successful relationships with third-party partners which are subject to a number of significant risks, including the following:
| · | the Company’s partners’ ability to execute their responsibilities in a timely, cost-efficient and compliant manner; |
| · | reduced control over delivery and manufacturing schedules; |
| · | price increases and product reliability; |
| · | manufacturing deviations from internal or regulatory specifications; |
| · | the failure of partners to perform their obligations for technical, market or other reasons; |
| · | misappropriation of the Company’s current or future product candidates; and |
| · | other risks in potentially meeting the Company’s current and future product commercialization schedule or satisfying the requirements of its end-users. |
| · | the Company cannot assure you that it will be able to establish or maintain third-party relationships in order to successfully develop and commercialize its product candidates. |
The Company relies completely on third-party contractors to supply, manufacture and distribute clinical drug supplies for its product candidates, which may include sole-source suppliers and manufacturers; the Company intends to rely on third parties for commercial supply, manufacturing and distribution if any of its product candidates receive regulatory approval; and the Company expects to rely on third parties for supply, manufacturing and distribution of preclinical, clinical and commercial supplies of any future product candidates.
The Company does not currently have, nor does it plan to acquire, the infrastructure or capability to supply, store, manufacture or distribute preclinical, clinical or commercial quantities of drug substances or products. Additionally, the Company has not entered into a long-term commercial supply agreement to provide it with such drug substances or products. As a result, the Company’s ability to develop its product candidates is dependent, and the Company’s ability to supply its products commercially will depend, in part, on the Company’s ability to obtain the active pharmaceutical ingredients (“APIs”) and other substances and materials used in its product candidates successfully from third parties and to have finished products manufactured by third parties in accordance with regulatory requirements and in sufficient quantities for preclinical and clinical testing and commercialization. If the Company fails to develop and maintain supply and other technical relationships with these third parties, it may be unable to continue to develop or commercialize its products and product candidates.
The Company does not have direct control over whether its contract suppliers and manufacturers will maintain current pricing terms, be willing to continue supplying the Company with APIs and finished products or maintain adequate capacity and capabilities to serve its needs, including quality control, quality assurance and qualified personnel. The Company is dependent on its contract suppliers and manufacturers for day-to-day compliance with applicable laws and cGMPs for production of both APIs and finished products. If the safety or quality of any product or product candidate or component is compromised due to a failure to adhere to applicable laws or for other reasons, the Company may not be able to commercialize or obtain regulatory approval for the affected product or product candidate successfully, and the Company may be held liable for injuries sustained as a result.
In order to conduct larger or late-stage clinical trials for its product candidates and supply sufficient commercial quantities of the resulting drug product and its components, if that product candidate is approved for sale, the Company’s contract manufacturers and suppliers will need to produce its drug substances and product candidates in larger quantities, more cost-effectively and, in certain cases, at higher yields than they currently achieve. If the Company’s third-party contractors are unable to scale up the manufacture of any of its product candidates successfully in sufficient quality and quantity and at commercially reasonable prices, or are shut down or put on clinical hold by government regulators, and the Company is unable to find one or more replacement suppliers or manufacturers capable of production at a substantially equivalent cost in substantially equivalent volumes and quality, and the Company is unable to transfer the processes successfully on a timely basis, the development of that product candidate and regulatory approval or commercial launch for any resulting products may be delayed, or there may be a shortage in supply, either of which could significantly harm its business, financial condition, operating results and prospects.
The Company expects to continue to depend on third-party contract suppliers and manufacturers for the foreseeable future. the Company’s supply and manufacturing agreements, if any, do not guarantee that a contract supplier or manufacturer will provide services adequate for its needs. Additionally, any damage to or destruction of the Company’s third-party manufacturer’s or suppliers’ facilities or equipment, even by force majeure, may significantly impair its ability to have its products and product candidates manufactured on a timely basis. The Company’s reliance on contract manufacturers and suppliers further exposes it to the possibility that they, or third parties with access to their facilities, will have access to and may misappropriate the Company’s trade secrets or other proprietary information. In addition, the manufacturing facilities of certain of the Company’s suppliers may be located outside of the United States. This may give rise to difficulties in importing the Company’s products or product candidates or their components into the United States or other countries.
Risks Related to the Company’s Financial Operations
The Company has expressed substantial doubt about its ability to continue as a going concern.
Management has determined that there is substantial doubt about the Company’s ability to continue as a going concern due to uncertainties that the Company’s cash flows generated from its operations will be sufficient to meet its current operating costs and the Company’s future financial statements may include a similar qualification about its ability to continue as a going concern. The Company’s year-end and interim financial statements were prepared assuming that it will continue as a going concern and do not include any adjustments that may result from the outcome of this uncertainty.
If the Company is unable to meet its current operating costs, the Company would need to seek additional financing or modify its operational plans. If the Company seeks additional financing to fund its business activities in the future and there remains substantial doubt about its ability to continue as a going concern, investors or other financing sources may be unwilling to provide additional funding to the Company on commercially reasonable terms or at all.
The Company may be adversely affected by natural disasters and other catastrophic events and by man-made problems such as terrorism that could disrupt its business operations, and its business continuity and disaster recovery plans may not adequately protect it from a serious disaster.
The Company’s headquarters and main research facility are located in the greater San Diego area, which in the past has experienced severe earthquakes and fires. If these earthquakes, fires, other natural disasters, health pandemics or epidemics, terrorism and similar unforeseen events beyond its control, including for example the ongoing COVID-19 pandemic, prevented it from using all or a significant portion of its headquarters or research facility, it may be difficult or, in certain cases, impossible for the Company to continue its business for a substantial period of time. The Company does not have a disaster recovery or business continuity plan in place and may incur substantial expenses as a result of the absence or limited nature of the Company’s internal or third party service provider disaster recovery and business continuity plans, which, particularly when taken together with its lack of earthquake insurance, could have a material adverse effect on its business. Furthermore, integral parties in the Company’s supply chain are operating from single sites, increasing their vulnerability to natural disasters or other sudden, unforeseen and severe adverse events. If such an event were to affect its supply chain, it could have a material adverse effect on the Company’s ability to conduct clinical trials, its development plans and its business.
The Company’s business and operations would suffer in the event of system failures, cyber-attacks or a deficiency in its cyber-security.
Despite the implementation of security measures, the Company’s internal computer systems and those of its current and future CROs and other contractors and consultants are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failures. The risk of a security breach or disruption, particularly through cyber-attacks or cyber-intrusion, including by computer hackers, foreign governments, and cyber-terrorists, has generally increased as the number, intensity and sophistication of attempted attacks and intrusions from around the world have increased. While the Company has not experienced any such material system failure, accident or security breach to date, if such an event were to occur and cause interruptions in the Company’s operations, it could result in a material disruption of its development programs and its business operations. In addition, since the Company sponsors clinical trials, any breach that compromises patient data and identities causing a breach of privacy could generate significant reputational damage and legal liabilities and costs to recover and repair, including affecting trust in the Company to recruit for future clinical trials. For example, the loss of clinical trial data from completed or future clinical trials could result in delays in the Company’s regulatory approval efforts and significantly increase its costs to recover or reproduce the data. To the extent that any disruption or security breach were to result in a loss of, or damage to, the Company’s data or applications or inappropriate disclosure of confidential or proprietary information, the Company could incur liability and the further development and commercialization of its products and product candidates could be delayed.
Failure to remediate a material weakness in internal accounting controls could result in material misstatements in the Company’s financial statements.
The Company’s management has identified a material weakness in its internal control over financial reporting. The material weakness was due to a lack of controls in the financial closing and reporting process, including a lack of segregation of duties and the documentation and design of formalized processes and procedures surrounding the creation and posting of journal entries and account reconciliations. If not remediated, or if the Company identifies further material weaknesses in its internal controls, the Company’s failure to establish and maintain effective disclosure controls and procedures and internal control over financial reporting could result in material misstatements in its financial statements and a failure to meet its reporting and financial obligations.
Risks Related to the Company’s Intellectual Property
The Company may not be able to obtain, maintain or enforce global patent rights or other intellectual property rights that cover its product candidates and technologies that are of sufficient breadth to prevent third parties from competing against the Company.
The Company’s success with respect to its product candidates will depend, in part, on its ability to obtain and maintain patent protection in both the United States and other countries, to preserve its trade secrets and to prevent third parties from infringing on its proprietary rights. The Company’s ability to protect its product candidates from unauthorized or infringing use by third parties depends in substantial part on its ability to obtain and maintain valid and enforceable patents around the world.
The patent application process, also known as patent prosecution, is expensive and time-consuming, and the Company and its current or future licensors and licensees may not be able to prepare, file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner in all the countries that are desirable. It is also possible that the Company or its current licensors, or any future licensors or licensees, will fail to identify patentable aspects of inventions made in the course of development and commercialization activities before it is too late to obtain patent protection on them. Therefore, these and any of the Company’s patents and applications may not be prosecuted and enforced in a manner consistent with the best interests of its business. Moreover, the Company’s competitors independently may develop equivalent knowledge, methods and know-how or discover workarounds to the Company patents that would not constitute infringement. Any of these outcomes could impair the Company’s ability to enforce the exclusivity of its patents effectively, which may have an adverse impact on its business, financial condition and operating results.
Due to legal standards relating to patentability, validity, enforceability and claim scope of patents covering pharmaceutical inventions, the Company’s ability to obtain, maintain and enforce patents is uncertain and involves complex legal and factual questions especially across countries. Accordingly, rights under any existing patents or any patents the Company might obtain or license may not cover its product candidates or may not provide the Company with sufficient protection for its product candidates to afford a sustainable commercial advantage against competitive products or processes, including those from branded, generic and over-the-counter pharmaceutical companies. In addition, the Company cannot guarantee that any patents or other intellectual property rights will issue from any pending or future patent or other similar applications owned by or licensed to the Company. Even if patents or other intellectual property rights have issued or will issue, the Company cannot guarantee that the claims of these patents and other rights are or will be held valid or enforceable by the courts, through injunction or otherwise, or will provide the Company with any significant protection against competitive products or otherwise be commercially valuable to the Company in every country of commercial significance that the Company may target.
The Company’s ability to obtain and maintain valid and enforceable patents depends on whether the differences between its technology and the prior art allow its technology to be patentable over the prior art. The Company does not have outstanding issued patents covering all of the recent developments in its technology and is unsure of the patent protection that it will be successful in obtaining, if any. Even if the patents do successfully issue, third parties may design around or challenge the validity, enforceability or scope of such issued patents or any other issued patents the Company owns or licenses, which may result in such patents being narrowed, invalidated or held unenforceable. If the breadth or strength of protection provided by the patents the Company holds or pursues with respect to its product candidates is challenged, it could dissuade companies from collaborating with the Company to develop or threaten its ability to commercialize or finance its product candidates.
The laws of some foreign jurisdictions do not provide intellectual property rights to the same extent or duration as in the United States, and many companies have encountered significant difficulties in acquiring, maintaining, protecting, defending and especially enforcing such rights in foreign jurisdictions. If the Company encounters such difficulties in protecting or are otherwise precluded from effectively protecting its intellectual property in foreign jurisdictions, its business prospects could be substantially harmed, especially internationally.
Proprietary trade secrets and unpatented know-how are also very important to the Company’s business. Although the Company has taken steps to protect its trade secrets and unpatented know-how by entering into confidentiality agreements with third parties, and intellectual property protection agreements with officers, directors, employees, and certain consultants and advisors, there can be no assurance that binding agreements will not be breached or enforced by courts, that the Company would have adequate remedies for any breach, including injunctive and other equitable relief, or that its trade secrets and unpatented know-how will not otherwise become known, inadvertently disclosed by the Company or its agents and representatives, or be independently discovered by its competitors. If trade secrets are independently discovered, the Company would not be able to prevent their use and if the Company and its agents or representatives inadvertently disclose trade secrets and/or unpatented know-how, the Company may not be allowed to retrieve this and maintain the exclusivity it previously enjoyed.
The Company may not be able to protect its intellectual property rights throughout the world.
Filing, prosecuting and defending patents on the Company’s product candidates does not guarantee exclusivity. The requirements for patentability differ in certain countries, particularly developing countries. In addition, the laws of some foreign countries do not protect intellectual property rights to the same extent as laws in the United States, especially when it comes to granting use and other kinds of patents and what kind of enforcement rights will be allowed, especially injunctive relief in a civil infringement proceeding. Consequently, the Company may not be able to prevent third parties from practicing its inventions in all countries outside the United States and even in launching an identical version of the Company’s product notwithstanding the Company has a valid patent in that country. Competitors may use the Company’s technologies in jurisdictions where it has not obtained patent protection to develop their own products, or produce copy products, and, further, may export otherwise infringing products to territories where the Company has patent protection but enforcement on infringing activities is inadequate or where the Company has no patents. These products may compete with the Company’s products, and the Company’s patents or other intellectual property rights may not be effective or sufficient to prevent them from competing.
Many companies have encountered significant problems in protecting and defending intellectual property rights in foreign jurisdictions. The legal systems of certain countries, particularly certain developing countries, do not favor the enforcement of patents and other intellectual property protection, particularly those relating to pharmaceuticals, and the judicial and government systems are often corrupt, which could make it difficult for the Company to stop the infringement of its patents or marketing of competing products in violation of its proprietary rights generally. Proceedings to enforce its patent rights in foreign jurisdictions could result in substantial costs and divert its efforts and attention from other aspects of its business, could put its global patents at risk of being invalidated or interpreted narrowly and its global patent applications at risk of not issuing, and could provoke third parties to assert claims against it. The Company may not prevail in any lawsuits that the Company initiates or infringement actions brought against the Company, and the damages or other remedies awarded, if any, may not be commercially meaningful when the Company is the plaintiff. When the Company is the defendant it may be required to post large bonds to stay in the market while it defends itself from an infringement action.
In addition, certain countries in Europe and certain developing countries have compulsory licensing laws under which a patent owner may be compelled to grant licenses to third parties, especially if the patent owner does not enforce or use its patents over a protracted period of time. In some cases, the courts will force compulsory licenses on the patent holder even when finding the patent holder’s patents are valid if the court believes it is in the best interests of the country to have widespread access to an essential product covered by the patent. In these situations, the royalty the court requires to be paid by the license holder receiving the compulsory license is not calculated at fair market value and can be inconsequential, thereby disaffecting the patentholder’s business. In these countries, the Company may have limited remedies if its patents are infringed or if the Company is compelled to grant a license to its patents to a third party, which could also materially diminish the value of those patents. This would limit its potential revenue opportunities. Accordingly, the Company’s efforts to enforce its intellectual property rights around the world may be inadequate to obtain a significant commercial advantage from the intellectual property that the Company owns or licenses, especially in comparison to what it enjoys from enforcing its intellectual property rights in the Unites States. Finally, the Company’s ability to protect and enforce its intellectual property rights may be adversely affected by unforeseen changes in both U.S. and foreign intellectual property laws, or changes to the policies in various government agencies in these countries, including but not limited to the patent office issuing patents and the health agency issuing pharmaceutical product approvals. Finally, many countries have large backlogs in patent prosecution, and in some countries in Latin America it can take years, even decades, just to get a pharmaceutical patent application reviewed notwithstanding the merits of the application.
Obtaining and maintaining the Company’s patent protection depends on compliance with various procedural, document submission, fee payment, and other requirements imposed by governmental patent agencies, and its patent protection could be reduced or eliminated for non-compliance with these requirements.
Periodic maintenance and annuity fees on any issued patent are due to be paid to the USPTO and foreign patent agencies in several stages over the lifetime of the patent. The USPTO and various foreign governmental patent agencies require compliance with a number of procedural, documentary, fee payment and other similar provisions during the patent application process. While an inadvertent lapse can, in many cases, be cured by payment of a late fee or by other means in accordance with the applicable rules, there are situations in which noncompliance can result in abandonment or lapse of the patent or patent application, resulting in partial or complete loss of patent rights in the relevant jurisdiction just for failure to know about and/or timely pay a prosecution fee. Non-compliance events that could result in abandonment or lapse of a patent or patent application include failure to respond to official actions within prescribed time limits, non-payment of fees in prescribed time periods, and failure to properly legalize and submit formal documents in the format and style the country requires. If the Company or its licensors fail to maintain the patents and patent applications covering its product candidates for any reason, the Company’s competitors might be able to enter the market, which would have an adverse effect on the Company’s business.
If the Company fails to comply with its obligations under its intellectual property license agreements, it could lose license rights that are important to its business. Additionally, these agreements may be subject to disagreement over contract interpretation, which could narrow the scope of its rights to the relevant intellectual property or technology or increase its financial or other obligations to its licensors.
The Company has entered into in-license arrangements with respect to certain of its product candidates. These license agreements impose various diligence, milestone, royalty, insurance and other obligations on the Company. If the Company fails to comply with these obligations, the respective licensors may have the right to terminate the license, in which event the Company may not be able to develop or market the affected product candidate. The loss of such rights could materially adversely affect its business, financial condition, operating results and prospects.
If the Company is sued for infringing intellectual property rights of third parties, such litigation could be costly and time consuming and could prevent or delay it from developing or commercializing its product candidates.
The Company’s commercial success depends on its ability to develop, manufacture, market and sell its product candidates and use its proprietary technologies without infringing the proprietary rights of third parties. The Company cannot assure that marketing and selling such candidates and using such technologies will not infringe existing or future patents. Numerous U.S.- and foreign-issued patents and pending patent applications owned by third parties exist in the fields relating to its product candidates. As the biotechnology and pharmaceutical industries expand and more patents are issued, the risk increases that others may assert that its product candidates, technologies or methods of delivery or use infringe their patent rights. Moreover, it is not always clear to industry participants, including us, which patents and other intellectual property rights cover various drugs, biologics, drug delivery systems or their methods of use, and which of these patents may be valid and enforceable. Thus, because of the large number of patents issued and patent applications filed in the Company’s fields across many countries, there may be a risk that third parties may allege they have patent rights encompassing the Company’s product candidates, technologies or methods.
In addition, there may be issued patents of third parties that are infringed or are alleged to be infringed by the Company’s product candidates or proprietary technologies notwithstanding patents the Company may possess. Because some patent applications in the United States may be maintained in secrecy until the patents are issued, because patent applications in the United States and many foreign jurisdictions are typically not published until 18 months after filing and because publications in the scientific literature often lag behind actual discoveries, the Company cannot be certain that others have not filed patent applications for technology covered by its own and in-licensed issued patents or its pending applications. the Company’s competitors may have filed, and may in the future file, patent applications covering the Company’s own product candidates or technology similar to the Company’s technology. Any such patent application may have priority over the Company’s own and in-licensed patent applications or patents, which could further require the Company to obtain rights to issued patents covering such technologies, which may mean paying significant licensing fees or the like. If another party has filed a U.S. patent application on inventions similar to those owned or in-licensed to us, the Company or, in the case of in-licensed technology, the licensor may have to participate, in the United States, in an interference proceeding to determine priority of invention.
The Company may be exposed to, or threatened with, future litigation by third parties having patent or other intellectual property rights alleging that its product candidates or proprietary technologies infringe such third parties’ intellectual property rights, including litigation resulting from filing under Paragraph IV of the Hatch-Waxman Act or other countries’ laws similar to the Hatch-Waxman Act. These lawsuits could claim that there are existing patent rights for such drug, and this type of litigation can be costly and could adversely affect its operating results and divert the attention of managerial and technical personnel, even if the Company does not infringe such patents or the patents asserted against the Company is ultimately established as invalid. There is a risk that a court would decide that the Company is infringing the third party’s patents and would order the Company to stop the activities covered by the patents. In addition, there is a risk that a court will order the Company to pay the other party significant damages for having violated the other party’s patents.
Because the Company relies on certain third-party licensors and partners and will continue to do so in the future, if one of its licensors or partners is sued for infringing a third party’s intellectual property rights, the Company’s business, financial condition, operating results and prospects could suffer in the same manner as if the Company were sued directly. In addition to facing litigation risks, the Company has agreed to indemnify certain third-party licensors and partners against claims of infringement caused by the Company’s proprietary technologies, and the Company has entered or may enter into cost-sharing agreements with some its licensors and partners that could require the Company to pay some of the costs of patent litigation brought against those third parties whether or not the alleged infringement is caused by its proprietary technologies. In certain instances, these cost-sharing agreements could also require the Company to assume greater responsibility for infringement damages than would be assumed just on the basis of its technology.
The occurrence of any of the foregoing could adversely affect the Company’s business, financial condition or operating results.
The Company may be subject to claims that its officers, directors, employees, consultants or independent contractors have wrongfully used or disclosed to us alleged trade secrets of their former employers or their former or current customers.
As is common in the biotechnology and pharmaceutical industries, certain of the Company’s employees were formerly employed by other biotechnology or pharmaceutical companies, including its competitors or potential competitors. Moreover, we engage the services of consultants to assist us in the development of our products and product candidates, many of whom were previously employed at, or may have previously been or are currently providing consulting services to, other biotechnology or pharmaceutical companies, including its competitors or potential competitors. We may be subject to claims that these employees and consultants or we have inadvertently or otherwise used or disclosed trade secrets or other proprietary information of their former employers or their former or current customers. Although we have no knowledge of any such claims being alleged to date, if such claims were to arise, litigation may be necessary to defend against any such claims. Even if we are successful in defending against any such claims, any such litigation could be protracted, expensive, a distraction to its management team, not viewed favorably by investors and other third parties, and may potentially result in an unfavorable outcome.
Other Risks Related to the Company
The Company will need to raise additional financing in the future to fund its operations, which may not be available to it on favorable terms or at all.
The Company will require substantial additional funds to conduct the costly and time-consuming clinical efficacy trials necessary to pursue regulatory approval of LB1148 and any other product candidates. The Company’s future capital requirements will depend upon a number of factors, including: the number and timing of future product candidates in the pipeline; progress with and results from preclinical testing and clinical trials; the ability to manufacture sufficient drug supplies to complete preclinical and clinical trials; the costs involved in preparing, filing, acquiring, prosecuting, maintaining and enforcing patent and other intellectual property claims; and the time and costs involved in obtaining regulatory approvals and favorable reimbursement or formulary acceptance. Raising additional capital may be costly or difficult to obtain and could significantly dilute stockholders’ ownership interests or inhibit the Company’s ability to achieve its business objectives. If the Company raises additional funds through public or private equity offerings, the terms of these securities may include liquidation or other preferences that adversely the rights of its common stockholders. Further, to the extent that the combined company raises additional capital through the sale of common stock or securities convertible or exchangeable into common stock, its stockholder’s ownership interest in the Company will be diluted. In addition, any debt financing may subject the Company to fixed payment obligations and covenants limiting or restricting its ability to take specific actions, such as incurring additional debt, making capital expenditures or declaring dividends. If the Company raises additional capital through marketing and distribution arrangements or other collaborations, strategic alliances or licensing arrangements with third parties, the Company may have to relinquish certain valuable intellectual property or other rights to its product candidates, technologies, future revenue streams or research programs or grant licenses on terms that may not be favorable to it. Even if the Company were to obtain sufficient funding, there can be no assurance that it will be available on terms acceptable to the Company or its stockholders.
The Company’s business could be adversely affected by the effects of health pandemics or epidemics, including the recent COVID-19 pandemic, in regions where it or third parties on which it relies have significant manufacturing facilities, concentrations of clinical trial sites or other business operations. The COVID-19 pandemic could materially affect the Company’s operations, including at its headquarters in California, which has been in the past, and could be in the future, subject to a county-wide stay-at-home order, and at clinical trial sites, as well as the business or operations of manufacturers, CROs or other third parties with whom the Company conducts business.
The Company’s business could be adversely affected by the effects of health pandemics or epidemics in regions where it has concentrations of clinical trial sites or other business operations, and could cause significant disruption in the operations of third-party manufacturers and CROs upon whom it relies. For example, in December 2019, a novel strain of coronavirus, SARS-CoV-2, causing a disease referred to as COVID-19, was reported to have surfaced in Wuhan, China. Since then, COVID-19 has spread to most countries, including the United States and many other countries. The Company’s headquarters is located in San Diego County, California, and many of the Company’s raw materials for manufacture of LB1148 are produced in foreign countries. In March 2020, the World Health Organization declared the COVID-19 outbreak a pandemic and the U.S. government imposed travel restrictions on travel between the United States and numerous other countries. Further, the President of the United States declared the COVID-19 pandemic a national emergency, invoking powers under the Stafford Act, the legislation that directs federal emergency disaster response. Similarly, the State of California declared a state of emergency related to the spread of COVID-19. Further, on March 19, 2020 the State of California declared a statewide stay at home order for an indefinite period of time (subject to certain exceptions to facilitate authorized necessary activities) to mitigate the impact of the COVID-19 pandemic. Due to the stay at home order, the Company implemented work-from-home policies for all of its employees. The stay at home order has since expired and currently not in effect. The effects of future stay-at-home orders and work-from-home policies may negatively impact productivity, disrupt business and delay clinical programs and timelines, the magnitude of which will depend, in part, on the length and severity of the restrictions and other limitations on its ability to conduct business in the ordinary course. These and similar, and perhaps more severe, disruptions in operations could negatively impact the Company’s business, operating results and financial condition.
Quarantines, stay at home and similar government orders, or the perception that such orders, shutdowns or other restrictions on the conduct of business operations could occur, related to COVID-19 or other infectious diseases, may impact personnel at third-party manufacturing facilities in the United States and other countries, or the availability or cost of materials, which could disrupt the Company’s supply chain. In particular, some of the Company’s suppliers of certain materials used in the production of the Company’s drug products are located in countries outside the United States, where there have been government-imposed quarantines. While many of these materials may be obtained by more than one supplier, restrictions resulting from the COVID-19 pandemic may disrupt the Company’s supply chain or limit its ability to obtain sufficient materials for its product candidates.
In addition, the Company’s clinical trials may be affected by the COVID-19 pandemic. Clinical site initiation and patient enrollment may be delayed due to prioritization of hospital resources toward the COVID-19 pandemic. Some patients may not be able or willing to comply with clinical trial protocols if quarantines interrupt healthcare services, particularly surgical services. Similarly, the Company’s ability to recruit and retain patients, principal investigators and site staff (who as healthcare providers may have heightened exposure to COVID-19) may be hindered, which would adversely affect clinical trial operations. In addition, the COVID-19 pandemic may cause interruption or delays in the operation of the FDA or other regulatory authorities which could negatively affect the Company’s planned clinical trials.
The spread of COVID-19, which has caused a broad impact globally, may materially affect the Company economically. While the potential economic impact brought by, and the duration of, the COVID-19 pandemic may be difficult to assess or predict, it is currently resulting in significant disruption of global financial markets. This disruption, if sustained or recurrent, could make it more difficult for the Company to access capital, which could in the future negatively affect its liquidity. In addition, a recession or market correction resulting from the spread of COVID-19 could materially affect the Company’s business and the value of its common stock.
The global pandemic of COVID-19 continues to rapidly evolve. The ultimate impact of the COVID-19 pandemic or a similar health pandemic or epidemic is highly uncertain and subject to change. The Company does not yet know the full extent of potential delays or impacts on its business, its clinical trials, healthcare systems or the global economy as a whole. These effects could have a material impact on the Company’s operations, and it will continue to monitor the COVID-19 situation closely. To the extent the COVID-19 pandemic adversely affects the Company’s operations, it may also have the effect of heightening many of the other risks described in this “Risk Factors” section.
The stock price of the Company may be highly volatile.
The market price of shares of the Company could be subject to significant fluctuations. Since the completion of the Merger on April 27, 2021, the Company’s stock price has already been subject to significant fluctuation. Market prices for securities of biotechnology and other life sciences companies historically have been particularly volatile subject even to large daily price swings. Some of the factors that may cause the market price of shares of the Company to fluctuate include, but are not limited to:
| | · | the ability of the Company to obtain timely regulatory approvals for LB1148 or future product candidates, and delays or failures to obtain such approvals; |
| | · | failure of LB1148 if approved, to achieve commercial success; |
| | · | issues in manufacturing LB1148 or future product candidates; |
| | · | the results of current and any future clinical trials of LB1148; |
| | · | failure of other Company product candidates, if approved, to achieve commercial success; |
| | · | the entry into, or termination of, or breach by partners of key agreements, including key commercial partner agreements; |
| | | |
| | · | the initiation of, material developments in, or conclusion of any litigation to enforce or defend any intellectual property rights or defend against the intellectual property rights of others; |
| | · | announcements of any dilutive equity financings; |
| | · | announcements by commercial partners or competitors of new commercial products, clinical progress or the lack thereof, significant contracts, commercial relationships or capital commitments; |
| | · | failure to elicit meaningful stock analyst coverage and downgrades of the company’s stock by analysts; and |
| | · | the loss of key employees. |
Moreover, the stock markets in general have experienced substantial volatility in the biotech industry that has often been unrelated to the operating performance of individual companies or a certain industry segment. These broad market fluctuations may also adversely affect the trading price of the Company’s shares.
In the past, following periods of volatility in the market price of a company’s securities, shareholders have often instituted class action securities litigation against those companies. Such litigation, if instituted, could result in substantial costs and diversion of management attention and resources, which could significantly harm the Company’s profitability and reputation. In addition, such securities litigation often has ensued after a reverse merger or other merger and acquisition activity of the type that the Company recently completed with LBS. Such litigation if brought could impact negatively the Company’s business.
The Company is expected to take advantage of reduced disclosure and governance requirements applicable to smaller reporting companies, which could result in its common stock being less attractive to investors.
As of the date of this Quarterly Report, the public float of the Company is less than $250 million and therefore qualifies as a smaller reporting company under the rules of the SEC. As a smaller reporting company, the Company will be able to take advantage of reduced disclosure requirements, such as simplified executive compensation disclosures and reduced financial statement disclosure requirements in its SEC filings. Decreased disclosures in the Company’s SEC filings due to its status as a smaller reporting company may make it harder for investors to analyze its results of operations and financial prospects. We cannot predict if investors will find the Company’s common stock less attractive if it relies on these exemptions. If some investors find its common stock less attractive as a result, there may be a less active trading market for its common stock and its stock price may be more volatile. The Company may take advantage of the reporting exemptions applicable to a smaller reporting company until it is no longer a smaller reporting company, which status would end once it has a public float greater than $250 million. In that event, the Company could still be a smaller reporting company if its annual revenues were below $100 million and it has a public float of less than $700 million.
The Company does not anticipate paying any dividends in the foreseeable future.
The current expectation is that the Company will retain its future earnings to fund the development and growth of the company’s business. As a result, capital appreciation, if any, of the shares of the Company will be your sole source of gain, if any, for the foreseeable future.
If the Company fails to attract and retain management and other key personnel, it may be unable to successfully develop or commercialize its product candidates or otherwise implement its business plan.
The biotech industry has experienced a high rate of turnover in recent years. The Company’s ability to compete in the highly competitive biopharmaceuticals industry depends upon the ability to attract, retain and motivate highly skilled and experienced personnel with scientific, medical, regulatory, manufacturing and management skills and experience. The Company will conduct its operations in the greater San Diego area, a region that is home to many other biopharmaceutical companies as well as many academic and research institutions, resulting in fierce competition for qualified personnel. The Company may not be able to attract or retain qualified personnel in the future due to the intense competition for a limited number of qualified personnel among biopharmaceutical companies. Many of the other biopharmaceutical companies against which the Company will compete have greater financial and other resources, different risk profiles and a longer history in the industry. The Company’s competitors may provide higher compensation, more diverse opportunities and/or better opportunities for career advancement. Any or all of these competing factors may limit the Company’s ability to continue to attract and retain high quality personnel, which could negatively affect its ability to successfully develop and commercialize our product candidates and to grow the business and operations as currently contemplated.
The Company’s ability to use NOL carryforwards and certain other tax attributes may be limited.
The Company has incurred substantial losses during its history and does not expect to become profitable in the near future, and it may never achieve profitability. Unused losses for the tax year ended December 31, 2017 and prior tax years will carry forward to offset future taxable income, if any, until such unused losses expire. Pursuant to U.S. federal tax legislation enacted in late 2017, informally referenced as the Tax Cuts and Jobs Act, as modified under the Coronavirus Aid, Relief and Economic Security Act, or CARES Act, unused federal losses generated after December 31, 2017 will not expire and may be carried forward indefinitely but will be only deductible to the extent of 80% of current year taxable income in any given year. However, the CARES Act temporarily repealed the 80% taxable income limitation for tax years beginning before January 1, 2021; NOL carryforwards generated from 2018 or later and carried forward to taxable years beginning after December 31, 2020 will be subject to the 80% limitation. Also, under the CARES Act, NOLs arising in 2018, 2019 and 2020 can be carried back five years. Many states have similar laws. In addition, both current and future unused losses and other tax attributes may be subject to limitation under Sections 382 and 383 of the Internal Revenue Code of 1986, as amended, or the Code, if we undergo an “ownership change,” generally defined as a greater than 50 percentage point change (by value) in our equity ownership by certain stockholders over a three-year period. The Company has not completed a Section 382 study to assess whether an ownership change has occurred or whether there have been multiple ownership changes since its formation due to the complexity and cost associated with such a study and the fact that there may be additional such ownership changes in the future. As a result, if the Company earns net taxable income, its pre-2018 NOL carryforwards may expire prior to being used, its NOL carryforwards generated in 2018 and thereafter will be subject to a percentage limitation after 2020 and, if the Company undergoes an ownership change (or if it previously underwent such an ownership change), its ability to use all of the pre-change NOL carryforwards, and other pre-change tax attributes (such as research tax credits) to offset post-change income or taxes may be limited. Similar provisions of state tax law may also apply to limit our use of accumulated state tax attributes. In addition, at the state level, there may be periods during which the use of NOLs is suspended or otherwise limited, which could accelerate or permanently increase state taxes owed. As a result, even if the Company or the Company attains profitability, it may be unable to use all or a material portion of its NOLs and other tax attributes, which could adversely affect future cash flows.
Changes in tax law could adversely affect the Company’s business.
The rules dealing with U.S. federal, state and local income taxation are constantly under review by the Internal Revenue Service, the U.S. Treasury Department and other governmental bodies. Changes to tax laws (which changes may have retroactive application) could adversely affect the Company or holders of its common stock. In recent years, many such changes have been made and changes are likely to continue to occur in the future. Future changes in tax laws could have a material adverse effect on the Company’s business, cash flow, financial condition or results of operations.
The Company will incur costs and demands upon management as a result of complying with the laws and regulations affecting public companies.
The Company will incur significant legal, accounting and other expenses that the Company did not incur as a private company prior to the Merger, including costs associated with public company reporting requirements. The Company also incurs costs associated with corporate governance requirements, including requirements under the Sarbanes-Oxley Act, as well as new implemented requirements by the SEC and Nasdaq. These rules and regulations are expected to increase the Company’s legal and financial compliance costs and to make some activities more time consuming and costly. For example, the Company’s management team consists of the executive officers of the Company prior to the Merger, some of whom have not previously managed and operated a public company. These executive officers and other personnel need to devote substantial time to gaining expertise regarding operations as a public company and compliance with applicable laws and regulations. These rules and regulations also may make it difficult and expensive for the Company to obtain directors’ and officers’ liability insurance. As a result, it may be more difficult for the Company to attract and retain qualified individuals to serve on the Company’s board of directors or as executive officers of the Company, which may adversely affect investor confidence in the Company and could cause its business or stock price to suffer.
Anti-takeover provisions in the Company’s charter documents and under Delaware law could make an acquisition of the Company more difficult and may prevent attempts by the Company stockholders to replace or remove the Company management.
Provisions in the Company’s certificate of incorporation and bylaws may delay or prevent an acquisition or a change in management. In addition, because the Company is incorporated in Delaware, it is governed by the provisions of Section 203 of the DGCL, which prohibits stockholders owning in excess of 15% of the outstanding Company voting stock from merging or combining with the Company. Although the Company believes these provisions collectively will provide for an opportunity to receive higher bids by requiring potential acquirors to negotiate with the Company’s board of directors, they would apply even if the offer may be considered beneficial by some stockholders. In addition, these provisions may frustrate or prevent any attempts by the Company’s stockholders to replace or remove then current management by making it more difficult for stockholders to replace members of the board of directors, which is responsible for appointing the members of management.
If equity research analysts do not publish research or reports, or publish unfavorable research or reports, about the Company, its business or its market, its stock price and trading volume could decline.
The trading market for the Company’s common stock is and will be influenced by the research and reports that equity research analysts publish about it and its business. Equity research analysts may elect not to provide research coverage of the Company’s common stock, and such lack of research coverage may adversely affect the market price of its common stock. In the event it does have equity research analyst coverage, the Company will not have any control over the analysts, or the content and opinions included in their reports. The price of the Company’s common stock could decline if one or more equity research analysts downgrade its stock or issue other unfavorable commentary or research. If one or more equity research analysts ceases coverage of the Company or fails to publish reports on it regularly, demand for its common stock could decrease, which in turn could cause its stock price or trading volume to decline.
If the Company fails to maintain proper and effective internal controls, its ability to produce accurate financial statements on a timely basis could be impaired.
The Company is subject to the reporting requirements of the Exchange Act, the Sarbanes-Oxley Act and the rules and regulations of Nasdaq. The Sarbanes-Oxley Act requires, among other things, that the Company maintain effective disclosure controls and procedures and internal control over financial reporting. The Company must perform system and process evaluation and testing of its internal control over financial reporting to allow management to report on the effectiveness of its internal controls over financial reporting in its Annual Report on Form 10-K filing for that year, as required by Section 404 of the Sarbanes-Oxley Act. This has required that the Company incur substantial professional fees and internal costs to expand its accounting and finance functions and that it expend significant management efforts. The Company may experience difficulty in meeting these reporting requirements in a timely manner.
The Company may discover weaknesses in its system of internal financial and accounting controls and procedures that could result in a material misstatement of its financial statements. Prior to the Merger, LBS’s management identified a material weakness in its internal control over financial reporting. The material weakness was due to a lack of controls in the financial closing and reporting process for LBS, including a lack of segregation of duties and the documentation and design of formalized processes and procedures surrounding the creation and posting of journal entries and account reconciliations. If the Company does not remediate this material weakness, or if the Company identifies further material weaknesses in its internal controls, the Company’s failure to establish and maintain effective internal financial and accounting controls and procedures could result in material misstatements in its financial statements and a failure to meet its reporting and financial obligations.
The Company’s internal control over financial reporting will not prevent or detect all errors and all fraud. A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be met. Because of the inherent limitations in all control systems, no evaluation of controls can provide absolute assurance that misstatements due to error or fraud will not occur or that all control issues and instances of fraud will be detected.
If the Company is not able to comply with the requirements of Section 404 of the Sarbanes-Oxley Act, or if it is unable to maintain proper and effective internal controls, the Company may not be able to produce timely and accurate financial statements. If that were to happen, the market price of its common stock could decline and it could be subject to sanctions or investigations by Nasdaq, the SEC or other regulatory authorities.
Our board of directors has broad discretion to issue additional securities, which might dilute the net tangible book value per share of our common stock for existing stockholders.
The Company is entitled under its certificate of incorporation to issue up to 300,000,000 shares of common stock and 7,000,000 “blank check” shares of preferred stock. Shares of the Company’s blank check preferred stock provide its board of directors with broad authority to determine voting, dividend, conversion, and other rights. The Company expects that significant additional capital may be needed in the future to continue its planned operations. To the extent the Company raises additional capital by issuing equity securities, its existing shareholders may experience substantial dilution. The Company may sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner the Company determines from time to time. If the Company sells common stock, convertible securities or other equity securities in more than one transaction, investors may be materially diluted by subsequent sales. These sales may also result in material dilution to the Company’s existing shareholders, and new investors could gain rights superior to existing shareholders. Pursuant to the Company’s equity incentive plans and employee stock purchase plan, management is authorized to grant stock options, restricted stock units and other equity-based awards to employees, directors and consultants, and to sell common stock to employees, respectively. Any increase in the number of shares outstanding as a result of the exercise of outstanding options, the vesting or settlement of outstanding stock awards, or the purchase of shares pursuant to the employee stock purchase plan will cause shareholders to experience additional dilution, which could cause the stock price to fall.
USE OF PROCEEDS
We will not receive any of the proceeds from the sale or other disposition of shares of our common stock held by the selling stockholder pursuant to this prospectus. We will bear the out-of-pocket costs, expenses and fees incurred in connection with the registration of shares of our common stock to be sold by the selling stockholder, including registration, listing and qualifications fees, printers and accounting fees, and fees and disbursements of counsel, or collectively, the Registration Expenses. Other than Registration Expenses, the selling stockholder will bear underwriting discounts, commissions, placement agent fees or other similar expenses payable with respect to sales of shares.
SELLING STOCKHOLDERS
We are registering the resale of 7,262,460 shares of common stock which are issuable upon the exercise of Warrants held by the selling stockholder identified below to permit such selling stockholder, or their permitted transferees or other successors-in-interest that may be identified in a supplement to this prospectus or, if required, a post-effective amendment to the registration statement of which this prospectus is a part, to resell or otherwise dispose of these shares in the manner contemplated under the section entitled “Plan of Distribution” in this prospectus (as may be supplemented and amended).
The selling stockholder may sell some, all or none of their shares. We do not know how long the selling stockholder will hold the shares before selling them, and we currently have no agreements, arrangements or understandings with the selling stockholder regarding the sale or other disposition of any of the shares. The shares covered hereby may be offered from time to time by the selling stockholder. As a result, we cannot estimate the number of shares of common stock each of the selling stockholder will beneficially own after termination of sales under this prospectus. In addition, the selling stockholder may have sold, transferred or otherwise disposed of all or a portion of its shares of common stock since the date on which it provided information for this table.
Beneficial ownership is determined in accordance with the rules of the SEC and includes voting or investment power with respect to our common stock. Generally, a person “beneficially owns” shares of our common stock if the person has or shares with others the right to vote those shares or to dispose of them, or if the person has the right to acquire voting or disposition rights within 60 days.
Under the terms of the Equity SPA, the selling stockholder may not release shares of our common stock from escrow to the extent such delivery would cause the selling stockholder, together with its affiliates, to beneficially own a number of shares of common stock which would exceed 9.99% of our then outstanding common stock following such delivery, excluding for purposes of such determination common stock held in escrow which have not been delivered to the selling stockholder. The number of shares in the second column reflect this limitation.
Under the terms of the Warrants, the selling stockholder may not exercise the Warrants to the extent such exercise would cause the selling stockholder, together with its affiliates, to beneficially own a number of shares of common stock which would exceed 4.99% of our then outstanding common stock following such exercise, excluding for purposes of such determination common stock issuable upon exercise of the Warrants which have not been exercised. The number of shares in the second and fourth columns and the percentage in the fourth column reflect this limitation.
The information in the table below and the footnotes thereto regarding shares of common stock to be beneficially owned after the offering assumes the exercise of the Warrants by the selling stockholder and sale of all shares being offered by the selling stockholder under this prospectus. The percentage of shares owned prior to and after the offering is based on 11,279,716 shares of common stock outstanding as of July 21, 2021.
| | | | Before Offering | | | | | | | After Offering | |
| Name and Address | | | Number of
Shares
Beneficially
Owned | | | | | Number of
Shares
Offered | | | | Number of Shares
Beneficially
Owned(1) | | | | Percentage
of Shares
Beneficially
Owned | |
Altium Growth Fund, LP(2) 152 West 57th Street, 20th Floor New York, NY 10019 | | | 1,126,843 | (3) | | | | 7,262,460 | (4) | | | 1,760,809 | (5) | | | 9.50 | % |
| (1) | Assumes the exercise of the Warrants and sale of all shares available for sale under this prospectus and no further acquisitions of shares by the selling stockholder. |
| (2) | Altium Capital Management, LP, the investment manager of Altium Growth Fund, LP (the "Altium Fund"), has voting and investment power over these securities. Jacob Gottlieb is the managing member of Altium Capital Growth GP, LLC, which is the general partner of the Altium Fund. Each of the Altium Fund and Jacob Gottlieb disclaims beneficial ownership over these shares. |
| (3) | The number of shares beneficially owned by the Altium Fund set forth in the second column consists of 1,126,843 shares of common stock and reflect (i) the 9.99% beneficial ownership limitation described above applicable to shares purchased by Altium pursuant to the Private Placement Agreement and exchanged for shares of Common Stock pursuant to the Merger Agreement but issued by us to an escrow agent and (ii) the 4.99% beneficial ownership limitation described above set forth in the Warrants held by the Altium Fund. |
| (4) | Consists of 7,262,460 shares of common stock issuable upon the exercise of Warrants held by the Altium Fund. |
| (5) | Consists of 1,760,809 shares of common stock held by the Altium Fund. |
Relationship with Selling Stockholder
As discussed in greater detail above under the section entitled “Prospectus Summary” we, or our subsidiary, entered into agreements with the selling stockholder pursuant to which it acquired the Warrants, and agreed with the selling stockholder to file a registration statement to enable the resale of the shares of common stock issuable upon the exercise of the Warrants.
Except as a result of the transactions with the selling stockholder described herein, the selling stockholder has not, within the prior three years, been the beneficial owner of more than 5% of our common stock. None of, nor any persons having control over, the selling stockholder has held any position or office with us or our affiliates within the last three years or has had a material relationship with us or any of our predecessors or affiliates within the past three years, other than as a result of the ownership of our shares or other debt or equity securities.
PLAN OF DISTRIBUTION
We are registering the shares of common stock issued and issuable upon exercise of the warrants to permit the resale of these shares of common stock by the holders of the common stock warrants from time to time after the date of this prospectus. We will not receive any of the proceeds from the sale by the selling stockholders of the shares of common stock. We will bear all fees and expenses incident to our obligation to register the shares of common stock.
The selling stockholders may sell all or a portion of the shares of common stock beneficially owned by them and offered hereby from time to time directly or through one or more underwriters, broker-dealers or agents. If the shares of common stock are sold through underwriters or broker-dealers, the selling stockholders will be responsible for underwriting discounts or commissions or agent's commissions. The shares of common stock may be sold in one or more transactions at fixed prices, at prevailing market prices at the time of the sale, at varying prices determined at the time of sale, or at negotiated prices. These sales may be effected in transactions, which may involve crosses or block transactions,
| · | on any national securities exchange or quotation service on which the securities may be listed or quoted at the time of sale; |
| · | in the over-the-counter market; |
| · | in transactions otherwise than on these exchanges or systems or in the over-the-counter market; |
| · | through the writing of options, whether such options are listed on an options exchange or otherwise; |
| · | ordinary brokerage transactions and transactions in which the broker-dealer solicits purchasers; |
| · | block trades in which the broker-dealer will attempt to sell the shares as agent but may position and resell a portion of the block as principal to facilitate the transaction; |
| · | purchases by a broker-dealer as principal and resale by the broker-dealer for its account; |
| · | an exchange distribution in accordance with the rules of the applicable exchange; |
| · | privately negotiated transactions; |
| · | sales pursuant to Rule 144; |
| · | broker-dealers may agree with the selling securityholders to sell a specified number of such shares at a stipulated price per share; |
| · | a combination of any such methods of sale; and |
| · | any other method permitted pursuant to applicable law. |
If the selling stockholders effect such transactions by selling shares of common stock to or through underwriters, broker-dealers or agents, such underwriters, broker-dealers or agents may receive commissions in the form of discounts, concessions or commissions from the selling stockholders or commissions from purchasers of the shares of common stock for whom they may act as agent or to whom they may sell as principal (which discounts, concessions or commissions as to particular underwriters, broker-dealers or agents may be in excess of those customary in the types of transactions involved). In connection with sales of the shares of common stock or otherwise, the selling stockholders may enter into hedging transactions with broker-dealers, which may in turn engage in short sales of the shares of common stock in the course of hedging in positions they assume. The selling stockholders may also sell shares of common stock short and deliver shares of common stock covered by this prospectus to close out short positions and to return borrowed shares in connection with such short sales. The selling stockholders may also loan or pledge shares of common stock to broker-dealers that in turn may sell such shares.
The selling stockholders may pledge or grant a security interest in some or all of the warrants or shares of common stock owned by them and, if they default in the performance of their secured obligations, the pledgees or secured parties may offer and sell the shares of common stock from time to time pursuant to this prospectus or any amendment to this prospectus under Rule 424(b)(3) or other applicable provision of the Securities Act of 1933, as amended, amending, if necessary, the list of selling stockholders to include the pledgee, transferee or other successors in interest as selling stockholders under this prospectus. The selling stockholders also may transfer and donate the shares of common stock in other circumstances in which case the transferees, donees, pledgees or other successors in interest will be the selling beneficial owners for purposes of this prospectus.
The selling stockholders and any broker-dealer participating in the distribution of the shares of common stock may be deemed to be "underwriters" within the meaning of the Securities Act, and any commission paid, or any discounts or concessions allowed to, any such broker-dealer may be deemed to be underwriting commissions or discounts under the Securities Act. At the time a particular offering of the shares of common stock is made, a prospectus supplement, if required, will be distributed which will set forth the aggregate amount of shares of common stock being offered and the terms of the offering, including the name or names of any broker-dealers or agents, any discounts, commissions and other terms constituting compensation from the selling stockholders and any discounts, commissions or concessions allowed or reallowed or paid to broker-dealers.
Under the securities laws of some states, the shares of common stock may be sold in such states only through registered or licensed brokers or dealers. In addition, in some states the shares of common stock may not be sold unless such shares have been registered or qualified for sale in such state or an exemption from registration or qualification is available and is complied with.
There can be no assurance that any selling stockholder will sell any or all of the shares of common stock registered pursuant to the registration statement, of which this prospectus forms a part.
The selling stockholders and any other person participating in such distribution will be subject to applicable provisions of the Securities Exchange Act of 1934, as amended, and the rules and regulations thereunder, including, without limitation, Regulation M of the Exchange Act, which may limit the timing of purchases and sales of any of the shares of common stock by the selling stockholders and any other participating person. Regulation M may also restrict the ability of any person engaged in the distribution of the shares of common stock to engage in market-making activities with respect to the shares of common stock. All of the foregoing may affect the marketability of the shares of common stock and the ability of any person or entity to engage in market-making activities with respect to the shares of common stock.
We will pay all expenses of the registration of the shares of common stock pursuant to the registration rights agreement, estimated to be $80,000 in total, including, without limitation, Securities and Exchange Commission filing fees and expenses of compliance with state securities or "blue sky" laws; provided, however, that a selling stockholder will pay all underwriting discounts and selling commissions, if any. We will indemnify the selling stockholders against liabilities, including some liabilities under the Securities Act, in accordance with the registration rights agreements, or the selling stockholders will be entitled to contribution. We may be indemnified by the selling stockholders against civil liabilities, including liabilities under the Securities Act, that may arise from any written information furnished to us by the selling stockholder specifically for use in this prospectus, in accordance with the related registration rights agreement, or we may be entitled to contribution.
Once sold under the registration statement, of which this prospectus forms a part, the shares of common stock will be freely tradable in the hands of persons other than our affiliates.
EXPERTS
The financial statements of Leading Biosciences, Inc. as of December 31, 2020 and 2019 and for the years then ended incorporated by reference in this prospectus and in the registration statement have been so incorporated in reliance upon the report of BDO USA LLP, an independent registered public accounting firm, incorporated herein by reference, given on the authority of said firm as experts in auditing and accounting. The report on the financial statements contains an explanatory paragraph regarding the Company’s ability to continue as a going concern.
The consolidated financial statements of Seneca Biopharma, Inc. at December 31, 2020 and 2019, and for each of the years then ended, incorporated by reference in this prospectus and registration statement have been audited by Dixon Hughes Goodman LLP, independent registered public accounting firm, as set forth in their report (which contains an explanatory paragraph describing conditions that raise substantial doubt about Seneca Biopharma, Inc.’s ability to continue as a going concern as described in Note 1 to the consolidated financial statements), incorporated herein by reference, and are included in reliance upon such report given on the authority of such firm as experts in accounting and auditing.
LEGAL MATTERS
Certain legal matters, including the validity of the shares of common stock offered pursuant to this registration statement, will be passed upon for us by Cooley LLP, San Diego, California.
WHERE YOU CAN FIND MORE INFORMATION
We must comply with the informational requirements of the Exchange Act, and we are required to file reports and proxy statements and other information with the SEC. You may read and copy these reports, proxy statements and other information at the Public Reference Room maintained by the SEC at 100 F Street, N.E., Washington, D.C. 20549. You may also obtain copies at the prescribed rates from the Public Reference Section of the Securities and Exchange Commission at its principal office in Washington, D.C. You may call the SEC at 1-800-SEC-0330 for further information about the public reference room. The SEC also maintains a website that contains reports, proxy and information statements and other information regarding issuers like us that file electronically with the SEC. You may access the SEC’s web site at http://www.sec.gov. We maintain a website at www.palisadebio.com. The information contained in, or that can be accessed through, our website is not incorporated by reference herein and is not part of this prospectus.
Statements contained in this prospectus as to the contents of any contract or other document are not necessarily complete, and in each instance we refer you to the copy of the contract or document filed as an exhibit to the registration statement, each such statement being qualified in all respects by such reference.
INCORPORATION OF CERTAIN INFORMATION BY REFERENCE
The SEC allows us to incorporate by reference in this prospectus the information that we file with it. Incorporation by reference means that we can disclose important information to you by referring you to other documents that are legally considered to be part of this prospectus. Later information that we file with the SEC will automatically update and supersede the information in this prospectus, any supplement and the documents listed below. Our SEC file number is 001-36912. We incorporate by reference the specific documents listed below and any future filings made with the SEC under Section 13(a), 13(c), 14, or 15(d) of the Exchange Act, as amended, until all of the shares of common stock covered by this prospectus are sold:
| · | our Annual Report on Form 10-K for the year ended December 31, 2020, filed with the SEC on March 22, 2021; |
| · | our Quarterly Report on Form 10-Q for the fiscal quarter ended March 31, 2021, filed with the SEC on May 14, 2021; |
| · | the description of our common stock in our registration statement on Form 8-A filed with the SEC on July 1, 2015, including any amendments or reports filed for the purpose of updating such description; and |
| · | all documents filed by us with the SEC pursuant to Sections 13(a), 13(c), 14 and 15(d) of the Exchange Act (other than Current Reports furnished under Item 2.02 or Item 7.01 of Form 8-K and exhibits filed on such forms that are related to such items) after the date of the initial registration statement of which this prospectus is a part and prior to effectiveness of such registration statement, provided that all documents “furnished” by the Company to the SEC and not “filed” are not deemed incorporated by reference herein. |
We will furnish without charge to each person, including any beneficial owner, to whom this prospectus is delivered, upon written or oral request, a copy of any document incorporated by reference. Requests should be addressed to 5800 Armada Drive, Suite 210, Carlsbad, CA 92008, Attn: Secretary or may be made telephonically at (858) 704-4900.
You should rely only on the information incorporated by reference or provided in this prospectus or any prospectus supplement. We have not authorized anyone to provide you with different information. You should not assume that the information contained in this prospectus or the accompanying prospectus supplement is accurate on any date subsequent to the date set forth on the front of the document or that any information we have incorporated by reference is correct on any date subsequent to the date of the document incorporated by reference, even though this prospectus and any accompanying prospectus supplement is delivered or securities are sold on a later date.
39