| ITEM 2: | MANAGEMENT’S DISCUSSION AND ANALYSIS OF FINANCIAL CONDITION AND RESULTS OF OPERATIONS |
The following discussion and analysis of our financial condition and results of operations should be read in conjunction with our unaudited interim condensed consolidated financial statements and related notes thereto appearing elsewhere in this Quarterly Report on Form 10-Q (this “Quarterly Report”) and with the audited financial statements and notes thereto of the Company as of and for the year ended December 31, 2020 on Form 10-K, filed with the Securities and Exchange Commission, or SEC, on March 18, 2021.
Cautionary Note Regarding Forward-Looking Statements
This Quarterly Report contains forward-looking statements that involve substantial risks and uncertainties. All statements other than statements of historical facts contained in this Quarterly Report, including statements regarding our future results of operations and financial position, strategy and plans, and our expectations for future operations, are forward-looking statements. In some cases, you can identify forward-looking statements by terminology such as “may,” “will,” “should,” “could,” “expects,” “intends,” “plans,” “anticipates,” “believes,” “estimates,” “predicts,” “potential,” “continue” or the negative of these terms or other comparable terminology. These forward-looking statements are subject to a number of risks, uncertainties and assumptions, including those described under the heading “Risk Factors” below. In light of these risks, uncertainties and assumptions, actual results could differ materially and adversely from those anticipated or implied in the forward-looking statements in this Quarterly Report and you should not place undue reliance on these forward-looking statements.
These forward-looking statements may include, but are not limited to, statements about:
| ● | the accuracy of estimates of our expenses, future revenue, capital requirements and our needs for additional financing; |
| ● | our ability to obtain funding for our operations in the event we determine the need to raise additional capital; |
| ● | our ability to retain key management personnel; |
| ● | the accuracy of our estimates regarding expenses, future revenues and capital requirements; |
| ● | Our ability to efficiently and effectively conduct our clinical trials; |
| ● | our ability to maintain our listing on the Nasdaq Stock Market; |
| ● | regulatory developments in the United States and foreign countries; |
| ● | unforeseen circumstances or other disruptions to normal business operations arising from or related to COVID-19; |
| ● | our expectations regarding the time during which we will be an emerging growth company under the Jumpstart Our Business Startups Act of 2012 (“JOBS Act”); and |
| ● | other risks and uncertainties, including those listed under Part II, Item 1A. Risk Factors. |
Any forward-looking statements in this Quarterly Report reflect our views and assumptions only as of the date that this report is signed with respect to future events or to our future financial performance and involve known and unknown risks, uncertainties and other factors that may cause our actual results, performance or achievements to be materially different from any future results, performance or achievements expressed or implied by these forward-looking statements. Given these uncertainties, you should not place undue reliance on these forward-looking statements. Except as required by law, we assume no obligation to update or revise these forward-looking statements for any reason, even if new information becomes available in the future.
We qualify all of our forward-looking statements by these cautionary statements. In addition, with respect to all of our forward-looking statements, we claim the protection of the safe harbor for forward-looking statements contained in the Private Securities Litigation Reform Act of 1995.
Company Overview
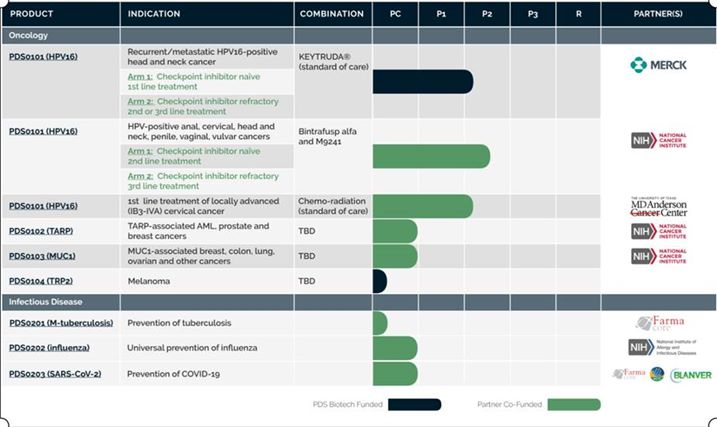
We are a clinical-stage immunotherapy company developing a growing pipeline of cancer immunotherapy and infectious disease vaccine candidates designed to overcome the limitations of current immunotherapy technologies. Our Versamune® technology is a proprietary T-cell activating platform designed to treat diseases such as cancer by training the immune system to better recognize, attack and destroy diseased cells. When paired with an antigen (which is a disease-related protein that is recognizable by the immune system), in pre-clinical studies, Versamune® has been shown to induce, in vivo, large quantities of high-quality, highly potent polyfunctional CD8+ killer T-cells, a specific sub-type of T-cell that is more effective at killing infected or targeted cancer cells. Versamune® has also been shown in pre-clinical studies to induce antigen-specific highly potent polyfunctional CD4+ helper T-cells as well as neutralizing antibodies. PDS0101 in its first human clinical trial provided data supporting the previously reported preclinical study results and demonstrated the unique in vivo induction of high levels of active HPV-specific CD8+ T-cells in humans. Positive preliminary clinical results of PDS0101 in combination with investigational immune-modulating agents Bintrafusp alfa (M7824), and NHS-IL12 (M9241), discussed in more detail below, suggest that PDS0101 induction of in vivo highly active tumor-attacking HPV16 killer (CD8+) T-cells even in advanced, refractory cancer patients who have failed multiple treatments, who may be immunologically limited patients presents the potential for effective disease reduction and prolonged survival.
We believe that the Versamune® platform has the potential to become an industry-leading immuno-oncology technology. It is currently being applied to the development of a robust pipeline of high-value, novel and first-in-class immunotherapies, potentially as single agents and also as part of combination therapies with other leading immuno-oncology technologies and standard of care therapies. To date, Versamune® based therapies have not been linked to any dose limiting toxicities in any human clinical trial. Based upon the promising efficacy data and the positive safety data so far, PDS is preparing to move at least one additional product into Phase 1/2 clinical trials in 2022. We believe our product candidates could be of interest for use in combination with other cancer therapies in the treatment of several advanced, metastatic, and recurrent cancers including HPV-associated cancers, colorectal, lung, breast and prostate cancers or potentially as monotherapies in earlier-stage disease.
PDS0101 in Combination with KEYTRUDA® Clinical Trial (VERSATILE-002):
In November 2020, our VERSATILE-002 Phase 2 clinical trial evaluating the combination of PDS0101 (Versamune® plus a mixture of HPV16 antigens) in combination with Merck’s anti-PD-1 therapy, KEYTRUDA® (pembrolizumab) for treatment of recurrent/ metastatic HPV16-positive head and neck cancer opened and is actively recruiting patients. The clinical trial is evaluating the efficacy and safety of this therapeutic combination as a treatment in patients with recurrent or metastatic head and neck cancer caused by high-risk HPV16 infection.
In this sponsored trial, patients whose cancer has returned or spread following initial treatment, will take this combination of two immuno-therapy drugs in lieu of chemotherapy. Enrolling patients with more functional immune systems earlier in disease recurrence that have not been compromised by extensive chemotherapy may allow improved efficacy of the combination. Patients in the study will receive a total of 5 cycles of combination therapy in the context of standard of care KEYTRUDA™ therapy administered every three weeks until disease progression or intolerance to therapy.
The original study design for this combination trial was restricted to patients who have not been treated with a checkpoint inhibitor and are therefore naïve to checkpoint inhibitor (CPI) therapy. PDS amended the protocol in May 2021 to expand this study to include patients with advanced head and neck cancer who have failed multiple treatments, including CPI therapy (CPI refractory patients), a treatment refractory population with extremely high unmet medical need. The amended study design is a Simon two stage design. The primary efficacy endpoint in both CPI naïve and CPI refractory subjects will be the best overall response (BOR) of confirmed complete response (CR) or confirmed partial response (PR) per Response Evaluation Criteria in Solid Tumors (RECIST) 1.1 of the combination of pembrolizumab and PDS0101. Objective response is measured by radiographic tumor responses according to RECIST 1.1. The lead-in cohort of the initial twelve (12) patients to assess the safety of the combination was successfully enrolled in September 2021. The Data Monitoring Committee assessed the safety data from 13 patients on September 15, 2021 and observed no dose limiting toxicities. On October 4, 2021, we announced that full enrollment of 17 patients was achieved in the first stage of the CPI naïve arm of the study. If an objective response is achieved among at least four (4) of these seventeen (17) patients, then the study will advance to the second stage of enrollment of the planned 54 patients in this arm. In the expansion CPI refractory arm, the first twenty-one (21) patients will be evaluated for efficacy with a minimum of two (2) or more patients with objective response required to progress to full enrollment of this arm.
If the endpoints are achieved, this study may serve as confirmation that PDS0101 enhances the therapeutic benefit of checkpoint inhibitors and could expand evaluation of Versamune®-based therapies in multiple cancer indications. Total enrollment of both study arms is expected to be approximately ninety-five (95) patients, essentially unchanged from the accrual target of ninety-six (96) patients in the original study design. Preliminary data is expected between the fourth quarter of 2021 and the first quarter of 2022.
PDS0101 in Combination with Bintrafusp alfa (M7824) and NHS-IL12 Clinical Trial:
In June 2020, the first patient was dosed under a PDS0101 Cooperative Research and Development Agreement, which we refer to as the CRADA, in a National Cancer Institute, or NCI, led Phase 2 clinical study evaluating PDS0101, in combination with two investigational immune-modulating agents Bintrafusp alfa (M7824), a bifunctional “trap” fusion protein targeting TGF-β and PD-L1, and NHS-IL12 (M9241), a tumor-targeting immune-cytokine. Both bintrafusp alfa and NHS-IL12 are owned by and are being developed by Merck KGaA, Darmstadt, Germany. The NCI-led trial is evaluating the novel triple combination for the treatment of both CPI naïve and refractory patients with advanced HPV-associated cancers that have progressed or returned after treatment. Almost all types of HPV-related cancers (anal, cervical, head and neck, vaginal and vulvar cancers) are represented among the study subjects to date. Objective response (tumor reduction > 30%) is measured by radiographic tumor responses according to RECIST 1.1. Full enrollment of a total of 56 patients is expected to be completed during the first quarter of 2022. The recruitment in the trial was temporarily suspended for administrative reasons on October 20, 2021. The pause in recruitment was unrelated to safety or efficacy of the combination and we continue to communicate with the NCI regarding enrollment. The treatment of already enrolled patients has continued without interruption. At this time, this pause is not expected to affect the timing of clinical data from the trial.
The study was accepted for oral presentation at the 2021 meeting of the American Society of Clinical Oncology (ASCO) held June 4-8, 2021. On May 20, 2021, we announced the abstract summarizing interim data of 14 patients from the study. Objective responses were observed in 83% (5 of 6) of HPV16-positive checkpoint inhibitor naïve patients with advanced disease. Objective responses were observed in 63% (5 of 8) of HPV16-positive advanced cancer patients who had also failed checkpoint inhibitor therapy.
On June 7, 2021, updated interim data (Abstract 2501) was presented at ASCO by the study’s Principal Investigator Dr. Julius Strauss. This data included an update on the initial 14 patients with an additional four (4) subsequently recruited CPI refractory HPV16-positive patients whose data became available after the abstract submission as well as an additional seven (7) HPV16-negative patients (patients whose cancer was not caused by HPV16 infection) who were not discussed in the abstract. Tumor reduction and objective response was seen in 83% (5/8) of CPI naïve patients with one (1) achieving a complete response. After a median of eight (8) months of follow-up, 100% (6/6) of these patients were alive, compared to the historical median survival of 7-11 months. In the CPI refractory arm, tumor reduction was observed in 58% (7/12) patients. These 12 patients included the initial 8 patients reported in the abstract where 5 of 8, or 63% had tumor reduction. Of the 4 additional patients in this updated data set, 2 patients already had ongoing tumor reduction but had not yet met the criteria for objective response. Objective response was achieved in 42% (5/12) patients with one (1) achieving a complete response in this refractory arm. The objective response rates reported in checkpoint refractory, advanced HPV-cancer patients are lower than those naïve to checkpoint inhibitors, generally only 5-12%. Importantly, 83% (10/12) of all checkpoint inhibitor refractory patients were still alive at median eight (8) months of follow-up; historic average median survival for this patient population is 3-4 months. Overall, tumor reduction occurred in 67% (12/18) of the HPV16-positive patients, including both CPI naïve and CPI refractory patients. Of note, in the 7 HPV16-negative patients- those with an HPV type other than HPV16 that do not express the molecular target of PDS0101, zero (0) of seven (7) patients experienced tumor reduction or an objective response, providing insight into the potential role of PDS0101 in the observed activity of the triple combination. These data suggest that HPV16-specific CD8+ and CD4+ T-cell induction by PDS0101 as demonstrated in preclinical studies and phase 1 clinical studies, may promote tumor reduction and enhanced clinical benefit of the triple combination.
Moreover, PDS0101 did not appear to compound the toxicities already reported to be induced by Bintrafusp alfa and NHS-IL12. This is an important consideration for combination oncology treatment regimens where a key goal is to avoid additional or excess toxicity associated with limited anti-tumor activity. As would be expected with both PDS0101 and M9241 being delivered subcutaneously, injection site reactions were seen in 20% of patients. No new or additional toxicities have been observed to date from the addition of PDS0101 to the combination of the other two immunotherapies.
PDS0101 in Combination with Chemo-Radiotherapy Clinical Trial (IMMUNOCERV):
In October 2020, a third PDS0101 Phase 2 clinical trial was initiated with The University of Texas MD Anderson Cancer Center and is actively recruiting patients. This clinical trial is investigating the safety and preliminary anti-tumor efficacy of PDS0101 in combination with standard-of-care chemo-radiotherapy, or CRT, and their correlation with critical immunological biomarkers in patients with locally advanced cervical cancer. The study goals include evaluation of the rate of regression in patients with a primary tumor that is greater than or equal to 5cm among approximately thirty-five (35) patients. The rate of complete metabolic response is measured by PET-CT at day 170 and the rate of > 90% tumor volume reduction evaluated by MRI at 30-40 days from the start of treatment. Preliminary data is expected from this study in 2022. We believe that Versamune®’s strong T-cell induction has the potential to meaningfully enhance efficacy of the current standard of care CRT treatment in this indication.
Other Versamune® -Based Products in Development:
PDS0102 – Versamune® + TARP antigens
The TARP antigen is over-expressed in acute myelogenous leukemia (AML), prostate and breast cancers. In the U.S. 450,000 patients are projected to be diagnosed with prostate or breast cancer this year and 19,900 patients are projected to be diagnosed with AML. One hundred percent of adult and pediatric AML overexpress the TARP tumor antigen while approximately 90% of prostate cancers and 50% of breast cancers overexpress TARP, providing a potentially significant therapeutic opportunity to impact these diseases. In an early human clinical study, the National Cancer Institute demonstrated that its proprietary TARP antigens (TARP mix) were recognized by the immune system in prostate cancer patients with PSA biochemical recurrence leading to a notable reduction in tumor growth rates. In preclinical studies, a dramatically enhanced TARP-specific CD8+ killer T-cell response was observed when our designed TARP antigens were combined with Versamune® (PDS0102). Preclinical development is ongoing. On November 5, 2021 PDS and NCI entered into a Patent License Agreement to develop and commercialize a product including TARP antigens. The patent for the TARP antigens extends through 2034. This license agreement is an important step to the further development and commercialization of PDS0102 and plans for a Phase 1/2 clinical trial to start in the second half of 2022.
PDS0103 – Versamune® + MUC1 antigens
In April 2020, the PDS-NCI CRADA was expanded beyond PDS0101 to include clinical and preclinical development of PDS0103. PDS0103 is an investigational immunotherapy owned by PDS and designed to treat cancers associated with the mucin-1, or MUC1, oncogenic protein. PDS0103 combines Versamune® with novel highly immunogenic agonist epitopes of MUC1 developed by the NCI and licensed by PDS. MUC1 is highly expressed in multiple solid tumors and has been shown to be associated with drug resistance and poor disease prognosis in breast, colorectal, lung and ovarian cancers, for which PDS0103 is being developed. In preclinical studies, and similarly to PDS0101 and PDS0102, PDS0103 demonstrated the ability to generate powerful MUC1-specific CD8+ killer T-cells. PDS0103 is currently in late preclinical development, and we requested a pre-IND meeting with the FDA to align on a clinical development plan. We are planning to initiate a Phase 2 clinical trial in mid- 2022.
PDS0104 – Versamune® + TRP-2 and melanoma antigens
Rates of melanoma have been rising rapidly over the past few decades and approximately 96,480 new melanomas will be diagnosed this year alone. More than 7,000 of these will prove fatal. PDS0104 combines Versamune ® with various melanoma antigens including the Tyrosinase-related protein 2 (TRP2) antigen which is highly expressed in melanoma. In pre-clinical B16 animal models of aggressive melanoma, PDS0104 has been shown to have unique and significant anti-tumor activity as a monotherapy and has also demonstrated strong anti-tumor synergy in combination with checkpoint inhibitors. As the first approval indications for checkpoint inhibitors occurred in melanoma, there is a growing unmet medical need to identify combination regimens with potential activity in checkpoint inhibitor refractory patients with this disease. Preclinical development is ongoing.
Infectious Diseases
Our focus remains on developing our oncology products and progressing the clinical trials described above. However, we are also developing preventive vaccines to address several infectious diseases. Based on the key characteristics of Versamune® we are progressing preclinical development of PDS0202, a universal influenza vaccine candidate, which combines Versamune® with COBRA (Computationally Optimized Broadly Reactive Antigen) antigens. These COBRA antigens were designed by renowned influenza expert Dr. Ted Ross at the University of Georgia. PDS0202 leverages Versamune®’s ability to induce the immune system to generate high levels of flu-specific neutralizing antibodies, CD4 helper and CD8 killer T-cells, as well as long-acting memory T-cells to potentially provide broad and long-term protection against multiple influenza strains. PDS0202 pre-clinical development is being supported by an agreement with the National Institute of Allergy and Infectious Diseases Collaborative Influenza Vaccine Innovation Centers, or CIVIC program. We have completed pre-clinical formulation and we have entered into an agreement to license COBRA antigens with the University of Georgia initially to progress into a human clinical trial in 2022.
PDS0203 is being designed with the goal to potentially provide long-term and broad protection against infection from COVID-19 and its potential mutations, based on the understood potential of Versamune® to prime the immune system to generate both antibodies for near term protection and T-cell responses for immune memory and long-term protection against pathogens. Farmacore Biotechnology has licensed Versamune® in Latin America to develop PDS0203 in Brazil. Farmacore is developing and manufacturing the antigen component of the vaccine, leads all regulatory and clinical trial efforts in Brazil and has selected a top clinical research organization to conduct clinical trials.
On March 11, 2021, we announced that the COVID-19 vaccine consortium consisting of PDS, Farmacore Biotechnology and Blanver Farmoquímica, received a commitment from the Secretary for Research and Scientific Training of The Ministry of Science, Technology and Innovation of Brazil (“MCTI”) to fund up to approximately US$60 million to support the clinical development and commercialization of a Versamune®-based COVID-19 vaccine in Brazil. All funding is contingent on the availability of financial resources within the MCTI and subject to negotiations among Farmacore, Blanver and MCTI. We are monitoring the evolving political and public health situation in Brazil that may pose challenges to developing PDS0203, such as the Brazilian Senate’s vote for compulsory licensing for COVID-19 vaccine technology (legislative process is ongoing).
MCTI indicated that it intends to make the funding available to perform a combined Phase 1/2 clinical trial, upon authorization by the Brazilian regulatory agency, Agência Nacional de Vigilância Sanitária (ANVISA) to initiate the proposed clinical program in Brazil. Additional capital planned to be provided by MCTI will fund the manufacturing process scale up, production and the Phase 3 trial, pending the results of the Phase 1/2 trial. We plan to contribute Versamune® scientific expertise and operational support. Blanver Farmoquímica is expected to manufacture, promote, distribute, and commercialize the Versamune®-based COVID-19 vaccine in Latin America. We have not received confirmation of the availability of financial resources within the MCTI to support the clinical development and commercialization of a Versamune®-based COVID-19 vaccine in Brazil. Clinical development and commercialization of a Versamune®-based COVID-19 vaccine in Brazil has not been initiated. As part of our strategic assessment of the program, Farmacore provided a comprehensive update on its efforts to develop a cGMP ready manufacturing plan and product for the antigen component of the vaccine. Farmacore has presented a proposal to meet certain development milestones in order to continue development of a COVID-19 vaccine containing Versamune®. We have performed a program review and are in discussions with Farmacore to define the upcoming milestones for further development.
Our current pipeline of Versamune®-based therapies is as follows:
Corporate Information
From the Company’s inception, it has devoted substantially all of its efforts to drug development, business planning, engaging regulatory, manufacturing and other technical consultants, acquiring operating assets, planning and executing clinical trials and raising capital. We currently operate the existing business of Private PDS (as defined below) as a publicly traded company under the name PDS Biotechnology Corporation. We were incorporated as Edge Therapeutics, Inc., or Edge, on January 22, 2009. On March 15, 2019, privately held PDS Biotechnology Corporation, or Private PDS, completed a reverse merger, or the Merger, with Edge, pursuant to which Private PDS survived as a wholly owned subsidiary of Edge. Under the terms of the Merger, Edge changed its name to PDS Biotechnology Corporation and Private PDS changed its name to PDS Biotechnology Corporation.
We have never been profitable and have incurred net losses in each year since inception. Our net losses were $10.6 million, and $10.8 million for the nine months ended September 30, 2021 and 2020, respectively. As of September 30, 2021, we had an accumulated deficit of $54.4 million. Substantially all of our net losses have resulted from costs incurred in connection with its research and development programs and from general and administrative costs associated with these operations.
As of September 30, 2021, we had $69.7 million in cash and cash equivalents.
Our future funding requirements will depend on many factors, including the following:
| ● | the timing and costs of our planned clinical trials; |
| ● | the timing and costs of our planned preclinical studies of our Versamune® platform; |
| ● | the outcome, timing and costs of seeking regulatory approvals; |
| ● | the terms and timing of any future collaborations, licensing, consulting or other arrangements that we may enter into; |
| ● | the amount and timing of any payments we may be required to make in connection with the licensing, filing, prosecution, maintenance, defense and enforcement of any patents or patent applications or other intellectual property rights; and |
| ● | the extent to which we license or acquire other products and technologies. |
KEY COMPONENTS OF OUR STATEMENT OF OPERATIONS
Revenue
We have not generated any revenues from commercial product sales and do not expect to generate any such revenue in the near future. We may generate revenue in the future from a combination of research and development payments, license fees and other upfront payments or milestone payments.
Research and Development Expenses
Research and development expenses include employee-related expenses, licensing fees to use certain technology in our research and development projects, costs of acquiring, developing and manufacturing clinical trial materials, as well as fees paid to consultants and various entities that perform certain research and testing on our behalf. Costs for certain development activities, such as clinical trials, are recognized based on an evaluation of the progress to completion of specific tasks using data such as patient enrollment, clinical site activations or information provided by vendors on their actual costs incurred. Payments for these activities are based on the terms of the individual arrangements, which may differ from the pattern of costs incurred, and are reflected in the consolidated financial statements as prepaid or accrued expenses. Costs incurred in connection with research and development activities are expensed as incurred.
We expect that our research and development expenses will increase significantly over the next several years as we advance our Versamune®-based immuno-oncology, or I-O, candidates into and through clinical trials, pursues regulatory approval of our investigational Versamune® candidates and prepare for a possible commercial launch, all of which will also require a significant investment in contract and internal manufacturing and inventory related costs.
The process of conducting human clinical trials necessary to obtain regulatory approval is costly and time consuming. We may never succeed in achieving marketing approval for our injectable I-O candidates. The probability of successful commercialization of our I-O candidates may be affected by numerous factors, including clinical data obtained in future trials, competition, manufacturing capability and commercial viability. As a result, we are unable to determine the duration and completion costs of our research and development projects or when and to what extent we will generate revenue from the commercialization and sale of any of our product candidates.
Results of Operations
The following table summarizes the results of our operations for the three months ended September 30, 2021 and 2020:
| | | Three Months Ended September 30, | | | Increase (Decrease) | |
| | | 2021 | | | 2020 | | | $ Amount | | | % | |
| | | (in thousands) | | | | | | | |
| Operating expenses: | | | | | | | | | | | | |
| Research and development expenses | | $ | 3,688 | | | $ | 2,061 | | | $ | 1,627 | | | | 79 | % |
| General and administrative expenses | | | 3,274 | | | | 1,846 | | | | 1,428 | | | | 77 | % |
| Total operating expenses | | | 6,962 | | | | 3,907 | | | | 3,055 | | | | 78 | % |
| Loss from operations | | | (6,962 | ) | | | (3,907 | ) | | | (3,055 | ) | | | 78 | % |
| Interest income | | | 1 | | | | 1 | | | | – | | | | – | |
| Net loss and comprehensive loss | | $ | (6,961 | ) | | $ | (3,906 | ) | | $ | (3,055 | ) | | | 78 | % |
Research and Development Expenses
Research and development expenses increased to $3.7 million for the three months ended September 30, 2021 from $2.1 million for the three months ended September 30, 2020. The increase of $1.6 million was primarily attributable to an increase of $0.7 million in personnel costs of which $0.5 million was stock compensation costs, and $0.9 million in costs related to clinical studies.
General and Administrative Expenses
General and administrative expenses increased to $3.2 million for the three months ended September 30, 2021 from $1.8 million for the three months ended September 30, 2020. The increase of $1.4 million is primarily attributable to an increase in personnel costs of $1.6 million, of which $1.0 million was stock compensation costs and $0.4 million was severance, partially offset by a decrease in professional fees of $0.2 million.
Comparison of the Nine Months Ended September 30, 2021 and 2020
The following table summarizes the results of our operations for the nine months ended September 30, 2021 and 2020:
| | | Nine months ended September 30, | | | Increase (Decrease) | |
| �� | | 2021 | | | 2020 | | | $ Amount | | | % | |
| | | (in thousands) | | | | | | | |
| Operating expenses: | | | | | | | | | | | | |
| Research and development expenses | | $ | 7,865 | | | $ | 5,447 | | | $ | 2,418 | | | | 44 | % |
| General and administrative expenses | | | 7,252 | | | | 5,428 | | | | 1,824 | | | | 34 | % |
| Total operating expenses | | | 15,117 | | | | 10,875 | | | | 4,242 | | | | 39 | % |
| Loss from operations | | | (15,117 | ) | | | (10,875 | ) | | | (4,242 | ) | | | 39 | % |
| Interest income | | | 3 | | | | 54 | | | | (51 | ) | | | (94 | )% |
| Benefit from income taxes | | | 4,516 | | | | - | | | | 4,516 | | | | 100 | % |
| Net loss and comprehensive loss | | $ | (10,598 | ) | | $ | (10,821 | ) | | $ | 223 | | | | (2 | )% |
Research and Development Expenses
Research and development expenses increased to $7.9 million for the nine months ended September 30, 2021 from $5.4 million for the same period in 2020. The increase of $2.5 million was primarily attributable to an increase in personnel costs of $0.9 million, of which 0.7 million was stock compensation costs, clinical studies of $2.1 million, regulatory fees of $0.2 million, facility expenses of $0.1 million offset by a decrease in manufacturing costs of $0.8 million.
General and Administrative Expenses
General and administrative expenses increased to $7.2 million for the nine months ended September 30, 2021 from $5.4 million for the same period in 2020. The $1.8 million increase was primarily attributable to an increase in personnel costs of $2.3 million, of which $1.4 million was stock compensation costs and $0.4 million was severance, facility expenses of $0.1 million offset by a decrease in professional services of $0.6 million.
Benefit from Income Taxes
Income tax benefit was $4.5 million for the nine months ended September 30, 2021 and $0 million for the nine months ended September 30, 2020. In the second quarter of 2021, we recognized approximately $4.5 million as a Benefit from Income Taxes from the sale of our NJ tax benefits pursuant to the New Jersey Technology Business Tax Certificate Transfer Net Operating Loss program. This transaction reduced our reported net loss and comprehensive income for the nine months ended September 30, 2021 to approximately $10.6 million, or approximately $0.2 million less than reported for the same period in 2020 which did not include a similar sale of NJ tax benefits.
Liquidity and Capital Resources
Our operations were initially financed from cash of $29.1 million from the consummation of the Merger in March 2019.
In July 2019, we entered into a common stock purchase agreement, or the Aspire Purchase Agreement, with Aspire Capital pursuant to which, we have the right, in our sole discretion, to present Aspire Capital Fund, LLC, or Aspire Capital, with a purchase notice, directing Aspire Capital (as principal) to purchase up to 100,000 shares of our common stock per business day, in an aggregate amount of up to $20.0 million of our common stock, or the Purchased Shares, over the term of the Aspire Purchase Agreement. We may sell an aggregate of 1,034,979 shares of our common stock (which represented 19.99% of our outstanding shares of common stock on the date of the Aspire Purchase Agreement) without stockholder approval. We may sell additional shares of our common stock above the 19.99% limit provided that (i) we obtain stockholder approval or (ii) stockholder approval has not been obtained at any time the 1,034,979 share limitation is reached and at all times thereafter the average price paid for all shares issued under the Aspire Purchase Agreement, is equal to or greater than $5.76, which was the consolidated closing bid price of our common stock on July 26, 2019. The minimum price at which we can sell shares under the Aspire Purchase Agreement is $0.50. On July 29, 2019, we issued 100,654 shares of our common stock to Aspire Capital, as consideration for entering into the Aspire Purchase Agreement, which we refer to as the Commitment Shares. We recorded the fair value of the shares at July 29, 2019 of $603,924 as an expense in the third quarter of 2019. Concurrently with the Aspire Purchase Agreement, we entered into a registration rights agreement with Aspire Capital, or the Registration Rights Agreement. In accordance with the Registration Rights Agreement, on August 20, 2019 we filed a Registration Statement on Form S-1 (File No. 333-232988) to cover the resale of the Commitment Shares and any Purchased Shares issuable to Aspire Capital under the Aspire Purchase Agreement. There is market uncertainty regarding the utilization of financing associated from the Aspire Purchase Agreement. As of September 30, 2021, no Purchase Shares have been sold to Aspire Capital under the Aspire Purchase Agreement.
In February 2020, we completed an underwritten public offering, in which we sold 10,000,000 shares of common stock at a public offering price of $1.30 per share. The shares sold included 769,230 shares issued upon the exercise by the underwriter of its option to purchase additional shares at the public offering price, minus underwriting discounts and commissions. We received gross proceeds of approximately $13 million and net proceeds of approximately $11.9 million after deducting underwriting discounts and commissions.
In July 2020, we filed a shelf registration statement, or the 2020 Shelf Registration Statement, with the SEC, for the issuance of common stock, preferred stock, warrants, rights, debt securities and units, which we refer to collectively as the Shelf Securities, up to an aggregate amount of $100 million. The 2020 Shelf Registration Statement was declared effective on July 31, 2020. On August 13, 2020, we sold 6,900,000 shares of its common stock at a public offering price of $2.75 per share pursuant to the 2020 Shelf Registration Statement, which includes 900,000 shares issued upon the exercise by the underwriter of its option to purchase additional shares at the public offering price, minus underwriting discounts and commissions. We received gross proceeds of approximately $19.0 million and net proceeds of approximately $17.1 million, after deducting underwriting discounts and offering expenses.
In May 2021, we received approximately $4.5 million from the net sale of tax benefits to an unrelated, profitable New Jersey corporation pursuant the Company’s participation in the New Jersey Technology Business Tax Certificate Transfer Net Operating Loss (NOL) program for State Fiscal Year 2020.
In June 2021, we sold 6,088,235 shares of our common stock at a public offering price of $8.50 per share pursuant to the 2020 Shelf Registration Statement, which includes 794,117 shares issued upon the exercise by the underwriter of its option to purchase additional shares at the public offering price, minus underwriting discounts and commissions. We received gross proceeds of approximately $51.7 million and net proceeds of approximately $48.5 million, after deducting underwriting discounts and offering expenses. Approximately $29,300,000 of Shelf Securities remain available for future sale under the 2020 Shelf Registration Statement.
As of September 30, 2021, we had $69.7 million in cash and cash equivalents. Our primary uses of cash are to fund operating expenses, primarily research and development expenditures. Cash used to fund operating expenses is impacted by the timing of when we pay these expenses, as reflected in the change in our outstanding accounts payable and accrued expenses.
We evaluated whether there are any conditions and events, considered in the aggregate, that raise substantial doubt about our ability to continue as a going concern within one year beyond the filing of this Quarterly Report on Form 10-Q. Based on such evaluation and our current plans, which are subject to change, management believes that our existing cash and cash equivalents as of September 30, 2021 will be sufficient to satisfy our operating cash needs for at least one year after the filing of this Quarterly Report on Form 10-Q.
We plan to continue to fund our operations and capital funding needs through equity and/or debt financings. However, we cannot be certain that additional financing will be available when needed or that, if available, financing will be obtained on terms favorable to us or our existing stockholders. We may also enter into government funding programs and consider selectively partnering for clinical development and commercialization. The sale of additional equity would result in additional dilution to our stockholders. Incurring debt financing would result in debt service obligations, and the instruments governing such debt could provide for operating and financing covenants that would restrict our operations. If we are unable to raise additional capital in sufficient amounts or on acceptable terms, we may be required to delay, limit, reduce, or terminate our product development or future commercialization efforts or grant rights to develop and market immunotherapies that we would otherwise prefer to develop and market ourselves. Any of these actions could harm our business, results of operations and prospects.
Cash Flows
The following table shows a summary of our cash flows for each of the periods indicated (in thousands):
| | | Nine Months Ended September 30, | |
| | | 2021 | | | 2020 | |
Net cash used in operating activities | | | | | | | | |
Net cash provided by investing activities | | | | | | | | |
Net cash provided by financing activities | | | | | | | | |
| | | | | | | | |
Net Cash Used in Operating Activities
Net cash used in operating activities was $8.0 million and $8.5 million for the nine months ended September 30, 2021 and 2020, respectively. The decrease in cash used in operating activities of $0.5 million includes the $4.5 million NOL sale. Excluding the NOL sale, net cash used in operating activities would have increased $4.0 million primarily due to the increase in the loss before income taxes of $4.3 million, the change in prepaid expenses and other assets of $2.0 million, accounts payables and accruals of $0.3 million partially offset by an increase in stock-based compensation of $2.1 million, and a decrease in restructuring reserve of $0.5 million.
Net Cash Provided by Financing Activities
Net cash provided by financing activities for the nine months ended September 30, 2021 was primarily due to the receipt of net proceeds from the issuance of common stock of $48.5 million.
Net cash provided by financing activities for the nine months ended September 30, 2020 was primarily due to the receipt of net proceeds from the issuance of common stock of $29.8 million.
Operating Capital Requirements
To date, we have not generated any product revenue. We do not know when, or if, we will generate any product revenue and we do not expect to generate significant product revenue unless and until we obtain regulatory approval and commercialize one of our current or future product candidates. We anticipate that we will continue to generate losses for the foreseeable future, and we expect the losses to increase as we continue the development of, and seek regulatory approvals for, our tablet vaccine candidates, and begin to commercialize any approved vaccine candidates. We are subject to all of the risks incident to the development of new products, and may encounter unforeseen expenses, difficulties, complications, delays and other unknown factors that may harm our business. We expect to incur additional costs associated with operating as a public company and anticipate that we will need substantial additional funding in connection with our continuing operations.
We evaluated whether there are any conditions and events, considered in the aggregate, that raise substantial doubt about our ability to continue as a going concern within one year beyond the filing of this Quarterly Report. Our budgeted cash requirements in 2021 and beyond include expenses related to continuing development and clinical studies. We believe that our existing cash and cash equivalents as of September 30, 2021 are sufficient to continue operations and research and development programs for at least the next 12 months from the date of this Quarterly Report. Until we can generate significant cash from our operations, we expect to continue to fund our operations with available financial resources. These financial resources may not be adequate to sustain our operations.
We have based our projections of operating capital requirements on assumptions that may prove to be incorrect and we may use all of our available capital resources sooner than we expect. Because of the numerous risks and uncertainties associated with research, development and commercialization of pharmaceutical products, we are unable to estimate the exact amount of our operating capital requirements. Our future funding requirements will depend on many factors, including, but not limited to:
| ● | the initiation, progress, timing, costs and results of our planned clinical trials; |
| ● | the effects of health epidemics, pandemics, or outbreaks of infectious diseases, including the recent COVID-19 pandemic, on our business operations, financial condition, results of operations and cash flows; |
| ● | the outcome, timing and cost of meeting regulatory requirements established by the U.S. Food and Drug Administration, or FDA, the European Medicines Agency, or EMA, and other comparable foreign regulatory authorities; |
| ● | the cost of filing, prosecuting, defending and enforcing our patent claims and other intellectual property rights; |
| ● | the cost of defending potential intellectual property disputes, including patent infringement actions brought by third parties against us now or in the future; |
| ● | the effect of competing technological and market developments; |
| ● | the cost of establishing sales, marketing and distribution capabilities in regions where we choose to commercialize our products on our own; and |
| ● | the initiation, progress, timing and results of our commercialization of our product candidates, if approved, for commercial sale. |
Please see the section titled “Risk Factors” elsewhere in the Quarterly Report and Annual Report for additional risks associated with our operations.
Purchase Commitments
We have no material non-cancelable purchase commitments with service providers as we have generally contracted on a cancelable, purchase order basis.
Critical Accounting Polices and Estimates
Our management’s discussion and analysis of our financial condition and results of operations is based on our financial statements, which have been prepared in accordance with U.S. GAAP. The preparation of these financial statements requires us to make estimates and assumptions that affect the reported amounts of assets and liabilities and the disclosure of contingent assets and liabilities at the date of the financial statements, as well as the reported revenue generated and expenses incurred during the reporting periods. Our estimates are based on our historical experience and on various other factors that we believe are reasonable under the circumstances, the results of which form the basis for making judgments about the carrying value of assets and liabilities that are not readily apparent from other sources. Actual results may differ from these estimates under different assumptions or conditions.
There have been no material changes to our critical accounting policies and estimates during the nine months ended September 30, 2021 from those disclosed in our Annual Report on - Form 10-K for the year ended December 31, 2020.
Impact of the CARES Act
On March 27, 2020, the Coronavirus Aid, Relief, and Economic Security Act, or the CARES Act, was enacted and signed into law, and GAAP requires recognition of the tax effects of new legislation during the reporting period that includes the enactment date. The CARES Act, among other things, includes changes to the tax provisions that benefits business entities and makes certain technical corrections to the 2017 Tax Cuts and Jobs Act, including, permitting net operating losses, or NOLs, carryovers and carrybacks to offset 100% of taxable income for taxable years beginning before 2021. In addition, the CARES Act allows NOLs incurred in 2018, 2019, and 2020 to be carried back to each of the five preceding taxable years to generate a refund of previously paid income taxes. The CARES Act provides other reliefs and stimulus measures. We have evaluated the impact of the CARES Act, and do not expect that any provision of the CARES Act would result in a material cash benefit to us or have a material impact on our financial statements or internal control over financial reporting.
Operations and Liquidity
While the potential economic impact brought by and over the duration of the COVID-19 pandemic may be difficult to assess or predict, the COVID-19 pandemic has resulted in significant disruption of global financial markets, which could in the future negatively affect our liquidity. In addition, a recession or market volatility resulting from the COVID-19 pandemic could affect our business. We have taken proactive, aggressive action throughout the COVID-19 pandemic to protect the health and safety of our employees and expect to continue to implement these measures until we determine that the COVID-19 pandemic is adequately contained for purposes of our business. We may take further actions as government authorities require or recommend or as we determine to be in the best interests of our employees. Given the nature and type of our short-term investments, we do not believe that the COVID-19 pandemic will have a material impact on our current investment liquidity.
Outlook and Impact of COVID-19 on our Business
In December 2019, a novel (new) coronavirus known as SARS-CoV-2 was first detected in Wuhan, Hubei Province, People’s Republic of China, causing outbreaks of the coronavirus disease, known as COVID-19, that has now spread globally. On January 30, 2020 the World Health Organization (WHO) declared COVID-19 a public health emergency. The Secretary of Health and Human Services declared a public health emergency on January 31, 2020, under section 319 of the Public Health Service Act (42 U.S.C. 247d), in response to the COVID-19 outbreak. On March 11, 2020, the WHO declared COVID-19 a pandemic. The full impact of the COVID-19 pandemic is unknown and rapidly evolving. The COVID-19 pandemic has and could continue to negatively affect the Company’s liquidity and operations. To date, two of the three currently planned PDS0101 clinical trials have been delayed, specifically as a result of the adverse impact the COVID-19 pandemic has had on clinical trial operations for cancer indications in the United States. Even though both of these studies have since been initiated despite the pandemic challenges, the evolving COVID-19 pandemic has also impacted the pace of enrollment in clinical trials and we may be affected by similar delays as patients may avoid or may not be able to travel to healthcare facilities and physicians’ offices unless due to a health emergency and clinical trial staff can no longer get to the clinic.
Although there is uncertainty related to the anticipated impact of the COVID-19 pandemic on our future results, we believe our current cash reserves, leave us well-positioned to manage our business through this crisis as it continues to unfold. However, the impacts of the COVID-19 pandemic are broad-reaching and continuing and the financial impacts associated with the COVID-19 pandemic are still uncertain.
Despite the economic uncertainty resulting from the COVID-19 pandemic, we intend to continue to focus on the development of our product candidates and we have expanded our infectious disease pipeline since the pandemic brought renewed resources and interest on technologies such as the Versamune® platform in the context of research and development in prevention of COVID-19. We licensed Versamune® to Farmacore in Brazil to develop PDS0203; a vaccine for the prevention of COVID-19. The Secretary for Research and Scientific Training of The Ministry of Science, Technology and Innovation of Brazil provided a commitment to fund up to approximately US$60 million to support the clinical development and commercialization of a Versamune®-based COVID-19 vaccine in Brazil. We have not received confirmation of the availability of financial resources within the MCTI to support the clinical development and commercialization of a Versamune®-based COVID-19 vaccine in Brazil. Clinical development and commercialization of a Versamune®-based COVID-19 vaccine in Brazil has not been initiated.
Off-Balance Sheet Arrangements
We did not have during the periods presented, and we do not currently have, any off-balance sheet arrangements, as defined in the rules and regulations of the SEC.
Smaller Reporting Company
As of January 1, 2021, we are no longer an “emerging growth company,” as defined in the Jumpstart Our Business Startups Act of 2012, or the JOBS Act. However, we remain a “smaller reporting company,” as defined in Rule 12b-2 under the Securities Exchange Act of 1934, as amended. We will cease to be a smaller reporting company if we have a non-affiliate public float in excess of $250 million and annual revenues in excess of $100 million, or a non-affiliate public float in excess of $700 million, determined on an annual basis. As a smaller reporting company, we are permitted and intend to rely on exemptions from certain disclosure requirements that are applicable to other public companies that are not smaller reporting companies. We will continue to take advantage of some or all of the available exemptions.
| ITEM 3. | QUANTITATIVE AND QUALITATIVE DISCLOSURE ABOUT MARKET RISK |
The primary objectives of our investment activities are to ensure liquidity and to preserve principal, while at the same time maximizing the income we receive from our cash and marketable securities without significantly increasing risk. As of September 30, 2021, we had cash equivalents of $69.7 million that were held in a non-interest-bearing money operating account and an institutional U.S. Treasury money market fund. Our primary exposure to market risk is interest rate sensitivity, which is affected by changes in the general level of U.S. interest rates. Due to the short-term maturities of our cash equivalents and the low risk profile of our investments, we do not believe that an immediate 100 basis point change in interest rates would have a material effect on the fair market value of our cash equivalents. To minimize the risk in the future, we intend to maintain our portfolio of cash equivalents and short-term investments in institutional market funds that are comprised of U.S. Treasury and Treasury backed repurchase agreements.
| ITEM 4. | CONTROLS AND PROCEDURES |
Evaluation of Disclosure Controls and Procedures
An evaluation was carried out, under the supervision of and with the participation of our management, including our Chief Executive Officer and our Chief Financial Officer, of the effectiveness of our disclosure controls and procedures (as defined in Rules 13a-15(e) and 15d-15 (e)) under the Securities Exchange Act of 1934, or the Exchange Act, as of the end of the period covered by this report. Based on the evaluation, our Chief Executive Officer and our Chief Financial Officer have concluded that our disclosure controls and procedures are effective to ensure that the information required to be disclosed by us in the reports we file or submit under the Exchange Act was recorded, processed, summarized and reported within the time periods specified in the SEC’s rules and forms.
Changes in Internal Control over Financial Reporting
There were no changes in our internal control over financial reporting (as such term is defined in Rule 13a-15(f) under the Exchange Act) identified in connection with the evaluation identified above that occurred during the quarter ended September 30, 2021 that have materially affected, or are reasonably likely to materially affect, our internal control over financial reporting.
| PART II. | OTHER INFORMATION |
On July 23, 2021, David R. Rosener, a purported stockholder of the Company, filed a putative class action and shareholder derivative complaint in the Court of Chancery of the State of Delaware (C.A. No. 2021-0644 JRS) against the Company and all of its directors and certain of its executive officers. The plaintiff named all current directors of PDS as defendants as well as PDS’s Chief Scientific Officer and PDS’s Chief Medical Officer and also named PDS as a nominal defendant. The plaintiff claims PDS’s bylaws required tabulation of broker non-votes on Proposal 3 at the Company’s 2021 annual stockholder meeting held on June 17, 2021, which sought shareholder approval of the Second Amended and Restated PDS Biotechnology Corporation 2014 Equity Incentive Plan (the “Restated Plan”). The complaint asserts claims for breach of fiduciary duties, declaratory judgment, waste of corporate assets and unjust enrichment in connection with the Restated Plan and the granting of an aggregate award of 1,040,700 stock options to certain executive officers pursuant to the Restated Plan. The plaintiff seeks unspecified monetary damages, seeks to have the Restated Plan declared void, and seeks recission of the grant of stock options as ultra vires. Given the early stage of this lawsuit, PDS is unable to reasonably estimate the costs associated with the lawsuit or predict the outcome. Each of the Company, its directors and the named aforementioned executives intend to defend this action vigorously. There can be no guarantee as to the timing of any resolution.
PDS may be involved, from time to time, in legal proceedings and claims arising in the ordinary course of its business. Such matters are subject to many uncertainties and outcomes are not predictable with assurance.
There have been no material changes from our risk factors as previously reported in our Annual Report on Form 10-K for the year ended December 31, 2020. However, any investment in our business involves a high degree of risk. Before making an investment decision, you should carefully consider the information we include in this Quarterly Report on Form 10-Q, including our unaudited interim condensed consolidated financial statements and accompanying notes, our Annual Report on Form 10-K for the year ended December 31, 2020 filed on March 18, 2021, including our financial statements and related notes contained therein, and the additional information in the other reports we file with the Securities and Exchange Commission. These risks may result in material harm to our business and our financial condition and results of operations. In this event, the market price of our common stock may decline and you could lose part or all of your investment. Additional risks that we currently believe are immaterial may also impair our business operations. Our business, financial conditions and future prospects and the trading price of our common stock could be harmed as a result of any of these risks.
| ITEM 2. | UNREGISTERED SALES OF EQUITY SECURITIES AND USE OF PROCEEDS |
There were no unregistered sales of the Company’s equity securities during the nine months ended September 30, 2021.
| ITEM 3. | DEFAULTS UPON SENIOR SECURITIES |
None.
| ITEM 4. | MINE SAFETY DISCLOSURES |
Not applicable.
On September 30, 2021, the Company delivered a notice of termination without cause to Seth Van Voorhees and removed him from his position as the Company’s Chief Financial Officer, principal financial officer and principal accounting officer. The Company and Mr. Van Voorhees are continuing to discuss the terms of Mr. Van Voorhees separation from the Company.
Exhibit Number | | Exhibit Description |
| | Executive Employment Agreement, dated October 4, 2021, by and between PDS Biotechnology Corporation and Matthew Hill (filed as Exhibit 10.1 to the Company’s Current Report on Form 8-K filed on October 6, 2021, and incorporated by reference herein). |
| | | |
| | Certification of Principal Executive Officer Pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
| | | |
| | Certification of Principal Financial Officer Pursuant to Rules 13a-14(a) and 15d-14(a) under the Securities Exchange Act of 1934, as adopted pursuant to Section 302 of the Sarbanes-Oxley Act of 2002. |
| | | |
| | Certification of Principal Executive Officer Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (furnished herewith). |
| | | |
| | Certification of Principal Financial Officer Pursuant to 18 U.S.C. Section 1350, as adopted pursuant to Section 906 of the Sarbanes-Oxley Act of 2002 (furnished herewith). |
| | | |
| 101.INS* | | Inline XBRL Instance Document (the instance document does not appear in the Interactive Data File because its XBRL tags are embedded within the Inline XBRL document) |
| | | |
| 101.SCH* | | Inline XBRL Taxonomy Extension Schema Document |
| | | |
| 101.CAL* | | Inline XBRL Taxonomy Extension Calculation Linkbase Document |
| | | |
| 101.DEF* | | Inline XBRL Taxonomy Extension Definition Linkbase Document |
| | | |
| 101.LAB* | | Inline XBRL Taxonomy Extension Label Linkbase Document |
| | | |
| 101.PRE* | | Inline XBRL Taxonomy Extension Presentation Linkbase Document |
| | | |
| 104 | | Cover Page Interactive Data File (formatted as inline XBRL and contained in Exhibit 101) |
| * | Filed herewith (unless otherwise noted as being furnished herewith) |
| ** | Certain portions of the Exhibit have been redacted pursuant to Item 601(b)(10)(iv) of Regulation S-K. |
Pursuant to the requirements of the Exchange Act, the registrant has duly caused this report to be signed on its behalf by the undersigned thereunto duly authorized.
| | PDS Biotechnology Corporation |
| | | |
| November 10, 2021 | By: | /s/ Frank Bedu-Addo |
| | | Frank Bedu-Addo |
| | | President and Chief Executive Officer (Principal Executive Officer) |
| | | |
| | | |
| November 10, 2021 | By: | /s/ Matthew Hill |
| | | Matthew Hill |
| | | Chief Financial Officer |
| | | (Principal Financial and Accounting Officer) |
31