Reata Pharmaceuticals, Inc.
Notes to Unaudited Consolidated Financial Statements (continued)
classified along with other income tax cash flows as an operating activity in the statement of cash flows; (3) in the area of forfeitures, an entity can still follow the current U.S. GAAP practice of making an entity-wide accounting policy election to estimate the number of awards that are expected to vest or may instead account for forfeitures when they occur; and (4) classification as a financing activity in the statement of cash flows of cash paid by an employer to the taxing authorities when directly withholding shares for tax withholding purposes. ASU 2016-09 is effective for annual periods beginning after December 15, 2016. The Company adopted ASU 2016-09 as of January 1, 2017, which resulted in an adjustment to retained earnings of $110,000 related to the cumulative effect of the accounting policy election to account for forfeitures of share-based awards when they occur, and an adjustment of $115,000 to recognize excess tax benefits as a component of the provision for income taxes on a prospective basis. For the six months ended June 30, 2017, the effect on the provision for income taxes included in the consolidated statement of operations was not significant.
In August 2016, the FASB issued ASU No. 2016-15, Statement of Cash Flows: Classification of Certain Cash Receipts and Cash Payments (Topic 230) (ASU 2016-15). This update addresses eight specific cash flow issues with the objective of reducing the existing diversity in practice. The ASU is effective for public companies for fiscal years, and interim periods within those fiscal years, beginning after December 15, 2017. The Company is currently evaluating this standard and has not yet determined what, if any, effect ASU 2016-15 will have on its consolidated results of operations or financial position.
In January 2017, the FASB issued ASU No. 2017-03, Accounting Changes and Error Corrections (Topic 250) and Investments—Equity Method and Joint Ventures (Topic 323) (ASU 2017-03). This ASU amends the disclosure requirements for ASU No. 2014-09, Revenue from Contracts with Customers (Topic 606), ASU No. 2016-02, Leases (Topic 842) and ASU No. 2016-13, Financial Instruments—Credit Losses (Topic 326): Measurement of Credit Losses on Financial Instruments. This ASU states that if a registrant does not know or cannot reasonably estimate the impact that the adoption of the above ASUs is expected to have on the financial statements, then in addition to making a statement to that effect, the registrant should consider additional qualitative financial statement disclosures to assist the reader in assessing the significance of the impact that the standard will have on the financial statements of the registrant when adopted. ASU 2017-03 was effective upon issuance. The adoption did not have a material impact on the Company’s financial statements.
In May 2017, the FASB issued ASU No. 2017-09, Compensation—Stock Compensation (Topic 718) (ASU 2017-09). This ASU provides guidance about which changes to the terms or conditions of a share-based payment award require an entity to apply modification accounting in Topic 718. ASU 2017-09 is effective for interim and annual periods beginning after December 15, 2017. Early adoption is permitted. We have adopted the standard as of June 30, 2017. The adoption did not have a material impact on the Company’s financial statements.
3. Term Loan
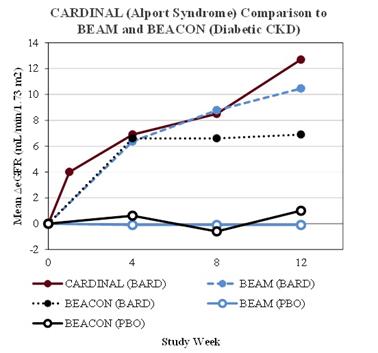
On March 31, 2017, the Company entered into a loan and security agreement (Loan Agreement) with Oxford Finance LLC and Silicon Valley Bank (collectively, Lenders), under which the Lenders agreed to lend the Company up to $35,000,000, issuable in two separate term loans of $20,000,000 (Term A Loan) and $15,000,000 (Term B Loan). On March 31, 2017, the Company borrowed $20,000,000 from the Term A Loan. Beginning July 1, 2017, the Company may, at its sole discretion, borrow $15,000,000 under Term B Loan following the achievement of first patient enrollment in either (a) the Phase 3 portion of the ongoing Phase 2/3 clinical trial of bardoxolone methyl in CKD caused by Alport syndrome or (b) Part 2 of the ongoing two-part clinical trial, or a separate Phase 3 clinical trial, of omaveloxolone in FA until the earlier of 90 days thereafter or March 31, 2018. On August 7, 2017, the Company enrolled the first patient in the Phase 3 portion of CARDINAL.
All outstanding Term Loans will mature on March 1, 2022. Under the Term A Loan, the Company will make interest-only payments for 18 months through October 1, 2018; however, if the Company draws the Term B Loan, the Company will make interest-only payments for 24 months through April 1, 2019. The interest-only payment period will be followed by 41 equal monthly payments, or 35 equal monthly payments if the Company draws the Term B Loan, of principal and interest payments. The Term Loans will bear interest at a floating per annum rate calculated as 7.40% plus the greater of the 30-day U.S. Dollar LIBOR rate reported in The Wall Street Journal on the last business day of the month that immediately precedes the month in which the interest will accrue or 0.75%, with a minimum rate of 8.15% and maximum rate of 10.15%.
The Company has the option to prepay all, but not less than all, of the borrowed amounts, provided that the Company will be obligated to pay a prepayment fee equal to (a) 3.0% of the outstanding principal balance of the applicable Term Loan if prepayment is made prior to the first anniversary of the applicable funding date of the Term Loan, (b) 2.0% of the outstanding principal balance of the applicable Term Loan if prepayment is made by the second anniversary of the applicable funding date of the Term Loan, or (c) 1.0%