Exhibit 99.2
MANAGEMENT’S DISCUSSION AND ANALYSIS
The following is a discussion and analysis of the operating results and financial position of Transition Therapeutics Inc. for the year ended June 30, 2015. This document should be read in conjunction with the Company’s audited consolidated financial statements and the accompanying notes, which have been prepared in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board (IFRS). This Management’s Discussion and Analysis (“MD&A”) provides a review of the performance of the Company for the year ended June 30, 2015 as compared to the year ended June 30, 2014. This review was performed by management with information available as of September 14, 2015.
Where “we”, “us”, “our”, “Transition” or the “Company” is used, it is referring to Transition Therapeutics Inc. and its wholly-owned subsidiaries, unless otherwise indicated. All amounts are in Canadian dollars, unless otherwise indicated.
Additional information relating to the Company, including the Company’s most recently filed Annual Information Form, can be found on SEDAR at www.sedar.com.
CAUTION REGARDING FORWARD-LOOKING STATEMENTS
This MD&A contains certain forward-looking statements within the meaning of applicable securities laws. Forward-looking information typically contains statements with words such as “anticipate”, “believe”, “expect”, “plan”, “estimate”, “intend”, “may” or similar words suggesting future outcomes. Forward-looking statements in this MD&A include, but are not limited to statements with respect to: the clinical study phases of the Company’s product candidates which the Company expects to complete in fiscal 2016 and beyond; the ability of the Company’s business model to maximize shareholder returns; the potential for ELND005 to slow the progression of Alzheimer’s disease and improve symptoms; the potential for ELND005 to be effective for the treatment of agitation and or aggression in patients with Alzheimer’s disease; the potential for ELND005 to be effective for the treatment of Down syndrome; the timing and manner of future clinical development of ELND005; the global population size of those affected by Alzheimer’s disease; the demand for a product that can slow or reverse the progression of Alzheimer’s disease; the demand for a product that can reduce the emergence and severity of neuropsychiatric symptoms like depression, anxiety, agitation and aggression in Alzheimer’s disease; the potential clinical benefit of ELND005 in the treatment of other disease indications; the development of TT401 and the series of preclinical compounds in-licensed from Eli Lilly and Company (“Lilly”) and their potential benefit in type 2 diabetes patients and obese individuals; the timing and manner of future clinical development of TT401 performed by Lilly; TT701 development plans and timelines for individuals with androgen deficiency or other disease indications; the potential clinical benefit of TT701 to increase lean body mass, improve functional and sexual outcomes or improve other symptoms associated with androgen deficiency; the engagement of third party manufacturers to produce the Company’s drug substances and products; the potential future in-licensing of additional drug candidates to expand the development pipeline; the intention of the Company to make collaborative arrangements for the marketing and distribution of its products and the impact of human capital on the growth and success of the Company.
This forward-looking information is subject to various risks and uncertainties, including those discussed below, that could cause actual results and experience to differ materially from the anticipated results or other expectations expressed. Readers are cautioned not to place undue reliance on this forward-looking information, which is provided as of the date of this MD&A unless otherwise stated, and the Company will not undertake any obligation to publicly update orrevise any forward-looking information, whether as a result of new information, future events, or otherwise, except as required by securities laws.
Some of the assumptions, risks and factors which could cause future outcomes to differ materially from those set forth in the forward-looking information include, but are not limited to: (i) the assumption that the Company will be able to obtain sufficient and suitable financing to support operations, clinical trials and commercialization of products, (ii) the risk that the Company may not be able to capitalize on partnering and acquisition opportunities, (iii) the assumption that the Company will obtain favourable clinical trial results in the expected timeframe, (iv) the assumption that the Company will be able to adequately protect proprietary information and technology from competitors, (v) the risks relating to the uncertainties of the regulatory approval process, (vi) the impact of competitive products and pricing and the assumption that the Company will be able to compete in the targeted markets, and (vii) the risk that the Company may be unable to retain key personnel or maintain third party relationships, including relationships with key collaborators.
By its nature, forward-looking information involves numerous assumptions, inherent risks and uncertainties, both general and specific, that contribute to the possibility that the predictions, forecasts, projections or other forward-looking statements will not occur. Prospective investors should carefully consider the information contained under the heading “RISKS AND UNCERTAINTIES” as described in the MD&A.
OVERVIEW
Transition is a biopharmaceutical development company, advancing novel therapeutics for CNS and metabolic disease indications. The Company’s wholly-owned subsidiary, Transition Therapeutics Ireland Limited (“TTIL”) is developing CNS drug candidate ELND005 for the treatment of Alzheimer’s disease (“AD”) and Down syndrome. Transition’s lead metabolic drug candidate is TT401 for the treatment of type 2 diabetes and accompanying obesity.
During the fiscal year ended June 30, 2015 and up to the date of this MD&A, the Company announced the following:
ELND005:
| · | June 24, 2015 – Transition announced resultsof Clinical Study of ELND005 in Agitation and Aggression in Patients with Alzheimer’s Disease.The Phase 2/3 clinical study of neuropsychiatric drug candidate ELND005 did not meet its primary efficacy endpoint. In the study, both the treatment and placebo groups showed a significant, but similar, reduction in agitation and aggression relative to baseline. There was a greater than expected reduction in agitation and aggression observed in the placebo group as measured in weeks 4, 8 and 12 in the study. The safety and tolerability profile of ELND005 was consistent with previous studies in AD at the 250mg bid dose; |
| · | March 26, 2015 – Transition announced results from two phase 1 clinical studies of neuropsychiatric drug candidate ELND005. These studies, an absorption-metabolism-excretion (“AME”) study and a renal clearance study, are specialized clinical pharmacology trials that are required by the United States Food and Drug Administration (“FDA”) for the approval of most drugs in development; |
| · | November 24, 2014 – Transition announced results from a thorough QT (tQT) study in which no QT effects were observed at supra-therapeutic single doses of neuropsychiatric drug candidate, ELND005. A tQT study is a specialized clinical trial required by the FDA for the approval of most drugs in development. From a safety perspective, drugs that have no QT prolongation effects are particularly desirable for administration to an elderly Alzheimer’s disease AD population; |
| · | November 20, 2014 – Transition announced the results of a clinical study of neuropsychiatric drug candidate ELND005 in young adults with Down syndrome. TTIL completed this first study in Down syndrome subjects without dementia to allow optimal dose selection for future larger studies. The study enrolled 23 Down syndrome subjects in three study arms over a four-week treatment period. At the doses evaluated, ELND005 was determined to have an acceptable safety and tolerability profile and there were no serious adverse events reported; |
| · | November 4, 2014 - Transition announced findings from a Phase 2 study of neuropsychiatric drug candidate, ELND005, as an adjunctive maintenance treatment for bipolar disorder type I patients (BPD). TTIL terminated the bipolar disorder Phase 2 study on April 7, 2014 for business reasons. TTIL has completed a review of the data from this bipolar disorder Phase 2 study. Overall, ELND005 had an acceptable safety and tolerability profile in the study, and showed numerical differences in the number of mood event recurrences favoring ELND005. |
TT401:
| · | In February 2015, development partner Lilly informed Transition that 420 type 2 diabetic subjects have been enrolled in the current Phase 2 study thereby completing the enrollment phase of the study.The randomized, double-blind, placebo-controlled study includes six study arms, four doses of TT401, a placebo arm and a once-weekly exenatide arm. The main efficacy outcome measures are the change in HbA1c, a measure of blood-glucose levels, at week 12 and 24 and change in body weight over the course of the study; |
| · | Transition has paid all three installment payments totaling US$14 millionto diabetes drug candidate development partner Lilly.Transition has no further financial obligations for the development and commercialization of TT401. In December, 2014, Lilly informed Transition that the 70% enrollment milestone had been achieved. |
Corporate Developments:
| · | June 16, 2015 - Transition announced that Carl Damiani has been appointed as President and Chief Operating Officer of Transition; |
| · | May 6, 2015 – Transition announced its wholly-owned subsidiary, TTIL has exclusively licensed worldwide rights to a novel small molecule drug candidate (“TT701”) from Eli Lilly and Company.Under the terms of the agreement, TTIL has acquired rights to develop and commercialize TT701. Lilly will receive upfront consideration of up to US$1 million. In addition, Lilly is eligible to receive up to US$100 million in commercial milestones and a mid-single digit royalty on sales of TT701 products should such products be successfully commercialized. TT701 is a selective androgen receptor modulator that has been shown in a Phase 2 study to significantly increase lean body mass and a measurement of muscle strength in male subjects. This completed 12-week, Phase 2 study of 350 subjects also demonstrated additional beneficial effects, including significant fat mass reduction with no significant change in prostate specific antigen (PSA) levels. TTIL is evaluating multiple development paths for TT701, including as a new therapeutic option for patients with androgen deficiency. TTIL is engaged with potential collaborators to rapidly commence a Phase 2 clinical study; |
| · | February 18, 2015 – Transition announced the closing of a public offering of US$23 million of common shares equivalent to an aggregate of 3,538,461 common shares at a price to the public of US$6.50 per share, including 461,538 common shares issued upon the exercise of the underwriters’ over-allotment option.Cowen and Company, LLC was the sole book-running manager and Canaccord Genuity Inc., H.C. Wainwright & Co., LLC, and LifeSci Capital LLC were the co-managers for the offering; |
STRATEGIC COLLABORATIONS
Perrigo Company plc(“Perrigo”)
In 2006, Transition exclusively licensed the ELND005 technology to Elan Pharma International Limited (“Elan”) for worldwide development and commercialization. Following amendment of that agreement in 2010, Elan held all development and commercialization rights to ELND005 and Transition became eligible to receive milestone and royalty payments with the successful advancement of ELND005. Transition has received US$40 million from Elan in upfront and achieved milestone payments. Perrigo acquired Elan in December 2013, including all Elan’s rights and obligations to the development of ELND005.
On February 28, 2014, Transition announced that after a series of transactions, Perrigo had transferred all of its ELND005 rights and assets under the collaboration agreement to the Company’s wholly owned subsidiary, TTIL. In parallel with this acquisition, Perrigo invested US$15 million and received 2,255,640 Transition common shares representing approximately a 7% ownership stake in Transition as of the date of the transaction. Perrigo will also be eligible to receive up to US$40 million in approval and commercial milestone payments and a 6.5% royalty on net sales of ELND005 products and sublicense fees received. Going forward, TTIL is responsible for all future development and commercialization activities of the ELND005 drug candidate.
Lilly
Diabetes
On March 3, 2010, Transition and Lilly entered into a licensing and collaboration agreement granting Transition the rights to a series of preclinical compounds in the area of diabetes. Under the licensing and collaboration agreement, Transition received exclusive worldwide rights to develop and potentially commercialize a class of compounds that, in preclinical models, showed potential to provide glycemic control and other beneficial effects including weight loss.
Under the terms of the agreement, Lilly received an up-front payment of $1,055,900 (US$1 million) which has been capitalized as a license acquired from Lilly and is being amortized over 20 years which represents the estimated life of the underlying compounds and patents.
In June 2013, Lilly exercised its option and assumed all development and commercialization rights to type 2 diabetes drug candidate TT401. In conjunction with this assumption of rights, Transition received a US$7 million milestone payment. Lilly has assumed all costs and will perform all future development and commercialization activities of TT401, and Transition paid US$14 million to Lilly in three separate installments during the Phase 2 clinical study; the first installment of US$6 million was paid during the three month period ended September 30, 2014 when the study achieved 20% patient enrollment. The remaining two installments totaling US$8 million were paid during the three month period ended December 31, 2014 when the study achieved both the 50% and 70% patient enrollment milestones. Transition has no additional funding obligations related to this clinical study or any other development or commercialization activities in the future.
Transition is eligible to receive up to approximately US$240 million in additional milestone payments plus double-digit royalties on sales of TT401 products and a low single digit royalty on sales of related compounds.
PROGRAMS
Transition is focused on developing innovative therapies in several distinct areas of opportunity. Transition’s vision is to build a company that has a strong foundation for growth based on multiple technologies and product opportunities, which reduces risk and enhances shareholder return. The Company’s technologies are as follows:
ELND005
Alzheimer’s Disease:
Alzheimer’s disease is a progressive brain disorder that gradually destroys a person’s memory and ability to learn, reason, make judgments, communicate and carry out daily activities. As Alzheimer’s disease progresses, individuals may also experience changes in personality and behavior, such as anxiety, suspiciousness or agitation, as well as delusions or hallucinations. Approximately 90% of Alzheimer’s disease patients develop neuropsychiatric symptoms, and up to 60% develop agitation/aggression over the course of their disease. Agitation/aggression are among the most disruptive neuropsychiatric symptoms in Alzheimer’s disease and are associated with increased morbidity and caregiver burden.
The disease mainly affects individuals over age 65 and it is estimated over 18 million people are suffering from Alzheimer’s disease worldwide. In the U.S., Alzheimer’s disease is the sixth leading cause of death and current direct/indirect costs of caring for an estimated 5.4 million Alzheimer’s disease patients are at least US$100 billion annually.
Current U.S. Food and Drug Administration approved Alzheimer’s disease medications may temporarily delay memory decline for some individuals, but none of the currently approved drugs are known to stop the underlying degeneration of brain cells. Certain drugs approved to treat other illnesses may sometimes help with the emotional and behavioral symptoms of Alzheimer’s disease. With an aging population, there is a great need for therapies to address Alzheimer’s disease patient’s neuropsychiatric symptoms and declines in cognitive ability.
Down Syndrome:
Down syndrome (DS, Trisomy 21), caused by an extra copy of chromosome 21, is the most common genetic form of intellectual disability with a prevalence of approximately 1 in 700 live births in the US. Children with DS exhibit developmental delay and various degrees of intellectual disability, while adults are at increased risk of Alzheimer’s dementia. There are currently no drugs approved for the treatment of cognitive dysfunction in DS.
Excess activity of genes on chromosome 21, such as amyloid precursor protein (APP) and sodium-myo-inositol active transporter (SMIT), are thought to play a role in the cognitive dysfunction of DS. Life-long exposure to increased amyloid and myo-inositol levels in the brain are thought to lead to synaptic dysfunction and cognitive disability. ELND005 may have the potential to improve cognition in DS by decreasing amyloid levels and regulating myo-inositol-dependent neuronal signaling.
Clinical Development Update
ELND005 for Neuropsychiatric Diseases
TTIL is developing neuropsychiatric drug candidate ELND005, (scyllo-inositol). ELND005 is an orally bioavailable small molecule that is being investigated for multiple neuropsychiatric indications on the basis of its proposed dual mechanism of action, which includes β-amyloid anti-aggregation and regulation of brain myo-inositol levels. An extensive clinical program of Phase 1 and Phase 2 studies have been completed with ELND005 to support clinical development. The Phase 2 study (ELND005-AD201) which evaluated ELND005 in more than 350 mild to moderate AD patients was published in the peer-reviewed journal,Neurology. TheNeurology article was entitled “A Phase 2 randomized trial of ELND005, scyllo-inositol, in mild-moderate Alzheimer’s disease”.
Currently, the use of ELND005 is being investigated in two clinical areas:
| (a) | Agitation and Aggression in Alzheimer’s Disease |
On November 27, 2012, the first patient was enrolled in a Phase 2 clinical trial of ELND005 for the treatment of agitation/aggression in patients with mild to severe Alzheimer’s disease. The objectives of the study are to evaluate the efficacy, safety and tolerability of ELND005 over 12 weeks of treatment in patients with mild to severe AD, who are experiencing at least moderate levels of agitation/aggression. Enrollment of this clinical study (AG201) known as the “Harmony AD” study (www.harmonyadstudy.com) was completed on March 2, 2015 with a total of 350 patients being enrolled.
On June 24, 2015, Transition announced that ELND005 did not meet its primary efficacy endpoints in the Phase 2/3 clinical study of ELND005 in agitation and aggression in patients with Alzheimer’s Disease. In the study, both the treatment and placebo groups showed a significant, but similar, reduction in agitation and aggression relative to baseline. There was a greater than expected reduction in agitation and aggression observed in the placebo group as measured in weeks 4, 8 and 12 in the study. The safety and tolerability profile of ELND005 was consistent with previous studies in AD at the 250mg bid dose.
As ELND005 did not meet its primary efficacy endpoint in the Phase 2/3 clinical study in agitation and aggression in Alzheimer’s disease, management performed an impairment test and noted there is no impairment of the ELND005 asset as at June 30, 2015. The Company is performing a thorough review of the data from the completed study in agitation and aggression. An external clinical advisory board is working with the Company to evaluate the data and consider potential future clinical development paths for ELND005.
On November 20, 2014, Transition announced the results of a clinical study of neuropsychiatric drug candidate ELND005 in young adults with Down syndrome. Transition’s wholly-owned subsidiary, TTIL completed this first study in Down syndrome subjects without dementia to allow optimal dose selection for future larger studies.
The study enrolled 23 Down syndrome subjects in three study arms over a four-week treatment period: placebo, 250 mg once daily; and 250 mg twice daily. At the doses evaluated, ELND005 was determined to have an acceptable safety and tolerability profile and there were no serious adverse events reported in the study. Treatment emergent adverse events were reported in seven of the subjects receiving ELND005 and all were deemed mild in severity. The two ELND005 doses achieved the plasma levels expected in pharmacokinetic modeling and will inform the selection of a higher dose in a larger Phase 2b study in Down syndrome subjects.
The ELND005 technology is claimed in multiple issued patents and pending patent applications in many jurisdictions throughout the world.
Expenditures for the ELND005 Program
On February 28, 2014, Transition announced that after a series of transactions, Perrigo has transferred all of its ELND005 rights and assets to the Company’s wholly owned subsidiary, TTIL. As a result, effective March 1, 2014, TTIL is responsible for all future development and commercialization activities of the ELND005 drug candidate.
During the years ended June 30, 2015 and 2014, the Company incurred direct research and development costs for this program as follows:
| ELND005 Program(1) | | Fiscal 2015 | | | Fiscal 2014 | |
| | | | | | | | | |
| Pre-clinical studies | | $ | - | | | $ | - | |
| Clinical studies | | | 20,154,069 | | | | 8,473,306 | |
| Manufacturing | | | 796,511 | | | | 270,241 | |
| Other direct research | | | 2,356,831 | | | | 955,608 | |
| TOTAL | | $ | 23,307,411 | | | $ | 9,699,155 | |
Note(1)These costs are direct research and development costs only and do not include patent costs, investment tax credits, salaries and benefits, amortization of intangible assets or an allocation of Company overhead.
Prior to the February, 2014 acquisition, Transition was not required to fund the development or commercialization of ELND005 and accordingly, development costs were nil during the first eight months of fiscal 2014.
TT401
Development of TT401 for Diabetes
Diabetes is a disease in which the body does not produce or properly use insulin. Insulin is a hormone released from islet cells located in the pancreas that is needed to convert sugar, starches and other food into energy needed for daily life. There are two primary forms of diabetes; type 1 diabetes and type 2 diabetes.
Type 2 diabetes usually begins as insulin resistance, a disorder in which the cells do not use insulin properly. As the need for insulin increases, the pancreas gradually loses its ability to produce it. Current treatments for type 2 diabetes include lifestyle changes, oral medications, incretin therapy and insulin therapy. Type 2 diabetes accounts for about 90-95% of all diagnosed cases of diabetes.
Clinical Development Update of TT401 (LY2944876)
On March 3, 2010, Transition announced that it had acquired the exclusive worldwide rights to develop and potentially commercialize a series of preclinical compounds from Lilly in the area of diabetes. In preclinical diabetes models, these compounds showed potential to provide glycemic control and other beneficial effects including weight loss.
On June 18, 2012, Transition announced the results of the Phase 1 clinical study of type 2 diabetes drug candidate, TT401. The Phase 1, double-blind, placebo-controlled randomized study enrolled 48 non-diabetic obese subjects in six cohorts evaluating six escalating subcutaneous single doses of TT401. TT401 demonstrated an acceptable safety and tolerability profile in non-diabetic obese subjects in the study. TT401 exhibited the expected pharmacological effect on glucose and pharmacodynamic biomarkers at doses that were safe and tolerable. The pharmacokinetic profile, assessed over 28 days, demonstrated a half-life consistent with once-weekly dosing.
On April 30, 2013, Transition announced the results of a five-week proof of concept clinical study of TT401 in type 2 diabetes and obese non-diabetic subjects. The study enrolled diabetic patients at five dosing levels and non-diabetic obese patients at one dose level. All dosing cohorts received five doses over a five week period. Diabetic patients were on stable doses of metformin.
At the end of the treatment period, TT401-treated patients in the 3 highest dose groups experienced statistically significant reductions in mean fasting plasma glucose relative to placebo. Statistically significant mean body weight reduction relative to baseline occurred in the three highest dose groups. A similar reduction in body weight was also observed in the obese non-diabetic cohort. TT401 demonstrated an acceptable safety and tolerability profile at all doses evaluated in diabetic and non-diabetic obese subjects. The most common adverse event noted in the study was decreased appetite. Some subjects in the highest three dose groups experienced mild nausea and vomiting, which are consistent with studies of other GLP-1 agonist drug candidates. The pharmacokinetic profile, assessed over the five week study, demonstrated a half-life consistent with once-weekly dosing.
On June 17, 2013, Lilly exercised its option to assume all development and commercialization rights to type 2 diabetes drug candidate TT401. In conjunction with this assumption of rights, Transition received a US$7 million milestone payment. Lilly and Transition have amended their agreement to address future development of TT401 and associated financial arrangements. Lilly has assumed all costs and will perform all future development and commercialization activities of TT401. In May, 2014, Transition announced the dosing of the first patient in a Phase 2 clinical study of TT401. The study is expected to enroll up to 375 type 2 diabetes subjects and will be performed by Transition’s development partner Lilly. The objectives of the study will be to evaluate the safety and effectiveness of TT401 compared to once-weekly exenatide extended release and placebo. In February 2015, Lilly informed Transition that 420 type 2 diabetic subjects had been enrolled in the current Phase 2 study, thereby completing the enrollment phase of the study.
Transition has made three separate installments to Lilly during the Phase 2 clinical study totaling US$14 million to Lilly. The first installment of US$6 million was paid in September 2014 when the study achieved 20% patient enrollment.The remaining two installments totaling US$8 million were paid during the three month period ended December 31, 2014 when the study achieved both the 50% and 70% patient enrollment milestones. There are no additional funding obligations related to this clinical study.
Expenditures for the TT401 Program
During the years ended June 30, 2015 and 2014, the Company incurred direct research and development costs for this program as follows:
| TT401/402 Program(1) | | Fiscal 2015 | | | | Fiscal 2014 | |
| Pre-clinical studies | | $ | - | | | | $ | 7,488 | |
| Clinical studies | | | - | | | | | 87,379 | |
| Manufacturing | | | - | | | | | (37,419 | ) |
| Other direct research | | | - | | | | | 37,803 | |
| Development payments to Lilly | | | 15,491,600 | | | | | - | |
| TOTAL | | $ | 15,491,600 | | | | $ | 95,251 | |
Note(1)These costs are direct research and development costs only and do not include patent costs, investment tax credits, salaries and benefits, amortization of intangible assets or an allocation of Company overhead.
TT701 for Androgen Deficiency
On May 6, 2015, TTIL exclusively licensed worldwide rights to a novel small molecule drug candidate TT701 from Lilly. TT701 is a selective androgen receptor modulator that has been shown in a Phase 2 study to significantly increase lean body mass and a measurement of muscle strength in male subjects.
Clinical Development of TT701
Since acquiring the exclusive worldwide rights to TT701 the Company has incurred drug development manufacturing costs as they prepare to move the drug candidate into a Phase 2 clinical trial. TTIL has been actively working with potential partners to collaborate on the clinical development of TT701 and it is expected that a Phase 2 study of TT701 will commence before the end of calendar 2015.
Expenditures for the TT701 Program
During the years ended June 30, 2015 and 2014, the Company incurred direct research and development costs for this program as follows:
| TT701 Program(1) | | Fiscal 2015 | | | Fiscal 2014 | |
| Pre-clinical studies | | $ | - | | | $ | - | |
| Clinical studies | | | - | | | | - | |
| Manufacturing | | | 253,729 | | | | - | |
| Other direct research | | | 41,034 | | | | - | |
| TOTAL | | $ | 294,763 | | | $ | - | |
Note(1)These costs are direct research and development costs only and do not include patent costs, investment tax credits, salaries and benefits, amortization of intangible assets or an allocation of Company overhead.
The Next Steps
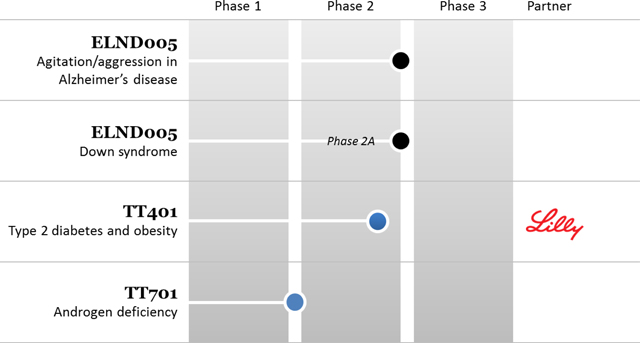
Transition’s goal for its programs is to achieve product approval and ultimately significant revenues or royalties. To achieve product approval, the Company and or its partners, must successfully complete clinical trials and achieve regulatory approval. The stages of development of the Company’s technologies are illustrated below:
OVERALL PERFORMANCE
During the year ended June 30, 2015, the Company recorded a net loss of $51,339,528 ($1.41 loss per common share) compared to a net loss of $21,782,255 ($0.72 loss per common share) for the year ended June 30, 2014.
During the fiscal year ended June 30, 2015, the Company reported an increase in net loss of $29,557,273 compared to the fiscal year ending June 30, 2014. The increase in net loss is due to the significant increase in research and development expenses resulting from the reacquisition of the rights to develop the ELND005 drug candidate, as well as the US$14 million milestone payments made to Lilly and increased general and administration expenses. The increase in net loss has been partially offset by the settlement of a pre-existing relationship recognized in connection with the re-acquisition of the ELND005 asset in February 2014, increased foreign exchange gains and the change in fair value of contingent consideration payable.
On February 18, 2015, the Company announced the closing of its underwritten public offering of an aggregate of 3,538,461 common shares at a price to the public of US$6.50 per share, including 461,538 common shares issued upon the exercise of the underwriters’ over-allotment option, raising gross proceeds of $28,561,400 (US$23.0 million). The Company incurred total share issuance costs of $2,492,010, resulting in net cash proceeds of $26,069,390.
At June 30, 2015, the Company has $40,510,758 in cash and a working capital of $32,026,606.
The Company’s current cash projection indicates that the current cash resources should enable the Company to execute its core business plan and meet its projected cash requirements beyond the next 12 months.
SELECTED ANNUAL INFORMATION
The following table is a summary of selected financial information from the audited consolidated financial statements of the Company for each of the three most recently completed financial years.
| | | June 30, | | | June 30, | | | June 30, | |
| | | 2015 | | | 2014 | | | 2013 | |
| | | $ | | | $ | | | $ | |
| Revenue | | | — | | | | — | | | | 17,933,500 | |
| Net income (loss) (1) | | | (51,339,528 | ) | | | (21,782,255 | ) | | | 23,297 | |
| Basic and diluted net income (loss) per common share | | | (1.41 | ) | | | (0.72 | ) | | | - | |
| Total assets | | | 49,649,085 | | | | 68,907,236 | | | | 37,807,955 | |
| Total long-term liabilities (2) | | | 3,503,344 | | | | 3,849,718 | | | | 1,457,821 | |
| Cash dividends declared per share | | | — | | | | — | | | | — | |
Note(1)Net income (loss) before discontinued operations and extraordinary items was equivalent to the net income (loss) for such periods.
Note(2)Total long-term liabilities represents contingent consideration payable as set forth in the Company’s audited consolidated financial statements for the year ended June 30, 2015. For the years ended June 30, 2014 and 2013, total long-term liabilities also includes leasehold inducement.
ANNUAL RESULTS – YEAR ENDED JUNE 30, 2015 COMPARED TO YEAR ENDED JUNE 30, 2014
RESULTS OF OPERATIONS
Research and Development
Research and development expenses increased $31,842,318 or 183% from $17,367,385 for the fiscal year ended June 30, 2014 to $49,209,703 for the fiscal year ended June 30, 2015.
The increases in research and development expenses are primarily due to increases in development costs related to ELND005. The increases are also attributed to increases in development costs associated with diabetes drug candidate TT401 as during fiscal 2015 the Company paid Lilly an aggregate of US$14 million upon the achievement of all three patient enrollment milestones. The increase in research and development costs have been partially offset by decreases in clinical development costs associated with the costs related to the TT601 program.
The Company anticipates that research and development expenses will decrease significantly during fiscal 2016 as the Company discontinues the safety extension trial AG251 in agitation and aggression in Alzheimer’s disease and has no further funding obligations to Lilly for the ongoing Phase 2 clinical study of diabetes drug candidate TT401. The decrease will be offset by incurring costs relating to the development of TT701, a novel small molecule drug candidate licensed from Lilly.
General and Administrative
General and administrative expenses increased by $787,698 or 17% from $4,726,574 for the fiscal year ended June 30, 2014 to $5,514,272 for the fiscal year ended June 30, 2015.
The increases in general and administrative expenses are primarily due to increases in compensation and overhead costs relating to the Company’s premises in San Mateo, California.
The Company anticipates general and administrative expenses will decrease during fiscal 2016 as all activities based out of the San Mateo, California location will be transferred to head office, which will result in a reduction in general and administrative expenses.
Settlement of a Pre-existing Relationship
During the comparative year ended June 30, 2014, the Company recognized an expense of $3,096,186 as a settlement of a pre-existing relationship relating to the collaboration agreement with Elan. The Company did not recognize a similar expense during the year ended June 30, 2015.
Change in Fair Value of Contingent Consideration Payable
Contingent consideration is required to be measured as a financial liability at fair value and re-measured at each reporting date. Management revisited the assumptions used in the valuation of the contingent consideration payable and accordingly, the Company has recognized an increase in the fair value of contingent consideration payable of $65,787 during the fiscal year ended June 30, 2015.
During the comparative year ended June 30, 2014, the Company recognized a change in fair value of contingent consideration payable of $2,911,218.
SUMMARY OF QUARTERLY RESULTS
The following table is a summary of selected quarterly consolidated financial information of the Company for each of the eight most recently completed quarters ending at June 30, 2015.
| | | First | | | Second | | | Third | | | Fourth | | | | |
| | | Quarter | | | Quarter | | | Quarter | | | Quarter | | | Total | |
| 2015 | | | | | | | | | | | | | | | | | | | | |
| Revenue | | | - | | | | - | | | | - | | | | - | | | | - | |
| Net income (loss)(1) | | $ | (15,695,324 | ) | | $ | (16,910,139 | ) | | $ | (4,748,096 | ) | | $ | (13,985,969 | ) | | $ | (51,339,528 | ) |
| Basic and diluted net income (loss) per common share | | $ | (0.45 | ) | | $ | (0.48 | ) | | $ | (0.13 | ) | | $ | (0.38 | ) | | $ | (1.41 | ) |
| 2014 | | | | | | | | | | | | | | | | | | | | |
| Revenue | | | - | | | | - | | | | - | | | | - | | | | - | |
| Net income (loss) (1) | | $ | (2,331,186 | ) | | $ | (1,253,772 | ) | | $ | (5,067,292 | ) | | $ | (13,130,005 | ) | | $ | (21,782,255 | ) |
| Basic and diluted net income (loss) per common share | | $ | (0.08 | ) | | $ | (0.04 | ) | | $ | (0.17 | ) | | $ | (0.43 | ) | | $ | (0.72 | ) |
Note(1) Net income (loss) before discontinued operations was equivalent to the net income (loss) for such periods. The net income (loss) represented in the chart excludes any amounts for items that may be subsequently reclassified as net income such as cumulative translation adjustment.
The fluctuations of Transition’s quarterly results are primarily due to milestone payments made to Lilly to help fund TT401 Phase 2 clinical development and changes in: activity levels of the clinical trials being performed by the Company and foreign exchange gains and losses.
FOURTH QUARTER RESULTS
The following table is a summary of selected information for the three month periods ended June 30, 2015 and June 30, 2014:
| | | 2015 | | | 2014 | |
| | | ($) | | | ($) | |
| Revenue – Licensing fees | | | - | | | | - | |
| Research and development, net | | | 12,381,651 | | | | 10,464,484 | |
| General and administrative | | | 1,736,460 | | | | 1,673,616 | |
| Impairment of intangible assets | | | - | | | | - | |
| Interest income | | | 49,522 | | | | 56,250 | |
| Net loss | | | 13,985,969 | | | | 13,130,005 | |
Review of Operations
For the three month period ended June 30, 2015, the Company’s net loss increased by $855,964 or 7% to $13,985,969 from $13,130,005 for the same period in fiscal 2014.
Research and development expenses increased by $1,917,167 or 18% to $12,381,651 compared to $10,464,484 for the same period in fiscal 2014. This increase was primarily due to an increase in clinical development costs related to the re-acquired rights to the drug candidate ELND005 and the development of the recently in-license TT701, which has been partially offset by decreases in clinical development costs associated with pre-clinical research on TT601.
General and administrative expenses increased by $62,844 or 4% to $1,736,460 from $1,673,616 for the same period in fiscal 2014. This increase was primarily due to increases in compensation and overhead costs relating to the Company’s premises in San Mateo, California.
Due to changes in assumptions relating to the development of ELND005, the Company has recognized a change in fair value of contingent consideration payable of $681,911 during the three month period ended June 30, 2015. During the comparative period in fiscal 2014, the company recognized a change in fair value of contingent consideration payable of $129,311 due to the passage of time.
CRITICAL ACCOUNTING ESTIMATES AND JUDGMENTS
The preparation of consolidated financial statements in accordance with IFRS requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results can differ from those estimates. We have identified the following areas which we believe require management’s most subjective estimates and judgments, often requiring the need to make estimates about the effects of matters that are inherently uncertain and may change in subsequent periods.
Valuation and Amortization of Intangible Assets
The Company’s intangible assets are comprised of purchased or licensed pharmaceutical compounds, technology and patents. The costs of the Company’s intangible assets are amortized over the estimated useful life of up to 20 years. Factors considered in estimating the useful life of the intangible asset include the expected use of the asset by the Company, legal, regulatory and contractual provisions that may limit the useful life, the effects of competition and other economic factors, and the level of expenditures required to obtain the expected future cash flows from the intangible asset. The Company re-evaluates the useful life when there has been a change in these factors. The Company assesses its intangible assets for recoverability whenever indicators of impairment exist. As ELND005 did not meet its primary efficacy endpoint in the Phase 2/3 clinical study in agitation and aggression in Alzheimer’s disease, management performed an impairment test and noted there is no impairment of the ELND005 asset as at June 30, 2015. The Company is performing a thorough review of the data from the completed study in agitation and aggression. An external clinical advisory board is working with the Company to evaluate the data and consider potential future clinical development paths for ELND005.
When the carrying value of an asset is greater than its recoverable amount, which is the higher of its value in use or fair value less costs to sell, an impairment loss is recognized.
Valuation of Contingent Consideration Payable
The contingent consideration is measured at fair value based on level 3 inputs. The contingent consideration is not based on observable inputs and is measured using a discounted cash flow analysis of expected payments in future periods. The significant estimates used in the fair value calculations are as follows:
| (a) | Management has estimated the timing of the milestone payments based on current expectations and plans for the development of ELND005. The milestone payments are assigned a probability based on industry statistics for the successful development of pharmaceutical products including regulatory approval and achievement of revenue targets. An increase of 10% applied to the probability assumptions, with all other variables held constant, will increase the contingent consideration payable by $1,428,951. Conversely a decrease of 10% applied to the probability assumptions, with all other variables held constant, would reduce the contingent consideration payable by $1,858,858; |
| (b) | The probability adjusted cash flows are discounted at a rate of 20% which is management’s best estimate of the Company’s cost of capital. An increase of 5% to the discount rate would decrease the contingent consideration payable by $1,080,299. Conversely, a decrease of 5% to the discount rate would increase the contingent consideration payable by $1,538,400. |
Management revisited the assumptions used in the valuation of the contingent consideration payable and accordingly, the Company has recognized a change in fair value of contingent consideration payable of $65,787 during the fiscal year ended June 30, 2015.
Share Based Payments and Warrants
When the Company issues stock options and warrants, an estimate of fair value is derived for the equity instrument using the Black-Scholes option pricing model. The application of this option pricing model requires management to make assumptions regarding several variables, including the period for which the instrument will be outstanding, the price volatility of the Company’s stock over a relevant timeframe, the determination of a relevant risk free interest rate and an assumption regarding the Company’s dividend policy in the future. If other assumptions are used, the value derived for the equity instruments could be significantly impacted.
Settlement of a Pre-Existing Relationship
The Company has determined that the transactions entered into with Perrigo on February 28, 2014 have resulted in the re-acquisition of the rights to the development and commercialization of ELND005 previously licensed to Elan which in accordance with IFRS must be accounted for as a settlement of a pre-existing relationship (the collaboration agreement between Waratah and Elan). Accordingly, the company expensed $3,096,186 in fiscal 2014 as the cost related to the settlement of the pre-existing relationship.
ACCOUNTING CHANGES
The following accounting policies have been adopted effective July 1, 2014:
IAS 36 – Impairment of Assets
IAS 36 has been amended to include limited scope amendments to the impairment disclosures. The amendments are effective for annual periods beginning on or after January 1, 2014. The adoption of IAS 36 did not significantly impact the Company’s consolidated financial statements;
IFRS 2 – Share Based Payments
IFRS 2 has been amended to clarify the definition of vesting conditions. The amendments are effective for annual periods beginning on or after July 1, 2014. The adoption of IFRS 2 did not significantly impact the Company’s consolidated financial statements.
IFRS ISSUED BUT NOT YET ADOPTED
IFRS 15 – Revenue from Contracts with Customers
IFRS 15 specifies how and when to recognize revenue as well as requiring entities to provide users of financial statements with some informative, relevant disclosures. The standard supersedes IAS 18, Revenue, IAS 11, Construction Contracts, and a number of revenue-related interpretations. Application of the standard is mandatory for all IFRS reporters and it applies to nearly all contracts with customers: the main exceptions are leases, financial instruments and insurance contracts. Currently IFRS 15 must be applied in an entity’s first annual IFRS financial statements for periods beginning on or after January 1, 2017 however the IASB has proposed to defer the date of adoption to periods beginning on or after January 1, 2018, with early adoption permitted. Management is evaluating the standard and has not yet determined the impact on its consolidated financial statements.
INTERNAL CONTROLS OVER FINANCIAL REPORTING
Internal controls over financial reporting are designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with IFRS.
Management’s Evaluation of Disclosure Controls and Procedures
Under the supervision and with the participation of management, including the Company’s CEO and CFO, the Company conducted an evaluation of the effectiveness of its disclosure controls and procedures as of June 30, 2015 as required by Canadian securities legislation. Disclosure controls and procedures (as defined in Rule 13a-15(e) under the Securities Exchange Act of 1934) are designed to ensure that the information required to be disclosed by the Company in the reports it files or submits under securities legislation is recorded, processed, summarized and reported on a timely basis and that such information is accumulated and reported to management, including the Company’s CEO and CFO, as appropriate, to allow required disclosures to be made in a timely fashion. Based on their evaluation, the CEO and CFO have concluded that as of June 30, 2015, the Company’s disclosure controls and procedures were effective.
Management’s Report on Internal Control over Financial Reporting
The Company’s management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) under the Securities Exchange Act of 1934). The Company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Company’s management assessed the effectiveness of the Company’s internal control over financial reporting as of June 30, 2015. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (2013) (“COSO”) in the updated Internal Control-Integrated Framework. The Company’s management, including the CEO and CFO, concluded that, as of June 30, 2015, the Company’s internal control over financial reporting was effective based on the criteria in Internal Control — Integrated Framework issued by COSO.
The effectiveness of the Company’s internal control over financial reporting as of June 30, 2015 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in its report in the Company’s audited consolidated financial statements for the year ended June 30, 2015.
LIQUIDITY AND CAPITAL RESOURCES
Overview
The Company commenced operations in July 1998, and has devoted its resources primarily to fund its research and development programs. All revenue to date has been generated from milestone payments and licensing fees. The Company has incurred a cumulative deficit to June 30, 2015 of $222,454,699. Losses are expected to continue for the next several years as the Company invests in research and development, preclinical studies, clinical trials, manufacturing and regulatory compliance.
Since inception, the Company has been financed primarily from public and private sales of equity, the exercise of warrants and stock options, interest earned on cash deposits and short term investments and revenues and reimbursements from partners.
The Company’s cash was $40,510,758 at June 30, 2015 as compared to cash and short term investments of $60,271,566 at June 30, 2014, resulting in a decrease of $19,760,808. The Company’s working capital position at June 30, 2015 decreased $22,751,265 from $54,777,871 at June 30, 2014 to $32,026,606, at June 30, 2015.
The decrease in the Company’s cash and short term investments as well as the decrease in working capital are primarily due to the expenditures incurred during the fiscal year ended June 30, 2015 which included three milestone payments totaling US$14 million paid to Lilly upon the achievement of all three patient enrollment milestones for the TT401 Phase 2 diabetes study. The decrease is offset by the February 18, 2015 public offering of 3,538,461 common shares which resulted in net proceeds of $26,069,390.
The Company’s current cash projection indicates that the current cash resources should enable the Company to execute its core business plan and meet its projected cash requirements for the next 12 months.
The success of the Company is dependent on its ability to bring its products to market, obtain the necessary regulatory approvals and achieve future profitable operations. The continuation of the research and development activities and the commercialization of its products are dependent on the Company’s ability to successfully complete these activities and to obtain adequate financing through a combination of financing activities, operations, and partnerships. It is not possible to predict either the outcome of future research and development programs or the Company’s ability to fund these programs going forward.
Financial Instruments
Financial instruments of the Company consist mainly of cash, short term investments, other receivables, accounts payable and accrued liabilities, and contingent consideration payable. Management’s primary investment objective is to maintain safety of principal and provide adequate liquidity to meet all current payment obligations and future planned expenditures.
The Company is exposed to market risks related to volatility in interest rates for the Company’s investment portfolio and foreign currency exchange rates related to cash and purchases of supplies and services made in U.S. dollars.
The Company is exposed to interest rate risk to the extent that the cash is held in deposit accounts which earn interest at variable rates. The Company’s maximum exposure to interest rate risk is based on the effective interest rate of the current carrying value of these assets. The Company does not speculate on interest rates and holds all deposits until their date of maturity.
Contractual Obligations
Minimum payments under our contractual obligations are as follows:
| | | Less than 1 Year | | | 1- 3 Years | | | 4 – 5 Years | | | After 5 Years | | | Total | |
| Operating leases | | $ | 238,179 | | | $ | 343,165 | | | $ | 307,453 | | | $ | - | | | $ | 888,797 | |
| Clinical and toxicity study agreements | | | 3,545,412 | | | | - | | | | - | | | | - | | | | 3,545,412 | |
| Manufacturing agreements | | | 214,857 | | | | - | | | | - | | | | - | | | | 214,857 | |
| Contingent Consideration Payable | | | - | | | | 2,847,759 | | | | - | | | | 58,028,760 | | | | 60,876,519 | |
| Other | | | 327,218 | | | | - | | | | - | | | | - | | | | 327,218 | |
| TOTAL | | $ | 4,325,666 | | | $ | 3,190,924 | | | $ | 307,453 | | | $ | 58,028,760 | | | $ | 65,852,803 | |
Contractual obligations denominated in US dollars have been translated to Canadian dollars using the exchange rate at June 30, 2015.
PROPOSED TRANSACTIONS
On July 19, 2013, the Company’s shelf registration statement filed with the United States Securities and Exchange Commission (“SEC”) on Form F-3 became effective. The shelf prospectus provides for the potential offering in the United States of up to an aggregate amount of US$50 million of Transition’s common shares, warrants, or a combination thereof, from time to time in one or more offerings until July 19, 2016.
On January 5, 2015, the Company filed with the SEC a prospectus supplemental to the shelf prospectus and a sales agreement with Cowen and Company, LLC or Cowen, relating to the sale of the Company’s common shares. In accordance with the terms of the sales agreement, the Company may offer and sell from time to time common shares having an aggregate offering price of up to US $25 million with Cowen acting as sales agent. After the closing of the February, 2015 US$23 million public offering, the Company can raise an additional US$27 million through the issuance of common shares, warrants or a combination thereof, from time to time in in one of more offerings until July 19, 2016.
Utilization of the US shelf prospectus is dependent upon meeting certain market capitalization thresholds at the time of financing.
RELATED PARTY TRANSACTIONS
During fiscal 2015, the Company paid legal fees to a law firm where the Company’s Secretary is a partner and to a corporation controlled by the Company’s Secretary. Total fees and disbursements charged to the Company by these companies was $45,346 for fiscal 2015 and $49,000 for fiscal 2014 and are included in general and administrative expenses. The balance owing at June 30, 2015 and 2014 is nil.
Members of the Company’s Board of Directors, management and employees participated in both the August, 2013 and June, 2014 private placements.
These transactions occurred in the normal cause of operations and were measured at the exchange amount, which is the amount of consideration established and agreed to by the related parties.
OUTSTANDING SHARE DATA
Authorized
The authorized share capital of the Company consists of an unlimited number of common shares.
Issued and Outstanding
The following details the issued and outstanding equity securities of the Company:
Common Shares
As at September 14, 2015 the Company has 38,878,879 common shares outstanding.
Stock Options
As at September 14, 2015 the Company has 2,408,381 stock options outstanding with exercise prices ranging from $2.09 to $10.19 and various expiry dates extending to June 14, 2025. At September 14, 2015, on an if-converted basis, these stock options would result in the issuance of 2,408,381 common shares in the capital of the Company at an aggregate exercise price of $11,430,633.
Warrants
As at September 14, 2015, the Company has a total of 1,949,250 warrants outstanding with a purchase price of US$7.10.
Each warrant entitles the holder, within two years of the June 23, 2013 issuance date, to purchase one additional common share in the capital of the Company.
RISKS AND UNCERTAINTIES
Investing in the Company’s securities involves a high degree of risk. Before making an investment decision, individuals should carefully consider the following risk factors, in addition to the other information provided in this MD&A and the Company’s other disclosure documents filed on www.sedar.com.
The Company will require significant additional financing and it may not have access to sufficient capital.
The Company anticipates that it will need additional financing in the future to fund its ongoing research and development programs and for general corporate requirements. The Company may choose to seek additional funding through public or private offerings, corporate collaborations or partnership arrangements. The amount of financing required will depend on many factors including the financial requirements of the Company to fund its research and clinical trials, and the ability of the Company to secure partnerships and achieve partnership milestones as well as to fund other working capital requirements. The Company’s ability to access the capital markets or to enlist partners is mainly dependent on the progress of its research and development and regulatory approval of its products. There is no assurance that additional funding will be available on acceptable terms, if at all.
The Company has a history of losses, and it has not generated any product revenue to date. It may never achieve or maintain profitability.
Since inception, the Company has incurred significant losses each year and expects to incur significant operating losses as the Company continues product research and development and clinical trials. There is no assurance that the Company will ever successfully commercialize or achieve revenues from sales of its therapeutic products if they are successfully developed or that profitability will ever be achieved or maintained. Even if profitability is achieved, the Company may not be able to sustain or increase profitability.
The Company is an early stage development company in an uncertain industry.
The Company is at an early stage of development. Preclinical and clinical trial work must be completed before our products could be ready for use within the markets we have identified. We may fail to develop any products, to obtain regulatory approvals, to enter clinical trials or to commercialize any products. The Company does not know whether any of our potential product development efforts will prove to be effective, meet applicable regulatory standards, obtain the requisite regulatory approvals or be capable of being manufactured at a reasonable cost. If the Company’s products are approved for sale, there can be no assurance that the products will gain market acceptance among consumers, physicians, patients and others in the medical community. A failure to gain market acceptance may adversely affect the revenues of the Company.
The Company is subject to a strict regulatory environment.
None of the Company’s product candidates have received regulatory approval for commercial sale.
Numerous statutes and regulations govern human testing and the manufacture and sale of human therapeutic products in Canada, the United States and other countries where the Company intends to market its products. Such legislation and regulation bears upon, among other things, the approval of protocols and human testing, the approval of manufacturing facilities, testing procedures and controlled research, review and approval of manufacturing, preclinical and clinical data prior to marketing approval including adherence to Good Manufacturing Practices (“GMP”) during production and storage as well as regulation of marketing activities including advertising and labelling.
The completion of the clinical testing of our product candidates and the obtaining of required approvals are expected to take years and require the expenditure of substantial resources. There can be no assurance that clinical trials will be completed successfully within any specified period of time, if at all. Furthermore, clinical trials may be delayed or suspended at any time by the Company or by regulatory authorities if it is determined at any time that patients may be or are being exposed to unacceptable health risks, including the risk of death, or that compounds are not manufactured under acceptable GMP conditions or with acceptable quality. Any failure or delay in obtaining regulatory approvals would adversely affect the Company’s ability to utilize its technology thereby adversely affecting operations. No assurance can be given that the Company’s product candidates or lead compounds will prove to be safe and effective in clinical trials or that they will receive the requisite protocol approval or regulatory approval. Furthermore, no assurance can be given that current regulations relating to regulatory approval will not change or become more stringent. There are no assurances the Company can scale-up, formulate or manufacture any compound in sufficient quantities with acceptable specifications for the regulatory agencies to grant approval or not require additional changes or additional trials be performed. The agencies may also require additional trials be run in order to provide additional information regarding the safety, efficacy or equivalency of any compound for which the Company seeks regulatory approval. Similar restrictions are imposed in foreign markets other than the United States and Canada. Investors should be aware of the risks, problems, delays, expenses and difficulties which may be encountered by the Company in light of the extensive regulatory environment in which the Company’s business operates.
Even if a product candidate is approved by the FDA or any other regulatory authority, the Company may not obtain approval for an indication whose market is large enough to recoup its investment in that product candidate. The Company may never obtain the required regulatory approvals for any of its product candidates.
The Company is faced with uncertainties related to its research.
The Company’s research programs are based on scientific hypotheses and experimental approaches that may not lead to desired results. In addition, the timeframe for obtaining proof of principle and other results may be considerably longer than originally anticipated, or may not be possible given time, resource, financial, strategic and collaborator scientific constraints. Success in one stage of testing is not necessarily an indication that the particular program will succeed in later stages of testing and development. It is not possible to predict, based upon studies in in-vitro models and in animals, whether any of the compounds made for these programs will prove to be safe, effective, and suitable for human use. Each compound will require additional research and development, scale-up, formulation and extensive clinical testing in humans. Decisions regarding future development activities may be based on results from completed studies or interim results from on-going studies or projections derived from interim or administrative analyses of studies not yet completed. Development of these compounds will require investigations into the mechanism of action of the molecules as these are not fully understood. Unsatisfactory results obtained from a particular study relating to a program may cause the Company to abandon its commitment to that program or to the lead compound or product candidate being tested. The discovery of unexpected toxicities, lack of sufficient efficacy, poor physiochemical properties, unacceptable ADME (absorption, distribution, metabolism and excretion) and DMPK (drug metabolism and pharmacokinetics), pharmacology, inability to increase scale of manufacture, market attractiveness, regulatory hurdles, competition, as well as other factors, may make the Company’s targets, lead compounds or product candidates unattractive or unsuitable for human use, and the Company may abandon its commitment to that program, target, lead compound or product candidate. In addition, preliminary results seen in animal and/or limited human testing may not be substantiated in larger controlled clinical trials.
If difficulties are encountered enrolling patients in the Company’s clinical trials, the Company’s trials could be delayed or otherwise adversely affected.
Clinical trials for the Company’s product candidates require that the Company identify and enrol a large number of patients with the disorder under investigation. The Company may not be able to enrol a sufficient number of patients to complete its clinical trials in a timely manner. Patient enrolment is a function of many factors including, but not limited to, design of the study protocol, size of the patient population, eligibility criteria for the study, the perceived risks and benefits of the therapy under study, the patient referral practices of physicians and the availability of clinical trial sites. If the Company has difficulty enrolling a sufficient number of patients to conduct the Company’s clinical trials as planned, it may need to delay or terminate ongoing clinical trials.
Even if regulatory approvals are obtained for the Company’s product candidates, the Company will be subject to ongoing government regulation.
Even if regulatory authorities approve any of the Company’s human therapeutic product candidates, the manufacture, marketing and sale of such products will be subject to strict and ongoing regulation. Compliance with such regulation may be expensive and consume substantial financial and management resources. If the Company, or any future marketing collaborators or contract manufacturers, fail to comply with applicable regulatory requirements, it may be subject to sanctions including fines, product recalls or seizures, injunctions, total or partial suspension of production, civil penalties, withdrawal of regulatory approvals and criminal prosecution. Any of these sanctions could delay or prevent the promotion, marketing or sale of the Company’s products.
The Company may not achieve its projected development goals in the time frames announced and expected.
The Company sets goals for and makes public statements regarding the timing of the accomplishment of objectives material to its success, such as the commencement and completion of clinical trials, anticipated regulatory submission and approval dates and time of product launch. The actual timing of these events can vary dramatically due to factors such as delays or failures in the Company’s clinical trials, the uncertainties inherent in the regulatory approval process and delays in achieving manufacturing or marketing arrangements sufficient to commercialize its products.
There can be no assurance that the Company’s clinical trials will be completed, that the Company will make regulatory submissions or receive regulatory approvals as planned. If the Company fails to achieve one or more of these milestones as planned, the price of the Common Shares would likely decline.
If the Company fails to obtain acceptable prices or adequate reimbursement for its human therapeutic products, its ability to generate revenues will be diminished.
The Company’s ability to successfully commercialize its human therapeutic products will depend significantly on its ability to obtain acceptable prices and the availability of reimbursement to the patient from third-party payers, such as government and private insurance plans. While the Company has not commenced discussions with any such parties, these third-party payers frequently require companies to provide predetermined discounts from list prices, and they are increasingly challenging the prices charged for pharmaceuticals and other medical products. The Company’s human therapeutic products may not be considered cost-effective, and reimbursement to the patient may not be available or sufficient to allow the Company to sell its products on a competitive basis. The Company may not be able to negotiate favourable reimbursement rates for its human therapeutic products.
In addition, the continuing efforts of third-party payers to contain or reduce the costs of healthcare through various means may limit the Company’s commercial opportunity and reduce any associated revenue and profits. The Company expects proposals to implement similar government control to continue. In addition, increasing emphasis on managed care will continue to put pressure on the pricing of pharmaceutical and biopharmaceutical products. Cost control initiatives could decrease the price that the Company or any current or potential collaborators could receive for any of its human therapeutic products and could adversely affect its profitability. In addition, in Canada and in many other countries, pricing and/or profitability of some or all prescription pharmaceuticals and biopharmaceuticals are subject to government control.
If the Company fails to obtain acceptable prices or an adequate level of reimbursement for its products, the sales of its products would be adversely affected or there may be no commercially viable market for its products.
The Company may not obtain adequate protection for its products through its intellectual property.
The Company’s success depends, in large part, on its ability to protect its competitive position through patents, trade secrets, trademarks and other intellectual property rights. The patent positions of pharmaceutical and biopharmaceutical firms, including the Company, are uncertain and involve complex questions of law and fact for which important legal issues remain unresolved. The patents issued or to be issued to the Company may not provide the Company with any competitive advantage. The Company’s patents may be challenged by third parties in patent litigation, which is becoming widespread in the biopharmaceutical industry. In addition, it is possible that third parties with products that are very similar to the Company’s will circumvent its patents by means of alternate designs or processes. The Company may have to rely on method of use protection for its compounds in development and any resulting products, which may not confer the same protection as compounds per se. The Company may be required to disclaim part of the term of certain patents. There may be prior applications of which the Company is not aware that may affect the validity or enforceability of a patent claim. There also may be prior applications which are not viewed by the Company as affecting the validity or enforceability of a claim, but which may, nonetheless ultimately be found to affect the validity or enforceability of a claim. No assurance can be given that the Company’s patents would, if challenged, be held by a court to be valid or enforceable or that a competitor’s technology or product would be found by a court to infringe the Company’s patents. Applications for patents and trademarks in Canada, the United States and in foreign markets have been filed and are being actively pursued by the Company. Pending patent applications may not result in the issuance of patents, and the Company may not develop additional proprietary products which are patentable.
Patent applications relating to or affecting the Company’s business have been filed by a number of pharmaceutical and biopharmaceutical companies and academic institutions. A number of the technologies in these applications or patents may conflict with the Company’s technologies, patents or patent applications, and such conflict could reduce the scope of patent protection which the Company could otherwise obtain. The Company could become involved in interference proceedings in the United States in connection with one or more of its patents or patent applications to determine priority of invention. The Company’s granted patents could also be challenged and revoked in opposition proceedings in certain countries outside the United States.
In addition to patents, the Company relies on trade secrets and proprietary know-how to protect its intellectual property. The Company generally requires its employees, consultants, outside scientific collaborators and sponsored researchers and other advisors to enter into confidentiality agreements. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with the Company is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of the Company’s employees, the agreements provide that all of the technology which is conceived by the individual during the course of employment is the Company’s exclusive property. These agreements may not provide meaningful protection or adequate remedies in the event of unauthorized use or disclosure of the Company’s proprietary information. In addition, it is possible that third parties could independently develop proprietary information and techniques substantially similar to those of the Company or otherwise gain access to the Company’s trade secrets.
The Company currently has the right to use certain technology under license agreements with third parties. The Company’s failure to comply with the requirements of material license agreements could result in the termination of such agreements, which could cause the Company to terminate the related development program and cause a complete loss of its investment in that program.
As a result of the foregoing factors, the Company may not be able to rely on its intellectual property to protect its products in the marketplace.
The Company may infringe the intellectual property rights of others.
The Company’s commercial success depends significantly on its ability to operate without infringing the patents and other intellectual property rights of third parties. There could be issued patents of which the Company is not aware that its products infringe or patents, that the Company believes it does not infringe, but that it may ultimately be found to infringe. Moreover, patent applications are in some cases maintained in secrecy until patents are issued. The publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made and patent applications were filed. Because patents can take many years to issue, there may be currently pending applications of which the Company is unaware that may later result in issued patents that its products infringe.
The biopharmaceutical industry has produced a proliferation of patents, and it is not always clear to industry participants, including the Company, which patents cover various types of products. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. The Company is aware of, and has reviewed, third party patents relating to the treatment of Alzheimer’s disease, diabetes and other relevant indication areas. In the event of infringement or violation of another party’s patent, the Company may not be able to enter into licensing arrangements or make other arrangements at a reasonable cost. Any inability to secure licenses or alternative technology could result in delays in the introduction of the Company’s products or lead to prohibition of the manufacture or sale of the products.
Patent litigation is costly and time consuming and may subject the Company to liabilities.
The Company’s involvement in any patent litigation, interference, opposition or other administrative proceedings will likely cause the Company to incur substantial expenses, and the efforts of its technical and management personnel will be significantly diverted. In addition, an adverse determination in litigation could subject the Company to significant liabilities.
The Company operates in a fiercely competitive business environment.
The biopharmaceutical industry is highly competitive. Competition comes from healthcare companies, pharmaceutical companies, large and small biotech companies, specialty pharmaceutical companies, universities, government agencies and other public and private companies. Research and development by others may render the Company’s technology or products non-competitive or obsolete or may result in the production of treatments or cures superior to any therapy the Company is developing or will develop. In addition, failure, unacceptable toxicity, lack of sales or disappointing sales or other issues regarding competitors’ products or processes could have a material adverse effect on the Company’s product candidates, including its clinical candidates or its lead compounds.
The market price of the Company’s Common Shares may experience a high level of volatility due to factors such as the volatility in the market for biotechnology stocks generally, and the short-term effect of a number of possible events.
The Company is a public growth company in the biotechnology sector. As frequently occurs among these companies, the market price for the Company’s Common Shares may experience a high level of volatility. Numerous factors, including many over which the Company has no control, may have a significant impact on the market price of Common Shares including, among other things, (i) clinical and regulatory developments regarding the Company’s products and product candidates and those of its competitors, (ii) arrangements or strategic partnerships by the Company, (iii) other announcements by the Company or its competitors regarding technological, product development, sales or other matters, (iv) patent or other intellectual property achievements or adverse developments, (v) arrivals or departures of key personnel; (vi) government regulatory action affecting the Company’s product candidates in the United States, Canada and foreign countries, (vii) actual or anticipated fluctuations in the Company’s revenues or expenses, (viii) general market conditions and fluctuations for the emerging growth and biopharmaceutical market sectors, (ix) reports of securities analysts regarding the expected performance of the Company, and (x) events related to threatened, new or existing litigation. Listing on NASDAQ and the TSX may increase share price volatility due to various factors including, (i) different ability to buy or sell the Company’s Common Shares, (ii) different market conditions in different capital markets; and (iii) different trading volume.
In addition, the stock market in recent years has experienced extreme price and trading volume fluctuations that often have been unrelated or disproportionate to the operating performance of individual companies. These broad market fluctuations may adversely affect the price of Common Shares, regardless of the Company’s operating performance. In addition, sales of substantial amounts of Common Shares in the public market after any offering, or the perception that those sales may occur, could cause the market price of Common Shares to decline.
Furthermore, shareholders may initiate securities class action lawsuits if the market price of the Company’s stock drops significantly, which may cause the Company to incur substantial costs and could divert the time and attention of its management.
The Company is highly dependent on third parties.
The Company is or may in the future be dependent on third parties for certain raw materials, product manufacture, marketing and distribution and, like other biotechnology and pharmaceutical companies, upon medical institutions to conduct clinical testing of its potential products. Although the Company does not anticipate any difficulty in obtaining any such materials and services, no assurance can be given that the Company will be able to obtain such materials and services.
The Company is subject to intense competition for its skilled personnel, and the loss of key personnel or the inability to attract additional personnel could impair its ability to conduct its operations.
The Company is highly dependent on its management and its clinical, regulatory and scientific staff, the loss of whose services might adversely impact its ability to achieve its objectives. Recruiting and retaining qualified management and clinical, scientific and regulatory personnel is critical to the Company’s success. Competition for skilled personnel is intense, and the Company’s ability to attract and retain qualified personnel may be affected by such competition.
The Company’s business involves the use of hazardous materials which requires the Company to comply with environmental regulation.
The Company’s discovery and development processes involve the controlled use of hazardous materials. The Company is subject to federal, provincial and local laws and regulations governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. The risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, the Company could be held liable for any damages that result, and any such liability could exceed the Company’s resources. The Company may not be adequately insured against this type of liability. The Company may be required to incur significant costs to comply with environmental laws and regulations in the future, and its operations, business or assets may be materially adversely affected by current or future environmental laws or regulations.
Legislative actions, potential new accounting pronouncements and higher insurance costs are likely to impact the Company’s future financial position or results of operations.
Compliance with changing regulations regarding corporate governance and public disclosure, notably with respect to internal controls over financial reporting, may result in additional expenses. Changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for companies such as ours, and insurance costs are increasing as a result of this uncertainty.
Future healthcare reforms may produce adverse consequences.
Healthcare reform and controls on healthcare spending may limit the price the Company can charge for any products and the amounts thereof that it can sell. In particular, in the United States, the federal government and private insurers have considered ways to change, and have changed, the manner in which healthcare services are provided. Potential approaches and changes in recent years include controls on healthcare spending and the creation of large purchasing groups. In the future, the U.S. government may institute further controls and different reimbursement schemes and limits on Medicare and Medicaid spending or reimbursement. These controls, reimbursement schemes and limits might affect the payments the Company could collect from sales of any of its products in the United States. Uncertainties regarding future health care reform and private market practices could adversely affect the Company’s ability to sell any products profitably in the United States. Election of new or different political or government officials in large market countries could lead to dramatic changes in pricing, regulatory approval legislation and reimbursement which could have material impact on product approvals and commercialization.
The Company faces an unproven market for its future products.
The Company believes that there will be many different applications for products successfully derived from its technologies and that the anticipated market for products under development will continue to expand. No assurance, however, can be given that these beliefs will prove to be correct due to competition from existing or new products and the yet to be established commercial viability of the Company’s products. Physicians, patients, formularies, third party payers or the medical community in general may not accept or utilize any products that the Company or its collaborative partners may develop.
The Company may be faced with future lawsuits related to secondary market liability.
Securities legislation in Canada has recently changed to make it easier for shareholders to sue. These changes could lead to frivolous law suits which could take substantial time, money, resources and attention or force the Company to settle such claims rather than seek adequate judicial remedy or dismissal of such claims.
The Company may encounter unforeseen emergency situations and information technology breaches.
Despite the implementation of security measures, any of the Company’s, its collaborators’ or its third party service providers’ internal computer systems are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failure. Any resulting system failure, accident or security breach could result in a material disruption of the Company’s operations. Likewise, data privacy or security breaches by employees and others with permitted access to our systems, including in some cases third-party service providers to which we may outsource certain business functions, may pose a risk that sensitive data, including intellectual property or personal information, may be exposed to unauthorized persons or to the public. While we have invested in the protection of data and information technology, there can be no assurance that our efforts will prevent service interruptions, or identify breaches in our systems, that could adversely affect our business and/or result in the loss of critical or sensitive information, which could result in financial, legal, business or reputational harm to us.
The Company’s technologies may become obsolete.
The pharmaceutical industry is characterized by rapidly changing markets, technology, emerging industry standards and frequent introduction of new products. The introduction of new products embodying new technologies, including new manufacturing processes, and the emergence of new industry standards may render the Company’s technologies obsolete, less competitive or less marketable.
Our product candidates may cause undesirable serious adverse events during clinical trials that could delay or prevent their regulatory authorization, approval or other permission to conduct further testing or commence commercialization.
Our product candidates in clinical development, including ELND005 can potentially cause adverse events. In 2010, together with our collaborator, Elan, we completed a Phase 2 study that evaluated three dose groups of ELND005 and a placebo group in mild to moderate Alzheimer’s disease patients. The study included four treatment arms: placebo, 250mg bid, 1000mg bid and 2000mg bid. The two high dose ELND005 groups were electively discontinued in 2009 by the companies due to an observed imbalance of serious adverse events, including deaths. No causal relationship could be determined between these higher doses and the events.
Of the 351 subjects who received study drug, a total of 171 subjects received either 250mg bid or placebo, the rest were in the two discontinued high dose groups. The overall incidence of adverse events in the 250mg bid and placebo groups was 87.5% versus 91.6%; and the incidence of withdrawals due to adverse events was 10.2% versus 9.6%, respectively. The incidence of serious adverse events in the 250mg bid and placebo groups was 21.6% versus 13.3%, but the incidence of serious adverse events that were considered drug related was 2.3% and 2.4%, respectively. The total number of deaths in the study was five and four in the 1000mg bid and 2000mg bid dose groups versus one and zero in the 250mg bid and placebo groups, respectively. These deaths occurred between August 2008 and November 2009. The study’s independent safety monitoring committee reviewed the final safety results and continued to conclude that a causal relationship between the deaths and drug could not be determined.
The most common adverse events in the 250mg bid group that were >5% in incidence and double the placebo rate were: falls (12.5% vs. placebo 6%), depression (11.4% vs. placebo 4.8%), and confusional state (8% vs. placebo 3.6%). Because our product candidates have been tested in relatively small patient populations and for limited durations, additional adverse events may be observed as their development progresses.
Adverse events caused by any of our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or other non-U.S. regulatory authorities for any or all targeted indications. This, in turn, could prevent the commercialization of our product candidates and the generation of revenues from their sale. In addition, if our product candidates receive authorization, marketing approval or other permission and we or others later identify adverse events caused by the product, the material adverse consequences that may arise, include, but are not limited to:
| • | regulatory authorities may withdraw their authorization, approval, or other permission to test or market the candidate product; |
| • | we may be required to recall the product, change the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
| • | a product may become less competitive and product sales may decrease; or |
| • | our reputation may suffer. |
Any one or a combination of these events could prevent us from achieving or maintaining market acceptance or could substantially increase the costs and expenses of commercializing the product candidate, which in turn could delay or prevent us from generating significant revenues from the sale of such products.
The Company may be subject to costly product liability claims and may not have adequate insurance.
The conduct of clinical trials in humans involves the potential risk that the use of our product candidates will result in adverse effects. The Company currently maintains product liability insurance for their clinical trials; however, such liability insurance may not be adequate to fully cover any liabilities that arise from clinical trials of our product candidates. The Company may not have sufficient resources to pay for any liabilities resulting from a claim excluded from, or beyond the limit of, our insurance coverage.
Clinical Study Results from our product candidates may not support further clinical development.
The clinical studies performed to evaluate the safety, tolerability and efficacy of our product candidates, including ELND005, can yield study results that may or may not support further clinical development. In June 2015, the Company announced that a Phase 2/3 study of neuropsychiatric drug candidate ELND005 did not meet its primary efficacy endpoint. In the study, both the treatment and placebo groups showed a significant, but similar, reduction in agitation and aggression relative to baseline. There was a greater than expected reduction in agitation and aggression observed in the placebo group as measured in weeks 4, 8 and 12 in the study. The safety and tolerability profile of ELND005 was consistent with previous studies in AD at the 250mg bid dose. The Phase 2/3 clinical study evaluated the efficacy, safety and tolerability of ELND005 over 12 weeks of treatment in patients with mild to severe AD, who were experiencing at least moderate levels of agitation/aggression. The randomized, double-blind, placebo-controlled study enrolled 350 AD patients (175 subjects per study arm). The primary efficacy endpoint of the study was the change from baseline in the Neuropsychiatric Inventory – Clinician (“NPI-C”) scale of agitation and aggression. An analysis of the full study dataset is being performed. A group of expert external clinical advisors is being consulted to determine any future development of ELND005. The results of this data analysis and clinical advisory interaction may or may not support the further development of ELND005. Future development may include evaluating ELND005 as a treatment of neuropsychiatric symptoms such as agitation and aggression in Alzheimer’s disease patients or potentially narrower patient populations or other disease indications. Any ELND005 development plan will be strategically focused to advance the drug candidate in the targeted patient population and therefore may differ from previously proposed development plans. Further, interactions with regulatory authorities including the FDA may or may not support proposed ELND005 clinical plans. Regulatory interactions may also result in modifications to the ELND005 development plan potentially increasing the time and cost of clinical development activities.