UNITED STATES
SECURITIES AND EXCHANGE COMMISSION
WASHINGTON, DC 20549
FORM 10-K
☒ ANNUAL REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the fiscal year ended: December 31, 2021
OR
☐ TRANSITION REPORT PURSUANT TO SECTION 13 OR 15(d) OF THE SECURITIES EXCHANGE ACT OF 1934
For the transition period from _________________ to ______________________
Commission file number: 001-34673
CORMEDIX INC.
(Exact name of Registrant as Specified in Its Charter)
| Delaware | | 20-5894890 |
(State or Other Jurisdiction of
Incorporation or Organization) | | (I.R.S. Employer
Identification No.) |
| 300 Connell Drive, Suite 4200, Berkeley Heights, NJ | | 07922 |
| (Address of Principal Executive Offices) | | (Zip Code) |
Registrant’s telephone number, including area code: (908) 517-9500
Securities registered pursuant to Section 12(b) of the Act:
| Title of each class | | Trading Symbol | | Name of each exchange on which registered |
| Common Stock, $0.001 Par Value | | CRMD | | Nasdaq Global Market |
Securities registered pursuant to Section 12(g) of the Act: none
Indicate by check mark if the registrant is a well-known seasoned issuer, as defined in Rule 405 of the Securities Act.
Yes ☐ No ☒
Indicate by check mark if the registrant is not required to file reports pursuant to Section 13 or Section 15(d) of the Act.
Yes ☐ No ☒
Indicate by check mark whether the registrant: (1) has filed all reports required to be filed by Section 13 or 15(d) of the Securities Exchange Act of 1934 during the preceding 12 months (or for such shorter period that the registrant was required to file such reports), and (2) has been subject to such filing requirements for the past 90 days.
Yes ☒ No ☐
Indicate by check mark whether the registrant has submitted electronically every Interactive Data File required to be submitted pursuant to Rule 405 of Regulation S-T during the preceding 12 months (or for such shorter period that the registrant was required to submit such files).
Yes ☒ No ☐
Indicate by check mark whether the registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer, a smaller reporting company, or an emerging growth company. See the definitions of “large accelerated filer,” “accelerated filer,” “smaller reporting company” and “emerging growth company” in Rule 12b-2 of the Exchange Act:
| Large accelerated filer ☐ | | Accelerated filer ☐ | |
| | | | |
| Non-accelerated filer ☒ | | Smaller reporting company ☒ | |
| | | | |
Emerging growth company ☐ | | | |
If an emerging growth company, indicate by check mark if the registrant has elected not to use the extended transition period for complying with any news or revised financial accounting standards provided pursuant to Section 13(a) of the Exchange Act. ☐
Indicate by check mark whether the registrant has filed a report on and attestation to its management’s assessment of the effectiveness of its internal control over financial reporting under Section 404(b) of the Sarbanes-Oxley Act (15 U.S.C. 7262(b)) by the registered public accounting firm that prepared or issued its audit report. ☐
Indicate by check mark whether the registrant is a shell company (as defined in Rule 12b-2 of the Act).
Yes ☐ No ☒
The aggregate market value of the registrant’s voting common equity held by non-affiliates of the registrant, based upon the closing price of the registrant’s common stock on the last business day of the registrant’s most recently completed second fiscal quarter was approximately $208.4 million. Solely for the purpose of this calculation, shares held by directors and executive officers of the registrant have been excluded.
The number of outstanding shares of the registrant’s common stock was 38,727,979 as of March 25, 2022.
DOCUMENTS INCORPORATED BY REFERENCE
None
CORMEDIX INC.
Neutrolin® is our registered trademark. DefenCath™ and CorMedix Inc. are our filed trademarks. All other trade names, trademarks and service marks appearing in this report are the property of their respective owners. We have assumed that the reader understands that all such terms are source-indicating. Accordingly, such terms, when first mentioned in this report, appear with the trade name, trademark or service mark notice and then throughout the remainder of this report without trade name, trademark or service mark notices for convenience only and should not be construed as being used in a descriptive or generic sense.
PART I
Forward-Looking Statements
This report contains “forward-looking statements” that involve risks and uncertainties, as well as assumptions that, if they never materialize or prove incorrect, could cause our results to differ materially from those expressed or implied by such forward-looking statements. The statements contained in this report that are not purely historical are forward-looking statements within the meaning of Section 27A of the Securities Act of 1933, as amended (the “Securities Act”), and Section 21E of the Securities Exchange Act of 1934, as amended (the “Exchange Act”). Forward-looking statements are often identified by the use of words such as, but not limited to, “anticipate,” “believe,” “can,” “continue,” “could,” “estimate,” “expect,” “intend,” “may,” “will,” “plan,” “project,” “seek,” “should,” “target,” “will,” “would,” and similar expressions or variations intended to identify forward-looking statements. These statements are based on the beliefs and assumptions of our management based on information currently available to management. Such forward-looking statements are subject to risks, uncertainties and other important factors that could cause actual results and the timing of certain events to differ materially from future results expressed or implied by such forward-looking statements. Factors that could cause or contribute to such differences include, but are not limited to, those identified below in the section titled “Item 1A. Risk Factors.” The impact of COVID-19 may also exacerbate these risks, any of which could have a material effect on us. Furthermore, such forward-looking statements speak only as of the date of this report. Except as required by law, we undertake no obligation to update any forward-looking statements to reflect events or circumstances after the date of such statements.
Item 1. Business
Overview
We are a biopharmaceutical company focused on developing and commercializing therapeutic products for the prevention and treatment of infectious and inflammatory diseases.
Our primary focus is on the development of our lead product candidate, DefenCath™, for potential commercialization in the United States, or U.S., and other key markets. We have in-licensed the worldwide rights to develop and commercialize DefenCath and Neutrolin®. The name DefenCath is the U.S. proprietary name conditionally approved by the U.S. Food and Drug Administration, or FDA. The name Neutrolin is currently used in the European Union, or EU, and other territories where the Company has received CE-Mark approval for the commercial distribution of Neutrolin as a catheter lock solution, or CLS, regulated as a medical device.
DefenCath/Neutrolin is a novel anti-infective solution (a formulation of taurolidine 13.5 mg/mL, and heparin 1000 USP Units/mL) intended for the reduction of catheter-related infections and thrombosis in patients requiring central venous catheters, or CVCs, in clinical settings such as hemodialysis and total parenteral nutrition. Infections and thrombosis represent key complications among hemodialysis patients with CVCs. These complications can lead to treatment delays and increased costs to the healthcare system when they occur due to hospitalizations, need for intravenous, or IV, antibiotic treatment, removal/replacement of the CVC, related treatment costs and increased mortality. We believe DefenCath addresses a significant unmet medical need and a potential large market opportunity.
DefenCath – United States
In late 2013, we met with the FDA, to determine the pathway for obtaining U.S. marketing approval of DefenCath as a new drug. In January 2015, the FDA designated DefenCath as a Qualified Infectious Disease Product, or QIDP, for prevention of catheter-related blood stream infections, or CRBSIs, in patients with end stage renal disease receiving hemodialysis through a CVC. CRBSIs can be life-threatening. The QIDP designation provides five years of market exclusivity in addition to the five years granted for a New Chemical Entity, or NCE, upon approval of a New Drug Application, or NDA. In addition, in January 2015 the FDA granted Fast Track designation to DefenCath Catheter Lock Solution, a designation intended to facilitate development and expedite review of drugs that treat serious and life-threatening conditions so that the approved drug can reach the market expeditiously. The Fast Track designation of DefenCath provides the Company with the opportunity to meet with the FDA on a more frequent basis during the development process, and also ensures eligibility to request priority review of the marketing application.
We launched the Phase 3 clinical trial in patients with hemodialysis catheters in the U.S. in December 2015. The clinical trial, named Phase 3 Prospective, Multicenter, Double-blind, Randomized, Active Control Study to Demonstrate Safety and Effectiveness of DefenCath in Preventing Catheter-related Bloodstream Infection in Subjects on Hemodialysis for End Stage Renal Disease, or LOCK-IT-100, was a prospective, multicenter, randomized, double-blind, active control trial which aimed to demonstrate the efficacy and safety of DefenCath in preventing CRBSIs, in subjects receiving hemodialysis therapy as treatment for end stage renal disease. The primary endpoint for the trial was time to CRBSI. The trial evaluated DefenCath relative to the active control heparin by documenting the incidence of CRBSI and the time until the occurrence of CRBSI for each study subject. Secondary endpoints were catheter patency, which was defined as required use of tissue plasminogen activating factor, or tPA, or removal of catheter due to dysfunction, and removal of catheter for any reason.
During the course of the study, in consultation with the FDA, we established the Clinical Adjudication Committee, or CAC, to critically and independently assess CRBSI while being blinded to treatment assignment. As announced in July 2018, the CAC reviewed potential cases of CRBSI in our LOCK-IT-100 study that occurred through early December 2017 and identified 28 such cases. As previously agreed with the FDA, an interim efficacy analysis was performed when the first 28 CRBSIs were identified. On July 25, 2018, we announced that the independent Data Safety Monitoring Board, or DSMB, had completed its review of the interim analysis of the data from the LOCK-IT-100 study. Based on the first 28 cases, there was a highly statistically significant 72% reduction in CRBSI relative to the control (p=0.0034). Because the pre-specified level of statistical significance was reached for the primary endpoint and efficacy had been demonstrated with no safety concerns, the DSMB recommended the study be terminated early.
Following discussions with the FDA, we proceeded with an orderly termination of LOCK-IT-100. In late January 2019, we announced the topline results of the full data set of the LOCK-IT-100 study. The study continued enrolling and treating subjects until study termination, and the final efficacy analysis was based on a total of 795 subjects.
The primary endpoint of the Phase 3 LOCK-IT-100 study was the reduction of the risk of occurrence of CRBSI by DefenCath relative to the active control of heparin. In the analysis of the full data set, a total of 41 CRBSI events were determined by the CAC. There was a 71% reduction in the risk of occurrence of CRBSIs compared with the active control of heparin, which was well in excess of the study’s assumed treatment effect size of a 55% reduction. In the DefenCath arm, the CRBSI event rate was 0.13 per 1000 catheter days, which is significantly lower than the event rate of 0.46 per 1000 catheter days in the control arm. The statistical significance of the primary endpoint in the full data set (p=0.0006) was even more impressive than that of the interim analysis (p=0.0034).
The FDA granted our request for a rolling submission and review of the New Drug Application, or NDA, that is designed to expedite the approval process for products being developed to address an unmet medical need. Although the FDA usually requires two pivotal clinical trials to provide substantial evidence of safety and effectiveness for approval of the NDA, the FDA will in some cases accept one adequate and well-controlled trial, where it is a large multicenter trial with a broad range of subjects and investigation sites with procedures to include trial quality that has demonstrated a clinically meaningful and statistically very persuasive effect on prevention of a disease with potentially serious outcome.
In March 2020, we began the modular submission process for the NDA for DefenCath for the prevention of CRBSI in hemodialysis patients, and in August 2020, the FDA accepted for filing the DefenCath NDA. The FDA also granted our request for priority review, which provides for a six-month review period instead of the standard ten-month review period. As we announced in March 2021, the FDA informed us in its Complete Response Letter (“CRL”) that it cannot approve the NDA for DefenCath in its present form. The FDA noted concerns at the third-party manufacturing facility after a review of records requested by the FDA and provided by the contract manufacturing organization, or CMO. Additionally, the FDA is requiring a manual extraction study to demonstrate that the labeled volume can be consistently withdrawn from the vials despite an existing in-process control to demonstrate fill volume within specifications.
In April 2021, we and the CMO met with the FDA to discuss proposed resolutions for the deficiencies identified in the CRL to us and the Post-Application Action Letter, or PAAL, received by the CMO from the FDA for the NDA for DefenCath. There was an agreed upon protocol for the manual extraction study identified in the CRL, which now has been successfully completed. Addressing the FDA’s concerns regarding the qualification of the filling operation necessitated adjustments in the process and generation of additional data on operating parameters for manufacture of DefenCath. We and the CMO determined that additional process qualification is needed with subsequent validation to address these issues. The FDA stated that the review timeline would be determined when the NDA resubmission is received. The FDA also stated that it expected all corrections to facility deficiencies to be complete at the time of resubmission so that all corrective actions may be verified during an onsite evaluation of the manufacturing facility in the next review cycle, if the FDA determines it will do an onsite evaluation.
CorMedix and the CMO worked closely to ensure that the identified deficiencies were resolved and on February 28, 2022, we announced that we resubmitted the NDA for DefenCath to address the CRL issued by the FDA. In parallel, our third-party manufacturer submitted responses to the deficiencies identified at the manufacturing facility in the PAAL issued by the FDA concurrently with the CRL. Satisfactory resolution of these issues is required for approval of the DefenCath NDA. If an onsite inspection is required, we may encounter delays in obtaining FDA approval because the FDA is currently facing a backlog due to the COVID-19 pandemic. The FDA issued a guidance document on its plan to use voluntary remote interactive evaluations at facilities, including for a pre-approval inspection to assess a marketing application. The FDA will request the manufacturing facility to participate in a voluntary remote interactive evaluation, if the FDA believes it is appropriate. A manufacturing facility cannot request the remote interaction. The FDA expects the use of remote interactive evaluations should help the FDA operate within normal timeframes in spite of the COVID-19 pandemic.
The FDA did not request additional clinical data and did not identify any deficiencies related to the data submitted on the efficacy or safety of DefenCath from LOCK-IT-100. In draft labeling discussed with the FDA, the FDA added that the initial approval will be for the limited population of patients with kidney failure receiving chronic hemodialysis through a central venous catheter. This is consistent with our request for approval pursuant to the Limited Population Pathway for Antibacterial and Antifungal Drugs, or LPAD. LPAD, passed as part of the 21st Century Cures Act, is a new program intended to expedite the development and approval of certain antibacterial and antifungal drugs to treat serious or life-threatening infections in limited populations of patients with unmet needs. LPAD provides for a streamlined clinical development program involving smaller, shorter, or fewer clinical trials and is intended to encourage the development of safe and effective products that address unmet medical needs of patients with serious bacterial and fungal infections. We believe that LPAD will provide additional flexibility for the FDA to approve DefenCath to reduce CRBSIs in the limited population of patients with kidney failure receiving hemodialysis through a central venous catheter.
In March 2020, we were granted a deferral by the FDA under the Pediatric Research Equity Act, or PREA, that requires sponsors to conduct pediatric studies for NDAs for a new active ingredient, such as taurolidine in DefenCath, unless a waiver or deferral is obtained from the FDA. A deferral acknowledges that a pediatric assessment is required but permits the applicant to submit the pediatric assessment after the submission of an NDA. We have made a commitment to conduct the pediatric study after approval of the NDA for use in adult hemodialysis patients. Pediatric studies for an approved product conducted under PREA may qualify for pediatric exclusivity, which if granted would provide an additional six months of marketing exclusivity. DefenCath would then have the potential to receive a total marketing exclusivity period of 10.5 years, including exclusivity pursuant to NCE and QIDP.
Neutrolin – International
In the European Union, or EU, Neutrolin is regulated as a Class 3 medical device. In July 2013, we received CE Mark approval for Neutrolin. In December 2013, we commercially launched Neutrolin in Germany for the prevention of CRBSI, and maintenance of catheter patency in hemodialysis patients using a tunneled, cuffed central venous catheter for vascular access. To date, Neutrolin is registered and may be sold in certain European Union countries for such treatment.
In September 2014, the TUV-SUD and The Medicines Evaluation Board of the Netherlands, or MEB, granted a label expansion for Neutrolin for these same expanded indications for the EU. In December 2014, we received approval from the Hessian District President in Germany to expand the label to include use in oncology patients receiving chemotherapy, IV hydration and IV medications via central venous catheters. The expansion also adds patients receiving medication and IV fluids via central venous catheters in intensive or critical care units (cardiac care unit, surgical care unit, neonatal critical care unit, and urgent care centers). An indication for use in total parenteral nutrition was also approved.
Additional Development Possibilities
In addition to developing the use of taurolidine as a catheter lock solution, we are sponsoring a pre-clinical research collaboration for the use of taurolidine as a possible treatment for rare pediatric tumors. In February 2018, the FDA granted orphan drug designation to taurolidine for the treatment of neuroblastoma in children. We may seek one or more strategic partners or other sources of capital to help us develop and commercialize taurolidine for the treatment of neuroblastoma in children. We are also evaluating opportunities for the possible expansion of taurolidine as a platform compound for use in certain medical devices. Patent applications have been filed in several indications, including wound closure, surgical meshes, and wound management. Based on initial feasibility work, we are advancing pre-clinical studies for taurolidine-infused surgical meshes, suture materials and hydrogels. We will seek to establish development/commercial partnerships as these programs advance.
The FDA regards taurolidine as a new chemical entity and therefore it is currently regulated as an unapproved new drug. We might in the future pursue product candidates that would involve devices impregnated with taurolidine, and we believe that at the current time such products would be combination products subject to both device premarket submission requirements and drug regulations. Consequently, given that there is no appropriate predicate medical device currently marketed in the U.S. on which a 510(k) clearance process could be based and that taurolidine is not yet approved in any application, we anticipate that we would be required to submit a premarket approval application, or PMA, for marketing authorization for any medical device indications that we may pursue for devices containing taurolidine. In the event that an NDA for DefenCath is approved by the FDA, the regulatory pathway for these medical device product candidates may be revisited with the FDA. Although there may be no appropriate predicate, de novo Class II designation can be proposed, based on a risk assessment and a reasonable assurance of safety and effectiveness.
DefenCath
Market Opportunity
Central venous catheters and peripherally inserted central catheters (“Central Catheters”) are an important and frequently used method for accessing the vasculature in hemodialysis (a form of dialysis where the patient’s blood is circulated through a dialysis filter), administering chemotherapy and basic fluids in cancer patients and for cancer chemotherapy, long term antibiotic therapy, and total parenteral nutrition (complete or partial dietary support via intravenous nutrients).
According to the 2015 United States Renal Disease System, there were 660,000 patients on hemodialysis in the U.S. Hemodialysis National Kidney Foundation has reported that patients requiring Central Catheters represent over 63 million catheter/dialysis treatment days per year.
One of the major and common complications for all patients requiring CVCs is CRBSI and the clinical complications associated with them. The total annual cost for treating CRBSI episodes and their related complications in the U.S. is up to $2.7 billion, with approximately 250,000 CRBSI episodes per year (Becker’s Hospital Review).
Biofilm build up is the pathogenesis of both infections and thrombotic complications in central venous catheters. Prevention of CRBSI and inflammatory complications requires both removal of pathogens from the internal surface of the catheter to prevent the systemic dissemination of organisms contained within the biofilm as well as an anticoagulant to retain blood flow during dialysis. Biofilm forms when bacteria adhere to surfaces in aqueous environments and begin to excrete a slimy, glue-like substance that can anchor them to various types of materials, including intravenous catheters. The presence of biofilm has many adverse effects, including the ability to release bacteria into the blood stream. The current standard of catheter care is to instill a heparin lock solution at a concentration of 1000 u/mL into each catheter lumen immediately following treatment, in order to prevent clotting between dialysis treatments. However, a heparin lock solution provides no protection from the risk of infection.
Currently, there are no pharmacologic agents approved in the U.S. for the prevention of CRBSI in CVCs. As noted above, we received the CE Mark approval for Neutrolin from the MEB of the EU in July 2013. We believe there is a significant need for prevention of CRBSI in the hemodialysis patient population as well as for other patient populations utilizing central venous catheters and peripherally inserted central catheters, such as oncology/chemotherapy, and total parenteral nutrition.
DefenCath is a broad-spectrum antibacterial, antifungal and anticoagulant combination that is active against common microbes including antibiotic-resistant strains and in addition may prevent biofilm formation. We believe that using DefenCath as an anti-infective solution will significantly reduce the incidence of life-threatening catheter-related blood stream infections, thus reducing the need for local and systemic antibiotics while prolonging catheter function.
Initially, we expect to sell DefenCath in the U.S. primarily to key operators of dialysis centers. We anticipate that Medicare reimbursement could be available for DefenCath in hemodialysis and other catheter indications, such as oncology patients and total parenteral nutrition patients through relevant hospital inpatient diagnosis-related groups, or DRGs, or outpatient ambulatory payment classifications, or APCs, the End-Stage Renal Disease Prospective Payment System, or ESRD PPS, base payment, or under the Durable Medical Equipment, Prosthetics, Orthotics, and Supplies, or DMEPOS, Fee Schedule, depending on the setting of care. We also plan to seek separate reimbursement as a drug, where available under Medicare, through mechanisms such as pass-through status under the Hospital Outpatient Prospective Payment System, the transitional drug add-on payment adjustment, or TDAPA, under the ESRD PPS, or reimbursement as a drug used with a DMEPOS infusion pump. We have engaged the U.S. Centers for Medicare & Medicaid Services, or CMS, in preliminary discussions concerning the reimbursement for DefenCath under TDAPA, however, qualifications cannot be determined until after FDA approval and CMS evaluates the request for coverage in a quarterly review. If approved under TDAPA, reimbursement of DefenCath would be calculated based on its average selling price. To be eligible for TDAPA, a new renal drug or biologic must be:
| ● | Approved by FDA pursuant to Section 505(b)(1) of the Federal Food, Drug, and Cosmetic Act |
| | | |
| ● | Assigned a Healthcare Common Procedure Coding System code |
| | | |
| ● | Identified as having an end action effect that treats or manages a condition or conditions associated with ESRD |
| | | |
| ● | Identified as not fitting into an established ESRD PPS functional category |
| | | |
| ● | Designated by CMS as a renal dialysis service. |
Although we cannot fully anticipate changes in reimbursement requirements and mechanisms in the coming years, we expect DefenCath would be eligible for and would obtain TDAPA. DefenCath meets the criterion of being a new renal dialysis product used to treat or manage a condition associated with ESRD, since infections are the second leading cause of death in patients with ESRD and CVCs are a significant risk factor for infection-associated mortality.
Furthermore, we anticipate that the CMS, and private payers will increasingly demand that manufacturers demonstrate the cost effectiveness of their product as part of the reimbursement review and approval process. With this in mind, we are performing health economic evaluations to support this review in the context of the prospective use of DefenCath in dialysis, and other settings. Our studies may not be sufficient to support coverage or reimbursement at levels that allow providers to use DefenCath.
Competitive Landscape
The drug and medical device industries are highly competitive and subject to rapid and significant technological change. DefenCath’s current and future competitors include large as well as specialty pharmaceutical and biotechnology companies and large and specialty medical device companies. Many of our competitors have substantially greater financial, technical and human resources than we do and significantly more experience in the development and commercialization of drugs and medical devices. Further, the development of new treatment methods could render DefenCath non-competitive or obsolete.
We believe that the key competitive factors that will affect the development and commercial success of DefenCath are efficacy and safety, as well as pricing and reimbursement. Given that there are no approved catheter lock solutions with antimicrobial properties in the U.S., and that the current standard of care is heparin, we believe there is an opportunity for DefenCath to become the new standard of care as a CLS in the U.S. market, if approved by FDA. We are not aware of any potentially competitive CLS which are approved or under development by other companies in the U.S. A development stage product from Citius is being studied for salvage of CVCs once a patient becomes diagnosed with a catheter related blood stream infection.
In the EU, several catheter lock solutions have received a CE Mark, in addition to Neutrolin. For example, TauoLock contains a combination of citrate 4% with (cyclo)-taurolidine and heparin or urokinase, but it is not approved for use in the U.S. Some device companies have launched antibiotic or antimicrobial-coated catheters as short-term prevention of catheter infection. We believe these are not effective for hemodialysis catheters due to the long-term use and high blood flow associated with hemodialysis.
Manufacturing/Supply Chain
We do not own or operate any manufacturing facilities related to the production of our products. All our manufacturing processes currently are, and we expect them to continue, to be outsourced to third parties. We rely on third-party manufacturers to produce sufficient quantities of drug product for use both commercially and in clinical trials. We intend to continue this practice in the future.
With regards to taurolidine, an active drug ingredient, or API, of DefenCath, we have a Drug Master File filed with the FDA. There is a master commercial supply agreement between the third-party manufacturer, and us in place from August 2018. We have two sources for the other key API, Heparin sodium.
We have utilized two drug product CMOs. One CMO manufactures for the EU and Middle East markets and the other is for U.S. production. In order to assure supply, we are in the process of identifying an additional CMO.
We are confident that these CMO’s have adequate capacity to produce the volumes needed, and that there exists a sufficient number of potential alternate sources for the drug substances required to produce our products, as well as third-party manufacturers, that we will be able to find alternate suppliers and third-party manufacturers in the event that our relationship with any supplier or third-party manufacturer deteriorates. The process for selecting and qualifying an alternative contract manufacturer and for completing the technology transfer to such a manufacturer to the point of enabling commercialization of the product would take several years.
United States Government Regulation
The research, development, testing, manufacture, labeling, promotion, advertising, distribution, and marketing, among other things, of our products are extensively regulated by governmental authorities in the U.S. and other countries. Our products may be classified by the FDA as a drug or a medical device depending upon the indications for use or claims. Because certain of our product candidates are considered as medical devices and others are considered as drugs for regulatory purposes, we intend to submit applications to regulatory agencies for approval or clearance of both medical devices and pharmaceutical product candidates.
In the U.S., the FDA regulates drugs and medical devices under the Federal Food, Drug, and Cosmetic Act (FDCA) and the Agency’s implementing regulations. If we fail to comply with the applicable U.S. requirements at any time during the product development process, clinical testing, and during the approval process or after approval, we may become subject to administrative or judicial sanctions. These sanctions could include the FDA’s refusal to approve pending applications, license suspension or revocation, withdrawal of an approval, warning letters, adverse publicity, product recalls, product seizures, total or partial suspension of production or distribution, injunctions, fines, civil penalties or criminal prosecution, among other actions. Any agency enforcement action and/or any related impact could have a material adverse effect on us.
Drug Approval Process
The research, development, and approval process in the United States and elsewhere is intensive and rigorous and generally takes many years to complete. The typical process required by the FDA before a therapeutic drug may be marketed in the United States includes:
| ● | Pre-clinical laboratory and animal tests performed under the FDA’s Good Laboratory Practices, or GLP, regulations; |
| | | |
| ● | submission to the FDA of an investigational new drug application, or IND, which must become effective before human clinical trials may commence; |
| | | |
| ● | human clinical studies to evaluate the drug’s safety and effectiveness for its intended uses; |
| | | |
| ● | FDA review of whether the facility in which the drug is manufactured, processed, packaged, or held meets standards designed to assure the product’s continued quality and FDA review of clinical trial sites to determine whether the clinical trials were conducted in accordance with Good Clinical Practices, or GCPs; and |
| | | |
| ● | submission of a new drug application, or NDA, to the FDA, and approval of the application by the FDA to allow sales of the drug. |
During pre-clinical testing, studies are performed with respect to the chemical and physical properties of candidate formulations. These studies are subject to GLP requirements. Biological testing is typically done in animal models to demonstrate the activity of the compound against the targeted disease or condition and to assess the apparent effects of the new product candidate on various organ systems, as well as its relative therapeutic effectiveness and safety. An IND application must be submitted to the FDA and become effective before studies in humans may commence.
Clinical trial programs in humans generally follow a three-phase process. Typically, Phase 1 studies are conducted in small numbers of healthy volunteers or, on occasion, in patients afflicted with the target disease. Phase 1 studies are conducted to determine the metabolic and pharmacological action of the product candidate in humans and the side effects associated with increasing doses, and, if possible, to gain early evidence of effectiveness. In Phase 2, studies are generally conducted in larger groups of patients having the target disease or condition in order to validate clinical endpoints, and to obtain preliminary data on the effectiveness of the product candidate and optimal dosing. This phase also helps determine further the safety profile of the product candidate. In Phase 3, large-scale clinical trials are generally conducted in patients having the target disease or condition to provide sufficient data for the statistical proof of effectiveness and safety of the product candidate as required by United States and foreign regulatory agencies. Typically, two Phase 3 trials are required for marketing approval.
In the case of products for certain serious or life-threatening diseases, the initial human testing may be done in patients with the disease rather than in healthy volunteers. Because these patients are already afflicted with the target disease or condition, it is possible that such studies will also provide results traditionally obtained in Phase 2 studies. These studies are often referred to as “Phase 1/2” studies. However, even if patients participate in initial human testing and a Phase 1/2 study is carried out, the sponsor is still responsible for obtaining all the data usually obtained in both Phase 1 and Phase 2 studies.
Before proceeding with a study, sponsors may seek a written agreement known as a Special Protocol Assessment, or SPA, from the FDA regarding the design, size, and conduct of a clinical trial. Among other things, SPAs can cover clinical studies for pivotal trials whose data will form the primary basis to establish a product’s efficacy. SPAs help establish up-front agreement with the FDA about the adequacy of a clinical trial design to support a regulatory approval, but the agreement is not binding on the FDA if new circumstances arise. An SPA may only be modified with the agreement of the FDA and the trial sponsor or if the director of the FDA reviewing division determines that a substantial scientific issue essential to determining the safety or efficacy of the drug was identified after the testing began. There is no guarantee that a study will ultimately be adequate to support an approval even if the study is subject to an SPA.
Additionally, some clinical trials are overseen by an independent group of qualified experts organized by the clinical trial sponsor, known as a data safety monitoring board or committee. This group regularly reviews accumulated data and advises the study sponsor regarding the continuing safety of trial subjects, and the continuing validity and scientific merit of the clinical trial. The data safety monitoring board receives special access to unblinded data during the clinical trial and may advise the sponsor to halt the clinical trial if it determined there is an unacceptable safety risk for subjects or on other grounds, such as no demonstration of efficacy. The committee can also stop a clinical trial for an overwhelming demonstration of efficacy, based on pre-defined, stringent statistical parameters and ethical considerations.
The manufacture of investigational drugs for the conduct of human clinical trials is subject to current Good Manufacturing Practice, or cGMP, requirements. Investigational drugs and active pharmaceutical ingredients imported into the United States are also subject to regulation by the FDA relating to their labeling and distribution. Further, the export of investigational drug products outside of the United States is subject to regulatory requirements of the receiving country as well as U.S. export requirements under the FDCA.
IND sponsors are required to submit a number of reports to the FDA during the course of a development program. For instance, sponsors are required to make annual reports to the FDA concerning the progress of their clinical trial programs as well as more frequent reports for certain serious adverse events. Sponsors must submit a protocol for each clinical trial, and any subsequent protocol amendments to the FDA. Investigators must also provide certain information to the clinical trial sponsors to allow the sponsors to make certain financial disclosures to the FDA. Information about certain clinical trials, including a description of the study and study results, must be submitted within specific timeframes to the National Institutes of Health, or NIH, for public dissemination on their clinicaltrials.gov website. Moreover, under the 21st Century Cures Act, manufacturers or distributors of investigational drugs for the diagnosis, monitoring, or treatment of one or more serious diseases or conditions must have a publicly available policy concerning expanded access to investigational drugs.
United States law requires that studies conducted to support approval for product marketing be “adequate and well controlled.” In general, this means that either a placebo or a product already approved for the treatment of the disease or condition under study must be used as a reference control. The recently passed 21st Century Cures Act, however, provides for FDA acceptance of new kinds of data such as patient experience data, real world evidence, and, for appropriate indications sought through supplemental marketing applications, data summaries. Studies must also be conducted in compliance with good clinical practice requirements, and informed consent must be obtained from all study subjects.
In addition, under the Pediatric Research Equity Act, or PREA, an NDA or supplement to an NDA for a new active ingredient, indication, dosage form, dosage regimen, or route of administration must contain data that are adequate to assess the safety and effectiveness of the drug for the claimed indications in all relevant pediatric subpopulations, and to support dosing and administration for each pediatric subpopulation for which the product is safe and effective. The FDA may, on its own initiative or at the request of the applicant, grant deferrals for submission of some or all pediatric data until after approval of the product for use in adults, or full or partial waivers from the pediatric data requirements.
The FDA also may require submission of a risk evaluation and mitigation strategy, or REMS, to ensure that the benefits of the drug outweigh the risks of the drug. The REMS plan could include medication guides, physician communication plans, and elements to assure safe use, such as restricted distribution methods, patient registries, or other risk minimization tools. An assessment of the REMS must also be conducted at set intervals. Following product approval, a REMS may also be required by the FDA if new safety information is discovered and the FDA determines that a REMS is necessary to ensure that the benefits of the drug outweigh the risks of the drug.
The clinical trial process for a new compound can take ten years or more to complete. The FDA may prevent clinical trials from beginning or may place clinical trials on hold at any point in this process if, among other reasons, it concludes that study subjects are being exposed to an unacceptable health risk. Trials may also be prevented from beginning or may be terminated by institutional review boards, or IRBs, who must review and approve all research involving human subjects and amendments thereto. The IRB must continue to oversee the clinical trial while it is being conducted. This includes the IRB receiving information concerning unanticipated problems involving risk to subjects. Side effects or adverse events that are reported during clinical trials can delay, impede, or prevent marketing authorization. Similarly, adverse events that are reported after marketing authorization can result in additional limitations being placed on a product’s use and, potentially, withdrawal of the product from the market.
Following the completion of a clinical trial, the data are analyzed by the sponsoring company to determine whether the trial successfully demonstrated safety and effectiveness and whether a product approval application may be submitted. In the United States, if the product is regulated as a new drug, an NDA must be submitted and approved by the FDA before commercial marketing may begin. The NDA must include a substantial amount of data and other information concerning the safety and effectiveness of the compound from laboratory, animal, and human clinical testing, as well as data and information on manufacturing, product quality and stability, and proposed product labeling.
Each domestic and foreign manufacturing establishment, including any contract manufacturers that we may decide to use, must be listed in the NDA and must be registered with the FDA. The application generally will not be approved until the FDA conducts a manufacturing inspection, approves the applicable manufacturing process for the drug product, and determines that the facility is in compliance with current cGMP requirements. Moreover, FDA will also typically inspect one or more clinical trial sites to confirm that the applicable clinical trials were conducted in accordance with GCPs.
Under the Prescription Drug User Fee Act (PDUFA), as amended, the FDA assesses and receives application user fees for reviewing an NDA, as well as annual program fees for commercial manufacturing establishments and for approved products. These fees can be significant. Fee waivers, reductions or refunds are available in certain circumstances. One basis for a waiver or refund of the application user fee is if the applicant is a “small business” generally defined as employing fewer than 500 employees, including employees of affiliates, no approved marketing application for a product that has been introduced or delivered for introduction into interstate commerce, and the applicant, including its affiliates, is submitting its first marketing application. Product candidates that are designated as orphan drugs, which are further described below, are also not subject to application user fees unless the application includes an indication other than the orphan indication. Under certain circumstances, orphan products may also be exempt from product and establishment fees.
Each NDA submitted for FDA approval is usually reviewed for administrative completeness and reviewability. Following this review, the FDA may request additional information rather than accept an NDA for filing. In this event, the application must be resubmitted with the additional information. The resubmitted application is also subject to review before the FDA accepts it for filing.
Once accepted for filing, the FDA’s review of an application may involve review and recommendations by an independent FDA advisory committee. The FDA must refer applications for drugs that contain active ingredients, including any ester or salt of the active ingredients that have not previously been approved by the FDA to an advisory committee or provide in an action letter a summary for not referring it to an advisory committee. The FDA may also refer drugs to advisory committees when it is determined that an advisory committee’s expertise would be beneficial to the regulatory decision-making process, including the evaluation of novel products and the use of new technology. An advisory committee is typically a panel that includes clinicians and other experts, which review, evaluate, and make a recommendation as to whether the application should be approved and under what conditions. The FDA is not bound by the recommendations of an advisory committee, but it considers such recommendations carefully when making decisions.
After evaluating the NDA and all related information, including the advisory committee recommendation, if any, and inspection reports regarding the manufacturing facilities and clinical trial sites, the FDA may issue an approval letter, or, in some cases, a Complete Response Letter, or CRL. If a CRL is issued, the applicant may either resubmit the NDA, addressing all the deficiencies identified in the letter; withdraw the application; or request an opportunity for a hearing. A CRL indicates that the review cycle of the application is complete, and the application is not ready for approval and describes all the specific deficiencies that the FDA identified in the NDA. A CRL generally contains a statement of specific conditions that must be met in order to secure final approval of the NDA and may require additional clinical or pre-clinical testing in order for the FDA to reconsider the application. The deficiencies identified may be minor, for example, requiring labeling changes; or major, for example, requiring additional clinical trials. Even with submission of this additional information, the FDA ultimately may decide that the application does not satisfy the regulatory criteria for approval. If and when those conditions have been met to the FDA’s satisfaction, the FDA may issue an approval letter. An approval letter authorizes commercial marketing of the drug with specific prescribing information for specific indications.
Even if the FDA approves a product, it may limit the approved therapeutic uses for the product as described in the product labeling, require that warning statements be included in the product labeling, require that additional studies be conducted following approval as a condition of the approval, impose restrictions and conditions on product distribution, prescribing, or dispensing in the form of a REMS or otherwise limit the scope of any approval.
Special FDA Expedited Review and Approval Programs
The FDA has various programs, including Fast Track designation, priority review and breakthrough designation, that are intended to expedite or simplify the process for the development and FDA review of certain drug products that are intended for the treatment of serious or life-threatening diseases or conditions, and demonstrate the potential to address unmet medical needs or present a significant improvement over existing therapy. The purpose of these programs is to provide important new drugs to patients earlier than under standard FDA review procedures.
To be eligible for a Fast Track designation, the FDA must determine, based on the request of a sponsor, that a product is intended to treat a serious or life-threatening disease or condition and demonstrates the potential to address an unmet medical need. The FDA will determine that a product will fill an unmet medical need if the product will provide a therapy where none exists or provide a therapy that may be potentially superior to existing therapy based on efficacy, safety, or public health factors. If Fast Track designation is obtained, drug sponsors may be eligible for more frequent development meetings and correspondence with the FDA. In addition, the FDA may initiate review of sections of an NDA before the application is complete. This “rolling review” is available if the applicant provides and the FDA approves a schedule for the remaining information. A Fast Track product is also eligible to apply for accelerated approval and priority review.
The FDA may give a priority review designation to drugs that are intended to treat serious conditions and, if approved, would provide significant improvements in the safety or effectiveness of the treatment, diagnosis, or prevention of serious conditions. A priority review means that the goal for the FDA is to review an application within six months, rather than the standard review of ten months under current PDUFA guidelines, of the 60-day filing date for new molecular entities.
Moreover, under the provisions of the Food and Drug Administration Safety and Innovation Act, or FDASIA, enacted in 2012, a sponsor can request designation of a product candidate as a “breakthrough therapy.” A breakthrough therapy is defined as a drug that is intended, alone or in combination with one or more other drugs, to treat a serious or life-threatening disease or condition, and preliminary clinical evidence indicates that the drug may demonstrate substantial improvement over existing therapies on one or more clinically significant endpoints, such as substantial treatment effects observed early in clinical development. Drugs designated as breakthrough therapies are eligible for the Fast Track designation features as described above, intensive guidance on an efficient drug development program beginning as early as Phase 1 trials, and a commitment from the FDA to involve senior managers and experienced review staff in a proactive collaborative, cross-disciplinary review.
Even if a product qualifies for one or more of these programs, the FDA may later decide that the product no longer meets the conditions for qualification or decide that the time period for FDA review or approval will not be shortened.
A final new program to expedite the development of drug products is the LPAD, which was passed as part of the 21st Century Cures Act. LPAD allows for the FDA’s determination of safety and effectiveness to reflect the risk-benefit profile of the drug in the intended limited population, taking into account the severity, rarity, or prevalence of the infection and the availability of alternative treatments in the limited population. Under LPAD, a sponsor may request drug approval for an antibacterial or antifungal drug if the drug is intended to treat a serious life-threatening infection in a limited population of patients with unmet needs. The drug may be approved for the limited population notwithstanding a lack of evidence to fully establish a favorable benefit-risk profile in a broader population. The FDA must provide prompt advice to sponsors seeking approval under LPAD to enable them to plan a development program. If approved under LPAD, certain post-marketing requirements would apply, such as required labeling and advertising statements and pre-distribution submission of promotional materials to FDA. If after approval for a limited population, a product receives a broader approval, the FDA may remove such post-marketing restrictions. While a drug may only be approved for a limited population under this program, the 21st Century Cures Act states that it is not intended to restrict the prescribing of antimicrobial drugs or other products by healthcare professionals.
Exclusivity
For approved drug products, market exclusivity provisions under the FDCA provide periods of regulatory exclusivity, which gives the holder of an approved NDA limited protection from new competition in the marketplace for the innovation represented by its approved drug.
Section 505 of the FDCA describes three types of marketing applications that may be submitted to the FDA to request marketing authorization for a new drug. A Section 505(b)(1) NDA is an application that contains full reports of investigations of safety and efficacy. A Section 505(b)(2) NDA is an application in which the applicant, in part, relies on investigations that were not conducted by or for the applicant and for which the applicant has not obtained a right of reference or use from the person by or for whom the investigations were conducted. Section 505(j) establishes an abbreviated approval process for a generic version of approved drug products through the submission of an Abbreviated New Drug Application, or ANDA. An ANDA provides for marketing of a generic drug product that has the same active ingredients, dosage form, strength, route of administration, labeling, performance characteristics, and intended use, among other things, to a previously approved product. Limited changes must be pre-approved by the FDA via a suitability petition.
Five years of exclusivity are available to New Chemical Entities, or NCEs. A NCE is a drug that contains no active moiety that has been approved by the FDA in any other NDA. An active moiety is the molecule or ion, excluding those appended portions of the molecule, that cause the drug to be an ester, salt, including a salt with hydrogen or coordination bonds, or other noncovalent derivatives, such as a complex, chelate, or clathrate, of the molecule, responsible for the therapeutic activity of the drug substance. During the exclusivity period, the FDA may not accept for review and make an ANDA or a 505(b)(2) NDA approval effective for an application submitted by another company that contains the previously approved active moiety. An ANDA or 505(b)(2) application, however, may be submitted one year before NCE exclusivity expires if the applicant submits a certification stating that the patents listed by the NCE sponsor in FDA’s list of Approved Drug Products with Therapeutic Equivalence Evaluations, or Orange Book, are invalid or will not be infringed by the manufacture, use, or sale of the drug product for which approval is sought. Five-year exclusivity will also not delay the submission or approval of a full NDA; however, an applicant submitting a full NDA would be required to conduct or obtain a right of reference to all the pre-clinical studies and adequate and well-controlled clinical trials necessary to demonstrate safety and efficacy.
Pediatric exclusivity is another type of non-patent marketing exclusivity in the United States and, if granted, provides for the attachment of an additional six months of marketing protection to the term of any existing regulatory exclusivity, including the non-patent exclusivity period described above. This six-month exclusivity may be granted if an NDA sponsor submits pediatric data that fairly respond to a written request from the FDA for such data. The data do not need to show the product to be effective in the pediatric population studied; rather, if the clinical trial is deemed to fairly respond to the FDA’s request, the additional protection is granted. If reports of requested pediatric studies are submitted to and accepted by the FDA within the required time frames, whatever statutory or regulatory periods of exclusivity or Orange Book listed patent protection cover the drug are extended by six months. Moreover, pediatric exclusivity attaches to all formulations, dosage forms, and indications for products with existing marketing exclusivity or patent life that contain the same active moiety as that which was studied.
The Orphan Drug Act also provides incentives for the development of drugs intended to treat rare diseases or conditions, which generally are diseases or conditions affecting fewer than 200,000 individuals annually in the United States, or affecting more than 200,000 in the United States and for which there is no reasonable expectation that the cost of developing and making the drug available in the United States will be recovered from sales in the United States. Additionally, sponsors must present a plausible hypothesis for clinical superiority to obtain orphan designation if there is a drug already approved by the FDA that is intended for the same indication and that is considered by the FDA to be the same drug as the already approved drug. This hypothesis must be demonstrated to obtain orphan drug exclusivity. If granted, prior to product approval, Orphan Drug Designation entitles a party to financial incentives such as opportunities for grant funding towards clinical study costs, tax advantages, and user-fee waivers. In addition, if a product receives FDA approval for the indication for which it has orphan designation, the product is generally entitled to orphan drug exclusivity, which means the FDA may not approve any other application to market the same drug for the same indication for a period of seven years, except in limited circumstances, such as a showing of clinical superiority over the product with orphan exclusivity.
For certain infectious disease products, the above discussed exclusivity periods may be further extended under the FDA’s qualified infectious disease product program. A qualified infectious disease product, or QIDP, is an antibacterial or antifungal drug for human use intended to treat serious or life-threatening infections, including those caused by an antibacterial or antifungal resistant pathogen, including novel or emerging infectious pathogens; or qualifying pathogens designated by the FDA that have the potential to pose a serious threat to public health. Subject to the specified statutory limitations, a drug that is designated as a QIDP and is approved for the use for which the QIDP designation was granted will receive a 5-year extension to any exclusivity for which the application qualifies upon approval. For example, if the FDA approves an NDA for a drug designated as a QIDP, the NCE exclusivity period is extended to ten years and the FDA may not accept applications for nine years. Moreover, if a product is designated as a QIDP and an orphan product, the orphan product exclusivity period is extended to twelve years. These extensions are in addition to any extension that an application may be entitled to under the pediatric exclusivity provisions. To receive a QIDP designation, the sponsor must request that the FDA designate the product as such prior to the submission of an NDA. This designation may not be withdrawn except if the FDA finds that the request for designation contained an untrue statement of material fact. QIDPs are also eligible for Fast Track status and priority review.
In March 2020, we were granted a deferral by the FDA under the PREA, that requires sponsors to conduct pediatric studies for NDAs for a new active ingredient, such as taurolidine in DefenCath, unless a waiver or deferral is obtained from the FDA. A deferral acknowledges that a pediatric assessment is required but permits the applicant to submit the pediatric assessment after the submission of an NDA. We have made a commitment to conduct the pediatric study after approval of the NDA for use in adult hemodialysis patients. Pediatric studies for an approved product conducted under PREA may qualify for pediatric exclusivity, which if granted would provide an additional six months of marketing exclusivity. DefenCath would then have the potential to receive a total marketing exclusivity period of 10.5 years, including exclusivity pursuant to NCE and QIDP.
Post Approval Requirements
Significant legal and regulatory requirements also apply after FDA approval to market under an NDA. These include, among other things, requirements related to adverse event and other reporting, product tracking and tracing, suspect and illegitimate product investigations and notifications, product advertising and promotion and ongoing adherence to cGMPs, as well as the need to submit appropriate new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling, or manufacturing process. The FDA also enforces the requirements of the Prescription Drug Marketing Act which, among other things, imposes various requirements in connection with the distribution of product samples to physicians. The FDA enforces these requirements through, among other ways, periodic announced and unannounced facility inspections.
The FDA also strictly regulates marketing, labeling, advertising, and promotion of products that are placed on the market. A company can make only those claims relating to safety and efficacy that are approved by the FDA. Physicians, in their independent professional medical judgment, may prescribe legally available products for unapproved indications that are not described in the product’s labeling and that differ from those tested and approved by the FDA. Pharmaceutical companies, however, are allowed to promote their drug products only for the approved indications and in accordance with the provisions of the approved label. The FDA and other agencies actively enforce the laws and regulations prohibiting the promotion of off-label uses, and a company that is found to have improperly promoted off-label uses may be subject to significant liability, including, but not limited to, criminal and civil penalties under the FDCA and the civil False Claims Act, or FCA, exclusion from participation in federal healthcare programs, mandatory compliance programs under corporate integrity agreements, debarment, and refusal of government contracts.
The regulatory framework applicable to the production, distribution, marketing, and/or sale, of our product candidates may change significantly from the current descriptions provided herein in the time that it may take for any of our product candidates to reach a point at which an NDA is approved. Moreover, individual states may have laws and regulations that we must comply with, such as laws and regulations concerning licensing, promotion, sampling, distribution, and reporting.
Overall research, development, and approval times depend on a number of factors, including the period of review at the FDA, the number of questions posed by the FDA during review, how long it takes to respond to the FDA’s questions, the severity or life-threatening nature of the disease in question, the availability of alternative treatments, the availability of clinical investigators and eligible patients, the rate of enrollment of patients in clinical trials, and the risks and benefits demonstrated in the clinical trials.
Medical Device Approval Process
In addition to our lead product candidate DefenCath, which is subject to regulation by the FDA as a drug, we may be developing other products that may be regulated as medical devices in the United States. The FDA considers a product to be a device, and subject to the FDA regulation, if it meets the definition of a medical device in the FDCA, which states that a device is an instrument, apparatus, implement, machine, contrivance, implant, in vitro reagent, or other similar or related article, including a component part, or accessory which is:
| ● | recognized in the official National Formulary, or the United States Pharmacopoeia, or any supplement to them, |
| | | |
| ● | intended for use in the diagnosis of disease or other conditions, or in the cure, mitigation, treatment, or prevention of disease, in man or other animals, or |
| | | |
| ● | intended to affect the structure or any function of the body of man or other animals, and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which does not achieve its primary intended purposes through chemical action within or on the body of man or other animals and which is not dependent upon being metabolized for the achievement of its primary intended purposes. |
The FDA regulates the design, development, clinical testing, manufacture, labeling, distribution, import and export, sale and promotion of medical devices. Unless an exemption applies or a product is a Class I device, all medical devices must receive either 510(k) clearance or an approved pre-market application, or PMA, from the FDA before they may be commercially distributed in the U.S. In addition, certain modifications made to marketed devices also may require 510(k) clearance or approval of a PMA supplement. Unlike approved drug products, there are no market exclusivity provisions under the FDCA for products regulated as medical devices.
To obtain a 510(k) clearance for a device, a pre-market notification to the FDA must be submitted demonstrating that the device is substantially equivalent to a legally marketed predicate device. For a new device to be found “substantially equivalent” to one or other legally marketed predicate devices, the new device must have: 1) the same intended use as a predicate; and 2) either a) the same technological characteristics as the predicate device or b) different technological characteristics, but the information submitted must not raise new questions of safety and effectiveness and must demonstrate substantial equivalence. The FDA attempts to respond to a 510(k) pre-market notification within 90 days of submission, but as a practical matter, pre-market clearance can take significantly longer, potentially up to one year or more.
The PMA process is much more demanding and uncertain than the 510(k) pre-market notification process and must be supported by extensive clinical, laboratory, technical and other information, including at least one adequate and well-controlled clinical investigation conducted under an investigational device exemption (IDE). The FDA has 180 days to review an accepted PMA, although the review generally occurs over a significantly longer period of time and can take up to several years.
The FDA has informed us that it regards taurolidine as a new chemical entity and therefore an unapproved new drug. Consequently, for any other products that we intend to develop as a medical device, there is currently no appropriate predicate device currently marketed in the U.S. on which a 510(k) approval process could be based. As a result, we will be required to submit a premarket approval application for marketing authorization for these indications. In the event that the NDA for DefenCath is approved by the FDA, the regulatory pathway for these taurolidine product candidates can be revisited with the FDA. Although there will presumably still be no appropriate predicate, de novo Class II designation can be proposed, a process that provides a pathway to classify novel medical device for which there is no legally marketed predicate device, based on a risk assessment and a reasonable assurance of safety and effectiveness.
After a device is placed on the market, numerous regulatory requirements apply, including:
| ● | Quality System Regulations, or QSRs, which require manufacturers to have a quality system for the design, manufacture, packaging, labeling, storage, installation, and servicing of finished medical devices; |
| | | |
| ● | labeling regulations, which govern product labels and labeling, prohibit the promotion of products for unapproved, or off-label, uses and impose other restrictions on labeling and promotional activities; |
| | | |
| ● | medical device listing and establishment registration; |
| | | |
| ● | post-approval restrictions or conditions, including post-approval study commitments; |
| | | |
| ● | post-market surveillance requirements; |
| | | |
| ● | medical device reporting, or MDR, regulations, which require that manufacturers evaluate and investigate potential adverse events and malfunctions, and report to the FDA if their device may have caused or contributed to a death or serious injury or malfunctioned in a way that would likely cause or contribute to a death or serious injury if it were to recur; |
| | | |
| ● | regulations requiring the reporting of any device corrections or removals if the correction or removal was initiated to reduce a risk to health posed by the device or remedy a violation of the FDCA which may present a risk to health; and |
| | | |
| ● | the FDA’s recall authority, whereby it can ask, or under certain conditions order, device manufacturers to recall from the market a product that is a risk to health. |
Our manufacturing facilities, as well as those of certain of our suppliers, are subject to periodic and for-cause inspections by the FDA and other governmental authorities to verify compliance with the QSR and other regulatory requirements.
Reimbursement and Pricing Controls
In many of the markets where we or the parties we collaborate with have targeted or will target DefenCath for sale, laws control the prices charged to certain purchasers of pharmaceutical products and the prices paid by drug reimbursement programs through varying price control mechanisms. Public and private health care payors control costs and influence drug pricing through a variety of mechanisms, including through negotiating rebates with the manufacturers, limiting the reimbursement rate paid to providers, and using tiered formularies, co-payment structures that incentivize beneficiaries to request lower cost alternatives, and other mechanisms that provide preferential access to certain drugs over others within a therapeutic class. Federal and commercial payors use competition for health plan coverage and market share as leverage to obtain rebates on products they reimburse, which impacts the manufacturer’s net realization on the sale of the products. These rebates may be paid on drugs sold at a mandatory discount. Additionally, federal and commercial health plans may choose to reimburse dialysis providers for dialysis services and drugs used in the provision of those services through a single bundled payment rate, which tends to make cost a more important factor for providers when making drug purchase decisions than it would otherwise be if the providers were reimbursed for drugs on a stand-alone basis. Payors also set other criteria to govern the uses of a drug that will be deemed medically appropriate and therefore reimbursed or otherwise covered. In particular, many public and private health care payors limit reimbursement and coverage to the uses of a drug that are either approved by the FDA or that are supported by other appropriate evidence (for example, published medical literature) and appear in a recognized drug compendium. Drug compendia are publications that summarize the available medical evidence for particular drug products and identify which uses of a drug are supported or not supported by the available evidence, whether or not such uses have been approved by the FDA.
Foreign Regulatory Requirements
We and our collaborative partners may be subject to widely varying foreign regulations, which may be quite different from those of the FDA, governing clinical trials, manufacture, product registration and approval, and pharmaceutical sales. Whether or not FDA approval has been obtained, we or our collaboration partners must obtain a separate approval for a product by the comparable regulatory authorities of foreign countries prior to the commencement of product marketing in those countries. In certain countries, regulatory authorities also establish pricing and reimbursement criteria. The approval process varies from country to country, and the time may be longer or shorter than that required for FDA approval. In addition, under current United States law, there are restrictions on the export of products not approved by the FDA, depending on the country involved and the status of the product in that country.
International sales of medical devices manufactured in the U.S. that are not approved by the FDA for use in the U.S., or are banned or deviate from lawful performance standards, are subject to FDA export requirements. Exported devices are subject to the regulatory requirements of each country to which the device is exported. Some countries do not have medical device regulations, but in most foreign countries, medical devices are regulated. Frequently, regulatory approval may first be obtained in a foreign country prior to application in the U.S. to take advantage of differing regulatory requirements. Most countries outside of the U.S. require that product approvals be recertified on a regular basis, generally every five years. The recertification process requires that we evaluate any device changes and any new regulations or standards relevant to the device and conduct appropriate testing to document continued compliance. Where recertification applications are required, they must be approved in order to continue selling our products in those countries.
In the European Union, in order for our product candidates to be marketed and sold, we are required to comply with the Medical Devices Directive and obtain CE Mark certification. The CE Mark certification encompasses an extensive review of our quality management system which is inspected by a notified body’s auditor as part of a Stage 1 and 2 International Organization for Standardization, or ISO, 13485:2003 audit, in accordance with worldwide recognized ISO standards and applicable European Medical Devices Directives for quality management systems for medical device manufacturers. Once the quality management system and design dossier has been successfully audited by a notified body and reviewed and approved by a competent authority, a CE certificate for the medical device will be issued. We are also required to comply with other foreign regulations such as the requirement that we obtain Ministry of Health, Labor and Welfare approval before we can launch new products in Japan. The time required to obtain these foreign approvals to market our products may vary from U.S. approvals, and requirements for these approvals may differ from those required by the FDA.
Medical device laws and regulations are in effect in many of the countries in which we may do business outside the United States. These laws and regulations range from comprehensive device approval requirements for our medical device product to requests for product data or certifications. The number and scope of these requirements can be complex and could increase. We may not be able to obtain or maintain regulatory approvals in such countries and we may be required to incur significant costs in obtaining or maintaining our foreign regulatory approvals. In addition, the export of certain of our products which have not yet been cleared for domestic commercial distribution may be subject to FDA export restrictions. Any failure to obtain product approvals in a timely fashion or to comply with state or foreign medical device laws and regulations may have a serious adverse effect on our business, financial condition or results of operations.
Intellectual Property
On January 30, 2008, we entered into a License and Assignment Agreement, or the NDP License Agreement, with ND Partners, LLC, or NDP. Pursuant to the NDP License Agreement, NDP granted us exclusive, worldwide licenses for certain antimicrobial catheter lock solutions, processes for treating and inhibiting infections, a biocidal lock system and a taurolidine delivery apparatus, and the corresponding United States and foreign patents and applications (the “NDP Technology”). We acquired such licenses and patents through our assignment and assumption of NDP’s rights under certain separate license agreements by and between NDP and Dr. Hans-Dietrich Polaschegg, Dr. Klaus Sodemann, and Dr. Johannes Reinmueller. NDP also granted us exclusive licenses, with the right to grant sublicenses, to use and display certain trademarks in connection with the NDP Technology. As consideration in part for the rights to the NDP Technology, we paid NDP an initial licensing fee of $325,000 and granted NDP an equity interest in our Company consisting of 73,107 shares of common stock as of December 31, 2010. In addition, we are required to make payments to NDP upon the achievement of certain regulatory and sales-based milestones. Certain of the milestone payments are to be made in the form of shares of common stock currently held in escrow for NDP, and other milestone payments are to be paid in cash. The maximum aggregate number of shares issuable upon achievement of milestones and the number of shares held in escrow is 29,109 shares of common stock. The maximum aggregate amount of cash payments upon achievement of milestones is $3,000,000 with $2,500,000 remaining at December 31, 2020. Events that trigger milestone payments include but are not limited to the reaching of various stages of regulatory approval processes and certain worldwide net sales amounts.
During the year ended December 31, 2013, a milestone payment of $500,000 was earned by NDP upon the first issuance of the CE Mark for Neutrolin. Under Article 6 of the NDP License Agreement, we were obligated to make a milestone payment of $500,000 to NDP upon the first issuance of a CE Mark for a licensed product, which payment was payable to NDP within 30 days after such issuance. On April 11, 2013, we entered into an amendment to the NDP License Agreement which extended the milestone payment from within 30 days after such issuance to within twelve months after the achievement of such issuance. As consideration for the amendment, we issued NDP a five-year warrant to purchase 25,000 shares of our common stock at an exercise price of $7.50 per share. The warrant was exercisable immediately upon issuance and expired in April 2018. In January 2014, the $500,000 milestone payment due to NDP was converted into 10,000 Series C-3 non-voting preferred stock and a warrant to purchase 50,000 shares of our common stock at an exercise price of $4.50 per share. The warrants expired during the year ended December 31, 2020.
During the year ended December 31, 2014, a certain milestone was achieved resulting in the release of 7,277 shares held in escrow. The number of shares held in escrow as of December 31, 2021 is 21,832 shares of common stock. There were no milestones achieved in 2021 or 2020.
The NDP License Agreement will expire on a country-by-country basis upon the earlier of (i) the expiration of the last patent claim under the NDP License Agreement in a given country, or (ii) the payment of all milestone payments and release of all shares of our common stock held in escrow under the NDP License Agreement. Upon the expiration of the NDP License Agreement in each country, we will have an irrevocable, perpetual, fully paid-up, royalty-free exclusive license to the NDP Technology in such country. The NDP License Agreement also may be terminated by NDP if we materially breach or default under the NDP License Agreement and that breach is not cured within 60 days following the delivery of written notice to us, or by us on a country-by-country basis upon 60 days prior written notice. If the NDP License Agreement is terminated by either party, our rights to the NDP Technology will revert back to NDP.
We believe that the patents and patent applications we have licensed pursuant to the NDP License Agreement cover effective solutions to the various medical problems discussed previously when using taurolidine in clinical applications, and specifically in hemodialysis applications. Our patent portfolio consists of 5 issued U.S. patents and 11 pending U.S. patent applications; 17 issued foreign patents and 51 pending foreign patent applications. Additional patent applications will be filed to cover any additional related subject matter developed. The patents cover additional applications using taurolidine in, among others, sutures, hydrogels, meshes, transdermal and biofilm products.
Employees and Human Capital Resources
As of March 15, 2022, we employed 29 full-time employees and one part-time employee, who work out of our corporate offices in Berkeley Heights NJ or work remotely in various locations throughout the United States and Europe. We are committed to diversity, equity and inclusion, regardless of gender or race/ethnicity, or any protected status, and conduct training to reflect our commitment as an organization and build awareness.
We invest in our workforce by offering competitive salaries and benefits. We endeavor to foster a strong sense of ownership by offering stock options under our stock incentive program. We also offer comprehensive and locally relevant benefits for all eligible employees. We recognize and support the growth and development of our employees and we provide performance feedback and conduct employee goal and development discussions.
We have implemented COVID-19 policies designed to ensure the safety and well-being of all employees and the people associated with them. As a result of the COVID-19 pandemic, to reduce risk, our employees have been asked to work remotely, and all employees have been asked to avoid all non-essential travel, adhere to recommended health and safety practices.
None of our employees are subject to a collective bargaining agreement. We emphasize organizational communication and consider our relationship with our employees to be strong.
Corporate Information
We were organized as a Delaware corporation on July 28, 2006 under the name “Picton Holding Company, Inc.” and we changed our corporate name to “CorMedix Inc.” on January 18, 2007. Our principal executive offices are located at 300 Connell Drive, Suite 4200, Berkeley Heights, New Jersey 07922. Our telephone number is (908) 517-9500.
On March 26, 2019, we effected a 1-for-5 reverse stock split of our issued and outstanding shares of common stock, par value $0.001, per share (“Common Stock”), by combining, reclassifying and changing each authorized and outstanding five shares of “old” common stock into one share of “new” common stock. No fractional shares were issued, and, in lieu thereof, where applicable, one whole share was issued. To reflect the reverse stock split, reclassification, combination and change, proportional adjustments were also made to the number of shares of our common stock issuable upon conversion of outstanding preferred shares and the convertible note payable, warrants and options and other equity awards. The reverse stock split did not affect the par value per share of our common stock (which remains at $0.001 per share) or the total number of shares of common stock that are authorized to be issued pursuant to our Amended and Restated Certificate of Incorporation, as amended, which remains at 160 million shares. All issued and outstanding share and per share amounts included in the accompanying consolidated financial statements and in this report have been adjusted to reflect the reverse stock split, reclassification, combination and change for all periods presented.
In April 2021, we received approximately $1.3 million, net of expenses, from the sale of most of our remaining unused New Jersey net operating losses (“NOL”) eligible for sale under the State of New Jersey’s Economic Development Authority’s New Jersey Technology Business Tax Certificate Transfer program (“NJEDA Program”). The NJEDA Program allowed us to sell approximately $1.3 million of our total $1.3 million in available NOL tax benefits for the state fiscal year 2019.
The NJEDA has approved our application to participate in the NJEDA Program for the state fiscal year 2021. The approval will allow us to sell approximately $0.6 million of the total $0.6 million in available tax benefits to an unrelated, profitable New Jersey corporation in return for approximately $0.6 million in cash. Closing is subject to NJEDA’s typical closing conditions, which are in process of completion.
In November 2020, we filed a registration statement, under which we could issue and sell up to an aggregate of $100.0 million of shares of our common stock, $0.001 par value per share. On November 27, 2020, we entered into an Amended and Restated At Market Issuance Sales Agreement (“Amended Sales Agreement”) with B. Riley and Needham & Company, LLC (“Needham”), together with B. Riley, acting as sales agents (“Sales Agent”). The Amended Sales Agreement relates to the sale of shares of up to $25.0 million of our common stock under our ATM program, of which we may issue and sell common stock from time to time through the Sales Agent, subject to limitations imposed by us and subject to Sales Agent’s acceptance, such as the number or dollar amount of shares registered under the registration statement to which the offering relates. Sales Agent is entitled to a commission of up to 3% of the gross proceeds from the sale of common stock sold under the ATM program. During the year ended December 31, 2020, we sold 832,676 shares of common stock under the Amended Sales Agreement at the weighted average price of $8.69 per share and realized net proceeds of approximately $7.0 million. At December 31, 2020, we had approximately $17.8 million available under the Amended Sales Agreement and $75.0 million available under our current shelf registration for the issuance of equity, debt or equity-linked securities unrelated to the Amended Sales Agreement. On February 5, 2021, we allocated to our ATM program an additional $25.0 million of the remaining $75.0 million available under our shelf registration statement. Giving effect to the additional $25.0 million, plus the $17.8 million available at December 31, 2020, we had a total of $42.8 million available under our ATM program, which were sold during January and February 2021, for an aggregate of 3,737,862 shares of our common stock and approximately $41.5 million in net proceeds.
On August 12, 2021, we entered into an At Market Issuance Sales Agreement with Truist Securities, Inc. and JMP Securities LLC, as sales agents, pursuant to which we may sell, from time to time, an aggregate of up to $50.0 million of our common stock through the sales agents under our ATM program, subject to limitations imposed by us and subject to the sales agent’s acceptance, such as the number or dollar amount of shares registered under the registration statement to which the offering relates. The sales agents are entitled to a commission of up to 3% of the gross proceeds from the sale of common stock sold under the ATM program. As of December 31, 2021, we have $50.0 million available under our ATM program relating to our shelf registration statement filed in November 2020 and we have $150.0 million available under our shelf registration statement filed on August 12, 2021 for the issuance of equity, debt or equity-linked securities.
We maintain a website at www.cormedix.com; however, the information on, or that can be accessed through, our website or certain information in our website is not part of this report. This report and all of our filings under the Exchange Act, including copies of annual reports on Form 10-K, quarterly reports on Form 10-Q, current reports on Form 8-K, and any amendments to those reports, are available free of charge through our website on the date we file those materials with, or furnish them to, the Securities and Exchange Commission (the “SEC”). Such filings are also available to the public on the internet at the SEC’s website at www.sec.gov.
Item 1A. Risk Factors
Summary Risk Factors
The following is a summary of material risks that could affect our business. This summary may not contain all of our material risks, and it is qualified in its entirety by the more detailed risk factors set forth below.
Risks Related to our Financial Position and Need for Additional Capital
| 1. | We have a history of operating losses, expect to incur additional operating losses in the future and may never be profitable. |
| 2. | Our cost of operations could increase significantly more than what we expect depending on the costs to complete our development program for DefenCath/Neutrolin. |
| 3. | We will need to finance our future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. Any additional funds that we obtain may not be on terms favorable to us or our stockholders and may require us to relinquish valuable rights. |
Risks Related to the Development and Commercialization of Our Product Candidates
| 1. | Defencath, our lead product candidate, has received Fast Track designation and Qualified Infectious Disease Product designation from FDA, but we cannot provide assurances that these designations will not be rescinded. |
| 2. | If the FDA requires a second clinical trial for DefenCath or imposes additional manufacturing requirements to approve the New Drug Application, the development of DefenCath will take longer and cost more to complete, and we will need significant additional funds to undertake a second trial, if required. |
| 3. | Our only product Neutrolin is only approved in Europe and is still in development in the United States. |
| 4. | Final approval by regulatory authorities of our product candidates for commercial use may be delayed, limited or prevented, any of which would adversely affect our ability to generate operating revenues. |
| 5. | Successful development and commercialization of our other products is uncertain. |
| 6. | If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could harm our business. |
| 7. | The successful commercialization of Neutrolin will depend on obtaining coverage and reimbursement for use of Neutrolin from third-party payors. |
| 8. | Physician and patients may not accept and use our products. |
| 9. | Changes in funding for the FDA and other government agencies or future government shutdowns or disruptions could cause delays in the submission and regulatory review of marketing applications, which could negatively impact our business or prospects. |
| 10. | The ongoing COVID-19 pandemic, or other pandemic, epidemic or outbreak of an infectious disease may materially and adversely impact our business, including our preclinical studies and clinical trials. |
| 11. | Clinical trials required for our product candidates may be expensive and time-consuming, and their outcome is uncertain. |
| 12. | If we fail to comply with international regulatory requirements, we could be subject to regulatory delays, fines or other penalties. |
| 13. | We do not have, and may never obtain, the regulatory approvals we need to market our product candidates outside of the European Union. |
| 14. | Even if approved, our products will be subject to extensive post-approval regulation. |
Risks Related to Our Business and Industry
| 1. | Competition and technological change may make our product candidates and technologies less attractive or obsolete. |
| 2. | Healthcare policy changes, including reimbursement policies for drugs and medical devices, may have an adverse effect on our business, financial condition and results of operations. |
| 3. | If we lose key management or scientific personnel, cannot recruit qualified employees, directors, officers, or other personnel or experience increases in compensation costs, our business may materially suffer. |
| 4. | If we are unable to hire additional qualified personnel, our ability to grow our business may be harmed. |
| 5. | We may not successfully manage our growth. |
| 6. | We face the risk of product liability claims and the amount of insurance coverage we hold now or in the future may not be adequate to cover all liabilities we might incur. |
| 7. | We may be exposed to liability claims associated with the use of hazardous materials and chemicals. |
| 8. | Negative U.S. and global economic conditions may pose challenges to our business strategy, which relies on funding from the financial markets or collaborators. |
Risks Related to Our Intellectual Property
| 1. | If we materially breach or default under any of our license agreements, the licensor party to such agreement will have the right to terminate the license agreement, which termination may materially harm our business. |
| 2. | If we and our licensors do not obtain protection for and successfully defend our respective intellectual property rights, competitors may be able to take advantage of our research and development efforts to develop competing products. |
| 3. | Ongoing and future intellectual property disputes could require us to spend time and money to address such disputes and could limit our intellectual property rights. |
| 4. | The decisions by the European and German patent offices may affect patent rights in other jurisdictions. |
| 5. | If we infringe the rights of third parties we could be prevented from selling products and forced to pay damages and defend against litigation. |
Risks Related to Dependence on Third Parties
| 1. | We currently have no internal marketing and sales organization and currently rely and intend to continue to rely on third parties to market, sell, and distribute Neutrolin outside of the U.S. We may seek a sales partner in the U.S. if DefenCath receives FDA approval or we may undertake marketing and sales of DefenCath in the U.S. on our own. If we are unable to enter into or maintain agreements with third parties to market and sell DefenCath or any other product after approval or are unable to find a sales partner or establish our own marketing and sales capabilities, we may not be able to generate significant or any product revenues. |
| 2. | If we or our collaborators are unable to manufacture our products in sufficient quantities or are unable to obtain regulatory approvals for a manufacturing facility, we may be unable to meet demand for our products and we may lose potential revenues. |
| 3. | Corporate and academic collaborators may take actions that delay, prevent, or undermine the success of our products. |
| 4. | Data provided by collaborators and others upon which we rely that has not been independently verified could turn out to be false, misleading, or incomplete. |
| 5. | We rely on third parties to conduct our clinical trials and pre-clinical studies. If those parties do not successfully carry out their contractual duties or meet expected deadlines, our product candidates may not advance in a timely manner or at all. |
| 6. | We will depend on third party suppliers and contract manufacturers for the manufacturing of our product candidates and have no direct control over the cost of manufacturing our product candidates. Increases in the cost of manufacturing our product candidates would increase our costs of conducting clinical trials and could adversely affect our future profitability. |
Risks Related to Our Common Stock
| 1. | We will need additional financing to fund our activities in the future, which likely will dilute our stockholders. |
| 2. | Our executive officers and directors may sell shares of their stock, and these sales could adversely affect our stock price. |
| 3. | Our common stock price has fluctuated considerably and is likely to remain volatile, in part due to the limited market for our common stock and you could lose all or a part of your investment. |
| 4. | A significant number of additional shares of our common stock may be issued at a later date, and their sale could depress the market price of our common stock. |
| 5. | Provisions in our corporate charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult. |
| 6. | If we fail to comply with the continued listing standards of the Nasdaq Global Market, it may result in a delisting of our common stock from the exchange. |
| 7. | Laws, rules and regulations relating to public companies may be costly and impact our ability to attract and retain directors and executive officers. |
| 8. | Our internal control over financial reporting and our disclosure controls and procedures may not prevent all possible errors that could occur. |
| 9. | Security breaches and other disruptions could compromise our information and expose us to liability, which would cause our business and reputation to suffer. |
| 10. | We do not intend to pay dividends on our common stock so any returns on our common stock will be limited to the value of our common stock. |
Risks Related to Our Financial Position and Need for Additional Capital
We have a history of operating losses, expect to incur additional operating losses in the future and may never be profitable.
Our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in the early stages of operation. We incurred net losses of approximately $28.2 million and $22.0 million for the years ended December 31, 2021 and 2020, respectively. As of December 31, 2021, we had an accumulated deficit of approximately $245.7 million. We expect to incur substantial additional operating expenses over the next several years as our research, development, pre-clinical testing, clinical trial and commercialization activities increase as we develop and commercialize DefenCath and our other product candidates. As a result, we expect to experience negative cash flow as we fund our operating losses and capital expenditures. The amount of future losses and when, if ever, we will achieve profitability are uncertain. Neutrolin was launched in December 2013 and is currently available for distribution in certain European Union countries. We have not generated any significant commercial revenue and do not expect to generate substantial revenues from DefenCath unless and until it is approved by the United States Food and Drug Administration (“FDA”) and launched in the United States (“U.S.”) market, and we might never generate significant revenues from the sale of DefenCath or any other products. Our ability to generate revenue and achieve profitability will depend on, among other things, the following: obtaining FDA approval of DefenCath for the prevention of catheter-related bloodstream infections (“CRBSIs”) in patients with kidney failure receiving hemodialysis through a central venous catheter; successfully launching and marketing DefenCath in the U.S., if approved by the FDA; successfully marketing Neutrolin in foreign countries in which it is approved for sale; obtaining necessary regulatory approvals for our other product candidates from the FDA and, if sought, international regulatory agencies; establishing manufacturing, sales, and marketing arrangements, either alone or with third parties; and raising sufficient funds to finance our activities. We might not succeed at any of these undertakings. If we are unsuccessful at some or all of these undertakings, our business, prospects, and results of operations may be materially adversely affected.
Our cost of operations could increase significantly more than what we expect depending on the costs to complete our development program for DefenCath.
Our operations are subject to a number of factors that can affect our operating results and financial condition. Such factors include, but are not limited to: the results of clinical testing and trial activities of our product candidates; the ability to obtain regulatory approval to market our products; ability to manufacture successfully; competition from products manufactured and sold or being developed by other companies; the price of, and demand for, our products; our ability to negotiate favorable licensing or other manufacturing and marketing agreements for our products; and our ability to raise capital to support our operations.
To date, our commercial operations have not generated sufficient revenues to enable profitability. As of December 31, 2021, we had an accumulated deficit of $245.7 million, and incurred net losses of $28.2 million for the year then ended. Based on the current development plans for DefenCath and Neutrolin in both the U.S. and foreign markets (including the concluded hemodialysis Phase 3 clinical trial in the U.S.) and our other operating requirements, management believes that the existing cash at December 31, 2021, after taking into consideration the costs for resubmission of the NDA and initial preparations for the commercial launch for DefenCath, will be sufficient to fund operations at least through the first half of 2023. We will need additional funding for the commercialization of DefenCath upon FDA approval and funding for the label expansion studies for DefenCath.
Our continued operations will ultimately depend on our ability to raise additional capital through various potential sources, such as equity and/or debt financings, strategic relationships, potential strategic transactions or out-licensing of our products in order to complete the development of DefenCath and until we achieve profitability, if ever. We can provide no assurances that such financing or strategic relationships will be available on acceptable terms, or at all. Without this funding, we could be required to delay, scale back or eliminate some or all of our research and development programs which would likely have a material adverse effect on our business.
We will need to finance our future cash needs through public or private equity offerings, debt financings or corporate collaboration and licensing arrangements. Any additional funds that we obtain may not be on terms favorable to us or our stockholders and may require us to relinquish valuable rights.
We have launched Neutrolin in certain European Union and Middle East countries, but to date have no other approved product on the market and have not generated significant product revenue from Neutrolin to date. Unless and until we receive applicable regulatory approval for DefenCath in the U.S., we cannot sell DefenCath in the U.S. Therefore, for the foreseeable future, we will have to fund all of our operations and capital expenditures from Neutrolin sales in Europe and other foreign markets, if approved, cash on hand, additional financings, licensing fees and grants.
We believe that our cash resources as of December 31, 2021, after taking into consideration the costs for resubmission of the NDA and initial preparations for the commercial launch for DefenCath, will be sufficient to fund operations at least through the first half of 2023. Nevertheless, we may need to raise additional funds through financings or strategic relationships if our costs exceed our expectations, as well as funds for our continued operations. We can provide no assurances that any financing or strategic relationships will be available to us on acceptable terms, or at all. We expect to continue to use significant cash to fund our operations as we seek FDA approval of DefenCath in the U.S., commercialize Neutrolin in Europe and other markets, pursue development of our medical devices and other business development activities, and incur additional legal costs to defend our intellectual property.
To raise needed capital, we may sell additional equity or debt securities, obtain a bank credit facility, or enter into a corporate collaboration or licensing arrangement. The sale of additional equity or debt securities, if convertible, could result in dilution to our stockholders. The incurrence of indebtedness would result in fixed obligations and could also result in covenants that would restrict our operations. Raising additional funds through collaboration or licensing arrangements with third parties may require us to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates, or to grant licenses on terms that may not be favorable to us or our stockholders.
Risks Related to the Development and Commercialization of Our Product Candidates
DefenCath, our lead product candidate, has received Fast Track designation and Qualified Infectious Disease Product designation from FDA, but we cannot provide assurances that these designations will not be rescinded.
DefenCath is being developed as a catheter lock solution for the reduction of CRBSIs in patients with kidney failure receiving hemodialysis through a central venous catheter. The FDA has determined that DefenCath will be regulated as a New Drug, because it contains the new chemical entity taurolidine as a novel antimicrobial agent. After we filed the Investigational New Drug Application (“IND”), FDA granted designations as Fast Track and a Qualified Infectious Disease Product (“QIDP”) in January 2015. Fast Track is designed to facilitate development of a drug that is intended to treat a serious or life-threatening condition and address an unmet medical need. Fast Track confers eligibility to request priority review of an NDA, with FDA’s decision regarding potential priority review to be made after receipt of a complete application. QIDP was established pursuant to the Generating Antibiotic Incentives Now (“GAIN”) Act and creates incentives for the development of antibacterial and antifungal drug products that treat serious or life-threatening infections. Subject to the specified statutory limitations, a drug that is designated as QIDP and is approved for the use for which the QIDP designation was granted will receive a 5-year extension to any exclusivity for which the application qualifies upon approval, such as the 5 year exclusivity for a new chemical entity. We cannot provide assurances that DefenCath will retain these designations and continue to receive the benefits conferred.
If the FDA requires a second clinical trial for DefenCath or imposes additional manufacturing requirements to approve the New Drug Application, the development of DefenCath will take longer and cost more to complete, and we will need significant additional funds to undertake a second trial, if required.
Although two pivotal clinical trials to demonstrate safety and effectiveness of DefenCath are generally required by the FDA to secure marketing approval in the U.S., FDA will in some cases accept one adequate and well-controlled trial, where it is a large multicenter trial with a broad range of subjects and investigation sites with procedures to include trial quality that has demonstrated a clinically meaningful and statistically very persuasive effect on prevention of a disease with potentially serious outcome. We discussed submission of the NDA with the FDA based on the data from LOCK-IT-100 and were granted our request for rolling submission and review of the NDA for DefenCath as a catheter lock solution for the prevention of CRBSIs in patients with end stage renal disease receiving hemodialysis through a central venous catheter. In August 2020, the FDA accepted the DefenCath NDA for filing and granted our request for priority review, with a PDUFA date of February 28, 2021. As we announced in March 2021, the FDA informed us in a Complete Response Letter that it will not approve the NDA in its present form, because of concerns at the third-party manufacturing facility and a requirement to conduct a manual extraction study to demonstrate that the labeled volume can be consistently withdrawn from the vials. The FDA did not request additional clinical data and did not identify any deficiencies related to the data submitted on the efficacy and safety of DefenCath from LOCK-IT-100. In draft labeling discussed with FDA, the FDA added that the initial approval will be for the limited population of patients with kidney failure receiving hemodialysis through a central venous catheter. This is consistent with our request for approval of the NDA pursuant to the Limited Population Pathway for Antibacterial and Antifungal Drugs (“LPAD”) pathway, which was passed as part of the 21st Century Cures Act. LPAD is intended to expedite the development and approval of certain antibacterial and antifungal drugs which meet three criteria: intended to treat serious or life-threatening infections; in limited populations of patients; and with unmet needs. The LPAD pathway provides for a streamlined clinical development program for a limited population that may involve smaller, shorter or fewer clinical trials. Labeling of an LPAD approved product will specify the use in the limited population. We have resubmitted the NDA after addressing the manufacturing issues, but until the NDA is approved, if FDA raises issues related to the clinical trial results, we may incur additional costs and delays in the trial, and may not be able to complete the clinical trial in a cost-effective or timely manner, which would have an adverse effect on our development program for DefenCath as a treatment for catheter-related bloodstream infections.
Our only product Neutrolin is only approved in Europe and is still in development in the United States.
Neutrolin currently and for at least the near future is our only current product as well as product candidate. Neutrolin has received CE Mark approval in Europe, and we started sales in Germany in December 2013. We also are pursuing development of DefenCath in the U.S. Our product commercialization and development efforts may not lead to commercially viable products for any of several reasons. For example, our product candidates may fail to be proven safe and effective in clinical trials, or we may have inadequate financial or other resources to pursue development efforts for our product candidates. Even if approved, our product may not be accepted in the marketplace. DefenCath will require approval by the FDA of an NDA and/or investment by us or our collaborators as we continue its commercialization, as will any other product candidates.
In April 2017, we entered into a commercial collaboration with Hemotech SAS covering France and certain overseas territories. We have an agreement with a South Korean company to market, sell and distribute Neutrolin in South Korea upon receipt of regulatory approval in that country, which requires approval by the U.S. FDA. We also have commercial sales in Germany and a distributor agreement for the United Arab Emirates, which is pursuant to the EU CE Mark. Consequently, we will be dependent on these companies and individuals for the success of sales in those countries and any other countries in which we receive regulatory approval and in which we contract with third parties for the marketing, sale and/or distribution of Neutrolin. If these companies or individuals do not perform for whatever reason, our business, prospects and results of operations will be adversely affected. Finding a suitable replacement organization or individual for these or any other companies or individuals with whom we might contract could be difficult, which would further harm our business, prospects and results of operations. The negotiation and consummation of collaboration agreements typically involve simultaneous discussions with multiple potential collaborators and require significant time and resources. In addition, in attracting the attention of pharmaceutical and biotechnology company collaborators, we compete with numerous other third parties with product opportunities as well as the collaborators’ own internal product opportunities. We may not be able to consummate collaborative agreements, or we may not be able to negotiate commercially acceptable terms for these agreements.
Final approval by regulatory authorities of our product candidates for commercial use may be delayed, limited or prevented, any of which would adversely affect our ability to generate operating revenues.
Our ability to generate operating revenue will be severely limited until we, a licensee, or a potential collaborator successfully commercializes DefenCath in the United States. We may experience unforeseen events during product development that may substantially delay or prevent product approval. For example, in the course of conducting a clinical trial, the FDA could order the temporary, or permanent, discontinuation at any time if it believes that the clinical trial either is not being conducted in accordance with FDA requirements or presents an unacceptable risk to the clinical trial patients. An Institutional Review Board (“IRB”) may also require the clinical trial at the site to be halted, either temporarily or permanently, for failure to comply with the IRB’s requirements or if the trial poses an unexpected serious harm to clinical trial patients. The FDA or an IRB may also impose conditions on the conduct of a clinical trial. Clinical trial sponsors may also choose to discontinue clinical trials as a result of risks to clinical trial patients, a lack of favorable results, or changing business priorities.
The clinical development, manufacturing, labeling, packaging, storage, recordkeeping, export, marketing, promotion and distribution, and other possible activities relating to our product candidates are subject to extensive regulation by the FDA and other regulatory agencies. Failure to comply with applicable regulatory requirements may, either before or after product approval, subject us to administrative or judicially imposed sanctions that may negatively impact the approval of one or more of our product candidates or otherwise negatively impact our business. Compliance with such regulations may consume substantial financial and management resources and expose us and our collaborators to the potential for other adverse circumstances. For example, a regulatory authority can place restrictions on the sale or marketing of a drug in order to manage the risks identified during initial clinical trials or after the drug is on the market. A regulatory authority can condition the approval for a drug on costly post-marketing follow-up studies. Based on these studies, if a regulatory authority does not believe that the drug demonstrates a clinical benefit to patients or an acceptable safety profile, it could limit the indications for which a drug may be sold or revoke the drug’s marketing approval. In addition, identification of certain side effects either during clinical trials or after a drug is on the market may result in reformulation of a drug, additional pre-clinical and clinical trials, labeling changes, termination of ongoing clinical trials or withdrawal of approval. Any of these events could delay or prevent us from generating revenue from the commercialization of these drugs and cause us to incur significant additional costs.
Neither collaborators, licensees nor we are permitted to market a product candidate in the United States until the particular product candidate is approved for marketing by the FDA. Specific pre-clinical data, chemistry, manufacturing and controls data, a proposed clinical trial protocol and other information must be submitted to the FDA as part of an IND application, and clinical trials may commence only after the IND application becomes effective. To market a new drug in the United States, we must submit to the FDA and obtain FDA approval of an NDA. An NDA must be supported by extensive clinical and pre-clinical data, as well as extensive information regarding chemistry, manufacturing and controls, to demonstrate the safety and effectiveness of the product candidate, and the FDA will also assess whether the manufacturing processes and facilities are suitable to support the application. Approval of an NDA may be delayed due to delays in FDA’s review of the manufacturing facility, which may require an onsite inspection.
Obtaining approval of an NDA can be a lengthy, expensive and uncertain process. Review time can be impacted by the quality of the information included in the application, FDA’s internal resources such as the availability of reviewers, or requests from the FDA for additional information. Regulatory approval of an NDA is not guaranteed. The number and types of pre-clinical studies and clinical trials that will be required for FDA approval varies depending on the product candidate, the disease or condition that the product candidate is designed to target and the regulations applicable to any particular product candidate. Despite the time and expense exerted in pre-clinical and clinical studies, failure can occur at any stage, and we could encounter problems that delay our product candidate development or that cause us to abandon clinical trials or to repeat or perform additional pre-clinical studies and clinical trials. The FDA can delay, limit or deny approval of a product candidate for many reasons, and product candidate development programs may be delayed or may not be successful for many reasons including but not limited to, the following:
| ● | The FDA or IRBs may not authorize us to commence, amend, or continue clinical studies; |
| | | |
| ● | we may not be able to enroll a sufficient number of qualified patients for clinical trials in a timely manner or at all, patients may drop out of our clinical trials or be lost to follow-up at a higher rate than we anticipate, patients may not follow the clinical trial procedures, or the number of patients required for clinical trials may be larger than we anticipate; |
| ● | the FDA may not accept an NDA or other submission due to, among other reasons, the content or formatting of the submission; |
| | | |
| ● | a product candidate may not be deemed adequately safe or effective for an intended use; |
| | | |
| ● | the FDA may not find the data from pre-clinical studies and clinical trials sufficient; |
| | | |
| ● | the FDA may require that we conduct additional pre-clinical or clinical studies, change our manufacturing process, or gather additional manufacturing information above what we currently have planned for; |
| | | |
| ● | the FDA’s interpretation and our interpretation of data from pre-clinical studies and clinical trials or chemistry, manufacturing and controls data may differ significantly; |
| | | |
| ● | the FDA may not agree with our intended indications, the design of our clinical or pre-clinical studies, or there may be a flaw in the design that does not become apparent until the studies are well advanced; |
| | | |
| ● | we may not be able to establish agreements with contractors or collaborators or they or we may fail to comply with applicable FDA and other regulatory requirements, including those identified in other risk factors; |
| | | |
| ● | the FDA may not accept aspects of our proposed labeling, or may impose specific limitations in the labeling and require post-marking commitments or Phase 4 clinical trials before the labeling can be expanded; |
| | | |
| ● | the FDA may determine that the manufacturing processes and facilities for our product candidate do not have sufficient good manufacturing practice (GMP) controls in place to support approval; or |
| | | |
| ● | the FDA may change its approval policies or adopt new regulations. |
Our pre-clinical and clinical data, other information and procedures relating to a product candidate may not be sufficient to support approval by the FDA or any other U.S. or foreign regulatory authority, or regulatory interpretation of these data and procedures may be unfavorable. Failure to conduct required post-approval studies, or confirm a clinical benefit, will allow the FDA to withdraw the drug from the market on an expedited basis. Our business and reputation may be harmed by any failure or significant delay in receiving regulatory approval for the sale of any drugs resulting from our product candidates. As a result, we cannot predict when or whether regulatory approval will be obtained for any drug we develop.
Additionally, other factors may serve to delay, limit or prevent the final approval by regulatory authorities of our product candidates for commercial use, including, but not limited to:
| ● | we or our licensees will need to conduct significant clinical testing and development work to demonstrate the quality, safety, and efficacy of these product candidates before applications for marketing can be filed with the FDA, or with the regulatory authorities of other countries; |
| | | |
| ● | development and testing of product formulation, including identification of suitable excipients, or chemical additives intended to facilitate delivery of our product candidates; |
| | ● | it may take us many years to complete the testing of our product candidates, and failure can occur at any stage of this process; |
| | ● | negative or inconclusive results or adverse medical events during a clinical trial could cause us to delay or terminate our development efforts; and |
| | | |
| | ● | inspection delay given the FDA’s current backlog of foreign inspections. |
The successful development of any of these product candidates is uncertain and, accordingly, we may never commercialize any of these product candidates or generate significant revenue.
Successful development and commercialization of our products is uncertain.
Our development and commercialization of current and future product candidates is subject to the risks of failure and delay inherent in the development of new pharmaceutical products, including but not limited to the following:
| ● | inability to produce positive data in pre-clinical and clinical trials; |
| | | |
| ● | delays in product development, pre-clinical and clinical testing, or manufacturing; |
| | | |
| ● | unplanned expenditures in product development, clinical testing, or manufacturing; |
| | | |
| ● | challenges with securing the heparin supply chain; |
| | | |
| ● | uncertainties relating to, or changes in FDA view of, the appropriate product approval pathway; |
| ● | failure to obtain treatment of a drug or application under expedited development and review programs or to obtain marketing exclusivities; |
| | | |
| ● | failure to receive or maintain regulatory approvals; |
| | | |
| ● | emergence of superior or equivalent products; |
| | | |
| ● | inability to manufacture our product candidates on a commercial scale on our own, or in collaboration with third parties; |
| | | |
| ● | failure to comply with a broad range of post-marketing requirements including those related to labeling, promotion and advertising, manufacturing and quality, pharmacovigilance and adverse event reporting, commercial distribution and supply chain requirements, and drug sample distribution requirements; and |
| | | |
| ● | failure to achieve market acceptance. |
Because of these risks, our development efforts may not result in any commercially viable products. If a significant portion of these development efforts are not successfully completed, required regulatory approvals are not obtained or any approved products are not commercialized successfully, our business, financial condition, and results of operations will be materially harmed.
If we fail to comply with environmental, health and safety laws and regulations, we could become subject to fines or penalties or incur costs that could harm our business.
From time to time and in the future, our operations may involve the use of hazardous and flammable materials, including chemicals and biological materials, and may also produce hazardous waste. Even if we contract with third parties for the disposal of these materials and waste, we cannot completely eliminate the risk of contamination or injury resulting from these materials. In the event of contamination or injury resulting from the use or disposal of our hazardous materials, we could be held liable for any resulting damages, and any liability could exceed our resources. We also could incur significant costs associated with civil or criminal fines and penalties for failure to comply with such laws and regulations.
In addition, we may incur substantial costs in order to comply with current or future environmental, health and safety laws and regulations. Current or future environmental laws and regulations may impair our research, development or production efforts. In addition, failure to comply with these laws and regulations may result in substantial fines, penalties or other sanctions.
The successful commercialization of DefenCath will depend on obtaining coverage and reimbursement for use of DefenCath from third-party payors.
Sales of pharmaceutical products largely depend on the reimbursement of patients’ medical expenses by government health care programs and/or private health insurers, both in the U.S. and abroad. Further, significant uncertainty exists as to the reimbursement status of newly approved health care products. We initially expect to sell DefenCath directly to hospitals and key dialysis center operators, but also plan to expand its usage into oncology and total parenteral nutrition patients requiring catheters. All of these potential customers are healthcare providers who depend upon reimbursement by government and commercial insurance payors for dialysis and other treatments. Depending on the treatment setting, we believe that DefenCath would be eligible for coverage under various reimbursement programs, such as the End Stage Renal Disease (“ESRD”) Prospective Payment System and ESRD Quality Incentive Program; however, coverage by any of these reimbursement programs is not assured, and even if coverage is granted, it could later be revoked or modified under future regulations. Further, the U.S. Centers for Medicare & Medicaid Services (“CMS”), which administers Medicare, and works with states to administer Medicaid, has adopted and will continue to adopt and/or amend rules governing reimbursement for specific treatments. We anticipate that CMS and private insurers will increasingly demand that manufacturers demonstrate the cost effectiveness of their products as part of the reimbursement review and approval process. Rising healthcare costs have also led many European and other foreign countries to adopt healthcare reform proposals and medical cost containment measures. Similar legislation could be introduced in the U.S. Any measures affecting the reimbursement programs of these governmental and private insurance payors, including any uncertainty in the medical community regarding their nature and effect on reimbursement programs, could have an adverse effect on purchasing decisions regarding DefenCath, as well as limit the prices we may charge for DefenCath. The failure to obtain or maintain reimbursement coverage for DefenCath or any other products could materially harm our operations.
In anticipation that the CMS and private payers will demand that we demonstrate the cost effectiveness of DefenCath as part of the reimbursement review and approval process, we will incorporate health economic evaluations into our clinical studies to support this review in the context of the prospective use of DefenCath in dialysis, oncology and total parenteral nutrition settings. However, our studies might not be sufficient to support coverage or reimbursement at levels that allow providers to use DefenCath.
Physicians and patients may not accept and use our products.
Even with the CE Mark approval of Neutrolin, and even if we receive FDA or other foreign regulatory approval for DefenCath/Neutrolin or other product candidates, physicians and patients may not accept and use our products. Acceptance and use of our products will depend upon a number of factors including the following:
| ● | perceptions by members of the health care community, including physicians, about the safety and effectiveness of our drug or device product; |
| | | |
| ● | prevalence of the disease to be treated; |
| | | |
| ● | prevalence and severity of any side effects; |
| | | |
| ● | cost-effectiveness of our product relative to competing products; |
| | | |
| ● | availability of coverage and reimbursement from government and other third-party payers; |
| | | |
| ● | timing of market introduction of our drugs and competitive drugs; |
| | | |
| ● | effectiveness of marketing and distribution efforts by us and our licensees and distributors, if any; |
| | | |
| ● | potential or perceived advantages or disadvantages over alternative treatments; |
| | | |
| ● | potential post-marketing commitments imposed by regulatory authorities, such as patient registries; |
| | | |
| ● | price of our future products, both in absolute terms and relative to alternative treatments; and |
| | | |
| ● | the effect of current and future healthcare laws and regulations on our product candidates. |
Because we expect sales of DefenCath to generate substantially all of our product revenues for the foreseeable future, the failure of DefenCath to find market acceptance would harm our business and would require us to seek additional financing.
Changes in funding for the FDA and other government agencies or future government shutdowns or disruptions could cause delays in the submission and regulatory review of marketing applications, which could negatively impact our business or prospects.
The ability of the FDA to review and approve new products can be affected by a variety of factors, including government budget and funding levels, ability to hire and retain key personnel and accept submission, applications, and the payment of user fees, and statutory, regulatory, and policy changes. In addition, government funding of other government agencies that fund research and development activities is subject to the political process, which is inherently fluid and unpredictable. The impact of global events, including terrorism, natural disasters and pandemics, including the ongoing COVID-19 pandemic or other health emergencies, may also cause disruptions in the normal functioning of the FDA or other government agencies.
Disruptions at the FDA and other agencies may also slow the time necessary for new drugs to be reviewed and/or approved by necessary government agencies, which would adversely affect our business. For example, over the last several years, including for 35 days beginning on December 22, 2018, the U.S. government has shut down several times and certain regulatory agencies, such as the FDA, had to furlough critical FDA employees and stop critical activities. In addition, in March 2020, the FDA announced the postponement of most foreign inspections due to the global impact of COVID-19, which has continued for more than a year. If a prolonged government shutdown or other disruption to the normal functioning of government agencies occurs, it could significantly impact the ability of the FDA to timely review and process our regulatory submissions, which could have a material adverse effect on our business or prospects. At this time, there is a backlog at FDA in conducting pre-approval inspections of manufacturing facilities, because of ongoing travel restrictions imposed by COVID-19. Such backlog has prevented the FDA from inspecting the facilities of our CMO for the manufacturing of DefenCath, which is located outside the United States. If FDA deems a pre-approval inspection to be necessary for approval of the DefenCath NDA, there will be a delay until FDA inspectors can resume travel.
The ongoing COVID-19 pandemic, or other pandemic, epidemic or outbreak of an infectious disease may materially and adversely impact our business, including our preclinical studies and clinical trials.
Global health concerns relating to the COVID-19 pandemic and related government actions to reduce the spread of the virus have had a significant impact, both direct and indirect, on businesses and commerce, as worker shortages have occurred; supply chains have been disrupted; facilities and production have been suspended; and demand for certain goods and services, such as medical services and supplies, has spiked, while demand for other goods and services, such as travel, has fallen. In response to the COVID-19 outbreak, governmental authorities implementing numerous measures to try to contain the virus, including travel bans and restrictions, quarantines, “shelter-in-place” orders, and business limitations and shutdowns across much of the United States, Europe and Asia, including in the locations of our offices, clinical trial sites, key vendors and partners. Such “shelter in place” orders were previously lifted, at least partially, in many locations. However, an increase in the spread of COVID-19 and variants, including the Delta and Omicron variants, which may affect the spread or severity of one or more successive waves of the virus, has led, and may continue to lead, to the re-imposition by many nations and the U.S. of quarantine requirements for travelers from other regions and may lead to the re-imposition of “shelter-in-place” or other similar orders. Although several vaccines for prevention or mitigation of the severity of the virus have been granted Emergency Use Authorization by the FDA and foreign regulatory authorities, the timely distribution, public acceptance and effectiveness thereof in reducing the pandemic remain uncertain. If an onsite inspection of the third-party manufacturing facility of our contract manufacturer is required for the satisfactory resolution of issues required for approval of the DefenCath NDA, the Company may encounter additional delays in obtaining FDA approval because the FDA is currently facing a backlog due to the COVID-19 pandemic. Our clinical development program timelines may be negatively affected by COVID-19, which could materially and adversely affect our business, financial condition and results of operations. Further, due to “shelter in place” orders and other public health guidance measures, we have implemented a work-from-home policy for all staff members excluding those necessary to maintain minimum basic operations. Our increased reliance on personnel working from home may negatively impact productivity, or disrupt, delay or otherwise adversely impact our business. Furthermore, a widespread return of in-person workplace or commercial activities could lead to a resurgence of the virus.
As a result of the COVID-19 outbreak, or similar pandemics, and related travel restrictions and “shelter in place” orders and other public health guidance measures, we have and may in the future experience disruptions that could materially and adversely impact our clinical trials, business, financial condition and results of operations. Potential disruptions include but are not limited to:
| ● | delays or difficulties at our third-party vendors on whom we are dependent for manufacturing activities; |
| | | |
| ● | delays or difficulties in enrolling patients in our clinical trials; |
| | | |
| ● | delays or difficulties in initiating or expanding clinical trials, including delays or difficulties with clinical site initiation and recruiting clinical site investigators and clinical site staff; |
| | | |
| ● | increased rates of patients withdrawing from our clinical trials following enrollment as a result of contracting COVID-19 or other health conditions or being forced to quarantine; |
| | | |
| ● | diversion of healthcare resources away from the conduct of clinical trials, including the diversion of hospitals serving as our clinical trial sites and hospital staff supporting the conduct of our clinical trials; |
| | | |
| ● | interruption of key clinical trial activities, such as clinical trial site data monitoring, due to limitations on travel imposed or recommended by federal or state governments, employers and others or interruption of clinical trial subject visits and study procedures, which may impact the integrity of subject data and clinical study endpoints; |
| | | |
| ● | interruption or delays in the operations of the FDA or other regulatory authorities, including a halt in on-site inspections, which may impact review and approval timelines for our NDA; |
| | | |
| ● | delays or disruptions in preclinical experiments and investigational new drug application-enabling studies due to restrictions of on-site staff and unforeseen circumstances at contract research organizations and vendors; |
| | | |
| ● | interruption of, or delays in receiving supplies of our product candidates from our contract manufacturing organizations due to staffing shortages, production slowdowns or stoppages and disruptions in delivery systems; |
| | | |
| ● | limitations on our ability to recruit and hire key personnel due to our inability to meet with candidates because of travel restrictions and “shelter in place” orders; |
| | | |
| ● | limitations on employee resources that would otherwise be focused on the conduct of our preclinical studies and clinical trials, including because of sickness of employees or their families or the desire of employees to avoid contact with large groups of people; and |
| | | |
| ● | interruption or delays to our sourced discovery and clinical activities. |
The COVID-19 pandemic continues to rapidly evolve. The extent to which the outbreak impacts our business, preclinical studies and clinical trials will depend on future developments, which are highly uncertain and cannot be predicted with confidence, such as the ultimate geographic spread of the disease, the severity of any new variations of the virus, the duration of the pandemic, travel restrictions and social distancing in the United States and other countries, business closures or business disruptions and the effectiveness of actions taken in the United States and other countries to contain and treat the disease. If we or any of the third parties with whom we engage were to experience shutdowns or other business disruptions, our ability to conduct our business in the manner and on the timelines presently planned could be materially and negatively impacted.
In addition, the trading prices for our common stock and other biopharmaceutical companies have been highly volatile as a result of the COVID-19 pandemic. As a result, we may face difficulties raising capital through sales of our common stock or such sales may be on unfavorable terms.
Clinical trials required for our product candidates may be expensive and time-consuming, and their outcome is uncertain.
In order to obtain FDA or foreign approval to market a new drug or device product, we must demonstrate proof of safety and effectiveness in humans. Foreign regulations and requirements are similar to those of the FDA. To meet FDA requirements, we must conduct “adequate and well-controlled” clinical trials. Conducting clinical trials is a lengthy, time-consuming, and expensive process. The length of time may vary substantially according to the type, complexity, novelty, and intended use of the product candidate, and often can be several years or more per trial. Delays associated with the DefenCath development program or the development plans for any other product candidates may cause us to incur additional operating expenses. The commencement and rate of completion of clinical trials may be delayed by many factors, including, for example:
| ● | inability to manufacture sufficient quantities of qualified materials under the FDA’s cGMP requirements for use in clinical trials; |
| | | |
| ● | slower than expected rates of patient recruitment; |
| | | |
| ● | failure to recruit a sufficient number of patients; |
| | | |
| ● | modification of clinical trial protocols; |
| | | |
| ● | changes in regulatory requirements for clinical trials; |
| | | |
| ● | lack of effectiveness during clinical trials; |
| | | |
| ● | emergence of unforeseen safety issues; |
| | | |
| ● | delays, suspension, or termination of clinical trials due to the IRB responsible for overseeing the study at a particular study site; and |
| | | |
| ● | government or regulatory delays or “clinical holds” requiring suspension or termination of the trials. |
Further, the results from early pre-clinical and clinical trials are not necessarily predictive of results to be obtained in later clinical trials. Accordingly, even if we obtain positive results from early pre-clinical or clinical trials, we may not achieve the same success in later clinical trials. Moreover, comparisons of results across different studies should be viewed with caution as such comparisons are limited by a number of factors, including differences in study designs and populations. Such comparisons also will not provide a sufficient basis for any comparative claims following product approval. Clinical results are frequently susceptible to varying interpretations that may delay, limit or prevent regulatory approvals or commercialization. Negative or inconclusive results or adverse medical events during a clinical trial could cause a clinical trial to be delayed, repeated or terminated, or a clinical program to be abandoned.
Our clinical trials may be conducted in patients with serious or life-threatening diseases for whom conventional treatments have been unsuccessful or for whom no conventional treatment exists, and in some cases, our product is expected to be used in combination with approved therapies that themselves have significant adverse event profiles. During the course of treatment, these patients could suffer adverse medical events or die for reasons that may or may not be related to our products. We cannot ensure that safety issues will not arise with respect to our products in clinical development.
Clinical trials may not demonstrate statistically significant safety and effectiveness to obtain the requisite regulatory approvals for product candidates. The failure of clinical trials to demonstrate safety and effectiveness for the desired indications could harm the development of our product candidates. Such a failure could cause us to abandon a product candidate and could delay development of other product candidates. Any delay in, or termination of, our clinical trials would delay the filing of any NDA or any Premarket Approval Application, or PMA, with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues. Any change in, or termination of, our clinical trials could materially harm our business, financial condition, and results of operations.
If we fail to comply with international regulatory requirements we could be subject to regulatory delays, fines or other penalties.
Regulatory requirements in foreign countries for international sales of medical devices often vary from country to country. The occurrence and related impact of the following factors would harm our business:
| ● | delays in receipt of, or failure to receive, foreign regulatory approvals or clearances; |
| | | |
| ● | the loss of previously obtained approvals or clearances; or |
| | | |
| ● | the failure to comply with existing or future regulatory requirements. |
The CE Mark is a mandatory conformity mark for products to be sold in the European Economic Area. Currently, 28 countries in Europe require products to bear CE Marking. To market in Europe, a product must first obtain the certifications necessary to affix the CE Mark. The CE Mark is an international symbol of adherence to the Medical Device Regulations, previously the Medical Device Directives, and the manufacturer’s declaration that the product complies with essential requirements. Compliance with these requirements is ascertained within a certified Quality Management System (QMS) pursuant to ISO 13485. In order to obtain and to maintain a CE Mark, a product must be in compliance with the applicable quality assurance provisions of the aforementioned ISO and obtain certification of its quality assurance systems by a recognized European Union notified body. We received CE Mark approval for Neutrolin on July 5, 2013. However, certain individual countries within the European Union require further approval by their national regulatory agencies. Additionally, implementation of the new European Union Medical Device Regulations may pose challenges in demonstrating continued conformity to the new medical device regulatory paradigm. Failure to receive or maintain these other requisite approvals could prohibit us from marketing and selling Neutrolin in the entire European Economic Area or elsewhere.
We do not have, and may never obtain, the regulatory approvals we need to market our product candidates outside of the European Union.
While we have received the CE Mark approval for Neutrolin in Europe, certain individual countries within the European Union require further approval by their national regulatory agencies. Failure to receive or maintain these other requisite approvals could prohibit us from marketing and selling Neutrolin in the entire European Economic Area. In addition, we will need regulatory approval to market and sell Neutrolin in foreign countries outside of Europe.
In the United States, we have not received the regulatory approvals required for the commercial sale of any of our product candidates. The NDA for DefenCath could not be approved by FDA in its present form and resolution of deficiencies at our third-party manufacturing facility is required. Additionally, the FDA is requiring a manual extraction study to demonstrate that the labeled volume can be consistently withdrawn from the vials despite an existing in-process control to demonstrate fill volume within specifications. We met with the FDA to discuss proposed resolutions to the deficiencies, but we may not be able to obtain regulatory approval for commercial distribution.
We also are pursuing development of taurolidine-based devices for several indications, including wound closure, surgical meshes, and wound management. The FDA regards taurolidine as a new chemical entity and therefore an unapproved new drug. Consequently, there is no appropriate predicate device currently marketed in the U.S. on which a 510(k) approval process for these devices could be based. As a result, we will be required to submit a premarket approval application for marketing authorization for these indications. In the event that the NDA for DefenCath is approved by the FDA, the regulatory pathway for these devices can be revisited with the FDA. Although there will presumably still be no appropriate predicate, de novo Class II designation can be proposed, based on a risk assessment and a reasonable assurance of safety and effectiveness.
It is possible that DefenCath will not receive any further approval or that any of our other product candidates will be approved for marketing. Failure to obtain regulatory approvals, or delays in obtaining regulatory approvals, would adversely affect the successful commercialization of DefenCath or any other drugs or products that we or our partners develop, impose additional costs on us or our collaborators, diminish any competitive advantages that we or our partners may attain, and/or adversely affect our cash flow, financial condition and results of operations.
Even if approved, our products will be subject to extensive post-approval regulation.
Once a product is approved, numerous post-approval requirements apply in the United States and abroad. These include, among other things, requirements related to pharmacovigilance and adverse event and other reporting, supply chain security requirements, suspect and illegitimate product investigations and notifications, limitations on product advertising and promotion and on the distribution of product samples, and ongoing adherence to cGMPs, as well as the need to submit appropriate new or supplemental applications and obtain FDA approval for certain changes to the approved product, product labeling, or manufacturing process. Establishing and maintaining systems and procedures for compliance with these requirements, and for training and monitoring personnel relative to their compliance, is expensive, time consuming, and an ongoing effort. Depending on the circumstances, failure to meet these post-approval requirements can result in criminal prosecution, fines, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product approvals, or refusal to allow us to enter into supply contracts, including government contracts. In addition, even if we comply with FDA, foreign and other requirements, new information regarding the safety or effectiveness of a product could lead the FDA or a foreign regulatory body to modify or withdraw product approval.
Risks Related to Our Business and Industry
Competition and technological change may make our product candidates and technologies less attractive or obsolete.
We compete with established pharmaceutical and medical device companies that are pursuing other forms of prevention or treatment for the same or similar indications we are pursuing and that have greater financial and other resources. Other companies may succeed in developing products earlier than we do, obtaining FDA or any other regulatory agency approval for products more rapidly, or developing products that are more effective than our product candidates. Research and development by others may render our technology or product candidates obsolete or noncompetitive, or result in processes, treatments or cures superior to any therapy we develop. We face competition from companies that internally develop competing technology or acquire competing technology from universities and other research institutions. As these companies develop their technologies, they may develop competitive positions that may prevent, make futile, or limit our product commercialization efforts, which would result in a decrease in the revenue we would be able to derive from the sale of any products.
There can be no assurance that DefenCath or any other product candidate will be accepted by the marketplace as readily as these or other competing treatments. Furthermore, if our competitors’ products are approved before ours, it could be more difficult for us to obtain approval from the FDA or any other regulatory agency. Even if our products are successfully developed and approved for use by all governing regulatory bodies, there can be no assurance that physicians and patients will accept any of our products as a treatment of choice.
Furthermore, the pharmaceutical and medical device industry is diverse, complex, and rapidly changing. By its nature, the business risks associated with the industry are numerous and significant. The effects of competition, intellectual property disputes, market acceptance, and FDA or other regulatory agency regulations preclude us from forecasting regulatory approval, product acceptance, revenues or income with certainty or even confidence.
Healthcare policy changes, including reimbursement policies for drugs and medical devices, may have an adverse effect on our business, financial condition and results of operations.
Our future revenues, profitability and access to capital will be affected by the continuing efforts of governmental and private third-party payors to manage, contain or reduce the costs of health care through various means, such as capping prices, limiting price increases, reducing reimbursement, and requiring rebates. Market acceptance and sales of DefenCath or any other product candidates that we develop will depend on reimbursement policies and may be affected by health care reform measures in the U.S. and abroad. Government authorities and other third-party payors, such as private health insurers, decide which drugs they will pay for and establish reimbursement levels. We cannot be sure that reimbursement will be available for DefenCath or any other product candidates that we develop. Also, we cannot be sure that the amount of reimbursement available, if any, will not reduce the demand for, or the price of, our products. If reimbursement is not available or is available only at limited levels, we may not be able to successfully commercialize DefenCath or any other product candidates that we develop.
In both the U.S. and certain foreign jurisdictions, there have been and we expect there will continue to be a number of legislative and regulatory changes to the health care system that could affect our ability to sell our approved products profitably. The U.S. government and other governments have shown significant interest in pursuing healthcare reform. In particular, the Medicare Modernization Act of 2003 revised the payment methodology for many products under the Medicare program in the United States. This has resulted in lower rates of reimbursement. In 2010, the Patient Protection and Affordable Care Act, as amended by the Health Care and Education Reconciliation Act (collectively, the “Affordable Care Act”), was enacted. The Affordable Care Act substantially changed the way healthcare is financed by both governmental and private insurers. Such government-adopted reform measures may adversely affect the pricing of healthcare products and services in the U.S. or internationally and the amount of reimbursement available from governmental agencies or other third-party payors.
In recent years, the U.S. Congress has sought to repeal and has significantly amended the Affordable Care Act. We expect that there will continue to be proposals by legislators at both the federal and state levels, regulators and third-party payors to keep healthcare costs down while expanding individual healthcare benefits. Certain of these changes could impose limitations on the prices we will be able to charge for any products that are approved or the amounts of reimbursement available for these products from governmental agencies or other third-party payors or may increase the tax requirements for life sciences companies such as ours. Any such legislation could have an adverse effect on our business, financial condition and results of operations.
There has been heightened governmental scrutiny over the manner in which manufacturers set prices for their marketed products, which have resulted in several recent Congressional inquiries and proposed and enacted bills by Congress and the states designed to, among other things, bring more transparency to product pricing, review the relationship between pricing and manufacturer patient programs, and reform government program reimbursement methodologies for products. In addition, the U.S. government, state legislatures, and foreign governments have shown significant interest in implementing cost containment programs, including price-controls, restrictions on reimbursement and requirements for substitution of generic products for branded prescription drugs to limit the growth of government paid health care costs. For example, the U.S. government has passed legislation requiring pharmaceutical manufacturers to provide rebates and discounts to certain entities and governmental payors to participate in federal healthcare programs. Further, Congress and the current administration have each indicated that it will continue to seek new legislative and/or administrative measures to control drug costs, and the current administration recently released a “Blueprint”, or plan, to reduce the cost of drugs. The current administration’s Blueprint contains certain measures that the U.S. Department of Health and Human Services is already working to implement. Individual states in the United States have also been increasingly passing legislation and implementing regulations designed to control pharmaceutical product pricing, including price or patient reimbursement constraints, discounts, restrictions on certain product access and marketing cost disclosure and transparency measures, and, in some cases, designed to encourage importation from other countries and bulk purchasing.
Health administration authorities in countries other than the U.S. may not provide reimbursement for Neutrolin or any of our other product candidates at rates sufficient for us to achieve profitability, or at all. Like the U.S., these countries could adopt health care reform proposals and could materially alter their government-sponsored health care programs by reducing reimbursement rates.
Any reduction in reimbursement rates under Medicare or private insurers or foreign health care programs could negatively affect the pricing of our products. If we are not able to charge a sufficient amount for our products, then our margins and our profitability will be adversely affected.
If we lose key management or scientific personnel, cannot recruit qualified employees, directors, officers, or other personnel or experience increases in compensation costs, our business may materially suffer.
We are highly dependent on the principal members of our management and scientific staff, specifically, Dr. Matthew David, our interim Chief Executive Officer, Executive Vice President and Chief Financial Officer, Phoebe Mounts, our Executive Vice President and General Counsel, Elizabeth Masson-Hurlburt, our Executive Vice President and Head of Clinical Operations and Thomas Nusbickel, our Chief Commercial Officer. Our future success will depend in part on our ability to identify, hire, and retain current and additional personnel. We experience intense competition for qualified personnel and may be unable to attract and retain the personnel necessary for the development of our business. Moreover, our work force is located in the New York metropolitan area, where competition for personnel with the scientific and technical skills that we seek is extremely high and is likely to remain high. Because of this competition, our compensation costs may increase significantly. In addition, we have only limited ability to prevent former employees from competing with us.
If we are unable to hire additional qualified personnel, our ability to grow our business may be harmed.
Over time, we expect to hire additional qualified personnel with expertise in government regulation, formulation and manufacturing, and sales and marketing, among others. We compete for qualified individuals with numerous pharmaceutical companies, universities and other research institutions. Competition for such individuals is intense, and we cannot be certain that our search for such personnel will be successful. Attracting and retaining such qualified personnel will be critical to our success.
We may not successfully manage our growth.
Our success will depend upon the expansion of our operations to commercialize DefenCath and the effective management of any growth, which could place a significant strain on our management and our administrative, operational and financial resources. To manage this growth, we may need to expand our facilities, augment our operational, financial and management systems and hire and train additional qualified personnel. If we are unable to manage our growth effectively, our business may be materially harmed.
We face the risk of product liability claims and the amount of insurance coverage we hold now or in the future may not be adequate to cover all liabilities we might incur.
Our business exposes us to the risk of product liability claims that are inherent in the development of drugs. If the use of one or more of our or our collaborators’ drugs or devices harms people, we may be subject to costly and damaging product liability claims brought against us by clinical trial participants, consumers, health care providers, pharmaceutical companies or others selling our products.
We currently carry product liability insurance. We cannot predict all of the possible harms or side effects that may result and, therefore, the amount of insurance coverage we hold may not be adequate to cover all liabilities we might incur. Our insurance covers bodily injury and property damage arising from our clinical trials, subject to industry-standard terms, conditions and exclusions. Our coverage also includes the sale of commercial products. We have expanded our insurance coverage to include the sale of commercial products due to the receipt of the CE Mark approval, but we may be unable to maintain such coverage or obtain commercially reasonable product liability insurance for any other products approved for marketing.
If we are unable to obtain insurance at an acceptable cost or otherwise protect against potential product liability claims, we may be exposed to significant liabilities, which may materially and adversely affect our business and financial position. If we are sued for any injury allegedly caused by our or our collaborators’ products and do not have sufficient insurance coverage, our liability could exceed our total assets and our ability to pay the liability. A successful product liability claim or series of claims brought against us would decrease our cash and could cause the value of our capital stock to decrease.
We may be exposed to liability claims associated with the use of hazardous materials and chemicals.
Our research, development and manufacturing activities and/or those of our third-party contractors may involve the controlled use of hazardous materials and chemicals. Although we believe that our safety procedures for using, storing, handling and disposing of these materials comply with federal, state and local, as well as foreign, laws and regulations, we cannot completely eliminate the risk of accidental injury or contamination from these materials. In the event of such an accident, we and the third-party could be held liable for any resulting damages and any liability could materially adversely affect our business, financial condition and results of operations. In addition, the federal, state and local, as well as foreign, laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous or radioactive materials and waste products may require us to incur substantial compliance costs that could materially adversely affect our business, financial condition and results of operations.
Negative U.S. and global economic conditions may pose challenges to our business strategy, which relies on funding from the financial markets or collaborators.
Negative conditions in the U.S. or global economy, including financial markets, may adversely affect our business and the business of current and prospective vendors, licensees and collaborators, and others with whom we do or may conduct business. The U.S. or global economy may experience disruptions as the result of international hostilities, natural disasters, pandemics, other international health emergencies, or weather-related or similar events (such as fires, hurricanes, earthquakes, floods, landslides and other natural conditions including the effects of climate change), political instability, labor strikes or turmoil, or terrorist attacks. In particular, countries around the world have experienced the spread of the COVID-19 pandemic, resulting in quarantines, supply chain disruptions, reduction in travel, increased demand for medical services and a general decline in economic activity and market confidence. Similar potential disruptions may occur in the future in any of the locations in which we or our collaborators do business. We continue to assess the potential impact on our counterparties and customers of such events, and what impact, if any, these events could have on our business.
The duration and severity of these conditions is uncertain. If negative economic conditions occur, we may be unable to secure funding on terms satisfactory to us to sustain our operations or to find suitable collaborators to advance our internal programs, even if we achieve positive results from our drug development programs.
Risks Related to Our Intellectual Property
If we materially breach or default under any of our license agreements, the licensor party to such agreement will have the right to terminate the license agreement, which termination may materially harm our business.
Our commercial success will depend in part on the maintenance of our license agreements. Each of our license agreements provides the licensor with a right to terminate the license agreement for our material breach or default under the agreement, including the failure to make any required milestone or other payments. Should the licensor under any of our license agreements exercise such a termination right, we would lose our right to the intellectual property under the respective license agreement, which loss may materially harm our business.
If we and our licensors do not obtain protection for and successfully defend our respective intellectual property rights, competitors may be able to take advantage of our research and development efforts to develop competing products.
Our commercial success will depend in part on obtaining further patent protection for our products, product candidates and other technologies and successfully defending any patents that we currently have or will obtain against third-party challenges. The patents which we currently believe are most material to our business are as follows:
| ● | U.S. Patent No. 8,541,393 (expiring November 2, 2024) (the “Prosl Patent”) - use of Neutrolin for preventing infection and maintenance of catheter patency in hemodialysis catheters; |
| | | |
| ● | U.S. Patent No. 9,339,036 (expiring November 2, 2024); |
| | | |
| ● | U.S. Patent No. 7,696,182 (expiring May 16, 2025); and |
| | | |
| ● | European Patent EP 1 814 562 B1 (expiring October 12, 2025) (the “Prosl European Patent”) - a low heparin catheter lock solution for maintaining and preventing infection in a hemodialysis catheter. The European Patent Office has found the Prosl European Patent to be invalid and has revoked it. An appeal to that decision is pending. |
We are currently seeking further patent protection for our compounds and methods of treating diseases. However, the patent process is subject to numerous risks and uncertainties, and there can be no assurance that we will be successful in protecting our products by obtaining and defending patents. These risks and uncertainties include the following:
| ● | patents that may be issued or licensed may be challenged, invalidated, or circumvented, or otherwise may not provide any competitive advantage; |
| | | |
| ● | our competitors, many of which have substantially greater resources than we have and many of which have made significant investments in competing technologies, may seek, or may already have obtained, patents that will limit, interfere with, or eliminate our ability to make, use, and sell our potential products either in the United States or in international markets; |
| ● | there may be significant pressure on the United States government and other international governmental bodies to limit the scope of patent protection both inside and outside the United States for treatments that prove successful as a matter of public policy regarding worldwide health concerns; and |
| | | |
| ● | countries other than the United States may have less restrictive patent laws than those upheld by United States courts, allowing foreign competitors the ability to exploit these laws to create, develop, and market competing products. |
In addition, the United States Patent and Trademark Office (“PTO”), and patent offices in other jurisdictions have often required that patent applications concerning pharmaceutical and/or biotechnology-related inventions be limited or narrowed substantially to cover only the specific innovations exemplified in the patent application, thereby limiting the scope of protection against competitive challenges. Thus, even if we or our licensors are able to obtain patents, the patents may be substantially narrower than anticipated. Additionally, the breadth of claims allowed in biotechnology and pharmaceutical patents or their enforceability cannot be predicted. We cannot be sure that, should any patents issue, we will be provided with adequate protection against potentially competitive products. Furthermore, we cannot be sure that should patents issue, they will be of commercial value to us, or that private parties, including competitors, will not successfully challenge our patents or circumvent our patent position in the U.S. or abroad.
The above-mentioned patents are exclusively licensed to us. To support our patent strategy, we have engaged in a review of patentability and certain freedom to operate issues, including performing certain searches. However, patentability and certain freedom to operate issues are inherently complex, and we cannot provide assurances that a relevant patent office and/or relevant court will agree with our conclusions regarding patentability issues or with our conclusions regarding freedom to operate issues, which can involve subtle issues of claim interpretation and/or claim liability. Furthermore, we may not be aware of all patents, published applications or published literature that may affect our business either by blocking our ability to commercialize our product candidates, preventing the patentability of our product candidates to us or our licensors, or covering the same or similar technologies that may invalidate our patents, limit the scope of our future patent claims or adversely affect our ability to market our product candidates. Additionally, it is also possible that prior art of which we are aware, but which we do not believe affects the validity or enforceability of a claim, may, nonetheless, ultimately be found by a court of law or an administration panel to affect the validity or enforceability of a claim. If a third party were to prevail on a legal assertion of invalidity and/or unenforceability, we would lose at least part, and perhaps all, of the patent protection on our product candidates. Such loss of patent protection could have a material adverse impact on our business. Additionally, since patent applications in the United States are maintained in secrecy until published or issued and as publication of discoveries in the scientific or patent literature often lag behind the actual discoveries, we cannot be certain that we were the first to make the inventions covered by the pending patent applications or issued patents referred to above or that we were the first to file patent applications for such inventions.
In addition to patents, we also rely on trade secrets and proprietary know-how. Although we take measures to protect this information by entering into confidentiality and inventions agreements with our employees, and some but not all of our scientific advisors, consultants, and collaborators, we cannot provide any assurances that these agreements will not be breached, that we will be able to protect ourselves from the harmful effects of disclosure or dispute ownership if they are breached, or that our trade secrets will not otherwise become known or be independently discovered by competitors. We may also be unsuccessful in executing such an agreement with each party who in fact develops intellectual property that we regard as our own, which may result in claims by or against us related to the ownership of such intellectual property. If any of these events occurs, or we otherwise lose protection for our trade secrets or proprietary know-how, the value of our intellectual property may be greatly reduced. Even if we are successful in prosecuting or defending against such claims, litigation could result in substantial costs and be a distraction to our senior management and scientific personnel.
Ongoing and future intellectual property disputes could require us to spend time and money to address such disputes and could limit our intellectual property rights.
The biotechnology and pharmaceutical industries have been characterized by extensive litigation regarding patents and other intellectual property rights, and companies have employed intellectual property litigation to gain a competitive advantage. We may initiate or become subject to infringement claims or litigation arising out of patents and pending applications of our competitors, or we may become subject to proceedings initiated by our competitors or other third parties or the PTO or applicable foreign bodies to reexamine the patentability of our licensed or owned patents. In addition, litigation may be necessary to enforce our issued patents, to protect our trade secrets and know-how, or to determine the enforceability, scope, and validity of the proprietary rights of others. If we are required to defend patent infringement actions brought by third parties, or if we sue to protect our own patent rights, we may be required to pay substantial litigation costs and managerial attention may be diverted from business operations even if the outcome is not adverse to us. In addition, any legal action that seeks damages or an injunction to stop us from carrying on our commercial activities relating to the affected technologies could subject us to monetary liability and require us or any third party licensors to obtain a license to continue to use the affected technologies. We cannot predict whether we would prevail in any of these types of actions or that any required license would be made available on commercially acceptable terms or at all. Furthermore, to the extent that we or our consultants or research collaborators use intellectual property owned by others in work performed for us, disputes may also arise as to the rights in such intellectual property or in resulting know-how and inventions. An adverse claim could subject us to significant liabilities to such other parties and/or require disputed rights to be licensed from such other parties.
We initiated court proceedings in Germany for patent infringement and unfair use of our proprietary information related to Neutrolin (as described below). We also have had opposition proceedings brought against the European Patent and the German utility model patent which are the basis of our infringement proceedings (as described below). The defense and prosecution of these ongoing and any future intellectual property suits, PTO or foreign proceedings, and related legal and administrative proceedings are costly and time-consuming to pursue, and their outcome is uncertain. An adverse determination in litigation or PTO or foreign proceedings to which we may become a party could subject us to significant liabilities, including damages, require us to obtain licenses from third parties, restrict or prevent us from selling our products in certain markets, or invalidate or render unenforceable our licensed or owned patents. Although patent and intellectual property disputes might be settled through licensing or similar arrangements, the costs associated with such arrangements may be substantial and could include our paying large fixed payments and ongoing royalties. Furthermore, the necessary licenses may not be available on satisfactory terms or at all.
On September 9, 2014, we filed in the District Court of Mannheim, Germany a patent infringement action against TauroPharm GmbH and Tauro-Implant GmbH as well as their respective CEOs, referred to as the Defendants claiming infringement of our European Patent EP 1 814 562 B1, which was granted by the EPO on January 8, 2014, or the Prosl European Patent. The Prosl European Patent covers a low dose heparin catheter lock solution for maintaining patency and preventing infection in a hemodialysis catheter. In this action, we claim that the Defendants infringe on the Prosl European Patent by manufacturing and distributing catheter locking solutions to the extent they are covered by the claims of the Prosl European Patent. We believe that our patent is sound and are seeking injunctive relief and raising claims for information, rendering of accounts, calling back, destruction and damages. Separately, TauroPharm has filed an opposition with the EPO against the Prosl European Patent alleging that it lacks novelty and inventive step. We cannot predict the ultimate outcome of either of these related matters. At present, the EPO has revoked the Prosl European Patent as invalid, and we have filed an appeal, which is currently pending.
In the same complaint against the same Defendants, we also alleged an infringement (requesting the same remedies) of NDP’s utility model DE 20 2005 022 124 U1, referred to as the Utility Model, which we believe is fundamentally identical to the Prosl European Patent in its main aspects and claims. The Court separated the two proceedings and the Prosl European Patent and the Utility Model claims were tried separately. TauroPharm has filed a cancellation action against the Utility Model before the German Patent and Trademark Office, or German PTO based on the similar arguments as those in the opposition against the Prosl European Patent.
The Court issued its decisions on May 8, 2015, staying both proceedings. In its decisions, the Court found that the commercialization by TauroPharm in Germany of its TauroLock catheter lock solutions Hep100 and Hep500 infringes both the Prosl European Patent and the Utility Model and further that there is no prior use right that would allow TauroPharm to continue to make, use or sell its product in Germany. However, the Court declined to issue an injunction in favor of us that would preclude the continued commercialization by TauroPharm based upon its finding that there is a sufficient likelihood that the EPO, in the case of the Prosl European Patent, or the German PTO, in the case of the Utility Model, may find that such patent or utility model is invalid. Specifically, the Court noted the possible publication of certain instructions for product use that may be deemed to constitute prior art. As such, the District Court determined that it will defer any consideration of the request by us for injunctive and other relief until such time as the EPO or the German PTO made a final decision on the underlying validity of the Prosl European Patent and the Utility Model.
The opposition proceeding against the Prosl European Patent before the EPO is ongoing. Oral proceedings before the Opposition Division at the EPO were held on November 25, 2015, at which the three-judge patent examiner panel considered arguments related to the validity of the Prosl European Patent. The hearing was adjourned due to the fact that the panel was of the view that Claus Herdeis, one of the managing directors of TauroPharm, had to be heard as a witness in a further hearing in order to close some gaps in the documentation presented by TauroPharm as regards the publication of prior art.
The German PTO held a hearing in the validity proceedings relating to the Utility Model on June 29, 2016, at which the panel affirmed its preliminary finding that the Utility Model was invalid based upon prior publication of a reference to the benefits that may be associated with adding heparin to a taurolidine based solution. We filed an appeal against the ruling on September 7, 2016. An oral hearing was held on September 17, 2019 in which the German Federal Patent Court affirmed the first instance decision that the Utility Model was invalid. The decision has only a declaratory effect, as the Utility Model had expired in November 2015. On April 28, 2020, we filed a withdrawal of the complaint on the German utility model, thereby waiving our claims on these proceedings.
On November 22, 2017, the EPO in Munich, Germany held a further oral hearing in this matter. At the hearing, the panel held that the Prosl European Patent would be invalidated because it did not meet the requirements of novelty based on a technical aspect of the European intellectual property law. We disagree with this decision and have appealed the decision. We continue to believe that the Prosl European Patent is indeed novel and that its validity should be maintained. There can be no assurance that we will prevail in this matter.
On January 16, 2015, we filed a complaint against TauroPharm GmbH and its managing directors in the District Court of Cologne, Germany. In the complaint, we allege violation of the German Unfair Competition Act by TauroPharm and that TauroPharm is improperly and unfairly using our proprietary information relating to the composition and manufacture of Neutrolin, in the manufacture and sale of TauroPharm’s products TauroLockTM, TauroLock-HEP100 and TauroLock-HEP500. We sought a cease and desist order against TauroPharm from continuing to manufacture and sell any product containing taurolidine (the API of Neutrolin) and citric acid in addition to possible other components, damages for any sales in the past and the removal of all such products from the market. Hearings in this matter were held in the District Court of Cologne, Germany on November 19, 2015, on November 15, 2016 and on November 20, 2018. A decision was rendered by the Court on December 11, 2018, dismissing the complaint in its entirety. We appealed in January 2019. An oral hearing was held on September 6, 2019. In view of new arguments brought forward in this hearing, the Court issued an evidentiary order on September 27, 2019 ordering an expert opinion. The expert opinion was not in our favor. In a supplementary expert opinion submitted after we had brought forward arguments against the first expert opinion, the expert confirmed his view. In an oral hearing held on June 18, 2021, the Court only heard from the expert, and the Court as well as both parties asked further questions to the expert around his expert opinion. At the end of the hearing and internal deliberation among the panel of judges, the Court indicated that it would dismiss our complaint if we did not withdraw the appeal. As there were no advantages to further pursuing the matter in view of the Court’s statements, we withdrew the appeal and the proceedings are therefore now closed. TauroPharm requested an increase of the value in dispute determined by the Court in order to receive a higher reimbursement of costs (as this is based on the value in dispute under German law) but the request was rejected in view of arguments brought forward against it by our legal counsel. We will have to reimburse costs in the amount of approximately $41,000 plus interest to TauroPharm.
The decisions by the European and German patent offices may affect patent rights in other jurisdictions.
The prior art on the basis of which the Prosl European Patent and the German Utility Model have been
found to be invalid may be used to challenge the validity of issued United States and/or other foreign patents that are directed to the same or similar subject matter, in a court action or in an administrative proceeding before the USPTO. Pending United States and/or foreign patent applications may be denied on that basis of that prior art as well. Such patents and patent applications include: US 7,696,182; US 8,541,393; US 9,339,036; US 17/176,718; and EP 14150248.4.
If we infringe the rights of third parties we could be prevented from selling products and forced to pay damages and defend against litigation.
If our products, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we may have to do one or more of the following:
| ● | obtain licenses, which may not be available on commercially reasonable terms, if at all; |
| | | |
| ● | abandon an infringing product candidate; |
| | | |
| ● | redesign our products or processes to avoid infringement; |
| | | |
| ● | stop using the subject matter claimed in the patents held by others; |
| | | |
| ● | defend litigation or administrative proceedings, which may be costly whether we win or lose, and which could result in a substantial diversion of our financial and management resources. |
Risks Related to Dependence on Third Parties
We currently have no internal marketing and sales organization and currently rely and intend to continue to rely on third parties to market, sell, and distribute Neutrolin outside of the U.S. We may seek a sales partner in the U.S. if DefenCath receives FDA approval or we may undertake marketing and sales of DefenCath in the U.S. on our own. If we are unable to enter into or maintain agreements with third parties to market and sell DefenCath or any other product after approval or are unable to find a sales partner or establish our own marketing and sales capabilities, we may not be able to generate significant or any product revenues.
We currently have no sales, marketing, or distribution infrastructure in the EU and have only started to build necessary functions in the U.S. Our business strategy for Neutrolin relies on collaborating with larger firms with experience in marketing and selling medical devices and pharmaceutical products; for other products we may also rely on such marketing collaborations or out-licensing of our product candidates. Specifically, for Neutrolin, we have a distributor agreement with each of an Emirati, and a South Korean company for sales and marketing (upon receipt of approval to market in the U.S., which is required for approval to market in South Korea). We have a commercial collaboration with Hemotech SAS covering France and certain overseas territories. Assuming we receive applicable regulatory approval for other markets, we plan to enter into distribution agreements with one or more third parties for the sale of Neutrolin in various European, Middle East and other markets. We will be dependent on the firms and individuals with whom we contract for the success of sales in the countries in which they operate. However, there can be no assurance that we will be able to successfully maintain those relationships or establish and maintain additional marketing, sales, or distribution relationships, nor can there be assurance that such relationships will be successful, or that we will be successful in gaining market acceptance for our products. If these firms or individuals do not perform for whatever reason, our business, prospects and results of operations may be materially adversely affected. Finding a new or replacement organization for sales and marketing could be difficult, which would further harm our business, prospects and results of operations. To the extent that we enter into any marketing, sales, or distribution arrangements with third parties, our product revenues will be lower than if we marketed and sold our products directly, and any revenues we receive will depend upon the efforts of such third parties.
If we are unable to establish and maintain such third-party sales and marketing relationships, or choose not to do so, we will have to establish our own in-house capabilities. To market any of our products directly, we would need to develop a marketing, sales, and distribution force that has both technical expertise and the ability to support a distribution capability. The establishment of a marketing, sales, and distribution capability would take time and significantly increase our costs, possibly requiring substantial additional capital. In addition, there is intense competition for proficient sales and marketing personnel, and we may not be able to attract individuals who have the qualifications necessary to market, sell, and distribute our products. There can be no assurance that we will be able to establish internal marketing, sales, or distribution capabilities. If we are unable to, or choose not to establish these capabilities, or if the capabilities we establish are not sufficient to meet our needs, we will be required to establish collaborative marketing, sales, or distribution relationships with third parties, which we might not be able to do on acceptable terms or at all. The failure to successfully develop our own marketing and sales infrastructure would have a negative adverse effect on our business and results of operations.
If we or our collaborators are unable to manufacture our products in sufficient quantities or are unable to obtain regulatory approvals for a manufacturing facility, we may be unable to meet demand for our products and we may lose potential revenues.
Completion of our clinical trials and commercialization of DefenCath and any other product candidate require access to, or development of, facilities to manufacture sufficient supplies. All of our manufacturing processes currently are, and we expect them to continue to be, outsourced to third parties. Specifically, we will rely on one or more manufacturers to supply us and/or our distribution partners with commercial quantities of DefenCath. If, for any reason, we become unable to rely on our current sources for the manufacture of DefenCath or any other product candidates or for active pharmaceutical ingredient (“API”), either for clinical trials or for commercial quantities, then we would need to identify and contract with additional or replacement third-party manufacturers to manufacture compounds for pre-clinical, clinical, and commercial purposes. We may not be successful in identifying such additional or replacement third-party manufacturers, or in negotiating acceptable terms with any that we do identify. Such third-party manufacturers must receive FDA or applicable foreign approval before they can produce clinical material or commercial product, and any that are identified may not receive such approval or may fail to maintain such approval. We were recently informed by FDA that the DefenCath NDA cannot be approved in its present form, because of concerns at the third-party manufacturing facility, which must be resolved to FDA’s satisfaction before the NDA can be approved. In addition, we may be in competition with other companies for access to these manufacturers’ facilities and may be subject to delays in manufacturing if the manufacturers give other clients higher priority than they give to us. If we are unable to secure and maintain third-party manufacturing capacity, the development and sales of our products and our financial performance may be materially adversely affected.
Before we could begin to commercially manufacture DefenCath or any other product candidate on our own, we must obtain regulatory approval of the manufacturing facility and process. The manufacture of drugs for clinical and commercial purposes must comply with cGMP and applicable non-U.S. regulatory requirements. The cGMP requirements govern quality control and documentation policies and procedures. Complying with cGMP and non-U.S. regulatory requirements would require that we expend time, money, and effort in production, recordkeeping, and quality control to assure that the product meets applicable specifications and other requirements. We would also have to pass a pre-approval inspection prior to FDA or non-U.S. regulatory agency approval. Failure to pass a pre-approval inspection may significantly delay regulatory approval of our products. If we fail to comply with these requirements, we would be subject to possible regulatory action and may be limited in the jurisdictions in which we are permitted to sell our products. As a result, our business, financial condition, and results of operations could be materially adversely affected.
Corporate and academic collaborators may take actions that delay, prevent, or undermine the success of our products.
Our operating and financial strategy for the development, clinical testing, manufacture, and commercialization of our product candidates is heavily dependent on our entering into collaborations with corporations, academic institutions, licensors, licensees, and other parties. Our current strategy assumes that we will successfully establish and maintain these collaborations or similar relationships. However, there can be no assurance that we will be successful establishing or maintaining such collaborations. Some of our existing collaborations, such as our licensing agreements, are, and future collaborations may be, terminable at the sole discretion of the collaborator in certain circumstances. Replacement collaborators might not be available on attractive terms, or at all.
In addition, the activities of any collaborator will not be within our control and may not be within our power to influence. There can be no assurance that any collaborator will perform its obligations to our satisfaction or at all, that we will derive any revenue or profits from such collaborations, or that any collaborator will not compete with us. If any collaboration is not pursued, we may require substantially greater capital to undertake on our own the development and marketing of our product candidates and may not be able to develop and market such products successfully, if at all. In addition, a lack of development and marketing collaborations may lead to significant delays in introducing product candidates into certain markets and/or reduced sales of products in such markets.
Data provided by collaborators and others upon which we rely that has not been independently verified could turn out to be false, misleading, or incomplete.
We rely on third-party vendors, scientists, and collaborators to provide us with significant data and other information related to our projects, clinical trials, and business. If such third parties provide inaccurate, misleading, or incomplete data, our business, prospects, and results of operations could be materially adversely affected.
We rely on third parties to conduct our clinical trials and pre-clinical studies. If those parties do not successfully carry out their contractual duties or meet expected deadlines, our product candidates may not advance in a timely manner or at all.
In the course of our pre-clinical testing and clinical trials, we rely on third parties, including laboratories, investigators, clinical contract research organizations (“CROs”), and manufacturers, to perform critical services for us. For example, we rely on third parties to conduct our clinical trials and many of our pre-clinical studies, which are required to be conducted consistent with regulations on Good Laboratory Practice (“GLP”). CROs and study sites are responsible for many aspects of the trials, including finding and enrolling subjects for testing and administering the trials. Although we rely on these third parties to conduct our pre-clinical and clinical trials, we are responsible for ensuring that each of our trials is conducted in accordance with its investigational plan and protocol and that the integrity of the studies and resulting data is protected. Moreover, the FDA and foreign regulatory authorities require us to comply with regulations and standards, commonly referred to as Good Clinical Practices (“GCPs”), for conducting, monitoring, recording, and reporting the results of clinical trials to ensure that the data and results are scientifically credible and accurate, and that the trial subjects are adequately informed of the potential risks of participating in clinical trials. Our reliance on third parties does not relieve us of these responsibilities and requirements. These third parties may not be available when we need them or, if they are available, may not comply with all regulatory and contractual requirements or may not otherwise perform their services in a timely or acceptable manner, and we may need to enter into new arrangements with alternative third parties and our clinical trials may be extended, delayed or terminated. These independent third parties may also have relationships with other commercial entities, some of which may compete with us. In addition, if such third parties fail to perform their obligations in compliance with our protocols or the applicable regulatory requirements, our trials may not meet regulatory requirements or may need to be repeated, we may not receive marketing approvals, or we or such third parties may face regulatory enforcement. As a result of our dependence on third parties, we may face delays, failures or cost increases outside of our direct control. These risks also apply to the development activities of collaborators, and we do not control their research and development, clinical trial or regulatory activities.
We will depend on third party suppliers and contract manufacturers for the manufacturing of our product candidates and have no direct control over the cost of manufacturing our product candidates. Increases in the cost of manufacturing our product candidates would increase our costs of conducting clinical trials and could adversely affect our future profitability.
We do not intend to manufacture our product candidates ourselves, and we will rely on third parties for our drug supplies both for clinical trials and for commercial quantities in the future. We have taken the strategic decision not to manufacture API for our product candidates, as these can be more economically supplied by third parties with particular expertise in this area. We have identified contract facilities that are registered with the FDA, have a track record of large-scale API manufacture, and have already invested in capital and equipment. We have no direct control over the manufacturing of our product candidates, or the cost thereof. If the contract manufacturers are unable to produce sufficient quantities of our product candidates, as a result of a lack of available materials or otherwise, our ability to complete product candidate development and our future profitability would be adversely affected. If the cost of manufacturing increases, or if the cost of the materials used increases, these costs will be passed on to us, making the cost of conducting clinical trials more expensive. For example, there could be issues securing the API heparin for our product as a result of the outbreak of African swine fever in China in 2019, which threatened the global heparin supply. The United States is largely dependent on China for its heparin, because almost half of the global pig supply, the main animal source for heparin, is in China. Increases in manufacturing costs could adversely affect our future profitability if we are unable to pass all of the increased costs along to our customers.
Further, we, along with our contract manufacturers, are required to comply with FDA requirements for cGMPs, related to product testing, quality assurance, manufacturing and documentation. Our contract manufacturers may not be able to comply with the applicable FDA regulatory requirements, which could result in delays to our product development programs, could result in adverse regulatory actions against them or us, and could prevent us from ultimately receiving product marketing approval. They also generally must pass an FDA preapproval inspection for conformity with cGMPs before we can obtain approval to manufacture our product candidates and will be subject to ongoing, periodic, unannounced inspection by the FDA and corresponding state agencies to ensure strict compliance with cGMP, and other applicable government regulations and corresponding foreign standards. If we and our contract manufacturers fail to achieve and maintain high manufacturing standards in compliance with cGMP, we may experience manufacturing errors resulting in defective products that could be harmful to patients, product recalls or withdrawals, delays or interruptions of production or failures in product testing or delivery, delay or prevention of filing or approval of marketing applications for our products, cost overruns or other problems that could seriously harm our business. Not complying with FDA requirements could result in a product recall or prevent commercialization of our product candidates and delay our business development activities. In addition, such failure could be the basis for the FDA to issue a warning or untitled letter or take other regulatory or legal enforcement action, including recall or seizure, total or partial suspension of production, suspension of ongoing clinical trials, refusal to approve pending applications or supplemental applications, and potentially civil and/or criminal penalties depending on the matter.
Risks Related to our Common Stock
We will need additional financing to fund our activities in the future, which likely will dilute our stockholders.
To date, our commercial operations have not generated sufficient revenues to enable profitability. As of December 31, 2021, we had an accumulated deficit of $245.7 million, and incurred net losses of $28.2 million for the year then ended. Based on the current development plans for DefenCath/Neutrolin in both the U.S. and foreign markets (including the resubmission of an NDA for DefenCath in hemodialysis catheters) and our other operating requirements, management believes that the existing cash at December 31, 2021, will be sufficient to fund operations at least through the first half of 2023, after taking into consideration the costs for resubmission of the NDA and initial preparations for the commercial launch for DefenCath. Further, we will need additional funding for DefenCath’s commercial launch. We anticipate that we will incur operating losses for the foreseeable future. Additionally, we will require substantial funds in the future to support our operations. Accordingly, we will need to obtain additional financing, including through issuances of equity securities.
To the extent we raise additional capital by issuing equity securities, our stockholders may experience substantial dilution. We may, as we have in the past, sell common stock, convertible securities or other equity securities in one or more transactions at prices and in a manner we determine from time to time. If we sell common stock, convertible securities or other equity securities in more than one transaction, investors may be further diluted by subsequent sales. Such sales may also result in material dilution to our existing stockholders, and new investors could gain rights superior to existing stockholders.
Our executive officers and directors may sell shares of their stock, and these sales could adversely affect our stock price.
Sales of our common stock by our executive officers and directors, or the perception that such sales may occur, could adversely affect the market price of our common stock. Our executive officers and directors may sell stock in the future, either as part, or outside, of trading plans under Rule 10b5-1 under the Securities Exchange Act of 1934, as amended (the “Exchange Act”).
Our common stock price has fluctuated considerably and is likely to remain volatile, in part due to the limited market for our common stock and you could lose all or a part of your investment.
During the period from the completion of our initial public offering (“IPO”), on March 30, 2010 through December 31, 2021, the high and low sales prices for our common stock were $52.00 and $0.75, respectively. There is a limited public market for our common stock and we cannot provide assurances that an active trading market will develop or continue. As a result of low trading volume in our common stock, the purchase or sale of a relatively small number of shares could result in significant share price fluctuations.
Additionally, the market price of our common stock may continue to fluctuate significantly in response to a number of factors, some of which are beyond our control, including the following:
| ● | the receipt of or failure to obtain additional regulatory approvals for DefenCath, including FDA approval in the U.S.; |
| | | |
| ● | market acceptance of Neutrolin in those markets in which it is approved for sale; |
| | | |
| ● | our need for additional capital; |
| | | |
| ● | results of clinical trials of our product candidates, including any other Phase 3 trial for DefenCath in the U.S., if required, or those of our competitors; |
| | | |
| ● | our entry into or the loss of a significant collaboration, or expiration or termination of licenses; |
| | | |
| ● | regulatory or legal developments in the United States and other countries, including changes in the healthcare payment systems; |
| | | |
| ● | changes in financial estimates or investment recommendations by securities analysts relating to our common stock; |
| | | |
| ● | future sales or anticipated sales of our securities by us or our stockholders; |
| | | |
| ● | announcements by our competitors of significant developments, technological innovations, strategic partnerships, joint ventures or capital commitments; |
| | | |
| ● | changes in key personnel; |
| | | |
| ● | variations in our financial results or those of companies that are perceived to be similar to us; |
| | | |
| ● | actual or anticipated variations in operating results; |
| | | |
| ● | market conditions in the pharmaceutical and medical device sectors and issuance of new or changed securities analysts’ reports or recommendations; |
| | | |
| ● | instability in the stock market as a result of current or future domestic and global events; |
| | | |
| ● | liquidity of any market for our securities; |
| | | |
| ● | threatened or actual delisting of our common stock from a national stock exchange; |
| | | |
| ● | general economic, industry and market conditions; |
| | | |
| ● | developments or disputes concerning patents or other proprietary rights; and |
| | | |
| ● | any other factors described in this “Risk Factors” section. |
In addition, the stock markets in general, and the stock of pharmaceutical and medical device companies in particular, have experienced extreme price and volume fluctuations that have often been unrelated or disproportionate to the operating performance of these companies. In addition, changes in economic conditions in the U.S., the European Union or globally, particularly in the context of current global events, could impact upon our ability to grow profitably. Adverse economic changes are outside our control and may result in material adverse impacts on our business or our results of operations. Broad market and industry factors may negatively affect the market price of our common stock, regardless of our actual operating performance. In the past, following periods of volatility in the market price of a company’s securities, securities class-action litigation has often been instituted against that company. Such litigation, if instituted against us, could cause us to incur substantial costs and divert management’s attention and resources.
For these reasons and others, an investment in our securities is risky and you should invest only if you can withstand wide fluctuations in and a significant or complete loss of the value of your investment.
A significant number of additional shares of our common stock may be issued at a later date, and their sale could depress the market price of our common stock.
As of December 31, 2021, we had outstanding the following securities that are convertible into or exercisable for shares of our common stock:
| ● | options to purchase an aggregate of 1,034,984 shares of our common stock issued to our officers, directors and non-employee consultants under our 2013 Stock Plan, with a weighted average exercise price of $9.47 per share; |
| | | |
| ● | options to purchase an aggregate of 2,323,147 shares of our common stock issued to our officers, directors and non-employee consultants under our 2019 Stock Plan, with a weighted average exercise price of $6.67 per share; |
| | | |
| ● | 2,000 shares of Series C-3 Preferred Stock, which are convertible into 4,000 shares of common stock; |
| | | |
| ● | 89,623 shares of Series E Preferred Stock, which are convertible into 391,953 shares of common stock; |
| | | |
| ● | 89,999 shares of Series G Preferred Stock, which are convertible into 5,004,069 shares of common stock; |
| | | |
| | ● | warrants to purchase an aggregate of 56,455 shares of common stock with a weighted average exercise price of $5.25 per share; and |
| | | |
| | ● | 48,909,shares of common stock issuable for payment of deferred board compensation. |
Additionally, there are 1,547,725 shares of common stock available for grants under the 2019 Stock Plan (adopted on November 26, 2019).
The possibility of the issuance of these shares, as well as the actual sale of such shares, could substantially reduce the market price for our common stock and impede our ability to obtain future financing.
Provisions in our corporate charter documents and under Delaware law could make an acquisition of us, which may be beneficial to our stockholders, more difficult.
Provisions in our Amended and Restated Certificate of Incorporation, as amended, and our Amended and Restated Bylaws, as well as provisions of the General Corporation Law of the State of Delaware, or DGCL, may discourage, delay or prevent a merger, acquisition or other change in control of our Company, even if such a change in control would be beneficial to our stockholders. These provisions include the following:
| ● | authorizing the issuance of “blank check” preferred stock, the terms of which may be established and shares of which may be issued without stockholder approval; |
| | | |
| ● | prohibiting our stockholders from fixing the number of our directors; and |
| | | |
| ● | establishing advance notice requirements for stockholder proposals that can be acted on at stockholder meetings and nominations to our Board of Directors. |
These provisions may frustrate or prevent any attempts by our stockholders to replace or remove our current management by making it more difficult for stockholders to replace members of our board of directors, which is responsible for appointing the members of our management. In addition, we are subject to Section 203 of the DGCL, which generally prohibits a Delaware corporation from engaging in any of a broad range of business combinations with an interested stockholder for a period of three years following the date on which the stockholder became an interested stockholder, unless such transactions are approved by the board of directors. This provision could have the effect of discouraging, delaying or preventing someone from acquiring us or merging with us, whether or not it is desired by, or beneficial to, our stockholders. Any provision of our Amended and Restated Certificate of Incorporation, as amended, or Amended and Restated Bylaws or Delaware law that has the effect of delaying or deterring a change in control could limit the opportunity for our stockholders to receive a premium for their shares of our common stock and could also affect the price that some investors are willing to pay for our common stock.
If we fail to comply with the continued listing standards of the Nasdaq Global Market, it may result in a delisting of our common stock from the exchange.
Our common stock is currently listed for trading on the Nasdaq Global Market under the symbol “CRMD”, and the continued listing of our common stock on the Nasdaq Global Market is subject to our compliance with a number of listing standards. If we fail to satisfy the continued listing requirements of The Nasdaq Capital Market such as the corporate governance requirements, the stockholder’s equity requirement or the minimum closing bid price requirement, The Nasdaq Capital Market may take steps to de-list our common stock. Such a de-listing or even notification of failure to comply with such requirements would likely have a negative effect on the price of our common stock and would impair your ability to sell or purchase our common stock when you wish to do so. In addition, the delisting of our common stock could materially adversely impact our ability to raise capital on acceptable terms or at all. Delisting from Nasdaq could also have other negative results, including the potential loss of confidence by our current or prospective third-party providers and collaboration partners, the loss of institutional investor interest, and fewer licensing and partnering. In the event of a de-listing, we would take actions to restore our compliance with The Nasdaq Capital Market’s listing requirements, but we can provide no assurance that any such action taken by us would allow our common stock to become listed again, stabilize the market price or improve the liquidity of our common stock.
If our common stock were no longer listed on the Nasdaq Global Market, investors might only be able to trade on one of the over-the-counter markets, including the OTC Bulletin Board ® or in the Pink Sheets ® (a quotation medium operated by Pink Sheets LLC). This would impair the liquidity of our common stock not only in the number of shares that could be bought and sold at a given price, which might be depressed by the relative illiquidity, but also through delays in the timing of transactions and reduction in media coverage.
Laws, rules and regulations relating to public companies may be costly and impact our ability to attract and retain directors and executive officers.
Laws and regulations affecting public companies, including rules adopted by the Securities and Exchange Commission (“SEC”) and by the Nasdaq Global Market, may result in increased costs to us. These laws, rules and regulations could make it more difficult or costly for us to obtain certain types of insurance, including director and officer liability insurance, and we may be forced to accept reduced policy limits and coverage or incur substantially higher costs to obtain the same or similar coverage. The impact of these events could also make it more difficult for us to attract and retain qualified persons to serve on our board of directors, on our board committees or as executive officers. We cannot estimate accurately the amount or timing of additional costs we may incur to respond to these laws, rules and regulations.
Our internal control over financial reporting and our disclosure controls and procedures may not prevent all possible errors that could occur.
Our management is responsible for establishing and maintaining adequate internal control over financial reporting to provide reasonable assurance regarding the reliability of our financial reporting and the preparation of financial statements for external purposes in accordance with accounting principles generally accepted in the United States of America (“U.S. GAAP”). Ensuring that we have adequate internal financial and accounting controls and procedures in place to produce accurate financial statements on a timely basis is a costly and time-consuming effort that needs to be re-evaluated frequently. Failure on our part to have effective internal financial and accounting controls would cause our financial reporting to be unreliable, could have a material adverse effect on our business, operating results, and financial condition, and could cause the trading price of our common stock to fall dramatically.
A control system, no matter how well designed and operated, can provide only reasonable, not absolute, assurance that the control system’s objectives will be satisfied. Internal control over financial reporting and disclosure controls and procedures are designed to give a reasonable assurance that they are effective to achieve their objectives. We cannot provide absolute assurance that all of our possible future control issues will be detected. These inherent limitations include the possibility that judgments in our decision making can be faulty, and that isolated breakdowns can occur because of simple human error or mistake. The design of our system of controls is based in part upon assumptions about the likelihood of future events, and there can be no assurance that any design will succeed absolutely in achieving our stated goals under all potential future or unforeseeable conditions. Because of the inherent limitations in a cost-effective control system, misstatements due to error could occur and not be detected. This and any future failures could cause investors to lose confidence in our reported financial information, which could have a negative impact on our financial condition and stock price.
In future periods, if the process required by Section 404 of the Sarbanes-Oxley Act reveals any material weaknesses or significant deficiencies, the correction of any such material weaknesses or significant deficiencies could require remedial measures which could be costly and time-consuming. In addition, in such a case, we may be unable to produce accurate financial statements on a timely basis. Any associated accounting restatement could create a significant strain on our internal resources and cause delays in our release of quarterly or annual financial results and the filing of related reports, increase our costs and cause management distraction. Any of the foregoing could cause investors to lose confidence in the reliability of our financial statements, which could cause the market price of our common stock to decline and make it more difficult for us to finance our operations and growth.
Security breaches and other disruptions could compromise our information and expose us to liability, which would cause our business and reputation to suffer.
In the ordinary course of our business, we collect and store sensitive data, including intellectual property, our proprietary business information and that of our suppliers, as well as personally identifiable information of clinical trial participants and employees. Similarly, our third-party providers possess certain of our sensitive protected health data. The secure maintenance of this information is critical to our operations and business strategy. Despite our security measures, our information technology and infrastructure may be vulnerable to attacks by hackers or breached due to employee error, malfeasance or other disruptions. Attacks of this nature are increasing in their frequency, levels of persistence, sophistication and intensity, and are being conducted by sophisticated and organized groups and individuals with a wide range of motives and expertise. Although we develop and maintain systems and controls designed to prevent these events from occurring, and we have a process to identify and mitigate threats, the development and maintenance of these systems, controls and processes is costly and requires ongoing monitoring and updating as technologies change and efforts to overcome security measures become more sophisticated, and such systems, controls and processes may not be successful in preventing a breach. Any such breach could compromise our networks and the information stored there could be accessed, publicly disclosed, lost or stolen. We could be required to expend significant amounts of money and other resources to repair or replace information systems or networks. In addition, our liability insurance may not be sufficient in type or amount to cover us against claims related to security breaches, cyberattacks and other related breaches.
The legislative and regulatory landscape for privacy and data protection continues to evolve, and there has been an increasing amount of focus on privacy and data protection issues with the potential to affect our business, including compliance with the Health Insurance Portability and Accountability Act of 1996 and recently enacted laws in a majority of states requiring security breach notification. The collection and use of personal health data of individuals in the European Union is also governed by strict data protection laws. In addition to existing laws, since May 25, 2018, the General Data Protection Regulation (“GDPR”) has imposed new obligations with respect to European Union data and substantial fines for breaches of the data protection rules. It will increase our responsibility and potential liability in relation to personal data that we process, and we will be required to put in place additional mechanisms ensuring compliance with the new European Union data protection rules. There is significant uncertainty related to the manner in which data protection authorities will seek to enforce compliance with GDPR. For example, it is not clear if the authorities will conduct random audits of companies doing business in the European Union, or if the authorities will wait for complaints to be filed by individuals who claim their rights have been violated. Enforcement uncertainty and the costs associated with ensuring GDPR compliance may be onerous and adversely affect our business, operating results, prospects and financial condition.
Additionally, California recently enacted legislation that has been dubbed the first “GDPR-like” law in the United States. Known as the California Consumer Privacy Act (“CCPA”), it creates new individual privacy rights for consumers (as that word is broadly defined in the law) and places increased privacy and security obligations on entities handling personal data of consumers or households. The CCPA, which went into effect on January 1, 2020, requires covered companies to provide new disclosures to California consumers, provide such consumers new ways to opt-out of certain sales of personal information, and allow for a new cause of action for data breaches. The CCPA may significantly impact our business activities and require substantial compliance costs that adversely affect business, operating results, prospects and financial condition.
Thus, any access, disclosure or other loss of information, including our data being breached at our partners or third-party providers, could result in legal claims or proceedings and liability under laws that protect the privacy of personal information, disrupt our operations and damage our reputation, which could adversely affect our business.
We do not intend to pay dividends on our common stock so any returns on our common stock will be limited to the value of our common stock.
We have never declared dividends on our common stock, and currently do not plan to declare dividends on shares of our common stock in the foreseeable future. Pursuant to the terms of our Series C-3, E and G Convertible Preferred Stock, we may not declare or pay any dividends or make any distributions on any of our shares or other equity securities as long as any of those preferred shares remain outstanding. We currently expect to retain future earnings, if any, for use in the operation and expansion of our business. The payment of cash dividends in the future, if any, will be at the discretion of our Board of Directors and will depend upon such factors as earnings levels, capital requirements, our overall financial condition and any other factors deemed relevant by our Board of Directors. Any return to holders of our common stock will be limited to the value of their common stock.
Item 1B. Unresolved Staff Comments
None.
Item 2. Properties
In March 2020, we entered into a seven-year operating lease agreement for an office space at 300 Connell Drive, Berkeley Heights, New Jersey 07922. The lease agreement, with a monthly average cost of approximately $17,000, commenced on September 16, 2020. Our sublease from our previous premises terminated on November 30, 2020.
Our subsidiary leases its offices in Fulda, Germany pursuant to a three-month lease agreement which commenced in June 2017, renewable every three months for a base monthly payment of €400.
We believe that our existing facilities are adequate to meet our current needs, and that suitable additional alternative spaces will be available in the future on commercially reasonable terms.
Item 3. Legal Proceedings
On October 13, 2021, the United States District Court for the District of New Jersey consolidated into In re CorMedix Inc. Securities Litigation, Case No. 2:21-cv014020-JXN-CLW, two putative class action lawsuits filed on or about July 22, 2021 and September 13, 2021, respectively, and appointed lead counsel and lead plaintiff, a purported stockholder. The lead plaintiff filed a consolidated amended class action complaint on December 14, 2021, alleging violations of Sections 10(b) and 20(a) of the Exchange Act, along with Rule 10b-5 promulgated thereunder, and Sections 11 and 15 of the Securities Act of 1933. The complaint names as defendants the Company, Khoso Baluch, Matthew David, Phoebe Mounts, John L. Armstrong, Robert Cook, Janet Dillione, Alan W. Dunton, Myron Kaplan, Steven Lefkowitz, Paulo F. Costa, and Greg Duncan, as well as two underwriters of the Company’s secondary stock offering, B. Riley Securities, Inc. and Needham & Company, LLC. The purported bases for these claims are alleged misstatements and omissions in connection with the NDA submitted to the FDA for DefenCath, and the subsequent notification by the FDA that the NDA could not be approved in its present form. The lead plaintiff purports to assert the Exchange Act claims on behalf of persons that purchased or otherwise acquired shares of our securities between October 16, 2019, and September 6, 2021, and purports to assert the Securities Act claims on behalf of persons that purchased shares of our securities pursuant or traceable to a secondary offering of stock that commenced on November 27, 2020. We intend to vigorously contest such claims and filed a motion to dismiss the current complaint in full, with prejudice, on February 21, 2022. As of this filing, the current schedule set by the Court requires us and the other defendants to refile their motion to dismiss on March 28, 2022, requires the lead plaintiff to file an opposition to our motion to dismiss on or before April 27, 2022 and requires that we file a reply on or before May 27, 2022.
On or about October 13, 2021, a purported shareholder, derivatively and on our behalf, filed a shareholder derivative complaint in the United States District Court for the District of New Jersey, in a case entitled Voter v. Baluch, et al., Case No. 2:21-cv-18493-JXN-LDW. The complaint names as defendants Khoso Baluch, Janet Dillione, Alan W. Dunton, Myron Kaplan, Steven Lefkowitz, Paulo F. Costa, Greg Duncan, Matthew David, and Phoebe Mounts, along with us as Nominal Defendant. The complaint alleges breaches of fiduciary duties, abuse of control, and waste of corporate assets against the defendants and a claim for contribution for purported violations of Sections 10(b) and 21D of the Exchange Act against certain defendants. We intend to vigorously contest such claims. On January 21, 2022, pursuant to a stipulation between the parties, the Court entered an order staying the case while the motion to dismiss the class action lawsuit described in the foregoing paragraph is pending. The stay may be terminated before the motion to dismiss is resolved according to certain circumstances described in the stipulation available on the Court’s public docket.
On September 9, 2014, we filed in the District Court of Mannheim, Germany a patent infringement action against TauroPharm GmbH and Tauro-Implant GmbH as well as their respective CEOs, referred to as the Defendants claiming infringement of our European Patent EP 1 814 562 B1, which was granted by the EPO on January 8, 2014, or the Prosl European Patent. The Prosl European Patent covers a low dose heparin catheter lock solution for maintaining patency and preventing infection in a hemodialysis catheter. In this action, we claim that the Defendants infringe on the Prosl European Patent by manufacturing and distributing catheter locking solutions to the extent they are covered by the claims of the Prosl European Patent. We believe that our patent is sound and are seeking injunctive relief and raising claims for information, rendering of accounts, calling back, destruction and damages. Separately, TauroPharm has filed an opposition with the EPO against the Prosl European Patent alleging that it lacks novelty and inventive step. We cannot predict the ultimate outcome of either of these related matters. At present, the EPO has revoked the Prosl European Patent as invalid, and we have filed an appeal, which is currently pending.
In the same complaint against the same Defendants, we also alleged an infringement (requesting the same remedies) of NDP’s utility model DE 20 2005 022 124 U1, referred to as the Utility Model, which we believe is fundamentally identical to the Prosl European Patent in its main aspects and claims. The Court separated the two proceedings and the Prosl European Patent and the Utility Model claims were tried separately. TauroPharm has filed a cancellation action against the Utility Model before the German Patent and Trademark Office, or German PTO based on the similar arguments as those in the opposition against the Prosl European Patent.
The Court issued its decisions on May 8, 2015, staying both proceedings. In its decisions, the Court found that the commercialization by TauroPharm in Germany of its TauroLock catheter lock solutions Hep100 and Hep500 infringes both the Prosl European Patent and the Utility Model and further that there is no prior use right that would allow TauroPharm to continue to make, use or sell its product in Germany. However, the Court declined to issue an injunction in favor of us that would preclude the continued commercialization by TauroPharm based upon its finding that there is a sufficient likelihood that the EPO, in the case of the Prosl European Patent, or the German PTO, in the case of the Utility Model, may find that such patent or utility model is invalid. Specifically, the Court noted the possible publication of certain instructions for product use that may be deemed to constitute prior art. As such, the District Court determined that it will defer any consideration of the request by us for injunctive and other relief until such time as the EPO or the German PTO made a final decision on the underlying validity of the Prosl European Patent and the Utility Model.
The opposition proceeding against the Prosl European Patent before the EPO is ongoing. Oral proceedings before the Opposition Division at the EPO were held on November 25, 2015, at which the three-judge patent examiner panel considered arguments related to the validity of the Prosl European Patent. The hearing was adjourned due to the fact that the panel was of the view that Claus Herdeis, one of the managing directors of TauroPharm, had to be heard as a witness in a further hearing in order to close some gaps in the documentation presented by TauroPharm as regards the publication of prior art.
The German PTO held a hearing in the validity proceedings relating to the Utility Model on June 29, 2016, at which the panel affirmed its preliminary finding that the Utility Model was invalid based upon prior publication of a reference to the benefits that may be associated with adding heparin to a taurolidine based solution. We filed an appeal against the ruling on September 7, 2016. An oral hearing was held on September 17, 2019 in which the German Federal Patent Court affirmed the first instance decision that the Utility Model was invalid. The decision has only a declaratory effect, as the Utility Model had expired in November 2015. On April 28, 2020, we filed a withdrawal of the complaint on the German utility model, thereby waiving our claims on these proceedings.
On November 22, 2017, the EPO in Munich, Germany held a further oral hearing in this matter. At the hearing, the panel held that the Prosl European Patent would be invalidated because it did not meet the requirements of novelty based on a technical aspect of the European intellectual property law. We disagree with this decision and have appealed the decision. We continue to believe that the Prosl European Patent is indeed novel and that its validity should be maintained. There can be no assurance that we will prevail in this matter.
On January 16, 2015, we filed a complaint against TauroPharm GmbH and its managing directors in the District Court of Cologne, Germany. In the complaint, we allege violation of the German Unfair Competition Act by TauroPharm and that TauroPharm is improperly and unfairly using our proprietary information relating to the composition and manufacture of Neutrolin, in the manufacture and sale of TauroPharm’s products TauroLockTM, TauroLock-HEP100 and TauroLock-HEP500. We sought a cease and desist order against TauroPharm from continuing to manufacture and sell any product containing taurolidine (the API of Neutrolin) and citric acid in addition to possible other components, damages for any sales in the past and the removal of all such products from the market. Hearings in this matter were held in the District Court of Cologne, Germany on November 19, 2015, on November 15, 2016 and on November 20, 2018. A decision was rendered by the Court on December 11, 2018, dismissing the complaint in its entirety. We appealed in January 2019. An oral hearing was held on September 6, 2019. In view of new arguments brought forward in this hearing, the Court issued an evidentiary order on September 27, 2019 ordering an expert opinion. The expert opinion was not in our favor. In a supplementary expert opinion submitted after we had brought forward arguments against the first expert opinion, the expert confirmed his view. In an oral hearing held on June 18, 2021, the Court only heard from the expert, and the Court as well as both parties asked further questions to the expert around his expert opinion. At the end of the hearing and internal deliberation among the panel of judges, the Court indicated that it would dismiss our complaint if we did not withdraw the appeal. As there were no advantages to further pursuing the matter in view of the Court’s statements, we withdrew the appeal and the proceedings are therefore now closed. TauroPharm requested an increase of the value in dispute determined by the Court in order to receive a higher reimbursement of costs (as this is based on the value in dispute under German law) but the request was rejected in view of arguments brought forward against it by our legal counsel. We will have to reimburse costs in the amount of approximately $41,000 plus interest to TauroPharm.
Item 4. Mine Safety Disclosures
Not applicable.
PART II
Item 5. Market for the Registrant’s Common Equity, Related Stockholder Matters and Issuer Purchases of Equity Securities
Market for Common Equity
Our common stock trades on the Nasdaq Global Market under the symbol “CRMD.”
Based upon information furnished by our transfer agent, at March 25, 2022, we had approximately 272 holders of record of our common stock.
A comparison of the performance of our common stock is found in Item 12 of the report under the heading “Stock Performance Graph.”
Dividend Policy
We have never declared dividends on our equity securities, and currently do not plan to declare dividends on shares of our common stock in the foreseeable future. We expect to retain our future earnings, if any, for use in the operation and expansion of our business. Further, pursuant to the terms of our Series C-3, E and G Non-Voting Convertible Preferred Stock, we may not declare or pay any dividends or make any distributions on any of our shares or other equity securities as long as any of those preferred shares remain outstanding. Subject to the foregoing, the payment of cash dividends in the future, if any, will be at the discretion of our Board of Directors and will depend upon such factors as earnings levels, capital requirements, our overall financial condition and any other factors deemed relevant by our Board of Directors.
Equity Compensation Plan Information
The following table provides information as of December 31, 2021 about our common stock that may be issued upon the exercise of options, warrants and rights under all of our existing equity compensation plans (including individual arrangements):
| Plan Category | | Number of securities
to be issued upon
exercise of outstanding
options, warrants and
rights
(a) | | | Weighted-average
exercise price of
outstanding options,
warrants and rights
(b) | | | Number of
securities remaining
available for future issuance
under equity compensation
plans (excluding securities
reflected in column (a)
(c) | |
| Equity compensation plans approved by security holders (1) | | | 3,358,131 | (2) | | $ | 7.53 | (3) | | | 1,547,725 | |
| (1) | Our 2013 Stock Incentive Plan was approved by our stockholders on July 30, 2013. Our 2019 Omnibus Stock Incentive Plan was approved by our stockholders on November 26, 2019. |
| (2) | Consist of underlying stock options. |
| (3) | Applicable to shares underlying outstanding stock options only. |
Item 6. [RESERVED]
Not applicable.
Item 7. Management’s Discussion and Analysis of Financial Condition and Results of Operations
You should read the following discussion and analysis together with our audited consolidated financial statements and the accompanying notes contained elsewhere in this report. This discussion contains forward-looking statements, within the meaning of Section 27A of Securities Act, Section 21E of the Exchange Act, and the Private Securities Litigation Reform Act of 1995, including statements regarding our expected financial condition, business and financing plans. These statements involve risks and uncertainties. Our actual results could differ materially from the results described in or implied by these forward-looking statements as a result of various factors, including those discussed below and elsewhere in this report, particularly under the heading “Risk Factors.”
Overview
CorMedix Inc., together with our wholly owned subsidiaries, (collectively referred to herein as “we,” “us,” “our” and the “Company”), is a biopharmaceutical company focused on developing and commercializing therapeutic products for the prevention and treatment of infectious and inflammatory diseases. In May 2020, we formed a wholly-owned Spanish subsidiary, CorMedix Spain, S.L.U.
Our primary focus is on the development of our lead product candidate, DefenCath™, for potential commercialization in the United States, or U.S., and other key markets as a catheter lock solution, or CLS. We have in-licensed the worldwide rights to develop and commercialize DefenCath and Neutrolin®. The name DefenCath is the U.S. proprietary name conditionally approved by the U.S. Food and Drug Administration, or FDA, while the name Neutrolin® is currently used in the European Union, or EU, and other territories where we received CE-Mark approval for the commercial distribution of Neutrolin as a CLS regulated as a medical device. DefenCath/Neutrolin is a novel anti-infective solution (a formulation of taurolidine 13.5 mg/mL and heparin 1000 USP Units/mL) intended for the reduction and prevention of catheter-related infections and thrombosis in patients requiring central venous catheters in clinical settings such as hemodialysis, total parenteral nutrition, and oncology. Infection and thrombosis represent key complications among hemodialysis, total parenteral nutrition and oncology patients with central venous catheters. These complications can lead to treatment delays and increased costs to the healthcare system when they occur due to hospitalizations, need for intravenous, or IV antibiotic treatment, long-term anticoagulation therapy, removal/replacement of the central venous catheter, related treatment costs and increased mortality. We believe DefenCath addresses a significant unmet medical need and a potential large market opportunity.
In January 2015, the FDA designated DefenCath as a Qualified Infectious Disease Product, or QIDP, for prevention of catheter-related blood stream infections in patients with end stage renal disease receiving hemodialysis through a central venous catheter. Catheter-related blood stream infections and clotting can be life-threatening. The QIDP designation provides five years of market exclusivity in addition to the five years granted for a New Chemical Entity upon approval of a New Drug Application, or NDA. In addition, in January 2015, the FDA granted Fast Track designation to DefenCath Catheter Lock Solution, a designation intended to facilitate development and expedite review of drugs that treat serious and life-threatening conditions so that the approved drug can reach the market expeditiously. The Fast Track designation of DefenCath provides us with the opportunity to meet with the FDA on a more frequent basis during the development process, and also ensures eligibility to request priority review of the marketing application.
In December 2015, we launched our Phase 3 Prospective, Multicenter, Double-blind, Randomized, Active Control Study to Demonstrate Safety & Effectiveness of DefenCath/Neutrolin in Preventing Catheter-related Bloodstream Infection in Subjects on Hemodialysis for End Stage Renal Disease, or LOCK-IT-100, in patients with hemodialysis catheters in the U.S. The clinical trial was designed to demonstrate the safety and effectiveness of DefenCath compared to the standard of care CLS, Heparin, in preventing CRBSIs. The primary endpoint for the trial assessed the incidence of CRBSI and time to CRBSI for each study subject. Secondary endpoints were catheter patency, which was defined as required use of tPA, or removal of catheter due to dysfunction, and removal of catheter for any reason.
As previously agreed with the FDA, an interim efficacy analysis was performed when the first 28 potential CRBSI cases were identified in our LOCK-IT-100 study that occurred through early December 2017. Based on these first 28 cases, there was a highly statistically significant 72% reduction in CRBSI by DefenCath relative to the active control of heparin (p=0.0034). Because the pre-specified level of statistical significance was reached for the primary endpoint and efficacy had been demonstrated with no safety concerns, the LOCK-IT-100 study was terminated early. The study continued enrolling and treating subjects until study termination, and the final analysis was based on a total of 795 subjects. In a total of 41 cases, there was a 71% reduction in CRBSI by DefenCath relative to heparin, which was highly statistically significant (p=0.0006), with a good safety profile.
The FDA granted our request for a rolling submission and review of the NDA, which is designed to expedite the approval process for products being developed to address an unmet medical need. Although the FDA usually requires two pivotal clinical trials to provide substantial evidence of safety and effectiveness for approval of an NDA, the FDA will in some cases accept one adequate and well-controlled trial, where it is a large multicenter trial with a broad range of subjects and study sites that has demonstrated a clinically meaningful and statistically very persuasive effect on a disease with potentially serious outcome.
In March 2020, we began the modular submission process for the NDA for DefenCath for the prevention of CRBSI in hemodialysis patients, and in August 2020, the FDA accepted for filing the DefenCath NDA. The FDA also granted our request for priority review, which provides for a six-month review period instead of the standard ten-month review period. As we announced in March 2021, the FDA informed in its Complete Response Letter, or CRL, to us that it cannot approve the NDA for DefenCath in its present form. The FDA noted concerns at the third-party manufacturing facility after a review of records requested by the FDA and provided by the contract manufacturing organization, or CMO. Additionally, the FDA is requiring a manual extraction study to demonstrate that the labeled volume can be consistently withdrawn from the vials despite an existing in-process control to demonstrate fill volume within specifications.
In April 2021, we and the CMO met with the FDA to discuss proposed resolutions for the deficiencies identified in the CRL to us and the Post-Application Action Letter, or PAAL, received by the CMO from the FDA for the NDA for DefenCath. There was an agreed upon protocol for the manual extraction study identified in the CRL, which now has been successfully completed. Addressing the FDA’s concerns regarding the qualification of the filling operation necessitated adjustments in the process and generation of additional data on operating parameters for manufacture of DefenCath. We and the CMO determined that additional process qualification is needed with subsequent validation to address these issues. The FDA did not request additional clinical data and did not identify any deficiencies related to the data submitted on the efficacy or safety of DefenCath from LOCK-IT-100. In draft labeling discussed with the FDA, the FDA added that the initial approval will be for the limited population of patients with kidney failure receiving chronic hemodialysis through a central venous catheter. This is consistent with our request for approval pursuant to the Limited Population Pathway for Antibacterial and Antifungal Drugs, or LPAD. LPAD, passed as part of the 21st Century Cures Act, is a new program intended to expedite the development and approval of certain antibacterial and antifungal drugs to treat serious or life-threatening infections in limited populations of patients with unmet needs. LPAD provides for a streamlined clinical development program involving smaller, shorter, or fewer clinical trials and is intended to encourage the development of safe and effective products that address unmet medical needs of patients with serious bacterial and fungal infections. We believe that LPAD will provide additional flexibility for the FDA to approve DefenCath to prevent CRBSIs in the limited population of patients with kidney failure receiving hemodialysis through a central venous catheter.
On February 28, 2022, we announced that we resubmitted the NDA for DefenCath to address the CRL issued by the FDA. In parallel, our third-party manufacturer submitted responses to the deficiencies identified at the manufacturing facility in the PAAL issued by the FDA concurrently with the CRL. FDA will evaluate the submission to accept for filing and determine the review timeline. FDA has stated that it expected all corrections to facility deficiencies to be complete at the time of resubmission so that all corrective actions may be verified during an onsite evaluation of the manufacturing facility in the next review cycle, if the FDA determines it will do an onsite evaluation. If an onsite inspection is required, we may encounter delays in obtaining FDA approval because the FDA is currently facing a backlog due to the COVID-19 pandemic. The FDA issued a guidance document on its plan to use voluntary remote interactive evaluations at facilities, including for a pre-approval inspection to assess a marketing application. The FDA will request the manufacturing facility to participate in a voluntary remote interactive evaluation, if the FDA believes it is appropriate. A manufacturing facility cannot request the remote interaction. The FDA expects the use of remote interactive evaluations should help the FDA operate within normal timeframes in spite of the COVID-19 pandemic.
We intend to pursue additional indications for DefenCath use as a CLS in populations with an unmet medical need that also represent potentially significant market opportunities. While we are continuing to assess these areas, potential future indications may include use as a CLS to reduce CRBSIs in total parenteral nutrition patients using a central venous catheter and in oncology patients using a central venous catheter.
In addition to DefenCath, we are sponsoring a pre-clinical research collaboration for the use of taurolidine as a possible treatment for rare orphan pediatric tumors. In February 2018, the FDA granted orphan drug designation to taurolidine for the treatment of neuroblastoma in children. We may seek one or more strategic partners or other sources of capital to help us develop and commercialize taurolidine for the treatment of neuroblastoma in children. We are also evaluating opportunities for the possible expansion of taurolidine as a platform compound for use in certain medical devices. Patent applications have been filed in several indications, including wound closure, surgical meshes, and wound management. Based on initial feasibility work, we are advancing pre-clinical studies for taurolidine-infused surgical meshes, suture materials and hydrogels. We will seek to establish development/commercial partnerships as these programs advance.
We were granted a deferral by the FDA under the Pediatric Research Equity Act, or PREA, that requires sponsors to conduct pediatric studies for NDAs for a new active ingredient, such as taurolidine in DefenCath, unless a waiver or deferral is obtained from the FDA. A deferral acknowledges that a pediatric assessment is required but permits the applicant to submit the pediatric assessment after the submission of an NDA. We have made a commitment to conduct the pediatric study after approval of the NDA for use in adult hemodialysis patients. Pediatric studies for an approved product conducted under PREA may qualify for pediatric exclusivity, which if granted would provide an additional six months of marketing exclusivity. DefenCath would then have the potential to receive a total marketing exclusivity period of 10.5 years, including exclusivity pursuant to NCE and QIDP.
The FDA regards taurolidine as a new chemical entity and therefore an unapproved new drug. Consequently, there is no appropriate predicate medical device currently marketed in the U.S. on which a 510(k) approval process could be based. As a result, we will be required to submit a premarket approval application, or PMA, for marketing authorization for any medical device indications that we may pursue. In the event that an NDA for DefenCath is approved by the FDA, the regulatory pathway for these medical device product candidates may be revisited with the FDA. Although there may be no appropriate predicate, de novo Class II designation can be proposed, based on a risk assessment and a reasonable assurance of safety and effectiveness.
In the European Union, or EU, Neutrolin is regulated as a Class 3 medical device. In July 2013, we received CE Mark approval for Neutrolin. In December 2013, we commercially launched Neutrolin in Germany for the prevention of CRBSI, and maintenance of catheter patency in hemodialysis patients using a tunneled, cuffed central venous catheter for vascular access. To date, Neutrolin is registered and may be sold in certain European Union countries for such treatment.
In September 2014, the TUV-SUD and The Medicines Evaluation Board of the Netherlands, or MEB, granted a label expansion for Neutrolin to include use in oncology patients receiving chemotherapy, intravenous, or IV, hydration and IV medications via CVC for the EU. In December 2014, we received approval from the Hessian District President in Germany to expand the label for these same expanded indications. The expansion also adds patients receiving medication and IV fluids via CVC in intensive or critical care units (cardiac care unit, surgical care unit, neonatal critical care unit, and urgent care centers). An indication for use in total parenteral nutrition was also approved.
In September 2019, our registration with the Saudi Arabia Food and Drug Administration, or the SFDA, expired. As a result, we cannot sell Neutrolin in Saudi Arabia. We intend to complete the documentation required to renew our registration with the SFDA, however, we cannot predict how long the renewal process will take. There is no assurance that the registration will be renewed by the SFDA.
The novel coronavirus has been declared a pandemic and has spread to multiple global regions. The outbreak and government measures taken in response have also had a significant impact, both direct and indirect, on businesses and commerce, as worker shortages have occurred; supply chains have been disrupted; facilities and production have been suspended; and demand for certain goods and services, such as medical services and supplies, has spiked, while demand for other goods and services, such as travel, has fallen. In response to the COVID-19 outbreak, “shelter in place” orders and other public health guidance measures have been implemented across much of the United States, Europe and Asia, including in the locations of our offices, clinical trial sites, key vendors and partners. Such “shelter in place” orders were previously lifted, at least partially, in many locations. However, an increase in the spread of COVID-19 and variants, including the Delta variant, which may affect the spread or severity of one or more successive waves of the virus, has led, and may continue to lead, to the re-imposition by many nations and U.S. of quarantine requirements for travelers from other regions and may lead to the re-imposition of “shelter-in-place” or other similar orders. Our program timelines may be negatively affected by COVID-19, which could materially and adversely affect its business, financial conditions and results of operations.
Since our inception, our operations have been primarily limited to conducting clinical trials and establishing manufacturing for our product candidates, licensing product candidates, business and financial planning, research and development, seeking regulatory approval for our products, initial commercialization activities for DefenCath in the U.S. and Neutrolin in the EU and other foreign markets, and maintaining and improving our patent portfolio. We have funded our operations primarily through debt and equity financings. We have generated significant losses to date, and we expect to use substantial amounts of cash for our operations as we prepare our pre-launch commercial activities for DefenCath for the U.S. market and commercialize Neutrolin in the EU and other foreign markets, pursue business development activities, and incur additional legal costs to defend our intellectual property. As of December 31, 2021, we had an accumulated deficit of approximately $245.7 million. We are unable to predict the extent of any future losses or when we will become profitable, if ever.
Financial Operations Overview
Revenue
We have not generated substantial revenue since our inception. Through December 31, 2021, we have funded our operations primarily through debt and equity financings.
Research and Development Expense
Research and development, or R&D, expense consists of: (i) internal costs associated with our development activities; (ii) payments we make to third party contract research organizations, contract manufacturers, investigative sites, and consultants; (iii) technology and intellectual property license costs; (iv) manufacturing development costs; (v) personnel related expenses, including salaries, stock–based compensation expense, benefits, travel and related costs for the personnel involved in drug development; (vi) activities relating to regulatory filings and the advancement of our product candidates through pre-clinical studies and clinical trials; (vii) facilities and other allocated expenses, which include direct and allocated expenses for rent, facility maintenance, as well as laboratory and other supplies; and (viii) costs related to the manufacturing of the product that could potentially be available to support the commercial launch prior to marketing approval. All R&D is expensed as incurred.
Conducting a significant amount of development is central to our business model. Product candidates in later-stage clinical development generally have higher development costs than those in earlier stages of development, primarily due to the significantly increased size and duration of the clinical trials.
The process of conducting pre-clinical studies and clinical trials necessary to obtain regulatory approval is costly and time consuming. The probability of success for each product candidate and clinical trial may be affected by a variety of factors, including, among others, the quality of the product candidate’s early clinical data, investment in the program, competition, manufacturing capabilities and commercial viability. As a result of the uncertainties associated with clinical trial enrollments and the risks inherent in the development process, we are unable to determine the duration and completion costs of current or future clinical stages of our product candidates or when, or to what extent, we will generate revenues from the commercialization and sale of any of our product candidates.
Development timelines, probability of success and development costs vary widely. We are currently focused on securing the marketing approval for DefenCath in the U.S. as well as on continuing sales in foreign markets where Neutrolin is approved. In December 2015, we signed an agreement with a clinical research organization, or CRO, to help us conduct our LOCK-IT-100 Phase 3 clinical trial in hemodialysis patients with central venous catheters to demonstrate the efficacy and safety of DefenCath in preventing catheter-related bloodstream infections and blood clotting in subjects receiving hemodialysis therapy as treatment for end stage renal disease. Our LOCK-IT-100 study was completed and all costs related to the agreement with the CRO has been paid.
We are pursuing additional opportunities to generate value from taurolidine, an active component of DefenCath. Based on initial feasibility work, we have completed an initial round of pre-clinical studies for taurolidine-infused surgical meshes, suture materials, and hydrogels, which require a PMA regulatory pathway for approval. We are also involved in a pre-clinical research collaboration for the use of taurolidine as a possible treatment for rare orphan pediatric tumors. In February 2018, the FDA granted orphan drug designation to taurolidine for the treatment of neuroblastoma in children. We may seek one or more strategic partners or other sources of capital to help us develop and commercialize taurolidine for the treatment of neuroblastoma in children.
Selling, General and Administrative Expense
Selling, general and administrative, or SG&A, expense includes costs related to commercial personnel, medical education professionals, marketing and advertising, salaries and other related costs, including stock-based compensation expense, for persons serving in our executive, sales, finance and accounting functions. Other SG&A expense includes facility-related costs not included in R&D expense, promotional expenses, costs associated with industry and trade shows, and professional fees for legal services and accounting services.
Foreign Currency Exchange Transaction Gain (Loss)
Foreign currency exchange transaction gain (loss) is the result of re-measuring transactions denominated in a currency other than our functional currency and is reported in the consolidated statement of operations as a separate line item within other income (expense). The intercompany loans outstanding between our Company based in New Jersey and our subsidiary based in Germany are not expected to be repaid in the foreseeable future and the nature of the funding advanced is of a long-term investment nature. As such, unrealized foreign exchange movements related to long-term intercompany loans are recorded in other comprehensive income (loss).
Interest Income
Interest income consists of interest earned on our cash equivalents and short-term investments.
Interest Expense
Interest expense consists of interest incurred on our convertible debt, amortization of debt discount and on financing of expenditures.
Results of Operations
Comparison of the Years Ended December 31, 2021 and 2020
The following is a tabular presentation of our consolidated operating results for the years ended December 31, 2021 and 2020 (in thousands):
| | | 2021 | | | 2020 | | | % of Change Increase
(Decrease) | |
| Revenue | | $ | 191 | | | $ | 239 | | | | (20 | )% |
| Cost of sales | | | (149 | ) | | | (205 | ) | | | (27 | )% |
| Gross profit | | | 42 | | | | 34 | | | | 22 | % |
| Operating Expenses: | | | | | | | | | | | | |
| Research and development | | | (13,133 | ) | | | (13,377 | ) | | | (2 | )% |
| Selling, general and administrative | | | (16,346 | ) | | | (13,878 | ) | | | 18 | % |
| Total operating expenses | | | (29,479 | ) | | | (27,255 | ) | | | 8 | % |
| Loss from operations | | | (29,437 | ) | | | (27,221 | ) | | | 8 | % |
| Interest income | | | 14 | | | | 116 | | | | (88 | )% |
| Foreign exchange transaction loss | | | (21 | ) | | | (59 | ) | | | (64 | )% |
| Interest expense, including amortization of debt discount | | | (16 | ) | | | (33 | ) | | | (52 | )% |
| Total other (expense) income | | | (23 | ) | | | 24 | | | | (196 | )% |
| Loss before income taxes | | | (29,460 | ) | | | (27,197 | ) | | | 8 | % |
| Tax benefit | | | 1,250 | | | | 5,169 | | | | (76 | )% |
| Net loss | | | (28,210 | ) | | | (22,028 | ) | | | 28 | % |
| Other comprehensive (loss) income | | | (15 | ) | | | 5 | | | | (413 | )% |
| Comprehensive loss | | $ | (28,225 | ) | | $ | (22,023 | ) | | | 28 | % |
Revenue. Revenue for the year ended December 31, 2021 was $191,000 as compared to $239,000 for the same period in 2020, a decrease of $48,000. The decrease was attributable to lower sales in the Middle East and European Union countries in 2021 as compared to the same period in 2020.
Cost of Sales. Cost of sales for the year ended December 31, 2021 was $149,000 as compared to $205,000 for the same period in 2020, a decrease of $56,000. The decrease was primarily attributable to the net decrease in cost of materials as a result of lower sales in 2021 as compared to the same period in 2020.
Research and Development Expense. R&D expense for the year ended December 31, 2021 was $13,133,000, a decrease of $244,000 from $13,377,000 for the same period in 2020. The decrease was driven by net decreases in costs related to the manufacturing of DefenCath prior to its potential marketing approval of $1,489,000 and a reduction in clinical trial expenses of $444,000, attributable to the closing of our LOCK-IT clinical trial. These decreases were partially offset, among others of lesser significance, by increases in non-cash charges for stock-based compensation of $704,000, an increase in consulting fees of $554,000, driven by fees related to the resubmission of the DefenCath NDA to the FDA, and an increase in personnel expenses of $487,000, as a result of additional hires during the fourth quarter of 2020 through the first half of 2021.
Selling, General and Administrative Expense. SG&A expense for the year ended December 31, 2021 was $16,346,000, an increase of $2,468,000 from $13,878,000 for the same period in 2020. The increase was primarily attributable to an increase in non-cash charges for stock-based compensation of $1,839,000, and an increase in personnel expenses of $1,237,000, as a result of additional hires during the fourth quarter of 2020 through the first half of 2021. These increases were partially offset, among others of lesser significance, by a decrease in consulting fees of $730,000, and reduced costs related to marketing research studies in preparation for the potential marketing approval of DefenCath of $202,000.
Interest Income. Interest income for the year ended December 31, 2021 was $14,000, a decrease of $102,000 from $116,000 for the same period in 2020. The decrease was attributable to lower interest rates this year as compared to the same period last year.
Foreign Exchange Transaction Loss. Foreign exchange transaction losses for the year ended December 31, 2021 and 2020 were due to the re-measuring of transactions denominated in a currency other than our functional currency.
Interest Expense. Interest expense for the year ended December 31, 2021 was $16,000 as compared to $33,000 for the same period in 2020. The decrease of $17,000 was due primarily to lower interest rates on expenses that were financed this year as compared to the same period last year.
Tax Benefit. Tax benefit for the years ended December 31, 2021 of $1,250,000 and December 31, 2020 of $5,169,000, represents income tax benefits due to the sale of our unused NOL for state fiscal year 2021 and 2020, respectively, through the NJEDA Technology Business Tax Certificate Transfer program.
Other Comprehensive Income (Loss). Unrealized foreign exchange movements related to long-term loans and the translation of the foreign affiliate financial statements to U.S. dollars and unrealized movements related to short term investment are recorded in other comprehensive income (loss) which resulted in a loss of $15,000 and a gain of $5,000 for the years ended December 31, 2021 and 2020, respectively.
Liquidity and Capital Resources
Sources of Liquidity
As a result of our cost of sales, R&D and SG&A expenditures and the lack of substantial product sales revenue, our ongoing operations have not been profitable since our inception. During the year ended December 31, 2021, we received net proceeds of $41,456,000 from the issuance of 3,737,862 shares of common stock under our at-the-market-issuance sales agreement as compared to $18,433,000 of net proceeds for the same period in 2020 from the issuance of 2,687,646 shares of common stock. Additionally, we also received $165,000 and $412,000 from the exercise of warrants during the years ended December 31, 2021 and 2020, respectively. We will continue to be reliant on external sources of cash for the foreseeable future until we are able to generate revenue.
In June 2021, we received approximately $1,250,000, net of expenses, from the sale of our unused New Jersey NOL eligible for sale under the NJEDA Program. The NJEDA Program allowed us to sell approximately $1,250,000 of our total $1,337,000 in available NOL tax benefits for the state fiscal year 2020.
The NJEDA has approved our application to participate in the NJEDA Program for the state fiscal year 2021. The approval will allow us to sell approximately $0.6 million of the total $0.6 million in available tax benefits to an unrelated, profitable New Jersey corporation in return for approximately $0.6 million in cash. Closing is subject to NJEDA’s typical closing conditions, which are in process of completion.
Net Cash Used in Operating Activities
Net cash used in operating activities for the year ended December 31, 2021 was $21,155,000 as compared to $21,968,000 in 2020, a decrease in net cash use of $813,000. The decrease was driven by an increase in accounts payable of $1,082,000 as compared to $103,000 for the same period in 2020, partially offset by an increase in net loss of $6,182,000 mainly attributable to lower cash received from the NOL sale of $1,250,000 as compared to $5,169,000 for the same period in 2020. In addition, the decrease was also offset by a decrease in prepaid expenses and other current assets for the year ended December 31, 2021 of $667,000, primarily due to a deposit on the equipment, compared to a $992,000 increase for the same period in 2020.
Net Cash (Used in) Provided by Investing Activities
Cash used in investing activities for the year ended December 31, 2021 was $9,135,000 as compared to $7,426,000 of cash provided in the same period in 2020. The net cash used during the year ended December 31, 2021 was mainly driven by the higher amount invested in short-term investments in addition to an increase in purchases of equipment offset by the lower amount of matured investments as compared to the same period in 2020.
Net Cash Provided by Financing Activities
Net cash provided by financing activities for the year ended December 31, 2021 was $41,758,000 as compared to $40,100,000 for the same period in 2020. During the year ended December 31, 2021, we generated net proceeds of $41,456,000 from the sale of our common stock in our at-the-market, or ATM program, $165,000 from the exercise of warrants and $137,000 from the exercise of stock options. In the same period in 2020, we generated net proceeds of $21,255,000 from the underwritten public offering of our common stock, $18,433,000 from the sale of our common stock in our ATM program, and $412,000 from the exercise of warrants.
Funding Requirements and Liquidity
Our total cash and cash equivalents and short-term investments as of December 31, 2021 and 2020, excluding restricted cash of $234,000 and $191,000, respectively, was $65,466,000 and $46,350,000, respectively. During the year ended December 31, 2021, we realized net proceeds of $41,456,000 from the sale of 3,737,862 shares of common stock under our ATM program. At December 31, 2021, we have $150,000,000 available under our shelf registration statement filed on August 12, 2021 for the issuance of equity, debt or equity-linked securities and $50,000,000 under our ATM program, filed on August 12, 2021.
Because our business has not generated positive operating cash flow, we will need to raise additional capital in order to continue to fund our research and development activities, as well as to fund operations generally. Our continued operations are focused primarily in activities leading to the pre-launch and commercialization for DefenCath and will depend on our ability to raise sufficient funds through various potential sources, such as equity, debt financings, and/or strategic relationships and potential strategic transactions. We can provide no assurances that financing or strategic relationships will be available on acceptable terms, or at all.
We expect to continue to fund operations from cash on hand and through capital raising sources as previously described, which may be dilutive to existing stockholders, through revenues from the licensing of our products, or through strategic alliances. We expect to continue to utilize our ATM program, if conditions allow, to support our ongoing funding requirements. Additionally, we may seek to sell additional equity or debt securities through one or more discrete transactions, or enter into a strategic alliance arrangement, but can provide no assurances that any such financing or strategic alliance arrangement will be available on acceptable terms, or at all. Moreover, the incurrence of indebtedness would result in increased fixed obligations and could contain covenants that would restrict our operations. Raising additional funds through strategic alliance arrangements with third parties may require significant time to complete and could force us to relinquish valuable rights to our technologies, future revenue streams, research programs or product candidates, or to grant licenses on terms that may not be favorable to us or our stockholders. Our actual cash requirements may vary materially from those now planned due to a number of factors, any change in the focus and direction of our research and development programs, any acquisition or pursuit of development of new product candidates, competitive and technical advances, the costs of commercializing any of our product candidates, and costs of filing, prosecuting, defending and enforcing any patent claims and any other intellectual property rights.
Sales of Neutrolin outside the U.S. are not expected to generate significant product revenues for the foreseeable future, and we expect to grow product sales for DefenCath in the U.S., should we receive FDA approval. In the absence of significant revenue, we are likely to continue generating operating cash flow deficits. We will continue to use cash as we increase other activities leading to the commercialization of DefenCath upon approval, pursue business development activities, and incur additional legal costs to defend our intellectual property.
We currently estimate that as of December 31, 2021, we have sufficient cash on hand to fund operations at least through the first half of 2023, after taking into consideration the costs for resubmission of the NDA and initial preparations for the commercial launch for DefenCath. Additional financing may be required to build out our commercial infrastructure following FDA approval and to continue our operations should we decide to market and sell DefenCath in the U.S. on our own. If we are unable to raise additional funds when needed, we may be forced to slow or discontinue our preparations for the commercial launch of DefenCath. We may also be required to delay, scale back or eliminate some or all of our research and development programs. Each of these alternatives would likely have a material adverse effect on our business.
Contractual Obligations
We entered into a seven-year operating lease agreement in March 2020 for an office space at 300 Connell Drive, Berkeley Heights, New Jersey 07922. The lease agreement, with a monthly average cost of approximately $17,000, commenced on September 16, 2020. Our sublease on our previous premises at 400 Connell Drive, Berkeley Heights, New Jersey 07922 terminated on November 30, 2020.
Critical Accounting Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our consolidated financial statements, which have been prepared in accordance with accounting principles generally accepted in the United States, or GAAP. The preparation of these consolidated financial statements requires us to make estimates and judgments that affect the reported amounts of assets, liabilities and expenses. On an ongoing basis, we evaluate these estimates and judgments, including those described below. We base our estimates on our historical experience and on various other assumptions that we believe to be reasonable under the circumstances. These estimates and assumptions form the basis for making judgments about the carrying values of assets and liabilities that are not readily apparent from other sources. Actual results and experiences may differ materially from these estimates.
While our significant accounting policies are more fully described in Note 3 to our financial statements included with this report, we believe that the following accounting policies are the most critical to aid you in fully understanding and evaluating our reported financial results and affect the more significant judgments and estimates that we use in the preparation of our financial statements.
Stock-Based Compensation
We account for stock options according to the Financial Accounting Standards Board (“FASB”) Accounting Standards Codification (“ASC”) No. 718, “Compensation — Stock Compensation” (“ASC 718”). Share-based compensation cost is measured at grant date, based on the estimated fair value of the award using a Black-Scholes option pricing model for options with service or performance-based conditions. Stock-based compensation cost is recognized as expense, over the requisite service period on a straight-line basis.
Valuations incorporate several variables, including expected term, expected volatility, expected dividend yield and a risk-free interest rate. We estimate the expected term of the options granted based on anticipated exercises in future periods. The expected stock price volatility for the Company’s stock options is calculated based on the historical volatility of the Company’s common stock. The expected dividend yield reflects our current and expected future policy for dividends on our common stock. To determine the risk-free interest rate, we utilize the U.S. Treasury yield curve in effect at the time of grant with a term consistent with the expected term of our awards which is 5 years for employees and 10 years for non-employees.
Recently Adopted Authoritative Pronouncements:
In December 2019, the FASB issued ASU 2019-12 which removes certain exceptions to the general principles of the accounting for income taxes and also improves consistent application of and simplification of other areas when accounting for income taxes. The guidance was effective for us beginning in the first quarter of fiscal year 2021. Early adoption was permitted. This adoption on January 1, 2021 did not have a material impact on our consolidated financial statements.
Item 7A. Quantitative and Qualitative Disclosures About Market Risk
Not applicable.
Item 8. Financial Statements and Supplementary Data
See the financial statements included at the end of this report beginning on page F-1.
Item 9. Changes in and Disagreements With Accountants on Accounting and Financial Disclosure
Not applicable.
Item 9A. Controls and Procedures
As of the end of the period covered by this Annual Report on Form 10-K, we carried out an evaluation, under the supervision and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, of the effectiveness of the design and operation of our disclosure controls and procedures (as defined in the Exchange Act Rules 13a-15(e) and 15d-15(e)) (the “Exchange Act”). Based on the foregoing evaluation, our Chief Executive Officer and Chief Financial Officer have concluded that our disclosure controls and procedures are effective to ensure that information required to be disclosed by us in the reports we file or submit under the Exchange Act is recorded, processed, summarized and reported within the time periods specified in the rules and forms of the SEC, and that such information is accumulated and communicated to our management, including our Chief Executive Officer and Chief Financial Officer, to allow timely decisions regarding required disclosures.
Changes in Internal Control Over Financial Reporting
Other than the appointment of our current Chief Financial Officer as interim Chief Executive Officer in addition to his role as our Chief Financial Officer on October 4, 2021, there were no changes in our internal control over financial reporting during our fourth quarter ended December 31, 2021, or in other factors that could significantly affect these controls, that materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
Management’s Annual Report on Internal Controls Over Financial Reporting
Our management is responsible for establishing and maintaining adequate internal control over financial reporting and for the assessment of the effectiveness of internal control over financial reporting. As defined by the Securities and Exchange Commission, internal control over financial reporting is a process designed by, or under the supervision of, our principal executive and principal financial officers and effected by our Board of Directors, management and other personnel, to provide reasonable assurance regarding the reliability of financial reporting and the preparation of the consolidated financial statements in accordance with U.S. generally accepted accounting principles.
Our internal control over financial reporting includes those policies and procedures that (1) pertain to the maintenance of records that, in reasonable detail, accurately and fairly reflect our transactions and dispositions of our assets; (2) provide reasonable assurance that transactions are recorded as necessary to permit preparation of the consolidated financial statements in accordance with generally accepted accounting principles, and that our receipts and expenditures are being made only in accordance with authorizations of our management and directors; and (3) provide reasonable assurance regarding prevention or timely detection of unauthorized acquisition, use or disposition of our assets that could have a material effect on the consolidated financial statements.
Because of its inherent limitations, internal control over financial reporting may not prevent or detect misstatements. Also, projections of any evaluation of effectiveness to future periods are subject to the risk that controls may become inadequate because of changes in conditions, or that the degree of compliance with the policies or procedures may deteriorate.
In connection with the preparation of our annual consolidated financial statements, management, including, our Principal Executive and Financial Officer, has undertaken an assessment of the effectiveness of our internal control over financial reporting as of December 31, 2021, based on the criterial established in Internal Control—Integrated Framework (2013) issued by the Committee of Sponsoring Organizations of the Treadway Commission (COSO). Management’s assessment included an evaluation of the design of our internal control over financial reporting and testing of the operational effectiveness of those controls.
Based on this evaluation, management has concluded that our internal control over financial reporting was effective as of December 31, 2021.
Item 9B. Other Information
Not applicable.
Item 9C. Disclosure Regarding Foreign Jurisdictions that Prevent Inspections
Not applicable.
PART III
Item 10. Directors, Executive Officers, and Corporate Governance
We have adopted a written Code of Conduct and Ethics that applies to our directors, executive officers and all employees. We intend to disclose any amendments to, or waivers from, our code of ethics and business conduct that are required to be publicly disclosed pursuant to rules of the SEC by filing such amendment or waiver with the SEC. This code of ethics and business conduct can be found in the “Investors - Corporate Governance” section of our website, www.cormedix.com.
Delinquent Section 16(a) Reports
Section 16(a) of the Exchange Act requires our directors, executive officers and holders of more than 10% of our common stock to file with the SEC initial reports of ownership and reports of changes in the ownership of our common stock and other equity securities. Such persons are required to furnish us copies of all Section 16(a) filings. To our knowledge, based solely on a review of the copies of such reports furnished to us and representations that no other reports were required, during the fiscal year ended December 31, 2021, all Section 16(a) filing requirements applicable to its officers, directors and greater than ten percent beneficial owners were complied with, except as to the following: (i) Dr, David was not timely in filing a Form 4 for changes in beneficial ownership that occurred on November 1, 2021, the changes in beneficial ownership were reported on November 10, 2021; and (ii) Mr. Kaplan was not timely in filing a Form 4 for changes in beneficial ownership that occurred on November 16, 2021, the changes in beneficial ownership were reported on November 19, 2021.
Directors
The following table sets forth the name, age and position of each of our directors as of March 25, 2022:
| Name | | Age | | Director Since | | Position(s) with CorMedix |
| Paulo F. Costa | | 71 | | September 2020 | | Director |
| Janet Dillione | | 62 | | August 2015 | | Director |
| Gregory Duncan | | 56 | | November 2020 | | Director |
| Alan W. Dunton | | 67 | | March 2019 | | Director |
| Myron Kaplan | | 76 | | April 2016 | | Director and Chairman of the Board |
| Steven Lefkowitz | | 65 | | June 2017 | | Director |
| Joseph Todisco | | 46 | | March 2022 | | Director |
Paulo F. Costa has been a director of CorMedix since September 2020. Mr. Costa previously served as President and Chief Executive Officer of Novartis U.S. Corporation, from October 2005 to August 2008. Prior to his work at Novartis U.S. Corporation, Mr. Costa was President and Chief Executive Officer of Novartis Pharmaceuticals, U.S. from July 1999 to September 2005. Prior to joining Novartis, Mr. Costa spent 30 years at Johnson & Johnson, including as President of Janssen Pharmaceutica, Inc. From August 2009 to August 2012, Mr. Costa served as Chairman of the Board of Amylin Pharmaceuticals Inc, a commercial stage biopharma company, until its sale to Bristol-Myers Squibb and AstraZeneca in a $7 billion transaction in 2012. Mr. Costa currently serves as Chairman of the Board of MacroGenics, Inc., a public late stage biopharma company focused on oncology. Mr. Costa received his undergraduate degree from São Paulo School of Business Administration and earned a master’s degree in business administration from Harvard Business School. Among other experience, qualifications, attributes and skills, Mr. Costa’s significant depth of experience in the pharmaceutical industry, including service as a director and executive of pharmaceutical companies, led to the conclusion of our Board that he should serve as a director of our Company in light of our business and structure.
Janet Dillione has been a director of CorMedix since August 2015. Since November 2020, Ms. Dillione currently serves as the Chief Executive Officer of Connect America, a nationally recognized leader in comprehensive telehealth and remote patient monitoring solutions. Prior to joining Connect America, she served as Chief Executive Officer of Bernoulli Enterprise, Inc. since May 2014, a real-time connected healthcare information technology company. Previously, she was at Nuance Communications, Inc., a leading provider of voice and language solutions for businesses and consumers around the world, having joined Nuance in April 2010 as Executive Vice President and General Manager of the Healthcare Division and serving as an executive officer from March 2010 until May 2014. From June 2000 to March 2010, Ms. Dillione held several senior level management positions at Siemens Medical Solutions, a global leader in medical imaging, laboratory diagnostics, and healthcare information technology, including President and CEO of the global healthcare IT division. Ms. Dillione currently serves as a director of Vizient, Inc., a private health care performance improvement company. Ms. Dillione received her B.A. from Brown University in 1981 and completed the Executive Program at The Wharton School of Business of the University of Pennsylvania in 1998. She has over 25 years of experience leading global teams in the development and delivery of healthcare technology and services. Among other qualifications, attributes and skills, Ms. Dillione’s financial expertise and significant executive management experience with medical device and healthcare companies led to the conclusion of our Board that she should serve as a director of our Company in light of our business and structure.
Gregory Duncan has been a director of CorMedix since November 2020. Mr. Duncan currently serves as the Chairman and CEO of Virios Therapeutics, a clinical-stage biopharmaceutical company developing and commercializing innovative antiviral therapies to treat diseases associated with a viral triggered abnormal immune response, such as fibromyalgia (FM), and has served since April 2020. From 2014 and prior to joining his current company earlier this year, Mr. Duncan served as President and CEO of Celtaxsys, a privately held biotechnology company focused on cystic fibrosis and other rare, inflammatory diseases. Mr. Duncan has spent the majority of his career in senior leadership roles in commercial stage pharmaceutical companies. From 2007 to 2013, he served as a senior executive at UCB, including as President of its North America business, as well as an executive committee member. Prior to his roles with UCB, Mr. Duncan spent approximately 17 years at Pfizer where he gained significant experience across sales and marketing functions including serving as SVP of US Marketing and later as President of Pfizer’s Latin America business from 2005 to 2007. Mr. Duncan received his undergraduate degree from the State University of New York, Albany, and earned an MBA degree from Emory University. Among other experience, qualifications, attributes and skills, Mr. Duncan’s significant depth of experience in the pharmaceutical industry led to the conclusion of our Board that he should serve as a director of our Company in light of our business and structure.
Alan W. Dunton, M.D. has been a director of CorMedix since March 2019. He is the founder and principal consultant of Danerius, LLC, a biotechnology and pharmaceutical consulting business which he started in 2006. From 1994, he served in senior positions in Research and Development in the Pharmaceutical Division of Johnson and Johnson including President and Managing Director of the Janssen, the major research, development and regulatory arm of the pharmaceuticals division at Johnson & Johnson. From January 2007 through March 2009, Dr. Dunton served as President and Chief Executive Officer of Panacos Pharmaceuticals, Inc. From November 2015 through March 2018, Dr. Dunton was the Head/Senior Vice President of Research, Development and Regulatory Affairs of Purdue Pharma L.P., a private pharmaceutical company. Dr. Dunton received his Bachelor of Science degree in biochemistry, magna cum laude, from State University of New York at Buffalo, and received his M.D. from New York University School of Medicine. In addition to CorMedix, Dr. Dunton currently serves on the boards of three public companies, as a Director at Palatin Technologies, Inc. and Oragenics, Inc. he chairs the Compensation Committees of both companies. He also serves as a member of the Audit Committees of these companies. Additionally, Dr. Dunton is a member of the board of Recce Pharma Ltd., an Australian public biotechnology company focused on developing novel anti-infectives for serious and life threatening diseases. Among other qualifications, Dr. Dunton’s significant depth of experience in the pharmaceutical industry, including service as a director of public pharmaceutical companies, led to the conclusion of our Board that he should serve as a director of our Company in light of our business and structure.
Myron Kaplan became a director of CorMedix in April 2016. He is a founding partner of Kleinberg, Kaplan, Wolff & Cohen, P.C., a New York City general practice law firm, where he has practiced corporate and securities law for more than forty years. In 2012, Mr. Kaplan became a trustee of the Lehman Brothers Plan Holding Trust. Previously, he served as a member of the board of directors of SAirGroup Finance (USA) Inc., a subsidiary of SAirGroup that had publicly issued debt securities, Trans World Airlines, Inc. and Kitty Hawk, Inc. Among his business and civic involvements, Mr. Kaplan currently serves on the boards of directors of a number of private companies and has been active for many years on the boards of trustees and various board committees of The Children’s Museum of Manhattan and JBI International (formerly The Jewish Braille Institute of America). Mr. Kaplan graduated from Columbia College and holds a Juris Doctor from Harvard Law School. Among other experience, qualifications, attributes and skills, Mr. Kaplan’s experience in a broad range of corporate and securities matters and service as a director of public companies led to the conclusion of our Board that he should serve as a director of our Company in light of our business and structure.
Steven Lefkowitz was a director of CorMedix from August 2011 to June 2016. He was reappointed to the Board in June 2017. He also served as our acting Chief Financial Officer from August 2013 to July 2014. Mr. Lefkowitz has been the President and Founder of Wade Capital Corporation, a financial advisory services company, since June 1990. Mr. Lefkowitz has been a director of both public and private companies. Mr. Lefkowitz received his A.B. from Dartmouth College in 1977 and his M.B.A. from Columbia University in 1985. Among other experience, qualifications, attributes and skills, Mr. Lefkowitz’s education, experience and financial expertise led to the conclusion of our Board that he should serve as a director of our Company in light of our business and structure.
Joseph Todisco became a director of CorMedix in March 2022. Prior to joining CorMedix, he was a senior executive at Amneal Pharmaceuticals, where for the past 11 years he has held various roles, most recently as Executive Vice President, Chief Commercial Officer where he was responsible for Amneal Specialty, a growing branded products business. During his tenure at Amneal, Mr. Todisco held roles overseeing corporate development and international operations, leading commercial teams in several international markets including the UK, Australia and Germany, as well as leading Amneal’s merger integration with Impax Laboratories in 2018. He was previously Co-Founder and managing executive of Gemini Laboratories, a specialty pharmaceutical company focused on the sales and marketing for niche branded products in the US Market. Gemini Laboratories was established as an affiliate of Amneal Pharmaceuticals and was subsequently acquired by Amneal in 2018. Prior to joining Amneal, Mr. Todisco was Vice President, Business Development & Licensing at Ranbaxy, Inc. where he was responsible for developing and executing Ranbaxy’s North American commercial business strategy. Prior to Ranbaxy, he held various roles at Par Pharmaceutical, and in his earlier career held positions at Oppenheimer & Company and Marsh & McLennan Companies. Mr. Todisco obtained his MBA in finance from Fordham Graduate School of Business and his BA in Economics from Georgetown University. Among other qualifications, attributes and skills, Mr. Todisco’s business expertise and significant executive management experience in the pharmaceutical industry led to the conclusion of our Board that he should serve as a director of our Company in light of our business and structure.
Board Independence
Our Board has undertaken a review of the independence of our directors and has determined that (i) all current directors are independent within the meaning of Section 5605(b) of the Nasdaq Marketplace Rules, (ii) all members of our Audit Committee meet the additional test for independence for audit committee members imposed by SEC regulation and Section 5605(c) of the Nasdaq Marketplace Rules, (iii) all of the members of our Compensation Committee are independent within the meaning of Section 5605(d) of the Nasdaq Marketplace Rules, and (iv) all of the members of our Nominating and Governance Committee are independent within the meaning of Section 5605(e) of the Nasdaq Marketplace Rules.
Board Committees
Our Board has established an Audit Committee, a Compensation Committee and a Nominating and Governance Committee. Our Audit Committee currently consists of Mr. Lefkowitz (Chair), Dr. Dunton and Mr. Duncan. Our Compensation Committee currently consists of Ms. Dillione (Chair), Dr. Dunton and Mr. Duncan. Our Nominating and Governance Committee currently consists of Mr. Costa (Chair), Mr. Kaplan and Ms. Dillione. The membership of these Committees may be changed after our next annual meeting.
Each of the above-referenced committees operates pursuant to a formal written charter. The charters for each committee, which have been adopted by our Board, contain a detailed description of the respective committee’s duties and responsibilities and are available on our website at www.cormedix.com under the “Investor Relations—Corporate Governance” tab.
Audit Committee
The Audit Committee monitors our corporate financial statements and reporting and our external audits, including, among other things, our internal controls and audit functions, the results and scope of the annual audit and other services provided by our independent registered public accounting firm and our compliance with legal matters that have a significant impact on our financial statements. The Audit Committee also consults with our management and our independent registered public accounting firm prior to the presentation of financial statements to stockholders and, as appropriate, initiates inquiries into aspects of our financial affairs. The Audit Committee is responsible for establishing procedures for the receipt, retention and treatment of complaints regarding accounting, internal accounting controls or auditing matters, and for the confidential, anonymous submission by our employees of concerns regarding questionable accounting or auditing matters. In addition, the Audit Committee is directly responsible for the appointment, retention, compensation and oversight of the work of our independent registered public accounting firm, including approving services and fee arrangements. All related party transactions will be approved by the Audit Committee before we enter into them.
Both our independent registered public accounting firm and internal financial personnel regularly meet with, and have unrestricted access to, the Audit Committee.
The Board has determined that each of Mr. Lefkowitz, Dr. Dunton and Mr. Duncan qualifies as an “audit committee financial expert” as that term is defined in the rules and regulations of the SEC. The designation of each of Mr. Lefkowitz, Dr. Dunton and Mr. Duncan as an “audit committee financial expert” does not impose on them any duties, obligations or liability that are greater than those that are generally imposed on them as a member of the Audit Committee and the Board, and their designation as an “audit committee financial expert” pursuant to this SEC requirement does not affect the duties, obligations or liability of any other member of the Audit Committee or the Board.
Compensation Committee
The Compensation Committee reviews and approves our compensation policies and all forms of compensation to be provided to our executive officers, including, among other things, annual salaries, bonuses, and other incentive compensation arrangements. In addition, the Compensation Committee administers our equity compensation plans, including granting stock options to our executive officers. The Compensation Committee also reviews and approves employment agreements with executive officers and other compensation policies and matters.
Since 2016, we have periodically engaged Frederic W. Cook & Co., an independent compensation consultant, for input on the compensation of our Named Executive Officers and directors. The Compensation Committee assessed the independence of Frederic W. Cook & Co., considering the factors required by the Nasdaq Global Market Listing Rules and concluded that no conflict of interest exists that would prevent Frederic W. Cook & Co. from independently representing our Company. In the future, we, or the Compensation Committee, may engage or seek the advice of Frederic W. Cook & Co., or another compensation consultant.
At our 2021 annual meeting of stockholders, our stockholders indicated their preference that we solicit a non-binding advisory vote on the compensation of the named executive officers, commonly referred to as a “Say-On-Pay” vote, every year. This vote is not intended to address any specific item of compensation, but rather the overall compensation of our named executive officers and the philosophy, policies and practices described in this Annual Report on Form 10-K. The Compensation Committee evaluates our executive compensation program in light of our benchmarking of peer companies with the advice of Frederic W. Cook as well as our shareholders’ views’ including the “Say-On-Pay” votes when making future decisions regarding executive compensation. The next “Say-On-Pay” vote will occur at the 2022 annual meeting of stockholders.
Each member of the Compensation Committee is a non-employee director, as defined pursuant to Rule 16b-3 promulgated under the Exchange Act.
Nominating and Governance Committee
The Nominating and Governance Committee identifies, evaluates and recommends nominees to the Board and committees of the Board, conducts searches for appropriate directors and evaluates the performance of the Board and of individual directors. The Nominating and Governance Committee also is responsible for reviewing developments in corporate governance practices, evaluating the adequacy of our corporate governance practices and reporting and making recommendations to the Board concerning corporate governance matters.
Executive Officers
The following table sets forth the name, age and position of each of our executive officers as of December 31, 2021:
| Name | | Age | | Position(s) with CorMedix |
| Matthew David | | 44 | | Interim Chief Executive Officer, Chief Financial Officer |
| Phoebe Mounts | | 71 | | Executive Vice President and General Counsel and Head of Regulatory, Compliance and Legal |
| Elizabeth Masson-Hurlburt | | 42 | | Executive Vice President and Head of Clinical Operations |
| Thomas Nusbickel | | 64 | | Executive Vice President and Chief Commercial Officer |
Matthew David became our Executive Vice President and Chief Financial Officer in May 2020 and is currently serving as interim Chief Executive Officer since the retirement of Mr. Baluch on October 4, 2021. Prior to joining us, he most recently served as Head of Strategy at Ovid Therapeutics Inc, a late-stage clinical biopharmaceutical company focused on developing treatments for rare neurological disorders, where he was responsible for financing strategy and investor relations, and joined in October 2018. Prior to Ovid, Dr. David was a Strategic Advisor to Frequency Therapeutics, advising on financing, investor relations and strategic initiatives from 2017 to early 2019. Prior to Frequency, Dr. David spent the majority of his career as an investment banker specialized in the life sciences sectors, including at Piper Jaffray, Thomas Weisel Partners, Ferghana Partners and most recently at Bank of America Merrill Lynch. As part of his experience as an investment banker, Dr. David has advised on a broad range of capital raising and strategic transactions. Earlier in his career, Dr. David was part of the equity research team at Lehman Brothers, focusing on Large Pharma. Dr. David began his career as a surgical resident at Beth Israel Hospital, after receiving an M.D. from NYU School of Medicine. Dr. David earned his Bachelor of Arts degree in Chemistry, magna cum laude, from Dartmouth College.
Phoebe Mounts became our Executive Vice President and General Counsel and Head of Regulatory, Compliance and Legal in May 2019. Prior to her employment with us, Dr. Mounts was a partner at Morgan, Lewis & Bockius LLP, where she provided legal counsel to life sciences companies for over 20 years. As part of her work at Morgan Lewis, Dr. Mounts had been providing us legal services as outside counsel since 2013, with responsibility for developing our FDA regulatory strategies for DefenCath. Prior to graduating from Georgetown University Law Center, Dr. Mounts was on the faculty of the Johns Hopkins University School of Public Health for 16 years, specializing in molecular biology and infectious disease. She received her Ph.D. in molecular biology from the University of Edinburgh in Scotland.
Elizabeth Masson-Hurlburt became our Executive Vice President and Head of Clinical Operations in March 2018. Prior to her employment, Ms. Masson-Hurlburt had been providing us clinical operations expertise as a consultant since late November 2017. Before she began her consulting career, she held several progressive management roles in clinical operations, most recently at Gemphire Therapeutics, as a Senior Director, Clinical Operations from April 2015 to October 2016, then as Vice President, Clinical Operations from October 2016 to March 2018. Ms. Masson-Hurlburt received her B.A. in Leadership and Organizational Management from Bay Path College.
Thomas Nusbickel became our Executive Vice President and Chief Commercial Officer in May 2021. Prior to his employment, Mr. Nusbickel held several leadership roles in the commercial strategy and renal disease space, most recently at Coherus Biosciences, as Vice President of Market Access and Government Affairs, Opko Inc., as Chief Commercial Officer, and served for more than two decades at Amgen. Mr. Nusbickel has an undergraduate degree from Eckerd College and an M.B.A. from Pepperdine University.
On October 1, 2021, the Company and Khoso Baluch came to a mutual agreement pursuant to which Mr. Baluch retired from his position as our Chief Executive Officer, effective October 4, 2021. Mr. Baluch also resigned from our Board of Directors. Dr. David is serving as interim Chief Executive Officer and Chief Financial Officer. The Board of Directors appointed Joseph Todisco as the Chief Executive Officer on March 16, 2022, commencing no later than May 16, 2022, and appointed Mr. Todisco to serve as a member of the Board on March 18, 2022. Dr. David will continue to serve as interim Chief Executive Officer and Chief Financial Officer until Mr. Todisco commences employment, after which Dr. David will continue to serve as our Chief Financial Officer.
On October 4, 2021, we and John L. Armstrong, Jr. came to a mutual agreement pursuant to which Mr. Armstrong retired from his position as our Executive Vice President, Technical Operations, effective October 4, 2021.
Item 11. Executive Compensation
DIRECTOR COMPENSATION
Director Compensation in Fiscal 2021
The following table shows the compensation earned by each non-employee director of our Company for the year ended December 31, 2021.
| Name | | Fees Earned
($) | | | Option
Awards (1) (2)
($) | | | Total
($) | |
| Paulo F. Costa | | | 84,000 | | | | 125,460 | | | | 209,460 | |
| Janet Dillione | | | 78,000 | | | | 188,190 | | | | 266,190 | |
| Gregory Duncan | | | 72,000 | | | | 125,460 | | | | 197,460 | |
| Alan W. Dunton | | | 72,000 | | | | 188,190 | | | | 260,190 | |
| Myron Kaplan | | | 120,000 | | | | 188,190 | | | | 308,190 | |
| Steven Lefkowitz | | | 93,000 | | | | 188,190 | | | | 281,190 | |
| | (1) | The amounts included in this column are the dollar amounts representing the full grant date fair value of each stock option award calculated in accordance with FASB ASC Topic 718 and do not represent the actual value that may be recognized by the directors upon option exercise. For information on the valuation assumptions used in calculating these amounts, see Note 7 to our audited financial statements included in this Annual Report on Form 10-K. |
| (2) | As of December 31, 2021, the number of shares underlying options held by each non-employee director was as follows: 43,750 shares for Mr. Costa; 105,000 shares for Ms. Dillione; 42,500 for Mr. Duncan; 72,500 shares for Dr. Dunton; 86,000 shares for Mr. Kaplan; and 83,000 shares for Mr. Lefkowitz. |
Director Compensation Plan
We maintain a Deferred Compensation Plan for Directors, pursuant to which our non-employee directors may defer all of their cash director fees and restricted stock units. Any cash fees due to a participating director will be converted into a number of shares of our common stock by dividing the dollar amount of fees payable by the closing price of our common stock on the date such fees would be payable, and the director’s unfunded account is credited with the shares. The shares that accumulate in a director’s account will be paid to the director on the tenth business day in January following the year in which the director’s service terminates for whatever reason, other than death, in which case the account will be paid within 30 days of the date of death to the designated beneficiary, as applicable. In the event of a change in control of our Company, the director would receive cash in an amount equal to the number of shares in the account multiplied by the fair market value of our common stock on the change in control date, and the payment would be accelerated to five business days after the effective date of the change in control.
In January 2021, the Board, following the recommendation of the Compensation Committee and based on advice of Frederic W. Cook & Co., determined that no adjustment was needed with regard to Board and committee cash compensation. Following a review of board compensation practices of the Company’s peer group, the Board made the following changes to equity compensation effective as of January 2021, (i) increased the annual grant of stock options to each non-employee director from 15,000 to 20,000 shares, (ii) increased the initial grant of stock options to new non-employee directors from 20,000 to 25,000 shares; and (iii) provided a one-time 10,000 share grant of stock options as of January 11, 2021 for non-employee directors who joined the Board prior to 2020.
The 2020 and 2021 compensation programs are set forth below in the table. All stock options are subject to continued service on the Board through the vesting date. The exercise price per share of each stock option granted to our non-employee directors is equal to the fair market value of our common stock as determined based upon the closing sales price for our stock on the date of grant.
On January 11, 2021, our Board amended outstanding stock options to purchase shares of our common stock held by the non-employee directors to extend the post-termination exercise period of such options such that each vested stock option held by a director as of the date of separation from service will remain exercisable for the 12-month period following the date of separation from service, but in no event later than the end of the term of the option.
| | | Effective January 1,
2020 | | | Effective January 1,
2021 | |
| | | Cash | | | Stock
Options | | | Cash | | | Stock
Options | |
| Annual Fee | | $ | 55,000 | | | | | | | $ | 55,000 | | | | | |
| First Election to Board | | | | | | | 20,000 | (1) | | | | | | | 25,000 | (1) |
| Annual Grant, Prorated in First Year Following Election to the Board | | | | | | | 15,000 | (2) | | | | | | | 20,000 | (2) |
| Additional Annual Fee - Board Chair | | $ | 45,000 | | | | | | | $ | 45,000 | | | | | |
| Additional Annual Fee - Audit Chair | | $ | 23,000 | | | | | | | $ | 23,000 | | | | | |
| Additional Annual Fee - Compensation Chair | | $ | 18,000 | | | | | | | $ | 18,000 | | | | | |
| Additional Annual Fee - Nomination and Governance Chair | | $ | 14,000 | | | | | | | $ | 14,000 | | | | | |
| Additional Annual Fee - Audit Committee Non-Chair Members | | $ | 10,000 | | | | | | | $ | 10,000 | | | | | |
| Additional Annual Fee - Compensation Committee Non-Chair Members | | $ | 7,000 | | | | | | | $ | 7,000 | | | | | |
| Additional Annual Fee – Nomination and Governance Committee Non-Chair Members | | $ | 5,000 | | | | | | | $ | 5,000 | | | | | |
| Additional Annual Fee – Strategic Committee Members | | $ | - | | | | | | | $ | 15,000 | | | | | |
| Additional Annual Fee – Strategic Finance Committee Two Co-Chairs | | $ | 20,000 | (3) | | | | | | | - | | | | | |
| (1) | Vest one third each on the date of grant and the first and second anniversary date of grant. |
| (2) | Vest monthly over one year after the grant date. |
| (3) | The Additional Annual Fee for the Strategic Finance Committee Co-Chairs ended on June 30, 2020. |
EXECUTIVE COMPENSATION
Components of Compensation
The key components of our executive compensation package are cash compensation (salary and annual bonuses), long-term equity incentive awards and change in control and other severance agreements. These components are administered with the goal of providing total compensation that recognizes meaningful differences in individual performance, is competitive, varies the opportunity based on individual and corporate performance, and is valued by our Named Executive Officers. During 2021, our Named Executive Officers were Khoso Baluch, Matthew David, John Armstrong, Phoebe Mounts, Elizabeth Masson-Hurlburt and Thomas Nusbickel. Mr. Baluch retired as our Chief Executive Officer effective October 4, 2021, and Mr. Armstrong retired as our Executive Vice President for Technical Operations effective October 4, 2021. Dr. David, who has served as our Executive Vice President and Chief Financial Officer since May 11, 2020, has from October 4, 2021, also served as our interim Chief Executive Officer in addition to his role as Chief Financial Officer.
Base Salary
It is the Compensation Committee’s objective to set a competitive rate of annual base salary for each Named Executive Officer. The Compensation Committee believes competitive base salaries are necessary to attract and retain top quality executives, since it is common practice for public companies to provide their named executive officers with a guaranteed annual component of compensation that is not subject to performance risk. The Compensation Committee, on its own or with outside consultants, may establish salary ranges for the Named Executive Officers, with minimum to maximum opportunities that cover the normal range of market variability. The actual base salary for each Named Executive Officer is then derived from those salary ranges based on his or her responsibility, tenure and past performance and market comparability. Annual base salaries for the Named Executive Officers are reviewed and approved by the Compensation Committee in the first quarter following the end of the previous performance year. Changes in base salary are based on the scope of an individual’s current job responsibilities, individual performance in the previous performance year, target pay position relative to the peer group, and our salary budget guidelines. The Compensation Committee reviews established goals and objectives, and determines an individual’s achievement of those goals and objectives and considers the recommendations provided by the Chief Executive Officer to assist it in determining appropriate salaries for the Named Executive Officers other than the Chief Executive Officer.
For the years ended December 31, 2020 and 2021, with the advice of outside consultants, including Frederic W. Cook & Co., the Compensation Committee increased the salaries of certain of our Named Executive Officers to account for adjustments in the market. See under the caption “Employment Agreements.”
In May 2020, March 2019, March 2021 and May 2021, respectively, we entered into an employment agreement with each of Matthew David, our Executive Vice President and Chief Financial Officer, Phoebe Mounts, our Executive Vice President and General Counsel and Head of Regulatory, Compliance and Legal, Elizabeth Masson-Hurlburt, our Executive Vice President and Head of Clinical Operations, and Thomas Nusbickel, our Chief Commercial Officer. These agreements provide for a salary for each Named Executive Officer and are described under the caption “Employment Agreements.”
Effective October 4, 2021, Matthew David, our Chief Financial Officer, began serving as interim Chief Executive Officer, until a new Chief Executive Officer is appointed. Dr. David’s new base salary as an interim Chief Executive Officer is described under the caption “Employment Agreements.” Dr. David’s new base salary was increased to account for the additional responsibilities associated with serving as interim Chief Executive Officer. Dr. David’s new base salary was increased from $330,000 to $425,000 to account for the additional responsibilities associated with serving as the interim Chief Executive Officer. Should Dr. David continue to serve as the interim Chief Executive for six months after October 4, 2021, the Board, or its Compensation Committee, will review such base salary to determine whether an increase is appropriate at that time. After Dr. David ceases to serve as interim Chief Executive Officer, and as he continues to serve as Chief Financial Officer, we will provide him with an annual base salary of $375,000, representing a $45,000 increase from his current salary level under the employment agreement.
The base salary information for our Named Executive Officers for 2020 and 2021 is set forth in the Summary Compensation Table below.
Annual Bonuses
As part of their compensation package, our Named Executive Officers generally have the opportunity to earn annual non-equity incentive bonuses. Annual non-equity bonuses are designed to reward superior executive performance while reinforcing our short-term strategic operating goals. The Board approves, based on the Compensation Committee’s recommendation, an annual corporate target award for the Named Executive Officers based on a percentage of base salary and any applicable terms in any individual employment agreements. Annual bonus targets as a percentage of base salary increase with executive rank so that for the more senior executives, a greater proportion of their total cash compensation is contingent upon annual performance. For 2021, Messrs. Baluch and Armstrong, Dr. Mounts, Ms. Masson-Hurlburt and Mr. Nusbickel were each eligible for an annual target bonus of 80%, 35%, 30%, 30% and 30% of base salary, respectively, of his or her base salary then in effect. Dr. David was eligible for an annual target bonus of 30% of base salary prior to serving as interim Chief Executive Officer and the target was increased to 60% of base salary while serving in that role. Dr. David’s new annual target bonus as interim Chief Executive Officer is described under the caption “Employment Agreements.”
On December 20, 2021, the Board adopted the CorMedix Inc. Executive Bonus Plan (the “Bonus Plan”), which will be used to grant annual and other performance bonuses to executives, including our Named Executive Officers. The Bonus Plan provides for bonuses based on achievement of performance objectives, as determined by the Compensation Committee for each performance period.
Participants may receive bonuses based on a target bonus amount, which may be a percentage of the participant’s base salary or such other amount as the Compensation Committee determines, and achievement of the applicable performance objectives. Bonuses are subject to continued employment through the end of the applicable performance period and compliance with restrictive covenant agreements. The Compensation Committee will set the performance periods, target bonuses and performance objectives and will select the eligible executives for each performance period. The performance metrics may include (but shall not be limited to) any of the following: (i) net earnings or net income (before or after taxes); (ii) earnings per share; (iii) net sales growth; (iv) net operating profit; (v) return measures (including, but not limited to, return on assets, capital, equity, or sales); (vi) cash flow (including, but not limited to, operating cash flow, free cash flow, and cash flow return on capital); (vii) cash flow per share; (viii) earnings before or after taxes, interest, depreciation, and/or amortization; (ix) gross or operating margins; (x) productivity ratios; (xi) share price (including, but not limited to, growth measures and total stockholder return); (xii) expense targets or ratios; (xiii) charge-off levels; (xiv) improvement in or attainment of revenue levels; (xv) margins; (xvi) operating efficiency; (xvii) operating expenses; (xviii) economic value added; (xix) improvement in or attainment of expense levels; (xx) improvement in or attainment of working capital levels; (xxi) debt reduction; (xxii) capital targets; (xxiii) regulatory, clinical, or manufacturing milestones; (xxiv) consummation of acquisitions, dispositions, projects or other events or transactions; (xxv) developing strategic plans, (xxvi) objectives related to product development, testing, product design, regulatory approval, product manufacturing and other business needs, and (xxvii) personal objectives for the participant. Bonuses are to be paid in a cash lump sum within 2 ½ months following the end of the applicable performance period (but no later than March 15 of the calendar year following the calendar year in which the performance period ends).
Effective as of the inception of the Bonus Plan, the Compensation Committee approved a special performance bonus opportunity under the Bonus Plan for Dr. Matthew David, interim Chief Executive Officer, Executive Vice President and Chief Financial Officer, Dr. Phoebe Mounts, Executive Vice President and General Counsel, and Ms. Liz Masson-Hurlburt, Executive Vice President and Head of Clinical Operations, to provide an incentive for the Company’s leadership team to accomplish specific performance objectives during a performance period beginning October 1, 2021 and ending March 31, 2022. The executives have an opportunity to earn a performance bonus of up to 30% of salary for Dr. Mounts and Ms. Masson-Hurlburt and up to 60% of salary for Dr. David based on attainment of key performance objectives, continued employment and compliance with restrictive covenants. With new leadership under Dr. David as interim Chief Executive Officer, the Compensation Committee determined that it was appropriate to provide specific targeted performance objectives tied to incentive payments to drive performance that is intended to support our long-term performance.
At the beginning of the performance year, the Board approves annual corporate goals and objectives, based on the recommendations of the Compensation Committee. The Board or Compensation Committee approves bonus awards, if any, for each Named Executive Officer based on the achievement of these pre-established corporate goals and such other factors as our Board or Compensation Committee deems appropriate, based on recommendations of the Compensation Committee. For any given performance year, proposed annual bonuses may range from 0% to 100% of target, or higher under certain circumstances. Corporate performance has a significant impact on the annual bonus amounts because the Compensation Committee and Board believe it is an appropriate measure of how the Named Executive Officer contributed to business results.
The Compensation Committee determined that it was appropriate to pay discretionary bonuses to Dr. David, Dr. Mounts, Ms. Masson-Hurlburt and Mr. Nusbickel based on individual performance and the challenges the Company had faced during 2021. Each of Dr. David, Dr. Mounts, Ms. Masson-Hurlburt and Mr. Nusbickel received discretionary bonuses of $75,900, $61,875, $37,800 and $75,000, respectively.
In 2021, we paid our Named Executive Officers annual bonuses equal to their target annual bonuses for 2020, and we paid Dr. Mounts and Ms. Masson-Hurlburt each a special bonus equal to two months of base salary on account of their work on the submission of the New Drug Application for DefenCath. The Board, based on the recommendation of the Compensation Committee, approved bonuses at these levels as a result of corporate and individual performance and the submission of the New Drug Application for DefenCath.
Long-Term Incentive Equity Awards
We believe that long-term performance is achieved through an ownership culture that encourages high performance by our Named Executive Officers through the use of stock-based awards. Our long-term incentive plans were established to provide our employees, including our Named Executive Officers, with incentives to help align employees’ interests with the interests of our stockholders. The Compensation Committee believes that the use of stock-based awards offers the best approach to achieving our long-term compensation goals. We have historically elected to use stock options as the primary long-term equity incentive vehicle; however, the Compensation Committee may in the future utilize other forms of equity grants as part of our long-term incentive program. We have selected the Black-Scholes method of valuation for share-based compensation. Due to the early stage of our business and our desire to preserve cash, we may provide a greater portion of total compensation to our Named Executive Officers through stock options and other equity grants than through cash-based compensation. The Compensation Committee generally oversees the administration of our equity plans.
Stock Options
Our 2019 Omnibus Stock Incentive Plan (the 2019 Plan), which was approved by the shareholders on November 26, 2019, authorizes us to grant options to purchase shares of our common stock and other equity awards to our employees, directors and consultants.
The Compensation Committee or the Board, based on Compensation Committee recommendations, makes stock option awards to Named Executive Officers based upon a review of competitive compensation data, its assessment of individual performance, a review of each Named Executive Officer’s existing long-term incentives, and retention considerations. Periodic stock option grants are made, or recommended to the Board, at the discretion of the Compensation Committee to eligible employees and, in appropriate circumstances, the Compensation Committee considers the recommendations of our Chief Executive Officer
Stock options granted to employees have an exercise price equal to the fair market value of our common stock on the day of grant, typically vest based on continued employment and, for performance-based grants, upon the achievement of certain performance-based milestones, and generally expire 10 years after the date of grant. The fair value of the options granted to the Named Executive Officers in the Summary Compensation Table is determined in accordance with the Black-Scholes method of valuation for share-based compensation. Incentive stock options also include certain other terms necessary to ensure compliance with the Code.
In 2021, the Board, based on the recommendation of the Compensation Committee, granted a mix of time-based and performance-based stock options to our Named Executive Officers. The time-based stock options vest annually in four increments while the executive remains employed by the Company. The performance-based stock options generally vest based upon achievement of performance milestones and continued employment. In January 2021, the Board granted 70,000 time-based stock options to Dr. Mounts, Mr. Armstrong, and Ms. Masson-Hurlburt, respectively, 40,000 time-based stock options to Dr. David, and 160,000 time-based stock options to Mr. Baluch, and granted the same number of performance-based stock options to each, respectively.
In November 2021, the Board, based on the recommendation of the Compensation Committee, granted additional stock options to Drs. David and Mounts, in order to recognize their increased responsibilities, including assuming the additional obligations associated with the interim Chief Executive Director role in the case of Dr. David, and overseeing the Company’s technical operations group, in the case of Dr. Mounts. Dr. David was granted a stock option with respect to 125,000 shares of our common stock and Dr. Mounts was granted a stock option with respect to 100,000 shares of our common stock, both with an exercise price of $5.56 per share, which was the closing price of our common stock on the Nasdaq Global Market on the date of grant. The options will vest over four years in four equal annual installments beginning on the date of grant, subject to Drs. David and Mounts’ continued employment, consistent with the terms of our standard form of option agreement.
In February 2021, the Compensation Committee amended outstanding time-based stock options held by our Named Executive Officers to extend the post-termination exercise periods with respect to such stock options that are vested as of the date of termination of employment: (i) from 90 days to 12 months following the date of termination in the event of an involuntary termination without Cause, a termination for Good Reason, death or disability and (ii) by implementing a new three-year post-termination exercise period following the date of termination of employment in the event of a termination by reason of retirement (i.e., termination after reaching age 62 with five years of continuous service or age 55 with ten years of continuous service), but not beyond the date of expiration of the term of the option in either case. The amendment did not apply to outstanding incentive stock option so as to not affect their tax status.
We expect to continue to use stock options as a long-term incentive vehicle because:
| ● | Stock options align the interests of our Named Executive Officers with those of our stockholders, supporting a pay-for performance culture, foster employee stock ownership, and focus the management team on increasing value for our stockholders. |
| | ● | Stock options are performance-based. All of the value received by the recipient of a stock option is based on the growth of the stock price. In addition, stock options can be issued with vesting based on the achievement of performance goals. |
| ● | Stock options help to provide balance to the overall executive compensation program as base salary and annual bonuses focus on short-term compensation, while the vesting of stock options increases stockholder value over the longer term. |
| ● | The vesting period of stock options encourages executive retention and the preservation of stockholder value. In determining the number of stock options to be granted to our Named Executive Officers, we take into account the individual’s position, scope of responsibility, ability to affect profits and stockholder value and the individual’s historic and recent performance and the value of stock options in relation to other elements of the individual Named Executive Officer’s total compensation. |
Executive Benefits and Perquisites
Our Named Executive Officers are parties to employment agreements as described below. In addition, consistent with our compensation philosophy, we intend to continue to maintain our current benefits for our Named Executive Officers, including medical, dental and life insurance and the ability to contribute to a 401(k) plan; however, the Compensation Committee in its discretion may revise, amend, or add to the officer’s executive benefits if it deems it advisable. We believe these benefits are currently comparable to benefit levels for comparable companies.
Employment Agreements
Employment Agreements with Current Named Executive Officers
On September 26, 2019, we entered into an employment agreement with Mr. Baluch, our former Chief Executive Officer. In connection with Mr. Baluch’s separation from service on October 4, 2021, we and Mr. Baluch entered into a separation agreement and release dated as of October 1, 2021 (the “Baluch Separation Agreement”). Mr. Baluch’s retirement was treated as a termination without Cause (as defined below) under the employment agreement, based on the circumstances of his retirement. Under the Baluch Separation Agreement, Mr. Baluch received the severance payments and benefits described in his employment agreement as follows: (i) lump sum payment of 60 days compensation, payment of any accrued compensation and any unpaid bonus for the prior year, as well as rights to indemnification and directors’ and officers’ liability insurance and any rights or privilege otherwise required by law; (ii) payment of base salary for a period of 12 months following October 4, 2021; (iii) payment on a prorated basis, if any, for the 2021 year, based on the actual achievement of the specified bonus objectives; (iv) if Mr. Baluch elected to continue health insurance coverage under COBRA, monthly payment of a portion of his COBRA premium for a period of 12 months following October 4, 2021 or until he became eligible for group health insurance coverage under another employer’s plan, whichever occurs first; and (v) all equity awards and stock options that are scheduled to vest on or before the next succeeding anniversary of the date of termination shall be accelerated and deemed to have vested as of the termination date, provided that any performance-based equity awards and stock options whose vesting requirements have not been successfully met as of the date of termination will not accelerate. Mr. Baluch met the eligibility requirements for retirement as of the date of his separation, so certain of Mr. Baluch’s vested stock options will be exercisable for up to three years after the date of his separation under the terms of the applicable grant agreements. The Baluch Separation Agreement provides this retirement treatment for all of Mr. Baluch’s outstanding vested options. We reimbursed Mr. Baluch for reasonable legal fees up to $20,000 incurred in connection with the review of the Baluch Separation Agreement. Mr. Baluch is bound by confidentiality, non-solicitation and non-competition covenants under his employment agreement, and an extended covenant not to solicit employees under the Baluch Separation Agreement, among other terms.
On April 17, 2020, we entered into an employment agreement with Mr. Armstrong, our former Executive Vice President for Technical Operations. In connection with Mr. Armstrong’s separation from service on October 4, 2021, we and Mr. Armstrong entered into a separation agreement and release dated as of October 4, 2021 (the “Armstrong Separation Agreement”). Mr. Armstrong’s retirement was treated as a termination without Cause under the employment agreement, based on the circumstances of his requirement. Under the Armstrong Separation Agreement, Mr. Armstrong received the severance payments and benefits described in his employment agreement are as follows: (i) lump sum payment of 60 days compensation, payment of any accrued compensation and any unpaid bonus for the prior year, as well as rights to indemnification and directors’ and officers’ liability insurance and any rights or privilege otherwise required by law; (ii) payment of base salary for a period of nine months following October 4, 2021; (iii) payment on a prorated basis, if any, for the 2021 year, based on the actual achievement of the specified bonus objectives; (iv) if Mr. Armstrong elected to continue health insurance coverage under COBRA, then monthly payment of a portion of his COBRA premium for a period of nine months following October 4, 2021 or until he became eligible for group health insurance coverage under another employer’s plan, whichever occurs first; and (v) all equity awards and stock options that are scheduled to vest on or before the next succeeding anniversary of the date of termination shall be accelerated and deemed to have vested as of the termination date, provided that any performance-based equity awards and stock options whose vesting requirements have not been successfully met as of the date of termination will not accelerate. Mr. Armstrong met the eligibility requirements for retirement as of the date of his separation, so certain of Mr. Armstrong’s vested stock options will be exercisable for up to three years after the date of his separation under the terms of the applicable grant agreements. The Armstrong Separation Agreement provides this retirement treatment for all outstanding vested options. We reimbursed Mr. Armstrong for reasonable legal fees up to $10,000 incurred in connection with the review of the Armstrong Separation Agreement. Mr. Armstrong is bound by confidentiality, non-solicitation and non-competition covenants under his employment agreement, and an extended covenant not to solicit employees under the Armstrong Separation Agreement, among other terms.
On March 10, 2021, we entered into a new employment agreement with Ms. Masson-Hurlburt to serve as our Executive Vice President and Head of Clinical Operations. On March 19, 2019, we entered into an employment agreement with Dr. Mounts to serve as our Executive Vice President and General Counsel and Head of Regulatory, Compliance and Legal, effective May 1, 2019. On April 29, 2021, we entered into an employment agreement with Mr. Nusbickel to serve as our Executive Vice President and Chief Commercial Officer, effective May 13, 2021. After the initial three-year term of each employment agreement, the term of the employment agreement will automatically renew for additional successive one-year periods, unless either party notifies the other in writing at least 90 days before the expiration of the then-current term that the term will not be renewed.
On May 11, 2020, we entered into an employment agreement with Dr. David to serve as our Chief Financial Officer. After the initial three-year term of the employment agreement, the term of the employment agreement will automatically renew for additional successive one-year periods, unless either party notifies the other in writing at least 90 days before the expiration of the then-current term that the term will not be renewed. On October 26, 2021, we entered into a letter agreement with Dr. David which modified certain terms of his employment agreement and provided other compensation as a result of Dr. David serving as our interim Chief Executive Officer effective as of October 4, 2021. Pursuant to the letter agreement, during the period in which Dr. David serves as interim Chief Executive Officer, his base salary was increased to $425,000 from $330,000, which is the amount set forth in his employment agreement. Should Dr. David continue to serve as the interim Chief Executive Officer for six months after October 4, 2021, the Board or Compensation Committee will review such base salary to determine whether an increase is appropriate at that time. After Dr. David ceases to serve as interim Chief Executive Officer, and as he continues to serve as Chief Financial Officer, we will provide him with an annual base salary of $375,000, representing a $45,000 increase from his current salary level under the employment agreement. The Board or Compensation Committee will review such base salary to determine whether an increase is appropriate in 2022 as part of the 2022 compensation review cycle and benchmarking review. Under the letter agreement, Dr. David’s target annual bonus with respect to the period during which he serves as interim Chief Executive Officer is increased to 60% from 30% of his base salary. After Dr. David ceases to serve as interim Chief Executive Officer, and as he continues to serve as Chief Financial Officer, his target annual bonus will increase to 40% of his base salary. Under the letter agreement, in the event Dr. David’s employment is terminated by us other than as a result of his death or disability or notice of nonrenewal of the employment agreement, and other than for Cause, or if he resigns for Good Reason, in either case during the period he serves as interim Chief Executive Officer, he will be eligible for severance equal to his base salary for a period of 12 months following his termination date, which is increased from nine months as is otherwise provided for in his employment agreement. Dr. David has agreed to waive any rights he may have under his employment agreement to a Good Reason termination as a result of his ceasing to serve as our interim Chief Executive Officer at a future date. In connection with Dr. David serving as interim Chief Executive Officer, the Board granted Dr. David a stock option with respect to 125,000 shares of our common stock with an exercise price of $5.56 per share, which was the closing price of our common stock on the Nasdaq Global Market on the date of grant. The option will vest over four years in four equal annual installments beginning on the date of grant, subject to Dr. David’s continued employment, consistent with the terms of our standard form of option agreement.
Pursuant to their respective employment agreements, Mr. Baluch received an annual salary of $425,000, Mr. Armstrong received an annual salary of $325,000, Ms. Masson-Hurlburt receives an annual salary of $315,000 (effective March 2021), Dr. Mounts receives an annual salary of $350,000 (amended to $375,000 in January 2021), Mr. Nusbickel receives and annual salary of $375,000 and Dr. David receives an annual salary of $330,000 (amended to $425,000 while he serves as interim Chief Executive Officer). Such salaries cannot be decreased unless all officers and/or members of our executive management team experience an equal or greater percentage reduction in base salary and/or total compensation, provided that any reduction in an executive’s salary may be no greater than 25%. Messrs. Baluch and Armstrong were eligible for an annual bonus, of up to 80% of his base salary for Mr. Baluch (the target amount is 80%, but the bonus could exceed that amount) and up to 35% of his base salary for Mr. Armstrong, as determined by our Board or the Compensation Committee. Each other executive will be eligible for an annual bonus of up to 30% for Ms. Masson-Hurlburt, up to 30% for Dr. Mounts, up to 30% for Mr. Nusbickel and up to 30% for Dr. David (and up to 60% while he serves as interim Chief Executive Officer), of his or her base salary then in effect, as determined by our Board or the Compensation Committee. In determining such bonus payment, our Board or the Compensation Committee will take into consideration the achievement of specified Company objectives, predetermined by our Board or the Compensation Committee and Chief Executive Officer, and such other factors as our Board or the Compensation Committee deems appropriate. Each executive generally must be employed through December 31 of a given year to be eligible to earn that year’s annual bonus.
The following provisions of the employment agreements with Dr. David, Dr. Mounts, Ms. Masson-Hurlburt and Messrs. Nusbickel, Baluch and Armstrong are identical except where noted.
If we terminate the executive’s employment for Cause, the executive will be entitled to receive only the accrued compensation due to him or her as of the date of such termination, rights to indemnification and directors’ and officers’ liability insurance, and as otherwise required by law, and certain equity awards will be forfeited.
If we terminate the executive’s employment other than for Cause, and other than for death, disability or notice of nonrenewal, or if the executive resigns for Good Reason (as defined below), the executive will receive the following benefits: (i) payment of any accrued compensation and any unpaid bonus relating to the completed prior year, as well as rights to indemnification and directors’ and officers’ liability insurance and any rights or privilege otherwise required by law; (ii) we will continue to pay the executive’s base salary for a period of twelve months in the case of Mr. Baluch, and in the case of Dr. David while he is serving as interim Chief Executive Officer, following termination of employment and nine months for the other executives, and for Dr. David while he is not serving as interim Chief Executive Officer, following termination of employment; (iii) payment on a prorated basis for any target bonus for the year of termination based on the actual achievement of the specified bonus objectives; (iv) if the executive timely elects continued health insurance coverage under COBRA, then we will pay the premium to continue such coverage for him or her and his or her eligible dependents in an amount equal to the portion paid for by us during the executive’s employment until the conclusion of the time when he or she is receiving continuation of base salary payments or until he or she becomes eligible for group health insurance coverage under another employer’s plan, whichever occurs first, provided however that we have the right to terminate such payment of COBRA premiums on behalf of the executive and instead pay him or her a lump sum amount equal to the COBRA premium times the number of months remaining in the specified period if we determine in our discretion that continued payment of the COBRA premiums is or may be discriminatory under Section 105(h) of the Code; and (v) unvested equity awards that are scheduled to vest on or before the next succeeding anniversary of the date of termination shall be accelerated and deemed to have vested as of the termination date; provided that any performance based equity awards or stock options whose vesting requirements have not been successfully met as of the date of termination of employment or resignation with Good Reason will not accelerate. In addition, the event of a termination by the Company without Cause or the executive’s resignation of employment for Good Reason, in either case within 24 months following a Corporate Transaction (as defined in the employment agreement), all equity awards and stock options shall become fully vested and exercisable, and vested stock options will remain exercisable for a specified period of time following termination or resignation or, if earlier, the expiration date of the stock option. The separation benefits set forth above are conditioned upon the executive executing a release of claims against us, our parents, subsidiaries, and affiliates, and each such entities’ officers, directors, employees, agents, successors, and assigns in a form acceptable to us, within a time specified therein, which release is not revoked within any time period allowed for revocation under applicable law.
For purposes of the agreement, “Cause” is defined as: (i) the willful failure, disregard, or refusal by the executive to perform his or her material duties or obligations under the employment agreement (other than as a result of executive’s mental incapacity or illness; (ii) any willful, intentional, or grossly negligent act by the executive having the effect of materially injuring (whether financially or otherwise) our business or reputation or any of our affiliates; (iii) executive’s conviction of any felony involving moral turpitude (including entry of a guilty or nolo contendere plea); (iv) the executive’s qualification as a “bad actor,” as defined by 17 CFR 230.506(a); (v) the good faith determination by the Board, after a reasonable and good-faith investigation by us that the executive engaged in some form of harassment or discrimination prohibited by law (including, without limitation, harassment on the basis of age, sex or race) unless the executive’s actions were specifically directed by the Board; (vi) any material misappropriation or embezzlement by the executive of our or our affiliates’ property (whether or not a misdemeanor or felony); or (vii) material breach by the executive of the employment agreement that is materially injurious to us and that is not cured, to the extent subject to cure, by executive to our reasonable satisfaction.
For purposes of the agreement, “Good Reason” is defined as any of the following without the executive’s consent: (i) any material breach of the employment agreement by us; (ii) any material diminution by us of the executive’s duties, responsibilities, or authority; (iii) a material reduction in the executive’s annual base salary unless all officers and/or members of our executive management team experience an equal or greater percentage reduction in annual base salary and/or total compensation, provided that any reduction may be no greater than 25%; (iv) a material reduction in the executive’s target bonus level unless all officers and/or members of our executive management team experience an equal or greater percentage reduction related to target bonus levels, provided that any reduction may be no greater than 25%.
If the executive terminates his or her employment by written notice of termination or if the executive or we terminate his or her employment by providing a notice of nonrenewal at least 90 days before the employment agreement is set to expire, the executive will not be entitled to receive any payments or benefits other than any accrued compensation, any unpaid prior year’s bonus, rights to indemnification and directors’ and officers’ liability insurance and as otherwise required by law.
If the executive’s employment is terminated as a result of his or her death or disability, we will pay the executive or the executive’s estate, as applicable, any accrued compensation and any unpaid prior year’s bonus.
Our employment agreements with Dr. David, Dr. Mounts, Ms. Masson-Hurlburt and Messrs. Nusbickel, Baluch and Armstrong each contain a non-compete provision that provides that during the employment and for a specified period immediately following the executive’s separation from employment for any reason, the executive is prohibited from engaging in any business involving the development or commercialization of a preventive anti-infective product that would be a direct competitor of Defencath/Neutrolin or a product containing taurolidine or any other product being actively developed or produced by us within the United States and the European Union (or in the case of Dr. David, Ms. Masson-Hurlburt and Mr. Nusbickel, worldwide) on the date of termination of his or her employment.
Tax and Accounting Considerations
U.S. federal income tax generally limits the tax deductibility of compensation we pay to our Named Executive Officers and certain other officers to $1.0 million each in the year the compensation becomes taxable to the executive officers. Although deductibility of compensation is preferred, tax deductibility is not a primary objective of our compensation programs. Rather, we seek to maintain flexibility in how we compensate our executive officers so as to meet a broader set of corporate and strategic goals and the needs of stockholders, and as such, we may be limited in our ability to deduct amounts of compensation from time to time. Accounting rules require us to expense the cost of our stock option grants. Because of option expensing and the impact of dilution on our stockholders, we pay close attention to, among other factors, the type of equity awards we grant and the number and value of the shares underlying such awards.
Pension Benefits
We do not maintain any qualified or nonqualified defined benefit pension plans. As a result, none of our Named Executive Officers participate in or have benefits under qualified or nonqualified defined benefit pension plans sponsored by us. Our Compensation Committee may elect to adopt qualified or nonqualified pension benefit plans in the future if it determines that doing so is in our best interests.
Nonqualified Deferred Compensation
None of our Named Executive Officers participate in nonqualified defined contribution plans or other nonqualified deferred compensation plans maintained by us. Our Compensation Committee may elect to provide our officers and other employees with nonqualified deferred compensation benefits in the future if it determines that doing so is in our best interests.
Summary Compensation Table
The following table sets forth information with respect to compensation earned by our Named Executive Officers in the years ended December 31, 2021 and 2020:
| Name and Principal Position | | Year | | | Salary
($) | | | Option
Awards
(1)
($) | | | Non-equity
Incentive Plan
Compensation
($)
(4) | | | All Other
Compensation
($) | | | Total
($) | |
| Matthew David (2) | | | 2021 | | | | 351,923 | | | | 951,895 | | | | 165,150 | | | | 43,584 | (5) | | | 1,512,552 | |
| Interim Chief Executive Officer and Chief Financial Officer | | | 2020 | | | | 209,423 | | | | 768,386 | | | | 99,000 | | | | 20,866 | (5) | | | 1,097,675 | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Phoebe Mounts | | | 2021 | | | | 375,000 | | | | 1,164,810 | | | | 101,250 | | | | 11,412 | (5) | | | 1,652,472 | |
| Executive Vice President and General Counsel and Head of Regulatory, Compliance and Legal | | | 2020 | | | | 350,000 | | | | 408,070 | | | | 163,333 | | | | 9,745 | (5) | | | 931,148 | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Elizabeth Masson-Hurlburt | | | 2021 | | | | 310,800 | | | | 742,910 | | | | 70,875 | | | | 44,818 | (5) | | | 1,169,403 | |
| Executive Vice President and Head of Clinical Operations | | | 2020 | | | | 291,792 | | | | 332,420 | | | | 136,500 | | | | 35,561 | (5) | | | 796,273 | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Thomas Nusbickel (3) | | | 2021 | | | | 233,654 | (3) | | | 1,540,265 | | | | 75,000 | | | | 27,663 | (5) | | | 1,876,582 | |
| Executive Vice President and Chief Commercial Officer | | | | | | | | | | | | | | | | | | | | | | | | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| Khoso Baluch (6) | | | 2021 | | | | 366,700 | | | | 1,698,080 | (6) | | | -- | | | | 531,393 | (7) | | | 2,596,173 | |
| Former Chief Executive Officer | | | 2020 | | | | 425,000 | | | | 428,583 | | | | 340,000 | | | | 40,088 | (10) | | | 1,233,671 | |
| | | | | | | | | | | | | | | | | | | | | | | | | |
| John Armstrong (8) | | | 2021 | | | | 272,500 | | | | 742,910 | (8) | | | -- | | | | 323,938 | (9) | | | 1,339,348 | |
| Former Executive Vice President for Technical Operations | | | 2020 | | | | 322,635 | | | | 332,420 | | | | 112,875 | | | | 19,388 | (11) | | | 787,318 | |
| (1) | The amounts included in this column are the dollar amounts representing the full grant date fair value of each award calculated in accordance with FASB ASC Topic 718 and do not represent the actual value that may be recognized by the Named Executive Officers upon option exercise. |
| (2) | Dr. David became our Executive Vice President and Chief Financial Officer on May 11, 2020 and is also serves as the interim Chief Executive Officer effective October 4, 2021. |
| (3) | Mr. Nusbickel became our Executive Vice President and Chief Commercial Officer on May 13, 2021. His salary does not include the sign-in bonus in cash amounting to $50,000, to be paid on the first anniversary of his employment, subject to his continued employment with us. |
| (4) | The non-equity incentive plan compensation are bonuses reflected in 2021 were for the performance for the year 2021 which were accrued in 2021 but will be paid in 2022. |
| (5) | Consists of health benefits and 401(k) employer match. |
| (6) | On October 1, 2021, the Company and Khoso Baluch came to a mutual agreement pursuant to which Mr. Baluch retired from his position as the Company’s Chief Executive Officer, effective October 4, 2021. Option awards include 240,000 options that were forfeited when he retired, with a grant date fair value of $1,196,240. |
| | (7) | Consists of health benefits, 401(k) employer match and severance pay of $495,833 of which $177,492 was paid in 2021 and the remaining balance of $318,341 will be paid in 2022. |
| (8) | On October 4, 2021, the Company and John Armstrong came to a mutual agreement pursuant to which Mr. Armstrong retired from his position as the Company’s Executive Vice President, Technical Operations, effective October 4, 2021. Option awards include 105,000 options that were forfeited when he retired, with a grant date fair value of $523,355. |
| | (9) | Consists of health benefits and severance pay of $297,917 of which $134,514 was paid in 2021 and the remaining balance of $163,403 will be paid in 2022. |
| (10) | Consists of health benefits, 401(k) employer match, and reimbursed commuter expenses |
| (11) | Consists of health benefits. |
Outstanding Equity Awards at Fiscal Year-End 2021
The following table contains certain information concerning unexercised options for the Named Executive Officers as of December 31, 2021.
| Name | | Number of
Shares
Underlying
Unexercised
Options (#) –
Exercisable | | | Number of
Shares
Underlying
Unexercised
Options (#) –
Unexercisable (1) | | | Equity
Incentive Plan
Awards:
Number of
Shares
Underlying
Unexercised
Unearned
Options # (2) | | | Option
Exercise
Price ($) | | | Option
Expiration
Date |
| Matthew David | | | 42,917 | | | | 62,250 | | | | 19,833 | | | | 5.63 | | | 05/11/2030 |
| | | | 42,917 | | | | 62,250 | | | | 19,833 | | | | 4.08 | | | 05/11/2030 |
| | | | 10,000 | | | | 30,000 | | | | 40,000 | | | | 8.32 | | | 01/10/2031 |
| | | | 31,250 | | | | 93,750 | | | | -- | | | | 5.56 | | | 10/31/2031 |
| | | | | | | | | | | | | | | | | | | |
| Phoebe Mounts | | | 39,000 | | | | 21,000 | | | | 10,000 | | | | 7.92 | | | 05/01/2029 |
| | | | 12,382 | | | | 12,382 | | | | -- | | | | 5.63 | | | 02/25/2030 |
| | | | 25,000 | | | | 25,000 | | | | -- | | | | 4.08 | | | 05/11/2030 |
| | | | 25,000 | | | | 25,000 | | | | -- | | | | 5.63 | | | 05/11/2030 |
| | | | 17,500 | | | | 52,500 | | | | 70,000 | | | | 8.32 | | | 01/10/2031 |
| | | | 25,000 | | | | 75,000 | | | | -- | | | | 5.56 | | | 10/31/2031 |
| | | | | | | | | | | | | | | | | | | |
| Elizabeth Masson-Hurlburt | | | 44,700 | | | | 9,300 | | | | -- | | | | 1.45 | | | 3/19/2028 |
| | | | 18,780 | | | | 2,100 | | | | -- | | | | 8.30 | | | 01/10/2029 |
| | | | 12,382 | | | | 12,382 | | | | -- | | | | 5.63 | | | 02/25/2030 |
| | | | 18,750 | | | | 18,750 | | | | -- | | | | 4.08 | | | 05/11/2030 |
| | | | 18,750 | | | | 18,750 | | | | -- | | | | 5.63 | | | 05/11/2030 |
| | | | 17,500 | | | | 52,500 | | | | 70,000 | | | | 8.32 | | | 01/10/2031 |
| | | | | | | | | | | | | | | | | | | |
| Thomas Nusbickel | | | -- | | | | 167,500 | | | | 100,000 | | | | 7.56 | | | 05/12/2031 |
| | | | | | | | | | | | | | | | | | | |
| Khoso Baluch | | | 310,000 | | | | -- | | | | -- | | | | 12.60 | | | 10/04/2024 |
| | | | 69,600 | | | | -- | | | | -- | | | | 8.30 | | | 10/04/2024 |
| | | | 90,000 | | | | -- | | | | -- | | | | 6.82 | | | 10/04/2024 |
| | | | 75,472 | | | | -- | | | | -- | | | | 5.63 | | | 10/04/2024 |
| | | | 80,000 | | | | -- | | | | -- | | | | 8.32 | | | 10/04/2024 |
| | | | | | | | | | | | | | | | | | | |
| John Armstrong | | | 2,000 | | | | -- | | | | -- | | | | 7.60 | | | 10/04/2024 |
| | | | 3,000 | | | | -- | | | | -- | | | | 16.25 | | | 10/04/2024 |
| | | | 40,000 | | | | -- | | | | -- | | | | 12.55 | | | 10/04/2024 |
| | | | 6,600 | | | | -- | | | | -- | | | | 10.90 | | | 10/04/2024 |
| | | | 7,830 | | | | -- | | | | -- | | | | 8.30 | | | 10/04/2024 |
| | | | 46,698 | | | | -- | | | | -- | | | | 5.63 | | | 10/04/2024 |
| | | | 28,125 | | | | -- | | | | -- | | | | 4.08 | | | 10/04/2024 |
| | | | 35,000 | | | | -- | | | | -- | | | | 8.32 | | | 10/04/2024 |
| (1) | Options vest based on continued employment over three or four years. |
| (2) | Options vest based on achievement of specific milestones and continued employment and become exercisable if and when a milestone is achieved. |
Option Repricings
We did not engage in any repricings or other modifications to any of our Named Executive Officers’ outstanding options during the year ended December 31, 2021.
Potential Payments on a Qualifying Termination
If the severance payments called for in our employment agreements for Dr. David, Dr. Mounts, Ms. Masson-Hurlburt and Mr. Nusbickel had been triggered on December 31, 2021, we would have been obligated to make the following payments:
| Name | | Cash Severance
Payment
($ per month) and
(# of months paid) | | | Severance Benefits
($ per month) and
(# of months paid) (1) | | | Number of Options
(# that would vest) and
($ market value) (2) | |
| Matthew David | | $ | 35,417 | (3) | | | 12 mos. | | | $ | 2,915 | | | | 12 mos. | | | | 62,250 | | | $ | 29,258 | |
| Phoebe Mounts | | $ | 31,250 | (4) | | | 9 mos. | | | $ | 0 | | | | 9 mos. | | | | 25,000 | | | $ | 11,750 | |
| Elizabeth Masson-Hurlburt | | $ | 26,250 | (5) | | | 9 mos. | | | $ | 2,915 | | | | 9 mos. | | | | 28,050 | | | $ | 37,643 | |
| Thomas Nusbickel | | $ | 31,250 | (6) | | | 9 mos. | | | $ | 2,882 | | | | 9 mos. | | | | 0 | | | $ | 0 | |
| (1) | Consists of COBRA payments. |
| (2) | The market value equals the difference between the fair market value of the shares that could be acquired based on the closing sale price per share of our common stock on the Nasdaq Global Market on December 31, 2021, which was $4.55, and the exercise prices of the applicable stock options. |
| (3) | Represents severance based on monthly base salary, payable for 12 months. Any bonus for the year of termination based on performance would also be paid. |
| (4) | Represents severance based on monthly base salary, payable for 9 months. Any bonus for the year of termination based on performance would also be paid. |
| (5) | Represents severance based on monthly base salary, payable for 9 months. Any bonus for the year of termination based on performance would also be paid. |
| (6) | Represents severance based on monthly base salary, payable for 9 months. Any bonus for the year of termination based on performance would also be paid. |
| (7) | Represents severance based on monthly base salary, payable for 9 months. Any bonus for the year of termination based on performance would also be paid. |
The severance payments called for in the employment agreements with Messrs. Baluch and Armstrong were both triggered on October 4, 2021, and the Company was obligated to make the following payments pursuant to their respective separation agreements:
| Name | | Cash Severance
Payment
($ per month) and
(# of months paid) | | | Severance Benefits
($ per month) and
(# of months paid) (1) | | | Number of Options
(# that vested) and
($ market value) (2 ) | |
| Khoso Baluch(3) | | $ | 35,417 | (5) | | | 12 mos. | | | $ | 2,016 | | | | 12 mos. | | | | 0 | | | $ | 0 | |
| John L. Armstrong, Jr.(4) | | $ | 27,083 | (5) | | | 9 mos. | | | $ | 2,081 | | | | 9 mos. | | | | 28,125 | | | $ | 13,219 | |
| (1) | Consists of COBRA payments. |
| (2) | The market value equals the difference between the fair market value of the shares that could be acquired based on the closing sale price per share of our common stock on the Nasdaq Global Market on December 31, 2021, which was $4.55, and the exercise prices of the applicable stock options. |
| (3) | We entered into a Separation Agreement with Mr. Baluch on October 4, 2021, and pursuant to that agreement, we paid Mr. Baluch an additional lump sum payment of $70,833 which represented base salary for 60 days as pay in lieu of notice. |
| (4) | We entered into a Separation Agreement with Mr. Armstrong on October 4, 2021, and pursuant to that agreement, we paid Mr. Armstrong an additional lump sum payment of $54,167 which represented base salary for 60 days as pay in lieu of notice. |
| (5) | Represents severance pay based on monthly base salary. |
Item 12. Security Ownership of Certain Beneficial Owners and Management and Related Stockholder Matters
The following table shows the number of shares of our common stock beneficially owned as of March 25, 2022 by:
| | ● | each person known by us to own beneficially more than 5% of the outstanding shares of our common stock; |
| | | |
| | ● | each director; |
| | | |
| | ● | each of our Named Executive Officers; and |
| | | |
| | ● | all of our current directors and executive officers as a group. |
This table is based upon the information supplied by our Named Executive Officers, directors and principal stockholders and from Schedules 13D and 13G filed with the SEC. Except as indicated in footnotes to this table, the persons named in this table have sole voting and investment power with respect to all shares of common stock shown, and their address is c/o CorMedix Inc., 300 Connell Drive, Suite 4200, Berkeley Heights, New Jersey 07922. As March 25, 2022 we had 38,727,979 shares of common stock outstanding. Beneficial ownership in each case also includes shares issuable upon exercise of outstanding options that can be exercised within 60 days after March 25, 2022 for purposes of computing the percentage of common stock owned by the person named. Options owned by a person are not included for purposes of computing the percentage owned by any other person.
| Name and Address of Beneficial Owner | | Common Stock
Beneficially Owned (1) | |
| | | Shares | | | % | |
| 5% or Greater Stockholders | | | | | | |
| Elliott Associates, L.P. (2) | | | 1,303,411 | | | | 4.99 | % |
| BlackRock, Inc. (3) | | | 2,500,721 | | | | 6.6 | % |
| Nomura Global Financial Products, Inc. (4) | | | 2,003,612 | | | | 5.3 | % |
| | | | | | | | | |
| Directors: | | | | | | | | |
| Paulo F. Costa (5) | | | 42,083 | | | | * | |
| Janet Dillione (6) | | | 163,473 | | | | * | |
| Gregory Duncan (7) | | | 40,833 | | | | * | |
| Alan W. Dunton (8) | | | 83,750 | | | | * | |
| Myron Kaplan (9) | | | 261,034 | | | | * | |
| Steven Lefkowitz (10) | | | 180,650 | | | | * | |
| Joseph Todisco | | | 0 | | | | * | |
| | | | | | | | | |
| Named Executive Officers: | | | | | | | | |
| Matthew David (11) | | | 206,734 | | | | * | |
| Phoebe Mounts (12) | | | 245,273 | | | | * | |
| Elizabeth Masson-Hurlburt (13) | | | 209,703 | | | | * | |
| Thomas Nusbickel (14) | | | 72,500 | | | | * | |
| Khoso Baluch (15) | | | 685,977 | | | | 1.7 | |
| John Armstrong (16) | | | 266,131 | | | | * | |
| | | | | | | | | |
| All Executive Officers and directors as a group (11 persons) (17) | | | 1,506,033 | | | | 3.8 | % |
* Less than 1%
| (1) | Based upon 38,727,979 shares of our common stock outstanding on March 25, 2022 and, with respect to each individual holder, rights to acquire our common stock exercisable within 60 days of March 25, 2022. |
| (2) | Based solely on information contained in Amendment No. 1 to the Statement on Schedule 13D filed with the SEC on February 11, 2021 by Elliott Associates, L.P. (“Elliott Associates”), Elliott International, L.P. (“Elliott International”) and Elliott International Capital Advisors Inc. (“Elliott International Capital Advisors”, and together with Elliott Associates and Elliott International, the “Elliott Reporting Entities”), the investment manager of Elliott International, and other information known to us. The Elliott Reporting Entities may be deemed to collectively beneficially own 7,136,979 shares of our common stock, including (i) 1,184,889 shares of common stock outstanding, (ii) 391,953 shares of common stock issuable upon conversion of the Series E preferred stock, and (iii) 5,560,137 shares of common stock issuable upon conversion of the Series G preferred stock. The number of shares of our common stock into which the Series E and Series G preferred stock are convertible into, as applicable, are limited pursuant to the terms of the convertible securities to that number of shares of our common stock which would result in the Elliott Reporting Entities having aggregate beneficial ownership of not more than 4.99% (calculated in accordance with Rule 13d-4 under the Exchange Act) of the total issued and outstanding shares of our common stock (the “Ownership Limitation”). The Elliott Reporting Entities disclaim beneficial ownership of any and all shares of our common stock issuable upon any conversion of the convertible securities if such conversion would cause the Elliott Reporting Entities aggregate beneficial ownership of our common stock to exceed or remain above the Ownership Limitation. Therefore, the Elliott Reporting Entities disclaim beneficial ownership of any shares of our common stock, issuable upon any conversion of the Series E preferred stock and the Series G preferred stock, which conversion would be prohibited by the Ownership Limitation. The Ownership Limitation does not prevent the Elliott Reporting Entities or their affiliates from voting the shares of Series E and Series G preferred stock held by Elliott Associates and Elliott International. Accordingly, the shares of Series E preferred stock and Series G preferred stock, as of the record date, will be entitled to an aggregate of 2,682,477 votes. The business address of Elliott Investment Management L.P., the investment manager of the Elliott Reporting Entities, is 40 West 57th Street, 30th Floor, New York, New York 10019. |
| (3) | Based solely on information contained in Amendment No. 1 to the Statement on Schedule 13G filed with the SEC on February 3, 2022 by BlackRock, Inc. (“BlackRock”). BlackRock has the sole voting power with respect to 2,450,933 shares of our common stock and the sole dispositive power with respect to 2,500,721 shares of our common stock. The business address of BlackRock is 55 East 52nd Street, New York, New York 10055. |
| (4) | Based solely on information contained on Schedule 13G filed with the SEC on February 14, 2022 by Nomura Global Financial Products, Inc. (“NGFP”). NGFP is a wholly owned subsidiary of Nomura Holdings, Inc., which accordingly may be deemed to beneficially own the shares beneficially owned by NGFP. NGFP has the shared voting power with respect to 2,003,612 shares of our common stock and the shared dispositive power with respect to 2,003,612 shares of our common stock. The business address of NGFP is Worldwide Plaza, 309 West 49th Street, New York, NY 10019. The business address of Nomura Holdings, Inc. is 13-1, Nihonbashi 1-chome, Chuo-ku, Tokyo 103-8645, Japan. |
| (5) | Consists of 42,083 shares of our common stock issuable upon exercise of stock options. |
| (6) | Consists of (i) 53,473 shares of our common stock, and (ii) 110,000 shares of our common stock issuable upon exercise of stock options. Ms. Dillione also holds 48,909 shares of common stock deferred under Director’s Compensation Plan, which is excluded for purposes of calculating the number of shares of our common stock beneficially owned as of March 25, 2022. |
| (7) | Consists of 40,833 shares of our common stock issuable upon exercise of stock options. |
| (8) | Consists of (i) 6,250 shares of our common stock, and (ii) 77,500 shares of our common stock issuable upon exercise of stock options. |
| (9) | Consists of (i) 140,034 shares of our common stock held directly, (ii) 30,000 shares of our common stock held by Mr. Kaplan’s wife, 20,000 of which are held by her individually and 10,000 of which are held as a custodian for two of Mr. Kaplan’s grandchildren, and (iii) 91,000 shares of our common stock issuable upon exercise of stock options. |
| (10) | Consists of (i) 60,498 shares of our common stock held directly, (ii) 2,000 shares of our common stock held by Mr. Lefkowitz’s wife, (iv) 30,152 shares of our common stock held by Wade Capital Corporation Money Purchase Plan, an entity for which Mr. Lefkowitz has voting and investment control, and (v) 88,000 shares of our common stock issuable upon exercise of stock options. |
| (11) | Consists of (i) 3,150 shares of our common stock, and (ii) 203,584 shares of our common stock issuable upon exercise of stock options. |
| (12) | Consists of (i) 7,200 shares of our common stock, and (ii) 238,073 shares of our common stock issuable upon exercise of stock options. |
| (13) | Consists of 209,703 shares of our common stock issuable upon exercise of stock options. |
| (14) | Consists of 72,500 shares of our common stock issuable upon exercise of stock options. |
| (15) | Consists of (i) 60,905 shares of our common stock, and (ii) 625,072 shares of our common stock issuable upon exercise of stock options. On October 1, 2021, we came to a mutual agreement with Mr. Baluch pursuant to which, Mr. Baluch retired from his position as our Chief Executive Officer, effective October 4, 2021. Mr. Baluch also resigned from our Board of Directors. |
| (16) | Consists of (i) 96,878 shares of our common stock, and (ii) 169,253 shares of our common stock issuable upon exercise of stock options. On October 4, 2021, we came to a mutual agreement with Mr. Armstrong pursuant to which Mr. Armstrong retired from his position as our Executive Vice President, Technical Operations, effective October 4, 2021. |
| (17) | Consists of the following held by our directors and executive officers (A) 302,757 shares of our common stock, and (B) 1,173,276 shares of our common stock issuable upon exercise of stock options. |
Stock Performance Graph
The following performance graph shall not be deemed to be “soliciting material” or “filed” or incorporated by reference in future filings with the SEC, or subject to the liabilities of Section 18 of the Exchange Act except as shall be expressly set forth by specific reference in such filing. The performance graph compares the performance of our common stock to the Russel 2000 Index and the NASDAQ Biotechnology Index. The graph covers the most recent five-year period ended December 31, 2021. The graph assumes that the value of the investment in our common stock and each index was $100.00 at December 31, 2016, and that all dividends are reinvested.
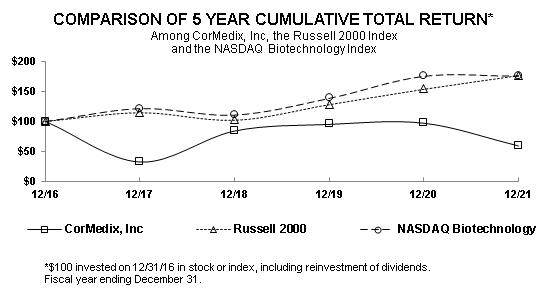
| | | Cumulative Total Return | |
| | | 12/2016 | | | 12/2017 | | | 12/2018 | | | 12/2019 | | | 12/2020 | | | 12/2021 | |
| CorMedix Inc. | | $ | 100.00 | | | $ | 32.81 | | | $ | 84.31 | | | $ | 95.16 | | | $ | 97.12 | | | $ | 59.48 | |
| Russell 2000 | | $ | 100.00 | | | $ | 114.65 | | | $ | 102.02 | | | $ | 128.06 | | | $ | 153.62 | | | $ | 176.39 | |
| NASDAQ Biotechnology | | $ | 100.00 | | | $ | 121.63 | | | $ | 110.85 | | | $ | 138.69 | | | $ | 175.33 | | | $ | 175.37 | |
Item 13. Certain Relationships and Related Transactions and Director Independence
Related Party Transactions
In February 2021, Manchester Securities Corp., Elliott Associates LP and Elliott International LP (collectively, “Elliott”), an existing institutional investor who collectively beneficially own the largest portion of the Company’s common stock, converted an aggregate of 10,001 Series G preferred shares into an aggregate of 556,069 shares of our common stock.
Procedures for Review and Approval of Transactions with Related Persons
Pursuant to the Audit Committee Charter, the Audit Committee is responsible for reviewing and approving all related party transactions as defined under Item 404 of Regulation S-K, after reviewing each such transaction for potential conflicts of interests and other improprieties. Our policies and procedures for review and approval of transactions with related persons are in writing in our Code of Conduct and Ethics available on our website at www.cormedix.com under the “Investor Relations—Corporate Governance” tab.
The information on Board independence is found in Item 10 of this Report under the heading “Board Independence.”
Item 14. Principal Accounting Fees and Services
Fees Paid to the Independent Registered Public Accounting Firm
The following table sets forth fees billed to us by Friedman LLP, our independent registered public accounting firm for the years ended December 31, 2021 and 2020, for services relating to: auditing our annual financial statements; reviewing our financial statements included in our quarterly reports on Form 10-Q; reviewing registration statements during 2021 and 2020; financing activities in 2021 and 2020; and services rendered in connection with tax compliance, tax advice and tax planning, and all other fees for services rendered.
| | | 2021 | | | 2020 | |
| Audit Fees | | $ | 155,000 | | | $ | 153,000 | |
| Audit Related Fees | | | 16,000 | | | | 37,000 | |
| Tax Fees | | | - | | | | - | |
| All Other Fees | | | - | | | | - | |
| Total | | $ | 171,000 | | | $ | 190,000 | |
Audit Committee Pre-Approval Policies and Procedures
Pursuant to its charter, the Audit Committee is responsible for reviewing and approving in advance any audit and any permissible non-audit engagement or relationship between us and our independent registered public accounting firm. The Audit Committee may delegate to one or more designated members of the Audit Committee the authority to grant pre-approvals, provided such approvals are presented to the Audit Committee at a subsequent meeting. If the Audit Committee elects to establish pre-approval policies and procedures regarding non-audit services, the Audit Committee must be informed of each non-audit service provided by our independent registered public accounting firm. Audit Committee pre-approval of audit and non-audit services will not be required if the engagement for the services is entered into pursuant to pre-approval policies and procedures, provided the policies and procedures are detailed as to the particular service, the Audit Committee is informed of each service provided and such policies and procedures do not include delegation of the Audit Committee’s responsibilities under the Exchange Act to our management. Audit Committee pre-approval of non-audit services (other than review and attestation services) also will not be required if such services fall within available exceptions established by the SEC. All services performed by our independent registered public accounting firm during 2021 were pre-approved by the Audit Committee.
PART IV
Item 15. Exhibits, Financial Statement Schedules
| (a) | List of documents filed as part of this report: |
The financial statements of the Company and the related reports of the Company’s independent registered public accounting firms thereon have been filed under Item 8 hereof.
| 2. | Financial Statement Schedules: |
None.
The following is a list of exhibits filed as part of this Form 10-K:
Exhibit
Number | | Description of Document | | Registrant’s
Form | | Dated | | Exhibit
Number | | Filed
Herewith |
| 1.1 | | At Market Issuance Sales Agreement, dated March 9, 2018, between CorMedix Inc. and B. Riley FBR, Inc. | | S-3 | | 3/09/2018 | | 1.1 | | |
| 1.2 | | Amended and Restated At Market Issuance Sales Agreement, dated November 27, 2020, by and among CorMedix Inc., B. Riley Securities, Inc. and Needham & Company LLC | | 8-K | | 11/27/2020 | | 1.1 | | |
| 1.3 | | At Market Issuance Sales Agreement, dated August 12, 2021, by and among CorMedix Inc., Truist Securities, Inc. and JMP Securities LLC. | | 8-K | | 08/12/2021 | | 1.1 | | |
| 3.1 | | Form of Amended and Restated Certificate of Incorporation. | | S-1/A | | 3/01/2010 | | 3.3 | | |
| 3.2 | | Certificate of Amendment to Amended and Restated Certificate of Incorporation, dated February 24, 2010. | | S-1/A | | 3/19/2010 | | 3.5 | | |
Exhibit
Number | | Description of Document | | Registrant’s
Form | | Dated | | Exhibit
Number | | Filed
Herewith |
| 3.3 | | Second Amended and Restated Bylaws as amended October 8, 2020 | | 8-K | | 10/14/2020 | | 3.1 | | |
| 3.4 | | Certificate of Amendment to Amended and Restated Certificate of Incorporation, dated December 3, 2012. | | 10-K | | 3/27/2013 | | 3.3 | | |
| 3.5 | | Certificate of Amendment to Amended and Restated Certificate of Incorporation, dated August 9, 2017. | | 8-K | | 8/10/2017 | | 3.1 | | |
| 3.6 | | Certificate of Amendment to Amended and Restated Certificate of Incorporation, dated March 25, 2019 | | 8-K | | 3/25/2019 | | 3.1 | | |
| 3.7 | | Amended and Restated Certificate of Designation of Series C-3 Non-Voting Convertible Preferred Stock of CorMedix Inc., filed with the Delaware Secretary of State on September 15, 2014. | | 8-K | | 9/16/2014 | | 3.16 | | |
| 3.8 | | Second Amended and Restated Certificate of Designation of Series E Convertible Preferred Stock of CorMedix Inc., filed with the Delaware Secretary of State on September 5, 2019. | | 8-K | | 9/11/2019 | | 3.2 | | |
| 3.9 | | Certificate of Designation of Series G Convertible Preferred Stock of CorMedix Inc., filed with the Delaware Secretary of State on September 5, 2019 | | 8-K | | 9/11/2019 | | 3.1 | | |
| 4.1 | | Specimen of Common Stock Certificate. | | S-1/A | | 3/19/2010 | | 4.1 | | |
| 4.2 | | Form of Warrant issued on January 8, 2014. | | 8-K | | 1/09/2014 | | 4.23 | | |
| 4.3 | | Form of Series B Warrant to Purchase Common Stock of CorMedix Inc. issued on May 3, 2017. | | 8-K | | 5/03/2017 | | 4.2 | | |
| 4.4 | | Form of Underwriter’s Warrant to Purchase Common Stock of CorMedix Inc., issued May 3, 2017. | | 8-K | | 5/03/2017 | | 4.3 | | |
| 4.5 | | Description of Capital Stock of CorMedix Inc. | | 10-K | | 3/16/2020 | | 4.5 | | |
| 10.1* | | License and Assignment Agreement, dated as of January 30, 2008, between the Company and ND Partners LLC. | | S-1/A | | 12/312009 | | 10.5 | | |
| 10.2 | | Escrow Agreement, dated as of January 30, 2008, among the Company, ND Partners LLC and the Secretary of the Company, as Escrow Agent. | | S-1 | | 11/25/2009 | | 10.6 | | |
| 10.3+ | | Form of Indemnification Agreement between the Company and each of its directors and executive officers. | | S-1/A | | 3/01/2010 | | 10.17 | | |
| 10.4+ | | 2013 Stock Incentive Plan | | 10-K | | 3/27/2013 | | 10.27 | | |
| 10.5+ | | Executive Employment Agreement, dated as of September 26, 2019, between CorMedix Inc. and Khoso Baluch | | 8-K | | 10/01/2019 | | 10.1 | | |
| 10.6+ | | Separation Agreement and Release, dated October 4, 2021, between CorMedix Inc. and Khoso Baluch. | | 10-Q | | 11/09/2021 | | 10.1 | | |
| 10.7**+ | | Executive Employment Agreement, dated and effective May 11, 2020, between CorMedix Inc. and Matthew David. | | 10-K | | | | 10.10 | | |
| 10.8+ | | Letter Agreement, dated and effective October 26, 2021, between CorMedix Inc. and Matthew David, M.D. | | 8-K | | 10/29/2021 | | 10.1 | | |
Exhibit
Number | | Description of Document | | Registrant’s
Form | | Dated | | Exhibit
Number | | Filed
Herewith |
| 10.9**+ | | Executive Employment Agreement, dated and effective April 17, 2020, between CorMedix Inc. and John Armstrong. | | 8-K | | 4/23/2020 | | 10.14 | | |
| 10.10+ | | Separation Agreement, dated October 4, 2021, between CorMedix Inc. and John K. Armstrong, Jr. | | 10-Q | | 11/09/2021 | | 10.2 | | |
| 10.11 | | Form of Securities Purchase Agreement, dated November 17, 2017, between CorMedix Inc. and the investors signatory thereto. | | 8-K | | 11/13/2017 | | 10.1 | | |
| 10.12 | | Backstop Agreement, dated November 9, 2017, between CorMedix Inc. and the investor named therein. | | 8-K | | 11/13/2017 | | 10.2 | | |
| 10.13 | | Form of Registration Rights Agreement, dated November 9, 2017, by and between CorMedix Inc. and the investor named therein. | | 8-K | | 11/13/2017 | | 10.3 | | |
| 10.14 | | Amendment No. 1, dated as of December 11, 2017, to Registration Rights Agreement, dated November 9, 2017, by and between CorMedix Inc. and the investor named therein. | | 8-K | | 12/11/2017 | | 10.1 | | |
| 10.15**+ | | Executive Employment Agreement, dated and effective March 10, 2021, between CorMedix Inc. and Elizabeth Masson-Hurlburt | | 8-K | | 3/12/2021 | | 10.1 | | |
| 10.16 | | Securities Purchase Agreement, dated December 31, 2018, between CorMedix Inc. and the investor named therein. | | 8-K | | 1/03/2019 | | 10.1 | | |
| 10.17* | | Employment Agreement, dated as of March 19, 2019, between CorMedix Inc. and Phoebe Mounts | | 10-Q | | 5/13/19 | | 10.1 | | |
| 10.18 | | Securities Exchange Agreement, dated August 14, 2019, by and among CorMedix Inc. and the Existing Security holders listed on the Schedule of Holders thereto. | | 8-K | | 8/15/2019 | | 10.1 | | |
| 10.19 | | Amended and Restated Registration Rights Agreement, dated as of September 6, 2019, by and among CorMedix Inc. and Manchester Securities Corp., and Elliot International, L.P. and Elliot Associates, L.P. | | 8-K | | 9/11/2019 | | 10.1 | | |
| 10.20 | | 2019 Omnibus Stock Incentive Plan | | 8-K | | 11/27/2019 | | 10.1 | | |
10.21**+ | | Executive Employment Agreement, dated April 29, 2021, between CorMedix Inc. and Thomas Nusbickel. | | 10-Q | | 08/21/2021 | | 10.1 | | |
| 10.22+ | | 2021 Executive Bonus Plan | | 8-K | | 12/23/2021 | | 10.1 | | |
10.23+ | | Executive Employment Agreement, dated March 16, 2022, between CorMedix Inc. and Joseph Todisco. | | 8-K | | 03/21/2022 | | 10.2 | | |
| 21.1 | | List of Subsidiaries. | | 10-K | | 3/27/2013 | | 21.1 | | |
| 23.1 | | Consent of Independent Registered Public Accounting Firm. | | | | | | | | X |
| 31.1 | | Certification of Principal Executive Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | | | | | | | | X |
31.2 | | Certification of Principal Financial Officer pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. | | | | | | | | X |
| 32.1 | | Certification of Principal Executive Officer and Principal Financial Officer pursuant to Section 906 of the Sarbanes-Oxley Act of 2002. | | | | | | | | X |
| 101 | | The following materials from CorMedix Inc. Form 10-K for the year ended December 31, 2021, formatted in Extensible Business Reporting Language (XBRL): (i) Balance Sheets at December 31, 2021 and 2020, (ii) Statements of Operations for the years ended December 31, 2021 and 2020, (iii) Statements of Changes in Stockholders’ Equity for the years ended December 31, 2021 and 2020, (iv) Statements of Cash Flows for the years ended December 31, 2021 and 2020 and (v) Notes to the Financial Statements.** | | | | | | | | X |
| 101.INS | | Inline XBRL Instance Document. |
| 101.SCH | | Inline XBRL Taxonomy Extension Schema Document. |
| 101.CAL | | Inline XBRL Taxonomy Extension Calculation Linkbase Document. |
| 101.DEF | | Inline XBRL Taxonomy Extension Definition Linkbase Document. |
| 101.LAB | | Inline XBRL Taxonomy Extension Label Linkbase Document. |
| 101.PRE | | Inline XBRL Taxonomy Extension Presentation Linkbase Document. |
| 104 | | Cover Page Interactive Data File (formatted as Inline XBRL and contained in Exhibit 101). |
| * | Confidential treatment has been granted for portions of this document. The omitted portions of this document have been filed separately with the SEC. |
| ** | Portions of the exhibit have been omitted in reliance on Item 601(b)(10)(iv) of Regulation S-K. |
| + | Indicates management contract or compensation plan. |
Item 16. Form 10-K Summary
Not applicable.
SIGNATURES
Pursuant to the requirements of the Securities Exchange Act of 1934, the Registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| | CORMEDIX INC. |
| | |
| March 29, 2022 | By: | /s/ Matthew David |
| | | Matthew David |
| | | Interim Chief Executive Officer and Chief Financial Officer |
| | | (Principal Executive Officer) |
| | | |
| March 29, 2022 | By: | /s/ Matthew David |
| | | Matthew David |
| | | Interim Chief Executive Officer and Chief Financial Officer |
| | | (Principal Financial and Accounting Officer) |
Pursuant to the requirements of the Securities Exchange Act of 1934, this report has been signed below by the following persons on behalf of the Registrant and in the capacities and on the dates indicated:
Signature | | Title | | Date |
| | | | | |
| /s/ Matthew David | | Interim Chief Executive Officer and Chief Financial Officer | | March 29, 2022 |
| Matthew David | | (Principal Executive Officer) | | |
| | | | | |
| /s/ Matthew David | | Interim Chief Executive Officer and Chief Financial Officer | | March 29, 2022 |
| Matthew David | | (Principal Financial and Accounting Officer) | | |
| | | | | |
| /s/ Myron Kaplan | | Director and Chairman of the Board | | March 29, 2022 |
| Myron Kaplan | | | | |
| | | | | |
| /s/ Paulo Costa | | Director | | March 29, 2022 |
| Paulo Costa | | | | |
| | | | | |
| /s/ Janet Dillione | | Director | | March 29, 2022 |
| Janet Dillione | | | | |
| | | | | |
| /s/ Gregory Duncan | | Director | | March 29, 2022 |
| Gregory Duncan | | | | |
| | | | | |
| /s/ Alan Dunton | | Director | | March 29, 2022 |
| Alan Dunton | | | | |
| | | | | |
| /s/ Steven Lefkowitz | | Director | | March 29, 2022 |
| Steven Lefkowitz | | | | |
| | | | | |
| /s/ Joseph Todisco | | Director | | March 29, 2022 |
| Joseph Todisco | | | | |
CORMEDIX INC. AND SUBSIDIARIES
FINANCIAL STATEMENTS
Financial Statements Index
REPORT OF INDEPENDENT REGISTERED PUBLIC ACCOUNTING FIRM
To the Board of Directors and
Stockholders of CorMedix Inc.
Opinion on the Consolidated Financial Statements
We have audited the accompanying consolidated balance sheets of CorMedix, Inc. and Subsidiaries (the “Company”) as of December 31, 2021 and 2020, and the related consolidated statements of operations and comprehensive income (loss), changes in stockholders’ equity, and cash flows for each of the years in the two-year period ended December 31, 2021, and the related notes (collectively referred to as the “consolidated financial statements”). In our opinion, the consolidated financial statements present fairly, in all material respects, the financial position of the Company as of December 31, 2021 and 2020, and the results of its operations and its cash flows for each of the years in the two-year period ended December 31, 2021, in conformity with accounting principles generally accepted in the United States of America.
Basis for Opinion
These consolidated financial statements are the responsibility of the Company’s management. Our responsibility is to express an opinion on the Company’s consolidated financial statements based on our audits. We are a public accounting firm registered with the Public Company Accounting Oversight Board (United States) (PCAOB) and are required to be independent with respect to the Company in accordance with the U.S. federal securities laws and the applicable rules and regulations of the Securities and Exchange Commission and the PCAOB.
We conducted our audits in accordance with the standards of the PCAOB. Those standards require that we plan and perform the audit to obtain reasonable assurance about whether the consolidated financial statements are free of material misstatement, whether due to error or fraud. The Company is not required to have, nor were we engaged to perform, an audit of its internal control over financial reporting. As part of our audits, we are required to obtain an understanding of internal control over financial reporting, but not for the purpose of expressing an opinion on the effectiveness of the Company’s internal control over financial reporting. Accordingly, we express no such opinion.
Our audits included performing procedures to assess the risks of material misstatement of the consolidated financial statements, whether due to error or fraud, and performing procedures that respond to those risks. Such procedures included examining, on a test basis, evidence regarding the amounts and disclosures in the consolidated financial statements. Our audits also included evaluating the accounting principles used and significant estimates made by management, as well as evaluating the overall presentation of the consolidated financial statements. We believe that our audits provide a reasonable basis for our opinion.
Critical Audit Matter
The critical audit matters communicated below are matters arising from the current period audit of the consolidated financial statements that were communicated or required to be communicated to the audit committee and that: (1) relate to accounts or disclosures that are material to the consolidated financial statements and (2) involved our especially challenging, subjective, or complex judgments. The communication of critical audit matters does not alter in any way our opinion on the consolidated financial statements, taken as a whole, and we are not, by communicating the critical audit matters below, providing separate opinions on the critical audit matters or on the accounts or disclosures to which they relate.
Critical Audit Matter Description
Stock Based Compensation
During the year ended December 31, 2021, the Company recorded stock-based compensation expense of approximately $5.0 million. As discussed in Note 7 to the consolidated financial statements, the Company issues various types of equity awards, including stock options and restricted stock units.
Auditing the Company’s accounting for stock-based compensation required complex auditor judgment due to the number and variety of equity awards outstanding, the inclusion of market and performance vesting criteria in certain awards, and the subjectivity of assumptions used to value stock-based awards. In particular, judgment was required to evaluate the nature of the performance conditions, as well as to assess the satisfaction of the performance targets.
How We Addressed the Matter in Our Audit
To test stock based-compensation expense, we performed audit procedures that included, among others, obtaining an understanding of the Company’s controls over stock-based compensation, assessing the completeness of the awards granted and evaluating the methodologies used to estimate the fair value of these awards. We also tested the accuracy of the data used in measuring the awards by agreeing the underlying inputs, such as grant date, grant price, performance targets and vesting terms, among others, back to source documents, such as compensation meeting minutes or award letters and testing the clerical accuracy of the calculation of the expense recorded. We determined whether milestone targets were satisfied in accordance with the contractual conditions and recalculated grant date fair value. We also evaluated the adequacy of the Company’s stock-based compensation disclosures included in Note 7 in relation to these matters.
| /s/ Friedman LLP | |
| | |
| We have served as the Company’s auditor since 2014. | |
| | |
| Marlton, NJ | |
| March 29, 2022 | |
CorMedix Inc. And Subsidiaries
CONSOLIDATED BALANCE SHEETS
December 31, 2021 and 2020
| | | December 31, | |
| | | 2021 | | | 2020 | |
| ASSETS | | | | | | |
| Current assets | | | | | | |
| Cash and cash equivalents | | $ | 53,317,405 | | | $ | 41,905,469 | |
| Restricted cash | | | 131,567 | | | | 191,314 | |
| Short-term investments | | | 12,149,003 | | | | 4,444,072 | |
| Trade receivables, net | | | 45,368 | | | | 3,357 | |
| Inventories | | | 3,008 | | | | 143,564 | |
| Prepaid research and development expenses | | | 51,993 | | | | 62,210 | |
| Security deposit | | | - | | | | 20,000 | |
| Other prepaid expenses and current assets | | | 770,485 | | | | 1,412,183 | |
| Total current assets | | | 66,468,829 | | | | 48,182,169 | |
| Property and equipment, net | | | 1,474,937 | | | | 111,499 | |
| Restricted cash, long term | | | 102,305 | | | | - | |
| Operating lease right-of-use assets | | | 899,505 | | | | 1,014,635 | |
| TOTAL ASSETS | | $ | 68,945,576 | | | $ | 49,308,303 | |
| | | | | | | | | |
| LIABILITIES AND STOCKHOLDERS’ EQUITY | | | | | | | | |
| Current liabilities | | | | | | | | |
| Accounts payable | | $ | 2,209,552 | | | $ | 1,128,104 | |
| Accrued expenses | | | 3,014,156 | | | | 2,924,351 | |
| Operating lease liabilities, short-term | | | 121,368 | | | | 109,128 | |
| Total current liabilities | | | 5,345,076 | | | | 4,161,583 | |
| Operating lease liabilities, net of current portion | | | 802,433 | | | | 923,708 | |
| TOTAL LIABILITIES | | | 6,147,509 | | | | 5,085,291 | |
| | | | | | | | | |
| COMMITMENTS AND CONTINGENCIES (Note 6) | | | | | | | | |
| | | | | | | | | |
| STOCKHOLDERS’ EQUITY | | | | | | | | |
| Preferred stock - $0.001 par value: 2,000,000 shares authorized; 181,622 and 241,623 shares issued and outstanding at December 31, 2021 and 2020, respectively | | | 182 | | | | 242 | |
| Common stock - $0.001 par value: 160,000,000 shares authorized at December 31, 2021 and 2020; 38,086,437 and 33,558,096 shares issued and outstanding at December 31, 2021 and 2020, respectively | | | 38,086 | | | | 33,558 | |
| Accumulated other comprehensive gain | | | 87,130 | | | | 102,006 | |
| Additional paid-in capital | | | 308,331,750 | | | | 261,536,061 | |
| Accumulated deficit | | | (245,659,081 | ) | | | (217,448,855 | ) |
| TOTAL STOCKHOLDERS’ EQUITY | | | 62,798,067 | | | | 44,223,012 | |
| TOTAL LIABILITIES AND STOCKHOLDERS’ EQUITY | | $ | 68,945,576 | | | $ | 49,308,303 | |
The accompanying notes are integral part of these consolidated financial statements.
CorMedix Inc. and Subsidiaries
CONSOLIDATED STATEMENTS OF OPERATIONS AND COMPREHENSIVE INCOME (LOSS)
Years Ended December 31, 2021 and 2020
| | | December 31, | |
| | | 2021 | | | 2020 | |
| Revenue: | | | | | | |
| Net sales | | $ | 190,936 | | | $ | 239,231 | |
| Cost of sales | | | (148,938 | ) | | | (204,846 | ) |
| Gross profit | | | 41,998 | | | | 34,385 | |
| | | | | | | | | |
| Operating Expenses: | | | | | | | | |
| Research and development | | | (13,132,982 | ) | | | (13,377,193 | ) |
| Selling, general and administrative | | | (16,346,601 | ) | | | (13,877,944 | ) |
| Total operating expenses | | | (29,479,583 | ) | | | (27,255,137 | ) |
| | | | | | | | | |
| Loss From Operations | | | (29,437,585 | ) | | | (27,220,752 | ) |
| Other Income (Expense): | | | | | | | | |
| Interest income | | | 14,403 | | | | 116,065 | |
| Foreign exchange transaction loss | | | (21,287 | ) | | | (59,165 | ) |
| Interest expense | | | (15,943 | ) | | | (33,226 | ) |
| Total other (expense) income | | | (22,827 | ) | | | 23,674 | |
| Net Loss Before Income Taxes | | | (29,460,412 | ) | | | (27,197,078 | ) |
| Tax benefit | | | 1,250,186 | | | | 5,169,395 | |
| Net Loss | | | (28,210,226 | ) | | | (22,027,683 | ) |
| Other Comprehensive Income (Loss): | | | | | | | | |
| Unrealized loss from investments | | | (4,655 | ) | | | (1,271 | ) |
| Foreign currency translation (loss) gain | | | (10,221 | ) | | | 6,020 | |
| Total other comprehensive (loss) income | | | (14,876 | ) | | | 4,749 | |
| Comprehensive Loss | | $ | (28,225,102 | ) | | $ | (22,022,934 | ) |
| Net Loss Per Common Share – Basic and Diluted | | $ | (0.75 | ) | | $ | (0.77 | ) |
| Weighted Average Common Shares Outstanding – Basic and Diluted | | | 37,666,081 | | | | 28,561,963 | |
The accompanying notes are integral part of these consolidated financial statements.
CORMEDIX INC. AND SUBSIDIARY
CONSOLIDATED STATEMENTS OF CHANGES IN STOCKHOLDERS’ EQUITY (DEFICIENCY)
Years Ended December 31, 2014 and 2013
| | | Common Stock | | | Preferred Stock –
Series C-2, C-3,
Series D, Series E,
Series F and Series G | | | Accumulated
Other
Comprehensive
Gain | | | Additional
Paid-in | | | Accumulated | | | Total
Stockholder’ | |
| | | Shares | | | Amount | | | Shares | | | Amount | | | (Loss) | | | Capital | | | Deficit | | | Equity | |
| Balance at December 31, 2019 | | | 25,665,350 | | | $ | 25,665 | | | | 241,623 | | | $ | 242 | | | $ | 97,257 | | | $ | 218,944,268 | | | $ | (195,421,172 | ) | | $ | 23,646,260 | |
| Stock issued in connection with public offering, net | | | 5,111,110 | | | | 5,111 | | | | - | | | | - | | | | - | | | | 21,250,059 | | | | - | | | | 21,255,170 | |
| Stock issued in connection with ATM sale of common stock, net | | | 2,687,646 | | | | 2,688 | | | | - | | | | - | | | | - | | | | 18,430,257 | | | | - | | | | 18,432,945 | |
| Stock issue in connection with warrants exercised | | | 91,500 | | | | 92 | | | | - | | | | - | | | | - | | | | 411,659 | | | | - | | | | 411,751 | |
| Issuance of vested restricted stock | | | 2,490 | | | | 2 | | | | - | | | | - | | | | - | | | | (2 | ) | | | - | | | | - | |
| Stock-based compensation | | | - | | | | - | | | | - | | | | - | | | | - | | | | 2,499,820 | | | | - | | | | 2,499,820 | |
| Other comprehensive income | | | - | | | | - | | | | - | | | | - | | | | 4,749 | | | | - | | | | - | | | | 4,749 | |
| Net loss | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | (22,027,683 | ) | | | (22,027,683 | ) |
| Balance at December 31, 2020 | | | 33,558,096 | | | $ | 33,558 | | | | 241,623 | | | $ | 242 | | | $ | 102,006 | | | $ | 261,536,061 | | | $ | (217,448,855 | ) | | $ | 44,223,012 | |
| Stock issued in connection with ATM sale of common stock, net | | | 3,737,862 | | | | 3,738 | | | | - | | | | - | | | | - | | | | 41,451,892 | | | | - | | | | 41,455,630 | |
| Stock issued in connection with warrants exercised, cash | | | 31,407 | | | | 31 | | | | - | | | | - | | | | - | | | | 164,855 | | | | - | | | | 164,886 | |
| Stock issued in connection with warrants exercised, cashless | | | 70,269 | | | | 70 | | | | - | | | | - | | | | - | | | | (70 | ) | | | - | | | | - | |
| Stock issued in connection with options exercised | | | 32,734 | | | | 33 | | | | - | | | | - | | | | - | | | | 137,002 | | | | - | | | | 137,035 | |
| Conversion of Series G preferred shares to common stock | | | 556,069 | | | | 556 | | | | (10,001 | ) | | | (10 | ) | | | - | | | | (546 | ) | | | - | | | | - | |
| Conversion of Series C-3 preferred shares to common stock | | | 100,000 | | | | 100 | | | | (50,000 | ) | | | (50 | ) | | | - | | | | (50 | ) | | | - | | | | - | |
| Stock-based compensation | | | - | | | | - | | | | - | | | | - | | | | - | | | | 5,042,606 | | | | - | | | | 5,042,606 | |
| Other comprehensive loss | | | - | | | | - | | | | - | | | | - | | | | (14,876 | ) | | | - | | | | - | | | | (14,876 | ) |
| Net loss | | | - | | | | - | | | | - | | | | - | | | | - | | | | - | | | | (28,210,226 | ) | | | (28,210,226 | ) |
| Balance at December 31, 2021 | | | 38,086,437 | | | $ | 38,086 | | | | 181,622 | | | $ | 182 | | | $ | 87,130 | | | $ | 308,331,750 | | | $ | (245,659,081 | ) | | $ | 62,798,067 | |
The accompanying notes are integral part of these consolidated financial statements.
CORMEDIX INC. AND SUBSIDIARIES
CONSOLIDATED STATEMENTS OF CASH FLOWS
Years Ended December 31, 2021 and 2020
| | | December 31, | |
| | | 2021 | | | 2020 | |
| CASH FLOWS FROM OPERATING ACTIVITIES: | | | | | | |
| Net loss | | $ | (28,210,226 | ) | | $ | (22,027,683 | ) |
| Adjustments to reconcile net loss to net cash used in operating activities: | | | | | | | | |
| Stock-based compensation | | | 5,042,606 | | | | 2,499,820 | |
| Change in right-of-use assets | | | 115,130 | | | | 34,368 | |
| Inventory reserve | | | - | | | | 44,006 | |
| Depreciation | | | 61,890 | | | | 127,964 | |
| Changes in operating assets and liabilities: | | | | | | | | |
| Change in operating lease liabilities | | | (109,035 | ) | | | (18,845 | ) |
| Increase in trade receivables | | | (44,080 | ) | | | (3,089 | ) |
| Decrease in inventory | | | 145,456 | | | | 149,597 | |
| Decrease (Increase) in prepaid expenses and other current assets | | | 666,628 | | | | (991,754 | ) |
| Increase in accounts payable | | | 1,082,129 | | | | 103,333 | |
| Decrease in accrued expenses | | | 94,279 | | | | (1,883,149 | ) |
| Decrease in deferred revenue | | | - | | | | (2,206 | ) |
| Net cash used in operating activities | | | (21,155,223 | ) | | | (21,967,638 | ) |
| CASH FLOWS FROM INVESTING ACTIVITIES: | | | | | | | | |
| Purchase of short-term investments | | | (15,289,586 | ) | | | (8,549,758 | ) |
| Maturity of short-term investments | | | 7,580,000 | | | | 16,088,572 | |
| Purchase of equipment | | | (1,425,329 | ) | | | (112,638 | ) |
| Net cash (used in) provided by investing activities | | | (9,134,915 | ) | | | 7,426,176 | |
| CASH FLOWS FROM FINANCING ACTIVITIES: | | | | | | | | |
| Proceeds from sale of common stock from at-the-market program, net | | | 41,455,630 | | | | 18,432,945 | |
| Proceeds from the public offering, net | | | - | | | | 21,255,170 | |
| Proceeds from exercise of warrants | | | 164,886 | | | | 411,751 | |
| Proceeds from exercise of stock options | | | 137,035 | | | | - | |
| Net cash provided by financing activities | | | 41,757,551 | | | | 40,099,866 | |
| Foreign exchange effects on cash | | | (12,919 | ) | | | 13,192 | |
| NET INCREASE IN CASH AND CASH EQUIVALENTS | | | 11,454,494 | | | | 25,571,596 | |
| CASH AND CASH EQUIVALENTS AND RESTRICTED CASH – BEGINNING OF YEAR | | | 42,096,783 | | | | 16,525,187 | |
| CASH AND CASH EQUIVALENTS AND RESTRICTED CASH – END OF YEAR | | $ | 53,551,277 | | | $ | 42,096,783 | |
| | | | | | | | | |
| Cash paid for interest | | $ | 15,943 | | | $ | 33,226 | |
| | | | | | | | | |
| Supplemental Disclosure of Non-Cash Financing and Investing Activities: | | | | | | | | |
| Conversion of Series G preferred stock to common stock | | $ | 10 | | | $ | - | |
| Conversion of Series C-3 preferred stock to common stock | | $ | 50 | | | $ | - | |
| Unrealized loss from investments | | $ | 4,655 | | | $ | 1,271 | |
| Deposit on equipment reclassified from prepaid expenses and current assets to property and equipment, net | | $ | 501,821 | | | $ | - | |
| Right-of-use assets obtained in exchange for lease liability | | $ | - | | | $ | 1,014,635 | |
| Issuance of common stock for vested restricted stock units | | $ | - | | | $ | 2 | |
The accompanying notes are integral part of these consolidated financial statements.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS
Note 1 — Organization, Business and Basis of Presentation:
Organization and Business:
CorMedix Inc. (“CorMedix” or the “Company”) was incorporated in the State of Delaware on July 28, 2006. The Company is a biopharmaceutical company focused on developing and commercializing therapeutic products for the prevention and treatment of infectious and inflammatory diseases. In 2013, the Company formed a wholly-owned subsidiary, CorMedix Europe GmbH and in May 2020, the Company formed a wholly-owned Spanish subsidiary, CorMedix Spain, S.L.U.
The Company’s primary focus is to develop its lead product candidate, DefenCath™, for potential commercialization in the United States (“U.S.”) and other key markets. The Company has in-licensed the worldwide rights to develop and commercialize DefenCath/Neutrolin®, which is a novel anti-infective solution (a formulation of taurolidine 13.5 mg/mL, and heparin 1000 USP Units/mL) intended for the reduction and prevention of catheter-related infections and thrombosis in patients requiring central venous catheters in clinical settings such as hemodialysis, total parenteral nutrition, and oncology. The name DefenCath is the U.S. proprietary name conditionally approved by the U.S. Food and Drug Administration (“FDA”), while the name Neutrolin is currently used in the European Union (“EU”) and other territories where the Company has received CE-Mark approval for the commercial distribution of Neutrolin as a catheter lock solution (“CLS”) regulated as a medical device.
In January 2015, the FDA designated DefenCath as a Qualified Infectious Disease Product (“QIDP”) for prevention of catheter-related blood stream infections in patients with end stage renal disease receiving hemodialysis through a central venous catheter. Catheter-related blood stream infections and clotting can be life-threatening. The QIDP designation provides five years of market exclusivity in addition to the five years granted for a New Chemical Entity upon approval of a New Drug Application (“NDA”). In addition, in January 2015, the FDA granted Fast Track designation to DefenCath Catheter Lock Solution, a designation intended to facilitate development and expedite review of drugs that treat serious and life-threatening conditions so that the approved drug can reach the market expeditiously. The Fast Track designation of DefenCath provides us with the opportunity to meet with the FDA on a more frequent basis during the development process, and also ensures eligibility to request priority review of the marketing application.
In December 2015, the Company launched its Phase 3 Prospective, Multicenter, Double-blind, Randomized, Active Control Study to Demonstrate Safety & Effectiveness of DefenCath/Neutrolin in Preventing Catheter-related Bloodstream Infection in Subjects on Hemodialysis for End Stage Renal Disease (“LOCK-IT-100”), in patients with hemodialysis catheters in the U.S. The clinical trial was designed to demonstrate the safety and effectiveness of DefenCath compared to the standard of care CLS, Heparin, in preventing CRBSIs. The primary endpoint for the trial assessed the incidence of CRBSI and time to CRBSI for each study subject. Secondary endpoints were catheter patency, which was defined as required use of tissue plasminogen activating factor, or tPA, or removal of catheter due to dysfunction, and removal of catheter for any reason.
As previously agreed with the FDA, an interim efficacy analysis was performed when the first 28 potential CRBSI cases were identified in our LOCK-IT-100 study that occurred through early December 2017. Based on these first 28 cases, there was a highly statistically significant 72% reduction in CRBSI by DefenCath relative to the active control of heparin (p=0.0034). Because the pre-specified level of statistical significance was reached for the primary endpoint and efficacy had been demonstrated with no safety concerns, the LOCK-IT-100 study was terminated early. The study continued enrolling and treating subjects until study termination, and the final analysis was based on a total of 795 subjects. In a total of 41 cases, there was a 71% reduction in CRBSI by DefenCath relative to heparin, which was highly statistically significant (p=0.0006), with a good safety profile.
The FDA granted the Company’s request for a rolling submission and review of the NDA which is designed to expedite the approval process for products being developed to address an unmet medical need. Although the FDA usually requires two pivotal clinical trials to provide substantial evidence of safety and effectiveness for approval of an NDA, the FDA will in some cases accept one adequate and well-controlled trial, where it is a large multicenter trial with a broad range of subjects and investigation sites with procedures to include trial quality that has demonstrated a clinically meaningful and statistically very persuasive effect on prevention of a disease with potentially serious outcome.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
In March 2020, the Company began the modular submission process for the NDA for DefenCath for the prevention of CRBSI in hemodialysis patients, and in August 2020, the FDA accepted for filing the DefenCath NDA. The FDA also granted the Company’s request for priority review, which provides for a six-month review period instead of the standard ten-month review period. As announced in March 2021, the FDA informed in its Complete Response Letter (“CRL”), to the Company that it cannot approve the NDA for DefenCath in its present form. The FDA noted concerns at the third-party manufacturing facility after a review of records requested by the FDA and provided by the contract manufacturing organization (“CMO”). Additionally, the FDA is requiring a manual extraction study to demonstrate that the labeled volume can be consistently withdrawn from the vials despite an existing in-process control to demonstrate fill volume within specifications.
In April 2021, the Company and the CMO met with the FDA to discuss proposed resolutions for the deficiencies identified in the CRL to the Company and the Post-Application Action Letter (“PAAL”), received by the CMO from the FDA for the NDA for DefenCath. There was an agreed upon protocol for the manual extraction study identified in the CRL, which now has been successfully completed. Addressing the FDA’s concerns regarding the qualification of the filling operation necessitated adjustments in the process and generation of additional data on operating parameters for manufacture of DefenCath. The Company and the CMO determined that additional process qualification is needed with subsequent validation to address these issues. The FDA did not request additional clinical data and did not identify any deficiencies related to the data submitted on the efficacy or safety of DefenCath from LOCK-IT-100. In draft labeling discussed with the FDA, the FDA added that the initial approval will be for the limited population of patients with kidney failure receiving chronic hemodialysis through a central venous catheter. This is consistent with the Company’s request for approval pursuant to the Limited Population Pathway for Antibacterial and Antifungal Drugs (“LPAD”). LPAD, passed as part of the 21st Century Cures Act, is a new program intended to expedite the development and approval of certain antibacterial and antifungal drugs to treat serious or life-threatening infections in limited populations of patients with unmet needs. LPAD provides for a streamlined clinical development program involving smaller, shorter, or fewer clinical trials and is intended to encourage the development of safe and effective products that address unmet medical needs of patients with serious bacterial and fungal infections. The Company believes that LPAD will provide additional flexibility for the FDA to approve DefenCath to prevent CRBSIs in the limited population of patients with kidney failure receiving hemodialysis through a central venous catheter.
On February 28, 2022, the Company announced that it resubmitted the NDA for DefenCath to address the CRL issued by the FDA. In parallel, the Company’s third-party manufacturer submitted responses to the deficiencies identified at the manufacturing facility in the PAAL issued by the FDA concurrently with the CRL. The FDA will evaluate the submission to accept for filing and determine the review timeline. The FDA has stated that it expected all corrections to facility deficiencies to be complete at the time of resubmission so that all corrective actions may be verified during an onsite evaluation of the manufacturing facility in the next review cycle, if the FDA determines it will do an onsite evaluation. If an onsite inspection is required, the Company may encounter delays in obtaining FDA approval because the FDA is currently facing a backlog due to the COVID-19 pandemic. The FDA issued a guidance document on its plan to use voluntary remote interactive evaluations at facilities, including for a pre-approval inspection to assess a marketing application. The FDA will request the manufacturing facility to participate in a voluntary remote interactive evaluation, if the FDA believes it is appropriate. A manufacturing facility cannot request the remote interaction. The FDA expects the use of remote interactive evaluations should help the FDA operate within normal timeframes in spite of the COVID-19 pandemic.
The Company intends to pursue additional indications for DefenCath use as a CLS in populations with an unmet medical need that also represent potentially significant market opportunities. While the Company is continuing to assess these areas, potential future indications may include use as a CLS to reduce CRBSIs in total parenteral nutrition patients using a central venous catheter and in oncology patients using a central venous catheter.
In addition to DefenCath, the Company is sponsoring a pre-clinical research collaboration for the use of taurolidine as a possible treatment for rare orphan pediatric tumors. In February 2018, the FDA granted orphan drug designation to taurolidine for the treatment of neuroblastoma in children. The Company may seek one or more strategic partners or other sources of capital to help develop and commercialize taurolidine for the treatment of neuroblastoma in children. The Company is also evaluating opportunities for the possible expansion of taurolidine as a platform compound for use in certain medical devices. Patent applications have been filed in several indications, including wound closure, surgical meshes, and wound management. Based on initial feasibility work, the Company is advancing pre-clinical studies for taurolidine-infused surgical meshes, suture materials and hydrogels. The Company will seek to establish development/commercial partnerships as these programs advance.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
The Company was granted a deferral by the FDA under the Pediatric Research Equity Act (“PREA”), that requires sponsors to conduct pediatric studies for NDAs for a new active ingredient, such as taurolidine in DefenCath, unless a waiver or deferral is obtained from the FDA. A deferral acknowledges that a pediatric assessment is required but permits the applicant to submit the pediatric assessment after the submission of an NDA. The Company has made a commitment to conduct the pediatric study after approval of the NDA for use in adult hemodialysis patients. Pediatric studies for an approved product conducted under PREA may qualify for pediatric exclusivity, which if granted would provide an additional six months of marketing exclusivity. DefenCath would then have the potential to receive a total marketing exclusivity period of 10.5 years, including exclusivity pursuant to NCE and QIDP.
The FDA regards taurolidine as a new chemical entity and therefore, it is currently an unapproved new drug. The Company might in the future pursue product candidates that would involve devices impregnated with taurolidine, and the Company believes that at the current time such products would be combination products subject to device premarket submission requirements (while subject also, under review by the FDA, to the standards for drug approvability). Consequently, given that there is no appropriate predicate medical device currently marketed in the U.S. on which a 510(k) approval process could be based and that taurolidine is not yet approved in any application, the Company anticipates that it would be required to submit a premarket approval application (“PMA”) for marketing authorization for any medical device indications that we may pursue for devices containing taurolidine. In the event that an NDA for DefenCath is approved by the FDA, the regulatory pathway for these medical device product candidates may be revisited with the FDA. Although there may be no appropriate predicate, de novo Class II designation can be proposed, based on a risk assessment and a reasonable assurance of safety and effectiveness.
In the European Union (“EU”), Neutrolin is regulated as a Class 3 medical device. In July 2013, the Company received CE Mark approval for Neutrolin. In December 2013, the Company commercially launched Neutrolin in Germany for the prevention of CRBSI, and maintenance of catheter patency in hemodialysis patients using a tunneled, cuffed central venous catheter for vascular access. To date, Neutrolin is registered and may be sold in certain European Union countries for such treatment.
In September 2014, the TUV-SUD and The Medicines Evaluation Board of the Netherlands (“MEB”), granted a label expansion for Neutrolin to include use in oncology patients receiving chemotherapy, intravenous (“IV”) hydration and IV medications via CVC for the EU. In December 2014, the Company received approval from the Hessian District President in Germany to expand the label for these same expanded indications. The expansion also adds patients receiving medication and IV fluids via CVC in intensive or critical care units (cardiac care unit, surgical care unit, neonatal critical care unit, and urgent care centers). An indication for use in total parenteral nutrition was also approved.
In September 2019, the Company’s registration with the Saudi Arabia Food and Drug Administration, or the SFDA, expired. As a result, the Company cannot sell Neutrolin in Saudi Arabia. The Company intends to complete the documentation required to renew its registration with the SFDA, however, the Company cannot predict how long the renewal process will take. There is no assurance that the registration will be renewed by the SFDA.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
Note 2 — Liquidity and Uncertainties:
The consolidated financial statements have been prepared in conformity with generally accepted accounting principles which contemplate continuation of the Company as a going concern. To date, the Company’s commercial operations have not generated sufficient revenues to enable profitability. As of December 31, 2021, the Company had an accumulated deficit of $245.7 million, and incurred net losses of $28.2 million and $22.0 million for the years ended December 31, 2021 and 2020, respectively. Based on the Company’s current development plans for DefenCath/Neutrolin in both the U.S. and foreign markets and its other operating requirements, the Company’s existing cash and cash equivalents and short-term investments at December 31, 2021 are expected to fund its operations at least through the first half of 2023, after taking into consideration the costs for resubmission of the NDA and initial preparations for the commercial launch for DefenCath.
The Company’s continued operations will depend on its ability to raise additional capital through various potential sources, such as equity and/or debt financings, strategic relationships, potential strategic transactions or out-licensing of its products in order to commercially launch DefenCath upon NDA approval and until profitability is achieved, if ever. Management can provide no assurances that such financing or strategic relationships will be available on acceptable terms, or at all. As of December 31, 2021, the Company has $50.0 million available under its At-the-Market Issuance Sales Agreement (the “ATM program”) and has $150.0 million available under its current shelf registration for the issuance of equity, debt or equity-linked securities (see Note 7).
The Company’s operations are subject to a number of other factors that can affect its operating results and financial condition. Such factors include, but are not limited to: the results of clinical testing and trial activities of the Company’s product candidates; the ability to obtain regulatory approval to market the Company’s products; ability to manufacture successfully; competition from products manufactured and sold or being developed by other companies; the price of, and demand for, Company products; the Company’s ability to negotiate favorable licensing or other manufacturing and marketing agreements for its products; and the Company’s ability to raise capital to support its operations.
The novel coronavirus has been declared a pandemic and has spread to multiple global regions. The outbreak and government measures taken in response have also had a significant impact, both direct and indirect, on businesses and commerce, as worker shortages have occurred; supply chains have been disrupted; facilities and production have been suspended; and demand for certain goods and services, such as medical services and supplies, has spiked, while demand for other goods and services, such as travel, has fallen. In response to the COVID-19 outbreak, “shelter in place” orders and other public health guidance measures have been implemented across much of the United States, Europe and Asia, including in the locations of the Company’s offices, clinical trial sites, key vendors and partners. The Company’s program timelines may be negatively affected by COVID-19, which could materially and adversely affect its business, financial conditions and results of operations.
Note 3 — Summary of Significant Accounting Policies:
Use of Estimates
The preparation of financial statements in conformity with accounting principles generally accepted in the United States of America requires management to make estimates and assumptions that affect the reported amounts of assets and liabilities and disclosure of contingent assets and liabilities at the date of the financial statements and reported amounts of revenue and expenses during the reporting period. Actual results could differ from those estimates.
Reclassifications
Certain reclassifications were made to the prior year’s amounts to conform to the 2021 presentation. Non-cash lease expense, as presented on the Company’s consolidated statement of cash flows for the year ended December 31, 2020, is now presented as change in right-of-use assets and change in operating lease liabilities.
Basis of Consolidation
The consolidated financial statements include the accounts of the Company, CorMedix Europe GmbH and CorMedix Spain, S.L.U. its wholly owned subsidiaries. All significant intercompany accounts and transactions have been eliminated in consolidation.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
Financial Instruments
Financial instruments that potentially subject the Company to concentrations of credit risk consist principally of cash and cash equivalents and short-term investments. The Company maintains its cash and cash equivalents in bank deposit and other interest-bearing accounts, the balances of which, at times, may exceed federally insured limits.
The following table is the reconciliation of the accounting standard that modifies certain aspects of the recognition, measurement, presentation and disclosure of financial instruments as shown on the Company’s consolidated statement of cash flows:
| | | December 31, | |
| | | 2021 | | | 2020 | |
| Cash and cash equivalents | | $ | 53,317,405 | | | $ | 41,905,469 | |
| Restricted cash, short-term and long-term | | | 233,872 | | | | 191,314 | |
| Total cash, cash equivalents and restricted cash | | $ | 53,551,277 | | | $ | 42,096,783 | |
The appropriate classification of marketable securities is determined at the time of purchase and reevaluated as of each balance sheet date. Investments in marketable debt and equity securities classified as available-for-sale are reported at fair value. Fair value is determined using quoted market prices in active markets for identical assets or liabilities or quoted prices for similar assets or liabilities or other inputs that are observable or can be corroborated by observable market data for substantially the full term of the assets or liabilities. Changes in fair value that are considered temporary are reported net of tax in other comprehensive income (loss). Realized gains and losses, amortization of premiums and discounts and interest and dividends earned are included in income (expense). For declines in the fair value of equity securities that are considered other-than-temporary, impairment losses are charged to other (income) expense, net. The Company considers available evidence in evaluating potential impairments of its investments, including the duration and extent to which fair value is less than cost. There were no deemed permanent impairments at December 31, 2021 or 2020.
The Company’s marketable securities are highly liquid and consist of U.S. government agency securities, high-grade corporate obligations and commercial paper with original maturities of more than 90 days. As of December 31, 2021 and 2020, all of the Company’s investments had contractual maturities which were less than one year. The following table summarizes the amortized cost, unrealized gains and losses and the fair value at December 31, 2021 and 2020:
| December 31, 2021: | | Amortized
Cost | | | Gross
Unrealized
Losses | | | Gross
Unrealized
Gains | | | Fair Value | |
| Money Market Funds and Cash Equivalents | | $ | 10,462,877 | | | $ | (23 | ) | | $ | - | | | $ | 10,462,854 | |
| U.S. Government Agency Securities | | | 2,806,597 | | | | (1,261 | ) | | | - | | | | 2,805,336 | |
| Corporate Securities | | | 7,548,493 | | | | (4,467 | ) | | | 1 | | | | 7,544,027 | |
| Commercial Paper | | | 1,799,548 | | | | - | | | | 92 | | | | 1,799,640 | |
| Subtotal | | | 12,154,638 | | | | (5,728 | ) | | | 93 | | | | 12,149,003 | |
| Total December 31, 2021 | | $ | 22,617,515 | | | $ | (5,751 | ) | | $ | 93 | | | $ | 22,611,857 | |
| December 31, 2020: | | | | | | | | | | | | | | | | |
| Money Market Funds and Cash Equivalents | | $ | 3,182,762 | | | $ | (81 | ) | | $ | 8 | | | $ | 3,182,689 | |
| Corporate Securities | | | 3,565,501 | | | | (1,005 | ) | | | 3 | | | | 3,564,499 | |
| Commercial Paper | | | 879,501 | | | | - | | | | 72 | | | | 879,573 | |
| Subtotal | | | 4,445,002 | | | | (1,005 | ) | | | 75 | | | | 4,444,072 | |
| Total December 31, 2020 | | $ | 7,627,764 | | | $ | (1,086 | ) | | $ | 83 | | | $ | 7,626,761 | |
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
Fair Value Measurements
The Company’s financial instruments recorded in the consolidated balance sheets include cash and cash equivalents, accounts receivable, investment securities, accounts payable and accrued expenses. The carrying value of certain financial instruments, primarily cash and cash equivalents, accounts receivable, accounts payable, and accrued expenses approximate their estimated fair values based upon the short-term nature of their maturity dates. The Company’s senior secured convertible note (prior to its extinguishment in August 2019) falls into the Level 3 category within the fair value level hierarchy. The fair value was determined using market data for valuation.
The Company categorizes its financial instruments into a three-level fair value hierarchy that prioritizes the inputs to valuation techniques used to measure fair value, which is set out below. The fair value hierarchy gives the highest priority to quoted prices in active markets for identical assets (Level 1) and the lowest priority to unobservable inputs (Level 3). If the inputs used to measure fair value fall within different levels of the hierarchy, the category level is based on the lowest priority level input that is significant to the fair value measurement of the instrument.
| ● | Level 1 inputs—Observable inputs that reflect quoted prices (unadjusted) for identical assets or liabilities in active markets. |
| ● | Level 2 inputs— Significant other observable inputs (e.g., quoted prices for similar items in active markets, quoted prices for identical or similar items in markets that are not active, inputs other than quoted prices that are observable such as interest rate and yield curves, and market-corroborated inputs). |
| ● | Level 3 inputs—Unobservable inputs for the asset or liability, which are supported by little or no market activity and are valued based on management’s estimates of assumptions that market participants would use in pricing the asset or liability. |
The following table provides the carrying value and fair value of the Company’s financial assets measured at fair value as of December 31, 2021 and 2020:
| December 31, 2021: | | Carrying Value | | | Level 1 | | | Level 2 | | | Level 3 | |
| Money Market Funds and Cash Equivalents | | $ | 10,462,854 | | | $ | 10,462,854 | | | $ | - | | | $ | - | |
| U.S. Government Agency Securities | | | 2,805,336 | | | | 2,805,336 | | | | - | | | | - | |
| Corporate Securities | | | 7,544,027 | | | | - | | | | 7,544,027 | | | | - | |
| Commercial Paper | | | 1,799,640 | | | | - | | | | 1,799,640 | | | | - | |
| Subtotal | | | 12,149,003 | | | | 2,805,336 | | | | 9,343,667 | | | | - | |
| Total December 31, 2021 | | $ | 22,611,857 | | | $ | 13,268,190 | | | $ | 9,343,667 | | | $ | - | |
| December 31, 2020: | | | | | | | | | | | | | | | | |
| Money Market Funds and Cash | | | | | | | | | | | | | | | | |
| Equivalents | | $ | 3,182,689 | | | | 3,182,689 | | | | - | | | | - | |
| Corporate Securities | | | 3,564,499 | | | | - | | | | 3,564,499 | | | | - | |
| Commercial Paper | | | 879,573 | | | | - | | | | 879,573 | | | | - | |
| Subtotal | | | 4,444,072 | | | | - | | | | 4,444,072 | | | | - | |
| Total December 31, 2020 | | $ | 7,626,761 | | | $ | 3,182,689 | | | $ | 4,444,072 | | | $ | - | |
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
Foreign Currency Translation and Transactions
The consolidated financial statements are presented in U.S. Dollars (USD), the reporting currency of the Company. For the financial statements of the Company’s foreign subsidiaries, whose functional currency is the EURO, foreign currency asset and liability amounts, if any, are translated into USD at end-of-period exchange rates. Foreign currency income and expenses are translated at average exchange rates in effect during the year. Translation gains and losses are included in other comprehensive income (loss). The Company had a foreign currency translation loss of $10,221 in 2021 and a gain of $6,020 in 2020.
Foreign currency exchange transaction gain (loss) is the result of re-measuring transactions denominated in a currency other than the functional currency of the entity recording the transaction.
Geographic Information
The following table summarizes the geographic information:
| | | December 31, | |
| | | 2021 | | | 2020 | |
| Reported revenues | | $ | 190,936 | | | $ | 239,231 | |
| Revenues attributable to European and Mideast operations, which are based in Germany | | | 190,936 | | | | 237,025 | |
| Total assets | | | 68,945,576 | | | | 49,308,303 | |
| Total assets located in the United States, with the remainder in the European Union | | $ | 68,558,413 | | | $ | 48,928,244 | |
Restricted Cash
As of December 31, 2021, and 2020 the Company has restricted cash in connection with the patent and utility model infringement proceedings against TauroPharm (see Note 7). The Company was required by the District Courts of Mannheim to provide security deposit to cover legal fees in the event TauroPharm is entitled to reimbursement of these costs. The Company furthermore had to provide a deposit for the first and second instances, respectively, in connection with the unfair competition proceedings in Cologne. During the year ended December 31, 2021, approximately $48,000 was released by the court for the reimbursement of legal fees and other costs which was removed from restricted cash. As of December 31, 2021 and 2020, restricted cash in connection with the patent and utility model infringement proceedings were $132,000 and $191,000, respectively.
As of December 31, 2021, the Company had $102,000 in long-term restricted cash for a lease security deposit.
Prepaid Research and Development and Other Prepaid Expenses
Prepaid expenses consist of payments made in advance to vendors relating to service contracts for clinical trial development, manufacturing, pre-clinical development and insurance policies. These advanced payments are amortized to expense either as services are performed or over the relevant service period using the straight-line method.
Inventories
Inventories are valued at the lower of cost or net realizable value on a first in, first out basis. Inventories consist of raw materials (including labeling and packaging), work-in-process, and finished goods, if any, for the DefenCath product. Inventories consist of the following:
| | | December 31, | |
| | | 2021 | | | 2020 | |
| Finished goods | | $ | 3,008 | | | $ | 143,564 | |
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
Property and Equipment
Property and equipment consist primarily of furnishings, fixtures, leasehold improvements, office equipment and computer equipment all of which are recorded at cost. Depreciation is provided for by the straight-line method over the estimated useful lives of the related assets. Leasehold improvements are amortized using the straight-line method over the remaining lease term or the life of the asset, whichever is shorter. Property and equipment, as of December 31, 2021 and 2020 were $1,474,937 and $111,499, respectively, net of accumulated depreciation of $365,169 and $303,279, respectively. Depreciation and amortization of property and equipment is included in selling, general and administrative expenses.
| Description | | Estimated Useful Life |
| Office equipment and furniture | | 5 years |
| Leasehold improvements | | 7 years |
| Computer equipment | | 5 years |
| Computer software | | 3 years |
Leases
The Company determines if an arrangement is a lease at inception. Operating leases are included in operating lease right-of-use (“ROU”) assets, current portion of operating lease liabilities (included in accrued expenses), and operating lease liabilities, net of current portion, on the consolidated balance sheet (see Note 10).
Operating lease ROU assets and operating lease liabilities are recognized based on the present value of the future minimum lease payments over the lease term at commencement date. As the Company’s leases do not provide an implicit rate, the Company uses its incremental borrowing rate based on the information available at commencement date in determining the present value of future payments. The Company’s lease terms may include options to extend or terminate the lease when it is reasonably certain that the Company will exercise that option. Lease expense for minimum lease payments is recognized on a straight-line basis over the lease term.
The Company has elected, as an accounting policy, not to apply the recognition requirements in ASC 842 to short-term leases. Short-term leases are leases that have a term of 12 months or less and do not include an option to purchase the underlying asset that the Company is reasonably certain to exercise. The Company recognizes the lease payments for short-term leases on a straight-line basis over the lease term.
The Company has also elected, as a practical expedient, by underlying class of asset, not to separate lease components from non-lease components and, instead, account for them as a single component.
Accrued Expenses
Accrued expenses consist of the following:
| | | December 31, | |
| | | 2021 | | | 2020 | |
| Professional and consulting fees | | $ | 311,408 | | | $ | 146,129 | |
| Accrued payroll and payroll taxes | | | 2,508,398 | | | | 2,490,441 | |
| Manufacturing development related | | | 99,614 | | | | 143,780 | |
| Other | | | 94,736 | | | | 144,001 | |
| Total | | $ | 3,014,156 | | | $ | 2,924,351 | |
Revenue Recognition
The Company uses Accounting Standards Codification (“ASC”) 606, “Revenue from Contracts with Customers,” issued by the Financial Accounting Standards Board (“FASB”), that prescribes a five-step model for recognizing revenue which includes (i) identifying contracts with customers; (ii) identifying performance obligations; (iii) determining the transaction price; (iv) allocating the transaction price; and (v) recognizing revenue.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
The Company recognizes net sales upon shipment of product to the dialysis centers and upon meeting the five-step model prescribed by ASC 606 outlined above.
Deferred Revenue
In August 2014, the Company entered into an exclusive distribution agreement (the “Wonik Agreement”) with Wonik Corporation, a South Korean company, to market, sell and distribute Neutrolin for hemodialysis and oncolytic patients upon receipt of regulatory approval in South Korea. Upon execution, Wonik paid the Company a non-refundable $50,000 payment and will pay an additional $50,000 upon receipt of the product registration necessary to sell Neutrolin in South Korea (the “Territory”). The term of the Wonik Agreement commenced on August 8, 2014 and will continue for three years after the first commercial sale of Neutrolin in the Territory. The non-refundable up-front payment has been recorded as deferred revenue and will be recognized as revenue on a straight-line basis over the contractual term of the Agreement. Deferred revenue related to this agreement was fully amortized at December 31, 2020.
Loss Per Common Share
Basic loss per common share excludes dilution and is computed by dividing net loss by the weighted average number of common shares outstanding during the period. Diluted loss per common share reflects the potential dilution that could occur if securities or other contracts to issue common stock were exercised or converted into common stock or resulted in the issuance of common stock that then shared in the earnings of the entity.
The Company's outstanding shares of Series E preferred stock entitle the holders to receive dividends on a basis equivalent to the dividends paid to holders of common stock. As a result, the Series E preferred stock meet the definition of participating securities requiring the application of the two-class method. Under the two-class method, earnings available to common shareholders, including both distributed and undistributed earnings, are allocated to each class of common stock and participating securities according to dividends declared and participating rights in undistributed earnings, which may cause diluted earnings per share to be more dilutive than the calculation using the treasury stock method. No loss has been allocated to these participating securities since they do not have contractual obligations that require participation in the Company’s losses.
Since the Company has only incurred losses, basic and diluted loss per share are the same as potentially dilutive shares have been excluded from the calculation of diluted net loss per share as their effect would be anti-dilutive. The shares outstanding at the end of the respective periods presented below were excluded from the calculation of diluted net loss per share due to their anti-dilutive effect:
| | | Number of Shares of
Common Stock Issuable At | |
| | | December 31, | |
| | | 2021 | | | 2020 | |
| Series C non-voting preferred stock | | | 4,000 | | | | 104,000 | |
| Series E voting preferred stock | | | 391,953 | | | | 391,953 | |
| Series G voting preferred stock | | | 5,004,069 | | | | 5,560,137 | |
| Shares issuable for payment of deferred board compensation | | | 48,909 | | | | 48,909 | |
| Shares underlying outstanding warrants | | | 56,455 | | | | 183,148 | |
| Shares underlying outstanding stock options | | | 3,358,131 | | | | 2,447,687 | |
| Total potentially dilutive shares | | | 8,863,517 | | | | 8,735,834 | |
Stock-Based Compensation
Share-based compensation cost is measured at grant date, based on the estimated fair value of the award using the Black-Scholes option pricing model for options with service or performance-based conditions. Stock-based compensation is recognized as expense over the requisite service period on a straight-line basis or when the achievement of the performance condition is probable. For options with market-based vesting, share-based compensation cost is measured at grant date using the Monte Carlo option pricing model and the expense is recognized over the derived service period.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
Research and Development
Research and development costs are charged to expense as incurred. Research and development include fees associated with operational consultants, contract clinical research organizations, contract manufacturing organizations, clinical site fees, contract laboratory research organizations, contract central testing laboratories, licensing activities, and allocated executive, human resources and facilities expenses. The Company accrues for costs incurred as the services are being provided by monitoring the status of the trial and the invoices received from its external service providers. As actual costs become known, the Company adjusts its accruals in the period when actual costs become known. Costs related to the acquisition of technology rights and patents for which development work is still in process are charged to operations as incurred and considered a component of research and development expense.
Income Taxes
Deferred tax assets and liabilities are recognized for the future tax consequences attributable to temporary differences between the financial statement carrying amounts of existing assets and liabilities and their respective tax bases. Deferred tax assets and liabilities are measured using enacted tax rates expected to apply to taxable income in the years in which those temporary differences are expected to be recovered or settled. The effect on deferred tax assets and liabilities of a change in tax rates is recognized in income in the period that includes the enactment date. Valuation allowances are established when it is more likely than not that some or all of the deferred tax assets will not be realized.
Recently Adopted Authoritative Pronouncements
In June 2016, the Financial Accounting Standards Board (“FASB”) issued new guidance which replaces the incurred loss impairment methodology in current GAAP with a methodology that reflects expected credit losses and requires consideration of a broader range of reasonable and supportable information to inform credit loss estimates. This adoption on January 1, 2020 did not have a material impact on the Company’s consolidated financial statements.
In August 2018, the FASB issued new guidance which modifies the disclosure requirements on fair value measurements. The guidance was effective for the Company beginning in the first quarter of fiscal year 2020. This adoption on January 1, 2020 did not have a material impact on the Company’s consolidated financial statements.
In November 2018, the FASB issued new guidance to clarify the interaction between the authoritative guidance for collaborative arrangements and revenue from contracts with customers. The new guidance clarifies that, when the collaborative arrangement participant is a customer in the context of a unit-of-account, revenue from contracts with customers guidance should be applied, adds unit-of-account guidance to collaborative arrangements guidance, and, in a transaction with a collaborative arrangement participant who is not a customer, precludes presenting the transaction together with revenue recognized under contracts with customers. The guidance was effective for the Company beginning in the first quarter of fiscal year 2020. This adoption on January 1, 2020 did not have a material impact on the Company’s consolidated financial statements.
In November 2019, the FASB issued new guidance which requires that an entity measure and classify share-based payment awards granted to a customer by applying the guidance in FASB ASC 718. The guidance was effective for the Company beginning in the first quarter of fiscal year 2020. This adoption on January 1, 2020 did not have a material impact on the Company’s consolidated financial statements.
In December 2019, the FASB issued ASU 2019-12 which removes certain exceptions to the general principles of the accounting for income taxes and also improves consistent application of and simplification of other areas when accounting for income taxes. The guidance was effective for the Company beginning in the first quarter of fiscal year 2021. Early adoption was permitted. This adoption on January 1, 2021 did not have a material impact on the Company’s consolidated financial statements.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
Note 4 — Related Party Transactions:
In February 2021, Manchester Securities Corp., Elliott Associates LP and Elliott International LP (collectively, “Elliott”), an existing institutional investor who collectively beneficially own the largest portion of the Company’s common stock, converted an aggregate of 10,001 Series G preferred shares into an aggregate of 556,069 shares of the Company’s common stock.
Note 5 — Income Taxes:
The Company’s U.S. and foreign loss before income taxes are set forth below:
| | | December 31, | |
| | | 2021 | | | 2020 | |
| United States | | $ | (29,031,585 | ) | | $ | (20,605,821 | ) |
| Foreign | | | (428,827 | ) | | | (591,257 | ) |
| Total | | $ | (29,460,412 | ) | | $ | (27,197,078 | ) |
There were no current or deferred income tax provision for the years ended December 31, 2021 and 2020 because the Company has incurred operating losses since inception.
The Company’s deferred tax assets consist of the following:
| | | December 31, | |
| | | 2021 | | | 2020 | |
| Net operating loss carryforwards – Federal | | $ | 44,085,000 | | | $ | 38,986,000 | |
| Net operating loss carryforwards – State | | | 3,717,000 | | | | 2,958,000 | |
| Net operating loss carryforwards – Foreign | | | 5,000 | | | | 2,455,000 | |
| Capitalized licensing fees | | | 449,000 | | | | 600,000 | |
| Stock-based compensation | | | 4,430,000 | | | | 3,358,000 | |
| Accrued compensation | | | 320,000 | | | | 102,000 | |
| Other | | | (17,000 | ) | | | 21,000 | |
| Totals | | | 52,989,000 | | | | 48,480,000 | |
| Less valuation allowance | | | (52,989,000 | ) | | | (48,480,000 | ) |
| Deferred tax assets | | $ | - | | | $ | - | |
The Company had the following potentially utilizable net operating loss tax carryforwards:
| | | December 31, | |
| | | 2021 | | | 2020 | |
| Federal | | $ | 209,930,000 | | | $ | 185,650,000 | |
| State | | $ | 52,280,000 | | | $ | 41,600,000 | |
| Foreign | | $ | 20,000 | | | $ | 8,185,000 | |
The net operating losses generated will start to expire in 2026 for Federal purposes whereas the operating losses for state purposes will begin expiring in 2038. The Tax Cuts and Jobs Act of 2017 (the “Act”) limits the net operating loss deduction to 80% of taxable income for losses arising in tax years beginning after December 31, 2017. However, the net operating losses now have an indefinite carryforward as opposed to the former 20-year carryforward. The foreign net operating loss tax carryforwards do not expire. Our federal and state operating loss carryforwards include windfall tax deductions from stock option exercises.
During 2021, the Company’s German subsidiary was audited by the German taxing authorities for the years 2013-2015. It was determined that the amount of German income was not sufficient, so the taxing authorities made adjustments accordingly. Further, amended returns were filed for the subsequent years to provide the German subsidiary sufficient income. As a result of these changes, the German NOL was fully utilized and no longer has a carryforward attribute. Since such adjustments are statutory adjustments in Germany for tax purposes, there is no material effect on the Company’s financial statements. The foreign net operating loss carryforward relates to the Company’s Spanish subsidiary.
The utilization of the Company’s net operating losses may be subject to a substantial limitation due to the “change of ownership provisions” under Section 382 of the Internal Revenue Code and similar state provisions. Such limitation may result in the expiration of the net operating loss carryforwards before their utilization.
The Company’s foreign earnings are derived from its German subsidiary. The Company does not expect any foreign earnings to be repatriated in the U.S. in the near future.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
The Company’s effective tax rate varied from the statutory rate as follows:
| | | December 31, | |
| | | 2021 | | | 2020 | |
| Statutory federal tax rate | | | 21.0 | % | | | 21.0 | % |
| State income tax rate (net of federal) | | | 3.5 | % | | | 4.3 | % |
| Effect of foreign operations | | | 0.0 | % | | | 0.7 | % |
| Change in foreign NOL | | | (8.3 | )% | | | 0.5 | % |
| NJ NOL adjustment | | | 4.2 | % | | | 2.9 | % |
| Other permanent differences | | | (0.9 | )% | | | (0.6 | )% |
| Effect of valuation allowance | | | (15.3 | )% | | | (9.8 | )% |
| Effective tax rate | | | 4.2 | % | | | 19.0 | % |
In assessing the realizability of deferred tax assets, management considers whether it is more-likely-than-not that some portion or all of the deferred tax assets will not be realized. The ultimate realization of deferred tax assets is dependent upon the generation of future taxable income of the appropriate character during the periods in which those temporary differences become deductible and the loss carryforwards are available to reduce taxable income. In making its assessment, the Company considered all sources of taxable income including carryback potential, future reversals of existing deferred tax liabilities, prudent and feasible tax planning strategies, and lastly, objectively verifiable projections of future taxable income exclusive of reversing temporary differences and carryforwards. At December 31, 2021 and 2020, the Company maintained a full valuation allowance against its net deferred tax assets. The Company will continue to assess all available evidence during future periods to evaluate the realization of its deferred tax assets.
The following table presents the changes in the deferred tax asset valuation allowance for the periods indicated:
| Year Ended | | Balance at
Beginning of
Year | | | Increase
(Decrease)
Charged
(Credited) to
Income Taxes
(Benefit) | | | Increase
(Decrease)
Charged
(Credited)
to OCI | | | Balance at
End of
Year | |
| December 31, 2021 | | $ | 48,480,000 | | | $ | 4,541,000 | | | $ | (32,000 | ) | | $ | 52,989,000 | |
| December 31, 2020 | | $ | 45,815,000 | | | $ | 2,696,000 | | | $ | (31,000 | ) | | $ | 48,480,000 | |
Accounting for uncertainty in income taxes requires uncertain tax positions to be classified as non-current income tax liabilities unless they are expected to be paid within one year. The Company has concluded that there are no uncertain tax positions requiring recognition in its consolidated financial statements as of December 31, 2021 and 2020. The Company recognizes interest and penalties related to uncertain tax positions if any as a component of income tax expense.
The Company files U.S. federal and state returns. The Company’s foreign subsidiary also files a local tax return in their local jurisdiction. From a U.S. federal, state and local perspective the years that remains open to examination are consistent with each jurisdiction’s statute of limitations. From a foreign perspective, tax years 2016 to 2020 remain open to examination.
During the years ended December 31, 2021 and 2020, the Company received net proceeds of $1,250,186 and $5,169,395, respectively, from the sale of most of its remaining unused New Jersey net operating losses (“NOL”) eligible for sale under the State of New Jersey’s Economic Development Authority’s New Jersey Technology Business Tax Certificate Transfer program (“NJEDA Program”). The NJEDA Program allowed the Company to sell $1,337,000 of its total $1,337,000 in available NOL tax benefits for the state fiscal year 2020 and $5,529,000 of its total $6,018,000 for the state fiscal year 2019.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
Note 6 — Commitments and Contingencies:
Contingency Matters
On October 13, 2021, the United States District Court for the District of New Jersey consolidated into In re CorMedix Inc. Securities Litigation, Case No. 2:21-cv014020-JXN-CLW, two putative class action lawsuits filed on or about July 22, 2021 and September 13, 2021, respectively, and appointed lead counsel and lead plaintiff, a purported stockholder of the Company. The lead plaintiff filed a consolidated amended class action complaint on December 14, 2021, alleging violations of Sections 10(b) and 20(a) of the Exchange Act, along with Rule 10b-5 promulgated thereunder, and Sections 11 and 15 of the Securities Act of 1933. The complaint names as defendants the Company, Khoso Baluch, Matthew David, Phoebe Mounts, John L. Armstrong, Robert Cook, Janet Dillione, Alan W. Dunton, Myron Kaplan, Steven Lefkowitz, Paulo F. Costa, and Greg Duncan, as well as two underwriters of the Company’s secondary stock offering, B. Riley Securities, Inc. and Needham & Company, LLC. The purported bases for these claims are alleged misstatements and omissions in connection with the NDA submitted to the FDA for DefenCath, and the subsequent notification by the FDA that the NDA could not be approved in its present form. The lead plaintiff purports to assert the Exchange Act claims on behalf of persons that purchased or otherwise acquired shares of the Company’s securities between October 16, 2019, and September 6, 2021, and purports to assert the Securities Act claims on behalf of persons that purchased shares of the Company’s securities pursuant or traceable to a secondary offering of stock that commenced on November 27, 2020. The Company intends to vigorously contest such claims and filed a motion to dismiss the current complaint in full, with prejudice, on February 21, 2022. As of this filing, the current schedule set by the Court requires the Company and the other defendants to refile their motion to dismiss on March 28, 2022, requires the lead plaintiff to file an opposition to the Company’s motion to dismiss on or before April 27, 2022 and requires that the Company file a reply on or before May 27, 2022.
On or about October 13, 2021, a purported shareholder, derivatively and on behalf of the Company, filed a shareholder derivative complaint in the United States District Court for the District of New Jersey, in a case entitled Voter v. Baluch, et al., Case No. 2:21-cv-18493-JXN-LDW. The complaint names as defendants Khoso Baluch, Janet Dillione, Alan W. Dunton, Myron Kaplan, Steven Lefkowitz, Paulo F. Costa, Greg Duncan, Matthew David, and Phoebe Mounts along with the Company as Nominal Defendant. The complaint alleges breaches of fiduciary duties, abuse of control, and waste of corporate assets against the defendants and a claim for contribution for purported violations of Sections 10(b) and 21D of the Exchange Act against certain defendants. The Company intends to vigorously contest such claims. On January 21, 2022, pursuant to a stipulation between the parties, the Court entered an order staying the case while the motion to dismiss the class action lawsuit described in the foregoing paragraph is pending. The stay may be terminated before the motion to dismiss is resolved according to certain circumstances described in the stipulation available on the Court’s public docket.
On September 9, 2014, the Company filed in the District Court of Mannheim, Germany, a patent infringement action against TauroPharm GmbH and Tauro-Implant GmbH as well as their respective CEOs (the “Defendants”) claiming infringement of the Company’s European Patent EP 1 814 562 B1, which was granted by the European Patent Office (the “EPO”) on January 8, 2014 (the “Prosl European Patent”). The Prosl European Patent covers the formulation of taurolidine and citrate with low dose heparin in a catheter lock solution for maintaining patency and preventing infection in hemodialysis catheters. In this action, the Company claims that the Defendants infringe on the Prosl European Patent by manufacturing and distributing catheter locking solutions to the extent they are covered by the claims of the Prosl European Patent. The Company believes that its patent is sound and is seeking injunctive relief and raising claims for information, rendering of accounts, calling back, destruction and damages. Separately, TauroPharm has filed an opposition with the EPO against the Prosl European Patent alleging that it lacks novelty and inventive step. The Company cannot predict the ultimate outcome of either of these related matters. At present, the EPO has revoked the Prosl European Patent as invalid, and the Company has filed an appeal, which is currently pending.
In the same complaint against the same Defendants, the Company also alleged an infringement (requesting the same remedies) of ND Partners’ utility model DE 20 2005 022 124 U1 (the “Utility Model”), which the Company believes is fundamentally identical to the Prosl European Patent in its main aspects and claims. The Court separated the two proceedings and the Prosl European Patent and the Utility Model claims were tried separately. TauroPharm has filed a cancellation action against the Utility Model before the German Patent and Trademark Office (the “German PTO”) based on the similar arguments as those in the opposition against the Prosl European Patent.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
The Court issued its decisions on May 8, 2015, staying both proceedings. In its decisions, the Court found that the commercialization by TauroPharm in Germany of its TauroLock catheter lock solutions Hep100 and Hep500 infringes both the Prosl European Patent and the Utility Model and further that there is no prior use right that would allow TauroPharm to continue to make, use or sell its product in Germany. However, the Court declined to issue an injunction in favor of the Company that would preclude the continued commercialization by TauroPharm based upon its finding that there is a sufficient likelihood that the EPO, in the case of the Prosl European Patent, or the German PTO, in the case of the Utility Model, may find that such patent or utility model is invalid. Specifically, the Court noted the possible publication of certain instructions for product use that may be deemed to constitute prior art. As such, the District Court determined that it will defer any consideration of the request by the Company for injunctive and other relief until such time as the EPO or the German PTO made a final decision on the underlying validity of the Prosl European Patent and the Utility Model.
The opposition proceeding against the Prosl European Patent before the EPO is ongoing. The EPO held a hearing in the opposition proceeding on November 25, 2015. However, the EPO did not issue a decision at the end of the hearing but adjourned the matter due to the fact that the panel was of the view that Claus Herdeis, one of the managing directors of TauroPharm, had to be heard as a witness in a further hearing in order to close some gaps in the documentation presented by TauroPharm as regards the publication of the prior art.
The German PTO held a hearing in the validity proceedings relating to the Utility Model on June 29, 2016, at which the panel affirmed its preliminary finding that the Utility Model was invalid based upon prior publication of a reference to the benefits that may be associated with adding heparin to a taurolidine based solution. The Company filed an appeal against the ruling on September 7, 2016. An oral hearing was held on September 17, 2019 in which the German Federal Patent Court affirmed the first instance decision that the Utility Model was invalid. The decision has only a declaratory effect, as the Utility Model had expired in November 2015. On April 28, 2020, the Company filed a withdrawal of the complaint on the German utility model, thereby waiving its claims on these proceedings. The proceedings were closed and during the year ended December 31, 2020, final reimbursement of approximately $30,000 for the costs in connection with the utility model infringement were paid to TauroPharm.
On November 22, 2017, the EPO in Munich, Germany held a further oral hearing in this matter. At the hearing, the panel held that the Prosl European Patent would be invalidated because it did not meet the requirements of novelty based on a technical aspect of the European intellectual property law. The Company disagrees with this decision and has appealed the decision. The Company continues to believe that the Prosl European Patent is indeed novel and that its validity should be maintained. There can be no assurance that the Company will prevail in this matter.
On January 16, 2015, the Company filed a complaint against TauroPharm GmbH and its managing directors in the District Court of Cologne, Germany. In the complaint, the Company alleged violation of the German Unfair Competition Act by TauroPharm and that TauroPharm is improperly and unfairly using its proprietary information relating to the composition and manufacture of Neutrolin, in the manufacture and sale of TauroPharm’s products TauroLockTM, TauroLock-HEP100 and TauroLock-HEP500. The Company sought a cease and desist order against TauroPharm from continuing to manufacture and sell any product containing taurolidine (the active pharmaceutical ingredient (“API”) of Neutrolin) and citric acid in addition to possible other components, damages for any sales in the past and the removal of all such products from the market. Hearings in this matter were held in the District Court of Cologne, Germany on November 19, 2015, on November 15, 2016 and on November 20, 2018. A decision was rendered by the court on December 11, 2018, dismissing the complaint in its entirety. The Company therefore appealed in January 2019. An oral hearing was held on September 6, 2019. In view of new arguments brought forward in this hearing, the Court issued an evidentiary order on September 27, 2019 ordering an expert opinion. The expert opinion was not in the Company’s favor. In a supplementary expert opinion submitted after the Company had brought forward arguments against the first expert opinion, the expert confirmed his view. In an oral hearing held on June 18, 2021, the Court only heard from the expert, and the Court, as well as both parties, asked further questions to the expert around his expert opinion. At the end of the hearing and internal deliberation among the panel of judges, the Court indicated that it would dismiss the complaint of the Company, if the Company did not withdraw the appeal. As there were no advantages to further pursuing the matter in view of the Court’s statements, the Company withdrew the appeal and the proceedings are therefore now closed. TauroPharm requested an increase of the value in dispute determined by the Court in order to receive a higher reimbursement of costs (as this is based on the value in dispute under German law) but the request was rejected in view of arguments brought forward against it by legal counsel of the Company. The Company will have to reimburse costs in the amount of approximately $41,000 plus interest to TauroPharm.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
In connection with the aforementioned patent and utility model infringement and unfair competition proceedings against TauroPharm, the Company was required by the District Courts of Mannheim and Cologne to provide security deposits to cover legal fees in the event TauroPharm is entitled to reimbursement of these costs. As of December 31, 2021, the aggregate deposit was approximately $132,000, which the Company recorded as restricted cash on the condensed consolidated balance sheets, after deducting approximately $48,000 released by the court to the Company during the year ended December 31, 2021.
Commitments
In-Licensing
In 2008, the Company entered into a License and Assignment Agreement (the “NDP License Agreement”) with ND Partners, LLP (“NDP”). Pursuant to the NDP License Agreement, NDP granted the Company exclusive, worldwide licenses for certain antimicrobial catheter lock solutions, processes for treating and inhibiting infections, a biocidal lock system and a taurolidine delivery apparatus, and the corresponding United States and foreign patents and applications (the “NDP Technology”). The Company acquired such licenses and patents through its assignment and assumption of NDP’s rights under certain separate license agreements by and between NDP and Dr. Hans-Dietrich Polaschegg, Dr. Klaus Sodemann and Dr. Johannes Reinmueller. As consideration in part for the rights to the NDP Technology, the Company paid NDP an initial licensing fee of $325,000 and granted NDP a 5% equity interest in the Company, consisting of 7,996 shares of the Company’s common stock.
The Company is required to make payments to NDP upon the achievement of certain regulatory and sales-based milestones. Certain of the milestone payments are to be made in the form of shares of common stock currently held in escrow for NDP, and other milestone payments are to be paid in cash. The maximum aggregate number of shares issuable upon achievement of milestones is 29,109 shares. In 2014, a certain milestone was achieved resulting in the release of 7,277 shares held in escrow. The number of shares held in escrow as of December 31, 2021 is 21,832 shares of common stock. The maximum aggregate amount of cash payments due upon achievement of milestones is $3,000,000 with the balance being $2,500,000 as of December 31, 2021 and 2020. Events that trigger milestone payments include but are not limited to the reaching of various stages of regulatory approval and upon achieving certain worldwide net sales amounts. There were no milestones achieved during the years ended December 31, 2021 and 2020.
The NDP License Agreement may be terminated by the Company on a country-by-country basis upon 60 days prior written notice. If the NDP License Agreement is terminated by either party, the Company’s rights to the NDP Technology will revert back to NDP.
Employment Agreements
On September 27, 2016, the Company entered into an employment agreement with Khoso Baluch, its former Chief Executive Officer, which upon its expiration in September 2019, was replaced with a new agreement, dated September 26, 2019. On October 4, 2021, Mr. Baluch retired from the Company and his employment agreement was terminated. In connection with his separation from service, the Company and Mr. Baluch entered into a separation agreement and release dated as of October 1, 2021 (the “Baluch Separation Agreement”). Mr. Baluch’s retirement was treated as a termination without Cause (as defined below) under the employment agreement. Under the Baluch Separation Agreement, Mr. Baluch received the severance payments and benefits described below with respect to a termination by the Company without Cause. Mr. Baluch met the eligibility requirements for retirement as of the date of his separation, so certain of Mr. Baluch’s vested stock options will be exercisable for up to three years after the date of his separation under the terms of the applicable grant agreements. The Baluch Separation Agreement provides this retirement treatment for all of Mr. Baluch’s outstanding vested options. The Company reimbursed Mr. Baluch for legal fees incurred in connection with the review of the Baluch Separation Agreement. Mr. Baluch is bound by confidentiality, non-solicitation and non-competition covenants under his employment agreement, and an extended covenant not to solicit employees under the Baluch Separation Agreement, among other terms. Total severance amount was $495,833 of which $177,492 was paid and $318,341 was accrued as of December 31, 2021.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
On March 1, 2017, the Company entered into an employment agreement with John Armstrong, its former Executive Vice President for Technical Operations, which upon its expiration in March 2020, was replaced with a new agreement dated April 17, 2020. On October 4, 2021, Mr. Armstrong retired from the Company and his employment agreement was terminated. In connection with his separation from service, the Company and Mr. Armstrong entered into a separation agreement and release dated as of October 4, 2021 (the “Armstrong Separation Agreement”). Mr. Armstrong’s retirement was treated as a termination without Cause under the employment agreement. Under the Armstrong Separation Agreement, Mr. Armstrong received the severance payments and benefits described below with respect to a termination by the Company without Cause. Mr. Armstrong met the eligibility requirements for retirement as of the date of his separation, so certain of Mr. Armstrong’s vested stock options will be exercisable for up to three years after the date of his separation under the terms of the applicable grant agreements. The Armstrong Separation Agreement provides this retirement treatment for all outstanding vested options. The Company reimbursed Mr. Armstrong for legal fees incurred in connection with the review of the Armstrong Separation Agreement. Mr. Armstrong is bound by confidentiality, non-solicitation and non-competition covenants under his employment agreement, and an extended covenant not to solicit employees under the Armstrong Separation Agreement, among other terms. Total severance amount was $297,917 of which $134,514 was paid and $163,403 was accrued as of December 31, 2021.
On March 19, 2018, the Company entered into an employment agreement with Elizabeth Masson-Hurlburt to serve as its Executive Vice President and Head of Clinical Operations, which upon its expiration in March 2021, was replaced with a new agreement dated March 10, 2021. On March 19, 2019, the Company entered into an employment agreement with Phoebe Mounts to serve as its Executive Vice President and General Counsel and Head of Regulatory, Compliance and Legal, effective May 1, 2019. On April 29, 2021, the Company entered into an employment agreement with Thomas Nusbickel to serve as its Executive Vice President and Chief Commercial Officer, effective May 13, 2021. After the initial three-year term of each employment agreement, the term of the employment agreement will automatically renew for additional successive one-year periods, unless either party notifies the other in writing at least 90 days before the expiration of the then-current term that the term will not be renewed.
On May 11, 2020, the Company entered into an employment agreement with Matthew David to serve as its Chief Financial Officer. After the initial three-year term of the employment agreement, the term of the employment agreement will automatically renew for additional successive one-year periods, unless either party notifies the other in writing at least 90 days before the expiration of the then-current term that the term will not be renewed. On October 26, 2021, the Company entered into a letter agreement with Dr. David which modified certain terms of his employment agreement and provided other compensation as a result of Dr. David serving as the Company’s interim Chief Executive Officer effective as of October 4, 2021. Pursuant to the letter agreement, during the period in which Dr. David serves as interim Chief Executive Officer, his base salary will increase to $425,000 from $330,000, which is the amount set forth in his employment agreement and will be reviewed and increased, if appropriate, by the Company’s Compensation Committee six months following October 4, 2021 if Dr. David continues to serve as interim Chief Executive Officer on such date. After Dr. David ceases to serve as interim Chief Executive Officer, and as he continues to serve as Chief Financial Officer, the Company will provide him with an annual base salary of $375,000, representing a $45,000 increase from his current salary level under the employment agreement. The Board, or the Company’s Compensation Committee, will review such base salary to determine whether an increase is appropriate in 2022 as part of the 2022 compensation review cycle and benchmarking review. Under the letter agreement, Dr. David’s target annual bonus with respect to the period during which he serves as interim Chief Executive Officer is increased to 60% from 30% of his base salary, which is otherwise set forth in his employment agreement. After Dr. David ceases to serve as interim Chief Executive Officer, and as he continues to serve as Chief Financial Officer, his target annual bonus will increase to 40% of his base salary. Under the letter agreement, in the event Dr. David’s employment is terminated by the Company other than as a result of his death or disability or notice of nonrenewal of the employment agreement, and other than for Cause, or if he resigns for Good Reason, in either case during the period he serves as interim Chief Executive Officer, he will be eligible for severance equal to his base salary for a period of 12 months following his termination date, which is increased from nine months as is otherwise provided for in his employment agreement. Dr. David has agreed to waive any rights he may have under his employment agreement to a Good Reason termination as a result of his ceasing to serve as our interim Chief Executive Officer at a future date. In connection with Dr. David serving as interim Chief Executive Officer, the Board granted Dr. David a stock option with respect to 125,000 shares of the Company’s common stock with an exercise price of $5.56 per share, which was the closing price of the Company’s common stock on the Nasdaq Global Market on the date of grant. The option vests over four years in four equal annual installments beginning on the date of grant, subject to Dr. David’s continued employment, consistent with the terms of the Company’s standard form of option agreement.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
Pursuant to their respective employment agreements, Mr. Baluch received an annual salary of $425,000, Mr. Armstrong received an annual salary of $325,000, Ms. Masson-Hurlburt receives an annual salary of $315,000 (effective March 2021), Dr. Mounts receives an annual salary of $350,000 (amended to $375,000 in January 2021), Mr. Nusbickel receives and annual salary of $375,000 and Dr. David receives an annual salary of $330,000 (amended to $425,000 while he serves as interim Chief Executive Officer). Such salaries cannot be decreased unless all officers and/or members of the Company’s executive management team experience an equal or greater percentage reduction in base salary and/or total compensation, provided that any reduction in an executive’s salary may be no greater than 25%. Each executive will be eligible for an annual bonus, which may equal up to 80% for Mr. Baluch (the target amount is 80%, but the bonus may exceed that amount), up to 35% for Mr. Armstrong, up to 30% for Ms. Masson-Hurlburt, up to 30% for Dr. Mounts, up to 30% for Mr. Nusbickel and up to 30% for Dr. David (and up to 60% while he serves as interim Chief Executive Officer), of his or her base salary then in effect, as determined by the Company’s Board or the Compensation Committee. In determining such bonus payment, the Company’s Board or the Compensation Committee will take into consideration the achievement of specified Company objectives, predetermined by the Company’s Board or the Compensation Committee and Chief Executive Officer, and such other factors as the Company’s Board or the Compensation Committee deems appropriate. Each executive must be employed through December 31 of a given year to be eligible to earn that year’s annual bonus.
The following provisions of the employment agreements with Dr. David, Dr. Mounts, Ms. Masson-Hurlburt and Mr. Nusbickel are identical except where noted.
If the Company terminates the executive’s employment for Cause, the executive will be entitled to receive only the accrued compensation due to him or her as of the date of such termination, rights to indemnification and directors’ and officers’ liability insurance, and as otherwise required by law, and certain equity awards will be forfeited.
If the Company terminates the executive’s employment other than for Cause, and other than for death, disability or notice of nonrenewal, or if the executive resigns for Good Reason (as defined below), the executive will receive the following benefits: (i) payment of any accrued compensation and any unpaid bonus relating to the completed prior year, as well as rights to indemnification and directors’ and officers’ liability insurance and any rights or privilege otherwise required by law; (ii) the Company will continue to pay his or her base salary for a period of twelve months in the case of Mr. Baluch and Dr. David while he is serving as interim Chief Executive Officer, and nine months for the other executives following the effective date of the termination of employment; (iii) payment on a prorated basis for any target bonus for the year of termination based on the actual achievement of the specified bonus objectives; (iv) if the executive timely elects continued health insurance coverage under COBRA, then the Company will pay the premium to continue such coverage for him or her and his or her eligible dependents in an amount equal to the portion paid for by the Company during the executive’s employment until the conclusion of the time when he or she is receiving continuation of base salary payments or until he or she becomes eligible for group health insurance coverage under another employer’s plan, whichever occurs first, provided however that the Company has the right to terminate such payment of COBRA premiums on behalf of the executive and instead pay him or her a lump sum amount equal to the COBRA premium times the number of months remaining in the specified period if the Company determines in its discretion that continued payment of the COBRA premiums is or may be discriminatory under Section 105(h) of the Code; and (v) unvested equity awards that are scheduled to vest on or before the next succeeding anniversary of the date of termination shall be accelerated and deemed to have vested as of the termination date; provided that any performance based equity awards or stock options whose vesting requirements have not been successfully met as of the date of termination of employment or resignation with Good Reason will not accelerate. In addition, the event of a termination by the Company without Cause or the executive’s resignation of employment for Good Reason, in either case within 24 months following a Corporate Transaction (as defined in the employment agreement), all equity awards and stock options shall become fully vested and exercisable, and vested stock options will remain exercisable for a specified period of time following termination or resignation or, if earlier, the expiration date of the stock option. The separation benefits set forth above are conditioned upon the executive executing a release of claims against us, our parents, subsidiaries, and affiliates, and each such entities’ officers, directors, employees, agents, successors, and assigns in a form acceptable to us, within a time specified therein, which release is not revoked within any time period allowed for revocation under applicable law.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
For purposes of the agreement, “Cause” is defined as: (i) the willful failure, disregard, or refusal by the executive to perform his or her material duties or obligations under the employment agreement (other than as a result of executive’s mental incapacity or illness; (ii) any willful, intentional, or grossly negligent act by the executive having the effect of materially injuring (whether financially or otherwise) our business or reputation or any of our affiliates; (iii) executive’s conviction of any felony involving moral turpitude (including entry of a guilty or nolo contendere plea); (iv) the executive’s qualification as a “bad actor,” as defined by 17 CFR 230.506(a); (v) the good faith determination by the Board, after a reasonable and good-faith investigation by the Company that the executive engaged in some form of harassment or discrimination prohibited by law (including, without limitation, harassment on the basis of age, sex or race) unless the executive’s actions were specifically directed by the Board; (vi) any material misappropriation or embezzlement by the executive of the Company or its affiliates’ property (whether or not a misdemeanor or felony); or (vii) material breach by the executive of the employment agreement that is materially injurious to the Company and that is not cured, to the extent subject to cure, by executive to our reasonable satisfaction.
For purposes of the agreement, “Good Reason” is defined as any of the following without the executive’s consent: (i) any material breach of the employment agreement by the Company; (ii) any material diminution by the Company of the executive’s duties, responsibilities, or authority; (iii) a material reduction in the executive’s annual base salary unless all officers and/or members of the Company’s executive management team experience an equal or greater percentage reduction in annual base salary and/or total compensation, provided that any reduction may be no greater than 25%; (iv) a material reduction in the executive’s target bonus level unless all officers and/or members of our executive management team experience an equal or greater percentage reduction related to target bonus levels, provided that any reduction may be no greater than 25%.
If the executive terminates his or her employment by written notice of termination or if the executive or the Company terminates his or her employment by providing a notice of nonrenewal at least 90 days before the employment agreement is set to expire, the executive will not be entitled to receive any payments or benefits other than any accrued compensation, any unpaid prior year’s bonus, rights to indemnification and directors’ and officers’ liability insurance and as otherwise required by law.
If the executive’s employment is terminated as a result of his or her death or disability, the Company will pay him or her or his or her estate, as applicable, any accrued compensation and any unpaid prior year’s bonus.
The Company’s employment agreements with Dr. David, Dr. Mounts, Ms. Masson-Hurlburt and Mr. Nusbickel each contain a non-compete provision that provides that during the employment and for a specified period immediately following the executive’s separation from employment for any reason, the executive is prohibited from engaging in any business involving the development or commercialization of a preventive anti-infective product that would be a direct competitor of Defencath/Neutrolin or a product containing taurolidine or any other product being actively developed or produced by the Company within the United States and the European Union (or in the case of Dr. David, Ms. Masson-Hurlburt and Mr. Nusbickel, worldwide) on the date of termination of his or her employment.
Other
The Company entered into a seven-year operating lease agreement in March 2020 for an office space at 300 Connell Drive, Berkeley Heights, New Jersey 07922. The lease agreement, with a monthly average of approximately $17,000 commenced on September 16, 2020. The Company’s sublease on its previous premises at 400 Connell Drive, Berkeley Heights, New Jersey 07922 terminated on November 30, 2020 (see Note 10).
Note 7 — Stockholders’ Equity:
Common Stock:
On July 30, 2020, the Company completed an underwritten public offering of its common stock, par value $0.001 per share, which yielded net proceeds of approximately $21.3 million. The public offering was made pursuant to an underwriting agreement with SunTrust Robinson Humphrey, Inc. and JMP Securities LLC (collectively, the “Underwriters”), relating to the issuance and sale of an aggregate of 5,111,110 shares of common stock, including 666,666 shares of common stock pursuant to the full exercise of the Underwriters’ option to purchase additional shares, at a public offering price of $4.50 per share. The offering was made pursuant to the Company’s effective registration statement on Form S-3 Registration Statement No. 333-223562 previously filed with and declared effective by the SEC and a prospectus supplement and accompanying prospectus filed with the SEC.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
The Company had a prior sales agreement with FBR Securities, Inc., (formerly known as B. Riley FBR, Inc.) (“B. Riley”) for its ATM program, which expired on April 16, 2018, under which the Company could issue and sell up to an aggregate of $60.0 million of shares of its common stock. On March 9, 2018, the Company entered into a new agreement with B. Riley for the sale of up to $14.7 million of the Company’s common stock under the ATM program, pursuant to a registration statement filed on March 9, 2018 for an aggregate of $70 million of the Company’s securities, which became effective on April 16, 2018. This new ATM agreement replaced a prior sales agreement with B. Riley that expired on April 16, 2018. The ATM program amount was increased by $25.0 million in November 2018. Under the ATM program, the Company may issue and sell common stock from time to time through B. Riley acting as agent, subject to limitations imposed by the Company and subject to B. Riley’s acceptance, such as the number or dollar amount of shares registered under the registration statement to which the offering relates. B. Riley is entitled to a commission of up to 3% of the gross proceeds from the sale of common stock sold under the ATM program. During the year ended December 31, 2020, the Company sold 1,854,970 shares of common stock under the new and expired ATM programs, and realized net proceeds of approximately $11.4 million. At December 31, 2020, this ATM program and the current shelf registration for the issuance of equity, debt or equity-linked securities has been exhausted.
In November 2020, the Company filed a new registration statement, under which the Company could issue and sell up to an aggregate of $100.0 million of shares of its common stock. On November 27, 2020, the Company entered into an Amended and Restated At Market Issuance Sales Agreement (“Amended Sales Agreement”) with B. Riley FBR Inc. and Needham & Company, LLC as sales agents. The Amended Sales Agreement relates to the sale of shares of up to $25.0 million of the Company’s common stock under its ATM program, of which the Company may issue and sell common stock from time to time through the sales agents, subject to limitations imposed by the Company and subject to the sales agents’ acceptance, such as the number or dollar amount of shares registered under the registration statement to which the offering relates. The sales agents are entitled to a commission of up to 3% of the gross proceeds from the sale of common stock sold under the ATM program. During the year ended December 31, 2020, the Company sold 832,676 shares of common stock under the Amended Sales Agreement and realized net proceeds of approximately $7.0 million. At December 31, 2020, the Company had approximately $17.8 million available under the Amended Sales Agreement and $75.0 million available under its shelf registration statement for the issuance of equity, debt or equity-linked securities unrelated to the Amended Sales Agreement. On February 5, 2021, the Company allocated to its ATM program an additional $25.0 million of the remaining $75.0 million available under its shelf registration statement. Giving effect to the additional $25.0 million, plus the $17.8 million available at December 31, 2020, the Company had a total of $42.8 million available under the ATM program at February 5, 2021. During the year ended December 31, 2021, the Company sold an aggregate of 3,737,862 shares of its common stock under the ATM program and realized net proceeds of approximately $41.5 million.
On August 12, 2021, the Company entered into an At Market Issuance Sales Agreement with Truist Securities, Inc. and JMP Securities LLC, as sales agents, pursuant to which the Company may sell, from time to time, an aggregate of up to $50.0 million of its common stock through the sales agents under its ATM program, subject to limitations imposed by the Company and subject to the sales agent’s acceptance, such as the number or dollar amount of shares registered under the registration statement to which the offering relates. The sales agents are entitled to a commission of up to 3% of the gross proceeds from the sale of common stock sold under the ATM program. As of December 31, 2021, the Company has $50.0 million available under its ATM program relating to its shelf registration statement filed in November 2020 and it has $150.0 million available under its new shelf registration statement filed on August 12, 2021 for the issuance of equity, debt or equity-linked securities.
During the year ended December 31, 2021, the Company issued an aggregate of 656,069 shares of its common stock upon conversion of 50,000 Series C-3 preferred shares by an unrelated party and 10,001 Series G preferred shares by a related party.
During the year ended December 31, 2021 and 2020, the Company issued an aggregate of 31,407 and 91,500 shares of its common stock, respectively, upon cash exercise of warrants, resulting in net proceeds to the Company of $165,000 and $412,000, respectively.
During the year ended December 31, 2021, the Company issued an aggregate of 70,269 shares of its common stock upon cashless exercise of 95,286 warrants.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
During the year ended December 31, 2021, there were no restricted stock units issued by the Company and for the year ended December 31, 2020, the Company issued an aggregate of 2,490 shares of its common stock upon the vesting of restricted stock units issued to the Company’s board of directors.
During the year ended December 31, 2021, the Company issued an aggregate of 32,734 shares of its common stock upon exercise of stock options, resulting in net proceeds to the Company of $137,000. No stock options were exercised during the year ended December 31, 2020.
Restricted Stock Units
During the years ended December 31, 2021 and 2020 the Company did not grant any restricted stock units (“RSUs”) to its officers and directors. During the year ended December 31, 2020, the compensation expense recorded for the 2,490 RSUs that vested was $11,000. At December 31, 2021 and 2020, there were no RSUs outstanding.
Preferred Stock
The Company is authorized to issue up to 2,000,000 shares of preferred stock in one or more series without stockholder approval. The Company’s board of directors has the discretion to determine the rights, preferences, privileges and restrictions, including voting rights, dividend rights, conversion rights, redemption privileges and liquidation preferences, of each series of preferred stock. Of the 2,000,000 shares of preferred stock authorized, the Company’s board of directors has designated (all with par value of $0.001 per share) the following:
| | | As of December 31, 2021 | | | As of December 31, 2020 | |
| | | Preferred
Shares
Outstanding | | | Liquidation
Preference
(Per Share) | | | Total
Liquidation
Preference | | | Preferred
Shares
Outstanding | | | Liquidation
Preference
(Per Share) | | | Total
Liquidation
Preference | |
| Series C-3 | | | 2,000 | | | $ | 10.00 | | | $ | 20,000 | | | | 52,000 | | | $ | 10.00 | | | $ | 520,000 | |
| Series E | | | 89,623 | | | $ | 49.20 | | | $ | 4,409,452 | | | | 89,623 | | | $ | 49.20 | | | $ | 4,409,452 | |
| Series G | | | 89,999 | | | $ | 187.36 | | | $ | 16,862,213 | | | | 100,000 | | | $ | 187.36 | | | $ | 18,736,452 | |
| Total | | | 181,622 | | | | | | | $ | 21,291,665 | | | | 241,623 | | | | | | | $ | 23,665,904 | |
During the year ended December 31, 2021, 50,000 Series C-3 preferred shares were converted into 100,000 shares of the Company’s common stock by an unrelated party and 10,001 Series G preferred shares were converted into 556,069 shares of the Company’s common stock by a related party.
The following rights, privileges, terms and condition apply to the outstanding preferred stock at December 31, 2021:
Series C-3 Non-Voting Preferred Stock
Rank. The Series C-3 non-voting preferred stock will rank senior to our common stock; senior to any class or series of capital stock created after the issuance of the Series C-3 non-voting preferred stock; and junior to the Series E voting convertible preferred stock in each case, as to dividends or distributions of assets upon our liquidation, dissolution or winding up whether voluntarily or involuntarily.
Conversion. Each share of Series C-3 preferred stock is convertible into 2 shares of our common stock (subject to adjustment in the event of stock dividends and distributions, stock splits, stock combinations, or reclassifications affecting our common stock) at a per share price of $5.00 at any time at the option of the holder, except that a holder will be prohibited from converting shares of Series C-3 preferred stock into shares of common stock if, as a result of such conversion, such holder, together with its affiliates, would beneficially own more than 9.99% of the total number of shares of our common stock then issued and outstanding.
Liquidation Preference. In the event of our liquidation, dissolution or winding up, holders of Series C-3 preferred stock will receive a payment equal to $10.00 per share of Series C-3 preferred stock before any proceeds are distributed to the holders of our common stock. After the payment of this preferential amount, and subject to the rights of holders of any class or series of our capital stock hereafter created specifically ranking by its terms senior to the Series C-3 preferred stock and holders of Series C-3 preferred stock will participate ratably in the distribution of any remaining assets with the common stock and any other class or series of our capital stock hereafter created that participates with the common stock in such distributions.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
Voting Rights. Shares of Series C-3 preferred stock will generally have no voting rights, except as required by law and except that the consent of holders of two thirds of the outstanding Series C-3 preferred Stock will be required to amend the terms of the Series C-3 preferred stock or the certificate of designation for the Series C-3 preferred stock.
Dividends. Holders of Series C-3 preferred stock are entitled to receive, and we are required to pay, dividends on shares of the Series C-3 preferred stock equal (on an as-if-converted-to-common-stock basis) to and in the same form as dividends (other than dividends in the form of common stock) actually paid on shares of the common stock when, as and if such dividends (other than dividends in the form of common stock) are paid on shares of the common stock.
Redemption. We are not obligated to redeem or repurchase any shares of Series C-3 preferred stock. Shares of Series C-3 preferred stock are not otherwise entitled to any redemption rights, or mandatory sinking fund or analogous fund provisions.
Listing. There is no established public trading market for the Series C-3 preferred stock, and we do not expect a market to develop. In addition, we do not intend to apply for listing of the Series C-3 preferred stock on any national securities exchange or trading system.
Fundamental Transactions. If, at any time that shares of Series C-3 preferred stock are outstanding, we effect a merger or other change of control transaction, as described in the certificate of designation and referred to as a fundamental transaction, then a holder will have the right to receive, upon any subsequent conversion of a share of Series C-3 preferred stock (in lieu of conversion shares) for each issuable conversion share, the same kind and amount of securities, cash or property as such holder would have been entitled to receive upon the occurrence of such fundamental transaction if such holder had been, immediately prior to such fundamental transaction, the holder of a share of common stock.
Series E Voting Convertible Preferred Stock
Rank. The Series E voting preferred stock will rank senior to our common stock; senior to any class or series of capital stock created after the issuance of the Series E voting convertible preferred stock; senior to the Series C-3 non-voting convertible preferred stock; and on parity with the Series G voting convertible preferred stock in each case, as to dividends or distributions of assets upon our liquidation, dissolution or winding up whether voluntarily or involuntarily.
Conversion. Each share of Series E preferred stock is convertible into 4.3733 shares of our common stock (subject to adjustment as provided in the certificates of designation for the Series E preferred stock) at a per share price of $3.75 at any time at the option of the holder, except that a holder will be prohibited from converting shares of Series E preferred stock into shares of common stock if, as a result of such conversion, such holder, together with its affiliates, would beneficially own more than 4.99% of the total number of shares of our common stock then issued and outstanding.
Liquidation Preference. In the event of our liquidation, dissolution or winding up, holders of Series E preferred stock will receive a payment equal to $49.20 per share of Series E preferred stock on parity with the payment of the liquidation preference due the Series G preferred stock, but before any proceeds are distributed to the holders of common stock, and the Series C-3 non-voting convertible preferred stock. After the payment of this preferential amount, holders of Series E preferred stock will participate ratably in the distribution of any remaining assets with the common stock and any other class or series of our capital stock that participates with the common stock in such distributions.
Voting Rights. Shares of Series E preferred stock are entitled to vote on an as-converted basis, based upon an assumed conversion price of $7.93.
Dividends. Holders of Series E preferred stock are entitled to receive, and we are required to pay, dividends on shares of the Series E preferred stock equal (on an as-if-converted-to-common-stock basis) to and in the same form as dividends (other than dividends in the form of common stock) actually paid on shares of the common stock when, as and if such dividends (other than dividends in the form of common stock) are paid on shares of the common stock.
Redemption. We are not obligated to redeem or repurchase any shares of Series E preferred stock. Shares of Series E preferred stock are not otherwise entitled to any redemption rights, or mandatory sinking fund or analogous fund provisions.
Listing. There is no established public trading market for the Series E preferred stock, and we do not expect a market to develop. In addition, we do not intend to apply for listing of the Series E preferred stock on any national securities exchange or trading system.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
Fundamental Transactions. If, at any time that shares of Series E preferred stock are outstanding, we effect a merger or other change of control transaction, as described in the certificate of designation and referred to as a fundamental transaction, then a holder will have the right to receive, upon any subsequent conversion of a share of Series E preferred stock (in lieu of conversion shares) for each issuable conversion share, the same kind and amount of securities, cash or property as such holder would have been entitled to receive upon the occurrence of such fundamental transaction if such holder had been, immediately prior to such fundamental transaction, the holder of a share of common stock.
Debt Restriction. As long as any of the Series E preferred stock is outstanding, we cannot create, incur, guarantee, assume or suffer to exist any indebtedness, other than (i) trade payables incurred in the ordinary course of business consistent with past practice, and (ii) up to $10 million aggregate principal amount of indebtedness with a maturity less than twelve months outstanding at any time, which amount may include up to $5 million of letters of credit outstanding at any time.
Other Covenants. In addition to the debt restrictions above, as long as any of the Series E preferred stock is outstanding, we cannot, among others things: create, incur, assume or suffer to exist any encumbrances on any of our assets or property; redeem, repurchase or pay any cash dividend or distribution on any of our capital stock (other than as permitted, which includes the dividends on the Series E preferred stock and Series G preferred stock); redeem, repurchase or prepay any indebtedness (other than as permitted); or engage in any material line of business substantially different from our current lines of business.
Purchase Rights. In the event we issue any options, convertible securities or rights to purchase stock or other securities pro rata to the holders of common stock, then a holder of Series E preferred stock will be entitled to acquire, upon the same terms a pro rata amount of such stock or securities as if the Series E preferred stock had been converted to common stock.
Series G Voting Convertible Preferred Stock
Rank. The Series G voting convertible preferred stock will rank senior to our common stock; senior to any class or series of capital stock created after the issuance of the Series G voting convertible preferred stock; junior to the Series C-3 non-voting convertible preferred stock, pending the consent of the holders of such series to the subordination thereof; and on parity with the Series E voting convertible preferred stock in each case, as to dividends or distributions of assets upon our liquidation, dissolution or winding up whether voluntarily or involuntarily.
Conversion. Each share of Series G preferred stock is convertible into approximately 55.5978 shares of our common stock (subject to adjustment as provided in the certificate of designation for the Series G preferred stock) at a per share price of $3.37 at any time at the option of the holder, except that a holder will be prohibited from converting shares of Series G preferred stock into shares of common stock if, as a result of such conversion, such holder, together with its affiliates, would beneficially own more than 4.99% of the total number of shares of our common stock then issued and outstanding.
Liquidation Preference. In the event of our liquidation, dissolution or winding up, holders of Series E preferred stock will receive a payment equal to $187.36452 per share of Series G preferred stock on parity with the payment of the liquidation preference due the Series E preferred stock, but before any proceeds are distributed to the holders of Series C-3 preferred stock (pending the consent of the holders of such series to the subordination thereof) and any proceeds are distributed to the holders of common stock. After the payment of this preferential amount, holders of Series G preferred stock will participate ratably in the distribution of any remaining assets with the common stock and any other class or series of our capital stock that participates with the common stock in such distributions.
Voting Rights. Shares of Series G preferred stock are entitled to vote on an as-converted basis, based upon an assumed conversion price of $7.93.
Dividends. Holders of Series G Preferred stock are entitled to receive, and we are required to pay, dividends on shares of the Series G preferred stock equal (on an as-if-converted-to-common-stock basis) to and in the same form as dividends (other than dividends in the form of common stock) actually paid on shares of the common stock when, as and if such dividends (other than dividends in the form of common stock) are paid on shares of the common stock.
Redemption. We are not obligated to redeem or repurchase any shares of Series G preferred stock. Shares of Series G preferred stock are not otherwise entitled to any redemption rights, or mandatory sinking fund or analogous fund provisions.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
Listing. There is no established public trading market for the Series G preferred stock, and we do not expect a market to develop. In addition, we do not intend to apply for listing of the Series G preferred stock on any national securities exchange or trading system.
Fundamental Transactions. If, at any time that shares of Series G preferred stock are outstanding, we effect a merger or other change of control transaction, as described in the certificate of designation and referred to as a fundamental transaction, then a holder will have the right to receive, upon any subsequent conversion of a share of Series G preferred stock (in lieu of conversion shares) for each issuable conversion share, the same kind and amount of securities, cash or property as such holder would have been entitled to receive upon the occurrence of such fundamental transaction if such holder had been, immediately prior to such fundamental transaction, the holder of a share of common stock.
Debt Restriction. As long as any of the Series G preferred stock is outstanding, we cannot create, incur, guarantee, assume or suffer to exist any indebtedness, other than (i) trade payables incurred in the ordinary course of business consistent with past practice, and (ii) up to $10 million aggregate principal amount of indebtedness with a maturity less than twelve months outstanding at any time, which amount may include up to $5 million of letters of credit outstanding at any time.
Other Covenants. In addition to the debt restrictions above, as long as any of the Series G preferred stock is outstanding, we cannot, among others things: create, incur, assume or suffer to exist any encumbrances on any of our assets or property; redeem, repurchase or pay any cash dividend or distribution on any of our capital stock (other than as permitted, which includes the dividends on the Series E preferred stock and the Series G preferred stock); redeem, repurchase or prepay any indebtedness (other than as permitted); or engage in any material line of business substantially different from our current lines of business.
Purchase Rights. In the event we issue any options, convertible securities or rights to purchase stock or other securities pro rata to the holders of common stock, then a holder of Series G preferred stock will be entitled to acquire, upon the same terms a pro rata amount of such stock or securities as if the Series G preferred stock had been converted to common stock.
Stock Options:
On November 26, 2019, the Company’s shareholders approved the CorMedix Inc. 2019 Omnibus Stock Incentive Plan (the “2019 Plan”). Pursuant to the 2019 Plan and subject to certain adjustments as described below, the Company may issue up to 3,000,000 shares of its common stock, plus any shares that remain available for grant under its 2013 Stock Incentive Plan (the “2013 Plan”) as of the effective date (up to a maximum carry-forward of 522,606 shares plus any outstanding options under the 2013 Plan that were canceled, forfeited and expired after the approval of the 2019 Plan), as long-term equity incentives to the Company’s employees, consultants, and directors. The long-term incentives may be in the form of stock options, stock appreciation rights, restricted stock, restricted stock units, dividend equivalent rights, or other rights or benefits (collectively, stock rights) to employees, consultants, and directors of the Company or a related entity (collectively, participants). The Company believes that the effective use of long- term equity incentives is essential to attract, motivate, and retain employees, consultants and directors, to further align participants’ interests with those of the Company’s stockholders, and to provide participants incentive compensation opportunities that are competitive with those offered by other companies in the same industry and locations as the Company.
The 2019 Plan is a new equity compensation plan for the Company’s employees, consultants, and directors which replaced the 2013 Plan. The 2013 Plan and the Amended and Restated 2006 Stock Incentive Plan are referred to collectively as the “Prior Plans”. No further awards will be granted under the Prior Plans after the approval of the 2019 Plan. Awards outstanding under the Prior Plans will remain outstanding in accordance with their terms and the Prior Plans.
During the years ended December 31, 2021 and 2020, the Company granted ten-year qualified and non-qualified stock options to its officers, directors, employees and consultants covering an aggregate of 1,664,700 and 1,111,984 shares of the Company’s common stock under the 2019 Plan, respectively. The weighted average exercise price of these options is $7.98 and $5.11 per share, respectively.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
During the years ended December 31, 2021 and 2020, total compensation expense for stock options issued to employees, directors, officers and consultants was $5,043,000 and $2,489,000, respectively. As of December 31, 2021, there was $5,516,000 total unrecognized compensation expense related to unvested stock options granted which expense is expected to be recognized over an expected remaining weighted average period of 1.6 years. All share-based awards are recognized on a straight-line method, assuming all awards granted will vest. Forfeitures of share-based awards are recognized in the period in which they occur.
The fair value at grants dates of the grants issued subject to service and performance-based vesting conditions were determined using the Black-Scholes option pricing model with the following assumptions:
| | | Year Ended December 31, |
| | | 2021 | | 2020 |
| Risk-free interest rate | | 0.5% - 1.26% | | 0.27% - 1.67% |
| Expected volatility | | 102.93% - 107.1% | | 102.7% - 107.9% |
| Expected term (years) | | 1.97 – 5 years | | 5-10 years |
| Expected dividend yield | | 0.0% | | 0.0% |
| Weighted-average grant date fair value of options granted during the period | | $5.56 | | $3.59 |
The Company estimated the expected term of the stock options granted based on anticipated exercises in future periods. The expected term of the stock options granted to consultants is based upon the full term of the respective option agreements. The expected stock price volatility for the Company’s stock options is calculated based on the historical volatility since the initial public offering of the Company’s common stock in March 2010. The expected dividend yield of 0.0% reflects the Company’s current and expected future policy for dividends on the Company’s common stock. To determine the risk-free interest rate, the Company utilized the U.S. Treasury yield curve in effect at the time of grant with a term consistent with the expected term of the Company’s awards which is 5 years for employees and 10 years for non-employees.
The following table summarizes the Company’s stock options activity and related information for the year ended December 31, 2021:
| | | Shares
Underlying
Stock
Options | | | Weighted-
Average
Exercise
Price | | | Weighted-
Average
Remaining
Contractual
Term
(Years) | | | Aggregate
Intrinsic
Value | |
| Outstanding at December 31, 2020 | | | 2,447,687 | | | $ | 7.22 | | | | 7.1 | | | $ | 552,030 | |
| Granted | | | 1,664,700 | | | $ | 7.98 | | | | | | | $ | - | |
| Exercised | | | (32,734 | ) | | $ | 4.19 | | | | | | | $ | 11,904 | |
| Expired/Canceled | | | (248,001 | ) | | $ | 7.53 | | | | | | | $ | 52,475 | |
| Forfeited | | | (473,521 | ) | | $ | 7.72 | | | | | | | $ | 4,406 | |
| Outstanding at December 31, 2021 | | | 3,358,131 | | | $ | 7.53 | | | | 6.8 | | | $ | 483,244 | |
| Vested at December 31, 2021 | | | 1,931,950 | | | $ | 8.04 | | | | 5.2 | | | $ | 332,831 | |
| Expected to vest in the future | | | 1,426,181 | | | $ | 6.85 | | | | 8.9 | | | $ | 150,413 | |
The aggregate intrinsic value is calculated as the difference between the exercise prices of the underlying options and the quoted closing price of the common stock of the Company at the end of the reporting period for those options that have an exercise price below the quoted closing price.
Warrants:
During the years ended December 31, 2021 and 2020, the Company issued an aggregate of 31,407 and 91,500 shares of its common stock, respectively, upon cash exercise of warrants, resulting in net proceeds to the Company of $165,000 and $412,000, respectively.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
During the year ended December 31, 2021, the Company issued an aggregate of 70,269 shares of its common stock upon cashless exercise of 95,286 warrants.
The following table is the summary of warrant activities:
| | | Shares
Underlying
Warrants | | | Weighted
Average
Exercise
Price | | | Weighted
Average
Remaining
Contractual
Life | |
| Outstanding at December 31, 2020 | | | 183,148 | | | $ | 4.96 | | | | 1.61 | |
| Exercised | | | (126,693 | ) | | $ | 4.83 | | | | - | |
| Outstanding at December 31, 2021 | | | 56,455 | | | $ | 5.25 | | | | 0.61 | |
Stock-based Deferred Compensation Plan for Non-Employee Directors
In 2014, the Company established an unfunded stock-based deferred compensation plan, providing non-employee directors the opportunity to defer up to 100 percent of fees and compensation, including restricted stock units. The amount of fees and compensation deferred by a non-employee director is converted into stock units, the number of which is determined based on the closing price of the Company’s common stock on the date such compensation would have otherwise been payable. At all times, the plan participants are 100 percent vested in their respective deferred compensation accounts. On the tenth business day of January in the year following a director’s termination of service, the director will receive a number of common shares equal to the number of stock units accumulated in the director’s deferred compensation account. The Company accounts for this plan as stock-based compensation under ASC 718. During the year ended December 31, 2021, no compensation was deferred under this plan and during the year ended December 31, 2020, the amount of compensation that was deferred under this plan was $62,250.
Note 8 — Concentrations:
At December 31, 2021 and 2020, one customer exceeded 10% of the Company’s accounts receivable (100% and 95%). During the year ended December 31, 2021, the Company had revenue from three customers that exceeded 10% of its total sales (60%, 14% and 10%) and the Company had revenue from two customers that exceeded 10% of its total sales (58%, 12%) for the year ended December 31, 2020.
Note 9 — Leases:
The Company entered into a seven-year operating lease agreement in March 2020 for an office space at 300 Connell Drive, Berkeley Heights, New Jersey 07922. The lease agreement, with a monthly average cost of approximately $17,000 commenced on September 16, 2020.
The Company’s sublease on its previous premises at 400 Connell Drive, Berkeley Heights, New Jersey 07922 terminated on November 30, 2020.
The Company entered into an operating lease for office space in Germany that began in July 2017. The rental agreement has a three-month term which automatically renews and includes a monthly cost of 400 Euros. The Company elected to apply the short-term practical expedient to the office lease. The Company also has an operating lease for office equipment.
Operating lease expense in the Company’s consolidated statements of operations and comprehensive loss for the year ended December 31, 2021 and 2020 was approximately $209,000 and $66,000, respectively, which includes costs associated with leases for which ROU assets have been recognized as well as short-term leases.
At December 31, 2021 and 2020, the Company has a total operating lease liability of $924,000 and $1,033,000, respectively. At December 31, 2021, approximately $122,000 and $802,000 were classified as operating lease liabilities, short-term and operating lease liabilities, net of current portion, respectively, on the consolidated balance sheet. Operating ROU assets as of December 31, 2021 and 2020 are $900,000 and $1,015,000, respectively.
CORMEDIX INC. AND SUBSIDIARIES
NOTES TO CONSOLIDATED FINANCIAL STATEMENTS, (Continued)
For the year ended December 31, 2021 and 2020, cash paid for amounts included in the measurement of lease liabilities in operating cash flows from operating leases was $195,000 and $48,000, respectively.
As of December 31, 2021 and 2020, the weighted average remaining lease term were 5.8 years and 6.8 years, respectively and the weighted average discount rate of 9% and 9% at December 31, 2021 and 2020, respectively.
As of December 31, 2021, maturities of lease liabilities were as follows:
| 2022 | | $ | 200,000 | |
| 2023 | | | 202,000 | |
| 2024 | | | 205,000 | |
| 2025 | | | 208,000 | |
| 2026 and thereafter | | | 380,000 | |
| Total future minimum lease payments | | | 1,195,000 | |
| Less imputed interest | | | (271,000 | ) |
| Total | | $ | 924,000 | |
Note 10 — Subsequent Events:
During the first quarter of 2022, the Company sold an aggregate of 641,542 shares of its common stock under the ATM program (see Note 7) and realized net proceeds of approximately $3.0 million. As of the filing of this Annual Report on Form 10-K, the Company has $46.9 million available balance under its ATM program and it has $150.0 million available under its current shelf registration for the issuance of equity, debt or equity-linked securities.
On March 16, 2022, the Company’s Board of Directors (the “Board”) appointed Joseph Todisco as the Company’s Chief Executive Officer, commencing on such date as mutually agreed by Mr. Todisco and the Board, but in no event later than May 16, 2022. Mr. Todisco was appointed to serve as a member of the Board on March 18, 2022. Mr. Todisco will receive an annual salary of $600,000, which may be adjusted from time to time. He will be eligible for an annual bonus, based on a target of 65% of his base salary, as determined by the Board or the Compensation Committee of the Board (“Compensation Committee”). In determining such bonus, the Board or Compensation Committee will take into consideration the achievement of specified company objectives and personal objectives. Mr. Todisco generally must be employed through December 31 of a given year to earn that year’s annual bonus. Solely with respect to the 2022 fiscal year, Mr. Todisco will be paid an annual bonus in an amount that is not less than $195,000 (equal to 50% of the 2022 target bonus amount). Effective as of the date Mr. Todisco’s employment with the Company commences (the “Start Date”), the Company will grant Mr. Todisco stock option to purchase 500,000 shares of the Company’s common stock, with an exercise price equal to the closing price of the Company’s stock on the date of grant. The option will vest over four years in four equal annual installments on the first four anniversaries of the Start Date, provided that Mr. Todisco remains an employee or consultant through the applicable vesting date. Mr. Todisco will also be granted 207,469 restricted stock units (“Initial RSUs”), which will vest as to 50% on the first anniversary of the Start Date, as to 30% on the second anniversary of the Start Date, and as to 20% on the third anniversary of the Start Date, provided that Mr. Todisco remains an employee or consultant through the applicable vesting date. Dr. Matthew David will continue to serve as interim Chief Executive Officer and Chief Financial Officer until Mr. Todisco commences employment, after which Dr. David will continue to serve as the Company’s Chief Financial Officer.
On March 28, 2022, the Company announced that the resubmission of the NDA for DefenCath has been accepted for filing by the FDA. The FDA considers the resubmission as a complete, Class 2 response with a six-month review cycle.
F-33
iso4217:USD xbrli:shares