Table of Contents
UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
Form 10-K
| | |
ý |
|
ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934: |
For the fiscal year ended December 31, 2008 |
o |
|
TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE
SECURITIES EXCHANGE ACT OF 1934: |
For the transition period from to
|
Commission file number 001-32966
Osiris Therapeutics, Inc.
(Exact name of registrant as specified in its charter)
| | |
Delaware
(State or other jurisdiction of
incorporation or organization) | | 71-0881115
(I.R.S. Employer Identification No.) |
7015 Albert Einstein Drive, Columbia, Maryland
(Address of principal executive offices) |
|
21046-1707
(Zip Code) |
443-545-1800
(Registrant's telephone number, including area code)
Securities registered pursuant to Section 12(b) of the Act:
| | |
| Title of Each Class | | Name of Each Exchange on with Registered |
|---|
| Common Stock, $0.001 par value | | NASDAQ Global Market |
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act. Yes o No ý
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yes o No ý
Indicate by check mark whether the registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days. Yes ý No o
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K is not contained herein, and will not be contained, to the best of registrant's knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. ý
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definitions of "large accelerated filer," "accelerated filer" and "smaller reporting company" in Rule 12b-2 of the Exchange Act. (Check one):
| | | | | | |
| Large accelerated filer o | | Accelerated filer ý | | Non-accelerated filer o | | Smaller reporting company o |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes o No ý
On June 30, 2008, the last business day of the registrant's most recently completed second fiscal quarter, the aggregate market value of voting Common Stock held by non-affiliates of registrant, based upon the last sale price of the Common Stock reported on the NASDAQ Global Market was approximately $278,226,000.
The number of shares of the registrant's Common Stock outstanding as of March 6, 2009 is 32,708,920.
Table of Contents
OSIRIS THERAPEUTICS, INC.
Annual Report on Form 10-K
Fiscal Year Ended December 31, 2008
INDEX
Table of Contents
PART I
ITEM 1. BUSINESS
Forward-Looking Information
This Annual Report on Form 10-K includes "forward-looking statements" within the meaning of Section 21E of the Securities Exchange Act of 1934, or the Exchange Act. Forward-looking statements include statements concerning our plans, objectives, goals, strategies, future events, future revenues or performance, capital expenditures, compensation arrangements, financing needs, plans or intentions relating to collaborations or business combinations, business trends and other information that is not historical information and may appear under the headings "Risk Factors" in this Part I—Item 1A, "Management's Discussion and Analysis of Financial Condition and Results of Operations" in Part II—Item 7, and elsewhere herein or in the other documents we file with the Securities and Exchange Commission, or SEC, including, among others, our quarterly reports on Form 10-Q and any amendments thereto.
When used in this Annual Report, the wordsestimates, expects, anticipates, projects, plans, intends, believes, forecasts and variations of such words or similar expressions are intended to identify forward-looking statements. Examples of forward-looking statements include, but are not limited to, statements regarding the following: our product development efforts; our clinical trials and anticipated regulatory requirements; the success of our product candidates in development; status of the regulatory process for our biologic drug candidates; implementation of our corporate strategy; our financial performance; our product research and development activities and projected expenditures, including our anticipated timeline and clinical strategy for mesenchymal stem cells (MSCs) and biologic drug candidates; our cash needs; patents, trademarks and other proprietary rights; ability of our potential products to treat disease; our ability to supply a sufficient amount of our products to meet regular and repeated demand; our costs to comply with governmental regulations; our relationship with collaborating partners; our ability to benefit from our collaborative arrangements; our ability to benefit from government contracts; our plans for sales and marketing; our plans regarding facilities; types of regulatory frameworks we expect will be applicable to our potential products; and results of our clinical trials and scientific research.
All forward-looking statements, including, without limitation, management's examination of historical operating trends, are based upon our current expectations and various assumptions. Our expectations, beliefs and projections are expressed in good faith and we believe there is a reasonable basis for them. However, there can be no assurance that management's expectations, beliefs and projections will result or be achieved.
There are a number of risks and uncertainties that could cause our actual results to differ materially from the forward-looking statements contained in this Annual Report. Important factors that could cause our actual results to differ materially from the forward-looking statements we make in this Annual Report are set forth in this report, including "Risk Factors." There may be other factors that may cause our actual results to differ materially from the forward-looking statements.
All forward-looking statements attributable to us or persons acting on our behalf apply only as of the date of this Annual Report and are expressly qualified in their entirety by the cautionary statements included herein. We undertake no obligation to publicly update or revise any forward-looking statements to reflect subsequent events or circumstances and do not intend to do so.
When we use the terms "Osiris," "we," "us," and "our" we mean Osiris Therapeutics, Inc., a Delaware corporation.
2
Table of Contents
Company Overview
We are a leading stem cell therapeutic company headquartered in Columbia, Maryland and focused on developing and marketing products to treat medical conditions in the inflammatory, orthopedic, and cardiovascular areas. We were incorporated in Delaware in April 2002. Our predecessor company was organized in 1992. Our lead biologic drug candidate, Prochymal®, is being evaluated in Phase III clinical trials for four indications, including acute and steroid refractory graft versus host disease ("GvHD"), Crohn's disease and for the repair of gastrointestinal injury resulting from radiation exposure, and is the only stem cell therapeutic currently granted both Orphan Drug and Fast Track status by the United States Food and Drug Administration ("FDA"). Prochymal is also being developed for the repair of heart tissue following a heart attack, for protection of pancreatic islet cells in patients with type 1 diabetes, and for the treatment of Chronic Obstructive Pulmonary Disease ("COPD"). Our pipeline of internally developed biologic drug candidates under evaluation also includes Chondrogen® for osteoarthritis in the knee.
In the fourth quarter of 2008, we entered into a collaboration agreement with Genzyme Corporation for the development and commercialization of Prochymal and Chondrogen. Under the terms of the agreement, we retained the rights to commercialize Prochymal and Chondrogen in the United States and Canada and Genzyme has been granted exclusive rights to commercialize Prochymal and Chondrogen in all other countries, except with respect to GvHD in Japan, where Prochymal has previously been licensed to another. The agreement provides for non-contingent, non-refundable upfront payments of $130 million, $75 million of which has been received with $55 million to be received on July 1, 2009. The agreement also provides for contingent milestone payments of up to $1.25 billion in the aggregate in addition to royalties on any sales by Genzyme to be paid by Genzyme to us.
We have also partnered with Genzyme Corporation to develop Prochymal as a medical countermeasure to nuclear terrorism and other radiological emergencies. In January 2008, we were awarded a contract from the U.S. Department of Defense, pursuant to which we, in partnership with Genzyme, are seeking to develop and stockpile Prochymal for the repair of gastrointestinal injury resulting from acute radiation exposure. Additionally, we have partnered with the Juvenile Diabetes Research Foundation ("JDRF") for the development of Prochymal as a treatment for the preservation of insulin production in patients with newly diagnosed type 1 diabetes mellitus.
In April 2008, we committed to a plan to sell our assets related to Osteocel®, a product that we have produced and marketed since July 2005, for regenerating bone in orthopedic indications. On May 8, 2008, we entered into an Asset Purchase Agreement to sell our entire product line relating to the processing, manufacturing, marketing and selling of Osteocel and Osteocel XO to NuVasive, Inc. The agreement provides for the sale to be effected at two closings—a "technology assets closing," at which technology and certain other business assets are transferred, and a "manufacturing assets closing," at which manufacturing assets and facilities are transferred. The technology assets closing, occurred on July 24, 2008, at which time we received an initial payment of $35.0 million and entered into a Manufacturing Agreement under which we will continue to manufacture Osteocel for up to eighteen months after the technology assets closing, and sell 100% of the manufactured product to NuVasive at specified prices. The Asset Purchase Agreement provides that the manufacturing assets closing, is to occur eighteen months after the date of the technology assets closing, or upon earlier termination of the Manufacturing Agreement. The agreements with NuVasive provide for contingent milestone payments of up to $50.0 million, $5.0 million of which was earned by us in the fourth quarter of 2008. The assets and operations related to Osteocel are reported as discontinued operations in the financial statements included in Item 8 to this Annual Report on Form 10-K for all periods.
We are a fully integrated company, having developed capabilities in research, development, manufacturing, marketing and distribution of stem cell products. We have developed an extensive
3
Table of Contents
intellectual property portfolio to protect our technology including 48 U.S. and 284 foreign patents owned or licensed.
Our two biologic drug candidates utilize human mesenchymal stem cells, or MSCs. MSCs can selectively differentiate, based on the tissue environment, into various tissue lineages, such as muscle, bone, cartilage, marrow stroma, tendon or fat. In addition, MSCs have anti-inflammatory properties and can prevent fibrosis or scarring. These characteristics give MSCs the potential to treat a wide variety of medical conditions. We believe that our biologic drug candidates have advantages over other stem cell therapeutics in development for the following reasons:
- •
- Stem Cell Source. Our stem cells are obtained from adult bone marrow, a readily available source. The cells are drawn from the hips of volunteer donors between the ages of 18 and 30 years, using a simple needle and syringe aspiration. Because the cells are obtained from consenting adult donors, we are able to largely avoid the ethical controversy surrounding embryonic and fetal stem cell research.
- •
- Ability to Mass Produce. Through our proprietary manufacturing methods, we can grow MSCs in a controlled fashion to produce up to 10,000 treatments of our biologic drug candidates from a single bone marrow donation. Our ability to produce a large quantity of treatments from one donation provides us with manufacturing efficiencies and product consistency that are essential to commercialization.
- •
- Universal Compatibility. Many stem cell therapies under development can elicit a rejection response in the recipient and therefore require donor-to-recipient matching or potentially harmful immunosuppression. This greatly reduces manufacturing efficiencies and creates a risk of mismatch which can result in an acute inflammatory response and, potentially, in death. Based on our clinical experience, we believe that our biologic drug candidates are not rejected by the patient's immune system and so, like type O negative blood, do not require matching. This universal compatibility allows us to produce a standardized product available to all patients in almost any medical setting.
- •
- Treatment on Demand. Our biologic drug candidates can be stored frozen at end-user medical facilities until they are needed. We anticipate that medical facilities will be able to prescribe and dispense these products in much the same way as conventional drugs. In contrast, other stem cell technologies under development require weeks to prepare after a patient's need is identified. This is a key feature of our technology, as many patients in the critical care setting require prompt treatment.
The following table summarizes key information about our biologic drug candidates.
| | | | |
Product/Candidate | | Indication | | Status |
|---|
| Prochymal | | Steroid Refractory Acute GvHD | | Phase III |
| | | First Line Treatment of Acute GvHD | | Phase III |
| | | Biologics Refractory Crohn's Disease | | Phase III |
| | | Type I Diabetes Mellitus | | Phase II |
| | | Acute Myocardial Infarction | | Phase II |
| | | Chronic Obstructive Pulmonary Disease | | Phase II |
| | | Acute Radiation Syndrome | | Phase III (Animal Rule) |
Chondrogen |
|
Osteoarthritis & Cartilage Protection |
|
Phase II |
Prochymal is our lead biologic drug candidate and is being evaluated in Phase III clinical trials for three indications, including the first line treatment of acute GvHD, steroid refractory acute GvHD and biologics refractory Crohn's disease and is the only stem cell therapeutic currently designated by the FDA as both an Orphan Drug and Fast Track product.
4
Table of Contents
GvHD is a life threatening immune system reaction that commonly affects the skin, gastrointestinal tract, and liver in patients who have received a bone marrow transplant. Although in the U.S. there are no drugs approved for treating GvHD, the disease is commonly treated off-label with steroids. GvHD that does not respond to this treatment is known as steroid refractory GvHD. The majority of steroid refractory GvHD patients die within six months. In our Phase II trial for treatment refractory GvHD, we enrolled patients that did not respond to treatment with steroids and at least one second line therapy. Of these patients, 59% responded to Prochymal. Prochymal has been granted Fast Track status by FDA and Orphan Drug status by FDA and the European Medicines Agency for GvHD.
Our Phase III trial to evaluate Prochymal as a treatment for steroid refractory GvHD is a randomized, double blind, placebo controlled study. The trial has completed enrollment with a total of 244 patients. The trial is investigating patient response to eight infusions of Prochymal administered twice per week for four consecutive weeks. The primary trial endpoint is complete resolution of GvHD for at least 28 day duration. Each patient will also be monitored for safety for up to 180 days after their first treatment with Prochymal. Six-month survival is a key secondary endpoint. This trial is being conducted in 72 centers in the United States, Canada, Europe and Australia.
We are also enrolling patients in a Phase III trial evaluating Prochymal as an add-on therapy to steroids for the first-line treatment of acute GvHD. Our Phase II trial for treatment of newly diagnosed acute GvHD, indicated that patients were twice as likely to have total clinical resolution of their disease when Prochymal was added to steroid therapy, compared to reported results for treatment with steroids alone. Twenty-nine of 31 patients, or 94%, responded after receiving two infusions of Prochymal, with 24 patients, or 77%, achieving a complete response, meaning the patients had experienced total clinical resolution of the disease. At six months, 61% of all patients treated with Prochymal still had a durable response requiring no additional immunosuppressive therapy, clinical intervention, or increased steroid use. Of these, 95% were alive at six months.
Our Phase III trial to evaluate Prochymal as a first line treatment for acute GvHD is a randomized, double blind, placebo controlled study designed to enroll up to 184 patients. The trial is investigating patient response to a total of six infusions during the first four weeks of the study. The primary trial endpoint is the proportion of patients who achieve a complete response at day 28 and who survive to day 90 without the addition of a second line therapy. The study is being conducted at approximately 50 centers in the United States and Canada.
Based on our clinical observations of the effects of Prochymal on the gastrointestinal symptoms of patients with GvHD, we are developing Prochymal for the treatment of Crohn's disease. Crohn's disease is a chronic condition that results in inflammation of the gastrointestinal tract. We completed patient enrollment in a Phase II trial for Crohn's disease under a separate Investigational New Drug application ("IND"). We received Fast Track designation from the FDA for the development of Prochymal for patients with moderate to severe Crohn's disease that is refractory to standard therapies, including biologics.
We are currently enrolling patients in a Phase III trial evaluating Prochymal for the treatment of moderate to severe Crohn's disease that is refractory to biological therapy. The study is designed to enroll 270 patients and is double blind, placebo controlled, and includes patients, 18 to 70 years of age, with a Crohn's Disease Activity Index ("CDAI") greater than 250. The primary endpoint of this trial is the proportion of patients with CDAI of less than 150 (clinical remission) at day 28. The study is being conducted at approximately 60 leading centers in the United States and Canada.
5
Table of Contents
Prochymal is also being evaluated for the repair of heart muscle in patients who have suffered a heart attack. Based on statistics published in 2005 by the American Stroke Association and the American Heart Association, approximately 700,000 individuals in the United States each year experience their first heart attack. According to these same statistics, approximately 20% of these patients suffer extensive damage to their heart muscle leading to heart failure within six years. In preclinical studies in animal models, Prochymal targeted the damaged area of the heart following a single intravenous infusion. These studies also indicate that Prochymal prevents scar formation that typically occurs after a heart attack and improves cardiac function eight to ten weeks after its administration.
We recently reported positive two-year data on the Phase I clinical trial for Prochymal to evaluate its safety and efficacy to restore heart function in patients experiencing a first time myocardial infarction (MI). In a 53-patient, double-blind, placebo-controlled study evaluating the safety and preliminary efficacy of the intravenous administration of Prochymal, MI patients receiving the therapy had significantly lower rates of adverse events, such as cardiac arrhythmias, as well as significant improvements in heart, lung and global function. Administration of Prochymal was found to be well tolerated at all dose levels. Based on these positive findings, we have received approval from the FDA to initiate a Phase II trial.
We recently initiated a Phase II clinical trial which is expected to include approximately 220 patients to be treated at approximately 40 sites in the United States and Canada.
We initiated a Phase II, 60 patient, placebo controlled study in the United States for the treatment of early onset type 1 diabetes in individuals 18 to 30 years old. We believe that based upon their mechanism of action, MSCs may home to the pancreas and inhibit the local immune and inflammatory responses, preventing the destruction of pancreatic islets and promoting the repair of pancreatic tissue damage. Patients must be enrolled within 2 to 16 weeks of being diagnosed with type 1 diabetes and will receive three infusions of Prochymal over the course of 60 days. Primary efficacy will be measured at one year—the primary endpoint is the marker of insulin response in response to glucose stimulation. We have entered into a collaborative agreement with the Juvenile Diabetes Research Foundation ("JDRF") for this study. This agreement provides for JDRF to fund $4.0 million of clinical study costs for 2008 through 2010. During 2008, we received $2.0 million from JDRF to fund clinical costs, and expect to receive the remaining $2.0 million during 2009 and 2010.
We recently completed the enrollment of 62 patients in the Phase II clinical trial evaluating the safety and efficacy of Prochymal in conjunction with standard of care for improving pulmonary function in patients with moderate to severe COPD. The clinical trial is a double-blind, placebo-controlled study. Patients were randomized to either Prochymal or placebo at a 1:1 ratio and received 4 infusions over the course of 90 days. Measurements used in the trial to detect potential improvements in subjects treated with Prochymal include pulmonary function tests, exercise capability, and quality of life assessments. In addition, exacerbations and hospitalizations due to COPD will be monitored for both safety and efficacy. Patients will be evaluated over the course of two years following initial Prochymal or placebo infusion.
In 2007, we partnered with Genzyme Corporation to develop Prochymal as a medical countermeasure to nuclear terrorism and other radiological emergencies. In January 2008, we were
6
Table of Contents
awarded a contract from the United States Department of Defense ("DoD") for the development and stockpiling of Prochymal for the treatment of acute radiation syndrome ("ARS"). The fully funded value of the contract, assuming FDA approval of Prochymal for ARS and exercise by DoD of all of its purchase options for up to 20,000 doses of Prochymal at a price of $10,000 per dose, is up to $224.7 million. We are carrying out this contract in collaboration with Genzyme.
Chondrogen is our biologic drug candidate for the treatment of osteoarthritis and the reduction of pain in the knee. According to a 2005 article in theAmerican Journal of Sports Medicine, approximately 1.0 million people have surgery to remove damaged or torn meniscus in the United States each year. As noted in a 1999 article in the journalSports Medicine, patients who have had this procedure are 10 to 15 times more likely to develop osteoarthritis, a highly debilitating orthopedic condition. There are currently no FDA approved products available to regenerate meniscal tissue. In several preclinical studies, Chondrogen regenerated meniscal tissue and prevented osteoarthritis in animal models. We completed enrollment in a Phase I/II clinical trial for Chondrogen to evaluate its safety and efficacy in patients following surgery to remove torn meniscus. A total of 55 patients were treated in the Phase I/II, double-blind study evaluating the safety and exploratory effectiveness of Chondrogen, a preparation of adult stem cells formulated for direct injection into the knee. At the twelve month time point, Chondrogen met its primary endpoint, demonstrating product safety. The data also showed improvements in joint condition that correlated with a clinically and statistically significant improvement in pain in patients with osteoarthritis (OA) who received Chondrogen as compared to those treated with the control, hyaluronic acid (HA).
We completed enrollment of a randomized double-blind, placebo controlled Phase I/II clinical trial evaluating Chondrogen for safety and preliminary efficacy based upon regeneration of meniscus at six-months. In November 2007, we reported one-year data for the Phase I/II Chondrogen trial. The data continued to show improvements in joint condition that correlated with a clinically and statistically significant improvement in pain in patients with osteoarthritis (OA) who received Chondrogen as compared to those treated with the control, hyaluronic acid (HA). Patients receiving the control were 3.5 times more likely to experience degenerative bone changes associated with OA as compared to those receiving Chondrogen. The effects were dose dependent and pain scores improved from six months to one year following treatment, suggesting Chondrogen caused a biological modification of patients' OA. Patients will be followed for safety and additional preliminary efficacy, such as cartilage damage and changes in the meniscus for two years under the current study protocol.
We expended approximately $69.9 million in fiscal 2008, $47.2 million in fiscal 2007, and $37.6 million in fiscal 2006, on research and development. Our research and development expenditures in 2007 and 2006 were entirely sponsored by us. In 2008, we were reimbursed for $2.5 million of research and development expenditures through our contract with the Department of Defense and received $2.0 million in funding from JDRF. For more detailed financial information, including information regarding our revenues, profit and loss, and total assets and research and development costs and expenses for the past three fiscal years, see our Financial Statements included in Item 8 to this Annual Report on Form 10-K for fiscal year 2008.
Scientific Background
Stem cells are a special class of cells that can self-replicate and differentiate into multiple tissue types. Different populations of stem cells, also called progenitor or precursor cells, reside within the body. These cells are generally classified according to their differentiation potential, or ability to become distinct cell types. Embryonic stem cells are recognized as being totipotent, or unlimited, in terms of the number of different cell types they can become. Other stem cells are either multipotent, meaning capable of becoming two or more cell types, or unipotent, meaning preprogrammed for a single final cell type. Multipotent stem cells include the hematopoietic stem cells responsible for
7
Table of Contents
generating cells associated with the circulatory and immune systems, mesenchymal stem cells responsible for the formation of connective tissue cells, and neuronal stem cells dedicated to producing the different nervous system cell types. Stem cells participate in embryological and fetal development and orchestrate tissue repair following disease or injury in the adult. Though the precise mechanism of their activity has not yet been determined, experimental work has provided empirical evidence of the therapeutic benefit of various types of stem cells administered to animal and human subjects.
The embryonic stem cell, or ESC, has the greatest differentiation potential and is capable of developing into all cell types found within the human body. ESCs must be harvested from human embryos, giving rise to ethical controversies surrounding the procurement of ESCs, which have hindered progress in ESC research. Also, technical difficulties in purifying and growing ESCs have prevented widespread experimental work capable of withstanding academic or regulatory scrutiny.
In adults, two major classes of stem cells exist in bone marrow: hematopoietic stem cells and mesenchymal stem cells. Throughout life, hematopoietic stem cells, or HSCs, located within the bone marrow give rise to most types of blood cells. HSC transplantation has served as the basis for a number of aggressive treatments for various types of cancer. However, therapies based on HSCs are largely limited to hematological disorders because HSCs can only differentiate into blood cells.
In contrast to HSCs, mesenchymal stem cells, or MSCs, are progenitor cells that differentiate into various connective tissues, such as bone, muscle, fat, tendon, ligament, cartilage and bone marrow stroma when they receive appropriate biochemical and biomechanical signals. Other biochemical stimuli cause MSCs to mobilize to areas of injury or inflammatory disease. Once there, MSCs coordinate tissue regeneration at a local level by producing tissue growth factors and by interacting with local cells to reduce inflammation and scarring. Importantly, MSCs do not express markers on the surface of cells, known as HLA class II antigens, which are responsible for recognition of the cells by the immune system. Also, the cell surface markers, CD40, CD80 and CD86, which are essential for activation of immune cells, are not present on MSCs. These characteristics allow MSCs to:
- •
- be transplanted into an unrelated patient without giving rise to an immune response;
- •
- regenerate connective tissues like bone and cartilage;
- •
- act as a potent anti-inflammatory agent; and
- •
- exhibit anti-fibrotic activity to limit tissue damage.
MSCs and HSCs are most readily isolated from bone marrow. Because MSCs represent a small fraction of bone marrow cells, they require amplification to be clinically useful. We have developed and optimized a proprietary process for isolating and expanding these cells using standardized cell culture methodologies. We can grow MSCs in a controlled fashion to produce up to 10,000 treatments of our biologic drug candidates from a single bone marrow donation.
Stem cells can be derived from either the patient, referred to as an autologous source, or from a donor, referred to as an allogeneic source. For many cell therapies, allogeneic sourcing is not possible due to the immune response that typically occurs following the injection of unrelated cells. The non-immunogenic nature of MSCs permits allogeneic cell sourcing and carries significant advantages over autologous sourcing. Allogeneic cell sourcing from a healthy donor population allows for specific quality control measures to select therapeutically optimal stem cells. For example, if a patient's cells are of poor quality due to advanced age, disease or metabolic state, the resulting product will likely be of similarly poor quality. We believe that allogeneic sources used in large scale production will enable us to utilize quality control practices to ensure that product potency is reproducible from treatment to treatment. We have developed quality standards for our biologic drug candidates, including potency assays directed to the specific indications for use. No patients participating in our clinical trials or who have used Osteocel to date have experienced an immunogenic response.
8
Table of Contents
Strategy
We are striving to be the first company to receive FDA marketing approval of a stem cell drug and to become the world's leading provider of stem cell therapies.
Successfully commercialize our lead stem cell therapy, Prochymal. We completed the enrollment of the first worldwide Phase III stem cell clinical trial in the fourth quarter of 2008, enrolling 244 patients at 72 leading bone marrow transplant centers across the United States, Canada, United Kingdom, Spain, Italy, Australia, Germany and Switzerland, and are currently enrolling patients in two additional Phase III clinical trials for Prochymal. Assuming marketing approval, we plan to develop a sales and marketing organization to promote Prochymal initially for the treatment of steroid refractory GvHD. Based on the small number of bone marrow transplantation hospitals treating patients with GvHD in the United States and Canada and the lack of effective treatments for this population, we believe we can successfully market Prochymal with a small, specialized sales force.
Expand our pipeline of biologic drug candidates where our stem cell technology has a therapeutic potential. We are continuously investing in our biologic drug candidate pipeline by evaluating our therapies in additional diseases and disorders where we believe MSCs may have therapeutic benefit. This will allow us to maintain our position as the leader in cellular therapeutics.
Exploit our MSC technology, manufacturing ability and proprietary know-how to advance our pipeline. We intend to leverage our preclinical research, safety data and manufacturing ability to rapidly and efficiently grow our biologic drug candidate pipeline. Because we utilize MSCs as the active agent for all of our biologic drug candidates, we believe the accumulated safety data will reduce the time and cost associated with early stage clinical trials for new indications.
Internally develop and commercialize future biologic drug candidates. We believe that we have the requisite experience to develop and commercialize any future biologic drug candidates with or without the help of a strategic partner. Due to our experience with Osteocel and our current pipeline candidates, we believe we have gained the clinical, regulatory, manufacturing and commercial capabilities to successfully develop and commercialize biologic drug candidates.
Clinical Programs
Prochymal
Prochymal is our biologic drug candidate that is being used to treat medical conditions in a variety of indications. Prochymal is being evaluated in Phase III clinical trials for three indications, including first line and steroid refractory acute graft versus host disease (GvHD) and Crohn's disease, and is the only stem cell therapeutic currently designated by the FDA as both an Orphan Drug and Fast Track product. Prochymal is also being developed for the repair of heart tissue following a heart attack and for the protection of pancreatic islet cells in patients with type 1 diabetes, for the treatment of Chronic Obstructive Pulmonary Disease ("COPD") and for the repair of gastrointestinal injury resulting from radiation exposure.
Graft versus Host Disease
GvHD is a life threatening immune system reaction that commonly affects the skin, gastrointestinal tract, and liver in patients who have received a bone marrow transplant. We estimate that there are approximately 3,000 instances of GvHD in the United States each year.
Bone marrow transplantation is a treatment of last resort for patients with certain cancers and some genetic diseases. This procedure can result in a particularly serious type of rejection referred to as acute GvHD. This condition gets its name because the bone marrow transplant, or the graft, begins to attack the recipient, or the host. As noted in an article published in the journalBiology of Blood and
9
Table of Contents
Marrow Transplantation in 2005, acute GvHD is one of the most common complications of allogeneic bone marrow or hematopoietic stem cell transplantation, affecting approximately 50% of transplant patients. Acute GvHD is graded for prognostic and treatment purposes on a four grade scale, with Grade I considered mild, Grade II moderate, and Grades III-IV considered severe and life-threatening. The onset of GvHD in patients who have received a bone marrow transplant leads to a poor prognosis because of the already weakened state of these patients. According to a 2002 article published inBiology of Blood and Marrow Transplantation, the estimated one-year survival rate for patients with acute GvHD decreases drastically with increasing disease severity, as illustrated below:
| | | | |
Acute GvHD | | Estimated One
Year Survival | |
|---|
Grade I | | | 65% | |
Grade II | | | 60% | |
Grade III | | | 39% | |
Grade IV | | | 22% | |
Typically, patients are treated aggressively with steroids when their GvHD reaches Grade II. A 2001 article published in the journalBlood noted that approximately 50% of these patients will not respond to treatment with steroids and approximately 50-80% of steroid refractory GvHD patients die of the disease.
The current treatments available for acute GvHD are inadequate in two primary ways. First, mortality in patients with acute GvHD is unacceptably high. Second, most treatments for acute GvHD work by suppressing or destroying the immune system. This leads to a number of debilitating side effects, including severe and life threatening infection. Unlike steroids or other immunosuppressant drugs, which have a systemic effect, Prochymal's mechanism of action is designed to specifically target areas of inflammation. Therefore, we believe the use of Prochymal will result in a lower rate of life threatening infection.
We are currently conducting two Phase III pivotal trials for acute GvHD and have been granted Fast Track status by FDA for both. The first Phase III trial is investigating Prochymal in patients with steroid refractory acute GvHD. Seventy two sites in the United States, Canada, Europe and Australia are participating in this trial and we recently completed enrollment of this trial. The second Phase III trial is evaluating Prochymal as a first line treatment for acute GvHD and is taking place in 50 leading centers in the United States and Canada.
Expanded access programs are also ongoing to provide Prochymal to pediatric and adult patients with life threatening GvHD. At the end of 2007, we reported data for a total of twelve pediatric patients between five months and fifteen years of age suffering from severe (Grade III/IV) acute GvHD. These patients had failed, on average, 3.2 lines of therapy prior to treatment with Prochymal. Patients received a median of eight infusions of Prochymal (range 3-21) at a dose of 2 million cells per kilogram of body weight. All patients (12/12) experienced an objective clinical response to therapy, with 58% (7/12) of patients achieving a complete resolution of their GvHD. The 100 day survival was also 58% (7/12) and directly correlated with response rate. There were no infusional toxicities associated with the administration of Prochymal. This pediatric compassionate use program for Prochymal is ongoing.
We completed a Phase II trial evaluating Prochymal as a first-line treatment in combination with steroids, for patients diagnosed with Grade II -IV acute GvHD. A total of 32 patients were treated with two infusions of Prochymal, administered 72 hours apart. The treatment commenced within 48 hours of GvHD diagnosis. In this study, we were evaluating safety, dose and response to treatment by day 28. When Prochymal was added to steroid therapy, patients were twice as likely to have total clinical resolution of their disease compared to reported results for treatment with steroids only. Twenty-nine of 31 patients, or 94%, responded after receiving two infusions of Prochymal, with 24 patients, or 77%, achieving a complete response, meaning the patients had experienced total clinical
10
Table of Contents
resolution of the disease. At six months, 61% of all patients treated with Prochymal still had a durable response requiring no additional use. Of these, 95% were alive at six months.
Beginning in 2004, several requests were made by physicians to use Prochymal in a compassionate use setting for patients with acute severe treatment refractory GvHD and no remaining treatment options. Both pediatric and adult patients that had failed to respond to steroids and other immunosuppressive agents were treated on an emergency-use basis, and clinical improvements were seen in gastrointestinal and skin GvHD. Patients were treated with Prochymal every 72 hours as needed for response, for a maximum of eight treatments. Fourteen patients were treated and had failed to respond to an average of 4.4 other drug therapies prior to treatment with Prochymal. A 59% Prochymal response rate was observed in this treatment refractory population, defined as an improvement in at least one affected organ by at least one full GvHD stage without disease progression in any other organ.
In 2003 we completed a Phase I trial to determine the safety of Prochymal in patients who received hematopoietic stem cell transplants. The trial investigated patient response to three different doses of Prochymal. No safety concerns related to the use of Prochymal were observed in the 46 subjects who were evaluated.
We obtained both Fast Track and Orphan Drug designation in 2005 for the use of Prochymal in GvHD patients. The FDA grants Fast Track designation to investigational drugs that have the potential to treat life-threatening diseases with unmet medical needs. Our Biologic License Application will be eligible for an expedited review process by the FDA as a result of this designation. Orphan Drug designation offers several benefits including eligibility for grants to fund studies, up to seven years of marketing exclusivity and a waiver of the Biologic License Application fee of approximately $900,000. Prochymal is the only stem cell therapy currently designated by the FDA as both an Orphan Drug and Fast Track product candidate.
Crohn's Disease
Based on our clinical observations of the effects of Prochymal on the gastrointestinal symptoms of patients with GvHD, we are developing Prochymal for the treatment of Crohn's disease. Crohn's disease is a chronic, life-long condition that features relapsing inflammation of the gastrointestinal tract. Severe Crohn's disease can cause intractable diarrhea and abdominal pain, undesirable changes in lifestyle, hospitalization, and unwanted side effects from required medications. Approximately 60% of Crohn's disease patients require at least one surgery to remove an affected portion of their intestine at some time during their lifetime, according to a 2002 article in the journalAlimentary Pharmacology & Therapeutics. This article further notes that there are over 500,000 cases of diagnosed Crohn's disease in the United States, and at any given time approximately 10% of these cases have a severe exacerbation or relapse that does not respond to traditional immunosuppressive treatments, including biologics. Standard treatments of steroids and other immune suppressants often cause secondary health problems. According to a 2003 article in theBritish Journal of Clinical Pharmacology, with current medical therapies about 50% of patients with severe Crohn's disease will relapse within one year.
We completed a Phase II trial studying Prochymal as a treatment for moderate to severe Crohn's disease that is refractory to steroids and other immune suppressants. We enrolled ten patients in this study and communicated the results during the October 2006 annual meeting of the American College of Gastroenterology. The trial was a prospective, randomized, open label trial, conducted at 4 leading centers in the United States. Patients with moderate to severe Crohn's disease, defined as having a Crohn's Disease Activity Index (CDAI) of at least 220, who had previously failed treatment with steroids and other immunosuppressive agents, were given two infusions of Prochymal seven days apart. A total of ten patients were treated and nine patients were evaluated through the 28 day follow-up. One patient elected to exit the trial prior to completion. Patients were assigned to one of two
11
Table of Contents
treatment groups and received Prochymal on an outpatient basis. In addition to safety parameters, patients were evaluated for changes in CDAI and improvement in the Inflammatory Bowel Disease Questionnaire (IBDQ). Prior to entering the trial, patients who had been treated with infliximab or other biological agents were required to complete a washout period of 90 days to preclude the possibility that response was the result of a previous treatment.
Entering this trial, the average CDAI score at baseline was 341. Patients entering this study had suffered from Crohn's disease for an average of 14.2 years, and 80% of the patients required prior surgical intervention to treat their Crohn's disease. In the study, one-third of the patients had a reduction of CDAI of greater than 100 points within 14 days of treatment. Each of these responders had failed previous treatment with infliximab (Remicade®). Mean IBDQ scores improved significantly from baseline to day 28 (113 to 146, p=0.008). One-third of the patients reported IBDQ scores of at least 170, indicating they had achieved clinical remission of their disease. Although not reaching statistical significance, there appeared to be correlation between dose and response. Patients receiving the high dose had a 72-point greater reduction in CDAI than those receiving low dose (CDAI reduction of 137 vs. 65). There were no infusional toxicities, and no treatment-related severe adverse events.
After review and consideration of this data, we commenced and are enrolling patients in a Phase III trial evaluating Prochymal for the treatment of moderate to severe Crohn's disease that is refractory to biological therapy. The double blind, placebo controlled study is designed to enroll 270 patients between the ages of 18-70 years with a Crohn's Disease Activity Index ("CDAI") greater than 250. The primary endpoint of this trial is the proportion of patients with CDAI of less than 150 (clinical remission) at day 28. The study is being conducted at 50 leading centers in the United States and Canada. We have received Fast Track designation from the FDA, which makes us eligible for expedited FDA review of Prochymal for this indication.
Acute Myocardial Infarction
We are also evaluating Prochymal for the repair of heart muscle in patients who have suffered a heart attack. Preclinical studies indicate that Prochymal prevents scar formation that typically occurs after a heart attack and improves cardiac function eight to ten weeks after its administration. As discussed further below, we completed enrollment in a Phase I clinical trial for Prochymal. This trial is designed to evaluate the safety and efficacy of Prochymal to restore heart function in patients experiencing a first time heart attack.
A heart attack, or acute myocardial infarction ("AMI"), occurs when coronary arteries become blocked with fatty deposits, depriving the heart muscle of oxygen and nutrients. Based on statistics published in 2005 by the American Stroke Association and the American Heart Association, in the United States approximately 700,000 individuals each year experience their first heart attack. According to these same statistics, approximately 20% of patients experiencing their first heart attack suffer extensive damage to their heart muscle, leading to heart failure within six years. Furthermore, we believe the statistics indicate that despite improvements in the standard of care, this progression from myocardial infarction to heart failure remains largely unavoidable in patients with AMIs.
Prochymal is being developed for the treatment of heart muscle damage following AMI. Its primary indication is to treat post-AMI complications and prevent the formation of scar tissue and associated cardiac dysfunction. Our preclinical studies indicate that the mechanism by which Prochymal improves myocardial function includes the prevention of pathological scarring of the heart muscle and the formation of new blood vessels. We are developing Prochymal as a therapy to be delivered through a standard intravenous line up to 10 days post-myocardial infarction.
In preclinical studies, Prochymal selectively targeted the damaged area of the heart when a single infusion was administered. These studies also indicated that Prochymal has the effect of retarding or
12
Table of Contents
stopping the progression of further cardiac tissue deterioration and limiting the damage caused by an AMI. Significant improvements in cardiac function as demonstrated by increased ejection fraction, reduced end diastolic pressures, and reduced wall stress were observed eight to ten weeks after administration of Prochymal. A preclinical study was performed to determine if an intravenous infusion of MSCs following myocardial infarction would result in an improvement in cardiac function. Significant improvement in cardiac function as indicated by left ventricular ejection fraction (LVEF) was observed three months after infarct in those animals receiving intravenous delivery of MSCs compared to control animals. MSCs were detected in the damaged area of the heart muscle of Prochymal treated animals, but not in the remote, undamaged regions.
In March 2006, we completed enrollment of a 53-patient Phase I randomized, double-blind, placebo-controlled clinical study to evaluate Prochymal in patients following AMI. The trial was designed to investigate patient response to three different doses of Prochymal or placebo. Exploratory efficacy endpoints included overall improvement in the function and remodeling of the heart muscle six months after treatment. A safety evaluation for each subject will be conducted two years after the subject is enrolled in the trial.
In March 2007, we reported six-month results in this trial. Heart attack patients receiving Prochymal had significantly lower rates of adverse events, such as cardiac arrhythmias, as well as significant improvements in heart, lung and global function. Administration of Prochymal was found to be well tolerated at all dose levels. Patients in the Prochymal group were four times less likely to experience an arrhythmic event compared to those receiving placebo (9% vs. 37%, p=0.025). Fewer patients experienced clinically significant premature ventricular contractions after receiving Prochymal as compared to placebo at the one month (6% vs. 32%, p <0.05) and two month (9% vs. 38%, p <0.05) time points. Patients with anterior wall myocardial infarctions had a statistically significant 7.0 point (24%) improvement in ejection fraction at three months and a 7.3 point (25%) improvement at six months over baseline (p <0.05). In comparison, placebo patients in this group did not have a significant increase. Patients receiving Prochymal had significantly improved pulmonary function as measured by improvement in FEV1% predicted values (17 point Prochymal vs. 6 point placebo, p <0.05). Patients receiving Prochymal had significantly improved pulmonary function as measured by improvement in FEV1% predicted values (17 point Prochymal vs. 6 point placebo, p <0.05). FEV1 or forced expiratory volume in one second is the amount of air that can forcibly be blown out in the first second of exhalation. FEV1% predicted values provides a measure of the severity of pulmonary disease by comparing the patient's FEV1 results with normal predicted values. Significantly more patients who received Prochymal experienced improvement in their overall condition at six months as compared to those receiving placebo (42% vs. 11%, p=0.027).
In February 2008, we reported one-year results in the Phase I trial. The trial continued to demonstrate Prochymal's strong safety profile as well as continued statistically significant improvement in heart function. One year magnetic resonance imaging (MRI) data on LVEF was collected and patients treated with Prochymal showed a statistically significant 5.2 point increase over baseline (p=0.021). Patients receiving placebo showed only a 1.8-point improvement over baseline, which was not statistically significant. Patients with more severe myocardial infarction, defined as a baseline LVEF of 45% or less, demonstrated even greater effects. The Prochymal treatment group showed a 6.5-point improvement one year post-treatment, compared to a 1.9-point increase in the placebo group. Prochymal treated patients continued to experience fewer adverse events at a rate of 6.1 per patient, compared to 8.0 per patient in the placebo group. This one-year interim analysis was performed as a part of the full two-year follow-up, and as a result, contains only limited data.
Data from the final two-year time point was reported in February 2009 and the benefits of treatment with Prochymal continued. The trial met its primary endpoint demonstrating the safety of Prochymal in the acute myocardial infarction setting. Patients receiving Prochymal continued to experience fewer adverse events than that of placebo (8 vs. 11 per patient). During the trial, patients
13
Table of Contents
receiving Prochymal experienced fewer arrhythmias. This effect was maintained for the duration of the study, with 47.4% of placebo patients experiencing cardiac arrhythmia compared to only 11.8% of Prochymal patients (p=0.006). Ventricular arrhythmias are associated tissue damage and scar formation in the heart resulting from infarction and can be a sign of poorer prognosis. Two year MRI demonstrated that there was statistically significant improvement in LVEF over baseline, 6.6 point in Prochymal relative to a 3.9 point improvement in placebo. For patients with more severe myocardial infarction, defined as a baseline LVEF of 45% or less, even greater effects were observed. The Prochymal group showed a significant 9.5 point improvement over baseline two years post-treatment (p <0.05). This compares favorably to the 3.1 point increase observed for the placebo group. No serious adverse events were attributed to Prochymal, and all-cause hospitalizations trended lower in the Prochymal group (38.2%) as compared to the placebo group (47.4%).
Based upon the positive results from the Phase I trial, we received approval from the FDA to initiate a Phase II trial. The Phase II double-blind, placebo-controlled trial will evaluate the safety and efficacy of Prochymal in conjunction with standard of care for improving heart function in patients who experienced a first heart attack. This trial focuses on patients who have suffered a severe myocardial infarction, defined as LVEF between 30% and 45% at baseline. The target enrollment is 220 patients. Patients will be randomized to either Prochymal or placebo at 1:1. Efficacy endpoints determined from cardiac MRI include end systolic volume, LVEF and the ability of Prochymal to preserve functional heart tissue, or limit scar formation following a heart attack. In addition, functional and quality of life assessments will be performed.
Type 1 Diabetes
We are also investigating Prochymal as a treatment for the preservation of insulin production in patients with newly diagnosed type 1 diabetes mellitus. Type 1 diabetes, commonly known as juvenile diabetes or insulin-dependent diabetes, is an autoimmune disorder that attacks and destroys insulin producing islet cells in the pancreas causing glucose accumulation in the blood. As a result, those suffering from type 1 diabetes must take insulin to regulate blood sugar levels. Over time, poorly controlled diabetes can lead to serious health conditions, including heart disease, stroke, blindness, amputations, kidney disease and nerve damage. Currently, there are no preventative measures for type 1 diabetes. In preclinical research, both animal and human bone marrow-derived mesenchymal stem cells (MSCs) were shown to preserve beta cell function in animal models of diabetes.
In October 2007, we reported the initiation of a Phase II trial evaluating the safety and efficacy of Prochymal in conjunction with standard of care in preserving insulin production in patients recently diagnosed with type 1 diabetes mellitus. The trial is double-blind, placebo-controlled and will target the enrollment of 60 patients. Patients in the study will receive three intravenous infusions of Prochymal over the course of sixty days. The primary endpoint of the trial is the measurement of C-peptide produced during a Mixed Meal Tolerance Test in patients treated with Prochymal, compared to those receiving placebo. This test is frequently used in diabetic patients to determine how much insulin is being produced by the pancreas in response to glucose stimulation. The patients in the trial will be followed for safety and efficacy for two years.
Chondrogen
Chondrogen is our biologic drug candidate for the treatment of osteoarthritis and the reduction of pain in the knee following meniscectomy. The meniscus is a crescent-shaped cushion in the knee joint that protects cartilage and enables the knee to move smoothly. Injury and tears to the meniscus are common and can be traumatic, arising from sports injury for example, or degenerative, due to daily wear and tear. An injured or torn meniscus is painful and typically requires surgical intervention. The current standard of care for significant injuries is partial meniscectomy surgery, in which the damaged portion of the meniscus is permanently removed. According to a 2005 article in theAmerican Journal of
14
Table of Contents
Sports Medicine, approximately 1.0 million people have surgery to remove damaged or torn meniscus in the United States each year. As noted in a 1999 article in the journalSports Medicine, patients who have had this procedure are 10 to 15 times more likely to develop osteoarthritis, a highly debilitating orthopedic condition.
There are currently no FDA approved products available to regenerate meniscal tissue. In several preclinical studies, Chondrogen, a preparation of adult stem cells formulated for direct injection into the knee, regenerated meniscus and prevented osteoarthritis in animal models. As described further below, at the end of the first quarter of 2006 we completed enrollment in a Phase I/II clinical trial for Chondrogen, designed to evaluate the safety and preliminary efficacy in patients following surgery to remove torn meniscus.
At the end of the first quarter of 2006, we completed a randomized double-blind, placebo-controlled Phase I/II clinical trial evaluating Chondrogen for safety and preliminary efficacy based upon regeneration of meniscus at six-months. We plan on evaluating each patient for safety two years after the patient enrolled in the trial. Participants in the trial received one of two doses of Chondrogen or placebo, and a total of 55 patients were treated. At the one-year time point, Chondrogen met its primary endpoint, demonstrating product safety. An initial review of the data showed that Chondrogen was well tolerated, was not associated with serious adverse events, did not result in any adverse hematological events, and did not result in the formation of any unwanted or ectopic tissue. There was no significant change in the volume of meniscus on MRI at six-months in patients that received Chondrogen compared to those patients receiving placebo. However, about 30% of patients treated with Chondrogen demonstrated an improvement in their baseline cartilage or joint condition, while no patients in the placebo group demonstrated similar improvement.
In November 2007, we reported one-year data for the Phase I/II Chondrogen trial. The data continued to show improvements in joint condition that correlated with a clinically and statistically significant improvement in pain in patients with osteoarthritis (OA) who received Chondrogen as compared to those treated with the control, hyaluronic acid (HA). Patients receiving the control were 3.5 times more likely to experience degenerative bone changes associated with OA as compared to those receiving Chondrogen. The effects were dose dependent and pain scores improved from six months to one year following treatment, suggesting Chondrogen caused a biological modification of patients' OA. Patients will be followed for safety and additional preliminary efficacy, such as cartilage damage and changes in the meniscus for two years under the current study protocol.
Collaborations
Genzyme Corporation—Collaboration Agreements
Prochymal and Chondrogen Development and Commercialization
In October 2008, we entered into a collaboration agreement with Genzyme Corporation for the development and commercialization of Prochymal and Chondrogen. Under the terms of the agreement, we retain the right to commercialize Prochymal and Chondrogen in the United States and Canada and Genzyme is provided the right to commercialize the treatments in all other countries, except with respect to GvHD in Japan where JCR Pharmaceuticals Co., Ltd. has these rights. This collaboration agreement provides for contingent milestone payments to us of up to $1.25 billion, in addition to royalties on any sales by Genzyme, as described below.
The Collaboration Agreement also provides for upfront non-contingent, non-refundable payments to us of $130.0 million ($75.0 million of which was received in November 2008 and $55.0 million to be paid to us on July 1, 2009), and up to $500 million in development and regulatory milestone payments for Prochymal related to GvHD, Crohn's disease and other potential additional indications that we and Genzyme develop together. Based upon sales in Genzyme territories, we are eligible to receive up to
15
Table of Contents
$250 million in sales milestones for Prochymal as follows: $100 million payable when annual sales reach $500 million in the Genzyme territories and $150 million payable when annual sales reach $1 billion.
Based on the results of the recently initiated Phase II clinical trial for Chondrogen, Genzyme may elect to opt-out of further Chondrogen development, at which point all rights to Chondrogen revert back to us with no further obligation by either company. If Genzyme elects to continue with Chondrogen development, we are eligible to receive up to $100 million in development and regulatory milestones based on the achievement of certain clinical trial results and regulatory approvals. Based on sales in Genzyme territories, we are eligible to receive up to $400 million in sales milestones for Chondrogen.
We are also eligible to receive significant escalating royalties on sales of Prochymal and Chondrogen within the Genzyme territories.
We are obligated to complete the on-going clinical trials for Prochymal and Chondrogen at our own cost and will also be responsible for the costs of any new clinical trials for agreed upon indications through Phase II trials. The costs for any subsequent Phase III clinical trials will be borne 60% by us and 40% by Genzyme.
Genzyme has also agreed to provide launch support to us for the sales and marketing of Prochymal in the United States and Canada upon receipt of marketing approval from regulatory authorities. We will pay Genzyme royalties for these services in amounts that we presently estimate to approximate their fair value.
Prochymal Development to Treat Acute Radiation Syndrome
In July 2007, we entered into an agreement with Genzyme for collaboration in the preparation and execution of development and purchase agreements with United States and Allied government agencies for countermeasures to nuclear terrorism and other radiological emergencies. In January 2008, we were awarded a contract from the United States Department of Defense ("DoD") for the development and stockpiling of Prochymal for the treatment of Acute Radiation Syndrome ("ARS"). Under the terms of the contract, the DoD will provide technology and product development funding to us up to $24.7 million.. The contract further provides for additional funding for activities leading to FDA approval of Prochymal for ARS and the scaling up of manufacturing processes, and provides the DoD with successive options for the purchase of up to 20,000 doses of Prochymal in the aggregate. The total value of the contract, assuming FDA approval and the exercise by the DoD of all of its options to purchase doses of Prochymal at $10,000 per dose, is up to $224.7 million. We will carry out this contract in partnership with Genzyme, with us contributing Prochymal and our corresponding safety and advocacy database to the effort, and with Genzyme lending its mass product development and large scale commercialization expertise. Our agreement with Genzyme provides for Genzyme to receive a royalty of 15% of net product sales, limited to those sales made under contracts with United States or Allied government agencies for emergency preparedness.
Juvenile Diabetes Research Foundation—Collaborative Agreement
In October 2007, we entered into a collaborative agreement with the Juvenile Diabetes Research Foundation ("JDRF") for the development of Prochymal as a treatment for the preservation of insulin production in patients with newly diagnosed type 1 diabetes. Under the terms of the agreement, JDRF has agreed to fund $4.0 million of the research costs, payable to us based upon the achievement of established milestones. We received $2.0 million in funding from JDRF during 2008, and expect to receive the remaining $2.0 million during 2009 and 2010. We are recognizing the revenue from the JDRF agreement over the estimated schedule of completing this research.
16
Table of Contents
JCR Pharmaceuticals Co., Ltd.—License Agreement
In August 2003, we entered into a license agreement with JCR Pharmaceuticals Co., Ltd. ("JCR"), pursuant to which we granted to JCR an exclusive right in Japan to our MSC technology for use in connection with the use of hematopoietic stem cells derived from peripheral blood, cord blood or bone marrow in the treatment of hematological malignancies.
The license agreement provided for a payment by JCR to us of an up-front license fee of $3.0 million and payment of an additional $0.5 million upon a certain technology transfer. In addition, if and when marketing approval is obtained in Japan, JCR is required to pay up to $7.0 million in pre-commercialization milestones per product and certain amounts for pre-determined thresholds of cumulative net sales. Lastly, JCR has an obligation to pay royalties to us, with such amount dependent upon the cumulative net sales. We received a $0.5 million milestone payment in 2007 when JCR filed an IND in Japan.
Under the terms of the collaborative arrangement, JCR will bear all costs associated with bringing the drug to market in Japan. JCR is obligated to use its reasonable best efforts to develop and commercialize in Japan products covered under the terms of the license, including conducting clinical trials and procuring regulatory and other approvals. The license expires with respect to specific products on the later of 15 years from the date of the first sale of the product in Japan or the date on which our last patent in Japan covering that product expires. Also, the license and the collaboration can be terminated unilaterally by JCR upon 180 days notice to us or by mutual agreement between us and JCR.
In conjunction with this collaboration, JCR made a $3.0 million investment in our preferred stock, which converted at the closing of our initial public offering into 136,363 shares of our common stock.
Intellectual Property
Our broad intellectual property portfolio originates from our pioneering scientific efforts. We have established a considerable patent position in adult stem cell technology and actively seek to protect proprietary technologies that we consider important to our business, including compositions of matter as well as methods of manufacture and methods of use. We believe that one particularly strong aspect of our patent estate is a claim that relates to the isolated mesenchymal stem cell regardless of its origin, type of expansion or ultimate use. Other layers of our patent protection relate to the application of our stem cell technologies to various organs (e.g. heart, lungs and therapeutic areas (e.g. auto-immune disorders, inflammatory disorders). Additionally we own patents related to methods of purifying MSCs that allow for the manufacture of a safe product. We currently own or have exclusive licenses to 48 issued U.S. patents; foreign counterparts to many of these patents have issued, and we own or hold licenses to 284 patents in Europe, South America, Australia, Japan, Canada, and other countries. We have approximately 15 U.S. patent applications pending and 85 foreign patent applications pending. We are committed to protecting our intellectual property position by continuously monitoring the competitive landscape and are prepared to act aggressively in the event that our strong market position is ever threatened by an infringing product.
We also rely upon trade secrets to protect our proprietary information. Through our experience with MSCs and MSC-based product development, we have developed expertise and know-how in this field. We manufacture clinical grade MSCs in-house and contract for the production through contract manufacturers. To protect this know-how, our policies require confidentiality agreements with our employees, consultants, contractors, manufacturers, outside collaborators, sponsored researchers, and advisors. These agreements generally provide for protection of confidential information, restrictions on the use of materials and assignment of inventions conceived during the course of performance for us. These agreements might not effectively prevent disclosure of our confidential information.
17
Table of Contents
We were founded on the basis of MSC technology obtained from Case Western Reserve University, or CWRU. In January 1993, we entered into a Technology Transfer and License Agreement with CWRU, which was subsequently amended in October 1999 and twice in October 2003. Pursuant to this license agreement certain patents were assigned to us and others were exclusively licensed to us, with the right to grant sublicenses. The exclusive license is subject to any rights of a governmental agency based on research funding by such an agency, and to CWRU's retained rights under the patents for non-clinical research, testing or educational purposes of CWRU.
With respect to the patents licensed to us, we are obligated to pay royalties to CWRU based on sales of products covered by granted licensed patents, and such royalties commence with respect to each such product on the third anniversary of the initial sale thereof. We are also obligated to pay minimum royalties under the agreement with CWRU and remain responsible for patent costs. The license is terminable by CWRU in the event that there is a material breach by us. Otherwise the license is for the life of the patents. Under certain circumstances, we are obligated to negotiate in good faith with a third party a sublicense under patents licensed from CWRU and under patents and know-how owned by us that are reasonably required by the third party to exercise the granted sublicense. We are not obligated to grant such a sublicense where it would have a potential adverse effect on a product being researched, developed or commercialized by us or by a licensee or sublicensee of ours.
Under terms of a Marketing, Collaboration and License Agreement with Lonza, we have licensed our MSC technology to Lonza to sell MSCs, the MSC descendants, cells produced from MSCs and materials used with MSCs for commercial and non-commercial research purposes. Under the terms of this agreement, Lonza is specifically precluded from selling the licensed products for use in humans. We receive royalties on any sales under this agreement.
Patent life determination depends on the date of filing of the application and other factors promulgated under patent law and regulatory laws including, for example, the United States Drug Price Competition and Patent Term Restoration Act of 1984, known as the Hatch-Waxman Act. The patent term restoration period is generally one-half the time between the effective date of an Investigational New Drug Application or IND and the submission date of a New Drug Application or NDA, plus the time between the submission date of an NDA and the approval of the drug. Only the earliest patent applicable to an approved drug is eligible for the extension. The United States Patent and Trademark Office, in consultation with the FDA, reviews and approves applications for patent term extension. We expect to apply for patent term extensions where eligible to bolster market exclusivity beyond nominal patent expiration dates.
Manufacturing
Production of Biologic Drug Candidates
We believe that we have differentiated ourselves from other stem cell companies through proprietary manufacturing methods that allow for the controlled growth of MSCs to produce up to 10,000 treatments of our biologic drug candidates from a single bone marrow donation. This is in contrast to most other stem cell technologies that are able to make only a single treatment from each donation.
We have been manufacturing MSCs for over ten years. The first material manufactured in-house was released in 1999. Since that time manufacturing has continued to expand to support our clinical trials. The current manufacturing process utilizes cell factories, a closed system of surfaces on which the cells adhere, for stem cell expansion. We have developed this technology into a reproducible process that can be scaled up and transferred to additional sites. A second manufacturing site was successfully qualified in 2003. In addition, JCR Pharmaceuticals, our partner in Japan, has successfully implemented our manufacturing technology in Japan. We and our contract manufacturers believe that we perform all
18
Table of Contents
of our manufacturing activities in compliance with the FDA's current Good Manufacturing Practice requirements.
Our manufacturing process begins with the collection of bone marrow aspirate from qualified volunteer donors, 18-30 years of age. Prior to donation, these individuals are screened and tested for a battery of diseases including HIV and hepatitis according to the FDA's donor suitability guidance. We purchase bone marrow aspirate from commercial sources. Since the mesenchymal stem cell is extremely rare, accounting for only one in every 100,000 cells in bone marrow, an initial purification process is required. Upon arrival at our facilities, MSCs are isolated and selectively removed from the bone marrow through a multi-step process. A beneficial feature of our stem cells is that they adhere to the surface of the cell factory and the other remaining cell populations do not adhere and are washed away throughout the process. Our stem cells are then expanded, harvested, packaged and cryopreserved as an in-process intermediate, and we conduct a second battery of quality testing. Each packaged intermediate is further expanded and formulated to produce the final product. Sterility and quality testing completes the process. This well-defined process has allowed for the development of a supply chain where material specifications have been established and vendors have been qualified.
The final product will be configured to allow for ease of storage, distribution and use in the clinic. We expect the product will be provided in ready-to-use patient dose quantities, shipped from the distribution center on dry ice, and stored in the freezer at the pharmacy.
Production of Osteocel
In May 2008, we entered into an Asset Purchase Agreement to sell our Osteocel business to NuVasive, Inc. The agreement provides for the sale to be effected at two closings—a technology assets closing, at which technology and certain other business assets are transferred, and a manufacturing assets closing at which manufacturing assets, including the right to all contracts with tissue recovery agencies related to Osteocel, are to be transferred. The technology assets closing occurred in July 2008, at which time we entered into a Manufacturing Agreement which provides for us to continue to manufacture Osteocel for the exclusive sale to NuVasive for approximately eighteen months or until the earlier occurrence of the manufacturing assets closing. In 2008, we expanded Osteocel production capacity by contracting with AlloSource, one of our tissue suppliers, to produce Osteocel.
Osteocel is a matrix of viable cancellous bone containing primary or unexpanded MSCs. Unlike our biologic drug candidates, the stem cells and cancellous bone used in Osteocel are obtained from organ and tissue donors. Additionally, the production of Osteocel is different from our biologic drug candidates in that it does not feature the expansion of MSCs.
Since its introduction into the marketplace in July 2005, we have been unable to produce Osteocel in quantities sufficient to meet our customer demand due to constraints in our manufacturing facility and the lack of sufficient quantities of marrow-rich bone. We contract with tissue recovery agencies for Osteocel source tissue. We currently have ten agencies under contract, including Allosource. These agencies in turn have contracts with federally designated Organ Procurement Organizations who notify the agencies of donor candidates in their areas. Once an initial qualification of the donor is performed, a surgical team is deployed to remove the tissue and send it to our processing center via overnight delivery. The agencies also compile the donor's medical records, perform a medical and social history evaluation, collect serum samples for serological testing and perform other donor screening services. These agencies operate on a fee-for-service basis, which varies depending upon the tissue type and transplant suitability.
The processing of Osteocel is in many ways more like the process of organ donation than standard tissue processing. This is because it is essential that the stem cells contained within Osteocel are kept in a living, healthy state. We overcome this challenge through a proprietary process that is designed to preserve the material, particularly the stem cells. Sterility cultures are performed on the final product from every lot according to United States Pharmacopeia standards. Following completion of quality control testing and quality assurance review, the product is released for distribution.
19
Table of Contents
Sales, Marketing and Distribution
We intend to self commercialize all of our biologic drug candidates in the United States and Canada upon FDA approval through the creation of sales and marketing capabilities in existing and new indications. In addition, under our collaborative agreement with Genzyme Corporation, Genzyme has agreed to provide us with launch support of our biologic drug candidates for a two-year period commencing approximately six months before the anticipated receipt of marketing approval from the FDA. Genzyme has expertise and extensive resources in the successful launch, sales and marketing of cellular therapies. We have also entered into a collaborative arrangement with JCR Pharmaceuticals Co., Ltd. for the distribution of Prochymal for GvHD in Japan following marketing approval.
Our biologic drug candidates are stored at -140 degrees Centigrade. Generally, we do not believe this will pose a significant problem for end-users as most hospitals and medical centers have freezers with these storage capabilities readily available. However, some facilities may not have this type of storage available and this may limit distribution. In an effort to mitigate this, we are performing studies to store the product at higher temperatures.
Assuming FDA approval of Prochymal for one or more GvHD indications, we expect to focus our sales and marketing efforts on the approximately 210 transplantation hospitals in the United States that are registered with the International Bone Marrow Transplantation Registry. We expect to employ a number of sales representatives, initially targeting the most active transplantation centers in a region. An important component of the sales strategy will be to gain the support of key opinion leaders, facilitating the adoption of Prochymal as the treatment strategy for GvHD. We have entered into a license agreement with JCR Pharmaceuticals that grants it the exclusive right to distribute Prochymal for the treatment of GvHD in Japan when it has been approved for marketing in that country.
Competition
Our industry is subject to rapid and intense technological change. We face, and will continue to face, intense competition from pharmaceutical, biopharmaceutical and biotechnology companies, as well as numerous academic and research institutions and governmental agencies engaged in drug discovery activities or funding, both in the United States and abroad. Some of these competitors are pursuing the development of drugs and other therapies that target the same diseases and conditions that we target in our commercial, clinical and preclinical programs.
Many of the companies competing against us have financial and other resources substantially greater than our own. In addition, many of our competitors have significantly greater experience in testing pharmaceutical and other therapeutic products, obtaining FDA and other regulatory approvals of products, and marketing and selling those products. Accordingly, our competitors may succeed more rapidly than we will in obtaining FDA approval for products and achieving widespread market acceptance. If we obtain necessary regulatory approval and commence significant commercial sales of our products, we will also be competing with respect to manufacturing efficiency and marketing capabilities, areas in which we have limited or no commercial-scale experience.
Our two biologic drug candidates, if approved, would compete with several marketed products and other future biologic drug candidates. For our existing product and each of our clinical-stage biologic drug candidates, the primary competitors include:
- •
- Prochymal. If approved, Prochymal will likely be the first drug indicated for the treatment of acute GvHD. The competitive landscape in Crohn's disease is more crowded and if approved for this indication, Prochymal will compete with Johnson & Johnson's Remicade®, Abbott's HUMIRA®, Biogen's Tysabri® and UCB's Cimzia®.
20
Table of Contents
- •
- Chondrogen. If approved, Chondrogen will compete with pain relievers such as acetaminophen, nonsteroidal anti-inflammatory drugs intra-articular injection of corticosteroid or hyaluronic acid. However, none of these have proven disease-modification, one of the goals of Chondrogen development.
We may face competition in the future from other companies that are researching and developing stem cell therapies. We are aware of many companies working in this area, including: Aastrom Biosciences, Advanced Cell Technology, Athersys, Cellerant Therapeutics, Cognate Therapeutics, Cytori Therapeutics, Gamida Cell, Geron, Mesoblast, MultiCell Technologies, Neuronyx, Theradigm, ViaCell and StemCells.
We expect to compete based upon, among other things, our intellectual property portfolio and the efficacy of our products. Our ability to compete successfully will depend on our continued ability to attract and retain skilled and experienced scientific, clinical development and executive personnel, to identify and develop viable biologic drug candidates and to exploit these products and compounds commercially before others are able to develop competitive products.
In addition, our stem cell therapies may be expensive as compared to other therapies and this may make it more difficult for us to compete with other pharmaceuticals.
Government Regulation
Regulation by governmental authorities in the United States and other countries is a significant factor in the development, manufacture, commercialization and reimbursement of our products and services. Virtually all of the products we develop will require marketing approval, or licensure, by governmental agencies prior to commercialization. In particular, human therapeutic products are subject to rigorous preclinical and clinical testing and other approval procedures of the U.S. Food and Drug Administration, or FDA, and similar regulatory authorities in other countries. Various governmental statutes and regulations also govern or influence testing, manufacturing, safety, labeling, storage and record keeping related to such products and their marketing. State, local and other authorities may also regulate pharmaceutical manufacturing facilities. The process of obtaining these approvals and the subsequent compliance with appropriate statutes and regulations require the expenditure of substantial time and money, and there can be no guarantee that approvals will be granted.
FDA Approval Process
Our biologic drug candidates will require approval from the FDA and corresponding agencies in other countries before they can be marketed. The FDA regulates human therapeutic products in one of three broad categories: biologics, drugs, or medical devices. Our biologic drug candidates will be regulated as biological products. The FDA generally requires the following steps for pre-market approval or licensure of a new biological product or new drug product:
- •
- preclinical laboratory and animal tests conducted in compliance with the FDA's Good Laboratory Practice, or GLP, requirements to assess a drug's biological activity and to identify potential safety problems, and to characterize and document the product's chemistry, manufacturing controls, formulation, and stability;
- •
- submission to the FDA of an Investigational New Drug or IND application, which must become effective before clinical testing in humans can begin;
- •
- obtaining approval of Institutional Review Boards, or IRBs, of research institutions or other clinical sites to introduce the biologic drug candidate into humans in clinical trials;
21
Table of Contents
- •
- adequate and well-controlled human clinical trials to establish the safety and efficacy of the product for its intended indication conducted in compliance with the FDA's Good Clinical Practice, or GCP, requirements;
- •
- compliance with current Good Manufacturing Practices, or cGMP regulations and standards;
- •
- submission to the FDA of a Biologics License Application, or BLA, or New Drug Application, or NDA, for marketing that includes adequate results of preclinical testing and clinical trials;
- •
- FDA review of the marketing application in order to determine, among other things, whether the product is safe, effective and potent for its intended uses; and
- •
- obtaining FDA approval of the BLA or NDA, including inspection and approval of the product manufacturing facility as compliant with cGMP requirements, prior to any commercial sale or shipment of the pharmaceutical agent.
Typically, clinical testing involves a three-phase process although the phases may overlap. In Phase I, clinical trials are conducted with a small number of healthy volunteers or patients and are designed to provide information about product safety and to evaluate the pattern of drug distribution and metabolism within the body. In Phase II, clinical trials are conducted with groups of patients afflicted with a specific disease in order to determine preliminary efficacy, optimal dosages and expanded evidence of safety. In some cases, an initial trial is conducted in diseased patients to assess both preliminary efficacy and preliminary safety and patterns of drug metabolism and distribution, in which case it is referred to as a Phase I/II trial. Phase III clinical trials are generally large-scale, multi-center, comparative trials conducted with patients afflicted with a target disease in order to provide statistically valid proof of efficacy, as well as safety and potency. In some circumstances, the FDA may require Phase IV or post-marketing trials if it feels that additional information needs to be collected about the drug after it is on the market. During all phases of clinical development, regulatory agencies require extensive monitoring and auditing of all clinical activities, clinical data and clinical trial investigators. An agency may, at its discretion, re-evaluate, alter, suspend, or terminate the testing based upon the data which have been accumulated to that point and its assessment of the risk/benefit ratio to the patient. Monitoring all aspects of the study to minimize risks is a continuing process. All adverse events must be reported to the FDA.
The results of the preclinical and clinical testing on a non-biologic drug and certain diagnostic drugs are submitted to the FDA in the NDA or BLA. In the case of vaccines or gene and cell therapies, the results of clinical trials are submitted as a BLA. In responding to the submission of a BLA or NDA, the FDA may grant marketing authority, request additional clinical data or deny approval if the FDA determines that the application does not satisfy its regulatory approval criteria. FDA review of a BLA or NDA typically takes one to three years, but may last longer, especially if the FDA asks for more information or clarification of information already provided. Further clinical trials may be required to gain approval to promote the use of the product for any additional indications. Such additional indications are obtained through the approval of a supplemental BLA or NDA.
The process of obtaining regulatory approval is lengthy, uncertain, and requires the expenditure of substantial resources. Each NDA or BLA must be accompanied by a user fee, pursuant to the requirements of the Prescription Drug User Fee Act, or PDUFA, and its amendments. The FDA adjusts the PDUFA user fees on an annual basis. According to the FDA's fee schedule, effective through September 30, 2008, the user fee for an application requiring clinical data, such as an NDA or BLA, is $1.2 million. PDUFA also imposes an annual product fee for prescription drugs and biologics ($0.1 million), and an annual establishment fee ($0.4 million) on facilities used to manufacture prescription drugs and biologics. Fee waivers or reductions are available in certain circumstances, including a waiver of the application fee for the first application filed by a small business. Additionally, no user fees are assessed on NDAs or BLAs for products designated as orphan drugs, unless the drug
22
Table of Contents
also includes a non-orphan indication, and if a contract manufacturer is used, the contract manufacturer is responsible for the establishment fee.
Before approving an NDA or BLA, all facilities and manufacturing techniques used for the manufacture of products must comply with applicable FDA regulations governing cGMP. A local field division of the FDA is responsible for completing this inspection and providing recommendation for or against approval. This effort is intended to assure appropriate facility and process design to avoid potentially lengthy delays in product approvals due to inspection deficiencies. Similarly, before approving a new drug or biologics application, the FDA may also conduct pre-licensing inspections of a company, its contract research organizations and/or its clinical trial sites to ensure that clinical, safety, quality control and other regulated activities are compliant with GCP. To assure such cGMP and GCP compliance, the applicants must incur significant time, money and effort in the area of training, record keeping, production, and quality control. Following approval, the manufacture, holding, and distribution of a product must continue to devote significant resources to maintain full compliance in these areas.
After FDA approval has been obtained, the FDA will require post-marketing reporting to monitor the side effects of the drug. Further studies may be required to provide additional data on the product's risks, benefits, and optimal use, and will be required to gain approval for the use of the product as a treatment for clinical indications other than those for which the product was initially tested. Results of post-marketing programs may limit or expand the further marketing of the product. Further, if there are any modifications to the drug, including changes in indication, labeling, or a change in the manufacturing process or manufacturing facility, an NDA or BLA supplement may be required to be submitted to the FDA.
Additionally, after the FDA has authorized a drug product to enter commercial distribution, numerous regulatory requirements apply. These include, among others, the cGMPs, which require manufacturers to follow extensive design, testing, control, documentation and other quality assurance procedures during the manufacturing process; labeling regulations; the FDA's general prohibition against promoting drug products for unapproved or off-label uses; and adverse event reporting regulations, which require that manufacturers report to the FDA if their drug may have caused or contributed to a death or serious injury. The FDA has broad post-market and regulatory and enforcement powers. Failure to comply with the applicable U.S. drug regulatory requirements could result in, among other things, warning letters, fines, injunctions, consent decrees, civil penalties, refunds, recalls or seizures of products (which would result in the cessation or reduction of production volume), total or partial suspension of production, withdrawals or suspensions of current product applications, and criminal prosecution. Adverse events related to a drug product in any existing or future markets could cause regulatory authorities to withdraw market approval for such product.
Fast Track and Orphan Drug Designations
Congress enacted the Food and Drug Administration Modernization Act of 1997 in part to ensure the availability of safe and effective drugs, biologics, and medical devices by expediting the FDA review process for new products. The Modernization Act establishes a statutory program for the approval of Fast Track products, including biologics. A Fast Track product is defined as a new drug or biologic intended for the treatment of a serious or life-threatening condition that demonstrates the potential to address unmet medical needs for this condition. Under the Fast Track program, the sponsor of a new drug or biologic may request the FDA to designate the drug or biologic as a Fast Track product at any time during the clinical development of the product. This designation assures access to FDA personnel for consultation throughout the development process and provides an opportunity to request priority review of a marketing application providing a six-month review timeline for the designated product. If a preliminary review of the clinical data suggests that a Fast Track product may be effective, the FDA may initiate review of sections of a marketing application for a Fast Track product before the sponsor completes the application. This rolling review is available if the applicant provides a schedule for
23
Table of Contents
submission of remaining information and pays applicable user fees. However, the time periods specified under PDUFA concerning timing goals to which the FDA has committed in reviewing an application do not begin until the sponsor submits the complete application. During the first quarter of 2005 the FDA designated Prochymal as a Fast Track product for the treatment of GvHD. Prochymal also received Fast Track designation from the FDA in January 2007 for the treatment of refractory Crohn's disease. We cannot predict whether this designation will impact the timing or likelihood of FDA approval of Prochymal.
The Orphan Drug Act provides incentives to manufacturers to develop and market drugs for rare diseases and conditions affecting fewer than 200,000 persons in the United States at the time of application for Orphan Drug designation. The first developer to receive FDA marketing approval for an Orphan Drug is entitled to a seven year exclusive marketing period in the United States for that product as well as a waiver of the BLA user fee. However, a drug that the FDA considers to be clinically superior to, or different from, another approved orphan drug, even though for the same indication, may also obtain approval in the United States during the seven year exclusive marketing period. The FDA granted Orphan Drug designation for Prochymal during the last quarter of 2005.
Legislation similar to the Orphan Drug Act has been enacted in countries other than the United States, including the European Union. The orphan legislation in the European Union is available for therapies addressing conditions that affect five or fewer out of 10,000 persons. The marketing exclusivity period is for ten years, although that period can be reduced to six years if, at the end of the fifth year, available evidence establishes that the product is sufficiently profitable not to justify maintenance of market exclusivity.
Privacy Law
Federal and state laws govern our ability to obtain and, in some cases, to use and disclose data we need to conduct research activities. Through the Health Insurance Portability and Accountability Act of 1996, or HIPAA, Congress required the Department of Health and Human Services to issue a series of regulations establishing standards for the electronic transmission of certain health information. Among these regulations were standards for the privacy of individually identifiable health information. Most health care providers were required to comply with the Privacy Rule as of April 14, 2003.
HIPAA does not preempt, or override, state privacy laws that provide even more protection for individuals' health information. These laws' requirements could further complicate our ability to obtain necessary research data from our collaborators. In addition, certain state privacy and genetic testing laws may directly regulate our research activities, affecting the manner in which we use and disclose individuals' health information, potentially increasing our cost of doing business, and exposing us to liability claims. In addition, patients and research collaborators may have contractual rights that further limit our ability to use and disclose individually identifiable health information. Claims that we have violated individuals' privacy rights or breached our contractual obligations, even if we are not found liable, could be expensive and time-consuming to defend and could result in adverse publicity that could harm our business.
Other Regulations
In addition to privacy law requirements and regulations enforced by the FDA, we also are subject to various local, state and federal laws and regulations relating to safe working conditions, laboratory and manufacturing practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances, including chemicals, micro-organisms and various radioactive compounds used in connection with our research and development activities. These laws include, but are not limited to, the Occupational Safety and Health Act, the Toxic Test Substances Control Act and the Resource Conservation and Recovery Act. Although we believe that our safety procedures for handling and disposing of these materials comply with the standards prescribed by state and federal
24
Table of Contents
regulations, we cannot assure you that accidental contamination or injury to employees and third parties from these materials will not occur. We may not have adequate insurance to cover claims arising from our use and disposal of these hazardous substances.
Foreign Regulation
We will most likely have to obtain approval for the manufacturing and marketing of each of our products from regulatory authorities in foreign countries prior to the commencement of marketing of the product in those countries. The approval procedure varies among countries, may involve additional preclinical testing and clinical trials, and the time required may differ from that required for FDA approval or licensure. Although there is now a centralized European Union approval mechanism in place, this applies only to certain specific medicinal product categories. In respect of all other medicinal products, each European country may impose certain of its own procedures and requirements in addition to those requirements set out in the appropriate legislation, many of which could be time-consuming and expensive. Although data requirements presently exist for gene therapy and somatic cell therapy medicinal products, additional European approval standards for cellular therapy are still under development, and consequently approval of cell therapy products in Europe may require additional data that we may not be able to satisfy.
Employees
As of December 31, 2008, our headcount was 163, comprised of 158 full-time employees and 5 full-time contract employees. Of this total, 94 were engaged in quality, manufacturing and distribution of Osteocel under our Manufacturing Agreement with NuVasive, 52 were engaged in research and development and clinical trials for our Biologic Drug products and 17 were engaged in administration, facilities and finance. All of our employees have entered into non-disclosure agreements with us regarding our intellectual property, trade secrets and other confidential information. None of our employees are represented by a labor union or covered under a collective bargaining agreement, nor have we experienced any work stoppages.
Executive Officers of the Registrant
Executive officers are appointed annually by the Board of Directors and, subject to the terms of any applicable employment agreement, serve at the discretion of the Board of Directors. Information regarding our executive officers is as follows:
| | | | | | | |
Name | | Age | | Position | | Other Offices or Positions Held
During the Past Five Years |
|---|
C. Randal Mills, Ph.D. | | | 37 | | President and Chief Executive Officer (since July 2004) | | Dr. Mills is also a member of the Board of Directors. Prior to joining Osiris, Dr. Mills was an executive officer of Regeneration Technologies, Inc. (NASDAQ—RTIX). Dr. Mills served in several leadership positions at RTI from its formation in 1998 until 2004, including Vice President of Business Development and Vice President of Operation and R&D and is credited with several key initiatives including the development and commercialization of RTI's core technology, BioCleanse®. |
25
Table of Contents
| | | | | | | |
Name | | Age | | Position | | Other Offices or Positions Held
During the Past Five Years |
|---|
Richard W. Hunt | | | 54 | | Chief Financial Officer (since July 2008) | | Mr Hunt has over 30 years of Finance and Healthcare Experience. Prior to joining Osiris, Mr. Hunt, served as the Senior Vice President and Chief Financial Officer of Global Healthcare Exchange, LLC, a health care e-commerce exchange venture from July 2007 through July 2008. From June 2006 through July 2007, he served as the Chief Financial Officer of HealthExtras, Inc. (NASDAQ—CHSI) a pharmacy benefit management services provider. From June 2004 through November 2005, Mr. Hunt was the Senior Vice President and CFO of NeighborCare, Inc. (NASDAQ—NCRX), a long-term care pharmaceutical services provider, and from July 2000 through February 2004, he served as the Senior Vice President and CFO of Global Healthcare Exchange, LLC. Mr. Hunt spent several years in progressive finance leadership positions with Baxter Healthcare/American Hospital Supply (NYSE—BAX) and GD Searle Pharmaceuticals. |
Harry E. Carmitchel | | |
58 | | Chief Operating Officer (since September 2004) | | Mr. Carmitchel has over 25 years of general management and operations experience in the medical field. Prior to joining Osiris, Mr. Carmitchel was a Principal with the Pacific Consulting Group for four years, where he specialized in corporate turnarounds. Prior to this time, Mr. Carmitchel was a General Manager with McQuay International, running a $410 million group, and spent eight years as President of the Medical Division for Stryker Corporation. |
Lode Debrabandere, Ph.D. | | |
43 | | Vice President and General Manager, Inflammatory Diseases (since July 2006) | | Prior to joining Osiris, Dr. Debrabandere served for over four years with Bristol-Myers Squibb as Vice President for Strategic Marketing for Neuroscience and Infectious Diseases. He led the Neuroscience Unit and was the Global Brand Leader for Abilify™. Previously, Dr. Debrabandere led the Marketing department of UCB Pharma Inc., focusing in the areas of allergy/respiratory (Zyrtec™) and neurology (Keppra™). |
Michelle LeRoux Williams, Ph.D. | | |
34 | | Vice President of Development (since May 2007) | | Dr. Williams joined Osiris in 2001 as the Director of Orthopedics and was responsible for the development of Osteocel from the initial concept through product launch in 2005. Dr. Williams also advanced the Chondrogen program from preclinical testing through the Phase I/II clinical trial. Prior to joining Osiris, Dr. Williams completed an NIH postdoctoral fellowship in tissue engineering at Columbia University, evaluating cellular constructs for the repair and regeneration of cartilage in arthritis patients. |
26
Table of Contents
| | | | | | | |
Name | | Age | | Position | | Other Offices or Positions Held
During the Past Five Years |
|---|
Philip R. Jacoby, Jr. | | | 56 | | Vice President of Finance (principal accounting officer) and Corporate Secretary (since October 2005) | | Mr. Jacoby has over 30 years of financial and management experience with public and privately held companies. Mr. Jacoby joined Osiris in April 2005 as our Corporate Controller and principal accounting officer in preparation for our initial public offering. Prior to joining Osiris, Mr. Jacoby was the Vice President and Corporate Controller for FTI Consulting, Inc. (NYSE—FCN) from 1999 through the first quarter of 2005. |
Available Information
Our website address iswww.osiris.com. Our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K and any amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934 are available on our website free of charge as soon as reasonably practicable after we electronically file such material with, or furnish it to, the U.S. Securities and Exchange Commission, or SEC. The public may read and copy these materials at the SEC's Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. The public may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC maintains a website that contains such reports, proxy and information statements and other information, and the Internet address ishttp://www.sec.gov. Information contained on our website is not and should not be deemed a part of this annual report or any other report or filing filed with the SEC.
ITEM 1A. RISK FACTORS
Risks Related To Our Business
We have a history of operating losses and may not achieve or sustain profitability.
We have incurred losses in each year since our inception, and may incur additional losses over the next several years. As of December 31, 2008, we had an accumulated deficit of $274.9 million. These losses resulted principally from costs incurred in our research and development programs and from our general and administrative expenses. These losses, among other things, have had and will continue to have an adverse effect on our stockholders' equity, total assets and working capital.
We expect to continue to incur significant operating expenses in the foreseeable future as we seek to:
- •
- complete our Phase III clinical trials for Prochymal for GvHD and Crohn's disease;
- •
- complete our Phase II clinical trial for Chondrogen, and, if supported by the Phase II clinical trial, initiate additional clinical trials;
- •
- complete our Phase II clinical trial for Prochymal for cardiac indications, and, if supported by the Phase II clinical trial, initiate Phase III clinical trials;
- •
- complete our Phase II clinical trial for Prochymal for type 1 diabetes, and, if supported by the Phase II clinical trial, initiate Phase III clinical trials;
- •
- complete our Phase II clinical trial for Prochymal for COPD, and, if supported by the Phase II clinical trial, initiate Phase III clinical trials;
- •
- complete our animal studies for Prochymal for acute radiation syndrome, and, if supported by the preclinical studies, initiate further studies;
- •
- maintain, expand and protect our intellectual property portfolio; and
27
Table of Contents
- •
- add operational, financial, accounting, facilities engineering and information systems personnel, consistent with expanding our operations and our status as a public company.
In addition, during 2008 we sold our Osteocel business unit, including our only commercially available product. While we expect to achieve commercialization of at least some of our other products, there can be no assurances when, or if, we will be able to do so.
The extent of our future operating losses or profits is highly uncertain, and we may not achieve or sustain profitability. If we are unable to achieve and then maintain profitability, the market value of our common stock will decline and you could lose part or all of your investment.
The current credit and financial market conditions may exacerbate certain risks affecting our business.
We rely upon third-parties for certain aspects of our business, including collaboration partners, wholesale distributors, contract clinical trial providers, contact manufacturers and third-party suppliers. Because of the recent tightening of global credit and the volatility in the financial markets, there may be a delay or disruption in the performance or satisfaction of commitments to us by these third-parties or our access to capital may be restricted or eliminated, any of which could adversely affect our continuing operations or business.
If we are not able to recruit and retain qualified management and scientific personnel, we may fail in developing our technologies and biologic drug candidates.
Our future success depends to a significant extent on the skills, experience and efforts of the principal members of our scientific, management and sales personnel. These members include C. Randal Mills, Ph.D., Richard W. Hunt, Harry E. Carmitchel, Michelle L. Williams, Ph.D., Philip R. Jacoby, Jr., and Lode Debrabandere Ph.D. The loss of any or all of these individuals could harm our business and might significantly delay or prevent the achievement of research, development or business objectives. We have entered into employment agreements with Dr. Mills, Messrs. Carmitchel and Hunt, and Dr. Debrabandere. The existence of an employment agreement does not, however, guarantee retention of these employees, and we may not be able to retain those individuals for the duration of or beyond the end of their respective terms. Except for Dr. Mills, Messrs. Carmitchel and Hunt, and Dr. Debrabandere, none of our employees is employed for a specified term. Competition for personnel is intense. We may be unable to retain our current personnel or attract or integrate other qualified management and scientific personnel in the future.
If the potential of our stem cell therapies to treat diseases is not realized, the value of our technology and our development programs could be significantly reduced.
The potential of our stem cell therapies to treat diseases is currently being explored by us. We have not proven in clinical trials that our stem cell therapies will be a safe and effective treatment for any disease. Our stem cell therapies are susceptible to various risks, including undesirable and unintended side effects, unintended immune system responses, inadequate therapeutic efficacy or other characteristics that may prevent or limit their marketing approval or commercial use. We have not yet completed all of the testing necessary to allow us to make a determination that serious unintended consequences will not occur. If the potential of our stem cell therapies to treat disease is not realized, the value of our technology and our development programs could be significantly reduced. Because our biologic drug candidates are based on MSCs, any negative developments regarding the therapeutic potential or side effects of MSCs could have a material adverse effect on our business, financial condition and results of operations.
28
Table of Contents
Our product development programs are based on novel technologies and are inherently risky.
We are subject to the risks of failure inherent in the development of products based on new technologies. The novel nature of our therapeutics creates significant challenges in regards to product development and optimization, manufacturing, government regulation, third-party reimbursement and market acceptance. For example, the FDA has relatively limited experience with stem cell therapies. None has been approved by the FDA for commercial sale, and the pathway to regulatory approval for our biologic drug candidates may accordingly be more complex and lengthy. Additionally, stem cells are subject to donor-to-donor variability, which can make standardization more difficult. As a result, the development and commercialization pathway for our therapies may be subject to increased uncertainty, as compared to the pathway for new conventional drugs.
There are no FDA approved treatments for some of the disease indications we are pursuing. This could complicate and delay FDA approval of our biologic drug candidates.
There are no drugs or therapies currently approved with stated indications for the first-line treatment of acute GvHD or the treatment of steroid refractory GvHD. As a result, the clinical efficacy endpoints, or the criteria to measure the intended results of treatment, for our biologic drug candidate Prochymal for the treatment of GvHD may be difficult to determine. In addition, patients battling GvHD and who, therefore, are candidates for treatment with Prochymal, are typically very ill as a result of an underlying genetic or oncologic condition. Due to the graveness of their underlying disease and the very serious complications and disorders that often accompany acute GvHD, many of these patients will die from causes other than GvHD prior to the completion of the study even if their GvHD responds favorably to treatment with Prochymal. The resulting reduction in the number of patients available for evaluation at the end of the study may make it more difficult for us to demonstrate efficacy, as necessary to obtain FDA approval to market Prochymal for commercial sale.
There are also no drugs or therapies currently approved with stated indications for the repair of heart muscle following heart attack. As a result, the clinical endpoints for our biologic drug candidate Prochymal for cardiac indications may be difficult to determine. In the case of Prochymal for the treatment of Crohn's disease, there are other products approved for the treatment of this disease, so it is expected that the clinical efficacy endpoints for Prochymal for this indication will be established by comparison with these already approved treatments. In order to obtain FDA approval for any indication, we will have to demonstrate, among other things, that our biologic drug candidate is safe and effective for that indication. The results of our clinical trials must be statistically significant, meaning that there must be sufficient data to indicate that it is unlikely the outcome occurred by chance. These challenges may prevent us from developing and commercializing products on a timely or profitable basis, or at all.
Our biologic drug candidates represent new classes of therapy that the marketplace may not understand or accept.
Even if we successfully develop and obtain regulatory approval for our biologic drug candidates, the market may not understand or accept them. We are developing biologic drug candidates that represent novel treatments and will compete with a number of more conventional products and therapies manufactured and marketed by others, including major pharmaceutical companies. The degree of market acceptance of any of our developed and potential products will depend on a number of factors, including:
- •
- the clinical safety and effectiveness of our products and their perceived advantage over alternative treatment methods;
29
Table of Contents
- •
- our ability to demonstrate that Prochymal can have a clinically significant effect, initially on steroid refractory GvHD and acute GvHD, and then also the other indications for which we seek approval;
- •
- our ability to separate ourselves from the ethical controversies associated with stem cell drug candidates derived from human embryonic or fetal tissue;
- •
- ethical controversies that may arise regarding the use of stem cells or human tissue of any kind, including adult stem cells, adult bone marrow and other adult tissues derived from donors;
- •
- adverse events involving our biologic drug candidates or the products or product candidates of others that are stem cell based;
- •
- our ability to supply a sufficient amount of our product to meet regular and repeated demand in order to develop a core group of medical professionals familiar with and committed to the use of our products; and
- •
- the cost of our products and the reimbursement policies of government and third-party payors.
If the health care community does not accept our potential products for any of the foregoing reasons, or for any other reason, it could affect our sales, having a material adverse effect on our business, financial condition and results of operations.
The successful commercialization of our biologic drug candidates, or any of our other potential stem cell therapeutics, will depend on obtaining reimbursement from third-party payors.
If we successfully develop and obtain necessary regulatory approvals, we intend to sell our biologic drug candidates initially in the United States and Canada. In the United States, the market for any pharmaceutical product is affected by the availability of reimbursement from third-party payors, such as government health administration authorities, private health insurers, health maintenance organizations and pharmacy benefit management companies. Stem cell therapies like Prochymal and Chondrogen may be expensive compared with standard pharmaceuticals, due to the higher cost and complexity associated with the research, development and production of stem cell therapies, the small size and large geographic diversity of the target patient population for some indications, and the complexity associated with distribution of stem cell therapies which require special handling, storage and shipment procedures and protocols. This, in turn, may make it more difficult for us to obtain adequate reimbursement from third-party payors, particularly if we cannot demonstrate a favorable cost-benefit relationship. Third-party payors may also deny coverage or offer inadequate levels of reimbursement for our potential products if they determine that the product has not received appropriate clearances from the FDA or other government regulators or is experimental, unnecessary or inappropriate. For example, patients battling GvHD and who, therefore, are candidates for treatment with Prochymal, are typically very ill as a result of an underlying genetic or oncologic condition. Because these patients have a low probability of survival (whether or not their GvHD is successfully treated), third-party payors may resist reimbursing the cost of treatment.
In some of the other countries in which we or other entities with which we collaborate, including Genzyme Corporation, may seek to market our products, the pricing of prescription pharmaceutical products and services and the level of government reimbursement are subject to governmental control. In these countries, pricing negotiations with governmental authorities can take six to twelve months or longer after the receipt of marketing approval for a product. To obtain reimbursement or pricing approval in some countries, we or our collaborators may be required to conduct one or more clinical trials that compares the cost effectiveness of our biologic drug candidates or products to other available therapies. Conducting one or more additional clinical trials would be expensive and result in delays in commercialization of our products.
30
Table of Contents
Managing and reducing health care costs has been a general concern of federal and state governments in the United States and of foreign governments. Although we do not believe that any recently enacted or presently proposed legislation should impact our business, we might be subject to future regulations or other cost-control initiatives that materially restrict the price we receive for our products. In addition, third-party payors are increasingly challenging the price and cost-effectiveness of medical products and services, and many limit reimbursement for newly approved health care products. In particular, third-party payors may limit the indications for which they will reimburse patients who use any products that we may develop. Cost control initiatives could decrease the price for products that we may develop, which would result in lower product revenues to us.
Our dependence upon a limited supply of bone marrow donors and biologics growth media may impact our ability to produce sufficient quantities of our biologic drug candidates as necessary to complete our clinical trials, and if our trials are successful, to meet product demand.
The population of acceptable bone marrow donors is limited to volunteers between the ages of 18 and 30. In addition, potential donors are prescreened for a variety of health conditions and are only allowed to donate bone marrow a total of six times in their lifetime, further limiting the total number of potential donors. The amount of bone marrow donated may be insufficient for us to mass produce our biologic drug candidates. In addition, the expansion of MSCs through our proprietary manufacturing methods utilizes biologic growth media which may be in limited supply. Future government regulation or health concerns may also reduce the number of donors or otherwise limit the amount of bone marrow available to us. If we cannot secure quantities of bone marrow or biologic growth media sufficient to meet the manufacturing demands for our clinical trials, we might not be able to complete our clinical trials and obtain marketing approval for our biologic drug candidates. Moreover, even if our clinical trials are successful and we obtain marketing approval for our biologic drug candidates, our inability to secure enough bone marrow to meet product demand would limit our potential revenues.
Osteocel and our biologic drug candidates are derived from human bone and bone marrow sources and therefore have the potential for disease transmission.
The utilization of donated bone and bone marrow creates the potential for transmission of communicable disease, including but not limited to human immunodeficiency virus, or HIV, viral hepatitis, syphilis, Creutzfeldt-Jakob disease, or the human form of "mad cow" disease, and other viral, fungal or bacterial pathogens. Although we are required to comply with federal and state regulations intended to prevent communicable disease transmission, and our suppliers of adult human bone and bone marrow are also required to comply with such regulations in connection with their collection, storage and supply to us:
- •
- we or our suppliers may fail to comply with such regulations;
- •
- even with compliance, our products might nevertheless be viewed by the public as being associated with transmission of disease; and
- •
- a patient that contracts an infectious disease might assert that the use of our products resulted in disease transmission, even if the patient became infected through another source.
Any actual or alleged transmission of communicable disease could result in patient claims, litigation, distraction of management's attention and potentially increased expenses. Further, any failure in screening, whether by us or other manufacturers of similar products, could adversely affect our reputation, the support we receive from the medical community and overall demand for our products. As a result, such actions or claims, whether or not directed at us, could have a material adverse effect on our reputation with our customers and our ability to market our products, which could have a material adverse effect on our business, financial condition and results of operations.
31
Table of Contents
We may not be able to manufacture our biologic drug candidates in quantities sufficient for later stage clinical studies or for commercial sale.
If we successfully obtain marketing approval for one of our biologic drug candidates, we may not be able to produce sufficient quantities of the product at an acceptable cost. Commercial-scale production of therapies made from live human mesenchymal stem cells involves production in small batches and strict adherence to complex manufacturing and storage protocols and procedures. Our biologic drug candidates are inherently more difficult to manufacture at commercial-scale than chemical pharmaceuticals, which are manufactured using precise chemical formulations and operational methods.
We use third-party collaborators to help us develop and commercialize our products, and our ability to commercialize such products may be impaired or delayed if collaborations are unsuccessful.
We have arrangements in place with third-party collaborators as a means to help us with research and development efforts or marketing and distribution. For example:
- •
- we are party to a Collaboration Agreement with Genzyme Corporation for the development and commercialization of Prochymal and Chondrogen outside the United States and Canada for certain indications, and with the potential for the development and commercialization of these product candidates for additional indications in the future;
- •
- we are party to a Manufacturing Agreement with NuVasive, Inc. for the manufacture of Osteocel on an interim basis and until the "Manufacturing Assets Closing" occurs under the Asset Purchase Agreement between us and NuVasive, pursuant to which we sold our Osteocel and Osteocel XO lines of business to NuVasive;
- •
- we have a collaboration with JCR Pharmaceuticals Co., Ltd. granting to JCR an exclusive right to Prochymal for the treatment of GvHD in Japan; and
- •
- we have a collaboration with Genzyme Corporation to develop effective countermeasures to nuclear terrorism and other radiological emergencies. The initial focus of the collaboration is to develop Prochymal to treat the potentially lethal complications of acute radiation syndrome.
We may enter into additional collaborations in the future. We are dependent upon the success of our current and any future collaborators in performing their responsibilities in connection with the relevant collaboration. If we fail to maintain these collaborative relationships for any reason, we would need to undertake on our own and at our own expense, or find other collaborators, to perform the activities we currently anticipate will be performed by our collaborators. This would substantially increase our cash requirements. We may not have the capability or financial capacity to undertake these activities on our own, or we may not be able to find other collaborators on acceptable terms, or at all. This may limit the programs we are able to pursue and result in significant delays in the development, sale and manufacture of our products, and may have a material adverse effect on our business.
We are subject to a number of risks associated with our dependence upon our collaborative relationships, including:
- •
- our collaborators may not cooperate with us or perform their obligations under our agreements with them;
- •
- we cannot control the quality, amount and timing of our collaborators' resources that will be devoted to performing their responsibilities under our agreements with them, and our collaborators may choose to pursue alternative technologies in preference to those being developed in collaboration with us;
- •
- refusal to or failure of our collaborators to perform their responsibilities in a timely manner, including breach;
32
Table of Contents
- •
- the right of the collaborator to terminate its collaboration agreement with us for reasons outside our control, and in some cases on limited notice;
- •
- business combinations and changes in a collaborator's business strategy may adversely affect the party's willingness or ability to complete its obligations;
- •
- loss of significant rights to our collaborative parties if we fail to meet our obligations;
- •
- disagreements as to ownership of clinical trial results or regulatory approvals;
- •
- withdrawal of support by a collaborator following development or acquisition by the collaborator of competing products; and
- •
- disagreements with a collaborator regarding the collaboration agreement or ownership of intellectual property or other proprietary rights.
In addition, the recent tightening of global credit and the volatility in the financial markets may result in or contribute to a delay or disruption in the performance or satisfaction of commitments to us by these third-parties.
Due to these factors and other possible events, we could suffer delays in the research, development or commercialization of our products or we may become involved in litigation or arbitration, which would be time consuming and expensive.
Two of our most significant collaborative arrangements are with Genzyme Corporation, and our ultimate success may depend upon performance on the part of Genzyme and the success of these collaborations.
We are party to two collaborative arrangements with Genzyme, one for the development and commercialization of Prochymal and Chondrogen outside the United States and Canada for certain indications, and the other to develop effective countermeasures to nuclear terrorism and other radiological emergencies. These collaborations are subject to all of the risks and uncertainties applicable to collaborative arrangements generally, including those described above. In addition, these collaborations are subject to a number of risks and uncertainties specific to the transactions and the parties.
Under our collaborative arrangement with Genzyme for commercialization of Prochymal and Chondrogen outside the United States, Genzyme is obligated to make two up front payments to us: an initial payment of $75.0 million, which has already been received, and an additional payment of $55.0 million scheduled to be paid on July 1, 2009. In addition, we have the opportunity to earn up to an additional $1.25 billion in milestone payments under this collaboration. Receipt of these additional milestone payments is conditioned upon the achievement of the applicable development, regulatory and sales milestones, all of which are subject to all of the risks and uncertainties otherwise applicable to our business, including the success of Prochymal and Chondrogen. Genzyme has the right to terminate the collaboration at any time after July 1, 2009. Genzyme also has the right to "opt-out" of further participation with regard to Chondrogen development, whereupon all rights to Chondrogen will revert to us, but our opportunity to earn Chondrogen-related development, regulatory and sales milestones of up to approximately $500.0 million will cease. The success of this collaboration for us will in part be dependent upon Genzyme, including determinations regarding the exercise of its termination and opt-out rights, and its success in obtaining timely regulatory approvals for the marketing of products outside of the United States, and ability to generate sales sufficient to trigger milestone and royalty payments to us.
Under our collaborative arrangement with Genzyme for the development of effective countermeasures to nuclear terrorism and other radiological emergencies, we were awarded in January 2008 a contract from the U.S. Department of Defense to develop and supply Prochymal for acute radiation syndrome. We are carrying out this contract in partnership with Genzyme, with us contributing Prochymal and our corresponding safety and advocacy database to the effort, and with Genzyme lending its mass product development and large scale commercialization expertise.
33
Table of Contents
Genzyme has significantly greater resources than we do, and these collaborations are not as core to its business, as they are to ours. We are dependent upon Genzyme's continued performance under these collaborations, and any determination by Genzyme not to proceed or perform, or any material adverse event that affects Genzyme's ability or desire to perform, under either of these collaborations may have a material adverse effect on our business.
We are currently dependent upon third-parties for services and raw materials needed for the manufacture of our biologic drug candidates, and if these products are successfully commercialized, may become dependent upon third-parties for their distribution. If any of these third parties fail or are unable to perform in a timely manner, our ability to manufacture and deliver could be compromised.
In order to produce our biologic drug candidates for use in clinical studies, and to produce any of our biologic drug candidates that may be approved for commercial sale, we require biological media, reagents and other highly specialized materials. This is in addition to the bone marrow aspirate used in the manufacture of our biologic drug candidates. These items must be manufactured and supplied to us in sufficient quantities and in compliance with current Good Manufacturing Practices, or cGMP. To meet these requirements, we have entered into supply agreements with firms that manufacture these components to cGMP standards. Our requirements for these items are expected to increase if and when we transition to the manufacture of commercial quantities of our biologic drug candidates.
In addition, as we proceed with our clinical trial efforts, we must be able to demonstrate to the FDA that we can manufacture our biologic drug candidates with consistent characteristics. Accordingly, we are materially dependent on these suppliers for supply of cGMP-grade components of consistent quality. Our ability to complete ongoing clinical trials may be negatively affected in the event that we are forced to seek and validate a replacement source for any of these critical components. If we are not able to obtain adequate supplies of these items of consistent quality from our third-party suppliers, it will also be more difficult to manufacture commercial quantities of our biologic drug candidates that are approved for commercial sale.
In addition, if commercial sale of our biologic drug candidates is approved, we intend to rely on third parties for their distribution. Proper shipping and distribution requires compliance with specific storage and shipment procedures. Failure to comply with these procedures or the occurrence of inadvertent damage to the shipping container will necessitate return and replacement, potentially resulting in additional cost and causing us to fail to meet supply requirements.
Use of third-party manufacturers may increase the risk that we will not have adequate quantities of our biologic drug candidates.
We use third-party manufacturers to supply our biologic drug candidates for clinical trials. Reliance on third-party manufacturers entails risks to which we would not be subject if we manufactured such components ourselves, including:
- •
- reliance on the third-party for regulatory compliance and quality assurance;
- •
- the possible breach of the manufacturing agreement by the third-party; and
- •
- the possible termination or nonrenewal of the agreement by the third-party, based on its own business priorities, at a time that is costly or inconvenient for us.
Our contract manufacturers are subject to all of the risks and uncertainties that we have when we manufacture on our own. Similar to us, they are subject to ongoing, periodic, unannounced inspection by the FDA and corresponding state and foreign agencies or their designees to ensure strict compliance with cGMP regulations and other governmental regulations and corresponding foreign standards. However, we do not control compliance by our contract manufacturers with these regulations and
34
Table of Contents
standards. Our present or future manufacturers might not be able to comply with these regulatory requirements. If our third-party manufacturers fail to comply with applicable regulations, the FDA or other regulatory authorities could impose sanctions on us, including fines, injunctions, civil penalties, denial of marketing approval of our biologic drug candidates, delays, suspension or withdrawal of approvals, license revocation, seizures or recalls of biologic drug candidates or our other products, operating restrictions and criminal prosecutions. Any of these actions could significantly and adversely affect supplies of our biologic drug candidates or other products and could have a material adverse effect on our business, financial condition and results of operations.
These manufacturers are also subject to many of the general business risks that we and are collaborators are faced with. For example, the recent tightening of global credit and the volatility in the financial markets may result in or contribute to a delay or disruption in the performance or satisfaction of commitments to us by these third parties.
We have contracted with Lonza to manufacture quantities of our stem cell drug candidates for our clinical trials. If Lonza is unable to increase production sufficiently, we may also not be able to meet anticipated market demand in the future.
If our processing and storage facility or our clinical manufacturing facilities are damaged or destroyed, our business and prospects would be negatively affected.
If our processing and storage facility or the equipment in the facility were to be significantly damaged or destroyed, we could suffer a loss of some or all of the stored units of our biologic drug candidates and it would force us to halt our clinical trial processes.
We lease approximately 61,203 square feet of space in Columbia, Maryland that houses essentially all of our corporate operations. Currently, we maintain insurance coverage totaling $19.4 million against damage to our property and equipment, an additional $4.0 million to cover business interruption and extra expenses, and $5.6 million to cover R&D restoration expenses. If we have underestimated our insurance needs, we will not have sufficient insurance to cover losses above and beyond the limits on our policies.
Ethical and other concerns surrounding the use of stem cell therapy or human tissue may negatively affect public perception of us or our products or biologic drug candidates, or may negatively affect regulatory approval of our products or biologic drug candidates, thereby reducing demand for our products and adversely affecting the market price for our common stock.
The commercial success of our biologic drug candidates will depend in part on general public acceptance of the use of stem cell therapy for the prevention or treatment of human diseases. The use of embryonic stem cells and fetal tissue for research and stem cell therapy has been the subject of substantial national and international debate regarding related ethical, legal and social issues. In the U.S., for example, until March 2009, federal government funding of embryonic stem cell research has been limited to specifically identified cell lines and is not otherwise available. We do not use embryonic stem cells or fetal tissue, but the public may not be able to, or may fail to, differentiate our use of adult stem cells from the use by others of embryonic stem cells or fetal tissue. This could result in a negative perception of our company or our products or biologic drug candidates.
We may obtain stem cells from volunteer adult bone marrow donors from non-profit organizations that collect and process tissue donations. Bone marrow donors receive payment, but ethical concerns have been raised by some about the use of donated human tissue in a for-profit setting, as we are doing.
35
Table of Contents
Future adverse events in the field of stem cell therapy or changes in public policy could also result in greater governmental regulation of our biologic drug candidates and potential regulatory delays relating to their testing or approval.
We may eventually compete with other companies for product sales. Many of these competitors have greater resources or capabilities than we have, or may succeed in developing better products or in developing products more quickly than we do, and we may not compete successfully with them.
In the marketplace, we compete or may eventually compete with other companies and organizations that are marketing or developing therapies for our targeted disease indications, based on traditional pharmaceutical, medical device or other, non-cellular therapy and technologies. These include: Novartis, the manufacturer of Neoral® for the prevention of organ rejection in transplant patients, which would compete with Prochymal for the treatment of GvHD; and Johnson & Johnson, the manufacturer of Remicade®, and Abbott, the manufacturer of Humira, which would compete with Prochymal for the treatment of Crohn's disease. In addition to those listed above, we have other potential competitors developing a variety of therapeutics.
We also face competition in the cell therapy field from academic institutions and governmental agencies. Many of our current and potential competitors have greater financial and human resources than we have, including more experience in research and development and more established sales, marketing and distribution capabilities.
We anticipate that competition in our industry will increase. In addition, the health care industry is characterized by rapid technological change, resulting in new product introductions and other technological advancements. Our competitors may develop and market products that render products now or in the future under development by us, or any products manufactured or marketed by us, non-competitive or otherwise obsolete.
The use in human subjects of our stem cell therapies or products produced by us may expose us to product liability claims, and we may not be able to obtain adequate insurance.
We face an inherent risk of product liability claims. None of our products or product candidates have been widely used over an extended period of time, and therefore our safety data are limited. We derive the raw materials for our products and product candidates from human donor sources, the manufacturing process is complex, and the handling requirements are specific, all of which increase the likelihood of quality failures and subsequent product liability claims. We will need to increase our insurance coverage if and when we begin commercializing our biologic drug candidates. We may not be able to obtain or maintain product liability insurance on acceptable terms with adequate coverage or at all. If we are unable to obtain insurance, or if claims against us substantially exceed our coverage, then our business could be adversely impacted. Whether or not we are ultimately successful in any product liability litigation, such litigation could consume substantial amounts of our financial and managerial resources and could result in, among other things:
- •
- significant awards against us;
- •
- substantial litigation costs;
- •
- recall of the product;
- •
- injury to our reputation;
- •
- withdrawal of clinical trial participants; and
- •
- adverse regulatory action.
36
Table of Contents
Any of these results could have a material adverse effect on our business, financial condition and results of operations.
Risk Factors Regarding the Sale of our Osteocel Business.
We may not receive all of the payments available to us under the terms of the asset purchase agreement for the sale of our Osteocel business, and accordingly, we may have less cash available to us to fund our remaining operations.
The terms of the asset purchase agreement for the sale by us of our Osteocel business provide for an initial payment of $35 million dollars in cash and allow for the prospect of additional milestone payments, of up to approximately an additional $50 million dollars in the aggregate. We earned, and have received, the initial payment of $35 million and the initial $5 million milestone payment under this agreement.
In addition, pursuant to the terms of a manufacturing agreement entered into concurrent with the initial closing under the asset purchase agreement, we have the ability to earn fee revenues related to the production of Osteocel for supply to NuVasive over a period of approximately eighteen months following the initial closing.
Our ability to earn the additional milestone payments and fee revenues is, however, subject to a number of conditions and uncertainties, and we have no assurances that these amounts will, in fact, be paid to or be received by us in full. If we do not receive these payments, we will have less cash available to fund our remaining operations and to support the continued development and pursuit of FDA approval for our biologic drug candidates, including Prochymal.
The manufacturing agreement provides for concessionary pricing on the sale of Osteocel, which could result in losses from the operation of discontinued operations. If we incur losses under the manufacturing agreement, we will have less cash available to fund our remaining operations and to support the continued development and pursuit of FDA approval for our biologic drug candidates, including Prochymal.
The asset purchase agreement for the sale of our Osteocel business exposes us to contingent liabilities which could adversely affect our ability to pursue our core business focused on the development and marketing approval for our biologic drug candidates, including Prochymal.
In the asset purchase agreement we have made customary representations and warranties and the parties have agreed to indemnify each other for breaches of representations, warranties and covenants contained in the asset purchase agreement, and we have agreed to indemnify NuVasive for certain excluded liabilities. Should we incur liability for breach of these representations or warranties, our ability to pursue our core business focused on the development and marketing approval for our biologic drug candidates, including Prochymal, could be materially and adversely affected.
By completing the sale, we sold the assets that produce our only currently commercialized product.
Pursuant to the asset purchase agreement, we sold our entire product line relating to the processing, manufacturing, marketing and selling of Osteocel and Osteocel XO. Although we generate revenues from a variety of other sources, including collaborative agreements and a government contract, the Osteocel business that we sold to Nuvasive included our only commercially available product.
37
Table of Contents
Our long term business prospects will depend primarily on the success of our biologic drug candidates business.
Although we expect to continue to manufacture Osteocel until the expiration of the manufacturing agreement approximately eighteen months after the initial closing under the asset purchase agreement, our biologic drug candidate business will be the primary focus of our business. Our long term business prospects will, therefore, be dependent almost solely on the success of our biologic drug candidate business. This business is based on novel technologies and involves significant risks and challenges in regards to product development and optimization, manufacturing, government regulation, intellectual property, third-party reimbursement and market acceptance, among the other risks disclosed by us.
Risks Related to Intellectual Property
If our patent position does not adequately protect our products, others could compete against us more directly, which would harm our business and have a material adverse effect on our financial condition and results of operations.
Our success depends, in large part, on our ability to obtain and maintain intellectual property protection for our biologic drug candidates. The patent position of biotechnology companies is generally highly uncertain, involves complex legal and factual questions and has been the subject of much litigation. Neither the U.S. Patent and Trademark Office nor the courts has a consistent policy regarding the breadth of claims allowed or the degree of protection afforded under many biotechnology patents.
The claims of our existing U.S. patents and those that may issue in the future, or those licensed to us, may not confer on us significant commercial protection against competing products. Third parties may challenge, narrow, invalidate or circumvent any patents we own or may obtain in the future. Our patents on MSC technology, in particular, are quite broad in that they cover mesenchymal stem cells and the therapeutic uses thereof. Patents with broad claims tend to be more vulnerable to challenge by other parties than patents with more narrowly written claims. Also, our pending patent applications may not issue, and we may not receive any additional patents. Further, the laws of foreign countries may not protect our intellectual property rights to the same extent as do the laws of the United States. Our patents might not contain claims that are sufficiently broad to prevent others from utilizing our technologies. Consequently, our competitors may independently develop competing products that do not infringe our patents or other intellectual property.
Because of the extensive time required for development, testing and regulatory review of a potential product, it is possible that, before any of our products can be commercialized, any related patent may expire or remain in force for only a short period following commercialization, thereby reducing any advantages of the patent. For instance, one of our patents related to our MSC technology will expire in 2013 if no extension is applied for and received. To the extent our biologic drug candidates based on that technology are not commercialized ahead of this date, to the extent we have no other patent protection on such products, or to the extent that regulatory or patent extensions are not granted, those products would not have the robust protection we currently expect to enjoy. The background technologies used in the development of our biologic drug candidates are known in the scientific community, and it is possible to duplicate the methods we use to create our biologic drug candidates.
If certain license agreements are terminated, our market exclusivity could be adversely affected.
We are a party to various agreements that give us rights to use specified technologies applicable to research, development and commercialization of our product candidates. If these agreements were voided or terminated, our product development, research and commercialization efforts may be altered or delayed. Certain aspects of our technology rely on patented inventions developed using university or
38
Table of Contents
third party resources. The universities or third parties may have certain rights, as defined by law or applicable agreements, in such patents, and may choose to exercise such rights. If we fail to comply with any terms or provisions of these agreements, our rights could be terminated. Currently, we are in compliance with the terms of all agreements, and we do not have any reason to believe that our rights might be terminated.
If we are unable to protect the confidentiality of our proprietary information, trade secrets and know-how, our competitive position would be impaired and our business, financial condition and results of operations could be adversely affected.
Some aspects of our technology, especially regarding manufacturing processes, is unpatented and is maintained by us as trade secrets. In an effort to protect these trade secrets, we require our employees, consultants, collaborators and advisors to execute confidentiality agreements upon the commencement of their relationships with us. These agreements require that all confidential information developed by the individual or made known to the individual by us during the course of the individual's relationship with us be kept confidential and not disclosed to third parties. These agreements, however, may not provide us with adequate protection against improper use or disclosure of confidential information, and these agreements may be breached. A breach of confidentiality could affect our competitive position. In addition, in some situations, these agreements may conflict with, or be subject to, the rights of third parties with whom our employees, consultants, collaborators or advisors have previous employment or consulting relationships. Also, others may independently develop substantially equivalent proprietary information and techniques or otherwise gain access to our trade secrets.
Adequate remedies may not exist in the event of unauthorized use or disclosure of our confidential information. The disclosure of our trade secrets would impair our competitive position and could have a material adverse effect on our business, financial condition and results of operations.
If we infringe or are alleged to infringe intellectual property rights of third parties, it will adversely affect our business, financial condition and results of operations.
Our research, development and commercialization activities, including any biologic drug candidates or products resulting from these activities, may infringe or be claimed to infringe patents owned by third parties and to which we do not hold licenses or other rights. There may be applications that have been filed but not published that, when issued, could be asserted against us. These third parties could bring claims against us that would cause us to incur substantial expenses and, if successful against us, could cause us to pay substantial damages. Further, if a patent infringement suit were brought against us, we could be enjoined from certain activities including a stop or delay in research, development, manufacturing or sales activities related to the product or biologic drug candidate that is the subject of the suit.
As a result of patent infringement claims, or in order to avoid potential claims, we may choose or be required to seek a license from the third party. These licenses may not be available on acceptable terms, or at all. Even if we are able to obtain a license, the license would likely obligate us to pay license fees or royalties or both, and the rights granted to us might be nonexclusive, which could result in our competitors gaining access to the same intellectual property. Ultimately, we could be prevented from commercializing a product, or be forced to cease some aspect of our business operations, if, as a result of actual or threatened patent infringement claims, we are unable to enter into licenses on acceptable terms. All of the issues described above could also impact our collaborators, which would also impact the success of the collaboration and therefore us.
There has been substantial litigation and other proceedings regarding patent and other intellectual property rights in the pharmaceutical and biotechnology industries. In addition to infringement claims against us, we may become a party to other patent litigation and other proceedings, including
39
Table of Contents
interference and reexamination proceedings declared by the United States Patent and Trademark Office and opposition proceedings before the patent offices for other countries (e.g. the European Patent Office), regarding intellectual property rights with respect to our products and technology. For example, a patent that was granted to us in Europe for human mesenchymal stem cells in the cardiac context was opposed in the European Patent Office by two different companies. In 2008 we prevailed in an opposition proceeding brought before the European Patent Office against one of our patents related to the cardiac indications of Prochymal. Though we were successful in that particular proceeding, the outcome of any future patent controversies is uncertain. The cost to us of any patent litigation or other proceeding, even if resolved in our favor, could be substantial. Some of our competitors may be able to sustain the costs of such litigation or proceedings more effectively than we can because of their greater financial resources. Patent litigation and other proceedings may also absorb significant management time. Uncertainties resulting from the initiation and continuation of patent litigation or other proceedings could have a material adverse effect on our ability to compete in the marketplace and, as a result, on our business, financial condition and results of operations. To the extent that our employees, consultants or contractors use intellectual property owned by others, disputes may arise as to the rights related to or resulting from the use of such intellectual property.
We may become involved in lawsuits to protect or enforce our patents or the patents of our collaborators or licensors, which could be expensive and time consuming.
Litigation may be necessary to enforce patents issued or licensed to us, to protect trade secrets or know-how, or to determine the scope and validity of the proprietary rights. Litigation, opposition or interference proceedings could result in substantial additional costs and diversion of management focus. If we are ultimately unable to protect our technology, trade secrets or know-how, we may be unable to operate profitably.
Competitors may infringe our patents or the patents of our collaborators or licensors. As a result, we may be required to file infringement claims to protect our proprietary rights. This can be expensive, particularly for a company of our size, and time-consuming. In addition, in an infringement proceeding, a court may decide that a patent of ours is invalid or is unenforceable, or may refuse to enjoin the other party from using the technology at issue. An adverse determination of any litigation or defense proceedings could put one or more of our patents at risk of being invalidated or interpreted narrowly. Interference proceedings brought by the U.S. Patent and Trademark Office may be necessary to determine the priority of inventions with respect to our patent applications or those of our collaborators or licensors. Litigation or interference proceedings may fail and, even if successful, may result in substantial costs and distraction to our management. We may not be able, alone or with our collaborators and licensors, to prevent misappropriation of our proprietary rights, particularly in countries where the laws may not protect such rights as fully as in the United States.
Furthermore, because of the substantial amount of discovery required in connection with intellectual property litigation, there is a risk that some of our confidential information could be compromised by disclosure during this type of litigation. In addition, during the course of this kind of litigation, there could be public announcements of the results of hearings, motions or other interim proceedings or developments. If investors perceive these results to be negative, the market price for our common stock could be significantly harmed.
The biotechnology industry, including our fields of therapeutic interest, is highly competitive and subject to significant and rapid technological change. Accordingly, our success will depend, in part, on our ability to respond quickly to such change through the development and introduction of new products. Our ability to compete successfully against currently existing and future alternatives to our product candidates and systems and competitors who compete directly with us in the biopharmaceutical industry will depend, in part, on our ability to: attract and retain skilled scientific and research personnel; develop technologically superior products; develop competitively priced products; obtain
40
Table of Contents
patent or other required regulatory approvals for our products; and be early entrants to the market; manufacture, market and sell our products, independently or through collaborations. If a third party were to commercialize a competitive product, there is no assurance that we would have a basis for initiating patent infringement proceedings or that, if initiated, we would prevail in such proceedings.
Risks Related to Regulatory Approval and Other Government Regulations
If we are not able to successfully develop and commercialize our biologic drug candidates and obtain the necessary regulatory approvals, we may not generate sufficient revenues to continue our business operations.
In order to generate sales revenue from our biologic drug candidates, we must conduct extensive preclinical studies and clinical trials to demonstrate that our biologic drug candidates are safe and effective and obtain required regulatory approvals. Our early stage biologic drug candidates may fail to perform as we expect. Moreover, our biologic drug candidates in later stages of development may fail to show the desired safety and efficacy traits despite having progressed successfully through preclinical or initial clinical testing. We will need to devote significant additional research and development, financial resources and personnel to develop commercially viable products and obtain the necessary regulatory approvals.
If our biologic drug candidates do not prove to be safe and efficacious in clinical trials, we will not obtain the required regulatory approvals. If we fail to obtain such approvals, we may not generate sufficient revenues to continue our business operations.
Even if we obtain regulatory approval of a product, that approval may be subject to limitations on the indicated uses for which it may be marketed. Even after granting regulatory approval, the FDA and regulatory agencies in other countries continue to review and inspect marketed products, manufacturers and manufacturing facilities, which may create additional regulatory burdens. Later discovery of previously unknown problems with a product, manufacturer or facility, may result in restrictions on the product or manufacturer, including a withdrawal of the product from the market. Further, regulatory agencies may establish additional regulations that could prevent or delay regulatory approval of our products.
We cannot market and sell our biologic drug candidates in the United States or in other countries if we fail to obtain the necessary regulatory approvals or licensure.
We cannot sell our biologic drug candidates until regulatory agencies grant marketing approval, or licensure. The process of obtaining regulatory approval is lengthy, expensive and uncertain. It is likely to take several years to obtain the required regulatory approvals for our lead stem cell biologic drug candidate, Prochymal, or we may never gain the necessary approvals. Any difficulties that we encounter in obtaining regulatory approval may have a substantial adverse impact on our operations and cause our stock price to decline significantly. Moreover, because our biologic drug candidates are all based on a single platform technology, MSCs, any adverse events in our clinical trials for one of our biologic drug candidates could negatively impact the clinical trials and approval process for our other biologic drug candidates.
To obtain marketing approvals in the United States for MSC products, for instance, we must, among other requirements, complete carefully controlled and well-designed clinical trials sufficient to demonstrate to the FDA that the biologic drug candidate is safe and effective for each disease for which we seek approval. Several factors could prevent completion or cause significant delay of these trials, including an inability to enroll the required number of patients or failure to demonstrate adequately that MSCs are safe, effective and potent for use in humans. Negative or inconclusive results from or adverse medical events during a clinical trial could cause the clinical trial to be repeated or a program to be terminated, even if other studies or trials relating to the program are successful. The
41
Table of Contents
FDA can place a clinical trial on hold if, among other reasons, it finds that patients enrolled in the trial are or would be exposed to an unreasonable and significant risk of illness or injury. If safety concerns develop, we or the FDA could stop our trials before completion. Some participants in our MSC clinical trial have experienced serious adverse events, seven of which have been determined to be possibly related to MSCs and one of which has been determined to be probably related. A serious adverse event is an event that results in significant medical consequences, such as hospitalization, disability or death, and must be reported to the FDA. We cannot assure you that safety concerns regarding MSCs will not develop.
The pathway to regulatory approval for MSCs may be more complex and lengthy than for approval of a new conventional drug. Similarly, to obtain approval to market our stem cell products outside of the United States, we, together with our collaborative partners, will need to submit clinical data concerning our products and receive regulatory approval from governmental agencies, which in certain countries includes approval of the price we intend to charge for our product. We may encounter delays or rejections if changes occur in regulatory agency policies during the period in which we develop a biologic drug candidate or during the period required for review of any application for regulatory agency approval. If we are not able to obtain regulatory approvals for use of our biologic drug candidates under development, we will not be able to commercialize such products, and therefore may not be able to generate sufficient revenues to support our business.
If we are not able to conduct our clinical trials properly and on schedule, marketing approval by FDA and other regulatory authorities may be delayed or denied.
The completion of our clinical trials may be delayed or terminated for many reasons, including, but not limited to, if:
- •
- the FDA does not grant permission to proceed and places the trial on clinical hold;
- •
- subjects do not enroll in our trials at the rate we expect;
- •
- subjects experience an unacceptable rate or severity of adverse side effects;
- •
- third-party clinical investigators do not perform our clinical trials on our anticipated schedule or consistent with the clinical trial protocol, Good Clinical Practice and regulatory requirements, or other third parties do not perform data collection and analysis in a timely or accurate manner;
- •
- inspections of clinical trial sites by the FDA or Institutional Review Boards (IRBs) of research institutions participating in our clinical trials find regulatory violations that require us to undertake corrective action, suspend or terminate one or more sites, or prohibit us from using some or all of the data in support of our marketing applications; or
- •
- one or more IRBs suspends or terminates the trial at an investigational site, precludes enrollment of additional subjects, or withdraws its approval of the trial.
Our development costs will increase if we have material delays in our clinical trials, or if we are required to modify, suspend, terminate or repeat a clinical trial. If we are unable to conduct our clinical trials properly and on schedule, marketing approval may be delayed or denied by the FDA.
We may not be able to secure and maintain research institutions to conduct our clinical trials.
We rely on research institutions to conduct our clinical trials. Specifically, the limited number of bone marrow transplant centers further heightens our dependence on such research institutions for our Phase III trials. Our reliance upon research institutions, including hospitals and clinics, provides us with less control over the timing and cost of clinical trials and the ability to recruit subjects. If we are unable to reach agreement with suitable research institutions on acceptable terms, or if any resulting agreement is terminated, we may be unable to quickly replace the research institution with another qualified institution on acceptable terms. We may not be able to secure and maintain suitable research institutions to conduct our clinical trials.
42
Table of Contents
Final marketing approval of our biologic drug candidates by the FDA or other regulatory authorities for commercial use may be delayed, limited, or denied, any of which would adversely affect our ability to generate operating revenues.
Any of the following factors may cause final marketing approval for our biologic drug candidates to be delayed, limited or denied:
- •
- our biologic drug candidates require significant clinical testing to demonstrate safety and effectiveness before applications for marketing approval can be filed with the FDA;
- •
- data obtained from preclinical and nonclinical animal testing and clinical trials can be interpreted in different ways, and the FDA may not agree with our interpretations;
- •
- it may take many years to complete the testing of our biologic drug candidates, and failure can occur at any stage of the process;
- •
- negative or inconclusive results or adverse side effects during a clinical trial could cause us to delay or terminate development efforts for a biologic drug candidate; and
- •
- commercialization may be delayed if the FDA requires us to expand the size and scope of the clinical trials.
If marketing approval for our biologic drug candidates is delayed, limited or denied, our ability to market products, and our ability to generate product sales, would be adversely affected.
Producing and marketing an approved drug or other medical product is subject to significant and costly post-approval regulation.
It is likely that Prochymal, if approved for GvHD based on our currently contemplated Phase III trial, will receive conditional approval by the FDA, and we will be required to conduct Phase IV clinical trials to obtain full approval. Even if we obtain full approval of a product, that approval is subject to limitations on the indicated uses for which we can market it. After granting marketing approval, the FDA and regulatory agencies in other countries continue to review and inspect marketed products, manufacturers and manufacturing facilities, creating additional regulatory burdens. Later discovery of previously unknown problems with a product, manufacturer or facility may result in restrictions on the product or manufacturer, including a withdrawal of the product from the market. Further, regulatory agencies may establish additional regulations that could prevent or delay marketing approval of our products.
Our business involves the use of hazardous materials that could expose us to environmental and other liability.
We have facilities in Maryland that are subject to various local, state and federal laws and regulations relating to safe working conditions, laboratory and manufacturing practices, the experimental use of animals and the use and disposal of hazardous or potentially hazardous substances, including chemicals, micro-organisms and various radioactive compounds used in connection with our research and development activities. In the United States, these laws include the Occupational Safety and Health Act, the Toxic Test Substances Control Act and the Resource Conservation and Recovery Act. We cannot assure you that accidental contamination or injury to our employees and third parties from hazardous materials will not occur. We do not have insurance to cover claims arising from our use and disposal of these hazardous substances other than limited clean-up expense coverage for environmental contamination due to an otherwise insured peril, such as fire.
43
Table of Contents
We may not be able to obtain or maintain Orphan Drug designation for our biologic drug candidates.
Some jurisdictions, including the European Union and the United States, may designate drugs for relatively small patient populations as orphan drugs. Although the FDA and its European counterpart, the European Medicines Agency ("EMEA") have designated Prochymal for the treatment of steroid refractory GvHD as an orphan drug, none of our other biologic drug candidates have received such designation. Orphan Drug designation does not convey any advantage in, or shorten the duration of, the regulatory review and approval process, but does make the product eligible for orphan drug exclusivity, reduced filing fees and specific tax credits. Generally, if a company receives the first marketing approval for a product with an Orphan Drug designation in the clinical indication for which it has such designation, the product is entitled to orphan drug exclusivity. Orphan drug exclusivity means that the health authorities will not approve another application to market the same drug for the same indication, except in limited circumstances, for a period of up to seven years in the United States and ten years in Europe. This exclusivity, however, could block the approval of our biologic drug candidates if a competitor obtains marketing approval before us. Even if we obtain orphan drug exclusivity for any of our biologic drug candidates, we may not be able to maintain it. For example, if a competitive product is shown to be clinically superior to our product, any orphan drug exclusivity we have will not block the approval of such competitive product.
The Fast Track designation for development of any of our products may not lead to a faster development or regulatory review or approval process, and it does not increase the likelihood the biologic drug candidate will receive marketing approval.
If a drug is intended for the treatment of a serious or life-threatening condition and the drug demonstrates the potential to address unmet medical needs for this condition, the drug sponsor may apply for FDA Fast Track designation for a particular indication. Marketing applications filed by sponsors of products in Fast Track development may qualify for priority review under the policies and procedures offered by the FDA, but the Fast Track designation does not assure any such qualification. Although we have obtained a Fast Track designation from the FDA for Prochymal for the treatment of GvHD and treatment refractory Crohn's disease, receipt of Fast Track designation may not result in a faster development process, review or approval compared to drugs considered for approval under conventional FDA procedures. In addition, the FDA may withdraw our Fast Track designation at any time. If we lose our Fast Track designation, the approval process may be delayed. In addition, our Fast Track designation does not guarantee that we will qualify for or be able to take advantage of the expedited review procedures and does not increase the likelihood that Prochymal will receive regulatory approval for the treatments of steroid refractory GvHD or Crohn's disease.
Risks Related to Government Contracts
Federal government spending priority or our relationships with the federal government may change in a manner that harms our business or prospects.
Our ability to successfully pursue and perform under development and purchase agreements with United States and Allied governmental agencies for countermeasures to nuclear terrorism and other radiological emergencies, including the contract awarded to us by the DoD for the development and stockpiling of Prochymal for the treatment of acute radiation syndrome ("ARS"), depends upon continued federal government expenditures on defense, emergency preparedness and other programs. These expenditures will likely fluctuate over time. While spending authorizations for defense and emergency preparedness related programs by the government have increased in recent years, and in particular after the 2001 terrorist attacks, future levels of expenditures and authorizations for these programs may decrease, remain constant or shift to program areas inapplicable to us. Our business, prospects, financial condition and/or operating results could be materially harmed by budgetary constraints affecting federal government spending generally, or specific departments or agencies in
44
Table of Contents
particular, and by changes in fiscal policies or available funding, or by changes in federal government programs or requirements or delays in government appropriations process. In addition, our business, prospects, financial condition and/or operating results could be materially harmed if we are suspended or disbarred from contracting with the federal government or a significant governmental agency, or our reputation or relationship with governmental entities is impaired, or the government otherwise declines to do business with us, or significantly decreases the amount of business it is willing to do with us.
Federal government contracts contain provisions that may be unfavorable to us.
Federal government contracts contain provisions, and are subject to laws and regulations, that give the government rights and remedies not typically found in commercial contracts. These provisions may allow the government to terminate existing contracts for convenience, as well as for default, to reduce or modify contracts or subcontracts, to cancel multi-year contracts or related purchase orders if funds for contract performance for any subsequent year become unavailable, to decline to exercise an option to renew a multi-year contract or to decline to purchase product pursuant to an option afforded under a contract. If the government terminates a contract for convenience, we may recover only our incurred or committed costs, settlement expenses and profit on the work completed prior to the termination. If the government terminates a contract for default, we may not recover even those amounts, and instead may be liable for excess costs incurred by the government in procuring undelivered items and services from another source.
Unfavorable federal government audit results could subject us to penalties or sanctions and could impair our ability to win new contracts.
The Defense Contract Audit Agency ("DCAA") and other government agencies routinely audit and investigate government contracts and systems. These agencies review a contractor's performance on its contract, cost structure and compliance with applicable laws, regulations and standards. The DCAA also reviews the adequacy of, and a contractor's compliance with, its internal control systems and policies, including the contractor's accounting, purchasing, property, estimating, compensation and managing information systems. Allegations of impropriety or deficient controls could harm our reputation and/or adversely influence the award of new contracts. Any costs founds to be improperly allocated to a specific contract will not be reimbursed, while such costs already reimbursed must be refunded. Therefore, a DCAA audit could result in a substantial adjustment to our revenue earned from federal government contracts.
The government may terminate our federal government contracts at any time.
Federal government contracts may span one or more base years and one or more option years, and may provide the government with one or more options in respect of continued performance by us thereunder. For example, our contract with the U.S. Department of Defense ("DoD") for the development and stockpiling of Prochymal for the treatment of ARS provides the DoD with successive options for the purchase of Prochymal, assuming receipt of FDA approval for its use in the treatment of ARS. Federal government agencies have no obligation to exercise these options unless determined to be in the best interest of the government. Additionally, federal government contracts typically contain provisions permitting the government to terminate the contract for its convenience. A decision not to exercise an option or a decision to terminate a contract could have a material adverse effect on our business and prospects.
If we fail to comply with complex procurement laws and regulations, we could incur various penalties or sanctions.
To the extent which we enter into contracts or other arrangements with the United States or other Allied governments, we must comply with the laws and regulations relating to the formation,
45
Table of Contents
administration and performance of those contracts. These laws and regulations affect how we conduct business with our government contracts. In complying with these laws and regulations, we may incur additional costs and delays, and non-compliance may also allow for the assignment of additional fines and penalties, including contractual damages. Among these laws and regulations are the Federal Acquisition Regulations, which comprehensively regulate the formation, administration and performance of United States federal government contracts, the Truth in Negotiations Act, which requires certification and disclosure of all costs and pricing data in connection with contract negotiations, and laws, regulations and executive orders restricting the use and dissemination of information classified for national security purposes, and restricting the export of certain products and technical data. We are subject to periodic review of our performance under and compliance with the terms of any federal government contracts to which we are a party. As a result of these reviews, we may learn that we are not in compliance with all of the terms of any such contracts and we could be subject to civil or criminal penalties or administrative sanctions for failure of compliance.
Risks Related to Our Common Stock
The trading price of the shares of our common stock is highly volatile, and purchasers of our common stock could incur substantial losses.
Our stock price is volatile. The stock market in general and the market for biotechnology companies in particular have experienced extreme volatility that has often been unrelated to the operating performance of particular companies. As a result of this volatility, investors may not be able to sell their common stock at or above the price they paid for it. The market price for our common stock may be influenced by many factors, including:
- •
- results of clinical trials of our biologic drug candidates or those of our competitors;
- •
- regulatory developments in the United States and foreign countries;
- •
- variations in our financial results or those of companies that are perceived to be similar to us;
- •
- changes in the structure of healthcare payment systems;
- •
- announcements by us of significant acquisitions, strategic partnerships, joint ventures or capital commitments;
- •
- market conditions in the pharmaceutical and biotechnology sectors and issuance of securities analysts' reports or recommendations;
- •
- sales of substantial amounts of our stock by existing stockholders;
- •
- sales of our stock by insiders and 5% stockholders;
- •
- general economic, industry and market conditions;
- •
- additions or departures of key personnel;
- •
- intellectual property, product liability or other litigation against us;
- •
- expiration or termination of our relationships with our collaborators; and
- •
- the other factors described in this "Risk Factors" section.
In addition, in the past, stockholders have initiated class action lawsuits against biotechnology and pharmaceutical companies following periods of volatility in the market prices of these companies' stock. Such litigation, if instituted against us, could cause us to incur substantial costs and divert management's attention and resources, which could have a material adverse effect on our business, financial condition and results of operations.
46
Table of Contents
Concentration of ownership of our common stock among our existing executive officers, directors and principal stockholders may prevent new investors from influencing significant corporate decisions.
Our executive officers, directors and beneficial owners of 5% or more of our common stock and their affiliates, in aggregate, beneficially own approximately 50% of our outstanding common stock as of December 31, 2008. These persons, acting together, will be able to significantly influence all matters requiring stockholder approval, including the election and removal of directors and any merger or other significant corporate transactions. The interests of this group of stockholders may not coincide with our interests or the interests of other stockholders.
Peter Friedli, our Chairman of the Board of Directors, and certain entities with which he is affiliated, beneficially own approximately 43% of our outstanding common stock as of December 31, 2008. Accordingly, Mr. Friedli currently has, and will continue to have, a significant influence over the outcome of all corporate actions requiring stockholder approval.
Certain provisions of Delaware law and of our charter and bylaws contain provisions that could delay and discourage takeover attempts and any attempts to replace our current management by stockholders.
Certain provisions of our certificate of incorporation and bylaws, and applicable provisions of Delaware corporate law, may make it more difficult for or prevent a third party from acquiring control of us or changing our Board of Directors and management. These provisions include:
- •
- the ability of our Board of Directors to issue preferred stock with voting or other rights or preferences;
- •
- the inability of stockholders to act by written consent;
- •
- a classified Board of Directors with staggered three-year terms;
- •
- requirements that special meetings of our stockholders may only be called by the chairman of our Board of Directors, upon request of stockholders holding at least 20% of our capital stock issued and outstanding, or upon a resolution adopted by, or an affirmative vote of, a majority of our Board of Directors; and
- •
- requirements that our stockholders comply with advance notice procedures in order to nominate candidates for election to our Board of Directors or to place stockholders' proposals on the agenda for consideration at meetings of stockholders.
We will also be afforded the protections of Section 203 of the Delaware General Corporation Law, which will prevent us from engaging in a business combination with a person who acquires at least 15% of our common stock for a period of three years from the date such person acquired such common stock, unless advance board or stockholder approval was obtained.
Any delay or prevention of a change of control transaction or changes in our Board of Directors or management could deter potential acquirers or prevent the completion of a transaction in which our stockholders could receive a substantial premium over the then current market price for their shares.
ITEM 1B. UNRESOLVED STAFF COMMENTS
Not Applicable.
ITEM 2. PROPERTIES
Our corporate headquarters are located in Columbia, Maryland, where we lease approximately 61,000 square feet, currently at a rent of approximately $1.0 million per annum. This lease expires in July 2016, and includes options to extend the term of the lease for two additional five year periods.
47
Table of Contents
Historically, we had leased approximately 126,000 square feet in Baltimore, Maryland where we housed our manufacturing operations. The lease expired in September 2008. As planned, we have vacated those facilities and consolidated our operations in our Columbia, Maryland facilities. The Asset Purchase Agreement with NuVasive, Inc. provides for the assignment of the lease on our Columbia, MD facility and the transfer of all other manufacturing assets at the conclusion of the manufacturing agreement, which is scheduled to occur on or before the end of December 2009. We are presently evaluating our future facilities requirements and expect to continue to be located in the Baltimore-Washington business district.
ITEM 3. LEGAL PROCEEDINGS
From time to time in the ordinary course of business, we are subject to claims, asserted or unasserted, or named as a party to lawsuits or investigations. Litigation, in general, and intellectual property and securities litigation in particular, can be expensive and disruptive to normal business operations. Moreover, the results of legal proceedings cannot be predicted with any certainty and in the case of more complex legal proceedings, such as intellectual property and securities litigation, the results are difficult to predict at all. We are not aware of any asserted or unasserted legal proceedings or claims that we believe would have a material adverse effect on our financial condition or results of our operations.
ITEM 4. SUBMISSION OF MATTERS TO A VOTE OF SECURITIES HOLDERS
No matters were submitted to a vote of our security holders during the fourth quarter of the year ended December 31, 2008.
48
Table of Contents
PART II
ITEM 5. MARKET FOR REGISTRANT'S COMMON EQUITY, RELATED STOCKHOLDER MATTERS AND ISSUER PURCHASES OF EQUITY SECURITIES
Market Information
Our common stock trades on the NADSAQ Global Market under the symbol "OSIR." The following table lists the high and low sale prices per share for our common stock based on the closing sales prices as reported on the NASDAQ Global Market for the periods indicated.
| | | | | | | | | | | | | |
| | 2008 | | 2007 | |
|---|
Quarter Ended | | High | | Low | | High | | Low | |
|---|
March 31 | | $ | 15.60 | | $ | 10.01 | | $ | 29.29 | | $ | 11.85 | |
June 30 | | | 14.30 | | | 11.50 | | | 19.05 | | | 10.60 | |
September 30 | | | 20.20 | | | 12.10 | | | 14.42 | | | 11.01 | |
December 31 | | | 21.65 | | | 10.80 | | | 14.57 | | | 9.98 | |
Stockholders
As of March 5, 2009, there were approximately 234 stockholders of record of our common stock and, according to our estimates, approximately 2,200 beneficial owners of our common stock.
Dividends
We have not paid dividends to our stockholders since our inception and we do not plan to pay cash dividends in the foreseeable future. We currently intend to retain earnings, if any, to finance our growth.
Unregistered Sales of Securities
In January 2008, we induced the conversion of $1.2 million of our 10% convertible promissory notes, together with accrued interest into 87,524 shares of common stock at the conversion price of $14.00 per share. The notes were held by institutional and accredited investors based outside of the U.S. The debt conversion was arranged for us by Friedli Corporate Finance, Inc., of which Peter Friedli, the Chairman of our Board of Directors and largest shareholder, is President and sole owner. The securities issued as a result of the debt conversion were issued pursuant to exemptions from registration as provided for under Regulation S and Regulation D promulgated under Securities Act of 1933, as amended (the "Securities Act"). In January 2008, we filed a Registration Statement on Form S-3 to register the resale of these shares by the respective holders.
In March and May 2008, we accepted subscription agreements for the sale of $10.5 million convertible promissory notes to non-U.S. investors in a private placement intended to qualify under Regulation S and Section 4(2) of the Securities Act of 1933, as amended. The notes were funded in March and May 2008 and bore interest at rates between 2% and 4% per annum, payable upon maturity on November 2008 or November 2009. The notes were convertible into shares of our common stock at the conversion price equal to the closing price of our common stock on the NASDAQ Global Market on the date the subscription agreements were accepted. These notes were convertible at prices ranging from $12.04–$13.18 per share. All the notes were converted into 851,914 shares of common stock in November 2008.
The private placements and the debt conversions were led by Friedli Corporate Finance, Inc., of which Peter Friedli, the Chairman of our Board of Directors and largest shareholder, is President and sole owner. Included among the purchasers of the convertible notes was Mr. Friedli, who individually invested $2.5 million in the March 2008 private placement. New Venturetec, Inc., a Swiss publicly
49
Table of Contents
traded company approximately 3% owned by Mr. Friedli who serves as its President, invested $2.5 million in the May 2008 private placement. Our Board of Directors, including all independent directors, but with Mr. Friedli abstaining, together with our Audit Committee, unanimously approved the private placements and note conversion transactions including to Mr. Friedli and New Venturetec, Inc., and the arrangements with Friedli Corporate Finance, Inc.
The securities issued as a result of the debt conversion have not been registered under the Securities Act or any state securities laws and may not be offered or sold within the United States or to U.S. Persons unless registered under the Securities Act and applicable state securities laws or unless an exemption from such registration is available. Pursuant to the terms of the private placements and debt conversions, the holders were provided "short form" demand registration rights.
Issuer Purchase of Equity Securities and Use of Proceeds
There were no repurchases by us of our securities during fiscal 2008 or 2007.
Stock Performance Graph
The following graph shows the cumulative total return, assuming the investment of $100 on August 4, 2006 (the date on which our initial public offering was declared effective and our common stock began trading on the NASDAQ Global Market), on an investment in each of our common stock, the NASDAQ Composite Index (U.S. and Foreign) and the NASDAQ Biotechnology Index. The comparisons in the table are required by the SEC and are not intended to forecast or be indicative of possible future performance of our common stock.
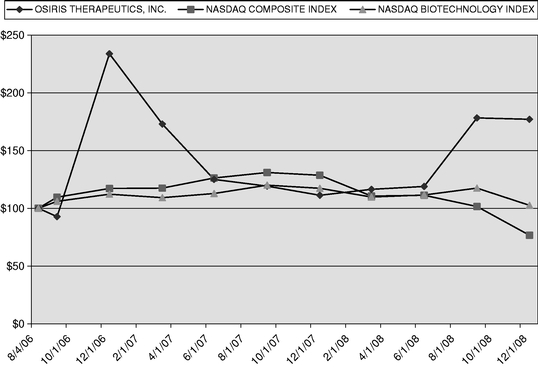
Securities Authorized for Issuance under Equity Compensation Plans
The information required by Item 201(d) of Regulation S-K, pursuant to paragraph (a) of this Item 5, is incorporated by reference to the information set forth under the caption "Equity Compensation Plan Information" in the Company's Proxy Statement for the 2009 Annual Meeting of Stockholders, which is anticipated to be filed pursuant to Regulation 14A no later than one hundred twenty (120) days following the end of the fiscal year reported on.
50
Table of Contents
ITEM 6. SELECTED FINANCIAL DATA
We derived the selected financial data presented below for the periods or dates indicated from our financial statements. Our financial statements for these periods were audited by Stegman & Company, an independent registered public accounting firm. You should read the data below in conjunction with our financial statements, related notes and other financial information appearing in "Item 7. Management's Discussion and Analysis of Financial Condition and Results of Operations" and "Item 8. Financial Statements and Supplementary Data."
| | | | | | | | | | | | | | | | | |
| | Year Ended December 31, | |
|---|
| | 2008 | | 2007 | | 2006 | | 2005 | | 2004 | |
|---|
| | (in thousands, except per share data)
| |
|---|
Statement of Operations Data: | | | | | | | | | | | | | | | | |
Revenue from collaborative research agreements, government contracts, and royalties | | $ | 10,044 | | $ | 2,048 | | $ | 1,181 | | $ | 3,013 | | $ | 3,911 | |
Operating expenses: | | | | | | | | | | | | | | | | |
| | Research and development | | | 69,897 | | | 47,140 | | | 37,590 | | | 16,508 | | | 11,888 | |
| | General and administrative and other expenses | | | 8,586 | | | 6,071 | | | 8,459 | | | 2,294 | | | 1,704 | |
| | | | | | | | | | | | |
Total operating expenses | | | 78,483 | | | 53,211 | | | 46,049 | | | 18,802 | | | 13,592 | |
| | | | | | | | | | | | |
Loss from operations | | | (68,439 | ) | | (51,163 | ) | | (44,868 | ) | | (15,789 | ) | | (9,681 | ) |
Interest expense, net | | | (978 | ) | | (6,695 | ) | | (4,685 | ) | | (4,300 | ) | | (847 | ) |
| | | | | | | | | | | | |
Loss from continuing operations | | | (69,417 | ) | | (57,858 | ) | | (49,553 | ) | | (20,089 | ) | | (10,528 | ) |
Income from discontinued operations | | | 35,925 | | | 3,937 | | | 4,594 | | | 94 | | | — | |
| | | | | | | | | | | | |
Net loss | | $ | (33,492 | ) | $ | (53,921 | ) | $ | (44,959 | ) | $ | (19,995 | ) | $ | (10,528 | ) |
| | | | | | | | | | | | |
Basic and diluted loss per share from continuing operations | | $ | (2.18 | ) | $ | (2.03 | ) | $ | (2.97 | ) | $ | (2.23 | ) | $ | (1.19 | ) |
| | | | | | | | | | | | |
Basic and diluted net loss per share | | $ | (1.05 | ) | $ | (1.89 | ) | $ | (2.70 | ) | $ | (2.23 | ) | $ | (1.19 | ) |
| | | | | | | | | | | | |
Weighted average shares of common stock used in computing basic and diluted net loss per share | | | 31,895 | | | 28,489 | | | 16,663 | | | 8,959 | | | 8,814 | |
| | | | | | | | | | | | |
| | | | | | | | | | | | | | | | |
| | At December 31, | |
|---|
| | 2008 | | 2007 | | 2006 | | 2005 | | 2004 | |
|---|
Balance Sheet Data: | | | | | | | | | | | | | | | | |
Cash and investments available for sale | | $ | 62,238 | | $ | 18,164 | | $ | 39,181 | | $ | 43,471 | | $ | 488 | |
Working capital | | | 70,599 | | | 7,247 | | | 33,166 | | | 38,103 | | | (5,459 | ) |
Total assets | | | 137,467 | | | 37,041 | | | 49,168 | | | 51,014 | | | 5,972 | |
Notes payable, less current portion | | | — | | | 1,200 | | | 25,000 | | | 47,411 | | | 7,519 | |
Mandatorily redeemable convertible preferred stock | | | — | | | — | | | — | | | 64,267 | | | — | |
Convertible preferred stock | | | — | | | — | | | — | | | 32,746 | | | 15,243 | |
Accumulated deficit | | | (274,916 | ) | | (241,424 | ) | | (187,503 | ) | | (142,544 | ) | | (122,549 | ) |
Total stockholders' (deficit) equity | | | (5,020 | ) | | 14,336 | | | 11,287 | | | (73,662 | ) | | (13,004 | ) |
51
Table of Contents
ITEM 7. MANAGEMENT'S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS
When reviewing the discussion below, you should keep in mind the substantial risks and uncertainties that characterize our business. In particular, we encourage you to review the risks and uncertainties described under "Risk Factors" included as Item 1A in this Annual Report on Form 10-K. These risks and uncertainties could cause actual results to differ materially from those forecasted in forward-looking statements or implied by past results and trends. Forward-looking statements are statements that attempt to project or anticipate future developments in our business; we encourage you to review the examples of forward-looking statements included in this Annual Report on Form 10-K under Item 1 at the beginning of this report. These statements, like all statements in this report, speak only as of the date of this report (unless another date is indicated), and we undertake no obligation to update or revise the statements n light of future developments.
The following is a discussion and analysis of our financial condition, results of operations, liquidity and capital resources for each of the three years in the period ended December 31, 2008 and significant factors that could affect our prospective financial condition and results of operations. You should read this discussion together with our financial statements and notes included in "Item 8. Financial Statements and Supplementary Data."
2008 Highlights
We believe cellular therapies have certain advantages over traditional medical approaches. For example, cell therapies can be targeted, avoiding many of the safety complications arising from systemic treatments. Cell therapies can also be responsive to their environment, turning on or off certain effects as conditions in the surrounding tissue change. Cell therapies can also be multifaceted. For example, the cells in Prochymal, our leading biologic drug candidate, have demonstrated the ability to not only down regulate inflammation, but also repair the damage caused by the inflammation.
We believe the combination of these unique properties will allow us to solve many of the more challenging questions facing medicine today. We have established ourselves as the leader in the emerging field of cell therapy. We are an industry leader in the field, having developed the only commercially available stem cell product on the market. We are continuing to make advances, with eight active clinical trials. Four of the trials have progressed to Phase III, each of which has been granted fast track status by the FDA.
During 2008, we:
- •
- Formed major strategic alliance with Genzyme Corporation pursuant to a collaboration agreement worth up to $1.4 billion, for the development and commercialization of Prochymal and Chondrogen in countries outside the United States and Canada;
- •
- Utilized the first $75.0 million of the non-contingent, non-refundable up front payment received under our collaboration agreement with Genzyme to extinguish all of our outstanding debt and now report cash, short-term investments and receivables of $123.5 million as of December 31, 2008. We are scheduled to receive an additional $55.0 million non-contingent payment under this agreement on July 1, 2009;
- •
- Sold our Osteocel business in a transaction worth up to $85.0 million. To date, we have earned and received the $35.0 million of initial purchase price and the first $5.0 million milestone under this transaction;
- •
- Awarded a contract by the United States Department of Defense to develop and stockpile Prochymal for acute radiation syndrome (ARS), fully valued at up to $224.7 million;
52
Table of Contents
- •
- Completed enrollment in first worldwide Phase III stem cell clinical trial for the treatment of steroid-refractory acute Graft versus Host Disease (GvHD);
- •
- Reached agreement with the FDA regarding the timing and content of the submission of the first marketing application for a stem cell drug;
- •
- Received approval to treat pediatric patients suffering from GvHD with Prochymal under the FDA's Expanded Access Program;
- •
- Received FDA clearance to broaden Prochymal Expanded Access Program to adults with GvHD;
- •
- Received approval to initiate Prochymal Expanded Access Program in Canada for pediatric patients suffering from life-threatening GvHD;
- •
- Completed enrollment of Phase II clinical trial evaluating Prochymal in patients with chronic obstructive pulmonary disease (COPD); and
- •
- Reported positive two-year data from the Phase I clinical trial evaluating Prochymal in heart attack patients.
Business Overview
We are a leading stem cell therapeutic company focused on developing and marketing products to treat medical conditions in the inflammatory, orthopedic and cardiovascular areas. Our biologic drug candidates utilize mesenchymal stem cells, or MSCs. We launched the only commercially available stem cell product in 2005. We currently have eight clinical trials ongoing, including four Phase III clinical trials. We are conducting Phase III trials for Prochymal, our lead biologic drug candidate, for (1) the treatment of steroid refractory acute GvHD, (2) first line treatment of acute GvHD, (3) biologics refractory Crohn's disease, and (4) acute radiation syndrome, which is under the Animal Rule.
We have received Fast Track Status from the FDA for each of these pivotal Phase III trials for Prochymal. We have initiated Phase II clinical trials for Prochymal for both type 1 diabetes and acute myocardial infarction.
We are a fully integrated company, having developed capabilities in research, development, manufacturing, marketing and distribution of stem cell products. We have developed an extensive intellectual property portfolio to protect our technology in the United States and a number of foreign countries, including 48 U.S. and 284 foreign patents owned or licensed. We believe that our biologic drug candidates have advantages over other stem cell therapeutics in development for at least the following reasons:
- •
- Stem Cell Source. Our stem cells are obtained from adult bone marrow, a readily available source. The cells are drawn from the hips of volunteer donors between the ages of 18 and 30 years, using a simple needle and syringe aspiration. Because the cells are obtained from consenting adult donors, we are able to largely avoid the ethical controversy surrounding embryonic and fetal stem cell research.
- •
- Ability to Mass Produce. Through our proprietary manufacturing methods, we can grow mesenchymal stem cells ("MSC") in a controlled fashion to produce up to 10,000 treatments of our biologic drug candidates from a single bone marrow donation. Our ability to produce a large quantity of treatments from one donation provides us with manufacturing efficiencies and product consistency that are essential to commercialization.
- •
- Universal Compatibility. Many stem cell therapies under development can elicit a rejection response in the recipient and therefore require donor-to-recipient matching or potentially harmful immunosuppression. This greatly reduces manufacturing efficiencies and creates a risk of mismatch which can result in an acute inflammatory response and, potentially, in death.
53
Table of Contents
Based on our clinical experience, we believe that our biologic drug candidates are not rejected by the patient's immune system and so, like type O negative blood, do not require donor-to-recipient matching. This universal compatibility allows us to produce a standardized product available to all patients in almost any medical setting.
- •
- Treatment on Demand. Our biologic drug candidates can be stored frozen at end-user medical facilities until they are needed. We anticipate that medical facilities will be able to prescribe and dispense these products in much the same way as conventional drugs. In contrast, other stem cell technologies under development require weeks to prepare after a patient's need is identified. This is a key feature of our technology, as many patients in the critical care setting require prompt treatment.
On October 31, 2008, we entered into a Collaboration Agreement with Genzyme Corporation for the global development and commercialization of Prochymal and Chondrogen. Under the terms of the Collaboration Agreement, we retain the right to commercialize Prochymal and Chondrogen in the United States and Canada and Genzyme is granted the exclusive right and license to commercialize the products in all other countries (except with respect to GvHD in Japan where Prochymal has previously been licensed to another pharmaceutical company).
In July 2007, we separately partnered with Genzyme for the development of effective countermeasures to nuclear terrorism and other radiological emergencies. In January 2008, we were awarded a contract from the U.S. Department of Defense, pursuant to which we, in partnership with Genzyme, are seeking to develop and stockpile Prochymal for the repair of gastrointestinal injury resulting from acute radiation exposure. Additionally, we have partnered with the JDRF for the development of Prochymal as a treatment for the preservation of insulin production in patients with newly diagnosed type 1 diabetes mellitus.
In April 2008, we committed to a plan to sell our assets related to Osteocel, a product that we have produced and marketed since July 2005, for regenerating bone in orthopedic indications. On May 8, 2008, we entered into an agreement to sell our entire Osteocel and Osteocel XO product line to NuVasive, Inc. The agreement provides for the sale to be effected at two closings—a "technology assets closing," at which technology and certain other business assets are transferred, and a "manufacturing assets closing," at which manufacturing assets and facilities are transferred. The technology assets closing, occurred on July 24, 2008, at which time we received an initial payment of $35.0 million and entered into a Manufacturing Agreement under which we will continue to manufacture Osteocel for up to eighteen months after the technology assets closing, and sell 100% of the product to NuVasive at specified prices. NuVasive has certain minimum purchase order obligations under the Manufacturing Agreement. The Asset Purchase Agreement provides that the manufacturing assets closing is to occur eighteen months after the technology assets closing, or upon earlier termination of the Manufacturing Agreement. As a result of these transactions, all of the activities related to our Osteocel product line have been reported as "discontinued operations" in the accompanying financial statements.
Financial Operations Overview
Revenue
In 2008, we entered into a Collaboration Agreement with Genzyme for the development and commercialization of Prochymal and Chondrogen that provides for non-contingent, non-refundable up front payments of $130.0 million and contingent milestone payments. This Collaboration Agreement has multiple deliverables, and consistent with our accounting policy for such transactions, we are amortizing these amounts into revenue on a straight-line basis over the estimated completion period of the deliverables, which extend through the first quarter of 2012. We recognized $6.7 million of revenue in 2008 related to this agreement. Contingent milestone payments earned and for which we have no
54
Table of Contents
continuing performance obligations, will be recognized as revenue upon achievement of the related milestone, while milestone payments for which we have a continuing performance obligation will be deferred when received and amortized to revenue over the term of the related performance obligations.
In 2007, we partnered with Genzyme to develop Prochymal as a medical countermeasure to nuclear terrorism and other radiological emergencies. In the first quarter of 2008, we were awarded a contract from the United States Department of Defense ("DoD") pursuant to which we, in partnership with Genzyme, are seeking to develop and stockpile Prochymal for the repair of gastrointestinal injury resulting from acute radiation exposure. Under the terms of the contract, the DoD will provide funding to us, and if we are successful in obtaining FDA approval for acute radiation syndrome, the contract provides for the additional purchase of up to 20,000 doses of Prochymal, at $10,000 per dose. We recognized $2.5 million in revenue under the terms of this contract in 2008.
�� In prior years, we have entered into strategic agreements with other companies for the development and commercialization of select stem cell biologic drug candidates for specific indications and geographic markets. In 2007, we entered into a collaborative agreement with the Juvenile Diabetes Research Foundation ("JDRF") to conduct a Phase II clinical trial evaluating Prochymal as a treatment for type 1 diabetes mellitus. This collaborative agreement provides for JDRF to provide up to $4.0 million of contingent milestone funding to support the development of Prochymal for the preservation of insulin production in patients with newly diagnosed type 1 diabetes mellitus. The contingent milestone payments under the agreement are amortized into revenue on a straight line basis over the duration of our obligations under the collaborative agreement as they are earned. We received $2.0 million in milestone payments from JDRF in 2008 and recognized $0.6 million in revenue during 2008 under this agreement.
In 2003, we entered into an agreement with a foreign pharmaceutical company granting it exclusive rights to Prochymal for the treatment of GvHD in Japan. We recognized $0.5 million of revenue during 2007 related to this agreement.
Also in 2003 we entered into an agreement with a major pharmaceutical company relating to the development of our cardiac biologic drug candidate, and we received a $5.0 million fee for licensing the use of our technology. This agreement was terminated in 2007 and as a result we regained the worldwide rights to Prochymal for cardiac indications. We recognized $1.4 million in license fee revenue in 2007 and $1.0 million in 2006 related to this agreement.
Research and Development Costs
Our research and development costs consist of expenses incurred in identifying, developing and testing biologic drug candidates. These expenses consist primarily of salaries and related expenses for personnel, fees paid to professional service providers for independent monitoring and analysis of our clinical trials, costs of contract research and manufacturing, costs of facilities, and the costs of manufacturing clinical batches of biologic drug candidates, quality control supplies and material to expand biologic drug candidates.
Consistent with our focus on the development of biologic drug candidates with potential uses in multiple indications, many of our costs are not attributable to a specifically identified product. We use our employee and infrastructure resources across several projects. Accordingly, we do not account for internal research and development costs on a project-by-project basis. From inception through December 31, 2008, we incurred aggregate research and development costs of approximately $304 million.
We expect our research and development expenses to continue to increase in the future, as we expand our clinical trial activity for our existing biologic drug candidates as they advance through the development cycle and as we invest in additional product opportunities and research programs. Clinical
55
Table of Contents
trials and preclinical studies are time-consuming and expensive. Our expenditures on current and future preclinical and clinical development programs are subject to many uncertainties. We test our products in several preclinical studies, and we then conduct clinical trials for those biologic drug candidates that we determine to be the most promising. As we obtain results from clinical trials, we may elect to discontinue or delay trials for some biologic drug candidates in order to focus our resources on more promising biologic drug candidates. Completion of clinical trials may take several years or more, but the length of time generally varies substantially according to the type, size of trial and intended use of a biologic drug candidate. The cost of clinical trials may vary significantly over the life of a project as a result of a variety of factors, including:
- •
- the number of patients who participate in the trials;
- •
- the number of sites included in the trials;
- •
- the length of time required to enroll trial participants;
- •
- the duration of patient treatment and follow-up;
- •
- the costs of producing supplies of the biologic drug candidates needed for clinical trials and regulatory submissions;
- •
- the efficacy and safety profile of the biologic drug candidate; and
- •
- the costs and timing of, and the ability to secure, regulatory approvals.
As a result of these uncertainties, we are unable to determine with any significant degree of certainty the duration and completion costs of our research and development projects or when and to what extent we will generate revenues from the commercialization and sale of any of our biologic drug candidates.
General and Administrative Expenses
General and administrative expenses consist primarily of the costs associated with our general management, including salaries, allocations of facilities and related costs, and professional fees such as legal and accounting expenses. We have increased our general and administrative expense for legal and accounting compliance costs, investor relations and other activities associated with operating as a publicly traded company and strengthened our administrative capabilities as we approach the commercial launch of Prochymal. Continued increases will also likely result from the hiring of additional operational, financial, accounting, facilities engineering and information systems personnel.
Interest Expense, Net
Interest income consists of interest earned on our cash and investments available for sale. Interest expense consists of interest incurred on capital leases and other debt financings. We pay interest on our promissory notes, capital leases and convertible long-term debt. We do not expect to incur material interest expense in the future as we have extinguished all of our outstanding debt as of December 31, 2008, and had invested the excess in investments available for sale.
Income Taxes
We have not recognized any deferred tax assets or liabilities in our financial statements since we cannot assure their future realization. Because realization of deferred tax assets is dependent upon future earnings, a full valuation allowance has been recorded on the net deferred tax assets, which relate primarily to net operating loss and research and development carry-forwards. In the event that we become profitable within the next several years, we have net deferred tax assets (before a 100% valuation allowance) of approximately $105.3 million that may be utilized prior to us having to
56
Table of Contents
recognize any income tax expense or make payments to the taxing authorities. Utilization of our net operating loss carry-forwards in any one year may be limited under IRC Section 382, and we could be subject to the alternative minimum tax, thereby potentially diminishing the value to us of this tax asset.
Critical Accounting Policies
General
Our discussion and analysis of our financial condition and results of operations are based on our financial statements, which we have prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities, revenues and expenses, and related disclosure of contingent assets and liabilities. On an ongoing basis, we evaluate our estimates, including those related to revenue recognition, inventory valuation, deferred tax assets, share-based compensation, and contingencies. We base our estimates on historical experience and on various other assumptions that we believe are reasonable. These results form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
We believe that the following critical accounting policies reflect our more significant judgments and estimates used in the preparation of our financial statements.
Revenue Recognition
We generate revenues from collaborative agreements, research licenses, and a government contract. Our revenue recognition policies are in accordance with the Securities and Exchange Commission's ("SEC") Staff Accounting Bulletin No. 101,Revenue Recognition in Financial Statements, as amended by SEC Staff Accounting Bulletin No. 104,Revenue Recognition.
Revenues from collaboration agreements are evaluated under Emerging Issues Task Force ("EITF") Issue No. 07-01, Draft Abstract,Accounting for Collaboration Agreements. Management evaluates revenues from agreements that have multiple elements to determine whether the components of the arrangement represent separate units of accounting as defined in EITF Issue No. 00-21,Revenue Arrangements with Multiple Deliverables. To recognize a delivered item in a multiple element arrangement, EITF Issue No. 00-21 requires that the delivered items have value to the customer on a stand alone basis, that there is objective and reliable evidence of fair value of the undelivered items and that delivery or performance is probable and within our control for any delivered items that have a right of return. The determination whether multiple elements of a collaboration agreement meet the criteria for separate units of accounting requires us to exercise judgments.
Revenues from research licenses and government contracts are recognized as earned upon either the incurring of reimbursable expenses directly related to the particular research plan or the completion of certain development milestones as defined within the terms of the agreement. Payments received in advance of research performed are designated as deferred revenue. Non-refundable upfront license fees and certain other related fees are recognized on a straight-line basis over the development periods of the contract deliverables. Fees associated with substantive at risk, performance based milestones are recognized as revenue upon their completion, as defined in the respective agreements. Incidental assignment of technology rights is recognized as revenue as it is earned and received.
In October 2008, we entered into a Collaboration Agreement with Genzyme Corporation for the development and commercialization of Prochymal and Chondrogen. Under the Agreement, Genzyme is obligated to pay to us non-contingent, non-refundable cash payments totaling $130.0 million, with $75.0 million paid during November 2008 and $55.0 million scheduled to be paid on July 1, 2009. The Agreement provides Genzyme with certain rights to intellectual property developed by us, and requires
57
Table of Contents
that we continue to perform certain development work related to the subject products. Management has evaluated the deliverables related to these payments under EITF 00-21, and concluded that the various deliverables represent a single unit of accounting. Accordingly, we have deferred the recognition of revenue related to the upfront payments, and are amortizing these amounts to revenue on a straight-line basis over the estimated delivery period of the required development services, which extend through the first quarter of 2012. The Agreement also provides for contingent milestone payments of up to $1.25 billion in the aggregate, as well as royalties to be paid to us on any sales by Genzyme. Consistent with our revenue recognition policies, we will recognize revenue from these contingent milestone payments for which we have no continuing performance obligations upon achievement of the related milestone. For any milestone payments for which we have a continuing performance obligation, the milestone payments will be deferred and recognized as revenue over the term of the related performance obligations.
In 2007, we partnered with Genzyme to develop Prochymal as a medical countermeasure to nuclear terrorism and other radiological emergencies. In January 2008, we were awarded a contract from the United States Department of Defense pursuant to which we, in partnership with Genzyme, are seeking to develop and stockpile Prochymal for the repair of gastrointestinal injury resulting from acute radiation exposure. We began recognizing revenue under this contract during the first quarter of 2008. Contract revenue is recognized as the related costs are incurred, in accordance with the terms of the contract.
In October 2007, we entered into a collaborative agreement with the Juvenile Diabetes Research Foundation ("JDRF") to conduct a Phase II clinical trial evaluating Prochymal as a treatment for type 1 diabetes mellitus. This collaborative agreement provides for JDRF to provide up to $4.0 million of contingent milestone funding to support the development of Prochymal for the preservation of insulin production in patients with newly diagnosed type 1 diabetes mellitus. The contingent milestone payments under the agreement are amortized to revenue on a straight line basis over the duration of our obligations under the collaborative agreement as they are earned.
In July 2005, we launched our first commercial product, Osteocel. Revenues on Osteocel sales are recognized when legal title to the product has passed to the customer. In April 2008, we committed to a plan to sell the Osteocel product line. Concurrent with the technology assets closing on July 24, 2008, we entered into a Manufacturing Agreement under which we will continue to manufacture Osteocel for up to 18-months and thereafter sell 100% of the product to NuVasive at specified prices. Osteocel operations are accounted for as discontinued operations in the accompanying financial statements, and the prior period financial statements have been restated for comparative purposes.
�� We have entered into several strategic agreements with other pharmaceutical companies focusing on the development and commercialization of its stem cell drug products. In 2003, we entered into such an agreement with Boston Scientific Corporation pertaining to our cardiac drug development and received a $5 million fee for licensing the use of our technology. We terminated the agreement with Boston Scientific Corporation in 2007 and recognized the remaining unamortized license fee. Also in 2003, we entered into a similar agreement with JCR Pharmaceuticals Co., Ltd. pertaining to our hematologic malignancies drugs for distribution in Japan, and recognize revenue upon the achievement of milestone events specified in the agreement.
We also earn royalties on the sale of human MSCs sold for research purposes and recognize the revenue on these sales as the sales are made.
Accounts Receivable
Accounts receivable are reported at their net realizable value. As of December 31, 2008 and 2007, there was no allowance for doubtful accounts related to accounts receivable from continuing operations, as we believe the reported amounts are fully collectible. Accounts receivable balances are not collateralized.
58
Table of Contents
Share-Based Compensation
In December 2004, the Financial Accounting Standards Board, ("FASB") issued Statement No. 123(R), "Share-Based Payment," which is a revision of Statement No. 123, "Accountingfor Stock Issued to Employees." Effective January 1, 2006, we adopted Statement No. 123(R) using the modified prospective method under which prior period amounts are not restated for comparative purposes. Under the modified prospective method, we recognized compensation cost for:
- •
- all share-based payments granted after January 1, 2006 based upon the requirements of Statement No. 123(R); and
- •
- all unvested awards granted prior to January 1, 2006 using the compensation cost calculated for pro forma disclosure purposes under Statement No. 123.
Under Statement No. 123(R), we have recognized all share-based payments to employees and non-employee directors in our financial statements based on their grant date fair values, calculated using the Black-Scholes option pricing model. Compensation expense related to share-based awards is recognized on a straight-line basis based on the value of share awards that are scheduled to vest during the requisite service period. Under Statement No. 123(R), share-based compensation expense is based on awards ultimately expected to vest and must be reduced for estimated forfeitures.
Significant New Accounting Pronouncements
Recent Accounting Pronouncements that may have a material impact on future consolidated financial statements
In June 2008, the FASB ratified Emerging Issue Task Force ("EITF") Issue No. 07-5, "Determining Whether an Instrument (or an Embedded Feature) is Indexed to an Entity's Own Stock" (EITF 07-5). This issue provides guidance for determining whether an equity-linked financial instrument (or embedded feature) is indexed to an entity's own stock. EITF 07-5 applies to any freestanding financial instrument or embedded feature that has all the characteristics of a derivative under paragraphs 6-9 of Statement of Financial Accounting Standards No. 133, "Accounting for Derivative Instruments and Hedging Activities," (SFAS 133) for purposes of determining whether that instrument or embedded feature qualifies for the first part of the scope exception under paragraph 11(a) of SFAS 133. EITF 07-5 also applies to any freestanding financial instrument that is potentially settled in an entity's own stock, regardless of whether the instrument has all the characteristics of a derivative under paragraphs 6-9 of SFAS 133, for purposes of determining whether the instrument is within the scope of EITF Issue 00-19, "Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company's Own Stock," (Issue 00-19) which provides accounting guidance for instruments that are indexed to, and potentially settled in, the issuer's own stock. EITF 07-5 is effective for fiscal years beginning after December 15, 2008. Early application is not permitted by entities that have previously adopted an alternative accounting policy. We are currently evaluating the requirements of EITF 07-5, but do not expect our adoption of this issue to have a material impact on our consolidated financial statements.
In May 2008, the FASB issued FASB Staff Position No. APB 14-1,Accounting for Convertible Debt Instruments That May Be Settled in Cash Upon Conversion (Including Partial Cash Settlement) ("FSP APB 14-1"). Under the new rules for convertible debt instruments that may be settled entirely or partially in cash upon conversion, an entity should separately account for the liability and equity components of the instrument in a manner that reflects the issuer's economic interest cost. Previous guidance provided for accounting of this type of convertible debt instruments entirely as debt. For instruments subject to the scope of FSP APB 14-1, higher interest expense may result through the accretion of the discounted carrying value of the convertible debt instruments to their face amount over their term. FSP APB 14-1 will be effective for fiscal years beginning after December 15, 2008, and for interim periods within those fiscal years, with retrospective application required. Early adoption is not
59
Table of Contents
permitted. As of December 31, 2008, we do not have any instruments outstanding that would be subject to FSP APB 14-1, but any instruments that we may issue in the future will be subject to this pronouncement.
In December 2007, the FASB issued FASB Statement No. 141 (Revised 2007) ("SFAS 141R"),Business Combinations. SFAS 141R will significantly change the accounting for business combinations. Under SFAS 141R, an acquiring entity will be required to recognize all the assets acquired and liabilities assumed in a transaction at the acquisition date at fair value with limited exceptions. SFAS 141R will change the accounting treatment for certain specific items, including: acquisition costs will be generally expensed as incurred, minority interests will be valued at fair value at the acquisition date, acquired contingent liabilities will be recorded at fair value at the acquisition date and subsequently measured at either the higher of such amount or the amount determined under existing guidance for non-acquired contingencies, in-process research and development will be recorded at fair value as an indefinite-lived intangible asset at the acquisition date, restructuring costs associated with a business combination will be generally expensed subsequent to the acquisition date, and changes in deferred tax asset valuation allowances and income tax uncertainties after the acquisition date generally will affect income tax expense. SFAS 141R also includes a substantial number of new disclosure requirements. SFAS 141R applies prospectively to business combinations for which the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2008. Earlier adoption is prohibited. We are required to record and disclose business combinations following existing GAAP until January 1, 2009.
In September 2006, the FASB issued SFAS No. 157,Fair Value Measurements ("SFAS 157"). SFAS 157 establishes a common definition for fair value to be applied to GAAP guidance requiring use of fair value, establishes a framework for measuring fair value, and expands disclosure about such fair value measurements. SFAS 157 applies to fair value measurements that are already required or permitted by other accounting standards, except for measurements of share-based payments and measurements that are similar to, but not intended to be, fair value. The FASB has previously concluded in those accounting pronouncements that fair value is the relevant measurement attribute. Accordingly, this Statement does not require any new fair value measurements. SFAS 157 was effective for fiscal years beginning after November 15, 2007. The effective date of SFAS 157 with regard to non-financial assets and liabilities is January 1, 2009. Our adoption of SFAS 157 with respect to financial assets and liabilities as of January 1, 2008 did not have a material impact on our consolidated financial statements.
Recent Accounting Pronouncements that did not have a material impact on our consolidated financial statements
In December 2007, the Financial Accounting Standards Board ratified Emerging Issue Task Force Issue No. 07-1 ("EITF 07-1"),Accounting for Collaborative Arrangements. The key elements of EITF 07-1 relate to: (a) the scope of the issue; (b) the income statement presentation of transactions with third parties; (c) the income statement presentation of payments between parties to the collaborative arrangement; (d) the disclosures about collaborative arrangements that should be required in the financial statements of the parties to the collaborative arrangements; and (e) the transition method. A contractual arrangement falls within the scope of EITF 07-1 if the arrangement requires the parties to be active participants and the arrangement exposes the parties to significant risks and rewards that are tied to the commercial success of the endeavor. Costs incurred and revenue generated on sales to third parties should be reported in the statement of operations based on the guidance in EITF Issue No. 99-19,Reporting Revenue Gross as a Principal versus Net as an Agent. The equity method of accounting should not be applied to a collaborative arrangement within the scope of this issue without the creation of a separate legal entity for the arrangement. Payments between parties to the collaborative arrangement should be presented in the statement of operations based on the nature
60
Table of Contents
of the arrangement and each entity's business operations, the contractual terms of the arrangement as well as if existing GAAP is applicable. EITF 07-1 requires companies to disclose the nature and purpose of the arrangement, its rights and obligations under the arrangement, the accounting policy applied to the arrangement, and the amounts attributable to transactions between other participants to the collaborative arrangement and where in the statement of operations these amounts have been classified. EITF 07-1 requires that companies comply in its first fiscal year beginning after December 15, 2008 and transition to the guidance in this issue by retrospectively applying the guidance to all periods presented for all arrangements existing at the effective date, unless it is impracticable to do so. The impracticability assessment should be made on an arrangement-by-arrangement basis and certain disclosures would be required if a company utilized the impracticability exception. Our adoption of the provisions of EITF 07-1, did not have a material impact on our consolidated financial statements.
In June 2007, the FASB ratified Emerging Issue Task Force Issue No. 07-3 ("EITF 07-3"),Accounting for Non-Refundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities, which requires nonrefundable advance payments for goods and services that will be used or rendered for future research and development activities to be deferred and capitalized. These amounts will be recognized as an expense in the period that the related goods are delivered or the related services are performed or when an entity does not expect the goods to be delivered or services to be rendered. EITF 07-3 is effective for the fiscal years beginning after December 31, 2007, including interim periods within those fiscal years. Our adoption of the provisions of EITF 07-3, beginning January 1, 2008 did not have a material impact on our consolidated financial statements.
In February 2007, the FASB issued SFAS No. 159The Fair Value Option for Financial Assets and Financial Liabilities—Including an Amendment of FASB Statement No. 115 ("SFAS 159"), which became effective for fiscal periods beginning after November 15, 2007. Under SFAS 159, companies may elect to measure specified financial assets and liabilities at fair value that are not otherwise measured at fair value, with changes in fair value recognized in earnings each subsequent reporting period. This election, called the "fair value option," will enable some companies to reduce volatility in reported earnings caused by measuring related assets and liabilities differently. SFAS 159 also establishes presentation and disclosure requirements designed to draw a comparison between the different measurement attributes a company elects for similar types of assets and liabilities. We did not elect the "fair value option" for any financial assets or liabilities and, therefore, the adoption of SFAS 159 did not have an impact on our financial statements.
Results of Operations
Year ended December 31, 2008 compared to December 31, 2007
Revenue
Total revenues increased to $10.0 million for the twelve months ended December 31, 2008 from $2.0 million in the corresponding period in 2007. The increase is the result of the Genzyme Collaboration Agreement and the DoD contract, each of which was entered during 2008, and the JDRF agreement entered into in 2007. Revenues in 2007 included $1.3 million of unamortized license fees that was recognized in 2007 upon the termination of the agreement with Boston Scientific Corporation in December 2007.
Research and Development Expenses
Research and development expenses were $69.9 million for the twelve months ended December 31, 2008 compared to $47.1 million in the prior year. The increase in research and development expenses in 2008 reflects the increased number of clinical trials in process versus the prior year and the resulting increases in clinical doses of our drug candidates. We also incurred additional research and
61
Table of Contents
development costs in connection with the development of our Biologics License Application ("BLA") as we approach the commercialization of Prochymal.
General and Administrative Expenses
General and administrative expenses were $8.3 million for the twelve months ended December 31, 2008 compared to $6.0 million in the prior year. The increase was attributable to additional personnel and related costs to support our expanded operations as we approach the anticipated commercialization of Prochymal.
Share Based Payments to Related Parties
We pay fees to members of our Board of Directors through grants of our common stock and/or a combination of stock and cash. In 2008, we issued 21,500 shares of common stock for services on the Board and valued these shares at the then current closing price of our stock on the NASDAQ Global Market, resulting in the $0.3 million charge. In 2007, we also issued 21,5000 shares of common stock for services on the Board and recorded a $0.1 million charge.
Interest Expense, Net
Interest expense, net was $1.0 million for the twelve months ended December 31, 2008 compared to $6.7 million in the prior year. Interest expense decreased significantly in 2008 due to a reduced amount of debt outstanding throughout the year. We did not have any debt outstanding as of December 31, 2008. Interest expense in 2008 included $0.5 million paid to related parties, as discussed more fully in Note 9 to the accompanying financial statements included in Item 8 to this Annual Report on Form 10-K. Interest expense in 2007 also includes a non-cash charge of $4.8 million resulting from the induced conversion of $18.8 million of our convertible promissory notes into common stock and $0.3 million of previously deferred debt financing fees.
Year ended December 31, 2007 compared to December 31, 2006
Revenue
Total revenues increased to $2.0 million for the twelve months ended December 31, 2007 from $1.2 million in the corresponding period in 2006. Our revenues in both years resulted primarily from licensing fees and royalties, and the increase in 2007 is due to the additional revenue recognized for the balance of the unamortized license fees under our collaborative agreement with Boston Scientific Corporation, which was terminated in December 2007.
Research and Development Expenses
Research and development expenses were $47.1 million for the twelve months ended December 31, 2007 compared to $37.6 million in the prior year. The increase in research and development expenses in 2007 reflects the increased number of clinical trials in process versus the prior year as well as higher costs associated with the Phase III clinical trials for Prochymal.
General and Administrative Expenses
General and administrative expenses were $6.0 million for the twelve months ended December 31, 2007 compared to $4.3 million in the prior year. The increase in 2007 was attributable to our increased operations and costs related to our status as a publicly traded company following our IPO during the third quarter of 2006.
62
Table of Contents
Related Party Expenses
Fees paid to related parties were $0.5 million for the twelve months ended December 31, 2006 and were made in connection with pre-IPO financings.
Share Based Payments to Related Parties
Share based payments to related parties were $0.1 million in 2007 for stock awards for services on our board of directors. In 2006, we recorded $3.5 million in non-cash charges related to warrants issued to Mr. Peter Friedli, the Chairman of our Board of Directors that were priced and vested upon completion of our initial public offering in addition to $0.2 million in share based compensation to Mr. Friedli for service on our board of directors.
Interest Expense, Net
Interest expense, net was $6.7 million for the twelve months ended December 31, 2007 compared to $4.7 million in the prior year. The increase in 2007 resulted from the non cash charge of $4.8 million resulting from the induced conversion of our convertible promissory notes and $0.3 million of previously deferred debt financing fees compared to 2006 charges of $2.7 million of previously deferred debt financing costs associated with debt that was converted in conjunction with our initial public offering.
Liquidity and Capital Resources
Liquidity
At December 31, 2008, we had $0.9 million in cash and $61.3 million in investments available for sale and $61.3 million of short-term receivables. In addition, we had extinguished all of our outstanding debt as of that date.
Cash Flow
| | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2008 | | 2007 | | 2006 | |
|---|
| | (amounts in thousands)
| |
|---|
Net cash provided by (used in) operating activities | | $ | 10,222 | | $ | (46,518 | ) | $ | (35,310 | ) |
Net cash (used in) provided by investing activities | | | (13,068 | ) | | 16,305 | | | 2,611 | |
Net cash provided by financing activities | | | 3,082 | | | 30,203 | | | 32,816 | |
We generated cash from operating activities for the first time since our inception during the year ended December 31, 2008. The cash provided by operating activities was primarily the result of the $75.0 million of up front fees received under the Collaboration Agreement with Genzyme. We used a portion of these proceeds to completely extinguish our outstanding debt, and made net purchases of investments available for sale of $43.6 million during the year with our excess cash. We have progressively increased our clinical trial activities since inception as we strive to bring our biologic drug candidates to market, and we expect this trend will continue in the foreseeable future as our existing biologic drug candidates continue in their development cycles and we develop additional product opportunities. Although there can be no assurances, based on current expectations, we believe that we have sufficient liquidity on hand as of December 31, 2008 to fund our operations through the commercialization of Prochymal for an initial indication. Prior to this year, we had historically financed our research and development activities through cash flows provided by financing activities.
Net cash provided by operating activities was $10.2 million for the twelve months ended December 31, 2008. During that period, our continuing operations used $2.7 million of cash, primarily reflecting our loss from continuing operations and cash used to fund operating assets, which was
63
Table of Contents
partially offset by the $75.0 million installment of up front payments received from Genzyme. The $55.0 million installment of the up front payments that we will receive on July 1, 2009 was a non cash increase in both accounts receivable and deferred revenue. Net cash provided by discontinued operations was $13.0 million for the year ended December 31, 2008, which was the result of the cash portion of the income from discontinued operations and the net change in the operating assets and liabilities of the Osteocel disposal group. Net cash used in operating activities was $46.5 million for the twelve months ended December 31, 2007, of which continuing operations used cash of $47.4 million. The cash used by continuing operations in 2007 primarily reflects our loss from continuing operations. The operating activities of our discontinued operations provided $0.8 million in cash during 2007.
Net cash used in investing activities was $13.1 million for the twelve months ended December 31, 2008. During that period, the investing activities of our continuing operations used $10.3 million of cash, utilizing our excess cash resources to make net purchases of investments available for sale, which was partially offset by the net proceeds from the sale of the Osteocel disposal group. Investing activities of discontinued operations used $2.8 million of cash for the purchases property and equipment during the period. Net cash provided by investing activities was $16.3 million for the twelve months ended December 31, 2007. Our continuing operations provided $20.2 million of cash during that period, primarily the result of the net sale of investments available for sale to fund our growth and operations. Discontinued operations used $3.9 million in purchase property and equipment.
Net cash provided by financing activities was $3.1 million for the twelve months ended December 31, 2008, all of which was attributable to our continuing operations. This cash represents net proceeds realized on the issuance and subsequent redemption of convertible and short-term notes during the period. As of December 31, 2008, we had no debt outstanding. Our financing activities provided $30.2 million of cash during the twelve months ended December 31, 2007, principally from the two private placements of our common stock that raised $31.7 million.
Capital Resources
Our future capital requirements will depend on many factors, including:
- •
- the scope and results of our research and preclinical development programs;
- •
- the scope and results of our clinical trials, particularly regarding the number of patients required for our Phase III trials;
- •
- the timing of and the costs involved in obtaining regulatory approvals for our biologic drug candidates, which could be more lengthy or complex than obtaining approval for a new conventional drug, given the FDA's limited experience with late-stage clinical trials and marketing approval for stem cell therapeutics;
- •
- the timing and achievement of contingent milestone payments under the NuVasive agreement and the Genzyme collaboration agreement.
- •
- the costs of maintaining, expanding and protecting our intellectual property portfolio, including possible litigation costs and liabilities; and
- •
- the costs of enlarging our work force consistent with expanding our business and operations and status as a public company.
As of December 31, 2008, we had completely extinguished our outstanding debt. As a result of our financial position and forecasts as of that date, we believe that we have sufficient liquidity on hand as of that date to fund our operations through the initial commercialization of our biological drug candidate.
64
Table of Contents
Off-Balance Sheet Arrangements
We have no off-balance sheet financing arrangements and we have not entered into any transactions involving unconsolidated subsidiaries or special purpose entities.
Future Contractual Obligations
The following table sets forth our estimates as to the amounts and timing of contractual payments for our most significant contractual obligations at December 31, 2008. The information in the table reflects future unconditional payments and is based on the terms of the relevant agreements, appropriate classification of item under accounting principles generally accepted in the United States and certain assumptions. Future events could cause actual payments to differ from these amounts.
| | | | | | | | | | | | | | | | |
| | Payment Due by Fiscal Year | |
|---|
Contractual Obligations | | Total | | Less Than
1-Year | | Years 1-3 | | Years 4-5 | | More Than
5-Years | |
|---|
| | (amounts in thousands)
| |
|---|
Operating lease—facilities | | $ | 8,224 | | $ | 978 | | $ | 3,247 | | $ | 3,123 | | $ | 876 | |
Contract Research Organizations | | | 44,734 | | | 19,172 | | | 25,562 | | | — | | | — | |
Clinical Manufacturing Services Agreement | | | 14,500 | | | 14,500 | | | — | | | — | | | — | |
Capital leases—equipment | | | 11 | | | 8 | | | 3 | | | — | | | — | |
Interest payments | | | 2 | | | 2 | | | — | | | — | | | — | |
| | | | | | | | | | | | |
Total contractual cash obligations | | $ | 67,471 | | $ | 34,660 | | $ | 28,812 | | $ | 3,123 | | $ | 876 | |
| | | | | | | | | | | | |
Contract Research Organizations. We utilize independent contract research organizations ("CROs") to perform the clinical trials of our biological drug candidates. Under the terms of these agreements, we design the protocol regarding the testing to be performed, and the CRO enrolls the patients and testing sites, administers the trial, performs statistical analysis of the results, and compiles the final report.
We pay fees directly to the CROs for their professional services, which may be payable upon specified trial milestones or as they provide services, depending on the structure of the contract. We are also responsible for reimbursing the CROs for certain pass through expenses they incur in administering the trial. Our agreements with CROs provide for cancellation for various reasons. In the event any of our agreements with CROs are cancelled our liablities will be reduced. The timing of our payments to the CROs is dependent upon the progress of the various trials, which is highly variable dependent upon the speed with which the CROs are able to enroll patients and testing sites. As such, we are unable to specifically predict the timing of future payments to CROs.
As of December 31, 2008, we had contracted with CROs to perform six clinical trials which were in varying stages of completion. The total contracted payments to CROs under these agreements were $85.7 million, of which we have incurred $40.1 million. Although we cannot specifically predict the timing of the remaining payments, based on our current estimates and assumptions, we currently expect to make payments to the CROs through 2011 as detailed above.
Clinical Manufacturing Services Agreement. We have contracted for lot screening services for the isolation, growth, and differentiation of the MSCs to be used in our biological drug candidates. Due to the long production cycle for MSCs, we are obligated to provide a twelve month future forecast production plan, under which the counterparty will produce our required MSC doses for a fixed price per dose. The term of the agreement is five years, provided that we continue to provide a forecast production plan. We have the right to terminate the contract with thirty days written notice. In the event we terminate the contract, we are obligated for any in-process production as of the termination date that has been initiated according to our production forecast. Accordingly, our minimum
65
Table of Contents
contractual commitment under this contract is limited to in-process production at any given time. Our obligation would not extend for a period longer than twelve months, the maximum outlook of the production forecast. Based on our current forecast production plan, we estimate that our future minimum contractual commitments under this contract were $14.5 million as of December 31, 2008.
Leases. As previously discussed, we have executed an Asset Purchase Agreement for the sale of our Osteocel asset disposal group. NuVasive has agreed to assume the lease to our Columbia facility concurrent with the manufacturing asset closing under that agreement, which we expect to occur during 2009. The amount of our future lease payments for the Columbia facility is dependent upon the occurrence and exact timing of the manufacturing assets closing. Accordingly, the table above includes all payments specified by the lease agreement.
Effect of Inflation.
Inflation and changing prices are not generally a material factor affecting our business. General operating expenses such as salaries, employee benefits and lease costs are, however, subject to normal inflationary pressures
ITEM 7A. QUANTITATIVE AND QUALITATIVE DISCLOSURES ABOUT MARKET RISK
Due to the short duration of our investment portfolio and the high quality of our investments, an immediate 10% change in interest rates would not have a material effect on the value of our portfolio. Therefore, we would not expect our operating results or cash flows to be affected to any material degree by the effect of a sudden change in market interest rates on our securities portfolio.
We believe that the interest rate risk related to our accounts receivable is not significant. We manage the risk associated with these accounts through periodic reviews of the carrying value for non-collectability and establishment of appropriate allowances in connection with our internal controls and policies.
We conduct clinical trial activities in areas that operate in a functional currency other than the United States dollar (USD). As a result, when the USD rises and falls against the functional currencies of these other nations, our costs will either increase or decrease by the relative change in the exchange rate. Foreign currency gains and losses were not significant during the three years ended December 31, 2008, and at the present time, we have elected not to hedge our exposure to foreign currency fluctuations.
We do not enter into hedging or derivative instrument arrangements.
66
Table of Contents
ITEM 8. FINANCIAL STATEMENTS AND SUPPLEMENTARY DATA
OSIRIS THERAPEUTICS, INC.
FINANCIAL STATEMENTS
INDEX
| | |
| | Page |
|---|
Management's Report on Internal Control Over Financial Reporting | |
68 |
Report of Independent Registered Public Accounting Firm | |
69 |
Balance Sheets at December 31, 2008 and 2007 | |
70 |
Statements of Operations for the years ended December 31, 2008, 2007 and 2006 | |
71 |
Statements of Stockholders' Equity (Deficit) for the years ended December 31, 2008, 2007 and 2006 | |
72 |
Statements of Cash Flows for the years ended December 31, 2008, 2007 and 2006 | |
73 |
Notes to Financial Statements | |
74 |
67
Table of Contents
MANAGEMENT'S REPORT ON INTERNAL CONTROL OVER FINANCIAL REPORTING
Our management is responsible for establishing and maintaining adequate internal control over financial reporting and for performing an assessment of the effectiveness of internal control over financial reporting as of December 31, 2008. Internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. Our system of internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of our assets; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with the authorization of our management and directors; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of our assets that could have a material effect on the financial statements. Under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer, we conducted an evaluation of the effectiveness of our internal control over financial reporting as of December 31, 2008 based on the framework inInternal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission ("COSO"). Based on that evaluation, our management concluded that our internal control over financial reporting was effective as of December 31, 2008.
Stegman & Company, the independent registered public accounting firm that audited our financial statements, has issued an attestation report on their assessment of internal controls over financial reporting, which is included elsewhere in this Annual Report.
Date: March 14, 2009
| |
| | /s/ C. RANDAL MILLS
C. Randal Mills, Ph.D.
President and Chief Executive Officer
(principal executive officer) |
|
/s/ RICHARD W. HUNT
Richard W. Hunt
Chief Financial Officer
(principal financial officer) |
68
Table of Contents

REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of Osiris Therapeutics, Inc.
We have audited the accompanying balance sheets of Osiris Therapeutics, Inc. (the "Company") as of December 31, 2008 and 2007, and the related statements of operations, stockholders' equity (deficit) and cash flows for each of the years in the three-year period ended December 31, 2008. We also have audited the Company's internal control over financial reporting as of December 31, 2008, based on criteria established inInternal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). The Company's management is responsible for these financial statements, for maintaining effective internal control over financial reporting, and for its assessment of the effectiveness of internal control over financial reporting, included in the accompanying Management's Report on Internal Control over Financial Reporting. Our responsibility is to express an opinion on these financial statements and an opinion on the company's internal control over financial reporting based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audits to obtain reasonable assurance about whether the financial statements are free of material misstatement and whether effective internal control over financial reporting was maintained in all material respects. Our audits of the financial statements included examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. Our audit of internal control over financial reporting included obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, and testing and evaluating the design and operating effectiveness of internal control based on the assessed risk. Our audits also included performing such other procedures as we considered necessary in the circumstances. We believe that our audits provide a reasonable basis for our opinions.
A company's internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company's internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company's assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Osiris Therapeutics, Inc. as of December 31, 2008 and 2007, and the results of its operations and its cash flows for each of the years in the three-year period ended December 31, 2008 in conformity with accounting principles generally accepted in the United States of America. Also in our opinion, Osiris Therapeutics, Inc. maintained, in all material respects, effective internal control over financial reporting as of December 31, 2008, based on criteria established inInternal Control—Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
Baltimore, Maryland
March 14, 2009

69
Table of Contents
OSIRIS THERAPEUTICS, INC.
BALANCE SHEETS
(amounts in thousands)
| | | | | | | | | |
| | December 31,
2008 | | December 31,
2007 | |
|---|
ASSETS | | | | | | | |
Current assets: | | | | | | | |
| | Cash | | $ | 940 | | $ | 704 | |
| | Investments available for sale | | | 61,298 | | | 17,460 | |
| | Accounts receivable | | | 61,287 | | | 549 | |
| | Prepaid expenses and other current assets | | | 2,060 | | | 1,583 | |
| | Current assets of discontinued operations | | | 3,223 | | | 8,445 | |
| | | | | | |
| | | Total current assets | | | 128,808 | | | 28,741 | |
Property and equipment, net | | |
394 | | |
2,020 | |
Restricted cash | | | 130 | | | 280 | |
Other assets | | | 615 | | | 1,404 | |
Long-term assets of discontinued operations | | | 7,520 | | | 4,596 | |
| | | | | | |
| | | Total assets | | $ | 137,467 | | $ | 37,041 | |
| | | | | | |
LIABILITIES AND STOCKHOLDERS' (DEFICIT) EQUITY | | | | | | | |
Current liabilities: | | | | | | | |
| | Accounts payable and accrued expenses | | $ | 10,513 | | $ | 11,535 | |
| | Deferred revenue, current portion | | | 40,471 | | | — | |
| | Notes payable, current portion | | | — | | | 6,521 | |
| | Capital lease obligations, current portion | | | 6 | | | 886 | |
| | Current liabilities of discontinued operations | | | 7,219 | | | 2,552 | |
| | | | | | |
| | | Total current liabilities | | | 58,209 | | | 21,494 | |
Deferred revenue, net of current portion | | |
84,275 | | |
— | |
Notes payable, net of current portion | | | — | | | 1,200 | |
Other long-term liabilities | | | 3 | | | 11 | |
Long-term liabilities of discontinued operations | | | — | | | — | |
| | | | | | |
| | | Total liabilities | | | 142,487 | | | 22,705 | |
| | | | | | |
Stockholders' (deficit) equity: | | | | | | | |
| | Common stock, $.001 par value, 90,000 shares authorized 32,676 and 31,648 shares outstanding in 2008 and 2007 | | | 33 | | | 32 | |
| | Additional paid-in-capital | | | 269,830 | | | 255,728 | |
| | Accumulated other comprehensive income | | | 33 | | | — | |
| | Accumulated deficit | | | (274,916 | ) | | (241,424 | ) |
| | | | | | |
| | | Total stockholders' (deficit) equity | | | (5,020 | ) | | 14,336 | |
| | | | | | |
| | | Total liabilities and stockholders' (deficit) equity | | $ | 137,467 | | $ | 37,041 | |
| | | | | | |
The accompanying notes are an integral part of these financial statements.
70
Table of Contents
OSIRIS THERAPEUTICS, INC.
STATEMENTS OF OPERATIONS
(amounts in thousands, except per share data)
| | | | | | | | | | | | | |
| | Years ended December 31, | |
|---|
| | 2008 | | 2007 | | 2006 | |
|---|
Revenue from collaborative research agreements, government contract, and royalties | | $ | 10,044 | | $ | 2,048 | | $ | 1,181 | |
| | Operating expenses: | | | | | | | | | | |
| | | Research and development | | | 69,897 | | | 47,140 | | | 37,590 | |
| | | General and administrative | | | 8,328 | | | 5,953 | | | 4,340 | |
| | | Fees paid to related parties | | | — | | | — | | | 451 | |
| | | Share based payments to related parties | | | 258 | | | 118 | | | 3,668 | |
| | | | | | | | |
| | | | Total operating expenses | | | 78,483 | | | 53,211 | | | 46,049 | |
| | | | | | | | |
| | | | Loss from operations | | | (68,439 | ) | | (51,163 | ) | | (44,868 | ) |
| | Interest income (expense) | | | | | | | | | | |
| | | Interest income | | | 416 | | | 1,321 | | | 2,069 | |
| | | Interest paid to related parties | | | (508 | ) | | — | | | — | |
| | | Interest expense | | | (886 | ) | | (8,016 | ) | | (6,754 | ) |
| | | | | | | | |
| | Total interest expense, net | | | (978 | ) | | (6,695 | ) | | (4,685 | ) |
| | | | Loss from continuing operations | | |
(69,417 |
) | |
(57,858 |
) | |
(49,553 |
) |
| | | | | | | | |
| | Discontinued operations: | | | | | | | | | | |
| | | Income from operations of discontinued operations | | | 5,525 | | | 3,937 | | | 4,594 | |
| | | Gain from sale of discontinued operations | | | 30,400 | | | — | | | — | |
| | | | | | | | |
| | | | Income from discontinued operations | | | 35,925 | | | 3,937 | | | 4,594 | |
| | | | | | | | |
| | | Net loss | | $ | (33,492 | ) | $ | (53,921 | ) | $ | (44,959 | ) |
| | | | | | | | |
Basic and diluted net loss per share: | | | | | | | | | | |
| | Loss from continuing operations | | $ | (2.18 | ) | $ | (2.03 | ) | $ | (2.97 | ) |
| | Income from discontinued operations | | | 1.13 | | | 0.14 | | | 0.27 | |
| | | | | | | | |
| | | Net loss | | $ | (1.05 | ) | $ | (1.89 | ) | $ | (2.70 | ) |
| | | | | | | | |
| | | Weighted Average Common Shares (basic and diluted) | | | 31,895 | | | 28,489 | | | 16,663 | |
| | | | | | | | |
The accompanying notes are an integral part of these financial statements.
71
Table of Contents
OSIRIS THERAPEUTICS, INC.
STATEMENTS OF STOCKHOLDERS' EQUITY (DEFICIT)
(amounts in thousands, except for share data)
| | | | | | | | | | | | | | | | | | | | | | | | | |
| | Convertible Preferred
Stock | |
| |
| |
| |
| |
| |
| |
|---|
| | Common Stock | |
| | Accumulated
Other
Comprehensive
Income | |
| | Total
Stockholders'
Equity
(Deficit) | |
|---|
| | Additional
Paid-in
Capital | | Accumulated
Deficit | |
|---|
| | Shares | | Amount | | Shares | | Amount | |
|---|
Balance at January 1, 2006 | | | 10,650,544 | | $ | 32,746 | | | 9,097,506 | | $ | 9 | | $ | 36,127 | | $ | — | | $ | (142,544 | ) | $ | (73,662 | ) |
Exercise of options to purchase common stock ($0.40–$0.80 per share) | | | — | | | — | | | 178,378 | | | — | | | 73 | | | — | | | — | | | 73 | |
Exercise of warrants to purchase common stock ($0.40 per share) | | | — | | | — | | | 875,000 | | | 1 | | | 349 | | | — | | | — | | | 350 | |
Share-based payment—director services ($6.84–$11.00 per share) | | | — | | | — | | | 22,807 | | | — | | | 198 | | | — | | | — | | | 198 | |
Share-based payment—consulting services ($6.84 per share)) | | | — | | | — | | | 6,250 | | | — | | | 43 | | | — | | | — | | | 43 | |
Initial public offering of common stock, net of offering costs | | | — | | | — | | | 3,500,000 | | | 3 | | | 34,399 | | | — | | | — | | | 34,402 | |
Conversion of convertible preferred stock into common stock | | | (10,650,544 | ) | | (32,746 | ) | | 2,833,914 | | | 3 | | | 32,743 | | | — | | | — | | | — | |
Conversion of mandatorily redeemable preferred stock into common stock | | | — | | | — | | | 8,033,388 | | | 8 | | | 64,259 | | | — | | | — | | | 64,267 | |
Conversion of convertible notes payable into common stock | | | — | | | — | | | 2,774,076 | | | 3 | | | 25,320 | | | — | | | — | | | 25,323 | |
Share-based payment—employee compensation | | | — | | | — | | | — | | | — | | | 1,725 | | | — | | | — | | | 1,725 | |
Share-based payment—related party | | | — | | | — | | | — | | | — | | | 3,527 | | | — | | | — | | | 3,527 | |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | (44,959 | ) | | (44,959 | ) |
| | | | | | | | | | | | | | | | | | |
Balance at December 31, 2006 | | | — | | | — | | | 27,321,319 | | | 27 | | | 198,763 | | | — | | | (187,503 | ) | | 11,287 | |
Exercise of options to purchase common stock ($0.40–$6.84 per share) | | | — | | | — | | | 141,312 | | | — | | | 60 | | | — | | | — | | | 60 | |
Share-based payment—director services ($23.62 per share) | | | — | | | — | | | 12,500 | | | — | | | 295 | | | — | | | — | | | 295 | |
Issuance of common stock in private placements to overseas investors ($11.38–$12.37 per share) | | | — | | | — | | | 2,707,469 | | | 3 | | | 31,698 | | | — | | | — | | | 31,701 | |
Induced conversion of convertible notes payable into common stock ($13.00 per share) | | | — | | | — | | | 1,465,837 | | | 2 | | | 23,867 | | | — | | | — | | | 23,869 | |
Share-based payment—employee compensation | | | — | | | — | | | — | | | — | | | 1,045 | | | — | | | — | | | 1,045 | |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | (53,921 | ) | | (53,921 | ) |
| | | | | | | | | | | | | | | | | | |
Balance at December 31, 2007 | | | — | | | — | | | 31,648,437 | | | 32 | | | 255,728 | | | — | | | (241,424 | ) | | 14,336 | |
Exercise of options to purchase common stock ($0.40–$12.50 per share) | | | — | | | — | | | 66,545 | | | — | | | 289 | | | — | | | — | | | 289 | |
Share-based payment—director services ($12.01 per share) | | | — | | | — | | | 21,500 | | | — | | | 258 | | | — | | | — | | | 258 | |
Conversion of convertible notes payable into common stock ($12.04–$13.18 per share) | | | — | | | — | | | 851,914 | | | 1 | | | 10,499 | | | — | | | — | | | 10,500 | |
Induced conversion of convertible notes payable into common stock ($14.00 per share) | | | — | | | — | | | 87,524 | | | — | | | 1,500 | | | — | | | — | | | 1,500 | |
Share-based payment—employee compensation | | | — | | | — | | | — | | | — | | | 1,556 | | | — | | | — | | | 1,556 | |
Comprehensive loss: | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | | — | | | — | | | — | | | — | | | — | | | — | | | (33,492 | ) | | (33,492 | ) |
Unrealized gain on investments available for sale | | | — | | | — | | | — | | | — | | | — | | | 33 | | | — | | | 33 | |
| | | | | | | | | | | | | | | | | | |
Total comprehensive loss | | | — | | | — | | | — | | | — | | | — | | | — | | | — | | | (33,459 | ) |
| | | | | | | | | | | | | | | | | | |
Balance at December 31, 2008 | | | — | | $ | — | | | 32,675,920 | | $ | 33 | | $ | 269,830 | | $ | 33 | | $ | (274,916 | ) | $ | (5,020 | ) |
| | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these financial statements.
72
Table of Contents
OSIRIS THERAPEUTICS, INC.
STATEMENTS OF CASH FLOWS
(amounts in thousands)
| | | | | | | | | | | | | | |
| | Years Ended December 31, | |
|---|
| | 2008 | | 2007 | | 2006 | |
|---|
Cash flows from operating activities: | | | | | | | | | | |
| | Continuing Operations | | | | | | | | | | |
| | | Loss from continuing operations | | $ | (69,417 | ) | $ | (57,858 | ) | $ | (49,553 | ) |
| | | Adjustments to reconcile loss from continuing operations to net cash used in operations | | | | | | | | | | |
| | | Depreciation and amortization | | | 1,633 | | | 1,928 | | | 1,469 | |
| | | Non cash share-based payments | | | 1,627 | | | 1,271 | | | 5,494 | |
| | | Non cash interest expense | | | 130 | | | 6,881 | | | 5,653 | |
| | | Changes in operating assets and liabilities: | | | | | | | | | | |
| | | | Accounts receivable | | | (60,738 | ) | | (422 | ) | | 681 | |
| | | | Prepaid expenses and other current assets | | | (477 | ) | | (618 | ) | | (700 | ) |
| | | | Other assets | | | 784 | | | (663 | ) | | (457 | ) |
| | | | Accounts payable and accrued expenses | | | (1,022 | ) | | 4,588 | | | 3,106 | |
| | | | Deferred revenue | | | 124,746 | | | (1,349 | ) | | (952 | ) |
| | | | Long-term interest and other liabilities | | | — | | | (1,120 | ) | | (1,896 | ) |
| | | | | | | | |
| | Net cash used in continuing operations | | | (2,734 | ) | | (47,362 | ) | | (37,154 | ) |
| | | | | | | | |
| | Discontinued Operations | | | | | | | | | | |
| | | Income from discontinued operations | | | 35,925 | | | 3,937 | | | 4,594 | |
| | | Adjustments to reconcile loss from discontinued operations to net cash provided by discontinued operations: | | | | | | | | | | |
| | | | Gain from sale of discontinued operations | | | (30,400 | ) | | — | | | — | |
| | | | Depreciation & amortization | | | 562 | | | 105 | | | 77 | |
| | | | Provision for bad debts | | | 29 | | | — | | | — | |
| | | | Non cash share-based payments | | | 187 | | | 69 | | | — | |
| | | | Changes in operating assets and liabilities | | | | | | | | | | |
| | | | | Accounts receivable | | | 2,779 | | | (2,855 | ) | | (1,303 | ) |
| | | | | Inventory and other current assets | | | 1,821 | | | (2,228 | ) | | (1,791 | ) |
| | | | | Accounts payable and accrued expenses | | | 2,053 | | | 1,816 | | | 268 | |
| | | | | | | | |
| | Net cash provided by discontinued operations | | | 12,956 | | | 844 | | | 1,844 | |
| | | | | | | | |
| | | Net cash provided by (used in) operating activities | | | 10,222 | | | (46,518 | ) | | (35,310 | ) |
| | | | | | | | |
Cash flows from investing activities : | | | | | | | | | | |
| | Investing activities of continuing operations: | | | | | | | | | | |
| | | | Purchases of property and equipment | | | (166 | ) | | (798 | ) | | (376 | ) |
| | | | Proceeds from the sale of property and equipment | | | 104 | | | — | | | — | |
| | | | Proceeds from sale of discontinued operations, net | | | 33,607 | | | — | | | — | |
| | | | Proceeds from sale of investments available for sale | | | 16,195 | | | 50,900 | | | 40,112 | |
| | | | Purchases of investments available for sale | | | (60,000 | ) | | (29,893 | ) | | (35,805 | ) |
| | | | | | | | |
| | Net cash (used in) provided by investing activities of continuing operations | | | (10,260 | ) | | 20,209 | | | 3,931 | |
| | | | | | | | |
| | Investing activities of discontinued operations | | | | | | | | | | |
| | | | Purchases of property and equipment of discontinued operations | | | (2,808 | ) | | (3,904 | ) | | (1,320 | ) |
| | | | | | | | |
| | Net cash used in discontinued operations | | | (2,808 | ) | | (3,904 | ) | | (1,320 | ) |
| | | | | | | | |
| | | Net cash (used in) provided by investing activities | | | (13,068 | ) | | 16,305 | | | 2,611 | |
| | | | | | | | |
Cash flows from financing activities: | | | | | | | | | | |
| | | | Principal payments on capital lease obligations and notes payable | | | (14,357 | ) | | (1,115 | ) | | (21,692 | ) |
| | | | Restricted cash | | | 150 | | | 17 | | | (107 | ) |
| | | | Proceeds from convertible and short-term notes payable | | | 17,000 | | | — | | | 20,000 | |
| | | | Proceeds from the issuance of preferred and common stock, net | | | 289 | | | 31,701 | | | 34,824 | |
| | | | Payment of debt financing fees | | | — | | | (400 | ) | | (209 | ) |
| | | | | | | | |
| | | Net cash provided by financing activities | | | 3,082 | | | 30,203 | | | 32,816 | |
| | | | | | | | |
| | | Net increase (decrease) in cash and cash equivalents | | | 236 | | | (10 | ) | | 117 | |
| | | Cash and cash equivalents at beginning of period | | | 704 | | | 714 | | | 597 | |
| | | | | | | | |
| | | Cash and cash equivalents at end of period | | | 940 | | | 704 | | | 714 | |
| | | | | | | | |
| | | Supplemental disclosure of cash flows information: | | | | | | | | | | |
| | | Cash paid for interest | | $ | 1,264 | | $ | 1,450 | | $ | 1,260 | |
| | | Supplemental schedule of non cash investing and financing activities: | | | | | | | | | | |
| | | Conversion of notes payable to common stock | | | 11,700 | | | 18,800 | | | 21,762 | |
| | | Conversion of accrued interest into common stock | | | 300 | | | 5,069 | | | 3,558 | |
| | | Common stock issued to directors for services rendered | | | 258 | | | 295 | | | 198 | |
| | | Share-based payment—related party for financing services | | | — | | | — | | | 3,527 | |
The accompanying notes are an integral part of these financial statements.
73
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
1. Description of Business and Significant Accounting Policies
Description of Business
Osiris Therapeutics, Inc. ("we," "our," or the "Company") is a Delaware corporation headquartered in Columbia, Maryland. We began operations on December 23, 1992. We are a leading stem cell therapeutic company focused on developing and marketing products to treat medical conditions in the inflammatory, orthopedic, and cardiovascular areas. Our biologic drug candidates utilize adult human mesenchymal stem cells, or MSCs, which can selectively differentiate, based on the tissue environment, into various tissue lineages, such as muscle, bone, cartilage, marrow stroma, tendon or fat. In addition, MSCs have anti-inflammatory properties and can prevent fibrosis or scarring, which gives MSCs the potential to treat a wide variety of medical conditions. Our operations consist primarily of research, development and clinical activities to bring our biologic drug candidates to the marketplace. We have several research collaboration agreements and a government contract for additional product development.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States requires management to make estimates and assumptions that affect the amounts reported in the financial statements and accompanying notes. Due to the inherent uncertainty involved in making those assumptions, actual results could differ from those estimates. We believe that the most significant estimates that affect the accompanying financial statements are those that relate to inventory valuation, deferred tax assets, and share-based compensation.
Investments Available for Sale and Other Comprehensive Income
Investments available for sale consist primarily of marketable securities with maturities varying between three months and one year. Investments available for sale are valued at cost, which approximates their fair value, with unrealized gains and losses reported as a separate component of stockholders' equity in accumulated other comprehensive income. Gains or losses on investments available for sale are reclassified to earnings when realized.
Investments available for sale are evaluated periodically to determine whether a decline in their value is "other than temporary." The term "other than temporary" is not intended to indicate a permanent decline in value. Rather, it means that the prospects for near term recovery of value are not necessarily favorable, or that there is a lack of evidence to support fair values equal to, or greater than, the carrying value of the security. Management reviews criteria such as the magnitude and duration of the decline, as well as the reasons for the decline, to predict whether the loss in value is other than temporary. If a decline in value is determined to be other than temporary, the value of the security is reduced and a corresponding charge to earnings is recognized.
Accounts Receivable
Accounts receivable are reported at their net realizable value. As of December 31, 2008 and 2007, there was no allowance for doubtful accounts related to accounts receivable from continuing
74
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
1. Description of Business and Significant Accounting Policies (Continued)
operations, as we believe the reported amounts are fully collectible. Accounts receivable balances are not collateralized.
Property and Equipment
Property and equipment, including improvements that extend useful lives, are valued at cost, while maintenance and repairs are charged to operations as incurred. Depreciation is calculated using the straight-line method based on estimated useful lives ranging from three to seven years for furniture, equipment and internal use software. Leasehold improvements and assets under capital leases are amortized over the shorter of the estimated useful life of the asset or the original term of the lease.
Valuation of Long-lived Assets
Management reviews long-lived assets for impairment whenever events or changes in circumstances indicate that the carrying amount of an asset or group of assets may not be fully recoverable. These events or changes in circumstances may include a significant deterioration of operating results, changes in business plans, or changes in anticipated future cash flows. If an impairment indicator is present, management evaluates recoverability by a comparison of the carrying amount of the assets to future undiscounted net cash flows expected to be generated by the assets. Assets are grouped at the lowest level for which there is identifiable cash flows that are largely independent of the cash flows generated by other asset groups. If the total of the expected undiscounted future cash flows is less than the carrying amount of the asset, an impairment loss is recognized for the difference between the fair value and carrying value of assets. Fair value is generally determined by estimates of discounted cash flows. The discount rate used in any estimate of discounted cash flows would be the rate required for a similar investment of like risk. There were no impairment losses recognized during the years 2008, 2007 or 2006.
Assets to be disposed of are reported at the lower of carrying values or fair values, less estimated costs of disposal.
Revenue Recognition
We generate revenues from collaborative agreements, research licenses, and a government contract. Our revenue recognition policies are in accordance with the Securities and Exchange Commission's ("SEC") Staff Accounting Bulletin No. 101,Revenue Recognition in Financial Statements, as amended by SEC Staff Accounting Bulletin No. 104,Revenue Recognition.
Revenues from collaboration agreements are evaluated under Emerging Issues Task Force ("EITF") Issue No. 07-01, Draft Abstract,Accounting for Collaboration Agreements. Management evaluates revenues from agreements that have multiple elements to determine whether the components of the arrangement represent separate units of accounting as defined in EITF Issue No. 00-21,Revenue Arrangements with Multiple Deliverables. To recognize a delivered item in a multiple element arrangement, EITF Issue No. 00-21 requires that the delivered items have value to the customer on a stand alone basis, that there is objective and reliable evidence of fair value of the undelivered items and that delivery or performance is probable and within our control for any delivered items that have a
75
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
1. Description of Business and Significant Accounting Policies (Continued)
right of return. The determination whether multiple elements of a collaboration agreement meet the criteria for separate units of accounting requires us to exercise judgments.
Revenues from research licenses and government contracts are recognized as earned upon either the incurring of reimbursable expenses directly related to the particular research plan or the completion of certain development milestones as defined within the terms of the agreement. Payments received in advance of research performed are designated as deferred revenue. Non-refundable upfront license fees and certain other related fees are recognized on a straight-line basis over the development periods of the contract deliverables. Fees associated with substantive at risk, performance based milestones are recognized as revenue upon their completion, as defined in the respective agreements. Incidental assignment of technology rights is recognized as revenue as it is earned and received.
In October 2008, we entered into a Collaboration Agreement with Genzyme Corporation ("Genzyme") for the development and commercialization of Prochymal and Chondrogen. Under the agreement, Genzyme is obligated to pay to us non-contingent, non-refundable cash payments totaling $130.0 million, with $75.0 million paid during November 2008 and $55.0 million to be paid on July 1, 2009. The agreement provides Genzyme with certain rights to intellectual property developed by Osiris, and requires that we continue to perform certain development work related to the subject products. Management has evaluated the deliverables related to these payments under EITF 00-21, and concluded that the various deliverables represent a single unit of accounting. Accordingly, we have deferred the recognition of revenue related to the upfront payments, and are amortizing these amounts to revenue on a straight-line basis over the estimated delivery period of the required development services, which extend through the first quarter of 2012. Accordingly, we recognized $6.7 million of revenue in 2008 related to the amortization of the upfront payments. The balance of these payments has been recorded as $40.0 million of current deferred revenue and $83.3 million of long term deferred revenue as of December 31, 2008. The agreement also provides for contingent milestone payments of up to $1.25 billion in the aggregate, as well as royalties to be paid to us on any sales by Genzyme. Consistent with our revenue recognition policies, we will recognize revenue from these contingent milestone payments for which we have no continuing performance obligations upon achievement of the related milestone. For any milestone payments for which we have a continuing performance obligation, the milestone payments will be deferred and recognized as revenue over the term of the related performance obligations.
In 2007, we partnered with Genzyme to develop Prochymal as a medical countermeasure to nuclear terrorism and other radiological emergencies. In January 2008, we were awarded a contract from the United States Department of Defense ("DoD") pursuant to which we, in partnership with Genzyme, are seeking to develop and stockpile Prochymal for the repair of gastrointestinal injury resulting from acute radiation exposure. We began recognizing revenue under this contract during the first quarter of 2008. Contract revenue is recognized as the related costs are incurred, in accordance with the terms of the contract. We recognized $2.5 million in revenue from the DoD contract during 2008.
In October 2007, we entered into a collaborative agreement with the Juvenile Diabetes Research Foundation ("JDRF") to conduct a Phase II clinical trial evaluating Prochymal as a treatment for type 1 diabetes mellitus. This collaborative agreement provides for JDRF to provide up to $4.0 million
76
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
1. Description of Business and Significant Accounting Policies (Continued)
of contingent milestone funding to support the development of Prochymal for the preservation of insulin production in patients with newly diagnosed type 1 diabetes mellitus. The contingent milestone payments under the agreement are amortized to revenue on a straight line basis over the duration of our obligations under the collaborative agreement as they are earned. We received $2.0 million of the contingent milestones during 2008 and expect to receive the remaining $2.0 million in 2009. We began amortizing the $2.0 million of funding received, resulting in $0.6 million of revenue during 2008 under the agreement with JDRF. The remainder of the payments received under this agreement has been recorded as $0.5 million of current deferred revenue and $0.9 million of long term deferred revenue as of December 31, 2008.
We have entered into several strategic agreements with other pharmaceutical companies focusing on the development and commercialization of its stem cell drug products. In 2003, we entered into such an agreement with Boston Scientific Corporation pertaining to our cardiac drug development and received a $5 million fee for licensing the use of our technology. We terminated the agreement with Boston Scientific Corporation in 2007 and recognized the remaining unamortized license fee of $1.3 million. Also in 2003, we entered into a similar agreement with JCR Pharmaceuticals Co., Ltd. ("JCR") pertaining to our hematologic malignancies drugs for distribution in Japan, and have recognized $500 of revenue in 2007 from JCR upon the achievement of milestone events specified in the agreement.
We also earn royalties on the sale of human mesenchymal stem cells sold for research purposes and recognize the revenue on these sales as the sales are made. Revenues in 2008 include $244 and $197 of royalty revenue during 2008 and 2007, respectively.
Research and Development Costs
We expense internal and external research and development ("R&D") costs, including costs of funded R&D arrangements and the manufacture of clinical batches of our biologic drug candidates used in clinical trials, in the period incurred.
Income Taxes
Deferred tax liabilities and assets are recognized for the estimated future tax consequences of temporary differences, income tax credits and net operating loss carry-forwards. Temporary differences are primarily the result of the differences between the tax bases of assets and liabilities and their financial reporting values. Deferred tax liabilities and assets are measured by applying the enacted statutory tax rates applicable to the future years in which deferred tax liabilities or assets are expected to be settled or realized. Valuation allowances are established when necessary to reduce deferred tax assets to the amount expected to be realized. Income tax expense, if any, consists of the taxes payable for the current period and the change during the period in deferred tax assets and liabilities. For all periods presented, valuation allowances have been provided for the full amount of net deferred tax assets and no income tax expense or benefit has been recognized.
77
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
1. Description of Business and Significant Accounting Policies (Continued)
Loss per Common Share
Basic loss per common share is calculated by dividing net loss by the weighted average number of common shares outstanding during the period. Diluted loss per common share refers to the adjustment to basic loss per share for the potentially dilutive effects of shares issuable under stock option plans and the conversion of preferred stock and convertible debt using the treasury stock method. All of the common equivalent shares from the conversion of preferred stock and convertible debt and the exercise of stock options and warrants are excluded from the computation of diluted loss per share for all periods presented as their effect is anti-dilutive. Since our initial public offering in August 2006, the market value of our common stock is determined based upon the closing price on the NASDAQ Global Market. Prior to August 2006, when our common stock started publicly trading, our Board of Directors determined the fair value of our common stock.
Share-Based Compensation
In December 2004, the Financial Accounting Standards Board, ("FASB") issued Statement No. 123(R), "Share-Based Payment," which is a revision of Statement No. 123, "Accounting for Stock Issued to Employees." Effective January 1, 2006, we adopted Statement No. 123(R) using the modified prospective method under which prior period amounts are not restated for comparative purposes. Under the modified prospective method, we recognized compensation cost for:
- •
- all share-based payments granted after January 1, 2006 based upon the requirements of Statement No. 123(R); and
- •
- all unvested awards granted prior to January 1, 2006 using the compensation cost calculated for pro forma disclosure purposes under Statement No. 123.
Under Statement No. 123(R), we have recognized all share-based payments to employees and non-employee directors in our financial statements based on their grant date fair values, calculated using the Black-Scholes option pricing model. Compensation expense related to share-based awards is recognized on a straight-line basis based on the value of share awards that are scheduled to vest during the requisite service period. Under Statement No. 123(R), share-based compensation expense is based on awards ultimately expected to vest and must be reduced for estimated forfeitures.
Concentration of Risk
We maintain cash and short-term investment balances in accounts that exceed federally insured limits, although we have not experienced any losses on such accounts. We also invest excess cash in investment grade securities, generally with maturities of three months or less.
We have historically provided credit in the normal course of business to contract counterparties and to the distributors of our product. Accounts receivable in the accompanying balance sheets consists primarily of amounts due from four contract counterparties. The balance as of December 31, 2008 includes the $55.0 million non-refundable payment due from Genzyme on July 1, 2009, and we expect these receivables to be fully collected. Receivables from distributors of the Osteocel product line have been classified as a portion of current assets of discontinued operations.
78
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
1. Description of Business and Significant Accounting Policies (Continued)
Significant New Accounting Pronouncements
Recent Accounting Pronouncements that may have a material impact on future consolidated financial statements
In June 2008, the Financial Accounting Standards Board ("FASB") ratified Emerging Issue Task Force ("EITF") Issue No. 07-5, "Determining Whether an Instrument (or an Embedded Feature) is Indexed to an Entity's Own Stock" (EITF 07-5). This issue provides guidance for determining whether an equity-linked financial instrument (or embedded feature) is indexed to an entity's own stock. EITF 07-5 applies to any freestanding financial instrument or embedded feature that has all the characteristics of a derivative under paragraphs 6-9 of Statement of Financial Accounting Standards No. 133, "Accounting for Derivative Instruments and Hedging Activities," (SFAS 133) for purposes of determining whether that instrument or embedded feature qualifies for the first part of the scope exception under paragraph 11(a) of SFAS 133. EITF 07-5 also applies to any freestanding financial instrument that is potentially settled in an entity's own stock, regardless of whether the instrument has all the characteristics of a derivative under paragraphs 6-9 of SFAS 133, for purposes of determining whether the instrument is within the scope of EITF Issue 00-19, "Accounting for Derivative Financial Instruments Indexed to, and Potentially Settled in, a Company's Own Stock," (Issue 00-19) which provides accounting guidance for instruments that are indexed to, and potentially settled in, the issuer's own stock. EITF 07-5 is effective for fiscal years beginning after December 15, 2008. Early application is not permitted by entities that have previously adopted an alternative accounting policy. We are currently evaluating the requirements of EITF 07-5, but do not expect our adoption of this issue to have a material impact on our consolidated financial statements.
In May 2008, the FASB issued FASB Staff Position No. APB 14-1, "Accounting for Convertible Debt Instruments That May Be Settled in Cash Upon Conversion (Including Partial Cash Settlement)" ("FSP APB 14-1"). Under the new rules for convertible debt instruments that may be settled entirely or partially in cash upon conversion, an entity should separately account for the liability and equity components of the instrument in a manner that reflects the issuer's economic interest cost. Previous guidance provided for accounting of this type of convertible debt instruments entirely as debt. For instruments subject to the scope of FSP APB 14-1, higher interest expense may result through the accretion of the discounted carrying value of the convertible debt instruments to their face amount over their term. FSP APB 14-1 will be effective for fiscal years beginning after December 15, 2008, and for interim periods within those fiscal years, with retrospective application required. Early adoption is not permitted. As of December 31, 2008, we do not have any instruments outstanding that would be subject to FSP APB 14-1, but any instruments that we may issue in the future will be subject to this pronouncement.
In December 2007, the FASB issued FASB Statement No. 141 (Revised 2007)Business Combinations, ("SFAS 141R"). SFAS 141R will significantly change the accounting for business combinations. Under SFAS 141R, an acquiring entity will be required to recognize all the assets acquired and liabilities assumed in a transaction at the acquisition date at fair value with limited exceptions. SFAS 141R will change the accounting treatment for certain specific items, including; acquisition costs will be generally expensed as incurred, minority interests will be valued at fair value at the acquisition date, acquired contingent liabilities will be recorded at fair value at the acquisition date
79
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
1. Description of Business and Significant Accounting Policies (Continued)
and subsequently measured at either the higher of such amount or the amount determined under existing guidance for non-acquired contingencies, in-process research and development will be recorded at fair value as an indefinite-lived intangible asset at the acquisition date, restructuring costs associated with a business combination will be generally expensed subsequent to the acquisition date, and changes in deferred tax asset valuation allowances and income tax uncertainties after the acquisition date generally will affect income tax expense. SFAS 141R also includes a substantial number of new disclosure requirements. SFAS 141R applies prospectively to business combinations for which the acquisition date is on or after the beginning of the first annual reporting period beginning on or after December 15, 2008. Earlier adoption is prohibited. We are required to record and disclose business combinations following existing GAAP until January 1, 2009.
In September 2006, the FASB issued SFAS No. 157,Fair Value Measurements ("SFAS 157"). SFAS 157 establishes a common definition for fair value to be applied to GAAP guidance requiring use of fair value, establishes a framework for measuring fair value, and expands disclosure about such fair value measurements. SFAS 157 applies to fair value measurements that are already required or permitted by other accounting standards, except for measurements of share-based payments and measurements that are similar to, but not intended to be, fair value. The FASB has previously concluded in those accounting pronouncements that fair value is the relevant measurement attribute. Accordingly, this Statement does not require any new fair value measurements. SFAS 157 was effective for fiscal years beginning after November 15, 2007. The effective date of SFAS 157 with regard to non-financial assets and liabilities is January 1, 2009. Our adoption of SFAS 157 with respect to financial assets and liabilities as of January 1, 2008 did not have a material impact on our consolidated financial statements.
Recent Accounting Pronouncements that did not have a material impact on our consolidated financial statements
In December 2007, the Financial Accounting Standards Board ratified Emerging Issue Task Force Issue No. 07-1 "Accounting for Collaborative Arrangements," ("EITF 07-1") . The key elements of EITF 07-1 relate to: (a) the scope of the issue; (b) the income statement presentation of transactions with third parties; (c) the income statement presentation of payments between parties to the collaborative arrangement; (d) the disclosures about collaborative arrangements that should be required in the financial statements of the parties to the collaborative arrangements; and (e) the transition method. A contractual arrangement falls within the scope of EITF 07-1 if the arrangement requires the parties to be active participants and the arrangement exposes the parties to significant risks and rewards that are tied to the commercial success of the endeavor. Costs incurred and revenue generated on sales to third parties should be reported in the statement of operations based on the guidance in EITF Issue No. 99-19,Reporting Revenue Gross as a Principal versus Net as an Agent. The equity method of accounting should not be applied to a collaborative arrangement within the scope of this issue without the creation of a separate legal entity for the arrangement. Payments between parties to the collaborative arrangement should be presented in the statement of operations based on the nature of the arrangement and each entity's business operations, the contractual terms of the arrangement as well as if existing accounting principles generally accepted in the United States ("GAAP") is applicable. EITF 07-1 requires companies to disclose the nature and purpose of the arrangement, its rights and
80
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
1. Description of Business and Significant Accounting Policies (Continued)
obligations under the arrangement, the accounting policy applied to the arrangement, and the amounts attributable to transactions between other participants to the collaborative arrangement and where in the statement of operations these amounts have been classified. EITF 07-1 requires that companies comply in its first fiscal year beginning after December 15, 2008 and transition to the guidance in this issue by retrospectively applying the guidance to all periods presented for all arrangements existing at the effective date, unless it is impracticable to do so. The impracticability assessment should be made on an arrangement-by-arrangement basis and certain disclosures would be required if a company utilized the impracticability exception. Our adoption of EITF 07-1 did not have a material impact on our consolidated financial statements.
In June 2007, the FASB ratified Emerging Issue Task Force Issue No. 07-3 "Accounting for Non-Refundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities" ("EITF 07-3"), which requires nonrefundable advance payments for goods and services that will be used or rendered for future research and development activities to be deferred and capitalized. These amounts will be recognized as an expense in the period that the related goods are delivered or the related services are performed or when an entity does not expect the goods to be delivered or services to be rendered. EITF 07-3 is effective for the fiscal years beginning after December 31, 2007, including interim periods within those fiscal years. Our adoption of the provisions of EITF 07-3, beginning January 1, 2008 did not have a material impact on our consolidated financial statements.
In February 2007, the FASB issued SFAS No. 159 "The Fair Value Option for Financial Assets and Financial Liabilities—Including an Amendment of FASB Statement No. 115" ("SFAS 159"), which became effective for fiscal periods beginning after November 15, 2007. Under SFAS 159, companies may elect to measure specified financial assets and liabilities at fair value that are not otherwise measured at fair value, with changes in fair value recognized in earnings each subsequent reporting period. This election, called the "fair value option," will enable some companies to reduce volatility in reported earnings caused by measuring related assets and liabilities differently. SFAS 159 also establishes presentation and disclosure requirements designed to draw a comparison between the different measurement attributes a company elects for similar types of assets and liabilities. We did not elect the "fair value option" for any financial assets or liabilities, and therefore our adoption of SFAS 159 did not have any impact on our consolidated financial statements.
2. Collaboration Agreements and Government Contract
Following is a detailed discussion of each of our material collaborative agreements and contracts. The accounting policies related to each of these contracts, including material impact on our financial statements, is included above under the "Revenue Recognition" section of Note 1, Description of Business and Significant Accounting Policies.
Collaboration Agreement with Genzyme Corporation On October 31, 2008, we entered into a Collaboration Agreement with Genzyme for the development and commercialization of Prochymal and Chondrogen. Under the terms of the Agreement, we will retain the rights to commercialize Prochymal and Chondrogen in the United States and Canada, and Genzyme has been granted the exclusive right and license to commercialize Prochymal in all other countries (the "Genzyme Territory"), except with respect to Graft vs. Host Disease ("GvHD") in Japan where it has previously been licensed to another
81
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
2. Collaboration Agreements and Government Contract (Continued)
pharmaceutical company. Genzyme has also been granted the option to elect to participate in any future development of Chondrogen at the completion of Phase II trial we are currently conducting for that product. In the event that Genzyme elects to participate in the future development efforts for Chondrogen, they would receive similar rights and licenses to our intellectual property related to that product in all countries outside the United States and Canada.
As partial consideration for the grant of these rights, the Collaboration Agreement provides for a non-contingent, non-refundable payment to us of $130.0 million cash from Genzyme, with $75.0 million paid in November 2008 and $55.0 million scheduled to be paid on July 1, 2009. The Collaboration Agreement also provides for contingent milestone payments of up to $1.25 billion in the aggregate, in addition to royalties on any sales by Genzyme, to be paid by Genzyme to us, as follows:
Prochymal: As respects Prochymal, we are eligible to receive up to $500.0 million in development and regulatory milestone payments and up to $250.0 million in sales based milestone payments, as follows:
- •
- Total development milestones related to GvHD of up to $50.0 million, with $25.0 million payable upon marketing approval from the United States Food & Drug Administration ("FDA"), and $25.0 million payable upon marketing approval from the European Medicines Agency ("EMEA").
- •
- Total development milestones of up to $180.0 million related to Crohn's disease and Ulcerative Colitis, with $50.0 million payable upon achieving statistically significant endpoint(s) in a Phase III clinical trial for Crohn's disease, $100.0 million payable upon marketing approval by the EMEA for Crohn's disease, $10.0 million payable upon achieving statistically significant endpoint(s) in a Phase II or Phase III clinical trial for Ulcerative Colitis, and $20.0 million payable upon achieving marketing approval for Ulcerative Colitis by the EMEA.
- •
- Total development milestones of up to $270.0 million related to the development of follow-on indications for Prochymal, with $20.0 million payable upon each success in a Phase II clinical trial for acute myocardial infarction, type 1 diabetes or other follow-on indications, as agreed to by the Company and Genzyme, and $40.0 million payable upon receipt of each marketing approval by the EMEA for Prochymal for chronic obstructive pulmonary disease ("COPD"), acute myocardial infarction, type 1 diabetes mellitus or other follow-on indications, as agreed to by the Company and Genzyme.
- •
- Total sales based milestones of up to $250.0 million for Prochymal, with $100.0 million payable when annual Prochymal sales reach $500.0 million in the Genzyme Territory, and $150.0 million payable when annual Prochymal sales reach $1.0 billion in the Genzyme Territory.
Chondrogen: Upon receipt of the results of the planned Phase II/III clinical trial of Chondrogen, Genzyme may elect to opt out on any further Chondrogen development, at which point all rights to Chondrogen will revert to us with no further obligations between the companies with regard to
82
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
2. Collaboration Agreements and Government Contract (Continued)
Chondrogen. If Genzyme does not opt-out, we are eligible to receive up to $500.0 million in development, regulatory and sales based milestone payments for Chondrogen, as follows:
- •
- Total development and regulatory milestones of up to $100.0 million, with $10.0 million payable if Genzyme does not opt-out, $10.0 million payable upon demonstration of disease modification in the current clinical trial program, $40.0 million payable upon marketing approval by either the FDA or EMEA for a pain reduction indication, and $40.0 million payable upon marketing approval by either the FDA or EMEA for a disease modification indication.
- •
- Total sales milestones of up to $400.0 million, with $100.0 million payable when annual Chondrogen sales reach $500.0 million in the Genzyme Territory, $150.0 million payable when annual Chondrogen sales reach $1.0 billion in the Genzyme territory, and $150.0 million payable when annual Chondrogen sales reach $2.0 billion in the Genzyme Territory.
We will be solely responsible for ongoing clinical trial costs and future clinical trial costs with respect to both Prochymal and Chondrogen through Phase II clinical trials. We and Genzyme will share all costs of future Phase III and Phase IV clinical trials for agreed-upon indications (assuming in the case of Chondrogen that Genzyme does not opts out), with us being responsible for 60% of such costs and Genzyme responsible for 40% of such costs.
Assuming successful development and marketing approval, we will receive escalating royalties on sales of Prochymal and Chondrogen within the Genzyme Territory.
Genzyme Partnership and the United States Department of Defense Contract. In 2007, we partnered with Genzyme to develop Prochymal as a medical countermeasure to nuclear terrorism and other radiological emergencies. In January 2008, we were awarded a contract from the DoD pursuant to which we, in partnership with Genzyme, are seeking to develop and stockpile Prochymal for the repair of gastrointestinal injury resulting from acute radiation exposure. Under the terms of the contract, the DoD will provide funding to us for the development of Prochymal for acute radiation syndrome ("ARS"). If we are successful in obtaining FDA approval for ARS, the contract provides for the purchase by the DoD of up to 20,000 doses of Prochymal, at $10,000 per dose, in four 5,000 dose increments. Under the terms of our partnership with Genzyme, we will contribute Prochymal and corresponding safety and efficacy data to the effort and Genzyme will lend its vast product development and large-scale commercialization expertise. The agreement provides for Genzyme to receive a royalty of 15% on sales of Prochymal, limited to those sales made under contract to U.S. or Allied governmental agencies for emergency preparedness.
Juvenile Diabetes Research Foundation Agreement. In 2007, we entered into a collaborative agreement with the JDRF which provides funding to support the development of Prochymal as a treatment for the preservation of insulin production in patients with newly diagnosed type 1 diabetes mellitus. This collaborative agreement provides for JDRF to provide up to $4.0 million of contingent milestone funding. We initiated a Phase II clinical trial evaluating Prochymal as a treatment for type 1 diabetes in the fourth quarter of 2007, and received $2.0 million of the contingent milestones during 2008 and expect to receive the remaining $2.0 million in 2009. Consistent with our revenue recognition policies for such contingent milestones, we began amortizing the $2.0 million of funding received during 2008 when received for a term over the length of our obligations under the agreement.
83
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
2. Collaboration Agreements and Government Contract (Continued)
JCR Pharmaceuticals Agreement. In 2003, we entered into a strategic alliance with JCR. Under the JCR agreement, we have granted JCR the exclusive right in Japan to use our technology in conjunction with the treatment of hematologic malignancies using hematopoietic stem cell transplants. The JCR agreement entitles us to a licensing fee and to royalties on any resulting revenue. Upon commencement of the agreement, JCR purchased 545,454 shares of our Series B Convertible Preferred Stock for $3.0 million. These shares were converted into 136,363 shares of our common stock concurrent with our initial public offering in August 2006. They have also paid us a total of $4.0 million in licensing fees under the agreement.
3. Segment Reporting and Discontinued Operations
In 2007, we began to manage our business in two reportable operating segments: the Biologic Drug Candidates segment and the Biologic Tissue Product segment. Our Biologic Drug Candidates segment focuses on developing and marketing products to treat medical conditions in the inflammatory, orthopedic and cardiovascular areas. Its operations have focused on clinical trials and discovery efforts to identify additional medical indications. Our Biologic Drug Candidates segment generates revenues from license fees, government contracts, and royalties from collaborative agreements.
Our Biologic Tissue Product segment includes the manufacture and sale of Osteocel, which we launched in July 2005 and is currently being used by orthopedic surgeons for focal bone repair. In April 2008, we committed to a plan to sell our biologic tissue product segment, including our entire product line relating to the processing, manufacturing, marketing and selling of Osteocel® and Osteocel® XO, an allograft material containing cancellous bone, used in spinal fusion and other surgical procedures. We refer to these assets as our Osteocel asset disposal group. Not included among the Osteocel asset disposal group is Osteocel® XC, our second generation product candidate under development for bone repair, utilizing culture expanded mesenchymal stem cells to create a synthetic version of Osteocel.
We eliminated the Osteocel asset disposal group from our ongoing operations as a result of the disposal transaction and have presented the results of the group's operations as a discontinued operation for all periods. Accordingly, our continuing operations now represent the business that was previously referred to as the Biologic Drug Candidates segment. Substantially all of our revenues and assets are attributed to and are received from entities located in the United States.
On May 8, 2008, we entered into an Asset Purchase Agreement to sell the Osteocel asset disposal group to NuVasive, Inc., a Delaware corporation. The agreement provides for the sale to be effected at two closings—a "technology assets closing," at which technology and certain other business assets are transferred, and a "manufacturing assets closing," at which manufacturing assets and facilities are transferred. On July 24, 2008, we held a Special Meeting of Stockholders at which our stockholders overwhelmingly approved the sale of the Osteocel business. The technology assets closing also occurred on that date, at which time we received an initial payment of $35.0 million and entered into a Manufacturing Agreement under which we will continue to manufacture Osteocel for up to eighteen months after the technology assets closing and sell 100% of the product to NuVasive at specified prices. The Asset Purchase Agreement provides that the manufacturing assets closing is to occur eighteen
84
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
3. Segment Reporting and Discontinued Operations (Continued)
months after the date of the technology assets closing, or upon earlier termination of the Manufacturing Agreement. NuVasive has certain minimum purchase order obligations under the Manufacturing Agreement. We could recognize approximately $40.0 million in revenue from discontinued operations under the Manufacturing Agreement, but do not expect to report significant profits, if any, from manufacturing the product under this Manufacturing Agreement. In September 2008, we entered into amendments to the Asset Purchase Agreement and the related Manufacturing Agreement to accommodate reduced shipments of Osteocel by NuVasive to Blackstone Medical, Inc.
We recognized a gain on the technology assets closing of $25,539 in the third quarter of 2008. We incurred transaction costs of $1,364, including legal, accounting and advisory fees. At that time, we also established reserves totaling $8,097 to recognize the concessionary pricing of Osteocel that we will produce under the Manufacturing Agreement. These reserves are being amortized into the income (loss) from operations of discontinued operations beginning in the fourth quarter of 2008 through the end of 2009, concurrent with our fulfillment of our obligations under the Manufacturing Agreement. At December 31, 2008, $3,618 of these reserves remain unamortized, and are included in current liabilities of discontinued operations.
The Asset Purchase Agreement, as amended, also provides for up to $50.0 million of contingent additional payments to us upon our achievement of milestone events; in addition to the $35.0 million initial payment, as follows:
| | | | | |
Milestone | | Amount | | Cut-Off Date |
|---|
Initial payment | | $ | 35,000 | | Received in July 2008 |
Cumulative sales to NuVasive of 75,000 units(1) | | | 5,000 | | Achieved in December 2008 |
Cumulative sales to NuVasive of 180,000 units | | | 5,000 | | January 2010 |
Net end-user sales of $35 million | | | 15,000 | | none |
Cumulative sales to NuVasive of 205,000 units | | | 5,000 | | January 2010 |
Cumulative sales to NuVasive of 308,300 units | | | 2,500 | | January 2010 |
Cumulative sales to NuVasive of 350,000 units | | | 2,500 | | January 2010 |
Cumulative sales to NuVasive of 350,000 units | | | 2,500 | | January 2010 |
Transfer of Manufacturing Assets | | | 12,500 | | January 2010 |
| | | | | |
Total possible purchase price | | $ | 85,000 | | |
| | | | | |
- (1)
- Each unit represents 1.0 cubic centimeter of Osteocel product.
We earned the first $5.0 million contingent milestone payment during December 2008. This milestone, net of $139 of additional expenses, was recorded as an increase to the gain from the sale of discontinued operations during the fourth quarter of 2008. If and when the requisite additional milestone events occur, we will likewise recognize the proceeds from those milestones as an increase to the gain from the sale of discontinued operations.
85
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
3. Segment Reporting and Discontinued Operations (Continued)
The net assets allocable to the Osteocel asset disposal group at December 31, 2008 and 2007 were as follows:
| | | | | | | | |
| | 2008 | | 2007 | |
|---|
Accounts receivable | | $ | 1,516 | | $ | 4,324 | |
Inventory | | | 1,502 | | | 3,983 | |
Other current assets | | | 205 | | | 138 | |
| | | | | | |
| | Current assets of discontinued operations | | | 3,223 | | | 8,445 | |
| | | | | | |
Property and equipment, net | | | 7,520 | | | 4,596 | |
Current liabilities of discontinued operations | | | (7,219 | ) | | (2,552 | ) |
| | | | | | |
Net assets of discontinued operations | | $ | 3,524 | | $ | 10,489 | |
| | | | | | |
Summarized operating results of the Osteocel asset disposal group for the years ended December 31, 2008, 2007, and 2006 are as follows:
| | | | | | | | | | |
| | 2008 | | 2007 | | 2006 | |
|---|
Product sales | | $ | 24,670 | | $ | 15,240 | | $ | 8,291 | |
Cost of goods sold | | | 17,455 | | | 6,955 | | | 3,697 | |
| | | | | | | | |
Gross profit | | | 7,215 | | | 8,285 | | | 4,594 | |
| | | | | | | | |
Operating expenses | | | 1,690 | | | 4,348 | | | — | |
| | | | | | | | |
Income from operations of discontinued operations | | $ | 5,525 | | $ | 3,937 | | $ | 4,594 | |
| | | | | | | | |
Inventory consists of tissue products in process and available for distribution, and is valued using the first-in, first-out method. Due to the nature of the Osteocel product, all of the costs to manufacture the product are incurred prior to completing the extensive testing and evaluation necessary to determine if the product can be released. Accordingly, we estimate the reserve for work-in-process inventory based upon historical experience.
Revenues on Osteocel sales are recognized when legal title to the product has passed to the customer.
Costs of goods sold related to the Osteocel product consist primarily of the costs to obtain the tissue and other chemicals and supplies, quality and sterility testing, plus labor and allocated overhead costs and the costs of operating the clean-room facilities.
Operating expenses for the year ended December 31, 2007 were unusually high due to costs associated with the expansion of the Osteocel manufacturing facility, as we experienced failed production qualification runs while expanding our capacity. The production issues were subsequently resolved in late March 2007. None of the corporate operating expenses have been allocated to the Osteocel asset disposal group for the year ended December 31, 2006, as the product had just recently been launched and did not constitute a material component of our corporate operations prior to the expansion of the manufacturing facility.
86
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
3. Segment Reporting and Discontinued Operations (Continued)
Prior to the execution of the Manufacturing Agreement, we sold Osteocel primarily to two customers, each of which represented greater than ten percent of our consolidated revenues for the years ended December 31, 2007 and 2006.
4. Initial Public Offering and Reverse Stock Split
On August 9, 2006, we consummated our initial public offering, consisting of 3,500,000 shares of common stock at a public offering price of $11.00, resulting in net proceeds to us of approximately $34.4 million (after deducting payment of underwriters' discounts and commissions, as well as offering expenses). Our common stock began trading on the NASDAQ Global Market on August 4, 2006.
In connection with the initial public offering, we affected a 1-for-4 reverse stock split of our issued and outstanding common stock. Information relating to our common stock and common stock-equivalents set forth in this report has been restated to reflect this split for all periods presented. Upon consummation of the initial public offering, all outstanding shares of the Class I, Series 2003, Series B, Series C, Series E and the Mandatorily Redeemable Series D convertible preferred stock were converted into an aggregate of 10,867,302 shares of our common stock. In addition, approximately $21.8 million of our convertible notes payable, together with accrued interest, were converted into an aggregate of 2,774,076 shares of our common stock.
Immediately following the initial public offering, we had 27,198,307 shares of common stock outstanding.
We incurred $7.0 million in non-cash charges relating to the completion of the initial public offering, including $0.8 million for stock-based compensation related to the accelerated vesting of certain employee stock options, a $3.5 million share-based payment to related party related to warrants that were priced upon the completion of the initial public offering, and $0.2 million in share-based compensation for stock awarded to our directors for service on our board. In addition, interest expense for the year ended December 31, 2006 includes $2.7 million in previously deferred financing costs and premiums that were expensed as a result of debt that was converted into common stock at the completion of the initial public offering.
5. Property and Equipment
Property and equipment consist of the following at December 31,
| | | | | | | |
| | 2008 | | 2007 | |
|---|
Laboratory and manufacturing equipment | | $ | 212 | | $ | 4,802 | |
Computer hardware, furniture and fixtures | | | 289 | | | 1,402 | |
Leased assets | | | 26 | | | 11,725 | |
Leasehold improvements | | | — | | | 4,518 | |
| | | | | | |
| | | 527 | | | 22,447 | |
Accumulated depreciation and amortization | | | (133 | ) | | (20,427 | ) |
| | | | | | |
Property and equipment, net | | $ | 394 | | $ | 2,020 | |
| | | | | | |
87
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
5. Property and Equipment
During the third quarter of 2008, we consolidated all of our corporate activities, including the production of Osteocel, in our Columbia, Maryland facility. Prior to that time, we had leased approximately 126,000 square feet of laboratory, production, warehouse and office space in Baltimore, Maryland under a lease agreement that expired in September 2008 which had originally been arranged by the Maryland Economic Development Corporation and the City of Baltimore. Upon the expiration of the Baltimore lease, we vacated that location and consolidated our operations in our Columbia facility.
6. Notes Payable and Capital Lease Obligations
| | | | | | | | |
| | December 31, | |
|---|
| | 2008 | | 2007 | |
|---|
Boston Scientific Corporation Term Note, 8%, due quarterly through January 2009 | | $ | — | | $ | 6,521 | |
Term Notes, 10%, convertible into common stock at $18 per share and under specified conditions | | | — | | | 1,200 | |
| | | | | | |
| | | — | | | 7,721 | |
| | Less current portion | | | — | | | (6,521 | ) |
| | | | | | |
Notes payable—long-term | | $ | — | | $ | 1,200 | |
| | | | | | |
Total capital lease obligations | | $ | 9 | | $ | 897 | |
| | Less current portion | | | (6 | ) | | (886 | ) |
| | | | | | |
Capital lease obligations, long-term | | $ | 3 | | $ | 11 | |
| | | | | | |
The majority of our debt incurred since 1993 has been provided by or arranged for by related parties, as discussed more fully in Note 9—Related Party Transactions.
In 2004, we borrowed $5.0 million on the $50.0 million line-of-credit entered into with Boston Scientific Corporation which was part of a collaborative arrangement for the development of our biologic drug candidate for cardiac indications. Under the terms of the original line-of-credit, this loan was to be repaid from the proceeds of future sales. In December 2007, the collaborative agreement was terminated. The line-of-credit was cancelled and the outstanding principal, together with accrued interest, was converted into a one-year term note, bearing interest at 8% and payable in quarterly installments starting in January 2008. We repaid this note, including accrued interest, during 2008.
In October 2006, we issued $20.0 million in convertible promissory notes in a private placement to several Swiss investors. The notes accrued interest at a rate of 10%, with semi-annual payments of accrued interest becoming due and payable on April 30 and October 30 of each calendar year, until maturity on April 30, 2009. The notes were convertible at the option of the respective holders at any time after February 9, 2007, into shares of common stock at the conversion price of $18.00 per share. The notes initially provided for automatic conversion into common stock at the same conversion price, if at any time after February 9, 2007, the closing price of the our common stock on the NASDAQ Global Market closed for ten consecutive trading days at $25.00 per share or greater. The notes also
88
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
6. Notes Payable and Capital Lease Obligations (Continued)
provided for redemption at any time at our option, with 30-day written notice. In December 2007, we induced the conversion of $18.8 million of the notes, together with accrued interest into 1,465,837 shares of common stock. In connection with this induced conversion, we recorded a non-cash charge of $4.8 million as interest expense. In January 2008, we induced the conversion the remaining $1.2 million of these notes, together with accrued interest into 87,524 shares of common stock at the conversion price of $14.00 per share, which resulted in a non-cash charge of $248 as interest expense.
In March and May 2008, we issued an aggregate of $10.5 million in convertible promissory notes to several non-U.S. investors pursuant to a private placement intended to qualify under Regulation S and Section 4(2) of the Securities Act of 1933, as amended. Three of these notes with an aggregate principal amount of $8.0 million bore interest at 2% and were due and payable on November 30, 2008. The fourth note with a principal amount of $2.5 million bore interest at 4% and was due and payable on November 30, 2009. The notes were convertible at the option of the respective holders at any time, into shares of our common stock at conversion prices ranging from $12.04 to $13.18 per share (the respective closing prices on the NASDAQ Global Market on the dates of the definitive agreements). The notes provided for redemption at any time at our option, with 30-days prior written notice. These notes, together with accrued interest, were converted into shares of our common stock at their respective conversion rates during the fourth quarter of 2008.
In June 2008, we issued an aggregate of $5.5 million in short-term notes, and in July 2008, we issued an aggregate of $1.0 million in short-term notes to several non-U.S. investors pursuant to a private placement intended to qualify under Regulation S and Section 4(2) of the Securities Act of 1933, as amended. These notes bore interest at 10% semi-annually and became due and payable in December 2008. In the third quarter of 2008, we repaid $2.5 million of these notes together with accrued interest, and repaid the remaining $4.0 million, with accrued interest, during the fourth quarter of 2008.
Future Maturities of Capital Lease Obligations
As of December 31, 2008, the current portion of our capital lease obligations was due in 2009 and the long term portion of our obligations are due in 2010.
89
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
7. Preferred Stock Conversion
All our then outstanding convertible preferred stock was converted into common stock upon the completion of our initial public offering in August 2006.
| | | | |
| | No. Shares of Common
Stock Issued Upon
Conversion at IPO
(Aggregate Liquidation
Preference / Conversion
Price) Per Share | |
|---|
Convertible preferred stock, Class I, Series 2003, $0.001 par value, 2,000,000 shares designated, issued and outstanding in 2005 | | | 500,000 | |
Convertible preferred stock, Series B, $0.001 par value, 750,000 shares designated, 545,454 shares issued and outstanding in 2005 | | | 136,364 | |
Convertible preferred stock, Series C, $0.001 par value, 3,500,000 shares designated, 548,090 shares issued and outstanding in 2005 | | | 308,300 | |
Convertible preferred stock, Series E, $0.001 par value, 8,000,000 shares designated, 7,557,000 shares issued and outstanding in 2005 | | | 1,889,250 | |
| | | | |
| | | 2,833,914 | |
| | | | |
We issued 2,000,000 shares of our Class I, Series 2003 convertible preferred stock to a collaborative partner as part of an agreement related to product development, clinical trials and FDA approval. These shares were converted into 500,000 shares of our common stock at the conversion price of $20.00 per share.
We issued 545,454 shares of our Series B convertible preferred stock in 2003, as part of a collaborative agreement. These shares were converted into 136,364 shares of our common stock at the conversion price of $22.00 per share.
We issued 548,090 shares of our Series C convertible preferred stock in 2004 at the price of $4.50 per share. These shares were converted into 308,300 shares of our common stock at the conversion price of $8.00 per share.
We issued 7,557,000 shares of our Series E convertible preferred stock in 2005 at a price of $2.50 per share. These shares were converted into 1,889,250 shares of our common stock at the conversion price of $10.00 per share.
Also in 2005, we issued 3,213,335 shares of Series D Mandatorily Redeemable Convertible Preferred Stock at a price of $2.00 per share. These shares were converted into 8,033,388 shares of our common stock at the conversion price of $0.80 per share.
8. Share-Based Compensation
In April 2006, we adopted our 2006 Omnibus Plan. We amended and restated this plan in 2008 to, among other things, increase the number of shares available for grant. In addition, we had previously established our Amended and Restated 1994 Stock Incentive Plan. Both Plans authorize the issuance of various forms of stock-based awards, including incentive and non-qualified stock options, stock
90
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
8. Share-Based Compensation (Continued)
purchase rights, stock appreciation rights and restricted and unrestricted stock awards. A total of 1,450,000 shares of our common stock have been reserved for issuance under the Amended and Restated 2006 Omnibus Plan, and 736,378 shares were reserved under our Amended and Restated 1994 Stock Incentive Plan. We ceased all grants under the Amended and Restated 1994 Stock Incentive Plan concurrent with our initial public offering in August 2006. As a result, no shares are currently available for future awards under the Amended and Restated 1994 Stock Incentive Plan. At December 31, 2008, there were 567,760 shares available for future awards under the Amended and Restated 2006 Omnibus Plan.
We generally issue stock option awards that vest over four years and have a ten-year life. We estimate the fair value of stock options using the Black-Scholes option-pricing model. Our common stock started trading on the public market in August 2006, and the historical data to determine volatility does not presently exist. We determine volatility by using the historical stock volatility of other companies with similar characteristics. Estimates of fair value are not intended to predict actual future events or the value ultimately realized by persons who receive equity awards. The fair value of stock options granted during each of the periods was estimated using the following assumptions:
| | | | | | | | | | |
| | Years ended December 31, | |
|---|
| | 2008 | | 2007 | | 2006 | |
|---|
Assumptions | | | | | | | | | | |
Weighted average risk-free interest rate | | | 3.83% | | | 4.61% | | | 4.75% | |
Dividend yield | | | 0.0% | | | 0.0% | | | 0.0% | |
Expected life of option grants | | | 6.25-years | | | 4.5-years | | | 5-years | |
Weighted average expected stock price volatility | | | 67.23% | | | 67.07% | | | 74.11% | |
91
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
8. Share-Based Compensation (Continued)
A summary of stock option activity for the years ended December 31, 2008, 2007 and 2006 is as follows:
| | | | | | | | | | | | | |
| | Number of
Shares | | Weighted Average
Exercise Price | | Weighted Average
Remaining
Contractual Life | | Aggregate
Intrinsic Value
(in thousands) | |
|---|
Outstanding at January 1, 2006 | | | 566,480 | | $ | 0.40 | | 8.4-years | | | | |
| | Granted | | | 314,500 | | $ | 4.06 | | | | | | |
| | Exercised | | | (178,378 | ) | $ | (0.41 | ) | | | $ | 1,827 | |
| | Forfeited or canceled | | | (6,687 | ) | $ | (1.36 | ) | | | | | |
| | | | | | | | | | | | |
Outstanding at December 31, 2006 | | | 695,915 | | $ | 2.05 | | 8.3-years | | | | |
| | Granted | | | 373,500 | | $ | 18.30 | | | | | | |
| | Exercised | | | (141,312 | ) | $ | (0.42 | ) | | | $ | 2,174 | |
| | Forfeited or canceled | | | (47,693 | ) | $ | (12.49 | ) | | | | | |
| | | | | | | | | | | | |
Balance, December 31, 2007 | | | 880,410 | | $ | 8.64 | | 8.3-years | | | | |
| | Granted | | | 552,000 | | $ | 13.56 | | | | | | |
| | Exercised | | | (66,545 | ) | $ | (4.36 | ) | | | $ | 663 | |
| | Forfeited or canceled | | | (154,688 | ) | $ | (12.23 | ) | | | | | |
| | | | | | | | | | | | |
Balance, December 31, 2008 | | | 1,211,177 | | $ | 10.66 | | 8.0-years | | $ | 10,952 | |
| | | | | | | | | | | | |
Exercisable at December 31, 2008 | | | 440,409 | | $ | 4.23 | | 6.5-years | | $ | 6,741 | |
| | | | | | | | | | | | |
A summary of stock options outstanding at December 31, 2008, by price range is as follows:
| | | | | | | | | | | | | | | | |
| | Options Outstanding | |
| |
| |
|---|
| | Options Exercisable | |
|---|
| |
| | Weighted-Average
Remaining
Contractual Life
(in years) | |
| |
|---|
Range of Exercise Prices | | Number
Outstanding | | Weighted-Average
Exercise Price | | Number
Outstanding | | Weighted-Average
Exercise Price | |
|---|
$0.40 to $1.00 | | | 339,239 | | | 6.1 | | $ | 0.40 | | | 317,917 | | $ | 0.40 | |
1.01 to 7.00 | | | 77,563 | | | 7.6 | | | 6.84 | | | 38,563 | | | 6.84 | |
7.01 to 12.50 | | | 340,125 | | | 9.0 | | | 12.06 | | | 23,932 | | | 12.04 | |
12.51 to 15.00 | | | 92,750 | | | 7.8 | | | 13.57 | | | 22,874 | | | 13.43 | |
15.01 to 20.00 | | | 214,500 | | | 9.6 | | | 15.90 | | | 375 | | | 17.27 | |
$20.01 to $28.56 | | | 147,000 | | | 8.1 | | | 23.63 | | | 36,748 | | | 23.63 | |
| | | | | | | | | | | | | | | |
| | | 1,211,177 | | | 8.0 | | | 10.66 | | | 440,409 | | | 4.23 | |
| | | | | | | | | | | | | | | |
The weighted fair value of options granted during the years ended December 31, 2008, 2007 and 2006 were $8.77, $10.61 and $7.11, respectively.
In connection with the stock options exercised during the year ended December 31, 2008, we received cash proceeds of $289. At December 31, 2008, there was $4.2 million of total unrecognized compensation costs related to non-vested stock options, which is expected to be recognized over a weighted average period of three years.
92
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
8. Share-Based Compensation (Continued)
The table below reflects the total share-based compensation expense recognized in our income statements for the years ended December 31, 2008, 2007 and 2006. FASB Statement No. 123(R) requires forfeitures to be estimated at the time an award is granted and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. Pre-vesting forfeitures were estimated to be between 0% and 20% based on historical experience. For the years ended December 31, 2008, 2007 and 2006, share-based compensation expense is based on awards ultimately expected to vest and has been reduced for estimated forfeitures.
| | | | | | | | | | |
| | Year ended December 31, | |
|---|
| | 2008 | | 2007 | | 2006 | |
|---|
Research and development | | $ | 652 | | $ | 608 | | $ | 755 | |
General and administrative | | | 975 | | | 663 | | | 4,738 | |
Discontinued operations | | | 187 | | | 69 | | | — | |
| | | | | | | | |
Share-based compensation | | $ | 1,814 | | $ | 1,340 | | $ | 5,493 | |
| | | | | | | | |
9. Related Party Transactions
General. Peter Friedli, the Chairman of our Board of Directors, or entities with which he is affiliated, have been responsible for procuring since 1993, an aggregate of approximately $270 million in debt and equity financing for us and our predecessor company. Mr. Friedli is the beneficial owner of approximately 43% of our common stock as of December 31, 2008. Of the shares beneficially owned by Mr. Friedli at December 31, 2008, 35,000 shares were received by him as Board compensation since 1996, 12,500 shares and warrants for 1,000,000 shares were granted in recognition of his fundraising efforts, as discussed below, and the remaining shares were acquired through investment or through purchase from third parties.
Consulting Agreement. Beginning in 1995, we and our predecessor company were party to a Consulting Agreement, originally with Friedli Corporate Finance AG, and subsequently Friedli Corporate Finance, Inc., or FCF, for the provision of business and advisory services to us. Mr. Friedli is the sole owner of FCF. Under this agreement, FCF provided general business, financial and investment advice to us, and served as a liaison between us and FCF clients who have invested in us, many of which are located in Switzerland. The Consulting Agreement between us and FCF was terminated upon the closing of our initial public offering in August 2006. The base compensation paid by us under this agreement was $47 in 2006. In addition, pursuant to this Consulting Agreement, we paid $350 in 2006, to or as directed by FCF.
Private Placement Financings During the Three Years Ended December 31, 2008. Separate from the Consulting Agreement, FCF served as our agent in Europe in connection with:
- •
- the issuance and sale in 2006 of $20 million of our Convertible Notes to non-U.S. investors, which were converted into an aggregate of 1,553,361 shares of our common stock in December 2007 and January 2008, at prices above the NASDAQ closing price on the dates of conversion. Mr. Friedli and an entity affiliated with Mr. Friedli purchased $8.5 million of these Notes, and
93
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
9. Related Party Transactions (Continued)
In October 2007, we also obtained a $30.0 million financing commitment from FCF. This financing commitment was for a twelve month term and provided for financing through the issuance by us of common stock at a price determined as the basis of market value, or the issuance by us of three-year promissory notes bearing interest at LIBOR plus 4%. We did not incur any fees in connection with the establishment of this financing commitment, nor did we draw any funds on this financing commitment, which has since expired.
Our Board of Directors, including all of our independent directors, but with Mr. Friedli abstaining, together with the audit committee for all transactions occurring following our initial public offering, unanimously approved each of these financings, including the participation of Mr. Friedli and entities affiliated with Mr. Friedli and the arrangements with FCF.
We did not pay any fees for any of the financings arranged in 2007 through Mr. Friedli or entities with which he is affiliated. In 2008, we accrued $150 for fees payable to Mr. Friedli or entities with which he is affiliated in connection with the $17 million of financing provided by him during 2008. These fees are classified as Interest Paid to Related Parties in the accompanying statement of operations.
94
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
9. Related Party Transactions (Continued)
Prior to 2007, we paid referral fees and costs of $3.4 million to accounts designated by Mr. Friedli, including accounts of unrelated third parties. In addition, we paid $600 to FCF, Inc. in connection with the issuance of the $20.0 million in Convertible Notes in 2006.
To facilitate borrowings and other financings prior to our initial public offering, and for commitments of consideration in respect of yet additional financing if needed, we issued warrants for an aggregate of 1,250,000 shares at an exercise price of $0.40 per share. Mr. Friedli subsequently arranged for the acquisition of those warrants and they have since been cancelled. In recognition of his efforts in procuring financing over the years and the cancellation of all of these warrants, in 2006, we issued a new warrant to Mr. Friedli, exercisable for up to 1,000,000 shares of our common stock at $11.00 per share, the price for which shares were sold in the initial public offering. This warrant expires in May 2011.
Lockup Agreement. On October 30, 2006, we entered into a Lockup Agreement with Mr. Friedli, Venturetec, Inc. and U.S. Venture 05, Inc. Pursuant to the Lockup Agreement, Mr. Friedli and such entities initially agreed with us, subject to limited exceptions, not to transfer our securities held by them without our approval, until January 30, 2008. The Lockup Agreement was amended in September 2007, by an Amendment to Lockup Agreement, pursuant to which Mr. Friedli and Venturetec. Inc agreed to extend the term of the Lockup Agreement as applicable to them, until January 30, 2009. The term of the Amended Lockup Agreement expired on January 30, 2009.
10. Warrants
At December 31, 2008, we had warrants to purchase our common stock outstanding as shown in the following table.
| | | | | | | |
| | Common Stock | |
|---|
| | # of Shares | | Weighted
Average Price | |
|---|
Warrants outstanding, January 1, 2006 | | | 2,125,000 | | $ | 0.40 | |
Warrants granted | | | 1,000,000 | | | 11.00 | |
Warrants exercised | | | (875,000 | ) | | 0.40 | |
Warrants cancelled | | | (1,250,000 | ) | | 0.40 | |
| | | | | | |
Warrants outstanding, December 31, 2006 | | | 1,000,000 | | $ | 11.00 | |
| | | | | | |
There was no additional activity related to the warrants outstanding during either of the years ended December 31, 2008 or 2007.
95
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
10. Warrants
Following is the summary of the status of outstanding warrants to purchase our common stock at December 31, 2008.
| | | | | | | | | | |
Warrant Price | | Common Shares | | Weighted Average
Remaining
Contractual Life | | Intrinsic Value | |
|---|
| | $11.00 | | | 1,000,000 | | 2.5 years | | $ | 1,000 | |
The warrants issued during 2006 have an exercise price of $11.00. We computed the value of this warrant using the Black-Scholes option pricing method, using a risk free interest rate of 4.80%, the expected life of 2.5-years and a stock volatility factor of 44.66%. Since the Company just recently completed its initial public offering at the time of the valuation, and previously its stock did not trade, we determined the volatility by selecting a comparable public company in the biotech industry and tracking its stock prices over the past 2.5-years. The value of this warrant was determined to be $3.5 million, which has been recorded in the accompanying statement of operations as General and Administrative expenses during the year ended December 31, 2006.
11. Income Taxes
The components of the Company's net deferred tax assets at December 31 are as follows:
| | | | | | | | |
| | 2008 | | 2007 | |
|---|
Deferred Tax Assets: | | | | | | | |
| | Net operating loss carry-forwards | | $ | 95,128 | | $ | 85,016 | |
| | Research and experimentation credit carry-forwards | | | 10,090 | | | 7,167 | |
| | Property and equipment | | | 40 | | | 1,432 | |
| | | | | | |
| | | 105,258 | | | 93,615 | |
Valuation allowance | | | (105,258 | ) | | (93,615 | ) |
| | | | | | |
Net deferred tax assets | | $ | — | | $ | — | |
| | | | | | |
Our deferred tax assets have been fully reserved in both 2008 and 2007 since their ultimate future realization cannot be assured. The valuation allowance increased by $11.6 million for the year ended December 31, 2008. We presently have available for federal income tax purposes, approximately $241.0 million of net operating loss carry-forwards and $10.0 million of research and experimentation credit carry-forwards, which expire beginning in 2009 through 2028. However, as a result of changes in our ownership since inception, the amount of these carry-forwards available to offset future taxable income and income taxes could be subject to annual limitations under IRC Section 382. We may be subject to the alternative minimum tax regardless of our net operating loss carry-forwards.
12. Defined Contribution Plan
We have a 401(k) plan that is available to all employees. Employee contributions are voluntary and are determined on an individual basis up to the amount allowable under federal regulations. Employer
96
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
12. Defined Contribution Plan (Continued)
contributions to the plan are at the discretion of the Board of Directors and vest over a seven year period beginning after the third year of eligibility. No employer contributions have been made to date.
13. Commitments and Contingencies
Contract Research Organizations. We utilize independent contract research organizations ("CROs") to perform the clinical trials of our biological drug candidates to utilize their testing expertise and to ensure the objectivity of the clinical results. Under the terms of these agreements, we design the protocol regarding the testing to be performed, and the CRO enrolls the patients and testing sites, administers the trial, performs statistical analysis of the results, and compiles the final report.
We pay fees directly to the CROs for their professional services, which may be payable upon specified trial milestones or as they provide services, depending on the structure of the contract. We are also responsible for reimbursing the CROs for certain pass thru expenses they incur in administering the trial. The timing of our payments to the CROs is dependent upon the progress of the various trials, which is highly variable dependent upon the speed with which the CROs are able to enroll patients and testing sites. As such, we are unable to specifically predict the timing of future payments to CROs.
As of December 31, 2008, we had contracted with CROs to perform six clinical trials which were in varying stages of completion. The total contracted payments to CROs under these agreements were $85.7 million, of which we had incurred $40.1 million as of that date. Although we cannot directly control the timing of the remaining payments, based on our estimates and assumptions as of December 31, 2008, we expect to make payments to CROs as follows:
| | | | |
| | Estimated CRO
Payments | |
|---|
2009 | | $ | 19,172 | |
2010 | | | 15,976 | |
2011 | | | 9,586 | |
| | | | |
| | $ | 44,734 | |
| | | | |
Clinical Manufacturing Services Agreement. In 2005, we contracted with Lonza Walkersville, Inc. ("Lonza") to provide lot screening services for the isolation, growth, and differentiation of the MSCs to be used in our biological drug candidates. Due to the long production cycle for MSCs, we are obligated to provide Lonza a twelve month future forecast production plan for our MSC requirements. Lonza then produces the MSC doses according to our production plan at a fixed price per dose. The price per dose is based upon our volume commitment level.
In June 2008, the Clinical Manufacturing Services Agreement with Lonza was amended such that the term of the agreement is five years, provided that we continue to provide a forecast production plan to Lonza. We have the right to terminate the contract with thirty days written notice. In the event we terminate the contract, we are obligated for any in-process production as of the termination date that has been initiated according to our production forecast. Accordingly, our minimum contractual
97
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
13. Commitments and Contingencies (Continued)
commitment under this contract is limited to in-process production at any given time. Our obligation would not extend for a period longer than twelve months, which is the maximum outlook of the production forecast that we provide to Lonza.
Our expenses under this agreement were $15.6 million during 2008. Based on our current forecast production plan, we estimate that our future minimum contractual commitments under this contract were $14.5 million as of December 31, 2008, all of which we expect to be incurred within 2009.
Leases. During 2006, we entered into a sublease agreement for approximately 61,000 square feet of laboratory, production, warehouse and office space in Columbia, Maryland. We have also entered into a direct lease with the owner of this facility that is effective upon the expiration of the sublease and expires in July 2016. As of December 31, 2008, we had an outstanding letter of credit of $130 that was used in lieu of a security deposit for this lease and which is fully collateralized by restricted cash. During 2009, following the expiration of the sublease agreement, we have increased the letter of credit to $591 according to the terms of the direct lease with the owner of the facility. The increased letter of credit has been fully collateralized by restricted cash.
The future minimum lease payments due under the operating lease for this facility are as follows:
| | | | |
| | Columbia
Facility | |
|---|
2009 | | $ | 978 | |
2010 | | | 1,056 | |
2011 | | | 1,082 | |
2012 | | | 1,109 | |
2013–2016 | | | 3,999 | |
| | | | |
| | $ | 8,224 | |
| | | | |
As discussed in Note 3, Segment Reporting and Discontinued Operations, we have executed an Asset Purchase Agreement for the sale of our Osteocel asset disposal group. Pursuant to the Asset Purchase Agreement, NuVasive, Inc has agreed to assume both the lease and sublease to the Columbia facility concurrent with the manufacturing asset closing under that agreement, which we expect to occur during 2009. The amount of our future lease payments for the Columbia facility is dependent upon the exact timing of the manufacturing assets closing.
We are currently evaluating various strategic alternatives for the facilities we will occupy following the manufacturing assets closing, but no decision has yet been made with respect to any such alternative.
98
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
13. Commitments and Contingencies (Continued)
We also have entered into various financing arrangements to lease laboratory and other equipment. The terms of these facilities and equipment leases are considered capitalized leases, and the following amounts are included in our balance sheets at December 31, 2008 and 2007:
| | | | | | | |
| | 2008 | | 2007 | |
|---|
Facilities leases | | $ | — | | $ | 8,568 | |
Equipment leases | | | 26 | | | 3,157 | |
| | | | | | |
| | | 26 | | | 11,725 | |
Less—accumulated amortization | | | (17 | ) | | (11,098 | ) |
| | | | | | |
Leased property and equipment, net | | $ | 9 | | $ | 627 | |
| | | | | | |
Future minimum lease payments under these capitalized facilities and equipment arrangements are as follows:
| | | | |
| | Equipment | |
|---|
2009 | | $ | 8 | |
2010 | | | 3 | |
| | | | |
| | | 11 | |
Less interest | | | (2 | ) |
| | | | |
Present value of minimum lease payments | | | 9 | |
Less long-term portion | | | (3 | ) |
| | | | |
Capital lease obligations, current | | $ | 6 | |
| | | | |
Technology Transfer and License Agreement. In 1994, we entered into a Technology Transfer and License Agreement with Case Western Reserve University ("CWRU") under which we purchased rights to certain mesenchymal stem cell and related technology and patents. We are required to pay royalties on revenues related to CWRU developed technology, with minimum royalties of $50 per year. We paid CWRU $64 in 2008 and $50 in both 2007 and 2006 under this agreement.
14. Fair Value
In September 2006, the FASB issued Statement No. 157, "Fair Value Measurements," or SFAS No. 157. SFAS No. 157 establishes a standard framework for measuring fair value in generally accepted accounting principles, clarifies the definition of "fair value" within that framework, and expands disclosures about the use of fair value measurements. We adopted SFAS No. 157 in the first quarter of 2008 with regard to all financial assets and liabilities in our financial statements going forward, and, consistent with FASB Staff Position 157-2,"Effective Date of FASB Statement No. 157," we have elected to delay the adoption of SFAS No. 157 for non-financial assets and liabilities not recognized or disclosed at fair value on a recurring basis until the first quarter of 2009. The adoption of SFAS No. 157 had no material impact on our financial statements.
99
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
14. Fair Value (Continued)
Fair value is defined as the price at which an asset could be exchanged or a liability transferred (an exit price) in an orderly transaction between knowledgeable, willing parties in the principal or most advantageous market for the asset or liability. Where available, fair value is based on observable market prices or parameters or derived from such prices or parameters. Where observable prices or inputs are not available, valuation models are applied.
Financial assets recorded at fair value in the accompanying financial statements are categorized based upon the level of judgment associated with the inputs used to measure their fair value. Hierarchical levels, defined by SFAS No. 157 and directly related to the amount of subjectivity associated with the inputs to fair valuation of these assets and liabilities, are as follows:
| | |
| |
|
|---|
| Level 1— | | Inputs are unadjusted, quoted prices in active markets for identical assets at the reporting date. Active markets are those in which transactions for the asset or liability occur in sufficient frequency and volume to provide pricing information on an ongoing basis. |
|
|
The fair valued assets we hold that are generally included in this category are money market securities where fair value is based on publicly quoted prices. |
Level 2— |
|
Inputs are other than quoted prices included in Level 1, which are either directly or indirectly observable for the asset or liability through correlation with market data at the reporting date and for the duration of the instrument's anticipated life. |
|
|
The fair valued assets we hold that are generally included in this category are investment grade auction rate certificates and commercial paper where fair value is based on valuation methodologies such as models using observable market inputs, such as benchmark yields, reported trades, broker/dealer quotes, bids, offers and other reference data. |
Level 3— |
|
Unobservable inputs that are supported by little or no market activity and that are significant to the fair value of the assets or liabilities and which reflect management's best estimate of what market participants would use in pricing the asset or liability at the reporting date. Consideration is given to the risk inherent in the valuation technique and the risk inherent in the inputs to the model. |
We carry no investments classified as Level 3.
Assets and liabilities measured at fair value on a recurring basis are summarized below:
| | | | | | | | | | | | | | |
| | Fair value measurements at December 31, 2008 using | |
|---|
| | December 31,
2008 | | Quoted prices in
active markets for
identical assets
(Level 1) | | Significant other
observable
inputs
(Level 2) | | Significant
unobservable
inputs
(Level 3) | |
|---|
Assets: | | | | | | | | | | | | | |
| | Overnight securities included in Cash | | $ | 929 | | $ | 929 | | $ | — | | $ | — | |
| | Investments available for sale | | | 61,298 | | | — | | | 61,298 | | | — | |
100
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
14. Fair Value (Continued)
Investments available for sale consisted of the following as of December 31, 2008 and 2007:
| | | | | | | | | | | | | | |
| | 2008 | | 2007 | |
|---|
| | Cost | | Fair Value | | Cost | | Book Value | |
|---|
Cash equivalents: | | | | | | | | | | | | | |
| | Money market funds | | $ | 33,436 | | $ | 33,436 | | $ | 4,760 | | $ | 4,760 | |
| | Commercial paper | | | 11,187 | | | 11,195 | | | — | | | — | |
| | | | | | | | | | |
| | | 44,623 | | | 44,631 | | | 4,760 | | | 4,760 | |
Short term investments: | | | | | | | | | | | | | |
| | Auction rate certificates | | | 3,100 | | | 3,100 | | | 12,700 | | | 12,700 | |
| | Corporate notes and bonds | | | 1,921 | | | 1,921 | | | — | | | — | |
| | US government agencies | | | 11,621 | | | 11,646 | | | — | | | — | |
| | | | | | | | | | |
| | | 16,642 | | | 16,667 | | | 12,700 | | | 12,700 | |
| | | | | | | | | | |
Total investments available for sale | | $ | 61,265 | | $ | 61,298 | | $ | 17,460 | | $ | 17,460 | |
| | | | | | | | | | |
| | | | | | | | | | | | | | |
| | 2008 | | 2007 | |
|---|
| | Cost | | Fair Value | | Cost | | Book Value | |
|---|
Maturities: | | | | | | | | | | | | | |
| | Within 3-months | | $ | 46,826 | | $ | 46,832 | | $ | 17,460 | | $ | 17,460 | |
| | Between 3–12 months | | | 10,208 | | | 10,190 | | | — | | | — | |
| | Between 1–2 years | | | 4,231 | | | 4,276 | | | — | | | — | |
| | | | | | | | | | |
| | Total investments available for sale | | $ | 61,265 | | $ | 61,298 | | $ | 17,460 | | $ | 17,460 | |
| | | | | | | | | | |
101
Table of Contents
OSIRIS THERAPEUTICS, INC.
NOTES TO FINANCIAL STATEMENTS (Continued)
YEARS ENDED DECEMBER 31, 2008, 2007 AND 2006
(amounts in thousands, except for share and per share data)
15. Quarterly Financial Data (Unaudited)
Following is a summary of our unaudited quarterly results for the years ended December 31, 2008 and 2007:
| | | | | | | | | | | | | | |
| | First
Quarter | | Second
Quarter | | Third
Quarter | | Fourth
Quarter | |
|---|
2008 | | | | | | | | | | | | | |
| | Revenues | | $ | 362 | | $ | 2,530 | | $ | 995 | | $ | 6,157 | |
| | Research and development expenses | | | 16,694 | | | 19,048 | | | 18,592 | | | 15,563 | |
| | General and administrative expenses and fees | | | 2,608 | | | 1,782 | | | 1,887 | | | 2,309 | |
| | Loss from continuing operations | | | (19,149 | ) | | (18,472 | ) | | (19,903 | ) | | (11,893 | ) |
| | Income (loss) from discontinued operations | | | 3,549 | | | 3,104 | | | 25,155 | | | 4,117 | |
| | Net (loss) income | | | (15,600 | ) | | (15,368 | ) | | 5,252 | | | (7,776 | ) |
**Loss per share from continuing operations, basic and diluted | | |
(0.60 |
) | |
(0.58 |
) | |
(0.63 |
) | |
(0.37 |
) |
**Income per share from discontinued operations, basic and diluted | | | 0.11 | | | 0.10 | | | 0.80 | | | 0.13 | |
**Net (loss) income per share, basic and diluted | | | (0.49 | ) | | (0.48 | ) | | 0.17 | | | (0.24 | ) |
2007 | | | | | | | | | | | | | |
| | Revenues | | $ | 279 | | $ | 295 | | $ | 294 | | $ | 1,180 | |
| | Research and development expenses | | | 8,494 | | | 10,440 | | | 10,842 | | | 17,364 | |
| | General and administrative expenses and fees | | | 1,506 | | | 1,501 | | | 1,294 | | | 1,770 | |
| | Loss from continuing operations | | | (10,064 | ) | | (12,064 | ) | | (12,172 | ) | | (23,558 | ) |
| | (Loss) income from discontinued operations | | | (1,437 | ) | | 1,606 | | | 1,782 | | | 1,986 | |
| | Net loss | | | (11,501 | ) | | (10,458 | ) | | (10,390 | ) | | (21,572 | ) |
**Loss per share from continuing operations, basic and diluted | | |
(0.37 |
) | |
(0.43 |
) | |
(0.42 |
) | |
(0.80 |
) |
**(Loss) income per share from discontinued operations, basic and diluted | | | (0.05 | ) | | 0.06 | | | 0.06 | | | 0.07 | |
**Net loss per share, basic and diluted | | | (0.42 | ) | | (0.37 | ) | | (0.36 | ) | | (0.73 | ) |
- **
- Loss per share is calculated on a quarterly basis and may not be additive to year-to-date amounts.
102
Table of Contents
PART III
Certain information required in Part III is omitted from this report, but is incorporated by reference from our definitive proxy statement for the 2009 Annual Meeting of Stockholders anticipated to be filed within 120 days after the end of our fiscal year ended December 31, 2008, pursuant to Regulation 14A with the Securities and Exchange Commission.
ITEM 9. CHANGES IN AND DISAGREEMENTS WITH ACCOUNTANTS ON ACCOUNTING AND FINANCIAL DISCLOSURE
None.
ITEM 9A. CONTROLS AND PROCEDURES
Evaluation of Disclosure Controls and Procedures. An evaluation of the effectiveness of the design and operation of our "disclosure controls and procedures" (as defined in Rule 13a-15(e) under the Securities Exchange Act of 1934, as amended), as of the end of the period covered by this Annual Report on Form 10-K was made under the supervision and with the participation of our management, including our Chief Executive Officer and Chief Financial Officer. Based upon this evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures (a) are effective to ensure that information required to be disclosed by us in reports filed or submitted under the Securities Exchange Act of 1934 is timely recorded, processed, summarized and reported and (b) include, without limitation, controls and procedures designed to ensure that information required to be disclosed by us in reports filed or submitted under the Securities Exchange Act of 1934 is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, as appropriate to allow timely decisions regarding required disclosure.
Management's Report on Internal Control over Financial Reporting. Management's report on internal control over financial reporting is included in "Item 8. Financial Statements and Supplementary Data."
Changes in Internal Control over Financial Reporting. There have not been any changes in our internal control over financial reporting that occurred during the year ended December 31, 2008 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
ITEM 9B. OTHER INFORMATION
None.
ITEM 10. DIRECTORS, EXECUTIVE OFFICERS AND CORPORATE GOVERNANCE
The information contained in our proxy statement under the captions "Information About the Board of Directors and Committees," "Corporate Governance," "Executive Officers and Compensation" and "Section 16(a) Beneficial Ownership Reporting Compliance" is incorporated herein by reference.
We have adopted the Osiris Therapeutics, Inc. Code of Business Conduct & Ethics for Members of the Board of Directors and Executive Officers and the Osiris Therapeutics, Inc. Code of Conduct which applies to all our employees and members of the Board of Directors. These policies are publicly available on our website athttp://www//investor.osiris.com/documents.cfm.
103
Table of Contents
ITEM 11. EXECUTIVE COMPENSATION
The information contained in our proxy statement under the caption "Executive Officers and Compensation" is incorporated herein by reference.
ITEM 12. SECURITY OWNERSHIP OF CERTAIN BENEFICIAL OWNERS AND MANAGEMENT AND RELATED STOCKHOLDER MATTERS
The information contained in our proxy statement under the captions "Security Ownership of Certain Beneficial Owners and Management" and this Annual Report on Form 10-K under the caption "Part II—Item 5. Market for the Registrant's Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities—Securities Authorized for Issuance under Equity Compensation Plans" is incorporated herein by reference.
ITEM 13. CERTAIN RELATIONSHIPS AND RELATED TRANSACTIONS, AND DIRECTOR INDEPENDENCE
The information contained in our proxy statement under the caption "Executive Officers and Compensation—Certain Relationships and Related Party Transactions," "Information About the Board of Directors and Committees" and "Corporate Governance" is incorporated herein by reference.
ITEM 14. PRINCIPAL ACCOUNTING FEES AND SERVICES
The information contained in our proxy statement under the caption "Auditor Services" is incorporated herein by reference.
104
Table of Contents
PART IV
ITEM 15. EXHIBITS, FINANCIAL STATEMENT SCHEDULES
- (a)
- The following documents are filed as part of this report:
- 1.
- The following financial statements are included in Item 8 of this Annual Report:
Management's Report on Internal Control over Financial Reporting
Report of Independent Registered Public Accounting Firm
Balance Sheets as of December 31, 2008 and 2007
Statements of Operations for the years ended December 31, 2008, 2007 and 2006
Statements of Stockholders' Equity (Deficit) for the years ended December 31, 2008, 2007 and 2006
Statements of Cash Flows for the years ended December 31, 2008, 2007 and 2006
Notes to Financial Statements
Schedules not listed above have been omitted because the information required to be set forth therein is not applicable.
| | |
Exhibit
Number | | Description of Exhibit |
|---|
| 3.1† | | Amended and Restated Certificate of Incorporation of the Registrant. |
| 3.2† | | Amended and Restated Bylaws of the Registrant. |
| 4.1† | | Form of Common Stock Certificate. |
| 10.1† | | Amended and Restated 1994 Stock Incentive Plan, as amended. |
| 10.2 | | Amended and Restated 2006 Omnibus Plan. (Incorporated herein by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q filed by the Registrant with the SEC on August 11, 2008). |
| 10.3† | | Director Compensation Policy. |
| 10.4† | | Employment Agreement by and between the Registrant and C. Randal Mills, Ph.D., dated as of May 15, 2004. |
| 10.6† | | Employment Agreement by and between the Registrant and Harry Carmitchel, dated as of September 1, 2004. |
| 10.16† | | Investor Rights Agreement by and between the Registrant and JCR Pharmaceuticals, Inc. dated August 26, 2003. |
| 10.17†* | | License Agreement by and between the Registrant and JCR Pharmaceuticals, Inc. dated August 26, 2003. |
| 10.19† | | Technology Transfer and License Agreement by and between the Registrant and Case Western University, dated as of January 1, 1993, as amended. |
| 10.31† | | Warrant to Purchase up to 1,000,000 shares of Common Stock granted by Registrant to Peter Friedli, dated May 24, 2006. |
| 10.34† | | Employment Agreement, dated July 31, 2006, by and between the Registrant and Lode Debrabandere. |
| 10.35 | | Letter Agreement, dated June 6, 2007, by and between Friedli Corporate Finance, Inc. and Osiris Therapeutics, Inc. (Incorporated herein by reference to Exhibit 10.1 to the Current Report on Form 8-K filed by the Registrant with the SEC on June 7, 2007). |
105
Table of Contents
| | |
Exhibit
Number | | Description of Exhibit |
|---|
| 10.36 | | Form of Subscription Agreement, dated June 6, 2007, by and between the Registrant and Private Placement Investor (Incorporated herein by reference to Exhibit 10.2 to the Current Report on Form 8-K filed by the Registrant with the SEC on June 7, 2007). |
| 10.37 | | Amendment to Lock-up Agreement, acknowledged September 20, 2007, by and between the Registrant and Peter Friedli and Venturetec, Inc., (Incorporated herein by reference to Exhibit 10.1 to the Current Report on Form 8-K filed by the Registrant with the SEC on September 20, 2007). |
| 10.38 | | Letter Agreement, dated October 5, 2007, between Friedli Corporate Finance, Inc. and the Registrant (together with forms of subscription agreements and promissory note), (Incorporated herein by reference to Exhibit 10.1 to the Current Report on Form 8-K filed by the Registrant with the SEC on October 10, 2007). |
| 10.39 | | Employment Separation Agreement and Release, dated November 23, 2007, by and between the Registrant and Cary J. Claiborne (Incorporated herein by reference to Exhibit 10.1 to the Current Report on Form 8-K filed by the Registrant with the SEC on November 23, 2007). |
| 10.40 | | Form of Agreement, dated December 19, 2007, by and between the Registrant and the holders of Convertible Promissory Notes (Incorporated herein by reference to Exhibit 10.3 to the Registration Statement on Form S-3 filed by the Registrant with the SEC on January 18, 2008). |
| 10.41 | | Form of Subscription Agreement, dated December 19, 2007, by and between the Registrant and Private Placement Investor (Incorporated herein by reference to the form of Subscription Agreement attached as Exhibit 10.2 to the Current Report on Form 8-K filed by the Registrant with the SEC on June 7, 2007). |
| 10.42 | | Termination Agreement, dated December 31, 2007, by and between the Registrant and Boston Scientific Corporation (Incorporated herein by reference to Exhibit 10.42 to the Annual Report on Form 10-K filed by the Registrant with the SEC on March 17, 2008). |
| 10.43 | | Promissory Note, dated December 31, 2007, made by the Registrant in favor of Boston Scientific Corporation (Incorporated herein by reference to Exhibit 10.43 to the Annual Report on Form 10-K filed by the Registrant with the SEC on March 17, 2008). |
| 10.44 | | Award/Contract, dated January 3, 2008, issued by the U.S. Army Space & Missile Defense Command to the Registrant (Incorporated herein by reference to Exhibit 10.1 to the Current Report on Form 8-K filed by the Registrant with the SEC on January 4, 2008). |
| 10.45 | | Form of Subscription Agreements, entered into on March 19 and March 24, 2008, by and between the Registrant and certain non-U.S. Purchasers in connection with the issuance and sale of $8.0 million in Convertible Promissory Notes in the aggregate (Incorporated herein by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q filed by the Registrant with the SEC on May 12, 2008). |
106
Table of Contents
| | |
Exhibit
Number | | Description of Exhibit |
|---|
| 10.46 | | Form of 2% Convertible Promissory Notes of the Registrant, dated March 19 and March 24, 2008, issued in the aggregate principal amount of $8 million to certain non-U.S. Purchasers (Incorporated herein by reference to Exhibit 10.2 to the Quarterly Report on Form 10-Q filed by the Registrant with the SEC on May 12, 2008). |
| 10.48 | | Asset Purchase Agreement, dated May 8, 2008, by and between the Registrant and NuVasive, Inc. (Incorporated herein by reference to Exhibit 10.1 to the Current Report on Form 8-K filed by the Registrant with the SEC on May 12, 2008). |
| 10.49 | | Form of Company Voting and Support Agreement, dated May 8, 2008, by and among each of Peter Friedli, Venturetec, Inc., U.S. Venture 05, Inc., Joyce, Ltd. and C. Randal Mills, Ph.D., and NuVasive, Inc. (Incorporated herein by reference to Exhibit 10.2 to the Current Report on Form 8-K filed by the Registrant with the SEC on May 12, 2008). |
| 10.50 | | Form of Subscription Agreements, dated June 12, 2008 and June 30, 2008, respectively, by and between the Registrant and certain non-U.S. purchasers in connection with the issuance and sale of Promissory Notes in the aggregate principal amount of $5.5 million (Incorporated herein by reference to Exhibit 10.4 to the Quarterly Report on Form 10-Q filed by the Registrant with the SEC on August 11, 2008). |
| 10.51 | | Form of Promissory Notes of the Registrant, dated June 12, 2008 and June 30, 2008, issued to certain non-U.S. purchasers in the aggregate principal amount of $5.5 million (Incorporated herein by reference to Exhibit 10.5 to the Quarterly Report on Form 10-Q filed by the Registrant with the SEC on August 11, 2008). |
| 10.52** | | Manufacturing Agreement, dated as of July 24, 2008, by and between the Registrant and NuVasive, Inc. (Incorporated herein by reference to Exhibit 10.1 to the Quarterly Report on Form 10-Q filed by the Registrant with the SEC on November 10, 2008). |
| 10.53** | | Amendment to Manufacturing Agreement, dated as of September 30, 2008, by and between Registrant and NuVasive, Inc. (Incorporated herein by reference to Exhibit 10.2 to the Quarterly Report on Form 10-Q filed by the Registrant with the SEC on November 10, 2008). |
| 10.54** | | Amendment to Asset Purchase Agreement, dated as of September 30, 2008, by and between Registrant and NuVasive, Inc. (Incorporated herein by reference to Exhibit 10.3 to the Quarterly Report on Form 10-Q filed by the Registrant with the SEC on November, 2008). |
| 10.55 | | Employment Agreement by and between Osiris Therapeutics, Inc. and Richard W. Hunt entered into as of July 23, 2008 (Incorporated herein by reference to Exhibit 10.1 to the Current Report on Form 8-K filed by the Registrant with the SEC on July 29, 2008). |
| 10.56*** | | Collaboration Agreement, dated October 31, 2008, by and between the Registrant and Genzyme Corporation (filed herewith). |
| 11.1.1 | | Statement re: Computation of Per Share Loss (included in Note 2 to Financial Statements included in Part II Item 8 herein). |
| 23.1.1 | | Consent of Independent Registered Public Accounting Firm (filed herewith). |
| 31.1.1 | | Rule 15d-14(a) Certification of C. Randal Mills, President and Chief Executive Officer (filed herewith). |
107
Table of Contents
| | |
Exhibit
Number | | Description of Exhibit |
|---|
| 31.2.1 | | Rule 15d-14(a) Certification of Richard W. Hunt , Chief Financial Officer (filed herewith). |
| 32.1.1 | | Section 1350 Certification of C. Randal Mills, Chief Executive Officer, and Richard W. Hunt., Chief Financial Officer (filed herewith). |
- †
- Incorporated herein by reference to corresponding Exhibit to the Registrant's Registration Statement on Form S-1, which was declared effective by the SEC on August 3, 2006.
- *
- Confidential treatment has been granted for certain portions thereof pursuant to an order of the United States Securities and Exchange Commission issued in response to a Confidential Treatment Application filed by the Registrant in connection with its Registration Statement on Form S-1, declared effective on August 3, 2006.
- **
- Confidential treatment has been granted for certain portions thereof pursuant to an order of the United States Securities and Exchange Commission issued in response to a Confidential Treatment Application filed by the Registrant in connection with its Quarterly Report on Form 10-Q, for the quarterly period ended September 30, 2008.
- ***
- Confidential portions omitted and filed separately with the United States Securities and Exchange Commission pursuant to Rule 24b-2 promulgated under the Securities Exchange Act of 1934, as amended.
108
Table of Contents
Signatures
Pursuant to the requirements of Section 13 or 15(d) of the Securities Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | |
| | | OSIRIS THERAPEUTICS, INC. |
March 16, 2009 |
|
By: |
|
/s/ C. RANDAL MILLS
C. Randal Mills, Ph.D.
President & Chief Executive Officer |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| | | | |
| |
| |
|
|---|
|
|
|
|
|
/s/ C. RANDAL MILLS
C. Randal Mills, Ph.D. | | President and Chief Executive Officer (principal executive officer) | | March 16, 2009 |
/s/ RICHARD W. HUNT
Richard W. Hunt |
|
Chief Financial Officer (principal financial officer) |
|
March 16, 2009 |
/s/ PHILIP R. JACOBY, JR.
Philip R. Jacoby, Jr. |
|
Vice President of Finance & Secretary (principal accounting officer) |
|
March 16, 2009 |
/s/ GREGORY H. BARNHILL
Gregory H. Barnhill |
|
Director |
|
March 16, 2009 |
/s/ PETER FRIEDLI
Peter Friedli |
|
Director |
|
March 16, 2009 |
/s/ FELIX GUTZWILLER
Felix Gutzwiller |
|
Director |
|
March16, 2009 |
/s/ JAY M. MOYES
Jay M. Moyes |
|
Director |
|
March 16, 2009 |
109