Exhibit 99.2
MANAGEMENT’S DISCUSSION AND ANALYSIS
The following information should be read in conjunction with the Company’s unaudited consolidated financial statements for the three and six month periods ended December 31, 2015 and the related notes, which are prepared in accordance with International Financial Reporting Standards as issued by the International Accounting Standards Board for interim financial statements, including IAS 34, Interim Reporting (IFRS), as well as the audited consolidated financial statements for the year ended June 30, 2015, including the notes thereto, prepared in accordance with IFRS, and the annual fiscal 2015 MD&A. This Management’s Discussion and Analysis (“MD&A”) provides a review of the performance of the Company for the three and six month periods ended December 31, 2015 as compared to the three and six month periods ended December 31, 2014. This review was performed by management with information available as of February 5, 2016.
Where “we”, “us”, “our”, “Transition” or the “Company” is used, it is referring to Transition Therapeutics Inc. and its wholly-owned subsidiaries, unless otherwise indicated. All amounts are in Canadian dollars, unless otherwise indicated.
Additional information relating to the Company, including the Company’s most recently filed Annual Information Form, can be found on SEDAR at www.sedar.com.
CAUTION REGARDING FORWARD-LOOKING STATEMENTS
This MD&A contains certain forward-looking statements within the meaning of applicable securities laws. Forward-looking information typically contains statements with words such as “anticipate”, “believe”, “expect”, “plan”, “estimate”, “intend”, “may” or similar words suggesting future outcomes. Forward-looking statements in this MD&A include, but are not limited to statements with respect to: the clinical study phases of the Company’s product candidates which the Company expects to complete in fiscal 2016 and beyond; the ability of the Company’s business model to maximize shareholder returns; the potential for ELND005 to slow the progression of Alzheimer’s disease and improve symptoms; the potential for ELND005 to be effective for the treatment of agitation and or aggression in patients with Alzheimer’s disease; the potential for ELND005 to be effective for the treatment of Down syndrome; the timing and manner of future clinical development of ELND005; the global population size of those affected by Alzheimer’s disease; the demand for a product that can slow or reverse the progression of Alzheimer’s disease; the demand for a product that can reduce the emergence and severity of neuropsychiatric symptoms like depression, anxiety, agitation and aggression in Alzheimer’s disease; the potential clinical benefit of ELND005 in the treatment of other disease indications; the development of TT401 and the series of preclinical compounds in-licensed from Eli Lilly and Company (“Lilly”) and their potential benefit in type 2 diabetes patients and obese individuals; the timing and manner of future clinical development of TT401 performed by Lilly; TT701 development plans and timelines for individuals with androgen deficiency or other disease indications; the potential clinical benefit of TT701 to increase lean body mass, improve functional and sexual outcomes or improve other symptoms associated with androgen deficiency; the engagement of third party manufacturers to produce the Company’s drug substances and products; the potential future in-licensing of additional drug candidates to expand the development pipeline; the intention of the Company to make collaborative arrangements for the marketing and distribution of its products and the impact of human capital on the growth and success of the Company.
This forward-looking information is subject to various risks and uncertainties, including those discussed below, that could cause actual results and experience to differ materially from the anticipated results or other expectations expressed. Readers are cautioned not to place undue reliance on this forward-looking information, which is provided as of the date of this MD&A unless otherwise stated, and the Company will not undertake any obligation to publicly update orrevise any forward-looking information, whether as a result of new information, future events, or otherwise, except as required by securities laws.
Some of the assumptions, risks and factors which could cause future outcomes to differ materially from those set forth in the forward-looking information include, but are not limited to: (i) the assumption that the Company will be able to obtain sufficient and suitable financing to support operations, clinical trials and commercialization of products, (ii) the risk that the Company may not be able to capitalize on partnering and acquisition opportunities, (iii) the assumption that the Company will obtain favourable clinical trial results in the expected timeframe, (iv) the assumption that the Company will be able to adequately protect proprietary information and technology from competitors, (v) the risks relating to the uncertainties of the regulatory approval process, (vi) the impact of competitive products and pricing and the assumption that the Company will be able to compete in the targeted markets, and (vii) the risk that the Company may be unable to retain key personnel or maintain third party relationships, including relationships with key collaborators.
By its nature, forward-looking information involves numerous assumptions, inherent risks and uncertainties, both general and specific, that contribute to the possibility that the predictions, forecasts, projections or other forward-looking statements will not occur. Prospective investors should carefully consider the information contained under the heading “RISKS AND UNCERTAINTIES” as described in the MD&A for the year ended June 30, 2015.
OVERVIEW
Transition is a biopharmaceutical development company, advancing novel therapeutics for CNS and metabolic disease indications. The Company’s wholly-owned subsidiary, Transition Therapeutics Ireland Limited (“TTIL”) is developing CNS drug candidate ELND005 for the treatment of Alzheimer’s disease (“AD”) and Down syndrome. Transition’s lead metabolic drug candidate is TT401 for the treatment of type 2 diabetes and accompanying obesity.
Highlights for the Company during the six month period ended December 31, 2015 and up to the date of this MD&A include the following:
ELND005:
| · | October 28, 2015 –Transition announced that data from the Phase 2/3 clinical study of ELND005 in Alzheimer’s disease patients with moderate and severe agitation and aggression was presented at the Clinical Trials in Alzheimer’s Disease (CTAD) meeting. A copy of the CTAD oral presentation is available on the Company website atwww.transitiontherapeutics.com; |
| · | October 15, 2015 - Transition announced that its subsidiary, TTIL, has completed a thorough review of the data related to the Phase 2/3 study of ELND005 in AD patients with moderate or severe agitation and aggression.The analysis identified a significant clinical benefit of ELND005 in AD patients with severe agitation and aggression, and will serve as the basis for patient selection in a Phase 3 clinical study. The review was performed in consultation with a group of key opinion leaders in the field of neuropsychiatry. |
TT401:
| · | February 1, 2016 – Transition announced the results of a Phase 2 clinical study of drug candidate TT401 (LY2944876) for the treatment of type 2 diabetes.TT401 is a once-weekly administered oxyntomodulin analog with dual GLP-1 and glucagon agonist activity. TT401 development collaborator Eli Lilly and Company performed the Phase 2 study enrolling 420 type 2 diabetes subjects into a 24 week study consisting of a 12-week randomized blinded stage followed by a 12-week open-label stage. The study included 4 once-weekly dose arms of TT401 (10mg, 15mg, 30mg, 50mg), a placebo arm, and an active comparator arm (exenatide extended release – 2mg). TT401 demonstrated HbA1c improvements of up to -1.43% (similar to the exenatide arm). All TT401 dose arms and the exenatide arm were statistically significant relative to the placebo arm at Weeks 12 and 24. TT401 also produced dose dependent weight loss (up to -3.3 kg). The weight loss observed in the highest dose arm (50mg of TT401) was statistically significant relative to both the placebo and exenatide arms at weeks 12 and 24. |
TT701 SARM:
| · | October 29, 2015 –Transition announced that its subsidiary, TTIL, has entered into an agreement withBrigham and Women’s Hospital (“BWH”) for an investigator-led clinical study of drug candidate TT701. TTIL will support a Phase 2 study to evaluate selective androgen receptor modulator (SARM) drug candidate TT701 as a therapy to improve the symptoms of androgen deficiency in men with prostate cancer that have undergone a radical prostatectomy procedure. |
STRATEGIC COLLABORATIONS
Perrigo Company plc(“Perrigo”)
In 2006, Transition exclusively licensed the ELND005 technology to Elan Pharma International Limited (“Elan”) for worldwide development and commercialization. Following amendment of that agreement in 2010, Elan held all development and commercialization rights to ELND005 and Transition became eligible to receive milestone and royalty payments with the successful advancement of ELND005. Transition has received US$40 million from Elan in upfront and achieved milestone payments. Perrigo acquired Elan in December 2013, including all Elan’s rights and obligations to the development of ELND005.
On February 28, 2014, Transition announced that after a series of transactions, Perrigo had transferred all of its ELND005 rights and assets under the collaboration agreement to the Company’s wholly owned subsidiary, TTIL. In parallel with this acquisition, Perrigo invested US$15 million and received 2,255,640 Transition common shares representing approximately a 7% ownership stake in Transition as of the date of the transaction. Perrigo will also be eligible to receive up to US$40 million in approval and commercial milestone payments and a 6.5% royalty on net sales of ELND005 products and sublicense fees received. Going forward, TTIL is responsible for all future development and commercialization activities of the ELND005 drug candidate.
Lilly
Diabetes
On March 3, 2010, Transition and Lilly entered into a licensing and collaboration agreement granting Transition the rights to a series of preclinical compounds in the area of diabetes. Under the licensing and collaboration agreement, Transition received exclusive worldwide rights to develop and potentially commercialize a class of compounds that, in preclinical models, showed potential to provide glycemic control and other beneficial effects including weight loss.
Under the terms of the agreement, Lilly received an up-front payment of $1,055,900 (US$1 million) which has been capitalized as a license acquired from Lilly and is being amortized over 20 years which represents the estimated life of the underlying compounds and patents.
In June 2013, Lilly exercised its option and assumed all development and commercialization rights to type 2 diabetes drug candidate TT401. In conjunction with this assumption of rights, Transition received a US$7 million milestone payment. Lilly has assumed all costs and will perform all future development and commercialization activities of TT401, and Transition paid US$14 million to Lilly in three separate installments during the Phase 2 clinical study in fiscal 2015. Transition has no additional funding obligations related to this clinical study or any other development or commercialization activities in the future.
Transition is eligible to receive up to approximately US$240 million in additional milestone payments plus double-digit royalties (less than 15%) on sales of TT401 products and a low single digit royalty on sales of related compounds.
PROGRAMS
Transition is focused on developing innovative therapies in several distinct areas of opportunity. Transition’s vision is to build a company that has a strong foundation for growth based on multiple technologies and product opportunities, which reduces risk and enhances shareholder return. The Company’s technologies are as follows:
ELND005
Alzheimer’s Disease:
Alzheimer’s disease is a progressive brain disorder that gradually destroys a person’s memory and ability to learn, reason, make judgments, communicate and carry out daily activities. As Alzheimer’s disease progresses, individuals may also experience changes in personality and behavior, such as anxiety, suspiciousness or agitation, as well as delusions or hallucinations. Approximately 90% of Alzheimer’s disease patients develop neuropsychiatric symptoms, and up to 60% develop agitation/aggression over the course of their disease. Agitation/aggression are among the most disruptive neuropsychiatric symptoms in Alzheimer’s disease and are associated with increased morbidity and caregiver burden.
The disease mainly affects individuals over age 65 and it is estimated over 18 million people are suffering from Alzheimer’s disease worldwide. In the U.S., Alzheimer’s disease is the sixth leading cause of death and current direct/indirect costs of caring for an estimated 5.4 million Alzheimer’s disease patients are at least US$100 billion annually.
Current U.S. Food and Drug Administration approved Alzheimer’s disease medications may temporarily delay memory decline for some individuals, but none of the currently approved drugs are known to stop the underlying degeneration of brain cells. Certain drugs approved to treat other illnesses may sometimes help with the emotional and behavioral symptoms of Alzheimer’s disease. With an aging population, there is a great need for therapies to address Alzheimer’s disease patient’s neuropsychiatric symptoms and declines in cognitive ability.
Down Syndrome:
Down syndrome (DS, Trisomy 21), caused by an extra copy of chromosome 21, is the most common genetic form of intellectual disability with a prevalence of approximately 1 in 700 live births in the U.S. Children with DS exhibit developmental delay and various degrees of intellectual disability, while adults are at increased risk of Alzheimer’s dementia. There are currently no drugs approved for the treatment of cognitive dysfunction in DS.
Excess activity of genes on chromosome 21, such as amyloid precursor protein (APP) and sodium-myo-inositol active transporter (SMIT), are thought to play a role in the cognitive dysfunction of DS. Life-long exposure to increased amyloid and myo-inositol levels in the brain are thought to lead to synaptic dysfunction and cognitive disability. ELND005 may have the potential to improve cognition in DS by decreasing amyloid levels and regulating myo-inositol-dependent neuronal signaling.
Clinical Development Update
ELND005 for Neuropsychiatric Diseases
TTIL is developing neuropsychiatric drug candidate ELND005, (scyllo-inositol). ELND005 is an orally bioavailable small molecule that is being investigated for multiple neuropsychiatric indications on the basis of its proposed dual mechanism of action, which includes β-amyloid anti-aggregation and regulation of brain myo-inositol levels. An extensive clinical program of Phase 1 and Phase 2 studies have been completed with ELND005 to support clinical development. The Phase 2 study (ELND005-AD201) which evaluated ELND005 in more than 350 mild to moderate AD patients was published in the peer-reviewed journal,Neurology. TheNeurology article was entitled “A Phase 2 randomized trial of ELND005, scyllo-inositol, in mild-moderate Alzheimer’s disease”.
Currently, the use of ELND005 is being investigated in two clinical areas:
(a) Agitation and Aggression in Alzheimer’s Disease
A Phase 2 clinical trial of ELND005 for the treatment of agitation/aggression in patients with Alzheimer’s disease was performed at 70 clinical sites in North America and Europe. The objectives of the study were to evaluate the efficacy, safety and tolerability of ELND005 over 12 weeks of treatment in AD patients who were experiencing moderate or severe levels of agitation/aggression. Enrollment of this clinical study (AG201) known as the “Harmony AD” study was completed on March 2, 2015 with a total of 350 patients being enrolled.
On June 24, 2015, Transition announced that ELND005 did not meet its primary efficacy endpoint in the Phase 2/3 clinical study of ELND005 in agitation and aggression in patients with AD. In the study, both the treatment and placebo groups showed a significant, but similar, reduction in agitation and aggression relative to baseline. There was a greater than expected reduction in agitation and aggression observed in the placebo group as measured in weeks 4, 8 and 12 in the study. The safety and tolerability profile of ELND005 was consistent with previous studies in AD at the 250mg bid dose.
The Company’s subsidiary, TTIL performed a thorough review of the data from the completed Phase 2/3 study in agitation and aggression and released the results of this review on October 14, 2015. Although the primary efficacy endpoint was not achieved in the overall study, ELND005 significantly (p value <0.05) improved agitation and aggression in a sub-population of Alzheimer’s disease patients with severe agitation and aggression. In this population, ELND005 demonstrated numerical improvement in 20 of 21 behavioral symptoms measured as part of the primary efficacy endpoint. ELND005 also demonstrated an acceptable safety and tolerability profile. TTIL intends to meet with regulators to seek guidance on an ELND005 Phase 3 program for AD patients with severe agitation and aggression.
(b) Down Syndrome
On November 20, 2014, Transition announced the results of a clinical study of neuropsychiatric drug candidate ELND005 in young adults with Down syndrome. Transition’s wholly owned subsidiary, TTIL completed this first study in Down syndrome subjects without dementia to allow optimal dose selection for future larger studies.
The study enrolled 23 Down syndrome subjects in three study arms over a four-week treatment period: placebo, 250 mg once daily; and 250 mg twice daily. At the doses evaluated, ELND005 was determined to have an acceptable safety and tolerability profile and there were no serious adverse events reported in the study. Treatment emergent adverse events were reported in seven of the subjects receiving ELND005 and all were deemed mild in severity. The two ELND005 doses achieved the plasma levels expected in pharmacokinetic modeling and will inform the selection of a higher dose in a larger Phase 2b study in Down syndrome subjects.
The ELND005 technology is claimed in multiple issued patents and pending patent applications in many jurisdictions throughout the world.
Expenditures for the ELND005 Program
On February 28, 2014, Transition announced that after a series of transactions, Perrigo has transferred all of its ELND005 rights and assets to the Company’s wholly owned subsidiary, TTIL. As a result, effective March 1, 2014, TTIL is responsible for all future development and commercialization activities of ELND005.
During the three and six month periods ended December 31, 2015 and 2014, the Company incurred direct research and development costs for this program as follows:
| ELND005 Program(1) | | Three month
period ended
December 31, 2015 | | | Three month period
ended December
31, 2014 | | | Six month period
ended December
31, 2015 | | | Six month period
ended December 31,
2014 | |
| Pre-clinical studies | | $ | - | | | $ | - | | | $ | - | | | $ | - | |
| Clinical studies | | | 123,839 | | | | 4,246,127 | | | | 2,900,538 | | | | 9,734,253 | |
| Manufacturing | | | 1,185 | | | | 253,238 | | | | 14,959 | | | | 471,016 | |
| Other direct research | | | 137,289 | | | | 457,346 | | | | 522,130 | | | | 1,152,669 | |
| TOTAL | | $ | 262,313 | | | $ | 4,956,711 | | | $ | 3,437,627 | | | $ | 11,357,938 | |
Note(1)These costs are direct research and development costs only and do not include patent costs, investment tax credits, salaries and benefits, amortization of intangible assets or an allocation of Company overhead.
TT401
Development of TT401 for Diabetes
Diabetes is a disease in which the body does not produce or properly use insulin. Insulin is a hormone released from islet cells located in the pancreas that is needed to convert sugar, starches and other food into energy needed for daily life. There are two primary forms of diabetes; type 1 diabetes and type 2 diabetes.
Type 2 diabetes usually begins as insulin resistance, a disorder in which the cells do not use insulin properly. As the need for insulin increases, the pancreas gradually loses its ability to produce it. Current treatments for type 2 diabetes include lifestyle changes, oral medications, incretin therapy and insulin therapy. Type 2 diabetes accounts for about 90-95% of all diagnosed cases of diabetes.
Clinical Development Update of TT401 (LY2944876)
On March 3, 2010, Transition announced that it had acquired the exclusive worldwide rights to develop and potentially commercialize a series of preclinical compounds from Lilly in the area of diabetes. In preclinical diabetes models, these compounds showed potential to provide glycemic control and other beneficial effects including weight loss.
��
On June 18, 2012, Transition announced the results of the Phase 1 clinical study of type 2 diabetes drug candidate, TT401. The Phase 1, double-blind, placebo-controlled randomized study enrolled 48 non-diabetic obese subjects in six cohorts evaluating six escalating subcutaneous single doses of TT401. TT401 demonstrated an acceptable safety and tolerability profile in non-diabetic obese subjects in the study. TT401 exhibited the expected pharmacological effect on glucose and pharmacodynamic biomarkers at doses that were safe and tolerable. The pharmacokinetic profile, assessed over 28 days, demonstrated a half-life consistent with once-weekly dosing.
On April 30, 2013, Transition announced the results of a five-week proof of concept clinical study of TT401 in type 2 diabetes and obese non-diabetic subjects. The study enrolled diabetic patients at five dosing levels and non-diabetic obese patients at one dose level. All dosing cohorts received five doses over a five week period. Diabetic patients were on stable doses of metformin.
At the end of the treatment period, TT401-treated patients in the 3 highest dose groups experienced statistically significant reductions in mean fasting plasma glucose relative to placebo. Statistically significant mean body weight reduction relative to baseline occurred in the three highest dose groups. A similar reduction in body weight was also observed in the obese non-diabetic cohort. TT401 demonstrated an acceptable safety and tolerability profile at all doses evaluated in diabetic and non-diabetic obese subjects. The most common adverse event noted in the study was decreased appetite. Some subjects in the highest three dose groups experienced mild nausea and vomiting, which are consistent with studies of other GLP-1 agonist drug candidates. The pharmacokinetic profile, assessed over the five week study, demonstrated a half-life consistent with once-weekly dosing.
On June 17, 2013, Lilly exercised its option to assume all development and commercialization rights to type 2 diabetes drug candidate TT401. In conjunction with this assumption of rights, Transition received a US$7 million milestone payment. Lilly and Transition have amended their agreement to address future development of TT401 and associated financial arrangements. Lilly has assumed all costs and will perform all future development and commercialization activities of TT401. In May, 2014, Transition announced the dosing of the first patient in a Phase 2 clinical study of TT401. The study was performed by Transition’s development partner Lilly. The objectives of the study were to evaluate the safety and effectiveness of TT401 compared to once-weekly exenatide extended release and placebo. In February 2015, Lilly informed Transition that 420 type 2 diabetic subjects had been enrolled in the current Phase 2 study, thereby completing the enrollment phase of the study.
In fiscal 2015, Transition made three separate installments to Lilly to support the Phase 2 clinical study totaling US$14 million. There are no additional funding obligations related to this clinical study or the TT401 development program.
On February 1, 2016, Transition announced the results of a Phase 2 clinical study of drug candidate TT401 (LY2944876) for the treatment of type 2 diabetes. TT401 is a once-weekly administered oxyntomodulin analog with dual GLP-1 and glucagon agonist activity. TT401 development collaborator Eli Lilly and Company performed the Phase 2 study enrolling 420 type 2 diabetes subjects into a 24 week study consisting of a 12-week randomized blinded stage followed by a 12-week open-label stage. The study included 4 once-weekly dose arms of TT401 (10mg, 15mg, 30mg, 50mg), a placebo arm, and an active comparator arm (exenatide extended release – 2mg).
TT401 demonstrated HbA1c improvements of up to -1.43% (similar to the exenatide arm). All TT401 dose arms and the exenatide arm were statistically significant relative to the placebo arm at Weeks 12 and 24.
TT401 also produced dose dependent weight loss (up to -3.3 kg). The weight loss observed in the highest dose arm (50mg of TT401) was statistically significant relative to both the placebo and exenatide arms at weeks 12 and 24.
In the study, TT401 appeared to have an acceptable safety and tolerability profile. There were a similar number of study discontinuations and serious adverse events between the TT401 dose arms and the exenatide arm. The most frequently observed adverse events were gastrointestinal; these were generally classified as mild to moderate and diminished over time.
The Phase 2 study data will be submitted for presentation at a future medical meeting.
Should Lilly continue TT401 development, Transition would be eligible to receive a US$15 million milestone payment as well as future milestone payments and royalties. A decision from Lilly is expected within the next 90 days. Otherwise, Transition may elect to assume development and commercialization rights to TT401. This option allows Transition to pursue TT401 development on its own or with a third party, subject to future royalty payments to Lilly.
TT701 for Androgen Deficiency
On May 6, 2015, TTIL exclusively licensed worldwide rights to a novel small molecule drug candidate TT701 from Lilly. TT701 is a selective androgen receptor modulator that has been shown in a Phase 2 study to significantly increase lean body mass and a measurement of muscle strength in male subjects.
Clinical Development of TT701
Since acquiring the exclusive worldwide rights to TT701 the Company has incurred drug development manufacturing costs as it prepares to move the drug candidate into a Phase 2 clinical trial.
On October 29, 2015, Transition announced that TTIL had entered into an agreement with Brigham and Women’s Hospital for an investigator-led clinical study of drug candidate TT701.
TTIL will support a Phase 2 study to evaluate selective androgen receptor modulator (SARM) drug candidate TT701 as a therapy to improve the symptoms of androgen deficiency in men with prostate cancer that have undergone a radical prostatectomy procedure. The Phase 2 clinical study is expected to enroll up to 125 subjects and will be performed at selected specialized clinical sites including Brigham and Women’s Hospital. The principal investigator for the Phase 2 study will be Dr. Shalender Bhasin, an internationally recognized endocrinologist with expertise in testosterone biology and men’s aging. The Company expects the Phase 2 study of TT701 to commence during the first quarter of calendar 2016.
Expenditures for the TT701 Program
During the three and six month periods ended December 31, 2015 and 2014, the Company incurred direct research and development costs for this program as follows:
| TT-701 Program (1) | | Three month
period ended
December 31, 2015 | | | Three month period
ended December
31, 2014 | | | Six month period
ended December
31, 2015 | | | Six month period
ended December 31,
2014 | |
| Pre-clinical studies | | $ | - | | | $ | - | | | $ | - | | | $ | - | |
| Clinical studies | | | - | | | | - | | | | - | | | | - | |
| Manufacturing | | | (414 | ) | | | - | | | | 153,954 | | | | - | |
| Other direct research | | | 4,017 | | | | - | | | | 19,121 | | | | - | |
| TOTAL | | $ | 3,603 | | | $ | - | | | $ | 173,075 | | | $ | - | |
Note(1)These costs are direct research and development costs only and do not include patent costs, investment tax credits, salaries and benefits, amortization of intangible assets or an allocation of Company overhead.
The Next Steps
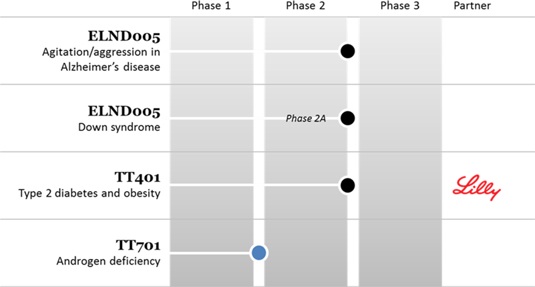
Transition’s goal for its programs is to achieve product approval and ultimately significant revenues or royalties. To achieve product approval, the Company and or its partners, must successfully complete clinical trials and achieve regulatory approval. The stages of development of the Company’s technologies are illustrated below:
RESULTS OF OPERATIONS
During the three month period ended December 31, 2015, the Company recorded a net loss of $2,005,780 ($0.05 loss per common share) compared to a net loss of $16,910,139 ($0.48 loss per common share) for the three month period ended December 31, 2014.
For the six month period ended December 31, 2015, the Company recorded a net loss of $6,497,236 ($0.17 loss per common share) compared to a net loss of $32,605,463 ($0.93 loss per common share) for the six month period ended December 31, 2014.
The decreases in net loss of $14,904,359 and $26,108,227 for the three and six month periods ended December 31, 2015 is due to the significant decrease in research and development expenses resulting from the Company having no further funding obligations to TT401 development partner Lilly as well as decreased clinical development costs relating to ELND005.
Research and Development
Research and development expenses decreased by $14,241,943 from $15,904,889 for the three month period ended December 31, 2014 to $1,662,946 for the three month period ended December 31, 2015. For the six month period ended December 31, 2015, research and development expenses decreased $25,551,354 to $6,388,426 from $31,939,780 for the same period in fiscal 2015.
The decreases in research and development expenses for both the three and six month periods ended December 31, 2015 are primarily due to a decrease in funding obligations relating to TT401 as the Company paid a US$6 million milestone payment to Lilly during the comparative three month period. The decrease in research and development expenses is also due to a decrease in clinical development costs related to ELND005 and reduced salary and related expenses which resulted from cost cutting efforts.
The Company anticipates research and development expenses for the third quarter of fiscal 2016 will remain fairly consistent with the second quarter as the Company prepares for Phase 3 development of ELND005 and commences research payments to Brigham and Women’s Hospital for the development of TT701.
General and Administrative
General and administrative expenses increased by $15,345 from $1,203,449 for the three month period ended December 31, 2014 to $1,218,794 for the three month period ended December 31, 2015. For the six month period ended December 31, 2015, general and administrative expenses increased $109,921 to $2,619,202 from $2,509,281 for the same period in fiscal 2015.
The increases in general and administrative expenses for both the three and six month periods ended December 31, 2015 are primarily due to inflationary increases in compensation costs which have been partially offset by reduced professional fees.
The Company anticipates that general and administrative expenses in the third quarter of fiscal 2016 will remain relatively consistent with the second quarter.
SUMMARY OF QUARTERLY RESULTS
The following table is a summary of selected quarterly consolidated financial information of the Company for each of the eight most recently completed quarters ending at December 31, 2015.
| | | First | | | Second | | | Third | | | Fourth | |
| | | Quarter | | | Quarter | | | Quarter | | | Quarter | |
| 2016 | | | | | | | | | | | | | | | | |
| Revenue | | | - | | | | - | | | | | | | | | |
| Net loss(1) | | $ | (4,491,456 | ) | | $ | (2,005,780 | ) | | | | | | | | |
| Basic and diluted net loss per common share | | $ | (0.12 | ) | | $ | (0.05 | ) | | | | | | | | |
| 2015 | | | | | | | | | | | | | | | | |
| Revenue | | | - | | | | - | | | | | | | | | |
| Net loss(1) | | $ | (15,695,324 | ) | | $ | (16,910,139 | ) | | $ | (4,748,096 | ) | | $ | (13,985,969 | ) |
| Basic and diluted net loss per common share | | $ | (0.45 | ) | | $ | (0.48 | ) | | $ | (0.13 | ) | | $ | (0.38 | ) |
| 2014 | | | | | | | | | | | | | | | | |
| Revenue | | | | | | | | | | | - | | | | - | |
| Net loss(1) | | | | | | | | | | $ | (5,067,292 | ) | | $ | (13,130,005 | ) |
| Basic and diluted net loss per common share | | | | | | | | | | $ | (0.17 | ) | | $ | (0.43 | ) |
Note(1) Net income (loss) before discontinued operations was equivalent to the net income (loss) for such periods.
The fluctuations of Transition’s quarterly results are primarily due to milestone payments made to Lilly to help fund TT401 Phase 2 clinical development and changes in activity levels of the clinical trials being performed by the Company and foreign exchange gains.
CRITICAL ACCOUNTING ESTIMATES AND JUDGMENTS
The preparation of consolidated financial statements in accordance with IFRS requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities, the disclosure of contingent assets and liabilities at the date of the consolidated financial statements and the reported amounts of revenue and expenses during the reporting period. Actual results can differ from those estimates. We have identified the following areas which we believe require management’s most subjective estimates and judgments, often requiring the need to make estimates about the effects of matters that are inherently uncertain and may change in subsequent periods.
(a) Estimates
Valuation and Amortization of Intangible Assets
The Company’s intangible assets are comprised of purchased or licensed pharmaceutical compounds, technology and patents. The costs of the Company’s intangible assets are amortized over the estimated useful life of up to 20 years. Factors considered in estimating the useful life of the intangible asset include the expected use of the asset by the Company, legal, regulatory and contractual provisions that may limit the useful life, the effects of competition and other economic factors, and the level of expenditures required to obtain the expected future cash flows from the intangible asset. The Company re-evaluates the useful life when there has been a change in these factors. The Company assesses its intangible assets for recoverability whenever indicators of impairment exist. As ELND005 did not meet its primary efficacy endpoint in the Phase 2/3 clinical study in agitation and aggression in Alzheimer’s disease, management performed an impairment test and noted there is no impairment of the ELND005 asset as at June 30, 2015.
When the carrying value of an asset is greater than its recoverable amount, which is the higher of its value in use or fair value less costs to sell, an impairment loss is recognized.
Valuation of Contingent Consideration Payable
The contingent consideration is measured at fair value based on level 3 inputs. The contingent consideration is not based on observable inputs and is measured using a discounted cash flow analysis of expected payments in future periods. The significant estimates used in the fair value calculations are as follows:
(a) Management has estimated the timing of the milestone payments based on current expectations and plans for the development of ELND005. The milestone payments are assigned a probability based on industry statistics for the successful development of pharmaceutical products including regulatory approval and achievement of revenue targets. An increase of 10% applied to the probability assumptions, with all other variables held constant, will increase the contingent consideration payable by $1,624,168. Conversely a decrease of 10% applied to the probability assumptions, with all other variables held constant, would reduce the contingent consideration payable by $1,710,592;
(b) The probability adjusted cash flows are discounted at a rate of 20% which is management’s best estimate of the Company’s cost of capital. An increase of 5% to the discount rate would decrease the contingent consideration payable by $1,191,416. Conversely, a decrease of 5% to the discount rate would increase the contingent consideration payable by $1,682,499.
There were no significant changes in the assumptions for the three and six month periods ended December 31, 2015. The Company has recognized a change in fair value of contingent consideration payable of $1,563 and $230,422 during the three and six month periods ended December 31, 2015 (three and six month periods ended December 31, 2014 - $245,658 and $470,959) mainly related to the passage of time.
Share Based Payments and Warrants
When the Company issues stock options and warrants, an estimate of fair value is derived for the equity instrument using the Black-Scholes option pricing model. The application of this option pricing model requires management to make assumptions regarding several variables, including the period for which the instrument will be outstanding, the price volatility of the Company’s stock over a relevant timeframe, the determination of a relevant risk free interest rate and an assumption regarding the Company’s dividend policy in the future. If other assumptions are used, the value derived for the equity instruments could be significantly impacted.
IFRS ISSUED BUT NOT YET ADOPTED
IFRS 15 – Revenue from Contracts with Customers
IFRS 15 specifies how and when to recognize revenue as well as requiring entities to provide users of financial statements with some informative, relevant disclosures. The standard supersedes IAS 18, Revenue, IAS 11, Construction Contracts, and a number of revenue-related interpretations. Application of the standard is mandatory for all IFRS reporters and it applies to nearly all contracts with customers: the main exceptions are leases, financial instruments and insurance contracts. Currently IFRS 15 must be applied in an entity’s first annual IFRS financial statements for periods beginning on or after January 1, 2018 with early adoption permitted. Management is evaluating the standard and has not yet determined the impact on its consolidated financial statements.
IFRS 16 – Leases
On January 13, 2016, the International Accounting Standards Board issued IFRS 16, Leases which replaces the current guidance in IAS 17, Leases. IFRS 16 requires lessees to recognize a lease liability reflecting future lease payments and a right of use asset for virtually all lease contracts. IFRS 16 must be applied to an entity’s first annual IFRS financial statements for periods beginning on or after January 1, 2019, with early adoption permitted. Management is evaluating the standard and has not yet determined the impact on its consolidated financial statements.
INTERNAL CONTROLS OVER FINANCIAL REPORTING
Internal controls over financial reporting are designed to provide reasonable assurance regarding the reliability of financial reporting and the preparation of financial statements for external purposes in accordance with IFRS.
There have been no substantive changes in the Company’s internal controls over financial reporting that have occurred during the most recent interim period beginning October 1, 2015 and ending December 31, 2015 that have materially affected, or are reasonably likely to materially affect, the Company’s internal controls over financial reporting.
LIQUIDITY AND CAPITAL RESOURCES
Overview
The Company commenced operations in July 1998, and has devoted its resources primarily to fund its research and development programs. All revenue to date has been generated from milestone payments and licensing fees. The Company has incurred a cumulative deficit to December 31, 2015 of $228,951,935. Losses are expected to continue for the next several years as the Company invests in research and development, preclinical studies, clinical trials, manufacturing and regulatory compliance.
Since inception, the Company has been financed primarily from public and private sales of equity, the exercise of warrants and stock options, interest earned on cash deposits and short term investments and revenues and reimbursements from partners.
The Company’s cash was $29,070,189 at December 31, 2015 as compared to $40,510,758 at June 30, 2015, resulting in a decrease of $11,440,569. The Company’s working capital position at December 31, 2015 decreased $5,155,903 from $32,026,606 at June 30, 2015 to $26,870,703, at December 31, 2015.
The decrease in the Company’s cash and working capital is primarily due to the expenditures incurred during the six month period ended December 31, 2015.
The Company’s current cash projection indicates that the current cash resources should enable the Company to execute its core business plan and meet its projected cash requirements beyond the next 12 months.
The success of the Company is dependent on its ability to bring its products to market, obtain the necessary regulatory approvals and achieve future profitable operations. The continuation of the research and development activities and the commercialization of its products are dependent on the Company’s ability to successfully complete these activities and to obtain adequate financing through a combination of financing activities, operations, and partnerships. It is not possible to predict either the outcome of future research and development programs or the Company’s ability to fund these programs going forward.
Financial Instruments
Financial instruments of the Company consist mainly of cash, other receivables, trade and other payables and contingent consideration payable. Management’s primary investment objective is to maintain safety of principal and provide adequate liquidity to meet all current payment obligations and future planned expenditures.
The Company is exposed to market risks related to volatility in interest rates for the Company’s investment portfolio and foreign currency exchange rates related to cash and purchases of supplies and services made in U.S. dollars.
The Company is exposed to interest rate risk to the extent that the cash is held in deposit accounts which earn interest at variable rates. The Company’s maximum exposure to interest rate risk is based on the effective interest rate of the current carrying value of these assets. The Company does not speculate on interest rates and holds all deposits until their date of maturity.
Contractual Obligations
Minimum payments under our contractual obligations are as follows:
| | | Less than 1 | | | | | | | | | | | | | |
| | | Year | | | 1- 3 Years | | | 4-5 Years | | | After 5 Years | | | Total | |
| Operating leases | | $ | 177,873 | | | $ | 335,690 | | | $ | 229,740 | | | $ | - | | | $ | 743,303 | |
| Clinical and toxicity study agreements | | | 996,557 | | | | 996,557 | | | | - | | | | - | | | | 1,993,114 | |
| Manufacturing agreements | | | 51,377 | | | | - | | | | - | | | | - | | | | 51,377 | |
| Contingent Consideration Payable | | | 2,847,759 | | | | - | | | | - | | | | 63,428,760 | | | | 66,276,519 | |
| Other | | | 155,875 | | | | - | | | | - | | | | - | | | | 155,875 | |
| TOTAL | | $ | 4,229,441 | | | $ | 1,332,247 | | | $ | 229,740 | | | $ | 63,428,760 | | | $ | 69,220,188 | |
Contractual obligations denominated in U.S. dollars have been translated to Canadian dollars using the exchange rate at December 31, 2015 of 1.3840.
The Company may also be required to pay commercial milestone payments of US$10,000,000 to Brigham and Women’s Hospital in respect of TT701.
PROPOSED TRANSACTIONS
On July 19, 2013, the Company’s shelf registration statement filed with the United States Securities and Exchange Commission (“SEC”) on Form F-3 became effective. The shelf prospectus provides for the potential offering in the United States of up to an aggregate amount of US$50 million of Transition’s common shares, warrants, or a combination thereof, from time to time in one or more offerings until July 19, 2016.
On January 5, 2015, the Company filed with the SEC a prospectus supplemental to the shelf prospectus and a sales agreement with Cowen and Company, LLC or Cowen, relating to the sale of the Company’s common shares. In accordance with the terms of the sales agreement, the Company may offer and sell from time to time common shares having an aggregate offering price of up to US $25 million with Cowen acting as sales agent. After the closing of the February 2015 US$23 million public offering, the Company can raise an additional US$27 million through the issuance of common shares, warrants or a combination thereof, from time to time in one of more offerings until July 19, 2016.
Utilization of the U.S. shelf prospectus is dependent upon meeting certain market capitalization thresholds at the time of financing.
OUTSTANDING SHARE DATA
Authorized
The authorized share capital of the Company consists of an unlimited number of common shares.
Issued and Outstanding
The following details the issued and outstanding equity securities of the Company:
Common Shares
As at February 5, 2016 the Company has 38,878,879 common shares outstanding.
Stock Options
As at February 5, 2016 the Company has 2,341,507 stock options outstanding with exercise prices ranging from $2.09 to $10.19 and various expiry dates extending to June 14, 2025. At February 5, 2016, on an if-converted basis, these stock options would result in the issuance of 2,341,507 common shares in the capital of the Company at an aggregate exercise price of $11,002,408.
Warrants
The warrants issued on August 15, 2013 expired unexercised and accordingly, the carrying value of the expired warrants of $2,025,839 was reclassified to contributed surplus during the three month period ending September 30, 2015.
As at February 5, 2016, the Company has a total of 1,949,250 warrants outstanding with a purchase price of US$7.10. Each warrant entitles the holder, within two years of the June 23, 2013 issuance date, to purchase one additional common share in the capital of the Company.
RISKS AND UNCERTAINTIES
The Company’s risks and uncertainties are as described in the Company’s annual MD&A, which can be found on SEDAR at www.SEDAR.com.