Exhibit 99.2
Throughout this Exhibit 99.2, unless the context indicates otherwise,the words “we,” “us,” “our,” the “Company,” and “SELLAS” refer to SELLAS Life Sciences Group, Inc. and its consolidated subsidiaries.The names “SELLAS Life Sciences Group, Inc.,” “SELLAS,” the SELLAS logo, and other trademarks or service marks of SELLAS Life Sciences Group, Inc. appearing in this Exhibit are the property of SELLAS Life Sciences Group, Inc. Other trademarks, service marks or trade names appearing herein are the property of their respective owners.
BUSINESS
Overview
We are a late-stage biopharmaceutical company focused on the development and commercialization of novel cancer immunotherapeutics for a broad range of indications. Our lead product candidate,galinpepimut-S, or GPS, is an immunotherapeutic agent licensed from Memorial Sloan Kettering Cancer Center, or MSK, which targets the Wilms tumor 1, or WT1, protein. WT1 has been shown to be present in 20 or more cancer types and is one of the most commonly expressed cancer antigens. GPS has been engineered to incorporate novel technology to preserveWT-1 antigenicity and overcome the tolerance the immune system commonly develops to tumor antigens, a major challenge in the development of effective immunotherapies for cancer. Based on its mechanism of action as a direct immunizing agent, GPS has potential as a monotherapy or in combination with other immunotherapeutic agents to address a broad spectrum of hematological malignanciesand solid tumor indications.
Phase 2 clinical trials for GPS are completed for two potential indications, acute myeloid leukemia, or AML, and malignant pleural mesothelioma, or MPM. We have planned Phase 3 clinical trials (pending funding availability) evaluating GPS as monotherapy for these two indications with AMLbeing our priority. GPS is also in early clinical development as a potential treatment for multiple myeloma, or MM, and epithelial ovarian cancer. We also plan to study GPS in up to four additional indications: as a combination therapy in small cell lung cancer, colorectal cancer, triple-negative breast cancer, or TNBC; and, as a monotherapy in chronic myelogenous leukemia, or CML. We received Orphan Drug Product designations for GPS from the U.S. Food and Drug Administration, or FDA, for AML, MPM and MM as well as Orphan Medicinal Product designations from the European Medicines Agency, or EMA, for AML and MPM, and Fast Track designation for AML and MPM from the FDA.
Pipeline
Our pipeline also includes the ongoing development programs of our predecessor company, including novel cancer immunotherapy programs for NeuVax(nelipepimut-S; a vaccine against the E75 peptide derived from the human epidermal growth factor 2, or HER2, protein),GALE-301 (a vaccine against the E39 peptide derived from the folate binding protein, or FBP),GALE-302 (a vaccine against the J65 peptide derived from FBP) andGALE-401 (a controlled release version of the approved drug anagrelide). NeuVax is currently in multiple investigator-sponsored Phase 2 clinical trials in breast cancer, including a prospective, randomized, single-blinded, controlled Phase 2b clinical trial of trastuzumab (Herceptin®) +/- NeuVax in HER2 1+/2+ breast cancer patients in the adjuvant setting to prevent recurrences.
On April 2, 2018, we announced that apre-specified interim analysis of safety and efficacy, conducted by an independent Data Safety Monitoring Board, or DSMB, for the investigator sponsored Phase 2b NeuVax + Herceptin study demonstrated a clinically meaningful difference in median disease-free survival, or DFS, in favor of the active arm , a primary endpoint of the study. The interim analysis further demonstrated a statistically significant and clinically meaningful improvement in DFS among a cohort of patients with TNBC associated with the NeuVax + Herceptin combination. Based on these results, and the DSMB’s recommendation, we plan to expeditiously seek regulatory guidance by the FDA for further development of NeuVax + Herceptin combination therapy in TNBC, a population of breast cancer patients with large unmet need. In June 2018, we announced that the sponsor-principal investigator of this study, after taking into account that key clinical development objectives were met as well as other regulatory considerations, and with our agreement, determined to terminate early the study. In addition, in late May 2018, we conducted two advisory meetings with global experts in regulatory affairs and breast cancer clinical development in order to determine the optimal path for further development of the NeuVax + Herceptin combination in TNBC in a pivotal setting and for engagement with the FDA and EMA.
GALE-301 andGALE-302, our E39 folate binding peptide vaccine product candidates, have completed early stage trials in ovarian, endometrial and breast cancers. Both candidates have received Orphan Drug Product designation by the FDA.
1
The following chart summarizes the current status of our clinical development pipeline:

Our Strategy
We seek to use our expertise and understanding of cancer immunotherapy and general cancer therapeutic product development to advance novel products that have the potential to transform the current standard of care. The key components of our strategy are as follows:
| | • | | Continue to rapidly advance our pipeline of product candidates through clinical development, including our potentialfirst-in-class, lead immunotherapy product candidate, GPS, which we are currently developing in both monotherapy and combination therapy settings.We intend to continue to execute a focused clinical development plan that takes our product candidates through approval by regulatory authorities. This includes developing GPS as both a monotherapy or in combination, in addition to exploring opportunities for the other product candidates in our pipeline. The entire GPS clinical program currently targets up to eight tumor types: AML, MPM, MM, ovarian cancer, small cell lung cancer, colorectal cancer, TNBC, and CML). We may pursue additional development of GPS for other indications, both as a monotherapy or in combination with other therapeutic agents. |
GPS monotherapy: GPS has completed Phase 2 clinical trials and has Phase 3 clinical trials planned (pending funding availability) for AML and MPM. There is also an ongoing Phase 2 clinical trial of GPS for MM as monotherapy. We also have plans to pursue additional clinical development programs for GPS as a monotherapy, including in CML and AML treated with hypomethylators.
GPS combination therapy: GPS has an ongoing Phase 1/2 clinical trial for ovarian cancer, in combination with nivolumab (Opdivo) (the clinical trial is independently sponsored by MSK). We plan
2
to test GPS in combination with other therapeutic agents for various solid and hematologic cancers. Our leading combination clinical program will be in collaboration with a Merck & Co., Inc., Kenilworth, N.J., USA subsidiary (known as MSD outside the United States and Canada), or Merck subsidiary. The purpose of the trials is to determine if the administration of GPS in combination with thePD-1 blocker pembrolizumab (Keytruda) has the potential to demonstrate clinical activity in the presence of macroscopic disease, where monotherapy with either agent would have a more limited effect.
| | • | | Address significant unmet need in patients with rare cancers allowing for the utilization of rare disease pathways and expedited approvals with the FDA and comparable foreign agencies.A component of our strategy is to focus on rare cancers where our immunotherapy product candidates may produce clinical benefit and where we can take advantage of regulatory programs intended to expedite drug development in rare indications. We received Orphan Drug Product designation from the FDA as well as Orphan Medicinal Product designation from the EMA for GPS in AML, MPM and MM, as well as Fast Track designation from the FDA for AML, MPM, and NeuVax. We plan to apply for Orphan Drug Product designation, Fast Track designation, Breakthrough Therapy designation and Priority Review from the FDA as well as Orphan Medicinal Product designation, Priority Medicines designations, and Conditional Authorizations from the EMA for any given indication, if applicable when pertinent data becomes available, to potentially reduce clinical trial expense and increase speed to commercialization. |
| | • | | Evaluate the potential for collaboration and license agreements with other biotechnology and pharmaceutical companies for the development of our current and other future product candidates. We seek out collaborations for additional opportunities and development of programs in our pipeline that require larger clinical trials or extensive commercial infrastructure. Specifically, we plan to advance the development of NeuVax through partnership or other strategic collaborations. We are also evaluating licensing and other strategic options forGALE-301,GALE-302 andGALE-401. |
| | • | | Selectively build focused commercial capabilities and establish commercial collaborations to maximize the value of our clinical development pipeline. We have not yet defined our sales, marketing or product distribution strategy for GPS or any future product candidates. Our future commercial strategy may include the use of strategic alliances, distributors, a contract sales force, or the establishment of our own commercial and specialty sales force to maximize the value of our pipeline. |
The Cancer Immunotherapy Industry
Overview
Cancer immunotherapies seek to stimulate a person’s own immune system to selectively attack cancer cells while keeping normal cells unaffected or delivering certain immune system components in order to inhibit the spread of cancer. Cancer immunotherapy drugs now constitute a new mode of cancer treatment, alongside more established options such as surgery, chemotherapy, targeted therapy and radiation therapy. A July 2016 report by Kelly Scientific Publications estimates that immunotherapies may eventually be used in as many as 60% of cases of advanced cancer. Furthermore, based on a recent Allied Market Research report on the estimated market value of oncology drugs in 2020, cancer immunotherapies could represent up to 71% of the total value of $160 billion. Either in mono or in combination therapies, immunotherapies may produce long-term remissions or even operational “cures” for cancers that have, until recently, been uniformly fatal. Thus, cancer immunotherapy is an important and rapidly emerging field, which has led to new clinical research studies and garnered the attention of investors, biotechnology and pharmaceutical companies, regulatory agencies, payors and hospital systems, cancer patients and their families and the general public at large.
3
Market
The global market for cancer drugs (including immunotherapy drugs) is expected to reach $161.3 billion by end of 2021, growing at a compound annual growth rate, or CAGR of approximately 7.4% from 2016 to 2021 (according to a December 2016 report by Zion Market Research). According to a September 2016 report by MarketsandMarkets, the global cancer immunotherapy market is expected to reach $119.4 billion by 2021 from $61.97 billion in 2016 at an estimated CAGR of 14.0%.
We expect that the first category ofFDA-approved immunotherapies, immune synapse modulators (which includes checkpoint inhibitors and immune synapseco-stimulators), is likely to reach and exceed 90% of the immunotherapy market in the coming years, leaving approximately 10% for the other three major categories, which include peptide cancer active immunizers such as our product candidate, GPS.
GPS targets malignancies and tumors characterized by an overexpression of the WT1 protein. The WT1 protein is one of the most widely expressed cancer proteins in multiple malignancies. A 2009 pilot project regarding the prioritization of cancer antigens conducted by the National Cancer Institute, or NCI, a division of the National Institutes of Health, or NIH, ranked the WT1 protein as a top priority for immunotherapy. WT1 is a protein that resides in the cell’s nucleus and participates in the process of cancer formation and progression. As such, it is classified as an “oncogene.” WT1 plays a key role in the development of the kidneys in fetal life, but then almost disappears from normal organs and tissues. In a wide variety of cancers (20 or more cancer types), WT1 becomes detectable again in the cells of these cancers. WT1 appears in large amounts (i.e., becomes “overexpressed”) in numerous hematological malignancies, including AML, MM and chronic myeloid leukemia, as well as in many solid malignancies such as MPM, gastrointestinal cancers (such as colorectal cancer), glioblastoma multiforme, triple-negative breast cancer, ovarian cancer and small-cell lung cancer. Overall, WT1 is expressed in at least 50% of tumor pathology specimens in 20 or more cancer types. The following figure shows the ratio of samples testing positive for WT1 to those testing negative for WT1 in a number of different malignancies.
4
WT1 EXPRESSION FREQUENCY ACROSS VARIOUS CANCERS
(Positive samples / Total samples)
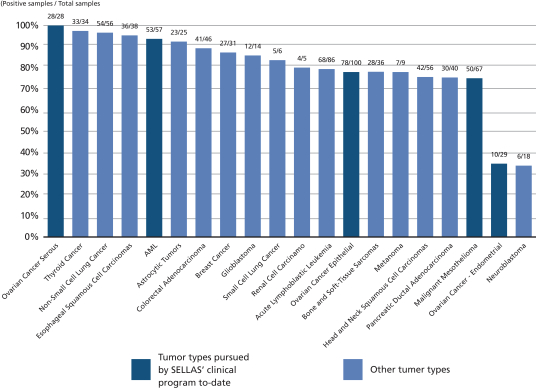
Data sampling overview from multiple studies in human tumor samples or cancer cell lines
The prevention of tumor recurrence is a critical component of overall cancer treatment. The clinical programs for our later stage immunizing agents, GPS and NeuVax, target patients with functional immunity in the adjuvant, post-surgical, complete remission, or minimal residual disease settings. In these patients, the presence of small collections of tumor cells, or micrometastasis, is no longer detectable by current radiographic scanning technology, but the risk of tumor recurrence remains significant.
While GPS and NeuVax are both cancer vaccines, they have some distinguishing features. GPS is tetravalent and Neuvax is monovalent. GPS is a direct immunogen emulsified into the clinically safe adjuvant Montanide, and administered subcutaneously after priming the immune system with recombinant human granulocyte macrophage-colony stimulating factor, orGM-CSF, Sargramostim. NeuVax, on the other hand, uses an immunodominant HER2 peptide combined withGM-CSF as the immune adjuvant, and is administered intradermally. Both GPS and NeuVax, however, work by harnessing the patient’s own immune system to seek out and attack any residual cancer cells. We believe using peptide immunogens has many potential clinical advantages, including a favorable safety profile, because these therapies may lack the toxicities typical of most cancer therapies. Peptide immunogens also have the potential to induce immunologic memory and provide long-lasting protection with convenient modes of delivery.
5
Galinpepimut-S
Overview
GPS is aWT1-targeting peptide-based cancer immunotherapeutic being developed as a monotherapy and in combination with other therapeutic agents to treat different types of cancers that result from uninhibited tumor cell growth.
Cancer immunotherapy harnesses the body’s natural immune system response to fight and/or prevent such tumor growth. An essential feature of the immune system is its ability to recognize foreign, ornon-self, threats, including cancerous growths, as distinct from normal, or self, cells. Despite originating from normal cells, tumor cells can be recognized asnon-self because of their capacity to elicit the production of tumor antigens. These antigens may be released in the interstitial tissues and, eventually, the bloodstream or remain on the surface of cognate cancer cells. Such tumor-associated antigens, or TAAs, have been identified in most human cancers. The WT1 protein is one of the most widely expressed TAAs in multiple malignances.
The immune system is a network of tissues, cells, and signaling molecules that work to protect the body by recognizing and attacking foreign cells, including cancer cells. Several of cell types play an important role in the development and maintenance of immune responses against cancer. The most important cell types with regards to immune response are antigen-presenting cells, or APCs, and lymphocytes. APCs include various subtypes, such as dendritic cells, monocytes and macrophages. Once a patient is exposed to a TAA (either by the presence of cancer itself or through active immunization through a vaccine type immunotherapeutic), this antigen gets recognized by the APC and becomes “processed” through digestion into smaller fragments within the APC. Subsequently, the APC “communicates” with a specific type of lymphocytes calledT-cells. InactiveT-cells search for TAAs by transiently binding to antigens presented by major histocompatibility complexes, or MHCs, on the APCs. Notably, there is great variability in the expression of different subtypes of MHCs in the human population. The MHC system expresses theso-called human leukocyte antigens, or HLAs, and there are dozens of subclasses that determine the vigor and duration of any givenT-cell response to a cancer among different patients. Consequently, active immunizers that work across many HLA types, such as GPS, are predicted to be more efficacious across larger segments of patient populations as compared to agents that act in the context of only one or few HLA types.
T-cells themselves also come in many variants. CD8 cells recognize the processed TAA fragment as foreign and respond. The CD8 cells also develop properties that can directly kill theTAA-expressing cancer cell by becoming “cytotoxic” CD8 cells. The CD8 cells, as well as the APCs, also activate CD4 cells, which are very important for the development of immunologic memory. Immunologic memory is developed when a host keeps a long-term trace of the TAA associated with the cancer and is a desirable result, as it allows the host to continue attacking the TAA associated with the cancer. Therefore, activation of CD4 cells helps avoid or mitigate immune “tolerance.” Immune tolerance is an undesirable result, as it dampens the host’s immune response against the cancer. This cascade of events is collectively called “cellular immunity” and is very important for anti-cancer activity of immunotherapeutic compounds such as GPS. Of note, onceT-cells are activated, another class of lymphocytes, calledB-cells, are also secondarily activated.B-cells are responsible for making antibodies against TAAs. These antibodies become expressed on the surface of theB-cells and are eventually secreted as soluble proteins in tissue fluids and blood. Such anti-cancer antibodies can be detected and have variable degree of activity against the cancer itself. This type of immunity is called “humoral immunity” and complements the actions and effects of the cellular immunity.
Key Features
GPS is a multi-peptide product that we have exclusively licensed from MSK, which has been modified to enhance the degree and duration of the immune response against the WT1 protein. GPS has been modified such that two of the four peptides in the peptide mixture comprising GPS are deliberately mutated in a single amino acid residue. These mutated peptides are recognized by the immune system asnon-self entities and are therefore
6
less likely to induce immune tolerance. After administration of these mutated peptides, the patient becomes immunized against the corresponding native versions of these peptides (which are expressed by the tumor cells), and thus, are able to cross-react against them. The aforementioned concept is referred to as the heteroclitic principle. The enhanced immunity and duration are largely independent of a patient’s HLA type. GPS also elicits both CD4 and CD8 immune responses. As described above, CD8 cells are extremely important, as their activation by GPS would lead to direct cancer cell killing, or cytotoxicity, and eventual establishment of immunologic memory against aWT1-expressing cancer. This occurs by two mechanisms, conversion of some of the activated CD8 cells to CD8 memory cells, and activation of CD4 cells and eventual creation of CD4 terminal effective memory cells.
The following table summarizes the key features of GPS:
| | |
Key features of an Optimal Cancer Active Immunizer Therapeutic | | GPS Properties and Clinical Strategy |
| Selecting the right target antigen and epitopes within that antigen | | Four peptides and 25 epitopes selected optimally to ensure: • optimal MHC complex presentation; • specificity across different HLA types; • production of both CD4 and CD8 activated cells; and • the ability to apply the heteroclitic principle, as described above, to overcome tolerance. |
| |
| OptimalT-cell engagement leading to cancer cell destruction | | Immune response data from the multiple myeloma clinical study of GPS in 12 evaluable patients that was presented at the Society of Hematologic Oncology Fifth Annual Meeting (Dr. Kohne et al.), showed 83.3% frequency of either CD8+ or CD4+ responses to anall-pool mixture ofWT1-derived antigens after completion of the 12 vaccinations per the study protocol. This evidence of multi-epitope, broad cross-reactivity along the full-length of the WT1 protein, is suggestive of epitope spreading, as it emerged across epitopes against which the patients were not specifically immunized. These data strongly suggest stimulation ofT-cells towards intracellular antigen fragments fromGPS-induced destruction of tumor cells, which effect is a hallmark of an effective vaccine, e.g., that it is targeting the right (e.g., chosen by design) epitopes. |
| |
| Overcoming the barriers of an adverse/immunosuppressive tumor micro-environment, or TME | | The GPS monotherapy clinical studies are in the setting of complete remission, or CRem, and minimal residual disease, whereby no bulky or measurable tumor deposits exist. This is typically seen after successful frontline therapy in select cancer types for which such debulking standard therapies exist (e.g., AML or MPM). In these settings, the TME is substantially absent. We are also pursuing combination therapy with checkpoint inhibitors in tumor settings whereby measurable disease exists, as contemporaneous checkpoint inhibition would abrogate the immunosuppressive effects of the TME. |
| |
| Overcoming or mitigating immune tolerance | | Heteroclitic peptides are those in which mutations have been deliberately introduced in the amino acid sequence. The use of heteroclitic peptide in an active immunizer, such as GPS, increases immunogenicity without changes in the antigenicity profile, as well as strengthens MHC binding of the peptide to produce cytotoxic CD8 cells that continue to recognize the corresponding native peptide sequence. This is a key factor differentiating GPS from essentially all |
7
| | |
Key features of an Optimal Cancer Active Immunizer Therapeutic | | GPS Properties and Clinical Strategy |
| | previously developed peptide vaccines, and applies a highly innovative technology platform, peptide heteroclicity, in a clinical late-stage cancer immunotherapeutic candidate product. |
| |
| Addressing the broadest possible patient population | | GPS has activity across multiple HLA types that could allow treatment of a vast majority of global patient populations harboringWT1-positive malignancies. |
Potential Key Differentiators
GPS’ potential key differentiators as compared to other active immunization or vaccine-type approaches, as well as compared to immunotherapy approaches more generally, are as follows:
| | • | | heteroclitic peptides may offer increased immune response and less potential for tolerance; |
| | • | | multivalent oligopeptide mixture potentially drives differentiated immunotherapeutic efficacy, targeting 25 key epitopes of WT1; |
| | • | | is potentially applicable to 20 or more cancer types worldwide and the vast majority of HLA types; |
| | • | | CRem or minimal residual disease status (after initial tumor debulking with preceding standard therapy) is the preferred setting for GPS monotherapy; |
| | • | | does not directly compete with current clinical standard of care therapies, but rather complements them in the maintenance setting; |
| | • | | has potential for combination approaches with other cancer immunotherapies, due to tolerable adverse event profile; |
| | • | | cost-effective manufacturing is anticipated; allogeneic,“off-the-shelf,” vialed subcutaneously administered drug that is not patient-specific; and |
| | • | | positive Phase 2 clinical data on effectiveness (based on overall survival, or OS, in AML and progression-free survival, or PFS, in MM) with good tolerability and an innocuous safety profile. |
Mechanism of Action
While the precise mechanism of action of GPS, and other immunizing agents is not fully understood, it generally involves direct activation of the patient’s immune system, and in the case of GPS, specifically and solely against the WT1 protein. Typically, patients harboringWT1-positive malignancies have very few or noT-lymphocytes specifically reactive or responsive to, and therefore activated by, WT1. WT1 is a “self” antigen, against which the immune system isnon-reactive, or said to be in a state of immune tolerance. Even if some patients have some innateT-cell responses naturally, these responses are weak and not adequate for any anti-cancer effect.
GPS is a WT1 peptide mixture. It cannot be administered to patients in a water-soluble form, and so it is given under the skin, or subcutaneously. If administered on its own, GPS would rapidly degrade and would not have the opportunity and the necessary time interval to activate the immune system. Therefore, GPS is mixed with Montanide, creating a dense emulsion. Additionally, prior to the administration of GPS, patients receive an adjuvant,GM-CSF tonon-specifically stimulate and activate APCs in the vicinity of the subcutaneous injection of GPS.
After subcutaneous injection, the WT1 peptides within GPS disperse locally underneath the injection site and at local lymph nodes, and are ingested by APCs. Digested peptide fragments are then presented on the surface of APCs to CD8 and CD4 lymphocytes while simultaneously associated on the cell membrane with MHC/HLA molecules. This process activates the CD4 and CD8 cells and sensitizes them to the key 25 epitopes of WT1, thus
8
initiating the process of short- and long-termT-cell-mediated immunity against WT1. CD8 cells then circulate around the lymphatic system and blood stream throughout the patient’s body targetingWT1-positive cancer cells. The stimulated CD8 cells transform into cytotoxicT-lymphocytes, or CTLs, which are able to attack and destroy specificallyWT1-positive cancer cells. Each CTL typically destroys oneWT1-positive cancer cell, but they have been shown to be able to kill up to 10 to 20WT1-positive cancer cells. Further, CD4 cells are stimulated to produceWT1-specific, helperT-cells, which are able to in turn activate CTLs andB-cells. TheB-cells “helped” by the helperT-cells produce antibodies to specific WT1 epitopes. The anti-cancer effect is considered to be a result of a combination of all of the above actions, as well as possible additional, less clear mechanisms involving other immune cell types (e.g., natural killer cells). The principles behind the above described mechanism of action of GPS are well established for the class of peptide-based active immunizing therapies of the vaccine type.
The following diagram illustrate GPS’ mechanism of action:
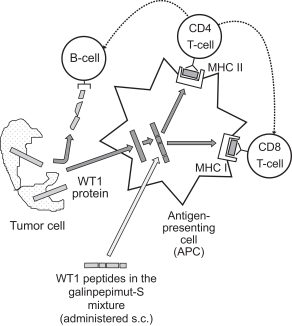
We are currently developing GPS for up to eight indications.
GPS monotherapy. GPS has completed Phase 2 clinical trials and has Phase 3 clinical trials planned (pending funding availability) for AML and MPM and is also in various phases of developmentas a potential treatment for MM and ovarian cancer. There is also an ongoing Phase 2 clinical trial of GPS for MM as monotherapy. We also have plans to pursue additional clinical development programs for GPS as a monotherapy, including in CML and AML treated with hypomethylators.
GPS combination therapy. In October 2017, we announced a clinical trial collaboration and supply agreement through a Merck subsidiary to conduct a combination clinical trial (using GPS along with thePD-1 blocker pembrolizumab (Keytruda)) targeting up to five cancer types, namely colorectal cancer, small cell lung cancer, triple negative breast cancer, ovarian cancer and AML treated with hypomethylators. We are preparing to start this clinical trial pending funding availability. Separately, a clinical trial of GPS in combination with nivolumab (Opdivo) in ovarian cancer is being conducted as an investigator-sponsored trial by MSK. Finally, we also have GPS delivery technology in preclinical development using licensed technology from Advaxis using a bacterial vector, Lm (which if successful, could lead to a second-generation product calledWT1-Lm).
9
Targeted Indications
GPS Monotherapy for Acute Myeloid Leukemia
AML is an aggressive and highly lethal blood cancer characterized by the rapid growth of abnormal white blood cells that build up in the bone marrow and interfere with the production of normal blood cells. Its symptoms include fatigue, shortness of breath, bruising and bleeding, and increased risk of infection. The cause of AML is unknown, and the disease is typically fatal within weeks or months if untreated. AML most commonly affects adults, and its incidence increases with age. Current treatments include chemotherapy, and some patients may receive a hematopoietic, or blood-forming, stem cell transplant, or HSCT. The goal of upfront therapy for AML is to achieve a state of CRem. CRem is defined per consensus criteria by the European Leukemia Net, whereby the hematologic and clinical features of the disease are no longer detected. In principle, an allogeneic HSCT is an immunotherapy used clinically and specifically in AML, which works in four stages:
| | • | | achievement of CRem with standard upfront therapy followed by additional very high-dose chemotherapy that completely destroys any remnant of the patient’s blood forming cells, including any residual AML malignant cells; |
| | • | | selection of a sufficiently genetically similar donor (usually one of the patient’s close relatives), called a histocompatible donor; |
| | • | | removal of blood-forming cells from the bloodstream of that donor; and |
| | • | | infusion of these donor cells into the patient for eventual engraftment onto the patient’s bone marrow and eventual creation of a completelyre-instituted blood-forming system to sustain life and long-term leukemia-free status for the patient. |
Barring the successful completion of an allogeneic HSCT in AML, no therapies have been proven to accord any meaningful long-term benefit after patients achieve a CRem status. Without allogeneic HSCT, once the disease relapses, second-line therapies can be given, but these have very limited positive clinical impact to date and their benefit is transitory; this means that eventually essentially all AML patients who do not undergo an allogeneic HSCT succumb to AML or complications associated with it.
The overall treatment landscape for AML has remained static for decades, as numerous (at the time, novel) targeted and antiproliferative agents failed to yield meaningful long-term clinical benefits, including increments in survival.
The AML indication was chosen forfirst-in-human clinical studies of GPS for the following reasons:
| | • | | AML presents a clinical setting in which CRem status can be achieved with standard upfront therapy; |
| | • | | the almost universal expression of WT1 in leukemic blasts, which are AML’s malignant cells, as well as leukemic stem cells, or LSCs, cells that are or become extremely resistant to standard chemotherapy or targeted agent approaches and which can be realistically eradicated only with immunotherapy methods (including allogeneic HSCT). LSCs have been shown to be susceptible to targeting by cytotoxicT-cells (CD8 and CD4 cells) stimulated against leukemia-associated antigens; |
| | • | | the fact that WT1 has been associated with the actual development of leukemia; |
| | • | | the positive correlation between the level of expression of WT1 and the prognosis in AML; |
| | • | | the fact that the level of expression of WT1 can be followed over time in patients during and after therapy, including immunotherapy, as a method of monitoring for minimal residual disease, or MRD; |
| | • | | early evidence from mouse models that vaccination with peptides against select WT1 antigenic epitopes leads to detection of immune response; |
| | • | | early evidence that human immunocytes sensitizedex-vivo to peptides contained in GPS were able to recognize naturally presented WT1 peptides on the surface of several leukemia cell lines; |
10
| | • | | early anecdotal (at the time) clinical data showing antileukemic activity of WT1 monovalent vaccines in the Japanese population (albeit restricted toHLA-A*2401 type), as well as a dendritic cell vaccine in the Netherlands (independent of HLA haplotype); |
| | • | | the high degree of unmet medical need in AML and the absence of an effective maintenance therapy over the decades after initial upfront induction until and immediately after achievement of CRem status, particularly in patients older than 60 years of age; |
| | • | | a predictive assumption of very low to negligible degree of clinical toxicity with aWT1-targeted immunotherapy such as GPS, due to the fact that WT1 in normal,non-cancerous, tissues is both expressed at extremely low levels and limited in number of organs and tissues, and also due to the fact that WT1 fragments, or peptide epitopes, in normal cells are presented to host APCs in a different manner than are WT1 fragments produced in cancer cells; and |
| | • | | the advent of modern immunotherapeutics in cancer and the promise of an innovative,off-the-shelf immunotherapy for AML, a disease that was associated with dearth of deep and sustained responses to checkpoint inhibitors. |
Clinical Data—AML
In an initial pilot clinical trial in AML, a total of nine adult patients of all ages with de novo AML were treated with upfront standard chemotherapy and were able to achieve their first complete remission, or CRem1. Administration of GPS resulted in a median overall survival, or OS, that was at least 35 months from the time of GPS administration. In this study, specifically for patients who were 60 yrs and older (n=5), median OS was at least 33 months from the time of GPS administration or approximately 43 months from the time of initial AML diagnosis. The mean time offollow-up was 30 months from the time of diagnosis at the time of this analysis for all patients. Of the eight patients tested for immunologic response, seven, or 87.5%, demonstrated aWT1-specific immune response.
In a subsequent Phase 2 clinical trial in AML, a total of 22 adult patients of all ages with de novo AML were treated with upfront standard chemotherapy and were able to achieve CRem1. Most patients also received one to four cycles of “consolidation” chemotherapy per standard AML treatment guidelines. GPS was then administered within three months from the completion of the consolidation chemotherapy regimen in up to 12 total doses: Six initial doses (priming immunization) followed by six additional “booster” immunizations over a total period of up to 15 months to qualifying patients (i.e., patients who were clinically stable and did not show disease recurrence after the first six injections). This Phase 2 clinical trial met its primary endpoint of an actual OS rate of at least 34%, measured three years into the clinical trial (i.e., percentage of patients alive after three years offollow-up). An actual OS rate of 47.4% was demonstrated at three yearspost-GPS treatment, exceeding historical published data of OS of 20% to 25% by2.4- to1.9-fold (or 240% to 190%), respectively.
GPS administration was also shown to improve OS in comparison to historical data in patients in CRem1. Administration of GPS resulted in a median OS that was at least 67.6 months from the time of initial AML diagnosis in patients of all ages, which represents a substantial improvement compared to best standard therapy. Only five of the 22 patients underwent allogeneic HSCT and an ad hoc statistical analysis failed to show a significant effect of the transplant upon OS (either in median survival times or survival rates at specific landmark time-points). GPS was well tolerated with no Grade 3 or worse systemic side effects observed in this patient population, whose median age was 64 years old. Moreover, GPS elicitedWT1-specific immune responses in 88% of patients, including CD4 and CD8T-cell responses. CD4 responses were seen across HLA Class II subtypes and there was no discernible effect of HLA allelic type expression on clinical outcomes. Immune responses were seen against the native version of the two mutated WT1 peptides within the GPS mixture, which we believe demonstrates its heteroclitic properties. The results showed a trend in improved clinical outcomes in patients who mounted an immune response with GPS compared to those patients who did not. Importantly, a preplanned subgroup analysis for the cohort of 13 patients within the clinical trial who were 60 years of age or
11
older demonstrated a median OS of 35.3 months from time of initial diagnosis. Comparable historical populations have a median OS ranging from 9.5 to 15.8 months from initial diagnosis.
An additional Phase 2 clinical trial of GPS was performed at the H. Lee Moffitt Cancer Center & Research Institute, or Moffitt. This Phase 2 trial included ten AML patients who had received first-line therapy for their disease, who then experienced relapse and were subsequently treated with second-line chemotherapy and achieved a second complete remission, or CRem2. This group of patients had a more advanced disease in comparison to those treated in the other Phase 2 clinical trials discussed above, and typically demonstrated a historical OS of less than approximately eight months, even with post-CRem2 allogeneic HSCT. In the Moffitt trial, the efficacy of GPS (measured as median OS from the time of administration of a maintenance therapy to immediately after achievement of CRem2) was compared with that of “watchful waiting” in a cohort of 15 contemporaneously treated (but not matched by randomization) broadly comparable patients treated by the same clinical team at Moffitt. GPS administration resulted in a median OS of 16.3 months (495 days) compared to 5.4 months (165 days) from the time of achievement of CRem2. This was a statistically significant difference (P=0.0175). Two of 14 AML patients demonstrated relapse-free survival of more than one year. Both such patients were in CRem2 at time of GPS administration, with duration of their remission exceeding duration of their CRem1, suggesting a potential benefit based on immune response mechanisms. GPS was well-tolerated in this clinical trial with a comparable side effect profile to that observed in the prior Phase 2 clinical trial.
Planned Phase 3 Clinical Trial—AML
We are planning a Phase 3 clinical trial for GPS in AML patients 60 years of age or older who have achieved CRem1 following upfront chemotherapy and up to two cycles of post-remission consolidation chemotherapy, but who will not undergo allogeneic HSCT. This clinical trial has been planned, a principal investigator and the majority of site investigators have been identified and our operational partners for the execution of the trial are in the process of being identified. After several meetings and correspondence exchanges, the FDA has indicated that the agency has no further comments on the clinical trial design, protocol or statistical analysis plan. In addition, agreement has been reached with members of an independent data monitoring committee, which will be established upon commencement of the clinical trial.
We currently plan to initiate this clinical trial, pending funding availability, in the second half of 2018.
The clinical trial is planned to include up to 180 centers in the United States, Canada, European Union, Australia, New Zealand, Asia, and other countries and an estimated total sample size of up to 390 patients. Randomization will be 2:1 (GPS:placebo) andon-trial treatment duration will be up to approximately 82 weeks (1.58 years). The primary endpoint of the clinical trial is OS, measured from the time of randomization (not initial diagnosis). No companion diagnostic will be used as AML universally expresses WT1. Randomization will be stratified by region (U.S. compared tonon-U.S.), cytogenetic risk at diagnosis (favorable compared to not favorable compared to unknown), and type of AML (de novo compared to secondary). Patients will provide historical cytogenetic analysis results from initial diagnosis, before the start of their original chemotherapy treatment, to assess National Comprehensive Cancer Network genetic risk category. The clinical trial is currently powered to declare a positive result if GPS provides a four-month OS advantage compared to placebo, namely increasing median OS from approximately nine months in the control arm to approximately 13 months in the active,GPS-treated arm with an 1-sided alpha of 2.5%. Three interim analyses are planned in addition to a final analysis. If we are able to commence this Phase 3 trial in the second half of 2018, we anticipate the first interim data analysis being available in the second half of 2019.
12
The following figure illustrates the AML Phase 3 GPS clinical trial schema described in the above paragraph.
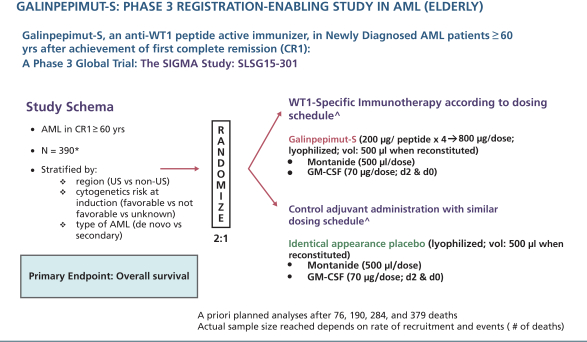
| * | maximum for final analysis, unless futility or efficacy thresholds are met in interim analyses prior to final analysis |
| ^ | All agents are administered subcutaneously |
GPS Monotherapy for Malignant Pleural Mesothelioma
MPM is an asbestos-related cancer that forms on the protective tissues that cover many of the internal organs. The most common area affected is the lining of the lungs and abdomen, though it can also form around the lining of the heart. Most cases are traced tojob-related exposures to asbestos and it can take approximately 40 years between exposure and cancer formation. Symptoms may include shortness of breath, a swollen abdomen, chest wall pain, cough, feeling tired, and weight loss. MPM is generally resistant to radiation and chemotherapy, and long-term survival is rare, even in cases where aggressive upfront debulking multimodality therapy (i.e., extirpative surgery, chemotherapy and in some cases radiotherapy, often described as “trimodality therapy” when used to treat MPM) are used.
Assuming absence of distant, systemic metastatic disease, MPM patients can initially present with a verydifficult-to-treat malignancy. The location, geometry, and origin of the tumor in the pleura (the external lining of the lungs and inner lining of the chest cage) present significant challenges for local and regional disease control. Extensive and complex surgery is initially considered. Patients without distant disease are broadly divided in two subgroups: (a) those who are in an inoperable status and (b) those who are operable. Patients in the former subgroup may be inoperable for two reasons: first, because they may be medically unfit for an extensive “definitive” surgery, most commonly due toco-morbidities (contemporaneously active diseases unrelated to their cancer) or, secondly, for technical reasons (location and/or bulk of tumor); the latter group of patients is defined as harboring “unresectable” disease. In general, approximately 35% to 40% of patients witha priori unresectable disease can be converted to technically resectable/marginally resectable, particularly if surgical expertise is high, after several cycles of upfront chemotherapy. This preoperative chemotherapy is termed “neoadjuvant” therapy. After the patient’s tumor becomes technically resectable, they receive extirpative surgery, often followed by more chemotherapy and sometimes radiotherapy. On the other hand, patients who area priori operable proceed
13
immediately with definitive surgery, resulting in either R0 or R1 resections, the degree between the two being assessed by surgical pathology review, with R0 corresponding to resection for “curative intent”, and R1 corresponding to microscopic residual tumor despite complete eradication by visual inspection at the time of surgery. After surgery, this subgroup of patients receives several cycles of “adjuvant” chemotherapy and sometimes radiotherapy.
In essence, all MPM patients who receive successful upfront trimodality therapy (upfront neoadjuvant chemotherapy, followed by definitive surgery, followed by possible further additional chemotherapy, or upfront definitive surgery followed by adjuvant chemotherapy) become free of residual detectable, macroscopic malignant deposits. Like AML patients who achieve CRem after upfront chemotherapy (in the absence of allogeneic HSCT), virtually all MPM patients will eventually relapse. Recurrent disease is unfortunately minimally responsive to second-line chemotherapy in MPM and typically these patients succumb to their disease or related complications within a few weeks to months after the emergence of clinically evident recurrent MPM. To date, there is no effective maintenance type of therapy to delay or prevent MPM relapse after initially successful upfront trimodality therapy. Typical median OS, even when following a fairly aggressive regimen when surgery is feasible, is between 12 and 16 months following diagnosis. Nonetheless, highly select patients who both undergo R0/R1 extensive surgery and complete a full course (6 cycles) of indicated chemotherapy (specifically those receiving the combination of pemetrexed with cisplatin, either in the neoadjuvant or adjuvant setting) can survive up to 21.0 to 24.8 months following initial diagnosis. These patients are typically younger, in excellent functional status, withoutco-morbidities and possibly having tumor-related factors related to better prognosis, such as intrinsically higher sensitivity of MPM cancer cells to chemotherapy-induced destruction.
Like AML, MPM represents a “model” type of solid tumor for testing the effects of GPS in clinical studies for the following reasons:
| | • | | MPM presents a clinical setting whereby minimal residual disease status can be achieved with standard upfront therapy; |
| | • | | WT1 is universally expressed in MPM malignant cells; in fact, WT1 expression is an established pathognomonic criterion for the actual diagnosis of MPM and its differentiation of other chest malignancies, for example, pulmonary adenocarcinoma; |
| | • | | the positive correlation between the level of expression of WT1 and prognosis in MPM; |
| | • | | preliminary evidence that WT1 expression could be involved in the MPM tumorigenesis and malignant growth promotion; |
| | • | | early evidence that human APCs sensitizedex-vivo to peptides contained in the GPS mixture were able to recognize naturally presented WT1 peptides from MPM cell lysates; |
| | • | | evidence that CD8 tumor-infiltrating lymphocytes predict favorable prognosis in MPM after resection (with the assumption that these CD8 cells are highly sensitized to tumor-associated antigens, including WT1); |
| | • | | the high degree of unmet medical need in MPM and the absence of an effective maintenance therapy; indeed, despite extensive research efforts and recent promising, yet preliminary, results with checkpoint inhibitors in second or third line therapy of MPM patients, few options are available for the treatment of MPM in the maintenance setting after successful debulking with upfront trimodality therapy (with the vast majority being managed with “watchful waiting” until the disease’s inexorable relapse) and its prognosis remains very poor; |
| | • | | a predictive assumption of very low to negligible degree of clinical toxicity with aWT1-targeted immunotherapy such as GPS, due to the fact that WT1 in normal,non-cancerous, tissues is both expressed at extremely low levels and limited in number of organs and tissues, but also due to the fact that WT1 fragments, or peptide epitopes, in normal cells are presented to host APCs in a different manner than are WT1 fragments produced in cancer cells; and |
14
| | • | | an initial preliminary clinical efficacy “signal” from the Phase 2 clinical trial of GPS at MSK in patients with MPM. |
Clinical Data—MPM
A randomized, double-blind, placebo-controlled Phase 2 clinical trial in MPM patients enrolled a total of 41 patients at MSK and M.D. Anderson Cancer Center. According to the Phase 2 MPM clinical trial data of GPS presented at the 2016 International Mesothelioma Interest Group and the 2016 Annual Meeting of the American Society of Clinical Oncology, as of May 2016, based on an initial analysis of 40 patients who were eligible at the time with a medianfollow-up of 16.3 months, a median OS of 24.8 months was recorded forGPS-treated MPM patients, compared to a median OS of 16.6 months for patients in the control arm, with a hazard ratio, or HR, of 0.51 in favor of GPS based on an initial analysis of 40 patients who were eligible at the time. Patients with an R0 tumor resection and subsequent treatment with GPS showed a significant survival benefit compared to those who received a placebo, with a median OS of 39.3 months compared to 24.8 months (HR: 0.415) in favor of GPS; this was a statistically significant difference (P<0.05). In a subsequent analysis of these endpoints for the entire cohort (n=41) in August 2016, with a medianfollow-up of 17.2 months, a median OS of 22.8 months was observed forGPS-treated MPM patients, compared to a median OS of 18.3 months for patients in the control arm, with an HR of 0.54 in favor of GPS. Furthermore, in the datasets from both of these analyses, GPS was shown to induceWT1-specific CD8 and CD4T-cell activation. GPS administration in the 19 MPM patients in the active arm of the aforementioned study was commonly associated with mild (grade 1 and 2) and self-limited injection site reactions. Clinically significant severe adverse events did not occur.
Planned Phase 3 Clinical Trial—MPM
We have planned a Phase 3 clinical trial in MPM, pending funding availability. The FDA has reviewed the clinical trial design in previous meetings and, following a formalend-of-phase 2 meeting, has indicated that the agency has no further comments on the clinical trial design, protocol or statistical analysis plan. We are currently evaluating the best strategy to develop GPS in this indication.
The planned Phase 3 clinical trial may include up to 120 centers in the United States, European Union, and other countries and an estimated total sample size ranging from 120 to 500 patients. The sample size is variable due to the Bayesian statistical design of the clinical trial. Randomization will be 1:1 (GPS:placebo) andon-trial treatment duration will be up to 13 to 18 months. The primary endpoint of the clinical trial is OS, measured from the time of randomization (not initial diagnosis). Secondary endpoints include leukemia free survival, safety/MRD and immune response. No companion diagnostic will be used as MPM universally expresses WT1. Randomization will be stratified by region (U.S. compared tonon-U.S.), timing of chemotherapy (neoadjuvant compared to adjuvant setting), and type of radiotherapyco-administered (intensity-modulated radiation therapy compared to other radiotherapy compared to none). The clinical trial will be adequately powered through a Bayesian adaptive approach to declare a positive result if certaina priori criteria are met, such as GPS providing an eight-month OS advantage compared to placebo, namely increasing median OS from approximately 16 months in the control arm to approximately 24 months in the active,GPS-treated arm with a2-sided alpha of 5%; the exact values of the OS in the control and active arms (as well as the difference between the two) may differ from the above estimates so long as the “two look” group sequential, adaptive statistical design would be able to deliver at least 90% power with anone-sided alpha of 5% at the time of the definitive “positive signal” analysis. Two interim analyses are planned (the second of the two, if positive for efficacy, would lead to main clinical trial early termination) in addition to a final analysis.
15
The following figure illustrates the MPM Phase 3 GPS clinical trial schema.
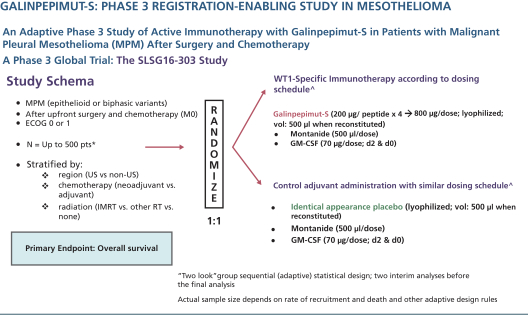
| * | Final N will be dependent on a priori rules from adaptive design implemented during the trial to ensure achievement of 90% power and1-sided alpha of 5% |
| ^ | All agents are administered subcutaneously |
GPS Monotherapy for Multiple Myeloma
MM is a cancer formed by malignant plasma cells, and its cause is unknown. The overgrowth of plasma cells in the bone marrow crowds out normal blood-forming cells, causing low blood counts and anemia (a shortage of red blood cells). MM can also cause a shortage of platelets (cells responsible for normal blood clotting) and lead to increased bleeding and bruising, along with problems fighting infections due to low white cell counts and/or lower levels of infection-fighting antibodies. MM causes a host of organ problems and symptoms, including fatigue, bone pain, fractures, circulatory problems (in small vessels of the brain, eye retina, heart, bowel, etc.) and kidney failure.
Treatment for MM includes chemotherapy, glucocorticoids, drugs that modulate the immune system (immunomodulatory drugs, or IMiDs), radiation and autologous stem cell transplants, or ASCTs. Recently, several novel targeted agents, such as proteasome inhibitors and immunotherapeutics have been introduced in the treatment paradigms for MM. Most therapies in MM are applied in combination, sometimes with usage of three to four or even five agents administered concomitantly or sequentially. This has led to a progressive increase in the number of “lines” of therapy that MM patients receive, which currently can reach up to five to six or even higher. Of note, ASCT can be used more than once, called tandem ASCTs, to debulk the disease and offer prolonged secondary remissions. Finally, allogeneic HSCT is rarely used in MM, but still has its use in selected high-risk patients who are or become refractory to antimyeloma therapies.
The prognosis in MM is highly variable and depends on numerous risk factors, some related to the biology of the disease, others to the host (e.g., age and functional status). Consequently, median survival can vary from up to at least 15 years innon-high-risk patients who achieve CRes, as defined by the International Myeloma Working Group, or IMWG, criteria, to approximately three years (from time of initial treatment) in patients with MM who achieve less than partial response, or PR, after ASCT. There are patients with MM who fare even more poorly, for example those aforementioned group who also have high-risk cytogenetics at baseline who may survive on
16
average less than three years. Similarly, patients who are ineligible for ASCT and are managed only with chemotherapy and long-term IMiD maintenance (with up to nine cycles of lenalidomide) who also achieve less than CRes and remain MRD(+) demonstrate a three-year OS rate of only about 55%; these landmark three-year OS rates decrease by approximately 40 to 50% in patients who also have high-risk cytogenetics at baseline. Despite significant therapeutic advances in the management of MM, the prognosis of patients with high risk cytogenetics at the time of diagnosis remains quite poor, even when they successfully complete an ASCT, particularly if such patients continue to have evidence of MRD.
GPS monotherapy for MM presents an opportunity to study both the clinical and immunologic effects of GPS in a hematologic malignancy. Therapeutic targeting of WT1 through immune pathways has largely not been pursued by others to date, and this indication presents an opportunity to target a malignancy that remains “incurable” in a strict sense, even in the face of significant advances that have accorded significant survival and freedom-from-active-disease benefit in standard risk patients. MM was chosen as a target indication for GPS for the following reasons:
| | • | | a clinical setting whereby MRD status can be achieved with standard upfront therapy. In this indication, with induction therapy using modern combination regimens followed by melphalan conditioning for myeloablation and a successful autotransplant, MM patients can achieve either CRes or very good partial response per IMWG criteria. This subgroup of patients would be optimal candidates for GPS therapy, even if they remain MRD(+) by flow cytometry or molecular markers; |
| | • | | the detectable expression of WT1 in MM cells (malignant plasmacytes). In the past, MM was considered not to be a tumor type with strong expression of WT1. This was due to the use of immunohistochemical staining analysis withanti-WT1 antibodies that had suboptimal diagnostic sensitivity. It has been recently shown that while WT1 is expressed at lower levels in MM compared to other hematologic and solid tumors, this expression is almost universally seen and is highly relevant from an immunobiological perspective, as the immune system is able to reliably raise vigorous and sustainedWT1-specific responses against malignant plasmacytes in the context of both MM and the rare, very aggressive variant of plasma-cell leukemia; |
| | • | | preliminary evidence that WT1 expression could be involved in the MM tumorigenesis and promotion; |
| | • | | early anecdotal (at the time) clinical data showing anti-myeloma activity of WT1 monovalent vaccines in Japanese patients (albeit restricted toHLA-A*2401 type); |
| | • | | the high degree of unmet medical need in MM patients with high-risk cytogenetics who also remain MRD(+) after frontline induction therapy and successful autotransplant, even when maintenance therapy is applied with either bortezomib or IMiDs (thalidomide); this has been shown in multiple studies, andto-date few options are available for addition of effective therapies in the maintenance setting to be added to agents such as lenalidomide (which is now standard of care in this setting); and |
| | • | | a predictive assumption of very low to negligible degree of clinical toxicity with aWT1-targeted immunotherapy such as GPS due to the fact that WT1 in normal,non-cancerous tissues is both expressed at extremely low levels and limited in number of organs and tissues, but also due to the fact that WT1 fragments, or peptide epitopes, in normal cells are presented to host APCs in a different manner than are WT1 fragments produced in cancer cells. |
Clinical Data—MM
We have reported comprehensive Phase 2 data for GPS in 19 patients with MM, which indicate promising clinical activity among MM patients with high-risk cytogenetics at initial diagnosis who also remain at least MRD(+) after successful frontline therapy (induction regimen followed by ASCT). This subgroup of MM patients, when serially assessed per IMWG criteria, typically relapse/progress within 12 to 14 months after ASCT, even when they receive maintenance therapy with IMiDs such as thalidomide or proteasome inhibitors such as bortezomib. Of note, 18 of the 19 patients received lenalidomide maintenance starting after the first three
17
GPS administrations following ASCT; the remaining single patient received bortezomib under the same schedule. All patients had evidence of at least MRD after ASCT, while 15 of the 19 also had high-risk cytogenetics at diagnosis. Combined, these characteristics typically result in low PFS rates that do not exceed 12 to 14 months following ASCT, even while on maintenance therapy with IMiDs or proteasome inhibitors, which are the current standards of care. As of June 2017, median PFS with GPS was 23.6 months, while median OS had not been reached. Our results compare favorably with an unmatched cohort of broadly comparable MM patients with high-risk cytogenetics published by the Spanish PETHEMA group from the PETHEMA Network No. 2005–001110–41 trial. Our GPS therapy demonstrated a1.87-fold increase in median PFS, as well as a1.34-fold increase in the PFS rate at 18 months compared to the aforementioned historical cohort, which included MM patients with high-risk cytogenetics and MRD(+) post-ASCT and on continuous intensive maintenance with thalidomide +/- bortezomib. Our Phase 2 clinical trial started in June 2014 and has enrolled a total of 20 patients of which 19 are currently evaluable. Patients are treated for up to nine months (12 GPS administrations) post-ASCT. The safety profile was devoid of grade 3/4/5 treatment-related adverse events. Allnon-progression events were confirmed and ongoing as of the time of the latest presentation (medianfollow-up at 20 months for survivors). Immune response data showed that up to 91% of patients had successfully developedT-cell (CD8 or CD4) reactivity to any of the 4 peptides within the GPS mixture, while up to 64% of patients demonstrated immune response positivity (CD4/CD8) against more than one WT1 peptide (multivalent responses). Moreover, multifunctional cross-epitopeT-cell reactivity was observed in 75% of patients to antigenic epitopes against which hosts were not specifically immunized, in a pattern akin to epitope spreading. Further, a link was shown between the evolution of immune responses and changes in clinical response status (achievement of CR/very good partial response clinical status per IMWG criteria) over time following treatment with GPS, with each patient being used as his or her own control for each longitudinal comparison. We believe the results offer mechanistic underpinnings for immune activation against WT1 in patients with aggressive, high-risk MM, and support the potential antimyeloma activity of GPS.
GPS Combination Therapy withPD-1 blocker (nivolumab) for Ovarian Cancer
Epithelial cancer of the ovary, or ovarian cancer, is a relatively common gynecologic cancer that develops insidiously, and hence is associated with vague or no symptoms that would urge patients to seek medical attention. Not surprisingly, most women with ovarian cancer present with advanced (at least locally or regionally, and often systemically spread) disease. Ovarian cancer is managed with initial surgical resection followed by platinum-based chemotherapy. During the past decade, incremental advances in chemotherapy, and the introduction of targeted therapies (such aspoly-ADP-ribose polymerase inhibitors and several others) and specially formulated compounds (such as liposomal anthracyclines) have resulted in improved survival and in more effective treatment of relapsed disease. In addition, a better understanding of genetic risk factors, along with aggressive screening, has permitted a tailored approach to preventive strategies, such as bilateral salpingo-oophorectomy in selected women along in specific patient populations genetically predisposed to this cancer (such as those harboring genetic alterations of the BRCA gene family). Although a complete clinical remission following initial chemotherapy can be anticipated for many patients, a review of “second-look” laparotomy, when it was often performed as a matter of routine care, indicates that less than 50% of patients are actually free of disease. Furthermore, nearly half of patients with a negative “second-look” procedure relapse and require additional treatment. Many patients will achieve a second complete clinical response with additional chemotherapy. However, almost all patients will relapse after a short remission interval of nine to 11 months. Effective strategies, such as introduction of novel immunotherapies, to prolong remission or to prevent relapse are required, as subsequent remissions are of progressively shorter duration until chemotherapy resistance broadly develops, leading to eventual disease-related demise.
18
Ovarian cancer represents an intriguing opportunity to study both the clinical and immunologic effects of GPS in another solid tumor. Additionally, therapeutic targeting of WT1 through immune pathways has largely not been pursued by others to date for this indication and ovarian cancer remains “incurable” once it advances and becomes disseminated, even in the face of significant advances in the field. Ovarian cancer was chosen as a target indication for the following reasons:
| | • | | ovarian cancer presents a clinical setting whereby MRD status can be achieved with standard upfront therapy both immediately after first line therapy, but also after effective debulking of the “first relapse.” The latter subgroup of patients (after successful second line treatment/first salvage, lacking demonstrable macroscopic residual disease) would be optimal candidates for GPS therapy, as no standard maintenance therapy exists for such patients and the subsequent relapse patterns and metrics are known and predictable; |
| | • | | the high levels of expression of WT1 in ovarian cancer cells. In fact, WT1 expression is so frequent that pathologists routinely use immunohistochemical stains for WT1 (with a standardized convention for describing expression and determining as “positive” or “negative”) to help distinguish epithelial ovarian cancers from other tumors; |
| | • | | preliminary evidence that WT1 expression may be linked to prognosis in ovarian cancer and that it may play an anti-apoptotic role in ovarian cancer cell lines; |
| | • | | the high degree of unmet medical need in ovarian cancer patients after first (or subsequent) successful “salvage” debulking therapy and the absence of effective therapies for such patients; and |
| | • | | a predictive assumption of very low to negligible degree of clinical toxicity with aWT1-targeted immunotherapy such as GPS due to the fact that WT1 in normal,non-cancerous tissues is both expressed at extremely low levels and limited in number of organs and tissues, but also due to the fact that WT1 fragments, or peptide epitopes, in normal cells are presented to host APCs in a different manner than are WT1 fragments produced in cancer cells. |
Clinical Data—Ovarian Cancer
GPS is being studied in combination with nivolumab, aPD-1 immune checkpoint inhibitor, in an open-label,non-randomized Phase 1 clinical trial, which is independently sponsored by MSK. The aim of the study is to evaluate the safety and efficacy of this combination in patients with recurrent ovarian, fallopian tube or primary peritoneal cancer who are in second or greater clinical remission (after their successful first or subsequent “salvage” therapy). This Phase 1 clinical trial was planned to enroll at least ten patients with recurrent ovarian cancer who are in second or greater clinical remission at MSK. Patients enrolled in the clinical trial received the combination therapy during the clinical trial’s14-week treatment period. Individuals who had not progressed by the end of this period also received a maintenance course of GPS. The primary endpoint of this clinical trial is the evaluation of the safety of repeated GPS administrations, for a total of six doses, in combination with seven infusions of nivolumab. This clinical trial addresses the safety of GPS whenco-administered with a checkpoint inhibitor, with the goal of possibly detecting an efficacy signal based on PFS and OS (versus historical data of monotherapy with nivolumab in this patient population), as well as documenting the pattern ofWT1-specific immune responsespost-GPS. Interim Phase 1 data from this study was presented at the 2018 American Society of Clinical Oncology, or ASCO, Annual Meeting held in June 2018. Exploratory efficacy interim data from the study showed that GPS, when combined with a PD-1 inhibitor, demonstrated PFS of 64% at one year in an intent to treat, or ITT, group of 11 evaluable patients with WT1+ ovarian cancer in second or greater remission. Among patients who received at least three doses of GPS in combination with nivolumab, PFS at one year was 70% (7/10). The historical rates with best standard treatment do not exceed 50% in this disease setting. The most common adverse events were Grade 1 or 2, including fatigue and injection site reactions. Dose limiting toxicity was observed in one patient, following the second dose of the combination. No additional adverse event burden was observed for the combination as compared to nivolumab monotherapy. The combination induced a high frequency of T- and B-cell immune responses. Pending the successful progress of this clinical trial a larger,follow-on, randomized clinical trial may be planned.
19
GPS Combination Therapy withPD-1 blocker pembrolizumab (Keytruda) for Other Cancers
In addition, given the potential immunobiologic and pharmacodynamic synergy between GPS and aPD-1 blocker, as well as the prevalent expression of WT1 in five select tumor types (colorectal cancer, triple-negative breast cancer, small cell lung cancer, ovarian cancer and AML), we entered into a clinical trial collaboration and supply agreement through a Merck subsidiary for the conduct of a combination clinical trial of GPS with pembrolizumab (Keytruda).
The purpose of thisfive-arm “basket” trial is to determine if the administration of GPS in combination with pembrolizumab (Keytruda) has the potential to demonstrate clinical activity in the presence of macroscopic disease, where monotherapy with either agent would have a more limited effect. The negative influence of tumor microenvironment factors on the immune response is predicted to be mitigated byPD-1 inhibition (by pembrolizumab) thus allowing the patients’ own immune cells to invade and destroy cancerous growth deposits specifically sensitized against WT1 (by concomitantly-administered GPS). The planned study will be open-label with multiple sites. The primary endpoints will be safety, overall rate of response (RECIST and iRECIST) and CR, MRD(-) status (for AML patients only) with exploratory endpoints including PFS, OS and immune response correlates. If we are able to commence this Phase 1/2 trial in the third quarter of 2018, we anticipate the first interim data analysis being available in the third quarter of 2019.
NeuVax™(nelipepimut-S)
Overview
NeuVax(nelipepimut-S) is a cancer immunotherapy targeting HER2 expressing cancers. NeuVax is the immunodominant nonapeptide derived from the extracellular domain of the HER2 protein, a well-established and validated target for therapeutic intervention in breast carcinomas. The NeuVax vaccine is combined withGM-CSF (Sargramostim) for injection in between the layers of the skin epidermis, i.e., intradermal administration. Data has shown that an increased presence of circulating tumor cells, or CTCs, may predict reduced disease free survival, or DFS, and OS, suggesting a presence of isolated micrometastases, not detectable clinically, but, over time, can lead to recurrence of cancer, most often in distant sites. After binding to the specific HLA molecules on antigen presenting cells, thenelipepimut-S sequence stimulates specific cytotoxic T lymphocytes, or CTLs, causing significant clonal expansion. These activated CTLs recognize, neutralize and destroy, through cell lysis, HER2 expressing cancer cells, including occult cancer cells and micrometastatic foci. The nelipepimut immune response can also generate CTLs to other immunogenic peptides through inter- and intra-antigenic epitope spreading.
Targeted Indication
NeuVax for Breast Cancer
According to NCI, over 230,000 women in the United States are diagnosed with breast cancer annually. While improved diagnostics and targeted therapies have decreased breast cancer mortality in the United States, metastatic breast cancer remains incurable. Approximately 75% to 80% of breast cancer patients have tissue test positive for some increased amount of the HER2 receptor, which is associated with disease progression and decreased survival. Only approximately 20% to 30% of all breast cancer patients-those with HER2 immunohistochemistry, or IHC, 3+ disease, or IHC 2+ and fluorescence in situ hybridization, or FISH, amplified-have a HER2 directed, approved treatment option available after their initial standard of care. This leaves the majority of breast cancer patients withlow-to-intermediate HER2 expression (IHC 1+, 2+; HER2 FISH,non-amplified), or approximately 15% of all breast cancer patients, with tumors that are not HER2-amplified by FISH ineligible for targeted therapy with trastuzumab and without an effective targeted treatment option to prevent cancer recurrence.
20
Clinical Trials
We currently have multiple investigator-sponsored trials ongoing with NeuVax in combination with trastuzumab (Herceptin; Genentech/Roche). The combination of trastuzumab and NeuVax has been shownpre-clinically and in a pilot study to be synergistic. Our Phase 2b trial is a multi-center, randomized, single-blinded, placebo-controlled trial in 275 HER2 1+/2+ breast cancer patients with positive nodes and/or TNBC. The study combines NeuVax and trastuzumab (Herceptin) in the adjuvant setting aiming to prevent recurrence or death. Tumors in these women show low levels of expression of HER2, as measured by IHC, i.e., at a level of either 1+ or 2+ and, hence, these patients are not considered candidates for Herceptin. Patients who are hormone receptor-negative and HER2 1+/2+ by IHC are currently defined as TNBC patients. Eligible patients are randomized to receive NeuVax +GM-CSF + trastuzumab or trastuzumab +GM-CSF alone. The primary endpoint of the study is DFS at 24 months. Genentech/Roche is providing the trastuzumab and partial funding for this trial. Data presented in October 2016 demonstrated that this novel combination of trastuzumab and NeuVax with HER2low-expressing patients is well tolerated and the cardiac effects of trastuzumab are not impacted by the addition of NeuVax. In February 2017, the DSMB reported that there were no safety concerns with the trial and the trial is not futile. The recommendation from the DSMB was to continue the trial with one revision to the statistical analysis plan regarding the timing of thepre-specified interim analysis. Given the lengthy duration of enrollment for the trial, the DSMB determined that thepre-specified interim efficacy analysis be moved up from 12 months to 6 months after the last patient is enrolled. Enrollment was completed and the interim efficacy analysis occurred in late March 2018.
The interim analysis, conducted by an independent DSMB of the efficacy and safety data for the study in an overall population of 275 patients as well as the two primary study target patient populations (node-positive and TNBC) after a medianfollow-up of 19 months, demonstrated a clinically meaningful difference in median DFS in favor of the active arm (NeuVax + Herceptin), a primary endpoint of the study, with hazard ratios of 0.67 and 0.61 in the intent to treat and modified intent to treat populations (i.e., those who received at least one dose of vaccine or control) as well as a 34.9% and 39.5% reduction in relative risk of recurrence in the active versus control arms in the intent to treat and modified intent to treat populations, respectively. A clinically meaningful and also statistically significant difference was found between the two arms in the cohort of patients (n= 98) with TNBC, with a hazard ratio of 0.26 and ap-value of 0.023 in favor of the NeuVax + Herceptin combination, with landmark DFS at 24 months showing a 70.4% reduction in relative risk of recurrence in the active arm versus control. Similarly, a clinically meaningful and statistically significant difference was found between the two arms in favor of the combination in the cohort of patients not receiving hormonal therapy (n = 110), with a hazard ratio of 0.24 and ap-value of 0.009. In vivo HER2- specific T-cell immune responses showed a time-dependent increase in IR potency compared to the earliest data point tested (p=0.000023) in the active arm, while no such increase was observed in the control arm. Thispre-specified interim analysis also showed an adverse event profile with no notable differences between treatment arms. This analysis confirmed the 2016 data showing that the addition of NeuVax to Herceptin did not result in any additional cardiotoxicity compared to Herceptin alone. Based on these results, and the DSMB’s recommendation, we plan to expeditiously seek regulatory guidance from the FDA for further development of the combination of NeuVax + Herceptin in TNBC, considering the statistically significant benefit of the combination therapy seen in this population with large unmet medical need. In June 2018, we announced that the sponsor-principal investigator of this study, after taking into account that key clinical development objectives were met as well as other regulatory considerations, and with our agreement, determined to terminate early the study. In addition, in late May 2018, we conducted two advisory meetings with global experts in regulatory affairs and breast cancer clinical development in order to determine the optimal path for further development of the NeuVax + Herceptin combination in TNBC in a pivotal setting and for engagement with the FDA and EMA. Data from this study have been submitted for presentation at a major medical conference that will take place during the second half of 2018.
Our second combination investigator sponsored trial is a Phase 2 in HER2 3+ breast cancer patients who have completed neoadjuvant therapy with an approved regimen that includes trastuzumab and failed to achieve a pathological complete response, meaning they have microscopic evidence of residual disease and are therefore at an increased risk of disease recurrence. This multi-center, prospective, randomized, single-blinded Phase 2
21
clinical trial has enrolled approximately 100 patients with a diagnosis of HER2 3+ breast cancer who are HLA A2+ or HLA A3+ and are determined to be at high-risk for recurrence. High-risk is defined as having received neoadjuvant therapy with an approved regimen that includes trastuzumab but not obtaining a pathological complete response at surgery, or those who undergo surgery as a first intervention and are found to be pathologically node-positive. These high-risk patients are known to have higher recurrence rates than other HER2 3+ breast cancer patients. Eligible patients will be randomized to receive NeuVax +GM-CSF + trastuzumab or trastuzumab +GM-CSF alone. The primary endpoint of the study is disease-free survival. Funding for this trial was awarded through the Congressionally Directed Medical Research Program, funded through the Department of Defense, via a breast cancer research program breakthrough award. In February 2017, the DSMB reported that there were no safety concerns with the trial and the trial is not futile. Thepre-specified interim safety analysis was also completed on n=50 patients and demonstrated that the agent is well tolerated with no increased cardiotoxicity associated with giving NeuVax in combination with trastuzumab. The recommendation from the DSMB was to continue the HER2 3+ trial unmodified.
A Phase 3 PRESENT (Prevention ofRecurrence inEarly-Stage, Node- Positive Breast Cancer with Low to Intermediate HER2Expression withNeuVax Treatment) study enrolled 758 HER2 1+/2+ patients who are node-positive and HLA A2 or A3 positive. On June 27, 2016, the independent data monitoring committee recommended that the Phase 3 PRESENT clinical trial be stopped for futility. The PRESENT trial was stopped, and we initiated an investigation into the causes of the recommendation. Our analysis of the data showed that there was a separation of the curves, albeit not statistically significant, with the control arm performing better than expected and the NeuVax arm performing consistent with our protocol assumptions for the control group. Because the study was deemed futile, we closed the PRESENT trial.
NeuVax for Ductal Carcinoma In Situ of the Breast
DCIS is defined by the NCI as a noninvasive condition in which abnormal cells are found in the lining of a breast duct and have not spread outside the duct to other tissues in the breast. DCIS is the most common type of breast neoplasm with malignant potential. In some cases, DCIS may become invasive cancer and spread to other tissues, and at this time, there is no way to know which lesions could become invasive. Current treatment options for DCIS include breast-conserving surgery and radiation therapy with or without tamoxifen, breast-conserving surgery without radiation therapy, or total mastectomy with or without tamoxifen. According to the American Cancer Society, in the United States, there were over 60,000 diagnoses of DCIS in 2015. We are supporting an independent investigator-sponsored, or IST, Phase 2 trial to evaluate women diagnosed with DCIS who areHLA-A2 positive, who express HER2 at IHC 1+, 2+, or 3+ levels, and who are pre or post menopausal. Patients will be randomized to one of two arms: NeuVax plusGM-CSF orGM-CSF alone. The clinical study name is VADIS: Phase 2 Trial ofNelipepimut-SVaccine in Women withDCIS of the Breast. The trial is sponsored and operationalized by the NCI, studying NeuVax’s potential clinical effects in earlier stage disease. The trial has an immunological endpoint evaluating NeuVax peptide-specific cytotoxic T lymphocyte (CTL; CD8+T-cell) response in vaccinated patients.
22
E39 Folate Binding Peptide Vaccine Program(GALE-301 andGALE-302)
Overview
GALE-301 andGALE-302, our E39 folate binding peptide vaccines, are cancer immunotherapies that target FBP receptor-alpha. FBP is a well-validated therapeutic target that is highly over-expressed in ovarian, endometrial and breast cancers, and is the source of immunogenic peptides that can stimulate CTLs to recognize and destroyFBP-expressing cancer cells. Current treatments after surgery for these diseases are principally with platinum-based chemotherapeutic agents. These patients suffer a high recurrence rate and most relapse with an extremely poor prognosis.GALE-301 andGALE-302 are immunogenic peptides that consist of a peptide derived from FBP combined withGM-CSF for the prevention of cancer recurrence in the adjuvant setting.GALE-301 is the E39 peptide, whileGALE-302 is an attenuated version of this peptide, known as E39’. Two early stage clinical trials have been completed with our FBP peptides in ovarian, endometrial, and breast cancers. In a Phase 1 study of E39 (both GALE 301 and GALE 302) in ovarian cancer and endometrial cancer that enrolled 59 patients with concomitantly enrolled controls, 24-month DFS was achieved in 55.5% of the patients receiving E39 as compared to 40% of the control group. For those patients receiving the highest dose of E39, the 24-month DFS rate was 77.9% (or p=0.013). A manuscript presenting the data from this study has been submitted for publication, with publication expected in the second half of 2018. In June 2016, the FDA granted two Orphan Drug Product designations for the treatment (including prevention of recurrence) of ovarian cancer: one forGALE-301 (E39) and one forGALE-302 (E39’).
GALE-301 andGALE-302 in Ovarian Cancer
According to the NCI’s Surveillance, Epidemiology, and End Results, or SEER Program, new cases of ovarian cancer occur at an annual rate of 11.9 per 100,000 women in the United States, with an estimated 22,280 new cases and 14,240 deaths in 2016. Only 46.2% of ovarian cancer patients are expected to survive five years after diagnosis. Approximately 1.3% of women will be diagnosed with ovarian cancer at some point during their lifetime (2011-2013 data). The prevalence data from 2013 showed an estimated 195,767 women living with ovarian cancer in the United States. Due to the lack of specific symptoms, the majority of ovarian cancer patients are diagnosed at later stages of the disease, with an estimated 80% of women presenting with advanced-stage (III or IV) disease. These patients have their tumors routinely surgically debulked to minimal residual disease, and then are treated with platinum- and/or taxane-based chemotherapy. While many patients respond to this treatment regimen and become clinicallyfree-of-disease, the majority of these patients will relapse. Depending upon their level of residual disease, the risk for recurrence after completion of primary therapy is approximately 70%. Unfortunately, for these women, once the disease recurs, treatment options are limited.
GALE-401 (anagrelide controlled release)
Overview
GALE-401 contains the active ingredient anagrelide, anFDA-approved product, for the treatment of patients with MPNs to lower abnormally elevated platelet levels. The currently available immediate release, or IR, version of anagrelide causes adverse events that are believed to be dose and plasma concentration dependent and may limit the use of the IR version of the drug. Therefore, reducing the maximum concentration, or C max, and increasing the half-life of the drug is hypothesized to reduce the side effects, while preserving the efficacy, potentially allowing a broader use of the drug.
GALE-401 in Essential Thrombocythemia
ET is a myeloproliferative blood disorder and is characterized by the overproduction of platelets in the bone marrow. Elevated platelets alter the normal process of blood coagulation and can lead to thromboembolic events. About a third of patients are asymptomatic at the time of diagnosis. However, many patients develop symptoms during the course of the disease that affect the quality of life.
23
Multiple Phase 1 studies in 98 healthy subjects have shownGALE-401 reduces the C max of anagrelide and increases the half-life following oral administration, appears to be well tolerated at the doses administered, and to be capable of reducing platelet levels effectively. The Phase 1 program provided the desired PK/PD (pharmacokinetic/pharmacodynamic) profile to enable the initiation of the Phase 2proof-of-concept trial. The Phase 2, open label, single arm,proof-of concept trial enrolled 18 patients in the United States for the treatment of thrombocytosis, or elevated platelet counts, in patients with MPNs. Final safety and efficacy data from this Phase 2 trial were presented in December 2015 and demonstrated a prolonged clinical benefit with a potentially improved safety profile.
We have analyzed our data and the treatment landscape for MPNs, with a current focus on ET. Subject to completion of the manufacturing of the new formulation and other internal work,GALE-401 would be poised to advance into a Phase 3 clinical trial in ET patients who are intolerant or resistant to hydroxyurea. This trial is designed to compareGALE-401 (drug arm) versus best available therapy to include a sizable population of patients treated with anagrelide IR. We plan to evaluateout-licensing and partnering opportunities forGALE-401.
Strategic Collaborations and License Agreements
Although we currently have a number of collaborations with corporate partners for the development of our product candidates in various territories worldwide, the following development collaborations are of the most significance from both a financial statement and ongoing collaboration perspective.
Exclusive License Agreement—Memorial Sloan Kettering Cancer Center
In September 2014, we entered into a license agreement with MSK, under which we were granted an exclusive license to develop and commercialize MSK’s WT1 peptide vaccine technology. The MSK original license agreement was first amended in October 2015, further amended in August 2016, amended and restated in May 2017 and again amended and restated in October 2017. In connection with the entry of the original license agreement and its amendments, MSK was issued or assigned an aggregate of 4,846 ordinary shares of Private SELLAS common stock for the year ended December 31, 2017. These common stock shares were converted into our common stock shares upon the Merger.
Under the terms of the current amended and restated MSK license agreement, we agreed to pay minimum royalty payments in the amount of $0.1 million each year commencing in 2015 and research funding costs of $0.2 million in each year and for three years commencing in January 2016. We also agreed to pay MSK amid-six digit amount over a one year period in exchange for MSK’s agreement to further amend and restate the MSK license agreement in October 2017, which resulted in the grant of rights to additional intellectual property to us and extension/relaxing of certain deadlines. In addition, to the extent certain development and commercial milestones are achieved, we also agreed to pay MSK up to $17.4 million in aggregate milestone payments for each licensed product, and for each additional patent licensed product, up to $2.8 million in additional milestone payments. We also agreed to pay MSK a tiered royalty in themid-single digits in the event of commercial sales of any licensed products. We also agreed to raise $25.0 million in gross proceeds no later than December 31, 2018. In the event we do not raise such amount by December 31, 2018, MSK may terminate the license agreement after complying with the notice and cure periods of the agreement, or MSK may elect to receive additional common stock shares in an amount equal to 1.5% of our then fully diluted share capital, which would stay the right to terminate for a period of time.
Unless terminated earlier in accordance with its terms, the MSK license agreement as amended and restated, will continue on acountry-by-country and licensedproduct-by-licensed product basis, until the later, of: (a) expiration of the last valid claim embracing such licensed product; (b) expiration of any market exclusivity period granted by law with respect to such licensed product; or (c) ten (10) years from the first commercial sale in such country.
24
Merck & Co., Inc. Clinical Trial Collaboration and Supply Agreement
In September 2017, we entered into a clinical trial collaboration and supply agreement through a Merck subsidiary, whereby we agreed with the Merck subsidiary to collaborate on a research program to evaluate GPS as it is administered in combination with theirPD-1 blocker pembrolizumab (Keytruda) in a Phase 1/2 clinical trial enrolling patients in up to five cancer indications, including both hematologic malignancies and solid tumors.
The Phase 1/2 clinical trial will utilize a combination of GPS plus pembrolizumab (Keytruda) in patients with WT1+ relapsed or refractory tumors. Specifically, the study is expected to explore the following cancer indications: colorectal (arm enriched in but not exclusive to patients with microsatelliteinstability-low), ovarian, small cell lung, triple-negative breast, and AML. This study will assess the efficacy and safety of the combination, comparing overall response rates and immune response markers achieved with the combination compared to prespecified rates based on those seen with pemrolizumab alone in comparable patient populations. The trial is anticipated to begin in the third quarter of 2018 (pending funding availability).
Advaxis, Inc. Research and Development Collaboration Agreement
In February 2017, we entered into a research and development collaboration agreement with Advaxis whereby we agreed to collaborate on a research program to evaluate, through a PoP trial, a clinical candidate comprised of the combination of Advaxis’ proprietaryLm-based antigen delivery technology and GPS. Unless terminated earlier in accordance with its terms, the Advaxis agreement will expire upon the earlier of: (a) completion of the PoP trial or (b) a decision by the parties to cease further development of the clinical candidate.
The Advaxis agreement provides for cost-sharing between the parties, with Advaxis being responsible for the costs of performing the research activities and filing any investigational new drug, or IND, cost-sharing for preparation of the IND, and we being responsible for the costs (exclusive of product costs) of conducting the PoP trial. Upon successful completion of the PoP trial, or otherwise at our election, we will become the sponsor under the IND and will control further development and commercialization of any product developed pursuant to the Advaxis agreement. We also agreed to make certainnon-refundable milestone payments to Advaxis having an aggregate amount of up to $108.0 million, upon meeting certain clinical, regulatory and commercial milestones. In addition, if net sales exceed certain targets, we agreed to makenon-refundable sales milestone payments up to $250.0 million and royalty payments based on specific royalty rates, with a maximum rate capped at a percentage rate in the low teens if net sales exceed $1.0 billion.
The University of Texas M. D. Anderson Cancer Center and The Henry M. Jackson Foundation for the Advancement of Military Medicine, Inc. License Agreement
In September 2006, we acquired rights and assumed obligations under a license agreement between Apthera and The University Texas M.D. Anderson Cancer Center, or MDACC, and The Henry M. Jackson Foundation, or HJF, which granted us exclusive worldwide rights to an United States patent covering thenelipepimut-S peptide and several United States and foreign patents and patent applications covering methods of using the peptide as a vaccine. Under the terms of this license, we are required to pay an annual maintenance fee, clinical milestone payments and royalty payments based on sales of NeuVax, or other therapeutic products developed from the licensed technologies.
Biovascular, Inc. Exclusive License Agreement
In December 2013, we acquired worldwide rights to anagrelide controlled release, or CR, formulation,GALE-401, through our acquisition of Mills, LLC, or Mills our wholly owned subsidiary.GALE-401 contains the active ingredient anagrelide, anFDA-approved product that has been in use since the late 1990s for the treatment of MPNs. Mills entered into an exclusive license agreement with BioVascular, Inc., or BioVascular.
25
The license agreement granted us an exclusive license to develop and commercialize anagrelide CR formulation. Under the terms of the license agreement and its amendments, Mills agreed to pay BioVascular, amid-to-low single digit royalty on net revenue from the sale of licensed products, as well as, future cash milestone payments based on the achievement of specified regulatory milestones. We are responsible for patent prosecution and maintenance.
In September 2017, Mills and BioVascular entered into an amendment to our exclusive license agreement to modify the certain terms of the license agreement, including but not limited to, (i) eliminating the 3% royalty rate on annual net sales of $50.0 million and the 4% royalty now applies to annual net sales of up to $100.0 million, (ii) making an advance payment of approximately $0.4 million for the milestone related to the initiation of the Phase 3 clinical trial payable in two tranches with the first payment of $0.2 million payable on or before October 31, 2017 and the second payment of approximately $0.2 million payable 30 days after the consummation of the Merger but no later than December 31, 2017, (iii) adding a payment for a sublicense by Mills to a third party of 25% of any cash received for upfront fees or milestone payments if the sublicense is executed prior to first patient enrolled in the Phase 3 clinical trial and 17.5% of any cash received for upfront fees or milestone payments if the sublicense is executed after the first patient is enrolled in the Phase 3 clinical trial, and (iv) if the first patient is not enrolled in the Phase 3 clinical trial by December 31, 2018, BioVascular shall have the right to terminate the license agreement and the advance payment shall not be repaid to Mills. Under the terms of a September 2017 consent between Comerica Bank, BioVascular and Mills, Comerica Bank shall receive $0.1 million of the approximately $0.4 million advance payment from Mills.
Manufacturing
We do not own or operate manufacturing facilities for the production of our product candidates nor do we have plans to develop our own manufacturing operations in the foreseeable future. We currently depend on third-party contract manufacturers for all of our required raw materials, active pharmaceutical ingredients, and finished product candidate for our clinical trials. We do not have any current contractual arrangements for the manufacture of commercial supplies of any product candidates. We currently employ internal resources and third-party consultants to manage our manufacturing contractors.
Sales and Marketing
We have not yet defined our sales, marketing or product distribution strategy for our product candidates or any future product candidates because they are still inpre-clinical or clinical development. Our future commercial strategy may include the use of strategic partners, distributors, a contract sale force, or the establishment of our own commercial and specialty sales force, as well as similar strategies for regions and territories outside the United States. We plan to further evaluate these alternatives as we approach approval for the use of our product candidates for one or more indications.
Intellectual Property
Our commercial success depends in part on our ability to avoid infringing the proprietary rights of third parties, our ability to obtain and maintain proprietary protection for our technologies where applicable and to prevent others from infringing our proprietary rights. We seek to protect our proprietary technologies by, among other methods, evaluating relevant patents, establishing defensive positions, monitoring European Union oppositions and pending intellectual property rights, preparing litigation strategies in view of the United States legislative framework and filing United States and international patent applications on technologies, inventions and improvements that are important to our business. Patents and other intellectual property rights are crucial to our success. It is our policy to protect our intellectual property rights through available means, including filing and prosecuting patent applications in the United States and other countries, protecting trade secrets, and utilizing regulatory protections such as data exclusivity. We also include restrictions regarding use and disclosure of our proprietary information in our contracts with third parties, and utilize customary confidentiality agreements with
26
our employees, consultants, clinical investigators and scientific advisors to protect our confidential information andknow-how. Together with our licensors, we also rely on trade secrets to protect our combined technology especially where we do not believe patent protection is appropriate or obtainable. It is our policy to operate without knowingly infringing on, or misappropriating, the proprietary rights of others.
An international patent law treaty, the Patent Cooperation Treaty, or PCT, provides a unified procedure for filing patent applications to protect inventions in each of its contracting states. Thus, a single PCT application can be converted into a national stage patent application in any of the more than 145 PCT contracting states, and is considered a simple, cost-effective means for seeking patent protection in numerous regions or countries. This nationalization (converting into an application in any of the contracting states) typically occurs 18 months after the PCT application filing date. We also rely on trade secrets,know-how and continuing technological innovation to develop and maintain our proprietary position.
The term of individual patents depends upon the legal term of the patents in countries in which they are obtained. In most countries, including the United States, the patent term is generally 20 years from the earliest date of filing anon-provisional patent application in the applicable country. In the United States, a patent’s term may, in certain cases, be lengthened by patent term adjustment, which compensates a patentee for administrative delays by the United States Patent and Trademark Office in examining and granting a patent or may be shortened if a patent is terminally disclaimed over a commonly owned patent or a patent naming a common inventor and having an earlier expiration date.
The following chart summarizes our intellectual property rights:
| | | | | | | | | | | | |
Product Candidate | | Product Candidate
Component | | Jurisdiction | | Indication | | Claims | | Scope | | Latest Estimated
Patent Exclusivity
Period |
GPS | | PeptideWT1-A1 | | United States | | Any | | Composition of Matter | | 1 issued | | 03/22/2026* |
GPS | | PeptideWT1-A1 | | Australia, Switzerland, Germany, Spain, France, Great Britain, Italy | | Any | | Composition of Matter and Method of Use | | 7 issued | | 11/30/2024 |
GPS | | PeptideWT1-A1 | | Canada | | Any | | Composition of Matter and Method of Use | | 1 issued | | 11/30/2024 |
GPS | | PeptidesWT1-427 long andWT1-331 long | | United States | | Any | | Composition of Matter | | 1 issued | | 10/26/2031* |
GPS | | PeptidesWT1-427 long andWT1-331 long | | United States | | WT1-expressing
cancer | | Method of Use | | 1 issued | | 10/17/2026 |
GPS | | Peptides WT1-427 long andWT1-331 long | | United States | | Any | | Composition of Matter and Method of Use | | 1 pending | | 10/17/2026** |
GPS | | Peptide WT1-427 long | | Switzerland, Germany, Spain, France, Great Britain, Ireland, Italy | | Any | | Composition of Matter and Method of Use | | 7 issued | | 10/17/2026 |
GPS | | PeptideWT1-331 long | | Switzerland, Germany, Spain, France, Great Britain, Ireland, Italy | | Any | | Composition of Matter and Method of Use | | 87 issued | | 10/17/2026 |
27
| | | | | | | | | | | | |
Product Candidate | | Product Candidate
Component | | Jurisdiction | | Indication | | Claims | | Scope | | Latest Estimated
Patent Exclusivity
Period |
GPS | | PeptideWT1-427 andWT1-331 long | | Australia, | | Any | | Composition of Matter and Method of Use | | 1 issued | | 10/17/2026 |
GPS | | PeptideWT1-427 long | | Canada | | Any | | Composition of Matter and Method of Use | | 1 issued | | 10/17/2026 |
GPS | | PeptidesWT1-331 long | | Canada | | Any | | Composition of Matter and Method of Use | | 1 pending | | 10/17/2026** |
GPS | | Non-product peptide | | United States | | Any | | Composition of Matter | | 1 issued | | 12/21/2026 |
GPS | | Peptide WT1-122A1 long | | United States | | Any | | Composition of Matter | | 1 issued | | 02/20/2033* |
GPS | | PeptideWT1-122A1 long | | United States | | Any | | Composition of Matter and Method of Use | | 1 pending | | 04/10/2027** |
GPS | | PeptideWT1-122A1 long | | Austria, Belgium, Switzerland, Germany, Spain, Finland, France, Great Britain, Greece, Ireland, Italy, Netherlands, Poland, Romania, Turkey | | Any | | Composition of Matter and Method of Use | | 15 issued | | 04/10/2027 |
GPS | | PeptideWT1-122A1 long | | Europe, Canada, Hong Kong | | Any | | Composition of Matter and Method of Use | | 3 pending | | 04/10/2027 |
| | | | | | | | | | | | | | |
Product
Candidate | | Product Candidate
Component | | Jurisdiction | | Indication | | Claims | | Scope | | Latest Estimated
Patent Exclusivity
Period | |
Not applicable | | Non-product peptide | | United States | | Any | | Composition of Matter and Method of Use | | 1 issued, 1 pending | | | 01/15/2034 | |
Not applicable | | Non-product peptide | | Australia, Canada, China, Europe, Japan | | Any | | Composition of Matter and Method of Use | | 5 pending | | | 01/15/2034 | ** |
Not applicable | | Not applicable | | United States | | Any | | Composition of Matter and Method of Use | | 1 pending | | | 06/30/2038 | *** |
| NeuVax™(nelipepimut-S) | | | | United States, Australia, Canada, China, Europe, Hong Kong, Japan, Korea
and Mexico | | Recurrence of cancers expressing low to intermediate levels of HER2/neu | | Methods of Use | | 6 pending and 10 issued | | | 2028 | |
| NeuVax™ in combination with trastuzumab | | | | United States and Australia | | HER2/neu expressing cancer | | Methods of Use | | 2 issued | | | 2026 | |
| GALE-401 | | | | United States | | Vaso-occlusive | | Method of Use | | 1 issued | | | 2020 | |
| GALE-401 (Anagrelide Controlled Release) | | | | United States, Europe, India, Japan and UK | | Platelet Lowering | | Anagrelide Controlled Release Formulations & Methods of Use | | 4 pending and 7 issued | | | 2029 | |
28
| | | | | | | | | | | | | | |
Product
Candidate | | Product Candidate
Component | | Jurisdiction | | Indication | | Claims | | Scope | | Latest Estimated
Patent Exclusivity
Period | |
| GALE-301 | | | | United States and PCT | | Cancers expressing low levels of FBP (IHC 0 or 1+) | | Dosage Regimen | | 2 pending | | | 2037 | |
| GALE-301 &GALE-302 Combination | | | | United States, Canada, Europe, and Japan | | Cancers expressing Folate Binding Protein (FBP) | | Compositions & Methods of Use | | 1 pending and 8 issued | | | 2022 | |
| GALE-301 &GALE-302 Combination | | | | United States | | Cancers expressing Folate Binding Protein (FBP) | | Combination Dosage Regimen | | 1 allowed | | | 2036 | |
| * | Includes patent term adjustment |
| ** | Projected expiration date of pending application, if granted |
| *** | Projected expiration date ofnon-provisional application to be filed from provisional application |
Each of the above-referenced pending or issued patents has been licensed by us. To our knowledge, there are no contested proceedings or third-party claims relating to any of the above pending or issued patents.
Competition
Cancer immunotherapy has become a significant growth area for the biopharmaceutical industry, attracting large pharmaceutical companies as well as small niche players. Generally, our principal competitors in the cancer immunotherapy market comprise both companies with currently approved products for various indications, such as manufactures of approved bispecific antibodies,CAR-T cells, and checkpoint inhibitors, as well as companies currently engaged in cancer immunotherapy clinical development. The competitors who have successfully obtained approval for cancer immunotherapy products include Bristol-Myers Squib Company, Merck & Co., Inc., Genentech, Inc. (a subsidiary of Roche Holding AG), AstraZeneca PLC, Celgene Corporation, Johnson & Johnson/Janssen Pharmaceuticals, Amgen, Novartis, Acerta Pharmaceuticals, Juno Therapeutics, Inc., Kite Pharm, Inc., a wholly-owned subsidiary of Gilead Sicencies, Inc. and Pfizer, Inc./EMD Serono, Inc.
Companies developing products with similar indications to those we are pursuing are expected to influence our ability to penetrate and maintain market share. Principal competitors for our AML indication include both companies with currently approved products in AML, such as Agios Pharmaceuticals, Inc. (the holder of U.S. rights to Idhifa), Novartis AG (the holder of rights to Rydapt), among others, as well as those with front-line chemotherapy drugs and maintenance therapies such as Jazz Pharmaceuticals plc (the holder of rights to Vyxeos), as well as Pfizer (the holder of rights to Mylotarg), among others, as well as companies with drugs currently in development in AML. Our principal competitors for the MPM indication include both companies with currently approved products in MPM, such as Eli Lilly and Co. (the holder of rights to Alimta), among others, as well as those with drugs currently in development in MPM. Our principal competitor for ET patients who are intolerant or resistant to hydroxyurea indication is Incyte Corporation in the United States (the holder of rights for Jakafi) and Novartis outside the United States (the holder of rights for JAKAVI).
For patients with MPNs, current treatment options include Agrylin (anagrelide hydrochloride) and its generic equivalents, hydorxyurea and interferon alpha. Agents currently being studied in patients with MPNs include investigational JAK2 inhibitors (e.g., LY2784544 (Eli Lilly), momelotinib (Gilead Sciences), ruxolitinib (Incyte), fedratinib (Impact Biomedicines/Celgene) and pegylated interferonalfa-2a (Pegasys, Genentech/Roche).
For patients with early stage breast cancer, adjuvant therapy is often given to prevent recurrence and increase the chance of long-term disease free survival. Adjuvant therapy for breast cancer can include chemotherapy,
29
hormonal therapy, radiation therapy, or combinations thereof. In addition, the HER2 targeted drug trastuzumab (Herceptin)—alone or in combination with pertuzumab (Perjeta), both manufactured and marketed by Roche/Genentech- may be given to patients with tumors with high expression of HER2 (IHC 3+), while immune-oncology agents focusing on other novel targets such as MUC1, may be useful in treating breast cancer.
There are a number of cancer vaccines in development for breast cancer, including but not limited toLapuleucel-T (Dendreon),AE-37 (Antigen Express), and Stimuvax (Merck KgA). While these development candidates are aimed at a number of different targets, andAE-37 has published data in the HER2 breast cancer patient population, there is no guarantee that any of the these compounds will not in the future be indicated for treatment oflow-to-intermediate HER2 breast cancer patients and become directly competitive with NeuVax.
A number of chemotherapeutic agents have demonstrated activity in gynecological carcinomas (ovarian and endometrial), particularly platinum-based regimens. New chemotherapy agents are being evaluated including trabectedin (Yondelis) and belotecan, as well as targeted agents such as bevacizumab (Avastin) and pazopanib (Votrient). Monoclonal antibodies are also being developed including farletuzumab and catumaxomab. We are not aware of any of these agents being evaluated in the adjuvant setting whereGALE-301 is being considered for further development. TPIV200 (TapImmune) is in development targeting FBP in ovarian cancer.
Many of our competitors, either alone or with their strategic partners, have substantially greater financial, technical and human resources than we do, and experience in obtaining FDA and other regulatory approvals of treatments and commercializing those treatments. Accordingly, our competitors may be more successful than us in obtaining approval for cancer immunotherapy products and achieving widespread market acceptance. Our competitors’ treatments may be more effectively marketed and sold than any products we may commercialize, thus causing limited market share before we can recover the expenses of developing and commercializing of our cancer immunotherapy product candidate.
Mergers and acquisitions in the biotechnology and pharmaceutical industries may result in even more resources being concentrated among a smaller number of our competitors. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies. These activities may lead to consolidated efforts that allow for more rapid development of cancer immunotherapy product candidates.
These competitors also compete with us in recruiting and retaining qualified scientific and management personnel, the ability to work with specific clinical contract organizations due to conflict of interest, and also the conduct of trials in the ability to recruit clinical trial sites and subjects for our clinical trials.
We expect any products that we develop and commercialize to compete on the basis of, among other things, efficacy, safety, price and the availability of coverage and reimbursement from government and other third-party payors. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are viewed as safer, more convenient or less expensive than any products that we may develop. Our competitors also may obtain FDA or other regulatory approval for their products more rapidly than we may obtain approval for our current product candidates or any other future product candidate, which could result in our competitors establishing a strong market position before we are able to enter the market.
Government Regulation
The FDA and other regulatory authorities at federal, state, and local levels, as well as in foreign countries, extensively regulate, among other things, the research, development, testing, manufacture, quality control, import, export, safety, effectiveness, labeling, packaging, storage, distribution, record keeping, approval, advertising, promotion, marketing, post-approval monitoring, and post-approval reporting of biologics such as those we are developing. Along with third-party contractors, we will be required to navigate the various preclinical, clinical and commercial approval requirements of the governing regulatory agencies of the countries
30
in which we wish to conduct studies or seek approval or licensure of its current or future product candidates. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources.
The process required by the FDA before biologic product candidates may be marketed in the United States generally involves the following:
| | • | | completion of preclinical laboratory tests and animal studies performed in accordance with the FDA’s current Good Laboratory Practices, or GLP, regulation; |
| | • | | submission to the FDA of an IND, which must become effective before clinical trials may begin and must be updated annually or when significant changes are made; |
| | • | | approval by an independent Institutional Review Board, or IRB, or ethics committee at each clinical site before the trial is begun; |
| | • | | performance of adequate and well-controlled human clinical trials to establish the safety, purity and potency of the proposed biologic product candidate for its intended purpose; |
| | • | | preparation of and submission to the FDA of a Biologics License Application, or BLA, after completion of all pivotal clinical trials; |
| | • | | satisfactory completion of an FDA Advisory Committee review, if applicable; |
| | • | | a determination by the FDA within 60 days of its receipt of a BLA to file the application for review; |
| | • | | satisfactory completion of an FDApre-approval inspection of the manufacturing facility or facilities at which the proposed product is produced to assess compliance with current Good Manufacturing Practices, or cGMP, and to assure that the facilities, methods and controls are adequate to preserve the biological product’s continued safety, purity and potency, and of selected clinical investigations to assess compliance with current Good Clinical Practices, or cGCP; and |
| | • | | FDA review and approval of the BLA to permit commercial marketing of the product for particular indications for use in the United States, which must be updated annually when significant changes are made. |
The testing and approval process requires substantial time, effort and financial resources, and, or cGCP, we cannot be certain that any approvals for our current or future product candidates will be granted on a timely basis, if at all. Prior to beginning the first clinical trial with a product candidate, we must submit an IND to the FDA. An IND is a request for authorization from the FDA to administer an investigational new drug product to humans. The central focus of an IND submission is on the general investigational plan and the protocol(s) for clinical studies. The IND also includes results of animal and in vitro studies assessing the toxicology, pharmacokinetics, pharmacology, and pharmacodynamic characteristics of the product; chemistry, manufacturing, and controls information; and any available human data or literature to support the use of the investigational product. An IND must become effective before human clinical trials may begin. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the30-day time period, raises safety concerns or questions about the proposed clinical trial. In such a case, the IND may be placed on clinical hold and the IND sponsor and the FDA must resolve any outstanding concerns or questions before the clinical trial can begin. Submission of an IND therefore may or may not result in FDA authorization to begin a clinical trial.
Clinical trials involve the administration of the investigational product to human subjects under the supervision of qualified investigators in accordance with cGCP, which include the requirement that all research subjects provide their informed consent for their participation in any clinical trial. Clinical trials are conducted under protocols detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated. A separate submission to the existing IND must
31
be made for each successive clinical trial conducted during product development and for any subsequent protocol amendments. Furthermore, an IRB for each site proposing to conduct the clinical trial must review and approve the plan for any clinical trial and its informed consent form before the clinical trial begins at that site and must monitor the clinical trial until completed. Regulatory authorities, the IRB or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the subjects are being exposed to an unacceptable health risk or that the trial is unlikely to meet its stated objectives. Some studies also include oversight by an independent group of qualified experts organized by the clinical trial sponsor, known as DSMB, which provides authorization for whether or not a clinical trial may move forward at designated check points based on access to certain data from the clinical trial and may halt the clinical trial if it determines that there is an unacceptable safety risk for subjects or other grounds, such as no demonstration of efficacy. There are also requirements governing the reporting of ongoing clinical studies and clinical trial results to public registries.
For purposes of BLA approval, human clinical trials are typically conducted in three sequential phases that may overlap.
| | • | | Phase 1—The investigational product is initially introduced into healthy human subjects or patients with the target disease or condition. These studies are designed to test the safety, dosage tolerance, absorption, metabolism and distribution of the investigational product in humans, the side effects associated with increasing doses, and, if possible, to gain early evidence on effectiveness. |
| | • | | Phase 2—The investigational product is administered to a limited patient population with a specified disease or condition to evaluate the preliminary efficacy, optimal dosages and dosing schedule and to identify possible adverse side effects and safety risks. Multiple Phase 2 clinical trials may be conducted to obtain information prior to beginning larger and more expensive Phase 3 clinical trials. |
| | • | | Phase 3—The investigational product is administered to an expanded patient population to further evaluate dosage, to provide statistically significant evidence of clinical efficacy and to further test for safety, generally at multiple geographically dispersed clinical trial sites. These clinical trials are intended to establish the overall risk/benefit ratio of the investigational product and to provide an adequate basis for product approval. |
| | • | | Phase 4—In some cases, the FDA may require, or companies may voluntarily pursue, additional clinical trials after a product is approved to gain more information about the product. Theseso-called Phase 4 studies may be made a condition to approval of the BLA. |
Phase 1, Phase 2 and Phase 3 testing may not be completed successfully within a specified period, if at all, and there can be no assurance that the data collected will support FDA approval or licensure of the product. Concurrent with clinical trials, companies may complete additional animal studies and develop additional information about the biological characteristics of the product candidate and must finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, must develop methods for testing the identity, strength, quality and purity of the final product, or for biologics, the safety, purity and potency. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
BLA Submission and Review by the FDA
Assuming successful completion of all required testing in accordance with all applicable regulatory requirements, the results of product development, nonclinical studies and clinical trials are submitted to the FDA as part of a BLA requesting approval to market the product for one or more indications. The BLA must include all relevant data available from pertinent preclinical and clinical studies, including negative or ambiguous results as well as positive findings, together with detailed information relating to the product’s chemistry, manufacturing, controls, and proposed labeling, among other things. Data can come from company-sponsored clinical studies intended to
32
test the safety and effectiveness of a use of the product, or from a number of alternative sources, including studies initiated by investigators. The submission of a BLA requires payment of a substantial user fee to FDA, and the sponsor of an approved BLA is also subject to annual product and establishment user fees. These fees are typically increased annually. A waiver of user fees may be obtained under certain limited circumstances.
Once a BLA has been submitted, the FDA’s goal is to review the application within ten months after it accepts the application for filing, or, if the application relates to an unmet medical need in a serious or life-threatening indication, six months after the FDA accepts the application for filing. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA reviews a BLA to determine, among other things, whether a product is safe, pure and potent and the facility in which it is manufactured, processed, packed, or held meets standards designed to assure the product’s continued safety, purity and potency. The FDA may convene an advisory committee to provide clinical insight on application review questions. Before approving a BLA, the FDA will typically inspect the facility or facilities where the product is manufactured. The FDA will not approve an application unless it determines that the manufacturing processes and facilities are in compliance with cGMP requirements and adequate to assure consistent production of the product within required specifications. Additionally, before approving a BLA, the FDA will typically inspect one or more clinical sites to assure compliance with cGCP. If the FDA determines that the application, manufacturing process or manufacturing facilities are not acceptable, it will outline the deficiencies in the submission and often will request additional testing or information. Notwithstanding the submission of any requested additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval.
The testing and approval process requires substantial time, effort and financial resources, and each may take several years to complete. The FDA may not grant approval on a timely basis, or at all, and we may encounter difficulties or unanticipated costs in its efforts to secure necessary governmental approvals, which could delay or preclude us from marketing its products. After the FDA evaluates a BLA and conducts inspections of manufacturing facilities where the investigational product and/or its drug substance will be produced, the FDA may issue an approval letter or a Complete Response Letter. An approval letter authorizes commercial marketing of the product with specific prescribing information for specific indications. A Complete Response Letter indicates that the review cycle of the application is complete and the application is not ready for approval. A Complete Response Letter may request additional information or clarification. The FDA may delay or refuse approval of a BLA if applicable regulatory criteria are not satisfied, require additional testing or information and/or require post-marketing testing and surveillance to monitor safety or efficacy of a product.
If regulatory approval of a product is granted, such approval may entail limitations on the indicated uses for which such product may be marketed. For example, the FDA may approve the BLA with a Risk Evaluation and Mitigation Strategy, or REMS, plan to mitigate risks, which could include medication guides, physician communication plans, or elements to assure safe use, such as restricted distribution methods, patient registries and other risk minimization tools. The FDA also may condition approval on, among other things, changes to proposed labeling or the development of adequate controls and specifications. Once approved, the FDA may withdraw the product approval if compliance withpre- and post-marketing regulatory standards is not maintained or if problems occur after the product reaches the marketplace. The FDA may require one or more Phase 4 post-market studies and surveillance to further assess and monitor the product’s safety and effectiveness after commercialization and may limit further marketing of the product based on the results of these post-marketing studies. In addition, new government requirements, including those resulting from new legislation, may be established, or the FDA’s policies may change, which could delay or prevent regulatory approval of our products under development.
A sponsor may seek approval of its product candidate under programs designed to accelerate FDA’s review and approval of new drugs and biological products that meet certain criteria. Specifically, new drugs and biological products are eligible for Fast Track designation if they are intended to treat a serious or life-threatening condition and demonstrate the potential to address unmet medical needs for the condition. For a product candidate with
33
Fast Track designation, the FDA may consider sections of the BLA for review on a rolling basis before the complete application is submitted if relevant criteria are met. A Fast Track designated product candidate may also qualify for priority review, under which the FDA sets the target date for FDA action on the BLA at six months after the FDA accepts the application for filing. Priority review is granted when there is evidence that the proposed product would be a significant improvement in the safety or effectiveness of the treatment, diagnosis, or prevention of a serious condition. If criteria are not met for priority review, the application is subject to the standard FDA review period of 10 months after FDA accepts the application for filing. Priority review designation does not change the scientific/medical standard for approval or the quality of evidence necessary to support approval. We have obtained Fast Track designation for GPS in AML, MPM, and NeuVax.
Under the Accelerated Approval program, the FDA may approve a BLA on the basis of either a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. Post-marketing studies or completion of ongoing studies after marketing approval are generally required to verify the biologic’s clinical benefit in relationship to the surrogate endpoint or ultimate outcome in relationship to the clinical benefit.
In addition, a sponsor may seek FDA designation of its product candidate as a Breakthrough Therapy, if the product candidate is intended, alone or in combination with one or more other drugs or biologics, to treat a serious or life-threatening disease or condition and preliminary clinical evidence indicates that the therapy may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. If the FDA designates a breakthrough therapy, it may take actions appropriate to expedite the development and review of the application. Breakthrough designation also allows the sponsor to file sections of the BLA for review on a rolling basis. we plan to seek designation as a breakthrough therapy for GPS in one or more indications.
Fast Track, Priority Review and Breakthrough Therapy designations do not change the standards for approval but may expedite the development or approval process.
Orphan Drugs
Under the Orphan Drug Act, the FDA may grant Orphan Drug Product designation to a drug or biologic intended to treat a rare disease or condition, defined as a disease or condition with a patient population of fewer than 200,000 individuals in the United States, or a patient population greater than 200,000 individuals in the United States and when there is no reasonable expectation that the cost of developing and making available the drug or biologic in the United States will be recovered from sales in the United States for that drug or biologic. Orphan Drug Product designation must be requested before submitting a BLA. After the FDA grants Orphan Drug Product designation, the generic identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA.
If a product that has Orphan Drug Product designation subsequently receives the first FDA approval for a particular active ingredient for the disease for which it has such designation, the product is entitled to Orphan Drug Product exclusivity, which means that the FDA may not approve any other applications, including a full BLA, to market the same biologic for the same indication for seven years, except in limited circumstances, such as a showing of clinical superiority to the product with Orphan Drug exclusivity or if FDA finds that the holder of the Orphan Drug exclusivity has not shown that it can assure the availability of sufficient quantities of the Orphan Drug to meet the needs of patients with the disease or condition for which the drug was designated. Orphan Drug exclusivity does not prevent the FDA from approving a different drug or biologic for the same disease or condition, or the same drug or biologic for a different disease or condition. Among the other benefits of Orphan Drug Product designation are tax credits for certain research and a waiver of the BLA application user fee.
34
A drug with Orphan Drug Product designation may not receive Orphan Drug exclusivity if it is approved for a use that is broader than the indication for which it received Orphan Drug Product designation. In addition, Orphan Drug exclusive marketing rights in the United States may be lost if the FDA later determines that the request for designation was materially defective or if the manufacturer is unable to assure sufficient quantities of the product to meet the needs of patients with the rare disease or condition. We plan to seek Orphan Drug Product designation for GPS in specific orphan indications in which there is a medically plausible basis for the use of GPS for such indications, if applicable. We have obtained Orphan Drug Product designation for GPS in AML, MPM and MM and forGALE-301 and one forGALE-302.
Post-Approval Requirements
Any products manufactured or distributed by us pursuant to FDA approvals are subject to pervasive and continuing regulation by the FDA, including, among other things, requirements relating to record-keeping, reporting of adverse experiences, periodic reporting, product sampling and distribution, and advertising and promotion of the product. After approval, most changes to the approved product, such as adding new indications or other labeling claims, are subject to prior FDA review and approval. There also are continuing, annual program user fee requirements for any marketed products, as well as new application fees for supplemental applications with clinical data. Biologic manufacturers and their subcontractors are required to register their establishments with the FDA and certain state agencies, and are subject to periodic unannounced inspections by the FDA and certain state agencies for compliance with cGMP, which impose certain procedural and documentation requirements upon our third-party manufacturers and us. Changes to the manufacturing process are strictly regulated, and, depending on the significance of the change, may require prior FDA approval before being implemented. FDA regulations also require investigation and correction of any deviations from cGMP and impose reporting requirements upon us and any third-party manufacturers that we may decide to use. Accordingly, manufacturers must continue to expend time, money and effort in the area of production and quality control to maintain compliance with cGMP and other aspects of regulatory compliance. We cannot be certain that we or our present or future suppliers will be able to comply with the cGMP regulations and other FDA regulatory requirements. If we or our present or future suppliers are not able to comply with these requirements, the FDA may, among other things, halt our clinical trials, require us to recall a product from distribution, or withdraw approval of the BLA.
We rely, and expect to continue to rely, on third parties for the production of clinical quantities of our product candidates and we expect to rely in the future on third parties for the production of commercial quantities. Future FDA and state inspections may identify compliance issues at the facilities of our contract manufacturers that may disrupt production, distribution, or require substantial resources to correct. In addition, discovery of previously unknown problems with a product or the failure to comply with applicable requirements may result in restrictions on a product, manufacturer or holder of an approved BLA, including withdrawal or recall of the product from the market or other voluntary,FDA-initiated or judicial action that could delay or prohibit further marketing. The FDA may withdraw approval if compliance with regulatory requirements and standards is not maintained or if problems occur after the product reaches the market. Later discovery of previously unknown problems with a product, including adverse events of unanticipated severity or frequency, or with manufacturing processes, or failure to comply with regulatory requirements, may result in revisions to the approved labeling to add new safety information; imposition of post-market studies or clinical studies to assess new safety risks; or imposition of distribution restrictions or other restrictions under a REMS program. Other potential consequences include, among other things:
| | • | | restrictions on the marketing or manufacturing of the product, complete withdrawal of the product from the market or product recalls; |
| | • | | fines, warning letters or holds on post-approval clinical studies; |
| | • | | refusal of the FDA to approve pending applications or supplements to approved applications, or suspension or revocation of product license approvals; |
35
| | • | | product seizure or detention, or refusal to permit the import or export of products; or |
| | • | | injunctions or the imposition of civil or criminal penalties. |
The FDA closely regulates the marketing, labeling, advertising and promotion of biologics. A company can make only those claims relating to safety and efficacy, purity and potency that are approved by the FDA and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion ofoff-label uses. Failure to comply with these requirements can result in, among other things, adverse publicity, warning letters, corrective advertising and potential civil and criminal penalties. Physicians may prescribe legally available products for uses that are not described in the product’s labeling and that differ from those tested by us and approved by the FDA. Suchoff-label uses are common across medical specialties. Physicians may believe that suchoff-label uses are the best treatment for many patients in varied circumstances. The FDA does not regulate the behavior of physicians in their choice of treatments. The FDA does, however, restrict manufacturers’ communications on the subject ofoff-label use of their products.
Other Healthcare Laws and Compliance Requirements
Our sales, promotion, medical education and other activities, including the research and development of our product candidates, will be subject to regulation by numerous regulatory and law enforcement authorities in the United States. Our business activities must comply with the federal Anti-Kickback Statute, the Foreign Corrupt Practices Act, the federal False Claims Act the Veterans Health Care Act, physician payment transparency laws, privacy laws, security laws, and additional state laws similar to the foregoing.
The federal Anti-Kickback Statute prohibits, among other things, the offer, receipt, or payment of remuneration in exchange for or to induce the referral of patients or the use of products or services that would be paid for in whole or part by Medicare, Medicaid or other federal health care programs. Remuneration has been broadly defined to include anything of value, including cash, improper discounts, and free or reduced price items and services. The government has enforced the federal Anti-Kickback Statute to reach large settlements with healthcare companies based on sham research or consulting and other financial arrangements with physicians. Further, a person or entity does not need to have actual knowledge of the statute or specific intent to violate it to have committed a violation. In addition, a claim for government payment including items or services resulting from a violation of the federal Anti-Kickback Statute constitutes a false or fraudulent claim for purposes of the federal False Claims Act. Many states have similar laws that apply to their state health care programs as well as private payors.
The federal False Claims Act, prohibits any individual or entity from, among other things, presenting or causing to be presented false or fraudulent claims for payment by a federal health care program. The federal False Claims Act has been used to prosecute persons submitting claims for payment that are inaccurate or fraudulent, that are for services not provided as claimed, or for services that are not medically necessary. Actions under the federal False Claims Act may be brought by the Attorney General or as a qui tam action by a private individual in the name of the government. The federal government is using the federal False Claims Act, and the accompanying threat of significant liability, in its investigation and prosecution of pharmaceutical and biotechnology companies throughout the country, for example, in connection with the promotion of products for unapproved uses and other sales and marketing practices. The government has obtained multi-million and multibillion dollar settlements under the federal False Claims Act in addition to individual criminal convictions under applicable criminal statutes. In addition, companies have been forced to implement extensive corrective action plans, and have often become subject to consent decrees or corporate integrity agreements, restricting the manner in which they conduct their business. Many states have their own false claims laws that apply to claims submitted for payment of state government funds.
The federal Health Insurance Portability and Accountability Act of 1996, or HIPAA, created additional federal criminal statutes that prohibit, among other things, knowingly and willfully executing a scheme to defraud any
36
healthcare benefit program, including private third-party payors and knowingly and willfully falsifying, concealing or covering up a material fact or making any materially false, fictitious or fraudulent statement in connection with the delivery of or payment for healthcare benefits, items or services. Given the significant size of actual and potential settlements, it is expected that the government will continue to devote substantial resources to investigating healthcare providers’ and manufacturers’ compliance with applicable fraud and abuse laws.
In addition, there has been a recent trend of increased federal and state regulation of payments made to physicians and other healthcare providers. The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act, or collectively, the Affordable Care Act, among other things, imposed new reporting requirements on drug manufacturers for payments or other transfers of value made by them to physicians and teaching hospitals, as well as ownership and investment interests held by physicians and their immediate family members. Failure to submit required information may result in significant civil monetary penalties. Certain states also mandate implementation of commercial compliance programs, impose restrictions on drug manufacturer marketing practices and/or require the tracking and reporting of gifts, compensation and other remuneration to physicians and other healthcare professionals.
We may also be subject to data privacy and security regulation by both the federal government and the states in which we conducts our business. HIPAA, as amended by the Health Information Technology for Economic and Clinical Health Act, or HITECH, and their respective implementing regulations, imposes specified requirements relating to the privacy, security and transmission of individually identifiable health information on entities subject to the law, such as certain healthcare providers, health plans, and healthcare clearinghouses, known as covered entities, and their respective business associates that perform services for them that involve the creation, use, maintenance or disclosure of, individually identifiable health information. Among other things, HITECH makes HIPAA’s privacy and security standards directly applicable to business associates. HITECH also increased the civil and criminal penalties that may be imposed against covered entities, business associates and possibly other persons, and gave state attorneys general new authority to file civil actions for damages or injunctions in federal courts to enforce the federal HIPAA laws and seek attorney’s fees and costs associated with pursuing federal civil actions. In addition, state laws govern the privacy and security of health information in certain circumstances, many of which differ from each other in significant ways and may not have the same effect.
If our operations are found to be in violation of any of such laws or any other governmental regulations that apply to it, we may be subject to significant penalties, including, without limitation, civil, criminal and administrative penalties, damages, fines, the curtailment or restructuring of our operations, exclusion from participation in federal and state healthcare programs, integrity oversight and reporting obligations, and imprisonment, any of which could adversely affect our ability to operate our business and our financial results.
Also, the U.S. Foreign Corrupt Practices Act and similar worldwide anti-bribery laws generally prohibit companies and their intermediaries from making improper payments to foreign officials for the purpose of obtaining or retaining business. We cannot assure you that our internal control policies and procedures will protect us from reckless or negligent acts committed by our employees, future distributors, partners, collaborators or agents. Violations of these laws, or allegations of such violations, could result in fines, penalties or prosecution and have a negative impact on our business, results of operations and reputation.
Coverage and Reimbursement
Sales of pharmaceutical products depend significantly on the availability of third-party coverage and reimbursement. Third-party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. Although we currently believe that third-party payors will provide coverage and reimbursement for our product candidates, if approved, these third-party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. We
37
may need to conduct expensive clinical studies to demonstrate the comparative cost-effectiveness of its products. GPS for the indications that we develop may not be considered cost-effective. It is time consuming and expensive for us to seek coverage and reimbursement from third-party payors. Additionally, no uniform policy for coverage and reimbursement exists in the United States, and coverage and reimbursement can differ significantly from payor to payor. Third-party payors often rely upon Medicare coverage policy and payment limitations in setting their own reimbursement rates, but also have their own methods and approval process apart from Medicare determinations. Moreover, a payor’s decision to provide coverage for a drug product does not imply that an adequate reimbursement rate will be approved. Reimbursement may not be available or sufficient to allow us to sell our products on a competitive and profitable basis.
Healthcare Reform
The United States and some foreign jurisdictions are considering or have enacted a number of legislative and regulatory proposals to change the healthcare system in ways that could affect our ability to sell our products profitably. Among policy makers and payors in the United States and elsewhere, there is significant interest in promoting changes in healthcare systems with the stated goals of containing healthcare costs, improving quality and/or expanding access. In the United States, the pharmaceutical industry has been a particular focus of these efforts and has been significantly affected by major legislative initiatives.
By way of example, in March 2010, the Affordable Care Act was signed into law which substantially changed the way healthcare is financed by both governmental and private insurers. The Affordable Care Act, among other things, (i) subjected therapeutic biologics to potential competition by lower-cost biosimilars by creating a licensure framework for follow on biologic products, (ii) proscribed a new methodology by which rebates owed by manufacturers under the Medicaid Drug Rebate Program are calculated for drugs and therapeutic biologics that are inhaled, infused, instilled, implanted or injected, (iii) increased the minimum Medicaid rebates owed by manufacturers under the Medicaid Drug Rebate Program and extended the rebate program to individuals enrolled in Medicaid managed care organizations, (iv) established annual fees and taxes on manufacturers of certain branded prescription drugs and therapeutic biologics, (v) established a new Medicare Part D coverage gap discount program, in which manufacturers must agree to offer 50%point-of-sale discounts off negotiated prices of applicable brand drugs and therapeutic biologics to eligible beneficiaries during their coverage gap period, as a condition for the manufacturer’s outpatient drugs and therapeutic biologics to be covered under Medicare Part D, (vi) expanded eligibility criteria for Medicaid programs by, among other things, allowing states to offer Medicaid coverage to additional individuals and by adding new mandatory eligibility categories for individuals with income at or below 133% of the federal poverty level, thereby potentially increasing manufacturers’ Medicaid rebate liability, (vii) expanded the entities eligible for discounts under the Public Health program (viii) created a new Patient-Centered Outcomes Research Institute to oversee, identify priorities in, and conduct comparative clinical effectiveness research, along with funding for such research, and (ix) established a Center for Medicare Innovation at the Centers for Medicare & Medicaid Services, or CMS, to test innovative payment and service delivery models to lower Medicare and Medicaid spending, potentially including prescription drug spending .
Some of the provisions of the Affordable Care Act have yet to be implemented, and there have been judicial and Congressional challenges to certain aspects of the Affordable Care Act, as well as recent efforts by the Trump administration to repeal or replace certain aspects of the Affordable Care Act. Since January 2017, President Trump has signed two Executive Orders and other directives designed to delay the implementation of certain provisions of the Affordable Care Act or otherwise circumvent some of the requirements for health insurance mandated by the Affordable Care Act. Concurrently, Congress has considered legislation that would repeal or repeal and replace all or part of the Affordable Care Act. While Congress has not passed comprehensive repeal legislation, two bills affecting the implementation of certain taxes under the Affordable Care Act have been signed into law. The Tax Act includes a provision repealing, effective January 1, 2019, thetax-based shared responsibility payment imposed by the Affordable Care Act on certain individuals who fail to maintain qualifying health coverage for all or part of a year that is commonly referred to as the “individual mandate”. Additionally, on January 22, 2018, President Trump signed a continuing resolution on appropriations for fiscal
38
year 2018 that delayed the implementation of certain Affordable CareAct-mandated fees, including theso-called “Cadillac” tax on certain high cost employer-sponsored insurance plans, the annual fee imposed on certain health insurance providers based on market share, and the medical device excise tax onnon-exempt medical devices. Further, the Bipartisan Budget Act of 2018, or the BBA, among other things, amends the Affordable Care Act, effective January 1, 2019, to increase from 50 percent to 70 percent thepoint-of-sale discount that is owed by pharmaceutical manufacturers who participate in Medicare Part D and to close the coverage gap in most Medicare drug plans, commonly referred to as the “donut hole”.
In addition, other legislative changes have been proposed and adopted since the Affordable Care Act was enacted. On August 2, 2011, the Budget Control Act of 2011, was signed into law, which, among other things, created the Joint Select Committee on Deficit Reduction to recommend to Congress proposals for spending reductions. The Joint Select Committee did not achieve a targeted deficit reduction of at least $1.2 trillion for the years 2013 through 2021, triggering the legislation’s automatic reduction to several government programs. This includes reductions to Medicare payments to providers of up to 2% per fiscal year, starting April 1, 2013, and, due to subsequent legislative amendments to the statute, including the BBA, will remain in effect through 2027 unless additional congressional action is taken. On January 2, 2013, the American Taxpayer Relief Act of 2012 was signed into law, which, among other things, further reduced Medicare payments to several providers, including hospitals and cancer treatment centers, and increased the statute of limitations period for the government to recover overpayments to providers from three to five years.
Recently there has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which has resulted in several Congressional inquiries and proposed and enacted federal and state legislation designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for drug products. At the federal level, the Trump administration’s budget proposal for fiscal year 2019 contains further drug price control measures that could be enacted during the 2019 budget process or in other future legislation, including, for example, measures to permit Medicare Part D plans to negotiate the price of certain drugs under Medicare Part B, to allow some states to negotiate drug prices under Medicaid, and to eliminate cost sharing for generic drugs forlow-income patients. Additionally, on May 11, 2018, President Trump laid out his administration’s “Blueprint” to reduce the cost of prescription drugs while preserving innovation and cures. HHS has already started the process of soliciting feedback on some of these measures and, at the same, is immediately implementing others under its existing authority. Although some of these, and other, proposals will require authorization through additional legislation to become effective, Congress and the Trump administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs. At the state level, legislatures are increasingly passing legislation and implementing regulations designed to control pharmaceutical and biological product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing. We expect that additional state and federal healthcare reform measures will be adopted in the future, any of which could limit the amounts that federal and state governments will pay for healthcare products and services, which could result in reduced demand for our product candidates or additional pricing pressures.
We expect that the Affordable Care Act, as well as other healthcare reform measures that may be adopted in the future, may result in more rigorous coverage criteria and lower reimbursement, and in additional downward pressure on the price that we receive for any approved product. Any reduction in reimbursement from Medicare or other government-funded programs may result in a similar reduction in payments from private payors. The implementation of cost containment measures or other healthcare reforms may prevent us from being able to generate revenue, attain profitability or commercialize our product candidates.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of its products to the extent we choose to develop or sell any
39
products outside of the United States. The approval process varies from country to country and the time may be longer or shorter than that required to obtain FDA approval. Additionally, the requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country. In the European Union, member states require both regulatory clearances by the national competent authority and a favorable ethics committee opinion prior to the commencement of a clinical trial. Under the European Union regulatory systems, marketing authorization applications may be submitted under either a centralized or decentralized procedure. The centralized procedure provides for the grant of a single marketing authorization that is valid for all European Union member states. It is compulsory for medicines produced by certain biotechnological processes. Because our products are produced in that way, we would be subject to the centralized procedure. Under the centralized procedure, pharmaceutical companies submit a single marketing authorization application to the EMA. Once granted by the European Commission, a centralized marketing authorization is valid in all European Union member states, as well as the European Economic Area countries Iceland, Liechtenstein and Norway. By law, a company can only start to market a medicine once it has received a marketing authorization.
Employees
As of June 30, 2018, we had 11 full-time employees. two of our employees have Ph.D. or M.D. degrees and one of our employees is engaged in research and development activities. None of our employees are represented by labor unions or covered by collective bargaining agreements. We consider our relationship with our employees to be good.
Facilities
We lease our facility containing our headquarters, which consists of approximately 2,033 square feet of office space located at 315 Madison Avenue, New York, New York. Our lease will terminate on July 31, 2018. We have entered into a new lease commencing on August 1, 2018 for approximately 3,700 square feet of office space located at 15 West 38th Street, New York, New York with a term through July 31, 2020. We believe that our facilities are adequate for our current needs and that suitable additional or substitute space would be available if needed.
Legal Proceedings
Our predecessor company, Galena was involved in multiple legal proceedings and administrative actions, including stockholder class actions, both state and federal, some of which are ongoing and to which we are now subject as a result of the Merger. They are as follows:
On December 16, 2015, we received a subpoena issued by the U.S. Attorney’s Office for the District of New Jersey, or USAO NJ, requesting the production of a broad range of documents pertaining to our marketing and promotional practices for Abstral, which we had sold to a third party in the fourth quarter of 2015. In September 2017, the Department of Justice announced that we were to pay more than $7.55 million to resolve allegations under the civil False Claims Act that we paid kickbacks to doctors to induce them to prescribe our fentanyl-based drug Abstral. On December 29, 2017, we fully paid the civil payment to resolve those allegations. Pursuant to a separate settlement agreement, we paid $0.2 million in attorneys’ fees.
On February 13, 2017, putative shareholder securities class action complaints were filed in federal court alleging, among other things, that we and certain of our former officers and directors and current employee failed to disclose that our promotional practices for Abstral (fentanyl) sublingual tablets were allegedly improper and that we may be subject to civil and criminal liability, and that these alleged failures rendered our statements about our business misleading. The actions were consolidated, a lead plaintiff was named by the court and an amended complaint was filed. We filed a motion to dismiss the amended complaint and the briefing was completed in April of 2018. The Court has taken the matter under advisement. It is not known when the Court will issue a ruling in this matter.
40
In March 2017, a derivative complaint was filed in U.S. District Court for the District of New Jersey against our former directors and us, as a nominal defendant. In July 2017, a derivative complaint was filed in California state court against our former directors and us, as a nominal defendant. In January 2018, a derivative complaint was filed in the U.S. District Court for the District of New Jersey against our former directors, officers and employees, and us as a nominal defendant. In June 2018, a derivative complaint was filed in U.S. District Court for the Northern District of California against our current and former directors, and us as a nominal defendant. These complaints purport to assert derivative claims for breach of fiduciary duty on our behalf against our former directors and, in certain of the complaints, our current directors and our former officers and former employees, based on substantially similar facts as alleged in the putative shareholder securities class action complaints mentioned above. The July 2017 lawsuit is currently stayed pending resolution of motions to dismiss in the referenced securities class action. The plaintiffs in the March 2017 and January 2018 lawsuits have sought to consolidate those actions; a motion to consolidate the proceedings is currently pending. It is anticipated that we and the individual defendants will respond to the June 2018 complaint through an appropriate pleading or motion and/or seek a stay of that proceeding pending further developments in the securities litigation.
In April 2017, a putative stockholder class action was filed in Delaware state court seeking relief under Section 225 of the DGCL and alleging breaches of fiduciary duties by our former board of directors and former interim chief executive officer regarding the proposals to amend Galena’s certificate of incorporation to increase the amount of authorized shares of common stock and effectuate a reverse stock split at the July 2016 and October 2016 stockholder meetings, respectively. On June 2, 2017, an amended verified complaint was filed along with a motion to expedite the proceedings. On June 5, 2017, Galena filed a verified petition under Section 205 of the DGCL and a motion to expedite the proceedings. On June 8, 2017, the Court denied a request by the plaintiff to schedule a preliminary injunction motion and ordered a prompt trial on both the plaintiff and Galena’s claims. On June 20, 2017, the Court consolidated the claims intoIn re Galena Biopharma, Inc., C. A. No.2017-0423-JTL. On July 10, 2017, the Court ordered that the trial of the claims be held on August 28, 30 and 31, 2017. On July 24, 2017, we entered into a binding settlement term sheet involving the payment of $50,000 in cash and $1,250,000 in unrestricted shares of our common stock. The Court enforced the settlement term sheet on November 30, 2017, over the objection of the plaintiff. On December 8, 2017, the Court set the hearing on the settlement for March 15, 2018. On December 11, 2017, the Court also granted an order validating the ratification votes at the special stockholder meeting held on July 6, 2017 and the certificate of amendments filed by the Company for the increase in authorized shares in 2011, 2013, 2015, and 2016 as well as for the reverse stock split in 2016. On February 22, 2018, the plaintiff filed his brief in support of the settlement as well as his request for attorneys’ fees and an incentive award. On February 28, 2018, the former directors and former interim chief executive officer requested the Court continue the date of the hearing to approve the settlement as we were working with the staff of the SEC to obtain theno-action letter required by the binding settlement term sheet. On March 15, 2018, the Court ruled in favor of a continuance and continued the settlement hearing for 90 days. On June 14, 2018, the Court entered an Order and Final Judgment approving the settlement and awarding attorneys’ fees to the plaintiff. On June 21, 2018, we issued 228,672 shares of our common stock and paid the balance of the cash portion of the settlement, which is full settlement of this litigation.
On April 10, 2017, the SEC issued a cease and desist order against us and the former chief executive officer, or CEO, Mark Ahn, requiring each of them to cease and desist from any future violations of Sections 5(a), 5(b), 5(c), 17(a), and 17(b) of the Securities Act of 1933, as amended, or the Securities Act, and Section 10(b), 13(a), and 13(b)(2)(A) of the Exchange Act, and various rules thereunder, which refer to as the SEC Order. Based upon the order, we made a $0.2 million penalty payment as well as a payment of approximately $0.75 million, which was the indemnification payment of our former CEO for the disgorgement and prejudgment interest payment that he was required to pay by the order. We made such indemnification payment after a special committee of the board of directors determined that we were required under Delaware law to indemnify our former CEO for the disgorgement and prejudgment interest payment. The former CEO also made a penalty payment of $0.6 million. As a result of the SEC Order, we were not able to use certain exemptions from registration under the federal securities laws, including Regulation A and Regulation D, as we were subject to a “bad actor” disqualification.
41
The SEC waived this disqualification on June 6, 2018. In addition, we are an “ineligible issuer” as the term is defined under Rule 405 promulgated under the Securities Act.
On or about April 9, 2018, JGB filed a lawsuit in the U.S. District Court for the Southern District of New York captionedJGB (Cayman) Newton, Ltd. v. Sellas Life Sciences Group, Inc., et al., Case1:18-cv-3095 (DLC), or the JGB Action. The complaint in the JGB Action asserts claims under state law and federal securities law against us, our Chief Executive Officer, Angelos M. Stergiou, M.D., ScD H.C, and our former Interim Chief Financial Officer, Aleksey N. Krylov (Mr. Krylov together with us and Dr. Stergiou, the Defendants). The complaint in the JGB Action alleges, among other things, that we breached a contractual obligation to deliver certain shares of our common stock to JGB and that, in the course of negotiations related to the senior secured debenture agreement, the Defendants fraudulently failed to disclose to JGB certain information regarding positive clinical trial results that was not then public. On or about May 2, 2018, JGB filed an amended complaint, adding as defendants Jane Wasman, Stephen Ghiglieri, David Scheinberg, M.D., Robert Van Nostrand and John Varian, each of whom is a member of our Board of Directors, or the Director Defendants. The amended complaint, in addition to the allegations in the original complaint, alleges, among other things, that the Director Defendants are liable as “control persons” for the purposes of the purported securities laws violations committed by us, Dr. Stergiou and Mr. Krylov. According to the amended complaint, JGB seeks to receive an unspecified number of shares of our common stock, a declaratory judgment in respect of the dispute underlying the litigation, damages, and costs and expenses incurred in the JGB action, among other things. On May 18, 2018, we filed counterclaims and an answer to the amended complaint filed by JGB. On May 25, 2018, we filed amended counterclaims and answer to the amended complaint. We are seeking, among other things, a dismissal of all claims, a declaratory judgment regarding the underlying contractual dispute between the parties, a declaratory judgment regarding a breach of the senior secured debenture agreement by JGB relating to JGB’s failure to release certain cash collateral owed to us for redemptions by JGB and interest on the collateral, damages, restitution and fees. On June 18, 2018, we filed a motion for judgment on the pleadings and a motion to dismiss the amended complaint. On July 6, 2018, JGB filed a motion for partial judgment on the pleadings and motion to dismiss all of our counterclaims. We intend to continue to defend vigorously against the claims in the JGB Action, all of which we continue to dispute.
42