EXHIBIT 99.1
DICERNA PHARMACEUTICALS, INC.
ANNUAL REPORT ON FORM 10-K
UPDATED BY THIS CURRENT REPORT ON FORM 8-K
For the Year Ended December 31, 2015
PART I
| Item 1. | Business |
We are a biopharmaceutical company focused on the discovery and development of innovative subcutaneously delivered RNA Interference (RNAi)-based pharmaceuticals using our GalXCTM RNAi platform for the treatment of rare diseases involving the liver and for other therapeutic areas involving the liver such as chronic liver diseases, cardiovascular diseases, and viral infectious diseases. Within these therapeutic areas, we believe our GalXC RNAi platform will allow us to build a broad pipeline with commercially attractive pharmaceutical properties, including a subcutaneous route of administration, infrequent dosing (e.g., dosing that is monthly or quarterly, and potentially even less frequent), high therapeutic index, and specificity to a single target gene. Within our therapeutic areas, we seek to focus our drug discovery and development efforts on target genes where the connection between that target gene and the disease is well understood and documented.
The GalXC RNAi platform supports Dicerna’s long-term strategy to retain a full or substantial ownership stake and invest internally for diseases with focused patient populations, such as certain rare diseases, as we see such diseases as representing opportunities that carry high probabilities of success, have easily identifiable patient populations and a limited number of Centers of Excellence to facilitate reaching these patients, and have the potential for more rapid clinical development programs. For more complex diseases with multiple gene dysfunctions and larger patient populations, we plan to pursue partnerships that can provide the enhanced scale, resources and commercial infrastructure required to maximize these prospects.
GalXC Development Programs
In choosing which development programs to advance, we apply scientific, clinical, and commercial criteria that we believe allow us to best leverage our GalXC RNAi platform and maximize value. In addition to DCR-PHXC, which is in preclinical development, Dicerna expects to launch two more GalXC programs in 2016: one will focus on cardiovascular disease by targeting the PCSK9 gene; the other will focus on an undisclosed rare disease. Dicerna has the capacity to launch up to three additional programs annually, with the intent to advance five programs into the clinic by the end of 2019. Our 2016 development programs are as follows:
| • | Primary Hyperoxaluria (PH). We are developing DCR-PHXC for the treatment of PH. PH is a rare inborn error of metabolism in which the liver produces excessive levels of oxalate, which in turn causes damage to the kidneys and to other tissues in the body. In preclinical models of PH, DCR-PHXC reduces oxalate production to near-normal levels, ameliorating the disease condition. DCR-PHXC is in preclinical development, and is advancing into IND-enabling studies. We plan to file an Investigational New Drug (IND) submission or Clinical Trial Application (CTA) for DCR-PHXC in late 2017 and commence human clinical trials shortly thereafter. |
| • | In the third quarter of 2016, we announced that we transitioned our PH program to DCR-PHXC from DCR-PH1, which was a lipid nanoparticle (LNP) formulated RNAi compound. The LNP delivery system was obtained through a licensing agreement with Arbutus Biopharma Corporation (formerly known as Tekmira Pharmaceuticals Corporation). DCR-PH1 was being studied in two clinical trials, DCR-PH1-101 in patients with primary hyperoxaluria type 1 (PH1) and DCR-PH1-102 in normal healthy volunteers (NHVs). We presented initial data from the NHV study at the 17th Congress of the International Pediatric Nephrology Association (IPNA) in Iguaçu, Brazil on Sept. 22, 2016. We believe these data provide the proof of concept for the pharmacological activity of RNAi-based therapy in PH. |
1
| • | Hypercholesterolemia (PCSK9 targeted therapy). We are using our GalXC RNAi platform to develop a therapeutic that targets the PCSK9 gene for the treatment of hypercholesterolemia. PCSK9 is a validated target for hypercholesterolemia, and there are FDA-approved therapies targeting PCSK9 that are based on monoclonal antibody (MAb) technology. Based on preclinical studies, we believe that our GalXC RNAi platform can produce a PCSK9-targeted therapy with more attractive commercial properties than existing MAb therapies, based on comparatively smaller subcutaneous injection volumes and less frequent dosing, while providing equal or superior control of serum cholesterol. |
| • | An undisclosed rare disease involving the liver.We are developing a GalXC-based therapeutic, targeting a liver-expressed gene involved in a serious rare disease. For competitive reasons we have not yet publicly disclosed the target gene or disease. We have selected this target gene and disease based on criteria that include having a strong therapeutic hypothesis, a readily-identifiable patient population, the availability of a potentially predictive biomarker, high unmet medical need, favorable competitive positioning, and what we believe is a rapid projected path to approval. |
| • | 2017 development programs. We are currently using our GalXC RNAi platform to evaluate a series of target genes in multiple disease states. In 2017, we expect to initiate programs targeting hepatitis B virus (HBV), as well as two additional programs from our core therapeutic areas. |
In addition to our GalXC development programs, we have partnered our early generation, non-GalXC RNAi technology against two targets, the KRAS oncogene and an additional undisclosed gene, with the global pharmaceutical company Kyowa Hakko Kirin Co., Ltd. (KHK) to use for development in oncology and formulated using KHK’s proprietary drug delivery system. KHK is responsible for all preclinical and clinical development activities, including the selection of patient population and disease indications for clinical trials. We are eligible to receive royalties on any worldwide net sales for these product candidates. In addition, we have the option to elect to co-promote, in the U.S., a therapeutic product targeting the KRAS gene for an equal share of the profits resulting from U.S. net sales of the product.
We have also developed a wholly-owned clinical candidate, DCR-BCAT, targeting the ß-catenin oncogene for tumors believed to be driven by activating mutations in ß catenin or other tumor-driving genes in the Wnt signaling pathway. In multiple animal models including both colorectal carcinoma and hepatocellular carcinoma tumor models, DCR-BCAT has shown anti-tumor efficacy in tumors driven by ß catenin and/or Wnt signaling pathway mutations. DCR-BCAT is based on an extended version of our earlier generation Dicer Substrate RNAi technology and is delivered by an advanced version of our lipid LNP tumor delivery system, EnCoreTM. We plan to out-license or spin-out the DCR-BCAT opportunity, given our focus on our GalXC platform-based programs.
Strategy
We are committed to delivering transformative therapies based on our GalXC RNAi platform to patients with rare inherited diseases involving the liver and for other therapeutic areas involving the liver such as chronic liver diseases, cardiovascular diseases, and viral infectious diseases. The key elements of our strategy are as follows.
| • | Create new programs in indication areas with high unmet medical need. We intend to continue to use our proprietary GalXC RNAi technology platform to create new, high value pharmaceutical programs. Our primary focus will remain: (1) rare inherited diseases involving the liver; and (2) other therapeutic areas in involving the liver such as chronic liver diseases, cardiovascular diseases, and viral infectious diseases. |
| • | Validate our product candidates and our platform in clinical proof-of-concept studies. We believe data from the DCR-PH1-102 clinical trial in NHVs provide the proof of concept for the pharmacological activity of RNAi-based therapy in PH. We intend to demonstrate clinical proof-of-concept for DCR-PHXC (which focuses on the treatment of PH as well) and for our other 2016 current development programs. Based on precedents in the RNAi field, we are optimistic that our preclinical data showing the significant knockdown of target messenger RNA (mRNA) activity and disease biomarker activity, may translate into clinical results for these programs. |
2
| • | Retain significant portions of the commercial rights for certain rare disease programs. We seek to retain a full or substantial ownership stake and invest internally for diseases with focused patient populations, such as certain rare diseases, as we see such diseases representing opportunities that carry high probabilities of success, have easily identifiable patient populations and a limited number of Centers of Excellence to facilitate reaching these patients, and have the potential for more rapid clinical development programs. For more complex diseases with multiple gene dysfunctions and larger patient populations, we plan to pursue partnerships that can provide the enhanced scale, resources and commercial infrastructure required to maximize these prospects. |
| • | Enter into additional partnerships with pharmaceutical companies either on our GalXC RNAi technology platform or specific indications or therapeutic areas. We may choose to establish partnerships with pharmaceutical companies across multiple programs or indication areas depending on the attractiveness of the opportunities. These partnerships may provide us with further validation of our technology platform, funding to advance our proprietary product candidates, and/or access to development, manufacturing and commercial capabilities. |
| • | Continue to invest in our RNAi technology platform and intellectual property. We plan to continue to invest in expanding and improving our GalXC RNAi platform technology. We believe we have a robust patent portfolio covering our proprietary GalXC RNAi platform and other RNAi technologies. As of October 10, 2016, our worldwide patent estate, not including the patents and patent applications we have licensed, included over 20 issued patents or allowed patent applications and over 100 pending patent applications supporting commercial development of our RNAi molecules and delivery technologies. |
| • | Leverage the experience and the expertise of our executive management team. To execute on our strategy, we have assembled an executive management team that has extensive experience in the biopharmaceutical industry. In addition, various members of our management team and our board of directors have contributed to the progress of the RNAi field through their substantial involvement in companies such as Cephalon Inc., Genta Inc., GlaxoSmithKline plc, Pfizer Inc., Sanofi, Sirna Therapeutics Inc., and other companies. Our co-founder and chief executive officer, Douglas M. Fambrough III, Ph.D., was a lead venture capital investor and board member of Sirna Therapeutics, an early RNAi company acquired by Merck & Co., Inc. in 2006 for $1.1 billion. |
Our GalXC RNAi Technology Platform
The RNAi Therapeutic Modality All of our GalXC drug discovery and development efforts are based on the therapeutic modality of RNAi, a highly potent and specific mechanism for silencing the activity of a targeted gene. In this naturally occurring biological process, double-stranded RNA molecules induce the enzymatic destruction of the mRNA of a target gene that contains sequences that are complementary to one strand of the therapeutic double-stranded RNA molecule. Our approach is to design proprietary double-stranded RNA molecules that have the potential to engage the enzyme Dicer and initiate an RNAi process to silence a specific target gene. We refer to these proprietary molecules generally as Dicer Substrate short-interfering RNAs (DsiRNAs). Our GalXC RNAi platform utilizes a particular Dicer Substrate structure configured for subcutaneous delivery to the liver. Due to the enzymatic nature of RNAi, a single GalXC molecule incorporated into the RNAi machinery can destroy hundreds or thousands of mRNAs from the targeted gene.
RNAi therapeutics represent a novel advance in drug development. Historically, the pharmaceutical industry has developed small molecules or antibodies to inhibit the activity of disease-causing proteins. This approach is effective for many diseases; nevertheless, many proteins cannot be inhibited by either small molecules or antibodies. Some proteins lack the binding pockets small molecules require for interaction. Other proteins are solely intracellular and therefore inaccessible to antibody-based therapeutics, which are limited to cell surface and extracellular proteins. The novel advantage of RNAi is that instead of targeting proteins, RNAi goes upstream to silence the genes themselves. Rather than seeking to inhibit a protein directly, the RNAi approach is to prevent its creation in the first place.
We believe our approach to RNAi drug development provides the following qualities and advantages compared to other methods of inducing RNAi.
3
| • | We initiate RNAi through the Dicer enzyme. Our GalXC molecules are structured to be processed by the enzyme Dicer, the initiation point for RNAi in the human cell cytoplasm. Unlike earlier generation RNAi molecules, which mimic the output product of Dicer processing, all our DsiRNAs, including GalXC molecules, enter the RNAi pathway prior to Dicer processing. This can result in preferential use of the correct strand of a double-stranded RNA molecule, and therefore increase the efficacy of the RNAi mechanism. We have found in animal tests that this benefit both increases the potency of our GalXC molecules relative to other RNAi-inducing molecules and enables more sequences to be used compared to other RNAi-inducing molecules. In addition, all our DsiRNAs, including GalXC molecules, have an extended structure relative to conventional RNAi inducing molecules. This extended region presents multiple sites for chemical modification and conjugation compared to earlier RNAi technologies. At these sites, we can use modifications that enhance the drug-like properties on our molecules. Specifically, we can employ modifications that enhance the pharmacokinetic profile and/or suppress immunostimulatory activity. |
| • | Our GalXC RNAi platform enables subcutaneous dosing for delivery to the liver. The GalXC RNAi platform is designed to enable convenient subcutaneous delivery for our emerging pipeline of liver-targeted RNAi investigational therapies. The GalXC RNAi platform does not involve LNPs or other formulation components that facilitate drug delivery, which simplifies the platform and eliminates liability associated with functional excipients. Instead, our GalXC molecules are stabilized by chemical modifications and utilize a four base sequence known as a tetraloop, where each base is conjugated to a simple sugar, N-acetylgalactosamine (GalNAc), that is specifically recognized by a receptor on the surface of hepatocyte liver cells. With the GalXC RNAi platform, we believe that a full human dose may be administered via a single subcutaneous injection. After injection, the GalXC molecules enter the bloodstream and are exposed to the liver hepatocytes expressing the GalNAc receptor. After binding to the receptor, the GalXC molecules are internalized by the hepatocyte, ultimately enabling the GalXC molecules to access the RNAi machinery inside the hepatocyte. To date, we have demonstratedin vivo gene silencing activity with GalXC molecules after subcutaneous administration against nearly three dozen disease-associated genes in the liver. |
GalXC Molecule Structure

The structure of GalXC molecules underlying Dicerna’s GalXC RNAi technology platform consist of two RNA strands: a shorter Guide strand with a two-base overhang on its 3’ end, and a longer Passenger strand with a four-based “tetraloop” structure that folds back to form a short stem section that abuts the end of the Guide strand. Attached to each of the four tetraloop bases, via a short linker, is a single GalNAc (N-acetylgalactosamine) molecule. The GalNAc sugars are recognized by a receptor on the surface of hepatocyte cells in the liver, which enables the GalXC molecule to enter the hepatocyte and engage the RNAi machinery, leading the silencing of the targeted gene.
Optimization of our GalXC molecules
For therapeutic use in humans, our GalXC molecules are optimized both with respect to base sequence and chemical modifications to increase stability and mask them from mechanisms that recognize foreign RNAs, inducing immune system stimulation. Our optimization process begins with an analysis of the target gene sequence using our proprietary GalXC prediction algorithm, which we have developed based on the results of testing thousands of sequences for RNAi activity. We select the sequences with the highest predicted RNAi activity and apply patterns of chemical modification, including a GalNAc-linked tetraloop stem-loop structure, which design-in enhanced stability and hepatocyte delivery specificity and engineers-out immunostimulatory activity. Our GalXC molecules routinely achieve high potencies, with EC50 values in the liver (the amount of material required to silence
4
a target gene by 50 percent) typically in the 0.1 to 1.0 milligram per kilogram bodyweight (mg/kg) range inin vivostudies in mice. We have routinely generated GalXC molecules of this potency within 30 days of doing the initial algorithmic gene sequence analysis, which allows us to explore a large number of potential target genes when selecting our programs.
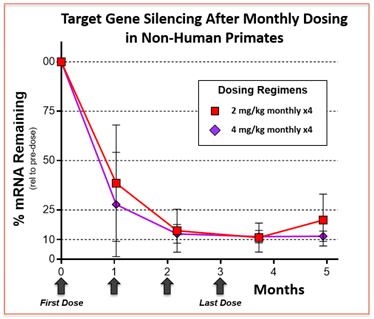
GalXC molecules yield high-potency gene silencing agents. The data are derived from a single GalXC molecule administered subcutaneously at two different dose levels, resulting in potent gene silencing of the target gene in the liver of monkeys. In this example a dose of either 2 or 4 milligrams per kilogram bodyweight (mg/kg) yields nearly 90% gene silencing after four monthly subcutaneous doses. At 4 mg/kg the full level of gene silencing was still present two months after the last dose.
Our Product Candidates
In choosing clinical programs to pursue using our GalXC technology, we apply the criteria listed below. We believe that our current development programs meet most or all of these criteria.
| • | Strength of therapeutic hypothesis. Our current product candidate gene targets, and those we intend to pursue in the future, are a well-understood part of the disease process where a therapeutic intervention is likely to have substantial benefit for the patient. |
| • | Readily-identified patient population. We seek disease indications where patients can be readily identified by the presence of characteristic genetic mutations or other readily-accessible disease features. In the case of genetic diseases, these are heritable genetic mutations that can be identified with available genetic tests. |
| • | Predictivity of biomarkers for early efficacy assessment. We seek disease indications where there is a clear relationship between the disease status and an associated biomarker that we can readily measure. This approach will allow us to determine in early stages of clinical development whether our GalXC molecules are likely to have the expected biological and clinical effects in patients. |
| • | Unmet medical need. We seek to provide patients with significant benefit and alleviation of disease. The indications we choose to approach have high unmet medical need, which is intended to enable us to better access patients and qualify for pricing and reimbursement that justify our development efforts. |
| • | Competitive positioning. We seek indications where we believe we have the opportunity to develop either a first-in-class product or a clearly differentiated therapy. |
| • | Rapid development path to approval. To reach commercialization expeditiously and to help ensure our ability to finance development of our product candidates, we have identified indications with the potential for rapid development through marketing approval. Specifically, we believe that certain of our product candidates have the potential to obtain Breakthrough Therapy Designation as well as accelerated review process from the U.S. Food and Drug Administration (FDA). |
5
DCR-PHXC for PH
During the second quarter of 2016, Dicerna announced the first GalXC clinical candidate, DCR-PHXC, which is in preclinical development. We plan to file an IND or CTA for DCR-PHXC in late 2017 and commence human clinical trials shortly thereafter.
PH is a family of rare, inherited autosomal recessive disorder of metabolism in the liver. The most common and severe form of PH is Primary Hyperoxaluria type 1 (PH1), which usually results in severe damage to the kidneys. PH1 is caused by the failure of the liver to metabolize a precursor of oxalate, a highly insoluble metabolic end-product in humans, resulting in excess oxalate production. This oxalate is formed during the metabolic breakdown of hydroxyproline, a naturally occurring component of collagen. In individuals with PH1, crystals of calcium oxalate form in the renal tubules, leading to chronic and painful cases of kidney stones and subsequent fibrosis, known as nephrocalcinosis. Despite the typical interventions of a large daily intake of water to dilute the oxalate and other interventions, many patients eventually develop kidney failure (end-stage renal disease, or ESRD) and require transplant. The median age for kidney failure in PH1 patients is 23 years old. While in ESRD, besides having to endure frequent dialysis, patients are afflicted with a build-up of oxalate in the bone, skin, heart, retina, and other tissues with concomitant debilitating complications, a condition known as systemic oxalosis. Some patients show partial disease amelioration with oral pyridoxine supplementation, although disease progression usually continues. Supportive care treatments are available, generally with only minor or no effect on disease progression. Currently, aside from dual liver and kidney organ transplantation, there are no highly efficacious therapeutic options for most patients with PH1. Dual liver and kidney transplantation presents a challenge in identifying a donor and is associated with high morbidity and mortality rates. Even in those U.S. patients treated with dual liver and kidney transplant, five-year post-transplant survival is 64 percent. For patients treated with kidney transplant alone, five-year survival is 45 percent.
While the true prevalence of PH1 is unknown, according to estimates recently published by theNew England Journal of Medicine, the prevalence of PH1 is at least one to three per million of population. Based on the frequency of occurrence of disease mutations in the population derived from genome sequence databases, the estimated genetic incidence is six and half per million of population, which we believe suggests that PH1 is under-diagnosed. Roughly consistent with the genetic incidence estimate, the disease is thought to have an incidence of one per 120,000 live births a year in Europe. Certain populations, for example in the Canary Islands (Spain) or Kuwait, have higher incidences due to founder effects or consanguinity. We believe over 1,000 patients total are currently in two distinct disease registries in North America and Europe, although these registries do not capture all afflicted patients. Incidence is believed to be similar in Asia. Given the severity of PH1, we believe this disease represents a significant market opportunity. The patient advocacy group, the Oxalosis and Hyperoxaluria Foundation, based in New York City, New York, seeks to represent patients with PH1.
We believe that there is a strong rationale for focusing our RNAi technology on the development of product candidates for the treatment of PH1. The hydroxyproline breakdown metabolic pathway that is disrupted in PH1 consists of a number of enzymes. The gene encoding the final enzyme in the pathway, alanine-glyoxylate aminotransferase 1 (AGT1), is mutated in patients with PH1. Under normal circumstances, AGT1 metabolizes oxalate precursors into the harmless amino acid glycine, which is then used by the body or excreted. But when AGT1 function is disrupted due to mutation, oxalate begins to build up, resulting in progressive loss of kidney function and, ultimately, kidney failure. DCR-PHXC is designed to block the production of oxalate in patients with PH1.
Using DCR-PHXC, and also other GalXC molecules synthesized during the discovery and optimization of DCR-PHXC, we have shown that RNAi can be used to block the production of oxalate in an animal model of PH1.
6
These studies employ mice in which the gene encoding AGT1 has been genetically deleted to create an animal model of PH1. Similar to human patients, these mice have elevated levels of oxalate in their urine. A single dose of DCR-PHXC of 5 mg/kg delivered subcutaneously in the animal model of PH1 silences target gene expression by greater than 90% and results normalization or near normalization of urinary and plasma oxalate levels. We believe these results, if achievable in patients with PH1, would be highly beneficial.
Additional programs under investigation involving the liver
We are investigating a number of other rare diseases and other therapeutic classes involving disease target genes expressed in the liver. We have selected these target genes and diseases based on our stated criteria, including having a strong therapeutic hypothesis, a readily-identifiable patient population, the availability of a potentially predictive biomarker, high unmet medical need, favorable competitive positioning, and what we believe is a rapid projected path to approval. In addition to DCR-PHXC, which is in preclinical development, Dicerna expects to launch two more GalXC programs in 2016: one will focus on cardiovascular disease by targeting the PCSK9 gene; the other will focus on an undisclosed rare disease. Dicerna has the capacity to launch up to three additional programs annually, with the intent to advance five programs into the clinic by the end of 2019.
Intellectual Property
We invest significant amounts in research and development. Our research and development expenses were approximately $44.0 million, $29.5 million and $11.6 million in 2015, 2014 and 2013, respectively.
We are seeking multifaceted protection for our intellectual property that includes licenses, confidentiality and non-disclosure agreements, copyrights, patents, trademarks and common law rights, such as trade secrets. We enter into confidentiality and proprietary rights agreements with our employees, consultants, collaborators, subcontractors and other third parties and generally control access to our documentation and proprietary information.
Patents and proprietary rights
We own U.S. patents and a number of pending patent applications with claims to methods and compositions of matters that cover various aspects of our RNAi technology and our discovery technologies, including our proprietary GalXC technology. These U.S. patents include U.S. 8,349,809 (issued in January 2013 with a projected expiration date of January 2030), U.S. 8,513,207 (issued in August 2013 with a projected expiration date of May 2030) and U.S. 8,927,705 (issued in January 2015 with a projected expiration date of July 2030). We also own numerous patents and patent applications covering specific DsiRNA sequences that drive activity against high value disease targets, including KRAS (U.S. 8,372,816; issued in February 2013, with projected expiration in April 2030), HAO1, CTNNB1 (ß catenin; U.S. 9,428,752; issued in August 2016, with projected expiration in July 2031), Androgen Receptor (US 8,927,515; issued in January 2015, with projected expiration in September 2031); Alpha-1-antitrypsin (US 9,458,457; issued October 4, 2016, with projected expiration in July 2034). Further, we own seven U.S. patents expiring by 2017 and numerous patent applications with claims to methods and compositions of matters related to our lipid delivery technology, such as lipid compositions and particle formulations and the EnCore formulation process. We have issued or pending claims to DsiRNA molecules, pharmaceutical compositions/formulations, methods of use, includingin vitroandin vivomethods of reducing target gene expression, methods of treatment, methods of inhibiting cell growth and methods of synthesis.
We jointly own with KHK U.S. and foreign patent applications pursuant to our research collaboration and license agreement claiming developments made in the course of the collaboration focused on delivery of KRAS specific DsiRNA molecules. Depending on the subject matter of future issued claims, we may also jointly own future patents issuing from patent applications filed under the research collaboration and license agreement with KHK.
Our strategy around protection of our proprietary technology, including any innovations and improvements, is to obtain worldwide patent coverage with a focus on jurisdictions that represent significant global pharmaceutical markets. Generally, patents have a term of 20 years from the earliest non-provisional priority date, assuming that all maintenance fees are paid, no portion of the patent has been terminally disclaimed and the patent has not been
7
invalidated. In certain jurisdictions, and in certain circumstances, patent terms can be extended or shortened. We are obtaining worldwide patent protection for at least novel molecules, composition of matter, pharmaceutical formulations, methods of use, including treatment of disease, methods of manufacture and other novel uses for the inventive molecules originating from our research and development efforts. We continuously assess whether it is strategically more favorable to maintain confidentiality for the “know-how” regarding a novel invention rather than pursue patent protection. For each patent application that is filed we strategically tailor our claims in accordance with the existing patent landscape around a particular technology. There can be no assurance that an issued patent will remain valid and enforceable in a court of law through the entire patent term. Should the validity of a patent be challenged, the legal process associated with defending the patent may be costly and time consuming. Issued patents can be subject to oppositions, interferences, post-grant proceedings, and other third party challenges that can result in the revocation of the patent or limit patent claims such that patent coverage lacks sufficient breadth to protect subject matter that is commercially relevant. Competitors may be able to circumvent our patents. Development and commercialization of pharmaceutical products can be subject to substantial delays and it is possible that at the time of commercialization any patent covering the product will have expired or will be in force for only a short period of time thereafter.
We cannot predict with any certainty if any third party U.S. or foreign patent rights, other proprietary rights, will be deemed infringed by the use of our technology. Nor can we predict with certainty which, if any, of these rights will or may be asserted against us by third parties. Should we need to defend ourselves and our partners against any such claims, substantial costs may be incurred. Furthermore, parties making such claims may be able to obtain injunctive or other equitable relief, which could effectively block our ability to develop or commercialize some or all of our products in the U.S. and abroad, and could result in the award of substantial damages. In the event of a claim of infringement, we or our partners may be required to obtain one or more licenses from a third party. There can be no assurance that we can obtain a license on a reasonable basis should we deem it necessary to obtain rights to an alternative technology that meets our needs. The failure to obtain a license may have a material adverse effect on our business, results of operations and financial condition.
We also rely on trade secret protection for our confidential and proprietary information. No assurance can be given that we can meaningfully protect our trade secrets on a continuing basis. Others may independently develop substantially equivalent confidential and proprietary information or otherwise gain access to our trade secrets.
See “— Risk Factors — Risks Related to Intellectual Property” for a more detailed discussion of the risks to our intellectual property.
It is our policy to require our employees and consultants, outside scientific collaborators, sponsored researchers and other advisors who receive confidential information from us, to execute confidentiality agreements upon the commencement of employment or consulting relationships. These agreements provide that all confidential information developed or made known to these individuals during the course of the individual’s relationship with us is to be kept confidential and is not to be disclosed to third parties except in specific circumstances. The agreements provide that all inventions conceived by an employee shall be our property. There can be no assurance, however, that these agreements will provide meaningful protection or adequate remedies for our trade secrets in the event of unauthorized use or disclosure of such information.
Our success will depend in part on our ability to obtain and maintain patent protection, preserve trade secrets, prevent third parties from infringing upon our proprietary rights and operate without infringing upon the proprietary rights of others, both in the U.S. and other territories worldwide.
Strategic Partnership
KHK research collaboration and license agreement
In December 2009, we entered into a research collaboration and license agreement (the collaboration agreement) with KHK for the research, development and commercialization of drug delivery platforms and DsiRNA molecules for therapeutic targets, primarily in oncology. Under the collaboration agreement, we engaged in the discovery of DsiRNA molecules against KRAS and other gene targets nominated by KHK. In 2011, KHK exercised its option for one additional target, the identity of which we have not publicly disclosed. As part of the research we
8
are conducting in the collaboration, we are using our specific RNAi-inducing double-stranded DsiRNA molecules with a lipid nanoparticle drug delivery platform proprietary to KHK. KHK is responsible for all costs it incurs to develop any compound that is directed against a target included in the collaboration that KHK designates for development, subject to our exercise of our co-promotion option with respect to that compound if that compound is directed against KRAS.
We have granted KHK an exclusive license to certain of our technology and patents relating to compounds resulting from the collaboration. KHK has granted us certain non-exclusive licenses in its technology as necessary for us to perform research and development activities as part of the research collaboration.
Under the terms of the collaboration agreement, we have received total payments of $17.5 million. We are entitled to receive up to an additional $110.0 million for each product candidate resulting from the collaboration of certain clinical, regulatory and commercialization milestones. KHK is also obligated to pay us royalties on worldwide net sales of products resulting from the research collaboration. The amount of royalty varies depending on the total worldwide net sales and range from percentages of net sales in the high single digits to the teens. None of the previously-paid milestones are subject to reimbursement.
We have the option to elect to co-promote the KRAS product in the U.S. for an equal share of the profits resulting from U.S. net sales of the product.
If we exercise our option to co-promote a KRAS product in the U.S., the collaboration agreement will remain in effect pursuant to its terms in the U.S. for as long as any product is being sold by either KHK or us in the U.S. For each country outside of the U.S., the collaboration agreement will remain in effect pursuant to its terms on a product-by-product and country-by-country basis until the later of the last to expire of any patent rights licensed under the agreement applicable to the manufacture, use or sale of the product or twelve years after the date of the first commercial sale of such product in the applicable country. In the event we do not exercise our option to co-promote a KRAS product in the U.S., the collaboration agreement will remain in effect pursuant to its terms on a product-by-product and country-by-country basis until the later of the last to expire of any patent rights licensed under the agreement applicable to the manufacture, use or sale of the product or twelve years after the date of the first commercial sale of such product in the applicable country.
KHK may terminate the agreement at any time upon prior written notice to us until such time as we exercise our option to co-promote under the agreement. We may terminate the agreement if KHK challenges the validity or enforceability of any patents licensed by us to KHK. Either we or KHK may terminate the agreement in the event of the bankruptcy or uncured material breach by the other party.
License Agreements
City of Hope license agreement
In September 2007, we entered into a license agreement with City of Hope (COH), an academic research and medical center, pursuant to which COH has granted to us an exclusive (subject to the exception described below), royalty-bearing, worldwide license under certain patent rights in relation to DsiRNA, including the core DsiRNA patent (U.S. 8,084,599), to manufacture, use, offer for sale, sell and import products covered by the licensed patent rights for the prevention and treatment of any disease in humans. COH is restricted from granting any additional rights to develop, manufacture, use, offer to sell, sell or import products covered by the licensed patent rights for the prevention and treatment of any disease in humans. Prior to entering into the license with us, COH had entered into a non-exclusive license with respect to such patent rights to manufacture, use, import, offer for sale and sell products covered by the licensed patent rights for the treatment or prevention of disease in humans (excluding viruses and delivery of products into the eye or ear). While that non-exclusive license has been terminated, a sublicensee to that non-exclusive license was permitted to enter into an equivalent non-exclusive license which, to our knowledge, is subsisting with Arrowhead Research Corporation, (Arrowhead) as successor to the non-exclusive license holder. In addition, COH has granted to us an exclusive, royalty-bearing, worldwide license under the licensed patent rights providing certain rights for up to 20 licensed products selected by us for human diagnostic uses, provided that COH has not granted or is not negotiating a license of rights to diagnostic uses for such licensed products to a third party.
9
The exclusive licenses granted by COH to us under the agreement are subject to any retained rights of the U.S. government in the licensed patent rights and a royalty-free right of COH to practice the licensed patent rights for educational, research and clinical uses. We have the right to sublicense the licensed patent rights to third parties with COH’s written consent. The core DsiRNA patent (U.S. 8,084,599), titled “methods and compositions for the specific inhibition of gene expression by double-stranded RNA,” describes RNA structures having a 25 to 30 nucleotides sense strand, a blunt end at the 3’ end of the sense strand and a one to four nucleotides overhang at the 3’ end of the antisense strand. The expiration date of this patent is July 17, 2027. The COH license is applicable to our DCR-MYC and KHK programs.
Pursuant to the terms of the agreement, we paid COH a one-time, non-refundable license fee and issued shares of our common stock to COH and a co-inventor of the core DsiRNA patent. COH is entitled to receive milestone payments in an aggregate amount of up to $5.25 million for each licensed product upon achievement of certain clinical and regulatory milestones. COH is further entitled to receive royalties at a low single-digit percentage of any net sale revenue of the licensed products sold by us and our sublicensees. If we sublicense the licensed patent rights to a third party, COH has the right to receive a double digit percentage of sublicense income, the percentage of which decreases after we have expended $12.5 million in development and commercialization costs. We are also obligated to pay COH an annual license maintenance fee, which may be credited against any royalties due to COH in the same year, and reimburse COH for expenses associated with the prosecution and maintenance of the license patent rights. Royalties shall be paid on a product-by-product and country-by-country basis until the expiration in each country of the last to expire of the licensed patent rights.
Under the agreement, we are obligated to use commercially reasonable efforts to develop and commercialize the licensed products in certain major markets. COH has the right to terminate the agreement in its entirety if we fail to enroll patients for clinical trials of one or more licensed products at various phases before certain specified deadlines unless we exercise the right to extend the deadlines in one-year increments by making a payment of $0.5 million to COH for each one-year extension. We have extended one milestone deadline for three one-year extensions, paying an aggregate of $1.5 million to COH for such extensions.
The agreement will remain in effect pursuant to its terms until all of the obligations under the agreement with respect to the payment of milestones or royalties related to licensed products have terminated or expired. Either party may terminate the license agreement for any uncured material breach by the other party. COH may terminate the agreement upon our bankruptcy or insolvency. We may terminate the agreement without cause upon written notice to COH.
Plant Bioscience Limited license agreement
In September 2013, we entered into a commercial license agreement with Plant Bioscience Limited (PBL), pursuant to which PBL has granted to us a nominated-target-limited, worldwide, non-exclusive, fee-bearing license to certain of its U.S. patents (the Baulcombe patent estate) and patent applications to research, discover, develop, manufacture, sell, import and export, for human diagnostic and therapeutic uses, products incorporating one or more short RNA molecules (SRMs) designed to target and modify the expression of a human gene or genes nominated by us from time to time. We are entitled to nominate multiple SRMs and have so far nominated one gene as the first SRM under the agreement. We are not obligated to nominate any additional genes.
We have paid PBL a one-time, non-refundable signature fee and will pay PBL a nomination fee for any additional SRMs nominated by us under the agreement. We are further obligated to pay PBL milestone payments in an aggregate amount of up to $3.85 million for each licensed product upon achievement of certain clinical and regulatory milestones. In addition, PBL is entitled to receive royalties at a low single-digit percentage of any net sale revenue of any licensed products sold by us. The agreement will expire on a country-by-country basis in each country where any licensed products are used, provided, manufactured or sold upon the date of the last to expire of applicable valid claim. Each party may terminate the agreement for any uncured material breach by the other party. We may terminate the agreement at any time for convenience upon prior written notice to PBL. The PBL license is applicable to our KHK programs.
10
Carnegie Institution of Washington license agreement
In January 2009, we entered into a license agreement with the Carnegie Institution of Washington (Carnegie), pursuant to which Carnegie has granted to us a worldwide, non-exclusive license under certain of its patents and patent applications (the Fire and Mello patent estate) relating to genetic inhibition by double-stranded RNA molecules for internal research, screening and development of product candidates for human and non-human diagnostic and therapeutic uses. We have paid Carnegie a one-time upfront fee and will in addition pay an annual license fee during the term of the agreement. We are further obligated to make two one-time additional payments in the aggregate amount of $100,000 upon achievement of the filing with the FDA of an NDA for a licensed product candidate and the first commercial sale of a licensed product candidate or licensed method. Carnegie is entitled to receive royalties on any net sale revenue from licensed product candidates sold by us, with the royalty rate to be further negotiated between Carnegie and us in good faith reflecting customary rates in the industry.
The agreement will terminate with respect to each licensed product candidate upon the last to expire of any valid claim within the licensed patent rights. Each party may terminate the agreement upon any uncured material breach by the other party. We may terminate the agreement at any time for any reason upon written notice to Carnegie. Any patents associated with this license will expire in 2018, removing any obligations.
Manufacturing and Supply
We do not currently own or operate manufacturing facilities for the production of preclinical, clinical or commercial quantities of any of our product candidates. For each product candidate, we currently contract with drug substance manufacturers and we expect to continue to do so to meet the preclinical and any clinical requirements of our product candidates. In June 2016, we entered into a supply agreement with a third party for supply of certain products and services. There is no minimum purchase requirement for the services provided by this third party.
Currently, some of our drug starting materials for our manufacturing activities are supplied by a single source supplier. We have agreements for the supply of such drug materials with manufacturers or suppliers that we believe have sufficient capacity to meet our demands. In addition, we believe that adequate alternative sources for such supplies exist. However, there is a risk that, if supplies are interrupted, it would materially harm our business. We typically order raw materials and services on a purchase order basis and do not enter into long-term dedicated capacity or minimum supply arrangements.
In November 2014, we entered into a development and supply agreement with Arbutus. Arbutus will perform, or subcontract, certain development and manufacture processes in accordance with the specifications in development and supply agreement. There is no minimum purchase requirement for the services provided by Arbutus.
KHK is responsible for all manufacturing under our collaboration agreement with KHK both for the KRAS DsiRNA and the oncology program selected by KHK for development under the agreement.
Manufacturing is subject to extensive regulations that impose various procedural and documentation requirements, which govern record keeping, manufacturing processes and controls, personnel, quality control and quality assurance, among others. Our contract manufacturing organizations manufacture our product candidates under current Good Manufacturing Practice (cGMP) conditions. cGMP is a regulatory standard for the production of pharmaceuticals that will be used in humans.
Competition
We believe that our scientific knowledge and expertise in RNAi-based therapies provide us with competitive advantages over the various companies and other entities that are attempting to develop similar treatments. However, we face competition at the technology platform and therapeutic indication levels from both large and small biopharmaceutical companies, academic institutions, governmental agencies and public and private research institutions. Many of our competitors have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel and establishing clinical trial sites and patient registration for clinical trials, as well as in acquiring technologies complementary to, or necessary for, our programs.
11
Our success will be based in part upon our ability to identify, develop and manage a portfolio of drugs that are safer and more effective than competing products in the treatment of our targeted patients. Our commercial opportunity could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, are more convenient or are less expensive than any products we may develop.
RNA-based therapeutics
To our knowledge, there are no other companies developing GalXC molecules for therapeutic use. However, there are several companies that are currently developing RNAi-based therapies for various indications. We believe that Arrowhead Pharmaceuticals, Alnylam and Arbutus through their company specific development or through various partnerships with the aforementioned companies are developing RNAi-based therapies that are competing against our current programs or potential future programs.
Among these, Alnylam, in partnership with Genzyme (a Sanofi company), is developing multiple genetic rare disease programs including its patisiran (ALN-TTR) program, which is an RNAi-based therapy for the treatment of hereditary transthyretin-mediated amyloidosis with polyneuropathy (hATTR-PN) and is currently in Phase 3 trials. Alnylam has announced the APOLLO study has completed enrollment of 225 patients at 44 sites in 19 countries, between December 2013 and January 2016 and it expects to announce top-line data from the study in mid-2017. In October 2016, Alnylam discontinued the development of revusiran (ALN-TTRsc), a potential treatment for hereditary ATTR amyloidosis with cardiomyopathy (hATTR-CM). Additional genetic rare disease programs are being developed by Alnylam in partnership with Genzyme including ALN-TTRsc02 for all forms of ATTR amyloidosis; fitusiran (ALN-AT3), for the treatment of hemophilia and rare bleeding disorders; ALN-GO1for the treatment of Primary Hyperoxaluria Type 1 (PH1); ALN-CC5 for the treatment of complement-mediated diseases; ALN-AS1, for the treatment of acute hepatic porphyrias; and ALN-AAT, for the treatment of AAT deficiency-associated liver disease; among others. In addition, Alnylam initiated a Phase 1/2 clinical trial with ALN-HBV for the treatment of hepatitis B virus (HBV) infection in mid-2016 and previously announced its intention to seek strategic partnerships for its hepatic infectious disease therapeutic area. The Medicines Company (MDCO) and their partner, Alnylam Pharmaceuticals, are advancing an experimental PCSK9 RNAi therapy (PCSK9si) for the treatment of hypercholesterolemia.
Arbutus is clinically investigating its RNAi molecules for use in treating serious human diseases, such as cancer and viral infections, including hepatitis B virus (HBV) and Ebola. ARB-1467 for the treatment of HBV is in a multi-dose Phase II study in chronic HBV patients that was initiated in December 2015 and Arbutus reported interim results from the first two cohorts of the ongoing study in September 2016. Arbutus has rights under Alnylam’s intellectual property to develop 13 RNAi therapeutic products.
Arrowhead Pharmaceuticals is developing multiple RNAi therapeutics including ARC-520 and ARC-521 for chronic hepatitis B (HBV);and ARC-AAT for the treatment of liver disease associated with alpha-1 antitrypsin deficiency (AATD).
In addition to RNAi therapies, there are other intracellular technologies focused on silencing the activity of specific genes by targeting mRNAs copied from them. Companies such as miRagen Therapeutics, Inc., Mirna Therapeutics, Inc., Regulus Therapeutics Inc. and Santaris Pharma A/S, which was acquired by Roche in 2014 and is now known as Roche Innovation Center Copenhagen (RICC), target or inhibit or replace microRNAs, which are approximately 22 nucleotides in length, short, non-coding RNAs, to alter mRNA expression levels. Wave Life Sciences is developing stereopure nucleic acid therapeutics spanning multiple modalities including antisense, exon-skipping, and single-stranded RNAi. Moderna and other companies are developing a new class of drugs made of messenger RNA (mRNA). This new drug modality may be able to direct the body’s cellular machinery to produce therapeutic proteins of interest that may have therapeutic benefit for the treatment of various diseases. The product candidates being developed by these companies are currently in preclinical and clinical trials for various indications. If our lead product candidates are approved for the indications for which we undertake clinical trials, they may compete with therapies that are either in development or currently marketed by our competitors.
12
Primary Hyperoxaluria Type 1
The current standard of care for treating PH1 is dual-organ transplant, namely a kidney and liver transplant in patients with PH1, which is often difficult to perform due to lack of donors and the threat of organ rejection. Other treatments include pyridoxine regimens and intensive dialysis, as well as treatments generally used in kidney stone disorders such as high-volume fluid intake and oral citrate. These other treatments do not halt disease progression. OxThera has a competing approach to PH1 treatment, currently in Phase 2 clinical trials, that is not RNAi-based. In January 2016, Alnylam announced their plans to start a Phase 1 clinical trial for ALN-GO1, an investigational RNAi therapeutic for the treatment of PH1. Alnylam presented initial Phase 1 clinical data from its NHV portion of the study in the third quarter of 2016 at the IPNA.
Hypercholesterolemia
Hypercholesterolemia is characterized by abnormally high blood serum levels of low-density lipoproteins (LDL) and is one of the key known risk factors for atherosclerosis and cardiovascular disease (CVD). Managing hypercholesterolemia by lowering LDL is one of the cornerstones of the strategy to reduce the risk of CVD morbidity and mortality.
The use of statins to lower LDL and reduce CVD morbidity and mortality has been successful although many patients may benefit from additional and alternative therapeutics that more aggressively lower LDL. It is estimated that 35 million U.S. patients are treated with statin therapy with approximately 12 million of these patients classified as suffering from cardiovascular disease (CVD) placing them at higher risk of CVD morbidity and mortality. Roughly 37%, or 4.5 million of these higher risk CVD patients, are not treated to their LDL goal with standard of care therapy: diet and statin drugs. Inhibition of the circulating protein proprotein convertase subtilisin/kexin type 9 (PCSK9) using anti-PCSK9 monoclonal antibodies (MAb’s) has been a strategy utilized to more aggressively lower serum LDL levels than with statin therapy alone.
PRALUENT® (alirocumab) was approved in July 2015 and launched in the U.S. as a second line treatment for adults with heterozygous familial hypercholesterolemia or clinical atherosclerotic cardiovascular disease whose LDL cholesterol is not adequately controlled by diet and statin treatment. Alirocumab was the first anti-PCSK9 MAb to receive FDA approval and was developed by Sanofi and Regeneron. Repatha® (evolocumab) was the second anti-PCSK9 MAb to receive FDA approval and was developed by Amgen. There are additional anti-PCSK9 MAb therapies in clinical development. Multiple cardiovascular outcome studies are being conducted with the anti-PCSK9 MAb therapies to determine if these higher risk patients will have superior cardiovascular outcomes vs. patients treated with standard of care. Results from these trials are expected in early 2017 and, if successful, may drive increased demand for anti-PCSK9 MAb therapies such as alirocumab and evolovumab.
The Medicines Company (MDCO) and their partner, Alnylam Pharmaceuticals, are advancing an experimental PCSK9 RNAi therapy (PCSK9si) that has a similar mechanism of action as Dicerna’s GalXC PCSK9. Recently, MDCO provided an update of their ongoing study of PCSK9si in a placebo-controlled, double-blind, randomized Phase II trial of single or multiple subcutaneous injections of PCSK9si in patients. The primary endpoint of the study is the percentage change in LDL from baseline at Day 180 and an interim analysis of Day 90 follow-up for all patients will be conducted and presented at the AHA Scientific Sessions on November 15, 2016. No safety concerns have been raised to date by the Independent Data Monitoring Committee (IDMC) monitoring the ongoing PCSK9si studies.
Government Regulation and Product Approval
Governmental authorities in the U.S., at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, promotion, storage, record-keeping, advertising, distribution, sampling, marketing, safety, post-approval monitoring and reporting, and export and import of products such as those we are developing. Our product candidates must be approved by the FDA through the NDA process before they may be legally marketed in the U.S. and will be subject to similar requirements in other countries prior to marketing in those countries. The process of obtaining regulatory approvals and the subsequent compliance with applicable federal, state, local and foreign statutes and regulations require the expenditure of substantial time and financial resources.
13
U.S. government regulation
NDA approval processes
In the U.S., the FDA regulates drugs under the Federal Food, Drug, and Cosmetic Act (the FDCA) and implementing regulations. Failure to comply with the applicable U.S. requirements at any time during the product development or approval process, or after approval, may result in a delay of approval or subject an applicant to administrative or judicial sanctions, any of which could have a material adverse effect on us. These sanctions could include:
| • | refusal to approve pending applications; |
| • | withdrawal of an approval; |
| • | imposition of a clinical hold; |
| • | issuance of warning or untitled letters; |
| • | product recalls; |
| • | product seizures; |
| • | refusals of government contracts; |
| • | total or partial suspension of production or distribution; or |
| • | injunctions, fines, restitution, disgorgement, civil penalties or criminal prosecution. |
The process required by the FDA before a drug may be marketed in the U.S. generally includes the following:
| • | completion of nonclinical laboratory tests, animal studies and formulation studies conducted according to Good Laboratory Practices (GLPs) or other applicable laws and regulations; |
| • | submission to the FDA of an investigational new drug application (IND), which must become effective before human clinical trials may begin; |
| • | approval by an institutional review board (IRB) at each clinical site before each trial may be initiated |
| • | performance and inspection of adequate and well-controlled human clinical trials and clinical data according to FDA regulations and Good Clinical Practices (GCP) to establish the safety and efficacy of the product candidate for its intended use; |
| • | submission of an NDA to FDA and FDA’s acceptance of the NDA for filing; |
| • | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the product candidate is produced to assess compliance with current Good Manufacturing Practices (cGMPs) to assure that the facilities, methods and controls are adequate to preserve the product candidate’s identity, strength, quality and purity; |
| • | satisfactory completion of an FDA inspection of the major investigational sites to ensure data integrity and assess compliance with good clinical practice (GCP) requirements; and |
| • | FDA review and approval of the NDA. |
Once a pharmaceutical candidate is identified for development, it enters the preclinical or nonclinical testing stage. Nonclinical tests include laboratory evaluations of product chemistry, stability, toxicity and formulation, as well as animal studies. An IND sponsor must submit the results of the nonclinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. Some nonclinical testing may continue even after the IND is submitted. In addition to including the results of the nonclinical studies, the IND will also include a protocol detailing, among other things, the objectives of the clinical trial, the parameters to be used in monitoring safety and the effectiveness criteria to be evaluated if the first phase lends itself to an efficacy determination. The IND automatically becomes effective 30 days after receipt by the FDA, unless the FDA, within the 30-day time period, places the IND on clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before clinical trials can begin. A clinical hold may occur at any time during the life of an IND and may affect one or more specific studies or all studies conducted under the IND.
14
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with FDA regulations and GCPs. They must be conducted under protocols detailing the objectives of the trial, dosing procedures, research subject selection and exclusion criteria and the safety and effectiveness criteria to be evaluated. Each protocol and protocol amendments must be submitted to the FDA as part of the IND, and progress reports detailing the status of the clinical trials must be submitted to the FDA annually. Sponsors also must timely report to FDA serious and unexpected adverse reactions, any clinically important increase in the rate of a serious suspected adverse reaction over that listed in the protocol or investigation brochure or any findings from other studies or animal orin vitrotesting that suggest a significant risk in humans exposed to the drug. All research subjects or their legally authorized representatives must provide their informed consent in writing prior to their participation in a clinical trial. An institutional review board (IRB) at each institution participating in the clinical trial must review and approve the protocol and the informed consent form before a clinical trial commences at that institution, monitor the study until completed and otherwise comply with IRB regulations. Information about most clinical trials must be submitted within specific timeframes to the National Institutes of Health (NIH) to be publicly posted on the ClinicalTrials.gov website.
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined.
| • | Phase 1—The product candidate is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and elimination. In the case of some product candidates for severe or life-threatening diseases, such as cancer, especially when the product candidate may be inherently too toxic to ethically administer to healthy volunteers, the initial human testing is often conducted in patients. |
| • | Phase 2—Clinical trials are performed on a limited patient population intended to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| • | Phase 3—Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These studies are intended to establish the overall risk-benefit ratio of the product and provide an adequate basis for product labeling. |
Human clinical trials are inherently uncertain and Phase 1, Phase 2 and Phase 3 testing may not be successfully completed. The FDA, the sponsor, or a data safety monitoring board, may suspend a clinical trial at any time for a variety of reasons, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its institution if the clinical trial is not being conducted in accordance with the IRB’s requirements or if the product candidate has been associated with unexpected serious harm to patients.
During the development of a new product candidate, sponsors are given opportunities to meet with the FDA at certain points. These points may be prior to the submission of an IND, at the end of Phase 2 and before an NDA is submitted. Meetings at other times may be requested. These meetings can provide an opportunity for the sponsor to share information about the data gathered to date and for the FDA to provide advice on the next phase of development. Sponsors typically use the meeting at the end of Phase 2 to discuss their Phase 2 clinical results and present their plans for the pivotal Phase 3 clinical trial that they believe will support the approval of an NDA. If a Phase 2 clinical trial is the subject of discussion at the end of Phase 2 meeting with the FDA, a sponsor may be able to request a Special Protocol Assessment (SPA), the purpose of which is to reach agreement with the FDA on the Phase 3 clinical trial protocol design and analysis that will form the primary basis of an efficacy claim.
Concurrent with clinical trials, sponsors usually complete additional animal safety studies and also develop additional information about the chemistry and physical characteristics of the product candidate and finalize a process for manufacturing commercial quantities of the product candidate in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and the manufacturer must develop methods for testing the safety, identity, strength, purity, and quality of the product candidate. Additionally, appropriate packaging must be selected and tested and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its proposed shelf-life. Before approving an NDA, the FDA will inspect the facility or facilities where the product is manufactured and tested and will not approve the product unless cGMP compliance is satisfactory. The FDA will also typically inspect one or more clinical sites to assure compliance with FDA regulations and GCPs.
15
The results of product development, nonclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests and other control mechanisms, proposed labeling and other relevant information are submitted to the FDA as part of an NDA requesting approval to market the product. The submission of an NDA is subject to the payment of user fees, but a waiver of such fees may be obtained under specified circumstances. The FDA reviews all NDAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. It may request additional information rather than accept an NDA for filing. In this event, the NDA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing.
Once the submission is accepted for filing, the FDA begins an in-depth review. The FDA reviews an NDA to determine, among other things, whether a product is safe and effective for its intended use and whether its manufacturing is cGMP-compliant. The FDA typically requires that an NDA include data from two adequate and well-controlled clinical trials, but approval may be based upon a single adequate and well-controlled clinical trial in certain circumstances. The FDA may refuse to approve an NDA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data. Even if such data are submitted, the FDA may ultimately decide that the NDA does not satisfy the criteria for approval. The FDA may refer the NDA to an advisory committee for review and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it generally follows such recommendations.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. Further, the FDA may require that certain contraindications, warnings or precautions be included in the product labeling. In addition, the FDA may condition approval on the completion of post approval studies. Such studies may involve clinical trials designed to further assess a product’s safety and effectiveness and may require testing and surveillance programs to monitor the safety of approved products that have been commercialized. If FDA determines that it is necessary to ensure the safe use of the drug, FDA may also condition approval on the implementation of a risk evaluation and mitigation strategy, or REMS. The REMS could include medication guides, physician communication plans or elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools.
Expedited review and approval
The FDA has various programs, including Fast Track, priority review, breakthrough, and accelerated approval, which are intended to expedite or simplify the process for reviewing product candidates. Generally, product candidates that are eligible for these programs are those for serious or life-threatening conditions, those with the potential to address unmet medical needs and those that offer meaningful benefits over existing treatments. A sponsor can request application of these programs either alone or in combination with each other, depending on the circumstances. Even if a product candidate qualifies for one or more of these programs, the FDA may later decide that the product candidate no longer meets the conditions for qualification or that the time period for FDA review or approval will be shortened. None of the expedited approval programs change the NDA approval standard applied to a product.
New drugs are eligible for Fast Track status if they are intended to treat a serious or life-threatening disease or condition and demonstrate the potential to address unmet medical needs for the disease or condition. Fast Track status entitles such a drug to expedited review and frequent contact with the FDA review division. Unlike other expedited review programs, Fast Track designation allows FDA to accept for review individual sections of the NDA on a rolling basis. The FDA may also grant a priority review designation to drugs that offer major advances in treatment, or provide a treatment where no adequate therapy exists. A priority review means that the goal for the FDA to review an application is six months from filing of an NDA, rather than the standard review of ten months from filing under current PDUFA guidelines. Most products that are eligible for fast track designation are also likely to be considered appropriate to receive a priority review.
16
Drug products studied for their safety and effectiveness in treating serious or life-threatening diseases or conditions may receive accelerated approval upon a determination that the product has an effect on a surrogate endpoint that is reasonably likely to predict clinical benefit, or on a clinical endpoint that can be measured earlier than irreversible morbidity or mortality, that is reasonably likely to predict an effect on irreversible morbidity or mortality or other clinical benefit, taking into account the severity, rarity, or prevalence of the condition and the availability or lack of alternative treatments. As a condition of approval, the FDA typically requires that a sponsor of a product candidate receiving accelerated approval conduct post-approval clinical trials. As an additional condition of approval, the FDA currently requires pre-approval of all promotional materials, which could adversely impact the timing of the commercial launch of the product.
The FDA may expedite the approval of a designated breakthrough therapy, which is a drug that is intended, to treat a serious or life-threatening disease or condition for which preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. A sponsor may request that a drug be designated as a breakthrough therapy at any time during the clinical development of the product. If FDA designates a drug as a breakthrough therapy, FDA must take actions appropriate to expedite the development and review of the application, which may include holding meetings with the sponsor and the review team throughout the development of the drug; providing timely advice to the sponsor regarding the development of the drug to ensure that the development program is as efficient as practicable; involving senior managers and experienced review staff, as appropriate, in a collaborative, cross-disciplinary review; and taking steps to ensure that the design of the clinical trials is as efficient as practicable.
Patent term restoration and marketing exclusivity
Depending upon the timing, duration and specifics of FDA approval of the use of our product candidates, some of our U.S. patents may be eligible for limited patent term extension under the Drug Price Competition and Patent Term Restoration Act of 1984, referred to as the Hatch-Waxman Act. The Hatch-Waxman Act permits a patent restoration term of up to five years as compensation for patent term lost during product development and the FDA regulatory review process. However, patent term restoration cannot extend the remaining term of a patent beyond a total of 14 years from the product candidate’s approval date. The patent term restoration period is generally one half of the time between the effective date of an IND and the submission date of an NDA, plus the time between the submission date of an NDA and the approval of that application. Only one patent applicable to an approved product candidate is eligible for the extension and the application for extension must be made prior to expiration of the patent. The U.S. Patent and Trademark Office, in consultation with the FDA, reviews and approves the application for any patent term extension or restoration. In the future, we intend to apply for restorations of patent term for some of our currently owned or licensed patents to add patent life beyond their current expiration date, depending on the expected length of clinical trials and other factors involved in the submission of the relevant NDA.
Market exclusivity provisions under the FDCA also can delay the submission or the approval of certain applications. The FDCA provides a five-year period of non-patent marketing exclusivity within the U.S. to the first applicant to gain approval of an NDA for a new chemical entity. A product candidate is a new chemical entity if the FDA has not previously approved any other new product candidate containing the same active moiety, which is the molecule or ion responsible for the action of the product candidate substance. During the exclusivity period, the FDA may not accept for review an abbreviated new drug application (ANDA) or a 505(b)(2) NDA submitted by another company for another version of such product candidate where the applicant does not own or have a legal right of reference to all the data required for approval. However, an application may be submitted after four years if it contains a certification of patent invalidity or non-infringement. The FDCA also provides three years of marketing exclusivity for an NDA, 505(b)(2) NDA or supplement to an approved NDA if new clinical investigations, other than bioavailability studies, that were conducted or sponsored by the applicant are deemed by the FDA to be essential to the approval of the application, for example, for new indications, dosages or strengths of an existing product candidate. This three-year exclusivity covers only the conditions associated with the new clinical investigations and does not prohibit the FDA from approving ANDAs for product candidates containing the original active agent. Five-year and three-year exclusivity will not delay the submission or approval of a full NDA. However, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all of the preclinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and effectiveness.
17
Orphan drug designation
Under the Orphan Drug Act, the FDA may grant orphan drug designation to product candidates intended to treat a rare disease or condition, which is generally a disease or condition that affects fewer than 200,000 individuals in the U.S. or more than 200,000 individuals in the U.S. and for which there is no reasonable expectation that the cost of developing and making available in the U.S. a product candidate for this type of disease or condition will be recovered from sales in the U.S. for that product candidate. Orphan drug designation must be requested before submitting an NDA. After the FDA grants orphan drug designation, the identity of the therapeutic agent and its potential orphan use are disclosed publicly by the FDA. Orphan drug designation does not convey any advantage in or shorten the duration of the regulatory review and approval process.
If a product candidate that has orphan drug designation subsequently receives the first FDA approval for the disease for which it has such designation, the product candidate is entitled to orphan product exclusivity, which means that the FDA may not approve any other applications including a full NDA to market the same product candidate for the same indication, except in very limited circumstances, for seven years. Orphan drug exclusivity, however, could also block the approval of one of our product candidates for seven years if a competitor obtains approval of the same product candidate as defined by the FDA prior to us, or if our product candidate is determined to be contained within the competitor’s approved orphan product candidate for the same indication or disease.
Pediatric exclusivity, pediatric use and rare pediatric disease priority review vouchers
Under the Best Pharmaceuticals for Children Act (BPCA), certain product candidates may obtain an additional six months of exclusivity if the sponsor submits information requested in writing by the FDA (a Written Request) relating to the use of the active moiety of the product candidate in children. The FDA may not issue a Written Request for studies on unapproved or approved indications or where it determines that information relating to the use of a product candidate in a pediatric population, or part of the pediatric population, may not produce health benefits in that population.
In addition, the Pediatric Research Equity Act (PREA) requires a sponsor to conduct pediatric studies for most product candidates and biologics, for a new active ingredient, new indication, new dosage form, new dosing regimen or new route of administration. Under PREA, original NDAs, biologics license application and supplements thereto must contain a pediatric assessment unless the sponsor has received a deferral or waiver. The required assessment must assess the safety and effectiveness of the product candidate for the claimed indications in all relevant pediatric subpopulations and support dosing and administration for each pediatric subpopulation for which the product candidate is safe and effective. The sponsor or FDA may request a deferral of pediatric studies for some or all of the pediatric subpopulations. A deferral may be granted for several reasons, including a finding that the product candidate or biologic is ready for approval for use in adults before pediatric studies are complete or that additional safety or effectiveness data needs to be collected before the pediatric studies begin. After April 2013, the FDA must send a noncompliance letter to any sponsor that fails to submit the required assessment, keep a deferral current or fails to submit a request for approval of a pediatric formulation. PREA does not apply to any drug for an indication for which orphan designation has been granted. However, if only one indication for a product has orphan designation, a pediatric assessment may still be required for any applications to market that same product for the non-orphan indication(s).
Currently, under section 529 of the FDCA, FDA will award priority review vouchers to sponsors of certain rare pediatric disease product applications. Section 529 will sunset December 31, 2016 unless Congress takes action to reauthorize the program.
Section 529 of the FDCA is intended to encourage development of new drug and biological products for prevention and treatment of certain rare pediatric diseases. Although there are existing incentive programs to encourage the development and study of drugs for rare diseases, pediatric populations, and unmet medical needs, section 529 provides an additional incentive for rare pediatric diseases, which may be used alone or in combination with other incentive programs. “Rare pediatric disease” is defined as a disease that:
| • | “primarily affects individuals aged from birth to 18 years, including age groups often called neonates, infants, children, and adolescents,” which is interpreted as meaning that greater than 50% of the affected population in the U.S. is aged 0 through 18 years; and |
| • | is “a rare disease or condition” as defined in FDCA, which includes diseases and conditions that affect fewer than 200,000 persons in the United States (U.S.) and diseases and conditions that affect a larger number of persons and for which there is no reasonable expectation that the costs of developing and making available the drug in the U.S. can be recovered from sales of the drug in the U.S. |
18
Under section 529, the sponsor of a human drug application for a rare pediatric disease drug product may be eligible for a voucher that can be used (or sold) to obtain a priority review for a subsequent human drug application submitted under section 505(b)(1) of the FDCA or section 351 of the Public Health Service (PHS) Act after the date of approval of the rare pediatric disease drug product. FDA has issued draft Guidance for Industry for Rare Pediatric Disease Priority Review Vouchers.
Post-approval requirements
Once an approval is granted, the FDA may withdraw the approval if compliance with regulatory requirements is not maintained or if problems occur after the product candidate reaches the market. Requirements for additional Phase 4 (post-approval marketing studies) to confirm safety and efficacy may be imposed as a condition of approval. Later discovery of previously unknown problems with a product candidate may result in restrictions on the product candidate, or REMS, or even complete withdrawal of the product candidate from the market. After approval, some types of changes to the approved product candidate, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval. In addition, the FDA may require testing and surveillance programs to monitor the effect of approved product candidates that have been commercialized, and the FDA has the power to prevent or limit further marketing of a product candidate based on the results of these post-marketing programs.
Any product candidates manufactured or distributed by us pursuant to FDA approvals are subject to continuing regulation by the FDA, including, among other things:
| • | record-keeping requirements; |
| • | reporting of adverse experiences with the product candidate; |
| • | submission of periodic reports; |
| • | providing the FDA with updated safety and efficacy information; |
| • | drug sampling, stability and distribution requirements; |
| • | notifying the FDA and gaining its approval of specified manufacturing or labeling changes; and |
| • | complying with statutory and regulatory requirements for promotion and advertising. |
Drug manufacturers and other entities involved in the manufacture and distribution of approved product candidates are required to register their establishments and provide product listing information to the FDA and certain state agencies and are subject to periodic unannounced inspections by the FDA and some state agencies for compliance with cGMPs and other laws.
Regulation outside of the U.S.
In addition to regulations in the U.S., we will be subject to regulations of other jurisdictions governing any clinical trials and commercial sales and distribution of our product candidates. Whether or not we obtain FDA approval for a product, we must obtain approval by the comparable regulatory authorities of countries outside of the U.S. before we can commence clinical trials in such countries, and approval of the regulators of such countries or
19
supranational areas, such as the European Union, before we may market products in those countries or areas. The approval process and requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from place to place, and the time may be longer or shorter than that required for FDA approval.
Under European Union regulatory systems, a company may submit marketing authorization applications either under a centralized or decentralized procedure. The centralized procedure, which is compulsory for certain medicines, including those produced by biotechnology or those intended to treat HIV, AIDS, cancer, neurodegenerative disorders, autoimmune and other immune dysfunctions, viral diseases or diabetes and is optional for those medicines which are a significant therapeutic, scientific or technical innovation or whose authorization would be in the interest of public health, provides for the grant of a single marketing authorization that is valid for all European Union member states. The decentralized procedure provides for mutual recognition of national approval decisions. Under this procedure, the holder of a national marketing authorization may submit an application to the remaining member states. Within 90 days of receiving the applications and assessment reports, each member state must decide whether to recognize the approval. If a member state does not recognize the marketing authorization, the disputed points are eventually referred to the European Commission, whose decision is binding on all member states.
As in the U.S., we may apply for designation of a product candidate as an orphan drug for the treatment of a specific indication in the European Union before the application for marketing authorization is made. Sponsors of orphan drugs in the European Union can enjoy economic and marketing benefits, including up to ten years of market exclusivity for the approved indication unless another applicant can show that its product is safer, more effective or otherwise clinically superior to the orphan-designated product.
Reimbursement
Sales of our products will depend, in part, on the extent to which the costs of our products will be covered by third-party payors, such as government health programs, commercial insurance and managed healthcare organizations. These third-party payors are increasingly challenging the prices charged for medical products and services. Additionally, the containment of healthcare costs has become a priority of federal and state governments and the prices of drugs have been a focus in this effort. The U.S. government, state legislatures and foreign governments have shown significant interest in implementing cost-containment programs, including price controls, restrictions on reimbursement and requirements for substitution of generic products. Adoption of price controls and cost-containment measures, and adoption of more restrictive policies in jurisdictions with existing controls and measures, could further limit our net revenue and results. If these third-party payors do not consider our products to be cost-effective compared to other therapies, they may not cover our products after approved as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The Medicare Prescription Drug, Improvement, and Modernization Act of 2003 (MMA) imposed new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities which will provide coverage of outpatient prescription drugs. Part D plans include both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary that identifies which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee. Government payment for some of the costs of prescription drugs may increase demand for our products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
20
The American Recovery and Reinvestment Act of 2009 provides funding for the federal government to compare the effectiveness of different treatments for the same illness. A plan for the research will be developed by the Department of Health and Human Services, the Agency for Healthcare Research and Quality and the National Institutes for Health, and periodic reports on the status of the research and related expenditures will be made to the U.S. Congress. Although the results of the comparative effectiveness studies are not intended to mandate coverage policies for public or private payors, it is not clear what effect, if any, the research will have on the sales of any product, if any such product or the condition that it is intended to treat is the subject of a study. It is also possible that comparative effectiveness research demonstrating benefits in a competitor’s product could adversely affect the sales of our product candidates. If third-party payors do not consider our products to be cost-effective compared to other available therapies, they may not cover our products as a benefit under their plans or, if they do, the level of payment may not be sufficient to allow us to sell our products on a profitable basis.
The Patient Protection and Affordable Care Act, as amended by the Health Care and Education Affordability Reconciliation Act of 2010, collectively referred to as the ACA, enacted in March 2010, is expected to have a significant impact on the health care industry. ACA is expected to expand coverage for the uninsured while at the same time containing overall healthcare costs. With regard to pharmaceutical products, among other things, ACA is expected to expand and increase industry rebates for drugs covered under Medicaid programs and make changes to the coverage requirements under the Medicare Part D program. We cannot predict the impact of ACA on pharmaceutical companies, as many of the ACA reforms require the promulgation of detailed regulations implementing the statutory provisions which has not yet occurred. In addition, although the U.S. Supreme Court upheld the constitutionality of most of the ACA, some states have indicated that they intend to not implement certain sections of the ACA, and some members of the U.S. Congress are still working to repeal parts of the ACA. These challenges add to the uncertainty of the legislative changes enacted as part of ACA.
In addition, in some non-U.S. jurisdictions, the proposed pricing for a product candidate must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our product candidates. Historically, product candidates launched in the European Union do not follow price structures of the U.S. and generally tend to be significantly lower.
Environment
Our third party manufacturers are subject to inspections by the FDA for compliance with cGMP and other U.S. regulatory requirements, including U.S. federal, state and local regulations regarding environmental protection and hazardous and controlled substance controls, among others. Environmental laws and regulations are complex, change frequently and have tended to become more stringent over time. We have incurred, and may continue to incur, significant expenditures to ensure we are in compliance with these laws and regulations. We would be subject to significant penalties for failure to comply with these laws and regulations.
Sales and Marketing
Our current focus is on the development of our existing portfolio, the completion of clinical trials and, if and where appropriate, the registration of our product candidates. We currently do not have marketing, sales and distribution capabilities. If we receive marketing and commercialization approval for any of our product candidates, we intend to market the product either directly or through strategic alliances and distribution agreements with third parties. The ultimate implementation of our strategy for realizing the financial value of our product candidates is dependent on the results of clinical trials for our product candidates, the availability of funds and the ability to negotiate acceptable commercial terms with third parties.
21
Scientific Advisors
We seek advice from our scientific advisory board, which consists of a number of leading scientists and physicians, on scientific and medical matters. We also seek advice on an as-needed basis from other leading scientists and physicians, who are not on our scientific advisory board, based on their particular knowledge and expertise. Our scientific advisory board meets periodically to assess:
| • | our research and development programs; |
| • | the design and implementation of our clinical programs; |
| • | our patent and publication strategies; |
| • | new technologies relevant to our research and development programs; and |
| • | specific scientific and technical issues relevant to our business. |
The current members of our scientific advisory board are as follows.
NAME | POSITION AND INSTITUTIONAL AFFILIATION | |
| Mark Behlke, M.D., Ph.D. | Chief Scientific Officer, Integrated DNA Technologies | |
| Frank McCormick, Ph.D., F.R.S., D.Sc. (Hon) | Director, University of California, San Francisco Helen Diller Family Comprehensive Cancer Center | |
| John Rossi, Ph.D. | Co-Founder of Dicerna and Professor and Dean of Irell and Manella Graduate School of Biological Sciences at City of Hope’s Beckman Research Institute | |
Employees
As of September 30, 2016, we had 48 full-time employees, of whom 38 are engaged in research and development and 10 in administration. None of our employees is represented by a labor union or covered by a collective bargaining agreement. Geographically, 46 employees are located in Massachusetts, one in Colorado and one in New Jersey. We consider our relationship with our employees to be good.
Corporate Information
We were incorporated in Delaware in 2006. We maintain our executive offices at 87 Cambridgepark Drive, Cambridge, MA 02140, and our main telephone number is (617) 621-8097. Our website is located at www.dicerna.com, which contains information about us. The information contained in, or that can be accessed through, our website is not part of, and is not incorporated in the documents we file with the Securities Exchange Commission (SEC).
We are an “emerging growth company” as defined in the Jumpstart Our Business Startups Act of 2012. We will remain an emerging growth company until the earlier of: (1) the last day of the fiscal year (a) following the fifth anniversary of the completion of our initial public offering on February 4, 2014, (b) in which we have total annual gross revenue of at least $1.0 billion, or (c) in which we are deemed to be a large accelerated filer, which means the market value of our common stock that is held by non-affiliates exceeds $700.0 million as of the prior June 30th, and (2) the date on which we have issued more than $1.0 billion in non-convertible debt during the prior three-year period. We refer to the Jumpstart Our Business Startups Act of 2012 herein as the “JOBS Act,” and references herein to “emerging growth company” shall have the meaning associated with it in the JOBS Act.
Our website address ishttp://www.dicerna.com. The information in, or that can be accessed through, our website is not part of this Annual Report on Form 10-K. Our annual reports on Form 10-K, quarterly reports on Form 10-Q and current reports onForm 8-K and amendments to those reports are available, free of charge, on or through our website as soon as reasonably practicable after we electronically file such material with, or furnish it to, the SEC. The public may read and copy any materials we file with the SEC at the SEC’s Public Reference Room at 100 F Street, NE, Washington, D.C. 20549. Information on the operation of the Public Reference Room can be obtained by calling 1-800-SEC-0330. The SEC maintains an Internet site that contains reports, proxy and information statements and other information regarding our filings atwww.sec.gov.
22