Exhibit 99.2
MANAGEMENT’S DISCUSSION AND ANALYSIS
The following information should be read in conjunction with the Company’s audited consolidated financial statements for the year ended June 30, 2008 and the related notes, which are prepared in accordance with Canadian generally accepted accounting principles. This Management’s Discussion and Analysis (“MD&A”) provides a review of the performance of the Company for the year ended June 30, 2008 as compared to the year ended June 30, 2007. Material differences between Canadian and U.S. generally accepted accounting principles are described in note 24 to the financial statements for the year ended June 30, 2008. This MD&A includes financial information derived from the annual audited consolidated financial statements and from the unaudited interim consolidated financial statements. This review was performed by management with information available as of September 15, 2008.
Where “we”, “us”, “our”, “Transition” or the “Company” is used, it is referring to Transition Therapeutics Inc. and its wholly-owned subsidiaries, unless otherwise indicated. All amounts are in Canadian dollars, unless otherwise indicated.
In July 2007, the Company completed the consolidation of its issued and outstanding common shares on the basis of one (1) post-consolidation common share for every nine (9) pre-consolidation common shares. As a result of this consolidation, the number of common shares, warrants and options, related exercise prices and basic and diluted loss per common share for all periods prior to July 9, 2007 have been retroactively adjusted to reflect the consolidation.
Additional information relating to the Company, including the Company’s most recently filed Annual Information Form, can be found on SEDAR at www.sedar.com.
CAUTION REGARDING FORWARD-LOOKING STATEMENTS
This MD&A contains certain forward-looking statements relating, but not limited to operations, anticipated financial performance, business prospects and strategies. This forward-looking information is subject to various risks and uncertainties, including those discussed below, that could cause actual results and experience to differ materially from the anticipated results or other expectations expressed. Readers are cautioned not to place undue reliance on this forward-looking information, which is provided as of the date of this MD&A unless otherwise stated, and the Company will not undertake any obligation to publicly update or revise any forward-looking information, whether as a result of new information, future events, or otherwise, except as required by securities laws.
Forward-looking information typically contains statements with words such as “anticipate”, “believe”, “expect”, “plan”, “estimate”, “intend”, “may” or similar words suggesting future outcomes or statements regarding an outlook on the estimated amounts and timing of capital expenditures, anticipated future debt levels and partnership revenues or other revenues or other expectations, beliefs, plans, objectives, assumptions, intentions or statements about future events or performance.
Factors which could cause future outcomes to differ materially from those set forth in the forward-looking information include, but are not limited to: (i) obtaining sufficient and suitable financing to support operations, clinical trials and commercialization of products, (ii) capitalizing on partnering and acquisition opportunities; (iii) clinical trial timing and results; (iv) adequately protecting proprietary information and technology from competitors; (v) regulatory approvals; (vi) successfully competing in the targeted markets; and (vii) maintaining third party relationships, including key personnel, and key collaborators.
By its nature, forward-looking information involves numerous assumptions, inherent risks and uncertainties, both general and specific, that contribute to the possibility that the predictions, forecasts, projections or other forward-looking statements will not occur. Prospective investors should carefully consider the information contained under the heading “RISKS AND UNCERTAINTIES” in this MD&A and all other information included in or incorporated by reference in this MD&A before making investment decisions with regard to the securities of the Company.
OVERVIEW
Transition is a product-focused biopharmaceutical company, developing novel therapeutics for disease indications with large markets. The Company’s lead products are: ELND005 (AZD-103) for the treatment of Alzheimer’s disease and TT-223 for the treatment of diabetes. The lead product for diabetes is Transition’s gastrin analogue, TT-223, formerly known as “G1”. Going forward GLP1-I.N.T.Ô will be referred to as TT-223 in combination with GLP1 analogues and E1-I.N.T.Ô will be referred to as TT-223 in combination with epidermal growth factor analogue. Transition also has an emerging pipeline of pre-clinical drug candidates acquired externally or developed internally using its proprietary drug discovery engine.
General Risk Factors for the Biotechnology Industry
Prospects for companies in the biopharmaceutical industry generally may be regarded as uncertain given the nature of the industry and, accordingly, investments in such companies should be regarded as highly speculative. It is not possible to predict, based upon studies in animals and early clinical data, whether a new therapeutic or device will prove to be safe and effective in humans or whether it will ultimately receive regulatory approval. In addition, there is no assurance that adequate funds or relationships required to continue product development such as those with employees, collaborators, or other third parties will be available and sustained.
If a product is ultimately approved for sale, there is no assurance that it will ever result in significant revenues or profitable operations. There are many factors such as competition, patent protection and the regulatory environment that can influence a product’s profitability potential.
In addition, due to the speculative nature of this industry, market prices for securities of biotechnology companies may be highly volatile and subject to significant fluctuation
2
and may not necessarily be related to the operating or other performances of such companies.
Recent Achievements
During fiscal 2008 and up to the date of this MD&A, the Company achieved the following significant milestones:
ELND005 (AZD-103) — Alzheimer’s Disease:
| • | | On January 22, 2008, the Company received a US$5,000,000 milestone payment from Elan. The milestone payment was triggered by the initiation of a Phase II clinical study of Alzheimer’s disease drug candidate, ELND005 (AZD-103) on December 21, 2007; |
| |
| • | | On December 21, 2007, Elan and Transition Dose First Patient in Phase II Clinical Study of ELND005 (AZD-103) in Alzheimer’s Disease.The study is a randomized, double-blind, placebo-controlled, dose-ranging, safety and efficacy study in approximately 340 patients with mild to moderate Alzheimer’s disease. Approximately 65 sites in North America are expected to participate in the program. The study will evaluate both cognitive and functional endpoints and each patient’s participation is planned to last approximately 18 months; |
| |
| • | | On October 26, 2007, the Company Received the Remaining US$7,500,000 Upfront Payment from Elan.The receipt of US$7,500,000 represents the second half of the US$15 million upfront payment under the Company’s global collaboration agreement with Elan, and; |
| |
| • | | On August 30, 2007, the Company Announced Completion of Multiple Phase I Clinical Studies with Alzheimer’s Disease Drug Candidate ELND005 (AZD-103).ELND005 (AZD-103) was safe and well-tolerated at all doses and dosing regimens examined.There were no severe or serious adverse events observed. ELND005 (AZD-103) was also shown to be orally bio-available, cross the blood-brain barrier and achieve levels in the human brain and cerebral spinal fluid (“CSF”) that were shown to be effective in animal models of Alzheimer’s disease. |
TT-223 — Diabetes:
| • | | On September 12, 2008, Transition announced that the first patient has been dosed in a Phase II clinical study of gastrin analogue, TT-223, in patients with type 2 diabetes.The study is a randomized, double-blind, placebo-controlled, dose-ranging study to evaluate the safety, tolerability and efficacy of daily TT-223 treatments for 12 weeks with a 6-month follow-up. Approximately 80 patients with type 2 diabetes are expected to be enrolled in the study and will receive a daily treatment of TT-223 in addition to their current regimen of oral glucose lowering agents (metformin and/or thiazolidinediones); |
| |
| • | | On March 13, 2008, Eli Lilly and Company (“Lilly”) and Transition announced that the two companies had entered into a licensing and collaboration agreement granting Lilly exclusive worldwide rights to develop and commercialize Transition’s gastrin based therapies, including the lead compound TT-223, which is currently in |
3
| | | early Phase II testing.Under the terms of the agreement, Transition received a US$7 million upfront payment on April 17, 2008, and may also receive up to US$130 million in potential development and sales milestones, as well as royalties on sales of gastrin based therapies if any product is successfully commercialized, and; |
| |
| • | | On November 5, 2007, the Company Announced an Update on the Clinical Development and Partnership activities for the Company’s diabetes program.Following negotiations, Novo Nordisk and Transition were not able to come to agreement for an exclusive license to the Company’s diabetes programs. Accordingly, Transition sent notice to Novo Nordisk terminating the agreement between the companies, returning to Transition all rights held by Novo Nordisk, relating to E1-I.N.T.TM |
Drug Discovery Initiatives:
| | • | | On August 18, 2008, the Company announced the acquisition of certain assets and the exclusive rights to selected drug discovery projects from Forbes Medi-Tech (Research) Inc., a wholly owned subsidiary of Forbes Medi-Tech Inc. (“Forbes”).In consideration for the acquisition of these assets and intellectual property rights, Forbes has received from Transition US$1 million, and will potentially receive up to an additional US$6 million in contingent consideration dependent on the successful achievement of certain developmental and regulatory milestones. These acquired discovery projects and other internal projects will be the focus of a small group of research scientists which shall operate through a newly formed United States-based subsidiary called Transition Therapeutics (USA) Inc. |
Corporate Developments:
| • | | On January 7, 2008, the Company’s common shares were approved for listing and commenced trading on the NASDAQ Global Market.Prior to this change, the Company’s common shares were listed on the NASDAQ Capital Market since August 20, 2007 under the symbol “TTHI”. The Company’s common shares continue to trade on the Toronto Stock Exchange in addition to the NASDAQ; |
| |
| • | | On October 31, 2007, the Company received the third anniversary payment of $650,000 from the sale of its subsidiary, Stem Cell Therapeutics (“SCT”).Total payments received to date amount to $1,850,000 with the final payment of $1,650,000 due in the first quarter of fiscal 2009; |
| |
| • | | On July 11, 2007, the Company completed a private placement financing issuing 1,736,107 common shares at a price of $14.40 per common share, raising gross proceeds of approximately $25,000,000 from a number of funds managed by Oracle Investment Management Inc., The Invus Group LLC, and a large Boston based investment management company.The Company incurred total share issuance costs of $1,031,433 resulting in net cash proceeds of $23,968,567, and; |
| |
| • | | On July 9, 2007, the Company completed a consolidation of its issued and outstanding common shares on the basis of one (1) post-consolidation common share for every nine (9) pre-consolidation common shares.The share consolidation was effected to satisfy the NASDAQ listing criteria regarding minimum bid price. |
4
STRATEGIC COLLABORATIONS
Elan Pharma International Limited
In September 2006, Transition announced a global collaboration with Elan to develop and commercialize ELND005 (AZD-103). Under the terms of the agreement, Transition has received an upfront payment of US$15 million in two separate tranches. The upfront payments received from Elan have been recorded as deferred revenue. On December 21, 2007, the Company and Elan jointly announced that the first patient had been dosed in the Phase II clinical study of ELND005 (AZD-103). As a result, the Company received a US$5 million milestone payment, which was triggered by the initiation of the Phase II clinical trial.
Dependent upon the successful development, regulatory and commercial launch of ELND005 (AZD-103), Transition will be eligible to receive additional milestone payments of up to US$180 million. Transition and Elan will share the costs of development and profits from commercialization. Each party’s cost share and ownership interest may vary throughout the term of the agreement dependant on certain elections that may be made during the development of ELND005 (AZD-103).
Eli Lilly and Company
On March 13, 2008, Lilly and the Company entered into a licensing and collaboration agreement granting Lilly exclusive worldwide rights to develop and commercialize Transition’s gastrin based therapies, including the lead compound TT-223, which is currently in early Phase II testing. Under the terms of the agreement, Transition has received a US$7 million upfront payment, and may also receive up to US$130 million in potential development and sales milestones, as well as royalties on sales of gastrin based therapies if any product is successfully commercialized. Transition and Lilly are both participating in the Phase II clinical trial with lead compound TT-223 in type 2 diabetes and under the terms of the agreement, Lilly will reimburse the Company up to US$3 million for development costs associated with this trial. In addition, the parties have established a joint development committee to coordinate and oversee activities relating to the TT-223 program. Upon completion of this trial, Lilly will be responsible for further development activities and the commercialization of all gastrin based therapeutic products worldwide.
On September 11, 2008, Transition dosed the first patient in a Phase II clinical study of gastrin analogue, TT-223, in patients with type 2 diabetes. Transition and Lilly are both funding this Phase II clinical trial. Upon completion of this trial, Lilly will be responsible for further development activities and the commercialization of all gastrin based therapeutic products worldwide.
5
PROGRAMS
Transition is focused on developing innovative therapies in several distinct areas of opportunity. Transition’s vision is to build a company that has a strong foundation for growth based on multiple technologies and product opportunities, which reduces risk and enhances return. The Company’s lead technologies are as follows:
ELND005 (AZD-103) for Alzheimer’s Disease
Alzheimer’s disease is a progressive brain disorder that gradually destroys a person’s memory and ability to learn, reason, make judgments, communicate and carry out daily activities. As Alzheimer’s disease progresses, individuals may also experience changes in personality and behavior, such as anxiety, suspiciousness or agitation, as well as delusions or hallucinations. In late stages of the disease, individuals need help with dressing, personal hygiene, eating and other basic functions. People with Alzheimer’s disease die an average of eight years after first experiencing symptoms, but the duration of the disease can vary from three to 20 years.
The disease mainly affects individuals over the age 65 and it is estimated over 18 million people are suffering from Alzheimer’s disease worldwide. The likelihood of developing late-onset Alzheimer’s approximately doubles every five years after age 65. By age 85, the risk reaches nearly 50 percent. In the U.S., Alzheimer’s disease is the fourth leading cause of death and current direct/indirect costs of caring for an estimated 4.5 million Alzheimer’s disease patients are at least US$100 billion annually.
Current FDA approved Alzheimer’s disease medications may temporarily delay memory decline for some individuals, but none of the currently approved drugs is known to stop the underlying degeneration of brain cells. Certain drugs approved to treat other illnesses may sometimes help with the emotional and behavioral symptoms of Alzheimer’s disease. With an aging population, there is a great need for disease-modifying compounds that can slow or reverse disease progression.
In March 2006, the Company announced the acquisition of all the remaining outstanding shares of Alzheimer’s focused Ellipsis Neurotherapeutics Inc. (“ENI”) that the Company did not already own. The key asset in the acquisition is the Alzheimer’s disease compound ELND005 (AZD-103), a disease modifying agent with the potential to both prevent and reduce disease progression, and improve symptoms such as cognitive function.
In April 2006, the Company received clearance from the Therapeutic Products Directorate of Health Canada to commence a Phase I clinical trial to evaluate the pharmacokinetics, safety and efficacy of escalating doses of ELND005 (AZD-103) in healthy volunteers. The study demonstrated that ELND005 (AZD-103) was well tolerated and no safety concerns or significant adverse events were observed in the study. In August 2006, the Company also received clearance from the FDA to commence a subsequent Phase I clinical trial evaluating higher doses of ELND005 (AZD-103).
6
In September 2006, Transition announced a global collaboration with Elan to develop and commercialize ELND005 (AZD-103).
In April 2007, Transition announced that the FDA granted Fast Track designation to the investigational drug candidate ELND005 (AZD-103) which is being developed in collaboration with Elan. Under the FDA Modernization Act of 1997, Fast Track designation is intended to facilitate the development and expedite the review of a drug or biologic if it is intended for the treatment of a serious or life-threatening condition, and it demonstrates the potential to address unmet medical needs for such a condition.
On August 30, 2007, the Company announced the completion of Phase I clinical studies with ELND005 (AZD-103). Transition and its development partner Elan have performed multiple Phase I studies evaluating the safety, tolerability and pharmacokinetic profile of ELND005 (AZD-103) in healthy volunteers. Approximately 110 subjects have been exposed to ELND005 (AZD-103) in multiple Phase I studies, including single and multiple ascending dosing; pharmacokinetic evaluation of levels in the brain; and CSF and plasma studies. ELND005 (AZD-103) was safe and well-tolerated at all doses and dosing regimens examined.There were no severe or serious adverse events observed. ELND005 (AZD-103) was also shown to be orally bio-available, cross the blood-brain barrier and achieve levels in the human brain and CSF that were shown to be effective in animal models for Alzheimer’s disease.
On December 21, 2007, Elan and Transition announced that the first patient had been dosed in a Phase II clinical study of ELND005 (AZD-103) in patients with Alzheimer’s disease. The study is a randomized, double-blind, placebo-controlled, dose-ranging, safety and efficacy study in approximately 340 patients with mild to moderate Alzheimer’s disease. Approximately 65 sites in North America are expected to participate in the program. The study will evaluate both cognitive and functional endpoints, and each patient’s participation is planned to last approximately 18 months. Patient enrolment of this study is on-going and its progress is on target.
On December 24, 2007, Transition announced that in connection with the initiation of the Phase II clinical study, the Company issued the former shareholders of ENI the first contingent consideration milestone in the form of 174,123 Transition common shares at a price of $10.86 per share. The shares issued had a fair value of $1,890,976 which represents additional consideration paid to acquire the technology, products and patents from ENI and accordingly, has been capitalized as intangible assets and will be amortized over the remaining useful life of the technology, products and patents.
Transition and its partner Elan are considering initiating clinical trials for other amyloid beta related indications including early/pre-Alzheimer’s disease.
Under the terms of the agreement, the Company can elect to participate in post Phase II development. The Company has 45 days after the receipt of the proof of concept data from the on-going Phase II clinical trial to make this election. Currently, certain post Phase II development costs are being incurred by Elan and these costs are being tracked by Elan for potential reimbursement by Transition should the Company elect to
7
participate in post Phase II development. If the Company elects to participate in the post Phase II development, based on the Company’s development percentage, the Company would owe Elan approximately US$1.1 million for post Phase II development costs incurred up to June 30, 2008. These costs have not been recorded as an expense or a liability at June 30, 2008 as the Company has not yet made a decision as to its participation.
Expenditures for the ELND005 (AZD-103) Program
During the year ended June 30, 2008, the Company incurred direct research and development costs for this program as follows:
| | | | | | | | | | | |
| ELND005 (AZD-103) | | | | | | | | |
| Program(1) | | | | Fiscal 2008 | | | Fiscal 2007 | |
| Pre-clinical studies | | | | $ | 4,596 | | | $ | 1,051,401 | |
| Clinical studies | | | | | 225,522 | | | | 1,327,796 | |
| Manufacturing | | | | | 111,643 | | | | 1,371,296 | |
| Other direct research | | | | | 36,305 | | | | 228,581 | |
| Due to (from) Elan | | | | | 4,501,034 | | | | (1,013,561 | ) |
| | | | | | | | | |
| TOTAL | | | | $ | 4,879,100 | | | $ | 2,965,513 | |
| | | | | | | | | |
| | |
| (1) | | These costs are direct research and development costs only and do not include patent costs, investment tax credits, salaries and benefits or an allocation of Company overhead. |
TT-223 for Diabetes
General
Diabetes is a disease in which the body does not produce or properly use insulin. Insulin is a hormone released from islet cells located in the pancreas that is needed to convert sugar, starches and other food into energy needed for daily life. There are two primary forms of diabetes; type 1 diabetes and type 2 diabetes.
Type 1 diabetes develops when the body’s immune system destroys pancreatic islet beta cells, the only cells in the body that make the hormone insulin that regulates blood glucose. To survive, people with type 1 diabetes must have insulin delivered by injection or pump. Type 1 diabetes accounts for 5-10% of all diagnosed cases of diabetes.
Type 2 diabetes usually begins as insulin resistance, a disorder in which the cells do not use insulin properly. As the need for insulin increases, the pancreas gradually loses its ability to produce it. Current treatments for type 2 diabetes include lifestyle changes, oral medications, incretin therapy and insulin therapy. Type 2 diabetes accounts for about 90-95% of all diagnosed cases of diabetes.
Transition has developed a patented diabetes therapy, which offers a new paradigm in the treatment of diabetes. Pre-clinical and clinical data suggest that gastrin plays an important
8
role in beta cell differentiation and function, capable of providing sustained glucose control in type 2 diabetes.
TT-223 in combination with EGF
Transition’s first diabetes therapy TT-223 in combination with EGF, a combination of Transition’s epidermal growth factor analogue and a gastrin analogue, has completed two Phase I clinical trials, in which it was shown that it was safe to administer. Transition received FDA clearance to initiate exploratory Phase IIa clinical trials for the drug candidatein both type 1 and type 2 diabetics. These two clinical trials evaluated the efficacy, safety and tolerability of a 28-day course of daily TT-223 in combination with EGF treatment with a six-month follow-up.
In March, 2007, the Company announced positive unblinded interim safety, tolerability and efficacy data from these exploratory Phase IIa trials for type 1 and type 2 diabetes patients. In the type 1 diabetes study, 6 of 11 (54%) patients responded to TT-223 in combination with EGF therapy, either by decreasing their average daily insulin usage by more than 20% or reducing their HbA1c levels by 1.2 to 2%. There were no responders among the placebo group.
On June 28, 2007, the Company announced final results from the exploratory Phase IIa clinical trial. A 4-week therapy with TT-223 in combination with EGF lead to sustained reductions in blood glucose levels for 6 months post-treatment in type 2 diabetes patients. In the treated group of patients, the mean HbA1c level was reduced by 0.94% to 1.21% vs. baseline levels in months 2 to 6 post-treatment. More specifically, the mean HbA1c level among treated patients was reduced 0.43%, 0.94% (p<0.05), 1.09% (p<0.05), 1.12% (p<0.05), 1.21% (p<0.05), and 1.14% in months 1, 2, 3, 4, 5, and 6 post-treatment, respectively. In contrast, the mean HbA1c levels of the placebo group ranged from a reduction of 0.1% to an increase of 1.0% over the same period. In addition to the HbA1c reductions, the data demonstrated decreases in fasting blood glucose levels as well as improvements in glucose tolerance over a six month period following treatment with TT-223 in combination with EGF. Trends in increased insulin levels as measured with an oral glucose tolerance test were also observed, particularly in patients where the HbA1c levels decreased over 1% with the TT-223 in combination with EGF therapy. These data are consistent with the increased glucose control observed in diabetes animal models where a short treatment with TT-223 in combination with EGF resulted in a sustained increase in beta cell mass and function. These clinical improvements, including HbA1c reductions greater than 1% in patients six month post-treatment, highlight the potential that TT-223 in combination with EGF therapy could provide patients significant clinical benefit in excess of six months.
TT-223 Clinical Development
These clinical data support the potential of the TT-223 gastrin analogue as a stand alone therapy and in combination with other diabetes therapies. On March 13, 2008, Lilly and the Company entered into a licensing and collaboration agreement granting Lilly
9
exclusive worldwide rights to develop and commercialize Transition’s gastrin based therapies, including the lead compound TT-223, which is currently in Phase II testing.
To support the Phase II clinical development program for TT-223, Transition has performed two Phase I studies to expand the dose ranges for TT-223. The first study, a single ascending dose study of TT-223 in healthy volunteers and the second study, a multiple ascending dose study of TT-223 have both been completed.
In August 2008, Transition and its collaboration partner Lilly initiated a Phase II trial evaluating TT-223 in type 2 diabetes patients receiving metformin and/or thiazolidinediones (TZDs). The companies are also in discussions regarding the timing and planning of another Phase II study with TT-223 in combination with a GLP1 analogue in type 2 patients. The next steps in the development of TT-223 in combination with epidermal growth factor analogue, will be evaluated following the review of data from the above proposed Phase II trials.
Juvenile Diabetes Research Foundation (“JDRF”)
In September 2006, the Company entered into an agreement with the JDRF to support the clinical development of TT-223 in combination with GLP1 analogues for the treatment of type 1 diabetes over a two year period. The clinical studies in type 1 diabetes patients will be disclosed at a later date.
Under the terms of the agreement, the Company is obligated to pay the JDRF a 5% royalty on license fees and milestone payments received in connection with the Company’s diabetes technology. Accordingly, the Company owes the JDRF $356,895 resulting from the US$7 million up-front payment received from Lilly. The obligation to the JDRF was included in accounts payable and accrued liabilities at June 30, 2008.
Licensing Agreement with Novo Nordisk
On November 5, 2007, the Company announced that following negotiations, Novo Nordisk and Transition were not able to come to agreement for an exclusive license to the Company’s diabetes programs. Accordingly, Transition sent notice terminating the agreement between the companies, returning to Transition all rights held by Novo Nordisk relating to the combination therapy TT-223 plus EGF.
Under the licensing agreement, the Company received $1,968,580 [US$1,500,000] in up-front payments that were recorded as deferred revenue and were recognized as licensing fee revenue over the term of the licensing agreement, which had been estimated as 15 years. In light of the agreement being terminated the Company recognized the remaining deferred amounts totaling $1,563,911 as licensing fee revenue during the second quarter of fiscal 2008.
Expenditures for the TT-223 Program
During the year ended June 30, 2008, the Company incurred direct research and development costs for this program as follows:
10
| | | | | | | | | |
| TT-223 Program(1) | | Fiscal 2008 | | | Fiscal 2007 | |
| Pre-clinical studies | | $ | 321,564 | | | $ | 1,691,070 | |
| Clinical studies | | | 1,460,709 | | | | 1,571,336 | |
| Manufacturing | | | 681,453 | | | | 504,703 | |
| Other direct research | | | 175,838 | | | | 95,862 | |
| Reimbursement from Lilly | | | (472,220 | ) | | | — | |
| Reimbursement from Novo Nordisk | | | — | | | | (805,353 | ) |
| Reimbursement from JDRF | | | — | | | | (564,800 | ) |
| | | | | | | |
| TOTAL | | $ | 2,167,344 | | | $ | 2,492,818 | |
| | | | | | | |
| | |
| (1) | | These costs are direct research and development costs only and do not include patent costs, investment tax credits, salaries and benefits or an allocation of Company overhead. |
Drug Discovery Initiatives
On August 18, 2008, the Company announced the acquisition of certain assets and the exclusive rights to selected drug discovery projects from Forbes Medi-Tech (Research) Inc., a wholly owned subsidiary of Forbes Medi-Tech Inc. (“Forbes”). In consideration for the acquisition of these assets and intellectual property rights, Forbes has received from Transition US$1 million, and will potentially receive up to an additional US$6 million in contingent consideration dependent on the successful achievement of certain developmental and regulatory milestones. These acquired discovery projects and other internal projects will be the focus of a small group of research scientists which shall operate through a newly formed United States-based subsidiary called Transition Therapeutics (USA) Inc.
In light of this acquisition, Transition has prioritized its drug discovery activities to accelerate the identification and optimization of novel lead molecules. The Company is pursuing a number of discovery programs to advance novel lead molecules into pre-clinical development.
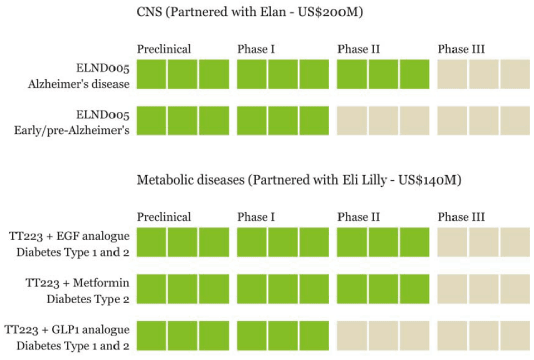
The Next Steps
Transition’s goal for its programs is to achieve product approval and ultimately significant revenues or royalties. To achieve product approval, the Company must successfully complete clinical trials and achieve regulatory approval. The stages of development of the Company’s technologies are illustrated below:
11
OVERALL PERFORMANCE
During fiscal 2008, the Company continued to advance its lead products through the clinic. The Company is collaborating with Elan to develop the Alzheimer’s disease drug candidate ELND-005 (AZD-103). Transition and Elan announced that the first patient had been dosed in a Phase II clinical study of ELND005 (AZD-103) in patients with Alzheimer’s disease. In addition, the Company and Lilly entered into a licensing and collaboration agreement granting Lilly exclusive worldwide rights to develop and commercialize Transition’s gastrin based therapies, including the lead compound TT-223, which is currently in Phase II testing.
During the year ended June 30, 2008, the Company strengthened its cash position by completing an offering for 1,736,107 common shares resulting in net cash proceeds of $23,968,567. The Company’s cash and cash equivalents and held-to-maturity investments were $63,663,630 at June 30, 2008. The Company currently believes that it has adequate financial resources for anticipated expenditures until the end of fiscal 2010.
The Company’s net loss for the year ended June 30, 2008 decreased by $842,588 or 5% to $16,119,202 from a loss of $16,961,790 reported in fiscal 2007. The decrease in net loss is due to decreased amortization expense resulting from the Waratah technology being fully amortized in the third quarter of fiscal 2007, recognition of the remaining unamortized upfront fee from Novo Nordisk, increased interest income resulting from higher cash and held-to-maturity investment balances, increased foreign exchange gains resulting from the Company holding US dollar investments and the receipt of the second milestone payment from SCT. The decrease in net loss was offset by the future income tax recovery of $2,729,422 that was recognized in the first quarter of fiscal 2007, and was
12
also offset by increases in research and development and general and administrative expenses.
In upcoming periods, the Company’s losses are expected to increase primarily as a result of increased clinical expenditures as the Company continues the clinical development of multiple products.
SELECTED ANNUAL INFORMATION
The following table is a summary of selected audited consolidated financial information of the Company for each of the three most recently completed financial years:
| | | | | | | | | | | | | |
| | | June 30, | | June 30, | | June 30, |
| | | 2008 | | 2007 | | 2006 |
| Revenue | | $ | 1,596,722 | | | $ | 683,894 | | | $ | 371,174 | |
Net loss(1) | | $ | 16,119,202 | | | $ | 16,961,790 | | | $ | 23,018,090 | |
| Basic and diluted net loss per common share | | $ | 0.70 | | | $ | 0.87 | | | $ | 1.53 | |
| Total assets | | $ | 94,875,961 | | | $ | 63,995,728 | | | $ | 44,128,024 | |
Total long-term liabilities(2) | | $ | 80,024 | | | $ | 91,456 | | | $ | 2,862,711 | |
| Cash dividends declared per share | | $ | — | | | $ | — | | | $ | — | |
| | |
| Notes: | | |
| |
| (1) | | Net loss before discontinued operations and extraordinary items was equivalent to the net loss for such periods. |
| |
| (2) | | Total long-term liabilities exclude deferred revenue, a non-financial liability. |
ANNUAL RESULTS — YEAR ENDED JUNE 30, 2008 COMPARED TO YEAR ENDED JUNE 30, 2007
Results of Operations
For the fiscal year ended June 30, 2008, the Company recorded a net loss of $16,119,202 ($0.70 per common share) compared to a net loss of $16,961,790 ($0.87 per common share) for the fiscal year ended June 30, 2007. This decrease in net loss of $842,588 or 5% is due to decreased amortization expense resulting from the Waratah technology being fully amortized in the third quarter of fiscal 2007, recognition of the remaining unamortized upfront fee from Novo Nordisk, increased interest income resulting from higher cash and held-to-maturity investment balances, increased foreign exchange gains resulting from the Company holding US dollar investments and the receipt of the second milestone payment from SCT. The decrease in net loss was offset by the future income tax recovery of $2,729,422 that was recognized in the first quarter of fiscal 2007, and was also offset by increases in research and development and general and administrative expenses.
13
Revenues
In fiscal 2008, the Company recorded licensing fees of $1,596,722. In fiscal 2007, the Company recognized milestone revenue and licensing fees of $683,894. The increase of $912,828 or 133% is due to the fact that the licensing agreement with Novo Nordisk was terminated during the second quarter of fiscal 2008 and all remaining deferred amounts were recognized as licensing fee revenue during the period.
During fiscal 2008, under the terms of the licensing and collaboration agreements, the Company received the second half of the up-front payment in the amount of $7,284,000 (US$7.5 million), as well as the milestone payment of $5,015,495 (US$5 million) from ELAN for the initiation of the Phase II clinical trial. In addition, the Company also received a $7,017,000 (US$7 million) upfront payment from Lilly. These payments are recorded as deferred revenue and will be recognized as income on a systematic basis once the profitability of the collaboration arrangements can be reasonably estimated.
Research and Development
Research and development expenses increased to $12,822,913 for the fiscal year ended June 30, 2008 from $9,839,170 for the fiscal year ended June 30, 2007. The increase, $2,983,743 or 30%, is primarily due to an increase in clinical development costs related to ELND005 (AZD-103), salaries and stock option expense. The increase was partially offset by decreases in direct clinical program expenses that related to the Company’s TT-223 and the completed I.E.T. clinical trials and reduced drug discovery expenses.
The Company anticipates that research and development expenses will continue to increase in fiscal 2009 as the Company incurs net development costs relating to the on-going ELND005 (AZD-103) Phase II clinical trials and clinical development costs associated with the TT‑223 Phase II clinical trials.
General and Administrative
General and administrative expenses increased to $5,820,864 for the fiscal year ended June 30, 2008 from $5,317,524 for the fiscal year ended June 30, 2007. The increase, $503,340 or 9%, primarily resulted from increased stock option expense, insurance, salaries, and investor relation costs, which were partially offset by a decrease in professional fees.
The Company anticipates that general and administrative expenses will increase in fiscal 2009 as the Company incurs additional corporate development and investor relation costs, in line with the Company’s strategy for its next stage of growth.
Amortization
Amortization for the fiscal year ended June 30, 2008 decreased by $4,075,516 or 60% to $2,747,743 as compared to $6,823,259 for the fiscal year ended June 30, 2007. The decrease is primarily due to the Waratah technology being fully amortized during the third quarter of fiscal 2007. This decrease was partially offset by the full period impact
14
of the amortization relating to the NeuroMedix technology acquired during the fourth quarter of fiscal 2007.
The Company anticipates that amortization expense will be consistent in fiscal 2009.
Recovery of Future Income taxes
Recovery of future income taxes was nil for the year ended June 30, 2008 compared to $2,729,422 for the same period in fiscal 2007. The decrease is due to the recognition of future income tax assets resulting from the amalgamation of Ellipsis Neurotherapeutics Inc., 1255205 Ontario Inc., 1255206 Ontario Inc. and Waratah Pharmaceuticals Inc. which occurred during the first quarter of fiscal 2007.
In the absence of additional acquisitions, the Company does not anticipate recording a future income tax recovery in fiscal 2009.
Interest Income, net
Interest income, net for the fiscal year ended June 30, 2008, was $2,417,537 as compared to $1,226,099 for the fiscal year ended June 30, 2007. The increase in interest income, net of $1,191,438 primarily resulted from increased cash balances due to the July 2007 private placement and the upfront and milestone payments received from Elan and Lilly.
In the absence of additional financing, interest income is expected to decrease in fiscal 2009.
SCT Anniversary Payment
On October 4, 2004, the Company signed a Share Purchase Agreement (the “Agreement”) to sell one of its wholly-owned subsidiaries, Stem Cell Therapeutics Inc. (“SCT”), whose only significant asset is technology. SCT is developing a series of regenerative therapies for the treatment of neurological diseases including stroke and Parkinson’s disease. The Agreement includes an upfront cash payment of $325,000, anniversary payments totaling $3.175 million that may be settled in either cash or shares at the option of the purchaser, and royalties on sales and other income.
This transaction was not recorded as a sale for accounting purposes as the risks and rewards of the ownership of SCT did not transfer to the purchaser under the terms of the Agreement. Therefore, on closing of the transaction, the Company classified the net carrying amount of the assets and liabilities of SCT as assets transferred under a contractual arrangement. Prior to July 1, 2007, the Company accounted for the assets transferred under contractual arrangement using the cost recovery method whereby the carrying value of the assets transferred under contractual arrangement have been reduced by [i] proceeds upon receipt, [ii] losses of SCT and [iii] amortization of the technology, resulting in a carrying value of nil as of the end of fiscal 2006. Any proceeds received subsequent to the assets being reduced to nil and June 30, 2007 have been included as a gain in the statement of loss.
15
During the year ended June 30, 2008, the Company received the third anniversary payment of $650,000 in cash which has been recorded as a gain in the statement of loss. As of June 30, 2008, total payments received amount to $1,850,000.
Effective July 1, 2007, the Company determined that the asset was a financial asset and has classified the asset transferred under contractual arrangement as a financial asset held for trading as described in note 2. The Company has estimated the fair value of this financial asset using a discounted cash flow method based on the contractual payments due to the Company. A change of 10% in the discount rates used would have resulted in an increase or decrease in net income of $67,000 or $57,000 respectively, for the year ended June 30, 2008 and a nominal change in total assets as at June 30, 2008. The change in fair value recognized by the Company during the year was $650,000. The final payment of $1,650,000 is due in the first quarter of fiscal 2009.
Financing Activities
During fiscal 2008, the Company closed on a private placement financing issuing 1,736,107 common shares at a price of $14.40 per common share, raising gross proceeds of approximately $25,000,000 from a number of funds managed by Oracle Investment Management Inc., The Invus Group LLC, and a large Boston based investment management company. The Company incurred total share issuance costs of $1,031,433, resulting in net cash proceeds of $23,968,567. The proceeds from the offering are planned to be used to fund Transition’s clinical studies, research and product development, working capital and general corporate purposes.
SUMMARY OF QUARTERLY RESULTS
The following table is a summary of selected quarterly consolidated financial information of the Company for each of the eight most recently completed quarters ending at June 30, 2008.
| | | | | | | | | | | | | | | | | | | | | |
| | | First | | Second | | Third | | Fourth | | |
| | | Quarter | | Quarter | | Quarter | | Quarter | | Year |
2008 | | | | | | | | | | | | | | | | | | | | |
| Revenue | | $ | 32,811 | | | $ | 1,563,911 | | | $ | — | | | $ | — | | | $ | 1,596,722 | |
| | | | | | | | | | | | | | | | | | | | | |
Net loss(1) | | $ | 4,098,978 | | | $ | 1,552,208 | | | $ | 4,977,020 | | | $ | 5,490,996 | | | $ | 16,119,202 | |
| Basic and diluted net loss per Common Share | | $ | 0.18 | | | $ | 0.07 | | | $ | 0.22 | | | $ | 0.23 | | | $ | 0.70 | |
2007 | | | | | | | | | | | | | | | | | | | | |
| Revenue | | $ | 585,461 | | | $ | 32,811 | | | $ | 32,811 | | | $ | 32,811 | | | $ | 683,894 | |
| | | | | | | | | | | | | | | | | | | | | |
Net loss(1) | | $ | 2,324,722 | | | $ | 4,858,107 | | | $ | 3,804,694 | | | $ | 5,974,267 | | | $ | 16,961,790 | |
| Basic and diluted net loss per Common Share | | $ | 0.13 | | | $ | 0.25 | | | $ | 0.19 | | | $ | 0.30 | | | $ | 0.87 | |
| | |
| Notes: | | |
| |
| (1) | | Net loss before discontinued operations and extraordinary items was equivalent to the net loss for such periods. |
16
The fluctuations of Transition’s quarterly results are primarily due to changes in activity levels of the clinical trials being performed by the Company, amortization of the technology relating to the assets acquired from Waratah, Protana, ENI, and NeuroMedix, recognition of upfront and licensing fees relating to the Novo Nordisk agreement, recognition of anniversary payments resulting from SCT, changes in the recovery of future income taxes, interest income, corporate development costs, and the growth of the Company’s management team.
FOURTH QUARTER RESULTS
The following table is a summary of selected information for the three month periods ended June 30, 2008 and June 30, 2007:
| | | | | | | | | |
| | | 2008 | | 2007 |
| Revenue — Licensing fees | | | — | | | $ | 32,811 | |
| Research and development, net | | $ | 3,796,562 | | | $ | 3,788,062 | |
| General and administrative | | $ | 1,526,176 | | | $ | 2,024,065 | |
| Amortization | | $ | 695,190 | | | $ | 586,062 | |
| Interest income, net | | $ | 489,547 | | | $ | 305,782 | |
| Net loss | | $ | 5,490,996 | | | $ | 5,974,267 | |
Review of Operations
For the three month period ended June 30, 2008, the Company’s net loss decreased by $483,271 or 8% to $5,490,996 compared to $5,974,267 for the same period in fiscal 2007.
Licensing fees decreased to nil from $32,811 for the same period in fiscal 2007. The decrease is due to the fact that the licensing agreement with Novo Nordisk was terminated during the second quarter of fiscal 2008 and all remaining deferred amounts were recognized at that time. Also, the licensing fees and the upfront and milestone payments received by the Company associated with the Elan and Lilly collaborations are being deferred until profitability of the arrangements can be reasonably estimated.
Research and development expenses increased by $8,500 or 0.2% to $3,796,562 compared to $3,788,062 for the same period in fiscal 2007. This increase was primarily due to an increase in clinical development costs related to ELND005 (AZD-103), salaries and option expense. These increases are partially offset by decreased TT-223 research and development costs resulting from the reimbursement by Lilly recorded in the fourth quarter and reduced drug discovery expenses.
17
General and administrative expenses decreased by $497,889 or 25% to $1,526,176 from $2,024,065 for the same period in fiscal 2007. This decrease was primarily due to decreased professional fees relating to the fiscal 2007 Nasdaq listing and amalgamation of various subsidiaries and decreased stock option expense. The decrease has been partially offset by an increase in salaries incurred to strengthen the finance and management teams, Directors and Officers’ insurance and investor relation related expense.
Amortization expense increased $109,128 or 19% to $695,190 from $586,062 for the same period in fiscal 2007. This increase is primarily due to the full quarter impact of the amortization relating to the NeuroMedix technology acquired during the fourth quarter of fiscal 2007.
Interest income, net, increased $183,765 or 60% to $489,547 from $305,782 for the same period in fiscal 2007. This increase primarily resulted from increased cash balances due to the July 2007 private placement and the upfront and milestone payments received from Elan and Lilly.
Financing Activities
There were no financing activities during the three-month period ended June 30, 2008.
CRITICAL ACCOUNTING ESTIMATES
The preparation of financial statements in accordance with Canadian generally accepted accounting principles requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results can differ from those estimates. We have identified the following areas which we believe require management’s most subjective judgments, often requiring the need to make estimates about the effects of matters that are inherently uncertain and may change in subsequent periods.
Valuation and Amortization of Intangible Assets
The Company’s intangible assets are comprised of purchased or licensed pharmaceutical technology, patents and workforce. The cost of the Company’s intangible assets are amortized over the estimated useful life ranging from 5 to 15 years. Factors considered in estimating the useful life of the intangible asset include the expected use of the asset by the Company, legal, regulatory and contractual provisions that may limit the useful life, the effects of competition and other economic factors, and the level of expenditures required to obtain the expected future cash flows from the intangible asset. The Company assesses its intangible assets for recoverability whenever indicators of impairment exist. When the carrying value of an asset is greater than its net recoverable value as determined on an undiscounted basis, an impairment loss is recognized to the extent that its fair value is below the asset’s carrying value.
18
Refundable Investment Tax Credits
The Company incurs research and development expenditures which are eligible for refundable investment tax credits in the province of Ontario. The investment tax credits recorded are based on our best estimates of amounts expected to be recovered. Actual investment tax credits received are based on the ultimate determination of the taxation authorities and, accordingly these amounts may vary from the amounts recorded.
Valuation Allowance for Future Tax Assets
The Company has recorded a valuation allowance on certain future tax assets primarily related to the carryforward of operating losses and qualifying research and development expenses. The Company has determined that it is more likely than not that some of these carryforward amounts will not be realized based on historical results and estimated future taxable income. The generation of future taxable income or the implementation of tax planning strategies could result in the realization of some or all of the carryforward amounts, which could result in a material change in our net income (loss) through the recovery of future income taxes. However, there is no assurance that the Company will be able to record future income tax recoveries in the future.
Equity Based Valuations
When the Company issues equity based instruments (i.e. stock options), an estimate of fair value is derived for the equity instrument using the Black-Scholes pricing model. The application of this pricing model requires management to make assumptions regarding several variables, including the period for which the instrument will be outstanding, the price volatility of the Company’s stock over a relevant timeframe, the determination of a relevant risk free interest rate and an assumption regarding the Company’s dividend policy in the future. If other assumptions are used, the value derived for the equity instruments could be significantly impacted.
Recognition of Deferred Revenue
As a result of the Company’s collaboration agreements with Elan and Lilly, the Company has recorded deferred revenue.
The up-front and milestone payments received from Elan and the up-front payment received from Lilly have been recorded as deferred revenue and will be recognized as income on a systematic basis once the profitability of the collaboration arrangement can be reasonably estimated. Actual results could differ materially from the estimates made by management.
As a result of the Novo Nordisk licensing agreement being terminated, the remaining deferred amounts were recognized as licensing fee revenue during the year ended June 30, 2008.
19
ADOPTION OF NEW ACCOUNTING POLICIES
Financial Instruments
Effective July 1, 2007, the Company has adopted the Canadian Institute of Chartered Accountants (“CICA”) Handbook Section 1530, Comprehensive Income, CICA Section 3855, Financial Instruments – Recognition and Measurement, CICA Section 3861, Financial Instruments – Disclosure and Presentation, and Handbook Section 3865, Hedges. These new Handbook Sections, which apply to fiscal years beginning on or after October 1, 2006, provide comprehensive requirements for the recognition and measurement of financial instruments, as well as standards on when or how hedge accounting may be applied. Handbook Section 1530 also establishes standards for reporting and disclosing comprehensive income (loss). Comprehensive income (loss) is defined as the change in equity from transactions and other events from non-owner sources. Other comprehensive income (loss) refers to items recognized in comprehensive income (loss) but that are excluded from net income (loss) calculated in accordance with Canadian generally accepted accounting principles.
Under the new standards, all financial instruments are classified into one of the following five categories: held-for-trading; held-to-maturity; loans and receivables; available-for-sale financial assets or other financial liabilities. All financial instruments, including derivatives, are included on the balance sheet and are measured at fair value with the exception of loans and receivables, investments held-to-maturity and other financial liabilities, which are measured at amortized cost. Subsequent measurement and recognition of changes in the carrying value of financial instruments depend on their initial classification.
Held-for-trading financial instruments are measured at fair value and all gains or losses are included in the results of operations in the period in which they arise. Available-for-sale financial instruments are measured at fair value with revaluation gains and losses included in other comprehensive income (loss) until the asset is removed from the balance sheet or an impairment occurs. As a result of the adoption of these standards, the Company has classified its cash equivalents and short-term investments as “held-to-maturity” which are measured at amortized cost using the effective interest method. The Company has classified the SCT receivable relating to the assets transferred under contractual arrangement, previously measured using a cost recovery basis, as held-for-trading and it is measured at fair value. The Company has also classified its accounts receivable as “Loans and receivables” and its accounts payable and accrued liabilities as “Other financial liabilities”, both of which are measured at amortized cost. The standard was adopted retroactively without restatement in accordance with the transitional provisions. As a result of the adoption of this standard, the Company has reclassified $423,628 from interest receivable to held-to-maturity investments to conform with the measurement basis recommended by the standard. In addition, the Company adjusted the carrying value of the SCT receivable relating to the assets transferred under a contractual arrangement by an amount of $1,650,000 resulting in a corresponding adjustment to the deficit. The adoption of the standard had no impact on previously reported earnings per share. Transaction costs that are directly attributable to the acquisition or issue of a financial asset or financial liability are added to the value of the instrument. The adoption had no other impact on the Company’s balance sheet at July 1, 2007.
20
Inventory
During the fourth quarter of fiscal 2007, the Company changed its accounting policy related to inventories to adopt CICA Handbook section 3031 — Inventories, effective July 1, 2006. As a result of the adoption, the net realizable value of the inventory is now measured at the estimated selling price of the inventory less estimated costs of completion and estimated costs to make the sale. Previously the Company measured net realizable value at the inventory’s replacement cost. The change in accounting policy was applied in accordance with the transitional provisions which permitted the Company to charge the difference in the measurement of opening inventory of $3,225,599 to the opening deficit for the year without restatement of prior years.
RECENT CANADIAN ACCOUNTING PRONOUNCEMENTS
CICA Section 1400, General Standards of Financial Statement Presentation
Under the amended section, management is required to make an assessment of an entity’s ability to continue as a going concern. In making its assessment, management must consider all available information about the future, which is at least, but is not limited to, twelve months from the balance sheet date. Financial statements must be prepared on a going concern basis unless management either intends to liquidate the entity, to cease trading or cease operations, or has no realistic alternative but to do so. Disclosure is required of material uncertainties related to events or conditions that may cast significant doubt upon the entity’s ability to continue as a going concern. When financial statements are not prepared on a going concern basis, that fact should be disclosed, together with the basis on which the financial statements are prepared and the reason the entity is not regarded as a going concern. The effective date of these amendments is for interim and annual financial statements relating to fiscal years beginning on or after January 1, 2008. The Company intends to adopt this standard for the three-month period ended September 30, 2008 and does not expect the adoption of this standard will have an impact on the disclosures in the financial statements.
CICA Section 1535, Capital Disclosures
This pronouncement establishes standards for disclosing information, both qualitative and quantitative, that enable users of financial statements to evaluate an entity’s objectives, policies and processes for management of capital. The Company has not yet assessed the impact this standard will have on the disclosures of the financial statements. The Company intends to adopt this standard for the three-month period ended September 30, 2008.
21
CICA Section 3064, Goodwill and Intangible Assets
This pronouncement replaces CICA 3062, “Goodwill and Other Intangible Assets” and CICA 3450, “Research and Development Costs”. The standard establishes standards for recognition, measurement, and disclosure of goodwill and intangibles. The changes relating to the definition and initial recognition of intangible assets, including internally generated intangible assets, are equivalent to the corresponding provisions of International Financial Reporting Standards. These changes are effective for years beginning on or after October 1, 2008, with early adoption encouraged. The Company is evaluating the effects of adopting this standard as to potential impact and the date at which the Company will adopt the new standard.
CICA Section 3862, Financial Instruments – Disclosures and CICA Section 3863, Financial Instruments – Presentation
These pronouncements revise and enhance disclosure requirements for financial instruments and carry forward unchanged the presentation requirements for financial instruments, respectively. The standards replace CICA Section 3861, Financial Instruments – Disclosure and Presentation. The new sections are effective for interim and annual financial statements for fiscal years beginning on and after October 1, 2007. The disclosure requirements cover the significance of financial instruments, fair value of financial instruments and exposures to risks from financial instruments. The Company has not yet assessed the impact this standard will have on the disclosures of the financial statements. The Company intends to adopt this standard for the three-month period ended September 30, 2008.
RECENT U.S. ACCOUNTING PRONOUNCEMENTS
On April 25, 2008, the FASB issued FASB Staff Position (“FSP”) No. FAS 142-3, Determination of the Useful Life of Intangible Assets. FSP FAS 142-3 amends the factors that should be considered in developing renewal or extension assumptions used to determine the useful life of a recognized intangible asset under the FASB Statement No. 142, Goodwill and Other Intangible Assets. The intent of this FSP is to improve the consistency between the useful life of a recognized intangible asset under SFAS 142 and the period of expected cash flows used to measure the fair value of the asset under SFAS 41, Business Combination, and other U.S. GAAP. FSP FAS 142-3 is effective for financial years beginning after December 15, 2008. Early adoption is prohibited. The guidance for determining the useful life of a recognized intangible asset of this FSP shall be applied prospectively to intangible assets acquired after the effective date. The disclosure requirements shall be applied prospectively to all intangible assets recognized as of, and subsequent to, the effective date. The Company has not yet assessed the impact the adoption of this new abstract is expected to have on its consolidated financial position or results of operations.
On December 12, 2007, the EITF ratified abstract: Issue 07-1, Accounting for Collaborative Arrangements Related to the Development and Commercialization of Intellectual Property. The abstract may impact the presentation or revenues and costs generated in a collaborative arrangement. The abstract is effective for years beginning on or after December 15, 2008. The Company has not yet assessed the impact the adoption
22
of this new abstract is expected to have on its consolidated financial position or results of operations.
On June 27, 2007, the EITF ratified abstract: Issue 07-3, Accounting for Non-refundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities. The abstract may impact the treatment of non-refundable advance payments for goods or services that will be used or rendered for research and development activities. The abstract is effective for years beginning on or after December 15, 2007. The Company has not yet assessed the impact the adoption of this new abstract is expected to have on its consolidated financial position or results of operations.
In September 2006, the FASB issued FASB Statement No. 157 (“SFAS 157”), Fair Value Measurements. SFAS 157 defines fair value, establishes a framework for measuring fair value in generally accepted accounting principles and expands disclosures about fair value measurements. SFAS 157 applies under other accounting pronouncements that require or permit fair value measurements, the FASB having previously concluded in those accounting pronouncements that fair value is the relevant measurement attribute. Accordingly, SFAS 157 does not require any new fair value measurements. SFAS 157 is effective for fiscal years beginning after November 15, 2007. The Company is currently evaluating the impact, if any, the adoption of SFAS 157 will have on its consolidated financial position and results of operations.
In December 2007, the FASB issued Statement No. 141R, Business Combinations (“FAS 141R”), and Statement No. 160, Noncontrolling Interests in Consolidated Financial Statements—an amendment of ARB No. 51 (“FAS 160”). FAS 141R expands the scope of acquisition accounting to all transactions under which control of a business is obtained. Among other things, FAS 141R requires that contingent consideration as well as contingent assets and liabilities be recorded at fair value on the acquisition date, that acquired in-process research and development be capitalized and recorded as intangible assets at the acquisition date, and also requires transaction costs and costs to restructure the acquired company be expensed. FAS 160 provides guidance for the accounting, reporting and disclosure of noncontrolling interests and requires, among other things, that noncontrolling interests be recorded as equity in the consolidated financial statements. FAS 141R and FAS 160 are both effective, on a prospective basis, July 1, 2009 with the exception of the presentation and disclosure requirements of FAS 160 which must be applied retrospectively. The Company is assessing the impacts of these standards on its financial position and results of operations.
In May 2008, the FASB issued Statement No. 162, The Hierarchy of Generally Accepted Accounting Principles (“FAS 162”). FAS 162 identifies the sources of accounting principles and the framework for selecting the principles used (order of authority) in the preparation of financial statements that are presented in conformity with generally accepted accounting standards in the United States. FAS 162 is effective 60 days following the Securities and Exchange Commission’s approval of the Public Company Accounting Oversight Board amendments to AU Section 411, The Meaning of Present Fairly in Conformity with Generally Accepted Accounting Principles. The Company
23
does not expect the adoption of FAS 162 to have a material impact on its financial statements.
DISCLOSURE CONTROLS AND PROCEDURES AND INTERNAL CONTROLS
As at June 30, 2008, Transition’s management evaluated the effectiveness of the design and operation of its disclosure controls and procedures. Based on their evaluation, the Chief Executive Officer and the Chief Financial Officer have concluded that Transition’s disclosure controls and procedures are effective.
There have been no significant changes in Transition’s internal control over financial reporting that have materially affected, or are reasonably likely to materially affect Transition’s internal control over financial reporting.
LIQUIDITY AND CAPITAL RESOURCES
Overview
The Company commenced operations in July 1998, and has devoted its resources primarily to fund its research and development programs. All revenue to date has been generated from interest income on surplus funds, management fees, milestone payments, and licensing fees. The Company has incurred a cumulative deficit to June 30, 2008 of $104,160,771. Losses are expected to continue for the next several years as the Company invests in research and development, pre-clinical studies, clinical trials, manufacturing and regulatory compliance.
Since inception, the Company has been financed primarily from public and private sales of equity, the exercise of warrants and stock options, interest earned on cash deposits, held-to-maturity investments and investment tax credits, revenues and reimbursements from partners, and proceeds from the sale of assets transferred under contractual arrangement.
The Company’s cash, cash equivalents and held-to-maturity investments and the Company’s working capital position were $63,663,630 and $64,360,685, respectively, at June 30, 2008, a significant increase from June 30, 2007 balances of $34,791,770 and $32,624,693, respectively. The increase is primarily the result of the net proceeds from the July 2007 private placement in the amount of $23,968,567 and $19,316,495 in partnership payments received, which were partially offset by expenditures incurred during the twelve-month period ended June 30, 2008. As a result, the Company currently believes it has adequate financial resources for anticipated expenditures until the end of fiscal 2010.
Financial instruments of the Company consist mainly of cash and cash equivalents, held-to-maturity investments, receivables, accounts payable and accrued liabilities and amounts due to/from Elan, SCT and Lilly. Management’s primary investment objective
24
is to maintain safety of principal and provide adequate liquidity to meet all current payment obligations and future planned expenditures.
The Company is exposed to market risks related to volatility in interest rates for the Company’s investment portfolio and foreign currency exchange rates related to purchases of supplies and services made in US dollars.
The success of the Company is dependent on its ability to bring its products to market, obtain the necessary regulatory approvals and achieve future profitable operations. The continuation of the research and development activities and the commercialization of its products are dependent on the Company’s ability to successfully complete these activities and to obtain adequate financing through a combination of financing activities, operations, and partnerships. It is not possible to predict either the outcome of future research and development programs or the Company’s ability to fund these programs going forward.
Financing Activities
On July 11, 2007, the Company completed a private placement financing issuing 1,736,107 common shares at a price of $14.40 per common share, raising gross proceeds of approximately $25,000,000 from a number of funds managed by Oracle Investment Management Inc., The Invus Group LLC, and a large Boston based investment management company. The Company incurred total share issuance costs of $1,031,433, resulting in net cash proceeds of $23,968,567.
The proceeds from these offerings are planned to be used to fund Transition’s clinical studies, research and product development, working capital and for general corporate purposes.
Contractual Obligations
Minimum payments under our contractual obligations as of June 30, 2008 are as follows:
| | | | | | | | | | | | | | | | | | | | | |
| | | Less than 1 | | | 1 - 3 | | | 4 – 5 | | | After 5 | | | | |
| | | Year | | | Years | | | Years | | | Years | | | Total | |
| Operating leases | | $ | 282,738 | | | $ | 571,569 | | | $ | 158,981 | | | $ | — | | | $ | 1,013,288 | |
| Collaboration agreements | | $ | 45,336 | | | $ | — | | | $ | — | | | $ | — | | | $ | 45,336 | |
| Clinical and toxicity study agreements | | $ | 5,867,877 | | | $ | — | | | $ | — | | | $ | — | | | $ | 5,867,877 | |
| Manufacturing agreements | | $ | 104,487 | | | $ | — | | | $ | — | | | $ | — | | | $ | 104,487 | |
| | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | |
TOTAL | | $ | 6,300,438 | | | $ | 571,569 | | | $ | 158,981 | | | $ | — | | | $ | 7,030,988 | |
| | | | | | | | | | | | | | | | |
25
RELATED PARTY TRANSACTIONS
During fiscal 2008, the Company paid legal fees to a law firm where the Company’s Secretary is a partner and to a corporation controlled by the Company’s Secretary. Total fees and disbursements charged to the Company by these companies during the year ended June 30, 2008 were $6,165 [2007 - $2,700] and are included in general and administrative expenses. The balance owing at June 30, 2008 and 2007 is nil. These transactions occurred in the normal course of operations and were measured at the exchange amount, which is the amount of consideration established and agreed to by the related parties.
OUTSTANDING SHARE DATA
Authorized
The authorized share capital of the Company consists of an unlimited number of common shares.
Issued and Outstanding
The following details the issued and outstanding equity securities of the Company:
Common Shares
As at September 15, 2008, the Company has 23,204,049 common shares outstanding.
Stock Options
As at September 15, 2008, the Company has 1,852,553 stock options outstanding with exercise prices ranging from $3.15 to $18.00 and expiry dates ranging from November 19, 2008 to June 30, 2013. At September 15, 2008, on an if-converted basis, these stock options would result in the issuance of 1,852,553 common shares at an aggregate exercise price of $21,912,911.
RISKS AND UNCERTAINTIES
Prospects for companies in the biopharmaceutical industry generally may be regarded as uncertain given the nature of the industry and, accordingly, investments in such companies should be regarded as highly speculative. The Company’s technologies are currently in either the research and development stage or early in the clinical development stage, which are both risky stages for a corporation in the biopharmaceutical industry. It is not possible to predict, based upon studies in animals and early clinical data, whether a new therapeutic treatment or device will prove to be safe and effective in humans.
The Company’s products will require additional development and testing, including extensive toxicity and other clinical testing, before the Company will be able to apply to
26
obtain regulatory approval to market the product commercially. To date, the Company has not introduced a product into the market and there is no assurance that research and development programs conducted by the Company will result in any commercially viable products. If a product is approved for sale, there is no assurance that the Company will generate adequate funds to continue development or will ever achieve profitable operations. There are many factors such as financial and human resources, competition, patent protection and the regulatory environment that can influence the Company’s ability to be profitable.
The Company will require significant additional financing and it may not have access to sufficient capital.
The Company anticipates that it will need additional financing in the future to fund its ongoing research and development programs and for general corporate requirements. The Company may choose to seek additional funding through public or private offerings, corporate collaborations or partnership arrangements. The amount of financing required will depend on many factors including the financial requirements of the Company to fund its research and clinical trials, and the ability of the Company to secure partnerships and achieve partnership milestones as well as to fund other working capital requirements. The Company’s ability to access the capital markets or to enlist partners is mainly dependent on the progress of its research and development and regulatory approval of its products. There is no assurance that additional funding will be available on acceptable terms, if at all.
To continue the Company’s research and development programs and to conduct future clinical trials, the Company will rely upon employees, collaborators and other third party relationships. There is no assurance that the Company will be able to maintain or establish these relationships as required.
The Company has a history of losses, and it has not generated any product revenue to date. It may never achieve or maintain profitability.
Since inception, the Company has incurred significant losses each year and expects to incur significant operating losses as the Company continues product research and development and clinical trials. There is no assurance that the Company will ever successfully commercialize or achieve revenues from sales of its therapeutic products if they are successfully developed or that profitability will ever be achieved or maintained. Even if profitability is achieved, the Company may not be able to sustain or increase profitability.
The Company is subject to a strict regulatory environment.
None of the Company’s product candidates have received regulatory approval for commercial sale.
Numerous statutes and regulations govern human testing and the manufacture and sale of human therapeutic products in Canada, the United States and other countries where the Company intends to market its products. Such legislation and regulation bears upon, among other things, the approval of protocols and human testing, the approval of manufacturing facilities, testing procedures and controlled research, review and approval of
27
manufacturing, preclinical and clinical data prior to marketing approval including adherence to GMP during production and storage as well as regulation of marketing activities including advertising and labelling.
The products and processes the Company is currently developing require significant development, testing and the investment of significant funds prior to their commercialization. There can be no assurance that any of such products or processes will actually be developed. The completion of clinical testing and the obtaining of required approvals are expected to take years and require the expenditure of substantial resources. There can be no assurance that clinical trials will be completed successfully within any specified period of time, if at all. Furthermore, clinical trials may be delayed or suspended at any time by the Company or by regulatory authorities if it is determined at any time that patients may be or are being exposed to unacceptable health risks, including the risk of death or compounds are not manufactured under acceptable GMP conditions or with acceptable quality. Any failure or delay in obtaining regulatory approvals would adversely affect the Company’s ability to utilize its technology thereby adversely affecting operations. No assurance can be given that the Company’s product candidates or lead compounds will prove to be safe and effective in clinical trials or that they will receive the requisite protocol approval or regulatory approval. Furthermore, no assurance can be given that current regulations relating to regulatory approval will not change or become more stringent. There are no assurances the Company can scale-up, formulate or manufacture any compound in sufficient quantities with acceptable specifications for the regulatory agencies to grant approval or not require additional changes or additional trials be performed. The agencies may also require additional trials be run in order to provide additional information regarding the safety, efficacy or equivalency of any compound for which the Company seeks regulatory approval. Similar restrictions are imposed in foreign markets other than the United States and Canada. Investors should be aware of the risks, problems, delays, expenses and difficulties which may be encountered by the Company in light of the extensive regulatory environment in which the Company’s business operates.
Even if a product candidate is approved by the FDA or any other regulatory authority, the Company may not obtain approval for an indication whose market is large enough to recoup its investment in that product candidate. The Company may never obtain the required regulatory approvals for any of its product candidates.
The Company is faced with uncertainties related to its research.
The Company’s research programs are based on scientific hypotheses and experimental approaches that may not lead to desired results. In addition, the timeframe for obtaining proof of principle and other results may be considerably longer than originally anticipated, or may not be possible given time, resource, financial, strategic and collaborator scientific constraints. Success in one stage of testing is not necessarily an indication that the particular program will succeed in later stages of testing and development. It is not possible to predict, based upon studies in in vitro models and in animals, whether any of the compounds made for these programs will prove to be safe, effective, and suitable for human use. Each compound will require additional research and development, scale-up, formulation and extensive clinical testing in humans.
28
Development of these compounds will require investigations into the mechanism of action of the molecules as these are not fully understood. Unsatisfactory results obtained from a particular study relating to a program may cause the Company to abandon its commitment to that program or to the lead compound or product candidate being tested. The discovery of unexpected toxicities, lack of sufficient efficacy, poor physiochemical properties, unacceptable ADME (absorption, distribution, metabolism and excretion) and DMPK (drug metabolism and pharmacokinetics), pharmacology, inability to increase scale of manufacture, market attractiveness, regulatory hurdles, competition, as well as other factors, may make the Company’s targets, lead compounds or product candidates unattractive or unsuitable for human use, and the Company may abandon its commitment to that program, target, lead compound or product candidate. In addition, preliminary results seen in animal and/or limited human testing may not be substantiated in larger controlled clinical trials.
The Company’s clinical trials may not yield results which will enable it to obtain regulatory approval for its human therapeutic products.
If clinical trials for a product candidate are unsuccessful, the Company will be unable to commercialize such product candidate. If one or more of the Company’s clinical trials is delayed, the Company will be unable to meet its anticipated development or commercialization timelines. Either circumstance could cause the price of the Common Shares to decline.
If difficulties are encountered enrolling patients in the Company’s clinical trials, the Company’s trials could be delayed or otherwise adversely affected.
Clinical trials for the Company’s product candidates require that the Company identify and enrol a large number of patients with the disorder under investigation. The Company may not be able to enrol a sufficient number of patients to complete its clinical trials in a timely manner. If the Company has difficulty enrolling a sufficient number of patients to conduct the Company’s clinical trials as planned, it may need to delay or terminate ongoing clinical trials.
Even if regulatory approvals are obtained for the Company’s product candidates, the Company will be subject to ongoing government regulation.
Even if regulatory authorities approve any of the Company’s human therapeutic product candidates, the manufacture, marketing and sale of such products will be subject to strict and ongoing regulation. Compliance with such regulation may be expensive and consume substantial financial and management resources.
If the Company’s products do not gain market acceptance, the Company may be unable to generate significant revenues.
Even if the Company’s products are approved for sale, they may not be successful in the marketplace. If the Company’s products do not gain market acceptance among consumers, physicians, patients and others in the medical community, the Company’s ability to generate significant revenues from the Company’s products would be limited.
29
The Company may not achieve its projected development goals in the time frames announced and expected.
There can be no assurance that the Company’s clinical trials will be completed, that the Company will make regulatory submissions or receive regulatory approvals as planned or that the Company will be able to adhere to its current schedule for the launch of any of its products. If the Company fails to achieve one or more of these milestones as planned, the price of the Common Shares would likely decline.
If the Company fails to obtain acceptable prices or adequate reimbursement for its human therapeutic products, its ability to generate revenues will be diminished.
If the Company fails to obtain acceptable prices or an adequate level of reimbursement for its products, the sales of its products would be adversely affected or there may be no commercially viable market for its products.
The Company may not obtain adequate protection for its products through its intellectual property.
The Company’s success depends, in large part, on its ability to protect its competitive position through patents, trade secrets, trademarks and other intellectual property rights. The patent positions of pharmaceutical biopharmaceutical firms, including the Company, are uncertain and involve complex questions of law and fact for which important legal issues remain unresolved. The patents issued or to be issued to the Company may not provide the Company with any competitive advantage. The Company’s patents may be challenged by third parties in patent litigation, which is becoming widespread in the biopharmaceutical industry. In addition, it is possible that third parties with products that are very similar to the Company’s will circumvent its patents by means of alternate designs or processes. The Company may have to rely on method of use protection for its compounds in development and any resulting products, which may not confer the same protection as compounds per se. The Company may be required to disclaim part of the term of certain patents. There may be prior applications of which the Company is not aware that may affect the validity or enforceability of a patent claim. There also may be prior applications of which the Company is aware, but which does not believe affects the validity or enforceability of a claim, which may, nonetheless ultimately be found to affect the validity or enforceability of a claim. No assurance can be given that the Company’s patents would, if challenged, be held by a court to be valid or enforceable or that a competitor’s technology or product would be found by a court to infringe the Company’s patents. Applications for patents and trademarks in Canada, the United States and in foreign markets have been filed and are being actively pursued by the Company. Pending patent applications may not result in the issuance of patents, and the Company may not develop additional proprietary products which are patentable.
Patent applications relating to or affecting the Company’s business have been filed by a number of pharmaceutical and biopharmaceutical companies and academic institutions. A number of the technologies in these applications or patents may conflict with the Company’s technologies, patents or patent applications, and such conflict could reduce the scope of patent protection which the Company could otherwise obtain. The Company could become involved in interference proceedings in the United States in connection
30
with one or more of its patents or patent applications to determine priority of invention. The Company’s granted patents could also be challenged and revoked in opposition proceedings in certain countries outside the United States.
In addition to patents, the Company relies on trade secrets and proprietary know-how to protect its intellectual property. The Company generally requires its employees, consultants, outside scientific collaborators and sponsored researchers and other advisors to enter into confidentiality agreements. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with the Company is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of the Company’s employees, the agreements provide that all of the technology which is conceived by the individual during the course of employment is the Company’s exclusive property. These agreements may not provide meaningful protection or adequate remedies in the event of unauthorized use or disclosure of the Company’s proprietary information. In addition, it is possible that third parties could independently develop proprietary information and techniques substantially similar to those of the Company or otherwise gain access to the Company’s trade secrets.
The Company currently has the right to use certain technology under license agreements with third parties. The Company’s failure to comply with the requirements of material license agreements could result in the termination of such agreements, which could cause the Company to terminate the related development program and cause a complete loss of its investment in that program.
As a result of the foregoing factors, the Company may not be able to rely on its intellectual property to protect its products in the marketplace.
The Company may infringe the intellectual property rights of others.
The Company’s commercial success depends significantly on its ability to operate without infringing the patents and other intellectual property rights of third parties. There could be issued patents of which the Company is not aware that its products infringe or patents, that the Company believes it does not infringe, but that it may ultimately be found to infringe. Moreover, patent applications are in some cases maintained in secrecy until patents are issued. The publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made and patent applications were filed. Because patents can take many years to issue, there may be currently pending applications of which the Company is unaware that may later result in issued patents that its products infringe.
The biopharmaceutical industry has produced a proliferation of patents, and it is not always clear to industry participants, including the Company, which patents cover various types of products. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. The Company is aware of, and has reviewed, third party patents relating to the treatment of Alzheimer’s disease and diabetes. In the event of infringement or violation of another party’s patent, the Company may not be able to enter into licensing arrangements or make other arrangements at a reasonable cost. Any inability to secure licenses or alternative technology could result in
31
delays in the introduction of the Company’s products or lead to prohibition of the manufacture or sale of products by it.
Patent litigation is costly and time consuming and may subject the Company to liabilities.
The Company’s involvement in any patent litigation, interference, opposition or other administrative proceedings will likely cause the Company to incur substantial expenses, and the efforts of its technical and management personnel will be significantly diverted. In addition, an adverse determination in litigation could subject the Company to significant liabilities.
The Company operates in a fiercely competitive business environment.
The biopharmaceutical industry is highly competitive. Competition comes from health care companies, pharmaceutical companies, large and small biopharmaceutical companies, specialty pharmaceutical companies, universities, government agencies and other public and private companies. Research and development by others may render the Company’s technology or products non-competitive or obsolete or may result in the production of treatments or cures superior to any therapy the Company is developing or will develop. In addition, failure, unacceptable toxicity, lack of sales or disappointing sales or other issues regarding competitors’ products or processes could have a material adverse effect on the Company’s product candidates, including its clinical candidates or its lead compounds.
The market price of the Company’s Common Shares experience a high level of volatility due to factors such as the volatility in the market for biotechnology stocks generally, and the short-term effect of a number of possible events.
The Company is a public growth company in the biotechnology sector. As frequently occurs among these companies, the market price for the Company’s Common Shares may experience a high level of volatility. Numerous factors, including many over which the Company has no control, may have a significant impact on the market price of Common Shares. Listing on NASDAQ and the TSX may increase share price volatility due to various factors.
In addition, the stock market in recent years has experienced extreme price and trading volume fluctuations that often have been unrelated or disproportionate to the operating performance of individual companies. These broad market fluctuations may adversely affect the price of Common Shares, regardless of the Company’s operating performance. In addition, sales of substantial amounts of Common Shares in the public market after any offering, or the perception that those sales may occur, could cause the market price of Common Shares to decline.
Furthermore, shareholders may initiate securities class action lawsuits if the market price of the Company’s stock drops significantly, which may cause the Company to incur substantial costs and could divert the time and attention of its management.
32
These factors, among others, could significantly depress the trading price of the Company’s securities.
Because the Company may experience a high level of volatility in its Common Shares, you should not invest in the Company’s stock unless you are prepared to absorb a significant loss of your capital. At any given time, you may not be able to sell your shares at a price that you think is acceptable.
The Company is highly dependent on third parties.
The Company is or may in the future be dependent on third parties for certain raw materials, product manufacture, marketing and distribution and, like other biotechnology and pharmaceutical companies, upon medical institutions to conduct clinical testing of its potential products. Although the Company does not anticipate any difficulty in obtaining any such materials and services, no assurance can be given that the Company will be able to obtain such materials and services.
The Company is subject to intense competition for its skilled personnel, and the loss of key personnel or the inability to attract additional personnel could impair its ability to conduct its operations.
The Company is highly dependent on its management and its clinical, regulatory and scientific staff, the loss of whose services might adversely impact its ability to achieve its objectives. Recruiting and retaining qualified management and clinical, scientific and regulatory personnel is critical to the Company’s success. Competition for skilled personnel is intense, and the Company’s ability to attract and retain qualified personnel may be affected by such competition.
The Company’s business involves the use of hazardous materials which requires the Company to comply with environmental regulation.
The Company’s discovery and development processes involve the controlled use of hazardous materials. The Company is subject to federal, provincial and local laws and regulations governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. The risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, the Company could be held liable for any damages that result, and any such liability could exceed the Company’s resources. The Company may not be adequately insured against this type of liability. The Company may be required to incur significant costs to comply with environmental laws and regulations in the future, and its operations, business or assets may be materially adversely affected by current or future environmental laws or regulations.
Legislative actions, potential new accounting pronouncements and higher insurance costs are likely to impact the Company’s future financial position or results of operations.
Compliance with changing regulations of corporate governance and public disclosure, notably with respect to internal controls over financial reporting, may result in additional
33
expenses. Changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for companies such as ours, and insurance costs are increasing as a result of this uncertainty.
The Company may experience difficulties in managing its future growth.
The Company may have difficulty managing its growth with the increased level of clinical development and potentially if it achieves commercialization.
Future health care reforms may produce adverse consequences.
Health care reform and controls on health care spending may limit the price the Company can charge for any products and the amounts thereof that it can sell. At present, the Company is not aware of any immediate changes in FDA or Canadian regulatory policies that would adversely affect its development programs.
The Company faces an unproven market for its future products.
The Company believes that there will be many different applications for products successfully derived from its technologies and that the anticipated market for products under development will continue to expand. No assurance can be given that these beliefs will prove to be correct due to competition from existing or new products and the yet to be established commercial viability of the Company’s products. Physicians, patients, formularies, third party payers or the medical community in general may not accept or utilize any products that the Company or its collaborative partners may develop.
The Company may be faced with future lawsuits related to secondary market liability.
Securities legislation in Canada has recently changed to make it easier for shareholders to sue. These changes could lead to frivolous law suits which could take substantial time, money, resources and attention or force the Company to settle such claims rather than seek adequate judicial remedy or dismissal of such claims.
The Company may encounter unforeseen emergency situations.
Despite the implementation of security measures, any of the Company’s, its collaborators’ or its third party service providers’, internal computer systems are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failure. Any system failure, accident or security breach that causes interruptions in that of the Company, in collaboration or in third party service vendors’ operations could result in a material disruption of the Company’s drug discovery programs.
The Company’s technologies may become obsolete
The pharmaceutical industry is characterized by rapidly changing markets, technology, emerging industry standards and frequent introduction of new products. The introduction of new products embodying new technologies, including new manufacturing processes,
34
and the emergence of new industry standards may render the Company’s technologies obsolete, less competitive or less marketable.
Other Risks
The Company is exposed to market risks related to volatility in interest rates for the Company’s investment portfolio and foreign currency exchange rates related to purchases of supplies and services made in U.S. dollars. The Company invests its cash in certain investment vehicles that provide a low risk rate of interest. If interest rates change, the interest earned by the Company as income will be affected. In addition, the Company’s share price is subject to equity market risk, which may result in significant speculation and volatility of trading due to the uncertainty inherent in the Company’s business and in the biotechnology industry in general. The expectations of the Company made by securities analysts could also have a significant impact on the trading price of the Company’s shares.
OTHER
Additional information relating to the Company, including the Company’s most recently filed Annual Information Form, can be found on SEDAR at www.sedar.com.
35