UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM 10-K
Q ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the Fiscal Year Ended: December 31, 2013
or
¨ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
| POWER OF THE DREAM VENTURES, INC. |
| (Exact name of registrant as specified in its charter) |
| Delaware | 001-52289 | 51-0597895 |
(State or other jurisdiction of incorporation or organization) | (Commission File Number) | (I.R.S. Employer Identification Number) |
| 1095 Budapest, Soroksari, ut 94-96, Hungary |
| (Address of principal executive offices, including zip code) |
| +36-1-456-6061 |
| (Registrant’s telephone number, including area code) |
Securities registered pursuant to Section 12(b) of the Act: None
Securities registered pursuant to Section 12(g) of the Act:Common Stock, $0.001 par value
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 the Securities Act. Yes¨ Noþ
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act. Yesþ No¨
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the last 90 days.
Yesþ No¨
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the registrant was required to submit and post such files). Yesþ No¨
Indicate by check mark if disclosure of delinquent filers pursuant to Item 405 of Regulation S-K (§229.405 of this chapter) is not contained herein, and will not be contained, to the best of registrant’s knowledge, in definitive proxy or information statements incorporated by reference in Part III of this Form 10-K or any amendment to this Form 10-K. Yes¨ Noþ
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, or a smaller reporting company. See definition of “large accelerated filer,” “accelerated filer” and “smaller reporting company” in Rule 12b-2 of the Exchange Act.
| Large Accelerated Filer¨ | Accelerated Filer¨ |
| | |
| Non-Accelerated Filer¨ | Smaller reporting companyþ |
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act). Yes¨ Noþ
Issuer’s revenues for its most recent fiscal year were approximately $0.
The aggregate market value of the voting and non-voting common equity held by non-affiliates of the registrant on June 29, 2013, based on a closing price of $0.10 was approximately $3,500,000 As of April 14, 2014, the registrant had 46,500,896 shares of its common stock, par value $0.001 per share, outstanding.
Documents Incorporated By Reference: None.
POWER OF THE DREAM VENTURES, INC.
FOR THE FISCAL YEAR ENDED
DECEMBER 31, 2012
TABLE OF CONTENTS
| | | Page |
| PART I | | |
| | | |
| Item 1. | Business. | 5 |
| Item 1A. | Risk Factors. | 36 |
| Item 1B. | Unresolved Staff Comments. | 56 |
| Item 2. | Properties. | 56 |
| Item 3. | Legal Proceedings. | 56 |
| Item 4. | Mine Safety Disclosures. | 56 |
| | |
| PART II | |
| | | |
| Item 5. | Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities. | 57 |
| Item 6. | Selected Financial Data. | 58 |
| Item 7. | Management’s Discussion and Analysis of Financial Condition and Results Of Operations. | 58 |
| Item 7A. | Quantitative And Qualitative Disclosures About Market Risk. | 61 |
| Item 8. | Financial Statements and Supplementary Data. | 61 |
| Item 9. | Changes in and Disagreements With Accountants on Accounting and Financial Disclosure. | 61 |
| Item 9A. | Controls and Procedures. | 61 |
| Item 9B. | Other Information. | 63 |
| | | |
| PART III |
| | | |
| Item 10. | Directors, Executive Officers and Corporate Governance. | 63 |
| Item 11. | Executive Compensation. | 64 |
| Item 12. | Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters. | 65 |
| Item 13. | Certain Relationships and Related Transactions, and Director Independence. | 66 |
| Item 14. | Principal Accounting Fees and Services. | 66 |
| | | |
| PART IV |
| | | |
| Item 15. | Exhibits, Financial Statement Schedules. | 67 |
FORWARD LOOKING STATEMENTS
Included in this Form 10-K are “forward-looking” statements, as well as historical information. Although we believe that the expectations reflected in these forward-looking statements are reasonable, we cannot assure you that the expectations reflected in these forward-looking statements will prove to be correct. Our actual results could differ materially from those anticipated in forward-looking statements as a result of certain factors, including matters described in the section titled “Risk Factors.” Forward-looking statements include those that use forward-looking terminology, such as the words “anticipate,” “believe,” “estimate,” “expect,” “intend,” “may,” “project,” “plan,” “will,” “shall,” “should,” and similar expressions, including when used in the negative. Although we believe that the expectations reflected in these forward-looking statements are reasonable and achievable, these statements involve risks and uncertainties and we cannot assure you that actual results will be consistent with these forward-looking statements. We undertake no obligation to update or revise these forward-looking statements, whether to reflect events or circumstances after the date initially filed or published, to reflect the occurrence of unanticipated events or otherwise.
PART I
Item 1. Business.
Power of the Dream Ventures, Inc., a Delaware corporation (“PDV”, “We” or the “Company”) is a Hungarian-based holding company focused on technology acquisition and development enabling the delivery of revolutionary concepts and ready to market products to the international market place. We develop, acquire, license, or co-develop technologies that typically originate in Hungary that are in prototype stage based on existing patents; in prototype stage prior to patenting; existing products that require expansion capital to commercialize; emerging science and high-technology research projects that require help in patenting, developing the product and marketing, university spin-off technologies and ideas from the very early stages of what represents “disruptive technologies.”
We were incorporated in Delaware on August 17, 2006, under the name Tia V, Inc. Since inception, and prior to our acquisition of Vidatech on April 10, 2007, we were engaged solely in organizational efforts and obtaining initial financing. Our sole business purpose was to identify, evaluate and complete a business combination with an operating company.
On April 10, 2007, we completed our acquisition of Vidatech, Kft. (also known as Vidatech Technological Research and Development LLC) a limited liability company formed under the laws of the Republic of Hungary. Vidatech is a company formed for the purpose of investing in, acquiring, developing, licensing, and commercializing technologies developed in Hungary. In furtherance of its business, Vidatech provides research and development services to the companies from which it acquires technologies or participation interests in such technologies.
Through Vidatech, we aim to provide pro-active support for idea, research, start-up and expansion-stage technology companies having rights to technologies or intellectual properties which we believe to be potentially commercially viable, by offering a range of services designed to encourage and protect the continuing development and eventual commercialization of those technologies.
On September 28, 2012, the Company entered into an Acquisition Agreement and Plan of Merger, by and among the Company, as parent, Power of the Dream Ventures Acquisition Corp., a Delaware corporation and wholly-owned subsidiary of Parent (the “Merger Sub”) and Genetic Immunity, Inc., a Delaware corporation (“Genetic Immunity”), whereby, the Merger Sub merged with Genetic Immunity, and Genetic Immunity shall continue as a wholly-owned subsidiary of the Company (the “Merger”). The Merger became effective upon the filing of the Certificate of Merger with the Secretary of the State of Delaware on October 2, 2012.
OVERVIEW
The Company currently focuses on the operations of its wholly-owned subsidiary, Genetic Immunity, which is a clinical-stage biotechnology company focusing on the discovery, development and commercialization of a new class of immunotherapeutic biologics ("Immune Therapies" or "Therapeutic Vaccines") for the treatment of chronic viral infections, cancer and allergy. Our Immune Therapies are designed to intensify or boost specific immune responses to modify or control these presently incurable diseases. The Company’s main focus is completing Genetic Immunity’s clinical trials program and on commercialization tasks related to the Company’s lead product candidate DermaVir therapeutic HIV vaccine.
PRODUCTS
Our Lead Product Candidate – DermaVir HIV-specific Immune Therapy
| 1. | Summary of DermaVir Development Milestones |
We have been developing DermaVir for the treatment of HIV/AIDS.DermaVir is a patented Immune Therapy which, based on the Food and Drug Administration (“FDA”) classification, is a combination of our original biologic product (DermaVir) and our medical device (DermaPrep).DermaVir is administered topically with DermaPrep. DermaVir’s Active Pharmaceutical Ingredient (“API”) is a pDNA expressing the broadest HIV antigen repertoires and Virus-like Particles (“VLP”).
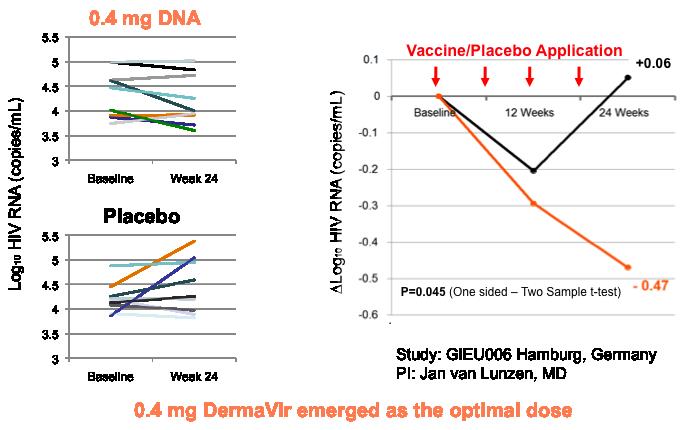
We demonstrated in Phase I and Phase II clinical tirals that DermaVir exhibits its specific pharmacologic effects by intensifying the patient’s own immune system to kill HIV-infected cells. Our clinical trials in approximate 70 HIV-infected patients demonstrated an excellent safety and tolerability in all doses. We found the optimal DermaVir dose for boosting HIV-specific T cell immune responses and demonstrated killing of HIV-infected cells by showing a statistically significant reduction of HIV-RNA in DermaVir treated patients compared to placebo.
DermaVir has a novel immunological mechanism of action different from antiretroviral drugs that kill the virus but cannot kill the infected cells. During effective Highly Active Antiretroviral Therapy (“HAART”), HIV-infected cells remain in the reservoirs. Even HAART intensification could not eliminate infected cells, decrease reservoirs or improve the immune system to fight against the virus. We believe that DermaVir treatments will reduce the amount of HIV-infected cells remaining in the reservoirs of HAART petients. This could provide additional treatment benefits by reducing the number of antiretroviral drugs that patients must take.
“Proof of concept” for the immunological and antiretroviral activities of our products was demonstrated in non-human primates with AIDS. This data showed that repeated DermaVir immunization resulted in a cumulative boosting of the HIV-specific cellular immunity without causing significant toxicities or adverse effects. After successful reconstitution of the monkey’s SIV-specific immune system, DermaVir immunized monkeys controlled their virus after interruption of HAART.
Among our animal studies, the non-human primate model is superior to other commonly used animal models (e.g., mice, rats, rabbits) because the disease progression and response to treatments in non-human primates is similar to the human disease. We believe that DermaVir has the potential to be the first Immune Therapeutic agent designed for the immune mediated control of viral replication by cytotoxic killing of HIV-infected cells. Currently, there are no approved Immune Therapy products on the market for HIV.
DermaVir was found to be safe and well tolerated at all doses in all clinical studies. A single treatment with DermaVir takes 20 minutes to apply, and left on the skin for three hours after which the patches are removed.. Our Phase I clinical results indicated an increase in the number of long-lived HIV-specific precursor T cells in all treated individuals in a dose-dependent manner. DermaVir-induced T cells remained in the body even after a one-year follow up, albeit at a lower level. These findings suggest that DermaVir boosted HIV-specific T cells similarly to that seen in primates. A separate Phase I/II study conducted by our collaborators in the US further confirmed the safety of repeated DermaVir vaccinations in combination with HAART (ACTG5176). We have recently completed Phase II clinical trial with DermaVir in Germany on a drug naïve patient population (36 HIV-infected individuals). These trials demonstrated excellent safety, immunogenicity and antiretroviral efficacy and have shown clinically significant median 70% viral load reduction in patients treated, as compared to placebo. This viral load reduction is the result of killing of HIV-infected cells that we could demonstrate even in the absence of HAART. Viral load suppression by DermaVir vaccinations occurs slowly, similarly to cancer vaccines as predicted by its mechanism of action, and that is different from antiretroviral drugs that kill the virus but do not kill HIV-infected cells. Consistent with our primate results, DermaVir immunizations alone did not suppress viral load to an undetectable level and did not increase CD4 T cell counts. These human clinical data appear to have confirmed and extended our pre-clinical results.
Although our clinical development program is not yet final, and the marketing approval has not yet been obtained, we believe that our preclinical and human studies performed to date support the Proof of Concept, which is a relationship between DermaVir-induced antiretroviral immune responses and clinical benefit. We believe that DermaVir may be most effective if it is initially administered every six weeks to intensify HIV-specific immunity then at every regular doctor’s visit (every three months) to maintain the immunity.
| 2. | Our DermaVir Family for Personalized Treatment |
DermaVir is based on HIV subtype-B that represents the majority of HIV infection within the major marketplaces including North America and Western Europe. In addition, we have developed an initial DermaVir Product Portfolio to provide optimal treatment for every HIV-infected patient by considering the diversity of the infected population.
HIV is a highly variable virus. We found that the therapeutic effectiveness of DermaVir depends on the HIV sequence replicating in the patient and the patient’s genetic background. DermaVir contains related HIV-specific antigen sequences characteristic for the different Clades (subtypes) of the virus. To ensure the optimal therapeutic benefit for every patient, the treating physician will enter the information on the patient’s HIV sequences and genetic background (HLA) into our eMINER software. After analyzing the data, eMINER will select the optimal DermaVir product from the DermaVir Product Family and also calculate the specific T cell epitopes that will play a role in the immune boosting of the individual patient. The treating physician can then make an educated decision for personalized treatment of the patient and could also prescribe the proper immune monitoring based on the predicted T cell epitopes.
The targeted epidemics and our initial Product Portfolio are summarized in the Table below. According to UNAIDS, subtype-B represents approximately four million individuals (Europe, North, Central and South America, Caribbean, Australia). Our other product candidates can provide optimized treatment options for an additional 24.7 million HIV-infected individuals (2009 UNAIDS Report and HIV-1 Global Distribution, IAVI report Aug 2003).
| Present DermaVir Product Family | Targeted epidemics | Infected population |
| DermaVir B | Europe, America, Caribbean, Australia | 4 million |
| DermaVir C | Sub-Saharan Africa, South-Asia | 22.5 million |
| DermaVir BC | China | 0.77 million |
| DermaVir BF | South-America | 1.4 million |
| DermaVir AB | Eastern Europe | 10,000 |
We are pursuing the development of a DermaVir Product Family based on subtypes for the following reasons:
| · | DermaVir Product Family provides an individually-optimized Immune Therapy to boost the immune system against the specific HIV replicating in the patients. The availability of subtype-specific products permits “personalized treatment” with DermaVir Product Family. HIV subtype of the infected individual is diagnosed with available sequencing of the virus. The patient’s genetic background (HLA type) can also be diagnosed with routine methods. The subtype or recombinant form (HIV diversity) plus the patient’s genetic background for antigen processing (patient diversity) together define the best-matching Product from the Family. Based on these data our eMINER can suggest the personalized treatment for the patient. |
| · | DermaVir Product Family also supports different pricing in the various markets. Differential pricing of drugs manufactured by the same company has resulted in the illegal re-importation of the cheaper products to North America and Western Europe. The subtype-optimized DermaVir products will, by medical necessity, differ in HIV subtypes by geographical markets, product supplied to developing countries should not be diverted to other markets. |
| · | We intend to seek additional patent protection for the API in our subtype-specific DermaVir Product Family and method for treatment, also within the target regions. This will allow the granting of manufacturing and marketing licenses to specific countries based on unmet medical needs while providing additional protection against re-importation of third country-manufactured products to the United States and E.U. |
| · | Based on our technology platform, manufacturing and control of the DermaVir Product Family will not require additional methods or technology. The same manufacturing technology, control methods and facility can be used for cGMP manufacturing of all our products. |
Prior to human trials, regulatory agencies require the demonstration of their quality, safety and efficacy initially in appropriate animal models. To demonstrate the mechanism of action, first we established in human primary cellsin vitro and in macaquesex vivo that DermaVir-expressing dendritic cells prime naïve T cells and induce both HIV-specific helper and cytotoxic T cells. Preclinical animal studies conducted in mice, rabbits and macaques consistently demonstrated thatin vivo DermaVir immunization targets the pDNA immunogen to the dendritic cells of the lymph nodes that express the pDNA-encoded antigens. Importantly, immune responses followingin vivo DermaVir immunization were shown to be similar toex vivo immunization with cultured dendritic cells “DCs”. Bothex vivo andin vivo DermaVir immunizations employed lymph node dendritic cells, expressed similar amounts of pDNA-derived antigens, and induced Th1-type antigen-specific CD4 T helper and CD8 cytotoxic T cells. The induction of Th1-type cellular immune responses by DermaVir was also confirmed by delayed-type hypersensitivity skin reaction (DTH) tests. Additional experiments showed thatin vivo DermaVir immunization leads to the induction of antigen-specific memory/precursor T cells. Recent animal studies have demonstrated that generation of highly protective immunity against smallpox required a live virus delivered via skin scarification. For DermaVir we used our platform technology, the modern version of the smallpox vaccination approach by employing a pDNA nanomedicine expressing complex VLP+ instead of a live virus and DermaPrep administration instead of skin scarification.
To study the safety of DermaVir vaccination, repeated immunization studies in good laboratory practice “GLP” were performed in swine and rabbits. These preclinical studies demonstrated that the major side effect of DermaVir vaccinations was erythema caused primarily by the DermaPrep administration. The erythema was transient and provided a mild inflammation, a desired side effect resulting in the activation of the Langerhans cells to seek and capture DermaVir nanoparticles applied under the patch.
To study the reproductive toxicity of DermaVir another repeated dose study GLP was performed in Wistar rats from the premating period through implantation. No significant differences between DermaVir treated and control animals were observed in body weights, food consumption, estrus cycle, fertility and mating index, or mating days until Day 0 postcoitus. Because no adverse effects were seen, the Maximum Tolerated Dose (MTD) cannot be determined, but is considered to be greater than 0.33 mg pDNA.
We have assessed thein vivo stability of our nanomedicine product. We could visualize the nanoparticles in the epidermis by two-photon microscopy over 120 minutes and demonstrate that DermaPrep skin preparation is essential for the effective penetration of the nanomedicine into the epidermis. The half-life of the nanomedicine in the lymph nodes was 11-12 days. The half-life of the nanoparticles in the lymph nodes depends on the migration of nanoparticle-containing Langerhans cells to the lymph nodes, intracellular degradation of pDNA, cytotoxic killing of antigen-presenting dendritic cells and clearance of dendritic cells.
The immunogenicity of DermaVir was investigated in uninfected and Simian Immunodeficiency Virus (“SIV”)-infected rhesus macaques with a DermaVir product formulated with a relevant plasmid DNA construct based on Simian Human Immunodeficiency Virus. The API of DermaVir used in all macaque studies expresses a VLP+ with the gag and pol genes homologous to SIV and an HIV env. The method for topical administration and the dose per skin surface in the macaque studies were similar to human trials. Proof of Concept efficacy studies in chronically SIV-infected macaques, some of them with AIDS, suggested that repeated DermaVir immunizations, alone or in combination with antiretroviral drugs, result in viral load reduction and survival benefit (see data in Lisziewicz et al AIDS 2005). DermaVir administered in combination with HAART boosted SIV-specific T cells that possessed significant antiretroviral activity demonstrated by maintenance of undetectable viral load after interruption of HAART in both chronically infected and late stage macaques. DermaVir treatment alone doubled the median survival time of infected macaques. These primate experiments provided the rationale to investigate repeated DermaVir immunizations in combination with HAART in HIV-infected human subjects.
| ii. | Summary of the Clinical Trials to Date |
As DermaVir associated clinical benefits were first seen in primates receiving HAART, the first two human studies were designed for the treatment of HIV-infected subjects receiving stable, fully suppressive HAART.
GIHU004 Trial to Evaluate the Tolerability and Safety of DermaVir in HIV-Infected Subject Currently Under Treatment with HAART (EudraCT Number 2004-001585-41, ClinicalTrials.gov ID NCT00712530).
The objective of the trial was to provide data on the safety, tolerability and immunogenicity of one DermaVir treatment in individuals with chronic HIV-1 infection treated with fully suppressive HAART.
This Phase I clinical study demonstrates that DermaVir was found to be safe and well tolerated at all doses. Vaccination did not affect HIV RNA (all subjects remained less than 50 copies/mL) and CD4+ counts. However, this therapeutic vaccination broadened and significantly increased the HIV-specific memory/precursor T cell pool, measured as the PHPC count, in a dose-dependent manner. The durability of vaccine-induced T cell responses was demonstrated after one year following a single vaccination in all treated individuals, albeit in significantly lower quantities (Lisziewicz et al. CROI2008 - Poster #715). These findings suggest that DermaVir boosted HIV-specific T cells similarly to that seen in primates.
Trial ACTG A5176. A Phase I/II, Randomized, Double-Blind Study to Evaluate the Safety, Tolerability, and Immunogenicity of DermaVir, in HIV-1-Infected Subjects Currently Under Treatment with HAART. (ClinicalTrials.gov IDNCT00270205 ).
The trial, conducted under a US Investigational New Drug “IND” application was sponsored by the US government (NIAID, NIH) enrolled 26 HIV-infected individuals on fully suppressive HAART to three cohorts in four leading HIV clinics located in the US. Based on the design, dose escalation to the last cohort implied that no subject in the current or lower dose cohort experienced a primary safety endpoint (i.e., no more than one subject in any cohort exhibited an adverse event greater than Grade 3).
The primary objective of this randomized, double-blind, placebo-controlled trial was to evaluate the safety and tolerability of multiple DermaVir treatments. Secondary objectives were to explore the immunogenicity of DermaVir for the treatment of individuals with chronic HIV-1 infection and HAART-induced durable reduction of viral replication.
The trial population comprised 26 HIV-infected men and women 18 to 50 years of age. HAART regimens were fully reduced within the 12 weeks prior to trial entry (Screening plasma HIV-1 level of less than 50 copies/mL,and CD4+ cell count greater than 350 cells/mm3 within 12 weeks prior to trial entry and nadir CD4+ cell count greater than 250 cells/mm3). The total treatment duration was 61 weeks. Each cohort’s treatment schedule was administered over a 13-week period, with an additional 48 weeks of follow-up for safety evaluations.
Twenty-six subjects were enrolled in the study. The primary endpoint was any possibly or definitely vaccine-related grade ≥3 adverse event (AE) appearing up to 28 days after the final study vaccination. No primary safety endpoints or AE-related study treatment changes / discontinuations have occurred. AE incidence was similar across groups (Rodriguez B. et al. XVIII International AIDS Conference, 2010 - Abstract # A-240-0111-10145).
DermaVir administration was associated with a trend toward greater HIV-specific, predominantly central memory T-cell responses. The intermediate DermaVir dose tended to show the greatest immunogenicity, consistent with previous studies in different HIV-infected patient populations.
Phase II Trial
Repeated DermaVir immunizations in chronically infected macaques in the absence of HAART transiently suppressed virus replication leading to improvement of median survival time from 18 to 38 weeks compared to no treatment. These primate experiments provided the rationale to investigate repeated DermaVir immunizations prior to initiation of HAART in HIV-infected individuals. As DermaVir immunizations in combination with HAART did not show any product- or administration-related AEs higher than grade 2, we developed a Phase II protocol to evaluate the safety and to test the immunogenicity and antiretroviral efficacy of repeated DermaVir immunizations.
GIEU-006: Randomized, Placebo-Controlled, Multi-Center Trial to Evaluate the Safety, Tolerability, Immunogenicity, and Antiretroviral Activity of DermaVir in Treatment-Naïve HIV-1-Infected Patients (EudraCT number: 2007-001955-20, ClinicalTrials.gov IDNCT00711230)
This study was conducted in Germany in accordance with European Medicines Agency (“EMA”) approval and Current Good Clinical Practices “cGCP”. GIEU-006, a Phase II randomized, placebo-controlled, dose-finding, double-blinded study was conducted to assess the safety, tolerability, immunogenicity, and antiretroviral activity of DermaVir therapeutic vaccine, for the treatment of antiretroviral therapy naïve adults with HIV-infection.
The study population comprised of 36 antiretroviral treatment naïve HIV-infected men and women 18-50 years of age. The patients were randomized into one of the six treatment arms to receive either DermaVir or placebo. Four immunizations were administered over an 18-week period with an identical follow up schedule continuing until week 24; patients were followed for an additional 24 weeks for safety and immunogenicity evaluations. An additional 234-week safety follow up is being performed (every 26 weeks) including virology, immunology, chemistry and hematology assessments and physical examinations.
Immunizations were done on days 0, 42, 84, and 126. The total number of patches that a patient received throughout the study is 8, 16, or 32 in the low, medium, and high dose arms, respectively. The same skin sites were used for all immunizations.
The primary endpoint of the study was safety at week 24. Adverse events solely attributed to DermaPrep and were analyzed separately in the Local Reaction Assessment.
Out of the 36, 34 subjects have reached week 24 (visit 10), the time point of the analysis of primary and secondary endpoints of the study. The data of the two subjects that did not reach visit 10 were used according to the statistical section of the protocol. One of the subjects got arrested after visit 7 and discontinued from the study; the other subject decided to start antiretroviral therapy despite a stable CD4 count greater than 600/µl.
We did not observe any greater than grade 2 adverse events including signs/symptoms, lab toxicities, and/or clinical events possibly or definitely related to study treatment any time from the first day of study treatment until 42 days after the last DermaVir administration. There was no premature discontinuation of immunizations at the patient’s request for reasons having to do with the effects, or the perceived effects, of DermaVir or the DermaPrep administration procedure.
No patients required termination from any further immunizations due to toxicity attributable to DermaVir product, or due to tolerability failure. No patients experienced Grade 3 or higher toxicity to be discussed regardless of study treatment attribution.
The overall adverse event profile of the treatment was found to be consisting of grade 1 and grade 2 events. Most of the events were judged by the investigators to be not related to the study treatment. Only one grade 2 adverse events judged as possibly related to treatment (Positive Gaenslen's Test) in the 0.2 mg DermaVir group. We did not find any significant differences between study arms and treatment groups.
The Figure above demonstrates the efficacy results of the Phase II trial
Phase IV trials following marketing approval will be necessary if we intend to further study the efficacy of DermaVir in different patient populations. Presentations and publications of such trials are essential to successful marketing of DermaVir. Therefore, our strategy will be to obtain marketing approval within the shortest period of time and consequently continue the clinical trials for the establishment of a large safety and efficacy database. Please note that such a database on DermaVir is expected to facilitate the approval of pipeline products since the differences between products will be only in the sequence of the plasmid DNA.
| iv. | Our DermaVir Product Development and Registration Strategy |
Our clinical development and market approval strategy is focusing on immune intensification with DermaVir in Conditional Marketing Approval. We believe that patients treated with such Immune Intensification will maintain an undetectable HIV-RNA level and have an increased HIV-specific T cell frequency. Achieving optimal T cell responses and elimination the majority of HIV-infected cells from the reservoir might lead to remission (functional cure) in some HIV-infected people. These people would stop their antiretroviral drug treatment (if any) and receive only immune boosting DermaVir treatments during regular doctor visits.
In addition to the foregoing reasons, we believe that the treatment for HIV/AIDS with DermaVir may be desirable to physicians and patients for the following reasons:
| · | Effectiveness based upon boosted natural immunity; |
| · | Delayed disease progression; |
| · | No interference with current or future drug-treatment options; |
| · | Infrequent administration of a patch (only for three hours) during regular office visits; |
| · | No fear of the side effects and acquiring resistance of anti-HIV drugs; and |
| · | Reimbursement by insurance. |
According to Datamonitor (Future of HIV Market 2011), some patients may prefer a once-monthly administration (even intravenous drug infusion in the doctor’s office) over daily oral HIV treatment, which makes topical DermaVir treatment very attractive.
We believe that DermaVir, when approved, may meet the needs of these market opportunities due to favorable safety, tolerability and efficacy characteristics in comparison to currently prescribed drugs. The major toxicities of DermaVir are not systemic, but limited to local skin reactions. It is conveniently administered during regular doctor visits. The efficacy is based on the induction of HIV specific precursor T cells that are long lasting. DermaVir is expected to be introduced to the market for the treatment of HIV in combination with the presently available drugs since none of them can reconstitute HIV-specific immunity.
DermaVir has the potential to be the first Immune Therapy registered for the treatment of HIV/AIDS. To reach this objective we will use a strategy combining the experiences of registration of antiretroviral drugs and biologics. We have worked closely with the FDA on studies required for both the Biologics Master File “BMF” and the Investigational New Drug “IND” applications and will continue this effort. The FDA has expressed the desire to work with the pharmaceutical industry in the development of useful immune-based therapies for HIV/AIDS that may contribute to the body’s own defense against HIV and improve clinical outcome over drug therapy alone. We intend to develop effective and safe Immune Therapy products to delay HIV disease in HIV infected individuals not treated with antiretroviral drugs.
We had a preliminary meeting with the European Medicines Agency (“EMA”) to discuss the clinical development plan of DermaVir. EMA ensured us to fully support the development of DermaVir and suggested to organize an official Scientific Advisory Meeting to obtain approval for our clinical development and Chemistry, Manufacturing and Controls (“CMC”) strategy. We intend to propose the development of DermaVir for immune intensification in frame of a Conditional Marketing Approval. During the treatments we intend to use eMINER to match the patient with the best vaccine selected from our DermaVir Product Portfolio. After the DermaVir induction phase (3 treatments every 6 weeks) we expect DermaVir boosting of HIV-specific immunity compared to placebo. This is followed by the DermaVir maintenance phase (4 treatments every year). We believe that the maintenance not only maintains the high immune responses in the patients but also decreases the number of infected cells in the reservoir. This will be clinically demonstrated by maintenance of undetectable HIV-RNA after decreasing the number of drugs used in the HAART regimen. The primary endpoint of the pivotal trial will be non-inferiority between DermaVir + 1 Drug and HAART (standard of care). Additional treatment benefits include less toxicity in the DermaVir arm and lower pill burden.
We believe that we will get approval for our DermaVir Product Family together with our biomarker strategy and that regulatory agencies will not ask us to conduct separate clinical development programs with the different members in our DermaVir Product Family. The size of DermaVir Product Family initially will be less than 10, which is less than we envision to be expanded after emergence of new HIV variants, because it will provide optimal immunogenicity for selected HIV-infected patients, no difference in toxicities and all members in the DermaVir Product Family is produced by the same manufacturing and control methods. The future of HIV therapy, similar to several other indications, is already moving to personalized treatment. Pharmacogenetics screening is already used prior to initiation of some drug treatment in the US and EU (Datamonitor, Future of HIV Market 2011). We envision that our personalized DermaVir Immune Therapy will combine the scientific knowledge and our product portfolio to select the safest and most effective treatment of all HIV-infected patients.
According to EMA regulations, investigational drugs treating HIV infection with appropriate phase II clinical results, a favorable toxicity profile and good rationale for efficacy may receive Conditional Marketing Approval. Based on our clinical results with DermaVir parallel to the Conditional Marketing Approval process, we are planning to apply for Breakthrough Therapy Designation at the FDA.
Our Immune Therapy Technology Platform
The human immune system is well prepared to fight against diseases and most of the time does it successfully. In people with chronic diseases the immune system fights against such diseases but often cannot prevail. To live a better and longer life people boost their immune systems with vitamins, dietary supplements and herbs. We designed our Immune Therapy technology to specifically boost the patients’ own the immune system to focus on the target disease and win the battle.
The disease-modifying efficacy of our Immune Therapeutic products is based on the intensification or boosting of Th1-type cellular immunity. Based on the available preclinical and clinical data, we expect from this type of immune intensification to provide a therapeutic benefit that cannot be achieved by traditional drugs in patients with chronic infectious diseases, cancer or allergies.
There are four principal components that comprise our comprehensive Immune Therapy platform technology which are designed to work in conjunction to treat and/or control chronic infectious diseases, cancer and allergy, as follows:
| · | Our proprietary Active Pharmaceutical Ingredients (“API”):We have been designing (ANTIGENeering) our APIs to be specific, safe and effective. It consists of a single plasmid DNA (pDNA) immunogen ANTIGENeered to express several antigens, to contain molecular safety features and to release Virus Like Particles (VLP+) in the body of the patients. API with these crucial properties is not feasible with protein or peptide antigens. |
| · | Nanomedicine - our proprietary product platform:Our products are referred here as the “nanomedicine” according to their unique biophysical and biological features. Our APIs are formulated with our novel polymer excipient to synthetic “pathogen-like” nanoparticles. The nanomedicine formulation is essential to achieve potent antigen expression from the pDNA and antigen presentation by dendritic cells. This is critical for the intensification of antigen-specific immune responses in people with chronic diseases. |
| · | DermaPrep - our topical administration platform: We have developed the DermaPrep medical device for targetedin vivo delivery of our nanomedicine products to the dendritic cells of the lymph nodes via the Langerhans cells. We obtained the marketing approval in the European Union (CE Mark) for our DermaPrep device. |
| · | IT - Applied information technology:IT innovations have been supporting the discovery, development, manufacturing, and personalized treatment processes of Genetic Immunity. These include the rational antigen design, clinical trial and data management and matching the patients with the optimal Immune Therapy Product. |
Each of these components, as well as our lead product DermaVir, which has been developed using this platform technology, is described below in detail.
Our Proprietary Active Pharmaceutical Ingredients (“API”)
We have been designing (ANTIGENeering) our APIs to be specific, safe and effective. It consists of a single plasmid DNA (pDNA) immunogen ANTIGENeered to express several antigens, to contain molecular safety features and to release Virus Like Particles (“VLP+”) in the body of the patients. API with these crucial properties is not feasible with protein or peptide antigens.
In all our products, the specificity of immune boosting is determined by the nucleotide sequence of the pDNA that encodes several antigens. These antigens are specific to the causative agent of the disease. For chronic infectious diseases the recombinant antigens are derived from the target virus or intracellular bacteria to boost the immune system to eliminate the infected cells. For cancer, the antigens are derived from genes that specifically expressed in the cancer cells to boost the immune system to kill the cancer cells. For allergy, the antigens are derived from recombinant allergens to boost the immune system and balance the pathogenic immune responses.
We refer to our proprietary process for designing, preparing and testing pDNA-encoded antigens as “ANTIGENeering”. The API discovery program with ANTIGENeering technology supports a large proprietary product portfolio with long patent life.
Our Immune Therapies elicit potent Th1-type antigen-specific CD8 and CD4 T cell responses. The broadest antigen repertoire needs to be ANTIGENeered to boost polyclonal antigen-specific T cells to successfully fight the target disease.
Main steps of the ANTIGENeering process are:
| · | Establishment of anin silico database by collecting relevant scientific and patent information for the target disease; |
| · | Safety profile design of the API to completely eliminate antigen-related dangerous features (e.g., viral integration and replication) by molecular modifications using our proprietary ANTIGENeering software; |
| · | Immunological profile design of the API to express broad specificity CD4 and CD8 T cell epitopes using our proprietary eMINER software; and |
| · | In vitro testing of the new API candidates for antigen expression and antigen characterization with our standardized methods. |
The first example is the API in our lead DermaVir product. It is a single pDNA immunogen representing the broadest antigen repertoire among HIV vaccine candidates. This pDNA was ANTIGENeered for the regulated expression of thirteen complete and two non-functional HIV protein antigens. These proteins self-assemble into VLP+ structurally resembling the wild type HIV. We have introduced multiple irreversible safety features by genetic modifications including the complete impairment of integration, reverse transcription, the function of Nef and the 3’LTR. Our epitope analysis, validated with experimentally proven epitopes, predicted that DermaVir Product could present over 3,000 high-affinity T cell epitopes.In silico prediction of high-affinity epitopes revealed the importance of encoding multiple antigens in the pDNA.
The Figure below illustrate the mechanism of action of our API in the DermaVir (from Somogyi et al. Vaccine 2011)

We developed anin silico antigen-specific T cell epitope prediction method for reliable modeling of the immunological potential of our API candidates. Our analysis demonstrated that HIV protein antigens have important differences in the ability to boost CD4 and CD8 T cell responses and revealed the importance of including both structural and regulatory proteins in an HIV vaccine. The other unique advantage of encoding the above mentioned broad antigen repertoire is the formation of VLP+ that further boosts the immune responses.
We have also developed the specification and validated quality control methods for the API. We have licensed a pDNA manufacturing process suitable for in house API manufacturing (1g scale) and also identified several contract manufacturers capable of producing cGMP quality API according to our specification. We have also built our own GMP certified facility to manufacture our API and excipients to mitigate the dependency from the Contract Manufacturing Organization (“CMO”) and to deliver the necessary material for preclinical and clinical trials.
Nanomedicine – Our Proprietary Product Platform
Our products are referred here as the “nanomedicines” according to their unique biophysical and biological features. Our APIs are formulated with a novel polymer excipient to synthetic “pathogen-like” nanoparticles. The nanomedicine formulation is essential to achieve potent antigen expression from the pDNA (“API”) and antigen presentation by dendritic cells. This is critical for the intensification of antigen-specific immune responses in people with chronic diseases.
Our nanomedicine products consist of a pDNA (API) core that covered with a synthetic polymer (polyethylenimine mannose, PEIm). These synthetic “pathogen-like” nanoparticles have the size and the shape of spherical viruses that naturally evolved to deliver nucleic acids to the cells. These nanoparticles deliver the pDNA to the cells similarly to viruses, but much safer because replication cannot occur. The nanoparticles mimic the size, surface properties, cellular entry by endocytosis, endosomal escape, and gene expression of pathogens (eg, viruses), thus allowing direct targeting of the antigen presenting cells of the immune system. The table below compares the properties of our nanoparticles to one of the pathogens (viruses):
| Features | Our “pathogen-like” nanomedicines | Pathogens (virus) |
| Structure | Synthetic polymer coat (PEIm) and pDNA | Protein coat and genetic material |
| Size | 70 to 300 nm | 50 to 500 nm |
| Surface properties | Sugar residues | Glycoproteins |
| Entry into the cell | Endocytosis | Endocytosis and other mechanisms |
Delivery of genetic material to the nucleus | 1. Efficient endosomal escape promoted by PEIm buffering capacity (acts as proton sponge) 2. Intra-cytoplasmic trafficking determined by the degree of association of PEIm and pDNA | Viral protein-mediated mechanisms 1. efficient endosomal escape 2. intra-cytoplasmic trafficking |
| Gene expression | Authentic protein expression profile, Virus-like Particle (VLP+) assembly | Authentic virus protein expression profile, virion assembly |
The formation of “pathogen-like” nanoparticles is required not only for the targeting of dendritic cells, but also to protect the API from cellular degradation. These properties are essential for the efficient expression of the pDNA in dendritic cells of the lymphoid organs, which is required for the boosting of antigen specific T-cells and directing the immune responses to Th1-type. A naked pDNA without formulation to nanoparticles is susceptible to degradation both in extra- and intracellular space. Therefore, the biological activity of our nanomedicines is superior to the technologies which use pDNA without protection. Furthermore, to increase the efficiency of our nanomedicines we have inserted in our technology some targeting elements to ensure that the nanomedicines are targeted to the Langerhans/dendritic cells (mannosylation) and the API arrives at the place of antigen expression (nuclear targeting).
We have identified the key steps of the mechanism of action, supported byin vitro andin vivo experiments and developed a nanomedicine formulation that is optimal for both the intracellular stability and shelf life of our products. We have discovered a new biophysical property, the degree of association (measured by hyperchromicity) that determines the ability of the nanomedicine to escape from the endosome and to release the pDNA at the nuclear site. This parameter, which is in direct relation with the biological activity of the nanomedicine, can be controlled by the means of CMC methods like choosing the optimal ionic strength or pH. Optimizing the structure of these nanomedicines was warranted to improve their biological activity, develop stable formulations, and design an efficient, reproducible manufacturing technology.
The Figure below illustrates the experimentally-proven mechanism of action of our nanomedicine products inside the cells.

We have performed the detailed physico-chemical characterization of the nanomedicine according to the regulatory guidelines using our validated assays, and systematically investigated the variability of nanomedicine components and their relationship with the structure,in vitro biological activity and stability of nanomedicine(Tőke et al. IJP 2010).
The potent antigen expression of the nanoparticles depends on their size, their entry through endocytosis, their ability to escape from the endosome and the release of the plasmid DNA cargo at the nucleus.
We have not only exploited this novel formulation but also implemented “Quality-by-Design” (“QbD”) in our biologic product development, manufacturing and control processes. The FDA encourages companies to implement the concept of QbD into their processes. The focus of QbD is that quality should be built into a product with thorough characterization of the product and understanding the processes by which it is developed and manufactured along with a knowledge of the risks involved in manufacturing and how best to mitigate those risks. We have been developing commercial manufacturing processes and validated quality control methods based on our QbD processes and thorough knowledge on our nanomedicine.
DermaPrep – Our Topical Administration Platform
We have developed the DermaPrep medical device for targetedin vivo delivery of our nanomedicine products to the dendritic cells of the lymph nodes via the Langerhans cells. We obtained the Marketing Approval in the European Union (CE Mark) for our DermaPrep medical device.
Provenge of Dendreon Corporation (“Provenge”) is the only antigen-specific Immune Therapy that is approved by the FDA. Provenge employs dendritic cells for antigen presentation to boost cellular immunity and eliminate prostate cancer cells. A prostate cancer-specific antigen is targeted to the patient’s own dendritic cells during theex vivo manufacturing processes that Provenge developed for personalized treatment. Earlier, we have successfully utilized a similarex vivo technology to target our nanomedicine into dendritic cells and boosted potent cellular immunity.
Dendritic cell targeting is very important for the efficacy of antigen-specific Immune Therapies. To improve the above described logistically cumbersomeex vivo immunization approach we developed DermaPrep the first dendritic cell-targeting administration device. Unlike thatex vivo technology, our Immune Therapy products can be manufactured in sizeable batches and administered topically with DermaPrep. One immunization procedure takes about 20 minutes and after three hours the patch is removed by the patient. Our filed patent application covers the new medical device for topical administration of liquid formulations. The figure below illustrates ourin vivo Immune Therapy compared to theex vivo technology of our competitors:
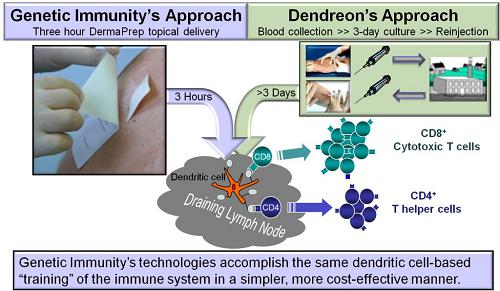
DermaPrep employs a skin preparation method that interrupts the stratum corneum facilitating the epidermal penetration of our nanomedicine product applied on the skin surface under our specially designed patch. The Langerhans cells, located in the skin under our patch, then look for pathogens and 800,000 of them has the chance to capture our “pathogen-like” nanomedicine and travel to the local lymph nodes. Here Langerhans cells mature to antigen-presenting dendritic cells. These cells secrete cytokines to induce Th1-polarized antigen-specific precursor/memory T cells that further differentiate into circulating effector or killer cells.
Several preclinical and clinical trials consistently demonstrated that DermaPrep administration was safe, well-tolerated, no greater than grade 2 events ever occurred. The main adverse event of DermaPrep vaccination is mild and transient erythema. The efficiency and reproducibility of the DermaPrep administration procedure was proven in the biodistribution study conducted on different cohorts of rabbits, where more than 50% of the absorbed nanomedicine was targeted to the lymph nodes.
Based on communication with the EMA and the FDA, we understood that there are currently no similar topical administration devices on the market. We obtained the Marketing Approval in the European Union (CE Mark) for our DermaPrep device.
IT - Applied Information Technology
IT innovations have been supporting the discovery, development, manufacturing, and personalized treatment processes of Genetic Immunity. These include the rational antigen design, clinical trial and data management, and matching the patients with the optimal Immune Therapy Product. We utilize the latest IT and physics including semantic web tools, ultrafast femtosecond laser technologies and embed the gathered knowledge in a broad portfolio of innovative, patient tailored therapeutics.
Our clinical development processes are supported by e-CRF solutions for managing Clinical Trials and eCTD conform solutions to optimize the clinical development processes, minimize the errors and time to submit different applications and reports to regulatory agencies word-wide. Our IT team also provides logistic support for the clinical trials by auditing the sites and identifying potential errors of measurements’ methods and errors in data collection and evaluation.
An electronic Labor Management System (“e-LMS”) supporting our processes in CMC provides a competitive advantage in product development by saving time and manpower. e-LMS supports our quality control tests using computerized Standard Operating Procedures (“SOP”). We electronically select the SOP and the required materials
and equipment from our storage. In some cases, experimental data are captured directly from the equipment and stored in our databases. Deviations from protocols are registered and the results are calculated and summarized in a table format. Detailed reports, required by regulatory agencies, can be either printed and filed or stored electronically. Our IT team continuously improves the software solutions to meet our emerging requirement.
The API discovery and documentation process is presently supported by two of our proprietary softwares. TheANTIGENeering software ‘collect and mine’ research data from different sources and publications to support the pre-decision making and to ensure safe and specific antigen design. TheeMINERsoftware is utilized for the determination of the immunological potential (high affinity epitopes) on different human leukocyte antigen (“HLA”) alleles. With greater than 85% probability, we canin silico predict the cellular immune responses of patients treated with our Immune Therapeutic products. We envision that the eMINER will help doctors to match patients with optimal Products and design specific immune diagnostic tools.
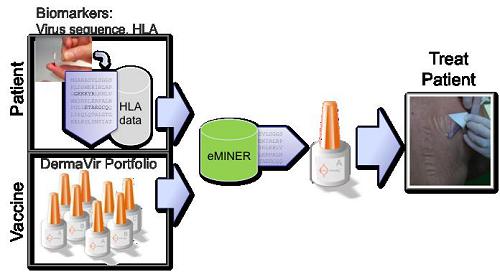
Using the above-described tools we have established a DermaVir Product Family to provide every HIV-infected individual an optimal Immune Therapy. HIV is a highly variable virus. We found that the therapeutic effectiveness of DermaVir depends on HIV sequence replicating in the patients and the patients’ genetic background. Our DermaVir Product Portfolio is a family of products containing related HIV-specific antigen sequences characteristic for the different Clades (subtypes) of the virus. To ensure the optimal therapeutic benefit for every patient the treating physician will enter the diagnostic information on the patient’s HIV sequences and genetic background (HLA) into our eMINER software. After analyzing the data the eMINER will select the optimal DermaVir product from the DermaVir Product Family and also calculate the specific T cell epitopes that will play a role in the immune boosting of the individual patient. This way the treating physician can then make an educated decision for selecting the optimal Product for personalized treatment for the patient and could also prescribe the immune monitoring.
Our competitive IT team consists of informaticians and mathematicians who are dedicated to support our business agenda. They are essential part of our patient-driven technology platform, providing us innovative solution to daily challenges including data capture, evaluation and storage of experimental data, website design, literature search, 3D visualization of the skin penetration of our nanomedicines based on fiber-integrated 2-photon microscopy and auditing the data management and data quality on clinical sites. We believe that our integration of bioinformatics and mathematics into our everyday workflow will significantly improve competitiveness because it provides efficient, accessible and high quality data and information collection and distribution throughout the entire research and development and commercialization processes. Our IT team develops project-based, customized services to improve upon the efficiency of our work being performed and to save time and eliminate errors caused by insufficient data management.
Pipeline Product Candidate
Our pipeline product candidate with animal proof of concept, for the treatment of Human Papilloma Virus (“HPV”) infection, is referred to as “HuPaDerm” and our allergen-specific immunotherapy is referred to as “DermAll”. We partnered with the German Cancer Research Center located in Heidelberg (“DKFZ”) to develop HuPaDerm. DKFZ is a world leading research center in tumor virology and Harald zur Hausen was awarded the Nobel Medicine Prize for his work on HPV caused cancer of the cervix. HuPaDerm animal trials are performed in Heidelberg. DermAll has decreased the allergic symptoms by balancing allergen-specific immune responses in mice experiments.
DermAll
Allergic diseases are caused by an immune response induced by different allergens. We believe that our DermAll products will suppress pathogenic immune responses in allergic people by boosting allergen-specific immune responses. This new mechanism is expected to restore a balanced immune response that is normally characteristic for people who do not have any allergic disease. The DermAll Product Portfolio will target several different allergies. The first target will be Egg Allergy (“EA”), since we have already collected preclinical animal data in this DermAll indication suggesting that DermAll-EA treatment can suppress allergic sneezing by balancing immunity. We expect clinical efficacy of specific Immune Therapy that not only suppress allergic rhinitis symptoms but also prevent progression to asthma or atopic dermatitis.
For allergic diseases Immune Therapy offers a vastly different treatment option with disease-modifying potential to traditional symptomatic treatments. In 2009, Grazax became the first Immune Therapy in Europe to gain approval as a ‘disease modifying treatment,’ representing a significant step towards the possibility of a cure. The major limitations of present Immune Therapy approaches include serious safety concerns and cost of treatment, therefore conventional symptomatic treatments continue to be the more popular option (Datamonitor 2010). DermAll Immune Therapy addresses both limitations using our proprietary API design to obtain products with clinically proven excellent safety features, and cost effective manufacturing technology.
HuPaDerm
Development of HuPaDerm Product Portfolio is targeting the treatment of the leading cause of cervical cancer as well as papillomatosis, genital warts and later cancer caused by the different HPVs. Our development strategy for the HuPaDerm Product Portfolio has already been discussed with the EMA. We partnered with DKFZ to develop the HuPaDerm product. DKFZ is a world leading research center in tumor virology. Harald zur Hausen was awarded the Nobel Medicine Prize for his work on HPV caused cancer of the cervix. Zur Hausen, former Scientific Director of DKFZ, is recognized for finding that cervical cancer is caused by viral infections. His research made it possible to develop a vaccine against one of the most frequent cancers in women. Zur Hausen shared the Nobel Prize for Medicine with Françoise Barré-Sinoussi and Luc Montagnier for discovering HIV, the virus that causes AIDS. HuPaDerm animal trials are performed in Heidelberg. DermAll decreased the allergic symptoms by balancing allergen-specific immune responses in mice experiments.
Immune Therapy for Cancer
ProstaDerm Immune Therapy against virus-based prostate cancer
We have also considered developing ProstaDerm Immune Therapy to provide anin vivo alternative to Provenge that is manufacturedex vivo from the patient’s blood. Xenotropic murine leukemia virus-related virus (“XMRV”) is an authentic, newly recognized human retrovirus first identified in prostate cancer tissue. Studies have detected XMRV at different rates in prostate cancer cases (up to 27%) and in patients with chronic fatigue syndrome (CFS; up to 67%) (Silverman et al. Nature 2010). Therefore a pDNA developed for expressing XMRV antigens might effectively be used for the treatment of XMRV-associated prostate cancer.
Immune Therapy against melanoma
Melanoma is a malignant tumor of melanocytes, pigment producing skin cells. Melanoma is less common than other skin cancers. However, it is much more dangerous and causes the majority (75%) of deaths related to skin cancer (Jerant et al. AAFP 2000). This cancer has well characterized and specific biomarker proteins (melanoma-associated antigens, MAGE) therefore melanoma is a possible target indication for our Immune Therapy Platform Technology. ANTIGENeering of a pDNA specific for these antigens could result in a product candidate for patients suffering from melanoma.
Immune Therapies for Emerging Infectious Diseases
A potential extension of our technology could be utilized to develop treatments for infectious diseases like Chlamydia, hepatitis C and B viruses and West Nile Virus where T-cell responses can be directed against cells harboring the intracellular pathogens. Opportunities are under consideration for initiating the development of some of these products at a later stage.
Immune Therapy against West Nile Virus
We are working together with a European Consortium (WINGs) to develop a prophylactic and therapeutic vaccine against the West Nile virus. The project is led by the Fraunhofer Institute IZI in Leipzig, Germany, and is funded from a $3.5M European Union grant.
Our role in the project is to provide the nanoformulation technology for the vaccine candidates and the transdermal delivery technology for the administration of the vaccine. As industrial partner of the consortium we are also responsible for the clinical development and the commercialization if the animal studies are successful.
GRANTS
In 2005 we received a $1 million grant from the EU, which funded Phase I clinical trials of DermaVir in Hungary. Since being awarded a $7 million grant from the Hungarian government in 2005, we have been applying the funds to develop product manufacturing technology in Hungary, build a pilot manufacturing facility and develop pipelines in collaboration with Hungarian academic institutions pursuant to a Consortium Agreement establishing the Vaccine Therapy Cluster, a collaboration between the Company and the University of Szeged. As a result, our product development, manufacturing, information technology, and clinical operations have been established in Budapest. The business, financial and commercial operations remain in the U.S. In 2009, the DVCLIN01 consortium we lead received an additional grant of $4.5 million to perform a Phase II clinical study with DermaVir in Germany. The results of this study were presented at the AIDS International Conference in Vienna July, 2010. In 2010, the FIBERSCN consortium led by R&D Ultrafast Lasers Kft. received a grant of $3 million to investigate DermaVir immunization technology with fiber integrated 2-photon microscopy. We investigated the penetration of DermaVir to the skin and uptake by epidermal Langerhans cells. The West Nile Shield project (“Wings”) consortium led by Fraunhofer Institute won a $3.5 million grant to develop novel approach for prophylactic and therapeutic vaccines against West Nile Virus. As an active member of this consortium we provide nanomedicine formulation and our DermaPrep device for transdermal delivery of potential West Nile Virus vaccine candidates.
Using the above funding, we have developed improved formulation and scalable manufacturing technology for our biological product portfolio; conducted preclinical animal studies to demonstrate that our proprietary technology is suitable for additional product development including DermAll and ChlamyDerm; developed regulatory strategy for obtaining Marketing Approval for our DermaVir Product Portfolio as personalized Immune Therapy; and designed an allergen-specific Immune Therapy Product Portfolio.
INTELLECTUAL PROPERTY
Protection of our intellectual property and proprietary technology is a strategic priority for our business. We rely on a combination of license agreements and patent, trademark, copyright and trade secret laws along with institutional know-how and continuing technological advancement to develop and maintain our competitive position. Our ability to protect and use our intellectual property rights in the continued development and commercialization of our technologies and products, operate without infringing the rights of others, and prevent others from infringing our rights, is crucial to our continued success. We will be able to protect our products and technologies from unauthorized use by third parties only to the extent that they are covered by valid and enforceable patents, trademarks or copyrights, or are effectively maintained as trade secrets, know-how or other proprietary information. Our policy is to seek U.S. and international patent protection for technological developments that we believe will enhance the market position of our product candidates and methods of using our product candidates.
Present patents protect DermaVir until 2020.Ourfilingsrepresent(1)ourHIVplasmidDNAmaterial;(2) theformulationofDermaVir;(3)thetopical deliveryofgenesintoantigen-presentingcellsoftheskin;and(4)thebroadertechnologyrelatedtoraisingan immuneresponseviaexpressionoftheDNA-encodedantigensinantigenpresentingcells.Thefirst ofourpatents expiresin2020.
For subtype-specific DermaVir Product Family we plan to file a new patent application for the API that would protect our DermaVir Products until 2031 when it is issued. Filing the new patent application on API designed with our ANTIGENeering technology is our strategy for all the pipeline products.
Tosupplementourpatentportfolio,wealsodependontheskill,knowledgeandexperienceofourscientific andtechnicalpersonnel,aswellasthatofouradvisors,consultants,andothercontractors.Tohelpprotectour proprietaryknow-howandinventionsforwhichpatentsmaybedifficulttoenforce,werelyon tradesecret protectionandconfidentialityagreementstoprotectourinterests.Werequireallemployees, consultants,advisors, andothercontractorstoenterintoconfidentialityagreementsthatprohibitdisclosureofconfidentialinformation and,whereapplicable,requiredisclosureandassignmenttousofideas,developments,discoveriesandinventions importanttoourbusiness.
WehaveaU.S.federaltrademarkregistrationfor“GeneticImmunity.”
MARKET OPPORTUNITIES
Therapeutic Vaccine Market for HIV/AIDS
According to the Joint United Nations Programme on HIV/AIDS (“UNAIDS”), about 33.3 million people are living with HIV/AIDS in the world, including 2.5 million children, with 2.3 million of them in North America and Europe. During 2009, 100,000 patients were newly infected and 35,000 patients were reported to have died from AIDS in North America and Europe. The rise in prevalence has been driven by a number of factors, including a growing number of heterosexual transmissions, immigration from countries with a high HIV prevalence, and a growth in high-risk sexual behavior. There is strong evidence of resurgent HIV epidemics among men who have sex with men in North America and in Western Europe. Immigrants living with HIV have become a growing feature of the epidemics in several countries in Europe. Heterosexual transmission accounts for about half of the people newly infected with HIV in Central Europe (2010 UNAIDS Report). There are significant emerging markets including Eastern Europe, Russia, India and China with significant income and are expected to take actions to slow down their fast growing HIV epidemics. Therefore, additional market opportunities for HIV/AIDS treatments are projected to emerge.
The patient population on HAART is already demanding simplification of treatment that can be achieved by combining several drugs into one pill. The development of novel original antiretroviral drugs will be more and more expensive and more difficult to get approved. In 2009, the HIV market generated sales of about $12 billion and is expected to grow up to $15.6 billion in 2015 (Datamonitor HIV market forecast (06/2010)). This growth has been driven by the launch of several new fixed-dose combinations such as Truvada and Epzicom™, and a new class of antiretrovirals known as the entry inhibitors. The combination of several drugs in one pill improves the tolerability of HAART.
When DermaVir enters the market, there will be greater than 28 generic antiretroviral drugs on the market providing treating physicians a diverse source of inexpensive and potent drugs for managing their patients. According to recent treatment recommendations, doctors will start treating their patients earlier with HAART and as a result, more HIV-infected patients will live a longer life. We expect that by 2016 a significantly larger patient population will be treated with HAART due to the inexpensive and potent generics. These patients will be eligible for DermaVir immune intensification and that might further increase the potential market for DermaVir.
Market opportunity for DermaVir
Market demand for DermaVir will be driven by the demand for (1) a cure and (2) simplified treatment options by HIV-infected patients. Such patients seek safe drug sparing treatment options that fully control their virus, decrease of toxicities, immune boosting and cure of HIV disease. We believe that the DermaVir immune intensification will be the first on the market offering significant therapeutic benefit over HAART and a potential cure (remission) for some of the patients who achieved the maximal immune intensification to control HIV disease. Due to decreasing costs and better access to generic HAART, the treated patient population will significantly grow. The new state of the art treatment will be DermaVir in combination with generic HAART. Doctors will use our eMINER to match patients with their optimally effective DermaVir selected from our product portfolio and prescribe the personalized diagnostic assay to monitor when the HAART can be simplified by dropping one, two or even all three components. We believe that the new, potent and expensive antiretroviral drugs will be very important for salvage and will be used for patients failing generic HAART.
We believe that after market entry, DermaVir Immune Therapy will be the new state of the art treatment. Since HIV drugs decrease immune responses and do not provide a cure, DermaVir immune boosting could be added to every HAART-treated patients. The minimum effect of DermaVir would be to boost the patients’ own immune system’s responses to eliminate infected cells from the body. But additional benefits expected for DermaVir treated patients are the decrease in the number of drugs and consequently such patient will suffer less toxicity. DermaVir treatment will provide a hope for cure; in some DermaVir-treated patients, immune boosting can result in remission allowing the patient to lead a healthy life in the absence of HIV drug treatment (for some years). If the HIV rebounds because the immune system cannot fight any longer, the patient resume to take the drugs and his/her life will not be in danger.
As a new state of the art immune boosting therapy, DermaVir treatment should be cost effective. Saving on drugs and cost of treatment could support insurance reimbursement. It is conservative to estimate that annual DermaVir treatment will be lower than the cost of HAART. We believe this can represent up to over $5 billion DermaVir sales for the seven major markets per year, based on 30% market penetration. In the event competitors for a cure enter into the market this figure will decrease. DermaVir is one of the best-characterized HIV therapeutic vaccine product candidates (solid randomized and controlled Phase II clinical data, commercial manufacturing methods and controls) and may have the highest chance to obtain first marketing approval.
We believe that in the not too distant future DermaVir will have significant sales outside of the major markets based on the high unmet need. These markets include the 22 million people, including children, in sub-Saharan Africa. However, developing countries such as Brazil, China, India and Russia could become significant markets.
The global HIV drug market size is expected to grow to $15.6 billion by 2015 with approximately 35 million people infected with the virus. Of this amount, Europe represents approximately 2.3 million HIV infected people. Russia, our next potential approval target, represents another 1 million infected people.
Initially we will aim to treat 5% of the European population, or approximately 130,000 individuals (of the 2.3 million total infected.) Current HAART based treatments cost approximately 12,000 EURO a year per patient. Based on our initial pricing model, and assuming a 5% market penetration, this represents a EURO 1 billion market opportunity per year for DermaVir in Europe alone.
Competition
In the field of HIV treatment, several immune therapies are under development because there is a substantial unmet medical need to find new treatment options for patients who have developed resistance to the current antiviral drugs and for patients who cannot tolerate life-long daily drug treatment. A description of such immune therapies follows:
| i. | Broad-Spectrum Immune Therapies |
The rationale of broad-spectrum Immune Therapy originated from the successful use of interferon-alpha for the treatment of another chronic infectious disease, hepatitis C. The best available current treatment for chronic hepatitis C (peg-interferon alpha plus ribavirin) leads to an overall sustained response rate of approximately 70%. In HIV infection, however, no viral load suppression after current drug treatment interruption can be achieved. The current, broad-spectrum immune therapies described below are not specific to any disease:
| · | Hemispherx Biopharma is evaluating the safety and activity of orally administered low dose interferon alfa to modulate general, nonspecific immune responses in subjects infected with HIV who have not yet demonstrated clinical symptoms of immune suppression. |
| · | Interleukin-2 (IL-2, Aldesleukin, Proleukin) is made by Novartis (Chiron) and Amgen. This drug is currently approved for the treatment of certain types of blood cell cancers (e.g., specific types of leukemia). It is considered an experimental drug for HIV-related therapy because the FDA has not yet evaluated it for the HIV indication. Two large efficacy trials in HIV patients (SILCAAT and ESPRIT) were conducted recently. However, despite a substantial and sustained increase in the CD4+ cell count, as compared with antiretroviral therapy alone, Interleukin-2 plus antiretroviral therapy yielded no clinical benefit in either study. |
| · | CYT107, Cytheris’s recombinant human Interleukin-7 developed as a general immune system booster. The company has four ongoing Phase I/II or Phase II studies with CYT107. According to the company’s latest press release, dated March 1, 2011, which reported the results of INSPIRE2 Phase IIa study, CYT107 was able to reconstitute CD4 T-cells in chronically HIV-1 infected patients whose CD4 T-cell counts remained low despite treatment with anti-retroviral-therapies (HAART). |
| · | Inovio has been developing a new DNA administration technology, called Electroporation, for the intracellular delivery of various immunotherapeutic DNA solutions. Resultant gene expression matched or exceeded that achieved using viral or lipid delivery methods, without side effects that hamper these approaches. The device is being tested in Phase I/II and Phase II trials. |
| ii. | HIV-Specific Immune Therapies |
The rationale for HIV-specific immune therapies is to amplify HIV-specific immune responses present during chronic infection to control viral replication. Such antiviral immune responses provide the balance between infection and the immune system and in some cases the immune system wins over the infection (e.g., most of the non-progressors), but in the majority of the cases, HIV overwhelms the immune system resulting in disease progression and AIDS. Immune Therapies have been developed for viral, parasitic, cancer, and allergic diseases. Antigen-specific immune therapeutic approaches are (1) based on antibodies that specifically bind the target antigens, and (2) T-cells that can eliminate HIV-infected cells from the body. Our competitors are the most promising therapeutic vaccines according to the Datamonitor report: R&D Trends: HIV (Reference Code: HC00083-007), published in March 2011. Each of our competitor’s products described below areex vivo or injected by needle and not topically delivered like DermaVir:
Phase II Immune Therapy candidates
| · | Vical, in collaboration with the NIH and Merck & Co., is developing a DNA vaccine, VRC DNA/rA, for the treatment and prevention of HIV using its naked DNA gene delivery technology. The vaccine is currently in Phase II clinical development (NCT00865566, 2011; www.clinicaltrials.gov) (Vical, 2011) |
| · | FIT-06 is a DNA vaccine consisting of the Clade B HIV nef gene, under development by FIT Biotech using its GTU nuclear anchoring technology, for the treatment and prophylaxis of HIV/AIDS. It has completed Phase II trials in South Africa (Fit Biotech, 2010) |
| · | AGS-004 is one of a series of autologous dendritic cell vaccines under development by Argos Therapeutics for the treatment of HIV infection. The vaccines are pulsed with amplified mRNA from infected cells to stimulate an immune response. |
| · | The Vaccine Research Center at the US NIAID is collaborating with GenVec on the development of an HIV DNA vaccine (VRC-HIVADV014-00VP). The vaccine consists of an adenovirus-5 vector expressing modified HIV-1 genes. It is being evaluated alone and in combination with a plasmid DNA vaccine prime.
Vaccine candidates are evaluated as therapeutic and prophylactic AIDS vaccines (GenVec, 2011a). It has been evaluated in several Phase I/II clinical trials (GenVec, 2011b) |
| · | In October 2010 Bionor Pharma reported that the primary endpoints of the placebo-controlled study of Vacc-4x (NCT 00659789, 2010, www.clinicaltrials.gov) had not been met. However, the decision was made to resume the development of the vaccine candidate due to a statistical significant treatment difference regarding viral load, the secondary endpoint (Bionor Pharma 2010). Bionor Pharma is currently seeking partnering opportunities. It is also developing a follow-up vaccine Vacc-C5, targeting immune activation associated with HIV infection that is expected to enter clinical trials in 2011 (Bionor Pharma 2011). |
| · | V-1 Immunitor (V1) is a therapeutic AIDS vaccine formulated as an oral pill comprising heat-inactivated HIV antigens derived from pooled blood of HIVpositive donors that is being developed in Russia and Thailand (Bourinbaiar et al., 2010) |
Phase I/II Immune Therapy candidates
| · | TUTI-16 is a water-soluble, fully synthetic, self-adjuvanting lipopeptide vaccine developed by Thymon (Goldstein and Chicca, 2010). A US Phase I/II clinical trial was completed in February 2011 (NCT00848211, 2011; www.clinicaltrials.gov). A second Phase I/II clinical trial, NCT01144026, is currently recruiting participants |
| · | A dendritic cell vaccine is developed by Esteve, consisting of autologous monocyte-derived dendritic cells pulsed with heat inactivated autologous HIV-1. Phase I/II results were reported in 2011 (Garcia et al., 2011) |
| · | Virax’s HIV program is focused on the development of a therapy for early stage HIV infection using an immunotherapeutic approach. VIR201 is a recombinant fowl pox-virus technology designed to co-express genes for immunogenic but highly conserved parts of the HIV in conjunction with interferon gamma. The objective is to amplify the T-cell-mediated immune response of the human immune system to the HIV virus. Results from a Phase I/IIa trial were reported in October 2010 (Virax, 2010) |
Phase I Immune Therapy Candidates
| · | PENNVAX-B is an HIV env clade B-specific/gag/pol DNA vaccine for the prevention and treatment of HIV/AIDS. The vaccine was licensed from the University of Pennsylvania. As part of the deal Inovio has worldwide commercialization rights for vaccine and delivery technology. Pennvax-B has completed a US Phase Ib study (NCT00775424, 2011; www.clinicaltrials.gov) (Inovio, 2011) |
| · | A pDNA vaccine delivered via electroporation in a co-formulation with a plasmid containing IL-12, which acts as a molecular adjuvant is developed by Profectus BioSciences. The vaccine is currently in a US Phase I clinical trial (NCT01266616, 2011; www.clinicaltrials.gov) |
| · | EP-1090 is a DNA vaccine developed by Inovio and VaxOnco in In Phase I clinical trial since 2007 (NCT00532974, 2007; www.clinicaltrials.gov) |
| · | GX-120 is a DNA vaccine developed by Dong-A Pharmaceuticals and Genexine. GX-120 contains four plasmids expressing gag, pol, env and a mutant IL-12 (IL-12N222L). It is in a Korean Phase I trial (Genexine, 2010) |
| · | NAcGM3/VSSP is a Ganglioside-based vaccine developed by the Center of Molecular Immunology Havanna, Cuba and Recombio. The N-acetylated ganglioside GM3 vaccine (NGcGM3) is combined with Neisseria OMP vesicle-based very small proteoliposomes (VSSP) as carrier. It is in two Phase I trials in Argentina and Cuba for the treatment of HIV/AIDS (Recombio, 2011) |
Advantages of DermaVir Over Antiretroviral Drugs
HAART requires the combination of three drugs selected from minimum two different classes. It has been effective in decreasing morbidity and mortality associated with HIV infection. However, full life expectancy of HIV-infected people cannot be restored even with optimal care. It has been demonstrated that HIV-infected people treated with optimal HAART live 12 years shorter than those without HIV.
HAART does not provide a cure for HIV/AIDS. Several recent studies demonstrated that HAART intensification with drugs of additional classes (e.g. integrase inhibtor) leads to no change of viremia or reservoirs measured by HIV DNA. In addition, long-term HAART decreases HIV-specific immune responses that could negatively affect the clearance of the reservoir. Viremia persist in the reservoirs even after 7 year of fully suppressive HAART (ABBOTT 720 study), therefore eradication of HIV with HAART does not seems to be feasible.
An alternative strategy to cure HIV disease would be to induce remission, similar to cancer model. A recently completed study has demonstrated (presented at CROI2010)an inverse correlation between the frequency of HIV-specific T cells in the gut and the size of the reservoir. This is the first evidence suggesting that boosting of the patients’ own immune system might decrease the number of HIV-infected cells in the reservoirs.
We have shown that DermaVir boosts cellular immunity to kill HIV-infected cells and functions differently from antiretroviral drugs that kill the virus. We believe that DermaVir treatment can reduce the amount of HIV-infected cells remaining in the reservoirs in patients treated by HAART. This novel activity of DermaVir could provide additional treatment benefits by reducing the number of antiretroviral drugs that patients must take daily. We envision that after successful reconstitution of the HIV-specific immune system by DermaVir, immune control of HIV might be achieved. DermaVir could change the HIV treatment paradigm by giving patients an option for a safe interruption of life-long drug treatment by boosting their own immune system.
Achieving optimal T cell responses and elimination the majority of HIV-infected cells from the reservoirs could lead to remission in some HIV-infected people. These people would be able to stop their antiretroviral drug treatment and receive immune boosting DermaVir treatments during regular doctor visits.
In contrast to antiretroviral drugs, DermaVir treatment provides a hope for patients for afunctional cure. We do not believe that eradication of integrated HIV can be achieved by any treatments. However, several viruses are successfully controlled by a healthy immune system, including HIV in some exceptional people; therefore remission (functional cure) is an achievable treatment objective. Based on our findings, we believe that DermaVir, by boosting HIV-specific cellular immunity in patients treated with HAART, could effectively kill HIV-infected cells and decrease the size of the reservoirs. After repeated DermaVir treatments a significant intensification of HIV-specific immunity and the elimination of the majority of infected cells from the reservoirs could be achieved. We believe that the hope for a cure that HAART cannot provide for HIV-infected people will be an option with DermaVir when used initially in combination with HAART to eliminate both the virus and the infected cells.
DermaVir sales could be favored and not limited by Generic Antiretroviral Drugs, which would be a significant competitive advantage compared to original or new antiretrovirals. These original drugs could be more expensive than the generics and as a result their sales could be limited to patients who are not responding to generic drugs. Therefore, effective generic treatment options could limit the profit of a new HIV drugs entering into the market. In contrast, we believe that DermaVir sales will be independent of original or generic drugs used in HAART.
We believe that the DermaVir Immune Therapy might be used for immune intensification of most of the HAART treated people. According to recent treatment recommendations, doctors will start treating their patients with HAART earlier and consequently more HIV-infected patients will have a longer life expectancy. We believe that a significantly larger patient population will be treated with HAART and DermaVir due to the market entry of inexpensive and potent generics.
An additional benefit of DermaVir will be what we believe to be a favorable price. We believe that the cost of DermaVir manufacturing will be manageable on a per dose level and that will allow us to sell the product for a competitive price. We estimate that the price of DermaVir in the seven major markets might be similar to original antiretrovirals (e.g. $12,000 per year). The cheapest price of DermaVir might be in sub-Saharan Africa (e.g. $500 per year), which is similar to generic HAART.
We believe that DermaVir immune intensification will be the first option on the market offering a significant therapeutic benefit over HAART and a potential functional cure (remission) for some of the patients who achieve maximal immune intensification to control the HIV disease. We believe DermaVir will be used in conjunction with generic HAART. We believe that doctors will use our eMINER application to match patients with their optimally effective DermaVir selected from our product portfolio and prescribe the personalized diagnostic assay to monitor when HAART can be simplified by dropping one, two or even all three components. We believe that the new, potent and expensive antiretroviral drugs will be very important for salvage therapy and will be used for patients failing generic HAART.
OUR BUSINESS STRATEGY
Our business objective is to develop and commercialize DermaVir and our pipeline products in order to become one of the leading Immune Therapy companies. We believe that our unique combination of product candidates, our preclinical and clinical Proof of Concept and our nanomedicine manufacturing technology that beside the patents provides additional long-term protection against genetic competitors, supports such an ambitious objective. It is our intention to obtain the funding required for the growth of the Company from the public market.
Up to date we have focused on R&D and proof of concept for our Technology Platform. From now on, besides adding to our team talents and experts essential to develop our products, we intend to expand our internal management and business development team.
SALES AND MARKETING
We currently have no internal sales or distribution capabilities. In order to successfully commercialize any of our products, we must either internally develop sales, marketing and distribution capabilities, or make arrangements with third parties to perform such services. For the foreseeable future, we may not be able to establish marketing and sales capabilities internally or hire a large enough number of sales personnel with appropriate expertise to market and sell our products, if the products are approved by appropriate regulatory authorities. Therefore, we may need to enter into an alliance with a major pharmaceutical company or other third party to market and sell our products, which we may be unable to do.
EFFECT OF EXISTING OR PROBABLE GOVERNMENTAL REGULATION
Weplanto marketourproductsfollowingapprovalonaglobalbasis.Theapprovalandsellingofour productsaresubjecttovariousregulatoryandgovernmentaloversightbodies.Dependingontheregulatorycategory ofeachproduct,suchasbiologicalproduct,medicaldeviceordrug,separateregulationsapplyandcouldvaryonamarket-by-marketbasis.
FDARegulation
Webelievethatourproductsaredefinedascombinationproductsconsistingof twoormoreregulated components,abiologicandamedicaldevice.IntheU.S.,acombinationproductusuallyisassignedby theFDAto oneof theagency’scenters,suchasthe Center for Biologics Evaluation & Research (“CBER”)ortheCenter for Device and Radiological Health (“CDRH”) ,withthechosencentertaking theleadinpre-marketing reviewandapprovalof thecombinationproduct.OtherFDAcentersalsomayreviewtheproductinregardto mattersthatarewithintheirexpertise.TheFDAselectstheleadcenterbasedonanassessmentofthecombination product’s“primarymodeofaction.”Someproductsalsomayrequireapprovalorclearancefrommorethanone FDAcenter.
TodeterminewhichFDAcenterorcenterswillreviewacombinationproductsubmission,companiesmay submitarequestforassignmentto theFDA.Thoserequestsmaybehandledformallyorinformally.Insomecases, jurisdictionmaybedeterminedinformallybasedonFDAexperiencewithsimilarproducts.However,informal jurisdictionaldeterminationsarenotbindingon theFDA.Companies alsomaysubmitaformalRequestfor Designation totheFDA’sOfficeofCombination Products.TheOfficeof Combination Productswillreviewthe requestandmakeitsjurisdictionaldeterminationwithin60daysofreceivingaRequestforDesignation. Weare currentlydiscussingthepotentialregulatory pathwayforDermaVirwiththeFDA. Basedonthose discussionstodate,webelievethattheproductwillbereviewedby the CBERandthattheDermaPrepmaybe reviewedby theCDRHeitherinconsultationwithCBERaspartof theBiologics License Application (“BLA”)orseparatelyasamedicaldevice.
DomesticRegulationofOurProductsandBusiness
Thetesting,manufacturing,andpotentiallabeling,advertising, promotion,distribution,importand marketingofourproductcandidatesaresubjecttoextensiveregulationbygovernmental authoritiesintheU.S.and inothercountries.Inthe U.S.,the FDA,underthe PublicHealthServiceAct, the Federal Food,DrugandCosmetic Act,and its implementingregulations, regulates biologicsandmedical deviceproducts.
Thelabeling,advertising,promotion,marketinganddistributionofbiopharmaceuticals,orbiologicsand medicaldevicesalsomustbeincompliancewiththeFDAandU.S.FederalTradeCommission(“FTC”) requirementswhichinclude,amongothers,standardsandregulationsforoff-labelpromotion,industrysponsored scientificandeducationalactivities,promotional activitiesinvolvingthe internet,anddirect-to-consumer advertising.TheFDAandFTChaveverybroadenforcementauthority,andfailuretoabidebytheseregulationscan resultinpenalties,includingtheissuanceofawarningletterdirectingus tocorrectdeviationsfromregulatory standardsandenforcementactionsthatcanincludeseizures,injunctionsandcriminal prosecution.Recently, promotionalactivitiesforFDA-regulated productsofothercompanieshavebeenthesubjectofenforcementaction broughtunderhealthcarereimbursement lawsandconsumerprotectionstatutes. Inaddition,underthefederal LanhamActandsimilarstatelaws,competitorsandotherscaninitiatelitigationrelatingtoadvertisingclaims. In addition,wearerequiredto meetregulatoryrequirementsincountriesoutsidetheU.S.,whichcanchangerapidly withrelativelyshortnotice.
TheFDAhasbroadpost-marketandregulatoryenforcementpowers.Manufacturersofbiologicsand medicaldevicesaresubjecttounannouncedinspectionsby theFDAtodeterminecompliancewithapplicable regulations,andtheseinspectionsmayincludethe manufacturingfacilitiesofsomeofoursubcontractors. Failure by manufacturers ortheirsupplierstocomplywithapplicableregulatoryrequirementscanresultinenforcement actionbytheFDAorotherregulatoryauthorities. PotentialFDAenforcementactionsinclude:
- warning letters;
- civilorcriminalpenalties,finesand/orinjunctions;
- productseizuresordetentions;
- importorexport bansor restrictions;
- voluntary ormandatory productrecallsandrelatedpublicityrequirements;
- suspensionorwithdrawalofregulatoryapprovals;
- totalorpartialsuspension ofproduction;and
- refusaltoapprovependingapplicationsformarketingapprovalofnewproductsorsupplements to approvedapplications.
Inaddition,othergovernmentauthoritiesinfluencethesuccessofourbusiness,includingtheavailabilityof adequatereimbursementfromthirdpartypayors,includinggovernmentprogramssuchasMedicareandMedicaid. MedicareandMedicaidreimbursementpoliciescanalsoinfluencecorrespondingpoliciesofprivateinsurersandmanagedcareproviders,whichcanfurtheraffectourbusiness.
BiologicsRegulation
BiologicalproductsmustsatisfytherequirementsofthePublicHealthServicesActanditsimplementing regulations.InorderforabiologicproducttobelegallymarketedintheU.S.,theproductmusthaveaBLA approvedbytheFDA.
TheBLAApprovalProcess
ThestepsforobtainingFDAapprovalofaBLAto marketabiopharmaceutical,orbiologicproductinthe U.S.include:
| · | completionofpreclinicallaboratorytests,animalstudiesandformulationstudiesundertheFDA’s GLPregulations; |
| · | submissionto theFDAofanINDapplication,forhumanclinicaltesting,whichmustbecomeeffective beforehumanclinicaltrialsmaybeginandwhichmustincludeInstitutional Review Board (“IRB”)approvalateachclinicalsite beforethe trialsmaybeinitiated; |
| · | performanceofadequateandwell-controlledclinicaltrialsinaccordancewithGCPtoestablishthe safety,purity,andpotencyoftheproductforeachindication; |
| · | submissionto theFDAofaBLA,whichcontainsdetailedinformationaboutthechemistry,manufacturingandcontrolsfortheproduct,reportsoftheoutcomesof theclinicaltrials,andproposed labelingandpackagingfortheproduct; |
| · | theFDA’sacceptanceoftheBLAforfiling; |
| · | foranybiologicalproductcontaininganactiveingredientnotpreviouslyapproved,automaticreferral toanappropriateadvisorycommitteeforreviewpriortoapproval,unlesstheFDAdecidesotherwise; |
| · | satisfactoryreviewof thecontentsof theBLAbytheFDA,includingthesatisfactoryresolutionofany questionsraisedduringtherevieworbytheadvisorycommittee,ifapplicable; |
| · | satisfactorycompletionofanFDAinspectionofthe manufacturingfacilityorfacilitiesatwhichthe productisproducedtoassesscompliancewithcGMPregulations,toassurethatthefacilities,methods andcontrolsareadequateto ensuretheproduct’sidentity,strength,qualityandpurity;and |
Preclinicalstudiesincludelaboratoryevaluationsofproductchemistry,toxicityandformulation,aswellas animalstudies.Wesubmittedtheresultsofthesepreclinicalstudies,togetherwithmanufacturinginformationand analyticaldatatotheFDAinourBiologicMasterFile(BB-MF12518),Title:HIVtypeIDNAPlasmid(expressingmultipleregulatoryandstructuralgenes,E.coli,LC002,DermaVir)Vaccine,TopicalAdministration;Dateof SubmissionJune15,2005.
AnINDwillautomaticallybecomeeffective30daysafterreceiptbytheFDA,unlessbeforethattimethe FDAraisesconcernsorquestionsaboutissuessuchastheconductof thetrialsasoutlinedintheIND.Inthatcase, theINDsponsorandtheFDAmustresolveanyoutstandingFDAconcernsorquestionsbeforeclinicaltrialscan proceed.
Clinicaltrialsaresubjecttoextensivemonitoring,recordkeepingandreportingrequirements.ClinicaltrialsmustbeconductedundertheoversightofanIRBfortherelevantclinicaltrialsitesandmustcomplywithFDA regulations,includingbutnotlimitedto thoserelatingtoGCP.Adverseeventsmustbereportedandinvestigated timely. Toconductaclinicaltrial,acompanyisalsorequiredtoobtainthepatients’informedconsentinformand substancethatcomplieswithbothFDArequirementsandstateandfederalprivacyandhumansubjectprotection regulations. Thesponsor,theFDAortheIRBcouldsuspendaclinicaltrialatanytimeforvariousreasons, includingabeliefthattheriskstotrialsubjectsoutweightheanticipatedbenefits. Aprotocolforeachclinicaltrial andanysubsequentprotocolamendmentsmustbesubmittedtotheFDAaspartoftheIND. Inaddition,anIRBat eachsiteatwhichthe trialisconductedmustapprovetheprotocolandanyamendments. Foreignstudiesperformed underanINDmustmeetthesamerequirementsthatapplytoU.S.studies.TheFDAwillacceptaforeignclinical trialnotconductedunderanINDonlyifthetrialiswell-designed, well-conducted, performedbyqualified investigatorsinaccordancewithinternationalprinciplesforGCP,andconformstotheethicalprinciplescontainedin theDeclarationofHelsinki,orwiththelawsandregulationsofthecountryinwhichtheresearchwasconducted, whicheverprovidesgreaterprotectionof thehumansubjects.TheFDA,however,hassubstantialdiscretionin decidingwhethertoacceptdatafromforeignnon-INDclinicaltrials.
Clinicaltrialsinvolvingbiopharmaceuticalproductsaretypicallyconductedinthreesequentialphases. Thephasesmayoverlaporbecombined.Afourth,orpost-approval,phasemayincludeadditionalclinicaltrials. Thesephasesgenerallyincludethefollowing:
| · | PhaseI.PhaseIclinicaltrialsinvolvetheinitialintroductionofthe medicineintohumansubjectsto determinetheadverseeffectsassociatedwithincreasingdoses. |
| · | PhaseII.PhaseIIclinicaltrialsusuallyinvolvestudiesinalimitedpatientpopulationtoevaluatethe efficacyof themedicineforspecific,targetedindications,todeterminedosagetoleranceandoptimal dosage,andtoidentifypossibleadverseeffectsandsafetyrisks. |
| · | PhaseIII.Ifthe medicineisfoundtobepotentiallyeffectiveandtohaveanacceptablesafetyprofile inPhaseII(orsometimesPhaseI)trials,theclinicaltrialprogramwillbeexpandedtofurther demonstrateclinicalefficacy,optimaldosageandsafetywithinanexpandedpatientpopulationat geographicallydispersedclinicaltrialsites. |
| · | Post-Approval(PhaseIV).Post-approvalclinicaltrialsarerequiredoforagreedtobyasponsorasa conditionof,orsubsequentto marketingapproval.Further,iftheFDAbecomesawareofnewsafety informationaboutanapprovedproduct,itisauthorizedtorequirepostapprovaltrialsofthebiological product.Thesetrialsareusedtogainadditionalexperiencefromthetreatmentofpatientsin the intendedtherapeuticindicationand todocumentaclinicalbenefitinthecaseofbiologicsapproved underacceleratedapprovalregulations.IftheFDAapproves aproductwhileacompanyhasongoing clinicaltrialsthatwerenotnecessaryforapproval,acompanymaybeabletousethedatafromthese clinicaltrialsto meetallorpartofanyPhaseIVclinicaltrialrequirement.Theseclinicaltrialsare oftenreferredtoasPhaseIII/IVpostapprovalclinicaltrials.FailuretopromptlyconductPhaseIV clinicaltrialscouldresultinwithdrawalofapprovalforproductsapprovedunderacceleratedapproval regulations. |
Clinicaltestingmaynotbecompletedsuccessfullywithinanyspecifiedtimeperiod,ifatall.TheFDA closelymonitorstheprogressofeachofthethreephasesofclinicaltrialsthatareconductedunderanINDandmay, atitsdiscretion,reevaluate,alter,suspend,orterminatethetestingbaseduponthedataaccumulatedto thatpointand theFDA’sassessmentoftherisk/benefitratio tothepatient.TheFDAorthesponsormaysuspendorterminate clinicaltrialsatanytimeforvariousreasons,includingafindingthatthesubjectsorpatientsarebeingexposedtoan unacceptablehealthrisk.TheFDAcanalsorequestthatadditionalpre-clinicalstudiesorclinicaltrialsbeconducted asaconditiontoproductapproval.Additionally,newgovernmentrequirementsmaybeestablishedthatcoulddelay orpreventregulatoryapprovalofourproductsunderdevelopment. Furthermore,IRBshavetheauthoritytosuspend clinicaltrialsintheirrespectiveinstitutionsatanytimeforavarietyofreasons,includingsafetyissues.
Certaininformationaboutclinicaltrials,includingadescriptionof thetrial,participationcriteria,location oftrialsites,andcontactinformation,isrequiredtobesenttotheNIHforinclusioninapublicly-assessable database.Sponsorsalsoaresubjecttocertainstatelawsimposingrequirementsto makepubliclyavailablecertain informationonclinicaltrialresults.Inaddition,theFDAAmendmentsActof2007directstheFDAtoissue regulationsthatwillrequiresponsorstosubmittotheNIHtheresultsofcertaincontrolledclinicaltrials,otherthan PhaseIstudies.
Assumingsuccessfulcompletionof therequiredclinicaltesting,theresultsofthepreclinicalstudiesandof theclinicaltrials,togetherwithotherdetailedinformation,includinginformationon thechemistry,manufactureand compositionoftheproduct,aresubmittedtotheFDAintheformofaBLArequestingapprovalto marketthe productforoneor moreindications.In mostcases,theBLAmustbeaccompaniedbyasubstantialuserfee.The FDAwillinitiallyreviewtheBLAforcompletenessbeforeitacceptstheBLAforfiling. Therecanbenoassurance thatthesubmissionwillbeacceptedforfilingorthattheFDAmaynotissueaRefuse to File (“RTF”). IfaRTFisissued,thereis opportunityfordialoguebetweenthesponsorandtheFDAinanefforttoresolveallconcerns.IftheBLA submissionis acceptedforfiling,theFDAwillbeginanin-depthreviewoftheBLAtodetermine,amongother things,whetheraproductissafeandeffectiveforitsintendeduseandwhethertheproductisbeingmanufacturedin accordancewithcGMPtoassureandpreservetheproduct’sidentity,strength,qualityandpurity. Ifthebiologicalproductcontainsanewactiveingredientnotpreviouslyapproved,theBLAautomaticallywillbereferredtoan appropriateadvisorycommitteeforreviewpriortoapprovalofthebiologicalproduct,unlesstheFDAdecides otherwiseandspecifiessuch reasonsinacompleteresponselettertothesponsor. TheFDA,however,isnotbound bytheopinionoftheadvisorycommittee.
Companiesalsomayseekfasttrackdesignationfor theirproducts.Fasttrackproductsarethosethatare intendedfor thetreatmentofa seriousorlife-threateningconditionandthatdemonstratethepotentialtoaddress unmetmedicalneedsforsuchacondition.Ifawarded,thefasttrackdesignationappliestotheproductonlyfor the indicationforwhichthedesignationwasreceived. Fasttrack productsareeligiblefor twomeansofpotentially expeditingproductdevelopmentandFDAreviewofBLAs. First,afasttrackproductmaybeapprovedonthebasis ofeitheraclinicalendpointorasurrogateendpointthatisreasonablylikelytopredictclinicalbenefit.Approvalsof thiskindmaybesubjecttorequirementsforappropriatepost-approvalstudiestovalidatethesurrogateendpointor otherwiseconfirmtheeffectontheclinicalendpoint,andtocertainotherconditions. Second,iftheFDAdetermines afterreviewofpreliminaryclinicaldatasubmittedbythesponsorthatafasttrackproductmaybeeffective,it may beginreviewofportionsofaBLAbeforethesponsorsubmitsthecompleteBLA,therebyacceleratingthedateon whichreviewofaportionoftheBLAcanbegin.Therecanbenoassurancethatanyofourotherproductcandidates willreceivedesignationasfasttrackproducts. Andeveniftheyaredesignatedasfasttrackproducts,wecannot assureyouthatourproductcandidateswillbereviewedorapprovedmoreexpeditiouslyfortheirfasttrack indicationsthanwouldotherwisehavebeenthecaseorwillbeapprovedpromptly,oratall. Furthermore,theFDA canrevokefasttrackstatusatanytime.
Inaddition, productsstudiedfortheirsafetyandeffectivenessintreating seriousorlife-threatening illnessesandthat providemeaningfultherapeutic benefitoverexistingtreatmentsmayreceiveacceleratedapproval andmaybeapprovedon thebasisof adequate andwell-controlledclinicaltrials establishingthatthedrugproduct hasaneffect onasurrogateendpoint thatisreasonablylikely topredict clinicalbenefit oronthebasis ofaneffect onaclinical endpointotherthan survival or irreversiblemorbidity. Asacondition ofapproval, theFDAmay requirethat a sponsor ofadrugreceiving acceleratedapproval perform adequateandwell-controlled post-approval clinicaltrialstoverify andfurtherdefine thedrug’sclinical benefitand safetyprofile. Therecan benoassurance thatanyofour productcandidates willreceiveacceleratedapproval. Evenifacceleratedapproval isgranted,the FDAmay withdrawsuchapprovalif thesponsorfailsto conduct therequired post-approvalclinicaltrials, orifthe post-approval clinicaltrialsfailto confirmtheearly benefits seenduring theaccelerated approvalprocess.
Fast-Trackdesignationandacceleratedapprovalshouldbedistinguishedfrompriorityreviewalthough productsawardedfasttrackstatusmayalsobeeligibleforpriorityreview.ProductsregulatedbytheCBERmay receivepriorityreviewiftheyprovidesignificantimprovementin thesafetyoreffectivenessof thetreatment, diagnosis,orpreventionofaseriousorlife-threatening disease.Productsawardedpriorityreviewaregiven abbreviatedreviewgoalsbytheagency. UnderthePrescriptionDrugUserFeeActof2007,theagencyhasagreed totheperformancegoalofreviewingproductsawardedpriorityreviewwithinsix months,whereasproductsunder standardreviewreceiveaten-monthtarget.Thereviewprocess,however,isoftensignificantlyextendedbyFDA requestsforadditionalinformationorclarificationregardinginformation alreadyprovidedinthesubmission. PriorityreviewisrequestedatthetimetheBLAissubmitted,andtheFDAmakesadecisionaspartoftheagency’s reviewof theapplicationforfiling.WeplantoseekpriorityreviewforDermaVirbutcannotguaranteethatthe FDAwillgrantthedesignationandcannotpredictifawarded,whatimpact,ifany,itwillhaveonthereviewtime forapprovalofourproduct.
Ifgranted,Fast-Trackdesignation,acceleratedapproval,andpriorityreviewmayexpeditetheapproval process,buttheydonotchangethestandardsforapproval.
BeforeapprovingaBLA,theFDAwillgenerallyinspectthefacilityor thefacilitiesatwhichthefinished productanditscomponentsare manufacturedtoensurecompliancewithcGMP.IftheFDAdeterminesthe application,manufacturingprocessor manufacturingfacilitiesarenotacceptable,itwilleitherissue“not approvable”letteroran“approvable”letter. A “notapprovable”lettermeansthattheFDArefusestoapprovethe applicationbecausetheBLAor manufacturingfacilitiesdonotsatisfytheregulatorycriteriaforapproval. An “approvable”lettermeansthattheFDAconsiderstheBLAandmanufacturingfacilitiestobefavorable,butthe letterwilloutlinethedeficienciesandprovidetheapplicantwithanopportunitytosubmitadditionalinformationor datatoaddressthedeficiencies. IfandwhenthoseconditionshavebeenmettotheFDA’ssatisfaction,theFDA willtypicallyissueanapprovalletter. Notwithstandingthesubmissionofanyrequestedadditionalinformation,the FDAultimatelymaydecidethattheapplicationdoesnotsatisfytheregulatorycriteriaforapproval. Separate approvalisrequiredforeachproposedindication. Ifwewanttoexpandtheuseofanapprovedproduct,wewill havetodesignadditionalclinicaltrials,submitthetrialdesignstotheFDAforreviewandcompletethosetrials successfully.
Thetestingandapprovalprocessrequiressubstantialtime,effortandfinancialresources,andeachmay takeseveralyearstocomplete.Dataobtainedfromclinicalactivitiesarenotalwaysconclusive,whichcoulddelay, limitorpreventregulatoryapproval.TheFDAmaynotgrantapprovalonatimelybasis,oratall.Wemay encounterdifficultiesorunanticipated costsinoureffortstosecurenecessarygovernmental approvals,whichcould delayorprecludeusfrommarketingourproducts.TheFDAmaylimittheindications foruseorplaceother conditions,suchaspostapprovalstudies,onanyapprovalsthatcouldrestrictthecommercialapplicationofthe products. Afterapproval,sometypesofchangestotheapprovedproduct, suchasaddingnewindications, manufacturing changesandadditionallabelingclaims,aresubjecttofurthertestingrequirementsandFDAreview andapproval.
Post-ApprovalRequirements
Afterregulatoryapprovalofaproductisobtained,companiesarerequired tocomplywithanumber of post-approvalrequirements relatingto manufacturing, labeling,packaging,adverseeventreporting,storage, advertising,promotion,distributionandrecordkeeping.Forexample, asaconditionofapprovalofaBLA,the FDA mayrequirepost-approvaltestingandsurveillanceto monitor theproduct’ssafety orefficacy. Inaddition,holders ofanapprovedBLAarerequiredtokeepextensive records,toreportcertainadversereactionsandproduction deviationsandproblemstotheFDA,toprovideupdatedsafetyandefficacyinformation andtocomply with requirementsconcerningadvertisingandpromotionallabeling fortheirproducts. Ifwefailtocomplywiththe regulatoryrequirementsoftheFDAandother applicableU.S.andforeignregulatoryauthorities, orpreviously unknownproblemswithanyapprovedcommercialproducts,manufacturers or manufacturing processesare discovered, wecouldbesubject toadministrative orjudicially imposedsanctions orother setbacks. Accordingly, manufacturersmust continue toexpend time,moneyandeffort in the areaofproductionand qualitycontrolto maintaincompliance withcGMPandotheraspectsofregulatorycompliance.
Specifically,ourproductscouldbesubjecttovoluntaryrecallifweortheFDAdetermine,foranyreason, thatourproductsposeariskofinjuryorareotherwisedefective.Moreover,theFDAcanorderamandatoryrecall ifthereisareasonableprobabilitythatourdevicewouldcauseseriousadversehealthconsequencesordeath.In addition,theFDAcouldsuspendthe marketingoforwithdrawapreviouslyapprovedproductfromthemarketupon receiptofnewlydiscoveredinformationregardingthedrug’ssafetyoreffectiveness.
MedicalDeviceRegulation
Withrespectto medicaldevices,theFDAhasimplementednumerousregulationstoensurethatmedical productsdistributeddomesticallyorinternationallyaresafeandeffectivefortheirintendeduses.Theseinclude:
| · | productlistingandestablishmentregistration,whichhelpsfacilitateFDAinspectionsandother regulatoryaction; |
| · | theFDA’sQSR,whichrequiresmanufacturers,includingthirdpartymanufacturers,tofollowstringent design,testing,control,documentationandotherqualityassuranceproceduresduringallaspectsofthemanufacturingprocess; |
| · | labelingregulationsandFDAprohibitionsagainstthepromotionofproductsforuncleared, unapprovedoroff-labeluseorindications; |
| · | clearanceorapprovalofproductmodificationsthatcouldsignificantlyaffectsafetyorefficacyorthat wouldconstituteamajorchangeinintendeduse; |
| · | medicaldevicereportingregulations,whichrequirethatmanufacturerscomplywithFDArequirements toreportiftheirdevicemayhavecausedorcontributedtoadeathorseriousinjury,orhas malfunctionedinawaythatwouldlikelycauseorcontributetoadeathorseriousinjuryifthemalfunctionofthedeviceorasimilardeviceweretorecur; |
| · | post-approvalrestrictionsorconditions,includingpost-approvaltrialcommitments; |
| · | post-marketsurveillanceregulations,whichapplywhennecessarytoprotectthepublichealthorto provideadditionalsafetyandeffectivenessdataforthedevice;and |
| · | noticesofcorrectionorremovalandrecallregulations. |
FDA’sPremarket Clearanceand ApprovalRequirements
Unlessanexemptionapplies,oriftheFDAdeterminesotherwise,beforewecancommerciallydistributemedicaldevicesin theU.S.,we mustobtain,dependingonthetypeofdevice,eitherprior510(k)clearanceorPremarket Approval (“PMA”) fromtheFDA.TheFDAclassifiesmedicaldevicesintooneofthreeclasses:
| · | ClassIdevices(e.g.,lowriskdevices),whicharesubjecttoonlygeneralregulatoryoversight(e.g., labeling,medicaldevicesreporting,andprohibitionsagainstadulterationandmisbranding)and,in somecases,tothe510(k)premarketclearancerequirements; |
| · | ClassIIdevices(e.g.,greaterriskdevices),generallyrequiring510(k)premarketclearancebeforethey maybecommerciallymarketedintheU.S.;and |
| · | ClassIIIdevices(e.g.,greatestriskdevices),consistingofdevicesdeemedbytheFDAtoposethe greatestrisk,suchaslife-sustaining,life-supportingorimplantabledevices,ordevicesdeemednot substantiallyequivalenttoapredicatedevice,generallyrequiringsubmissionofaPMAsupportedby clinicaltrialdata. |
Allofourproductcandidatesarecurrentlyinthedevelopmentstage.DermaVir,ourlead candidate,is currentlyundergoingclinicalevaluation.Wearecurrentlydevelopingourregulatorystrategieswith respecttowhichregulatorypathwaywillbenecessarytoobtainclearanceorapprovaloftheDermaPrepcomponent, ifmedicaldeviceclearanceorapprovalisrequiredatall.
510(k)ClearancePathway
Whena510(k)clearanceisrequired,we mustsubmitapremarketnotificationdemonstratingthatour proposeddeviceissubstantiallyequivalenttoapreviouslycleared510(k)deviceoradevicethatwasincommercial distributionbeforeMay28,1976forwhichtheFDAhasnotyetcalledfor thesubmissionofPMAs.Byregulation, theFDAisrequiredtoclearordenya510(k)premarketnotificationwithin90daysofsubmissionof theapplication. Asapracticalmatter,clearancemaytakelonger.TheFDAmayrequirefurtherinformation,includingclinicaldata, to makeadeterminationregardingsubstantialequivalence.
Anymodificationtoa510(k)-cleareddevicethatwouldconstituteamajorchangeinitsintendeduse,or anychangethatcouldsignificantlyaffectthesafetyoreffectivenessofthedevice,requiresanew510(k)clearance andmayeven,insomecircumstances,requireaPMA,ifthechangeraisescomplexornovelscientificissuesorthe producthasanewintendeduse.TheFDArequireseverymanufacturertomakethedeterminationregardingthe needforanew510(k)submissionin thefirstinstance,buttheFDAmayreviewanymanufacturer’sdecision. We havemodifiedourdevicessincetheyreceivedtheFDAclearance. IftheFDAweretodisagreewithanyofour determinationsthatchangesdidnotrequireanew510(k),itcouldrequireustoceasemarketinganddistribution and/orrecallthe modifieddeviceuntil510(k)clearanceorPMAapprovalisobtained. IftheFDArequiresus to seek510(k)clearanceorPMAapprovalforanymodifications,we mayberequiredtoceasemarketingand/orrecall the modifieddevice,ifalreadyindistribution, until510(k)clearanceorPMAapprovalisobtainedandwecouldbe subjecttosignificantregulatoryfinesorpenalties.
ThereisnoguaranteethattheFDAwillgrant510(k)clearanceorPMAapprovalofourfutureproducts,if necessary,andfailuretoobtainnecessaryclearancesorapprovalsforourfutureproductswouldadverselyaffectour abilitytogrowourbusiness.Delaysinreceiptorfailuretoreceiveclearancesorapprovals,thelossofpreviously receivedclearancesorapprovals,orthefailuretocomplywithexistingorfutureregulatoryrequirementscould reduceoursales,profitabilityandfuturegrowthprospects.
PMAPathway
APMAmustbesubmittedtotheFDAifthedevicecannotbeclearedthroughthe510(k)process.A PMAmustbesupportedbyextensivedata,includingbutnotlimitedto,technical,preclinical,clinicaltrials,manufacturingandlabelingtodemonstratetotheFDA’ssatisfactionthesafetyandeffectivenessofthedevicefor its intendeduse.Nodevicethatweare marketingtodatehasrequiredpremarketapproval. Duringthereviewperiod, theFDAwilltypicallyrequestadditionalinformationorclarificationoftheinformationalreadyprovided. Also,an advisorypanelofexpertsfromoutsidetheFDAmaybeconvenedtoreviewandevaluatetheapplicationand providerecommendations totheFDAas to theapprovabilityofthedevice. TheFDAmayor maynotacceptthe panel’srecommendation. Inaddition,theFDAwillgenerallyconductapre-approvalinspectionof the manufacturingfacilityorfacilitiestoensurecompliancewiththeFDA’sQSR.
NewPMAsorPMAsupplementsarerequiredfor modificationsthataffectthesafetyoreffectivenessofthe device,including,forexample,certaintypesof modificationstothedevice’sindicationforuse,manufacturing process,labelinganddesign.PMAsupplementsoftenrequiresubmissionofthesametypeofinformationasa PMA,exceptthatthesupplementislimited toinformationneededtosupportanychangesfromthedevicecovered bytheoriginalPMAandmaynotrequireasextensiveclinicaldataor theconveningofanadvisorypanel.Thereis noguaranteethattheFDAwillgrantPMAapprovalofourfutureproducts,ifnecessary,andfailuretoobtain necessaryapprovalsforourfutureproductswouldadversely affectourabilitytogrowourbusiness. Delaysin receiptorfailuretoreceiveapprovals,thelossofpreviouslyreceivedapprovals,or thefailuretocomplywith existingorfutureregulatoryrequirementscouldreduceoursales,profitabilityandfuturegrowthprospects.
ClinicalTrials
ClinicaltrialsaregenerallyrequiredtosupportaPMAapplicationandaresometimesrequiredfor510(k) clearance.Suchtrialsgenerally requirean investigational deviceexemptionapplication(“IDE”), approvedin advancebytheFDA foraspecified numberofpatientsandtrialsites,unlesstheproductisdeemedanonsignificant riskdeviceeligiblefor more abbreviatedIDErequirements. Clinicaltrialsaresubjectto extensivemonitoring, recordkeepingandreportingrequirements. Clinicaltrialsmust beconductedundertheoversightofanIRBforthe relevant clinicaltrialsitesandmust comply withFDAregulations,includingbutnotlimited to thoserelatingto GCPs. Toconductaclinicaltrial,wearealso requiredto obtainthe patients’informed consentin formand substancethat complieswithbothFDA requirementsandstate andfederal privacyandhumansubject protection regulations.We,the FDAor the IRBcouldsuspenda clinicaltrialatany timefor various reasons,includingabelief thattherisksto trialsubjectsoutweighthe anticipatedbenefits. Evenif atrialis completed,the resultsof clinical testingmaynot adequatelydemonstrate the safetyandefficacy of thedevice or mayotherwisenotbesufficientto obtainFDA approval to marketthe product in the U.S.Similarly,in Europethe clinicaltrial must be approvedby a localethicscommittee andin somecases,including studieswithhigh-risk devices,by theministry of healthin the applicablecountry.
PervasiveandContinuingRegulation
Afteradeviceisplacedon the market,medicaldevicecompaniesarerequiredtocomplywithregulations relatingto manufacturing,labeling,packaging,adverseeventreporting,storage,advertising,promotion,distribution andrecordkeeping.IfwefailtocomplywiththeregulatoryrequirementsoftheFDAandotherapplicableU.S.and foreignregulatoryauthorities,orpreviouslyunknownproblemswithanyapprovedcommercialproducts,manufacturersor manufacturingprocessesarediscovered,we couldbesubjecttoadministrative orjudicially imposedsanctionsorothersetbacks. Accordingly,manufacturersmustcontinuetoexpendtime,moneyandeffort intheareaofproductionandqualitycontrolto maintaincompliancewithQSRandotheraspectsofregulatory compliance.
Specifically,ourproductscouldbesubjecttovoluntaryrecallifweortheFDAdetermine,foranyreason, thatourproductsposeariskofinjuryorareotherwisedefective.Moreover,theFDAcanorderamandatoryrecall ifthereisareasonableprobabilitythatourdevicecouldcauseseriousadversehealthconsequencesordeath.In addition,theFDAcouldsuspendthe marketingoforwithdrawapreviouslyclearedorapprovedproductfromthemarketuponreceiptofnewlydiscoveredinformationregardingthedrug’ssafetyoreffectiveness.
HealthCareRegulation
FraudandAbuse
Othergovernmentauthoritiesinfluencethesuccessofourbusiness,includingtheavailabilityofadequate reimbursementfromthirdpartypayors,includinggovernmentprogramssuchasMedicareandMedicaid.Medicare andMedicaidreimbursementpoliciescanalsoinfluencecorrespondingpoliciesofprivateinsurersandmanaged careproviders,whichcanfurtheraffectourbusiness.
Wemaydirectlyor indirectlybesubjecttovariousfederalandstatelawspertainingtohealthcarefraudand abuse,includinganti-kickbacklaws.Inparticular,thefederalhealthcareprogramsanti-kickbackstatuteprohibits personsfromknowinglyandwillfullysoliciting,offering,receivingorprovidingremuneration,directlyor indirectly,inexchangeforortoinduceeitherthereferralofan individual foragoodorservice,orthefurnishing, arrangingfororrecommendingagoodorservice,forwhichpaymentmaybe madeinwholeorpartunderfederal healthcareprograms,suchastheMedicareandMedicaidprograms.Theanti-kickbackstatuteisbroadandprohibits orrestrictsmanyarrangementsandpracticesthatarelawfulinbusinessesoutsideof thehealthcareindustry. In implementingthestatute,theU.S.DepartmentofHealthandHumanServices’OfficeofInspectorGeneralhas issuedaseriesofregulations,knownas the“safeharbors,”whichbeganinJuly1991. Thesesafeharborssetforth provisionsthat,ifalltheirapplicablerequirementsaremet,willassurehealthcareprovidersandotherpartiesthat theywillnotbeprosecutedunderthefederalanti-kickbackstatute.Thefailureofatransactionorarrangementtofit preciselywithinoneor moresafeharborsdoesnotnecessarilymeanthatitisillegalorthatprosecutionwillbe pursued. However,conductandbusinessarrangementsthatdonotfullysatisfyallrequirementsofanapplicable safeharbormay resultinincreasedscrutinybygovernmentenforcementauthoritiessuchastheOfficeofInspector General. Penaltiesforviolationsofthefederalanti-kickbackstatuteincludecriminalpenaltiesandcivilsanctions suchasfines,imprisonmentandpossibleexclusionfromMedicare,Medicaidandotherfederalhealthcareprograms.
TheFederal FalseClaims Actprohibitspersons fromknowingly filingorcausingtobefiledafalseclaim to,ortheknowinguseoffalsestatementstoobtainpayment from,thefederalgovernment. Certaintypesoflawsuits filedundertheFalseClaimsAct,knownas“quitam”actions,canbebroughtbyany individualonbehalfofthe government.Theseindividuals,sometimes knownas“relators”or,more commonly,as“whistleblowers,”may shareinanyamountspaidbytheentitytothegovernmentinfinesorsettlement.Thenumberoffilingsofquitam actionshasincreased significantlyinrecentyears,causingmore healthcarecompaniestohavetodefend aFalse Claimaction. Ifanentityisdeterminedtohaveviolated the federalFalseClaims Act,itmaybe requiredto payup to threetimesthe actualdamagessustained by the government,pluscivil penaltiesranging from$5,500to $11,000 for each separate falseclaim.Various stateshavealsoenacted similarlawsmodeledafter the federal FalseClaims Actwhich applyto itemsandservices reimbursedunder Medicaidandotherstate programs,or, in several states, applyregardless of thepayor.
HIPAAcreatedtwonewfederalcrimes:healthcarefraudandfalsestatementsrelatingtohealthcare matters.Thehealthcarefraudstatuteprohibitsknowinglyandwillfullyexecutingaschemetodefraudany healthcarebenefitprogram,includingprivatepayors.Aviolationofthisstatuteisafelonyandmayresultinfines, imprisonmentorexclusionfromgovernment sponsoredprograms. Thefalsestatements statuteprohibitsknowingly andwillfullyfalsifying,concealingorcoveringupamaterialfactor makinganymateriallyfalse,fictitiousor fraudulentstatementinconnectionwiththedeliveryoforpaymentforhealthcarebenefits,items,orservices.A violationof thisstatuteisafelonyandmayresultinfinesorimprisonment.
If anyofouroperationsarefoundtohaveviolatedorbe inviolationofanyofthelawsdescribedaboveand otherapplicablestateandfederalfraudandabuselaws,wemaybesubjecttopenalties,amongthembeingciviland criminalpenalties,damages,fines,exclusionfromgovernmenthealthcareprograms,andthecurtailment or restructuringofouroperations.
HealthInformation
U.S.federalandstatelawsprotecttheconfidentialityofcertainhealthinformation,inparticular individuallyidentifiableinformationsuchas medicalrecords,andrestricttheuseanddisclosureofthatprotected information.Atthefederallevel,HIPAAprotectshealthinformationbyregulatingitsuseanddisclosure,including forresearchpurposes.FailureofaHIPAA“coveredentity”(suchasahospitaloracademicmedicalcenter)to complywithHIPAAcouldconstituteaviolationoffederallaw,subjecttocivilandcriminalpenalties.Wearenot directlysubjecttotheHIPAArulesasa“coveredentity,”however,andunderHIPAAwewouldbepermittedto obtaininformationfromourcustomersundercertainconditions,suchaswhenrelevanttoourresponsibilitiesfor overseeingthequality,safety,oreffectivenessofourproducts. Nevertheless,becauseconductbyapersonthatmay notbeprosecuteddirectlyunderHIPAA’scriminalprovisionscouldpotentiallybeprosecutedunderaidingand abettingorconspiracylaws,weareunabletodeterminewhetherouractionscouldbesubjecttoprosecutionin the eventofanimpermissible disclosureofdatatous.
Inaddition,manystatelawsapplytotheuseanddisclosureofhealthinformation,whichcouldaffectthemannerinwhichweconductourresearchanddevelopment,aswellasotheraspectsofouroperations.Moreover, suchlawsarenotnecessarilypreemptedbyHIPAAanditsrules,inparticularthosestatelawsthataffordgreater privacyprotectiontotheindividualthan HIPAAand/orthatprotectcertaincategoriesofinformation,suchas HIV/AIDS-relatedinformation. Suchstatehealthinformationconfidentiality lawstypicallyhavetheirownpenaltyprovisions,whichcouldbeappliedintheeventofanunlawfulactionregardinghealthinformation.
International GovernmentRegulation
Internationalsalesofbiologicalsubstancesandmedicaldevicesaresubjecttoforeigngovernment regulations,whichvarysubstantiallyfromcountrytocountry.Thetimerequiredtoobtainapprovalbyaforeign countrymaybelongerorshorterthanthatrequiredforFDAlicensureclearanceorapproval,andtherequirementsmaydiffer.
TheE.U.,whichconsistsof25ofthe majorcountriesinEurope, hasadoptednumerous directivesand standardsregulatingthedesign,manufacture,clinicaltrials,labeling, andadverseeventreportingfor medical devices.Other countries,such asSwitzerland,havevoluntarily adoptedlawsand regulationsthatmirrorthoseof the E.U.with respectto medicaldevices.Devicesthatcomplywiththerequirements ofarelevantdirectivewillbe entitledtobearCEconformitymarkingand,accordingly,can becommercially distributedthroughoutthe member statesoftheE.U.,andothercountriesthatcomply withormirror thesedirectives.Themethodofassessing conformity variesdepending onthetypeandclassofthe product,butnormallyinvolves acombinationofself- assessment bythe manufacturerandathirdpartyassessmentbya “NotifiedBody,”anindependentand neutral institutionappointedtoconductconformity assessment.This thirdpartyassessmentconsistsofanaudit ofthe manufacturer’s qualitysystemandtechnicalreviewandtestingof the manufacturer’s product. Anassessmentbya NotifiedBodyinonecountrywithinthe E.U.isrequired inorderforamanufacturer tocommercially distributethe productthroughout theE.U. Inaddition,compliancewithvoluntaryharmonizing standardsInternationalStandards Organization9001andInternationalStandards Organization13845 issuedbytheInternational Organization for Standardsestablishesthepresumption ofconformity withtheessential requirements foraCEmarking.
ESTIMATE OF THE AMOUNT SPENT ON RESEARCH AND DEVELOPMENT
Research and development expenses were $0 and $0 in 2013 and 2012, respectively. We are focused on regulatory issues, including the deliver of data from all clinical trials to data. Currently no research and development is required.
COSTS AND EFFECTS OF ENVIRONMENTAL COMPLIANCE
The Company has not spent any sums on environmental compliance and does not expect to be required to spend any sums on environmental compliance in the future.
EMPLOYEES
We currently have eight (8) employees, of which five (5) are full-time and three (3) are part-time.
AVAILABLE INFORMATION
We file electronically with the U.S. Securities and Exchange Commission (SEC) our annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934. We make available on our website at www.powerofthedream.com, free of charge, copies of these reports as soon as reasonably practicable after filing these reports with, or furnishing them to, the SEC. The public can also obtain materials that we file with the SEC through the SEC’s website at http://www.sec.gov or at the SEC’s Public Reference Room at 100 F Street, NE, Washington, DC 20549. Information on the operation of the Public Reference Room is available by calling the SEC at 800-SEC-0330.
Item 1A. Risk Factors.
You should carefully consider the risks described below, together with all of the other information included in this report, in considering our business and prospects. The risks and uncertainties described below are not the only ones facing the Company. Additional risks and uncertainties not presently known to us or that we currently deem immaterial also may impair our business operations. The occurrence of any of the following risks could harm our business, financial condition or results of operations.
Risks Related to the Our Business and Industry
WE HAVE A LIMITED OPERATING HISTORY, WHICH MAKES YOUR EVALUATION OF OUR BUSINESS DIFFICULT. WE HAVE INCURRED LOSSES IN RECENT PERIODS FOR START-UP EFFORTS AND MAY INCUR LOSSES IN THE FUTURE.
Our future is dependent upon our ability to obtain financing and upon future profitable operations from the commercialization of our product candidates. Oursuccessandabilitytogeneraterevenueorbe profitablewilldependonourabilityto,amongotherthings,undertakepre-clinicaldevelopmentstudiesandclinical trials;applyforandreceiveregulatoryapprovalsforconductofclinicaltrialsandformarketingofourproducts whendevelopmenthasbeencompleted;formulateandmanufactureproducts;andconductsalesandmarketing activities. These factors raise substantial doubt that we will be able to continue as a going concern. From April 26, 2006 (inception) through December 31, 2013, we incurred aggregate losses of $12,817,575 and anticipate incurring additional losses for at least the next twelve (12) months.
Our auditors have expressed substantial doubt about our ability to continue as a going concern.
As of December 31, 2013, we had $22,922 of cash and $8,240,680 in current obligations. In addition, we anticipate our new operating expenses to be between $2,000,000 - $3,000,000 for year 2014. We do not have sufficient resources to satisfy our current obligations nor to fund our planned operations for the next 12 months. We will require additional financing to sustain our operations and without it we will not be able to continue operations.
Our ability to obtain additional funding will determine our ability to continue as a going concern. Our financial statements do not include any adjustments that might result from the outcome of this uncertainty.
Management believes the Company has enough existing cash and cash equivalent and can raise adequate capital to keep the Company functioning through December 31, 2014. However, the need may arise, to raise additional capital if we want to supportour futuredevelopmentprograms,including thecompletionof additionalclinicaltrials.Ourfundingrequirementsmayalso increaseorchangeasaresultof manyfactors,includingdelaysindevelopmentactivities,underestimatesofbudget items,unanticipatedcashrequirements,limitationofdevelopmentofnewpotentialproducts,futureproduct opportunitieswithcollaborators,futurelicensingopportunitiesandfuturebusinesscombinations. No assurance can be given that the Company can obtain additional working capital, or if obtained, that such funding will not cause substantial dilution to shareholders of the Company. If the Company is unable to raise additional funds, if needed, it maynotbeableto(i)complete plannedpre-clinicalstudiesandclinicaltrials,(ii)completeconstructionofamanufacturingfacility,or(iii)obtain conditional marketingapproval for DermaVirfromthe EMAand/orotherregulatoryauthorities. Being a development stage company, the Company is subject to all the risks inherent in the establishment of a new enterprise and the marketing and manufacturing of a new product, many of which risks are beyond the control of the Company. All of the factors discussed above raise substantial doubt about the Company’s ability to continue as a going concern.
Our future is dependent upon our ability to obtain financing. If we do not obtain such financing, we maybeforcedtodiscontinueproductdevelopment,curtailoperations,reduceorforegosalesandmarketingefforts andloseattractivebusinessopportunities. We have no arrangements or agreements with any person regarding any potential future financings.
WE CURRENTLY DO NOT HAVE, AND MAY NEVER DEVELOP OR ACQUIRE ANY COMMERCIALIZED PRODUCTS FOR DISTRIBUTION AND SALE.
We currently do not have any commercialized product(s) or any significant source of revenue. We havedevoted significantfinancialresourcestoresearchanddevelopmentofourproductcandidates,buthavenotyet commercializedanyof them.Asaresult,wehavenevergeneratedanyrevenuefromproductsales.Untiland unlesswereceiveapprovalfromthe EMA,FDAandforeignregulatoryauthoritiesto marketaproductcandidate,we cannotsellanyproductandwillhavenoproduct revenues.
PhysiciansandpatientsmaynotacceptanduseourImmuneTherapies.
Evenifthe EMA/FDAand/orforeignagenciesapproveoneofourImmuneTherapiesfor marketing,physiciansand patientsmaynotacceptanduseit.Acceptanceanduseofourproductswilldependuponanumberoffactors, includingperceptionsbythehealthcarecommunity,includingphysicians,abouttheirsafetyandeffectiveness,their cost-effectivenessrelativetocompetingproducts,availabilityofreimbursementforourproductsfromgovernment orotherhealthcarepayors,andtheeffectivenessof marketinganddistributioneffortsbyus,ourlicenseesand distributors,ifany.
Becauseweexpectsalesofourcurrentleadproductcandidate,if approvedfor marketing,togenerate substantiallyallofourproductrevenuesfor theforeseeablefuture,thefailureofthisproducttofindmarket acceptancewouldharmourbusinessandcouldrequireus toseekadditionalfinancingortakeothermeasures, possiblyretrenching.
Ourabilitytogenerateproductrevenueswillbediminishedifourproductssellforinadequatepricesor patientsareunabletoobtainadequatereimbursement.
Ourabilitytocommercializeourproducts,aloneorwithcollaborators,willdependinpartontheextentto whichreimbursementwillbeavailablefromgovernmentandhealthadministrationauthorities,privatehealthmaintenanceorganization,healthinsurers,andotherpayors.
Significantuncertaintyexistsasto thereimbursementstatusofnewlyapprovedhealthcareproducts. Healthcarepayors,includingMedicare,routinelychallengepricescharged.Governmentandotherhealthcare payorsincreasinglyattempttocontainhealthcarecostsbylimitingbothcoverageandreimbursement.IntheU.S., therehavebeenanumberof legislativeandregulatoryproposalstochangethehealthcaresysteminwaysthatcould affectourabilityto marketandsellourproductcandidatesprofitably. Althoughwecannotpredictthefulleffecton ourbusinessoftheimplementation ofanylegislation,webelievethatlegislationthatreducesreimbursementforour productscouldadverselyimpacthowmuchorunderwhatcircumstanceshealthcareproviderswillprescribeor administerourproducts. Thiscouldmateriallyandadverselyimpactourbusinessbyreducingourabilityto generaterevenue,raisecapital,obtainadditionalcollaboratorsandmarketourproducts.Inaddition,webelievethe increasingemphasison managedcarein theU.S.hasandwillcontinuetoputpressureonthepriceandusageof pharmaceuticalproducts,whichmayadverselyimpactproductsales.
Ourproductdevelopmentprogramsdependuponthirdpartyresearchersandwecannotcontrolthe amountoftimingorresourcestheydevotetoourprogramsorthequalityoftheirperformance.
Wedependin largepartuponindependentinvestigatorsandcollaborators,suchasuniversities,medical institutions,andclinicalresearchorganizations,toconductourbasicresearch,animalstudies,pre-clinicalstudies andclinicaltrialsand,incertaincases,todevelopaproductforitstargetindication.Althoughwecontractwith thesecollaborators,theyarenotouremployees,andwecannotcontroltheamountortimingofresourcesthey devotetoourprograms. Theymaynotassignasgreataprioritytoourprogramsorpursuethemasdiligentlyaswe wouldifwewereundertakingthemourselves.Ifoutsidecollaboratorsfailtodevotesufficienttimeandresourcesto ourprograms,oriftheirperformanceissubstandard,theapprovalofourFDAandEMAapplications,andour introductionofnewproducts,ifany,willbedelayed. Thesecollaboratorsmayalsohaverelationshipswithother commercialentities,someofwhommaycompetewithus. Ifourcollaboratorsweretoassistourcompetitorsatour expense,ourcompetitivepositionwouldbeharmed.
Wecurrentlyrelyonthirdpartiestomanufactureandsupplyuswiththerawmaterialsthat weassembletomakeupourProductcandidates,andanyinterruptionsinthemanufactureorsupplyoftheseraw materialscouldharmourbusiness.
Wecurrentlyrelyonalimitednumberofvendorstosupplyrawmaterialsforourproduct candidates,andthelossofoneofthesepartiescouldsignificantlyharmourbusiness.For the clinical trials we have conducted up to date wereliedonAlthea Technologies,Inc.,aDNAmanufacturingcompanylocatedintheU.S.,asthesourceforoursupply ofpDNA,theAPIinDermaVir.We havealsoreliedonPolyPlusTransfectionSA,amanufacturerin France,as thesourceforour clinical supply of the raw material polyethylenimine mannose (“PEIm”) used in DermaVir. DermaPrep was manufactured by Tapemark, a medical devicemanufacturingcompanylocatedintheU.S.The finished DermaVir product was manufactured at the clinical pharmacy prior to DermaVir immunization of patients.
In order to obtain the raw materials in the required quality and quantity we contacted several alternative CMOs. We tested the quality of pDNAs contract manufactured by Aldevron and WGX, two DNAmanufacturingcompanieslocatedintheU.S. Both pDNA materials could fulfill our quality criteria. To mitigate the risk on manufacturing CMOs (1) we licensed pDNA manufacturing technology from Aldevron for the high quality purification of our pDNA and (2) developed our own chemical synthesis for the manufacturing of PEIm. We also developed a commercial manufacturing method for our finished products. Therefore, based on our own technology our cGMP manufacturing facility could be built and we could be independent from any CMO to produce enough material for clinical trials. However, to build and staff such facility will be costly and time consuming. Therefore, if either of our selected CMO fails to provide us with sufficient quantities, we may not be able to obtain an alternative supplyonatimelyorcommerciallyacceptablebasis.Evenifweareabletoreachan agreementwithback-upsuppliers,achangeinsuppliermayrequirecomparabilitytesting,includingclinicaltests,to demonstratethatthenewrawmaterialwillnotsubstantiallychangethefinishedProduct.Furthermore,evenifan alternativesourceis available,therecanbenoassurancethatsuchalternativesourcewouldbecapableofproducing therawmaterialsinsufficientquantitiestomeetourtotalrequirements. Anysuchinterruptionwoulddisruptour abilityto manufacturetheDermaVirandcouldhaveamaterialadverseeffectonourbusiness.
We identified Stokvis Tapes Group a Hungarian manufacturer with worldwide presence specialized for patches for health industry. Stokvis presented us a sample batch of DermaPrep to demonstrate their capability to manufacture our DermaPrep medical device. The sample batch fulfilled our quality criteria. We plan to contract Stokvis to deliver DermaPrep for clinical trials. This company would also have the capability and capacity to produce our device also in commercial quantity.
If thereisasignificantincreaseinthepriceofrawmaterials or DermaPrep,ourbusinesscouldbeseriouslyharmed.Wecurrently havenosupplycontractsbeyondcurrentclinicaltrials.Wemaynotbeabletocontractwith thirdpartiestoprovidetheAPIand DermaPrepbeyondcurrentclinicaltrialsonacceptabletermsandatcommercially reasonableprices,ifatall.Thenumberofthirdpartieswiththeexpertise,requiredregulatoryapprovals,and facilitiestosupplytheAPIandthe finished Productonacommercialscaleislimited.Evenifwe locatethirdpartiesthatcan providecommercialsupplies,theFDAandcomparableforeignregulatorsmustapprovethesemanufacturers’ facilitiesandprocessespriortoouruse,whichwouldrequirenewtestingandcompliance inspections. Anyof these factorscouldcauseus todelayorsuspendregulatorysubmissions, requiredapprovalsorcommercialization of DermaVir.Theycouldalsoentailhighercosts,andcouldresultinourbeingunabletocommercialize DermaVirsuccessfully. Furthermore,ifourcontractmanufacturers failtodelivertherequiredcommercial quantitiesofbulkrawmaterials onatimelybasisandatcommercially reasonable prices,andifwewereunableto findoneor morereplacementmanufacturers capableofproductionata substantially equivalentcost,insubstantially equivalentvolumesandquality,andonatimelybasis,wearelikelytobeunableto meetthedemandforDermaVir,ifmarketingapprovalisobtained,andwewouldlosepotentialrevenue.
WE DO NOT CURRENTLY HAVE SALES, MARKETING AND DISTRIBUTION CAPABILITIES, AND WE MAY BE UNABLE TO EFFECTIVELY SELL, MARKET AND DISTRIBUTE OUR PRODUCT CANDIDATES IN THE FUTURE, WHICH WOULD HAVE AN ADVERSE EFFECT ON OUR BUSINESS.
Wecurrentlyhavenointernalsalesordistributioncapabilities.Inordertosuccessfullycommercializeany ofourproductcandidates,wemusteitherinternallydevelopsales,marketinganddistributioncapabilitiesor make arrangementswiththirdpartiestoperformtheseservices.Fortheforeseeablefuture,we maynotbeableto establishmarketingandsalescapabilitiesinternallyorhireasufficientnumberofsalespersonnelwithappropriate expertiseto marketandsellourproductswhentheproductsareapprovedbyappropriateregulatoryauthorities. Therefore,wemayneedtoenterintoanalliancewithamajorpharmaceuticalcompanyorotherthirdpartyto marketandsellourproducts,whichwe maybeunabletodo.Evenifweareabletoidentifyoneor moreacceptable collaboratorstoperformtheseservicesforus,wemaynotbeabletoenterintocollaborativearrangements on favorableterms,oratall.
Ifweenterintocollaborativearrangementsforthe marketingorsaleofourproducts,ourproductrevenues arelikelytobelowerthanifwedirectlymarketedandsoldourproducts.Inaddition,anyrevenueswereceive woulddependupontheeffortsofourcollaborators,whichmaynotbeadequateduetolackofattentionorresource commitments,managementturnover,and/orchangeofstrategicfocus,businesscombinationsorotherfactors outsideourcontrol.Dependingon thetermsofourcollaboration,theremedieswehaveagainstanunderperforming collaboratormaybelimited. Ifweweretoterminatetherelationship,it couldbedifficultorimpossibletofind a replacementcollaboratoronacceptableterms,ifatall.
Ifwecannotcompetesuccessfullyformarketshareagainstotherdrugcompanies,wemaynotachieve sufficientproductrevenues,andourbusinesswillsuffer.
TheHIV/AIDSmarketforourleadproduct,DermaVir,ischaracterizedbyintensecompetition andrapidtechnologicaladvances.Ifanyofourproductcandidatesreceivemarketingapproval,itwillcompetewith anumberofexistingandfuturedrugsandImmuneTherapiesdeveloped,manufacturedandmarketedbyothers.The extenttowhichDermaViroranyofourother Productcandidatesachievesmarketacceptancewilldepend inpartonourabilitytocompetewiththeseothercompanies.Competition inthepharmaceuticalandbiotechnology industriesisintenseandhasbeenaccentuated bytherapidpaceoftechnologydevelopment. Ifourcompetitors developandcommercializeproductsfasterthanwedo,ordevelopandcommercializeproducts thataresuperiorto ourproductcandidates,ourcommercialopportunitieswillbereducedoreliminated. Existingorfuturecompeting productsmayprovidegreatertherapeutic convenienceorclinicalorotherbenefitsfora specificindicationthanour products,or mayoffercomparable performance atalowercost.Ifourproductsfailtocaptureandmaintainmarket share,we maynotachievesufficientproductrevenues,andourbusinesswillsuffer.
Wecompeteagainstlarger,fullyintegratedpharmaceuticalcompanies,smallercompaniesthatcollaborate withlargerpharmaceuticalcompanies,academicinstitutions,governmentagenciesandotherpublicandprivate researchorganizations.CompaniesthatcurrentlysellHIV/AIDSproductsincludeBristol-MyersSquibbCompany, F.Hoffmann-LaRocheLtd.,GileadSciences,Inc.,GlaxoSmithKlineplc.,Merck& Co., Inc.,Novartis AG,PfizerInc.,Sanofi-AventisU.S.LLC. Alternativetechnologiesarebeingdeveloped toimproveuponorreplacecurrentproductsforthetreatmentofHIV/AIDS,severalofwhichare inclinicaltrialsor areawaitingmarketingapproval. Inaddition,companiespursuingdifferentbutrelatedfieldsrepresentsubstantial competition.Manyofthesecompetitors,eitheraloneortogetherwiththeircollaborative partners,operatelarger researchanddevelopmentprogramsandhavesubstantiallygreaterfinancialresourcesthanwedo. Additionally, theyhavesignificantlygreaterexperienceindevelopingdrugs,undertakingpre-clinicalstudiesandhumanclinical trials,obtaining EMA andFDAandotherregulatoryapprovalsandformulating,manufacturing,launching,marketingand sellingdrugs. Theseorganizationsalsocompete withustoattractqualifiedpersonnel,licenseproprietary technologythatiscompetitivewiththetechnologywearedeveloping,attractfunding,andattractpartiesfor acquisitions,jointventuresorothercollaborations.
Wemaybeexposedtoliabilityclaimsassociatedwithusinghazardousmaterialsandchemicals.
Ourresearchanddevelopmentactivitiesinvolvethecontrolleduseofhazardousmaterialsandchemicals. Althoughwebelieveoursafetyproceduresforusing,storing,handling,anddisposingofthesematerialscomply withapplicableenvironmentallawsandregulations,wecannoteliminatetheriskofaccidentalinjuryor contaminationfromthesematerials.Intheeventofsuchanaccident,wecouldbeheldliableforresultingdamages, whichcouldmateriallyadverselyaffectourbusiness,financialconditionandresultsofoperations.Inaddition,laws andregulationsgoverningtheuse,manufacture,storage,handling,anddisposalofhazardousorradioactivematerialsandwasteproductsmayrequireusto incursubstantialcompliancecoststhatcouldmateriallyadversely affectourbusiness,financial conditionandresultsofoperations.
Wemayincursubstantialliabilitiesandberequiredtolimitcommercializationofourproductsin responsetoproductliabilitylawsuits.
Ourproposedproductscouldbe thesubjectofproductliabilityclaims.Afailureofourproductcandidates tofunctionasanticipated,whetherasaresultofthedesignoftheseproducts,unanticipatedhealthconsequencesor sideeffects,ormisuseor mishandlingbythirdpartiesofsuchproducts,couldresultin injury.Wemaybeheld liableifseriousadversereactionsfromtheuseofourproductcandidatesoccureitherinclinicaltrialsorin subsequentmarketing.Ifwecannotsuccessfully defendourselvesagainstproductliabilityclaims,wemayincur substantialliabilitiesorberequiredtolimitcommercialization ofourproductcandidates. Ourinabilitytoobtain sufficientproductliabilityinsuranceatacceptablecostagainstclaimscouldpreventorinhibitthecommercialization ofbiopharmaceutical productswedevelop,alone orwithcollaborators. Wecurrentlycarryclinicaltrialinsurance butdonotcarryproductliabilityinsurance. We,oranycollaborators,maynotbeabletoobtaininsuranceat reasonablecost,ifatall. Agreements withfuturecollaborators entitlingustoindemnification againstlossesmaynot beadequateifclaimsarise. Wemayalsoberequiredto indemnify collaborators againstlossesarisingoutof productliabilityclaims.
THERE ARE RISKS ASSOCIATED WITH OUR PROPOSED OPERATIONS IN HUNGARY.
Special risks may be associated with our efforts to undertake operations in the Republic of Hungary. Such operations will be subject to political, economic and other uncertainties, including among other things, import, export and transportation regulations, tariffs, taxation policy, including royalty and tax increases and retroactive tax claims, exchange controls, currency fluctuations and other uncertainties arising out of the Republic of Hungary’s sovereignty over our operations.
FLUCTUATION IN THE VALUE OF THE HUNGARIAN FORINT RELATIVE TO OTHER CURRENCIES MAY HAVE A MATERIAL ADVERSE EFFECT ON OUR BUSINESS AND/OR AN INVESTMENT IN OUR SHARES.
We maintain our books in local currency: U.S. dollars for Power of the Dream and for Genetic Immunity, Inc. in the United States and the Hungarian Forint for Vidatech and Genetic Immunity, Kft in Hungary. Our operations are conducted primarily outside of the United States through Vidatech and Genetic Immunity, Kft., our wholly-owned subsidiaries. As a result, fluctuations in currency exchange rates may significantly affect our sales, profitability and financial position when the foreign currencies, primarily the Hungarian Forint, of our international operations are translated into U.S. dollars for financial reporting. During 2013, the Hungarian Forint has fluctuated between HUF 240 and HUF 205 to the U.S. dollar. In addition, we are also subject to currency fluctuation risk with respect to certain foreign currency denominated receivables and payables. Although we cannot predict the extent to which currency fluctuations may or will affect our business and financial position, there is a risk that such fluctuations will have an adverse impact on our sales, profits and financial position. Because differing portions of our revenues and costs are denominated in foreign currency, movements could impact our margins by, for example, decreasing our foreign revenues when the dollar strengthens and not correspondingly decreasing our expenses. We do not currently hedge our currency exposure. In the future, we may engage in hedging transactions to mitigate foreign exchange risk.
OUR MANAGEMENT TEAM HAS LIMITED EXPERIENCE IN PUBLIC COMPANY MATTERS, WHICH COULD IMPAIR OUR ABILITY TO COMPLY WITH LEGAL AND REGULATORY REQUIREMENTS.
Our management team has had limited experience managing a U.S. public company, which could impair our ability to comply with legal and regulatory requirements, such as the Sarbanes-Oxley Act of 2002 and applicable federal securities laws, including filing on a timely basis required reports and other required information. Our management may not be able to implement programs and policies in an effective and timely manner that adequately responds to increased legal or regulatory compliance and reporting requirements imposed by such laws and regulations. Our failure to comply with such laws and regulations could lead to the imposition of fines and penalties and further result in the deterioration of our business.
INVESTOR CONFIDENCE AND THE MARKET PRICE OF OUR SHARES MAY BE ADVERSELY IMPACTED IF WE ARE UNABLE TO ISSUE AN UNQUALIFIED OPINION ON THE ADEQUACY OF OUR INTERAL CONTROLS OVER OUR FINANCIAL REPORTING.
We are subject to the reporting requirements of the U.S. Securities and Exchange Commission, or SEC. The SEC, as directed by Section 404 of the U.S. Sarbanes-Oxley Act of 2002, adopted rules requiring public companies to include a report of management on their internal control structure and procedures for financial reporting in their annual reports on Form 10-K that contain an assessment by management of the effectiveness of their internal controls over financial reporting. These requirements first applied to our annual report on Form 10-KSB for the fiscal year ending on December 31, 2007. Our management may conclude that our internal controls over financial reporting are not effective. Any failure to implement effective controls, or difficulties encountered in their implementation, could harm our operating results or cause us to fail to meet our reporting obligations. Even if our management concludes that our internal controls over financial reporting are effective, our independent registered public accounting firm may issue a report that is qualified if it is not satisfied with our controls or the level at which our controls are documented, designed, operated or reviewed, or if it interprets the relevant requirements differently from us. In the event that we are unable to have effective internal controls, investors and others may lose confidence in the reliability of our financial statements and our ability to obtain equity or debt financing as needed could suffer.
CONCENTRATION OF OWNERSHIP AMONG OUR DIRECTORS, EXECUTIVE OFFIERS AND PRINCIAPL SHAREHOLDERS MAY PRESENT NEW INVESTORS FROM INFLUENCE SIGNIFICANT CORPORATE DECISIONS.
Based upon beneficial ownership as of December 31, 2013, our directors, executive officers and holders of more than 5% of our common stock, alone or together with their affiliates own, in the aggregate, approximately 12.% of our outstanding shares of common stock. Our executive officer, Viktor Rozsnyay, and Peter Boros, Chief Executive Officer of iGlue, Inc., own 100% of our Series A Preferred Stock, which gives them majority voting power. As a result, these shareholders, will be able to exercise a controlling influence over matters requiring shareholder approval, including the election of directors and approval of significant corporate transactions, and will have significant control over our management and policies. Some of these persons or entities may have interests that are different from yours. For example, these shareholders may support proposals and actions with which you may disagree or which are not in your interests. The concentration of ownership could delay or prevent a change in control of our company or otherwise discourage a potential acquirer from attempting to obtain control of our company, which in turn could reduce the price of our common stock. In addition, these shareholders, some of whom have representatives sitting on our Board of Directors, could use their voting influence to maintain our existing management and directors in office, delay or prevent changes of control of our company, or support or reject other management and board proposals that are subject to stockholder approval, such as amendments to our employee stock plans and approvals of significant financing transactions.
WE DEPEND ON KEY EMPLOYEES, AND IF WE FAIL TO ATTRACT AND RETAIN EMPLOYEES WITH EXPERTISE REQUIRED FOR OUR BUSINESS, WE CANNOT GROW OR ACHIEVE PROFITABILITY.
Wearedependentonourexecutiveofficers and key employees,primarily Viktor Rozsnyay, our Chief Executive Officer of Power of the Dream, Ildiko Rozsa, Chief Financial Officer of Power of the Dream and Peter Boros a member of the board of directors.Wedonothave“keyperson”lifeinsurancepoliciesforanyof ourofficers.Thelossofthe technicalknowledgeandmanagementandindustryexpertiseofanyofourkey personnelcouldresultindelaysinproductdevelopment,lossofcustomersand sales,if any,anddiversionofmanagementresources,whichcouldadverselyaffectouroperatingresults. Inaddition,werelyonclinicalandregulatoryadvisorstoassistus informulatingthedevelopmentstrategy. Ouradvisorshaveotherjobsandcommitments,andmaybesubjecttonon-disclosureobligationsthatmaylimit theiravailabilitytoworkwithus.
Ifweareunabletohirequalifiedpersonnel,ourabilitytogrowourbusinessmaybeharmed.Wemust hireandretainskilledemployeesinatightlabormarket.
Attractingandretainingqualifiedpersonneliscriticaltooursuccess.Weneedtohireadditionalqualified personnelwithexpertiseinbasicandclinicalresearch,governmentregulation,formulationandmanufacturing,and salesandmarketing.Wewillrequireadditionalscientificpersonnelin manyfields,someofwhichareaddressedby relativelyfewcompanies. Asaresult,dependinguponthesuccessandthetimingofclinicaltrials,wemay experiencedifficultyinhiringandretaininghighlyskilledemployees.
Wecompeteforqualifiedindividualswithnumerouspharmaceuticalandbiotechnologycompanies, universitiesandotherresearchinstitutions,someofwhichare moreestablishedthanweareandhavetheabilityto paymorecashcompensationthanwedo.Competitionforsuchindividuals,particularlyin Budapest,Hungary,isintense,andwecannot becertainthatoursearchforsuchpersonnelwillbe successful. Ifweareunabletohireandretainskilledscientists,ourbusiness,financialcondition,operatingresults andfuture prospectscouldbemateriallyadverselyaffected.
Ifweareunabletoobtainadequateinsurance,ourfinancialconditioncouldbeadverselyaffectedinthe eventofuninsuredorinadequatelyinsuredlossordamage.Ourabilitytoeffectivelyrecruitandretainqualified officersanddirectorscouldalsobeadverselyaffectedifweexperiencedifficultyinobtainingadequatedirectors’ andofficers’liabilityinsurance.
Wemaynotbeabletoobtaininsurancepoliciesontermsaffordabletousthatwouldadequatelyinsureour businessandpropertyagainstdamage,lossorclaimsbythirdparties.Totheextentourbusinessorpropertysuffers anydamages,lossesorclaimsbythirdparties,whicharenotcoveredoradequatelycoveredbyinsurance, our financialconditionmaybemateriallyadverselyaffected.
Wemaybeunableto maintainsufficientinsuranceasapubliccompanytocoverliabilityclaimsmade againstourofficersanddirectors.Ifweareunabletoadequatelyinsureourofficersanddirectors,wemaynotbe abletoretainorrecruitqualifiedofficersanddirectorsto manageus.
OUR COMPLIANCE WITH CHANGING LAWS AND RULES REGARDING CORPORATE GOVERNANCE AND PUBLIC DISCLOSURE MAY RESULT IN ADDITIONAL EXPENSES TO US WHICH, IN TURN, MAY ADVERSELY AFFECT OUR ABILITY TO CONTINUE OUR OPERATIONS.
Keeping abreast of, and in compliance with, changing laws, regulations and standards relating to corporate governance and public disclosure, including the Sarbanes-Oxley Act of 2002, new SEC regulations and, in the event we are ever approved for listing on either NASDAQ or a registered exchange, NASDAQ and stock exchange rules, will require an increased amount of management attention and external resources. We intend to continue to invest all reasonably necessary resources to comply with evolving standards, which may result in increased general and administrative expenses and a diversion of management time and attention from revenue-generating activities to compliance activities. This could have a materially adverse effect on our ongoing operations.
Risks Related to Regulatory Environment
NONE OF OUR PRODUCT CANDIDATES HAVE BEEN APPROVED BY E.U., U.S., AND/OR WORLDWIDE REGULATORY AUTHORITIES FOR COMMERCIAL SALE, AND WE MAY NEVER RECEIVE SUCH APPROVALS. OBTAINING AND MAINTAINING THE NECESSARY E.U. OR U.S. AND/OR WORLDWIDE MARKETING APPROVALS FOR OUR PRODUCT CANDIDATES MAY BE TIME CONSUMING, DIFFICULT, COMPLEX AND COSTLY, PARTICULARLY SINCE OUR LEAD PRODUCT AND OTHER PRODUCT CANDIDATES ARE COMBINATION PRODUCTS BASED ON NOVEL MECHANISMS OF ACTION. IF WE FAIL TO OBTAIN AND MAINTAIN THE NECESSARY APPROVALS, WE WILL BE UNABLE TO COMMERCIALIZE OUR PRODUCT CANDIDATES.
Noneofourproductcandidateshavebeenapprovedby the E.U. orU.S.orworldwideregulatoryauthoritiesfor commercialsale,andwe mayneverreceivesuchapprovals.Allofourproductcandidatesare inpre-commercial stages,andwedonotexpectanyofourproductcandidatestobecommerciallyavailableforseveralyears,ifatall. Governmentregulationsinthe E.U. or theU.S.andothercountrieshaveasignificantimpactonourbusinessandaffectresearch anddevelopment,manufactureand marketingofourproducts.Wewillneed EMA andFDAapprovaltocommercialize our productcandidatesinthe E.U, theU.S.,andapprovalsfromsimilarforeignregulatoryauthoritiestocommercialize our productcandidatesinothersuchjurisdictions.Inordertoobtain EMA andFDAmarketingapprovalofaproductcandidate, we mustsubmittothe EMA and theFDAaBiologicsLicenseApplication(a“BLA”)demonstrating, tothesatisfactionof the EMA and the FDAandotherregulatoryauthorities,thattheproductcandidateissafeforhumansandeffectiveforits intended use. Thisdemonstrationrequiressignificantlaboratoryresearchandanimal tests,whicharereferredtoaspre- clinicalstudies,aswellasadequateandwell-controlled humantests,whicharereferredtoasclinicaltrials. These studiesandtrialsmaybetimeconsuming,difficultandcostly,andwecannotpredictwhetherourresearchand clinicalapproacheswillresultinproductsthe EMA, theFDAandotherregulatoryauthoritiesconsidersafeforhumansand effectiveforindicateduses. The EMA and theFDAhassubstantialdiscretion,tobeexercisedinthepublicinterest,inthe approvalprocess,andmayeitherrefusetoacceptourapplication,or maydecideafterreviewthatourdataare insufficienttoallowapproval.
CertainclinicaltrialsforDermaVirwillbeconductedinEurope. AlthoughtheFDAmayacceptaforeignclinicaltrialtosupportapprovalofaBLA,thetrialmustbewell- designed,well-conducted,performedbyqualifiedinvestigatorsinaccordancewithinternationalprinciplesforGood ClinicalPractice(“GCP”),andmustconformtotheethicalprinciplescontainedin theDeclarationofHelsinki,or withthelawsandregulationsofthecountryinwhichtheresearchwasconducted,whicheverprovidesgreater protectionof thehumansubjects.The EMA and theFDAhassubstantialdiscretionindecidingwhethertoacceptdatafromour foreignclinicaltrials,andiftheagencydoesnotacceptsuchdata,ourproductdevelopmentand commercializationeffortsfor theDermaVirorourotherproductcandidatescouldbecurtailedorsignificantly delayed.
OncewesubmitourBLA,therecanbenoassurancethattheBLAwillbeacceptedforfilingorthatthe FDAmaynotissueaRefusaltoFile(“RTF”),ifitbelievesthefilingisinadequateorincomplete.EveniftheFDA acceptstheBLAforfiling,dataobtainedfrompre-clinicalstudiesandclinicaltrialsaresusceptibletovarying interpretations.IftheFDAdisagreeswithourinterpretationanddoesnotacceptorapproveourapplication,itmay requireustoconductadditionalpre-clinicalstudies,clinicaltrials,or manufacturingstudiesandsubmitthatdata beforeitwillreconsiderourapplication,orrequireustoperformpost-approvalstudies. Delaysinobtaining regulatoryapprovalswillmostlikelydelaycommercialization of,andourabilitytoderiveproductrevenuesfrom, ourproductcandidates,impose costlyproceduresonus,anddiminishthecompetitiveadvantagesthatwe may otherwiseenjoy.
Ourleadproductcandidate,DermaVir,andourotherproductcandidates,are immunetherapies, whichutilizeanovelmechanismofaction.Therefore,regulatoryagenciesmaylackexperiencewiththem,whichmaylengthentheregulatoryreviewprocess,increaseourdevelopmentcostsanddelayorpreventour commercializationofDermaVirandourotherImmuneTherapyProductsunderdevelopment.Todate,the EMA nor the FDAhasnotapprovedforcommercialsaleintheU.S.anyImmuneTherapyproductdesignedtostimulatean immuneresponseagainstHIV. Consequently,thereisnoprecedentforthesuccessfulcommercialization of productsbasedonourtechnologiesin thisarea.
Forourleadcandidate,we intendtoseekfasttrackdesignation, and conditional marketingapproval,and priorityreviewdesignation,allofwhicharevariousregulatoryprocessesintendedtoacceleratedrugdevelopment, review,orapprovalforanyProductcandidates.Fasttrackdesignationisaregulatorymechanismintendedto facilitatethedevelopmentandexpeditethereviewofproductsintendedforthetreatmentofseriousor life- threateningconditionsandthatdemonstratethepotentialtoaddressanunmetmedicalneedforsuchacondition. Acceleratedmarketingapprovalisreservedforaproductthatdemonstratesameaningfultherapeuticadvantageover existingtreatmentsorshowsthepotentialtoaddressanunmetmedicalneedinaseriousorlife-threatening condition. Insomecases,aproductreceivingacceleratedapprovalmaybeapprovedonthebasisofasurrogate measureofbenefit,inwhichcasefurtherclinicaltrials(aspost-approvalcommitments)aregenerallyrequiredto furtherdefinethesafetyandefficacyof theproduct.PriorityreviewdesignationwouldallowDermaVirto bereviewedinashortertimeframe. Therecanbenoassurance,however,thattheFDAwillgrantDermaVir fasttrackdesignationoracceleratedapprovalorwillawardtheBLApriorityreview. Further,theseregulatoryprocessesdonotchangethestandardsforapprovalanddonotguaranteethatourproductwillbeapproved.
WehavelimitedexperienceindevelopingourImmuneTherapyproducts,whichconsistofnanoparticles assembledfromplasmidDNAandapolymerwithspecialproperties.Duetotheirspecialproperties,nanomedicine formulationsmayposedifferentissuesthannon-nanoscaleproducts.Itisunknownwhetherthenanoscalesizeof theseproductsmay magnifysafetyandeffectivenessissues.Wealsodonotknowwhethertheuseofnanoscale materialsmaychangetheregulatorystatusofourproducts.TheFDAmayrequireustoconductadditional toxicologytestsandclinicaltrialstoconfirmthesafetyandeffectivenessofournanomedicineProductcandidates, whichwouldimpactourdevelopmentplan.
Evenifwecomplywithall EMA andFDArequestsfordataandinformation,the EMA or theFDAmayultimatelydetermine, underitsstatutoryauthority,torejectoneormoreofourBLAs.Wecannotbecertainthatwewilleverobtain regulatoryapprovalorclearanceforanyproductcandidate.Failuretoobtain EMA and/orFDAapprovalorclearanceofanyof ourprincipalproductcandidateswillseverelyundermineourbusinessbyreducingournumberofsaleableproducts and,therefore,correspondingproductrevenues. Evenifourproductcandidatesobtainmarketingapproval,the EMA and theFDAmayimposerestrictionsontheapprovedindicationsordistributionoftheproduct. Also,the EMA and theFDAmightapprove oneor moreofourproductcandidatesfor marketingbutmayalsoapprovecompetitors’productswith characteristics thatoffertheirowntreatmentadvantages.
Ourleadproductalsoisacombinationproductandincludesanewmedicaldevicethatmayrequiremedical deviceclearanceorapprovalseparatefromthebiologicsapproval.Thisclearanceorapprovalwillneedtobe obtainedfromagroupwithinthe EMA and theFDA,whichmay bedifferentfromthegroupactingontheBLA.Thismakesthe approvalprocessmorecomplexandcoulddelayapproval.
Inforeignjurisdictions,approvalfromtheappropriateregulatory,pricingandreimbursementauthoritiesmustbeobtainedbeforecommercializationandmarketingofourProductscanbegin.Theseprocessesgenerally includeallof therisksassociatedwith EMA andFDAprocedures. Pursuingforeignapprovalswillbetime-consuming and expensive. Regulationsvaryamongcountries,andforeignauthoritiesmayrequiredifferentoradditionalclinical trialsthanthose requiredtoobtain EMA andFDAapproval.
OurProductcandidates,includingourleadProductcandidate,arecombinationproductscomprisedofabiologicandmedicaldevicecomponent.WecannotbesurehowtheEMA and theFDAwillregulateourproductcandidates. The EMA and theFDAmayrequireus toobtainmarketingclearanceandapprovalfromtwodifferent EMA andFDAcenters.The reviewofcombinationproductsisoftenmorecomplexandmoretimeconsumingthanthereviewofproduct candidatesunderthejurisdictionofonlyonecenterwithinthe EMA and theFDA.
Ourproductcandidatesrequire EMA andFDAauthorizationby meansofanapprovalorclearancepriorto marketing. Someofourproductcandidates,includingDermaVir,willlikelyberegulatedascombinationproducts. Foracombinationproduct,the EMA and theFDAmustdeterminewhichcenterorcenterswithinthe EMA and theFDAwillreviewtheproduct candidateandunderwhatlegalauthoritytheproductcandidate willbereviewed.Wearecurrentlydeveloping our regulatorystrategieswithrespecttowhichregulatorypathwaywillbenecessarytoobtainclearanceorapprovalof theDermaPrepcomponentofDermaVir,ifmedicaldeviceclearanceorapprovalisrequiredatall. Based ondiscussionswiththeFDAwebelievethatthebiologiccomponentofDermaVirwillbereviewedby the CenterforBiologicsEvaluationandResearch(“CBER”)andthattheDermaPrepdevicemaybereviewedbythe CenterforDevicesandRadiologicalHealth(“CDRH”)eitherinconsultationwithCBERaspartof the BLAor separatelyasamedicaldevice.TheprocessofobtainingFDAmarketingclearanceorapprovalislengthy, expensive,anduncertain,andwecannotbesurethatourbiologic-devicecombinationproductcandidates,orany otherproductcandidates,willbeclearedorapprovedinatimelyfashion,oratall. Inaddition,thereviewof combinationproductsisoftenmorecomplexandmoretimeconsumingthanthereviewofaproductcandidateunder thejurisdictionofonlyonecenterwithintheFDA.Wecannotbe surethattheFDAwillnotselecttohaveour combinationproductsreviewedandregulatedbyonlyoneFDAcenterand/ordifferentlegalauthority,inwhichcase thepathtoregulatoryapprovalwouldbedifferentandcouldbe morelengthyandcostly. IftheFDAdoesnot approveorclearourproductcandidatesinatimelyfashionoratall,ourbusinessandfinancialconditionwillbe adverselyaffected.
Wehaveonlylimitedexperienceinregulatoryaffairs,andsomeofourproductcandidatesarebasedon novelmechanismsofaction.Thesefactorsmayaffectourabilityorthetimewerequiretoobtainnecessary marketingapprovals.
Wehaveonlylimitedexperienceinpreparingandfilingtheapplicationsnecessarytogainmarketing approvals.Moreover,someofourproductcandidatesarebasedonnovelmechanismsofactionthathavenotbeen extensivelytestedinhumans.Theregulatoryrequirementsgoverningthesetypesofproductcandidatesmaybeless welldefinedormorerigorousthanforconventionalproducts.Asaresult,we mayexperiencealongerregulatory processinconnectionwithobtainingmarketingapprovalsofanyproductsthatwedevelop.
Ourproductcandidateswillremainsubjecttoongoingregulatoryrequirementseveniftheyreceive marketingapproval,andifwefailtocomplywiththeserequirements,wecouldlosetheseapprovals,andthesales ofanyapprovedcommercialproductscouldbesuspended.
Evenifwereceiveapprovalto marketaparticular productcandidate,theproductwillremainsubjectto extensive regulatoryrequirements,including requirementsrelatingto manufacturing,labeling,packaging,adverse eventreporting,storage,advertising, promotion, productdeviation reporting,distributionand recordkeeping.Even ifmarketing approvalisgranted,itmaycontain requirementsforcostly post-approvaltestingandsurveillance tomonitorthesafety orefficacy oftheproduct, whichcould negatively impact usorourcollaboration partnersby reducing revenues orincreasing expenses,and causetheapprovedproduct candidatenotto becommerciallyviable. Inaddition, asclinicalexperience withabiologic, drugormedicaldevice expandsafterapprovalor clearance, typicallybecauseitis usedbya greaternumberandmore diverse groupofpatients afterapprovalthan during clinicaltrials,side effectsand otherproblemsmay beobservedafterapproval thatwere notseenoranticipated duringpre-approval clinicaltrialsorother studies. Anyadverseeffectsobservedaftermarketing ofaproductcould resultinlimitations on the useoforwithdrawal ofsuch productfromthemarketplace. Absence oflong-termsafety datamay alsolimit theapproved usesofour products,ifany. Ifwefail tocomply withthe regulatoryrequirements ofthe EMA and theFDAand otherapplicable E.U., U.S.andforeign regulatory authorities, orpreviously unknownproblems withany approvedcommercial products,manufacturers ormanufacturing processes arediscovered, wecould besubjectto administrative orjudicially imposed sanctions orother setbacks,including thefollowing:
| · | restrictionsontheproducts,manufacturersor manufacturingprocess; |
| · | civilorcriminalpenalties,finesand/orinjunctions; |
| · | unanticipatedexpenditurestoaddressordefendsuchactions; |
| · | productseizuresordetentions; |
| · | ordersforphysiciannotificationordevicerepair,replacementorrefund; |
| · | importorexportbansorrestrictions; |
| · | voluntaryormandatoryproductrecallsandrelatedpublicityrequirements; |
| · | suspensionorwithdrawalofregulatoryclearancesandapprovals; |
| · | totalorpartialsuspensionofproduction;and |
| · | refusaltoapprovependingapplicationsformarketingapprovalorclearanceofnewproductsor supplementstoapprovedapplications. |
If anyoftheseactionsweretooccurit wouldharmourreputationandcauseourproductsalesand profitabilitytosufferandmightpreventusfromgeneratingrevenue.Furthermore,ourkeycomponentsuppliers maynotcurrentlybeor mightnotcontinuetobeincompliancewithallapplicableregulatoryrequirements,which couldresultinourfailuretoproduceourproductsonatimelybasisandintherequiredquantities,ifatall.
Inaddition,ifthe EMA and theFDAbecomesawareofnewpost-approvalsafetyinformation,the EMA andFDAisauthorizedto requireustoconductpostapprovalstudies,includingclinicaltrialstoinvestigateknownseriousrisksorsignalsof seriousrisks,oridentifyingunexpectedseriousrisks.Ifwearerequiredtoconductsuchstudies,wemustsubmit periodicstatusreportsto the EMA andFDA.Failuretoconductsuchpost-approvalstudiesinatimelymannermayresultin substantialcivilfines.
Ifweorourcollaboratorsareslowtoadapt,orareunabletoadapt,tochangesinexistingregulatory requirementsoradoptionofnewregulatoryrequirementsorpolicies,marketingapprovalforourproductcandidates maybelostorceasetobeachievable,resultingindecreasedrevenuefrommilestones,productsalesorroyalties, whichwouldhaveamaterialadverseeffect onourresultsofoperations.
Wemaybesubjecttofines,penaltiesorinjunctionsifwearedeterminedtobepromotingtheuseofour productsforunapprovedor“off-label”uses.
Ourpromotionalmaterialsandtrainingmethodsmustcomplywith EMA andFDAandotherapplicablelawsand regulations.Ifthe EMA andFDAdeterminesthatourpromotionalmaterialsortrainingconstitutespromotionofan unapproveduseofourproductcandidates,itcouldrequestthatwemodifyourtrainingorpromotionalmaterialsor subjectus toregulatoryorenforcementactions,includingtheissuanceofanuntitledletter,awarningletter, injunction,seizure,civilfineandcriminalpenalties.Itisalsopossiblethatotherfederal,stateorforeign enforcementauthoritiesmighttakeactioniftheyconsiderourpromotionalortrainingmaterialstoconstitutepromotionofanunapproveduse,whichcouldresultinsignificantfinesorpenalties underotherstatutoryauthorities, suchaslawsprohibitingfalseclaimsforreimbursement.Inthatevent,ourreputationcouldbedamagedand adoptionoftheproductswouldbeimpaired.
Ifthethirdpartiesonwhichwerelytoconductourclinicaltrialsandtoassistuswithpre-clinical developmentdonotperformascontractuallyrequiredorexpected,wemaynotbeabletoobtainregulatory approvalfororcommercializeourproducts.
Wedonothavetheabilitytoindependentlyconductsomeofourpre-clinicalstudiesandclinicaltrialsfor ourproductsandwe mustrelyonthirdparties,contractresearchorganizations,medicalinstitutions,clinical investigatorsandcontractlaboratoriestoconductsuchstudies.Althoughwerelyon thirdpartiestoconductourpre- clinicalstudiesandclinicaltrials,wearenotrelievedfromtheresponsibilityofensuringthatsuchstudiesandtrials areconductedinaccordancewithregulatoryrequirements.Somepre-clinicalstudiesmustcomplywithcurrent GoodLaboratoryPractice(“cGLP”)requirements.Clinicaltrialsmustbeconductedinaccordancewiththe investigational planandprotocol,aswellascurrentGoodClinicalPractice(“cGCP”)requirementsforconducting,monitoring,maintainingrecords,and reportingadverseeventsandtheresultsofclinicaltrialstoensure,among otherthings,thatthedataandresultsarescientificallycredibleandaccurateandthatthe trialsubjectsareadequately informedofthepotentialrisksofparticipatinginclinicaltrials. Ourrelianceonthirdpartiestoconductpre-clinical studiesandclinicaltrialsdoesnotrelieveusoftheresponsibilityforcomplyingwithregulatoryobligations.Ifthese thirdpartiesdonotsuccessfullycarryouttheircontractualdutiesorregulatoryobligationsor meetexpected deadlines,ifthesethirdpartiesneedtobereplaced,orifthequalityoraccuracyofthedatatheyobtainis compromiseddueto thefailuretoadheretoourclinicalprotocolsorregulatoryrequirementsorfor otherreasons, ourpre-clinicaldevelopmentactivitiesorclinicaltrialsmaybeextended,delayed,suspendedorterminated,andwe maynotbeabletoobtainregulatoryapprovalfor,orsuccessfully commercialize, ourproductsonatimelybasis,if atall,andourbusiness,operatingresultsandprospectsmaybeadverselyaffected.Furthermore,ourthirdparty clinicaltrialinvestigatorsmaybedelayedinconductingourclinicaltrialsforreasonsoutsideof theircontrol.
Weandourcontractmanufacturersaresubjecttosignificantregulationwithrespecttomanufacturing ofourproducts.
Allofthoseinvolvedin thepreparationofabiologicor medicaldeviceforclinicaltrialsorcommercial sale,includingourexistingthirdpartysuppliersandmanufacturers,aresubjecttoextensiveregulation.Thefinishedproductanditscomponentsusedforcommercialsaleorinclinicaltrialsmustbe manufacturedin accordancewithcurrentGoodManufacturingPractices(“cGMP”),a seriesofcomplexregulationsand recommendations inguidancedocuments.ThemedicaldevicecGMPsarecodifiedin theQualitySystem Regulation(“QSR”)promulgatedby theFDA.ThecGMPandQSRrequirementsgovernmanufacturing processes andproceduresandtheimplementation andoperationofqualitysystemstocontrolandassurethequalityof investigational productsandproductsapprovedforcommercialdistribution. Ourfacilitiesandqualitysystemsand thefacilitiesandqualitysystemsofourthirdpartysuppliersandmanufacturersmaybesubjecttoapre-approval inspectionforcompliancewiththeapplicableregulationsasaconditionofFDAapprovalofDermaViror anyofourotherpotentialproducts. Inaddition, theFDAortheEuropean MedicinesAgency(“EMA”)may,atanytime,auditorinspectamanufacturing facilityinvolvedwiththepreparationofour products,itscomponents,orourotherproductcandidatesortheassociatedqualitysystemsforcompliancewiththe regulationsapplicabletotheactivitiesbeingconducted.Ifanysuchinspectionorauditidentifiesafailuretocomply withapplicableregulationsorifaviolationofourproductspecificationsorapplicableregulationoccursindependent ofsuchan inspectionoraudit,weortheFDAmayrequireremedialmeasuresthatmaybecostlyand/ortime consumingforusorathirdpartytoimplementandthatmayincludethetemporaryorpermanentsuspensionofa clinicaltrialorcommercialsalesorthetemporaryorpermanentclosureofafacility. MaterialviolationsofcGMP orQSRrequirementscouldresultinregulatorysanctionsand,inseverecases,couldresultinamandatedclosureof ourorourthirdparty’sfacilities. Anysuchremedialmeasures imposeduponusor thirdpartieswithwhomwe contractcouldharmourbusiness.
Ourproductsmayinthefuturebesubjecttoproductrecallsthatcouldharmourreputation,business andfinancialresults.
The EMA,FDAandsimilarforeigngovernmental authoritieshavetheauthority torequiretherecallof commercialized productsin theevent of materialdeficiencies,safetyissues,ordefectsindesignor manufacture. Manufacturersmay, undertheirowninitiative,recallaproductif anymaterial deficiency,orsafetyissueina productisfound.Recallsofanyofourproductswoulddivertmanagerialandfinancialresourcesandhavean adverseeffect onourfinancial conditionand resultsofoperations. Afuturerecallannouncement couldharmour reputationwithcustomersandnegatively affectour sales.In addition,the EMA and FDAcould take enforcementaction againstusifsuchrecall istheresultoffailure(s) tocomply withapplicableregulatoryrequirements, orifwefail to reportthe recallswithinrequired timeframes.
FAILURE TO OBTAIN MARKETING APPROVAL IN ADDITIONAL FOREIGN JURISDICTIONS WILL PREVENT US FROM EXPANDING THE COMMERCIALIZATION OF OUR PRODUCTS ABROAD.
Weintendtomarketourproductsinanumberofinternationalmarkets.Inorderto marketourproductsin foreignjurisdictionswe mustobtainseparateregulatoryapprovals.Theapprovalprocedurevariesamong jurisdictionsandcaninvolvesubstantialadditionaltesting.Approvalorclearanceby the EMA and theFDAdoesnotensure approvalbyregulatoryauthoritiesinotherjurisdictions,andapprovalbyoneforeignregulatoryauthoritydoesnot ensureapproval byregulatoryauthoritiesinotherforeignjurisdictions orbytheFDA. Theforeignregulatory approvalprocessmayincludealloftherisksassociatedwithobtaining EMA orFDAapprovalinadditiontootherrisks. In addition,thetimerequiredtoobtainforeignapprovalmaydifferfromthatrequiredtoobtain EMA andFDAapprovalandwemaynotobtainforeignregulatoryapprovalsonatimelybasis,ifatall.
FEDERAL REGULATORY REFORMS MAY ADVERSELY AFFECT OUR ABILITY TO SELL OUR PRODUCTS PROFITABLY.
Fromtimetotime,legislationisdraftedandintroducedinthe European Union and in theUnited StatesCongress(“Congress”)thatcould significantlychangethestatutoryprovisionsgoverningthemarketingapproval,manufactureandmarketingofa regulatedproduct.Inaddition, EMA andFDAregulationsandguidanceareoftenrevisedorreinterpretedbytheagencyin waysthatmaysignificantlyaffectourbusinessandourproducts.Itisimpossibletopredictwhetherlegislative changeswillbeenactedor EMA andFDAregulations,guidanceor interpretations changed,andwhattheimpactofsuch changes,if any,maybe.
Withoutlimitingthegeneralityof theforegoing,Congresshasrecentlyenacted,andthePresidenthas signedintolaw,theFoodandDrugAdministrationAmendmentsActof2007(the“FDAAmendments”).TheFDA Amendmentsrequirecertaininformationaboutclinicaltrialsforbiologicalproducts,including adescriptionofthe trial,participationcriteria,locationoftrialsites,andcontactinformation,tobesenttotheNationalInstitutesof Health(“NIH”)forinclusioninapublicly-assessable database. WealsowillberequiredtosubmittotheNIHthe resultsofcertainclinicaltrials,otherthanPhaseIstudies,forourbiologicalproducts. Onceimplemented, compliancewiththoseregulationsmayrequireus totakeadditionalstepsinthedevelopmentandmanufactureof ourproductsandlabeling. Thesestepsmayrequireadditionalresourcesandcouldbecostly.
Inaddition,Congresscouldtakeactiontocreateanabbreviatedregulatorypathwayforbiologicalproducts. Ifthisweretooccur,thenourProductsmaybesubjecttocompetitionfromfollow-onversions.
Theregulatoryclimateforfollow-onversionsofbiologicalproductsapprovedunderaBLAintheU.S. remainsuncertain.Currently,thereisnoestablishedstatutoryorregulatorypathwayfortheabbreviatedapprovalof follow-onor“generic”versionsofbiologicalproductsapprovedunderaBLA.However,membersof Congress recentlyintroducedlegislationtoestablishalegalpathwaytoapprovefollow-onversionsofapprovedBLA products.Weareuncertainastowhenanysuchprocessmaybeadoptedorhowsuchaprocesswouldrelatetoour intellectualpropertyrights,butanysuchprocesscouldhaveamaterialeffectontheprospectsofourproducts.
Risks Related to Biologics Regulation
TheDermaViristheonlyProductwearecurrentlytestinginclinicaltrialsanditmayneverbe successfullycommercializediftheseclinicaltrialsarenotsuccessful.
Ourleadproductcandidate,DermaVir,istheonlyproductwearetestinginclinicaltrials anditmayneverbesuccessfullycommercializediftheseclinicaltrialsarenotsuccessful.Theclinicaltrials requiredbythe EMA and theFDAforDermaVirarelongandcomplex.Currently,thisproductcandidateis completed Phase IIclinicaltestingonalimitednumberofsubjectsasdescribedbelow.
ItalsoispossibletheFDAortheEMAwillrequireustoconductmoreextensivenon-clinicalstudiesthan wecurrentlyanticipatebeforetheywillconsiderourproductsfor marketingapproval.Someofourfuturetrialsalsomayinvolvetreatmentfordurationsthataresignificantlylongerthanthose wehavetestedthusfar.Thelonger-termtrialscouldrevealsafetyorotherissuesthatcouldadverselyaffect marketingapproval.Wemayneedtocommitsubstantialtimeandadditionalresourcesinordertoconductfurther clinicaltrialsbeforewecansubmitaBLAwithrespecttoanyof theseproductcandidates.Wecannotpredictwith anycertaintyiforwhenwe mightsubmitaBLAforregulatoryapproval ofanyofourproductcandidates.
CLINICAL TRIALS ARE TIME-CONSUMING, DIFFICULT AND COSTLY TO DESIGN AND IMPLEMENT.
Beforewecanobtainmarketingapprovalforthecommercialsaleofanyofourproductcandidates,weand ourcollaboratorsarerequiredtocompleteextensivehumanclinicaltrialstodemonstrate,tothesatisfactionof the EMA, the FDAandotherregulatoryauthorities,thatourproductcandidatesaresafeandeffectiveforuseinhumans.In addition,ifwechooseto makeclaimsofsuperiorityovercurrentlymarketed competitiveproducts,wemust substantiatethoseclaimswithscientificevidencefromprospectivelydesignedhead-to-headclinicaltrials.Human clinicaltrialsareexpensiveanddifficulttodesignandimplement,inpartbecausethesciencebehindthemis complexandtheyarethereforesubjecttorigorousregulatoryrequirements.In theeventwe incorrectlydesignor carryoutclinicaltrialsorthoseclinicaltrialsfailtodemonstratestatisticallyclinicalsignificance,wearenotlikely tobeabletoobtain EMA andFDAapprovalforourproductcandidates.Further,the medical,regulatoryandcommercial environmentforbiopharmaceutical products,orbiologics,changesquicklyandofteninwayswemaynotbeableto accuratelypredict. Theclinicaltrialprocessisalsotime-consuming. Weestimatethatclinicaltrialsofourlead productcandidate,theDermaVir,willtakeatleastseveralmoreyearstocomplete. Furthermore,asfailure canoccurat anystage,wecouldencounterproblemsthatcauseus toabandonorrepeatclinicaltrials.The commencement andcompletionofclinicaltrialsmaybedelayedorsuspendedby thesponsor,regulatoryauthorities, andethicscommitteesbasedon manyfactors,includingthefollowing:
| · | changes in regulatory requirements; |
| · | delays in obtaining regulatory authorizations to commence a clinical trial; |
| · | delays in identifying and reaching agreement on acceptable terms with prospective clinical trial sites; |
| · | delay or failure in obtaining Institutional Review Board (“IRB”) review and approval of the clinical trial protocol; |
| · | unforeseen safety issues and side effects; |
| · | determination of dosing issues; |
| · | lack of effectiveness in clinical trials; |
| · | slower than expected patient recruitment; |
| · | inability to monitor patients adequately during or after treatment; |
| · | inability or unwillingness of medical investigators to follow our clinical protocols and current cGCP requirements; |
| · | inability to maintain a sufficient supply of the investigational product to support the trials; |
| · | suspension or termination of clinical trials for noncompliance with regulatory requirements; and |
| · | changes in clinical care protocols and standards of care within the institutions in which our trials take place. |
Anumberofcompaniesin thebiologicsandmedicaldeviceindustryhavesufferedsignificantsetbacksin advancedclinicaltrialsdespitepromisingresultsinearliertrials.Forthesamereason,we maybeunabletodevelopmarketableproducts.
Delaysinpatientenrollmentforclinicaltrialscouldincreasecostsanddelayanyregulatoryapprovals.
Therateofcompletion ofourclinical trialswilldepend ontherateofpatientenrollment. Theremaybe substantialcompetition toenrollpatientsinclinicaltrialsforourproducts indevelopment. Thiscompetition has delayedclinicaltrialsofother biopharmaceuticalanddrugdevelopmentcompanies. Inaddition,recentimprovements inexistingdrugtherapy, particularlyformedical productsforthetreatment of HIV/AIDS,maymake itmore difficultfor us to enrollpatientsin ourclinical trials asthe patientpopulationmay chooseto (i) remainwith existingdrugtherapies, (ii)enroll inclinicaltrials sponsoredby other companies, or (iii)switchtoalternative therapies.Delays in patientenrollment canresult in increaseddevelopment costsand delaysinregulatory approvals.
TheresultsofourclinicaltrialsmaynotsupportourProductcandidateclaimsormayresultinthe discoveryofadversesideeffects.
Evenifourclinicaltrialsarecompletedasplanned,wecannotbecertainthattheirresultswillsupportour Productcandidateclaimsorthatthe EMA,FDAorforeignauthoritieswillagreewithourconclusionsregardingthem. Successinpre-clinicalstudiesandearlyclinicaltrialsdoesnotensurethatlaterclinicaltrialswillbesuccessful,and wecannotbesurethatthelatertrialswillreplicatetheresults ofpriortrialsandpre-clinicalstudies.Theclinical trialprocessmayfailtodemonstratethatourproductcandidatesaresafeforhumansor thattheyareeffectivefor the proposedindicateduses,whichcouldcauseus toabandonaproductcandidateandmaydelaydevelopmentof others.Anydelayorterminationofourclinicaltrialswilldelaythefilingof ourBLAsand,ultimately,ourabilityto commercializeourproductcandidatesandgeneraterevenues.Itisalsopossiblethatpatientsenrolledinclinical trialswillexperienceadversesideeffectsthatarenotcurrentlypartof theproductcandidate’sprofile.Inaddition, ourclinicaltrialsfor theDermaVirinvolvearelativelysmallpatientpopulation.Becauseofthesmallsample size,theirresultsmay notbe indicativeoffutureresults.
Inanydrugdevelopmentprogram,itistypicallyduringthepivotallargePhaseIIIclinicaltrials,whenlarge numbersofpatientsaretested,thatthemostreliableinformationonsideeffectsis collected.Wehavenotreached thisdevelopmentstageyet,andwecannotaccuratelypredictifpivotalclinicaltrialswithourleadProduct DermaVir,willrevealunexpectedsideeffects.Occurrenceofanyside effectcoulddelayorterminatefurtherdevelopmentandhamperorpreventregulatoryapprovalormarketingofourproduct.
WewillneedtodemonstratethattheDermaVir PRODUCTmanufacturedatourownfacilityiscomparableto theProductusedinclinicaltrials.
Inadditionto increasedproductionefforts,we may makemanufacturingchangesto thecomponentsor to the manufacturingprocessforDermaVir.Thesechangescouldresultindelaysinthedevelopmentor marketingapprovaloftheDermaVirandinreductionorinterruptionofcommercialsales,iftheproductis approved,anyofwhichcouldmateriallyharmourbusiness. Wewillberequiredtodemonstrateproduct comparabilityforeachmanufacturingsite. TheFDAandEMAmayrequireadditionaltestingbeyondwhatwe propose.
Weintendtorelyonresultsofpre-clinicalstudiesandclinicaltrialsperformedusingtheformofthe Productcandidateproducedusingthepriorformulationorproductionmethodoratthepriorscale.Dependingupon thetypeanddegreeofdifferencesbetweenmanufacturingprocessesorcomponentsubstitutionsforaProduct candidate,wemayberequiredto conductadditionalstudiesorclinicaltrialstodemonstratethatthenewmethod(s), substitutecomponentorproductcandidateissufficientlysimilartothepreviouslyproducedmaterial.
Wehavenocommercialorotherlarge-scalemanufacturingexperience,andifweareunsuccessfulin developingcommercially viabletechnologiestoenableus tomanufacturethefinishedProductourselvesinsufficient qualitiesandquantitiesatacompetitivepricetomeetprojectedcommercialdemand,ourbusinesswouldbe harmed.
Tobesuccessful,ourproductcandidatesmust becapable ofbeingmanufactured insufficientquantities,in compliance withregulatoryrequirements andatamarketablecost.Wehavenocommercialorotherlarge-scalemanufacturing experience.Wecurrentlyrelyonthird partiesforcertainaspectsof theclinical trialmanufacture of DermaViranditscomponentsandfor ourotherProductcandidates. Weareintheprocessofdeveloping andimplementingmanufacturingmethodsandbuilding apilotcGMPcompliantfacilityinHungaryto manufacture largerquantitiesofourImmune TherapyProducts. Wemust hireandtrain asignificantnumber ofemployeesand complywithapplicableregulations forourfacilities,whichareextensive. Therecurrentlyarenocontractmanufacturers that have already manufacturedthe finalDermaVirproduct orany otherproductcandidate. Therefore,ifweencounter delaysordifficulties withoursuppliersand/or cannotmanufacturethe finishedProduct ourselves,we may not beable toconductclinicaltrials asplanned orto meetdemand forDermaVir,ifitis approved, anyofwhich could harmourbusiness.
The EMA,FDAandregulatoryagenciesinothercountriesperiodicallyinspectmanufacturingfacilities,including thirdpartieswhomanufactureourrawmaterialsforus.TheFDAorEMAmaybelievethatneitherwenorour suppliershaveenoughexperiencemakingtheseProductsor therawmaterials,andmaysubjectthosemanufacturers and/orusto increasedscrutiny.Biopharmaceuticalmanufacturing facilitiesmustcomplywithcGMPstandards, requiringusuallyaninvestmentofsubstantialfunds,timeandefforttoensurefullcompliance withthesestandards andmakequalityproducts. Wedonothavecontroloveroursuppliers’compliancewiththeserequirements. Failure tocomplywithregulatoryrequirements canresultinsanctions,fines,delays,suspensionsofapprovals,seizuresor recallsofproducts,operatingrestrictions,manufacturinginterruptions, costlycorrectiveactions,injunctions,adverse publicityagainstusandourproducts,andcriminalprosecution.
Ifanyofourproductcandidatesreceivesmarketingapproval,weexpectto manufacturethefinalproduct usingourtechnologyandourproductionfacilityinHungary.IfweareunabletocomplywithcGMPstandardsand othergovernmentregulations,thequalificationofourfacilitycouldbealengthyprocess,andtheremaynotbe adequatealternativesto meetourneeds.Asaresult,we maynotbeableto obtainthenecessaryrawmaterialsor finalproductinthefutureonatimelybasis,ifat all.Thiswouldnegativelyaffectourbusiness.
Risks Related to Medical Device Regulations
Ifwefailtoobtainregulatoryapprovalsandclearances,orexperiencesignificantdelaysinobtaining, FDAclearancesorapprovalsforfutureproductsorproductenhancements,ourabilitytocommerciallydistribute andmarkettheseproductscouldsuffer.
Ifrequiredby theFDAtoobtaina510(k)clearanceor approvalofaPremarketApproval(“PMA”),the processofobtainingregulatoryclearancesorapprovalstomarketamedicaldevicecanbecostlyandtime consuming,andwe maynotbeabletoobtaintheseclearancesorapprovalsonatimelybasis,ifatall.Inparticular, theFDApermitscommercialdistributionofanewmedicaldeviceonlyafterthedevicehasreceivedclearanceunder Section510(k)oftheFederalFood,DrugandCosmeticAct,oristhesubjectofanapprovedPMAunlessthedevice isspecificallyexemptfromthoserequirements.TheFDAwillclearmarketingofalowerriskmedicaldevice throughthe510(k)processifthe manufacturerdemonstrates thatthenewproductissubstantiallyequivalenttoother 510(k)-clearedproducts. Highriskdevicesdeemedtoposethegreatestrisk,suchaslife-sustaining,life-supporting, orimplantabledevices,ordevicesnotdeemedsubstantiallyequivalenttoapreviouslycleareddevice,requirethe approvalofa PMA. ThePMAprocessismorecostly,lengthyanduncertainthanthe510(k)clearanceprocess. A PMAapplicationmustbesupported byextensivedata,including,butnotlimitedto,technical,preclinical,clinical trial,manufacturingandlabelingdata,todemonstratetotheFDA’ssatisfactionthesafetyandefficacyofthedevice foritsintendeduse.
Wearecurrentlydevelopingourregulatorystrategieswithrespecttowhichregulatorypathwaywillbe necessarytoobtainclearanceorapprovaloftheDermaPrepcomponentofDermaVir,ifmedicaldevice clearanceorapprovalisrequiredatall.DermaVir,aswellasourotherproductcandidates,areImmune Therapies,whichutilizeanovelmechanismofaction.Todate,theFDAhasnotclearedorapprovedforcommercial saleintheU.S.anyImmuneTherapyproductdesignedtostimulateanimmuneresponseagainstHIV.Consequently, thereisnoprecedentfor thesuccessfulcommercialization ofproductsbasedonourtechnologiesin thisarea.Ifthe FDArequiresus tosubmita510(k)orPMAforDermaPrep,we mayexperiencesignificantdelaysinobtaining marketingapprovalofourproductcandidates,ifatall. Ifwefailtoobtainsuchapprovals,orexperiencesignificantdelaysinobtaining,suchapprovalsforfutureproductsorproductenhancements, ourabilitytocommerciallydistributeandmarkettheseproductscouldsuffer.
If ourmedicaldeviceproductscauseorcontribute toadeath oraserious injury,or malfunctionin certainways,wewillbesubjecttomedicaldevicereportingregulations, whichcanresultinvoluntarycorrective actionsoragencyenforcementactions.
Under theFDA’smedicaldevicereportingregulations,medicaldevicemanufacturers arerequired toreport totheFDAinformation thatadevice has ormayhave caused orcontributed toadeathor seriousinjuryor has malfunctionedinaway thatwould likely cause or contribute to death or serious injury ifthemalfunctionof the deviceor one ofour similar deviceswereto recur.If wefailto reportthese eventstothe FDA within the required timeframes, or atall,theFDA could takeenforcement actionagainst us.Anysuchadverseeventinvolving our productsalsocould result infuture voluntary correctiveactions,such asrecalls orcustomer notifications,oragency action,such asinspectionorenforcement action.Anycorrective action, whethervoluntary orinvoluntary, aswellas defendingourselves inalawsuit,will requirethededication ofourtime andcapital,distractmanagement from operatingour business,andmayharmour reputationandfinancial results.
Risks Related to Health Care Regulations
WE MAY BE SUBJECT, DIRECTLY OR INDIRECTLY, TO FEDERAL AND STATE HEALTHCARE FRAUD AND ABSUSE LAWS AND REGULATIONS AND COULD FACE SUBSTANTIAL PENALITIES IF WE ARE UNABLE TO FULLY COMPLY WITH SUCH LAWS.
WhilewedonotcontrolreferralsofhealthcareservicesorbilldirectlytoMedicare,Medicaidorotherthird partypayors,manyhealthcarelawsandregulationsapplytoourbusiness.Forexample,wecouldbesubjectto healthcarefraudandabuseandpatientprivacyregulationandenforcementbyboththefederalgovernmentandthe statesinwhichweconductourbusiness.Thehealthcarelawsandregulationsthatmayaffectourabilitytooperate include:
| · | thefederalhealthcareprograms’Anti-KickbackLaw,whichprohibits,amongotherthings,personsor entitiesfromsoliciting,receiving,offeringorprovidingremuneration,directlyor indirectly,inreturn fororto induceeitherthereferralofanindividualfor,orthepurchaseorderorrecommendationof, anyitemorservicesforwhichpaymentmaybe madeunderafederalhealthcareprogramsuchasthe MedicareandMedicaidprograms;
|
| · | federalfalseclaimslawswhichprohibit,amongotherthings,individualsorentitiesfromknowingly presenting,orcausingtobepresented,claimsforpaymentfromMedicare,Medicaid,orotherthird partypayorsthatarefalseorfraudulent,andwhichmayapplytoentitieslikeus totheextentthatour interactionswithcustomersmayaffecttheirbillingorcodingpractices;
|
| · | thefederalHealthInsurancePortabilityandAccountabilityActof1996(“HIPAA”)whichestablished newfederalcrimesforknowinglyandwillfullyexecutingaschemetodefraudanyhealthcarebenefit programor makingfalsestatementsinconnectionwiththedeliveryoforpaymentforhealthcare benefits,itemsorservices,aswellasimposingcertainrequirementsrelatingto theprivacy,security andtransmissionof individuallyidentifiablehealthinformation;and
|
| · | statelawequivalentsofeachofthefederallawsmentionedontheprecedingpage,suchasanti- kickbackandfalseclaimslawswhichmayapplytoitemsorservicesreimbursedbyanythirdparty payor,includingcommercialinsurers,andstatelawsgoverningtheprivacyofhealthinformationin certaincircumstances,manyofwhichdifferfromeachotherinsignificantwaysandoftenarenot preemptedbyHIPAA,thuscomplicatingcomplianceefforts. |
Thebiopharmaceuticalsindustryis,andinrecentyearshasbeen,underheightenedscrutinyasthe subject of governmentinvestigationsandenforcementactionsinvolvingmanufacturerswhoallegedlyofferedunlawful inducementstopotentialorexistingcustomersinanattempttoprocuretheirbusiness,includingarrangementswith physicianconsultants.Ifouroperationsarefoundtobe inviolationofanyofthelaws describedaboveoranyother governmentalregulationsthatapplytous,we maybesubjecttopenalties,includingcivilandcriminalpenalties, damages,fines,exclusionfromtheMedicareandMedicaidprograms,andthecurtailmentorrestructuringofour operations. Anypenalties,damages,fines,exclusions,curtailmentorrestructuringofouroperationscouldadversely affectourabilitytooperateourbusinessandourfinancialresults. Theriskofourbeingfoundinviolationofthese lawsisincreasedbythefactthatmanyof theselawsarebroadandtheirprovisionsareopentoavarietyof interpretations. Anyactionagainstusforviolationof theselaws,evenifwesuccessfullydefendagainst it,could causeus toincursignificantlegalexpensesanddivertourmanagement’sattentionfromtheoperationof our business.
Risks Related to Intellectual Property
Ifwe,orthethirdpartiesfromwhomwelicenseintellectualproperty,areunabletosecureandmaintain patentorotherintellectualpropertyprotectionfortheintellectualpropertycontainedinourProducts,ourability tocompetewillbeharmed.
Ourcommercialsuccessdepends,inpart,onobtainingpatentandotherintellectualpropertyprotectionfor ourproducts,methods,processesandothertechnologies.Thepatentpositionsofbiopharmaceuticalcompanies, includingours,canbehighlyuncertainandinvolvecomplexandevolvinglegalandfactualquestions.If we,orthe thirdpartiesfromwhomwe licenseintellectualproperty,failtoobtainadequatepatentorotherintellectualproperty protectionforintellectualpropertycontainedinourproducts,methods,processesandothertechnologiesorifany protectionisreducedoreliminated,otherscoulduseourintellectualproperty,resultinginharmtoourcompetitive businessposition.Inaddition,patentandotherintellectualpropertyprotectionmaynotprovideuswitha competitiveadvantageagainstcompetitorsthatdevisewaysof makingcompetitiveproductswithoutinfringingany patentsthatweownorhaverightsto.
Asof December 31,2012,wehavefiled28patentapplications,includingU.S.andE.U.patentapplications, aswellasrights underforeign patentsandpatent applications,andhave20issuedpatents, includingU.S.andEuropeanPatent Officepatent.U.S.patents andpatentapplicationsmaybesubjectto interference proceedingsandU.S.patentsmaybe subject toreexaminationproceedingsintheU.S.Patent andTrademark Office. Foreignpatentsmaybesubjecttooppositionorcomparable proceedingsin thecorrespondingforeignpatentoffices. Anyoftheseproceedingscouldresult ineitherloss of thepatentordenialofthepatentapplication,orlossor reductioninthescopeofoneor more oftheclaims of thepatentorpatentapplication.Changesineitherpatentlaws orin interpretations ofpatentlawsmayalsodiminishthevalueofourintellectual property ornarrow thescopeof ourprotection.Interference, reexamination andoppositionproceedingsmaybecostlyandtime-consuming, andwe, orthe thirdpartiesfromwhomwelicenseintellectualproperty,maybeunsuccessfulindefendingagainst such proceedings. Thus,any patentsthatweownorlicensemayprovidelimited ornoprotectionagainstcompetitors.In addition,ourpendingpatentapplicationsandthosewe mayfileinthefuturemayhaveclaims narrowedduring prosecution ormaynotresultinpatentsbeingissued. Evenif anyofourpendingorfuture applicationsareissued, theymaynotprovideuswithadequateprotectionorany competitive advantages. Ourability todevelop additional patentableproducts,methods, processesand othertechnologies isalsouncertain.
Non-paymentordelayinpaymentofpatentfeesorannuities,whetherintentionalorunintentional,may alsoresultinthelossofpatentsorpatentrightsimportanttoourbusiness.Manycountries,includingcertain countriesinEurope,havecompulsorylicensinglawsunderwhichapatentownermaybecompelledtogrant licensesto thirdparties.Inaddition,manycountrieslimit theenforceabilityofpatentsagainstthirdparties, includinggovernmentagenciesorgovernmentcontractors.Inthesecountries,thepatentownermayhavelimited remedies,whichcouldmateriallydiminishthevalueofthepatent. Inaddition,thelawsofsomeforeigncountries donotprotectintellectualpropertyrightsto thesameextentasdothelawsoftheU.S.,particularlyinthefieldof biopharmaceuticals.
Ifweareunabletopreventunauthorizeduseordisclosureofourproprietarytradesecretsand unpatentedknow-how,ourabilitytocompetewillbeharmed.
Proprietarytradesecrets,copyrights, trademarksandunpatented know-howarealsoveryimportant toour business. Werelyonacombination oftradesecrets,copyrights,trademarks, confidentialityagreements andother contractual provisionsandmeasures toprotectcertainaspects ofourproducts,methods,processesandother technologies,especially where wedonotbelievethatpatentprotectionisappropriateorobtainable.To this end,we requireallofouremployees, consultants,advisorsandcontractorstoenterintoconfidentiality and,where applicable,grant-backagreements. However,thesemeasuresmaynotbeadequateto safeguardourproprietary intellectualproperty. Ouremployees,consultants,advisorsandcontractorsmayunintentionally orwillfullydisclose ourconfidential informationtocompetitors. Inaddition,confidentialityagreementsmaybeunenforceableor may notprovideanadequateremedyin theevent ofunauthorized disclosure.Enforcingaclaim thatathirdparty illegallyobtained andisusingour trade-secrets isexpensive andtime consuming, andtheoutcome isunpredictable. Moreover,ourcompetitorsmayindependently developequivalent knowledge,methodsand know-how. Unauthorizedpartiesmayalsoattempttocopyorreverseengineer certainaspectsofproductsdeveloped byour employees,consultants,advisorsandcontractorsthatweconsiderproprietary. Asaresult, thirdpartiesmaybeable touseourproprietary employees,consultants, advisorsand contractors,andourability tocompete inthe market would beharmed.
Wecouldbecomesubjecttopatentandotherintellectualpropertylitigationthatcouldbecostly,resultin thediversionofmanagement’sattention,requireustopaydamagesandforceus todiscontinuesellingour products.
Thebiopharmaceuticalindustryischaracterizedbycompetingintellectualpropertyanda substantial amount oflitigationoverpatentandotherintellectualpropertyrights.Determiningwhetheraproductinfringesa patentinvolvescomplexlegalandfactualissues,andtheoutcomeofapatentlitigationactionisoftenuncertain.Wehavenotconductedanextensivesearchofpatentsissuedtothirdparties,andnoassurancecanbegiventhatthird partypatentscontainingclaimscoveringourproducts,partsofourproducts,technologyor methodsdonotexist, havenotbeenfiled,orcouldnotbefiledorissued.Becauseofthenumberofpatentsissuedandpatentapplications filedinourareas,ourcompetitors orotherthirdpartiesmayassertthatourproductsandthe methodsweemployin theuseofourproductsarecoveredbyU.S.orforeignpatentsheldbythem. Inaddition,becausepatentapplications cantakemanyyearstoissueandbecausepublicationschedulesforpendingapplications varybyjurisdiction,theremaybeapplications nowpendingofwhichweareunaware, andwhichmayresultinissuedpatentswhichour currentorfutureproductsinfringe. Also,becausetheclaimsofpublishedpatentapplicationscanchangebetween publicationandpatentgrant,theremaybepublishedpatentapplicationsthat mayultimatelyissuewithclaimsthat weinfringe.Therecouldalsobeexisting patentsthatoneormoreofourproductsorpartsmayinfringeandof whichweareunaware. Incertainsituations,weorthirdpartiesfromwhomwelicenseintellectualpropertymay determinethatitisinourbestinterestsortheirbestintereststovoluntarilychallengeathirdparty’sproductsor patentsinlitigationorotherproceedings,includingpatentinterferencesorreexaminations. Asaresult,we may becomeinvolvedinunwantedlitigationsandactionsthatcouldbecostly,resultindiversionof managements’ attention,requireustopaydamagesandforceustodiscontinuesellingourproducts.
Infringementactionsandotherintellectualpropertyclaimsandproceedings,whetherwithorwithoutmerit, maycauseustoincursubstantialcostsandcouldplaceasignificantstrainonourfinancialresources,divertthe attentionof managementfromourbusinessandharmourreputation.Someofourcompetitorsmaybeableto sustainthecosts ofcomplexpatentorintellectualpropertylitigationmoreeffectivelythanwecanbecausetheyhave substantiallygreaterresources.
Wecannotbecertainthatwewillsuccessfullydefendagainstallegationsofinfringementofthirdparty patentsandintellectualpropertyrights.In theeventthatwebecomesubjecttoapatentinfringementorother intellectualpropertylawsuitandiftheotherparty’spatentsorotherintellectualpropertywereupheldasvalidand enforceableandwewere foundto infringetheotherparty’spatentsorviolatethetermsofalicensetowhichweare aparty,wecouldberequiredtopaydamages.Wecouldalsobepreventedfromsellingourproductsunlesswe couldobtainalicensetousetheproducts,methods,processesorothertechnologiescoveredbysuchpatentsor were abletoredesignourproducts,methods,processesandothertechnologiesto avoidinfringement.Alicensemaynot beavailableatalloroncommerciallyreasonabletermsorwe maynotbeabletoredesignourproducts,methods, processesandothertechnologiestoavoidinfringement.Modificationofourproductsordevelopment ofnew productscouldrequireustoconductclinicaltrialsandtoreviseourfilingswiththeFDAandotherregulatory bodies,whichwouldbetime-consumingandexpensive.Inthesecircumstances, wemaybeunabletosellour productsatcompetitivepricesoratall,ourbusinessandoperatingresultscouldbeharmedandourstockpricemay decline.Inaddition,anyuncertaintiesresultingfromtheinitiationandcontinuation ofanylitigationcouldhavea materialadverseeffectonourabilitytoraisethefundsnecessarytocontinueouroperations.
Wemaybesubjecttodamagesresultingfromclaimsthatouremployeesorwehavewrongfullyusedor disclosedallegedtradesecretsoftheirformeremployers.
Some ofouremployees werepreviouslyemployedatuniversities orother enterprises.Wecould,in the future,besubjecttoclaimsthattheseemployees,orwe,haveinadvertently orotherwise usedordisclosedtrade secretsorother proprietaryinformation of theirformeremployers. Litigationmaybenecessarytodefendagainst theseclaims. Ifwefailindefending againstsuch claims,acourtcould orderustopaysubstantialdamagesand prohibitusfromusingproducts,methods, processesand othertechnologies thatareessential toourproducts, ifsuch products,methods, processesandother technologiesarefoundtoincorporate orbederived fromthetrade secrets or otherproprietaryinformation oftheformer employers.Inaddition, we maylose valuable intellectualproperty rights orpersonnel. Aloss ofkey researchpersonnel ortheir work productcould hamper orpreventourability to commercializecertainpotential products,whichcould severely harmourbusiness. Evenifwe aresuccessfulindefending against theseclaims,such litigation could resultinsubstantial costsandbe adistractiontomanagement.
Wemaybeunabletoprotecttheintellectualpropertyrightsofthethirdpartiesfromwhomwelicense certainofourintellectualpropertyorwithwhomwehaveenteredintootherstrategicrelationships.
Certainofourintellectualpropertyrights,includingtheHIVclade“B”plasmidDNAutilizedinDermaVirforthetreatmentofHIV/AIDS,arecurrentlylicensedfromRIGHTand,inthefuture,we may licenseadditionalintellectualpropertyfromRIGHTand/orotherkeystrategicpartners.Weare,andwillcontinue tobe,reliantuponsuchthirdpartiestoprotecttheirintellectualpropertyrightstoanylicensedtechnology.Such thirdpartiesmaydeterminenottoprotecttheintellectualpropertyrightsthatwelicensefromthemandwe maybe unabletodefendsuchintellectualpropertyrightsonourownorwe mayhavetoundertakecostlylitigationtodefend theintellectualpropertyrightsofsuchthirdparties.Therecanbenoassurancesthatwewillcontinuetohaverights toanyof theintellectualpropertythatwelicensefromsuchthirdpartiesorotherwisehavetherighttousethrough similarstrategicrelationships. Anylossorlimitationsonusewithrespecttoourrighttousesuchintellectual propertylicensedfromthirdpartiesorotherwiseobtainedfromthirdpartieswithwhomwehaveenteredinto strategicrelationshipscouldhaveamaterialadverseeffectonourbusiness,operatingresultsandfinancialcondition.
Risks Related to Our Common Stock
Our common stock is quoted on the OTCBB which may have an unfavorable impact on our stock price and liquidity.
Our common stock is quoted on the OTCBB. The OTCBB is a significantly more limited market than the New York Stock Exchange or the NASDAQ Stock Market. The quotation of our shares on the OTCBB may result in a less liquid market available for existing and potential stockholders to trade shares of our common stock, could depress the trading price of our common stock and could have a long-term adverse impact on our ability to raise capital in the future.
BECAUSE OUR BUSINESS ASSETS, DIRECTORS AND OFFICERS ARE LOCATED OUTSIDE OF THE UNITED STATES, OUR SHAREHOLDERS MAY BE LIMITED IN ABILITY TO ENFORCE CIVIL ACTIONS AGAINST OUR ASSETS OR OUR DIRECTORS AND OFFICERS.
We are incorporated under the laws of Delaware but because we are headquartered in Hungary, all of our officers and members of the Board reside in Hungary. Therefore our shareholders may have difficulty enforcing civil liabilities under the U.S. federal securities laws against our officers and directors. Because some of our assets are located outside the U.S., it may be difficult for an investor to succeed in an action, for any reason, against us or any of our directors or officers through U.S. jurisdictions. If an investor was able to obtain a judgment against us or any of our directors or officers in a U.S. court based on U.S. securities or other laws, it may be difficult to enforce such judgment in Hungary. We are uncertain as to the enforceability, in original actions in Hungarian courts, of liability based upon the U.S. federal securities laws and as to the enforceability in Hungarian courts of judgments of U.S. courts obtained in actions based upon the civil liability provisions of the U.S. federal securities laws.
WE MAY CONDUCT FURTHER OFFERINGS IN THE FUTURE, IN WHICH CASE YOUR PERCENTAGE INTEREST IN OUR COMPANY WILL BE DILUTED.
Since inception, we have relied on sales of our common stock to fund our operations. We may conduct further offerings in the future to finance our current projects or to finance subsequent projects that we decide to undertake. If common stock is issued in return for additional funds, the price per share could be lower than that paid by our current shareholders. We anticipate continuing to rely on equity sales of our common stock in order to fund our business operations. If we issue additional stock, your percentage interest in us will be diluted and the value of your stock could be reduced.
WE MAY ISSUE PREFERRED STOCK WHICH MAY HAVE GREATER RIGHTS THAN OUR COMMON STOCK.
We are permitted in our Certificate of Incorporation to issue up to 10,000,000 shares of preferred stock. We currently have 2,000,000 shares of Series A Preferred Shares, 1,000,000 shares of Series B Preferred Shares, 1,000,000 shares of Series C Preferred Shares, 1,000,000 shares of Series D Preferred Shares and 250,000 shares of Series E Preferred Shares issued and outstanding; however, we can issue shares of our preferred stock in one or more series and can set the terms of the preferred stock without seeking any further approval from our common shareholders. Any preferred stock that we issue may rank ahead of our common stock in terms of dividend priority or liquidation premiums and may have greater voting rights than our common stock. In addition, such preferred stock may contain provisions allowing it to be converted into shares of common stock, which could dilute the value of common stock to current shareholders and could adversely affect the market price, if any, of our common stock.
THE APPLICATION OF THE “PENNY STOCK” RULES COULD ADVERSELY AFFECT THE MARKET PRICE OF OUR COMMON SHARES AND INCREASE YOUR TRANSACTION COSTS TO SELL THOSE SHARES.
The Securities and Exchange Commission (the “SEC”) has adopted rule 3a51-1 which establishes the definition of a “penny stock,” for the purposes relevant to us, as any equity security that has a market price of less than $5.00 per share or with an exercise price of less than $5.00 per share, subject to certain exceptions. For any transaction involving a penny stock, unless exempt, Rule 15g-9 requires:
- that a broker or dealer approve a person’s account for transactions in penny stocks;
- the broker or dealer receive from the investor a written agreement to the transaction, setting forth the identity and quantity of the penny stock to be purchased; and
- that a broker or dealer approve a person’s account for transactions in penny stocks.
In order to approve a person’s account for transactions in penny stocks, the broker or dealer must:
- obtain financial information and investment experience objectives of the person; and
- make a reasonable determination that the transactions in penny stocks are suitable for that person and the person has sufficient knowledge and experience in financial matters to be capable of evaluating the risks of transactions in penny stocks.
The broker or dealer must also deliver, prior to any transaction in a penny stock, a disclosure schedule prescribed by the SEC relating to the penny stock market, which, in highlight form:
- sets forth the basis on which the broker or dealer made the suitability determination; and
- that the broker or dealer received a signed, written agreement from the investor prior to the transaction.
Generally, brokers may be less willing to execute transactions in securities subject to the “penny stock” rules. This may make it more difficult for investors to dispose of our common stock and cause a decline in the market value of our stock.
AS AN ISSUER OF “PENNY STOCK,” THE PROTECTION PROVIDED BY THE FEDERAL SECURITIES LAWS RELATING TO FORWARD LOOKING STATEMENTS DOES NOT APPLY TO US.
Although federal securities laws provide a safe harbor for forward-looking statements made by a public company that files reports under the federal securities laws, this safe harbor is not available to issuers of penny stocks. As a result, the Company will not have the benefit of this safe harbor protection in the event of any legal action based upon a claim that the material provided by the Company contained a material misstatement of fact or was misleading in any material respect because of the Company’s failure to include any statements necessary to make the statements not misleading. Such an action could hurt our financial condition.
WE HAVE NOT PAID DIVIDENDS IN THE PAST AND DO NOT EXPECT TO PAY DIVIDENDS FOR THE FORESEEABLE FUTURE. ANY RETURN ON INVESTMENT MAY BE LIMITED TO THE VALUE OF OUR COMMON STOCK.
No cash dividends have been paid on the Company’s common stock. We expect that any income received from operations will be devoted to our future operations and growth. The Company does not expect to pay cash dividends in the near future. Payment of dividends would depend upon our profitability at the time, cash available for those dividends, and other factors as the Company’s board of directors may consider relevant. If the Company does not pay dividends, the Company’s common stock may be less valuable because a return on an investor’s investment will only occur if the Company’s stock price appreciates.
Item 1B. Unresolved Staff Comments.
Not applicable.
Item 2. Property.
The Company currently leases a 1,000 square foot office locatedat1095 Budapest, Soroksari ut 94-96, Hungary, for approximately $1,100 per month. We also lease research and development facilities for Genetic Immunity, consisting of approximately 6,500 square feet, for approximately $12,500 a month, located at 1045 Budapest, Berlini ut 47-49.
Item 3. Legal Proceedings.
The Company’s wholly-owned subsidiary, Genetic Immunity, has initiated a lawsuit against the National Development Agency of Hungary. In 2009, Genetic Immunity successfully applied for and won an 800 million Hungarian forint (approximately $4,000,000) research grant to conduct dust mite allergy vaccine research. Subsequently, the agency refused to sign the grant and denied payment of funds. The company initiated a lawsuit to request that the court reinstate the contract that resulted in a grant award. On February 13, 2013, an appeals court ruled that the court could not reinstate the contract between Genetic Immunity and the National Development Agency as the contract has been validly in existence since its original execution in 2009. As such Genetic Immunity was ordered to pay 52,500,000 HUF (approximately $250,000) in lawsuit costs. However, simultaneously, the judgment stated that the company is entitled to receive the original grant amount, interest payment and/or damages since the original grant contract is valid. On February 18, 2013, we filed a new petition to have the courts establish the damages and awards that Genetic Immunity can receive based on the appeals court’s ruling. We are currently seeking 4,800,000,000 HUF (approximately $20 million) for damages sustained. ON November 18, 2013 we have entered into a six month suspension of said lawsuit, as agreed with the government, in hopes of negotiating an out of court settlement. If these negotiations are unsuccessful we will resume the law suite in the court of law on or before May 18, 2014.
Other than the matter described above, we are not currently involved in any litigation that we believe could have a materially adverse effect on our financial condition or results of operations. There is no action, suit, proceeding, inquiry or investigation before or by any court, public board, government agency, self-regulatory organization or body pending or, to the knowledge of the executive officers of our Company or any of our subsidiaries, threatened against or affecting our Company, our common stock, any of our subsidiaries or of our Company’s or our Company’s subsidiaries’ officers or directors in their capacities as such, in which an adverse decision could have a material adverse effect.
Item 4. Mine Safety Disclosures.
Not applicable.
PART II
Item 5. Market for Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities.
(a) Market Information
The Company’s Common Stock is quoted on the OTCBB under the under the symbol “PWRV.” The following table sets forth the quarterly high and low sale prices for our common shares for the last two completed fiscal years and the subsequent interim periods. Quotations on the OTCBB reflect bid and ask quotations, may reflect inter-dealer prices, without retail markup, markdown or commission, and may not represent actual transactions.
| Quarter ended | | High | | Low |
| March 31, 2014 | | $ | 0.10 | | | $ | 0.05 | |
| | | | | | | | | |
| December 31, 2013 | | $ | 0.12 | | | $ | 0.04 | |
| September 30, 2013 | | $ | 0.10 | | | $ | 0.04 | |
| June 30, 2013 | | $ | 0.16 | | | $ | 0.05 | |
| March 31, 2013 | | $ | 0.33 | | | $ | 0.10 | |
| | | | | | | | | |
| December 31, 2012 | | $ | 0.35 | | | $ | 0.12 | |
| September 30, 2012 | | $ | 0.16 | | | $ | 0.06 | |
| June 30, 2012 | | $ | 0.21 | | | $ | 0.06 | |
| March 31, 2012 | | $ | 0.25 | | | $ | 0.08 | |
(b) Holders
As of April 14, 2014, there were approximately 90 holders of record of our common stock. The common stock figure does not take into account those shareholders whose certificates are held in the name of broker-dealers or other nominees.
(c) Dividends
The Company issued a dividend to our shareholders of record on December 23, 2011, in the form of restricted common stock of iGlue, Inc. (“iGlue”). Each shareholder received 0.05 shares of restricted common stock of iGlue. A total of 3,115,015 shares of iGlue’s common stock were distributed on February 15, 2012. The Company still holds 2,884,986 shares of iGlue’s common stock and warrants to purchase shares of iGlue’s common stock.
We have never paid any cash dividends on our common shares, and we do not anticipate that we will pay any dividends with respect to those securities in the foreseeable future. Our current business plan is to retain any future earnings to finance the expansion development of our business.
(d) Securities Authorized for Issuance under Equity Compensation Plan
We currently have no Equity Compensation Plan in effect.
Transfer Agent
Our transfer agent is Fidelity Transfer Company, 8915 S. 700 E. Suite 102, Sandy, UT 84070.
Recent Sales of Unregistered Securities
On November 1, 2013, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 1,000,000 shares of its Series D preferred stock to one affiliated private investor, Peter Boros, Director of the company, for aggregate proceeds of $337,653. This amount was previously provided to the company by Mr. Boros as a loan, and on November 1 Mr. Boros elected to convert this loan into aforementioned preferred stock.
On October 17, 2013, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 250,000 shares of its Series C Preferred Stock to one unaffiliated private investors for aggregate proceeds of $75,000.
On September 26, 2013, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 650,000 shares of its Series C Preferred Stock to two unaffiliated private investors for aggregate proceeds of $195,000.
On September 25, 2013, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 1,000,000 shares of its common stock to one unaffiliated private investors for aggregate proceeds of $80,000.
Other than the above, there are no recent sales of unregistered securities that werenot previously disclosed in a Current Report on Form 8-K or a Quarterly Report on Form 10-Q.
Rule 10B-18 Transactions
During the years ended December 31, 2012, there were no repurchases of the Company’s common stock by the Company.
Item 6. Selected Financial Data.
Not applicable.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations.
THE FOLLOWING DISCUSSION OF OUR PLAN OF OPERATION AND RESULTS OF OPERATIONS SHOULD BE READ IN CONJUNCTION WITH THE FINANCIAL STATEMENTS AND RELATED NOTES TO THE FINANCIAL STATEMENTS INCLUDED ELSEWHERE IN THIS REPORT. THIS DISCUSSION CONTAINS FORWARD-LOOKING STATEMENTS THAT RELATE TO FUTURE EVENTS OR OUR FUTURE FINANCIAL PERFORMANCE. THESE STATEMENTS INVOLVE KNOWN AND UNKNOWN RISKS, UNCERTAINTIES AND OTHER FACTORS THAT MAY CAUSE OUR ACTUAL RESULTS, LEVELS OF ACTIVITY, PERFORMANCE OR ACHIEVEMENTS TO BE MATERIALLY DIFFERENT FROM ANY FUTURE RESULTS, LEVELS OF ACTIVITY, PERFORMANCE OR ACHIEVEMENTS EXPRESSED OR IMPLIED BY THESE FORWARD-LOOKING STATEMENTS. THESE RISKS AND OTHER FACTORS INCLUDE, AMONG OTHERS, THOSE LISTED UNDER “FORWARD-LOOKING STATEMENTS” AND “RISK FACTORS” AND THOSE INCLUDED ELSEWHERE IN THIS REPORT.
Overview
Power of the Dream Ventures, Inc. is one of Hungary’s leading technology holding companies. We identify, invest in and acquire technologies originating in Hungary we deem to be internationally marketable. We currently hold equity interest in iGlue, Inc. a US public company. Through our wholly owned subsidiary, Genetic Immunity, a biotechnology company we acquired in October of 2012, we now focus on the commercialization tasks related to Genetic Immunity’s lead product candidate therapeutic HIV vaccine called DermaVir.
We are looking to generate revenue through the sale of Genetic Immunity’s therapeutic vaccines upon US, European and global marketing approvals, and through the sale of our equity interest in iGlue.
Since inception through December 31, 2013, we had a deficit accumulated during the development stage of $12,817,575 and net cash used in operations of $2,486,088. To date all of our funding has been provided through the sale of our common and preferred stock. We believe we can raise sufficient additional financing through the sale of our common stock to allow us to continue operations and execute our business plan to the end of 2014. The Company also anticipates receiving a minimum of $4 million and a maximum of $22 million from a lawsuit initiated against the National Development Agency in Hungary for breach of contract for a previously awarded grant. Should the need arise, we plan to raise additional funds, both from U.S. and international investors. We believe that through the acquisition of Genetic Immunity and the company’s lead product candidate DermaVir, we will be in a position to start generating revenue in 2015. We believe that our ownership of one of the world’s first, clinically proven, therapeutic HIV vaccine will prove attractive to both private and institutional investors, making fundraising a less strenuous process than before. We also believe that as Hungary’s only US public company that is actively engaged in Hungarian intellectual property acquisition (like Genetic Immunity and iGlue,) we are an even more attractive investment candidate.
If necessary, we will raise additional capital in a manner that is the least dilutive to our shareholders yet at the same time serves our development, commercialization and operating needs. We anticipate one round of fundraising in 2014 as we move closer to commercializing DermaVir. By the end of 2014, we will seek conditional marketing approval for DermaVir in the European Union.
Even though we believe our public status will allow us to raise additional capital, if needed, no assurance can be given that we can in fact obtain additional working capital, or if obtained, that such funding will not cause substantial dilution to shareholders of the Company. If we are unable to raise additional funds, we may be forced to change or delay our contemplated marketing and business plan.
In 2014 we anticipate spending approximately $2,000,000 - $3.000.000 on DermaVir’s regulatory approval, commercialization and general administrative expenses. We believe these funds will become available to us from proceeds obtained from our lawsuit against the Hungarian Development Agency as described above in Item 3.
Being a development stage company, we are subject to all the risks inherent in the establishment of a new enterprise and the marketing and manufacturing of new products, many of which risks are beyond the control of the Company.
Over the course of the next 12 months we will focus on accelerating the development and potential commercialization of Genetic Immunity’s DermaVir HIV vaccine. We plan on applying for a Conditional Marketing Authorization (“CMA”) with the EMA, and pursuing similar available venues in the United States. We believe given DermaVir’s safety and efficacy profile and recent changes in the regulatory landscape, the time is ripe for these initiatives.
Although we believe that DermaVir can become a significant new product in the treatment of HIV, there can be no assurance that we will be able to complete the required regulatory processes. If we are unable to complete regulatory permits within the next 12 months we will be required to raise additional capital to support our operations until such time when we can maintain operations from revenue(s) realized upon marketing authorization.
Upon regulatory approval of DermaVir, we anticipate making substantial equipment purchases to expand our GMP manufacturing faculty and to increase our work force to fully implement commercialization tasks.
If we are unable to raise additional financing there is substantial doubt about our ability to continue as a going concern. The condensed consolidated financial statements included elsewhere in this prospectus do not include any adjustments relating to the recoverability of recorded asset amounts that might be necessary as a result of the above uncertainty.
Results of Operations
For the Year Ended December 31, 2013 Compared to the Year Ended December 31, 2012
| | | For the Year Ended December 31, |
| | | 2013 | | 2012 |
| Net sales | | $ | — | | | | — | |
| Gross profit | | $ | — | | | | — | |
| General and administrative expenses | | $ | 738,975 | | | | 797,189 | |
| Loss from operations | | $ | (1,164,332 | ) | | | (1,625,141 | ) |
| Interest Expenses and Exchange Gains | | $ | (629,625 | ) | | | (51,927 | ) |
| Net loss | | $ | (1,794,718 | ) | | | (1,679,052 | ) |
Revenues
During the years ended December 31, 2013 and 2012, respectively, the Company had no revenue.
Research and development
For the year ended December 31, 2013, research and development expenses were $41,812 as compared to $0 for the year ended December 31, 2012. The increase of $41,812 in research and development expenses is attributed to the Fiberscan grant activities.
General, selling and administrative expenses
For the year ended December 31, 2013, general, selling and administrative expenses were $738,975 as compared to $797,189 for the year ended December 31, 2012. The decrease in general, selling and administrative expenses is attributable to lower stock based employee compensation.
Loss from Operations
Loss from operations for the year ended December 31, 2013, was $1,794,718, as compared to $1,679,052 for the year ended December 31, 2012. The primary increase in loss from operations is attributable to the acquisition of Genetic Immunity and fulfillment of Grant related expenditures.
Net Loss
Net loss for year ended December 31, 2013, was $1,794,718 or loss per share of $0.04, as compared to $1,679,052 or loss per share of $0.04, for the comparable year ended December 31, 2012.
Liquidity and Capital Resources
The following table summarizes total current assets, liabilities and working capital at December 31, 2013, compared to December 31, 2012:
| | | December 31,
2013 | | December 31,
2012 | | Increase/Decrease |
| Current Assets | | $ | 262,992 | | | $ | 419,974 | | | $ | (156,982 | ) |
| Current Liabilities | | $ | 8,240,680 | | | $ | 7,761,171 | | | $ | 479,509 | |
| Working Capital (Deficit) | | $ | (7,977,688 | ) | | $ | (7,341,197 | ) | | $ | (636,491 | ) |
At December 31, 2013, we had a working capital deficit of $7,977,688, as compared to a working capital deficit of $7,341,197, at December 31, 2012, an increase of $636,491. The increase in deficit is mainly attributed due to the increase in accrued interest.
Net cash used in operating activities for the year ended December 31, 2013 and 2012 was $(175,521) and $7,772, respectively. The net loss for the years ended December 31, 2013 and 2012 was $1,794,718 and $1,679,052, respectively. The Company’s cash used in operations increased due to financing from accounts payable.
Net cash obtained through all financing activities for the years ended December 31, 2013 was $500,326, as compared to $79,841 for the year ended December 31, 2012. The company raised $500,326 through private placements of our common and preferred stock.
Off-Balance Sheet Arrangements
We have no significant known off balance sheet arrangements.
Critical Accounting Policies
We prepare our consolidated financial statements in accordance with accounting principles generally accepted in the United States of America. The preparation of these financial statements require the use of estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amount of revenues and expenses during the reporting period. Our management periodically evaluates the estimates and judgments made. Management bases its estimates and judgments on historical experience and on various factors that are believed to be reasonable under the circumstances. Actual results may differ from these estimates as a result of different assumptions or conditions.
The methods, estimates, and judgment we use in applying our most critical accounting policies have a significant impact on the results we report in our financial statements. The SEC has defined “critical accounting policies” as those accounting policies that are most important to the portrayal of our financial condition and results, and require us to make our most difficult and subjective judgments, often as a result of the need to make estimates of matters that are inherently uncertain. Based upon this definition, our most critical estimates relate to the fair value of warrant liabilities. We also have other key accounting estimates and policies, but we believe that these other policies either do not generally require us to make estimates and judgments that are as difficult or as subjective, or it is less likely that they would have a material impact on our reported results of operations for a given period. For additional information see Note 2, “Summary of Significant Accounting Policies” in the notes to our reviewed financial statements appearing elsewhere in this report. Although we believe that our estimates and assumptions are reasonable, they are based upon information presently available, and actual results may differ significantly from these estimates.
Going Concern
We have limited working capital and no revenues from sales of products, services, or licensing. During the fiscal year ended December 31, 2013, our operating expenses continued to be greater than our revenues. These factors have caused our accountants to express substantial doubt about our ability to continue as a going concern. The accompanying financial statements do not include any adjustment that might be necessary if we are unable to continue as a going concern.
Our ability to continue as a going concern has caused the Board of Directors to continue to look for sources of investment capital, and investigate merger and acquisition opportunities. We will look to further diversify our holdings and sources of cash flow until we can support our operation from revenue.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk.
We do not hold any derivative instruments and do not engage in any hedging activities.
Item 8. Financial Statements and Supplementary Data.
Our financial statements are contained in pages F-1 through F-24 which appear at the end of this Annual Report.
Item 9. Changes in and Disagreements with Accountants on Accounting and Financial Disclosure.
None.
Item 9A. Controls and Procedures.
(a) Evaluation of Disclosure and Control Procedures
Based on their evaluation as of the end of the period covered by this Annual Report on Form 10-K, our principal executive officer and principal financial officer have concluded that our disclosure controls and procedures (as defined in Rules 13a-15(c) and 15d-15(e) under the Exchange Act) are not effective to ensure that information required to be disclosed by us in report that we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the U.S. Securities and Exchange Commission’s rules and forms and to ensure that information required to be disclosed by us in the reports that we file or submit under the Exchange Act is accumulated and communicated to our management, including our chief executive officer and chief financial officer, as appropriate to allow timely decisions regarding required disclosure due to material weakness in our internal control over financial reporting discussed more fully below...
(b) Management’s Assessment of Internal Control over Financial Reporting
Management is responsible for establishing and maintaining adequate internal control over financial reporting, as such term is defined in the Exchange Act Rules 13a-15(f). A system of internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles.
Under the supervision and with the participation of management, including the principal executive officer and the principal financial officer, the Company’s management has evaluated the effectiveness of its internal control over financial reporting as of December 31, 2013, based on the criteria established in a report entitled “Internal Control - Integrated Framework issued by the Committee of Sponsoring Organizations of the Treadway Commission” and the interpretive guidance issued by the Commission in Release No. 34-55929. Based on this evaluation, the Company’s management has evaluated and concluded that as of December 31, 2013, the Company’s internal control over financial reporting was not effective due to the existence of material weakness as described below.
A material weakness is a control deficiency, or combination of control deficiencies, in internal control over financial reporting such that there is a reasonable possibility that a material misstatement of our annual or interim financial statements would not be prevented or detected on a timely basis. As of December 31, 2013, we have concluded that our internal control over financial reporting was ineffective. The Company’s assessment identified certain material weaknesses which are set forth below:
Functional Controls and Segregation of Duties
Because of the company’s limited resources, there are limited controls over information processing, and no internal controls over the accuracy, completeness and authorization of transactions.
There is an inadequate segregation of duties consistent with control objectives. Our Company’s management is composed of a small number of individuals resulting in a situation where limitations on segregation of duties exist. In order to remedy this situation we would need to hire additional staff to provide greater segregation of duties. Currently, it is not feasible to hire additional staff to obtain optimal segregation of duties. Management will reassess this matter in the following year to determine whether improvement in segregation of duty is feasible.
There is a lack of top level reviews in place to review targets, product development, joint ventures or financing. All major business decisions are carried out by the officers with board of director approval when needed.
Accordingly, as the result of identifying the above material weaknesses we have concluded that these control deficiencies resulted in a reasonable possibility that a material misstatement of the annual or interim financial statements will not be prevented or detected on a timely basis by the Company’s internal controls.
Management believes that the material weaknesses set forth above were the result of the scale of our operations and are intrinsic to our small size. Management believes these weaknesses did not have a material effect on our financial results and intends to take remedial actions upon receiving funding for the Company’s business operations.
This annual report does not include an attestation report of our registered public accounting firm regarding internal control over financial reporting. Management’s report was not subject to attestation by our registered public accounting firm pursuant to temporary rules of the SEC that permit the Company to provide only management’s report herein.
(c) Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting, as defined in Rules 13a-15(f) and 15d-15(f) under the Exchange Act, during our most recently completed fiscal quarter that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Item 9B. Other Information.
None.
PART III
Item 10. Directors, Executive Officers, and Corporate Governance.
The following table and biographical summaries set forth information, including principal occupation and business experience, about our directors and executive officers at April 15, 2013:
| Name | | Age | | Position | | Officer and/or Director Since |
| | | | | | | |
| Viktor Rozsnyay | | 43 | | Chairman, Chief Executive Officer, President | | April 2007 |
| | | | | | | |
| Ildiko Rozsa | | 37 | | Chief Financial Officer | | October 2007 |
| | | | | | | |
| Peter Boros | | 37 | | Director | | May 2013 |
Viktor Rozsnyay, age 43
Viktor Rozsnyayhas served as the Chairman, President and Chief Executive Officer of the Company since April 5, 2007. From April 2006 to present, Mr. Rozsnyay has been a Manager of Vidatech Kft., a Hungarian company focused on the acquisition of technologies developed in Hungary, of which he was a founder; Vidatech is now our wholly owned Hungarian subsidiary. From 2004 to 2006 Mr. Rozsnyay was engaged in researching and establishing the foundation of Vidatech. From 2001 to 2004, he was the Founder and Chief Operating Officer of 10Charge, Inc., an ISSO R&D spin-off company formed to commercialize 10 minute battery charging technology, a product Mr. Rozsnyay and his former partners invented and patented. 10Charge, Inc. was a full SEC reporting company until it was acquired by US investors. Mr. Rozsnyay was the primarily person responsible for organizing and completing a reverse merger for 10Charge, Inc. Prior thereto, Mr. Rozsnyay was the Founder and Managing Director of ISSO R&D Kft., an aerospace research company based in the Republic of Hungary, pursuing advanced aerospace propulsion research. Mr. Rozsnyay attended the Jozsef Katona Technical College in Budapest, graduating in 1989.
Mr. Rozsnyay’s unique expertise in Hungary of US public company operations and requirements and as our Founder and CEO make him the perfect and only choice to fulfill the Chairman of the Board position.
Ildiko Rozsa, age 37
Ildiko Rozsa has served as the Chief Financial Officer of the Company since October 24, 2007. Mrs. Rozsa is a qualified statutory accountant, currently completing her PhD studies at the Budapest Technical and Economic University in Business Science. Ms. Rozsa graduated at the College of Finance and Accountancy, later obtaining her mastered degree (MBA) at the Budapest University of Economics. From 1995 through 1996 she was audit assistant at Price Waterhouse’s Budapest Audit Department, where she gained experience in IAS and HAS audits. From 1997 through 2002 Ms. Rozsa was Finance and Accounting Director at Vivendi Telecom Hungary. Ms. Rozsa was responsible for the production of Vivendi Group Consolidated Financial Statements for 23 companies including foreign companies in accordance with IFRS, US GAAP and the related statutory requirements; definition of the accounting policy and procedures of the group, and controlling of their application. Management of the related audits and Managing the Accounting Department, the Reporting and Consolidation Department and the Payroll Department of Vivendi Group which consists of approximately 60 persons. From 2002 through 2004 Ms. Rozsa was Chief Financial Officer at Bacardi-Martini Hungary Kft., where she was responsible for Managing the finance and administration team of the company. Responsible for financial, accounting, controlling, IT, customs and payroll activities. In 2004 she founded and became Managing Director, RIBZ Consulting where she works for multinational clients on IFRS and US GAAP projects, on privatization engagements. Ildiko is a tutor at the Budapest Technical University, Economic Faculty.
Peter Boros, age 37
Mr. Peter Boros, age 37, currently serves as Chief Executive Officer of iGlue, Inc., a Hungarian-based development stage software company, where he is responsible for the day to day operations of the company. Since July 1, 2007, Mr. Boros has served as the Commercial Director of Co-Op Inc., a retail chain in Hungary. From 2000 to 2007, Mr. Boros was the manager of the Sales and Marketing Department of Co-Op Inc. Additionally, since 2006, Mr. Boros was the owner and operator of Co-Op Gyor, Inc., a grocery chain which was sold in 2011. Mr. Boros currently serves as member of the board of directors of iGlue, Inc.
Family Relationships
There are no family relationships among our directors, executive officers, or persons nominated or chosen by the Company to become directors or executive officers. None of our directors or executive officers or their respective immediate family members or affiliates are indebted to us.
Committees of the Board of Directors
We have not established any committees, including an audit committee, a compensation committee or a nominating committee.The Board currently acts as our audit committee. At the present time, we believe that our Board is capable of analyzing and evaluating our financial statements and understanding internal controls and procedures for financial reporting.
Legal Proceedings
There are no material proceedings to which any director or officer, or any associate of any such director or officer, is a party that is adverse to our Company or any of our subsidiaries or has a material interest adverse to our Company or any of our subsidiaries. No director or executive officer has been a director or executive officer of any business which has filed a bankruptcy petition or had a bankruptcy petition filed against it during the past ten years. No director or executive officer has been convicted of a criminal offense or is the subject of a pending criminal proceeding during the past ten years. No director or executive officer has been the subject of any order, judgment or decree of any court permanently or temporarily enjoining, barring, suspending or otherwise limiting his involvement in any type of business, securities or banking activities during the past ten years. No director or officer has been found by a court to have violated a federal or state securities or commodities law during the past ten years.
Compliance with Section 16(A) of the Exchange Act
Section 16(a) of the Exchange Act requires the Company’s directors, executive officers and persons who beneficially own 10% or more of a class of securities registered under Section 12 of the Exchange Act to file reports of beneficial ownership and changes in beneficial ownership with the SEC. Directors, executive officers and greater than 10% stockholders are required by the rules and regulations of the SEC to furnish the Company with copies of all reports filed by them in compliance with Section 16(a).
Based solely on our review of certain reports filed with the Securities and Exchange Commission pursuant to Section 16(a) of the Securities Exchange Act of 1934, as amended, the reports required to be filed with respect to transactions in our common stock during the fiscal year ended December 31, 2012, were not timely.
Code of Ethics
We have not yet adopted a code of ethics because we wanted to complete our constitution of the Board prior to adopting such code of ethics to allow the entire Board to review and approve our code of ethics.
Item 11. Executive Compensation.
The following summary compensation table sets forth all compensation awarded to, earned by, or paid to the named executive officers paid by us during the periods ended December 31, 2013, 2012 and 2011.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Name And
Principal Position | | | Year | | | | Salary
($) | | | | Bonus
($) | | | | Stock
Awards
($) | | | | Option
Awards
($) | | | | Non-Equity
Incentive
Plan
Compen-
sation ($) | | | | All Other Compen- sation ($) | | | | Total ($) | |
| Viktor Rozsnyay | | | 2013 | | | $ | 8,400 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 8,400 | |
| Chief Executive Officer & President | | | 2012 | | | $ | 8,400 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 8,400 | |
| | | | 2011 | | | $ | 8,400 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 8,400 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Ildiko Rozsa | | | 2012 | 3 | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | | | $ | 0 | |
| Chief Financial Officer | | | 2011 | 2 | | | 0 | | | | 0 | | | | 0 | | | | 0 | | | | 0 | | | | 0 | | | $ | 0 | |
| | | | 2010 | 1 | | | 0 | | | | 0 | | | | 15,000 | | | | 0 | | | | 0 | | | | 0 | | | $ | 15,000 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Outstanding Equity Awards at December 31, 2013
As of December 31, 2013, there were no outstanding equity awards.
Director Compensation
During the year, ended December 31, 2013, the Company did not pay any cash fees or expenses to our directors for serving on the Board.
Employment Agreements
We currently do not have any employee agreements.
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters.
The following table sets forth, as of April 14, 2014, the number of shares of our common stock owned by (i) each person who is known by us to own of record or beneficially five percent (5%) or more of our outstanding shares, (ii) each of our directors, (iii) each of our executive officers and (iv) all of our directors and executive officers as a group. Unless otherwise indicated, each of the persons listed below has sole voting and investment power with respect to the shares of our common stock beneficially owned.
| Name and Address | Beneficial Relationship to Company | | Outstanding Common Stock | | | Percentage of Ownership of Common Stock (1) | |
| | | | | | | | |
| | | | | | | | | | |
| Directors and Named Executive Officers | | | | | | | | | |
| Viktor Rozsnyay(3) | Chief Executive Officer, President, Chairman | | | 1,874,300 | | | | 4 | %(2) |
| | | | | | | | | | |
| | | | | | | | | | |
| Ildiko Rozsa (4) | Chief Financial Officer | | | 570,000 | | | | 1.22 | % |
| | | | | | | | | | |
| | | | | | | | | | |
| Peter Boros (5) | Director | | | 1,159,907 | | | | 2.5 | % |
| | | | | | | | | | |
| Officers and Directors as a Group (3 persons) | | | | 2,444,300 | | | | 8.4 | % |
* less than 1%
| | (1) | Based on 46,500,896 shares of common stock outstanding as of April 14, 2014. |
| | (2) | This percentage does not include shares of Preferred Stock. |
| | (3) | Mr. Rozsnyay owns 1,000,000 shares of our Series A Preferred Stock and 1,874,300 shares of our common stock. His mailing address is 2049 Diosd, Ligetszepe ut 54, Hungary. |
| | (4) | Ms. Rozsa owns 570,000 shares of our common stock. Ms. Rozsa’s mailing address is 1066 Budapest, Terez Krt. 22, Hungary. |
| | (5) | Peter Boros owns 1,000,000 shares of our Series A Preferred stock, 1,000,000 shares of our Series D Preferred stock and 1,159,907 shares of our common stock. Mr. Boros’s address is Gyor, Ybl seteny 23, Hungary. |
Securities Authorized for Issuance under Equity Compensation Plans
The Company currently does not have an equity compensation plans in effect.
Changes in Control
We are not aware of any arrangements that may result in “changes in control” as that term is defined by the provisions of Item 403(c) of Regulation S-K.
Item 13. Certain Relationships and Related Transactions, and Director Independence.
Related Transactions
During the year ended December 31, 2013, there were no related transactions required to be reported under Item 404 of Regulation S-K.
Director Independence
On an annual basis, each director and executive officer will be obligated to disclose any transactions with the Company in which a director or executive officer, or any member of his or her immediate family, have a direct or indirect material interest in accordance with Item 407(a) of Regulation S-K. Following completion of these disclosures, the Board will make an annual determination as to the independence of each director using the current standards for “independence” that satisfy both the criteria for the Nasdaq and the American Stock Exchange.
As of December 31, 2013, the Board determined that none of our directors are independent under these standards.
Item 14. Principal Accounting Fees and Services.
The following table sets forth the aggregate fees billed for each of the last two fiscal years for professional services rendered by the principal accountant for the audit of the Company's annual financial statements and review of financial statements included in the Company's Form 10-Q quarterly reports or services that are normally provided by the accountant in connection with statutory and regulatory filings or engagements for those fiscal years.
| | | 2013 | | 2012 |
| | | | | | | | | |
| Audit Fees | | $ | 40,290 | | | $ | 34,227 | |
| Audit-Related Fees | | $ | 0 | | | $ | 0 | |
| Tax Fees | | $ | 0 | | | $ | 0 | |
| All Other Fees | | $ | 0 | | | $ | 0 | |
| TOTAL | | $ | 40,290 | | | $ | 34,227 | |
| | | | | | | | | |
We do not have an audit committee. Our entire board of directors pre-approves all services provided by our independent auditors.
PART IV
Item 15. Exhibits, Financial Statement Schedules.
| Exhibit No. | | Description |
| | | |
| 23.1 | | Consent of BDO Hungary, Ltd.* |
| | | |
| 31.1 | | Certification by the Principal Executive Officer of Registrant pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (Rule 13a-14(a) or Rule 15d-14(a)). * |
| | | |
| 31.2 | | Certification by the Principal Financial Officer of Registrant pursuant to Section 302 of the Sarbanes-Oxley Act of 2002 (Rule 13a-14(a) or Rule 15d-14(a)). * |
| | | |
| 32.1 | | Certification by the Principal Executive Officer pursuant to 18 U.S.C. 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. * |
| | | |
| 32.2 | | Certification by the Principal Executive Officer pursuant to 18 U.S.C. 1350 as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. * |
| | | |
| | | |
| 101.INS | | XBRL Instance Document ** |
| | | |
| 101.SCH | | XBRL Taxonomy Extension Schema ** |
| | | |
| 101.CAL | | XBRL Taxonomy Extension Calculation Linkbase ** |
| | | |
| 101.DEF | | XBRL Taxonomy Extension Definition Linkbase ** |
| | | |
| 101.LAB | | XBRL Taxonomy Extension Label Linkbase ** |
| | | |
| 101.PRE | | XBRL Taxonomy Extension Presentation Linkbase ** |
| | | |
* filed herewith
** In accordance with Regulation S-T, the XBRL-related information on Exhibit No. 101 to this Annual Report on Form 10-K shall be deemed “furnished” herewith and not “filed.”
SIGNATURES
Pursuant to the requirements of Section 13 or 15(d) of the Exchange Act of 1934, the registrant has duly caused this report to be signed on its behalf by the undersigned, thereunto duly authorized.
| | | | POWER OF THE DREAM VENTURES, INC. |
| | | | | | |
| | | | | | |
| Date: April 14, 2014 | | By: | /s/ Viktor Rozsnyay | |
| | | | | Name: Viktor Rozsnyay | |
| | | | | Title: Chief Executive Officer
(Principal Executive Officer) | |
| Date: April 14, 2014 | | By: | /s/ Ildiko Rozsa | |
| | | | | Name: Ildiko Rozsa | |
| | | | | Title: Chief Financial Officer
(Principal Financial Officer) | |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the registrant and in the capacities and on the dates indicated.
| Name | | Position | | Date |
| | | | | |
| /s/ Viktor Rozsnyay | | Chief Executive Officer, President and Chairman | | April 14, 2014 |
| Viktor Rozsnyay | | | | |
| | | | | |
| /s/ Ildiko Rozsa | | Chief Financial Officer | | April 14, 2014 |
| Ildiko Rozsa | | | | |
| | | | | |
| /s/ Peter Boros | | Director | | April 14, 2014 |
| Peter Boros | | | | |
| | | | | |
| | | | | |
Index to Financial Statements
| | Page No. |
| Report of Independent Registered Public Accounting Firm | F-2 |
| Consolidated Balance Sheet at December 31, 2013 | F-3 to F-4 |
| Consolidated Statements of Operations and Comprehensive Income for the years ended December 31, 2013 and 2012 | F-5 |
| Consolidated Statements of Deficiency in Stockholders’ Equity for the two years ended December 31, 2013 | F-6 to F-11 |
| Consolidated Statements of Cash Flows for the years ended December 31, 2013 and 2012 | F-12 |
| Notes to Consolidated Financial Statements | F-13 to F-41 |
Report of Independent Public Accounting Firm
Board of Directors and Stockholders
Power of the Dream Ventures, Inc. (formerly Tia V)
(a development stage company)
Budapest, Hungary
We have audited the accompanying balance sheets of Power of the Dream Ventures, Inc. (formerly Tia V), a development stage company, as of December 31, 2013 and 2012 and the related statements of operations, stockholders’ equity, and cash flows for each of the two years in the period then ended and the period from inception (April 26, 2006) to December 31, 2013. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. Our audits included consideration of internal control over financial reporting as a basis for designing audit procedures that are appropriate in the circumstances, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion. An audit also includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the financial statements referred to above present fairly, in all material respects, the financial position of Power of the Dream Ventures, Inc. (formerly Tia V) at December 31, 2013 and 2012, and the results of its operations and its cash flows for each of the two years in the period then ended and the period from inception (April 26, 2006) through December 31, 2013, in conformity with accounting principles generally accepted in the United States of America.
The accompanying financial statements have been prepared assuming that the Company will continue as a going concern. As discussed in Note 1 to the financial statements, the Company has suffered recurring losses from operations and has a net capital deficiency that raise substantial doubt about its ability to continue as a going concern. Management’s plans in regard to these matters are also described in Note 1. The financial statements do not include any adjustments that might result from the outcome of this uncertainty.
April 14, 2014
BDO Hungary Audit Ltd.
1103 Budapest, Kőér utca 2/A
Registration number: 002387
| | | |
| Zsuzsanna Nagy | | Ferenc Baumgartner |
| Managing Director | | Certified Auditor Chamber registration No.: 002955 |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED BALANCE SHEET
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
Amounts in USD
| | | | | |
| | | December 31, 2013 | | December 31, 2012 |
| | | | | | | | | |
| ASSETS | | | | | | | | |
| | | | | | | | | |
| Current Assets | | | | | | | | |
| Cash | | $ | 22,922 | | | $ | 63,986 | |
| Other receivables | | | 217,419 | | | | 332,018 | |
| Inventories | | | 22,651 | | | | 23,970 | |
| Total Current Assets | | | 262,992 | | | | 419,974 | |
| | | | | | | | | |
| Intangibles | | | 30,055,381 | | | | 30,092,841 | |
| Fixed assets, net | | | 189,194 | | | | 503,169 | |
| Total Assets | | | 30,507,567 | | | | 31,015,984 | |
| | | | | | | | | |
| LIABILITIES | | | | | | | | |
| | | | | | | | | |
| Current Liabilities | | | | | | | | |
| Accounts payable and accrued and other liabilities | | $ | 4,083,294 | | | $ | 2,867,804 | |
| Grants received | | | 1,432,042 | | | | 1,546,774 | |
| Capital leases payable, current portion | | | 55,784 | | | | 56,284 | |
| Short term loans from related parties | | | 86,899 | | | | 91,884 | |
| Liability from warrants | | | 87,502 | | | | 608,540 | |
| Note payable | | | 154,000 | | | | 154,000 | |
| Short term loans | | | 2,341,159 | | | | 2,435,885 | |
| Total Current Liabilities | | | 8,240,680 | | | | 7,761,171 | |
| | | | | | | | | |
| Long term liabilities | | | | | | | | |
| Capital leases payable, less current portion | | | 35,692 | | | | 75,199 | |
| Total Long Term Liabilities | | | 35,692 | | | | 75,199 | |
| | | | | | | | | |
| | | | | | | | | |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED BALANCE SHEET
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
Amounts in USD
| Stockholders’ Equity | | | | | | | | |
Preferred stock, $0.001 par value,
10,000,000 shares authorized,
4,900,000 and 3,000,000 issued | | | | | | | | |
| of which Series “A” | | | 2,000 | | | | 2,000 | |
| of which Series “B” | | | 1,000 | | | | 1,000 | |
| of which Series “C” | | | 900 | | | | — | |
| of which Series “D” | | | 1,000 | | | | — | |
| | | | | | | | | |
Common stock, $.001 par value;
100,000,000 shares authorized, 46,500,896 and 42,495,244 shares issued and outstanding | | | 46,501 | | | | 42,495 | |
Additional Paid-In Capital
Deficit accumulated during development stage
Other Comprehensive Income | | | 35,403,285 (13,018,339) (205,152) | | | | 34,480,163 (11,223,621) (45,499) | |
| Unearned Compensation | | | — | | | | (76,924 | ) |
| Total Stockholders’ Equity | | | 22,231,195 | | | | 23,179,614 | |
| | | | | | | | | |
| Total liabilities and stockholders’ equity | | | 30,507,567 | | | | 31,015,984 | |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF OPERATIONS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
Amounts in USD
| | Notes | | For the year ended December 31, 2013 | | For the year ended December 31, 2012 | | For the Period from April 26, 2006 (date of inception) to December 31, 2013 | |
| | | | | | | | | |
| Net Sales | | | $ - | | $ - | | $ 5,833 | |
| Cost of Sales | | | - | | - | | (3,711) | |
| | | | | | | | | |
| Gross margin | | | - | | - | | 2,122 | |
| Material expenses | | | 34,073 | | 17,437 | | 145,674 | |
| General administration | 12 | | 738,975 | | 797,189 | | 8,895,232 | |
| Research and development | 4 | | 41,812 | | - | | 866,619 | |
| Personnel expenses | | | 271,114 | | 213,823 | | 965,604 | |
| Depreciation and amortization | 5 | | 145,219 | | 108,099 | | 707,281 | |
| Grants received | 10 | | (150,196) | | (126,783) | | (276,979) | |
| Other expenses, net | 13 | | 83,335 | | 615,376 | | 713,958 | |
| Operating expenses | | | 1,164,332 | | 1,625,141 | | 12,017,389 | |
| Loss from operations | | | (1,164,332) | | (1,625,141) | | (12,015,267) | |
| Interest expense and losses | | | (629,625) | | (51,927) | | (798,356) | |
Loss before income taxes | | | (1,793,957) | | (1,677,068) | | (12,813,623) | |
| Income taxes | | | (761) | | (1,984) | | (3,952) | |
Net loss | | | (1,794,718) | | (1,679,052) | | $ (12,817,575) | |
Basic loss per share Diluted loss per share Weighted average number of shares outstanding – Basic Weighted average number of shares outstanding – Diluted | | | $(0.04) $(0.04) 44,909,013 44,909,013 | | $(0.04) $(0.04) 39,018,009 39,018,009 | | | |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| | | Common
Shares | | Stocks
Amount | | Accumulated Deficit During Developmental Stage | | Additional
Paid In
Capital | | Other Comprehensive Income | | Unearned Compensation | | Total | | Comprehensive Income/ (Loss) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Issuance of common stock | | | 33,300,000 | | | $ | 3,330 | | | | | | | $ | 10,670 | | | | | | | | | | | $ | 14,000 | | | | | |
| Contributed Capital | | | | | | | | | | | | | | | 96,100 | | | | | | | | | | | | 96,100 | | | | | |
| Currency Translation Adjustment | | | | | | | | | | | | | | | | | | $ | 4,151 | | | | | | | | 4,151 | | | | 4,151 | |
| Net loss for the period | | | | | | | | | | $ | (35,100 | ) | | | | | | | | | | | | | | | (35,100 | ) | | | (35,100 | ) |
| Balance at December 31, 2006 | | | 33,300,000 | | | | 3,330 | | | | (35,100 | ) | | | 106,770 | | | | 4,151 | | | | — | | | | 79,151 | | | | (30,949 | ) |
| Contributed Capital | | | | | | | | | | | | | | | 53,735 | | | | | | | | | | | | 53,735 | | | | | |
Recapitalization upon Reverse Merger
on April 10, 2007 (See Note 1) | | | 2,500,000 | | | | 250 | | | | (250,763 | ) | | | | | | | | | | | | | | | (250,513 | ) | | | | |
Private placement of shares at $0.34 per share
(See Note 10) | | | 2,250,000 | | | | 225 | | | | | | | | 764,775 | | | | | | | | | | | | 765,000 | | | | | |
Shares issued for services at $0.34 per
share (See Note 10) | | | 1,875,000 | | | | 188 | | | | | | | | 637,313 | | | | | | | $ | (467,501 | ) | | | 170,000 | | | | | |
| Shares issued for research and development at $0.34 per share (See Note 10) | | | 100,000 | | | | 10 | | | | | | | | 33,990 | | | | | | | | | | | | 34,000 | | | | | |
| Private placement at $2.5 per share (See Note 10) | | | 104,000 | | | | 10 | | | | | | | | 259,990 | | | | | | | | | | | | 260,000 | | | | | |
| Shares issued for stock based compensation at $2.5 per share (See Note 10) | | | 1,036,000 | | | | 104 | | | | | | | | 2,589,896 | | | | | | | | (2,590,000 | ) | | | — | | | | | |
| Amortization of Unearned Compensation | | | | | | | | | | | | | | | | | | | | | | | 1,124,932 | | | | 1,124,932 | | | | | |
| Currency Translation Adjustment | | | | | | | | | | | | | | | | | | | (14,001 | ) | | | | | | | (14,001 | ) | | | (14,001 | ) |
| Net loss for the period | | | | | | | | | | | (1,992,472 | ) | | | | | | | | | | | | | | | (1,992,472 | ) | | | (1,992,472 | ) |
| Balance at December 31, 2007 | | | 41,165,000 | | | $ | 4,117 | | | $ | (2,278,335 | ) | | $ | 4,446,469 | | | $ | (9,850 | ) | | $ | (1,932,569 | ) | | $ | 229,832 | | | $ | (2,006,473 | ) |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| | | Common
Shares | | Stocks
Amount | | Accumulated Deficit During Developmental Stage | | Additional
Paid In
Capital | | Other Comprehensive Income | | Unearned Compensation | | Total | | Comprehensive Income/ (Loss) |
| Balance at December 31, 2007 | | | 41,165,000 | | | $ | 4,117 | | | $ | (2,278,335 | ) | | $ | 4,446,469 | | | $ | (9,850 | ) | | $ | (1,932,569 | ) | | $ | 229,832 | | | | | |
| Private placement of shares at $3.25 per share (See Note 10) | | | 32,500 | | | | 3 | | | | | | | | 105,622 | | | | | | | | | | | | 105,625 | | | | | |
Shares issued for services at $0.7 per
share (See Note 10) | | | 306,570 | | | | 31 | | | | | | | | 214,568 | | | | | | | | (214,599 | ) | | | — | | | | | |
Shares issued for services at $0.75 per
share (See Note 10) | | | 1,500,000 | | | | 150 | | | | | | | | 1,124,850 | | | | | | | | (1,125,000 | ) | | | — | | | | | |
Shares issued for services at $1.35 per
share (See Note 10) | | | 111,111 | | | | 11 | | | | | | | | 149,989 | | | | | | | | (150,000 | ) | | | — | | | | | |
Private placement of shares at $0.4 per
share (See Note 10) | | | 2,500,000 | | | | 250 | | | | | | | | 999,750 | | | | | | | | | | | | 1,000,000 | | | | | |
Shares issued for Standby Equity
Distribution Agreement at $0.4 per share | | | 2,000,000 | | | | 200 | | | | | | | | (200 | ) | | | | | | | | | | | — | | | | | |
| Amortization of Unearned Compensation | | | | | | | | | | | | | | | | | | | | | | | 3,018,710 | | | | 3,018,710 | | | | | |
| Currency Translation Adjustment | | | | | | | | | | | | | | | | | | | 36,465 | | | | | | | | 36,465 | | | | 36,465 | |
| Net loss for the period | | | | | | | | | | | (3,958,212 | ) | | | | | | | | | | | | | | | (3,958,212 | ) | | | (3,958,212 | ) |
| Balance at December 31, 2008 | | | 47,615,181 | | | $ | 4,762 | | | $ | (6,236,547 | ) | | $ | 7,041,048 | | | $ | 26,615 | | | $ | (403,458 | ) | | $ | 432,420 | | | $ | (3,921,747 | ) |
| Shares issued for stock based compensation at $0.4 per share (See Note 8) | | | 700,000 | | | | 70 | | | | | | | | 279,930 | | | | | | | | (280,000 | ) | | | — | | | | | |
| Private placement for $0.20 per share | | | 111,110 | | | | 11 | | | | | | | | 22,211 | | | | | | | | | | | | 22,222 | | | | | |
| Private placement for $0.16 per share | | | 175,000 | | | | 18 | | | | | | | | 27,982 | | | | | | | | | | | | 28,000 | | | | | |
| Shares issued for services at $0.4 per share (See Note 8 | | | 250,000 | | | | 25 | | | | | | | | 99,975 | | | | | | | | (60,000 | ) | | | 40,000 | | | | | |
| Amortization of Unearned Compensation | | | | | | | | | | | | | | | | | | | | | | | 728,458 | | | | 728,458 | | | | | |
| Currency Translation Adjustment | | | | | | | | | | | | | | | | | | | 17,823 | | | | | | | | 17,823 | | | | 17,823 | |
| Net loss for the period | | | | | | | | | | | (1,518,077 | ) | | | | | | | | | | | | | | | (1,518,077 | ) | | | (1,518,077 | ) |
| Balance at December 31, 2009 | | | 48,851,291 | | | $ | 4,886 | | | $ | (7,754,624 | ) | | $ | 7,471,146 | | | $ | 44,438 | | | $ | (15,000 | ) | | $ | (249,154 | ) | | $ | (1,500,254 | ) |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| | | Common
Shares | | Stocks
Amount | | Accumulated Deficit During Developmental Stage | | Additional
Paid In
Capital | | Other Comprehensive Income | | Unearned Compensation | | Total | | Comprehensive Income/ (Loss) |
| Balance at December 31, 2009 | | | 48,851,291 | | | $ | 4,886 | | | $ | (7,754,624 | ) | | $ | 7,471,146 | | | $ | 44,438 | | | $ | (15,000 | ) | | $ | (249,154 | ) | | | | |
| Private placement for $0.12 and $0.1 per share | | | 1,597,500 | | | | 159 | | | | | | | | 159,542 | | | | | | | | | | | | 159,701 | | | | | |
| Private placement for $0.5 per share (See Note 8) | | | 70,000 | | | | 7 | | | | | | | | 34,993 | | | | | | | | | | | | 35,000 | | | | | |
| Shares issued for services at $0.5 per share (See Note 8) | | | 1,027,000 | | | | 100 | | | | | | | | 513,400 | | | | | | | | (513,500 | ) | | | — | | | | | |
Shares issued for stock based compensation at $0.1 per
share (See Note 8) | | | 540,000 | | | | 54 | | | | | | | | 53,946 | | | | | | | | (54,000 | ) | | | — | | | | | |
Shares issued for services at $0.1 per share (See Note 8) | | | 20,000 | | | | 2 | | | | | | | | 1,998 | | | | | | | | (2,000 | ) | | | — | | | | | |
| Exercise of warrant | | | 3,226 | | | | 3 | | | | | | | | 1,613 | | | | | | | | | | | | 1,616 | | | | | |
| Amortization of Unearned Compensation | | | | | | | | | | | | | | | | | | | | | | | 200,528 | | | | 200,528 | | | | | |
| Currency Translation Adjustment | | | | | | | | | | | | | | | | | | | (38,874 | ) | | | | | | | (38,874 | ) | | | (38,874 | ) |
| Net loss for the period | | | | | | | | | | | (588,910 | ) | | | | | | | | | | | | | | | (588,910 | ) | | | (588,910 | ) |
| Balance at December 31, 2010 | | | 52,109,017 | | | $ | 5,211 | | | $ | (8,343,534 | ) | | | 8,236,638 | | | $ | 5,564 | | | $ | (383,972 | ) | | $ | (480,093 | ) | | $ | (627,784 | ) |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| | | Preferred
Shares | | Common
Shares | | Stocks
Amount | | Accumulated Deficit During Developmental Stage | | Additional
Paid In
Capital | | Other Comprehensive Income | | Unearned Compensation | | Total |
| Balance at December 31, 2010 | | | — | | | | 52,109,017 | | | $ | 5,211 | | | $ | (8,343,534 | ) | | | 8,236,638 | | | $ | 5,564 | | | $ | (383,972 | ) | | $ | (480,093 | ) | | $ | (627,784 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Private placement at $0.29 per share (see
Note 8) | | | | | | | 196,489 | | | | 20 | | | | | | | | 56,962 | | | | | | | | | | | | 56,982 | | | | | |
| Conversion of note at $0.29 per share | | | | | | | 2,400,000 | | | | 240 | | | | | | | | 69,760 | | | | | | | | | | | | 70,000 | | | | | |
Private placement at $0.16 per share (see
Note 8) | | | | | | | 1,727,013 | | | | 172 | | | | | | | | 276,150 | | | | | | | | | | | | 276,322 | | | | | |
Private placement at $0.12 per share (see
Note 8) | | | | | | | 17,825 | | | | 2 | | | | | | | | 2,135 | | | | | | | | | | | | 2,137 | | | | | |
Shares issued for services per share
(See Note 8) | | | | | | | 1,970,000 | | | | 197 | | | | | | | | 457,691 | | | | | | | | (457,888 | ) | | | — | | | | | |
| Shares issued for stock based compensation at $0.15 per share (See Note 8) | | | | | | | 540,000 | | | | 54 | | | | | | | | 80,946 | | | | | | | | (81,000 | ) | | | — | | | | | |
Private placement at $0.16 per share (see
Note 8) | | | | | | | 27,700 | | | | 3 | | | | | | | | 4,429 | | | | | | | | | | | | 4,432 | | | | | |
| Conversion of note at $0.01 per share | | | | | | | 3,500,000 | | | | 350 | | | | | | | | 19,650 | | | | | | | | | | | | 20,000 | | | | | |
Cancellation of common stock (and issue of preferred stock) | | | 2,000,000 | | | | (24,000,000 | ) | | | | | | | | | | | | | | | | | | | | | | | — | | | | | |
| Amortization of Unearned Compensation | | | | | | | | | | | | | | | | | | | | | | | | | | | 781,458 | | | | 781,458 | | | | | |
| Currency Translation Adjustment | | | | | | | | | | | | | | | | | | | | | | | (13,791 | ) | | | | | | | (13,791 | ) | | | (13,791 | ) |
| Net loss for the period | | | | | | | | | | | | | | | (1,251,035 | ) | | | | | | | | | | | | | | | (1,251,035 | ) | | | (1,251,035 | ) |
| Balance at December 31, 2011 | | | 2,000,000 | | | | 38,488,044 | | | $ | 6,249 | | | $ | (9,594,569 | ) | | | 9,204,361 | | | $ | (8,227 | ) | | $ | (141,402 | ) | | $ | (533,588 | ) | | $ | (1,264,826 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| | | Preferred
Shares | | Common
Shares | | Stocks
Amount | | Accumulated Deficit During Developmental Stage | | Additional
Paid In
Capital | | Other Comprehensive Income | | Unearned Compensation | | Total |
| Balance at December 31, 2011 | | | 2,000,000 | | | | 38,488,044 | | | $ | 6,249 | | | $ | (9,594,569 | ) | | | 9,204,361 | | | $ | (8,227 | ) | | $ | (141,402 | ) | | $ | (533,588 | ) | | $ | (1,264,826 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Conversion of common shares to preferred shares (change in value) | | | | | | | | | | | (2,200 | ) | | | | | | | 2,200 | | | | | | | | | | | | — | | | | | |
| Shares issued for services | | | | | | | 135,000 | | | | 14 | | | | | | | | 21,586 | | | | | | | | | | | | 21,600 | | | | | |
Private placement at $0.30 per share (see
Note 8) | | | | | | | 42,200 | | | | 4 | | | | | | | | 12,654 | | | | | | | | | | | | 12,658 | | | | | |
| Amortization of Unearned Compensation | | | | | | | | | | | | | | | | | | | | | | | | | | | 197,328 | | | | 197,328 | | | | | |
| Change of par value from $0.0001 to $0.001 | | | | | | | | | | | 36,598 | | | | | | | | (36,598 | ) | | | | | | | | | | | — | | | | | |
| Shares issued for services | | | | | | | 1,930,000 | | | | 1,930 | | | | | | | | 178,240 | | | | | | | | (132,850 | ) | | | 47,320 | | | | | |
| Conversion of loan to stocks | | | | | | | 1,900,000 | | | | 1,900 | | | | | | | | 74,100 | | | | | | | | | | | | 76,000 | | | | | |
| Acquisition of Genetic Immunity | | | 1,000,000 | | | | | | | | 1,000 | | | | 50,000 | | | | 25,023,620 | | | | | | | | | | | | 25,074,620 | | | | | |
| Currency Translation Adjustment | | | | | | | | | | | | | | | | | | | | | | | (37,272 | ) | | | | | | | (37,272 | ) | | | (37,272 | ) |
| Net loss for the period | | | | | | | | | | | | | | | (1,679,052 | ) | | | | | | | | | | | | | | | (1,679,052 | ) | | | (1,679,052 | ) |
| Balance at December 31, 2012 | | | 3,000,000 | | | | 42,495,244 | | | $ | 45,495 | | | $ | (11,223,621 | ) | | | 34,480,163 | | | $ | (45,499 | ) | | $ | (76,924 | ) | | $ | 23,179,614 | | | $ | (1,716,324 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF STOCKHOLDERS’ EQUITY AND COMPREHENSIVE LOSS
Amounts in USD
| | | Preferred
Shares | | Common
Shares | | Stocks
Amount | | Accumulated Deficit During Developmental Stage | | Additional
Paid In
Capital | | Other Comprehensive Income | | Unearned Compensation | | Total |
| Balance at December 31, 2012 | | | 3,000,000 | | | | 42,495,244 | | | $ | 45,495 | | | $ | (11,223,621 | ) | | | 34,480,163 | | | $ | (45,499 | ) | | $ | (76,924 | ) | | $ | 23,179,614 | | | $ | (1,716,324 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Cancellation of shares for services | | | | | | | (250,000 | ) | | | (250 | ) | | | | | | | (34,775 | ) | | | | | | | 35,025 | | | | — | | | | | |
| Amortization of Unearned Compensation | | | | | | | | | | | | | | | | | | | | | | | | | | | 41,899 | | | | 41,899 | | | | | |
| Shares issued for services | | | | | | | 100,000 | | | | 100 | | | | | | | | 19,800 | | | | | | | | — | | | | 19,900 | | | | | |
| Shares issued to prior owner | | | | | | | 1,000,000 | | | | 1,000 | | | | | | | | 159,000 | | | | | | | | | | | | 160,000 | | | | | |
| Issuance of shares to settle payable | | | | | | | 655,652 | | | | 656 | | | | | | | | 43,344 | | | | | | | | | | | | 44,000 | | | | | |
| Private placement | | | | | | | 2,500,000 | | | | 2,500 | | | | | | | | 130,000 | | | | | | | | | | | | 132,500 | | | | | |
| Private placement | | | 900,000 | | | | | | | | 900 | | | | | | | | 269,100 | | | | | | | | | | | | 270,000 | | | | | |
| Private Placement | | | 1,000,000 | | | | | | | | 1000 | | | | | | | | 336,653 | | | | | | | | | | | | 337,653 | | | | | |
| Currency Translation Adjustment | | | | | | | | | | | | | | | | | | | | | | | (159,653 | ) | | | | | | | (159,653 | ) | | | (159,653 | ) |
| Net profit for the period | | | | | | | | | | | | | | | (1,794,718 | ) | | | | | | | | | | | | | | | (1,794,718 | ) | | | (1,794,718 | ) |
| Balance at December 31, 2013 | | | 4,900,000 | | | | 46,500,896 | | | $ | 51,401 | | | $ | (13,018,339 | ) | | | 35,403,285 | | | $ | (205,152 | ) | | $ | — | | | $ | 22,231,195 | | | $ | (1,954,371 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
CONSOLIDATED STATEMENT OF CASH FLOWS
Amounts in USD
| | | For the year ended December 31, 2013 | | For the year ended December 31, 2012 | | Cumulative from April 26, 2006 (date of inception) to
December 31,
2013 |
| CASH FLOWS FROM OPERATING ACTIVITIES | | | | | | |
| Net loss | | $(1,794,718) | | $(1,679,052) | | $(12,817,575) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | |
| Amortization of stock-based compensation | | 256,824 | | 197,328 | | 6,308,238 |
| Recapitalization under reverse merger | | - | | - | | (250,763) |
| Issue of shares for services | | - | | - | | 210,000 |
| Issue of shares for research and development | | - | | - | | 34,000 |
| Liability from warrant | | (521,038) | | 608,540 | | 87,502 |
| Reversal of grant accrual | | (114,732) | | (130,574) | | (245,306) |
| Depreciation and amortization | | 145,219 | | 108,099 | | 707,281 |
| | | (2,028,445) | | (895,659) | | (5,966,623) |
| Changes in operating assets and liabilities: | | | | | | |
| (Increase) Decrease in other current assets | | 114,599 | | (31,016) | | (217,419) |
(Decrease) Increase in accounts payable and accrued liabilities | | 1,842,136 | | 934,447 | | 3,676,631 |
| | | | | | | |
| Net cash provided by (used in) operating activities | | (71,710) | | 7,772 | | (2,507,411) |
| | | | | | | |
| CASH FLOWS FROM INVESTING ACTIVITIES | | | | | | |
| Purchase of fixed assets | | (206,216) | | (15,303) | | (702,413) |
| Net cash used in investing activities | | (206,216) | | (15,303) | | (702,413) |
| | | | | | | |
| CASH FLOWS FROM FINANCING ACTIVITIES | | | | | | |
| Proceeds from stockholders | | (4,985) | | 67,183 | | 212,033 |
| Exercise of warrant | | - | | - | | 1,616 |
| Proceeds from sale of preferred stock | | 269,000 | | | | 269,000 |
| Proceeds from sale of common stock | | 132,500 | | 12,658 | | 2,885,249 |
| Proceeds from issuance of notes | | - | | - | | 70,000 |
| Net cash from financing activities | | 396,515 | | 79,841 | | 3,437,898 |
| | | | | | | |
| Effect of exchange rate changes on cash | | (159,653) | | (37,272) | | (205,152) |
| | | | | | | |
| Net (decrease) increase in cash | | (41,064) | | 35,038 | | 22,922 |
| Cash at beginning of period | | 63,986 | | 28,948 | | - |
| Cash at end of period | | $22,922 | | $63,986 | | $22,922 |
| | | | | | | |
| Supplemental disclosure of cash flow information: | | | | | | |
| Non-cash investing and financing transactions | | | | | | |
| Issuance of shares for services | | $19,900 | | $201,770 | | $4,256,158 |
Issuance of shares for liabilities assumed under reverse merger | | - | | - | | $250,513 |
| Issuance of stock based compensation shares | | - | | - | | $2,590,000 |
| Purchase of fixed assets through the assumption of capital lease obligations | | 406,558 | | 406,558 | | $406,558 |
| | | | | | | |
| Cash paid for: | | | | | | |
| Interest | | - | | - | | 40,980 |
| Taxes | | - | | - | | - |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 1 - GENERAL INFORMATION
Power of the Dream Ventures, Inc., f/k/a “Tia V, Inc.” (“PDV” or the “Company”) was incorporated in Delaware on August 17, 2006, with the objective to acquire, or merge with, an operating business.
Reverse merger
PDV entered into and consummated a Securities Exchange Agreement (“Exchange Agreement”) on April 10, 2007. Under the terms of the Exchange Agreement, PDV acquired all the outstanding equity interests of Vidatech, Kft. (also known as Vidatech Technological Research and Development LLC) a limited liability company formed under the laws of the Republic of Hungary, (“Vidatech”) in exchange for 33,300,000 shares of PDV’s common stock, and Vidatech thereby became a wholly-owned Hungarian subsidiary of PDV. PDV is governed by the law of the State of Delaware, and its wholly-owned subsidiary, Vidatech, is governed by the law of the Republic of Hungary. PDV and Vidatech are herein collectively referred to as the “Company.”
Following the acquisition the former stockholders of Vidatech owned a majority of the issued and outstanding common stock of PDV and the management of Vidatech controlled the Board of Directors of PDV and its wholly-owned Hungarian subsidiary Vidatech. Therefore the acquisition has been accounted for as a reverse merger (the “Reverse Merger”) with Vidatech as the accounting acquirer of PDV. The accompanying consolidated financial statements of the Company reflect the historical results of Vidatech, and the consolidated results of operations of PDV subsequent to the acquisition date. In connection with the Exchange Agreement, PDV adopted the fiscal year end of Vidatech as December 31.
All reference to shares and per share amounts in the accompanying consolidated financial statements have been restated to reflect the aforementioned shares exchange.
Genetic Immunity
On October 2, 2012 the Company acquired Genetic Immunity, Inc., a US corporation domiciled in the state of Delaware. Genetic Immunity is an immunotherapeutic vaccine company whose lead product candidate DermaVir has passed Phase II clinical trials.
Through the acquisitions Genetic Immunity became 100% wholly owned subsidiary. The acquisition was completed for 1,000,000 shares of Series B Preferred stock, convertible into 40 million shares of common stock beginning on January 1, 2014.
The Company filed an 8-K with the SEC disclosing this transaction on October 4, 2012.
The acquisition of Genetic Immunity, Inc. has been accounted for as a business combination whereby the purchase price was allocated to intangible assets based on their fair values of the acquisition date. A summary of the purchase price consideration and related purchase price allocation are shown below:
Purchase price (at par value) $1.000
Fair value adjustment of shares 25,072,621
Total purchase price 25,073,621
The fair value of the shares equals the fair value of Genetic Immunity, Inc. at the acquisition date. The fair value of the company was estimated by an independent valuation company and the valuation was carried out on a discounted cash flow basis. The Company issued 1,000,000 shares of Series B Preferred stock which is not traded publicly and no market price could be estimated. As a result of this, the fair value of the shares was evaluated by a valuation company. The value of the shares is $25.07362 per share.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 1 - GENERAL INFORMATION (Continued)
Purchase price allocation
| | |
| Cash | $357,276 |
| Inventories | 13,843 |
| Other receivables | 306,731 |
| Intangibles | 30,111,909 |
| Fixed assets | 418,483 |
| Accounts payable and accrued liabilities | (1,893,833) |
| Grants received | (1,677,348) |
| Short term loans to related parties | (154,631) |
| Bridge loan | (2,334,326) |
| Other liabilities | (73,483) |
| Total purchase price | 25,074,621 |
| | |
| | |
Unaudited pro forma operation results for the year ended December 31, 2012 as though the Company acquired Genetic Immunity on January 1, 2012 are set forth below:
| | | |
| Net Sales | | $ - |
| Cost of Sales | | - |
| | | |
| Gross margin | | - |
| Material expenses | | 68,406 |
| General administration | | 1,482,366 |
| Research and development | | - |
| Personnel expenses | | 724,264 |
| Depreciation and amortization | | 191,427 |
| Grants received | | (518,631) |
| Other expenses, net | | 347,448 |
| Operating expenses | | 2,295,280 |
| Loss from operations | | (2,295,280) |
| Interest income and exchange gains | | (279,982) |
Loss before income taxes | | (2,575,262) |
| Income taxes | | (1,985) |
Net loss | | (2,577,247) |
On February 23, 2009 the Company entered into a consultation agreement with Genetic Immunity, a Hungarian biotechnology research and development company working on immune amplification nanomedicine products, including a HIV vaccine currently in Phase 2 clinical trials.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
According to the terms of the agreement PDV will provide Genetic Immunity business development expertise, will facilitate the publication, via internationally distributed press releases, of Genetic Immunity’s past, present and future news items, and will advise Genetic Immunity on going public in the United States based on Power of the Dream Ventures’ own experience in achieving public states.
In addition, PDV was granted rights to acquire equity in Genetic Immunity in one or several tranches for total consideration of USD $10 million in exchange for 20% equity in the Company, if all options are exercised.
Exact terms of this equity purchase option were: PDV is to acquire 2% of the Company via a USD 1,000,000 investment by April 30, 2009 in exchange for 72 units of the Genetic Immunity’s Class B stock; Company is to acquire an additional 2% of the Genetic Immunity via a USD 1,000,000 investment by August 30, 2009 in exchange for 72 units of the Genetic Immunity’s Class B stock. PDV is also granted an option to acquire an additional 16% of the Genetic Immunity via an $8,000,000 investment by February 20, 2010, in tranches or in whole, in exchange for 578 units of the Genetic Immunity’s Class B stock. If the Company misses the first deadline of April 30, 2009 this agreement shall immediately terminate. If the Company completes the first investment but missed the second date of August 30, 2009 this agreement shall terminate, but the Company will retain the Class B units already acquired. Any portion of the optional 16% equity purchase that is not exercised and closed by February 23, 2010 shall terminate. The option was extended for an unlimited period.
On March 7, 2011 we entered into an extension of this agreement for the purpose of assisting Genetic Immunity in becoming a publicly traded corporation in the United States. The new agreement gives the Company rights to invest up to $1,400,000 into Genetic Immunity, to be completed by December 31, 2011 in one or more payment. The investment buys Genetic Immunity LLC Series B units that are convertible into common stock once the company becomes publicly traded in the United States. As of June 30, 2011 we have invested $50,000 of this amount. We anticipate raising additional capital to complete the entire investment. In exchange for the investment we are to receive 14% of Genetic Immunity once $500,000 dollars has been invested, and warrant to purchase Genetic Immunity common stock, including one million shares at $20 dollars, one million shares at $30 dollars and one million shares at $40 dollars once Genetic immunity becomes a public company.
On September 28, 2011 we signed a termination letter with Genetic Immunity whereby our option to invest in and receive warrant in Genetic Immunity have been terminated. In consideration for the $50,000 we have invested in Genetic Immunity we have received 10 Series B Units of Genetic Immunity. These units were accounted for in the Genetic Immunity acquisition in 2012.
Business
The Company is engaged in the acquisition, development, licensing and commercialization of and the investment in, directly or through business acquisitions, technologies developed in Hungary. In furtherance of its business, the Company provides research and development services to the companies, inventors from whom it acquires technologies or participation interests in technologies. A goal of the Company is to support research and development activities and to sell the products of inventions to the technological market.
From inception through December 31, 2013, the Company primarily focused on the raising of capital. As of December 31, 2013, the Company originally managed seven technologies: RiverPower, the Buresch Inventions (Desalination and H2O gas technology,) the Kalmar inventions (FireSAFE fire-proofing liquid; technology for utilizing communal waste as a concrete additive; technology for repairing potholes with the use of recycled plastics; technology for neutralizing red mud; biodegradable deicing solution and PVC shielded electric cable recycling technology), and an equity interest in ‘iGlue, Inc (Ticker: IGLU)’, a company formed to develop next generation semantic internet based search engine and content organizer applications. As of December 31, 2013 the Company has discontinued efforts to develop the Buresch inventions, the Kalmar inventions, RiverPower and the Toth Telescope. (see Note 4). As of December 31, 2013, the Company has only realized limited revenues from the now discontinued TothTelescope project and has not realized any revenues from the other, now discontinued inventions. As a result, the accompanying consolidated financial statements have been presented on a development stage basis.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
The Kalmar Inventions: FireSAFE, HardCrete concrete from communal waste additive; Recycled plastic pothole filler; PVC covered electrical cable and radial tire recycling technology; red mud neutralization; and environmentally friendly salt free de-icing solution.
As of December 31, 2011 the Company elected to discontinue development work on all of these technologies. The rational behind this decision rests with availability of resources, the time that would be needed to develop said inventions, the potential return the Company could realize on a partial or full exit from these investments and international completion.
TothTelescope
On July 15, 2009 the Company’s exclusive distributor agreement expired with Attila Toth, Inventor of the TothTelescope. After extensive review of the technology, the available market and hurdles associated with manufacturing and distributing the TothTelescope, we have elected not to renew the distribution agreement and discontinue the project. Since inception only 10 telescopes were sold. Overall invested capital was returned from commissions earned on these sales.
Yorkville SEDA
On October 8, 2008, Power of the Dream Ventures, Inc. (the “Company”) entered into a Standby Equity Distribution Agreement (the “Standby Equity Distribution Agreement”) with YA Global Investments, L.P. (the “Investor”). Pursuant to the terms of the Standby Equity Distribution Agreement, the Company (a) agreed to issue and sell to the Investor up to $5,000,000 of shares of the Company’s common stock, par value $0.0001 per share (the “Common Stock”) in tranches of equity, based upon a specified discount to the market price of the Common Stock, calculated over the five trading days following notice by the Company of an election to sell shares; and (b) issued to the Investor a warrant (the “Warrant”) to purchase 4,027,386 shares of Common Stock at the exercise price per share of $0.29. The Warrant is not part of the commitment shares issued by the company to the investor. The investor must purchase the shares underlying the Warrant. The Warrant price was determined based upon the highest Bid price on the day of the closing of the agreement.
In connection with the Standby Equity Distribution Agreement, the Company entered into a Registration Rights Agreement with the Investor (the “Registration Rights Agreement”) pursuant to which the Company agreed to register for resale the shares of Common Stock that may be purchased by the Investor pursuant to the Standby Equity Distribution Agreement, the shares of Common Stock issuable upon exercise of the Warrant and 2,000,000 shares of Common Stock (the “Commitment Shares”) issued to the Investor as a commitment fee pursuant to the terms of the Standby Equity Distribution Agreement. The commitment fee is recorded by decreasing additional paid in capital.
Financing from this transaction will be used by the Company for the continued development of its current technologies, commercialization of same, the acquisition of new technologies and for general corporate expenses.
The Company filed the details of this transaction on Form 8-K with the Commission on October 14, 2008.
On September 24, 2010 the Company notified YA Global Investments in writing of its intention to terminate the Standby Equity Distribution Agreement (SEDA).
In response to this letter YA Global informed the company in a letter dated October 5, 2010 that the SEDA agreement will terminate on or about October 15, 2010. YA Global also informed the company that even though the SEDA agreement is terminated its associated Warrant to purchase 4,027,386 shares of Our common stock remains in effect until it expires on October 8, 2013.
On October 18, 2010 the company received notice from YA global for the exercise of 50,000 warrant shares on a cashless basis. This cashless exercise resulted in the issuance of 3,226 shares of Common stock to YA Global. Following this exercise YA Global has 3,977,386 warrant shares available for purchase.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2012 AND 2011
iGlue Software via Equity position in iGlue, Inc.
From 2007 to 2011 we funded the development of iGlue, the cornerstone of the current technology portfolio. The financial commitment to in4, Ltd, the company behind iGlue, amounted to a total of approximately $600,000 for years 2007 through 2010. The total chronological events of iGlue are:
- On August 2, 2007 Vidatech Kft., the Company’s wholly owned Hungarian subsidiary, entered into a joint development agreement with the inventors/creators of iGlue, an online content organization and search application based on semantic web technology. In the transaction Vidatech received 30% equity in the newly formed in4 Kft (http://www.in4.hu), the owner and exclusive developer of iGlue (http://www.iglue.com), for HUF 900 thousand (approximately USD 5,000). Up until May 31, 2009, Vidatech was expected to provide up to HUF 48 million (approximately USD 271,000) in member loans to in4 Kft. to cover the development expense of iGlue.
- As of December 31, 2008 the Company has provided this amount to in4, Ltd.
- As part of the agreement, Vidatech had the option of converting its HUF 48 million member loan into an additional 10% equity in in4 Kft. on or before the end of May, 2009. As of January 8, 2009 the Company has elected to covert our member loan into the additional 10% equity in in4, Ltd., resulting in Vidatech owning 40% of equity.
- On May 17th, 2010, the Company transferred its equity ownership from its Hungarian subsidiary Vidatech, Kft. to Power of the Dream Ventures. The transfer was initiated to prepare for its eventual exit from iGlue.
- Also on May 17th, 2010, the Company arranged for a new investor to provide further funds for the continued development of iGlue, in the amount of HUF 110 million (approximately USD 550,000.) Prior to this event, up to May 17th, 2010 the Company has provided an additional HUF 64million ($310,000) as member loan to in4 Ltd. to finance development work, bringing our total investment to date to HUF 112,000,000 (approximately $600,000). As part of the new round of fund raising the Company agreed to convert its HUF 60 million in member loan into 3% equity. Following this conversion the new investor received 10% in in4, Ltd., of which the Company provided 1.5% and Peter Vasko, Founder of in4 limited provided 8.5%. Following the fund raising the new equity positions in in4 changed to Peter Vasko owning 51.5% (down from the pre-transaction 60%), Power of the Dream Ventures owning 38.5% (down from the pre-transaction 40%) and the new investor owning 10% equity.
- On May 19 of 2011 a new member joined in4, Ltd, Zoltan Siklosi who received 3% equity in the company for services rendered. Following his entry the new equity structure of the company changed to Peter Vasko owning 48.5% percent, Gyorgy Markos owning 10%, Zoltan Siklosi owning 3% and Power of the Dream Ventures owning 38.5%.
- Subsequently, to take iGlue to the next level, the Company organized a going public roadmap for the company. As part of this process, on November 3, 2011, in4, Ltd signed a Definitive Share Exchange Agreement to merge with Hardwired Interactive (HDWR) a fully reporting public company. As part of this agreement all holders of in4, Ltd equity received common and preferred stock in the newly public entity. As part of the transaction we received 600,000 shares of Series B Hardwired Interactive common stock convertible into 6,000,000 shares of common stock upon a reverse stock split. The Company also received warrants to purchase a total of three million shares of post split common stock, one million shares each at $5 dollars, $7 dollars and $9 dollars per share.
- On November 23, 2011 the Company announced plans to provide a dividend, in the form of iGlue common stock, to PWRV shareholders of record on December 23, 2011. Each PWRV share was to receive 0.05 share of iGlue common stock in the dividend. Upon approval from FINRA this dividend was paid out to PWRV shareholders on February 15, 2012. Following this distribution the Company still holds 2,884,986 shares of iGlue common stock and warrants to purchase a total of three million addition shares.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
Basis of presentation
The accompanying consolidated financial statements have been prepared by the Company pursuant to the rules and regulations of the SEC. Certain information and footnote disclosures normally included in financial statements prepared in accordance with accounting principles generally accepted in the United States of America for financial information have been condensed or omitted pursuant to such rules and regulations. In the opinion of management, the accompanying financial statements include all adjustments (consisting of normal recurring accruals) considered necessary to make the financial statements not misleading as of and for the period ended December 31, 2013 and for the period from April 26, 2006 (date of inception) to December 31, 2013.
Going Concern and Management’s Plan
The accompanying consolidated financial statements have been prepared in conformity with accounting principles generally accepted in the United States of America, which contemplate the continuation of the Company as a going concern and assume realization of assets and the satisfaction of liabilities in the normal course of business. The Company has incurred losses from operations since inception. Management anticipates incurring additional losses in 2014. Further, the Company may incur additional losses thereafter, depending on its ability to generate revenues from the licensing or sale of its technologies and products, or to enter into any or a sufficient number of joint ventures. The Company has minimal revenue to date. There is no assurance that the Company can successfully commercialize any of its technologies and products and realize any revenues therefore. The Company’s technologies and products have never been utilized on a large-scale commercial basis and there is no assurance that any of its technologies or products will receive market acceptance. There is no assurance that the Company can continue to identify and acquire new technologies.
Since inception through December 31, 2013, the Company had an accumulated deficit of $13,018,339 and net cash used in operations of $2,507,411. However, management of the Company believes that future funding from the private placement of the Company’s common shares will allow them to continue operations and execute its business plan.
During 2013 we raised a total of $401,500 through private placements of common and preferred stock. These funds were used to finance operations, to reduce Genetic Immunity debt, and to finance Genetic Immunity’s regulatory process towards a conditional marketing approval. Additional funds used for these tasks, in the amount of $337,657 were previously provided in the prior years by the director, Peter Boros, who elected in November of 2013 to convert said loan into 1,000,000 shares of our Series D Preferred Stock.
On March 16, 2011 the Company entered into a Convertible Promissory Note with Infinite Funding LLC whereby Infinite Funding provided $76,000 dollars in loans to the Company. The note bears interest at 10%, is due December 5, 2011, and is convertible into shares of common stock at a conversion price of $0.04. On December 4, 2011 we received an extension to this note to December 31, 2012. The note was converted to 1,900,000 common shares at September 30, 2012.
On July 19, 2011 the Company entered into a Convertible Promissory Note with Infinite Funding LLC whereby Infinite Funding provided $49,000 dollars in loans to the Company. The note bears interest at 12%.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
Management believes the Company has adequate capital to keep the Company functioning through December 31, 2014. As discussed in contingencies, the Company anticipates receiving funds from the Hungarian state pursuant to the lawsuit it has filed to recover the awards granted to the Company plus damages. Also, additional fund raisings are in progress. However, the Company cannot guarantee that any amount from the lawsuit will be recoverable nor that any fund raising activities will be successful. The need may arise, in the normal course of business, to raise additional capital if we want to accelerate development work, for the acquisition of additional technologies, or to meet unforeseen financial needs. No assurance can be given that the Company can obtain additional working capital, or if obtained, that such funding will not cause substantial dilution to shareholders of the Company. If the Company is unable to raise additional funds, if needed, it may be forced to change or delay its contemplated marketing and business plan. Being a development stage company, the Company is subject to all the risks inherent in the establishment of a new enterprise and the marketing and manufacturing of a new product, many of which risks are beyond the control of the Company. All of the factors discussed above raise substantial doubt about the Company’s ability to continue as a going concern.
These consolidated financial statements do not include any adjustments relating to the recoverability of recorded asset amounts that might be necessary as a result of the above uncertainty.
NOTE 2 – SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES
The significant accounting policies adopted in preparation of the consolidated financial statements are set out below.
Principles of Consolidation
The consolidated financial statements include the accounts of PDV and its wholly-owned Hungarian subsidiary, Vidatech. All significant intercompany accounts and transactions have been eliminated in consolidation.
Use of Estimates:
The preparation of the financial statements in conformity with GAAP requires management to make estimates, judgments and assumptions that affect amounts reported herein. Management believes that such estimates, judgments and assumptions are reasonable and appropriate. However, due to the inherent uncertainty involved, actual results may differ from those based upon management’s judgments, estimates and assumptions. Critical accounting policies requiring the use of estimates are depreciation and amortization and share-based payments
Revenue Recognition:
Sales are recognized when there is evidence of a sales agreement, the delivery of the goods or services has occurred, the sales price is fixed or determinable and collectability is reasonably assured, generally upon shipment of product to customers and transfer of title under standard commercial terms. Sales are measured based on the net amount billed to a customer. Generally there are no formal customer acceptance requirements or further obligations. Customers do not have a general right of return on products shipped therefore no provisions are made for return.
Accounts Receivable and Allowance for Doubtful Accounts:
Accounts receivable are stated at historical value, which approximates fair value. The Company does not require collateral for accounts receivable. Accounts receivable are reduced by an allowance for amounts that may be uncollectible in the future. This estimated allowance is determined by considering factors such as length of time accounts are past due, historical experience of write offs, and customers’ financial condition.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
Inventories:
Inventories are stated at the lower of cost, determined based on weighted average cost or market. Inventories are reduced by an allowance for excess and obsolete inventories based on management’s review of on-hand inventories compared to historical and estimated future sales and usage.
Fixed assets:
Fixed assets are stated at cost or fair value for impaired assets. Depreciation and amortization is computed principally by the straight-line method. Asset amortization charges are recorded for long lived assets. In the related periods, no asset impairment charges were accounted for. Depreciation is recorded commencing the date the assets are placed in service and is calculated using the straight line basis over their estimated useful lives.
The estimated useful lives of the various classes of long-lived assets are approximately 3-7 years.
Pensions and Other Post-retirement Employee benefits:
In Hungary, pensions are guaranteed and paid by the government or by pension funds, therefore no pensions and other post-retirement employee benefit costs or liabilities are to be calculated and accounted by the Company.
Product warranty:
The Company accrues for warranty obligations for products sold based on management estimates, with support from sales, quality and legal functions, of the amount that eventually will be required to settle such obligations. At December 31, 2013, the Company had no warranty obligations in connection with products sold.
Advertising costs:
Advertising and sales promotion expenses are expensed as incurred.
Research and development and Investment and Advances to Non-Consolidated Entities:
In accordance with ASC 730-10-25 “Accounting for Research and Development Costs,” all research and development (“R&D”) costs are expensed when they are incurred, unless they are reimbursed under specific contracts. Assets used in R&D activity, such as machinery, equipment, facilities and patents that have alternative future use either in R&D activities or otherwise are capitalized. In connection with investments and advances in development-stage technology entities in which the company owns or controls less than a 50% voting interest, (see Note 4) where repayment from such entity is based on the results of the research and development having future economic benefit, the investment and advances are accounted for as costs incurred by the Company as research and development in accordance with ASC 730-20-25 “Research and Development Arrangements”.
Income taxes:
The Company accounts for income taxes in accordance with ASC 740-10-25, “Accounting for Income Taxes”. Deferred tax assets and liabilities are recognized for the future tax consequences attributable to differences between financial statement carrying amounts of existing assets and liabilities and their respective tax bases and operating loss and tax credit carry forwards. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. Valuation allowances are provided against deferred tax assets to the extent that it is more likely than not that the deferred tax assets will not be realized.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
Comprehensive Income (Loss):
ASC 220-10-25, “Accounting for Comprehensive Income,” establishes standards for reporting and disclosure of comprehensive income and its components (including revenues, expenses, gains and losses) in a full set of general-purpose financial statements. The items of other comprehensive income that are typically required to be disclosed are foreign currency items, minimum pension liability adjustments, and unrealized gains and losses on certain investments in debt and equity securities. Accumulated other comprehensive loss, at December 31, 2013 is $205,152.
Translation of Foreign Currencies:
The U.S. dollar is the functional currency for all of the Company’s businesses, except its operations in Hungary. Foreign currency denominated assets and liabilities for this unit is translated into U.S. dollars based on exchange rates prevailing at the end of each period presented, and revenues and expenses are translated at average exchange rates during the period presented. The effects of foreign exchange gains and losses arising from these translations of assets and liabilities are included as a component of equity, under other comprehensive income.
Loss per Share:
Under ASC 260-10-45, “Earnings Per Share”, basic loss per common share is computed by dividing the loss applicable to common stockholders by the weighted average number of common shares assumed to be outstanding during the period of computation. Diluted loss per common share is computed using the weighted average number of common shares and, if dilutive, potential common shares outstanding during the period. Accordingly, the weighted average number of common shares outstanding for the years ended December 31, 2013 and 2012, respectively, is the same for purposes of computing both basic and diluted net income per share for such years. For the years ended December 31, 2013 and 2012, all potential shares of common stock were excluded from diluted EPS as their effect would be anti-dilutive.
The Company currently has the following convertible securities issued and outstanding:
| - | 1,000,000 Shares of our Series B Preferred Stock used for the Genetic Immunity acquisition, convertible into 40 shares of common stock per each Series B preferred, or a total of 40,000,000 common shares on a fully converted basis, beginning on January 1, 2014. |
| - | 900,000 Shares of our Series C Preferred Stock convertible into 20 shares of common stock per each Series C preferred, or a total of 18,000,000 common stock on a fully converted basis. |
| - | 1,000,000 Shares of our Series D Preferred Stock convertible into 2 shares of common stock per each Series D preferred, or a total of 2,000,000 common stock on a fully converted basis. |
| - | And we have one Warrant outstanding, convertible into 3,977,386 Shares of our common stock on a fully converted basis. |
Business Segment:
ASC 280-10-45, “Disclosures About Segments of an Enterprise and Related Information,” establishes standards for the way public enterprises report information about operating segments in annual consolidated financial statements and requires reporting of selected information about operating segments in interim financial statements regarding products and services, geographical areas and major customers. The Company has determined that under ASC 280-10-45, there are no operating segments since substantially all business operations, assets and liabilities are in Hungarian geographic segment.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
Share-Based Payments:
In accordance with ASC 718-10 “Share-Based Payment” all forms of share-based payment (“SBP”) awards including shares issued under employee stock purchase plans, stock options, restricted stock and stock appreciation rights result in a cost that is measured at fair value on the awards’ grant date, based on the estimated number of awards that are expected to vest. As the Company did not issue any employee SBP prior to September 30, 2007, there is no compensation cost recognized in the accompanied consolidated financial statements.
The Company accounts for equity instruments issued to non-employees in accordance with the provisions of ASC 505-50, “Accounting for Equity Instruments That Are Issued to Other Than Employees for Acquiring, or in Conjunction with Selling, Goods or Services,” which requires that such equity instruments are recorded at their fair value on the measurement date. The measurement of stock-based compensation is subject to periodic adjustment as the underlying equity instrument vests. Non-employee stock-based compensation charges are amortized over the vesting period or period of performance of the services.
Recent Accounting Pronouncements:
In September 2011, the FASB issued Accounting Standards Update 2011-08 Intangibles—Goodwill and Other (Topic 350), Testing Goodwill for Impairment. The objective of this update is to simplify how entities, both public and nonpublic, test goodwill for impairment. The amendments in the update permit an entity to first assess qualitative factors to determine whether it is more likely than not that the fair value of a reporting unit is less than its carrying amount as a basis for determining whether it is necessary to perform the two-step goodwill impairment test described in Topic 350. The more-likely-than-not threshold is defined as having a likelihood of more than 50 percent. The amendments in this update are effective for fiscal years, and interim periods within those fiscal years, beginning on or after December 15, 2011. The adoption of this standard did not have a material impact on its financial position, results of operations or cash flows.
In June 2011, the FASB issued Accounting Standards Update 2011-05 - Comprehensive Income (Topic 220): Presentation of Comprehensive Income. Under the amendments to Topic 220, Comprehensive Income, an entity has the option to present the total of comprehensive income, the components of net income, and the components of other comprehensive income either in a single continuous statement of comprehensive income or in two separate but consecutive statements. The amendments do not change the option for an entity to present components of other comprehensive income either net of related tax effects or before related tax effects, with one amount shown for the aggregate income tax expense or benefit related to the total of other comprehensive income items. In both cases, the tax effect for each component must be disclosed in the notes to the financial statements or presented in the statement in which other comprehensive income is presented. The amendments in this update are effective for fiscal years, and interim periods within those fiscal years, beginning on or after December 15, 2011. The adoption of this standard did not have a material impact on its financial position, results of operations or cash flows.
In May 2011, the FASB issued Accounting Standards Update 2011-04—Fair Value Measurement (Topic 820): Amendments to Achieve Common Fair Value Measurement and Disclosure Requirements in U.S. GAAP and IFRSs. The amendments in this update result in common fair value measurement and disclosure requirements in U.S. GAAP and IFRSs. Consequently, the amendments change the wording used to describe many of the requirements in U.S. GAAP for measuring fair value and for disclosing information about fair value measurements. The FASB does not intend for the amendments in this update to result in a change in the application of the requirements in Topic 820. Some of the amendments clarify the application of existing fair value measurement requirements. Other amendments change a particular principle or requirement for measuring fair value or for disclosing information about fair value measurements. The amendments in this update are effective for fiscal years, and interim periods within those fiscal years, beginning on or after December 15, 2011. The adoption of this standard did not have a material impact on its financial position, results of operations or cash flows.
There were various other updates recently issued, most of which represented technical corrections to the accounting literature or application to specific industries and are not expected to a have a material impact on the Company's consolidated financial position, results of operations or cash flows.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 3 - OTHER RECEIVABLES
| | | December 31,
2013 | | December 31,
2012 |
| | | | | |
| Taxes reclaimable | | $ | 10,940 | | | $ | 37,418 | |
| Advances given | | | 178,075 | | | | 264,389 | |
| Other | | | 28,404 | | | | 30,211 | |
| Total | | $ | 217,419 | | | $ | 332,018 | |
Given advances contain advances to lawyers on the legal case of Genetic Immunity on its legal case in progress. The Company accounted for impairment on advances given in the amount of $92,721 during 2013.
NOTE 4 - RESEARCH AND DEVELOPMENT (“R&D”)
In August, 2008, the Company entered into an agreement with a Hungarian individual to establish FireLESS Kft (FireLESS). FireLESS’s business is focused on acquiring the appropriate licenses and certificates to internationally market FireSAFE and will work with local and international fire agencies to test and establish usability baselines for FireSAFE. The Company is a minority shareholder in FireLESS with 30% voting rights, which operates under independent management.
In August, 2007, the Company entered into an agreement with two Hungarian individuals to establish In4 Kft (“in4). in4’s business is focused on software development and information technology purposes. Originally the Company was a minority shareholder in in4 with 30% voting rights. In4 Ltd. operates under independent management.
In August, 2007, the Company also entered into a loan commitment agreement with in4. According to the agreement the Company has committed a loan of approximately $271,000 to in4. The loan amount has been provided by May 31, 2009. In November of 2008 the Company transferred to in4 the entire loan amount, upon which the Company elected to covert the loan into an additional 10% equity in in4. As of January 15, 2009 the Company maintains a minority 40% equity in in4 Ltd. Since capitalization of the loan to equity, as of December 31, 2009 the Company has provided an additional $310,000 loan to in4. During 2010 a new investor entered in4 Ltd, and the share of the Company decreased to 38.5%.
Since the repayment of loans, advances and other investment is contingent on the results of the R&D of iGlue having future economic benefit, management has expensed the Company's investment in in4 and in FireLESS of approximately $5,000 and $900, respectively, and loans to in4 of approximately $310,000 as R&D in the accompanying condensed consolidated statements of operations, in accordance with ASC 730-10-25 "Research and Development Arrangements".
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 5 - INTANGIBLES AND FIXED ASSETS
Intangibles and Net property and equipment consisted of the followings at December 31, 2013 and 2012:
| | | | Intangibles | | | | Machinery and other equipments | | | | Vehicles | | | | Total Net property and equipment | |
| Gross book value | | | | | | | | | | | | | | | | |
| at December 31, 2011 | | | — | | | | 166,284 | | | | 377,139 | | | | 543,423 | |
| Genetic Immunity acquisition | | | 30,111,909 | | | | 418,483 | | | | — | | | | 418,483 | |
| Additions | | | 274,986 | | | | — | | | | — | | | | — | |
| Disposals | | | — | | | | — | | | | (90,369 | ) | | | (90,369 | ) |
| Change in FX rates | | | (271,780 | ) | | | 14,998 | | | | 33,063 | | | | 48,061 | |
| | | | | | | | | | | | | | | | | |
| at December 31, 2012 | | | 30,115,115 | | | | 599,765 | | | | 319,833 | | | | 919,598 | |
| Corrections | | | 480,491 | | | | 376,769 | | | | | | | | 376,769 | |
| Additions | | | 11,329 | | | | 1,471 | | | | — | | | | 1,471 | |
| Disposals | | | — | | | | (324,837 | ) | | | — | | | | (324,837 | ) |
| Change in FX rates | | | 5,251 | | | | 24,266 | | | | 7,948 | | | | 32,214 | |
| | | | | | | | | | | | | | | | | |
| at December 31, 2013 | | | 30,612,186 | | | | 677,434 | | | | 327,781 | | | | 1,005,215 | |
| | | | | | | | | | | | | | | | | |
| Accumulated depreciation and amortization | | | | | | | | | | | | | | | | |
| at December 31, 2011 | | | — | | | | 127,002 | | | | 220,650 | | | | 347,652 | |
| Additions | | | 22,268 | | | | 36,881 | | | | 48,950 | | | | 85,831 | |
| Disposals | | | — | | | | — | | | | (58,608 | ) | | | (58,608 | ) |
| Change in FX rates | | | 6 | | | | 8,839 | | | | 32,715 | | | | 41,554 | |
| | | | | | | | | | | | | | | | | |
| at December 31, 2012 | | | 22,274 | | | | 172,722 | | | | 243,707 | | | | 416,429 | |
| Corrections | | | 480,491 | | | | 376,770 | | | | | | | | 376,770 | |
| Additions | | | 50,914 | | | | 79,744 | | | | 18,489 | | | | 98,233 | |
| Disposals | | | — | | | | (95,121 | ) | | | — | | | | (95,121 | ) |
| Change in FX rates | | | 3,126 | | | | 13,654 | | | | 6,056 | | | | 19,710 | |
| | | | | | | | | | | | | | | | | |
| at December 31, 2013 | | | 556,805 | | | | 547,769 | | | | 268,252 | | | | 816,021 | |
| | | | | | | | | | | | | | | | | |
| Net book values at | | | | | | | | | | | | | | | | |
| December 31, 2011 | | | — | | | | 39,282 | | | | 156,489 | | | | 195,771 | |
| December 31, 2012 | | | 30,092,841 | | | | 427,043 | | | | 76,126 | | | | 503,169 | |
| December 31, 2013 | | | 30,055,381 | | | | 129,665 | | | | 59,529 | | | | 189,194 | |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 5 - INTANGIBLES AND FIXED ASSETS
The net book value of fixed assets under capital lease amount to $59,529 and $76,126 at December 31, 2013 and at December 31, 2012, respectively.
The Company owns the following patents as intangible assets:
- Re: PCT US97/02933 publication No. WO9731119A1, corresp to USSN 08/803,484 'Methods and Compositions for Therapeutic and Genetic Immunization' filed 20 February 1997 by J. Lisziewicz and F. Lori, Atty. Docket No. RGT 100A PCT
- Re: PCT/US98/19498 publication No. WO9913915A1 'Method of Delivering Genes into Antigen Presenting Cells of the skin' corresp. to USSN 09/153,198 filed 15 September 1998 by Lisziewicz & Lori, Atty Docket No. RGT 9771 PCT
- Re: PCT US 04/000746 publication No. WO 2004/065575, corresp. to USSN 10/757,343 “Plasmid DNA and Uses therefor” filed 14 January 2004, based on prov’l appln 60/440,245 filed 15 January, 2003, by Lisziewicz, et al., Atty Docket No. RGT 7033
- Above the patents, the following trademark is owned by:
- Trademark “Genetic Immunity” Serial No. 75153267, filed January 22, 1999, 1st use at least Jan 13, 1999, Reg. No. 2657301 on December 3, 2002
During the acquisition of Genetic Immunity, the Company has received the valuation of the patents from an independent valuation company. According to the valuation, the Company entered the acquired intangibles to the consolidated balance sheet. The acquired intangibles have indefinite useful life and annual impairment test is carried out on a discounted cash flow basis. According to the latest valuation carried out at the end of December 2013, no impairment losses are to be recognized.
NOTE 6 - NOTE PAYABLE
Mary Passalaqua
On October 20, 2011 the Company was presented with an Amended Convertible Promissory Note by Mary Passalaqua. In the revised agreement Ms. Passalaqua requested that the outstanding portion of the Promissory Note be paid in shares in such quantity that will satisfy the debt of $125,000 owned. As second step in this conversion process Mary Passalaqua requested the conversion of $20,000 dollars into shares of common stock. To satisfy this request the Company instructed its transfer agent to issue Mary Passalaqua 3,500,000 shares of common. Upon conversion of the $20,000 our outstanding Promissory Note balance will be $105,000 plus accrued interest.
On August 5, 2011 the company was presented with an Amended Convertible Promissory Note by Mary Passalaqua. In the revised agreement Ms. Passalaqua requested that the outstanding portion of the Promissory Note be paid in shares in such quantity that will satisfy the debt of $195,000 owned. As first step in this conversion process Mary Passalaqua requested the conversion of $70,000 dollars into shares of common stock. To satisfy this request the Company instructed its transfer agent to issue Mary Passalaqua 2,400,000 shares of common. Upon conversion of the $70,000 outstanding Promissory Note balance will be $125,000 plus accrued interest.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 6 - NOTE PAYABLE (Continued)
On October 20, 2011 the Company was presented with an Amended Convertible Promissory Note by Mary Passalaqua. In the revised agreement Ms. Passalaqua requested that the outstanding portion of the Promissory Note be paid in shares in such quantity that will satisfy the debt of $125,000 owned. As second step in this conversion process Mary Passalaqua requested the conversion of $20,000 dollars into shares of common stock. To satisfy this request the Company instructed its transfer agent to issue Mary Passalaqua 3,500,000 shares of common. Upon conversion of the $20,000 our outstanding Promissory Note balance will be $105,000 plus accrued interest.
On April 10, 2007, in connection with reverse merger (See Note 1), the Company assumed a note payable of $250,000 to a former stockholder, Mary Passalaqua originally with one year maturity at April 5, 2008. The note has been expanded by one year to April 5, 2009 with the same conditions. As such note payable was issued immediately prior to the reverse merger, such issuance was recorded as additional compensation by the Company prior to the reverse merger. Accordingly, such compensation is reflected in the accompanying consolidated balance sheet as the accumulated deficit of the Company, and will not be reflected in the Statement of operations, as such compensation expense was structured as an expense prior to the recapitalization.
In November, 2008 the Company settled $35,000 from the outstanding liability, and in May of 2009 the Company settled another $20,000 from the liability. The note payable bears interest at the prime rate (3.25% at December 31, 2013). Interest expense in connection with such note amounted to $52,417 and $49,004 for the year ended at December 31, 2013 and at December 31, 2012, respectively, and was accrued and included in accounts payable and accrued liabilities in the accompanying consolidated balance sheet.
Infinite Funding
On July 19, 2011 the Company entered into a Promissory Note with Infinite Funding, Inc whereby Infinite Funding provided $49,000 dollars in loans to the Company. The Note bears interest at 12% per annum.
On March 16, 2011 the Company entered into a Convertible Promissory Note with Infinite Funding LLC whereby Infinite Funding provided $76,000 dollars in loans to the Company. The note bears interest at 10%, is due December 5, 2011, and is convertible into shares of common stock at a conversion price of $0.05. On December 4, 2011 the Company received an extension to this note to December 31, 2012. On September 30, 2012, the loan was converted into 1,900,000 common stocks at a conversion price of $0.04 per share.
NOTE 7 - CAPITAL LEASES PAYABLE
In August, 2007, the Company entered into capital lease agreements on 3 vehicles for management purposes. The maturity of the lease is 60 months and is denominated in CHF. Installments and interest is due on a monthly basis. In December, 2007, the Company entered into additional capital lease agreements on 2 vehicles for management purposes. The maturity of the lease is 72 months and is denominated in CHF. Installments and interest is due on a monthly basis.
In December 2008, the Company settled the capital lease agreements on 2 vehicles and replaced them with new agreements for additional 2 vehicles, totaling 5 capital lease agreements at December 31, 2008. The maturity of the new leases varies from 60 to 72 months and are denominated in EUR and CHF. Installments and interests are due on a monthly basis.
In September 2009, the Company closed a capital lease agreement for one vehicle. The related asset has been sold.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 7 - CAPITAL LEASE PAYABLE (Continued)
The following is a schedule by years of future minimum lease payments under capital leases together with the present value of the net minimum lease payments as of December 31, 2013:
| For the year ending December 31, 2013 | | Amounts |
| | | |
| 2014 | | 71,897 |
| 2015 | | 23,058 |
| | | |
| Total minimum lease payments | | 94,955 |
| | | |
| Less: amounts representing interest | | 3,479 |
| | | |
| Present value of net minimum lease payments | | 91,476 |
| | | |
| Less: current portion | | 55,784 |
| | | |
| Long term liability | | $35,692 |
NOTE 8 - STOCKHOLDERS’ EQUITY
On October 2, 2012 the Company acquired Genetic Immunity, Inc., a US corporation domiciled in the state of Delaware. Genetic Immunity is an immunotherapeutic vaccine company whose lead product candidate DermaVir has passed Phase II clinical trials.
Through the acquisitions Genetic Immunity became 100% wholly owned subsidiary. The acquisition was completed for 1,000,000 shares of Series B Preferred stock, convertible into 40 million shares of common stock beginning on January 1, 2014.
On October 26, 2011 the Company entered into an equity exchange agreement with each of Messrs. Viktor Rozsnyay and Daniel Kun, Jr. Mr. Rozsnyay is President, CEO and Chairman of the Board of Directors of the Company and Mr. Kun is its Vice-President. Each Agreement is identical and provides in summary form as follows: each of Messrs. Rozsnyay and Kun will deliver twelve million (12,000,000) shares of their shares of common stock of the Company to the Company for cancellation immediately. The Agreement provides that the Exchange Shares so delivered will be returned to the treasury of the Company as unauthorized shares of common stock and will become available for subsequent issuance by the Company from time to time. In return for the surrender of the Exchange Shares, the Company will issue and deliver two million (2,000,000) restricted shares of a new class of Series A Preferred shares, 1,000,000 shares each to Mr. Rozsnyay and Mr. Kun. The Company attached as an Exhibit the Series A preferred agreements.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
Private placements
On November 1, 2013, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 1,000,000 shares of its Series D preferred stock to one affiliated private investor, Peter Boros, Director of the company, for aggregate proceeds of $337,653. This amount was previously provided to the company by Mr. Boros as a loan, and on November 1 Mr. Boros elected to convert this loan into aforementioned preferred stock. The Series D preferred stock is convertible into 2,000,000 shares of our common stock on a fully converted basis. No special redemption rights.
On October 17, 2013, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 250,000 shares of its Series C Preferred Stock to one unaffiliated private investors for aggregate proceeds of $75,000.
On September 26, 2013, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 650,000 shares of its Series C Preferred Stock to two unaffiliated private investors for aggregate proceeds of $195,000.
On September 25, 2013, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 1,000,000 shares of its common stock to one unaffiliated private investors for aggregate proceeds of $80,000.
Series C Preferred Stocks can be converted with a completed form of conversion notice into 20 shares of common stock. The holders of Series C Preferred shall be entitled to receive dividends on a fully converted basis. Each one share of Series C Preferred shall be entitled to the dividend granted to each one share of common stock multiplied by twenty. Dividends on Series C Preferred must be delivered and paid to the holders not later than five business days after each specified payment date of the dividend.
On May 9, 2013, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 100,000 shares of its common stock at $0.035 per share to an unaffiliated private investor for aggregate proceeds of $3,500.
On April 3, 2013, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 1,400,000 shares of its common stock at $0.035 per share to an unaffiliated private investor for aggregate proceeds of $49,000.
On June 13, 2012, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 42,200 shares of its common stock at $0.3 per share to an unaffiliated private investor for aggregate proceeds of $12,658.
During the first half of 2012, the Company received stock advance from an unaffiliated private investor for aggregate proceeds of $67,153.
On November 14, 2011, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 27,700 shares of its common stock at $0.16 per share to an unaffiliated private investor for aggregate proceeds of $4,432.
On September 2, 2011, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 1,727,013 shares of its common stock at $0.16 per share to two unaffiliated private investors for aggregate proceeds of $276,322.
On August 29, 2011, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 17,825 shares of its common stock at $0.12 per share to one unaffiliated private investor for aggregate proceeds of $2,137.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
During the second quarter of 2011, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 196,489 shares of its common stock at $0.29 for aggregate proceeds of $56,983.
During the third quarter of 2010, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 70,000 shares of its common stock at $0.50 for aggregate proceeds of $35,000.
During the first half of 2010, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 1,470,000 shares of its common stock at $0.10 for aggregate proceeds of $147,000, and sold 62,500 shares of its common stock at $0.08 for proceeds of $5,000.
In February 2010, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 65,000 shares of its common stock at $0.12 for aggregate proceeds of $7,701.
In November 2009, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 175,000 shares of its common stock at $0.16 and 111,110 shares of its common stock at $0.20 per share for aggregate proceeds of $50,222.
In October 2008, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 2,500,000 shares of its common stock at $0.4 per share for aggregate proceeds of $1,000,000.
In February of 2008, shares of common stock of the Company have been approved by FINRA for quotation and trading on the Over The Counter Bulletin Board (OTCBB) under the ticker symbol PWRV. Trading commenced in the Company’s securities on the OTCBB beginning on February 21, 2008.
In January 2008, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 32,500 shares of its common stock at $3.25 per share for aggregate proceeds of $105,625.
In October 2007, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 104,000 shares of its common stock at $2.50 per share for aggregate proceeds of $260,000. The Company also entered into a Registration Rights Agreement, pursuant to which it agreed that as soon as practicable from the Offering Termination Date, as defined in the Registration Rights Agreement, it would file a registration statement with the SEC covering the resale of the shares of the Company’s common stock that are issuable pursuant to this private placement. There are no stipulated damages outlined in the Registration Rights Agreement for failure to file within the agreed upon time frame. Under such agreement, the holder is entitled to exercise all rights granted by law, including recovery of damages under this agreement.
The Company filed Form SB-2 a registration statement with SEC on November 14, 2007, which was approved on January 30, 2008.
Consulting agreements
During November 2012, the Company entered into agreements with four unaffiliated professionals for one year business consulting services. According to the agreement the professionals provide consulting and rental services to the Company for 12 months. In connection with these services, the Company issued to them 1,930,000 shares of the Company’s common stock. These share issuances were recorded at the fair value of contracting date which is the market price on the commitment date $0.06 and $0.19 per share in the total amount of $180,170 and the related expense was recorded under general administration.
On April 11, 2012 the Company entered into a Consultation Services agreement with Stockvest for IR services and issued a total of 135,000 shares. As of May 17, 2012 this agreement has been terminated.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
During October 2011, the Company entered into agreements with three unaffiliated professionals for one year business consulting services. According to the agreement the professionals provide consulting services to the Company for 12 months. In connection with these services, the Company issued to them 640,000 shares of the Company’s common stock. These share issuances were recorded at the fair value of contracting date which is the market price on the commitment date $0.11 and $0.09 per share in the total amount of $61,200 and the related expense was recorded under general administration.
On September 27, 2011, the Company entered into agreements with two unaffiliated professionals for one year business consulting services. According to the agreement the professionals provide consulting services to the Company for 12 months. In connection with these services, the Company issued to them 100,000 shares of the Company’s common stock. These share issuances were recorded at the fair value of contracting date which is the market price on the commitment date $0.14 per share in the total amount of $14,000 and the related expense was recorded under general administration.
During April and May of 2011, the Company entered into agreements with five unaffiliated professionals for one year business consulting services. According to the agreement the professionals provide consulting services to the Company for 12 months. In connection with these services, the Company issued to them 505,000 shares of the Company’s common stock.
These share issuances were recorded at the fair value of contracting date which is the market price on the commitment date, ranging from $0.34 to $0.48 per share in the total amount of $190,900 and the related expense was recorded under general administration.
On February 7, 2011 the Company entered into a one year business consultation agreement with an unaffiliated person who is to provide general business development and incubation consultation. In exchange for his services the Company issued a onetime payment of 215,000 shares of our common stock, which vested immediately. These share issuances were recorded at $0.3 per share in the total amount of $64,500 and the related expense was recorded under general administration.
On February 9, 2011 the Company entered into a one year business consultation agreement with an unaffiliated person who is to provide services in term of potential business opportunities in Poland. In exchange for his services we issued a onetime payment of 435,000 shares of our common stock, which vested immediately. These share issuances were recorded at $0.24 per share in the total amount of $104,400 and the related expense was recorded under general administration.
On February 24, 2011 the Company entered into a one year business consultation agreement with an unaffiliated person who is to provide general business development and incubation consultation. In exchange for his services we issued a onetime payment of 75,000 shares of our common stock, which vested immediately. These share issuances were recorded at $0.3 per share in the total amount of $22,500 and the related expense was recorded under general administration.
The consulting agreements are entered for a 12 months period. According to the agreements the consultants will provide general business consulting services. As consideration for such services, the Company issued an aggregate of 1,970,000 shares of the Company’s common stock. These share issuances were recorded at the fair value of commitment date in the total amount of $457,888 in accordance with measurement date principles prescribed under ASC 505-50 and ASC 718-10. The Company is amortizing the fair value of the shares over the term of the agreement to stock-based compensation expense, which amounted to $316,098 for the period ended December 31, 2013, respectively and $316,098 for the period from April 26, 2006 (date of inception) to December 31, 2013, in accordance with ASC 505-50 and ASC 718-10.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
During the October and November 2010, the Company entered into agreements with several unaffiliated professionals for consulting services. According to the agreement the professionals provide consulting services to the Company in 2010. In connection with these services, the Company issued to them 874,000 shares of the Company’s common stock. These share issuances were recorded at $0.5 per share in the total amount of $437,000 and the related expense was recorded under general administration.
In September 2010, the Company entered into an agreement with four unaffiliated professionals for consulting services. According to the agreement the professionals provided consulting services to the Company in 2010. In connection with these services, the Company issued to them 153,000 shares of the Company’s common stock. These share issuances were recorded at $0.5 per share in the total amount of $76,500 and the related expense was recorded under general administration.
The consulting agreements are entered for a 12 months period. According to the agreements the consultants will provide general business consulting services. As consideration for such services, the Company issued an aggregate of 1,027,000 shares of the Company’s common stock. These share issuances were recorded at the fair value of commitment date ($0.5 per share) in the total amount of $513,500 in accordance with measurement date principles prescribed under ASC 505-50 and ASC 718-10. The Company is amortizing the fair value of the shares over the term of the agreement to stock-based compensation expense, which amounted to $312,972 and $200,528 for the periods ended December 31, 2013 and 2012, respectively and $513,500 for the period from April 26, 2006 (date of inception) to December 31, 2013, in accordance with ASC 505-50 and ASC 718-10.
In June 2010, the Company entered into an agreement with two unaffiliated professionals for consulting services. According to the agreement the professionals provided consulting services to the Company in 2010. In connection with these services, the Company issued to them 20,000 shares of the Company’s common stock. These share issuances were recorded at $0.1 per share in the total amount of $2,000 and the related expense was recorded under general administration.
In April 2009, the Company entered into an agreement with two unaffiliated professionals for consulting services. According to the agreement the professionals provided consulting services to the Company in 2009. In connection with these services, the Company issued to them 250,000 shares of the Company’s common stock. These share issuances were recorded at $0.4 per share in the total amount of $100,000 and the related expense was recorded under general administration.
In June 2007, pursuant to a private placement under Regulation S of the Securities Act of 1933, as amended, the Company sold 2,250,000 shares of its common stocks at $0.34 per share for a total subscription receivable of $765,000. The Company also entered into a Registration Rights Agreement, pursuant to which it agreed that as soon as practicable from the Offering Termination Date, as defined in the Registration Rights Agreement, it would file a registration statement with the SEC covering the resale of the shares of the Company’s common stock that are issuable pursuant to this private placement. There are no stipulated damages outlined in the Registration Rights Agreement for failure to file within the agreed upon time frame. Under such agreement, the holder is entitled to exercise all rights granted by law, including recovery of damages under this agreement. In June 2007, the Company entered into five consulting agreements with five consultants for 12 to 24 month periods. According to the agreements the consultants will provide general business consulting services. As consideration for such services, the Company issued an aggregate of 1,375,000 shares of the Company’s common stock. These share issuances were recorded at $0.34 per share in the total amount of $467,501 in accordance with measurement date principles prescribed under ASC 505-50 and ASC 718-10. The Company is amortizing the fair value of the shares over the term of the agreement to stock-based compensation expense, which amounted to $0 for the period ended December 31, 2013 and 2012, respectively and $467,501 for the period from April 26, 2006 (date of inception) to December 31, 2013, in accordance with ASC 505-50 and ASC 718-10.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
In April 2007, the Company entered into an agreement with two professionals for legal services. According to the agreement the professionals provided legal services to the Company in 2007. In connection with these services, the Company issued to them 500,000 shares of the Company’s common stock. These share issuances were recorded at $0.34 per share in the total amount of $170,000 and the related expense was recorded under general administration.
In connection with the ITA in 2007 (See Note 4), the Company issued 100,000 shares of the Company’s common stock to the Inventors. These shares issuance were recorded at fair value of $0.34 per share in the total amount of $34,000. The cost of the related invention was recorded as research and development expense.
Other agreements
On October 8, 2008, Power of the Dream Ventures, Inc. (the “Company”) entered into a Standby Equity Distribution Agreement (the “Standby Equity Distribution Agreement”) with YA Global Investments, L.P. (the “Investor”). Pursuant to the terms of the Standby Equity Distribution Agreement, the Company (a) agreed to issue and sell to the Investor up to $5,000,000 of shares of the Company’s common stock, par value $0.0001 per share (the “Common Stock”) in tranches of equity, based upon a 7% discount to the market price of the Common Stock, calculated over the five trading days following notice by the Company of an election to sell shares; and (b) issued to the Investor a warrant (the “Warrant”) to purchase 4,027,386 shares of Common Stock at the exercise price per share of $0.29. The Warrant is not part of the commitment shares issued by the company to the investor.
The investor must purchase the shares underlying the Warrant. The Warrant price was determined based upon the highest Bid price on the day of the closing of the agreement. The YA Global is required to exercise the warrant upon notice by PDV of an election to have the warrants exercised at a fix price of $0.29 per common stock.
In connection with the Standby Equity Distribution Agreement, the Company entered into a Registration Rights Agreement with the Investor (the “Registration Rights Agreement”) pursuant to which the Company agreed to register for resale the shares of Common Stock that may be purchased by the Investor pursuant to the Standby Equity Distribution Agreement, the shares of Common Stock issuable upon exercise of the Warrant and 2,000,000 shares of Common Stock (the “Commitment Shares”) issued to the Investor as a commitment fee pursuant to the terms of the Standby Equity Distribution Agreement. The 2,000,000 shares issued as a commitment fee were valued at $0.4 per share or $800,000 based on the fair value at issuance date. The $800,000 commitment fee has been debited against additional paid in capital in accordance with the provisions of Staff Accounting Bulletin Topic 5A .
On September 24, 2010 the Company notified YA Global Investments in writing of its intention to terminate the Standby Equity Distribution Agreement (SEDA).
In response to this letter YA Global informed the company in a letter dated October 5, 2010 that the SEDA agreement will terminate on or about October 15, 2010. YA Global also informed the company that even though the SEDA agreement is terminated its associated Warrant to purchase 4,027,386 shares of common stock remains in effect until it expires on October 8, 2013.
On October 18, 2010 the company received notice from YA global for the exercise of 50,000 warrant shares on a cashless basis. This cashless exercise resulted in the issuance of 3,226 shares of Common stock to YA Global. Following this exercise YA Global has 3,977,386 warrant shares available for purchase.
On September 10, 2012 YA Global notifed the company that according to the terms of the Warrant Agreement there should have been a proportional reduction in the Warrant’s exercise price to the the price of a private placement that was completed at a lower price than the $0.29 exercise price. YA Global and the company have agreed to modify the Warrant exercise price from the original $0.29 cents to $0.10 cents.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
At the same time YA Global also notified the company of their intention to sell the Warrant in a private transaction to an unaffiliated accredited investor. This transaction was contemplated on September 10, 2012. The new owner, Bluestar Consulting, now has the right to acquire, on or before October 8, 2013, 3,977,386 share of common stock at a per share exercise price of $0.10. The terms of the warrants are such that the exercise price was lowered due to a subsequent round of financing, from $0.29 per share to the price the subsequent investors paid, $0.10 per share. This down round protection feature causes the warrant to fail the criteria in ASC 815-40-15-7 (EITF 07-5) which would permit them to be classified as equity. Consequently, the warrants are recorded at fair value and classified as liabilities and marked to fair value each period. The warrant is extended to October 8, 2018.
The fair value of the liability using Black-Scholes valuation at December 31, 2013 is $87,502, which is classified at short term liabilities. The fair value of the liability is established as a Level 3 fair value measurement. The following table shows the movement in fair value along with the change of warrant conditions:
| December 31, 2013 | Book value | Fair value |
| | | |
| Financial liabilities | | |
| Liabilities measured at fair value 87,502 | 87,502 |
| December 31, 2012 | Book value | Fair value |
| | | |
| Financial liabilities | | |
| Liabilities measured at fair value 608,540 | 608,540 |
| Fair value of warrant per share | December 31, 2013 | December 31, 2012 |
| | | |
| Per share | $0.022 | $0.153 |
The inputs used in fair value estimation are:
current share price at December 31, 2013: $0.65
exercise price $0.1
expected time to expiry (years): 4.75
risk free rate 3.25 (FED prime rate)
expected dividend yield: 0
volatility: 50%
The change in the amount of warrant liability is accounted as other expense.
On May 17, 2008 the Company entered into an agreement with Wakabayashi Fund LLC in order to arrange financing for working capital as an intermediary. Wakabayashi Fund LLC provided capital funding services including serving as an investment banking liaison and acted as capital consultant for a six month period. The Company issued 111,111 shares of restricted common stock upfront at $1.35 per share, the market price of the stock on the commitment date of the agreement. Additionally, the Company agreed to pay for the capital funding services 7% success fee. These share issuances were recorded at $1.35 per share in the total amount of $150,000 in accordance with measurement date principles prescribed under ASC 505-50. The Company is amortizing the fair value of the shares in general and administration expenses over the term of the agreement to stock-based compensation expense, which amounted to $0 for the period ended December 31, 2013 and $150,000 for the period from April 26, 2006 (date of inception) to December 31, 2013, in accordance with ASC 505-50. As of November 17, 2008 this agreement has been terminated without any funds raised.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
On April 18, 2008 the Company entered an agreement with RedChip Companies Inc. and Partner Media4Equity Inc. for an investor relationship program for a period of 12 months. The Company secured and delivered 306,570 restricted common shares with a market price of $0.70 for a 12 months period in connection with RedChip investor relationship services. The compensation for Media4Equity services was the delivery of 1,500,000 restricted common shares. These share issuances were recorded at $0.75 per share, the market price of the stock on the commitment date of the agreement, for a total amount of $1,125,000 in accordance with measurement date principles prescribed under ASC 505-50. The Company is amortizing the fair value of the shares in general and administration expenses over the term of the agreement to stock-based compensation expense, which amounted to $0 for the periods ended December 31, 2013 and December 31, 2012, respectively and $1,339,599 for the period from April 26, 2006 (date of inception) to December 31, 2013, in accordance with ASC 505-50.
Stock based compensations
On January 1, 2011, the Company entered into a restricted stock agreement with Ildiko Rozsa, who is to serve as the Chief Financial Officer of the Company. As part of the agreement Ms. Rozsa was granted 100,000 shares of the Company’s restricted common stock of which 25,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Rozsa is employed by the Company.
On January 1, 2011, the Company entered into a restricted stock agreement with Szilvia Toth, the Chief Accounting Officer of the Company. As part of the agreement Ms. Toth was granted 100,000 shares of restricted common stock, of which 25,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Toth is employed by the Company.
On January 1, 2011, the Company entered into a restricted stock agreement with Imre Eotvos, the Technology Assistant of the Company. As part of the agreement Mr. Eotvos was granted 120,000 shares of restricted common stock, which will vest on equal installments of 30,000 shares quarterly, at the end of each quarter, so long as Mr. Eotvos is employed by the Company.
On January 1, 2011, the Company entered into a restricted stock agreement with Daniel Kun, Jr., who is to serve as Secretary and Vice President of the company on a going forward basis. As part of the agreement Mr. Kun was granted 120,000 shares of restricted common stock, which will vest on equal installments of 30,000 shares quarterly, at the end of each quarter, so long as Mr. Kun is employed by the Company.
On January 1, 2011, the Company entered into a restricted stock agreement with Gene Guhne, who is to serve as Director of the company on a going forward basis. As part of the agreement Mr. Guhne was granted 100,000 shares of restricted common stock, which will vest on equal installments of 25,000 shares quarterly, at the end of each quarter, so long as Mr. Guhne is employed by the Company.
As consideration for the above services, the Company issued an aggregate of 540,000 shares of the Company’s common stock. These share issuances were recorded at $0.15 per share in the total amount of $81,000 in accordance with measurement date principles prescribed under FAS 123 (R).
During the second quarter of 2010, the Company entered into a restricted stock agreement with Ildiko Rozsa, who is to serve as the Chief Financial Officer of the Company. As part of the agreement Ms. Rozsa was granted 100,000 shares of the Company’s restricted common stock of which 25,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Rozsa is employed by the Company.
During the second quarter of 2010, the Company entered into a restricted stock agreement with Szilvia Toth, the Chief Accounting Officer of the Company. As part of the agreement Ms. Toth was granted 100,000 shares of restricted common stock, of which 25,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Toth is employed by the Company.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
During the second quarter of 2010, the Company entered into a restricted stock agreement with Imre Eotvos, the Technology Assistant of the Company. As part of the agreement Mr. Eotvos was granted 120,000 shares of restricted common stock, which will vest on equal installments of 30,000 shares quarterly, at the end of each quarter, so long as Mr. Eotvos is employed by the Company.
During the second quarter of 2010, the Company entered into a restricted stock agreement with Daniel Kun, Jr., who is to serve as Secretary and Vice President of the company on a going forward basis. As part of the agreement Mr. Kun was granted 120,000 shares of restricted common stock, which will vest on equal installments of 30,000 shares quarterly, at the end of each quarter, so long as Mr. Kun is employed by the Company.
During the second quarter of 2010, the Company entered into a restricted stock agreement with Gene Guhne, who is to serve as Director of the company on a going forward basis. As part of the agreement Mr. Guhne was granted 100,000 shares of restricted common stock, which will vest on equal installments of 25,000 shares quarterly, at the end of each quarter, so long as Mr. Guhne is employed by the Company.
As consideration for the above services, the Company issued an aggregate of 540,000 shares of the Company’s common stock. These share issuances were recorded at $0.1 per share in the total amount of $54,000 in accordance with measurement date principles prescribed under FAS 123 (R).
On February 5, 2009, the Company entered into a restricted stock agreement with Ildiko Rozsa, who is to serve as the Chief Financial Officer of the Company. As part of the agreement Ms. Rozsa was granted 120,000 shares of the Company’s restricted common stock of which 30,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Rozsa is employed by the Company.
On February 5, 2009, the Company entered into a restricted stock agreement with Szilvia Toth, the Chief Accounting Officer of the Company. As part of the agreement Ms. Toth was granted 120,000 shares of restricted common stock, of which 30,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Toth is employed by the Company.
On February 5, 2009, the Company entered into a restricted stock agreement with Mihaly Zala, the Chief Technology Officer of the Company. As part of the agreement Mr. Zala was granted 120,000 shares of restricted common stock, which will vest on equal installments of 30,000 shares quarterly, at the end of each quarter, so long as Mr. Zala is employed by the Company. As of July 15, 2009 Mr. Zala is no longer employed by the Company, therefore 60,000 shares of the 120,000 granted to him were cancelled and returned to the authorized and unissued stock of the company.
On February 5, 2009, the Company entered into a restricted stock agreement with Imre Eotvos, the Technology Assistant of the Company. As part of the agreement Mr. Eotvos was granted 200,000 shares of restricted common stock, which will vest on equal installments of 50,000 shares quarterly, at the end of each quarter, so long as Mr. Eotvos is employed by the Company.
On February 5, 2009, the Company entered into a restricted stock agreement with Daniel Kun, Jr., who is to serve as Secretary and Vice President of the company on a going forward basis. As part of the agreement Mr. Kun was granted 200,000 shares of restricted common stock, which will vest on equal installments of 50,000 shares quarterly, at the end of each quarter, so long as Mr. Kun is employed by the Company.
As consideration for the above services, the Company issued an aggregate of 700,000 shares of the Company’s common stock. These share issuances were recorded at $0.4 per share in the total amount of $280,000 in accordance with measurement date principles prescribed under FAS 123 (R).
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
On October 24, 2007, the Company entered into a restricted stock agreement with Ildiko Rozsa, who is to serve as the Chief Financial Officer of the Company. As part of the agreement Ms. Rozsa was granted 250,000 shares of the Company’s restricted common stock of which 100,000 shares are vested upon grant and 30,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Rozsa is employed by the Company.
On October 1, 2007, the Company entered into a restricted stock agreement with Szilvia Toth, the Chief Accounting Officer of the Company. As part of the agreement Ms. Toth was granted 100,000 shares of restricted common stock of which 50,000 shares are vested upon grant and 10,000 shares will vest quarterly, at the end of each quarter, so long as Ms. Toth is employed by the Company.
On October 24, 2007, the Company entered into a restricted stock agreement with Mihaly Zala, the Chief Technology Officer of the Company. As part of the agreement Mr. Zala was granted 150,000 shares of restricted common stock, which will vest on equal installments of 30,000 shares quarterly, at the end of each quarter, so long as Mr. Zala is employed by the Company.
On October 24, 2007, the Company entered into a restricted stock agreement with Imre Eotvos, the Technology Assistant of the Company. As part of the agreement Mr. Eotvos was granted 25,000 shares of restricted common stock, which will vest on equal installments of 5,000 shares quarterly, at the end of each quarter, so long as Mr. Eotvos is employed by the Company.
On October 1, 2007, the Company entered into a restricted stock agreement with Sandorne Juhasz, who provides payroll accounting services to the Company on a subcontracting basis. As part of the agreement Ms. Juhasz was granted 11,000 shares of restricted common stock, of which 8,000 is will vest upon grant and 750 shares will vest quarterly, at the end of each quarter, so long as Ms. Juhasz is employed by the Company.
On October 24, 2007, the Company entered into a restricted stock agreement with Daniel Kun, Jr., who is to serve as Secretary and Vice President of the company on a going forward basis. As part of the agreement Mr. Kun was granted 250,000 shares of restricted common stock, which will vest on equal installments of 50,000 shares quarterly, at the end of each quarter, so long as Mr. Kun is employed by the Company.
On October 24, 2007, the Company entered into a restricted stock agreement with Viktor Rozsnyay, who is serving as President and Chief Executive Officer of the Company. As part of the agreement Mr. Rozsnyay was granted 250,000 shares of restricted common stock, which will vest on equal installments of 50,000 shares quarterly, at the end of each quarter, so long as Mr. Rozsnyay is employed by the Company.
As consideration for the above services for the employment of the above 7 persons, the Company issued an aggregate of 1,036,000 shares of the Company’s common stock. These share issuances were recorded at $2.5 per share in the total amount of $2,590,000 in accordance with measurement date principles prescribed under ASC 505-50 and ASC 718-10. The Company is amortizing the fair value of the shares over the term of the agreement to stock-based compensation expense, which amounted to $0 for the period ended December 31, 2013 and $2,590,000 for the period from April 26, 2006 (date of inception) to December 31, 2013, in accordance with ASC 505-50 and ASC 718-10.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 8 - STOCKHOLDERS’ EQUITY (Continued)
Reverse merger
On August 5, 2011 the company was presented with an Amended Convertible Promissory Note by Mary Passalaqua. In the revised agreement Ms. Passalaqua requested that the outstanding portion of the Promissory Note be paid in shares in such quantity that will satisfy the debt of $195,000 owned. As first step in this conversion process Mary Passalaqua requested the conversion of $70,000 dollars into shares of common stock. To satisfy this request the Company instructed its transfer agent to issue Mary Passalaqua 2,400,000 shares of common. On October 20, 2011, Mary Passalaqua requested the conversion of additional $20,000 dollars into shares of common stock. To satisfy this request the Company instructed its transfer agent to issue Mary Passalaqua 3,600,000 shares of common.
On October 20, 2011 the Company was presented with an Amended Convertible Promissory Note by Mary Passalaqua. In the revised agreement Ms. Passalaqua requested that the outstanding portion of the Promissory Note be paid in shares in such quantity that will satisfy the debt of $125,000 owned. As second step in this conversion process Mary Passalaqua requested the conversion of $20,000 dollars into shares of common stock. To satisfy this request the Company instructed its transfer agent to issue Mary Passalaqua 3,500,000 shares of common. Upon conversion of the $20,000 our outstanding Promissory Note balance will be $105,000 plus accrued interest.
On April 10, 2007, PDV entered into a reverse merger transaction with Vidatech. In connection with the merger 2,500,000 shares of PDV common stock remained outstanding and PDV issued 33,300,000 shares of its common stock for all the outstanding common stock of Vidatech. As a result of this transaction, the former stockholders of Vidatech became the controlling stockholders of PDV. Accordingly, the reverse merger has been accounted for as a recapitalization of Vidatech.
In May, 2006, the Company entered into a short term loan agreement with its Chief Executive Officer, Viktor Rozsnyay, for approximately $96,100 with a maturity of April 30, 2007. On December 28, 2006 Mr. Rozsnyay elected to convert the loan into equity, which is recorded as additional paid in capital.
In March, 2007, the Company entered into a short term loan agreement with its Chief Financial Officer, Daniel Kun Jr., for approximately $53,735 with a maturity of March 31, 2007. On March 31, 2007 Mr. Kun elected to convert the loan into equity, which is recorded as additional paid in capital.
At December 31, 2013 there are no non-vested restricted stock awards.
The weighted average grant-date fair value of restricted stock awards granted for the year ended December 31, 2013 and 2012 were $0. The total fair value of shares vested during the year ended December 31, 2013 and 2012 was $0.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 9 - ACCOUNTS PAYABLE, ACCRUED AND OTHER LIABILITIES
| | | December 31,
2013 | | December 31,
2012 |
| | | | | |
| Accounts payable and accrued expenses | | $ | 1,868,534 | | | $ | 1,662,172 | |
| Interest accruals | | | 2,214,760 | | | | 1,205,632 | |
| Total | | $ | 4,083,294 | | | $ | 2,867,804 | |
In 2007, Genetic Immunity conducted a US$2.0 million convertible debenture bridge in anticipation of a reverse merger which was not consummated due to then current economic and market conditions. The debenture was due August 2009. The US$1.1 million in face value of debentures was purchased by a fund operating under the Small Business Investment Company (“SBIC”) which has since been put into receivership with the U.S. Small Business Administration (the “SBA”). The SBA is expected to execute a forbearance agreement. The remaining US$900,000 of face value of the debenture is held by individuals who are considering whether to sign the forbearance agreement. Conrad Mir, the Company’s Chairman of the Board of Directors, is in dialogue with them. The debenture holders have liens on certain intellectual property of the Company. Because there is significant know how associated with the intellectual property, the involvement of the Company's management team and scientific team is necessary to commercialize the intellectual property. As of the printing of this document, the Company received written consent from the SBA to proceed with the forbearance agreement and is awaiting similar approval from the aforementioned individuals. If said individuals elect to not accept the terms of the forbearance agreement as approved by the Company and the SBA, thus keeping the Company in default on its obligations, there is the potential for litigation by the holders of the debentures.
In 2013, the Company accounted for the late payment interest for the Bridge loan (18%) which increased the accrued interest liability significantly, along with the interest expenses.
NOTE 10 - GRANTS RECEIVED
During 2007 and 2008, Genetic Immunity gained several government grants from the Hungarian state in order to finance the research and development projects. The grants are payable in arrears to 4 projects of the Company: DERMAVI_, DVCLIN01, FIBERSC2 and WINGS. The Company accrued for the grants received and it is reversed to the income statement along with the occurrence of the related expenses. Grants are payable in accordance with the milestone, not annually. Audited financial statements had to be disclosed to State Office as settlement of each milestone. Theses audits have been performed by the statutory auditor of the Hungarian Subsidiary. WINGS is an EU FP7 international consortium grant financed by the European Union.
DERMAVI_ is successfully closed in 2009. DVCLIN01 supported the Phase II clinical development of DermaVir. This grant was closed at the end of 2013.. FIBERSC2 supports the investigation of DermaVir mechanism of action with novel fiber integrated microscopy. Originally scheduled for completion in February of 2014, this grant has been extended to August of 2`14.. WINGS supports the development of West Nile Virus vaccine with DermaVir technology. The grant ended in February of 2`14, we are no in the process of filing the required closing documents. We expect full closure to this grant in Q3 of 2014.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 11 - SHORT TERM LOANS
| | | December 31,
2013 | | December 31,
2012 |
| | | | | |
| Bridge Loan | | $ | 2,000,000 | | | $ | 2,000,000 | |
| Bridge loan payable provision | | | — | | | | 188,465 | |
| RIGHT loan | | | 311,159 | | | | 217,420 | |
| Bryan Sanders loan | | | 30,000 | | | | 30,000 | |
| Total | | | 2,341,159 | | | | 2,435,885 | |
Bridge Loan
On August 29, 2007, the Company entered into a Bridge Loan agreement as a Senior Secured Convertible Debenture for an amount of $2,000,000. The loan providers are the followings:
| Name | | Bridge Amount |
| | | | | |
| SBIC Investors | | | | |
| Trident | | $ | 1,100,000 | |
| Non-SBIC Investors | | | | |
| Andy Browder | | | 25,000 | |
| Proto Investments | | | 250,000 | |
| Ford Sasser | | | 25,000 | |
| Danny Vela | | | 25,000 | |
| Steve Walton | | | 150,000 | |
| Hilton Wilson | | | 25,000 | |
| Will Wilson | | | 250,000 | |
| Rodman & Renshaw | | | 150,000 | |
| | | $ | 2,000,000 | |
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 11 - LONG TERM LIABILITIES (Continued)
The loan bears an annual interest rate of 12%. According to the loan agreement, the Company settles the loan principal on the earlier of August 29, 2009 or consummation of a qualifying transaction other than reverse merger effected by the Company in completion of or in connection with a Public Offering. In case of late payment, the Company is liable to pay 18% late interest fee. The accumulated interest payable, along with the late payment interest is accrued in accrued expenses (see Note 6).
As of the printing of this document, the US$1.1 million in face value of debentures was purchased by a fund operating under the SBIC, which has since been put into receivership with the U.S. SBA. The SBA is expected to execute a forbearance agreement. The remaining US$900,000.00 of face value of the debenture is held by individuals who are considering whether to sign the forbearance agreement. Conrad Mir, the Company’s Chairman of the Board of Directors, is in dialogue with them. The debenture holders have liens on certain intellectual property of the Company. Because there is significant know how associated with the intellectual property, the involvement of the Company's management team and scientific team is necessary to commercialize the intellectual property. Most recently, the Company received written consent from the SBA to proceed with the forbearance agreement and is awaiting similar approval from the aforementioned individuals. If said individuals elect to not accept the terms of the forbearance agreement as approved by the Company and the SBA, thus keeping the Company in default on its obligations, there is the potential for litigation by the holders of the debentures.
NOTE 12 - GENERAL AND ADMINISTRATION
General and administration expenses consisted of the followings at December 31, 2013 and at December 31, 2012:
| | | December 31,
2013 | | December 31,
2012 |
| | | | | |
| Stock based compensation | | | — | | | | — | |
| Stock based Consultant services | | | 201,899 | | | | 201,770 | |
| Other | | | 537,076 | | | | 595,419 | |
| Total | | | 738,975 | | | | 797,189 | |
General and administration expenses include $92,721 on impairment of advances given.
POWER OF THE DREAMS VENTURES, INC. (formerly TIA V)
(A DEVELOPMENT STAGE COMPANY)
NOTES TO THE CONSOLIDATED FINANCIAL STATEMENTS
FOR THE YEARS ENDED DECEMBER 31, 2013 AND 2012
NOTE 13 - OTHER EXPENSES
Other expenses include the revaluation of liability for warrants for Bluestar Consulting in the amount of $521,038.
NOTE 14 - SUBSEQUENT EVENTS
On April 7, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 50,000 shares of its Series E Preferred Stock to one unaffiliated private investors for aggregate proceeds of $50,000.
On February 19, 2014, pursuant to a private placement under Regulation D of the Securities Act of 1933, as amended, the Company sold a total of 200,000 shares of its Series E Preferred Stock to two unaffiliated private investors for aggregate proceeds of $200,000.
NOTE 15 - CONTINGENCIES
The Company’s wholly-owned subsidiary, Genetic Immunity, has initiated a lawsuit against the National Development Agency of Hungary. In 2009, Genetic Immunity successfully applied for and won an 800 million Hungarian forint (approximately $4,000,000) research grant to conduct dust mite allergy vaccine research. Subsequently, the agency refused to sign the grant and denied payment of funds. The company initiated a lawsuit to request that the court reinstate the contract that resulted in a grant award. On February 13, 2013, an appeals court ruled that the court could not reinstate the contract between Genetic Immunity and the National Development Agency as the contract has been validly in existence since its original execution in 2009. As such Genetic Immunity was ordered to pay 52,500,000 HUF (approximately $250,000) in lawsuit costs. However, simultaneously, the judgment stated that the company is entitled to receive the original grant amount, interest payment and/or damages since the original grant contract is valid. On February 18, 2013, the Company filed a new petition to have the courts establish the damages and awards that Genetic Immunity can receive based on the appeals court’s ruling. The Company is currently seeking 4,800,000,000 HUF (approximately $20 million) for damages sustained. On November 18, 2013, the Company has entered into a six month suspension of said lawsuit, as agreed with the government, in hopes of negotiating an out of court settlement. If these negotiations are unsuccessful the Company will resume the law suite in the court of law on or before May 18, 2014.