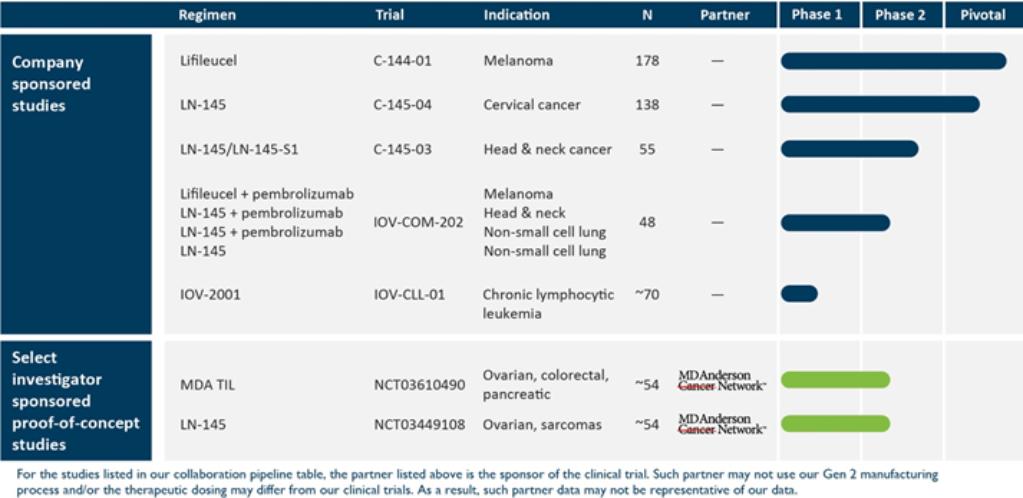
for violations of the federal securities laws (Leonard DeSilvio v. Lion Biotechnologies, Inc., et al., case no. 3:17cv2086) against our company and three of our former officers and directors. On April 19, 2017, a second class action complaint (Amra Kuc vs. Lion Biotechnologies, Inc., et al., case no. 3:17-cv-2188) was filed in the same court. Both complaints allege, among other things, that the defendants violated the federal securities laws by making materially false and misleading statements, or by failing to make certain disclosures, regarding the actions taken by Manish Singh, our former CEO, and our former investor relations firm that were the subject of the In the Matter of Certain Stock Promotions investigation. On July 20, 2017, the plaintiff in the Kuc case filed a notice to voluntarily dismiss that case. The court entered an order dismissing the Kuc complaint on July 21, 2017. On July 26, 2017, the court appointed a movant as lead plaintiff. On September 8, 2017, the lead plaintiff filed an amended complaint (Jay Rabkin v. Lion Biotechnologies, Inc., et al., case no. 3:17-cv-2086) seeking class action status that alleges, among other things, that the defendants violated federal securities laws by making materially false and misleading statements, or by failing to make certain disclosures, regarding the actions taken by Manish Singh and our former investor relations firm that were the subject of the In the Matter of Certain Stock Promotions SEC investigation. On February 5, 2018, the court entered an order dismissing two of plaintiff’s six claims. As the result of mediation, on September 28, 2018, lead plaintiff filed an unopposed motion for settlement, the cost of which was expected to be borne by our insurance carrier and would result in no loss to us. The court gave preliminary approval to the proposed settlement on November 30, 2018. A final hearing was held on April 12, 2019 to determine whether the proposed settlement was fair, reasonable, and adequate, and whether the claims should be dismissed. On April 17, 2019, the court approved the final settlement, involving a payment of $3,250,000 by our insurance carrier to a settlement fund, awarded attorney’s fees and costs to be paid to plaintiff’s counsel from the settlement fund, approved the plan of allocation for settlement class members, and ordered that the claims against us should be dismissed with prejudice. We do not expect to incur any costs or expenses in connection with this settlement.
Derivative Lawsuits. On December 15, 2017, a purported stockholder derivative complaint was filed by plaintiff Kevin Fong against us, as nominal defendant, and certain of our current and former officers and directors, and others, as defendants, in the U.S. District Court for the District of Delaware (case no. 1:17-cv-1806). The complaint alleges breaches of fiduciary duties, unjust enrichment, and violations of Section 14(a) of the Securities Exchange Act of 1934 and Rule 14a-9 promulgated thereunder arising from the SEC’s investigation in the In the Matter of Certain Stock Promotions investigation and our April 10, 2017 settlement thereof, and seeks unspecified damages on behalf of our company and injunctive relief. On March 28, 2018, a purported stockholder derivative complaint was filed by plaintiff Nazeer Khaleeluddin on our behalf, against us, as nominal defendant, and certain of our current and former officers and directors, and others, as defendants, in the U.S. District Court for the District of Delaware (case no. 1:18-cv-00469). The complaint alleges, among other things, violations of securities law, breach of fiduciary duty, aiding and abetting, waste of corporate assets, and unjust enrichment. The complaint is based on claims arising from the SEC’s investigation in the In the Matter of Certain Stock Promotions investigation and our April 10, 2017 settlement thereof and seeks unspecified damages on behalf of our company and injunctive relief. On May 1, 2018, the court consolidated this case with the aforementioned purported stockholder derivative case filed by plaintiff Kevin Fong. The consolidated case is titled In re Iovance Biotherapeutics, Inc. Stockholder Derivative Litigation (lead case no. 17-cv-1806). On January 28, 2020, we reached a proposed settlement with the plaintiffs, which is currently awaiting preliminary approval by the court. The terms of the settlement will be disseminated to shareholders as part of the notice process for the settlement if preliminary approval is granted. If the proposed settlement is given final approval by the court, we do not expect to incur any significant costs or expenses in connection with this settlement. Based on the current stage of the litigation, and the uncertainty of final approval by the court, it is not possible to estimate the amount or range of possible loss that might result from an adverse judgment or a settlement of these matters.
Solomon Capital, LLC. On April 8, 2016, a lawsuit, or the First Solomon Suit, titled Solomon Capital, LLC, Solomon Capital 401(K) Trust, Solomon Sharbat and Shelhav Raff v. Lion Biotechnologies, Inc. was filed by Solomon Capital, LLC, Solomon Capital 401(k) Trust, Solomon Sharbat and Shelhav Raff, which we refer to as the Solomon Plaintiffs, against us in the Supreme Court of the State of New York, County of New York (index no. 651881/2016). The Solomon Plaintiffs allege that, between June and November 2012, they provided to us $0.1 million and that they advanced and paid on our behalf an additional $0.2 million. The complaint further alleges that we agreed to (i) provide them with promissory notes totaling $0.2 million, plus interest, (ii) issue a total of 1,110 shares to the Solomon Plaintiffs (after the 1-for-100 reverse split of our common stock effected in March 2013), and (iii) allow the Solomon Plaintiffs to convert the foregoing funds into our securities in the next transaction. The Solomon Plaintiffs allege that they should have been able to convert their advances and payments into shares of our common stock in the restructuring that took effect in May 2013. Based on the foregoing, the Solomon Plaintiffs allege causes for breach of contract and unjust enrichment and demand judgment against us in an unspecified amount exceeding $1.5 million, plus interest. On June 3, 2016, we filed an answer and counterclaims in the lawsuit. We have asserted counterclaims for fraudulent inducement, fraudulent misrepresentation, fraudulent concealment, breach of fiduciary duty, and breach of contract, alleging principally that the counterclaim defendants misrepresented their qualifications and failed to disclose that Solomon Sharbat was the subject of an investigation by the Financial Industry Regulatory Authority, or FINRA, that resulted in the loss of his FINRA license. In our counterclaims, we are seeking damages in an amount exceeding $0.5 million and