As filed with the Securities and Exchange Commission on August 13, 2007
Registration No. 333-
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM S-1
REGISTRATION STATEMENT
UNDER
THE SECURITIES ACT OF 1933
BIOLEX, INC.
(Exact name of registrant as specified in its charter)
| | | | |
| Delaware | | 2834 | | 56-2065137 |
| (State or other jurisdiction of incorporation or organization) | | (Primary Standard Industrial Classification Code Number) | | (IRS Employer Identification No.) |
158 Credle Street
Pittsboro, North Carolina 27312
(919) 542-9901
(Address, including zip code, and telephone number,
including area code, of registrant’s principal executive offices)
Jan Turek, President and Chief Executive Officer
158 Credle Street
Pittsboro, North Carolina 27312
(919) 542-9901
(Name, address, including zip code, and telephone number,
including area code, of agent for service)
With copies to:
| | |
Jonathan L. Kravetz, Esq. Brian P. Keane, Esq. Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C. One Financial Center Boston, MA 02111 (617) 542-6000 | | Todd W. Eckland, Esq. David G. Odrich, Esq. Pillsbury Winthrop Shaw Pittman LLP 1540 Broadway New York, NY 10036-4039 (212) 858-1000 |
Approximate date of commencement of proposed sale to public: As soon as practicable after this Registration Statement becomes effective.
If any of the securities being registered on this Form are being offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act, check the following box. ¨
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier registration statement for the same offering. ¨
If this Form is a post-effective amendment filed pursuant to Rule 462(d) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier registration statement for the same offering. ¨
CALCULATION OF REGISTRATION FEE
| | | | |
| |
| Title of each class of securities to be registered | | Proposed Maximum
Aggregate Offering Price(1) | | Amount of Registration Fee(2) |
Common Stock, $0.001 par value per share | | $70,000,000 | | $2,149 |
| |
| (1) | Estimated solely for the purpose of calculating the amount of registration fee pursuant to Rule 457(o) under the Securities Act. Includes shares of common stock subject to the underwriters’ option to purchase additional shares of common stock. |
| (2) | Calculated pursuant to Rule 457(o) based on an estimate of the proposed maximum aggregate offering price. |
The Registrant hereby amends this Registration Statement on such date or dates as may be necessary to delay its effective date until the Registrant shall file a further amendment which specifically states that this Registration Statement shall thereafter become effective in accordance with Section 8(a) of the Securities Act or until the Registration Statement shall become effective on such date as the Securities and Exchange Commission, acting pursuant to Section 8(a), may determine.
The information in this preliminary prospectus is not complete and may be changed. We may not sell these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities and it is not soliciting an offer to buy these securities in any jurisdiction where the offer or sale is not permitted.
| | |
PROSPECTUS | | |
Subject to Completion, dated August 13, 2007 |
Shares

Common Stock
Biolex, Inc. is offering shares of its common stock in an underwritten public offering. This is the initial public offering of shares of our common stock, for which no public market currently exists.
We have applied to have our common stock listed on The NASDAQ Global Market under the symbol “BLEX.” We anticipate that the initial public offering price will be between $ and $ per share.
Investing in our common stock involves risks. See “Risk Factors” beginning on page 9 of this prospectus.
| | | | | | |
| | | Per Share | | Total |
Price to the public | | $ | | | $ | |
Underwriting discounts and commissions | | $ | | | $ | |
Proceeds to us (before expenses) | | $ | | | $ | |
We have granted the underwriters a 30-day option to purchase up to an additional shares of common stock from us on the same terms and conditions as set forth above if the underwriters sell more than shares of common stock in this offering.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The underwriters expect to deliver the shares to purchasers on or about , 2007.
| | |
| LEHMAN BROTHERS | | DEUTSCHE BANK SECURITIES |
LEERINK SWANN & COMPANY
, 2007
TABLE OF CONTENTS
Until , 2007 (25 days after the commencement of this offering), all dealers that effect transactions in shares of our common stock, whether or not participating in this offering, may be required to deliver a prospectus. This delivery requirement is in addition to the obligation of dealers to deliver a prospectus when acting as underwriters and with respect to their unsold allotments or subscriptions.
You should rely only on the information contained in this prospectus or in any free writing prospectus we may authorize to be delivered to you. We have not, and the underwriters have not, authorized anyone to provide you with additional or different information. We are offering to sell, and seeking offers to buy, shares of our common stock only in jurisdictions where offers and sales are permitted. The information in this prospectus is accurate only as of the date of this prospectus, regardless of the time of delivery of this prospectus or any sale of shares of our common stock.
For investors outside the United States: Neither we nor any of the underwriters have done anything that would permit this offering or possession or distribution of this prospectus in any jurisdiction where action for that purpose is required, other than in the United States. Persons outside the United States who come into possession of this prospectus must inform themselves about, and observe any restrictions relating to, the offering of shares of common stock and the distribution of this prospectus outside of the United States, except as may otherwise be described in this prospectus.
i
PROSPECTUS SUMMARY
This summary highlights information contained elsewhere in this prospectus and may not contain all of
the information that may be important to you in making an investment decision. You should read this summary together with the more detailed information, including our consolidated financial statements and the related notes, elsewhere in this prospectus. You should carefully consider, among other things, the matters discussed in “Risk Factors” beginning on page 9.Unless otherwise stated or the context requires otherwise, references in this prospectus to the “Company,” “Biolex,” “Biolex Therapeutics,” “we,” “us” and “our” refer to Biolex, Inc. and our subsidiaries.
BIOLEX, INC.
We are a clinical-stage biopharmaceutical company that uses our patented LEX System to develop hard-to-make therapeutic proteins and optimized monoclonal antibodies. The LEX System is a novel technology that genetically transforms the aquatic plantLemna to enable the production of biologic product candidates. Our proprietary product candidates are designed to provide superior efficacy/tolerability profiles and to address large, proven pharmaceutical markets.
Our lead product candidate, Locteron, is in Phase 2 clinical trials and is the only controlled-release interferon alfa known to be currently in clinical development for the treatment of chronic hepatitis C. Locteron is designed to improve upon the current standard of patient care, pegylated interferon alfa administered weekly in combination with the antiviral drug ribavirin, which is associated with significant side effects. In a 32-patient Phase 2a clinical trial of Locteron administered once every two weeks in combination with ribavirin, an early virologic response, or EVR, was achieved by 100% (8/8) of hepatitis C patients treated with the 480 microgram, or 480 µg, dose of Locteron. Achievement of EVR, which is a specified reduction in viral load, has been broadly established to be a prerequisite for long-term response in hepatitis C patients. In addition, reported side effects were fewer and less severe than previously reported with other interferon products currently marketed or in development.
We have also developed two other product candidates that capitalize on the benefits of the LEX System, which we are advancing toward clinical trials: BLX-155, a direct-acting thrombolytic designed to dissolve blood clots in patients; and BLX-301, an anti-CD20 antibody we are optimizing for the treatment of non-Hodgkin’s B-cell lymphoma and other diseases. We believe that all of our product candidates have reduced clinical and regulatory risk profiles because they are based on well-understood proteins with proven mechanisms of action or clinical validation, and well-established regulatory pathways.
Our Product Candidates
We have focused our pipeline of product candidates on biologics that we believe can be enhanced by the LEX System. The LEX System facilitates the development of proteins whose complexity and other properties limit their commercial viability in traditional production systems. It also enables us to produce monoclonal antibodies that can be optimized with the objective of improving their potency and efficacy. By capitalizing on the significant benefits of the LEX System over traditional production systems, we are developing the following pipeline of product candidates.
Locteron
The World Health Organization estimates that as many as 130 million people worldwide are chronically infected with hepatitis C and that an additional three to four million people are infected each year. It is estimated that annual worldwide sales of interferon for the treatment of hepatitis C will increase from $2.8 billion in 2005 to
1
more than $5 billion by 2014. Under the current standard of care, interferon alfa is injected weekly for up to 48 weeks and is associated with significant side effects, including fatigue, flu-like symptoms, depression and anemia, resulting in high rates of patient non-compliance and discontinuation of therapy. Future treatment is expected to continue to consist of interferon in combination with one or more antiviral agents, which could heighten patient tolerability concerns, because the antiviral agents currently under development have been reported to cause additional side effects.
Locteron is designed to improve patient care by providing a more favorable side-effect profile and more convenient dosing. Locteron incorporates PolyActive, a controlled-release technology patented by our co-development partner OctoPlus N.V., which provides a gradual release of interferon alfa to patients without the high initial blood levels associated with the significant side effects experienced by patients today. Locteron is also formulated to allow dosing once every two weeks, a substantial improvement in patient convenience compared to currently marketed pegylated interferon alfa products that require dosing every week.
We are conducting a 12-week, Phase 2a clinical trial (SELECT-1) in Europe to evaluate four different doses of Locteron administered once every two weeks in combination with ribavirin in 32 treatment-naïve hepatitis C patients. In this trial, an EVR was achieved by 100% (8/8) of patients treated with the 480mg dose of Locteron and by 88% (7/8) of patients treated with the 320mg dose. In addition, patients receiving Locteron experienced fewer and less severe side effects than those previously reported for pegylated interferons and Albuferon, an albumin-fused interferon product currently under development. Accordingly, the objective of this trial has been met as we have identified a range of doses with an appropriate combination of viral reduction and favorable tolerability to proceed to a Phase 2b trial.
In the first half of 2008, we plan to initiate a 72-week, 100-patient Phase 2b clinical trial (SELECT-2) in Europe comparing three doses of Locteron in combination with ribavirin against a control arm treated with pegylated interferon alfa in combination with ribavirin. Patients will be treated for 48 weeks with a 24-week follow-up period. The results after 12 weeks of treatment with Locteron will be evaluated and will be the basis for dose selection for Phase 3 clinical trials. The overlap between the Phase 2 and Phase 3 trials is designed to expedite the development path of Locteron, with dosing in Phase 3 trials expected to commence in 2009. We are developing Locteron under an exclusive collaboration agreement with OctoPlus, the developer of PolyActive.
BLX-155
BLX-155 is a direct-acting thrombolytic that we are developing to dissolve blood clots in patients with certain diseases or conditions such as acute peripheral arterial disease, deep vein thrombosis and hemodialysis graft thrombosis. Thrombosis, the formation or presence of a blood clot, is a leading cause of morbidity and mortality and, according to the National Heart Lung and Blood Institute, is estimated to affect over 50 million people in the developed world. BLX-155 is the recombinant form of plasmin, a naturally occurring human enzyme responsible for dissolving fibrin, a key component of blood clots. Plasmin has been studied for a number of decades as a drug candidate for dissolving blood clots, but recombinant full-length human plasmin has not been successfully produced at commercially viable levels in traditional protein production systems.
In July 2007, our researchers presented results demonstrating the ability of the LEX System to produce full-length recombinant plasmin at commercially viable levels. This achievement provides us with a unique opportunity to pursue development and commercialization of a therapeutic protein with a known mechanism of action for which prior development historically had been prevented due to production challenges. As a direct-acting thrombolytic, we believe BLX-155 has the potential to provide efficacy and safety advantages over
2
plasminogen activators that dissolve blood clots indirectly, including tissue-type plasminogen activators, or t-PAs, which are the current standard of care. BLX-155 is currently in preclinical development, and we expect to commence a Phase 1 clinical trial in hemodialysis graft thrombosis patients in the first half of 2008.
BLX-301
BLX-301 is an anti-CD20 antibody that we are optimizing for the treatment of non-Hodgkin’s B-cell lymphoma and other diseases. BLX-301 is based on the capability of the LEX System to optimize the glycosylation structure of monoclonal antibodies to increase their potency and efficacy. In May 2007, we announced preclinical results highlighting the ability of the LEX System to produce an anti-CD20 antibody with enhanced cytotoxicity and potentially lower side effects than Rituxan, the top selling treatment for non-Hodgkin’s B-cell lymphoma, which is also used to treat rheumatoid arthritis. BLX-301 is currently in preclinical development, and we expect to commence a Phase 1 clinical trial in non-Hodgkin’s B-cell lymphoma patients in 2009.
The LEX System
The market for therapeutic proteins is projected to grow at twice the rate of the market for small molecule drugs, or synthetic pharmaceutical compounds, and is expected to increase from $51 billion in 2005 to $87 billion in 2010, according to Kalorama Information. Therapeutic proteins have traditionally been produced primarily inmicrobial and mammalian systems. These traditional production systems, however, have a number of inherent drawbacks, including the need for significant capital investments, long lead times to establish manufacturing facilities, and high costs of production. In some instances, the commercialization of proteins has been prevented because production at commercially viable levels is not achievable in traditional systems or the use of traditional systems to produce certain proteins is blocked by existing process patents.
In addition, monoclonal antibodies are the fastest growing class of products in the pharmaceutical industry, with total sales growing from $4 billion in 2001 to $17 billion in 2005, and projected to reach $34 billion in 2010. Demand for improved production techniques results from recent research suggesting that the efficacy and potency of monoclonal antibodies can be enhanced by modifying their sugar-chain components, or glycosylation structures.
We have applied advanced genetic engineering to create our recombinant protein production system, theLemna Expression System, or LEX System, that addresses many of the drawbacks of traditional production systems. The LEX System facilitates the development of proteins whose complexity and other properties limit their commercial viability in traditional production systems. It also enables us to produce monoclonal antibodies that can be optimized with the objective of improving potency and efficacy. The strengths of the LEX System include:
| | • | | Production of Complex Proteins. We have not encountered a class of proteins that cannot be expressed by the LEX System. To date, as part of our research and development activities, we have produced more than 35 different proteins using the LEX System, including complex proteins that are challenging to produce in traditional production systems. For instance, the protein plasmin has been studied for several decades as a drug candidate for resolving blood clots, but production of full-length human plasmin at commercially viable levels has not been reported with any traditional recombinant production system. Our product candidate BLX-155, a recombinant full-length plasmin, can be produced at commercially viable levels using the LEX System. |
| | • | | Complex Protein Folding.Lemna is able to correctly produce proteins requiring folding or other complex processing, which is key to the efficient production of many proteins, including interferon alfa. Traditional production systems are unable to express interferon alfa that is correctly folded, therefore |
3
| | requiring a series of unfolding and refolding steps during purification that results in substantial yield loss and contributes to the high cost of production in these systems. |
| | • | | Optimization of Monoclonal Antibodies. The LEX System has demonstrated the capability to express monoclonal antibodies in which the glycosylation structure has been optimized to enhance the efficacy and potency of the antibodies. |
| | • | | Access to Product Candidates. TheLemna-based protein production system is a novel technology that we believe will provide us with multiple opportunities to produce proteins without conflicting with patent claims relating to the production of certain proteins in traditional production systems. For example, the LEX System allows us to produce the interferon alfa used in Locteron without conflicting with the patent claims covering the production of interferon alfa in a bacteria-based system. |
| | • | | Reduced Safety Risk.The LEX System contains no human or animal components, which can carry a risk of viral or other infectious agent contamination. |
| | • | | Reduced Capital Cost and Facility Timelines. We believe that the simplicity and other characteristics of the LEX System will result in capital investment requirements and timelines for implementation of manufacturing facilities that are substantially less than those associated with traditional systems. |
Our Strategy
Our objective is to become a leading biopharmaceutical company by developing and commercializing a diverse portfolio of best-in-class biologic products for large proven markets. The key elements of our strategy to achieve this objective are:
| | • | | leverage our extensive biopharmaceutical experience and the power of the LEX System to source and develop high-value product candidates; |
| | • | | continue to develop our current product candidates, Locteron, BLX-155 and BLX-301; |
| | • | | position Locteron as the interferon of choice for the treatment of chronic hepatitis C; |
| | • | | position BLX-155 to address multiple large-market indications involving blood clots; |
| | • | | establish an effective commercial infrastructure as we progress toward regulatory approval; and |
| | • | | continue to strategically partner with leading pharmaceutical and biotechnology companies. |
Risk Factors
Our business is subject to numerous risks, as more fully described in the section entitled “Risk Factors” immediately following this prospectus summary, including the following:
| | • | | We are a development stage company and have incurred significant losses since our inception. We had an accumulated deficit of $75.4 million as of March 31, 2007. |
| | • | | We expect to continue to incur significant losses for the foreseeable future, and we expect these losses to increase substantially as we conduct larger-scale trials for our drug candidates, commercialize any approved product candidates, hire additional personnel, and add operational, financial and management information systems. |
| | • | | The FDA has not previously approved products developed in a plant-based expression system, such as the LEX System, and our product candidates are still in the early stages of development and remain subject to clinical testing and regulatory approval. If we are unable to successfully develop and test our product candidates, we will not be successful. |
4
| | • | | Our success is largely dependent on the success of our lead product candidate, Locteron, which has been tested only in a limited number of patients over a short duration, and we cannot be certain that we will be able to obtain regulatory approval for or successfully commercialize this product candidate. |
| | • | | Even if any of our product candidates receives regulatory approval, if the approved product does not achieve broad market acceptance, the revenues that we generate from sales of the product will be limited. |
| | • | | If we are unable to protect our intellectual property and operate our business without infringing upon the intellectual property rights of others, our ability to commercialize our product candidates would be harmed. |
| | • | | We have never manufactured a product candidate on a commercial scale, and we will need to establish commercial-scale manufacturing capabilities if any of our or our collaboration partners’ product candidates receives regulatory approval. |
Corporate Information
We were incorporated in Delaware in September 1997. Our principal executive offices are located at 158 Credle Street, Pittsboro, North Carolina 27312, and our telephone number is (919) 542-9901. Our website address is www.biolex.com. The information on our website is not part of this prospectus. We have included our website address as a factual reference and do not intend it to be an active link to our website.
Our trademarks and service marks include Biolex, Biolex Therapeutics, the LEX System, theLemna Expression System, Locteron and our logo. This prospectus also includes trademarks and service marks of other persons.
5
The Offering
Common stock offered by us | shares |
Common stock to be outstanding after this offering | shares |
Underwriters’ option to purchase additional shares | shares |
Use of proceeds | We estimate that our net proceeds from this offering will be approximately $ million, assuming an initial public offering price of $ per share, the midpoint of the range listed on the cover page of this prospectus, and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. We intend to use the net proceeds from this offering to fund clinical trials, preclinical testing and other research and development activities, and for general and administrative expenses, working capital needs and other general corporate purposes. For a more complete description of our intended use of the proceeds, see “Use of Proceeds.” |
Proposed NASDAQ Global Market symbol | BLEX |
Except as otherwise indicated, throughout this prospectus the number of shares of common stock to be outstanding after this offering is based on the number of shares outstanding as of August 1, 2007, and excludes:
| | • | | 9,456,007 shares of common stock issuable upon the exercise of stock options outstanding as of August 1, 2007, at a weighted average exercise price of $0.28 per share; |
| | • | | 93,640 shares of common stock issuable upon the exercise of warrants outstanding as of August 1, 2007, at a weighted average exercise price of $1.09 per share; and |
| | • | | 1,814,658 shares of common stock reserved for future awards under our stock plan. |
Unless otherwise indicated, all information contained in this prospectus assumes that the underwriters do not exercise their option to purchase up to additional shares of our common stock in this offering and also reflects:
| | • | | the conversion of all outstanding shares of our Class B non-voting common stock and our preferred stock into shares of our Class A voting common stock and the reclassification of our Class A voting common stock into common stock upon completion of this offering; |
| | • | | a -for- split of our common stock to be effected prior to the completion of this offering; and |
| | • | | the adoption of our amended and restated certificate of incorporation and restated bylaws upon the completion of this offering. |
6
Summary Consolidated Financial Data
(in thousands, except per share amounts)
The summary consolidated financial data set forth below should be read in conjunction with our consolidated financial statements and the related notes, “Selected Consolidated Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this prospectus. Our historical results are not necessarily indicative of the results to be expected in any future period.
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | Year Ended December 31, | | | Three Months Ended March 31, | | | Period From
Sept. 19,
1997
(Inception)
Through
March 31, 2007 | |
| | | 2004 | | | 2005 | | | 2006 | | | 2006 | | | 2007 | | |
| | | | | | as restated | | | | | | (unaudited) | | | (unaudited) | |
Statement of Operations Data: | | | | | | | | | | | | | | | | | | | | | | | | |
Revenues | | $ | 719 | | | $ | 3,008 | | | $ | 5,017 | | | $ | 1,327 | | | $ | 724 | | | $ | 9,752 | |
Operating expenses: | | | | | | | | | | | | | | | | | | | | | | | | |
Research and development | | | 8,666 | | | | 14,448 | | | | 19,196 | | | | 5,084 | | | | 5,292 | | | | 60,632 | |
General and administrative | | | 2,400 | | | | 4,126 | | | | 4,370 | | | | 1,096 | | | | 1,114 | | | | 19,687 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Total operating expenses | | | 11,066 | | | | 18,574 | | | | 23,566 | | | | 6,180 | | | | 6,406 | | | | 80,319 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Loss from operations | | | (10,347 | ) | | | (15,566 | ) | | | (18,549 | ) | | | (4,853 | ) | | | (5,682 | ) | | | (70,567 | ) |
Interest (expense) income, net | | | (57 | ) | | | (421 | ) | | | 774 | | | | 152 | | | | (35 | ) | | | (1,734 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Loss before income taxes | | | (10,404 | ) | | | (15,987 | ) | | | (17,774 | ) | | | (4,701 | ) | | | (5,717 | ) | | | (72,301 | ) |
Income tax benefit | | | — | | | | 170 | | | | 277 | | | | 31 | | | | 35 | | | | 482 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | | (10,404 | ) | | | (15,817 | ) | | | (17,497 | ) | | | (4,670 | ) | | | (5,682 | ) | | | (71,819 | ) |
Less: accretion of mandatorily redeemable convertible preferred stock | | | (1,165 | ) | | | (3,034 | ) | | | (5,600 | ) | | | (1,369 | ) | | | (1,495 | ) | | | (16,772 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Net loss attributable to common stockholders | | $ | (11,569 | ) | | $ | (18,851 | ) | | $ | (23,097 | ) | | $ | (6,039 | ) | | $ | (7,177 | ) | | $ | (88,591 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Basic and diluted net loss attributable to common stockholders per share | | $ | (42.91 | ) | | $ | (65.25 | ) | | $ | (68.52 | ) | | $ | (20.18 | ) | | $ | (14.93 | ) | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Shares used in the calculation of basic and diluted net loss per share | | | 270 | | | | 289 | | | | 337 | | | | 299 | | | | 481 | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Pro forma basic and diluted net loss per share (unaudited)(1) | | | | | | | | | | $ | (0.29 | ) | | | | | | $ | (0.09 | ) | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Shares used to compute pro forma basic and diluted net loss per share (unaudited)(1) | | | | | | | | | | | 60,145 | | | | | | | | 60,288 | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | |
(1) | The pro forma basic and diluted net loss per share gives effect to the conversion of all outstanding shares of preferred stock into common stock upon completion of this offering. |
The following summary balance sheet data as of March 31, 2007 is presented:
| | • | | on a pro forma basis to give effect to: |
| | • | | the sale by us of 25,663,339 shares of our Series CC preferred stock on May 18, 2007 at $1.18 per share in exchange for $25.0 million in cash and the conversion of a $5.0 million convertible note and accrued interest payable to a related party, less transaction expenses; and |
7
| | • | | the conversion of all outstanding shares of our Class B non-voting common stock and our preferred stock into shares of our Class A voting common stock and the reclassification of our Class A voting common stock into common stock upon completion of this offering; and |
| | • | | on a pro forma as adjusted basis to give further effect to our sale of shares of common stock in this offering at an assumed initial public offering price of $ per share, the midpoint of the range listed on the cover page of this prospectus, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
| | | | | | | | | | | |
| | | As of March 31, 2007 |
| | | Actual | | | Pro Forma | | | Pro Forma As Adjusted(1) |
| | | (unaudited) |
Balance Sheet Data: | | | | | | | | | | | |
Cash and cash equivalents | | $ | 8,708 | | | $ | 32,542 | | | $ | |
Working capital | | | 2,505 | | | | 26,622 | | | | |
Total assets | | | 19,172 | | | | 43,007 | | | | |
Long-term liabilities | | | 10,690 | | | | 5,690 | | | | |
Total liabilities | | | 17,921 | | | | 12,638 | | | | |
Mandatorily redeemable convertible preferred stock | | | 73,070 | | | | — | | | | |
Additional paid-in-capital | | | 3,635 | | | | 105,736 | | | | |
Deficit accumulated during development stage | | | (75,421 | ) | | | (75,421 | ) | | | |
Total stockholders’ equity (deficit) | | | (71,819 | ) | | | 30,369 | | | | |
(1) | A $1.00 increase (decrease) in the assumed initial public offering price of $ per share would increase (decrease) each of the pro forma as adjusted cash and cash equivalents, working capital, total assets, additional paid-in capital and total stockholders’ equity by $ million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. The pro forma as adjusted information discussed above is illustrative only and following the completion of this offering will be adjusted based on the actual public offering price and other terms of this offering determined at pricing. |
8
RISK FACTORS
An investment in our common stock involves a high degree of risk. You should carefully read and consider the risks and uncertainties described below together with all of the other information contained in this prospectus, including the consolidated financial statements and the related notes appearing at the end of this prospectus, before deciding to invest in our common stock. If any of these risks actually occur, our business, business prospects, financial condition, results of operations or cash flows could be materially harmed. In any such case, the trading price of our common stock could decline, and you could lose all or part of your investment.
Risks Related to Our Financial Position
We are a development stage company and have incurred significant losses since our inception, and we expect to incur losses for the foreseeable future and may never reach profitability.
We are a development stage company and have been engaged in designing and developing product candidates since 1997. Since our inception we have incurred significant operating losses and, as of March 31, 2007, we had an accumulated deficit of $75.4 million. We have not generated any revenue from the sale of our product candidates to date. We had net losses of $10.4 million, $15.8 million, $17.5 million and $5.7 million for the three years ended December 31, 2006 and the three months ended March 31, 2007, respectively. Since our inception, we have incurred $60.6 million of research and development expenses. We expect to continue to incur research and development and other significant operating expenses and capital expenditures and anticipate that our expenses and losses will increase substantially in the foreseeable future as we:
| | • | | initiate additional Phase 2 trials of Locteron for the treatment of chronic hepatitis C, including a Phase 2b clinical trial that we expect to initiate in the first half of 2008; |
| | • | | complete preclinical development of BLX-155 and initiate clinical trials, if supported by positive preclinical data; |
| | • | | initiate Phase 3 clinical development of Locteron, if supported by the Phase 2 results; |
| | • | | begin to establish commercial manufacturing facilities and establish sales and marketing functions; |
| | • | | continue preclinical development of BLX-301 and initiate clinical trials, if supported by positive preclinical data; |
| | • | | identify additional product candidates and acquire rights from third parties to product candidates through licenses, acquisitions or other means; |
| | • | | commercialize any approved product candidates; |
| | • | | hire additional clinical, scientific, manufacturing/quality and management personnel; and |
| | • | | add operational, financial and management information systems and personnel. |
We must generate significant revenue to achieve and maintain profitability. Even if we succeed in developing and commercializing one or more of our product candidates, we may not be able to generate sufficient revenue and we may never be able to achieve or maintain profitability.
9
If we fail to obtain the capital necessary to fund our operations, we will be unable to successfully develop and commercialize our product candidates.
Although we have raised substantial capital to date, we will require substantial future capital in order to complete the clinical development of and to commercialize our lead product candidate, Locteron, and to complete the preclinical and clinical development of and to commercialize our two preclinical stage product candidates, BLX-155 and BLX-301. Our future capital requirements will depend on many factors that are currently unknown to us, including:
| | • | | the timing of initiation, progress, results and costs of our planned Phase 2 clinical trials of Locteron for the treatment of chronic hepatitis C; |
| | • | �� | the timing of initiation, progress, results and costs of any Phase 3 clinical trials of Locteron we may initiate based on the results of our Phase 2 trials, which will be affected by interactions with the FDA and similar foreign regulatory authorities and our continuing evaluation of third-party intellectual property relevant to our commercialization plans and other factors; |
| | • | | the results of preclinical studies of our two preclinical product candidates, BLX-155 and BLX-301, and the timing of initiation, progress, results and costs of any clinical trials of these product candidates that we may initiate based on the preclinical results; |
| | • | | the costs of establishing commercial manufacturing facilities and of establishing sales and marketing functions; |
| | • | | the scope, progress, results, and costs of preclinical development, clinical trials, and regulatory review of any new product candidates for which we may initiate development; |
| | • | | the costs of preparing, filing, and prosecuting patent applications and maintaining, enforcing, and defending intellectual property-related claims; |
| | • | | our ability to establish strategic collaborations and licensing or other arrangements on terms favorable to us; |
| | • | | the costs to satisfy our obligations under potential future collaborations; and |
| | • | | the timing, receipt, and amount of sales or royalties, if any, from any approved product candidates. |
We cannot assure you that additional funds will be available when we need them on terms that are acceptable to us, or at all. If adequate funds are not available on a timely basis, we may be required to:
| | • | | terminate or delay clinical trials or other development for one or more of our product candidates; |
| | • | | delay our establishment of sales and marketing capabilities, commercial manufacturing capabilities, or other activities that may be necessary to commercialize our product candidates; or |
| | • | | curtail significant drug development programs that are designed to identify new product candidates. |
We believe that the proceeds we receive from this offering and our existing cash and investment securities will be sufficient to support our current operating plan into 2009. However, our operating plan may change as a result of many factors currently unknown to us, and we may need additional funds sooner than planned. In addition, we may seek additional capital due to favorable market conditions or strategic considerations even if we believe we have sufficient funds for our current or future operating plans.
10
Risks Related to the Development and Regulatory Approval of Our Product Candidates
Our success is largely dependent on the success of our lead product candidate, Locteron, and we cannot be certain that we will be able to obtain regulatory approval for or successfully commercialize this product candidate.
We have invested a significant portion of our time and financial resources in the development of our lead product candidate, Locteron, for the treatment of chronic hepatitis C. We anticipate that our success will depend largely on the receipt of regulatory approval and successful commercialization of this product candidate, which will depend on several factors, including the following:
| | • | | our ability to provide acceptable evidence of its safety and efficacy, and the receipt of marketing approval from the FDA and any similar foreign regulatory authorities, particularly given that the FDA has not previously approved a product developed in a plant-based expression system, such as the LEX System; |
| | • | | establishing commercial-scale manufacturing capabilities; |
| | • | | establishing an internal sales force or collaborating with pharmaceutical companies or contract sales organizations to market and sell Locteron, if approved; and |
| | • | | acceptance of Locteron, if approved, in the medical community and by patients and third-party payors. |
Many of these factors are beyond our control. Accordingly, we cannot assure you that we will ever be able to generate revenues through the sale of Locteron.
Our product candidates are still in the early stages of development and remain subject to clinical testing and regulatory approval. If we are unable to successfully develop and test our product candidates, we will not be successful.
To date, we have not marketed, distributed or sold any product candidates. The success of our business depends substantially upon our ability to develop and commercialize our product candidates successfully. We currently have only three product candidates, all of which are in the early stages of development: Locteron is currently in Phase 2 clinical trials and both BLX-155 and BLX-301 are in preclinical development. To date we have not completed any Phase 2 or Phase 3 clinical trials. Our product candidates are prone to the risks of failure inherent in drug development. Before obtaining regulatory approvals for the commercial sale of Locteron, BLX-155, BLX-301 or any other product candidate for a target indication, we must demonstrate with substantial evidence gathered in well-controlled clinical trials, and, with respect to approval in the United States, to the satisfaction of the FDA and, with respect to approval in other countries, similar regulatory authorities in those countries, that the product candidate is safe and effective for use for that target indication. Satisfaction of these and other regulatory requirements is costly, time consuming, uncertain, and subject to unanticipated delays. Despite our efforts, our product candidates may not:
| | • | | offer improvement over existing, comparable drugs; |
| | • | | be proven safe and effective in clinical trials; |
| | • | | meet applicable regulatory standards; or |
| | • | | be successfully commercialized. |
Positive results in preclinical studies of a product candidate may not be predictive of similar results in humans during clinical trials, and promising results from early clinical trials of a product candidate may not be replicated in later clinical trials. Interim results of a clinical trial do not necessarily predict final results. A number of companies in the pharmaceutical and biotechnology industries have suffered significant setbacks in late-stage clinical trials even after achieving promising results in early-stage development. Accordingly, the results from the completed preclinical studies and clinical trials and ongoing clinical trials for Locteron and the
11
completed and ongoing preclinical studies for BLX-155 and BLX-301 may not be predictive of the results we may obtain in later stage trials or studies. Our preclinical studies or clinical trials may produce negative or inconclusive results, and we may decide, or regulators may require us, to conduct additional preclinical studies or clinical trials, or to discontinue clinical trials altogether. We do not expect any of our product candidates to be commercially available for at least several years and some or all may never become commercially available.
If clinical trials for our product candidates are prolonged or delayed, we may be unable to commercialize our product candidates on a timely basis, which would require us to incur additional costs and delay our receipt of any revenue from potential product sales.
We cannot predict whether we will encounter problems with any of our completed, ongoing or planned clinical trials that will cause us or any regulatory authority to delay or suspend those clinical trials or delay the analysis of data derived from them. A number of events, including any of the following, could delay the completion of our ongoing and planned clinical trials and negatively impact our ability to obtain regulatory approval for, and to market and sell, a particular product candidate, including our clinical-stage product candidate Locteron:
| | • | | conditions imposed on us or our collaborators by the FDA or any foreign regulatory authority regarding the scope or design of our clinical trials; |
| | • | | delays in obtaining, or our inability to obtain, required approvals from institutional review boards, or IRBs, or other reviewing entities at clinical sites selected for participation in our clinical trials; |
| | • | | insufficient supply or deficient quality of our product candidates or other materials necessary to conduct our clinical trials; |
| | • | | delays in obtaining regulatory agency agreement for the conduct of our clinical trials; |
| | • | | lower than anticipated enrollment and retention rate of subjects in clinical trials for a variety of reasons, including size of patient population, nature of trial protocol, the availability of approved effective treatments for the relevant disease and competition from other clinical trial programs for similar indications; |
| | • | | serious and unexpected drug-related side effects experienced by patients in clinical trials; or |
| | • | | failure of our third-party contractors to meet their contractual obligations to us in a timely manner. |
Clinical trials may also be delayed or terminated as a result of ambiguous or negative interim results. In addition, a clinical trial may be suspended or terminated by us, the FDA, the IRBs at the sites where the IRBs are overseeing a trial, or a data safety monitoring board, or DSMB, overseeing the clinical trial at issue, or other regulatory authorities due to a number of factors, including:
| | • | | failure to conduct the clinical trial in accordance with regulatory requirements or our clinical protocols; |
| | • | | inspection of the clinical trial operations or trial sites by the FDA or other regulatory authorities resulting in the imposition of a clinical hold; |
| | • | | varying interpretation of data by the FDA or similar foreign regulatory authorities; |
| | • | | failure to achieve primary or secondary endpoints or other failure to demonstrate efficacy; |
| | • | | unforeseen safety issues; or |
| | • | | lack of adequate funding to continue the clinical trial. |
Additionally, changes in regulatory requirements and guidance may occur and we may need to amend clinical trial protocols to reflect these changes. Amendments may require us to resubmit our clinical trial protocols to IRBs for reexamination, which may impact the cost, timing or successful completion of a clinical trial.
12
We do not know whether our clinical trials will begin as planned, will need to be restructured or will be completed on schedule, if at all. Delays in our clinical trials will result in increased development costs for our product candidates. In addition, if we experience delays in completion of, or if we terminate, any of our clinical trials, the commercial prospects for our product candidates may be harmed and our ability to generate product revenues will be delayed. Furthermore, many of the factors that cause, or lead to, a delay in the commencement or completion of clinical trials may also ultimately lead to the denial of regulatory approval of a product candidate.
Unanticipated side effects during the clinical development of Locteron may result in an interruption, delay or halt in clinical trials and could result in the denial of regulatory approval by the FDA or other regulatory authorities.
The active agent in our product candidate Locteron is interferon alfa, variants of which are currently marketed in the United States and elsewhere for treatment of chronic hepatitis C. Treatment of patients with interferon alfa has historically been associated with significant adverse side effects, including severe flu-like symptoms, depression, suicidal thoughts and attempts, fatigue, alopecia, bone marrow toxicity (thrombocytopenia, neutropenia), endocrine disorders, hepatotoxicity, cardiovascular and pulmonary disorders, pancreatitis, worsening of autoimmune disorders, sleep disorders, arthralgia, myalgia and ophthalmologic disorders. A number of participants in our Phase 2a clinical trial for Locteron have experienced adverse events adjudicated or determined by trial investigators to be potentially attributable to Locteron, although most of these adverse events have been rated as mild and only one such adverse event was rated as severe. We cannot assure you, however, that the favorable safety and tolerability profile suggested by Locteron in our Phase 2a clinical trial will be replicated in future trials that will be required prior to regulatory approval. In addition, these future clinical trials may demonstrate that the gradual release of interferon alfa does not reduce the frequency, duration or severity of side effects commonly experienced by patients treated with currently marketed pegylated interferons and with Albuferon.
Because Locteron has only been tested in a limited number of patients over a short duration, we cannot assure you that Locteron’s efficacy or safety and tolerability profile suggested to date will be replicated in the future.
To date, Locteron has only been tested in a very limited number of patients, including a total of 32 patients (eight patients per dose cohort) in our European multi-center, open-label Phase 2a clinical trial. Furthermore, most of the adverse side effects observed in our Phase 2a trial were based on symptoms that are measured subjectively, as they are subject to both varying perceptions by the patients and varying interpretations by our clinical investigators, including by virtue of their characterization as mild, moderate, severe or serious and their varying effect on diverse patient populations. Accordingly, and because observed significant adverse effects have been associated with currently marketed variants of interferon alfa, we cannot assure you that the favorable safety and tolerability profile suggested by Locteron in our completed early-stage clinical trials will be replicated in larger, later-stage trials. In addition, a definitive comparison of Locteron with currently marketed drugs or other drugs in clinical development requires a controlled, head-to-head clinical trial. To date, the longest a patient has been dosed with Locteron has been for 12 weeks. The anticipated dosing period for Locteron, if approved, is expected to be 48 weeks in the most prevalent genotype of hepatitis C patients. Accordingly, we cannot assure you that the results indicated by Locteron in early trials will be replicated in longer trials that will be required prior to regulatory approval. Unanticipated side effects could cause us, the FDA or other regulatory authorities or IRBs to interrupt, delay or halt clinical trials and could result in the denial of regulatory approval by the FDA or other regulatory authorities of Locteron.
13
If side effects are identified after our product candidates are approved and on the market, we may be required to change the labeling of any such products, or withdraw any such products from the market, any of which would hinder or preclude our ability to generate revenues.
Even if Locteron or any of our other product candidates receives marketing approval, we or others may later identify undesirable side effects. As greater numbers of patients use a drug following its approval, side effects and other problems may be observed after approval that were not seen or anticipated during pre-approval clinical trials. In that event a number of potentially significant negative consequences could result, including:
| | • | | regulatory authorities may withdraw their approval of the product; |
| | • | | regulatory authorities may require the addition of labeling statements, such as warnings or contraindications; |
| | • | | we may be required to change the way the product is administered, conduct additional clinical trials or change the labeling of the product; |
| | • | | we could be sued and held liable for harm caused to patients; and |
| | • | | our reputation may suffer. |
Even if our product candidates receive regulatory approval in the United States, we may never receive approval or commercialize our products outside of the United States.
In order to market any products outside of the United States, we must establish and comply with numerous and varying regulatory requirements of other countries regarding safety and efficacy. Approval procedures vary among countries and can involve additional product testing and additional administrative review periods. The time required to obtain approval in other countries might differ from that required to obtain FDA approval. The regulatory approval process in other countries may include all of the risks detailed above regarding FDA approval in the United States as well as other risks. Regulatory approval in one country does not ensure regulatory approval in another, but a failure or delay in obtaining regulatory approval in one country may have a negative effect on the regulatory process in others. Failure to obtain regulatory approval in other countries or any delay or setback in obtaining such approval would impair our ability to develop foreign markets for our product candidates.
Both before and after marketing approval, our product candidates are subject to ongoing regulatory requirements, and if we fail to comply with these continuing requirements, we could be subject to a variety of sanctions and the sale of any approved commercial products could be suspended.
Both before and after regulatory approval to market a particular product candidate, the manufacturing, labeling, packaging, adverse event reporting, storage, advertising, promotion and record keeping related to the product are subject to extensive regulatory requirements. If we fail to comply with the regulatory requirements of the FDA and other applicable U.S. and foreign regulatory authorities, we could be subject to administrative or judicially imposed sanctions, including:
| | • | | restrictions on the products or manufacturing processes; |
| | • | | untitled or warning letters; |
| | • | | civil or criminal penalties; |
| | • | | product seizures or detentions; |
14
| | • | | voluntary or mandatory product recalls and related publicity requirements; |
| | • | | suspension or withdrawal of regulatory approvals; |
| | • | | total or partial suspension of production; and |
| | • | | refusal to approve pending applications for marketing approval of new products or supplements to approved applications. |
We deal with hazardous materials and must comply with environmental laws and regulations, which can be expensive and restrict how we do business.
Certain of our drug development activities involve the controlled storage, use, and disposal of hazardous materials. We are subject to federal, state, and local laws and regulations governing the use, manufacture, storage, handling, and disposal of these hazardous materials. Although we believe that our safety procedures for the handling and disposing of these materials comply with the standards prescribed by these laws and regulations, we cannot eliminate the risk of accidental contamination or injury from these materials.
In the event of an accident, state or federal authorities may curtail our use of these materials, and we could be liable for any civil damages that result, which may exceed our financial resources and may seriously harm our business. Because we believe that our laboratory and materials handling policies and practices sufficiently mitigate the likelihood of materials liability or third-party claims, we currently carry no insurance covering such claims. An accident could damage, or force us to shut down, our operations.
Risks Related to the Commercialization of Our Product Candidates
Even if any of our product candidates receives regulatory approval, if the approved product does not achieve broad market acceptance, the revenues that we generate from sales of the product will be limited.
Even if Locteron, BLX-155 or BLX-301, or any other product candidates we may develop or acquire in the future, obtain regulatory approval, they may not gain broad market acceptance among physicians, healthcare payors, patients, and the medical community. The degree of market acceptance for any approved product candidate will depend on a number of factors, including:
| | • | | timing of market introduction of competitive products; |
| | • | | demonstration of clinical safety and efficacy compared to other products; |
| | • | | prevalence and severity of adverse side effects; |
| | • | | availability of reimbursement from managed care plans and other third-party payors; |
| | • | | convenience and ease of administration; |
| | • | | other potential advantages of alternative treatment methods; and |
| | • | | ineffective marketing and distribution support of our products. |
If our approved drugs fail to achieve broad market acceptance, we may not be able to generate significant revenue and our business would suffer.
15
If we are unable to establish sales and marketing capabilities or enter into agreements with third parties to market and sell our product candidates, we may be unable to generate product revenue.
We do not currently have an organization for the sales, marketing and distribution of pharmaceutical products. In order to market any products that may be approved by the FDA, we must build our sales, marketing, managerial and other non-technical capabilities or make arrangements with third parties to perform these services. If we are unable to establish adequate sales, marketing and distribution capabilities, whether independently or with third parties, we may not be able to generate product revenue and may not become profitable.
If government and third-party payors fail to provide adequate coverage and reimbursement rates for any of our product candidates that receive regulatory approval, our revenue and prospects for profitability will be harmed.
In both domestic and foreign markets, our sales of any future products will depend in part upon the availability of reimbursement from third-party payors. Such third-party payors include government health programs such as Medicare, managed care providers, private health insurers, and other organizations. These third-party payors are increasingly attempting to contain healthcare costs by demanding price discounts or rebates and limiting both coverage and the amounts that they will pay for new drugs, and, as a result, they may not cover or provide adequate payment for our product candidates. We might need to conduct post-marketing studies in order to demonstrate the cost-effectiveness of any future products to such payors’ satisfaction. Such studies might require us to commit a significant amount of management time and financial and other resources. Our future products might not ultimately be considered cost-effective. Adequate third-party reimbursement might not be available to enable us to maintain price levels sufficient to realize an appropriate return on investment in product development.
U.S. and foreign governments continue to propose and pass legislation designed to reduce the cost of healthcare. For example, in some foreign markets, the government controls the pricing and profitability of prescription pharmaceuticals. In the United States, we expect that there will continue to be federal and state proposals to implement similar governmental controls. In addition, recent changes in the Medicare program and increasing emphasis on managed care in the United States will continue to put pressure on pharmaceutical product pricing. Cost control initiatives could decrease the price that we would receive for any products in the future, which would limit our revenue and profitability. Accordingly, legislation and regulations affecting the pricing of pharmaceuticals might change before our product candidates are approved for marketing. Adoption of such legislation could further limit reimbursement for pharmaceuticals.
For example, the Medicare Prescription Drug Improvement and Modernization Act of 2003, or MMA, changes the way Medicare will cover and pay for pharmaceutical products. The legislation expanded Medicare coverage for drug purchases by the elderly and disabled and introduced new reimbursement methodologies, based on average sales prices for drugs that are administered in an in-patient setting or by physicians. In addition, this legislation provides authority for limiting the number of drugs that will be covered in any therapeutic class. Although we do not know what impact the new reimbursement methodologies will have on the prices of new drugs, we expect that there will be added pressure to contain and reduce costs. These cost reduction initiatives and other provisions of this legislation could decrease the coverage and price that we receive for any approved products and could seriously harm our business. While the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own reimbursement rates, and any reduction in reimbursement that results from the MMA may result in a similar reduction in payments from private payors.
16
Recent federal legislation and actions by state and local governments may permit re-importation of drugs from foreign countries into the United States, including foreign countries where the drugs are sold at lower prices than in the United States, which could materially adversely affect our operating results and our overall financial condition.
We may face competition for any of our product candidates, once approved, from lower priced products from foreign countries that have placed price controls on pharmaceutical products. The MMA contains provisions that may change U.S. importation laws and expand pharmacists’ and wholesalers’ ability to import lower priced versions of our product candidates and competing products from Canada, where there are government price controls. These changes to U.S. importation laws will not take effect unless and until the Secretary of Health and Human Services certifies that the changes will pose no additional risk to the public’s health and safety and will result in a significant reduction in the cost of products to consumers. The Secretary of Health and Human Services has not yet announced any plans to make this required certification. As directed by Congress, a task force on drug importation conducted a comprehensive study regarding the circumstances under which drug importation could be safely conducted and the consequences of importation on the health, medical costs and development of new medicines for U.S. consumers. The task force issued its report in December 2004, finding that there are significant safety and economic issues that must be addressed before importation of prescription drugs is permitted. However, a number of federal legislative proposals have been made to implement the changes to the U.S. importation laws without any certification, and to broaden permissible imports in other ways. Even if the changes do not take effect, and other changes are not enacted, imports from Canada and elsewhere may continue to increase due to market and political forces, and the limited enforcement resources of the FDA, the U.S. Customs Service and other government agencies. For example, Public Law No. 109-295, which was signed into law in October 2006 and provides appropriations for the Department of Homeland Security for the 2007 fiscal year, expressly prohibits the U.S. Customs Service from using funds to prevent individuals from importing from Canada less than a 90-day supply of a prescription drug for personal use, when the drug otherwise complies with the Federal Food, Drug, and Cosmetic Act, or FDCA. Further, several states and local governments have implemented importation schemes for their citizens, and, in the absence of federal action to curtail such activities, we expect other states and local governments to launch importation efforts. The importation of foreign products that compete with any of our approved product candidates could negatively impact our profitability.
The markets for our product candidates are subject to intense competition. If we are unable to compete effectively, our product candidates may be rendered noncompetitive or obsolete.
Many large pharmaceutical and biotechnology companies, academic institutions, governmental agencies, and other public and private research organizations are pursuing the development of novel drugs that target the same indications we are targeting with our product candidates. We face, and expect to continue to face, intense and increasing competition as new products enter the market and advanced technologies become available. If approved, we would expect Locteron, BLX-155 and BLX-301 to compete with approved drugs and product candidates currently under development, including the following:
| | • | | Locteron. If approved, we would expect Locteron to compete with currently approved forms of interferon alfa, including PEG-Intron, currently marketed by Schering-Plough Corporation, and Pegasys, currently marketed by Hoffmann-La Roche. We would also expect Locteron to compete with any product candidate for the treatment of chronic hepatitis C currently under development that is successful in obtaining approval, including Albuferon, currently under development in Phase 3 by Human Genome Sciences and Novartis. |
| | • | | BLX-155. If approved, we would expect BLX-155 to compete with Activase, currently marketed by Genentech, and presently used off-label in the potential indications targeted by us. We would also expect BLX-155 to compete with any direct-acting thrombolytic product candidate currently under development that is successful in obtaining approval for clot lysis, including micro-plasmin, human-derived plasmin, under development by Thrombogenics, Talecris, and Nuvelo, respectively. |
17
| | • | | BLX-301. If approved, we would expect BLX-301 to compete with Rituxan, which is currently marketed by Genentech, Biogen-Idec and Hoffmann-La Roche. We would also expect BLX-301 to compete with any anti-CD20 monoclonal antibody product candidate currently under development that is successful in obtaining approval for non-Hodgkins lymphoma, including HuMax-CD20, under development by GenMab A/S and GlaxoSmithKline, and TRU-015, under development by Trubion Pharmaceuticals and Wyeth. |
Many of our competitors have:
| | • | | significantly greater financial, technical and human resources than we have and may be better equipped to discover, develop, manufacture and commercialize product candidates; |
| | • | | more extensive experience in preclinical testing and clinical trials, obtaining regulatory approvals and manufacturing and marketing pharmaceutical products; |
| | • | | product candidates that have been approved or are in late-stage clinical development; or |
| | • | | collaborative arrangements in our target markets with leading companies and research institutions. |
Competitive products may render our products obsolete or noncompetitive before we can recover the expenses of developing and commercializing our product candidates. Furthermore, the development of new treatment methods or the widespread adoption or increased utilization of any vaccine or development of other products or treatments for the diseases we are targeting could render our product candidates noncompetitive, obsolete or uneconomical. If we successfully develop and obtain approval for our product candidates, we will face competition based on the safety and effectiveness of our product candidates, the timing of their entry into the market in relation to competitive products in development, the availability and cost of supply, marketing and sales capabilities, reimbursement coverage, price, patent position and other factors. If we successfully develop product candidates but those product candidates do not achieve and maintain market acceptance, our business will not be successful.
If a successful product liability claim or series of claims is brought against us for uninsured liabilities or in excess of insured liabilities, we could incur substantial liability.
The use of our product candidates in clinical trials and the sale of any products for which we obtain marketing approval expose us to the risk of product liability claims. Product liability claims might be brought against us by consumers, health care providers or others selling or otherwise coming into contact with our products. If we cannot successfully defend ourselves against product liability claims, we could incur substantial liabilities. In addition, regardless of merit or eventual outcome, product liability claims may result in:
| | • | | decreased demand for any approved product candidates; |
| | • | | impairment of our business reputation; |
| | • | | withdrawal of clinical trial participants; |
| | • | | costs of related litigation; |
| | • | | distraction of management’s attention from our primary business; |
| | • | | substantial monetary awards to patients or other claimants; |
| | • | | the inability to successfully commercialize any approved product candidates. |
We have obtained product liability insurance coverage for our clinical trials with a $5.0 million annual aggregate coverage limit. However, our insurance coverage may not be sufficient to reimburse us for any expenses or losses we may suffer. Moreover, insurance coverage is becoming increasingly expensive, and, in the future, we may not be able to maintain insurance coverage at a reasonable cost or in sufficient amounts to protect
18
us against losses due to liability. If and when we obtain marketing approval for any of our product candidates, we intend to expand our insurance coverage to include the sale of commercial products; however, we may be unable to obtain this product liability insurance on commercially reasonable terms. On occasion, large judgments have been awarded in class action lawsuits based on drugs that have had unanticipated side effects. A successful product liability claim or series of claims brought against us could cause our stock price to decline and, if judgments exceed our insurance coverage, could decrease our cash and adversely affect our business.
Risks Related to Our Dependence on Third Parties
We rely on third parties to conduct our clinical trials, and those third parties may not perform satisfactorily, including failing to meet established deadlines for the completion of such clinical trials.
We do not have the ability to independently conduct clinical trials for our product candidates, and we rely on third parties, such as contract research organizations, medical institutions, and clinical investigators, to perform this function. Our reliance on these third parties for clinical development activities reduces our control over these activities. Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. Although we have, in the ordinary course of business, entered into agreements with these third parties, we continue to be responsible for confirming that each of our clinical trials is conducted in accordance with its general investigational plan and protocol. Moreover, the FDA requires us to comply with regulations and standards, commonly referred to as good clinical practices, for conducting, recording and reporting the results of clinical trials to assure that data and reported results are credible and accurate and that the trial participants are adequately protected. Our reliance on third parties does not relieve us of these responsibilities and requirements. If these third parties do not successfully carry out their contractual duties or meet expected deadlines, we may be delayed in obtaining regulatory approvals for our product candidates and may be delayed in our efforts to successfully commercialize our product candidates for targeted diseases.
We rely heavily upon our collaboration with OctoPlus for the development of Locteron, and the loss of OctoPlus as a partner, or any adverse developments in the collaboration, would materially harm our business.
In February 2005, we entered into a co-development agreement with OctoPlus to develop and commercialize Locteron. Locteron combines our proprietary BLX-883 interferon alfa with OctoPlus’ PolyActive drug delivery technology. We are subject to a number of risks associated with our dependence on this collaboration with OctoPlus, including:
| | • | | We and OctoPlus could disagree as to development plans, including clinical trials or regulatory approval strategy, or as to which additional indications for Locteron should be pursued. Disputes regarding the collaboration agreement that delay or terminate the development, receipt of regulatory approvals or commercialization of Locteron would harm our business and could result in significant litigation or arbitration. |
| | • | | OctoPlus could fail to devote sufficient resources towards the development and approval of Locteron. After the time periods stated in the collaboration agreement, OctoPlus could shift its research and development resources to other product opportunities. |
| | • | | OctoPlus may not be able to provide sufficient resources to ensure the efficient and timely production of Locteron. |
| | • | | OctoPlus may not be able to scale up its manufacturing process for the production of Locteron to produce quantities suitable for Phase 3 clinical supply or commercialization. |
Furthermore, OctoPlus may terminate our collaboration agreement upon 60 days notice upon the occurrence of certain events, subject to the survival of certain obligations, or upon our material breach of the collaboration agreement. If OctoPlus does not perform its obligations under, or devote sufficient resources to, our collaboration, or if we and OctoPlus do not work effectively together, Locteron may not be successfully developed and
19
commercialized. Although our agreement provides for non-exclusive access to the OctoPlus controlled-release technology in the event that OctoPlus opts out of further development or terminates for convenience, we would need to establish an alternative arrangement for production and may not be able to do so in a timely manner, on acceptable terms, or at all.
Conflicts may arise with our collaborators, resulting in renegotiation or termination of, or litigation related to, our agreements with them, which would adversely affect our revenues.
Conflicts could arise between us and our collaborators, including OctoPlus, as to royalty rates, milestone payments or other commercial terms. Similarly, the parties may disagree as to which party owns or has the right to commercialize intellectual property that is developed during the course of the relationship or as to other non-commercial terms. If such a conflict were to arise, our collaborators might attempt to compel renegotiation of certain terms of their agreements or terminate their agreements entirely, and we might lose the benefits of the agreements. Either we or our collaborators might initiate litigation to determine commercial obligations, establish intellectual property rights or resolve other disputes under the related agreements. Such litigation could be costly to us and require substantial attention of management. If we were unsuccessful in such litigation, we could lose the commercial benefits of the agreements, be liable for other financial damages and suffer losses of intellectual property or other rights that are the subject of dispute. Any of these adverse outcomes could cause our business strategy to fail.
If we do not establish additional collaborations, we may have to alter our development plans.
Our drug development programs and potential commercialization of our product candidates will require substantial additional cash to fund expenses. Our strategy includes potentially collaborating with leading pharmaceutical and biotechnology companies to assist us in furthering development and potential commercialization of some of our product candidates, including Locteron, in some or all geographies. It may be difficult to enter into one or more of such collaborations in the future. We face significant competition in seeking appropriate collaborators and these collaborations are complex and time-consuming to negotiate and document. We may not be able to negotiate collaborations on acceptable terms, or at all. If that were to occur, we may have to curtail the development of a particular product candidate, reduce or delay its development program or one or more of our other development programs, delay its potential commercialization or reduce the scope of our sales or marketing activities, or increase our expenditures and undertake development or commercialization activities at our own expense. If we elect to increase our expenditures to fund development or commercialization activities on our own, we may need to obtain additional capital, which may not be available to us on acceptable terms, or at all. If we do not have sufficient funds, we will not be able to bring our product candidates to market and generate product revenue.
Risks Related to Our Intellectual Property
If we are unable to adequately protect the intellectual property relating to our product candidates, or if we infringe the rights of others, our ability to successfully commercialize our product candidates will be harmed.
As of August 1, 2007, we own or hold exclusive licenses to a total of 22 U.S. issued patents and 22 U.S. pending patent applications, as well as 15 foreign patents and 20 pending PCT applications and foreign counterparts. While we own or have exclusive licenses to issued patents covering out platform technology, we only have pending patent applications directed specifically to our product candidates including Locteron, BLX-883 (Locteron’s active agent), BLX-155 and BLX-301, and we cannot ensure that we will succeed in obtaining and preserving patent coverage of any of our product candidates.
Our success depends in part on our ability to obtain patent protection both in the United States and in other countries for our product candidates. Our ability to protect our product candidates from unauthorized or infringing use by third parties depends in substantial part on our ability to obtain and maintain valid and enforceable patents. Due to evolving legal standards relating to the patentability, validity and enforceability of patents covering pharmaceutical inventions and the scope of claims made under these patents, our ability to
20
obtain, maintain and enforce patents is uncertain and involves complex legal and factual questions. Accordingly, rights under any issued patents may not provide us with sufficient protection for our product candidates or provide sufficient protection to afford us a commercial advantage against competitive products or processes.
In addition, we cannot guarantee that any patents will issue from any pending or future patent applications owned by or licensed to us. Even if patents have issued or will issue, we cannot guarantee that the claims of these patents are or will be valid or enforceable or will provide us with any significant protection against competitive products or otherwise be commercially valuable to us. Patent applications in the United States are maintained in confidence for up to 18 months after their filing. In some cases, however, patent applications remain confidential in the U.S. Patent and Trademark Office, which we refer to as the U.S. Patent Office, for the entire time prior to issuance as a U.S. patent. Similarly, publication of discoveries in the scientific or patent literature often lag behind actual discoveries. Consequently, we cannot be certain that we or our licensors or co-owners were the first to invent, or the first to file patent applications on, our product candidates or their use as drugs. In the event that a third party has also filed a U.S. patent application relating to our product candidates or a similar invention, we may have to participate in interference proceedings declared by the U.S. Patent Office to determine priority of invention in the United States. The costs of these proceedings could be substantial and it is possible that our efforts would be unsuccessful, resulting in a loss of our U.S. patent position. Furthermore, we may not have identified all U.S. and foreign patents or published applications that affect our business either by blocking our ability to commercialize our drugs or by covering similar technologies.
The laws of some foreign jurisdictions do not protect intellectual property rights to the same extent as in the United States and many companies have encountered significant difficulties in protecting and defending such rights in foreign jurisdictions. If we encounter such difficulties in protecting or are otherwise precluded from effectively protecting our intellectual property rights in foreign jurisdictions, our business prospects could be substantially harmed.
We license patent rights from third-party owners. If such owners do not properly maintain or enforce the patents underlying such licenses, our competitive position and business prospects will be harmed.
We are party to a number of licenses that give us rights to third-party intellectual property that is necessary or useful for our business. In particular, we have obtained exclusive licenses from North Carolina State University, or NCSU, Yeda Research and Development Company Limited, or Yeda, and The Scripps Research Institute, or TSRI, relating to technologies used in our LEX System. We may also enter into additional licenses to third-party intellectual property in the future. Our success will depend in part on the ability of our licensors to obtain, maintain (including making periodic filings and payments) and enforce patent protection for their intellectual property, in particular, those patents to which we have secured exclusive rights. Under our license with NCSU, NCSU has sole responsibility for filing, prosecuting and maintaining all patents that are subject to the license. Under our license with Yeda, Yeda controls the filing, prosecution and maintenance of all patents. Under our license agreement with TSRI, TSRI controls the filing, prosecution and maintenance of all patents. Our licensors may not successfully prosecute the patent applications to which we are licensed. Even if patents issue in respect of these patent applications, our licensors may fail to maintain these patents, may determine not to pursue litigation against other companies that are infringing these patents, or may pursue such litigation less aggressively than we would. Without protection for the intellectual property we license, other companies might be able to offer substantially identical products for sale, which could adversely affect our competitive business position and harm our business prospects.
Our licenses may be subject to early termination if we fail to comply with our obligations in our licenses with third parties.
Our licensors may terminate their agreements with us in the event we breach the applicable license agreement and fail to cure the breach within a specified period of time. Under our existing license agreements we
21
are obligated to pay the licensor fees, which may include annual license fees, royalties, a percentage of revenues associated with the licensed technology and a percentage of sublicensing revenue. In addition, under our existing license agreements, we are required to diligently pursue the development of products using the licensed technology. If we breach any of the terms of our licenses, the licensors may terminate the agreements. The termination of any of our licensing agreements would result in the loss of patent protection for, and freedom to operate with regard to, the technology under license, which could adversely affect our business.
Our success will depend on our ability to operate without infringing the proprietary rights of others, which we cannot assure. If third parties claim that we are infringing their patents and we become involved in patent litigation, it could be very expensive and time consuming, could cause delays in bringing our products to market, and could limit the markets for our products.
Our success will depend in part on our ability to operate without infringing the proprietary rights of third parties. Although we are not currently aware of any litigation or other proceedings or third-party claims of intellectual property infringement related to our therapeutic protein candidates or the LEX System, the pharmaceutical industry is characterized by extensive litigation regarding patents and other intellectual property rights. Other parties may in the future allege that our activities infringe their patents or that we are employing their proprietary technology without authorization.
For example, we are aware of U.S. patents and corresponding European patents with claims directed to the treatment of hepatitis C, as well as U.S. patents directed to mature human leukocyte interferon, which includes interferon alfa, and DNA encoding mature human leukocyte interferon. Some of these patents include claims directed to the treatment of specific subgroups of hepatitis C patients with a combination of interferon alfa and ribavirin for prescribed periods. These patents are relevant to our development and commercialization plans for Locteron. In addition, a wide range of new antiviral therapies and new interferon products are currently under development for the treatment of chronic hepatitis C by third parties. Each of the new therapies currently advancing in clinical trials is being studied in combination with interferon alfa, and third parties may be successful in prosecuting and maintaining patents encompassing interferon compositions and the use of antiviral drugs in combination with interferon. Any such patents could also be relevant to our development and commercialization plans for Locteron.
As another example, we are aware of a U.S. patent including claims directed to transgenic plants coding for an inactive proenzyme form of a protease. This patent is relevant to the production in the LEX System of our product candidate, BLX-155, which is a protease. In addition, various thrombolytics are under development by third parties, and third parties may be successful in prosecuting and maintaining patents encompassing the use, production or purification of plasmin for thrombolytic therapy. Any such patents could also be relevant to our development and commercialization plans for BLX-155.
As yet another example, we are aware of a U.S. patent including claims directed to antibody compositions with complex sugar chains wherein 100% of the molecules comprising the Fc region of the sugar chains do not contain fucose bound to the N-acetylglucosamines. This patent is relevant to our glycosylation optimization technology. In addition, various third parties are developing antibodies having enhanced antibody dependent cell-mediated cytotoxicity, or ADCC, and methods of optimizing the glycosylation structure of proteins such as antibodies, including the use of RNA interference for such optimization. Third parties may be successful in prosecuting and maintaining patents directed at ADCC enhanced anti-CD20 antibodies or optimizing the glycosylation structure of antibodies, including methods of RNA interference. Any such patents could also be relevant to our development and commercialization plans for BLX-301 and other optimized monoclonal antibodies.
We believe that, if we receive regulatory approval, we will be able to commercialize our product candidates without infringing any valid and enforceable issued patent of which we are currently aware, as described below. In each case where there exist third-party patents that are potentially relevant to our product candidates or
22
processes for producing them, we believe that the patents are invalid or we have identified approaches to avoid infringement. In the case of non-infringement approaches, we believe that each of the approaches will enable us to market the relevant products to large patient populations, although some of the approaches could significantly reduce the patient populations to whom we market specific products in particular geographic regions. With regard to Locteron, the extent to which any existing patent that is held to be valid might limit our potential market will depend on the specific drug combination, treatment duration, patient characteristics, and geographic region under which Locteron is used. In the most restrictive non-infringement approach we evaluated, for approximately five years after anticipated launch, we estimate that our product labeling, or approved target market, for Locteron would likely be limited to a market of approximately half of chronic hepatitis C patients in the United States and approximately 80% of hepatitis C patients in Europe. We would be required to obtain licenses under such patents to avoid limitation on the markets for the affected products. There is no assurance that we would be able to obtain such licenses on commercially reasonable terms, or at all.
While we have investigated the patent positions of certain competitors with respect to the manufacture and commercialization of our proprietary form of interferon alfa (BLX-883) and believe that we have freedom to operate with respect to BLX-883, with respect to the PolyActive component of Locteron and its combination with BLX-883, we have principally relied upon our collaborator OctoPlus with respect to the search for, and evaluation of, possibly adverse third party intellectual property. Thus, while we believe that we have freedom to operate with respect to Locteron, we cannot guarantee that we or OctoPlus have adequately searched for competitive intellectual property, nor can we guarantee that new patents which could impair our freedom to operate will not issue in the future.
In addition, third parties may challenge or infringe our existing or future patents. Under our license agreements with NCSU, Yeda and TSRI, we have the right, but not the obligation, to bring actions against an infringing third party. If we do not bring an action within a specified number of days, the licensor may bring an action against the infringing party.
Proceedings involving our patents or patent applications or those of others under which we have been granted licenses could result in adverse decisions regarding:
| | • | | the patentability of our inventions relating to our product candidates or processes; and/or |
| | • | | the enforceability, validity or scope of protection offered by our owned or licensed patents relating to our product candidates or processes. |
Even if we are successful in these proceedings, we may incur substantial costs and divert management time and attention in pursuing these proceedings, which could have a material adverse effect on us. Similarly, if we are unable to avoid infringing the patent rights of others, we may be required to seek a license, defend an infringement action or challenge the validity of the patents in court. Patent litigation is costly and time consuming. We may not have sufficient resources to bring or defend these actions to a successful conclusion. In cases where third parties allege our infringement of their patents, if we do not obtain a license, develop or obtain non-infringing technology, defend an infringement action successfully, or have infringed patents declared invalid, we may:
| | • | | incur substantial monetary damages; |
| | • | | encounter significant delays in bringing our product candidates to market; and/or |
| | • | | be precluded from participating in the manufacture, use or sale of our product candidates or methods of treatment requiring licenses. |
We may be unable to adequately prevent disclosure of trade secrets and other proprietary information.
We rely on trade secrets to protect our proprietary technologies, especially where we do not believe patent protection is appropriate or obtainable. However, trade secrets are difficult to protect. We rely in part on
23
confidentiality agreements with our employees, consultants, outside scientific collaborators, sponsored researchers, and other advisors to protect our trade secrets and other proprietary information. These agreements may not effectively prevent disclosure of confidential information and may not provide an adequate remedy in the event of unauthorized disclosure of confidential information. In addition, others may independently discover our trade secrets and proprietary information. Costly and time-consuming litigation could be necessary to enforce and determine the scope of our proprietary rights, and failure to obtain or maintain trade secret protection could adversely affect our competitive business position.
Risks Related to Our Manufacturing Activities
No product manufactured using a plant-based production system, including our LEX System, has received regulatory approval.
Each of our product candidates and our collaboration agreements are based on our proprietary LEX System, a plant-based production systemthat is intended to serve as an alternative to the mammalian and bacterial based production systems commonly used today. To date, no products produced in a plant-based production system have reached the stage of development where they could be evaluated for approvability, and therefore none have been approved for sale in the United States or any other jurisdiction by any regulatory authority. If unforeseen technological, regulatory or other challenges associated with plant-based production systems, such as our LEX System, materialize, our ability to develop and commercialize our product candidates and achieve value from our collaborations will be significantly disrupted.
Our manufacturing facilities are subject to extensive regulatory requirements.
Before approving a new drug or biologic product, the FDA requires that the facilities at which the product will be manufactured are in compliance with current good manufacturing practices, or cGMP requirements. To successfully develop our product candidates, we must demonstrate adequate compliance with the applicable cGMP requirements, which include quality control and quality assurance requirements as well as the maintenance of extensive records and documentation. Our manufacturing facilities will also be subject to ongoing periodic inspection by the FDA and corresponding foreign and state authorities, including unannounced inspections. In addition, cGMP requirements are constantly evolving, and new or different requirements may apply in the future. As a result of these challenges, we may not be able to comply with the applicable regulations. After regulatory approvals are obtained, the subsequent discovery of previously unknown problems, or the failure to maintain compliance with existing or new regulatory requirements, may result in restrictions on the marketing of a product, withdrawal of the product from the market, seizures, the shutdown of manufacturing facilities, injunctions, monetary fines or civil or criminal sanctions.
We have never manufactured a product candidate on a commercial scale, and we will need to establish commercial-scale manufacturing capabilities if any of our or our collaboration partners’ product candidates receives regulatory approval.
To be successful, our product candidates and those of our partners must be manufactured for development and, following approval, in commercial quantities, in compliance with regulatory requirements and at acceptable costs. Although we have manufactured product candidates for use in clinical trials, we cannot assure you that we can successfully manufacture our products under applicable cGMP and applicable laws and regulations in sufficient quantities for commercial launch, or in a timely or economical manner. We have not yet produced any product candidates on a large scale or in commercial quantities and have not implemented the facilities and staff required for commercial production of any products. While we believe our current facilities are adequate for the production of product candidates for clinical trials, additional facilities will be required to produce sufficient quantities of any products for commercial sale. The establishment of additional manufacturing facilities will require additional capital outlays and an increase in future operating expenditures. A delay in the implementation
24
of these facilities will seriously disrupt our ability to develop and commercialize our drug candidates as well as our ability to realize value from our collaborations.
If we are unable to provide commercial quantities of our product candidates, we will have to successfully engage a third party to manufacture them. Engaging a third-party manufacturer would require us to conduct comparative studies or use other means to determine bioequivalence between product candidates manufactured by a third-party manufacturer and those previously manufactured by us, if any, which could delay or prevent our ability to commercialize our product candidates. If we are unable to increase our manufacturing capacity or we are unable to establish alternative arrangements on a timely basis or on acceptable terms, the development and commercialization of our product candidates may be delayed or there may be a shortage in supply.
Risks Related to Employee Matters and Managing Growth
Our future success depends on our ability to retain our Chief Executive Officer and other key executives and to attract, retain, and motivate qualified personnel.
We are highly dependent on Jan Turek, our President and Chief Executive Officer, and the other principal members of our executive and scientific teams listed under “Management.” All of the agreements with the principal members of our executive and scientific teams provide that employment may be terminated by the employee at any time upon two weeks’ written notice to us. Although we do not have any reason to believe that we may lose the services of any of these persons in the foreseeable future, the loss of the services of any of these persons might impede the achievement of our research, development, and commercialization objectives. Recruiting and retaining qualified scientific personnel and possibly sales and marketing personnel will also be critical to our success. We may not be able to attract and retain these personnel on acceptable terms given the competition among numerous pharmaceutical and biotechnology companies for similar personnel. We also experience competition for the hiring of scientific personnel from universities and research institutions. In addition, we rely on consultants and advisors, including scientific and clinical advisors, to assist us in formulating our research and development and commercialization strategy. Our consultants and advisors may be employed by employers other than us and may have commitments under consulting or advisory contracts with other entities that may limit their availability to us.
We expect to expand our development, clinical research, manufacturing and marketing capabilities, and as a result, we may encounter difficulties in managing our growth, which could disrupt our operations.
Our future success depends on our continuing ability to identify, hire, develop, motivate and retain qualified management, clinical and scientific personnel for all areas of our organization. We expect to experience significant growth in the number of our employees and the scope of our operations. In anticipation of increased development activities, we are currently planning to increase the total number of our full-time employees. As a result, we expect personnel costs to increase in the future. To manage our anticipated future growth, we must continue to implement and improve our managerial, operational, and financial systems, expand our facilities, and continue to recruit and train additional qualified personnel. Due to our limited resources, we may not be able to effectively manage the expansion of our operations or recruit and train additional qualified personnel. The physical expansion of our operations may lead to significant costs and may divert our management and business development resources. Any inability to manage growth could delay the execution of our business plans or disrupt our operations.
If we make strategic acquisitions, we will incur a variety of costs and might never realize the anticipated benefits.
Currently we are not a party to any acquisition agreements, nor do we have any understanding or commitment with respect to any such acquisition. If appropriate opportunities become available, however, we might attempt to acquire approved products, additional product candidates or businesses that we believe are a strategic fit with our
25
business. If we pursue any transaction of that sort, the process of negotiating the acquisition and integrating an acquired product, product candidate or business might result in operating difficulties and expenditures and might require significant management attention that would otherwise be available for ongoing development of our business, whether or not any such transaction is ever consummated. Moreover, we might never realize the anticipated benefits of any acquisition. Future acquisitions could result in potentially dilutive issuances of equity securities, the incurrence of debt, contingent liabilities or impairment expenses related to goodwill and impairment or amortization expenses related to other intangible assets, which could harm our financial condition.
Risks Related to Our Common Stock and this Offering
Our stock price is likely to be volatile and the market price of our common stock after this offering may drop below the price you pay.
You should consider an investment in our common stock as risky and invest only if you can withstand a significant loss and wide fluctuations in the market value of your investment. Prior to this offering, there was not a public market for our stock. We will negotiate and determine the initial public offering price with the representatives of the underwriters based on several factors. This price may vary from the market price of our common stock after this offering. You may be unable to sell your shares of common stock at or above the initial offering price due to fluctuations in the market price of our common stock arising from changes in our operating performance or prospects. In addition, the stock market has recently experienced significant volatility, particularly with respect to pharmaceutical, biotechnology and other life sciences company stocks. The volatility of pharmaceutical, biotechnology and other life sciences company stocks often does not relate to the operating performance of the companies represented by the stock. Some of the factors that may cause the market price of our common stock to fluctuate include:
| | • | | progress in and results from our planned Phase 2 clinical trials of Locteron for the treatment of chronic hepatitis C or any other future clinical trials of Locteron we may initiate; |
| | • | | failure or delays in advancing our preclinical product candidates, BLX-155 and BLX-301, or other product candidates we may develop in the future, into clinical trials; |
| | • | | progress in and results from any clinical trials of BLX-155, BLX-301 or any other future product candidate that we may initiate in the future; |
| | • | | results of clinical trials conducted by others on drugs that would compete with our product candidates; |
| | • | | issues in manufacturing our product candidates or approved products; |
| | • | | regulatory developments or enforcement in the United States and foreign countries; |
| | • | | developments or disputes concerning patents or other proprietary rights; |
| | • | | introduction of technological innovations or new commercial products by us or our competitors; |
| | • | | changes in estimates or recommendations by securities analysts, if any cover our common stock; |
| | • | | public concern over our product candidates or any approved products; |
| | • | | future sales of our common stock; |
| | • | | general market conditions; |
| | • | | changes in the structure of healthcare payment systems; |
| | • | | failure of any of our product candidates, if approved, to achieve commercial success; |
| | • | | economic and other external factors or other disasters or crises; |
26
| | • | | period-to-period fluctuations in our financial results; and |
| | • | | overall fluctuations in U.S. equity markets. |
These and other external factors may cause the market price and demand for our common stock to fluctuate substantially, which may limit or prevent investors from readily selling their shares of common stock and may otherwise negatively affect the liquidity of our common stock. In addition, in the past, when the market price of a stock has been volatile, holders of that stock have instituted securities class action litigation against the company that issued the stock. If any of our stockholders brought a lawsuit against us, we could incur substantial costs defending the lawsuit. Such a lawsuit could also divert the time and attention of our management.
An active, liquid trading market for our common stock may not develop or be sustained after this offering.
There is currently no established trading market for our common stock. Although we have applied to have our common stock listed on The NASDAQ Global Market, there is no guarantee that an active trading market for our common stock will develop or be sustained after this offering on The NASDAQ Global Market or any other exchange. If a trading market does not develop or is not maintained, you may experience difficulty in reselling, or an inability to sell, your shares quickly or at the latest market price.
Raising additional capital may cause dilution to existing stockholders, restrict our operations or require us to relinquish rights.
We may seek the additional capital necessary to fund our operations through public or private equity offerings, debt financings, and collaborative and licensing arrangements. To the extent that we raise additional capital through the sale of equity or convertible debt securities, your ownership interest will be diluted and the terms may include liquidation or other preferences that adversely affect your rights as a stockholder. Debt financing, if available, may involve agreements that include covenants limiting or restricting our ability to take specific actions such as incurring additional debt, making capital expenditures, or declaring dividends. If we raise additional funds through collaboration and licensing arrangements with third parties, we may have to relinquish valuable rights to our technologies or product candidates, or grant licenses on terms that are not favorable to us.
Investors in this offering will pay a much higher price than the book value of our common stock and therefore you will incur immediate and substantial dilution of your investment.
If you purchase common stock in this offering, you will incur immediate and substantial dilution of $ per share, representing the difference between our pro forma as adjusted net tangible book value per share and the assumed initial public offering price of $ ��per share, which is the midpoint of the price range listed on the cover page of this prospectus. In addition, investors purchasing common stock in this offering will contribute approximately % of the total amount invested by stockholders since inception, but will only own approximately % of the shares of common stock outstanding. In the past, we issued options and warrants to acquire common stock at prices significantly below the assumed initial public offering price. To the extent these outstanding options or warrants are ultimately exercised, you will sustain further dilution.
We will incur significant increased costs as a result of operating as a public company, and our management will be required to devote substantial time to new compliance initiatives.
As a public company, we will incur significant legal, accounting and other expenses that we did not incur as a private company. In addition, the Sarbanes-Oxley Act of 2002, as well as rules subsequently implemented by the SEC and The NASDAQ Stock Market, have imposed various new requirements on public companies, including establishment and maintenance of effective disclosure and financial controls and changes in corporate governance practices. Our management and other personnel will need to devote a substantial amount of time to these new compliance initiatives. Moreover, these rules and regulations will increase our legal and financial compliance costs and will make some activities more time-consuming and costly. For example, we expect these
27
rules and regulations to make it more difficult and more expensive for us to obtain director and officer liability insurance, and we may be required to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. As a result, it may be more difficult for us to attract and retain qualified people to serve on our board of directors, our board committees or as executive officers.
Under the corporate governance standards of The NASDAQ Stock Market, a majority of our board of directors and each member of our audit committee must be an independent director no later than the first anniversary of the completion of this offering. Although we intend to replace Messrs. Hegele and Dougherty as members of our audit committee prior to this time to comply with these standards, we may encounter difficulty in attracting qualified persons to serve on our audit committee. In addition, if vacancies on our board or our audit committee occur that need to be filled by independent directors, we may encounter difficulty in attracting qualified persons to serve on our board and, in particular, our audit committee. If we fail to attract and retain the required number of independent directors we may be subject to SEC enforcement proceedings and delisting of our common stock from The NASDAQ Global Market.
The Sarbanes-Oxley Act of 2002 requires, among other things, that we maintain effective internal controls for financial reporting and disclosure. In particular, commencing in fiscal 2008, we must perform system and process evaluation and testing of our internal controls over financial reporting to allow management and our independent registered public accounting firm to report on the effectiveness of our internal controls over financial reporting, as required by Section 404 of the Sarbanes-Oxley Act of 2002. Our testing, or the subsequent testing by our independent registered public accounting firm, may reveal deficiencies in our internal controls over financial reporting that are deemed to be material weaknesses. We expect to incur significant expense and devote substantial management effort toward ensuring compliance with Section 404. We currently do not have an internal audit function, and we will need to hire additional accounting and financial staff with appropriate public company experience and technical accounting knowledge. Moreover, if we are not able to comply with the requirements of Section 404 in a timely manner, or if we or our independent registered public accounting firm identifies deficiencies in our internal controls that are deemed to be material weaknesses, the market price of our stock could decline and the ability of registered broker-dealers to make a market in our stock could be adversely affected and we could be subject to sanctions or investigations by The NASDAQ Stock Market, the SEC or other regulatory authorities, which would entail expenditure of additional financial and management resources.
Insiders will continue to have substantial control over us which could delay or prevent a change in corporate control or result in the entrenchment of management or our board of directors.
After this offering, our directors, executive officers, together with their affiliates and related persons, and stockholders owning more than 10% of our common stock will beneficially own, in the aggregate, approximately % of our outstanding common stock. As a result, these stockholders, if acting together, may have the ability to determine the outcome of matters submitted to our stockholders for approval, including the election and removal of directors and any merger, consolidation or sale of all or substantially all of our assets. In addition, these persons, acting together, may have the ability to control the management and affairs of our company. Accordingly, this concentration of ownership may harm the market price of our common stock by:
| | • | | delaying, deferring or preventing a change in control; |
| | • | | entrenching our management or our board of directors; |
| | • | | impeding a merger, consolidation, takeover or other business combination involving us; or |
| | • | | discouraging a potential acquirer from making a tender offer or otherwise attempting to obtain control of us. |
28
Future sales of common stock by our existing stockholders may cause our stock price to fall.
Sales of a substantial number of shares of our common stock in the public market could occur at any time. These sales, or the perception in the market that the holders of a large number of shares intend to sell shares, could reduce the market price of our common stock. After this offering, we will have outstanding shares of common stock. This includes the shares that we are selling in this offering, which may be resold in the public market immediately. The remaining shares are currently restricted as a result of securities laws or lock-up agreements but will be able to be sold in the near future as follows:
| | |
Number of Shares | | Date |
| | On the date of this prospectus. |
| | At 90 days from the date of this prospectus. |
| | At various times between the 90 days and 180 days* from the date of this prospectus. |
| | At or after 180 days* from the date of this prospectus (subject, in some cases, to volume limitations). |
| * | This 180-day period corresponds to the end of the lock-up period described in “Shares Eligible for Future Sale—Lock-Up Agreements.” This lock-up period may be extended under certain circumstances as described in that section. |
Moreover, beginning after the lock-up period described in “Shares Eligible for Future Sale—Lock-Up Agreements” expires, the holders of 85,471,096 shares of our common stock will have rights, subject to some conditions, to require us to file registration statements covering their shares or to include their shares in registration statements that we may file for ourselves or other stockholders. We also intend to register all shares of common stock that we may issue under our stock plans. Once we register these shares, they will be freely tradable, subject to the lock-up agreements. See “Shares Eligible for Future Sale” for a more detailed description of sales of our common stock that may occur in the future.
We have broad discretion in the use of the net proceeds from this offering and may not use them effectively.
Management will retain broad discretion over the use of the net proceeds from this offering. Stockholders may not agree with such uses, and our use of the proceeds may not yield a significant return or any return at all for our stockholders. The failure by our management to apply these funds effectively could have a material adverse effect on our business.
We intend to use the proceeds from this offering for clinical trials, clinical supply manufacturing, preclinical testing and other research and development activities, general and administrative expenses, working capital needs and other general corporate purposes. Because of the number and variability of factors that will determine our use of the proceeds from this offering, their ultimate use may vary substantially from their currently intended use. For a further description of our intended use of the proceeds of the offering, see “Use of Proceeds.”
29
Provisions of our charter and bylaws and Delaware law may make an acquisition of us or a change in our management more difficult.
Certain provisions of our amended and restated certificate of incorporation and restated bylaws that will be in effect upon the completion of this offering could discourage, delay, or prevent a merger, acquisition, or other change in control that stockholders may consider favorable, including transactions in which you might otherwise receive a premium for your shares. These provisions also could limit the price that investors might be willing to pay in the future for shares of our common stock, thereby depressing the market price of our common stock. Stockholders who wish to participate in these transactions may not have the opportunity to do so. Furthermore, these provisions could prevent or frustrate attempts by our stockholders to replace or remove our management. These provisions:
| | • | | allow the authorized number of directors to be changed only by resolution of our board of directors; |
| | • | | establish a classified board of directors, providing that not all members of our board be elected at one time; |
| | • | | authorize our board of directors to issue without stockholder approval blank check preferred stock that, if issued, could operate as a “poison pill” to dilute the stock ownership of a potential hostile acquirer to prevent an acquisition that is not approved by our board of directors; |
| | • | | require that stockholder actions must be effected at a duly called stockholder meeting and prohibit stockholder action by written consent; |
| | • | | establish advance notice requirements for stockholder nominations to our board of directors or for stockholder proposals that can be acted on at stockholder meetings; |
| | • | | limit who may call stockholder meetings; and |
| | • | | require the approval of the holders of 80% of the outstanding shares of our capital stock entitled to vote in order to amend certain provisions of our amended and restated certificate of incorporation and restated bylaws. |
In addition, because we are incorporated in Delaware, we are governed by the provisions of Section 203 of the Delaware General Corporation Law, which may, unless certain criteria are met, prohibit large stockholders, in particular those owning 15% or more of our outstanding voting stock, from merging or combining with us for a prescribed period of time.
We do not anticipate paying cash dividends, and accordingly, stockholders must rely on stock appreciation for any return on their investment.
We have never declared or paid any cash dividend on our stock and do not currently intend to do so for the foreseeable future. We currently anticipate that we will retain future earnings for the development, operation and expansion of our business and do not anticipate declaring or paying any cash dividends for the foreseeable future. Therefore, the success of an investment in shares of our common stock will depend upon any future appreciation in their value. There is no guarantee that shares of our common stock will appreciate in value or even maintain the price at which our stockholders have purchased their shares.
30
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
This prospectus contains forward-looking statements. The forward-looking statements are contained principally in the sections entitled “Prospectus Summary,” “Risk Factors,” “Use of Proceeds,” “Management’s Discussion and Analysis of Financial Condition and Results of Operations,” and “Business.” These statements involve known and unknown risks, uncertainties, and other factors which may cause our actual results, performance, or achievements to be materially different from any future results, performance, or achievements expressed or implied by the forward-looking statements. Forward-looking statements include statements about:
| | • | | the anticipated progress of our research, development, and clinical programs, including the timing of current and future clinical trials; |
| | • | | our ability to succeed in obtaining FDA clearance for Locteron, BLX-155, or BLX-301 or for any future product candidates; |
| | • | | our ability to market, commercialize, and achieve market acceptance for our drug candidates that we may develop or acquire; |
| | • | | our ability to protect our intellectual property and operate our business without infringing upon the intellectual property rights of others; |
| | • | | our anticipated use of the proceeds of this offering; and |
| | • | | estimates regarding the sufficiency of our cash resources. |
In some cases, you can identify forward-looking statements by terms such as “anticipates,” “believes,” “could,” “estimates,” “expects,” “intends,” “may,” “plans,” “potential,” “predicts,” “projects,” “should,” “will,” “would,” and similar expressions intended to identify forward-looking statements. Forward-looking statements reflect our current views with respect to future events and are based on assumptions and subject to risks and uncertainties. We discuss many of these risks in this prospectus in greater detail under the heading “Risk Factors.” Given these uncertainties, you should not place undue reliance on these forward-looking statements. Also, forward-looking statements represent our estimates and assumptions only as of the date of this prospectus. You should read this prospectus and the documents that we have filed as exhibits to the registration statement, of which this prospectus is a part, completely and with the understanding that our actual future results may be materially different from what we expect.
Except as required by law, we assume no obligation to update any forward-looking statements publicly or to update the reasons actual results could differ materially from those anticipated in any forward-looking statements, even if new information becomes available in the future.
This prospectus contains market data and industry forecasts that were obtained from industry publications. These publications generally indicate that this information has been obtained from sources believed to be reliable but do not guarantee the accuracy or completeness of this information. Although we believe that the reports are reliable, we have not independently verified any of this information.
31
USE OF PROCEEDS
We estimate that our net proceeds from the sale of shares of our common stock in this offering will be approximately $ million, or approximately $ million if the underwriters exercise their option to purchase additional shares in full, assuming an initial public offering price of $ per share, the midpoint of the range listed on the cover page of this prospectus, and after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. A $1.00 increase (decrease) in the assumed initial public offering price of $ per share would increase (decrease) the net proceeds to us from this offering by $ million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
The principal purposes of this offering are to obtain additional working capital to fund anticipated operating losses, establish a public market for our common stock, and facilitate future access to the public markets. We estimate that we will use the proceeds of this offering as follows:
| | • | | approximately $ to $ million of these net proceeds to fund the continued clinical development of Locteron, including ; |
| | • | | approximately $ to $ million of these net proceeds to fund the continued development of BLX-155, including ; and |
| | • | | approximately $ million to fund general research and development, general and administrative expenses, working capital needs, and other general corporate purposes. |
We may also use a portion of the proceeds for the potential acquisition of, or investment in, technologies, products, or companies that complement our business, although we have no current understandings, commitments, or agreements to do so.
As of the date of this prospectus, we cannot predict with certainty all of the particular uses for the proceeds from this offering, or the amounts that we will actually spend on the uses set forth above. The amounts and timing of our actual expenditures will depend upon numerous factors, including the progress of our research, development, and commercialization efforts, the progress of our clinical trials, and our operating costs and expenditures. Accordingly, our management will have significant flexibility in applying the net proceeds of this offering.
The costs and timing of drug development and regulatory approval, particularly conducting clinical trials, are highly uncertain, are subject to substantial risks, and can often change. Accordingly, we may change the allocation of use of these proceeds as a result of contingencies such as the progress and results of our clinical trials and other research and development activities, the establishment of collaborations, the results of our commercialization efforts, our manufacturing requirements and regulatory or competitive developments. In addition, assuming our current clinical programs proceed further to the next stage of clinical development, we do not expect our existing capital resources and the net proceeds from this offering to be sufficient to enable us to fund the completion of all such clinical development programs through commercial introduction. Accordingly, we expect we will need to raise additional funds.
Pending use of the proceeds from this offering as described above or otherwise, we intend to invest the net proceeds in short-term interest-bearing, investment grade securities.
DIVIDEND POLICY
We have never paid or declared any cash dividends on our common stock, and we do not anticipate paying any cash dividends on our common stock in the foreseeable future. We intend to retain all available funds and any future earnings to fund the development and expansion of our business.
32
CAPITALIZATION
The following table sets forth our capitalization as of March 31, 2007:
| | • | | on a pro forma basis to give effect to: |
| | • | | the sale by us of 25,663,339 shares of our Series CC preferred stock on May 18, 2007 at $1.18 per share in exchange for $25.0 million in cash and the conversion of a $5.0 million convertible note and accrued interest payable to a related party, less transaction expenses; and |
| | • | | the conversion of all outstanding shares of our Class B non-voting common stock and our preferred stock into shares of our Class A voting common stock and the reclassification of our Class A voting common stock into common stock upon completion of this offering; and |
| | • | | on a pro forma as adjusted basis to give further effect to our sale of shares of common stock in this offering at an assumed initial public offering price of $ per share, the midpoint of the range listed on the cover page of this prospectus, after deducting estimated underwriting discounts and commissions and estimated offering expenses payable by us. |
This table should be read with “Selected Consolidated Financial Data” and “Management’s Discussion and Analysis of Financial Condition and Results of Operations” and our consolidated financial statements and the related notes appearing elsewhere in this prospectus.
| | | | | | |
| | | As of March 31, 2007 |
| | | Actual | | Pro
Forma | | Pro Forma
as Adjusted(1) |
| | | (unaudited) |
| | | (in thousands, except share and per share amounts) |
Long-term debt and capital lease obligations, excluding current portion | | 7,319 | | 2,319 | | |
| | | | | | |
Mandatorily redeemable convertible preferred stock: | | | | | | |
Series AA1 preferred stock, $.001 par value; 5,092,701 shares authorized and outstanding, actual; no shares authorized and outstanding, pro forma and pro forma as adjusted (liquidation preference of $5,092 at March 31, 2007) | | 5,910 | | — | | |
Series AA2 preferred stock, $.001 par value; 22,594,864 shares authorized and outstanding, actual; no shares authorized and outstanding, pro forma and pro forma as adjusted (liquidation preference of $22,594 at March 31, 2007) | | 25,905 | | — | | |
Series BB preferred stock, $.001 par value; 33,902,000 shares authorized and 32,120,192 shares outstanding, actual; no shares authorized and outstanding, pro forma and pro forma as adjusted (liquidation preference of $35,974 at March 31, 2007) | | 41,255 | | — | | |
Series CC preferred stock, $.001 par value; no shares authorized and outstanding, actual; no shares authorized and outstanding, pro forma and pro forma as adjusted (liquidation preference of $30,282 at March 31, 2007 on a pro forma basis) | | — | | — | | |
| | | | | | |
| | 73,070 | | 2,319 | | |
| | | | | | |
33
| | | | | | | | | | |
| | | As of March 31, 2007 |
| | | Actual | | | Pro
Forma | | | Pro Forma
as Adjusted(1) |
| | | (unaudited) |
| | | (in thousands, except share and per share amounts) |
Stockholders’ equity (deficit): | | | | | | | | | | |
Common stock, $.001 par value; no shares authorized and outstanding, actual; 110,000,000 shares authorized and 62,087,644 shares outstanding, pro forma; shares authorized and shares outstanding, pro forma as adjusted | | | — | | | | 88 | | | |
Class A common stock, $.001 par value; 80,000,000 shares authorized and 494,258 shares outstanding, actual; no shares authorized and outstanding, pro forma and pro forma as adjusted | | | — | | | | — | | | |
Class B common stock, $.001 par value; 3,821 shares authorized and outstanding, actual; no shares authorized and outstanding, pro forma and pro forma as adjusted | | | — | | | | — | | | |
Additional paid-in-capital | | | 3,635 | | | | 105,736 | | | |
Accumulated other comprehensive loss | | | (33 | ) | | | (33 | ) | | |
Deficit accumulated during development stage | | | (75,421 | ) | | | (75,421 | ) | | |
| | | | | | | | | | |
Total stockholders equity (deficit) | | | (71,819 | ) | | | (30,468 | ) | | |
| | | | | | | | | | |
Total capitalization | | $ | 8,570 | | | $ | 32,787 | | | |
| | | | | | | | | | |
(1) | A $1.00 increase (decrease) in the assumed initial public offering price of $ per share would increase (decrease) each of the pro forma as adjusted additional paid-in capital, total stockholders’ equity and total capitalization by $ million, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us. The pro forma as adjusted information discussed above is illustrative only and following the completion of this offering will be adjusted based on the actual public offering price and other terms of this offering determined at pricing. |
The outstanding share information excludes:
| | • | | 8,545,630 shares of common stock issuable upon the exercise of stock options outstanding as of March 31, 2007 at a weighted average exercise price of $0.17 per share; |
| | • | | 93,640 shares of common stock issuable upon the exercise of warrants outstanding as of March 31, 2007 at a weighted average exercise price of $1.09 per share; and |
| | • | | 2,826,732 shares of common stock reserved for future awards under our stock plan as of March 31, 2007. |
34
DILUTION
If you invest in our common stock, your interest will be diluted to the extent of the difference between the initial public offering price per share of our common stock and the pro forma as adjusted net tangible book value per share of our common stock after this offering. We calculate net tangible book value per share of common stock by dividing the net tangible book value (tangible assets less total liabilities) by the number of outstanding shares of common stock.
Our historical net tangible book value at March 31, 2007 was $ million, or $ per share of common stock, based on shares of common stock outstanding at March 31, 2007. Our pro forma net tangible book value as of March 31, 2007 was $ million, or $ per share of common stock, based on shares of common stock outstanding after giving effect to the conversion of all outstanding shares of our Class B non-voting common stock and preferred stock into shares of our Class A voting common stock and the reclassification of our Class A voting common stock into common stock upon completion of this offering. After giving further effect to the sale of shares of common stock by us in this offering at an assumed initial public offering price of $ per share, the midpoint of the range listed on the cover page of this prospectus, less the estimated underwriting discounts and commissions and the estimated offering expenses payable by us, our pro forma as adjusted net tangible book value at March 31, 2007, would be $ million, or $ per share. This represents an immediate increase in the pro forma net tangible book value of $ per share to existing stockholders and an immediate dilution of $ per share to new investors purchasing shares in this offering. The following table illustrates this per share dilution:
| | | | | | |
Assumed initial public offering price per share | | | | | $ | |
Historical net tangible book value per share as of March 31, 2007 | | $ | | | | |
Pro forma increase per share attributable to conversion of the preferred stock | | | | | | |
| | | | | | |
Pro forma net tangible book value per share as of March 31, 2007 | | | | | | |
Increase per share attributable to this offering | | | | | | |
| | | | | | |
Pro forma as adjusted net tangible book value per share after this offering | | | | | | |
| | | | | | |
Dilution per share to new investors in this offering | | | | | | $ |
| | | | | | |
A $1.00 increase (decrease) in the assumed initial public offering price of $ per share would increase (decrease) the pro forma as adjusted net tangible book value by $ million, the pro forma as adjusted net tangible book value per share after this offering by $ per share and the dilution per share to new investors in this offering by $ per share, assuming that the number of shares offered by us, as set forth on the cover page of this prospectus, remains the same and after deducting the estimated underwriting discounts and commissions and estimated offering expenses payable by us.
If the underwriters exercise their option to purchase additional shares in full, the pro forma as adjusted net tangible book value per share after this offering would be $ per share, the increase in the pro forma net tangible book value per share attributable to this offering would be $ per share and the dilution to new investors purchasing shares in this offering would be $ per share.
The following table shows at March 31, 2007, on a pro forma as adjusted basis as described above, the difference between the number of shares of common stock purchased from us, the total consideration paid to us and the average price paid per share by existing stockholders and by new investors purchasing common stock in this offering:
| | | | | | | | | | | | | | |
| | | Shares Purchased | | | Total Consideration | | | Average
Price
Per Share |
| | | Number | | Percent | | | Amount | | Percent | | |
Existing stockholders | | | | % | | | $ | | | % | | | $ | |
New investors | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | |
Total | | | | 100.0 | % | | $ | | | 100.0 | % | | | |
| | | | | | | | | | | | | | |
35
Assuming the underwriters’ option to purchase additional shares is exercised in full, sales by us in this offering will reduce the percentage of shares held by existing stockholders to % and will increase the number of shares held by new investors to , or %.
The information set forth above is based on shares outstanding as of March 31, 2007 and excludes:
| | • | | 8,545,630 shares of common stock issuable upon the exercise of stock options outstanding as of March 31, 2007 at a weighted average exercise price of $0.17 per share; |
| | • | | 93,640 shares of common stock issuable upon the exercise of warrants outstanding as of March 31, 2007 at a weighted average exercise price of $1.09 per share; and |
| | • | | 2,826,732 shares of common stock reserved for future awards under our stock plan as of March 31, 2007. |
To the extent outstanding options and warrants are exercised, there will be further dilution to the new investors. We may grant more options or warrants in the future.
In addition, we may raise additional capital through public or private equity or debt offerings, subject to market conditions, to complete our clinical trials or for other corporate purposes and may also choose to raise additional capital because of strategic considerations even if we believe we have sufficient funds for our future operating plans. To the extent that additional capital is raised through the sale of equity or convertible debt securities, the issuance of these securities could result in further dilution to our stockholders.
36
SELECTED CONSOLIDATED FINANCIAL DATA
(in thousands, except per share amounts)
The selected consolidated financial data presented below have been derived from financial statements that have been prepared in accordance with generally accepted accounting principles and should be read with our consolidated financial statements and the related notes and with “Management’s Discussion and Analysis of Financial Condition and Results of Operations” included elsewhere in this prospectus. The selected statement of operations data for each of the years ended December 31, 2004, 2005 and 2006, and the selected balance sheet data as of December 31, 2005 and 2006, have been derived from our audited financial statements included elsewhere in this prospectus, which have been audited by PricewaterhouseCoopers LLP, independent registered public accounting firm. The selected statement of operations data for the years ended December 31, 2002 and 2003, and the selected balance sheet data as of December 31, 2002, 2003 and 2004, have been derived from our audited financial statements not included in this prospectus. The selected statement of operations data for the three months ended March 31, 2006 and 2007, and the selected balance sheet data as of March 31, 2007, have been derived from our unaudited financial statements included elsewhere in this prospectus. Our unaudited financial statements include, in the opinion of our management, all adjustments that management considers necessary for a fair statement of the financial information set forth in those statements. Our historical results are not necessarily indicative of results to be expected for any future period, and our results for the three months ended March 31, 2007 are not necessarily indicative of results to be expected for the full fiscal year.
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Year Ended December 31, | | | Three Months
Ended March 31, | | | Period From
Sept. 19, 1997
(Inception)
Through
March 31, 2007 | |
| | | 2002 | | | 2003 | | | 2004 | | | 2005 | | | 2006 | | | 2006 | | | 2007 | | |
| | | | | | | | | | | | as restated | | | | | | (unaudited) | | | (unaudited) | |
Statement of Operations Data: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Revenues | | $ | 25 | | | $ | 235 | | | $ | 719 | | | $ | 3,008 | | | $ | 5,017 | | | $ | 1,327 | | | $ | 724 | | | $ | 9,752 | |
Operating expenses: | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Research and development | | | 4,141 | | | | 5,049 | | | | 8,666 | | | | 14,448 | | | | 19,196 | | | | 5,084 | | | | 5,292 | | | | 60,632 | |
General and administrative | | | 1,372 | | | | 1,820 | | | | 2,400 | | | | 4,126 | | | | 4,370 | | | | 1,096 | | | | 1,114 | | | | 19,687 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Total operating expenses | | | 5,513 | | | | 6,869 | | | | 11,066 | | | | 18,574 | | | | 23,566 | | | | 6,180 | | | | 6,406 | | | | 80,319 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Loss from operations | | | (5,488 | ) | | | (6,634 | ) | | | (10,347 | ) | | | (15,566 | ) | | | (18,549 | ) | | | (4,853 | ) | | | (5,682 | ) | | | (70,567 | ) |
Interest (expense) income, net | | | (1,619 | ) | | | (388 | ) | | | (57 | ) | | | (421 | ) | | | 775 | | | | 152 | | | | (35 | ) | | | (1,734 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Loss before income taxes | | | (7,107 | ) | | | (7,022 | ) | | | (10,404 | ) | | | (15,987 | ) | | | (17,774 | ) | | $ | (4,701 | ) | | $ | (5,717 | ) | | | (72,301 | ) |
Income tax benefit | | | — | | | | — | | | | — | | | | 170 | | | | 277 | | | | 31 | | | | 35 | | | | 482 | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | | (7,107 | ) | | | (7,022 | ) | | | (10,404 | ) | | | (15,817 | ) | | | (17,497 | ) | | | (4,670 | ) | | | (5,682 | ) | | | (71,819 | ) |
Less: accretion of mandatorily redeemable convertible preferred stock | | | (1,826 | ) | | | (1,648 | ) | | | (1,165 | ) | | | (3,034 | ) | | | (5,600 | ) | | | (1,369 | ) | | | (1,495 | ) | | | (16,772 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss attributable to common stockholders | | $ | (8,933 | ) | | $ | (8,670 | ) | | $ | (11,569 | ) | | $ | (18,851 | ) | | $ | (23,097 | ) | | $ | (6,039 | ) | | $ | (7,177 | ) | | $ | (88,591 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Basic and diluted net loss attributable to common stockholders per share | | $ | (33.79 | ) | | $ | (32.64 | ) | | $ | (42.91 | ) | | $ | (65.25 | ) | | $ | (68.52 | ) | | $ | (20.18 | ) | | $ | (14.93 | ) | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Shares used in the calculation of basic and diluted net loss per share | | | 264 | | | | 266 | | | | 270 | | | | 289 | | | | 337 | | | | 299 | | | | 481 | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Pro forma basic and diluted net loss per share (unaudited)(1) | | | | | | | | | | | | | | | | | | $ | (0.29 | ) | | | | | | $ | (0.09 | ) | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Shares used to compute pro forma basic and diluted net loss per share (unaudited)(1) | | | | | | | | | | | | | | | | | | | 60,145 | | | | | | | | 60,288 | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
(1) | The pro forma basic and diluted net loss per share gives effect to the conversion of all outstanding shares of preferred stock into common stock upon completion of this offering. |
37
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | As of December 31, | | | As of
March 31,
2007 | |
| | | 2002 | | | 2003 | | | 2004 | | | 2005 | | | 2006 | | |
| | | | | | | | | | | | as restated | | | | | | (unaudited) | |
Balance Sheet Data: | | | | | | | | | | | | | | | | | | | | | | | | |
Cash and cash equivalents | | $ | 249 | | | $ | 7,265 | | | $ | 367 | | | $ | 27,468 | | | $ | 14,553 | | | $ | 8,708 | |
Working capital (deficit) | | | (6,005 | ) | | | 6,298 | | | | (1,702 | ) | | | 20,699 | | | | 8,456 | | | | 2,505 | |
Total assets | | | 1,591 | | | | 8,723 | | | | 3,901 | | | | 38,096 | | | | 24,906 | | | | 19,172 | |
Long-term liabilities | | | 125 | | | | 329 | | | | 623 | | | | 6,238 | | | | 11,316 | | | | 10,690 | |
Total liabilities | | | 6,407 | | | | 1,353 | | | | 3,103 | | | | 13,697 | | | | 17,980 | | | | 17,921 | |
Mandatorily redeemable convertible preferred stock | | | 12,955 | | | | 18,783 | | | | 21,617 | | | | 65,976 | | | | 71,576 | | | | 73,070 | |
Additional paid-in-capital | | | 1 | | | | 14,612 | | | | 13,653 | | | | 10,668 | | | | 5,118 | | | | 3,635 | |
Deficit accumulated during development stage | | | (17,759 | ) | | | (26,021 | ) | | | (36,424 | ) | | | (52,242 | ) | | | (69,739 | ) | | | (75,421 | ) |
Total stockholders’ deficit | | | (17,771 | ) | | | (11,414 | ) | | | (22,820 | ) | | | (41,577 | ) | | | (64,649 | ) | | | (71,819 | ) |
38
MANAGEMENT’S DISCUSSION AND ANALYSIS OF
FINANCIAL CONDITION AND RESULTS OF OPERATIONS
You should read the following discussion and analysis of our financial condition and results of operations together with “Selected Consolidated Financial Data” and our consolidated financial statements and the related notes appearing elsewhere in this prospectus. This discussion and analysis contains forward-looking statements, including, but not limited to, those set forth under “Special Note Regarding Forward-Looking Statements,” that involve risks, uncertainties and assumptions. The actual results may differ materially from those anticipated in these forward-looking statements as a result of certain factors, including, but not limited to, those set forth under “Risk Factors” and elsewhere in this prospectus.
Overview
We are a clinical-stage biopharmaceutical company that uses our patented LEX System to develop hard-to-make therapeutic proteins and optimized monoclonal antibodies. The LEX System is a novel technology that genetically transforms the aquatic plantLemna to enable the production of biologic product candidates. Our proprietary product candidates are designed to provide superior efficacy/tolerability profiles and to address large, proven pharmaceutical markets.
Our lead product candidate, Locteron, is in Phase 2 clinical trials and is the only controlled-release interferon alfa known to be currently in clinical development for the treatment of chronic hepatitis C. We have developed two additional product candidates that also capitalize on the benefits of the LEX System, which we are advancing toward clinical trials: BLX-155, a direct-acting thrombolytic designed to dissolve blood clots in patients; and BLX-301, an anti-CD20 antibody we are optimizing for the treatment of non-Hodgkin’s B-cell lymphoma and other diseases. We believe that all of our product candidates have reduced clinical and regulatory risk profiles because they are based on understood proteins with proven mechanisms of action or clinical validation, and well-established regulatory pathways.
In addition to enabling our internally generated product candidates, the LEX System has provided us with opportunities to form strategic alliances with leading pharmaceutical and biotechnology companies such as Centocor, Genmab A/S, Medarex and Merial Limited. Under these collaborations, we are applying the LEX System to protein and antibody candidates held by our partners, including the optimization of antibody candidates and the development ofLemna plant lines to enable commercially viable production of these therapeutic proteins. To date, we have received limited revenue pursuant to these collaborations, and there is no certainty that we and any of our partners will agree to move any of the product candidates that are the subject of these collaborations into more advanced development or that such product candidates will ever be successfully commercialized.
We have generated significant losses to date and expect to continue to generate significant losses as we continue the clinical development of Locteron and undertake research activities required to support the advancement of BLX-155 and BLX-301 into clinical development. From our inception in September 1997 through March 31, 2007, we have incurred net losses of $71.8 million and have accumulated a deficit of $75.4 million. Our net losses were $10.4 million, $15.8 million and $17.5 million for the years ended December 31, 2004, 2005 and 2006, respectively, and $4.7 million and $5.7 million for the three months ended March 31, 2006 and 2007, respectively. We have devoted and are continuing to devote substantially all of our efforts toward product research and development. From our inception through March 31, 2007, we have incurred research and development expenses of $60.6 million. Research and development expenses were $8.7 million, $14.4 million and $19.2 million for the years ended December 31, 2004, 2005 and 2006, respectively, and $5.1 million and $5.3 million for the three months ended March 31, 2006 and 2007, respectively.
39
We are a development-stage company, and through March 31, 2007, we have funded operations primarily through:
| | • | | approximately $72.3 million of gross proceeds from private placements of our preferred stock and our common stock and the exercise of options; |
| | • | | approximately $9.8 million in revenue from inception through March 31, 2007, consisting of research funding, milestone payments and recognition of up-front fees from collaborations related to our LEX System; |
| | • | | $5.0 million in gross proceeds from a convertible note payable to a related party; and |
| | • | | $3.7 million in proceeds, net of repayments, from notes payable to financial institutions. |
In addition, in May 2007, we raised approximately $25.0 million in gross cash proceeds from the private placement of 25,663,339 shares of our Series CC preferred stock in a transaction that also included the conversion of a $5.0 million convertible note and accrued interest payable to a related party.
Because we do not currently generate revenue from any of our product candidates, our losses will continue as we continue our research and development activities. We expect these activities to expand over time and require further resources if we are to be successful. As a result, our operating losses are likely to be substantial over the next several years. We will need substantial additional funding to seek regulatory approvals for our product candidates, fund operating losses, establish commercial manufacturing and sales and, if deemed appropriate, establish marketing capabilities, which we may seek to raise through public or private equity or debt financings, collaborative or other arrangements with third parties or through other sources of funding. There can be no assurance that such funds will be available on terms favorable to us, if at all. We expect that our operating results will fluctuate for the foreseeable future. Accordingly, period-to-period comparisons should not be relied upon as predictive of the results for future periods.
Restatement of Previously Issued Consolidated Financial Statements
We have restated our previously issued consolidated financial statements and related footnotes as of and for the year ended December 31, 2005. The restatement was to:
| | • | | correct an error in accounting for the revenue recognition of our selected protein development and manufacturing alliance agreement with Centocor; and |
| | • | | to correct an error in the accounting for income taxes in connection with the purchase price allocation related to the LemnaGene acquisition. |
The error we are correcting in the restatement related to revenue originated in the first quarter of 2005 and continued throughout 2005. The identification of this error occurred as a result of our reevaluation of the assumptions we used underEITF 00-21, in accounting for arrangements with multiple deliverables that require significant judgment and estimates.
We reassessed our obligations under our agreement with Centocor and determined that certain obligations that were previously accounted for as separate units of accounting needed to be combined with other, undelivered items and accounted for as a single unit of accounting. We determined that our obligations during the protein development phase of the agreement should be accounted for as a single unit of accounting. Future manufacturing and training obligations under the alliance agreement with Centocor are considered separate units of accounting. In addition, we determined that the substantive milestone method of revenue recognition we had been using for certain payments was not appropriate as those payments did not meet the substantive milestone criteria. We determined a time-based proportional performance model would be more appropriate to account for these payments. Accordingly, in the restated consolidated financial statements for the year ended December 31, 2005, we decreased revenue and increased deferred revenue by $2.0 million.
40
In addition, we have revised our consolidated financial statements to correct an error in the accounting for income taxes in connection with purchase price allocation for the LemnaGene acquisition, which occurred on July 7, 2005. We changed the associated income tax accounting related to this acquisition to be in accordance with EITF Issue No. 98-11,Accounting for Acquired Temporary Differences in Certain Purchase Transactions That Are Not Accounted for as Business Combinations.We determined that we should have assigned values for the differences between the recorded assets and the tax bases of the assets acquired. Accordingly, in our restated consolidated financial statements for the year ended December 31, 2005, we increased intangible assets by $0.7 million, increased deferred tax asset by $0.4 million and increased deferred tax liabilities by $1.0 million. In addition, amortization expense related to the intangible asset increased by $0.3 million and a deferred tax benefit of $0.2 million was realized during the year ended December 31, 2005.
All data included in this discussion and analysis for the year ended December 31, 2005 is derived from our restated financial statements for those periods. For additional information regarding this restatement, see note 2 to our consolidated financial statements.
Financial Operations
Revenue
We have generated approximately $9.8 million in revenue from inception through March 31, 2007, consisting of research funding, milestones and recognition of up-front fees from collaborations related to our LEX System. During 2006 and the three months ended March 31, 2007, we recognized revenue of $5.0 million and $0.7 million, respectively, from our collaborations. Approximately 82% and 44% of the revenues for 2006 and the three months ended March 31, 2007, respectively, resulted from our collaboration with one partner, Centocor. $2.3 million of the 2006 revenue related to the funding of a research program that ended December 31, 2006. There is no material revenue guaranteed under any of our existing collaboration agreements. Future collaboration revenues are contingent upon our existing partners, or new partners, undertaking additional product development activities using our LEX System, or completion of partnerships or other agreements related to our product candidates.
Research and Development Expense
The majority of our operating expenses to date have been for research and development activities. Research and development expenses consist of costs associated with the development of the LEX System and related manufacturing capabilities, support for our collaborations, and our product development efforts, including development of manufacturing processes, preclinical studies, clinical trials and manufacturing of preclinical and clinical supplies. Research and development expenses, including those paid to third parties, are recognized as incurred. Research and development expenses include:
| | • | | employee and consultant-related expenses, which include salaries and benefits; |
| | • | | third-party supplier expenses, including supplies used in manufacturing within the LEX System; |
| | • | | external research and development expenses incurred pursuant to agreements with contract research organizations, and clinical investigational sites; and |
| | • | | facilities, depreciation and other allocated expenses, which include direct and allocated expenses for rent and maintenance of facilities, depreciation of leasehold improvements and equipment and laboratory and other supplies. |
Our research and development expenses originate within three groups: research, operations, and development. At any time, we have a number of research and development projects and collaborative activities that draw upon the same resources, particularly within our research group. Our internal resources, employees and infrastructure within our research group are typically deployed across multiple projects. We do not allocate the
41
expenses of our research group on a program-specific basis as we do not believe that such an allocation would reflect the actual incremental cost of a project.
During 2006 and the three months ended March 31, 2007, the two product development projects encompassing the greatest use of resources within our development and operations groups were our Locteron and BLX-155 programs. A summary of costs directly attributable to Locteron and BLX-155 within our development and operations groups, including the cost of conducting and supporting preclinical studies and clinical trials, and the cost of manufacturing product candidates for use in process development, preclinical studies and clinical tests are summarized below, as are the costs for all other research and development activities.
| | | | | | | | | | | | | | | |
| | | Years Ended December 31, | | Three Months Ended March 31, |
| | | 2004 | | 2005 | | 2006 | | 2006 | | 2007 |
| | | | | | | (in thousands) |
Costs associated with Locteron preclinical and clinical development borne by us | | $ | 3,291 | | $ | 5,461 | | $ | 8,981 | | $ | 2,603 | | $ | 931 |
Costs associated with BLX-155 preclinical development | | | — | | | — | | | 636 | | | 32 | | | 2,109 |
Other research and development activities, including unallocated, shared cost centers | | | 5,375 | | | 8,988 | | | 9,579 | | | 2,449 | | | 2,252 |
| | | | | | | | | | | | | | | |
Total | | $ | 8,666 | | $ | 14,449 | | $ | 19,196 | | $ | 5,084 | | $ | 5,292 |
| | | | | | | | | | | | | | | |
Costs associated with Locteron and BLX-155 preclinical and clinical development include third-party research laboratories associated with preclinical studies, clinical trial site and investigator fees and contract research organization fees associated with clinical trials, and cost of manufacturing, including personnel costs, materials and supplies, manufacturing facilities costs and other related costs within our development and operations groups. These costs associated with Locteron increased in 2005 and 2006 due to the progression of product development activities, including the conduct of the Phase 1 clinical trial of BLX-883, the active agent in Locteron, in 2005, the conduct of the Phase 1 Locteron trial in 2006, and the manufacturing associated with the Phase 2a Locteron trial undertaken in 2006. Under our agreement with OctoPlus, the third-party preclinical and clinical costs associated with Locteron are shared equally between our two companies. The costs associated with BLX-155 increased in 2006 and 2007 due to the development of manufacturing processes, the manufacturing associated with preclinical studies, and the commencement of preclinical studies. Other research and development costs include earlier-stage research associated with Locteron and BLX-155, the advancement of our glycosylation optimization technology, the build-up of a manufacturing and research infrastructure and capabilities, support of our corporate collaborations, and shared departments within our research group whose resources are utilized in multiple projects and product candidates.
All of our research and development programs are at an early stage and may not result in any approved products. Product candidates that may appear promising at early stages of development may not reach the market for a variety of reasons. Product candidates may be found to be ineffective or to cause harmful side effects during clinical trials, may take longer to pass through clinical trials than had been anticipated, may fail to receive necessary regulatory approvals and may prove impracticable to manufacture in commercial quantities at reasonable cost and with acceptable quality. As part of our business strategy, we may enter into collaborative arrangements with third parties to complete the development and commercialization of our product candidates and it is uncertain which of our product candidates may be subject to future collaborative arrangements. The participation of a collaborative partner may accelerate the time to completion and reduce the cost to us of a product candidate or it may delay the time to completion and increase the cost to us due to the alteration of our existing strategy.
42
As a result of the uncertainties discussed above, the uncertainty associated with clinical trial enrollments, and the risks inherent in the development process, we are unable to determine the duration and completion costs of the current or future clinical stages of our product candidates or when, or to what extent or at all, we will generate revenue from the commercialization and sale of any of our product candidates. Development timelines, probability of success and development costs vary widely. Under our collaboration with OctoPlus, we are jointly responsible for completing the Phase 2a and Phase 2b clinical trials of Locteron for chronic hepatitis C with the future development and commercial strategy to be determined. The extent to which we elect to retain development and commercialization responsibilities will greatly determine our future costs. We anticipate developing additional product candidates, which will also increase our research and development expenses in future periods. We do not expect any of our current product candidates to be commercially available in major markets before 2012, if at all.
General and Administrative Expenses
General and administrative expenses consist principally of salaries and related costs for personnel in executive, finance, accounting, business development, information technology, legal and human resources functions. Other general and administrative expenses include facility costs not otherwise included in research and development expenses and professional fees for legal, consulting and accounting services. After completion of this offering, we anticipate that general and administrative expenses will increase significantly as we operate as a public company. These increases will likely include increased costs for director and officer liability insurance, costs related to the hiring of additional personnel and increased fees for outside consultants, lawyers and accountants. We also expect to incur significant costs to comply with the corporate governance, internal controls and similar requirements applicable to public companies.
Interest Income and Interest Expense
Interest income consists of interest earned on our cash and cash equivalents and marketable securities. Interest expense consists of interest incurred on our notes payable, convertible notes payable to a related party, and capital lease facilities.
Accretion of Mandatorily Redeemable Convertible Preferred Stock
Our mandatorily redeemable convertible preferred stock is carried at its cost and is increased by periodic accretions so that the carrying amount will equal the redemption amount at August 22, 2010, the date that redemption first becomes available.
Critical Accounting Policies and Significant Judgments and Estimates
Our management’s discussion and analysis of our financial condition and results of operations are based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States. The preparation of these financial statements requires us to make estimates and judgments that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as reported revenues and expenses during the reporting periods. We base our estimates on historical experience and on various other factors that we believe are reasonable under the circumstances. The SEC considers an accounting policy to be critical if it is important to a company’s financial condition and results of operations, and if it requires the exercise of significant judgment and the use of estimates on the part of management in its application. We have discussed the selection and development of the critical accounting policies with the audit committee of our board of directors, and the audit committee has reviewed our related disclosures in this prospectus. Although we believe that our judgments and estimates are appropriate, actual results may differ from those estimates. If actual results or events differ materially from those contemplated by us in making these estimates, our reported financial condition and results of operations for future periods could be materially affected. See “Risk Factors” for certain matters that may affect our future results of operations or financial condition.
43
We believe the following to be our critical accounting policies because they are both important to the portrayal of our financial condition and results of operations and they require critical management judgment and estimates about matters that are uncertain:
| | • | | stock-based compensation; and |
| | • | | value and useful life of acquired intangible assets. |
Revenue Recognition
We recognize revenue in accordance with the SEC’s Staff Accounting Bulletin No. 104,Revenue Recognition in Financial Statements.When evaluating multiple element arrangements, we consider whether the components of the arrangement represent separate units of accounting as defined in Emerging Issues Task Force, or EITF, Issue No. 00-21,Revenue Arrangements with Multiple Deliverables, or EITF 00-21. To recognize a delivered item in a multiple element arrangement, EITF 00-21 requires that the delivered items have value to the customer on a standalone basis, that there is objective and reliable evidence of fair value of the undelivered items and that delivery or performance is probable and within our control for any delivered items that have a right of return. Application of this standard requires subjective determinations and requires management to make judgments about the fair value of the individual elements and whether such elements are separable from the other aspects of the contractual relationship.
We have entered into a collaboration agreement with Centocor which includes multiple deliverables. We account for our obligations during the protein-development phase of this agreement as a single unit of accounting. Future manufacturing and training obligations under this agreement are considered separate units of accounting. We use a time-based proportional performance model to account for cash payments received under this agreement. Amounts resulting from payments received in advance of revenue recognized are recorded as deferred revenue and recognized over the time period in which we fulfill our contractual obligations under this agreement.
Accrued Expenses
As part of the process of preparing our financial statements, we are required to estimate accrued expenses. This process involves identifying services that have been performed on our behalf and estimating the level of service performed and the associated cost incurred for the service when we have not yet been invoiced or otherwise notified of actual cost. The majority of our service providers invoice us monthly in arrears for services performed. We make estimates of our accrued expenses as of each balance sheet date in our financial statements based on facts and circumstances known to us. Examples of estimated accrued expenses include:
| | • | | fees paid to contract research organizations and contract laboratories in connection with preclinical studies; |
| | • | | fees paid to contract research organizations, clinical sites and contract laboratories in connection with clinical trials; |
| | • | | fees paid to contract manufacturers in connection with certain elements of production, such as filling and labeling of vials, of drug supplies for preclinical studies and clinical trials; |
| | • | | fees owed for professional services; and |
| | • | | unpaid salaries, wages, and benefits. |
44
We record accruals for such preclinical study and clinical trial expenses based upon the estimated amount of work completed. The financial terms of agreements vary from contract to contract and may result in uneven payment flows. All such costs are included in research and development expenses based on these estimates. Costs related to setting up the preclinical study or clinical trial, patient enrollment and treatment are accrued and expensed immediately as the related services are provided throughout the duration of the study. We monitor patient enrollment levels and related activities to the extent possible through internal reviews, correspondence and discussions with research institutions and organizations. However, if we have incomplete or inaccurate information, we may underestimate or overestimate activity levels associated with various preclinical studies and clinical trials at a given point in time. To date, we have not made any material adjustments to our estimates of preclinical study and clinical test expenses.
Stock-Based Compensation
On January 1, 2006, we adopted the fair value recognition provisions of the Financial Accounting Standards Board, or FASB, Statement No. 123R,Share-Based Payment, or SFAS 123R, which requires the measurement and recognition of compensation expense for all future share-based payments made to employees and directors be based on estimated fair values. SFAS 123R supersedes our previous accounting for employee stock options in accordance with Accounting Principles Board Opinion No. 25,Accounting for Stock Issued to Employees, or APB 25, FASB Interpretation, or FIN, No. 44,Accounting for Certain Transactions Involving Stock Compensation, an interpretation of APB 25, and related to interpretations, and the disclosure-only provisions of SFAS No. 123,Accounting for Stock-Based Compensation, or SFAS 123, as amended by SFAS 148,Accounting for Stock-Based Compensation—Transition and Disclosure. In March 2005, the SEC issued Staff Accounting Bulletin No. 107, or SAB 107, relating to SFAS 123R. We have applied the provisions of SAB 107 in our adoption of SFAS 123R.
We adopted SFAS 123R using the prospective transition method. Under this method, compensation costs recognized during the year ended December 31, 2006 and the three months ended March 31, 2007 include: (a) compensation costs for all share-based payment awards granted prior to, but not yet vested as of January 1, 2006, based on the intrinsic value in accordance with the original provisions of APB 25 and (b) compensation costs for all share-based payment awards granted or modified subsequent to January 1, 2006, based on the grant-date fair value estimated in accordance with the provisions of SFAS 123R.
As stock-based compensation expense recognized in our statement of operations for the year ended December 31, 2006 and the three months ended March 31, 2007 is based on options ultimately expected to vest, it has been reduced for estimated forfeitures. SFAS 123R requires forfeitures to be estimated at the time of grant and revised, if necessary, in subsequent periods if actual forfeitures differ from those estimates. In the pro forma information required under SFAS 123 for the periods prior to fiscal 2006, we accounted for forfeitures as they occurred.
Under SFAS 123R, we also had a choice of two attribution methods for allocating compensation cost: the “straight-line” method, which allocates expense on a straight-line basis over the requisite service period of the last separately vesting portion of an award, or the “graded vesting attribution method,” which allocates expense on a straight-line basis over the requisite service period for each separately vesting portion of the award as if the award was, in-substance, multiple awards. We chose the straight-line method.
We also chose to continue utilizing the Black-Scholes model as our chosen option-pricing model. We concluded that this was the most appropriate method with which to value our share-based payment arrangements, but if any share-based payment instruments should be granted for which the Black-Scholes method does not meet the measurement objective as stated within SFAS 123R, we will utilize a more appropriate method for valuing that instrument. However, we do not believe that any instruments granted to date and accounted for under SFAS 123R would require a method other than Black-Scholes.
45
Our determination of the fair market value of share-based payment awards on the grant date using option valuation models requires the input of highly subjective assumptions, including the expected price volatility and option life. As we have been operating as a private company, we are unable to use actual price volatility or option life data as input assumptions within our Black-Scholes valuation model.
For the calculation of expected volatility, because we are a private company, and therefore lack company specific historical and implied volatility information, we based our estimate of expected volatility on the volatility of an industry index, the AMEX Biotechnology Index. We intend to continue to consistently apply this process using the same index until a sufficient amount of historical information regarding the volatility of our own share price becomes available.
In regards to the calculation of expected term, we chose to utilize the “simplified” method for “plain vanilla” options as discussed within SAB 107. We believe that all factors listed within SAB 107 as pre-requisites for utilizing the simplified method are true for us and our share-based payment arrangements. We currently intend to utilize the simplified method through December 31, 2008, at which point we anticipate that more detailed information about exercise behavior will be more widely available.
In accordance with the prospective transition method, our financial statements for prior periods have not been restated to reflect, and do not include, the impact of SFAS 123R. Total employee stock-based compensation expense recognized under SFAS 123R for the year ended December 31, 2006 and the three months ended March 31, 2007 was $33,513 and $7,036, respectively. Of these amounts, we allocated $22,529 and $4,187 to research and development and $10,984 and $2,849 to general administration expenses for the year ended December 31, 2006 and the three months ended March 31, 2007, respectively. As a result of the adoption of SFAS 123R, our net loss increased by approximately $33,513 and $7,036 for the year ended December 31, 2006 and the three months ended March 31, 2007, respectively, each of which were less than $0.01 per share. As of March 31, 2007, total compensation related to nonvested options not yet recognized in the financial statements is approximately $30,000 and the weighted-average period over which it is expected to be recognized is approximately 2.3 years. We have not recognized, and do not expect to recognize in the near future, any tax benefit related to employee stock-based compensation costs as a result of the full valuation allowance on our net deferred tax assets and our net operating loss carryforwards. We expect quarterly stock-based compensation expense to increase for the remainder of 2007.
Prior to this offering there has been no public market for our common stock, and in connection with our issuance of stock options, the fair market value for our common stock has been determined by our board of directors, with input from management. Our board of directors exercised judgment in determining the estimated fair market value of our common stock and believes the determinations of the fair market value of our common stock to be reasonable and consistent with its understanding of how similarly situated companies in our industry are valued. Given the absence of an active market for our common stock, our board of directors determined the fair market value of our common stock on the date of grant of stock options based on several factors, including:
| | • | | our stage of development and business strategy; |
| | • | | the prices at which our preferred stock was issued to investors in arms-length transactions and the rights, preferences and privileges of the preferred stock relative to the common stock; |
| | • | | important developments relating to the progression of our research and development programs; and |
| | • | | the likelihood of achieving a liquidity event. |
In April 2006, in connection with our annual compensation review of our employees, we granted options to purchase an aggregate of 468,500 shares of common stock at an exercise price, determined by our board of directors based on the factors described above to be the fair market value of the common stock, of $0.22 per share. From April 2006 through May 2007, we did not grant any stock options. On May 18, 2007, we completed a private placement of 21,186,441 shares of our Series CC preferred stock at $1.18 per share. The purchase price
46
of the Series CC preferred stock was the fair market value as determined by arms-length negotiations between sophisticated investors and our management and board of directors, based on factors such as our stage of development and valuations of similarly situated private biopharmaceutical companies. In May 2007, at the same time as the closing of the Series CC preferred stock financing, we granted options to purchase an aggregate of 1,064,500 shares of common stock, at an exercise price, determined by our board of directors based on the factors described above to be the fair market value of the common stock, equal to the Series CC preferred stock sale price of $1.18 per share.
The following factors contributed to the increase in the fair market value of our common stock between April 2006 and May 2007:
| | • | | the completion of the Phase 1 clinical trial of Locteron in late April 2006 with favorable results; |
| | • | | publication in November 2006 of research demonstrating the potential ability of the LEX System to increase the potency and efficacy of antibodies by optimizing their glycosylation structures; and |
| | • | | the initiation of the Locteron Phase 2a clinical trial in January 2007. |
Acquired Intangible Assets
Intangible assets are comprised of licenses and intellectual property rights, net of accumulated amortization, acquired as a result of the purchase in 2004 of substantially all of the assets of Epicyte Pharmaceutical, Inc., or Epicyte, and the purchase in 2005 of all of the outstanding capital stock of LemnaGene S.A., or LemnaGene, each as described more fully below. The assets are being amortized over the shorter of the underlying patent life or the estimated useful life.
On April 30, 2004 we acquired substantially all of the assets of Epicyte and assumed certain liabilities of Epicyte. Epicyte was a privately held company that had terminated the majority of its operations prior to the acquisition. The primary asset of Epicyte consisted of a portfolio of patents, patent applications and licenses related to the production of monoclonal antibodies in plants. The total purchase price was $1.0 million, comprised of $0.3 million in cash, and the estimated value of 700,000 shares of our Series AA2 preferred stock issued as consideration in the purchase. The estimated fair value of the license and patents acquired was $1.0 million and is being amortized using the straight-line method until April 2010, the expiration date of the primary patent acquired.
On July 7, 2005 we acquired all of the outstanding capital stock of LemnaGene. LemnaGene was a privately held company operating in France and was engaged in early stage research regarding the production of proteins, including human and animal vaccines, in plants. The primary asset of LemnaGene was a license to a portfolio of patents and patent applications. The total purchase price was $2.4 million, consisting of $2.1 million in cash and transaction costs of $0.3 million. The estimated fair value of the license and underlying patents acquired was adjusted to $3.2 million as a result of applying EITF Issue No. 98-11 to record a deferred tax liability. The estimated fair value of the license and underlying patents and the related deferred tax liability are both being amortized using the straight-line method over ten years, the estimated useful life of the license.
Amortization expense was $0.4 million and $0.1 million for the year ended December 31, 2006 and the three months ended March 31, 2007, respectively.
Results of Operations
Three Months Ended March 31, 2007 Compared to Three Months Ended March 31, 2006
Revenue.Revenue for the three months ended March 31, 2007 was $0.7 million representing a decrease of $0.6 million, or 46%, from $1.3 million for the period ending March 31, 2006. The decrease was attributable to a completion of the research and development program with Centocor.
47
Research and Development Expense.Research and development expense was $5.3 million for the three months ended March 31, 2007, consistent with the $5.1 million in expense for the three months ended March 31, 2006.
General and Administrative Expense.General and administrative expense was $1.1 million for the three months ended March 31, 2007, consistent with the $1.1 million in expense for the three months ended March 31, 2006.
Interest Income and Interest Expense.Interest income was $0.1 million for the three months ended March 31, 2007, compared to $0.3 million for the three months ended March 31, 2006. The decrease in interest income resulted from lower cash balances during 2007. Interest expense was $0.2 million for the three months ended March 31, 2007, compared to $0.1 million for the three months ended March 31, 2006. The increase in interest expense is attributable to interest on a convertible note executed in June 2006.
Year Ended December 31, 2006 Compared to Year Ended December 31, 2005
Revenue.Revenue was $5.0 million in 2006, an increase of $2.0 million, or 67%, from the $3.0 million in revenue in 2005. The primary source of revenue was our collaborations, and the increase was primarily related to the advancement of the research and development program with Centocor. Our collaboration with Centocor accounted for $4.1 million, or 82%, and $2.6 million, or 87%, of our revenue in 2006 and 2005, respectively.
Research and Development Expense.Research and development expense was $19.2 million in 2006, an increase of $4.8 million, or 33%, from the $14.4 million expense in 2005. The increase was primarily attributable increased efforts related to the development of Locteron and BLX-155, including manufacturing costs related to the production of drug substance for preclinical studies, clinical testing and process development, and preclinical study and clinical trial costs including the Phase 1 clinical trial of Locteron. Among the specific cost increases associated with these efforts included salaries related to increased headcount, materials associated with manufacturing, contract research organization and other third party costs associated with preclinical studies and clinical trials, additional manufacturing facility costs, and patent expenses.
General and Administrative Expense.General and administrative expense was $4.4 million in 2006, an increase of $0.3 million, or 6%, from the $4.1 million expense in 2005. The increase resulted principally from an increase in salaries and general operating expenses.
Interest Income and Interest Expense.Interest income was $0.9 million in 2006, compared to $0.5 million in 2005. The increase in interest income resulted from higher cash balances due to the proceeds of our Series BB preferred stock financing in August 2005. Interest expense was $1.0 million in 2005 and $0.2 million in 2006. The interest expense in 2005 included interest for warrants issued upon the conversion of the $8.0 million note into Series BB preferred stock.
Year Ended December 31, 2005 Compared to Year Ended December 31, 2004
Revenue.Revenue was $3.0 million in 2005, an increase of $2.3 million, or 329%, from the $0.7 million in revenue from 2004. The primary increase in revenue was related to a new collaboration with Centocor entered into at the end of 2004. Our collaboration with Centocor accounted for $2.6 million, or 87%, of our revenue in 2005 and $0.7 million, or 100%, of our revenue in 2004.
Research and Development Expense.Research and development expense was $14.4 million in 2005, an increase of $5.7 million, or 67%, from the $8.7 million expense in 2004. The increase resulted primarily from the advancement of the Locteron program, including the Phase 1 clinical study of BLX-883, Locteron’s active agent, and support related to the increased collaboration activity. The primary area of increase was salaries related to increased headcount as a result of hiring additional staff in manufacturing, research, operations, and support functions.
48
General and Administrative Expense.General and administrative expense was $4.1 million in 2005, an increase of $1.7 million, or 71%, from the $2.4 million in expense in 2004. This increase is primarily attributable to an increase in headcount in finance and business development.
Interest Income and Interest Expense.Interest income was $0.5 million in 2005, compared to less than $0.1 million in 2004. The increase in interest income resulted from a higher average cash balance due to our Series BB preferred stock financing, which was completed in August 2005. Interest expense was $1.0 million in 2005, compared to $0.1 million in 2004. The increase in interest expense resulted from an increase in loans payable related to the financing of additional manufacturing and research equipment and leasehold improvements added in 2005.
Liquidity and Capital Resources
Sources of Liquidity
As a result of our significant research and development expenditures and the absence of product sales revenue as our product candidates have not reached the approval stage, we have not been profitable and have generated operating losses since we were incorporated in 1997. Since inception through March 31, 2007, we have funded our operations principally with: approximately $72.3 million in gross proceeds from the sale of preferred stock and common stock and the exercise of options; approximately $9.8 million in revenue, consisting of research funding, milestone payments and recognition of up-front fees from collaborations related to our LEX System; $5.0 million in gross proceeds from a convertible note payable to a related party; and $3.7 million, net of repayments, under capital equipment leases. The gross proceeds we have received from stock issuances are as follows.
| | | | | | | |
Issue | | Year | | Number of
Shares | | Gross
Proceeds |
Series AA1 preferred stock | | 2003 | | 5,092,701 | | $ | 7,734,500 |
Series AA2 preferred stock | | 2003, 2004, 2005 | | 19,354,901 | | | 19,354,801 |
Series BB preferred stock | | 2005 | | 32,120,192 | | | 35,974,615 |
Prior mandatorily redeemable preferred stock series, converted to common stock in 2003 | | 1998 to 2000 | | 261,178 | | | 9,153,858 |
Sale of common stock and exercise of stock options | | 1998 to 2007 | | 233,080 | | | 51,106 |
| | | | | | | |
| | | | 62,695,388 | | $ | 72,268,880 |
| | | | | | | |
As of March 31, 2007, we had cash and cash equivalents of $8.7 million. In addition, in May 2007, we sold 21,186,441 shares of our Series CC preferred stock at $1.18 per share resulting in net proceeds to us of approximately $23.8 million. We hold our cash and investment balances primarily in interest-bearing obligations of the United States Treasury. We invest cash in excess of our immediate requirements with an objective of maintaining liquidity and capital preservation. Wherever possible, we seek to minimize the potential effects of concentration and degrees of risk. Also, we maintain cash balances with financial institutions, and at times deposits with the financial institution exceeded the amount insured by the Federal Deposit Insurance Company. We do not anticipate any losses with respect to such cash balances.
49
Cash Flows
The following table provides information regarding our cash flows and our capital expenditures for the years ended December 31, 2004, 2005 and 2006, and the three months ended March 31, 2006 and 2007:
| | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31, | | | Three Months Ended
March 31, | |
| | | 2004 | | | 2005 | | | 2006 | | | 2006 | | | 2007 | |
| | | | | | as restated | | | (in thousands) | |
Cash provided by (used in): | | | | | | | | | | | | | | | | | | | | |
Operating activities | | $ | (8,166 | ) | | $ | (7,487 | ) | | $ | (16,152 | ) | | $ | (4,515 | ) | | $ | (5,325 | ) |
Investing activities | | | 4,178 | | | | (7,643 | ) | | | (1,935 | ) | | | (714 | ) | | | (163 | ) |
Financing activities | | | 3,590 | | | | 42,230 | | | | 5,167 | | | | 796 | | | | (358 | ) |
Capital expenditures (included in investing activities above) | | | 2,022 | | | | 4,960 | | | | 1,935 | | | | 714 | | | | 163 | |
Net cash used in operations was $16.2 million, $7.5 million and $8.2 million for the years ended December 31, 2006, 2005 and 2004, respectively. The net losses for the year ended December 31, 2006 of $17.5 million were partially offset by non-cash charges for depreciation and amortization of $2.0 million. The net losses for the years ended December 31, 2005 and 2004 of $15.8 million and $10.4 million, respectively, were partially offset by non-cash charges for depreciation and amortization of $2.0 million and $1.3 million, respectively, and a reduction in cash used due to changes in operating assets and liabilities of $5.7 million and $0.8 million, respectively. Net cash used in operations was $5.3 million and $4.5 million for the three months ended March 31, 2007 and 2006, respectively. The net losses for the three months ended March 31, 2007 and 2006 of $5.7 million and $4.7 million, respectively, were partially offset by non-cash charges for depreciation and amortization of $0.5 million in each period, and were increased due to changes in operating assets and liabilities of $0.3 million for the three months ended March 31, 2006. The net loss, and net cash used in operations, for each of these periods was primarily due to our investment in research and development.
Net cash used in investing activities was $1.9 million for the year ended December 31, 2006 and was attributable to the purchase of property and equipment. Net cash used in investing activities was $7.6 million in the year ended December 31, 2005 and was attributable primarily to $5.0 million in purchases of property and equipment and $2.4 million related to the acquisition of LemnaGene. Net cash provided by investing activities was $4.2 million in the year ended December 31, 2004 and was attributable to $6.5 million of net sales of available-for-sale securities offset by $2.0 million in purchases of property and equipment and $0.3 million related to the purchase of the assets of Epicyte. Net cash used in investing activities was $0.2 million and $0.7 million for the three months ended March 31, 2007 and 2006, respectively and was attributable to the purchase of property and equipment in each of these periods.
Net cash provided by financing activities was $5.2 million for the year ended December 31, 2006 and consisted of $5.0 million in proceeds from a convertible note payable to a related party, and $0.2 million in proceeds from notes payable, net of repayments. Net cash provided by financing activities was $42.2 million for the year ended December 31, 2005 and consisted of $31.3 million in net proceeds from the sale of preferred stock, $8 million in proceeds from a convertible note payable to a related party, and $2.9 million in proceeds from notes payable, net of repayments. Net cash provided by financing activities was $3.6 million for the year ended December 31, 2004 and consisted of $3.0 million in net proceeds from the sale of preferred stock and $0.6 million in proceeds from notes payable, net of repayments. Net cash used in financing activities for the three months ended March 31, 2007 was $0.4 million, consisting primarily of payments of a note payable and capital lease obligations. Net cash provided by financing activities for the three months ended March 31, 2006 was $0.8 million, consisting of $0.8 million in proceeds from notes payable, net of repayments.
50
Funding Requirements
We expect to incur losses from operations for the foreseeable future. We expect to incur increasing research and development expenses, including expenses related to the planned advancement of Locteron in clinical development and the commencement of clinical testing of BLX-155 and BLX-301, and the related hiring of personnel and expansion of facilities. We also expect that our general and administrative expenses will also increase as we expand our finance and administrative staff, add infrastructure, and incur additional costs related to being a public company.
We believe that the net proceeds from this offering, together with our existing cash and cash equivalents will be sufficient to enable us to fund our operating expenses and capital expenditure requirements into 2009. We have based this estimate on assumptions that may prove to be wrong, and we could use our available capital resources sooner than we currently expect. Because of the numerous risks and uncertainties associated with the development and commercialization of our product candidates, we are unable to estimate the amounts of increased capital outlays and operating expenditures associated with our current and anticipated clinical trials.
Our future capital requirements will depend on many factors that are currently unknown to us, including:
| | • | | the timing of initiation, progress, results and costs of our planned Phase 2 clinical trials of Locteron for the treatment of chronic hepatitis C; |
| | • | | the timing of initiation, progress, results and costs of any Phase 3 clinical trials of Locteron that we may initiate based on the results of our Phase 2 trials, which will be affected by interactions with the FDA and similar foreign regulatory authorities and our continuing evaluation of third-party patents relevant to our commercialization plans and other factors; |
| | • | | the results of preclinical studies of our two preclinical product candidates, BLX-155 and BLX-301, and the timing of initiation, progress, results and costs of any clinical trials of these product candidates that we may initiate based on the preclinical results; |
| | • | | the costs of establishing commercial manufacturing facilities and of establishing sales and marketing functions; |
| | • | | the scope, progress, results, and costs of preclinical development, clinical trials, and regulatory review of any new product candidates for which we may initiate development; |
| | • | | the costs of preparing, filing, and prosecuting patent applications and maintaining, enforcing, and defending intellectual property-related claims; |
| | • | | our ability to establish strategic collaborations and licensing or other arrangements on terms favorable to us; |
| | • | | the costs to satisfy our obligations under potential future collaborations; and |
| | • | | the timing, receipt, and amount of sales or royalties, if any, from any approved product candidates. |
We do not anticipate that we will generate product revenue for at least the next several years. In the absence of additional funding, we expect our continuing operating losses to result in increases in our cash used in operations over the next several quarters and years. We will need to finance our future cash needs through public or private equity offerings, debt, financings or corporate collaboration and licensing arrangements. We do not currently have any commitments for future external funding. We may need to raise additional funds more quickly if one or more of our assumptions prove to be incorrect or if we choose to expand our product development efforts more rapidly than we presently anticipate, and we may decide to raise additional funds even before we need them if the conditions for raising capital are favorable. We may seek to sell additional equity or debt securities or obtain a bank credit facility. The sale of additional equity or debt securities, if convertible, could result in dilution to our stockholders. The incurrence of indebtedness would result in increased fixed obligations and could also result in covenants that would restrict our operations. Additional equity or debt financing, grants,
51
or corporate collaboration and licensing arrangements may not be available on acceptable terms, if at all. If adequate funds are not available, we may be required to delay, reduce the scope of or eliminate our research and development programs, reduce our planned commercialization efforts or obtain funds through arrangements with collaborators or others that may require us to relinquish rights to certain product candidates that we might otherwise seek to develop or commercialize independently.
Based on our current business plan, our requirement to fund capital expenditures for 2007, 2008 and 2009 is not expected to exceed $8 million per year. We have leased a building designed to house a second cGMP manufacturing facility to accommodate the production of product for the planned Phase 3 clinical trials and potential commercial launch of Locteron as well as manufacturing requirements for other product candidates advancing into later-stage clinical development. We expect that construction of the tenant improvements for this facility will commence before the end of 2007. Under the terms of the lease agreement for the facility, we expect that the lessor will fund most of the cost of the tenant improvements.
Contractual Obligations
The following table summarizes our significant contractual obligations and commercial commitments at March 31, 2007 and the effects such obligations are expected to have on our liquidity and cash flows in future periods.
| | | | | | | | | | | | | | | |
| | | Total | | Less Than
1 Year | | 1-3 Years | | 3-5 Years | | After 5 Years |
| | | (in thousands) |
Long-term debt obligations(1),(2) | | $ | 8,754 | | $ | 1,412 | | $ | 2,314 | | $ | 28 | | $ | 5,000 |
Operating lease obligations(1) | | | 8,196 | | | 1,166 | | | 1,917 | | | 1,567 | | | 3,546 |
Capital lease obligations(1) | | | 2 | | | 2 | | | — | | | — | | | — |
License obligations(3) | | | 1,325 | | | 175 | | | 350 | | | 250 | | | 550 |
| | | | | | | | | | | | | | | |
Total fixed contractual obligation | | $ | 18,277 | | $ | 2,755 | | $ | 4,581 | | $ | 1,845 | | $ | 9,096 |
| | | | | | | | | | | | | | | |
(1) | Excludes amounts, if any, that may be committed in the future to construct and equip additional facilities. |
(2) | Includes $5.0 million convertible note payable to a related party that was converted into Series CC preferred stock on May 18, 2007. |
(3) | Includes annual payments under license agreements. Excludes royalty and milestone payments to third parties as the amounts, timing and the likelihood of any such payments are not known. |
The above amounts exclude contracts that are entered into in the ordinary course of business which are not material in the aggregate in any period presented above.
In August 2005, we renewed a five-year, non-cancelable operating lease agreement for manufacturing, laboratory and office space in Pittsboro, North Carolina that expires in August 2010. In December 2005, we entered into a lease agreement for manufacturing space in Research Triangle Park, North Carolina that expires in December 2015.
In September 2005, we entered an agreement to borrow up to $5.0 million to fund purchases of equipment and other qualified expenditures. The loan commitment was drawn down in its entirety under three separate notes payable in 2005 and 2006. In connection with the term notes payable, warrants were granted to the financial institution to purchase a total 66,967 shares of Series BB preferred stock at $1.12 per share.
In June 2006, we borrowed $5.0 million under a convertible note payable to a related party. The note and accrued interest were converted into Series CC preferred stock on May 18, 2007.
We have no other lines of credit or other committed sources of capital. To the extent our capital resources are insufficient to meet future capital requirements, we will need to raise additional capital or incur indebtedness to fund our operations. We cannot assure you that additional debt or equity financing will be available on acceptable terms, if at all.
52
Effects of Inflation
Inflation generally affects us by increasing our cost of labor and clinical trial costs. We do not believe that inflation has had a material effect on our results of operations during 2004, 2005, 2006 or through March 31, 2007.
Off-Balance Sheet Arrangements
We do not have any off-balance sheet arrangements or relationships with unconsolidated entities or financial partnerships, such as entities often referred to as structured finance or special-purpose entities.
Recent Accounting Pronouncements
In July 2006, FASB issued FASB Interpretation No. 48,Accounting for Uncertainty in Income Taxes—an Interpretation of FASB Statement No. 109, or FIN 48, which clarifies the accounting for uncertainty in tax positions. This Interpretation prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. It also provides guidance on derecognition, clarification, interest and penalties, accounting in interim periods, disclosures and transitions. The provisions of FIN 48 are effective as of the beginning of our 2007 fiscal year, with the cumulative effect, if any, of the change in accounting principle recorded as an adjustment to opening retained earnings. We adopted FIN 48 effective January 1, 2007.
Upon adoption of FIN 48 and through March 31, 2007, we had no unrecognized tax benefits. As of the date of adoption, there were no tax positions for which it is reasonably possible that the total amounts of unrecognized tax benefits will significantly increase or decrease within twelve months from the date of adoption of FIN 48 or from March 31, 2007. As of March 31, 2007, we were subject to federal and state income tax in the United States. Since we are in a loss carryforward position, we are generally subject to U.S. federal and state income tax examinations by tax authorities for all years for which a loss carryforward is available. Thus, upon adoption of FIN 48, our open tax years extend back to 2003. In the event that we conclude that we are subject to interest or penalties arising from uncertain tax positions, we will record interest and penalties as a component of other income and expense. No amounts of interest or penalties were recognized in our consolidated financial statements upon adoption of FIN 48 or as of and for the three months ended March 31, 2007.
In September 2006, the FASB issued SFAS No. 157,Fair Value Measurement, or SFAS No. 157. SFAS No. 157 defines fair value, establishes a framework for measuring fair value and enhances disclosures about fair value measures required under other accounting pronouncements, but does not change existing guidance as to whether or not an instrument is carried at fair value. SFAS No. 157 is effective as of the beginning of our 2008 fiscal year. We are currently reviewing the provisions of SFAS No. 157 to determine the impact.
In June 2007, the EITF of the FASB reached consensus on Issue No. 07-3,Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities, or EITF Issue No. 07-3. EITF Issue No. 07-3 addresses the issue of when to record nonrefundable advance payments for goods or services that will be used or rendered for research and development activities as expenses. EITF Issue No. 07-3 has concluded that nonrefundable advance payments for future research and development activities should be deferred and recognized as an expense as the goods are delivered or the related services are performed. EITF Issue No. 07-3 is effective for fiscal years beginning after December 15, 2007. We have assessed the impact of EITF Issue No. 07-3 and expect no impact to our financial statements upon adoption of this guidance.
In February 2007, FASB issued Statement No. 159,The Fair Value Option for Financial Assets and Financial Liabilities, or FAS 159. FAS 159 provides companies with an option to report selected financial assets and liabilities at fair value. The objective of FAS 159 is to reduce both complexity in accounting for financial
53
instruments and the volatility in earnings caused by measuring related assets and liabilities differently. Generally accepted accounting principles have required different measurement attributes for different assets and liabilities that can create artificial volatility in earnings. The FASB has indicated it believes that FAS 159 helps to mitigate this type of accounting-induced volatility by enabling companies to report related assets and liabilities at fair value, which would likely reduce the need for companies to comply with detailed rules for hedge accounting.
FAS 159 also establishes presentation and disclosure requirements designed to facilitate comparisons between companies that choose different measurement attributes for similar types of assets and liabilities. For example, FAS 159 requires a company to provide additional information that will help investors and other users of financial statements to more easily understand the effect of the company’s choice to use fair value on its earnings. It also requires entities to display the fair value of those assets and liabilities for which the company has chosen to use fair value on the face of the balance sheet. FAS 159 does not eliminate disclosure requirements included in other accounting standards, including requirements for disclosures about fair value measurements included in FASB Statement No. 157,Fair Value Measurements, or FAS 157, and FASB Statement No. 107,Disclosures about Fair Value of Financial Instruments. FAS 159 is effective as of the beginning of a company’s first fiscal year beginning after November 15, 2007. We are currently evaluating the effects of FAS 159 on its consolidated financial statements.
Quantitative and Qualitative Disclosures about Market Risk
The primary objective of our investment activities is to preserve our capital to fund operations. We also seek to maximize income from our investments without assuming significant risk. To achieve our objectives, we maintain a portfolio of cash equivalents and investments in a variety of securities of high credit quality. As of March 31, 2007, we had cash and cash equivalents of $8.7 million. A portion of our investments may be subject to interest rate risk and could fall in value if market interest rates increase. However, because our investments are short-term in duration, we believe that our exposure to interest rate risk is not significant and a 1% movement in market interest rates would not have a significant impact on the total value of our portfolio. We actively monitor changes in interest rates.
54
BUSINESS
Overview
We are a clinical-stage biopharmaceutical company that uses our patented LEX System to develop hard-to-make therapeutic proteins and optimized monoclonal antibodies. The LEX System is a novel technology that genetically transforms the aquatic plantLemna to enable the production of biologic product candidates. Our proprietary product candidates are designed to provide superior efficacy/tolerability profiles and to address large, proven pharmaceutical markets.
Our lead product candidate, Locteron, is in Phase 2 clinical trials and is the only controlled-release interferon alfa known to be currently in clinical development for the treatment of chronic hepatitis C. Locteron is designed to improve upon the current standard of patient care, pegylated interferon alfa administered weekly in combination with the antiviral drug ribavirin, which is associated with significant side effects. In a 32-patient Phase 2a clinical trial of Locteron administered once every two weeks in combination with ribavirin, an early virologic response, or EVR, was achieved by 100% (8/8) of patients treated with the 480 microgram, or 480 µg, dose of Locteron. Achievement of EVR, which is a specified reduction in viral load, has been broadly established to be a prerequisite for long-term response in hepatitis C patients. In addition, reported side effects were fewer and less severe than previously reported with other interferon products currently marketed or in development.
We have also developed two other product candidates that capitalize on the benefits of the LEX System, which we are advancing toward clinical trials: BLX-155, a direct-acting thrombolytic designed to dissolve blood clots in patients; and BLX-301, an anti-CD20 antibody we are optimizing for the treatment of non-Hodgkin’s B-cell lymphoma and other diseases. We believe that all of our product candidates have reduced clinical and regulatory risk profiles because they are based on well understood proteins with proven mechanisms of action or clinical validation and well-established regulatory pathways.
We have focused our pipeline of product candidates on biologics that we believe can be enhanced by the LEX System. The LEX System facilitates the development of proteins whose complexity and other properties limit their commercial viability in traditional production systems. It also enables us to produce monoclonal antibodies that can be optimized with the objective of improving potency and efficacy. Additional benefits provided by the LEX System over traditional production systems include the potential for reduced capital investment, shorter lead times to establish manufacturing facilities, and the ability to produce certain proteins without conflicting with process patents covering production in traditional systems. By capitalizing on the significant benefits of the LEX System over traditional production systems, we are developing the following pipeline of product candidates:
| | • | | Locteron. The World Health Organization estimates that as many as 130 million people worldwide are chronically infected with hepatitis C and that an additional three to four million people are infected each year. It is estimated that annual worldwide sales of interferon for the treatment of hepatitis C will increase from $2.8 billion in 2005 to more than $5 billion by 2014. Under the current standard of care, interferon alfa is injected weekly for up to 48 weeks and is associated with significant side effects, including fatigue, flu-like symptoms, depression and anemia, resulting in high rates of patient non-compliance and discontinuation of therapy. Future treatment is expected to continue to be comprised of interferon in combination with one or more antiviral agents, which could heighten patient tolerability concerns, because the antiviral agents currently under development have been reported to cause additional side effects. |
We are developing Locteron to improve patient care by providing a more favorable side-effect profile and more convenient dosing. Locteron incorporates PolyActive, a patented controlled-release technology which provides a gradual release of interferon alfa to patients without the high initial blood levels associated with the significant side effects experienced by patients today. Locteron is also formulated to allow dosing once every two weeks, a substantial improvement in patient convenience compared to currently marketed pegylated interferon alfa products that require dosing every week.
55
We are currently conducting a 12-week, Phase 2a clinical trial in Europe to evaluate four different doses of Locteron administered once every two weeks in combination with ribavirin in treatment-naïve hepatitis C patients. Results for the first three dosing cohorts suggested that 12 weeks of treatment with the combination of Locteron and ribavirin was effective in reducing hepatitis C virus levels. Patients receiving Locteron also experienced fewer and less severe side effects than those reported for pegylated interferons and Albuferon, an albumin-fused interferon product currently under development. Accordingly, the objective of the trial has been met as we have identified a range of doses with an appropriate combination of viral reduction and favorable tolerability to proceed to the next step of clinical development. We currently expect to commence a Phase 2b clinical trial of Locteron in the first half of 2008 and to enter Phase 3 trials in 2009. We are developing Locteron under an exclusive collaboration agreement with OctoPlus, the developer of PolyActive.
| | • | | BLX-155. BLX-155 is a direct-acting thrombolytic that we are developing to dissolve blood clots in patients with certain diseases or conditions such as acute peripheral arterial disease, deep vein thrombosis and hemodialysis graft thrombosis. Thrombosis, the formation or presence of a blood clot, is a leading cause of morbidity and mortality and, according to the National Heart Lung and Blood Institute, is estimated to affect over 50 million people in the developed world. BLX-155 is the recombinant form of plasmin, a naturally occurring human enzyme responsible for dissolving fibrin, a key component of blood clots. Plasmin has been studied for a number of decades as a drug candidate for dissolving blood clots, but recombinant full-length human plasmin has not been successfully produced at commercially viable levels in traditional protein production systems. |
In July 2007, our researchers presented results demonstrating the ability of the LEX System to produce full-length recombinant plasmin at commercially viable levels. This achievement provides us with a unique opportunity to pursue development and commercialization of a therapeutic protein with a known mechanism of action for which prior development historically has been prevented due to production challenges. As a direct-acting thrombolytic, we believe BLX-155 has the potential to provide efficacy and safety advantages over plasminogen activators, including tissue-type plasminogen activators, or t-PAs, the current standard of care which dissolve blood clots indirectly. BLX-155 is currently in preclinical development, and we expect to commence a Phase 1 clinical trial in hemodialysis graft thrombosis patients in the first half of 2008.
| | • | | BLX-301. BLX-301 is an anti-CD20 antibody that we are optimizing for the treatment of non-Hodgkin’s B-cell lymphoma and other diseases. BLX-301 is based on the capability of the LEX System to optimize the sugar chain component, or glycosylation structure, of monoclonal antibodies to increase their potency and efficacy. In May 2007, we announced preclinical results highlighting the ability of the LEX System to produce an anti-CD20 antibody with enhanced cytotoxicity and potentially lower side effects than Rituxan, the top selling treatment for non-Hodgkin’s B-cell lymphoma, which is also used to treat rheumatoid arthritis. BLX-301 is currently in preclinical development, and we expect to commence a Phase 1 clinical trial in non-Hodgkin’s B-cell lymphoma patients in 2009. |
Our Strategy
Our objective is to become a leading biopharmaceutical company by developing and commercializing a diverse portfolio of best-in-class biologic products for large proven markets. The key elements of our strategy to achieve this objective are:
| | • | | Leverage our extensive biopharmaceutical experience and the power of the LEX System to source and develop high-value product candidates. We are led by Jan Turek, our President and Chief Executive Officer, who has worked in biotechnology for over 14 years and for five years headed the $1.2 billion Biological Products business unit of Bayer Corporation. Mr. Turek has built a senior management team with extensive experience in the biopharmaceutical industry. We will continue to focus on the commercialization of proteins and antibodies where we can leverage the strengths of the LEX System with the experience of our senior management to facilitate the development of product |
56
| | candidates with best-in-class efficacy/tolerability profiles. A key criterion that we have and will continue to use in developing our pipeline of product candidates is the identification of proteins and antibodies that have proven mechanisms of actions that we can build upon to reduce clinical risk and provide a straightforward regulatory pathway. |
| | • | | Continue to develop our current product candidates. We have commenced development of three product candidates that meet the strategic criteria outlined above and which we believe represent substantial commercial opportunities. We have staged these programs to provide for concurrent development of each opportunity, with Locteron currently in Phase 2 trials, BLX-155 expected to enter clinical development in early 2008, and BLX-301 expected to enter clinical development in 2009. Each of these products derive key benefits from our LEX System, and we expect to allocate a substantial portion of our research and development resources, and are currently implementing an expansion of our manufacturing capabilities, to allow us to aggressively pursue in parallel the clinical development of these product candidates. |
| | • | | Position Locteron as the interferon of choice. We plan to use our positive Phase 2a clinical trial results as a basis for more advanced clinical testing of Locteron in Phase 2b and Phase 3 trials. Our core strategy will be to pursue regulatory approval of Locteron in combination with ribavirin for the treatment of hepatitis C. We also plan to position Locteron to benefit from the expected evolution in the standard of care by initiating clinical trials to support the use of Locteron with emerging antiviral therapies currently under development. Our goal is to provide clinical support for our hypotheses that Locteron provides key efficacy/tolerability advantages over other interferons and should be considered as the interferon of choice for any treatment combination used in the future in the treatment of hepatitis C. |
| | • | | Position BLX-155 to address multiple large-market indications. As a recombinant form of plasmin, the body’s native protein for dissolving clots, BLX-155 offers the potential opportunity to address medical needs in a series of diseases and medical conditions involving blood clots. We have two clinical trials planned for 2008, a Phase 1 proof of concept trial in hemodialysis graft thrombosis and a Phase 2a clinical trial in deep vein thrombosis, each of which represent medical needs that we may elect to target in advanced clinical development. These clinical trials will have the additional benefits of testing a broad range of doses of BLX-155 that can potentially be used to support the development of the product candidate in other indications with high-value markets such as acute peripheral arterial occlusive disease. |
| | • | | Establish a commercial infrastructure upon regulatory approval. We plan to establish a commercial infrastructure and targeted specialty sales force to market certain of our product candidates in North America as we progress toward regulatory approval. We believe that the collective experience of our management team in the commercial launch and sales of biologic products, coupled with the specialty nature of the markets currently targeted by our pipeline of product candidates, will allow us to directly commercialize our product candidates in instances where we believe doing so will maximize our potential return. |
| | • | | Partner selectively. The benefits provided by our proprietary LEX System provide us with opportunities to form strategic alliances with leading pharmaceutical and biotechnology companies. We currently have early stage collaborations with companies that include Centocor, Genmab A/S, Medarex, and Merial Limited. We have entered into these collaborations primarily to obtain non-dilutive revenue, expand our core competencies and establish our platform infrastructure. We will continue to evaluate the initiation of selected collaborations, or the advancement of our existing collaborations, with companies where we can employ the LEX System to enhance the development or clinical attributes of their proteins and antibodies in exchange for participating in the commercial upside of our partners’ product candidates. |
The LEX System
Background and Market Opportunity
The market for therapeutic proteins, including recombinant human proteins and monoclonal antibodies, is projected to grow at twice the rate of the market for small molecule drugs, or synthetic pharmaceutical
57
compounds, and is expected to increase from $51 billion in 2005 to $87 billion in 2010, according to Kalorama Information. Therapeutic proteins have traditionally been produced primarily in microbial systems, such asE. coli and yeast systems, or mammalian systems, such as Chinese hamster ovary, or CHO, systems. These traditional production systems are employed to produce a wide range of therapeutic proteins and protein candidates, although each system has a number of inherent drawbacks, including the need for significant capital investments, long lead times to establish manufacturing facilities, and high costs of production. In some instances, the commercialization of proteins has been prevented because production at commercially viable levels is not achievable in traditional systems. In other instances, use of traditional systems to produce certain proteins is blocked by existing process patents.
In addition, recent research has suggested that the therapeutic potential of monoclonal antibodies can be enhanced by modifying their sugar-chain components, or glycosylation structures, to improve clinical attributes. Monoclonal antibodies are currently being used and developed primarily to treat cancers, autoimmune diseases, infectious diseases and inflammatory diseases, and according to Kalorama Information, are the fastest growing class of products in the pharmaceutical industry. The effectiveness of monoclonal antibodies, particularly those targeting oncology indications, is greatly impacted by the glycosylation structure of one section of the antibody, known as the Fc region. Research has shown that if the Fc region of the antibody is modified to create an optimized glycosylation structure, the ability of the antibody to bind to immune cells, and to stimulate those immune cells to destroy diseased cells, can be greatly enhanced. There are a limited number of technologies that have the potential to modify the glycosylation structures of antibodies to improve Fc function. Each of these technologies is subject to the limitations of existing production systems, such as the expression of a heterogeneous mix of glycosylation structures or a historic lack of utility in the production of monoclonal antibodies.
The LEX System Solution
We use our patented LEX System to develop hard-to-make therapeutic proteins and optimized monoclonal antibodies. The LEX System is a novel technology that genetically transforms the aquatic plantLemna to enable the production, or expression, of biologic product candidates.Lemna doubles its body mass every 36 hours and has a very high protein content, making it ideal for the expression of recombinant proteins. We have applied advanced genetic engineering toLemna to create our recombinant protein production system, theLemna Expression System, or LEX System, that addresses many of the drawbacks of traditional production systems. Our proprietary technology for the genetic manipulation ofLemna enables the gene sequence for a protein of interest to be transferred to the DNA ofLemna. After transformation, the clonal nature ofLemna,meaning its ability to reproduce asexually, helps ensure that the plant line will produce the protein candidate in a repeatable manner regardless of production scale. The LEX System has been incorporated into our contained and controlled manufacturing facilities to accommodate cGMP production.
The LEX System has demonstrated the ability to produce cytokines and other proteins traditionally produced inE. coli systems, and monoclonal antibodies produced today in mammalian-cell systems. The strengths of the LEX System include:
| | • | | Production of Complex Proteins. We have not encountered a class of proteins that cannot be expressed by the LEX System. To date, as part of our research and development activities, we have produced more than 35 different proteins using the LEX System, including complex proteins that are challenging to produce in traditional production systems. For instance, the protein plasmin has been studied for several decades as a drug candidate for resolving blood clots, but production of full-length human plasmin at commercially viable levels has not been reported with any traditional recombinant production system. Our product candidate BLX-155, a recombinant full-length plasmin, can be produced at commercially viable levels using the LEX System. |
| | • | | Complex Protein Folding.Lemna is categorized as a higher plant, meaning it has a vascular system and is considered to be higher on the evolutionary scale.Lemna is able to correctly produce proteins requiring folding or other complex processing. Interferon alfa historically has been produced inE. coli |
58
| | expression systems.E. coli systems are unable to express interferon alfa that is correctly folded, therefore requiring a series of unfolding and refolding steps during purification that results in substantial yield loss and contributes to the high cost of production in these systems. In contrast, the LEX System produces correctly folded interferon alfa with no refolding steps required. This capability of the LEX System is key to the efficient production of our lead drug candidate Locteron. |
| | • | | Optimization of Monoclonal Antibodies. The LEX System has demonstrated the capability to express monoclonal antibodies in which the glycosylation structure has been optimized to enhance the efficacy and potency of the antibodies. This capability has enabled us to undertake the development of BLX-301, a humanized and optimized anti-CD20 antibody for the treatment of non-Hodgkin’s B-cell lymphoma and other diseases. |
| | • | | Access to Product Candidates. TheLemna-based protein expression system is a novel technology that we believe will provide us with multiple opportunities to produce proteins without conflicting with patent claims relating to the production of certain proteins in traditional production systems. For example, the LEX System allows us to produce the interferon alfa used in Locteron without conflicting with the patent claims covering the production of interferon alfa in a bacteria-based system. |
| | • | | Reduced Safety Risk.The LEX System contains no human or animal components, which can carry a risk of viral or other infectious agent contamination. The production of proteins in traditional production systems, whether bacterial or mammalian systems, rely upon human or animal components that risk contaminating the product with viruses and other infectious agents such as prions (the infectious agent of bovine spongiform encephalopathy, or mad cow disease). Protein purification steps have been developed to maximize product safety in these other systems, however these steps are quite costly and do not eliminate all risks. In the LEX System,Lemna are grown in non-organic media, and the U.S. Food and Drug Administration, or FDA, has confirmed that expensive viral inactivation and removal processes inherent with traditional systems are not required. |
| | • | | Reduced Capital Cost and Facility Timelines. We believe that the simplicity and other characteristics of the LEX System will result in capital investment requirements and timelines for implementation of manufacturing facilities that are substantially less than those associated with traditional systems, based on our experience with cGMP production in our existing facility and third-party engineering evaluations. Among the factors contributing to these reductions are the expected efficiency of the LEX System as well as the robustness of theLemna platform, which eliminates the need for the initial phase of protein production to be performed in a costly, sterile environment. |
Our Product Candidates
Locteron
Locteron is the only controlled-release interferon alfa known to be currently in clinical development for the treatment of chronic hepatitis C. We are developing Locteron with the objective of improving patient care through a more favorable side-effect profile and more convenient dosing. According to Kalorama Information, total worldwide annual sales of pegylated interferons for the treatment of hepatitis C were $2.8 billion in 2005. Decision Resources estimates that worldwide sales of interferon products for the treatment of hepatitis C will exceed $5 billion by 2014. Interferon alfa serves as the foundation of current combination therapy for hepatitis C patients, and all major hepatitis C therapeutic candidates currently in clinical trials are being studied in combination with interferon alfa. The currently marketed pegylated interferon products, Pegasys and PEG-Intron, and Albuferon, an interferon product under development, are associated with significant side effects, including fatigue, flu-like symptoms, depression and anemia.
Locteron incorporates PolyActive, a controlled-release technology patented by our co-development partner OctoPlus, which provides a gradual release of interferon alfa to patients without the high initial blood levels associated with the significant side effects experienced by patients today. This controlled-release mechanism is designed to reduce the frequency, duration and severity of side effects, including flu-like symptoms, commonly
59
experienced by patients treated with currently marketed pegylated interferons and with Albuferon. Locteron is also configured to allow dosing once every two weeks, a substantial improvement in patient convenience compared to currently marketed pegylated interferon alfa products that require dosing every week. In a 32-patient Phase 2a clinical trial of Locteron administered once every two weeks in combination with ribavirin, an EVR was achieved by 100% (8/8) of hepatitis C patients treated with the 480 µg dose of Locteron and by 88% (7/8) of patients treated with the 320 µg dose. In addition, reported side effects were fewer and less severe than those previously reported in clinical trials for other interferon products.
Chronic Hepatitis C Virus Infection and Current Treatment
Hepatitis C is a viral infection that causes chronic inflammation in the liver. Disease progression occurs over a period of 20 to 30 years, during which patients generally have no symptoms. However, if left untreated, chronic hepatitis C can lead to permanent liver damage, resulting in the development of liver cancer, liver failure or death. The World Health Organization estimates that as many as 130 million people worldwide are chronically infected with hepatitis C and that an additional three to four million are infected each year. Decision Resources estimates that approximately 11 million people in the United States, Japan and the five major European markets (the United Kingdom, Spain, France, Italy and Germany) are chronically infected with hepatitis C. Hepatitis C can be classified into one of six major sub-populations, or genotypes. Genotype-1 is the most common single variant of the hepatitis C virus, representing 70% of the estimated four million infected patients in the United States, and is the most difficult of the genotypes to treat.
The current standard treatment for hepatitis C patients is the combination of pegylated interferon alfa administered subcutaneously once per week and the antiviral drug ribavirin administered orally daily, with patients treated for up to 48 weeks. This treatment combination is associated with significant side effects, including fatigue, flu-like symptoms, depression and anemia. According to published studies, approximately 50% of hepatitis C patients fail to attain a sustained response to treatment. Among the reasons for treatment failure in many patients is the inability to tolerate the side effects of the treatment, resulting in termination of therapy or reduction of the dose level administered to a patient. Recent studies indicate that adverse events associated with interferon therapy result in up to 20% of hepatitis C patients dropping out of treatment when undergoing 48 weeks of therapy.
Interferon alfa is an immune-stimulating agent naturally produced by the body in response to a viral infection. In hepatitis C, the administration of interferon alfa-based products is believed to elicit multiple mechanisms, including stimulating natural killer cells to enhance their killing activity against the virally infected cells and inhibiting viral replication. The two primary interferon alfa products marketed today in the United States and Europe for the treatment of hepatitis C are PEG-Intron marketed by Schering-Plough Corporation and Pegasys marketed by Hoffmann-La Roche. These products are “pegylated” by attaching polyethylene glycol, a large inert molecule, to interferon alfa to slow its clearance from the body, thereby increasing its half-life and facilitating administration to patients once per week. Albuferon, a product candidate under development by Human Genome Sciences, similarly fuses recombinant human serum albumin to interferon to increase its half life.
The pegylation and albumin-fusion technologies cause these interferon products to circulate longer in the body by slowing their clearance. However, unlike a controlled-release mechanism, under both of these technologies the full complement of interferon administered to the patient is available immediately after injection, resulting in high initial blood levels of interferon that are believed to be responsible for many of the side effects suffered by the patients. These side effects include flu-like symptoms such as fever, headache, muscle aches and joint aches. As many patients with chronic hepatitis C infection experience no hepatitis C-related symptoms, they may be even more likely to discontinue therapy when confronted with the severe side effects of the current standard of care or may decline to even initiate treatment. Less than 4% of the currently diagnosed hepatitis C-infected population is treated each year.
60
A wide range of new therapies is currently under development for the treatment of hepatitis C, all of which are being studied in combination with interferon and ribavirin. Included among these are protease inhibitors such as Vertex Pharmaceuticals, Inc.’s VX-950, designed to inhibit the replication of the virus. A second class of drugs under development consists of polymerase inhibitors, which are also designed to inhibit viral replication. Each of these novel antivirals has demonstrated a tendency to provoke the development of rapid resistance in some patients when administered as a monotherapy. The likelihood of resistance appears to be greatly reduced when these agents are used in combination with interferon. The addition of interferon has also resulted in stronger antiviral effects in comparison to the use of these agents by themselves. Regardless of the success of these candidates or the exact composition of future treatment combinations, we believe that interferon alfa will continue to form the foundation for hepatitis C treatment.
As a consequence, all major programs currently advancing in clinical trials are being studied in combination with interferon alfa and ribavirin. We believe that the advent of these new agents will serve to expand the overall market for the treatment of hepatitis C, as new treatment combinations will provide an opportunity to treat the growing population of non-responders, patients who failed to achieve a sustained response when undergoing prior treatment. However, because each of these new agents has been shown in clinical trials to cause additional substantial side effects, we believe that the eventual adoption of these agents will create a greater need for an improved interferon alfa product with reduced side effects.
The Benefits of Locteron
We believe that Locteron is the only controlled-release interferon alfa currently in clinical development for the treatment of hepatitis C. Locteron is formulated through the combination of BLX-883, our proprietary interferon alfa produced in the LEX System, and the PolyActive microspheres developed by our co-development partner OctoPlus. We have an exclusive co-development agreement to use the PolyActive controlled-release technology in combination with interferon alfa. Locteron is designed to be dosed once every two weeks in patients and to gradually release active interferon alfa over a 14-day period.
PolyActive is comprised of biodegradable microspheres consisting of polybutylene terephthalate and polyethylene glycol. The PolyActive polymer has been used in medical devices for orthopedic and wound repair applications for more than a decade. In Locteron, interferon alfa molecules are suspended throughout the microspheres and after injection in patients are released by diffusion in a continuous manner as the microspheres degrade. We and OctoPlus selected a once-every-two-week dosing regimen for Locteron after an assessment of clinical practice, although other durations of release could be developed as follow-on products. Locteron is formulated so that it can be administered to patients using the same or similar gauge needle and syringe volume as pegylated interferon alfa.
By releasing interferon alfa into circulation in a gradual manner, Locteron avoids the high initial blood levels of the active interferon that characterize pegylated interferon alfa and Albuferon. Locteron is designed to provide at least the same therapeutic benefit to hepatitis C patients as the aforementioned products with fewer and less severe side effects. We believe that an improved side-effect profile has the potential to lead to enhanced patient compliance, reduce patient drop outs, and provide the opportunity for improved therapeutic outcomes. We also believe that a more tolerable side-effect profile could help encourage patients who currently delay or refuse treatment to commence treatment.
Potential advantages of Locteron compared to pegylated interferon alfas and Albuferon are as follows:
| | • | | More Convenient Dosing Schedule.The controlled-release formulation of Locteron has been engineered to allow a once-every-two-week dosing regimen, reducing the patient burden associated with the currently marketed pegylated products that require dosing once per week. |
| | • | | Reduction in Frequency, Duration and Severity of Side Effects.By releasing gradually over a two-week period, Locteron avoids the initial high peak blood concentrations of interferon that are |
61
| | associated with administration of pegylated interferon alfa and Albuferon. We believe that avoiding these initial high peak plasma levels has the potential to reduce the frequency, duration and severity of certain side effects. |
| | • | | Locteron Releases Native Interferon.Unlike currently marketed pegylated interferons and Albuferon, Locteron’s interferon is not chemically modified by conjugation with polyethylene glycol or by fusion to human albumin. Accordingly, the interferon released by Locteron most closely resembles native interferon alfa produced by the human body. As a result, we believe that Locteron will achieve a higher volume of distribution than the pegylated and albumin-fused interferons, thereby reaching more compartments within the body where the hepatitis C virus may reside. We also expect that the Locteron will have a more stable dose profile compared to other interferon products in terms of the trough, or lowest interferon levels, between doses. |
The number of potential competitors of Locteron in the United States is limited by patents encompassing the production of interferon alfa in bacteria-based systems, the method traditionally used for the production of interferon products. In addition to our patent activities regarding the production of interferon alpha in the LEX System, patents encompassing the use of PolyActive for the delivery of therapeutic proteins are exclusively held by our co-development partner OctoPlus. The use of PolyActive for the administration of any interferon alfa agent for the treatment of hepatitis C is subject to an exclusive co-development agreement between us and OctoPlus.
Clinical Trial Summary
The following table summarizes the status of our completed and currently planned clinical trials for Locteron:
| | | | | | |
| Phase/Name | | Trial Objective | | Protocol | | Status |
1 | | Evaluate safety of BLX-883, the active agent in Locteron, and compare safety and bio-activity to Intron-A | | Single-dose, controlled trial in 24 healthy volunteers of BLX-883 versus Intron-A | | Complete |
| | | | |
1 | | Evaluate safety of Locteron and compare safety and bio-activity to PEG-Intron | | Single-dose, controlled trial in 27 healthy volunteers of Locteron versus PEG-Intron | | Complete |
| | | | |
2a/ SELECT-1 | | Evaluate safety, tolerability and viral response of Locteron in treatment-naïve hepatitis C patients | | 12-week, 32-patient trial of four doses of Locteron in combination with ribavirin in the treatment of genotype-1, treatment-naïve hepatitis C patients. | | Ongoing; three of four dose cohorts completed |
| | | | |
2b/ SELECT-2 | | Evaluate viral response, safety and tolerability of Locteron in treatment-naïve hepatitis C patients Results after 12 weeks of treatment to be used as basis for selecting dose for Phase 3 clinical trials. | | Controlled, 72-week, 100 patient trial of three doses of Locteron in combination with ribavirin in the treatment of genotype-1, treatment-naïve hepatitis C patients compared with control arm of pegylated interferon alfa in combination with ribavirin. | | Plan to initiate first half of 2008 |
Phase 1 Clinical Trials
In 2005, we conducted a controlled Phase 1 clinical trial of 24 healthy volunteers in Europe under a clinical trial application, or CTA, as required by the European Union’s Clinical Trial Directive, and an investigational new drug application, or IND, submitted to the FDA. In this trial, BLX-883, the interferon alfa produced in our
62
LEX System and the active agent in Locteron, was compared to Intron A, Schering-Plough Corporation’s interferon alfa product. The objective of the trial was to evaluate the safety of BLX-883, the first drug produced in the LEX System, and to evaluate the biologic activity of BLX-883 to determine if it would serve as the appropriate backbone of a controlled-release interferon product. The results of the trial suggested that BLX-883 was equivalent to Intron A in terms of biologic activity, pharmacokinetics, and tolerability and supported the use of BLX-883 as the active agent in a controlled-release interferon.
In April 2006, we announced the results of a randomized, double-blind, placebo-controlled Phase 1 clinical trial of Locteron in 27 healthy volunteers conducted in Europe under Dutch regulations at the annual meeting of the European Association for the Study of the Liver. Four of the participants were included in each of three different dose cohorts of Locteron (20 µg, 80 µg, or 320 µg), six participants were included in the PEG-Intron control cohort, while the remainder were included in cohorts that were administered either a placebo or the PolyActive vehicle. The Phase 1 trial supported the safety and tolerability of Locteron given as a single dose at 20 µg, 80 µg, or 320 µg, while pharmacokinetic and pharmacodynamic results supported dosing every two weeks. The pharmacokinetic results also indicated that the administration of Locteron resulted in a gradual release of interferon without the high initial blood concentration levels.
Flu-like symptoms among the groups receiving Locteron in the Phase 1 trial were reported to be fewer, less severe and of shorter duration than in the subjects receiving PEG-Intron at 80 µg, the approved monotherapy dose of PEG-Intron which served as a control. Only one of the 12 participants, or 8%, administered Locteron experienced a fever, compared to five of the six participants, or 83%, administered PEG-Intron. Additionally, the Phase 1 trial measured biomarkers such as 2’, 5’ oligoadenylate synthetase, an enzyme commonly associated with the biological effects of interferon alfa. The administration of Locteron resulted in biomarker levels for the 80 µg and 320 µg dose cohorts that were equal to or greater than those measured in the PEG-Intron arm of the trial and that were sustained for a two-week period, supporting the progression of Locteron to Phase 2 trials.
SELECT-1 Phase 2a Clinical Trial
In January 2007, clinical researchers commenced dosing hepatitis C patients in our Phase 2a randomized clinical trial of Locteron, known as SELECT-1 (Safety andEfficacy ofLocteron:EuropeanClinicalTrial-1) under a CTA. This multi-center, open-label trial was designed to evaluate a range of up to four doses of Locteron administered once every two weeks in combination with ribavirin in 32 treatment-naïve hepatitis C patients in Europe with the geneotype-1 variant of the virus. The SELECT-1 clinical trial protocol calls for patients to be treated for 12 weeks with the Locteron/ribavirin combination, and the repeat-dose study will assess safety, tolerability and viral response. Results from the SELECT-1 trial will be used to choose the doses of Locteron that will be studied in the later phase clinical trials. Under the protocol for the SELECT-1 trial, dosing was commenced in January 2007 for the first three eight-patient cohorts of the trial, at doses of 160 µg, 320 µg and 480 µg, respectively. Dosing of the eight-patient 640 µg cohort was to be triggered based on an assessment of safety and tolerability through four weeks of treatment for the first three cohorts. Based on a favorable safety review of the results for the first three cohorts, the dosing of patients in the 640 µg cohort commenced in May 2007.
A full 12 weeks of treatment have been completed for the 160 µg, 320 µg and 480 µg cohorts of the trial. Results for the first three cohorts of the Phase 2a trial indicated that the combination of Locteron and ribavirin was effective in reducing the hepatitis C virus through 12 weeks of treatment. Importantly, patients receiving Locteron experienced fewer and less severe side effects than previously reported for the currently marketed pegylated interferons and with Albuferon. The results of our Phase 2a clinical trial are not necessarily predictive of results that may be observed in larger and/or longer future clinical trials. In addition, although we believe the comparison of our clinical results of Locteron with previously reported results for marketed drugs or other drug candidates currently under development provides a reasonable basis for comparison, a definitive comparison of our drug candidate with such currently marketed drugs or other drugs in clinical development requires a controlled, head-to-head clinical trial.
63
SELECT-1: Viral Response Results
The results regarding viral response were as follows at the conclusion of the 12 weeks of treatment for the 160 µg, 320 µg, and 480 µg cohorts:
| | • | | A dose response was observed in the trial, as illustrated in Figure 1 below, with patients treated with the 320 µg and 480 µg doses of Locteron achieving a greater reduction in hepatitis C virus than the 160 µg dose at all measurement times. |
| | • | | The percentage of patients who achieved early virologic response, or EVR, defined as at least a two-log reduction in hepatitis C virus after 12 weeks of treatment, was 87.5% and 100% in the 320 µg and 480 µg dose cohorts, respectively, compared to 37.5% in the 160 µg dose cohort. |
| | • | | Average viral reduction after 12 weeks of treatment for the 320 µg and 480 µg doses was 4.48 and 4.22 logs, respectively, compared to 1.83 logs in the lowest dose of 160 µg. |
| | • | | After 12 weeks of treatment, 62.5% (5/8) and 62.5% (5/8) of the patients had undetectable levels of hepatitis C virus, measured by plasma RNA < 28 IU/ml, in the 320 µg and 480 µg dose cohorts, respectively, compared to 12.5% (1/8) in the lowest dose of 160 µg. |
Figure 1—Response to Treatment with Locteron and Ribavirin
by Dose in SELECT-1 Phase 2a Trial
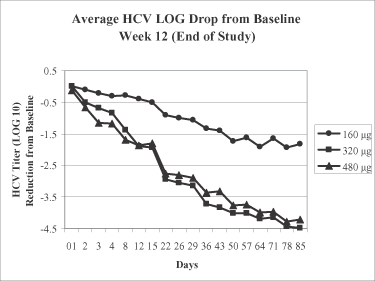
The SELECT-1 12-week EVR results suggest a dose response, and the EVR results for the two Locteron doses that were most effective, the 320 µg and 480 µg doses, compare favorably with the EVR results reported for the currently marketed pegylated interferon alfa products and for Albuferon as illustrated in Table 1 below. Achievement of EVR has been broadly established to be a prerequisite for long-term response.
64
Table 1—Locteron SELECT-1 Phase 2a Trial 12-Week EVR Results
and Published 12-Week EVR Results for Comparator Products
| | | | | | | | | | | | | | | | | | | | | |
| | | Locteron SELECT-1
Phase 2a Trial(2) | | | | | | Albuferon(5) | |
Results After 12 Weeks of Treatment | | 160 µg | | | 320 µg | | | 480 µg | | | PEG-Intron(3) | | | Pegasys(4) | | | 900 µg | | | 1200 µg | |
| % of Patients with Early Virologic Response (EVR)(1) | | 38 | % | | 88 | % | | 100 | % | | 74 | % | | 81 | % | | 84 | % | | 90 | % |
(1) | EVR is defined as at least a two-log reduction in hepatitis C virus. |
(2) | Includes results for eight patients in each of the 160 µg, 320 µg and 480 µg dose cohorts. |
(3) | Davis, et al, J Hepatol 2003.50364. Includes studies encompassing a total of 511 patients. |
(4) | Ferenci, et al, J Hepatol 2005.40.009. Includes studies encompassing a total of 298 patients. |
(5) | Zeuzem, et al., 2006 Annual European Association for the Study of the Liver meeting. Includes 110 patients in the 900 µg dose cohort and 118 patients in the 1200 µg dose cohort. |
SELECT-1: Safety Results
Patient tolerability was evaluated by rating adverse events using the following definitions: “mild,” any symptom of which the patient is aware, but which is easily tolerated; “moderate,” any symptom that causes sufficient discomfort to interfere with the patient’s usual activities; “severe,” any symptom that is incapacitating and prevents the patient from engaging in work or usual activities. A “serious adverse event” is a patient reaction during treatment that results in death, a life-threatening situation, or a need for hospitalization. The frequency of these adverse events is measured by the percentage of patients who experienced adverse events, as opposed to the number of these adverse events experienced by any particular patient.
The following results were observed regarding Locteron side effects and patient tolerability at the conclusion (12 weeks of treatment) of the SELECT-1 Phase 2a trial for the 160 µg, 320 µg and 480 µg cohorts.
| | • | | Locteron was safe and well tolerated at 160 µg, 320 µg, and 480 µg. |
| | • | | There were no serious adverse events in the 160 µg, 320 µg, and 480 µg cohorts. |
| | • | | The vast majority (over 90%) of the adverse events that were experienced were rated as mild, with only one adverse event rated as severe, which occurred in the 320 µg cohort. |
| | • | | The incidence of fever in the 160 µg, 320 µg, and 480 µg cohorts was limited to one patient (4%) in the 320 µg cohort. |
| | • | | Dose reductions were limited to one patient each in the 320 µg and 480 µg cohorts, with none in the 160 µg cohort. |
We believe that the 12-week results provide a meaningful basis for evaluating the potential tolerability of Locteron, as any side effects experienced by patients through a full course of interferon treatment will generally be experienced at least once during the first four weeks of therapy. The Locteron patient tolerability through 12 weeks of treatment compare favorably with results reported for the currently marketed pegylated interferon alfa products and for Albuferon. Figure 2 below includes a comparison of severe adverse events reported in the SELECT-1 Locteron clinical trial and in trials of comparator products presented at the 2006 Annual European Association for the Study of the Liver meeting.
65
Figure 2—Locteron SELECT-1 Phase 2a Trial Adverse Events Rated
as Severe and Results for Comparator Products
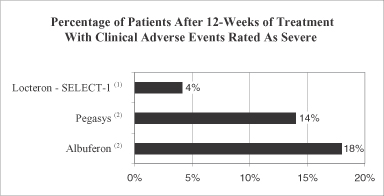
(1) | SELECT-1 Phase 2a clinical trial results for the 24 patients in the 160 µg, 320 µg and 480 µg cohorts. |
(2) | Zeuzem, et al., 2006 Annual European Association for the Study of the Liver meeting. Patients included in trial were 114 for Pegasys and 110 and 118 for Albuferon 900 and 1200 µg dose cohorts, respectively. Adverse events that were reported as severe were defined as “interferon related,” therefore certain severe adverse events determined not to be interferon related may have been excluded. Adverse events rated as severe after 48 weeks of treatment as reported in Zeuzem, et al., 2007 Annual European Association for the Study of the Liver meeting were 27%, 31% and 41% for the Pegasys and Albuferon 900 and 1200 µg dose cohorts, respectively. |
An objective point of comparison in evaluating the tolerability of Locteron is fever, a marker for the family of adverse events characterized as flu-like syndrome. Fever, characterized by a temperature reading of at least 38ºC (100.4ºF), occurred in only one (4%) of the Locteron patients in SELECT-1, notably lower than for results previously reported for other interferon products as illustrated in Figure 3 below.
Figure 3—Incidence of Fever in Locteron Clinical Trials
and in Published Results for Comparator Products
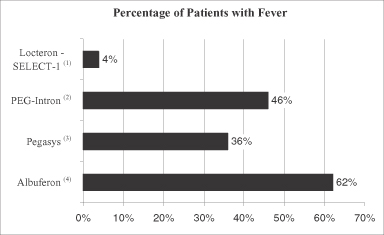
(1) | Results through 12 weeks of treatment in the SELECT-1 Phase 2a clinical trial results for the 24 patients in the 160 µg, 320 µg and 480 µg cohorts. |
(2) | Results through 48 weeks of treatment reported in PEG-Intron package insert. |
(3) | Results through 48 weeks of treatment reported in Pegasys package insert. |
(4) | Results through four weeks of treatment reported in Bain, et al.,Journal of Hepatology (2006) 1-8. Patients included in trial were 12 and 14 for Albuferon 900 and 1200 µg dose cohorts, respectively. |
66
Other adverse events reported for the Locteron patients in the SELECT-1 trial after four weeks and 12 weeks of treatment are compared to published four-week results for Albuferon in Table 2 below. Reported side effects for Locteron were fewer and less severe than those reported for Albuferon.
Table 2—Incidence of Other Adverse Events in SELECT-1 Clinical Trial
and in Published Results for Comparator Product(1)
| | | | | | | | | |
Adverse Events | | Albuferon Bain Publication Four Weeks(2) | | | Locteron
SELECT-1 Phase 2a Trial Four Weeks(2) | | | Locteron
SELECT-1 Phase 2a Trial 12 Weeks(2) | |
| | (n=26 | ) | | (n=24 | ) | | (n=24 | ) |
Abdominal pain | | 15 | % | | 4 | % | | 13 | % |
Arthralgia | | 58 | | | 46 | | | 50 | |
Chills | | 81 | | | 4 | | | 4 | |
Diarrhea | | 23 | | | 4 | | | 4 | |
Dizziness | | 46 | | | 4 | | | 4 | |
Fatigue / weakness | | 77 | | | 42 | | | 50 | |
Headache | | 92 | | | 25 | | | 33 | |
Irritability | | 31 | | | 13 | | | 13 | |
Myalgia | | 58 | | | 38 | | | 38 | |
Nausea | | 31 | | | 4 | | | 8 | |
Sleep disorder(3) | | 15 | | | 25 | | | 33 | |
(1) | Comparable published data for Pegasys and PEG-Intron is not available. |
(2) | Results for Locteron are presented for both 12 weeks of treatment and four weeks of treatment. The results for Albuferon are based on four weeks of treatment as published 12-week results for Albuferon are not available. |
(3) | All incidences of sleep disorder in the Locteron SELECT-1 Phase 2a trial were rated as mild. |
Other adverse events reported for Locteron that exceeded 10% of the patients in the SELECT-1 trial but are excluded from Table 2 above because they were not reported for Albuferon were dry cough (17%), dry mouth (21%), dry skin (42%), and pruritis, or itchiness (13%), all of which are side effects that are typically attributable to ribavirin.
We currently expect that data from the 640 µg cohort will be available in the fourth quarter of 2007.
Clinical Development and Commercialization
Based on the results for the first three dosing cohorts, the objective of the SELECT-1 Phase 2a trial has been met as we have identified a range of doses with an appropriate combination of viral reduction and favorable tolerability to proceed to the next stage of clinical development. Accordingly, after assessment of the results from the 640 µg cohort in the fourth quarter of 2007, we plan to commence a Phase 2b trial of Locteron in Europe under a CTA, with dosing expected to commence in the first half of 2008. We expect that the Phase 2b trial will be a multi-center trial that will evaluate three doses of Locteron, in combination with weight-based ribavirin, in treatment-naïve patients with the genotype-1 variant of the virus. The three Locteron/ribavirin arms will be compared against each other on a double-blinded basis and against a control arm consisting of a pegylated interferon alfa in combination with weight-based ribavirin. We currently expect to enroll approximately 100 patients with approximately 25 patients in each arm. Patients will be treated for 48 weeks, but the results at 12-weeks will be evaluated and will be the basis for dose selection for the commencement of Phase 3 clinical trials. This overlap between the Phase 2 and Phase 3 trials is designed to expedite the development path of Locteron, with dosing in Phase 3 trials expected to commence in 2009. We would expect patients in Phase 3 trials to be dosed for 48 weeks, with a 24-week follow-up period to evaluate sustained response. The specific design of the Phase 3 clinical trials, including the number of patients and patient characteristics, will be determined in advance of the commencement of these clinical trials based on interactions with the FDA and other regulatory agencies, our continuing evaluation of intellectual property as described in this prospectus, as well as other factors.
67
Given our current stage of development, we have not established marketing, sales or distribution capabilities. In order to commercialize Locteron, if approved, we would need to develop these capabilities internally or through collaboration with third parties. Together with our co-development partner OctoPlus, we currently intend to evaluate the formation of one or more collaborations with pharmaceutical companies under which the partner would assume responsibility for Phase 3 clinical trials and registration of Locteron. We may, however, elect to initiate such trials independently. In addition, we or OctoPlus may also decide to retain certain rights for the promotion or co-promotion of Locteron in certain geographic areas.
BLX-155—Direct-Acting Thrombolytic
BLX-155 is a direct-acting thrombolytic designed to break up the blood clots that form in certain diseases such as acute peripheral arterial disease, deep vein thrombosis and hemodialysis graft thrombosis. BLX-155 is the recombinant form of plasmin, the naturally occurring human enzyme responsible for dissolving fibrin, a key component of blood clots. Plasmin has been proposed for a number of decades as a drug candidate for resolving blood clots, but recombinant full-length human plasmin has not been successfully produced at commercially viable levels in traditional production systems. In July 2007, our researchers presented results at a scientific conference demonstrating the ability of the LEX System to produce recombinant plasmin at commercially viable levels. This achievement provides us the opportunity to pursue development and commercialization of a therapeutic protein with a recognized mechanism of action for which prior development within the pharmaceutical industry had been blocked by production challenges. As a direct-acting thrombolytic, we believe BLX-155 has the potential to provide efficacy and safety advantages over plasminogen activators, which dissolve blood clots indirectly. Plasminogen activators include t-PAs, which are the current standard of care.
Market Need for Direct-Acting Thrombolytic
Thrombosis, the formation or presence of a blood clot, is a leading cause of morbidity and mortality and is estimated to affect over 50 million people in the developed world. Thrombotic events are caused by the formation of blood clots inside a blood vessel or in an intravascular catheter. These blood clots are induced by the clumping of blood platelets followed by the formation of a fibrin network. We are focusing the initial development of BLX-155 on the treatment of clots present in blood vessels, particularly in blood vessels in the extremities of the body. We have chosen a strategy of focusing initially on blood clots in the extremities because the current standard of care, t-PA, is of limited effectiveness in these types of clots and these indications have the potential to require less costly and lengthy clinical trials compared to other diseases associated with thrombosis. Current treatment for these blood clots varies from disease to disease and upon the severity of the disease. In general, treatment ranges from the use of anti-coagulants or anti-platelet agents to prevent clot formation to the use of plasminogen activators in combination with catheters and surgery to resolve established blood clots.
We believe a key potential target indication for BLX-155 is acute peripheral arterial occlusive disease, or aPAO, a condition caused by a sudden reduction in blood flow to the extremities due to progressive clot accumulation over an atherosclerotic plaque in a diseased artery or in a previously placed synthetic graft. More than 100,000 cases of aPAO are reported annually in the United States and the disease is a significant cause of nerve and muscle damage and, in severe cases, amputation or death. Although no drugs are currently approved for the treatment of aPAO, patients are typically treated with a combination that includes the use of catheters, the delivery of plasminogen activators such as t-PA to the site of the clot, and surgical intervention. Treatment of aPAO with plasminogen activators often requires intensive care stays of 24 to 36 hours and may result in bleeding complications at sites remote from the clot.
Plasminogen activators have been relatively unsuccessful in the dissolution of the long, older and more compacted blood clots associated with aPAO. Plasminogen activators like t-PA work indirectly by converting plasminogen in the clot into the active protein plasmin which in turn directly dissolves the clot. Plasminogen activators therefore rely upon the presence of adequate quantities of plasminogen in the clot to effectively dissolve the clot. This property allows plasminogen activators to work in the dissolution of relatively small,
68
newer clots found in coronary arteries. However, in aPAO, blood clots as old a just a week often have inadequate quantities of plasminogen available for activation by plasminogen activators, and research suggests that administration of plasminogen activators has only limited effectiveness in aPAO. Importantly, as the quantity of plasminogen at the site of these established clots is limited, the unused plasminogen activator can circulate systemically and is available to convert plasminogen to plasmin wherever present and thus randomly dissolve clots throughout the body. Treatment with plasminogen activators for aPAO has been found to cause intracranial hemorrhage in up to 2.9% of patients. As a result of these efficacy and safety deficiencies, we believe that there is a need for a direct-acting recombinant plasmin agent, such as BLX-155, that does not rely upon the presence of plasminogen within the site of the clot to treat aPAO.
Another potential target indication for BLX-155 is hemodialysis graft thrombosis, or HGT. Among the greatest challenges facing physicians and other health care professionals who care for patients with chronic renal failure is the preservation of vascular access for hemodialysis. HGT, the blockage of vascular access due to blood clots, is a cause of considerable morbidity, discomfort, inconvenience and cost for patients who must undergo routine hemodialysis. Currently, there are more than 175,000 patients undergoing hemodialysis in the United States, and half or more will have at least one episode of HGT each year. Approximately 20% to 30% of all hospital admissions for patients with end-stage renal disease are related to complications of HGT.
Deep vein thrombosis, or DVT, is also a potential target indication for BLX-155. Each year approximately 600,000 patients are diagnosed with DVT in the United States. DVT is characterized by blood clots in the venous system of limbs, typically the legs. The consequences of DVT include pain and swelling of the affected limb and, in relatively rare circumstances, pulmonary embolism which can result in death. Similar to aPAO, research suggests that more established blood clots in DVT are often deficient in plasminogen and therefore the administration of plasminogen activators is only partially effective and can result in the activators dissolving clots in random locations within the body.
Scientific Basis for Development of BLX-155 / Full-Length Plasmin as a Thrombolytic
BLX-155 is full-length recombinant human plasmin produced in the LEX System. Plasmin is the key enzyme in the human body that dissolves the fibrin component of blood clots, a process known as lysis. In fact, dissolution of blood clots, whether it is accomplished naturally within the body or through indirect treatment with existing drugs like t-PA, is ultimately accomplished by plasmin. We believe that plasmin’s accepted role in dissolving clots makes it the logical basis for a direct-acting thrombolytic.
69
Plasmin works by cutting, or cleaving, the fibrin network that supports blood clots, thereby releasing the cells trapped within the network and dissolving the blood clot. As illustrated in Figure 4 below, full-length plasmin is a complex protein with an amalgamation of structures that are key to its mechanism. The serine protease component of plasmin is responsible for cleaving fibrin. Natural plasmin also includes five structural sequences, known as kringle domains, that as a group have a high level of affinity for fibrin. Plasmin has been shown to be more effective at dissolving blood clots than t-PA in bothin vitro andin vivo preclinical models. We have recently conductedin vivo tests in pig models showing rapid cleaving of clots with BLX-155.
Figure 4—Three-Dimensional Illustration of BLX-155
(Full-Length, Recombinant Human Plasmin)
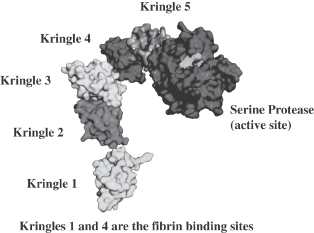
In contrast, truncated forms of plasmin such as microplasmin currently under development lack the full Kringle domains illustrated above, and therefore lack the binding affinity for fibrin believed to be critical to provide an effective therapy. Furthermore, another drug currently under development, alfimeprase, lacks any of the Kringle domains and is comprised of a protease site that is a recombinant form of a protein from snake venom, and therefore lacks the well-described mechanism associated with full-length plasmin.
Additionally, the safety mechanisms inherent in the body provide the potential to use recombinant plasmin as a direct-acting thrombolytic and greatly reduce the bleeding risks associated with plasminogen activators. Native plasmin is regulated by a number of inhibitors within the body that exist in high quantities and serve to rapidly inactivate plasmin that circulates beyond the immediate site of the clot. Within the body, free plasmin is not normally found circulating in the blood and the inactivation of plasmin is carefully regulated to prevent degradation of important circulating procoagulant proteins, such as fibrinogen and factor VIII. Plasmin is rapidly inactivated naturally within the body by alpha-2 antiplasmin, alpha-2 macroglobulin, and certain serine protease inhibitors. We believe these safety mechanisms also have the potential to enable the safe delivery of higher doses of BLX-155 relative to other therapies, along with the resulting potential for a higher therapeutic index. In addition, inin vivo preclinical re-bleed models in animals, plasmin resulted in a substantially lower risk of bleeding compared to t-PA.
Production of BLX-155 Using the LEX System
Full-length, active plasmin has been proposed as a potential thrombolytic agent for several decades. However, in addition to the complexity of the full-length plasmin protein, active plasmin has the propensity to degrade itself during all stages of production, making production on a large scale difficult. To date, there have been no reports of the commercially viable production of full-length plasmin using any traditional recombinant production system. The limitations of existing production systems have resulted in research regarding truncated
70
versions of plasmin, such as micro-plasmin, that lack the five kringle domains of native plasmin and therefore lack the natural affinity for binding to fibrin. These production challenges have also resulted in full-length plasmin only being derived from human donor plasma, a source that is limited and carries the potential for transmitting human pathogens.
In July 2007, our researchers presented results at the XXIst Congress of The International Society on Thrombosis and Haemostasis demonstrating the successful production of full-length recombinant human plasmin using the LEX System, a milestone never reported with traditional protein expression systems.Results presented at the conference demonstrated that BLX-155, recombinant full-length plasmin produced using the LEX System, is indistinguishable from human plasmin in characterization and activity. We believe that the development of BLX-155 using the LEX System allows exploitation of the natural binding, efficacy and safety attributes of native plasmin without the limitations or risks associated with plasminogen activators or product candidates under development such as truncated plasmin, plasma-derived plasmin, and alfimeprase, a recombinant form of a snake venom protease under development.
Benefits of BLX-155
The potential advantages of BLX-155, compared to plasminogen activators like t-PA and to products known to be under development, are as follows:
| | • | | Direct Acting.BLX-155 is direct acting and does not require the presence of plasminogen at the site of blood clots to be effective. By contrast, plasminogen activators such as t-PA do not directly cause dissolution of clots but must first find and activate plasminogen in the vasculature near the site of a clot. As the long, established clots found in aPAO and in DVT are often deficient in plasminogen, plasminogen activators are of limited effectiveness. |
| | • | | High-Binding Affinity.We believe that full-length plasmin’s high affinity for binding to fibrin may result in a therapy that is more effective than other direct-acting thrombolytic agents in development such as alfimeprase and micro-plasmin, both of which lack the five kringle domains of full-length plasmin. |
| | • | | Rapid Action. The direct mode of action and high, specific fibrin binding affinity lead to the strong potential for a more rapid therapeutic option for longer, established clots containing little plasminogen. Currently, treatment of aPAO with plasminogen activators often requires intensive care stays of 24 to 36 hours. |
| | • | | Safety Screen.As a native protein, BLX-155 is regulated by a number of inhibitors within the body that exist in high quantities and serve to rapidly inactivate any plasmin that circulates beyond the immediate site of the clot. This safety mechanism may decrease the risk of bleeding complications associated with the therapeutic administration of plasminogen activators and highlighted in recent clinical trials of alfimeprase. |
| | • | | Known Mechanism. Plasmin is the key enzyme in the human body that dissolves the fibrin component of blood clots. Other direct thrombolytic agents under development rely upon either substantial modification of full-length plasmin, such as micro-plasmin, or unproven mechanisms such as alfimeprase. Each approach lacks the body of natural evidence supporting the use of full-length plasmin. |
Clinical Development and Commercialization
BLX-155 is currently in preclinical development, and we expect to commence a Phase 1 clinical trial in the first half of 2008. Our planned Phase 1 and Phase 2a clinical trials are designed to support the development of BLX-155 regardless of the specific indication or indications ultimately selected for potential regulatory approval. Our planned Phase 1 clinical trial will be a dose-escalation trial conducted in hemodialysis graft thrombosis patients. The trial is designed to provide data regarding the safety of BLX-155 while allowing for an assessment of clot dissolution in this medical condition. Our planned Phase 2a clinical trial will be conducted in deep vein
71
thrombosis patients and is expected to commence in the second half of 2008 after evaluation of the initial safety results from the Phase 1 trial. These two trials are designed to facilitate the exploration of a wide range of doses for BLX-155, including the higher doses expected to be evaluated if aPAO is selected as an indication for development.
We currently intend to retain the rights to develop, market and sell BLX-155 in the United States and potentially other selected markets. We may enter into one or more collaborations with pharmaceutical companies for the right to market BLX-155 in major markets outside the United States or in specific indications outside of our primary areas of focus. Given our current stage of development, we have not established marketing, sales or distribution capabilities. In order to commercialize BLX-155, if approved, we would need to develop these capabilities.
BLX-301
Optimized Monoclonal Antibodies
The LEX System has demonstrated the capability to express monoclonal antibodies in which the sugar-chain component, known as the glycosylation structure, has been optimized to enhance the efficacy and potency of the antibody. Monoclonal antibodies are currently being used and developed primarily to treat cancers, autoimmune diseases, infectious diseases and inflammatory diseases. According to Kalorama Information, monoclonal antibodies are the fastest growing class of products in the pharmaceutical industry, with total sales growing from $4 billion in 2001 to $17 billion in 2005, and projected to reach $34 billion in 2010. The effectiveness of monoclonal antibodies, particularly those targeting oncology indications, are greatly impacted by the glycosylation structure of one section of the antibody, known as the Fc region. Research has shown that if the Fc region of the antibody is modified to create an optimized glycosylation structure, the ability of the antibody to bind to immune cells, and to stimulate those immune cells to destroy diseased cells, can be greatly enhanced. This modification can be described as improving the “Fc functionality” of the antibody.
In December 2006, results were published inNature Biotechnologydemonstrating the ability of the LEX System to produce monoclonal antibodies with an optimized glycosylation structure. Specifically,in vitro studies suggested that a monoclonal antibody under development as an anti-CD30 candidate was more potent and efficacious when produced in the LEX System compared to the same antibody produced in a traditional mammalian system. The publication also described the LEX System’s ability to produce antibodies with homogeneous glycosylation structures, one of the major advantages of the LEX System compared to expression systems commonly used today and a key to reducing production risks, regulatory risks and costs.
Our optimization of monoclonal antibodies is achieved by engineering the LEX System to silence two genes responsible for adding two specific sugars, fucose and xylose, to the glycosylation structure of the antibody, resulting in a fully human glycosylation structure completely devoid of any detectable presence of fucose or xylose. Fucose is a sugar typically present in the glycosylation structure of monoclonal antibodies produced in traditional expression systems, and xylose is typically associated with antibodies produced in plant-based systems. The ability to engineer the LEX System to accomplish the elimination of fucose and xylose results in improved Fc functionality. Monoclonal antibodies produced in the LEX System also have a G0 glycosylation structure, which means they are absent of galactose. We believe G0 glycosylation structure will potentially provide the additional benefit of reducing side effects in certain diseases. We have successfully produced more than ten monoclonal antibodies, and the ability of the LEX System to produce optimized antibodies with fully human glycosylation, meaning they have no fucose or xylose, has been demonstrated with multiple corporate partners, including Medarex, and in multiple proteins, highlighting the potential robustness and consistency of the system.
72
BLX-301—Optimized Anti-CD20 Candidate
We are currently optimizing BLX-301, a humanized anti-CD20 candidate. BLX-301 will be designed to incorporate the potential benefits indicated in the proof-of-concept optimized rituximab study described below, including the potential for improved potency and efficacy and reduced side effects, into a humanized antibody. We currently expect to complete selection of an antibody candidate in early 2008.
To provide proof-of-concept support for the potential benefits of developing an anti-CD20 antibody optimized in the LEX System, we evaluated an optimized version of rituximab produced in the LEX System. Rituximab, marketed under the trade name Rituxan, is an anti-CD20 antibody used today in the treatment of non-Hodgkin’s B-cell lymphoma, or NHL, and rheumatoid arthritis. Rituximab is a chimeric antibody, which means that it is a combination of mouse derived and human derived components. In May 2007, our researchers announced results at a scientific conference highlighting the ability of the LEX System to produce an anti-CD20 antibody with enhanced cytotoxicity and potentially lower side effects than rituximab. As reported at the conference, rituximab was produced in the LEX System with an optimized glycosylation structure in which fucose and xylose was absent. Optimized rituximab produced in the LEX System had antibody dependent, cell-mediated cytotoxicity, or ADCC, that was 20 to 200 times higher than rituximab produced in a CHO, or traditional mammalian, system as illustrated in Figure 5 below. In addition, optimized rituximab produced in the LEX System with a G0 glycosylation structure had complement dependent cytotoxicity that was ten-fold lower than Rituxan. Complement dependent cytotoxicity has been reported to be a key contributor to serious side effects associated with rituximab treatment.
Figure 5—Comparison of Optimized Rituximab Produced in LEX System
To Rituxan in Preclinical ADCC (Cell-Killing) Assay
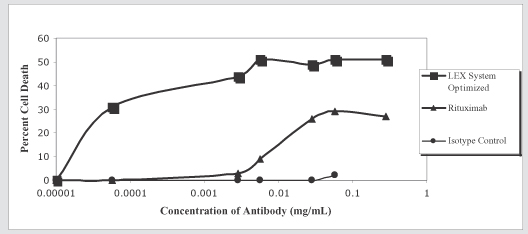
We believe that the above results support the advancement of BLX-301, which is designed to be differentiated from rituximab and other antibodies under development by combining the favorable attributes of humanization, optimized glycosylation, and reduced complement dependent cytotoxicity. Beyond BLX-301, we expect that antibody glycosylation optimization will be one of the focal points of future expansion of our product pipeline.
Intellectual Property
We actively seek to protect the proprietary technologies that we consider important to our business, including methods, compositions, cultures, sequences, and assemblies for the expression and manufacture of proteins in the LEX System, including interferon alfa and BLX-155, and for optimizing the glycosylation
73
structures of monoclonal antibodies. We also rely on trade secrets, know-how, continuing technological innovation and in-licensing opportunities to develop and maintain our proprietary position. We currently own or hold exclusive rights to 22 patents issued in the United States and 22 patent applications pending in the United States. Corresponding patents and patent applications are held in major international markets outside the United States.
We consider the following U.S. patents that we own or license to be most important to the protection of our most advanced product candidates.
| | • | | Genetic Manipulation of Lemna.We have three U.S. patents and three patent applications relating to the genetic manipulation of theLemna family of plants to enable the expression of proteins. These patents and applications encompass the methods and compositions for transformingLemna to produce proteins, enhanced expression levels, and transgenic plant regeneration, growth and recovery. The issued U.S. patents expire between 2017 and 2018. |
| | • | | Industrialization of Lemna Technology.We have three U.S. patents and five patent applications relating to the industrialization of theLemna technology. These patents and applications encompass processes ranging from the transformation, selection and high-throughput screening methodologies employed in the development ofLemna plant lines, as well as the methods and assemblies used in production, including a bioreactor for growing biologic materials supported on a liquid surface. The issued U.S. patents expire between 2022 and 2024. |
| | • | | Production of Monoclonal Antibodies in Plants.We have nine U.S. patents and two patent applications encompassing compositions and methods for the production of monoclonal antibodies in any plant-based system. The issued U.S. patents expire between 2009 and 2014. |
| | • | | Production of Interferon Alfa.We have one U.S. patent and four patent applications encompassing the methods, sequences, and cultures enabling the production of interferon alfa and other polypeptides inLemna. Interferon alfa serves as the active agent in our lead product candidate Locteron. The issued U.S. patent expires in 2021. |
| | • | | Production of BLX-155.We have one U.S. patent application encompassing the production of recombinant plasminogen, the precursor to recombinant plasmin or BLX-155, inLemna. If a U.S. patent related to this application issues, it will expire in 2025. |
| | • | | Glycosylation Optimization.We have three U.S. patent applications related to the optimization of the glycosylation structure of monoclonal antibodies inLemna. |
In each of the above areas of focus, corresponding patents and patent applications are held in major international markets outside the United States.
We have a co-development agreement with OctoPlus which encompasses the development and commercialization of Locteron. The rights to PolyActive, the controlled-release component of Locteron, are exclusively held by OctoPlus. PolyActive provides a gradual release of interferon alfa to patients without the high initial blood levels associated with the significant side effects experienced by patients today. We and OctoPlus have agreed to the commercial use of PolyActive for the administration of interferon agents solely under this agreement. OctoPlus holds a U.S. patent for the use of PolyActive as a drug carrier that expires in 2016. OctoPlus also has patent applications relating to the use of PolyActive to deliver interferon, which if issued will expire in 2026. OctoPlus and we jointly hold a U.S. patent application covering the specific controlled-release formulation of Locteron, its release profile, and methods of treatment, which if issued will expire in 2028. Corresponding patents and patent applications are held in major international markets.
The actual protection afforded by a patent varies from country to country and depends upon many factors, including the type of patent, the scope of its coverage and the availability of legal remedies in a particular
74
country. The life of individual patents extend for varying periods depending on the date of filing of the patent application or the date of patent issuance and the legal term of patents in the countries in which they are obtained. In the United States, The United States Drug Price Competition and Patent Term Restoration Act of 1984, known as the Hatch-Waxman Act, permits a patent extension term of up to five years as compensation for patent term lost during the FDA regulatory review process. Patent extension cannot extend the remaining term of a patent beyond a total of 14 years. The patent term restoration period is generally one-half the time between the effective date of an IND and the submission date of a Biologics License Application, or BLA, plus the time between the submission date of a BLA and the approval of that application. Only one patent applicable to an approved drug is eligible for the extension, and the extension must be applied for prior to expiration of the patent. The U.S. Patent Office, in consultation with the FDA, reviews and approves applications for patent term extension. We expect to consider applying for patent term extensions for some of our current patents, depending on the expected length of clinical trials and other factors involved in the filing of a new drug application.
In addition to patents, we rely upon unpatented trade secrets and know-how and continuing technological innovation to develop and maintain our competitive position. We seek to protect our proprietary information, in part, using confidentiality agreements with our commercial partners, collaborators, employees and consultants and invention assignment agreements with our employees. We also have confidentiality agreements or invention assignment agreements with some of our commercial partners and consultants. These agreements are designed to protect our proprietary information and, in the case of the invention assignment agreements, to grant us ownership of technologies that are developed through a relationship with a third party.
License Agreements
The rights to certain of our patents have been acquired through license agreements with universities, research institutions or other corporations. In addition to expanding our patent protection, we have also entered into exclusive and non-exclusive licenses in an attempt to ensure that we have freedom to operate within the areas of commercial interest to us. Three license agreements that we consider of particular importance to our business are summarized below.
North Carolina State University
In 1998 we entered into a license agreement with North Carolina State University, or NCSU, under which we hold an exclusive worldwide license to patents and patent applications owned by NCSU for use in the development and commercialization of genetically engineeredLemna. Under the agreement, we are obligated to pay to NCSU royalties on net sales of products subject to the license. Such royalties may be reduced by up to 50% if we are obligated to pay a royalty to a third party in order to make, use or sell such products. As partial consideration for the license we granted 3,821 shares of Class B Non-Voting Common Stock to NCSU. To date, we have paid no license fees or royalties to NCSU under this agreement.
NCSU is responsible for filing, prosecuting and maintaining the licensed patent rights and we are required to cooperate with NCSU in the prosecution of patent applications to ensure that such applications reflect all items of commercial, scientific and technical interest and importance. We are required to reimburse NCSU’s out-of-pocket costs for the prosecution of licensed patents. We have the right, but not the obligation, to enforce the patent rights against infringement.
We are obligated to indemnify NCSU against any liabilities resulting from utilization of the licensed patent rights. We are also required to maintain product liability insurance if and when we commercialize a product that is subject to the license.
Under the terms of the license, we are required to diligently pursue the development of the technology underlying this license, and if we fail to do so the agreement may be terminated by NCSU at its option. Currently, virtually all of our development efforts are directly related to the development and commercialization
75
of this fundamental technology. We may terminate the agreement at any time upon 90-days written notice. If not terminated earlier, the agreement will terminate upon expiration of the last to expire of the licensed patent rights, which will occur in 2018 based on the last U.S. patent issued to date.
YEDA Research and Development Company Limited
In 2005 we entered into a license agreement with YEDA Research and Development Company Limited, or YEDA, under which we hold an exclusive worldwide license to patents and patent applications owned by YEDA for use in the development and commercialization of transgenic lemnaceae. Under the agreement, we are obligated to pay to YEDA:
| | • | | royalties on net sales ofLemna-based products; |
| | • | | a percentage of manufacturing service revenues and other revenues associated withLemna-based production of proteins; and |
| | • | | a percentage of sublicensing revenues. |
Should royalties payable in any year not reach a stipulated minimum, we are obliged to pay the balance as a supplemental royalty. To date, we have paid YEDA an aggregate of $300,000 under this agreement.
YEDA controls the filing, prosecution and maintenance of the licensed patent rights and we have the right to comment on and advise YEDA with respect to such matters. We are required to pay all expenses related to filing, prosecution and maintenance of the licensed patent rights. After consulting with YEDA, we have the right, but not the obligation, to enforce the patent rights against infringement.
We bear full responsibility and liability for costs of any person or entity arising out of the utilization of the licensed patent rights by us or any sublicensee. We are also required to maintain general liability insurance, product liability insurance, or clinical trial insurance, as applicable, throughout the term of the license.
Under the terms of the license, we are required to diligently pursue the commercialization ofLemna-based products, and if we fail to do so the agreement may be terminated by YEDA at its option. Currently, virtually all of our development efforts are directly related to the development and commercialization ofLemna-based products. We may terminate the license on 60 days’ notice to YEDA. If not terminated earlier, the agreement will terminate upon expiration of the last to expire of the licensed patent rights which will occur in 2017 based on the last U.S. patent issued to date.
The Scripps Research Institute
As a result of our 2004 acquisition of substantially all of the assets of Epicyte, we have a license agreement with The Scripps Research Institute, or TSRI, under which we hold an exclusive worldwide license to patents and patent applications owned by TSRI for the development and commercialization of technology encompassing the expression of antibodies and other immunoglobulin molecules in plants. Under the agreement, we are obligated to pay to TSRI:
| | • | | a percentage of sublicensing revenues; and |
| | • | | royalties on net sales of products subject to the license. |
Royalties may be reduced if we are obligated to pay a royalty to a third party in order to make, use or sell such products. A minimum royalty is payable annually. To date, we have paid TSRI an aggregate of $75,000 under this agreement.
TSRI controls the filing, prosecution and maintenance of the licensed patent rights and we have full rights of consultation on such matters. We are required to pay all expenses related to filing, prosecution and maintenance of the licensed patent rights. We have the first option to enforce the patent rights against infringement.
76
We are obligated to indemnify TSRI against any liability or expense arising from any claims arising out of the use of the licensed patent rights. We are also required to use our best efforts to have TSRI and its affiliates named as additional insured parties on any product liability insurance policies that we maintain.
We are required to diligently pursue the development of commercially viable products using the licensed technology, and if we fail to do so the agreement may be terminated by TSRI at its option. However, we believe that currently we are actively engaged in efforts to develop and commercialize products using the licensed technology. We may terminate the license on 90 days’ notice to TSRI. If not terminated earlier, the agreement will terminate upon expiration of the last to expire of the licensed patent rights, which will occur in 2014 based on the last U.S. patent issued to date.
Collaborations
Co-Development Agreement With OctoPlus
In February 2005 we entered into an exclusive co-development agreement with OctoPlus Technologies B.V., a wholly owned subsidiary of OctoPlus for the development of Locteron. Locteron combines BLX-883, the interferon alfa produced in our proprietary LEX System, with OctoPlus’ PolyActive drug delivery technology. Pursuant to this agreement, unless a party elects not to continue further development, all third-party development costs are shared on a substantially equal basis and we share any profits from the commercialization of Locteron on an equal basis.
A steering committee comprised of an equal number of representatives from Biolex and OctoPlus make decisions with respect to the development and commercialization of Locteron. In the event of a deadlock, decisions may be submitted to non-binding mediation, but Biolex has a right to determine the final resolution of any deadlock concerning development or commercialization of Locteron as related solely to North America, and OctoPlus has a right to determine the final resolution of any deadlock concerning development or commercialization of Locteron as related solely to Europe. Currently, we and OctoPlus intend to seek one or more development and commercialization partners for this product candidate in advance of the commencement of Phase 3 clinical trials. However, we may initiate Phase 3 clinical trials independently if circumstances allow.
Both we and OctoPlus may file and own patents and patent applications covering our respective sole inventions. Patent applications on joint inventions may be filed only if we and OctoPlus agree to jointly file them, in which case all expenses incurred in connection with the filing of joint patents are shared equally. Neither party has the right to transfer its interest in a joint patent to a third party unless the other party consents to such transfer. In the event of infringement on a joint patent or patents, we and OctoPlus will cooperate with one another in determining the course of action necessary to stop the infringement and shall equally share all costs and expenses related thereto. In the event that either we or OctoPlus makes use of a joint invention or joint patent outside the scope of the agreement, such party shall be solely responsible for the cost and expenses incurred to enforce the patent rights against infringement.
We and OctoPlus are required to indemnify one another from damages arising out of our respective intellectual property that is the subject of the agreement, a material breach of our respective representations, warranties, covenants or obligations set forth in the agreement, or arising out of willful misconduct, gross negligence, or a knowing violation of the law with regard to activities conducted under the agreement, or third party claims of patent infringement by our respective intellectual property that is the subject of the agreement. We are obligated to establish and maintain general liability insurance.
Our agreement with OctoPlus provides both parties with a right to opt out of further development. In the event that one party decides to opt out of further development, the party that has opted out no longer may participate in decision making with respect to development and commercialization of Locteron, and residual obligations and rights may exist for that party, including its respective manufacturing obligations for the Locteron product or granting a royalty-free license for such manufacture. Either party may terminate the
77
agreement upon 60-days notice if both parties have opted out of the development or upon the occurrence of certain other events, including a material breach of the agreement or insolvency of the other party, in each case subject to the survival of certain obligations.
Collaborations Relating to the LEX System
In addition to enabling our internally generated product candidates, our LEX System has provided us with opportunities to form strategic alliances with leading pharmaceutical and biotechnology companies such as Centocor, Genmab A/S, Medarex, and Merial Limited. Under these collaborations, we are applying the LEX System to protein and antibody candidates held by our partners, including the optimization of antibody candidates and the development ofLemna plant lines to enable commercially viable production of these therapeutic proteins. Corporate partnerships have provided a number of key benefits during the earlier-stage development of our LEX System and internal capabilities, including:
| | • | | Serving as a source of non-dilutive revenue to offset a portion of the costs associated with the development of our platform technology. For example, much of the research that led to our glycosylation optimization capabilities as well as the development of our scaled-up production processes was funded by corporate partners despite the fact that we retained all patent rights to these advancements. |
| | • | | Strengthening our organizational expertise as a result of our work with more than ten novel antibodies and a series of other proteins under our collaborations. |
We will continue to evaluate the initiation of selected collaborations, or the advancement of our existing collaborations, with companies where we can employ the LEX System to enhance the development or clinical attributes of their proteins and antibodies in exchange for participating in the commercial upside of our partners’ product candidates. However, there is no certainty that we and any of our partners will agree to move any of the product candidates that are the subject of these collaborations into more advanced development or achieve successful commercialization.
Manufacturing and Supply
We have the capability to manufacture therapeutic proteins and monoclonal antibody drug candidates for use in clinical development and other research activities in our manufacturing facility that is designed for cGMP production. These proteins are produced and purified by us in our existing facility of approximately 40,000 square feet that houses our manufacturing facility and research operations. The current facility accommodates the production of pharmaceutical-grade and research-grade proteins and antibodies for our own development programs as well as the programs underlying our corporate collaborations. We do not expect to produce any products for commercial use in this facility.
We have leased and are currently building out a second facility of approximately 48,000 square feet to accommodate cGMP production for later-stage clinical development and commercial launch. The first phase of the build-out is expected to be complete in time to accommodate the production of clinical trial materials for the planned Phase 3 clinical trials of Locteron. Under the terms of the lease agreement for this facility, we expect that the lessor will fund most of the cost of the tenant improvements.
In addition to our activities, we rely upon third-party manufacturers and partners to complete the manufacturing of our product candidates. In the case of Locteron, the interferon alfa produced in our facility is transferred to OctoPlus to be combined with the PolyActive microspheres, and OctoPlus formulates the final drug product at its facilities. In the future, we expect to contract with additional third-party manufacturers to perform specific manufacturing activities such as formulation and filling operations.
78
Sales and Marketing
We do not have any marketed products. However, we have personnel who devote a substantial portion of their time to strategic marketing. The objective of our strategic marketing efforts is to identify opportunities within our product candidate portfolio, analyze the competitive environment, patient needs and customer preferences, and help guide our product development strategies.
If we are successful in developing products that can be marketed, we intend to market the products either independently or together with collaborators or strategic partners. If we decide to market any products, either independently or together with partners, we will incur significant additional expenditures and commit significant additional management resources to establish a sales and marketing organization. For any products that we market together with partners, we will rely, in whole or in part, on the marketing capabilities of those parties. We may also enter into collaborations with pharmaceutical companies to market certain of our products.
Competition
The biotechnology and pharmaceutical industries are characterized by rapidly advancing technologies, intense competition and a strong emphasis on proprietary products. While we believe that our technologies, knowledge, experience and scientific resources provide us with competitive advantages, we face potential competition from many different sources, including commercial biotechnology and pharmaceutical enterprises, academic institutions, government agencies and private and public research institutions. Any product candidates that we successfully develop and commercialize will compete with existing therapies and new therapies that may become available in the future.
Many of our competitors may have significantly greater financial resources and expertise in research and development, manufacturing, preclinical testing, conducting clinical trials, obtaining regulatory approvals and marketing approved products than we do. These competitors also compete with us in recruiting and retaining qualified scientific and management personnel, as well as in acquiring technologies complementary to, or necessary for, our programs. Smaller or early stage companies may also prove to be significant competitors, particularly through collaborative arrangements with large and established companies.
Our commercial opportunities could be reduced or eliminated if our competitors develop and commercialize products that are safer, more effective, have fewer side effects, are more convenient or are less expensive than products that we may develop. In addition, our ability to compete may be affected because in some cases insurers or other third-party payors seek to encourage the use of generic products. This may have the effect of making branded products less attractive to buyers.
Our major competitors include pharmaceutical and biotechnology companies in the United States and abroad that have approved therapies or therapies in development for diseases targeted by our pipeline of product candidates, including our lead programs Locteron and BLX-155. The key competitive factors affecting the success of our product candidates are likely to be their efficacy, safety, tolerability, convenience and price.
Locteron
The market for the treatment of hepatitis C is large and growing, factors that have attracted a wide range of companies to pursue the development of new drug candidates for this disease. We expect that if approved, Locteron will have to compete with the two pegylated interferon alfa products currently marketed today, PEG-Intron from Schering-Plough Corporation and Pegasys from Hoffmann-La Roche. Although we believe that Locteron has the potential to provide a number of benefits compared to these two products, including patient tolerability, both of these companies have substantial resources and the two products currently dominate the market. Another potentially competitive product candidate is Albuferon, currently in Phase 3 development by Human Genome Sciences and Novartis. Although we believe that Locteron has the potential to provide a number of benefits compared to Albuferon, including patient tolerability, Albuferon is currently at a more advanced stage
79
of development. We are aware of other forms of interferon that are at earlier stages of development for the potential treatment of hepatitis C, however we are not aware of any to date that have demonstrated in clinical testing any advantage over the current pegylated interferons.
Much of the drug development underway today in hepatitis C involves new antiviral drug candidates. Among the more advanced candidates are Vertex Pharmaceuticals, Inc.’s VX-950 protease inhibitor. All of these new antiviral drug candidates are currently being developed in combination with interferon alfa, and therefore we believe that they represent opportunities for potential combination use with Locteron. However, if any of these candidates, or combination treatment encompassing these candidates, are shown to be efficacious without the need to be combined with interferon alfa, the commercial opportunity for Locteron could be reduced or eliminated.
BLX-155
We expect that if BLX-155 is approved in one or more of its target indications, it will likely have to compete against plasminogen activators such as Genentech’s Activase. Despite the fact that Activase is not currently approved for the treatment of diseases such as aPAO and DVT, it is often used by physicians on an off-label basis. Although we believe that BLX-155 has the potential to provide a number of benefits compared to Activase, including efficacy and safety, Genentech has substantial resources and Activase has been used in various indications for twenty years. Other potentially competitive product candidates are micro-plasmin from Thrombogenics, human-plasma-derived plasmin from Talecris, and alfimeprase from Nuvelo. Although we believe that BLX-155 has the potential to provide a number of benefits compared to each of these product candidates, and the specific diseases to potentially be targeted by each is uncertain, each represents potential competition for BLX-155.
BLX-301
If BLX-301 is approved in one or more of its target indications, it will likely have to compete against monoclonal antibodies targeting CD20, such as Rituxan which is currently marketed by Genentech, Biogen-Idec and Hoffmann-La Roche, each of whom have substantial resources. Other potentially competitive product candidates under development are HuMax-CD20, under development by GenMab A/S and GlaxoSmithKline, and TRU-015, under development by Trubion Pharmaceuticals and Wyeth.
Government Regulation and Product Approvals
Government authorities in the United States, at the federal, state and local level, and other countries extensively regulate, among other things, the research, development, testing, manufacture, quality control, approval, labeling, packaging, storage, record-keeping, promotion, advertising, distribution, marketing and export and import of products such as those we are developing. The process of obtaining regulatory approvals and the subsequent compliance with appropriate federal, state, local, and foreign statutes and regulations require the expenditure of substantial time and financial resources. Failure to comply with the applicable United States requirements at any time during the product development process, approval process or after approval, may subject an applicant to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, suspension or withdrawal of an approval, a clinical hold, warning letters, product recalls, product seizures, total or partial suspension of production or distribution injunctions, fines, refusals of government contracts, restitution, disgorgement, or civil or criminal penalties.
United States Government Regulation
In the United States, new drugs, biologics, and medical devices generally require FDA marketing authorization before they can be marketed. None of our products have received such authorization. The kind of authorization required depends on the product. A new drug will follow the new drug approval, or NDA route, and
80
a new biologic will follow the biologics license application, or BLA, route. Medical devices are approved or cleared for marketing through either the premarket approval application, or PMA, process, or the 510(k) clearance process. Drugs or biologics that are combined with medical devices may be regulated as combination products through one or more of the FDA’s regulatory pathways. The FDA has preliminarily advised us that Locteron will be regulated as a combination biologic/medical device product that will be reviewed under the FDA’s authority to regulate biologics.
BLA Approval Processes
The FDA regulates drugs, biologics, and medical devices under the Federal Food, Drug and Cosmetic Act, or FDCA, and, in the case of biologics, also under the Public Health Service Act, and implementing regulations. To obtain authorization to market a biologic, a company generally must complete the following steps:
| | • | | completion of preclinical laboratory tests, animal studies and formulation studies according to Good Laboratory Practices or other applicable regulations; |
| | • | | submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may begin; |
| | • | | performance of adequate and well-controlled human clinical trials according to Good Clinical Practices to evaluate the safety and efficacy of the proposed drug for its intended use; |
| | • | | submission to the FDA of a BLA; |
| | • | | satisfactory completion of an FDA inspection of the manufacturing facility or facilities at which the drug is produced to assess compliance with current good manufacturing practice, or cGMP, to assure that each facility meets standards designed to ensure the biologic’s continued safety, purity and potency; and |
| | • | | FDA review and approval of the BLA. |
Once a pharmaceutical candidate is identified for development it enters the preclinical testing stage. Preclinical tests include laboratory evaluations of product chemistry, toxicity and formulation, as well as animal studies. An IND sponsor must submit the results of the preclinical tests, together with manufacturing information and analytical data, to the FDA as part of the IND. The sponsor must also include a protocol detailing, among other things, the objectives of the first phase of the clinical trial, the parameters to be used in monitoring safety, and the effectiveness criteria to be evaluated, if the first phase lends itself to an efficacy evaluation. Some preclinical testing may continue even after the IND is submitted. The IND automatically becomes effective 30 days after receipt by the FDA, unless, within the 30-day time period, the FDA raises concerns or questions about issues such as the conduct of trials outlined in the IND and places the trial on a clinical hold. In such a case, the IND sponsor and the FDA must resolve any outstanding concerns before the clinical trial can begin. There can be no assurances that the submission of an IND will result in FDA authorization to commence clinical trials. Clinical holds also may be imposed by the FDA at any time before or during studies due to safety concerns or non-compliance.
All clinical trials must be conducted under the supervision of one or more qualified investigators in accordance with good clinical practice regulations. These regulations include the requirement that all research subjects provide informed consent. Further, an institutional review board, or IRB, at each site participating in the clinical trial must review and approve the plan for any clinical trial before it commences at that site. An IRB considers, among other things, whether the risks to individuals participating in the trials are minimized and are reasonable in relation to anticipated benefits. The IRB also must approve the information regarding the trial and the consent form that must be provided to each trial subject or his or her legal representative and must monitor the study until completed. Each new clinical protocol must be submitted to the IND for FDA review, and to the IRBs for approval. Protocols detail, among other things, the objectives of the study, dosing procedures, subject selection and exclusion criteria, and the parameters to be used to monitor subject safety.
81
Human clinical trials are typically conducted in three sequential phases that may overlap or be combined:
| | • | | Phase 1: The drug is initially introduced into healthy human subjects and tested for safety, dosage tolerance, absorption, metabolism, distribution and excretion. In the case of some products for severe or life-threatening diseases, especially when the product may be too inherently toxic to ethically administer to healthy volunteers, the initial human testing may be conducted in patients. |
| | • | | Phase 2: Involves studies in a limited patient population to identify possible adverse effects and safety risks, to preliminarily evaluate the efficacy of the product for specific targeted diseases and to determine dosage tolerance and optimal dosage. |
| | • | | Phase 3: Clinical trials are undertaken to further evaluate dosage, clinical efficacy and safety in an expanded patient population at geographically dispersed clinical study sites. These studies are intended to establish the overall risk-benefit ratio of the product and provide, if appropriate, an adequate basis for product labeling, if the product is approved for marketing. |
Progress reports detailing the results of the clinical trials must be submitted at least annually to the FDA and safety reports must be submitted to the FDA and the investigators for serious and unexpected adverse events. Phase 1, Phase 2, and Phase 3 testing may not be completed successfully within any specified period, if at all. The FDA or the sponsor may suspend a clinical trial at any time on various grounds, including a finding that the research subjects or patients are being exposed to an unacceptable health risk. Similarly, an IRB can suspend or terminate approval of a clinical trial at its site if, among other things, the clinical trial is not being conducted in accordance with the IRB’s requirements or if the drug has been associated with unexpected serious harm to patients.
Concurrent with clinical trials, companies usually complete additional animal studies and must also develop additional information about the chemistry and physical characteristics of the drug and finalize a process for manufacturing the product in commercial quantities in accordance with cGMP requirements. The manufacturing process must be capable of consistently producing quality batches of the product candidate and, among other things, the manufacturer must develop methods for testing the identity, strength, quality and purity of the final product. Additionally, appropriate packaging must be selected and tested, and stability studies must be conducted to demonstrate that the product candidate does not undergo unacceptable deterioration over its shelf life.
The results of product development, preclinical studies and clinical trials, along with descriptions of the manufacturing process, analytical tests conducted on the chemistry of the drug, proposed labeling, and other relevant information are submitted to the FDA as part of a BLA requesting approval to market the product for one or more indications. The FDA reviews a BLA to determine, among other things, whether a product is sufficiently safe and effective for its intended use and whether its manufacturing is cGMP-compliant. The submission of a BLA is subject to the payment of user fees; a waiver of such fees may be obtained under certain limited circumstances.
The FDA reviews BLAs submitted to ensure that they are sufficiently complete for substantive review before it accepts them for filing. If not, the FDA will issue a refusal to file letter and may request additional information rather than accept a BLA for filing. In this event, the BLA must be resubmitted with the additional information. The resubmitted application also is subject to review before the FDA accepts it for filing. Once the submission is accepted for filing, the FDA begins an in-depth substantive review. The review process is often significantly extended by FDA requests for additional information or clarification. The FDA may refer the BLA to an advisory committee for review, evaluation and recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendation of an advisory committee, but it often follows such recommendations. The approval process is lengthy and difficult and the FDA may refuse to approve a BLA if the applicable regulatory criteria are not satisfied or may require additional clinical or other data and information. Even if such data and information is submitted, the FDA may ultimately decide that the BLA does not satisfy the criteria for approval. Data obtained from clinical trials are not always conclusive and the FDA may interpret data differently than we interpret the same data. The FDA may issue an approvable
82
letter, which may require additional clinical or other data or impose other conditions that must be met in order to secure final approval of the BLA. Before approving a BLA, the FDA will inspect the facility or facilities where the product is manufactured and must find them to be acceptable under cGMP requirements.
If a product receives regulatory approval, the approval may be significantly limited to specific diseases and dosages or the indications for use may otherwise be limited, which could restrict the commercial value of the product. In addition, the FDA may require us to conduct Phase 4 testing which involves clinical trials designed to further assess a drug’s safety and effectiveness after BLA approval, and may require testing and surveillance programs to monitor the safety of approved products which have been commercialized or impose other restrictions on marketing or distribution.
Combination Product Regulation
Products that are a combination of more than one jurisdictional product type, for example, biologic/medical device combinations that are integrated as a whole or combined by copackaging or colabeling, are typically regulated as a combination product and may be subject to review by more than one center at the FDA. The FDA determines the primary mode of action of the product and assigns the primary responsibility for review of that product to the center at the FDA that regulates that type of product. The FDA has preliminarily advised us that the primary mode of action of Locteron will be related to its status as a therapeutic biologic, so the FDA’s Center for Drug Evaluation and Research, or CDER, will be the lead center.
Post-Approval Requirements
Both before and after FDA approval, we and our partners are subject to continuing regulation by the FDA, including, among other things, record-keeping requirements, reporting of adverse experiences with the drug, providing the FDA with updated safety and efficacy information, drug sampling and distribution requirements, complying with cGMP, and complying with FDA promotion and advertising requirements. The FDA strictly regulates labeling, advertising, promotion and other types of information on products that are placed on the market. Drugs may be promoted only for the approved indications and in accordance with the provisions of the approved label. Quality control and manufacturing practices must conform to cGMP and FDA requirements. Additionally, certain state agencies periodically inspect manufacturing facilities to evaluate compliance with cGMP and other requirements.
Later discovery of previously unknown problems with a product may result in restrictions on the product or even complete withdrawal of the product from the market. After approval, some types of changes to the approved product, such as adding new indications, manufacturing changes and additional labeling claims, are subject to further FDA review and approval.
From time to time, legislation is drafted, introduced and passed in Congress that could significantly change the statutory provisions governing the approval, manufacturing and marketing of products regulated by the FDA. In addition, FDA regulations and guidance are often revised or reinterpreted by the agency in ways that may significantly affect our business and our products. It is impossible to predict whether legislative changes will be enacted, or whether FDA regulations, guidance or interpretations will have changed or what the impact of such changes, if any, may be.
Foreign Regulation
In addition to regulations in the United States, we will be subject to a variety of foreign regulations governing clinical trials and commercial sales and distribution of our products. Whether or not we obtain FDA approval for a product, we must obtain approval of a product by the comparable regulatory authorities of foreign countries before we can commence clinical trials or marketing of the product in those countries. The approval process varies from country to country and the time may be longer or shorter than that required for FDA
83
approval. The requirements governing the conduct of clinical trials, product licensing, pricing and reimbursement vary greatly from country to country.
Under European Union, or EU, regulatory systems, we must submit a marketing authorization application and receive approval before we can market our products in the EU. If we plan to perform clinical trials in any EU Member State, we must submit a clinical trial application, or CTA, in the country in which we intend to perform the trial. While the informational requirements are set forth in an EU Directive that applies to all Member States, some Member States may have additional requirements. We may not begin the trial until the CTA has been accepted without conditions by the responsible authority in the relevant country. Before submitting the CTA request, we must also obtain a EudraCT number which is a unique reference that must be included in all CTA requests and a favorable opinion from an ethics committee.
Reimbursement
Sales of pharmaceutical products depend in significant part on the availability of third-party reimbursement. Third-party payors include government health administrative authorities, managed care providers, private health insurers and other organizations. We anticipate third-party payors will provide reimbursement for our products. However, these third-party payors are increasingly challenging the price and examining the cost-effectiveness of medical products and services. In addition, significant uncertainty exists as to the reimbursement status of newly approved healthcare products. We may need to conduct expensive pharmacoeconomic studies in order to demonstrate the cost-effectiveness of our products. Our product candidates may not be considered cost-effective. It is time consuming and expensive for us to seek reimbursement from third-party payors. Reimbursement may not be available or sufficient to allow us to sell our products on a competitive and profitable basis.
The passage of the Medicare Prescription Drug, Improvement, and Modernization Act of 2003, or the MMA, imposes new requirements for the distribution and pricing of prescription drugs for Medicare beneficiaries, and includes a major expansion of the prescription drug benefit under a new Medicare Part D. Medicare Part D went into effect on January 1, 2006. Under Part D, Medicare beneficiaries may enroll in prescription drug plans offered by private entities which will provide coverage of outpatient prescription drugs. Part D plans include both stand-alone prescription drug benefit plans and prescription drug coverage as a supplement to Medicare Advantage plans. Unlike Medicare Part A and B, Part D coverage is not standardized. Part D prescription drug plan sponsors are not required to pay for all covered Part D drugs, and each drug plan can develop its own drug formulary in which it indicates which drugs it will cover and at what tier or level. However, Part D prescription drug formularies must include drugs within each therapeutic category and class of covered Part D drugs, though not necessarily all the drugs in each category or class. Any formulary used by a Part D prescription drug plan must be developed and reviewed by a pharmacy and therapeutic committee.
It is not clear what effect the MMA will have on the prices paid for currently approved drugs and the pricing options for new drugs approved after January 1, 2006. Government payment for some of the costs of prescription drugs may increase demand for products for which we receive marketing approval. However, any negotiated prices for our products covered by a Part D prescription drug plan will likely be lower than the prices we might otherwise obtain. Moreover, while the MMA applies only to drug benefits for Medicare beneficiaries, private payors often follow Medicare coverage policy and payment limitations in setting their own payment rates. Any reduction in payment that results from the MMA may result in a similar reduction in payments from non-governmental payors.
84
We expect that there will continue to be a number of federal and state proposals to implement governmental pricing controls and limit the growth of healthcare costs, including the cost of prescription drugs. At the present time, Medicare is prohibited from negotiating directly with pharmaceutical companies for drugs. However, Congress is currently considering passing legislation that would lift the ban on federal negotiations. While we cannot predict whether such legislative or regulatory proposals will be adopted, the adoption of such proposals could have a material adverse effect on our business, financial condition and profitability.
In addition, in some foreign countries, the proposed pricing for a drug must be approved before it may be lawfully marketed. The requirements governing drug pricing vary widely from country to country. For example, the European Union provides options for its member states to restrict the range of medicinal products for which their national health insurance systems provide reimbursement and to control the prices of medicinal products for human use. A member state may approve a specific price for the medicinal product or it may instead adopt a system of direct or indirect controls on the profitability of the company placing the medicinal product on the market. There can be no assurance that any country that has price controls or reimbursement limitations for pharmaceutical products will allow favorable reimbursement and pricing arrangements for any of our products.
Employees
As of June 30, 2007, we had 105 employees, including 18 who hold Ph.D. or M.D. degrees. We had 94 employees engaged in research and development, including manufacturing operations, and our remaining employees are management or administrative staff. None of our employees is subject to a collective bargaining agreement. We believe that we have good relations with our employees.
Property and Facilities
We lease approximately 40,000 square feet of laboratory, manufacturing and office space in Pittsboro, North Carolina under a lease agreement that terminates in August 2010. We also lease approximately 48,000 square feet in a building located in Research Triangle Park, North Carolina, that we intend to build out as manufacturing space. This lease expires in December 2016. We also lease temporary office space under smaller, short-term leases. We believe that our facilities adequately meet our present needs.
Legal Proceedings
We are not currently a party to any material legal proceedings.
85
MANAGEMENT
Executive Officers, Key Employees and Directors
The following table sets forth certain information concerning our executive officers, key employees and directors as of August 1, 2007:
| | | | |
Name | | Age | | Position |
Executive Officers and Key Employees | | | | |
Jan Turek | | 53 | | President and Chief Executive Officer and Director |
John Irick | | 60 | | Chief Business Officer and Senior Vice President |
Dale Sander | | 47 | | Chief Financial Officer and Senior Vice President, Finance |
David Spencer, Ph.D. | | 54 | | Chief Operating Officer and Senior Vice President, Research and Development |
Glen Williams | | 49 | | Senior Vice President, Operations |
| | |
Directors | | | | |
Chris Hegele(1)(2) | | 57 | | Chairman of the Board |
W. Thomas Amick(2) | | 64 | | Director |
David L. Castaldi(1) | | 67 | | Director |
Dennis Dougherty(1) | | 59 | | Director |
P. Sherrill Neff(2) | | 55 | | Director |
Sunny Sharma, M.D. | | 32 | | Director |
(1) | Member of our Audit Committee |
(2) | Member of our Compensation Committee |
(3) | Member of our Nominating and Corporate Governance Committee |
Jan Turekhas been our President and Chief Executive Officer and a member of our board of directors since June 2001. From 1996 to 2001, Mr. Turek served as Senior Vice President and General Manager for the Biological Products business unit of Bayer Corporation, a unit dedicated to therapeutic proteins. Mr. Turek has also held positions at Bristol Myers Squibb Co. and Sandoz Inc. Mr. Turek currently serves on the board of directors of Southeast BIO and is Co-Chair of the Emerging Companies Forum of the North Carolina Biosciences Organization. Mr. Turek received his B.S. in Biochemistry from McGill University in Montreal, Canada.
John Irickhas served as our Chief Business Officer and Senior Vice President since August 2004 and prior to that served as our Senior Vice President, Corporate and Business Development since December 2001. From 1998 to 2001, Mr. Irick was Executive Vice President and Director of Merix Bioscience, Inc., an immunotherapy company which he co-founded. Mr. Irick is a Director and Chairman Emeritus of the North Carolina Biosciences Organization and serves on the board of directors of the North Carolina Biotechnology Center. Mr. Irick received his A.B. in Mathematical Economics from Brown University and his M.B.A. from The University of Chicago.
Dale Sander has served as our Chief Financial Officer and Senior Vice President, Finance since January 2005. From October 1996 to April 2004, Mr. Sander served as Chief Financial Officer and Vice President of Corporate Development of Maxim Pharmaceuticals, Inc., a public biopharmaceutical company. From 1993 to 1996, Mr. Sander held Chief Financial Officer positions at Xytronyx, Inc., a public biotechnology company, and Tactyl Technologies, Inc., a medical device manufacturer. Mr. Sander is a Certified Public Accountant and was a Senior Manager with Ernst & Young LLP. He received his B.S. in Business Administration from San Diego State University.
86
David Spencer, Ph.D. has served as our Chief Operating Officer since October 2001 and has served as our Senior Vice President, Research and Development since August 2004. From October 2001 through August 2004, he served as our Vice President of Research and Development. From 1983 to 2001, Dr. Spencer held several positions of increasing responsibility, including Vice President, Respiratory/Critical Care Life Cycle Management in the Biological Products Business Unit at Bayer Corporation. Dr. Spencer received his B.A. in Psychobiology from the University of California at Los Angeles, his M.A. in Physiological Psychology from California State University at Fullerton, and his Ph.D. in Physiological Psychology from Indiana University.
Glen Williamshas served as our Senior Vice President, Operations since November 2005. From January 1996 to October 2005, Mr. Williams served as Vice President of Manufacturing and General Manager for Biogen Idec, Inc’s Research Triangle Park facilities in North Carolina. Mr. Williams received his B.S. in Mechanical Engineering from The Ohio State University and his M.B.A. from Central Michigan University.
Chris Hegele has been a member of our board of directors and has served as Chairman since October 1998. Mr. Hegele has served as a General Partner with Intersouth Partners, a venture capital firm, since April 2003. Mr. Hegele has also served as a General Partner with Kitty Hawk Capital, Inc., a venture capital firm, since June 1984. Mr. Hegele received his B.S. in Economics from the University of North Carolina at Wilmington and his M.B.A. from the University of North Carolina at Chapel Hill.
W. Thomas Amickhas been a member of our board of directors since March 2004. Mr. Amick currently serves as Chairman of the board of Aldagen, Inc., a privately held biotech company. From February 1974 to February 2004, Mr. Amick served in a series of positions with Johnson & Johnson, including Vice President, Business Development of the Johnson & Johnson Development Corporation, President of Ortho Biotech Europe, President of Janssen-Ortho, and Vice President of the Oncology Franchise for Ortho Biotech Products, L.P. Mr. Amick currently serves as a member of the advisory board for Intersouth Partners and Quaker BioVentures and is on the boards of directors of Argolyn Pharmaceuticals and Transport Pharmaceuticals and is Chairman of the board of Discovery Laboratories, Inc. Mr. Amick received his B.S. in Business Administration from Elon University.
David L. Castaldi has been a member of our board of directors since May 2004. Mr. Castaldi has worked as an independent consultant for the biotechnology and pharmaceutical industry since December 1994. Mr. Castaldi currently serves as a director of Nabi Biopharmaceuticals. Mr. Castaldi received his B.A. in Business from the University of Notre Dame and his M.B.A. from the Harvard Business School.
Dennis Doughertyhas been a member of our board of directors since October 1998. Mr. Dougherty is the founding general partner of Intersouth Partners, a venture capital firm, which was founded in 1985. Mr. Dougherty earned his B.A. in Business from Oklahoma City University.
P. Sherrill Neff has been a member of our board of directors since March 2004. Mr. Neff is a founding partner of and has served as a managing partner of Quaker BioVentures, a venture capital firm, since 2002. From 1994 to 2002, Mr. Neff was President, Chief Operating Officer, and a director of Neose Technologies, Inc., a publicly traded life sciences company. Mr. Neff currently serves as a director of Resource Capital Corp. and Amicus Therapeutics, Inc. Mr. Neff received his B.A. in Religion from Wesleyan University and his J.D. from the University of Michigan Law School.
Sunny Sharma, M.D.has been a member of our board of directors since May 2007. Dr. Sharma currently serves as a Vice President of Investor Growth Capital, Inc., an investment advisory service company, where he has been employed since since April 2006. Previously, Dr. Sharma served as a Managing Director of Easton Hunt Capital Partners, a venture capital fund, where he held several positions of increasing responsibility from October 2002 to April 2006. Dr. Sharma received his M.B.B.S. from the Christian Medical College and Hospital and his M.B.A. from IIM Bangalor.
Development Advisory Board
As we moved our products from research into development, we established a development advisory board to provide us with outside industry expertise. Our development advisory board includes experts in protein
87
manufacturing, process development, clinical development and regulatory affairs and consists of the following individuals:
| | |
Name | | Expertise |
| |
Carl E. Brooks | | Process development, manufacturing, general management |
| |
Richard V. McCloskey, M.D. | | Clinical development |
| |
James E. Pennington, M.D. | | Clinical development |
| |
Kathryn E Stein, Ph.D. | | Regulatory affairs |
| |
Mark F. Witcher, Ph.D. | | Process development, manufacturing |
Board of Directors
Board Composition
Our amended and restated certificate of incorporation and restated bylaws provide that the authorized number of directors may be changed only by resolution of our board of directors. Seven directors are currently authorized.
In accordance with our amended and restated certificate of incorporation, immediately upon the closing of this offering, our board of directors will be divided into three classes with staggered three-year terms. At each annual meeting of stockholders following the offering, the successors to the directors whose terms then expire will be elected to serve until the third annual meeting following the election. At the closing of this offering, our directors will be divided among the three classes as follows:
| | • | | The Class I directors will be , and their terms will expire at the annual meeting of stockholders to be held in 2008; |
| | • | | The Class II directors will be , and their terms will expire at the annual meeting of stockholders to be held in 2009; and |
| | • | | The Class III directors will be , and their terms will expire at the annual meeting of stockholders to be held in 2010. |
Any additional directorships resulting from an increase in the number of directors will be distributed among the three classes so that, as nearly as possible, each class will consist of one-third of the directors.
Director Independence
Our board of directors has reviewed the materiality of any relationship that each of our directors has with Biolex, either directly or indirectly. Based on this review, our board has determined that Messrs. Hegele, Amick, Castaldi, Dougherty, and Neff and Dr. Sharma are “independent directors” as defined by NASDAQ.
Committees of our Board of Directors
Our board of directors has an audit committee, a compensation committee and a nominating and governance committee, each of which has the composition and responsibilities described below.
Audit Committee
Our audit committee is currently comprised of Messrs. Hegele, Castaldi and Dougherty. qualifies as an “audit committee financial expert” for purposes of the Securities Exchange Act of 1934, as amended, or the Exchange Act. Under the applicable NASDAQ rules, a company listing in connection with its initial public offering is permitted to phase in its compliance with the independent audit committee requirements
88
set forth in Marketplace Rule 4350(d)(2) on the same schedule as it is permitted to phase in its compliance with the independent audit committee requirement pursuant to Rule 10A-3 under the Exchange Act. Pursuant to Rule 10A-3, a newly listed company must have (1) one independent member at the time of listing; (2) a majority of independent members within 90 days of listing; and (3) all independent members within one year of listing. Mr. Castaldi is currently the only member of the audit committee who qualifies as independent under Rule 10A-3. Within 90 days of our listing on The NASDAQ Global Market, we expect that either Mr. Hegele or Mr. Dougherty will resign from our audit committee and be replaced with a new director that is independent under Rule 10A-3 and within one year of our listing, we expect that the remaining non-independent director will resign from our audit committee and be replaced with a new director that is independent. Our audit committee is authorized to:
| | • | | approve and retain the independent auditors to conduct the annual audit of our books and records; |
| | • | | review the proposed scope and results of the audit; |
| | • | | review and pre-approve the independent auditor’s audit and non-audit services rendered; |
| | • | | approve the audit fees to be paid; |
| | • | | review accounting and financial controls with the independent auditors and our financial and accounting staff; |
| | • | | review and approve transactions between us and our directors, officers and affiliates; |
| | • | | recognize and prevent prohibited non-audit services; |
| | • | | establish procedures for complaints received by us regarding accounting matters; |
| | • | | oversee internal audit functions; and |
| | • | | prepare the report of the audit committee that SEC rules require to be included in our annual meeting proxy statement. |
Compensation Committee
Our compensation committee is currently comprised of Messrs. Hegele, Amick and Neff and is authorized to:
| | • | | review and recommend the compensation arrangements for management, including the compensation for our President and Chief Executive Officer; |
| | • | | establish and review general compensation policies with the objective to attract and retain superior talent, to reward individual performance and to achieve our financial goals; |
| | • | | administer our stock incentive plan; |
| | • | | review the Compensation Discussion and Analysis, or CD&A, prepared by management, discuss the CD&A with management and, based on such review and discussions, recommend to our board of directors that the CD&A be included in our Annual Report on Form 10-K, proxy statement, or any other applicable filing as required by the SEC; and |
| | • | | prepare the report of the compensation committee that SEC rules require to be included in our annual meeting proxy statement. |
Nominating and Governance Committee
Our nominating and governance committee is currently comprised of and is authorized to:
| | • | | identify and nominate members of our board of directors; |
89
| | • | | develop and recommend to our board of directors a set of corporate governance principles applicable to our company; and |
| | • | | oversee the evaluation of our board of directors and management. |
Compensation Committee Interlocks and Insider Participation
Our compensation committee is currently comprised of Messrs. Hegele, Amick and Neff. No member of our compensation committee has at any time been an employee of ours. None of our executive officers serve as a member of our board of directors or compensation committee of any entity that has one or more executive officers serving as a member of our board of directors or compensation committee.
Each of Mr. Hegele and Mr. Neff and affiliates of theirs have participated in transactions with us. For a detailed description of these transactions, see “Certain Relationships and Related Person Transactions.”
90
EXECUTIVE COMPENSATION
Compensation Discussion and Analysis
Objectives and Philosophy of Executive Compensation
The primary objective of our compensation program, as established by the compensation committee of our board of directors, is to attract, retain and motivate the best possible executive talent. Our overall philosophy is to link a combination of short and long-term cash and equity incentives to the achievement by our executives of measurable corporate and individual performance objectives. A related objective is to align the incentives provided to our executives with the creation of long-term value for our stockholders. The role of the compensation committee is to oversee our compensation and benefit plans and policies, administer our equity incentive plans, and review and approve annually all compensation decisions relating to all executive officers. Specifically, our compensation programs are designed to:
| | • | | attract and retain executives of superior ability, industry experience and managerial skills; |
| | • | | ensure executive compensation is aligned with our corporate strategies, business objectives and the long-term interests of our stockholders; |
| | • | | increase the likelihood of achieving key strategic, timeline and financial performance measures by linking the incentive award opportunities provided to our executives to their achievement of performance goals in these areas; and |
| | • | | use long-term incentives to maximize stockholder value, as well as promote retention of key individuals, by providing a portion of total compensation opportunities for executives in the form of direct ownership in our company. |
To achieve the above objectives, the compensation committee strives to implement and maintain compensation plans that tie a substantial portion of overall compensation for each executive to the achievement of corporate and individual performance objectives. Increases in base salaries and performance bonus payments are tied primarily to these corporate and individual objectives, while the size of equity awards are primarily tied to a combination of corporate and individual objectives and those levels determined to be appropriate to promote long-term retention.
Corporate and Individual Objectives
Corporate objectives are established at the beginning of each year and are the basis for evaluating overall corporate performance for the year. Individual objectives for each executive officer are also established at the beginning of each year by our Chief Executive Officer and are approved by the compensation committee. These objectives represent significant milestones that must be met by each executive officer, along with dates for achieving the milestones. Objectives are established based on the executive officer’s principal areas of responsibility.
Performance Evaluations
After the completion of each fiscal year, we evaluate individual and corporate performance against stated goals for the year. Consistent with our overall compensation philosophy, each executive undergoes an annual performance evaluation that includes input from each of them as to his or her own performance. The performance of our Chief Executive Officer is evaluated by our compensation committee with opportunity for input from each member of our board of directors. The performance of our other executive officers is evaluated by the chief executive officer and then reviewed by the compensation committee. This process leads to a recommendation for annual salary increases, bonuses and equity awards, if any, which are then reviewed and approved by our compensation committee. Any annual base salary increases, bonuses, and equity awards, to the extent granted, are generally implemented during the first half of the following year.
91
The compensation committee evaluates individual executive performance with the goal of setting compensation at levels the committee believes are competitive for executives in companies of similar size and stage of development operating in the biotechnology industry, taking into account our relative performance and our own strategic goals. In order to ensure that we continue to remunerate our executives appropriately and consistent with market information, we will participate in, and review data from, certain compensation surveys, and may in the future confer with outside compensation consultants.
Elements of Executive Compensation
Executive compensation consists of the following elements:
Base Salary
Base salaries for our executives are established by the compensation committee based on the scope of their responsibilities, taking into account competitive market compensation paid by other companies for similar positions using data from the Radford Biotechnology Survey, an analysis of compensation in private and public biotechnology companies, as a primary reference point. As with total executive compensation, we believe that our executive base salaries should be targeted in the range of salaries for executives in similar positions and with similar responsibilities in comparable biotechnology companies. In general, base salaries are reviewed annually and adjusted to realign salaries with market levels and to reflect the performance evaluation of the executive.
Annual Performance Bonus
The compensation committee has the authority to award annual performance bonuses to our executives. Bonuses are determined by two factors, corporate performance and individual performance. Each of our executive officers is eligible to receive an annual performance bonus based upon a targeted percentage of base salary. The targeted bonus level as a percentage of base salary is 30% for the chief executive officer and 25% for the other executive officers.
If the company or the executive officer exceeds the objectives established at the beginning of the year, or if our performance or the performance of the executive officer is extraordinary, then the bonus payable to the executive officer could exceed the targeted percentage of base salary. If the company’s performance or the executive officer’s performance does not completely meet each of the objectives established for the year, then the bonus payable to the executive officer will be less than the targeted percentage. Performance levels and bonuses are at the sole discretion of the compensation committee.
At the beginning of 2006, the following corporate objectives were established:
| | • | | initiation and advancement of Locteron Phase 2a clinical trial; |
| | • | | expansion of corporate partnerships; and |
| | • | | advancement of preclinical development of BLX-155. |
Individual objectives were also established for each executive officer, with the objectives for Mr. Turek paralleling the overall corporate objectives. The objectives for Dr. Spencer related to the support of the overall corporate objectives with emphasis on the management of our efforts related to the Locteron Phase 2a trial and the preclinical development of BLX-155. The objectives for Mr. Irick related to the support of the overall corporate objectives with emphasis on the management of our efforts to expand our corporate partnerships. The objectives for Mr. Sander related to the support of the overall corporate objectives and management of our fund raising activities.
In April 2007, our compensation committee awarded bonuses to the executive officers based upon the achievement of certain corporate objectives, including (i) the initiation of the Locteron Phase 2a clinical study,
92
(ii) completion of additional early stage collaborations, (iii) advancement of preclinical development of BLX-155, including the production of the product candidate at commercially viable levels, and (iv) successful demonstration of the ability of the LEX System to optimize the glycosylation structures of monoclonal antibodies, including publication of the results inNature Biotechnology. Based on the compensation committee’s assessment of the extent to which each of the aforementioned objectives were achieved, and each executive officer’s achievement of his individual objectives, bonuses were awarded to the executive officers in amounts equal to 68% of targeted bonus levels for Mr. Turek and Dr. Spencer and 65% of targeted bonus levels for Mr. Sander and Mr. Irick.
Stock Options—Long-Term Incentive Program
We believe that long-term performance will be enhanced through equity awards that reward our executives and other employees for maximizing stockholder value over time and that align the interests of our executives and other employees with those of stockholders. The compensation committee believes that the use of equity awards offers the best approach to achieving our compensation goals because equity ownership ties a significant portion of an individual’s compensation to the performance of our stock. We have historically elected to use stock options as our long-term equity incentive vehicle.
Our 2003 Equity Incentive Plan, or the 2003 Plan, authorized us to grant options to purchase shares of common stock to our employees, directors and consultants. Our compensation committee oversees the administration of our stock options. Stock option grants are made at the commencement of employment and additional grants are considered as part of our annual performance evaluation process. We have historically made option grants on a company wide basis and expect to make company wide grants in the future. Option grants to the broad employee base, including those associated with annual performance evaluations, are made at the discretion of the compensation committee with the input from our Chief Executive Officer and other members of management.
The exercise price of our options is the fair market value of our common stock as determined by our board of directors on the date of grant. Our stock options typically vest over a four-year period with 25% vesting on the anniversary of the date of grant and 6.25% vesting every quarter thereafter, and generally expire ten years after the date of grant.
The compensation committee considers and approves stock option awards to executive officers based upon a review of competitive compensation data, an assessment of corporate and individual performance, and retention considerations. In September 2005, stock options were granted to each of our executive officers in conjunction with the completion of our Series BB preferred stock financing so as to maintain their potential ownership interests in the company. As a result, our compensation committee determined that an additional grant of stock options to our named executive officers during 2006 was not necessary to further the objectives of our compensation program. The compensation committee did, however, establish target levels of grants to be considered in 2007 for each of the named executive officers based on achievement of the 2006 corporate objectives outlined above. In April 2007, our compensation committee approved the award of stock option grants in May 2007 to our executive officers based upon the achievement of certain corporate objectives. The grants included options to purchase 215,000 shares for Mr. Turek, 80,000 shares for Mr. Sander, 175,000 shares for Dr. Spencer, and 120,000 shares for Mr. Irick.
We expect to continue to use stock options as a long-term incentive vehicle because we believe that:
| | • | | stock options and the related vesting provisions attract and retain executives; |
| | • | | stock options are inherently performance based, as all of the value received by the recipient of a stock option is based on the growth of our stock price, stock options enhance the individual’s incentive to increase our stock price and maximize stockholder value; and |
93
| | • | | stock options help to provide a balance to our overall executive compensation program, as base salary and our annual performance bonus program focus on short-term compensation, while stock options reward executives for increases in stockholder value over the longer term. |
Termination Based Change of Control Compensation
Upon termination of employment under certain circumstances, our executive officers are entitled to receive varying types of compensation. Elements of this compensation include payments based upon a number of months of base salary, bonus amounts, acceleration of vesting of stock options, and health and other similar benefits. We also have granted severance and acceleration of vesting of equity benefits to our executives in the event of a change of control if the executive is terminated within a certain period of time of the change of control. The specific provisions of each executive officer’s arrangement are described in further detail below. We believe that our termination-based compensation and acceleration of vesting of equity arrangements are in line with severance packages offered to executive officers of other similar companies, based upon the market information available.
Relationship of Elements of Compensation
Our compensation structure is primarily weighted toward base salary, annual performance bonus, and stock options. We utilize stock options as a substantial component of compensation because we currently have no revenue or earnings and expect this to be the case for the foreseeable future. Our mix of cash and non-cash compensation balances our need to limit cash expenditures with the expectations of those we hope to recruit and retain as employees. In the future, we may adjust the mix of cash and non-cash compensation if required by competitive market conditions for attracting and retaining skilled personnel.
We manage the expected impact of salary increases and performance bonuses by requiring that the size of such salary increases and bonuses be tied to the attainment of corporate and individual objectives. For example, the size of each executive’s bonus is determined not only by individual performance, but also by whether we have met our corporate objectives.
Employment Agreements
On March 8, 2007, we entered into employment agreements with our executive officers. The agreements remain in effect until terminated by either party. Pursuant to these agreements, each executive officer receives a base salary, which is reviewed annually and may be adjusted at the discretion of the compensation committee. The employment agreements provide for, among other things, eligibility to receive annual performance-based merit bonuses for fully meeting and exceeding expectations, payable at the discretion of the compensation committee of our board of directors based on the achievement of certain performance milestones set by the compensation committee each fiscal year. Each executive officer is eligible to participate in our employee benefit plans, to the extent he is eligible for those plans, on the same terms as other employees. Pursuant to these agreements, the annual base salary and target bonus percentage for Messrs. Turek, Sander, Spencer and Irick was $318,000 and 30%, $243,800 and 25%, $265,000 and 25% and $254,400 and 25%, respectively. On April 26, 2007, our compensation committee increased the annual base salary, effective April 1, 2007, for Messrs. Turek, Sander, Spencer and Irick to $340,260, $258,428, $283,550 and $269,664, respectively.
In each of these agreements, if employment is terminated by reason of the executive’s death or disability, the executive, his estate or his personal representative is entitled to severance compensation of six months base salary. If we terminate the executive without cause, he is entitled to receive six months as severance compensation and continuation of benefits for six months. Additionally, following a change of control, if the executive terminates employment for good reason or if we terminate his employment without cause, he is entitled to receive six months as severance compensation, a prorated bonus, continuation of benefits for six months and a
94
lump sum payment of any amounts contributed to a pension or retirement plan which the executive is entitled to under the terms of any such plan through the date of termination. The definition of change of control as set forth in the agreements includes a sale, transfer or other disposition of all or substantially all of our assets to another person or entity, a merger or consolidation into another person or entity, other than the merger of one of our subsidiaries into us or the merger of us into a subsidiary or a recapitalization, stock sale or other transaction or series of transactions if, after such transaction or series of transactions, our stockholders immediately before such transaction or the first in such series of transactions own less than 50% of us or the surviving entity, other than an equity financing transaction approved by a majority of the members of our board of directors. The definition of good reason as set forth in the agreements includes an adverse change in the executive’s title, authority or responsibility, a reduction in salary, relocation, or a bankruptcy filing by us. The definition of cause as set forth in the agreements includes the executive’s gross misconduct that is injurious to us, dishonesty, fraud or misrepresentation in connection with his job responsibilities and designed to enrich the executive, material violation of laws applicable to us, or his knowing failure or refusal to substantially perform his duties.
Summary Compensation Table
The following table sets forth certain information with respect to compensation for the year ended December 31, 2006 earned or paid to our Chief Executive Officer and our Chief Financial Officer, and to our other two executive officers. We refer to these individuals as our named executive officers.
| | | | | | | | | | | | | | | | | |
Name and Principal Position | | Year | | Salary($) | | Bonus(1) | | Option Awards($)(2) | | | All Other Compensation | | Total($) |
Jan P. Turek | | 2006 | | $ | 313,500 | | $ | 65,254 | | $ | 25,455 | (3) | | — | | $ | 404,209 |
President and Chief Executive Officer | | | | | | | | | | | | | | | | | |
Dale A. Sander | | 2006 | | $ | 240,350 | | $ | 39,496 | | $ | 6,792 | (3) | | — | | $ | 286,638 |
Chief Financial Officer and Senior Vice President, Finance | | | | | | | | | | | | | | | | | |
David G. Spencer, Ph.D. | | 2006 | | $ | 261,250 | | $ | 45,315 | | $ | 8,485 | (3) | | — | | $ | 315,050 |
Chief Operating Officer and Senior Vice President, Research and Development | | | | | | | | | | | | | | | | | |
John A. Irick | | 2006 | | $ | 250,800 | | $ | 41,213 | | $ | 8,485 | (3) | | — | | $ | 300,498 |
Chief Business Officer and Senior Vice President | | | | | | | | | | | | | | | | | |
(1) | Amounts are for performance during the year ended December, 31, 2006 and were paid in May 2007. These amounts represent bonuses earned upon achievement of objectives established by our compensation committee. |
(2) | See Note 10 to our consolidated financial statements included elsewhere in this prospectus for details as to the assumptions used to determine the fair value of the option awards. See also our discussion of stock-based compensation under “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Stock-Based Compensation.” |
(3) | Consists of the compensation expense incurred by us in fiscal year 2006, calculated in accordance with SFAS 123R. |
As of December 31, 2006, we did not have formal employment agreements with any of our named executive officers, however certain elements of the executive officers’ compensation and other employment arrangements are set forth in letter agreements that we executed with each of them at the time their employment with us commenced. The letter agreements provide for, among other things, the executive officer’s initial annual base salary, eligibility to receive annual performance-based bonuses in the 20%-30% range for fully meeting and exceeding expectations, and an initial stock option award. As a condition to their employment, each executive officer has also entered into a non-disclosure, inventions, and non-competition agreement pursuant to which each officer has agreed not to compete with us or to solicit customers or employees of ours for a period of 12 months after the termination of employment. Since the date of the letter agreements, the compensation paid to each of these executive officers has been increased, additional equity awards have been awarded and the amount of bonuses has increased. As of December 31, 2006, base salaries for Messrs. Turek, Sander, Spencer and Irick were $318,000, $243,800, $265,000 and $254,400, respectively. On March 8, 2007, we entered into employment
95
agreements with our named executive officers. See “Compensation Discussion and Analysis—Employment Agreements” for a discussion of these new agreements.
All of the stock option awards issued under the 2003 Plan were granted with an exercise price per share equal to the fair market value of our common stock on the date of grant, as determined by our board of directors. Subject to the specific terms of the option agreements, all of the options granted under the 2003 Plan vest as to 25% of the shares on the first anniversary date of the grant and as to 6.25% of the shares per three month period thereafter. Pursuant to option agreements with all of our employees, if, in connection with or within 12 months of a change of control, we terminate an option holder’s employment without “cause” or the option holder resigns for “good reason” (in each case as defined in the applicable option agreement), any unvested portion of the option shall immediately be fully vested and exercisable. See “—Potential Payments Upon Termination or Change of Control.”
Grants of Plan-Based Awards
No equity or non-equity incentive plan awards were made to our named executive officers during the fiscal year ended December 31, 2006.
Outstanding Equity Awards at 2006 Fiscal Year End
The following table presents the outstanding equity awards held by each of our named executive officers as of December 31, 2006.
| | | | | | | | | | |
Name and Principal Position | | Number of
Securities
Underlying
Unexercised
Options (#)
Exercisable | | Number of
Securities
Underlying
Unexercised
Options (#)
Unexercisable | | | Option
Exercise
Price ($) | | Option
Expiration
Date |
Jan P. Turek | | 1,045,145 | | 211,948 | (1) | | $ | 0.10 | | 8/8/2013 |
President and Chief Executive Officer | | 982,144 | | 1,022,638 | (2) | | | 0.22 | | 9/14/2015 |
| | | | |
Dale A. Sander | | 200,272 | | 120,164 | (3) | | | 0.20 | | 1/24/2015 |
Chief Financial Officer and Senior Vice President, Finance | | 176,935 | | 227,490 | (2) | | | 0.22 | | 9/14/2015 |
| | | | |
David G. Spencer, Ph.D. | | 415,609 | | 20,441 | (1) | | | 0.10 | | 8/8/2013 |
Chief Operating Officer and Senior Vice President, Research and Development | | 284,917 | | 366,325 | (2) | | | 0.22 | | 9/14/2015 |
| | | | |
John A. Irick | | 415,609 | | 20,441 | (1) | | | 0.10 | | 8/8/2013 |
Chief Business Officer and Senior Vice President | | 284,917 | | 366,325 | (2) | | | 0.22 | | 9/14/2015 |
(1) | The options vested as to 25% of the shares on August 8, 2004 and as to an additional 6.25% at the end of each three month period thereafter. |
(2) | The options vested as to 25% of the shares on September 14, 2006 and as to an additional 6.25% at the end of each three month period thereafter. |
(3) | The options vested as to 25% of the shares on January 24, 2006 and as to an additional 6.25% at the end of each three month period thereafter. |
Option Exercises and Stock Vested
There were no exercises of stock options or vesting of shares of restricted stock held by our named executive officers during the fiscal year ended December 31, 2006.
96
Pension Benefits
We do not have any qualified or non-qualified defined benefit plans.
Nonqualified Deferred Compensation
We do not have any non-qualified defined contribution plans or other deferred compensation plans.
Potential Payments Upon Termination or Change of Control
The employment arrangements with our named executive officers that were in effect as of December 31, 2006 provided certain benefits upon the termination of employment. Furthermore, pursuant to the terms of the 2003 Plan, the vesting of shares subject to purchase under stock options held by our named executive officers may accelerate upon a change of control. A change of control includes (1) a sale, transfer or other disposition of all or substantially all of our assets to another person or entity, (2) a merger or consolidation into another person or entity, other than the merger of one of our subsidiaries into us or the merger of us into one of our subsidiaries, or (3) a recapitalization, stock sale or other transaction or series of transactions if, after such transaction or series of transactions, our stockholders immediately before such transaction or the first in such series of transactions own less than 50% of us or the surviving entity, other than an equity financing transaction approved by a majority of our board of directors. In the event of a change of control in which the consideration received by our stockholders is all or substantially all cash, then all unvested shares underlying the stock options held by our named executive officers shall immediately vest and become exercisable. In the event of a change of control in which the consideration received by our stockholders is all or substantially all capital stock of the acquiring or successor company or its parent, then 50% of all unvested stock options held by our named executive officers shall immediately vest and become exercisable (in addition to any portion of the option that was previously vested and exercisable), and the remaining 50% shall continue to vest and become exercisable on the earlier of the date the option would otherwise have become fully vested pursuant to the original vesting term or 12 months from the effective date of the change of control.
Additionally, pursuant to option agreements with all of our executives, if, in connection with or within 12 months following a change of control, we or our successor terminates the executive without “cause” or the executive resigns for “good reason”, any unvested portion of the option shall immediately be fully vested and exercisable. The definition of cause as set forth in our option agreements includes (1) a willful act by the employee that constitutes gross misconduct and that is injurious to us, (2) a willful act of personal dishonesty, fraud or misrepresentation by the employee in connection with his responsibilities to us that is intended to result in substantial gain or personal enrichment at our expense, (3) a material and willful violation of a federal or state law or regulation applicable to our business or (4) the employee’s willful and continued failure to substantially perform his principal duties and obligations to us, which failure is not remedied in a reasonable period of time after receipt of written notice from us. The definition of good reason as set forth in the option agreements includes (1) the assignment or a reduction of duties, either of which results in a diminution of the employee’s title, position or responsibilities with us as in effect immediately prior to such assignment or reduction, (2) a reduction in the employee’s overall cash compensation or overall benefits package or (3) the relocation of the employee to a facility or a location more than 30 miles from his then present location.
The following sets forth, as of December 31, 2006, the payments that would have been made to each of our named executive officers in the event of their termination without cause or as a result of a change of control.
Jan P. Turek
Pursuant to a letter agreement dated June 19, 2001 between us and Mr. Turek, we agreed to employ Mr. Turek as Chief Executive Officer beginning on June 25, 2001. If we were to have terminated Mr. Turek’s employment without cause during the time this agreement was in effect, he would have been entitled to receive his base salary for four months, to be paid monthly, as severance compensation. The definition of cause as set
97
forth in the agreement with Mr. Turek includes (1) any material breach of the terms of any agreement with us by the executive officer, or failure of the executive officer to diligently and properly perform his duties for us, (2) any material failure by the executive officer to comply with the reasonable policies and/or directives of our board of directors, (3) any action by the executive officer that is illegal, (4) any failure by the executive officer to fully disclose any material conflict of interest he may have with us in a transaction between us and any third party which is detrimental to our interest and well-being or (5) any adverse act or omission by the executive officer which would be required to be disclosed pursuant to public securities laws or which would limit our ability or any entity affiliated with us to sell securities under any federal or state law or which would disqualify us or any affiliated entity from any exemption otherwise available to us. If Mr. Turek’s employment had been terminated without cause on December 31, 2006, his severance compensation would have been $106,000.
| | | | | | | | | |
Acceleration of Vesting of Options on Termination | | Change of Control With Cash as
Consideration | | Change of Control With
Acquiring
Company
Stock as
Consideration | | Termination Without Cause
or Resignation for Good Reason
After Change of Control |
Number of Option Shares | | | 1,234,586 | | | 617,293 | | | 1,234,586 |
Value Upon Termination(1) | | $ | | | $ | | | $ | |
(1) | There was no public market for our common stock on December 31, 2006. We have estimated the value of the unvested stock based on the assumed initial public offering price of $ per share, the midpoint of the range listed on the cover page of this prospectus. |
Dale A. Sander
Pursuant to a letter agreement dated January 12, 2005 between us and Mr. Sander, we agreed to employ Mr. Sander as Senior Vice President of Finance and Chief Financial Officer beginning on January 24, 2005. If we would have terminated Mr. Sander’s employment without cause or following a change of control during the time this agreement was in effect, he would have been entitled to receive his base salary and continuation of employee benefits for six months to be paid monthly as severance compensation. The agreement with Mr. Sander does not define cause.
| | | |
Executive Benefits Upon Termination | | Termination
Without Cause or
Following a
Change of Control |
Base Salary | | $ | 121,900 |
Continuation of Benefits(1) | | $ | 6,193 |
| | | | | | | | | |
Acceleration of Vesting of Options on Termination | | Change of
Control With Cash as
Consideration | | Change of Control With Acquiring
Company Stock as
Consideration | | Termination Without Cause or Resignation for Good Reason After Change of Control |
Number of Option Shares | | | 347,654 | | | 173,827 | | | 347,654 |
Value Upon Termination(2) | | $ | | | $ | | | $ | |
(1) | Includes continuation of standard employee benefits, including health insurance and dental insurance, for six months. |
(2) | There was no public market for our common stock on December 31, 2006. We have estimated the value of the unvested stock based on the assumed initial public offering price of $ per share, the midpoint of the range listed on the cover page of this prospectus. |
David G. Spencer, Ph.D.
Pursuant to a letter agreement dated September 25, 2001 between us and Dr. Spencer, we agreed to employ Dr. Spencer as Chief Operating Officer and Vice President of Research and Development beginning on October 25, 2001. If we were to have terminated Dr. Spencer’s employment without cause during the time this agreement was in effect, he would have been entitled to receive his base salary for two months, to be paid monthly, as severance compensation. The definition of cause as set forth in the agreement with Dr. Spencer includes (1) any material breach of the terms of any agreement with us by the executive officer, or failure of the
98
executive officer to diligently and properly perform his duties for us, (2) any material failure by the executive officer to comply with the reasonable policies and/or directives of our board of directors, (3) any action by the executive officer that is illegal, (4) any failure by the executive officer to fully disclose any material conflict of interest he may have with us in a transaction between us and any third party which is detrimental to our interest and well-being or (5) any adverse act or omission by the executive officer which would be required to be disclosed pursuant to public securities laws or which would limit our ability or any entity affiliated with us to sell securities under any federal or state law or which would disqualify us or any affiliated entity from any exemption otherwise available to us. If Dr. Spencer’s employment had been terminated without cause on December 31, 2006, his severance compensation would have been $44,167.
| | | | | | | | | |
Acceleration of Vesting of Options on Termination | | Change of
Control With Cash as
Consideration | | Change of Control With Acquiring
Company Stock as
Consideration | | Termination Without Cause or
Resignation for Good Reason After Change of Control |
Number of Option Shares | | | 386,766 | | | 193,383 | | | 386,766 |
Value Upon Termination(1) | | $ | | | $ | | | $ | |
(1) | There was no public market for our common stock on December 31, 2006. We have estimated the value of the unvested stock based on the assumed initial public offering price of $ per share, the midpoint of the range listed on the cover page of this prospectus. |
John A. Irick
Pursuant to a letter agreement dated December 17, 2001 between us and Mr. Irick, we agreed to employ Mr. Irick as Senior Vice President of Corporate and Business Development beginning on December 17, 2001. If we were to have terminated Mr. Irick’s employment without cause during the time this agreement was in effect, he would have been entitled to receive his base salary for three months, to be paid monthly, as severance compensation. The definition of cause as set forth in the agreement with Mr. Irick includes (1) any material breach of the terms of any agreement with us by the executive officer, or failure of the executive officer to diligently and properly perform his duties for us, (2) any material failure by the executive officer to comply with the reasonable policies or directives of our board of directors, (3) any action by the executive officer that is illegal, (4) any failure by the executive officer to fully disclose any material conflict of interest he may have with us in a transaction between us and any third party which is detrimental to our interest and well-being or (5) any adverse act or omission by the executive officer which would be required to be disclosed pursuant to public securities laws or which would limit our ability or any entity affiliated with us to sell securities under any federal or state law or which would disqualify us or any affiliated entity from any exemption otherwise available to us. If Mr. Irick’s employment had been terminated without cause on December 31, 2006, his severance compensation would have been $63,600.
| | | | | | | | | |
Acceleration of Vesting of Options on Termination | | Change of
Control With Cash as
Consideration | | Change of Control With Acquiring
Company Stock as
Consideration | | Termination Without Cause or Resignation for Good Reason
After Change of Control |
Number of Option Shares | | | 386,766 | | | 193,383 | | | 386,766 |
Value Upon Termination(1) | | $ | | | $ | | | $ | |
(1) | There was no public market for our common stock on December 31, 2006. We have estimated the value of the unvested stock based on the assumed initial public offering price of $ per share, the midpoint of the range listed on the cover page of this prospectus. |
Director Compensation
We reimburse each member of our board of directors who is not an employee for reasonable travel and other expenses in connection with attending meetings of our board of directors or committees thereof. In addition, prior to the completion of this offering, our policy with respect to compensation of directors is that each director who is a non-employee and is not serving as a representative of any of our stockholders receives an annual fee of
99
$10,000. We intend to adopt a new compensation policy for non-employee directors to be effective upon completion of this offering.
The following table provides information regarding the compensation that we paid to each of our directors during the fiscal year ended December 31, 2006, other than those directors included in the Summary Compensation Table above.
| | | | | | | | | | |
Name | | Fees Earned or
Paid in Cash ($)(1) | | Option
Awards ($)(2) | | | Total ($) |
W. Thomas Amick | | $ | 10,000 | | $ | 980 | (3) | | $ | 10,980 |
David L. Castaldi | | $ | 10,000 | | $ | 980 | (4) | | $ | 10,980 |
Dennis Dougherty | | | — | | | — | | | | — |
Chris Hegele | | | — | | | — | | | | — |
P. Sherrill Neff | | | — | | | — | | | | — |
Sunny Sharma, M.D. | | | — | | | — | | | | — |
(1) | Represents annual fees paid. |
(2) | The dollar amounts in this column represent the compensation expense incurred by us in fiscal 2006, calculated in accordance with SFAS 123R. See Note 10 to our consolidated financial statements included elsewhere in this prospectus for details as to the assumptions used to determine the fair value of the option awards. See also our discussion of stock-based compensation under “Management’s Discussion and Analysis of Financial Condition and Results of Operations—Stock-Based Compensation.” |
(3) | As of December 31, 2006, Mr. Amick held options to purchase 70,000 shares of common stock, 40,000 of which were granted on April 6, 2006 and have a grant date fair value of $0.09 per share. |
(4) | As of December 31, 2006, Mr. Castaldi held options to purchase 70,000 shares of common stock, 40,000 of which were granted on April 6, 2006 and have a grant date fair value of $0.09 per share. |
Stock and Benefit Plans
2003 Equity Incentive Plan
Our 2003 Plan was adopted by the compensation committee of our board of directors and approved by our stockholders in September 2003. In August 2005, our board of directors and stockholders approved amendments to the 2003 Plan. A maximum of 11,571,246 shares of common stock are authorized for issuance under the 2003 Plan.
As of August 1, 2007, 300,764 shares have been issued upon the exercise of options granted under this plan, 9,456,007 shares are subject to outstanding options under the 2003 Plan, and 1,814,658 shares are available for future grant under this plan. After completion of this offering, the 2003 Plan will terminate, and we will grant no further options or other awards under this plan. All outstanding options granted under the 2003 Plan as of the date of termination will remain outstanding and subject to their respective terms and the terms of the 2003 Plan.
The 2003 Plan is administered by our board of directors. Our board of directors has the authority to delegate full power and authority to a committee of our board to select the individuals to whom awards will be granted, to make any combination of awards to participants, to accelerate the exercisability or vesting of any award, to provide substitute awards, and to determine the specific terms and conditions of each award, subject to the provisions of the 2003 Plan.
The 2003 Plan permits us to make grants of incentive stock options, non-qualified stock options, restricted stock awards and unrestricted stock awards, and stock appreciation rights to officers, employees, directors, consultants and other key persons. Stock options granted under the 2003 Plan have a maximum term of ten years from the date of grant and incentive stock options have an exercise price of no less than the fair market value of our common stock on the date of grant.
100
Pursuant to the terms of the 2003 Plan, in the event of a recapitalization, forward or reverse split, reorganization, merger, consolidation, incorporation, spin-off, combination, repurchase, exchange of common stock or other securities, dividend or distribution of common stock or other special and nonrecurring dividend or distribution (whether in the form of cash, securities or other property), liquidation, dissolution, sale or purchase of assets or other similar transactions which affects the common stock, our compensation committee will make such appropriate adjustment to any terms and conditions of options, stock appreciation rights and restricted stock awards so as to prevent dilution or enlargement of the plan participants. In connection with one or more of the aforementioned events, our compensation committee may also make adjustments in the terms and conditions of unvested shares of restricted stock options and stock appreciation rights in recognition of unusual or nonrecurring events or in response to changes in applicable laws, regulations or accounting principles.
In addition, in the event of a change of control, which is defined as (i) a sale, transfer or other disposition of all or substantially all of our assets to another person or entity, (ii) a merger or consolidation into another person or entity, other than the merger of one of our subsidiaries into us or the merger of us into one of our subsidiaries, or (iii) a recapitalization, stock sale or other transaction or series of transactions where, after such transaction or series of transactions, our stockholders immediately before such transaction or the first in such series of transactions own less than 50% of us or the surviving entity, other than an equity financing transaction approved by a majority of our board of directors, restricted stock held by plan participants shall become vested and outstanding options and stock appreciation rights held by plan participants shall become vested and exercisable, in the following manner:
| | • | | where the consideration received in the transaction is all or substantially all cash, all restricted stock awards shall vest, and all options and stock appreciation rights will fully vest and become exercisable; |
| | • | | where the consideration received in the transaction is all or substantially all shares of capital stock of the acquiring or successor corporation or its parent, 50% of all unvested shares of restricted stock shall vest, and 50% of all unvested options and stock appreciation rights will vest and become exercisable (in each case, in addition to any portion of each that was previously vested and exercisable), and the remaining unvested shares of restricted stock and unvested options and stock appreciation rights shall continue to vest pursuant to the agreement with the plan participant and, if not fully vested earlier, such remaining unvested portion will fully vest 12 months from the effective date of the change of control; and |
| | • | | where the consideration is neither all nor substantially all cash or stock, our compensation committee shall determine the extent to which unvested shares of restricted stock and unvested options and stock appreciation awards shall vest. |
2007 Employee, Director and Consultant Stock Plan
Our 2007 Plan was adopted by our board of directors in 2007 and approved by our stockholders in 2007 and will become effective as of the date of this prospectus. The 2007 Plan will provide for the grant of incentive stock options, nonqualified stock options, restricted and unrestricted stock awards and other stock-based awards. As of , 2007, shares of common stock were reserved for issuance under the 2007 Plan. In addition, the 2007 Plan contains an “evergreen provision” which allows for an annual increase in the number of shares available for issuance under the plan on the first day of each of our fiscal years during the period beginning in fiscal year 2008 and ending on the second day of fiscal year 2016. The annual increase in the number of shares shall be equal to the lowest of:
| | • | | 5% of our outstanding shares on the first day of the fiscal year; and |
| | • | | an amount determined by our board of directors. |
Under this provision, no annual increase shall be made to the extent that the number of shares of common stock available for issuance under the 2007 Plan and all other employee or director stock plans would exceed % of our outstanding shares on the first day of the applicable fiscal year.
101
In accordance with the terms of the 2007 Plan, our board of directors has authorized our compensation committee to administer our 2007 Plan. However, our full board shall retain authority to make grants to our executive officers and members of our board of directors. In accordance with the provisions of the 2007 Plan, our board of directors or compensation committee will determine the terms of options and other awards, including:
| | • | | the determination of which employees, directors and consultants will be granted options and other awards; |
| | • | | the number of shares subject to options and other awards; |
| | • | | the exercise price of each option which may not be less than fair market value on the date of grant; |
| | • | | the schedule upon which options become exercisable; |
| | • | | the termination or cancellation provisions applicable to options; the terms and conditions of other awards, including conditions for repurchase, termination or cancellation, issue price and repurchase price; and |
| | • | | all other terms and conditions upon which each award may be granted in accordance with the 2007 Plan. |
No participant may receive awards for over shares of common stock in any fiscal year.
In addition, our board of directors or any committee to which our board of directors delegates authority may, with the consent of the affected plan participants, reprice or otherwise amend outstanding awards consistent with the terms of the 2007 Plan.
Upon a merger or other reorganization event, our board of directors, may, in their sole discretion, take any one or more of the following actions pursuant to our 2007 Plan, as to some or all outstanding awards:
| | • | | provide that all options shall be assumed or substituted by the successor corporation; |
| | • | | upon written notice to a participant, provide that the participant’s unexercised options will become exercisable in full and will terminate immediately prior to the consummation of such transaction unless exercised by the participant; |
| | • | | in the event of a merger pursuant to which holders of our common stock will receive a cash payment for each share surrendered in the merger, make or provide for a cash payment to the participants equal to the difference between the merger price times the number of shares of our common stock subject to such outstanding options (at prices not in excess of the merger price), and the aggregate exercise price of all such outstanding options (all options being made fully vested and immediately exercisable prior to their termination), in exchange for the termination of such options; and |
| | • | | provide that outstanding awards shall be assumed or substituted by the successor corporation, become realizable or deliverable, or restrictions applicable to an award will lapse, in whole or in part, prior to or upon the merger or reorganization event. |
Limitation of Officers’ and Directors’ Liability and Indemnification
The Delaware General Corporation Law authorizes corporations to limit or eliminate, subject to certain conditions, the personal liability of directors to corporations and their stockholders for monetary damages for breach of their fiduciary duties. Our amended and restated certificate of incorporation and restated bylaws limit the liability of our directors to the fullest extent permitted by Delaware law.
We have obtained director and officer liability insurance to cover liabilities our directors and officers may incur in connection with their services to us, including matters arising under the Securities Act of 1933, as amended. Our amended and restated certificate of incorporation and restated bylaws also provide that we will indemnify any of our directors and officers who, by reason of the fact that he or she is one of our officers or
102
directors, is involved in a legal proceeding of any nature. We will repay certain expenses incurred by a director or officer in connection with any civil or criminal action or proceeding, specifically including actions by us or in our name (derivative suits). Such indemnifiable expenses include, to the maximum extent permitted by law, attorneys’ fees, judgments, civil or criminal fines, settlement amounts and other expenses customarily incurred in connection with legal proceedings. A director or officer will not receive indemnification if he or she is found not to have acted in good faith and in a manner he or she reasonably believed to be in, or not opposed to, our best interest. We may enter into agreements to indemnify our directors and officers. These agreements would, among other things, indemnify our directors and officers for certain expenses, including attorneys’ fees, judgments, fines, and settlement amounts incurred by any such person in any action or proceeding, including any action by us arising out of such person’s services as our director or officer, any of our subsidiaries from time to time or any other company or enterprise to which the person provides services at our request.
Such limitation of liability and indemnification does not affect the availability of equitable remedies. In addition, we have been advised that in the opinion of the SEC, indemnification for liabilities arising under the Securities Act is against public policy as expressed in the Securities Act and is therefore unenforceable.
There is no pending litigation or proceeding involving any of our directors, officers, employees or agents in which indemnification will be required or permitted. We are not aware of any threatened litigation or proceeding that may result in a claim for such indemnification.
103
CERTAIN RELATIONSHIPS AND RELATED PERSON TRANSACTIONS
The following is a description of the transactions we have engaged in since January 1, 2004 with our directors and officers and beneficial owners of more than 5% of our voting securities and their affiliates.
Sales of Convertible Preferred Stock
The following table summarizes our sales of Series AA2 preferred stock, Series BB preferred stock, and Series CC preferred stock to our officers, directors and security holders who beneficially own more than 5% of any class of our voting securities since January 1, 2004. The purchase price was the fair market value as determined by arms-length negotiations between sophisticated investors and our management and board of directors, based on factors such as our stage of development and valuations of similarly situated private biopharmaceutical companies.
| | | | | | | | | | |
Name | | Type of Preferred
Shares | | Number of Shares | | Aggregate Purchase Price | | | Date of Purchase |
5% Stockholders | | | | | | | | | | |
Funds affiliated with Intersouth Partners V, L.P.(1) | | Series AA2 Series AA2 Series AA2 Series BB Series CC | | 922,511
1,098,139
786,217
6,942,858
1,758,475 | | $ $ $ $ $ | 922,511 1,098,139 786,217 7,776,001 2,074,731 | (2) | | 9/13/04
1/11/05
8/22/05
8/22/05
5/18/07 |
| | | | |
Funds affiliated with Investor Growth Capital Limited(3) | | Series CC | | 8,474,408 | | $ | 10,000,000 | | | 5/18/07 |
| | | | |
Johnson & Johnson Development Corporation | | Series AA2 Series AA2 Series AA2 Series AA2 Series BB Series CC | | 351,433
57,026
418,339
249,302
7,142,858
4,476,898 | | $ $ $ $ $ $ | 351,433 57,026 418,339 249,302 8,000,001 5,282,740 | (4) (2) (5) (5) | | 9/13/04
4/30/04
1/11/05
8/22/05
8/22/05
5/18/07 |
| | | | |
Mitsui & Co. Venture Partners II, L.P. | | Series AA2 Series AA2 Series AA2 Series BB Series CC | | 351,433
418,339
245,535
1,670,551
690,678 | | $ $ $ $ $ | 351,433 418,339 245,535 1,871,017 815,000 | (2) | | 9/13/04
1/11/05
8/22/05
8/22/05
5/18/07 |
| | | | |
Funds affiliated with Polaris Venture Partners IV, L.P.(6) | | Series BB Series CC | | 7,250,000 2,576,472 | | $ $ | 8,120,000 3,040,237 | | | 8/22/05 5/18/07 |
| | | | |
Quaker BioVentures, L.P.(7) | | Series AA2 Series AA2 Series AA2 Series BB Series CC | | 746,795
888,969
521,763
5,357,143
2,648,306 | | $ $ $ $ $ | 746,795 888,969 521,763 6,000,000 3,125,001 | (2) | | 9/13/04
1/11/05
8/22/05
8/22/05
5/18/07 |
(1) | Includes 2,063,210 shares of Series AA2 preferred stock, 4,672,132 shares of Series BB preferred stock and 1,031,856 shares of Series CC preferred stock held by Intersouth Partners V, L.P.; 94,318 shares of Series AA2 preferred stock, 213,583 shares of Series BB preferred stock and 47,171 shares of Series CC preferred stock held by Intersouth Affiliates V, L.P.; and 649,339 shares of Series AA2 preferred stock, 2,057,143 shares of Series BB preferred stock and 679,448 shares of Series CC preferred stock held by Intersouth Partners IV, L.P. Chris Hegele, the Chairman of our board of directors, is a partner (member) of Intersouth Associates V, LLC, the general partner of Intersouth Partners V, L.P. and Intersouth Affiliates V, L.P. Dennis Dougherty, a member of our board of directors, is a member manager of Intersouth Associates IV, LLC, the general partner of Intersouth Partners II, L.P., and a member manager of Intersouth Associates V, LLC, the general partner of Intersouth Partners V, L.P. and Intersouth Affiliates V, L.P. |
104
(2) | Represents dividends paid in kind on August 22, 2005 in the form of Series AA2 preferred stock. In connection with the sale of Series BB preferred stock on August 22, 2005, our board of directors declared a dividend payable to holders of record of Series AA2 preferred stock as of July 31, 2005. |
(3) | Includes 5,932,204 shares of Series CC preferred stock held by Investor Growth Capital Limited and 2,542,373 shares of Series CC preferred stock held by Investor Group, L.P. Dr. Sunny Sharma, a member of our board of directors, is a Vice President of Investor Growth Capital, Inc. |
(4) | Represents consideration paid in connection with our acquisition of Epicyte Pharmaceutical, Inc., a privately held company that held a portfolio of patents, patent applications and licenses related to the production of monoclonal antibodies in plants. |
(5) | Represents payment in the form of the conversion of a convertible promissory note. |
(6) | Includes 7,116,549 shares of Series BB preferred stock and 2,529,060 shares of Series CC preferred stock held by Polaris Venture Partners IV, L.P. and 133,451 shares of Series BB preferred stock and 47,412 shares of Series CC preferred stock held by Polaris Venture Partners Entrepreneurs’ Fund IV, L.P. |
(7) | P. Sherrill Neff, a member of our board of directors, is managing partner of Quaker BioVentures, L.P. |
Acquisition of Epicyte Pharmaceutical, Inc.
On April 30, 2004, we acquired substantially all of the assets of Epicyte Pharmaceutical, Inc., a privately held company that held a portfolio of patents, patent applications and licenses related to the production of monoclonal antibodies in plants, for consideration of approximately $1.0 million, consisting of 700,000 shares of Series AA2 preferred stock, at a per share value of $1.00 per share, and $300,000 in cash.
As part of this transaction, Johnson & Johnson Development Corporation, or JJDC, a beneficial owner of more than 5% of our voting securities, received 57,026 shares of Series AA2 preferred stock. The consideration paid in, and the terms of, this transaction were determined through negotiation between the stockholders of Epicyte and our management and independent directors, based on factors such as the value of intellectual property and technologies to be acquired. We believe this transaction was entered into on terms no less favorable to us than we could have obtained from unrelated third parties.
Collaboration Agreement, Security Agreement and Issuance of Convertible Promissory Notes and Warrant
On December 31, 2004, we entered into, and on November 27, 2006, amended and restated, a Selected Protein Development and Manufacturing Alliance Agreement with Centocor Research and Development, Inc., or Centocor, an affiliate of JJDC, which is one of our principal stockholders. The agreement provides a framework under which we may collaborate with Centocor, at its discretion, on the development of commercial-scale manufacturing processes for Centocor protein candidates based upon our LEX System, the clinical and commercial manufacture of such protein candidates, the development and construction by us of manufacturing facilities for the further implementation of our production systems and the transfer of manufacturing processes, technology and know-how for such protein candidates to Centocor. In 2006, 2005 and 2004, we recognized $4,097,387, $2,606,667 and $719,375 in revenue, respectively, from Centocor.
In connection with entering into this agreement with Centocor, on January 7, 2005, we entered into a Security Agreement, a Note and Warrant Purchase Agreement with JJDC in the amount of $8,000,000 and a Note Purchase Agreement in the amount of $17,000,000, whereby we could draw upon a secured promissory note in the principal amount of $5,000,000 at our election and, upon certain conditions relating to our agreement with Centocor, we could draw upon a secured promissory note in the principal amount of $12,000,000. On January 7, 2005, we borrowed $8,000,000 from JJDC pursuant to a convertible promissory note with an interest rate of 6%. On August 22, 2005, the convertible promissory note converted into 7,142,858 shares of Series BB preferred stock as a result of our Series BB private placement and 714,286 warrants to purchase shares of Series BB preferred stock at an exercise price of $1.40 per share were issued to JJDC. On June 1, 2006, we borrowed $5,000,000 from JJDC pursuant to a convertible promissory note with an interest rate of 6%. On May 18, 2007, this promissory noted converted into 4,476,898 shares of Series CC preferred stock as a result of our Series CC private placement. The promissory note in the principal amount of $12,000,000 has not been issued.
105
Investor Rights Agreement
Following the expiration of the lock-up period described below in “Shares Eligible for Future Sale—Lock-Up Agreements,” pursuant to an amended and restated investor rights agreement dated May 18, 2007, the holders of 85,471,096 shares of our common stock are entitled to registration rights with respect to the shares of common stock held by them. These stockholders include the following beneficial owners of more than 5% of our voting securities, and their affiliates:
| | |
Name of Holder | | Number of
Registrable Shares |
Intersouth Partners V, L.P. | | 17,497,131 |
Investor Growth Capital Limited | | 8,474,408 |
Johnson & Johnson Development Corporation | | 13,926,084 |
Mitsui & Co. Venture Partners II, L.P. | | 4,606,764 |
Polaris Venture Partners IV, L.P. | | 9,826,472 |
Quaker BioVentures, L.P. | | 12,777,212 |
David L. Castaldi | | 35,033 |
| | |
Total: | | 67,143,273 |
| | |
See “Description of Capital Stock—Registration Rights” for a more detailed description of these registration rights. Other than the registration rights set forth above, there are no provisions of the amended and restated investor rights agreement that will remain in effect after completion of this offering.
Policy for Approval of Related Person Transactions
Pursuant to the written charter of our audit committee, the audit committee is responsible for reviewing and approving, prior to our entry into any such transaction, all transactions in which we are a participant and in which any of the following persons has or will have a direct or indirect material interest:
| | • | | our executive officers; |
| | • | | the beneficial owners of more than 5% of our securities; |
| | • | | the immediate family members of any of the foregoing persons; and |
| | • | | any other persons whom our board determines may be considered related persons. |
For purposes of this policy, “immediate family members” means any child, stepchild, parent, stepparent, spouse, sibling, mother-in-law, father-in-law, son-in-law, daughter-in-law, brother-in-law, or sister-in-law, and any person (other than a tenant or employee) sharing the household with the executive officer, director or 5% beneficial owner.
In reviewing and approving such transactions, our audit committee shall obtain, or shall direct our management to obtain on its behalf, all information that the committee believes to be relevant and important to a review of the transaction prior to its approval. Following receipt of the necessary information, a discussion shall be held of the relevant factors if deemed to be necessary by the committee prior to approval. If a discussion is not deemed to be necessary, approval may be given by written consent of the committee. This approval authority may also be delegated to the chair of the audit committee in some circumstances. No related person transaction shall be entered into prior to the completion of these procedures.
Our audit committee or its chair, as the case may be, shall approve only those related person transactions that are determined to be in, or not inconsistent with, the best interests of us and our stockholders, taking into account all available facts and circumstances as the committee or the chair determines in good faith to be
106
necessary. These facts and circumstances will typically include, but not be limited to, the benefits of the transaction to Biolex; the impact on a director’s independence in the event the related person is a director, an immediate family member of a director or an entity in which a director is a partner, stockholder or executive officer; the availability of other sources for comparable products or services; the terms of the transaction; and the terms of comparable transactions that would be available to unrelated third parties or to employees generally. No member of our audit committee shall participate in any review, consideration or approval of any related person transaction with respect to which the member or any of his or her immediate family members is the related person.
107
PRINCIPAL STOCKHOLDERS
The following table sets forth certain information regarding the beneficial ownership of our common stock as of August 1, 2007, and as adjusted to reflect the sale of our common stock offered by this prospectus by:
| | • | | the executive officers named in the summary compensation table; |
| | • | | all of our current directors and executive officers as a group; and |
| | • | | each stockholder known by us to own beneficially more than 5% of our common stock. |
Beneficial ownership is determined in accordance with the rules of the SEC and includes voting or investment power with respect to the securities. Shares of common stock that may be acquired by an individual or group within 60 days of August 1, 2007, pursuant to the exercise of options or warrants, are deemed to be outstanding for the purpose of computing the percentage ownership of such individual or group, but are not deemed to be outstanding for the purpose of computing the percentage ownership of any other person shown in the table. Percentage of ownership is based on 86,036,859 shares of common stock outstanding on August 1, 2007, which assumes the conversion of all outstanding shares of our Class B non-voting common stock and preferred stock into shares of our Class A voting common stock and the reclassification of our Class A voting common stock into our common stock upon completion of this offering.
Except as indicated in footnotes to this table, we believe that the stockholders named in this table have sole voting and investment power with respect to all shares of common stock shown to be beneficially owned by them, based on information provided to us by such stockholders. Unless otherwise indicated, the address for each director and executive officer listed is: c/o Biolex, Inc., 158 Credle Street, Pittsboro, North Carolina, 27312.
| | | | | | | |
Beneficial Owner | | Number of
Shares
Beneficially
Owned | | Percentage of
Common Stock
Beneficially Owned |
| | | Before
Offering | | | After
Offering |
Directors and Executive Officers | | | | | | | |
Jan Turek(1) | | 2,199,890 | | 2.5 | % | | |
Dale Sander(2) | | 402,484 | | * | | | |
David Spencer(3) | | 761,670 | | * | | | |
John Irick(4) | | 761,670 | | * | | | |
Chris Hegele(5) | | 20,986,655 | | 24.4 | % | | |
W. Thomas Amick(6) | | 42,500 | | * | | | |
David L. Castaldi(7) | | 77,533 | | * | | | |
Dennis Dougherty(8) | | 17,497,131 | | 20.3 | % | | |
P. Sherrill Neff(9) | | 12,777,212 | | 14.9 | % | | |
Sunny Sharma, M.D.(10) | | 2,542,373 | | 3.0 | % | | |
All current executive officers and directors as a group (10 persons)(11) | | 40,551,987 | | 44.9 | % | | |
| | | |
5% Stockholders | | | | | | | |
Funds affiliated with Intersouth Partners V, L.P.(12) | | 17,497,131 | | 20.3 | % | | |
406 Blackwell Street, Suite 200 | | | | | | | |
Durham, North Carolina 27701 | | | | | | | |
Johnson & Johnson Development Corporation(13) | | 13,926,084 | | 16.2 | % | | |
410 George Street | | | | | | | |
New Brunswick, New Jersey 08901 | | | | | | | |
108
| | | | | | | |
Beneficial Owner | | Number of
Shares
Beneficially
Owned | | Percentage of
Common Stock
Beneficially Owned |
| | | Before
Offering | | | After
Offering |
Quaker BioVentures, L.P.(14) | | 12,777,212 | | 14.9 | % | | |
Two Greenville Crossing | | | | | | | |
4005 Kennett Pike, Suite 220 | | | | | | | |
Greenville, Delaware 19807 | | | | | | | |
Funds affiliated with Polaris Venture Partners IV, L.P.(15) | | 9,826,472 | | 11.4 | % | | |
1000 Winter Street, Suite 3350 | | | | | | | |
Waltham, Massachusetts 02451 | | | | | | | |
Investor Growth Capital Limited(16) | | 8,474,577 | | 9.9 | % | | |
Canada Court, Upland Road | | | | | | | |
St. Peter Port, Guernsey GY1 3BQ | | | | | | | |
Channel Islands | | | | | | | |
Mitsui & Co. Venture Partners(17) | | 4,606,764 | | 5.4 | % | | |
200 Park Avenue, 36th Floor | | | | | | | |
New York, New York 10166 | | | | | | | |
| * | Represents beneficial ownership of less than 1% of the shares of common stock. |
(1) | Consists of 2,199,890 shares of common stock issuable upon the exercise of options exercisable within 60 days of August 1, 2007 held by Mr. Turek. |
(2) | Consists of 402,484 shares of common stock issuable upon the exercise of options exercisable within 60 days of August 1, 2007 held by Mr. Sander. |
(3) | Consists of 761,670 shares of common stock issuable upon the exercise of options exercisable within 60 days of August 1, 2007 held by Dr. Spencer. |
(4) | Consists of 761,670 shares of common stock issuable upon the exercise of options exercisable within 60 days of August 1, 2007 held by Mr. Irick. |
(5) | Consists of the shares described in footnote 12 below, 55,640 shares of Class A common stock held by Kitty Hawk Capital Limited Part IV and shares of common stock issuable upon the conversion of 1,138,710 shares of Series AA1 preferred stock, 966,246 shares of Series AA2 preferred stock and 1,328,928 shares of Series BB preferred stock held by Kitty Hawk Capital Limited Part IV. Mr. Hegele disclaims beneficial ownership of the shares owned by the Intersouth funds and Kitty Hawk Capital Limited Part IV except to the extent of his pecuniary interest therein. |
(6) | Consists of 42,500 shares of common stock issuable upon the exercise of options exercisable within 60 days of August 1, 2007 held by Mr. Amick. |
(7) | Consists of the shares of common stock issuable upon conversion of 22,321 shares of Series BB preferred stock and 12,717 Series CC preferred stock owned of record by and 42,500 shares of common stock issuable upon the exercise of options exercisable within 60 days of August 1, 2007 held by Mr. Castaldi. |
(8) | Consists of the shares described in footnote 12 below. Mr. Dougherty disclaims beneficial ownership of the shares owned by the Intersouth funds except to the extent of his pecuniary interest therein. |
(9) | Consists of the shares described in footnote 14 below. Mr. Neff disclaims beneficial ownership of the shares owned by the Quaker BioVentures, L.P. except to the extent of his pecuniary interest therein. |
(10) | Consists of shares held by Investor Group, L.P. See footnote 16 below. Dr. Sharma is a limited partner of Investor Group, L.P. Dr. Sharma disclaims beneficial ownership of the shares owned by Investor Group, L.P. except to the extent of his pecuniary interest therein. |
(11) | Consists of shares set forth in footnotes 1 through 10. |
(12) | Consists of the shares of common stock issuable upon the reclassification of 85,132 shares of Class A common stock held by Intersouth Partners V, L.P. and shares of common stock issuable upon the conversion of 4,563,162 shares of Series AA2 preferred stock, 4,672,132 shares of Series BB preferred stock and 1,031,856 shares of Series CC preferred stock held by Intersouth Partners V, L.P.; 208,602 shares of Series AA2 preferred stock, 213,583 shares of Series BB preferred stock and 47,171 shares of Series CC preferred stock held by Intersouth Affiliates V, L.P.; and 1,813,174 shares of Series AA1 preferred stock, 2,125,728 shares of Series AA2 preferred stock, 2,057,143 shares of Series BB preferred stock and 679,448 shares of Series CC preferred stock held by Intersouth Partners IV, L.P. |
(13) | Consists of shares of common stock issuable upon the conversion of 2,306,328 shares of Series AA2 preferred stock, 7,142,858 shares of Series BB preferred stock and 4,476,898 shares of Series CC preferred stock. |
(14) | Consists of shares of common stock issuable upon the conversion of 4,771,763 shares of Series AA2 preferred stock, 5,357,143 shares of Series BB preferred stock and 2,648,306 shares of Series CC preferred stock. |
(15) | Consists of shares of common stock issuable upon the conversion of 7,116,549 shares of Series BB preferred stock and 2,529,060 shares of Series CC preferred stock held by Polaris Venture Partners IV, L.P. and 133,451 shares of Series BB preferred stock and 47,412 shares of Series CC preferred stock held by Polaris Venture Partners Entrepreneurs’ Fund IV, L.P. |
(16) | Consists of shares of common stock issuable upon the conversion of 5,932,204 shares of Series CC preferred stock held by Investor Growth Capital Limited (Investor Growth), an indirectly wholly-owned subsidiary of Investor AB, and 2,542,373 shares of Series CC preferred stock held by Investor Group, L.P., a limited partnership of which Investor AB serves as the ultimate General Partner. Investor AB and Investor Growth share voting and dispositive power over the shares and Investor AB may be deemed the beneficial owner of the shares. |
(17) | Consists of shares of common stock issuable upon the conversion of 2,245,535 shares of Series AA2 preferred stock, 1,670,551 shares of Series BB preferred stock and 690,678 shares of Series CC preferred stock. |
109
DESCRIPTION OF CAPITAL STOCK
Upon completion of this offering, we will be authorized to issue shares of common stock, $0.001 par value per share, and 5,000,000 shares of preferred stock, $0.001 par value per share, and there will be shares of common stock and no shares of preferred stock outstanding.
As of August 1, 2007, after giving effect to the conversion of all outstanding shares of our Class B non-voting common stock and preferred stock into shares of our Class A voting common stock and the reclassification of our Class A voting common stock into common stock upon completion of this offering, we would have had 86,036,859 shares of common stock outstanding held of record by 63 stockholders. In addition, as of August 1, 2007, there were outstanding options to purchase 9,456,007 shares of common stock and outstanding warrants to purchase 93,640 shares of common stock
Common Stock
Holders of common stock are entitled to one vote for each share held of record on all matters submitted to a vote of the stockholders, and do not have cumulative voting rights. Subject to preferences that may be applicable to any outstanding shares of preferred stock, holders of common stock are entitled to receive ratably such dividends, if any, as may be declared from time to time by our board of directors out of funds legally available for dividend payments. All outstanding shares of common stock are fully paid and nonassessable, and the shares of common stock to be issued upon completion of this offering will be fully paid and nonassessable. The holders of common stock have no preferences or rights of conversion, exchange, pre-emption or other subscription rights. There are no redemption or sinking fund provisions applicable to the common stock. In the event of any liquidation, dissolution or winding-up of our affairs, holders of common stock will be entitled to share ratably in our assets that are remaining after payment or provision for payment of all of our debts and obligations and after liquidation payments to holders of outstanding shares of preferred stock, if any.
Preferred Stock
The preferred stock, if issued, would have priority over the common stock with respect to dividends and other distributions, including the distribution of assets upon liquidation. Our board of directors has the authority, without further stockholder authorization, to issue from time to time shares of preferred stock in one or more series and to fix the terms, limitations, relative rights and preferences and variations of each series. Although we have no present plans to issue any shares of preferred stock, the issuance of shares of preferred stock, or the issuance of rights to purchase such shares, could decrease the amount of earnings and assets available for distribution to the holders of common stock, could adversely affect the rights and powers, including voting rights, of the common stock, and could have the effect of delaying, deterring or preventing a change in control of us or an unsolicited acquisition proposal.
Warrants
As of August 1, 2007, we had warrants outstanding for the number of shares of common stock, at the exercise prices and expiration dates set forth below:
| | | | |
Number of Shares | | Exercise Price | | Expiration Date |
5,493(1) | | $1.00 | | Upon closing of this offering |
6,981(1) | | $1.00 | | Upon closing of this offering |
3,649(1) | | $1.00 | | Upon closing of this offering |
7,424(1) | | $1.00 | | Upon closing of this offering |
3,126(1) | | $1.00 | | Upon closing of this offering |
48,066(1) | | $1.12 | | Earlier of QPO(2) and September 28, 2013 |
13,898(1) | | $1.12 | | Earlier of QPO(2) and March 15, 2014 |
5,003(1) | | $1.12 | | Earlier of QPO(2) and March 15, 2014 |
110
(1) | These warrants have net exercise provisions under which the holder may, in lieu of payment of the exercise price in cash, surrender the warrant and receive a net amount of shares based on the fair market value of our convertible preferred stock, or the fair market value of the underlying common stock, at the time of exercise of the warrant after deduction of the aggregate exercise price. |
(2) | A QPO is a fully-marketed underwritten public offering of our common stock with a price per share of at least $6.72. |
Registration Rights
Following the expiration of the lock-up period described below in “Shares Eligible for Future Sale—Lock-Up Agreements,” the holders of 85,471,096 shares of our common stock (assuming full exercise of outstanding warrants to purchase our stock) are entitled to certain registration rights with respect to these securities as set forth in an agreement between us and the holders of these securities. We are generally required to pay all expenses incurred in connection with registrations effected in connection with the following rights, excluding underwriting discounts and commissions, and fees and expenses of counsel to the registering security holders. All registration rights described below shall terminate within five years after the closing of this offering
Demand rights. At any time after 180 days following the completion of this offering but before May 18, 2011, subject to specified limitations, the holders of not less than 50% of these registrable securities may require that we register all or a portion of these securities for sale under the Securities Act, if the anticipated aggregate offering price of such securities is at least $5,000,000. We may be required to effect up to two such registrations. Stockholders with these registration rights who are not part of an initial registration demand are entitled to notice and are entitled to include their shares of common stock in the registration. Under certain circumstances, the underwriters, if any, may limit the number of shares included in any such registration.
Form S-3 rights. If we become eligible to file registration statements on Form S-3, subject to specified limitations, a holder of these registrable securities can require us to register all or a portion of its registrable securities on Form S-3, if the anticipated aggregate offering price of such securities is at least $1,000,000. We may not be required to effect more than two such registrations in any rolling 12-month period. Stockholders with these registration rights who are not part of an initial registration demand are entitled to notice and are entitled to include their shares of common stock in the registration. Under certain circumstances, the underwriters, if any, may limit the number of shares included in any such registration.
Piggyback rights. If we propose to register any of our equity securities under the Securities Act, other than in connection with (i) a registration relating solely to our employee benefit plans; (ii) a registration relating solely to a business combination or merger involving us; or (iii) a registration pursuant to an exercise of demand rights or S-3 rights described above, the holders of these registrable securities are entitled to notice of such registration and are entitled to include their shares of common stock in the registration. Under certain circumstances, the underwriters, if any, may limit the number of shares included in any such registration.
Anti-Takeover Provisions
The provisions of (1) Delaware law, (2) our amended and restated certificate of incorporation to be effective upon completion of this offering, and (3) our restated bylaws to be effective upon completion of this offering discussed below could discourage or make it more difficult to accomplish a proxy contest or other change in our management or the acquisition of control by a holder of a substantial amount of our voting stock. It is possible that these provisions could make it more difficult to accomplish, or could deter, transactions that stockholders may otherwise consider to be in their best interests or our best interests. These provisions are intended to enhance the likelihood of continuity and stability in the composition of our board of directors and in the policies formulated by our board of directors and to discourage certain types of transactions that may involve an actual or
111
threatened change of control of us. These provisions are designed to reduce our vulnerability to an unsolicited acquisition proposal. The provisions also are intended to discourage certain tactics that may be used in proxy fights. Such provisions also may have the effect of preventing changes in our management.
Delaware Statutory Business Combinations Provision. We are subject to the anti-takeover provisions of Section 203 of the Delaware General Corporation Law. In general, Section 203 prohibits a publicly held Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder, unless the business combination is, or the transaction in which the person became an interested stockholder was, approved in a prescribed manner or another prescribed exception applies. For purposes of Section 203, a “business combination” is defined broadly to include a merger, asset sale or other transaction resulting in a financial benefit to the interested stockholder, and, subject to certain exceptions, an “interested stockholder” is a person who, together with his or her affiliates and associates, owns (or within three years prior, did own) 15% or more of the corporation’s voting stock.
Classified Board of Directors; Removal of Directors for Cause. Our amended and restated certificate of incorporation and restated bylaws provide that upon completion of this offering, our board of directors will be divided into three classes, with the term of office of the first class to expire at the first annual meeting of stockholders following the initial classification of directors, the term of office of the second class to expire at the second annual meeting of stockholders following the initial classification of directors, and the term of office of the third class to expire at the third annual meeting of stockholders following the initial classification of directors. At each annual meeting of stockholders, directors elected to succeed those directors whose terms expire will be elected for a three-year term of office. All directors elected to our classified board of directors will serve until the election and qualification of their respective successors or their earlier resignation or removal. Our board of directors is authorized to create new directorships and to fill such positions so created and is permitted to specify the class to which any such new position is assigned. The person filling such position would serve for the term applicable to that class. Our board of directors (or its remaining members, even if less than a quorum) is also empowered to fill vacancies on our board of directors occurring for any reason for the remainder of the term of the class of directors in which the vacancy occurred. Members of our board of directors may only be removed for cause and only by the affirmative vote of 80% of our outstanding voting stock. These provisions are likely to increase the time required for stockholders to change the composition of our board of directors. For example, in general, at least two annual meetings will be necessary for stockholders to effect a change in a majority of the members of our board of directors.
Advance Notice Provisions for Stockholder Proposals and Stockholder Nominations of Directors. Our restated bylaws provide that, for nominations to our board of directors or for other business to be properly brought by a stockholder before a meeting of stockholders, the stockholder must first have given timely notice of the proposal in writing to our Secretary. For an annual meeting, a stockholder’s notice generally must be delivered not less than 45 days nor more than 75 days prior to the anniversary of the mailing date of the proxy statement for the previous year’s annual meeting. Detailed requirements as to the form of the notice and information required in the notice are specified in the restated bylaws. If it is determined that business was not properly brought before a meeting in accordance with our bylaws, such business will not be conducted at the meeting.
Special Meetings of Stockholders. Special meetings of the stockholders may be called only by our board of directors pursuant to a resolution adopted by a majority of the total number of directors.
No Stockholder Action by Written Consent. Our amended and restated certificate of incorporation and restated bylaws do not permit our stockholders to act by written consent. As a result, any action to be effected by our stockholders must be effected at a duly called annual or special meeting of the stockholders.
Super-Majority Stockholder Vote Required for Certain Actions. The Delaware General Corporation Law provides generally that the affirmative vote of a majority of the shares entitled to vote on any matter is required
112
to amend a corporation’s certificate of incorporation or bylaws, unless the corporation’s certificate of incorporation or bylaws, as the case may be, requires a greater percentage. Our amended and restated certificate of incorporation requires the affirmative vote of the holders of at least 80% of our outstanding voting stock to amend or repeal any of the provisions discussed in this section of this prospectus entitled “Anti-Takeover Provisions.” This 80% stockholder vote would be in addition to any separate class vote that might in the future be required pursuant to the terms of any preferred stock that might then be outstanding. In addition, an 80% vote is also required for any amendment to, or repeal of, our restated bylaws by the stockholders. Our restated bylaws may be amended or repealed by a vote of a majority of the total number of directors.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock will be , with offices at .
NASDAQ Global Market Listing
We have applied to have our common stock listed on The NASDAQ Global Market under the symbol “BLEX.”
113
SHARES ELIGIBLE FOR FUTURE SALE
Prior to this offering, there has been no market for our common stock. Future sales of substantial amounts of our common stock in the public market could adversely affect market prices prevailing from time to time. Furthermore, because only a limited number of shares will be available for sale shortly after this offering due to existing contractual and legal restrictions on resale as described below, there may be sales of substantial amounts of our common stock in the public market after the restrictions lapse. This may adversely affect the prevailing market price and our ability to raise equity capital in the future.
Upon completion of this offering, we will have shares of common stock outstanding, assuming the conversion of all outstanding shares of our Class B non-voting common stock and preferred stock into shares of our Class A voting common stock and the reclassification of our Class A voting common stock into our common stock upon completion of this offering and no exercise of any options and warrants outstanding as of August 1, 2007. Of these shares, the shares sold in this offering will be freely transferable without restriction or registration under the Securities Act, except for any shares purchased by any of our existing “affiliates,” as that term is defined in Rule 144 under the Securities Act. The remaining shares of common stock existing are “restricted securities” as defined in Rule 144. Restricted securities may be sold in the public market only if registered or if they qualify for an exemption from registration under Rules 144 or 701 of the Securities Act, as described below. As a result of the contractual 180-day lock-up period described below and the provisions of Rules 144 and 701, these shares will be available for sale in the public market as follows:
| | | | |
Number of Shares | | | | Date |
| | | | On the date of this prospectus. |
| | | | At 90 days from the date of this prospectus. |
| | | | At various times between the 90 days and 180 days* from the date of this prospectus. |
| | | | At or after 180 days* from the date of this prospectus (subject, in some cases, to volume limitations). |
| * | This 180-day period corresponds to the end of the lock-up period described below in “—Lock-Up Agreements.” This lock-up period may be extended as described below. |
Rule 144
In general, under Rule 144 as currently in effect, beginning 90 days after this offering, a person, or persons whose shares are aggregated, who owns shares that were purchased from us, or any affiliate, at least one year previously, is entitled to sell within any three-month period a number of shares that does not exceed the greater of (1) 1% of our then-outstanding shares of common stock, which will equal approximately shares immediately after this offering, and (2) the average weekly trading volume of our common stock on The NASDAQ Global Market during the four calendar weeks preceding the filing of a notice of the sale on Form 144. Sales under Rule 144 are also subject to manner of sale provisions, notice requirements and the availability of current public information about us. We are unable to estimate the number of shares that will be sold under rule 144 since this will depend on the market price for our common stock, the personal circumstances of the stockholder and other factors.
Rule 144(k)
Under Rule 144(k), a person who is not deemed to have been one of our affiliates at any time during the three months preceding a sale, and who owns shares within the definition of “restricted securities” under Rule 144 that were purchased from us, or any affiliate, at least two years previously, would be entitled to sell shares under Rule 144(k) without regard to the volume limitations, manner of sale provisions, public information requirements or notice requirements described above.
114
Rule 701
In general, under Rule 701, any of our employees, directors, officers, consultants or advisors who purchases shares from us in connection with a compensatory stock or option plan or other written agreement before the effective date of this offering is entitled to resell such shares 90 days after the effective date of this offering in reliance on Rule 144, without having to comply with some of the restrictions, including the holding period requirement.
The SEC has indicated that Rule 701 will apply to typical stock options granted by an issuer before it becomes subject to the reporting requirements of the Securities Exchange Act, along with the shares acquired upon exercise of such options, including exercises after the date of this prospectus. Securities issued in reliance on Rule 701 are restricted securities and, subject to the contractual restrictions described below, beginning 90 days after the date of this prospectus, may be sold by persons other than “affiliates,” as defined in Rule 144, subject only to the manner of sale provisions of Rule 144 and by “affiliates” under Rule 144 without compliance with its one-year minimum holding period requirement.
Registration Rights
Upon expiration of the lock-up period described below in “—Lock-Up Agreements,” the holders of approximately 85,471,096 shares of common stock will be entitled to various rights with respect to the registration of these shares under the Securities Act. Registration of these shares under the Securities Act would result in these shares becoming freely tradable without restriction under the Securities Act immediately upon the effectiveness of the registration, except for shares held by affiliates.
Stock Options
As of August 1, 2007, options to purchase a total of 9,456,007 shares of common stock were outstanding. An additional 1,814,658 shares of common stock were available for future option grants under our 2003 Plan.
Upon completion of this offering, we intend to file a registration statement on Form S-8 under the Securities Act covering all shares of common stock subject to outstanding options or issuable pursuant to our stock plans. Subject to the volume limitations under Rule 144 applicable to affiliates, shares registered under any registration statements will be available for sale in the open market, except to the extent that the shares are subject to vesting restrictions with us or the contractual restrictions described below.
Lock-Up Agreements
We, all of our directors and executive officers, and holders of substantially all of our outstanding stock have agreed that, subject to certain exceptions, without the prior written consent of Lehman Brothers Inc., we and they will not directly or indirectly, (1) offer for sale, sell, pledge, or otherwise dispose of (or enter into any transaction or device that is designed to, or could be expected to, result in the disposition by any person at any time in the future of) any shares of common stock (including, without limitation, shares of common stock that may be deemed to be beneficially owned by us or them in accordance with the rules and regulations of the SEC and shares of common stock that may be issued upon exercise of any options or warrants) or securities convertible into or exercisable or exchangeable for common stock, (2) enter into any swap or other derivatives transaction that transfers to another, in whole or in part, any of the economic consequences of ownership of the common stock, or (3) publicly disclose the intention to do any of the foregoing for a period of 180 days after the date of this prospectus. In addition, during this 180-day period, we have agreed not to file or cause to be filed a registration statement, including any amendments thereto, with respect to the registration of any shares of common stock or securities convertible, exercisable or exchangeable into common stock or any of our other securities, and our directors and executive officers, and holders of all of our outstanding stock have agreed not to exercise any rights they may have with respect to registration of common stock or securities convertible, exercisable or exchangeable into common stock or any of our other securities.
115
The 180-day restricted period described in the preceding paragraph will be extended if:
| | • | | during the last 17 days of the 180-day restricted period we issue an earnings release or material news or a material event relating to us occurs; or |
| | • | | prior to the expiration of the 180-day restricted period, we announce that we will release earnings results during the 16-day period beginning on the last day of the 180-day period, |
in which case the restrictions described in the preceding paragraph will continue to apply until the expiration of the 18-day period beginning on the issuance of the earnings release or the announcement of the material news or occurrence of a material event, unless such extension is waived in writing by Lehman Brothers Inc.
The lock-up agreements do not prevent a security holder from transferring such securities (1) by bona fide gift, (2) to a trust for the benefit of the security holder or a member of the immediate family of the security holder, (3) by will or intestate succession, (4) to any affiliate of the security holder, (5) if the security holder is a corporation or similar entity, to any wholly-owned subsidiaries, (6) if the security holder is a partnership or limited liability company, to partners or members of the security holder, or (7) if the security holder is a trust, to any beneficiary of the security holder, provided that each such transferee agrees to be bound by the terms of the lock-up agreement. In addition, the lock-up agreements do not prevent a security holder from establishing a trading plan pursuant to Rule 10b5-1 under the Exchange Act, provided that no sales pursuant to such trading plan may be made during the lock-up period. Furthermore, the lock-up provisions described above do not limit our ability to (a) issue securities pursuant to employee benefit plans, qualified stock option plans or other employee compensation plans existing on the date of this prospectus, (b) issue securities pursuant to currently outstanding options, warrants or rights, or (c) file a registration statement on Form S-8 to register securities issuable under our stock plans.
Lehman Brothers Inc., in its sole discretion, may release the common stock and other securities subject to the lock-up agreements described above in whole or in part at any time with or without notice. When determining whether or not to release common stock and other securities from lock-up agreements, Lehman Brothers Inc. will consider, among other factors, the holder’s reasons for requesting the release, the number of shares of common stock and other securities for which the release is being requested and market conditions at the time.
116
MATERIAL U.S. FEDERAL TAX CONSEQUENCES
FOR NON-U.S. HOLDERS
The following discussion is a general summary of the material U.S. federal income and estate tax consequences of the ownership and disposition of our common stock applicable to “Non-U.S. Holders.” As used herein, a Non-U.S. Holder means a beneficial owner of our common stock that is neither a U.S. person nor a partnership for U.S. federal income tax purposes, and that will hold shares of our common stock as capital assets. For U.S. federal income tax purposes, a U.S. person includes:
| | • | | an individual who is a citizen or resident of the United States; |
| | • | | a corporation (or other business entity treated as a corporation for U.S. federal income tax purposes) created or organized in the United States or under the laws of the United States, any state thereof or the District of Columbia; |
| | • | | an estate the income of which is includible in gross income regardless of source; or |
| | • | | a trust that (A) is subject to the primary supervision of a court within the United States and the control of one or more U.S. persons, or (B) otherwise has validly elected to be treated as a U.S. domestic trust for U.S. federal income tax purposes. |
If a partnership (including an entity treated as a partnership for U.S. federal income tax purposes) holds shares of our common stock, the U.S. federal income tax treatment of each partner generally will depend on the status of the partner and the activities of the partnership and the partner. Partnerships acquiring our common stock, and partners in such partnerships, should consult their own tax advisors with respect to the U.S. federal income tax consequences of the ownership and disposition of our common stock.
This summary does not consider specific facts and circumstances that may be relevant to a particular Non-U.S. Holder’s tax position and does not consider U.S. state and local or non-U.S. tax consequences. It also does not consider Non-U.S. Holders subject to special tax treatment under the U.S. federal income tax laws (including partnerships or other pass-through entities, banks and insurance companies, dealers in securities, holders of our common stock held as part of a “straddle,” “hedge,” “conversion transaction” or other risk-reduction transaction, controlled foreign corporations, passive foreign investment companies, companies that accumulate earnings to avoid U.S. federal income tax, foreign tax-exempt organizations, former U.S. citizens or residents, persons who hold or receive common stock as compensation and persons subject to the alternative minimum tax). This summary is based on provisions of the Internal Revenue Code, applicable final, temporary and proposed U.S. Treasury regulations, administrative pronouncements of the U.S. Internal Revenue Service, or the IRS, and judicial decisions, all as in effect on the date hereof, and all of which are subject to change, possibly on a retroactive basis, and different interpretations.
This summary is included herein as general information only. Accordingly, each prospective Non-U.S. Holder is urged to consult its own tax advisor with respect to the U.S. federal, state, local and non-U.S. income, estate and other tax consequences of owning and disposing of our common stock.
U.S. Trade or Business Income
For purposes of this discussion, dividend income and gain on the sale or other taxable disposition of our common stock will be considered to be “U.S. trade or business income” if such income or gain is (i) effectively connected with the conduct by a Non-U.S. Holder of a trade or business within the United States and (ii) in the case of a Non-U.S. Holder that is eligible for the benefits of an income tax treaty with the United States, attributable to a permanent establishment (or, for an individual, a fixed base) maintained by the Non-U.S. Holder in the United States. Generally, U.S. trade or business income is not subject to U.S. federal withholding tax (provided the Non-U.S. Holder complies with applicable certification and disclosure requirements); instead, U.S. trade or business income is subject to U.S. federal income tax on a net income basis at regular U.S. federal
117
income tax rates in the same manner as a U.S. person, unless an applicable income tax treaty provides otherwise. Any U.S. trade or business income received by a corporate Non-U.S. Holder may be subject to an additional “branch profits tax” at a 30% rate or such lower rate as may be specified by an applicable income tax treaty.
Dividends
Distributions of cash or property that we pay will constitute dividends for U.S. federal income tax purposes to the extent paid from our current or accumulated earnings and profits (as determined under U.S. federal income tax principles). A Non-U.S. Holder generally will be subject to U.S. federal withholding tax at a 30% rate, or, if the Non-U.S. Holder is eligible, at a reduced rate prescribed by an applicable income tax treaty, on any dividends received in respect of our common stock. If the amount of a distribution exceeds our current and accumulated earnings and profits, such excess first will be treated as a tax-free return of capital to the extent of the Non-U.S. Holder’s tax basis in our common stock (with a corresponding reduction in such Non-U.S. Holder’s tax basis in our common stock), and thereafter will be treated as capital gain. In order to obtain a reduced rate of U.S. federal withholding tax under an applicable income tax treaty, a Non-U.S. Holder will be required to provide a properly executed IRS Form W-8BEN certifying under penalties of perjury its entitlement to benefits under the treaty. Special certification requirements and other requirements apply to certain Non-U.S. Holders that are entities rather than individuals. A Non-U.S. Holder of our common stock that is eligible for a reduced rate of U.S. federal withholding tax under an income tax treaty may obtain a refund or credit of any excess amounts withheld by filing an appropriate claim for a refund with the IRS on a timely basis. A Non-U.S. Holder should consult its own tax advisor regarding its possible entitlement to benefits under an income tax treaty and the filing of a U.S. tax return for claiming a refund of U.S. federal withholding tax.
The U.S. federal withholding tax does not apply to dividends that are U.S. trade or business income, as defined and discussed above, of a Non-U.S. Holder who provides a properly executed IRS Form W-8ECI, certifying under penalties of perjury that the dividends are effectively connected with the Non-U.S. Holder’s conduct of a trade or business within the United States.
U.S. Federal Estate Tax
The estates of non-resident alien individuals are generally subject to U.S. federal estate tax on property with a U.S. situs. Since we are a U.S. corporation, our common stock will be U.S. situs property and therefore will be included in the taxable estate of a non-resident alien decedent. The U.S. federal estate tax liability of the estate of a non-resident alien may be affected by a tax treaty between the United States and the decedent’s country of residence.
Dispositions of Our Common Stock
A Non-U.S. Holder generally will not be subject to U.S. federal income or withholding tax in respect of any gain on a sale or other disposition of our common stock unless:
| | • | | the gain is U.S. trade or business income, as defined and discussed above; |
| | • | | the Non-U.S. Holder is an individual who is present in the United States for 183 or more days in the taxable year of the disposition and meets other conditions; or |
| | • | | we are or have been a “U.S. real property holding corporation,” or a USRPHC, under section 897 of the Internal Revenue Code at any time during the shorter of the five year period ending on the date of disposition and the Non-U.S. Holder’s holding period for our common stock. |
In general, a corporation is a USRPHC if the fair market value of its “U.S. real property interests” (as defined in the Internal Revenue Code and applicable Treasury regulations) equals or exceeds 50% of the sum of the fair market value of its worldwide real property interests and its other assets used or held for use in a trade or business. If we are determined to be a USRPHC, the U.S. federal income and withholding taxes relating to
118
interests in USRPHCs nevertheless will not apply to gains derived from the sale or other disposition of our common stock by a Non-U.S. Holder whose shareholdings, actual and constructive, at all times during the applicable period, amount to 5% or less of our common stock, provided that our common stock is regularly traded on an established securities market, within the meaning of the applicable Treasury regulations. We are not currently a USRPHC, and we do not anticipate becoming a USRPHC in the future. However, no assurance can be given that we will not be a USRPHC, or that our common stock will be considered regularly traded on an established securities market, when a Non-U.S. Holder sells its shares of our common stock.
Information Reporting and Backup Withholding Requirements
We must annually report to the IRS and to each Non-U.S. Holder any dividend income that is subject to U.S. federal withholding tax, or that is exempt from such withholding tax pursuant to an income tax treaty. Copies of these information returns also may be made available under the provisions of a specific treaty or agreement to the tax authorities of the country in which the Non-U.S. Holder resides. Under certain circumstances, the Internal Revenue Code imposes a backup withholding obligation (currently at a rate of 28%) on certain reportable payments. Dividends paid to a Non-U.S. Holder of our common stock generally will be exempt from backup withholding if the Non-U.S. Holder provides a properly executed IRS Form W-8BEN or otherwise establishes an exemption.
The payment of the proceeds from the disposition of our common stock to or through the U.S. office of any broker, U.S. or foreign, will be subject to information reporting and possible backup withholding unless the holder certifies as to its non-U.S. status under penalties of perjury or otherwise establishes an exemption, provided that the broker does not have actual knowledge or reason to know that the holder is a U.S. person or that the conditions of any other exemption are not, in fact, satisfied. The payment of the proceeds from the disposition of our common stock to or through a non-U.S. office of a non-U.S. broker is one that will not be subject to information reporting or backup withholding unless the non-U.S. broker has certain types of relationships with the United States (a “U.S. related person”). In the case of the payment of the proceeds from the disposition of our common stock to or through a non-U.S office of a broker that is either a U.S. person or a U.S. related person, the Treasury regulations require information reporting (but not backup withholding) on the payment unless the broker has documentary evidence in its files that the holder is a Non-U.S. Holder and the broker has no knowledge to the contrary. Non-U.S. Holders should consult their own tax advisors on the application of information reporting and backup withholding to them in their particular circumstances (including upon their disposition of our common stock).
Backup withholding is not an additional tax. Any amounts withheld under the backup withholding rules from a payment to a Non-U.S. Holder will be refunded or credited against the Non-U.S. Holder’s U.S. federal income tax liability, if any, if the Non-U.S. Holder provides the required information to the IRS on a timely basis. Non-U.S. Holders should consult their own tax advisors regarding the filing of a U.S. tax return for claiming a refund of such backup withholding.
119
UNDERWRITING
Lehman Brothers Inc. and Deutsche Bank Securities Inc. are acting as the representatives of the underwriters and the joint book-running managers of this offering. Under the terms of an underwriting agreement, which will be filed as an exhibit to the registration statement, each of the underwriters named below has severally agreed to purchase from us respective number of common stock shown opposite its name below:
| | |
Underwriters | | Number of
Shares |
Lehman Brothers Inc. | | |
Deutsche Bank Securities Inc. | | |
Leerink Swann & Co., Inc. | | |
| | |
| |
Total | | |
| | |
The underwriting agreement provides that the underwriters’ obligation to purchase shares of common stock depends on the satisfaction of the conditions contained in the underwriting agreement including:
| | • | | the obligation to purchase all of the shares of common stock offered hereby (other than those shares of common stock covered by their option to purchase additional shares as described below), if any of the shares are purchased; |
| | • | | the representations and warranties made by us to the underwriters are true; |
| | • | | there is no material change in our business or the financial markets; and |
| | • | | we deliver customary closing documents to the underwriters. |
Commissions and Expenses
The following table summarizes the underwriting discounts and commissions we will pay to the underwriters. These amounts are shown assuming both no exercise and full exercise of the underwriters’ option to purchase additional shares. The underwriting fee is the difference between the initial price to the public and the amount the underwriters pay to us for the shares.
| | | | | | |
| | | No Exercise | | Full Exercise |
Per share | | $ | | | $ | |
Total | | $ | | | $ | |
The representatives of the underwriters have advised us that the underwriters propose to offer the shares of common stock directly to the public at the public offering price on the cover of this prospectus and to selected dealers, which may include the underwriters, at such offering price less a selling concession not in excess of $ per share. After the offering, the representatives may change the offering price and other selling terms.
The expenses of the offering that are payable by us are estimated to be $ (excluding underwriting discounts and commissions).
Option to Purchase Additional Shares
We have granted the underwriters an option exercisable for 30 days after the date of this prospectus, to purchase, from time to time, in whole or in part, up to an aggregate of shares at the public offering price less underwriting discounts and commissions. This option may be exercised if the underwriters sell more than shares in connection with this offering. To the extent that this option is exercised, each underwriter will be obligated, subject to certain conditions, to purchase its pro rata portion of these additional shares based on the underwriter’s underwriting commitment in the offering as indicated in the table at the beginning of this “Underwriting” section.
120
Lock-Up Agreements
We and holders of more than % of our outstanding stock, including all of our directors and executive officers, have agreed that, without the prior written consent of Lehman Brothers Inc., we and they will not, directly or indirectly, (1) offer for sale, sell, pledge or otherwise dispose of (or enter into any transaction or device that is designed to, or could be expected to, result in the disposition by any person at any time in the future of) any shares of common stock (including, without limitation, shares of common stock that may be deemed to be beneficially owned by us or them in accordance with the rules and regulations of the SEC and shares of common stock that may be issued upon exercise of any options or warrants) or securities convertible into or exercisable or exchangeable for common stock, (2) enter into any swap or other derivatives transaction that transfers to another, in whole or in part, any of the economic consequences of ownership of the common stock, (3) make any demand for or exercise any right or file or cause to be filed a registration statement, including any amendments thereto, with respect to the registration of any shares of common stock or securities convertible, exercisable or exchangeable into common stock or any of our other securities or (4) publicly disclose the intention to do any of the foregoing for a period of 180 days after the date of this prospectus.
The 180-day restricted period described in the preceding paragraph will be extended if:
| | • | | during the last 17 days of the 180-day restricted period we issue an earnings release or material news or a material event relating to us occurs; or |
| | • | | prior to the expiration of the 180-day restricted period, we announce that we will release earnings results during the 16-day period beginning on the last day of the 180-day period, |
in which case the restrictions described in the preceding paragraph will continue to apply until the expiration of the 18-day period beginning on the issuance of the earnings release or the announcement of the material news or occurrence of a material event, unless such extension is waived in writing by Lehman Brothers Inc.
The lock-up agreements do not prevent a security holder from transferring such securities (1) by bona fide gift, (2) to a trust for the benefit of the security holder or a member of the immediate family of the security holder, (3) by will or intestate succession, (4) to any affiliate of the security holder, (5) if the security holder is a corporation or similar entity, to any wholly-owned subsidiaries, (6) if the security holder is a partnership or limited liability company, to partners or members of the security holder, or (7) if the security holder is a trust, to any beneficiary of the security holder, provided that each such transferee agrees to be bound by the terms of the lock-up agreement. In addition, the lock-up agreements do not prevent a security holder from establishing a trading plan pursuant to Rule 10b5-1 under the Exchange Act, provided that no sales pursuant to such trading plan may be made during the lock-up period. Furthermore, the lock-up provisions described above do not limit our ability to (a) issue securities pursuant to employee benefit plans, qualified stock option plans or other employee compensation plans existing on the date of this prospectus, (b) issue securities pursuant to currently outstanding options, warrants or rights, or (c) file a registration statement on Form S-8 to register securities issuable under our stock plans.
Lehman Brothers Inc., in its sole discretion, may release the common stock and other securities subject to the lock-up agreements described above in whole or in part at any time with or without notice. When determining whether or not to release common stock and other securities from lock-up agreements, Lehman Brothers Inc. will consider, among other factors, the holder’s reasons for requesting the release, the number of shares of common stock and other securities for which the release is being requested and market conditions at the time.
Offering Price Determination
Prior to this offering, there has been no public market for our common stock. The initial public offering price will be negotiated between the representatives and us. In determining the initial public offering price of our common stock, the representatives will consider:
| | • | | the history and prospects for the industry in which we compete; |
121
| | • | | our financial information; |
| | • | | the ability of our management and our business potential and earning prospects; |
| | • | | the prevailing securities markets at the time of this offering; and |
| | • | | the recent market prices of, and the demand for, publicly traded shares of generally comparable companies. |
Indemnification
We have agreed to indemnify the underwriters against certain liabilities, including liabilities under the Securities Act, and to contribute to payments that the underwriters may be required to make for these liabilities.
Stabilization, Short Positions and Penalty Bids
The representatives may engage in stabilizing transactions, short sales and purchases to cover positions created by short sales, and penalty bids or purchases for the purpose of pegging, fixing or maintaining the price of the common stock, in accordance with Regulation M under the Securities Exchange Act of 1934:
| | • | | Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specified maximum. |
| | • | | A short position involves a sale by the underwriters of shares in excess of the number of shares the underwriters are obligated to purchase in the offering, which creates the syndicate short position. This short position may be either a covered short position or a naked short position. In a covered short position, the number of shares involved in the sales made by the underwriters in excess of the number of shares they are obligated to purchase is not greater than the number of shares that they may purchase by exercising their option to purchase additional shares. In a naked short position, the number of shares involved is greater than the number of shares in their option to purchase additional shares. The underwriters may close out any short position by either exercising their option to purchase additional shares or purchasing shares in the open market. In determining the source of shares to close out the short position, the underwriters will consider, among other things, the price of shares available for purchase in the open market as compared to the price at which they may purchase shares through their option to purchase additional shares. A naked short position is more likely to be created if the underwriters are concerned that there could be downward pressure on the price of the shares in the open market after pricing that could adversely affect investors who purchase in the offering. |
| | • | | Syndicate covering transactions involve purchases of the common stock in the open market after the distribution has been completed in order to cover syndicate short positions. |
| | • | | Penalty bids permit the representatives to reclaim a selling concession from a syndicate member when the common stock originally sold by the syndicate member is purchased in a stabilizing or syndicate covering transaction to cover syndicate short positions. |
These stabilizing transactions, syndicate covering transactions and penalty bids may have the effect of raising or maintaining the market price of our common stock or preventing or retarding a decline in the market price of the common stock. As a result, the price of the common stock may be higher than the price that might otherwise exist in the open market. These transactions may be effected on The NASDAQ Global Market or otherwise and, if commenced, may be discontinued at any time.
Neither we nor any of the underwriters make any representation or prediction as to the direction or magnitude of any effect that the transactions described above may have on the price of the common stock. In addition, neither we nor any of the underwriters make representation that the representatives will engage in these stabilizing transactions or that any transaction, once commenced, will not be discontinued without notice.
122
Electronic Distribution
A prospectus in electronic format may be made available on the Internet sites or through other online services maintained by one or more of the underwriters or selling group members participating in this offering, or by their affiliates. In those cases, prospective investors may view offering terms online and, depending upon the particular underwriter or selling group member, prospective investors may be allowed to place orders online. The underwriters may agree with us to allocate a specific number of shares for sale to online brokerage account holders. Any such allocation for online distributions will be made by the representatives on the same basis as other allocations.
Other than the prospectus in electronic format, the information on any underwriter’s or selling group member’s website and any information contained in any other website maintained by an underwriter or selling group member is not part of the prospectus or the registration statement of which this prospectus forms a part, has not been approved or endorsed by us or any underwriter or selling group member in its capacity as underwriter or selling group member and should not be relied upon by investors.
The NASDAQ Global Market
We have applied to have our common stock listed on The NASDAQ Global Market under the symbol “BLEX.”
Discretionary Sales
The underwriters have informed us that they do not intend to confirm sales to discretionary accounts that exceed 5% of the total number of shares offered by them.
Stamp Taxes
If you purchase shares of common stock offered in this prospectus, you may be required to pay stamp taxes and other charges under the laws and practices of the country of purchase, in addition to the offering price listed on the cover page of this prospectus.
Relationships
The underwriters may in the future perform investment banking and advisory services for us from time to time for which they may in the future receive customary fees and expenses.
Foreign Selling Restrictions
United Kingdom
This prospectus is only being distributed to, and is only directed at, persons in the United Kingdom that are qualified investors within the meaning of Article 2(1)(e) of the Prospectus Directive (“Qualified Investors”) that are also (i) investment professionals falling within Article 19(5) of the Financial Services and Markets Act 2000 (Financial Promotion) Order 2005 (the “Order”) or (ii) high net worth entities, and other persons to whom it may lawfully be communicated, falling within Article 49(2)(a) to (d) of the Order (all such persons together being referred to as “relevant persons”). This prospectus and its contents are confidential and should not be distributed, published or reproduced (in whole or in part) or disclosed by recipients to any other persons in the United Kingdom. Any person in the United Kingdom that is not a relevant persons should not act or rely on this document or any of its contents.
123
European Economic Area
In relation to each member state of the European Economic Area that has implemented the Prospectus Directive (each, a relevant member state), with effect from and including the date on which the Prospectus Directive is implemented in that relevant member state (the relevant implementation date), an offer of common stock described in this prospectus may not be made to the public in that relevant member state prior to the publication of a prospectus in relation to the common stock that has been approved by the competent authority in that relevant member state or, where appropriate, approved in another relevant member state and notified to the competent authority in that relevant member state, all in accordance with the Prospectus Directive, except that, with effect from and including the relevant implementation date, an offer of securities may be offered to the public in that relevant member state at any time:
| | • | | to any legal entity that is authorized or regulated to operate in the financial markets or, if not so authorized or regulated, whose corporate purpose is solely to invest in securities; |
| | • | | to any legal entity that has two or more of (1) an average of at least 250 employees during the last financial year; (2) a total balance sheet of more than €43,000,000 and (3) an annual net turnover of more than €50,000,000, as shown in its last annual or consolidated accounts; or |
| | • | | in any other circumstances that do not require the publication of a prospectus pursuant to Article 3 of the Prospectus Directive. |
Each purchaser of common stock described in this prospectus located within a relevant member state will be deemed to have represented, acknowledged and agreed that it is a “qualified investor” within the meaning of Article 2(1)(e) of the Prospectus Directive.
For purposes of this provision, the expression an “offer to the public” in any relevant member state means the communication in any form and by any means of sufficient information on the terms of the offer and the securities to be offered so as to enable an investor to decide to purchase or subscribe the securities, as the expression may be varied in that member state by any measure implementing the Prospectus Directive in that member state, and the expression “Prospectus Directive” means Directive 2003/71/EC and includes any relevant implementing measure in each relevant member state.
We have not authorized and do not authorize the making of any offer of common stock through any financial intermediary on our behalf, other than offers made by the underwriters with a view to the final placement of any shares of common stock as contemplated in this prospectus. Accordingly, no purchaser of common stock, other than the underwriters, is authorized to make any further offer of any shares of common stock on behalf of us or the underwriters.
124
LEGAL MATTERS
The validity of the issuance of the common stock offered by us in this offering will be passed upon for us by Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C., Boston, Massachusetts. The underwriters are being represented by Pillsbury Winthrop Shaw Pittman LLP, New York, New York.
EXPERTS
The consolidated financial statements as of December 31, 2006 and December 31, 2005 and for each of the three years in the period ended December 31, 2006 included in this prospectus have been so included in reliance on the report of PricewaterhouseCoopers LLP, an independent registered public accounting firm, given on the authority of said firm as experts in auditing and accounting.
The profit and loss statement and cash flow statement of LemnaGene S.A. for the period from January 1, 2005 to July 7, 2005 included in this prospectus have been so included in reliance on the report of Frédéric Brejon, statutory auditor in France, given on the authority of said auditor as an expert in auditing and accounting.
WHERE YOU CAN FIND ADDITIONAL INFORMATION
We have filed with the SEC a registration statement on Form S-1 (File No. 333- ) under the Securities Act, with respect to the common stock offered by this prospectus. This prospectus, which is part of the registration statement, omits certain information, exhibits, schedules and undertakings set forth in the registration statement. For further information pertaining to us and our common stock, reference is made to the registration statement and the exhibits and schedules to the registration statement. Statements contained in this prospectus as to the contents or provisions of any documents referred to in this prospectus are not necessarily complete, and in each instance where a copy of the document has been filed as an exhibit to the registration statement, reference is made to the exhibit for a more complete description of the matters involved.
You may read and copy all or any portion of the registration statement without charge at the public reference room of the SEC at 100 F Street, N.E., Washington, D.C. 20549. Copies of the registration statement may be obtained from the SEC at prescribed rates from the public reference room of the SEC at such address. You may obtain information regarding the operation of the public reference room by calling 1-800-SEC-0330. In addition, registration statements and certain other filings made with the SEC electronically are publicly available through the SEC’s website at http://www.sec.gov. The registration statement, including all exhibits and amendments to the registration statement, has been filed electronically with the SEC.
Upon completion of this offering, we will become subject to the information and periodic reporting requirements of the Exchange Act and, accordingly, will file annual reports containing financial statements audited by our independent registered public accounting firm, quarterly reports containing unaudited financial data, current reports, proxy statements and other information with the SEC. You will be able to inspect and copy such periodic reports, proxy statements and other information at the SEC’s public reference room, and the website of the SEC referred to above.
125
INDEX TO FINANCIAL STATEMENTS
Biolex, Inc.
(A Development Stage Company)
Years ended December 31, 2004, 2005 and 2006, three months ended March 31, 2006 and 2007
and the period from September 19, 1997 (inception) through March 31, 2007
F-1
Report of Independent Registered Public Accounting Firm
To the Board of Directors and
Stockholders of Biolex, Inc. and its subsidiaries
In our opinion, the accompanying consolidated balance sheets and the related consolidated statements of operations, of changes in stockholders’ deficit and of cash flows present fairly, in all material respects, the financial position of Biolex, Inc. and its subsidiaries (a development stage enterprise) at December 31, 2006 and 2005, and the results of their operations and their cash flows for each of the three years in the period ended December 31, 2006 in conformity with accounting principles generally accepted in the United States of America. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits. We conducted our audits of these statements in accordance with the standards of the Public Company Accounting Oversight Board (United States). Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements, assessing the accounting principles used and significant estimates made by management, and evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
As discussed in Note 3 and Note 10 to the consolidated financial statements, the Company changed the manner in which it accounts for share-based compensation in 2006.
As discussed in Note 3 and Note 9 to the consolidated financial statements, the Company changed the manner in which it accounts for preferred stock warrants in 2005.
As discussed in Note 2 to the consolidated financial statements, the Company has restated its 2005 consolidated financial statements.
/s/ PricewaterhouseCoopers LLP
Raleigh, North Carolina
August 13, 2007
F-2
Biolex, Inc.
(A Development Stage Company)
Consolidated Balance Sheets
| | | | | | | | | | | | | | | | |
| | | December 31, | | | March 31, 2007 | | | Pro Forma
March 31,
2007 | |
| | | 2005 | | | 2006 | | | |
| | | as restated
(see Note 2) | | | | | | (unaudited) | | | (unaudited) | |
Assets | | | | | | | | | | | | | | | | |
Current assets | | | | | | | | | | | | | | | | |
Cash and cash equivalents | | $ | 27,468,138 | | | $ | 14,552,779 | | | $ | 8,707,698 | | | $ | 8,707,698 | |
Accounts receivable | | | 190,259 | | | | 70,764 | | | | 494,826 | | | | 494,826 | |
Other current assets and prepaid expenses | | | 498,969 | | | | 496,581 | | | | 533,766 | | | | 532,766 | |
| | | | | | | | | | | | | | | | |
Total current assets | | | 28,157,366 | | | | 15,120,124 | | | | 9,736,290 | | | | 9,736,290 | |
Property and equipment, net | | | 5,496,909 | | | | 5,826,763 | | | | 5,590,828 | | | | 5,590,828 | |
Deposits and other assets | | | 267,094 | | | | 268,534 | | | | 229,637 | | | | 229,637 | |
Intangible assets, net | | | 3,796,199 | | | | 3,312,602 | | | | 3,237,531 | | | | 3,237,531 | |
Restricted cash | | | 378,000 | | | | 378,000 | | | | 378,000 | | | | 378,000 | |
| | | | | | | | | | | | | | | | |
Total assets | | $ | 38,095,568 | | | $ | 24,906,023 | | | $ | 19,172,286 | | | $ | 19,172,286 | |
| | | | | | | | | | | | | | | | |
Liabilities, Mandatorily Redeemable Convertible Preferred Stock and Stockholders’ Equity (Deficit) | | | | | | | | | | | | | | | | |
Current liabilities | | | | | | | | | | | | | | | | |
Accounts payable | | $ | 1,136,109 | | | $ | 850,033 | | | $ | 905,240 | | | $ | 905,240 | |
Accrued expenses | | | 2,407,039 | | | | 2,181,116 | | | | 2,579,172 | | | | 2,579,172 | |
Deferred revenue | | | 2,906,667 | | | | 2,227,550 | | | | 2,354,915 | | | | 2,354,915 | |
Notes payable | | | 981,813 | | | | 1,403,445 | | | | 1,392,148 | | | | 1,392,148 | |
Capital lease obligations | | | 27,005 | | | | 1,967 | | | | — | | | | — | |
| | | | | | | | | | | | | | | | |
Total current liabilities | | | 7,458,633 | | | | 6,664,111 | | | | 7,231,475 | | | | 7,231,475 | |
Deferred revenue | | | 2,762,643 | | | | 2,676,144 | | | | 2,450,345 | | | | 2,450,345 | |
Notes payable, less current portion | | | 2,912,491 | | | | 2,668,136 | | | | 2,319,230 | | | | 2,319,230 | |
Convertible note payable to related party | | | — | | | | 5,000,000 | | | | 5,000,000 | | | | 5,000,000 | |
Deferred tax liability | | | 562,958 | | | | 285,471 | | | | 251,228 | | | | 251,228 | |
Deferred lease obligation | | | — | | | | 685,878 | | | | 668,697 | | | | 668,697 | |
Commitments and contingencies (Note 11) | | | | | | | | | | | | | | | | |
Mandatorily redeemable convertible preferred stock | | | | | | | | | | | | | | | | |
Series AA1 preferred stock, $0.001 par value, 5,092,701 shares authorized and outstanding at December 31, 2005 and 2006 and March 31, 2007 (unaudited) and no shares outstanding, pro forma; liquidation preference of $5,092,701 at December 31, 2006 and March 31, 2007 (unaudited) | | | 5,737,052 | | | | 5,784,750 | | | | 5,910,324 | | | | — | |
Series AA2 preferred stock, $0.001 par value, 22,594,864 shares authorized and outstanding at December 31, 2005 and 2006 and March 31, 2007 (unaudited) and no shares outstanding, pro forma; liquidation preference of $22,594,864 at December 31, 2006 and March 31, 2007 (unaudited) | | | 23,233,104 | | | | 25,376,149 | | | | 25,904,571 | | | | — | |
Series BB preferred stock, $0.001 par value, 33,902,000 shares authorized and 32,120,192 shares outstanding at December 31, 2005 and 2006 and March 31, 2007 (unaudited) and no shares outstanding, pro forma; liquidation preference of $35,974,615 at December 31, 2006 and March 31, 2007 (unaudited) | | | 37,005,711 | | | | 40,414,797 | | | | 41,255,393 | | | | — | |
Stockholders’ equity (deficit) | | | | | | | | | | | | | | | | |
Common Stock, $.001 par value, no shares outstanding at December 31, 2005 and 2006 and March 31, 2007 (unaudited) 110,000,000 shares authorized and 62,087,644 shares outstanding, pro forma | | | — | | | | — | | | | — | | | | 62,088 | |
Class A common stock, $0.001 par value, 80,000,000 shares authorized, 295,194, 460,064, and 494,258 shares issued and outstanding at December 31, 2005 and 2006 and March 31, 2007 (unaudited), and no shares outstanding, proforma | | | 296 | | | | 461 | | | | 495 | | | | — | |
Class B common stock, $0.001 par value, 3,821 shares authorized, 3,821 shares issued and outstanding at December 31, 2005 and 2006 and March 31, 2007 (unaudited), and no shares outstanding proforma | | | 4 | | | | 4 | | | | 4 | | | | — | |
Additional paid-in capital | | | 10,667,536 | | | | 5,118,063 | | | | 3,635,153 | | | | 76,643,852 | |
Deferred compensation | | | (10,748 | ) | | | — | | | | — | | | | — | |
Accumulated other comprehensive income (loss) | | | 7,573 | | | | (29,412 | ) | | | (33,725 | ) | | | (33,725 | ) |
Deficit accumulated during development stage | | | (52,241,685 | ) | | | (69,738,529 | ) | | | (75,420,904 | ) | | | (75,420,904 | ) |
| | | | | | | | | | | | | | | | |
Total stockholders' equity (deficit) | | | (41,577,024 | ) | | | (64,649,413 | ) | | | (71,818,977 | ) | | | 1,251,311 | |
| | | | | | | | | | | | | | | | |
Total liabilities, mandatorily redeemable convertible preferred stock and stockholders' equity (deficit) | | $ | 38,095,568 | | | $ | 24,906,023 | | | $ | 19,172,286 | | | $ | 19,172,286 | |
| | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-3
Biolex, Inc.
(A Development Stage Company)
Consolidated Statements of Operations
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31, | | | Three Months Ended
March 31, | | | Period from
September 19, 1997
(date of inception) to
March 31, 2007 | |
| | | 2004 | | | 2005 | | | 2006 | | | 2006 | | | 2007 | | |
| | | | | | as restated (see Note 2) | | | | | | (unaudited) | | | (unaudited) | |
Revenues | | $ | 719,375 | | | $ | 3,007,991 | | | $ | 5,016,896 | | | $ | 1,326,913 | | | $ | 724,146 | | | $ | 9,752,653 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Operating expenses | | | | | | | | | | | | | | | | | | | | | | | | |
Research and development | | | 8,665,838 | | | | 14,448,625 | | | | 19,195,615 | | | | 5,083,750 | | | | 5,292,221 | | | | 60,632,635 | |
General and administrative | | | 2,400,039 | | | | 4,125,559 | | | | 4,370,027 | | | | 1,095,844 | | | | 1,114,112 | | | | 19,686,775 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Total operating expenses | | | 11,065,877 | | | | 18,574,184 | | | | 23,565,642 | | | | 6,179,594 | | | | 6,406,333 | | | | 80,319,410 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Loss from operations | | | (10,346,502 | ) | | | (15,566,193 | ) | | | (18,548,746 | ) | | | (4,852,681 | ) | | | (5,682,187 | ) | | | (70,566,757 | ) |
Interest income | | | 33,689 | | | | 544,172 | | | | 935,169 | | | | 252,968 | | | | 142,236 | | | | 1,836,367 | |
Interest expense | | | (90,958 | ) | | | (965,423 | ) | | | (160,754 | ) | | | (101,610 | ) | | | (176,667 | ) | | | (3,570,469 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Loss before income taxes | | | (10,403,771 | ) | | | (15,987,444 | ) | | | (17,774,331 | ) | | | (4,701,323 | ) | | | (5,716,618 | ) | | | (72,300,859 | ) |
Income tax benefit | | | — | | | | 170,159 | | | | 277,487 | | | | 31,142 | | | | 34,243 | | | | 481,889 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | | (10,403,771 | ) | | | (15,817,285 | ) | | | (17,496,844 | ) | | | (4,670,181 | ) | | | (5,682,375 | ) | | | (71,818,970 | ) |
Less: Accretion of mandatorily redeemable convertible preferred stock | | | (1,165,523 | ) | | | (3,033,747 | ) | | | (5,599,829 | ) | | | (1,369,019 | ) | | | (1,494,592 | ) | | | (16,771,986 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Net loss attributable to common stockholders | | $ | (11,569,294 | ) | | $ | (18,851,032 | ) | | $ | (23,096,673 | ) | | $ | (6,039,200 | ) | | $ | (7,176,967 | ) | | $ | (88,590,956 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Basic and diluted net loss attributable to common stockholders per share | | $ | (42.91 | ) | | $ | (65.25 | ) | | $ | (68.52 | ) | | $ | (20.18 | ) | | $ | (14.93 | ) | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Basic and diluted weighted average shares outstanding | | | 269,595 | | | | 288,890 | | | | 337,078 | | | | 299,212 | | | | 480,573 | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Pro forma net loss per share (unaudited) | | | | | | | | | | $ | (0.29 | ) | | | | | | $ | (0.09 | ) | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Shares used to compute pro forma basic and diluted net loss per share (unaudited) | | | | | | | | | | | 60,144,836 | | | | | | | | 60,288,331 | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-4
Biolex, Inc.
(A Development Stage Company)
Consolidated Statements of Changes in Stockholders’ Deficit
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Class A Common Stock | | | Class B Common Stock | | Additional
Paid-in Capital | | | Deferred Compensation | | | Deficit
Accumulated
during
Development Stage | | | Accumulated
Other
Comprehensive Income (Loss) | | Total
Stockholders’
Equity (Deficit) | |
| | | Shares | | | Amount | | | Shares | | Amount | | | | | |
Balance as of September 19, 1997 (date of inception) | | — | | | $ | — | | | — | | $ | — | | $ | — | | | $ | — | | | $ | — | | | $ | — | | $ | — | |
Net loss | | — | | | | — | | | — | | | — | | | — | | | | — | | | | (4,650 | ) | | | — | | | (4,650 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance as of December 31, 1997 | | — | | | | — | | | — | | | — | | | — | | | | — | | | | (4,650 | ) | | | — | | | (4,650 | ) |
Sale of shares of common stock | | 31,457 | | | | 31 | | | — | | | — | | | 729 | | | | — | | | | — | | | | — | | | 760 | |
Issuance of shares of common stock in exchange for license | | — | | | | — | | | 2,940 | | | 3 | | | 6,588 | | | | — | | | | — | | | | — | | | 6,591 | |
Accretion of preferred stock | | — | | | | — | | | — | | | — | | | (6,520 | ) | | | — | | | | (10,284 | ) | | | — | | | (16,804 | ) |
Net loss | | — | | | | — | | | — | | | — | | | — | | | | — | | | | (149,511 | ) | | | — | | | (149,511 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance as of December 31, 1998 | | 31,457 | | | | 31 | | | 2,940 | | | 3 | | | 797 | | | | — | | | | (164,445 | ) | | | — | | | (163,614 | ) |
Repurchase of shares of restricted common stock | | (3,622 | ) | | | (3 | ) | | — | | | — | | | (85 | ) | | | — | | | | — | | | | — | | | (88 | ) |
Value of warrants issued in connection with notes payable | | — | | | | — | | | — | | | — | | | 17,141 | | | | — | | | | — | | | | — | | | 17,141 | |
Deferred compensation relating to stock options | | — | | | | — | | | — | | | — | | | 17,635 | | | | (17,635 | ) | | | — | | | | — | | | — | |
Amortization of deferred compensation | | — | | | | — | | | — | | | — | | | — | | | | 355 | | | | — | | | | — | | | 355 | |
Accretion of preferred stock | | — | | | | — | | | — | | | — | | | (34,776 | ) | | | — | | | | (93,641 | ) | | | — | | | (128,417 | ) |
Net loss | | — | | | | — | | | — | | | — | | | — | | | | — | | | | (994,443 | ) | | | — | | | (994,443 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance as of December 31, 1999 | | 27,835 | | | | 28 | | | 2,940 | | | 3 | | | 712 | | | | (17,280 | ) | | | (1,252,529 | ) | | | — | | | (1,269,066 | ) |
Issuance of shares of common stock in exchange for license | | — | | | | — | | | 881 | | | 1 | | | 20 | | | | — | | | | (21 | ) | | | — | | | — | |
Repurchase of shares of restricted common stock | | (2,717 | ) | | | (3 | ) | | — | | | — | | | (62 | ) | | | — | | | | — | | | | — | | | (65 | ) |
Amortization of deferred compensation | | — | | | | — | | | — | | | — | | | — | | | | 2,583 | | | | — | | | | — | | | 2,583 | |
Accretion of preferred stock | | — | | | | — | | | — | | | — | | | — | | | | — | | | | (445,057 | ) | | | — | | | (445,057 | ) |
Net loss | | — | | | | — | | | — | | | — | | | — | | | | — | | | | (2,362,447 | ) | | | — | | | (2,362,447 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance as of December 31, 2000 | | 25,118 | | | | 25 | | | 3,821 | | | 4 | | | 670 | | | | (14,697 | ) | | | (4,060,054 | ) | | | — | | | (4,074,052 | ) |
The accompanying notes are an integral part of these consolidated financial statements.
(continued)
F-5
Biolex, Inc.
(A Development Stage Company)
Consolidated Statements of Changes in Stockholders’ Deficit—(Continued)
| | | | | | | | | | | | | | | | | | | | | | |
| | | Class A Common Stock | | Class B
Common Stock | | Additional
Paid-in Capital | | | Deferred Compensation | | | Deficit
Accumulated
during
Development Stage | | | Accumulated
Other
Comprehensive Income (Loss) | | Total
Stockholders’
Equity (Deficit) | |
| | | Shares | | Amount | | Shares | | Amount | | | | | |
Balance as of December 31, 2000 | | 25,118 | | 25 | | 3,821 | | 4 | | 670 | | | (14,697 | ) | | (4,060,054 | ) | | — | | (4,074,052 | ) |
Issuance of shares of common stock for exercise of options | | 2,119 | | 2 | | — | | — | | 22,118 | | | — | | | — | | | — | | 22,120 | |
Value of warrants issued in connection with notes payable | | — | | — | | — | | — | | 282,966 | | | — | | | — | | | — | | 282,966 | |
Amortization of deferred compensation | | — | | — | | — | | — | | — | | | 6,145 | | | — | | | — | | 6,145 | |
Accretion of preferred stock | | — | | — | | — | | — | | (305,035 | ) | | — | | | (1,108,900 | ) | | — | | (1,413,935 | ) |
Net loss | | — | | — | | — | | — | | — | | | — | | | (4,779,075 | ) | | — | | (4,779,075 | ) |
| | | | | | | | | | | | | | | | | | | | | | |
Balance as of December 31, 2001 | | 27,237 | | 27 | | 3,821 | | 4 | | 719 | | | (8,552 | ) | | (9,948,029 | ) | | — | | (9,955,831 | ) |
Issuance of shares of common stock for exercise of options | | 948 | | 1 | | — | | — | | 3,169 | | | — | | | — | | | — | | 3,170 | |
Value of warrants issued in connection with notes payable | | — | | — | | — | | — | | 1,110,580 | | | — | | | — | | | — | | 1,110,580 | |
Deferred compensation relating to stock options | | — | | — | | — | | — | | 8,241 | | | (8,241 | ) | | — | | | — | | — | |
Amortization of deferred compensation | | — | | — | | — | | — | | — | | | 3,785 | | | — | | | — | | 3,785 | |
Accretion of preferred stock | | — | | — | | — | | — | | (1,121,968 | ) | | — | | | (704,094 | ) | | — | | (1,826,062 | ) |
Net loss | | — | | — | | — | | — | | — | | | — | | | (7,106,844 | ) | | — | | (7,106,844 | ) |
| | | | | | | | | | | | | | | | | | | | | | |
Balance as of December 31, 2002 | | 28,185 | | 28 | | 3,821 | | 4 | | 741 | | | (13,008 | ) | | (17,758,967 | ) | | — | | (17,771,202 | ) |
Issuance of shares of common stock for exercise of options | | 4,576 | | 5 | | — | | — | | 453 | | | — | | | — | | | — | | 458 | |
Value of warrants issued in connection with notes payable | | — | | — | | — | | — | | 829,655 | | | — | | | — | | | — | | 829,655 | |
Conversion of preferred stock to common stock | | 232,993 | | 233 | | — | | — | | 14,194,473 | | | — | | | — | | | — | | 14,194,706 | |
Deferred compensation relating to stock options | | — | | — | | — | | — | | (5,445 | ) | | 5,445 | | | — | | | — | | — | |
Amortization of deferred compensation | | — | | — | | — | | — | | — | | | 2,071 | | | — | | | — | | 2,071 | |
Accretion of preferred stock | | — | | — | | — | | — | | (408,082 | ) | | — | | | (1,239,937 | ) | | — | | (1,648,019 | ) |
Net loss | | — | | — | | — | | — | | — | | | — | | | (7,021,725 | ) | | — | | (7,021,725 | ) |
| | | | | | | | | | | | | | | | | | | | | | |
Balance as of December 31, 2003 | | 265,754 | | 266 | | 3,821 | | 4 | | 14,611,795 | | | (5,492 | ) | | (26,020,629 | ) | | — | | (11,414,056 | ) |
The accompanying notes are an integral part of these consolidated financial statements.
(continued)
F-6
Biolex, Inc.
(A Development Stage Company)
Consolidated Statements of Changes in Stockholders’ Deficit—(Continued)
| | | | | | | | | | | | | | | | | | | | | | | |
| | | Class A Common Stock | | Class B Common Stock | | Additional
Paid-in Capital | | | Deferred Compensation | | | Deficit
Accumulated
during
Development Stage | | | Accumulated
Other
Comprehensive Income (Loss) | | | Total
Stockholders’ Deficit | |
| | | Shares | | Amount | | Shares | | Amount | | | | | |
Balance as of December 31, 2003 | | 265,754 | | 266 | | 3,821 | | 4 | | 14,611,795 | | | (5,492 | ) | | (26,020,629 | ) | | — | | | (11,414,056 | ) |
Issuance of shares of common stock for exercise of options | | 7,500 | | 8 | | — | | — | | 742 | | | — | | | — | | | — | | | 750 | |
Value of warrants issued in connection with notes payable | | — | | — | | — | | — | | 23,697 | | | — | | | — | | | — | | | 23,697 | |
Deferred compensation relating to stock options | | — | | — | | — | | — | | 182,116 | | | (182,116 | ) | | — | | | — | | | — | |
Amortization of deferred compensation | | — | | — | | — | | — | | — | | | 139,320 | | | — | | | — | | | 139,320 | |
Accretion of preferred stock | | — | | — | | — | | — | | (1,165,523 | ) | | — | | | — | | | — | | | (1,165,523 | ) |
Net loss | | — | | — | | — | | — | | — | | | — | | | (10,403,771 | ) | | — | | | (10,403,771 | ) |
| | | | | | | | | | | | | | | | | | | | | | | |
Balance as of December 31, 2004 | | 273,254 | | 274 | | 3,821 | | 4 | | 13,652,827 | | | (48,288 | ) | | (36,424,400 | ) | | — | | | (22,819,583 | ) |
Issuance of shares of common stock for exercise of options | | 21,940 | | 22 | | — | | — | | 2,172 | | | — | | | — | | | — | | | 2,194 | |
Value of warrants issued in connection with notes payable | | — | | — | | — | | — | | 46,284 | | | — | | | — | | | — | | | 46,284 | |
Other comprehensive income foreign currency translation adjustment | | — | | — | | — | | — | | — | | | — | | | — | | | 7,573 | | | 7,573 | |
Amortization of deferred compensation | | — | | — | | — | | — | | — | | | 37,540 | | | — | | | — | | | 37,540 | |
Accretion of preferred stock | | — | | — | | — | | — | | (3,033,747 | ) | | — | | | — | | | — | | | (3,033,747 | ) |
Net loss, as restated (see Note 2) | | — | | — | | — | | — | | — | | | — | | | (15,817,285 | ) | | — | | | (15,817,285 | ) |
| | | | | | | | | | | | | | | | | | | | | | | |
Balance as of December 31, 2005 as restated (see Note 2) | | 295,194 | | 296 | | 3,821 | | 4 | | 10,667,536 | | | (10,748 | ) | | (52,241,685 | ) | | 7,573 | | | (41,577,024 | ) |
Issuance of shares of common stock for exercise of options | | 164,870 | | 165 | | — | | — | | 16,809 | | | — | | | — | | | — | | | 16,974 | |
Value of warrants issued in connection with notes payable | | — | | — | | — | | — | | 10,782 | | | — | | | — | | | — | | | 10,782 | |
Other comprehensive income foreign currency translation adjustment | | — | | — | | — | | — | | — | | | — | | | — | | | (36,985 | ) | | (36,985 | ) |
Accretion of preferred stock | | — | | — | | — | | — | | (5,599,829 | ) | | — | | | — | | | — | | | (5,599,829 | ) |
Deferred compensation relating to stock options and amortization of deferred compensation | | — | | — | | — | | — | | 22,765 | | | 10,748 | | | — | | | — | | | 33,513 | |
Net loss | | — | | — | | — | | — | | — | | | — | | | (17,496,844 | ) | | — | | | (17,496,844 | ) |
| | | | | | | | | | | | | | | | | | | | | | | |
Balance as of December 31, 2006 | | 460,064 | | 461 | | 3,821 | | 4 | | 5,118,063 | | | — | | | (69,738,529 | ) | | (29,412 | ) | | (64,649,413 | ) |
The accompanying notes are an integral part of these consolidated financial statements.
(continued)
F-7
Biolex, Inc.
(A Development Stage Company)
Consolidated Statements of Changes in Stockholders’ Deficit—(Continued)
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
| | | Class A Common Stock | | Class B Common Stock | | Additional
Paid-in Capital | | | Deferred Compensation | | Deficit
Accumulated
during
Development Stage | | | Accumulated
Other
Comprehensive Income (Loss) | | | Total
Stockholders’ Deficit | |
| | | Shares | | Amount | | Shares | | Amount | | | | | |
Balance as of December 31, 2006 | | 460,064 | | | 461 | | 3,821 | | | 4 | | | 5,118,063 | | | | — | | | (69,738,529 | ) | | | (29,412 | ) | | | (64,649,413 | ) |
Issuance of shares of common stock for exercise of options (unaudited) | | 34,194 | | | 34 | | — | | | — | | | 4,646 | | | | — | | | — | | | | — | | | | 4,680 | |
Other comprehensive income foreign currency translation adjustment (unaudited) | | — | | | — | | — | | | — | | | — | | | | — | | | — | | | | (4,313 | ) | | | (4,313 | ) |
Accretion of preferred stock (unaudited) | | — | | | — | | — | | | — | | | (1,494,592 | ) | | | — | | | — | | | | — | | | | (1,494,592 | ) |
Stock based compensation (unaudited) | | — | | | — | | — | | | — | | | 7,036 | | | | — | | | — | | | | — | | | | 7,036 | |
Net loss (unaudited) | | — | | | — | | — | | | — | | | — | | | | — | | | (5,682,375 | ) | | | — | | | | (5,682,375 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
Balance as of March 31, 2007 (unaudited) | | 494,258 | | $ | 495 | | 3,821 | | $ | 4 | | $ | 3,635,153 | | | $ | — | | $ | (75,420,904 | ) | | $ | (33,725 | ) | | $ | (71,818,977 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-8
Biolex, Inc.
(A Development Stage Company)
Consolidated Statements of Cash Flows
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31, | | | Three Months Ended
March 31, | | | Period from
September 19,
1997 (date of
inception) to
March 31, 2007 | |
| | | 2004 | | | 2005 | | | 2006 | | | 2006 | | | 2007 | | |
| | | | | | as restated
(see Note 2) | | | | | | (unaudited) | | | (unaudited) | |
Cash flows from operating activities | | | | | | | | | | | | | | | | | | | | | | | | |
Net loss | | $ | (10,403,771 | ) | | $ | (15,817,285 | ) | | $ | (17,496,844 | ) | | $ | (4,670,181 | ) | | $ | (5,682,375 | ) | | $ | (71,818,970 | ) |
Adjustments to reconcile net loss to net cash used in operating activities | | | | | | | | | | | | | | | | | | | | | | | | |
Depreciation and amortization expense | | | 1,287,687 | | | | 2,007,379 | | | | 2,016,754 | | | | 475,114 | | | | 520,012 | | | | 7,055,644 | |
Noncash compensation expense | | | 139,320 | | | | 37,527 | | | | 33,513 | | | | 3,970 | | | | 7,036 | | | | 232,336 | |
Deferred tax liability | | | — | | | | 562,958 | | | | (277,489 | ) | | | (31,142 | ) | | | (34,243 | ) | | | 251,226 | |
Noncash interest expense | | | 4,185 | | | | 8,754 | | | | 10,782 | | | | 6,351 | | | | — | | | | 2,308,874 | |
Impairment of fixed assets and loss on disposal of property and equipment | | | 52,671 | | | | 5,548 | | | | 74,158 | | | | 7,332 | | | | 40 | | | | 182,058 | |
Other | | | — | | | | — | | | | — | | | | — | | | | — | | | | 6,591 | |
Changes in operating assets and liabilities | | | | | | | | | | | | | | | | | | | | | | | | |
Accounts receivable | | | (357,308 | ) | | | 167,049 | | | | 134,127 | | | | 105,643 | | | | (438,693 | ) | | | (494,825 | ) |
Other current assets and prepaid expenses | | | 3,872 | | | | (489,659 | ) | | | 1,050 | | | | (63,208 | ) | | | (28,941 | ) | | | (541,114 | ) |
Deposits and other assets | | | (41,413 | ) | | | (752,970 | ) | | | 19,690 | | | | 6,668 | | | | (4,481 | ) | | | (818,954 | ) |
Accounts payable | | | 572,742 | | | | 23,714 | | | | (301,921 | ) | | | 268,579 | | | | 53,944 | | | | 555,212 | |
Accrued expenses | | | 295,078 | | | | 1,479,706 | | | | 401,240 | | | | (363,830 | ) | | | 381,314 | | | | 3,891,230 | |
Deferred revenue | | | 280,625 | | | | 5,280,738 | | | | (767,273 | ) | | | (259,801 | ) | | | (98,432 | ) | | | 4,780,033 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Net cash used in operating activities | | | (8,166,312 | ) | | | (7,486,540 | ) | | | (16,152,213 | ) | | | (4,514,506 | ) | | | (5,324,819 | ) | | | (54,410,659 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
Cash flows from investing activities | | | | | | | | | | | | | | | | | | | | | | | | |
Purchases of available for sale securities | | | (22,000,000 | ) | | | — | | | | — | | | | — | | | | — | | | | (49,000,000 | ) |
Proceeds from sale of available for sale securities | | | 28,500,000 | | | | — | | | | — | | | | — | | | | — | | | | 49,000,000 | |
Purchases of short term restricted cash | | | — | | | | (378,000 | ) | | | — | | | | — | | | | — | | | | (378,000 | ) |
Purchases of property and equipment | | | (2,021,940 | ) | | | (4,960,122 | ) | | | (1,934,580 | ) | | | (713,503 | ) | | | (163,212 | ) | | | (11,643,901 | ) |
Purchase of intangible assets | | | (300,000 | ) | | | — | | | | — | | | | | | | | | | | | (315,000 | ) |
Purchase of subsidiary | | | — | | | | (2,436,454 | ) | | | — | | | | — | | | | — | | | | (2,436,454 | ) |
Cash acquired from purchase of subsidiary | | | — | | | | 131,336 | | | | — | | | | — | | | | — | | | | 131,336 | |
Proceeds from sale of property and equipment | | | — | | | | — | | | | — | | | | — | | | | — | | | | 21,995 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Net cash provided by (used in) investing activities | | | 4,178,060 | | | | (7,643,240 | ) | | | (1,934,580 | ) | | | (713,503 | ) | | | (163,212 | ) | | | (14,620,024 | ) |
| | | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
(continued)
F-9
Biolex, Inc.
(A Development Stage Company)
Consolidated Statements of Cash Flows—(Continued)
| | | | | | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31, | | | Three Months Ended
March 31, | | | Period from
September 19,
1997 (date of
inception) to
March 31, 2007 | |
| | | 2004 | | | 2005 | | | 2006 | | | 2006 | | | 2007 | | |
| | | | | | as restated
(see Note 2) | | | | | | (unaudited) | | | (unaudited) | |
Cash flows from financing activities | | | | | | | | | | | | | | | | | | | | | | | | |
Proceeds from issuance of mandatorily redeemable convertible preferred stock, net of issuance costs | | | 2,968,017 | | | | 31,325,694 | | | | — | | | | — | | | | — | | | | 51,960,244 | |
Proceeds from issuance of notes payable and warrants | | | 922,807 | | | | 3,588,930 | | | | 1,411,209 | | | | 1,037,682 | | | | — | | | | 15,637,114 | |
Payments of notes payable | | | (273,494 | ) | | | (656,864 | ) | | | (1,236,194 | ) | | | (236,800 | ) | | | (360,387 | ) | | | (2,809,531 | ) |
Payment of capital lease obligations | | | (28,225 | ) | | | (29,833 | ) | | | (25,038 | ) | | | (6,088 | ) | | | (1,967 | ) | | | (106,205 | ) |
Proceeds from sale of common stock and exercise of stock options | | | 750 | | | | 2,194 | | | | 16,974 | | | | 975 | | | | 4,680 | | | | 50,953 | |
Proceeds from convertible note payable | | | — | | | | 8,000,000 | | | | 5,000,000 | | | | — | | | | — | | | | 13,000,000 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Net cash provided by (used in) financing activities | | | 3,589,855 | | | | 42,230,121 | | | | 5,166,951 | | | | 795,769 | | | | (357,674 | ) | | | 77,732,575 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Effect of foreign currency exchange rates on cash and cash equivalents | | | — | | | | 699 | | | | 4,483 | | | | 1,008 | | | | 624 | | | | 5,806 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Net increase (decrease) in cash and cash equivalents | | | (398,397 | ) | | | 27,101,040 | | | | (12,915,359 | ) | | | (4,421,358 | ) | | | (5,845,081 | ) | | | 8,707,698 | |
Cash and cash equivalents, beginning of period | | | 765,495 | | | | 367,098 | | | | 27,468,138 | | | | 27,468,138 | | | | 14,552,779 | | | | — | |
| | | | | | | | | | | | | | | | | | | | | | | | |
Cash and cash equivalents, end of period | | $ | 367,098 | | | $ | 27,468,138 | | | $ | 14,552,779 | | | $ | 23,036,907 | | | $ | 8,707,698 | | | $ | 8,707,698 | |
| | | | | | | | | | | | | | | | | | | | | | | | |
The accompanying notes are an integral part of these consolidated financial statements.
F-10
Biolex, Inc.
(A Development Stage Company)
Consolidated Statements of Cash Flows—(Continued)
| | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31, | | Three Months Ended
March 31, | | Period from
September 19,
1997 (date of
inception) to
March 31, 2007 | |
| | | 2004 | | | 2005 | | 2006 | | 2006 | | 2007 | |
| | | | | | as restated
(see Note 2) | | | | (unaudited) | | (unaudited) | |
Supplemental disclosures of cash flow information | | | | | | | | | | | | | | | | | | | | |
Cash paid for interest | | $ | 71,830 | | | $ | 621,654 | | $ | 408,689 | | $ | 71,314 | | $ | 98,961 | | $ | 1,247,887 | |
Supplemental disclosures of non-cash investing and financing activities | | | | | | | | | | | | | | | | | | | | |
Accretion of mandatorily redeemable preferred stock | | | 1,165,523 | | | | 3,033,747 | | | 5,599,829 | | | 1,369,019 | | | 1,494,592 | | | 16,450,981 | |
Issuance of Series AA2 preferred stock for intangible assets | | | 700,000 | | | | — | | | — | | | — | | | — | | | 700,000 | |
Issuance of warrants in connection with financing | | | 23,697 | | | | 46,284 | | | 10,782 | | | 6,351 | | | — | | | 2,965,796 | |
Deferred compensation adjustment related to variable accounting for stock options | | | (182,116 | ) | | | — | | | — | | | — | | | — | | | (187,561 | ) |
Conversion of mandatorily redeemable preferred stock into Class A common stock | | | — | | | | — | | | — | | | — | | | — | | | 14,194,706 | |
Conversion of notes payable and accrued interest into mandatorily redeemable preferred stock | | | — | | | | 8,000,000 | | | — | | | — | | | — | | | 17,833,362 | |
Lease obligations entered into for property and equipment | | | — | | | | — | | | — | | | — | | | — | | | 149,981 | |
The accompanying notes are an integral part of these consolidated financial statements.
F-11
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
1. Nature of Business and Operating Environment
Biolex, Inc. (the “Company”) was incorporated in the State of Delaware on September 19, 1997. The Company was formed to commercialize biologic drugs using its LEX System, a process for developing and manufacturing therapeutic proteins based on a genetically engineered plant (Lemna). Subsequent to incorporation, the Company completed two acquisitions to expand its intellectual property holdings. In 2004, the Company formed Plantibodies Corporation to acquire substantially all of the assets of Epicyte Pharmaceuticals, Inc. (“Epicyte”) and in 2005, the Company acquired LemnaGene S.A. (“LemnaGene”).
The Company’s primary activities since incorporation have been research and development, establishment and development of proprietary product candidates, corporate alliances, and establishment of manufacturing facilities. Accordingly, the Company is in the development stage and, in order to continue as a going concern, will need to raise additional capital to fund its research and development activities and expand its manufacturing and other capabilities.
The accompanying financial statements have been prepared on a basis which assumes that the Company will continue as a going concern and which contemplates the realization of assets and the satisfaction of liabilities in the normal course of business. The Company has incurred losses from operations since its inception and has a deficit accumulated during the development stage of $75,420,904 at March 31, 2007 (unaudited). Management’s future plans include continued development of its product candidates, the collaboration with pharmaceutical partners, as well as seeking additional sources of financing. However, there is no assurance that the Company will be successful in commercializing its product candidates, in obtaining sufficient collaboration fees, or in obtaining adequate financing to fund planned operations.
2. Restatement of Previously Issued Consolidated Financial Statements
The Company has restated its previously issued consolidated financial statements and related footnotes as of and for the year ended December 31, 2005, as set forth in these consolidated financial statements. The restatement was (1) to correct an error in accounting for the revenue recognition of the selected protein development and manufacturing alliance agreement with Centocor, Inc. (“Centocor”) and (2) to correct an error in the accounting for income taxes in connection with the purchase price allocation related to the LemnaGene acquisition. All amounts in these consolidated financial statements have been updated to reflect this restatement.
Description of Restatement
The Company identified an error that originated in the first quarter of 2005 and continued throughout 2005. The identification of this error occurred as a result of the Company evaluating its assumptions under Emerging Issues Task Force (“EITF”) 00-21 “Revenue Arrangements with Multiple Deliverables” (“EITF 00-21”), in accounting for arrangements with multiple deliverables that require significant judgment and estimates.
The Company reassessed its obligations under its agreement with Centocor and determined that certain obligations that were previously accounted for as separate units of accounting needed to be combined with other, undelivered items and accounted for as a single unit of accounting. The Company determined that its obligations during the protein development phase of the arrangement should be accounted for as a single unit of accounting. Future manufacturing and training obligations under the alliance agreement with Centocor are considered separate units of accounting. In addition, the Company determined that the substantive milestone method of revenue recognition which had been used for certain payments was not appropriate as those payments did not meet the substantive milestone criteria. The Company determined a time-based proportional performance model
F-12
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
would be more appropriate to account for these payments. Accordingly, in the restated consolidated financial statements for the year ended December 31, 2005, the Company decreased revenue by $2.0 million and increased deferred revenue by $2.0 million.
In addition, the Company has revised its financial statements to correct an error in the accounting for income taxes in connection with the purchase price allocation for the LemnaGene acquisition, which was completed on July 7, 2005. The Company has changed the associated income tax accounting related to this acquisition to be in accordance with EITF Issue No. 98-11, “Accounting for Acquired Temporary Differences in Certain Purchase Transactions That Are Not Accounted for as Business Combinations.” The Company determined that it should have assigned values for the differences between the recorded assets and the tax bases of the assets acquired. Accordingly, in the restated consolidated financial statements for the year ended December 31, 2005, the Company increased its intangible assets by $0.7 million, increased its deferred tax asset by $0.4 million and increased deferred tax liabilities by $1.0 million. In addition, amortization expense related to the intangible asset increased by $0.1 million and a deferred tax benefit of $0.2 million was realized during the year ended December 31, 2005.
The following tables present the effects of the restatement adjustments on the affected line items in the previously reported consolidated statement of operations for the year ended December 31, 2005 and consolidated balance sheet as of December 31, 2005. The restatement adjustments did not affect the overall cash (used in) provided by operating, investing or financing activities or the effect of exchange rates on cash and cash equivalents in the consolidated statements of cash flows for the year ended December 31, 2005.
Impact to consolidated statement of operations
| | | | | | | | | | | | |
Year Ended December 31, 2005 | | As Reported | | | Adjustment | | | Restated | |
Revenues | | $ | 5,017,991 | | | $ | (2,010,000 | ) | | $ | 3,007,991 | |
Research and development | | | 14,414,879 | | | | 33,746 | | | | 14,448,625 | |
Total operating expenses | | | 18,540,438 | | | | 33,746 | | | | 18,574,184 | |
Loss from operations | | | (13,522,447 | ) | | | (2,043,746 | ) | | | (15,566,193 | ) |
Income tax benefit | | | — | | | | 170,159 | | | | 170,159 | |
Net loss | | | (13,943,698 | ) | | | (1,873,587 | ) | | | (15,817,285 | ) |
Basic and diluted earnings per share | | | (58.76 | ) | | | (6.49 | ) | | | (65.26 | ) |
Impact to consolidated balance sheet
| | | | | | | | |
Year Ended December 31, 2005 | | As Reported | | Adjustment | | Restated |
Intangible assets | | 3,096,828 | | | 699,371 | | | 3,796,199 |
Total assets | | 37,396,197 | | | 699,371 | | | 38,095,568 |
Deferred revenue | | 3,659,310 | | | 2,010,000 | | | 5,669,310 |
Total current liabilities | | 5,448,633 | | | 2,010,000 | | | 7,458,633 |
Deferred tax liability | | — | | | 562,958 | | | 562,958 |
Total liabilities, mandatorily redeemable preferred stock and stockholders’ equity (deficit) | | 37,396,197 | | $ | 699,371 | | $ | 38,095,568 |
F-13
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
3. Summary of Significant Accounting Policies
Unaudited Interim Financial Information
The accompanying interim balance sheet as of March 31, 2007, the statements of operations and cash flows for the three months ended March 31, 2006 and 2007 and the statement of changes in stockholders’ deficit for the three months ended March 31, 2007 are unaudited. The unaudited interim financial statements have been prepared in accordance with U.S. generally accepted accounting principles. The unaudited interim financial statements have been prepared on the same basis as the audited financial statements. In the opinion of management, the unaudited interim financial statements include all adjustments, consisting of normal recurring adjustments, necessary for the fair statement of the Company’s financial position at March 31, 2007 and the Company’s results of operations and cash flows for the three months ended March 31, 2006 and 2007. The results for the three months ended March 31, 2007 are not necessarily indicative of the results to be expected for the year ending December 31, 2007 or for any future period. All references to March 31 in these footnotes are also unaudited.
Pro Forma Balance Sheet (Unaudited)
The Company’s pro forma balance sheet as March 31, 2007 gives effect to the automatic conversion of all Series AA1, AA2 and BB mandatorily redeemable convertible preferred stock into an aggregate of 59,807,757 shares of Class A common stock, the automatic conversion of all Class B common stock into Class A common stock and the reclassification of all Class A common stock into shares of common stock, upon consummation of the Company’s initial public offering.
Principles of Consolidation
The consolidated financial statements include the financial statements of Biolex, Inc. and its wholly owned subsidiaries, Plantibodies Corporation, Biolex Clinical Development Limited and LemnaGene S.A., located in France. All intercompany balances and transactions have been eliminated in consolidation.
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and the reported amounts of revenues and expenses during the reporting period. Actual results could differ from those estimates.
Cash and Cash Equivalents
The Company considers all highly liquid investments purchased with a remaining maturity of three months or less at the date of purchase to be cash equivalents. Cash equivalents held at December 31, 2005 and 2006 and at March 31, 2007 (unaudited) consist of a money market account comprised of direct obligations of the United States Treasury.
Restricted Cash
At December 31, 2005 and 2006 and at March 31, 2007 (unaudited), the Company has restricted cash of $378,000, which serves as collateral for an operating lease commitment.
F-14
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
Concentration of Credit Risk
Financial instruments which potentially subject the Company to concentrations of credit risk consist principally of cash and cash equivalents. The Company maintains its cash and cash equivalents primarily in securities comprised of direct obligations of the United States Treasury. Operating cash is deposited with a federally insured financial institution. Based on the Company’s daily liquidity requirements, deposits with the financial institution exceeded the amount insured by the Federal Deposit Insurance Company.
Foreign Currency
The Company’s foreign subsidiaries, LemnaGene S.A. and Biolex Clinical Development Limited, have designated the Euro and the U.S. dollar, respectively, as their functional currencies. Financial statements of LemnaGene S.A. are translated to U.S. dollars for consolidation purposes using current rates of exchange for assets and liabilities. Equity is translated using historical exchange rates and revenue and expense amounts are translated using the average exchange rate for the period. Net unrealized gains and losses resulting from foreign currency translation are included in “Accumulated other comprehensive income (loss),” a separate component of stockholders’ deficit.
Property and Equipment
Property and equipment are recorded at cost and depreciated over their estimated useful lives using the straight-line method. Property and equipment held under capital leases and leasehold improvements are amortized over the estimated useful life of the related asset which is shorter than the life of the lease. The estimated useful lives of the assets are as follows:
| | |
Laboratory equipment | | 5 years |
Computer hardware and software | | 3 years |
Office furniture and equipment | | 5 -7 years |
Leasehold improvements | | 1 -5 years |
Upon retirement or sale, the cost of assets disposed of and the related accumulated depreciation and amortization are removed from the accounts and any resulting gain or loss is credited or charged to operations. Repairs and maintenance costs are expensed as incurred.
Impairment of Long-Lived Assets
The Company evaluates the recoverability of its property and equipment, and other long-lived assets, in accordance with Statement of Financial Accounting Standards No. 144, “Accounting for the Impairment or Disposal of Long-Lived Assets” (“SFAS 144”). SFAS 144 requires recognition of impairment of long-lived assets in the event the net book value of such assets exceeds the estimated future undiscounted cash flows attributable to such assets. During the year ended December 31, 2004, the Company recorded a write-off of $50,000 for capitalized equipment which had been removed from operations. No impairments were recognized by the Company during the years ended December 31, 2005 and 2006 or during the three months ended March 31, 2007 (unaudited).
F-15
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
Intangible Assets
Intangible assets are comprised of license and intellectual property rights, net of accumulated amortization, acquired as a result of the purchase of assets of Epicyte Pharmaceutical, Inc. in 2004 and the purchase of LemnaGene in 2005. The assets are being amortized over their estimated useful life. Amortization expense was $111,111, $320,444 and $483,417 for the years ended December 31, 2004, 2005 and 2006, respectively, and $120,906 and $120,904 for the three months ended March 31, 2006 and 2007, respectively (unaudited). Accumulated amortization was $431,565, $881,226 and $934,838 at December 31, 2005 and 2006 and March 31, 2007 (unaudited), respectively. Amortization expense will be approximately $416,000 per year through April 2010 and thereafter will be approximately $249,000 per year through July 2015.
Income Taxes
Income taxes are computed using the asset and liability approach that requires the recognition of deferred tax assets and liabilities for the expected future tax consequences of events that have been recognized in the Company’s financial statements. In estimating future tax consequences, the Company generally considers all expected future events other than enactment of changes in tax laws or rates. A valuation allowance is recorded, if necessary, to reduce net deferred tax assets to their realizable values if management does not believe it is more likely than not that the net deferred tax assets will be realized.
Revenue Recognition
The Company recognizes revenue in accordance with the Securities and Exchange Commission’s Staff Accounting Bulletin No. 104, “Revenue Recognition in Financial Statements.” When evaluating multiple element arrangements, the Company considers whether the components of the arrangement represent separate units of accounting as defined in Emerging Issues Task Force (“EITF”) Issue No. 00-21, “Revenue Arrangements with Multiple Deliverables” (“EITF 00-21”). To recognize a delivered item in a multiple element arrangement, EITF Issue No. 00-21 requires that the delivered items have value to the customer on a standalone basis, that there is objective and reliable evidence of fair value of the undelivered items that have a right of return. Application of this standard requires subjective determinations and requires management to make judgments about the fair value of the individual elements and whether such elements are separable from the other aspects of the contractual relationship.
The Company has entered into a collaboration agreement (the “Agreement”) with Centocor which includes multiple deliverables. The Company accounts for its obligations during the protein development phase of the arrangement as a single unit of accounting. Future manufacturing and training obligations under the Agreement are considered separate units of accounting. The Company utilizes a time-based proportional performance model to account for cash payments received under the Agreement. Amounts resulting from payments received in advance of revenue recognized are recorded as deferred revenue and recognized over the time period in which the Company fulfills its contractual obligations under the Agreement.
Research and Development
Research and development costs include all direct costs, including compensation expense, supplies, preclinical study and clinical trial expenses, facilities and third-party contractor costs, related to the development of the Company’s product candidates and underlying technology. These costs are charged to operating expense as incurred.
F-16
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
Mandatorily Redeemable Convertible Preferred Stock
The Company is accounting for its mandatorily redeemable preferred stock under the requirements of SFAS No. 150 (“FAS 150”) and Accounting Series Release No. 268 (“ASR 268”). “Accounting for Certain Financial Instruments with Characteristics of Liabilities and Equity,” which establishes standards for how an issuer classifies and measures certain financial instruments with characteristics of both liabilities and equity. Commencing with the completion of the private placement of Series BB preferred stock and the related amendment of the Certificate of Incorporation on August 22, 2005 (see Note 9), the Company determined that the mandatorily redeemable convertible preferred stock is no longer within the scope of FAS 150 and therefore adopted the provisions of Accounting Series Release No. 268.
The carrying value of the mandatorily redeemable convertible preferred stock is increased by periodic accretions, so that the carrying amount will equal the redemption amount at August 22, 2010, the date that redemption first became available. Prior to the Series BB transaction and the related modification to the Certificate of Incorporation, the mandatorily redeemable preferred stock was increased by periodic accretions, calculated using the interest method. These increases were affected through charges against income available to common stockholders to the extent available, additional paid-in capital to the extent available, then against the deficit accumulated during the development stage.
Accounting for Stock-Based Compensation
The Company adopted Financial Accounting Standards Board Statement (“FASB”) No. 123(R), “Share-Based Payment” (“FAS 123(R)”), as of January 1, 2006. FAS 123(R) revises Financial Accounting Standards Board Statement No. 123, “Accounting for Stock-Based Compensation” (“FAS 123”), supersedes Accounting Principles Board (“APB”) Opinion No. 25, “Accounting for Stock Issued to Employees” (“APB 25”), and amends Financial Accounting Standards Board Statement No. 95, “Statement of Cash Flows” (“FAS 95”). FAS 123(R) requires companies to expense the fair value of employee stock options over the employees’ service periods. Compensation cost is measured at the fair value of the award using the Black Scholes model at the grant date, including estimated forfeitures, and is adjusted to reflect actual forfeitures and the outcomes of certain conditions.
In accordance with FASB No. 148, “Accounting for Stock-Based Compensation, Transition and Disclosure” (“FAS 148”), for periods prior to January 1, 2006 the Company adopted the disclosure-only provisions of FAS 123 and also applied APB 25 and related interpretations in accounting for all stock awards granted to employees. Under APB 25, for periods prior to January 1, 2006 the Company measured compensation expense for its employee stock-based compensation using the intrinsic value method and provided pro forma disclosures of net loss as if the fair value method had been applied in measuring compensation expense. Under the intrinsic value method of accounting for stock-based compensation, when the exercise price of options granted to employees is less than the estimated fair value of the underlying stock on the date of the grant, deferred compensation is recognized and amortized to compensation expense over the applicable vesting period. For all periods the Company has followed the provisions of FAS 123R and FAS 123, as applicable, for valuing and recording options and warrants.
Segments
Operating segments are defined as components of an enterprise about which separate financial information is available that is evaluated regularly by the chief operating decision maker, or decision making group, in deciding how to allocate resources and in assessing performance of the Company. The Company has concluded that it operates in one segment and, accordingly, has provided only the required enterprise-wide disclosures. In addition, all of the Company’s sales and expenses are incurred in the United States. There are no assets held by its foreign subsidiary at December 31, 2005 or 2006.
F-17
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
Net Loss per Share
The Company calculates net loss per share in accordance with SFAS No. 128,Earnings Per Share. Basic and diluted net loss per common share was determined by dividing net loss by the weighted average common shares outstanding during the period. The Company’s potentially dilutive shares, which include mandatorily redeemable convertible preferred stock options and warrants, have not been included in the computation of diluted net loss per share for all periods as the result would be anti-dilutive.
| | | | | | | | | | | | | | | | | | | | |
| | | Years Ended December 31, | | | Three Months Ended
March 31, | |
| | | 2004 | | | 2005 | | | 2006 | | | 2006 | | | 2007 | |
| As reported: | | | | | | | | | | | (unaudited) | |
Net income (loss) | | $ | (10,403,771 | ) | | $ | (15,817,285 | ) | | $ | (17,496,844 | ) | | $ | (4,670,181 | ) | | $ | (5,682,375 | ) |
Accretion of mandatorily redeemable convertible preferred stock | | | (1,165,523 | ) | | | (3,033,747 | ) | | | (5,599,829 | ) | | | (1,369,019 | ) | | | (1,494,592 | ) |
| | | | | | | | | | | | | | | | | | | | |
Net loss attributable to common stockholders | | $ | (11,569,294 | ) | | $ | (18,851,032 | ) | | $ | (23,096,673 | ) | | $ | (6,039,200 | ) | | $ | (7,176,967 | ) |
| | | | | | | | | | | | | | | | | | | | |
Weighted average common shares outstanding, basic and diluted | | | 269,595 | | | | 288,890 | | | | 337,078 | | | | 299,212 | | | | 480,573 | |
| | | | | | | | | | | | | | | | | | | | |
Net loss per share attributable to common stockholders, basic and diluted | | $ | (42.91 | ) | | $ | (65.26 | ) | | $ | (68.52 | ) | | $ | (20.18 | ) | | $ | (14.94 | ) |
| | | | | | | | | | | | | | | | | | | | |
The following potentially dilutive securities have been excluded from the computation of diluted weighted average shares outstanding as of December 31, 2004, 2005 and 2006 and March 31, 2006 and 2007, as they would be anti-dilutive.
| | | | | | | | | | |
| | | Years Ended December 31, | | Three Months Ended
March 31, |
| | | 2004 | | 2005 | | 2006 | | 2006 | | 2007 |
| | | | | | | | | (unaudited) |
Mandatorily redeemable convertible preferred stock | | 21,576,361 | | 59,807,757 | | 59,807,757 | | 59,807,757 | | 59,807,757 |
Stock options outstanding | | 2,669,536 | | 8,463,630 | | 8,630,083 | | 8,468,320 | | 8,539,255 |
Warrants outstanding | | 26,673 | | 837,091 | | 93,640 | | 855,992 | | 93,640 |
F-18
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
Unaudited pro forma net loss per share assuming the conversion of all mandatorily redeemable convertible preferred stock, which converts automatically upon the completion of the initial public offering, is as follows:
| | | | | | | | |
| | | Year Ended
December 31, 2006 | | | Three Months
Ended March 31, 2007 | |
| | | | | | (unaudited) | |
Net loss, as reported | | $ | (17,496,844 | ) | | $ | (5,682,375 | ) |
Weighted-average common shares outstanding | | | 337,078 | | | | 480,573 | |
Weighted-average number of common shares assuming conversion of all redeemable convertible preferred stock at the original date of issuance | | | 59,807,757 | | | | 59,807,757 | |
Weighted-average common shares used in computing pro forma net loss per share | | | 60,144,835 | | | | 60,288,330 | |
Pro forma net loss per share—basic and diluted | | $ | (0.29 | ) | | $ | (0.09 | ) |
Recent Accounting Pronouncements
In February 2007, FASB issued Statement No. 159, “The Fair Value Option for Financial Assets and Financial Liabilities” (“FAS 159”). FAS 159 provides companies with an option to report selected financial assets and liabilities at fair value. The objective of FAS 159 is to reduce both complexity in accounting for financial instruments and the volatility in earnings caused by measuring related assets and liabilities differently. Generally accepted accounting principles have required different measurement attributes for different assets and liabilities that can create artificial volatility in earnings. The FASB has indicated it believes that FAS 159 helps to mitigate this type of accounting-induced volatility by enabling companies to report related assets and liabilities at fair value, which would likely reduce the need for companies to comply with detailed rules for hedge accounting.
FAS 159 also establishes presentation and disclosure requirements designed to facilitate comparisons between companies that choose different measurement attributes for similar types of assets and liabilities. For example, FAS 159 requires companies to provide additional information that will help investors and other users of financial statements to more easily understand the effect of the company’s choice to use fair value on its earnings. It also requires entities to display the fair value of those assets and liabilities for which the company has chosen to use fair value on the face of the balance sheet. FAS 159 does not eliminate disclosure requirements included in other accounting standards, including requirements for disclosures about fair value measurements included in FASB Statement No. 157, “Fair Value Measurements” (“FAS 157”), and FASB Statement No. 107, “Disclosures about Fair Value of Financial Instruments.” FAS 159 is effective as of the beginning of a company’s first fiscal year beginning after November 15, 2007.
In September 2006, the FASB issued Statement No. 157 (“FAS 157”). FAS 157 provides guidance for using fair value to measure assets and liabilities and requires additional disclosure about the use of fair value measures, the information used to measure fair value, and the effect fair-value measurements have on earnings. FAS 157 does not require any new fair value measurements. FAS 157 will be effective for the Company beginning January 1, 2008. The Company is currently evaluating the effects of FAS 159 and FAS 157 on its consolidated financial statements.
In June 2007, the EITF of the FASB reached consensus on Issue No. 07-3, “Accounting for Nonrefundable Advance Payments for Goods or Services to Be Used in Future Research and Development Activities” or (“EITF Issue No. 07-3”). EITF Issue No. 07-3 addresses the issue of when to record nonrefundable advance payments
F-19
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
for goods or services that will be used or rendered for research and development activities as expenses. The EITF has concluded that nonrefundable advance payments for future research and development activities should be deferred and recognized as an expense as the goods are delivered or the related services are performed. EITF Issue No. 07-3 is effective for fiscal years beginning after December 15, 2007. The Company has assessed the impact of EITF Issue No. 07-3 and expects no impact to its financial statements upon adoption of this guidance.
In June 2006, the FASB issued FASB Interpretation 48 (“FIN 48”), “Accounting for Uncertainty in Income Taxes -an interpretation of FASB Statement No 109”. This interpretation prescribes a recognition threshold and measurement attribute for the financial statement recognition and measurement of a tax position taken or expected to be taken in a tax return. The interpretation also provides guidance on derecognition, classification and other matters. The statement was effective for the fiscal year 2007 and the Company adopted the Interpretation at that time.
4. Property and Equipment
Property and equipment consist of the following:
| | | | | | | | | | | | |
| | | December 31, | | | March 31, 2007 | |
| | | 2005 | | | 2006 | | |
| | | | | | | | | (unaudited) | |
Laboratory equipment | | $ | 4,642,315 | | | $ | 5,743,433 | | | $ | 5,891,805 | |
Office furniture and equipment | | | 130,200 | | | | 161,279 | | | | 161,279 | |
Computer hardware and software | | | 294,893 | | | | 333,031 | | | | 345,475 | |
Leasehold improvements | | | 4,268,790 | | | | 4,789,620 | | | | 4,789,620 | |
| | | | | | | | | | | | |
| | | 9,336,198 | | | | 11,027,363 | | | | 11,188,179 | |
Less accumulated depreciation and amortization | | | (3,839,289 | ) | | | (5,200,600 | ) | | | (5,597,351 | ) |
| | | | | | | | | | | | |
Property and equipment, net | | $ | 5,496,909 | | | $ | 5,826,763 | | | $ | 5,590,828 | |
| | | | | | | | | | | | |
At December 31, 2005 and 2006 and March 31, 2007 (unaudited), property and equipment includes equipment under a capital lease of $69,870, net of accumulated depreciation of $38,817, $56,672 and $60,165, respectively. Depreciation expense for the years ended December 31, 2004, 2005, 2006, and for the three months ended March 31, 2007 (unaudited) was $370,823, $704,072, $1,086,216, and $282,091, respectively.
5. Purchase of Intangible Assets
On July 7, 2005 the Company acquired all of the outstanding capital stock of LemnaGene S.A. (“LemnaGene”). LemnaGene was a privately-held company operating in France and was engaged in early stage research regarding the production of proteins, including human and animal vaccine, in plants. The primary asset of LemnaGene was a license to a portfolio of patents and patent applications. The total purchase price was $2,436,454, consisting of $2,072,263 in cash and transaction costs of $364,191. The estimated fair value of the license and underlying patents acquired was adjusted to $3,169,571 as a result of applying EITF Issue No. 98-11. The estimated fair value and a related deferred tax liability of $733,117 are being amortized using the straight-line method over ten years, the estimated useful life of the license.
On April 30, 2004 the Company acquired substantially all of the assets of Epicyte Pharmaceuticals, Inc. (“Epicyte”) and assumed certain liabilities of Epicyte. Epicyte was a privately held company that had terminated the majority of its operations prior to the acquisition. The primary asset of Epicyte consisted of a portfolio of
F-20
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
patents, patent applications and licenses related to the production of monoclonal antibodies in plants. The total purchase price was $1,000,000, comprised of $300,000 in cash, and the estimated value of 700,000 shares of Series AA2 preferred stock of the Company issued as consideration in the purchase (valued at $1.00 per share). The estimated fair value of the license and patents acquired was $1,000,000 and is being amortized using the straight-line method until April 2010, the expiration date of the primary patent acquired.
6. Notes Payable
Notes payable consists of the following:
| | | | | | | | | | | | |
| | | December 31, | | | March 31, | |
| | | 2005 | | | 2006 | | | 2007 | |
| | | | | | | | | (unaudited) | |
Convertible promissory note payable to a related party, payable in June 2013 including interest at 6%, collateralized by equipment and inventory | | $ | — | | | $ | 5,000,000 | | | $ | 5,000,000 | |
Promissory note due September 2009, payable in monthly installments including interest at 9.71%, collateralized by equipment | | | 3,313,180 | | | | 2,591,571 | | | | 2,383,410 | |
Promissory notes due March 2010 and June 2010, payable in monthly installments including interest at 10.44% and 10.74% respectively, collateralized by equipment | | | — | | | | 1,199,070 | | | | 1,122,454 | |
Promissory notes due January 2008 through September 2008, payable in monthly installments including interest ranging from 8.89% to 9.73%, collateralized by equipment | | | 615,969 | | | | 310,965 | | | | 231,954 | |
Promissory notes due July 2009, payable in monthly installments including interest at 4.70% | | | 21,920 | | | | 18,011 | | | | 16,556 | |
| | | | | | | | | | | | |
| | | 3,951,069 | | | | 9,119,617 | | | | 8,754,374 | |
Less current portion | | | (981,813 | ) | | | (1,403,445 | ) | | | (1,392,148 | ) |
Less debt discount | | | (56,765 | ) | | | (48,036 | ) | | | (42,996 | ) |
| | | | | | | | | | | | |
| | $ | 2,912,491 | | | $ | 7,668,136 | | | $ | 7,319,230 | |
| | | | | | | | | | | | |
Maturities of notes payable at December 31, 2006 and March 31, 2007 are as follows :
| | | | | | |
| | | December 31, 2006 | | March 31, 2007 |
| | | | | (unaudited) |
2007 | | $ | 1,419,319 | | $ | 1,054,076 |
2008 | | | 1,393,422 | | | 1,393,423 |
2009 | | | 1,173,920 | | | 1,173,919 |
2010 | | | 132,956 | | | 132,956 |
2011 | | | — | | | — |
2012 | | | — | | | — |
Thereafter | | | 5,000,000 | | | 5,000,000 |
| | | | | | |
| | $ | 9,119,617 | | $ | 8,754,374 |
| | | | | | |
F-21
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
In June 2006, the Company borrowed $5,000,000 from Johnson and Johnson Development Corp. (“JJDC”) (see Note 15) under a convertible promissory note with an interest rate of 6%. The note was secured by certain assets of the Company, including equipment and inventory. Interest accrued on the note at December 31, 2006 was $175,068.
In September 2005 the Company entered into an agreement with a financial institution under which the Company could borrow up to $5.0 million to fund equipment purchases and other qualified expenditures. On September 28, 2005, the Company borrowed $3,588,930 from the financial institution under a promissory note that bears interest at 9.71% and matures in September 2009. On March 15, 2006 and June 9, 2006 the Company borrowed $1,037,682 and $373,527, respectively, from the financial institution under promissory notes that bear interest rates at 10.44% and 10.74% and mature in March 2010 and June 2010. The Company also granted the financial institution coincidental with issuance of the notes a series of warrants to purchase a total of 66,967 shares of Series BB preferred stock at a price of $1.12 per share. The total amount allocated to the warrants using the Black-Scholes pricing model was $57,536 and was recorded as a debt discount. The debt discount is being amortized to interest expense over the 48-month term of the notes.
From December 2003 to August 2004 the Company borrowed a total of $1,066,904 from a financial institution under a series of promissory notes that bear interest ranging from 8.89% to 9.73%. In addition, the Company issued a series of warrants to purchase a total of 26,663 shares of Class A Voting common stock at an exercise price of $1.00 per share. The total amount allocated to the warrants was $24,236, determined using the Black-Scholes option pricing model, and was recorded as a debt discount. The debt discount is being amortized to interest expense over the 48-month terms of the notes.
7. Accrued Expenses and Other Liabilities
Accrued expenses and other liabilities consists of the following:
| | | | | | | | | |
| | | December 31, | | March 31, |
| | | 2005 | | 2006 | | 2007 |
| | | | | | | (unaudited) |
Compensation and benefits | | $ | 1,026,515 | | $ | 841,686 | | $ | 1,120,659 |
Clinical cost | | | 165,242 | | | 253,774 | | | 514,614 |
Research contracts | | | — | | | 450,601 | | | 464,506 |
Interest | | | 403,212 | | | 206,554 | | | 279,198 |
Other | | | 812,070 | | | 428,501 | | | 200,195 |
| | | | | | | | | |
Total accrued expenses and other liabilities | | $ | 2,407,039 | | $ | 2,181,116 | | $ | 2,579,172 |
| | | | | | | | | |
8. Common Stock
As of December 31, 2006 and March 31, 2007 (unaudited), the Company had 494,258 shares of Class A Voting and 3,821 shares of Class B Nonvoting common stock outstanding.
Dividend Rights
The holders of common stock are entitled to receive dividends from time to time as may be declared by the Board of Directors. No cash dividend may be declared or paid to common stockholders until paid on each series of preferred stock in accordance with its terms.
F-22
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
Liquidation
After payment of any preference due to the preferred stockholders (Note 9), holders of common stock are entitled to share ratably in all remaining assets of the Company.
Voting
The holders of Class A Voting common stock are entitled to one vote for each share held with respect to all matters voted on by the stockholders of the Company, with the exception of certain items for which only specific series preferred stock are entitled to vote as described below. The holders of Class B Non-Voting common stock are not entitled to vote, except as otherwise required by applicable law.
Warrants to Purchase Common Stock
At December 31, 2006 and March 31, 2007 (unaudited), warrants issued to a financial institution are outstanding to purchase 26,663 shares of Class A Voting common stock at an exercise price of $1.00 per share. The warrants expire at various dates between December 2013 and August 2014.
9. Mandatorily Redeemable Convertible Preferred Stock
Preferred stock outstanding at December 31, 2006 and March 31, 2007 (unaudited), in order of preference upon the occurrence of a liquidating event, as defined below, is as follows:
| | | | | | | | |
Description | | Number of
Shares | | Amount | | Liquidation
Value |
Series BB | | 32,120,192 | | $ | 35,974,615 | | $ | 35,974,615 |
Series AA2 | | 22,594,864 | | | 25,306,248 | | | 22,594,864 |
Series AA1 | | 5,092,701 | | | 5,703,825 | | | 5,092,701 |
| | | | | | | | |
Total | | 59,807,757 | | $ | 66,984,688 | | $ | 63,662,180 |
| | | | | | | | |
The Company has increased the carrying value of its preferred stock by periodic accretions so that the carrying amount will equal the redemption amount at August 22, 2010, the date that redemption first becomes available.
Dividend Rights
The holders of the preferred shares are entitled to receive dividends, when and if declared by the Company’s Board of Directors. No dividends shall be paid on the common stock unless an equivalent dividend is declared and paid on the Series AA1, Series AA2 and Series BB preferred stock.
In connection with the Series BB transaction, the Board of Directors of the Company declared a dividend in the amount of $2,540,063 payable to holders of record of the Series AA2 preferred stock as of July 31, 2005. The dividends were paid in kind on August 22, 2005 in the form of 2,540,063 shares of Series AA2 preferred stock. No other dividends were declared during the period from September 19, 1997 (date of inception) to December 31, 2006.
Under the Certificate of Incorporation, there are no mandatory dividends on any series of preferred stock. Through July 31, 2005, each holder of Series AA2 preferred stock was entitled to cumulative annual dividends at
F-23
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
a rate of 8% of the original purchase price (defined as $1.00). The Series AA2 mandatory dividends were to compound annually and accrue whether or not earned or declared. As a result of the adoption of the Certificate of Incorporation, the Series AA2 mandatory dividend was terminated effective July 31, 2005, and all dividends accrued through that date were paid by the Company on August 22, 2005 as described above.
Liquidation
In the event of any liquidation, dissolution or winding up of the Company (“Liquidating Event”), the holders of the preferred shares are entitled to receive the greater of (i) the respective amounts summarized in the table above, to be paid prior to and in preference to any distribution to the holders of common stock, or (ii) the amount that such holder would have received had all of the holder’s shares been converted to Class A Voting common stock prior to such Liquidating Event. Thereafter, holders of the common stock will receive any remaining assets of the Company. The Certificate of Incorporation defines the meaning of a Liquidating Event that would initiate a distribution.
Conversion
Each share of preferred stock, at the option of the holder, is convertible into shares of the Company’s Class A Voting common stock at a fixed conversion ratio, which as of December 31, 2006, was one share of Class A Voting common stock for each share of preferred stock. The conversion ratio is subject to certain adjustments for stock splits, stock dividends, and recapitalizations as defined in the Certificate of Incorporation. In addition, the conversion price of the Series BB and Series AA2 preferred stock will be adjusted to prevent dilution if additional equity securities are issued at a purchase price less than the applicable conversion price. Conversion of all preferred stock is automatic in the event of (i) an underwritten public offering with a minimum per share price of $6.72 or, (ii) an affirmative vote by a majority of the Series BB and Series AA2 preferred stockholders voting together as a separate class. Lastly, in the event of a consolidation or merger of the Company, or the sale of all or substantially all of the assets of the Company, the preferred shares will be convertible into the class and amount of securities that would have been received had the preferred shares been converted into Class A Voting common stock prior to the transaction.
Voting Rights
The holders of the Series BB preferred stock, voting separately as a single class, shall be entitled to elect one director of the Company. The holders of the Series AA2 preferred stock, voting separately as a single class, shall be entitled to elect two directors of the Company. The holders of the Series AA1 preferred stock, voting separately as a single class, shall be entitled to elect one director of the Company. The remaining directors shall be elected by all of the Company’s stockholders, voting together as a single class. Except with respect to the election of directors, and except with respect to certain protective provisions that may be waived only by the holders of a majority of the Series BB and Series AA2 preferred stock, holders of preferred stock vote together with the holders of common stock on an as-if-converted basis.
Redemption
The Company could be required to redeem the preferred shares from the stockholders upon receipt of written request from a majority of the holders of the then outstanding shares of Series BB and Series AA2 preferred stock at any time after August 22, 2010 or under certain specified put option rights as documented in the Certificate of Incorporation. The redemption price is the Series AA1, Series AA2 and Series BB liquidation amounts plus simple interest of 10% annually, accrued from August 22, 2005 through the date of redemption.
F-24
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
Warrants to Purchase Mandatorily Redeemable Convertible Preferred Stock
At December 31, 2006 and March 31, 2007 (unaudited), warrants to purchase 66,967 shares of Series BB preferred stock are outstanding. In 2005 a warrant to purchase 714,286 shares of Series BB preferred stock was issued to JJDC in connection with the issuance of an $8.0 million convertible note payable (see Note 15). The warrant had an exercise price of $1.12 per share and expired on August 22, 2006, one year from the date of conversion of the promissory note. The remaining warrants to purchase 66,967 shares of Series BB preferred stock were issued to a financial institution in connection with the issuance of promissory notes payable. The warrants have an exercise price of $1.12 per share and expire in September 2013 through June 2014. At March 31, 2007 (unaudited), none of these warrants have been exercised.
On June 29, 2005, the FASB issued Staff Position 150-5,Issuer’s Accounting under FASB Statement No. 150 for Freestanding Warrants and Other Similar Instruments on Shares That Are Redeemable, or FSP 150-5. FSP 150-5 requires us to classify outstanding preferred stock warrants as liabilities on the balance sheet and record adjustments to the value of preferred stock warrants in the statement of operations to reflect the fair value at each reporting period. The Company previously accounted for such warrants in accordance with APB 14,Accounting for Convertible Debt and Debt Issued with Stock Purchase Warrants .
The Company adopted FSP 150-5 during 2005 which resulted in $386,000 of additional expense. On August 22, 2006, upon the expiration of the warrants, the Company reversed the expense previously recorded.
Private Placements of Preferred Stock
On August 22, 2005, the Company completed a private placement of Series BB preferred stock in which it issued 32,120,192 shares at a price of $1.12 per share. The Company received net proceeds after issuance costs of $27,782,176 in cash and $8,000,000 in the form of the conversion of a convertible note payable to JJDC (see Note 15). On January 11, 2005 the Company completed a private placement of Series AA2 preferred stock in which it issued 3,571,141 shares at a price of $1.00 per share. The Company received net proceeds of $3,543,519 after issuance costs. On September 13, 2004, the Company completed a private placement of Series AA2 preferred stock in which it issued 3,000,000 shares at a price of $1.00 per share. The Company received net proceeds of $2,969,476 after issuance costs. The January 2005 and September 2004 private placements comprise the Second Tranche Purchase described below. On April 30, 2004 the Company issued 700,000 shares of Series AA2 preferred stock in consideration for its purchase of substantially all of the assets of Epicyte (see Note 5).
Effective August 22, 2005, in conjunction with the Series BB transaction, the Board of Directors and stockholders of the Company approved an amended and restated certificate of incorporation (the “Certificate of Incorporation”). Under the terms of the Certificate of Incorporation, the authorized capital shares of the Company were increased to include 80,000,000 shares of Class A Voting common stock, 3,821 shares of Class B Nonvoting common stock, and 61,589,565 shares of preferred stock. The authorized shares of preferred stock are designated as 5,092,701, 22,594,864, and 33,902,000 shares of Series AA1, Series AA2, and Series BB, respectively. All shares have a par value of $0.001 per share.
On August 8, 2003 the Company completed a private placement in which it issued a total of 12,783,660 shares of Series AA2 preferred stock and 5,092,701 shares of Series AA1 preferred stock in exchange for cash and the cancellation of debt and accrued interest. For the private placement of the Series AA2 preferred, 10,501,860 shares were issued in exchange for cash of $1.00 per share, and the Company received net proceeds of $10,263,804 after issuance costs. Under the terms of the private placement, the purchasers also committed to
F-25
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
purchase an additional 6,571,141 shares of Series AA2 shares at a price of $1.00 per share in cash upon the achievement of certain operational milestones (the “Second Tranche Purchase”). The Company also issued 2,281,800 shares of Series AA2 preferred stock in exchange for the cancellation of $2,281,800 of convertible notes and $92,742 of accrued interest payable to certain stockholders. Furthermore, the Company issued 5,092,701 shares of Series AA1 preferred stock in exchange for the cancellation of $5,127,800 of convertible notes and $609,251 of accrued interest. The convertible debt resulted from a series of borrowings the Company had made from stockholders between December 2001 and June 2003. Under these loan transactions, the stockholders providing the loans also received warrants to purchase a total of 1,444,000 shares of the then outstanding Series C preferred stock. The warrants had an estimated fair value of $1,389,244 as of the date of grant as determined using the Black-Scholes pricing model. The warrants were canceled under the terms of the private placement on August 8, 2003.
Under the terms of the August 8, 2003 private placement, the Company undertook a recapitalization under which the then outstanding preferred shares, comprised of Series A preferred stock, Series B preferred stock and Series C preferred stock, were converted into Class A Voting common stock, followed immediately by a reverse stock split whereby each 24.1604 shares of the then outstanding shares of Class A Voting common stock and Class B Non-Voting common stock were converted into one share of the same class of common stock. All common stock share amounts presented herein, have been adjusted for this split.
10. Common Stock Options
Stock Option Plan
In 2003, the Company adopted the 2003 Stock Option Plan (the “Plan”) that provides for the grant of either incentive stock options or nonqualified stock options to employees and other persons providing services to the Company for the purchase of the Company’s common stock. Under the Plan, as amended, options may be granted to purchase up to 11,571,429 shares of the Company’s Class A Voting Common Stock. Options that are granted have a vesting period of four years and have a maximum term of ten years. When an option holder ceases to be an employee or service provider to the Company, such options that are vested and not exercised terminate after 90 days of said termination.
F-26
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
The following table summarizes stock option activity for the Plan:
| | | | | | | | | | | | |
| | | Number | | | Weighted-
Average
Exercise Price
Per Share | | Exercise Price
Per Share
Range | | Aggregate
Intrinsic
Value |
Outstanding, January 1, 2004 | | 2,599,478 | | | $ | 0.10 | | $ | 0.10 | | $ | — |
Granted | | 116,858 | | | | 0.10 | | | 0.10 | | | |
Exercised | | (7,500 | ) | | | 0.10 | | | 0.10 | | | |
Cancelled | | (39,300 | ) | | | 0.10 | | | 0.10 | | | |
| | | | | | | | | | | | |
Outstanding, December 31, 2004 | | 2,669,536 | | | | 0.10 | | | 0.10 | | | — |
Granted | | 5,882,450 | | | | 0.22 | | | 0.22 | | | |
Exercised | | (21,936 | ) | | | 0.10 | | | 0.10 | | | |
Cancelled | | (66,420 | ) | | | 0.13 | | | 0.10-0.20 | | | |
| | | | | | | | | | | | |
Outstanding, December 31, 2005 | | 8,463,630 | | | | 0.17 | | | 0.12 | | | — |
Granted | | 519,500 | | | | 0.22 | | | 0.22 | | | |
Exercised | | (164,873 | ) | | | 0.10 | | | 0.10-0.22 | | | |
Cancelled | | (188,174 | ) | | | 0.15 | | | 0.10-0.22 | | | |
| | | | | | | | | | | | |
Outstanding, December 31, 2006 | | 8,630,083 | | | | 0.17 | | | 0.17 | | | 5,930,693 |
Granted (unaudited) | | — | | | | — | | | — | | | |
Exercised (unaudited) | | (34,194 | ) | | | 0.14 | | | .10-.22 | | | |
Cancelled (unaudited) | | (56,634 | ) | | | 0.20 | | | .10-.22 | | | |
| | | | | | | | | | | | |
Outstanding, March 31, 2007 (unaudited) | | 8,539,255 | | | | 0.17 | | | 0.17 | | $ | 8,601,321 |
| | | | | | | | | | | | |
During the year ended December 31, 2006 and the three months ended March 31, 2007 (unaudited), the Company granted 519,500 common stock options at a weighted average exercise price of $0.22, the fair market value, on April 6, 2006. There were no other grants during these periods.
The following table summarizes information about the Company’s outstanding stock options at December 31, 2006 and March 31, 2007 (unaudited):
| | | | | | | | | | | | |
| Options Outstanding | | Options Exercisable |
| Number | | Weighted
Average
Remaining
Contractual Life | | Weighted-
Average
Exercise Price
Per Share | | Number | | Weighted
Average
Remaining
Contractual Life | | Weighted-
Average
Exercise
Price
Per Share |
| December 31, 2006 | | | | | | | | | | | | |
| 3,283,263 | | 6.47 | | $ | 0.10 | | 2,822,407 | | | | $ | 0.10 |
| 672,436 | | 7.55 | | | 0.20 | | 284,750 | | | | | 0.20 |
| 4,674,384 | | 8.58 | | | 0.22 | | 1,233,509 | | | | | 0.22 |
| | | | | | | | | | | | |
| 8,630,083 | | 7.70 | | $ | 0.17 | | 4,340,666 | | 7.71 | | $ | 0.14 |
| | | | | | | | | | | | |
| March 31 2007 | | | | | | | | | | | | |
| 3,256,968 | | 6.28 | | $ | 0.10 | | 2,948,143 | | | | $ | 0.10 |
| 639,998 | | 7.66 | | | 0.20 | | 316,217 | | | | | 0.20 |
| 4,642,289 | | 8.40 | | | 0.22 | | 1,502,174 | | | | | 0.22 |
| | | | | | | | | | | | |
| 8,539,255 | | 7.53 | | $ | 0.17 | | 4,766,534 | | 7.05 | | $ | 0.14 |
| | | | | | | | | | | | |
F-27
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
Stock-Based Compensation
The Company adopted Financial Accounting Standards Board Statement No. 123(R), “Share-Based Payment” (“FAS 123(R)”), as of January 1, 2006. FAS 123(R) requires companies to expense the fair value of employee stock options over the employees’ service periods. Compensation cost is measured at the fair value of the award at the grant date, including estimated forfeitures, and is adjusted to reflect actual forfeitures and the outcomes of certain conditions.
During the year ended December 31, 2006 and the three months ended March 31, 2007 (unaudited), the Company recorded $33,513 and $7,036 (unaudited), respectively, of stock-based compensation expense as a result of the adoption of FAS 123(R). Of these amounts, the Company allocated $22,529 and $4,187 to research and development and $10,984 and $2,849 to general administration expenses for the year ended December 31, 2006 and the three months ended March 31, 2007 (unaudited), respectively, based on the department to which the associated employee reported. No related tax benefits of the stock-based compensation expense have been recognized since the inception of the Company.
The Black-Scholes option-pricing model was developed for use in estimating the fair value of traded options, which have no vesting restrictions and are fully transferable. In addition, the Black-Scholes option-pricing model requires the input of subjective assumptions, including the expected stock price volatility. The following table shows the assumptions used to compute stock-based compensation expense for the stock options granted during the year ended December 31, 2006 using the Black-Scholes option pricing model. There were no option grants in the period ended March 31, 2007 (unaudited).
| | | |
| | | Year Ended December 31, 2006 | |
Risk-free interest rate | | 4.86 | % |
Common Stock dividend yield | | — | |
Weighted average expected life of options (years) | | 6 | |
Expected volatility | | 37.0 | % |
The risk-free interest rate assumption was based on the United States Treasury’s rates for U.S. Treasury zero-coupon bonds with maturities similar to those of the expected term of the award being valued. The assumed dividend yield was based on the Company’s expectation of not paying dividends in the foreseeable future. The weighted average expected life of options was calculated using the simplified method as prescribed in Staff Accounting Bulletin No. 107,Share-Based Payment (“SAB 107”). This decision was based on the lack of relevant historical data due to the Company’s limited history. In addition, due to the Company’s limited historical data, the estimated volatility also reflects the application of SAB 107, incorporating the historical volatility of an industry index of companies whose share prices are publicly available.
Based on these assumptions, the weighted average fair values of stock options granted during the year ended December 31, 2006 was $0.098 per share. As of December 31, 2006 and March 31, 2007 (unaudited), the total unrecognized compensation expense related to stock options was approximately $36,584 and $25,768, respectively, and the related weighted-average period over which it is expected to be recognized approximately 2.5 and 2.32 years, respectively.
F-28
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
The fair value of options granted to employees was estimated using the following assumptions for the years ended December 31, 2005 and 2004.
| | | | | | |
| | | December 31, | |
| | | 2004 | | | 2005 | |
Expected dividend yield | | 0.00 | % | | 0.00 | % |
Expected stock price volatility | | 0.00 | % | | 0.00 | % |
Risk-free interest rate | | 3.60 | % | | 4.00 | % |
Expected life of options | | 5 years | | | 5 years | |
These assumptions resulted in weighted average fair values at the date of grant for options granted during the year ended December 31, 2005 and 2004 of $0.07 and $0.02 respectively. For purposes of the above pro forma disclosures, the estimated fair values of the options are amortized to expense over their respective vesting period.
11. Commitments
The Company leases its office facilities and certain equipment under operating lease agreements. Future minimum lease payments under noncancelable operating leases at December 31, 2006 and March 31, 2007 are as follows:
| | | | | | |
| | | December 31, 2006 | | March 31, 2007 |
| | | | | (unaudited) |
2007 | | $ | 1,197,369 | | $ | 894,325 |
2008 | | | 1,036,236 | | | 1,036,236 |
2009 | | | 926,973 | | | 926,973 |
2010 | | | 843,556 | | | 843,556 |
2011 | | | 759,717 | | | 759,717 |
2012 | | | 759,717 | | | 759,717 |
Thereafter | | | 2,975,558 | | | 2,975,558 |
| | | | | | |
Total minimum lease payments | | $ | 8,499,126 | | $ | 8,196,082 |
| | | | | | |
Rent expense for the years ended December 31, 2004, 2005 and 2006 and for the three months ended March 31 2007 (unaudited), and the period from September 19, 1997 (inception) to March 31, 2007 (unaudited) was $362,567, $465,441, $1,019,643, $465,441, $268,294 and $3,479,721, respectively.
12. Income Taxes
The following summarizes the components of the income tax benefit:
| | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2004 | | 2005 | | | 2006 | |
Current provision | | $ | — | | $ | — | | | $ | — | |
Deferred provision (benefit) | | | — | | | (170,159 | ) | | | (277,487 | ) |
| | | | | | | | | | | |
Total provision (benefit) | | $ | — | | $ | (170,159 | ) | | $ | (277,487 | ) |
| | | | | | | | | | | |
F-29
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
No current provision for federal or state income taxes has been recorded as the Company has incurred net operating losses since inception.
Significant components of the Company’s deferred tax assets and liabilities consist of the following:
| | | | | | | | |
| | | December 31, | |
| | | 2005 | | | 2006 | |
Deferred tax assets: | | | | | | | | |
Domestic net operating loss carryforwards | | $ | 14,730,739 | | | $ | 21,518,885 | |
Fixed assets | | | 608,400 | | | | 619,100 | |
Comprehensive accruals | | | 186,900 | | | | 386,200 | |
Deferred revenue | | | 2,174,450 | | | | 1,889,383 | |
Intangible assets | | | 62,100 | | | | 100,600 | |
Research and development credits | | | 1,331,300 | | | | 1,610,600 | |
| | | | | | | | |
Gross deferred tax assets | | | 19,093,889 | | | | 26,124,768 | |
Valuation allowance for deferred assets | | | (18,653,250 | ) | | | (25,512,283 | ) |
| | | | | | | | |
Net deferred tax assets | | | 440,639 | | | | 612,485 | |
| | | | | | | | |
Deferred tax liabilities: | | | | | | | | |
Intangible assets | | | 1,003,597 | | | | 897,956 | |
| | | | | | | | |
Total deferred tax liabilities | | | 1,003,597 | | | | 897,956 | |
| | | | | | | | |
Net deferred tax liabilities | | $ | 562,958 | | | $ | 285,471 | |
At December 31, 2005 and 2006, the Company provided a full valuation allowance against its net deferred tax assets since realization of these benefits could not be reasonably assured. The increase in valuation allowance in each period resulted primarily from the additional net operating loss carryforward generated and the deferred revenue.
As of December 31, 2006 the Company had federal and state net operating loss carryforwards of approximately $55,415,000 and $56,818,000, respectively. These net operating loss carryforwards begin to expire in 2017 for federal and state purposes. The research credit carryforward at December 31, 2006 of $1,608,000 begins to expire in 2018. The utilization of the federal net operating loss carryforwards may be subject to limitation under the rules regarding a change in stock ownership as determined by the Internal Revenue Code.
Taxes computed at the statutory federal income tax rate of 34% are reconciled to the provision for income taxes as follows:
| | | | | | | | | | | | |
| | | Year ended December 31, | |
| | | 2004 | | | 2005 | | | 2006 | |
United States Federal tax at statutory rate | | $ | (3,537,282 | ) | | $ | (5,307,891 | ) | | $ | (6,204,433 | ) |
State taxed (net of Federal benefit) | | | (473,372 | ) | | | (594,500 | ) | | | (792,900 | ) |
Change in valuation allowance | | | 4,472,929 | | | | 5,806,811 | | | | 6,859,033 | |
Research and development credits | | | (322,690 | ) | | | (170,800 | ) | | | (279,300 | ) |
Other nondeductible credit | | | (139,585 | ) | | | 92,800 | | | | 134,200 | |
Foreign rate differential | | | | | | | 3,421 | | | | 5,913 | |
| | | | | | | | | | | | |
Provision for income taxes | | $ | — | | | $ | (170,159 | ) | | $ | (277,487 | ) |
| | | | | | | | | | | | |
F-30
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
Adoption of FIN 48 (unaudited)
The Company adopted FIN 48 effective January 1, 2007. Upon adoption of FIN 48 and through March 31, 2007, the Company had no unrecognized tax benefits. As of the date of adoption, there were no tax positions for which it is reasonably possible that the total amounts of unrecognized tax benefits will significantly increase or decrease within twelve months from the date of adoption of FIN 48 or from March 31, 2007. As of March 31, 2007, the Company is subject to federal and state income tax in the United States. Since the Company is in a loss carryforward position, the Company is generally subject to U.S. federal and state income tax examinations by tax authorities for all years for which a loss carryforward is available. Thus, upon adoption of FIN 48, the Company’s open tax years extend back to 2003. In the event that the Company concludes that it is subject to interest and/or penalties arising from uncertain tax positions, the Company will record interest and penalties as a component of other income and expense. No amounts of interest or penalties were recognized in the Company’s Consolidated Statements of Operations or Consolidated Balance Sheets upon adoption of FIN 48 or as of and for the three months ended March 31, 2007.
13. Employee Benefit Plan
The Company has a retirement plan qualified under Section 401(k) of the Internal Revenue Code. Participants may elect to contribute a portion of their annual compensation to the plan. Under the terms of the plan, the Company is not obligated to make any contributions to the plan, and the Company has not made any contributions since inception.
14. License and Research Agreements
The Company holds licenses from universities, commercial enterprises and other third parties for certain technologies, patents and patent applications (collectively “Licensed Technology”). These licenses may obligate the Company to pay a royalty on net sales of products or other commercial revenues dependent upon the use of specific Licensed Technology in developing such product or in generating such revenues. The Company is also obligated to undertake or provide reimbursement for the cost of prosecuting, maintaining and defending patents underlying the Licensed Technology. Total royalty and license expense was $46,492, $175,000, $122,917 and $83,332 for the years ended December 31, 2004, 2005 and 2006, and the three months ended March 31, 2007 (unaudited), respectively. Future minimum annual royalties due under all licenses is approximately $175,000 per year through 2017.
15. Related Party Transactions
The Company has had a series of collaboration agreements with Centocor, an affiliate of Johnson & Johnson Development Corporation (“JJDC”), a holder of approximately 16% of the Company’s outstanding shares at March 31, 2007 (unaudited) on an as-converted basis. The Company recognized $719,375, $2,606,667, $4,097,387 and $319,394, or 100%, 87%, 82% and 44%, of its revenue from Centocor for the years ended December 31, 2004, 2005 and 2006, and the three months ended March 31, 2007 (unaudited), respectively. At December 31, 2005, the Company had $100,000 of accounts receivable from Centocor. The Company had no accounts receivable from Centocor at December 31, 2006 and March 31, 2007 (unaudited). At December 31, 2006 and 2005 and March 31, 2007 (unaudited), the Company had $5,213,334, $3,625,944, and $3,343,783 (unaudited) of deferred revenue, respectively, from Centocor. At March 31, 2007 (unaudited), future minimum revenue related to the current collaboration agreement with Centocor is $4,025,944 (unaudited).
On June 1, 2006, the Company borrowed $5,000,000 from JJDC under a convertible promissory note with interest at 6%. During the year ended December 31, 2006 and the three months ended March 31, 2007
F-31
Biolex, Inc.
(A Development Stage Company)
Notes to Consolidated Financial Statements
(unaudited), the Company recorded $175,068 and $73,973 (unaudited), respectively, in interest expense related to the note. On May 18, 2007, the Company completed a private placement that resulted in the conversion of the note payable and $282,740 of accrued interest into Series CC preferred stock.
On January 7, 2005, the Company borrowed $8,000,000 from JJDC under a convertible promissory note with interest at 6%. On August 22, 2005, the note was converted into 7,142,858 shares of Series BB preferred stock as a result of the private placement described in Note 9. As part of the convertible debt transaction, the Company issued to JJDC a warrant to purchase 714,286 shares of Series BB preferred stock at an exercise price of $1.12 per share that expired August 22, 2006. The amount allocated to the warrant was $473,751, determined using the Black-Scholes option pricing model, was recorded as debt discount. The Company recorded $358,467 in interest expense related to the note through the date of conversion, $260,313 of which was paid to JJDC in cash and $98,211 of which represented the amortization of debt discount.
16. Subsequent Event
On May 18, 2007, the Company completed a private placement of Series CC preferred stock in which it issued 25,663,339 shares at a price of $1.18 per share. The Company received net proceeds after issuance costs of $23,834,719 in cash and $5,282,740 in the form of the conversion of a convertible note payable and accrued interest to JJDC (see Note 15).
F-32
LEMNAGENE S.A.
REPORT OF STATUTORY AUDITOR
We have audited the accompanying profit and loss statement and cash flow statement of LemnaGene for the period from January 1, 2005 to July 7, 2005. These financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on these financial statements based on our audits.
We conducted our audits in accordance with auditing standards generally accepted in the United States of America. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the financial statements are free of material misstatement. An audit includes examining, on a test basis, evidence supporting the amounts and disclosures in the financial statements. An audit also includes assessing the accounting principles used and significant estimates made by management, as well as evaluating the overall financial statement presentation. We believe that our audits provide a reasonable basis for our opinion.
In our opinion, the income statement and cash flow statement referred to above present fairly, in all material respects, the results of LemnaGene operations and their cash flows for the period then ended in conformity with accounting principles generally accepted in France.
/s/ Frédéric Brejon
Dardilly, France
31 July 2007
F-33
LemnaGene S.A.
Profit and Loss Statement
January 1, 2005 through July 7, 2005
| | | | |
Revenues | | | | |
Net sales | | € | 137,806 | |
Accrued revenue not billed | | | (48,000 | ) |
Government grant | | | 120,000 | |
| | | | |
Total revenue | | | 209,806 | |
| |
Operating Expenses | | | | |
External services, supplies, charges | | | 201,278 | |
Salaries | | | 117,916 | |
Payroll taxes | | | 21,132 | |
Amortization | | | 6,512 | |
Other taxes | | | 1,523 | |
Other charges | | | 8,458 | |
| | | | |
Total operating expenses | | | 356,819 | |
Other income, net | | | 364 | |
| | | | |
Net loss | | € | (146,649 | ) |
| | | | |
See notes to financial statements
F-34
LemnaGene S.A.
Statement of Cash Flow
January 1, 2005 through July 7, 2005
| | | | |
Operating Activities | | | | |
Net loss | | € | (146,649 | ) |
Depreciation | | | 6,512 | |
Other | | | 1,461 | |
| | | | |
Net operating activities | | | (138,677 | ) |
Changes in current liabilities: | | | | |
Accounts payable | | | 79,327 | |
Accrued expenses | | | 5,000 | |
Current account | | | (90,000 | ) |
Other payables | | | (2,336 | ) |
Deferred revenue | | | (30,000 | ) |
Changes in current assets: | | | | |
Inventory | | | 48,000 | |
Other receivables | | | 60,328 | |
Accounts receivable | | | (28,825 | ) |
Prepaid expenses | | | 880 | |
Capital expenditures | | | (5,281 | ) |
| |
Increase in share capital | | | 109,999 | |
| | | | |
Change in cash | | € | 8,416 | |
| | | | |
See notes to financial statements
F-35
LemnaGene S.A.
Notes to Financial Statements
January 1, 2005 through July 7, 2005
1. Nature of Business
LemnaGene S.A. (the “Company” or “LemnaGene”) is a French Limited Company that was incorporated October 10, 2003 and is based in Lyon, France. The Company was formed for the purpose of research, development and production of molecules of commercial interest based on the plant lemnaceae. The Company’s primary activities since incorporation have been research and development and the establishment of corporate collaborations.
On July 7, 2005, 100% of the outstanding shares of LemnaGene were acquired by Biolex, Inc., a United States-based biopharmaceutical company.
2. Summary of Significant Accounting Policies
LemnaGene applies accounting principles that comply with French law for its financial statements. The difference between these accounting principles and the generally accepted accounting principles (GAAP) in the United States America that have a material impact on the financial statements of LemnaGene are described in note 3.
The financial statements of LemnaGene reflect the operations and cash flows for the period commencing January 1, 2005 and ending July 7, 2005, the date of acquisition by Biolex, Inc. The financial statements are presented in Euro, the functional currency of LemnaGene
Research and Development
Research and development expenditures are charged to expense as incurred.
Revenue Recognition
Revenue is recorded when delivery has occurred or services have been rendered, and collection is reasonably assured.
3. Significant Differences Between French and U.S. GAAP
LemnaGene’s financial statements have been prepared in accordance with French GAAP, which differs in some respects from U.S. GAAP. The effects of the application of U.S. GAAP to net loss are described below:
| | | | |
Net loss under French GAAP | | € | (146,649 | ) |
Adjustments to reflect U.S. GAAP: | | | | |
Recognition of grant revenues in accordance with Staff Accounting Bulletin No. 101 | | | (120,000 | ) |
Record liabilities that are probable and measurable under Financial Accounting Standard No. 5 | | | (70,347 | ) |
Adjustment to recognize pro rata share of research and development tax credits earned in 2005 | | | 62,673 | |
Record patent application costs as research and Development expense | | | (820 | ) |
| | | | |
Net loss under U.S. GAAP | | € | (275,143 | ) |
| | | | |
F-36
LemnaGene S.A.
Notes to Financial Statements
January 1, 2005 through July 7, 2005
The effects of the application of U.S. GAAP to cash flows are described below:
| | | | |
Change in cash under French GAAP | | € | 8,416 | |
Adjustments to reflect U.S. GAAP: | | | | |
Adjustments to net loss to reflect U.S. GAAP | | | (128,494 | ) |
Increase in accrued expenses | | | 190,347 | |
Increase in accounts receivable | | | (62,673 | ) |
Decrease in capitalized patent costs | | | 820 | |
| | | | |
Change in cash under U.S. GAAP | | € | 8,416 | |
| | | | |
F-37

Shares
Common Stock
PROSPECTUS
, 2007
Joint Book-Running Managers
LEHMAN BROTHERS
DEUTSCHE BANK SECURITIES
LEERINK SWANN & COMPANY
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
| Item 13. | Other Expenses of Issuance and Distribution. |
The following table sets forth an itemization of the various costs and expenses, all of which we will pay, in connection with the issuance and distribution of the securities being registered. All of the amounts shown are estimated except the SEC Registration Fee, The NASDAQ Global Market Listing Fee and the NASD Filing Fee.
| | | |
SEC Registration Fee | | $ | 2,149 |
The NASDAQ Global Market Listing Fee | | | * |
NASD Filing Fee | | | 7,500 |
Printing and Engraving Fees | | | * |
Legal Fees and Expenses | | | * |
Accounting Fees and Expenses | | | * |
Blue Sky Fees and Expenses | | | * |
Miscellaneous | | | * |
| | | |
Total | | $ | * |
| | | |
| * | To be provided by amendment. |
| Item 14. | Indemnification of Directors and Officers. |
Our amended and restated certificate of incorporation provides that we shall indemnify, to the fullest extent authorized by the Delaware General Corporation Law, each person who is involved in any litigation or other proceeding because such person is or was a director or officer of Biolex or is or was serving as an officer or director of another entity at our request, against all expense, loss or liability reasonably incurred or suffered in connection therewith. Our amended and restated certificate of incorporation provides that the right to indemnification includes the right to be paid expenses incurred in defending any proceeding in advance of its final disposition, provided, however, that such advance payment will only be made upon delivery to us of an undertaking, by or on behalf of the director or officer, to repay all amounts so advanced if it is ultimately determined that such director is not entitled to indemnification. If we do not pay a proper claim for indemnification in full within 60 days after we receive a written claim for such indemnification, our amended and restated certificate of incorporation and our restated bylaws authorize the claimant to bring an action against us and prescribe what constitutes a defense to such action.
Section 145 of the Delaware General Corporation Law permits a corporation to indemnify any director or officer of the corporation against expenses (including attorney’s fees), judgments, fines and amounts paid in settlement actually and reasonably incurred in connection with any action, suit or proceeding brought by reason of the fact that such person is or was a director or officer of the corporation, if such person acted in good faith and in a manner that he reasonably believed to be in, or not opposed to, the best interests of the corporation, and, with respect to any criminal action or proceeding, if he or she had no reason to believe his or her conduct was unlawful. In a derivative action, (i.e., one brought by or on behalf of the corporation), indemnification may be provided only for expenses actually and reasonably incurred by any director or officer in connection with the defense or settlement of such an action or suit if such person acted in good faith and in a manner that he or she reasonably believed to be in, or not opposed to, the best interests of the corporation, except that no indemnification shall be provided if such person shall have been adjudged to be liable to the corporation, unless and only to the extent that the court in which the action or suit was brought shall determine that the defendant is fairly and reasonably entitled to indemnity for such expenses despite such adjudication of liability.
Pursuant to Section 102(b)(7) of the Delaware General Corporation Law, Article Ninth of our amended and restated certificate of incorporation eliminates the liability of a director to us or our stockholders for monetary damages for such a breach of fiduciary duty as a director, except for liabilities arising:
| | • | | from any breach of the director’s duty of loyalty to us or our stockholders; |
II-1
| | • | | from acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law; |
| | • | | under Section 174 of the Delaware General Corporation Law; and |
| | • | | from any transaction from which the director derived an improper personal benefit. |
We carry insurance policies insuring our directors and officers against certain liabilities that they may incur in their capacity as directors and officers. In addition, we may enter into agreements to indemnify our directors and officers.
Additionally, reference is made to the Underwriting Agreement filed as Exhibit 1.1 hereto, which provides for indemnification by the underwriters of Biolex, our directors and officers who sign the registration statement and persons who control Biolex, under certain circumstances.
| Item 15. | Recent Sales of Unregistered Securities. |
In the three years preceding the filing of this registration statement, we have sold the following securities that were not registered under the Securities Act. The following information gives effect to a - -for- split of our common stock to be effected prior to the completion of this offering.
| | (a) | Issuances of Capital Stock and Warrants |
| | 1. | Between August 8, 2003 and August 22, 2005, we issued and sold 22,594,864 shares of our Series AA2 preferred stock at a purchase price per share of $1.00 to 18 accredited investors for an aggregate purchase price of $22,594,864. |
| | 2. | On January 7, 2005, in connection with the issuance of an $8.0 million convertible note, we issued a warrant to Johnson & Johnson Development Corporation to purchase 714,286 shares of Series BB preferred stock at an exercise price of $1.40 per share. This warrant expired without exercise on August 22, 2006, one year from the date of the conversion of the $8.0 million promissory note. |
| | 3. | On August 22, 2005, we issued and sold 32,120,192 shares of our Series BB preferred stock at a purchase price per share of $1.12 to 24 accredited investors for an aggregate purchase price of $35,974,615. |
| | 4. | On September 28, 2005, in connection with a capital line borrowing, we issued a warrant to Oxford Finance Corporation to purchase 48,066 shares of Series BB preferred stock at an exercise price of $1.12 per share. |
| | 5. | On March 15, 2006, in connection with a capital line borrowing, we issued a warrant to Oxford Finance Corporation to purchase 13,898 shares of Series BB preferred stock at an exercise price of $1.12 per share. |
| | 6. | On June 9, 2006, in connection with a capital line borrowing, we issued a warrant to Oxford Finance Corporation to purchase 5,003 shares of Series BB preferred stock at an exercise price of $1.12 per share. |
| | 7. | On May 18, 2007, we issued and sold 21,186,441 shares of our Series CC preferred stock at a purchase price per share of $1.18 to 22 accredited investors for an aggregate purchase price of $25,000,000 and issued an additional 4,476,898 shares of our Series CC preferred stock upon the conversion of a $5.0 million convertible note and accrued interest payable thereon to Johnson & Johnson Development Corporation. |
No underwriters were used in the foregoing transactions. The sales of securities described above were deemed to be exempt from registration pursuant to Section 4(2) of the Securities Act as transactions by an issuer not involving a public offering. All of the purchasers in these transactions represented to us in connection with
II-2
their purchase that they were acquiring the shares for investment and not distribution, and that they could bear the risks of the investment and could hold the securities for an indefinite period of time. Such purchasers received written disclosures that the securities had not been registered under the Securities Act and that any resale must be made pursuant to a registration or an available exemption from such registration. All of the foregoing securities are deemed restricted securities for the purposes of the Securities Act.
| | (b) | Certain Grants and Exercises of Stock Options |
Pursuant to our 2003 Plan, we have issued options to purchase an aggregate of 10,194,485 shares of common stock. Of these options:
| | • | | options to purchase 437,714 shares of common stock have been canceled or lapsed without being exercised; |
| | • | | options to purchase 300,764 shares of common stock have been exercised; and |
| | • | | options to purchase a total of 9,456,007 shares of common stock are currently outstanding, at a weighted average exercise price of $0.28 per share. |
The sale and issuance of the securities described above were deemed to be exempt from registration under the Securities Act in reliance on Rule 701 promulgated under Section 3(b) of the Securities Act, as transactions by an issuer not involving a public offering or transactions pursuant to compensatory benefit plans and contracts relating to compensation as provided under Rule 701.
| Item 16. | Exhibits and Financial Statement Schedules. |
| | |
Exhibit Number | | Description of Exhibit |
| *1.1 | | Form of Underwriting Agreement. |
| |
| 3.1 | | Amended and Restated Certificate of Incorporation of the Registrant. |
| |
| *3.2 | | Amended and Restated Certificate of Incorporation of the Registrant to be filed and effective upon completion of this offering. |
| |
| 3.3 | | Bylaws of the Registrant. |
| |
| *3.4 | | Restated Bylaws of the Registrant to be effective upon completion of this offering. |
| |
| *4.1 | | Form of Common Stock Certificate. |
| |
| 4.2 | | Fourth Amended and Restated Investor Rights Agreement, dated May 18, 2007. |
| |
| *5.1 | | Opinion of Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C., counsel to the Registrant, with respect to the legality of securities being registered. |
| |
| 10.1 | | Master Security Agreement between the Registrant and General Electric Capital Corporation, dated October 28, 2003, as amended. |
| |
| 10.2 | | Master Security Agreement between the Registrant and Oxford Finance Corporation, dated September 21, 2005, as amended. |
| |
| 10.3 | | Lease Agreement between the Registrant and ARE-7030 Kit Creek, LLC, dated December 22, 2005. |
| |
| 10.4 | | Commercial Lease Agreement between the Registrant and Harold J. Milholen and William F. Milholen, dated August 9, 2000, as amended. |
| |
| *#10.5 | | License Agreement between the Registrant and North Carolina State University, dated April 30, 1998. |
II-3
| | |
Exhibit Number | | Description of Exhibit |
| *#10.6 | | License Agreement between the Registrant, LemnaGene S.A. and Yeda Research and Development Company Limited, dated July 7, 2005. |
| |
| *#10.7 | | License Agreement between Epicyte Pharmaceutical, Inc. and The Scripps Research Institute, dated October 9, 1997. |
| |
| *#10.8 | | Collaboration Agreement between the Registrant and OctoPlus Technologies B.V., dated February 11, 2005, as amended. |
| |
| 10.9 | | 2003 Equity Incentive Plan. |
| |
| *10.10 | | 2007 Employee, Director and Consultant Stock Plan. |
| |
| *10.10.1 | | Form of Incentive Stock Option Agreement under the 2007 Employee, Director and Consultant Stock Plan. |
| |
| *10.10.2 | | Form of Non-Qualified Stock Option Agreement under the 2007 Employee, Director and Consultant Stock Plan. |
| |
| *10.10.3 | | Form of Restricted Stock Agreement under the 2007 Employee, Director and Consultant Stock Plan. |
| |
| 10.11 | | Executive Employment Agreement between the Registrant and Jan Turek, dated March 8, 2007. |
| |
| 10.12 | | Executive Employment Agreement between the Registrant and Dale Sander, dated March 8, 2007. |
| |
| 10.13 | | Executive Employment Agreement between the Registrant and David Spencer, dated March 8, 2007. |
| |
| 10.14 | | Executive Employment Agreement between the Registrant and John Irick, dated March 8, 2007. |
| |
| 10.15 | | Letter Agreement between the Registrant and W. Thomas Amick, effective March 4, 2004. |
| |
| 10.16 | | Letter Agreement between the Registrant and David Castaldi, dated May 19, 2004. |
| |
| *10.17 | | Director Compensation Policy. |
| |
| 21.1 | | Subsidiaries of the Registrant. |
| |
| 23.1 | | Consent of PricewaterhouseCoopers LLP. |
| |
| 23.2 | | Consent of Frédéric Brejon. |
| |
| *23.3 | | Consent of Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C. (see Exhibit 5.1). |
| |
| 24.1 | | Powers of Attorney (See signature page). |
| * | To be filed by amendment. |
| # | Confidential treatment has been requested for portions of this exhibit. |
| (b) | Financial Statement Schedules |
Financial Statement Schedules are omitted because the information is included in our financial statements or notes to those financial statements.
The undersigned registrant hereby undertakes to provide to the underwriters at the closing specified in the Underwriting Agreement certificates in such denominations and registered in such names as required by the underwriters to permit prompt delivery to each purchaser.
Insofar as indemnification for liabilities arising under the Securities Act may be permitted to directors, officers and controlling persons of the registrant pursuant to the provisions described under Item 14 above, or
II-4
otherwise, the registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Securities Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the registrant of expenses incurred or paid by a director, officer or controlling person of the registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Securities Act and will be governed by the final adjudication of such issue.
The undersigned registrant hereby undertakes that:
| | (1) | For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective. |
| | (2) | For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. |
II-5
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the registrant certifies that it has duly caused this registration statement to be signed on its behalf by the undersigned, thereunto duly authorized, in Pittsboro, North Carolina, on August 13, 2007.
| | |
| BIOLEX, INC. |
| |
| By: | | /s/ Jan Turek |
| | Jan Turek |
| | President and Chief Executive Officer |
POWER OF ATTORNEY
We the undersigned officers and directors of Biolex, Inc., hereby severally constitute and appoint Jan Turek and Dale Sander, and each of them singly (with full power to each of them to act alone), our true and lawful attorneys-in-fact and agents, with full power of substitution and resubstitution in each of them for him and in his name, place and stead, and in any and all capacities, to sign any and all amendments (including post-effective amendments) to this registration statement (or any other registration statement for the same offering that is to be effective upon filing pursuant to Rule 462(b) under the Securities Act of 1933), and to file the same, with all exhibits thereto and other documents in connection therewith, with the Securities and Exchange Commission, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite or necessary to be done in and about the premises, as full to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that said attorneys-in-fact and agents or any of them or their or his substitute or substitutes may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act of 1933, this registration statement has been signed by the following persons in the capacities held on the dates indicated.
| | | | |
Signature | | Title | | Date |
| | |
/s/ Jan Turek Jan Turek | | President, Chief Executive Officer and Director (principal executive officer) | | August 13, 2007 |
| | |
/s/ Dale Sander Dale Sander | | Chief Financial Officer and Senior Vice President, Finance (principal financial and accounting officer) | | August 13, 2007 |
| | |
/s/ Chris Hegele Chris Hegele | | Chairman of the Board | | August 13, 2007 |
| | |
/s/ W. Thomas Amick W. Thomas Amick | | Director | | August 13, 2007 |
| | |
/s/ David L. Castaldi David L. Castaldi | | Director | | August 13, 2007 |
| | |
/s/ Dennis Dougherty Dennis Dougherty | | Director | | August 13, 2007 |
| | |
/s/ P. Sherrill Neff P. Sherrill Neff | | Director | | August 13, 2007 |
| | |
/s/ Sunny Sharma Sunny Sharma, M.D. | | Director | | August 13, 2007 |
II-6
EXHIBIT INDEX
| | |
Exhibit Number | | Description of Exhibit |
| *1.1 | | Form of Underwriting Agreement. |
| |
| 3.1 | | Amended and Restated Certificate of Incorporation of the Registrant. |
| |
| *3.2 | | Amended and Restated Certificate of Incorporation of the Registrant to be filed and effective upon completion of this offering. |
| |
| 3.3 | | Bylaws of the Registrant. |
| |
| *3.4 | | Restated Bylaws of the Registrant to be effective upon completion of this offering. |
| |
| *4.1 | | Form of Common Stock Certificate. |
| |
| 4.2 | | Fourth Amended and Restated Investor Rights Agreement, dated May 18, 2007. |
| |
| *5.1 | | Opinion of Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C., counsel to the Registrant, with respect to the legality of securities being registered. |
| |
| 10.1 | | Master Security Agreement between the Registrant and General Electric Capital Corporation, dated October 28, 2003, as amended. |
| |
| 10.2 | | Master Security Agreement between the Registrant and Oxford Finance Corporation, dated September 21, 2005, as amended. |
| |
| 10.3 | | Lease Agreement between the Registrant and ARE-7030 Kit Creek, LLC, dated December 22, 2005. |
| |
| 10.4 | | Commercial Lease Agreement between the Registrant and Harold J. Milholen and William F. Milholen, dated August 9, 2000, as amended. |
| |
| *#10.5 | | License Agreement between the Registrant and North Carolina State University, dated April 30, 1998. |
| |
| *#10.6 | | License Agreement between the Registrant, LemnaGene S.A. and Yeda Research and Development Company Limited, dated July 7, 2005. |
| |
| *#10.7 | | License Agreement between Epicyte Pharmaceutical, Inc. and The Scripps Research Institute, dated October 9, 1997. |
| |
| *#10.8 | | Collaboration Agreement between the Registrant and OctoPlus Technologies B.V., dated February 11, 2005, as amended. |
| |
| 10.9 | | 2003 Equity Incentive Plan. |
| |
| *10.10 | | 2007 Employee, Director and Consultant Stock Plan. |
| |
| *10.10.1 | | Form of Incentive Stock Option Agreement under the 2007 Employee, Director and Consultant Stock Plan. |
| |
| *10.10.2 | | Form of Non-Qualified Stock Option Agreement under the 2007 Employee, Director and Consultant Stock Plan. |
| |
| *10.10.3 | | Form of Restricted Stock Agreement under the 2007 Employee, Director and Consultant Stock Plan. |
| |
| 10.11 | | Executive Employment Agreement between the Registrant and Jan Turek, dated March 8, 2007. |
| |
| 10.12 | | Executive Employment Agreement between the Registrant and Dale Sander, dated March 8, 2007. |
| |
| 10.13 | | Executive Employment Agreement between the Registrant and David Spencer, dated March 8, 2007. |
| |
| 10.14 | | Executive Employment Agreement between the Registrant and John Irick, dated March 8, 2007. |
| |
| 10.15 | | Letter Agreement between the Registrant and W. Thomas Amick, effective March 4, 2004. |
| |
| 10.16 | | Letter Agreement between the Registrant and David Castaldi, dated May 19, 2004. |
| |
| *10.17 | | Director Compensation Policy. |
| |
| 21.1 | | Subsidiaries of the Registrant. |
| |
| 23.1 | | Consent of PricewaterhouseCoopers LLP. |
| | |
Exhibit Number | | Description of Exhibit |
| 23.2 | | Consent of Frédéric Brejon. |
| |
| *23.3 | | Consent of Mintz, Levin, Cohn, Ferris, Glovsky and Popeo, P.C. (see Exhibit 5.1). |
| |
| 24.1 | | Powers of Attorney (See signature page). |
| * | To be filed by amendment. |
| # | Confidential treatment has been requested for portions of this exhibit. |