As filed with the Securities and Exchange Commission on November 26, 2015
Registration Statement No. 333-______
SECURITIES AND EXCHANGE COMMISSION
Washington, D.C. 20549
FORM S-3
REGISTRATION STATEMENT UNDER
THE SECURITIES ACT OF 1933
ASSEMBLY BIOSCIENCES, INC.
(Exact name of registrant as specified in its charter)
Delaware (State or other jurisdiction of incorporation or organization) | 20-8729264 (I.R.S. Employer Identification No.) |
99 Hudson Street, 5th Floor
New York, New York 10013
Telephone: (646) 706-5208
(Address, including zip code, and telephone number, including area code, of registrant’s principal executive offices)
RUSSELL H. ELLISON
Chief Executive Officer
Assembly Biosciences, Inc.
99 Hudson Street, 5th Floor
New York, New York 10013
Telephone: (646) 706-5208
(Name, address, including zip code, and telephone number, including area code, of agent for service)
Copies to:
ALEXANDER M. DONALDSON
Wyrick Robbins Yates & Ponton LLP
4101 Lake Boone Trail, Suite 300
Raleigh, North Carolina 27607
Telephone: (919) 781-4000
Fax (919) 781-4865
Approximate date of commencement of proposed sale to the public: From time to time after the effective date of this Registration Statement.
If the only securities being registered on this Form are being offered pursuant to dividend or interest reinvestment plans, please check the following box.¨
If any of the securities being registered on this Form are to be offered on a delayed or continuous basis pursuant to Rule 415 under the Securities Act of 1933, other than securities offered only in connection with dividend or interest reinvestment plans, check the following box.x
If this Form is filed to register additional securities for an offering pursuant to Rule 462(b) under the Securities Act, please check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering.¨
If this Form is a post-effective amendment filed pursuant to Rule 462(c) under the Securities Act, check the following box and list the Securities Act registration statement number of the earlier effective registration statement for the same offering.¨
If this Form is a registration statement pursuant to General Instruction I.D. or a post-effective amendment thereto that shall become effective upon filing with the Commission pursuant to Rule 462(c) under the Securities Act, check the following box.¨
If this Form is a post-effective amendment filed pursuant to General Instruction I.D. filed to register additional securities or additional classes of securities pursuant to Rule 413(b) under the Securities Act, check the following box.¨
Indicate by check mark whether the Registrant is a large accelerated filer, an accelerated filer, a non-accelerated filer or a smaller reporting company. See the definitions of “accelerated filer”, “large accelerated filer” and “smaller reporting company” (as defined in Rule 12b-2 of the Act) (Check one):
| Large accelerated filer¨ | Accelerated filer¨ |
| Non-accelerated filer¨(Do not check if smaller reporting company) | Smaller reporting companyx |
CALCULATION OF REGISTRATION FEE
| Title of each class of securities to be registered | Amount to be registered (1) | Proposed maximum aggregate offering price per unit (2) | Proposed maximum aggregate offering price | Amount of registration fee | ||||||||||||
| Common stock, $0.001 par value per share | — | $ | — | $ | — | $ | — | |||||||||
| Preferred stock, $0.001 par value per share | — | — | — | — | ||||||||||||
| Warrants | — | — | — | — | ||||||||||||
| Debt Securities | — | — | — | — | ||||||||||||
| Units | — | — | — | — | ||||||||||||
| Total | — | — | $ | 120,000,000 | $ | 13,944 | (3) | |||||||||
| (1) | There are being registered hereunder such indeterminate number of shares of common stock and preferred stock, such indeterminate principal amount of debt securities, such indeterminate number of warrants to purchase common stock, preferred stock or debt securities, and such indeterminate number of units as shall have an aggregate initial offering price not to exceed $120,000,000, less the aggregate dollar amount of all securities previously issued hereunder. If any debt securities are issued at an original issued discount, then the offering price of such debt securities shall be in such greater principal amount as shall result in an aggregate offering price not to exceed $120,000,000, less the aggregate dollar amount of all securities previously issued hereunder. Any securities registered hereunder may be sold separately or as units with the other securities registered hereunder. The proposed maximum offering price per unit will be determined, from time to time, by the Registrant in connection with the issuance by the Registrant of the securities registered hereunder. The securities registered hereunder also include such indeterminate number of shares of common stock and preferred stock and amount of debt securities as may be issued upon conversion of or exchange for preferred stock or debt securities that provide for conversion or exchange, upon exercise of warrants or pursuant to the antidilution provisions of any of such securities. In addition, pursuant to Rule 416 under the Securities Act, the shares being registered hereunder include such indeterminate number of shares of common stock and preferred stock as may be issuable with respect to the shares being registered hereunder as a result of stock splits, stock dividends or similar transaction. | |
| (2) | The proposed maximum offering price per unit will be determined from time to time by the Registrant in connection with, and at the time of, the issuance of the securities and is not specified as to each class of security pursuant to General Instruction II.D. of Form S-3, as amended. | |
| (3) | Calculated pursuant to Rule 457(o) under the Securities Act of 1933, as amended, based on the proposed maximum aggregate offering price of all securities listed. |
THE REGISTRANT HEREBY AMENDS THIS REGISTRATION STATEMENT ON SUCH DATE OR DATES AS MAY BE NECESSARY TO DELAY ITS EFFECTIVE DATE UNTIL THE REGISTRANT SHALL FILE A FURTHER AMENDMENT WHICH SPECIFICALLY STATES THAT THIS REGISTRATION STATEMENT SHALL THEREAFTER BECOME EFFECTIVE IN ACCORDANCE WITH SECTION 8(a) OF THE SECURITIES ACT OF 1933 OR UNTIL THE REGISTRATION STATEMENT SHALL BECOME EFFECTIVE ON SUCH DATE AS THE COMMISSION, ACTING PURSUANT TO SAID SECTION 8(a), MAY DETERMINE.
The information in this prospectus is not complete and may be changed. We may not sell these securities or accept an offer to buy these securities until the registration statement filed with the Securities and Exchange Commission is effective. This prospectus is not an offer to sell these securities, and we are not soliciting an offer to buy these securities in any state where the offer or sale is not permitted.
Subject to completion, dated November 26, 2014
Prospectus

$120,000,000 of
Common Stock,
Preferred Stock,
Warrants,
Debt Securities and/or
Units
From time to time, we may offer up to $120,000,000 of any combination of the securities described in this prospectus, either individually or in units,in one or more offerings in amounts, at prices and on the terms that we will determine at the time of offering. We may also offer common stock or preferred stock upon conversion of debt securities, common stock upon conversion of preferred stock, or common stock, preferred stock or debt securities upon the exercise of warrants.
Each time we sell securities, we will provide specific terms of the securities offered in a supplement to this prospectus. The prospectus supplement may also add, update or change information contained in this prospectus. We will specify in any accompanying prospectus supplement the terms of any offering. You should read this prospectus and the applicable prospectus supplement, as well as any documents incorporated by reference in this prospectus and any prospectus supplement, carefully before you invest in any securities.This prospectus may not be used by us to consummate a sale of securities unless accompanied by the applicable prospectus supplement.
We will sell these securities directly to our stockholders or to other purchasers or through agents on our behalf or through underwriters or dealers as designated from time to time. If any agents or underwriters are involved in the sale of any of these securities, the applicable prospectus supplement will provide the names of the agents or underwriters and any applicable fees, commissions or discounts.
Our common stock trades on the NASDAQ Capital Market under the trading symbol “ASMB.” On November 25, 2014, the reported closing price of our common stock was $8.60 per share. We recommend that you obtain current market quotations for our common stock prior to making an investment decision.
As of November 24, 2014, the aggregate market value of our outstanding common stock held by non-affiliates was $69,758,495 based on 10,647,059 shares of our common stock outstanding on October 20, 2014, of which 7,708,121 shares were held by non-affiliates, and a price of $9.05 per share, the closing price for our common stock on October 20, 2014. During the 12 calendar months prior to and including the date of this prospectus, we have sold securities with an aggregate market value of $15,989,639 pursuant to General Instruction I.B.6 of Form S-3.
You should carefully read this prospectus, the prospectus supplement relating to any specific offering of securities and all information incorporated by reference herein and therein.
Investing in our securities involves a high degree of risk. These risks are discussed in this prospectus under “Risk Factors” beginning on page 8 and in the documents incorporated by reference into this prospectus.
Neither the Securities and Exchange Commission nor any state securities commission has approved or disapproved of these securities or determined if this prospectus is truthful or complete. Any representation to the contrary is a criminal offense.
The date of this prospectus is ___ , 201__
TABLE OF CONTENTS
| i |
This prospectus is part of a registration statement that we filed with the Securities and Exchange Commission, or SEC, utilizing a “shelf” registration process.Under this shelf registration process, we may offer shares of our common stock and preferred stock, various series of debt securities and/or warrants to purchase any of such securities, either individually or in units, in one or more offerings, up to a total dollar amount of $120,000,000.This prospectus provides you with a general description of the securities we may offer. Each time we offer a type or series of securities under this prospectus, we will provide a prospectus supplement that will contain specific information about the terms of that offering.
This prospectus does not contain all of the information included in the registration statement. For a more complete understanding of the offering of the securities, you should refer to the registration statement, including its exhibits. Prospectus supplements may also add, update or change information contained or incorporated by reference in this prospectus. However, no prospectus supplement will fundamentally change the terms that are set forth in this prospectus or offer a security that is not registered and described in this prospectus at the time of its effectiveness. This prospectus, together with the applicable prospectus supplements and the documents incorporated by reference into this prospectus, includes all material information relating to this offering. You should carefully read this prospectus, the applicable prospectus supplement, the information and documents incorporated herein by reference and the additional information under the heading “Where You Can Find More Information” before making an investment decision.
You should rely only on the information we have provided or incorporated by reference in this prospectus or any prospectus supplement. We have not authorized anyone to provide you with information different from that contained or incorporated by reference in this prospectus. No dealer, salesperson or other person is authorized to give any information or to represent anything not contained or incorporated by reference in this prospectus. You must not rely on any unauthorized information or representation. This prospectus is an offer to sell only the securities offered hereby, but only under circumstances and in jurisdictions where it is lawful to do so. You should assume that the information in this prospectus or any prospectus supplement is accurate only as of the date on the front of the document and that any information we have incorporated herein by reference is accurate only as of the date of the document incorporated by reference, regardless of the time of delivery of this prospectus or any sale of a security.
To the extent there are inconsistencies between any prospectus supplement, this prospectus and any documents incorporated by reference, the document with the most recent date will control.
This prospectus may not be used to consummate sales of our securities, unless it is accompanied by a prospectus supplement.
Unless the context otherwise requires, “Assembly,” the “company,” “we,” “us,” “our” and similar names refer to Assembly Biosciences, Inc.
| 1 |
The following summary is qualified in its entirety by, and should be read together with, the more detailed information and financial statements and related notes thereto appearing elsewhere or incorporated by reference in this prospectus and any prospectus supplement. Before you decide to invest in our securities, you should read the entire prospectus and the prospectus supplement carefully, including the risk factors and the financial statements and related notes included or incorporated by reference in this prospectus and the prospectus supplement.
Business Overview
We are a biopharmaceutical company that aspires to scientific leadership in the field of infectious diseases by:
| · | discovering treatments for patients with Hepatitis B virus, or HBV; and |
| · | addressing diseases associated with the disruption of normal gut flora, such as clostridium difficile associated diarrhea or CDAD. |
We are developing two proprietary platforms to achieve our goals: (i) an HBV-cure platform focused on oral drugs with novel direct-acting mechanisms, and (ii) an orally-delivered microbiome therapeutics platform for treating CDAD, which we refer to as our microbiome program.
The HBV-Cure Platform
HBV is an underappreciated global epidemic with twice as many people infected (over 350 million globally) and a higher mortality and morbidity rate than Hepatitis C and HIV, combined — over 600,000 people die every year from HBV-related causes. Less than 5% are diagnosed and treated today, and there is a low cure rate with current chronic therapies.
The current therapies for HBV, such as the reverse transcriptase inhibitors entecavir or tenofovir, potently suppress virus production (commonly referred to as “viral load”), but they continue to allow for a living virus and production of viral proteins and specifically viral antigens such as HBsAg. Current therapies must therefore be taken chronically, submitting patients to their associated significant and treatment limiting side effects.
By comparison, modulation of key aspects of the viral replication reservoir in other viruses, such as the new therapies for Hepatitis C for instance, has shown to be key for increasing the rates of curing patients. The viral reservoir for HBV is the intra-nuclear covalently closed circular DNA, or cccDNA. We believe that a “functional cure” is possible by developing drugs that target multiple aspects of the viral life cycle, and specifically the HBV viral reservoir.
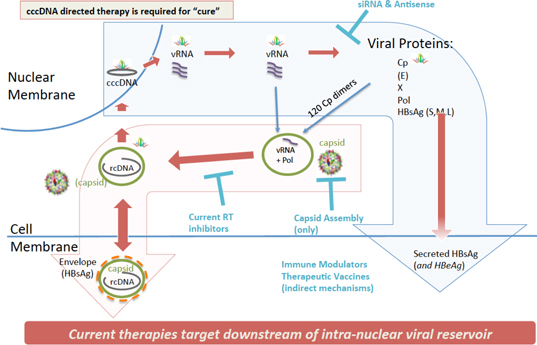
| 2 |
Figure 1 – The HBV life cycle with emphasis on the role of core protein and cccDNA relative to existing other approaches.
Other HBV treatments in development include immunomodulatory approaches, such as the TLR (Toll-like receptors: a class of proteins that play a key role in the innate immune system) programs and therapeutic vaccine programs that rely on the body’s immune system to clear virus; and siRNA (small interfering Ribo Nucleic Acid: a class of double-stranded RNA) approaches that target and rely on the degradation of the viral RNA, reducing RNA-to-protein translation, and also rely on the body’s immune system to potentially clear the virus. While these approaches are differentiated from the current standard of care therapies, they do not directly target the viral reservoir (see Figure 1).
We have discovered a series of new compounds based on the seminal research of our co-founder Dr. Adam Zlotnick focusing on HBV core protein – a unique viral protein that is required for HBV lifecycle, and has no human homologue, meaning the HBV core protein is unique from any human protein. Our molecules, known as Core protein Allosteric Modulators, or CpAMs, are capable of targeting and altering certain key proteins of HBV. The HBV core protein is a pleiotropic protein, or it is a protein that has more than one effect, and it flexes into multiple conformations required for several steps of the HBV lifecycle including interaction with the HBV intra-nuclear reservoir: cccDNA (see Figures 1 and 2).
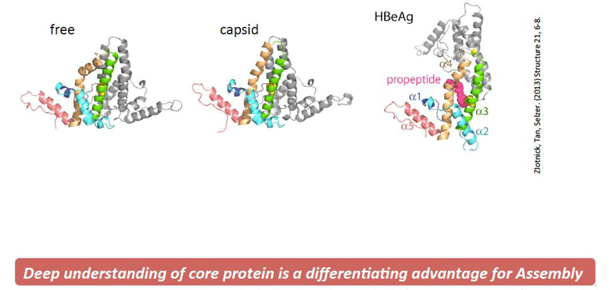
Figure 2 – HBV core protein is pleiotropic, which allows it to have multiple function in the HBV life cycle, including being required for cccDNA activity.
Modulation of these specific forms of HBV core proteins with our CpAMs has demonstrated preclinical proof of principle. We have shown in multiple cell models that our CpAMs can selectively reduce the production of viral antigens such as HBsAg and HBeAg—viral proteins which reflect the activity of cccDNA— and can reduce viral load. We believe that our CpAMs are the first of this novel class of molecules to show such effects in preclinical models.
We believe the key advantages of our approach will be:
| · | Convenience – we plan to offer a convenient oral pill formulation; |
| · | Combination therapy – we believe our product candidate may be used as either a single agent or in combination with existing therapies; |
| · | Immune response – our product candidate should not require an immune response, but may still benefit from this if reducing viral antigen and pre-genomic RNA supports anti HBV immunity; |
| · | Efficacy – since our CpAMs could target multiple aspects of the viral life cycle they may demonstrate an efficacy advantage over other approaches; and |
| 3 |
| · | Safety – since HBV core protein is a unique viral protein with no human homologue, there could be safety advantages due to a potentially reduced risk of off target activity. |
We are planning to select in mid-2015 a first generation lead molecule for development, and to initiate clinical trials in the first half of 2016. Our CpAM platform offers a multi-generation pipeline and we plan to advance second and third generation HBV-targeted molecules into clinical development rapidly behind the first program.
Our in-licensed intellectual property portfolio includes multiple patents filed in generally two categories: (i) platform patent applications, which include multiple applications filed and others in process that cover mechanisms of action, methods of treatment, and assays among other platform-related claims; and (ii) composition of matter applications for our several chemical series.
The CDAD Microbiome Platform
Our second program is based on a novel coating and encapsulation technology that allows for targeted delivery of complex agents to select regions of the gastrointestinal, or GI, tract.
We are currently focusing our technology on delivering beneficial specificbacteria to treat CDAD. CDAD is a major health problem with more than a five-fold increase in CDAD-associated deaths between 1999 and 2007, and CDAD is the leading cause of death associated with gastroenteritis in the U.S. This translates to an estimated 14,000 Americans that die from causes linked to CDAD each year.
There has been considerable experience reported in the literature of treating CDAD with fecal material transplant, or FMT. In addition, there are preliminary studies using selected bacterial strains and of bacterial spores from processed FMT. These reports have demonstrated a significant and growing precedent of successful cures in patients, and provide an excellent path for potentially curative therapy using a targeted and specific microbiome therapy.
We believe that by providing the benefits found in FMT therapy, in an oral capsule that contains specific bacteria rather than whole or processed feces, clinical adoption could be increased.
Our current research plan includes collaborations with key leaders in the microbiome field, while diligently developing the chemistry, manufacturing, and controls systems for supporting U.S. Food and Drug Administration regulated clinical trials and eventual commercial supply. We anticipate a clinical proof of principle for the delivery technology in the second quarter of 2015. Additionally, we expect to select our specific microbiome strain leads for development in mid-2015 and to initiate clinical trials in early 2016.
Our Strategy
We plan to build a multi-product company, based on our expertise in HBV and microbiome therapeutics, that discovers, develops, and commercializes first-in-class medicines to treat HBV, CDAD, and potentially other viruses and infectious diseases. Key elements of our strategy include:
| · | Aggressively pursuing the development of novel medicines to increase the cure rates in HBV, CDAD, and other infectious diseases of high unmet medical need; |
| · | Maintaining our leadership and competitive advantage in the fields of direct-acting HBV therapies and orally deliverable microbiome therapies for CDAD; |
| · | Continuing to build a multi-product platform for HBV, CDAD and other infectious diseases to generate medicines to increase cure rates; and |
| · | Maintaining a commitment to the advancement of innovative science in drug development. |
Our Guiding Principles
We and our employees are committed to increasing cure rates for patients with serious infections. This commitment drives our efforts for applying cutting edge science toward the development and commercialization of novel approaches to treating HBV and CDAD. We pledge to:
| 4 |
| · | Follow the science and do what is right for patients; |
| · | Maintain a culture of open communication and decision-making based on a deep understanding of our science and an appropriate allocation of resources; |
| · | Leverage strategic relationships with academic and corporate partners where and when appropriate; and |
| · | Execute relentlessly. |
Company History
On July 11, 2014, we merged with Assembly Pharmaceuticals Inc. in a reverse triangular merger with our wholly owned subsidiary, which we refer to as the Assembly Merger. In connection with the Assembly Merger, on July 11, 2014, we changed our name from Ventrus Biosciences, Inc. to Assembly Biosciences, Inc. We were incorporated in Delaware in October 2005 under the name South Island Biosciences, Inc. (which was changed to Ventrus Biosciences, Inc. in April 2007).
Corporate Information
Our executive offices are at 99 Hudson Street in New York, NY, 10013, and our research facility is at 953 Indiana Street, in San Francisco, CA 94103. Our telephone number is (646) 706-5208, and our website address iswww.assemblybio.com. The information contained on our website is not a part of, and should not be construed as being incorporated by reference into, this prospectus supplement.
As used in this prospectus supplement, unless the context otherwise requires, references to “Assembly,” “we,” “us,” “our” and similar references refer to Assembly Biosciences, Inc. and our wholly owned subsidiary Assembly Pharmaceuticals, Inc.
| 5 |
Offerings Under This Prospectus
We may offer shares of our common stock and preferred stock, various series of debt securities and/or warrants to purchase any of such securities, either individually or in units, with a total value of up to $120,000,000 from time to time under this prospectus at prices and on terms to be determined by market conditions at the time of any offering. This prospectus provides you with a general description of the securities we may offer. Each time we offer a type or series of securities under this prospectus, we will provide a prospectus supplement that will describe the specific amounts, prices and other important terms of the securities.
The prospectus supplement also may add, update or change information contained in this prospectus or in documents we have incorporated by reference into this prospectus. However, no prospectus supplement will fundamentally change the terms that are set forth in this prospectus or offer a security that is not registered and described in this prospectus at the time of its effectiveness.
This prospectus may not be used to consummate a sale of any securities unless it is accompanied by a prospectus supplement.
We may sell the securities directly to investors or to or through agents, underwriters or dealers. We, and our agents or underwriters, reserve the right to accept or reject all or part of any proposed purchase of securities. If we offer securities through agents or underwriters, we will include in the applicable prospectus supplement:
| · | the names of those agents or underwriters; |
| · | applicable fees, discounts and commissions to be paid to them; |
| · | details regarding over-allotment options, if any; and |
| · | the net proceeds to us. |
Common Stock
We may issue shares of our common stock from time to time. The holders of common stock are entitled to one vote per share on all matters to be voted upon by stockholders. Subject to preferences that may be applicable to any outstanding preferred stock, the holders of common stock are entitled to receive ratably any dividends that may be declared from time to time by our board of directors out of funds legally available for that purpose. In the event of our liquidation, dissolution or winding up, the holders of common stock are entitled to share ratably in all assets remaining after payment of liabilities, subject to prior distribution rights of any preferred stock then outstanding.
Preferred Stock
We may issue shares of our preferred stock from time to time, in one or more series. Our board of directors will determine the rights, preferences, privileges and restrictions of the preferred stock, including dividend rights, conversion rights, voting rights, terms of redemption, liquidation preferences, sinking fund terms and the number of shares constituting any series or the designation of such series, without any further vote or action by stockholders. Convertible preferred stock will be convertible into our common stock or exchangeable for our other securities. Conversion may be mandatory or at your option or both and would be at prescribed conversion rates.
If we sell any series of preferred stock under this prospectus and applicable prospectus supplements, we will fix the rights, preferences, privileges and restrictions of the preferred stock of such series in the certificate of designation relating to that series. We will file as an exhibit to the registration statement of which this prospectus is a part, or will incorporate by reference from reports that we file with the SEC, the form of any certificate of designation that describes the terms of the series of preferred stock we are offering before the issuance of the related series of preferred stock. We urge you to read the applicable prospectus supplement related to the series of preferred stock being offered, as well as the complete certificate of designation that contains the terms of the applicable series of preferred stock.
| 6 |
Warrants
We may issue warrants for the purchase of common stock, preferred stock and/or debt securities in one or more series. We may issue warrants independently or together with common stock, preferred stock and/or debt securities, and the warrants may be attached to or separate from these securities. We will evidence each series of warrants by warrant certificates that we will issue under a separate agreement. We may enter into warrant agreements with a bank or trust company that we select to be our warrant agent. We will indicate the name and address of the warrant agent in the applicable prospectus supplement relating to a particular series of warrants.
In this prospectus, we have summarized certain general features of the warrants. We urge you, however, to read the applicable prospectus supplement related to the particular series of warrants being offered, as well as the warrant agreements and warrant certificates that contain the terms of the warrants. We will file as exhibits to the registration statement of which this prospectus is a part, or will incorporate by reference from reports that we file with the SEC, the form of warrant agreement or warrant certificate containing the terms of the warrants we are offering before the issuance of the warrants.
Debt Securities
We may offer debt securities from time to time, in one or more series, as either senior or subordinated debt or as senior or subordinated convertible debt. The senior debt securities will rank equally with any other unsecured and unsubordinated debt. The subordinated debt securities will be subordinate and junior in right of payment, to the extent and in the manner described in the instrument governing the debt, to all of our senior indebtedness. Convertible debt securities will be convertible into or exchangeable for our common stock or our other securities. Conversion may be mandatory or at your option or both and would be at prescribed conversion rates.
With respect to any debt securities that we issue, we will issue such debt securities under an indenture, which we would enter into with the trustee named in the indenture. Any indenture would be qualified under the Trust Indenture Act of 1939.
Units
We may issue units consisting of common stock, preferred stock, debt securities and/or warrants for the purchase of common stock, preferred stock and/or debt securities in one or more series. In this prospectus, we have summarized certain general features of the units. We urge you, however, to read the applicable prospectus supplement related to the series of units being offered, as well as the unit agreements that contain the terms of the units. We will file as exhibits to the registration statement of which this prospectus is a part, or will incorporate by reference reports that we file with the SEC, the form of unit agreement and any supplemental agreements that describe the terms of the series of units we are offering before the issuance of the related series of units.
| 7 |
Investing in our common stock involves risk. Before deciding whether to invest in our common stock, you should consider carefully the risks and uncertainties described below. You should also consider the risks, uncertainties and assumptions discussed under the heading “Risk Factors” included in our most recent annual report on Form 10-K which is on file with the SEC and is incorporated herein by reference, and which has been and may be amended, supplemented or superseded from time to time by other reports we have filed and may file with the SEC in the future. There may be other unknown or unpredictable economic, business, competitive, regulatory or other factors that could have material adverse effects on our future results. If any of these risks actually occurs, our business, business prospects, financial condition or results of operations could be seriously harmed. This could cause the trading price of our common stock to decline, resulting in a loss of all or part of your investment. Please also read carefully the section above entitled “Cautionary Statement Regarding Forward-Looking Statements.”
Risks Related to Our Business
We have no approved products and currently are dependent on the success of our HBV and microbiome therapies.
To date, we have no approved product on the market and have generated no product revenues. Our prospects are substantially dependent on our ability to develop and commercialize our HBV and microbiome therapies. Unless and until we receive approval from the U.S. Food and Drug Administration, or FDA, and other regulatory authorities for our product candidates, we cannot sell our product candidates and will not have product revenues. We will have to fund all of our operations and capital expenditures from cash on hand, any licensing fees and any future securities offerings or debt financings.
We have a limited operating history and a history of operating losses, and expect to incur significant additional operating losses.
We were established in October 2005, began active operations in the spring of 2007 and have only a limited operating history. In addition, we have terminated our programs related to our three prior product candidates. Therefore, there is limited historical financial information upon which to base an evaluation of our performance. Our prospects must be considered in light of the uncertainties, risks, expenses, and difficulties frequently encountered by companies in their early stages of operations. We have generated losses since we began operations and, as of September 30, 2014, we had an accumulated deficit of $129.3 million. We expect to incur substantial additional losses over the next several years as we continue to pursue our research, development, preclinical studies and clinical trial activities. The amount of future losses and when, if ever, we will achieve profitability are uncertain. We have no products that have generated any commercial revenue, do not expect to generate revenues from the commercial sale of products unless and until our HBV or microbiome therapies or any other product candidate is approved by the FDA for sale, and we might never generate revenues from the sale of products.
We are not currently profitable and might never become profitable.
We have a history of losses and expect to incur significant operating and capital expenditures and resultant substantial losses and negative operating cash flow for the next several years, and beyond if we do not successfully launch and commercialize our HBV therapy or our microbiome program. We might never achieve or maintain profitability. We anticipate that our expenses will continue to be substantial in the foreseeable future as we:
| · | continue to undertake research and development to identify potential product candidates; |
| · | continue to undertake preclinical studies and clinical trials for our product candidates; and |
| · | seek regulatory approvals for our product candidates. |
As a result, we will need to generate significant revenues in order to achieve and maintain profitability. Our ability to generate revenue and achieve profitability will depend on, among other things:
| · | successful completion of research, preclinical studies and clinical trials for our product candidates; |
| 8 |
| · | obtaining necessary regulatory approvals from the FDA and international regulatory agencies for our product candidates; |
| · | establishing manufacturing, sales, and marketing arrangements with third parties for any approved products; and |
| · | raising sufficient funds to finance our activities, if and when needed. |
We might not succeed at any of these undertakings. If we are unsuccessful at some or all of these undertakings, our business, prospects, and results of operations might be materially adversely affected.
Pre-clinical testing and clinical trials involve a lengthy and expensive process with an uncertain outcome, and results of earlier studies and trials may not be predictive of future trial results.
Preclinical studies and clinical testing are expensive, can take many years to complete and their outcome is highly uncertain. Failure can occur at any time during the preclinical study and clinical trial processes due to inadequate performance of a drug candidate or inadequate adherence by patients or investigators to clinical trial protocols. In addition, the results of preclinical studies and early clinical trials of product candidates may not be predictive of the results of later-stage clinical trials. For example, in late June 2012, we reported that our Phase III randomized, double-blind, placebo-controlled clinical trial of iferanserin in patients with hemorrhoidal disease did not meet its endpoints, despite favorable Phase II trial results. We also reported in February 2014 that our second Phase III clinical trial for the treatment of anal fissures diltiazem demonstrated no significant improvement compared to placebo despite favorable results in a prior Phase III trial. These same risks apply to our planned development of our current and any other product candidates.
We are an early stage company and might not be able to commercialize any product candidates.
We are an early stage company and have not demonstrated our ability to perform the functions necessary for the successful commercialization of any product candidates. The successful commercialization of any product candidates will require us to perform a variety of functions, including:
| · | continuing to undertake research and development and preclinical studies and clinical trials; |
| · | participating in regulatory approval processes; |
| · | formulating and manufacturing products; and |
| · | conducting sales, marketing and distribution activities. |
Our development of our product candidates is subject to the risks of failure and delay inherent in the development of new pharmaceutical products and products based on new technologies, including:
| · | delays in product development, preclinical and clinical testing; |
| · | unplanned expenditures in product development, preclinical and clinical testing; |
| · | failure of a product candidate to demonstrate acceptable safety and efficacy; |
| · | failure to receive regulatory approvals; |
| · | emergence of superior or equivalent products; |
| · | inability to manufacture and sell on our own, or through any others, product candidates on a commercial scale or at a financially viable cost; and |
| · | failure to achieve market acceptance. |
| 9 |
Because of these risks, our research and development efforts might not result in any commercially viable products. If we do not successfully complete a significant portion of these development efforts, obtain required regulatory approvals, and have commercial success with any approved products, our business, financial condition and results of operations will be materially harmed.
The results of earlier studies and trials for any of our product candidates might not be predictive of the results in any future studies or trials.
The results of any earlier study or trial for any of our product candidates may not be predictive of the results for any future studies or trials. Further, the results of any study or trial for any of our product candidates may not be as positive as the results for any prior studies or trials, if at all. In addition, unforeseen safety issues could emerge in any future study or trial, which could severely hamper the likelihood of FDA or other regulatory approval of any product candidate. If any of these events were to occur, the development of any product candidate could be significantly delayed and more expensive than anticipated, and could lead us to abandon our development efforts entirely, any of which would have a significant adverse effect on our business.
We may need additional financing to complete the development of any product candidate and fund our activities in the future.
We anticipate that we will incur operating losses for the next several yearsas we continue to develop our HBV therapy and our microbiome program as well as initiate any development of any other product candidate and will require substantial funds during that time to support our operations. We expect that our current resources will provide us with sufficient capital to fund our operations into the second quarter of 2016. However, we might consume our available capital before that time if, for example, we are not efficient in managing our resources or if we encounter unforeseen costs, delays or other issues or if regulatory requirements change. If that happens, we may need additional financing to continue the development of our HBV therapy and our microbiome program. Thereafter, we will need additional capital to fund our operations in the future. However, there is no assurance that we will be successful in raising any necessary additional capital on terms that are acceptable to us, or at all. If such event or other unforeseen circumstances occurred and we were unable to raise capital, we could be forced to discontinue product development, sacrifice attractive business opportunities, cease operations entirely and sell or otherwise transfer all or substantially all of our remaining assets.
We are dependent on a license relationship for each of our HBV therapy and our microbiome program.
Our license agreement with Indiana University Research and Technology Corporation, or IURTC, from whom we have licensed our HBV therapy, requires us to make milestone payments based upon the successful accomplishment of clinical and regulatory milestones related to our HBV therapy. The total amount of all potential future milestone payments at September 30, 2014 is $825,000. We also are obligated to pay IURTC royalty payments based on net sales of the licensed technology, which increase if we sublicense our rights to a non-affiliate third party. We are also obligated to pay diligence maintenance fees ($25,000-$100,000) each year to the extent that the royalty, sublicensing, and milestone payments to IURTC are less than the diligence maintenance fee for that year. Our license with Therabiome, LLC, from whom we have licensed our microbiome program, also requires us to pay regulatory and clinical milestones as well as royalty payments to Therabiome. If we breach any of these obligations, we could lose our rights to our microbiome program. If we fail to comply with similar obligations to any other licensor, it would have the right to terminate the license, in which event we would not be able to commercialize drug candidates or technologies that were covered by the license. Also, the milestone and other payments associated with licenses will make it less profitable for us to develop our drug candidates than if we owned the technology ourselves.
We depend on our collaboration with Adam Zlotnick, the scientific founder of our HBV therapy. If that collaboration is not maintained, we may not be able to capitalize on the market potential of our HBV therapy.
Dr. Adam Zlotnick is the founder of our HBV therapy. We have entered into a three-year consulting agreement with Dr. Zlotnick pursuant to which he serves as the Chairman of our Scientific Advisory Board and provides consulting services as we request. Dr. Zlotnick could refuse to extend the agreement after its three-year term expires or we could terminate the consulting agreement for cause or no cause. Although Dr. Zlotnick assigned to us any rights to intellectual property related to our HBV therapy that arise during the term of the consulting agreement, and while the consulting agreement contains a non-compete during the term of the agreement, the loss of Dr. Zlotnick’s services could materially impair our ability to further the development of our HBV therapy.
| 10 |
Failure to integrate Assembly Pharmaceutical, Inc. into our operations successfully could adversely affect our business.
On July 11, 2014, we effected the Assembly Merger whereby Assembly Pharmaceuticals, Inc. became our wholly owned subsidiary. Our integration of the operations and personnel of Assembly may require significant efforts, including significant amounts of management’s time, and result in additional expenses. Factors that will affect the success of the merger include the strength of our combined technology, our ability to execute our business strategy, our ability to adequately fund research and development and retain key employees, and results of clinical trials, regulatory approvals and reimbursement levels of any approved product. Our failure to successfully manage the Assembly Merger could have a material adverse impact on our business. In addition, we cannot be certain that Assembly Pharmaceuticals’ technology will be successfully developed or, if approved, become profitable or remain so.
Corporate and academic collaborators might take actions to delay, prevent, or undermine the success of our product candidates.
Our operating and financial strategy for the development, preclinical and clinical testing, manufacture, and commercialization of drug candidates heavily depends on collaborating with corporations, academic institutions, licensors, licensees, and other parties. However, there can be no assurance that we will successfully establish these collaborations. In addition, should a collaboration be terminated, replacement collaborators might not be available on attractive terms, or at all. The activities of any collaborator will not be within our control and might not be within our power to influence. There can be no assurance that any collaborator will perform its obligations to our satisfaction or at all, that we will derive any revenue or profits from these collaborations, or that any collaborator will not compete with us. If any collaboration is not successful, we might require substantially greater capital to undertake development and marketing of our proposed products and might not be able to develop and market these products effectively, if at all. In addition, a lack of development and marketing collaborations might lead to significant delays in introducing proposed products into certain markets and/or reduced sales of proposed products in such markets.
We rely on data provided by our collaborators and others that has not been independently verified and could prove to be false, misleading, or incomplete.
We rely on third-party vendors, scientists, and collaborators to provide us with significant data and other information related to our projects, preclinical studies and clinical trials, and our business. If these third parties provide inaccurate, misleading, or incomplete data, our business, prospects, and results of operations could be materially adversely affected.
Preclinical and clinical testing required for our product candidates is expensive and time-consuming, and the outcome is uncertain.
In order to obtain FDA approval to market a new drug product, we must demonstrate safety and effectiveness in humans. To meet these requirements, we must conduct extensive preclinical testing and sufficient adequate and well-controlled clinical trials. Conducting clinical trials is a lengthy, time consuming, and expensive process. The length of time might vary substantially according to the type, complexity, novelty, and intended use of the product candidate, and often can be several years or more per trial. Delays associated with product candidates for which we are directly conducting preclinical studies or clinical trials might cause us to incur additional operating expenses. The commencement and rate of completion of clinical trials might be delayed by many factors, including, for example:
| · | the lack of effectiveness during clinical trials; |
| · | the emergence of unforeseen safety issues; |
| 11 |
| · | inability to manufacture sufficient quantities of qualified materials under current Good Manufacturing Practices, or cGMPs, for use in clinical trials; |
| · | slower than expected rates of patient recruitment; |
| · | failure to recruit a sufficient number of patients; |
| · | modification of clinical trial protocols; |
| · | changes in regulatory requirements for clinical trials; |
| · | delays, suspension, or termination of clinical trials by the institutional review board or ethics committee responsible for overseeing the study at a particular study site; and |
| · | government, institutional review board, ethics committee, or other regulatory delays or clinical holds requiring suspension or termination of the trials. |
The results from preclinical testing and early clinical trials are not necessarily predictive of results obtained in later clinical trials. Accordingly, even if we obtain or have obtained positive results from preclinical studies or early clinical trials, we might not achieve the same success in future clinical trials. For example, positive results were observed in earlier clinical trials of each of our two prior product candidates, but the subsequent clinical trials were not successful. Further, clinical trials might not provide statistically significant data supporting a product candidate’s safety and effectiveness to meet the requisite regulatory approvals.
We have used and intend to continue to rely on one or more contract research organizations, or CROs, to conduct our preclinical studies and clinical trials. We are highly dependent on these CROs to conduct our trials in accordance with the requirements of the FDA and good scientific practice. In the event the CROs fail to perform their duties in such a fashion, we may not obtain regulatory approval for any of our product candidates.
The failure of preclinical studies and clinical trials to demonstrate safety and effectiveness for the desired indications could harm the development of that product candidate and other product candidates. This failure could cause us to abandon a product candidate and could delay development of other product candidates. Any delay in, or termination of, our preclinical studies or clinical trials would delay the filing of our New Drug Applications, or NDAs, with the FDA and, ultimately, our ability to commercialize our product candidates and generate product revenues. Any change in, or termination of, our clinical trials could materially harm our business, financial condition, and results of operation.
Unforeseen safety issues could hinder the development of our product candidates and their adoption, if approved.
Safety issues could arise during development of our product candidates, which might delay testing or prevent further development entirely. We have not yet tested our HBV therapy or our microbiome therapy and safety issues could arise during that planned testing or testing of any other product candidates. If a product is approved, any limitation on use that might be necessary could hinder its adoption in the marketplace. In addition, if any product is approved, it could be used against any instructions that we publish that limit its use, which could subject us to litigation.
We lack suitable facilities for certain preclinical and clinical testing and expect to rely on third parties to conduct some of our research and preclinical testing and our clinical trials and those third parties may not perform satisfactorily, including failing to meet deadlines for the completion of such research, testing or trials.
We do not have sufficient facilities to conduct all of our anticipated preclinical and clinical testing. As a result, we expect to contract with third parties to conduct most or all preclinical and clinical testing required for regulatory approval for our product candidates. We currently plan to outsource all clinical testing to third parties and are reliant on the services of these third parties to conduct studies on our behalf. If we are unable to continue with or retain third parties for these purposes on acceptable terms, we may be unable to successfully develop our product candidates. In addition, any failures by third parties to adequately perform their responsibilities may delay the submission of our product candidates for regulatory approval, which would impair our financial condition and business prospects.
| 12 |
Our reliance on these third parties for research and development activities also reduces our control over these activities but will not relieve us of our responsibilities. For example, we are responsible for ensuring that each of our studies is conducted in accordance with the applicable protocol, legal, regulatory and scientific requirements and standards, and our reliance on third parties does not relieve us of our regulatory responsibilities. Furthermore, these third parties may also have relationships with other entities, some of which may be our competitors. In addition, these third parties are not our employees, and except for remedies available to us under our agreements with such third parties, we cannot control whether or not they devote sufficient time and resources to our clinical, nonclinical and preclinical programs. If these third parties do not successfully carry out their contractual duties or obligations or meet expected deadlines, if they need to be replaced or if the quality or accuracy of the clinical data they obtain is compromised due to the failure to adhere to our clinical protocols, regulatory requirements or for other reasons, our research, preclinical studies or clinical trials may be extended, delayed or terminated and we may not be able to obtain, or may be delayed in obtaining, regulatory approvals for our product candidates. As a result, our results of operations and business prospects would be harmed, our costs could increase and our ability to generate revenues could be delayed.
We will rely exclusively on third parties to formulate and manufacture our product candidates.
We do not have and do not intend to establish our own manufacturing facilities. Consequently, we lack the physical plant to formulate and manufacture our own product candidates. If any product candidate we might develop or acquire in the future receives FDA approval, we will rely on one or more third-party contractors to manufacture our products. If, for any reason, we become unable to rely on any future source to manufacture our product candidates, either for clinical trials or, at some future date, for commercial quantities, then we would need to identify and contract with additional or replacement third-party manufacturers to manufacture compounds for preclinical, clinical and commercial purposes. We might not be successful in identifying additional or replacement third-party manufacturers, or in negotiating acceptable terms with any that we do identify. If we are unable to secure and maintain third-party manufacturing capacity, the development and sales of our products and our financial performance might be materially affected.
In addition, before any of our collaborators can begin to commercially manufacture our product candidates, each manufacturing facility and process is subject to regulatory review. Manufacturing of drugs for clinical and commercial purposes must comply with the FDA’s cGMPs, and applicable non-U.S. regulatory requirements. The cGMP requirements govern quality control and documentation policies and procedures. Complying with cGMP and non-U.S. regulatory requirements will require that we expend time, money, and effort in production, recordkeeping, and quality control to assure that the product meets applicable specifications and other requirements. Any contracted manufacturing facility must also pass a pre-approval inspection prior to FDA approval. Failure to pass a pre-approval inspection might significantly delay FDA approval of our product candidates. If any of our future collaborators fails to comply with these requirements, it would be subject to possible regulatory action which could limit the jurisdictions in which we are permitted to sell our products, if approved. As a result, our business, financial condition, and results of operations might be materially harmed.
Our reliance on third-party manufacturers exposes us to the following risks:
| · | We might be unable to identify manufacturers for commercial supply on acceptable terms or at all because the number of potential manufacturers is limited and the FDA must approve any replacement contractor. This approval would generally require compliance inspections. In addition, a new manufacturer would have to be educated in, or develop substantially equivalent processes for, production of our products after receipt of FDA approval, if any. |
| · | Our third-party manufacturers might be unable to formulate and manufacture our product candidates in the volume and of the quality required to meet our clinical and, if approved, commercial needs. |
| · | Our contract manufacturers might not perform as agreed or might not remain in the contract manufacturing business for the time required to supply our clinical trials or to successfully produce, store and distribute our products. |
| 13 |
| · | One or more of our contract manufacturers could be foreign, which increases the risk of shipping delays and adds the risk of import restrictions. |
| · | Drug manufacturers are subject to ongoing periodic unannounced inspection by the FDA and corresponding state agencies to ensure strict compliance with cGMP and other government regulations and corresponding foreign requirements. We would not have complete control over third-party manufacturers’ compliance with these regulations and requirements. |
| · | If any third-party manufacturer makes improvements in the manufacturing process for our product candidates, we might not own, or might have to share, the intellectual property rights to the innovation with our licensors. |
| · | We might compete with other companies for access to these manufacturers’ facilities and might be subject to manufacturing delays if the manufacturers give other clients higher priority than us. |
Each of these risks could delay our development efforts, preclinical studies and clinical trials or the approval, if any, of our product candidates by the FDA or the commercialization of our product candidates and could result in higher costs or deprive us of potential product revenues. As a result, our business, financial condition, and results of operations might be materially harmed.
If we cannot compete successfully for market share against other drug companies, we might not achieve sufficient product revenues and our business will suffer.
If our product candidates receive FDA approval, they will compete with a number of existing and future drugs and therapies developed, manufactured and marketed by others. Existing or future competing drugs might provide greater therapeutic convenience or clinical or other benefits for a specific indication than our product candidates, or might offer comparable performance at a lower cost. If our product candidates fail to capture and maintain market share, we might not achieve sufficient product revenues and our business will suffer.
We might compete against fully integrated pharmaceutical companies and smaller companies that are collaborating with larger pharmaceutical companies, academic institutions, government agencies and other public and private research organizations. Many of these competitors, either alone or together with their collaborative partners, operate larger research and development programs or have substantially greater financial resources than we do, as well as significantly greater experience in:
| · | developing drugs; |
| · | undertaking preclinical testing and human clinical trials; |
| · | obtaining FDA and other regulatory approvals of drugs; |
| · | formulating and manufacturing drugs; and |
| · | launching, marketing and selling drugs. |
We might not obtain the same resources and experience as our competitors. If we are unable to perform these tasks effectively and efficiently, our results of operations might be materially adversely affected.
Developments by competitors might render our product candidates or technologies obsolete or non-competitive.
The pharmaceutical and biotechnology industries are intensely competitive. In addition, the clinical and commercial landscape for HBV and CDAD is rapidly changing; we expect new data from commercial and clinical-stage products to continue to emerge. We will compete with organizations that have existing treatments and that are or will be developing treatments for the indications that our product candidates target. If our competitors develop effective treatments for HBV, CDAD or any other indication or field we might pursue, and successfully commercialize those treatments, our business and prospects might be materially harmed, due to intense competition in these markets.
| 14 |
If we are not able to develop collaborative marketing relationships with licensees or partners, or create an effective internal sales, marketing, and distribution capability, we might be unable to market our products successfully.
To market our product candidates, if approved, we will have to establish our own marketing and sales force or out-license our product candidates to, or collaborate with, larger firms with experience in marketing and selling pharmaceutical products. There can be no assurance that we will be able to successfully establish our own marketing capabilities or establish marketing, sales, or distribution relationships with third parties; that such relationships, if established, will be successful; or that we will be successful in gaining market acceptance for our product candidates. To the extent that we enter into any marketing, sales, or distribution arrangements with third parties, our product revenues will be lower than if we marketed and sold our products directly, and any revenues we receive will depend upon the efforts of such third parties. If we are unable to establish such third-party sales and marketing relationships, or choose not to do so, we will have to establish our own in-house capabilities. We, as a company, have no experience in marketing or selling pharmaceutical products and currently have no sales, marketing, or distribution infrastructure. To market any of our products directly, we would need to develop a marketing, sales, and distribution force that both has technical expertise and the ability to support a distribution capability. To establish our own marketing, sales, and distribution capacity would significantly increase our costs, and require substantial additional capital. In addition, there is intense competition for proficient sales and marketing personnel, and we might not be able to attract individuals who have the qualifications necessary to market, sell, and distribute our products. There can be no assurance that we will be able to establish internal marketing, sales, or distribution capabilities.
Physicians and patients might not accept and use our drugs.
Even if the FDA approves one of our product candidates, physicians and patients might not accept and use it. Acceptance and use of our products will depend upon a number of factors, including:
| · | perceptions by members of the health care community, including physicians, about the safety and effectiveness of our product; |
| · | cost-effectiveness of our product relative to competing products or therapies; |
| · | availability of reimbursement for our product from government or other healthcare payors; and |
| · | effective marketing and distribution efforts by us and our licensees and distributors, if any. |
If our current product candidates are approved, we expect sales to generate substantially all of our revenues for the foreseeable future, and as a result, the failure of these products to find market acceptance would harm our business and would require us to seek additional financing.
Our ability to generate product revenues will be diminished if our products sell for inadequate prices or patients are unable to obtain adequate levels of reimbursement.
Our ability to commercialize our product candidates, if approved, alone or with collaborators, will depend in part on the extent to which reimbursement will be available from:
| · | government and health administration authorities; |
| · | private health maintenance organizations and health insurers; and |
| · | other healthcare payors. |
Significant uncertainty exists as to the reimbursement status of newly approved healthcare products. Healthcare payors, including Medicare, are challenging the prices charged for medical products and services. Government and other healthcare payors increasingly attempt to contain healthcare costs by limiting both coverage and the level of reimbursement for drugs. Even if our product candidates are approved by the FDA, insurance coverage might not be available, and reimbursement levels might be inadequate, to cover our products. If government and other healthcare payors do not provide adequate coverage and reimbursement levels for our products, once approved, market acceptance of such products could be reduced.
| 15 |
Proposals to modify the current health care system in the U.S. to improve access to health care and control its costs are continually being considered by the federal and state governments. In March 2010, the U.S. Congress passed landmark healthcare reform legislation, of which the coverage and reimbursement provisions went into effect in late 2013. We cannot predict what impact on federal reimbursement policies and regulatory compliance landscape this legislation will have in general or on our business specifically. We expect continued judicial and legislative review and assessment of this legislation and possibly alternative health care reform proposals. We cannot predict judicial results or whether new proposals will be made or adopted, when they may be adopted or what impact they may have on us if they are adopted.
Health administration authorities in countries other than the U.S. may not provide reimbursement for our product candidates at rates sufficient for us to achieve profitability, or at all. Like the U.S., these countries could adopt health care reform proposals and could materially alter their government-sponsored health care programs by reducing reimbursement rates.
Any reduction in reimbursement rates under Medicare or private insurers or foreign health care programs could negatively affect the pricing of our product candidates. If we are not able to charge a sufficient amount for our products, then our margins and our profitability will be adversely affected.
If we lose key management or scientific personnel, cannot recruit qualified employees, directors, officers, or other significant personnel or experience increases in our compensation costs, our business might materially suffer.
We are highly dependent on the services of our Chairman and Chief Executive Officer, Dr. Russell H. Ellison, our President and Chief Operating Officer, Derek Small, our Chief Medical Officer and Vice President of Research and Development, Dr. Uri Lopatin, our Chief Scientific Officer, Dr. Lee D. Arnold, and our Chief Financial Officer, David J. Barrett. Our employment agreements with Dr. Ellison, Mr. Small, Dr. Lopatin, Dr. Arnold and Mr. Barrett do not ensure their retention. This is also true for our other management team members, both present and future.
Furthermore, our future success also depends, in part, on our ability to identify, hire, and retain additional management team members as our operations grow. We expect to experience intense competition for qualified personnel and might be unable to attract and retain the personnel necessary for the development of our business. Finally, we do not currently maintain, nor do we intend to obtain in the future, “key man” life insurance that would compensate us in the event of the death or disability of any of the members of our management team.
If we cannot enforce non-compete and confidentiality provisions applicable to our employees and consultants, our business might materially suffer.
We include a non-compete provision in any employment agreement we enter into with an employee, including those for Messrs. Small and Barrett and Drs. Ellison and Arnold, that runs during the term of the agreement and for a period of time after termination, depending on the individual.
We include a confidentiality provision in any employment or consulting agreement we enter into with an employee or a consultant. The confidentiality provision runs during the term of the agreement and thereafter without limit.
For future employees with whom we do not enter into an employment agreement, we will enter into a confidentiality agreement with the same provisions described above.
To be able to enforce these non-compete and confidentiality provisions we would need to know of any breach and have sufficient funds to enforce the provisions. We cannot assure you that we would know of or be able to afford enforcement of any breach. In addition, such provisions are subject to state law and interpretation by courts, which could limit the scope and duration of these provisions. Any limitation on or non-enforcement of these non-compete and confidentiality provisions could have an adverse effect on our business.
| 16 |
If we are unable to hire additional qualified personnel, our ability to grow our business might be harmed.
At October 31, 2014, we had 18 employees, 10 consultants and multiple contract research organizations with whom we have contracted. We will need to hire or contract with additional qualified personnel with expertise in clinical research and testing, government regulation, formulation and manufacturing and sales and marketing to commercialize our HBV therapy and our microbiome program or any other product candidate we may seek to develop. We compete for qualified individuals with numerous biopharmaceutical companies, universities and other research institutions. Competition for these individuals is intense, and we cannot be certain that our search for such personnel will be successful. Attracting and retaining qualified personnel will be critical to our success.
We might not successfully manage our growth.
Our success will depend upon the expansion of our operations and the effective management of our growth, which will place a significant strain on our current and future management and other administrative and operational resources. To manage this growth, we must expand our facilities, augment our operational, financial and management systems and hire and train additional qualified personnel. If we are unable to manage our growth effectively, our business would be harmed.
We might seek to develop our business through acquisitions of or investment in new or complementary businesses, products or technologies, and the failure to manage these acquisitions or investments, or the failure to integrate them with our existing business, could have a material adverse effect on us.
We might consider opportunities to acquire or invest in other technologies, products and businesses that might enhance our capabilities or complement our current product candidates. Potential and completed acquisitions and strategic investments involve numerous risks, including potential problems or issues associated with the following:
| · | assimilating the purchased technologies, products or business operations; |
| · | maintaining uniform standards, procedures, controls and policies; |
| · | unanticipated costs associated with the acquisition or investment; |
| · | diversion of our management’s attention from our preexisting business; |
| · | maintaining or obtaining the necessary regulatory approvals or complying with regulatory standards; and |
| · | adverse effects on existing business operations. |
We have no current commitments with respect to any acquisition or investment in other technologies or businesses. We do not know if we will identify suitable acquisitions, whether we will be able to successfully complete any acquisitions, or whether we will be able to successfully integrate any acquired product, technology or business into our business or retain key personnel, suppliers or collaborators.
Our ability to successfully develop our business through acquisitions would depend on our ability to identify, negotiate, complete and integrate suitable target businesses or technologies and obtain any necessary financing. These efforts could be expensive and time consuming and might disrupt our ongoing operations. If we are unable to efficiently integrate any acquired business, technology or product into our business, our business and financial condition might be adversely affected.
| 17 |
Risks Related to Our Regulatory and Legal Environment
We are subject to extensive and costly government regulation.
Product candidates employing our technology are subject to extensive and rigorous domestic government regulation including regulation by the FDA, the Centers for Medicare and Medicaid Services, other divisions of the U.S. Department of Health and Human Services, the U.S. Department of Justice, state and local governments, and their respective foreign equivalents. The FDA regulates the research, development, preclinical and clinical testing, manufacture, safety, effectiveness, record-keeping, reporting, labeling, storage, approval, advertising, promotion, sale, distribution, import, and export of pharmaceutical products. The FDA regulates small molecule chemical entities, whether administered orally, topically or by injection, as drugs, subject to an NDA, under the Federal Food, Drug, and Cosmetic Act. If products employing our technologies are marketed abroad, they will also be subject to extensive regulation by foreign governments, whether or not they have obtained FDA approval for a given product and its uses. Such foreign regulation might be equally or more demanding than corresponding U.S. regulation.
Government regulation substantially increases the cost and risk of researching, developing, manufacturing, and selling our product candidates. The regulatory review and approval process, which includes preclinical testing and clinical trials of each product candidate, is lengthy, expensive, and uncertain. We or our collaborators must obtain and maintain regulatory authorization to conduct clinical trials and approval for each product we intend to market, and the manufacturing facilities used for the products must be inspected and meet legal requirements. Securing regulatory approval requires submitting extensive preclinical and clinical data and other supporting information for each proposed therapeutic indication in order to establish the product’s safety and efficacy for each intended use. The development and approval process might take many years, requires substantial resources, and might never lead to the approval of a product.
Even if we are able to obtain regulatory approval for a particular product, the approval might limit the intended medical uses for the product, limit our ability to promote, sell, and distribute the product, require that we conduct costly post-marketing surveillance, and/or require that we conduct ongoing post-marketing studies. Material changes to an approved product, such as, for example, manufacturing changes or revised labeling, might require further regulatory review and approval. Once obtained, any approvals might be withdrawn, including, for example, if there is a later discovery of previously unknown problems with the product, such as a previously unknown safety issue.
If we, our collaborators, or our contract manufacturers fail to comply with applicable regulatory requirements at any stage during the regulatory process, such noncompliance could result in, among other things, delays in the approval of applications or supplements to approved applications; refusal of a regulatory authority, including the FDA, to review pending market approval applications or supplements to approved applications; untitled letters or warning letters; fines; import and export restrictions; product recalls or seizures; injunctions; total or partial suspension of production; civil penalties; withdrawals of previously approved marketing applications or licenses; recommendations by the FDA or other regulatory authorities against governmental contracts; and/or criminal prosecutions.
We might not obtain the necessary U.S. or worldwide regulatory approvals to commercialize any product candidate.
We cannot assure you that we will receive the approvals necessary to commercialize for sale any of our product candidates, or any product candidate we acquire or develop in the future. We will need FDA approval to commercialize our product candidates in the U.S. and approvals from the FDA-equivalent regulatory authorities in foreign jurisdictions to commercialize our product candidates in those jurisdictions. In order to obtain FDA approval of any product candidate, we must submit to the FDA an NDA demonstrating that the product candidate is safe for humans and effective for its intended use. This demonstration requires significant research, preclinical studies, and clinical trials. Satisfaction of the FDA’s regulatory requirements typically takes many years, depends upon the type, complexity and novelty of the product candidate and requires substantial resources for research, development and testing. We cannot predict whether our research and clinical approaches will result in drugs that the FDA considers safe for humans and effective for their indicated uses. The FDA has substantial discretion in the drug approval process and might require us to conduct additional preclinical and clinical testing, perform post-marketing studies or otherwise limit or impose conditions on any approval we obtain. For example, the FDA proposed that we include an additional treatment arm in our pivotal Phase III trial for our former product candidate iferanserin, which increased the cost of that trial.
The approval process might also be delayed by changes in government regulation, future legislation or administrative action or changes in FDA policy that occur prior to or during our regulatory review. Delays in obtaining regulatory approvals might:
| 18 |
| · | delay commercialization of, and our ability to derive product revenues from, our product candidates; |
| · | impose costly procedures on us; and |
| · | diminish any competitive advantages that we might otherwise enjoy. |
Even if we comply with all FDA requests, the FDA might ultimately reject one or more of our NDAs. We cannot be sure that we will ever obtain regulatory approval for our product candidates. Failure to obtain FDA approval of our product candidates will severely undermine our business by leaving us without a saleable product, and therefore without any source of revenues, until another product candidate could be developed or obtained. There is no guarantee that we will ever be able to develop an existing, or acquire another, product candidate.
In foreign jurisdictions, we must receive approval from the appropriate regulatory authorities before we can commercialize any product candidates. The risks associated with foreign regulatory approval processes are similar to the risks associated with the FDA approval procedures described above. We cannot assure you that we will receive the approvals necessary to commercialize our product candidates for sale outside the U.S.
Even if approved, our product candidates will be subject to extensive post-approval regulation.
Once a product candidate is approved, numerous post-approval requirements apply. Among other things, the holder of an approved NDA is subject to ongoing FDA oversight monitoring and reporting obligations, including obligations to monitor and report adverse events and instances of the failure of a product to meet the specifications in the NDA. Application holders must submit new or supplemental applications and obtain FDA approval for changes to the approved product, product labeling, or manufacturing process. Application holders also must submit advertising and other promotional material to the FDA and report on ongoing clinical trials. The FDA also has the authority to require changes in the labeling of approved drug products and to require post-marketing studies.
Advertising and promotional materials must comply with FDA rules in addition to other applicable federal and state laws. The distribution of product samples to physicians must comply with the requirements of the Prescription Drug Marketing Act. Manufacturing facilities remain subject to FDA inspection and must continue to adhere to the FDA’s cGMP requirements. Application holders must obtain FDA approval for product, manufacturing, and labeling changes, depending on the nature of the change. Sales, marketing, and scientific/educational grant programs, among other activities, must comply with the anti-fraud and abuse provisions of the Social Security Act, the False Claims Act, and similar state laws, each as amended. Pricing and rebate programs must comply with the Medicaid rebate requirements of the Omnibus Budget Reconciliation Act of 1990 and the Veteran’s Health Care Act of 1992, each as amended. If products are made available to authorized users of the Federal Supply Schedule of the General Services Administration, additional laws and requirements apply. All of these activities are also potentially subject to federal and state consumer protection and unfair competition laws.
Depending on the circumstances, failure to meet these post-approval requirements can result in criminal prosecution, fines, injunctions, recall or seizure of products, total or partial suspension of production, denial or withdrawal of pre-marketing product approvals, or refusal to allow us to enter into supply contracts, including government contracts. In addition, even if we comply with FDA and other requirements, new information regarding the safety or effectiveness of a product could lead the FDA to modify or withdraw product approval.
We face the risk of product liability claims and might not be able to obtain insurance.
Our business exposes us to the risk of product liability claims that are inherent in the development of drugs. If the use of one or more of our or our collaborators’ drugs harms people, we might be subject to costly and damaging product liability claims brought against us by clinical trial participants, consumers, health care providers, pharmaceutical companies or others selling our products. Our inability to obtain sufficient product liability insurance at an acceptable cost to protect against potential product liability claims could prevent or inhibit the commercialization of pharmaceutical products we develop. We expect to obtain clinical trial insurance for our product candidates prior to beginning clinical trials. We cannot predict all of the possible harms or side effects that might result and, therefore, the amount of insurance coverage we obtain, if any, in the future might not be adequate to cover all liabilities we might incur. We intend to expand our insurance coverage to include product liability insurance covering the sale of commercial products if we obtain marketing approval for our drug candidates in development, but we might be unable to obtain commercially reasonable product liability insurance for any products approved for marketing. If we are unable to obtain insurance at an acceptable cost or otherwise protect against potential product liability claims, we will be exposed to significant liabilities, which might materially and adversely affect our business and financial position. If we are sued for any injury allegedly caused by our or our collaborators’ products, our liability could exceed our total assets and our ability to pay the liability. A successful product liability claim or series of claims brought against us would decrease our cash and could cause the value of our common stock to decrease.
| 19 |
We might be exposed to liability claims associated with the use of hazardous materials and chemicals.
Our research, development and manufacturing activities and/or those of our third-party contractors might involve the controlled use of hazardous materials and chemicals. Although we will strive to have our safety procedures, and those of our contractors, for using, storing, handling and disposing of these materials comply with federal, state and local laws and regulations, we cannot completely eliminate the risk of accidental injury or contamination from these materials. In the event of such an accident, we could be held liable for any resulting damages, and any liability could materially adversely affect our business, financial condition and results of operations. In addition, the federal, state and local laws and regulations governing the use, manufacture, storage, handling and disposal of hazardous or radioactive materials and waste products might require us to incur substantial compliance costs that could materially adversely affect our business, financial condition and results of operations. We currently do not carry hazardous materials liability insurance. We intend to obtain such insurance in the future if necessary, but cannot give assurance that we could obtain such coverage.
Risks Related to Our Intellectual Property
Our business depends on protecting our intellectual property.
If we and our licensors IURTC and Therabiome do not obtain protection for our respective intellectual property rights, our competitors might be able to take advantage of our research and development efforts to develop competing drugs. Our success, competitive position and future revenues, if any, depend in part on our ability and the abilities of our licensors to obtain and maintain patent protection for our products, methods, processes and other technologies, to preserve our trade secrets, to prevent third parties from infringing on our proprietary rights and to operate without infringing the proprietary rights of third parties.
We seek to protect our proprietary position by filing patent applications in the United States and abroad related to our novel technologies and medicines that are important to our business. To date, although our licensors have filed patent applications, we do not own or have any rights to any issued patents that cover any of our product candidates, and we cannot be certain that we will secure any rights to any issued patents with claims that cover any of our proprietary medicines and technologies. The patent prosecution process is expensive and time-consuming and we may not be able to file and prosecute all necessary or desirable patent applications at a reasonable cost or in a timely manner. It is also possible that we will fail to identify patentable aspects of our research and development output before it is too late to obtain patent protection.
The patent process also is subject to numerous risks and uncertainties, and there can be no assurance that we will be successful in protecting our products by obtaining and defending patents. These risks and uncertainties include the following:
| · | Any patent rights, if obtained, might be challenged, invalidated, or circumvented, or otherwise might not provide any competitive advantage; |
| · | Our competitors, many of which have substantially greater resources than we do and many of which might make significant investments in competing technologies, might seek, or might already have obtained, patents that will limit, interfere with, or eliminate our ability to make, use, and sell our potential products either in the U.S. or in international markets; |
| · | As a matter of public policy regarding worldwide health concerns, there might be significant pressure on the U.S. government and other international governmental bodies to limit the scope of patent protection both inside and outside the U.S. for disease treatments that prove successful; and |
| 20 |
| · | Countries other than the U.S. might have patent laws that provide less protection than those governing U.S. courts, allowing foreign competitors the ability to exploit these laws to create, develop, and market competing products. |
In addition, the U.S. Patent and Trademark Office and patent offices in other jurisdictions have often required that patent applications concerning pharmaceutical and/or biotechnology-related inventions be limited or narrowed substantially to cover only the specific innovations exemplified in the patent application, thereby limiting the scope of protection against competitive challenges. Thus, even if we or our licensors are able to obtain patents, the patents might be substantially narrower than anticipated.
Patent and other intellectual property protection is crucial to the success of our business and prospects, and there is a substantial risk that such protections, if obtained, will prove inadequate. Our business and prospects will be harmed if we fail to obtain these protections or they prove insufficient.
We might be involved from time to time in litigation to determine the enforceability, scope and validity of our proprietary rights. Any such litigation could result in substantial cost and divert management’s attention from our operations.
We rely on trade secret protections through confidentiality agreements with our employees, customers and other parties, and the breach of these agreements could adversely affect our business and prospects.
We rely on trade secrets and proprietary know-how, which we seek to protect, in part, through confidentiality, invention, and non-disclosure agreements with our employees, scientific advisors, consultants, collaborators, suppliers, and other parties. There can be no assurance that these agreements will not be breached, that we would have adequate remedies for any such breach or that our trade secrets will not otherwise become known to or independently developed by our competitors. If any of these events occurs, or we otherwise lose protection for our trade secrets or proprietary know-how, the value of this information may be greatly reduced.
If we infringe the rights of third parties we might have to forgo selling our future products, pay damages, or defend against litigation.
If our product candidates, methods, processes and other technologies infringe the proprietary rights of other parties, we could incur substantial costs and we might have to:
| · | obtain licenses, which might not be available on commercially reasonable terms, if at all; |
| · | abandon an infringing product candidate; |
| · | redesign our products or processes to avoid infringement; |
| · | stop using the subject matter claimed in the patents held by others; |
| · | pay damages; and/or |
| · | defend litigation or administrative proceedings which might be costly whether we win or lose, and which could result in a substantial diversion of our financial and management resources. |
Any of these events could substantially harm our earnings, financial condition and operations.
Risks Related to Our Common Stock
We might not be able to maintain the listing of our common stock on The NASDAQ Capital Market.
Our common stock is listed on The NASDAQ Capital Market under the symbol “ASMB.” We might not be able to maintain the listing standards of that exchange. If we fail to maintain the listing requirements, our common stock might trade on the OTC Bulletin Board or in the “pink sheets” maintained by Pink OTC Markets, Inc. These alternative markets are generally considered to be markets that are less efficient and less broad than The NASDAQ Capital Market.
| 21 |
The price of our common stock might fluctuate significantly, and you could lose all or part of your investment.
Since we went public on December 22, 2010 and through November 25, 2014, the closing price of our common stock has fluctuated between $5.35 and $85.75 (after giving effect to the 1-for-5 reverse stock split effected on July 11, 2014), with significant volatility after we announced on June 25, 2012 that iferanserin failed to meet the endpoints of our Phase III trial, and after we announced in February 2014 that diltiazem demonstrated no significant improvement compared to placebo. Continued volatility in the market price of our common stock might prevent you from being able to sell your shares of our common stock at or above the price you paid for such shares. The trading price of our common stock might be volatile and subject to wide price fluctuations in response to various factors, including:
| · | the receipt or loss of required regulatory approvals for our product candidates; |
| · | results of our preclinical studies and clinical trials and other studies involving our product candidates; |
| · | availability of capital; |
| · | future sales of our common stock; |
| · | sale of shares of our common stock by our significant stockholders or members of our management; |
| · | additions or departures of key personnel; |
| · | investor perceptions of us and the pharmaceutical industry; |
| · | issuance of new or changed securities analysts’ reports or recommendations, or the announcement of any changes to our credit rating; |
| · | success or failure of our product candidates; |
| · | introduction of new products or announcements of significant contracts, acquisitions or capital commitments by us or our competitors; |
| · | threatened or actual litigation and government investigations; |
| · | legislative, political or regulatory developments; |
| · | the overall performance of the equity markets; |
| · | actual or anticipated fluctuations in our quarterly financial and operating results; |
| · | general economic conditions; |
| · | changes in interest rates; and |
| · | changes in accounting standards, policies, guidance, interpretations or principles. |
These and other factors might cause the market price of our common stock to fluctuate substantially, which might limit or prevent investors from readily selling their shares of our common stock and might otherwise negatively affect the liquidity of our common stock. In addition, in recent years, the stock market has experienced significant price and volume fluctuations. This volatility has had a significant impact on the market price of securities issued by many companies across many industries. The changes frequently appear to occur without regard to the operating performance of the affected companies. Accordingly, the price of our common stock could fluctuate based upon factors that have little or nothing to do with our company, and these fluctuations could materially reduce our share price.
| 22 |
We do not intend to pay dividends for the foreseeable future and our stock may not appreciate in value.
We currently intend to retain our future earnings, if any, to finance the operation and growth of our business and do not expect to pay any cash dividends in the foreseeable future. As a result, the success of an investment in shares of our common stock will depend upon any future appreciation in its value. There is no guarantee that shares of our common stock will appreciate in value or that the price at which our stockholders have purchased their shares will be able to be maintained.
The requirements of being a public company adds to our operating costs and might strain our resources and distract our management.
As a public company, we face increased legal, accounting, administrative and other costs and expenses not faced by private companies. We are subject to the reporting requirements of the Securities Exchange Act of 1934, which requires that we file annual, quarterly and current reports with respect to our business and financial condition, and the rules and regulations implemented by the SEC, the Sarbanes-Oxley Act of 2002, and The NASDAQ Capital Market, each of which imposes additional reporting and other obligations on public companies. These rules and regulations increase our legal and financial compliance costs and make some activities more time-consuming and costly, although we are currently unable to estimate these costs with any degree of certainty. Complying with these requirements might divert management’s attention from other business concerns, which could have a material adverse effect on our prospects, business, and financial condition.
Additionally, the expenses incurred by public companies generally for reporting and corporate governance purposes have been increasing. These increased costs will require us to divert a significant amount of money that we could otherwise use to develop our product candidates or otherwise expand our business. If we are unable to satisfy our obligations as a public company, we could be subject to delisting of our common stock, fines, sanctions and other regulatory action and potentially civil litigation.
Several provisions of the Delaware General Corporation Law and our Amended and Restated Certificate of Incorporation and Bylaws could discourage, delay or prevent a merger or acquisition, which could adversely affect the market price of our securities.
Several provisions of the Delaware General Corporation Law and our Amended and Restated Certificate of Incorporation and Bylaws could discourage, delay or prevent a merger or acquisition that stockholders may consider favorable, and the market price of our securities could be reduced as a result. These provisions include:
| · | “blank check” preferred stock; |
| · | prohibiting us from engaging in a “business combination” with an “interested stockholder” for a period of three years after the date of the transaction in which the person became an interested stockholder unless certain provisions are met; |
| · | prohibiting cumulative voting in the election of directors; |
| · | limiting the persons who may call special meetings of stockholders; |
| · | establishing advance notice requirements for nominations for election to our board of directors or for proposing matters that can be acted on by stockholders at stockholder meetings; and |
| · | the ability of our board of directors to increase its size and fill vacancies. |
If securities analysts downgrade our stock or cease coverage of us, the price of our stock could decline.
The trading market for our common stock relies in part on the research and reports that industry or financial analysts publish about us or our business. Currently, two financial analysts publish reports about us and our business. We do not control these or any other analysts. Furthermore, there are many large, well-established, publicly traded companies active in our industry and market, which may mean that it is less likely that we will receive widespread analyst coverage. If any of the analysts who cover us downgrade our stock, our stock price would likely decline rapidly. If these analysts cease coverage of our company, we could lose visibility in the market, which in turn could cause our stock price to decline.
| 23 |
Substantial future sales of our common stock in the public market may depress our stock price and make it difficult for you to recover the full value of your investment in our shares of securities.
As of November 24, 2014, we had 10,647,059 shares of common stock outstanding. Substantially all of these shares are available for public sale, subject in some cases to volume and other limitations or delivery of a prospectus. The market price of our common stock may decline if our common stockholders sell a large number of shares of our common stock in the public market, or the market perceives that such sales may occur. In addition, at November 24, 2014, we had outstanding options and warrants to purchase an aggregate of 3,267,651 shares and 271,501 shares, respectively, of our common stock. If these options or warrants are exercised and the shares issued upon exercise are sold, the market price of our securities may also decline. These factors also could impair our ability to raise needed capital by depressing the price at which we could sell our securities.
| 24 |
SPECIAL NOTE REGARDING FORWARD-LOOKING STATEMENTS
The SEC encourages companies to disclose forward-looking information so that investors can better understand a company’s future prospects and make informed investment decisions. This prospectus and the documents we have filed with the SEC that are incorporated herein by reference contain such “forward-looking statements” within the meaning of the Private Securities Litigation Reform Act of 1995.
Words such as “may,” “might,” “should,” “anticipate,” “estimate,” “expect,” “projects,” “intends,” “plans,” “believes” and words and terms of similar substance used in connection with any discussion of future operating or financial performance, identify forward-looking statements. Forward-looking statements represent management’s current judgment regarding future events and are subject to a number of risks and uncertainties that could cause actual results to differ materially from those described in the forward-looking statements. These risks include, but are not limited to: risks related to costs, timing, regulatory review and results of our pre-clinical studies and clinical trials; our ability to obtain FDA and foreign approval of our product candidates; our anticipated capital expenditures, our estimates regarding our capital requirements, and our need for future capital; our liquidity and working capital requirements; our expectations regarding our revenues, expenses and other results of operations; the unpredictability of the size of the markets for, and market acceptance of, any of our product candidates; our need to obtain additional funding and our ability to obtain future funding on acceptable terms, or at all; our ability to retain and hire necessary employees and to staff our operations appropriately; our ability to compete in our industry and innovation by our competitors; our ability to stay abreast of and comply with new or modified laws and regulations that currently apply or become applicable to our business; estimates and estimate methodologies used in preparing our financial statements; and the future trading prices of our common stock and the impact of securities analysts' reports on these prices. Please also see the discussion of risks and uncertainties under “Risk Factors” above and contained in any supplements to this prospectus, and in our most recent annual report on Form 10-K, as revised or supplemented by our most recent quarterly report on Form 10-Q, as well as any amendments thereto, as filed with the SEC and which are incorporated herein by reference.
In light of these assumptions, risks and uncertainties, the results and events discussed in the forward-looking statements contained in this prospectus or in any document incorporated herein by reference might not occur. Investors are cautioned not to place undue reliance on the forward-looking statements, which speak only as of the date of this prospectus or the date of the document incorporated by reference in this prospectus. We are not under any obligation, and we expressly disclaim any obligation, to update or alter any forward-looking statements, whether as a result of new information, future events or otherwise. All subsequent forward-looking statements attributable to us or to any person acting on our behalf are expressly qualified in their entirety by the cautionary statements contained or referred to in this section.
| 25 |
We cannot assure you that we will receive any proceeds in connection with securities offered by us pursuant to this prospectus. Unless otherwise provided in the applicable prospectus supplement, we intend to use the net proceeds from the sale of our securities by us under this prospectus for general corporate purposes, including preclinical and clinical trials, research and development expenses, and general and administrative expenses. We will set forth in the applicable prospectus supplement our intended use for the net proceeds received from the sale of any securities by us. Pending the application of the net proceeds, we intend to invest the net proceeds generally in high quality, short-term, interest-bearing securities.
We may sell the securities from time to time pursuant to underwritten public offerings, negotiated transactions, block trades or a combination of these methods. We may sell the securities to or through underwriters or dealers, through agents, or directly to one or more purchasers. We may distribute securities from time to time in one or more transactions:
| · | at a fixed price or prices, which may be changed; |
| · | at market prices prevailing at the time of sale; |
| · | at prices related to such prevailing market prices; or |
| · | at negotiated prices. |
A prospectus supplement or supplements (and any related free writing prospectus that we may authorize to be provided to you) will describe the terms of the offering of the securities, including, to the extent applicable:
| · | the name or names of the underwriters, if any; |
| · | the purchase price of the securities or other consideration therefor, and the proceeds, if any, we will receive from the sale; |
| · | any over-allotment options under which underwriters may purchase additional securities from us; |
| · | any agency fees or underwriting discounts and other items constituting agents’ or underwriters’ compensation; |
| · | any public offering price; |
| · | any discounts or concessions allowed or reallowed or paid to dealers; and |
| · | any securities exchange or market on which the securities may be listed. |
Only underwriters named in the prospectus supplement will be underwriters of the securities offered by the prospectus supplement.
If underwriters are used in the sale, they will acquire the securities for their own account and may resell the securities from time to time in one or more transactions at a fixed public offering price or at varying prices determined at the time of sale. The obligations of the underwriters to purchase the securities will be subject to the conditions set forth in the applicable underwriting agreement. We may offer the securities to the public through underwriting syndicates represented by managing underwriters or by underwriters without a syndicate. Subject to certain conditions, the underwriters will be obligated to purchase all of the securities offered by the prospectus supplement, other than securities covered by any over-allotment option. Any public offering price and any discounts or concessions allowed or reallowed or paid to dealers may change from time to time. We may use underwriters with whom we have a material relationship. We will describe in the prospectus supplement, naming the underwriter, the nature of any such relationship.
| 26 |
We may sell securities directly or through agents we designate from time to time. We will name any agent involved in the offering and sale of securities and we will describe any commissions we will pay the agent in the prospectus supplement. Unless the prospectus supplement states otherwise, our agent will act on a best-efforts basis for the period of its appointment.
We may authorize agents or underwriters to solicit offers by certain types of institutional investors to purchase securities from us at the public offering price set forth in the prospectus supplement pursuant to delayed delivery contracts providing for payment and delivery on a specified date in the future. We will describe the conditions to these contracts and the commissions we must pay for solicitation of these contracts in the prospectus supplement.
We may provide agents and underwriters with indemnification against civil liabilities, including liabilities under the Securities Act, or contribution with respect to payments that the agents or underwriters may make with respect to these liabilities. Agents and underwriters may engage in transactions with, or perform services for, us in the ordinary course of business.
All securities we may offer, other than common stock, will be new issues of securities with no established trading market. Any underwriters may make a market in these securities, but will not be obligated to do so and may discontinue any market making at any time without notice. We cannot guarantee the liquidity of the trading markets for any securities.
Any underwriter may engage in over-allotment, stabilizing transactions, short-covering transactions and penalty bids in accordance with Regulation M under the Exchange Act. Over-allotment involves sales in excess of the offering size, which create a short position. Stabilizing transactions permit bids to purchase the underlying security so long as the stabilizing bids do not exceed a specified maximum price. Syndicate-covering or other short-covering transactions involve purchases of the securities, either through exercise of the over-allotment option or in the open market after the distribution is completed, to cover short positions. Penalty bids permit the underwriters to reclaim a selling concession from a dealer when the securities originally sold by the dealer are purchased in a stabilizing or covering transaction to cover short positions. Those activities may cause the price of the securities to be higher than it would otherwise be. If commenced, the underwriters may discontinue any of the activities at any time.
Any underwriters that are qualified market makers on the NASDAQ Capital Market may engage in passive market making transactions in the common stock on the NASDAQ Capital Market in accordance with Regulation M under the Exchange Act, during the business day prior to the pricing of the offering, before the commencement of offers or sales of the common stock. Passive market makers must comply with applicable volume and price limitations and must be identified as passive market makers. In general, a passive market maker must display its bid at a price not in excess of the highest independent bid for such security; if all independent bids are lowered below the passive market maker’s bid, however, the passive market maker’s bid must then be lowered when certain purchase limits are exceeded. Passive market making may stabilize the market price of the securities at a level above that which might otherwise prevail in the open market and, if commenced, may be discontinued at any time.
Pursuant to our certificate of incorporation, we are authorized to issue 50,000,000 shares of common stock, $0.001 par value per share. As of November 24, 2014, we had 10,647,059 shares of common stock outstanding and 122 stockholders of record.
The following summary of certain provisions of our common stock does not purport to be complete. You should refer to our certificate of incorporation and our bylaws, both of which are included as exhibits to the registration statement we have filed with the SEC in connection with this offering. The summary below is also qualified by provisions of applicable law.
| 27 |
General
The holders of our common stock are entitled to one vote per share on all matters to be voted on by the stockholders, and there are no cumulative voting rights. Generally, all matters to be voted on by stockholders must be approved by a majority (or, in the case of election of directors, by a plurality) of the votes entitled to be cast by all shares of common stock present in person or represented by proxy, subject to any voting rights granted to holders of any preferred stock.
The holders of common stock are entitled to receive ratable dividends, if any, payable in cash, in stock or otherwise if, as and when declared from time to time by our board of directors out of funds legally available for the payment of dividends, subject to any preferential rights that may be applicable to any outstanding preferred stock. In the event of a liquidation, dissolution, or winding up of our company, after payment in full of all outstanding debts and other liabilities, the holders of common stock are entitled to share ratably in all remaining assets, subject to prior distribution rights of preferred stock, if any, then outstanding. No shares of common stock have preemptive rights or other subscription rights to purchase additional shares of common stock. There are no redemption or sinking fund provisions applicable to the common stock. All outstanding shares of common stock are fully paid and nonassessable, and the shares of common stock included in this registration statement will be fully paid and nonassessable. The rights, preferences and privileges of holders of our common stock will be subject to, and might be adversely affected by, the rights of holders of any preferred stock that we may issue in the future. All shares of common stock that are acquired by us shall be available for reissuance by us at any time.
Transfer Agent and Registrar
The transfer agent and registrar for our common stock is VStock Transfer, LLC, Inc. The transfer agent’s address is 18 Lafayette Place, Woodmere, New York 11598 and its telephone number is (212) 828-8436.
NASDAQ Capital Market
Our common stock is listed for quotation on the NASDAQ Capital Market under the symbol “ASMB.” On November 25, 2014, the reported closing price of our common stock was $8.60 per share.
DESCRIPTION OF PREFERRED STOCK
Our board of directors has the authority, without further action by the stockholders, to issue up to 5,000,000 shares of preferred stock in one or more series and to fix the rights, preferences, privileges and restrictions thereof, including dividend rights, conversion rights, voting rights, terms of redemption, liquidation preferences, sinking fund terms and the number of shares constituting any series or the designation of such series, without any further vote or action by our stockholders. As of the date of this prospectus, no shares of preferred stock were outstanding. The issuance of preferred stock could adversely affect the voting power of holders of common stock and the likelihood that such holders will receive dividend payments and payments upon liquidation and could have the effect of delaying, deferring or preventing a change in control of our company.
We will fix the rights, preferences, privileges and restrictions of the preferred stock of each series in the certificate of designation relating to that series. We will file as an exhibit to the registration statement of which this prospectus is a part, or will incorporate by reference from reports that we file with the SEC, the form of any certificate of designation that describes the terms of the series of preferred stock we are offering before the issuance of the related series of preferred stock. This description will include any or all of the following, as required:
| · | the title and stated value; |
| · | the number of shares we are offering; |
| · | the liquidation preference per share; |
| · | the purchase price; |
| · | the dividend rate, period and payment date and method of calculation for dividends; |
| 28 |
| · | whether dividends will be cumulative or non-cumulative and, if cumulative, the date from which dividends will accumulate; |
| · | the procedures for any auction and remarketing, if any; |
| · | the provisions for a sinking fund, if any; |
| · | the provisions for redemption or repurchase, if applicable, and any restrictions on our ability to exercise those redemption and repurchase rights; |
| · | any listing of the preferred stock on any securities exchange or market; |
| · | whether the preferred stock will be convertible into our common stock, and, if applicable, the conversion price, or how it will be calculated, and the conversion period; |
| · | whether the preferred stock will be exchangeable into debt securities, and, if applicable, the exchange price, or how it will be calculated, and the exchange period; |
| · | voting rights, if any, of the preferred stock; |
| · | preemptive rights, if any; |
| · | restrictions on transfer, sale or other assignment, if any; |
| · | whether interests in the preferred stock will be represented by depositary shares; |
| · | a discussion of any material or special United States federal income tax considerations applicable to the preferred stock; |
| · | the relative ranking and preferences of the preferred stock as to dividend rights and rights if we liquidate, dissolve or wind up our affairs; |
| · | any limitations on issuance of any class or series of preferred stock ranking senior to or on a parity with the series of preferred stock as to dividend rights and rights if we liquidate, dissolve or wind up our affairs; and |
| · | any other specific terms, preferences, rights or limitations of, or restrictions on, the preferred stock. |
If we issue shares of preferred stock under this prospectus, the shares will be fully paid and non-assessable.
The General Corporation Law of the State of Delaware, the state of our incorporation, provides that the holders of preferred stock will have the right to vote separately as a class on any proposal involving fundamental changes in the rights of holders of that preferred stock. This right is in addition to any voting rights that may be provided for in the applicable certificate of designation.
Our board of directors may authorize the issuance of preferred stock with voting or conversion rights that could adversely affect the voting power or other rights of the holders of our common stock. Preferred stock could be issued quickly with terms designed to delay or prevent a change in control of our company or make removal of management more difficult. Additionally, the issuance of preferred stock may have the effect of decreasing the market price of our common stock.
DESCRIPTION OF DEBT SECURITIES
The following description, together with the additional information we include in any applicable prospectus supplement, summarizes the material terms and provision of any debt securities that we may offer under this prospectus. While the terms we have summarized below will apply generally to any future debt securities we may offer, we will describe the particular terms of any debt securities that we may offer in more detail in the applicable prospectus supplement. The terms of any debt securities we may offer under a prospectus supplement may differ from the terms described below. For any debt securities that we may offer, an indenture (and any relevant supplemental indenture) will contain additional important terms and provisions and will be incorporated by reference as an exhibit to the registration statement that includes this prospectus, or as an exhibit to reports that we file with the SEC and incorporated by reference in this prospectus.
| 29 |
With respect to any debt securities that we issue, we will issue such debt securities under an indenture, which we would enter into with the trustee named in the indenture. Any indenture would be qualified under the Trust Indenture Act of 1939.
With respect to any debt securities that we issue, we will describe in each prospectus supplement the following terms relating to a series of debt securities:
| · | the title; |
| · | the principal amount being offered, and if a series, the total amount authorized and the total amount outstanding; |
| · | any limit on the amount that may be issued; |
| · | whether or not we will issue the series of debt securities in global form, and if so, the terms and who the depository will be; |
| · | the maturity date; |
| · | the principal amount due at maturity; |
| · | whether and under what circumstances, if any, we will pay additional amounts on any debt securities held by a person who is not a United States person for tax purposes, and whether we can redeem the debt securities if we have to pay such additional amounts; |
| · | the annual interest rate, which may be fixed or variable, or the method for determining the rate and the date interest will begin to accrue, the dates interest will be payable and the regular record dates for interest payment dates or the method for determining such dates; |
| · | whether or not the debt securities will be convertible into shares of our common stock or our preferred stock and, if so, the terms of such conversion; |
| · | whether or not the debt securities will be secured or unsecured by some or all of our assets, and the terms of any secured debt; |
| · | the terms of the subordination of any series of subordinated debt; |
| · | the place where payments will be payable; |
| · | restrictions on transfer, sale or other assignment, if any; |
| · | our right, if any, to defer payment or interest and the maximum length of any such deferral period; |
| · | the date, if any, after which and the conditions upon which, and the price at which, we may, at our option, redeem the series of debt securities pursuant to any optional or provisional redemption provisions and the terms of those redemptions provisions; |
| · | the date, if any, on which, and the price at which we are obligated, pursuant to any mandatory sinking fund or analogous fund provisions or otherwise, to redeem, or at the holder’s option to purchase, the series of debt securities and the currency or currency unit in which the debt securities are payable; |
| · | whether the indenture will restrict our ability to pay dividends, or will require us to maintain any asset ratios or reserves; |
| · | whether we will be restricted from incurring any additional indebtedness, issuing additional securities, or entering into a merger, consolidation or sale of our business; |
| · | a discussion of any material or special United States federal income tax considerations applicable to the debt securities; |
| · | information describing any book-entry features; |
| · | any provisions for payment of additional amounts for taxes; |
| 30 |
| · | whether the debt securities are to be offered at a price such that they will be deemed to be offered at an “original issue discount” as defined in paragraph (a) of Section 1273 of the Internal Revenue Code of 1986, as amended; |
| · | the denominations in which we will issue the series of debt securities, if other than denominations of $1,000 and any integral multiple thereof; |
| · | events of default; |
| · | whether we and/or the debenture trustee may change an indenture without the consent of any holders; |
| · | the form of debt security and how it may be exchanged and transferred; |
| · | description of the debenture trustee and paying agent, and the method of payments; and |
| · | any other specified terms, preferences, rights or limitations of, or restrictions on, the debt securities and any terms that may be required by us or advisable under applicable laws or regulations. |
The following description, together with the additional information we may include in any applicable prospectus supplement, summarizes the material terms and provisions of any warrants that we may offer under this prospectus and the related warrant agreements and warrant certificates. While the terms summarized below will apply generally to any warrants that we may offer, we will describe the particular terms of any series of warrants in more detail in the applicable prospectus supplement. The terms of any warrants offered under a prospectus supplement may differ from the terms described below. With respect to any warrants that we offer, specific warrant agreements will contain additional important terms and provisions and will be incorporated by reference as an exhibit to the registration statement that includes this prospectus or as an exhibit to reports that we file with the SEC and incorporated by reference in this prospectus:
| · | the specific designation and aggregate number of, and the price at which we will issue, the warrants; |
| · | the currency or currency units in which the offering price, if any, and the exercise price are payable; |
| · | if applicable, the exercise price for shares of our common stock or preferred stock and the number of shares of common stock or preferred stock to be received upon exercise of the warrants; |
| · | in the case of warrants to purchase debt securities, the principal amount of debt securities purchasable upon exercise of one warrant and the price at, and currency in which, this principal amount of debt securities may be purchased upon such exercise; |
| · | the date on which the right to exercise the warrants will begin and the date on which that right will expire or, if you may not continuously exercise the warrants throughout that period, the specific date or dates on which you may exercise the warrants; |
| · | whether the warrants will be issued in fully registered form or bearer form, in definitive or global form or in any combination of these forms, although, in any case, the form of a warrant included in a unit will correspond to the form of the unit and of any security included in that unit; |
| · | any applicable material U.S. federal income tax consequences; |
| · | the identity of the warrant agent for the warrants and of any other depositaries, execution or paying agents, transfer agents, registrars or other agents; |
| · | the proposed listing, if any, of the warrants or the common stock issuable upon exercise of the warrants on any securities exchange; |
| · | if applicable, the date from and after which the warrants and the common stock will be separately transferable; |
| 31 |
| · | if applicable, the minimum or maximum amount of the warrants that may be exercised at any one time; |
| · | information with respect to book-entry procedures, if any; |
| · | the anti-dilution provisions of the warrants, if any; |
| · | any redemption or call provisions; |
| · | whether the warrants are to be sold separately or with other securities as parts of units; and |
| · | any additional terms of the warrants, including terms, procedures and limitations relating to the exchange and exercise of the warrants. |
Before exercising their warrants, holders of warrants will not have any of the rights of holders of the securities purchasable upon such exercise, including:
| · | in the case of warrants to purchase debt securities, the right to receive payments of principal of, or premium, if any, or interest on, the debt securities purchasable upon exercise or to enforce covenants in the applicable indenture; or |
| · | in the case of warrants to purchase common stock or preferred stock, the right to receive dividends, if any, or, payments upon our liquidation, dissolution or winding up or to exercise voting rights, if any. |
Transfer Agent and Registrar
The transfer agent and registrar for any warrants will be set forth in the applicable prospectus supplement.
We might issue units comprised of one or more debt securities, shares of common stock, shares of preferred stock and warrants in any combination. Each unit will be issued so that the holder of the unit is also the holder of each security included in the unit. Thus, the holder of a unit will have the rights and obligations of a holder of each included security. The unit agreement under which a unit is issued may provide that the securities included in the unit may not be held or transferred separately, at any time or at any time before a specified date. We will file as exhibits to the registration statement of which this prospectus is a part, or will incorporate by reference from reports that we file with the SEC, the form of unit agreement, warrant and any supplemental agreements that describe the terms of the series of units we are offering before the issuance of the related series of units.
We may choose to evidence each series of units by unit certificates that we would issue under a separate agreement. If we choose to evidence the units by unit certificates, we will enter into the unit agreements with a unit agent and will indicate the name and address of the unit agent in the applicable prospectus supplement relating to the particular series of units.
CERTAIN PROVISIONS OF DELAWARE LAW AND OF THE COMPANY’S CERTIFICATE OF
INCORPORATION AND BYLAWS
Certain provisions of Delaware law and our certificate of incorporation and bylaws discussed below may have the effect of making more difficult or discouraging a tender offer, proxy contest or other takeover attempt. These provisions are expected to encourage persons seeking to acquire control of our company to first negotiate with our board of directors. We believe that the benefits of increasing our ability to negotiate with the proponent of an unfriendly or unsolicited proposal to acquire or restructure our company outweigh the disadvantages of discouraging these proposals because negotiation of these proposals could result in an improvement of their terms.
| 32 |
Delaware anti-takeover law
We are subject to Section 203 of the Delaware General Corporation Law, an anti-takeover law. In general, Section 203 prohibits a publicly held Delaware corporation from engaging in a “business combination” with an “interested stockholder” for a period of three years following the date the person became an interested stockholder, unless:
| · | the board of directors approves the transaction in which the stockholder became an interested stockholder prior to the date the interested stockholder attained that status; |
| · | when the stockholder became an interested stockholder, he or she owned at least 85% of the voting stock of the corporation outstanding at the time the transaction commenced, excluding shares owned by persons who are directors and also officers and certain shares owned by employee benefits plans; or |
| · | on or subsequent to the date the business combination is approved by the board of directors, the business combination is authorized by the affirmative vote of at least 66 2/3% of the voting stock of the corporation at an annual or special meeting of stockholders. |
Generally, a “business combination” includes a merger, asset or stock sale, or other transaction resulting in a financial benefit to the interested stockholder. Generally, an “interested stockholder” is a person who, together with affiliates and associates, owns, or is an affiliate or associate of the corporation and within three years prior to the determination of interested stockholder status did own, 15% or more of a corporation’s voting stock.
The existence of Section 203 of the Delaware General Corporation Law would be expected to have an anti-takeover effect with respect to transactions not approved in advance by our board of directors, including discouraging attempts that might result in a premium over the market price for the shares of our common stock.
Charter Documents
Our certificate of incorporation and bylaws include a number of provisions that may have the effect of deterring hostile takeovers or delaying or preventing changes in control or management of our company. First, the Bylaws provide that all stockholder action must be effected at a duly called meeting of stockholders and not by a consent in writing. Further, our bylaws limit who may call special meetings of the stockholders. Our certificate of incorporation does not include a provision for cumulative voting for directors. Under cumulative voting, a minority stockholder holding a sufficient percentage of a class of shares may be able to ensure the election of one or more directors. Our bylaws provide that the number of directors on our board, which may range from three to nine directors, shall be exclusively fixed by our board, which has set the number of directors at five. Newly created directorships resulting from any increase in our authorized number of directors and any vacancies in our board resulting from death, resignation, retirement, disqualification or other cause (including removal from office by a vote of the shareholders) will be filled by a majority of our board then in office. Finally, our bylaws establish procedures, including advance notice procedures, with regard to the nomination of candidates for election as directors and stockholder proposals. These and other provisions of our certificate of incorporation and bylaws and Delaware law could discourage potential acquisition proposals and could delay or prevent a change in control or management of our company.
The validity of the securities being offered hereby will be passed upon by Wyrick Robbins Yates & Ponton, LLP, Raleigh, North Carolina.
The balance sheets of Ventrus Biosciences, Inc., a development stage company (now Assembly Biosciences, Inc.) as of December 31, 2013 and 2012 and the related statements of operations, changes in stockholders’ equity, and cash flows for each of the years in the two-year period ended December 31, 2013 and 2012 and for the period from October 7, 2005 (inception) to December 31, 2013 have been audited by EisnerAmper LLP, independent registered public accounting firm, as stated in their report which is incorporated herein by reference. Such financial statements have been incorporated herein by reference in reliance on the report of EisnerAmper LLP given upon their authority as experts in accounting and auditing.
| 33 |
WHERE YOU CAN FIND MORE INFORMATION
We are subject to the reporting requirements of the Securities Exchange Act of 1934, as amended, and file annual, quarterly and current reports, proxy statements and other information with the SEC. You may read and copy these reports, proxy statements and other information at the SEC’s public reference facilities at 100 F Street, N.E., Room 1580, Washington, D.C. 20549. You can request copies of these documents by writing to the SEC and paying a fee for the copying cost. Please call the SEC at 1-800-SEC-0330 for more information about the operation of the public reference facilities. SEC filings are also available at the SEC’s web site at http://www.sec.gov. Our common stock is listed on the NASDAQ Capital Market, and you can read and inspect our filings at the offices of the National Association of Securities Dealers, Inc. at 1735 K Street, Washington, D.C. 20006.
This prospectus is only part of a registration statement on Form S-3 that we have filed with the SEC under the Securities Act of 1933, as amended, and therefore omits certain information contained in the registration statement. We have also filed exhibits and schedules with the registration statement that are excluded from this prospectus, and you should refer to the applicable exhibit or schedule for a complete description of any statement referring to any contract or other document. You may inspect a copy of the registration statement, including the exhibits and schedules, without charge, at the public reference room or obtain a copy from the SEC upon payment of the fees prescribed by the SEC.
INCORPORATION OF DOCUMENTS BY REFERENCE
The SEC allows us to “incorporate by reference” information that we file with them. Incorporation by reference allows us to disclose important information to you by referring you to those other documents. The information incorporated by reference is an important part of this prospectus, and information that we file later with the SEC will automatically update and supersede this information. We filed a registration statement on Form S-3 under the Securities Act of 1933, as amended, with the SEC with respect to the securities being offered pursuant to this prospectus. This prospectus omits certain information contained in the registration statement, as permitted by the SEC. You should refer to the registration statement, including the exhibits, for further information about us and the securities being offered pursuant to this prospectus. Statements in this prospectus regarding the provisions of certain documents filed with, or incorporated by reference in, the registration statement are not necessarily complete and each statement is qualified in all respects by that reference. Copies of all or any part of the registration statement, including the documents incorporated by reference or the exhibits, may be obtained upon payment of the prescribed rates at the offices of the SEC listed above in “Where You Can Find More Information.” The documents we are incorporating by reference are:
| · | our Annual Report on Form 10-K for the fiscal year ended December 31, 2013, filed with the SEC on March 31, 2014; |
| · | our Quarterly Report on Form 10-Q for the quarter ended March 31, 2014 filed with the SEC on May 5, 2014; |
| · | our Quarterly Report on Form 10-Q for the quarter ended June 30, 2014, filed with the SEC on August 14, 2014; |
| · | our Quarterly Report on Form 10-Q for the quarter ended September 30, 2014 filed with the SEC November 17, 2014; |
| · | our Current Reports on Form 8-K filed with the SEC on January 8, January 16, February 12, April 4, May 19, July 11, July 14, July 30, August 13, September 17 (Form 8-K/A) and October 1, 2014; |
| · | our definitive proxy solicitation materials filed with the SEC on June 9, 2014; |
| · | the description of our common stock contained in our registration statement on Form 8-A (File No. 001-35005) filed with the SEC on December 10, 2010, including any amendment or report filed for the purpose of updating such description; and |
| · | all of the filings pursuant to the Securities Exchange Act of 1934, as amended, after the date of the filing of the original registration statement and prior to the effectiveness of the registration statement. |
| 34 |
In addition, all documents subsequently filed by us pursuant to Section 13(a), 13(c), 14 or 15(d) of the Securities Exchange Act of 1934, as amended, before the date our offering is terminated or completed are deemed to be incorporated by reference into, and to be a part of, this prospectus.
Any statement contained in this prospectus or in a document incorporated or deemed to be incorporated by reference into this prospectus will be deemed to be modified or superseded for purposes of this prospectus to the extent that a statement contained in this prospectus or any other subsequently filed document that is deemed to be incorporated by reference into this prospectus modifies or supersedes the statement. Any statement so modified or superseded will not be deemed, except as so modified or superseded, to constitute a part of this prospectus.
We will furnish without charge to you, on written or oral request, a copy of any or all of the documents incorporated by reference, including exhibits to these documents. You should direct any requests for documents to Assembly Biosciences, Inc., Attention: David J. Barrett, 99 Hudson Street, 5th Floor, New York, New York 10013, (646) 706-5208.
You should rely only on information contained in, or incorporated by reference into, this prospectus and any prospectus supplement. We have not authorized anyone to provide you with information different from that contained in this prospectus or incorporated by reference in this prospectus. We are not making offers to sell the securities in any jurisdiction in which such an offer or solicitation is not authorized or in which the person making such offer or solicitation is not qualified to do so or to anyone to whom it is unlawful to make such offer or solicitation.
| 35 |
PART II
INFORMATION NOT REQUIRED IN PROSPECTUS
Item 14. Other Expenses of Issuance and Distribution.
We estimate that expenses payable by us in connection with the offering described in this registration statement will be as follows:
| SEC registration fee | $ | 13,944 | ||
| Legal fees and expenses | $ | 15,000 | * | |
| Accounting fees and expenses | $ | 15,000 | * | |
| Printing expenses | $ | 5,000 | * | |
| Miscellaneous | $ | 6,056 | * | |
| Total | $ | 55,000 | * |
*Estimated as permitted under Rule 511 of Regulation S-K.
Item 15. Indemnification of Directors and Officers.
Section 145 of the Delaware General Corporation Law, or DGCL, permits a corporation, under specified circumstances, to indemnify its directors, officers, employees or agents against expenses (including attorneys’ fees), judgments, fines and amounts paid in settlements actually and reasonably incurred by them in connection with any action, suit or proceeding brought by third parties by reason of the fact that they were or are directors, officers, employees or agents of the corporation, if such directors, officers, employees or agents acted in good faith and in a manner they reasonably believed to be in or not opposed to the best interests of the corporation and, with respect to any criminal action or proceeding, had no reason to believe their conduct was unlawful. In a derivative action, that is one by or in the right of the corporation, indemnification may be made only for expenses actually and reasonably incurred by directors, officers, employees or agents in connection with the defense or settlement of an action or suit, and only with respect to a matter as to which they will have acted in good faith and in a manner they reasonably believed to be in or not opposed to the best interests of the corporation, except that no indemnification will be made if such person will have been adjudged liable to the corporation, unless and only to the extent that the court in which the action or suit was brought will determine upon application that the defendant directors, officers, employees or agents are fairly and reasonably entitled to indemnity for such expenses despite such adjudication of liability.
Pursuant to the DGCL, our Amended and Restated Certificate of Incorporation provides that no director will be personally liable to our company or our stockholders for monetary damages for breach of fiduciary duty as a director, except for liability (i) for any breach of the director’s duty of loyalty to our company or our stockholders, (ii) for acts or omissions not in good faith or which involve intentional misconduct or a knowing violation of law, (iii) under Section 174 of the DGCL, or (iv) for any transaction from which the director derived any improper personal benefit. Our amended and restated bylaws provide that we will generally indemnify our directors, officers, employees or agents to the fullest extent permitted by the law against all losses, claims, damages or similar events. We have obtained liability insurance for each director and officer for certain losses arising from claims or charges made against them while acting in their capacities as directors or officers of our company.
| II-1 |
Item 16. Exhibits.
(a) The following exhibits are filed as part of this Registration Statement:
Exhibit Number |
Description of Document | Registrant’s Form |
Dated | Exhibit Number | Filed Herewith | |||||
| 1.1* | Form of Underwriting Agreement | |||||||||
| 4.1 | Specimen of Common Stock Certificate. | S-1 | 10/29/10 | 4.1 | ||||||
| 4.2* | Specimen Preferred Stock Certificate and Form of Certificate of Designation of Preferred Stock. | |||||||||
| 4.3 | Form of Indenture. | X | ||||||||
| 4.4* | Form of Note. | |||||||||
| 4.5* | Form of Common Stock Warrant Agreement and Warrant Certificate. | |||||||||
| 4.6* | Form of Preferred Stock Warrant Agreement and Warrant Certificate. | |||||||||
| 4.7* | Form of Debt Securities Warrant Agreement and Warrant Certificate. | |||||||||
| 4.8* | Form of Unit Agreement. | |||||||||
| 5.1 | Opinion of Wyrick Robbins Yates & Ponton LLP. | X | ||||||||
| 12.1* | Computation of Ratio of Earnings to Fixed Charges. | |||||||||
| 23.1 | Consent of EisnerAmper LLP, Independent Registered Public Accounting Firm. | X | ||||||||
| 23.2 | Consent of Wyrick Robbins Yates & Ponton LLP (included as part of Exhibit 5.1). | X | ||||||||
| 24.1 | Power of Attorney (included in the signature pages hereto). | X | ||||||||
| 25.1* | Statement of Eligibility of Trustee. |
| * | To be filed by amendment or as an exhibit to a Current Report on Form 8-K and incorporated herein by reference, if applicable. |
(b) None.
Item 17. Undertakings
(a) The undersigned Registrant hereby undertakes:
(1) To file, during any period in which offers or sales are being made, a post-effective amendment to this Registration Statement:
(i) To include any prospectus required by Section 10(a)(3) of the Securities Act of 1933;
(ii) To reflect in the prospectus any facts or events arising after the effective date of the registration statement (or the most recent post-effective amendment thereof) which, individually or in the aggregate, represent a fundamental change in the information set forth in the registration statement. Notwithstanding the foregoing, any increase or decrease in volume of securities offered (if the total dollar value of securities offered would not exceed that which was registered) and any deviation from the low or high end of the estimated maximum offering range may be reflected in the form of prospectus filed with the SEC pursuant to Rule 424(b) if, in the aggregate, the changes in volume and price represent no more than a 20% change in the maximum aggregate offering price set forth in the “Calculation of Registration Fee” table in the effective registration statement;
| II-2 |
(iii) To include any material information with respect to the plan of distribution not previously disclosed in the registration statement or any material change to such information in the registration statement;
provided, however, that paragraphs (1)(i), (1)(ii) and (1)(iii) above do not apply if the information required to be included in a post-effective amendment by those paragraphs is contained in reports filed with or furnished to the Commission by the registrant pursuant to Section 13 and Section 15(d) of the Securities Exchange Act of 1934 that are incorporated by reference in the registration statement, or is contained in a form of prospectus filed pursuant to Rule 424(b) that is part of the registration statement.
(2) That, for the purpose of determining any liability under the Securities Act of 1933, each such post-effective amendment shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(3) To remove from registration by means of a post-effective amendment any of the securities being registered which remain unsold at the termination of the offering.
(4) That, for the purpose of determining liability under the Securities Act of 1933 to any purchaser:
(i) Each prospectus filed by the Registrant pursuant to Rule 424(b)(3) shall be deemed to be part of the registration statement as of the date the filed prospectus was deemed part of and included in the registration statement; and
(ii) Each prospectus required to be filed pursuant to Rule 424(b)(2), (b)(5) or (b)(7) as part of a registration statement in reliance on Rule 430B relating to an offering made pursuant to Rule 415(a)(1)(i), (vii) or (x) for the purpose of providing the information required by Section 10(a) of the Securities Act of 1933 shall be deemed to be part of and included in the registration statement as of the earlier of the date such form of prospectus is first used after effectiveness or the date of the first contract of sale of securities in the offering described in the prospectus. As provided in Rule 430B, for liability purposes of the issuer and any person that is at that date an underwriter, such date shall be deemed to be a new effective date of the registration statement relating to the securities in the registration statement to which that prospectus relates, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof. Provided, however, that no statement made in a registration statement or prospectus that is part of the registration statement or made in a document incorporated or deemed incorporated by reference into the registration statement or prospectus that is part of the registration statement will, as to a purchaser with a time of contract of sale prior to such effective date, supersede or modify any statement that was made in the registration statement or prospectus that was part of the registration statement or made in any such document immediately prior to such effective date.
(5) That, for the purpose of determining liability of the Registrant under the Securities Act of 1933 to any purchaser in the initial distribution of the securities:
The undersigned Registrant undertakes that in a primary offering of securities of the undersigned Registrant pursuant to this Registration Statement, regardless of the underwriting method used to sell the securities to the purchaser, if the securities are offered or sold to such purchaser by means of any of the following communications, the undersigned Registrant will be a seller to the purchaser and will be considered to offer or sell such securities to such purchaser:
(i) Any preliminary prospectus or prospectus of the undersigned Registrant relating to the offering required to be filed pursuant to Rule 424;
(ii) Any free writing prospectus relating to the offering prepared by or on behalf of the undersigned Registrant or used or referred to by the undersigned Registrant;
(iii) The portion of any other free writing prospectus relating to the offering containing material information about the undersigned Registrant or its securities provided by or on behalf of the undersigned Registrant; and
| II-3 |
(iv) Any other communication that is an offer in the offering made by the undersigned Registrant to the purchaser.
(b) The undersigned Registrant hereby undertakes that, for purposes of determining any liability under the Securities Act of 1933, each filing of the Registrant’s annual report pursuant to Section 13(a) or Section 15(d) of the Securities Exchange Act of 1934 (and, where applicable, each filing of an employee benefit plan’s annual report pursuant to Section 15(d) of the Securities Exchange Act of 1934) that is incorporated by reference in the registration statement shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(c) Insofar as indemnification for liabilities arising under the Securities Act of 1933 may be permitted to directors, officers and controlling persons of the Registrant pursuant to the foregoing provisions, or otherwise, the Registrant has been advised that in the opinion of the Securities and Exchange Commission such indemnification is against public policy as expressed in the Act and is, therefore, unenforceable. In the event that a claim for indemnification against such liabilities (other than the payment by the Registrant of expenses incurred or paid by a director, officer or controlling person of the Registrant in the successful defense of any action, suit or proceeding) is asserted by such director, officer or controlling person in connection with the securities being registered, the Registrant will, unless in the opinion of its counsel the matter has been settled by controlling precedent, submit to a court of appropriate jurisdiction the question whether such indemnification by it is against public policy as expressed in the Act and will be governed by the final adjudication of such issue.
(d) The undersigned Registrant hereby undertakes that:
(1) For purposes of determining any liability under the Securities Act of 1933, the information omitted from the form of prospectus filed as part of this registration statement in reliance upon Rule 430A and contained in a form of prospectus filed by the Registrant pursuant to Rule 424(b)(1) or (4) or 497(h) under the Securities Act shall be deemed to be part of this registration statement as of the time it was declared effective; and
(2) For the purpose of determining any liability under the Securities Act of 1933, each post-effective amendment that contains a form of prospectus shall be deemed to be a new registration statement relating to the securities offered therein, and the offering of such securities at that time shall be deemed to be the initial bona fide offering thereof.
(e) To file an application for the purpose of determining the eligibility of the trustee to act under subsection (a) of section 310 of the Trust Indenture Act (“Act”) in accordance with the rules and regulations prescribed by the Commission under section 305(b)(2) of the Act.
| II-4 |
SIGNATURES
Pursuant to the requirements of the Securities Act of 1933, the Registrant certifies that it has reasonable grounds to believe that it meets all of the requirements for filing on Form S-3 and has duly caused this Registration Statement on Form S-3 to be signed on its behalf by the undersigned, thereunto duly authorized, in the City of New York, State of New York, on November 26, 2014.
| ASSEMBLY BIOSCIENCES, INC. | ||
| By: | /s/ Russell H. Ellison | |
| Russell H. Ellison | ||
| Chief Executive Officer | ||
POWER OF ATTORNEY
We, the undersigned officers and directors of Assembly Biosciences, Inc., do hereby constitute and appoint Russell H. Ellison, Derek A. Small and David J. Barrett, or either of them, our true and lawful attorneys-in-fact and agents, each with full power of substitution and resubstitution, for him and in his name, place and stead, in any and all capacities, to sign any and all amendments to this Registration Statement, and to file the same, with exhibits thereto, and other documents in connection therewith, with the SEC, granting unto said attorneys-in-fact and agents, and each of them, full power and authority to do and perform each and every act and thing requisite are necessary to be done in and about the premises, as fully to all intents and purposes as he might or could do in person, hereby ratifying and confirming all that each of said attorney-in-fact and agents, or his substitute or substitutes, may lawfully do or cause to be done by virtue hereof.
Pursuant to the requirements of the Securities Act, this Registration Statement on Form S-3 has been signed by the following persons in the capacities and on the dates indicated.
| Signature | Capacity | Date | ||
| /s/ Russell H. Ellison | Chief Executive Officer and Director (Principal Executive Officer) | November 26, 2014 | ||
| Russell H. Ellison | ||||
| /s/ David J. Barrett | Chief Financial Officer (Principal Financial and Accounting Officer) | November 26, 2014 | ||
| David J. Barrett | ||||
| /s/ Derek A. Small | President and Director | November 26, 2014 | ||
Derek A. Small | ||||
| /s/ Anthony E. Altig | ||||
| Anthony E. Altig | Director | November 26, 2014 | ||
/s/ Mark Auerbach | ||||
| Mark Auerbach | Director | November 26, 2014 | ||
| /s/ Myron Z. Holubiak | ||||
| Myron Z. Holubiak | Director | November 26, 2014 | ||
| /s/ William Ringo | ||||
| William Ringo | Director | November 26, 2014 | ||
| /s/ Richard DiMarchi | ||||
| Richard DiMarchi | Director | November 26, 2014 |
| S-1 |