UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, D.C. 20549
FORM 20-F
[Mark One]
| ¨ | REGISTRATION STATEMENT PURSUANT TO SECTION 12 OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | |
| OR |
| | |
| x | ANNUAL REPORT PURSUANT TO SECTION 13(a) OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934 |
| | |
| | For the fiscal year ended: June 30, 2015 | Commission File Number: 001-33514 |
TRANSITION THERAPEUTICS INC.
(Exact name of Registrant as specified in its charter)
Ontario, Canada
(Jurisdiction of incorporation or organization)
101 College Street, Suite 220
Toronto, Ontario, Canada
M5G 1L7
(416) 260-7770
(Address and telephone number of Registrant’s principal executive offices)
Nicole Rusaw
Chief Financial Officer
101 College Street, Suite 220
Toronto, Ontario, Canada
M5G 1L7
(416) 260-7770 Tel.
(416) 260-2886 Fax
(Name, Telephone, E-mail and/or Facsimile number and Address of Company Contact Person)
Securities registered or to be registered pursuant to Section 12(b) of the Act:
| Title of each class | | Name of each exchange on which registered |
| Common Shares, no par value | | The NASDAQ Stock Market LLC |
Securities registered or to be registered pursuant to Section 12(g) of the Act: None
Securities for which there is a reporting obligation pursuant to Section 15(d) of the Act: None
Indicate the number of outstanding shares of each of the issuer’s classes of capital or common stock as of the close of the period covered by the annual report: As of June 30, 2015: 38,878,879 shares of Common Shares were outstanding.
Indicate by check mark if the Registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.¨ Yesx No
If this report is an annual or transition report, indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934.¨ Yesx No
Note – Checking the box above will not relieve any registrant required to file reports pursuant to Section 13 or 15(d) of the Securities Exchange Act of 1934 from their obligations under those Sections.
Indicate by check mark whether the Registrant (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports) and (2) has been subject to such filing requirements for the past 90 days.x Yes¨ No
Indicate by check mark whether the registrant has submitted electronically and posted on its corporate Web site, if any, every Interactive Data File required to be submitted and posted pursuant to Rule 405 of Regulation S-T (§232.405 of this chapter) during the preceding 12 months (or for such shorter period that the Registrant was required to submit and post such files).¨ Yes¨ No
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, or a non-accelerated filer. See definition of “accelerated filer and large accelerated filer” in Rule 12b-2 of the Exchange Act. (Check one):
| Large accelerated filer ¨ | Accelerated filer x | Non-accelerated filer ¨ |
Indicate by check mark which basis of accounting the registrant has used to prepare the financial statements included in this filing:
| U.S. GAAP ¨ | International Financial Reporting Standards as issued by
the International Accounting Standards Board x | Other ¨ |
If “Other” has been checked in response to the previous question, indicate by check mark which financial statement item the registrant has elected to follow.¨ Item 17¨ Item 18
If this is an annual report, indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Exchange Act).¨ Yesx No
(APPLICABLE ONLY TO ISSUERS INVOLVED IN BANKRUPTCY PROCEEDINGS DURING THE PAST
FIVE YEARS)
Indicate by check mark whether the registrant has filed all documents and reports required to be filed by Sections 12, 13 or 15(d) of the Securities Exchange Act of 1934 subsequent to the distribution of securities under a plan confirmed by acourt.¨ Yes¨ No
Table of Contents
Introduction
In this annual report, where “we”, “us”, “our”, “Transition”, “Corporation” or the “Company” is used, it is referring to Transition Therapeutics Inc. and its wholly-owned subsidiaries, unless otherwise indicated. All amounts are in Canadian dollars, unless otherwise indicated.
Additional information relating to the Corporation, including the Corporation’s most recently filed Annual Information Form, can be found on SEDAR at www.sedar.com.
Presentation of Financial Information
Unless we indicate otherwise, financial information in this annual report has been prepared in accordance with International Financial Reporting Standards, or IFRS, as issued by the International Accounting Standards Board. IFRS differs in some respects from United States generally accepted accounting principles, or United States GAAP, and thus our financial statements may not be comparable to the financial statements of United States companies.
Percentages and some amounts in this annual report have been rounded for ease of presentation. Any discrepancies between totals and the sums of the amounts listed are due to rounding.
Currency Translation
We present our historical financial statements in Canadian dollars, which is the reporting currency of the Corporation. All figures reported in this annual report are in Canadian dollars, except where we indicate otherwise, and are referenced as “CAD$,” “$” and “dollars”. This annual report contains a translation of some Canadian dollar amounts into United States dollars at specified exchange rates solely for your convenience. See “Exchange Rate Data” below for certain information about the rates of exchange between Canadian dollars and United States dollars.
Exchange Rate Data
The following table sets forth, for each period indicated, the low and high exchange rates for Canadian dollars expressed in United States dollars and the average of such exchange rates on the last day of each month during such period, based on the inverse of the noon buying rate in the City of New York for cable transfers in Canadian dollars as certified for customs purposes by the Federal Reserve Bank of New York. The source of the exchange rate data is the H.10 statistical release of the Federal Reserve Board. The exchange rates set forth below demonstrate trends in exchange rates, but the actual exchange rates used throughout this Annual Report may vary.
| | | Exchange Rate | |
| | | High | | | Average (1) | | | Low | |
| | | (US$ per CAD$1.00) | |
| Last Five Fiscal Years | | | | | | | | | | | | |
| Fiscal Year Ended June 30, 2011 | | | 1.0542 | | | | 1.0013 | | | | 0.9392 | |
| Fiscal Year Ended June 30, 2012 | | | 1.0584 | | | | 0.9977 | | | | 0.9430 | |
| Fiscal Year Ended June 30, 2013 | | | 1.03 | | | | 0.9920 | | | | 0.9495 | |
| Fiscal Year Ended June 30, 2014 | | | 0.9769 | | | | 0.9336 | | | | 0.8888 | |
| Fiscal Year Ended June 30, 2015 | | | 0.9388 | | | | 0.8438 | | | | 0.7811 | |
| | | | | | | | | | | | | |
| Last Six Months | | | | | | | | | | | | |
| March 2015 | | | 0.8039 | | | | | | | | 0.7811 | |
| April 2015 | | | 0.8365 | | | | | | | | 0.7930 | |
| May 2015 | | | 0.8368 | | | | | | | | 0.8011 | |
| June 2015 | | | 0.8193 | | | | | | | | 0.7970 | |
| July 2015 | | | 0.7962 | | | | | | | | 0.7658 | |
| August 2015 | | | 0.7708 | | | | | | | | 0.7518 | |
Notes:
| (1) | For the years indicated, the average exchange rates are determined by averaging the exchange rates on the last business day of each month during the relevant period. |
Cautionary Statement Concerning Forward-Looking Statements
This annual report contains and incorporates by reference certain forward looking statements within the meaning of applicable securities laws. Forward-looking information typically contains statements with words such as “anticipate”, “believe”, “expect”, “plan”, “estimate”, “intend”, “may” or similar words suggesting future outcomes.
Forward-looking statements in this annual report include, but are not limited to statements with respect to: the clinical study phases of the Company’s product candidates which the Company expects to complete in fiscal 2016 and beyond; the ability of the Company’s business model to maximize shareholder returns; the potential for ELND005 to slow the progression of Alzheimer’s disease and improve symptoms; the potential for ELND005 to be effective for the treatment of agitation and or aggression in patients with Alzheimer’s disease; the potential for ELND005 to be effective for the treatment of Down syndrome; the timing and manner of future clinical development, if any, of ELND005; the global population size of those affected by Alzheimer’s disease; the demand for a product that can slow or reverse the progression of Alzheimer’s disease; the demand for a product that can reduce the emergence or severity of neuropsychiatric symptoms like depression, anxiety, agitation and aggression in Alzheimer’s disease; the potential clinical benefit of ELND005 in the treatment of other disease indications; the development of TT401 and the series of preclinical compounds in-licensed from Eli Lilly and Company (“Lilly”) and their potential benefit in type 2 diabetes patients and obese individuals; the timing and manner of future clinical development of TT401 performed by Lilly; TT701 development plans and timelines for individuals with androgen deficiency or other disease indications; the potential clinical benefit of TT701 to increase lean body mass, improve functional and sexual outcomes or improve other symptoms associated with androgen deficiency; the engagement of third party manufacturers to produce the Company’s drug substances and products; the potential future in-licensing of additional drug candidates to expand the development pipeline; the intention of the Company to make collaborative arrangements for the marketing and distribution of its products and the impact of human capital on the growth and success of the Company.
Some of the assumptions, risks and factors which could cause future outcomes to differ materially from those set forth in the forward-looking information include, but are not limited to: (i) the assumption that the Company will be able to obtain sufficient and suitable financing to support operations, clinical trials and commercialization of products, (ii) the risk that the Company may not be able to capitalize on partnering and acquisition opportunities, (iii) the assumption that the Company will obtain favourable clinical trial results in the expected timeframe, (iv) the assumption that the Company will be able to adequately protect proprietary information and technology from competitors, (v) the risks relating to the uncertainties of the regulatory approval process, (vi) the impact of competitive products and pricing and the assumption that the Company will be able to compete in the targeted markets, and (vii) the risk that the Company may be unable to retain key personnel or maintain third party relationships, including relationships with key collaborators.
By its nature, forward looking information involves numerous assumptions, inherent risks and uncertainties, both general and specific, that contribute to the possibility that the predictions, forecasts, projections or other forward looking statements will not occur. Prospective investors should carefully consider the information contained under the heading “Risk Factors” and all other information included in or incorporated by reference in this annual report before making investment decisions with regard to the securities of the Corporation.
The section entitled “Risks Factors” discusses risks, uncertainties and factors that our management believes could cause actual results or events to differ materially from the forward-looking statements. Although we have attempted to identify important risks, uncertainties and other factors that could cause actual results or events to differ materially from those expressed or implied in the forward-looking information, there may be other factors that cause actual results or events to differ from those expressed or implied in the forward-looking information.
Forward-looking statements contained in this annual report are made as of the dates hereof, and such forward-looking statements are based on the beliefs, expectations and opinions of our management as of such date. We disclaim any obligation to update any forward-looking statements.
Part I
| Item 1. | Identity of Directors, Senior Management and Advisers |
Not applicable.
| Item 2. | Offer Statistics and Expected Timetable |
Not applicable.
| A. | Selected Financial Data |
The following information should be read in conjunction with the Corporation’s audited consolidated financial statements for the year ended June 30, 2015 and the related notes, which are prepared in accordance with IFRS. The selection of financial information includes financial information derived from the annual audited consolidated financial statements.
The following table is a summary of selected audited consolidated financial information of the Corporation for each of the five most recently completed financial years. The information presented is presented in accordance with IFRS:
| | | June 30, 2015 | | | June 30, 2014 | | | June 30, 2013 | | | June 30, 2012 | | | June 30, 2011 | |
| Revenue | | $ | — | | | $ | — | | | $ | 17,933,500 | | | $ | — | | | $ | 10,251,394 | |
| Net income (loss)(1) | | | (51,339,528 | ) | | | (21,782,255 | ) | | | 23,297 | | | | (12,269,846 | ) | | | (5,689,613 | ) |
| Basic and diluted net income ( loss) per share | | | (1.41 | ) | | | (0.72 | ) | | | 0.00 | | | | (0.48 | ) | | | (0.25 | ) |
| Total assets | | | 49,649,085 | | | | 68,907,236 | | | | 37,807,955 | | | | 37,093,030 | | | | 43,179,488 | |
| Total long-term liabilities | | | 3,503,344 | | | | 3,849,718 | | | | 1,457,821 | | | | 1,469,253 | | | | 1,480,685 | |
| Shareholders’ equity | | | 36,737,589 | | | | 59,094,260 | | | | 33,154,612 | | | | 32,123,489 | | | | 38,432,070 | |
| Common shares outstanding | | | 38,878,879 | | | | 35,303,913 | | | | 26,930,634 | | | | 26,921,302 | | | | 23,217,599 | |
| Cash dividends declared per share | | | — | | | | — | | | | — | | | | — | | | | — | |
Notes:
| (1) | Net income (loss) before discontinued operations and extraordinary items was equivalent to the net income (loss) for such periods. |
| B. | Capitalization and Indebtedness |
Not applicable.
| C. | Reasons for the Offer and Use of Proceeds |
Not applicable.
Investing in our securities involves a high degree of risk. Before making an investment decision, you should carefully consider the following risk factors, in addition to the other information provided in this annual report and the Corporation’s other disclosure documents filed with the U.S. Securities and Exchange Commission onwww.sec.gov.
The Company will require significant additional financing and it may not have access to sufficient capital.
The Company anticipates that it will need additional financing in the future to fund its ongoing research and development programs and for general corporate requirements. The Company may choose to seek additional funding through public or private offerings, corporate collaborations or partnership arrangements. The amount of financing required will depend on many factors including the financial requirements of the Company to fund its research and clinical trials, and the ability of the Company to secure partnerships and achieve partnership milestones as well as to fund other working capital requirements. The Company’s ability to access the capital markets or to enlist partners is mainly dependent on the progress of its research and development and regulatory approval of its products. There is no assurance that additional funding will be available on acceptable terms, if at all.
The Company has a history of losses, and it has not generated any product revenue to date. It may never achieve or maintain profitability.
Since inception, the Company has incurred significant losses each year and expects to incur significant operating losses as the Company continues product research and development and clinical trials. There is no assurance that the Company will ever successfully commercialize or achieve revenues from sales of its therapeutic products if they are successfully developed or that profitability will ever be achieved or maintained. Even if profitability is achieved, the Company may not be able to sustain or increase profitability.
The Company is an early stage development company in an uncertain industry.
The Company is at an early stage of development. Preclinical and clinical trial work must be completed before our products could be ready for use within the markets we have identified. We may fail to develop any products, to obtain regulatory approvals, to enter clinical trials or to commercialize any products. The Company does not know whether any of our potential product development efforts will prove to be effective, meet applicable regulatory standards, obtain the requisite regulatory approvals or be capable of being manufactured at a reasonable cost. If the Company’s products are approved for sale, there can be no assurance that the products will gain market acceptance among consumers, physicians, patients and others in the medical community. A failure to gain market acceptance may adversely affect the revenues of the Company.
The Company is subject to a strict regulatory environment.
None of the Company’s product candidates have received regulatory approval for commercial sale.
Numerous statutes and regulations govern human testing and the manufacture and sale of human therapeutic products in Canada, the United States and other countries where the Company intends to market its products. Such legislation and regulation bears upon, among other things, the approval of protocols and human testing, the approval of manufacturing facilities, testing procedures and controlled research, review and approval of manufacturing, preclinical and clinical data prior to marketing approval including adherence to Good Manufacturing Practices (“GMP”) during production and storage as well as regulation of marketing activities including advertising and labelling.
The completion of the clinical testing of our product candidates and the obtaining of required approvals are expected to take years and require the expenditure of substantial resources. There can be no assurance that clinical trials will be completed successfully within any specified period of time, if at all. Furthermore, clinical trials may be delayed or suspended at any time by the Company or by regulatory authorities if it is determined at any time that patients may be or are being exposed to unacceptable health risks, including the risk of death, or that compounds are not manufactured under acceptable GMP conditions or with acceptable quality. Any failure or delay in obtaining regulatory approvals would adversely affect the Company’s ability to utilize its technology thereby adversely affecting operations. No assurance can be given that the Company’s product candidates or lead compounds will prove to be safe and effective in clinical trials or that they will receive the requisite protocol approval or regulatory approval. Furthermore, no assurance can be given that current regulations relating to regulatory approval will not change or become more stringent. There are no assurances the Company can scale-up, formulate or manufacture any compound in sufficient quantities with acceptable specifications for the regulatory agencies to grant approval or not require additional changes or additional trials be performed. The agencies may also require additional trials be run in order to provide additional information regarding the safety, efficacy or equivalency of any compound for which the Company seeks regulatory approval. Similar restrictions are imposed in foreign markets other than the United States and Canada. Investors should be aware of the risks, problems, delays, expenses and difficulties which may be encountered by the Company in light of the extensive regulatory environment in which the Company’s business operates.
Even if a product candidate is approved by the FDA or any other regulatory authority, the Company may not obtain approval for an indication whose market is large enough to recoup its investment in that product candidate. The Company may never obtain the required regulatory approvals for any of its product candidates.
The Company is faced with uncertainties related to its research.
The Company’s research programs are based on scientific hypotheses and experimental approaches that may not lead to desired results. In addition, the timeframe for obtaining proof of principle and other results may be considerably longer than originally anticipated, or may not be possible given time, resource, financial, strategic and collaborator scientific constraints. Success in one stage of testing is not necessarily an indication that the particular program will succeed in later stages of testing and development. It is not possible to predict, based upon studies in in-vitro models and in animals, whether any of the compounds made for these programs will prove to be safe, effective, and suitable for human use. Each compound will require additional research and development, scale-up, formulation and extensive clinical testing in humans. Decisions regarding future development activities may be based on results from completed studies or interim results from on-going studies or projections derived from interim or administrative analyses of studies not yet completed. Development of these compounds will require investigations into the mechanism of action of the molecules as these are not fully understood. Unsatisfactory results obtained from a particular study relating to a program may cause the Company to abandon its commitment to that program or to the lead compound or product candidate being tested. The discovery of unexpected toxicities, lack of sufficient efficacy, poor physiochemical properties, unacceptable ADME (absorption, distribution, metabolism and excretion) and DMPK (drug metabolism and pharmacokinetics), pharmacology, inability to increase scale of manufacture, market attractiveness, regulatory hurdles, competition, as well as other factors, may make the Company’s targets, lead compounds or product candidates unattractive or unsuitable for human use, and the Company may abandon its commitment to that program, target, lead compound or product candidate. In addition, preliminary results seen in animal and/or limited human testing may not be substantiated in larger controlled clinical trials.
If difficulties are encountered enrolling patients in the Company’s clinical trials, the Company’s trials could be delayed or otherwise adversely affected.
Clinical trials for the Company’s product candidates require that the Company identify and enroll a large number of patients with the disorder under investigation. The Company may not be able to enroll a sufficient number of patients to complete its clinical trials in a timely manner. Patient enrolment is a function of many factors including, but not limited to, design of the study protocol, size of the patient population, eligibility criteria for the study, the perceived risks and benefits of the therapy under study, the patient referral practices of physicians and the availability of clinical trial sites. If the Company has difficulty enrolling a sufficient number of patients to conduct the Company’s clinical trials as planned, it may need to delay or terminate ongoing clinical trials.
Even if regulatory approvals are obtained for the Company’s product candidates, the Company will be subject to ongoing government regulation.
Even if regulatory authorities approve any of the Company’s human therapeutic product candidates, the manufacture, marketing and sale of such products will be subject to strict and ongoing regulation. Compliance with such regulation may be expensive and consume substantial financial and management resources. If the Company, or any future marketing collaborators or contract manufacturers, fail to comply with applicable regulatory requirements, it may be subject to sanctions including fines, product recalls or seizures, injunctions, total or partial suspension of production, civil penalties, withdrawal of regulatory approvals and criminal prosecution. Any of these sanctions could delay or prevent the promotion, marketing or sale of the Company’s products.
The Company may not achieve its projected development goals in the time frames announced and expected.
The Company sets goals for and makes public statements regarding the timing of the accomplishment of objectives material to its success, such as the commencement and completion of clinical trials, anticipated regulatory submission and approval dates and time of product launch. The actual timing of these events can vary dramatically due to factors such as delays or failures in the Company’s clinical trials, the uncertainties inherent in the regulatory approval process and delays in achieving manufacturing or marketing arrangements sufficient to commercialize its products.
There can be no assurance that the Company’s clinical trials will be completed, that the Company will make regulatory submissions or receive regulatory approvals as planned. If the Company fails to achieve one or more of these milestones as planned, the price of the Common Shares would likely decline.
If the Company fails to obtain acceptable prices or adequate reimbursement for its human therapeutic products, its ability to generate revenues will be diminished.
The Company’s ability to successfully commercialize its human therapeutic products will depend significantly on its ability to obtain acceptable prices and the availability of reimbursement to the patient from third-party payers, such as government and private insurance plans. While the Company has not commenced discussions with any such parties, these third-party payers frequently require companies to provide predetermined discounts from list prices, and they are increasingly challenging the prices charged for pharmaceuticals and other medical products. The Company’s human therapeutic products may not be considered cost-effective, and reimbursement to the patient may not be available or sufficient to allow the Company to sell its products on a competitive basis. The Company may not be able to negotiate favourable reimbursement rates for its human therapeutic products.
In addition, the continuing efforts of third-party payers to contain or reduce the costs of healthcare through various means may limit the Company’s commercial opportunity and reduce any associated revenue and profits. The Company expects proposals to implement similar government control to continue. In addition, increasing emphasis on managed care will continue to put pressure on the pricing of pharmaceutical and biopharmaceutical products. Cost control initiatives could decrease the price that the Company or any current or potential collaborators could receive for any of its human therapeutic products and could adversely affect its profitability. In addition, in Canada and in many other countries, pricing and/or profitability of some or all prescription pharmaceuticals and biopharmaceuticals are subject to government control.
If the Company fails to obtain acceptable prices or an adequate level of reimbursement for its products, the sales of its products would be adversely affected or there may be no commercially viable market for its products.
The Company may not obtain adequate protection for its products through its intellectual property.
The Company’s success depends, in large part, on its ability to protect its competitive position through patents, trade secrets, trademarks and other intellectual property rights. The patent positions of pharmaceutical and biopharmaceutical firms, including the Company, are uncertain and involve complex questions of law and fact for which important legal issues remain unresolved. The patents issued or to be issued to the Company may not provide the Company with any competitive advantage. The Company’s patents may be challenged by third parties in patent litigation, which is becoming widespread in the biopharmaceutical industry. In addition, it is possible that third parties with products that are very similar to the Company’s will circumvent its patents by means of alternate designs or processes. The Company may have to rely on method of use protection for its compounds in development and any resulting products, which may not confer the same protection as compounds per se. The Company may be required to disclaim part of the term of certain patents. There may be prior applications of which the Company is not aware that may affect the validity or enforceability of a patent claim. There also may be prior applications which are not viewed by the Company as affecting the validity or enforceability of a claim, but which may, nonetheless ultimately be found to affect the validity or enforceability of a claim. No assurance can be given that the Company’s patents would, if challenged, be held by a court to be valid or enforceable or that a competitor’s technology or product would be found by a court to infringe the Company’s patents. Applications for patents and trademarks in Canada, the United States and in foreign markets have been filed and are being actively pursued by the Company. Pending patent applications may not result in the issuance of patents, and the Company may not develop additional proprietary products which are patentable.
Patent applications relating to or affecting the Company’s business have been filed by a number of pharmaceutical and biopharmaceutical companies and academic institutions. A number of the technologies in these applications or patents may conflict with the Company’s technologies, patents or patent applications, and such conflict could reduce the scope of patent protection which the Company could otherwise obtain. The Company could become involved in interference proceedings in the United States in connection with one or more of its patents or patent applications to determine priority of invention. The Company’s granted patents could also be challenged and revoked in opposition proceedings in certain countries outside the United States.
In addition to patents, the Company relies on trade secrets and proprietary know-how to protect its intellectual property. The Company generally requires its employees, consultants, outside scientific collaborators and sponsored researchers and other advisors to enter into confidentiality agreements. These agreements provide that all confidential information developed or made known to the individual during the course of the individual’s relationship with the Company is to be kept confidential and not disclosed to third parties except in specific circumstances. In the case of the Company’s employees, the agreements provide that all of the technology which is conceived by the individual during the course of employment is the Company’s exclusive property. These agreements may not provide meaningful protection or adequate remedies in the event of unauthorized use or disclosure of the Company’s proprietary information. In addition, it is possible that third parties could independently develop proprietary information and techniques substantially similar to those of the Company or otherwise gain access to the Company’s trade secrets.
The Company currently has the right to use certain technology under license agreements with third parties. The Company’s failure to comply with the requirements of material license agreements could result in the termination of such agreements, which could cause the Company to terminate the related development program and cause a complete loss of its investment in that program.
As a result of the foregoing factors, the Company may not be able to rely on its intellectual property to protect its products in the marketplace.
The Company may infringe the intellectual property rights of others.
The Company’s commercial success depends significantly on its ability to operate without infringing the patents and other intellectual property rights of third parties. There could be issued patents of which the Company is not aware that its products infringe or patents, that the Company believes it does not infringe, but that it may ultimately be found to infringe. Moreover, patent applications are in some cases maintained in secrecy until patents are issued. The publication of discoveries in the scientific or patent literature frequently occurs substantially later than the date on which the underlying discoveries were made and patent applications were filed. Because patents can take many years to issue, there may be currently pending applications of which the Company is unaware that may later result in issued patents that its products infringe.
The biopharmaceutical industry has produced a proliferation of patents, and it is not always clear to industry participants, including the Company, which patents cover various types of products. The coverage of patents is subject to interpretation by the courts, and the interpretation is not always uniform. The Company is aware of, and has reviewed, third party patents relating to the treatment of Alzheimer’s disease, diabetes and other relevant indication areas. In the event of infringement or violation of another party’s patent, the Company may not be able to enter into licensing arrangements or make other arrangements at a reasonable cost. Any inability to secure licenses or alternative technology could result in delays in the introduction of the Company’s products or lead to prohibition of the manufacture or sale of the products.
Patent litigation is costly and time consuming and may subject the Company to liabilities.
The Company’s involvement in any patent litigation, interference, opposition or other administrative proceedings will likely cause the Company to incur substantial expenses, and the efforts of its technical and management personnel will be significantly diverted. In addition, an adverse determination in litigation could subject the Company to significant liabilities.
The Company operates in a fiercely competitive business environment.
The biopharmaceutical industry is highly competitive. Competition comes from healthcare companies, pharmaceutical companies, large and small biotech companies, specialty pharmaceutical companies, universities, government agencies and other public and private companies. Research and development by others may render the Company’s technology or products non-competitive or obsolete or may result in the production of treatments or cures superior to any therapy the Company is developing or will develop. In addition, failure, unacceptable toxicity, lack of sales or disappointing sales or other issues regarding competitors’ products or processes could have a material adverse effect on the Company’s product candidates, including its clinical candidates or its lead compounds.
The market price of the Company’s Common Shares may experience a high level of volatility due to factors such as the volatility in the market for biotechnology stocks generally, and the short-term effect of a number of possible events.
The Company is a public growth company in the biotechnology sector. As frequently occurs among these companies, the market price for the Company’s Common Shares may experience a high level of volatility. For example, in the last year, the Company’s stock price has ranged from a low of US$1.77 to a high of US$9.30. Numerous factors, including many over which the Company has no control, may have a significant impact on the market price of Common Shares including, among other things, (i) clinical and regulatory developments regarding the Company’s products and product candidates and those of its competitors, (ii) arrangements or strategic partnerships by the Company, (iii) other announcements by the Company or its competitors regarding technological, product development, sales or other matters, (iv) patent or other intellectual property achievements or adverse developments, (v) arrivals or departures of key personnel; (vi) government regulatory action affecting the Company’s product candidates in the United States, Canada and foreign countries, (vii) actual or anticipated fluctuations in the Company’s revenues or expenses, (viii) general market conditions and fluctuations for the emerging growth and biopharmaceutical market sectors, (ix) reports of securities analysts regarding the expected performance of the Company, and (x) events related to threatened, new or existing litigation. Listing on NASDAQ and the TSX may increase share price volatility due to various factors including, (i) different ability to buy or sell the Company’s Common Shares, (ii) different market conditions in different capital markets; and (iii) different trading volume.
In addition, the stock market in recent years has experienced extreme price and trading volume fluctuations that often have been unrelated or disproportionate to the operating performance of individual companies. These broad market fluctuations may adversely affect the price of Common Shares, regardless of the Company’s operating performance. In addition, sales of substantial amounts of Common Shares in the public market after any offering, or the perception that those sales may occur, could cause the market price of Common Shares to decline.
Furthermore, shareholders may initiate securities class action lawsuits if the market price of the Company’s stock drops significantly, which may cause the Company to incur substantial costs and could divert the time and attention of its management.
The Company is highly dependent on third parties.
The Company is or may in the future be dependent on third parties for certain raw materials, product manufacture, marketing and distribution and, like other biotechnology and pharmaceutical companies, upon medical institutions to conduct clinical testing of its potential products. Although the Company does not anticipate any difficulty in obtaining any such materials and services, no assurance can be given that the Company will be able to obtain such materials and services.
The Company is subject to intense competition for its skilled personnel, and the loss of key personnel or the inability to attract additional personnel could impair its ability to conduct its operations.
The Company is highly dependent on its management and its clinical, regulatory and scientific staff, the loss of whose services might adversely impact its ability to achieve its objectives. Recruiting and retaining qualified management and clinical, scientific and regulatory personnel is critical to the Company’s success. Competition for skilled personnel is intense, and the Company’s ability to attract and retain qualified personnel may be affected by such competition.
The Company’s business involves the use of hazardous materials which requires the Company to comply with environmental regulation.
The Company’s discovery and development processes involve the controlled use of hazardous materials. The Company is subject to federal, provincial and local laws and regulations governing the use, manufacture, storage, handling and disposal of such materials and certain waste products. The risk of accidental contamination or injury from these materials cannot be completely eliminated. In the event of such an accident, the Company could be held liable for any damages that result, and any such liability could exceed the Company’s resources. The Company may not be adequately insured against this type of liability. The Company may be required to incur significant costs to comply with environmental laws and regulations in the future, and its operations, business or assets may be materially adversely affected by current or future environmental laws or regulations.
Legislative actions, potential new accounting pronouncements and higher insurance costs are likely to impact the Company’s future financial position or results of operations.
Compliance with changing regulations regarding corporate governance and public disclosure, notably with respect to internal controls over financial reporting, may result in additional expenses. Changing laws, regulations and standards relating to corporate governance and public disclosure are creating uncertainty for companies such as ours, and insurance costs are increasing as a result of this uncertainty.
Future healthcare reforms may produce adverse consequences.
Healthcare reform and controls on healthcare spending may limit the price the Company can charge for any products and the amounts thereof that it can sell. In particular, in the United States, the federal government and private insurers have considered ways to change, and have changed, the manner in which healthcare services are provided. Potential approaches and changes in recent years include controls on healthcare spending and the creation of large purchasing groups. In the future, the U.S. government may institute further controls and different reimbursement schemes and limits on Medicare and Medicaid spending or reimbursement. These controls, reimbursement schemes and limits might affect the payments the Company could collect from sales of any of its products in the United States. Uncertainties regarding future health care reform and private market practices could adversely affect the Company’s ability to sell any products profitably in the United States. Election of new or different political or government officials in large market countries could lead to dramatic changes in pricing, regulatory approval legislation and reimbursement which could have material impact on product approvals and commercialization.
The Company faces an unproven market for its future products.
The Company believes that there will be many different applications for products successfully derived from its technologies and that the anticipated market for products under development will continue to expand. No assurance, however, can be given that these beliefs will prove to be correct due to competition from existing or new products and the yet to be established commercial viability of the Company’s products. Physicians, patients, formularies, third party payers or the medical community in general may not accept or utilize any products that the Company or its collaborative partners may develop.
The Company may be faced with future lawsuits related to secondary market liability.
Securities legislation in Canada has recently changed to make it easier for shareholders to sue. These changes could lead to frivolous law suits which could take substantial time, money, resources and attention or force the Company to settle such claims rather than seek adequate judicial remedy or dismissal of such claims.
The Company may encounter unforeseen emergency situations and information technology breaches.
Despite the implementation of security measures, any of the Company’s, its collaborators’ or its third party service providers’ internal computer systems are vulnerable to damage from computer viruses, unauthorized access, natural disasters, terrorism, war and telecommunication and electrical failure. Any resulting system failure, accident or security breach could result in a material disruption of the Company’s operations. Likewise, data privacy or security breaches by employees and others with permitted access to our systems, including in some cases third-party service providers to which we may outsource certain business functions, may pose a risk that sensitive data, including intellectual property or personal information, may be exposed to unauthorized persons or to the public. While we have invested in the protection of data and information technology, there can be no assurance that our efforts will prevent service interruptions, or identify breaches in our systems, that could adversely affect our business and/or result in the loss of critical or sensitive information, which could result in financial, legal, business or reputational harm to us.
The Company’s technologies may become obsolete.
The pharmaceutical industry is characterized by rapidly changing markets, technology, emerging industry standards and frequent introduction of new products. The introduction of new products embodying new technologies, including new manufacturing processes, and the emergence of new industry standards may render the Company’s technologies obsolete, less competitive or less marketable.
Our product candidates may cause undesirable serious adverse events during clinical trials that could delay or prevent their regulatory authorization, approval or other permission to conduct further testing or commence commercialization.
Our product candidates in clinical development, including ELND005 can potentially cause adverse events. In 2010, together with our collaborator, Elan, we completed a Phase 2 study that evaluated three dose groups of ELND005 and a placebo group in mild to moderate Alzheimer’s disease patients. The study included four treatment arms: placebo, 250mg bid, 1000mg bid and 2000mg bid. The two high dose ELND005 groups were electively discontinued in 2009 by the companies due to an observed imbalance of serious adverse events, including deaths. No causal relationship could be determined between these higher doses and the events.
Of the 351 subjects who received study drug, a total of 171 subjects received either 250mg bid or placebo, the rest were in the two discontinued high dose groups. The overall incidence of adverse events in the 250mg bid and placebo groups was 87.5% versus 91.6%; and the incidence of withdrawals due to adverse events was 10.2% versus 9.6%, respectively. The incidence of serious adverse events in the 250mg bid and placebo groups was 21.6% versus 13.3%, but the incidence of serious adverse events that were considered drug related was 2.3% and 2.4%, respectively. The total number of deaths in the study was five and four in the 1000mg bid and 2000mg bid dose groups versus one and zero in the 250mg bid and placebo groups, respectively. These deaths occurred between August 2008 and November 2009. The study’s independent safety monitoring committee reviewed the final safety results and continued to conclude that a causal relationship between the deaths and drug could not be determined.
The most common adverse events in the 250mg bid group that were >5% in incidence and double the placebo rate were: falls (12.5% vs. placebo 6%), depression (11.4% vs. placebo 4.8%), and confusional state (8% vs. placebo 3.6%). Because our product candidates have been tested in relatively small patient populations and for limited durations, additional adverse events may be observed as their development progresses.
Adverse events caused by any of our product candidates could cause us or regulatory authorities to interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or other non-U.S. regulatory authorities for any or all targeted indications. This, in turn, could prevent the commercialization of our product candidates and the generation of revenues from their sale. In addition, if our product candidates receive authorization, marketing approval or other permission and we or others later identify adverse events caused by the product, the material adverse consequences that may arise, include, but are not limited to:
| • | regulatory authorities may withdraw their authorization, approval, or other permission to test or market the candidate product; |
| • | we may be required to recall the product, change the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
| • | a product may become less competitive and product sales may decrease; or |
| • | our reputation may suffer. |
Any one or a combination of these events could prevent us from achieving or maintaining market acceptance or could substantially increase the costs and expenses of commercializing the product candidate, which in turn could delay or prevent us from generating significant revenues from the sale of such products.
The Company may be subject to costly product liability claims and may not have adequate insurance.
The conduct of clinical trials in humans involves the potential risk that the use of our product candidates will result in adverse effects. The Company currently maintains product liability insurance for their clinical trials; however, such liability insurance may not be adequate to fully cover any liabilities that arise from clinical trials of our product candidates. The Company may not have sufficient resources to pay for any liabilities resulting from a claim excluded from, or beyond the limit of, our insurance coverage.
Clinical Study Results from our product candidates may not support further clinical development.
The clinical studies performed to evaluate the safety, tolerability and efficacy of our product candidates, including ELND005, can yield study results that may or may not support further clinical development. In June 2015, the Company announced that a Phase 2/3 study of neuropsychiatric drug candidate ELND005 did not meet its primary efficacy endpoint. In the study, both the treatment and placebo groups showed a significant, but similar, reduction in agitation and aggression relative to baseline. There was a greater than expected reduction in agitation and aggression observed in the placebo group as measured in weeks 4, 8 and 12 in the study. The safety and tolerability profile of ELND005 was consistent with previous studies in AD at the 250mg bid dose. The Phase 2/3 clinical study evaluated the efficacy, safety and tolerability of ELND005 over 12 weeks of treatment in patients with mild to severe AD, who were experiencing at least moderate levels of agitation/aggression. The randomized, double-blind, placebo-controlled study enrolled 350 AD patients (175 subjects per study arm). The primary efficacy endpoint of the study was the change from baseline in the Neuropsychiatric Inventory – Clinician (“NPI-C”) scale of agitation and aggression. An analysis of the full study dataset is being performed. A group of expert external clinical advisors is being consulted to determine any future development of ELND005. The results of this data analysis and clinical advisory interaction may or may not support the further development of ELND005. Future development may include evaluating ELND005 as a treatment of neuropsychiatric symptoms such as agitation and aggression in Alzheimer’s disease patients or potentially narrower patient populations or other disease indications. Any ELND005 development plan will be strategically focused to advance the drug candidate in the targeted patient population and therefore may differ from previously proposed development plans. Further, interactions with regulatory authorities including the FDA may or may not support proposed ELND005 clinical plans. Regulatory interactions may also result in modifications to the ELND005 development plan potentially increasing the time and cost of clinical development activities.
U.S. holders of our Common Shares may suffer adverse tax consequences if we are characterized as a Passive Foreign Investment Company (“PFIC”).
There is a risk that we will be classified as a PFIC for U.S. federal income tax purposes. Our status as a PFIC could result in a reduction in the after-tax return to U.S. Holders of our Common Shares and may cause a reduction in the value of such shares. We will be classified as a PFIC for any taxable year in which (i) at least 75% of our gross income is passive income or (ii) at least 50% of the average value of all of our assets produce or are held for the production of passive income. For this purpose, passive income includes dividends, interest, and royalties and rents that are not derived in the active conduct of a trade or business, as well as gains from the sale of assets that produce passive income. Based on the composition of our income and valuation of our assets, we do not believe we were a PFIC for the taxable year ended June 30, 2015. However, there is no assurance that we will not be classified as a PFIC in subsequent taxable years. If we are classified as a PFIC, U.S. Holders of our common shares could be subject to greater U.S. income tax liability than might otherwise apply, imposition of U.S. income tax in advance of when tax would otherwise apply, and detailed tax filing requirements that would not otherwise apply. Subject to certain exceptions, once a U.S. Holder’s shares are treated as shares in a PFIC, they remain shares in a PFIC. Dividends received by a U.S. Holder from a PFIC will not constitute qualified dividend income qualifying for lower tax rates. The PFIC rules are complex and U.S. Holders of our common shares are urged to consult their own tax advisors regarding the possible application of the PFIC rules to them in their particular circumstances. See “Taxation—United States Federal Income Taxation.”
| Item 4. | Information on the Corporation |
Name, Address and Incorporation
Transition Therapeutics Inc. was incorporated pursuant to the Business Corporations Act (Ontario) on July 6, 1998 as “Transition Therapeutics and Diagnostics Inc.” The Corporation filed articles of amendment on October 12, 2000 and on October 19, 2000 to create a class of non-voting shares (the “Class B Shares”) and to amend certain attributes of its Common Shares. On November 2, 2000, the Corporation filed articles of amendment to delete its private company restrictions. On December 14, 2000, the Corporation filed articles of amendment to change its name to “Transition Therapeutics Inc.” and effect a split of its issued and outstanding Common Shares on the basis of 3.25649 Common Shares for each previously issued and outstanding Common Share. On December 14, 2004, the Corporation filed articles of amendment to eliminate the Class B Shares from its authorized capital. In July 2007, the Corporation completed the consolidation of its issued and outstanding Common Shares on the basis of one (1) post-consolidation Common Share for every nine (9) pre-consolidation Common Shares.
The Corporation’s principal and registered office is located at 101 College Street, Suite 220, Toronto, Ontario, Canada, M5G 1L7, and its telephone number is (416) 260-7770. The Corporation’s agent for service of process in the United States is CT Corporation System located at 111 Eighth Avenue, New York, NY 10011.
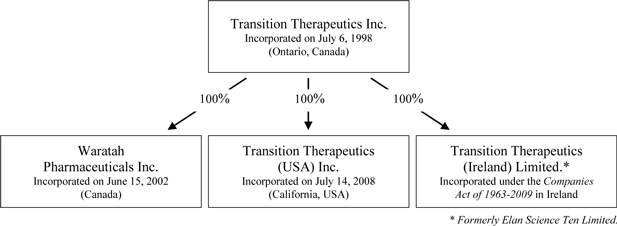
Intercorporate Relationships
The Corporation has three wholly-owned material subsidiaries: Waratah Pharmaceuticals Inc. (“Waratah”), which is incorporated under the Canada Business Corporations Act, Transition Therapeutics Ireland Limited (“Transition Ireland”) (formerly Elan Science Ten Limited) which is incorporated under the Companies Acts of 1963 to 2009 in Dublin, Ireland and Transition Therapeutics (USA) Inc. (“Transition USA”), which is incorporated under the laws of the State of California.
The chart below illustrates the corporate structure:
General Development of the Business
Three Year History
On August 30, 2012, the Corporation announced that Elan Pharma International Limited (“Elan”) had dosed the first patient in a Phase 2 clinical study of ELND005 in bipolar disorder. The study is a placebo-controlled, safety and efficacy study of oral ELND005 as an adjunctive maintenance treatment in patients with Bipolar 1 Disorder to delay the time to occurrence of mood episodes. As the first patient has been dosed in the study, the Corporation received a milestone payment of US$11 million from Elan.
On November 28, 2012, Transition announced that Elan had enrolled the first patient in a Phase 2 study of ELND005 for the treatment of agitation/aggression in patients with moderate to severe Alzheimer's disease.
On April 30, 2013, Transition announced the results of a five-week proof of concept clinical study of TT401 in type 2 diabetic and obese non-diabetic subjects. In the study, TT401, a once-weekly administered peptide, demonstrated significant improvements in glycemic control and reductions in body weight.
On June 17, 2013, Transition announced that Eli Lilly and Company (“Lilly”) had exercised its option to assume all development and commercialization rights to type 2 diabetes drug candidate TT401. In conjunction with this assumption of rights, Transition received a US$7 million milestone payment from Lilly.
On July 17, 2013, Transition announced that the US Food and Drug Administration (“FDA”) has granted Fast Track Designation to the development program for ELND005 which was submitted for the treatment of Neuropsychiatric Symptoms (“NPS”) in Alzheimer's disease (“AD”). The FDA concluded that the development program for ELND005 for the treatment of NPS in AD meets their criteria for Fast Track Designation.
On July 23, 2013, Transition announced the exclusive licensing of worldwide rights to a novel small molecule transcriptional regulator ("TT601") from Lilly for the treatment of osteoarthritis ("OA") pain.
On August 15, 2013, the Corporation announced the closing of its private placement financing issuing 2,625,300 units of the Corporation to certain existing shareholders, board members and management at a price of US$4.19 per unit, raising gross proceeds of US$11.0 million. Each unit consists of (i) one common share, (ii) 0.325 Common Share purchase warrant with a purchase price of US$4.60 per whole warrant and (iii) 0.4 Common Share purchase warrant with a purchase price of US$6.50 per whole warrant. Each whole warrant will entitle the holder, within two years of the closing date, to purchase one additional common share in the capital of the Corporation. If and when all of the warrants are exercised, the Corporation may realize up to an additional US$10.7 million in proceeds.
On September 4, 2013, Transition announced the first patient was dosed in a Phase 2a study of ELND005 in Down syndrome. Study ELND005-DS201 will evaluate the safety and pharmacokinetics of two doses of ELND005 and placebo in young adults with Down syndrome without dementia, and will also include select cognitive and behavioural measures.
On December 18, 2013 Perrigo Company plc “(Perrigo”) completed its acquisition of Elan Company plc and all of its subsidiaries. With this acquisition, Perrigo acquired all of the rights and obligations of Elan under the collaboration agreement (“ELND005 Agreement”) between Waratah, a wholly-owned subsidiary of the Corporation, and Elan for the development and commercialization of ELND005.
On February 28, 2014 Transition announced the acquisition of an Irish domiciled company, the holder of all the development and commercialization rights of neuropsychiatric drug candidate, ELND005. Going forward, Transition’s wholly owned subsidiary, Transition Therapeutics Ireland Limited, will be responsible for all future development and commercialization activities of the ELND005 drug candidate. In parallel with this acquisition, Perrigo has invested US$15 million and received 2,255,640 Transition common shares representing approximately a 7% ownership stake in Transition. Perrigo will also be eligible to receive up to US$40 million in approval and commercial milestone payments and a 6.5% royalty on net sales of ELND005 products and sublicense fees received. As a result of the transaction, Perrigo transferred its rights under the ELND005 Agreement to Transition Ireland.
On April 7, 2014, Transition provided a clinical development update and announced the decision to focus ELND005 development on the completion of current Phase 2 clinical studies in Agitation and Aggression in Alzheimer’s disease and a Phase 2a study in Down syndrome. A decision was also made to discontinue the clinical study of bipolar subjects following a commercial assessment of the size and length of the bipolar study, and costs and timelines for its completion. This decision was not based on any analysis of efficacy data and there were no adverse safety findings that contributed to this decision. Transition also announced that a Phase 2 study of TT401 is in the final preparation stage with dosing expected to commence in calendar Q2 2014 and that there would be no further development of osteoarthritis preclinical candidate, TT601. This decision was made after expanded toxicology study data and regulatory interactions revealed the development plan for TT601 would be restricted and timelines delayed.
On May 15, 2014, Transition announced the dosing of the first patient in a Phase 2 clinical study of TT401 (LY2944876), a drug candidate for the treatment of type 2 diabetes. The study is expected to enroll up to 375 type 2 diabetes subjects and will be performed by Transition's development partner, Lilly. The objectives of the study will be to evaluate the safety and effectiveness of TT401 compared to once-weekly exenatide extended release and placebo.
On June 23, 2014, the Corporation announced the closing of its private placement financing through which 3,195,487 units of the Corporation were purchased by certain existing shareholders, board members and management of the Corporation at a price of US$5.32 per unit for gross proceeds of $18,319,000 (US$17.0 million). Each unit consisted of one common share and 0.61 Common Share purchase warrant with a purchase price of US$7.10 per whole warrant. Each whole warrant will entitle the holder, within two years of the closing date, to purchase one additional common share in the capital of the Corporation. If and when all of the warrants are exercised, the Corporation will realize an additional US$13.8 million in proceeds.
On July 11, 2014, the Corporation announced that Carl Damiani has been appointed Chief Operating Officer of Transition.
On November 4, 2014, Transition announced findings from a Phase 2 study of neuropsychiatric drug candidate, ELND005, as an adjunctive maintenance treatment for bipolar disorder type I patients (BPD). Overall, ELND005 had an acceptable safety and tolerability profile in the study, and showed numerical differences in the number of mood event recurrences favoring ELND005.
On November 20, 2014, the Corporation announced the results of a clinical study of neuropsychiatric drug candidate ELND005 in young adults with Down syndrome. TTIL completed this first study in Down syndrome subjects without dementia to allow optimal dose selection for future larger studies. The study enrolled 23 Down syndrome subjects in three study arms over a four-week treatment period. At the doses evaluated, ELND005 was determined to have an acceptable safety and tolerability profile and there were no serious adverse events reported.
On November 24, 2014, Transition announced results from a thorough QT (tQT) study in which no QT effects were observed at supra-therapeutic single doses of neuropsychiatric drug candidate, ELND005. A tQT study is a specialized clinical trial required by the FDA for the approval of most drugs in development. From a safety perspective, drugs that have no QT prolongation effects are particularly desirable for administration to an elderly Alzheimer’s disease (“AD”) population.
In December, 2014, TT401 diabetes drug candidate development partner Lilly informed Transition that the 70% patient enrollment milestone had been achieved triggering the payment of the third and final milestone payment which in aggregate totalled US$14 million. The Corporation has no additional funding obligations related to this Phase 2 clinical study.
On February 18, 2015, the Corporation announced the closing of a public offering of US$23 million of common shares equivalent to an aggregate of 3,538,461 common shares at a price to the public of US$6.50 per share, including 461,538 common shares issued upon the exercise of the underwriters’ over-allotment option. Cowen and Company, LLC was the sole book-running manager and Canaccord Genuity Inc., H.C. Wainwright & Co., LLC, and LifeSci Capital LLC were the co-managers for the offering.
In February, 2015, development partner Lilly informed Transition that 420 type 2 diabetic subjects have been enrolled in the current Phase 2 study thereby completing the enrollment phase of the study.
On March 2, 2015, the Corporation announced that its wholly owned subsidiary, Transition Ireland completed enrolment of 350 patients in the Phase 2 clinical study evaluating neuropsychiatric drug candidate ELND005 as a treatment for agitation and aggression in patients with Alzheimer’s disease (“AD”). The objectives of the Phase 2 clinical study (“Harmony AD Study”) are to evaluate the efficacy, safety and tolerability of ELND005 over 12 weeks of treatment in patients with mild to severe AD, who are experiencing at least moderate levels of agitation/aggression.
On March 26, 2015, Transition announced results from two phase 1 clinical studies of neuropsychiatric drug candidate ELND005. These studies, an absorption-metabolism-excretion (“AME”) study and a renal clearance study, are specialized clinical pharmacology trials that are required by the United States Food and Drug Administration (“FDA”) for the approval of most drugs in development.
On May 6, 2015, the Corporation announced its wholly-owned subsidiary, Transition Ireland has exclusively licensed worldwide rights to a novel small molecule drug candidate (“TT701”) from Lilly.
On June 16, 2015, Transition announced that Carl Damiani has been appointed as President and Chief Operating Officer of Transition.
On June 24, 2015, the Corporation announced results of Clinical Study of ELND005 in Agitation and Aggression in Patients with Alzheimer’s Disease. The Phase 2/3 clinical study of neuropsychiatric drug candidate ELND005 did not meet its primary efficacy endpoint. In the study, both the treatment and placebo groups showed a significant, but similar, reduction in agitation and aggression relative to baseline. There was a greater than expected reduction in agitation and aggression observed in the placebo group as measured in weeks 4, 8 and 12 in the study. The safety and tolerability profile of ELND005 was consistent with previous studies in Alzheimer’s disease at the 250mg bid dose.
Business of the Corporation
Market sizes appearing in this annual report are estimates of potential markets only. The Corporation makes no claim that such figures represent sales figures actually anticipated should the Corporation successfully develop and receive approval for any of its product candidates.
General
The Corporation’s principal business activity is the researching and developing of therapeutic agents.
The Corporation has two entities (Transition Ireland and Waratah) that are developing novel pharmaceuticals for disease indications with large markets. The Corporation’s other two entities, (Transition Therapeutics Inc. and Transition Therapeutics USA) provide development services to support the clinical and non-clinical activities of Transition Ireland and Waratah. The Corporation’s entities each perform different activities and have different business models.
Transition Ireland is developing two drug candidates; neuropsychiatric candidate ELND005 and androgen deficiency candidate TT701. In the recently completed Phase 2/3 study in Alzheimer’s disease patients, ELND005 did not meet the study’s primary efficacy endpoint. Transition Ireland is currently conducting a thorough review of all data from the studies with external clinical advisors to determine the potential future development path for this asset. Transition Ireland is also developing TT701 toward a Phase 2 clinical study expected to commence in fiscal 2016. TT701 is a selective androgen receptor modulator (“SARM”) that has been shown in a Phase 2 study to significantly increase lean body mass and a measurement of muscle strength in male subjects.
Waratah’s lead development asset is diabetes drug candidate TT401. Currently, Waratah’s development partner Lilly, is performing a 420 patient, Phase 2 clinical study of TT401 in type 2 diabetes individuals.
Transition Therapeutics Inc. and Transition USA provide development services in support of the clinical and non-clinical activities of Transition Ireland and Waratah. The Corporation operates in one operating segment, the research and development of therapeutic agents.
The Corporation’s strategic focus is on building shareholder value. To effectively achieve this, the Corporation has established a core business model based on the following steps: 1) identifying attractive early stage technologies targeting large markets; 2) moving these products through the clinic to provide validation; 3) considering additional product opportunities; 4) identifying partners with the infrastructure and resources to complete late stage clinical development and product commercialization; and 5) identifying new product opportunities to expand the Corporation’s product pipeline.
In addition to this business model, Transition Ireland has a separate and distinct business. Transition Ireland acquired the development and commercialization rights to the late stage neuropsychiatric asset, ELND005. Transition Ireland’s business model is to perform later stage clinical development, including late stage Phase 2 and Phase 3 clinical studies for this asset and potentially other drug candidates, including TT701 which was acquired from Lilly in May, 2015. Transition Ireland has contracted development services from contract research organizations, Transition Therapeutics Inc., Transition Therapeutics USA and third parties to perform the development activities of ELND005. The development of un-partnered assets, such as ELND005, will allow Transition Ireland to access the economics associated with advancing assets to regulatory approval.
Technology
The Corporation’s technologies in development are as follows: (i) Transition Ireland is developing ELND005 for the treatment of Alzheimer’s disease and Down syndrome as well as TT701 for the treatment of androgen deficiencies and (ii) Waratah is developing TT401for the treatment of diabetes.
ELND005 for Alzheimer`s Disease
The Corporation’s subsidiary, Transition Ireland, is developing small molecule therapeutics that act by preventing the formation of and breaking down amyloid beta peptide aggregates. The accumulation of amyloid beta has been connected to several diseases including Alzheimer’s disease, AA Amyloidosis and others.
The Product
ELND005, scyllo-inositol, is an orally bioavailable small molecule that is being investigated for neuropsychiatric indications on the basis of its proposed dual mechanism of action, which includes β-amyloid anti-aggregation and regulation of brain myo-inositol levels. An extensive clinical program of Phase 1 and Phase 2 studies have been completed with ELND005. The Phase 2 study (ELND005-AD201) which evaluated ELND005 in more than 350 mild to moderate AD patients was published in the peer-reviewed journal,Neurology. TheNeurology article was entitled “A Phase 2 randomized trial of ELND005, scyllo-inositol, in mild-moderate Alzheimer’s disease”.
On July 17, 2013, the FDA granted Fast Track Designation to the development program for ELND005 which was submitted for the treatment of Neuropsychiatric Symptoms (“NPS”) in Alzheimer's disease. The FDA concluded that the development program for ELND005 for the treatment of NPS in AD meets their criteria for Fast Track Designation.
On June 24, 2015, the Corporation reported that a Phase 2/3 study of ELND005 in Alzheimer’s disease patients with at least moderate levels of agitation/aggression did not meet its primary endpoint. In the study, both the treatment and placebo groups showed a significant, but similar, reduction in agitation and aggression relative to baseline. There was a greater than expected reduction in agitation and aggression observed in the placebo group as measured in weeks 4, 8 and 12 in the study. The safety and tolerability profile of ELND005 was consistent with previous studies in AD at the 250mg bid dose. The Company is performing a thorough review of the data from the completed study in agitation and aggression. An external clinical advisory board is working with the Company to evaluate the data and consider potential future clinical development paths for ELND005.
Alzheimer’s Disease – The Disease and the Market Opportunity
Alzheimer’s disease is a progressive brain disorder that gradually destroys a person’s memory and ability to learn, reason, make judgments, communicate and carry out daily activities. As Alzheimer’s disease progresses, individuals may also experience changes in personality and behaviour, such as anxiety, suspiciousness or agitation, as well as delusions or hallucinations. In late stages of the disease, individuals need help with dressing, personal hygiene, eating and other basic functions. People with Alzheimer’s disease die an average of eight years after first experiencing symptoms, but the duration of the disease can vary from three to 20 years.
The disease mainly affects individuals over the age of 65 and it is estimated over 18 million people are suffering from Alzheimer’s disease worldwide. The likelihood of developing late-onset Alzheimer’s approximately doubles every five years after age 65. By age 85, the risk reaches nearly 50 percent. In the United States, Alzheimer’s disease is the sixth leading cause of death and current direct/indirect costs of caring for an estimated 5.4 million Alzheimer’s disease patients are at least US$100 billion annually. Scientists have so far discovered one gene that increases risk for late-onset of the disease.
Current FDA approved Alzheimer’s disease medications may temporarily delay memory decline for some individuals, but none of the currently approved drugs are known to stop the underlying degeneration of brain cells. With no approved therapies for neuropsychiatric symptoms of Alzheimer’s disease, certain drugs approved to treat other illnesses may sometimes help with the emotional and behavioural symptoms of Alzheimer’s disease.
Neuropsychiatric symptoms associated with Alzheimer’s disease are a significant problem for Alzheimer’s disease patients and their caregivers. Approximately 90% of Alzheimer’s disease patients develop neuropsychiatric symptoms, and up to 60% develop agitation/aggression over the course of their disease. Agitation/aggression are among the most disruptive neuropsychiatric symptoms in Alzheimer’s disease and are associated with increased morbidity and caregiver burden. With an aging population, there is a great need for new therapies that can effectively reduce or delay the neuropsychiatric symptoms associated with Alzheimer’s disease.
ELND005 for Down Syndrome
On November 20, 2014, the Corporation announced the results of a clinical study of neuropsychiatric drug candidate ELND005 in young adults with Down syndrome. Transition’s wholly-owned subsidiary, Transition Therapeutics Ireland Limited (“TTIL”) completed this first study in Down syndrome subjects without dementia to allow optimal dose selection for future larger studies.
The study enrolled 23 Down syndrome subjects in three study arms over a four-week treatment period: placebo (n=6), 250mg once daily (QD) (n=5), and 250mg twice daily (BID) (n=12). ELND005, at the doses evaluated, was determined to have an acceptable safety and tolerability profile and there were no serious adverse events reported in the study. Treatment emergent adverse events were reported in seven of the subjects receiving ELND005 and all were deemed to be mild in severity. The two ELND005 doses achieved the plasma levels expected in pharmacokinetic modeling and will inform the selection of a higher dose in a future clinical study.
The Product
Excess activity of genes on chromosome 21, such as amyloid precursor protein (APP) and sodium-myo-inositol active transporter (SMIT), are thought to play a role in the cognitive dysfunction of DS. Life-long exposure to increased amyloid and myo-inositol levels in the brain are thought to lead to synaptic dysfunction and cognitive disability. ELND005 may have the potential to improve cognition in DS by decreasing amyloid levels and regulating myo-inositol-dependent neuronal signaling.
Down Syndrome – The Disease and the Market Opportunity
Down syndrome (DS, Trisomy 21), caused by an extra copy of chromosome 21, is the most common genetic form of intellectual disability with a prevalence of approximately 1 in 700 live births in the US. Children with DS exhibit developmental delay and various degrees of intellectual disability, while adults are at increased risk of Alzheimer’s dementia. There are currently no drugs approved for the treatment of cognitive dysfunction in DS.
TT401 for Type 2 Diabetes
In March, 2010, the Corporation acquired the rights to a series of preclinical compounds from Lilly in the area of diabetes. Under the licensing and collaboration agreement, the Corporation receives exclusive worldwide rights to develop and potentially commercialize a class of compounds.
The Product
In preclinical diabetes models, the lead compound, TT401 has shown potential to provide glycemic control and other beneficial effects including weight loss. In June, 2012, the Corporation announced the results of the Phase 1 clinical study of type 2 diabetes drug candidate, TT401. The Phase 1, double-blind, placebo-controlled randomized study enrolled 48 non-diabetic obese subjects in six cohorts evaluating six escalating subcutaneous single doses of TT401. TT401 demonstrated an acceptable safety and tolerability profile in non-diabetic obese subjects in the study. TT401 exhibited the expected pharmacological effect on glucose and pharmacodynamic biomarkers at doses that were safe and tolerable. The pharmacokinetic profile, assessed over 28 days, demonstrated a half-life consistent with once-weekly dosing.
In April, 2013, Transition announced the results of a five-week proof of concept clinical study of TT401 in type 2 diabetes and obese non-diabetic subjects. The study enrolled diabetic patients at five dosing levels and non-diabetic obese patients at one dose level. All dosing cohorts received five doses over a five week period. Diabetic patients were on stable doses of metformin.
At the end of the treatment period, TT401-treated patients in the 3 highest dose groups experienced statistically significant reductions in mean fasting plasma glucose relative to placebo. Statistically significant mean body weight reduction relative to baseline occurred in the three highest dose groups. A similar reduction in body weight was also observed in the obese non-diabetic cohort. TT401 demonstrated an acceptable safety and tolerability profile at all doses evaluated in diabetic and non-diabetic obese subjects. The most common adverse event noted in the study was decreased appetite. Some subjects in the highest three dose groups experienced mild nausea and vomiting, which are consistent with studies of other GLP-1 agonist drug candidates. The pharmacokinetic profile, assessed over the five week study, demonstrated a half-life consistent with once-weekly dosing.
In June, 2013, Lilly exercised its option to assume all development and commercialization rights to type 2 diabetes drug candidate TT401. In conjunction with this assumption of rights, Transition received a US$7 million milestone payment. Lilly and Transition have amended their agreement to address future development of TT401 and associated financial arrangements. Lilly assumed all costs and will perform all future development and commercialization activities of TT401. Transition made payments totalling US$14 million to Lilly in three separate installments during the Phase 2 clinical study.
On May 15, 2014, Transition announced the dosing of the first patient in a Phase 2 clinical study of TT401 for the treatment of type 2 diabetes. The study enrolled 420 type 2 diabetes subjects and is being performed by Transition's development partner Lilly. The main efficacy outcome measures is the change in HbA1c (a measure of blood-glucose levels) at week 12 and 24 and change in body weight over the course of the study.
Diabetes – The Disease and the Market Opportunity
Diabetes is a disease in which the body does not produce or properly use insulin. Insulin is a hormone released from islet cells located in the pancreas that is needed to convert sugar, starches and other food into energy needed for daily life. There are two primary forms of diabetes; type 1 diabetes and type 2 diabetes.
Type 1 diabetes develops when the body’s immune system destroys pancreatic islet beta cells, the only cells in the body that make the hormone insulin that regulates blood glucose. To survive, people with type 1 diabetes must have insulin delivered by injection or pump. Type 1 diabetes accounts for 5-10% of all diagnosed cases of diabetes.
Type 2 diabetes usually begins as insulin resistance, a disorder in which the cells do not use insulin properly. As the need for insulin increases, the pancreas gradually loses its ability to produce it. Current treatments for type 2 diabetes include lifestyle changes, oral medications, incretin therapy and insulin therapy. Type 2 diabetes accounts for about 90-95% of all diagnosed cases of diabetes.
With the insulins and anti-diabetic medications markets having global sales of over US$9 billion and US$15 billion, respectively, the market opportunity for an effective therapeutic is significant.
TT701 for Androgen Deficiencies
The Product
TT701 is a selective androgen receptor modulator that has been shown in a Phase 2 study to significantly increase lean body mass and a measurement of muscle strength in male subjects. This completed 12-week, Phase 2 study of 350 subjects also demonstrated additional beneficial effects, including significant fat mass reduction with no significant change in prostate specific antigen (PSA) levels. Transition Ireland is evaluating multiple development paths for TT701, including as a new therapeutic option for patients with androgen deficiency. Transition Ireland is engaged with potential collaborators to rapidly commence a Phase 2 clinical study.
Androgen Deficiencies – The Disease and the Market Opportunity
Androgens are a group of hormones that play a role in male traits and reproductive activity. Present in both males and females, the principle androgens are testosterone and androstenedione. Men experience a modest and gradual drop in sex hormone levels over a long period of time, from the age of about 30. However, androgen deficiency is when the body has lower levels of male sex hormones, particularly testosterone, than is needed for health. This deficiency can be caused by a number of conditions leading to reduced libido, reduced lean body mass, increased fat mass and other undesired outcomes.
Transition Ireland is actively engaged with potential partners to collaborate on the development of TT701 for specific medical populations. These target medical populations are individuals who can benefit from androgen therapy, but their background condition would preclude the use of testosterone replacement therapy. The markets that Transition Ireland is targeting for the TT701 drug candidate do not have a product approved for androgen replacement therapy.
Product Pipeline
Below is a diagram illustrating the Corporation’s product pipeline for its lead technologies, and the current stage of development for each product:
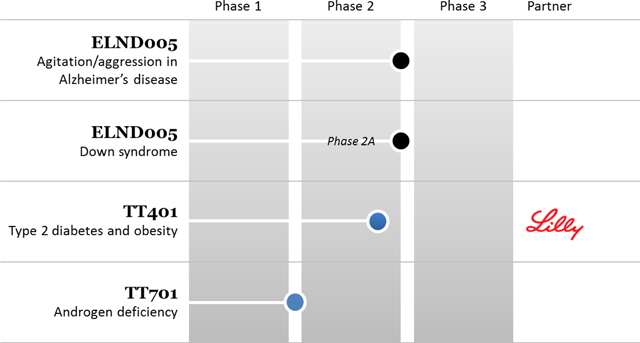
Regulatory Approval Process for Therapeutic Drugs
The development of new pharmaceuticals is strongly influenced by a country’s regulatory environment. The drug approval process in Canada is regulated by Health Canada. In the United States, the regulatory body is the FDA. Similar processes are conducted in other countries by similar regulatory bodies. Regulations in each jurisdiction require that licenses be obtained from regulatory agencies for drug manufacturing facilities and also mandate strict research and product testing standards in order to ensure quality in respect of the manufacturing of therapeutic products, “Good Manufacturing Practices” (“GMP”). Companies must establish that the production of their products comply with GMP and the clinical development be conducted with Good Clinical Practices in order to demonstrate the safety and effectiveness of the therapeutic. While the Corporation will pursue the approval of any product that it develops, success in acquiring regulatory approval for any such product is not assured. See “Risks and Uncertainties”.
In order to market its pharmaceutical products in Canada and the United States, the Corporation must successfully satisfy the requirements of each of the following stages of the regulatory approval process and drug development:
Pre–Clinical Studies: Pre–clinical studies involve extensive testing in laboratory animals to determine if a potential therapeutic product has utility in an in vivo disease model and has any adverse toxicological effects in animals. The conduct and results of these studies are reported to regulatory agencies in an Investigational New Drug (“IND”) application in the United States and a Clinical Trial Application(“CTA”)in Canada, to gain approval to commence clinical trials of the product in human subjects or patients, depending on the indication for use.
Phase 1 Clinical Trials:Phase 1 clinical trials are designed to determine the pharmacokinetics, metabolism and pharmacologic actions of the drug in humans, the side effects associated with increasing doses and the maximum tolerated dose. These studies, usually short in duration, are typically conducted with healthy volunteers.
Phase 2 Clinical Trials:Phase 2 studies are conducted to evaluate the safety of the drug in the intended patient population with the disease or condition under study and to determine the common short-term side effects and risks associated with the drug. Phase 2 studies are typically well controlled, closely monitored and conducted in a relatively small number of patients. These studies are usually designed to gain early evidence of the effectiveness of the therapeutic, along with its safety.
Phase 3 Clinical Trials:Phase 3 studies are expanded studies performed after preliminary evidence suggesting effectiveness of the drug is obtained. Phase 3 studies gather additional information about effectiveness and safety that is required to evaluate the overall benefit-risk profile of the drug and to provide adequate basis for physician labelling. Phase 3 trials usually involve several hundred to several thousand patients.
After all three phases of clinical trials have been completed the results are then submitted to the respective health authority for marketing approval in the respective countries. If and when marketing approval is granted by Health Canada or the FDA, as the case may be, the product is then approved for commercial sale in the respective jurisdiction.
In addition to the approval of the drug itself, Health Canada and the FDA each require that the manufacturer of a therapeutic drug be in full compliance with the current GMPs in effect in Canada or the United States, respectively. A similar process for therapeutic drug approval is followed in most other countries with sophisticated regulatory bodies that have appropriate regulations and oversight.
Manufacturing
The Corporation relies on third party manufacturers to supply all of its drug substances and the finished dosage form for its preclinical and clinical products. Similarly, it will rely on third party manufacturers to manufacture its products for sale. As such, the commercial success of such products may be outside of the Corporation’s control. See “Risk Factors”.
The preclinical and clinical products are produced in compliance with current GMPs as established by applicable regulatory authorities, and the manufacturer is responsible for ensuring compliance to the set standard, and biosafety testing, with full characterization being the responsibility of the Corporation.
Thus far, Transition Ireland’s neuropsychiatric drug candidate ELND005, the active pharmaceutical ingredient, has been produced by Albany Molecular Research Inc. (New York), Abbott Laboratories (Illinois) and Lonza Sales Limited (Czech Republic). The final drug product for all the Phase 1 and Phase 2 studies was manufactured by Patheon (Canada) and Alkermes Pharma Ireland Limited (Ireland), third party contract manufacturers for clinical use. For the recently completed ELND005 Phase 2 studies and any future studies, the final drug product is prepared in a tablet form by Alkermes Pharma Ireland Limited (Ireland) and packaged by Catalent CTS (Kansas City) LLC (Missouri).
The active pharmaceutical ingredient for diabetes drug candidate TT401 was manufactured by PolyPeptide Laboratories, Inc. (California). The final injectable drug product for the Phase 1 studies was manufactured by AAIPharma Services Corp. Lilly is responsible for manufacturing the final drug product that is currently being used in the ongoing Phase 2 clinical trial and beyond.
Product Marketing Strategy
The markets for the products being developed by the Corporation are large and may require substantial sales and marketing capability. Before successful completion of the development of the Corporation’s various products, the Corporation hopes to enter into one or more strategic partnerships or other collaborative arrangements with pharmaceutical or other companies that have marketing and distribution expertise to address these needs. If appropriate, the Corporation will establish arrangements with various partners for different geographical areas.
Transition Ireland’s business strategy of developing products towards obtaining regulatory approval may lead Transition Ireland to acquire a commercialization partner at or near the regulatory approval stage of its development assets, including, but not limited to ELND005.
Specialized Skills and Knowledge
As of June 30, 2015, the Corporation had 18 full-time employees, who possess the skills and knowledge that the Corporation requires to implement its current strategy.
The Corporation believes that investing in human capital is fundamental to its continued growth and success. The Corporation depends on its people for constant innovation and research and development. The Corporation intends to implement a practice of aggressively recruiting high calibre personnel and retaining such personnel by offering appropriate compensation incentives.
Competitive Conditions
There are a number of treatments in various stages of development which may compete with the Corporation’s therapeutic programs in each of its selected therapeutic disease-specific applications. The following is a summary of the principal therapeutic treatments which the Corporation understands are currently being developed by others for therapeutic areas in which the Corporation is currently focusing its clinical efforts. This summary is not necessarily an exhaustive list of such competing therapeutic treatments. Competition may have an adverse effect on the Corporation. See “Risk Factors”.
Alzheimer’s Disease
Currently, all of the approved Alzheimer’s therapies and many of the clinical candidates seek to reduce Alzheimer’s related symptoms rather than treating the underlying disease. These products include cholinesterase inhibitors and glutamate receptor antagonists. The emerging classes of compounds in clinical development are immunotherapies and secretase enzyme inhibitors. Each class seeks to slow or even to reverse the Alzheimer’s disease process by targeting a mechanism to reduce the occurrence of beta-amyloid plaques, a hallmark pathology of the disease.
The treatment of neuropsychiatric symptoms associated with AD is emerging as an important new focus for the AD clinical research community. There are currently no approved drugs for the treatment of agitation and aggression associated with AD. As diagnostics and clinical measures have evolved for this type of neuropsychiatric symptom, there are now multiple drug candidates being developed to address agitation and aggression in AD. The leading candidates are novel anti-depressant therapies and therapies that are approved for other neuropsychiatric indications.
Diabetes
The following is a brief summary of the principal therapeutic strategies, of which the Corporation is aware, currently being developed for the treatment of diabetes.
Intensive Insulin Therapy - The goal of intensive insulin therapy is to more accurately control hyperglycemia by increasing the frequency of insulin injection. This type of insulin therapy, however, increases the risk of hypoglycemia and demands more frequent blood sugar monitoring which can be painful and time consuming. This approach only addresses type 1 diabetics and the subset of type 2 diabetics that requires constant insulin injections and it only manages the disease, it does not offer a long-term solution.
Oral Antidiabetic Agents are FDA approved for use in conjunction with diet and exercise to enhance glycemic control in type 2 diabetic patients.
Oral sulfonylureas reduce blood glucose by stimulating insulin from pancreatic beta-cells as well as increasing responsiveness in insulin-sensitive tissues.
Metformin is an oral hypoglycemic agent that improves glycemic control by decreasing hepatic glucose production and intestinal glucose absorption as well as improving insulin sensitivity through increased peripheral glucose uptake and utilization.
Thiazolidinediones are potent agonists of peroxisome proliferator-activated receptor-gamma (“PPAR-gamma”), receptors important for insulin action which are located in adipose tissue, liver and skeletal muscle. Activation of these receptors affects the transcription of genes responsible for control of glucose and lipid metabolism. These agents, in the presence of insulin, decrease insulin resistance in the liver and at peripheral sites and improve insulin-dependent glucose disposal and reduce hepatic glucose output.
Non-Insulin Dependent Therapies -SGLT2 inhibitors are the most recent class of oral drugs approved for the treatment of type 2 diabetes mellitus. They inhibit glucose re-absorption in the proximal renal tubules providing an insulin independent mechanism to lower blood glucose. Their use in clinical practice is associated with improved glycemic control, marginal weight loss and a low risk of hypoglycemia.
Incretin Therapies - The incretin hormones, GLP-1 and glucose-dependent insulinotropic peptide (“GIP”), have been identified as important factors in glucose homeostasis. Released from the gut following a meal, GLP-1 and GIP stimulate insulin secretion from pancreatic beta cells in response to normal or elevated blood glucose concentrations. GLP-1 also lowers glucagon excretion from pancreatic beta cells, which results in reduced hepatic glucose production, and also reduces appetite, slows gastric emptying, and improves β-cell function. When administered intravenously or subcutaneously, GLP-1 is effective in treating type 2 diabetes. Unfortunately, both GLP-1 and GIP are rapidly degraded by dipeptidyl peptidase IV (“DPP-IV”); therefore, research has been focused on preventing degradation by inhibiting the DPP-IV enzyme. Sitagliptin was the first novel agent with this unique mechanism of action: the drug acts as a DPP-IV inhibitor which reduces inactivation of incretin hormones and improves glycemic control in type 2 diabetic patients. Additional DPP-IV inhibitors have been approved for use in the treatment of type 2 diabetes.
Intellectual Property
The Corporation’s intellectual property practice is to file patent and trademark applications to protect proprietary inventions, technologies, improvements and trademarks that are considered to be important to the development of the Corporation’s business and consistent with the Corporation’s strategic focus. The Corporation also depends upon trade secrets and licensing opportunities to expand and maintain its competitive position.
The Corporation possesses a strong intellectual property position for its platform technologies. The Corporation, through all its entities, currently holds the rights to ten patent families relating to its drug development programs. To date, the Corporation possesses or exclusively licenses fourteen issued patents, with the remainder in varying stages of the patent application process. Current Canadian and U.S. patent laws provide that the Corporation’s patents are protected for a period of 20 years after their filing dates.
Patent Protection
The Corporation’s patent portfolio provides protection for its areas of technology focus:
ELND005
The Corporation’s subsidiary, Transition Ireland, has a number of U.S., Canadian and foreign patent applications for the ELND005 technology. This portfolio has been expanded with the filing of additional patent applications based on findings from clinical and preclinical studies.
U.S. Patent Number 7,521,481, issued on April 21, 2009, covers treatment of Alzheimer's Disease with ELND005. European Patent No. 1608350 granted on June 3, 2009 and European Patent No. 2311445 granted on March 26, 2014 cover ELND005 for treatment or prevention of a condition of the central or peripheral nervous system or systemic organ associated with a disorder in protein folding aggregation, or amyloid formation, deposition accumulation or persistence. The U.S. Patent will expire in the year 2026 or later due to any patent term extensions. The Corporation’s subsidiary, Transition Ireland, is actively prosecuting patent application families in multiple jurisdictions throughout the world.
TT401 (LY2944876)
In March 2010, the Corporation acquired the rights to a series of preclinical compounds from Lilly in the area of diabetes. In June 2013, Lilly exercised its exclusive option to re-acquire all development and commercialization rights to TT401 and TT402. With this assumption of rights by Lilly, Transition’s license to develop and commercialize TT401 and TT402 was terminated. The Corporation, through its subsidiary Waratah Pharmaceuticals, has the right to receive milestone payments and royalties from the development and commercialization of TT401 and TT402 into the future. Patent applications covering the composition of matter for drug candidate TT401 have been filed worldwide, including in the United States, Australia, Canada, China, Europe and Japan. Patents have been issued in the US, Europe, Australia and a number of other jurisdictions and will expire in 2030. This patent expiry date does not include any possible patent term extensions.
Property, Plant and Equipment
The Corporation’s head office is located at Suite 220, 101 College Street, Toronto, Ontario, Canada,, M5G 1L7. The Corporation occupies a total of 5,716 square feet of the building which represents approximately 3.1% of the total facility.
Item 4A. Unresolved Staff Comments
None.
Item 5. Operating and Financial Review and Prospects
MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITIONS AND RESULTS OF OPERATIONS
The following information should be read in conjunction with the Corporation’s audited consolidated financial statements for the year ended June 30, 2015 and the related notes, which are prepared in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board (IFRS). This Management’s Discussion and Analysis (“MD&A”) provides a review of the performance of the Corporation for the year ended June 30, 2015 as compared to the year ended June 30, 2014. This MD&A includes financial information derived from the annual audited consolidated financial statements.
Overview
Transition is a biopharmaceutical development company, advancing novel therapeutics for CNS and metabolic disease indications. The Company’s wholly-owned subsidiary, Transition Therapeutics Ireland Limited (“TTIL”) is developing CNS drug candidate ELND005 for the treatment of Alzheimer’s disease (“AD”) and Down syndrome. Transition’s lead metabolic drug candidate is TT401 for the treatment of type 2 diabetes and accompanying obesity.
During the fiscal year ended June 30, 2015 and up to the date of this MD&A, the Company announced the following:
ELND005:
| · | June 24, 2015 – Transition announced resultsof Clinical Study of ELND005 in Agitation and Aggression in Patients with Alzheimer’s Disease.The Phase 2/3 clinical study of neuropsychiatric drug candidate ELND005 did not meet its primary efficacy endpoint. In the study, both the treatment and placebo groups showed a significant, but similar, reduction in agitation and aggression relative to baseline. There was a greater than expected reduction in agitation and aggression observed in the placebo group as measured in weeks 4, 8 and 12 in the study. The safety and tolerability profile of ELND005 was consistent with previous studies in AD at the 250mg bid dose; |
| · | March 26, 2015 – Transition announced results from two phase 1 clinical studies of neuropsychiatric drug candidate ELND005.These studies, an absorption-metabolism-excretion (“AME”) study and a renal clearance study, are specialized clinical pharmacology trials that are required by the United States Food and Drug Administration (“FDA”) for the approval of most drugs in development; |
| · | November 24, 2014 – Transition announced results from a thorough QT (tQT) study in which no QT effects were observed at supra-therapeutic single doses of neuropsychiatric drug candidate, ELND005. A tQT study is a specialized clinical trial required by the FDA for the approval of most drugs in development. From a safety perspective, drugs that have no QT prolongation effects are particularly desirable for administration to an elderly Alzheimer’s disease AD; |
| · | November 20, 2014 – Transition announced the results of a clinical study of neuropsychiatric drug candidate ELND005 in young adults with Down syndrome. TTIL completed this first study in Down syndrome subjects without dementia to allow optimal dose selection for future larger studies. The study enrolled 23 Down syndrome subjects in three study arms over a four-week treatment period. At the doses evaluated, ELND005 was determined to have an acceptable safety and tolerability profile and there were no serious adverse events reported; |
| · | November 4, 2014 - Transition announced findings from a Phase 2 study of neuropsychiatric drug candidate, ELND005, as an adjunctive maintenance treatment for bipolar disorder type I patients (BPD). TTIL terminated the bipolar disorder Phase 2 study on April 7, 2014 for business reasons. TTIL has completed a review of the data from this bipolar disorder Phase 2 study. Overall, ELND005 had an acceptable safety and tolerability profile in the study, and showed numerical differences in the number of mood event recurrences favoring ELND005. |
TT401:
| · | In February 2015, development partner Lilly informed Transition that 420 type 2 diabetic subjects have been enrolled in the current Phase 2 study thereby completing the enrollment phase of the study.The randomized, double-blind, placebo-controlled study includes six study arms, four doses of TT401, a placebo arm and a once-weekly exenatide arm. The main efficacy outcome measures are the change in HbA1c, a measure of blood-glucose levels, at week 12 and 24 and change in body weight over the course of the study; |
| · | Transition has paid all three installment payments totaling US$14 millionto diabetes drug candidate development partner Lilly.Transition has no further financial obligations for the development and commercialization of TT401. In December, 2014, Lilly informed Transition that the 70% enrollment milestone had been achieved. |
Corporate Developments:
| · | June 16, 2015 - Transition announced that Carl Damiani has been appointed as President and Chief Operating Officer of Transition; |
| · | May 6, 2015 – Transition announced its wholly-owned subsidiary, TTIL has exclusively licensed worldwide rights to a novel small molecule drug candidate (“TT701”) from Eli Lilly and Company.Under the terms of the agreement, TTIL has acquired rights to develop and commercialize TT701. Lilly will receive upfront consideration of up to US$1 million. In addition, Lilly is eligible to receive up to US$100 million in commercial milestones and a mid-single digit royalty on sales of TT701 products should such products be successfully commercialized. TT701 is a selective androgen receptor modulator that has been shown in a Phase 2 study to significantly increase lean body mass and a measurement of muscle strength in male subjects. This completed 12-week, Phase 2 study of 350 subjects also demonstrated additional beneficial effects, including significant fat mass reduction with no significant change in prostate specific antigen (PSA) levels. TTIL is evaluating multiple development paths for TT701, including as a new therapeutic option for patients with androgen deficiency. TTIL is engaged with potential collaborators to rapidly commence a Phase 2 clinical study; |
| · | February 18, 2015 – Transition announced the closing of a public offering of US$23 million of common shares equivalent to an aggregate of 3,538,461 common shares at a price to the public of US$6.50 per share, including 461,538 common shares issued upon the exercise of the underwriters’ over-allotment option.Cowen and Company, LLC was the sole book-running manager and Canaccord Genuity Inc., H.C. Wainwright & Co., LLC, and LifeSci Capital LLC were the co-managers for the offering. |
Strategic Collaborations
Perrigo Company plc (“Perrigo”)
In 2006, Transition exclusively licensed the ELND005 technology to Elan for worldwide development and commercialization. Following amendment of that agreement in 2010, Elan held all development and commercialization rights to ELND005 and Transition became eligible to receive milestone and royalty payments with the successful advancement of ELND005. Transition has received US$40 million from Elan in upfront and achieved milestone payments. Perrigo acquired Elan in December 2013, including all Elan’s rights and obligations to the development of ELND005.
On February 28, 2014, Transition announced that after a series of transactions,Perrigo had transferred all of its ELND005 rights and assets under the collaboration agreement to the Company’s wholly owned subsidiary, TTIL. In parallel with this acquisition, Perrigo invested US$15 million and received 2,255,640 Transition common shares representing approximately a 6.4% ownership stake in Transition as of the date of the transaction. Perrigo will also be eligible to receive up to US$40 million in approval and commercial milestone payments and a 6.5% royalty on net sales of ELND005 products and sublicense fees received. Going forward, TTIL is responsible for all future development and commercialization activities of the ELND005 drug candidate.
Lilly
Diabetes
On March 3, 2010, Transition and Lilly entered into a licensing and collaboration agreement granting Transition the rights to a series of preclinical compounds in the area of diabetes. Under the licensing and collaboration agreement, Transition received exclusive worldwide rights to develop and potentially commercialize a class of compounds that, in preclinical models, showed potential to provide glycemic control and other beneficial effects including weight loss.
Under the terms of the agreement, Lilly received an up-front payment of $1,055,900 (US$1 million) which has been capitalized as a license acquired from Lilly and is being amortized over 20 years which represents the estimated life of the underlying compounds and patents.
In June 2013, Lilly exercised its option and assumed all development and commercialization rights to type 2 diabetes drug candidate TT401. In conjunction with this assumption of rights, Transition received a US$7 million milestone payment. Lilly has assumed all costs and will perform all future development and commercialization activities of TT401, and Transition paid US$14 million to Lilly in three separate installments during the Phase 2 clinical study; the first installment of US$6 million was paid during the three month period ended September 30, 2014 when the study achieved 20% patient enrollment. The remaining two installments totaling US$8 million were paid during the three month period ended December 31, 2014 when the study achieved both the 50% and 70% patient enrollment milestones. Transition has no additional funding obligations related to this clinical study or any other development or commercialization activities in the future.
Transition is eligible to receive up to approximately US$240 million in additional milestone payments plus double-digit royalties on sales of TT401 products and a low single digit royalty on sales of related compounds.
Programs
Transition is focused on developing innovative therapies in several distinct areas of opportunity. Transition’s vision is to build a company that has a strong foundation for growth based on multiple technologies and product opportunities, which reduces risk and enhances shareholder return. The Company’s technologies are as follows:
ELND005
Alzheimer’s Disease:
Alzheimer’s disease is a progressive brain disorder that gradually destroys a person’s memory and ability to learn, reason, make judgments, communicate and carry out daily activities. As Alzheimer’s disease progresses, individuals may also experience changes in personality and behavior, such as anxiety, suspiciousness or agitation, as well as delusions or hallucinations. Approximately 90% of Alzheimer’s disease patients develop neuropsychiatric symptoms, and up to 60% develop agitation/aggression over the course of their disease. Agitation/aggression are among the most disruptive neuropsychiatric symptoms in Alzheimer’s disease and are associated with increased morbidity and caregiver burden.
The disease mainly affects individuals over age 65 and it is estimated over 18 million people are suffering from Alzheimer’s disease worldwide. In the U.S., Alzheimer’s disease is the sixth leading cause of death and current direct/indirect costs of caring for an estimated 5.4 million Alzheimer’s disease patients are at least US$100 billion annually.
Current U.S. Food and Drug Administration approved Alzheimer’s disease medications may temporarily delay memory decline for some individuals, but none of the currently approved drugs are known to stop the underlying degeneration of brain cells. Certain drugs approved to treat other illnesses may sometimes help with the emotional and behavioral symptoms of Alzheimer’s disease. With an aging population, there is a great need for therapies to address Alzheimer’s disease patient’s neuropsychiatric symptoms and declines in cognitive ability.
Down Syndrome:
Down syndrome (DS, Trisomy 21), caused by an extra copy of chromosome 21, is the most common genetic form of intellectual disability with a prevalence of approximately 1 in 700 live births in the US. Children with DS exhibit developmental delay and various degrees of intellectual disability, while adults are at increased risk of Alzheimer’s dementia. There are currently no drugs approved for the treatment of cognitive dysfunction in DS.
Excess activity of genes on chromosome 21, such as amyloid precursor protein (APP) and sodium-myo-inositol active transporter (SMIT), are thought to play a role in the cognitive dysfunction of DS. Life-long exposure to increased amyloid and myo-inositol levels in the brain are thought to lead to synaptic dysfunction and cognitive disability. ELND005 may have the potential to improve cognition in DS by decreasing amyloid levels and regulating myo-inositol-dependent neuronal signaling.
Clinical Development Update
ELND005 for Neuropsychiatric Diseases
TTIL is developing neuropsychiatric drug candidate ELND005, (scyllo-inositol). ELND005 is an orally bioavailable small molecule that is being investigated for multiple neuropsychiatric indications on the basis of its proposed dual mechanism of action, which includes β-amyloid anti-aggregation and regulation of brain myo-inositol levels. An extensive clinical program of Phase 1 and Phase 2 studies have been completed with ELND005 to support clinical development. The Phase 2 study (ELND005-AD201) which evaluated ELND005 in more than 350 mild to moderate AD patients was published in the peer-reviewed journal, Neurology. The Neurology article was entitled “A Phase 2 randomized trial of ELND005, scyllo-inositol, in mild-moderate Alzheimer’s disease”.
Currently, the use of ELND005 is being investigated in two clinical areas:
| (a) | Agitation and Aggression in Alzheimer’s Disease |
On November 27, 2012, the first patient was enrolled in a Phase 2 clinical trial of ELND005 for the treatment of agitation/aggression in patients with mild to severe Alzheimer’s disease. The objectives of the study are to evaluate the efficacy, safety and tolerability of ELND005 over 12 weeks of treatment in patients with mild to severe AD, who are experiencing at least moderate levels of agitation/aggression. Enrollment of this clinical study (AG201) known as the “Harmony AD” study (www.harmonyadstudy.com) was completed on March 2, 2015 with a total of 350 patients being enrolled.
On June 24, 2015, Transition announced that ELND005 did not meet its primary efficacy endpoints in the Phase 2/3 clinical study of ELND005 in agitation and aggression in patients with Alzheimer’s Disease. In the study, both the treatment and placebo groups showed a significant, but similar, reduction in agitation and aggression relative to baseline. There was a greater than expected reduction in agitation and aggression observed in the placebo group as measured in weeks 4, 8 and 12 in the study. The safety and tolerability profile of ELND005 was consistent with previous studies in AD at the 250mg bid dose.
As ELND005 did not meet its primary efficacy endpoint in the Phase 2/3 clinical study in agitation and aggression in Alzheimer’s disease, management performed an impairment test and noted there is no impairment of the ELND005 asset as at June 30, 2015. The Company is performing a thorough review of the data from the completed study in agitation and aggression. An external clinical advisory board is working with the Company to evaluate the data and consider potential future clinical development paths for ELND005.
On November 20, 2014, Transition announced the results of a clinical study of neuropsychiatric drug candidate ELND005 in young adults with Down syndrome. Transition’s wholly-owned subsidiary, TTIL completed this first study in Down syndrome subjects without dementia to allow optimal dose selection for future larger studies.
The study enrolled 23 Down syndrome subjects in three study arms over a four-week treatment period: placebo, 250 mg once daily; and 250 mg twice daily. At the doses evaluated, ELND005 was determined to have an acceptable safety and tolerability profile and there were no serious adverse events reported in the study. Treatment emergent adverse events were reported in seven of the subjects receiving ELND005 and all were deemed mild in severity. The two ELND005 doses achieved the plasma levels expected in pharmacokinetic modeling and will inform the selection of a higher dose in a larger Phase 2b study in Down syndrome subjects.
The ELND005 technology is claimed in multiple issued patents and pending patent applications in many jurisdictions throughout the world.
Expenditures for the ELND005 Program
On February 28, 2014, Transition announced that after a series of transactions, Perrigo has transferred all of its ELND005 rights and assets to the Company’s wholly owned subsidiary, TTIL. As a result, effective March 1, 2014, TTIL is responsible for all future development and commercialization activities of the ELND005 drug candidate.
During the years ended June 30, 2015 and 2014, the Company incurred direct research and development costs for this program as follows:
| ELND005 Program (1) | | Fiscal 2015 | | | Fiscal 2014 | |
| | | | | | | |
| Pre-clinical studies | | $ | - | | | $ | - | |
| Clinical studies | | | 20,154,069 | | | | 8,473,306 | |
| Manufacturing | | | 796,511 | | | | 270,241 | |
| Other direct research | | | 2,356,831 | | | | 955,608 | |
| TOTAL | | $ | 23,307,411 | | | $ | 9,699,155 | |
Note(1)These costs are direct research and development costs only and do not include patent costs, investment tax credits, salaries and benefits, amortization of intangible assets or an allocation of Company overhead.
Prior to the February, 2014 acquisition, Transition was not required to fund the development or commercialization of ELND005 and accordingly, development costs were nil during the first eight months of fiscal 2014.
TT401 / TT402
Development of TT401 and TT402 for Diabetes
Diabetes is a disease in which the body does not produce or properly use insulin. Insulin is a hormone released from islet cells located in the pancreas that is needed to convert sugar, starches and other food into energy needed for daily life. There are two primary forms of diabetes; type 1 diabetes and type 2 diabetes.
Type 2 diabetes usually begins as insulin resistance, a disorder in which the cells do not use insulin properly. As the need for insulin increases, the pancreas gradually loses its ability to produce it. Current treatments for type 2 diabetes include lifestyle changes, oral medications, incretin therapy and insulin therapy. Type 2 diabetes accounts for about 90-95% of all diagnosed cases of diabetes.
Clinical Development Update of TT401 (LY2944876)
On March 3, 2010, Transition announced that it had acquired the exclusive worldwide rights to develop and potentially commercialize a series of preclinical compounds from Lilly in the area of diabetes. In preclinical diabetes models, these compounds showed potential to provide glycemic control and other beneficial effects including weight loss.
On June 18, 2012, Transition announced the results of the Phase 1 clinical study of type 2 diabetes drug candidate, TT401. The Phase 1, double-blind, placebo-controlled randomized study enrolled 48 non-diabetic obese subjects in six cohorts evaluating six escalating subcutaneous single doses of TT401. TT401 demonstrated an acceptable safety and tolerability profile in non-diabetic obese subjects in the study. TT401 exhibited the expected pharmacological effect on glucose and pharmacodynamic biomarkers at doses that were safe and tolerable. The pharmacokinetic profile, assessed over 28 days, demonstrated a half-life consistent with once-weekly dosing.
On April 30, 2013, Transition announced the results of a five-week proof of concept clinical study of TT401 in type 2 diabetes and obese non-diabetic subjects. The study enrolled diabetic patients at five dosing levels and non-diabetic obese patients at one dose level. All dosing cohorts received five doses over a five week period. Diabetic patients were on stable doses of metformin.
At the end of the treatment period, TT401-treated patients in the 3 highest dose groups experienced statistically significant reductions in mean fasting plasma glucose relative to placebo. Statistically significant mean body weight reduction relative to baseline occurred in the three highest dose groups. A similar reduction in body weight was also observed in the obese non-diabetic cohort. TT401 demonstrated an acceptable safety and tolerability profile at all doses evaluated in diabetic and non-diabetic obese subjects. The most common adverse event noted in the study was decreased appetite. Some subjects in the highest three dose groups experienced mild nausea and vomiting, which are consistent with studies of other GLP-1 agonist drug candidates. The pharmacokinetic profile, assessed over the five week study, demonstrated a half-life consistent with once-weekly dosing.
On June 17, 2013, Lilly exercised its option to assume all development and commercialization rights to type 2 diabetes drug candidate TT401. In conjunction with this assumption of rights, Transition received a US$7 million milestone payment. Lilly and Transition have amended their agreement to address future development of TT401 and associated financial arrangements. Lilly has assumed all costs and will perform all future development and commercialization activities of TT401. In May, 2014, Transition announced the dosing of the first patient in a Phase 2 clinical study of TT401. The study is expected to enroll up to 375 type 2 diabetes subjects and will be performed by Transition’s development partner Lilly. The objectives of the study will be to evaluate the safety and effectiveness of TT401 compared to once-weekly exenatide extended release and placebo. In February 2015, Lilly informed Transition that 420 type 2 diabetic subjects had been enrolled in the current Phase 2 study, thereby completing the enrollment phase of the study.
Transition has made three separate installments to Lilly during the Phase 2 clinical study totaling US$14 million to Lilly. The first installment of US$6 million was paid in September 2014 when the study achieved 20% patient enrollment.The remaining two installments totaling US$8 million were paid during the three month period ended December 31, 2014 when the study achieved both the 50% and 70% patient enrollment milestones. There are no additional funding obligations related to this clinical study.
Expenditures for the TT401/402 Program
During the years ended June 30, 2015 and 2014, the Company incurred direct research and development costs for this program as follows:
| TT401/402 Program(1) | | Fiscal 2015 | | | Fiscal 2014 | |
| Pre-clinical studies | | $ | - | | | $ | 7,488 | |
| Clinical studies | | | - | | | | 87,379 | |
| Manufacturing | | | - | | | | (37,419 | ) |
| Other direct research | | | - | | | | 37,803 | |
| Development payments to Lilly | | | 15,491,600 | | | | - | |
| TOTAL | | $ | 15,491,600 | | | $ | 95,251 | |
Note(1)These costs are direct research and development costs only and do not include patent costs, investment tax credits, salaries and benefits, amortization of intangible assets or an allocation of Company overhead.
TT701 for Androgen Deficiency
On May 6, 2015, TTIL exclusively licensed worldwide rights to a novel small molecule drug candidate TT701 from Lilly. TT701 is a selective androgen receptor modulator that has been shown in a Phase 2 study to significantly increase lean body mass and a measurement of muscle strength in male subjects.
Clinical Development of TT701
Since acquiring the exclusive worldwide rights to TT701 the Company has incurred drug development manufacturing costs as they prepare to move the drug candidate into a Phase 2 clinical trial. TTIL has been actively working with potential partners to collaborate on the clinical development of TT701 and it is expected that a Phase 2 study of TT701 will commence before the end of calendar 2015.
Expenditures for the TT701 Program
During the years ended June 30, 2015 and 2014, the Company incurred direct research and development costs for this program as follows:
| TT701 Program(1) | | Fiscal 2015 | | | Fiscal 2014 | |
| Pre-clinical studies | | $ | - | | | $ | - | |
| Clinical studies | | | - | | | | - | |
| Manufacturing | | | 253,729 | | | | - | |
| Other direct research | | | 41,034 | | | | - | |
| TOTAL | | $ | 294,763 | | | $ | - | |
Note(1)These costs are direct research and development costs only and do not include patent costs, investment tax credits, salaries and benefits, amortization of intangible assets or an allocation of Company overhead.
The Next Steps
Transition’s goal for its programs is to achieve product approval and ultimately significant revenues or royalties. To achieve product approval, the Company and or its partners, must successfully complete clinical trials and achieve regulatory approval. The stages of development of the Company’s technologies are illustrated below:
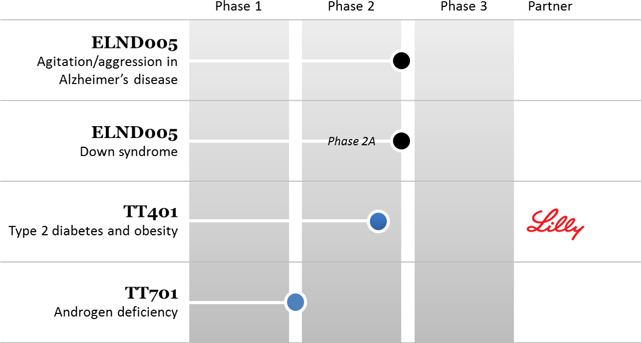
Overall Performance
During the year ended June 30, 2015, the Company recorded a net loss of $51,339,528 ($1.41 loss per common share) compared to a net loss of $21,782,255 ($0.72 loss per common share) for the year ended June 30, 2014.
During the fiscal year ended June 30, 2015, the Company reported an increase in net loss of $29,557,273 compared to the fiscal year ending June 30, 2014. The increasein net loss is due to the significant increase in research and development expenses resulting from the reacquisition of the rights to develop the ELND005 drug candidate, as well as the US$14 million milestone payments made to Lilly and increased general and administration expenses. The increase in net loss has been partially offset by the settlement of a pre-existing relationship recognized in connection with the re-acquisition of the ELND005 asset in February 2014, increased foreign exchange gains and the change in fair value of contingent consideration payable.
On February 18, 2015, the Company announced the closing of its underwritten public offering of an aggregate of 3,538,461 common shares at a price to the public of US$6.50 per share, including 461,538 common shares issued upon the exercise of the underwriters’ over-allotment option, raising gross proceeds of $28,561,400 (US$23.0 million). The Company incurred total share issuance costs of $2,492,010, resulting in net cash proceeds of $26,069,390.
At June 30, 2015, the Company has $40,510,758 in cash and a working capital of $32,026,606.
The Company’s current cash projection indicates that the current cash resources should enable the Company to execute its core business plan and meet its projected cash requirements beyond the next 12 months.
Selected Annual Information
The following table is a summary of selected financial information from the audited consolidated financial statements of the Company for each of the three most recently completed financial years.
| | | June 30,
2015
$ | | | June 30,
2014
$ | | | June 30,
2013
$ | |
| Revenue | | | — | | | | — | | | | 17,933,500 | |
| Net income (loss) (1) | | | (51,339,528 | ) | | | (21,782,255 | ) | | | 23,297 | |
| Basic and diluted net income (loss) per common share | | | (1.41 | ) | | | (0.72 | ) | | | - | |
| Total assets | | | 49,649,085 | | | | 68,907,236 | | | | 37,807,955 | |
| Total long-term liabilities (2) | | | 3,503,344 | | | | 3,849,718 | | | | 1,457,821 | |
| Cash dividends declared per share | | | — | | | | — | | | | — | |
Note(1)Net income (loss) before discontinued operations and extraordinary items was equivalent to the net income (loss) for such periods.
Note(2)Total long-term liabilities represents contingent consideration payable as set forth in the Company’s audited consolidated financial statements for the year ended June 30, 2015. For the years ended June 30, 2014 and 2013, total long-term liabilities also includes leasehold inducement.
ANNUAL RESULTS – YEAR ENDED JUNE 30, 2015 COMPARED TO YEAR ENDED JUNE 30, 2014
Results of Operations
Research and Development
Research and development expenses increased $31,842,318 or 183% from $17,367,385 for the fiscal year ended June 30, 2014 to $49,209,703 for the fiscal year ended June 30, 2015.
The increases in research and development expenses are primarily due to increases in development costs related to ELND005. The increases are also attributed to increases in development costs associated with diabetes drug candidate TT401 as during fiscal 2015 the Company paid Lilly an aggregate of US$14 million upon the achievement of all three patient enrollment milestones. The increase in research and development costs have been partially offset by decreases in clinical development costs associated with the costs related to the TT601 program.
The Company anticipates that research and development expenses will decrease significantly during fiscal 2016 as the Company discontinues the safety extension trial AG251 in agitation and aggression in Alzheimer’s disease and has no further funding obligations to Lilly for the ongoing Phase 2 clinical study of diabetes drug candidate TT401. The decrease will be offset by incurring costs relating to the development of TT701, a novel small molecule drug candidate licensed from Lilly.
General and Administrative
General and administrative expenses increased by $787,698 or 17% from $4,726,574 for the fiscal year ended June 30, 2014 to $5,514,272 for the fiscal year ended June 30, 2015.
The increases in general and administrative expenses are primarily due to increases in compensation and overhead costs relating to the Company’s premises in San Mateo, California.
The Company anticipates general and administrative expenses will decrease during fiscal 2016 as all activities based out of the San Mateo, California location will be transferred to head office, which will result in a reduction in general and administrative expenses.
Settlement of a Pre-existing Relationship
During the comparative year ended June 30, 2014, the Company recognized an expense of $3,096,186 as a settlement of a pre-existing relationship relating to the collaboration agreement with Elan. The Company did not recognize a similar expense during the year ended June 30, 2015.
Change in Fair Value of Contingent Consideration Payable
Contingent consideration is required to be measured as a financial liability at fair value and re-measured at each reporting date. Management revisited the assumptions used in the valuation of the contingent consideration payable and accordingly, the Company has recognized an increase in the fair value of contingent consideration payable of $65,787 during the fiscal year ended June 30, 2015.
During the comparative year ended June 30, 2014, the Company recognized a change in fair value of contingent consideration payable of $2,911,218.
ANNUAL RESULTS – YEAR ENDED JUNE 30, 2014 COMPARED TO YEAR ENDED JUNE 30, 2013
Results of Operations
Revenue
Revenue is nil in the year ended June 30, 2014 compared to $17,933,500 for the year ended June 30, 2013.
In fiscal 2013, the Company recognized $17,933,500 as revenue which is comprised of the milestone payment of $10,815,200 (US$11,000,000) received from Elan upon their commencement of the next ELND005 clinical trial and the milestone payment of $7,118,300 (US$7 million) received from Lilly upon exercising its option to assume all development and commercialization rights to type 2 diabetes drug candidate TT401.
Research and Development
Research and development expenses increased $8,504,513 or 96% from $8,862,872 for the fiscal year ended June 30, 2013 to $17,367,385 for the fiscal year ended June 30, 2014.
The increases in research and development expenses are primarily due to increases in clinical development costs related to the re-acquired rights to the drug candidate ELND005 as well as the costs associated with the pre-clinical research on TT601. The increase in research and development costs have been partially offset by decreases in clinical development costs associated with diabetes drug candidate TT401/TT402 as well as decreased amortization resulting from the write off of the TT301/302 technology.
The Company anticipates that research and development expenses will increase significantly during fiscal 2015 as the Company continues to fund the clinical development of the ongoing Phase 2 clinical trial of ELND005 in agitation and aggression in Alzheimer’s disease and will also pay US$14 million to Lilly in three separate installments to help fund the ongoing Phase 2 clinical study of diabetes drug candidate TT401.
General and Administrative
General and administrative expenses increased by $1,168,782 or 33% from $3,557,792 for the fiscal year ended June 30, 2013 to $4,726,574 for the fiscal year ended June 30, 2014.
The increases in general and administrative expenses are primarily due to increases in legal and professional fees as well as increased business and corporate development activities.
The Company anticipates that general and administrative expenses will remain relatively consistent during fiscal 2015.
Impairment of Intangible Assets
Impairment of intangible assets is nil for the year ended June 30, 2014 compared to $6,545,821 for the year ended June 30, 2013.
During fiscal 2013, the Company decided to no longer develop TT301 and TT302, the compounds acquired from NMX. Accordingly, the Company recognized an impairment loss of $6,545,821.
Settlement of a Pre-existing Relationship
During the year ended June 30, 2014, the Company recognized an expense of $3,096,186 as a settlement of a pre-existing relationship relating to the collaboration agreement with Elan. The Company did not recognize a settlement during the comparative year ended June 30, 2013.
Change in Fair Value of Contingent Consideration Payable
The contingent consideration is required to be measured as a financial liability at fair value and re-measured at each reporting date. On February 28, 2014, the Company became responsible for the development of ELND005 and accordingly has re-evaluated the development program timelines and adjusted the estimate relating to the timing of the milestone payments. Accordingly, the Company has recognized a change in fair value of contingent consideration payable of $2,911,218 during the year ended June 30, 2014. There was no change in fair value recognized during the comparative period ended June 30, 2013.
Summary of Quarterly Results
The following table is a summary of selected quarterly consolidated financial information of the Company for each of the eight most recently completed quarters ending at June 30, 2015.
| | | First | | | Second | | | Third | | | Fourth | | | | |
| | | Quarter | | | Quarter | | | Quarter | | | Quarter | | | Total | |
| 2015 | | | | | | | | | | | | | | | | | | | | |
| Revenue | | | - | | | | - | | | | - | | | | - | | | | - | |
| Net income (loss)(1) | | $ | (15,695,324 | ) | | $ | (16,910,139 | ) | | $ | (4,748,096 | ) | | $ | (13,985,969 | ) | | $ | (51,339,528 | ) |
| Basic and diluted net income (loss) per common share | | $ | (0.45 | ) | | $ | (0.48 | ) | | $ | (0.13 | ) | | $ | (0.38 | ) | | $ | (1.41 | ) |
| 2014 | | | | | | | | | | | | | | | | | | | | |
| Revenue | | | - | | | | - | | | | - | | | | - | | | | - | |
| Net income (loss) (1) | | $ | (2,331,186 | ) | | $ | (1,253,772 | ) | | $ | (5,067,292 | ) | | $ | (13,130,005 | ) | | $ | (21,782,255 | ) |
| Basic and diluted net income (loss) per common share | | $ | (0.08 | ) | | $ | (0.04 | ) | | $ | (0.17 | ) | | $ | (0.43 | ) | | $ | (0.72 | ) |
Note(1) Net income (loss) before discontinued operations was equivalent to the net income (loss) for such periods.
The fluctuations of Transition’s quarterly results are primarily due to milestone payments made to Lilly to help fund TT401 Phase 2 clinical development and changes in: activity levels of the clinical trials being performed by the Company and foreign exchange gains and losses.
Critical Accounting Estimates and Judgements
The preparation of consolidated financial statements in accordance with IFRS requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results can differ from those estimates. We have identified the following areas which we believe require management’s most subjective estimates and judgments, often requiring the need to make estimates about the effects of matters that are inherently uncertain and may change in subsequent periods.
(a) Estimates
Valuation and Amortization of Intangible Assets
The Company’s intangible assets are comprised of purchased or licensed pharmaceutical compounds, technology and patents. The costs of the Company’s intangible assets are amortized over the estimated useful life of up to 20 years. Factors considered in estimating the useful life of the intangible asset include the expected use of the asset by the Company, legal, regulatory and contractual provisions that may limit the useful life, the effects of competition and other economic factors, and the level of expenditures required to obtain the expected future cash flows from the intangible asset. The Company re-evaluates the useful life when there has been a change in these factors. The Company assesses its intangible assets for recoverability whenever indicators of impairment exist. As ELND005 did not meet its primary efficacy endpoint in the Phase 2/3 clinical study in agitation and aggression in Alzheimer’s disease, management performed an impairment test and noted there is no impairment of the ELND005 asset as at June 30, 2015. The Company is performing a thorough review of the data from the completed study in agitation and aggression. An external clinical advisory board is working with the Company to evaluate the data and consider potential future clinical development paths for ELND005.
When the carrying value of an asset is greater than its recoverable amount, which is the higher of its value in use or fair value less costs to sell, an impairment loss is recognized.
Valuation of Contingent Consideration Payable
The contingent consideration is measured at fair value based on level 3 inputs. The contingent consideration is not based on observable inputs and is measured using a discounted cash flow analysis of expected payments in future periods. The significant estimates used in the fair value calculations are as follows:
| (a) | Management has estimated the timing of the milestone payments based on current expectations and plans for the development of ELND005. The milestone payments are assigned a probability based on industry statistics for the successful development of pharmaceutical products including regulatory approval and achievement of revenue targets. An increase of 10% applied to the probability assumptions, with all other variables held constant, will increase the contingent consideration payable by $1,428,951. Conversely a decrease of 10% applied to the probability assumptions, with all other variables held constant, would reduce the contingent consideration payable by $1,858,858; |
| (b) | The probability adjusted cash flows are discounted at a rate of 20% which is management’s best estimate of the Company’s cost of capital. An increase of 5% to the discount rate would decrease the contingent consideration payable by $1,080,299. Conversely, a decrease of 5% to the discount rate would increase the contingent consideration payable by $1,538,400. |
Management revisited the assumptions used in the valuation of the contingent consideration payable and accordingly, the Company has recognized a change in fair value of contingent consideration payable of $65,787 during the fiscal year ended June 30, 2015.
Share Based Payments and Warrants
When the Company issues stock options and warrants, an estimate of fair value is derived for the equity instrument using the Black-Scholes option pricing model. The application of this option pricing model requires management to make assumptions regarding several variables, including the period for which the instrument will be outstanding, the price volatility of the Company’s stock over a relevant timeframe, the determination of a relevant risk free interest rate and an assumption regarding the Company’s dividend policy in the future. If other assumptions are used, the value derived for the equity instruments could be significantly impacted.
Settlement of a Pre-Existing Relationship
The Company has determined that the transactions entered into with Perrigo on February 28, 2014 have resulted in the re-acquisition of the rights to the development and commercialization of ELND005 previously licensed to Elan which in accordance with IFRS must be accounted for as a settlement of a pre-existing relationship (the collaboration agreement between Waratah and Elan). Accordingly, the company expensed $3,096,186 in fiscal 2014 as the cost related to the settlement of the pre-existing relationship.
Accounting Changes
The following accounting policies have been adopted effective July 1, 2014:
IAS 36 – Impairment of Assets
IAS 36 has been amended to include limited scope amendments to the impairment disclosures. The amendments are effective for annual periods beginning on or after January 1, 2014. The adoption of IAS 36 did not significantly impact the Company’s consolidated financial statements;
IFRS 2 – Share Based Payments
IFRS 2 has been amended to clarify the definition of vesting conditions. The amendments are effective for annual periods beginning on or after July 1, 2014. The adoption of IFRS 2 did not significantly impact the Company’s consolidated financial statements.
IFRS Issued But Not Yet Adopted
IFRS 15 – Revenue from Contracts with Customers
IFRS 15 specifies how and when to recognize revenue as well as requiring entities to provide users of financial statements with some informative, relevant disclosures. The standard supersedes IAS 18, Revenue, IAS 11, Construction Contracts, and a number of revenue-related interpretations. Application of the standard is mandatory for all IFRS reporters and it applies to nearly all contracts with customers: the main exceptions are leases, financial instruments and insurance contracts. Currently IFRS 15 must be applied in an entity’s first annual IFRS financial statements for periods beginning on or after January 1, 2017 however the IASB has proposed to defer the date of adoption to periods beginning on or after January 1, 2018, with early adoption permitted. Management is evaluating the standard and has not yet determined the impact on its consolidated financial statements.
Outstanding Share Data
Authorized
The authorized share capital of the Company consists of an unlimited number of common shares.
Issued and Outstanding
The following details the issued and outstanding equity securities of the Company:
Common Shares
As at September 14, the Company has 38,878,879 common shares outstanding.
Stock Options
As at September 14, 2015 the Company has 2,408,381 stock options outstanding with exercise prices ranging from $2.09 to $10.19 and various expiry dates extending to June 14, 2025. At September 14, 2015, on an if-converted basis, these stock options would result in the issuance of 2,408,381 common shares in the capital of the Company at an aggregate exercise price of $11,430,633.
Warrants
As at September 14, 2015, the Company has a total of 1,949,250 warrants outstanding with a purchase price of US$7.10.
Each warrant entitles the holder, within two years of the June 23, 2014 issuance date, to purchase one additional common share in the capital of the Company.
LIQUIDITY AND CAPITAL RESOURCES
Overview
The Company commenced operations in July 1998, and has devoted its resources primarily to fund its research and development programs. All revenue to date has been generated from milestone payments and licensing fees. The Company has incurred a cumulative deficit to June 30, 2015 of $222,454,699. Losses are expected to continue for the next several years as the Company invests in research and development, preclinical studies, clinical trials, manufacturing and regulatory compliance.
Since inception, the Company has been financed primarily from public and private sales of equity, the exercise of warrants and stock options, interest earned on cash deposits and short term investments and revenues and reimbursements from partners.
The Company’s cash was $40,510,758 at June 30, 2015 as compared to cash and short term investments of $60,271,566 at June 30, 2014, resulting in a decrease of $19,760,808. The Company’s working capital position at June 30, 2015 decreased $22,751,265 from $54,777,871 at June 30, 2014 to $32,026,606, at June 30, 2015.
The decrease in the Company’s cash and short term investments as well as the decrease in working capital are primarily due to the expenditures incurred during the fiscal year ended June 30, 2015 which included three milestone payments totaling US$14 million paid to Lilly upon the achievement of all three patient enrollment milestones for the TT401 Phase 2 diabetes study. The decrease is offset by the February 18, 2015 public offering of 3,538,461 common shares which resulted in net proceeds of $26,069,390.
The Company’s current cash projection indicates that the current cash resources should enable the Company to execute its core business plan and meet its projected cash requirements for the next 12 months.
The success of the Company is dependent on its ability to bring its products to market, obtain the necessary regulatory approvals and achieve future profitable operations. The continuation of the research and development activities and the commercialization of its products are dependent on the Company’s ability to successfully complete these activities and to obtain adequate financing through a combination of financing activities, operations, and partnerships. It is not possible to predict either the outcome of future research and development programs or the Company’s ability to fund these programs going forward.
On July 19, 2013, the Company’s shelf registration statement filed with the United States Securities and Exchange Commission (“SEC”) on Form F-3 became effective. The shelf prospectus provides for the potential offering in the United States of up to an aggregate amount of US$50 million of Transition’s common shares, warrants, or a combination thereof, from time to time in one or more offerings until July 19, 2016.
On January 5, 2015, the Company filed with the SEC a prospectus supplemental to the shelf prospectus and a sales agreement with Cowen and Company, LLC or Cowen, relating to the sale of the Company’s common shares. In accordance with the terms of the sales agreement, the Company may offer and sell from time to time common shares having an aggregate offering price of up to US $25 million with Cowen acting as sales agent. After the closing of the February, 2015 US$23 million public offering, the Company can raise an additional US$27 million through the issuance of common shares, warrants or a combination thereof, from time to time in in one of more offerings until July 19, 2016.
Utilization of the US shelf prospectus is dependent upon meeting certain market capitalization thresholds at the time of financing.
Financial Instruments
Financial instruments of the Company consist mainly of cash, short term investments, other receivables, accounts payable and accrued liabilities, and contingent consideration payable. Management’s primary investment objective is to maintain safety of principal and provide adequate liquidity to meet all current payment obligations and future planned expenditures.
The Company is exposed to market risks related to volatility in interest rates for the Company’s investment portfolio and foreign currency exchange rates related to cash and purchases of supplies and services made in U.S. dollars.
The Company is exposed to interest rate risk to the extent that the cash is held in deposit accounts which earn interest at variable rates. The Company’s maximum exposure to interest rate risk is based on the effective interest rate of the current carrying value of these assets. The Company does not speculate on interest rates and holds all deposits until their date of maturity.
Off-Balance Sheet Arrangements
The Company’s off-balance sheet arrangements include operating leases for the Company’s premises and certain office equipment. The minimum payments under these leases are included in the contractual obligation table below.
Contractual Obligations
Minimum payments under our contractual obligations are as follows:
| | | Less than 1 Year | | | 1- 3 Years | | | 4 – 5 Years | | | After 5 Years | | | Total | |
| Operating leases | | $ | 238,179 | | | $ | 343,165 | | | $ | 307,453 | | | $ | - | | | $ | 888,797 | |
| Clinical and toxicity study agreements | | | 2,904,125 | | | | - | | | | - | | | | - | | | | 2,904,125 | |
| Manufacturing agreements | | | 214,857 | | | | - | | | | - | | | | - | | | | 214,857 | |
| Contingent Consideration Payable | | | - | | | | 2,847,759 | | | | - | | | | 58,028,760 | | | | 60,876,519 | |
| Other | | | 327,218 | | | | - | | | | - | | | | - | | | | 327,218 | |
| TOTAL | | $ | 3,684,379 | | | $ | 3,190,924 | | | $ | 307,453 | | | $ | 58,028,760 | | | $ | 65,211,516 | |
Contractual obligations denominated in US dollars have been translated to Canadian dollars using the exchange rate at June 30, 2015.
| Item 6. | Directors, Senior Management and Employees |
A. Directors and Senior Managers. The following table sets forth the name and province or state and country of residence of each director and executive officer of the Corporation as well as their position(s) and offices held with the Corporation and their principal occupations during the five preceding years for each as of September 14, 2015.
Name and Province/
State and
Country of
Residence | | Position(s) Held
with the
Corporation | | Principal Occupations within the
Five Proceeding Years | | Director
Since (4) | | Age |
| | | | | | | | | |
Dr. Tony F. Cruz
Ontario, Canada | | Chief Executive Officer, Director and Chairman of the Board of Directors | | Chief Executive Officer of the Corporation. | | January 1999; | | 62 |
| | | | | | | | | |
Carl Damiani
Ontario, Canada | | President and Chief Operating Office | | President and Chief Operating Officer of the Corporation since June 16, 2015; prior thereto, Chief Operating Officer of the Corporation since July 11, 2014; prior thereto, VP of Business Development of the Corporation since December 2007, prior thereto, Director of Business Development of the Corporation since October 2003. | | N/A | | 43 |
Name and Province/
State and
Country of
Residence | | Position(s) Held
with the
Corporation | | Principal Occupations within the
Five Proceeding Years | | Director
Since (4) | | Age |
Nicole Rusaw
Ontario, Canada | | Chief Financial Officer | | Chief Financial Officer of the Corporation since December 2011; prior thereto, VP, Finance of the Corporation since July 2008, prior thereto, Director of Finance of the Corporation since June 2005. | | N/A | | 42 |
| | | | | | | | | |
Dr. Aleksandra Pastrak
Ontario, Canada | | VP of Clinical Development and Medical Officer | | VP of Clinical Development and Medical Officer since September 2011, prior thereto, VP Research and Medical Officer of the Corporation since July 2007, prior thereto, VP, Research of the Corporation since May 2005. | | N/A | | 49 |
| | | | | | | | | |
Bruce Connop
Ontario, Canada | | VP of Non-Clinical and Pharmaceutical Development | | VP of Non-Clinical and Pharmaceutical Development of the Corporation since July 2012, prior thereto, Senior Director Non-Clinical Drug Development since December 2006. | | N/A | | 46 |
| | | | | | | | | |
Michael Ashton(1)(2)(3)
Connecticut, USA | | Lead Director | | Director of Hikma Pharmaceuticals PLC since 2005 and Independent consultant to the pharmaceutical industry since March 2006; prior thereto, former Chief Executive Officer of Puricore plc; prior thereto, Chief Executive Officer of SkyePharma PLC, a U.K. based drug delivery company. | | December 2002; | | 69 |
| | | | | | | | | |
Paul Baeher(1)(2)(3)
Quebec, Canada | | Director | | President, Chief Executive Officer and Chairman of IBEX Technologies Inc., a publicly traded biotechnology company. | | December 2002; | | 72 |
Christopher Henley(1)(2)(3)
Ontario, Canada | | Director | | President of Henley Capital Corporation, an exempt market dealer and portfolio manager. | | October 2000; | | 55 |
| | | | | | | | | |
Dr. Gary W. Pace(1)(2)(3)
California, USA | | Director | | Currently a Director of ResMed Inc. and Pacira Pharmaceuticals, Inc.; prior thereto, Co-founder, Chairman and Chief Executive Officer of QRxPharma from 2002 to 2013. | | January 2002; | | 67 |
Notes:
| (1) | Member of the Audit Committee. |
| (2) | Member of the Compensation Committee. |
| (3) | Member of the Corporate Governance Committee. |
| (4) | The term of each current director’s appointment will expire at the Corporation’s Annual Meeting of shareholders which is scheduled for December 10, 2015. It is anticipated that each current director will be nominated by management for re-appointment at the Corporation’s Annual Meeting. |
Family Relationships
There are no family relationships among our directors and executive officers.
Arrangement or understanding with major shareholders, customers, suppliers or others
There are no arrangements or understandings with major shareholders, customers, suppliers or others, pursuant to which any person referred to above was selected as a director or member of senior management.
B. Executive Compensation
Compensation of Executives
Summary Compensation Table
The following table provides a summary of compensation earned during the most recently completed fiscal year by the Corporation’s Chief Executive Officer, Chief Financial Officer, and for the next three most highly compensated executive officers of the Corporation other than the Chief Executive Officer and the Chief Financial Officer (the “Named Executive Officers” as the term is used in National Instrument Form 51-102 – Statement of Executive Compensation), whose total salary and bonus exceeded $150,000.
| | | | | | | | | | | | | | | Non-equity incentive
plan compensation
($) | | | | | | | | | | |
Name and principal position | | Year | | | Salary
($) | | | Share-
based
awards
($) | | | Option-
based
awards
($) | | | Annual
incentive
plans | | | Long-
term
incentive
plans | | | Pension
value
($) | | | All other
compensation
($) | | | Total
compensation | |
Tony Cruz,
Chief Executive Officer | | | 2015 | | | | 413,860 | | | | — | | | | 426,755 | | | | 124,158 | | | | — | | | | — | | | | 1,350 | (3) | | | 966,123 | |
| | | | 2014 | | | | 401,806 | | | | — | | | | 664,500 | | | | 120,542 | | | | — | | | | — | | | | 1,174 | (3) | | | 1,188,022 | |
| | | | 2013 | | | | 390,103 | | | | — | | | | 198,000 | | | | 117,031 | | | | — | | | | — | | | | 1,174 | (3) | | | 706,308 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Nicole Rusaw
Chief Financial Officer | | | 2015 | | | | 204,000 | | | | — | | | | 243,860 | | | | 40,800 | | | | — | | | | — | | | | — | | | | 488,660 | |
| | | | 2014 | | | | 190,962 | | | | — | | | | 221,500 | | | | 38,192 | | | | — | | | | — | | | | 1,174 | (3) | | | 451,828 | |
| | | | 2013 | | | | 185,400 | | | | — | | | | 92,400 | | | | 37,080 | | | | — | | | | — | | | | 1,174 | (3) | | | 316,054 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Sawsan Abushakra
D5 Program Lead and Chief Medical Officer(1) | | | 2015 | | | | 481,258 | | | | — | | | | 274,343 | | | | 192,503 | | | | — | | | | — | | | | — | | | | 948,104 | |
| | | | 2014 | | | | 36,456 | | | | — | | | | 398,691 | | | | — | | | | — | | | | — | | | | — | | | | 435,147 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Earvin Liang
Senior Director Clinical Pharmacology(2) | | | 2015 | | | | 304,014 | | | | — | | | | 91,448 | | | | 91,204 | | | | — | | | | — | | | | — | | | | 486,666 | |
| | | | 2014 | | | | 23,029 | | | | — | | | | 66,449 | | | | — | | | | — | | | | — | | | | — | | | | 89,478 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Aleksandra Pastrak
Vice President Clinical Development & Medical Officer | | | 2015 | | | | 266,053 | | | | — | | | | 213,378 | | | | 53,211 | | | | — | | | | — | | | | 1,350 | (3) | | | 533,992 | |
| | | | 2014 | | | | 258,304 | | | | — | | | | 221,500 | | | | 51,661 | | | | — | | | | — | | | | 1,174 | (3) | | | 532,639 | |
| | | | 2013 | | | | 250,781 | | | | — | | | | 105,600 | | | | 50,156 | | | | — | | | | — | | | | 1,174 | (3) | | | 407,711 | |
Notes:
| (1) | Ms. Abushakra joined the Corporation on June 1, 2014 and was appointed ELND005 Program Lead and Chief Medical Officer. Ms. Abushakra’s employment with the Corporation was terminated on July 24, 2015 and as of that date, Ms. Abushakra owned 30,898 common shares of the Corporation. |
| (2) | Mr. Liang joined the Corporation on June 1, 2014 as the Senior Director Clinical Pharmacology. Mr. Liang’s employment with the Corporation was terminated on July 24, 2015 and as of that date, Mr. Liang owned 23,600 common shares of the Corporation. |
| (3) | Dr. Cruz, Dr. Pastrak and Ms. Rusaw receive subsidized parking from the Corporation and the subsidy represents the full amount of all other compensation disclosed. |
Outstanding Option-based Awards
The following table sets forth information with respect to stock option grants exercisable into Common Shares made to the Named Executive Officers that were outstanding at June 30, 2015:
| | | Option-based Awards | |
| Name | | Number of
securities
underlying
unexercised
Options
(#) | | | Option
exercise
price ($) | | | Option expiration
date | | Value of
unexercised
in-the-money
options ($)(1) | |
| Tony Cruz | | | 150,000 | | | | 3.22 | | | May 26, 2021 | | | — | |
| | | | 250,000 | | | | 2.10 | | | June 1, 2022 | | | 112,500 | |
| | | | 75,000 | | | | 3.66 | | | June 19, 2023 | | | — | |
| | | | 150,000 | | | | 6.00 | | | June 12, 2024 | | | — | |
| | | | 70,000 | | | | 8.73 | | | March 31, 2025 | | | — | |
| | | | | | | | | | | | | | | |
| Nicole Rusaw | | | 16,000 | | | | 3.22 | | | May 26, 2021 | | | — | |
| | | | 50,000 | | | | 2.10 | | | June 1, 2022 | | | 22,500 | |
| | | | 35,000 | | | | 3.66 | | | June 19, 2023 | | | — | |
| | | | 50,000 | | | | 6.00 | | | June 12, 2024 | | | — | |
| | | | 40,000 | | | | 8.73 | | | March 31, 2025 | | | — | |
| | | | | | | | | | | | | | | |
| Susan Abushakra | | | 90,000 | | | | 6.00 | | | June 12, 2024 | | | — | |
| | | | 45,000 | | | | 8.73 | | | March 31, 2025 | | | — | |
| | | | | | | | | | | | | | | |
| Earvin Liang | | | 15,000 | | | | 6.00 | | | June 12, 2024 | | | — | |
| | | | 15,000 | | | | 8.73 | | | March 31, 2025 | | | — | |
| | | | | | | | | | | | | | | |
| Aleksandra Pastrak | | | 85,000 | | | | 3.22 | | | May 26, 2021 | | | — | |
| | | | 100,000 | | | | 2.10 | | | June 1, 2022 | | | 45,500 | |
| | | | 40,000 | | | | 3.66 | | | June 19, 2023 | | | — | |
| | | | 50,000 | | | | 6.00 | | | June 12, 2024 | | | — | |
| | | | 35,000 | | | | 8.73 | | | March 31, 2025 | | | — | |
Notes:
| (1) | Calculated based on the difference between the closing price of the Common Shares on the TSX on June 30, 2015 of $2.55 and the exercise price of the options. |
Incentive Plan Awards – Value Vested or Earned during the Year
The following table sets forth for option-based awards, the aggregate dollar value that would have been realized if the options under the option-based award had been exercised on the vesting date.
| Name | | Option-based awards – Value
vested during the year
($)(1) | |
| Tony Cruz | | | 37,500 | |
| Nicole Rusaw | | | 12,750 | |
| Sawsan Abushakra | | | — | |
| Earvin Liang | | | — | |
| Aleksandra Pastrak | | | 15,000 | |
Notes:
| (1) | Calculated based on the difference between the closing price of the Common Shares on the TSX on June 30, 2015 of $2.55 and the exercise price of the options which were vested as of June 30, 2015. |
Compensation of Directors
The following table details the total compensation earned by each non-employee director during the year ended June 30, 2015:
| Name | | Fees
earned
($) | | | Option-
based
awards
($) | | | All other
compensation
($) | | | Total
($) | |
| Michael Ashton | | | 53,000 | | | | 109,482 | | | | — | | | | 162,482 | |
| Paul Baehr | | | 51,250 | | | | 109,482 | | | | — | | | | 160,732 | |
| Christopher Henley | | | 52,250 | | | | 109,482 | | | | — | | | | 161,732 | |
| Gary W. Pace | | | 43,000 | | | | 109,482 | | | | — | | | | 152,482 | |
Dr. Tony Cruz, Chief Executive Officer and a director of the Corporation, does not receive any compensation as a director of the Corporation, but receives compensation as an executive officer of the Corporation as detailed under the heading “Chief Executive Officer’s Compensation”. The remaining directors are not employees of the Corporation. Non-employee directors have been remunerated in the following manner.
Standard Arrangements
The Corporation has standard arrangements for its non-employee directors, which include the following:
| · | Board member annual retainer in the amount of $25,000 and an annual grant of stock options; |
| · | Committee Chair annual retainers – the Audit Committee Chair is paid $10,000 annually and the Corporate Governance and Nominating Committee and Compensation Committee Chairs are each paid $6,000 annually; |
| · | Board and Committee meeting fees are paid in the amount of $1,500 for each meeting attended and $750 for each conference call attended; |
| · | Travel fees of $1,000 for each meeting are paid to all non-employee directors who traveled from outside the Greater Toronto area to attend in person; and |
| · | All reasonable out of pocket expenses incurred by the non-employee directors in respect of their duties as directors are reimbursed by the Corporation. |
There are no contracts or arrangements with non-employee directors providing for benefits upon termination of service.
Directors – Outstanding Option-based Awards
The following table sets forth information with respect to stock option grants exercisable into Common Shares made to the directors that were outstanding at June 30, 2015.
| | | Option-based Awards | |
Name | | Number of
securities
underlying
unexercised
options (#) | | | Option
exercise price
($) | | | Option expiration
date | | Value of unexercised
in-the-money
options ($) (1) | |
| Michael Ashton | | | 15,000 | | | | 3.00 | | | June 30, 2021 | | | — | |
| | | | 15,000 | | | | 2.09 | | | June 30, 2022 | | | 6,900 | |
| | | | 15,000 | | | | 3.55 | | | June 30, 2023 | | | — | |
| | | | 15,000 | | | | 7.47 | | | June 30, 2024 | | | — | |
| | | | 15,000 | | | | 10.19 | | | June 14, 2025 | | | — | |
| | | | | | | | | | | | | | | |
| Paul Baehr | | | 15,000 | | | | 3.00 | | | June 30, 2021 | | | — | |
| | | | 15,000 | | | | 2.09 | | | June 30, 2022 | | | 6,900 | |
| | | | 15,000 | | | | 3.55 | | | June 30, 2023 | | | — | |
| | | | 15,000 | | | | 7.47 | | | June 30, 2024 | | | — | |
| | | | 15,000 | | | | 10.19 | | | June 14, 2025 | | | — | |
| | | | | | | | | | | | | | | |
| Christopher Henley | | | 15,000 | | | | 3.00 | | | June 30, 2021 | | | — | |
| | | | 15,000 | | | | 2.09 | | | June 30, 2022 | | | 6,900 | |
| | | | 15,000 | | | | 3.55 | | | June 30, 2023 | | | — | |
| | | | 15,000 | | | | 7.47 | | | June 30, 2024 | | | — | |
| | | | 15,000 | | | | 10.19 | | | June 14, 2025 | | | — | |
| | | | | | | | | | | | | | | |
| Gary W. Pace | | | 15,000 | | | | 3.00 | | | June 30, 2021 | | | — | |
| | | | 15,000 | | | | 2.09 | | | June 30, 2022 | | | 6,900 | |
| | | | 15,000 | | | | 3.55 | | | June 30, 2023 | | | — | |
| | | | 15,000 | | | | 7.47 | | | June 30, 2024 | | | — | |
| | | | 15,000 | | | | 10.19 | | | June 14, 2025 | | | — | |
Notes:
| (1) | Calculated based on the difference between the closing price of the Common Shares on the TSX on June 30, 2015 of $2.55 and the exercise price of the options which were vested as of June 30, 2015. |
Directors – Incentive Plan Awards – Value Vested or Earned during the Year
The following table sets forth (i) for option-based awards, the aggregate dollar value that would have been realized if the options under the option-based award had been exercised on the vesting.
| Name | | Option-based awards – Value
vested during the year
($) (1) | |
| Michael Ashton | | | — | |
| Paul Baehr | | | — | |
| Christopher Henley | | | — | |
| Gary W. Pace | | | — | |
Notes:
| (1) | All options that vested during the year were out of the money based on the closing price of Common Shares on the TSX on June 30, 2015 of $2.55. |
Audit Committee
The Corporation has a separately-designated standing Audit Committee established in accordance with section 3(a)(58) of the Exchange Act. The members of the Audit Committee are: Mr. Christopher Henley (chair), Mr. Paul Baehr, Mr. Michael Ashton and Dr. Gary Pace, each of whom is independent under the TSX and NASDAQ listing standards and financially literate.
Audit Committee Charter
The charter of the Corporation’s audit committee is attached as Appendix A to our Annual Information Form available at www.sedar.com.
Relevant Education and Experience
Mr. Christopher Henley (Chair):
Mr. Henley has a B.A. from Memorial University and an MBA from Dalhousie University. He has completed the Institute of Corporate Directors Education Program at the Rotman School of Management, University of Toronto and holds the professional designation ICD.D. He is currently founder and President of Henley Capital Corporation, an exempt market dealer and portfolio manager.
Mr. Henley has been an investment banker for over 25 years and previously was the head of investment banking in Toronto at what was then Canada’s largest independent employee-owned investment dealer where he also ran the firm’s High Technology and Communications practice. Mr. Henley is a former board member of the Exempt Market Dealers Association of Canada, a former director and Chair of the Toronto Port Authority, a former member of the Board and Audit Committee of Ontario Transportation Capital Corporation, a Government of Ontario Crown Corporation that, through a public partnership, developed Highway 407 in Toronto, Ontario, the first all-electronic toll highway in the world. Mr. Henley is also a founding member of the National Angel Organization in Canada and the Ministers’ Technology Advisory Group (“MTAG”) for the Province of Ontario. He is also a former Chair of the MTAG Task Force on Access to Capital, a former member of the Advisory Board, Faculty of Business Administration, Memorial University of Newfoundland and is an active member of the Institute of Corporate Directors. Mr. Henley is currently Chair of West Park Healthcare Centre, a member of the Governance Committee of the Ontario Cancer Research Ethics Board, and is actively involved in many philanthropic organizations including acting as Chair of the Board of the McMichael Canadian Art Foundation.
Mr. Paul Baehr:
Mr. Baehr has a B.A. from the University of British Columbia. He is currently President, Chief Executive Officer and Chairman of IBEX Technologies Inc., a biotechnology company listed on the TSX under the symbol “IBT”. Previously, Mr. Baehr was an Executive Vice President at Sterling Winthrop and prior thereto a Senior Vice President at CIBA-GEIGY Pharmaceuticals, both of which are large pharmaceutical companies. In addition, Mr. Baehr currently sits on the board of directors of IBEX Technologies Inc., a public company.
Mr. Michael Ashton:
In June, 2015, Mr. Ashton retired as the Chief Executive Officer of Puricore PLC. Mr. Ashton has been an independent consultant to the pharmaceutical industry since March 2006. Prior thereto, he was the Chief Executive Officer of SkyePharma PLC. He has over 30 years of experience in the pharmaceutical industry having worked for Merck, Pfizer, Purepac and Faulding, where Mr. Ashton was Chairman, President and CEO. Mr. Ashton is a member of the Board of Directors of Hikma Pharmaceuticals PLC. Mr. Ashton has a Bachelor of Pharmacy degree from Sydney University and his MBA degree from Rutgers University, New Jersey.
Dr. Gary Pace:
Dr. Pace has more than 40 years of experience in the development and commercialization of advanced technologies, spanning biotechnology, pharmaceuticals, medical devices and food industries. He is a serial entrepreneur and has held senior positions in small and large-scale life sciences ventures and companies in Australia, USA, Europe and Canada. Dr. Pace has long-term board level experience with multi-billion and small cap companies with an in-depth knowledge of all aspects of technical operations, compensation, and corporate governance. Dr. Pace has contributed to the development of the biotechnology industry through honorary university appointments and industry and government committees. He is an elected Fellow of the Australian Academy of Technological Sciences and Engineering, author and co-author of several research papers, reviews and patents. In 2003, Dr. Pace was awarded a Centenary Medal by the Australian Government “for service to Australian society in research and development” as well as “Corporate Governance 2011 Director of the Year” by the San Diego Directors Forum. Dr Pace currently sits on boards of directors of public companies Pacira Pharmaceutical Inc. and ResMed Inc. and several private companies. Dr. Pace has a B.Sc. from the University of New South Wales and a Ph.D. from the Massachusetts Institute of Technology.
Pre-Approval Policies and Procedures
The Corporation’s audit committee is responsible for the oversight of the work of the external auditor. As part of this responsibility, the audit committee is required to pre-approve all audit, audit related, tax services and non-audit services performed by the external auditor in order to assure that they do not impair the external auditor’s independence from the Corporation. Accordingly, the audit committee has adopted a pre-approval policy, which sets forth the procedures and the conditions pursuant to which services proposed to be performed by the external auditor may be pre-approved.
Under the pre-approval policy, the Corporation’s audit committee annually reviews and pre-approves specific audit, audit-related and tax services that may be provided by the external auditor without obtaining specific pre-approval from the audit committee, as well as maximum fees for such services. All services that are not pre-approved or exceed the pre-approved maximum fees require specific pre-approval by the audit committee before the service can be performed by the external auditor. The pre-approval policy also includes a list of prohibited services.
Compensation Committee
Compensation of the executive officers is determined by the Board of Directors upon recommendations made by the Compensation Committee. The following individuals comprise the Compensation Committee of the Board of Directors: Mr. Michael Ashton (Chair); Mr. Paul Baehr; Mr. Christopher Henley and Dr. Gary Pace.
The Compensation Committee of the Board of Directors (the “Compensation Committee”) exercises general responsibility regarding overall compensation of employees and executive officers of the Corporation. It annually reviews and recommends to the Board (i) executive compensation policies, practices and overall compensation philosophy, (ii) total compensation packages for all employees who receive aggregate annual compensation in excess of $150,000, (iii) bonuses and grants of options under the Corporation’s Stock Option Plan, and (iv) major changes in benefit plans. Final approval of all compensation items rests with the full Board.
As of June 30, 2015, the Corporation had 18 full-time employees.
Please see Items 6.B and 7.A.
| Item 7A. | Major Shareholders and Related Party Transactions |
Securities Ownership of Certain
Beneficial Owners and Management
The following table sets forth information with respect to the beneficial ownership of our Common Shares, by (1) each of the Corporation’s shareholders who is a beneficial owner of more than 5% of the Corporation’s Common Shares, (2) each of the Corporation’s directors and executive officers, individually, as of August 31, 2015.
| Name of Beneficial Owner | | Amount and Nature of
Beneficial Ownership | | | Percent of
Class | |
| Jack Schuler (1) | | | 7,428,678 | | | | 18.8 | % |
| Larry N Feinberg (2) | | | 5,724,399 | | | | 14.4 | % |
| Renate Schuler (1) | | | 4,642,668 | | | | 11.8 | % |
| Schuler Family Foundation (1) | | | 4,605,668 | | | | 11.7 | % |
| Fidelity Research and Management Company | | | 4,103,435 | | | | 10.6 | % |
| Elan Corporation | | | 2,255,640 | | | | 5.8 | % |
| Louis Alexopoulos | | | 36,003 | | | | * | |
| Michael Ashton (3) | | | 86,390 | | | | * | |
| Tony Cruz (4) | | | 1,450,949 | | | | 3.7 | % |
| Paul Baehr (3) | | | 88,512 | | | | * | |
| Christopher Henley (3) | | | 122,754 | | | | * | |
| Gary Pace (3) | | | 133,393 | | | | * | |
| Aleksandra Pastrak (5) | | | 285,166 | | | | * | |
| Nicole Rusaw (6) | | | 142,244 | | | | * | |
| Carl Damiani (7) | | | 324,976 | | | | * | |
| Bruce Connop (8) | | | 111,180 | | | | * | |
Notes:
| (1) | Includes the 3,986,117 shares beneficially owned by the Schuler Family Foundation over which Mr. Schuler and Mrs. Renate Schuler share dispositive power. The number of shares beneficially owned includes 619,551 warrants to purchase common shares which are exercisable within 60 days of August 31, 2015 and which are beneficially owned by the Schuler Family Foundation. The warrants provide that they are not exercisable to the extent that the exercise would cause the holder to own or have control over voting securities which constitute 20% or more of the total issued and outstanding voting securities of the Company, unless both the shareholders of the Company and the TSX have approved such exercise. Mr. Schuler and Mrs. Schuler disclaim any beneficial interest in the shares owned by the Schuler Family Foundation. Mr. Schuler disclaims any beneficial interest in the shares owned by Mrs. Schuler. Mrs. Schuler disclaims any beneficial interest in the shares owned by Mr. Schuler. |
| (2) | The shares reported to be beneficially owned by Mr. Larry Feinberg includes the 3,378,205 shares beneficially owned by Oracle Associates, LLC, which include 343,985 warrants to purchase common shares which are exercisable within 60 days of August 31, 2015, and 2,346,194 shares for which beneficial ownership is shared by Oracle Ten Fund Master, L.P. and Oracle Investment Management Inc., which include 519,093 warrants to purchase common shares which are exercisable within 60 days of August 31, 2015. The 3,378,205 shares beneficially owned by Oracle Associates, LLC, include 478,802 shares beneficially owned by Oracle Institutional Partners, L.P., which include 51,598 warrants to purchase common shares which are exercisable within 60 days of August 31, 2015, and 2,899,403 shares owned by Oracle Partners, L.P., which include 292,387 warrants to purchase common shares which are exercisable within 60 days of August 31, 2015. |
| (3) | Includes 57,501 options exercisable within 60 days of August 31, 2015. Gary Pace also has 2,293 warrants to purchase common shares which are exercisable within 60 days of August 31, 2015. |
| (4) | Includes 17,199 warrants to purchase common shares and 593,603 options exercisable within 60 days of August 31, 2015. |
| (5) | Includes 285,139 options exercisable within 60 days of August 31, 2015. |
| (6) | Includes 1,147 warrants to purchase common shares and 138,217 options exercisable within 60 days of August 31, 2015. |
| (7) | Includes 5,733 warrants to purchase common shares and 246,379 options exercisable within 60 days of August 31, 2015. |
| (8) | Includes 110,830 options exercisable within 60 days of August 31, 2015. |
The Corporation had 19 registered shareholders in the U.S. who held17,278,090 shares, representing approximately 44.4% of the Corporation’s common shares outstanding as of August 31, 2015.
Certain Relationships and Related Transactions
During fiscal 2015, the Company paid legal fees to a law firm where the Company’s Secretary is a partner and to a corporation controlled by the Company’s Secretary. Total fees and disbursements charged to the Company by these companies was $45,346 and $49,000 in fiscal 2015 and 2014 respectively and are included in general and administrative expenses. The balance owing at June 30, 2015 and 2014 is nil.
Members of the Company’s Board of Directors, management and employees participated in the June, 2014 private placements.
On June 23, 2014, the Company announced the closing of a private placement financing issuing 3,195,487 common shares of the Company at a price of US$5.32 per common share, together with warrants to purchase approximately 1,949,250 common shares of the Company. A portion of such shares and warrants were purchased in the private placement by Gary Pace and Paul Baehr, Directors of the Company, as well as Tony Cruz, Chief Executive Officer and Chairman of the Board of Directors of the Company, Carl Damiani, President and Chief Operating Officer of the Company and Nicole Rusaw, Chief Financial Officer of the Company.
The Company executed subscription agreements with these individuals in connection with such purchases. The form of such subscription agreements is filed as Exhibit 4.7 to this annual report on Form 20-F. These transactions occurred in the normal cause of operations and were measured at the exchange amount, which is the amount of consideration established and agreed to by the related parties.
| Item 8. | Financial Information |
| A. | Consolidated Statements and Other Information |
Please see our audited consolidated financial statements filed as a part of this annual report on page F-1.
Legal Proceedings
As of June 30, 2015, no litigation, arbitration or claim of material importance was pending and there were no material proceedings in which any director, member of our senior management or any of our affiliates was a party adverse to us or in which such persons or entities had a material interest that was adverse to us or any of our subsidiaries.
Dividend Policy
The Corporation has not declared any dividends to date and does not currently anticipate paying any dividends in the foreseeable future. Any future determination to pay dividends will be at the discretion of the board of directors of the Corporation and will depend upon the Corporation’s need to finance growth, its financial condition, results of operations, capital requirements and other factors which the Board of Directors may consider appropriate in the circumstances.
Except as disclosed elsewhere in this annual report, we have not experienced any significant changes since the date of our audited consolidated financial statements included in this annual report.
| Item 9. | The Offer and Listing |
Market Price
The Common Shares are listed and posted for trading on the TSX under the symbol “TTH” and as of January 7, 2008 on the NASDAQ Global Market under the symbol “TTHI”.
The following table sets forth, for the periods indicated, the high and low sales prices for our Common Shares on the NASDAQ (in U.S.$) and TSX (in CAN$).
| | | NASDAQ | | | TSX | |
| | | High | | | Low | | | High | | | Low | |
| Yearly Highs and Lows for the Year Ending June 30, | | | | | | | | | | | | | | | | |
| 2015 | | | 9.30 | | | | 2.23 | | | | 11.50 | | | | 2.50 | |
| 2014 | | | 8.22 | | | | 2.81 | | | | 9.25 | | | | 2.62 | |
| 2013 | | | 3.84 | | | | 2.81 | | | | 3.85 | | | | 1.74 | |
| 2012 | | | 3.23 | | | | 1.15 | | | | 3.00 | | | | 1.19 | |
| 2011 | | | 5.49 | | | | 1.81 | | | | 5.28 | | | | 1.82 | |
| | | | | | | | | | | | | | | | | |
| Quarterly Highs and Lows | | | | | | | | | | | | | | | | |
| 2015 | | | | | | | | | | | | | | | | |
| First Quarter (July-September 2014) | | | 7.50 | | | | 5.53 | | | | 8.15 | | | | 6.00 | |
| Second Quarter (October-December 2014) | | | 6.94 | | | | 6.05 | | | | 8.08 | | | | 6.10 | |
| Third Quarter (January-March 2015) | | | 7.25 | | | | 6.29 | | | | 9.47 | | | | 7.30 | |
| Fourth Quarter (April-June 2015) | | | 9.30 | | | | 2.02 | | | | 11.50 | | | | 2.50 | |
| 2014 | | | | | | | | | | | | | | | | |
| First Quarter (July-September 2013) | | | 4.99 | | | | 2.81 | | | | 4.98 | | | | 2.62 | |
| Second Quarter (October-December 2013) | | | 5.91 | | | | 4.00 | | | | 6.77 | | | | 4.16 | |
| Third Quarter (January-March 2014) | | | 8.16 | | | | 5.67 | | | | 9.25 | | | | 6.00 | |
| Fourth Quarter (April-June 2014) | | | 8.22 | | | | 4.36 | | | | 9.00 | | | | 4.80 | |
| | | | | | | | | | | | | | | | | |
| Monthly Highs and Lows | | | | | | | | | | | | | | | | |
| March 2015 | | | 7.12 | | | | 6.40 | | | | 9.05 | | | | 8.10 | |
| April 2015 | | | 8.50 | | | | 6.75 | | | | 10.29 | | | | 8.00 | |
| May 2015 | | | 7.76 | | | | 6.01 | | | | 9.60 | | | | 7.16 | |
| June 2015 | | | 9.30 | | | | 2.02 | | | | 11.50 | | | | 2.50 | |
| July 2015 | | | 2.53 | | | | 1.95 | | | | 3.30 | | | | 2.50 | |
| August 2015 | | | 2.72 | | | | 1.77 | | | | 3.87 | | | | 2.37 | |
| Item 10. | Additional Information |
Not applicable.
| B. | Memorandum and Articles of Association |
Articles of Incorporation
We are governed by our amended articles of incorporation (the “Articles”) under the Business Corporations Act (Ontario) (the “Act”) and by our by-laws (the “By-laws”). Our Ontario corporation number is 1303782. Our Articles provide that there are no restrictions on the business we may carry on or on the powers we may exercise.
Directors
Subject to certain exceptions, including in respect of voting on any resolution to approve a contract that relates primarily to the director’s remuneration, directors may not vote on resolutions to approve a material contract or material transaction if the director is a party to such contract or transaction or is a director or an officer of or has a material interest in any person who is a party to such contract or transaction. The directors are entitled to remuneration as shall from time to time be determined by the Board of Directors with no requirement for a quorum of independent directors. The directors have the ability under the Act to exercise our borrowing power, without authorization of the shareholders. The Act permits shareholders to restrict this authority through a company’s articles or by-laws (or through a unanimous shareholder agreement), but no such restrictions are in place for us. Our Articles and By-laws do not require directors to hold shares for qualification.
Rights, Preferences and Dividends Attaching to Shares
The holders of common shares have the right to receive dividends if and when declared by the Board of Directors. Each holder of common shares, as of the record date prior to a meeting, is entitled to attend and to cast one vote for each common share held as of such record date at such annual and/or special meeting, including with respect to the election or re-election of directors. The numbers of our Board of Directors are not replaced at staggered intervals but are elected annually.
In the event of the liquidation, dissolution or winding up of the Corporation or other distribution of assets of the Corporation among its shareholders for the purpose of winding up its affairs, the holders of common shares shall have a right to receive their pro rata share of such distribution, subject to the rights of the holders of any other class of shares of the Corporation entitled to receive the assets of the Corporation upon such a distribution in priority to the common shares. There are no sinking fund or redemption provisions in respect of the common shares. Our shareholders have no liability to further capital calls as all shares issued and outstanding are fully paid and non-assessable.
No other classes of shares are currently permitted to be issued.
Action Necessary to Change the Rights of Shareholders
In order to change the rights of our shareholders, we would need to amend our articles of incorporation to effect the change. Such an amendment would require approval by a special resolution passed by at least two-thirds of the votes cast at a duly called special meeting. For certain amendments such as those creating of a class of preferred shares, a shareholder is entitled to dissent in respect of such a resolution amending our articles and, if the resolution is adopted and the Corporation implements such changes, demand payment of the fair value of its shares.
Annual and Special Meetings of Shareholders
An annual meeting of shareholders is held each year for the purpose of receiving the financial statements and report of the auditor, electing directors, appointing the auditor and for the transaction of other business as may be brought before the meeting. The Board of Directors has the power to call a special meeting of shareholders at any time. Under the Act and our Bylaws, we are required to mail a Notice of Meeting and Management Information Circular to registered shareholders not less than 21 days and not more than 50 days prior to the date of the meeting. Such materials must be filed concurrently with the applicable securities regulatory authorities in Canada and the U.S. Subject to certain provisions of the Bylaws, a quorum of one or more shareholders in person or represented by proxy holding or representing by proxy not less than five (5%) percent of the total number of issued and outstanding shares enjoying voting rights at such meeting is required to properly constitute a meeting of shareholders. Shareholders and their duly appointed proxies and corporate representatives are entitled to be admitted to our annual and/or special meetings.
Limitations on the Rights to Own Shares
The Articles do not contain any limitations on the rights to own shares. Except as described below, there are currently no limitations imposed by Canadian federal or provincial laws on the rights of non-resident or foreign owners of Canadian securities to hold or vote the securities held. There are also no such limitations imposed by the Articles and By-laws with respect to our common shares.
Investment Canada Act
Under the Investment Canada Act, transactions exceeding certain financial thresholds, and which involve the acquisition of control of a Canadian business by a non-Canadian, may be subject to review and cannot be implemented unless the Minister of Industry and/or, in the case of a Canadian business engaged in cultural activities, the Minister of Canadian Heritage, are satisfied that the transaction is likely to be of “net benefit to Canada”. If a transaction is subject to review (a “Reviewable Transaction”), an application for review must be filed with the Investment Review Division of Industry Canada and/or the Department of Canadian Heritage and, generally speaking, approval must be obtained prior to the implementation of the Reviewable Transaction. The responsible Minister is, upon the filing of the application, required to determine whether the Reviewable Transaction is likely to be of net benefit to Canada taking into account, among other things, certain factors specified in the Investment Canada Act and any written undertakings that may have been given by the applicant. The Investment Canada Act contemplates an initial review period of up to 45 days after filing; however, if the responsible Minister has not completed the review by that date, the Minister may unilaterally extend the review period by up to 30 days (or such longer period as may be agreed to by the applicant and the Minister) to permit completion of the review. At the completion of his or her review, the responsible Minister will either approve or reject the proposed acquisition of control. Direct acquisitions of control of most Canadian businesses by or from World Trade Organization (“WTO”) investors who are not state-owned enterprises are reviewable under the Investment Canada Act only if the enterprise value of the Canadian business (including its worldwide assets) exceeds C$600 million (this threshold will increase to C$800 million in 2017, then to C$1 billion in 2019). The threshold applicable to WTO investors which are state-owned applies to the value of the worldwide assets of the Canadian business and is exceeded if the total value of such assets reflected on the balance sheet exceed C$369 million (adjusted annually to reflect inflation). Indirect acquisitions (e.g., an acquisition of a U.S. corporation with a Canadian subsidiary) of control of such businesses by or from WTO investors are not subject to review, regardless of the value of the Canadian businesses’ assets. Significantly lower review thresholds apply where neither the investor nor the Canadian business is WTO investor controlled or where the Canadian business is engaged in certain cultural businesses.
Even if the transaction is not reviewable because it does not meet or exceed the applicable threshold, the non-Canadian investor must still give notice to Industry Canada and, in the case of a Canadian business engaged in cultural activities, Canadian Heritage, of its acquisition of control of a Canadian business within 30 days of its implementation. Finally, the Government of Canada has the power, under the Investment Canada Act, to review and challenge any acquisition by a non-Canadian of all or part of a Canadian business if the government believes the transaction could be injurious to Canada’s national security.
Disclosure of Share Ownership
Under applicable securities regulation in Canada, notice is required to be given to the market in the event of an acquisition of equity or voting securities representing 10% (5% where a take-over bid has already been made) of a class of securities of a target (including shares beneficially owned, controlled or directed by the purchaser and its joint actors). The purchaser must give this notice to the market by issuing a press release forthwith and filing, within two business days, an “early warning” report in the prescribed form (which must include disclosure relating to the purpose of the purchaser in undertaking the transaction and of any intention on the part of the purchaser to acquire additional securities of the class). A press release is required to be issued and an additional report is required to be filed if there is a change in a material fact contained in a prior report, or upon the acquisition of a further 2% of the class of securities or securities convertible into an additional 2% of such class of securities.
An alternative monthly reporting system is available to eligible institutional investors, which requires only the filing of a report within 10 days after the end of the month in which the security holding percentage of the eligible institutional investor in a class of voting or equity securities of the issuer, as at the end of the month, increased to 10% or more or increased or decreased past thresholds that are products of whole numbers multiplied by 2.5% of the outstanding securities of that class and that are in excess of 10% or decreased to less than 10% or there has been a change in a material fact contained in the report of the eligible institutional investor most recently filed.
In addition, in Canada, a person or company (other than an eligible institutional investor) who beneficially owns, directly or indirectly, voting securities of a reporting issuer or who exercises control or direction over voting securities of a reporting issuer or a combination of both, carrying more than 10% of the voting rights attached to all the reporting issuer’s outstanding voting securities is a reporting insider and must, within 10 days of becoming a reporting insider, file an initial report in the required form effective the date on which the person became a reporting insider. The initial report must disclose any direct or indirect beneficial ownership of, or control or direction over, securities of the reporting issuer and any interest in, or right or obligation associated with, a related financial instrument involving a security of the reporting issuer. Additionally, securities regulation in Canada provides for the filing of a subsequent report by a reporting insider if there is any change in the information contained in the initial report, which subsequent report must be filed within 5 days from the day on which the change takes place.
The rules in the U.S. governing the ownership threshold above which shareholder ownership must be disclosed are more stringent than those discussed above. Section 13 of the Exchange Act imposes reporting requirements on persons who acquire beneficial ownership (as such term is defined in Rule 13d-3 under the Exchange Act) of more than 5% of a class of an equity security registered under Section 12 of the Securities Exchange Act of 1934 (the “Exchange Act”). In general, such persons must file, within 10 days after such acquisition, a report of beneficial ownership with the Securities and Exchange Commission containing the information prescribed by the regulations under Section 13 of the Exchange Act. This information is also required to be sent to the issuer of the securities and to each exchange where the securities are traded.
Other Provisions of Articles and By-laws
There are no provisions of our Articles or Bylaws that would have an effect of delaying, deferring or preventing a change in control of the Corporation and that would operate only with respect to a merger, acquisition or corporate restructuring involving the Corporation. Our Bylaws do not contain a provision governing the ownership threshold above which shareholder ownership must be disclosed. There are no provisions in our Articles or Bylaws governing changes in capital, where such provisions are more stringent than those required by law.
Other than those listed in the paragraphs below, we have not entered into any material contracts other than in the ordinary course of business or other than those described in Item 4 “Information on the Corporation” and elsewhere in this annual report.
The Corporation has entered into the following material contracts, which were not in the ordinary course of business, in the two-year period immediately preceding that filing date of this annual report:
Perrigo
On February 28, 2014, after a series of transactions, Perrigo has transferred all of its ELND005 rights and assets to Transition Ireland. Transition Therapeutics Inc. acquired Transition Ireland, which is now a 100% wholly-owned subsidiary. Transition Ireland is a party to a Milestone, Royalty and Sublicense Fee agreement with a subsidiary of Perrigo. Under this agreement, the subsidiary of Perrigo will be eligible to receive from Transition Ireland up to US$40 million in approval and commercial milestone payments and a 6.5% royalty on net sales of ELND005 products and sublicense fees received.
The agreement continues through the expiration of all payment obligations under the agreement.
Eli Lilly
TT401
On March 3, 2010, the Corporation and Lilly entered into a licensing and collaboration agreement granting the Corporation the rights to a series of preclinical compounds in the area of diabetes. Under the licensing and collaboration agreement, the Corporation will receive exclusive worldwide rights to develop and potentially commercialize a class of compounds that, in preclinical models showed potential to provide glycemic control and other beneficial effects including weight loss.
Under the terms of the agreement, Lilly received an up-front payment of US$1 million and retained the option to reacquire the rights to the compounds at a later date.
In June 2013, Lilly assumed all development and commercialization rights to type 2 diabetes drug candidate TT401. In conjunction with this assumption of rights, Transition received a US$7 million milestone payment. Lilly assumed all costs and will perform all future development and commercialization activities of TT401, and Transition paid US$14 million to Lilly in three separate installments during the Phase 2 clinical study. In return, Transition is eligible to receive up to approximately US$240 million in additional milestone payments. Transition will also be eligible to receive a low double-digit royalty (less than 15%) on sales of TT401 products and a low single digit royalty on related compounds.
The US$14 million was paid to Lilly in three instalments in the second half of calendar 2014. The Corporation has no additional funding obligations related to the ongoing clinical study or any other development or commercialization activities in the future under the Lilly agreement.
The agreement continues until the expiration of all payment obligations under the agreement. If Transition commercializes the product, Transition can terminate the agreement on 90 days notice. Either party may terminate for material breach.
TT701
On May 6, 2015, the Company, through its wholly owned subsidiary, Transition Ireland, exclusively licensed worldwide rights to a novel small molecule drug candidate, TT701 from Lilly. Under the terms of the agreement, the Corporation has acquired rights to develop and commercialize TT701. Transition will pay Lilly upfront consideration of up to US$1 million, US$500,000 of which has already been paid as of June 30, 2015. Lilly will also be eligible to receive up to approximately US$100 million in additional commercial milestone payments and a mid-single digit royalty on annual worldwide net sales.
There are currently no limitations imposed by Canadian federal or provincial laws on the rights of non-resident or foreign owners of our securities to hold or vote the securities held. There are also no such limitations imposed by our Articles and By-Laws with respect to our Common Shares.
United States Federal Income Taxation
The following discussion describes the material U.S. federal income tax consequences to U.S. Holders (defined below) under present law of an investment in our Common Shares. This summary applies only to investors that hold our Common Shares as capital assets and that have the U.S. dollar as their functional currency. This discussion assumes that we are not a “controlled foreign corporation” for U.S. federal income tax purposes. This discussion is based on the tax laws of the United States as in effect on the date of this annual report and on U.S. Treasury regulations in effect or, in some cases, proposed, as of the date of this annual report, as well as judicial and administrative interpretations thereof available on or before such date. All of the foregoing authorities are subject to change, which change could apply retroactively and could affect the tax consequences described below.
The following discussion does not deal with the tax consequences to any particular investor or to persons in special tax situations such as:
| · | certain financial institutions; |
| · | regulated investment companies; |
| · | real estate investment trusts; |
| · | traders that elect to mark to market; |
| · | persons liable for alternative minimum tax; |
| · | persons liable for the Medicare tax on investment income; |
| · | persons holding a Common Share as part of a straddle, hedging, constructive sale, conversion or integrated transaction; |
| · | persons who acquired Common Shares pursuant to the exercise of any employee share option or otherwise as compensation; or |
| · | persons holding Common Shares through partnerships or other pass-through entities for U.S. federal income tax purposes. |
U.S. HOLDERS ARE URGED TO CONSULT THEIR TAX ADVISORS ABOUT THE APPLICATION OF THE U.S. FEDERAL TAX RULES TO THEIR PARTICULAR CIRCUMSTANCES AS WELL AS THE STATE, LOCAL AND FOREIGN TAX CONSEQUENCES TO THEM OF THE PURCHASE, OWNERSHIP AND DISPOSITION OF COMMON SHARES.
A “U.S. Holder” includes:
| · | an individual who is a citizen or resident of the United States; |
| · | a corporation (or other entity taxable as a corporation for U.S. federal income tax purposes) created or organized in or under the laws of the United States, any State or the District of Columbia; |
| · | an estate whose income is subject to U.S. federal income taxation regardless of its source; or |
| · | a trust that (1) is subject to the supervision of a court within the United States and the control of one or more U.S. persons or (2) has a valid election in effect under applicable U.S. Treasury regulations to be treated as a U.S. person. |
Distributions
The gross amount of any distribution received by a U.S. Holder with respect to our Common Shares (including amounts withheld to pay Canadian withholding taxes) will be included in the gross income of such U.S. Holder as a dividend to the extent attributable to our current or accumulated earnings and profits, as determined under U.S. federal income tax principles. To the extent that the amount of any distribution exceeds our current and accumulated earnings and profits, such distribution will be treated first as a tax-free return of capital to the extent of a U.S. Holder’s adjusted tax basis in its Common Shares, causing a reduction in the adjusted basis of such Common Shares. To the extent that such distribution exceeds a U.S. Holder’s adjusted tax basis in its Common Shares, the distribution will be treated as gain from the sale or exchange of such Common Shares. Provided that we are not treated as a PFIC, as described below, we believe that we are considered to be a “qualified foreign corporation,” and therefore distributions, if any, to non-corporate U.S. Holders (including individuals) that are treated as dividends should qualify for a reduced rate of tax. If we are a PFIC under the rules discussed below, distributions treated as dividends will be taxable at the higher ordinary income tax rates. Dividends on our Common Shares paid to corporate shareholders will not be eligible for the dividends received deduction allowed with respect to dividends from domestic corporations under the Code.
Generally, the amount of any dividend paid in Canadian dollars (including amounts withheld to pay Canadian withholding taxes) will equal the U.S. dollar value of the Canadian dollars calculated by reference to the spot rate in effect on the date the dividend is received by the U.S. Holder, regardless of whether the Canadian dollars are converted into U.S. dollars. If the Canadian dollars received as a dividend are converted into U.S. dollars on the date of receipt, the U.S. Holder generally should not be required to recognize foreign currency gain or loss in respect of the dividend income. If the Canadian dollars received as a dividend are not converted into U.S. dollars on the date of receipt, a U.S. Holder will have a tax basis in the Canadian dollars equal to their U.S. dollar value on the date of receipt. Any gain or loss realized on a subsequent conversion or other disposition of the Canadian dollars by a U.S. Holder will generally be treated as U.S.-source ordinary income or loss (or foreign source ordinary income or loss if the U.S. Holder is an individual with a tax home located outside the United States).
A U.S. Holder may be entitled to deduct or credit the amount of Canadian withholding tax imposed on dividends paid to such U.S. Holder, subject to applicable limitations in the Code. For purposes of calculating the foreign tax credit, dividends paid on our Common Shares generally will be treated as income from foreign sources and generally will constitute “passive category income.” The rules governing the foreign tax credit are complex. U.S. Holders are urged to consult their own tax advisors regarding the availability of the foreign tax credit in their particular circumstances.
Sale, Exchange or Other Taxable Disposition
A U.S. Holder will recognize gain or loss on the sale, exchange or other taxable disposition of our Common Shares in an amount equal to the difference between the amount realized for our Common Shares and the U.S. Holder’s adjusted tax basis in our Common Shares. Subject to the discussion below under “Passive Foreign Investment Company Rules,” the gain or loss will be capital gain or loss. Capital gains of non-corporate U.S. Holders derived with respect to capital assets held for more than one year are eligible for reduced rates of taxation. The deductibility of capital losses is subject to limitations. Any capital gain or loss recognized by a U.S. Holder generally will be treated as U.S.-source gain or loss for U.S. foreign tax credit purposes.
Passive Foreign Investment Company Rules
Adverse U.S. federal income tax rules generally apply to U.S. Holders owning shares of a PFIC. A non-U.S. corporation generally will be classified as a PFIC for U.S. federal income tax purposes in any taxable year in which, after applying relevant look-through rules with respect to the income and assets of subsidiaries, either at least 75% of its gross income is “passive income” (the “income test”), or on average at least 50% of the gross value of its assets is attributable to assets that produce passive income or are held for the production of passive income (the “asset test”). For this purpose, passive income generally includes, among other things, dividends, interest, certain rents and royalties, certain gains from the sales of commodities, and gains from the disposition of passive assets.
Based on the composition of our income and the valuation of our assets, we do not believe we were a PFIC for the taxable year ended June 30, 2015. However, because the PFIC determination is made annually at the close of the taxable year in question on the basis of facts and circumstances that may be beyond our control and because the principles and methodology for applying the PFIC tests are not entirely clear, there can be no assurance that we will not be a PFIC in subsequent taxable years or that the IRS will not challenge our determination concerning our PFIC status.
If we were a PFIC in any taxable year during a U.S. Holder’s holding period for our Common Shares, the U.S. Holder generally would be subject to special rules with respect to “excess distributions” made by us on our Common Shares and with respect to gain from the sale, exchange or disposition of our Common Shares. An “excess distribution” generally is defined as the excess of distributions with respect to the Common Shares received by a U.S. Holder in any taxable year over 125% of the average annual distributions the U.S. Holder has received from us during the shorter of the three preceding taxable years or the U.S. Holder’s holding period for the Common Shares. Generally, a U.S. Holder would be required to allocate any excess distribution or gain from the sale, exchange or disposition of our Common Shares ratably over its holding period for the Common Shares. The amounts allocated to the taxable year of the sale, exchange or disposition and to any year before we became a PFIC would be taxed as ordinary income. The amount allocated to each other taxable year would be subject to tax at the highest tax rate in effect for individuals or corporations, as appropriate, for the other taxable year, and an interest charge would be imposed on the amount allocated to the taxable year. These rules would apply to a U.S. Holder that held our Common Shares during any year in which we were a PFIC, even if we were not a PFIC in the year in which the U.S. Holder sold our Common Shares or received an excess distribution in respect of its Common Shares.
If we were a PFIC in any taxable year, then, provided certain requirements were met, a U.S. Holder might be able to make a mark-to-market election to alleviate certain of the tax consequences referred to above. A “qualified electing fund” election would not be available to U.S. Holders, because we do not intend to provide the necessary information to allow U.S. Holders to make such an election for any tax year in which we are a PFIC. If we were a PFIC in any taxable year, special IRS reporting requirements would apply to a U.S. Holder in respect of its Common Shares. U.S. Holders are urged to consult their own tax advisors regarding the tax consequences and reporting obligations that would arise if we were treated as a PFIC for any year as well as the availability of any elections to mitigate the adverse tax consequences to a U.S. Holder if we were a PFIC.
Information Reporting and Backup Withholding
Information reporting requirements will apply to the payment of dividends on our Common Shares or the proceeds received on the sale, exchange, or other taxable disposition of Common Shares paid within the U.S. (and in certain cases, outside the U.S.) to U.S. Holders other than certain exempt recipients (such as corporations). In addition, a backup withholding tax may apply to such amounts if the U.S. Holder fails to timely provide an accurate taxpayer identification number or is notified by the IRS that it has failed to report dividends or interest required to be shown on its U.S. federal income tax returns. Backup withholding is not an additional tax. The amount of any backup withholding from a payment to a U.S. Holder generally will be allowed as a credit against the U.S. Holder’s U.S. federal income tax liability, and may entitle the U.S. Holder to a refund, provided that the required information is provided to the IRS in a timely manner.
Information Reporting with Respect to Foreign Financial Assets
U.S. individuals that own “specified foreign financial assets” with an aggregate value in excess of US$50,000 (or such higher dollar amount as prescribed by the Secretary of Treasury) are generally required to file an information report with respect to such assets with their tax returns. “Specified foreign financial assets” include any financial accounts maintained by foreign financial institutions, as well as any of the following, but only if they are not held in accounts maintained by financial institutions: (i) stocks and securities issued by non-U.S. persons; (ii) financial instruments and contracts held for investment that have non-U.S. issuers or counterparties; and (iii) interests in foreign entities. Under these rules, our Common Shares may be treated as “specified foreign financial assets”. U.S. Holders are urged to consult their own tax advisors regarding the possible implications of the reporting rules described above.
The above summary is not intended to constitute a complete analysis of all tax consequences relating to the purchase, ownership and disposition of our Common Shares. Each prospective investor should consult with its own tax advisor concerning the particular tax consequences with regard to its particular circumstances.
Material Canadian Federal Tax Considerations
General
The following is a summary of the material Canadian federal tax implications applicable to a holder (a “U.S. Holder”) who holds our Common Shares (the “Common Shares”) and who, at all relevant times, for purposes of the Income Tax Act (Canada) (the “Canadian Tax Act”) (i) has not been, is not and will not be resident (or deemed resident) in Canada at any time while such U.S. Holder has held or holds the Common Shares; (ii) holds the Common Shares as capital property and as beneficial owner; (iii) deals at arm’s length with and is not affiliated with us; (iv) does not use or hold, and is not deemed to use or hold, the Common Shares in the course of carrying on a business in Canada; (v) did not acquire the Common Shares in respect of, in the course of or by virtue of employment with our company; (vi) is not a financial institution, specified financial institution, partnership or trust as defined in the Canadian Tax Act; (vii) is a resident of the United States for purposes of the Canada-United States Income Tax Convention (1980), as amended (the “Convention”) who is fully entitled to the benefits of the Convention; and (viii) has not, does not and will not have a fixed base or permanent establishment in Canada within the meaning of the Convention at any time while such U.S. Holder has held or holds the Common Shares. Special rules, which are not addressed in this summary, may apply to a U.S. Holder that is a “registered non-resident insurer” or “authorized foreign bank”, as defined in the Canadian Tax Act, carrying on business in Canada and elsewhere.
This summary is based on the current provisions of the Canadian Tax Act, and the regulations thereunder, the Convention, and counsel’s understanding of the published administrative practices and policies of the Canada Revenue Agency all in effect as of the date of this annual report on Form 20-F. This summary takes into account all specific proposals (the “Proposals”) to amend the Canadian Tax Act and the regulations thereunder publicly announced by or on behalf of the Minister of Finance (Canada) prior to the date of this annual report on Form 20-F. No assurances can be given that such Proposals will be enacted in the form proposed, or at all. This summary is not exhaustive of all potential Canadian federal tax consequences to a U.S. Holder and does not take into account or anticipate any other changes in law or administrative practices, whether by judicial, governmental or legislative action or decision, nor does it take into account provincial, territorial or foreign tax legislation or considerations, which may differ from the Canadian federal tax considerations described herein.
This summary assumes that we are a resident of Canada for purposes of the Canadian Tax Act.
TAX MATTERS ARE VERY COMPLICATED AND THE CANADIAN FEDERAL TAX CONSEQUENCES OF PURCHASING, OWNING AND DISPOSING OF COMMON SHARES WILL DEPEND ON THE SHAREHOLDER’S PARTICULAR SITUATION. THIS SUMMARY IS NOT INTENDED TO BE A COMPLETE ANALYSIS OF OR DESCRIPTION OF ALL POTENTIAL CANADIAN FEDERAL TAX CONSEQUENCES, AND SHOULD NOT BE CONSTRUED TO BE, LEGAL, BUSINESS OR TAX ADVICE DIRECTED AT ANY PARTICULAR HOLDER OR PROSPECTIVE PURCHASER OF COMMON SHARES. ACCORDINGLY, HOLDERS OR PROSPECTIVE PURCHASERS OF COMMON SHARES SHOULD CONSULT THEIR OWN TAX ADVISORS FOR ADVICE WITH RESPECT TO THE CANADIAN FEDERAL TAX CONSEQUENCES OF AN INVESTMENT IN COMMON SHARES BASED ON THEIR PARTICULAR CIRCUMSTANCES.
Dividends
Amounts paid or credited, or deemed under the Canadian Tax Act to be paid or credited, on account of, in lieu of payment of, or in satisfaction of, dividends to a U.S. Holder will be subject to Canadian non-resident withholding tax at the reduced rate of 15% under the Convention. This rate is further reduced to 5% in the case of a U.S. Holder that is a company for purposes of the Convention that owns at least 10% of our voting shares at the time the dividend is paid or deemed to be paid. Under the Convention, dividends paid or credited to certain religious, scientific, literary, educational or charitable organizations and certain pension organizations that are resident in the United States and that have complied with certain administrative procedures may be exempt from Canadian withholding tax.
Disposition of Our Common Shares
A U.S. Holder will not be subject to tax under the Canadian Tax Act in respect of any capital gain realized on the disposition or deemed disposition of the Common Shares unless, at the time of disposition, the Common Shares constitute “taxable Canadian property” of the U.S. Holder for the purposes of the Canadian Tax Act and the U.S. Holder is not otherwise entitled to an exemption under the Convention.
Provided that the Common Shares are, at the time of disposition, listed on a designated stock exchange for purposes of the Canadian Tax Act (which currently includes Nasdaq and the TSX), and under the Canadian Tax Act currently in force, the Common Shares will not constitute “taxable Canadian property” to a U.S. Holder unless; (i) at any time during the 60-month period immediately preceding the disposition of the Common Shares the U.S. Holder, persons with whom the U.S. Holder did not deal at arm’s length, partnerships in which the U.S. Holder or a person with whom the U.S. Holder did not deal at arm’s length holds a membership interest (either directly or indirectly through one or more partnerships), or the U.S. Holder together with such persons, owned 25% or more of the issued shares of any class or series of our capital stock; and at any time during the 60-month period immediately preceding the disposition of the Common Shares more than 50% of the fair market value of the Common Shares was derived directly or indirectly, from one or any combination of real or immovable property situated in Canada, Canadian resource property, timber resource property, or any option in respect of, or interest in, such properties, as such terms are used for purposes of the Canadian Tax Act; or (ii) if the Common Shares are otherwise deemed under the Canadian Tax Act to be taxable Canadian property.
Pursuant to the Convention, even if the Common Shares constitute “taxable Canadian property” of a particular U.S. Holder, any capital gain realized on the disposition of the Common Shares by the U.S. Holder generally will be exempt from tax under the Canadian Tax Act, unless, at the time of disposition, the Common Shares derive their value principally from real property situated in Canada within the meaning of the Convention. U.S. Holders whose Common Shares may constitute taxable Canadian property should consult their own tax advisors.
| F. | Dividends and Paying Agents |
Not applicable.
Not applicable.
Additional information relating to the Corporation may be found on SEDAR atwww.sedar.com and on the SEC’s website at www.sec.gov.
Specifically, additional information, including directors’ and officers’ remuneration and indebtedness, principal holders of securities of the Corporation and securities authorized for issuance under the Corporation’s stock option plan is contained in the Corporation’s Management Information Circular for its most recent annual meeting of securities holders that involved the election of directors dated November 4, 2014.
Our corporate Internet address is http://www.transitiontherapeutics.com. We make available free of charge on or through our website our annual reports, current reports, and amendments to those reports filed or furnished pursuant to Section 13(a) or 15(d) of the Securities Exchange Act of 1934, as amended (the “Exchange Act”) as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. We may from time to time provide important disclosures to investors by posting them in the investor relations section of our website, as allowed by Securities and Exchange Commission (“SEC”) rules. Information contained on our website is not part of this report or any other report filed with the SEC. You may read and copy any public reports we filed with the SEC at the SEC’s Public Reference Room at 100 F Street, N.E., Washington, D.C. 20549. You may obtain information on the operation of the Public Reference Room by calling the SEC at 1-800-SEC-0330. The SEC also maintains an Internet site http://www.sec.gov that contains reports, proxy and information statements, and other information that we filed electronically.
For a listing of our subsidiaries, see “Item 4. Information on the Corporation” and Exhibit 8.1.
| Item 11. | Quantitative and Qualitative Disclosures About Market Risk. |
Qualitative and Quantitative Disclosures about Market Risk
We are exposed to various types of market risks in the normal course of business, including foreign exchange risk and interest rate risk. We have not in the past used derivatives to manage our exposure to interest rate risk or foreign exchange risk. For additional information please see Note 4.2 of the Corporation’s audited consolidated financial statements for the year ended June 30, 2015.
Foreign Exchange Risk
The Company operates in Canada and has relationships with entities in other countries. Foreign exchange risk arises from purchase transactions, as well as recognized financial assets and liabilities denominated in foreign currencies, mainly the US dollar. The Company does not enter into hedging or other contracts to mitigate its exposure to foreign exchange risk and maintains sufficient US dollars to meet the Company’s planned US dollar expenses.
Interest Rate Risk
The Corporation is exposed to interest rate risk to the extent that the short term investments are at a fixed rate of interest and their fair value can vary with the change in market interest rates. The Corporation’s maximum exposure to interest rate risk is based on the effective interest rate of the current carrying value of these assets. The Corporation does not speculate on interest rates and holds all deposits until their date of maturity.
| Item 12. | Description of Securities Other than Equity Securities. |
Not applicable.
Part II
| Item 13. | Defaults, Dividend Arrearages and Delinquencies. |
Not applicable.
| Item 14. | Material Modifications to the Rights of Security Holders and Use of Proceeds. |
Not applicable.
| Item 15. | Controls and Procedures. |
Management’s Evaluation of Disclosure Controls and Procedures
Under the supervision and with the participation of management, including the Company’s CEO and CFO, the Company conducted an evaluation of the effectiveness of its disclosure controls and procedures as of June 30, 2015 as required by Canadian securities legislation. Disclosure controls and procedures (as defined in Rule 13a-15(e) under the Securities Exchange Act of 1934) are designed to ensure that the information required to be disclosed by the Company in the reports it files or submits under securities legislation is recorded, processed, summarized and reported on a timely basis and that such information is accumulated and reported to management, including the Company’s CEO and CFO, as appropriate, to allow required disclosures to be made in a timely fashion. Based on their evaluation, the CEO and CFO have concluded that as of June 30, 2015, the Company’s disclosure controls and procedures were effective.
Management’s Report on Internal Control over Financial Reporting
The Company’s management is responsible for establishing and maintaining adequate internal control over financial reporting (as defined in Rule 13a-15(f) under the Securities Exchange Act of 1934). The Company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
The Company’s management assessed the effectiveness of the Company’s internal control over financial reporting as of June 30, 2015. In making this assessment, management used the criteria set forth by the Committee of Sponsoring Organizations of the Treadway Commission (2013) (“COSO”) in the updated Internal Control-Integrated Framework. The Company’s management, including the CEO and CFO, concluded that, as of June 30, 2015, the Company’s internal control over financial reporting was effective based on the criteria in Internal Control — Integrated Framework issued by COSO.
The effectiveness of the Company’s internal control over financial reporting as of June 30, 2015 has been audited by PricewaterhouseCoopers LLP, an independent registered public accounting firm, as stated in its report in the Company’s audited consolidated financial statements for the year ended June 30, 2015.
Changes in Internal Control over Financial Reporting.
There was no change in the Corporation’s internal control over financial reporting that occurred during the period covered by this report that has materially affected, or is reasonably likely to materially affect, the Corporation’s internal control over financial reporting.
The design of any system of controls and procedures is based in part upon certain assumptions about the likelihood of future events. There can be no assurance that any design will succeed in achieving its stated goals under all potential future conditions, regardless of how remote.
| Item 16A. | Audit committee financial expert. |
The Corporation’s Board of Directors has determined that it has one “audit committee financial expert” (as such term is defined in Form 20-F) serving on its Audit Committee. Such audit committee financial expert is Mr. Christopher Henley. Mr. Henley is “independent” as defined in NASDAQ Rule 5605(a)(2). Mr. Henley’s business experience is included under the heading “Audit Committee – Relevant Education and Experience” in this Annual Report on Form 20-F. The Securities and Exchange Commission (the “Commission”) has indicated that the designation of Mr. Henley as an audit committee financial expert doesnot make Mr. Henley an “expert” for any purpose, impose any duties, obligations or liability on Mr. Henley that are greater than those imposed on members of the Audit Committee and the Corporation’s Board of Directors who do not carry this designation or affect the duties, obligations or liability of any other member of the Audit Committee.
The Corporation has adopted a “code of ethics” (as that term is defined in Form 20-F) that applies to its principal executive officer, principal financial officer, principal accounting officer or controller, or persons performing similar functions (the “Ethics Code”). A copy of the Ethics Code entitled “Code of Business Ethics” is available for viewing on the Corporation’ website atwww.transitiontherapeutics.com. A copy of the Ethics Code may be obtained free of charge upon request to the Corporation in writing at the Corporation’s address at 101 College Street, Suite 220, Toronto, Ontario, Canada M5G1L7, Attention: Secretary.
During the fiscal year ended June 30, 2015, there were no amendments to the Ethics Code or waivers, including implicit waivers, from any provision of the Ethics Code.
| Item 16C. | Principal Accountant Fees and Services. |
Audit Fees
The aggregate fees billed for each of the last two fiscal years for professional services rendered by PricewaterhouseCoopers LLP for the audit of the Corporation’s annual consolidated financial statements, and the effectiveness of internal controls over financial reporting is included under the heading “Audit Fees” below.
Audit-Related Fees
The aggregate fees billed for each of the last two fiscal years for professional services rendered by PricewaterhouseCoopers LLP for audit-related services is included under the heading “Audit Related Fees” below.
Tax Fees
The aggregate fees billed for each of the last two fiscal years for professional services rendered by PricewaterhouseCoopers LLP for tax compliance, tax advice and tax planning is included under the heading “Tax Fees” below.
All Other Fees
The aggregate fees billed for each of the last two fiscal years for professional services rendered by PricewaterhouseCoopers LLP for services provided other than the services described above is included under the heading “Non-Audit” below.
| | | During the Year Ended
June 30, 2015
$ | | | During the Year Ended
June 30, 2014
$ | |
| Audit Fees(1) | | | 304,775 | | | | 238,150 | |
| Audit Related Fees(2) | | | 80,000 | | | | 40,000 | |
| Tax Fees(3) | | | 167,202 | | | | 114,914 | |
| Non-Audit(4) | | | 4,763 | | | | 4,346 | |
| Total | | | 556,740 | | | | 397,410 | |
Notes:
| (1) | During the years ended June 30, 2015 and 2014, Audit Fees” include fees for the annual audit, quarterly reviews, accounting consultations related to accounting, financial reporting or disclosure matters and assistance with understanding and implementing new accounting and financial reporting guidance from regulatory authorities. |
| (2) | During the years ended June 30, 2015 and 2014, “Audit Related Fees” include fees relating to the Corporations February 2015 public offering and August 15, 2013 and June 23, 2014 private placements. |
| (3) | During the years ended June 30, 2015 and 2014 “Tax Fees” include fees for assistance in the preparation and review of tax returns and related items, assistance with tax audits, general tax planning and advice relating to tax items such as withholding tax, SR&ED eligibility, and US and Ireland tax considerations. |
| (4) | For the years ended June 30, 2015 and 2014, the category “Non-Audit” includes a charge from the Corporation’s external auditors for a levy from the Canadian Public Accountability Board |
Pre-Approval Policies and Procedures
The Corporation’s audit committee is responsible for the oversight of the work of the external auditor. As part of this responsibility, the audit committee is required to pre-approve all audit, audit related, tax services and non-audit services performed by the external auditor in order to assure that they do not impair the external auditor’s independence from the Corporation. Accordingly, the audit committee has adopted a pre-approval policy, which sets forth the procedures and the conditions pursuant to which services proposed to be performed by the external auditor may be pre-approved.
Under the pre-approval policy, the Corporation’s audit committee annually reviews and pre-approves specific audit, audit-related and tax services that may be provided by the external auditor without obtaining specific pre-approval from the audit committee, as well as maximum fees for such services. All services that are not pre-approved or exceed the pre-approved maximum fees require specific pre-approval by the audit committee before the service can be performed by the external auditor. The pre-approval policy also includes a list of prohibited services.
| Item 16D. | Exemptions from the Listing Standards for Audit Committees. |
We have not been granted an exemption from the applicable listing standards for the audit committee of our board of directors.
| Item 16E. | Purchases of Equity Securities by the Issuer and Affiliated Purchasers. |
None.
| Item 16F. | Change in Registrant’s Certifying Accountant. |
None.
| Item 16G. | Corporate Governance. |
The Corporation is a foreign private issuer and its common shares are listed on the NASDAQ Stock Market (“NASDAQ”).
NASDAQ Rule 5615(a)(3) permits a foreign private issuer to follow its home country practice in lieu of the requirements of the Rule 5600 Series, the requirement to distribute annual and interim reports set forth in Rule 5250(d), and the Direct Registration Program requirement set forth in Rules 5210(c) and 5255, provided, however, that such a company shall: comply with the Notification of Material Noncompliance requirement ( Rule 5625), the Voting Rights requirement ( Rule 5640), have an audit committee that satisfies Rule 5605(c)(3), and ensure that such audit committee’s members meet the independence requirement in Rule 5605(c)(2)(A)(ii). The Corporation does not follow Rule 5620(c) (shareholder quorum) but instead follows its home country practice, as described below.
Shareholder Meeting Quorum Requirements: The NASDAQ minimum quorum requirement under Rule 5620(c) for a shareholder meeting is 33 1/3% of the outstanding shares of common stock. In addition, a registrant listed on NASDAQ is required to state its quorum requirement in its by laws. The Corporation’s quorum requirement is set forth in its by laws. A quorum for a meeting of shareholders of the Corporation is shareholders or proxyholders holding five percent of the issued and outstanding shares entitled to be voted at the meeting.
The foregoing is consistent with the laws, customs and practices in Canada.
| Item 16H. | Mine Safety Disclosure. |
Not applicable.
Part III
| Item 17. | Financial Statements. |
Not applicable.
| Item 18. | Financial Statements. |
Not applicable.
Exhibit
Number | | Description |
| 1.1 | | Articles of Incorporation of Transition Therapeutics Inc., as amended (incorporated by reference to Exhibit 99.110 of Form 40-F filed on June 5, 2007, File No. 001-33514). |
| | | |
| 1.2 | | Bylaws of Transition Therapeutics Inc., as amended (incorporated by reference to Exhibit 99.111 of Form 40-F filed on June 5, 2007, File No. 001-33514). |
| | | |
| 2.1 | | Form of Warrant. |
| | | |
| 4.1 | | Share Purchase Agreement, dated February 24, 2006, among Registrant, 1255205 Ontario Inc., Ellipsis Neurotherapeutics Inc., and other parties named therein (incorporated by reference to Exhibit 99.47 of Form 40-F filed on June 5, 2007, File No. 001-33514). |
| | | |
| 4.2 | | License Agreement dated May 28, 2003, by and between Ellipsis Biotherapeutics Corp. and JoAnne McLaurin, PHD, as amended on April 1, 2005. (incorporated by reference to Exhibit 99.2 of Form 6-K filed on March 17, 2008, File No. 001-33514). |
| | | |
| 4.3 | | Consent to License – JoAnne McLaurin Novel Treatment for Alzheimer’s Disease and Amyloid Disorders using Inositol-Based Compounds dated May 29, 2003, by the University of Toronto. (incorporated by reference to Exhibit 99.3 of Form 6-K filed on March 17, 2008, File No. 001-33514). |
| | | |
| 4.4 | | Consent to Assignment dated November 2, 2004, by and between JoAnne McLaurin, Ellipsis Biotherapeutics Corp. and Ellipsis Neurotherapeutics Inc. (incorporated by reference to Exhibit 99.4 of Form 6-K filed on March 17, 2008, File No. 001-33514). |
| | | |
| 4.5 | | Collaboration and License Agreement effective as of February 25, 2010, among Waratah Pharmaceuticals Inc. and Eli Lilly and Company (incorporated by reference to Exhibit 99.2 of Form 6-K filed on March 12, 2010, File No. 001-33514). |
| | | |
| 4.6 | | Collaboration and License Agreement effective as of July 23, 2013, between Transition Therapeutics Inc. and Eli Lilly and Company (incorporated by reference to Exhibit 99.2 of Form 6-K filed on July 26, 2013, File No. 001-33514). |
| | | |
| 4.7 | | Form of Subscription Agreement (Canada) dated June 23, 2014 between Transition Therapeutics Inc. and certain directors and officers of the Company. |
| | | |
| 4.8 | | Form of Subscription Agreement (U.S.) dated June 23, 2014 between Transition Therapeutics Inc. and certain shareholders of the Company. |
| | | |
| 4.9 | | Milestone, Royalty and Sublicensing Fee Deed effective as of February 27, 2014 between Transition Therapeutics Ireland Limited (formerly Elan Science Ten Limited) and Elan Pharma International Limited. |
| | | |
| 4.10 | | Purchase and Sale Agreement, dated February 28, 2014, by and among Elan Pharma International Limited, the Company and Perrigo Company PLC. |
Exhibit
Number | | Description |
| 4.11 | | License Agreement effective as of May 5, 2015 between Transition Therapeutics Inc. and Eli Lilly and Company (incorporated by reference to Exhibit 99.1 of Form 6-K filed on May 8, 2015, File No. 001-33514). |
| | | |
| 8.1 | | Subsidiaries |
| | | |
| 12.1 | | Certification of the Chief Executive Officer pursuant to Rule 13a-14(a) of the Securities Exchange Act of 1934. |
| | | |
| 12.2 | | Certification of the Chief Financial Officer pursuant to Rule 13a-14(a) of the Securities Exchange Act of 1934. |
| | | |
| 13.1 | | Section 1350 Certification of the Chief Executive Officer. |
| | | |
| 13.2 | | Section 1350 Certification of the Chief Financial Officer. |
| | | |
| 15.1 | | Consent of PricewaterhouseCoopers LLP. |
Signatures
The registrant hereby certifies that it meets all of the requirements for filing on Form 20-F and that it has duly caused and authorized the undersigned to sign this annual report on its behalf.
| | TRANSITION THERAPEUTICS INC. |
| | |
| | By | /s/ Nicole Rusaw | |
| | Name: | Nicole Rusaw | |
| | Title: | Chief Financial Officer | |
Date: September 15, 2015
MANAGEMENT’S RESPONSIBILITY FOR FINANCIAL STATEMENTS
The accompanying consolidated financial statements ofTransition Therapeutics Inc.have been prepared by management and have been approved by the Board of Directors. Management is responsible for the information and representation contained in these consolidated financial statements.
The consolidated financial statements have been prepared in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board and include some amounts that are based on best estimates and judgments.
Management, to meet its responsibility for integrity and objectivity of the data in the consolidated financial statements, has developed and maintains a system of internal accounting controls. Management believes that this system of internal accounting controls provides reasonable assurance that the financial records are reliable and form a proper basis for preparation of the consolidated financial statements, and that the assets are properly accounted for and safeguarded.
The Audit Committee reviews the consolidated financial statements, adequacy of internal controls, audit process and financial reporting with management. The Audit Committee, which consists of four directors not involved in the daily operations of the Company, reports to the Board of Directors prior to their approval of the audited consolidated financial statements for publication.
The shareholders’ auditors have full access to the Audit Committee, with and without management being present, to discuss the consolidated financial statements and to report their findings from the audit process. The consolidated financial statements have been examined by the shareholders’ independent auditors, PricewaterhouseCoopers LLP Chartered Professional Accountants, and their report is provided herein.
 |  |
| | |
| Tony Cruz | Nicole Rusaw |
| Chief Executive Officer | Chief Financial Officer |
September 11, 2015
Independent Auditor’s Report
To the Shareholders of
Transition Therapeutics Inc.
We have completed integrated audits of Transition Therapeutics Inc. and its subsidiaries’ June 30, 2015 and June 30, 2014 consolidated financial statements and their internal control over financial reporting as at June 30, 2015 and an audit of their June 30, 2013 consolidated financial statements. Our opinions, based on our audits, are presented below.
Report on the consolidated financial statements
We have audited the accompanying consolidated financial statements of Transition Therapeutics Inc. and its subsidiaries, which comprise the consolidated balance sheets as at June 30, 2015 and June 30, 2014 and the consolidated statements of income (loss) and comprehensive income (loss), shareholders’ equity and cash flows for each of the three years in the period ended June 30, 2015, and the related notes, which comprise a summary of significant accounting policies and other explanatory information.
Management’s responsibility for the consolidated financial statements
Management is responsible for the preparation and fair presentation of these consolidated financial statements in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Boardand for such internal control as management determines is necessary to enable the preparation of consolidated financial statements that are free from material misstatement, whether due to fraud or error.
Auditor’s responsibility
Our responsibility is to express an opinion on these consolidated financial statements based on our audits. We conducted our audits as at June 30, 2015 and June 30, 2014 and for each of the three years in the period ended June 30, 2015 in accordance with Canadian generally accepted auditing standards and the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free from material misstatement. Canadian generally accepted auditing standards also require that we comply with ethical requirements.
An audit involves performing procedures to obtain audit evidence, on a test basis, about the amounts and disclosures in the consolidated financial statements. The procedures selected depend on the auditor’s judgment, including the assessment of the risks of material misstatement of the consolidated financial statements, whether due to fraud or error. In making those risk assessments, the auditor considers internal control relevant to the company’s preparation and fair presentation of the consolidated financial statements in order to design audit procedures that are appropriate in the circumstances. An audit also includes evaluating the appropriateness of accounting principles and policies used and the reasonableness of accounting estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements.
We believe that the audit evidence we have obtained in our audits is sufficient and appropriate to provide a basis for our audit opinion on the consolidated financial statements.
Opinion
In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of Transition Therapeutics Inc. and its subsidiaries as at June 30, 2015 and June 30, 2014 and their financial performance and their cash flows for each of the three years in the period ended June 30, 2015 in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board.
Report on internal control over financial reporting
We have also audited Transition Therapeutics Inc. and its subsidiaries’ internal control over financial reporting as at June 30, 2015, based on criteria established in Internal Control - Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO).
Management’s responsibility for internal control over financial reporting
Management is responsible for maintaining effective internal control over financial reporting and for its assessment of the effectiveness of internal control over financial reporting appearing in the 2015 Annual Report to Shareholders in the section entitled “Item 15 - Controls and Procedures”.
Auditor’s responsibility
Our responsibility is to express an opinion on the Company’s internal control over financial reporting based on our audit. We conducted our audit of internal control over financial reporting in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether effective internal control over financial reporting was maintained in all material respects.
An audit of internal control over financial reporting includes obtaining an understanding of internal control over financial reporting, assessing the risk that a material weakness exists, testing and evaluating the design and operating effectiveness of internal control based on the assessed risk, and performing such other procedures as we consider necessary in the circumstances.
We believe that our audit provides a reasonable basis for our audit opinion on the Company’s internal control over financial reporting.
Definition of internal control over financial reporting
A company’s internal control over financial reporting is a process designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with generally accepted accounting principles. A company’s internal control over financial reporting includes those policies and procedures that: (i) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect the transactions and dispositions of the assets of the company; (ii) provide reasonable assurance that transactions are recorded as necessary to permit preparation of financial statements in accordance with generally accepted accounting principles, and that receipts and expenditures of the company are being made only in accordance with authorizations of management and directors of the company; and (iii) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use, or disposition of the company’s assets that could have a material effect on the financial statements.
Inherent limitations
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions or that the degree of compliance with the policies or procedures may deteriorate.
Opinion
In our opinion, Transition Therapeutics Inc. and its subsidiaries maintained, in all material respects, effective internal control over financial reporting as at June 30, 2015, based on criteria established in Internal Control - Integrated Framework (2013) issued by COSO.
/s/PricewaterhouseCoopers LLP
Chartered Professional Accountants, Licensed Public Accountants
Toronto, Ontario
September 14, 2015
Audited Consolidated Financial Statements
Transition Therapeutics Inc.
For the years ended June 30, 2015, 2014 and 2013
Transition Therapeutics Inc.
CONSOLIDATED BALANCE SHEETS
| In Canadian Dollars | | Note | | June 30, 2015 | | | June 30, 2014 | |
| | | | | | | | | |
| Assets | | | | | | | | | | |
| Current assets | | | | | | | | | | |
| Cash | | | | | 40,510,758 | | | | 57,212,004 | |
| Short term investments | | 5 | | | - | | | | 3,059,562 | |
| Other receivables | | | | | 265,189 | | | | 220,514 | |
| Income tax and investment tax credits receivable | | | | | 399,668 | | | | 212,393 | |
| Prepaid expenses and deposits | | | | | 259,143 | | | | 36,656 | |
| | | | | | 41,434,758 | | | | 60,741,129 | |
| | | | | | | | | | | |
| Non-current assets | | | | | | | | | | |
| Property and equipment | | | | | 191,944 | | | | 158,926 | |
| Intangible assets | | 6 | | | 8,022,383 | | | | 8,007,181 | |
| Total assets | | | | | 49,649,085 | | | | 68,907,236 | |
| | | | | | | | | | | |
| Liabilities | | | | | | | | | | |
| Current liabilities | | | | | | | | | | |
| Trade and other payables | | 7 | | | 8,549,895 | | | | 5,963,258 | |
| Contingent consideration payable | | 8 | | | 858,257 | | | | - | |
| | | | | | 9,408,152 | | | | 5,963,258 | |
| Non-current liabilities | | | | | | | | | | |
| Contingent consideration payable | | 8 | | | 3,503,344 | | | | 3,838,286 | |
| Leasehold inducement | | | | | - | | | | 11,432 | |
| Total liabilities | | | | | 12,911,496 | | | | 9,812,976 | |
| | | | | | | | | | | |
| Equity attributable to owners of the Company | | | | | | | | | | |
| Share capital | | 10 | | | 233,633,493 | | | | 207,374,493 | |
| Warrants | | 10 | | | 5,176,397 | | | | 5,176,397 | |
| Contributed surplus | | | | | 14,771,907 | | | | 14,768,221 | |
| Share-based payment reserve | | 10 | | | 5,892,305 | | | | 2,866,292 | |
| Accumulated other comprehensive income | | | | | (281,814 | ) | | | 24,028 | |
| Deficit | | | | | (222,454,699 | ) | | | (171,115,171 | ) |
| Total equity | | | | | 36,737,589 | | | | 59,094,260 | |
| | | | | | | | | | | |
| Total liabilities and equity | | | | | 49,649,085 | | | | 68,907,236 | |
| | | | | | | | | | | |
| Contingencies and commitments | | 15 | | | | | | | | |
The notes are an integral part of these consolidated financial statements
On behalf of the Board:
 |  |
| | |
| Tony Cruz | Christopher Henley |
| Director | Director |
Transition Therapeutics Inc.
CONSOLIDATED STATEMENTS OF INCOME (LOSS) AND COMPREHENSIVE INCOME (LOSS)
For the years ended June 30, 2015, 2014 and 2013
| In Canadian Dollars | | Note | | 2015 | | | 2014 | | | 2013 | |
| | | | | | | | | | | | |
| Revenues | | | | | | | | | | | | | | |
| Licensing fees | | 9 | | | - | | | | - | | | | 17,933,500 | |
| | | | | | | | | | | | | | | |
| Expenses | | | | | | | | | | | | | | |
| Research and development | | 13 | | | 49,209,703 | | | | 17,367,385 | | | | 8,862,872 | |
| Selling, general and administrative expenses | | 13 | | | 5,514,272 | | | | 4,726,574 | | | | 3,557,792 | |
| Impairment of intangible assets | | 6 | | | | | | | | | | | 6,545,821 | |
| Change in fair value of contingent consideration payable | | 8 | | | 65,787 | | | | (2,911,218 | ) | | | - | |
| Settlement of pre-existing relationship | | 8 | | | - | | | | 3,096,186 | | | | - | |
| | | | | | | | | | | | | | | |
| Operating loss | | | | | (54,789,762 | ) | | | (22,278,927 | ) | | | (1,032,985 | ) |
| Interest income | | | | | 196,073 | | | | 220,119 | | | | 146,209 | |
| Foreign exchange gain | | | | | 3,331,026 | | | | 284,523 | | | | 910,073 | |
| Loss on disposal of property and equipment | | | | | (76,865 | ) | | | (7,970 | ) | | | - | |
| Net income (loss) for the year | | | | | (51,339,528 | ) | | | (21,782,255 | ) | | | 23,297 | |
| Other comprehensive income (loss) for the year | | | | | | | | | | | | | | |
| Items that may be subsequently reclassified to net income: | | | | | | | | | | | | | | |
| Cumulative translation adjustment | | | | | (305,842 | ) | | | 24,028 | | | | - | |
| | | | | | | | | | | | | | | |
| Comprehensive income (loss) for the year | | | | | (51,645,370 | ) | | | (21,758,227 | ) | | | 23,297 | |
| Basic and diluted net income (loss) per common share | | 14 | | | (1.41 | ) | | | (0.72 | ) | | | 0.00 | |
The notes are an integral part of these consolidated financial statements.
Transition Therapeutics Inc.
CONSOLIDATED STATEMENTS OF SHAREHOLDERS’ EQUITY
For the years ended June 30, 2015, 2014 and 2013
| | | | | | | | Attributable to equity holders of the Company | | | | | | | | | | |
| In Canadian Dollars | | Note | | Number of
common
shares | | | Share
capital | | | Warrants | | | Contributed
surplus | | | Share-based
payment
reserve | | | Accumulated
Other
Comprehensive
Income | | | Deficit | | | Total equity | |
| | | | | # | | | $ | | | $ | | | $ | | | $ | | | $ | | | $ | | | $ | |
| Balance, July 1, 2014 | | | | | 35,303,913 | | | | 207,374,493 | | | | 5,176,397 | | | | 14,768,221 | | | | 2,866,292 | | | | 24,028 | | | | (171,115,171 | ) | | | 59,094,260 | |
| Net loss for the year | | | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | (51,339,528 | ) | | | (51,339,528 | ) |
| Cumulative translation adjustment | | | | | - | | | | - | | | | - | | | | - | | | | - | | | | (305,842 | ) | | | - | | | | (305,842 | ) |
| Issued pursuant to public offering, net | | 10 | | | 3,538,461 | | | | 26,069,390 | | | | - | | | | - | | | | - | | | | - | | | | - | | | | 26,069,390 | |
| Share options exercised, expired or cancelled | | 10 | | | 36,505 | | | | 189,610 | | | | - | | | | 3,686 | | | | (81,524 | ) | | | - | | | | - | | | | 111,772 | |
| Share-based payment compensation expense | | 10 | | | - | | | | - | | | | - | | | | - | | | | 3,107,537 | | | | - | | | | - | | | | 3,107,537 | |
| Balance, June 30, 2015 | | | | | 38,878,879 | | | | 233,633,493 | | | | 5,176,397 | | | | 14,771,907 | | | | 5,892,305 | | | | (281,814 | ) | | | (222,454,699 | ) | | | 36,737,589 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance, July 1, 2013 | | | | | 26,930,634 | | | | 165,367,524 | | | | - | | | | 14,768,002 | | | | 2,352,002 | | | | - | | | | (149,332,916 | ) | | | 33,154,612 | |
| Net loss for the year | | | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | (21,782,255 | ) | | | (21,782,255 | ) |
| Cumulative translation adjustment | | | | | - | | | | - | | | | - | | | | - | | | | - | | | | 24,028 | | | | - | | | | 24,028 | |
| Issued pursuant to private placements, net | | 10 | | | 8,076,427 | | | | 40,317,595 | | | | 5,176,397 | | | | - | | | | - | | | | - | | | | - | | | | 45,493,992 | |
| Share options exercised, expired or cancelled | | 10 | | | 296,852 | | | | 1,689,374 | | | | - | | | | 219 | | | | (623,836 | ) | | | - | | | | - | | | | 1,065,757 | |
| Share-based payment compensation expense | | 10 | | | - | | | | - | | | | - | | | | - | | | | 1,138,126 | | | | - | | | | - | | | | 1,138,126 | |
| Balance, June 30, 2014 | | | | | 35,303,913 | | | | 207,374,493 | | | | 5,176,397 | | | | 14,768,221 | | | | 2,866,292 | | | | 24,028 | | | | (171,115,171 | ) | | | 59,094,260 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| Balance, July 1, 2012 | | | | | 26,921,302 | | | | 165,334,259 | | | | - | | | | 13,168,411 | | | | 2,977,032 | | | | - | | | | (149,356,213 | ) | | | 32,123,489 | |
| Net income and comprehensive income for the year | | | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | 23,297 | | | | 23,297 | |
| Share options exercised, expired or cancelled | | 10 | | | 9,332 | | | | 33,265 | | | | - | | | | 1,599,591 | | | | (1,613,259 | ) | | | - | | | | - | | | | 19,597 | |
| Share-based payment compensation expense | | 10 | | | - | | | | - | | | | - | | | | - | | | | 988,229 | | | | - | | | | - | | | | 988,229 | |
| Balance, June 30, 2013 | | | | | 26,930,634 | | | | 165,367,524 | | | | - | | | | 14,768,002 | | | | 2,352,002 | | | | - | | | | (149,332,916 | ) | | | 33,154,612 | |
The notes are an integral part of these consolidated financial statements.
Transition Therapeutics Inc.
CONSOLIDATED STATEMENTS OF CASH FLOWS
For the years ended June 30, 2015, 2014 and 2013
| In Canadian Dollars | | Note | | 2015 | | | 2014 | | | 2013 | |
| | | | | | | | | | | | |
| Cash flows from (used in) operating activities | | | | | | | | | | | | | | |
| Net income (loss) for the year | | | | | (51,339,528 | ) | | | (21,782,255 | ) | | | 23,297 | |
| Adjustments for: | | | | | | | | | | | | | | |
| Change in fair value of contingent consideration payable | | | | | 65,787 | | | | (2,911,218 | ) | | | - | |
| Settlement of a pre-existing relationship | | | | | - | | | | 3,096,186 | | | | - | |
| Depreciation and amortization | | | | | 652,253 | | | | 946,897 | | | | 1,820,101 | |
| Share-based payment compensation expense | | | | | 3,107,537 | | | | 1,138,126 | | | | 988,229 | |
| Loss on disposal of property and equipment | | | | | 76,865 | | | | 7,970 | | | | - | |
| Accrued interest | | | | | 34,562 | | | | 5,140 | | | | 524 | |
| Unrealized foreign exchange gain | | | | | (1,774,842 | ) | | | (491,535 | ) | | | (410,226 | ) |
| Change in working capital | | 16 | | | 1,373,886 | | | | 5,257,439 | | | | (278,479 | ) |
| Impairment of intangible assets | | | | | - | | | | - | | | | 6,545,821 | |
| Net cash used in operating activities | | | | | (47,803,480 | ) | | | (14,733,250 | ) | | | 8,689,267 | |
| | | | | | | | | | | | | | | |
| Cash flows from (used in) investing activities | | | | | | | | | | | | | | |
| Maturity of short term investments | | | | | 3,025,000 | | | | 5,018,000 | | | | 9,023,910 | |
| Purchase of short term investments | | | | | - | | | | (3,025,000 | ) | | | (8,024,872 | ) |
| Purchase of intangible assets | | | | | (624,500 | ) | | | - | | | | - | |
| Purchase of property and equipment | | | | | (164,270 | ) | | | (34,697 | ) | | | (10,772 | ) |
| Proceeds on disposal of property and equipment | | | | | - | | | | 9,000 | | | | 5,500 | |
| Net cash provided by investing activities | | | | | 2,236,230 | | | | 1,967,303 | | | | 993,766 | |
| | | | | | | | | | | | | | | |
| Cash flows from financing activities | | | | | | | | | | | | | | |
| Net proceeds from issuance of common shares and warrants | | 10 | | | 26,069,390 | | | | 45,493,992 | | | | - | |
| Net proceeds from exercise of options | | | | | 111,772 | | | | 1,065,757 | | | | 19,597 | |
| Net cash provided by financing activities | | | | | 26,181,162 | | | | 46,559,749 | | | | 19,597 | |
| | | | | | | | | | | | | | | |
| Foreign exchange gains on cash | | | | | 2,684,842 | | | | 350,265 | | | | 410,226 | |
| | | | | | | | | | | | | | | |
| Net increase (decrease) in cash | | | | | (16,701,246 | ) | | | 34,144,067 | | | | 10,112,856 | |
| Cash and cash equivalents, beginning of year | | | | | 57,212,004 | | | | 23,067,937 | | | | 12,955,081 | |
| Cash at end of year | | | | | 40,510,758 | | | | 57,212,004 | | | | 23,067,937 | |
The notes are an integral part of these consolidated financial statements.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
| 1. | GENERAL INFORMATION AND NATURE OF OPERATIONS |
Transition Therapeutics Inc. and its subsidiaries (together the Company or Transition) was incorporated by Articles of Incorporation under the Business Corporations Act (Ontario) on July 6, 1998. The Company is a public company with common shares listed on both the NASDAQ and Toronto Stock Exchange and is incorporated and domiciled in Canada. The address of its registered office is 101 College Street, Suite 220, Toronto, Ontario, Canada.
The Company is a product-focused biopharmaceutical company developing therapeutics for disease indications with large markets. The Company’s lead technologies are focused on the treatment of agitation and aggression in Alzheimer’s disease and diabetes.
The success of the Company is dependent on bringing its products to market, obtaining the necessary regulatory approvals and achieving future profitable operations. The continuation of the research and development activities and the commercialization of its products are dependent on the Company’s ability to successfully complete these activities and to obtain adequate financing through a combination of financing activities and operations. It is not possible to predict either the outcome of future research and development programs or the Company’s ability to fund these programs going forward.
| 2. | SUMMARY OF SIGNIFICANT ACCOUNTING POLICIES |
The principal accounting policies applied in the preparation of these consolidated financial statements are set out below. These policies have been consistently applied to all periods presented.
These consolidated financial statements have been prepared in accordance with International Financial Reporting Standards (IFRS) as issued by the International Accounting Standards Board (IASB). The consolidated financial statements have been prepared using the historical cost convention except for the revaluation of contingent consideration payable to fair value.
The preparation of financial statements in conformity with IFRS requires use of certain critical accounting estimates. It also requires management to exercise its judgment in the process of applying the Company’s accounting policies. The areas involving a higher degree of judgment or complexity, or areas where assumptions and estimates are significant to the consolidated financial statements are disclosed in Note 3.
The consolidated financial statements were approved for issuance by the Board of Directors on September 11, 2015.
These consolidated financial statements incorporate the assets and liabilities of Transition and its wholly owned subsidiaries: Transition Therapeutics Leaseholds Inc., Waratah Pharmaceuticals Inc., Transition Therapeutics (USA) Inc. and Transition Therapeutics Ireland Limited. Intercompany transactions, balances and unrealized gains/losses on transactions between group companies are eliminated.
Subsidiaries are all those entities over which the Company has power over the investee, is exposed or has rights to variable returns from its involvement with the investee and has the ability to affect those returns through its power over the investee. Subsidiaries are fully consolidated from the date on which control is transferred to the Company and de-consolidated from the date that control ceases.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
The purchase method of accounting is used to account for the acquisition of subsidiaries. The cost of an acquisition is measured as the fair value of the assets given, equity instruments issued and liabilities assumed at the date of exchange. Identifiable assets acquired and liabilities and contingent liabilities assumed in a business combination are measured initially at their fair values at the acquisition date, irrespective of the extent of any minority interest. The excess of the cost of acquisition over the fair value of the Company’s share of the identifiable net assets acquired is recorded as goodwill. If the cost of acquisition is less than the fair value of the net assets of the subsidiary acquired, the difference is recognized directly in the consolidated statement of comprehensive income (loss).
| 2.3 | Foreign currency translation |
| (i) | Functional and presentation currency |
Items included in the consolidated financial statements of each entity of the Company are measured using the currency of the primary economic environment in which the entity operates (the functional currency). These consolidated financial statements are presented in Canadian dollars, which is the Company’s functional currency.
The Company has determined that its foreign operations located in the United States and Ireland have a functional currency of U.S. dollars. Consequently, revenue and expenses of these foreign operations are recorded using the rate of exchange in effect at the dates of the transactions and the translation of assets and liabilities uses the rates of exchange in effect at the period-end date, with the resulting net unrealized gains and losses arising from the translation of these foreign operations included as part of the currency translation adjustment in other comprehensive income (loss).
| (ii) | Transactions and balances |
Foreign currency transactions are translated into the functional currency using the rate of exchange in effect at the dates of the transactions. Foreign exchange gains and losses arising from translating monetary foreign currency balances are included in foreign exchange gain.
| 2.4 | Property and equipment |
Property and equipment is recorded at historical cost less depreciation. Historical cost includes expenditures that are directly attributable to the acquisition of the asset. Subsequent costs are included in the asset’s carrying amount or recognized as a separate asset, as appropriate, only when it is probable that future economic benefits associated with the item will flow to the Company and the cost of the item can be measured reliably. The carrying amount of a replaced asset is derecognized when it is replaced. Repairs and maintenance costs are charged to the consolidated statement of comprehensive income (loss) during the period in which they are incurred. Depreciation of property and equipment is calculated using either the straight-line or diminishing balance methods to allocate the cost of each item over its estimated useful life, as follows:
| Asset class | | Percentage | | Method |
| Computer equipment | | 30% - 45% | | Diminishing balance |
| Office equipment and furniture | | 20% | | Diminishing balance |
| Laboratory equipment | | 20% | | Diminishing balance |
| Leasehold improvements | | Term of lease plus one renewal period | | Straight-line |
The assets’ residual values and useful lives are reviewed, and adjusted if appropriate, at the end of each reporting period.
On disposal of items of property and equipment, the cost and related accumulated depreciation and impairments are removed from the consolidated balance sheet and the net amount, less any proceeds, is taken to the consolidated statement of comprehensive income (loss).
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
Intangible assets consist of intellectual property in the form of technology, patents, licenses and compounds. Separately acquired intangible assets are recorded at historical cost. Intangible assets acquired in a business combination are recognized at fair value at the acquisition date. All intangible assets have a finite useful life and are carried at cost less accumulated amortization. Amortization is calculated using the straight-line method to allocate the cost of the intangible assets over their estimated useful lives of up to 20 years.
| 2.6 | Impairment of non-financial assets |
Property and equipment and intangible assets that are subject to amortization or depreciation are reviewed for impairment whenever events or changes in circumstances indicate that the carrying amount may not be recoverable. An impairment loss is recognized for the amount by which the asset’s carrying amount exceeds its recoverable amount. The recoverable amount is the higher of an asset’s fair value less costs to sell and value in use. For the purposes of assessing impairment, assets are grouped at the lowest levels for which there are separately identifiable cash flows (cash generating units). Non-financial assets that suffered impairment are reviewed for possible reversal of the impairment at each reporting date.
| 2.7 | Financial Instruments: Classification and Measurement |
IFRS 9 was issued in November, 2009 and replaces parts of IAS 39 that relate to the classification and measurement of financial assets. IFRS 9 (2009) requires financial assets to be classified into two measurement categories: those measured at fair value and those measured at amortized cost. The classification depends on the purpose for which the financial assets were acquired. Management determines the classification of its financial assets at initial recognition. The Company has adopted IFRS 9 from July 1, 2010 as well as the related consequential amendments to other IFRSs, because this new accounting policy provides reliable and more relevant information for users to assess the amounts, timing and uncertainty of future cash flows.
The Company has assessed the financial assets held by the Company at July 1, 2010, the date of initial application of IFRS 9. Financial assets and liabilities are recognized when the Company becomes a party to the contractual provisions of the instrument. Financial assets are derecognized when the rights to receive cash flows from the assets have expired or have been transferred and the Company has transferred substantially all risks and rewards of ownership.
Financial assets and liabilities are offset and the net amount is reported in the balance sheet when there is a legally enforceable right to offset the recognized amounts and there is an intention to settle on a net basis, or realize the asset and settle the liability simultaneously.
Financial assets measured at amortized cost
Cash and cash equivalents, short term investments and trade and other receivables meet the requirements of IFRS 9 (2009) and are measured at amortized cost as these assets are non-derivative financial assets with fixed or determinable payments that are not quoted in an active market and have fixed maturities that the Company intends to hold until maturity. They are included in current assets, except for maturities greater than 12 months after the end of the reporting period. These are classified as non-current assets.
Financial liabilities measured at fair value
The Company’s contingent consideration payable is measured at fair value at each reporting period with changes in the fair value being recorded in the consolidated statement of comprehensive income (loss). The estimate of fair value is based on management’s best estimate of the timing and probability of having to make the contingent payments, discounted at the Company’s weighted average cost of capital.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
Fair Value Hierarchy
The Company categorizes its financial assets and liabilities that are recognized at fair value in the consolidated financial statements into one of three different levels. The hierarchy prioritizes the inputs into three levels based on the extent to which inputs used in measuring fair value are observable in the market. Each fair value measurement is reported in one of the three levels, which is determined by the lowest level input that is significant to the fair value measurement in its entirety. These levels are:
Level 1 – inputs are based upon unadjusted quoted prices for identical instruments traded in active markets;
Level 2 – inputs are based upon quoted prices for similar instruments in active markets, quoted prices for identical or similar instruments in markets that are not active, and model-based valuation techniques for which all significant assumptions are observable in the market or can be corroborated by observable market data for substantially the full term of the assets or liabilities;
Level 3 – inputs are generally unobservable and typically reflect management’s estimates of assumptions that market participants would use in pricing the asset or liability. The fair values are therefore determined using model-based techniques that include option pricing models, discounted cash flow models, and similar techniques.
| 2.8 | Impairment of financial assets |
The Company assesses at the end of each reporting period whether there is objective evidence that a financial asset or group of financial assets is impaired. A financial asset or group of financial assets is impaired and impairment losses are incurred only if there is objective evidence of impairment as a result of one or more events that occurred after the initial recognition of the asset (a loss event) and that the loss event has an impact on the estimated future cash flows of the financial asset or group of financial assets that can be reliably estimated.
The amount of the loss is measured as the difference between the asset’s carrying amount and the present value of estimated future cash flows discounted at the financial asset’s original effective interest rate. The asset’s carrying amount is reduced and the amount of the loss is recognized in the consolidated statement of comprehensive income (loss).
If, in a subsequent period, the amount of the impairment loss decreases and the decrease can be related objectively to an event occurring after the impairment was recognized, the reversal of the previously recognized impairment is recognized in the consolidated statement of comprehensive income (loss).
| 2.9 | Investment tax credits |
Investment tax credits (ITCs) are accounted for as government assistance and are accrued when qualifying expenditures are made and there is reasonable assurance that the credits will be realized. Government assistance is accounted for using the cost reduction method, whereby they are netted against the related research and development expenses or capital expenditures to which they relate.
Trade and other receivables are amounts due for services performed in the ordinary course of business. If collection is expected in one year or less, they are classified as current assets. If not, they are presented as non-current assets.
Trade and other receivables are initially recognized at fair value and subsequently measured at amortized cost using the effective interest method, less provision for impairment.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
| 2.11 | Cash and cash equivalents |
Cash and cash equivalents include cash on hand, deposits held with banks and other short-term highly liquid investments with original maturities of three months or less.
Common shares are classified as equity. Incremental costs directly attributable to the issuance of new shares, warrants or options are shown in equity as a deduction, net of income tax, from the proceeds received.
| 2.13 | Trade and other payables |
Trade and other payables are obligations to pay for goods or services that have been acquired in the ordinary course of business from suppliers. Trade and other payables are classified as current liabilities if payment is due within one year or less. If not, they are presented as non-current liabilities.
| 2.14 | Current and deferred income tax |
The income tax expense for the period comprises current and deferred tax. Income tax is recognized in the consolidated statement of comprehensive income (loss) except to the extent that it relates to items recognized directly in equity, in which case the income tax is also recognized directly in equity.
The current income tax charge is calculated on the basis of the tax laws enacted or substantively enacted at the balance sheet date in the countries where the Company and its subsidiaries operate and generate taxable income. Management periodically evaluates positions taken in tax returns with respect to situations in which applicable tax regulation is subject to interpretation. It establishes provisions where appropriate on the basis of amounts expected to be paid to the tax authorities.
Deferred income tax is recognized, using the liability method, on temporary differences arising between the tax bases of assets and liabilities and their carrying amounts in the consolidated financial statements. However, the deferred income tax is not accounted for if it arises from initial recognition of an asset or liability in a transaction other than a business combination that at the time of the transaction affects either accounting, taxable profit or loss. Deferred income tax is determined using tax rates and laws that have been enacted or substantially enacted by the balance sheet date and are expected to apply when the related deferred income tax asset is realized or the deferred income tax liability is settled.
Deferred income tax assets are recognized only to the extent that it is probable that the assets can be recovered.
Deferred income tax assets and liabilities are offset when there is a legally enforceable right to offset current tax assets against current tax liabilities and when the deferred income tax assets and liabilities relate to income taxes levied by the same taxation authority on either the taxable entity or different taxable entities where there is an intention to settle the balances on a net basis.
The Company has a stock option plan which is an equity settled, share-based payment compensation plan, under which the Company receives services from employees or consultants as consideration for equity instruments of the Company. The stock option plan is open to directors, officers, employees, members of the Scientific Advisory Board and consultants of the Company. The fair value of the employees or consultants services received in exchange for the grant of the options is recognized as an expense over the service period using the graded vesting method.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
The fair value of stock options is estimated using the Black-Scholes option pricing model. This model requires the input of a number of assumptions, including expected dividend yield, expected share price volatility, expected time until exercise and risk-free interest rates. Although the assumptions used reflect management’s best estimates, they involve inherent uncertainties based on conditions outside of the Company’s control. Changes in these assumptions could significantly impact share-based payment compensation.
The share-based payment reserve, included in equity, is reduced as the options are exercised or when the options expire unexercised. If the share options are exercised, cancelled or forfeited, the amount initially recorded for the options in share-based payment reserve is credited to common shares or contributed surplus, along with the proceeds received on the exercise. If the share options expire unexercised, the amount initially recorded for the options in the share based payment reserve is credited to contributed surplus.
Revenue comprises the fair value of consideration received or receivable for the sale of services in the ordinary course of the Company’s activities. The Company recognizes revenue when the amount of revenue can be reliably measured, it is probable that future economic benefits will flow to the entity and when specific criteria have been met for each of the Company’s activities as described below.
The Company generally enters into two types of revenue producing arrangements with pharmaceutical companies: licensing arrangements and collaboration / co-development arrangements (“collaborations”).
Licensing arrangements
Under a licensing arrangement the Company transfers the rights of a compound or series of compounds to a counterparty who directs the development, manufacture and commercialization of the product. The Company’s additional involvement is limited to involvement in a joint steering committee which the Company generally considers protective in nature. In return, the Company will generally receive an upfront fee, additional payments based on specifically defined developmental, regulatory, and commercial milestones, and a royalty based on a percentage of future sales of the product.
Revenue related to up-front payments received in licensing arrangements are deferred and amortized into income over the estimated term of the arrangement. Revenue from milestone payments are recognized when the milestones are achieved.
Collaboration arrangements
Under a collaboration arrangement the Company participates in the development by paying a fixed share of the development and commercialization costs in return for a fixed percentage of the product’s future profits. For contributing rights to the intellectual property the co-collaborator will pay the Company an upfront fee and additional payments based on specifically defined developmental and regulatory milestones. Collaboration agreements generally require the Company to participate in joint steering committees and to participate actively in the research and development of the product.
The Company accounts for collaboration arrangements using the percentage of completion model. Under this method, revenue is recorded as related costs are incurred, on the basis of the proportion of actual costs incurred to date, related to the estimated total costs to be incurred under the arrangement. The cumulative impact of any revisions in cost and earnings estimates are reflected in the period in which the need for a revision becomes known. In the event that there are significant uncertainties with respect to the outcome of the contract, the Company uses a zero profit model whereby revenue will be recognized equal to direct costs incurred, but not in excess of cash received or receivable. Losses on these contracts are recorded in the period in which management has determined that a loss is expected.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
The Company uses an input based measure, primarily direct costs or other appropriate inputs, to determine the percent complete because the Company believes that the inputs are representative of the value being conveyed through the research and development activities. The Company believes that using direct costs as the unit of measure of percentage complete also most closely reflects the level of effort related to the Company's performance under the arrangement. Direct costs are those costs that directly result in the culmination of an earnings process for which the counterparty to the arrangement receives a direct benefit. The nature of these costs are third party and internal costs associated with conducting clinical trial activities, allocated payroll related costs for representatives participating on the joint steering committee and sales and marketing costs during the co-commercialization period. Direct costs specifically exclude costs that are of a general and administrative nature.
Amounts resulting from payments received in advance of revenue recognized are recorded as deferred revenue.
The Company is required to assess the profitability of the overall arrangement on a periodic basis throughout the life of the arrangement when events or circumstances indicate a potential change in facts. Such assessment is based on estimates to determine the most likely outcome based on available facts and circumstances at each assessment date. The estimates include the consideration of factors such as the progress and timing of clinical trials, competition in the market, the development progress of other potential competitive therapies, drug related serious adverse events and other safety issues in the clinical trials, pricing reimbursement in relevant markets and historical costs incurred compared to original estimates. When the periodic assessment or other events or circumstances indicate a loss will result from performance under the arrangement, the entire amount of the loss is charged to the statement of comprehensive consolidated income (loss) in the period in which the determination is made.
| 2.17 | Research and development |
Research and development expenses include salaries, share-based payments, clinical trial costs, manufacturing and research inventory. Research and development expenditure is charged to the consolidated statement of comprehensive income (loss) in the period in which it is incurred. Development expenditure is capitalized when the criteria for recognizing an asset are met.
Research inventories
Inventories consist of materials that are used in future studies and clinical trials, and are measured at the lower of cost and net realizable value. Net realizable value is measured at the estimated selling price of the inventory less estimated costs of completion and estimated costs to make the sale. The amount of the write-down of inventories is included in research and development expense in the period the loss occurs, which is currently at the time the inventory is acquired since the Company does not intend to sell the material used in studies and clinical trials.
Leases in which a significant portion of the risks and rewards of ownership are retained by the lessor are classified as operating leases. Payments made under operating leases are expensed on a straight-line basis over the term of the lease.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
| 2.19 | IFRS issued but not yet adopted |
IFRS 15 – Revenue from Contracts with Customers
IFRS 15 specifies how and when to recognize revenue as well as requiring entities to provide users of financial statements with some informative, relevant disclosures. The standard supersedes IAS 18, Revenue, IAS 11, Construction Contracts, and a number of revenue-related interpretations. Application of the standard is mandatory for all IFRS reporters and it applies to nearly all contracts with customers: the main exceptions are leases, financial instruments and insurance contracts. Currently IFRS 15 must be applied in an entity’s first annual IFRS financial statements for periods beginning on or after January 1, 2017, however the IASB has proposed to defer the date of adoption to periods beginning on or after January 1, 2018, with early adoption permitted. Management is evaluating the standard and has not yet determined the impact on its consolidated financial statements.
| 3. | CRITICAL ACCOUNTING ESTIMATES AND JUDGMENTS |
The preparation of the consolidated financial statements in accordance with IFRS requires management to make estimates and assumptions that affect the reported amounts of assets, liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results can differ from those estimates. We have identified the following areas which we believe require management’s most subjective estimates and judgments, often requiring the need to make estimates about the effects of matters are inherently uncertain and may change in subsequent periods.
Valuation and Amortization of Intangible Assets
The Company’s intangible assets are comprised of purchased or licensed pharmaceutical compounds, technology and patents. The costs of the Company’s intangible assets are amortized over the estimated useful life of up to 20 years. Factors considered in estimating the useful life of the intangible asset include the expected use of the asset by the Company, legal, regulatory and contractual provisions that may limit the useful life, the effects of competition and other economic factors, and the level of expenditures required to obtain the expected future cash flows from the intangible asset. The Company re-evaluates the useful life when there has been a change in these factors. See note 6 for additional information on charges in useful life and impairment testing. The Company assesses its intangible assets for recoverability whenever indicators of impairment exist. When the carrying value of an asset is greater than its recoverable amount, which is the higher of its value in use or fair value less costs to sell, an impairment loss is recognized.
Valuation of Contingent Consideration Payable
The contingent consideration is measured at fair value based on level 3 inputs. The contingent consideration is not based on observable inputs and is measured using a discounted cash flow analysis of expected payments in future periods. The significant estimates used in the fair value calculations are as follows:
| (a) | Management has estimated the timing of the milestone payments based on current expectations and plans for the development of ELND005. The milestone payments are assigned a probability based on industry statistics for the successful development of pharmaceutical products including regulatory approval and achievement of revenue targets. An increase of 10% applied to the probability assumptions, with all other variables held constant, will increase the contingent consideration payable by $1,428,951. Conversely a decrease of 10% applied to the probability assumptions, with all other variables held constant, would reduce the contingent consideration payable by $1,858,858; |
| (b) | The probability adjusted cash flows are discounted at a rate of 20% which is management’s best estimate of the Company’s cost of capital. An increase of 5% to the discount rate would decrease the contingent consideration payable by $1,080,299. Conversely, a decrease of 5% to the discount rate would increase the contingent consideration payable by $1,538,400. |
Management revisited the assumptions used in the valuation of the contingent consideration payable and accordingly, the Company has recognized a change in fair value of contingent consideration payable of $65,787 during the fiscal year ended June 30, 2015.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
Share Based Payments and Warrants
When the Company issues stock options and warrants, an estimate of fair value is derived for the equity instrument using the Black-Scholes option pricing model. The application of this option pricing model requires management to make assumptions regarding several variables, including the period for which the instrument will be outstanding, the price volatility of the Company’s stock over a relevant timeframe, the determination of a relevant risk free interest rate and an assumption regarding the Company’s dividend policy in the future. If other assumptions are used, the value derived for the equity instruments could be significantly impacted. Assumptions used to estimate the fair value of stock options granted and warrants issued are disclosed in notes 11 and 10, respectively.
Settlement of a Pre-Existing Relationship
The Company has determined that the transactions entered into with Perrigo Company plc on February 28, 2014 resulted in the re-acquisition of the rights for the development and commercialization of ELND005 previously licensed to Elan Pharmaceuticals plc (“Elan”) which in accordance with IFRS must be accounted for as a settlement of a pre-existing relationship (the collaboration agreement between Waratah and Elan). Accordingly, the company has expensed $3,096,186 during the year ended June 30, 2014 as the cost related to the settlement of the pre-existing relationship.
| 4. | FINANCIAL RISK MANAGEMENT |
| 4.1 | Categories of financial assets and liabilities |
All financial instruments are measured at amortized cost except for the contingent consideration payable which is at fair value. The following table outlines the Company’s financial instruments, their classification, carrying value and fair value.
| Financial Instruments as at June 30, 2015 | | Classification | | Carrying
Value ($) | | | Fair Value
($) | |
| Cash | | Loans and receivables | | | 40,510,758 | | | | 40,510,758 | |
| Other receivables | | Loans and receivables | | | 265,189 | | | | 265,189 | |
| Accounts payable and accrued liabilities | | Other liabilities | | | 8,549,895 | | | | 8,549,895 | |
| Contingent consideration payable | | Fair value through profit and loss | | | 4,361,601 | | | | 4,361,601 | |
| Financial Instruments as at June 30, 2014 | | Classification | | Carrying
Value ($) | | | Fair Value
($) | |
| Cash | | Loans and receivables | | | 57,212,004 | | | | 57,212,004 | |
| Short term investments | | Loans and receivables | | | 3,059,562 | | | | 3,059,562 | |
| Other receivables | | Loans and receivables | | | 220,574 | | | | 220,574 | |
| Accounts payable and accrued liabilities | | Other liabilities | | | 5,963,258 | | | | 5,963,258 | |
| Contingent consideration payable | | Fair value through profit and loss | | | 3,838,286 | | | | 3,838,286 | |
The Company has determined the estimated fair values of its financial instruments based on appropriate valuation methodologies; however, considerable judgment is required to develop these estimates. Fair value of short term investments is determined based on a valuation model that uses daily pricing reports to determine the amount the holder would receive if the instrument were sold on that day. The fair value of the contingent consideration payable is determined using a valuation model as discussed in note 3.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
| 4.2 | Financial risk factors |
The Company’s activities expose it to a variety of financial risks: market risk (including foreign exchange and interest rate risks), credit risk and liquidity risk. Risk management is the responsibility of the Company’s finance function which identifies, evaluates and where appropriate, mitigates financial risks.
The Company operates in Canada and has relationships with entities in other countries. Foreign exchange risk arises from purchase transactions, as well as recognized financial assets and liabilities denominated in foreign currencies, mainly the US dollar. The Company does not enter into hedging or other contracts to mitigate its exposure to foreign exchange risk and maintains sufficient US dollars to meet the Company’s planned US dollar expenses.
Financial instruments in foreign currencies at June 30, 2015 and 2014 are approximately:
| | | 2015 | | | 2014 | |
| | | US$ | | | US$ | |
| Cash | | | 30,544,014 | | | | 48,722,203 | |
| Trade and other payables | | | (102,464 | ) | | | (711,490 | ) |
| | | | 30,441,550 | | | | 48,010,713 | |
Fluctuations in the US dollar exchange rate could potentially have a significant impact on the Company’s results. At June 30, 2015, if the Canadian dollar weakened 10% against the US dollar, with all other variables held constant, comprehensive income for the year ended June 30, 2015 would have increased by approximately $1,551,000. Conversely, if the Canadian dollar strengthened 10% against the US dollar, with all other variables held constant, comprehensive income for the year ended June 30, 2015 would have decreased by approximately $1,551,000.
Interest rate risk is the risk that the future cash flows of a financial instrument will fluctuate because of changes in market interest rates. The Company’s short term investments are at a fixed rate of interest and accordingly are not exposed to changes in market interest rates, however, their fair value can vary with the change in market interest rates. The Company’s cash is exposed to changes in market interest rates. An increase (decrease) in the market interest rate of 1% would decrease (increase) net loss by $435,632 and ($196,035) respectively.
Although the Company monitors market interest rates, the Company’s investment policies are designed to maintain safety of principal and provide adequate liquidity to meet all current payment obligations and future planned expenditures. The Company does not speculate on interest rates and holds all deposits until their date of maturity.
Interest income from cash, cash equivalents and short term investments was $196,104 for the year ended June 30, 2015 (2014 - $219,273; 2013 - $144,432).
Credit risk is the risk of a financial loss to the Company if a customer or counterparty to a financial instrument fails to meet its contractual obligations.
The Company’s exposure to credit risk at the period end is the carrying value of its cash and short term investments. The Company manages credit risk by maintaining bank accounts with financial institutions of high creditworthiness. Short term investments consist of bankers’ acceptances and other debentures maturing in less than 12 months and ratings of R-1 or higher.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
Liquidity risk is the risk that the Company will not be able to meet its obligations as they become due.
The Company’s investment policies are designed to maintain safety of principal and provide sufficient readily available cash in order to meet liquidity requirements. The Company manages its liquidity risk by forecasting cash flows from operations and anticipated investing and financing activities. All cash and short term investments have maturities less than one year.
At June 30, 2015 the Company’s financial liabilities which include trade and other payables are current and are expected to be repaid within 1 to 3 months of the period end date.
The contingent consideration payable is expected to be paid as follows:
| Fiscal year ending June 30, 2016 | | $ | 2,847,759 | |
| Fiscal year ending June 30, 2021 | | $ | 3,797,096 | |
| Fiscal year ending June 30, 2022 | | $ | 16,761,664 | |
| Fiscal year ending June 30, 2023 | | $ | 18,735,000 | |
| Fiscal year ending June 30, 2024 | | $ | 18,735,000 | |
| 4.3 | Capital risk management |
The Company’s primary objective when managing capital is to ensure its ability to continue as a going concern in order to pursue the development of its drug candidates and the out-license of these drug candidates to pharmaceutical companies. The Company attempts to maximize return to shareholders by minimizing shareholder dilution and, when possible, utilizing non-dilutive arrangements such as interest income and collaborative partnership arrangements.
The Company includes equity comprised of issued share capital, warrants, contributed surplus and deficit in the definition of capital. The Company has financed its capital requirements primarily through share issuances since inception and collaborative partnership agreements.
The Company manages its capital structure and makes adjustments to it in light of changes in economic conditions and risk characteristics of the underlying assets. The Company monitors its cash requirements and market conditions to anticipate the timing of requiring additional capital to finance the development of its drug candidates. The Company is not subject to externally imposed capital requirements and there has been no change with respect to the overall capital management strategy during the year ended June 30, 2015 from the year ended June 30, 2014.
The Company’s current cash projection indicates that the current cash resources should enable the Company to execute its core business plan and meet its projected cash requirements beyond the next 12 months. However, the Company’s working capital may not be sufficient to meet its stated business objectives in the event of unforeseen circumstances or a change in the strategic direction of the Company. When, or if, the Company requires additional capital, there can be no assurance that the Company will be able to obtain further financing on favourable terms, if at all.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
At June 30, 2014, short term investments consisted of two medium term note debentures totaling $3,059,562 with ratings of R1 or higher that matured on October 25, 2014 and November 28, 2014. There were no gains or losses realized on the disposal of the short term investments during the years ended June 30, 2015 and 2014 as all the financial assets were held to their redemption date. The maximum exposure to credit risk at the reporting date is the carrying amount of cash and short term investments.
Intangible assets consist of the following:
| | | Technology
acquired
(ELND005) | | | Lilly
Licenses
acquired
(TT401/402) | | | Lilly
SARM
License
acquired
(TT701)
(note 9a) | | | Total | |
| | | $ | | | $ | | | $ | | | $ | |
| | | | | | | | | | | | | |
| As at July 1, 2014 | | | | | | | | | | | | | | | | |
| Cost | | | 20,547,993 | | | | 1,055,900 | | | | - | | | | 21,603,893 | |
| Accumulated amortization | | | (13,367,489 | ) | | | (229,223 | ) | | | - | | | | (13,596,712 | ) |
| Net book value | | | 7,180,504 | | | | 826,677 | | | | - | | | | 8,007,181 | |
| | | | | | | | | | | | | | | | | |
| As at June 30, 2015 | | | | | | | | | | | | | | | | |
| Cost | | | 20,547,993 | | | | 1,055,900 | | | | 624,500 | | | | 22,228,393 | |
| Accumulated amortization | | | (13,919,829 | ) | | | (282,019 | ) | | | (4,162 | ) | | | (14,206,010 | ) |
| Net book value June 30, 2015 | | | 6,628,164 | | | | 773,881 | | | | 620,338 | | | | 8,022,383 | |
| | | | | | | | | | | | | | | | | |
| Year ended June 30, 2015 | | | | | | | | | | | | | | | | |
| Opening net book value | | | 7,180,504 | | | | 826,677 | | | | - | | | | 8,007,181 | |
| Acquisition of intangible assets | | | - | | | | - | | | | 624,500 | | | | 624,500 | |
| Amortization charge | | | (552,340 | ) | | | (52,796 | ) | | | (4,162 | ) | | | (609,298 | ) |
| Net book value June 30, 2015 | | | 6,628,164 | | | | 773,881 | | | | 620,338 | | | | 8,022,383 | |
| | | Technology
acquired
(ELND005) | | | Lilly
Licenses
acquired
(TT401/402) | | | Total | |
| | | $ | | | $ | | | $ | |
| | | | | | | | | | |
| As at July 1, 2013 | | | | | | | | | | | | |
| Cost | | | 20,547,993 | | | | 1,055,900 | | | | 21,603,893 | |
| Accumulated amortization | | | (12,488,792 | ) | | | (176,427 | ) | | | (12,665,219 | ) |
| Net book value | | | 8,059,201 | | | | 879,473 | | | | 8,938,674 | |
| | | | | | | | | | | | | |
| As at June 30, 2014 | | | | | | | | | | | | |
| Cost | | | 20,547,993 | | | | 1,055,900 | | | | 21,603,893 | |
| Accumulated amortization | | | (13,367,489 | ) | | | (229,223 | ) | | | (13,596,712 | ) |
| Net book value June 30, 2014 | | | 7,180,504 | | | | 826,677 | | | | 8,007,181 | |
| | | | | | | | | | | | | |
| Year ended June 30, 2014 | | | | | | | | | | | | |
| Opening net book value | | | 8,059,201 | | | | 879,473 | | | | 8,938,674 | |
| Amortization charge | | | (878,697 | ) | | | (52,796 | ) | | | (931,493 | ) |
| Net book value June 30, 2014 | | | 7,180,504 | | | | 826,677 | | | | 8,007,181 | |
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
As ELND005 did not meet its primary efficacy endpoint in the Phase 2/3 clinical study in agitation and aggression in Alzheimer’s disease, management performed an impairment test and noted there is no impairment of the ELND005 asset as at June 30, 2015. The Company is performing a thorough review of the data from the completed study in agitation and aggression. An external clinical advisory board is working with the Company to evaluate the data and consider potential future clinical development paths for ELND005.
In light of the series of agreements the Company entered into with Perrigo Company plc in fiscal 2014 relating to the ELND005 technology, management reviewed the estimate of the remaining useful life of the ELND005 technology and extended the remaining useful life to 12 years. Accordingly, the change in estimate resulted in a decrease in amortization expense of $108,774 being recognized during the year ended June 30, 2014.
During the year ended June 30, 2013, the Company decided to no longer develop TT-301 and TT-302, the compounds acquired from NMX. As the Company no longer expects to receive any benefits from the technology, the Company assessed the compounds for impairment and determined that the recoverable amount of the compounds was nil at June 30, 2013. Accordingly, the Company has recognized an impairment loss of $6,545,821. The Company has terminated the licensing agreement with Northwestern University and has no further commitments relating to this technology.
The amortization of all intangible assets relates to the research and development efforts of the Company and has therefore been included in the “research and development” line in the consolidated statement of comprehensive loss.
7. TRADE AND OTHER PAYABLES
Trade and other payables consist of the following:
| | | June 30, 2015 | | | June 30, 2014 | |
| | | $ | | | $ | |
| Accounts payable | | | 2,594 | | | | 1,591,128 | |
| Accrued expenses: | | | | | | | | |
| Clinical trials and manufacturing | | | 7,769,521 | | | | 3,320,992 | |
| Salaries and benefits | | | 398,017 | | | | 224,879 | |
| Professional fees and services | | | 235,477 | | | | 628,827 | |
| Other | | | 144,286 | | | | 197,432 | |
| | | | 8,547,301 | | | | 4,372,130 | |
| | | | 8,549,895 | | | | 5,963,258 | |
| 8. | CONTINGENT CONSIDERATION PAYABLE |
| (a) | Under the terms of the ENI step-acquisition agreement, the Company is committed to pay the former shareholders of ENI contingent clinical milestones potentially totaling $10.9 million payable in cash or Transition common shares at the then market price and a royalty of up to 1% on net sales of the ELND005 product. On February 28, 2014, the Company became responsible for the development of ELND005 and accordingly has re-evaluated the development program timelines and adjusted the estimate relating to the timing of the milestone payments. Accordingly, the Company has recognized a liability as at June 30, 2015 of $1,429,884 (June 30, 2014 - $1,030,775) which represents the fair value of the contingent consideration payable to the former shareholders of ENI. |
| (b) | Under the terms of the ELND005 milestone and royalty agreement, the Company is committed to pay Perrigo contingent approval and commercialization milestones potentially totaling US$40 million and a royalty of up to 6.5% on net sales of the ELND005 product. Accordingly, the Company has recognized a liability as at June 30, 2015 of $2,931,717 (June 30, 2014 - $2,807,511) which represents the fair value of the contingent consideration payable to Perrigo. |
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
| | | Payable to
ENI | | | Payable to
Perrigo | | | Total | |
| Contingent Consideration Payable | | $ | | | $ | | | $ | |
| Balance at July 1, 2013 | | | 3,756,331 | | | | - | | | | 3,756,331 | |
| Settlement of pre-existing relationship (note 3) | | | - | | | | 3,096,186 | | | | 3,096,186 | |
| Change in contingent consideration payable | | | (2,725,556 | ) | | | (185,662 | ) | | | (2,911,218 | ) |
| Foreign exchange | | | - | | | | (103,013 | ) | | | (103,013 | ) |
| Balance at June 30, 2014 | | | 1,030,775 | | | | 2,807,511 | | | | 3,838,286 | |
| Change in contingent consideration payable | | | 399,109 | | | | (333,322 | ) | | | 65,787 | |
| Foreign exchange | | | - | | | | 457,528 | | | | 457,528 | |
| Balance at June 30, 2015 | | | 1,429,884 | | | | 2,931,717 | | | | 4,361,601 | |
Significant assumptions and the sensitivity of changes to these assumptions are discussed in Note 3.
| 9. | LICENSING AND COLLABORATION AGREEMENTS WITH ELI LILLY AND COMPANY |
| (a) | On March 3, 2010, Transition and Eli Lilly and Company (“Lilly”) entered into a licensing and collaboration agreement granting Transition the rights to a series of preclinical compounds in the area of diabetes. Under the licensing and collaboration agreement, Transition will receive exclusive worldwide rights to develop and potentially commercialize a class of compounds that, in preclinical models showed potential to provide glycemic control and other beneficial effects including weight loss. |
Under the terms of the agreement, Lilly received an up-front payment of US$1 million and retained the option to reacquire the rights to the compounds at a later date. The up-front payment of $1,055,900 (US$1 million) has been capitalized as a license acquired from Lilly and will be amortized over 20 years which represents the estimated remaining life of the underlying compounds and patents.
In June 2013, Lilly exercised their option and assumed all development and commercialization rights to type 2 diabetes drug candidate TT401. In conjunction with this assumption of rights, Transition received a milestone payment of $7,118,300 (US$7 million) which has been recognized as revenue during the year ended June 30, 2013. Lilly has assumed all costs and will perform all future development and commercialization activities of TT401. In fiscal 2015, Transition has paid US$14 million ($15,491,600) to Lilly in three separate installments during the Phase 2 clinical study. In return, Transition is eligible to receive up to approximately US$240 million in additional milestone payments and will also be eligible to receive a double-digit royalty on sales of TT401 products and a low single digit royalty on related compounds. The Company has no further funding obligations under the Agreement.
| (b) | On May 6, 2015, the Company, through its wholly owned subsidiary TTIL, exclusively licensed worldwide rights to a novel small molecule drug candidate, TT701 from Lilly. Under the terms of the agreement, TTIL has acquired the rights to develop and commercialize TT701. Transition will pay Lilly upfront consideration of up to US$1 million. As of June 30, 2015, Transition has paid Lilly $624,500 (US$500,000) of the upfront consideration and this payment has been capitalized as a license acquired from Lilly and will be amortized over the estimated remaining life of 12.5 years. The remaining upfront payment of US$500,000 is due upon first patient enrollment in a clinical trial and is expected to be paid in fiscal 2016 once the milestone is achieved. |
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
At June 30, 2015, the authorized share capital of the Company consists of an unlimited number of no par value common shares. The common shares are voting and are entitled to dividends if, as and when declared by the Board of Directors.
| [b] | Common shares issued and outstanding during the period |
On February 18, 2015, the Company announced the closing of its underwritten public offering of an aggregate of 3,538,461 common shares at a price to the public of US$6.50 per share, including 461,538 common shares issued upon the exercise of the underwriters’ over-allotment option, raising gross proceeds of $28,561,400 (US$23.0 million). The Company incurred total share issuance costs of $2,492,010, resulting in net cash proceeds of $26,069,390.
On June 23, 2014, the Company announced the closing of its private placement financing issuing 3,195,487 units of the Company to existing shareholders, board members and management at a price of US$5.32 per unit, raising gross proceeds of $18,319,000 (US$17.0 million). Each unit consists of one common share and 0.61 Common Share purchase warrant with a purchase price of US$7.10 per whole warrant. The Company incurred total share issuance costs of $106,000, resulting in net cash proceeds of approximately $18,213,000.
On February 28, 2014, the Company issued 2,255,640 common shares to a subsidiary of Perrigo for gross proceeds of $16,422,000 (US$15.0 million). The Company incurred total share issuance costs of $59,000, resulting in net cash proceeds of approximately $16,363,000.
On August 15, 2013, the Company announced the closing of its private placement financing issuing 2,625,300 units of the Company to existing shareholders, board members and management at a price of US$4.19 per unit, raising gross proceeds of $11,439,000 (US$11.0 million). Each unit consists of (i) one common share, (ii) 0.325 Common Share purchase warrant with a purchase price of US$4.60 per whole warrant and (iii) 0.4 Common Share purchase warrant with a purchase price of US$6.50 per whole warrant. The Company incurred total share issuance costs of $521,000, resulting in net cash proceeds of approximately $10,918,000.
At June 30, 2015, there were 38,878,879 common shares issued and outstanding [June 30, 2014 – 35,303,913].
Warrants
Details of whole warrants outstanding at June 30, 2015 are as follows:
| | | | | | Fair
Value at Date
of Issuance | | | Expiry Date |
| Warrants | | # | | | $ | | | |
| US$4.60 Warrants issued at August 15, 2013 | | | 853,223 | | | | 1,108,107 | | | August 15, 2015 |
| US$6.50 Warrants issued August 15, 2013 | | | 1,050,118 | | | | 917,732 | | | August 15, 2015 |
| US$7.10 Warrants issued June 23, 2014 | | | 1,949,250 | | | | 3,150,558 | | | June 23, 2016 |
| Warrants outstanding June 30, 2015 and 2014 | | | 3,852,591 | | | | 5,176,397 | | | |
The outstanding warrants at June 30, 2015 have a total fair value at date of issuance of $5,176,397 which was calculated using the Black-Scholes pricing model with the following assumptions:
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
| Warrants Issued: | | August 15, 2013 | | | June 23, 2014 | |
| | | | | | | |
| Risk free interest rate | | | 1.18 | % | | | 1.03 | % |
| Expected dividend yield | | | 0 | % | | | 0 | % |
| Stock price volatility | | | 0.6348 | | | | 0.6694 | |
| Expected life of warrants | | | 2.0 years | | | | 2.0 years | |
Subsequent to year end, the warrants issued on August 15, 2013 expired unexercised and accordingly, the carrying value of the expired warrants of $2,025,839 will be reclassified to contributed surplus during the three month period ending September 30, 2015.
If and when all of the remaining warrants are exercised, the Company may realize up to an additional US$13.8 million in proceeds.
| | | | | | | | | Weighted Average
Exercise Price | |
| Stock options | | # | | | $ | | | $ | |
| Stock options outstanding, July 1, 2014 | | | 2,305,589 | | | | 2,866,292 | | | | 3.91 | |
| Stock options issued [i] | | | 518,500 | | | | - | | | | 8.80 | |
| Stock options exercised [ii] | | | (36,505 | ) | | | (77,838 | ) | | | 3.07 | |
| Stock options expired [iii] | | | (832 | ) | | | (3,686 | ) | | | 6.00 | |
| Stock options forfeited or cancelled [iv] | | | (30,988 | ) | | | - | | | | 5.75 | |
| Stock based compensation expense | | | - | | | | 3,107,537 | | | | - | |
| Stock options outstanding, June 30, 2015 | | | 2,755,764 | | | | 5,892,305 | | | | 4.82 | |
| | | | | | | | | Weighted Average
Exercise Price | |
| Stock options | | # | | | $ | | | $ | |
| Stock options outstanding, July 1, 2013 | | | 1,872,000 | | | | 2,352,002 | | | | 2.97 | |
| Stock options issued [i] | | | 742,000 | | | | - | | | | 6.12 | |
| Stock options exercised [ii] | | | (296,852 | ) | | | (623,617 | ) | | | 3.59 | |
| Stock options forfeited or cancelled [iv] | | | (11,559 | ) | | | (219 | ) | | | 2.82 | |
| Stock based compensation expense | | | - | | | | 1,138,126 | | | | - | |
| Stock options outstanding, June 30, 2014 | | | 2,305,589 | | | | 2,866,292 | | | | 3.91 | |
| | | | | | | | | Weighted Average
Exercise Price | |
| Stock options | | # | | | $ | | | $ | |
| Stock options outstanding, July 1, 2012 | | | 1,949,919 | | | | 2,977,032 | | | | 4.10 | |
| Stock options issued [i] | | | 325,000 | | | | - | | | | 3.64 | |
| Stock options exercised [ii] | | | (9,332 | ) | | | (13,668 | ) | | | 2.10 | |
| Stock options expired [iii] | | | (210,920 | ) | | | (1,190,334 | ) | | | 13.62 | |
| Stock options forfeited or cancelled [iv] | | | (182,667 | ) | | | (409,257 | ) | | | 3.56 | |
| Stock based compensation expense | | | - | | | | 988,229 | | | | - | |
| Stock options outstanding, June 30, 2013 | | | 1,872,000 | | | | 2,352,002 | | | | 2.97 | |
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
| [i] | The fair value of the stock options issued during the year ended June 30, 2015 was $3,211,700 [2014 - $3,346,000; 2013 - $853,800]. |
| [ii] | During the year ended June 30, 2015, 36,505 stock options were exercised. These stock options had a fair value of $77,838 at the grant date and resulted in cash proceeds to the Company of $111,772. |
| | | During the year ended June 30, 2014, 296,852 stock options were exercised. These stock options had a fair value of $623,617 at the grant date and resulted in cash proceeds to the Company of $1,065,757. |
| | | During the year ended June 30, 2013, 9,332 stock options were exercised. These stock options had a fair value of $13,668 at the grant date and resulted in cash proceeds to the Company of $19,597. |
| [iii] | During the year ended June 30, 2015, 832 stock options expired unexercised. These stock options had a fair value of $3,686 which was reclassified to contributed surplus. No stock options expired unexercised during the year ended June 30, 2014. |
| | | During the year ended June 30, 2013, 210,920 stock options expired unexercised. These stock options had a fair value of $1,190,334 which has been reclassified to contributed surplus. |
| [iv] | During the year ended June 30, 2015, 30,988 stock options were forfeited or cancelled. These options had a fair value of $131,363 and were unvested at the date of forfeit. |
| | | In the year ended June 30, 2014, 11,559 stock options were forfeited or cancelled, of which 83 were fully vested. The vested options had a fair value of $219 which has been reclassified to contributed surplus. |
| | | During the year ended June 30, 2013, 182,667 stock options were forfeited or cancelled, of which 178,000 were fully vested. The vested options had a fair value of $409,257 which has been reclassified to contributed surplus. |
| [v] | The maximum possible cash proceeds to the Company from the exercise of the stock options outstanding at June 30, 2015 are $13,274,428 [June 30, 2014 - $9,005,578]. |
| 11. | STOCK-BASED COMPENSATION PLANS |
The Company’s stock option plan is designed to attract and retain key individuals and recognize individual and overall corporate performance. In terms of performance, the Company’s policy is to establish annual goals with respect to business strategy and the individual’s area of direct responsibility. The Company grants options to its employees at the time when they join the organization and then subsequent grants are issued at the discretion of the Board of Directors. Grants issued are based on the level of the position that the employee is hired for and their overall experience and subsequent grants are based on the level of position, the Company’s performance, and the employee’s performance. Stock option grants are approved by the Board of Directors. The Board of Directors considers the amount and the terms of outstanding options when determining whether and how many new option grants will be made.
Options granted to employees generally vest monthly or annually over a 3 to 4 year period. The exercise price of the options is equal to the greater of (1) the closing price the day prior to the grant; (2) the weighted average trading price for five trading days prior to grant; and (3) the price determined by the Board of Directors at the time of the grant. All grants expire 10 years after the grant date or generally terminate 3 to 6 months after the employee leaves the Company depending on the circumstances of their departure.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
The fair value of each option award is estimated on the date of the grant using the Black-Scholes option pricing model. The expected volatilities have been computed based on trailing 8 year historical share price trading data of week ending closing prices. The risk-free rate is based on the 8 year Government of Canada marketable bond rates in effect at the time of the grants. The expected life of the option is estimated to be 8 years based on historical option exercising patterns.
All stock options granted under the Plan must be exercised within a maximum period of ten years following the grant date thereof (5 years for options granted prior to December 7, 2010). Options expiring during a blackout period are extended until 10 trading days after the blackout period is lifted. The maximum number of common shares that may be issued pursuant to stock options granted under the Plan shall not exceed 10% of the issued and outstanding common shares.
As at June 30, 2015, there are 1,132,124 options available for issuance under the Plan. The maximum number of common shares that may be issued to any individual pursuant to stock options granted under the Plan will not exceed 5% of the outstanding common shares and the total number of common shares that may be issued to consultants pursuant to stock options granted under the Plan will not exceed 2% of the issued and outstanding common shares in any twelve month period. The vesting period is determined at the time of each option grant but must not exceed five years.
A summary of options outstanding as at June 30, 2015 under the plans are presented below:
| Outstanding | | | Exercisable | |
Range of
exercise
prices
$ | | Number of
options
# | | | Weighted
average
remaining
contractual
life
[years] | | | Weighted
average
exercise price
$ | | | Number of
options
# | | | Weighted
average
remaining
contractual
life
[years] | | | Weighted
average
exercise price
$ | |
| 2.09-3.00 | | | 702,601 | | | 6.85 | | | 2.18 | | | | 702,601 | | | 6.85 | | | 2.18 | |
| 3.22-3.66 | | | 824,663 | | | 5.47 | | | 3.44 | | | | 739,951 | | | 5.18 | | | 3.41 | |
| 6.00-7.70 | | | 755,000 | | | 8.98 | | | 6.21 | | | | 269,322 | | | 8.99 | | | 6.27 | |
| 8.73-10.19 | | | 473,500 | | | 9.79 | | | 8.93 | | | | 34,069 | | | 9.76 | | | 8.73 | |
| | | | 2,755,764 | | | | | | | | | | 1,745,943 | | | | | | | |
A summary of options outstanding as at June 30, 2014 under the plans are presented below:
| Outstanding | | | Exercisable | |
Range of
exercise
prices
$ | | Number of
options
# | | | Weighted
average
remaining
contractual
life
[years] | | | Weighted
average exercise
price
$ | | | Number of
options
# | | | Weighted
average
remaining
contractual
life
[years] | | | Weighted
average
exercise price
$ | |
| 2.09-2.10 | | | 658,174 | | | 7.93 | | | 2.10 | | | | 455,544 | | | 7.94 | | | 2.10 | |
| 3.00-3.22 | | | 403,187 | | | 6.92 | | | 3.19 | | | | 315,610 | | | 6.93 | | | 3.18 | |
| 3.42-3.66 | | | 502,228 | | | 6.08 | | | 3.58 | | | | 299,524 | | | 4.11 | | | 3.54 | |
| 6.00-7.67 | | | 742,000 | | | 9.96 | | | 6.12 | | | | - | | | - | | | - | |
| | | | 2,305,589 | | | | | | | | | | 1,070,678 | | | | | | | |
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
A summary of options outstanding as at June 30, 2013 under the plans are presented below:
| Outstanding | | Exercisable |
Range of
exercise
prices
$ | | Number of
options
# | | | Weighted
average
remaining
contractual
life
[years] | | Weighted
average exercise
price
$ | | Number of
options
# | | | Weighted
average
remaining
contractual
life
[years] | | Weighted
average
exercise price
$ |
| 2.09-2.10 | | | 730,000 | | | 8.93 | | 2.10 | | | 267,403 | | | 8.93 | | 2.10 |
| 3.00-3.22 | | | 442,000 | | | 7.92 | | 3.19 | | | 258,909 | | | 7.93 | | 3.17 |
| 3.42-3.66 | | | 510,000 | | | 7.12 | | 3.58 | | | 151,232 | | | 2.09 | | 3.48 |
| 4.15-4.29 | | | 190,000 | | | 0.97 | | 4.18 | | | 190,000 | | | 0.97 | | 4.18 |
| | | | 1,872,000 | | | | | | | | 867,544 | | | | | |
For the year ended June 30, 2015, total stock based compensation expense was $3,107,537 [2014 - $1,138,126; 2013 - $988,229], split between general and administrative expense of $1,329,909 [2014 - $636,531; 2013 - $579, 291] and research and development of $1,777,628 [2014 - $501,595; 2013 - $408,938].
The fair value of options granted during fiscal 2015 is $3,211,700 [2014 - $3,346,000; 2013 - $853,800]. The fair value of the options at the date of grant for the year ended June 30, 2015 was estimated using the Black-Scholes option pricing model based on the following assumptions: expected option life of 8 years [2014 and 2013 - 8 years], volatility between 0.7324 and 0.7673 [2014 – between 0.7679 and 0.7691; 2013 – 0.743], risk free interest rate between 0.95 and 1.75% [2014 –1.79%; 2013 – 1.75%] and a dividend yield of 0% [2014 and 2013 - 0%].
The weighted average grant date fair value of options granted during the year ended June 30, 2015 was $6.19 [2014 - $4.51; 2013 - $2.63].
As at June 30, 2015, 2014 and 2013, total compensation cost related to non-vested awards not yet recognized is $2,354,038, $3,282,863 and $1,202,255, respectively. The weighted average period over which it is expected to be recognized is 27, 32 and 28 months, respectively.
For fiscal 2015, the weighted average exercise price and the weighted average remaining contractual life of the outstanding stock options are $4.82 and 7.53 years [2014 - $3.91 and 8.01 years; 2013 $2.97 and 7.39 years]. The weighted average exercise price and the weighted average remaining contractual life of the exercisable stock options are $3.46 and 6.53 years [2014 - $2.82 and 6.57 years; 2013 - $3.11 and 5.70 years].
The intrinsic value of options exercised during fiscal 2015 is $328,169 [2014 - $506,341; 2013 - $17,596] and the intrinsic value of options granted for fiscal 2015, 2014 and 2013 is nil.
| [a] | As at June 30, 2015, the Company has total non-capital losses of approximately $78,156,000 [2014- $64,969,000] available for carry forward to reduce future taxable income in Canada, the United States of America and Ireland. The non-capital losses will begin to expire as follows: |
| | | $ | |
| 2026 | | | 1,168,000 | |
| 2027 | | | 5,239,000 | |
| 2028 | | | 4,470,000 | |
| 2029 | | | 5,481,000 | |
| 2030 | | | 7,006,000 | |
| 2031 | | | 5,677,000 | |
| 2032 | | | 6,565,000 | |
| 2033 | | | 925,000 | |
| 2034 | | | 6,722,000 | |
| 2035 | | | 34,903,000 | |
| | | | 78,156,000 | |
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
As at June 30, 2015, the Company also has approximately $41,445,000 [2014 - $40,243,000] in Canadian scientific research and experimental development expenditures which can be carried forward indefinitely to reduce future years’ taxable income. During fiscal 2015 the Company recorded nil [2014 - $193,000] refundable provincial ITCs which was recorded as a reduction to research and development, net. The Company has approximately $9,035,000 [2014 - $9,044,000] in federal ITCs and $865,000 [2014 - $700,000] of non-refundable Ontario Research Development Tax Credits that can be carried forward for up to twenty years and used to reduce the Company’s taxes payable.
| [b] | Significant components of the Company’s unrecognized deferred tax assets and deferred tax liabilities are as follows: |
| | | 2015 $ | | | 2014 $ | |
| Deferred tax assets not recognized | | | | | | | | |
| Capital and intangible assets | | | 7,054,767 | | | | 2,098,064 | |
| Non-capital loss carryforwards | | | 15,192,929 | | | | 16,470,897 | |
| Canadian scientific research and experimental development expenditures | | | 10,982,806 | | | | 10,664,292 | |
| Investment tax credits | | | 7,601,282 | | | | 7,321,900 | |
| Contingent consideration payable | | | 745,384 | | | | 624,094 | |
| Financing and share issuance costs | | | 652,472 | | | | 175,602 | |
| Loss on disposal of SCT shares | | | 33,681 | | | | 33,681 | |
| Total deferred tax assets not recognized | | | 42,263,321 | | | | 37,388,530 | |
| | | | | | | | | |
| Deferred tax assets and liabilities | | | | | | | | |
| Intangible assets | | | - | | | | (284,718 | ) |
| Leasehold inducement | | | - | | | | (3,028 | ) |
| Non-capital loss carryforwards | | | - | | | | 287,746 | |
| Net deferred tax liability | | | - | | | | - | |
| [c] | The reconciliation of income tax attributable to continuing operations computed at the statutory tax rates to income tax recovery is as follows: |
| | | 2015 | | | 2014 | | | 2013 | |
| | | $ | | | $ | | | $ | |
| Tax expense (recovery) at combined federal and provincial rates of 26.5% (2014 – 26.5%) | | | (13,604,975 | ) | | | (5,772,298 | ) | | | 6,174 | |
| Non-deductible permanent differences: | | | | | | | | | | | | |
| Stock-based compensation | | | 823,498 | | | | 301,603 | | | | 261,881 | |
| Other permanent and non-deductible items | | | 28,969 | | | | 6,831 | | | | 8,576 | |
| Difference in foreign tax rates | | | 4,873,673 | | | | 889,481 | | | | - | |
| Deferred tax assets (recognized) not recognized for accounting | | | 7,878,835 | | | | 4,574,383 | | | | (276,631 | ) |
| | | | - | | | | - | | | | - | |
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
| | | 2015 | | | 2014 | | | 2013 | |
| | | $ | | | $ | | | $ | |
| Research and development | | | | | | | | | | | | |
| Clinical trials and manufacturing | | | 41,810,031 | | | | 13,327,761 | | | | 5,084,737 | |
| Salaries and benefits | | | 4,202,745 | | | | 2,432,519 | | | | 1,506,136 | |
| Stock compensation expense | | | 1,777,628 | | | | 501,595 | | | | 408,938 | |
| Amortization | | | 617,167 | | | | 937,441 | | | | 1,803,037 | |
| Facility lease costs and utilities | | | 290,440 | | | | 196,307 | | | | 176,153 | |
| Insurance | | | 176,800 | | | | 85,825 | | | | 90,475 | |
| General laboratory supplies and materials | | | 334,892 | | | | 132,493 | | | | 86,288 | |
| Ontario investment tax credits | | | - | | | | (246,556 | ) | | | (292,892 | ) |
| | | | 49,209,703 | | | | 17,367,385 | | | | 8,862,872 | |
| | | | | | | | | | | | | |
| Selling, general and administrative expenses | | | | | | | | | | | | |
| Salaries and benefits | | | 2,039,328 | | | | 1,601,891 | | | | 1,569,777 | |
| Stock compensation expense | | | 1,329,909 | | | | 636,531 | | | | 579,291 | |
| Professional fees and services | | | 775,597 | | | | 987,997 | | | | 394,549 | |
| Insurance | | | 261,173 | | | | 223,943 | | | | 250,252 | |
| Facility lease costs and utilities | | | 156,852 | | | | 151,192 | | | | 149,046 | |
| Business development, corporate communication and investor relations | | | 505,297 | | | | 798,954 | | | | 354,511 | |
| Regulatory and stock transfer fees | | | 137,412 | | | | 138,975 | | | | 94,481 | |
| Office and related expenses | | | 274,495 | | | | 177,635 | | | | 148,821 | |
| Amortization | | | 34,209 | | | | 9,456 | | | | 17,064 | |
| | | | 5,514,272 | | | | 4,726,574 | | | | 3,557,792 | |
| 14. | EARNINGS (LOSS) PER SHARE |
Basic and diluted loss per share is calculated by dividing the profit attributable to equity holders of the Company by the weighted average number of common shares outstanding during the year. Outstanding options to purchase common shares of 2,755,764 [June 30, 2014 – 2,305,589; June 30, 2013 - $1,872,000] and the warrants to purchase 1,903,341 common shares [June 30, 2014 – 853,223; June 30, 2013 - nil] are not included in the calculation of diluted earnings per share as the effect is anti-dilutive due to the losses incurred in the period.
For the year ended June 30, 2015, 2014 and 2013, 79,908 contingently returnable common shares were excluded from the basic and diluted net loss per common share calculation. The contingently returnable common shares relate to employment contracts and will be released from escrow based on the achievement of certain corporate milestones.
| | | 2015 | | | 2014 | | | 2013 | |
| Income (Loss) attributable to equity holders of the Company | | | (51,339,528 | ) | | | (21,782,255 | ) | | $ | 23,297 | |
| | | | | | | | | | | | | |
| Weighted average number of common shares outstanding | | | 36,523,897 | | | | 30,094,825 | | | | 26,841,528 | |
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
| 15. | CONTINGENCIES AND COMMITMENTS |
| [a] | As at June 30, 2015, the Company is committed to aggregate expenditures of nil [2014 -$14,976,412] under its collaboration agreements. In addition, at June 30, 2015, the Company is committed to aggregate expenditures of approximately $3,541,000 [2014 - $13,613,000] for clinical and toxicity studies to be completed during fiscal 2016, approximately $215,000 [2014 - $128,049] for manufacturing agreements and approximately $327,000 for consulting and other agreements [2013 – $482,000]. |
| [b] | The Company leases premises under an operating lease which originally expired on June 30, 2011 but has been extended to 2020. The Company also sub-leases premises under an operating lease which expires on December 31, 2015. In addition, the Company leases photocopiers under operating leases that expire on various dates to August 2018. Future minimum annual lease payments under these operating leases, in aggregate and over the next five years are as follows: |
| | | $ | |
| | | | |
| 2016 | | | 238,179 | |
| 2017 | | | 173,651 | |
| 2018 | | | 169,514 | |
| 2019 | | | 154,293 | |
| 2020 | | | 153,160 | |
| | | | 888,797 | |
During the year, the rental expense for the various premises under operating leases was $273,071 [2014 - $187,762; 2013- $163,660].
| [c] | The Company’s technology related commitments are as follows: |
| [i] | ELND005 Technology License: |
The Company has a worldwide exclusive license to intellectual property relating to ELND005 with the inventor, an Alzheimer’s disease researcher at the University of Toronto. Under the agreement, the inventor may receive milestone payments of up to $150,000. For therapeutic products, a royalty of 2.5% will be due on the first $100,000,000 of revenues received by the Company and 1.5% of revenues thereafter. For diagnostic products, a royalty of 10% will be due on the first $100,000,000 of revenues received by the Company and 7% of revenues thereafter. Also, the inventor may receive up to $25,000 for additional patent applications under this license. The agreement remains in force until the expiration of the last to expire patent.
In addition, under the terms of the ENI acquisition agreement, the Company is committed to pay the former shareholders of ENI contingent clinical milestones potentially totaling $10.9 million payable in cash or Transition common shares at the then market price and a royalty of up to 1% on net sales of ELND005 product (see note 8a).
In light of the series of transactions entered into on February 28, 2014, the Company is also committed to pay Perrigo Company plc up to US$40,000,000 in approval and commercial milestone payments and 6.5% royalties on net sales of ELND005 products and sublicense fees received (see note 8b).
| [ii] | TT701 Selective Androgen Receptor Molecule (“SARM”) |
On May 6, 2015, the Company exclusively licensed worldwide rights to a novel small molecule drug candidate TT701 from Lilly. TT701 is a selective androgen receptor modulator that has been shown in a Phase 2 study to significantly increase lean body mass and a measurement of muscle strength in male subjects. Under the terms of the agreement, TTIL has acquired rights to develop and commercialize TT701. Lilly will receive contingent upfront consideration of up to US$1 million of which US$500,000 ($624,500) has been paid as at June 30, 2015. In addition, Lilly is eligible to receive up to US$100 million in commercial milestones and a mid-single digit royalty on sales of TT701 products should such products be successfully commercialized.
Notes to the Audited Consolidated Financial Statements
June 30, 2015
(In Canadian Dollars)
| 16. | CHANGE IN WORKING CAPITAL |
The change in working capital consists of the following:
| | | 2015 | | | 2014 | | | 2013 | |
| | | $ | | | $ | | | $ | |
| Trade and other receivables | | | (44,675 | ) | | | (186,493 | ) | | | 7,866 | |
| Income tax and investment tax credits receivable | | | (187,275 | ) | | | (31,741 | ) | | | 61,299 | |
| Prepaid expenses and deposits | | | (222,487 | ) | | | 322,508 | | | | (42,878 | ) |
| Trade and other payables | | | 1,828,323 | | | | 5,153,165 | | | | (304,766 | ) |
| | | | 1,373,886 | | | | 5,257,439 | | | | (278,479 | ) |
| 17. | RELATED PARTY TRANSACTIONS |
Key management compensation
Key management includes the Company’s directors, and members of the senior management team. The compensation paid or payable to key management for employee services is shown below:
| | | 2015 | | | 2014 | | | 2013 | |
| | | $ | | | $ | | | $ | |
| Salaries and other short-term employee benefits | | | 2,525,182 | | | | 1,849,886 | | | | 1,714,325 | |
| Stock-compensation expenses | | | 1,856,849 | | | | 964,237 | | | | 860,897 | |
| | | | 4,382,031 | | | | 2,814,123 | | | | 2,575,222 | |
During fiscal 2015, the Company paid legal fees to a law firm where the Company’s Secretary is a partner and to a corporation controlled by the Company’s Secretary. Total fees and disbursements charged to the Company by these companies was $45,346 [2014 – $49,000; 2013 - $45,000] and are included in general and administrative expenses. The balance owing at June 30, 2015, 2014 and 2013 is nil.
Members of the Company’s Board of Directors, management and employees participated in both the August, 2013 and June, 2014 private placements (see note 10).
These transactions occurred in the normal course of operations and were measured at the exchange amount, which is the amount of consideration established and agreed to by the related parties.
The Company indemnifies its directors and officers against any and all claims or losses reasonably incurred in the performance of their service to the Company to the extent permitted by law. The Company has acquired and maintains liability insurance for its directors and officers.
The Company operates in one operating segment, the research and development of therapeutic agents. Total revenue recognized during the year ended June 30, 2013 amounted to $17,933,500. The Company received $10,815,200 from Elan, a company based in Ireland and received the balance of $7,118,300 from Eli Lilly and Company, a company based in the United States of America.
EXHIBIT INDEX
Exhibit
Number | Description |
| 1.1 | Articles of Incorporation of Transition Therapeutics Inc., as amended (incorporated by reference to Exhibit 99.110 of Form 40-F filed on June 5, 2007, File No. 001-33514). |
| | |
| 1.2 | Bylaws of Transition Therapeutics Inc., as amended (incorporated by reference to Exhibit 99.111 of Form 40-F filed on June 5, 2007, File No. 001-33514). |
| | |
| 2.1 | Form of Warrant. |
| | |
| 4.1 | Share Purchase Agreement, dated February 24, 2006, among Registrant, 1255205 Ontario Inc., Ellipsis Neurotherapeutics Inc., and other parties named therein (incorporated by reference to Exhibit 99.47 of Form 40-F filed on June 5, 2007, File No. 001-33514). |
| | |
| 4.2 | License Agreement dated May 28, 2003, by and between Ellipsis Biotherapeutics Corp. and JoAnne McLaurin, PHD, as amended on April 1, 2005. (incorporated by reference to Exhibit 99.2 of Form 6-K filed on March 17, 2008, File No. 001-33514). |
| | |
| 4.3 | Consent to License – JoAnne McLaurin Novel Treatment for Alzheimer’s Disease and Amyloid Disorders using Inositol-Based Compounds dated May 29, 2003, by the University of Toronto. (incorporated by reference to Exhibit 99.3 of Form 6-K filed on March 17, 2008, File No. 001-33514). |
| | |
| 4.4 | Consent to Assignment dated November 2, 2004, by and between JoAnne McLaurin, Ellipsis Biotherapeutics Corp. and Ellipsis Neurotherapeutics Inc. (incorporated by reference to Exhibit 99.4 of Form 6-K filed on March 17, 2008, File No. 001-33514). |
| | |
| 4.5 | Collaboration and License Agreement effective as of February 25, 2010, among Waratah Pharmaceuticals Inc. and Eli Lilly and Company (incorporated by reference to Exhibit 99.2 of Form 6-K filed on March 12, 2010, File No. 001-33514). |
| | |
| 4.6 | Collaboration and License Agreement effective as of July 23, 2013, between Transition Therapeutics Inc. and Eli Lilly and Company (incorporated by reference to Exhibit 99.2 of Form 6-K filed on July 26, 2013, File No. 001-33514). |
| | |
| 4.7 | Form of Subscription Agreement (Canada) dated June 23, 2014 between Transition Therapeutics Inc. and certain directors and officers of the Company. |
| | |
| 4.8 | Form of Subscription Agreement (U.S.) dated June 23, 2014 between Transition Therapeutics Inc. and certain shareholders of the Company. |
| | |
| 4.9 | Milestone, Royalty and Sublicensing Fee Deed effective as of February 27, 2014 between Transition Therapeutics Ireland Limited (formerly Elan Science Ten Limited) and Elan Pharma International Limited. |
| | |
| 4.10 | Purchase and Sale Agreement, dated February 28, 2014, by and among Elan Pharma International Limited, the Company and Perrigo Company PLC. |
| | |
| 4.11 | License Agreement effective as of May 5, 2015 between Transition Therapeutics Inc. and Eli Lilly and Company (incorporated by reference to Exhibit 99.1 of Form 6-K filed on May 8, 2015, File No. 001-33514). |
| | |
| 8.1 | Subsidiaries |
Exhibit
Number | Description |
| 12.1 | Certification of the Chief Executive Officer pursuant to Rule 13a-14(a) of the Securities Exchange Act of 1934. |
| | |
| 12.2 | Certification of the Chief Financial Officer pursuant to Rule 13a-14(a) of the Securities Exchange Act of 1934. |
| | |
| 13.1 | Section 1350 Certification of the Chief Executive Officer. |
| | |
| 13.2 | Section 1350 Certification of the Chief Financial Officer. |
| | |
| 15.1 | Consent of PricewaterhouseCoopers LLP. |